User login
Beneath the surface: Derm clues to underlying disorders
• When evaluating patients with suspected cutaneous lupus erythematosus, use multiple criteria—including histologic and immunofluorescent biopsy findings and American College of Rheumatology criteria—to rule out systemic disease. C
• Cancer screening with a careful history and physical examination is recommended for all adult patients whom you suspect of having dermatomyositis. C
• Suspect mixed connective tissue disease in patients with skin findings characteristic of varying auto-immune disorders appearing sequentially over several months or years. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Many systemic conditions are accompanied by skin manifestations. This is especially true for connective tissue disorders, for which dermatologic findings are often the key to diagnosis.
In this review, we describe the dermatologic findings of some well-known connective tissue disorders. The text and photographs in the pages that follow will help you hone your diagnostic skills, leading to earlier treatment and, possibly, better outcomes.
Lupus erythematosus: Cutaneous and systemic disease often overlap
Lupus erythematosus (LE), a chronic, inflammatory autoimmune condition that primarily affects women in their 20s and 30s, may initially present as a systemic disease or in a purely cutaneous form. However, most patients with systemic LE have some skin manifestations, and those with cutaneous LE often have—or subsequently develop—systemic involvement.1 Thus, recognizing the cutaneous manifestations of LE will not only aid in diagnosis, but will help you identify patients at risk for systemic disease.
Cutaneous LE has 4 subtypes
There are 4 subcategories of cutaneous LE—acute, subacute, chronic, and intermittent.2 Each is differentiated by the appearance of the lesions (TABLE 1), histology, and serological markers.1 Photosensitivity is common to all the subcategories to varying degrees.
Acute cutaneous lupus erythematosus (ACLE) is typically characterized by the classic malar “butterfly” rash, an erythematous eruption of macules or edematous papules over the bridge of the nose and cheek.3 Although this presentation is most common, there are variations—1 in which the lesions cover other exposed areas (commonly including the “V” of the chest, the extensor surface of the arms, and the hands), and a rare form in which toxic epidermal necrolysis-like blistering occurs.1,4 These skin changes—which generally last anywhere from a few hours to several weeks—typically resolve without scarring, although pigment changes can occur.5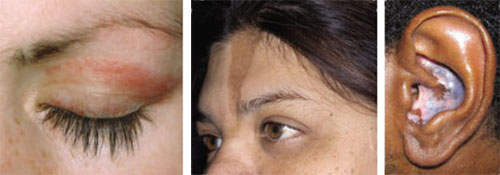
Left: Dermatomyositis is the underlying cause of the heliotrope discoloration on this patient’s upper eyelid.
Center: Linear morphea is associated with the lesion on this patient’s face—called en coup de sabre because it resembles the mark caused by the stroke of a sword in a duel.
Right: Discoid lupus erythematosus causes hypopigmentation and scarring.Patients with ACLE have a predisposition to systemic LE; unlike those with other forms of cutaneous LE, 40% to 90% will have double-stranded DNA (dsDNA) autoantibodies.3,6
Subacute cutaneous lupus erythematosus (SCLE), which usually affects middle-aged Caucasian women, is characterized by erythematous papulosquamous (psoriasis-like) eruptions or annular lesions with raised red borders and central clearing—or both. These lesions, which are nonscarring, lack in-duration, and rarely affect the scalp or face, appear suddenly, usually after exposure to sunlight (FIGURE 1)5,7-9 or certain drugs. Hydrochlorothiazide, terbinafine, calcium channel blockers, and angiotensin-converting enzyme inhibitors are common offenders.1,6
FIGURE 1
Annular lesions in subacute cutaneous lupus erythematosus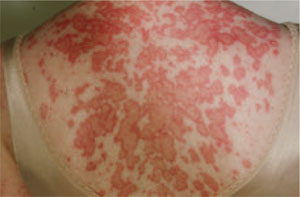
Ring-like lesions with raised red borders and central clearing on the back of a patient with subacute cutaneous lupus erythematosus.
SCLE is often associated with extracutaneous symptoms such as arthritis and myalgias,1,8 but patients are at relatively low risk for severe systemic manifestations.5,8,10 Serology is often notable, with anti-Ro (SS-A) antibodies present in 70% to 90% of patients and anti-La (SS-B) autoantibodies found in 30% to 50%.1,11
Chronic cutaneous lupus erythematosus (CCLE) also occurs predominantly in females, at a ratio as high as 5 to 1.12 There are 3 variations of CCLE: discoid lupus erythematosus (DLE), LE profundus, and chilblain LE (TABLE 1). DLE, characterized by alopecia, skin atrophy, and dyspigmentation, is the most common and affects patients of all ages and ethnic groups.9,13,14 (See image above.)
DLE lesions typically begin as erythematous papules and plaques with scale. As the disorder progresses, the lesions spread, causing follicular plugging, peripheral hyperpigmentation and central hypopigmentation, telangiectasia, and atrophy.9,15 In some cases, patients develop thickened, scarred skin and permanent scarring alopecia.15 Prompt recognition of DLE is particularly important, as early referral and treatment may reduce the likelihood of permanent scarring alopecia and pigment changes.11
Intermittent cutaneous lupus erythematosus (ICLE), a relatively new subtype of cutaneous LE, is represented by a rare condition—lupus erythematosus tumidus (LET)—reported in <100 cases worldwide. LET is characterized by succulent, erythematous, and edematous plaques on sun-exposed parts of the body.1,16
TABLE 1
Cutaneous lupus erythematosus: Recognizing the subtypes1
| Acute cutaneous lupus erythematosus (ACLE) |
|---|
| Localized: erythematous macules or papules over the bridge of the nose and cheek, with sparing of the nasolabial folds (“butterfly rash”) |
| Generalized: similar erythematous lesions over other photodistributed parts of the body, including the neck, chest, arms, and hands |
| Toxic epidermal necrolysis-like (TEN-like): blistering and epidermal cleavage in photodistributed parts of the body |
| Subacute cutaneous lupus erythematosus (SCLE) |
| Erythematous papulosquamous (psoriasis-like) eruptions or annular (ring-like) lesions with raised red borders and central clearing, occurring symmetrically and suddenly after sunlight exposure on photo-distributed body parts; the scalp and face are rarely affected |
| Chronic cutaneous lupus erythematosus (CCLE) |
| Discoid lupus erythematosus: erythematous papules and plaques with associated scale, spreading centrifugally with follicular plugging, pigment change, telangiectasia, and atrophy; scarring alopecia can occur |
| Lupus erythematosus profundus: tender, erythematous nodules and plaques, usually involving the proximal extremities, trunk, breasts, buttocks, and face |
| Chilblain lupus erythematosus: tender, erythematous nodules and plaques, occurring in acral areas often in response to cold |
| Intermittent cutaneous lupus erythematosus (ICLE) |
| Lupus erythematosus tumidus: succulent, erythematous, and edematous plaques found on photodistributed parts of the body |
Cutaneous LE diagnosis and treatment: Start with ACR criteria
When evaluating patients with suspected cutaneous LE, it is important not only to identify the subtype, but also to rule out systemic disease using criteria established by the American College of Rheumatology (ACR).17 Notably, 4 of the 11 diagnostic criteria for systemic disease involve visual clues, including malar rash, discoid rash, photosensitivity, and oral ulcerations. Laboratory evidence of systemic disease may include a positive antinuclear antibody, anti-dsDNA, or anti-Sm autoantibody test results, as well as hematologic abnormalities described in the ACR guidelines.
Ultimately, a diagnosis of cutaneous LE should be based on the patient’s history and physical exam, autoantibody profile, and histologic and immunofluorescent biopsy findings. A rheumatologic evaluation may help to determine which patients have systemic disease, as the ACR criteria may overdiagnose systemic LE in those with predominantly skin changes.6
Treatment of cutaneous LE is based on the subtype and extent of disease, with potent topical corticosteroids, in combination with antimalarial agents, being the primary therapies. ACLE skin lesions generally respond best to systemic corticosteroids and immunosuppressive agents (such as azathioprine or cyclophosphamide) that are used to control underlying systemic disease. SCLE can be managed with topical corticosteroids; however, patients typically also require systemic treatment, often with hydroxychloroquine, for optimal control. DLE, the most common form of CCLE, is managed in a similar fashion, with a greater role for intralesional corticosteroid injections. Lesions associated with ICLE, which often resolve spontaneously, may be treated with topical corticosteroids and antimalarials.16 It is also important to advise all patients with cutaneous LE to use a broad-spectrum sunscreen, as ultraviolet (UV) exposure can induce or exacerbate the lesions.9
Dermatomyositis: Rare but serious
Dermatomyositis is an idiopathic inflammatory myopathy characterized by chronic muscle inflammation, symmetric proximal muscle weakness, and distinct cutaneous findings. In addition to possible cardiac and pulmonary complications, dysphagia, and joint contractures, dermatomyositis is associated with cancer, with up to 25% of affected adults having an underlying occult malignancy.18
Dermatomyositis is a rare condition, with a prevalence of only 1 to 10 cases per million adults and 3.2 cases per million children.19 It has a bimodal age distribution, with most juvenile cases affecting children between the ages of 5 and 14 years and most adult cases developing in the fifth and sixth decades of life. Women are affected twice as often as men.18
Suspect dermatomyositis when you see any of the following signs:
- A heliotrope rash: violaceous macules and patches, with or without edema, symmetrically on the periorbital skin, present early in the disease course in 30% to 60% of patients.19 (See image)
- Gottron’s papules: violaceous papules on the dorsal interphalangeal and metacarpophalangeal joints of the hands, elbows, and knees, occurring in as many as 70% of patients (FIGURE 2).19,20
- Gottron’s sign: nonscaling, violaceous erythematous macules and plaques occurring symmetrically in the same distribution as Gottron’s papules, but with sparing of the interphalangeal spaces.20
- Periungual erythema and telangiectasias: redness and dilation of the blood vessels in the skin surrounding the nail plate.
- The shawl and V-signs: erythematous macular eruptions occurring in a “shawl” pattern on the shoulders, arms, and upper back and in a V-shaped pattern on the anterior neck and chest.
- Mechanic’s hand: extensive scaling, fissuring, and roughening of the palmar aspect of the hand.20,21
- Poikiloderma vasculare atrophicans: circumscribed violaceous erythema with thinning of the skin, prominent telangiectasias, and a mottled pattern of hypo- and hyperpigmentation, typically occurring on the anterior neck, chest, posterior shoulders, back, and buttocks, years after onset of the disease.21-24
- Cutaneous calcification (calcinosis cutis): usually on the buttocks, elbows, knees, and traumatized areas, affecting 30% to 70% of children with dermatomyositis, but only 10% of adults with the disorder.20,25-27
FIGURE 2
Gottron’s papules in dermatomyositis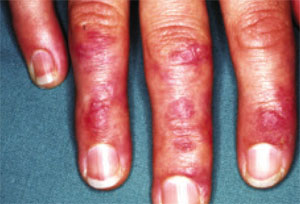
Violaceous papules on the dorsal interphalangeal and metacarpophalangeal joints of the hands are a common manifestation of dermatomyositis.
Bohan and Peter criteria help with dermatomyositis diagnosis
A diagnosis of dermatomyositis can be established using the following criteria developed by Bohan and Peter in 1975: (1) symmetric muscle weakness; (2) elevation of muscle enzymes, most notably creatinine phosphokinase; (3) evidence of inflammation on muscle biopsy; (4) electromyographic features of myositis; and (5) characteristic dermatologic signs, including a heliotrope rash and Gottron’s papules.28,29 A definitive diagnosis can be made when the patient meets 3 of the first 4 criteria, as well as the fifth.
Corticosteroids are the mainstay of treatment for dermatomyositis, although the dose and duration are subject to debate. Cutaneous manifestations of dermatomyositis are commonly treated with topical corticosteroids and oral hydroxychloroquine, as well as emollients and antipruritic agents.
Cancer screening. All adults with signs and symptoms of dermatomyositis should undergo cancer screening. The most common malignancies are ovarian cancer, gastric cancer, and lymphoma.21
Photoprotection. As with cutaneous LE, UV exposure can exacerbate dermatomyositis, and patients should use a broad-spectrum sunscreen.
Scleroderma: Localized and systemic disease
The major cutaneous manifestation of scleroderma is that of thickened, leathery, bound-down skin, seen in both localized and systemic disease. In both cases, the lesions typically evolve through 3 characteristic stages: the initial inflammatory and edematous stage; a fibrotic stage, during which the skin lesions appear hard, tight, and hidebound; and the final, atrophic stage. However, not all lesions progress to this final stage.8
Localized scleroderma (also known as morphea) affects roughly 1 in 100,000 people. It is more prevalent in females than males, with a ratio as high as 3 to 1.30-33 Cutaneous findings vary, and numerous clinical presentations are possible. The most widely used classification system, the Mayo Clinic Classification, recognizes 5 subtypes: plaque morphea (the most common), generalized morphea, linear morphea, deep morphea, and bullous morphea (TABLE 2).34 The mean age of onset depends on the subtype, and ranges from 12 years for linear morphea to 45 years for deep morphea.35
While systemic involvement is not common with localized scleroderma, extracutaneous manifestations have been reported in up to 25% of cases.31,36,37 In a 2005 multicenter study of 750 pediatric patients with localized scleroderma, the most frequently reported extracutaneous symptom was arthritis, found in 12% of patients.36 Less frequent findings included neurologic symptoms such as seizures and headaches, vascular changes such as deep vein thrombosis, and gastrointestinal, cardiac, and renal conditions. Although the precise etiology of localized scleroderma is unknown, it is thought to be associated with trauma, prior infection by Borrelia burgdorferi, chronic venous insufficiency, and irradiation for breast cancer.30,38,39
TABLE 2
Localized scleroderma: What to look for8,34
|
Systemic scleroderma is a heterogenous disorder
Commonly called systemic sclerosis (SSc), systemic scleroderma is characterized by proliferative vascular lesions; fibrosis of internal organs, including the lungs, heart, kidneys, and gastrointestinal tract; and distinct cutaneous manifestations.3 SSc affects about 240 people per million adults, mostly between the ages of 30 and 50 years, with women affected 3 times as often as men.40-42
The cause of SSc is unknown, although a genetic predisposition is likely. Environmental factors likely play a role in triggering the disease, with possible causative factors including cytomegalovirus and other viral infections and exposure to certain chemicals.43
As in localized scleroderma, cutaneous manifestations are a prominent part of SSc and typically develop early on. Raynaud’s phenomenon (RP) (FIGURE 3), which occurs in 90% to 98% of patients with SSc,44 can develop in association with other cutaneous findings or precede additional skin changes by months or years.
Other early skin changes include nonpitting edema of the fingers and toes, which creates a sausage-like appearance. This is typically followed by hardening and thickening of the skin in these areas, which can result in highly disabling sclerodactyly (FIGURE 4).45 Patients can also develop painful ulcerations on their fingertips and knuckles (rat bite necroses) due to local ischemia and vascular insufficiency. This may be complicated by secondary bacterial infection, gangrene, and acroosteolysis, leading to articular deformities and dissolution of terminal phalanges (FIGURE 5).45-48
On the face, SSc is characterized early on by periorbital edema and later by the development of a beaked nose, a reduction in the size of the mouth (microstomia) with radial furrowing, thinning of the lips, and telangiectasias.45,49 As the sclerosis worsens, patients are frequently left with expressionless, mask-like faces.48
Other common cutaneous manifestations of SSc include a “salt and pepper” appearance, with alternating areas of hypo- and hyperpigmented skin. Additionally, patients may suffer from a loss of hair follicles, severely dry skin, and pruritus.45,48
There are 2 major subsets of SSc—limited cutaneous SSc and diffuse cutaneous SSc. While they can be differentiated based on the history of symptoms, the appearance and extent of cutaneous involvement, and certain serological makers, the most important difference is the speed at which the disease progresses and its severity: Limited cutaneous SSc typically progresses slowly, while diffuse cutaneous SSc is characterized by a relatively rapid onset of disease, with skin and internal organ involvement likely to be severe.49
Scleroderma diagnosis and treatment: Vascular changes are an important clue
Skin changes seen in localized scleroderma and SSc can be clinically and histologically similar, making it difficult to arrive at a definitive diagnosis.
One clinical clue is that in localized scleroderma, vascular changes, such as RP and periungual nailfold telangiectasia, are typically absent. In addition, cutaneous changes in the hands and fingers, such as sclerodactyly, are more characteristic of systemic disease.10
SSc can be diagnosed using criteria proposed by the ACR, which have been shown to be highly sensitive (97%) and specific (98%).50 The major criterion is proximal scleroderma—symmetric thickening, tightening, and induration of the skin of the fingers and areas proximal to the metacarpophalangeal or metatarsophalangeal joints, which may include the trunk, neck, and face. Minor criteria include (1) sclerodactyly, (2) digital pitting scars of fingertips or loss of substance of the distal finger pad, and (3) bilateral basilar pulmonary fibrosis. Diagnosis is based on the presence of either the major criterion or 2 of the 3 minor criteria.50
Numerous therapies are available for localized scleroderma, including topical, intralesional, and systemic corticosteroids, topical tacrolimus, hydroxychloroquine, topical and systemic calcipotriol, penicillamine, sulfasalazine, interferon-[H9253], methotrexate, phototherapy with UV light, and imiquimod.
SSc skin manifestations, which can be severe and disabling, can be treated with a wide range of therapies. Topical or systemic corticosteroids; topical calcineurin inhibitors; systemic immunosuppressive agents such as methotrexate, cyclophosphamide, cyclosporine, and D-penicillamine; and phototherapy have all had varying success at reducing hardening of the skin.51-54 Other skin manifestations, including RP, dryness and itching, pigment changes, digital ulcerations, calcifications, and telangiectasias, should be managed as needed, with various supplemental treatment options available.45 These may include emollients, antihistamines, and topical corticosteroids for dryness and itching; laser therapy for telangiectasias; and corticosteroid injection, laser therapy, or surgery for calcifications. However, there is limited evidence of efficacy for most of these options.45
FIGURE 3
An attack of Raynaud’s phenomenon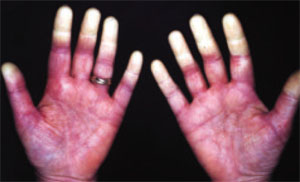
Raynaud’s phenomenon, characterized by blanching of the distal fingertips, is shown here in a patient with systemic sclerosis.
FIGURE 4
Sclerodactyly in a patient with systemic sclerosis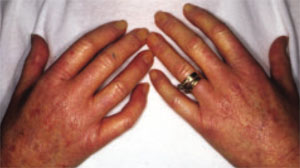
Hardening and thickening of the skin can result in highly disabling sclerodactyly in patients with systemic sclerosis.
FIGURE 5
Dissolution of terminal phalanges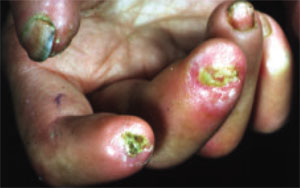
Bony resorption and ulceration have led to the loss of distal phalanges in this patient with systemic sclerosis.
Mixed connective tissue disease has features of several disorders
Mixed connective tissue disease (MCTD) is an apparently distinct rheumatologic condition characterized by a combination of clinical features of systemic LE, dermatomyositis, scleroderma, polymyositis, and rheumatoid arthritis. The presence of high titers of a unique autoantibody, anti-U1-RNP,54-57 aids in diagnosis.
Although precise prevalence data are lacking, MCTD is thought to occur in about 1 in 10,000 people.58 The disease is much more common in women, with a female-to-male ratio as high as 7 to 1, and generally occurs in the second or third decade of life.58-60
The clinical manifestations of MCTD typically evolve, with overlapping features of various autoimmune disorders appearing sequentially over several months to years.57 Early in the course of MCTD, patients commonly experience fatigue, polyarthritis, hand edema, and RP. In time, virtually every organ system may be involved. Pulmonary hypertension is a major cause of death.59,61
Unlike many other connective tissue disorders, MCTD lacks any distinct cutaneous findings. In addition to those mentioned earlier, prominent skin findings include swelling of the fingers, sclerodactyly, and the acute malar eruptions and discoid plaques typically associated with LE.56,57,62 Cutaneous manifestations associated with dermatomyositis and scleroderma may also be seen, particularly juxta-articular calcinosis. The mucous membranes may be involved, as well, resulting in nasal perforation, buccal and urogenital ulcerations, and sicca complex.58,63,64
MCTD diagnosis and treatment: Look for these serology and clinical findings
Diagnosing MCTD can be clinically challenging, as signs and symptoms of the disease commonly evolve over time. The Alarcon-Segovia criteria—largely regarded as the best diagnostic tool for MCTD65,66—include 1 serologic finding (elevated anti-U1-RNP [titer ≥1:1600]) and 5 clinical findings (RP, edema of the hands, synovitis, myositis, and acrosclerosis). Diagnosis requires the presence of the serologic criterion and ≥3 of the 5 clinical criteria.
Treatment of the cutaneous manifestations should be based on the effectiveness of therapies for similar skin findings seen in other disorders. In treating MCTD (or any other connective tissue disorder), a team that includes nurses, physical and occupational therapists, primary care physicians, and specialists in dermatology and rheumatology is essential for an optimal outcome.
CORRESPONDENCE Christian R. Halvorson, MD, Mercy Medical Center, Department of Medicine, 301 St. Paul Place, Baltimore, MD 21202; crhalvor@gmail.com
1. Renner R, Sticherling M. The different faces of cutaneous lupus erythematosus. G Ital Dermatol Venereol. 2009;144:135-147.
2. Kuhn A, Ruzicka T. Classification of cutaneous lupus erythematosus. In: Kuhn A, Lehmann P, Ruzicka T, eds. Cutaneous Lupus Erythematosus. New York: Springer; 2004:53–57.
3. Costner MI, Grau RH. Update on connective tissue diseases in dermatology. Semin Cutan Med Surg. 2006;25:207-220.
4. Ting W, Stone MS, Racila D, et al. Toxic epidermal necrolysis-like acute cutaneous lupus erythematosus and the spectrum of the acute syndrome of apoptotic pan-epidermolysis (ASAP): a case report, concept review and proposal for new classification of lupus erythematosus vesiculobullous skin lesions. Lupus. 2004;13:941-950.
5. Jorizzo JL, Carroll CL, Sangueza OP. Lupus erythematosus. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. Spain: Elsevier Limited; 2008.
6. Kuhn A, Sticherling M, Bonsmann G. Clinical manifestations of cutaneous lupus erythematosus. J Dtsch Dermatol Ges. 2007;5:1124-1137.
7. Sontheimer RD, Thomas JR, Gilliam JN. Subacute cutaneous lupus erythematosus: a cutaneous marker for a distinct lupus erythematosus subset. Arch Dermatol. 1979;115:1409-1415.
8. Sontheimer RD. Skin manifestations of systemic autoimmune connective tissue disease: diagnostics and therapeutics. Best Pract Res Clin Rheumatol. 2004;18:429-462.
9. Panjwani S. Early diagnosis and treatment of discoid lupus erythematosus. J Am Board Fam Med. 2009;22:206-213.
10. Sontheimer RD, Provost TT. Lupus erythematosus. In: Sontheimer RD, Provost TT, eds. Cutaneous Manifestations of Rheumatic Diseases. Baltimore: Williams & Wilkins; 1996.
11. Mond CB, Peterson MG, Rothfield NF. Correlation of antiro antibody with photosensitivity rash in systemic lupus erythematosus patients. Arthritis Rheum. 1989;32:202-204.
12. Jacyk WK, Damisah M. Discoid lupus erythematosus in the Nigerians. Br J Dermatol. 1979;100:131-135.
13. Callen JP. Cutaneous lupus erythematosus: A personal approach to management. Australas J Dermatol. 2006;47:13-27.
14. Gilliam JN, Sontheimer RD. Distinctive cutaneous subsets in the spectrum of lupus erythematosus. J Am Acad Dermatol. 1981;4:471-475.
15. Farley-Loftus R, Mahlberg M, Merola J, et al. Generalized discoid lupus erythematosus. Dermatol Online J. 2009;15:18.-
16. Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus—a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
17. American College of Rheumatology. The 1982 revised criteria for classification of systemic lupus erythematosus. Available at: http://www.rheumatology.org/practice/clinical/classification/SLE/sle.asp. Accessed September 21, 2010.
18. Na SJ, Kim SM, Sunwoo IN, et al. Clinical characteristics and outcomes of juvenile and adult dermatomyositis. J Korean Med Sci. 2009;24:715-721.
19. Kovacs SO, Kovacs SC. Dermatomyositis. J Am Acad Dermatol. 1998;39:899-922.
20. Koler RA, Montemarano A. Dermatomyositis. Am Fam Physician. 2001;64:1565-1572.
21. Sunkureddi P, Nguyen-Oghalai TU, Jarvis JL, et al. Signs of dermatomyositis. Hosp Physician. 2005;41:41-44.
22. Iorizzo LJ, 3rd, Jorizzo JL. The treatment and prognosis of dermatomyositis: an updated review. J Am Acad Dermatol. 2008;59:99-112.
23. Jorizzo JL, Carroll CL, Sangueza OP. Dermatomyositis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. Spain: Elsevier Limited; 2008:575–583.
24. Sontheimer RD. Dermatomyositis. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 5th ed. New York: McGraw-Hill; 1999:2009–2020.
25. Halbert AR. Juvenile dermatomyositis. Australas J Dermatol. 1996;37:106-108.
26. Orlow SJ, Watsky KL, Bolognia JL. Skin and bones II. J Am Acad Dermatol. 1991;25:447-462.
27. Bowyer SL, Blane CE, Sullivan DB, et al. Childhood dermatomyositis: factors predicting functional outcome and development of dystrophic calcification. J Pediatr. 1983;103:882-888.
28. Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. 1975;292:403-407.
29. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344-347.
30. Vancheeswaran R, Black CM, David J, et al. Childhood-onset scleroderma: is it different from adult-onset disease? Arthritis Rheum. 1996;39:1041-1049.
31. Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: Clinical and epidemiological features in 750 children. An international study. Rheumatology (Oxford). 2006;45:614-620.
32. Marzano AV, Menni S, Parodi A, et al. Localized scleroderma in adults and children. clinical and laboratory investigations on 239 cases. Eur J Dermatol. 2003;13:171-176.
33. Uziel Y, Krafchik BR, Silverman ED, et al. Localized scleroderma in childhood: a report of 30 cases. Semin Arthritis Rheum. 1994;23:328-340.
34. Peterson LS, Nelson AM, Su WP. Classification of morphea (localized scleroderma). Mayo Clin Proc. 1995;70:1068-1076.
35. Peterson LS, Nelson AM, Su WP, et al. The epidemiology of morphea (localized scleroderma) in Olmsted County 1960-1993. J Rheumatol. 1997;24:73-80.
36. Zulian F, Vallongo C, Woo P, et al. Localized scleroderma in childhood is not just a skin disease. Arthritis Rheum. 2005;52:2873-2881.
37. Dehen L, Roujeau JC, Cosnes A, et al. Internal involvement in localized scleroderma. Medicine (Baltimore). 1994;73:241-245.
38. Ludwig RJ, Werner RJ, Winker W, et al. Chronic venous insufficiency - a potential trigger for localized scleroderma. J Eur Acad Dermatol Venereol. 2006;20:96-99.
39. Aberer E, Neumann R, Stanek G. Is localised scleroderma a borrelia infection? Lancet. 1985;2:278.-
40. Krishnan E, Furst DE. Systemic sclerosis mortality in the United States: 1979-1998. Eur J Epidemiol. 2005;20:855-861.
41. Silman AJ. Scleroderma—demographics and survival. J Rheumatol Suppl. 1997;48:58-61.
42. Medsger TA,, Jr, Masi AT. Epidemiology of systemic sclerosis (scleroderma). Ann Intern Med. 1971;74:714-721.
43. Charles C, Clements P, Furst DE. Systemic sclerosis: Hypothesis-driven treatment strategies. Lancet. 2006;367:1683-1691.
44. Maricq HR, Harper FE, Khan MM, et al. Microvascular abnormalities as possible predictors of disease subsets in Raynaud phenomenon and early connective tissue disease. Clin Exp Rheumatol. 1983;1:195-205.
45. Krieg T, Takehara K. Skin disease: A cardinal feature of systemic sclerosis. Rheumatology (Oxford). 2009;48 (suppl 3):S14-S18.
46. Ingraham KM, Steen VD. Morbidity of digital tip ulcerations in scleroderma. Arthritis Rheum. 2006;54(9 suppl.):F78.-
47. Ferri C, Valentini G, Cozzi F, et al. Systemic sclerosis: Demographic, clinical, and serologic features and survival in 1,012 italian patients. Medicine (Baltimore). 2002;81:139-153.
48. Haustein UF. Systemic sclerosis - scleroderma. Dermatol Online J. 2002;8.-
49. Fitzpatrick TB, Johnson RA, Klaus W, et al. In Colour Atlas and Synopsis of Clinical Dermatology. 4th ed. New York: McGraw-Hill; 2001:368–369.
50. Preliminary criteria for the classification of systemic sclerosis (scleroderma). subcommittee for scleroderma criteria of the American Rheumatism Association diagnostic and therapeutic criteria committee. Arthritis Rheum. 1980;23:581-590.
51. Valentini G, Paone C, La Montagna G, et al. Low-dose intravenous cyclophosphamide in systemic sclerosis: an open prospective efficacy study in patients with early diffuse disease. Scand J Rheumatol. 2006;35:35-38.
52. Steen VD, Medsger TA, Jr. Improvement in skin thickening in systemic sclerosis associated with improved survival. Arthritis Rheum. 2001;44:2828-2835.
53. Basso M, Filaci G, Cutolo M, et al. Long-term treatment of patients affected by systemic sclerosis with cyclosporin A. Ann Ital Med Int. 2001;16:233-239.
54. Morton SJ, Powell RJ. Cyclosporin and tacrolimus: their use in a routine clinical setting for scleroderma. Rheumatology (Oxford). 2000;39:865-869.
55. Kasukawa R. Mixed connective tissue disease. Intern Med. 1999;38:386-393.
56. Bennett RM, O’Connell DJ. Mixed connective tisssue disease: A clinicopathologic study of 20 cases. Semin Arthritis Rheum. 1980;10:25-51.
57. Sharp GC, Irvin WS, Tan EM, et al. Mixed connective tissue disease—an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA). Am J Med. 1972;52:148-159.
58. Venables PJ. Mixed connective tissue disease. Lupus. 2006;15:132-137.
59. Farhey Y, Hess EV. Mixed connective tissue disease. Arthritis Care Res. 1997;10:333-342.
60. Alarcon-Segovia D. Mixed connective tissue disease - a decade of growing pains. J Rheumatol. 1981;8:535-540.
61. Ueda N, Mimura K, Maeda H, et al. Mixed connective tissue disease with fatal pulmonary hypertension and a review of literature. Virchows Arch A Pathol Anat Histopathol. 1984;404:335-340.
62. Bennett RM. Mixed connective tissue disease and other overlap syndromes. In: Kelley WN, Harris EDJ, Ruddy SH, et al, eds. Textbook of Rheumatology. 4th ed. Philadelphia: WB Saunders; 1993:1061.
63. Nakashima M, Suzuki K, Okada M, et al. Panniculitis in a patient with mixed connective tissue disease. Mod Rheumatol. 2004;14:250-253.
64. Magro CM, Crowson AN, Regauer S. Mixed connective tissue disease. A clinical, histologic, and immunofluorescence study of eight cases. Am J Dermatopathol. 1997;19:206-213.
65. Amigues JM, Cantagrel A, Abbal M, et al. Comparative study of 4 diagnosis criteria sets for mixed connective tissue disease in patients with anti-RNP antibodies. autoimmunity group of the hospitals of toulouse. J Rheumatol. 1996;23:2055-2062.
66. Alarcon-Segovia D, Cardiel MH. Comparison between 3 diagnostic criteria for mixed connective tissue disease. study of 593 patients. J Rheumatol. 1989;16:328-334.
• When evaluating patients with suspected cutaneous lupus erythematosus, use multiple criteria—including histologic and immunofluorescent biopsy findings and American College of Rheumatology criteria—to rule out systemic disease. C
• Cancer screening with a careful history and physical examination is recommended for all adult patients whom you suspect of having dermatomyositis. C
• Suspect mixed connective tissue disease in patients with skin findings characteristic of varying auto-immune disorders appearing sequentially over several months or years. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Many systemic conditions are accompanied by skin manifestations. This is especially true for connective tissue disorders, for which dermatologic findings are often the key to diagnosis.
In this review, we describe the dermatologic findings of some well-known connective tissue disorders. The text and photographs in the pages that follow will help you hone your diagnostic skills, leading to earlier treatment and, possibly, better outcomes.
Lupus erythematosus: Cutaneous and systemic disease often overlap
Lupus erythematosus (LE), a chronic, inflammatory autoimmune condition that primarily affects women in their 20s and 30s, may initially present as a systemic disease or in a purely cutaneous form. However, most patients with systemic LE have some skin manifestations, and those with cutaneous LE often have—or subsequently develop—systemic involvement.1 Thus, recognizing the cutaneous manifestations of LE will not only aid in diagnosis, but will help you identify patients at risk for systemic disease.
Cutaneous LE has 4 subtypes
There are 4 subcategories of cutaneous LE—acute, subacute, chronic, and intermittent.2 Each is differentiated by the appearance of the lesions (TABLE 1), histology, and serological markers.1 Photosensitivity is common to all the subcategories to varying degrees.
Acute cutaneous lupus erythematosus (ACLE) is typically characterized by the classic malar “butterfly” rash, an erythematous eruption of macules or edematous papules over the bridge of the nose and cheek.3 Although this presentation is most common, there are variations—1 in which the lesions cover other exposed areas (commonly including the “V” of the chest, the extensor surface of the arms, and the hands), and a rare form in which toxic epidermal necrolysis-like blistering occurs.1,4 These skin changes—which generally last anywhere from a few hours to several weeks—typically resolve without scarring, although pigment changes can occur.5
Left: Dermatomyositis is the underlying cause of the heliotrope discoloration on this patient’s upper eyelid.
Center: Linear morphea is associated with the lesion on this patient’s face—called en coup de sabre because it resembles the mark caused by the stroke of a sword in a duel.
Right: Discoid lupus erythematosus causes hypopigmentation and scarring.Patients with ACLE have a predisposition to systemic LE; unlike those with other forms of cutaneous LE, 40% to 90% will have double-stranded DNA (dsDNA) autoantibodies.3,6
Subacute cutaneous lupus erythematosus (SCLE), which usually affects middle-aged Caucasian women, is characterized by erythematous papulosquamous (psoriasis-like) eruptions or annular lesions with raised red borders and central clearing—or both. These lesions, which are nonscarring, lack in-duration, and rarely affect the scalp or face, appear suddenly, usually after exposure to sunlight (FIGURE 1)5,7-9 or certain drugs. Hydrochlorothiazide, terbinafine, calcium channel blockers, and angiotensin-converting enzyme inhibitors are common offenders.1,6
FIGURE 1
Annular lesions in subacute cutaneous lupus erythematosus
Ring-like lesions with raised red borders and central clearing on the back of a patient with subacute cutaneous lupus erythematosus.
SCLE is often associated with extracutaneous symptoms such as arthritis and myalgias,1,8 but patients are at relatively low risk for severe systemic manifestations.5,8,10 Serology is often notable, with anti-Ro (SS-A) antibodies present in 70% to 90% of patients and anti-La (SS-B) autoantibodies found in 30% to 50%.1,11
Chronic cutaneous lupus erythematosus (CCLE) also occurs predominantly in females, at a ratio as high as 5 to 1.12 There are 3 variations of CCLE: discoid lupus erythematosus (DLE), LE profundus, and chilblain LE (TABLE 1). DLE, characterized by alopecia, skin atrophy, and dyspigmentation, is the most common and affects patients of all ages and ethnic groups.9,13,14 (See image above.)
DLE lesions typically begin as erythematous papules and plaques with scale. As the disorder progresses, the lesions spread, causing follicular plugging, peripheral hyperpigmentation and central hypopigmentation, telangiectasia, and atrophy.9,15 In some cases, patients develop thickened, scarred skin and permanent scarring alopecia.15 Prompt recognition of DLE is particularly important, as early referral and treatment may reduce the likelihood of permanent scarring alopecia and pigment changes.11
Intermittent cutaneous lupus erythematosus (ICLE), a relatively new subtype of cutaneous LE, is represented by a rare condition—lupus erythematosus tumidus (LET)—reported in <100 cases worldwide. LET is characterized by succulent, erythematous, and edematous plaques on sun-exposed parts of the body.1,16
TABLE 1
Cutaneous lupus erythematosus: Recognizing the subtypes1
| Acute cutaneous lupus erythematosus (ACLE) |
|---|
| Localized: erythematous macules or papules over the bridge of the nose and cheek, with sparing of the nasolabial folds (“butterfly rash”) |
| Generalized: similar erythematous lesions over other photodistributed parts of the body, including the neck, chest, arms, and hands |
| Toxic epidermal necrolysis-like (TEN-like): blistering and epidermal cleavage in photodistributed parts of the body |
| Subacute cutaneous lupus erythematosus (SCLE) |
| Erythematous papulosquamous (psoriasis-like) eruptions or annular (ring-like) lesions with raised red borders and central clearing, occurring symmetrically and suddenly after sunlight exposure on photo-distributed body parts; the scalp and face are rarely affected |
| Chronic cutaneous lupus erythematosus (CCLE) |
| Discoid lupus erythematosus: erythematous papules and plaques with associated scale, spreading centrifugally with follicular plugging, pigment change, telangiectasia, and atrophy; scarring alopecia can occur |
| Lupus erythematosus profundus: tender, erythematous nodules and plaques, usually involving the proximal extremities, trunk, breasts, buttocks, and face |
| Chilblain lupus erythematosus: tender, erythematous nodules and plaques, occurring in acral areas often in response to cold |
| Intermittent cutaneous lupus erythematosus (ICLE) |
| Lupus erythematosus tumidus: succulent, erythematous, and edematous plaques found on photodistributed parts of the body |
Cutaneous LE diagnosis and treatment: Start with ACR criteria
When evaluating patients with suspected cutaneous LE, it is important not only to identify the subtype, but also to rule out systemic disease using criteria established by the American College of Rheumatology (ACR).17 Notably, 4 of the 11 diagnostic criteria for systemic disease involve visual clues, including malar rash, discoid rash, photosensitivity, and oral ulcerations. Laboratory evidence of systemic disease may include a positive antinuclear antibody, anti-dsDNA, or anti-Sm autoantibody test results, as well as hematologic abnormalities described in the ACR guidelines.
Ultimately, a diagnosis of cutaneous LE should be based on the patient’s history and physical exam, autoantibody profile, and histologic and immunofluorescent biopsy findings. A rheumatologic evaluation may help to determine which patients have systemic disease, as the ACR criteria may overdiagnose systemic LE in those with predominantly skin changes.6
Treatment of cutaneous LE is based on the subtype and extent of disease, with potent topical corticosteroids, in combination with antimalarial agents, being the primary therapies. ACLE skin lesions generally respond best to systemic corticosteroids and immunosuppressive agents (such as azathioprine or cyclophosphamide) that are used to control underlying systemic disease. SCLE can be managed with topical corticosteroids; however, patients typically also require systemic treatment, often with hydroxychloroquine, for optimal control. DLE, the most common form of CCLE, is managed in a similar fashion, with a greater role for intralesional corticosteroid injections. Lesions associated with ICLE, which often resolve spontaneously, may be treated with topical corticosteroids and antimalarials.16 It is also important to advise all patients with cutaneous LE to use a broad-spectrum sunscreen, as ultraviolet (UV) exposure can induce or exacerbate the lesions.9
Dermatomyositis: Rare but serious
Dermatomyositis is an idiopathic inflammatory myopathy characterized by chronic muscle inflammation, symmetric proximal muscle weakness, and distinct cutaneous findings. In addition to possible cardiac and pulmonary complications, dysphagia, and joint contractures, dermatomyositis is associated with cancer, with up to 25% of affected adults having an underlying occult malignancy.18
Dermatomyositis is a rare condition, with a prevalence of only 1 to 10 cases per million adults and 3.2 cases per million children.19 It has a bimodal age distribution, with most juvenile cases affecting children between the ages of 5 and 14 years and most adult cases developing in the fifth and sixth decades of life. Women are affected twice as often as men.18
Suspect dermatomyositis when you see any of the following signs:
- A heliotrope rash: violaceous macules and patches, with or without edema, symmetrically on the periorbital skin, present early in the disease course in 30% to 60% of patients.19 (See image)
- Gottron’s papules: violaceous papules on the dorsal interphalangeal and metacarpophalangeal joints of the hands, elbows, and knees, occurring in as many as 70% of patients (FIGURE 2).19,20
- Gottron’s sign: nonscaling, violaceous erythematous macules and plaques occurring symmetrically in the same distribution as Gottron’s papules, but with sparing of the interphalangeal spaces.20
- Periungual erythema and telangiectasias: redness and dilation of the blood vessels in the skin surrounding the nail plate.
- The shawl and V-signs: erythematous macular eruptions occurring in a “shawl” pattern on the shoulders, arms, and upper back and in a V-shaped pattern on the anterior neck and chest.
- Mechanic’s hand: extensive scaling, fissuring, and roughening of the palmar aspect of the hand.20,21
- Poikiloderma vasculare atrophicans: circumscribed violaceous erythema with thinning of the skin, prominent telangiectasias, and a mottled pattern of hypo- and hyperpigmentation, typically occurring on the anterior neck, chest, posterior shoulders, back, and buttocks, years after onset of the disease.21-24
- Cutaneous calcification (calcinosis cutis): usually on the buttocks, elbows, knees, and traumatized areas, affecting 30% to 70% of children with dermatomyositis, but only 10% of adults with the disorder.20,25-27
FIGURE 2
Gottron’s papules in dermatomyositis
Violaceous papules on the dorsal interphalangeal and metacarpophalangeal joints of the hands are a common manifestation of dermatomyositis.
Bohan and Peter criteria help with dermatomyositis diagnosis
A diagnosis of dermatomyositis can be established using the following criteria developed by Bohan and Peter in 1975: (1) symmetric muscle weakness; (2) elevation of muscle enzymes, most notably creatinine phosphokinase; (3) evidence of inflammation on muscle biopsy; (4) electromyographic features of myositis; and (5) characteristic dermatologic signs, including a heliotrope rash and Gottron’s papules.28,29 A definitive diagnosis can be made when the patient meets 3 of the first 4 criteria, as well as the fifth.
Corticosteroids are the mainstay of treatment for dermatomyositis, although the dose and duration are subject to debate. Cutaneous manifestations of dermatomyositis are commonly treated with topical corticosteroids and oral hydroxychloroquine, as well as emollients and antipruritic agents.
Cancer screening. All adults with signs and symptoms of dermatomyositis should undergo cancer screening. The most common malignancies are ovarian cancer, gastric cancer, and lymphoma.21
Photoprotection. As with cutaneous LE, UV exposure can exacerbate dermatomyositis, and patients should use a broad-spectrum sunscreen.
Scleroderma: Localized and systemic disease
The major cutaneous manifestation of scleroderma is that of thickened, leathery, bound-down skin, seen in both localized and systemic disease. In both cases, the lesions typically evolve through 3 characteristic stages: the initial inflammatory and edematous stage; a fibrotic stage, during which the skin lesions appear hard, tight, and hidebound; and the final, atrophic stage. However, not all lesions progress to this final stage.8
Localized scleroderma (also known as morphea) affects roughly 1 in 100,000 people. It is more prevalent in females than males, with a ratio as high as 3 to 1.30-33 Cutaneous findings vary, and numerous clinical presentations are possible. The most widely used classification system, the Mayo Clinic Classification, recognizes 5 subtypes: plaque morphea (the most common), generalized morphea, linear morphea, deep morphea, and bullous morphea (TABLE 2).34 The mean age of onset depends on the subtype, and ranges from 12 years for linear morphea to 45 years for deep morphea.35
While systemic involvement is not common with localized scleroderma, extracutaneous manifestations have been reported in up to 25% of cases.31,36,37 In a 2005 multicenter study of 750 pediatric patients with localized scleroderma, the most frequently reported extracutaneous symptom was arthritis, found in 12% of patients.36 Less frequent findings included neurologic symptoms such as seizures and headaches, vascular changes such as deep vein thrombosis, and gastrointestinal, cardiac, and renal conditions. Although the precise etiology of localized scleroderma is unknown, it is thought to be associated with trauma, prior infection by Borrelia burgdorferi, chronic venous insufficiency, and irradiation for breast cancer.30,38,39
TABLE 2
Localized scleroderma: What to look for8,34
|
Systemic scleroderma is a heterogenous disorder
Commonly called systemic sclerosis (SSc), systemic scleroderma is characterized by proliferative vascular lesions; fibrosis of internal organs, including the lungs, heart, kidneys, and gastrointestinal tract; and distinct cutaneous manifestations.3 SSc affects about 240 people per million adults, mostly between the ages of 30 and 50 years, with women affected 3 times as often as men.40-42
The cause of SSc is unknown, although a genetic predisposition is likely. Environmental factors likely play a role in triggering the disease, with possible causative factors including cytomegalovirus and other viral infections and exposure to certain chemicals.43
As in localized scleroderma, cutaneous manifestations are a prominent part of SSc and typically develop early on. Raynaud’s phenomenon (RP) (FIGURE 3), which occurs in 90% to 98% of patients with SSc,44 can develop in association with other cutaneous findings or precede additional skin changes by months or years.
Other early skin changes include nonpitting edema of the fingers and toes, which creates a sausage-like appearance. This is typically followed by hardening and thickening of the skin in these areas, which can result in highly disabling sclerodactyly (FIGURE 4).45 Patients can also develop painful ulcerations on their fingertips and knuckles (rat bite necroses) due to local ischemia and vascular insufficiency. This may be complicated by secondary bacterial infection, gangrene, and acroosteolysis, leading to articular deformities and dissolution of terminal phalanges (FIGURE 5).45-48
On the face, SSc is characterized early on by periorbital edema and later by the development of a beaked nose, a reduction in the size of the mouth (microstomia) with radial furrowing, thinning of the lips, and telangiectasias.45,49 As the sclerosis worsens, patients are frequently left with expressionless, mask-like faces.48
Other common cutaneous manifestations of SSc include a “salt and pepper” appearance, with alternating areas of hypo- and hyperpigmented skin. Additionally, patients may suffer from a loss of hair follicles, severely dry skin, and pruritus.45,48
There are 2 major subsets of SSc—limited cutaneous SSc and diffuse cutaneous SSc. While they can be differentiated based on the history of symptoms, the appearance and extent of cutaneous involvement, and certain serological makers, the most important difference is the speed at which the disease progresses and its severity: Limited cutaneous SSc typically progresses slowly, while diffuse cutaneous SSc is characterized by a relatively rapid onset of disease, with skin and internal organ involvement likely to be severe.49
Scleroderma diagnosis and treatment: Vascular changes are an important clue
Skin changes seen in localized scleroderma and SSc can be clinically and histologically similar, making it difficult to arrive at a definitive diagnosis.
One clinical clue is that in localized scleroderma, vascular changes, such as RP and periungual nailfold telangiectasia, are typically absent. In addition, cutaneous changes in the hands and fingers, such as sclerodactyly, are more characteristic of systemic disease.10
SSc can be diagnosed using criteria proposed by the ACR, which have been shown to be highly sensitive (97%) and specific (98%).50 The major criterion is proximal scleroderma—symmetric thickening, tightening, and induration of the skin of the fingers and areas proximal to the metacarpophalangeal or metatarsophalangeal joints, which may include the trunk, neck, and face. Minor criteria include (1) sclerodactyly, (2) digital pitting scars of fingertips or loss of substance of the distal finger pad, and (3) bilateral basilar pulmonary fibrosis. Diagnosis is based on the presence of either the major criterion or 2 of the 3 minor criteria.50
Numerous therapies are available for localized scleroderma, including topical, intralesional, and systemic corticosteroids, topical tacrolimus, hydroxychloroquine, topical and systemic calcipotriol, penicillamine, sulfasalazine, interferon-[H9253], methotrexate, phototherapy with UV light, and imiquimod.
SSc skin manifestations, which can be severe and disabling, can be treated with a wide range of therapies. Topical or systemic corticosteroids; topical calcineurin inhibitors; systemic immunosuppressive agents such as methotrexate, cyclophosphamide, cyclosporine, and D-penicillamine; and phototherapy have all had varying success at reducing hardening of the skin.51-54 Other skin manifestations, including RP, dryness and itching, pigment changes, digital ulcerations, calcifications, and telangiectasias, should be managed as needed, with various supplemental treatment options available.45 These may include emollients, antihistamines, and topical corticosteroids for dryness and itching; laser therapy for telangiectasias; and corticosteroid injection, laser therapy, or surgery for calcifications. However, there is limited evidence of efficacy for most of these options.45
FIGURE 3
An attack of Raynaud’s phenomenon
Raynaud’s phenomenon, characterized by blanching of the distal fingertips, is shown here in a patient with systemic sclerosis.
FIGURE 4
Sclerodactyly in a patient with systemic sclerosis
Hardening and thickening of the skin can result in highly disabling sclerodactyly in patients with systemic sclerosis.
FIGURE 5
Dissolution of terminal phalanges
Bony resorption and ulceration have led to the loss of distal phalanges in this patient with systemic sclerosis.
Mixed connective tissue disease has features of several disorders
Mixed connective tissue disease (MCTD) is an apparently distinct rheumatologic condition characterized by a combination of clinical features of systemic LE, dermatomyositis, scleroderma, polymyositis, and rheumatoid arthritis. The presence of high titers of a unique autoantibody, anti-U1-RNP,54-57 aids in diagnosis.
Although precise prevalence data are lacking, MCTD is thought to occur in about 1 in 10,000 people.58 The disease is much more common in women, with a female-to-male ratio as high as 7 to 1, and generally occurs in the second or third decade of life.58-60
The clinical manifestations of MCTD typically evolve, with overlapping features of various autoimmune disorders appearing sequentially over several months to years.57 Early in the course of MCTD, patients commonly experience fatigue, polyarthritis, hand edema, and RP. In time, virtually every organ system may be involved. Pulmonary hypertension is a major cause of death.59,61
Unlike many other connective tissue disorders, MCTD lacks any distinct cutaneous findings. In addition to those mentioned earlier, prominent skin findings include swelling of the fingers, sclerodactyly, and the acute malar eruptions and discoid plaques typically associated with LE.56,57,62 Cutaneous manifestations associated with dermatomyositis and scleroderma may also be seen, particularly juxta-articular calcinosis. The mucous membranes may be involved, as well, resulting in nasal perforation, buccal and urogenital ulcerations, and sicca complex.58,63,64
MCTD diagnosis and treatment: Look for these serology and clinical findings
Diagnosing MCTD can be clinically challenging, as signs and symptoms of the disease commonly evolve over time. The Alarcon-Segovia criteria—largely regarded as the best diagnostic tool for MCTD65,66—include 1 serologic finding (elevated anti-U1-RNP [titer ≥1:1600]) and 5 clinical findings (RP, edema of the hands, synovitis, myositis, and acrosclerosis). Diagnosis requires the presence of the serologic criterion and ≥3 of the 5 clinical criteria.
Treatment of the cutaneous manifestations should be based on the effectiveness of therapies for similar skin findings seen in other disorders. In treating MCTD (or any other connective tissue disorder), a team that includes nurses, physical and occupational therapists, primary care physicians, and specialists in dermatology and rheumatology is essential for an optimal outcome.
CORRESPONDENCE Christian R. Halvorson, MD, Mercy Medical Center, Department of Medicine, 301 St. Paul Place, Baltimore, MD 21202; crhalvor@gmail.com
• When evaluating patients with suspected cutaneous lupus erythematosus, use multiple criteria—including histologic and immunofluorescent biopsy findings and American College of Rheumatology criteria—to rule out systemic disease. C
• Cancer screening with a careful history and physical examination is recommended for all adult patients whom you suspect of having dermatomyositis. C
• Suspect mixed connective tissue disease in patients with skin findings characteristic of varying auto-immune disorders appearing sequentially over several months or years. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Many systemic conditions are accompanied by skin manifestations. This is especially true for connective tissue disorders, for which dermatologic findings are often the key to diagnosis.
In this review, we describe the dermatologic findings of some well-known connective tissue disorders. The text and photographs in the pages that follow will help you hone your diagnostic skills, leading to earlier treatment and, possibly, better outcomes.
Lupus erythematosus: Cutaneous and systemic disease often overlap
Lupus erythematosus (LE), a chronic, inflammatory autoimmune condition that primarily affects women in their 20s and 30s, may initially present as a systemic disease or in a purely cutaneous form. However, most patients with systemic LE have some skin manifestations, and those with cutaneous LE often have—or subsequently develop—systemic involvement.1 Thus, recognizing the cutaneous manifestations of LE will not only aid in diagnosis, but will help you identify patients at risk for systemic disease.
Cutaneous LE has 4 subtypes
There are 4 subcategories of cutaneous LE—acute, subacute, chronic, and intermittent.2 Each is differentiated by the appearance of the lesions (TABLE 1), histology, and serological markers.1 Photosensitivity is common to all the subcategories to varying degrees.
Acute cutaneous lupus erythematosus (ACLE) is typically characterized by the classic malar “butterfly” rash, an erythematous eruption of macules or edematous papules over the bridge of the nose and cheek.3 Although this presentation is most common, there are variations—1 in which the lesions cover other exposed areas (commonly including the “V” of the chest, the extensor surface of the arms, and the hands), and a rare form in which toxic epidermal necrolysis-like blistering occurs.1,4 These skin changes—which generally last anywhere from a few hours to several weeks—typically resolve without scarring, although pigment changes can occur.5
Left: Dermatomyositis is the underlying cause of the heliotrope discoloration on this patient’s upper eyelid.
Center: Linear morphea is associated with the lesion on this patient’s face—called en coup de sabre because it resembles the mark caused by the stroke of a sword in a duel.
Right: Discoid lupus erythematosus causes hypopigmentation and scarring.Patients with ACLE have a predisposition to systemic LE; unlike those with other forms of cutaneous LE, 40% to 90% will have double-stranded DNA (dsDNA) autoantibodies.3,6
Subacute cutaneous lupus erythematosus (SCLE), which usually affects middle-aged Caucasian women, is characterized by erythematous papulosquamous (psoriasis-like) eruptions or annular lesions with raised red borders and central clearing—or both. These lesions, which are nonscarring, lack in-duration, and rarely affect the scalp or face, appear suddenly, usually after exposure to sunlight (FIGURE 1)5,7-9 or certain drugs. Hydrochlorothiazide, terbinafine, calcium channel blockers, and angiotensin-converting enzyme inhibitors are common offenders.1,6
FIGURE 1
Annular lesions in subacute cutaneous lupus erythematosus
Ring-like lesions with raised red borders and central clearing on the back of a patient with subacute cutaneous lupus erythematosus.
SCLE is often associated with extracutaneous symptoms such as arthritis and myalgias,1,8 but patients are at relatively low risk for severe systemic manifestations.5,8,10 Serology is often notable, with anti-Ro (SS-A) antibodies present in 70% to 90% of patients and anti-La (SS-B) autoantibodies found in 30% to 50%.1,11
Chronic cutaneous lupus erythematosus (CCLE) also occurs predominantly in females, at a ratio as high as 5 to 1.12 There are 3 variations of CCLE: discoid lupus erythematosus (DLE), LE profundus, and chilblain LE (TABLE 1). DLE, characterized by alopecia, skin atrophy, and dyspigmentation, is the most common and affects patients of all ages and ethnic groups.9,13,14 (See image above.)
DLE lesions typically begin as erythematous papules and plaques with scale. As the disorder progresses, the lesions spread, causing follicular plugging, peripheral hyperpigmentation and central hypopigmentation, telangiectasia, and atrophy.9,15 In some cases, patients develop thickened, scarred skin and permanent scarring alopecia.15 Prompt recognition of DLE is particularly important, as early referral and treatment may reduce the likelihood of permanent scarring alopecia and pigment changes.11
Intermittent cutaneous lupus erythematosus (ICLE), a relatively new subtype of cutaneous LE, is represented by a rare condition—lupus erythematosus tumidus (LET)—reported in <100 cases worldwide. LET is characterized by succulent, erythematous, and edematous plaques on sun-exposed parts of the body.1,16
TABLE 1
Cutaneous lupus erythematosus: Recognizing the subtypes1
| Acute cutaneous lupus erythematosus (ACLE) |
|---|
| Localized: erythematous macules or papules over the bridge of the nose and cheek, with sparing of the nasolabial folds (“butterfly rash”) |
| Generalized: similar erythematous lesions over other photodistributed parts of the body, including the neck, chest, arms, and hands |
| Toxic epidermal necrolysis-like (TEN-like): blistering and epidermal cleavage in photodistributed parts of the body |
| Subacute cutaneous lupus erythematosus (SCLE) |
| Erythematous papulosquamous (psoriasis-like) eruptions or annular (ring-like) lesions with raised red borders and central clearing, occurring symmetrically and suddenly after sunlight exposure on photo-distributed body parts; the scalp and face are rarely affected |
| Chronic cutaneous lupus erythematosus (CCLE) |
| Discoid lupus erythematosus: erythematous papules and plaques with associated scale, spreading centrifugally with follicular plugging, pigment change, telangiectasia, and atrophy; scarring alopecia can occur |
| Lupus erythematosus profundus: tender, erythematous nodules and plaques, usually involving the proximal extremities, trunk, breasts, buttocks, and face |
| Chilblain lupus erythematosus: tender, erythematous nodules and plaques, occurring in acral areas often in response to cold |
| Intermittent cutaneous lupus erythematosus (ICLE) |
| Lupus erythematosus tumidus: succulent, erythematous, and edematous plaques found on photodistributed parts of the body |
Cutaneous LE diagnosis and treatment: Start with ACR criteria
When evaluating patients with suspected cutaneous LE, it is important not only to identify the subtype, but also to rule out systemic disease using criteria established by the American College of Rheumatology (ACR).17 Notably, 4 of the 11 diagnostic criteria for systemic disease involve visual clues, including malar rash, discoid rash, photosensitivity, and oral ulcerations. Laboratory evidence of systemic disease may include a positive antinuclear antibody, anti-dsDNA, or anti-Sm autoantibody test results, as well as hematologic abnormalities described in the ACR guidelines.
Ultimately, a diagnosis of cutaneous LE should be based on the patient’s history and physical exam, autoantibody profile, and histologic and immunofluorescent biopsy findings. A rheumatologic evaluation may help to determine which patients have systemic disease, as the ACR criteria may overdiagnose systemic LE in those with predominantly skin changes.6
Treatment of cutaneous LE is based on the subtype and extent of disease, with potent topical corticosteroids, in combination with antimalarial agents, being the primary therapies. ACLE skin lesions generally respond best to systemic corticosteroids and immunosuppressive agents (such as azathioprine or cyclophosphamide) that are used to control underlying systemic disease. SCLE can be managed with topical corticosteroids; however, patients typically also require systemic treatment, often with hydroxychloroquine, for optimal control. DLE, the most common form of CCLE, is managed in a similar fashion, with a greater role for intralesional corticosteroid injections. Lesions associated with ICLE, which often resolve spontaneously, may be treated with topical corticosteroids and antimalarials.16 It is also important to advise all patients with cutaneous LE to use a broad-spectrum sunscreen, as ultraviolet (UV) exposure can induce or exacerbate the lesions.9
Dermatomyositis: Rare but serious
Dermatomyositis is an idiopathic inflammatory myopathy characterized by chronic muscle inflammation, symmetric proximal muscle weakness, and distinct cutaneous findings. In addition to possible cardiac and pulmonary complications, dysphagia, and joint contractures, dermatomyositis is associated with cancer, with up to 25% of affected adults having an underlying occult malignancy.18
Dermatomyositis is a rare condition, with a prevalence of only 1 to 10 cases per million adults and 3.2 cases per million children.19 It has a bimodal age distribution, with most juvenile cases affecting children between the ages of 5 and 14 years and most adult cases developing in the fifth and sixth decades of life. Women are affected twice as often as men.18
Suspect dermatomyositis when you see any of the following signs:
- A heliotrope rash: violaceous macules and patches, with or without edema, symmetrically on the periorbital skin, present early in the disease course in 30% to 60% of patients.19 (See image)
- Gottron’s papules: violaceous papules on the dorsal interphalangeal and metacarpophalangeal joints of the hands, elbows, and knees, occurring in as many as 70% of patients (FIGURE 2).19,20
- Gottron’s sign: nonscaling, violaceous erythematous macules and plaques occurring symmetrically in the same distribution as Gottron’s papules, but with sparing of the interphalangeal spaces.20
- Periungual erythema and telangiectasias: redness and dilation of the blood vessels in the skin surrounding the nail plate.
- The shawl and V-signs: erythematous macular eruptions occurring in a “shawl” pattern on the shoulders, arms, and upper back and in a V-shaped pattern on the anterior neck and chest.
- Mechanic’s hand: extensive scaling, fissuring, and roughening of the palmar aspect of the hand.20,21
- Poikiloderma vasculare atrophicans: circumscribed violaceous erythema with thinning of the skin, prominent telangiectasias, and a mottled pattern of hypo- and hyperpigmentation, typically occurring on the anterior neck, chest, posterior shoulders, back, and buttocks, years after onset of the disease.21-24
- Cutaneous calcification (calcinosis cutis): usually on the buttocks, elbows, knees, and traumatized areas, affecting 30% to 70% of children with dermatomyositis, but only 10% of adults with the disorder.20,25-27
FIGURE 2
Gottron’s papules in dermatomyositis
Violaceous papules on the dorsal interphalangeal and metacarpophalangeal joints of the hands are a common manifestation of dermatomyositis.
Bohan and Peter criteria help with dermatomyositis diagnosis
A diagnosis of dermatomyositis can be established using the following criteria developed by Bohan and Peter in 1975: (1) symmetric muscle weakness; (2) elevation of muscle enzymes, most notably creatinine phosphokinase; (3) evidence of inflammation on muscle biopsy; (4) electromyographic features of myositis; and (5) characteristic dermatologic signs, including a heliotrope rash and Gottron’s papules.28,29 A definitive diagnosis can be made when the patient meets 3 of the first 4 criteria, as well as the fifth.
Corticosteroids are the mainstay of treatment for dermatomyositis, although the dose and duration are subject to debate. Cutaneous manifestations of dermatomyositis are commonly treated with topical corticosteroids and oral hydroxychloroquine, as well as emollients and antipruritic agents.
Cancer screening. All adults with signs and symptoms of dermatomyositis should undergo cancer screening. The most common malignancies are ovarian cancer, gastric cancer, and lymphoma.21
Photoprotection. As with cutaneous LE, UV exposure can exacerbate dermatomyositis, and patients should use a broad-spectrum sunscreen.
Scleroderma: Localized and systemic disease
The major cutaneous manifestation of scleroderma is that of thickened, leathery, bound-down skin, seen in both localized and systemic disease. In both cases, the lesions typically evolve through 3 characteristic stages: the initial inflammatory and edematous stage; a fibrotic stage, during which the skin lesions appear hard, tight, and hidebound; and the final, atrophic stage. However, not all lesions progress to this final stage.8
Localized scleroderma (also known as morphea) affects roughly 1 in 100,000 people. It is more prevalent in females than males, with a ratio as high as 3 to 1.30-33 Cutaneous findings vary, and numerous clinical presentations are possible. The most widely used classification system, the Mayo Clinic Classification, recognizes 5 subtypes: plaque morphea (the most common), generalized morphea, linear morphea, deep morphea, and bullous morphea (TABLE 2).34 The mean age of onset depends on the subtype, and ranges from 12 years for linear morphea to 45 years for deep morphea.35
While systemic involvement is not common with localized scleroderma, extracutaneous manifestations have been reported in up to 25% of cases.31,36,37 In a 2005 multicenter study of 750 pediatric patients with localized scleroderma, the most frequently reported extracutaneous symptom was arthritis, found in 12% of patients.36 Less frequent findings included neurologic symptoms such as seizures and headaches, vascular changes such as deep vein thrombosis, and gastrointestinal, cardiac, and renal conditions. Although the precise etiology of localized scleroderma is unknown, it is thought to be associated with trauma, prior infection by Borrelia burgdorferi, chronic venous insufficiency, and irradiation for breast cancer.30,38,39
TABLE 2
Localized scleroderma: What to look for8,34
|
Systemic scleroderma is a heterogenous disorder
Commonly called systemic sclerosis (SSc), systemic scleroderma is characterized by proliferative vascular lesions; fibrosis of internal organs, including the lungs, heart, kidneys, and gastrointestinal tract; and distinct cutaneous manifestations.3 SSc affects about 240 people per million adults, mostly between the ages of 30 and 50 years, with women affected 3 times as often as men.40-42
The cause of SSc is unknown, although a genetic predisposition is likely. Environmental factors likely play a role in triggering the disease, with possible causative factors including cytomegalovirus and other viral infections and exposure to certain chemicals.43
As in localized scleroderma, cutaneous manifestations are a prominent part of SSc and typically develop early on. Raynaud’s phenomenon (RP) (FIGURE 3), which occurs in 90% to 98% of patients with SSc,44 can develop in association with other cutaneous findings or precede additional skin changes by months or years.
Other early skin changes include nonpitting edema of the fingers and toes, which creates a sausage-like appearance. This is typically followed by hardening and thickening of the skin in these areas, which can result in highly disabling sclerodactyly (FIGURE 4).45 Patients can also develop painful ulcerations on their fingertips and knuckles (rat bite necroses) due to local ischemia and vascular insufficiency. This may be complicated by secondary bacterial infection, gangrene, and acroosteolysis, leading to articular deformities and dissolution of terminal phalanges (FIGURE 5).45-48
On the face, SSc is characterized early on by periorbital edema and later by the development of a beaked nose, a reduction in the size of the mouth (microstomia) with radial furrowing, thinning of the lips, and telangiectasias.45,49 As the sclerosis worsens, patients are frequently left with expressionless, mask-like faces.48
Other common cutaneous manifestations of SSc include a “salt and pepper” appearance, with alternating areas of hypo- and hyperpigmented skin. Additionally, patients may suffer from a loss of hair follicles, severely dry skin, and pruritus.45,48
There are 2 major subsets of SSc—limited cutaneous SSc and diffuse cutaneous SSc. While they can be differentiated based on the history of symptoms, the appearance and extent of cutaneous involvement, and certain serological makers, the most important difference is the speed at which the disease progresses and its severity: Limited cutaneous SSc typically progresses slowly, while diffuse cutaneous SSc is characterized by a relatively rapid onset of disease, with skin and internal organ involvement likely to be severe.49
Scleroderma diagnosis and treatment: Vascular changes are an important clue
Skin changes seen in localized scleroderma and SSc can be clinically and histologically similar, making it difficult to arrive at a definitive diagnosis.
One clinical clue is that in localized scleroderma, vascular changes, such as RP and periungual nailfold telangiectasia, are typically absent. In addition, cutaneous changes in the hands and fingers, such as sclerodactyly, are more characteristic of systemic disease.10
SSc can be diagnosed using criteria proposed by the ACR, which have been shown to be highly sensitive (97%) and specific (98%).50 The major criterion is proximal scleroderma—symmetric thickening, tightening, and induration of the skin of the fingers and areas proximal to the metacarpophalangeal or metatarsophalangeal joints, which may include the trunk, neck, and face. Minor criteria include (1) sclerodactyly, (2) digital pitting scars of fingertips or loss of substance of the distal finger pad, and (3) bilateral basilar pulmonary fibrosis. Diagnosis is based on the presence of either the major criterion or 2 of the 3 minor criteria.50
Numerous therapies are available for localized scleroderma, including topical, intralesional, and systemic corticosteroids, topical tacrolimus, hydroxychloroquine, topical and systemic calcipotriol, penicillamine, sulfasalazine, interferon-[H9253], methotrexate, phototherapy with UV light, and imiquimod.
SSc skin manifestations, which can be severe and disabling, can be treated with a wide range of therapies. Topical or systemic corticosteroids; topical calcineurin inhibitors; systemic immunosuppressive agents such as methotrexate, cyclophosphamide, cyclosporine, and D-penicillamine; and phototherapy have all had varying success at reducing hardening of the skin.51-54 Other skin manifestations, including RP, dryness and itching, pigment changes, digital ulcerations, calcifications, and telangiectasias, should be managed as needed, with various supplemental treatment options available.45 These may include emollients, antihistamines, and topical corticosteroids for dryness and itching; laser therapy for telangiectasias; and corticosteroid injection, laser therapy, or surgery for calcifications. However, there is limited evidence of efficacy for most of these options.45
FIGURE 3
An attack of Raynaud’s phenomenon
Raynaud’s phenomenon, characterized by blanching of the distal fingertips, is shown here in a patient with systemic sclerosis.
FIGURE 4
Sclerodactyly in a patient with systemic sclerosis
Hardening and thickening of the skin can result in highly disabling sclerodactyly in patients with systemic sclerosis.
FIGURE 5
Dissolution of terminal phalanges
Bony resorption and ulceration have led to the loss of distal phalanges in this patient with systemic sclerosis.
Mixed connective tissue disease has features of several disorders
Mixed connective tissue disease (MCTD) is an apparently distinct rheumatologic condition characterized by a combination of clinical features of systemic LE, dermatomyositis, scleroderma, polymyositis, and rheumatoid arthritis. The presence of high titers of a unique autoantibody, anti-U1-RNP,54-57 aids in diagnosis.
Although precise prevalence data are lacking, MCTD is thought to occur in about 1 in 10,000 people.58 The disease is much more common in women, with a female-to-male ratio as high as 7 to 1, and generally occurs in the second or third decade of life.58-60
The clinical manifestations of MCTD typically evolve, with overlapping features of various autoimmune disorders appearing sequentially over several months to years.57 Early in the course of MCTD, patients commonly experience fatigue, polyarthritis, hand edema, and RP. In time, virtually every organ system may be involved. Pulmonary hypertension is a major cause of death.59,61
Unlike many other connective tissue disorders, MCTD lacks any distinct cutaneous findings. In addition to those mentioned earlier, prominent skin findings include swelling of the fingers, sclerodactyly, and the acute malar eruptions and discoid plaques typically associated with LE.56,57,62 Cutaneous manifestations associated with dermatomyositis and scleroderma may also be seen, particularly juxta-articular calcinosis. The mucous membranes may be involved, as well, resulting in nasal perforation, buccal and urogenital ulcerations, and sicca complex.58,63,64
MCTD diagnosis and treatment: Look for these serology and clinical findings
Diagnosing MCTD can be clinically challenging, as signs and symptoms of the disease commonly evolve over time. The Alarcon-Segovia criteria—largely regarded as the best diagnostic tool for MCTD65,66—include 1 serologic finding (elevated anti-U1-RNP [titer ≥1:1600]) and 5 clinical findings (RP, edema of the hands, synovitis, myositis, and acrosclerosis). Diagnosis requires the presence of the serologic criterion and ≥3 of the 5 clinical criteria.
Treatment of the cutaneous manifestations should be based on the effectiveness of therapies for similar skin findings seen in other disorders. In treating MCTD (or any other connective tissue disorder), a team that includes nurses, physical and occupational therapists, primary care physicians, and specialists in dermatology and rheumatology is essential for an optimal outcome.
CORRESPONDENCE Christian R. Halvorson, MD, Mercy Medical Center, Department of Medicine, 301 St. Paul Place, Baltimore, MD 21202; crhalvor@gmail.com
1. Renner R, Sticherling M. The different faces of cutaneous lupus erythematosus. G Ital Dermatol Venereol. 2009;144:135-147.
2. Kuhn A, Ruzicka T. Classification of cutaneous lupus erythematosus. In: Kuhn A, Lehmann P, Ruzicka T, eds. Cutaneous Lupus Erythematosus. New York: Springer; 2004:53–57.
3. Costner MI, Grau RH. Update on connective tissue diseases in dermatology. Semin Cutan Med Surg. 2006;25:207-220.
4. Ting W, Stone MS, Racila D, et al. Toxic epidermal necrolysis-like acute cutaneous lupus erythematosus and the spectrum of the acute syndrome of apoptotic pan-epidermolysis (ASAP): a case report, concept review and proposal for new classification of lupus erythematosus vesiculobullous skin lesions. Lupus. 2004;13:941-950.
5. Jorizzo JL, Carroll CL, Sangueza OP. Lupus erythematosus. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. Spain: Elsevier Limited; 2008.
6. Kuhn A, Sticherling M, Bonsmann G. Clinical manifestations of cutaneous lupus erythematosus. J Dtsch Dermatol Ges. 2007;5:1124-1137.
7. Sontheimer RD, Thomas JR, Gilliam JN. Subacute cutaneous lupus erythematosus: a cutaneous marker for a distinct lupus erythematosus subset. Arch Dermatol. 1979;115:1409-1415.
8. Sontheimer RD. Skin manifestations of systemic autoimmune connective tissue disease: diagnostics and therapeutics. Best Pract Res Clin Rheumatol. 2004;18:429-462.
9. Panjwani S. Early diagnosis and treatment of discoid lupus erythematosus. J Am Board Fam Med. 2009;22:206-213.
10. Sontheimer RD, Provost TT. Lupus erythematosus. In: Sontheimer RD, Provost TT, eds. Cutaneous Manifestations of Rheumatic Diseases. Baltimore: Williams & Wilkins; 1996.
11. Mond CB, Peterson MG, Rothfield NF. Correlation of antiro antibody with photosensitivity rash in systemic lupus erythematosus patients. Arthritis Rheum. 1989;32:202-204.
12. Jacyk WK, Damisah M. Discoid lupus erythematosus in the Nigerians. Br J Dermatol. 1979;100:131-135.
13. Callen JP. Cutaneous lupus erythematosus: A personal approach to management. Australas J Dermatol. 2006;47:13-27.
14. Gilliam JN, Sontheimer RD. Distinctive cutaneous subsets in the spectrum of lupus erythematosus. J Am Acad Dermatol. 1981;4:471-475.
15. Farley-Loftus R, Mahlberg M, Merola J, et al. Generalized discoid lupus erythematosus. Dermatol Online J. 2009;15:18.-
16. Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus—a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
17. American College of Rheumatology. The 1982 revised criteria for classification of systemic lupus erythematosus. Available at: http://www.rheumatology.org/practice/clinical/classification/SLE/sle.asp. Accessed September 21, 2010.
18. Na SJ, Kim SM, Sunwoo IN, et al. Clinical characteristics and outcomes of juvenile and adult dermatomyositis. J Korean Med Sci. 2009;24:715-721.
19. Kovacs SO, Kovacs SC. Dermatomyositis. J Am Acad Dermatol. 1998;39:899-922.
20. Koler RA, Montemarano A. Dermatomyositis. Am Fam Physician. 2001;64:1565-1572.
21. Sunkureddi P, Nguyen-Oghalai TU, Jarvis JL, et al. Signs of dermatomyositis. Hosp Physician. 2005;41:41-44.
22. Iorizzo LJ, 3rd, Jorizzo JL. The treatment and prognosis of dermatomyositis: an updated review. J Am Acad Dermatol. 2008;59:99-112.
23. Jorizzo JL, Carroll CL, Sangueza OP. Dermatomyositis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. Spain: Elsevier Limited; 2008:575–583.
24. Sontheimer RD. Dermatomyositis. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 5th ed. New York: McGraw-Hill; 1999:2009–2020.
25. Halbert AR. Juvenile dermatomyositis. Australas J Dermatol. 1996;37:106-108.
26. Orlow SJ, Watsky KL, Bolognia JL. Skin and bones II. J Am Acad Dermatol. 1991;25:447-462.
27. Bowyer SL, Blane CE, Sullivan DB, et al. Childhood dermatomyositis: factors predicting functional outcome and development of dystrophic calcification. J Pediatr. 1983;103:882-888.
28. Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. 1975;292:403-407.
29. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344-347.
30. Vancheeswaran R, Black CM, David J, et al. Childhood-onset scleroderma: is it different from adult-onset disease? Arthritis Rheum. 1996;39:1041-1049.
31. Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: Clinical and epidemiological features in 750 children. An international study. Rheumatology (Oxford). 2006;45:614-620.
32. Marzano AV, Menni S, Parodi A, et al. Localized scleroderma in adults and children. clinical and laboratory investigations on 239 cases. Eur J Dermatol. 2003;13:171-176.
33. Uziel Y, Krafchik BR, Silverman ED, et al. Localized scleroderma in childhood: a report of 30 cases. Semin Arthritis Rheum. 1994;23:328-340.
34. Peterson LS, Nelson AM, Su WP. Classification of morphea (localized scleroderma). Mayo Clin Proc. 1995;70:1068-1076.
35. Peterson LS, Nelson AM, Su WP, et al. The epidemiology of morphea (localized scleroderma) in Olmsted County 1960-1993. J Rheumatol. 1997;24:73-80.
36. Zulian F, Vallongo C, Woo P, et al. Localized scleroderma in childhood is not just a skin disease. Arthritis Rheum. 2005;52:2873-2881.
37. Dehen L, Roujeau JC, Cosnes A, et al. Internal involvement in localized scleroderma. Medicine (Baltimore). 1994;73:241-245.
38. Ludwig RJ, Werner RJ, Winker W, et al. Chronic venous insufficiency - a potential trigger for localized scleroderma. J Eur Acad Dermatol Venereol. 2006;20:96-99.
39. Aberer E, Neumann R, Stanek G. Is localised scleroderma a borrelia infection? Lancet. 1985;2:278.-
40. Krishnan E, Furst DE. Systemic sclerosis mortality in the United States: 1979-1998. Eur J Epidemiol. 2005;20:855-861.
41. Silman AJ. Scleroderma—demographics and survival. J Rheumatol Suppl. 1997;48:58-61.
42. Medsger TA,, Jr, Masi AT. Epidemiology of systemic sclerosis (scleroderma). Ann Intern Med. 1971;74:714-721.
43. Charles C, Clements P, Furst DE. Systemic sclerosis: Hypothesis-driven treatment strategies. Lancet. 2006;367:1683-1691.
44. Maricq HR, Harper FE, Khan MM, et al. Microvascular abnormalities as possible predictors of disease subsets in Raynaud phenomenon and early connective tissue disease. Clin Exp Rheumatol. 1983;1:195-205.
45. Krieg T, Takehara K. Skin disease: A cardinal feature of systemic sclerosis. Rheumatology (Oxford). 2009;48 (suppl 3):S14-S18.
46. Ingraham KM, Steen VD. Morbidity of digital tip ulcerations in scleroderma. Arthritis Rheum. 2006;54(9 suppl.):F78.-
47. Ferri C, Valentini G, Cozzi F, et al. Systemic sclerosis: Demographic, clinical, and serologic features and survival in 1,012 italian patients. Medicine (Baltimore). 2002;81:139-153.
48. Haustein UF. Systemic sclerosis - scleroderma. Dermatol Online J. 2002;8.-
49. Fitzpatrick TB, Johnson RA, Klaus W, et al. In Colour Atlas and Synopsis of Clinical Dermatology. 4th ed. New York: McGraw-Hill; 2001:368–369.
50. Preliminary criteria for the classification of systemic sclerosis (scleroderma). subcommittee for scleroderma criteria of the American Rheumatism Association diagnostic and therapeutic criteria committee. Arthritis Rheum. 1980;23:581-590.
51. Valentini G, Paone C, La Montagna G, et al. Low-dose intravenous cyclophosphamide in systemic sclerosis: an open prospective efficacy study in patients with early diffuse disease. Scand J Rheumatol. 2006;35:35-38.
52. Steen VD, Medsger TA, Jr. Improvement in skin thickening in systemic sclerosis associated with improved survival. Arthritis Rheum. 2001;44:2828-2835.
53. Basso M, Filaci G, Cutolo M, et al. Long-term treatment of patients affected by systemic sclerosis with cyclosporin A. Ann Ital Med Int. 2001;16:233-239.
54. Morton SJ, Powell RJ. Cyclosporin and tacrolimus: their use in a routine clinical setting for scleroderma. Rheumatology (Oxford). 2000;39:865-869.
55. Kasukawa R. Mixed connective tissue disease. Intern Med. 1999;38:386-393.
56. Bennett RM, O’Connell DJ. Mixed connective tisssue disease: A clinicopathologic study of 20 cases. Semin Arthritis Rheum. 1980;10:25-51.
57. Sharp GC, Irvin WS, Tan EM, et al. Mixed connective tissue disease—an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA). Am J Med. 1972;52:148-159.
58. Venables PJ. Mixed connective tissue disease. Lupus. 2006;15:132-137.
59. Farhey Y, Hess EV. Mixed connective tissue disease. Arthritis Care Res. 1997;10:333-342.
60. Alarcon-Segovia D. Mixed connective tissue disease - a decade of growing pains. J Rheumatol. 1981;8:535-540.
61. Ueda N, Mimura K, Maeda H, et al. Mixed connective tissue disease with fatal pulmonary hypertension and a review of literature. Virchows Arch A Pathol Anat Histopathol. 1984;404:335-340.
62. Bennett RM. Mixed connective tissue disease and other overlap syndromes. In: Kelley WN, Harris EDJ, Ruddy SH, et al, eds. Textbook of Rheumatology. 4th ed. Philadelphia: WB Saunders; 1993:1061.
63. Nakashima M, Suzuki K, Okada M, et al. Panniculitis in a patient with mixed connective tissue disease. Mod Rheumatol. 2004;14:250-253.
64. Magro CM, Crowson AN, Regauer S. Mixed connective tissue disease. A clinical, histologic, and immunofluorescence study of eight cases. Am J Dermatopathol. 1997;19:206-213.
65. Amigues JM, Cantagrel A, Abbal M, et al. Comparative study of 4 diagnosis criteria sets for mixed connective tissue disease in patients with anti-RNP antibodies. autoimmunity group of the hospitals of toulouse. J Rheumatol. 1996;23:2055-2062.
66. Alarcon-Segovia D, Cardiel MH. Comparison between 3 diagnostic criteria for mixed connective tissue disease. study of 593 patients. J Rheumatol. 1989;16:328-334.
1. Renner R, Sticherling M. The different faces of cutaneous lupus erythematosus. G Ital Dermatol Venereol. 2009;144:135-147.
2. Kuhn A, Ruzicka T. Classification of cutaneous lupus erythematosus. In: Kuhn A, Lehmann P, Ruzicka T, eds. Cutaneous Lupus Erythematosus. New York: Springer; 2004:53–57.
3. Costner MI, Grau RH. Update on connective tissue diseases in dermatology. Semin Cutan Med Surg. 2006;25:207-220.
4. Ting W, Stone MS, Racila D, et al. Toxic epidermal necrolysis-like acute cutaneous lupus erythematosus and the spectrum of the acute syndrome of apoptotic pan-epidermolysis (ASAP): a case report, concept review and proposal for new classification of lupus erythematosus vesiculobullous skin lesions. Lupus. 2004;13:941-950.
5. Jorizzo JL, Carroll CL, Sangueza OP. Lupus erythematosus. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. Spain: Elsevier Limited; 2008.
6. Kuhn A, Sticherling M, Bonsmann G. Clinical manifestations of cutaneous lupus erythematosus. J Dtsch Dermatol Ges. 2007;5:1124-1137.
7. Sontheimer RD, Thomas JR, Gilliam JN. Subacute cutaneous lupus erythematosus: a cutaneous marker for a distinct lupus erythematosus subset. Arch Dermatol. 1979;115:1409-1415.
8. Sontheimer RD. Skin manifestations of systemic autoimmune connective tissue disease: diagnostics and therapeutics. Best Pract Res Clin Rheumatol. 2004;18:429-462.
9. Panjwani S. Early diagnosis and treatment of discoid lupus erythematosus. J Am Board Fam Med. 2009;22:206-213.
10. Sontheimer RD, Provost TT. Lupus erythematosus. In: Sontheimer RD, Provost TT, eds. Cutaneous Manifestations of Rheumatic Diseases. Baltimore: Williams & Wilkins; 1996.
11. Mond CB, Peterson MG, Rothfield NF. Correlation of antiro antibody with photosensitivity rash in systemic lupus erythematosus patients. Arthritis Rheum. 1989;32:202-204.
12. Jacyk WK, Damisah M. Discoid lupus erythematosus in the Nigerians. Br J Dermatol. 1979;100:131-135.
13. Callen JP. Cutaneous lupus erythematosus: A personal approach to management. Australas J Dermatol. 2006;47:13-27.
14. Gilliam JN, Sontheimer RD. Distinctive cutaneous subsets in the spectrum of lupus erythematosus. J Am Acad Dermatol. 1981;4:471-475.
15. Farley-Loftus R, Mahlberg M, Merola J, et al. Generalized discoid lupus erythematosus. Dermatol Online J. 2009;15:18.-
16. Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus—a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
17. American College of Rheumatology. The 1982 revised criteria for classification of systemic lupus erythematosus. Available at: http://www.rheumatology.org/practice/clinical/classification/SLE/sle.asp. Accessed September 21, 2010.
18. Na SJ, Kim SM, Sunwoo IN, et al. Clinical characteristics and outcomes of juvenile and adult dermatomyositis. J Korean Med Sci. 2009;24:715-721.
19. Kovacs SO, Kovacs SC. Dermatomyositis. J Am Acad Dermatol. 1998;39:899-922.
20. Koler RA, Montemarano A. Dermatomyositis. Am Fam Physician. 2001;64:1565-1572.
21. Sunkureddi P, Nguyen-Oghalai TU, Jarvis JL, et al. Signs of dermatomyositis. Hosp Physician. 2005;41:41-44.
22. Iorizzo LJ, 3rd, Jorizzo JL. The treatment and prognosis of dermatomyositis: an updated review. J Am Acad Dermatol. 2008;59:99-112.
23. Jorizzo JL, Carroll CL, Sangueza OP. Dermatomyositis. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. Spain: Elsevier Limited; 2008:575–583.
24. Sontheimer RD. Dermatomyositis. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 5th ed. New York: McGraw-Hill; 1999:2009–2020.
25. Halbert AR. Juvenile dermatomyositis. Australas J Dermatol. 1996;37:106-108.
26. Orlow SJ, Watsky KL, Bolognia JL. Skin and bones II. J Am Acad Dermatol. 1991;25:447-462.
27. Bowyer SL, Blane CE, Sullivan DB, et al. Childhood dermatomyositis: factors predicting functional outcome and development of dystrophic calcification. J Pediatr. 1983;103:882-888.
28. Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. 1975;292:403-407.
29. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344-347.
30. Vancheeswaran R, Black CM, David J, et al. Childhood-onset scleroderma: is it different from adult-onset disease? Arthritis Rheum. 1996;39:1041-1049.
31. Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: Clinical and epidemiological features in 750 children. An international study. Rheumatology (Oxford). 2006;45:614-620.
32. Marzano AV, Menni S, Parodi A, et al. Localized scleroderma in adults and children. clinical and laboratory investigations on 239 cases. Eur J Dermatol. 2003;13:171-176.
33. Uziel Y, Krafchik BR, Silverman ED, et al. Localized scleroderma in childhood: a report of 30 cases. Semin Arthritis Rheum. 1994;23:328-340.
34. Peterson LS, Nelson AM, Su WP. Classification of morphea (localized scleroderma). Mayo Clin Proc. 1995;70:1068-1076.
35. Peterson LS, Nelson AM, Su WP, et al. The epidemiology of morphea (localized scleroderma) in Olmsted County 1960-1993. J Rheumatol. 1997;24:73-80.
36. Zulian F, Vallongo C, Woo P, et al. Localized scleroderma in childhood is not just a skin disease. Arthritis Rheum. 2005;52:2873-2881.
37. Dehen L, Roujeau JC, Cosnes A, et al. Internal involvement in localized scleroderma. Medicine (Baltimore). 1994;73:241-245.
38. Ludwig RJ, Werner RJ, Winker W, et al. Chronic venous insufficiency - a potential trigger for localized scleroderma. J Eur Acad Dermatol Venereol. 2006;20:96-99.
39. Aberer E, Neumann R, Stanek G. Is localised scleroderma a borrelia infection? Lancet. 1985;2:278.-
40. Krishnan E, Furst DE. Systemic sclerosis mortality in the United States: 1979-1998. Eur J Epidemiol. 2005;20:855-861.
41. Silman AJ. Scleroderma—demographics and survival. J Rheumatol Suppl. 1997;48:58-61.
42. Medsger TA,, Jr, Masi AT. Epidemiology of systemic sclerosis (scleroderma). Ann Intern Med. 1971;74:714-721.
43. Charles C, Clements P, Furst DE. Systemic sclerosis: Hypothesis-driven treatment strategies. Lancet. 2006;367:1683-1691.
44. Maricq HR, Harper FE, Khan MM, et al. Microvascular abnormalities as possible predictors of disease subsets in Raynaud phenomenon and early connective tissue disease. Clin Exp Rheumatol. 1983;1:195-205.
45. Krieg T, Takehara K. Skin disease: A cardinal feature of systemic sclerosis. Rheumatology (Oxford). 2009;48 (suppl 3):S14-S18.
46. Ingraham KM, Steen VD. Morbidity of digital tip ulcerations in scleroderma. Arthritis Rheum. 2006;54(9 suppl.):F78.-
47. Ferri C, Valentini G, Cozzi F, et al. Systemic sclerosis: Demographic, clinical, and serologic features and survival in 1,012 italian patients. Medicine (Baltimore). 2002;81:139-153.
48. Haustein UF. Systemic sclerosis - scleroderma. Dermatol Online J. 2002;8.-
49. Fitzpatrick TB, Johnson RA, Klaus W, et al. In Colour Atlas and Synopsis of Clinical Dermatology. 4th ed. New York: McGraw-Hill; 2001:368–369.
50. Preliminary criteria for the classification of systemic sclerosis (scleroderma). subcommittee for scleroderma criteria of the American Rheumatism Association diagnostic and therapeutic criteria committee. Arthritis Rheum. 1980;23:581-590.
51. Valentini G, Paone C, La Montagna G, et al. Low-dose intravenous cyclophosphamide in systemic sclerosis: an open prospective efficacy study in patients with early diffuse disease. Scand J Rheumatol. 2006;35:35-38.
52. Steen VD, Medsger TA, Jr. Improvement in skin thickening in systemic sclerosis associated with improved survival. Arthritis Rheum. 2001;44:2828-2835.
53. Basso M, Filaci G, Cutolo M, et al. Long-term treatment of patients affected by systemic sclerosis with cyclosporin A. Ann Ital Med Int. 2001;16:233-239.
54. Morton SJ, Powell RJ. Cyclosporin and tacrolimus: their use in a routine clinical setting for scleroderma. Rheumatology (Oxford). 2000;39:865-869.
55. Kasukawa R. Mixed connective tissue disease. Intern Med. 1999;38:386-393.
56. Bennett RM, O’Connell DJ. Mixed connective tisssue disease: A clinicopathologic study of 20 cases. Semin Arthritis Rheum. 1980;10:25-51.
57. Sharp GC, Irvin WS, Tan EM, et al. Mixed connective tissue disease—an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA). Am J Med. 1972;52:148-159.
58. Venables PJ. Mixed connective tissue disease. Lupus. 2006;15:132-137.
59. Farhey Y, Hess EV. Mixed connective tissue disease. Arthritis Care Res. 1997;10:333-342.
60. Alarcon-Segovia D. Mixed connective tissue disease - a decade of growing pains. J Rheumatol. 1981;8:535-540.
61. Ueda N, Mimura K, Maeda H, et al. Mixed connective tissue disease with fatal pulmonary hypertension and a review of literature. Virchows Arch A Pathol Anat Histopathol. 1984;404:335-340.
62. Bennett RM. Mixed connective tissue disease and other overlap syndromes. In: Kelley WN, Harris EDJ, Ruddy SH, et al, eds. Textbook of Rheumatology. 4th ed. Philadelphia: WB Saunders; 1993:1061.
63. Nakashima M, Suzuki K, Okada M, et al. Panniculitis in a patient with mixed connective tissue disease. Mod Rheumatol. 2004;14:250-253.
64. Magro CM, Crowson AN, Regauer S. Mixed connective tissue disease. A clinical, histologic, and immunofluorescence study of eight cases. Am J Dermatopathol. 1997;19:206-213.
65. Amigues JM, Cantagrel A, Abbal M, et al. Comparative study of 4 diagnosis criteria sets for mixed connective tissue disease in patients with anti-RNP antibodies. autoimmunity group of the hospitals of toulouse. J Rheumatol. 1996;23:2055-2062.
66. Alarcon-Segovia D, Cardiel MH. Comparison between 3 diagnostic criteria for mixed connective tissue disease. study of 593 patients. J Rheumatol. 1989;16:328-334.
6 skin disorders of pregnancy: A management guide
The dermatoses of pregnancy are a poorly understood group of conditions. Their only common feature is a tendency to appear during pregnancy.
Only three of these conditions are considered unique to pregnancy, however; the others are probably exacerbations of preexisting conditions triggered by pregnancy. There isn’t even complete agreement on what to call them. To make management even more complex, two patients—mother and fetus—need to be considered in decisions about care.
Who manages these patients is another matter. These conditions fall into overlapping areas of health care, where family physicians, obstetricians, and dermatologists all might have some share in responsibility for diagnosis and treatment. You need to be sufficiently familiar with these conditions so that you can differentiate those that can be treated symptomatically and those that require referral to a specialist. This review and the handy TABLE, will help you toward that end.
TABLE
Skin disorders of pregnancy: What you’ll see, how to treat
| Disorder | Lesions | Diagnosis and sequelae | Treatment | Recurrence |
|---|---|---|---|---|
| Pemphigoid gestationis3,5 | Erythematous papules that progress to vesicles and bullae, in a periumbilical distribution that spares the face, palms, and soles |
|
| Frequent; skips a pregnancy 8% of the time |
| Pruritic urticarial papules and plaques of pregnancy8-10 | Urticarial papules and plaques on the abdomen, legs, arms, buttocks, chest, and back |
| Topical corticosteroids and antihistamines | Uncommon |
| Intrahepatic cholestasis of pregnancy14,17,19-22 | No primary lesions; secondary excoriations in any area that the patient can reach |
| Ursodeoxycholic acid, 450-1,200 mg/d | Frequent |
| Eczema of pregnancy/pruritus of pregnancy4,10,24 | Grouped, crusted erythematous papules, patches, and plaques, most often on extensor surfaces of the arms and legs or on the abdomen |
| Symptomatic treatment with topical corticosteroids or antihistamines | Frequent |
| Acute pustular psoriasis of pregnancy26-28 | Erythematous plaques and pustules that start on the inner thighs and groin and spread to the trunk and extremities |
|
| Unknown |
| Pruritic folliculitis of pregnancy24,28 | Papules and pustules concentrated around hair follicles, often beginning on the abdomen and spreading to the extremities |
| Topical corticosteroids | Unknown |
DERMATOSES UNIQUE TO PREGNANCY
1. Pemphigoid gestationis
Years ago, this disorder was referred to as herpes gestationis, because the lesions are herpetiform. Pemphigoid gestationis (PG) has an incidence of approximately 1 in 10,000 pregnancies.1,2 Time of onset is usually about the 21st week of gestation, although, in about 20% of cases, the eruption appears immediately postpartum.3
Presentation. The disease usually begins with urticarial papules and plaques around the umbilicus and extremities. Bullous lesions tend to develop as the disease progresses, and are often not present on first presentation (FIGURE 1). Lesions of PG tend to spare the face, palms, and soles. Mucosal surfaces are involved in fewer than 20% of cases. In about 75% of cases, PG flares around the time of delivery, regressing spontaneously after the baby is born.4
FIGURE 1 Pemphigoid gestationis
As the disease progresses, bullous lesions tend to develop.
Pathophysiology. The pathophysiology of PG is nearly identical to that of bullous pemphigoid, a blistering skin disorder seen more often in elderly patients.5 Pemphigoid disorders are immune processes, involving an immunoglobulin G (IgG) immune response directed at a 180-kDa hemidesmosome transmembrane glycoprotein. This protein is the common target in several subepidermal blistering diseases.
Differential diagnosis. Disorders that may have some of the same features as PG include pruritic urticarial papules and plaques of pregnancy (PUPPP), erythema multiforme, intrahepatic cholestasis of pregnancy (ICP), contact dermatitis, and drug reactions.
Diagnosis. A biopsy is necessary for definitive diagnosis. Direct immunofluorescence (DIF) microscopy of a sample of perilesional skin can show tissue-bound immunoreactants. Linear deposition of the complement component protein C3 along the basement membrane zone is diagnostic for PG. IgG is also deposited about 40% of the time.3
Serum enzyme-linked immunosorbent assay (ELISA) studies are also helpful in diagnosis. They have excellent sensitivity and specificity, as well as the capacity to monitor levels of antibody, which correlate with the severity of disease.1
Treatment. Oral corticosteroids are the first-line treatment for PG, typically 20 to 60 mg/d of prednisone. Oral corticosteroids are generally most effective at ameliorating symptoms. Prednisone at a dosage of 40 to 80 mg/d for a short time has not been associated with congenital abnormalities.6 PG patients can also be treated successfully with intravenous immunoglobulin (IVIG) and cyclosporine in refractory cases.7
Pruritus associated with this condition can interfere with day-to-day activities and with the patient’s ability to sleep. Patients may also complain that the rash is painful, particularly if bullae rupture, leading to superficial ulcerations. Fortunately, the patient’s quality of life can be dramatically improved with systemic corticosteroids—with no significant risk to the fetus.
Sequelae. PG uniformly resolves within a few weeks, but the mother’s autoantibodies can be passively transferred to the fetus, causing vesicles and bullae in the newborn.8 An increased incidence of small-for-gestational age (SGA) infants has also been noted in PG, although no lasting morbidity or mortality in the offspring has been noted.5 The disease tends to recur in future pregnancies.
2. Pruritic urticarial papules and plaques of pregnancy
This condition is known by many names besides its acronym PUPPP: polymorphic eruption of pregnancy, toxemic erythema of pregnancy, and late prurigo of pregnancy.1 It is a pruritic, inflammatory skin disorder that has been variously estimated to occur in anywhere from 1 in 120 to 1 in 240 pregnancies.8 PUPPP is second only to eczema as the most common dermatosis of pregnancy.
Presentation. As the name suggests, the lesions of PUPPP are itchy, red papules that often coalesce into plaques (FIGURE 2). Lesions usually occur in primigravidas after the 34th week of gestation, although they may be seen at any time from the first trimester through the postpartum period.9
Lesions are classically found on the abdomen, sparing the umbilical area, and are found primarily in the striae. This distribution helps you to differentiate PUPPP from PG, in which lesions typically cluster around the umbilicus. Most PUPPP lesions (80% in one study) are dispersed on the abdomen, legs, arms, buttocks, chest, and back. Another 17% appear only on the abdomen and proximal thighs, and the remaining 3% on the limbs.10 Nearly 50% of the time, lesions also include discrete vesicles.11 There are no reported cases of mucosal involvement.
Patients with this condition are often very uncomfortable. The associated pruritus is severe enough to interfere with sleep. Despite the itching, however, lesions are seldom excoriated.

FIGURE 2 Pruritic urticarial papules and plaques of pregnancy
The itchy, red papules of PUPP often coalesce into plaques.
Pathophysiology. The disorder has been strongly associated with maternal weight gain and multiple gestations. One working hypothesis is that rapid abdominal distention observed in the third trimester leads to damage of the connective tissue, which then releases antigenic molecules, causing an inflammatory reaction.12 Another hypothesis is that increased levels of fetal DNA that have been detected in the skin of PUPPP patients may contribute to the pathology. One study detected male DNA in six of 10 PUPPP sufferers, but found none in any of 26 controls—pregnant women without PUPPP pathology.5 There is some evidence that patients with atopy may be predisposed to PUPPP, as well as patients who are hypertensive or obese.10,13
Differential diagnosis. Initially, PUPPP lesions can be difficult to differentiate from urticarial PG lesions. The distribution of the lesions is the best clue: PG lesions cluster around the umbilicus, whereas PUPPP lesions uniformly spare the umbilical area. Additional disorders in the PUPPP differential are atopic dermatitis, superficial urticarial allergic eruption, viral exanthema, and contact or irritant dermatitis.
Diagnosis. PUPPP can be diagnosed only by clinical observation. None of the available laboratory tests—immunofluorescence, histology, serology—yield findings specific for PUPPP, although histology and immunofluorescence can readily differentiate between this condition and PG.
Treatment. Because the disease holds no real danger for mother or fetus, treatment can be aimed solely at symptomatic relief. Mild-to-potent topical corticosteroids (consider triamcinolone or fluocinonide) should relieve pruritus within 48 to 72 hours.8 Antihistamines and, occasionally, low-dose systemic corticosteroids may also be used. Consider hydroxyzine, although diphenhydramine has the more proven safety profile in pregnancy.
Nonpharmaceutical treatments such as oil baths and emollients should also be considered. If the condition appears classic for PUPPP, it can be managed symptomatically. If there is any question about the diagnosis, however, referral to a dermatologist is prudent.
Sequelae. No increase in maternal or fetal morbidity or mortality is associated with PUPPP. Recurrence is fairly uncommon, as the disease primarily affects women during their first pregnancy.
3. Intrahepatic cholestasis of pregnancy
This condition is also called recurrent or idiopathic jaundice of pregnancy, obstetric cholestasis, and pruritus gravidarum. Intrahepatic cholestasis of pregnancy (ICP) is caused by disruption of hepatic bile flow during pregnancy. It has been recorded at a rate of approximately 10 to 150 of every 10,000 pregnancies in Europe and 70 of every 10,000 in the United States.12 In 80% of patients, time of onset is after the 30th week.14
Although this disorder is not primarily a dermatosis of pregnancy, it is a pruritic condition that often presents with excoriations in pregnant women and is associated with fetal morbidity and mortality. It’s important to be able to identify this disease early to minimize sequelae.
Presentation. There are no primary lesions with ICP. The primary presenting symptom is a generalized pruritus affecting the palms and soles, and sometimes extending to the legs and abdomen (FIGURE 3). This itching is often so severe that it leads to chronic insomnia. You may see secondary skin lesions, such as erythema and excoriations. Observable jaundice occurs in 10% to 20% of patients.3 These patients do not develop the encephalopathy that is associated with cholestasis in the nonpregnant state, however.14
FIGURE 3 Intrahepatic cholestasis of pregnancy
ICP lacks primary lesions. Shown here are the secondary erythema and excoriations that results from scratching the intense pruritis.
Pathophysiology. The genesis of this condition is thought to be a combination of genetic and environmental factors. A family history of the disorder is present in one half of cases; cases with a familial component tend to be more severe.15 ICP may be an exaggerated response to increased estrogen levels in pregnancy, but the mechanism of this response is unknown.16
Differential diagnosis. Other conditions that must be considered in making the diagnosis are viral hepatitis, gallbladder disease, PG, PUPPP, drug hepatotoxicity, primary biliary cirrhosis, and uremia.
Diagnosis. Laboratory values are the definitive diagnostic tool in this condition. Increased levels of serum bile acids are the single most sensitive test. Average levels of serum bile acids in pregnancy are 6.6 µmol/L, with an upper limit of 11 µmol/L. The average value in women who have ICP is 47 µmol/L.17
Although serum bile acids remain the gold standard, a recent study showed that elevated urine bile acids have 100% sensitivity and 83% specificity for ICP.18 In 55% to 60% of cases, the liver enzymes aspartate aminotransferase (AST) and alanine aminotransferase (ALT) are mildly increased. Steatorrhea is often noted by the patient, and is followed by vitamin K deficiency.17
Treatment. The current standard of care for ICP is treatment with ursodeoxycholic acid (UDCA). In four controlled trials, UDCA caused a sustained decrease in serum bile acids.19-22 The dosage used in these trials ranged from 450 to 1,200 mg/d.
Before UDCA treatment was available, ICP was treated with cholestyramine, which could bring about a 70% rate of response. The drawback to cholestyramine treatment is that it precipitates vitamin K, which is already compromised by the disease process. Further, the onset of action of cholestyramine is slow.3
Elective delivery is indicated for ICP, particularly in patients who have a significant clinical presentation.12 Delivery for ICP should be performed around weeks 37 to 38, as stillbirths tend to cluster around weeks 37 to 39.14 Given the significant fetal mortality associated with ICP (see the next paragraph), the condition should be managed by a clinician experienced with the disease—likely, a gastroenterologist.
Sequelae. The impact of this maternal disorder on the fetus can be disastrous: a 10% to 15% rate of perinatal death, and a 30% to 40% rate of premature labor.14 Fortunately, the rate of preterm labor correlates strongly with the level of bile acids, so that as bile acid levels are reduced with UDCA treatment, the rate of preterm labor also falls. Management of ICP has reduced the rate of perinatal death to 3.5%. No evidence of fetal growth retardation has been noted.14
DERMATOSES TRIGGERED BY PREGNANCY
4. Eczema of pregnancy/prurigo of pregnancy
Eczema of pregnancy/prurigo of pregnancy (EP/PP) may not actually be correlated with the pregnant state. Both conditions manifest as eczematous lesions in an atopic distribution. Although they have been described in the literature as separate entities, the lack of clinical distinction between them led Ambros-Rudolph and colleagues to combine them under the umbrella term, atopic eruption of pregnancy.23
In some patients, at least, EP/PP may be preexisting conditions that are exacerbated by pregnancy. One study of 255 patients with the condition found that 20% had had the lesions before they became pregnant.23 The tendency of the condition to be made markedly worse by pregnancy, however, leads us to include it here.
PP has an estimated incidence of 1 in 450 pregnancies.11 Although many authorities consider EP to be the most common dermatosis of pregnancy, no clear estimation of its prevalence has been established.23,24 Taken together, the two conditions have the highest prevalence of all pregnancy-induced dermatoses.
PP is also known as popular dermatitis of Spangler, Nurse’s early prurigo of pregnancy, and linear IgM disease of pregnancy.3,4,23
Presentation. The typical presentation is grouped, crusted, erythematous papules, patches, and plaques—frequently with excoriations. Lesions typically present on the extensor surfaces of the arms and legs or on the abdomen (FIGURE 4).4 Recurrence in later pregnancies is common.
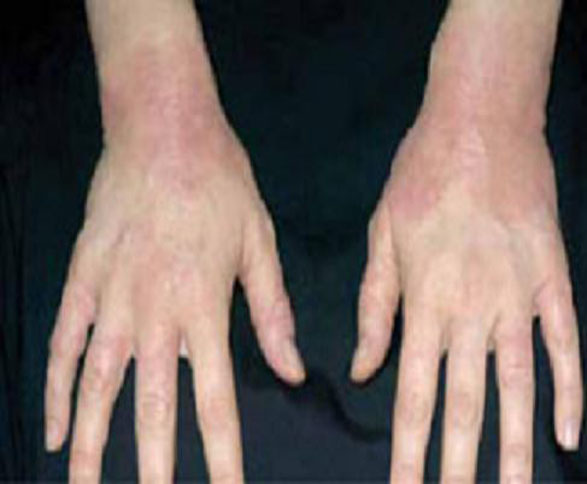
FIGURE 4 Eczema of pregnancy
EP/PP typically manifests as grouped, crusted, erythematous papules, patches, and plaques, often with excoriations.
Pathophysiology. The pathophysiology of EP/PP is not understood. Many patients who develop EP/PP have a history of atopy.10
Differential diagnosis. Conditions that need to be considered in making the diagnosis include Tinea infection, scabies, contact dermatitis, ICP, pruritic folliculitis of pregnancy (PFP), and PG.
Diagnosis. The history and the physical examination determine the diagnosis. Serology, histopathology, and immunofluorescence are nonspecific. Correlation of EP/PP with increased IgE is marginal, at best.24,25
Treatment. These conditions are treated symptomatically with topical corticosteroids or systemic antihistamines.
Sequelae. No increase in maternal or fetal morbidity or mortality is associated with EP/PP.
5. Acute pustular psoriasis of pregnancy
Whether or not acute pustular psoriasis of pregnancy (APPP) is actually a pregnancy-induced dermatosis is subject to debate.
There is evidence that APPP is not unique to pregnancy but simply a manifestation of ordinary psoriasis. Clinically and histologically, APPP is indistinguishable from pustular psoriasis. Unlike most cases of acute psoriasis, however, APPP often appears in pregnancy without any personal or family history of psoriasis and usually ceases when the pregnancy ends. This fact, combined with reports of increased fetal and maternal morbidity and mortality associated with APPP, leads us to include it here.26
Presentation. APPP is a rare condition that may have an onset at any point in pregnancy. Characteristic lesions begin as erythematous plaques with pustules on the inner thighs, flexural areas, and groin and spread to the trunk and extremities. As plaques enlarge, their center becomes eroded and crusted.
The nails may become onycholytic. Hands, feet, and face are usually spared. Oral and esophageal erosions can occur. Pruritus is typically mild, although the lesions are often painful. Flu-like symptoms are often present.27
Pathophysiology. The pathophysiology of APPP is unknown.
Differential diagnosis. Conditions with similar presentations include an adverse drug reaction, pityriasis rosea, lichen simplex chronicus, eczema, lupus, and pityriasis rubra pilaris.
Diagnosis. The clinical history and an association with systemic illness are the basis for a diagnosis of APPP. Cultures of pustules are negative for any infective pathology, although, as the disease progresses, pustules may become superinfected. Laboratory testing may show an increased erythrocyte sedimentation rate, hypocalcemia, and a low level of vitamin D.
Treatment. Prednisone, 15 to 60 mg/d, is often sufficient to control the disease.27 Cyclosporine, 100 mg twice daily, has also been shown to be useful.28 Cyclosporine in pregnancy is a Category C drug. Data on fetal malformation associated with cyclosporine therapy are limited, but risk appears minimal.6
Maternal hypocalcemia should be monitored and treated appropriately. If disease progression is judged serious enough, early induction of labor is indicated, because delivery will almost always lead to swift resolution.
Sequelae. A number of case reports link APPP to serious sequelae, including fetal growth retardation, hypocalcemia, and stillbirth.27,29,30 The condition is too rare, however, for good data on specific sequelae. Although APPP does give significant cause for concern, it appears that some of the traditional apprehension comes from older publications reporting a rate of maternal mortality of 70% to 90%.31 This statistic has not been borne out in practice. It does appear that the mother will frequently suffer systemic symptoms, including fever and malaise.
6. Pruritic folliculitis of pregnancy
Accounts of the prevalence of pruritic folliculitis of pregnancy (PFP) vary widely. Some sources report fewer than 30 cases in all of the literature; others indicate that the prevalence is equivalent to that of PG—one in every 10,000 pregnancies.3,11 PFP most often presents in the third trimester. It often resolves before delivery, but uniformly clears within 2 weeks of delivery.
Presentation. PFP presents as papules and pustules concentrated around hair follicles (FIGURE 5). Often, lesions begin on the abdomen and spread to the extremities.24,28 The condition is often, but not always, pruritic. Patients are more likely to be concerned about what the condition means for their health than distressed by the symptoms.
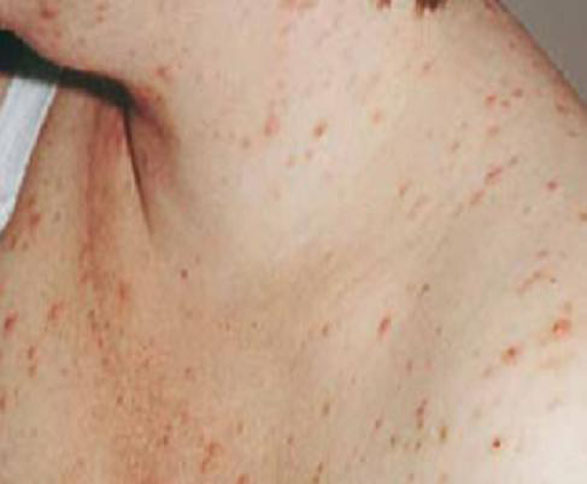
FIGURE 5 Pruritic folliculitis of pregnancy
The papules and pustules of PFP are concentrated around hair follicles.
Pathophysiology. Like many other dermatoses of pregnancy, the pathophysiology of PFP is unknown. There is little evidence that the condition is immunologically or hormonally mediated, and there is no evidence of an infectious component.24,28
Differential diagnosis. PFP must be distinguished from infectious folliculitis, acneiform disorders, HIV-associated eosinophilic folliculitis, and a drug reaction.
Diagnosis. The clinical diagnosis is based on presenting symptoms and third-trimester onset. No specific laboratory or histologic analysis can be used to make a definitive diagnosis.
Treatment. As the condition is, by definition, a nonmicrobial folliculitis, the most effective therapy tends to be with a low- or midpotency topical corticosteroid, such as triamcinolone or desonide. A benzoyl peroxide wash can also be effective.
Sequelae. One study reports an increased incidence of low birth weight, but no associated morbidity or mortality has been reported in recent studies.24
- Pemphigoid gestationis is best managed with oral prednisone at doses from 20 to 60 mg per day to control symptoms
- The pruritus associated with pruritic urticarial papules and plaques of pregnancy can be safely and effectively managed with topical corticosteroids and oral antihistamines
- Treat intrahepatic cholestasis of pregnancy with ursodeoxycholic acid, which likely reduces serum bile acids as well as associated fetal morbidity and mortality
1. Sitaru C, Powell J, Messer G, Bröcker EB, Wojnarowska F, Zillikens D. Immunoblotting and enzyme linked immunosorbent assay for the diagnosis of pemphigoid gestationis. Obstet Gynecol. 2004;103(4):757-763.
2. Engineer L, Bohl K, Ahmed AR. Pemphigoid gestationis: a review. Am J Obstet Gynecol. 2000;183(2):483-491.
3. Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence-based systematic review. Am J Obstet Gynecol. 2003;188(4):1083-1092.
4. Shornick JK. Dermatoses of pregnancy. Semin Cutan Med Surg. 1998;17(3):172-181.
5. Shornick JK, Bangert JL, Freeman RG, Gilliam JN. Herpes gestationis: clinical and histological features in 28 cases. J Am Acad Dermatol. 1983;8(2):214-224.
6. Leachman SA, Reed BR. The use of dermatologic drugs in pregnancy and lactation. Dermatol Clin. 2006;24(2):167-197, vi.
7. Hern S, Harman K, Bhogal BS, Black MM. A severe persistent case of pemphigoid gestationis treated with intravenous immunoglobulins and cyclosporine. Clin Exp Dermatol. 1998;23(4):185-188.
8. Fitzpatrick TP. Diseases in pregnancy. Color Atlas and Synopsis of Clinical Dermatology. New York, NY: McGraw Hill; 1997:414–419.
9. Aractingi D, Berkane N, Bertheau P, et al. Fetal DNA in skin of polymorphic eruptions of pregnancy. Lancet. 1998;352(9144):1898-1901.
10. Rudolph CM, Al-Fares S, Vaughan-Jones SA, et al. Polymorphic eruption of pregnancy: clinicopathology and potential trigger factors in 181 patients. Br J Dermatol. 2006;154(1):54-60.
11. Roger D, Vaillant L, Fignon A, et al. Specific pruritic diseases of pregnancy. A prospective study of 3192 pregnant women. Arch Dermatol. 1994;130(6):734-739.
12. McDonald JA. Cholestasis of pregnancy. J Gastroenterol Hepatol. 1999;14(6):515-518.
13. Ohel I, Levy A, Silberstein T, Holcberg G, Sheiner E. Pregnancy outcome of patients with pruritic urticarial papules and plaques of pregnancy. J Maternal Fetal Neonat Med. 2006;19(5):305-308.
14. Geenes V, Williamson C. Intrahepatic cholestasis of pregnancy. World J Gastroenterol. 2009;15(17):2049-2066.
15. Shaw D, Frohlich J, Wittmann BA, Willms M. A prospective study of 18 patients with cholestasis of pregnancy. Am J Obstet Gynecol. 1982;142(6 Pt 1):621-625.
16. Reyes H. The spectrum of liver and gastrointestinal disease seen in cholestasis of pregnancy. Gastroenterol Clin North Am. 1992;21(4):905-921.
17. Lammert F, Marschall H, Glantz A, Matern S. Intrahepatic cholestasis of pregnancy; molecular pathogenesis, diagnosis and management. J Hepatol. 2000;33(6):1012-1021.
18. Huang WM, Seubert DE, Donnelly JG, Liu M, Javitt NB. Intrahepatic cholestasis of pregnancy: detection with urinary bile acid assays. J Perinat Med. 2007;35(6):486-491.
19. Palma J, Reyes H, Ribalta J, et al. Ursodeoxycholic acid in the treatment of intrahepatic cholestasis of pregnancy: a randomized, double-blind study controlled with placebo. J Hepatol. 1997;27(6):1022-1028.
20. Diaferia A, Nicastri PL, Tartagni M, et al. Ursodeoxycholic acid therapy in pregnant women with cholestasis. Int J Gynaecol Obstet. 1996;52:133-140.
21. Nicastri PL, Diaferia A, Tartagni M, Loizzi P, Fanelli M. A randomised placebo-controlled trial of ursodeoxycholic acid and S-adenosylmethionine in the treatment of intrahepatic cholestasis of pregnancy. Br J Obstet Gynaecol. 1998;105(11):1205-1207.
22. Glantz A, Marschall HU, Lammert F, Mattsson LA. Intrahepatic cholestasis in pregnancy: a randomized controlled trial comparing dexamethasone and ursodeoxycholic acid. Hepatology. 2005;42(6):1399-1405.
23. Ambros-Rudolph C, Müllegger M, Vaughan-Jones S, et al. The specific dermatoses of pregnancy revisited and reclassified: results of a retrospective two-center study on 505 pregnant patients. J Am Acad Dermatol. 2006;54(3):395-404.
24. Vaughan Jones SA, Hern S, Nelson-Piercy C, Seed PT, Black MM. A prospective study of 200 women with dermatoses of pregnancy correlating clinical findings with hormonal and immunopathological profiles. Br J Dermatol. 1999;141(1):71-81.
25. Holmes RC, Black MM. The specific dermatoses of pregnancy. J Am Acad Dermatol. 1983;8(3):405-412.
26. Bukhari IA. Impetigo herpetiformis in a primagravida: successful treatment with etretinate. J Drugs Dermatol. 2004;3(4):449-451.
27. Oumeish OY, Parish JL. Impetigo herpetiformis. Clin Dermatol. 2006;24(2):101-104.
28. Kroumpouzos G, Cohen LM. Pruritic folliculitis of pregnancy. J Am Acad Dermatol. 2000;43(1 Pt 1):132-134.
29. Sahin HG, Sahin HA, Metin A, Zeteroglu S, Ugras S. Recurrent impetigo herpetiformis in a pregnant adolescent: case report. Eur J Obstet Gynecol Reprod Biol. 2002;101:201-203.
30. Aka N, Kuscu NK, Yazicioglu E. Impetigo herpetiformis at the 36th week of gestation. Int J Gynecol Obstet. 2000;69(2):153-154.
31. Wade T, Wade S, Jones H. Skin changes and diseases associated with pregnancy. Obstet Gynecol. 1978;52(2):233-242.
The dermatoses of pregnancy are a poorly understood group of conditions. Their only common feature is a tendency to appear during pregnancy.
Only three of these conditions are considered unique to pregnancy, however; the others are probably exacerbations of preexisting conditions triggered by pregnancy. There isn’t even complete agreement on what to call them. To make management even more complex, two patients—mother and fetus—need to be considered in decisions about care.
Who manages these patients is another matter. These conditions fall into overlapping areas of health care, where family physicians, obstetricians, and dermatologists all might have some share in responsibility for diagnosis and treatment. You need to be sufficiently familiar with these conditions so that you can differentiate those that can be treated symptomatically and those that require referral to a specialist. This review and the handy TABLE, will help you toward that end.
TABLE
Skin disorders of pregnancy: What you’ll see, how to treat
| Disorder | Lesions | Diagnosis and sequelae | Treatment | Recurrence |
|---|---|---|---|---|
| Pemphigoid gestationis3,5 | Erythematous papules that progress to vesicles and bullae, in a periumbilical distribution that spares the face, palms, and soles |
|
| Frequent; skips a pregnancy 8% of the time |
| Pruritic urticarial papules and plaques of pregnancy8-10 | Urticarial papules and plaques on the abdomen, legs, arms, buttocks, chest, and back |
| Topical corticosteroids and antihistamines | Uncommon |
| Intrahepatic cholestasis of pregnancy14,17,19-22 | No primary lesions; secondary excoriations in any area that the patient can reach |
| Ursodeoxycholic acid, 450-1,200 mg/d | Frequent |
| Eczema of pregnancy/pruritus of pregnancy4,10,24 | Grouped, crusted erythematous papules, patches, and plaques, most often on extensor surfaces of the arms and legs or on the abdomen |
| Symptomatic treatment with topical corticosteroids or antihistamines | Frequent |
| Acute pustular psoriasis of pregnancy26-28 | Erythematous plaques and pustules that start on the inner thighs and groin and spread to the trunk and extremities |
|
| Unknown |
| Pruritic folliculitis of pregnancy24,28 | Papules and pustules concentrated around hair follicles, often beginning on the abdomen and spreading to the extremities |
| Topical corticosteroids | Unknown |
DERMATOSES UNIQUE TO PREGNANCY
1. Pemphigoid gestationis
Years ago, this disorder was referred to as herpes gestationis, because the lesions are herpetiform. Pemphigoid gestationis (PG) has an incidence of approximately 1 in 10,000 pregnancies.1,2 Time of onset is usually about the 21st week of gestation, although, in about 20% of cases, the eruption appears immediately postpartum.3
Presentation. The disease usually begins with urticarial papules and plaques around the umbilicus and extremities. Bullous lesions tend to develop as the disease progresses, and are often not present on first presentation (FIGURE 1). Lesions of PG tend to spare the face, palms, and soles. Mucosal surfaces are involved in fewer than 20% of cases. In about 75% of cases, PG flares around the time of delivery, regressing spontaneously after the baby is born.4
FIGURE 1 Pemphigoid gestationis
As the disease progresses, bullous lesions tend to develop.
Pathophysiology. The pathophysiology of PG is nearly identical to that of bullous pemphigoid, a blistering skin disorder seen more often in elderly patients.5 Pemphigoid disorders are immune processes, involving an immunoglobulin G (IgG) immune response directed at a 180-kDa hemidesmosome transmembrane glycoprotein. This protein is the common target in several subepidermal blistering diseases.
Differential diagnosis. Disorders that may have some of the same features as PG include pruritic urticarial papules and plaques of pregnancy (PUPPP), erythema multiforme, intrahepatic cholestasis of pregnancy (ICP), contact dermatitis, and drug reactions.
Diagnosis. A biopsy is necessary for definitive diagnosis. Direct immunofluorescence (DIF) microscopy of a sample of perilesional skin can show tissue-bound immunoreactants. Linear deposition of the complement component protein C3 along the basement membrane zone is diagnostic for PG. IgG is also deposited about 40% of the time.3
Serum enzyme-linked immunosorbent assay (ELISA) studies are also helpful in diagnosis. They have excellent sensitivity and specificity, as well as the capacity to monitor levels of antibody, which correlate with the severity of disease.1
Treatment. Oral corticosteroids are the first-line treatment for PG, typically 20 to 60 mg/d of prednisone. Oral corticosteroids are generally most effective at ameliorating symptoms. Prednisone at a dosage of 40 to 80 mg/d for a short time has not been associated with congenital abnormalities.6 PG patients can also be treated successfully with intravenous immunoglobulin (IVIG) and cyclosporine in refractory cases.7
Pruritus associated with this condition can interfere with day-to-day activities and with the patient’s ability to sleep. Patients may also complain that the rash is painful, particularly if bullae rupture, leading to superficial ulcerations. Fortunately, the patient’s quality of life can be dramatically improved with systemic corticosteroids—with no significant risk to the fetus.
Sequelae. PG uniformly resolves within a few weeks, but the mother’s autoantibodies can be passively transferred to the fetus, causing vesicles and bullae in the newborn.8 An increased incidence of small-for-gestational age (SGA) infants has also been noted in PG, although no lasting morbidity or mortality in the offspring has been noted.5 The disease tends to recur in future pregnancies.
2. Pruritic urticarial papules and plaques of pregnancy
This condition is known by many names besides its acronym PUPPP: polymorphic eruption of pregnancy, toxemic erythema of pregnancy, and late prurigo of pregnancy.1 It is a pruritic, inflammatory skin disorder that has been variously estimated to occur in anywhere from 1 in 120 to 1 in 240 pregnancies.8 PUPPP is second only to eczema as the most common dermatosis of pregnancy.
Presentation. As the name suggests, the lesions of PUPPP are itchy, red papules that often coalesce into plaques (FIGURE 2). Lesions usually occur in primigravidas after the 34th week of gestation, although they may be seen at any time from the first trimester through the postpartum period.9
Lesions are classically found on the abdomen, sparing the umbilical area, and are found primarily in the striae. This distribution helps you to differentiate PUPPP from PG, in which lesions typically cluster around the umbilicus. Most PUPPP lesions (80% in one study) are dispersed on the abdomen, legs, arms, buttocks, chest, and back. Another 17% appear only on the abdomen and proximal thighs, and the remaining 3% on the limbs.10 Nearly 50% of the time, lesions also include discrete vesicles.11 There are no reported cases of mucosal involvement.
Patients with this condition are often very uncomfortable. The associated pruritus is severe enough to interfere with sleep. Despite the itching, however, lesions are seldom excoriated.
FIGURE 2 Pruritic urticarial papules and plaques of pregnancy
The itchy, red papules of PUPP often coalesce into plaques.
Pathophysiology. The disorder has been strongly associated with maternal weight gain and multiple gestations. One working hypothesis is that rapid abdominal distention observed in the third trimester leads to damage of the connective tissue, which then releases antigenic molecules, causing an inflammatory reaction.12 Another hypothesis is that increased levels of fetal DNA that have been detected in the skin of PUPPP patients may contribute to the pathology. One study detected male DNA in six of 10 PUPPP sufferers, but found none in any of 26 controls—pregnant women without PUPPP pathology.5 There is some evidence that patients with atopy may be predisposed to PUPPP, as well as patients who are hypertensive or obese.10,13
Differential diagnosis. Initially, PUPPP lesions can be difficult to differentiate from urticarial PG lesions. The distribution of the lesions is the best clue: PG lesions cluster around the umbilicus, whereas PUPPP lesions uniformly spare the umbilical area. Additional disorders in the PUPPP differential are atopic dermatitis, superficial urticarial allergic eruption, viral exanthema, and contact or irritant dermatitis.
Diagnosis. PUPPP can be diagnosed only by clinical observation. None of the available laboratory tests—immunofluorescence, histology, serology—yield findings specific for PUPPP, although histology and immunofluorescence can readily differentiate between this condition and PG.
Treatment. Because the disease holds no real danger for mother or fetus, treatment can be aimed solely at symptomatic relief. Mild-to-potent topical corticosteroids (consider triamcinolone or fluocinonide) should relieve pruritus within 48 to 72 hours.8 Antihistamines and, occasionally, low-dose systemic corticosteroids may also be used. Consider hydroxyzine, although diphenhydramine has the more proven safety profile in pregnancy.
Nonpharmaceutical treatments such as oil baths and emollients should also be considered. If the condition appears classic for PUPPP, it can be managed symptomatically. If there is any question about the diagnosis, however, referral to a dermatologist is prudent.
Sequelae. No increase in maternal or fetal morbidity or mortality is associated with PUPPP. Recurrence is fairly uncommon, as the disease primarily affects women during their first pregnancy.
3. Intrahepatic cholestasis of pregnancy
This condition is also called recurrent or idiopathic jaundice of pregnancy, obstetric cholestasis, and pruritus gravidarum. Intrahepatic cholestasis of pregnancy (ICP) is caused by disruption of hepatic bile flow during pregnancy. It has been recorded at a rate of approximately 10 to 150 of every 10,000 pregnancies in Europe and 70 of every 10,000 in the United States.12 In 80% of patients, time of onset is after the 30th week.14
Although this disorder is not primarily a dermatosis of pregnancy, it is a pruritic condition that often presents with excoriations in pregnant women and is associated with fetal morbidity and mortality. It’s important to be able to identify this disease early to minimize sequelae.
Presentation. There are no primary lesions with ICP. The primary presenting symptom is a generalized pruritus affecting the palms and soles, and sometimes extending to the legs and abdomen (FIGURE 3). This itching is often so severe that it leads to chronic insomnia. You may see secondary skin lesions, such as erythema and excoriations. Observable jaundice occurs in 10% to 20% of patients.3 These patients do not develop the encephalopathy that is associated with cholestasis in the nonpregnant state, however.14
FIGURE 3 Intrahepatic cholestasis of pregnancy
ICP lacks primary lesions. Shown here are the secondary erythema and excoriations that results from scratching the intense pruritis.
Pathophysiology. The genesis of this condition is thought to be a combination of genetic and environmental factors. A family history of the disorder is present in one half of cases; cases with a familial component tend to be more severe.15 ICP may be an exaggerated response to increased estrogen levels in pregnancy, but the mechanism of this response is unknown.16
Differential diagnosis. Other conditions that must be considered in making the diagnosis are viral hepatitis, gallbladder disease, PG, PUPPP, drug hepatotoxicity, primary biliary cirrhosis, and uremia.
Diagnosis. Laboratory values are the definitive diagnostic tool in this condition. Increased levels of serum bile acids are the single most sensitive test. Average levels of serum bile acids in pregnancy are 6.6 µmol/L, with an upper limit of 11 µmol/L. The average value in women who have ICP is 47 µmol/L.17
Although serum bile acids remain the gold standard, a recent study showed that elevated urine bile acids have 100% sensitivity and 83% specificity for ICP.18 In 55% to 60% of cases, the liver enzymes aspartate aminotransferase (AST) and alanine aminotransferase (ALT) are mildly increased. Steatorrhea is often noted by the patient, and is followed by vitamin K deficiency.17
Treatment. The current standard of care for ICP is treatment with ursodeoxycholic acid (UDCA). In four controlled trials, UDCA caused a sustained decrease in serum bile acids.19-22 The dosage used in these trials ranged from 450 to 1,200 mg/d.
Before UDCA treatment was available, ICP was treated with cholestyramine, which could bring about a 70% rate of response. The drawback to cholestyramine treatment is that it precipitates vitamin K, which is already compromised by the disease process. Further, the onset of action of cholestyramine is slow.3
Elective delivery is indicated for ICP, particularly in patients who have a significant clinical presentation.12 Delivery for ICP should be performed around weeks 37 to 38, as stillbirths tend to cluster around weeks 37 to 39.14 Given the significant fetal mortality associated with ICP (see the next paragraph), the condition should be managed by a clinician experienced with the disease—likely, a gastroenterologist.
Sequelae. The impact of this maternal disorder on the fetus can be disastrous: a 10% to 15% rate of perinatal death, and a 30% to 40% rate of premature labor.14 Fortunately, the rate of preterm labor correlates strongly with the level of bile acids, so that as bile acid levels are reduced with UDCA treatment, the rate of preterm labor also falls. Management of ICP has reduced the rate of perinatal death to 3.5%. No evidence of fetal growth retardation has been noted.14
DERMATOSES TRIGGERED BY PREGNANCY
4. Eczema of pregnancy/prurigo of pregnancy
Eczema of pregnancy/prurigo of pregnancy (EP/PP) may not actually be correlated with the pregnant state. Both conditions manifest as eczematous lesions in an atopic distribution. Although they have been described in the literature as separate entities, the lack of clinical distinction between them led Ambros-Rudolph and colleagues to combine them under the umbrella term, atopic eruption of pregnancy.23
In some patients, at least, EP/PP may be preexisting conditions that are exacerbated by pregnancy. One study of 255 patients with the condition found that 20% had had the lesions before they became pregnant.23 The tendency of the condition to be made markedly worse by pregnancy, however, leads us to include it here.
PP has an estimated incidence of 1 in 450 pregnancies.11 Although many authorities consider EP to be the most common dermatosis of pregnancy, no clear estimation of its prevalence has been established.23,24 Taken together, the two conditions have the highest prevalence of all pregnancy-induced dermatoses.
PP is also known as popular dermatitis of Spangler, Nurse’s early prurigo of pregnancy, and linear IgM disease of pregnancy.3,4,23
Presentation. The typical presentation is grouped, crusted, erythematous papules, patches, and plaques—frequently with excoriations. Lesions typically present on the extensor surfaces of the arms and legs or on the abdomen (FIGURE 4).4 Recurrence in later pregnancies is common.
FIGURE 4 Eczema of pregnancy
EP/PP typically manifests as grouped, crusted, erythematous papules, patches, and plaques, often with excoriations.
Pathophysiology. The pathophysiology of EP/PP is not understood. Many patients who develop EP/PP have a history of atopy.10
Differential diagnosis. Conditions that need to be considered in making the diagnosis include Tinea infection, scabies, contact dermatitis, ICP, pruritic folliculitis of pregnancy (PFP), and PG.
Diagnosis. The history and the physical examination determine the diagnosis. Serology, histopathology, and immunofluorescence are nonspecific. Correlation of EP/PP with increased IgE is marginal, at best.24,25
Treatment. These conditions are treated symptomatically with topical corticosteroids or systemic antihistamines.
Sequelae. No increase in maternal or fetal morbidity or mortality is associated with EP/PP.
5. Acute pustular psoriasis of pregnancy
Whether or not acute pustular psoriasis of pregnancy (APPP) is actually a pregnancy-induced dermatosis is subject to debate.
There is evidence that APPP is not unique to pregnancy but simply a manifestation of ordinary psoriasis. Clinically and histologically, APPP is indistinguishable from pustular psoriasis. Unlike most cases of acute psoriasis, however, APPP often appears in pregnancy without any personal or family history of psoriasis and usually ceases when the pregnancy ends. This fact, combined with reports of increased fetal and maternal morbidity and mortality associated with APPP, leads us to include it here.26
Presentation. APPP is a rare condition that may have an onset at any point in pregnancy. Characteristic lesions begin as erythematous plaques with pustules on the inner thighs, flexural areas, and groin and spread to the trunk and extremities. As plaques enlarge, their center becomes eroded and crusted.
The nails may become onycholytic. Hands, feet, and face are usually spared. Oral and esophageal erosions can occur. Pruritus is typically mild, although the lesions are often painful. Flu-like symptoms are often present.27
Pathophysiology. The pathophysiology of APPP is unknown.
Differential diagnosis. Conditions with similar presentations include an adverse drug reaction, pityriasis rosea, lichen simplex chronicus, eczema, lupus, and pityriasis rubra pilaris.
Diagnosis. The clinical history and an association with systemic illness are the basis for a diagnosis of APPP. Cultures of pustules are negative for any infective pathology, although, as the disease progresses, pustules may become superinfected. Laboratory testing may show an increased erythrocyte sedimentation rate, hypocalcemia, and a low level of vitamin D.
Treatment. Prednisone, 15 to 60 mg/d, is often sufficient to control the disease.27 Cyclosporine, 100 mg twice daily, has also been shown to be useful.28 Cyclosporine in pregnancy is a Category C drug. Data on fetal malformation associated with cyclosporine therapy are limited, but risk appears minimal.6
Maternal hypocalcemia should be monitored and treated appropriately. If disease progression is judged serious enough, early induction of labor is indicated, because delivery will almost always lead to swift resolution.
Sequelae. A number of case reports link APPP to serious sequelae, including fetal growth retardation, hypocalcemia, and stillbirth.27,29,30 The condition is too rare, however, for good data on specific sequelae. Although APPP does give significant cause for concern, it appears that some of the traditional apprehension comes from older publications reporting a rate of maternal mortality of 70% to 90%.31 This statistic has not been borne out in practice. It does appear that the mother will frequently suffer systemic symptoms, including fever and malaise.
6. Pruritic folliculitis of pregnancy
Accounts of the prevalence of pruritic folliculitis of pregnancy (PFP) vary widely. Some sources report fewer than 30 cases in all of the literature; others indicate that the prevalence is equivalent to that of PG—one in every 10,000 pregnancies.3,11 PFP most often presents in the third trimester. It often resolves before delivery, but uniformly clears within 2 weeks of delivery.
Presentation. PFP presents as papules and pustules concentrated around hair follicles (FIGURE 5). Often, lesions begin on the abdomen and spread to the extremities.24,28 The condition is often, but not always, pruritic. Patients are more likely to be concerned about what the condition means for their health than distressed by the symptoms.
FIGURE 5 Pruritic folliculitis of pregnancy
The papules and pustules of PFP are concentrated around hair follicles.
Pathophysiology. Like many other dermatoses of pregnancy, the pathophysiology of PFP is unknown. There is little evidence that the condition is immunologically or hormonally mediated, and there is no evidence of an infectious component.24,28
Differential diagnosis. PFP must be distinguished from infectious folliculitis, acneiform disorders, HIV-associated eosinophilic folliculitis, and a drug reaction.
Diagnosis. The clinical diagnosis is based on presenting symptoms and third-trimester onset. No specific laboratory or histologic analysis can be used to make a definitive diagnosis.
Treatment. As the condition is, by definition, a nonmicrobial folliculitis, the most effective therapy tends to be with a low- or midpotency topical corticosteroid, such as triamcinolone or desonide. A benzoyl peroxide wash can also be effective.
Sequelae. One study reports an increased incidence of low birth weight, but no associated morbidity or mortality has been reported in recent studies.24
- Pemphigoid gestationis is best managed with oral prednisone at doses from 20 to 60 mg per day to control symptoms
- The pruritus associated with pruritic urticarial papules and plaques of pregnancy can be safely and effectively managed with topical corticosteroids and oral antihistamines
- Treat intrahepatic cholestasis of pregnancy with ursodeoxycholic acid, which likely reduces serum bile acids as well as associated fetal morbidity and mortality
The dermatoses of pregnancy are a poorly understood group of conditions. Their only common feature is a tendency to appear during pregnancy.
Only three of these conditions are considered unique to pregnancy, however; the others are probably exacerbations of preexisting conditions triggered by pregnancy. There isn’t even complete agreement on what to call them. To make management even more complex, two patients—mother and fetus—need to be considered in decisions about care.
Who manages these patients is another matter. These conditions fall into overlapping areas of health care, where family physicians, obstetricians, and dermatologists all might have some share in responsibility for diagnosis and treatment. You need to be sufficiently familiar with these conditions so that you can differentiate those that can be treated symptomatically and those that require referral to a specialist. This review and the handy TABLE, will help you toward that end.
TABLE
Skin disorders of pregnancy: What you’ll see, how to treat
| Disorder | Lesions | Diagnosis and sequelae | Treatment | Recurrence |
|---|---|---|---|---|
| Pemphigoid gestationis3,5 | Erythematous papules that progress to vesicles and bullae, in a periumbilical distribution that spares the face, palms, and soles |
|
| Frequent; skips a pregnancy 8% of the time |
| Pruritic urticarial papules and plaques of pregnancy8-10 | Urticarial papules and plaques on the abdomen, legs, arms, buttocks, chest, and back |
| Topical corticosteroids and antihistamines | Uncommon |
| Intrahepatic cholestasis of pregnancy14,17,19-22 | No primary lesions; secondary excoriations in any area that the patient can reach |
| Ursodeoxycholic acid, 450-1,200 mg/d | Frequent |
| Eczema of pregnancy/pruritus of pregnancy4,10,24 | Grouped, crusted erythematous papules, patches, and plaques, most often on extensor surfaces of the arms and legs or on the abdomen |
| Symptomatic treatment with topical corticosteroids or antihistamines | Frequent |
| Acute pustular psoriasis of pregnancy26-28 | Erythematous plaques and pustules that start on the inner thighs and groin and spread to the trunk and extremities |
|
| Unknown |
| Pruritic folliculitis of pregnancy24,28 | Papules and pustules concentrated around hair follicles, often beginning on the abdomen and spreading to the extremities |
| Topical corticosteroids | Unknown |
DERMATOSES UNIQUE TO PREGNANCY
1. Pemphigoid gestationis
Years ago, this disorder was referred to as herpes gestationis, because the lesions are herpetiform. Pemphigoid gestationis (PG) has an incidence of approximately 1 in 10,000 pregnancies.1,2 Time of onset is usually about the 21st week of gestation, although, in about 20% of cases, the eruption appears immediately postpartum.3
Presentation. The disease usually begins with urticarial papules and plaques around the umbilicus and extremities. Bullous lesions tend to develop as the disease progresses, and are often not present on first presentation (FIGURE 1). Lesions of PG tend to spare the face, palms, and soles. Mucosal surfaces are involved in fewer than 20% of cases. In about 75% of cases, PG flares around the time of delivery, regressing spontaneously after the baby is born.4
FIGURE 1 Pemphigoid gestationis
As the disease progresses, bullous lesions tend to develop.
Pathophysiology. The pathophysiology of PG is nearly identical to that of bullous pemphigoid, a blistering skin disorder seen more often in elderly patients.5 Pemphigoid disorders are immune processes, involving an immunoglobulin G (IgG) immune response directed at a 180-kDa hemidesmosome transmembrane glycoprotein. This protein is the common target in several subepidermal blistering diseases.
Differential diagnosis. Disorders that may have some of the same features as PG include pruritic urticarial papules and plaques of pregnancy (PUPPP), erythema multiforme, intrahepatic cholestasis of pregnancy (ICP), contact dermatitis, and drug reactions.
Diagnosis. A biopsy is necessary for definitive diagnosis. Direct immunofluorescence (DIF) microscopy of a sample of perilesional skin can show tissue-bound immunoreactants. Linear deposition of the complement component protein C3 along the basement membrane zone is diagnostic for PG. IgG is also deposited about 40% of the time.3
Serum enzyme-linked immunosorbent assay (ELISA) studies are also helpful in diagnosis. They have excellent sensitivity and specificity, as well as the capacity to monitor levels of antibody, which correlate with the severity of disease.1
Treatment. Oral corticosteroids are the first-line treatment for PG, typically 20 to 60 mg/d of prednisone. Oral corticosteroids are generally most effective at ameliorating symptoms. Prednisone at a dosage of 40 to 80 mg/d for a short time has not been associated with congenital abnormalities.6 PG patients can also be treated successfully with intravenous immunoglobulin (IVIG) and cyclosporine in refractory cases.7
Pruritus associated with this condition can interfere with day-to-day activities and with the patient’s ability to sleep. Patients may also complain that the rash is painful, particularly if bullae rupture, leading to superficial ulcerations. Fortunately, the patient’s quality of life can be dramatically improved with systemic corticosteroids—with no significant risk to the fetus.
Sequelae. PG uniformly resolves within a few weeks, but the mother’s autoantibodies can be passively transferred to the fetus, causing vesicles and bullae in the newborn.8 An increased incidence of small-for-gestational age (SGA) infants has also been noted in PG, although no lasting morbidity or mortality in the offspring has been noted.5 The disease tends to recur in future pregnancies.
2. Pruritic urticarial papules and plaques of pregnancy
This condition is known by many names besides its acronym PUPPP: polymorphic eruption of pregnancy, toxemic erythema of pregnancy, and late prurigo of pregnancy.1 It is a pruritic, inflammatory skin disorder that has been variously estimated to occur in anywhere from 1 in 120 to 1 in 240 pregnancies.8 PUPPP is second only to eczema as the most common dermatosis of pregnancy.
Presentation. As the name suggests, the lesions of PUPPP are itchy, red papules that often coalesce into plaques (FIGURE 2). Lesions usually occur in primigravidas after the 34th week of gestation, although they may be seen at any time from the first trimester through the postpartum period.9
Lesions are classically found on the abdomen, sparing the umbilical area, and are found primarily in the striae. This distribution helps you to differentiate PUPPP from PG, in which lesions typically cluster around the umbilicus. Most PUPPP lesions (80% in one study) are dispersed on the abdomen, legs, arms, buttocks, chest, and back. Another 17% appear only on the abdomen and proximal thighs, and the remaining 3% on the limbs.10 Nearly 50% of the time, lesions also include discrete vesicles.11 There are no reported cases of mucosal involvement.
Patients with this condition are often very uncomfortable. The associated pruritus is severe enough to interfere with sleep. Despite the itching, however, lesions are seldom excoriated.
FIGURE 2 Pruritic urticarial papules and plaques of pregnancy
The itchy, red papules of PUPP often coalesce into plaques.
Pathophysiology. The disorder has been strongly associated with maternal weight gain and multiple gestations. One working hypothesis is that rapid abdominal distention observed in the third trimester leads to damage of the connective tissue, which then releases antigenic molecules, causing an inflammatory reaction.12 Another hypothesis is that increased levels of fetal DNA that have been detected in the skin of PUPPP patients may contribute to the pathology. One study detected male DNA in six of 10 PUPPP sufferers, but found none in any of 26 controls—pregnant women without PUPPP pathology.5 There is some evidence that patients with atopy may be predisposed to PUPPP, as well as patients who are hypertensive or obese.10,13
Differential diagnosis. Initially, PUPPP lesions can be difficult to differentiate from urticarial PG lesions. The distribution of the lesions is the best clue: PG lesions cluster around the umbilicus, whereas PUPPP lesions uniformly spare the umbilical area. Additional disorders in the PUPPP differential are atopic dermatitis, superficial urticarial allergic eruption, viral exanthema, and contact or irritant dermatitis.
Diagnosis. PUPPP can be diagnosed only by clinical observation. None of the available laboratory tests—immunofluorescence, histology, serology—yield findings specific for PUPPP, although histology and immunofluorescence can readily differentiate between this condition and PG.
Treatment. Because the disease holds no real danger for mother or fetus, treatment can be aimed solely at symptomatic relief. Mild-to-potent topical corticosteroids (consider triamcinolone or fluocinonide) should relieve pruritus within 48 to 72 hours.8 Antihistamines and, occasionally, low-dose systemic corticosteroids may also be used. Consider hydroxyzine, although diphenhydramine has the more proven safety profile in pregnancy.
Nonpharmaceutical treatments such as oil baths and emollients should also be considered. If the condition appears classic for PUPPP, it can be managed symptomatically. If there is any question about the diagnosis, however, referral to a dermatologist is prudent.
Sequelae. No increase in maternal or fetal morbidity or mortality is associated with PUPPP. Recurrence is fairly uncommon, as the disease primarily affects women during their first pregnancy.
3. Intrahepatic cholestasis of pregnancy
This condition is also called recurrent or idiopathic jaundice of pregnancy, obstetric cholestasis, and pruritus gravidarum. Intrahepatic cholestasis of pregnancy (ICP) is caused by disruption of hepatic bile flow during pregnancy. It has been recorded at a rate of approximately 10 to 150 of every 10,000 pregnancies in Europe and 70 of every 10,000 in the United States.12 In 80% of patients, time of onset is after the 30th week.14
Although this disorder is not primarily a dermatosis of pregnancy, it is a pruritic condition that often presents with excoriations in pregnant women and is associated with fetal morbidity and mortality. It’s important to be able to identify this disease early to minimize sequelae.
Presentation. There are no primary lesions with ICP. The primary presenting symptom is a generalized pruritus affecting the palms and soles, and sometimes extending to the legs and abdomen (FIGURE 3). This itching is often so severe that it leads to chronic insomnia. You may see secondary skin lesions, such as erythema and excoriations. Observable jaundice occurs in 10% to 20% of patients.3 These patients do not develop the encephalopathy that is associated with cholestasis in the nonpregnant state, however.14
FIGURE 3 Intrahepatic cholestasis of pregnancy
ICP lacks primary lesions. Shown here are the secondary erythema and excoriations that results from scratching the intense pruritis.
Pathophysiology. The genesis of this condition is thought to be a combination of genetic and environmental factors. A family history of the disorder is present in one half of cases; cases with a familial component tend to be more severe.15 ICP may be an exaggerated response to increased estrogen levels in pregnancy, but the mechanism of this response is unknown.16
Differential diagnosis. Other conditions that must be considered in making the diagnosis are viral hepatitis, gallbladder disease, PG, PUPPP, drug hepatotoxicity, primary biliary cirrhosis, and uremia.
Diagnosis. Laboratory values are the definitive diagnostic tool in this condition. Increased levels of serum bile acids are the single most sensitive test. Average levels of serum bile acids in pregnancy are 6.6 µmol/L, with an upper limit of 11 µmol/L. The average value in women who have ICP is 47 µmol/L.17
Although serum bile acids remain the gold standard, a recent study showed that elevated urine bile acids have 100% sensitivity and 83% specificity for ICP.18 In 55% to 60% of cases, the liver enzymes aspartate aminotransferase (AST) and alanine aminotransferase (ALT) are mildly increased. Steatorrhea is often noted by the patient, and is followed by vitamin K deficiency.17
Treatment. The current standard of care for ICP is treatment with ursodeoxycholic acid (UDCA). In four controlled trials, UDCA caused a sustained decrease in serum bile acids.19-22 The dosage used in these trials ranged from 450 to 1,200 mg/d.
Before UDCA treatment was available, ICP was treated with cholestyramine, which could bring about a 70% rate of response. The drawback to cholestyramine treatment is that it precipitates vitamin K, which is already compromised by the disease process. Further, the onset of action of cholestyramine is slow.3
Elective delivery is indicated for ICP, particularly in patients who have a significant clinical presentation.12 Delivery for ICP should be performed around weeks 37 to 38, as stillbirths tend to cluster around weeks 37 to 39.14 Given the significant fetal mortality associated with ICP (see the next paragraph), the condition should be managed by a clinician experienced with the disease—likely, a gastroenterologist.
Sequelae. The impact of this maternal disorder on the fetus can be disastrous: a 10% to 15% rate of perinatal death, and a 30% to 40% rate of premature labor.14 Fortunately, the rate of preterm labor correlates strongly with the level of bile acids, so that as bile acid levels are reduced with UDCA treatment, the rate of preterm labor also falls. Management of ICP has reduced the rate of perinatal death to 3.5%. No evidence of fetal growth retardation has been noted.14
DERMATOSES TRIGGERED BY PREGNANCY
4. Eczema of pregnancy/prurigo of pregnancy
Eczema of pregnancy/prurigo of pregnancy (EP/PP) may not actually be correlated with the pregnant state. Both conditions manifest as eczematous lesions in an atopic distribution. Although they have been described in the literature as separate entities, the lack of clinical distinction between them led Ambros-Rudolph and colleagues to combine them under the umbrella term, atopic eruption of pregnancy.23
In some patients, at least, EP/PP may be preexisting conditions that are exacerbated by pregnancy. One study of 255 patients with the condition found that 20% had had the lesions before they became pregnant.23 The tendency of the condition to be made markedly worse by pregnancy, however, leads us to include it here.
PP has an estimated incidence of 1 in 450 pregnancies.11 Although many authorities consider EP to be the most common dermatosis of pregnancy, no clear estimation of its prevalence has been established.23,24 Taken together, the two conditions have the highest prevalence of all pregnancy-induced dermatoses.
PP is also known as popular dermatitis of Spangler, Nurse’s early prurigo of pregnancy, and linear IgM disease of pregnancy.3,4,23
Presentation. The typical presentation is grouped, crusted, erythematous papules, patches, and plaques—frequently with excoriations. Lesions typically present on the extensor surfaces of the arms and legs or on the abdomen (FIGURE 4).4 Recurrence in later pregnancies is common.
FIGURE 4 Eczema of pregnancy
EP/PP typically manifests as grouped, crusted, erythematous papules, patches, and plaques, often with excoriations.
Pathophysiology. The pathophysiology of EP/PP is not understood. Many patients who develop EP/PP have a history of atopy.10
Differential diagnosis. Conditions that need to be considered in making the diagnosis include Tinea infection, scabies, contact dermatitis, ICP, pruritic folliculitis of pregnancy (PFP), and PG.
Diagnosis. The history and the physical examination determine the diagnosis. Serology, histopathology, and immunofluorescence are nonspecific. Correlation of EP/PP with increased IgE is marginal, at best.24,25
Treatment. These conditions are treated symptomatically with topical corticosteroids or systemic antihistamines.
Sequelae. No increase in maternal or fetal morbidity or mortality is associated with EP/PP.
5. Acute pustular psoriasis of pregnancy
Whether or not acute pustular psoriasis of pregnancy (APPP) is actually a pregnancy-induced dermatosis is subject to debate.
There is evidence that APPP is not unique to pregnancy but simply a manifestation of ordinary psoriasis. Clinically and histologically, APPP is indistinguishable from pustular psoriasis. Unlike most cases of acute psoriasis, however, APPP often appears in pregnancy without any personal or family history of psoriasis and usually ceases when the pregnancy ends. This fact, combined with reports of increased fetal and maternal morbidity and mortality associated with APPP, leads us to include it here.26
Presentation. APPP is a rare condition that may have an onset at any point in pregnancy. Characteristic lesions begin as erythematous plaques with pustules on the inner thighs, flexural areas, and groin and spread to the trunk and extremities. As plaques enlarge, their center becomes eroded and crusted.
The nails may become onycholytic. Hands, feet, and face are usually spared. Oral and esophageal erosions can occur. Pruritus is typically mild, although the lesions are often painful. Flu-like symptoms are often present.27
Pathophysiology. The pathophysiology of APPP is unknown.
Differential diagnosis. Conditions with similar presentations include an adverse drug reaction, pityriasis rosea, lichen simplex chronicus, eczema, lupus, and pityriasis rubra pilaris.
Diagnosis. The clinical history and an association with systemic illness are the basis for a diagnosis of APPP. Cultures of pustules are negative for any infective pathology, although, as the disease progresses, pustules may become superinfected. Laboratory testing may show an increased erythrocyte sedimentation rate, hypocalcemia, and a low level of vitamin D.
Treatment. Prednisone, 15 to 60 mg/d, is often sufficient to control the disease.27 Cyclosporine, 100 mg twice daily, has also been shown to be useful.28 Cyclosporine in pregnancy is a Category C drug. Data on fetal malformation associated with cyclosporine therapy are limited, but risk appears minimal.6
Maternal hypocalcemia should be monitored and treated appropriately. If disease progression is judged serious enough, early induction of labor is indicated, because delivery will almost always lead to swift resolution.
Sequelae. A number of case reports link APPP to serious sequelae, including fetal growth retardation, hypocalcemia, and stillbirth.27,29,30 The condition is too rare, however, for good data on specific sequelae. Although APPP does give significant cause for concern, it appears that some of the traditional apprehension comes from older publications reporting a rate of maternal mortality of 70% to 90%.31 This statistic has not been borne out in practice. It does appear that the mother will frequently suffer systemic symptoms, including fever and malaise.
6. Pruritic folliculitis of pregnancy
Accounts of the prevalence of pruritic folliculitis of pregnancy (PFP) vary widely. Some sources report fewer than 30 cases in all of the literature; others indicate that the prevalence is equivalent to that of PG—one in every 10,000 pregnancies.3,11 PFP most often presents in the third trimester. It often resolves before delivery, but uniformly clears within 2 weeks of delivery.
Presentation. PFP presents as papules and pustules concentrated around hair follicles (FIGURE 5). Often, lesions begin on the abdomen and spread to the extremities.24,28 The condition is often, but not always, pruritic. Patients are more likely to be concerned about what the condition means for their health than distressed by the symptoms.
FIGURE 5 Pruritic folliculitis of pregnancy
The papules and pustules of PFP are concentrated around hair follicles.
Pathophysiology. Like many other dermatoses of pregnancy, the pathophysiology of PFP is unknown. There is little evidence that the condition is immunologically or hormonally mediated, and there is no evidence of an infectious component.24,28
Differential diagnosis. PFP must be distinguished from infectious folliculitis, acneiform disorders, HIV-associated eosinophilic folliculitis, and a drug reaction.
Diagnosis. The clinical diagnosis is based on presenting symptoms and third-trimester onset. No specific laboratory or histologic analysis can be used to make a definitive diagnosis.
Treatment. As the condition is, by definition, a nonmicrobial folliculitis, the most effective therapy tends to be with a low- or midpotency topical corticosteroid, such as triamcinolone or desonide. A benzoyl peroxide wash can also be effective.
Sequelae. One study reports an increased incidence of low birth weight, but no associated morbidity or mortality has been reported in recent studies.24
- Pemphigoid gestationis is best managed with oral prednisone at doses from 20 to 60 mg per day to control symptoms
- The pruritus associated with pruritic urticarial papules and plaques of pregnancy can be safely and effectively managed with topical corticosteroids and oral antihistamines
- Treat intrahepatic cholestasis of pregnancy with ursodeoxycholic acid, which likely reduces serum bile acids as well as associated fetal morbidity and mortality
1. Sitaru C, Powell J, Messer G, Bröcker EB, Wojnarowska F, Zillikens D. Immunoblotting and enzyme linked immunosorbent assay for the diagnosis of pemphigoid gestationis. Obstet Gynecol. 2004;103(4):757-763.
2. Engineer L, Bohl K, Ahmed AR. Pemphigoid gestationis: a review. Am J Obstet Gynecol. 2000;183(2):483-491.
3. Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence-based systematic review. Am J Obstet Gynecol. 2003;188(4):1083-1092.
4. Shornick JK. Dermatoses of pregnancy. Semin Cutan Med Surg. 1998;17(3):172-181.
5. Shornick JK, Bangert JL, Freeman RG, Gilliam JN. Herpes gestationis: clinical and histological features in 28 cases. J Am Acad Dermatol. 1983;8(2):214-224.
6. Leachman SA, Reed BR. The use of dermatologic drugs in pregnancy and lactation. Dermatol Clin. 2006;24(2):167-197, vi.
7. Hern S, Harman K, Bhogal BS, Black MM. A severe persistent case of pemphigoid gestationis treated with intravenous immunoglobulins and cyclosporine. Clin Exp Dermatol. 1998;23(4):185-188.
8. Fitzpatrick TP. Diseases in pregnancy. Color Atlas and Synopsis of Clinical Dermatology. New York, NY: McGraw Hill; 1997:414–419.
9. Aractingi D, Berkane N, Bertheau P, et al. Fetal DNA in skin of polymorphic eruptions of pregnancy. Lancet. 1998;352(9144):1898-1901.
10. Rudolph CM, Al-Fares S, Vaughan-Jones SA, et al. Polymorphic eruption of pregnancy: clinicopathology and potential trigger factors in 181 patients. Br J Dermatol. 2006;154(1):54-60.
11. Roger D, Vaillant L, Fignon A, et al. Specific pruritic diseases of pregnancy. A prospective study of 3192 pregnant women. Arch Dermatol. 1994;130(6):734-739.
12. McDonald JA. Cholestasis of pregnancy. J Gastroenterol Hepatol. 1999;14(6):515-518.
13. Ohel I, Levy A, Silberstein T, Holcberg G, Sheiner E. Pregnancy outcome of patients with pruritic urticarial papules and plaques of pregnancy. J Maternal Fetal Neonat Med. 2006;19(5):305-308.
14. Geenes V, Williamson C. Intrahepatic cholestasis of pregnancy. World J Gastroenterol. 2009;15(17):2049-2066.
15. Shaw D, Frohlich J, Wittmann BA, Willms M. A prospective study of 18 patients with cholestasis of pregnancy. Am J Obstet Gynecol. 1982;142(6 Pt 1):621-625.
16. Reyes H. The spectrum of liver and gastrointestinal disease seen in cholestasis of pregnancy. Gastroenterol Clin North Am. 1992;21(4):905-921.
17. Lammert F, Marschall H, Glantz A, Matern S. Intrahepatic cholestasis of pregnancy; molecular pathogenesis, diagnosis and management. J Hepatol. 2000;33(6):1012-1021.
18. Huang WM, Seubert DE, Donnelly JG, Liu M, Javitt NB. Intrahepatic cholestasis of pregnancy: detection with urinary bile acid assays. J Perinat Med. 2007;35(6):486-491.
19. Palma J, Reyes H, Ribalta J, et al. Ursodeoxycholic acid in the treatment of intrahepatic cholestasis of pregnancy: a randomized, double-blind study controlled with placebo. J Hepatol. 1997;27(6):1022-1028.
20. Diaferia A, Nicastri PL, Tartagni M, et al. Ursodeoxycholic acid therapy in pregnant women with cholestasis. Int J Gynaecol Obstet. 1996;52:133-140.
21. Nicastri PL, Diaferia A, Tartagni M, Loizzi P, Fanelli M. A randomised placebo-controlled trial of ursodeoxycholic acid and S-adenosylmethionine in the treatment of intrahepatic cholestasis of pregnancy. Br J Obstet Gynaecol. 1998;105(11):1205-1207.
22. Glantz A, Marschall HU, Lammert F, Mattsson LA. Intrahepatic cholestasis in pregnancy: a randomized controlled trial comparing dexamethasone and ursodeoxycholic acid. Hepatology. 2005;42(6):1399-1405.
23. Ambros-Rudolph C, Müllegger M, Vaughan-Jones S, et al. The specific dermatoses of pregnancy revisited and reclassified: results of a retrospective two-center study on 505 pregnant patients. J Am Acad Dermatol. 2006;54(3):395-404.
24. Vaughan Jones SA, Hern S, Nelson-Piercy C, Seed PT, Black MM. A prospective study of 200 women with dermatoses of pregnancy correlating clinical findings with hormonal and immunopathological profiles. Br J Dermatol. 1999;141(1):71-81.
25. Holmes RC, Black MM. The specific dermatoses of pregnancy. J Am Acad Dermatol. 1983;8(3):405-412.
26. Bukhari IA. Impetigo herpetiformis in a primagravida: successful treatment with etretinate. J Drugs Dermatol. 2004;3(4):449-451.
27. Oumeish OY, Parish JL. Impetigo herpetiformis. Clin Dermatol. 2006;24(2):101-104.
28. Kroumpouzos G, Cohen LM. Pruritic folliculitis of pregnancy. J Am Acad Dermatol. 2000;43(1 Pt 1):132-134.
29. Sahin HG, Sahin HA, Metin A, Zeteroglu S, Ugras S. Recurrent impetigo herpetiformis in a pregnant adolescent: case report. Eur J Obstet Gynecol Reprod Biol. 2002;101:201-203.
30. Aka N, Kuscu NK, Yazicioglu E. Impetigo herpetiformis at the 36th week of gestation. Int J Gynecol Obstet. 2000;69(2):153-154.
31. Wade T, Wade S, Jones H. Skin changes and diseases associated with pregnancy. Obstet Gynecol. 1978;52(2):233-242.
1. Sitaru C, Powell J, Messer G, Bröcker EB, Wojnarowska F, Zillikens D. Immunoblotting and enzyme linked immunosorbent assay for the diagnosis of pemphigoid gestationis. Obstet Gynecol. 2004;103(4):757-763.
2. Engineer L, Bohl K, Ahmed AR. Pemphigoid gestationis: a review. Am J Obstet Gynecol. 2000;183(2):483-491.
3. Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence-based systematic review. Am J Obstet Gynecol. 2003;188(4):1083-1092.
4. Shornick JK. Dermatoses of pregnancy. Semin Cutan Med Surg. 1998;17(3):172-181.
5. Shornick JK, Bangert JL, Freeman RG, Gilliam JN. Herpes gestationis: clinical and histological features in 28 cases. J Am Acad Dermatol. 1983;8(2):214-224.
6. Leachman SA, Reed BR. The use of dermatologic drugs in pregnancy and lactation. Dermatol Clin. 2006;24(2):167-197, vi.
7. Hern S, Harman K, Bhogal BS, Black MM. A severe persistent case of pemphigoid gestationis treated with intravenous immunoglobulins and cyclosporine. Clin Exp Dermatol. 1998;23(4):185-188.
8. Fitzpatrick TP. Diseases in pregnancy. Color Atlas and Synopsis of Clinical Dermatology. New York, NY: McGraw Hill; 1997:414–419.
9. Aractingi D, Berkane N, Bertheau P, et al. Fetal DNA in skin of polymorphic eruptions of pregnancy. Lancet. 1998;352(9144):1898-1901.
10. Rudolph CM, Al-Fares S, Vaughan-Jones SA, et al. Polymorphic eruption of pregnancy: clinicopathology and potential trigger factors in 181 patients. Br J Dermatol. 2006;154(1):54-60.
11. Roger D, Vaillant L, Fignon A, et al. Specific pruritic diseases of pregnancy. A prospective study of 3192 pregnant women. Arch Dermatol. 1994;130(6):734-739.
12. McDonald JA. Cholestasis of pregnancy. J Gastroenterol Hepatol. 1999;14(6):515-518.
13. Ohel I, Levy A, Silberstein T, Holcberg G, Sheiner E. Pregnancy outcome of patients with pruritic urticarial papules and plaques of pregnancy. J Maternal Fetal Neonat Med. 2006;19(5):305-308.
14. Geenes V, Williamson C. Intrahepatic cholestasis of pregnancy. World J Gastroenterol. 2009;15(17):2049-2066.
15. Shaw D, Frohlich J, Wittmann BA, Willms M. A prospective study of 18 patients with cholestasis of pregnancy. Am J Obstet Gynecol. 1982;142(6 Pt 1):621-625.
16. Reyes H. The spectrum of liver and gastrointestinal disease seen in cholestasis of pregnancy. Gastroenterol Clin North Am. 1992;21(4):905-921.
17. Lammert F, Marschall H, Glantz A, Matern S. Intrahepatic cholestasis of pregnancy; molecular pathogenesis, diagnosis and management. J Hepatol. 2000;33(6):1012-1021.
18. Huang WM, Seubert DE, Donnelly JG, Liu M, Javitt NB. Intrahepatic cholestasis of pregnancy: detection with urinary bile acid assays. J Perinat Med. 2007;35(6):486-491.
19. Palma J, Reyes H, Ribalta J, et al. Ursodeoxycholic acid in the treatment of intrahepatic cholestasis of pregnancy: a randomized, double-blind study controlled with placebo. J Hepatol. 1997;27(6):1022-1028.
20. Diaferia A, Nicastri PL, Tartagni M, et al. Ursodeoxycholic acid therapy in pregnant women with cholestasis. Int J Gynaecol Obstet. 1996;52:133-140.
21. Nicastri PL, Diaferia A, Tartagni M, Loizzi P, Fanelli M. A randomised placebo-controlled trial of ursodeoxycholic acid and S-adenosylmethionine in the treatment of intrahepatic cholestasis of pregnancy. Br J Obstet Gynaecol. 1998;105(11):1205-1207.
22. Glantz A, Marschall HU, Lammert F, Mattsson LA. Intrahepatic cholestasis in pregnancy: a randomized controlled trial comparing dexamethasone and ursodeoxycholic acid. Hepatology. 2005;42(6):1399-1405.
23. Ambros-Rudolph C, Müllegger M, Vaughan-Jones S, et al. The specific dermatoses of pregnancy revisited and reclassified: results of a retrospective two-center study on 505 pregnant patients. J Am Acad Dermatol. 2006;54(3):395-404.
24. Vaughan Jones SA, Hern S, Nelson-Piercy C, Seed PT, Black MM. A prospective study of 200 women with dermatoses of pregnancy correlating clinical findings with hormonal and immunopathological profiles. Br J Dermatol. 1999;141(1):71-81.
25. Holmes RC, Black MM. The specific dermatoses of pregnancy. J Am Acad Dermatol. 1983;8(3):405-412.
26. Bukhari IA. Impetigo herpetiformis in a primagravida: successful treatment with etretinate. J Drugs Dermatol. 2004;3(4):449-451.
27. Oumeish OY, Parish JL. Impetigo herpetiformis. Clin Dermatol. 2006;24(2):101-104.
28. Kroumpouzos G, Cohen LM. Pruritic folliculitis of pregnancy. J Am Acad Dermatol. 2000;43(1 Pt 1):132-134.
29. Sahin HG, Sahin HA, Metin A, Zeteroglu S, Ugras S. Recurrent impetigo herpetiformis in a pregnant adolescent: case report. Eur J Obstet Gynecol Reprod Biol. 2002;101:201-203.
30. Aka N, Kuscu NK, Yazicioglu E. Impetigo herpetiformis at the 36th week of gestation. Int J Gynecol Obstet. 2000;69(2):153-154.
31. Wade T, Wade S, Jones H. Skin changes and diseases associated with pregnancy. Obstet Gynecol. 1978;52(2):233-242.
The skin disorders of pregnancy: A family physician’s guide
• Pemphigoid gestationis is best managed with oral prednisone at doses from 20 to 60 mg per day to control symptoms. B
• The pruritus associated with pruritic urticarial papules and plaques of pregnancy can be safely and effectively managed with topical corticosteroids and oral antihistamines. A
• Treat intrahepatic cholestasis of pregnancy with ursodeoxycholic acid, which likely reduces serum bile acids as well as associated fetal morbidity and mortality. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The dermatoses of pregnancy are a poorly understood group of dermatologic conditions. The only thing they have in common is a tendency to appear during pregnancy.
Only 3 are considered unique to pregnancy, however; the others are probably exacerbations of preexisting conditions triggered by pregnancy. There isn’t even complete agreement on what to call them. And to make management even more complex, 2 patients—the mother and the fetus—need to be considered.
Who will manage these patients is another matter. These conditions fall into an overlapping area of health care, where family physicians, obstetricians, and dermatologists all have some share in the responsibility for diagnosis and treatment. As a family physician who probably cares for any number of pregnant patients on a weekly basis, you need to be sufficiently familiar with these conditions so that you can differentiate those that can be treated symptomatically and those that require a referral to a specialist. This review and handy TABLE will help you toward that end.
TABLE
Skin disorders of pregnancy: What you’ll see, how to treat
| Disorder | Lesions | Diagnosis and sequelae | Treatment | Recurrence |
|---|---|---|---|---|
| PG3,5 | Erythematous papules, progressing to vesicles, bullae; periumbilical distribution, sparing face, palms, and soles | Mean onset at 21 weeks; postpartum in 20% of cases. Direct immunofluorescence microscopy shows linear C3 deposition. Newborn may be small for gestational age, but no associated morbidity or mortality | Oral corticosteroids 20-60 mg/d, IVIG, or cyclosporine in refractory cases | Frequent. Skips a pregnancy 8% of the time |
| PUPPP8-10 | Urticarial papules and plaques on abdomen, legs, arms, buttocks, chest, and back | Usually present after 34th week, but can present at any stage. Diagnosis is clinical. No increase in fetal morbidity or mortality | Topical steroids and antihistamines | Uncommon |
| ICP14,17,19-22 | No primary lesions; secondary excoriations in any area patient can reach | Onset after 30th week in 80% of patients. Strongly indicated by serum bile acid >11 mcmol/L. Increased fetal mortality | Ursodeoxycholic acid 450-1200 mg/d | Frequent |
| EP/PP4,10,24 | Grouped, crusted erythematous papules, patches, and plaques, most commonly on extensor surfaces of arms and legs or on abdomen | Onset at any point in pregnancy. Clinical diagnosis. No increase in fetal morbidity or mortality | Symptomatic treatment with topical steroids or antihistamines | Frequent |
| APPP27-29 | Erythematous plaques and pustules starting on inner thighs and groin and spreading to trunk and extremities | Onset at any point in pregnancy. Clinical diagnosis by appearance of lesions and association with systemic illness. Increased incidence of miscarriage and stillbirth, and maternal mortality | Prednisone 15-60 mg/d, cyclosporine 100 mg twice daily in refractory cases, management of associated hypocalcemia | Unknown |
| PFP24,29 | Papules and pustules concentrated around hair follicles, often beginning on abdomen and spreading to extremities | Onset most often in third trimester. Clinical diagnosis. No associated fetal morbidity or mortality | Topical steroids | Unknown |
| APPP, acute pustular psoriasis of pregnancy; EP/PP, eczema of pregnancy/pruritus of pregnancy; ICP, intrahepatic cholestasis of pregnancy; IVIG, intravenous immunoglobulin; PFP, pruritic folliculitis of pregnancy; PG, pemphigoid gestationis; PUPPP, pruritic urticarial papules and plaques of pregnancy. | ||||
Dermatoses unique to pregnancy
Pemphigoid gestationis
Years ago, this disorder was referred to as herpes gestationis, because the lesions are herpetiform. Pemphigoid gestationis (PG) has an incidence of approximately 1 in 10,000 pregnancies.1,2 The time of onset is usually about the 21st week of gestation, although in about 20% of cases, the eruption appears immediately postpartum.3
Presentation. The disease usually begins with urticarial papules and plaques around the umbilicus and extremities. Bullous lesions tend to develop as the disease progresses, and are often not present on first presentation (FIGURE 1). PG lesions tend to spare the face, palms, and soles. Mucosal surfaces are involved in less than 20% of cases. In about 75% of cases, PG flares around the time of delivery, regressing spontaneously after the baby is born.4
Pathophysiology. The pathophysiology is nearly identical to that of bullous pemphigoid, a blistering skin disorder more often seen in elderly patients.5 Pemphigoid disorders are immune processes, involving an immunoglobulin G (IgG) immune response directed at a 180-kDa hemidesmosome transmembrane glycoprotein. This protein is the common target of several subepidermal blistering diseases.
Differential. Disorders that may have some of the same features as PG include pruritic urticarial papules and plaques of pregnancy (PUPPP), erythema multiforme, intrahepatic cholestasis of pregnancy (ICP), contact dermatitis, and drug reactions.
Diagnosis. A biopsy is necessary for the definitive diagnosis. Direct immunofluorescence (DIF) microscopy of a sample of perilesional skin can show tissue-bound immunoreactants. Linear deposition of the complement component protein C3 along the basement membrane zone is diagnostic for PG. IgG is also deposited about 40% of the time.3 Serum enzyme-linked immunosorbent assay (ELISA) studies are also helpful in diagnosis. They have excellent sensitivity and specificity, as well as the capacity to monitor levels of antibody, which correlate with the severity of disease.1
Treatment. Oral corticosteroids are the first-line treatment for PG, typically 20 to 60 mg per day of prednisone. Oral corticosteroids are typically most effective in ameliorating the patient’s symptoms. Prednisone at doses of 40 to 80 mg per day for a short time has not been associated with congenital abnormalities.6 PG patients can also be treated successfully with intravenous immunoglobulin (IVIG) and cyclosporine in refractory cases.7
Pruritus associated with this condition can interfere with day-to-day activity and with the patient’s ability to sleep. Patients may also complain that the rash is painful, particularly if bullae rupture, leading to superficial ulcerations. Fortunately, the patient’s quality of life can be dramatically improved with systemic corticosteroids—with no significant risk to the fetus.
Sequelae. PG uniformly resolves within a few weeks, but the mother’s autoantibodies can passively be transferred to the fetus, causing vesicles and bullae in the newborn.8 An increased incidence of small-for-gestational age (SGA) infants has also been noted in PG, although no lasting morbidity or mortality in the offspring has been noted.5 This disease tends to recur with future pregnancies, but will skip a pregnancy 8% of the time.5
FIGURE 1
Pemphigoid gestationis
Pruritic urticarial papules and plaques of pregnancy
This condition is known by many names besides PUPPP: polymorphic eruption of pregnancy, toxemic erythema of pregnancy, and late prurigo of pregnancy.1 It is a pruritic, inflammatory skin disorder that has been variously estimated to occur in anywhere from 1 in 120 to 1 in 240 pregnancies.8 PUPPP is second only to eczema as the most common dermatosis of pregnancy.
Presentation. As the name implies, the lesions of PUPPP are itchy, red papules that often coalesce into plaques (FIGURE 2). The lesions usually occur in primigravidas after the 34th week of gestation, although they may be seen at any time from the first trimester through the postpartum period.9
Lesions are classically found on the abdomen, sparing the umbilical area, and are found primarily in the striae. This distribution helps to differentiate PUPPP from PG, where the lesions typically cluster around the umbilicus. Most PUPPP lesions (80% in 1 study) are dispersed on the abdomen, legs, arms, buttocks, chest, and back. Another 17% appear only on the abdomen and proximal thighs, and the remaining 3% on the limbs.10 Nearly 50% of the time, lesions also include discrete vesicles.11 There are no reported cases of mucosal involvement.
Patients with this condition are often very uncomfortable. The associated pruritus is severe enough to interfere with sleep. Despite the itching, however, lesions are seldom excoriated.
Pathophysiology. The disorder has been strongly associated with maternal weight gain and multiple gestations. One working hypothesis is that rapid abdominal distention observed in the third trimester leads to damage of the connective tissue, which then releases antigenic molecules, causing an inflammatory reaction.12 Another hypothesis is that increased levels of fetal DNA that have been detected in the skin of PUPPP patients may contribute to the pathology. One study detected male DNA in 6 of 10 PUPPP sufferers, but found none in any of 26 controls— pregnant women without PUPPP pathology.5 There is some evidence that patients with atopy may be predisposed to PUPPP, as well as patients who are hypertensive or obese.10,13
Differential. Initially, PUPPP lesions can be difficult to differentiate from urticarial PG lesions. The distribution of the lesions is the best clue: PG lesions cluster around the umbilicus, whereas PUPPP lesions uniformly spare the umbilical area. Additional disorders in the PUPPP differential are atopic dermatitis, superficial urticarial allergic eruption, viral exanthema, and contact or irritant dermatitis.
Diagnosis. PUPPP can only be diagnosed through clinical observation. None of the available laboratory tests—immunofluorescence, histology, serology—yield findings specific for PUPPP, although histology and immunofluorescence can readily differentiate between this condition and PG.
Treatment. Because the disease holds no real danger for mother or fetus, treatment can be aimed solely at symptomatic relief. Mild to potent topical steroids (consider triamcinolone or fluocinonide) should relieve pruritus within 48 to 72 hours.8 Antihistamines and occasionally low-dose systemic steroids may also be used. Consider hydroxyzine, although diphenhydramine has the more proven safety profile in pregnancy.
Nonpharmacologic treatments such as oil baths and emollients should also be considered. If the condition appears classic for PUPPP, it can be managed symptomatically. However, if there is any question about the diagnosis, referral to a dermatologist is prudent.
Sequelae. No increase in maternal or fetal morbidity or mortality is associated with PUPPP. Recurrence is fairly uncommon, as the disease primarily affects women during their first pregnancy.
FIGURE 2
Pruritic urticarial papules and plaques of pregnancy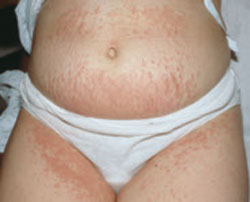
Intrahepatic cholestasis of pregnancy
This condition is also called recurrent or idiopathic jaundice of pregnancy, obstetric cholestasis, and pruritus gravidarum. ICP is caused by disruption of hepatic bile flow during pregnancy. It has been recorded at a rate of approximately 10 to 150 per 10,000 pregnancies in Europe and 70 per 10,000 in the United States.12 In 80% of patients, the time of onset is after the 30th week.14 Although this disorder is not primarily a dermatosis of pregnancy, it is a pruritic condition that often presents with excoriations in pregnant women and is associated with fetal morbidity and mortality. It’s important to be able to identify this disease early to minimize sequelae.
Presentation. There are no primary lesions with ICP. The primary presenting symptom is a generalized pruritus affecting the palms and soles, and sometimes extending to the legs and abdomen (FIGURE 3). This itching is often so severe that it leads to chronic insomnia. You may see secondary skin lesions, such as erythema and excoriations. Observable jaundice occurs in 10% to 20% of patients.3 These patients do not develop the encephalopathy that is associated with cholestasis in the nonpregnant state, however.14
Pathophysiology. The genesis of this condition is thought to be a combination of genetic and environmental factors. A family history of the disorder is present in half the cases, and cases with a familial component tend to be more severe.15 ICP may be an exaggerated response to increased estrogen levels in pregnancy, but the mechanism of this response is unknown.16
Differential. Other conditions that must be considered in making the diagnosis are viral hepatitis, gallbladder disease, PG, PUPPP, drug hepatotoxicity, primary biliary cirrhosis, and uremia.
Diagnosis. Laboratory values are the definitive diagnostic tool in this condition. Increased serum bile acids are the single most sensitive test. Average levels of serum bile acids in pregnancy are 6.6 mcmol/L, with an upper limit of 11. The average value in women with ICP is 47 mcmol/L.17
While serum acids remain the gold standard, a recent study showed elevated urine bile acids to have 100% sensitivity and 83% specificity for ICP.18 In 55% to 60% of cases, the liver enzymes aspartate aminotransferase (AST) and alanine aminotransferase (ALT) are mildly increased. Steatorrhea is often noted by the patient, and is followed by vitamin K deficiency.17
Treatment. The current standard of care for ICP is treatment with ursodeoxycholic acid (UDCA). In 4 controlled trials, UDCA showed a sustained decrease in serum bile acids.19-22 Doses used in these trials have varied between 450 and 1200 mg per day.
Before UDCA treatment was available, the disorder was treated with cholestyramine, which could bring about a 70% rate of response. The drawback to cholestyramine treatment is that it precipitates vitamin K, which is already compromised by the disease process. Further, its onset of action is slow.3
Elective delivery is indicated for ICP, particularly in patients with significant clinical presentations.12 Delivery for ICP should be performed around week 37 to 38, as stillbirths tend to cluster around weeks 37 to 39.14 Given the significant fetal mortality associated with this condition (see below), ICP should be managed by a clinician experienced with the disease, likely a gastroenterologist.
Sequelae. The impact of this maternal disorder on the fetus can be disastrous: a 10% to 15% rate of perinatal death, and a 30% to 40% rate of premature labor have been seen with ICP.14 Fortunately, rates of preterm labor are strongly correlated with levels of bile acids, so that as bile acid levels are reduced with UDCA treatment, rates of preterm labor also go down. Currently, management of the condition has reduced rates of perinatal death to 3.5%. There is no evidence of fetal growth retardation.14
FIGURE 3
Intrahepatic cholestasis of pregnancy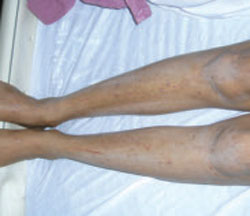
Dermatoses triggered by pregnancy
Eczema of pregnancy/prurigo of pregnancy
Eczema of pregnancy/prurigo of pregnancy (EP/PP) may not actually be correlated with the pregnant state. Both conditions manifest as eczematous lesions in an atopic distribution. Although they have been described in the literature as separate entities, the lack of clinical distinction between them led Ambros-Rudolph and colleagues to combine them under the umbrella term, atopic eruption of pregnancy.23 In some patients, at least, they may be preexisting conditions that pregnancy exacerbates. One study of 255 patients with the condition found that 20% had had the lesions before they became pregnant.23 However, the tendency of the condition to be markedly worsened by pregnancy leads us to include it here.
PP has an estimated incidence of 1 in 450 pregnancies.11 But while many authorities consider EP to be the most common dermatosis of pregnancy, no clear estimation of its prevalence has been established.23,24 Taken together, these 2 conditions have the highest prevalence of all pregnancy-induced dermatoses. PP is also known as popular dermatitis of Spangler, Nurse’s early prurigo of pregnancy, and linear IgM disease of pregnancy.3,23,25
Presentation. The typical presentation consists of grouped, crusted, erythematous papules, patches, and plaques—frequently with excoriations. The lesions typically present on the extensor surfaces of the arms and legs or on the abdomen (FIGURE 4).4 Recurrence in later pregnancies is common.
Pathophysiology. The pathophysiology of EP/PP is not understood. Many patients who present with EP/PP have a history of atopy.10
Differential. Conditions that need to be considered in making the diagnosis include tinea infection, scabies, contact dermatitis, ICP, pruritic folliculitis of pregnancy (PFP), and PG.
Diagnosis. History and physical examination determine the diagnosis. Serology, histopathology, and immunofluorescence are not specific, and correlation with increased IgE is marginal, at best.24,26
Treatment. These conditions are treated symptomatically with topical steroids or systemic antihistamines.
Sequelae. No maternal or fetal increase in morbidity or mortality is associated with these conditions.
FIGURE 4
Eczema of pregnancy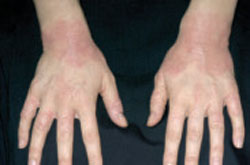
Acute pustular psoriasis of pregnancy
Whether or not APPP is actually a pregnancyinduced dermatosis is subject to debate.
There is evidence that APPP is not unique to pregnancy, but is simply a manifestation of ordinary psoriasis. Clinically and histologically, APPP is indistinguishable from pustular psoriasis. Unlike most cases of acute psoriasis, however, APPP often appears in pregnancy without any personal or family history of psoriasis, and usually ceases when the pregnancy is concluded. This fact, combined with reports of increased fetal and maternal morbidity and mortality associated with APPP, lead us to include it here.27
Presentation. APPP is a rare condition that may have an onset at any point in pregnancy. The characteristic lesions begin as erythematous plaques with pustules on the inner thighs, flexural areas, and groin and spread to the trunk and extremities. As the plaques enlarge, the center becomes eroded and crusted. Nails may become onycholytic. The hands, feet, and face are usually spared. Oral and esophageal erosions can occur. Pruritus is typically mild, although the lesions are often painful and flu-like symptoms are often present.28
Pathophysiology. The pathophysiology of this disease is unknown.
Differential. Conditions with similar presentations include an adverse drug reaction, pityriasis rosea, lichen simplex chronicus, eczema, lupus, and pityriasis rubra pilaris.
Diagnosis. Clinical history and association with systemic illness are the basis for a diagnosis of APPP. Cultures of the pustules are negative for any infective pathology, though as the disease progresses, pustules may become superinfected. Lab tests may show an increased erythrocyte sedimentation rate (ESR), hypocalcemia, and low levels of vitamin D.
Treatment. Prednisone 15 to 60 mg per day is often sufficient to control the disease.28 Cyclosporine 100 mg twice daily has also been shown to be useful.29 Cyclosporine in pregnancy is a category C drug. Data on fetal malformation associated with cyclosporine therapy are limited, but the risk appears to be minimal.6 Maternal hypocalcemia should be monitored and treated appropriately. If disease progression is judged serious enough, early induction of labor is indicated, since delivery will almost always lead to swift resolution.
Sequelae. There have been a number of case reports that link APPP to serious sequelae, including fetal growth retardation, hypocalcemia, and stillbirth.28,30,31 The condition is too rare, however, for good data on specific sequelae. While the disease does give significant cause for concern, it would appear that some of the traditional apprehension comes from older publications reporting a rate of maternal mortality of 70% to 90%.32 This statistic has not been borne out in clinical practice. It does appear that the mother will frequently suffer from systemic symptoms, including fever and malaise.
Pruritic folliculitis of pregnancy
Accounts of PFP’s prevalence vary widely: Some sources report fewer than 30 cases in all of the literature, while others indicate that the prevalence is equivalent to that of PG, 1 in 10,000.3,11 PFP most commonly presents in the third trimester. It often resolves before delivery, but uniformly clears within 2 weeks of delivery.
Presentation. PFP presents as papules and pustules concentrated around hair follicles (FIGURE 5). Often lesions begin on the abdomen and spread to the extremities.24,29 The condition is often, but not always, pruritic. Patients are more likely to be concerned about what the condition means for their health, rather than being distressed by the symptoms.
Pathophysiology. Like so many of these conditions, the pathophysiology of PFP is unknown. There is little evidence that the condition is immunologically or hormonally mediated, and there is no evidence of an infectious component.24,29
Differential. PFP must be distinguished from infectious folliculitis, acneiform disorders, HIV-associated eosinophilic folliculitis, and a drug reaction.
Diagnosis. The clinical diagnosis is based on presenting symptoms and third trimester onset. No specific lab or histological analysis can be used to make a definitive diagnosis.
Treatment. As the condition is, by definition, a nonmicrobial folliculitis, the most effective therapy tends to be with mid- to lowpotency topical steroids such as triamcinolone or desonide. Additionally, benzoyl peroxide wash can be effective.
Sequelae. One study reports an increased incidence of low birth weight, but currently no associated morbidity or mortality has been reported.24
FIGURE 5
Pruritic folliculitis of pregnancy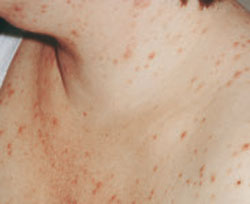
CORRESPONDENCE
Matthew Bremmer, MD, 419 W Redwood Street, Department of Dermatology, Baltimore, MD 21230; Mbrem001@umaryland.edu
1. Cassian S, Powell J, Messer G, et al. Immunoblotting and enzymelinked immunosorbent assay for the diagnosis of pemphigoid gestationis. Obstet Gynecol. 2004;103:757-763.
2. Engineer L, Bohl K, Ahmed AR. Pemphigoid gestationis: a review. Am J Obstet Gynecol. 2000;183:483-491.
3. Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence based systematic review. Am J Obstet Gynecol, 2003;188:1083-1092.
4. Shornick JD. Dermatoses of pregnancy. Semin Cutan Med Surg. 1998;17:172-181.
5. Shornick JK, Bangert JL, Freeman RG, et al. Herpes gestationis: clinical and histological features in 28 cases. J Am Acad Dermatol. 1983;8:214-224.
6. Leachman S, Reed B. The use of dermatologic drugs in pregnancy and lactation. Dermatol Clin. 2004;24:167-197.
7. Hern S, Harman K, Bhogal BS, et al. A severe persistent case of pemphigoid gestationis treated with intravenous immunoglobulins and cyclosporine. Clin Exp Dermatol. 1998;23:185-188.
8. Fitzpatrick TP. Diseases in pregnancy. Color Atlas and Synopsis of Clinical Dermatology. New York, NY: McGraw Hill; 1997:414–419.
9. Aractingi D, Berkane N, Bertheau P, et al. Fetal DNA in skin of polymorphic eruptions of pregnancy. Lancet. 1998;352:1898-1901.
10. Rudolph CM, Al-Fares S, Vaughan-Jones SA, et al. Polymorphic eruption of pregnancy: clinicopathology and potential trigger factors in 181 patients. Clin Lab Invest. 2006;154:54-60.
11. Roger D, Vaillant L, Fignon A, et al. Specific pruritic diseases of pregnancy. A prospective study of 3192 pregnant women. Arch Dermatol. 1994;130:734-739.
12. McDonald J. Cholestasis of pregnancy. J Gastroenterol Hepatol. 1999;14:515-518.
13. Ohel I, Levy A, Silberstein T, et al. Pregnancy outcome of patients with pruritic urticarial papules and plaques of pregnancy. J Maternal-Fetal Neonat Med. 2006;19:305-308.
14. Geenes V, Williamson C. Intrahepatic cholestasis of pregnancy. World J Gastroenterol. 2009;15:2049-2066.
15. Shaw D, Frohlich J, Wittmann BA, et al. A prospective study of 18 patients with cholestasis of pregnancy. Am J Obstet Gynecol. 1982;142:621-625.
16. Reyes H. The spectrum of liver and gastrointestinal disease seen in cholestasis of pregnancy. Gastroenterol Clin North Am. 1992;21:905-921.
17. Lammert F, Marschall H, Glantz A, et al. Intrahepatic cholestasis of pregnancy; molecular pathogenesis, diagnosis and management. J Hepatol. 2000;33:1012-1021.
18. Huang WM, Seubert DE, Donnelly JG, et al. Intrahepatic cholestasis of pregnancy: detection with urinary bile acid assays. J Perinat Med. 2007;35:486-491.
19. Palma J, Reyes H, Ribalta J, et al. Ursodeoxycholic acid in the treatment of intrahepatic cholestasis of pregnancy: a randomized, double blind study controlled with placebo. J Hepatol. 1997;27:1022-1028.
20. Diaferia A, Nicastri PL, Tartagni M, et al. Ursodeoxycholic acid therapy in pregnant women with cholestasis. Int J Gynaecol Obstet. 1996;52:133-140.
21. Nicastri PL, Diaferia A, Tartagni M, et al. A randomised placebocontrolled trial of ursodeoxycholic acid and S-adenosylmethionine in the treatment of intrahepatic cholestasis of pregnancy. Br J Obstet Gynaecol. 1998;105:1205-1207.
22. Glantz A, Marschall HU, Lammert F, et al. Intrahepatic cholestasis in pregnancy: a randomized controlled trial comparing dexamethasone and ursodeoxycholic acid. Hepatology. 2005;42:1399-1405.
23. Ambros-Rudolph CM, Mullegger MM, Vaughan-Jones SA, et al. The specific dermatoses of pregnancy revisited and reclassified: results of a retrospective two-center study on 505 pregnant patients. J Am Acad Dermatol. 2006;54:395-404.
24. Vaughan-Jones SA, Hern S, Nelson-Piercy C, et al. A prospective study of 200 women with dermatoses of pregnancy correlating clinical findings with hormonal and immunopathological profiles. Br J Dermatol. 1999;141:71-81.
25. Shornick JK. Dermatoses of pregnancy. Semin Cutan Med Surg. 1998;17:172-181.
26. Holmes RC, Black MM. The specific dermatoses of pregnancy. J Am Acad Dermatol. 1983;8:405-412.
27. Bukhari IA. Impetigo herpetiformis in a primagravida: successful treatment with etretinate. J Drugs Dermatol. 2004;3:449-451.
28. Oumeish OY, Parish JL. Impetigo herpetiformis. Clin in Dermatol. 2006;24:101-104.
29. Kroumpouzos G, Cohen LM. Pruritic folliculitis of pregnancy. J Am Acad Dermatol. 2000;43:132-134.
30. Sahin HG, Hasin HA, Metin A, et al. Recurrent impetigo herpetiformis in a pregnant adolescent: a case report. Eur J Obstet Gynecol Reprod Endocrinol. 2002;101:201-203.
31. Aka N, Kuscu NK, Yazicioglu E. Impetigo herpetiformis at the 36th week of gestation. Int J Gynecol Obstet. 2000;69:153-154.
32. Wade TR, Wade SL, Jones HE. Skin changes and diseases associated with pregnancy. Obstet Gynecol. 1978;52:233-242.
• Pemphigoid gestationis is best managed with oral prednisone at doses from 20 to 60 mg per day to control symptoms. B
• The pruritus associated with pruritic urticarial papules and plaques of pregnancy can be safely and effectively managed with topical corticosteroids and oral antihistamines. A
• Treat intrahepatic cholestasis of pregnancy with ursodeoxycholic acid, which likely reduces serum bile acids as well as associated fetal morbidity and mortality. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The dermatoses of pregnancy are a poorly understood group of dermatologic conditions. The only thing they have in common is a tendency to appear during pregnancy.
Only 3 are considered unique to pregnancy, however; the others are probably exacerbations of preexisting conditions triggered by pregnancy. There isn’t even complete agreement on what to call them. And to make management even more complex, 2 patients—the mother and the fetus—need to be considered.
Who will manage these patients is another matter. These conditions fall into an overlapping area of health care, where family physicians, obstetricians, and dermatologists all have some share in the responsibility for diagnosis and treatment. As a family physician who probably cares for any number of pregnant patients on a weekly basis, you need to be sufficiently familiar with these conditions so that you can differentiate those that can be treated symptomatically and those that require a referral to a specialist. This review and handy TABLE will help you toward that end.
TABLE
Skin disorders of pregnancy: What you’ll see, how to treat
| Disorder | Lesions | Diagnosis and sequelae | Treatment | Recurrence |
|---|---|---|---|---|
| PG3,5 | Erythematous papules, progressing to vesicles, bullae; periumbilical distribution, sparing face, palms, and soles | Mean onset at 21 weeks; postpartum in 20% of cases. Direct immunofluorescence microscopy shows linear C3 deposition. Newborn may be small for gestational age, but no associated morbidity or mortality | Oral corticosteroids 20-60 mg/d, IVIG, or cyclosporine in refractory cases | Frequent. Skips a pregnancy 8% of the time |
| PUPPP8-10 | Urticarial papules and plaques on abdomen, legs, arms, buttocks, chest, and back | Usually present after 34th week, but can present at any stage. Diagnosis is clinical. No increase in fetal morbidity or mortality | Topical steroids and antihistamines | Uncommon |
| ICP14,17,19-22 | No primary lesions; secondary excoriations in any area patient can reach | Onset after 30th week in 80% of patients. Strongly indicated by serum bile acid >11 mcmol/L. Increased fetal mortality | Ursodeoxycholic acid 450-1200 mg/d | Frequent |
| EP/PP4,10,24 | Grouped, crusted erythematous papules, patches, and plaques, most commonly on extensor surfaces of arms and legs or on abdomen | Onset at any point in pregnancy. Clinical diagnosis. No increase in fetal morbidity or mortality | Symptomatic treatment with topical steroids or antihistamines | Frequent |
| APPP27-29 | Erythematous plaques and pustules starting on inner thighs and groin and spreading to trunk and extremities | Onset at any point in pregnancy. Clinical diagnosis by appearance of lesions and association with systemic illness. Increased incidence of miscarriage and stillbirth, and maternal mortality | Prednisone 15-60 mg/d, cyclosporine 100 mg twice daily in refractory cases, management of associated hypocalcemia | Unknown |
| PFP24,29 | Papules and pustules concentrated around hair follicles, often beginning on abdomen and spreading to extremities | Onset most often in third trimester. Clinical diagnosis. No associated fetal morbidity or mortality | Topical steroids | Unknown |
| APPP, acute pustular psoriasis of pregnancy; EP/PP, eczema of pregnancy/pruritus of pregnancy; ICP, intrahepatic cholestasis of pregnancy; IVIG, intravenous immunoglobulin; PFP, pruritic folliculitis of pregnancy; PG, pemphigoid gestationis; PUPPP, pruritic urticarial papules and plaques of pregnancy. | ||||
Dermatoses unique to pregnancy
Pemphigoid gestationis
Years ago, this disorder was referred to as herpes gestationis, because the lesions are herpetiform. Pemphigoid gestationis (PG) has an incidence of approximately 1 in 10,000 pregnancies.1,2 The time of onset is usually about the 21st week of gestation, although in about 20% of cases, the eruption appears immediately postpartum.3
Presentation. The disease usually begins with urticarial papules and plaques around the umbilicus and extremities. Bullous lesions tend to develop as the disease progresses, and are often not present on first presentation (FIGURE 1). PG lesions tend to spare the face, palms, and soles. Mucosal surfaces are involved in less than 20% of cases. In about 75% of cases, PG flares around the time of delivery, regressing spontaneously after the baby is born.4
Pathophysiology. The pathophysiology is nearly identical to that of bullous pemphigoid, a blistering skin disorder more often seen in elderly patients.5 Pemphigoid disorders are immune processes, involving an immunoglobulin G (IgG) immune response directed at a 180-kDa hemidesmosome transmembrane glycoprotein. This protein is the common target of several subepidermal blistering diseases.
Differential. Disorders that may have some of the same features as PG include pruritic urticarial papules and plaques of pregnancy (PUPPP), erythema multiforme, intrahepatic cholestasis of pregnancy (ICP), contact dermatitis, and drug reactions.
Diagnosis. A biopsy is necessary for the definitive diagnosis. Direct immunofluorescence (DIF) microscopy of a sample of perilesional skin can show tissue-bound immunoreactants. Linear deposition of the complement component protein C3 along the basement membrane zone is diagnostic for PG. IgG is also deposited about 40% of the time.3 Serum enzyme-linked immunosorbent assay (ELISA) studies are also helpful in diagnosis. They have excellent sensitivity and specificity, as well as the capacity to monitor levels of antibody, which correlate with the severity of disease.1
Treatment. Oral corticosteroids are the first-line treatment for PG, typically 20 to 60 mg per day of prednisone. Oral corticosteroids are typically most effective in ameliorating the patient’s symptoms. Prednisone at doses of 40 to 80 mg per day for a short time has not been associated with congenital abnormalities.6 PG patients can also be treated successfully with intravenous immunoglobulin (IVIG) and cyclosporine in refractory cases.7
Pruritus associated with this condition can interfere with day-to-day activity and with the patient’s ability to sleep. Patients may also complain that the rash is painful, particularly if bullae rupture, leading to superficial ulcerations. Fortunately, the patient’s quality of life can be dramatically improved with systemic corticosteroids—with no significant risk to the fetus.
Sequelae. PG uniformly resolves within a few weeks, but the mother’s autoantibodies can passively be transferred to the fetus, causing vesicles and bullae in the newborn.8 An increased incidence of small-for-gestational age (SGA) infants has also been noted in PG, although no lasting morbidity or mortality in the offspring has been noted.5 This disease tends to recur with future pregnancies, but will skip a pregnancy 8% of the time.5
FIGURE 1
Pemphigoid gestationis
Pruritic urticarial papules and plaques of pregnancy
This condition is known by many names besides PUPPP: polymorphic eruption of pregnancy, toxemic erythema of pregnancy, and late prurigo of pregnancy.1 It is a pruritic, inflammatory skin disorder that has been variously estimated to occur in anywhere from 1 in 120 to 1 in 240 pregnancies.8 PUPPP is second only to eczema as the most common dermatosis of pregnancy.
Presentation. As the name implies, the lesions of PUPPP are itchy, red papules that often coalesce into plaques (FIGURE 2). The lesions usually occur in primigravidas after the 34th week of gestation, although they may be seen at any time from the first trimester through the postpartum period.9
Lesions are classically found on the abdomen, sparing the umbilical area, and are found primarily in the striae. This distribution helps to differentiate PUPPP from PG, where the lesions typically cluster around the umbilicus. Most PUPPP lesions (80% in 1 study) are dispersed on the abdomen, legs, arms, buttocks, chest, and back. Another 17% appear only on the abdomen and proximal thighs, and the remaining 3% on the limbs.10 Nearly 50% of the time, lesions also include discrete vesicles.11 There are no reported cases of mucosal involvement.
Patients with this condition are often very uncomfortable. The associated pruritus is severe enough to interfere with sleep. Despite the itching, however, lesions are seldom excoriated.
Pathophysiology. The disorder has been strongly associated with maternal weight gain and multiple gestations. One working hypothesis is that rapid abdominal distention observed in the third trimester leads to damage of the connective tissue, which then releases antigenic molecules, causing an inflammatory reaction.12 Another hypothesis is that increased levels of fetal DNA that have been detected in the skin of PUPPP patients may contribute to the pathology. One study detected male DNA in 6 of 10 PUPPP sufferers, but found none in any of 26 controls— pregnant women without PUPPP pathology.5 There is some evidence that patients with atopy may be predisposed to PUPPP, as well as patients who are hypertensive or obese.10,13
Differential. Initially, PUPPP lesions can be difficult to differentiate from urticarial PG lesions. The distribution of the lesions is the best clue: PG lesions cluster around the umbilicus, whereas PUPPP lesions uniformly spare the umbilical area. Additional disorders in the PUPPP differential are atopic dermatitis, superficial urticarial allergic eruption, viral exanthema, and contact or irritant dermatitis.
Diagnosis. PUPPP can only be diagnosed through clinical observation. None of the available laboratory tests—immunofluorescence, histology, serology—yield findings specific for PUPPP, although histology and immunofluorescence can readily differentiate between this condition and PG.
Treatment. Because the disease holds no real danger for mother or fetus, treatment can be aimed solely at symptomatic relief. Mild to potent topical steroids (consider triamcinolone or fluocinonide) should relieve pruritus within 48 to 72 hours.8 Antihistamines and occasionally low-dose systemic steroids may also be used. Consider hydroxyzine, although diphenhydramine has the more proven safety profile in pregnancy.
Nonpharmacologic treatments such as oil baths and emollients should also be considered. If the condition appears classic for PUPPP, it can be managed symptomatically. However, if there is any question about the diagnosis, referral to a dermatologist is prudent.
Sequelae. No increase in maternal or fetal morbidity or mortality is associated with PUPPP. Recurrence is fairly uncommon, as the disease primarily affects women during their first pregnancy.
FIGURE 2
Pruritic urticarial papules and plaques of pregnancy
Intrahepatic cholestasis of pregnancy
This condition is also called recurrent or idiopathic jaundice of pregnancy, obstetric cholestasis, and pruritus gravidarum. ICP is caused by disruption of hepatic bile flow during pregnancy. It has been recorded at a rate of approximately 10 to 150 per 10,000 pregnancies in Europe and 70 per 10,000 in the United States.12 In 80% of patients, the time of onset is after the 30th week.14 Although this disorder is not primarily a dermatosis of pregnancy, it is a pruritic condition that often presents with excoriations in pregnant women and is associated with fetal morbidity and mortality. It’s important to be able to identify this disease early to minimize sequelae.
Presentation. There are no primary lesions with ICP. The primary presenting symptom is a generalized pruritus affecting the palms and soles, and sometimes extending to the legs and abdomen (FIGURE 3). This itching is often so severe that it leads to chronic insomnia. You may see secondary skin lesions, such as erythema and excoriations. Observable jaundice occurs in 10% to 20% of patients.3 These patients do not develop the encephalopathy that is associated with cholestasis in the nonpregnant state, however.14
Pathophysiology. The genesis of this condition is thought to be a combination of genetic and environmental factors. A family history of the disorder is present in half the cases, and cases with a familial component tend to be more severe.15 ICP may be an exaggerated response to increased estrogen levels in pregnancy, but the mechanism of this response is unknown.16
Differential. Other conditions that must be considered in making the diagnosis are viral hepatitis, gallbladder disease, PG, PUPPP, drug hepatotoxicity, primary biliary cirrhosis, and uremia.
Diagnosis. Laboratory values are the definitive diagnostic tool in this condition. Increased serum bile acids are the single most sensitive test. Average levels of serum bile acids in pregnancy are 6.6 mcmol/L, with an upper limit of 11. The average value in women with ICP is 47 mcmol/L.17
While serum acids remain the gold standard, a recent study showed elevated urine bile acids to have 100% sensitivity and 83% specificity for ICP.18 In 55% to 60% of cases, the liver enzymes aspartate aminotransferase (AST) and alanine aminotransferase (ALT) are mildly increased. Steatorrhea is often noted by the patient, and is followed by vitamin K deficiency.17
Treatment. The current standard of care for ICP is treatment with ursodeoxycholic acid (UDCA). In 4 controlled trials, UDCA showed a sustained decrease in serum bile acids.19-22 Doses used in these trials have varied between 450 and 1200 mg per day.
Before UDCA treatment was available, the disorder was treated with cholestyramine, which could bring about a 70% rate of response. The drawback to cholestyramine treatment is that it precipitates vitamin K, which is already compromised by the disease process. Further, its onset of action is slow.3
Elective delivery is indicated for ICP, particularly in patients with significant clinical presentations.12 Delivery for ICP should be performed around week 37 to 38, as stillbirths tend to cluster around weeks 37 to 39.14 Given the significant fetal mortality associated with this condition (see below), ICP should be managed by a clinician experienced with the disease, likely a gastroenterologist.
Sequelae. The impact of this maternal disorder on the fetus can be disastrous: a 10% to 15% rate of perinatal death, and a 30% to 40% rate of premature labor have been seen with ICP.14 Fortunately, rates of preterm labor are strongly correlated with levels of bile acids, so that as bile acid levels are reduced with UDCA treatment, rates of preterm labor also go down. Currently, management of the condition has reduced rates of perinatal death to 3.5%. There is no evidence of fetal growth retardation.14
FIGURE 3
Intrahepatic cholestasis of pregnancy
Dermatoses triggered by pregnancy
Eczema of pregnancy/prurigo of pregnancy
Eczema of pregnancy/prurigo of pregnancy (EP/PP) may not actually be correlated with the pregnant state. Both conditions manifest as eczematous lesions in an atopic distribution. Although they have been described in the literature as separate entities, the lack of clinical distinction between them led Ambros-Rudolph and colleagues to combine them under the umbrella term, atopic eruption of pregnancy.23 In some patients, at least, they may be preexisting conditions that pregnancy exacerbates. One study of 255 patients with the condition found that 20% had had the lesions before they became pregnant.23 However, the tendency of the condition to be markedly worsened by pregnancy leads us to include it here.
PP has an estimated incidence of 1 in 450 pregnancies.11 But while many authorities consider EP to be the most common dermatosis of pregnancy, no clear estimation of its prevalence has been established.23,24 Taken together, these 2 conditions have the highest prevalence of all pregnancy-induced dermatoses. PP is also known as popular dermatitis of Spangler, Nurse’s early prurigo of pregnancy, and linear IgM disease of pregnancy.3,23,25
Presentation. The typical presentation consists of grouped, crusted, erythematous papules, patches, and plaques—frequently with excoriations. The lesions typically present on the extensor surfaces of the arms and legs or on the abdomen (FIGURE 4).4 Recurrence in later pregnancies is common.
Pathophysiology. The pathophysiology of EP/PP is not understood. Many patients who present with EP/PP have a history of atopy.10
Differential. Conditions that need to be considered in making the diagnosis include tinea infection, scabies, contact dermatitis, ICP, pruritic folliculitis of pregnancy (PFP), and PG.
Diagnosis. History and physical examination determine the diagnosis. Serology, histopathology, and immunofluorescence are not specific, and correlation with increased IgE is marginal, at best.24,26
Treatment. These conditions are treated symptomatically with topical steroids or systemic antihistamines.
Sequelae. No maternal or fetal increase in morbidity or mortality is associated with these conditions.
FIGURE 4
Eczema of pregnancy
Acute pustular psoriasis of pregnancy
Whether or not APPP is actually a pregnancyinduced dermatosis is subject to debate.
There is evidence that APPP is not unique to pregnancy, but is simply a manifestation of ordinary psoriasis. Clinically and histologically, APPP is indistinguishable from pustular psoriasis. Unlike most cases of acute psoriasis, however, APPP often appears in pregnancy without any personal or family history of psoriasis, and usually ceases when the pregnancy is concluded. This fact, combined with reports of increased fetal and maternal morbidity and mortality associated with APPP, lead us to include it here.27
Presentation. APPP is a rare condition that may have an onset at any point in pregnancy. The characteristic lesions begin as erythematous plaques with pustules on the inner thighs, flexural areas, and groin and spread to the trunk and extremities. As the plaques enlarge, the center becomes eroded and crusted. Nails may become onycholytic. The hands, feet, and face are usually spared. Oral and esophageal erosions can occur. Pruritus is typically mild, although the lesions are often painful and flu-like symptoms are often present.28
Pathophysiology. The pathophysiology of this disease is unknown.
Differential. Conditions with similar presentations include an adverse drug reaction, pityriasis rosea, lichen simplex chronicus, eczema, lupus, and pityriasis rubra pilaris.
Diagnosis. Clinical history and association with systemic illness are the basis for a diagnosis of APPP. Cultures of the pustules are negative for any infective pathology, though as the disease progresses, pustules may become superinfected. Lab tests may show an increased erythrocyte sedimentation rate (ESR), hypocalcemia, and low levels of vitamin D.
Treatment. Prednisone 15 to 60 mg per day is often sufficient to control the disease.28 Cyclosporine 100 mg twice daily has also been shown to be useful.29 Cyclosporine in pregnancy is a category C drug. Data on fetal malformation associated with cyclosporine therapy are limited, but the risk appears to be minimal.6 Maternal hypocalcemia should be monitored and treated appropriately. If disease progression is judged serious enough, early induction of labor is indicated, since delivery will almost always lead to swift resolution.
Sequelae. There have been a number of case reports that link APPP to serious sequelae, including fetal growth retardation, hypocalcemia, and stillbirth.28,30,31 The condition is too rare, however, for good data on specific sequelae. While the disease does give significant cause for concern, it would appear that some of the traditional apprehension comes from older publications reporting a rate of maternal mortality of 70% to 90%.32 This statistic has not been borne out in clinical practice. It does appear that the mother will frequently suffer from systemic symptoms, including fever and malaise.
Pruritic folliculitis of pregnancy
Accounts of PFP’s prevalence vary widely: Some sources report fewer than 30 cases in all of the literature, while others indicate that the prevalence is equivalent to that of PG, 1 in 10,000.3,11 PFP most commonly presents in the third trimester. It often resolves before delivery, but uniformly clears within 2 weeks of delivery.
Presentation. PFP presents as papules and pustules concentrated around hair follicles (FIGURE 5). Often lesions begin on the abdomen and spread to the extremities.24,29 The condition is often, but not always, pruritic. Patients are more likely to be concerned about what the condition means for their health, rather than being distressed by the symptoms.
Pathophysiology. Like so many of these conditions, the pathophysiology of PFP is unknown. There is little evidence that the condition is immunologically or hormonally mediated, and there is no evidence of an infectious component.24,29
Differential. PFP must be distinguished from infectious folliculitis, acneiform disorders, HIV-associated eosinophilic folliculitis, and a drug reaction.
Diagnosis. The clinical diagnosis is based on presenting symptoms and third trimester onset. No specific lab or histological analysis can be used to make a definitive diagnosis.
Treatment. As the condition is, by definition, a nonmicrobial folliculitis, the most effective therapy tends to be with mid- to lowpotency topical steroids such as triamcinolone or desonide. Additionally, benzoyl peroxide wash can be effective.
Sequelae. One study reports an increased incidence of low birth weight, but currently no associated morbidity or mortality has been reported.24
FIGURE 5
Pruritic folliculitis of pregnancy
CORRESPONDENCE
Matthew Bremmer, MD, 419 W Redwood Street, Department of Dermatology, Baltimore, MD 21230; Mbrem001@umaryland.edu
• Pemphigoid gestationis is best managed with oral prednisone at doses from 20 to 60 mg per day to control symptoms. B
• The pruritus associated with pruritic urticarial papules and plaques of pregnancy can be safely and effectively managed with topical corticosteroids and oral antihistamines. A
• Treat intrahepatic cholestasis of pregnancy with ursodeoxycholic acid, which likely reduces serum bile acids as well as associated fetal morbidity and mortality. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The dermatoses of pregnancy are a poorly understood group of dermatologic conditions. The only thing they have in common is a tendency to appear during pregnancy.
Only 3 are considered unique to pregnancy, however; the others are probably exacerbations of preexisting conditions triggered by pregnancy. There isn’t even complete agreement on what to call them. And to make management even more complex, 2 patients—the mother and the fetus—need to be considered.
Who will manage these patients is another matter. These conditions fall into an overlapping area of health care, where family physicians, obstetricians, and dermatologists all have some share in the responsibility for diagnosis and treatment. As a family physician who probably cares for any number of pregnant patients on a weekly basis, you need to be sufficiently familiar with these conditions so that you can differentiate those that can be treated symptomatically and those that require a referral to a specialist. This review and handy TABLE will help you toward that end.
TABLE
Skin disorders of pregnancy: What you’ll see, how to treat
| Disorder | Lesions | Diagnosis and sequelae | Treatment | Recurrence |
|---|---|---|---|---|
| PG3,5 | Erythematous papules, progressing to vesicles, bullae; periumbilical distribution, sparing face, palms, and soles | Mean onset at 21 weeks; postpartum in 20% of cases. Direct immunofluorescence microscopy shows linear C3 deposition. Newborn may be small for gestational age, but no associated morbidity or mortality | Oral corticosteroids 20-60 mg/d, IVIG, or cyclosporine in refractory cases | Frequent. Skips a pregnancy 8% of the time |
| PUPPP8-10 | Urticarial papules and plaques on abdomen, legs, arms, buttocks, chest, and back | Usually present after 34th week, but can present at any stage. Diagnosis is clinical. No increase in fetal morbidity or mortality | Topical steroids and antihistamines | Uncommon |
| ICP14,17,19-22 | No primary lesions; secondary excoriations in any area patient can reach | Onset after 30th week in 80% of patients. Strongly indicated by serum bile acid >11 mcmol/L. Increased fetal mortality | Ursodeoxycholic acid 450-1200 mg/d | Frequent |
| EP/PP4,10,24 | Grouped, crusted erythematous papules, patches, and plaques, most commonly on extensor surfaces of arms and legs or on abdomen | Onset at any point in pregnancy. Clinical diagnosis. No increase in fetal morbidity or mortality | Symptomatic treatment with topical steroids or antihistamines | Frequent |
| APPP27-29 | Erythematous plaques and pustules starting on inner thighs and groin and spreading to trunk and extremities | Onset at any point in pregnancy. Clinical diagnosis by appearance of lesions and association with systemic illness. Increased incidence of miscarriage and stillbirth, and maternal mortality | Prednisone 15-60 mg/d, cyclosporine 100 mg twice daily in refractory cases, management of associated hypocalcemia | Unknown |
| PFP24,29 | Papules and pustules concentrated around hair follicles, often beginning on abdomen and spreading to extremities | Onset most often in third trimester. Clinical diagnosis. No associated fetal morbidity or mortality | Topical steroids | Unknown |
| APPP, acute pustular psoriasis of pregnancy; EP/PP, eczema of pregnancy/pruritus of pregnancy; ICP, intrahepatic cholestasis of pregnancy; IVIG, intravenous immunoglobulin; PFP, pruritic folliculitis of pregnancy; PG, pemphigoid gestationis; PUPPP, pruritic urticarial papules and plaques of pregnancy. | ||||
Dermatoses unique to pregnancy
Pemphigoid gestationis
Years ago, this disorder was referred to as herpes gestationis, because the lesions are herpetiform. Pemphigoid gestationis (PG) has an incidence of approximately 1 in 10,000 pregnancies.1,2 The time of onset is usually about the 21st week of gestation, although in about 20% of cases, the eruption appears immediately postpartum.3
Presentation. The disease usually begins with urticarial papules and plaques around the umbilicus and extremities. Bullous lesions tend to develop as the disease progresses, and are often not present on first presentation (FIGURE 1). PG lesions tend to spare the face, palms, and soles. Mucosal surfaces are involved in less than 20% of cases. In about 75% of cases, PG flares around the time of delivery, regressing spontaneously after the baby is born.4
Pathophysiology. The pathophysiology is nearly identical to that of bullous pemphigoid, a blistering skin disorder more often seen in elderly patients.5 Pemphigoid disorders are immune processes, involving an immunoglobulin G (IgG) immune response directed at a 180-kDa hemidesmosome transmembrane glycoprotein. This protein is the common target of several subepidermal blistering diseases.
Differential. Disorders that may have some of the same features as PG include pruritic urticarial papules and plaques of pregnancy (PUPPP), erythema multiforme, intrahepatic cholestasis of pregnancy (ICP), contact dermatitis, and drug reactions.
Diagnosis. A biopsy is necessary for the definitive diagnosis. Direct immunofluorescence (DIF) microscopy of a sample of perilesional skin can show tissue-bound immunoreactants. Linear deposition of the complement component protein C3 along the basement membrane zone is diagnostic for PG. IgG is also deposited about 40% of the time.3 Serum enzyme-linked immunosorbent assay (ELISA) studies are also helpful in diagnosis. They have excellent sensitivity and specificity, as well as the capacity to monitor levels of antibody, which correlate with the severity of disease.1
Treatment. Oral corticosteroids are the first-line treatment for PG, typically 20 to 60 mg per day of prednisone. Oral corticosteroids are typically most effective in ameliorating the patient’s symptoms. Prednisone at doses of 40 to 80 mg per day for a short time has not been associated with congenital abnormalities.6 PG patients can also be treated successfully with intravenous immunoglobulin (IVIG) and cyclosporine in refractory cases.7
Pruritus associated with this condition can interfere with day-to-day activity and with the patient’s ability to sleep. Patients may also complain that the rash is painful, particularly if bullae rupture, leading to superficial ulcerations. Fortunately, the patient’s quality of life can be dramatically improved with systemic corticosteroids—with no significant risk to the fetus.
Sequelae. PG uniformly resolves within a few weeks, but the mother’s autoantibodies can passively be transferred to the fetus, causing vesicles and bullae in the newborn.8 An increased incidence of small-for-gestational age (SGA) infants has also been noted in PG, although no lasting morbidity or mortality in the offspring has been noted.5 This disease tends to recur with future pregnancies, but will skip a pregnancy 8% of the time.5
FIGURE 1
Pemphigoid gestationis
Pruritic urticarial papules and plaques of pregnancy
This condition is known by many names besides PUPPP: polymorphic eruption of pregnancy, toxemic erythema of pregnancy, and late prurigo of pregnancy.1 It is a pruritic, inflammatory skin disorder that has been variously estimated to occur in anywhere from 1 in 120 to 1 in 240 pregnancies.8 PUPPP is second only to eczema as the most common dermatosis of pregnancy.
Presentation. As the name implies, the lesions of PUPPP are itchy, red papules that often coalesce into plaques (FIGURE 2). The lesions usually occur in primigravidas after the 34th week of gestation, although they may be seen at any time from the first trimester through the postpartum period.9
Lesions are classically found on the abdomen, sparing the umbilical area, and are found primarily in the striae. This distribution helps to differentiate PUPPP from PG, where the lesions typically cluster around the umbilicus. Most PUPPP lesions (80% in 1 study) are dispersed on the abdomen, legs, arms, buttocks, chest, and back. Another 17% appear only on the abdomen and proximal thighs, and the remaining 3% on the limbs.10 Nearly 50% of the time, lesions also include discrete vesicles.11 There are no reported cases of mucosal involvement.
Patients with this condition are often very uncomfortable. The associated pruritus is severe enough to interfere with sleep. Despite the itching, however, lesions are seldom excoriated.
Pathophysiology. The disorder has been strongly associated with maternal weight gain and multiple gestations. One working hypothesis is that rapid abdominal distention observed in the third trimester leads to damage of the connective tissue, which then releases antigenic molecules, causing an inflammatory reaction.12 Another hypothesis is that increased levels of fetal DNA that have been detected in the skin of PUPPP patients may contribute to the pathology. One study detected male DNA in 6 of 10 PUPPP sufferers, but found none in any of 26 controls— pregnant women without PUPPP pathology.5 There is some evidence that patients with atopy may be predisposed to PUPPP, as well as patients who are hypertensive or obese.10,13
Differential. Initially, PUPPP lesions can be difficult to differentiate from urticarial PG lesions. The distribution of the lesions is the best clue: PG lesions cluster around the umbilicus, whereas PUPPP lesions uniformly spare the umbilical area. Additional disorders in the PUPPP differential are atopic dermatitis, superficial urticarial allergic eruption, viral exanthema, and contact or irritant dermatitis.
Diagnosis. PUPPP can only be diagnosed through clinical observation. None of the available laboratory tests—immunofluorescence, histology, serology—yield findings specific for PUPPP, although histology and immunofluorescence can readily differentiate between this condition and PG.
Treatment. Because the disease holds no real danger for mother or fetus, treatment can be aimed solely at symptomatic relief. Mild to potent topical steroids (consider triamcinolone or fluocinonide) should relieve pruritus within 48 to 72 hours.8 Antihistamines and occasionally low-dose systemic steroids may also be used. Consider hydroxyzine, although diphenhydramine has the more proven safety profile in pregnancy.
Nonpharmacologic treatments such as oil baths and emollients should also be considered. If the condition appears classic for PUPPP, it can be managed symptomatically. However, if there is any question about the diagnosis, referral to a dermatologist is prudent.
Sequelae. No increase in maternal or fetal morbidity or mortality is associated with PUPPP. Recurrence is fairly uncommon, as the disease primarily affects women during their first pregnancy.
FIGURE 2
Pruritic urticarial papules and plaques of pregnancy
Intrahepatic cholestasis of pregnancy
This condition is also called recurrent or idiopathic jaundice of pregnancy, obstetric cholestasis, and pruritus gravidarum. ICP is caused by disruption of hepatic bile flow during pregnancy. It has been recorded at a rate of approximately 10 to 150 per 10,000 pregnancies in Europe and 70 per 10,000 in the United States.12 In 80% of patients, the time of onset is after the 30th week.14 Although this disorder is not primarily a dermatosis of pregnancy, it is a pruritic condition that often presents with excoriations in pregnant women and is associated with fetal morbidity and mortality. It’s important to be able to identify this disease early to minimize sequelae.
Presentation. There are no primary lesions with ICP. The primary presenting symptom is a generalized pruritus affecting the palms and soles, and sometimes extending to the legs and abdomen (FIGURE 3). This itching is often so severe that it leads to chronic insomnia. You may see secondary skin lesions, such as erythema and excoriations. Observable jaundice occurs in 10% to 20% of patients.3 These patients do not develop the encephalopathy that is associated with cholestasis in the nonpregnant state, however.14
Pathophysiology. The genesis of this condition is thought to be a combination of genetic and environmental factors. A family history of the disorder is present in half the cases, and cases with a familial component tend to be more severe.15 ICP may be an exaggerated response to increased estrogen levels in pregnancy, but the mechanism of this response is unknown.16
Differential. Other conditions that must be considered in making the diagnosis are viral hepatitis, gallbladder disease, PG, PUPPP, drug hepatotoxicity, primary biliary cirrhosis, and uremia.
Diagnosis. Laboratory values are the definitive diagnostic tool in this condition. Increased serum bile acids are the single most sensitive test. Average levels of serum bile acids in pregnancy are 6.6 mcmol/L, with an upper limit of 11. The average value in women with ICP is 47 mcmol/L.17
While serum acids remain the gold standard, a recent study showed elevated urine bile acids to have 100% sensitivity and 83% specificity for ICP.18 In 55% to 60% of cases, the liver enzymes aspartate aminotransferase (AST) and alanine aminotransferase (ALT) are mildly increased. Steatorrhea is often noted by the patient, and is followed by vitamin K deficiency.17
Treatment. The current standard of care for ICP is treatment with ursodeoxycholic acid (UDCA). In 4 controlled trials, UDCA showed a sustained decrease in serum bile acids.19-22 Doses used in these trials have varied between 450 and 1200 mg per day.
Before UDCA treatment was available, the disorder was treated with cholestyramine, which could bring about a 70% rate of response. The drawback to cholestyramine treatment is that it precipitates vitamin K, which is already compromised by the disease process. Further, its onset of action is slow.3
Elective delivery is indicated for ICP, particularly in patients with significant clinical presentations.12 Delivery for ICP should be performed around week 37 to 38, as stillbirths tend to cluster around weeks 37 to 39.14 Given the significant fetal mortality associated with this condition (see below), ICP should be managed by a clinician experienced with the disease, likely a gastroenterologist.
Sequelae. The impact of this maternal disorder on the fetus can be disastrous: a 10% to 15% rate of perinatal death, and a 30% to 40% rate of premature labor have been seen with ICP.14 Fortunately, rates of preterm labor are strongly correlated with levels of bile acids, so that as bile acid levels are reduced with UDCA treatment, rates of preterm labor also go down. Currently, management of the condition has reduced rates of perinatal death to 3.5%. There is no evidence of fetal growth retardation.14
FIGURE 3
Intrahepatic cholestasis of pregnancy
Dermatoses triggered by pregnancy
Eczema of pregnancy/prurigo of pregnancy
Eczema of pregnancy/prurigo of pregnancy (EP/PP) may not actually be correlated with the pregnant state. Both conditions manifest as eczematous lesions in an atopic distribution. Although they have been described in the literature as separate entities, the lack of clinical distinction between them led Ambros-Rudolph and colleagues to combine them under the umbrella term, atopic eruption of pregnancy.23 In some patients, at least, they may be preexisting conditions that pregnancy exacerbates. One study of 255 patients with the condition found that 20% had had the lesions before they became pregnant.23 However, the tendency of the condition to be markedly worsened by pregnancy leads us to include it here.
PP has an estimated incidence of 1 in 450 pregnancies.11 But while many authorities consider EP to be the most common dermatosis of pregnancy, no clear estimation of its prevalence has been established.23,24 Taken together, these 2 conditions have the highest prevalence of all pregnancy-induced dermatoses. PP is also known as popular dermatitis of Spangler, Nurse’s early prurigo of pregnancy, and linear IgM disease of pregnancy.3,23,25
Presentation. The typical presentation consists of grouped, crusted, erythematous papules, patches, and plaques—frequently with excoriations. The lesions typically present on the extensor surfaces of the arms and legs or on the abdomen (FIGURE 4).4 Recurrence in later pregnancies is common.
Pathophysiology. The pathophysiology of EP/PP is not understood. Many patients who present with EP/PP have a history of atopy.10
Differential. Conditions that need to be considered in making the diagnosis include tinea infection, scabies, contact dermatitis, ICP, pruritic folliculitis of pregnancy (PFP), and PG.
Diagnosis. History and physical examination determine the diagnosis. Serology, histopathology, and immunofluorescence are not specific, and correlation with increased IgE is marginal, at best.24,26
Treatment. These conditions are treated symptomatically with topical steroids or systemic antihistamines.
Sequelae. No maternal or fetal increase in morbidity or mortality is associated with these conditions.
FIGURE 4
Eczema of pregnancy
Acute pustular psoriasis of pregnancy
Whether or not APPP is actually a pregnancyinduced dermatosis is subject to debate.
There is evidence that APPP is not unique to pregnancy, but is simply a manifestation of ordinary psoriasis. Clinically and histologically, APPP is indistinguishable from pustular psoriasis. Unlike most cases of acute psoriasis, however, APPP often appears in pregnancy without any personal or family history of psoriasis, and usually ceases when the pregnancy is concluded. This fact, combined with reports of increased fetal and maternal morbidity and mortality associated with APPP, lead us to include it here.27
Presentation. APPP is a rare condition that may have an onset at any point in pregnancy. The characteristic lesions begin as erythematous plaques with pustules on the inner thighs, flexural areas, and groin and spread to the trunk and extremities. As the plaques enlarge, the center becomes eroded and crusted. Nails may become onycholytic. The hands, feet, and face are usually spared. Oral and esophageal erosions can occur. Pruritus is typically mild, although the lesions are often painful and flu-like symptoms are often present.28
Pathophysiology. The pathophysiology of this disease is unknown.
Differential. Conditions with similar presentations include an adverse drug reaction, pityriasis rosea, lichen simplex chronicus, eczema, lupus, and pityriasis rubra pilaris.
Diagnosis. Clinical history and association with systemic illness are the basis for a diagnosis of APPP. Cultures of the pustules are negative for any infective pathology, though as the disease progresses, pustules may become superinfected. Lab tests may show an increased erythrocyte sedimentation rate (ESR), hypocalcemia, and low levels of vitamin D.
Treatment. Prednisone 15 to 60 mg per day is often sufficient to control the disease.28 Cyclosporine 100 mg twice daily has also been shown to be useful.29 Cyclosporine in pregnancy is a category C drug. Data on fetal malformation associated with cyclosporine therapy are limited, but the risk appears to be minimal.6 Maternal hypocalcemia should be monitored and treated appropriately. If disease progression is judged serious enough, early induction of labor is indicated, since delivery will almost always lead to swift resolution.
Sequelae. There have been a number of case reports that link APPP to serious sequelae, including fetal growth retardation, hypocalcemia, and stillbirth.28,30,31 The condition is too rare, however, for good data on specific sequelae. While the disease does give significant cause for concern, it would appear that some of the traditional apprehension comes from older publications reporting a rate of maternal mortality of 70% to 90%.32 This statistic has not been borne out in clinical practice. It does appear that the mother will frequently suffer from systemic symptoms, including fever and malaise.
Pruritic folliculitis of pregnancy
Accounts of PFP’s prevalence vary widely: Some sources report fewer than 30 cases in all of the literature, while others indicate that the prevalence is equivalent to that of PG, 1 in 10,000.3,11 PFP most commonly presents in the third trimester. It often resolves before delivery, but uniformly clears within 2 weeks of delivery.
Presentation. PFP presents as papules and pustules concentrated around hair follicles (FIGURE 5). Often lesions begin on the abdomen and spread to the extremities.24,29 The condition is often, but not always, pruritic. Patients are more likely to be concerned about what the condition means for their health, rather than being distressed by the symptoms.
Pathophysiology. Like so many of these conditions, the pathophysiology of PFP is unknown. There is little evidence that the condition is immunologically or hormonally mediated, and there is no evidence of an infectious component.24,29
Differential. PFP must be distinguished from infectious folliculitis, acneiform disorders, HIV-associated eosinophilic folliculitis, and a drug reaction.
Diagnosis. The clinical diagnosis is based on presenting symptoms and third trimester onset. No specific lab or histological analysis can be used to make a definitive diagnosis.
Treatment. As the condition is, by definition, a nonmicrobial folliculitis, the most effective therapy tends to be with mid- to lowpotency topical steroids such as triamcinolone or desonide. Additionally, benzoyl peroxide wash can be effective.
Sequelae. One study reports an increased incidence of low birth weight, but currently no associated morbidity or mortality has been reported.24
FIGURE 5
Pruritic folliculitis of pregnancy
CORRESPONDENCE
Matthew Bremmer, MD, 419 W Redwood Street, Department of Dermatology, Baltimore, MD 21230; Mbrem001@umaryland.edu
1. Cassian S, Powell J, Messer G, et al. Immunoblotting and enzymelinked immunosorbent assay for the diagnosis of pemphigoid gestationis. Obstet Gynecol. 2004;103:757-763.
2. Engineer L, Bohl K, Ahmed AR. Pemphigoid gestationis: a review. Am J Obstet Gynecol. 2000;183:483-491.
3. Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence based systematic review. Am J Obstet Gynecol, 2003;188:1083-1092.
4. Shornick JD. Dermatoses of pregnancy. Semin Cutan Med Surg. 1998;17:172-181.
5. Shornick JK, Bangert JL, Freeman RG, et al. Herpes gestationis: clinical and histological features in 28 cases. J Am Acad Dermatol. 1983;8:214-224.
6. Leachman S, Reed B. The use of dermatologic drugs in pregnancy and lactation. Dermatol Clin. 2004;24:167-197.
7. Hern S, Harman K, Bhogal BS, et al. A severe persistent case of pemphigoid gestationis treated with intravenous immunoglobulins and cyclosporine. Clin Exp Dermatol. 1998;23:185-188.
8. Fitzpatrick TP. Diseases in pregnancy. Color Atlas and Synopsis of Clinical Dermatology. New York, NY: McGraw Hill; 1997:414–419.
9. Aractingi D, Berkane N, Bertheau P, et al. Fetal DNA in skin of polymorphic eruptions of pregnancy. Lancet. 1998;352:1898-1901.
10. Rudolph CM, Al-Fares S, Vaughan-Jones SA, et al. Polymorphic eruption of pregnancy: clinicopathology and potential trigger factors in 181 patients. Clin Lab Invest. 2006;154:54-60.
11. Roger D, Vaillant L, Fignon A, et al. Specific pruritic diseases of pregnancy. A prospective study of 3192 pregnant women. Arch Dermatol. 1994;130:734-739.
12. McDonald J. Cholestasis of pregnancy. J Gastroenterol Hepatol. 1999;14:515-518.
13. Ohel I, Levy A, Silberstein T, et al. Pregnancy outcome of patients with pruritic urticarial papules and plaques of pregnancy. J Maternal-Fetal Neonat Med. 2006;19:305-308.
14. Geenes V, Williamson C. Intrahepatic cholestasis of pregnancy. World J Gastroenterol. 2009;15:2049-2066.
15. Shaw D, Frohlich J, Wittmann BA, et al. A prospective study of 18 patients with cholestasis of pregnancy. Am J Obstet Gynecol. 1982;142:621-625.
16. Reyes H. The spectrum of liver and gastrointestinal disease seen in cholestasis of pregnancy. Gastroenterol Clin North Am. 1992;21:905-921.
17. Lammert F, Marschall H, Glantz A, et al. Intrahepatic cholestasis of pregnancy; molecular pathogenesis, diagnosis and management. J Hepatol. 2000;33:1012-1021.
18. Huang WM, Seubert DE, Donnelly JG, et al. Intrahepatic cholestasis of pregnancy: detection with urinary bile acid assays. J Perinat Med. 2007;35:486-491.
19. Palma J, Reyes H, Ribalta J, et al. Ursodeoxycholic acid in the treatment of intrahepatic cholestasis of pregnancy: a randomized, double blind study controlled with placebo. J Hepatol. 1997;27:1022-1028.
20. Diaferia A, Nicastri PL, Tartagni M, et al. Ursodeoxycholic acid therapy in pregnant women with cholestasis. Int J Gynaecol Obstet. 1996;52:133-140.
21. Nicastri PL, Diaferia A, Tartagni M, et al. A randomised placebocontrolled trial of ursodeoxycholic acid and S-adenosylmethionine in the treatment of intrahepatic cholestasis of pregnancy. Br J Obstet Gynaecol. 1998;105:1205-1207.
22. Glantz A, Marschall HU, Lammert F, et al. Intrahepatic cholestasis in pregnancy: a randomized controlled trial comparing dexamethasone and ursodeoxycholic acid. Hepatology. 2005;42:1399-1405.
23. Ambros-Rudolph CM, Mullegger MM, Vaughan-Jones SA, et al. The specific dermatoses of pregnancy revisited and reclassified: results of a retrospective two-center study on 505 pregnant patients. J Am Acad Dermatol. 2006;54:395-404.
24. Vaughan-Jones SA, Hern S, Nelson-Piercy C, et al. A prospective study of 200 women with dermatoses of pregnancy correlating clinical findings with hormonal and immunopathological profiles. Br J Dermatol. 1999;141:71-81.
25. Shornick JK. Dermatoses of pregnancy. Semin Cutan Med Surg. 1998;17:172-181.
26. Holmes RC, Black MM. The specific dermatoses of pregnancy. J Am Acad Dermatol. 1983;8:405-412.
27. Bukhari IA. Impetigo herpetiformis in a primagravida: successful treatment with etretinate. J Drugs Dermatol. 2004;3:449-451.
28. Oumeish OY, Parish JL. Impetigo herpetiformis. Clin in Dermatol. 2006;24:101-104.
29. Kroumpouzos G, Cohen LM. Pruritic folliculitis of pregnancy. J Am Acad Dermatol. 2000;43:132-134.
30. Sahin HG, Hasin HA, Metin A, et al. Recurrent impetigo herpetiformis in a pregnant adolescent: a case report. Eur J Obstet Gynecol Reprod Endocrinol. 2002;101:201-203.
31. Aka N, Kuscu NK, Yazicioglu E. Impetigo herpetiformis at the 36th week of gestation. Int J Gynecol Obstet. 2000;69:153-154.
32. Wade TR, Wade SL, Jones HE. Skin changes and diseases associated with pregnancy. Obstet Gynecol. 1978;52:233-242.
1. Cassian S, Powell J, Messer G, et al. Immunoblotting and enzymelinked immunosorbent assay for the diagnosis of pemphigoid gestationis. Obstet Gynecol. 2004;103:757-763.
2. Engineer L, Bohl K, Ahmed AR. Pemphigoid gestationis: a review. Am J Obstet Gynecol. 2000;183:483-491.
3. Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence based systematic review. Am J Obstet Gynecol, 2003;188:1083-1092.
4. Shornick JD. Dermatoses of pregnancy. Semin Cutan Med Surg. 1998;17:172-181.
5. Shornick JK, Bangert JL, Freeman RG, et al. Herpes gestationis: clinical and histological features in 28 cases. J Am Acad Dermatol. 1983;8:214-224.
6. Leachman S, Reed B. The use of dermatologic drugs in pregnancy and lactation. Dermatol Clin. 2004;24:167-197.
7. Hern S, Harman K, Bhogal BS, et al. A severe persistent case of pemphigoid gestationis treated with intravenous immunoglobulins and cyclosporine. Clin Exp Dermatol. 1998;23:185-188.
8. Fitzpatrick TP. Diseases in pregnancy. Color Atlas and Synopsis of Clinical Dermatology. New York, NY: McGraw Hill; 1997:414–419.
9. Aractingi D, Berkane N, Bertheau P, et al. Fetal DNA in skin of polymorphic eruptions of pregnancy. Lancet. 1998;352:1898-1901.
10. Rudolph CM, Al-Fares S, Vaughan-Jones SA, et al. Polymorphic eruption of pregnancy: clinicopathology and potential trigger factors in 181 patients. Clin Lab Invest. 2006;154:54-60.
11. Roger D, Vaillant L, Fignon A, et al. Specific pruritic diseases of pregnancy. A prospective study of 3192 pregnant women. Arch Dermatol. 1994;130:734-739.
12. McDonald J. Cholestasis of pregnancy. J Gastroenterol Hepatol. 1999;14:515-518.
13. Ohel I, Levy A, Silberstein T, et al. Pregnancy outcome of patients with pruritic urticarial papules and plaques of pregnancy. J Maternal-Fetal Neonat Med. 2006;19:305-308.
14. Geenes V, Williamson C. Intrahepatic cholestasis of pregnancy. World J Gastroenterol. 2009;15:2049-2066.
15. Shaw D, Frohlich J, Wittmann BA, et al. A prospective study of 18 patients with cholestasis of pregnancy. Am J Obstet Gynecol. 1982;142:621-625.
16. Reyes H. The spectrum of liver and gastrointestinal disease seen in cholestasis of pregnancy. Gastroenterol Clin North Am. 1992;21:905-921.
17. Lammert F, Marschall H, Glantz A, et al. Intrahepatic cholestasis of pregnancy; molecular pathogenesis, diagnosis and management. J Hepatol. 2000;33:1012-1021.
18. Huang WM, Seubert DE, Donnelly JG, et al. Intrahepatic cholestasis of pregnancy: detection with urinary bile acid assays. J Perinat Med. 2007;35:486-491.
19. Palma J, Reyes H, Ribalta J, et al. Ursodeoxycholic acid in the treatment of intrahepatic cholestasis of pregnancy: a randomized, double blind study controlled with placebo. J Hepatol. 1997;27:1022-1028.
20. Diaferia A, Nicastri PL, Tartagni M, et al. Ursodeoxycholic acid therapy in pregnant women with cholestasis. Int J Gynaecol Obstet. 1996;52:133-140.
21. Nicastri PL, Diaferia A, Tartagni M, et al. A randomised placebocontrolled trial of ursodeoxycholic acid and S-adenosylmethionine in the treatment of intrahepatic cholestasis of pregnancy. Br J Obstet Gynaecol. 1998;105:1205-1207.
22. Glantz A, Marschall HU, Lammert F, et al. Intrahepatic cholestasis in pregnancy: a randomized controlled trial comparing dexamethasone and ursodeoxycholic acid. Hepatology. 2005;42:1399-1405.
23. Ambros-Rudolph CM, Mullegger MM, Vaughan-Jones SA, et al. The specific dermatoses of pregnancy revisited and reclassified: results of a retrospective two-center study on 505 pregnant patients. J Am Acad Dermatol. 2006;54:395-404.
24. Vaughan-Jones SA, Hern S, Nelson-Piercy C, et al. A prospective study of 200 women with dermatoses of pregnancy correlating clinical findings with hormonal and immunopathological profiles. Br J Dermatol. 1999;141:71-81.
25. Shornick JK. Dermatoses of pregnancy. Semin Cutan Med Surg. 1998;17:172-181.
26. Holmes RC, Black MM. The specific dermatoses of pregnancy. J Am Acad Dermatol. 1983;8:405-412.
27. Bukhari IA. Impetigo herpetiformis in a primagravida: successful treatment with etretinate. J Drugs Dermatol. 2004;3:449-451.
28. Oumeish OY, Parish JL. Impetigo herpetiformis. Clin in Dermatol. 2006;24:101-104.
29. Kroumpouzos G, Cohen LM. Pruritic folliculitis of pregnancy. J Am Acad Dermatol. 2000;43:132-134.
30. Sahin HG, Hasin HA, Metin A, et al. Recurrent impetigo herpetiformis in a pregnant adolescent: a case report. Eur J Obstet Gynecol Reprod Endocrinol. 2002;101:201-203.
31. Aka N, Kuscu NK, Yazicioglu E. Impetigo herpetiformis at the 36th week of gestation. Int J Gynecol Obstet. 2000;69:153-154.
32. Wade TR, Wade SL, Jones HE. Skin changes and diseases associated with pregnancy. Obstet Gynecol. 1978;52:233-242.