To the Editor:
A 78-year-old man presented with erythematous circular skin papules that were widely scattered over the trunk. He denied recent contact with ill individuals and denied any systemic symptoms indicating internal involvement or malignancy leading to possible paraneoplastic presentation. Physical examination showed erythematous, circular, slightly elevated plaques of varying sizes scattered over the trunk (Figure 1) and right axilla.
|
| Figure 1. Nontender, erythematous, brown nodules scattered over the trunk. |
|
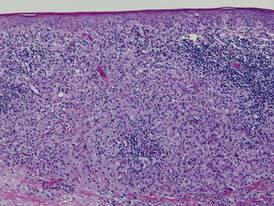
| Figure 2. Light microscopy revealed Langerhans cells filling the superficial dermis, abutting the epidermis, and extending into the deep dermis with surrounding inflammatory infiltrates (CD1a, original magnification ×100). |
|
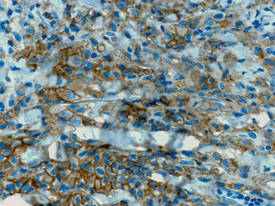
| Figure 3. Langerhans cells appeared strongly positive for CD1a (original magnification ×400). |
Biopsies of lesions were taken and stained with immunoperoxidase. On light microscopy there was a reticular and papillary dermal dense infiltrate of cells with indented nuclei (Figure 2). At higher magnification, cells appeared strongly positive for CD1a (Figure 3) and S-100 protein, which was histologically consistent with adult-type Langerhans cell histiocytosis (ALCH).
Computed tomography of the head, chest, abdomen, and pelvis were ordered to rule out spread of ALCH to other organ sites. Results were clear of evidence of systemic spread. Additionally, a complete blood cell count and comprehensive metabolic panel were within reference range.
He was started on topical tacrolimus; however, most of the lesions resolved on their own. As a result, tacrolimus was discontinued due to its propensity to cause skin irritation and lack of change in disease progression. At 3-month follow-up, he was prescribed triamcinolone acetonide cream 0.1% for minor outbreaks. After 2 years, he was completely clear of all skin signs of ALCH.
Adult-type Langerhans cell histiocytosis is characterized as a group of disorders associated with abnormal spread and proliferation of dendritic cells of the epidermis. The disease primarily affects children aged 1 to 4 years. It is estimated that only 1 to 2 cases of ALCH per million occur.1 The pathophysiology of ALCH is unknown; it is speculated that it may be associated with a reactive inflammatory process triggered by proliferation of Langerhans-type dendritic cells. It is possible that the release of multiple cytokines by dendritic cells and T cells in ALCH lesions leads to erythematous eruptions and can contribute to spontaneous remission of the disorder.2 Various cases of ALCH have reported high serum levels of IL-17 and IL-10 proinflammatory cytokines, supporting the theory of an inflammatory etiology of ALCH.3
Comparative genomic hybridization with loss of heterozygosity of pulmonary lesions has provided further evidence to suggest that chromosomal aberrations also may contribute to the pathophysiology of ALCH.4 One study evaluated 14 cases of pulmonary ALCH for loss of heterozygosity and found allelic loss of 1p, 1q, 3p, 5p, 17p, and 22q.5 In addition, allelic loss of 1 or more tumor suppressor genes was identified in 19 of 24 specimens, suggesting a neoplastic type of pathology through uncontrolled cellular proliferation.6
Lesions of ALCH can be broad but typically present as red-brown maculopapular lesions with petechiae that erupt over the trunk, axilla, and perivulvar or retroauricular regions.7 The papules may unify to form an erythematous, weeping, or crusted eruption that appears similar to seborrheic dermatitis. Typically the lesions remit on their own; however, lesions can recur with the same or decreased severity as the primary eruption. Complications have been noted with lesions, particularly secondary infection and ulceration.7
Systemic involvement has been noted in adults, particularly in the lungs. Patients typically present with chronic cough, dyspnea, and chest pain with evidence of a solitary nodular lesion on radiologic testing. In addition, bone involvement has been noted as eosinophilic granulomas that can produce osteolytic lesions that lead to spontaneous fractures. Use of corticosteroids and immunosuppressive agents, as opposed to just observation, is warranted in cases of systemic involvement, according to the National Cancer Institute.7
Exact treatment modalities have not yet been elucidated due to the ambiguity of pathogenesis. In addition, ALCH is known to remit and relapse in patients, which increases the difficulty in evaluating the efficacy of particular treatments. Trials conducted by the Histiocyte Society have shown that treatment regimens should be tailored to disease severity. Epidermal involvement of ALCH typically responds to corticosteroid creams, whereas patients with systemic involvement respond well to strong chemotherapeutic agents such as vincristine and prednisone with mercaptopurine.8 However, as demonstrated in our case, lesions may remit on their own and use of corticosteroids and immunosuppressive agents may lead to further detriment without treating disease progression.
Because of a low prevalence among adults, ALCH is difficult to recognize and diagnose, and the uncertainty of the pathogenesis of ALCH limits treatment alternatives. Further study into proper treatment modalities is warranted given that the remitting and relapsing course of the disease and cosmetic quandaries are detrimental to patient well-being. Our case illustrates that it is appropriate to simply monitor lesions for cases limited to cutaneous involvement. Systemic agents may be used when there are signs of organ involvement outside the skin, but providers must proceed to do so with caution.