User login
Imaging reveals different clinico-pathologic patterns in Takayasu’s, giant cell arteritis
a retrospective cohort study has found.
“Clinical symptoms were not sensitive markers of underlying vascular pathology but were specific when present,” Despina Michailidou, MD, PhD, and colleagues wrote in Annals of the Rheumatic Diseases. “Vascular imaging should be considered in the management of these patients since reliance on the presence of clinical symptoms may not be sensitive to detect vascular pathology within an acceptable window to prevent or minimize damage.”
Dr. Michailidou and coauthors in the Systemic Autoimmunity Branch of the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) examined the relationships between clinical presentation and imaging findings in 110 patients involved in an ongoing observational cohort study at the National Institutes of Health, including 56 with Takayasu’s arteritis (TAK) and 54 with giant cell arteritis (GCA). The study included data from 270 visits. Dr. Michailidou conducted the study while she was a research fellow at NIAMS, and she is now a rheumatology fellow at the University of Washington, Seattle.
The team looked at 11 symptoms (lightheadedness, positional lightheadedness, carotidynia, arm claudication vertigo, frontotemporal and posterior headache, posterior neck pain, blurred vision, vision loss, and major CNS events, including stroke, transient ischemic attack, or syncope). These were related to findings on MR angiography (MRA) and 18F-fluorodeoxyglucose PET (FDG-PET).
There were no significant between-group differences in six of the symptoms. However, those with TAK had significantly higher rates of carotidynia (21% vs. 0%), lightheadedness (30% vs. 9%), positional lightheadedness (29% vs. 5%), major CNS events (25% vs. 9%), and arm claudication (52% vs. 28%). Arm claudication was the most common symptom in those with TAK (52%), and blurred vision the most common in patients with GCA (37%).
On the day of evaluation, 8% of patients with TAK reported carotidynia; none of the GCA patients reported this. On FDG-PET, carotidynia was more strongly associated with inflammation of the carotid artery than with damage of the carotid artery on MRA.
The sensitivity of this association was low, which indicates “that an absence of carotidynia could still be associated with imaging abnormalities in the carotid artery, particularly on MRA compared with FDG-PET,” the authors wrote. But specificity was high for both FDG-PET and MRA, suggesting that carotidynia was strongly associated with corresponding carotid artery abnormalities on both FDG-PET and MRA.
More of those with GCA than those with TAK reported posterior neck pain (18% vs. 7%). It was significantly associated with vertebral artery inflammation in those with GCA, but not in those with TAK. There was no significant association with vertebral artery damage in either group.
While sensitivity was low for posterior neck pain and imaging abnormalities, specificity was very high in both groups, which indicates “the presence of posterior neck pain was strongly associated with corresponding vertebral artery abnormalities on both FDG-PET and MRA.”
Posterior headache was present in 5% of GCA patients and was significantly associated with vertebral artery damage, but it was not associated with such damage in patients with TAK.
“While posterior headaches in the occipital region are uncommon in patients with GCA, this study emphasizes that presence of a posterior headache should alert the clinician to the likelihood of associated vascular inflammation and damage in the corresponding vertebral artery,” the researchers wrote.
About 6% of patients with TAK and 10% of those with GCA reported frontotemporal headache. The headache was not associated with carotid PET activity or damage in either group of patients.
“While frontotemporal headaches frequently occur in patients with TAK, and are a cardinal feature of GCA, headaches in this region may reflect inflammation in smaller branches of cranial arteries, rather than the corresponding larger arteries of the neck,” the investigators wrote.
Arm claudication was the most commonly reported symptom overall, present in 52% of those with TAK and 28% of those with GCA. It was more strongly associated with subclavian artery damage on MRA than with inflammation on FDG-PET.
The investigators also assessed the association between specific clinical symptoms and the number of affected neck arteries. Patients with large vessel vasculitis and an increased number of damaged neck arteries on MRA were significantly more likely to experience lightheadedness (odds ratio, 2.61), positional lightheadedness (OR, 3.51), or a major CNS event (OR, 3.23). But those with large vessel vasculitis and inflamed neck arteries on FDG-PET were more likely to experience posterior headache (OR, 2.84).
The study isn’t intended to dictate how MRA and FDG-PET should be employed with these patients, the authors noted.
“Rather, these findings may help clinicians predict imaging pathology in specific vascular territories based on patient-reported symptoms and may inform which type of imaging modality would be the most useful to obtain in certain clinical scenarios, recognizing that additional sequences to detect wall morphology may augment the ability of MR-based assessments to detect vascular inflammation in addition to luminal damage.”
The Division of Intramural Research at NIAMS funded the research. The authors had no financial disclosures.
SOURCE: Michailidou D et al. Ann Rheum Dis. 2019 Oct 24. doi: 10.1136/annrheumdis-2019-216145.
a retrospective cohort study has found.
“Clinical symptoms were not sensitive markers of underlying vascular pathology but were specific when present,” Despina Michailidou, MD, PhD, and colleagues wrote in Annals of the Rheumatic Diseases. “Vascular imaging should be considered in the management of these patients since reliance on the presence of clinical symptoms may not be sensitive to detect vascular pathology within an acceptable window to prevent or minimize damage.”
Dr. Michailidou and coauthors in the Systemic Autoimmunity Branch of the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) examined the relationships between clinical presentation and imaging findings in 110 patients involved in an ongoing observational cohort study at the National Institutes of Health, including 56 with Takayasu’s arteritis (TAK) and 54 with giant cell arteritis (GCA). The study included data from 270 visits. Dr. Michailidou conducted the study while she was a research fellow at NIAMS, and she is now a rheumatology fellow at the University of Washington, Seattle.
The team looked at 11 symptoms (lightheadedness, positional lightheadedness, carotidynia, arm claudication vertigo, frontotemporal and posterior headache, posterior neck pain, blurred vision, vision loss, and major CNS events, including stroke, transient ischemic attack, or syncope). These were related to findings on MR angiography (MRA) and 18F-fluorodeoxyglucose PET (FDG-PET).
There were no significant between-group differences in six of the symptoms. However, those with TAK had significantly higher rates of carotidynia (21% vs. 0%), lightheadedness (30% vs. 9%), positional lightheadedness (29% vs. 5%), major CNS events (25% vs. 9%), and arm claudication (52% vs. 28%). Arm claudication was the most common symptom in those with TAK (52%), and blurred vision the most common in patients with GCA (37%).
On the day of evaluation, 8% of patients with TAK reported carotidynia; none of the GCA patients reported this. On FDG-PET, carotidynia was more strongly associated with inflammation of the carotid artery than with damage of the carotid artery on MRA.
The sensitivity of this association was low, which indicates “that an absence of carotidynia could still be associated with imaging abnormalities in the carotid artery, particularly on MRA compared with FDG-PET,” the authors wrote. But specificity was high for both FDG-PET and MRA, suggesting that carotidynia was strongly associated with corresponding carotid artery abnormalities on both FDG-PET and MRA.
More of those with GCA than those with TAK reported posterior neck pain (18% vs. 7%). It was significantly associated with vertebral artery inflammation in those with GCA, but not in those with TAK. There was no significant association with vertebral artery damage in either group.
While sensitivity was low for posterior neck pain and imaging abnormalities, specificity was very high in both groups, which indicates “the presence of posterior neck pain was strongly associated with corresponding vertebral artery abnormalities on both FDG-PET and MRA.”
Posterior headache was present in 5% of GCA patients and was significantly associated with vertebral artery damage, but it was not associated with such damage in patients with TAK.
“While posterior headaches in the occipital region are uncommon in patients with GCA, this study emphasizes that presence of a posterior headache should alert the clinician to the likelihood of associated vascular inflammation and damage in the corresponding vertebral artery,” the researchers wrote.
About 6% of patients with TAK and 10% of those with GCA reported frontotemporal headache. The headache was not associated with carotid PET activity or damage in either group of patients.
“While frontotemporal headaches frequently occur in patients with TAK, and are a cardinal feature of GCA, headaches in this region may reflect inflammation in smaller branches of cranial arteries, rather than the corresponding larger arteries of the neck,” the investigators wrote.
Arm claudication was the most commonly reported symptom overall, present in 52% of those with TAK and 28% of those with GCA. It was more strongly associated with subclavian artery damage on MRA than with inflammation on FDG-PET.
The investigators also assessed the association between specific clinical symptoms and the number of affected neck arteries. Patients with large vessel vasculitis and an increased number of damaged neck arteries on MRA were significantly more likely to experience lightheadedness (odds ratio, 2.61), positional lightheadedness (OR, 3.51), or a major CNS event (OR, 3.23). But those with large vessel vasculitis and inflamed neck arteries on FDG-PET were more likely to experience posterior headache (OR, 2.84).
The study isn’t intended to dictate how MRA and FDG-PET should be employed with these patients, the authors noted.
“Rather, these findings may help clinicians predict imaging pathology in specific vascular territories based on patient-reported symptoms and may inform which type of imaging modality would be the most useful to obtain in certain clinical scenarios, recognizing that additional sequences to detect wall morphology may augment the ability of MR-based assessments to detect vascular inflammation in addition to luminal damage.”
The Division of Intramural Research at NIAMS funded the research. The authors had no financial disclosures.
SOURCE: Michailidou D et al. Ann Rheum Dis. 2019 Oct 24. doi: 10.1136/annrheumdis-2019-216145.
a retrospective cohort study has found.
“Clinical symptoms were not sensitive markers of underlying vascular pathology but were specific when present,” Despina Michailidou, MD, PhD, and colleagues wrote in Annals of the Rheumatic Diseases. “Vascular imaging should be considered in the management of these patients since reliance on the presence of clinical symptoms may not be sensitive to detect vascular pathology within an acceptable window to prevent or minimize damage.”
Dr. Michailidou and coauthors in the Systemic Autoimmunity Branch of the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) examined the relationships between clinical presentation and imaging findings in 110 patients involved in an ongoing observational cohort study at the National Institutes of Health, including 56 with Takayasu’s arteritis (TAK) and 54 with giant cell arteritis (GCA). The study included data from 270 visits. Dr. Michailidou conducted the study while she was a research fellow at NIAMS, and she is now a rheumatology fellow at the University of Washington, Seattle.
The team looked at 11 symptoms (lightheadedness, positional lightheadedness, carotidynia, arm claudication vertigo, frontotemporal and posterior headache, posterior neck pain, blurred vision, vision loss, and major CNS events, including stroke, transient ischemic attack, or syncope). These were related to findings on MR angiography (MRA) and 18F-fluorodeoxyglucose PET (FDG-PET).
There were no significant between-group differences in six of the symptoms. However, those with TAK had significantly higher rates of carotidynia (21% vs. 0%), lightheadedness (30% vs. 9%), positional lightheadedness (29% vs. 5%), major CNS events (25% vs. 9%), and arm claudication (52% vs. 28%). Arm claudication was the most common symptom in those with TAK (52%), and blurred vision the most common in patients with GCA (37%).
On the day of evaluation, 8% of patients with TAK reported carotidynia; none of the GCA patients reported this. On FDG-PET, carotidynia was more strongly associated with inflammation of the carotid artery than with damage of the carotid artery on MRA.
The sensitivity of this association was low, which indicates “that an absence of carotidynia could still be associated with imaging abnormalities in the carotid artery, particularly on MRA compared with FDG-PET,” the authors wrote. But specificity was high for both FDG-PET and MRA, suggesting that carotidynia was strongly associated with corresponding carotid artery abnormalities on both FDG-PET and MRA.
More of those with GCA than those with TAK reported posterior neck pain (18% vs. 7%). It was significantly associated with vertebral artery inflammation in those with GCA, but not in those with TAK. There was no significant association with vertebral artery damage in either group.
While sensitivity was low for posterior neck pain and imaging abnormalities, specificity was very high in both groups, which indicates “the presence of posterior neck pain was strongly associated with corresponding vertebral artery abnormalities on both FDG-PET and MRA.”
Posterior headache was present in 5% of GCA patients and was significantly associated with vertebral artery damage, but it was not associated with such damage in patients with TAK.
“While posterior headaches in the occipital region are uncommon in patients with GCA, this study emphasizes that presence of a posterior headache should alert the clinician to the likelihood of associated vascular inflammation and damage in the corresponding vertebral artery,” the researchers wrote.
About 6% of patients with TAK and 10% of those with GCA reported frontotemporal headache. The headache was not associated with carotid PET activity or damage in either group of patients.
“While frontotemporal headaches frequently occur in patients with TAK, and are a cardinal feature of GCA, headaches in this region may reflect inflammation in smaller branches of cranial arteries, rather than the corresponding larger arteries of the neck,” the investigators wrote.
Arm claudication was the most commonly reported symptom overall, present in 52% of those with TAK and 28% of those with GCA. It was more strongly associated with subclavian artery damage on MRA than with inflammation on FDG-PET.
The investigators also assessed the association between specific clinical symptoms and the number of affected neck arteries. Patients with large vessel vasculitis and an increased number of damaged neck arteries on MRA were significantly more likely to experience lightheadedness (odds ratio, 2.61), positional lightheadedness (OR, 3.51), or a major CNS event (OR, 3.23). But those with large vessel vasculitis and inflamed neck arteries on FDG-PET were more likely to experience posterior headache (OR, 2.84).
The study isn’t intended to dictate how MRA and FDG-PET should be employed with these patients, the authors noted.
“Rather, these findings may help clinicians predict imaging pathology in specific vascular territories based on patient-reported symptoms and may inform which type of imaging modality would be the most useful to obtain in certain clinical scenarios, recognizing that additional sequences to detect wall morphology may augment the ability of MR-based assessments to detect vascular inflammation in addition to luminal damage.”
The Division of Intramural Research at NIAMS funded the research. The authors had no financial disclosures.
SOURCE: Michailidou D et al. Ann Rheum Dis. 2019 Oct 24. doi: 10.1136/annrheumdis-2019-216145.
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point: The associations between findings on MR angiography (MRA) and 18F-fluorodeoxyglucose (FDG) PET and differing clinical symptom presentations in patients with Takayasu’s arteritis and those with giant cell arteritis may be used to predict imaging pathology.
Major finding: Arm claudication was the most commonly reported symptom overall, present in 52% of those with Takayasu’s arteritis and 28% of those with giant cell arteritis. It was more strongly associated with subclavian artery damage on MRA than with inflammation on FDG-PET.
Study details: The cohort comprised 56 patients with Takayasu’s arteritis and 54 with giant cell arteritis who together had made 270 visits.
Disclosures: The Division of Intramural Research at the National Institute of Arthritis and Musculoskeletal and Skin Diseases funded the research. The investigators had no financial disclosures.
Source: Michailidou D et al. Ann Rheum Dis. 2019 Oct 24. doi: 10.1136/annrheumdis-2019-216145.
Psoriasis risk rises with TNF inhibitor use in children with inflammatory disorders
, a retrospective cohort study has determined.
“The incidence rate and risk factors of psoriasis in children with IBD [inflammatory bowel disease], JIA [juvenile idiopathic arthritis], or CNO [chronic nonbacterial osteomyelitis] who are exposed to TNFi [tumor necrosis factor inhibitors] are unknown. Additionally, there is a well-established association between these inflammatory conditions and psoriasis development. Yet, as TNFi can both treat and trigger psoriasis, it is not clear how TNFi exposure affects this relationship,” wrote Lisa H. Buckley, MD, of Children’s Hospital at Vanderbilt, Nashville, Tenn., and colleagues. Their report is in Arthritis Care & Research.
The team examined the relationship in children who were treated for an inflammatory disorder at Children’s Hospital of Philadelphia during 2008-2018. IBD was most common at 74%, followed by JIA at 24% and CNO at 2%.
Among 4,111 children with those inflammatory disorders, the psoriasis incidence was 12.3 per 1,000 person-years in exposed children and 3.8 per 1,000 person-years in unexposed. This significant difference equated to a hazard ratio of 3.84 for developing psoriasis after TNFi exposure.
“These data reflect the established association between inflammatory conditions and psoriasis development and suggest that TNFi exposure further increases the risk of psoriasis,” Dr. Buckley and coauthors wrote.
The median duration of follow-up in this study was about 2.5 years for patients exposed to TNFi and 2 years for those unexposed. Among the entire cohort, 39% had been exposed to a TNFi, with 4,705 person-years of follow-up. Among the unexposed children (61%), there were 6,604 person-years of follow-up.
In all, 83 cases of psoriasis developed: 58 in the exposed group and 25 in the unexposed group. Psoriasis incidence varied by disorder. Exposed children with IBD had a higher incidence than did unexposed children (10.9 vs. 2.6 per 1,000 person-years; HR = 4.52). Exposed children with JIA also had a higher incidence than did unexposed children (14.7 vs. 5.5 per 1,000 person-years; HR = 2.90). Among those with CNO, incidences were similar for exposed and unexposed children (33.5 and 38.9 per 1,000 person-years).
A family history of psoriasis significantly increased the risk of psoriasis with a hazard ratio of 3.11, the authors noted. But none of the other covariates (age, sex, race, obesity, methotrexate exposure, and underlying diagnosis) exerted a significant additional risk.
The study had no outside funding source. The authors had no financial disclosures. Dr. Buckley conducted the research when she was a pediatric rheumatology fellow at Children’s Hospital of Philadelphia.
SOURCE: Buckley LH et al. Arthritis Care Res. 2019 Oct 23. doi: 10.1002/ACR.24100
, a retrospective cohort study has determined.
“The incidence rate and risk factors of psoriasis in children with IBD [inflammatory bowel disease], JIA [juvenile idiopathic arthritis], or CNO [chronic nonbacterial osteomyelitis] who are exposed to TNFi [tumor necrosis factor inhibitors] are unknown. Additionally, there is a well-established association between these inflammatory conditions and psoriasis development. Yet, as TNFi can both treat and trigger psoriasis, it is not clear how TNFi exposure affects this relationship,” wrote Lisa H. Buckley, MD, of Children’s Hospital at Vanderbilt, Nashville, Tenn., and colleagues. Their report is in Arthritis Care & Research.
The team examined the relationship in children who were treated for an inflammatory disorder at Children’s Hospital of Philadelphia during 2008-2018. IBD was most common at 74%, followed by JIA at 24% and CNO at 2%.
Among 4,111 children with those inflammatory disorders, the psoriasis incidence was 12.3 per 1,000 person-years in exposed children and 3.8 per 1,000 person-years in unexposed. This significant difference equated to a hazard ratio of 3.84 for developing psoriasis after TNFi exposure.
“These data reflect the established association between inflammatory conditions and psoriasis development and suggest that TNFi exposure further increases the risk of psoriasis,” Dr. Buckley and coauthors wrote.
The median duration of follow-up in this study was about 2.5 years for patients exposed to TNFi and 2 years for those unexposed. Among the entire cohort, 39% had been exposed to a TNFi, with 4,705 person-years of follow-up. Among the unexposed children (61%), there were 6,604 person-years of follow-up.
In all, 83 cases of psoriasis developed: 58 in the exposed group and 25 in the unexposed group. Psoriasis incidence varied by disorder. Exposed children with IBD had a higher incidence than did unexposed children (10.9 vs. 2.6 per 1,000 person-years; HR = 4.52). Exposed children with JIA also had a higher incidence than did unexposed children (14.7 vs. 5.5 per 1,000 person-years; HR = 2.90). Among those with CNO, incidences were similar for exposed and unexposed children (33.5 and 38.9 per 1,000 person-years).
A family history of psoriasis significantly increased the risk of psoriasis with a hazard ratio of 3.11, the authors noted. But none of the other covariates (age, sex, race, obesity, methotrexate exposure, and underlying diagnosis) exerted a significant additional risk.
The study had no outside funding source. The authors had no financial disclosures. Dr. Buckley conducted the research when she was a pediatric rheumatology fellow at Children’s Hospital of Philadelphia.
SOURCE: Buckley LH et al. Arthritis Care Res. 2019 Oct 23. doi: 10.1002/ACR.24100
, a retrospective cohort study has determined.
“The incidence rate and risk factors of psoriasis in children with IBD [inflammatory bowel disease], JIA [juvenile idiopathic arthritis], or CNO [chronic nonbacterial osteomyelitis] who are exposed to TNFi [tumor necrosis factor inhibitors] are unknown. Additionally, there is a well-established association between these inflammatory conditions and psoriasis development. Yet, as TNFi can both treat and trigger psoriasis, it is not clear how TNFi exposure affects this relationship,” wrote Lisa H. Buckley, MD, of Children’s Hospital at Vanderbilt, Nashville, Tenn., and colleagues. Their report is in Arthritis Care & Research.
The team examined the relationship in children who were treated for an inflammatory disorder at Children’s Hospital of Philadelphia during 2008-2018. IBD was most common at 74%, followed by JIA at 24% and CNO at 2%.
Among 4,111 children with those inflammatory disorders, the psoriasis incidence was 12.3 per 1,000 person-years in exposed children and 3.8 per 1,000 person-years in unexposed. This significant difference equated to a hazard ratio of 3.84 for developing psoriasis after TNFi exposure.
“These data reflect the established association between inflammatory conditions and psoriasis development and suggest that TNFi exposure further increases the risk of psoriasis,” Dr. Buckley and coauthors wrote.
The median duration of follow-up in this study was about 2.5 years for patients exposed to TNFi and 2 years for those unexposed. Among the entire cohort, 39% had been exposed to a TNFi, with 4,705 person-years of follow-up. Among the unexposed children (61%), there were 6,604 person-years of follow-up.
In all, 83 cases of psoriasis developed: 58 in the exposed group and 25 in the unexposed group. Psoriasis incidence varied by disorder. Exposed children with IBD had a higher incidence than did unexposed children (10.9 vs. 2.6 per 1,000 person-years; HR = 4.52). Exposed children with JIA also had a higher incidence than did unexposed children (14.7 vs. 5.5 per 1,000 person-years; HR = 2.90). Among those with CNO, incidences were similar for exposed and unexposed children (33.5 and 38.9 per 1,000 person-years).
A family history of psoriasis significantly increased the risk of psoriasis with a hazard ratio of 3.11, the authors noted. But none of the other covariates (age, sex, race, obesity, methotrexate exposure, and underlying diagnosis) exerted a significant additional risk.
The study had no outside funding source. The authors had no financial disclosures. Dr. Buckley conducted the research when she was a pediatric rheumatology fellow at Children’s Hospital of Philadelphia.
SOURCE: Buckley LH et al. Arthritis Care Res. 2019 Oct 23. doi: 10.1002/ACR.24100
FROM ARTHRITIS RESEARCH & CARE
Dr. Paul Aisen Q&A: Aducanumab for Alzheimer’s
In the wake of Biogen and Eisai’s Oct. 22 announcement about plans to apply to the Food and Drug Administration next year for the regulatory approval of the investigational monoclonal antibody aducanumab as a treatment for Alzheimer’s disease, we spoke with Paul Aisen, MD, the founding director of the Alzheimer’s Therapy Research Institute at the University of Southern California, Los Angeles, for his views on the news. He has been a consultant for Biogen and is a member of the aducanumab steering committee.

Q: What was your first reaction when you heard about the plan to submit an application for aducanumab to the FDA?
A: My initial reaction is that this provides terrific support for the amyloid hypothesis, and is consistent with the early aducanumab studies showing significant reductions in brain amyloid with resulting clinical improvement.
My next thought was that these data are going to be very, very challenging to analyze because both of these trials were stopped early, and one was clearly negative. We really need to scrutinize the data, but even at this point I would say this strongly supports targeting amyloid. The scrutiny will begin in detail at the Clinical Trials in Alzheimer’s Disease conference in December, when Biogen will likely release detailed data. A lot of people will analyze it, and I think that’s great. It’s beneficial to bring different perspectives.
We have had a terribly frustrating series of disappointments in the field. After the futility analysis of aducanumab and the multiple failures of BACE [beta-secretase] inhibitors, many were convinced we were barking up the wrong tree. I think these results, although complicated, should resurrect the enthusiasm for targeting amyloid.
Q: What is different about aducanumab from other antibodies tested – and rejected – in Alzheimer’s drug development?
A: There are lots of antibodies that have been tested in clinical trials. They all differ in terms of their affinity for amyloid beta. Some target monomers of the protein. Some target dimers. Some target fibrils. Some tie up amyloid and some reduce it. Aducanumab directly attacks brain plaques, reducing the plaque load in the brain. It carries a liability of amyloid-related imaging abnormalities [ARIA], but it also allows us to assess the impact that removing plaques might have on downstream events, including biomarkers. Overall, these data show that aducanumab did remove brain plaques and that removing them had a beneficial effect on cognition and function, and also a favorable effect on downstream biomarkers.
But again, we must be cautious because this is a complex data set taken from a post hoc analysis of two different terminated trials.
Q: We see some statistically significant differences in cognitive and functional outcomes. What would that mean for patients on an everyday basis?
A: Well, everyone is different, so that’s hard to say. A 25% slowing of functional decline on the Clinical Dementia Rating Scale sum of boxes (CDR-SB) might mean that, at the end of a year, there’s not a significant change in memory, or that there’s better social function. If both trials had been completed and if people had 18 months of high-dose aducanumab, the slowing of functional decline on the CDR-SB might in fact be greater than reported. Again, we’re having to draw conclusions from interrupted trials.
Q: This suggestion you make of a potentially continuous slowing of decline – are you suggesting that aducanumab might slow decline to the point of stopping it altogether? If an elderly patient has little or no progression until death would that, in effect, be considered a “cure?”
A: I don’t think it is possible to cure AD once the disease is clinically evident. These are studies of people with early AD, late mild cognitive impairment, and mild dementia. At that stage, there’s already a loss of synapses that won’t come back, and these studies don’t suggest that aducanumab can cure that. But what if people took it earlier, when the brain is still functioning normally? Some of us have argued for many years that earlier intervention is the way to go. And since we can now identify people [with brain plaques] before they become symptomatic, there is the possibility that if we removed them, we could stop progression.
Q: Are there any plans to study aducanumab as a preventive agent?
A: A grant has been awarded for this, but it was put on hold after the futility analysis. I don’t know when or if that will go forward.
(Editor’s note: The National Institutes of Health previously awarded Banner Health a $32 million, 5-year grant to examine this. The 2-year prevention study of aducanumab is aimed at cognitively unimpaired 65- to 80-year-old patients with PET-confirmed amyloid brain plaques. It was to be a multicenter, double-blind, placebo-controlled trial using Alzheimer’s biomarker endpoints as primary outcomes, along with cognitive and clinical changes, safety, and tolerability. The study was put on hold after Biogen discontinued the aducanumab development program in March. Investigators are considering whether to resurrect plans considering the new data. The study is intended to be a public-private partnership, with additional unspecified funding from Biogen plus $10 million from philanthropic sources. It has three intended goals: To find an approved prevention therapy as early as 2023, ahead of the National Plan to Address Alzheimer’s Disease’s goal of an effective prevention strategy by 2025; to advance the use of surrogate biomarkers to rapidly test and support accelerated approval of prevention therapies in almost everyone at biomarker or genetic risk, even in earlier preclinical Alzheimer’s stages when some treatments may have their greatest benefit; and to help make it possible to conduct prevention trials in at-risk persons even before they have extensive amyloid plaques, when some treatments may have their greatest benefit.)
Q: It seems like rolling this out to an enormous population of patients is going to be difficult, if not impossible. Are people really going to be able to commit to what could be a lifetime of monthly intravenous infusions of a medicine that could be expensive, as therapeutic antibodies generally are?
A: I would say, nothing about this disease is easy. It’s devastating and horrible. And if someone is diagnosed at this stage, I would think that individual would embrace any opportunity to treat it. My hope is that we will be able to prescreen people with an effective blood test for amyloid that would be part of a regular testing protocol once they reach a certain age. Those with positive results would be referred for more testing, including amyloid brain imaging.
In the wake of Biogen and Eisai’s Oct. 22 announcement about plans to apply to the Food and Drug Administration next year for the regulatory approval of the investigational monoclonal antibody aducanumab as a treatment for Alzheimer’s disease, we spoke with Paul Aisen, MD, the founding director of the Alzheimer’s Therapy Research Institute at the University of Southern California, Los Angeles, for his views on the news. He has been a consultant for Biogen and is a member of the aducanumab steering committee.
Q: What was your first reaction when you heard about the plan to submit an application for aducanumab to the FDA?
A: My initial reaction is that this provides terrific support for the amyloid hypothesis, and is consistent with the early aducanumab studies showing significant reductions in brain amyloid with resulting clinical improvement.
My next thought was that these data are going to be very, very challenging to analyze because both of these trials were stopped early, and one was clearly negative. We really need to scrutinize the data, but even at this point I would say this strongly supports targeting amyloid. The scrutiny will begin in detail at the Clinical Trials in Alzheimer’s Disease conference in December, when Biogen will likely release detailed data. A lot of people will analyze it, and I think that’s great. It’s beneficial to bring different perspectives.
We have had a terribly frustrating series of disappointments in the field. After the futility analysis of aducanumab and the multiple failures of BACE [beta-secretase] inhibitors, many were convinced we were barking up the wrong tree. I think these results, although complicated, should resurrect the enthusiasm for targeting amyloid.
Q: What is different about aducanumab from other antibodies tested – and rejected – in Alzheimer’s drug development?
A: There are lots of antibodies that have been tested in clinical trials. They all differ in terms of their affinity for amyloid beta. Some target monomers of the protein. Some target dimers. Some target fibrils. Some tie up amyloid and some reduce it. Aducanumab directly attacks brain plaques, reducing the plaque load in the brain. It carries a liability of amyloid-related imaging abnormalities [ARIA], but it also allows us to assess the impact that removing plaques might have on downstream events, including biomarkers. Overall, these data show that aducanumab did remove brain plaques and that removing them had a beneficial effect on cognition and function, and also a favorable effect on downstream biomarkers.
But again, we must be cautious because this is a complex data set taken from a post hoc analysis of two different terminated trials.
Q: We see some statistically significant differences in cognitive and functional outcomes. What would that mean for patients on an everyday basis?
A: Well, everyone is different, so that’s hard to say. A 25% slowing of functional decline on the Clinical Dementia Rating Scale sum of boxes (CDR-SB) might mean that, at the end of a year, there’s not a significant change in memory, or that there’s better social function. If both trials had been completed and if people had 18 months of high-dose aducanumab, the slowing of functional decline on the CDR-SB might in fact be greater than reported. Again, we’re having to draw conclusions from interrupted trials.
Q: This suggestion you make of a potentially continuous slowing of decline – are you suggesting that aducanumab might slow decline to the point of stopping it altogether? If an elderly patient has little or no progression until death would that, in effect, be considered a “cure?”
A: I don’t think it is possible to cure AD once the disease is clinically evident. These are studies of people with early AD, late mild cognitive impairment, and mild dementia. At that stage, there’s already a loss of synapses that won’t come back, and these studies don’t suggest that aducanumab can cure that. But what if people took it earlier, when the brain is still functioning normally? Some of us have argued for many years that earlier intervention is the way to go. And since we can now identify people [with brain plaques] before they become symptomatic, there is the possibility that if we removed them, we could stop progression.
Q: Are there any plans to study aducanumab as a preventive agent?
A: A grant has been awarded for this, but it was put on hold after the futility analysis. I don’t know when or if that will go forward.
(Editor’s note: The National Institutes of Health previously awarded Banner Health a $32 million, 5-year grant to examine this. The 2-year prevention study of aducanumab is aimed at cognitively unimpaired 65- to 80-year-old patients with PET-confirmed amyloid brain plaques. It was to be a multicenter, double-blind, placebo-controlled trial using Alzheimer’s biomarker endpoints as primary outcomes, along with cognitive and clinical changes, safety, and tolerability. The study was put on hold after Biogen discontinued the aducanumab development program in March. Investigators are considering whether to resurrect plans considering the new data. The study is intended to be a public-private partnership, with additional unspecified funding from Biogen plus $10 million from philanthropic sources. It has three intended goals: To find an approved prevention therapy as early as 2023, ahead of the National Plan to Address Alzheimer’s Disease’s goal of an effective prevention strategy by 2025; to advance the use of surrogate biomarkers to rapidly test and support accelerated approval of prevention therapies in almost everyone at biomarker or genetic risk, even in earlier preclinical Alzheimer’s stages when some treatments may have their greatest benefit; and to help make it possible to conduct prevention trials in at-risk persons even before they have extensive amyloid plaques, when some treatments may have their greatest benefit.)
Q: It seems like rolling this out to an enormous population of patients is going to be difficult, if not impossible. Are people really going to be able to commit to what could be a lifetime of monthly intravenous infusions of a medicine that could be expensive, as therapeutic antibodies generally are?
A: I would say, nothing about this disease is easy. It’s devastating and horrible. And if someone is diagnosed at this stage, I would think that individual would embrace any opportunity to treat it. My hope is that we will be able to prescreen people with an effective blood test for amyloid that would be part of a regular testing protocol once they reach a certain age. Those with positive results would be referred for more testing, including amyloid brain imaging.
In the wake of Biogen and Eisai’s Oct. 22 announcement about plans to apply to the Food and Drug Administration next year for the regulatory approval of the investigational monoclonal antibody aducanumab as a treatment for Alzheimer’s disease, we spoke with Paul Aisen, MD, the founding director of the Alzheimer’s Therapy Research Institute at the University of Southern California, Los Angeles, for his views on the news. He has been a consultant for Biogen and is a member of the aducanumab steering committee.
Q: What was your first reaction when you heard about the plan to submit an application for aducanumab to the FDA?
A: My initial reaction is that this provides terrific support for the amyloid hypothesis, and is consistent with the early aducanumab studies showing significant reductions in brain amyloid with resulting clinical improvement.
My next thought was that these data are going to be very, very challenging to analyze because both of these trials were stopped early, and one was clearly negative. We really need to scrutinize the data, but even at this point I would say this strongly supports targeting amyloid. The scrutiny will begin in detail at the Clinical Trials in Alzheimer’s Disease conference in December, when Biogen will likely release detailed data. A lot of people will analyze it, and I think that’s great. It’s beneficial to bring different perspectives.
We have had a terribly frustrating series of disappointments in the field. After the futility analysis of aducanumab and the multiple failures of BACE [beta-secretase] inhibitors, many were convinced we were barking up the wrong tree. I think these results, although complicated, should resurrect the enthusiasm for targeting amyloid.
Q: What is different about aducanumab from other antibodies tested – and rejected – in Alzheimer’s drug development?
A: There are lots of antibodies that have been tested in clinical trials. They all differ in terms of their affinity for amyloid beta. Some target monomers of the protein. Some target dimers. Some target fibrils. Some tie up amyloid and some reduce it. Aducanumab directly attacks brain plaques, reducing the plaque load in the brain. It carries a liability of amyloid-related imaging abnormalities [ARIA], but it also allows us to assess the impact that removing plaques might have on downstream events, including biomarkers. Overall, these data show that aducanumab did remove brain plaques and that removing them had a beneficial effect on cognition and function, and also a favorable effect on downstream biomarkers.
But again, we must be cautious because this is a complex data set taken from a post hoc analysis of two different terminated trials.
Q: We see some statistically significant differences in cognitive and functional outcomes. What would that mean for patients on an everyday basis?
A: Well, everyone is different, so that’s hard to say. A 25% slowing of functional decline on the Clinical Dementia Rating Scale sum of boxes (CDR-SB) might mean that, at the end of a year, there’s not a significant change in memory, or that there’s better social function. If both trials had been completed and if people had 18 months of high-dose aducanumab, the slowing of functional decline on the CDR-SB might in fact be greater than reported. Again, we’re having to draw conclusions from interrupted trials.
Q: This suggestion you make of a potentially continuous slowing of decline – are you suggesting that aducanumab might slow decline to the point of stopping it altogether? If an elderly patient has little or no progression until death would that, in effect, be considered a “cure?”
A: I don’t think it is possible to cure AD once the disease is clinically evident. These are studies of people with early AD, late mild cognitive impairment, and mild dementia. At that stage, there’s already a loss of synapses that won’t come back, and these studies don’t suggest that aducanumab can cure that. But what if people took it earlier, when the brain is still functioning normally? Some of us have argued for many years that earlier intervention is the way to go. And since we can now identify people [with brain plaques] before they become symptomatic, there is the possibility that if we removed them, we could stop progression.
Q: Are there any plans to study aducanumab as a preventive agent?
A: A grant has been awarded for this, but it was put on hold after the futility analysis. I don’t know when or if that will go forward.
(Editor’s note: The National Institutes of Health previously awarded Banner Health a $32 million, 5-year grant to examine this. The 2-year prevention study of aducanumab is aimed at cognitively unimpaired 65- to 80-year-old patients with PET-confirmed amyloid brain plaques. It was to be a multicenter, double-blind, placebo-controlled trial using Alzheimer’s biomarker endpoints as primary outcomes, along with cognitive and clinical changes, safety, and tolerability. The study was put on hold after Biogen discontinued the aducanumab development program in March. Investigators are considering whether to resurrect plans considering the new data. The study is intended to be a public-private partnership, with additional unspecified funding from Biogen plus $10 million from philanthropic sources. It has three intended goals: To find an approved prevention therapy as early as 2023, ahead of the National Plan to Address Alzheimer’s Disease’s goal of an effective prevention strategy by 2025; to advance the use of surrogate biomarkers to rapidly test and support accelerated approval of prevention therapies in almost everyone at biomarker or genetic risk, even in earlier preclinical Alzheimer’s stages when some treatments may have their greatest benefit; and to help make it possible to conduct prevention trials in at-risk persons even before they have extensive amyloid plaques, when some treatments may have their greatest benefit.)
Q: It seems like rolling this out to an enormous population of patients is going to be difficult, if not impossible. Are people really going to be able to commit to what could be a lifetime of monthly intravenous infusions of a medicine that could be expensive, as therapeutic antibodies generally are?
A: I would say, nothing about this disease is easy. It’s devastating and horrible. And if someone is diagnosed at this stage, I would think that individual would embrace any opportunity to treat it. My hope is that we will be able to prescreen people with an effective blood test for amyloid that would be part of a regular testing protocol once they reach a certain age. Those with positive results would be referred for more testing, including amyloid brain imaging.
Topical benzyl benzoate–based treatment reduced Demodex in patients with rosacea
Daily treatment with benzyl benzoate (BB) cream reduced Demodex densities in patients with and without rosacea, and was associated with improvement in clinical signs, according to F.M.N. Forton, MD, of the Dermatology Clinic, Brussels, and his coauthor in the Journal of the European Academy of Dermatology and Venereology.
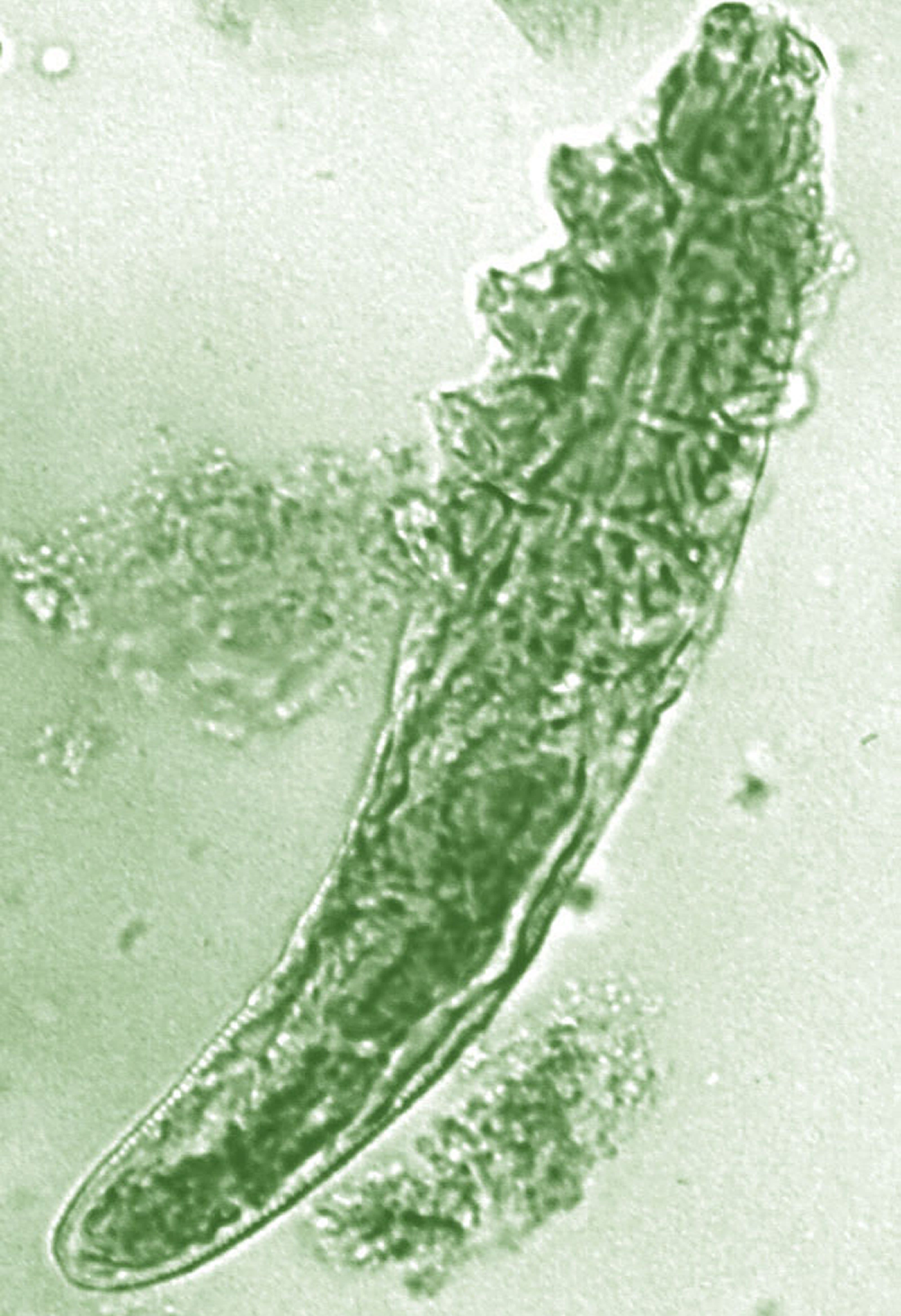
The retrospective study comprised 394 patients treated between 2002 and 2010; 117 of them had rosacea with papulopustules and the remainder only demodicosis. Their mean age was 49 years; most (278) were women. They had been treated with one of three doses of BB cream with crotamiton 10% cream: crotamiton applied in the morning, and BB 12% plus crotamiton in the evening; BB 12% plus crotamiton applied twice daily; and BB 20%-24% plus crotamiton applied once in the evening. Demodex densities (Dds) were measured with two consecutive standardized skin surface biopsies and deep biopsies at baseline and follow-up. Symptoms were measured with an investigator global assessment (IGA).
The authors said they had previously found that BB had acaricidal effects on Demodex, as did crotamiton “to a lesser extent,” but that the two treatments have not been well studied. They also referred to the increasing evidence that Demodex has a role in papulopustular rosacea, and that ivermectin, which is acaricidal, is recommended for topical treatment of papulopustular rosacea.
In the study, a mean of 2.7 months after starting treatment, mean Dds were significantly lower for the entire cohort, decreasing by 72.4% (plus or minus 2.6%) from baseline. Dds had normalized in 35% of patients, and in 31% of patients, symptoms had cleared.
Treatment was considered effective in 46% of patients and curative in 20%. Men responded slightly better, with clearance in 34% vs. 20% of women. The two regimens using the higher dose of BB were more effective than those using the lower dose and were associated with better compliance. Compliance overall was 77%.
After a mean of nearly 3 months of treatment, “topical application of BB (with crotamiton) was effective at reducing Dds and clearing clinical symptoms, not only in demodicosis but also in rosacea with papulopustules, indirectly supporting a key role of the mite in the pathophysiology of rosacea,” the authors concluded.
Neither of these products are approved in the United States for treating rosacea.
Dr. Forton disclosed that he occasionally works as a consultant for Galderma; the second author had no disclosures. The study had no funding source.
Source: Forton FMN et al. J Eur Acad Dermatol Venereol. 2019 Sep 7. doi: 10.1111/jdv.15938.
Daily treatment with benzyl benzoate (BB) cream reduced Demodex densities in patients with and without rosacea, and was associated with improvement in clinical signs, according to F.M.N. Forton, MD, of the Dermatology Clinic, Brussels, and his coauthor in the Journal of the European Academy of Dermatology and Venereology.
The retrospective study comprised 394 patients treated between 2002 and 2010; 117 of them had rosacea with papulopustules and the remainder only demodicosis. Their mean age was 49 years; most (278) were women. They had been treated with one of three doses of BB cream with crotamiton 10% cream: crotamiton applied in the morning, and BB 12% plus crotamiton in the evening; BB 12% plus crotamiton applied twice daily; and BB 20%-24% plus crotamiton applied once in the evening. Demodex densities (Dds) were measured with two consecutive standardized skin surface biopsies and deep biopsies at baseline and follow-up. Symptoms were measured with an investigator global assessment (IGA).
The authors said they had previously found that BB had acaricidal effects on Demodex, as did crotamiton “to a lesser extent,” but that the two treatments have not been well studied. They also referred to the increasing evidence that Demodex has a role in papulopustular rosacea, and that ivermectin, which is acaricidal, is recommended for topical treatment of papulopustular rosacea.
In the study, a mean of 2.7 months after starting treatment, mean Dds were significantly lower for the entire cohort, decreasing by 72.4% (plus or minus 2.6%) from baseline. Dds had normalized in 35% of patients, and in 31% of patients, symptoms had cleared.
Treatment was considered effective in 46% of patients and curative in 20%. Men responded slightly better, with clearance in 34% vs. 20% of women. The two regimens using the higher dose of BB were more effective than those using the lower dose and were associated with better compliance. Compliance overall was 77%.
After a mean of nearly 3 months of treatment, “topical application of BB (with crotamiton) was effective at reducing Dds and clearing clinical symptoms, not only in demodicosis but also in rosacea with papulopustules, indirectly supporting a key role of the mite in the pathophysiology of rosacea,” the authors concluded.
Neither of these products are approved in the United States for treating rosacea.
Dr. Forton disclosed that he occasionally works as a consultant for Galderma; the second author had no disclosures. The study had no funding source.
Source: Forton FMN et al. J Eur Acad Dermatol Venereol. 2019 Sep 7. doi: 10.1111/jdv.15938.
Daily treatment with benzyl benzoate (BB) cream reduced Demodex densities in patients with and without rosacea, and was associated with improvement in clinical signs, according to F.M.N. Forton, MD, of the Dermatology Clinic, Brussels, and his coauthor in the Journal of the European Academy of Dermatology and Venereology.
The retrospective study comprised 394 patients treated between 2002 and 2010; 117 of them had rosacea with papulopustules and the remainder only demodicosis. Their mean age was 49 years; most (278) were women. They had been treated with one of three doses of BB cream with crotamiton 10% cream: crotamiton applied in the morning, and BB 12% plus crotamiton in the evening; BB 12% plus crotamiton applied twice daily; and BB 20%-24% plus crotamiton applied once in the evening. Demodex densities (Dds) were measured with two consecutive standardized skin surface biopsies and deep biopsies at baseline and follow-up. Symptoms were measured with an investigator global assessment (IGA).
The authors said they had previously found that BB had acaricidal effects on Demodex, as did crotamiton “to a lesser extent,” but that the two treatments have not been well studied. They also referred to the increasing evidence that Demodex has a role in papulopustular rosacea, and that ivermectin, which is acaricidal, is recommended for topical treatment of papulopustular rosacea.
In the study, a mean of 2.7 months after starting treatment, mean Dds were significantly lower for the entire cohort, decreasing by 72.4% (plus or minus 2.6%) from baseline. Dds had normalized in 35% of patients, and in 31% of patients, symptoms had cleared.
Treatment was considered effective in 46% of patients and curative in 20%. Men responded slightly better, with clearance in 34% vs. 20% of women. The two regimens using the higher dose of BB were more effective than those using the lower dose and were associated with better compliance. Compliance overall was 77%.
After a mean of nearly 3 months of treatment, “topical application of BB (with crotamiton) was effective at reducing Dds and clearing clinical symptoms, not only in demodicosis but also in rosacea with papulopustules, indirectly supporting a key role of the mite in the pathophysiology of rosacea,” the authors concluded.
Neither of these products are approved in the United States for treating rosacea.
Dr. Forton disclosed that he occasionally works as a consultant for Galderma; the second author had no disclosures. The study had no funding source.
Source: Forton FMN et al. J Eur Acad Dermatol Venereol. 2019 Sep 7. doi: 10.1111/jdv.15938.
FROM JEADV
Fluoxetine tied to lower obsessive-compulsive scores among children with ASDs
Impact of the SSRI on those behaviors falls short in multiple secondary analyses
Fluoxetine appeared to lower scores for obsessive-compulsive behaviors among a group of children with autism spectrum disorders (ASDs), but the positive finding fell apart during multiple secondary analyses, Dinah S. Reddihough, MD, and colleagues have reported.
At 16 weeks, children and adolescents randomized to receive the SSRI had about a 2-point improvement on the Children’s Yale-Brown Obsessive Compulsive Scale, modified for pervasive developmental disorder (CYBOCS-PDD), compared with those taking placebo. But the finding lost significance in a multivariate analysis that accounted for a between-group difference in baseline scores – an uncontrollable variable that occurred during randomization, wrote Dr. Reddihough, of the Royal Children’s Hospital in Victoria, Australia, and coauthors.
“Moreover, repeating the analyses with multiple imputation to handle the missing data, arguably a preferable analysis, also failed to show evidence of benefit of fluoxetine compared with placebo irrespective of adjustment for the baseline imbalance,” the team noted. The study was published in JAMA.
Despite the null findings of the additional adjusted analyses, the authors held out hope for fluoxetine.
“Although cautious interpretation of the results from the primary analysis is warranted, all analyses of the primary outcome yielded 95% confidence intervals that extended well above the minimum clinically important difference of 2 points, indicating that fluoxetine may reduce the frequency and severity of obsessive-compulsive behaviors in children and adolescents with ASDs. Given the large amount of missing data, the study may have been underpowered to detect the minimum clinically important difference of 2 points.”
The study comprised 146 children (mean age, 11 years) recruited through three large practices in Australia. Children were randomized to fluoxetine or placebo for 16 weeks. Fluoxetine was weight-dosed and then titrated every week for the first month to a maximum of 20 mg/day.
The primary outcome was the difference between groups in the total score on the CYBOCS-PDD at 16 weeks. Secondary endpoints included changes on the Repetitive Behavior Scale–Revised, the Spence Children’s Anxiety Scale Aberrant Behavior Checklist–Community Version, the Clinical Global Impression Scale–Global Improvement and Efficacy Index, and a Disruptiveness Assessment.
Of the cohort, 85% were male, and 30% had an intellectual disability. The placebo group had higher scores on the Repetitive Behavior Scale–Revised and the Aberrant Behavior Checklist lethargy scale than did the fluoxetine groups.
There was a very high rate of nonadherence to study protocol, with 41% of those in the active group and 30% in the placebo group not completing the treatment regimen. The most often cited reasons for treatment discontinuation included parent decision to drop out (20 fluoxetine, 12 placebo), adverse events (5 fluoxetine, 4 placebo), and clinician decision (2 fluoxetine, 2 placebo).
The primary analysis found that scores on the CYBOCS-PDD were significantly lower in the fluoxetine group at 16 weeks; the fluoxetine group had decreased its score from 12.80 to 9.02, while the placebo group went from 13.13 to 10.89. This mean 2-point difference was statistically significant and, the authors wrote, met the minimum threshold for a clinically significant difference.
But the mean between-group difference decreased to a nonsignificant 1.17 points in the sensitivity analysis that controlled for sex, verbal ability, baseline CYBOCS-PDD, and imbalances found at baseline in some of the measures.
“Moreover, repeating the analyses with multiple imputation to handle the missing data, arguably a preferable analysis, also failed to show evidence of benefit of fluoxetine compared with placebo irrespective of adjustment for the baseline imbalance,” the team said.
There were no significant differences on any of the secondary measures.
Adverse events were similar in the active and placebo groups (45% and 42%, respectively). These included mood disturbances particularly irritability (9 fluoxetine, 12 placebo), gastrointestinal problems such as nausea and diarrhea (10 fluoxetine, 7 placebo), and sleep disorders (13 fluoxetine, 16 placebo). Two patients in the placebo group and none in the active group experienced suicidality.
Dr. Reddihough and coauthors cited the study’s high dropout rate as one of its limitations.
The study was supported by a federal grant from the Australian government. Dr. Reddihough had no financial disclosures.
SOURCE: Reddihough DS et al. JAMA. 2019;322(16):1561-9.
One could certainly take issue with the suggestion that this was presented as a positive trial on two levels: One, the prespecified primary outcome was not met, and two, the clinical significance (as distinct from statistical significance) of a 2-point change on that scale is problematic given the wide range of baseline scores allowed into the study.
The other thing that gets mixed up in this study is: Exactly what are obsessive-compulsive symptoms as distinct from repetitive behaviors? That question is a real challenge in this field when it comes to clinical trials for this target in autism, which tend to lump together heterogeneous repetitive behaviors.
The fact that there was absolutely no signal, or at least not a very strong one, is very challenging, considering how frequently this class of drugs is prescribed in autism.
This is not the first SSRI study for autism that’s come up empty. In this case, though, a negative study is still important because it confirms other negative studies. Another recently published study – the SOFIA fluoxetine study (J Autism Dev Disord. 2019 Jul 2. doi: 10:1007/s10803-019-04120-y) – also came up negative. SOFIA randomized 158 children to 14 weeks of fluoxetine or placebo. There were no significant differences on the primary endpoint, the Children’s Yale-Brown Obsessive-Compulsive Scale, and the placebo response rate was 41%.
However, it was clearly a heroic effort by Dr. Reddihough et al. to get this current study done: It took 7 years to get it over the finish line. This is probably because fluoxetine is so easily available. Why would a parent take a 50% chance of their child not getting a drug that might have some benefit – and that they could get without much trouble? And if it takes 7 years to complete a clinical trial, and we’re sitting around waiting for a definitive one, we are literally looking at potentially decades before we have some real answers that would inform your clinical practice in terms of this commonly prescribed drug.
As far as nailing the coffin shut on fluoxetine, I don’t think that will ever happen because some kids clearly improve. The placebo response in this population is very high. In our citalopram study (JAMA Pediatr. 2013 Nov;167[11]:1045-52), it was close to 33%. The improvement is dramatic and real, no less than any other response. If you see that response as a clinician and parent, it is very difficult to walk away from. Moreover, the population in clinical practice may be different from the population that shows up in a clinical trial specifically focused on restricted, repetitive behaviors.
One reason we may see a response in some is because SSRIs can help with anxiety, which is a common, arguably core symptom of autism. It appears to be part of the reason kids have catastrophic meltdowns when there are any changes in things they have come to expect, like a different route to school or a delay in their favorite TV show coming on. And if anxiety drives that, and an SSRI helps with anxiety, the child might be able to cope with something that would otherwise feel like the end of the world. Maybe that starts a positive feedback loop instead of a negative one, and maybe it propels more changes as the child and family experience success.
So, what are clinicians to do? The answer is still the same – they should use their best judgment about each child’s symptoms and about the risks and benefits that might occur with that individual. The fact that these trials are coming up negative for this indication in autism doesn’t mean that SSRIs might not be helpful for anxiety or depression, just as they are in the general population. I think we are back to basics. Clinicians need to use their best medical judgment according to each child’s unique needs.
These comments were adapted from an interview with Bryan H. King, MD, MBA. Dr. King is a professor of psychiatry at the University of California, San Francisco. He reported receiving personal fees from Genentech. Dr. King also commented on the study in an accompanying editorial (JAMA. 2019;322[16]:1557-8).
Impact of the SSRI on those behaviors falls short in multiple secondary analyses
Impact of the SSRI on those behaviors falls short in multiple secondary analyses
One could certainly take issue with the suggestion that this was presented as a positive trial on two levels: One, the prespecified primary outcome was not met, and two, the clinical significance (as distinct from statistical significance) of a 2-point change on that scale is problematic given the wide range of baseline scores allowed into the study.
The other thing that gets mixed up in this study is: Exactly what are obsessive-compulsive symptoms as distinct from repetitive behaviors? That question is a real challenge in this field when it comes to clinical trials for this target in autism, which tend to lump together heterogeneous repetitive behaviors.
The fact that there was absolutely no signal, or at least not a very strong one, is very challenging, considering how frequently this class of drugs is prescribed in autism.
This is not the first SSRI study for autism that’s come up empty. In this case, though, a negative study is still important because it confirms other negative studies. Another recently published study – the SOFIA fluoxetine study (J Autism Dev Disord. 2019 Jul 2. doi: 10:1007/s10803-019-04120-y) – also came up negative. SOFIA randomized 158 children to 14 weeks of fluoxetine or placebo. There were no significant differences on the primary endpoint, the Children’s Yale-Brown Obsessive-Compulsive Scale, and the placebo response rate was 41%.
However, it was clearly a heroic effort by Dr. Reddihough et al. to get this current study done: It took 7 years to get it over the finish line. This is probably because fluoxetine is so easily available. Why would a parent take a 50% chance of their child not getting a drug that might have some benefit – and that they could get without much trouble? And if it takes 7 years to complete a clinical trial, and we’re sitting around waiting for a definitive one, we are literally looking at potentially decades before we have some real answers that would inform your clinical practice in terms of this commonly prescribed drug.
As far as nailing the coffin shut on fluoxetine, I don’t think that will ever happen because some kids clearly improve. The placebo response in this population is very high. In our citalopram study (JAMA Pediatr. 2013 Nov;167[11]:1045-52), it was close to 33%. The improvement is dramatic and real, no less than any other response. If you see that response as a clinician and parent, it is very difficult to walk away from. Moreover, the population in clinical practice may be different from the population that shows up in a clinical trial specifically focused on restricted, repetitive behaviors.
One reason we may see a response in some is because SSRIs can help with anxiety, which is a common, arguably core symptom of autism. It appears to be part of the reason kids have catastrophic meltdowns when there are any changes in things they have come to expect, like a different route to school or a delay in their favorite TV show coming on. And if anxiety drives that, and an SSRI helps with anxiety, the child might be able to cope with something that would otherwise feel like the end of the world. Maybe that starts a positive feedback loop instead of a negative one, and maybe it propels more changes as the child and family experience success.
So, what are clinicians to do? The answer is still the same – they should use their best judgment about each child’s symptoms and about the risks and benefits that might occur with that individual. The fact that these trials are coming up negative for this indication in autism doesn’t mean that SSRIs might not be helpful for anxiety or depression, just as they are in the general population. I think we are back to basics. Clinicians need to use their best medical judgment according to each child’s unique needs.
These comments were adapted from an interview with Bryan H. King, MD, MBA. Dr. King is a professor of psychiatry at the University of California, San Francisco. He reported receiving personal fees from Genentech. Dr. King also commented on the study in an accompanying editorial (JAMA. 2019;322[16]:1557-8).
One could certainly take issue with the suggestion that this was presented as a positive trial on two levels: One, the prespecified primary outcome was not met, and two, the clinical significance (as distinct from statistical significance) of a 2-point change on that scale is problematic given the wide range of baseline scores allowed into the study.
The other thing that gets mixed up in this study is: Exactly what are obsessive-compulsive symptoms as distinct from repetitive behaviors? That question is a real challenge in this field when it comes to clinical trials for this target in autism, which tend to lump together heterogeneous repetitive behaviors.
The fact that there was absolutely no signal, or at least not a very strong one, is very challenging, considering how frequently this class of drugs is prescribed in autism.
This is not the first SSRI study for autism that’s come up empty. In this case, though, a negative study is still important because it confirms other negative studies. Another recently published study – the SOFIA fluoxetine study (J Autism Dev Disord. 2019 Jul 2. doi: 10:1007/s10803-019-04120-y) – also came up negative. SOFIA randomized 158 children to 14 weeks of fluoxetine or placebo. There were no significant differences on the primary endpoint, the Children’s Yale-Brown Obsessive-Compulsive Scale, and the placebo response rate was 41%.
However, it was clearly a heroic effort by Dr. Reddihough et al. to get this current study done: It took 7 years to get it over the finish line. This is probably because fluoxetine is so easily available. Why would a parent take a 50% chance of their child not getting a drug that might have some benefit – and that they could get without much trouble? And if it takes 7 years to complete a clinical trial, and we’re sitting around waiting for a definitive one, we are literally looking at potentially decades before we have some real answers that would inform your clinical practice in terms of this commonly prescribed drug.
As far as nailing the coffin shut on fluoxetine, I don’t think that will ever happen because some kids clearly improve. The placebo response in this population is very high. In our citalopram study (JAMA Pediatr. 2013 Nov;167[11]:1045-52), it was close to 33%. The improvement is dramatic and real, no less than any other response. If you see that response as a clinician and parent, it is very difficult to walk away from. Moreover, the population in clinical practice may be different from the population that shows up in a clinical trial specifically focused on restricted, repetitive behaviors.
One reason we may see a response in some is because SSRIs can help with anxiety, which is a common, arguably core symptom of autism. It appears to be part of the reason kids have catastrophic meltdowns when there are any changes in things they have come to expect, like a different route to school or a delay in their favorite TV show coming on. And if anxiety drives that, and an SSRI helps with anxiety, the child might be able to cope with something that would otherwise feel like the end of the world. Maybe that starts a positive feedback loop instead of a negative one, and maybe it propels more changes as the child and family experience success.
So, what are clinicians to do? The answer is still the same – they should use their best judgment about each child’s symptoms and about the risks and benefits that might occur with that individual. The fact that these trials are coming up negative for this indication in autism doesn’t mean that SSRIs might not be helpful for anxiety or depression, just as they are in the general population. I think we are back to basics. Clinicians need to use their best medical judgment according to each child’s unique needs.
These comments were adapted from an interview with Bryan H. King, MD, MBA. Dr. King is a professor of psychiatry at the University of California, San Francisco. He reported receiving personal fees from Genentech. Dr. King also commented on the study in an accompanying editorial (JAMA. 2019;322[16]:1557-8).
Fluoxetine appeared to lower scores for obsessive-compulsive behaviors among a group of children with autism spectrum disorders (ASDs), but the positive finding fell apart during multiple secondary analyses, Dinah S. Reddihough, MD, and colleagues have reported.
At 16 weeks, children and adolescents randomized to receive the SSRI had about a 2-point improvement on the Children’s Yale-Brown Obsessive Compulsive Scale, modified for pervasive developmental disorder (CYBOCS-PDD), compared with those taking placebo. But the finding lost significance in a multivariate analysis that accounted for a between-group difference in baseline scores – an uncontrollable variable that occurred during randomization, wrote Dr. Reddihough, of the Royal Children’s Hospital in Victoria, Australia, and coauthors.
“Moreover, repeating the analyses with multiple imputation to handle the missing data, arguably a preferable analysis, also failed to show evidence of benefit of fluoxetine compared with placebo irrespective of adjustment for the baseline imbalance,” the team noted. The study was published in JAMA.
Despite the null findings of the additional adjusted analyses, the authors held out hope for fluoxetine.
“Although cautious interpretation of the results from the primary analysis is warranted, all analyses of the primary outcome yielded 95% confidence intervals that extended well above the minimum clinically important difference of 2 points, indicating that fluoxetine may reduce the frequency and severity of obsessive-compulsive behaviors in children and adolescents with ASDs. Given the large amount of missing data, the study may have been underpowered to detect the minimum clinically important difference of 2 points.”
The study comprised 146 children (mean age, 11 years) recruited through three large practices in Australia. Children were randomized to fluoxetine or placebo for 16 weeks. Fluoxetine was weight-dosed and then titrated every week for the first month to a maximum of 20 mg/day.
The primary outcome was the difference between groups in the total score on the CYBOCS-PDD at 16 weeks. Secondary endpoints included changes on the Repetitive Behavior Scale–Revised, the Spence Children’s Anxiety Scale Aberrant Behavior Checklist–Community Version, the Clinical Global Impression Scale–Global Improvement and Efficacy Index, and a Disruptiveness Assessment.
Of the cohort, 85% were male, and 30% had an intellectual disability. The placebo group had higher scores on the Repetitive Behavior Scale–Revised and the Aberrant Behavior Checklist lethargy scale than did the fluoxetine groups.
There was a very high rate of nonadherence to study protocol, with 41% of those in the active group and 30% in the placebo group not completing the treatment regimen. The most often cited reasons for treatment discontinuation included parent decision to drop out (20 fluoxetine, 12 placebo), adverse events (5 fluoxetine, 4 placebo), and clinician decision (2 fluoxetine, 2 placebo).
The primary analysis found that scores on the CYBOCS-PDD were significantly lower in the fluoxetine group at 16 weeks; the fluoxetine group had decreased its score from 12.80 to 9.02, while the placebo group went from 13.13 to 10.89. This mean 2-point difference was statistically significant and, the authors wrote, met the minimum threshold for a clinically significant difference.
But the mean between-group difference decreased to a nonsignificant 1.17 points in the sensitivity analysis that controlled for sex, verbal ability, baseline CYBOCS-PDD, and imbalances found at baseline in some of the measures.
“Moreover, repeating the analyses with multiple imputation to handle the missing data, arguably a preferable analysis, also failed to show evidence of benefit of fluoxetine compared with placebo irrespective of adjustment for the baseline imbalance,” the team said.
There were no significant differences on any of the secondary measures.
Adverse events were similar in the active and placebo groups (45% and 42%, respectively). These included mood disturbances particularly irritability (9 fluoxetine, 12 placebo), gastrointestinal problems such as nausea and diarrhea (10 fluoxetine, 7 placebo), and sleep disorders (13 fluoxetine, 16 placebo). Two patients in the placebo group and none in the active group experienced suicidality.
Dr. Reddihough and coauthors cited the study’s high dropout rate as one of its limitations.
The study was supported by a federal grant from the Australian government. Dr. Reddihough had no financial disclosures.
SOURCE: Reddihough DS et al. JAMA. 2019;322(16):1561-9.
Fluoxetine appeared to lower scores for obsessive-compulsive behaviors among a group of children with autism spectrum disorders (ASDs), but the positive finding fell apart during multiple secondary analyses, Dinah S. Reddihough, MD, and colleagues have reported.
At 16 weeks, children and adolescents randomized to receive the SSRI had about a 2-point improvement on the Children’s Yale-Brown Obsessive Compulsive Scale, modified for pervasive developmental disorder (CYBOCS-PDD), compared with those taking placebo. But the finding lost significance in a multivariate analysis that accounted for a between-group difference in baseline scores – an uncontrollable variable that occurred during randomization, wrote Dr. Reddihough, of the Royal Children’s Hospital in Victoria, Australia, and coauthors.
“Moreover, repeating the analyses with multiple imputation to handle the missing data, arguably a preferable analysis, also failed to show evidence of benefit of fluoxetine compared with placebo irrespective of adjustment for the baseline imbalance,” the team noted. The study was published in JAMA.
Despite the null findings of the additional adjusted analyses, the authors held out hope for fluoxetine.
“Although cautious interpretation of the results from the primary analysis is warranted, all analyses of the primary outcome yielded 95% confidence intervals that extended well above the minimum clinically important difference of 2 points, indicating that fluoxetine may reduce the frequency and severity of obsessive-compulsive behaviors in children and adolescents with ASDs. Given the large amount of missing data, the study may have been underpowered to detect the minimum clinically important difference of 2 points.”
The study comprised 146 children (mean age, 11 years) recruited through three large practices in Australia. Children were randomized to fluoxetine or placebo for 16 weeks. Fluoxetine was weight-dosed and then titrated every week for the first month to a maximum of 20 mg/day.
The primary outcome was the difference between groups in the total score on the CYBOCS-PDD at 16 weeks. Secondary endpoints included changes on the Repetitive Behavior Scale–Revised, the Spence Children’s Anxiety Scale Aberrant Behavior Checklist–Community Version, the Clinical Global Impression Scale–Global Improvement and Efficacy Index, and a Disruptiveness Assessment.
Of the cohort, 85% were male, and 30% had an intellectual disability. The placebo group had higher scores on the Repetitive Behavior Scale–Revised and the Aberrant Behavior Checklist lethargy scale than did the fluoxetine groups.
There was a very high rate of nonadherence to study protocol, with 41% of those in the active group and 30% in the placebo group not completing the treatment regimen. The most often cited reasons for treatment discontinuation included parent decision to drop out (20 fluoxetine, 12 placebo), adverse events (5 fluoxetine, 4 placebo), and clinician decision (2 fluoxetine, 2 placebo).
The primary analysis found that scores on the CYBOCS-PDD were significantly lower in the fluoxetine group at 16 weeks; the fluoxetine group had decreased its score from 12.80 to 9.02, while the placebo group went from 13.13 to 10.89. This mean 2-point difference was statistically significant and, the authors wrote, met the minimum threshold for a clinically significant difference.
But the mean between-group difference decreased to a nonsignificant 1.17 points in the sensitivity analysis that controlled for sex, verbal ability, baseline CYBOCS-PDD, and imbalances found at baseline in some of the measures.
“Moreover, repeating the analyses with multiple imputation to handle the missing data, arguably a preferable analysis, also failed to show evidence of benefit of fluoxetine compared with placebo irrespective of adjustment for the baseline imbalance,” the team said.
There were no significant differences on any of the secondary measures.
Adverse events were similar in the active and placebo groups (45% and 42%, respectively). These included mood disturbances particularly irritability (9 fluoxetine, 12 placebo), gastrointestinal problems such as nausea and diarrhea (10 fluoxetine, 7 placebo), and sleep disorders (13 fluoxetine, 16 placebo). Two patients in the placebo group and none in the active group experienced suicidality.
Dr. Reddihough and coauthors cited the study’s high dropout rate as one of its limitations.
The study was supported by a federal grant from the Australian government. Dr. Reddihough had no financial disclosures.
SOURCE: Reddihough DS et al. JAMA. 2019;322(16):1561-9.
FROM JAMA
Trials examine T2T strategy in axial spondyloarthritis
Three international clinical trials in Europe are examining the effectiveness of treat-to-target (T2T) therapeutic regimens in patients with axial spondyloarthritis (axSpA), including two that will be the first randomized trial evidence to support or refute the T2T strategy for patients ranging from those with nonradiographic disease to patients with ankylosing spondylitis.
T2T has proved before to work in patients with rheumatoid arthritis and psoriatic arthritis with evidence from the TICORA (Tight Control of Rheumatoid Arthritis) and TICOPA (Tight Control in Psoriatic Arthritis) trials.
Two T2T trials in axSpA are still in the recruiting phase, and one has completed enrollment, with no results available yet.
Tight control in spondyloarthritis (TICOSPA)
TICOSPA is a 1-year, ongoing, multinational, cluster-randomized, prospective cohort study that has enrolled 163 patients with a diagnosis of active axial spondyloarthritis to evaluate the potential benefit of a T2T strategy in which the rheumatologist will agree to monitor very closely – at least every 4 weeks – and treat patients in accordance with a predefined strategy. The T2T strategy is compared with usual care as given by the treating rheumatologist. Prior to the trial, patients were on nonoptimal NSAID treatment.
“Tight control” in this study refers to the time from treatment initiation to adequate assessment of efficacy and safety, which for efficacy should be at 2-4 weeks for NSAIDs and 12-16 weeks for tumor necrosis factor inhibitors but can be a very short time frame for evaluating safety, “based on the occurrence of adverse events,” according to the study description at clinicaltrials.gov.
The primary endpoint is change on the Assessment of SpondyloArthritis international Society (ASAS) Health Index-Numerical Rating Score over the 1 year of follow-up.
There are 11 secondary endpoints, including:
- Percentage reaching major improvement in the Ankylosing Spondylitis Disease Activity Scale score (ASDAS).
- Percentage reaching 50% improvement of the initial Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score at 1 year.
- Change in the ASAS-NSAID score over 1 year.
- Change in the Work Productivity and Activity Impairment questionnaire.
The study is being conducted at 18 centers in Belgium, France, and the Netherlands and is sponsored by the Association de Recherche Clinique en Rhumatologie.
AScalate: Treat-to-target in axial spondyloarthritis
The Novartis-sponsored AScalate study seeks to enroll 300 patients with active disease despite NSAID therapy. The 36-week, randomized, parallel-group, open-label, multicenter trial will be conducted at seven sites in Germany.
The study will randomize patients to either of two arms: An active group will receive T2T therapy with secukinumab as a first-line biologic in escalating doses of 150-300 mg, determined by patient response until the T2T goal had been reached. Patients who don’t respond to secukinumab will be switched to an adalimumab biosimilar. The comparator group will receive standard-of-care therapy up to the maximum recommended dose at the discretion of the investigator.
The primary endpoint is the percentage of patients in each group who meet ASAS 40 response criteria by 24 weeks.
There are 11 secondary endpoints, including:
- Percentage achieving an ASAS40 response at 12 weeks.
- Percentage achieving ASAS20 and ASAS partial response at 12 and 24 weeks.
- Proportion of patients meeting the ASDAS definition of inactive disease, ASDAS clinically important and major improvement, and ASDAS low disease activity.
- Proportion of patients achieving 50% improvement of the initial BASDAI score.
Treat-to-target with secukinumab in axial spondyloarthritis (TRACE)
TRACE is a Novartis-sponsored phase 4 study examining reductions of inflammation seen on MRI of sacroiliac joints and spine at 16-24 weeks in patients who achieve ASDAS remission (score of less than 1.3) on 150 mg secukinumab by 16 weeks. The comparator group will be patients who are not in remission by week 16 and need a dose increase to 300 mg. The Danish trial seeks 88 participants with high disease activity and MRI signs of inflammation in the sacroiliac joints and/ or the spine.
After an initial four weekly doses of secukinumab 150 mg, patients will receive monthly secukinumab 150-mg doses out to week 16. Nonresponders at week 16 will escalate to 300 mg. If by 24 weeks these patients do not respond, they will be switched to a TNF inhibitor.
The primary outcome is the proportion of patients with a positive change in MRI-inflammation as measured by the sum of the Spondyloarthritis Research Consortium of Canada (SPARCC) sacroiliac joint and spine inflammation indices.
Three international clinical trials in Europe are examining the effectiveness of treat-to-target (T2T) therapeutic regimens in patients with axial spondyloarthritis (axSpA), including two that will be the first randomized trial evidence to support or refute the T2T strategy for patients ranging from those with nonradiographic disease to patients with ankylosing spondylitis.
T2T has proved before to work in patients with rheumatoid arthritis and psoriatic arthritis with evidence from the TICORA (Tight Control of Rheumatoid Arthritis) and TICOPA (Tight Control in Psoriatic Arthritis) trials.
Two T2T trials in axSpA are still in the recruiting phase, and one has completed enrollment, with no results available yet.
Tight control in spondyloarthritis (TICOSPA)
TICOSPA is a 1-year, ongoing, multinational, cluster-randomized, prospective cohort study that has enrolled 163 patients with a diagnosis of active axial spondyloarthritis to evaluate the potential benefit of a T2T strategy in which the rheumatologist will agree to monitor very closely – at least every 4 weeks – and treat patients in accordance with a predefined strategy. The T2T strategy is compared with usual care as given by the treating rheumatologist. Prior to the trial, patients were on nonoptimal NSAID treatment.
“Tight control” in this study refers to the time from treatment initiation to adequate assessment of efficacy and safety, which for efficacy should be at 2-4 weeks for NSAIDs and 12-16 weeks for tumor necrosis factor inhibitors but can be a very short time frame for evaluating safety, “based on the occurrence of adverse events,” according to the study description at clinicaltrials.gov.
The primary endpoint is change on the Assessment of SpondyloArthritis international Society (ASAS) Health Index-Numerical Rating Score over the 1 year of follow-up.
There are 11 secondary endpoints, including:
- Percentage reaching major improvement in the Ankylosing Spondylitis Disease Activity Scale score (ASDAS).
- Percentage reaching 50% improvement of the initial Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score at 1 year.
- Change in the ASAS-NSAID score over 1 year.
- Change in the Work Productivity and Activity Impairment questionnaire.
The study is being conducted at 18 centers in Belgium, France, and the Netherlands and is sponsored by the Association de Recherche Clinique en Rhumatologie.
AScalate: Treat-to-target in axial spondyloarthritis
The Novartis-sponsored AScalate study seeks to enroll 300 patients with active disease despite NSAID therapy. The 36-week, randomized, parallel-group, open-label, multicenter trial will be conducted at seven sites in Germany.
The study will randomize patients to either of two arms: An active group will receive T2T therapy with secukinumab as a first-line biologic in escalating doses of 150-300 mg, determined by patient response until the T2T goal had been reached. Patients who don’t respond to secukinumab will be switched to an adalimumab biosimilar. The comparator group will receive standard-of-care therapy up to the maximum recommended dose at the discretion of the investigator.
The primary endpoint is the percentage of patients in each group who meet ASAS 40 response criteria by 24 weeks.
There are 11 secondary endpoints, including:
- Percentage achieving an ASAS40 response at 12 weeks.
- Percentage achieving ASAS20 and ASAS partial response at 12 and 24 weeks.
- Proportion of patients meeting the ASDAS definition of inactive disease, ASDAS clinically important and major improvement, and ASDAS low disease activity.
- Proportion of patients achieving 50% improvement of the initial BASDAI score.
Treat-to-target with secukinumab in axial spondyloarthritis (TRACE)
TRACE is a Novartis-sponsored phase 4 study examining reductions of inflammation seen on MRI of sacroiliac joints and spine at 16-24 weeks in patients who achieve ASDAS remission (score of less than 1.3) on 150 mg secukinumab by 16 weeks. The comparator group will be patients who are not in remission by week 16 and need a dose increase to 300 mg. The Danish trial seeks 88 participants with high disease activity and MRI signs of inflammation in the sacroiliac joints and/ or the spine.
After an initial four weekly doses of secukinumab 150 mg, patients will receive monthly secukinumab 150-mg doses out to week 16. Nonresponders at week 16 will escalate to 300 mg. If by 24 weeks these patients do not respond, they will be switched to a TNF inhibitor.
The primary outcome is the proportion of patients with a positive change in MRI-inflammation as measured by the sum of the Spondyloarthritis Research Consortium of Canada (SPARCC) sacroiliac joint and spine inflammation indices.
Three international clinical trials in Europe are examining the effectiveness of treat-to-target (T2T) therapeutic regimens in patients with axial spondyloarthritis (axSpA), including two that will be the first randomized trial evidence to support or refute the T2T strategy for patients ranging from those with nonradiographic disease to patients with ankylosing spondylitis.
T2T has proved before to work in patients with rheumatoid arthritis and psoriatic arthritis with evidence from the TICORA (Tight Control of Rheumatoid Arthritis) and TICOPA (Tight Control in Psoriatic Arthritis) trials.
Two T2T trials in axSpA are still in the recruiting phase, and one has completed enrollment, with no results available yet.
Tight control in spondyloarthritis (TICOSPA)
TICOSPA is a 1-year, ongoing, multinational, cluster-randomized, prospective cohort study that has enrolled 163 patients with a diagnosis of active axial spondyloarthritis to evaluate the potential benefit of a T2T strategy in which the rheumatologist will agree to monitor very closely – at least every 4 weeks – and treat patients in accordance with a predefined strategy. The T2T strategy is compared with usual care as given by the treating rheumatologist. Prior to the trial, patients were on nonoptimal NSAID treatment.
“Tight control” in this study refers to the time from treatment initiation to adequate assessment of efficacy and safety, which for efficacy should be at 2-4 weeks for NSAIDs and 12-16 weeks for tumor necrosis factor inhibitors but can be a very short time frame for evaluating safety, “based on the occurrence of adverse events,” according to the study description at clinicaltrials.gov.
The primary endpoint is change on the Assessment of SpondyloArthritis international Society (ASAS) Health Index-Numerical Rating Score over the 1 year of follow-up.
There are 11 secondary endpoints, including:
- Percentage reaching major improvement in the Ankylosing Spondylitis Disease Activity Scale score (ASDAS).
- Percentage reaching 50% improvement of the initial Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score at 1 year.
- Change in the ASAS-NSAID score over 1 year.
- Change in the Work Productivity and Activity Impairment questionnaire.
The study is being conducted at 18 centers in Belgium, France, and the Netherlands and is sponsored by the Association de Recherche Clinique en Rhumatologie.
AScalate: Treat-to-target in axial spondyloarthritis
The Novartis-sponsored AScalate study seeks to enroll 300 patients with active disease despite NSAID therapy. The 36-week, randomized, parallel-group, open-label, multicenter trial will be conducted at seven sites in Germany.
The study will randomize patients to either of two arms: An active group will receive T2T therapy with secukinumab as a first-line biologic in escalating doses of 150-300 mg, determined by patient response until the T2T goal had been reached. Patients who don’t respond to secukinumab will be switched to an adalimumab biosimilar. The comparator group will receive standard-of-care therapy up to the maximum recommended dose at the discretion of the investigator.
The primary endpoint is the percentage of patients in each group who meet ASAS 40 response criteria by 24 weeks.
There are 11 secondary endpoints, including:
- Percentage achieving an ASAS40 response at 12 weeks.
- Percentage achieving ASAS20 and ASAS partial response at 12 and 24 weeks.
- Proportion of patients meeting the ASDAS definition of inactive disease, ASDAS clinically important and major improvement, and ASDAS low disease activity.
- Proportion of patients achieving 50% improvement of the initial BASDAI score.
Treat-to-target with secukinumab in axial spondyloarthritis (TRACE)
TRACE is a Novartis-sponsored phase 4 study examining reductions of inflammation seen on MRI of sacroiliac joints and spine at 16-24 weeks in patients who achieve ASDAS remission (score of less than 1.3) on 150 mg secukinumab by 16 weeks. The comparator group will be patients who are not in remission by week 16 and need a dose increase to 300 mg. The Danish trial seeks 88 participants with high disease activity and MRI signs of inflammation in the sacroiliac joints and/ or the spine.
After an initial four weekly doses of secukinumab 150 mg, patients will receive monthly secukinumab 150-mg doses out to week 16. Nonresponders at week 16 will escalate to 300 mg. If by 24 weeks these patients do not respond, they will be switched to a TNF inhibitor.
The primary outcome is the proportion of patients with a positive change in MRI-inflammation as measured by the sum of the Spondyloarthritis Research Consortium of Canada (SPARCC) sacroiliac joint and spine inflammation indices.
Biogen plans to submit application to FDA for Alzheimer’s drug aducanumab
Biogen aims to file with the Food and Drug Administration for regulatory approval of aducanumab, an antibody under investigation for Alzheimer’s disease, in 2020 following largely positive results of a secondary analysis of two failed phase 3 trials, ENGAGE and EMERGE, the company announced Oct. 22.
Biogen’s plans reverse its March 21, 2019, decision with codeveloper Eisai to discontinue work on the drug after a futility analysis of the trials determined aducanumab was unlikely to yield significant benefit. Biogen announced the plan to file a biologic drug application following a new analysis of additional data that became available after the data lock on the futility analysis. But while primary and secondary endpoints were nearly all positive for EMERGE in the secondary analysis of the larger dataset, the same could not be said for the twin trial, ENGAGE, which had negative results for most of its endpoints. However, Biogen said that “results from a subset of patients in the phase 3 ENGAGE study who received sufficient exposure to high-dose aducanumab support the findings from EMERGE.”
Both the Alzheimer’s Association and researchers interpreted the announcement with a measured tone, saying it offered a hopeful sign for a field continually stymied in its quest for an effective treatment. More than 100 clinical trials have failed over the last 20 years.
“This really is very encouraging news,” Rebecca M. Edelmayer, PhD, director of scientific engagement at the Alzheimer’s Association, said in an interview. The secondary combined analyses showed “the largest reductions in clinical and functional decline we have seen. It’s an important moment for the patients with AD and their families, and for researchers all around the world. This deserves to be discussed and considered by the research community, but we really need to dig deep into the data. We expect to see more of them at the Clinical Trials on Alzheimer’s Disease conference,” which is set for early December.
Paul Aisen, MD, a consultant for Biogen and director of the Alzheimer’s Therapeutic Research Institute at the University of Southern California, Los Angeles, was similarly measured.
“There is an enormous amount of data here and they will be very challenging to interpret, especially since both trials were stopped in a futility analysis,” he said in an interview. “We’re now interpreting data that continue to be collected after the initial data lock. But I do believe there is evidence that supports the amyloid hypothesis and the development of aducanumab.”
A deep data dive is in order before the field completely embraces aducanumab’s advancement, agreed Michael Wolfe, PhD, the Mathias P. Mertes Professor of Medicinal Chemistry at the University of Kansas, Lawrence.
“We would have to see exactly how they came up with this new data set and analysis. I have felt for many years now that these companies would try to parse and shuffle the data around until they got a statistically significant result, just to get approval. They would make billions per year but not really make a difference in people’s lives. That being said, if aducanumab truly slows the decline in activities of daily living, keeping people independent longer, that would be a worthwhile clinical result.”
Still to be considered is whether aducanumab could confer enough benefits to be worth monthly, potentially lifelong, infusions of a pricey medication that still won’t stop disease progression.
“Whether the clinical impact will be worth the anticipated cost remains to be seen,” Richard J. Caselli, MD, said in an interview. “This is likely to be a very expensive treatment for a subgroup of individuals with the hope of slowing decline, but – unless there is a huge upside surprise in data yet to be released – it is not going to halt progression and will certainly increase the cost of care dramatically.”
Dr. Caselli, clinical core director of the Alzheimer’s Disease Center at the Mayo Clinic in Phoenix, continued: “I imagine if approved, there will be a number of insurance obstacles to overcome regarding who qualifies, for how long, etc. Scientifically, this certainly supports the long-held view that beta amyloid is important in the pathophysiology of Alzheimer’s disease. But, again, unless the impact is unexpectedly huge, I don’t think this quiets those who feel there is more to the story than only a gain of beta amyloid toxicity, though this does support the idea that it plays a role, which should not surprise anyone.”
Dr. Edelmayer said the Alzheimer’s Association has raised the issue of access and cost with Biogen.
“We have had many discussions about their capacity to roll this out,” if aducanumab is approved, she said. “Those who were in the studies will be the first recipients. But Biogen is making plans to move this into the general population if approved, to all patients who meet the diagnostic criteria.”
“There is precedent out there when it comes to doing infusion medicines, and those centers are part of the planning process. In terms of pricing, this is a problem we would be happy to see, because it would mean that we have a treatment. But we’ll cross that bridge when we come to it.”
Secondary analysis results
The new analysis comprised 2,066 patients who had the opportunity to complete the full 18-month trials by March 20, 2019. The full intent-to-treat population of the trials comprised 3,285 patients with mild cognitive impairment caused by Alzheimer’s disease or mild Alzheimer’s disease dementia.
In the secondary analysis of EMERGE’s intent-to-treat population, patients who took the highest dose of aducanumab (10 mg/kg every month) showed 23% lower functional and cognitive decline on the trial’s primary endpoint of Clinical Dementia Rating–Sum of Boxes (CDR-SB) at 18 months when compared with placebo. The rate of decline was slowed by a nonsignificant 14% among users of the lower dose (6 mg/kg monthly).Secondary endpoints for the high-dose group in EMERGE showed 27% slower cognitive decline on the 13-item cognitive subscale of the AD Assessment Scale (ADAS-Cog13) and 40% lower decline in function among patients with mild cognitive impairment based on the MCI version of the AD Cooperative Study–Activities of Daily Living Inventory (ADCS-ADL-MCI).
However, data from the ENGAGE trial, which had the same primary endpoint, were not positive. CDR-SB scores worsened 2% more among high-dose aducanumab users but low-dose users slowed decline by a nonsignificant 12% when compared with placebo.“Exposure to high-dose aducanumab was important for efficacy,” the company noted in a slide set presented at an Oct. 22 investors webcast. “Differences in exposure to high-dose aducanumab largely explain the different results between the futility analysis and the new analysis of this larger dataset, as well as the different results between the two studies.”
In EMERGE, changes in secondary endpoints among patients who had the opportunity to complete the full 18-month trials included:
- Mini Mental State Exam (MMSE): Significant 23% decrease in rate of decline in the high-dose group and nonsignificant 3% increase in decline in the low-dose group.
- ADAS-Cog13: Significant 25% decrease for high-dose users and nonsignificant 10% decrease for low dose.
- ADCS-ADL-MCI: Significant decreases of 46% for high-dose and 20% for low-dose users.
The ENGAGE secondary endpoints of high- vs. low-dose patients who completed the full trials were:
- MMSE: Significant 13% increase, nonsignificant 3% decrease.
- ADAS-Cog13: Nonsignificant 2% decrease, nonsignificant 1% decrease.
- ADCS-ADL-MCI: Significant 12% declines in both dosage groups.
All of the positive cognitive and functional results tracked along with results of amyloid PET imaging and CSF biomarkers. In EMERGE, amyloid plaque binding declined about 27% in the high-dose group and about 16% in the low-dose group, reflecting plaque clearance. Phosphorylated tau in CSF decreased by about 17 pg/mL and 10 pg/mL, respectively, and total tau decreased by 160 pg/mL and 120 pg/mL. Tau decreases indicate a slowing of neuronal damage.
In ENGAGE, amyloid plaque binding decreased by about 24% in the high-dose group and by about 16% in the low-dose group. Phosphorylated tau dropped by about 10 pg/mL and 11 pg/mL, respectively. But total tau dropped more in the low-dose group than in the high-dose group (about –100 pg/mL vs. –20 pg/mL).
Biogen said the amyloid PET imaging biomarker results and CDR-SB scores in both studies were consistent with each other in a subset of patients with “sufficient exposure to 10 mg/kg,” which was defined as “10 or more uninterrupted 10-mg/kg dosing intervals at steady-state.”
The most common adverse events were amyloid related imaging abnormalities–edema (ARIA-E), which occurred in 35%, and headache in 20%. The majority of patients who experienced ARIA-E (74%) were asymptomatic; episodes generally resolved within 4-16 weeks, typically without long-term sequelae.
Dr. Aisen is a consultant for Biogen and on the aducanumab steering committee. None of the other sources in this article have any financial relationship with Biogen or Eisai.
This article was updated 10/23/19.
Biogen aims to file with the Food and Drug Administration for regulatory approval of aducanumab, an antibody under investigation for Alzheimer’s disease, in 2020 following largely positive results of a secondary analysis of two failed phase 3 trials, ENGAGE and EMERGE, the company announced Oct. 22.
Biogen’s plans reverse its March 21, 2019, decision with codeveloper Eisai to discontinue work on the drug after a futility analysis of the trials determined aducanumab was unlikely to yield significant benefit. Biogen announced the plan to file a biologic drug application following a new analysis of additional data that became available after the data lock on the futility analysis. But while primary and secondary endpoints were nearly all positive for EMERGE in the secondary analysis of the larger dataset, the same could not be said for the twin trial, ENGAGE, which had negative results for most of its endpoints. However, Biogen said that “results from a subset of patients in the phase 3 ENGAGE study who received sufficient exposure to high-dose aducanumab support the findings from EMERGE.”
Both the Alzheimer’s Association and researchers interpreted the announcement with a measured tone, saying it offered a hopeful sign for a field continually stymied in its quest for an effective treatment. More than 100 clinical trials have failed over the last 20 years.
“This really is very encouraging news,” Rebecca M. Edelmayer, PhD, director of scientific engagement at the Alzheimer’s Association, said in an interview. The secondary combined analyses showed “the largest reductions in clinical and functional decline we have seen. It’s an important moment for the patients with AD and their families, and for researchers all around the world. This deserves to be discussed and considered by the research community, but we really need to dig deep into the data. We expect to see more of them at the Clinical Trials on Alzheimer’s Disease conference,” which is set for early December.
Paul Aisen, MD, a consultant for Biogen and director of the Alzheimer’s Therapeutic Research Institute at the University of Southern California, Los Angeles, was similarly measured.
“There is an enormous amount of data here and they will be very challenging to interpret, especially since both trials were stopped in a futility analysis,” he said in an interview. “We’re now interpreting data that continue to be collected after the initial data lock. But I do believe there is evidence that supports the amyloid hypothesis and the development of aducanumab.”
A deep data dive is in order before the field completely embraces aducanumab’s advancement, agreed Michael Wolfe, PhD, the Mathias P. Mertes Professor of Medicinal Chemistry at the University of Kansas, Lawrence.
“We would have to see exactly how they came up with this new data set and analysis. I have felt for many years now that these companies would try to parse and shuffle the data around until they got a statistically significant result, just to get approval. They would make billions per year but not really make a difference in people’s lives. That being said, if aducanumab truly slows the decline in activities of daily living, keeping people independent longer, that would be a worthwhile clinical result.”
Still to be considered is whether aducanumab could confer enough benefits to be worth monthly, potentially lifelong, infusions of a pricey medication that still won’t stop disease progression.
“Whether the clinical impact will be worth the anticipated cost remains to be seen,” Richard J. Caselli, MD, said in an interview. “This is likely to be a very expensive treatment for a subgroup of individuals with the hope of slowing decline, but – unless there is a huge upside surprise in data yet to be released – it is not going to halt progression and will certainly increase the cost of care dramatically.”
Dr. Caselli, clinical core director of the Alzheimer’s Disease Center at the Mayo Clinic in Phoenix, continued: “I imagine if approved, there will be a number of insurance obstacles to overcome regarding who qualifies, for how long, etc. Scientifically, this certainly supports the long-held view that beta amyloid is important in the pathophysiology of Alzheimer’s disease. But, again, unless the impact is unexpectedly huge, I don’t think this quiets those who feel there is more to the story than only a gain of beta amyloid toxicity, though this does support the idea that it plays a role, which should not surprise anyone.”
Dr. Edelmayer said the Alzheimer’s Association has raised the issue of access and cost with Biogen.
“We have had many discussions about their capacity to roll this out,” if aducanumab is approved, she said. “Those who were in the studies will be the first recipients. But Biogen is making plans to move this into the general population if approved, to all patients who meet the diagnostic criteria.”
“There is precedent out there when it comes to doing infusion medicines, and those centers are part of the planning process. In terms of pricing, this is a problem we would be happy to see, because it would mean that we have a treatment. But we’ll cross that bridge when we come to it.”
Secondary analysis results
The new analysis comprised 2,066 patients who had the opportunity to complete the full 18-month trials by March 20, 2019. The full intent-to-treat population of the trials comprised 3,285 patients with mild cognitive impairment caused by Alzheimer’s disease or mild Alzheimer’s disease dementia.
In the secondary analysis of EMERGE’s intent-to-treat population, patients who took the highest dose of aducanumab (10 mg/kg every month) showed 23% lower functional and cognitive decline on the trial’s primary endpoint of Clinical Dementia Rating–Sum of Boxes (CDR-SB) at 18 months when compared with placebo. The rate of decline was slowed by a nonsignificant 14% among users of the lower dose (6 mg/kg monthly).Secondary endpoints for the high-dose group in EMERGE showed 27% slower cognitive decline on the 13-item cognitive subscale of the AD Assessment Scale (ADAS-Cog13) and 40% lower decline in function among patients with mild cognitive impairment based on the MCI version of the AD Cooperative Study–Activities of Daily Living Inventory (ADCS-ADL-MCI).
However, data from the ENGAGE trial, which had the same primary endpoint, were not positive. CDR-SB scores worsened 2% more among high-dose aducanumab users but low-dose users slowed decline by a nonsignificant 12% when compared with placebo.“Exposure to high-dose aducanumab was important for efficacy,” the company noted in a slide set presented at an Oct. 22 investors webcast. “Differences in exposure to high-dose aducanumab largely explain the different results between the futility analysis and the new analysis of this larger dataset, as well as the different results between the two studies.”
In EMERGE, changes in secondary endpoints among patients who had the opportunity to complete the full 18-month trials included:
- Mini Mental State Exam (MMSE): Significant 23% decrease in rate of decline in the high-dose group and nonsignificant 3% increase in decline in the low-dose group.
- ADAS-Cog13: Significant 25% decrease for high-dose users and nonsignificant 10% decrease for low dose.
- ADCS-ADL-MCI: Significant decreases of 46% for high-dose and 20% for low-dose users.
The ENGAGE secondary endpoints of high- vs. low-dose patients who completed the full trials were:
- MMSE: Significant 13% increase, nonsignificant 3% decrease.
- ADAS-Cog13: Nonsignificant 2% decrease, nonsignificant 1% decrease.
- ADCS-ADL-MCI: Significant 12% declines in both dosage groups.
All of the positive cognitive and functional results tracked along with results of amyloid PET imaging and CSF biomarkers. In EMERGE, amyloid plaque binding declined about 27% in the high-dose group and about 16% in the low-dose group, reflecting plaque clearance. Phosphorylated tau in CSF decreased by about 17 pg/mL and 10 pg/mL, respectively, and total tau decreased by 160 pg/mL and 120 pg/mL. Tau decreases indicate a slowing of neuronal damage.
In ENGAGE, amyloid plaque binding decreased by about 24% in the high-dose group and by about 16% in the low-dose group. Phosphorylated tau dropped by about 10 pg/mL and 11 pg/mL, respectively. But total tau dropped more in the low-dose group than in the high-dose group (about –100 pg/mL vs. –20 pg/mL).
Biogen said the amyloid PET imaging biomarker results and CDR-SB scores in both studies were consistent with each other in a subset of patients with “sufficient exposure to 10 mg/kg,” which was defined as “10 or more uninterrupted 10-mg/kg dosing intervals at steady-state.”
The most common adverse events were amyloid related imaging abnormalities–edema (ARIA-E), which occurred in 35%, and headache in 20%. The majority of patients who experienced ARIA-E (74%) were asymptomatic; episodes generally resolved within 4-16 weeks, typically without long-term sequelae.
Dr. Aisen is a consultant for Biogen and on the aducanumab steering committee. None of the other sources in this article have any financial relationship with Biogen or Eisai.
This article was updated 10/23/19.
Biogen aims to file with the Food and Drug Administration for regulatory approval of aducanumab, an antibody under investigation for Alzheimer’s disease, in 2020 following largely positive results of a secondary analysis of two failed phase 3 trials, ENGAGE and EMERGE, the company announced Oct. 22.
Biogen’s plans reverse its March 21, 2019, decision with codeveloper Eisai to discontinue work on the drug after a futility analysis of the trials determined aducanumab was unlikely to yield significant benefit. Biogen announced the plan to file a biologic drug application following a new analysis of additional data that became available after the data lock on the futility analysis. But while primary and secondary endpoints were nearly all positive for EMERGE in the secondary analysis of the larger dataset, the same could not be said for the twin trial, ENGAGE, which had negative results for most of its endpoints. However, Biogen said that “results from a subset of patients in the phase 3 ENGAGE study who received sufficient exposure to high-dose aducanumab support the findings from EMERGE.”
Both the Alzheimer’s Association and researchers interpreted the announcement with a measured tone, saying it offered a hopeful sign for a field continually stymied in its quest for an effective treatment. More than 100 clinical trials have failed over the last 20 years.
“This really is very encouraging news,” Rebecca M. Edelmayer, PhD, director of scientific engagement at the Alzheimer’s Association, said in an interview. The secondary combined analyses showed “the largest reductions in clinical and functional decline we have seen. It’s an important moment for the patients with AD and their families, and for researchers all around the world. This deserves to be discussed and considered by the research community, but we really need to dig deep into the data. We expect to see more of them at the Clinical Trials on Alzheimer’s Disease conference,” which is set for early December.
Paul Aisen, MD, a consultant for Biogen and director of the Alzheimer’s Therapeutic Research Institute at the University of Southern California, Los Angeles, was similarly measured.
“There is an enormous amount of data here and they will be very challenging to interpret, especially since both trials were stopped in a futility analysis,” he said in an interview. “We’re now interpreting data that continue to be collected after the initial data lock. But I do believe there is evidence that supports the amyloid hypothesis and the development of aducanumab.”
A deep data dive is in order before the field completely embraces aducanumab’s advancement, agreed Michael Wolfe, PhD, the Mathias P. Mertes Professor of Medicinal Chemistry at the University of Kansas, Lawrence.
“We would have to see exactly how they came up with this new data set and analysis. I have felt for many years now that these companies would try to parse and shuffle the data around until they got a statistically significant result, just to get approval. They would make billions per year but not really make a difference in people’s lives. That being said, if aducanumab truly slows the decline in activities of daily living, keeping people independent longer, that would be a worthwhile clinical result.”
Still to be considered is whether aducanumab could confer enough benefits to be worth monthly, potentially lifelong, infusions of a pricey medication that still won’t stop disease progression.
“Whether the clinical impact will be worth the anticipated cost remains to be seen,” Richard J. Caselli, MD, said in an interview. “This is likely to be a very expensive treatment for a subgroup of individuals with the hope of slowing decline, but – unless there is a huge upside surprise in data yet to be released – it is not going to halt progression and will certainly increase the cost of care dramatically.”
Dr. Caselli, clinical core director of the Alzheimer’s Disease Center at the Mayo Clinic in Phoenix, continued: “I imagine if approved, there will be a number of insurance obstacles to overcome regarding who qualifies, for how long, etc. Scientifically, this certainly supports the long-held view that beta amyloid is important in the pathophysiology of Alzheimer’s disease. But, again, unless the impact is unexpectedly huge, I don’t think this quiets those who feel there is more to the story than only a gain of beta amyloid toxicity, though this does support the idea that it plays a role, which should not surprise anyone.”
Dr. Edelmayer said the Alzheimer’s Association has raised the issue of access and cost with Biogen.
“We have had many discussions about their capacity to roll this out,” if aducanumab is approved, she said. “Those who were in the studies will be the first recipients. But Biogen is making plans to move this into the general population if approved, to all patients who meet the diagnostic criteria.”
“There is precedent out there when it comes to doing infusion medicines, and those centers are part of the planning process. In terms of pricing, this is a problem we would be happy to see, because it would mean that we have a treatment. But we’ll cross that bridge when we come to it.”
Secondary analysis results
The new analysis comprised 2,066 patients who had the opportunity to complete the full 18-month trials by March 20, 2019. The full intent-to-treat population of the trials comprised 3,285 patients with mild cognitive impairment caused by Alzheimer’s disease or mild Alzheimer’s disease dementia.
In the secondary analysis of EMERGE’s intent-to-treat population, patients who took the highest dose of aducanumab (10 mg/kg every month) showed 23% lower functional and cognitive decline on the trial’s primary endpoint of Clinical Dementia Rating–Sum of Boxes (CDR-SB) at 18 months when compared with placebo. The rate of decline was slowed by a nonsignificant 14% among users of the lower dose (6 mg/kg monthly).Secondary endpoints for the high-dose group in EMERGE showed 27% slower cognitive decline on the 13-item cognitive subscale of the AD Assessment Scale (ADAS-Cog13) and 40% lower decline in function among patients with mild cognitive impairment based on the MCI version of the AD Cooperative Study–Activities of Daily Living Inventory (ADCS-ADL-MCI).
However, data from the ENGAGE trial, which had the same primary endpoint, were not positive. CDR-SB scores worsened 2% more among high-dose aducanumab users but low-dose users slowed decline by a nonsignificant 12% when compared with placebo.“Exposure to high-dose aducanumab was important for efficacy,” the company noted in a slide set presented at an Oct. 22 investors webcast. “Differences in exposure to high-dose aducanumab largely explain the different results between the futility analysis and the new analysis of this larger dataset, as well as the different results between the two studies.”
In EMERGE, changes in secondary endpoints among patients who had the opportunity to complete the full 18-month trials included:
- Mini Mental State Exam (MMSE): Significant 23% decrease in rate of decline in the high-dose group and nonsignificant 3% increase in decline in the low-dose group.
- ADAS-Cog13: Significant 25% decrease for high-dose users and nonsignificant 10% decrease for low dose.
- ADCS-ADL-MCI: Significant decreases of 46% for high-dose and 20% for low-dose users.
The ENGAGE secondary endpoints of high- vs. low-dose patients who completed the full trials were:
- MMSE: Significant 13% increase, nonsignificant 3% decrease.
- ADAS-Cog13: Nonsignificant 2% decrease, nonsignificant 1% decrease.
- ADCS-ADL-MCI: Significant 12% declines in both dosage groups.
All of the positive cognitive and functional results tracked along with results of amyloid PET imaging and CSF biomarkers. In EMERGE, amyloid plaque binding declined about 27% in the high-dose group and about 16% in the low-dose group, reflecting plaque clearance. Phosphorylated tau in CSF decreased by about 17 pg/mL and 10 pg/mL, respectively, and total tau decreased by 160 pg/mL and 120 pg/mL. Tau decreases indicate a slowing of neuronal damage.
In ENGAGE, amyloid plaque binding decreased by about 24% in the high-dose group and by about 16% in the low-dose group. Phosphorylated tau dropped by about 10 pg/mL and 11 pg/mL, respectively. But total tau dropped more in the low-dose group than in the high-dose group (about –100 pg/mL vs. –20 pg/mL).
Biogen said the amyloid PET imaging biomarker results and CDR-SB scores in both studies were consistent with each other in a subset of patients with “sufficient exposure to 10 mg/kg,” which was defined as “10 or more uninterrupted 10-mg/kg dosing intervals at steady-state.”
The most common adverse events were amyloid related imaging abnormalities–edema (ARIA-E), which occurred in 35%, and headache in 20%. The majority of patients who experienced ARIA-E (74%) were asymptomatic; episodes generally resolved within 4-16 weeks, typically without long-term sequelae.
Dr. Aisen is a consultant for Biogen and on the aducanumab steering committee. None of the other sources in this article have any financial relationship with Biogen or Eisai.
This article was updated 10/23/19.
Native Americans appear to be at increased risk for AFib
Over 4 years, atrial fibrillation (AFib) developed significantly more often in a group of Native Americans men than it did among other racial and ethnic groups, a large longitudinal cohort study has found.

The overall incidence among Native Americans was 7.49 per 1,000 person-years – significantly higher than the incidence in a comparator cohort of black, white, Asian, and Hispanic men, Gregory M. Marcus, MD, of the University of California, San Francisco, and colleagues wrote in a research letter published in Circulation.
“We were surprised to find that American Indians experienced a higher risk of atrial fibrillation, compared to every other racial and ethnic group,” Dr. Marcus said in a press release that accompanied the study. “Understanding the mechanisms and factors by which American Indians experience this higher risk may help investigators better understand the fundamental causes of atrial fibrillation that prove useful to everyone at risk for AFib, regardless of their race or ethnicity.”
The team plumbed the Healthcare Cost and Utilization Project (HCUP) California state databases for information on more than 16 million cases of AFib that occurred during 2005-2011. Native Americans comprised just 0.6% of the cohort. Most of the patients (57.2%) were white; 8% were black, 25.6% Hispanic, and 8.6% Asian. After targeting only new-onset cases, there were 344,469 incident AFib episodes over a median follow-up of 4.1 years.
The overall incidence of AFib in Native Americans was 7.49 per 1,000 person-years, significantly higher than the 6.89 per 1000 person-years observed in the rest of the cohort ( P less than .0001). The difference remained significant even after the team controlled for age, sex, income, and heart and other diseases. Nor was it altered by a sensitivity analysis that controlled for place of presentation and patients who were aged at least 35 years with at least two encounters with medical facilities.
In an interaction analysis, the increased risk appeared to be driven by higher rates of diabetes and chronic kidney disease, the authors wrote.
“This may suggest that the presence of these two processes contributes some pathophysiology related to AF[ib] risk that may be similar to the heightened risk inherent among American Indians,” they wrote. “It is also important to note that there was no evidence of any other statistically significant interactions despite the inclusion of millions of patients.”
Supporting data for these associations were not included in the research letter.
The authors noted some limitations of their study. Race or ethnicity were self-reported and could not be independently confirmed, so there was no way to tease out the effects in multiracial patients. Also, the database didn’t record outpatient encounters, which might result in some selection bias.
“Last, because this was an observational study, these results should not be interpreted as evidence of causal effect,” they noted.
“In conclusion, we observed that American Indians had a higher risk of atrial fibrillation, compared with all other racial and ethnic group. The heightened risk … in American Indians persisted after multivariable adjustment for known conventional confounders and mediators, suggesting that an unidentified characteristic, including possible genetic or environmental factors, may be responsible,” the investigators wrote.
The HCUP database is supported by the Agency for Healthcare Research and Quality. Dr. Marcus reported receiving research support from Jawbone, Medtronic, Eight, and Baylis Medical, and is a consultant for and holds equity in InCarda Therapeutics. The other authors reported no conflicts of interest.
SOURCE: Marcus GM et al. Circulation. 2019 Oct 21. doi:10.1161/CIRCULATIONAHA.119.042882.
Over 4 years, atrial fibrillation (AFib) developed significantly more often in a group of Native Americans men than it did among other racial and ethnic groups, a large longitudinal cohort study has found.
The overall incidence among Native Americans was 7.49 per 1,000 person-years – significantly higher than the incidence in a comparator cohort of black, white, Asian, and Hispanic men, Gregory M. Marcus, MD, of the University of California, San Francisco, and colleagues wrote in a research letter published in Circulation.
“We were surprised to find that American Indians experienced a higher risk of atrial fibrillation, compared to every other racial and ethnic group,” Dr. Marcus said in a press release that accompanied the study. “Understanding the mechanisms and factors by which American Indians experience this higher risk may help investigators better understand the fundamental causes of atrial fibrillation that prove useful to everyone at risk for AFib, regardless of their race or ethnicity.”
The team plumbed the Healthcare Cost and Utilization Project (HCUP) California state databases for information on more than 16 million cases of AFib that occurred during 2005-2011. Native Americans comprised just 0.6% of the cohort. Most of the patients (57.2%) were white; 8% were black, 25.6% Hispanic, and 8.6% Asian. After targeting only new-onset cases, there were 344,469 incident AFib episodes over a median follow-up of 4.1 years.
The overall incidence of AFib in Native Americans was 7.49 per 1,000 person-years, significantly higher than the 6.89 per 1000 person-years observed in the rest of the cohort ( P less than .0001). The difference remained significant even after the team controlled for age, sex, income, and heart and other diseases. Nor was it altered by a sensitivity analysis that controlled for place of presentation and patients who were aged at least 35 years with at least two encounters with medical facilities.
In an interaction analysis, the increased risk appeared to be driven by higher rates of diabetes and chronic kidney disease, the authors wrote.
“This may suggest that the presence of these two processes contributes some pathophysiology related to AF[ib] risk that may be similar to the heightened risk inherent among American Indians,” they wrote. “It is also important to note that there was no evidence of any other statistically significant interactions despite the inclusion of millions of patients.”
Supporting data for these associations were not included in the research letter.
The authors noted some limitations of their study. Race or ethnicity were self-reported and could not be independently confirmed, so there was no way to tease out the effects in multiracial patients. Also, the database didn’t record outpatient encounters, which might result in some selection bias.
“Last, because this was an observational study, these results should not be interpreted as evidence of causal effect,” they noted.
“In conclusion, we observed that American Indians had a higher risk of atrial fibrillation, compared with all other racial and ethnic group. The heightened risk … in American Indians persisted after multivariable adjustment for known conventional confounders and mediators, suggesting that an unidentified characteristic, including possible genetic or environmental factors, may be responsible,” the investigators wrote.
The HCUP database is supported by the Agency for Healthcare Research and Quality. Dr. Marcus reported receiving research support from Jawbone, Medtronic, Eight, and Baylis Medical, and is a consultant for and holds equity in InCarda Therapeutics. The other authors reported no conflicts of interest.
SOURCE: Marcus GM et al. Circulation. 2019 Oct 21. doi:10.1161/CIRCULATIONAHA.119.042882.
Over 4 years, atrial fibrillation (AFib) developed significantly more often in a group of Native Americans men than it did among other racial and ethnic groups, a large longitudinal cohort study has found.
The overall incidence among Native Americans was 7.49 per 1,000 person-years – significantly higher than the incidence in a comparator cohort of black, white, Asian, and Hispanic men, Gregory M. Marcus, MD, of the University of California, San Francisco, and colleagues wrote in a research letter published in Circulation.
“We were surprised to find that American Indians experienced a higher risk of atrial fibrillation, compared to every other racial and ethnic group,” Dr. Marcus said in a press release that accompanied the study. “Understanding the mechanisms and factors by which American Indians experience this higher risk may help investigators better understand the fundamental causes of atrial fibrillation that prove useful to everyone at risk for AFib, regardless of their race or ethnicity.”
The team plumbed the Healthcare Cost and Utilization Project (HCUP) California state databases for information on more than 16 million cases of AFib that occurred during 2005-2011. Native Americans comprised just 0.6% of the cohort. Most of the patients (57.2%) were white; 8% were black, 25.6% Hispanic, and 8.6% Asian. After targeting only new-onset cases, there were 344,469 incident AFib episodes over a median follow-up of 4.1 years.
The overall incidence of AFib in Native Americans was 7.49 per 1,000 person-years, significantly higher than the 6.89 per 1000 person-years observed in the rest of the cohort ( P less than .0001). The difference remained significant even after the team controlled for age, sex, income, and heart and other diseases. Nor was it altered by a sensitivity analysis that controlled for place of presentation and patients who were aged at least 35 years with at least two encounters with medical facilities.
In an interaction analysis, the increased risk appeared to be driven by higher rates of diabetes and chronic kidney disease, the authors wrote.
“This may suggest that the presence of these two processes contributes some pathophysiology related to AF[ib] risk that may be similar to the heightened risk inherent among American Indians,” they wrote. “It is also important to note that there was no evidence of any other statistically significant interactions despite the inclusion of millions of patients.”
Supporting data for these associations were not included in the research letter.
The authors noted some limitations of their study. Race or ethnicity were self-reported and could not be independently confirmed, so there was no way to tease out the effects in multiracial patients. Also, the database didn’t record outpatient encounters, which might result in some selection bias.
“Last, because this was an observational study, these results should not be interpreted as evidence of causal effect,” they noted.
“In conclusion, we observed that American Indians had a higher risk of atrial fibrillation, compared with all other racial and ethnic group. The heightened risk … in American Indians persisted after multivariable adjustment for known conventional confounders and mediators, suggesting that an unidentified characteristic, including possible genetic or environmental factors, may be responsible,” the investigators wrote.
The HCUP database is supported by the Agency for Healthcare Research and Quality. Dr. Marcus reported receiving research support from Jawbone, Medtronic, Eight, and Baylis Medical, and is a consultant for and holds equity in InCarda Therapeutics. The other authors reported no conflicts of interest.
SOURCE: Marcus GM et al. Circulation. 2019 Oct 21. doi:10.1161/CIRCULATIONAHA.119.042882.
FROM CIRCULATION
Closed-loop delivery system increases time in target glucose range
Patients with type 1 diabetes who used a closed-loop insulin delivery system spent a greater percentage of time in their target blood glucose range, compared with patients using a sensor-augmented insulin pump.
The significant, between-group, mean-adjusted difference of 11 percentage points between the two groups translated into the closed-loop patients spending an additional 2.6 hr/day in the target range of 70-180 mg/dL, Susan A. Brown, MD, and colleagues wrote in the New England Journal of Medicine.
Most of the benefit occurred in the early morning hours, at 5 am, when 89% of patients using the closed-loop system remained in the target range, compared with 62% of those using the pump system, said Dr. Brown of the University of Virginia, Charlottesville, and colleagues.
The randomized study comprised 168 patients with a mean age of 33 years, although the age range was wide (14-71 years). The patients had a mean disease duration of 16 years. Their baseline glycated hemoglobin level ranged between 5.4% and 10.6%. At enrollment, 79% of patients used insulin pumps, and 21% used multiple daily insulin injections; 70% were using continuous glucose monitoring, of whom 86% were using pumps. Patients in both groups had follow-up visits at 2, 6, 13, and 26 weeks.
There were no dropouts in this study – 100% of clinical and telephone follow-ups were completed.
During the 6-month trial, the mean percentage of time spent in the glucose target range rose from 61% at baseline to 71% in the closed-loop group, but remained unchanged at 59% in the pump group. The difference became apparent very early in the study and remained consistent over its course.
“The mean percentage of time that the glucose level was in the target range was 70% in the closed-loop group and 59% in the control [pump] group during the daytime (6 a.m. to midnight) and 76% and 59%, respectively, during the nighttime (midnight to 6 am) ... and the greatest differences in the mean glucose level occurred at 5 a.m. and 6 a.m. [139 mg/dL in the closed-loop group vs. 166 mg/dL in the control group at both time points]. This diurnal pattern is a result of the increased aggressiveness of the algorithm to meet a lower glucose target during the second half of the night,” the authors noted.
The closed-loop system was also better than the pump system on all secondary endpoints, including the following:
- Glycated hemoglobin at 26 weeks: mean difference, –0.33 percentage points.
- Percentage of time with glucose higher than 180 mg/dL: mean difference, –10 percentage points (a difference of 2.4 hr/day).
- Percentage of time with glucose less than 70 mg/dL: mean difference, –0.88 percentage points (a difference of 13 min/day).
The other secondary endpoints – mean glucose level and mean glycated hemoglobin level – were also significantly better in those using the closed-loop system.
The benefits “consistently favored the closed-loop system across a broad range of baseline characteristics, including age, sex, body mass index, income, educational level, insulin pump or infection use, previous use of continuous glucose monitor, and glycated hemoglobin,” the authors said.
There were 17 adverse events in 16 patients in the closed-loop group, and 2 events in 2 patients in the pump group, but no incidents of severe hypoglycemia. One person in the closed-loop system experienced ketoacidosis because of a failure in the pump infusion set. There were 13 hyperglycemic or ketosis episodes in 12 patients in the closed-loop group, and 2 in 2 patients the pump group, but none of them met the criteria for diabetic ketoacidosis. All of these episodes were deemed related to infusion set failures.
There were three serious adverse events in the closed-loop group, and none related to the device. Blood ketones exceeding 1 mmol/L occurred in 11 closed-loop patients and 8 pump patients.
The results should be interpreted with consideration of potential group bias, the authors noted. “In our trial, 70% of the patients were using a continuous glucose monitor, and 79% were using an insulin pump at the time of enrollment, percentages that are substantially higher than the reported usage in the general population of type 1 diabetes. These data may reflect an interest in and willingness to use a closed-loop system among patients who were already using devices as part of diabetes management.”
Dr. Brown reported receiving grant support from Tandem Diabetes Care, Dexcom, and Roche Diagnostics. Other authors reported a range of support from numerous pharmaceutical and medical technology companies. Several reported patents on diabetes-related devices.
SOURCE: Brown SA et al. New Engl J Med. 2019 Oct 16. doi: 10.1056/NEJMoa1907863.
Patients with type 1 diabetes who used a closed-loop insulin delivery system spent a greater percentage of time in their target blood glucose range, compared with patients using a sensor-augmented insulin pump.
The significant, between-group, mean-adjusted difference of 11 percentage points between the two groups translated into the closed-loop patients spending an additional 2.6 hr/day in the target range of 70-180 mg/dL, Susan A. Brown, MD, and colleagues wrote in the New England Journal of Medicine.
Most of the benefit occurred in the early morning hours, at 5 am, when 89% of patients using the closed-loop system remained in the target range, compared with 62% of those using the pump system, said Dr. Brown of the University of Virginia, Charlottesville, and colleagues.
The randomized study comprised 168 patients with a mean age of 33 years, although the age range was wide (14-71 years). The patients had a mean disease duration of 16 years. Their baseline glycated hemoglobin level ranged between 5.4% and 10.6%. At enrollment, 79% of patients used insulin pumps, and 21% used multiple daily insulin injections; 70% were using continuous glucose monitoring, of whom 86% were using pumps. Patients in both groups had follow-up visits at 2, 6, 13, and 26 weeks.
There were no dropouts in this study – 100% of clinical and telephone follow-ups were completed.
During the 6-month trial, the mean percentage of time spent in the glucose target range rose from 61% at baseline to 71% in the closed-loop group, but remained unchanged at 59% in the pump group. The difference became apparent very early in the study and remained consistent over its course.
“The mean percentage of time that the glucose level was in the target range was 70% in the closed-loop group and 59% in the control [pump] group during the daytime (6 a.m. to midnight) and 76% and 59%, respectively, during the nighttime (midnight to 6 am) ... and the greatest differences in the mean glucose level occurred at 5 a.m. and 6 a.m. [139 mg/dL in the closed-loop group vs. 166 mg/dL in the control group at both time points]. This diurnal pattern is a result of the increased aggressiveness of the algorithm to meet a lower glucose target during the second half of the night,” the authors noted.
The closed-loop system was also better than the pump system on all secondary endpoints, including the following:
- Glycated hemoglobin at 26 weeks: mean difference, –0.33 percentage points.
- Percentage of time with glucose higher than 180 mg/dL: mean difference, –10 percentage points (a difference of 2.4 hr/day).
- Percentage of time with glucose less than 70 mg/dL: mean difference, –0.88 percentage points (a difference of 13 min/day).
The other secondary endpoints – mean glucose level and mean glycated hemoglobin level – were also significantly better in those using the closed-loop system.
The benefits “consistently favored the closed-loop system across a broad range of baseline characteristics, including age, sex, body mass index, income, educational level, insulin pump or infection use, previous use of continuous glucose monitor, and glycated hemoglobin,” the authors said.
There were 17 adverse events in 16 patients in the closed-loop group, and 2 events in 2 patients in the pump group, but no incidents of severe hypoglycemia. One person in the closed-loop system experienced ketoacidosis because of a failure in the pump infusion set. There were 13 hyperglycemic or ketosis episodes in 12 patients in the closed-loop group, and 2 in 2 patients the pump group, but none of them met the criteria for diabetic ketoacidosis. All of these episodes were deemed related to infusion set failures.
There were three serious adverse events in the closed-loop group, and none related to the device. Blood ketones exceeding 1 mmol/L occurred in 11 closed-loop patients and 8 pump patients.
The results should be interpreted with consideration of potential group bias, the authors noted. “In our trial, 70% of the patients were using a continuous glucose monitor, and 79% were using an insulin pump at the time of enrollment, percentages that are substantially higher than the reported usage in the general population of type 1 diabetes. These data may reflect an interest in and willingness to use a closed-loop system among patients who were already using devices as part of diabetes management.”
Dr. Brown reported receiving grant support from Tandem Diabetes Care, Dexcom, and Roche Diagnostics. Other authors reported a range of support from numerous pharmaceutical and medical technology companies. Several reported patents on diabetes-related devices.
SOURCE: Brown SA et al. New Engl J Med. 2019 Oct 16. doi: 10.1056/NEJMoa1907863.
Patients with type 1 diabetes who used a closed-loop insulin delivery system spent a greater percentage of time in their target blood glucose range, compared with patients using a sensor-augmented insulin pump.
The significant, between-group, mean-adjusted difference of 11 percentage points between the two groups translated into the closed-loop patients spending an additional 2.6 hr/day in the target range of 70-180 mg/dL, Susan A. Brown, MD, and colleagues wrote in the New England Journal of Medicine.
Most of the benefit occurred in the early morning hours, at 5 am, when 89% of patients using the closed-loop system remained in the target range, compared with 62% of those using the pump system, said Dr. Brown of the University of Virginia, Charlottesville, and colleagues.
The randomized study comprised 168 patients with a mean age of 33 years, although the age range was wide (14-71 years). The patients had a mean disease duration of 16 years. Their baseline glycated hemoglobin level ranged between 5.4% and 10.6%. At enrollment, 79% of patients used insulin pumps, and 21% used multiple daily insulin injections; 70% were using continuous glucose monitoring, of whom 86% were using pumps. Patients in both groups had follow-up visits at 2, 6, 13, and 26 weeks.
There were no dropouts in this study – 100% of clinical and telephone follow-ups were completed.
During the 6-month trial, the mean percentage of time spent in the glucose target range rose from 61% at baseline to 71% in the closed-loop group, but remained unchanged at 59% in the pump group. The difference became apparent very early in the study and remained consistent over its course.
“The mean percentage of time that the glucose level was in the target range was 70% in the closed-loop group and 59% in the control [pump] group during the daytime (6 a.m. to midnight) and 76% and 59%, respectively, during the nighttime (midnight to 6 am) ... and the greatest differences in the mean glucose level occurred at 5 a.m. and 6 a.m. [139 mg/dL in the closed-loop group vs. 166 mg/dL in the control group at both time points]. This diurnal pattern is a result of the increased aggressiveness of the algorithm to meet a lower glucose target during the second half of the night,” the authors noted.
The closed-loop system was also better than the pump system on all secondary endpoints, including the following:
- Glycated hemoglobin at 26 weeks: mean difference, –0.33 percentage points.
- Percentage of time with glucose higher than 180 mg/dL: mean difference, –10 percentage points (a difference of 2.4 hr/day).
- Percentage of time with glucose less than 70 mg/dL: mean difference, –0.88 percentage points (a difference of 13 min/day).
The other secondary endpoints – mean glucose level and mean glycated hemoglobin level – were also significantly better in those using the closed-loop system.
The benefits “consistently favored the closed-loop system across a broad range of baseline characteristics, including age, sex, body mass index, income, educational level, insulin pump or infection use, previous use of continuous glucose monitor, and glycated hemoglobin,” the authors said.
There were 17 adverse events in 16 patients in the closed-loop group, and 2 events in 2 patients in the pump group, but no incidents of severe hypoglycemia. One person in the closed-loop system experienced ketoacidosis because of a failure in the pump infusion set. There were 13 hyperglycemic or ketosis episodes in 12 patients in the closed-loop group, and 2 in 2 patients the pump group, but none of them met the criteria for diabetic ketoacidosis. All of these episodes were deemed related to infusion set failures.
There were three serious adverse events in the closed-loop group, and none related to the device. Blood ketones exceeding 1 mmol/L occurred in 11 closed-loop patients and 8 pump patients.
The results should be interpreted with consideration of potential group bias, the authors noted. “In our trial, 70% of the patients were using a continuous glucose monitor, and 79% were using an insulin pump at the time of enrollment, percentages that are substantially higher than the reported usage in the general population of type 1 diabetes. These data may reflect an interest in and willingness to use a closed-loop system among patients who were already using devices as part of diabetes management.”
Dr. Brown reported receiving grant support from Tandem Diabetes Care, Dexcom, and Roche Diagnostics. Other authors reported a range of support from numerous pharmaceutical and medical technology companies. Several reported patents on diabetes-related devices.
SOURCE: Brown SA et al. New Engl J Med. 2019 Oct 16. doi: 10.1056/NEJMoa1907863.
FROM NEW ENGLAND JOURNAL OF MEDICINE
Poor neonatal outcomes tied to excessive, insufficient weight gain during twin pregnancies
Lisa M. Bodnar, PhD, and colleagues determined.

The risks of cesarean section and neonatal death were elevated for those mothers who were overweight before pregnancy and then gained too much. But infants of underweight women who didn’t gain enough faced risks as well, wrote Dr. Bodnar of the University of Pittsburgh and associates in Obstetrics & Gynecology.
Among the most severely overweight women (obesity grade 2 or 3) who gained the most weight (43 kg) at 37 weeks’ gestation, there were 6 fewer small-for-gestational-age (SGA) infants per 100 births, but 14 more large-for-gestational-age (LGA) infants, 4 more cesarean deliveries, and 2 more neonatal deaths per 100 births. By contrast, among the most severely underweight women who gained the least amount of weight (9 kg), there were 18 more SGA infants, 3 fewer LGA infants, and 11 fewer cesareans, but 6 more preterm births before 32 weeks’ gestation.
The same U-shaped pattern also occurred within the individual weight categories. For example, compared with the outcomes among the most underweight women who gained least, among underweight women who gained the most (37 kg), there were eight fewer SGA infants, but four more LGA infants, 16 excess preterm births, and 9 excess infant deaths.
“If the associations we observed are even partially reflective of causality, targeted modification of pregnancy weight gain in women carrying twins might improve pregnancy outcomes,” wrote Dr. Bodnar and her team. “Data on a wide range of short- and long-term outcomes and information on the relative seriousness of these outcomes are needed to determine optimal gestational weight gain ranges for twin pregnancies.”
The cohort comprised 54,836 live-born twins from 27,723 twin pregnancies who were included in the MOMs database maintained by the University of Pennsylvania, Philadelphia. The population-based study tracks maternal obesity, gestational weight gain, and adverse birth outcomes. The information came from infant birth and death vital statistics records from 2003 to 2013.
However, this very source puts the findings in some degree of uncertainty, Ozhan Turan, MD, said in an interview.
“It’s a very nice study, and the statistics are very well done,” said Dr. Turan, who is the director of fetal therapy and complex obstetric surgery at the University of Maryland School of Medicine. “But that kind of data has pitfalls that are unavoidable. For example, they don’t have access to maternal medical comorbidities which are mostly related to the outcome, particularly gestational diabetes and preeclampsia. They also don’t have the information on chorionicity – and we know that monochorionic twins face much greater risk for these outcomes than dichorionic twins.”
The investigators calculated total gestational weight gain by subtracting prepregnancy weight from maternal weight at delivery. The analysis controlled for race and ethnicity, education, neonatal care, level of birth facility, parity, payment at delivery, smoking during pregnancy, marital status, year of birth, height, maternal age, preexisting diabetes or hypertension, infertility treatment, neonatal sex, and racial composition of neighborhood, as a proxy of neighborhood-level socioeconomic status. Approximately 16% of mothers received infertility treatment.
Of the cohort, 3% were underweight, 48% were normal weight, 24% were overweight, 13% were grade 1 obese, 7% grade 2 obese, and 5% grade 3 obese.
“Pregnancy weight gain was negatively associated with SGA and positively associated with LGA and cesarean delivery in all [body mass index] groups. For example, among normal-weight women, compared with a pregnancy weight gain equivalent to 20 kg at 37 weeks’ of gestation, a weight gain of 27 kg at 37 weeks’ of gestation was associated with 2.2 fewer cases of SGA but 2.9 more cases of LGA and 3.7 more cases of cesarean delivery,” Dr. Bodnar and associates wrote.
The investigators found that “weight gains well above or well below the [Institute of Medicine] provisional guidelines (less than 14 kg or more than 27 kg in underweight or normal-weight women, less than 11 kg or more than 28 kg in overweight women, and less than 6.4 kg or more than 26 kg in women with obesity) were associated with the highest risk of adverse outcomes.”
“I would not say this is practice-changing information,” said Dr. Turan. “We already know all this. What would be very helpful is an algorithm to tell us, if a patient is pregnant with twins, this is the amount of weight you have to gain.”
For overweight patients, Dr. Turan tries to impart the key message of moderate or slight weight gain, according to prepregnancy body mass index. For underweight patients, the picture is a bit more complex.
“There are not that many who are underweight before pregnancy, so first thing I look for is the reason a woman is underweight. Is she just not eating properly? Is there a drug dependence issue, alcohol dependence, HIV? Is there smoking? A gut problem that causes malnutrition. You can’t just say ‘eat more.’ That does not solve the problem. We need to find out why she is underweight and fix that first,” said Dr. Turan.
Neither Dr. Bodnar nor Dr. Turan had any relevant financial disclosures. One coauthor disclosed her institution received funds from the University of Pittsburgh. The study was funded by National Institutes of Health grants.
SOURCE: Bodnar LM et al. Obstet Gynecol. 2019;134:1075-86.
Lisa M. Bodnar, PhD, and colleagues determined.
The risks of cesarean section and neonatal death were elevated for those mothers who were overweight before pregnancy and then gained too much. But infants of underweight women who didn’t gain enough faced risks as well, wrote Dr. Bodnar of the University of Pittsburgh and associates in Obstetrics & Gynecology.
Among the most severely overweight women (obesity grade 2 or 3) who gained the most weight (43 kg) at 37 weeks’ gestation, there were 6 fewer small-for-gestational-age (SGA) infants per 100 births, but 14 more large-for-gestational-age (LGA) infants, 4 more cesarean deliveries, and 2 more neonatal deaths per 100 births. By contrast, among the most severely underweight women who gained the least amount of weight (9 kg), there were 18 more SGA infants, 3 fewer LGA infants, and 11 fewer cesareans, but 6 more preterm births before 32 weeks’ gestation.
The same U-shaped pattern also occurred within the individual weight categories. For example, compared with the outcomes among the most underweight women who gained least, among underweight women who gained the most (37 kg), there were eight fewer SGA infants, but four more LGA infants, 16 excess preterm births, and 9 excess infant deaths.
“If the associations we observed are even partially reflective of causality, targeted modification of pregnancy weight gain in women carrying twins might improve pregnancy outcomes,” wrote Dr. Bodnar and her team. “Data on a wide range of short- and long-term outcomes and information on the relative seriousness of these outcomes are needed to determine optimal gestational weight gain ranges for twin pregnancies.”
The cohort comprised 54,836 live-born twins from 27,723 twin pregnancies who were included in the MOMs database maintained by the University of Pennsylvania, Philadelphia. The population-based study tracks maternal obesity, gestational weight gain, and adverse birth outcomes. The information came from infant birth and death vital statistics records from 2003 to 2013.
However, this very source puts the findings in some degree of uncertainty, Ozhan Turan, MD, said in an interview.
“It’s a very nice study, and the statistics are very well done,” said Dr. Turan, who is the director of fetal therapy and complex obstetric surgery at the University of Maryland School of Medicine. “But that kind of data has pitfalls that are unavoidable. For example, they don’t have access to maternal medical comorbidities which are mostly related to the outcome, particularly gestational diabetes and preeclampsia. They also don’t have the information on chorionicity – and we know that monochorionic twins face much greater risk for these outcomes than dichorionic twins.”
The investigators calculated total gestational weight gain by subtracting prepregnancy weight from maternal weight at delivery. The analysis controlled for race and ethnicity, education, neonatal care, level of birth facility, parity, payment at delivery, smoking during pregnancy, marital status, year of birth, height, maternal age, preexisting diabetes or hypertension, infertility treatment, neonatal sex, and racial composition of neighborhood, as a proxy of neighborhood-level socioeconomic status. Approximately 16% of mothers received infertility treatment.
Of the cohort, 3% were underweight, 48% were normal weight, 24% were overweight, 13% were grade 1 obese, 7% grade 2 obese, and 5% grade 3 obese.
“Pregnancy weight gain was negatively associated with SGA and positively associated with LGA and cesarean delivery in all [body mass index] groups. For example, among normal-weight women, compared with a pregnancy weight gain equivalent to 20 kg at 37 weeks’ of gestation, a weight gain of 27 kg at 37 weeks’ of gestation was associated with 2.2 fewer cases of SGA but 2.9 more cases of LGA and 3.7 more cases of cesarean delivery,” Dr. Bodnar and associates wrote.
The investigators found that “weight gains well above or well below the [Institute of Medicine] provisional guidelines (less than 14 kg or more than 27 kg in underweight or normal-weight women, less than 11 kg or more than 28 kg in overweight women, and less than 6.4 kg or more than 26 kg in women with obesity) were associated with the highest risk of adverse outcomes.”
“I would not say this is practice-changing information,” said Dr. Turan. “We already know all this. What would be very helpful is an algorithm to tell us, if a patient is pregnant with twins, this is the amount of weight you have to gain.”
For overweight patients, Dr. Turan tries to impart the key message of moderate or slight weight gain, according to prepregnancy body mass index. For underweight patients, the picture is a bit more complex.
“There are not that many who are underweight before pregnancy, so first thing I look for is the reason a woman is underweight. Is she just not eating properly? Is there a drug dependence issue, alcohol dependence, HIV? Is there smoking? A gut problem that causes malnutrition. You can’t just say ‘eat more.’ That does not solve the problem. We need to find out why she is underweight and fix that first,” said Dr. Turan.
Neither Dr. Bodnar nor Dr. Turan had any relevant financial disclosures. One coauthor disclosed her institution received funds from the University of Pittsburgh. The study was funded by National Institutes of Health grants.
SOURCE: Bodnar LM et al. Obstet Gynecol. 2019;134:1075-86.
Lisa M. Bodnar, PhD, and colleagues determined.
The risks of cesarean section and neonatal death were elevated for those mothers who were overweight before pregnancy and then gained too much. But infants of underweight women who didn’t gain enough faced risks as well, wrote Dr. Bodnar of the University of Pittsburgh and associates in Obstetrics & Gynecology.
Among the most severely overweight women (obesity grade 2 or 3) who gained the most weight (43 kg) at 37 weeks’ gestation, there were 6 fewer small-for-gestational-age (SGA) infants per 100 births, but 14 more large-for-gestational-age (LGA) infants, 4 more cesarean deliveries, and 2 more neonatal deaths per 100 births. By contrast, among the most severely underweight women who gained the least amount of weight (9 kg), there were 18 more SGA infants, 3 fewer LGA infants, and 11 fewer cesareans, but 6 more preterm births before 32 weeks’ gestation.
The same U-shaped pattern also occurred within the individual weight categories. For example, compared with the outcomes among the most underweight women who gained least, among underweight women who gained the most (37 kg), there were eight fewer SGA infants, but four more LGA infants, 16 excess preterm births, and 9 excess infant deaths.
“If the associations we observed are even partially reflective of causality, targeted modification of pregnancy weight gain in women carrying twins might improve pregnancy outcomes,” wrote Dr. Bodnar and her team. “Data on a wide range of short- and long-term outcomes and information on the relative seriousness of these outcomes are needed to determine optimal gestational weight gain ranges for twin pregnancies.”
The cohort comprised 54,836 live-born twins from 27,723 twin pregnancies who were included in the MOMs database maintained by the University of Pennsylvania, Philadelphia. The population-based study tracks maternal obesity, gestational weight gain, and adverse birth outcomes. The information came from infant birth and death vital statistics records from 2003 to 2013.
However, this very source puts the findings in some degree of uncertainty, Ozhan Turan, MD, said in an interview.
“It’s a very nice study, and the statistics are very well done,” said Dr. Turan, who is the director of fetal therapy and complex obstetric surgery at the University of Maryland School of Medicine. “But that kind of data has pitfalls that are unavoidable. For example, they don’t have access to maternal medical comorbidities which are mostly related to the outcome, particularly gestational diabetes and preeclampsia. They also don’t have the information on chorionicity – and we know that monochorionic twins face much greater risk for these outcomes than dichorionic twins.”
The investigators calculated total gestational weight gain by subtracting prepregnancy weight from maternal weight at delivery. The analysis controlled for race and ethnicity, education, neonatal care, level of birth facility, parity, payment at delivery, smoking during pregnancy, marital status, year of birth, height, maternal age, preexisting diabetes or hypertension, infertility treatment, neonatal sex, and racial composition of neighborhood, as a proxy of neighborhood-level socioeconomic status. Approximately 16% of mothers received infertility treatment.
Of the cohort, 3% were underweight, 48% were normal weight, 24% were overweight, 13% were grade 1 obese, 7% grade 2 obese, and 5% grade 3 obese.
“Pregnancy weight gain was negatively associated with SGA and positively associated with LGA and cesarean delivery in all [body mass index] groups. For example, among normal-weight women, compared with a pregnancy weight gain equivalent to 20 kg at 37 weeks’ of gestation, a weight gain of 27 kg at 37 weeks’ of gestation was associated with 2.2 fewer cases of SGA but 2.9 more cases of LGA and 3.7 more cases of cesarean delivery,” Dr. Bodnar and associates wrote.
The investigators found that “weight gains well above or well below the [Institute of Medicine] provisional guidelines (less than 14 kg or more than 27 kg in underweight or normal-weight women, less than 11 kg or more than 28 kg in overweight women, and less than 6.4 kg or more than 26 kg in women with obesity) were associated with the highest risk of adverse outcomes.”
“I would not say this is practice-changing information,” said Dr. Turan. “We already know all this. What would be very helpful is an algorithm to tell us, if a patient is pregnant with twins, this is the amount of weight you have to gain.”
For overweight patients, Dr. Turan tries to impart the key message of moderate or slight weight gain, according to prepregnancy body mass index. For underweight patients, the picture is a bit more complex.
“There are not that many who are underweight before pregnancy, so first thing I look for is the reason a woman is underweight. Is she just not eating properly? Is there a drug dependence issue, alcohol dependence, HIV? Is there smoking? A gut problem that causes malnutrition. You can’t just say ‘eat more.’ That does not solve the problem. We need to find out why she is underweight and fix that first,” said Dr. Turan.
Neither Dr. Bodnar nor Dr. Turan had any relevant financial disclosures. One coauthor disclosed her institution received funds from the University of Pittsburgh. The study was funded by National Institutes of Health grants.
SOURCE: Bodnar LM et al. Obstet Gynecol. 2019;134:1075-86.
FROM OBSTETRICS & GYNECOLOGY