Article
Recalcitrant Folliculitis Decalvans Treatment Outcomes With Biologics and Small Molecule Inhibitors
Tumor necrosis factor inhibitors, Janus kinase inhibitors, phosphodiesterase 4 inhibitors, and monoclonal antibodies have shown success in the...
Article
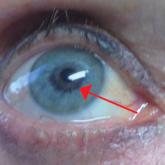
Bimatoprost-Induced Iris Hyperpigmentation: Beauty in the Darkened Eye of the Beholder
Iris hyperpigmentation can occur when bimatoprost eye drops are applied to the eyes for treatment of ocular hypertension and glaucoma, but reports...
Article
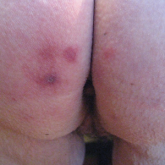
Ice Pack–Induced Perniosis: A Rare and Underrecognized Association
Perniosis, or chilblain, is characterized by skin lesions that occur as an abnormal reaction to exposure to cold and damp conditions. Ice pack–...
Article
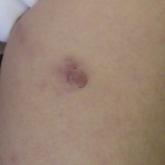
A Rare Case of Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type
Primary cutaneous diffuse large B-cell lymphoma, leg type (DLBCLLT) is a rare, intermediately aggressive form of primary cutaneous B-cell lymphoma...
