User login
To the Marrow
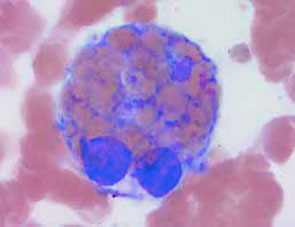
A44-year-old male presented with ecthyma gangrenosum and Pseudomonas aeruginosa bacteremia after a two-year history of fever of unknown origin, pancytopenia, hypertriglyceridemia, and splenomegaly. A bone marrow aspirate was performed, as shown.
Based on the bone marrow aspirate, the most likely diagnosis is:
- Acute leukemia
- Myelofibrosis with myeloid metaplasia
- Multiple myeloma
- Myelodysplastic syndrome
- Hemophagocytic syndrome.
Discussion
The answer is e, hemophagocytic syndrome. The bone marrow aspirate shown demonstrates macrophage hemophagocytosis of non-nucleated red blood cells, consistent with hemophagocytic syndrome (HPS). The hemophagocytic syndromes may be classified as either primary or secondary.
Primary HPS is an autosomal recessive disorder most commonly seen in children and characterized by the polyclonal accumulation of T-lymphocytes and activated macrophages. Many of these patients have null mutations in the gene coding for the cytolytic protein perforin.
In contrast, secondary HPS is characterized by the polyclonal accumulation of activated macrophages in patients with underlying infectious, malignant, or rheumatologic diseases. Patients commonly present with fever, splenomegaly, and complications related to pancytopenia. Hypertriglyceridemia (>160 mg/dL) and an elevated serum ferritin (>10,000 ng/mL) are all sensitive and specific (>0.75) for HPS in the appropriate clinical setting, though histologic demonstration of hemophagocytosis (ingestion of red blood cells by cytokine-activated macrophages) in the bone marrow is diagnostic.
Macrophage activation in these disorders may be attributed to dysregulation of cytokines such as IL-1, IL-6, IFN-y and TNF-a. While therapy with corticosteroids, immunosuppressants, intravenous immunoglobulin, and chemotherapeutic agents have provided conflicting results, future therapeutic strategies employing cytokine-specific antagonists (e.g., etanercept) are promising.1,2
In this case, the patient was noted to have a relative lymphocytosis comprising clonal CD16+CD56+ large granular lymphocytes. These large granular lymphocytes stained positive for Epstein-Barr virus (EBV)-encoded RNA by in situ hybridization. This patient with EBV-associated natural killer (NK) cell lymphoma complicated by hemophagocytic syndrome failed to recover, despite treatment with broad spectrum antibiotics, neutrophil transfusions, intravenous immunoglobulin, fludarabine, and cyclophosphamide.
The clinical and laboratory features of HPS, including fever of unknown origin, anemia, and splenomegaly, often mimic other disorders common in hospitalized patients—many of which may be associated with secondary HPS. As this case illustrates, secondary HPS is associated with significant morbidity and mortality, particularly in those patients in which the diagnosis is delayed. Therefore, prompt diagnosis requires a high index of suspicion among hospital-based physicians caring for patients with underlying infectious, rheumatologic or malignant conditions commonly associated with secondary HPS. TH
References
- Ravelli A. Macrophage activation syndrome. Curr Opin Rheumatol. 2002;14:548-552.
- Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991;18:29.
A44-year-old male presented with ecthyma gangrenosum and Pseudomonas aeruginosa bacteremia after a two-year history of fever of unknown origin, pancytopenia, hypertriglyceridemia, and splenomegaly. A bone marrow aspirate was performed, as shown.
Based on the bone marrow aspirate, the most likely diagnosis is:
- Acute leukemia
- Myelofibrosis with myeloid metaplasia
- Multiple myeloma
- Myelodysplastic syndrome
- Hemophagocytic syndrome.
Discussion
The answer is e, hemophagocytic syndrome. The bone marrow aspirate shown demonstrates macrophage hemophagocytosis of non-nucleated red blood cells, consistent with hemophagocytic syndrome (HPS). The hemophagocytic syndromes may be classified as either primary or secondary.
Primary HPS is an autosomal recessive disorder most commonly seen in children and characterized by the polyclonal accumulation of T-lymphocytes and activated macrophages. Many of these patients have null mutations in the gene coding for the cytolytic protein perforin.
In contrast, secondary HPS is characterized by the polyclonal accumulation of activated macrophages in patients with underlying infectious, malignant, or rheumatologic diseases. Patients commonly present with fever, splenomegaly, and complications related to pancytopenia. Hypertriglyceridemia (>160 mg/dL) and an elevated serum ferritin (>10,000 ng/mL) are all sensitive and specific (>0.75) for HPS in the appropriate clinical setting, though histologic demonstration of hemophagocytosis (ingestion of red blood cells by cytokine-activated macrophages) in the bone marrow is diagnostic.
Macrophage activation in these disorders may be attributed to dysregulation of cytokines such as IL-1, IL-6, IFN-y and TNF-a. While therapy with corticosteroids, immunosuppressants, intravenous immunoglobulin, and chemotherapeutic agents have provided conflicting results, future therapeutic strategies employing cytokine-specific antagonists (e.g., etanercept) are promising.1,2
In this case, the patient was noted to have a relative lymphocytosis comprising clonal CD16+CD56+ large granular lymphocytes. These large granular lymphocytes stained positive for Epstein-Barr virus (EBV)-encoded RNA by in situ hybridization. This patient with EBV-associated natural killer (NK) cell lymphoma complicated by hemophagocytic syndrome failed to recover, despite treatment with broad spectrum antibiotics, neutrophil transfusions, intravenous immunoglobulin, fludarabine, and cyclophosphamide.
The clinical and laboratory features of HPS, including fever of unknown origin, anemia, and splenomegaly, often mimic other disorders common in hospitalized patients—many of which may be associated with secondary HPS. As this case illustrates, secondary HPS is associated with significant morbidity and mortality, particularly in those patients in which the diagnosis is delayed. Therefore, prompt diagnosis requires a high index of suspicion among hospital-based physicians caring for patients with underlying infectious, rheumatologic or malignant conditions commonly associated with secondary HPS. TH
References
- Ravelli A. Macrophage activation syndrome. Curr Opin Rheumatol. 2002;14:548-552.
- Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991;18:29.
A44-year-old male presented with ecthyma gangrenosum and Pseudomonas aeruginosa bacteremia after a two-year history of fever of unknown origin, pancytopenia, hypertriglyceridemia, and splenomegaly. A bone marrow aspirate was performed, as shown.
Based on the bone marrow aspirate, the most likely diagnosis is:
- Acute leukemia
- Myelofibrosis with myeloid metaplasia
- Multiple myeloma
- Myelodysplastic syndrome
- Hemophagocytic syndrome.
Discussion
The answer is e, hemophagocytic syndrome. The bone marrow aspirate shown demonstrates macrophage hemophagocytosis of non-nucleated red blood cells, consistent with hemophagocytic syndrome (HPS). The hemophagocytic syndromes may be classified as either primary or secondary.
Primary HPS is an autosomal recessive disorder most commonly seen in children and characterized by the polyclonal accumulation of T-lymphocytes and activated macrophages. Many of these patients have null mutations in the gene coding for the cytolytic protein perforin.
In contrast, secondary HPS is characterized by the polyclonal accumulation of activated macrophages in patients with underlying infectious, malignant, or rheumatologic diseases. Patients commonly present with fever, splenomegaly, and complications related to pancytopenia. Hypertriglyceridemia (>160 mg/dL) and an elevated serum ferritin (>10,000 ng/mL) are all sensitive and specific (>0.75) for HPS in the appropriate clinical setting, though histologic demonstration of hemophagocytosis (ingestion of red blood cells by cytokine-activated macrophages) in the bone marrow is diagnostic.
Macrophage activation in these disorders may be attributed to dysregulation of cytokines such as IL-1, IL-6, IFN-y and TNF-a. While therapy with corticosteroids, immunosuppressants, intravenous immunoglobulin, and chemotherapeutic agents have provided conflicting results, future therapeutic strategies employing cytokine-specific antagonists (e.g., etanercept) are promising.1,2
In this case, the patient was noted to have a relative lymphocytosis comprising clonal CD16+CD56+ large granular lymphocytes. These large granular lymphocytes stained positive for Epstein-Barr virus (EBV)-encoded RNA by in situ hybridization. This patient with EBV-associated natural killer (NK) cell lymphoma complicated by hemophagocytic syndrome failed to recover, despite treatment with broad spectrum antibiotics, neutrophil transfusions, intravenous immunoglobulin, fludarabine, and cyclophosphamide.
The clinical and laboratory features of HPS, including fever of unknown origin, anemia, and splenomegaly, often mimic other disorders common in hospitalized patients—many of which may be associated with secondary HPS. As this case illustrates, secondary HPS is associated with significant morbidity and mortality, particularly in those patients in which the diagnosis is delayed. Therefore, prompt diagnosis requires a high index of suspicion among hospital-based physicians caring for patients with underlying infectious, rheumatologic or malignant conditions commonly associated with secondary HPS. TH
References
- Ravelli A. Macrophage activation syndrome. Curr Opin Rheumatol. 2002;14:548-552.
- Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin Oncol. 1991;18:29.