User login
The psychiatry workforce pool is shrinking. What are we doing about it?
The dilemma of a diminishing workforce pool might seem more the province of medical school deans, psychiatry department chairs, and psychiatry residency training directors, but our ability to recruit and retain psychiatrists is, in reality, everyone’s concern—including hospitals, clinics, and, especially, patients and their families. Even without knowledge of the specialty or any numerical appraisal, for example, it is common knowledge that we have a dire shortage of child and adolescent and geriatric psychiatrists—a topic of widespread interest and great consequence for access to mental health care.
Tracking a decline
The very title of a recent provocative paper1 in Health Affairs says it all: “Population of US practicing psychiatrists declined 2003-13, which may help explain poor access to mental health care.” In an elegant analysis, the authors expose (1) a 10% decline in the number of psychiatrists for every 100,000 people and (2) wide regional variability in the availability of psychiatrists. In stark contrast, the number of neurologists increased by >15% and the primary care workforce remained stable, with a 1.3% increase in the number of physicians, over the same 10 years.
At the beginning of the psychiatry workforce pipeline, the number of medical students who choose psychiatry remains both small (typically, slightly more than 4% of graduating students) and remarkably stable over time. Wilbanks et al,2 in a thoughtful analysis of the 2011 to 2013 Medical School Graduation Questionnaire of the Association of American Medical Colleges, affirm and, in part, explain this consistent pattern. They note that the 4 most important considerations among students who select psychiatry are:
- personality fit
- specialty content
- work–life balance
- role model influences.
Some of these considerations also overlap with those of students in other specialties; the authors also note that older medical students and women are more likely to choose psychiatry.
Here is what we must do to erase the shortage
It does appear that, despite scientific advances in brain and behavior, expanding therapeutic options, and unique patient interactions that, taken together, should make a career in psychiatry exciting and appealing, there are simply not enough of us to meet the population’s mental health needs. This is a serious problem. It is our professional obligation—all of us—that we take on this shortage and develop solutions to it.
At its zenith, only about 7% of medical students chose psychiatry. We need to proactively prime the pump for our specialty by encouraging more observerships and promoting mental health careers through community outreach to high school students.
We must be diligent and effective mentors to medical students; mentorship is a powerful catalyst for career decision-making.
We need to make psychiatry clerkships exciting, to show off the best of what our specialty has to offer, and to cultivate sustained interest among our students in the brain and its psychiatric disorders.
We need to highlight the momentous advances in knowledge, biology, and treatments that now characterize our psychiatric profession. We need to advocate for more of these accomplishments.
We must be public stigma-busters! (Our patients need us to do this, too.)
And there is more to do:
Collaborate. In delivering psychiatric health care, we need to expand our effectiveness to achieve more collaboration, greater extension of effect, and broader outreach. Collaborative care has come of age as a delivery model; it should be embraced more broadly. We need to continue our efforts to bridge the many sister mental health disciplines—psychology, nursing, social work, counseling—that collectively provide mental health care.
Unite. Given the inadequate workforce numbers and enormous need, we will diminish ourselves by “guild infighting” and, consequently, weaken our legislative advocacy and leverage. We need to embrace and support all medical specialties and have them support us as well. We need to grow closer to primary care and support this specialty as the true front line of mental health. We also need to bridge the gap between addiction medicine and psychiatry, especially given the high level of addiction comorbidity in many psychiatric disorders.
Foster innovation. The deficit of psychiatric workers might be buffered by innovations in how we leverage our expertise. Telepsychiatry, for example, is clearly advancing, and brings psychiatry to remote areas where psychiatrists are scarce. Mobile health also has great potential for mental health. As one of us (H.A.N.) highlighted recently,3 as genetics become more molecular, what has been the potential of clinically applicable pharmacogenomics might become reality. Psychiatry needs to make progress toward personalized medicine because the disorders we treat are extremely heterogeneous in their etiology, phenomenology, treatment response, and outcomes.
The appeal of working with mind and brain
The extent to which we can convey unfettered optimism about the role of psychiatry in medicine and the relentless progress in neurobiological research, together, will go a long way toward attracting the best and brightest newly minted physicians to our specialty. The brain is the last frontier in medicine; psychiatry is intimately tethered to its unfolding complexity. With millennials placing a higher premium on work–life issues, the enviable balance and quality of life of a psychiatric career might now be particularly opportune, enhancing the quantity and quality of professionals that we can attract to psychiatry.
1. Bishop TF, Seirup JK, Pincus HA, et al. Population of US practicing psychiatrist declined, 2003-13, which may help explain poor access to mental health care. Health Aff (Millwood). 2016;35(7):1271-1277.
2. Wilbanks L, Spollen J, Messias E. Factors influencing medical school graduates toward a career in psychiatry: analysis from the 2011-2013 Association of American Medical Colleges Graduation Questionnaire. Acad Psychiatry. 2016;40(2):255-260.
3. Nasrallah HA. ‘Druggable’ genes, promiscuous drugs, repurposed medications. Current Psychiatry. 2016;15(5):23,41.

Consulting Editor

Editor-in-Chief
Consulting Editor
Editor-in-Chief
Consulting Editor
Editor-in-Chief
The dilemma of a diminishing workforce pool might seem more the province of medical school deans, psychiatry department chairs, and psychiatry residency training directors, but our ability to recruit and retain psychiatrists is, in reality, everyone’s concern—including hospitals, clinics, and, especially, patients and their families. Even without knowledge of the specialty or any numerical appraisal, for example, it is common knowledge that we have a dire shortage of child and adolescent and geriatric psychiatrists—a topic of widespread interest and great consequence for access to mental health care.
Tracking a decline
The very title of a recent provocative paper1 in Health Affairs says it all: “Population of US practicing psychiatrists declined 2003-13, which may help explain poor access to mental health care.” In an elegant analysis, the authors expose (1) a 10% decline in the number of psychiatrists for every 100,000 people and (2) wide regional variability in the availability of psychiatrists. In stark contrast, the number of neurologists increased by >15% and the primary care workforce remained stable, with a 1.3% increase in the number of physicians, over the same 10 years.
At the beginning of the psychiatry workforce pipeline, the number of medical students who choose psychiatry remains both small (typically, slightly more than 4% of graduating students) and remarkably stable over time. Wilbanks et al,2 in a thoughtful analysis of the 2011 to 2013 Medical School Graduation Questionnaire of the Association of American Medical Colleges, affirm and, in part, explain this consistent pattern. They note that the 4 most important considerations among students who select psychiatry are:
- personality fit
- specialty content
- work–life balance
- role model influences.
Some of these considerations also overlap with those of students in other specialties; the authors also note that older medical students and women are more likely to choose psychiatry.
Here is what we must do to erase the shortage
It does appear that, despite scientific advances in brain and behavior, expanding therapeutic options, and unique patient interactions that, taken together, should make a career in psychiatry exciting and appealing, there are simply not enough of us to meet the population’s mental health needs. This is a serious problem. It is our professional obligation—all of us—that we take on this shortage and develop solutions to it.
At its zenith, only about 7% of medical students chose psychiatry. We need to proactively prime the pump for our specialty by encouraging more observerships and promoting mental health careers through community outreach to high school students.
We must be diligent and effective mentors to medical students; mentorship is a powerful catalyst for career decision-making.
We need to make psychiatry clerkships exciting, to show off the best of what our specialty has to offer, and to cultivate sustained interest among our students in the brain and its psychiatric disorders.
We need to highlight the momentous advances in knowledge, biology, and treatments that now characterize our psychiatric profession. We need to advocate for more of these accomplishments.
We must be public stigma-busters! (Our patients need us to do this, too.)
And there is more to do:
Collaborate. In delivering psychiatric health care, we need to expand our effectiveness to achieve more collaboration, greater extension of effect, and broader outreach. Collaborative care has come of age as a delivery model; it should be embraced more broadly. We need to continue our efforts to bridge the many sister mental health disciplines—psychology, nursing, social work, counseling—that collectively provide mental health care.
Unite. Given the inadequate workforce numbers and enormous need, we will diminish ourselves by “guild infighting” and, consequently, weaken our legislative advocacy and leverage. We need to embrace and support all medical specialties and have them support us as well. We need to grow closer to primary care and support this specialty as the true front line of mental health. We also need to bridge the gap between addiction medicine and psychiatry, especially given the high level of addiction comorbidity in many psychiatric disorders.
Foster innovation. The deficit of psychiatric workers might be buffered by innovations in how we leverage our expertise. Telepsychiatry, for example, is clearly advancing, and brings psychiatry to remote areas where psychiatrists are scarce. Mobile health also has great potential for mental health. As one of us (H.A.N.) highlighted recently,3 as genetics become more molecular, what has been the potential of clinically applicable pharmacogenomics might become reality. Psychiatry needs to make progress toward personalized medicine because the disorders we treat are extremely heterogeneous in their etiology, phenomenology, treatment response, and outcomes.
The appeal of working with mind and brain
The extent to which we can convey unfettered optimism about the role of psychiatry in medicine and the relentless progress in neurobiological research, together, will go a long way toward attracting the best and brightest newly minted physicians to our specialty. The brain is the last frontier in medicine; psychiatry is intimately tethered to its unfolding complexity. With millennials placing a higher premium on work–life issues, the enviable balance and quality of life of a psychiatric career might now be particularly opportune, enhancing the quantity and quality of professionals that we can attract to psychiatry.
The dilemma of a diminishing workforce pool might seem more the province of medical school deans, psychiatry department chairs, and psychiatry residency training directors, but our ability to recruit and retain psychiatrists is, in reality, everyone’s concern—including hospitals, clinics, and, especially, patients and their families. Even without knowledge of the specialty or any numerical appraisal, for example, it is common knowledge that we have a dire shortage of child and adolescent and geriatric psychiatrists—a topic of widespread interest and great consequence for access to mental health care.
Tracking a decline
The very title of a recent provocative paper1 in Health Affairs says it all: “Population of US practicing psychiatrists declined 2003-13, which may help explain poor access to mental health care.” In an elegant analysis, the authors expose (1) a 10% decline in the number of psychiatrists for every 100,000 people and (2) wide regional variability in the availability of psychiatrists. In stark contrast, the number of neurologists increased by >15% and the primary care workforce remained stable, with a 1.3% increase in the number of physicians, over the same 10 years.
At the beginning of the psychiatry workforce pipeline, the number of medical students who choose psychiatry remains both small (typically, slightly more than 4% of graduating students) and remarkably stable over time. Wilbanks et al,2 in a thoughtful analysis of the 2011 to 2013 Medical School Graduation Questionnaire of the Association of American Medical Colleges, affirm and, in part, explain this consistent pattern. They note that the 4 most important considerations among students who select psychiatry are:
- personality fit
- specialty content
- work–life balance
- role model influences.
Some of these considerations also overlap with those of students in other specialties; the authors also note that older medical students and women are more likely to choose psychiatry.
Here is what we must do to erase the shortage
It does appear that, despite scientific advances in brain and behavior, expanding therapeutic options, and unique patient interactions that, taken together, should make a career in psychiatry exciting and appealing, there are simply not enough of us to meet the population’s mental health needs. This is a serious problem. It is our professional obligation—all of us—that we take on this shortage and develop solutions to it.
At its zenith, only about 7% of medical students chose psychiatry. We need to proactively prime the pump for our specialty by encouraging more observerships and promoting mental health careers through community outreach to high school students.
We must be diligent and effective mentors to medical students; mentorship is a powerful catalyst for career decision-making.
We need to make psychiatry clerkships exciting, to show off the best of what our specialty has to offer, and to cultivate sustained interest among our students in the brain and its psychiatric disorders.
We need to highlight the momentous advances in knowledge, biology, and treatments that now characterize our psychiatric profession. We need to advocate for more of these accomplishments.
We must be public stigma-busters! (Our patients need us to do this, too.)
And there is more to do:
Collaborate. In delivering psychiatric health care, we need to expand our effectiveness to achieve more collaboration, greater extension of effect, and broader outreach. Collaborative care has come of age as a delivery model; it should be embraced more broadly. We need to continue our efforts to bridge the many sister mental health disciplines—psychology, nursing, social work, counseling—that collectively provide mental health care.
Unite. Given the inadequate workforce numbers and enormous need, we will diminish ourselves by “guild infighting” and, consequently, weaken our legislative advocacy and leverage. We need to embrace and support all medical specialties and have them support us as well. We need to grow closer to primary care and support this specialty as the true front line of mental health. We also need to bridge the gap between addiction medicine and psychiatry, especially given the high level of addiction comorbidity in many psychiatric disorders.
Foster innovation. The deficit of psychiatric workers might be buffered by innovations in how we leverage our expertise. Telepsychiatry, for example, is clearly advancing, and brings psychiatry to remote areas where psychiatrists are scarce. Mobile health also has great potential for mental health. As one of us (H.A.N.) highlighted recently,3 as genetics become more molecular, what has been the potential of clinically applicable pharmacogenomics might become reality. Psychiatry needs to make progress toward personalized medicine because the disorders we treat are extremely heterogeneous in their etiology, phenomenology, treatment response, and outcomes.
The appeal of working with mind and brain
The extent to which we can convey unfettered optimism about the role of psychiatry in medicine and the relentless progress in neurobiological research, together, will go a long way toward attracting the best and brightest newly minted physicians to our specialty. The brain is the last frontier in medicine; psychiatry is intimately tethered to its unfolding complexity. With millennials placing a higher premium on work–life issues, the enviable balance and quality of life of a psychiatric career might now be particularly opportune, enhancing the quantity and quality of professionals that we can attract to psychiatry.
1. Bishop TF, Seirup JK, Pincus HA, et al. Population of US practicing psychiatrist declined, 2003-13, which may help explain poor access to mental health care. Health Aff (Millwood). 2016;35(7):1271-1277.
2. Wilbanks L, Spollen J, Messias E. Factors influencing medical school graduates toward a career in psychiatry: analysis from the 2011-2013 Association of American Medical Colleges Graduation Questionnaire. Acad Psychiatry. 2016;40(2):255-260.
3. Nasrallah HA. ‘Druggable’ genes, promiscuous drugs, repurposed medications. Current Psychiatry. 2016;15(5):23,41.
1. Bishop TF, Seirup JK, Pincus HA, et al. Population of US practicing psychiatrist declined, 2003-13, which may help explain poor access to mental health care. Health Aff (Millwood). 2016;35(7):1271-1277.
2. Wilbanks L, Spollen J, Messias E. Factors influencing medical school graduates toward a career in psychiatry: analysis from the 2011-2013 Association of American Medical Colleges Graduation Questionnaire. Acad Psychiatry. 2016;40(2):255-260.
3. Nasrallah HA. ‘Druggable’ genes, promiscuous drugs, repurposed medications. Current Psychiatry. 2016;15(5):23,41.

Which antipsychotic do I choose next?
CATIE phase 2 offers insights on efficacy an tolerability
After nearly 3 out of 4 phase 1 patients stopped taking their assigned antipsychotics within 18 months, researchers in the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) braced themselves for phase 2.February 2006)
CATIE’s eligibility criteria are broad and include schizophrenia patients with comorbid conditions such as substance abuse and mood disorders. The primary outcome measure is all-cause treatment discontinuation, which incorporates efficacy, safety, tolerability, patient choice, and clinician choice (Table 1).
Phase 1 compared the efficacy and safety of four second-generation antipsychotics (SGA) and one first-generation antipsychotic (FGA).3 Nasrallah concluded that—despite the high discontinuation rate in that phase—there were “no winners or losers” among the five antipsychotics. The results, Nasrallah concluded:
- provide a compelling rationale for clinicians to match medication profiles to individual patients
- support the need for clinicians to have choices among medications when treating patients with schizophrenia.4
Table 1
Drug discontinuation patterns in CATIE phase 1
| Measures | Findings after 18 months | |
|---|---|---|
| % of patients who discontinued medication for any reason | Olanzapine (64%) | Ziprasidone (79%) |
| Risperidone (74%) | Quetiapine (82%) | |
| Perphenazine (75%) | ||
| Time to discontinuation for any reason | Longest (most favorable) with olanzapine, but not statistically longer with olanzapine than with ziprasidone or perphenazine | |
| No statistical difference among risperidone, quetiapine, ziprasidone, and perphenazine | ||
| Time to discontinuation for lack of efficacy* | Longer with olanzapine; no statistical difference among risperidone, quetiapine, ziprasidone, and perphenazine | |
| Time to discontinuation for intolerable side effects | No statistical difference among agents | |
| Rate of discontinuation for intolerable side effects | Highest (19%) with olanzapine (primarily because of weight gain or metabolic effects with this medication) | |
| Rate of discontinuation for extrapyramidal effects | Highest (8%) with perphenazine | |
| Rate of discontinuation for intolerability (overall) | Lowest with risperidone (10%) | |
| * Nonequivalent dosing in CATIE phase 1 is an ongoing debate. | ||
What to do next?
When an initial antipsychotic proves inadequate or causes intolerable side effects, how do you choose a more efficacious or tolerable medication? Phase 2 offered CATIE patients and their clinicians two choices—an efficacy and a tolerability pathway (Figure).1,2
CATIE phase 2: Distribution of patients in efficacy and tolerability pathways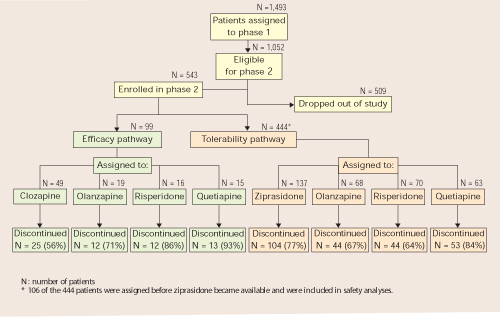
Efficacy pathway. Patients who chose the efficacy pathway were randomly assigned to clozapine (50%) or olanzapine, risperidone, or quetiapine.1 Researchers selected clozapine as the major efficacy comparator because of its robust effects in treatment-refractory schizophrenia. Clozapine was given open-label because of its safety monitoring requirements; other treatments were double-blind.
As in phase 1, the primary outcome measure was time until discontinuation for any reason. Secondary outcome measures included time to discontinuation because of side effects, patient choice, or lack of efficacy.
Tolerability pathway. Patients who chose the tolerability pathway were randomly assigned to double-blind treatment with ziprasidone, olanzapine, risperidone, or quetiapine.2 Ziprasidone was the major comparator because of clinical data showing a favorable tolerability profile.
The primary outcome measure was time to discontinuation for any reason. Secondary outcomes included reason for discontinuation (as determined by the study clinician), symptomatic ratings, and evaluations of adverse effects.
Trial duration. No patients in either pathway received the same antipsychotics they had taken in phase 1. All patients could continue treatment through the 18 months of the CATIE trial or until they completed 6 months in phase 2.
Efficacy pathway results
Discontinuation. Consistent with literature about its efficacy in treatment-refractory schizophrenia, clozapine showed a robust clinical effect. Overall, more patients receiving clozapine stayed on treatment and for longer periods, compared with patients receiving olanzapine, risperidone, or quetiapine (Table 2).
On secondary measures, discontinuation for lack of efficacy was significantly lower with clozapine (11%) than with:
- olanzapine (35%)
- risperidone or quetiapine (each at 43%).
Discontinuation rates because of adverse effects or by patient choice were the same across all medications (Table 3). Patients on clozapine achieved better ratings in overall psychotic symptoms, positive symptoms, and general function, but not in negative symptoms.
Weight gain. On average, patients gained more weight while taking olanzapine (+1.1 lb/mo) than with:
- risperidone (+0.5 lb/mo)
- clozapine (+0.5 lb/mo)
- quetiapine (+0.5 lb/mo)
Differences in weight gain—or in metabolic parameters or other adverse effects—were not statistically significant, however.
Table 2
Phase 2 efficacy pathway: Discontinuation for any reason
| Measure | Clozapine | Olanzapine | Risperidone | Quetiapine |
|---|---|---|---|---|
| How many patients discontinued | 25 of 49 (56%) | 12 of 19 (71%) | 12 of 16 (86%) | 13 of 15 (93%) |
| Median time to discontinuation | 10.5 months | 2.7 months | 2.8 months | 3.3 months |
Table 3
Reasons patients stopped taking their medications in CATIE phase 2
| Reason | Efficacy pathway | Tolerability pathway |
|---|---|---|
| All cause | 69% | 74% |
| Lack of efficacy | 26% | 29% |
| Lack of tolerability | 10% | 15% |
| Patient choice | 26% | 24% |
Tolerability pathway results
Discontinuation. Patients in the tolerability pathway took olanzapine or risperidone significantly longer—median 6.3 and 7 months, respectively— compared with ziprasidone (4 months) or quetiapine (2.8 months).
- Time to discontinuation during phase 2 was the same across all drugs among patients who entered phase 2 because of intolerable side effects in phase 1.
- Time to discontinuation because of side effects also was similar whether patients discontinued phase 1 for lack of efficacy or intolerable side effects. Patients stopped treatment in the efficacy and tolerability pathways for similar reasons (Table 3).
Weight gain. Patients taking olanzapine gained more weight (average +1.3 lb/mo) than did those taking the other drugs. Patients taking ziprasidone lost weight (average –1.7 lb/mo). Among 61 patients who gained weight during phase 1, 42% of those switched to ziprasidone lost weight in phase 2, as did:
- 20% of those switched to risperidone
- 7% of those switched to quetiapine.
Among those switched to olanzapine in phase 2, no one lost weight and 2% gained weight.
Metabolic effects. Some parameters changed, depending on drug assignment:
- prolactin increased in patients switched to risperidone
- cholesterol and triglycerides increased in patients switched to olanzapine or quetiapine but decreased in those switched to risperidone or ziprasidone
- QTc interval measurements showed no difference across all drugs.
Methodologic caveats
When considering how CATIE’s phase 2 findings might apply to clinical practice, keep in mind four caveats about the study’s design.
Clozapine was given open-label, yet quetiapine, olanzapine, and risperidone were given double-blind in the efficacy pathway. This pathway’s findings are consistent with what we know about clozapine and other SGAs in treatment-refractory schizophrenia, but how the open-label design affected clozapine therapy outcomes is unclear.
Were patients who knew they were taking clozapine more willing to “stay the course” than were patients in the pathway’s double-blind arm?
Discontinuation rates remained high. The 74% “overall discontinuation rate” in phase 1 surprised many psychiatrists because of the perceived high rate at which patients did not adhere to the first medications they received. To some extent, the word “discontinuation” is imprecise, however, because this group includes patients who did not drop out of treatment altogether but chose to move on to phase 2.
It is important to note, however, that nearly one-half of phase 1 patients who were eligible to enter phase 2 (509 of 1,052) did not. This group represents the true drop-out rate, which is substantial. The high rates of discontinuation seen in phase 1 also occurred in both phase 2 pathways (Table 3).
Few patients entered the efficacy pathway. In an approach designed to reflect routine clinical practice, the researchers recommended the efficacy pathway to patients who discontinued phase 1 because of lack of efficacy and the tolerability pathway to those who discontinued phase 1 because of intolerability. Many patients did not follow the recommendations, however, and seemed to choose their pathways based on whether they wanted a chance to receive clozapine or ziprasidone in phase 2.
Thus, among the 543 phase 1 patients who enrolled in phase 2, 99 (18%) entered the efficacy pathway, and 444 (82%) entered the tolerability pathway. The efficacy pathway included 85 patients who discontinued phase 1 for lack of efficacy and 5 for lack of tolerability. The tolerability pathway included 184 patients who discontinued phase 1 for lack of efficacy and 168 for lack of tolerability.
Dosages may not have been equivalent. SGAs’ dosing equivalency is unknown,5,6 which impedes our ability to interpret comparative studies such as CATIE. The study’s designers developed the its dosing ranges by careful consideration, including recommendations from each SGA’s manufacturer. As Nasrallah described,4 the trial’s dosages were not universally consistent with FDA-approved ranges or usual clinical practice (Table 4). In phase 2, for example, ziprasidone dosages were less than psychiatrists usually use, and quetiapine dosages were greater than usual.
Fortunately, studies are underway to determine each SGA’s optimum dosing. This work will help us understand what we can expect when we increase an antipsychotic’s dosage—a key step towards understanding dosing equivalency.
Table 4
Mean modal antipsychotic dosages (mg/d) in CATIE phase 2 pathways*
| Clozapine | Ziprasidone | Olanzapine | Risperidone | Quetiapine | |
|---|---|---|---|---|---|
| Efficacy pathway | 332 | — | 23.4 | 4.8 | 642.9 |
| Tolerability pathway | — | 115.9 | 20.5 | 4.15 | 65.2 |
| * 800 mg/d of quetiapine and 160 mg/d of ziprasidone are generally regarded as therapeutically equivalent to 20 mg/d of olanzapine. | |||||
What clinicians can expect
A recent analysis helps put CATIE’s findings in perspective. Citrome and Stroup7 quantified the results of phase 1 and 2 with respect to:
- number needed to treat (NNT)—how many patients a clinician needs to treat with drug A to see one additional benefit, compared with drug B
- number needed to harm (NNH)—how many patients a clinician needs to treat with drug A to see a given adverse effect, compared with drug B.
In this analysis, the NNT for olanzapine (5.5 to 10) was lowest among the drugs compared in phase 1, and the NNT for clozapine (3) was lowest among those compared in phase 2. A lower number means that, overall, clinicians can expect a more robust treatment response.
On the other hand, the NNH for olanzapine in weight gain and metabolic disturbances (12.4 to 17.7) was the lowest in phase 1, indicating that clinicians can expect more weight gain and metabolic effects with olanzapine than with other SGAs. Ziprasidone had the highest NNH (106 to 208) among the agents in phase 2 for avoiding discontinuation because of weight gain or metabolic disturbances. In other words, ziprasidone appears less likely than other SGAs to cause metabolic problems.
These risk-attribution measures demonstrate the dilemma clinicians face when trying to match schizophrenia patients with antipsychotics. CATIE was “an N of 1,493” subjects, whereas each patient we see in clinical practice is “an N of 1.” One patient may need a more-robust response; another may need improved tolerability.
We strive for balance, seeking to optimize efficacy—often by raising the dosage—while minimizing adverse effects.
What to tell patients
CATIE phases 1 and 2 provide a compelling rationale for individualized treatment, which should be standard clinical practice for schizophrenia:
- All drugs used in phases 1 and 2 worked.
- All showed noteworthy adverse effects that were different for each drug.
- Different patients responded differently to each drug.
Using our clinical judgment and available information, we must match—as best we can—the individual patient’s characteristics with the antipsychotics’ risk: benefit profiles. CATIE phases 1 and 2 provide independent information on the comparative efficacy and tolerability of each medication.
The CATIE investigators and NIMH have done a great service to our field in providing a rich repository of timely information to inform clinical practice. But the CATIE study was not designed to answer all our questions about treating schizophrenia.8,9 Clinicians and patients need to look elsewhere for guidance on the roles of:
- psychosocial treatments
- recovery and the therapeutic alliance in maximizing outcomes
- long-acting SGA formulations
- aripiprazole (addressed in CATIE phase 3)
- SGAs in first-episode schizophrenia
- FGAs when a patient does not adequately respond to an initial SGA.
Related resources
- Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE). www.CATIE.unc.edu.
- Lieberman JA. What the CATIE study means in clinical practice. Psychiatr Serv 2006;57(8):1075.
Drug brand names
- Aripiprazole • Abilify
- Clozapine • Clozaril
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosures
Dr. Buckley receives research/grant support from AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly & Co., Janssen Pharmaceutica, Pfizer, and Solvay Pharmaceuticals, and is a consultant to Abbott Laboratories, Alamo Pharmaceuticals, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly & Co., Janssen Pharmaceutica, Merck & Co., and Pfizer.
Acknowledgement
The author thanks Del Miller, MD, for comments given on a draft of this paper.
1. Stroup TS, Lieberman JA, McEvoy JP, et al for the CATIE investigators. Effectiveness of olanzapine, quetiapine, risperidone, and ziprasidone in patients with chronic schizophrenia following discontinuation of a previous atypical antipsychotic. Am J Psychiatry 2006;163:611-22.
2. McEvoy JP, Lieberman JA, Stroup TS, et al for the CATIE investigators. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry 2006;163:600-10.
3. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 2005;353:1209-23.
4. Nasrallah HA. CATIE’s surprises: In antipsychotics’ square-off, were there winners or losers? Current Psychiatry 2006;5(2):52-65.
5. Buckley PF. Dosing equivalency of second-generation antipsychotics. J Clin Psychopharmacol 2005;25(5):501-2.
6. Davis JM. The choice of drugs for schizophrenia. N Engl J Med 2006;354(5):518-20.
7. Citrome L, Stroup TS. Schizophrenia clinical antipsychotic trials intervention effectiveness and number needed to treat: How can CATIE inform clinicians? Int J Clin Pract 2006 (in press).
8. Ragins M. Should the CATIE study be a wake-up call? Psychiatr Serv 2005;56:1489.-
9. Lieberman JA, Hsiao J. Interpreting the results of the CATIE study. Psychiatr Serv 2006;57:139.-
CATIE phase 2 offers insights on efficacy an tolerability
After nearly 3 out of 4 phase 1 patients stopped taking their assigned antipsychotics within 18 months, researchers in the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) braced themselves for phase 2.February 2006)
CATIE’s eligibility criteria are broad and include schizophrenia patients with comorbid conditions such as substance abuse and mood disorders. The primary outcome measure is all-cause treatment discontinuation, which incorporates efficacy, safety, tolerability, patient choice, and clinician choice (Table 1).
Phase 1 compared the efficacy and safety of four second-generation antipsychotics (SGA) and one first-generation antipsychotic (FGA).3 Nasrallah concluded that—despite the high discontinuation rate in that phase—there were “no winners or losers” among the five antipsychotics. The results, Nasrallah concluded:
- provide a compelling rationale for clinicians to match medication profiles to individual patients
- support the need for clinicians to have choices among medications when treating patients with schizophrenia.4
Table 1
Drug discontinuation patterns in CATIE phase 1
| Measures | Findings after 18 months | |
|---|---|---|
| % of patients who discontinued medication for any reason | Olanzapine (64%) | Ziprasidone (79%) |
| Risperidone (74%) | Quetiapine (82%) | |
| Perphenazine (75%) | ||
| Time to discontinuation for any reason | Longest (most favorable) with olanzapine, but not statistically longer with olanzapine than with ziprasidone or perphenazine | |
| No statistical difference among risperidone, quetiapine, ziprasidone, and perphenazine | ||
| Time to discontinuation for lack of efficacy* | Longer with olanzapine; no statistical difference among risperidone, quetiapine, ziprasidone, and perphenazine | |
| Time to discontinuation for intolerable side effects | No statistical difference among agents | |
| Rate of discontinuation for intolerable side effects | Highest (19%) with olanzapine (primarily because of weight gain or metabolic effects with this medication) | |
| Rate of discontinuation for extrapyramidal effects | Highest (8%) with perphenazine | |
| Rate of discontinuation for intolerability (overall) | Lowest with risperidone (10%) | |
| * Nonequivalent dosing in CATIE phase 1 is an ongoing debate. | ||
What to do next?
When an initial antipsychotic proves inadequate or causes intolerable side effects, how do you choose a more efficacious or tolerable medication? Phase 2 offered CATIE patients and their clinicians two choices—an efficacy and a tolerability pathway (Figure).1,2
CATIE phase 2: Distribution of patients in efficacy and tolerability pathways
Efficacy pathway. Patients who chose the efficacy pathway were randomly assigned to clozapine (50%) or olanzapine, risperidone, or quetiapine.1 Researchers selected clozapine as the major efficacy comparator because of its robust effects in treatment-refractory schizophrenia. Clozapine was given open-label because of its safety monitoring requirements; other treatments were double-blind.
As in phase 1, the primary outcome measure was time until discontinuation for any reason. Secondary outcome measures included time to discontinuation because of side effects, patient choice, or lack of efficacy.
Tolerability pathway. Patients who chose the tolerability pathway were randomly assigned to double-blind treatment with ziprasidone, olanzapine, risperidone, or quetiapine.2 Ziprasidone was the major comparator because of clinical data showing a favorable tolerability profile.
The primary outcome measure was time to discontinuation for any reason. Secondary outcomes included reason for discontinuation (as determined by the study clinician), symptomatic ratings, and evaluations of adverse effects.
Trial duration. No patients in either pathway received the same antipsychotics they had taken in phase 1. All patients could continue treatment through the 18 months of the CATIE trial or until they completed 6 months in phase 2.
Efficacy pathway results
Discontinuation. Consistent with literature about its efficacy in treatment-refractory schizophrenia, clozapine showed a robust clinical effect. Overall, more patients receiving clozapine stayed on treatment and for longer periods, compared with patients receiving olanzapine, risperidone, or quetiapine (Table 2).
On secondary measures, discontinuation for lack of efficacy was significantly lower with clozapine (11%) than with:
- olanzapine (35%)
- risperidone or quetiapine (each at 43%).
Discontinuation rates because of adverse effects or by patient choice were the same across all medications (Table 3). Patients on clozapine achieved better ratings in overall psychotic symptoms, positive symptoms, and general function, but not in negative symptoms.
Weight gain. On average, patients gained more weight while taking olanzapine (+1.1 lb/mo) than with:
- risperidone (+0.5 lb/mo)
- clozapine (+0.5 lb/mo)
- quetiapine (+0.5 lb/mo)
Differences in weight gain—or in metabolic parameters or other adverse effects—were not statistically significant, however.
Table 2
Phase 2 efficacy pathway: Discontinuation for any reason
| Measure | Clozapine | Olanzapine | Risperidone | Quetiapine |
|---|---|---|---|---|
| How many patients discontinued | 25 of 49 (56%) | 12 of 19 (71%) | 12 of 16 (86%) | 13 of 15 (93%) |
| Median time to discontinuation | 10.5 months | 2.7 months | 2.8 months | 3.3 months |
Table 3
Reasons patients stopped taking their medications in CATIE phase 2
| Reason | Efficacy pathway | Tolerability pathway |
|---|---|---|
| All cause | 69% | 74% |
| Lack of efficacy | 26% | 29% |
| Lack of tolerability | 10% | 15% |
| Patient choice | 26% | 24% |
Tolerability pathway results
Discontinuation. Patients in the tolerability pathway took olanzapine or risperidone significantly longer—median 6.3 and 7 months, respectively— compared with ziprasidone (4 months) or quetiapine (2.8 months).
- Time to discontinuation during phase 2 was the same across all drugs among patients who entered phase 2 because of intolerable side effects in phase 1.
- Time to discontinuation because of side effects also was similar whether patients discontinued phase 1 for lack of efficacy or intolerable side effects. Patients stopped treatment in the efficacy and tolerability pathways for similar reasons (Table 3).
Weight gain. Patients taking olanzapine gained more weight (average +1.3 lb/mo) than did those taking the other drugs. Patients taking ziprasidone lost weight (average –1.7 lb/mo). Among 61 patients who gained weight during phase 1, 42% of those switched to ziprasidone lost weight in phase 2, as did:
- 20% of those switched to risperidone
- 7% of those switched to quetiapine.
Among those switched to olanzapine in phase 2, no one lost weight and 2% gained weight.
Metabolic effects. Some parameters changed, depending on drug assignment:
- prolactin increased in patients switched to risperidone
- cholesterol and triglycerides increased in patients switched to olanzapine or quetiapine but decreased in those switched to risperidone or ziprasidone
- QTc interval measurements showed no difference across all drugs.
Methodologic caveats
When considering how CATIE’s phase 2 findings might apply to clinical practice, keep in mind four caveats about the study’s design.
Clozapine was given open-label, yet quetiapine, olanzapine, and risperidone were given double-blind in the efficacy pathway. This pathway’s findings are consistent with what we know about clozapine and other SGAs in treatment-refractory schizophrenia, but how the open-label design affected clozapine therapy outcomes is unclear.
Were patients who knew they were taking clozapine more willing to “stay the course” than were patients in the pathway’s double-blind arm?
Discontinuation rates remained high. The 74% “overall discontinuation rate” in phase 1 surprised many psychiatrists because of the perceived high rate at which patients did not adhere to the first medications they received. To some extent, the word “discontinuation” is imprecise, however, because this group includes patients who did not drop out of treatment altogether but chose to move on to phase 2.
It is important to note, however, that nearly one-half of phase 1 patients who were eligible to enter phase 2 (509 of 1,052) did not. This group represents the true drop-out rate, which is substantial. The high rates of discontinuation seen in phase 1 also occurred in both phase 2 pathways (Table 3).
Few patients entered the efficacy pathway. In an approach designed to reflect routine clinical practice, the researchers recommended the efficacy pathway to patients who discontinued phase 1 because of lack of efficacy and the tolerability pathway to those who discontinued phase 1 because of intolerability. Many patients did not follow the recommendations, however, and seemed to choose their pathways based on whether they wanted a chance to receive clozapine or ziprasidone in phase 2.
Thus, among the 543 phase 1 patients who enrolled in phase 2, 99 (18%) entered the efficacy pathway, and 444 (82%) entered the tolerability pathway. The efficacy pathway included 85 patients who discontinued phase 1 for lack of efficacy and 5 for lack of tolerability. The tolerability pathway included 184 patients who discontinued phase 1 for lack of efficacy and 168 for lack of tolerability.
Dosages may not have been equivalent. SGAs’ dosing equivalency is unknown,5,6 which impedes our ability to interpret comparative studies such as CATIE. The study’s designers developed the its dosing ranges by careful consideration, including recommendations from each SGA’s manufacturer. As Nasrallah described,4 the trial’s dosages were not universally consistent with FDA-approved ranges or usual clinical practice (Table 4). In phase 2, for example, ziprasidone dosages were less than psychiatrists usually use, and quetiapine dosages were greater than usual.
Fortunately, studies are underway to determine each SGA’s optimum dosing. This work will help us understand what we can expect when we increase an antipsychotic’s dosage—a key step towards understanding dosing equivalency.
Table 4
Mean modal antipsychotic dosages (mg/d) in CATIE phase 2 pathways*
| Clozapine | Ziprasidone | Olanzapine | Risperidone | Quetiapine | |
|---|---|---|---|---|---|
| Efficacy pathway | 332 | — | 23.4 | 4.8 | 642.9 |
| Tolerability pathway | — | 115.9 | 20.5 | 4.15 | 65.2 |
| * 800 mg/d of quetiapine and 160 mg/d of ziprasidone are generally regarded as therapeutically equivalent to 20 mg/d of olanzapine. | |||||
What clinicians can expect
A recent analysis helps put CATIE’s findings in perspective. Citrome and Stroup7 quantified the results of phase 1 and 2 with respect to:
- number needed to treat (NNT)—how many patients a clinician needs to treat with drug A to see one additional benefit, compared with drug B
- number needed to harm (NNH)—how many patients a clinician needs to treat with drug A to see a given adverse effect, compared with drug B.
In this analysis, the NNT for olanzapine (5.5 to 10) was lowest among the drugs compared in phase 1, and the NNT for clozapine (3) was lowest among those compared in phase 2. A lower number means that, overall, clinicians can expect a more robust treatment response.
On the other hand, the NNH for olanzapine in weight gain and metabolic disturbances (12.4 to 17.7) was the lowest in phase 1, indicating that clinicians can expect more weight gain and metabolic effects with olanzapine than with other SGAs. Ziprasidone had the highest NNH (106 to 208) among the agents in phase 2 for avoiding discontinuation because of weight gain or metabolic disturbances. In other words, ziprasidone appears less likely than other SGAs to cause metabolic problems.
These risk-attribution measures demonstrate the dilemma clinicians face when trying to match schizophrenia patients with antipsychotics. CATIE was “an N of 1,493” subjects, whereas each patient we see in clinical practice is “an N of 1.” One patient may need a more-robust response; another may need improved tolerability.
We strive for balance, seeking to optimize efficacy—often by raising the dosage—while minimizing adverse effects.
What to tell patients
CATIE phases 1 and 2 provide a compelling rationale for individualized treatment, which should be standard clinical practice for schizophrenia:
- All drugs used in phases 1 and 2 worked.
- All showed noteworthy adverse effects that were different for each drug.
- Different patients responded differently to each drug.
Using our clinical judgment and available information, we must match—as best we can—the individual patient’s characteristics with the antipsychotics’ risk: benefit profiles. CATIE phases 1 and 2 provide independent information on the comparative efficacy and tolerability of each medication.
The CATIE investigators and NIMH have done a great service to our field in providing a rich repository of timely information to inform clinical practice. But the CATIE study was not designed to answer all our questions about treating schizophrenia.8,9 Clinicians and patients need to look elsewhere for guidance on the roles of:
- psychosocial treatments
- recovery and the therapeutic alliance in maximizing outcomes
- long-acting SGA formulations
- aripiprazole (addressed in CATIE phase 3)
- SGAs in first-episode schizophrenia
- FGAs when a patient does not adequately respond to an initial SGA.
Related resources
- Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE). www.CATIE.unc.edu.
- Lieberman JA. What the CATIE study means in clinical practice. Psychiatr Serv 2006;57(8):1075.
Drug brand names
- Aripiprazole • Abilify
- Clozapine • Clozaril
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosures
Dr. Buckley receives research/grant support from AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly & Co., Janssen Pharmaceutica, Pfizer, and Solvay Pharmaceuticals, and is a consultant to Abbott Laboratories, Alamo Pharmaceuticals, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly & Co., Janssen Pharmaceutica, Merck & Co., and Pfizer.
Acknowledgement
The author thanks Del Miller, MD, for comments given on a draft of this paper.
CATIE phase 2 offers insights on efficacy an tolerability
After nearly 3 out of 4 phase 1 patients stopped taking their assigned antipsychotics within 18 months, researchers in the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) braced themselves for phase 2.February 2006)
CATIE’s eligibility criteria are broad and include schizophrenia patients with comorbid conditions such as substance abuse and mood disorders. The primary outcome measure is all-cause treatment discontinuation, which incorporates efficacy, safety, tolerability, patient choice, and clinician choice (Table 1).
Phase 1 compared the efficacy and safety of four second-generation antipsychotics (SGA) and one first-generation antipsychotic (FGA).3 Nasrallah concluded that—despite the high discontinuation rate in that phase—there were “no winners or losers” among the five antipsychotics. The results, Nasrallah concluded:
- provide a compelling rationale for clinicians to match medication profiles to individual patients
- support the need for clinicians to have choices among medications when treating patients with schizophrenia.4
Table 1
Drug discontinuation patterns in CATIE phase 1
| Measures | Findings after 18 months | |
|---|---|---|
| % of patients who discontinued medication for any reason | Olanzapine (64%) | Ziprasidone (79%) |
| Risperidone (74%) | Quetiapine (82%) | |
| Perphenazine (75%) | ||
| Time to discontinuation for any reason | Longest (most favorable) with olanzapine, but not statistically longer with olanzapine than with ziprasidone or perphenazine | |
| No statistical difference among risperidone, quetiapine, ziprasidone, and perphenazine | ||
| Time to discontinuation for lack of efficacy* | Longer with olanzapine; no statistical difference among risperidone, quetiapine, ziprasidone, and perphenazine | |
| Time to discontinuation for intolerable side effects | No statistical difference among agents | |
| Rate of discontinuation for intolerable side effects | Highest (19%) with olanzapine (primarily because of weight gain or metabolic effects with this medication) | |
| Rate of discontinuation for extrapyramidal effects | Highest (8%) with perphenazine | |
| Rate of discontinuation for intolerability (overall) | Lowest with risperidone (10%) | |
| * Nonequivalent dosing in CATIE phase 1 is an ongoing debate. | ||
What to do next?
When an initial antipsychotic proves inadequate or causes intolerable side effects, how do you choose a more efficacious or tolerable medication? Phase 2 offered CATIE patients and their clinicians two choices—an efficacy and a tolerability pathway (Figure).1,2
CATIE phase 2: Distribution of patients in efficacy and tolerability pathways
Efficacy pathway. Patients who chose the efficacy pathway were randomly assigned to clozapine (50%) or olanzapine, risperidone, or quetiapine.1 Researchers selected clozapine as the major efficacy comparator because of its robust effects in treatment-refractory schizophrenia. Clozapine was given open-label because of its safety monitoring requirements; other treatments were double-blind.
As in phase 1, the primary outcome measure was time until discontinuation for any reason. Secondary outcome measures included time to discontinuation because of side effects, patient choice, or lack of efficacy.
Tolerability pathway. Patients who chose the tolerability pathway were randomly assigned to double-blind treatment with ziprasidone, olanzapine, risperidone, or quetiapine.2 Ziprasidone was the major comparator because of clinical data showing a favorable tolerability profile.
The primary outcome measure was time to discontinuation for any reason. Secondary outcomes included reason for discontinuation (as determined by the study clinician), symptomatic ratings, and evaluations of adverse effects.
Trial duration. No patients in either pathway received the same antipsychotics they had taken in phase 1. All patients could continue treatment through the 18 months of the CATIE trial or until they completed 6 months in phase 2.
Efficacy pathway results
Discontinuation. Consistent with literature about its efficacy in treatment-refractory schizophrenia, clozapine showed a robust clinical effect. Overall, more patients receiving clozapine stayed on treatment and for longer periods, compared with patients receiving olanzapine, risperidone, or quetiapine (Table 2).
On secondary measures, discontinuation for lack of efficacy was significantly lower with clozapine (11%) than with:
- olanzapine (35%)
- risperidone or quetiapine (each at 43%).
Discontinuation rates because of adverse effects or by patient choice were the same across all medications (Table 3). Patients on clozapine achieved better ratings in overall psychotic symptoms, positive symptoms, and general function, but not in negative symptoms.
Weight gain. On average, patients gained more weight while taking olanzapine (+1.1 lb/mo) than with:
- risperidone (+0.5 lb/mo)
- clozapine (+0.5 lb/mo)
- quetiapine (+0.5 lb/mo)
Differences in weight gain—or in metabolic parameters or other adverse effects—were not statistically significant, however.
Table 2
Phase 2 efficacy pathway: Discontinuation for any reason
| Measure | Clozapine | Olanzapine | Risperidone | Quetiapine |
|---|---|---|---|---|
| How many patients discontinued | 25 of 49 (56%) | 12 of 19 (71%) | 12 of 16 (86%) | 13 of 15 (93%) |
| Median time to discontinuation | 10.5 months | 2.7 months | 2.8 months | 3.3 months |
Table 3
Reasons patients stopped taking their medications in CATIE phase 2
| Reason | Efficacy pathway | Tolerability pathway |
|---|---|---|
| All cause | 69% | 74% |
| Lack of efficacy | 26% | 29% |
| Lack of tolerability | 10% | 15% |
| Patient choice | 26% | 24% |
Tolerability pathway results
Discontinuation. Patients in the tolerability pathway took olanzapine or risperidone significantly longer—median 6.3 and 7 months, respectively— compared with ziprasidone (4 months) or quetiapine (2.8 months).
- Time to discontinuation during phase 2 was the same across all drugs among patients who entered phase 2 because of intolerable side effects in phase 1.
- Time to discontinuation because of side effects also was similar whether patients discontinued phase 1 for lack of efficacy or intolerable side effects. Patients stopped treatment in the efficacy and tolerability pathways for similar reasons (Table 3).
Weight gain. Patients taking olanzapine gained more weight (average +1.3 lb/mo) than did those taking the other drugs. Patients taking ziprasidone lost weight (average –1.7 lb/mo). Among 61 patients who gained weight during phase 1, 42% of those switched to ziprasidone lost weight in phase 2, as did:
- 20% of those switched to risperidone
- 7% of those switched to quetiapine.
Among those switched to olanzapine in phase 2, no one lost weight and 2% gained weight.
Metabolic effects. Some parameters changed, depending on drug assignment:
- prolactin increased in patients switched to risperidone
- cholesterol and triglycerides increased in patients switched to olanzapine or quetiapine but decreased in those switched to risperidone or ziprasidone
- QTc interval measurements showed no difference across all drugs.
Methodologic caveats
When considering how CATIE’s phase 2 findings might apply to clinical practice, keep in mind four caveats about the study’s design.
Clozapine was given open-label, yet quetiapine, olanzapine, and risperidone were given double-blind in the efficacy pathway. This pathway’s findings are consistent with what we know about clozapine and other SGAs in treatment-refractory schizophrenia, but how the open-label design affected clozapine therapy outcomes is unclear.
Were patients who knew they were taking clozapine more willing to “stay the course” than were patients in the pathway’s double-blind arm?
Discontinuation rates remained high. The 74% “overall discontinuation rate” in phase 1 surprised many psychiatrists because of the perceived high rate at which patients did not adhere to the first medications they received. To some extent, the word “discontinuation” is imprecise, however, because this group includes patients who did not drop out of treatment altogether but chose to move on to phase 2.
It is important to note, however, that nearly one-half of phase 1 patients who were eligible to enter phase 2 (509 of 1,052) did not. This group represents the true drop-out rate, which is substantial. The high rates of discontinuation seen in phase 1 also occurred in both phase 2 pathways (Table 3).
Few patients entered the efficacy pathway. In an approach designed to reflect routine clinical practice, the researchers recommended the efficacy pathway to patients who discontinued phase 1 because of lack of efficacy and the tolerability pathway to those who discontinued phase 1 because of intolerability. Many patients did not follow the recommendations, however, and seemed to choose their pathways based on whether they wanted a chance to receive clozapine or ziprasidone in phase 2.
Thus, among the 543 phase 1 patients who enrolled in phase 2, 99 (18%) entered the efficacy pathway, and 444 (82%) entered the tolerability pathway. The efficacy pathway included 85 patients who discontinued phase 1 for lack of efficacy and 5 for lack of tolerability. The tolerability pathway included 184 patients who discontinued phase 1 for lack of efficacy and 168 for lack of tolerability.
Dosages may not have been equivalent. SGAs’ dosing equivalency is unknown,5,6 which impedes our ability to interpret comparative studies such as CATIE. The study’s designers developed the its dosing ranges by careful consideration, including recommendations from each SGA’s manufacturer. As Nasrallah described,4 the trial’s dosages were not universally consistent with FDA-approved ranges or usual clinical practice (Table 4). In phase 2, for example, ziprasidone dosages were less than psychiatrists usually use, and quetiapine dosages were greater than usual.
Fortunately, studies are underway to determine each SGA’s optimum dosing. This work will help us understand what we can expect when we increase an antipsychotic’s dosage—a key step towards understanding dosing equivalency.
Table 4
Mean modal antipsychotic dosages (mg/d) in CATIE phase 2 pathways*
| Clozapine | Ziprasidone | Olanzapine | Risperidone | Quetiapine | |
|---|---|---|---|---|---|
| Efficacy pathway | 332 | — | 23.4 | 4.8 | 642.9 |
| Tolerability pathway | — | 115.9 | 20.5 | 4.15 | 65.2 |
| * 800 mg/d of quetiapine and 160 mg/d of ziprasidone are generally regarded as therapeutically equivalent to 20 mg/d of olanzapine. | |||||
What clinicians can expect
A recent analysis helps put CATIE’s findings in perspective. Citrome and Stroup7 quantified the results of phase 1 and 2 with respect to:
- number needed to treat (NNT)—how many patients a clinician needs to treat with drug A to see one additional benefit, compared with drug B
- number needed to harm (NNH)—how many patients a clinician needs to treat with drug A to see a given adverse effect, compared with drug B.
In this analysis, the NNT for olanzapine (5.5 to 10) was lowest among the drugs compared in phase 1, and the NNT for clozapine (3) was lowest among those compared in phase 2. A lower number means that, overall, clinicians can expect a more robust treatment response.
On the other hand, the NNH for olanzapine in weight gain and metabolic disturbances (12.4 to 17.7) was the lowest in phase 1, indicating that clinicians can expect more weight gain and metabolic effects with olanzapine than with other SGAs. Ziprasidone had the highest NNH (106 to 208) among the agents in phase 2 for avoiding discontinuation because of weight gain or metabolic disturbances. In other words, ziprasidone appears less likely than other SGAs to cause metabolic problems.
These risk-attribution measures demonstrate the dilemma clinicians face when trying to match schizophrenia patients with antipsychotics. CATIE was “an N of 1,493” subjects, whereas each patient we see in clinical practice is “an N of 1.” One patient may need a more-robust response; another may need improved tolerability.
We strive for balance, seeking to optimize efficacy—often by raising the dosage—while minimizing adverse effects.
What to tell patients
CATIE phases 1 and 2 provide a compelling rationale for individualized treatment, which should be standard clinical practice for schizophrenia:
- All drugs used in phases 1 and 2 worked.
- All showed noteworthy adverse effects that were different for each drug.
- Different patients responded differently to each drug.
Using our clinical judgment and available information, we must match—as best we can—the individual patient’s characteristics with the antipsychotics’ risk: benefit profiles. CATIE phases 1 and 2 provide independent information on the comparative efficacy and tolerability of each medication.
The CATIE investigators and NIMH have done a great service to our field in providing a rich repository of timely information to inform clinical practice. But the CATIE study was not designed to answer all our questions about treating schizophrenia.8,9 Clinicians and patients need to look elsewhere for guidance on the roles of:
- psychosocial treatments
- recovery and the therapeutic alliance in maximizing outcomes
- long-acting SGA formulations
- aripiprazole (addressed in CATIE phase 3)
- SGAs in first-episode schizophrenia
- FGAs when a patient does not adequately respond to an initial SGA.
Related resources
- Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE). www.CATIE.unc.edu.
- Lieberman JA. What the CATIE study means in clinical practice. Psychiatr Serv 2006;57(8):1075.
Drug brand names
- Aripiprazole • Abilify
- Clozapine • Clozaril
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosures
Dr. Buckley receives research/grant support from AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly & Co., Janssen Pharmaceutica, Pfizer, and Solvay Pharmaceuticals, and is a consultant to Abbott Laboratories, Alamo Pharmaceuticals, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly & Co., Janssen Pharmaceutica, Merck & Co., and Pfizer.
Acknowledgement
The author thanks Del Miller, MD, for comments given on a draft of this paper.
1. Stroup TS, Lieberman JA, McEvoy JP, et al for the CATIE investigators. Effectiveness of olanzapine, quetiapine, risperidone, and ziprasidone in patients with chronic schizophrenia following discontinuation of a previous atypical antipsychotic. Am J Psychiatry 2006;163:611-22.
2. McEvoy JP, Lieberman JA, Stroup TS, et al for the CATIE investigators. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry 2006;163:600-10.
3. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 2005;353:1209-23.
4. Nasrallah HA. CATIE’s surprises: In antipsychotics’ square-off, were there winners or losers? Current Psychiatry 2006;5(2):52-65.
5. Buckley PF. Dosing equivalency of second-generation antipsychotics. J Clin Psychopharmacol 2005;25(5):501-2.
6. Davis JM. The choice of drugs for schizophrenia. N Engl J Med 2006;354(5):518-20.
7. Citrome L, Stroup TS. Schizophrenia clinical antipsychotic trials intervention effectiveness and number needed to treat: How can CATIE inform clinicians? Int J Clin Pract 2006 (in press).
8. Ragins M. Should the CATIE study be a wake-up call? Psychiatr Serv 2005;56:1489.-
9. Lieberman JA, Hsiao J. Interpreting the results of the CATIE study. Psychiatr Serv 2006;57:139.-
1. Stroup TS, Lieberman JA, McEvoy JP, et al for the CATIE investigators. Effectiveness of olanzapine, quetiapine, risperidone, and ziprasidone in patients with chronic schizophrenia following discontinuation of a previous atypical antipsychotic. Am J Psychiatry 2006;163:611-22.
2. McEvoy JP, Lieberman JA, Stroup TS, et al for the CATIE investigators. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry 2006;163:600-10.
3. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 2005;353:1209-23.
4. Nasrallah HA. CATIE’s surprises: In antipsychotics’ square-off, were there winners or losers? Current Psychiatry 2006;5(2):52-65.
5. Buckley PF. Dosing equivalency of second-generation antipsychotics. J Clin Psychopharmacol 2005;25(5):501-2.
6. Davis JM. The choice of drugs for schizophrenia. N Engl J Med 2006;354(5):518-20.
7. Citrome L, Stroup TS. Schizophrenia clinical antipsychotic trials intervention effectiveness and number needed to treat: How can CATIE inform clinicians? Int J Clin Pract 2006 (in press).
8. Ragins M. Should the CATIE study be a wake-up call? Psychiatr Serv 2005;56:1489.-
9. Lieberman JA, Hsiao J. Interpreting the results of the CATIE study. Psychiatr Serv 2006;57:139.-
Psychotic prodrome: Are antipsychotics effective? Ethical?
Because 40% of individuals with a psychotic prodrome develop schizophrenia, detecting and preventing this transition could improve many patients’ lives. Unfortunately:
- psychotic prodrome lacks clear-cut symptoms and is difficult to identify
- little evidence exists to help clinicians select psychotropics and decide how long to use them
- treating all prodromal patients would expose those who never develop psychosis to the risk of psychotropics’ side effects.
How, then, can psychiatrists help patients who present with possible prodromal symptoms? Based on research and our experience, this article describes the psychotic prodrome and offers a pragmatic, evidence-based approach to diagnosis and treatment.
WHAT CAUSES PSYCHOTIC CONVERSION?
Reduced gray matter volumes in certain brain regions may be associated with conversion to psychosis (Box 1). Stress also may play a role; elevated stress-reactive cortisol levels are associated with positive symptom severity in the prodrome.1 Other factors being investigated include obstetric complications at birth, maternal age >30, premorbid schizotypal personality disorder, and impaired olfaction.
Symptoms. Nearly 80% of patients with schizophrenia experience a psychotic prodrome that lasts a few months to several years.2 Common features include:
- gradual worsening of perceptual disturbance
- referential thinking
- paranoia
- mild cognitive deficits
- mood lability
- impulsivity
- suicidality
- declining social function and academic performance.3,4
A premorbid phase often precedes the prodrome, with symptoms such as impaired attention, soft neurologic signs, and subtle social deficits. These changes may be harbingers of the prodrome but are too nonspecific to be diagnostic. Other functional impairments—including anxiety, depression, drug abuse, and psychosocial factors such as school stress—may mimic schizophrenic prodrome.
Prognosis. Studies of patients’ first schizophrenia episodes suggest that prodrome duration may predict outcome. A longer prodrome is thought to indicate a poor prognosis,6 such as in patients who wait a year before seeking treatment.7 A review of 22 studies of first-episode psychosis found early psychosocial and pharmacologic interventions improved long-term prognosis, and medication discontinuation predicted more-severe and chronic disease.8
Reduced gray matter volumes in certain brain regions may be associated with conversion to psychosis. Imaging studies have found medial temporal lobe changes—specifically, hippocampal volume alterations—in persons with schizophrenia, genetic high-risk groups, and those thought to be at risk for imminent psychosis.11
MRI imaging of patients with prodromal signs has shown less gray matter in the right medial temporal, lateral temporal, inferior frontal cortex, and bilateral cingulate regions in those who have developed psychosis, compared with those who have not. In the psychotic patients, 12-month longitudinal follow-up has found reduced gray matter in the left hippocampal, fusiform, orbitofrontal, cerebellar cortices, and cingulate gyrus.12
Brain structure is related to genetic liability for schizophrenia in high-risk patients, who seem to have smaller right and left prefrontal lobes and smaller right and left thalami. These findings are consistent with the prodrome’s neurocognitive deficits, which are less than those reported in schizophrenia and greater than those seen in healthy subjects.
Pioneering work by McGorry et al10 identified an “ultra high-risk group” with a psychotic conversion rate of 40% to 60%. These patients present with three symptom patterns:
- attenuated positive symptoms
- brief intermittent psychotic episodes
- genetic risk and recent deterioration syndrome (Table 1).
3 patient groups considered at ‘ultra high risk’ to develop schizophrenia
| Patients with… | Symptoms |
|---|---|
| Attenuated psychotic symptoms | Overvalued ideas, perceptual disorders |
| Present at least 1 week; not >5 years | |
| At least 1 symptom several times a week | |
| Brief intermittent psychotic episodes | Frank psychotic features |
| Resolve spontaneously within 7 days | |
| Can be drug-induced | |
| Genetic risk and recent deterioration syndrome | Psychotic disorder in a first-degree relative |
| Schizotypal personality disorder | |
| Present at least 1 month; not >5 years | |
| Significant functional decline | |
| Source: Adapted from reference 10 | |
The Edinburgh High Risk Study of 162 individuals ages 16 to 25 showed more marked psychopathology in those with at least two close relatives with schizophrenia, compared with control groups. A direct correlation was seen between genetic liability and poor neurocognitive performance.11
PRODROME RATING SCALES
Researchers are using outcome measures to diagnose prodromal symptoms and assess their severity. Operational, validated assessment tools include:
- Bonn Scale for the Assessment of Basic Symptoms (BSABS): captures subtle changes in thinking, feeling, and perception.
- Schizophrenia Prediction Instrument for Adults (SPI-A): defines prepsychotic deviations and rates symptoms that are subjectively experienced by the patient.
- Comprehensive Assessment of At Risk Mental State (CAARMS): defines ultra high-risk criteria and incorporates eight dimensions of psychopathology.
- Scale of Prodromal Symptoms (SOPS): rates psychosis severity. When embedded within the Structured Interview for Prodromal Syndromes (SIPS), the SOPS determines the presence or absence of psychosis and predicts progression to psychopathology.
- Criteria for Prodromal Symptoms (COPS): defines ultra high-risk categories.
- Presence of Psychosis Scale (POPS): rates severity, intensity, and duration of positive prodromal symptoms.12
These instruments may identify prodromal symptoms in psychiatric practice, but further validation of clinical criteria is needed before they could be recommended for routine patient assessment.
PROPHYLACTIC ANTIPSYCHOTICS?
Atypical antipsychotics may be the standard of care for patients with a first psychotic episode, but this intervention is based on few double-blind controlled trials. Not surprisingly, only a handful of studies have examined antipsychotic therapy for the prodrome’s less clear-cut symptoms.
Risperidone. An 8- to 12-week open-label study in adolescents with first- and second-degree relatives with schizophrenia13 included four prodromal and six first-episode psychosis patients who met criteria for a cluster A personality disorder. Risperidone, 1.0 mg/d and 1.8 mg/d, respectively, improved thought disorder and attention symptoms, as measured with the Child Behavior Checklist. Verbal memory improved minimally, and no medication side effects were reported.
An open-label observational study14 identified four middle-aged subjects with a genetic risk of schizophrenia who reported negative symptoms and neurocognitive deficits. Risperidone, started at 0.25 mg/d and gradually increased to a maximum of 2 mg/d, improved negative symptoms, attention, and working memory. Mild side effects including tremors, sedation, dry mouth, and anxiety symptoms were reported.
An open-label, randomized, comparator trial15 examined psychotic transition rates in 59 subjects (mean age, 20) who met ultra high-risk criteria. They received:
- a needs-based intervention (NBI) comprising case management, psychotropics excluding antipsychotics, and supportive psychotherapy
- or a specific preventive intervention (SPI) that included risperidone, 1 to 2 mg/d, and a modified cognitive-behavioral therapy (CBT).
Should you intervene with patients in suspected psychotic prodrome?
| Arguments for: |
|
| Arguments against: |
|
At 12-months’ follow-up, another 3 SPI patients who had been partially adherent or non-adherent to antipsychotic therapy had converted to psychosis. For adherent SPI patients, protection against conversion appeared to persist for 6 months after risperidone therapy ended. All medication side effects were mild and transient.
Table 3
Psychotic prodrome: Unanswered clinical questions
|
In the first year, 11 of 29 placebo-group patients and 5 of 31 receiving olanzapine converted to psychosis. Among patients receiving no treatment in the second year, 2 of 8 former placebo patients and 3 of 9 former olanzapine patients converted to psychosis.
Discontinuation rates were 35% and 28%, respectively. Compared with the placebo group, patients taking olanzapine experienced greater weight gain, suggesting that risks associated with antipsychotic therapy may exceed unproven benefits in this population.
Discussion. Little information exists on using quetiapine, ziprasidone, or aripiprazole in prodromal patients. As cited above, preliminary studies with risperidone and olanzapine suggest that these agents may improve several domains of psychotic prodrome. The evidence does not support firm conclusions, however, given the trials’ small sample sizes and brief duration.
The prevalence of obesity and metabolic syndrome in patients with schizophrenia and the added metabolic risks associated with atypical antipsychotics make their use during the prodrome controversial. Weighing the potential advantages and disadvantages (Table 2), we consider antipsychotics to be the last resort after psychosocial interventions have failed to improve prodromal symptoms.
Low-dose atypical antipsychotics may be warranted for some patients, but their use requires stringent monitoring of:
- weight and waist circumference
- vital signs
- metabolic parameters such as fasting blood glucose and lipid profile
- abnormal involuntary movements
- prolactin elevations.
OTHER THERAPIES
Antidepressants. Researchers are also exploring the efficacy of using antidepressants and anxiolytics in the prodromal phase. The only published naturalistic study of adolescents found antidepressants alone or in combination with mood stabilizers or anxiolytics to be as effective as atypical antipsychotics in treating prodromal symptoms.17 A more substantial study is ongoing.
Psychotherapy. For patients with a suspected psychotic prodrome, nondrug strategies may help minimize functional and cognitive impairments, ease distress, and improve coping skills.
CBT has been shown to reduce psychotic progression over 12 months.18 Use CBT to help patients cope with the illness while focusing on:
- symptom monitoring
- premorbid and present functioning
- establishing a therapeutic alliance
- assessing the patient’s experience of psychosis and any thought distortions.
‘REAL WORLD’ EARLY INTERVENTION
Patients with prodromal symptoms are often referred to psychiatrists by family members, primary care physicians, or other mental health professionals. They tend to be young adults, and a few may present in their teens. Most are experiencing behavioral changes such as social isolation, feeling suspicious, perceptual disturbances, depression, and/or anxiety symptoms that seem abnormal but fall short of DSM-IV criteria for schizophrenia diagnosis.
Many clinical questions about schizophrenia’s prodromal phase remain unanswered (Table 3). Our primary aim is to adequately assess these patients and provide treatment and follow-up, taking into account:
- the individual’s presentation
- risks and benefits of available interventions.
- Provide patients and families information and emotional support; a strong therapeutic alliance may help keep the patient in treatment if schizophrenia develops
- Offer early psychosocial interventions such as vocational training, relapse prevention, substance abuse treatment, family therapy, supportive and CBT
- Explore using low-dose atypical antipsychotics as a last resort for patients with pronounced prodromal symptoms; explain risks of weight gain and other metabolic changes, obtain consent, and document need for such interventions
- Consider referral, if feasible, to a center specializing in psychotic prodrome diagnosis, treatment, and research
Consider atypical antipsychotics for patients with distressing psychotic symptoms, rapidly deteriorating function, increased agitation, and safety risks. Consider antidepressant and/or anxiolytic therapy for depression and anxiety, respectively.
Discuss at length with patients and families the risks and benefits of pharmacologic treatments. When clinically appropriate, cautiously discontinue or taper any medication with patients’ consent, while monitoring for side effects and symptoms.
- Issue devoted to early prodrome research. SchizophrBull 2003;29(4):621-879.
- Diagnostic and therapeutic intervention during psychotic prodrome. CNS Spectrums 2004;9(8):578-606.
- PRIME (Prevention through Risk Identification, Management & Education) Research Clinic. Department of Psychiatry, Yale University. http://info.med.yale.edu/psych/prime/pintro.html.
- Youth Mental Health Update. Schizophrenia: New strategies for early detection and treatment. RAPP Clinic, Zucker Hillside Hospital, Glen Oaks, NY. http://schoolnet.lij.edu/eshare/files/rapp.html
- Aripiprazole • Abilify
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Dr. Narasimhan receives research support from Eli Lilly and Co. and Janssen Pharmaceutica and is a speaker or consultant for Eli Lilly and Co., Pfizer Inc., and Abbott Laboratories.
Dr. Buckley receives research support from AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly and Co., Janssen Pharmaceutica, Novartis Pharmaceuticals Corp., Pfizer Inc., and Solvay Pharmaceuticals. He is a consultant to and/or speaker for Abbott Laboratories, Alamo Pharmaceuticals, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly and Co., Janssen Pharmaceutica, Novartis Pharmaceuticals Corp., Pfizer Inc., and Pharmstar.
1. Corcoran C, Walker E, Huot R, et al. The stress cascade and schizophrenia: etiology and onset. Schizophr Bull 2003;29(4):671-92.
2. Klosterkotter J. Diagnosing schizophrenia in the initial prodromal phase. Arch Gen Psychiatry 2001;58:158-64.
3. Yung AR. The initial prodrome in psychosis: the prodromal phase of first-episode psychosis: past and current conceptualizations. Schizophr Bull 1996;30:587-99.
4. Perkins DO. Evaluating and treating the prodromal stage of schizophrenia. Current Psychiatry Reports 2004;6:289-95.
5. Wood S W, Miller TJ, McGlashan TH. The “prodromal” patient: both symptomatic and at risk. CNS Spectrums 2001;6(3):223-32.
6. Keshavan MS, Haas G, Miewald J, et al. Prolonged untreated illness duration from prodromal onset predicts outcome in first episode psychosis. Schizoph Bull 2003;29(4):757-69.
7. Loebel AD, Lieberman JA, Alvir JM, et al. Duration of psychosis and outcome in first-episode schizophrenia. Am J Psychiatry 1992;149:1183-8.
8. Wyatt RJ, Green MF, Tuma AH. Long-term morbidity associated with delayed treatment of first admission schizophrenic patients: a re-analysis of the Camarillo State Hospital data. Psychol Med 1997;27:261-8.
9. Cornblatt BA, Lencz T, Smith CW, et al. The schizophrenia prodrome revisited: a neuro developmental perspective. Schizophr Bull 2003;29(4):633-51.
10. McGorry PD, Yung AR, Phillips LJ. The “close-in” or ultra high-risk model: a safe and effective strategy for research and clinical intervention in prepsychotic mental disorder. Schizophr Bull 2003;29(4):771-90.
11. Johnstone EC, Lawrie SM, Cosway R. What does the Edinburgh high-risk study tell us about schizophrenia? Am J Med Genet 2002;114(8):906-12.
12. Miller TJ, Clashing TH, Rosen JL, et al. Prodromal assessment with the Structured Interview for Prodromal Syndromes and the Scale of Prodromal Syndromes: predictive validity, interrater reliability, and training to reliability. Schizophr Bull 2003;29(4):703-15.
13. Cannon TD, Huttunen MO, Dahlstrom M, et al. Antipsychotic drug treatment in the prodromal phase in schizophrenia. Am J Psychiatry 2002;159:1230-2.
14. Tsuang MT, Stone WS, Faraone SV. Treatment of nonpsychotic relatives of patients with schizophrenia: four case studies. Biol Psychiatry 1999;45:1412-18.
15. McGorry PD, Yung AR, Phillips LJ, et al. Randomized controlled trial of intervention designed to reduce the risk of progression to first-episode psychosis in a clinical sample with subthreshold symptoms. Arch Gen Psychiatry 2002;59:921-8.
16. McGlashan TH, Zipursky R, Perkins DO, et al. The PRIME North America randomized double-blind clinical trial of olanzapine versus placebo in patients at risk of being prodromally symptomatic for psychosis. I. Study rationale and design. Schizophr Res 2003;61:7-18.
17. Cornblatt B, Lencz T, Correll C, et al. Treating the prodrome: naturalistic findings from the RAP program. Acta Psychiatr Scand 2002;106(suppl):44.-
18. Morrison AO, French P, Walford L, et al. Cognitive therapy for the prevention of psychosis in people at ultra-high risk: randomized controlled trial. Br J Psychiatry 2004;185:291-7.
19. Wentzell B. This family experience in a supportive first episode program (abstract S-25-03). Davos, Switzerland: Schizophrenia Research 67(1) 11th Biennial Winter Workshop on Schizophrenia. Feb. 7-13, 2004.
20. Stahl SM. Prophylactic antipsychotics: do they keep you from catching schizophrenia? J Clin Psychiatry 2004;65(11):1445-6.
Because 40% of individuals with a psychotic prodrome develop schizophrenia, detecting and preventing this transition could improve many patients’ lives. Unfortunately:
- psychotic prodrome lacks clear-cut symptoms and is difficult to identify
- little evidence exists to help clinicians select psychotropics and decide how long to use them
- treating all prodromal patients would expose those who never develop psychosis to the risk of psychotropics’ side effects.
How, then, can psychiatrists help patients who present with possible prodromal symptoms? Based on research and our experience, this article describes the psychotic prodrome and offers a pragmatic, evidence-based approach to diagnosis and treatment.
WHAT CAUSES PSYCHOTIC CONVERSION?
Reduced gray matter volumes in certain brain regions may be associated with conversion to psychosis (Box 1). Stress also may play a role; elevated stress-reactive cortisol levels are associated with positive symptom severity in the prodrome.1 Other factors being investigated include obstetric complications at birth, maternal age >30, premorbid schizotypal personality disorder, and impaired olfaction.
Symptoms. Nearly 80% of patients with schizophrenia experience a psychotic prodrome that lasts a few months to several years.2 Common features include:
- gradual worsening of perceptual disturbance
- referential thinking
- paranoia
- mild cognitive deficits
- mood lability
- impulsivity
- suicidality
- declining social function and academic performance.3,4
A premorbid phase often precedes the prodrome, with symptoms such as impaired attention, soft neurologic signs, and subtle social deficits. These changes may be harbingers of the prodrome but are too nonspecific to be diagnostic. Other functional impairments—including anxiety, depression, drug abuse, and psychosocial factors such as school stress—may mimic schizophrenic prodrome.
Prognosis. Studies of patients’ first schizophrenia episodes suggest that prodrome duration may predict outcome. A longer prodrome is thought to indicate a poor prognosis,6 such as in patients who wait a year before seeking treatment.7 A review of 22 studies of first-episode psychosis found early psychosocial and pharmacologic interventions improved long-term prognosis, and medication discontinuation predicted more-severe and chronic disease.8
Reduced gray matter volumes in certain brain regions may be associated with conversion to psychosis. Imaging studies have found medial temporal lobe changes—specifically, hippocampal volume alterations—in persons with schizophrenia, genetic high-risk groups, and those thought to be at risk for imminent psychosis.11
MRI imaging of patients with prodromal signs has shown less gray matter in the right medial temporal, lateral temporal, inferior frontal cortex, and bilateral cingulate regions in those who have developed psychosis, compared with those who have not. In the psychotic patients, 12-month longitudinal follow-up has found reduced gray matter in the left hippocampal, fusiform, orbitofrontal, cerebellar cortices, and cingulate gyrus.12
Brain structure is related to genetic liability for schizophrenia in high-risk patients, who seem to have smaller right and left prefrontal lobes and smaller right and left thalami. These findings are consistent with the prodrome’s neurocognitive deficits, which are less than those reported in schizophrenia and greater than those seen in healthy subjects.
Pioneering work by McGorry et al10 identified an “ultra high-risk group” with a psychotic conversion rate of 40% to 60%. These patients present with three symptom patterns:
- attenuated positive symptoms
- brief intermittent psychotic episodes
- genetic risk and recent deterioration syndrome (Table 1).
3 patient groups considered at ‘ultra high risk’ to develop schizophrenia
| Patients with… | Symptoms |
|---|---|
| Attenuated psychotic symptoms | Overvalued ideas, perceptual disorders |
| Present at least 1 week; not >5 years | |
| At least 1 symptom several times a week | |
| Brief intermittent psychotic episodes | Frank psychotic features |
| Resolve spontaneously within 7 days | |
| Can be drug-induced | |
| Genetic risk and recent deterioration syndrome | Psychotic disorder in a first-degree relative |
| Schizotypal personality disorder | |
| Present at least 1 month; not >5 years | |
| Significant functional decline | |
| Source: Adapted from reference 10 | |
The Edinburgh High Risk Study of 162 individuals ages 16 to 25 showed more marked psychopathology in those with at least two close relatives with schizophrenia, compared with control groups. A direct correlation was seen between genetic liability and poor neurocognitive performance.11
PRODROME RATING SCALES
Researchers are using outcome measures to diagnose prodromal symptoms and assess their severity. Operational, validated assessment tools include:
- Bonn Scale for the Assessment of Basic Symptoms (BSABS): captures subtle changes in thinking, feeling, and perception.
- Schizophrenia Prediction Instrument for Adults (SPI-A): defines prepsychotic deviations and rates symptoms that are subjectively experienced by the patient.
- Comprehensive Assessment of At Risk Mental State (CAARMS): defines ultra high-risk criteria and incorporates eight dimensions of psychopathology.
- Scale of Prodromal Symptoms (SOPS): rates psychosis severity. When embedded within the Structured Interview for Prodromal Syndromes (SIPS), the SOPS determines the presence or absence of psychosis and predicts progression to psychopathology.
- Criteria for Prodromal Symptoms (COPS): defines ultra high-risk categories.
- Presence of Psychosis Scale (POPS): rates severity, intensity, and duration of positive prodromal symptoms.12
These instruments may identify prodromal symptoms in psychiatric practice, but further validation of clinical criteria is needed before they could be recommended for routine patient assessment.
PROPHYLACTIC ANTIPSYCHOTICS?
Atypical antipsychotics may be the standard of care for patients with a first psychotic episode, but this intervention is based on few double-blind controlled trials. Not surprisingly, only a handful of studies have examined antipsychotic therapy for the prodrome’s less clear-cut symptoms.
Risperidone. An 8- to 12-week open-label study in adolescents with first- and second-degree relatives with schizophrenia13 included four prodromal and six first-episode psychosis patients who met criteria for a cluster A personality disorder. Risperidone, 1.0 mg/d and 1.8 mg/d, respectively, improved thought disorder and attention symptoms, as measured with the Child Behavior Checklist. Verbal memory improved minimally, and no medication side effects were reported.
An open-label observational study14 identified four middle-aged subjects with a genetic risk of schizophrenia who reported negative symptoms and neurocognitive deficits. Risperidone, started at 0.25 mg/d and gradually increased to a maximum of 2 mg/d, improved negative symptoms, attention, and working memory. Mild side effects including tremors, sedation, dry mouth, and anxiety symptoms were reported.
An open-label, randomized, comparator trial15 examined psychotic transition rates in 59 subjects (mean age, 20) who met ultra high-risk criteria. They received:
- a needs-based intervention (NBI) comprising case management, psychotropics excluding antipsychotics, and supportive psychotherapy
- or a specific preventive intervention (SPI) that included risperidone, 1 to 2 mg/d, and a modified cognitive-behavioral therapy (CBT).
Should you intervene with patients in suspected psychotic prodrome?
| Arguments for: |
|
| Arguments against: |
|
At 12-months’ follow-up, another 3 SPI patients who had been partially adherent or non-adherent to antipsychotic therapy had converted to psychosis. For adherent SPI patients, protection against conversion appeared to persist for 6 months after risperidone therapy ended. All medication side effects were mild and transient.
Table 3
Psychotic prodrome: Unanswered clinical questions
|
In the first year, 11 of 29 placebo-group patients and 5 of 31 receiving olanzapine converted to psychosis. Among patients receiving no treatment in the second year, 2 of 8 former placebo patients and 3 of 9 former olanzapine patients converted to psychosis.
Discontinuation rates were 35% and 28%, respectively. Compared with the placebo group, patients taking olanzapine experienced greater weight gain, suggesting that risks associated with antipsychotic therapy may exceed unproven benefits in this population.
Discussion. Little information exists on using quetiapine, ziprasidone, or aripiprazole in prodromal patients. As cited above, preliminary studies with risperidone and olanzapine suggest that these agents may improve several domains of psychotic prodrome. The evidence does not support firm conclusions, however, given the trials’ small sample sizes and brief duration.
The prevalence of obesity and metabolic syndrome in patients with schizophrenia and the added metabolic risks associated with atypical antipsychotics make their use during the prodrome controversial. Weighing the potential advantages and disadvantages (Table 2), we consider antipsychotics to be the last resort after psychosocial interventions have failed to improve prodromal symptoms.
Low-dose atypical antipsychotics may be warranted for some patients, but their use requires stringent monitoring of:
- weight and waist circumference
- vital signs
- metabolic parameters such as fasting blood glucose and lipid profile
- abnormal involuntary movements
- prolactin elevations.
OTHER THERAPIES
Antidepressants. Researchers are also exploring the efficacy of using antidepressants and anxiolytics in the prodromal phase. The only published naturalistic study of adolescents found antidepressants alone or in combination with mood stabilizers or anxiolytics to be as effective as atypical antipsychotics in treating prodromal symptoms.17 A more substantial study is ongoing.
Psychotherapy. For patients with a suspected psychotic prodrome, nondrug strategies may help minimize functional and cognitive impairments, ease distress, and improve coping skills.
CBT has been shown to reduce psychotic progression over 12 months.18 Use CBT to help patients cope with the illness while focusing on:
- symptom monitoring
- premorbid and present functioning
- establishing a therapeutic alliance
- assessing the patient’s experience of psychosis and any thought distortions.
‘REAL WORLD’ EARLY INTERVENTION
Patients with prodromal symptoms are often referred to psychiatrists by family members, primary care physicians, or other mental health professionals. They tend to be young adults, and a few may present in their teens. Most are experiencing behavioral changes such as social isolation, feeling suspicious, perceptual disturbances, depression, and/or anxiety symptoms that seem abnormal but fall short of DSM-IV criteria for schizophrenia diagnosis.
Many clinical questions about schizophrenia’s prodromal phase remain unanswered (Table 3). Our primary aim is to adequately assess these patients and provide treatment and follow-up, taking into account:
- the individual’s presentation
- risks and benefits of available interventions.
- Provide patients and families information and emotional support; a strong therapeutic alliance may help keep the patient in treatment if schizophrenia develops
- Offer early psychosocial interventions such as vocational training, relapse prevention, substance abuse treatment, family therapy, supportive and CBT
- Explore using low-dose atypical antipsychotics as a last resort for patients with pronounced prodromal symptoms; explain risks of weight gain and other metabolic changes, obtain consent, and document need for such interventions
- Consider referral, if feasible, to a center specializing in psychotic prodrome diagnosis, treatment, and research
Consider atypical antipsychotics for patients with distressing psychotic symptoms, rapidly deteriorating function, increased agitation, and safety risks. Consider antidepressant and/or anxiolytic therapy for depression and anxiety, respectively.
Discuss at length with patients and families the risks and benefits of pharmacologic treatments. When clinically appropriate, cautiously discontinue or taper any medication with patients’ consent, while monitoring for side effects and symptoms.
- Issue devoted to early prodrome research. SchizophrBull 2003;29(4):621-879.
- Diagnostic and therapeutic intervention during psychotic prodrome. CNS Spectrums 2004;9(8):578-606.
- PRIME (Prevention through Risk Identification, Management & Education) Research Clinic. Department of Psychiatry, Yale University. http://info.med.yale.edu/psych/prime/pintro.html.
- Youth Mental Health Update. Schizophrenia: New strategies for early detection and treatment. RAPP Clinic, Zucker Hillside Hospital, Glen Oaks, NY. http://schoolnet.lij.edu/eshare/files/rapp.html
- Aripiprazole • Abilify
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Dr. Narasimhan receives research support from Eli Lilly and Co. and Janssen Pharmaceutica and is a speaker or consultant for Eli Lilly and Co., Pfizer Inc., and Abbott Laboratories.
Dr. Buckley receives research support from AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly and Co., Janssen Pharmaceutica, Novartis Pharmaceuticals Corp., Pfizer Inc., and Solvay Pharmaceuticals. He is a consultant to and/or speaker for Abbott Laboratories, Alamo Pharmaceuticals, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly and Co., Janssen Pharmaceutica, Novartis Pharmaceuticals Corp., Pfizer Inc., and Pharmstar.
Because 40% of individuals with a psychotic prodrome develop schizophrenia, detecting and preventing this transition could improve many patients’ lives. Unfortunately:
- psychotic prodrome lacks clear-cut symptoms and is difficult to identify
- little evidence exists to help clinicians select psychotropics and decide how long to use them
- treating all prodromal patients would expose those who never develop psychosis to the risk of psychotropics’ side effects.
How, then, can psychiatrists help patients who present with possible prodromal symptoms? Based on research and our experience, this article describes the psychotic prodrome and offers a pragmatic, evidence-based approach to diagnosis and treatment.
WHAT CAUSES PSYCHOTIC CONVERSION?
Reduced gray matter volumes in certain brain regions may be associated with conversion to psychosis (Box 1). Stress also may play a role; elevated stress-reactive cortisol levels are associated with positive symptom severity in the prodrome.1 Other factors being investigated include obstetric complications at birth, maternal age >30, premorbid schizotypal personality disorder, and impaired olfaction.
Symptoms. Nearly 80% of patients with schizophrenia experience a psychotic prodrome that lasts a few months to several years.2 Common features include:
- gradual worsening of perceptual disturbance
- referential thinking
- paranoia
- mild cognitive deficits
- mood lability
- impulsivity
- suicidality
- declining social function and academic performance.3,4
A premorbid phase often precedes the prodrome, with symptoms such as impaired attention, soft neurologic signs, and subtle social deficits. These changes may be harbingers of the prodrome but are too nonspecific to be diagnostic. Other functional impairments—including anxiety, depression, drug abuse, and psychosocial factors such as school stress—may mimic schizophrenic prodrome.
Prognosis. Studies of patients’ first schizophrenia episodes suggest that prodrome duration may predict outcome. A longer prodrome is thought to indicate a poor prognosis,6 such as in patients who wait a year before seeking treatment.7 A review of 22 studies of first-episode psychosis found early psychosocial and pharmacologic interventions improved long-term prognosis, and medication discontinuation predicted more-severe and chronic disease.8
Reduced gray matter volumes in certain brain regions may be associated with conversion to psychosis. Imaging studies have found medial temporal lobe changes—specifically, hippocampal volume alterations—in persons with schizophrenia, genetic high-risk groups, and those thought to be at risk for imminent psychosis.11
MRI imaging of patients with prodromal signs has shown less gray matter in the right medial temporal, lateral temporal, inferior frontal cortex, and bilateral cingulate regions in those who have developed psychosis, compared with those who have not. In the psychotic patients, 12-month longitudinal follow-up has found reduced gray matter in the left hippocampal, fusiform, orbitofrontal, cerebellar cortices, and cingulate gyrus.12
Brain structure is related to genetic liability for schizophrenia in high-risk patients, who seem to have smaller right and left prefrontal lobes and smaller right and left thalami. These findings are consistent with the prodrome’s neurocognitive deficits, which are less than those reported in schizophrenia and greater than those seen in healthy subjects.
Pioneering work by McGorry et al10 identified an “ultra high-risk group” with a psychotic conversion rate of 40% to 60%. These patients present with three symptom patterns:
- attenuated positive symptoms
- brief intermittent psychotic episodes
- genetic risk and recent deterioration syndrome (Table 1).
3 patient groups considered at ‘ultra high risk’ to develop schizophrenia
| Patients with… | Symptoms |
|---|---|
| Attenuated psychotic symptoms | Overvalued ideas, perceptual disorders |
| Present at least 1 week; not >5 years | |
| At least 1 symptom several times a week | |
| Brief intermittent psychotic episodes | Frank psychotic features |
| Resolve spontaneously within 7 days | |
| Can be drug-induced | |
| Genetic risk and recent deterioration syndrome | Psychotic disorder in a first-degree relative |
| Schizotypal personality disorder | |
| Present at least 1 month; not >5 years | |
| Significant functional decline | |
| Source: Adapted from reference 10 | |
The Edinburgh High Risk Study of 162 individuals ages 16 to 25 showed more marked psychopathology in those with at least two close relatives with schizophrenia, compared with control groups. A direct correlation was seen between genetic liability and poor neurocognitive performance.11
PRODROME RATING SCALES
Researchers are using outcome measures to diagnose prodromal symptoms and assess their severity. Operational, validated assessment tools include:
- Bonn Scale for the Assessment of Basic Symptoms (BSABS): captures subtle changes in thinking, feeling, and perception.
- Schizophrenia Prediction Instrument for Adults (SPI-A): defines prepsychotic deviations and rates symptoms that are subjectively experienced by the patient.
- Comprehensive Assessment of At Risk Mental State (CAARMS): defines ultra high-risk criteria and incorporates eight dimensions of psychopathology.
- Scale of Prodromal Symptoms (SOPS): rates psychosis severity. When embedded within the Structured Interview for Prodromal Syndromes (SIPS), the SOPS determines the presence or absence of psychosis and predicts progression to psychopathology.
- Criteria for Prodromal Symptoms (COPS): defines ultra high-risk categories.
- Presence of Psychosis Scale (POPS): rates severity, intensity, and duration of positive prodromal symptoms.12
These instruments may identify prodromal symptoms in psychiatric practice, but further validation of clinical criteria is needed before they could be recommended for routine patient assessment.
PROPHYLACTIC ANTIPSYCHOTICS?
Atypical antipsychotics may be the standard of care for patients with a first psychotic episode, but this intervention is based on few double-blind controlled trials. Not surprisingly, only a handful of studies have examined antipsychotic therapy for the prodrome’s less clear-cut symptoms.
Risperidone. An 8- to 12-week open-label study in adolescents with first- and second-degree relatives with schizophrenia13 included four prodromal and six first-episode psychosis patients who met criteria for a cluster A personality disorder. Risperidone, 1.0 mg/d and 1.8 mg/d, respectively, improved thought disorder and attention symptoms, as measured with the Child Behavior Checklist. Verbal memory improved minimally, and no medication side effects were reported.
An open-label observational study14 identified four middle-aged subjects with a genetic risk of schizophrenia who reported negative symptoms and neurocognitive deficits. Risperidone, started at 0.25 mg/d and gradually increased to a maximum of 2 mg/d, improved negative symptoms, attention, and working memory. Mild side effects including tremors, sedation, dry mouth, and anxiety symptoms were reported.
An open-label, randomized, comparator trial15 examined psychotic transition rates in 59 subjects (mean age, 20) who met ultra high-risk criteria. They received:
- a needs-based intervention (NBI) comprising case management, psychotropics excluding antipsychotics, and supportive psychotherapy
- or a specific preventive intervention (SPI) that included risperidone, 1 to 2 mg/d, and a modified cognitive-behavioral therapy (CBT).
Should you intervene with patients in suspected psychotic prodrome?
| Arguments for: |
|
| Arguments against: |
|
At 12-months’ follow-up, another 3 SPI patients who had been partially adherent or non-adherent to antipsychotic therapy had converted to psychosis. For adherent SPI patients, protection against conversion appeared to persist for 6 months after risperidone therapy ended. All medication side effects were mild and transient.
Table 3
Psychotic prodrome: Unanswered clinical questions
|
In the first year, 11 of 29 placebo-group patients and 5 of 31 receiving olanzapine converted to psychosis. Among patients receiving no treatment in the second year, 2 of 8 former placebo patients and 3 of 9 former olanzapine patients converted to psychosis.
Discontinuation rates were 35% and 28%, respectively. Compared with the placebo group, patients taking olanzapine experienced greater weight gain, suggesting that risks associated with antipsychotic therapy may exceed unproven benefits in this population.
Discussion. Little information exists on using quetiapine, ziprasidone, or aripiprazole in prodromal patients. As cited above, preliminary studies with risperidone and olanzapine suggest that these agents may improve several domains of psychotic prodrome. The evidence does not support firm conclusions, however, given the trials’ small sample sizes and brief duration.
The prevalence of obesity and metabolic syndrome in patients with schizophrenia and the added metabolic risks associated with atypical antipsychotics make their use during the prodrome controversial. Weighing the potential advantages and disadvantages (Table 2), we consider antipsychotics to be the last resort after psychosocial interventions have failed to improve prodromal symptoms.
Low-dose atypical antipsychotics may be warranted for some patients, but their use requires stringent monitoring of:
- weight and waist circumference
- vital signs
- metabolic parameters such as fasting blood glucose and lipid profile
- abnormal involuntary movements
- prolactin elevations.
OTHER THERAPIES
Antidepressants. Researchers are also exploring the efficacy of using antidepressants and anxiolytics in the prodromal phase. The only published naturalistic study of adolescents found antidepressants alone or in combination with mood stabilizers or anxiolytics to be as effective as atypical antipsychotics in treating prodromal symptoms.17 A more substantial study is ongoing.
Psychotherapy. For patients with a suspected psychotic prodrome, nondrug strategies may help minimize functional and cognitive impairments, ease distress, and improve coping skills.
CBT has been shown to reduce psychotic progression over 12 months.18 Use CBT to help patients cope with the illness while focusing on:
- symptom monitoring
- premorbid and present functioning
- establishing a therapeutic alliance
- assessing the patient’s experience of psychosis and any thought distortions.
‘REAL WORLD’ EARLY INTERVENTION
Patients with prodromal symptoms are often referred to psychiatrists by family members, primary care physicians, or other mental health professionals. They tend to be young adults, and a few may present in their teens. Most are experiencing behavioral changes such as social isolation, feeling suspicious, perceptual disturbances, depression, and/or anxiety symptoms that seem abnormal but fall short of DSM-IV criteria for schizophrenia diagnosis.
Many clinical questions about schizophrenia’s prodromal phase remain unanswered (Table 3). Our primary aim is to adequately assess these patients and provide treatment and follow-up, taking into account:
- the individual’s presentation
- risks and benefits of available interventions.
- Provide patients and families information and emotional support; a strong therapeutic alliance may help keep the patient in treatment if schizophrenia develops
- Offer early psychosocial interventions such as vocational training, relapse prevention, substance abuse treatment, family therapy, supportive and CBT
- Explore using low-dose atypical antipsychotics as a last resort for patients with pronounced prodromal symptoms; explain risks of weight gain and other metabolic changes, obtain consent, and document need for such interventions
- Consider referral, if feasible, to a center specializing in psychotic prodrome diagnosis, treatment, and research
Consider atypical antipsychotics for patients with distressing psychotic symptoms, rapidly deteriorating function, increased agitation, and safety risks. Consider antidepressant and/or anxiolytic therapy for depression and anxiety, respectively.
Discuss at length with patients and families the risks and benefits of pharmacologic treatments. When clinically appropriate, cautiously discontinue or taper any medication with patients’ consent, while monitoring for side effects and symptoms.
- Issue devoted to early prodrome research. SchizophrBull 2003;29(4):621-879.
- Diagnostic and therapeutic intervention during psychotic prodrome. CNS Spectrums 2004;9(8):578-606.
- PRIME (Prevention through Risk Identification, Management & Education) Research Clinic. Department of Psychiatry, Yale University. http://info.med.yale.edu/psych/prime/pintro.html.
- Youth Mental Health Update. Schizophrenia: New strategies for early detection and treatment. RAPP Clinic, Zucker Hillside Hospital, Glen Oaks, NY. http://schoolnet.lij.edu/eshare/files/rapp.html
- Aripiprazole • Abilify
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Dr. Narasimhan receives research support from Eli Lilly and Co. and Janssen Pharmaceutica and is a speaker or consultant for Eli Lilly and Co., Pfizer Inc., and Abbott Laboratories.
Dr. Buckley receives research support from AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly and Co., Janssen Pharmaceutica, Novartis Pharmaceuticals Corp., Pfizer Inc., and Solvay Pharmaceuticals. He is a consultant to and/or speaker for Abbott Laboratories, Alamo Pharmaceuticals, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly and Co., Janssen Pharmaceutica, Novartis Pharmaceuticals Corp., Pfizer Inc., and Pharmstar.
1. Corcoran C, Walker E, Huot R, et al. The stress cascade and schizophrenia: etiology and onset. Schizophr Bull 2003;29(4):671-92.
2. Klosterkotter J. Diagnosing schizophrenia in the initial prodromal phase. Arch Gen Psychiatry 2001;58:158-64.
3. Yung AR. The initial prodrome in psychosis: the prodromal phase of first-episode psychosis: past and current conceptualizations. Schizophr Bull 1996;30:587-99.
4. Perkins DO. Evaluating and treating the prodromal stage of schizophrenia. Current Psychiatry Reports 2004;6:289-95.
5. Wood S W, Miller TJ, McGlashan TH. The “prodromal” patient: both symptomatic and at risk. CNS Spectrums 2001;6(3):223-32.
6. Keshavan MS, Haas G, Miewald J, et al. Prolonged untreated illness duration from prodromal onset predicts outcome in first episode psychosis. Schizoph Bull 2003;29(4):757-69.
7. Loebel AD, Lieberman JA, Alvir JM, et al. Duration of psychosis and outcome in first-episode schizophrenia. Am J Psychiatry 1992;149:1183-8.
8. Wyatt RJ, Green MF, Tuma AH. Long-term morbidity associated with delayed treatment of first admission schizophrenic patients: a re-analysis of the Camarillo State Hospital data. Psychol Med 1997;27:261-8.
9. Cornblatt BA, Lencz T, Smith CW, et al. The schizophrenia prodrome revisited: a neuro developmental perspective. Schizophr Bull 2003;29(4):633-51.
10. McGorry PD, Yung AR, Phillips LJ. The “close-in” or ultra high-risk model: a safe and effective strategy for research and clinical intervention in prepsychotic mental disorder. Schizophr Bull 2003;29(4):771-90.
11. Johnstone EC, Lawrie SM, Cosway R. What does the Edinburgh high-risk study tell us about schizophrenia? Am J Med Genet 2002;114(8):906-12.
12. Miller TJ, Clashing TH, Rosen JL, et al. Prodromal assessment with the Structured Interview for Prodromal Syndromes and the Scale of Prodromal Syndromes: predictive validity, interrater reliability, and training to reliability. Schizophr Bull 2003;29(4):703-15.
13. Cannon TD, Huttunen MO, Dahlstrom M, et al. Antipsychotic drug treatment in the prodromal phase in schizophrenia. Am J Psychiatry 2002;159:1230-2.
14. Tsuang MT, Stone WS, Faraone SV. Treatment of nonpsychotic relatives of patients with schizophrenia: four case studies. Biol Psychiatry 1999;45:1412-18.
15. McGorry PD, Yung AR, Phillips LJ, et al. Randomized controlled trial of intervention designed to reduce the risk of progression to first-episode psychosis in a clinical sample with subthreshold symptoms. Arch Gen Psychiatry 2002;59:921-8.
16. McGlashan TH, Zipursky R, Perkins DO, et al. The PRIME North America randomized double-blind clinical trial of olanzapine versus placebo in patients at risk of being prodromally symptomatic for psychosis. I. Study rationale and design. Schizophr Res 2003;61:7-18.
17. Cornblatt B, Lencz T, Correll C, et al. Treating the prodrome: naturalistic findings from the RAP program. Acta Psychiatr Scand 2002;106(suppl):44.-
18. Morrison AO, French P, Walford L, et al. Cognitive therapy for the prevention of psychosis in people at ultra-high risk: randomized controlled trial. Br J Psychiatry 2004;185:291-7.
19. Wentzell B. This family experience in a supportive first episode program (abstract S-25-03). Davos, Switzerland: Schizophrenia Research 67(1) 11th Biennial Winter Workshop on Schizophrenia. Feb. 7-13, 2004.
20. Stahl SM. Prophylactic antipsychotics: do they keep you from catching schizophrenia? J Clin Psychiatry 2004;65(11):1445-6.
1. Corcoran C, Walker E, Huot R, et al. The stress cascade and schizophrenia: etiology and onset. Schizophr Bull 2003;29(4):671-92.
2. Klosterkotter J. Diagnosing schizophrenia in the initial prodromal phase. Arch Gen Psychiatry 2001;58:158-64.
3. Yung AR. The initial prodrome in psychosis: the prodromal phase of first-episode psychosis: past and current conceptualizations. Schizophr Bull 1996;30:587-99.
4. Perkins DO. Evaluating and treating the prodromal stage of schizophrenia. Current Psychiatry Reports 2004;6:289-95.
5. Wood S W, Miller TJ, McGlashan TH. The “prodromal” patient: both symptomatic and at risk. CNS Spectrums 2001;6(3):223-32.
6. Keshavan MS, Haas G, Miewald J, et al. Prolonged untreated illness duration from prodromal onset predicts outcome in first episode psychosis. Schizoph Bull 2003;29(4):757-69.
7. Loebel AD, Lieberman JA, Alvir JM, et al. Duration of psychosis and outcome in first-episode schizophrenia. Am J Psychiatry 1992;149:1183-8.
8. Wyatt RJ, Green MF, Tuma AH. Long-term morbidity associated with delayed treatment of first admission schizophrenic patients: a re-analysis of the Camarillo State Hospital data. Psychol Med 1997;27:261-8.
9. Cornblatt BA, Lencz T, Smith CW, et al. The schizophrenia prodrome revisited: a neuro developmental perspective. Schizophr Bull 2003;29(4):633-51.
10. McGorry PD, Yung AR, Phillips LJ. The “close-in” or ultra high-risk model: a safe and effective strategy for research and clinical intervention in prepsychotic mental disorder. Schizophr Bull 2003;29(4):771-90.
11. Johnstone EC, Lawrie SM, Cosway R. What does the Edinburgh high-risk study tell us about schizophrenia? Am J Med Genet 2002;114(8):906-12.
12. Miller TJ, Clashing TH, Rosen JL, et al. Prodromal assessment with the Structured Interview for Prodromal Syndromes and the Scale of Prodromal Syndromes: predictive validity, interrater reliability, and training to reliability. Schizophr Bull 2003;29(4):703-15.
13. Cannon TD, Huttunen MO, Dahlstrom M, et al. Antipsychotic drug treatment in the prodromal phase in schizophrenia. Am J Psychiatry 2002;159:1230-2.
14. Tsuang MT, Stone WS, Faraone SV. Treatment of nonpsychotic relatives of patients with schizophrenia: four case studies. Biol Psychiatry 1999;45:1412-18.
15. McGorry PD, Yung AR, Phillips LJ, et al. Randomized controlled trial of intervention designed to reduce the risk of progression to first-episode psychosis in a clinical sample with subthreshold symptoms. Arch Gen Psychiatry 2002;59:921-8.
16. McGlashan TH, Zipursky R, Perkins DO, et al. The PRIME North America randomized double-blind clinical trial of olanzapine versus placebo in patients at risk of being prodromally symptomatic for psychosis. I. Study rationale and design. Schizophr Res 2003;61:7-18.
17. Cornblatt B, Lencz T, Correll C, et al. Treating the prodrome: naturalistic findings from the RAP program. Acta Psychiatr Scand 2002;106(suppl):44.-
18. Morrison AO, French P, Walford L, et al. Cognitive therapy for the prevention of psychosis in people at ultra-high risk: randomized controlled trial. Br J Psychiatry 2004;185:291-7.
19. Wentzell B. This family experience in a supportive first episode program (abstract S-25-03). Davos, Switzerland: Schizophrenia Research 67(1) 11th Biennial Winter Workshop on Schizophrenia. Feb. 7-13, 2004.
20. Stahl SM. Prophylactic antipsychotics: do they keep you from catching schizophrenia? J Clin Psychiatry 2004;65(11):1445-6.
Aripiprazole: What the researchers say
A typical antipsychotics have enhanced outcomes in schizophrenia while helping patients avert the troublesome motor effects associated with older agents. Some side effects, such as weight gain and prolactin elevation, have remained a concern, however.
Aripiprazole, a novel antipsychotic recently FDA-approved for treating schizophrenia, exhibited efficacy and tolerability in preclinical and clinical trials.
How aripiprazole works
Aripiprazole’s mechanism of action is important to our understanding of the dopamine hypothesis of antipsychotic effect.1
The dopamine hypothesis remains the predominant explanation of how antipsychotics work.2 However, the evolution of antipsychotic therapy has led to further refinement of the dopamine hypothesis, including selective dopamine (D4) antagonism, rapid dissociation from dopamine receptors, dopamine-serotonin receptor system interactions, dopamine-GABA system interactions, and now (with aripiprazole) partial agonist effects at dopamine (and selective serotonin) receptors.1,2
Table
ARIPIPRAZOLE: FASTFACTS
| Drug brand name: Abilify |
| Class: Atypical antipsychotic |
| FDA-approved indication: Schizophrenia |
| Approval date: Nov. 15, 2002 |
| Manufacturer: Bristol-Myers Squibb Co. & Otsuka America Pharmaceutical |
| Dosing forms: 10 mg, 15 mg, 20 mg, and 30 mg tablets |
| Recommended dosage: Start at 10 to 15 mg/d for all age groups. Maintenance dosage may be the same as initial dosage or may increase over time. Maximum recommended dosage is 30 mg/d. Cross-titration with prior treatment is recommended. |
Table 1
ARIPIPRAZOLE’S RECEPTOR-BINDING PROFILE
| Receptor type | Effect |
|---|---|
| Dopamine D2 | Partial agonism |
| Serotonin 5HT1A | Partial agonism |
| Serotonin 5HT2A | Antagonism |
| Alpha 1A | Minimal antagonism |
| Muscarinic | Minimal antagonism |
| Histaminergic | Minimal antagonism |
Table 2
POTENTIAL DRUG-DRUG INTERACTIONS WITH ARIPIPRAZOLE
| Drug | Effect on plasma concentration of aripiprazole |
|---|---|
| Quinidine | Increase |
| Ketoconazole | Increase |
| Carbamazepine | Decrease |
| Fluoxetine | Increase |
| Paroxetine | Increase |
Unlike other antipsychotics, which appear to act through dopamine receptor antagonism, aripiprazole is a potent partial agonist at both the dopamine (D2) and serotonin (5HT1A) receptors.1,2Table 1 describes the agent’s receptor-binding profile.
The agent offers 78% bioavailability. It is metabolized through the hepatic microenzyme system, specifically the cytochrome P450 enzymes 2D6 and 3A4. Use of nicotine does not alter the agent’s plasma levels. Its active moiety is aripiprazole with minor contributions from the derivative dehydro-aripiprazole.
Aripiprazole therapy can be started at 10 or 15 mg/d; the starting dosage—15 mg/d in most cases—may also suffice as maintenance therapy for many patients. If the patient does not respond, it is prudent to wait several weeks before increasing the dosage beyond 15 mg/d.
The FDA-approved maximum dosage for aripiprazole is 30 mg/d. However, information from clinical trials indicates that increasing the dosage from 15 to 30 mg/d does not enhance the antipsychotic’s efficacy.3,4 Because of its absorption properties, the agent can be taken with or without food.
Aripiprazole has a relatively long half-life (75 hours), so it can be administered once daily. This provides an advantage when switching treatments. Some information suggests that patients may be switched directly to aripiprazole,5 although cross-titration is recommended.3
Although data in clinical populations are insufficient, studies in normal volunteers suggest that aripiprazole can be given at regular dosages to older patients and to those with renal or hepatic impairment.3Table 2 highlights potential drug-drug interactions with agents that can influence the hepatic microenzyme system.
Efficacy
Aripiprazole has demonstrated efficacy in clinical studies of patients with schizophrenia and schizoaffective disorder.3-8
- In a placebo-controlled, 4-week trial, patients who received aripiprazole, 15 to 30 mg/d, or haloperidol, 10 mg/d, reported similar improvements in positive and negative symptoms, psychopathology, and overall function.6
- A placebo-controlled, 4-week trial of aripiprazole, 20 and 30 mg/d, compared with risperidone, 6 mg/d, revealed similar efficacy with respect to symptom improvement and overall functioning.7
- Aripiprazole and olanzapine demonstrated comparable efficacy in a 28-week study. However, patients in the aripiprazole group showed greater improvement at 8 weeks and sustained improvement through 28 weeks in a measure of verbal memory.8
Researchers have not yet compared aripiprazole with clozapine, quetiapine, or ziprasidone. Also, information on the dosing, efficacy, and tolerability of aripiprazole in patients with first-episode or treatment-refractory schizophrenia is limited. According to the manufacturers’ prescription information, aripiprazole’s long-term efficacy in schizophrenia treatment has not been established. Data on 1-year treatment with aripiprazole appear encouraging.3
Preliminary data suggest that aripiprazole may help treat nonpsychotic conditions, although which ones has yet to be determined. A 3-week, placebo-controlled study demonstrated that aripiprazole, 30 mg/d, helped ameliorate symptoms of mania.9
Tolerability
Aripiprazole’s side-effect profile, revealed in preclinical and clinical trials, suggests that the drug could be well tolerated among a broad range of patients.10
In the 4-week, placebo-controlled comparison with haloperidol, rates of extrapyramidal symptoms (EPS) among aripiprazole-treated patients were much lower than those in the haloperidol group and similar to those in the placebo group.6 There is no evidence that higher dosages of aripiprazole lead to increased EPS. It is also not known whether aripiprazole will cause EPS in children and in patients older than 65, who are more susceptible than other age groups to antipsychotic-induced motor side effects.
Aripiprazole is believed to be less likely than typical antipsychotics to induce tardive dyskinesia, but more long-term information is needed.3
Studies have associated aripiprazole use with some weight gain, but (marginally) less than risperidone,7 less than haloperidol,6 and substantially less than olanzapine.8 Direct comparisons with other atypicals are not yet available.
Aripiprazole’s effect on glucose metabolism has not been determined, but early information suggests a favorable profile with respect to metabolic indices. Aripiprazole does not appear to elevate prolactin or cause cardiac QTc prolongation. Sedation appears to be the most pronounced side effect; this effect also appears to increase with higher dosages.
As has happened with the other atypicals, the pattern of use for aripiprazole will unfold over time as clinicians gain experience with using this agent in distinct patient groups.
Related resources
- Jordan S, Koprivica V, Chen R, et al. The antipsychotic aripiprazole is a potent, partial agonist at the human 5HT(1A) receptor. Eur J Pharmacol 2002;50:873-83.
- Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry 2002;63:763-71.
Drug brand names
- Carbamazepine • Tegretol
- Clozapine • Clozaril
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Ketoconazole • Nizoral
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosure
Dr. Buckley receives grant support from, is a consultant to, and is a speaker for AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly and Co., Janssen Pharmaceutica, and Novartis Pharmaceuticals Corp.
Dr. Sinha is a consultant to Bristol-Myers Squibb Co.
Dr. Sebastian is a consultant to Eli Lilly and Co.
Ms. Stirewalt reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stahl SM. Dopamine system stabilizers, aripiprazole, and the next generation of antipsychotics, part 1: “Goldilocks” actions at dopamine receptors. J Clin Psychiatry 2001;62:841-2.
2. Kapur S, Remington G. Dopamine D2 receptors and their role in atypical antipsychotic action: still necessary and maybe even sufficient. Biol Psychiatry 2001;50:873-83.
3. Buckley PF. Aripiprazole: efficacy and tolerability profile of a novel-acting atypical antipsychotic. Drugs of Today 2003 (in press).
4. Kujawa M, Carson WH, Stock E, et al. Meta-analysis of the efficacy of aripiprazole (abstract). Schizophr Res 2002;53.:
5. Casey D, Kujawa M, et al. Switching from other antipsychotics to aripiprazole (abstract). Int J Neuropsychopharmacol 2002;5(1):S187.-
6. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry 2002;63:763-71.
7. Potkin SG, et al. A comparison of aripiprazole versus risperidone in patients with schizophrenia. Arch Gen Psychiatry (in press)
8. Cornblatt B, Creen MF, et al. A comparison of cognitive performance on aripiprazole versus olanzapine (abstract). Int J Neuropsychopharmacol 2002;5(1):S185.-
9. Kujawa M, et al. A 52 week comparison of aripiprazole versus haloperidol in the treatment of schizophrenia (abstract). Int J Neuropsychopharmacol 2002;5(1):S186-
10. Keck P, Carson WH, Saha AR. A placebo controlled trial of aripiprazole for the treatment of mania. American Psychiatric Association annual meeting, Philadelphia, May 2002.
A typical antipsychotics have enhanced outcomes in schizophrenia while helping patients avert the troublesome motor effects associated with older agents. Some side effects, such as weight gain and prolactin elevation, have remained a concern, however.
Aripiprazole, a novel antipsychotic recently FDA-approved for treating schizophrenia, exhibited efficacy and tolerability in preclinical and clinical trials.
How aripiprazole works
Aripiprazole’s mechanism of action is important to our understanding of the dopamine hypothesis of antipsychotic effect.1
The dopamine hypothesis remains the predominant explanation of how antipsychotics work.2 However, the evolution of antipsychotic therapy has led to further refinement of the dopamine hypothesis, including selective dopamine (D4) antagonism, rapid dissociation from dopamine receptors, dopamine-serotonin receptor system interactions, dopamine-GABA system interactions, and now (with aripiprazole) partial agonist effects at dopamine (and selective serotonin) receptors.1,2
Table
ARIPIPRAZOLE: FASTFACTS
| Drug brand name: Abilify |
| Class: Atypical antipsychotic |
| FDA-approved indication: Schizophrenia |
| Approval date: Nov. 15, 2002 |
| Manufacturer: Bristol-Myers Squibb Co. & Otsuka America Pharmaceutical |
| Dosing forms: 10 mg, 15 mg, 20 mg, and 30 mg tablets |
| Recommended dosage: Start at 10 to 15 mg/d for all age groups. Maintenance dosage may be the same as initial dosage or may increase over time. Maximum recommended dosage is 30 mg/d. Cross-titration with prior treatment is recommended. |
Table 1
ARIPIPRAZOLE’S RECEPTOR-BINDING PROFILE
| Receptor type | Effect |
|---|---|
| Dopamine D2 | Partial agonism |
| Serotonin 5HT1A | Partial agonism |
| Serotonin 5HT2A | Antagonism |
| Alpha 1A | Minimal antagonism |
| Muscarinic | Minimal antagonism |
| Histaminergic | Minimal antagonism |
Table 2
POTENTIAL DRUG-DRUG INTERACTIONS WITH ARIPIPRAZOLE
| Drug | Effect on plasma concentration of aripiprazole |
|---|---|
| Quinidine | Increase |
| Ketoconazole | Increase |
| Carbamazepine | Decrease |
| Fluoxetine | Increase |
| Paroxetine | Increase |
Unlike other antipsychotics, which appear to act through dopamine receptor antagonism, aripiprazole is a potent partial agonist at both the dopamine (D2) and serotonin (5HT1A) receptors.1,2Table 1 describes the agent’s receptor-binding profile.
The agent offers 78% bioavailability. It is metabolized through the hepatic microenzyme system, specifically the cytochrome P450 enzymes 2D6 and 3A4. Use of nicotine does not alter the agent’s plasma levels. Its active moiety is aripiprazole with minor contributions from the derivative dehydro-aripiprazole.
Aripiprazole therapy can be started at 10 or 15 mg/d; the starting dosage—15 mg/d in most cases—may also suffice as maintenance therapy for many patients. If the patient does not respond, it is prudent to wait several weeks before increasing the dosage beyond 15 mg/d.
The FDA-approved maximum dosage for aripiprazole is 30 mg/d. However, information from clinical trials indicates that increasing the dosage from 15 to 30 mg/d does not enhance the antipsychotic’s efficacy.3,4 Because of its absorption properties, the agent can be taken with or without food.
Aripiprazole has a relatively long half-life (75 hours), so it can be administered once daily. This provides an advantage when switching treatments. Some information suggests that patients may be switched directly to aripiprazole,5 although cross-titration is recommended.3
Although data in clinical populations are insufficient, studies in normal volunteers suggest that aripiprazole can be given at regular dosages to older patients and to those with renal or hepatic impairment.3Table 2 highlights potential drug-drug interactions with agents that can influence the hepatic microenzyme system.
Efficacy
Aripiprazole has demonstrated efficacy in clinical studies of patients with schizophrenia and schizoaffective disorder.3-8
- In a placebo-controlled, 4-week trial, patients who received aripiprazole, 15 to 30 mg/d, or haloperidol, 10 mg/d, reported similar improvements in positive and negative symptoms, psychopathology, and overall function.6
- A placebo-controlled, 4-week trial of aripiprazole, 20 and 30 mg/d, compared with risperidone, 6 mg/d, revealed similar efficacy with respect to symptom improvement and overall functioning.7
- Aripiprazole and olanzapine demonstrated comparable efficacy in a 28-week study. However, patients in the aripiprazole group showed greater improvement at 8 weeks and sustained improvement through 28 weeks in a measure of verbal memory.8
Researchers have not yet compared aripiprazole with clozapine, quetiapine, or ziprasidone. Also, information on the dosing, efficacy, and tolerability of aripiprazole in patients with first-episode or treatment-refractory schizophrenia is limited. According to the manufacturers’ prescription information, aripiprazole’s long-term efficacy in schizophrenia treatment has not been established. Data on 1-year treatment with aripiprazole appear encouraging.3
Preliminary data suggest that aripiprazole may help treat nonpsychotic conditions, although which ones has yet to be determined. A 3-week, placebo-controlled study demonstrated that aripiprazole, 30 mg/d, helped ameliorate symptoms of mania.9
Tolerability
Aripiprazole’s side-effect profile, revealed in preclinical and clinical trials, suggests that the drug could be well tolerated among a broad range of patients.10
In the 4-week, placebo-controlled comparison with haloperidol, rates of extrapyramidal symptoms (EPS) among aripiprazole-treated patients were much lower than those in the haloperidol group and similar to those in the placebo group.6 There is no evidence that higher dosages of aripiprazole lead to increased EPS. It is also not known whether aripiprazole will cause EPS in children and in patients older than 65, who are more susceptible than other age groups to antipsychotic-induced motor side effects.
Aripiprazole is believed to be less likely than typical antipsychotics to induce tardive dyskinesia, but more long-term information is needed.3
Studies have associated aripiprazole use with some weight gain, but (marginally) less than risperidone,7 less than haloperidol,6 and substantially less than olanzapine.8 Direct comparisons with other atypicals are not yet available.
Aripiprazole’s effect on glucose metabolism has not been determined, but early information suggests a favorable profile with respect to metabolic indices. Aripiprazole does not appear to elevate prolactin or cause cardiac QTc prolongation. Sedation appears to be the most pronounced side effect; this effect also appears to increase with higher dosages.
As has happened with the other atypicals, the pattern of use for aripiprazole will unfold over time as clinicians gain experience with using this agent in distinct patient groups.
Related resources
- Jordan S, Koprivica V, Chen R, et al. The antipsychotic aripiprazole is a potent, partial agonist at the human 5HT(1A) receptor. Eur J Pharmacol 2002;50:873-83.
- Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry 2002;63:763-71.
Drug brand names
- Carbamazepine • Tegretol
- Clozapine • Clozaril
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Ketoconazole • Nizoral
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosure
Dr. Buckley receives grant support from, is a consultant to, and is a speaker for AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly and Co., Janssen Pharmaceutica, and Novartis Pharmaceuticals Corp.
Dr. Sinha is a consultant to Bristol-Myers Squibb Co.
Dr. Sebastian is a consultant to Eli Lilly and Co.
Ms. Stirewalt reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
A typical antipsychotics have enhanced outcomes in schizophrenia while helping patients avert the troublesome motor effects associated with older agents. Some side effects, such as weight gain and prolactin elevation, have remained a concern, however.
Aripiprazole, a novel antipsychotic recently FDA-approved for treating schizophrenia, exhibited efficacy and tolerability in preclinical and clinical trials.
How aripiprazole works
Aripiprazole’s mechanism of action is important to our understanding of the dopamine hypothesis of antipsychotic effect.1
The dopamine hypothesis remains the predominant explanation of how antipsychotics work.2 However, the evolution of antipsychotic therapy has led to further refinement of the dopamine hypothesis, including selective dopamine (D4) antagonism, rapid dissociation from dopamine receptors, dopamine-serotonin receptor system interactions, dopamine-GABA system interactions, and now (with aripiprazole) partial agonist effects at dopamine (and selective serotonin) receptors.1,2
Table
ARIPIPRAZOLE: FASTFACTS
| Drug brand name: Abilify |
| Class: Atypical antipsychotic |
| FDA-approved indication: Schizophrenia |
| Approval date: Nov. 15, 2002 |
| Manufacturer: Bristol-Myers Squibb Co. & Otsuka America Pharmaceutical |
| Dosing forms: 10 mg, 15 mg, 20 mg, and 30 mg tablets |
| Recommended dosage: Start at 10 to 15 mg/d for all age groups. Maintenance dosage may be the same as initial dosage or may increase over time. Maximum recommended dosage is 30 mg/d. Cross-titration with prior treatment is recommended. |
Table 1
ARIPIPRAZOLE’S RECEPTOR-BINDING PROFILE
| Receptor type | Effect |
|---|---|
| Dopamine D2 | Partial agonism |
| Serotonin 5HT1A | Partial agonism |
| Serotonin 5HT2A | Antagonism |
| Alpha 1A | Minimal antagonism |
| Muscarinic | Minimal antagonism |
| Histaminergic | Minimal antagonism |
Table 2
POTENTIAL DRUG-DRUG INTERACTIONS WITH ARIPIPRAZOLE
| Drug | Effect on plasma concentration of aripiprazole |
|---|---|
| Quinidine | Increase |
| Ketoconazole | Increase |
| Carbamazepine | Decrease |
| Fluoxetine | Increase |
| Paroxetine | Increase |
Unlike other antipsychotics, which appear to act through dopamine receptor antagonism, aripiprazole is a potent partial agonist at both the dopamine (D2) and serotonin (5HT1A) receptors.1,2Table 1 describes the agent’s receptor-binding profile.
The agent offers 78% bioavailability. It is metabolized through the hepatic microenzyme system, specifically the cytochrome P450 enzymes 2D6 and 3A4. Use of nicotine does not alter the agent’s plasma levels. Its active moiety is aripiprazole with minor contributions from the derivative dehydro-aripiprazole.
Aripiprazole therapy can be started at 10 or 15 mg/d; the starting dosage—15 mg/d in most cases—may also suffice as maintenance therapy for many patients. If the patient does not respond, it is prudent to wait several weeks before increasing the dosage beyond 15 mg/d.
The FDA-approved maximum dosage for aripiprazole is 30 mg/d. However, information from clinical trials indicates that increasing the dosage from 15 to 30 mg/d does not enhance the antipsychotic’s efficacy.3,4 Because of its absorption properties, the agent can be taken with or without food.
Aripiprazole has a relatively long half-life (75 hours), so it can be administered once daily. This provides an advantage when switching treatments. Some information suggests that patients may be switched directly to aripiprazole,5 although cross-titration is recommended.3
Although data in clinical populations are insufficient, studies in normal volunteers suggest that aripiprazole can be given at regular dosages to older patients and to those with renal or hepatic impairment.3Table 2 highlights potential drug-drug interactions with agents that can influence the hepatic microenzyme system.
Efficacy
Aripiprazole has demonstrated efficacy in clinical studies of patients with schizophrenia and schizoaffective disorder.3-8
- In a placebo-controlled, 4-week trial, patients who received aripiprazole, 15 to 30 mg/d, or haloperidol, 10 mg/d, reported similar improvements in positive and negative symptoms, psychopathology, and overall function.6
- A placebo-controlled, 4-week trial of aripiprazole, 20 and 30 mg/d, compared with risperidone, 6 mg/d, revealed similar efficacy with respect to symptom improvement and overall functioning.7
- Aripiprazole and olanzapine demonstrated comparable efficacy in a 28-week study. However, patients in the aripiprazole group showed greater improvement at 8 weeks and sustained improvement through 28 weeks in a measure of verbal memory.8
Researchers have not yet compared aripiprazole with clozapine, quetiapine, or ziprasidone. Also, information on the dosing, efficacy, and tolerability of aripiprazole in patients with first-episode or treatment-refractory schizophrenia is limited. According to the manufacturers’ prescription information, aripiprazole’s long-term efficacy in schizophrenia treatment has not been established. Data on 1-year treatment with aripiprazole appear encouraging.3
Preliminary data suggest that aripiprazole may help treat nonpsychotic conditions, although which ones has yet to be determined. A 3-week, placebo-controlled study demonstrated that aripiprazole, 30 mg/d, helped ameliorate symptoms of mania.9
Tolerability
Aripiprazole’s side-effect profile, revealed in preclinical and clinical trials, suggests that the drug could be well tolerated among a broad range of patients.10
In the 4-week, placebo-controlled comparison with haloperidol, rates of extrapyramidal symptoms (EPS) among aripiprazole-treated patients were much lower than those in the haloperidol group and similar to those in the placebo group.6 There is no evidence that higher dosages of aripiprazole lead to increased EPS. It is also not known whether aripiprazole will cause EPS in children and in patients older than 65, who are more susceptible than other age groups to antipsychotic-induced motor side effects.
Aripiprazole is believed to be less likely than typical antipsychotics to induce tardive dyskinesia, but more long-term information is needed.3
Studies have associated aripiprazole use with some weight gain, but (marginally) less than risperidone,7 less than haloperidol,6 and substantially less than olanzapine.8 Direct comparisons with other atypicals are not yet available.
Aripiprazole’s effect on glucose metabolism has not been determined, but early information suggests a favorable profile with respect to metabolic indices. Aripiprazole does not appear to elevate prolactin or cause cardiac QTc prolongation. Sedation appears to be the most pronounced side effect; this effect also appears to increase with higher dosages.
As has happened with the other atypicals, the pattern of use for aripiprazole will unfold over time as clinicians gain experience with using this agent in distinct patient groups.
Related resources
- Jordan S, Koprivica V, Chen R, et al. The antipsychotic aripiprazole is a potent, partial agonist at the human 5HT(1A) receptor. Eur J Pharmacol 2002;50:873-83.
- Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry 2002;63:763-71.
Drug brand names
- Carbamazepine • Tegretol
- Clozapine • Clozaril
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Ketoconazole • Nizoral
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosure
Dr. Buckley receives grant support from, is a consultant to, and is a speaker for AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly and Co., Janssen Pharmaceutica, and Novartis Pharmaceuticals Corp.
Dr. Sinha is a consultant to Bristol-Myers Squibb Co.
Dr. Sebastian is a consultant to Eli Lilly and Co.
Ms. Stirewalt reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stahl SM. Dopamine system stabilizers, aripiprazole, and the next generation of antipsychotics, part 1: “Goldilocks” actions at dopamine receptors. J Clin Psychiatry 2001;62:841-2.
2. Kapur S, Remington G. Dopamine D2 receptors and their role in atypical antipsychotic action: still necessary and maybe even sufficient. Biol Psychiatry 2001;50:873-83.
3. Buckley PF. Aripiprazole: efficacy and tolerability profile of a novel-acting atypical antipsychotic. Drugs of Today 2003 (in press).
4. Kujawa M, Carson WH, Stock E, et al. Meta-analysis of the efficacy of aripiprazole (abstract). Schizophr Res 2002;53.:
5. Casey D, Kujawa M, et al. Switching from other antipsychotics to aripiprazole (abstract). Int J Neuropsychopharmacol 2002;5(1):S187.-
6. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry 2002;63:763-71.
7. Potkin SG, et al. A comparison of aripiprazole versus risperidone in patients with schizophrenia. Arch Gen Psychiatry (in press)
8. Cornblatt B, Creen MF, et al. A comparison of cognitive performance on aripiprazole versus olanzapine (abstract). Int J Neuropsychopharmacol 2002;5(1):S185.-
9. Kujawa M, et al. A 52 week comparison of aripiprazole versus haloperidol in the treatment of schizophrenia (abstract). Int J Neuropsychopharmacol 2002;5(1):S186-
10. Keck P, Carson WH, Saha AR. A placebo controlled trial of aripiprazole for the treatment of mania. American Psychiatric Association annual meeting, Philadelphia, May 2002.
1. Stahl SM. Dopamine system stabilizers, aripiprazole, and the next generation of antipsychotics, part 1: “Goldilocks” actions at dopamine receptors. J Clin Psychiatry 2001;62:841-2.
2. Kapur S, Remington G. Dopamine D2 receptors and their role in atypical antipsychotic action: still necessary and maybe even sufficient. Biol Psychiatry 2001;50:873-83.
3. Buckley PF. Aripiprazole: efficacy and tolerability profile of a novel-acting atypical antipsychotic. Drugs of Today 2003 (in press).
4. Kujawa M, Carson WH, Stock E, et al. Meta-analysis of the efficacy of aripiprazole (abstract). Schizophr Res 2002;53.:
5. Casey D, Kujawa M, et al. Switching from other antipsychotics to aripiprazole (abstract). Int J Neuropsychopharmacol 2002;5(1):S187.-
6. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry 2002;63:763-71.
7. Potkin SG, et al. A comparison of aripiprazole versus risperidone in patients with schizophrenia. Arch Gen Psychiatry (in press)
8. Cornblatt B, Creen MF, et al. A comparison of cognitive performance on aripiprazole versus olanzapine (abstract). Int J Neuropsychopharmacol 2002;5(1):S185.-
9. Kujawa M, et al. A 52 week comparison of aripiprazole versus haloperidol in the treatment of schizophrenia (abstract). Int J Neuropsychopharmacol 2002;5(1):S186-
10. Keck P, Carson WH, Saha AR. A placebo controlled trial of aripiprazole for the treatment of mania. American Psychiatric Association annual meeting, Philadelphia, May 2002.