User login
Congenital Self-healing Reticulohistiocytosis: An Underreported Entity
Langerhans cell histiocytosis (LCH), also known as histiocytosis X, is a general term that describes a group of rare disorders characterized by the proliferation of Langerhans cells.1 Central to immune surveillance and the elimination of foreign substances from the body, Langerhans cells are derived from bone marrow progenitor cells and found in the epidermis but are capable of migrating from the skin to the lymph nodes. In LCH, these cells congregate on bone tissue, particularly in the head and neck region, causing a multitude of problems.2
The spectrum of LCH includes 4 variants: congenital self-healing reticulohistiocytosis (CSHR)(also known as Hashimoto-Pritzker disease), Letterer-Siwe disease, Hand-Schüller-Christian disease, and eosinophilic granuloma (also known as pulmonary histiocytosis X)(Table). Despite the various clinical presentations and levels of severity, all variants are caused by the proliferation of Langerhans cells. We present a case of CSHR in a 6-month-old male infant that was initially diagnosed as molluscum contagiosum. We believe the actual incidence of CSHR may be underreported due to its spontaneous regression and low rate of clinical recognition.
Case Report
A 6-month-old male infant was referred to our clinic by his pediatrician with a generalized cutaneous eruption of 3 weeks’ duration. The eruption, which followed a recent viral upper respiratory tract infection, was characterized by multiple flesh-colored to erythematous, umbilicated papules distributed along the postauricular region, scalp (Figure 1A), abdomen (Figure 1B), and anterior aspect of the neck. Due to his recent illness, the patient was diagnosed with molluscum contagiosum by the referring pediatrician that was treated symptomatically with hydrocortisone lotion, Schamberg’s cream formulated in our office (a compound mixture of zinc oxide, menthol, calcium hydroxide solution, and olive oil), and pediatric diphenhydramine as needed. During a subsequent visit 2 weeks later, a more potent topical corticosteroid and a low-dose systemic corticosteroid was prescribed for 1 week due to development of new lesions and exacerbation of existing lesions. On follow-up 1 week later, the lesions on the trunk had improved, but the patient had developed new lesions on the scalp that differed from prior findings in that they were darker (more erythematous to brown) and firmer (papules and nodules).
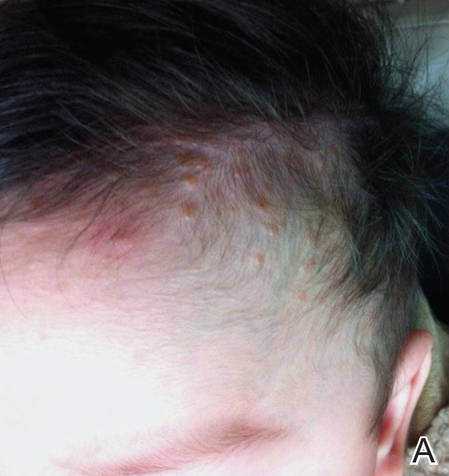 | 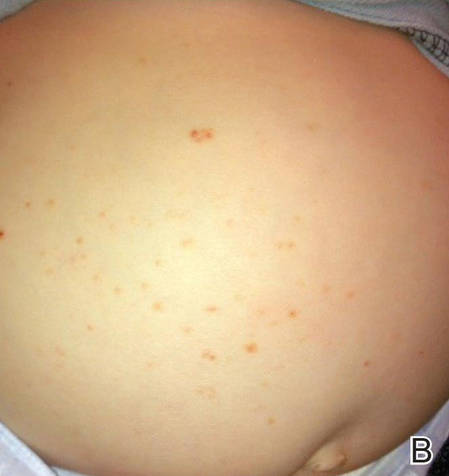 | |
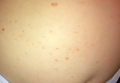
Figure 1. Multiple fleshcolored to erythematous, umbilicated papules on the frontal scalp (A) and erythematous papules on the abdomen (B). | ||
A shave biopsy was obtained from the frontal scalp to rule out LCH. Histologic examination and culture of the biopsy specimen revealed an atypical cellular infiltrate effacing the dermoepidermal junction and extensive epidermotropism. Focal erosion of the epidermis and an acute inflammatory exudate were visible. The nuclei of the cellular infiltrate were enlarged and hyperchromatic with a characteristic reniform appearance and indistinct nucleoli (Figure 2). The cells were admixed with scattered eosinophils and extravasated red blood cells.
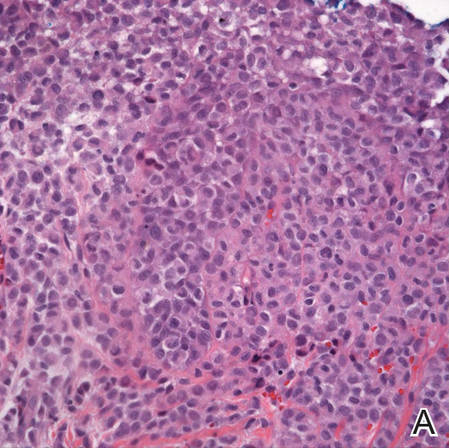 | 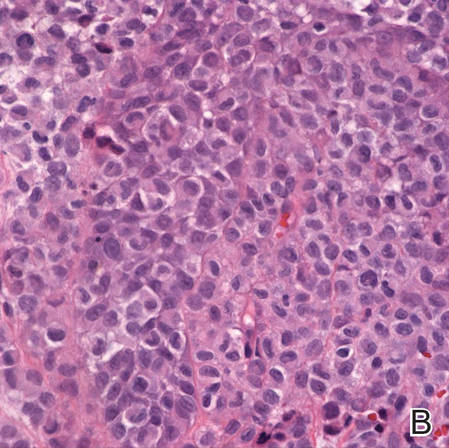 | |
Figure 2. Low-power view of dermal mononuclear cells with reniform nuclei (A)(H&E, original magnification ×100), and high-power view of enlarged and hyperchromatic nuclei with a characteristic reniform appearance admixed with eosinophils and extravasated red blood cells (B) (H&E, original magnification ×400). | ||
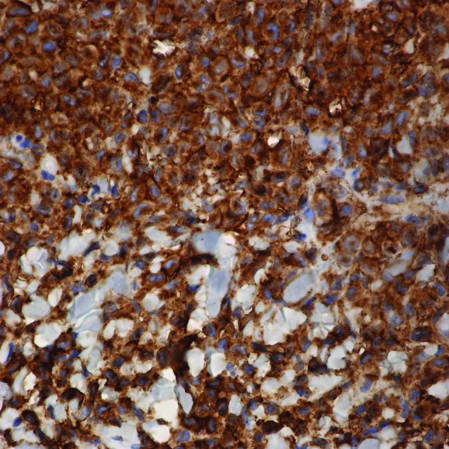
Immunohistochemical staining of the biopsy specimen was strongly positive for both CD1a and S-100 expression (Figure 3). Histopathologic findings were consistent with LCH. Clinicopathologic correlation strongly favored the diagnosis of CSHR.
Comment
Congenital self-healing reticulohistiocytosis is a rare, benign, congenital variant of LCH that spontaneously resolves with no systemic involvement. The more aggressive forms typically manifest at birth or during the first 2 months of life and regress within 3 to 4 months.5 Since CSHR was first described in 1973 by Hashimoto and Pritzker,5 more than 100 cases have been reported, but the true incidence is believed to be higher than reported given the high rate of spontaneous resolution and the low rate of clinical recognition.2 The first reported case of CSHR occurred in a female infant who presented at birth with multiple, diffusely distributed, red-brown papules that were 2 to 4 mm in diameter. Although the patient received no treatment, the exanthem completely resolved within 3.5 months without recurrence at 14-year follow-up.5 Most often, CSHR presents as multiple papules or nodules with occasional disseminated crusting and is followed within a few months by a dramatic and spontaneous regression. Lesions may heal with mild postinflammatory hyperpigmentation. Pseudo-Darier sign, the propensity to urticate from physical manipulation, has been reported in some lesions with an increased number of mast cells.6 Extensive superficial nasal and oral mucosal erosions have been reported in 2 cases.7 Solitary lesions have been reported in 25% of cases.8
The etiology of CSHR remains unknown, though neoplastic, viral, and immunologic origins have been suggested. There have been reports that human herpesvirus 6 may contribute to the development of LCH.9 It may be postulated that our patient’s presentation of CSHR was potentiated by his recent upper respiratory tract illness. In the literature, CSHR is distributed equally among males and females. Prevalence is higher in the white population than in other racial groups.5
Although CSHR is a benign cutaneous variant of LCH, there have been reports of patients with disseminated and extracutaneous involvement. In 1 rare case, CSHR reportedly involved the eyes, producing multiple, bilateral, well-circumscribed, diffuse, yellow-white lesions of the retinal pigment epithelium throughout the posterior pole of the eyes.10 The retinal lesions spontaneously regressed along with the skin manifestations. Additionally, it was reported that a neonate in Thailand presented with CSHR at birth and 1 month later developed multiple lung cysts that had completely regressed 11 months later.11 One study reported that initial diagnoses of LCH in 18 patients with only cutaneous involvement eventually progressed to systemic LCH, requiring further management.12 When LCH is suspected, a thorough physical examination, including hematologic and coagulation evaluation, liver function tests, musculoskeletal examination, and consultation with specialists if necessary, is recommended.13
There are 3 additional variants of LCH. Letterer-Siwe disease is an acute form of LCH that accounts for 10% of all LCH cases and typically presents in children younger than 2 years. It involves multiple organs, including the bones, lungs, liver, and lymph nodes.14 Affected patients usually present with fever; hepatosplenomegaly; anemia; lymphadenopathy; extensive lytic skull lesions; and a generalized cutaneous eruption, appearing as a maculopapular scaling rash with underlying purpura on the scalp, neck, axilla, and trunk.3 Letterer-Siwe disease is inherited in an autosomal-recessive pattern. Diagnosis is confirmed by skin biopsy demonstrating a thinning of the epidermis and a collection of reticulum cells in the dermis.3 Letterer-Siwe disease is treated with radiation and chemotherapy; if left untreated, the disease is fatal.4
Hand-Schüller-Christian disease, a chronic form of LCH, is most commonly seen in children aged 2 to 6 years and accounts for 15% to 20% of all LCH cases. This LCH variant presents with a classic triad of diabetes insipidus (resulting from erosion into the sella turcica), lytic bone lesions, and exophthalmos.15 Hand-Schüller-Christian disease also affects the oral cavity, producing nodular ulcerations of the hard palate, trouble swallowing, and halitosis.4 The involvement of lytic bone lesions of the mastoid process and petrous portions of the temporal bones may cause recurrent or chronic otitis media and otitis externa. Hand-Schüller-Christian disease is treated with a combination of chemotherapy, radiation, and surgical excision. The mortality rate is 30%.4
Eosinophilic granuloma is the most prevalent variant of LCH, accounting for 60% to 80% of all cases. Characterized by Langerhans cell granulomatous infiltration of the lungs and painful cystic bone lesions, eosinophilic granuloma primarily presents in the third or fourth decades of life.16 Some studies suggest an epidemiologic association with tobacco use.17 In the preliminary stages of this disease, Langerhans cells, eosinophils, lymphocytes, and fibroblasts infiltrate and form nodules on the terminal bronchioles in the upper and middle lung zones, damaging the airway walls.18 Fibrotic scarring progresses, ultimately resulting in alveolar destruction.10 The common signs and symptoms of eosinophilic granuloma are a nonproductive cough, dyspnea, weight loss, spontaneous pneumothorax, fever, peripheral edema, and a tricuspid regurgitation murmur.14 The prognosis of eosinophilic granuloma is variable. Although some patients progress to end-stage fibrotic lung disease requiring lung transplant, there have been reports of complete remission following cessation of cigarette smoking.17
Langerhans cells travel from the bone marrow to the epidermis where they express the CD1a protein on the surface of the antigen-presenting cell. Elevated levels of cytokines, such as tumor necrosis factor α, IFN-γ, granulocyte-macrophage colony-stimulating factor, and interleukins have been seen in patients with LCH.1 Their role in the pathogenesis of this disease remains unknown, but the elevated levels of cytokines may indicate the lack of an efficient immune system.
Histologically, hematoxylin and eosin–stained sections demonstrate an infiltrate of histiocytes, neutrophils, eosinophils, and an increased number of mast cells involving the papillary and reticular dermis. Infiltrating Langerhans cells have concave reniform nuclei18 and stain positive for CD1a, S-100, and CD68 antigens.15 In 10% to 30% of CSHR cases, Birbeck granules can be seen on electron microscopy and tend to transform into laminated dense bodies, signifying the degenerative changes seen in CSHR.15 The various forms of LCH exhibit no significant differences in the expression of the epithelial cadherin, the phosphorylated histone H3, and the Ki-67 proteins, indicating that they are simply different forms of the same disease represented on a spectrum.15
Conclusion
The actual incidence of CSHR may be notably underreported due to its spontaneous regression and low rate of clinical recognition. A subtype of LCH, CSHR is a diagnosis of exclusion. Although CSHR generally follows a benign clinical course, a thorough workup and evaluation for systemic disease with close follow-up is recommended after diagnosis due to the potential of LCH to involve multiple organs and to relapse at a later date after apparent regression.
1. Hussein MR. Skin-limited Langerhans’ cell histiocytosis in children. Cancer Invest. 2009;27:504-511.
2. Nakahigashi K, Ohta M, Sakai R, et al. Late-onset self-healing reticulohistiocytosis: pediatric case of Hashimoto-Pritzker type Langerhans cell histiocytosis. J Dermatol. 2007;34:205-209.
3. Pant C, Madonia P, Bahna SL, et al. Langerhans cell histiocytosis, a case of Letterer Siwe disease. J La State Med Soc. 2009;161:211-212.
4. Ferreira LM, Emerich PS, Diniz LM, et al. Langerhans cell histiocytosis: Letterer-Siwe disease–the importance of dermatological diagnosis in two cases [in Portuguese]. An Bras Dermatol. 2009;84:405-409.
5. Hashimoto K, Pritzker MS. Electron microscopic study of reticulohistiocytoma. an unusual case of congenital, self-healing reticulohistiocytosis. Arch Dermatol. 1973;107:263-270.
6. Kapur P, Erickson C, Rakheja D, et al. Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease): ten-year experience at Dallas Children’s Medical Center. J Am Acad Dermatol. 2007;56:290-294.
7. Le Bidre E, Lorette G, Delage M, et al. Extensive, erosive congenital self-healing cell histiocytosis [published online December 22, 2008]. J Eur Acad Dermatol Venereol. 2009;23:835-836.
8. Weiss T, Weber L, Scharffetter-Kochanek K, et al. Solitary cutaneous dendritic cell tumor in a child: role of dendritic cell markers for the diagnosis of skin Langerhans cell histiocytosis. J Am Acad Dermatol. 2005;53:838-844.
9. Csire M, Mikala G, Jákó J, et al. Persistent long-term human herpesvirus 6 (HHV-6) infection in a patient with Langerhans cell histiocytosis [published online July 3, 2007]. Pathol Oncol Res. 2007;13:157-160.
10. Zaenglein AL, Steele MA, Kamino H, et al. Congenital self-healing reticulohistiocytosis with eye involvement. Pediatr Dermatol. 2001;18:135-137.
11. Chunharas A, Pabunruang W, Hongeng S. Congenital self-healing Langerhans cell histiocytosis with pulmonary involvement: spontaneous regression. J Med Assoc Thai. 2002;85(suppl 4):S1309-S1313.
12. Minkov M, Prosch H, Steiner M, et al. Langerhans cell histiocytosis in neonates. Pediatr Blood Cancer. 2005;45:802-807.
13. Satter EK, High WA. Langerhans cell histiocytosis: a review of the current recommendations of the Histiocyte Society. Pediatr Dermatol. 2008;25:291-295.
14. Stacher E, Beham-Schmid C, Terpe HJ, et al. Pulmonary histiocytic sarcoma mimicking pulmonary Langerhans cell histiocytosis in a young adult presenting with spontaneous pneumothorax: a potential diagnostic pitfall [published online June 27, 2009]. Virchows Arch. 2009;455:187-190.
15. Scolozzi P, Lombardi T, Monnier P, et al. Multisystem Langerhans’ cell histiocytosis (Hand-Schüller-Christian disease) in an adult: a case report and review of the literature [published online October 10, 2003]. Eur Arch Otorhinolaryngol. 2004;261:326-330.
16. Noonan V, Kabani S, Alibhai K. Langerhans cell histiocytosis (eosinophilic granuloma). J Mass Dent Soc. 2011;60:35.
17. Podbielski FJ, Worley TA, Korn JM, et al. Eosinophilic granuloma of the lung and rib. Asian Cardiovasc Thorac Ann. 2009;17:194-195.
18. Rosso DA, Ripoli MF, Roy A, et al. Serum levels of interleukin-1 receptor antagonist and tumor necrosis factor-alpha are elevated in children with Langerhans cell histiocytosis. J Pediatr Hematol Oncol. 2003;25:480-483.
Langerhans cell histiocytosis (LCH), also known as histiocytosis X, is a general term that describes a group of rare disorders characterized by the proliferation of Langerhans cells.1 Central to immune surveillance and the elimination of foreign substances from the body, Langerhans cells are derived from bone marrow progenitor cells and found in the epidermis but are capable of migrating from the skin to the lymph nodes. In LCH, these cells congregate on bone tissue, particularly in the head and neck region, causing a multitude of problems.2
The spectrum of LCH includes 4 variants: congenital self-healing reticulohistiocytosis (CSHR)(also known as Hashimoto-Pritzker disease), Letterer-Siwe disease, Hand-Schüller-Christian disease, and eosinophilic granuloma (also known as pulmonary histiocytosis X)(Table). Despite the various clinical presentations and levels of severity, all variants are caused by the proliferation of Langerhans cells. We present a case of CSHR in a 6-month-old male infant that was initially diagnosed as molluscum contagiosum. We believe the actual incidence of CSHR may be underreported due to its spontaneous regression and low rate of clinical recognition.
Case Report
A 6-month-old male infant was referred to our clinic by his pediatrician with a generalized cutaneous eruption of 3 weeks’ duration. The eruption, which followed a recent viral upper respiratory tract infection, was characterized by multiple flesh-colored to erythematous, umbilicated papules distributed along the postauricular region, scalp (Figure 1A), abdomen (Figure 1B), and anterior aspect of the neck. Due to his recent illness, the patient was diagnosed with molluscum contagiosum by the referring pediatrician that was treated symptomatically with hydrocortisone lotion, Schamberg’s cream formulated in our office (a compound mixture of zinc oxide, menthol, calcium hydroxide solution, and olive oil), and pediatric diphenhydramine as needed. During a subsequent visit 2 weeks later, a more potent topical corticosteroid and a low-dose systemic corticosteroid was prescribed for 1 week due to development of new lesions and exacerbation of existing lesions. On follow-up 1 week later, the lesions on the trunk had improved, but the patient had developed new lesions on the scalp that differed from prior findings in that they were darker (more erythematous to brown) and firmer (papules and nodules).
| | |
Figure 1. Multiple fleshcolored to erythematous, umbilicated papules on the frontal scalp (A) and erythematous papules on the abdomen (B). | ||
A shave biopsy was obtained from the frontal scalp to rule out LCH. Histologic examination and culture of the biopsy specimen revealed an atypical cellular infiltrate effacing the dermoepidermal junction and extensive epidermotropism. Focal erosion of the epidermis and an acute inflammatory exudate were visible. The nuclei of the cellular infiltrate were enlarged and hyperchromatic with a characteristic reniform appearance and indistinct nucleoli (Figure 2). The cells were admixed with scattered eosinophils and extravasated red blood cells.
| | |
Figure 2. Low-power view of dermal mononuclear cells with reniform nuclei (A)(H&E, original magnification ×100), and high-power view of enlarged and hyperchromatic nuclei with a characteristic reniform appearance admixed with eosinophils and extravasated red blood cells (B) (H&E, original magnification ×400). | ||
Immunohistochemical staining of the biopsy specimen was strongly positive for both CD1a and S-100 expression (Figure 3). Histopathologic findings were consistent with LCH. Clinicopathologic correlation strongly favored the diagnosis of CSHR.
Comment
Congenital self-healing reticulohistiocytosis is a rare, benign, congenital variant of LCH that spontaneously resolves with no systemic involvement. The more aggressive forms typically manifest at birth or during the first 2 months of life and regress within 3 to 4 months.5 Since CSHR was first described in 1973 by Hashimoto and Pritzker,5 more than 100 cases have been reported, but the true incidence is believed to be higher than reported given the high rate of spontaneous resolution and the low rate of clinical recognition.2 The first reported case of CSHR occurred in a female infant who presented at birth with multiple, diffusely distributed, red-brown papules that were 2 to 4 mm in diameter. Although the patient received no treatment, the exanthem completely resolved within 3.5 months without recurrence at 14-year follow-up.5 Most often, CSHR presents as multiple papules or nodules with occasional disseminated crusting and is followed within a few months by a dramatic and spontaneous regression. Lesions may heal with mild postinflammatory hyperpigmentation. Pseudo-Darier sign, the propensity to urticate from physical manipulation, has been reported in some lesions with an increased number of mast cells.6 Extensive superficial nasal and oral mucosal erosions have been reported in 2 cases.7 Solitary lesions have been reported in 25% of cases.8
The etiology of CSHR remains unknown, though neoplastic, viral, and immunologic origins have been suggested. There have been reports that human herpesvirus 6 may contribute to the development of LCH.9 It may be postulated that our patient’s presentation of CSHR was potentiated by his recent upper respiratory tract illness. In the literature, CSHR is distributed equally among males and females. Prevalence is higher in the white population than in other racial groups.5
Although CSHR is a benign cutaneous variant of LCH, there have been reports of patients with disseminated and extracutaneous involvement. In 1 rare case, CSHR reportedly involved the eyes, producing multiple, bilateral, well-circumscribed, diffuse, yellow-white lesions of the retinal pigment epithelium throughout the posterior pole of the eyes.10 The retinal lesions spontaneously regressed along with the skin manifestations. Additionally, it was reported that a neonate in Thailand presented with CSHR at birth and 1 month later developed multiple lung cysts that had completely regressed 11 months later.11 One study reported that initial diagnoses of LCH in 18 patients with only cutaneous involvement eventually progressed to systemic LCH, requiring further management.12 When LCH is suspected, a thorough physical examination, including hematologic and coagulation evaluation, liver function tests, musculoskeletal examination, and consultation with specialists if necessary, is recommended.13
There are 3 additional variants of LCH. Letterer-Siwe disease is an acute form of LCH that accounts for 10% of all LCH cases and typically presents in children younger than 2 years. It involves multiple organs, including the bones, lungs, liver, and lymph nodes.14 Affected patients usually present with fever; hepatosplenomegaly; anemia; lymphadenopathy; extensive lytic skull lesions; and a generalized cutaneous eruption, appearing as a maculopapular scaling rash with underlying purpura on the scalp, neck, axilla, and trunk.3 Letterer-Siwe disease is inherited in an autosomal-recessive pattern. Diagnosis is confirmed by skin biopsy demonstrating a thinning of the epidermis and a collection of reticulum cells in the dermis.3 Letterer-Siwe disease is treated with radiation and chemotherapy; if left untreated, the disease is fatal.4
Hand-Schüller-Christian disease, a chronic form of LCH, is most commonly seen in children aged 2 to 6 years and accounts for 15% to 20% of all LCH cases. This LCH variant presents with a classic triad of diabetes insipidus (resulting from erosion into the sella turcica), lytic bone lesions, and exophthalmos.15 Hand-Schüller-Christian disease also affects the oral cavity, producing nodular ulcerations of the hard palate, trouble swallowing, and halitosis.4 The involvement of lytic bone lesions of the mastoid process and petrous portions of the temporal bones may cause recurrent or chronic otitis media and otitis externa. Hand-Schüller-Christian disease is treated with a combination of chemotherapy, radiation, and surgical excision. The mortality rate is 30%.4
Eosinophilic granuloma is the most prevalent variant of LCH, accounting for 60% to 80% of all cases. Characterized by Langerhans cell granulomatous infiltration of the lungs and painful cystic bone lesions, eosinophilic granuloma primarily presents in the third or fourth decades of life.16 Some studies suggest an epidemiologic association with tobacco use.17 In the preliminary stages of this disease, Langerhans cells, eosinophils, lymphocytes, and fibroblasts infiltrate and form nodules on the terminal bronchioles in the upper and middle lung zones, damaging the airway walls.18 Fibrotic scarring progresses, ultimately resulting in alveolar destruction.10 The common signs and symptoms of eosinophilic granuloma are a nonproductive cough, dyspnea, weight loss, spontaneous pneumothorax, fever, peripheral edema, and a tricuspid regurgitation murmur.14 The prognosis of eosinophilic granuloma is variable. Although some patients progress to end-stage fibrotic lung disease requiring lung transplant, there have been reports of complete remission following cessation of cigarette smoking.17
Langerhans cells travel from the bone marrow to the epidermis where they express the CD1a protein on the surface of the antigen-presenting cell. Elevated levels of cytokines, such as tumor necrosis factor α, IFN-γ, granulocyte-macrophage colony-stimulating factor, and interleukins have been seen in patients with LCH.1 Their role in the pathogenesis of this disease remains unknown, but the elevated levels of cytokines may indicate the lack of an efficient immune system.
Histologically, hematoxylin and eosin–stained sections demonstrate an infiltrate of histiocytes, neutrophils, eosinophils, and an increased number of mast cells involving the papillary and reticular dermis. Infiltrating Langerhans cells have concave reniform nuclei18 and stain positive for CD1a, S-100, and CD68 antigens.15 In 10% to 30% of CSHR cases, Birbeck granules can be seen on electron microscopy and tend to transform into laminated dense bodies, signifying the degenerative changes seen in CSHR.15 The various forms of LCH exhibit no significant differences in the expression of the epithelial cadherin, the phosphorylated histone H3, and the Ki-67 proteins, indicating that they are simply different forms of the same disease represented on a spectrum.15
Conclusion
The actual incidence of CSHR may be notably underreported due to its spontaneous regression and low rate of clinical recognition. A subtype of LCH, CSHR is a diagnosis of exclusion. Although CSHR generally follows a benign clinical course, a thorough workup and evaluation for systemic disease with close follow-up is recommended after diagnosis due to the potential of LCH to involve multiple organs and to relapse at a later date after apparent regression.
Langerhans cell histiocytosis (LCH), also known as histiocytosis X, is a general term that describes a group of rare disorders characterized by the proliferation of Langerhans cells.1 Central to immune surveillance and the elimination of foreign substances from the body, Langerhans cells are derived from bone marrow progenitor cells and found in the epidermis but are capable of migrating from the skin to the lymph nodes. In LCH, these cells congregate on bone tissue, particularly in the head and neck region, causing a multitude of problems.2
The spectrum of LCH includes 4 variants: congenital self-healing reticulohistiocytosis (CSHR)(also known as Hashimoto-Pritzker disease), Letterer-Siwe disease, Hand-Schüller-Christian disease, and eosinophilic granuloma (also known as pulmonary histiocytosis X)(Table). Despite the various clinical presentations and levels of severity, all variants are caused by the proliferation of Langerhans cells. We present a case of CSHR in a 6-month-old male infant that was initially diagnosed as molluscum contagiosum. We believe the actual incidence of CSHR may be underreported due to its spontaneous regression and low rate of clinical recognition.
Case Report
A 6-month-old male infant was referred to our clinic by his pediatrician with a generalized cutaneous eruption of 3 weeks’ duration. The eruption, which followed a recent viral upper respiratory tract infection, was characterized by multiple flesh-colored to erythematous, umbilicated papules distributed along the postauricular region, scalp (Figure 1A), abdomen (Figure 1B), and anterior aspect of the neck. Due to his recent illness, the patient was diagnosed with molluscum contagiosum by the referring pediatrician that was treated symptomatically with hydrocortisone lotion, Schamberg’s cream formulated in our office (a compound mixture of zinc oxide, menthol, calcium hydroxide solution, and olive oil), and pediatric diphenhydramine as needed. During a subsequent visit 2 weeks later, a more potent topical corticosteroid and a low-dose systemic corticosteroid was prescribed for 1 week due to development of new lesions and exacerbation of existing lesions. On follow-up 1 week later, the lesions on the trunk had improved, but the patient had developed new lesions on the scalp that differed from prior findings in that they were darker (more erythematous to brown) and firmer (papules and nodules).
| | |
Figure 1. Multiple fleshcolored to erythematous, umbilicated papules on the frontal scalp (A) and erythematous papules on the abdomen (B). | ||
A shave biopsy was obtained from the frontal scalp to rule out LCH. Histologic examination and culture of the biopsy specimen revealed an atypical cellular infiltrate effacing the dermoepidermal junction and extensive epidermotropism. Focal erosion of the epidermis and an acute inflammatory exudate were visible. The nuclei of the cellular infiltrate were enlarged and hyperchromatic with a characteristic reniform appearance and indistinct nucleoli (Figure 2). The cells were admixed with scattered eosinophils and extravasated red blood cells.
| | |
Figure 2. Low-power view of dermal mononuclear cells with reniform nuclei (A)(H&E, original magnification ×100), and high-power view of enlarged and hyperchromatic nuclei with a characteristic reniform appearance admixed with eosinophils and extravasated red blood cells (B) (H&E, original magnification ×400). | ||
Immunohistochemical staining of the biopsy specimen was strongly positive for both CD1a and S-100 expression (Figure 3). Histopathologic findings were consistent with LCH. Clinicopathologic correlation strongly favored the diagnosis of CSHR.
Comment
Congenital self-healing reticulohistiocytosis is a rare, benign, congenital variant of LCH that spontaneously resolves with no systemic involvement. The more aggressive forms typically manifest at birth or during the first 2 months of life and regress within 3 to 4 months.5 Since CSHR was first described in 1973 by Hashimoto and Pritzker,5 more than 100 cases have been reported, but the true incidence is believed to be higher than reported given the high rate of spontaneous resolution and the low rate of clinical recognition.2 The first reported case of CSHR occurred in a female infant who presented at birth with multiple, diffusely distributed, red-brown papules that were 2 to 4 mm in diameter. Although the patient received no treatment, the exanthem completely resolved within 3.5 months without recurrence at 14-year follow-up.5 Most often, CSHR presents as multiple papules or nodules with occasional disseminated crusting and is followed within a few months by a dramatic and spontaneous regression. Lesions may heal with mild postinflammatory hyperpigmentation. Pseudo-Darier sign, the propensity to urticate from physical manipulation, has been reported in some lesions with an increased number of mast cells.6 Extensive superficial nasal and oral mucosal erosions have been reported in 2 cases.7 Solitary lesions have been reported in 25% of cases.8
The etiology of CSHR remains unknown, though neoplastic, viral, and immunologic origins have been suggested. There have been reports that human herpesvirus 6 may contribute to the development of LCH.9 It may be postulated that our patient’s presentation of CSHR was potentiated by his recent upper respiratory tract illness. In the literature, CSHR is distributed equally among males and females. Prevalence is higher in the white population than in other racial groups.5
Although CSHR is a benign cutaneous variant of LCH, there have been reports of patients with disseminated and extracutaneous involvement. In 1 rare case, CSHR reportedly involved the eyes, producing multiple, bilateral, well-circumscribed, diffuse, yellow-white lesions of the retinal pigment epithelium throughout the posterior pole of the eyes.10 The retinal lesions spontaneously regressed along with the skin manifestations. Additionally, it was reported that a neonate in Thailand presented with CSHR at birth and 1 month later developed multiple lung cysts that had completely regressed 11 months later.11 One study reported that initial diagnoses of LCH in 18 patients with only cutaneous involvement eventually progressed to systemic LCH, requiring further management.12 When LCH is suspected, a thorough physical examination, including hematologic and coagulation evaluation, liver function tests, musculoskeletal examination, and consultation with specialists if necessary, is recommended.13
There are 3 additional variants of LCH. Letterer-Siwe disease is an acute form of LCH that accounts for 10% of all LCH cases and typically presents in children younger than 2 years. It involves multiple organs, including the bones, lungs, liver, and lymph nodes.14 Affected patients usually present with fever; hepatosplenomegaly; anemia; lymphadenopathy; extensive lytic skull lesions; and a generalized cutaneous eruption, appearing as a maculopapular scaling rash with underlying purpura on the scalp, neck, axilla, and trunk.3 Letterer-Siwe disease is inherited in an autosomal-recessive pattern. Diagnosis is confirmed by skin biopsy demonstrating a thinning of the epidermis and a collection of reticulum cells in the dermis.3 Letterer-Siwe disease is treated with radiation and chemotherapy; if left untreated, the disease is fatal.4
Hand-Schüller-Christian disease, a chronic form of LCH, is most commonly seen in children aged 2 to 6 years and accounts for 15% to 20% of all LCH cases. This LCH variant presents with a classic triad of diabetes insipidus (resulting from erosion into the sella turcica), lytic bone lesions, and exophthalmos.15 Hand-Schüller-Christian disease also affects the oral cavity, producing nodular ulcerations of the hard palate, trouble swallowing, and halitosis.4 The involvement of lytic bone lesions of the mastoid process and petrous portions of the temporal bones may cause recurrent or chronic otitis media and otitis externa. Hand-Schüller-Christian disease is treated with a combination of chemotherapy, radiation, and surgical excision. The mortality rate is 30%.4
Eosinophilic granuloma is the most prevalent variant of LCH, accounting for 60% to 80% of all cases. Characterized by Langerhans cell granulomatous infiltration of the lungs and painful cystic bone lesions, eosinophilic granuloma primarily presents in the third or fourth decades of life.16 Some studies suggest an epidemiologic association with tobacco use.17 In the preliminary stages of this disease, Langerhans cells, eosinophils, lymphocytes, and fibroblasts infiltrate and form nodules on the terminal bronchioles in the upper and middle lung zones, damaging the airway walls.18 Fibrotic scarring progresses, ultimately resulting in alveolar destruction.10 The common signs and symptoms of eosinophilic granuloma are a nonproductive cough, dyspnea, weight loss, spontaneous pneumothorax, fever, peripheral edema, and a tricuspid regurgitation murmur.14 The prognosis of eosinophilic granuloma is variable. Although some patients progress to end-stage fibrotic lung disease requiring lung transplant, there have been reports of complete remission following cessation of cigarette smoking.17
Langerhans cells travel from the bone marrow to the epidermis where they express the CD1a protein on the surface of the antigen-presenting cell. Elevated levels of cytokines, such as tumor necrosis factor α, IFN-γ, granulocyte-macrophage colony-stimulating factor, and interleukins have been seen in patients with LCH.1 Their role in the pathogenesis of this disease remains unknown, but the elevated levels of cytokines may indicate the lack of an efficient immune system.
Histologically, hematoxylin and eosin–stained sections demonstrate an infiltrate of histiocytes, neutrophils, eosinophils, and an increased number of mast cells involving the papillary and reticular dermis. Infiltrating Langerhans cells have concave reniform nuclei18 and stain positive for CD1a, S-100, and CD68 antigens.15 In 10% to 30% of CSHR cases, Birbeck granules can be seen on electron microscopy and tend to transform into laminated dense bodies, signifying the degenerative changes seen in CSHR.15 The various forms of LCH exhibit no significant differences in the expression of the epithelial cadherin, the phosphorylated histone H3, and the Ki-67 proteins, indicating that they are simply different forms of the same disease represented on a spectrum.15
Conclusion
The actual incidence of CSHR may be notably underreported due to its spontaneous regression and low rate of clinical recognition. A subtype of LCH, CSHR is a diagnosis of exclusion. Although CSHR generally follows a benign clinical course, a thorough workup and evaluation for systemic disease with close follow-up is recommended after diagnosis due to the potential of LCH to involve multiple organs and to relapse at a later date after apparent regression.
1. Hussein MR. Skin-limited Langerhans’ cell histiocytosis in children. Cancer Invest. 2009;27:504-511.
2. Nakahigashi K, Ohta M, Sakai R, et al. Late-onset self-healing reticulohistiocytosis: pediatric case of Hashimoto-Pritzker type Langerhans cell histiocytosis. J Dermatol. 2007;34:205-209.
3. Pant C, Madonia P, Bahna SL, et al. Langerhans cell histiocytosis, a case of Letterer Siwe disease. J La State Med Soc. 2009;161:211-212.
4. Ferreira LM, Emerich PS, Diniz LM, et al. Langerhans cell histiocytosis: Letterer-Siwe disease–the importance of dermatological diagnosis in two cases [in Portuguese]. An Bras Dermatol. 2009;84:405-409.
5. Hashimoto K, Pritzker MS. Electron microscopic study of reticulohistiocytoma. an unusual case of congenital, self-healing reticulohistiocytosis. Arch Dermatol. 1973;107:263-270.
6. Kapur P, Erickson C, Rakheja D, et al. Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease): ten-year experience at Dallas Children’s Medical Center. J Am Acad Dermatol. 2007;56:290-294.
7. Le Bidre E, Lorette G, Delage M, et al. Extensive, erosive congenital self-healing cell histiocytosis [published online December 22, 2008]. J Eur Acad Dermatol Venereol. 2009;23:835-836.
8. Weiss T, Weber L, Scharffetter-Kochanek K, et al. Solitary cutaneous dendritic cell tumor in a child: role of dendritic cell markers for the diagnosis of skin Langerhans cell histiocytosis. J Am Acad Dermatol. 2005;53:838-844.
9. Csire M, Mikala G, Jákó J, et al. Persistent long-term human herpesvirus 6 (HHV-6) infection in a patient with Langerhans cell histiocytosis [published online July 3, 2007]. Pathol Oncol Res. 2007;13:157-160.
10. Zaenglein AL, Steele MA, Kamino H, et al. Congenital self-healing reticulohistiocytosis with eye involvement. Pediatr Dermatol. 2001;18:135-137.
11. Chunharas A, Pabunruang W, Hongeng S. Congenital self-healing Langerhans cell histiocytosis with pulmonary involvement: spontaneous regression. J Med Assoc Thai. 2002;85(suppl 4):S1309-S1313.
12. Minkov M, Prosch H, Steiner M, et al. Langerhans cell histiocytosis in neonates. Pediatr Blood Cancer. 2005;45:802-807.
13. Satter EK, High WA. Langerhans cell histiocytosis: a review of the current recommendations of the Histiocyte Society. Pediatr Dermatol. 2008;25:291-295.
14. Stacher E, Beham-Schmid C, Terpe HJ, et al. Pulmonary histiocytic sarcoma mimicking pulmonary Langerhans cell histiocytosis in a young adult presenting with spontaneous pneumothorax: a potential diagnostic pitfall [published online June 27, 2009]. Virchows Arch. 2009;455:187-190.
15. Scolozzi P, Lombardi T, Monnier P, et al. Multisystem Langerhans’ cell histiocytosis (Hand-Schüller-Christian disease) in an adult: a case report and review of the literature [published online October 10, 2003]. Eur Arch Otorhinolaryngol. 2004;261:326-330.
16. Noonan V, Kabani S, Alibhai K. Langerhans cell histiocytosis (eosinophilic granuloma). J Mass Dent Soc. 2011;60:35.
17. Podbielski FJ, Worley TA, Korn JM, et al. Eosinophilic granuloma of the lung and rib. Asian Cardiovasc Thorac Ann. 2009;17:194-195.
18. Rosso DA, Ripoli MF, Roy A, et al. Serum levels of interleukin-1 receptor antagonist and tumor necrosis factor-alpha are elevated in children with Langerhans cell histiocytosis. J Pediatr Hematol Oncol. 2003;25:480-483.
1. Hussein MR. Skin-limited Langerhans’ cell histiocytosis in children. Cancer Invest. 2009;27:504-511.
2. Nakahigashi K, Ohta M, Sakai R, et al. Late-onset self-healing reticulohistiocytosis: pediatric case of Hashimoto-Pritzker type Langerhans cell histiocytosis. J Dermatol. 2007;34:205-209.
3. Pant C, Madonia P, Bahna SL, et al. Langerhans cell histiocytosis, a case of Letterer Siwe disease. J La State Med Soc. 2009;161:211-212.
4. Ferreira LM, Emerich PS, Diniz LM, et al. Langerhans cell histiocytosis: Letterer-Siwe disease–the importance of dermatological diagnosis in two cases [in Portuguese]. An Bras Dermatol. 2009;84:405-409.
5. Hashimoto K, Pritzker MS. Electron microscopic study of reticulohistiocytoma. an unusual case of congenital, self-healing reticulohistiocytosis. Arch Dermatol. 1973;107:263-270.
6. Kapur P, Erickson C, Rakheja D, et al. Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease): ten-year experience at Dallas Children’s Medical Center. J Am Acad Dermatol. 2007;56:290-294.
7. Le Bidre E, Lorette G, Delage M, et al. Extensive, erosive congenital self-healing cell histiocytosis [published online December 22, 2008]. J Eur Acad Dermatol Venereol. 2009;23:835-836.
8. Weiss T, Weber L, Scharffetter-Kochanek K, et al. Solitary cutaneous dendritic cell tumor in a child: role of dendritic cell markers for the diagnosis of skin Langerhans cell histiocytosis. J Am Acad Dermatol. 2005;53:838-844.
9. Csire M, Mikala G, Jákó J, et al. Persistent long-term human herpesvirus 6 (HHV-6) infection in a patient with Langerhans cell histiocytosis [published online July 3, 2007]. Pathol Oncol Res. 2007;13:157-160.
10. Zaenglein AL, Steele MA, Kamino H, et al. Congenital self-healing reticulohistiocytosis with eye involvement. Pediatr Dermatol. 2001;18:135-137.
11. Chunharas A, Pabunruang W, Hongeng S. Congenital self-healing Langerhans cell histiocytosis with pulmonary involvement: spontaneous regression. J Med Assoc Thai. 2002;85(suppl 4):S1309-S1313.
12. Minkov M, Prosch H, Steiner M, et al. Langerhans cell histiocytosis in neonates. Pediatr Blood Cancer. 2005;45:802-807.
13. Satter EK, High WA. Langerhans cell histiocytosis: a review of the current recommendations of the Histiocyte Society. Pediatr Dermatol. 2008;25:291-295.
14. Stacher E, Beham-Schmid C, Terpe HJ, et al. Pulmonary histiocytic sarcoma mimicking pulmonary Langerhans cell histiocytosis in a young adult presenting with spontaneous pneumothorax: a potential diagnostic pitfall [published online June 27, 2009]. Virchows Arch. 2009;455:187-190.
15. Scolozzi P, Lombardi T, Monnier P, et al. Multisystem Langerhans’ cell histiocytosis (Hand-Schüller-Christian disease) in an adult: a case report and review of the literature [published online October 10, 2003]. Eur Arch Otorhinolaryngol. 2004;261:326-330.
16. Noonan V, Kabani S, Alibhai K. Langerhans cell histiocytosis (eosinophilic granuloma). J Mass Dent Soc. 2011;60:35.
17. Podbielski FJ, Worley TA, Korn JM, et al. Eosinophilic granuloma of the lung and rib. Asian Cardiovasc Thorac Ann. 2009;17:194-195.
18. Rosso DA, Ripoli MF, Roy A, et al. Serum levels of interleukin-1 receptor antagonist and tumor necrosis factor-alpha are elevated in children with Langerhans cell histiocytosis. J Pediatr Hematol Oncol. 2003;25:480-483.
Practice Points
- Langerhans cell histiocytosis (LCH) is believed to occur in 1:200,000 children and tends to be underdiagnosed, as some patients may have no symptoms while others have symptoms that are misdiagnosed as other conditions.
- Patients with LCH usually should have long-term follow-up care to detect progression or complications of the disease or treatment.