User login
Severe Hypertriglyceridemia: A Review
The patient with a markedly high serum triglyceride (TG) level poses an interesting challenge for hospitalists. Hypertriglyceridemia (HTG) is defined as a fasting plasma TG level that is above the 95th percentile for age and sex.1 TG levels are commonly classified into categories according to Adult Treatment Panel III guidelines with desirable levels <150 mg/dL (1.7 mmol/L), borderline levels 150199, high levels 200499 mg/dL, and very high levels >500 mg/dL (5.6 mmol/L).2 A TG level exceeding an arbitrary threshold of >1000 mg/dL (11.3 mmol/L) is referred to as severe HTG. The Lipid Research Clinics Program Prevalence Study found that 1.79 per 10,000 outpatients (<0.02%) had TG levels > 2000 mg/dL.3 Chylomicronemia syndrome occurs when severe HTG is accompanied by 1 or more of the following: symptoms of abdominal pain or acute pancreatitis or physical examination findings such as eruptive xanthomas or lipemia retinalis. There is no TG level above which pancreatitis invariably occurs, making the decision to hospitalize difficult. The goal of this review is to discuss the causes of severe HTG; the clinical assessment, including criteria for hospitalization; and the available treatment options for this infrequent but serious condition. We begin with a clinical case of severe HTG.
CASE PRESENTATION
A 47‐year‐old woman with a history of chronic myelogenous leukemia was admitted to the hospital with a serum triglyceride level of 17,393 mg/dL. Two years prior to admission, she underwent allogenic stem cell transplantation for chronic myelogenous leukemia and has since remained in remission. Six months prior to admission, severe diarrhea from intestinal graft‐versus‐host disease required the use of total parenteral nutrition (TPN) and immunosuppressive therapy consisting of prednisone 20 mg/day, mycophenolate mofetil 250 mg thrice daily, and sirolimus 0.3 mg/day. During treatment with steroids, she developed diabetes mellitus requiring insulin, with a subsequent hemoglobin A1c level of 7.7% (normal, <7%). The serum TG level prior to transplantation was unknown but was 343 mg/dL prior to TPN initiation. One month prior to admission, the diarrhea resolved and TPN was stopped. The TG level was 7463 mg/dL 1 week prior to admission, and despite use of fenofibrate, it rose to 17,393 mg/dL. The patient denied abdominal pain, and did not have abdominal tenderness or eruptive xanthomas. She denied a family history of dyslipidemia or recent medication changes. Given the extreme TG elevation, the lack of response to outpatient treatment and the concern for developing acute pancreatitis, the patient was admitted to the hospital for inpatient TG‐lowering treatment.
Upon admission, serum lipase and amylase were within normal limits, but the blood glucose level was 243 mg/dL. Insulin infusion and oral fenofibrate 145 mg/day was started, and the patient was kept non per os (NPO). Six hours later, despite insulin infusion, the TG level rose to 26,250 mg/dL. Therapeutic plasma exchange (TPE) was performed on 2 consecutive days with a resultant decrease in TG level to 530 mg/dL. The patient was later discharged home on fenofibrate and omega‐3 ethyl esters, her same immunosuppressive and insulin regimen, and instructions for a very low‐fat diet. In the next 3 months, her serum TG level did not rise above 530 mg/dL. The cause of our patient's extreme TG elevation was likely a combination of genetic factors exacerbated by immunosuppressive and glucocorticoid therapy.
This case featured dramatic elevations in serum TG levels that the managing doctors believed merited a hospital admission. Management of patients with severe HTG first requires an understanding of TG metabolism.
ETIOLOGY
Serum TGs produced by the liver are carried by very low‐density lipoproteins (VLDLs), whereas TGs derived from dietary fat are carried by chylomicrons. Both chylomicrons and VLDLs are hydrolyzed by the same enzymelipoprotein lipase (LPL). TGs are hydrolyzed into fatty acids for uptake by muscle and adipose tissue, whereas remnants of VLDL and chylomicrons are removed by the liver. More details on TG pathophysiology may be found in a recent review.4 When LPL is saturated with VLDL, ingestion of a fatty meal may cause chylomicrons to linger in circulation for days instead of hours. Asking the laboratory to spin down the blood of a patient with severe HTG and keep the test tube upright at 4C may reveal a large creamy supernatant layer demonstrating chylomicronemia.
A fasting TG level drawn 12 hours after the last meal reflects hepatic TG production. Although a nonfasting TG level may reflect postprandial chylomicrons, values above 1000 mg/dL strongly suggest true HTG, particularly in the setting of acute pancreatitis. Treatment should not be delayed to obtain a fasting TG level.
HTG may result from increased VLDL production, reduced VLDL/chylomicron clearance, or more likely a combination of the two. The causes of these metabolic derangements are classified as primary (genetic) or secondary (acquired) (Table 1). In adult patients, HTG is usually the result of a combination of primary and secondary causes. A study of 123 patients with TG levels >2000 mg/dL found that all patients had a primary metabolic defect and 110/123 had a coexistent secondary cause.3 An underlying genetic lipoprotein metabolism derangement is often clinically silent until coupled with a secondary cause of HTG that together raise TG levels high enough to cause the chylomicronemia syndrome.
|
| Primary |
| Familial lipid disorders |
| Lipoprotein pattern type I |
| Familial chylomicronemia |
| Deficiency in LPL and/or apo‐CII |
| Autosomal recessive; presents in childhood |
| Rare functional disorders in LPL |
| Lipoprotein pattern type III |
| Familial dysbetalipoproteinemia |
| Inadequate VLDL clearance from apo‐E2 |
| Autosomal recessive; presents in adulthood |
| Lipoprotein pattern type IV |
| Familial HTG: increased VLDL |
| Autosomal dominant; presents in adulthood |
| Familial combined hyperlipidemia |
| Multiple phenotypes seen; increased apo‐B levels |
| Lipoprotein pattern type V |
| Mixed HTG: increased VLDL and chylomicrons; presents in adulthood |
| Secondary |
| Disease |
| Poorly controlled diabetes mellitus; hypothyroid; SLE; Cushing syndrome; HIV infection; sarcoid multiple myeloma; obesity; renal disease (nephrotic) |
| Disorder of metabolism |
| Pregnancy |
| Diet |
| Excessive alcohol, especially with high‐fat diet |
| Drugs |
| Estrogen; tamoxifen; glucocorticoids; protease inhibitors; nonselective beta‐blockers; propofol; isotretinoin; some antipsychotic medications (clozapine, olanzapine); tacrolimus; sirolimus; cyclosporine; bexarotene; all‐trans retinoic acid; L‐asparaginase; interferon‐ |
The most common primary cause of HTG in adults is familial HTG, an autosomal dominant condition with a population prevalence ranging from 1%2% to 5%10% and age‐dependent penetrance.5, 6 Other genetic causes are much rarer, such as LPL deficiency (1 in 1 million patients), apolipoprotein‐CII, and other mutations resulting in impaired binding to LPL.79 Primary causes of HTG are often listed as Fredrickson phenotypes (Table 1). Recent genome‐wide association studies reveal a complex polygenic basis to the Fredrickson categories and suggest additional undefined genes or nongenetic factors may significantly contribute to the final phenotype.10 Diagnosis of familial lipid disorders requires an accurate family history that may be difficult to obtain.
Secondary causes of HTG can be categorized using a four‐D mnemonic: Diseases, Diet, Disorder of Metabolism, and Drugs.11 The most common condition associated with HTG is obesity.6 The mechanism between obesity and HTG is complex and likely involves an increase in fatty acid flux from adipose tissue to other tissues and insulin resistance.12 Asking whether a patient's current weight is close to the heaviest lifetime weight is a clue to diagnosing obesity‐driven HTG. One case series of hypertriglyceridemic pancreatitis found that diabetes or excessive alcohol intake account for the majority of secondary causes of HTG.13 The cause of HTG among patients with diabetes is multifactorial: insulin deficiency reduces LPL levels (insulin is required for synthesis of LPL), whereas insulin resistance attenuates the ability of insulin to decrease hepatic cholesterol synthesis and thus increases hepatic secretion of VLDL.14 Alcohol impairs lipolysis and increases VLDL production that can lead to severe HTG, particularly in those patients with an underlying functional deficiency in LPL. Other secondary etiologies of severe HTG may be elicited through careful attention to medical and medication history.
CLINICAL ASSESSMENT
Table 2 proposes a reasonable initial assessment of the history and physical and laboratory tests in patients with severe HTG.
|
| History |
| Family history of lipid disorders |
| Maximal weight and when achieved |
| Detailed medication history (including those recently stopped) |
| Alcohol consumption |
| Diabetes mellitus |
| Possible physical examination findings |
| Eruptive xanthomas |
| Lipemia retinalis |
| Hepatomegaly |
| Lymphadenopathy |
| Laboratory tests |
| Basic Metabolic Panel* with glucose |
| Lipid panel |
| Thyroid‐stimulating hormone, free T4 |
| Liver function, amylase, lipase |
| Hemoglobin A1c |
| Urinalysis |
Distinguishing physical examination findings may arise when serum TG levels exceed 1000 mg/dL. Eruptive xanthoma form when large amounts of TG are sequestered in cutaneous histiocytes, resulting in small yellow‐orange papules with an erythematous base. This finding is seen in a minority of HTG patients and may be missed altogether without a careful examination of the extensor surfaces of arms, legs, back, and buttocks.15 Effective TG‐lowering treatment will result in resolution of these xanthomas. An ophthalmologic examination may reveal lipemia retinalis, a condition that occurs when retinal vessels appear white from lipemic serum and contrast against a pale salmon‐colored retina. Although a dramatic finding, these changes do not result in vision impairment. Hepatomegaly from fatty infiltration of the liver occurs frequently,16 and diffuse lymphadenopathy may also be found.17
Laboratory tests are also essential in the clinical assessment of severe HTG. Important tests include thyroid‐stimulating hormone, creatinine, serum urea nitrogen, and a urinalysis. A hemoglobin A1c test provides information on the level of glycemic control and is now recognized by the American Diabetes Association to diagnose diabetes mellitus.18 Liver function tests commonly reveal a transaminase elevation from underlying steatohepatitis and also provide a baseline value prior to initiating any lipid‐lowering medications. Additional diagnostic tests may be useful in selected patients: HIV testing, serum protein electrophoresis, and urine protein electrophoresis to help diagnose paraproteinemias such as multiple myeloma, and an antinuclear antibody and double‐stranded DNA for systemic lupus erythematosus.
Severe HTG may interfere with the result of 2 commonly obtained laboratory tests. The sodium concentration can be falsely low (pseudohyponatremia) due to the high levels of TG displacing sodium containing water from the plasma.19 Due to interference by plasma lipids, amylase levels may be near normal in up to 50% of patients with hypertriglyceridemic pancreatitis at the time of admission.20 Thus, if the suspicion for pancreatitis is high, it is reasonable to proceed to imaging if amylase or lipase levels are not confirmatory. Abdominal imaging with computed tomography or magnetic resonance imaging may be used to diagnose acute pancreatitis.
Behind excessive alcohol consumption and gallstone disease, HTG is the third leading cause of pancreatitis, accounting for up to 10% of cases in the general population.21 The exact mechanism by which HTG causes pancreatitis is unclear. One theory is that elevated plasma TG levels are hydrolyzed in the pancreas to cause an increase in local free fatty acids, which in turn may cause inflammation and overt pancreatitis.22 Another theory proposes that elevated levels of chylomicrons lead to plasma hyperviscosity, which causes ischemia and local acidosis in pancreatic capillaries.23 Whatever the cause, it is unclear why only some patients with severe HTG develop acute pancreatitis. One study of 129 patients with severe HTG found mean serum TG levels to be higher in patients with acute pancreatitis than in those without (4470 versus 2450 mg/dL), suggesting the threshold to develop acute pancreatitis is higher than previously thought.24 Without a firm TG threshold above which patients develop pancreatitis, the decision to hospitalize can be difficult.
WHEN TO HOSPITALIZE?
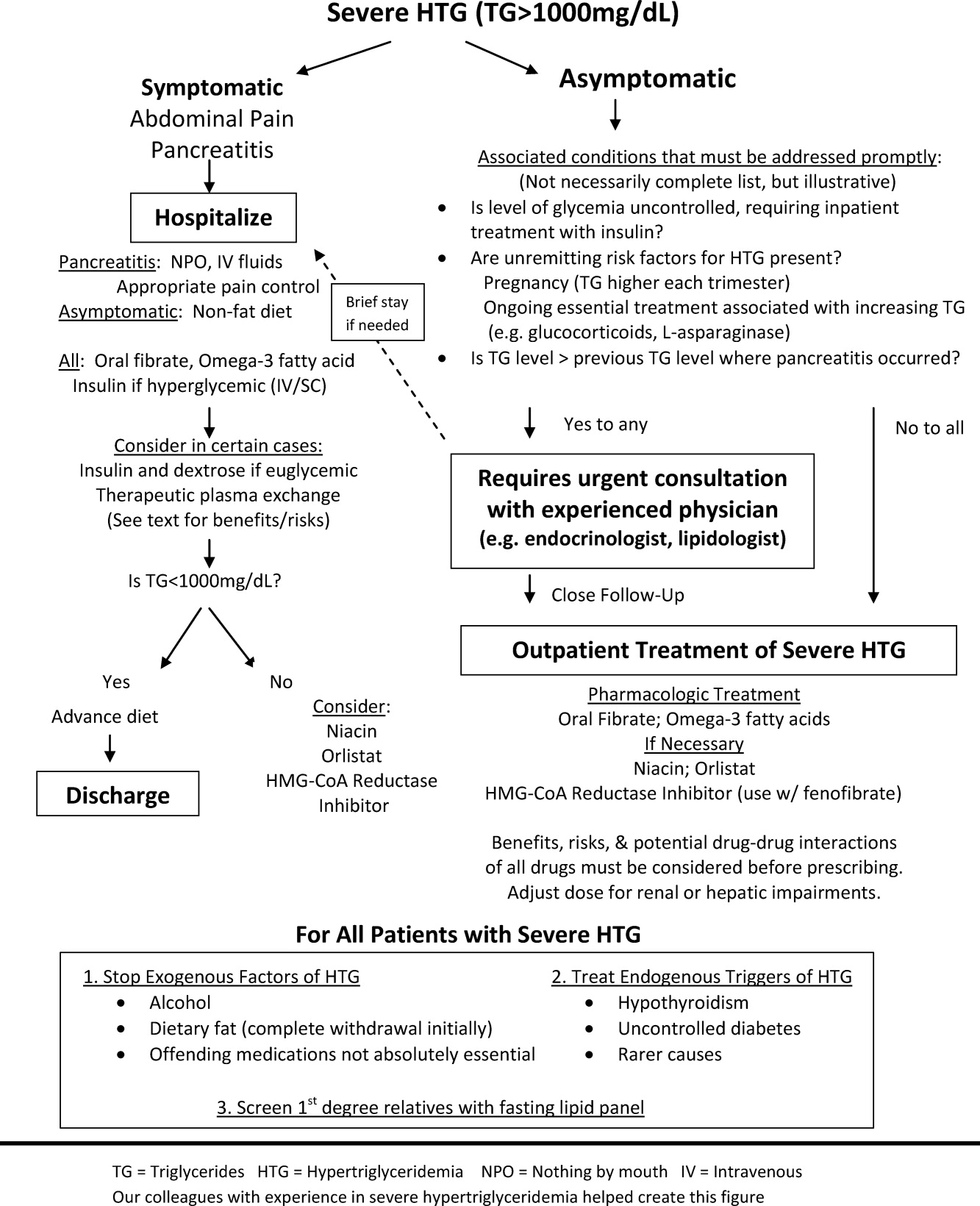
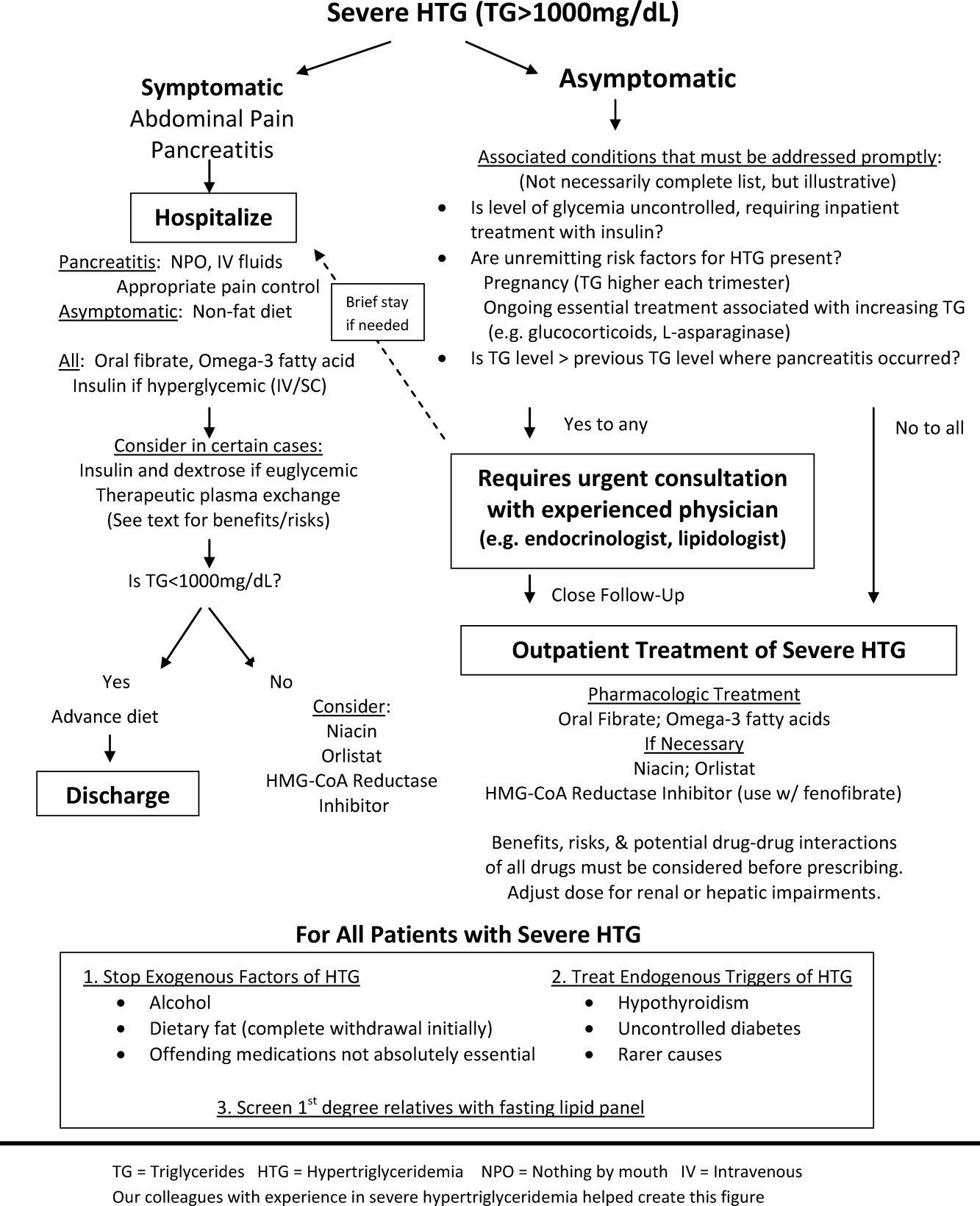
The choice of whether to hospitalize a patient with severe HTG is first based on the presence or absence of abdominal pain and/or acute pancreatitis. Figure 1 diagrams a suggested admission and treatment algorithm. If abdominal pain is present, the patient should be hospitalized and assessed for possible triggers with prompt initiation of pharmacologic treatment. In the absence of abdominal pain, the decision to admit the patient with severe HTG requires clinical judgment. In these cases, prompt consultation with a physician experienced in the management of lipid disorders is recommended. In our experience, admission is usually driven by factors such as (1) severe hyperglycemia requiring inpatient insulin therapy; (2) severe HTG at or near a level where pancreatitis has occurred in the past in a patient for whom adherence is suspect (mindful of the great variability at the levels where patients develop pancreatitis); (3) unremitting triggers of severe HTG such as ongoing use of essential medications also known to exacerbate HTG (such as some forms of chemotherapy) or pregnancy in the third trimester. TG levels rise continuously throughout pregnancy and peak during the third trimester, when hypertriglyceridemic pancreatitis most often occurs. Asymptomatic patients with severe HTG not requiring hospitalization need close outpatient follow‐up to prevent the onset of chylomicronemia syndrome.
INPATIENT MANAGEMENT
No professional recommendations exist regarding a standardized treatment plan for severe HTG. The treatment regimen is first based on the presence or absence of symptoms. Treatment of hypertriglyceridemic pancreatitis should target a serum TG level <1000 mg/dL and resolution of abdominal pain. The initial goal for asymptomatic patients is a TG level <1000 mg/dL, as this level represents a significant reduction in the risk of developing chylomicronemia syndrome. In either case, the first 2 components of the treatment regimen are dietary changes and oral medications.
Dietary Changes
Patients with hypertriglyceridemic pancreatitis should be made NPO, with the exception of necessary medications taken only with water to provide bowel rest and eliminate fat intake. As chylomicron production in the intestine falls, TG levels will fall dramatically within 12 days of NPO status regardless of other treatments. Once TG levels approach 1000 mg/dL and there is no residual abdominal pain, a no‐fat diet can be given. Patients with persistent abdominal pain requiring a prolonged fast (>57 days) may require nutrition through alternate means such as an enteral formula through a feeding tube or the use of TPN. If a feeding tube is required, we suggest beginning with an elemental, peptide‐based, fat‐free formula, with help from a nutrition consult to assist with individual tube‐feeding options.25 Enteral formula can be supplemented with medium‐chain triglyceride oils (found in coconut and palm kernel) to provide some additional nutritional support. MCTs do not raise serum TG levels, as they are absorbed directly into the portal vein for prompt oxidation by the liver, whereas long‐chain TGs are converted into chylomicrons for peripheral transport. One case report describes a dramatic therapeutic response to medium‐chain triglyceride oils in a patient with familial chylomicronemia,26 although we do not routinely recommend these oils as therapy given lack of long‐term safety data. Lastly, if TPN is required, it is crucial to avoid lipid emulsions to prevent a rise in serum TG levels.
Asymptomatic patients with severe HTG can be fed upon admission, but should be placed on a fat‐free diet. Fat is added back into the diet when TG levels fall below 1000 mg/dL and is slowly increased to a target fat content of 10% of the total calories, usually not exceeding 25 g/day.
Oral Medications
Oral medications should be initiated to lower TG levels for both symptomatic and asymptomatic patients. Table 3 lists the different classes of medications. In our experience, oral fibrates are a recommended first‐line treatment, with other agents used as adjunctive therapy. Through complex mechanisms, fibrates reduce hepatic VLDL secretion and increase serum lipolysis of TG.27 In patients who do not have diabetes and are at low risk for coronary heart disease (CHD), either gemfibrozil or fenofibrate may be used to lower serum TG levels. However, in patients with diabetes, CHD, or a CHD risk equivalent, use of fenofibrate is preferred as an HMG‐CoA reductase inhibitor (a statin) and will almost always be necessary to reach low‐density lipoprotein (LDL)‐cholesterol goals. Fenofibrate, unlike gemfibrozil, does not interfere with the glucuronidation of statins by the liver.28
| Drug | Usual Dose | TG Reduction | Cautions or Contraindications | Comments |
|---|---|---|---|---|
| ||||
| Fenofibrate | 130200 mg/day | 50% | Hepatic or renal insufficiency | Best fibrate to use with HMG‐CoA reductase Inhibitors |
| Gemfibrozil | 600 mg BID | 50% | Hepatic or renal insufficiency | Avoid combination with statins |
| HMG‐CoA reductase inhibitors or statins | Variable; the more potent LDL lowering, the more TG lowering | 25% | Decompensated cirrhosis; end‐stage renal disease | Not the primary treatment for patients with TG levels >1000 mg/dL; some statins such as atorvastatin and fluvastatin are favored for patients with renal insufficiency due to less renal excretion than other statins |
| Omega‐3 fatty acids | 2 g BID Lovaza (840 mg DHA/EPA per dose) | 25%50% with monotherapy (the higher the TG level, the greater the reduction); 30% with combination therapy | Allergy to fish | Fishy aftertaste; may cause flatulence; may increase serum glucose and LDL; low risk of clinical bleeding |
| Nicotinic acid | 12 g/day ER; up‐titrate from lowest dose | 15%35% | Active liver disease; active peptic ulcer disease; arterial bleeding | Can increase blood sugar levels by increasing insulin resistance |
| Orlistat (Xenical) | 120 mg TID | 15%35% | Cases of serious liver dysfunction have been reported | Can interfere with drug absorption, especially fat‐soluble vitamins; oily rectal discharge |
| Insulin | IV 0.10.3 U/kg/hr; titrate to serum glucose 140180 mg/dL OR basal/bolus subcutaneously | Variable; >50% in some cases | Hypoglycemia | Useful in patients who have diabetes |
If a fibrate fails to achieve an acceptable serum TG level, we recommend adjunctive therapy with omega‐3 fatty acid esters. Omega‐3 fatty acids lower serum TG levels by decreasing VLDL production and can lower TG levels by as much as 45% in cases of severe HTG.29 This medication is typically the first adjunctive medication chosen due to its low side effect profile.
If additional TG lowering is needed, niacin or nicotinic acid (vitamin B3) may be added next. This medication decreases VLDL production, lowers LDL, and increases high‐density lipoprotein, but invariably with initiation, patients exhibit prominent skin flushing, burning, or itching. This prostaglandin‐mediated effect may be prevented or at least reduced in severity by taking 325 mg aspirin 1 hour before niacin administration. If the TG level is still not at goal, orlistat, a lipase or fat blocker, may be useful.30 Orlistat improves postprandial lipemia through reduction of dietary fat absorption. Finally, although potent statins such as atorvastatin and rosuvastatin can lower TGs derived from VLDL substantially,31 their use should be prompted by the patient's risk for atherosclerotic cardiovascular disease. Because all of the hypotriglyceridemic medications can affect the liver, regular liver function testing is prudent, as is a periodic re‐evaluation of the ongoing need for these medications. Statin use causing mild transaminase elevation (up to 3 times the upper limit of normal) may be safely tolerated.32
Additional Inpatient Management
Insulin
Insulin increases lipoprotein lipase activity, thereby accelerating chylomicron degradation.33 Insulin (along with glucose if necessary to maintain euglycemia) is therefore a useful adjunctive TG‐lowering medication to oral medications, even in nondiabetic patients. Insulin administered intravenously should follow a titration protocol with hourly monitoring of blood glucose. The goal of the insulin protocol with severe HTG is not maintaining strict euglycemia but rather maintenance of LPL activation by exogenous insulin with avoidance of hypoglycemia. In hospitals where an insulin infusion protocol for diabetic ketoacidosis or postsurgical hyperglycemia already exists, the protocol can be applied for HTG management with minor modifications: introduce dextrose‐containing fluids at higher blood glucoses (180 mg/dL or less) and eliminate insulin boluses. A suggested dose is a continuous intravenous insulin drip at 0.10.3 U/kg/hr with glucose to maintain blood glucose levels between 140 and 180 mg/dL, although there are no guidelines from professional societies. Subcutaneous insulin has also been used to successfully lower TG levels.34, 35 The major limitation of subcutaneous administration is the inability to rapidly adjust the dosing when needed, which is particularly concerning when treating patients who do not have diabetes. We prefer to use subcutaneous basal insulin in patients requiring long‐term use of insulin after a significant TG reduction with intravenous insulin. Subcutaneous bolus prandial insulin should not be used until the patient has resumed a solid diet, because a liquid diet may not reliably contain enough carbohydrates for bolus therapy.
Intravenous Heparin
Heparin has been used in case reports as adjunctive treatment for hypertriglyceridemic pancreatitis.36, 37 Although heparin may increase circulating LPL levels, this effect is short‐lived and is quickly followed by increased hepatic LPL degradation.38 Therefore, the use of heparin to treat severe HTG cannot be routinely recommended.
Therapeutic Plasma Exchange
First used in 1978, therapeutic plasma exchange (TPE) has been demonstrated to quickly and dramatically lower serum TG levels.39 Since its first use, TPE has been used in several small case studies.4042 Without data from larger studies, the optimal frequency and duration of TPE remains unclear. One review suggests the use of TPE as first‐line therapy provided the patient is euglycemic, apheresis can be started within 48 hours of diagnosis, and the patient can tolerate the central venous access.43 On the other hand, guidelines from the American Society of Apheresis incorporating low‐quality evidence do not recommend TPE as routine first‐ or second‐line treatment for hypertriglyceridemic pancreatitis, but rather suggest the use of TPE on a case‐by‐case basis.44 When TPE is needed, this society recommends daily treatment for 13 days until an adequate postapheresis TG level is obtained. Although TPE rapidly lowers TG levels, it is also aggressive (requires placement of a pheresis catheter), expensive, and may not be readily available. We believe it remains an option for patients with severe HTG who do not respond readily to fat restriction, glycemic control with insulin, and pharmacologic treatment with a fibrate and omega‐3 fatty acids. In our judgment, routine use of TPE cannot be recommended without data from a randomized clinical trial examining the value of immediate lowering of TG levels with TPE versus the usually prompt fall in TG levels with less aggressive measures.
Discharge Planning
Typically, asymptomatic patients are discharged once their TG levels approach 1000 mg/dL. Patients recovering from hypertriglyceridemic pancreatitis may be discharged once they tolerate a no‐fat diet without recurrence of abdominal pain and without a significant TG increase above 1000 mg/dL.
Discharge Diet and Activity
At discharge, the diet should be high in fiber, fruits, vegetables, and lean protein, with fat intake restricted to approximately 10% of total calories. Insulin‐resistant patients and patients with diabetes should avoid sugar‐sweetened foods and drinks. Specifically, the daily amount of fructose intake should be no more than 50 mg to avoid a dose‐dependent increase in plasma TG levels compared with other sugars.4 At least 2 servings per week of marine foods naturally rich in omega‐3 fatty acids (fatty fish such as salmon or trout) are recommended. Nonmarine forms of omega‐3 fatty acids (walnuts, flaxseed), which have not demonstrated consistent reductions in TG, cannot be routinely recommended.4 In addition, alcohol consumption should be eliminated.
Once patients maintain a TG level near 500 mg/dL, we allow for dietary flexibility by slowly increasing the amount of dietary unsaturated fat. For patients with TG levels <500 mg/dL, Adult Treatment Panel III advocated restriction of daily dietary saturated fat levels to <7% and keeping the total fat level between 25% and 35%.2 A range in total fat was provided so that unsaturated fat could be increased to limit dietary carbohydrates if glycemic control was needed. In addition to dietary changes, counseling patients about the importance of physical activity and weight loss is crucial for long‐term management of severe HTG. TG lowering in response to diet and weight loss varies, but typically approximates 25%.45
Outpatient Medications
Patients without significant contraindications should be discharged on a fibrate and omega‐3 fatty acids. As mentioned, niacin, orlistat, and/or a statin may be used as adjunctive therapy. Despite use of these hypotriglyceridemic medications, secondary causes of HTG should be modified (such as removal of aggravating medications or appropriately treating uncontrolled diabetes) to yield lasting improvements in TG levels.
CONCLUSIONS
As the prevalence of obesity and diabetes continues to rise, so too does the clinical importance of proper management of severe HTG. Recognizing chylomicronemia syndromeone of the most dramatic consequences of lipid disordersand the underlying primary and secondary causes of HTG is required before starting treatment. Patients with severe HTG may require hospitalization for immediate reduction in TG levels and relief of abdominal pain, if present. Treatment involves modifying secondary causes, if possible, and eliminating dietary fat intake. Although use of medications such as an oral fibrate, omega‐3 fatty acids, and insulin are routine, the use of a more invasive procedure such as TPE should be considered on a case‐by‐case basis and may be limited by availability. Upon hospital discharge, careful follow‐up should promote lifestyle changes and medication adherence to prevent recurrence of severe HTG.
- ,,,.Pathophysiology of triglyceride‐rich lipoproteins in atherothrombosis: clinical aspects.Clin Cardiol.1999;22:II15–II20.
- Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III).JAMA.2001;285:2486–2497.
- ,.Chylomicronemia syndrome. Interaction of genetic and acquired hypertriglyceridemia.Med Clin North Am.1982;66:455–468.
- ,,, et al.Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association.Circulation.2011;123:2292–2333.
- .Familial lipoprotein lipase deficiency and other causes of the chylomicronemia syndrome. In: Scriver C, Beaudet A, Sly W, Valle D, eds.The Metabolic and Molecular Basis of Inherited Disease.7th ed.New York:McGraw‐Hill;1995:1913–1932.
- ,,.Hypertriglyceridemia: its etiology, effects and treatment.CMAJ.2007;176:1113–1120.
- ,,, et al.Chylomicronemia with a mutant GPIHBP1 (Q115P) that cannot bind lipoprotein lipase.Arterioscler Thromb Vasc Biol.2009;29:956–962.
- ,,, et al.A mutation in the human lipoprotein lipase gene as the most common cause of familial chylomicronemia in French Canadians.N Engl J Med.1991;324:1761–1766.
- ,,, et al.Inherited apolipoprotein A‐V deficiency in severe hypertriglyceridemia.Arterioscler Thromb Vasc Biol.2005;25:411–417.
- ,,, et al.A polygenic basis for four classical Fredrickson hyperlipoproteinemia phenotypes that are characterized by hypertriglyceridemia.Hum Mol Genet.2009;18:4189–4194.
- .Secondary causes of hyperlipidemia.Med Clin North Am.1994;78:117–141.
- ,,.Hypertriglyceridemic hyperapob: the unappreciated atherogenic dyslipoproteinemia in type 2 diabetes mellitus.Ann Intern Med.2001;135:447–459.
- .Hyperlipidemic pancreatitis.Gastroenterol Clin North Am.1990;19:783–791.
- ,,, et al.Effects of insulin on cholesterol synthesis in type II diabetes patients.Diabetes Care.1995;18:1362–1369.
- ,,,.Evidence for the chylomicron origin of lipids accumulating in diabetic eruptive xanthomas: a correlative lipid biochemical, histochemical, and electron microscopic study.J Clin Invest.1970;49:2172–2187.
- .Dyslipidaemia.Lancet.2003;362:717–731.
- ,,.Lymphadenopathy associated with severe hypertriglyceridemia.JAMA.1990;264:727–728.
- Diagnosis and classification of diabetes mellitus.Diabetes Care.2010;33(suppl 1):S62–S69.
- ,.Pseudohyponatremia in acute hyperlipemic pancreatitis. A potential pitfall in therapy.Arch Surg.1985;120:1053–1055.
- ,,.Suppression of amylase activity by hypertriglyceridemia.JAMA.1973;225:1331–1334.
- ,,,.Dyslipidaemic pancreatitis clinical assessment and analysis of disease severity and outcomes.Pancreatology.2009;9:252–257.
- .Pathogenesis, differentiation and management of hypertriglyceridemia.Adv Intern Med.1969;15:117–154.
- ,.Role of hypertriglyceridemia in the pathogenesis of experimental acute pancreatitis in rats.Int J Pancreatol.1996;20:177–184.
- ,,, et al.Acute pancreatitis in a cohort of 129 patients referred for severe hypertriglyceridemia.Pancreas.2008;37:13–12.
- ,,, et al.ESPEN Guidelines on Enteral Nutrition: pancreas.Clin Nutr.2006;25:275–284.
- ,,, et al.Therapeutic response to medium‐chain triglycerides and omega‐3 fatty acids in a patient with the familial chylomicronemia syndrome.Arterioscler Thromb Vasc Biol.1997;17:1400–1406.
- ,,,,,.Mechanism of action of fibrates on lipid and lipoprotein metabolism.Circulation.1998;98:2088–2093.
- ,,.Drug interactions with lipid‐lowering drugs: mechanisms and clinical relevance.Clin Pharmacol Ther.2006;80:565–81.
- ,,, et al.Safety and efficacy of Omacor in severe hypertriglyceridemia.J Cardiovasc Risk.1997;4:385–391.
- ,,.Usefulness of Orlistat in the treatment of severe hypertriglyceridemia.Am J Cardiol.2002;89:229–231.
- ,,, et al.Effects of intensive atorvastatin and rosuvastatin treatment on apolipoprotein B‐48 and remnant lipoprotein cholesterol levels.Atherosclerosis.2009;205:197–201.
- ,,,.Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force.Am J Cardiol.2006;97:89C–94C.
- .Lipoprotein lipase. A multifunctional enzyme relevant to common metabolic diseases.N Engl J Med.1989;320:1060–1068.
- ,,.Insulin therapy for a non‐diabetic patient with severe hypertriglyceridemia.J Am Coll Nutr.1998;17:458–461.
- ,,,,.Treatment of severe hypertriglyceridemia in nondiabetic patients with insulin.Am J Emerg Med.2005;23:415–417.
- ,.Decreasing the plasma triglyceride level in hypertriglyceridemia‐induced pancreatitis in pregnancy: a case report.Am J Obstet Gynecol.2002;187:241–242.
- ,,,,,.Pancreatitis may occur with a normal amylase concentration in hypertriglyceridaemia.BMJ.1996;313:1265.
- ,,,.Lipoprotein lipase during continuous heparin infusion: tissue stores become partially depleted.J Lab Clin Med.2001;138:206–13.
- ,,,,,.Treatment of severe diabetic hypertriglyceridaemia by plasma exchange.Lancet.1978;1:1368.
- ,,,.Therapeutic plasma exchange in patients with hyperlipidemic pancreatitis.World J Gastroenterol.2004;10:2272–2274.
- ,,.Plasma exchange in severe hypertriglyceridemia a clinical study.Transfus Apher Sci.2006;34:253–257.
- ,,, et al.Management of acute severe hyperlipidemic pancreatitis.Digestion.2006;73:259–264.
- ,,,,.Hypertriglyceridemic pancreatitis: presentation and management.Am J Gastroenterol.2009;104:984–991.
- ,,, et al.Guidelines on the use of therapeutic apheresis in clinical practice—evidence‐based approach from the Apheresis Applications Committee of the American Society for Apheresis.J Clin Apher.2010;25:83–177.
- ,,,,,.Effects of a low‐fat diet compared with those of a high‐monounsaturated fat diet on body weight, plasma lipids and lipoproteins, and glycemic control in type 2 diabetes.Am J Clin Nutr.2004;80:668–673.
The patient with a markedly high serum triglyceride (TG) level poses an interesting challenge for hospitalists. Hypertriglyceridemia (HTG) is defined as a fasting plasma TG level that is above the 95th percentile for age and sex.1 TG levels are commonly classified into categories according to Adult Treatment Panel III guidelines with desirable levels <150 mg/dL (1.7 mmol/L), borderline levels 150199, high levels 200499 mg/dL, and very high levels >500 mg/dL (5.6 mmol/L).2 A TG level exceeding an arbitrary threshold of >1000 mg/dL (11.3 mmol/L) is referred to as severe HTG. The Lipid Research Clinics Program Prevalence Study found that 1.79 per 10,000 outpatients (<0.02%) had TG levels > 2000 mg/dL.3 Chylomicronemia syndrome occurs when severe HTG is accompanied by 1 or more of the following: symptoms of abdominal pain or acute pancreatitis or physical examination findings such as eruptive xanthomas or lipemia retinalis. There is no TG level above which pancreatitis invariably occurs, making the decision to hospitalize difficult. The goal of this review is to discuss the causes of severe HTG; the clinical assessment, including criteria for hospitalization; and the available treatment options for this infrequent but serious condition. We begin with a clinical case of severe HTG.
CASE PRESENTATION
A 47‐year‐old woman with a history of chronic myelogenous leukemia was admitted to the hospital with a serum triglyceride level of 17,393 mg/dL. Two years prior to admission, she underwent allogenic stem cell transplantation for chronic myelogenous leukemia and has since remained in remission. Six months prior to admission, severe diarrhea from intestinal graft‐versus‐host disease required the use of total parenteral nutrition (TPN) and immunosuppressive therapy consisting of prednisone 20 mg/day, mycophenolate mofetil 250 mg thrice daily, and sirolimus 0.3 mg/day. During treatment with steroids, she developed diabetes mellitus requiring insulin, with a subsequent hemoglobin A1c level of 7.7% (normal, <7%). The serum TG level prior to transplantation was unknown but was 343 mg/dL prior to TPN initiation. One month prior to admission, the diarrhea resolved and TPN was stopped. The TG level was 7463 mg/dL 1 week prior to admission, and despite use of fenofibrate, it rose to 17,393 mg/dL. The patient denied abdominal pain, and did not have abdominal tenderness or eruptive xanthomas. She denied a family history of dyslipidemia or recent medication changes. Given the extreme TG elevation, the lack of response to outpatient treatment and the concern for developing acute pancreatitis, the patient was admitted to the hospital for inpatient TG‐lowering treatment.
Upon admission, serum lipase and amylase were within normal limits, but the blood glucose level was 243 mg/dL. Insulin infusion and oral fenofibrate 145 mg/day was started, and the patient was kept non per os (NPO). Six hours later, despite insulin infusion, the TG level rose to 26,250 mg/dL. Therapeutic plasma exchange (TPE) was performed on 2 consecutive days with a resultant decrease in TG level to 530 mg/dL. The patient was later discharged home on fenofibrate and omega‐3 ethyl esters, her same immunosuppressive and insulin regimen, and instructions for a very low‐fat diet. In the next 3 months, her serum TG level did not rise above 530 mg/dL. The cause of our patient's extreme TG elevation was likely a combination of genetic factors exacerbated by immunosuppressive and glucocorticoid therapy.
This case featured dramatic elevations in serum TG levels that the managing doctors believed merited a hospital admission. Management of patients with severe HTG first requires an understanding of TG metabolism.
ETIOLOGY
Serum TGs produced by the liver are carried by very low‐density lipoproteins (VLDLs), whereas TGs derived from dietary fat are carried by chylomicrons. Both chylomicrons and VLDLs are hydrolyzed by the same enzymelipoprotein lipase (LPL). TGs are hydrolyzed into fatty acids for uptake by muscle and adipose tissue, whereas remnants of VLDL and chylomicrons are removed by the liver. More details on TG pathophysiology may be found in a recent review.4 When LPL is saturated with VLDL, ingestion of a fatty meal may cause chylomicrons to linger in circulation for days instead of hours. Asking the laboratory to spin down the blood of a patient with severe HTG and keep the test tube upright at 4C may reveal a large creamy supernatant layer demonstrating chylomicronemia.
A fasting TG level drawn 12 hours after the last meal reflects hepatic TG production. Although a nonfasting TG level may reflect postprandial chylomicrons, values above 1000 mg/dL strongly suggest true HTG, particularly in the setting of acute pancreatitis. Treatment should not be delayed to obtain a fasting TG level.
HTG may result from increased VLDL production, reduced VLDL/chylomicron clearance, or more likely a combination of the two. The causes of these metabolic derangements are classified as primary (genetic) or secondary (acquired) (Table 1). In adult patients, HTG is usually the result of a combination of primary and secondary causes. A study of 123 patients with TG levels >2000 mg/dL found that all patients had a primary metabolic defect and 110/123 had a coexistent secondary cause.3 An underlying genetic lipoprotein metabolism derangement is often clinically silent until coupled with a secondary cause of HTG that together raise TG levels high enough to cause the chylomicronemia syndrome.
|
| Primary |
| Familial lipid disorders |
| Lipoprotein pattern type I |
| Familial chylomicronemia |
| Deficiency in LPL and/or apo‐CII |
| Autosomal recessive; presents in childhood |
| Rare functional disorders in LPL |
| Lipoprotein pattern type III |
| Familial dysbetalipoproteinemia |
| Inadequate VLDL clearance from apo‐E2 |
| Autosomal recessive; presents in adulthood |
| Lipoprotein pattern type IV |
| Familial HTG: increased VLDL |
| Autosomal dominant; presents in adulthood |
| Familial combined hyperlipidemia |
| Multiple phenotypes seen; increased apo‐B levels |
| Lipoprotein pattern type V |
| Mixed HTG: increased VLDL and chylomicrons; presents in adulthood |
| Secondary |
| Disease |
| Poorly controlled diabetes mellitus; hypothyroid; SLE; Cushing syndrome; HIV infection; sarcoid multiple myeloma; obesity; renal disease (nephrotic) |
| Disorder of metabolism |
| Pregnancy |
| Diet |
| Excessive alcohol, especially with high‐fat diet |
| Drugs |
| Estrogen; tamoxifen; glucocorticoids; protease inhibitors; nonselective beta‐blockers; propofol; isotretinoin; some antipsychotic medications (clozapine, olanzapine); tacrolimus; sirolimus; cyclosporine; bexarotene; all‐trans retinoic acid; L‐asparaginase; interferon‐ |
The most common primary cause of HTG in adults is familial HTG, an autosomal dominant condition with a population prevalence ranging from 1%2% to 5%10% and age‐dependent penetrance.5, 6 Other genetic causes are much rarer, such as LPL deficiency (1 in 1 million patients), apolipoprotein‐CII, and other mutations resulting in impaired binding to LPL.79 Primary causes of HTG are often listed as Fredrickson phenotypes (Table 1). Recent genome‐wide association studies reveal a complex polygenic basis to the Fredrickson categories and suggest additional undefined genes or nongenetic factors may significantly contribute to the final phenotype.10 Diagnosis of familial lipid disorders requires an accurate family history that may be difficult to obtain.
Secondary causes of HTG can be categorized using a four‐D mnemonic: Diseases, Diet, Disorder of Metabolism, and Drugs.11 The most common condition associated with HTG is obesity.6 The mechanism between obesity and HTG is complex and likely involves an increase in fatty acid flux from adipose tissue to other tissues and insulin resistance.12 Asking whether a patient's current weight is close to the heaviest lifetime weight is a clue to diagnosing obesity‐driven HTG. One case series of hypertriglyceridemic pancreatitis found that diabetes or excessive alcohol intake account for the majority of secondary causes of HTG.13 The cause of HTG among patients with diabetes is multifactorial: insulin deficiency reduces LPL levels (insulin is required for synthesis of LPL), whereas insulin resistance attenuates the ability of insulin to decrease hepatic cholesterol synthesis and thus increases hepatic secretion of VLDL.14 Alcohol impairs lipolysis and increases VLDL production that can lead to severe HTG, particularly in those patients with an underlying functional deficiency in LPL. Other secondary etiologies of severe HTG may be elicited through careful attention to medical and medication history.
CLINICAL ASSESSMENT
Table 2 proposes a reasonable initial assessment of the history and physical and laboratory tests in patients with severe HTG.
|
| History |
| Family history of lipid disorders |
| Maximal weight and when achieved |
| Detailed medication history (including those recently stopped) |
| Alcohol consumption |
| Diabetes mellitus |
| Possible physical examination findings |
| Eruptive xanthomas |
| Lipemia retinalis |
| Hepatomegaly |
| Lymphadenopathy |
| Laboratory tests |
| Basic Metabolic Panel* with glucose |
| Lipid panel |
| Thyroid‐stimulating hormone, free T4 |
| Liver function, amylase, lipase |
| Hemoglobin A1c |
| Urinalysis |
Distinguishing physical examination findings may arise when serum TG levels exceed 1000 mg/dL. Eruptive xanthoma form when large amounts of TG are sequestered in cutaneous histiocytes, resulting in small yellow‐orange papules with an erythematous base. This finding is seen in a minority of HTG patients and may be missed altogether without a careful examination of the extensor surfaces of arms, legs, back, and buttocks.15 Effective TG‐lowering treatment will result in resolution of these xanthomas. An ophthalmologic examination may reveal lipemia retinalis, a condition that occurs when retinal vessels appear white from lipemic serum and contrast against a pale salmon‐colored retina. Although a dramatic finding, these changes do not result in vision impairment. Hepatomegaly from fatty infiltration of the liver occurs frequently,16 and diffuse lymphadenopathy may also be found.17
Laboratory tests are also essential in the clinical assessment of severe HTG. Important tests include thyroid‐stimulating hormone, creatinine, serum urea nitrogen, and a urinalysis. A hemoglobin A1c test provides information on the level of glycemic control and is now recognized by the American Diabetes Association to diagnose diabetes mellitus.18 Liver function tests commonly reveal a transaminase elevation from underlying steatohepatitis and also provide a baseline value prior to initiating any lipid‐lowering medications. Additional diagnostic tests may be useful in selected patients: HIV testing, serum protein electrophoresis, and urine protein electrophoresis to help diagnose paraproteinemias such as multiple myeloma, and an antinuclear antibody and double‐stranded DNA for systemic lupus erythematosus.
Severe HTG may interfere with the result of 2 commonly obtained laboratory tests. The sodium concentration can be falsely low (pseudohyponatremia) due to the high levels of TG displacing sodium containing water from the plasma.19 Due to interference by plasma lipids, amylase levels may be near normal in up to 50% of patients with hypertriglyceridemic pancreatitis at the time of admission.20 Thus, if the suspicion for pancreatitis is high, it is reasonable to proceed to imaging if amylase or lipase levels are not confirmatory. Abdominal imaging with computed tomography or magnetic resonance imaging may be used to diagnose acute pancreatitis.
Behind excessive alcohol consumption and gallstone disease, HTG is the third leading cause of pancreatitis, accounting for up to 10% of cases in the general population.21 The exact mechanism by which HTG causes pancreatitis is unclear. One theory is that elevated plasma TG levels are hydrolyzed in the pancreas to cause an increase in local free fatty acids, which in turn may cause inflammation and overt pancreatitis.22 Another theory proposes that elevated levels of chylomicrons lead to plasma hyperviscosity, which causes ischemia and local acidosis in pancreatic capillaries.23 Whatever the cause, it is unclear why only some patients with severe HTG develop acute pancreatitis. One study of 129 patients with severe HTG found mean serum TG levels to be higher in patients with acute pancreatitis than in those without (4470 versus 2450 mg/dL), suggesting the threshold to develop acute pancreatitis is higher than previously thought.24 Without a firm TG threshold above which patients develop pancreatitis, the decision to hospitalize can be difficult.
WHEN TO HOSPITALIZE?
The choice of whether to hospitalize a patient with severe HTG is first based on the presence or absence of abdominal pain and/or acute pancreatitis. Figure 1 diagrams a suggested admission and treatment algorithm. If abdominal pain is present, the patient should be hospitalized and assessed for possible triggers with prompt initiation of pharmacologic treatment. In the absence of abdominal pain, the decision to admit the patient with severe HTG requires clinical judgment. In these cases, prompt consultation with a physician experienced in the management of lipid disorders is recommended. In our experience, admission is usually driven by factors such as (1) severe hyperglycemia requiring inpatient insulin therapy; (2) severe HTG at or near a level where pancreatitis has occurred in the past in a patient for whom adherence is suspect (mindful of the great variability at the levels where patients develop pancreatitis); (3) unremitting triggers of severe HTG such as ongoing use of essential medications also known to exacerbate HTG (such as some forms of chemotherapy) or pregnancy in the third trimester. TG levels rise continuously throughout pregnancy and peak during the third trimester, when hypertriglyceridemic pancreatitis most often occurs. Asymptomatic patients with severe HTG not requiring hospitalization need close outpatient follow‐up to prevent the onset of chylomicronemia syndrome.
INPATIENT MANAGEMENT
No professional recommendations exist regarding a standardized treatment plan for severe HTG. The treatment regimen is first based on the presence or absence of symptoms. Treatment of hypertriglyceridemic pancreatitis should target a serum TG level <1000 mg/dL and resolution of abdominal pain. The initial goal for asymptomatic patients is a TG level <1000 mg/dL, as this level represents a significant reduction in the risk of developing chylomicronemia syndrome. In either case, the first 2 components of the treatment regimen are dietary changes and oral medications.
Dietary Changes
Patients with hypertriglyceridemic pancreatitis should be made NPO, with the exception of necessary medications taken only with water to provide bowel rest and eliminate fat intake. As chylomicron production in the intestine falls, TG levels will fall dramatically within 12 days of NPO status regardless of other treatments. Once TG levels approach 1000 mg/dL and there is no residual abdominal pain, a no‐fat diet can be given. Patients with persistent abdominal pain requiring a prolonged fast (>57 days) may require nutrition through alternate means such as an enteral formula through a feeding tube or the use of TPN. If a feeding tube is required, we suggest beginning with an elemental, peptide‐based, fat‐free formula, with help from a nutrition consult to assist with individual tube‐feeding options.25 Enteral formula can be supplemented with medium‐chain triglyceride oils (found in coconut and palm kernel) to provide some additional nutritional support. MCTs do not raise serum TG levels, as they are absorbed directly into the portal vein for prompt oxidation by the liver, whereas long‐chain TGs are converted into chylomicrons for peripheral transport. One case report describes a dramatic therapeutic response to medium‐chain triglyceride oils in a patient with familial chylomicronemia,26 although we do not routinely recommend these oils as therapy given lack of long‐term safety data. Lastly, if TPN is required, it is crucial to avoid lipid emulsions to prevent a rise in serum TG levels.
Asymptomatic patients with severe HTG can be fed upon admission, but should be placed on a fat‐free diet. Fat is added back into the diet when TG levels fall below 1000 mg/dL and is slowly increased to a target fat content of 10% of the total calories, usually not exceeding 25 g/day.
Oral Medications
Oral medications should be initiated to lower TG levels for both symptomatic and asymptomatic patients. Table 3 lists the different classes of medications. In our experience, oral fibrates are a recommended first‐line treatment, with other agents used as adjunctive therapy. Through complex mechanisms, fibrates reduce hepatic VLDL secretion and increase serum lipolysis of TG.27 In patients who do not have diabetes and are at low risk for coronary heart disease (CHD), either gemfibrozil or fenofibrate may be used to lower serum TG levels. However, in patients with diabetes, CHD, or a CHD risk equivalent, use of fenofibrate is preferred as an HMG‐CoA reductase inhibitor (a statin) and will almost always be necessary to reach low‐density lipoprotein (LDL)‐cholesterol goals. Fenofibrate, unlike gemfibrozil, does not interfere with the glucuronidation of statins by the liver.28
| Drug | Usual Dose | TG Reduction | Cautions or Contraindications | Comments |
|---|---|---|---|---|
| ||||
| Fenofibrate | 130200 mg/day | 50% | Hepatic or renal insufficiency | Best fibrate to use with HMG‐CoA reductase Inhibitors |
| Gemfibrozil | 600 mg BID | 50% | Hepatic or renal insufficiency | Avoid combination with statins |
| HMG‐CoA reductase inhibitors or statins | Variable; the more potent LDL lowering, the more TG lowering | 25% | Decompensated cirrhosis; end‐stage renal disease | Not the primary treatment for patients with TG levels >1000 mg/dL; some statins such as atorvastatin and fluvastatin are favored for patients with renal insufficiency due to less renal excretion than other statins |
| Omega‐3 fatty acids | 2 g BID Lovaza (840 mg DHA/EPA per dose) | 25%50% with monotherapy (the higher the TG level, the greater the reduction); 30% with combination therapy | Allergy to fish | Fishy aftertaste; may cause flatulence; may increase serum glucose and LDL; low risk of clinical bleeding |
| Nicotinic acid | 12 g/day ER; up‐titrate from lowest dose | 15%35% | Active liver disease; active peptic ulcer disease; arterial bleeding | Can increase blood sugar levels by increasing insulin resistance |
| Orlistat (Xenical) | 120 mg TID | 15%35% | Cases of serious liver dysfunction have been reported | Can interfere with drug absorption, especially fat‐soluble vitamins; oily rectal discharge |
| Insulin | IV 0.10.3 U/kg/hr; titrate to serum glucose 140180 mg/dL OR basal/bolus subcutaneously | Variable; >50% in some cases | Hypoglycemia | Useful in patients who have diabetes |
If a fibrate fails to achieve an acceptable serum TG level, we recommend adjunctive therapy with omega‐3 fatty acid esters. Omega‐3 fatty acids lower serum TG levels by decreasing VLDL production and can lower TG levels by as much as 45% in cases of severe HTG.29 This medication is typically the first adjunctive medication chosen due to its low side effect profile.
If additional TG lowering is needed, niacin or nicotinic acid (vitamin B3) may be added next. This medication decreases VLDL production, lowers LDL, and increases high‐density lipoprotein, but invariably with initiation, patients exhibit prominent skin flushing, burning, or itching. This prostaglandin‐mediated effect may be prevented or at least reduced in severity by taking 325 mg aspirin 1 hour before niacin administration. If the TG level is still not at goal, orlistat, a lipase or fat blocker, may be useful.30 Orlistat improves postprandial lipemia through reduction of dietary fat absorption. Finally, although potent statins such as atorvastatin and rosuvastatin can lower TGs derived from VLDL substantially,31 their use should be prompted by the patient's risk for atherosclerotic cardiovascular disease. Because all of the hypotriglyceridemic medications can affect the liver, regular liver function testing is prudent, as is a periodic re‐evaluation of the ongoing need for these medications. Statin use causing mild transaminase elevation (up to 3 times the upper limit of normal) may be safely tolerated.32
Additional Inpatient Management
Insulin
Insulin increases lipoprotein lipase activity, thereby accelerating chylomicron degradation.33 Insulin (along with glucose if necessary to maintain euglycemia) is therefore a useful adjunctive TG‐lowering medication to oral medications, even in nondiabetic patients. Insulin administered intravenously should follow a titration protocol with hourly monitoring of blood glucose. The goal of the insulin protocol with severe HTG is not maintaining strict euglycemia but rather maintenance of LPL activation by exogenous insulin with avoidance of hypoglycemia. In hospitals where an insulin infusion protocol for diabetic ketoacidosis or postsurgical hyperglycemia already exists, the protocol can be applied for HTG management with minor modifications: introduce dextrose‐containing fluids at higher blood glucoses (180 mg/dL or less) and eliminate insulin boluses. A suggested dose is a continuous intravenous insulin drip at 0.10.3 U/kg/hr with glucose to maintain blood glucose levels between 140 and 180 mg/dL, although there are no guidelines from professional societies. Subcutaneous insulin has also been used to successfully lower TG levels.34, 35 The major limitation of subcutaneous administration is the inability to rapidly adjust the dosing when needed, which is particularly concerning when treating patients who do not have diabetes. We prefer to use subcutaneous basal insulin in patients requiring long‐term use of insulin after a significant TG reduction with intravenous insulin. Subcutaneous bolus prandial insulin should not be used until the patient has resumed a solid diet, because a liquid diet may not reliably contain enough carbohydrates for bolus therapy.
Intravenous Heparin
Heparin has been used in case reports as adjunctive treatment for hypertriglyceridemic pancreatitis.36, 37 Although heparin may increase circulating LPL levels, this effect is short‐lived and is quickly followed by increased hepatic LPL degradation.38 Therefore, the use of heparin to treat severe HTG cannot be routinely recommended.
Therapeutic Plasma Exchange
First used in 1978, therapeutic plasma exchange (TPE) has been demonstrated to quickly and dramatically lower serum TG levels.39 Since its first use, TPE has been used in several small case studies.4042 Without data from larger studies, the optimal frequency and duration of TPE remains unclear. One review suggests the use of TPE as first‐line therapy provided the patient is euglycemic, apheresis can be started within 48 hours of diagnosis, and the patient can tolerate the central venous access.43 On the other hand, guidelines from the American Society of Apheresis incorporating low‐quality evidence do not recommend TPE as routine first‐ or second‐line treatment for hypertriglyceridemic pancreatitis, but rather suggest the use of TPE on a case‐by‐case basis.44 When TPE is needed, this society recommends daily treatment for 13 days until an adequate postapheresis TG level is obtained. Although TPE rapidly lowers TG levels, it is also aggressive (requires placement of a pheresis catheter), expensive, and may not be readily available. We believe it remains an option for patients with severe HTG who do not respond readily to fat restriction, glycemic control with insulin, and pharmacologic treatment with a fibrate and omega‐3 fatty acids. In our judgment, routine use of TPE cannot be recommended without data from a randomized clinical trial examining the value of immediate lowering of TG levels with TPE versus the usually prompt fall in TG levels with less aggressive measures.
Discharge Planning
Typically, asymptomatic patients are discharged once their TG levels approach 1000 mg/dL. Patients recovering from hypertriglyceridemic pancreatitis may be discharged once they tolerate a no‐fat diet without recurrence of abdominal pain and without a significant TG increase above 1000 mg/dL.
Discharge Diet and Activity
At discharge, the diet should be high in fiber, fruits, vegetables, and lean protein, with fat intake restricted to approximately 10% of total calories. Insulin‐resistant patients and patients with diabetes should avoid sugar‐sweetened foods and drinks. Specifically, the daily amount of fructose intake should be no more than 50 mg to avoid a dose‐dependent increase in plasma TG levels compared with other sugars.4 At least 2 servings per week of marine foods naturally rich in omega‐3 fatty acids (fatty fish such as salmon or trout) are recommended. Nonmarine forms of omega‐3 fatty acids (walnuts, flaxseed), which have not demonstrated consistent reductions in TG, cannot be routinely recommended.4 In addition, alcohol consumption should be eliminated.
Once patients maintain a TG level near 500 mg/dL, we allow for dietary flexibility by slowly increasing the amount of dietary unsaturated fat. For patients with TG levels <500 mg/dL, Adult Treatment Panel III advocated restriction of daily dietary saturated fat levels to <7% and keeping the total fat level between 25% and 35%.2 A range in total fat was provided so that unsaturated fat could be increased to limit dietary carbohydrates if glycemic control was needed. In addition to dietary changes, counseling patients about the importance of physical activity and weight loss is crucial for long‐term management of severe HTG. TG lowering in response to diet and weight loss varies, but typically approximates 25%.45
Outpatient Medications
Patients without significant contraindications should be discharged on a fibrate and omega‐3 fatty acids. As mentioned, niacin, orlistat, and/or a statin may be used as adjunctive therapy. Despite use of these hypotriglyceridemic medications, secondary causes of HTG should be modified (such as removal of aggravating medications or appropriately treating uncontrolled diabetes) to yield lasting improvements in TG levels.
CONCLUSIONS
As the prevalence of obesity and diabetes continues to rise, so too does the clinical importance of proper management of severe HTG. Recognizing chylomicronemia syndromeone of the most dramatic consequences of lipid disordersand the underlying primary and secondary causes of HTG is required before starting treatment. Patients with severe HTG may require hospitalization for immediate reduction in TG levels and relief of abdominal pain, if present. Treatment involves modifying secondary causes, if possible, and eliminating dietary fat intake. Although use of medications such as an oral fibrate, omega‐3 fatty acids, and insulin are routine, the use of a more invasive procedure such as TPE should be considered on a case‐by‐case basis and may be limited by availability. Upon hospital discharge, careful follow‐up should promote lifestyle changes and medication adherence to prevent recurrence of severe HTG.
The patient with a markedly high serum triglyceride (TG) level poses an interesting challenge for hospitalists. Hypertriglyceridemia (HTG) is defined as a fasting plasma TG level that is above the 95th percentile for age and sex.1 TG levels are commonly classified into categories according to Adult Treatment Panel III guidelines with desirable levels <150 mg/dL (1.7 mmol/L), borderline levels 150199, high levels 200499 mg/dL, and very high levels >500 mg/dL (5.6 mmol/L).2 A TG level exceeding an arbitrary threshold of >1000 mg/dL (11.3 mmol/L) is referred to as severe HTG. The Lipid Research Clinics Program Prevalence Study found that 1.79 per 10,000 outpatients (<0.02%) had TG levels > 2000 mg/dL.3 Chylomicronemia syndrome occurs when severe HTG is accompanied by 1 or more of the following: symptoms of abdominal pain or acute pancreatitis or physical examination findings such as eruptive xanthomas or lipemia retinalis. There is no TG level above which pancreatitis invariably occurs, making the decision to hospitalize difficult. The goal of this review is to discuss the causes of severe HTG; the clinical assessment, including criteria for hospitalization; and the available treatment options for this infrequent but serious condition. We begin with a clinical case of severe HTG.
CASE PRESENTATION
A 47‐year‐old woman with a history of chronic myelogenous leukemia was admitted to the hospital with a serum triglyceride level of 17,393 mg/dL. Two years prior to admission, she underwent allogenic stem cell transplantation for chronic myelogenous leukemia and has since remained in remission. Six months prior to admission, severe diarrhea from intestinal graft‐versus‐host disease required the use of total parenteral nutrition (TPN) and immunosuppressive therapy consisting of prednisone 20 mg/day, mycophenolate mofetil 250 mg thrice daily, and sirolimus 0.3 mg/day. During treatment with steroids, she developed diabetes mellitus requiring insulin, with a subsequent hemoglobin A1c level of 7.7% (normal, <7%). The serum TG level prior to transplantation was unknown but was 343 mg/dL prior to TPN initiation. One month prior to admission, the diarrhea resolved and TPN was stopped. The TG level was 7463 mg/dL 1 week prior to admission, and despite use of fenofibrate, it rose to 17,393 mg/dL. The patient denied abdominal pain, and did not have abdominal tenderness or eruptive xanthomas. She denied a family history of dyslipidemia or recent medication changes. Given the extreme TG elevation, the lack of response to outpatient treatment and the concern for developing acute pancreatitis, the patient was admitted to the hospital for inpatient TG‐lowering treatment.
Upon admission, serum lipase and amylase were within normal limits, but the blood glucose level was 243 mg/dL. Insulin infusion and oral fenofibrate 145 mg/day was started, and the patient was kept non per os (NPO). Six hours later, despite insulin infusion, the TG level rose to 26,250 mg/dL. Therapeutic plasma exchange (TPE) was performed on 2 consecutive days with a resultant decrease in TG level to 530 mg/dL. The patient was later discharged home on fenofibrate and omega‐3 ethyl esters, her same immunosuppressive and insulin regimen, and instructions for a very low‐fat diet. In the next 3 months, her serum TG level did not rise above 530 mg/dL. The cause of our patient's extreme TG elevation was likely a combination of genetic factors exacerbated by immunosuppressive and glucocorticoid therapy.
This case featured dramatic elevations in serum TG levels that the managing doctors believed merited a hospital admission. Management of patients with severe HTG first requires an understanding of TG metabolism.
ETIOLOGY
Serum TGs produced by the liver are carried by very low‐density lipoproteins (VLDLs), whereas TGs derived from dietary fat are carried by chylomicrons. Both chylomicrons and VLDLs are hydrolyzed by the same enzymelipoprotein lipase (LPL). TGs are hydrolyzed into fatty acids for uptake by muscle and adipose tissue, whereas remnants of VLDL and chylomicrons are removed by the liver. More details on TG pathophysiology may be found in a recent review.4 When LPL is saturated with VLDL, ingestion of a fatty meal may cause chylomicrons to linger in circulation for days instead of hours. Asking the laboratory to spin down the blood of a patient with severe HTG and keep the test tube upright at 4C may reveal a large creamy supernatant layer demonstrating chylomicronemia.
A fasting TG level drawn 12 hours after the last meal reflects hepatic TG production. Although a nonfasting TG level may reflect postprandial chylomicrons, values above 1000 mg/dL strongly suggest true HTG, particularly in the setting of acute pancreatitis. Treatment should not be delayed to obtain a fasting TG level.
HTG may result from increased VLDL production, reduced VLDL/chylomicron clearance, or more likely a combination of the two. The causes of these metabolic derangements are classified as primary (genetic) or secondary (acquired) (Table 1). In adult patients, HTG is usually the result of a combination of primary and secondary causes. A study of 123 patients with TG levels >2000 mg/dL found that all patients had a primary metabolic defect and 110/123 had a coexistent secondary cause.3 An underlying genetic lipoprotein metabolism derangement is often clinically silent until coupled with a secondary cause of HTG that together raise TG levels high enough to cause the chylomicronemia syndrome.
|
| Primary |
| Familial lipid disorders |
| Lipoprotein pattern type I |
| Familial chylomicronemia |
| Deficiency in LPL and/or apo‐CII |
| Autosomal recessive; presents in childhood |
| Rare functional disorders in LPL |
| Lipoprotein pattern type III |
| Familial dysbetalipoproteinemia |
| Inadequate VLDL clearance from apo‐E2 |
| Autosomal recessive; presents in adulthood |
| Lipoprotein pattern type IV |
| Familial HTG: increased VLDL |
| Autosomal dominant; presents in adulthood |
| Familial combined hyperlipidemia |
| Multiple phenotypes seen; increased apo‐B levels |
| Lipoprotein pattern type V |
| Mixed HTG: increased VLDL and chylomicrons; presents in adulthood |
| Secondary |
| Disease |
| Poorly controlled diabetes mellitus; hypothyroid; SLE; Cushing syndrome; HIV infection; sarcoid multiple myeloma; obesity; renal disease (nephrotic) |
| Disorder of metabolism |
| Pregnancy |
| Diet |
| Excessive alcohol, especially with high‐fat diet |
| Drugs |
| Estrogen; tamoxifen; glucocorticoids; protease inhibitors; nonselective beta‐blockers; propofol; isotretinoin; some antipsychotic medications (clozapine, olanzapine); tacrolimus; sirolimus; cyclosporine; bexarotene; all‐trans retinoic acid; L‐asparaginase; interferon‐ |
The most common primary cause of HTG in adults is familial HTG, an autosomal dominant condition with a population prevalence ranging from 1%2% to 5%10% and age‐dependent penetrance.5, 6 Other genetic causes are much rarer, such as LPL deficiency (1 in 1 million patients), apolipoprotein‐CII, and other mutations resulting in impaired binding to LPL.79 Primary causes of HTG are often listed as Fredrickson phenotypes (Table 1). Recent genome‐wide association studies reveal a complex polygenic basis to the Fredrickson categories and suggest additional undefined genes or nongenetic factors may significantly contribute to the final phenotype.10 Diagnosis of familial lipid disorders requires an accurate family history that may be difficult to obtain.
Secondary causes of HTG can be categorized using a four‐D mnemonic: Diseases, Diet, Disorder of Metabolism, and Drugs.11 The most common condition associated with HTG is obesity.6 The mechanism between obesity and HTG is complex and likely involves an increase in fatty acid flux from adipose tissue to other tissues and insulin resistance.12 Asking whether a patient's current weight is close to the heaviest lifetime weight is a clue to diagnosing obesity‐driven HTG. One case series of hypertriglyceridemic pancreatitis found that diabetes or excessive alcohol intake account for the majority of secondary causes of HTG.13 The cause of HTG among patients with diabetes is multifactorial: insulin deficiency reduces LPL levels (insulin is required for synthesis of LPL), whereas insulin resistance attenuates the ability of insulin to decrease hepatic cholesterol synthesis and thus increases hepatic secretion of VLDL.14 Alcohol impairs lipolysis and increases VLDL production that can lead to severe HTG, particularly in those patients with an underlying functional deficiency in LPL. Other secondary etiologies of severe HTG may be elicited through careful attention to medical and medication history.
CLINICAL ASSESSMENT
Table 2 proposes a reasonable initial assessment of the history and physical and laboratory tests in patients with severe HTG.
|
| History |
| Family history of lipid disorders |
| Maximal weight and when achieved |
| Detailed medication history (including those recently stopped) |
| Alcohol consumption |
| Diabetes mellitus |
| Possible physical examination findings |
| Eruptive xanthomas |
| Lipemia retinalis |
| Hepatomegaly |
| Lymphadenopathy |
| Laboratory tests |
| Basic Metabolic Panel* with glucose |
| Lipid panel |
| Thyroid‐stimulating hormone, free T4 |
| Liver function, amylase, lipase |
| Hemoglobin A1c |
| Urinalysis |
Distinguishing physical examination findings may arise when serum TG levels exceed 1000 mg/dL. Eruptive xanthoma form when large amounts of TG are sequestered in cutaneous histiocytes, resulting in small yellow‐orange papules with an erythematous base. This finding is seen in a minority of HTG patients and may be missed altogether without a careful examination of the extensor surfaces of arms, legs, back, and buttocks.15 Effective TG‐lowering treatment will result in resolution of these xanthomas. An ophthalmologic examination may reveal lipemia retinalis, a condition that occurs when retinal vessels appear white from lipemic serum and contrast against a pale salmon‐colored retina. Although a dramatic finding, these changes do not result in vision impairment. Hepatomegaly from fatty infiltration of the liver occurs frequently,16 and diffuse lymphadenopathy may also be found.17
Laboratory tests are also essential in the clinical assessment of severe HTG. Important tests include thyroid‐stimulating hormone, creatinine, serum urea nitrogen, and a urinalysis. A hemoglobin A1c test provides information on the level of glycemic control and is now recognized by the American Diabetes Association to diagnose diabetes mellitus.18 Liver function tests commonly reveal a transaminase elevation from underlying steatohepatitis and also provide a baseline value prior to initiating any lipid‐lowering medications. Additional diagnostic tests may be useful in selected patients: HIV testing, serum protein electrophoresis, and urine protein electrophoresis to help diagnose paraproteinemias such as multiple myeloma, and an antinuclear antibody and double‐stranded DNA for systemic lupus erythematosus.
Severe HTG may interfere with the result of 2 commonly obtained laboratory tests. The sodium concentration can be falsely low (pseudohyponatremia) due to the high levels of TG displacing sodium containing water from the plasma.19 Due to interference by plasma lipids, amylase levels may be near normal in up to 50% of patients with hypertriglyceridemic pancreatitis at the time of admission.20 Thus, if the suspicion for pancreatitis is high, it is reasonable to proceed to imaging if amylase or lipase levels are not confirmatory. Abdominal imaging with computed tomography or magnetic resonance imaging may be used to diagnose acute pancreatitis.
Behind excessive alcohol consumption and gallstone disease, HTG is the third leading cause of pancreatitis, accounting for up to 10% of cases in the general population.21 The exact mechanism by which HTG causes pancreatitis is unclear. One theory is that elevated plasma TG levels are hydrolyzed in the pancreas to cause an increase in local free fatty acids, which in turn may cause inflammation and overt pancreatitis.22 Another theory proposes that elevated levels of chylomicrons lead to plasma hyperviscosity, which causes ischemia and local acidosis in pancreatic capillaries.23 Whatever the cause, it is unclear why only some patients with severe HTG develop acute pancreatitis. One study of 129 patients with severe HTG found mean serum TG levels to be higher in patients with acute pancreatitis than in those without (4470 versus 2450 mg/dL), suggesting the threshold to develop acute pancreatitis is higher than previously thought.24 Without a firm TG threshold above which patients develop pancreatitis, the decision to hospitalize can be difficult.
WHEN TO HOSPITALIZE?
The choice of whether to hospitalize a patient with severe HTG is first based on the presence or absence of abdominal pain and/or acute pancreatitis. Figure 1 diagrams a suggested admission and treatment algorithm. If abdominal pain is present, the patient should be hospitalized and assessed for possible triggers with prompt initiation of pharmacologic treatment. In the absence of abdominal pain, the decision to admit the patient with severe HTG requires clinical judgment. In these cases, prompt consultation with a physician experienced in the management of lipid disorders is recommended. In our experience, admission is usually driven by factors such as (1) severe hyperglycemia requiring inpatient insulin therapy; (2) severe HTG at or near a level where pancreatitis has occurred in the past in a patient for whom adherence is suspect (mindful of the great variability at the levels where patients develop pancreatitis); (3) unremitting triggers of severe HTG such as ongoing use of essential medications also known to exacerbate HTG (such as some forms of chemotherapy) or pregnancy in the third trimester. TG levels rise continuously throughout pregnancy and peak during the third trimester, when hypertriglyceridemic pancreatitis most often occurs. Asymptomatic patients with severe HTG not requiring hospitalization need close outpatient follow‐up to prevent the onset of chylomicronemia syndrome.
INPATIENT MANAGEMENT
No professional recommendations exist regarding a standardized treatment plan for severe HTG. The treatment regimen is first based on the presence or absence of symptoms. Treatment of hypertriglyceridemic pancreatitis should target a serum TG level <1000 mg/dL and resolution of abdominal pain. The initial goal for asymptomatic patients is a TG level <1000 mg/dL, as this level represents a significant reduction in the risk of developing chylomicronemia syndrome. In either case, the first 2 components of the treatment regimen are dietary changes and oral medications.
Dietary Changes
Patients with hypertriglyceridemic pancreatitis should be made NPO, with the exception of necessary medications taken only with water to provide bowel rest and eliminate fat intake. As chylomicron production in the intestine falls, TG levels will fall dramatically within 12 days of NPO status regardless of other treatments. Once TG levels approach 1000 mg/dL and there is no residual abdominal pain, a no‐fat diet can be given. Patients with persistent abdominal pain requiring a prolonged fast (>57 days) may require nutrition through alternate means such as an enteral formula through a feeding tube or the use of TPN. If a feeding tube is required, we suggest beginning with an elemental, peptide‐based, fat‐free formula, with help from a nutrition consult to assist with individual tube‐feeding options.25 Enteral formula can be supplemented with medium‐chain triglyceride oils (found in coconut and palm kernel) to provide some additional nutritional support. MCTs do not raise serum TG levels, as they are absorbed directly into the portal vein for prompt oxidation by the liver, whereas long‐chain TGs are converted into chylomicrons for peripheral transport. One case report describes a dramatic therapeutic response to medium‐chain triglyceride oils in a patient with familial chylomicronemia,26 although we do not routinely recommend these oils as therapy given lack of long‐term safety data. Lastly, if TPN is required, it is crucial to avoid lipid emulsions to prevent a rise in serum TG levels.
Asymptomatic patients with severe HTG can be fed upon admission, but should be placed on a fat‐free diet. Fat is added back into the diet when TG levels fall below 1000 mg/dL and is slowly increased to a target fat content of 10% of the total calories, usually not exceeding 25 g/day.
Oral Medications
Oral medications should be initiated to lower TG levels for both symptomatic and asymptomatic patients. Table 3 lists the different classes of medications. In our experience, oral fibrates are a recommended first‐line treatment, with other agents used as adjunctive therapy. Through complex mechanisms, fibrates reduce hepatic VLDL secretion and increase serum lipolysis of TG.27 In patients who do not have diabetes and are at low risk for coronary heart disease (CHD), either gemfibrozil or fenofibrate may be used to lower serum TG levels. However, in patients with diabetes, CHD, or a CHD risk equivalent, use of fenofibrate is preferred as an HMG‐CoA reductase inhibitor (a statin) and will almost always be necessary to reach low‐density lipoprotein (LDL)‐cholesterol goals. Fenofibrate, unlike gemfibrozil, does not interfere with the glucuronidation of statins by the liver.28
| Drug | Usual Dose | TG Reduction | Cautions or Contraindications | Comments |
|---|---|---|---|---|
| ||||
| Fenofibrate | 130200 mg/day | 50% | Hepatic or renal insufficiency | Best fibrate to use with HMG‐CoA reductase Inhibitors |
| Gemfibrozil | 600 mg BID | 50% | Hepatic or renal insufficiency | Avoid combination with statins |
| HMG‐CoA reductase inhibitors or statins | Variable; the more potent LDL lowering, the more TG lowering | 25% | Decompensated cirrhosis; end‐stage renal disease | Not the primary treatment for patients with TG levels >1000 mg/dL; some statins such as atorvastatin and fluvastatin are favored for patients with renal insufficiency due to less renal excretion than other statins |
| Omega‐3 fatty acids | 2 g BID Lovaza (840 mg DHA/EPA per dose) | 25%50% with monotherapy (the higher the TG level, the greater the reduction); 30% with combination therapy | Allergy to fish | Fishy aftertaste; may cause flatulence; may increase serum glucose and LDL; low risk of clinical bleeding |
| Nicotinic acid | 12 g/day ER; up‐titrate from lowest dose | 15%35% | Active liver disease; active peptic ulcer disease; arterial bleeding | Can increase blood sugar levels by increasing insulin resistance |
| Orlistat (Xenical) | 120 mg TID | 15%35% | Cases of serious liver dysfunction have been reported | Can interfere with drug absorption, especially fat‐soluble vitamins; oily rectal discharge |
| Insulin | IV 0.10.3 U/kg/hr; titrate to serum glucose 140180 mg/dL OR basal/bolus subcutaneously | Variable; >50% in some cases | Hypoglycemia | Useful in patients who have diabetes |
If a fibrate fails to achieve an acceptable serum TG level, we recommend adjunctive therapy with omega‐3 fatty acid esters. Omega‐3 fatty acids lower serum TG levels by decreasing VLDL production and can lower TG levels by as much as 45% in cases of severe HTG.29 This medication is typically the first adjunctive medication chosen due to its low side effect profile.
If additional TG lowering is needed, niacin or nicotinic acid (vitamin B3) may be added next. This medication decreases VLDL production, lowers LDL, and increases high‐density lipoprotein, but invariably with initiation, patients exhibit prominent skin flushing, burning, or itching. This prostaglandin‐mediated effect may be prevented or at least reduced in severity by taking 325 mg aspirin 1 hour before niacin administration. If the TG level is still not at goal, orlistat, a lipase or fat blocker, may be useful.30 Orlistat improves postprandial lipemia through reduction of dietary fat absorption. Finally, although potent statins such as atorvastatin and rosuvastatin can lower TGs derived from VLDL substantially,31 their use should be prompted by the patient's risk for atherosclerotic cardiovascular disease. Because all of the hypotriglyceridemic medications can affect the liver, regular liver function testing is prudent, as is a periodic re‐evaluation of the ongoing need for these medications. Statin use causing mild transaminase elevation (up to 3 times the upper limit of normal) may be safely tolerated.32
Additional Inpatient Management
Insulin
Insulin increases lipoprotein lipase activity, thereby accelerating chylomicron degradation.33 Insulin (along with glucose if necessary to maintain euglycemia) is therefore a useful adjunctive TG‐lowering medication to oral medications, even in nondiabetic patients. Insulin administered intravenously should follow a titration protocol with hourly monitoring of blood glucose. The goal of the insulin protocol with severe HTG is not maintaining strict euglycemia but rather maintenance of LPL activation by exogenous insulin with avoidance of hypoglycemia. In hospitals where an insulin infusion protocol for diabetic ketoacidosis or postsurgical hyperglycemia already exists, the protocol can be applied for HTG management with minor modifications: introduce dextrose‐containing fluids at higher blood glucoses (180 mg/dL or less) and eliminate insulin boluses. A suggested dose is a continuous intravenous insulin drip at 0.10.3 U/kg/hr with glucose to maintain blood glucose levels between 140 and 180 mg/dL, although there are no guidelines from professional societies. Subcutaneous insulin has also been used to successfully lower TG levels.34, 35 The major limitation of subcutaneous administration is the inability to rapidly adjust the dosing when needed, which is particularly concerning when treating patients who do not have diabetes. We prefer to use subcutaneous basal insulin in patients requiring long‐term use of insulin after a significant TG reduction with intravenous insulin. Subcutaneous bolus prandial insulin should not be used until the patient has resumed a solid diet, because a liquid diet may not reliably contain enough carbohydrates for bolus therapy.
Intravenous Heparin
Heparin has been used in case reports as adjunctive treatment for hypertriglyceridemic pancreatitis.36, 37 Although heparin may increase circulating LPL levels, this effect is short‐lived and is quickly followed by increased hepatic LPL degradation.38 Therefore, the use of heparin to treat severe HTG cannot be routinely recommended.
Therapeutic Plasma Exchange
First used in 1978, therapeutic plasma exchange (TPE) has been demonstrated to quickly and dramatically lower serum TG levels.39 Since its first use, TPE has been used in several small case studies.4042 Without data from larger studies, the optimal frequency and duration of TPE remains unclear. One review suggests the use of TPE as first‐line therapy provided the patient is euglycemic, apheresis can be started within 48 hours of diagnosis, and the patient can tolerate the central venous access.43 On the other hand, guidelines from the American Society of Apheresis incorporating low‐quality evidence do not recommend TPE as routine first‐ or second‐line treatment for hypertriglyceridemic pancreatitis, but rather suggest the use of TPE on a case‐by‐case basis.44 When TPE is needed, this society recommends daily treatment for 13 days until an adequate postapheresis TG level is obtained. Although TPE rapidly lowers TG levels, it is also aggressive (requires placement of a pheresis catheter), expensive, and may not be readily available. We believe it remains an option for patients with severe HTG who do not respond readily to fat restriction, glycemic control with insulin, and pharmacologic treatment with a fibrate and omega‐3 fatty acids. In our judgment, routine use of TPE cannot be recommended without data from a randomized clinical trial examining the value of immediate lowering of TG levels with TPE versus the usually prompt fall in TG levels with less aggressive measures.
Discharge Planning
Typically, asymptomatic patients are discharged once their TG levels approach 1000 mg/dL. Patients recovering from hypertriglyceridemic pancreatitis may be discharged once they tolerate a no‐fat diet without recurrence of abdominal pain and without a significant TG increase above 1000 mg/dL.
Discharge Diet and Activity
At discharge, the diet should be high in fiber, fruits, vegetables, and lean protein, with fat intake restricted to approximately 10% of total calories. Insulin‐resistant patients and patients with diabetes should avoid sugar‐sweetened foods and drinks. Specifically, the daily amount of fructose intake should be no more than 50 mg to avoid a dose‐dependent increase in plasma TG levels compared with other sugars.4 At least 2 servings per week of marine foods naturally rich in omega‐3 fatty acids (fatty fish such as salmon or trout) are recommended. Nonmarine forms of omega‐3 fatty acids (walnuts, flaxseed), which have not demonstrated consistent reductions in TG, cannot be routinely recommended.4 In addition, alcohol consumption should be eliminated.
Once patients maintain a TG level near 500 mg/dL, we allow for dietary flexibility by slowly increasing the amount of dietary unsaturated fat. For patients with TG levels <500 mg/dL, Adult Treatment Panel III advocated restriction of daily dietary saturated fat levels to <7% and keeping the total fat level between 25% and 35%.2 A range in total fat was provided so that unsaturated fat could be increased to limit dietary carbohydrates if glycemic control was needed. In addition to dietary changes, counseling patients about the importance of physical activity and weight loss is crucial for long‐term management of severe HTG. TG lowering in response to diet and weight loss varies, but typically approximates 25%.45
Outpatient Medications
Patients without significant contraindications should be discharged on a fibrate and omega‐3 fatty acids. As mentioned, niacin, orlistat, and/or a statin may be used as adjunctive therapy. Despite use of these hypotriglyceridemic medications, secondary causes of HTG should be modified (such as removal of aggravating medications or appropriately treating uncontrolled diabetes) to yield lasting improvements in TG levels.
CONCLUSIONS
As the prevalence of obesity and diabetes continues to rise, so too does the clinical importance of proper management of severe HTG. Recognizing chylomicronemia syndromeone of the most dramatic consequences of lipid disordersand the underlying primary and secondary causes of HTG is required before starting treatment. Patients with severe HTG may require hospitalization for immediate reduction in TG levels and relief of abdominal pain, if present. Treatment involves modifying secondary causes, if possible, and eliminating dietary fat intake. Although use of medications such as an oral fibrate, omega‐3 fatty acids, and insulin are routine, the use of a more invasive procedure such as TPE should be considered on a case‐by‐case basis and may be limited by availability. Upon hospital discharge, careful follow‐up should promote lifestyle changes and medication adherence to prevent recurrence of severe HTG.
- ,,,.Pathophysiology of triglyceride‐rich lipoproteins in atherothrombosis: clinical aspects.Clin Cardiol.1999;22:II15–II20.
- Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III).JAMA.2001;285:2486–2497.
- ,.Chylomicronemia syndrome. Interaction of genetic and acquired hypertriglyceridemia.Med Clin North Am.1982;66:455–468.
- ,,, et al.Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association.Circulation.2011;123:2292–2333.
- .Familial lipoprotein lipase deficiency and other causes of the chylomicronemia syndrome. In: Scriver C, Beaudet A, Sly W, Valle D, eds.The Metabolic and Molecular Basis of Inherited Disease.7th ed.New York:McGraw‐Hill;1995:1913–1932.
- ,,.Hypertriglyceridemia: its etiology, effects and treatment.CMAJ.2007;176:1113–1120.
- ,,, et al.Chylomicronemia with a mutant GPIHBP1 (Q115P) that cannot bind lipoprotein lipase.Arterioscler Thromb Vasc Biol.2009;29:956–962.
- ,,, et al.A mutation in the human lipoprotein lipase gene as the most common cause of familial chylomicronemia in French Canadians.N Engl J Med.1991;324:1761–1766.
- ,,, et al.Inherited apolipoprotein A‐V deficiency in severe hypertriglyceridemia.Arterioscler Thromb Vasc Biol.2005;25:411–417.
- ,,, et al.A polygenic basis for four classical Fredrickson hyperlipoproteinemia phenotypes that are characterized by hypertriglyceridemia.Hum Mol Genet.2009;18:4189–4194.
- .Secondary causes of hyperlipidemia.Med Clin North Am.1994;78:117–141.
- ,,.Hypertriglyceridemic hyperapob: the unappreciated atherogenic dyslipoproteinemia in type 2 diabetes mellitus.Ann Intern Med.2001;135:447–459.
- .Hyperlipidemic pancreatitis.Gastroenterol Clin North Am.1990;19:783–791.
- ,,, et al.Effects of insulin on cholesterol synthesis in type II diabetes patients.Diabetes Care.1995;18:1362–1369.
- ,,,.Evidence for the chylomicron origin of lipids accumulating in diabetic eruptive xanthomas: a correlative lipid biochemical, histochemical, and electron microscopic study.J Clin Invest.1970;49:2172–2187.
- .Dyslipidaemia.Lancet.2003;362:717–731.
- ,,.Lymphadenopathy associated with severe hypertriglyceridemia.JAMA.1990;264:727–728.
- Diagnosis and classification of diabetes mellitus.Diabetes Care.2010;33(suppl 1):S62–S69.
- ,.Pseudohyponatremia in acute hyperlipemic pancreatitis. A potential pitfall in therapy.Arch Surg.1985;120:1053–1055.
- ,,.Suppression of amylase activity by hypertriglyceridemia.JAMA.1973;225:1331–1334.
- ,,,.Dyslipidaemic pancreatitis clinical assessment and analysis of disease severity and outcomes.Pancreatology.2009;9:252–257.
- .Pathogenesis, differentiation and management of hypertriglyceridemia.Adv Intern Med.1969;15:117–154.
- ,.Role of hypertriglyceridemia in the pathogenesis of experimental acute pancreatitis in rats.Int J Pancreatol.1996;20:177–184.
- ,,, et al.Acute pancreatitis in a cohort of 129 patients referred for severe hypertriglyceridemia.Pancreas.2008;37:13–12.
- ,,, et al.ESPEN Guidelines on Enteral Nutrition: pancreas.Clin Nutr.2006;25:275–284.
- ,,, et al.Therapeutic response to medium‐chain triglycerides and omega‐3 fatty acids in a patient with the familial chylomicronemia syndrome.Arterioscler Thromb Vasc Biol.1997;17:1400–1406.
- ,,,,,.Mechanism of action of fibrates on lipid and lipoprotein metabolism.Circulation.1998;98:2088–2093.
- ,,.Drug interactions with lipid‐lowering drugs: mechanisms and clinical relevance.Clin Pharmacol Ther.2006;80:565–81.
- ,,, et al.Safety and efficacy of Omacor in severe hypertriglyceridemia.J Cardiovasc Risk.1997;4:385–391.
- ,,.Usefulness of Orlistat in the treatment of severe hypertriglyceridemia.Am J Cardiol.2002;89:229–231.
- ,,, et al.Effects of intensive atorvastatin and rosuvastatin treatment on apolipoprotein B‐48 and remnant lipoprotein cholesterol levels.Atherosclerosis.2009;205:197–201.
- ,,,.Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force.Am J Cardiol.2006;97:89C–94C.
- .Lipoprotein lipase. A multifunctional enzyme relevant to common metabolic diseases.N Engl J Med.1989;320:1060–1068.
- ,,.Insulin therapy for a non‐diabetic patient with severe hypertriglyceridemia.J Am Coll Nutr.1998;17:458–461.
- ,,,,.Treatment of severe hypertriglyceridemia in nondiabetic patients with insulin.Am J Emerg Med.2005;23:415–417.
- ,.Decreasing the plasma triglyceride level in hypertriglyceridemia‐induced pancreatitis in pregnancy: a case report.Am J Obstet Gynecol.2002;187:241–242.
- ,,,,,.Pancreatitis may occur with a normal amylase concentration in hypertriglyceridaemia.BMJ.1996;313:1265.
- ,,,.Lipoprotein lipase during continuous heparin infusion: tissue stores become partially depleted.J Lab Clin Med.2001;138:206–13.
- ,,,,,.Treatment of severe diabetic hypertriglyceridaemia by plasma exchange.Lancet.1978;1:1368.
- ,,,.Therapeutic plasma exchange in patients with hyperlipidemic pancreatitis.World J Gastroenterol.2004;10:2272–2274.
- ,,.Plasma exchange in severe hypertriglyceridemia a clinical study.Transfus Apher Sci.2006;34:253–257.
- ,,, et al.Management of acute severe hyperlipidemic pancreatitis.Digestion.2006;73:259–264.
- ,,,,.Hypertriglyceridemic pancreatitis: presentation and management.Am J Gastroenterol.2009;104:984–991.
- ,,, et al.Guidelines on the use of therapeutic apheresis in clinical practice—evidence‐based approach from the Apheresis Applications Committee of the American Society for Apheresis.J Clin Apher.2010;25:83–177.
- ,,,,,.Effects of a low‐fat diet compared with those of a high‐monounsaturated fat diet on body weight, plasma lipids and lipoproteins, and glycemic control in type 2 diabetes.Am J Clin Nutr.2004;80:668–673.
- ,,,.Pathophysiology of triglyceride‐rich lipoproteins in atherothrombosis: clinical aspects.Clin Cardiol.1999;22:II15–II20.
- Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III).JAMA.2001;285:2486–2497.
- ,.Chylomicronemia syndrome. Interaction of genetic and acquired hypertriglyceridemia.Med Clin North Am.1982;66:455–468.
- ,,, et al.Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association.Circulation.2011;123:2292–2333.
- .Familial lipoprotein lipase deficiency and other causes of the chylomicronemia syndrome. In: Scriver C, Beaudet A, Sly W, Valle D, eds.The Metabolic and Molecular Basis of Inherited Disease.7th ed.New York:McGraw‐Hill;1995:1913–1932.
- ,,.Hypertriglyceridemia: its etiology, effects and treatment.CMAJ.2007;176:1113–1120.
- ,,, et al.Chylomicronemia with a mutant GPIHBP1 (Q115P) that cannot bind lipoprotein lipase.Arterioscler Thromb Vasc Biol.2009;29:956–962.
- ,,, et al.A mutation in the human lipoprotein lipase gene as the most common cause of familial chylomicronemia in French Canadians.N Engl J Med.1991;324:1761–1766.
- ,,, et al.Inherited apolipoprotein A‐V deficiency in severe hypertriglyceridemia.Arterioscler Thromb Vasc Biol.2005;25:411–417.
- ,,, et al.A polygenic basis for four classical Fredrickson hyperlipoproteinemia phenotypes that are characterized by hypertriglyceridemia.Hum Mol Genet.2009;18:4189–4194.
- .Secondary causes of hyperlipidemia.Med Clin North Am.1994;78:117–141.
- ,,.Hypertriglyceridemic hyperapob: the unappreciated atherogenic dyslipoproteinemia in type 2 diabetes mellitus.Ann Intern Med.2001;135:447–459.
- .Hyperlipidemic pancreatitis.Gastroenterol Clin North Am.1990;19:783–791.
- ,,, et al.Effects of insulin on cholesterol synthesis in type II diabetes patients.Diabetes Care.1995;18:1362–1369.
- ,,,.Evidence for the chylomicron origin of lipids accumulating in diabetic eruptive xanthomas: a correlative lipid biochemical, histochemical, and electron microscopic study.J Clin Invest.1970;49:2172–2187.
- .Dyslipidaemia.Lancet.2003;362:717–731.
- ,,.Lymphadenopathy associated with severe hypertriglyceridemia.JAMA.1990;264:727–728.
- Diagnosis and classification of diabetes mellitus.Diabetes Care.2010;33(suppl 1):S62–S69.
- ,.Pseudohyponatremia in acute hyperlipemic pancreatitis. A potential pitfall in therapy.Arch Surg.1985;120:1053–1055.
- ,,.Suppression of amylase activity by hypertriglyceridemia.JAMA.1973;225:1331–1334.
- ,,,.Dyslipidaemic pancreatitis clinical assessment and analysis of disease severity and outcomes.Pancreatology.2009;9:252–257.
- .Pathogenesis, differentiation and management of hypertriglyceridemia.Adv Intern Med.1969;15:117–154.
- ,.Role of hypertriglyceridemia in the pathogenesis of experimental acute pancreatitis in rats.Int J Pancreatol.1996;20:177–184.
- ,,, et al.Acute pancreatitis in a cohort of 129 patients referred for severe hypertriglyceridemia.Pancreas.2008;37:13–12.
- ,,, et al.ESPEN Guidelines on Enteral Nutrition: pancreas.Clin Nutr.2006;25:275–284.
- ,,, et al.Therapeutic response to medium‐chain triglycerides and omega‐3 fatty acids in a patient with the familial chylomicronemia syndrome.Arterioscler Thromb Vasc Biol.1997;17:1400–1406.
- ,,,,,.Mechanism of action of fibrates on lipid and lipoprotein metabolism.Circulation.1998;98:2088–2093.
- ,,.Drug interactions with lipid‐lowering drugs: mechanisms and clinical relevance.Clin Pharmacol Ther.2006;80:565–81.
- ,,, et al.Safety and efficacy of Omacor in severe hypertriglyceridemia.J Cardiovasc Risk.1997;4:385–391.
- ,,.Usefulness of Orlistat in the treatment of severe hypertriglyceridemia.Am J Cardiol.2002;89:229–231.
- ,,, et al.Effects of intensive atorvastatin and rosuvastatin treatment on apolipoprotein B‐48 and remnant lipoprotein cholesterol levels.Atherosclerosis.2009;205:197–201.
- ,,,.Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force.Am J Cardiol.2006;97:89C–94C.
- .Lipoprotein lipase. A multifunctional enzyme relevant to common metabolic diseases.N Engl J Med.1989;320:1060–1068.
- ,,.Insulin therapy for a non‐diabetic patient with severe hypertriglyceridemia.J Am Coll Nutr.1998;17:458–461.
- ,,,,.Treatment of severe hypertriglyceridemia in nondiabetic patients with insulin.Am J Emerg Med.2005;23:415–417.
- ,.Decreasing the plasma triglyceride level in hypertriglyceridemia‐induced pancreatitis in pregnancy: a case report.Am J Obstet Gynecol.2002;187:241–242.
- ,,,,,.Pancreatitis may occur with a normal amylase concentration in hypertriglyceridaemia.BMJ.1996;313:1265.
- ,,,.Lipoprotein lipase during continuous heparin infusion: tissue stores become partially depleted.J Lab Clin Med.2001;138:206–13.
- ,,,,,.Treatment of severe diabetic hypertriglyceridaemia by plasma exchange.Lancet.1978;1:1368.
- ,,,.Therapeutic plasma exchange in patients with hyperlipidemic pancreatitis.World J Gastroenterol.2004;10:2272–2274.
- ,,.Plasma exchange in severe hypertriglyceridemia a clinical study.Transfus Apher Sci.2006;34:253–257.
- ,,, et al.Management of acute severe hyperlipidemic pancreatitis.Digestion.2006;73:259–264.
- ,,,,.Hypertriglyceridemic pancreatitis: presentation and management.Am J Gastroenterol.2009;104:984–991.
- ,,, et al.Guidelines on the use of therapeutic apheresis in clinical practice—evidence‐based approach from the Apheresis Applications Committee of the American Society for Apheresis.J Clin Apher.2010;25:83–177.
- ,,,,,.Effects of a low‐fat diet compared with those of a high‐monounsaturated fat diet on body weight, plasma lipids and lipoproteins, and glycemic control in type 2 diabetes.Am J Clin Nutr.2004;80:668–673.