User login
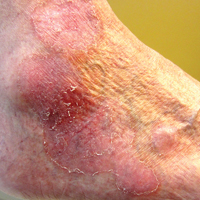
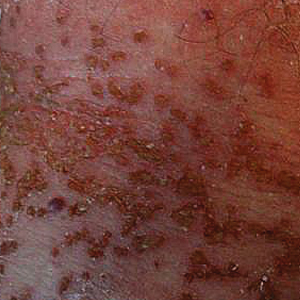
Annular Erythematous Plaques With Central Hypopigmentation on Sun-Exposed Skin
A biopsy showed a markedly elastotic dermis consisting of a palisading granulomatous inflammatory infiltrate and numerous multinucleated histiocytes (Figure). These histopathologic findings along with the clinical presentation confirmed a diagnosis of annular elastolytic granuloma (AEG). Treatment consisting of 3 months of oral minocycline, 2 months of oral doxycycline, and clobetasol ointment all failed. At that point, oral hydroxychloroquine was recommended. Our patient was lost to follow-up by dermatology, then subsequently was placed on hydroxychloroquine by rheumatology to treat both the osteoarthritis and AEG. A follow-up appointment with dermatology was planned for 3 months to monitor hydroxychloroquine treatment and monitor treatment progress; however, she did not follow-up or seek further treatment.

Annular elastolytic granuloma clinically is similar to granuloma annulare (GA), with both presenting as annular plaques surrounded by an elevated border.1 Although AEG clinically is distinct with hypopigmented atrophied plaque centers,2 a biopsy is required to confirm the lack of elastic tissue in zones of atrophy and the presence of multinucleated histiocytes.1,3 Lesions most commonly are seen clinically on sun-exposed areas in middle-aged White women; however, they rarely have been seen on frequently covered skin.4 Our case illustrates the striking photodistribution of AEG, especially on the posterior neck area. The clinical diagnoses of AEG, annular elastolytic giant cell granuloma, and GA in sun-exposed areas are synonymous and can be used interchangeably.5,6
Pathologies considered in the diagnosis of AEG include but are not limited to tinea corporis, annular lichen planus, erythema annulare centrifugum, and necrobiosis lipoidica. Scaling typically is absent in AEG, while tinea corporis presents with hyphae within the stratum corneum of the plaques.7 Papules along the periphery of annular lesions are more typical of annular lichen planus than AEG, and they tend to have a more purple hue.8 Erythema annulare centrifugum has annular erythematous plaques similar to those found in AEG but differs with scaling on the inner margins of these plaques. Histopathology presenting with a lymphocytic infiltrate surrounding vasculature and no indication of elastolytic degradation would further indicate a diagnosis of erythema annulare centrifugum.9 Histopathology showing necrobiosis, lipid depositions, and vascular wall thickenings is indicative of necrobiosis lipoidica.10
Similar to GA,11 the cause of AEG is idiopathic.2 Annular elastolytic granuloma and GA differ in the fact that elastin degradation is characteristic of AEG compared to collagen degradation in GA. It is suspected that elastin degradation in AEG patients is caused by an immune response triggering phagocytosis of elastin by multinucleated histiocytes.2 Actinic damage also is considered a possible cause of elastin fiber degradation in AEG.12 Granuloma annulare can be ruled out and the diagnosis of AEG confirmed with the absence of elastin fibers and mucin on pathology.13
Although there is no established first-line treatment of AEG, successful treatment has been achieved with antimalarial drugs paired with topical steroids.14 Treatment recommendations for AEG include minocycline, chloroquine, hydroxychloroquine, tranilast, and oral retinoids, as well as oral and topical steroids. In clinical cases where AEG occurs in the setting of a chronic disease such as diabetes mellitus, vascular occlusion, arthritis, or hypertension, treatment of underlying disease has been shown to resolve AEG symptoms.14
Although light therapy is not common for AEG, UV light radiation has demonstrated success in treating AEG.15,16 One study showed complete clearance of granulomatous papules after narrowband UVB treatment.15 Another study showed that 2 patients treated with psoralen plus UVA therapy reached complete clearance of AEG lasting at least 3 months after treatment.16
1. Lai JH, Murray SJ, Walsh NM. Evolution of granuloma annulare to mid-dermal elastolysis: report of a case and review of the literature. J Cutan Pathol. 2014;41:462-468. doi:10.1111/cup.12292 2. Klemke CD, Siebold D, Dippel E, et al. Generalised annular elastolytic giant cell granuloma. Dermatology. 2003;207:420-422. doi:10.1159/000074132 3. Limas C. The spectrum of primary cutaneous elastolytic granulomas and their distinction from granuloma annulare: a clinicopathological analysis. Histopathology. 2004;44:277-282. doi:10.1111/j.0309-0167.2004.01755.x 4. Revenga F, Rovira I, Pimentel J, et al. Annular elastolytic giant cell granuloma—actinic granuloma? Clin Exp Dermatol. 1996;21:51-53. 5. Hawryluk EB, Izikson L, English JC 3rd. Non-infectious granulomatous diseases of the skin and their associated systemic diseases: an evidence-based update to important clinical questions. Am J Clin Dermatol. 2010;11:171-181. doi:10.2165/11530080-000000000-00000 6. Berliner JG, Haemel A, LeBoit PE, et al. The sarcoidal variant of annular elastolytic granuloma. J Cutan Pathol. 2013;40:918-920. doi:10.1111/cup.12237 7. Pflederer RT, Ahmed S, Tonkovic-Capin V, et al. Annular polycyclic plaques on the chest and upper back [published online April 24, 2018]. JAAD Case Rep. 2018;4:405-407. doi:10.1016/j.jdcr.2017.07.022 8. Trayes KP, Savage K, Studdiford JS. Annular lesions: diagnosis and treatment. Am Fam Physician. 2018;98:283-291. 9. Weyers W, Diaz-Cascajo C, Weyers I. Erythema annulare centrifugum: results of a clinicopathologic study of 73 patients. Am J Dermatopathol. 2003;25:451-462. doi:10.1097/00000372-200312000-00001 10. Dowling GB, Jones EW. Atypical (annular) necrobiosis lipoidica of the face and scalp. a report of the clinical and histological features of 7 cases. Dermatologica. 1967;135:11-26. doi:10.1159/000254156 11. Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479. doi:10.1016/j.jaad.2015 .03.055 12. O’Brien JP, Regan W. Actinically degenerate elastic tissue is the likely antigenic basis of actinic granuloma of the skin and of temporal arteritis [published correction appears in J Am Acad Dermatol. 2000; 42(1 pt 1):148]. J Am Acad Dermatol. 1999;40(2 pt 1):214-222. doi:10.1016/s0190-9622(99)70191-x 13. Rencic A, Nousari CH. Other rheumatologic diseases. In: Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. 2nd ed. Elsevier Limited; 2008:600-601. 14. Burlando M, Herzum A, Cozzani E, et al. Can methotrexate be a successful treatment for unresponsive generalized annular elastolytic giant cell granuloma? case report and review of the literature. Dermatol Ther. 2021;34:E14705. doi:10.1111/dth.14705 15. Takata T, Ikeda M, Kodama H, et al. Regression of papular elastolytic giant cell granuloma using narrow-band UVB irradiation. Dermatology. 2006;212:77-79. doi:10.1159/000089028 16. Pérez-Pérez L, García-Gavín J, Allegue F, et al. Successful treatment of generalized elastolytic giant cell granuloma with psoralenultraviolet A. Photodermatol Photoimmunol Photomed. 2012;28:264-266. doi:10.1111/j.1600-0781.2012.00680.x
A biopsy showed a markedly elastotic dermis consisting of a palisading granulomatous inflammatory infiltrate and numerous multinucleated histiocytes (Figure). These histopathologic findings along with the clinical presentation confirmed a diagnosis of annular elastolytic granuloma (AEG). Treatment consisting of 3 months of oral minocycline, 2 months of oral doxycycline, and clobetasol ointment all failed. At that point, oral hydroxychloroquine was recommended. Our patient was lost to follow-up by dermatology, then subsequently was placed on hydroxychloroquine by rheumatology to treat both the osteoarthritis and AEG. A follow-up appointment with dermatology was planned for 3 months to monitor hydroxychloroquine treatment and monitor treatment progress; however, she did not follow-up or seek further treatment.
Annular elastolytic granuloma clinically is similar to granuloma annulare (GA), with both presenting as annular plaques surrounded by an elevated border.1 Although AEG clinically is distinct with hypopigmented atrophied plaque centers,2 a biopsy is required to confirm the lack of elastic tissue in zones of atrophy and the presence of multinucleated histiocytes.1,3 Lesions most commonly are seen clinically on sun-exposed areas in middle-aged White women; however, they rarely have been seen on frequently covered skin.4 Our case illustrates the striking photodistribution of AEG, especially on the posterior neck area. The clinical diagnoses of AEG, annular elastolytic giant cell granuloma, and GA in sun-exposed areas are synonymous and can be used interchangeably.5,6
Pathologies considered in the diagnosis of AEG include but are not limited to tinea corporis, annular lichen planus, erythema annulare centrifugum, and necrobiosis lipoidica. Scaling typically is absent in AEG, while tinea corporis presents with hyphae within the stratum corneum of the plaques.7 Papules along the periphery of annular lesions are more typical of annular lichen planus than AEG, and they tend to have a more purple hue.8 Erythema annulare centrifugum has annular erythematous plaques similar to those found in AEG but differs with scaling on the inner margins of these plaques. Histopathology presenting with a lymphocytic infiltrate surrounding vasculature and no indication of elastolytic degradation would further indicate a diagnosis of erythema annulare centrifugum.9 Histopathology showing necrobiosis, lipid depositions, and vascular wall thickenings is indicative of necrobiosis lipoidica.10
Similar to GA,11 the cause of AEG is idiopathic.2 Annular elastolytic granuloma and GA differ in the fact that elastin degradation is characteristic of AEG compared to collagen degradation in GA. It is suspected that elastin degradation in AEG patients is caused by an immune response triggering phagocytosis of elastin by multinucleated histiocytes.2 Actinic damage also is considered a possible cause of elastin fiber degradation in AEG.12 Granuloma annulare can be ruled out and the diagnosis of AEG confirmed with the absence of elastin fibers and mucin on pathology.13
Although there is no established first-line treatment of AEG, successful treatment has been achieved with antimalarial drugs paired with topical steroids.14 Treatment recommendations for AEG include minocycline, chloroquine, hydroxychloroquine, tranilast, and oral retinoids, as well as oral and topical steroids. In clinical cases where AEG occurs in the setting of a chronic disease such as diabetes mellitus, vascular occlusion, arthritis, or hypertension, treatment of underlying disease has been shown to resolve AEG symptoms.14
Although light therapy is not common for AEG, UV light radiation has demonstrated success in treating AEG.15,16 One study showed complete clearance of granulomatous papules after narrowband UVB treatment.15 Another study showed that 2 patients treated with psoralen plus UVA therapy reached complete clearance of AEG lasting at least 3 months after treatment.16
A biopsy showed a markedly elastotic dermis consisting of a palisading granulomatous inflammatory infiltrate and numerous multinucleated histiocytes (Figure). These histopathologic findings along with the clinical presentation confirmed a diagnosis of annular elastolytic granuloma (AEG). Treatment consisting of 3 months of oral minocycline, 2 months of oral doxycycline, and clobetasol ointment all failed. At that point, oral hydroxychloroquine was recommended. Our patient was lost to follow-up by dermatology, then subsequently was placed on hydroxychloroquine by rheumatology to treat both the osteoarthritis and AEG. A follow-up appointment with dermatology was planned for 3 months to monitor hydroxychloroquine treatment and monitor treatment progress; however, she did not follow-up or seek further treatment.
Annular elastolytic granuloma clinically is similar to granuloma annulare (GA), with both presenting as annular plaques surrounded by an elevated border.1 Although AEG clinically is distinct with hypopigmented atrophied plaque centers,2 a biopsy is required to confirm the lack of elastic tissue in zones of atrophy and the presence of multinucleated histiocytes.1,3 Lesions most commonly are seen clinically on sun-exposed areas in middle-aged White women; however, they rarely have been seen on frequently covered skin.4 Our case illustrates the striking photodistribution of AEG, especially on the posterior neck area. The clinical diagnoses of AEG, annular elastolytic giant cell granuloma, and GA in sun-exposed areas are synonymous and can be used interchangeably.5,6
Pathologies considered in the diagnosis of AEG include but are not limited to tinea corporis, annular lichen planus, erythema annulare centrifugum, and necrobiosis lipoidica. Scaling typically is absent in AEG, while tinea corporis presents with hyphae within the stratum corneum of the plaques.7 Papules along the periphery of annular lesions are more typical of annular lichen planus than AEG, and they tend to have a more purple hue.8 Erythema annulare centrifugum has annular erythematous plaques similar to those found in AEG but differs with scaling on the inner margins of these plaques. Histopathology presenting with a lymphocytic infiltrate surrounding vasculature and no indication of elastolytic degradation would further indicate a diagnosis of erythema annulare centrifugum.9 Histopathology showing necrobiosis, lipid depositions, and vascular wall thickenings is indicative of necrobiosis lipoidica.10
Similar to GA,11 the cause of AEG is idiopathic.2 Annular elastolytic granuloma and GA differ in the fact that elastin degradation is characteristic of AEG compared to collagen degradation in GA. It is suspected that elastin degradation in AEG patients is caused by an immune response triggering phagocytosis of elastin by multinucleated histiocytes.2 Actinic damage also is considered a possible cause of elastin fiber degradation in AEG.12 Granuloma annulare can be ruled out and the diagnosis of AEG confirmed with the absence of elastin fibers and mucin on pathology.13
Although there is no established first-line treatment of AEG, successful treatment has been achieved with antimalarial drugs paired with topical steroids.14 Treatment recommendations for AEG include minocycline, chloroquine, hydroxychloroquine, tranilast, and oral retinoids, as well as oral and topical steroids. In clinical cases where AEG occurs in the setting of a chronic disease such as diabetes mellitus, vascular occlusion, arthritis, or hypertension, treatment of underlying disease has been shown to resolve AEG symptoms.14
Although light therapy is not common for AEG, UV light radiation has demonstrated success in treating AEG.15,16 One study showed complete clearance of granulomatous papules after narrowband UVB treatment.15 Another study showed that 2 patients treated with psoralen plus UVA therapy reached complete clearance of AEG lasting at least 3 months after treatment.16
1. Lai JH, Murray SJ, Walsh NM. Evolution of granuloma annulare to mid-dermal elastolysis: report of a case and review of the literature. J Cutan Pathol. 2014;41:462-468. doi:10.1111/cup.12292 2. Klemke CD, Siebold D, Dippel E, et al. Generalised annular elastolytic giant cell granuloma. Dermatology. 2003;207:420-422. doi:10.1159/000074132 3. Limas C. The spectrum of primary cutaneous elastolytic granulomas and their distinction from granuloma annulare: a clinicopathological analysis. Histopathology. 2004;44:277-282. doi:10.1111/j.0309-0167.2004.01755.x 4. Revenga F, Rovira I, Pimentel J, et al. Annular elastolytic giant cell granuloma—actinic granuloma? Clin Exp Dermatol. 1996;21:51-53. 5. Hawryluk EB, Izikson L, English JC 3rd. Non-infectious granulomatous diseases of the skin and their associated systemic diseases: an evidence-based update to important clinical questions. Am J Clin Dermatol. 2010;11:171-181. doi:10.2165/11530080-000000000-00000 6. Berliner JG, Haemel A, LeBoit PE, et al. The sarcoidal variant of annular elastolytic granuloma. J Cutan Pathol. 2013;40:918-920. doi:10.1111/cup.12237 7. Pflederer RT, Ahmed S, Tonkovic-Capin V, et al. Annular polycyclic plaques on the chest and upper back [published online April 24, 2018]. JAAD Case Rep. 2018;4:405-407. doi:10.1016/j.jdcr.2017.07.022 8. Trayes KP, Savage K, Studdiford JS. Annular lesions: diagnosis and treatment. Am Fam Physician. 2018;98:283-291. 9. Weyers W, Diaz-Cascajo C, Weyers I. Erythema annulare centrifugum: results of a clinicopathologic study of 73 patients. Am J Dermatopathol. 2003;25:451-462. doi:10.1097/00000372-200312000-00001 10. Dowling GB, Jones EW. Atypical (annular) necrobiosis lipoidica of the face and scalp. a report of the clinical and histological features of 7 cases. Dermatologica. 1967;135:11-26. doi:10.1159/000254156 11. Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479. doi:10.1016/j.jaad.2015 .03.055 12. O’Brien JP, Regan W. Actinically degenerate elastic tissue is the likely antigenic basis of actinic granuloma of the skin and of temporal arteritis [published correction appears in J Am Acad Dermatol. 2000; 42(1 pt 1):148]. J Am Acad Dermatol. 1999;40(2 pt 1):214-222. doi:10.1016/s0190-9622(99)70191-x 13. Rencic A, Nousari CH. Other rheumatologic diseases. In: Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. 2nd ed. Elsevier Limited; 2008:600-601. 14. Burlando M, Herzum A, Cozzani E, et al. Can methotrexate be a successful treatment for unresponsive generalized annular elastolytic giant cell granuloma? case report and review of the literature. Dermatol Ther. 2021;34:E14705. doi:10.1111/dth.14705 15. Takata T, Ikeda M, Kodama H, et al. Regression of papular elastolytic giant cell granuloma using narrow-band UVB irradiation. Dermatology. 2006;212:77-79. doi:10.1159/000089028 16. Pérez-Pérez L, García-Gavín J, Allegue F, et al. Successful treatment of generalized elastolytic giant cell granuloma with psoralenultraviolet A. Photodermatol Photoimmunol Photomed. 2012;28:264-266. doi:10.1111/j.1600-0781.2012.00680.x
1. Lai JH, Murray SJ, Walsh NM. Evolution of granuloma annulare to mid-dermal elastolysis: report of a case and review of the literature. J Cutan Pathol. 2014;41:462-468. doi:10.1111/cup.12292 2. Klemke CD, Siebold D, Dippel E, et al. Generalised annular elastolytic giant cell granuloma. Dermatology. 2003;207:420-422. doi:10.1159/000074132 3. Limas C. The spectrum of primary cutaneous elastolytic granulomas and their distinction from granuloma annulare: a clinicopathological analysis. Histopathology. 2004;44:277-282. doi:10.1111/j.0309-0167.2004.01755.x 4. Revenga F, Rovira I, Pimentel J, et al. Annular elastolytic giant cell granuloma—actinic granuloma? Clin Exp Dermatol. 1996;21:51-53. 5. Hawryluk EB, Izikson L, English JC 3rd. Non-infectious granulomatous diseases of the skin and their associated systemic diseases: an evidence-based update to important clinical questions. Am J Clin Dermatol. 2010;11:171-181. doi:10.2165/11530080-000000000-00000 6. Berliner JG, Haemel A, LeBoit PE, et al. The sarcoidal variant of annular elastolytic granuloma. J Cutan Pathol. 2013;40:918-920. doi:10.1111/cup.12237 7. Pflederer RT, Ahmed S, Tonkovic-Capin V, et al. Annular polycyclic plaques on the chest and upper back [published online April 24, 2018]. JAAD Case Rep. 2018;4:405-407. doi:10.1016/j.jdcr.2017.07.022 8. Trayes KP, Savage K, Studdiford JS. Annular lesions: diagnosis and treatment. Am Fam Physician. 2018;98:283-291. 9. Weyers W, Diaz-Cascajo C, Weyers I. Erythema annulare centrifugum: results of a clinicopathologic study of 73 patients. Am J Dermatopathol. 2003;25:451-462. doi:10.1097/00000372-200312000-00001 10. Dowling GB, Jones EW. Atypical (annular) necrobiosis lipoidica of the face and scalp. a report of the clinical and histological features of 7 cases. Dermatologica. 1967;135:11-26. doi:10.1159/000254156 11. Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479. doi:10.1016/j.jaad.2015 .03.055 12. O’Brien JP, Regan W. Actinically degenerate elastic tissue is the likely antigenic basis of actinic granuloma of the skin and of temporal arteritis [published correction appears in J Am Acad Dermatol. 2000; 42(1 pt 1):148]. J Am Acad Dermatol. 1999;40(2 pt 1):214-222. doi:10.1016/s0190-9622(99)70191-x 13. Rencic A, Nousari CH. Other rheumatologic diseases. In: Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. 2nd ed. Elsevier Limited; 2008:600-601. 14. Burlando M, Herzum A, Cozzani E, et al. Can methotrexate be a successful treatment for unresponsive generalized annular elastolytic giant cell granuloma? case report and review of the literature. Dermatol Ther. 2021;34:E14705. doi:10.1111/dth.14705 15. Takata T, Ikeda M, Kodama H, et al. Regression of papular elastolytic giant cell granuloma using narrow-band UVB irradiation. Dermatology. 2006;212:77-79. doi:10.1159/000089028 16. Pérez-Pérez L, García-Gavín J, Allegue F, et al. Successful treatment of generalized elastolytic giant cell granuloma with psoralenultraviolet A. Photodermatol Photoimmunol Photomed. 2012;28:264-266. doi:10.1111/j.1600-0781.2012.00680.x
A 67-year-old White woman presented to our dermatology clinic with pruritic annular erythematous plaques with central hypopigmentation on the forearms, dorsal aspect of the hands, neck, and fingers of 3 to 4 months’ duration. The patient rated the severity of pruritus an 8 on a 10-point scale. A review of symptoms was positive for fatigue, joint pain, and headache. The patient had a history of type 2 diabetes mellitus, osteoarthritis, thyroid disease, and stage 3 renal failure. A punch biopsy from the left forearm was performed.


Eczema Herpeticum in a Patient With Hailey-Hailey Disease Confounded by Coexistent Psoriasis
To the Editor:
Hailey-Hailey disease (HHD), also known as benign familial pemphigus, is an uncommon autosomal-dominant skin disease.1 Defects in the ATPase type 2C member 1 gene, ATP2C1, result in abnormal intracellular epidermal adherence, and patients experience recurring blisters in skin folds. Longitudinal white streaks of the fingernails also may be present.1 The illness does not appear until puberty and is heightened by the second or third decade of life. Family history often suggests the presence of disease.2 Misdiagnosis of HHD occurs because of a wide spectrum of presentations. The presence of superimposed infections and carcinomas may both obscure and exacerbate this disease.2
Herpes simplex viruse types 1 and 2 (HSV-1 and HSV-2) are DNA viruses that cause common recurrent diseases. Usually, HSV-1 is associated with infection of the mouth and HSV-2 is associated with infection of the genitalia.3 Longitudinal cutaneous lesions manifest as grouped vesicles on an erythematous base. Tzanck smear of herpetic vesicles will reveal the presence of multinucleated giant cells. A direct fluorescent antibody technique also may be used to confirm the diagnosis.3
Erythrodermic HHD disease is a rare condition; moreover, there are only a few reported cases with coexistence of HHD and HSV in the literature.3-6 We report a rare presentation of erythrodermic HHD and coexistent psoriasis with HSV superinfection.
A 69-year-old man presented to an outpatient dermatology clinic for evaluation and treatment of a rash on the scalp, face, back, and lower legs. The patient confirmed a dandruff diagnosis on the scalp and face as well as psoriasis on the trunk and extremities for the last 45 years. He described a history of successful treatment with topical agents and UV light therapy. A family history revealed that the patient’s father and 1 of 2 siblings had a similar rash and “skin problems.” The patient had a medical history of thyroid cancer treated with radiation treatment and a partial thyroidectomy 35 years prior to the current presentation as well as incompletely treated chronic hepatitis C.
A search of medical records revealed a punch biopsy from the posterior neck that demonstrated an acantholytic dyskeratosis with suprabasal acantholysis. Clinicians were unable to differentiate if it was Darier disease (DAR) or HHD. Treatment of the patient’s seborrheic dermatitis and acantholytic disorder was successful at that time with ketoconazole shampoo, ketoconazole cream, desonide cream, and triamcinolone cream. The patient remained stable for 5 years before presenting again to the dermatology clinic for worsening rash despite topical therapies.
At the current presentation, physical examination at the outpatient dermatology clinic revealed few scaly, erythematous, eroded papules distributed on the mid-back; erythematous greasy scaling on the scalp, face, and chest; and pink scaly plaques with white-silvery scale on the anterior lower legs. Histopathology of a specimen from the right mid-back demonstrated acantholysis with suprabasal clefting, hyperkeratosis, and parakeratosis with no dyskeratotic cells identified. The pathologic differential diagnosis included primary acantholytic processes including Grover disease, DAR, HHD, and pemphigus. Pathology from the right shin demonstrated acanthosis, confluent parakeratosis with associated decreased granular cell layer and collections of neutrophils within the stratum corneum, spongiosis, and superficial dermal perivascular chronic inflammation with focal exocytosis and dilated blood vessels in the papillary dermis. The clinical and pathological diagnosis on the lower legs was consistent with psoriasis. Diagnoses of seborrheic dermatitis, psoriasis on the lower legs, and HHD vs DAR on the back and chest were made. The patient was instructed to continue ketoconazole shampoo, ketoconazole cream, and desonide for seborrheic dermatitis; fluocinonide ointment 0.05% to the lower legs for psoriasis; and triamcinolone cream and a bland moisturizer to the back and chest for HHD.
Over the ensuing months, the rash worsened with erythema and scaling affecting more than half of the body surface area. Topical corticosteroids and bland emollients resulted in minimal success. Biologics and acitretin were considered for the psoriasiform dermatitis but avoided due to the patient’s medical history of thyroid cancer and chronic hepatitis C infection. Because the patient described prior success with UV light therapy for psoriasis, he requested light therapy. A subsequent trial of narrowband UVB light therapy initially improved some of the psoriasiform dermatitis on the trunk and extremities; however, after 4 weeks of treatment, the patient described pain in some of the skin and felt he was burned by minimal exposure to light therapy on one particular visit, which caused him to stop light therapy.
Approximately 2 weeks later, the patient presented to the emergency department stating his psoriasis was infected; he was diagnosed with psoriasis with secondary cellulitis and received intravenous vancomycin and piperacillin-tazobactam, with bacterial cultures demonstrating Corynebacterium and methicillin-resistant Staphylococcus aureus. Some improvement was noted in the patient’s skin after antibiotics were initiated, but he continued to describe worsening “burning and pain” throughout the psoriasis lesions. The patient’s care was transferred to the Veterans Affairs hospital where a dermatology inpatient consultation was placed.
Our initial dermatologic examination revealed generalized scaly erythroderma on the neck, trunk, and extremities, sparing the face, palms, and soles (Figure 1). Multiple crusted and intact vesicles also were present overlying the erythematous plaques on the chest, back, and proximal extremities, most grouped in clusters. The patient endorsed new symptoms of pain and burning. Tzanck smear from the abdomen along with shave biopsies from the left flank and right abdomen were performed, and intravenous acyclovir was initiated immediately after these procedures.
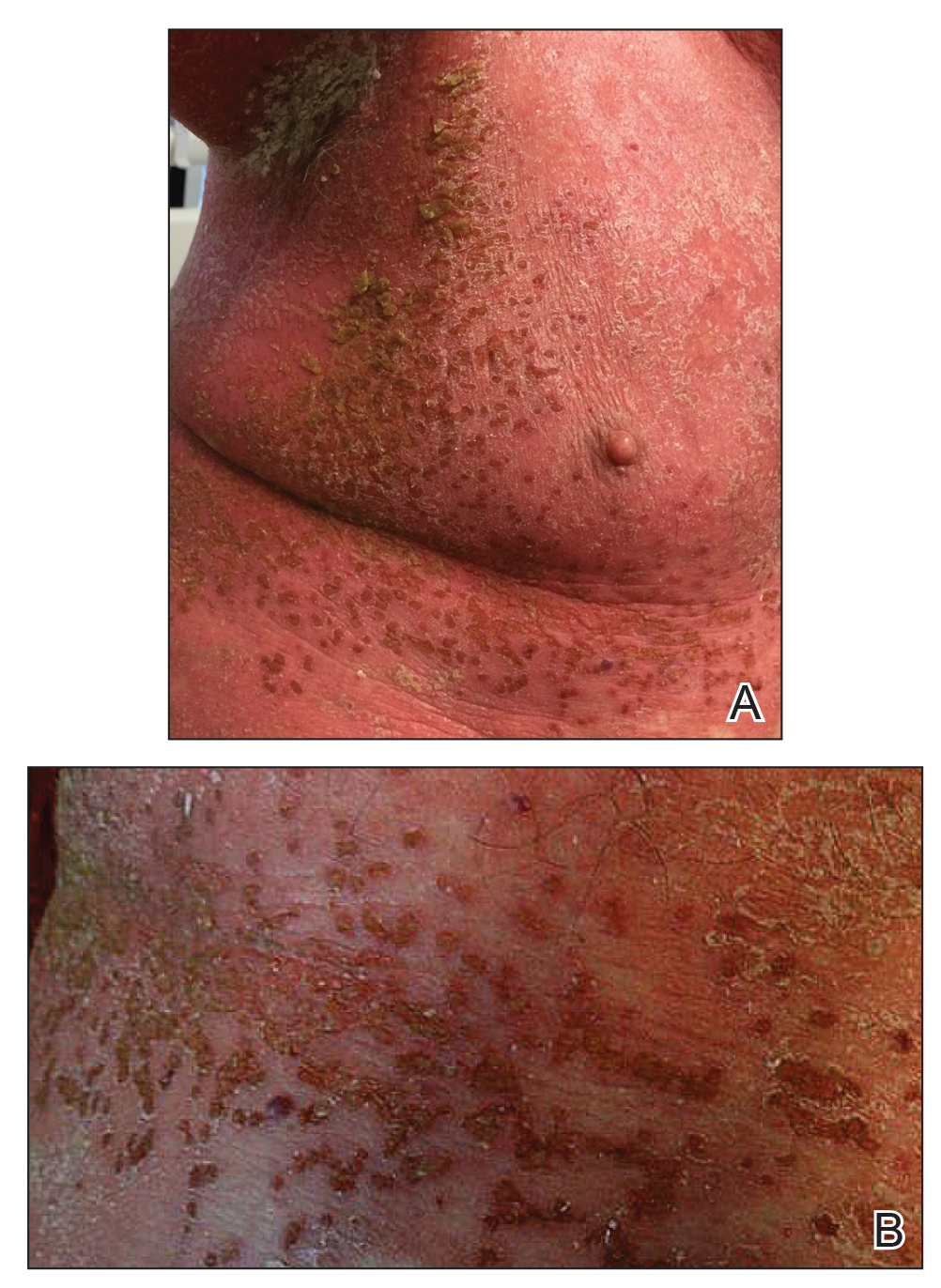
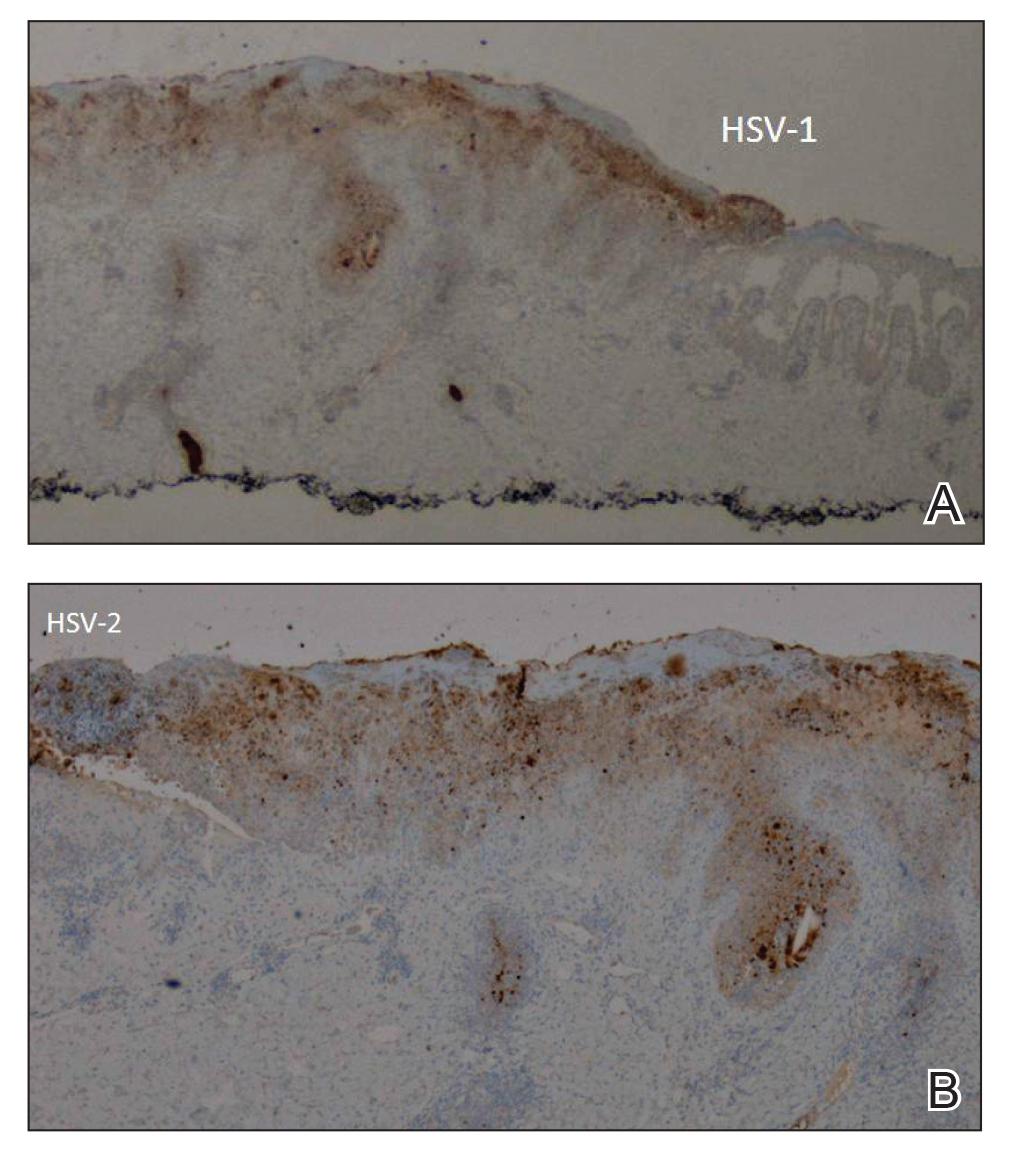
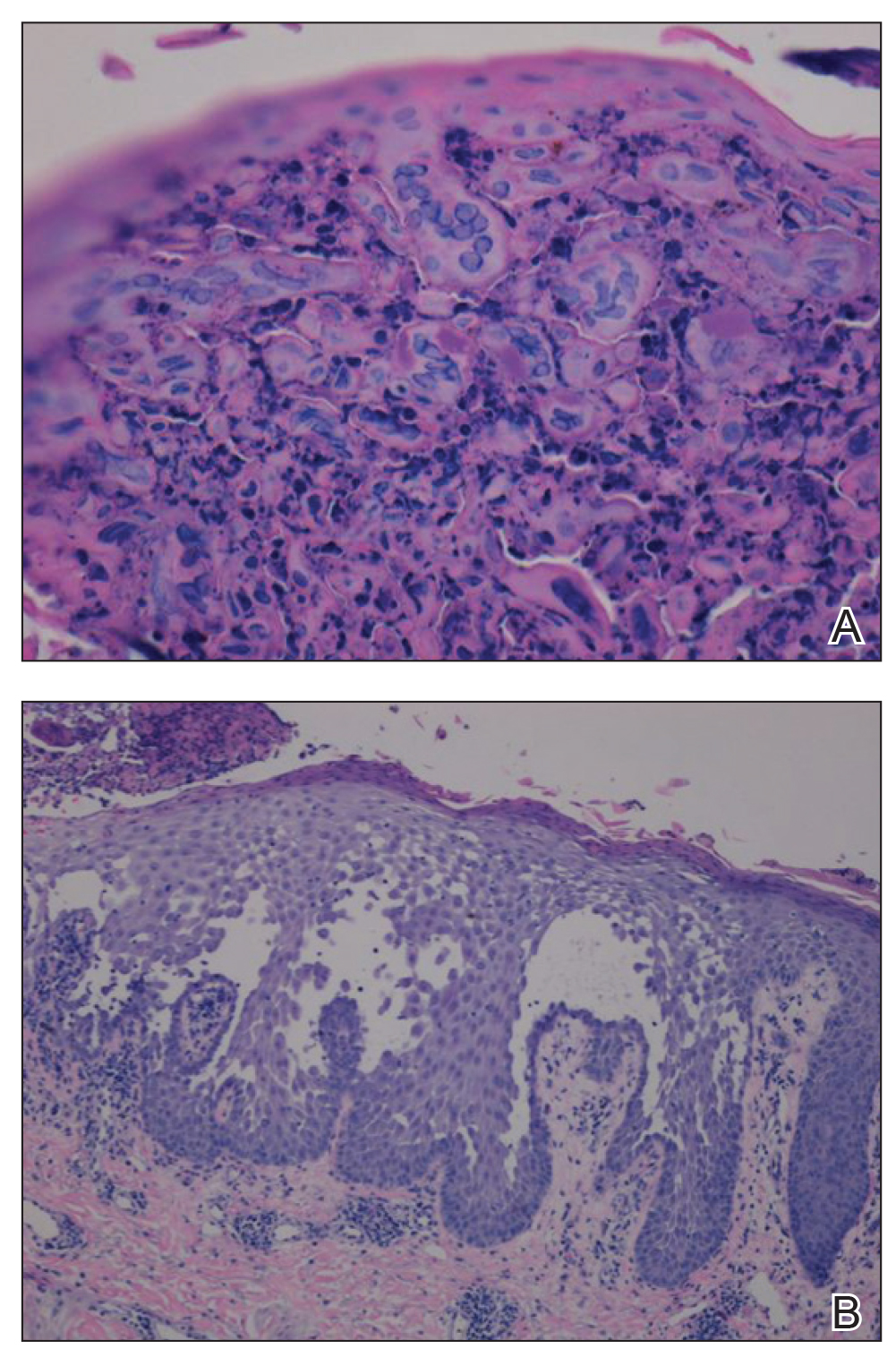
Viral cultures were taken but were incorrectly processed by the laboratory. Tzanck smear showed severe acute inflammation with numerous neutrophils, multinucleated giant cells with viral nuclear changes, and positive immunostaining for HSV and negative immunostaining for herpes zoster. Both pathology specimens revealed an intense acute mixed, mainly neutrophilic, inflammatory infiltrate extending into the deeper dermis as well as distorted and necrotic hair follicles, some of which displayed multinucleated epithelial cells with margination of chromatin that were positive for both HSV-1 and HSV-2 and negative for herpes zoster (Figure 2). The positivity of both HSV strains might represent co-infection or could be a cross-reaction of antibodies used in immunohistochemistry to the HSV antigens. There was acantholysis surrounding the ulceration and extending through the full thickness of the epidermis with a dilapidated brick wall pattern (Figure 3) as well as negative immunohistochemical staining for HSV-1 and HSV-2 antigens. The clinical and histological picture together, along with prior clinical and pathological reports, confirmed the diagnoses of acute erythrodermic HHD with HSV superinfection.
The patient’s condition and pain improved within 24 hours on intravenous acyclovir. On the third day, his lesions were resolving and symptoms improved, so he was transitioned to oral acyclovir and discharged from the hospital. Follow-up in the dermatology outpatient clinic 1 week later revealed that all vesicles and papules had cleared, but the patient was still erythrodermic. Because HHD cannot always be distinguished histologically from other forms of pemphigus but yields a negative immunofluorescence, direct immunofluorescence and indirect immunofluorescence were obtained upon patient follow-up in the clinic and were both negative. Hepatitis C viral loads were undetectable. Consultations to gastroenterology and oncology teams were placed for consideration of systemic agents, and the patient was initiated on oral acitretin 25 mg daily, along with clobetasol as adjuvant therapy for any residual skin plaques. The laboratory results were closely monitored. Within 4 weeks after starting acitretin, the patient’s erythroderma had completely resolved. The patient has remained stable since then, except for one episode of secondary Staphylococcus infection that cleared on oral antibiotics. The patient remains stable and clear on oral acitretin 25 mg daily, with concomitant desonide cream and fluocinonide ointment as needed.
Hailey-Hailey disease is characterized by recurrent episodes of erythema, blisters, and plaques localized to intertriginous and perianal areas.1,2 Patients display a spectrum of lesions that vary in severity.8 Typical histologic examination reveals a dilapidated brick wall appearance. Pathology of well-developed lesions will show suprabasal acantholysis with minimal dyskeratosis.2
The generalized form of HHD is an extremely rare variant of the disease.10 Generalized HHD may resemble acute hypersensitivity reaction, erythema multiforme, and toxic epidermal necrolysis.1 Chronic diseases, such as psoriasis (as in this patient), also may contribute to a clinically confusing picture.8 Hailey-Hailey disease and psoriasis are thought to occasionally koebnerize (isomorphic response) to areas of trauma.16 Our patient experienced widespread erythematous papules and plaques not restricted to skin folds. His skin lesions continued to worsen over several months progressing to erythroderma. The presence of suprabasal acantholysis in a dilapidated brick wall pattern, along with the patient’s history, prior pathology reports, clinical picture, and negative direct immunofluorescence and indirect immunofluorescence studies helped to confirm the diagnosis of erythrodermic HHD.
Hailey-Hailey disease is caused by heterozygous mutations in the ATP2C1 gene on chromosome 3q21-24 coding for a Golgi ATPase called SPCA1 (secretory pathway calcium/manganese-ATPase).9 Subsequent disturbances in cytosolic-Golgi calcium concentrations interfere with epidermal keratinocyte adherence resulting in acantholytic disease. Studies of interfamilial and intrafamilial mutations fail to pinpoint a common mutation pattern among patients with generalized phenotypes,9 which further supports theories that intrinsic or extrinsic factors such as friction, heat, radiation, contact allergens, and infection affect the severity of HHD disease and not the type of mutation.3,9
Generalization of HHD is likely caused by nonspecific triggers in an already genetically disturbed epidermis.10 Interrupted epithelial function exposes skin to infections that exacerbate the underlying disease. Superimposing bacterial infections are commonly reported in HHD. Staphylococcus, Streptococcus, and Candida species colonize the skin and aggravate the disease.11 Much less commonly, HSV superinfection can complicate HHD.3-7 No data are currently available about the frequency or incidence of Herpesviridae in HHD.7 Some studies suggest that UVB light therapy can be an exacerbating factor in DAR and some but not all HHD patients,12,13 while other case reports14,15 document clinically improved responses using phototherapy for patients with HHD. Clinicians should remain suspicious and evaluate for HSV infection in refractory or sudden exacerbation of HHD.7 Furthermore, coexistent psoriasis and HHD also is a rare entity but has been described,8 which illustrates the importance of not attributing all skin manifestations to a previously diagnosed disorder but instead keeping an open mind in case new dermatologic conditions present themselves at a later time.
We present a rare case of erythrodermic HHD and coexistent psoriasis with HSV superinfection. We hope to draw awareness to this association of generalized HHD with both HSV and psoriasis to help clinicians make the correct diagnosis promptly in similar cases in the future.
- Chave TA, Milligan A. Acute generalized Hailey-Hailey disease. Clin Exp Dermatol. 2002;27:290-292.
- Mohr MR, Erdag G, Shada Al, et al. Two patients with Hailey-Hailey disease, multiple primary melanomas, and other cancers. Arch Dermatol. 2011;147:211-215.
- Lee GM, Kim YM, Lee SY, et al. A case of eczema herpeticum with Hailey-Hailey Disease. Ann Dermatol. 2009;21:311-314.
- Zaim MT, Bickers DR. Herpes simplex associated with Hailey-Hailey disease. J Am Acad Dermatol. 1987;17:701-702.
- Peppiatt T, Keefe M, White JE. Hailey-Hailey disease-exacerbation by herpes simplex virus and patch tests. Clin Exp Dermatol. 2006;17:201-202.
- Almeida L, Grossman ME. Benign familial pemphigus complicated by herpes simplex virus. Cutis. 1989;44:261-262.
- Nikkels AF, Delvenne P, Herfs M, et al. Occult herpes simplex virus colonization of bullous dermatitides. Am J Clin Dermatol. 2008;9:163-168.
- Chao SC, Lee JY, Wu MC, et al. A novel splice mutation in the ATP2C1 gene in a woman with concomitant psoriasis vulgaris and disseminated Hailey-Hailey disease. Int J Dermatol. 2012;51:947-951.
- Ikeda S, Shigihara T, Mayuzumi N, et al. Mutations of ATP2C1 in Japanese patients with Hailey-Hailey disease: intrafamilial and interfamilial phenotype variations and lack of correlation with mutation patterns. J Invest Dermatol. 2001;117:1654-1656.
- Marsch W, Stuttgen G. Generalized Hailey-Hailey disease. Br J Dermatol. 1978;99:553-559.
- Friedman-Birnbaum R, Haim S, Marcus S. Generalized familial benign chronic pemphigus. Dermatologica. 1980;161:112-115.
- Richard G, Linse R, Harth W. Hailey-Hailey disease. early detection of heterozygotes by an ultraviolet provocation tests—clinical relevance of the method. Hautarzt. 1993;44:376-379.
- Mayuzumi N, Ikeda S, Kawada H, et al. Effects of ultraviolet B irradiation, proinflammatory cytokines and raised extracellular calcium concentration on the expression of ATP2A2 and ATP2C1. Br J Dermatol. 2005;152:697-701.
- Vanderbeck KA, Giroux L, Murugan NJ, et al. Combined therapeutic use of oral alitretinoin and narrowband ultraviolet-B therapy in the treatment of Hailey-Hailey disease. Dermatol Rep. 2014;6:5604.
- Mizuno K, Hamada T, Hasimoto T, et al. Successful treatment with narrow-band UVB therapy for a case of generalized Hailey-Hailey disease with a novel splice-site mutation in ATP2C1 gene. Dermatol Ther. 2014;27:233-235.
- Thappa DM. The isomorphic phenomenon of Koebner. Indian J Dermatol Venereol Leprol. 2004;70:187-189.
To the Editor:
Hailey-Hailey disease (HHD), also known as benign familial pemphigus, is an uncommon autosomal-dominant skin disease.1 Defects in the ATPase type 2C member 1 gene, ATP2C1, result in abnormal intracellular epidermal adherence, and patients experience recurring blisters in skin folds. Longitudinal white streaks of the fingernails also may be present.1 The illness does not appear until puberty and is heightened by the second or third decade of life. Family history often suggests the presence of disease.2 Misdiagnosis of HHD occurs because of a wide spectrum of presentations. The presence of superimposed infections and carcinomas may both obscure and exacerbate this disease.2
Herpes simplex viruse types 1 and 2 (HSV-1 and HSV-2) are DNA viruses that cause common recurrent diseases. Usually, HSV-1 is associated with infection of the mouth and HSV-2 is associated with infection of the genitalia.3 Longitudinal cutaneous lesions manifest as grouped vesicles on an erythematous base. Tzanck smear of herpetic vesicles will reveal the presence of multinucleated giant cells. A direct fluorescent antibody technique also may be used to confirm the diagnosis.3
Erythrodermic HHD disease is a rare condition; moreover, there are only a few reported cases with coexistence of HHD and HSV in the literature.3-6 We report a rare presentation of erythrodermic HHD and coexistent psoriasis with HSV superinfection.
A 69-year-old man presented to an outpatient dermatology clinic for evaluation and treatment of a rash on the scalp, face, back, and lower legs. The patient confirmed a dandruff diagnosis on the scalp and face as well as psoriasis on the trunk and extremities for the last 45 years. He described a history of successful treatment with topical agents and UV light therapy. A family history revealed that the patient’s father and 1 of 2 siblings had a similar rash and “skin problems.” The patient had a medical history of thyroid cancer treated with radiation treatment and a partial thyroidectomy 35 years prior to the current presentation as well as incompletely treated chronic hepatitis C.
A search of medical records revealed a punch biopsy from the posterior neck that demonstrated an acantholytic dyskeratosis with suprabasal acantholysis. Clinicians were unable to differentiate if it was Darier disease (DAR) or HHD. Treatment of the patient’s seborrheic dermatitis and acantholytic disorder was successful at that time with ketoconazole shampoo, ketoconazole cream, desonide cream, and triamcinolone cream. The patient remained stable for 5 years before presenting again to the dermatology clinic for worsening rash despite topical therapies.
At the current presentation, physical examination at the outpatient dermatology clinic revealed few scaly, erythematous, eroded papules distributed on the mid-back; erythematous greasy scaling on the scalp, face, and chest; and pink scaly plaques with white-silvery scale on the anterior lower legs. Histopathology of a specimen from the right mid-back demonstrated acantholysis with suprabasal clefting, hyperkeratosis, and parakeratosis with no dyskeratotic cells identified. The pathologic differential diagnosis included primary acantholytic processes including Grover disease, DAR, HHD, and pemphigus. Pathology from the right shin demonstrated acanthosis, confluent parakeratosis with associated decreased granular cell layer and collections of neutrophils within the stratum corneum, spongiosis, and superficial dermal perivascular chronic inflammation with focal exocytosis and dilated blood vessels in the papillary dermis. The clinical and pathological diagnosis on the lower legs was consistent with psoriasis. Diagnoses of seborrheic dermatitis, psoriasis on the lower legs, and HHD vs DAR on the back and chest were made. The patient was instructed to continue ketoconazole shampoo, ketoconazole cream, and desonide for seborrheic dermatitis; fluocinonide ointment 0.05% to the lower legs for psoriasis; and triamcinolone cream and a bland moisturizer to the back and chest for HHD.
Over the ensuing months, the rash worsened with erythema and scaling affecting more than half of the body surface area. Topical corticosteroids and bland emollients resulted in minimal success. Biologics and acitretin were considered for the psoriasiform dermatitis but avoided due to the patient’s medical history of thyroid cancer and chronic hepatitis C infection. Because the patient described prior success with UV light therapy for psoriasis, he requested light therapy. A subsequent trial of narrowband UVB light therapy initially improved some of the psoriasiform dermatitis on the trunk and extremities; however, after 4 weeks of treatment, the patient described pain in some of the skin and felt he was burned by minimal exposure to light therapy on one particular visit, which caused him to stop light therapy.
Approximately 2 weeks later, the patient presented to the emergency department stating his psoriasis was infected; he was diagnosed with psoriasis with secondary cellulitis and received intravenous vancomycin and piperacillin-tazobactam, with bacterial cultures demonstrating Corynebacterium and methicillin-resistant Staphylococcus aureus. Some improvement was noted in the patient’s skin after antibiotics were initiated, but he continued to describe worsening “burning and pain” throughout the psoriasis lesions. The patient’s care was transferred to the Veterans Affairs hospital where a dermatology inpatient consultation was placed.
Our initial dermatologic examination revealed generalized scaly erythroderma on the neck, trunk, and extremities, sparing the face, palms, and soles (Figure 1). Multiple crusted and intact vesicles also were present overlying the erythematous plaques on the chest, back, and proximal extremities, most grouped in clusters. The patient endorsed new symptoms of pain and burning. Tzanck smear from the abdomen along with shave biopsies from the left flank and right abdomen were performed, and intravenous acyclovir was initiated immediately after these procedures.
Viral cultures were taken but were incorrectly processed by the laboratory. Tzanck smear showed severe acute inflammation with numerous neutrophils, multinucleated giant cells with viral nuclear changes, and positive immunostaining for HSV and negative immunostaining for herpes zoster. Both pathology specimens revealed an intense acute mixed, mainly neutrophilic, inflammatory infiltrate extending into the deeper dermis as well as distorted and necrotic hair follicles, some of which displayed multinucleated epithelial cells with margination of chromatin that were positive for both HSV-1 and HSV-2 and negative for herpes zoster (Figure 2). The positivity of both HSV strains might represent co-infection or could be a cross-reaction of antibodies used in immunohistochemistry to the HSV antigens. There was acantholysis surrounding the ulceration and extending through the full thickness of the epidermis with a dilapidated brick wall pattern (Figure 3) as well as negative immunohistochemical staining for HSV-1 and HSV-2 antigens. The clinical and histological picture together, along with prior clinical and pathological reports, confirmed the diagnoses of acute erythrodermic HHD with HSV superinfection.
The patient’s condition and pain improved within 24 hours on intravenous acyclovir. On the third day, his lesions were resolving and symptoms improved, so he was transitioned to oral acyclovir and discharged from the hospital. Follow-up in the dermatology outpatient clinic 1 week later revealed that all vesicles and papules had cleared, but the patient was still erythrodermic. Because HHD cannot always be distinguished histologically from other forms of pemphigus but yields a negative immunofluorescence, direct immunofluorescence and indirect immunofluorescence were obtained upon patient follow-up in the clinic and were both negative. Hepatitis C viral loads were undetectable. Consultations to gastroenterology and oncology teams were placed for consideration of systemic agents, and the patient was initiated on oral acitretin 25 mg daily, along with clobetasol as adjuvant therapy for any residual skin plaques. The laboratory results were closely monitored. Within 4 weeks after starting acitretin, the patient’s erythroderma had completely resolved. The patient has remained stable since then, except for one episode of secondary Staphylococcus infection that cleared on oral antibiotics. The patient remains stable and clear on oral acitretin 25 mg daily, with concomitant desonide cream and fluocinonide ointment as needed.
Hailey-Hailey disease is characterized by recurrent episodes of erythema, blisters, and plaques localized to intertriginous and perianal areas.1,2 Patients display a spectrum of lesions that vary in severity.8 Typical histologic examination reveals a dilapidated brick wall appearance. Pathology of well-developed lesions will show suprabasal acantholysis with minimal dyskeratosis.2
The generalized form of HHD is an extremely rare variant of the disease.10 Generalized HHD may resemble acute hypersensitivity reaction, erythema multiforme, and toxic epidermal necrolysis.1 Chronic diseases, such as psoriasis (as in this patient), also may contribute to a clinically confusing picture.8 Hailey-Hailey disease and psoriasis are thought to occasionally koebnerize (isomorphic response) to areas of trauma.16 Our patient experienced widespread erythematous papules and plaques not restricted to skin folds. His skin lesions continued to worsen over several months progressing to erythroderma. The presence of suprabasal acantholysis in a dilapidated brick wall pattern, along with the patient’s history, prior pathology reports, clinical picture, and negative direct immunofluorescence and indirect immunofluorescence studies helped to confirm the diagnosis of erythrodermic HHD.
Hailey-Hailey disease is caused by heterozygous mutations in the ATP2C1 gene on chromosome 3q21-24 coding for a Golgi ATPase called SPCA1 (secretory pathway calcium/manganese-ATPase).9 Subsequent disturbances in cytosolic-Golgi calcium concentrations interfere with epidermal keratinocyte adherence resulting in acantholytic disease. Studies of interfamilial and intrafamilial mutations fail to pinpoint a common mutation pattern among patients with generalized phenotypes,9 which further supports theories that intrinsic or extrinsic factors such as friction, heat, radiation, contact allergens, and infection affect the severity of HHD disease and not the type of mutation.3,9
Generalization of HHD is likely caused by nonspecific triggers in an already genetically disturbed epidermis.10 Interrupted epithelial function exposes skin to infections that exacerbate the underlying disease. Superimposing bacterial infections are commonly reported in HHD. Staphylococcus, Streptococcus, and Candida species colonize the skin and aggravate the disease.11 Much less commonly, HSV superinfection can complicate HHD.3-7 No data are currently available about the frequency or incidence of Herpesviridae in HHD.7 Some studies suggest that UVB light therapy can be an exacerbating factor in DAR and some but not all HHD patients,12,13 while other case reports14,15 document clinically improved responses using phototherapy for patients with HHD. Clinicians should remain suspicious and evaluate for HSV infection in refractory or sudden exacerbation of HHD.7 Furthermore, coexistent psoriasis and HHD also is a rare entity but has been described,8 which illustrates the importance of not attributing all skin manifestations to a previously diagnosed disorder but instead keeping an open mind in case new dermatologic conditions present themselves at a later time.
We present a rare case of erythrodermic HHD and coexistent psoriasis with HSV superinfection. We hope to draw awareness to this association of generalized HHD with both HSV and psoriasis to help clinicians make the correct diagnosis promptly in similar cases in the future.
To the Editor:
Hailey-Hailey disease (HHD), also known as benign familial pemphigus, is an uncommon autosomal-dominant skin disease.1 Defects in the ATPase type 2C member 1 gene, ATP2C1, result in abnormal intracellular epidermal adherence, and patients experience recurring blisters in skin folds. Longitudinal white streaks of the fingernails also may be present.1 The illness does not appear until puberty and is heightened by the second or third decade of life. Family history often suggests the presence of disease.2 Misdiagnosis of HHD occurs because of a wide spectrum of presentations. The presence of superimposed infections and carcinomas may both obscure and exacerbate this disease.2
Herpes simplex viruse types 1 and 2 (HSV-1 and HSV-2) are DNA viruses that cause common recurrent diseases. Usually, HSV-1 is associated with infection of the mouth and HSV-2 is associated with infection of the genitalia.3 Longitudinal cutaneous lesions manifest as grouped vesicles on an erythematous base. Tzanck smear of herpetic vesicles will reveal the presence of multinucleated giant cells. A direct fluorescent antibody technique also may be used to confirm the diagnosis.3
Erythrodermic HHD disease is a rare condition; moreover, there are only a few reported cases with coexistence of HHD and HSV in the literature.3-6 We report a rare presentation of erythrodermic HHD and coexistent psoriasis with HSV superinfection.
A 69-year-old man presented to an outpatient dermatology clinic for evaluation and treatment of a rash on the scalp, face, back, and lower legs. The patient confirmed a dandruff diagnosis on the scalp and face as well as psoriasis on the trunk and extremities for the last 45 years. He described a history of successful treatment with topical agents and UV light therapy. A family history revealed that the patient’s father and 1 of 2 siblings had a similar rash and “skin problems.” The patient had a medical history of thyroid cancer treated with radiation treatment and a partial thyroidectomy 35 years prior to the current presentation as well as incompletely treated chronic hepatitis C.
A search of medical records revealed a punch biopsy from the posterior neck that demonstrated an acantholytic dyskeratosis with suprabasal acantholysis. Clinicians were unable to differentiate if it was Darier disease (DAR) or HHD. Treatment of the patient’s seborrheic dermatitis and acantholytic disorder was successful at that time with ketoconazole shampoo, ketoconazole cream, desonide cream, and triamcinolone cream. The patient remained stable for 5 years before presenting again to the dermatology clinic for worsening rash despite topical therapies.
At the current presentation, physical examination at the outpatient dermatology clinic revealed few scaly, erythematous, eroded papules distributed on the mid-back; erythematous greasy scaling on the scalp, face, and chest; and pink scaly plaques with white-silvery scale on the anterior lower legs. Histopathology of a specimen from the right mid-back demonstrated acantholysis with suprabasal clefting, hyperkeratosis, and parakeratosis with no dyskeratotic cells identified. The pathologic differential diagnosis included primary acantholytic processes including Grover disease, DAR, HHD, and pemphigus. Pathology from the right shin demonstrated acanthosis, confluent parakeratosis with associated decreased granular cell layer and collections of neutrophils within the stratum corneum, spongiosis, and superficial dermal perivascular chronic inflammation with focal exocytosis and dilated blood vessels in the papillary dermis. The clinical and pathological diagnosis on the lower legs was consistent with psoriasis. Diagnoses of seborrheic dermatitis, psoriasis on the lower legs, and HHD vs DAR on the back and chest were made. The patient was instructed to continue ketoconazole shampoo, ketoconazole cream, and desonide for seborrheic dermatitis; fluocinonide ointment 0.05% to the lower legs for psoriasis; and triamcinolone cream and a bland moisturizer to the back and chest for HHD.
Over the ensuing months, the rash worsened with erythema and scaling affecting more than half of the body surface area. Topical corticosteroids and bland emollients resulted in minimal success. Biologics and acitretin were considered for the psoriasiform dermatitis but avoided due to the patient’s medical history of thyroid cancer and chronic hepatitis C infection. Because the patient described prior success with UV light therapy for psoriasis, he requested light therapy. A subsequent trial of narrowband UVB light therapy initially improved some of the psoriasiform dermatitis on the trunk and extremities; however, after 4 weeks of treatment, the patient described pain in some of the skin and felt he was burned by minimal exposure to light therapy on one particular visit, which caused him to stop light therapy.
Approximately 2 weeks later, the patient presented to the emergency department stating his psoriasis was infected; he was diagnosed with psoriasis with secondary cellulitis and received intravenous vancomycin and piperacillin-tazobactam, with bacterial cultures demonstrating Corynebacterium and methicillin-resistant Staphylococcus aureus. Some improvement was noted in the patient’s skin after antibiotics were initiated, but he continued to describe worsening “burning and pain” throughout the psoriasis lesions. The patient’s care was transferred to the Veterans Affairs hospital where a dermatology inpatient consultation was placed.
Our initial dermatologic examination revealed generalized scaly erythroderma on the neck, trunk, and extremities, sparing the face, palms, and soles (Figure 1). Multiple crusted and intact vesicles also were present overlying the erythematous plaques on the chest, back, and proximal extremities, most grouped in clusters. The patient endorsed new symptoms of pain and burning. Tzanck smear from the abdomen along with shave biopsies from the left flank and right abdomen were performed, and intravenous acyclovir was initiated immediately after these procedures.
Viral cultures were taken but were incorrectly processed by the laboratory. Tzanck smear showed severe acute inflammation with numerous neutrophils, multinucleated giant cells with viral nuclear changes, and positive immunostaining for HSV and negative immunostaining for herpes zoster. Both pathology specimens revealed an intense acute mixed, mainly neutrophilic, inflammatory infiltrate extending into the deeper dermis as well as distorted and necrotic hair follicles, some of which displayed multinucleated epithelial cells with margination of chromatin that were positive for both HSV-1 and HSV-2 and negative for herpes zoster (Figure 2). The positivity of both HSV strains might represent co-infection or could be a cross-reaction of antibodies used in immunohistochemistry to the HSV antigens. There was acantholysis surrounding the ulceration and extending through the full thickness of the epidermis with a dilapidated brick wall pattern (Figure 3) as well as negative immunohistochemical staining for HSV-1 and HSV-2 antigens. The clinical and histological picture together, along with prior clinical and pathological reports, confirmed the diagnoses of acute erythrodermic HHD with HSV superinfection.
The patient’s condition and pain improved within 24 hours on intravenous acyclovir. On the third day, his lesions were resolving and symptoms improved, so he was transitioned to oral acyclovir and discharged from the hospital. Follow-up in the dermatology outpatient clinic 1 week later revealed that all vesicles and papules had cleared, but the patient was still erythrodermic. Because HHD cannot always be distinguished histologically from other forms of pemphigus but yields a negative immunofluorescence, direct immunofluorescence and indirect immunofluorescence were obtained upon patient follow-up in the clinic and were both negative. Hepatitis C viral loads were undetectable. Consultations to gastroenterology and oncology teams were placed for consideration of systemic agents, and the patient was initiated on oral acitretin 25 mg daily, along with clobetasol as adjuvant therapy for any residual skin plaques. The laboratory results were closely monitored. Within 4 weeks after starting acitretin, the patient’s erythroderma had completely resolved. The patient has remained stable since then, except for one episode of secondary Staphylococcus infection that cleared on oral antibiotics. The patient remains stable and clear on oral acitretin 25 mg daily, with concomitant desonide cream and fluocinonide ointment as needed.
Hailey-Hailey disease is characterized by recurrent episodes of erythema, blisters, and plaques localized to intertriginous and perianal areas.1,2 Patients display a spectrum of lesions that vary in severity.8 Typical histologic examination reveals a dilapidated brick wall appearance. Pathology of well-developed lesions will show suprabasal acantholysis with minimal dyskeratosis.2
The generalized form of HHD is an extremely rare variant of the disease.10 Generalized HHD may resemble acute hypersensitivity reaction, erythema multiforme, and toxic epidermal necrolysis.1 Chronic diseases, such as psoriasis (as in this patient), also may contribute to a clinically confusing picture.8 Hailey-Hailey disease and psoriasis are thought to occasionally koebnerize (isomorphic response) to areas of trauma.16 Our patient experienced widespread erythematous papules and plaques not restricted to skin folds. His skin lesions continued to worsen over several months progressing to erythroderma. The presence of suprabasal acantholysis in a dilapidated brick wall pattern, along with the patient’s history, prior pathology reports, clinical picture, and negative direct immunofluorescence and indirect immunofluorescence studies helped to confirm the diagnosis of erythrodermic HHD.
Hailey-Hailey disease is caused by heterozygous mutations in the ATP2C1 gene on chromosome 3q21-24 coding for a Golgi ATPase called SPCA1 (secretory pathway calcium/manganese-ATPase).9 Subsequent disturbances in cytosolic-Golgi calcium concentrations interfere with epidermal keratinocyte adherence resulting in acantholytic disease. Studies of interfamilial and intrafamilial mutations fail to pinpoint a common mutation pattern among patients with generalized phenotypes,9 which further supports theories that intrinsic or extrinsic factors such as friction, heat, radiation, contact allergens, and infection affect the severity of HHD disease and not the type of mutation.3,9
Generalization of HHD is likely caused by nonspecific triggers in an already genetically disturbed epidermis.10 Interrupted epithelial function exposes skin to infections that exacerbate the underlying disease. Superimposing bacterial infections are commonly reported in HHD. Staphylococcus, Streptococcus, and Candida species colonize the skin and aggravate the disease.11 Much less commonly, HSV superinfection can complicate HHD.3-7 No data are currently available about the frequency or incidence of Herpesviridae in HHD.7 Some studies suggest that UVB light therapy can be an exacerbating factor in DAR and some but not all HHD patients,12,13 while other case reports14,15 document clinically improved responses using phototherapy for patients with HHD. Clinicians should remain suspicious and evaluate for HSV infection in refractory or sudden exacerbation of HHD.7 Furthermore, coexistent psoriasis and HHD also is a rare entity but has been described,8 which illustrates the importance of not attributing all skin manifestations to a previously diagnosed disorder but instead keeping an open mind in case new dermatologic conditions present themselves at a later time.
We present a rare case of erythrodermic HHD and coexistent psoriasis with HSV superinfection. We hope to draw awareness to this association of generalized HHD with both HSV and psoriasis to help clinicians make the correct diagnosis promptly in similar cases in the future.
- Chave TA, Milligan A. Acute generalized Hailey-Hailey disease. Clin Exp Dermatol. 2002;27:290-292.
- Mohr MR, Erdag G, Shada Al, et al. Two patients with Hailey-Hailey disease, multiple primary melanomas, and other cancers. Arch Dermatol. 2011;147:211-215.
- Lee GM, Kim YM, Lee SY, et al. A case of eczema herpeticum with Hailey-Hailey Disease. Ann Dermatol. 2009;21:311-314.
- Zaim MT, Bickers DR. Herpes simplex associated with Hailey-Hailey disease. J Am Acad Dermatol. 1987;17:701-702.
- Peppiatt T, Keefe M, White JE. Hailey-Hailey disease-exacerbation by herpes simplex virus and patch tests. Clin Exp Dermatol. 2006;17:201-202.
- Almeida L, Grossman ME. Benign familial pemphigus complicated by herpes simplex virus. Cutis. 1989;44:261-262.
- Nikkels AF, Delvenne P, Herfs M, et al. Occult herpes simplex virus colonization of bullous dermatitides. Am J Clin Dermatol. 2008;9:163-168.
- Chao SC, Lee JY, Wu MC, et al. A novel splice mutation in the ATP2C1 gene in a woman with concomitant psoriasis vulgaris and disseminated Hailey-Hailey disease. Int J Dermatol. 2012;51:947-951.
- Ikeda S, Shigihara T, Mayuzumi N, et al. Mutations of ATP2C1 in Japanese patients with Hailey-Hailey disease: intrafamilial and interfamilial phenotype variations and lack of correlation with mutation patterns. J Invest Dermatol. 2001;117:1654-1656.
- Marsch W, Stuttgen G. Generalized Hailey-Hailey disease. Br J Dermatol. 1978;99:553-559.
- Friedman-Birnbaum R, Haim S, Marcus S. Generalized familial benign chronic pemphigus. Dermatologica. 1980;161:112-115.
- Richard G, Linse R, Harth W. Hailey-Hailey disease. early detection of heterozygotes by an ultraviolet provocation tests—clinical relevance of the method. Hautarzt. 1993;44:376-379.
- Mayuzumi N, Ikeda S, Kawada H, et al. Effects of ultraviolet B irradiation, proinflammatory cytokines and raised extracellular calcium concentration on the expression of ATP2A2 and ATP2C1. Br J Dermatol. 2005;152:697-701.
- Vanderbeck KA, Giroux L, Murugan NJ, et al. Combined therapeutic use of oral alitretinoin and narrowband ultraviolet-B therapy in the treatment of Hailey-Hailey disease. Dermatol Rep. 2014;6:5604.
- Mizuno K, Hamada T, Hasimoto T, et al. Successful treatment with narrow-band UVB therapy for a case of generalized Hailey-Hailey disease with a novel splice-site mutation in ATP2C1 gene. Dermatol Ther. 2014;27:233-235.
- Thappa DM. The isomorphic phenomenon of Koebner. Indian J Dermatol Venereol Leprol. 2004;70:187-189.
- Chave TA, Milligan A. Acute generalized Hailey-Hailey disease. Clin Exp Dermatol. 2002;27:290-292.
- Mohr MR, Erdag G, Shada Al, et al. Two patients with Hailey-Hailey disease, multiple primary melanomas, and other cancers. Arch Dermatol. 2011;147:211-215.
- Lee GM, Kim YM, Lee SY, et al. A case of eczema herpeticum with Hailey-Hailey Disease. Ann Dermatol. 2009;21:311-314.
- Zaim MT, Bickers DR. Herpes simplex associated with Hailey-Hailey disease. J Am Acad Dermatol. 1987;17:701-702.
- Peppiatt T, Keefe M, White JE. Hailey-Hailey disease-exacerbation by herpes simplex virus and patch tests. Clin Exp Dermatol. 2006;17:201-202.
- Almeida L, Grossman ME. Benign familial pemphigus complicated by herpes simplex virus. Cutis. 1989;44:261-262.
- Nikkels AF, Delvenne P, Herfs M, et al. Occult herpes simplex virus colonization of bullous dermatitides. Am J Clin Dermatol. 2008;9:163-168.
- Chao SC, Lee JY, Wu MC, et al. A novel splice mutation in the ATP2C1 gene in a woman with concomitant psoriasis vulgaris and disseminated Hailey-Hailey disease. Int J Dermatol. 2012;51:947-951.
- Ikeda S, Shigihara T, Mayuzumi N, et al. Mutations of ATP2C1 in Japanese patients with Hailey-Hailey disease: intrafamilial and interfamilial phenotype variations and lack of correlation with mutation patterns. J Invest Dermatol. 2001;117:1654-1656.
- Marsch W, Stuttgen G. Generalized Hailey-Hailey disease. Br J Dermatol. 1978;99:553-559.
- Friedman-Birnbaum R, Haim S, Marcus S. Generalized familial benign chronic pemphigus. Dermatologica. 1980;161:112-115.
- Richard G, Linse R, Harth W. Hailey-Hailey disease. early detection of heterozygotes by an ultraviolet provocation tests—clinical relevance of the method. Hautarzt. 1993;44:376-379.
- Mayuzumi N, Ikeda S, Kawada H, et al. Effects of ultraviolet B irradiation, proinflammatory cytokines and raised extracellular calcium concentration on the expression of ATP2A2 and ATP2C1. Br J Dermatol. 2005;152:697-701.
- Vanderbeck KA, Giroux L, Murugan NJ, et al. Combined therapeutic use of oral alitretinoin and narrowband ultraviolet-B therapy in the treatment of Hailey-Hailey disease. Dermatol Rep. 2014;6:5604.
- Mizuno K, Hamada T, Hasimoto T, et al. Successful treatment with narrow-band UVB therapy for a case of generalized Hailey-Hailey disease with a novel splice-site mutation in ATP2C1 gene. Dermatol Ther. 2014;27:233-235.
- Thappa DM. The isomorphic phenomenon of Koebner. Indian J Dermatol Venereol Leprol. 2004;70:187-189.
Practice Points
- Misdiagnosis of Hailey-Hailey disease (HHD) occurs because of a wide spectrum of presentations.
- Hailey-Hailey disease and psoriasis are thought to occasionally koebnerize (isomorphic response) to areas of trauma.
- Clinicians should remain suspicious and evaluate for herpes simplex virus infection in refractory or sudden exacerbation of HHD.
Phytophotodermatitis in a Butterfly Enthusiast Induced by Common Rue
To the Editor:
Phytophotodermatitis is common in dermatology during the summer months, especially in individuals who spend time outdoors; however, identification of the offending plant can be challenging. We report a case of phytophotodermatitis in which the causative plant, common rue, was not identified until it was revealed that the patient was a butterfly enthusiast.
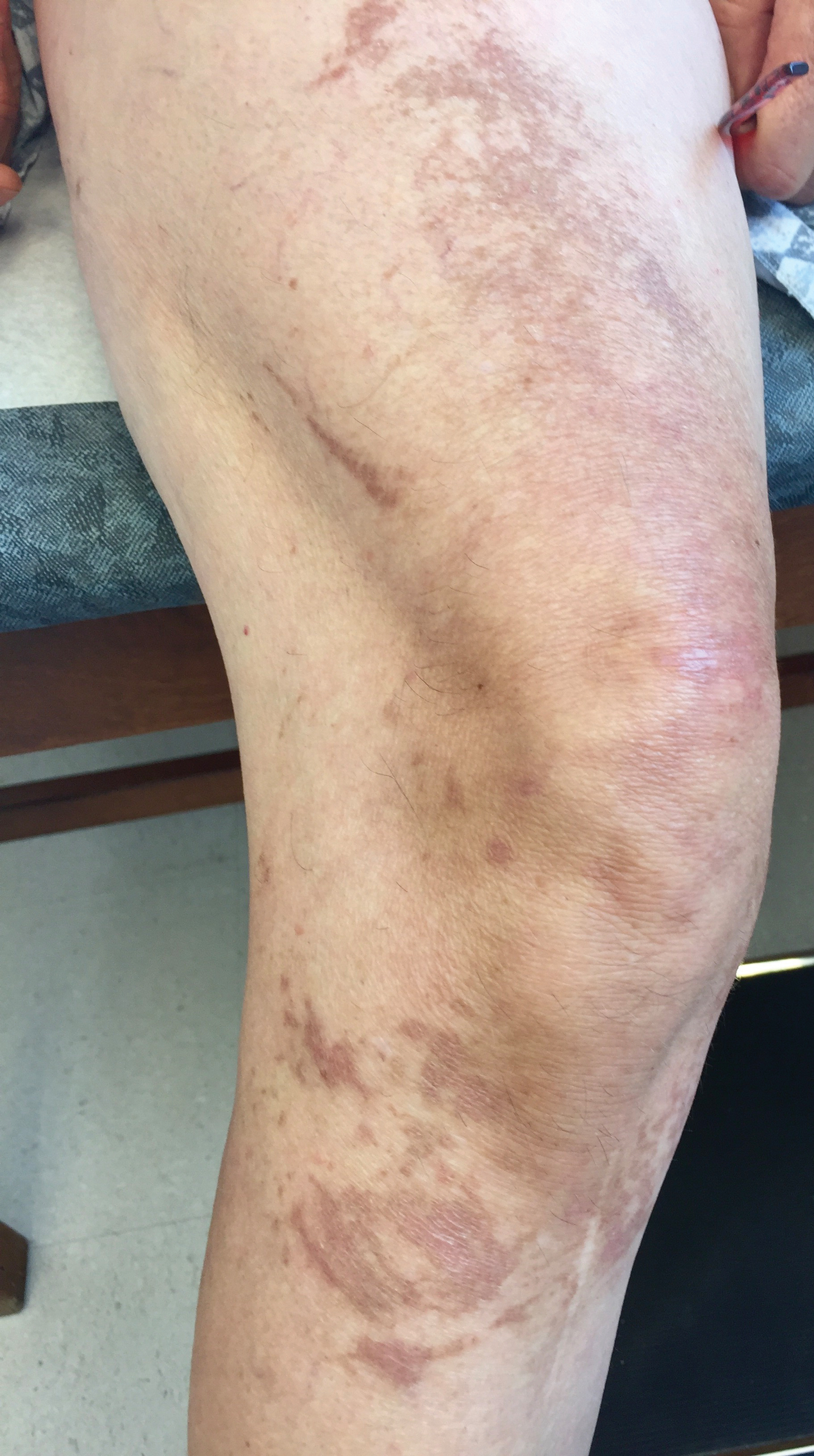
A 60-year-old woman presented to the outpatient dermatology clinic in late summer for a routine skin examination. An eruption was noted over the right thigh and knee that had first appeared approximately 2 weeks prior. The rash started as pruritic blisters but gradually progressed to erythema and then eventually to brown markings, which were observed at the current presentation. Physical examination revealed hyperpigmented, brown, streaky, linear patches and plaques over the right thigh, knee, and lower leg (Figure). When asked about her hobbies, the patient reported an affinity for butterflies and noted that she attracts them with specific species of plants in her garden. She recalled recently planting the herb of grace, or common rue, to attract the giant swallowtail butterfly (Papilio cresphontes). Upon further inquiry, she remembered working in the garden on her knees and digging up roots near the common rue plant while wearing shorts approximately 2 weeks prior to the current presentation. Given the streaky linear pattern of the eruption along with recent sun exposure and exposure to the common rue plant, a diagnosis of phytophotodermatitis was made. No further treatment was sought, as the eruption was not bothersome to her. She was intrigued that the common rue plant had caused the dermatitis and planned on taking proper precautions when working near the plant in the future.
In this case, the observed phototoxic skin findings resulted from exposure to common rue (Ruta graveolens),a pungently scented evergreen shrub native to the Mediterranean region and a member of the Rutaceae family. Extracts have been used in homeopathic practices for bruises, sprains, headache, neck stiffness, rheumatologic pain, neuralgia, stomach problems, and phlebitis, as well as in seasonings, soaps, creams, and perfumes.1 The most commonly encountered plants known to cause phytophotodermatitis belong to the Apiaceae and Rutaceae families.2 Members of Apiaceae include angelica, celery, dill, fennel, hogweed, parsley, and parsnip. Aside from the common rue plant, the Rutaceae family also includes bergamot orange, bitter orange, burning bush (or gas plant), grapefruit, lemon, and lime. Other potential offending agents are fig, mustard, buttercup, St. John’s wort, and scurfpea. The phototoxic properties are due to furocoumarins, which include psoralens and angelicins. They are inert until activated by UVA radiation, which inflicts direct cellular damage, causing vacuolization and apoptosis of keratinocytes, similar to a sunburn.3 Clinical findings typically present 24 hours after sun exposure with erythema, edema, pain, and occasionally vesicles or bullae in severe cases. Unlike sunburn, lesions often present in linear, streaky, or bizarre patterns, reflective of the direct contact with the plant. The lesions eventually transition to hyperpigmentation, which may take months to years to resolve.
Other considerations in cases of suspected phytophotodermatitis include polymorphic light eruption, actinic prurigo, hydroa vacciniforme, chronic actinic dermatitis, solar urticaria, drug reactions, porphyria, Smith-Lemli-Opitz syndrome, lupus erythematosus, and dermatomyositis.4 Clinicians should suspect phytophotodermatitis with phototoxic findings in bartenders, citrus farm workers, gardeners, chefs, and kitchen workers, especially those handling limes and celery. As in our case, phytophotodermatitis also should be considered in butterfly enthusiasts trying to attract the giant swallowtail butterfly. The caterpillars feed on the leaves of the common rue plant, one of a select few plants that giant swallowtail butterflies use as a host due to its bitter leaves that aid in avoiding predators.5
This case illustrates a unique perspective of phytophotodermatitis, as butterfly enthusiasm is not commonly reported in association with the common rue plant with respect to phytophotodermatitis. This case underscores the importance of inquiring about patients’ professions and hobbies, both in dermatology and other specialties.
- Atta AH, Alkofahi A. Anti-nociceptive and anti-inflammatory effects of some Jordanian medicinal plant extracts. J Ethnopharmacol. 1998;60:117-124.
- McGovern TW. Dermatoses due to plants. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. Edinburgh, Scotland: Mosby; 2007:265-283.
- Hawk JLM, Calonje E. The photosensitivity disorders. In: Elder DE, ed. Lever’s Histopathology of the Skin. 9th ed. Philadelphia, Pennsylvania: Lippincott Williams and Wilkins; 2005:345-353.
- Lim HW. Abnormal responses to ultraviolet radiation: photosensitivity induced by exogenous agents. In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012:1066-1074.
- McAuslane H. Giant swallowtail. University of Florida Department of Entomology and Nematology Featured Creatures website. http://entnemdept.ufl.edu/creatures/citrus/giantswallowtail.htm. Revised January 2018. Accessed April 10, 2020.
To the Editor:
Phytophotodermatitis is common in dermatology during the summer months, especially in individuals who spend time outdoors; however, identification of the offending plant can be challenging. We report a case of phytophotodermatitis in which the causative plant, common rue, was not identified until it was revealed that the patient was a butterfly enthusiast.
A 60-year-old woman presented to the outpatient dermatology clinic in late summer for a routine skin examination. An eruption was noted over the right thigh and knee that had first appeared approximately 2 weeks prior. The rash started as pruritic blisters but gradually progressed to erythema and then eventually to brown markings, which were observed at the current presentation. Physical examination revealed hyperpigmented, brown, streaky, linear patches and plaques over the right thigh, knee, and lower leg (Figure). When asked about her hobbies, the patient reported an affinity for butterflies and noted that she attracts them with specific species of plants in her garden. She recalled recently planting the herb of grace, or common rue, to attract the giant swallowtail butterfly (Papilio cresphontes). Upon further inquiry, she remembered working in the garden on her knees and digging up roots near the common rue plant while wearing shorts approximately 2 weeks prior to the current presentation. Given the streaky linear pattern of the eruption along with recent sun exposure and exposure to the common rue plant, a diagnosis of phytophotodermatitis was made. No further treatment was sought, as the eruption was not bothersome to her. She was intrigued that the common rue plant had caused the dermatitis and planned on taking proper precautions when working near the plant in the future.
In this case, the observed phototoxic skin findings resulted from exposure to common rue (Ruta graveolens),a pungently scented evergreen shrub native to the Mediterranean region and a member of the Rutaceae family. Extracts have been used in homeopathic practices for bruises, sprains, headache, neck stiffness, rheumatologic pain, neuralgia, stomach problems, and phlebitis, as well as in seasonings, soaps, creams, and perfumes.1 The most commonly encountered plants known to cause phytophotodermatitis belong to the Apiaceae and Rutaceae families.2 Members of Apiaceae include angelica, celery, dill, fennel, hogweed, parsley, and parsnip. Aside from the common rue plant, the Rutaceae family also includes bergamot orange, bitter orange, burning bush (or gas plant), grapefruit, lemon, and lime. Other potential offending agents are fig, mustard, buttercup, St. John’s wort, and scurfpea. The phototoxic properties are due to furocoumarins, which include psoralens and angelicins. They are inert until activated by UVA radiation, which inflicts direct cellular damage, causing vacuolization and apoptosis of keratinocytes, similar to a sunburn.3 Clinical findings typically present 24 hours after sun exposure with erythema, edema, pain, and occasionally vesicles or bullae in severe cases. Unlike sunburn, lesions often present in linear, streaky, or bizarre patterns, reflective of the direct contact with the plant. The lesions eventually transition to hyperpigmentation, which may take months to years to resolve.
Other considerations in cases of suspected phytophotodermatitis include polymorphic light eruption, actinic prurigo, hydroa vacciniforme, chronic actinic dermatitis, solar urticaria, drug reactions, porphyria, Smith-Lemli-Opitz syndrome, lupus erythematosus, and dermatomyositis.4 Clinicians should suspect phytophotodermatitis with phototoxic findings in bartenders, citrus farm workers, gardeners, chefs, and kitchen workers, especially those handling limes and celery. As in our case, phytophotodermatitis also should be considered in butterfly enthusiasts trying to attract the giant swallowtail butterfly. The caterpillars feed on the leaves of the common rue plant, one of a select few plants that giant swallowtail butterflies use as a host due to its bitter leaves that aid in avoiding predators.5
This case illustrates a unique perspective of phytophotodermatitis, as butterfly enthusiasm is not commonly reported in association with the common rue plant with respect to phytophotodermatitis. This case underscores the importance of inquiring about patients’ professions and hobbies, both in dermatology and other specialties.
To the Editor:
Phytophotodermatitis is common in dermatology during the summer months, especially in individuals who spend time outdoors; however, identification of the offending plant can be challenging. We report a case of phytophotodermatitis in which the causative plant, common rue, was not identified until it was revealed that the patient was a butterfly enthusiast.
A 60-year-old woman presented to the outpatient dermatology clinic in late summer for a routine skin examination. An eruption was noted over the right thigh and knee that had first appeared approximately 2 weeks prior. The rash started as pruritic blisters but gradually progressed to erythema and then eventually to brown markings, which were observed at the current presentation. Physical examination revealed hyperpigmented, brown, streaky, linear patches and plaques over the right thigh, knee, and lower leg (Figure). When asked about her hobbies, the patient reported an affinity for butterflies and noted that she attracts them with specific species of plants in her garden. She recalled recently planting the herb of grace, or common rue, to attract the giant swallowtail butterfly (Papilio cresphontes). Upon further inquiry, she remembered working in the garden on her knees and digging up roots near the common rue plant while wearing shorts approximately 2 weeks prior to the current presentation. Given the streaky linear pattern of the eruption along with recent sun exposure and exposure to the common rue plant, a diagnosis of phytophotodermatitis was made. No further treatment was sought, as the eruption was not bothersome to her. She was intrigued that the common rue plant had caused the dermatitis and planned on taking proper precautions when working near the plant in the future.
In this case, the observed phototoxic skin findings resulted from exposure to common rue (Ruta graveolens),a pungently scented evergreen shrub native to the Mediterranean region and a member of the Rutaceae family. Extracts have been used in homeopathic practices for bruises, sprains, headache, neck stiffness, rheumatologic pain, neuralgia, stomach problems, and phlebitis, as well as in seasonings, soaps, creams, and perfumes.1 The most commonly encountered plants known to cause phytophotodermatitis belong to the Apiaceae and Rutaceae families.2 Members of Apiaceae include angelica, celery, dill, fennel, hogweed, parsley, and parsnip. Aside from the common rue plant, the Rutaceae family also includes bergamot orange, bitter orange, burning bush (or gas plant), grapefruit, lemon, and lime. Other potential offending agents are fig, mustard, buttercup, St. John’s wort, and scurfpea. The phototoxic properties are due to furocoumarins, which include psoralens and angelicins. They are inert until activated by UVA radiation, which inflicts direct cellular damage, causing vacuolization and apoptosis of keratinocytes, similar to a sunburn.3 Clinical findings typically present 24 hours after sun exposure with erythema, edema, pain, and occasionally vesicles or bullae in severe cases. Unlike sunburn, lesions often present in linear, streaky, or bizarre patterns, reflective of the direct contact with the plant. The lesions eventually transition to hyperpigmentation, which may take months to years to resolve.
Other considerations in cases of suspected phytophotodermatitis include polymorphic light eruption, actinic prurigo, hydroa vacciniforme, chronic actinic dermatitis, solar urticaria, drug reactions, porphyria, Smith-Lemli-Opitz syndrome, lupus erythematosus, and dermatomyositis.4 Clinicians should suspect phytophotodermatitis with phototoxic findings in bartenders, citrus farm workers, gardeners, chefs, and kitchen workers, especially those handling limes and celery. As in our case, phytophotodermatitis also should be considered in butterfly enthusiasts trying to attract the giant swallowtail butterfly. The caterpillars feed on the leaves of the common rue plant, one of a select few plants that giant swallowtail butterflies use as a host due to its bitter leaves that aid in avoiding predators.5
This case illustrates a unique perspective of phytophotodermatitis, as butterfly enthusiasm is not commonly reported in association with the common rue plant with respect to phytophotodermatitis. This case underscores the importance of inquiring about patients’ professions and hobbies, both in dermatology and other specialties.
- Atta AH, Alkofahi A. Anti-nociceptive and anti-inflammatory effects of some Jordanian medicinal plant extracts. J Ethnopharmacol. 1998;60:117-124.
- McGovern TW. Dermatoses due to plants. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. Edinburgh, Scotland: Mosby; 2007:265-283.
- Hawk JLM, Calonje E. The photosensitivity disorders. In: Elder DE, ed. Lever’s Histopathology of the Skin. 9th ed. Philadelphia, Pennsylvania: Lippincott Williams and Wilkins; 2005:345-353.
- Lim HW. Abnormal responses to ultraviolet radiation: photosensitivity induced by exogenous agents. In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012:1066-1074.
- McAuslane H. Giant swallowtail. University of Florida Department of Entomology and Nematology Featured Creatures website. http://entnemdept.ufl.edu/creatures/citrus/giantswallowtail.htm. Revised January 2018. Accessed April 10, 2020.
- Atta AH, Alkofahi A. Anti-nociceptive and anti-inflammatory effects of some Jordanian medicinal plant extracts. J Ethnopharmacol. 1998;60:117-124.
- McGovern TW. Dermatoses due to plants. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. Edinburgh, Scotland: Mosby; 2007:265-283.
- Hawk JLM, Calonje E. The photosensitivity disorders. In: Elder DE, ed. Lever’s Histopathology of the Skin. 9th ed. Philadelphia, Pennsylvania: Lippincott Williams and Wilkins; 2005:345-353.
- Lim HW. Abnormal responses to ultraviolet radiation: photosensitivity induced by exogenous agents. In: Wolff K, Goldsmith LA, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012:1066-1074.
- McAuslane H. Giant swallowtail. University of Florida Department of Entomology and Nematology Featured Creatures website. http://entnemdept.ufl.edu/creatures/citrus/giantswallowtail.htm. Revised January 2018. Accessed April 10, 2020.
Practice Points
- It is important to inquire about patients’ professions and hobbies, which may lead to the diagnosis, as in this case of a butterfly enthusiast trying to attract the giant swallowtail butterfly with the common rue plant.
- One should suspect phytophotodermatitis with phototoxic findings in bartenders, citrus farm workers, gardeners, chefs, and kitchen workers, especially those handling limes and celery

Cutaneous Metastases From Esophageal Adenocarcinoma on the Scalp
To the Editor:
A 59-year-old man presented with a lesion on the right frontal scalp of 4 months’ duration and a lesion on the left frontal scalp of 1 month’s duration. Both lesions were tender, bleeding, nonhealing, and growing in size. The patient reported no improvement with the use of triple antibiotic ointment. He denied any associated symptoms or trauma to the affected areas. He had a history of stage IV esophageal adenocarcinoma that initially had been surgically removed 6 years prior but metastasized to the lungs and bone. The patient subsequently underwent treatment with FOLFOX (folinic acid, fluorouracil, oxaliplatin), trastuzumab, and radiation therapy.
Physical examination revealed a hyperkeratotic pink nodule with a central erosion and crust on the right frontal scalp measuring 1.5×2 cm in diameter (Figure 1A). The left frontal scalp lesion was a smooth pearly papule measuring 5×5 mm in diameter (Figure 1B). The differential diagnosis included basal cell carcinoma, squamous cell carcinoma, and cutaneous metastases from esophageal adenocarcinoma. Shave biopsies were taken of both scalp lesions.
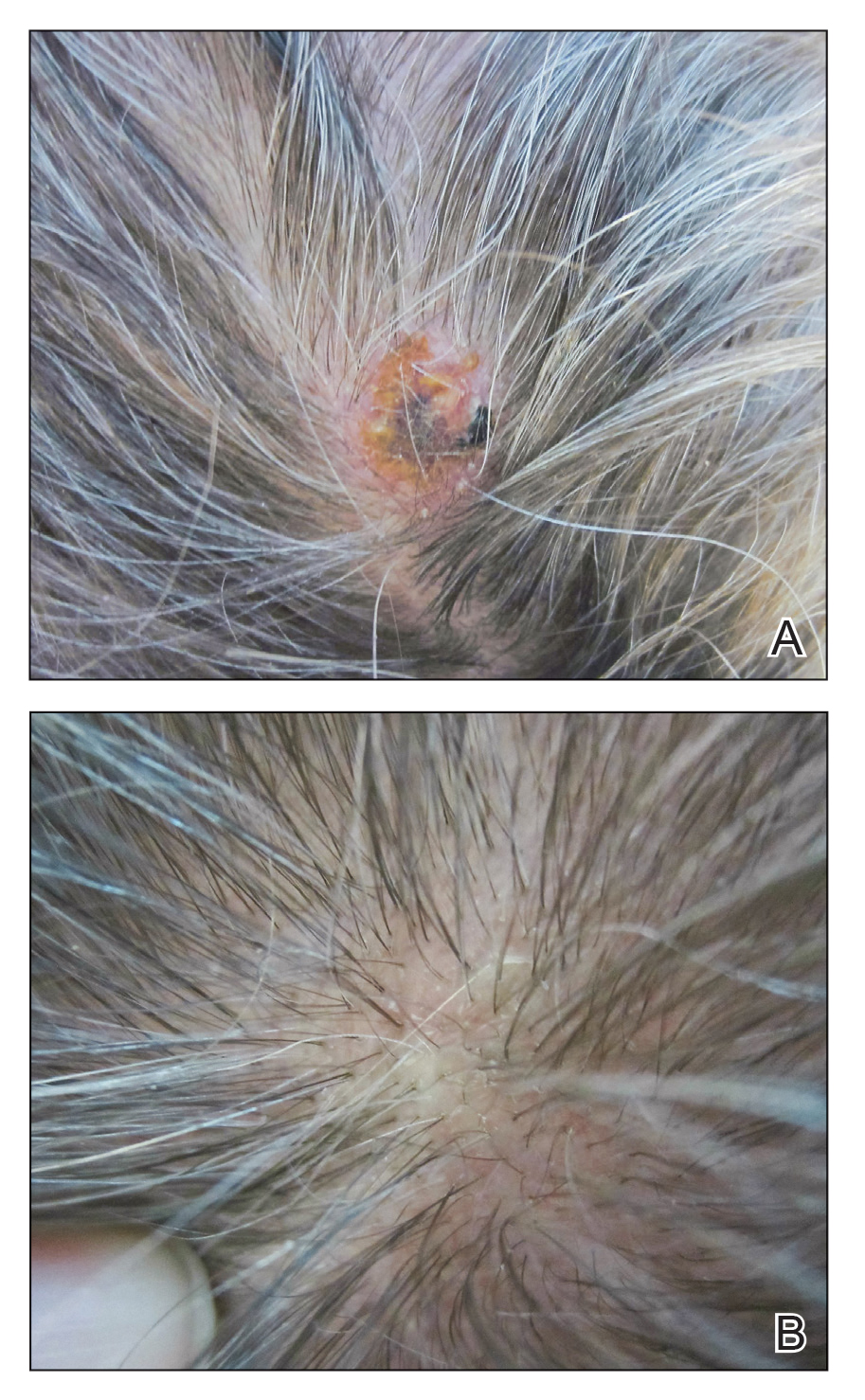
Histologic examination of both scalp lesions demonstrated a dermal gland-forming neoplasm with an infiltrative distribution that was comprised of irregular cribriform glands containing cellular debris (Figure 2). The cells of interest were enlarged and contained pleomorphic crowded nuclei that formed aberrant mitotic division figures. Both biopsies were positive for cytokeratin 7 and negative for cytokeratin 20 and CDX2. The final diagnosis for both scalp lesions was poorly differentiated adenocarcinoma, which was most suggestive of cutaneous metastases of the patient’s known esophageal adenocarcinoma. Given further metastasis, the patient was ultimately switched to ramucirumab and paclitaxel per oncology.
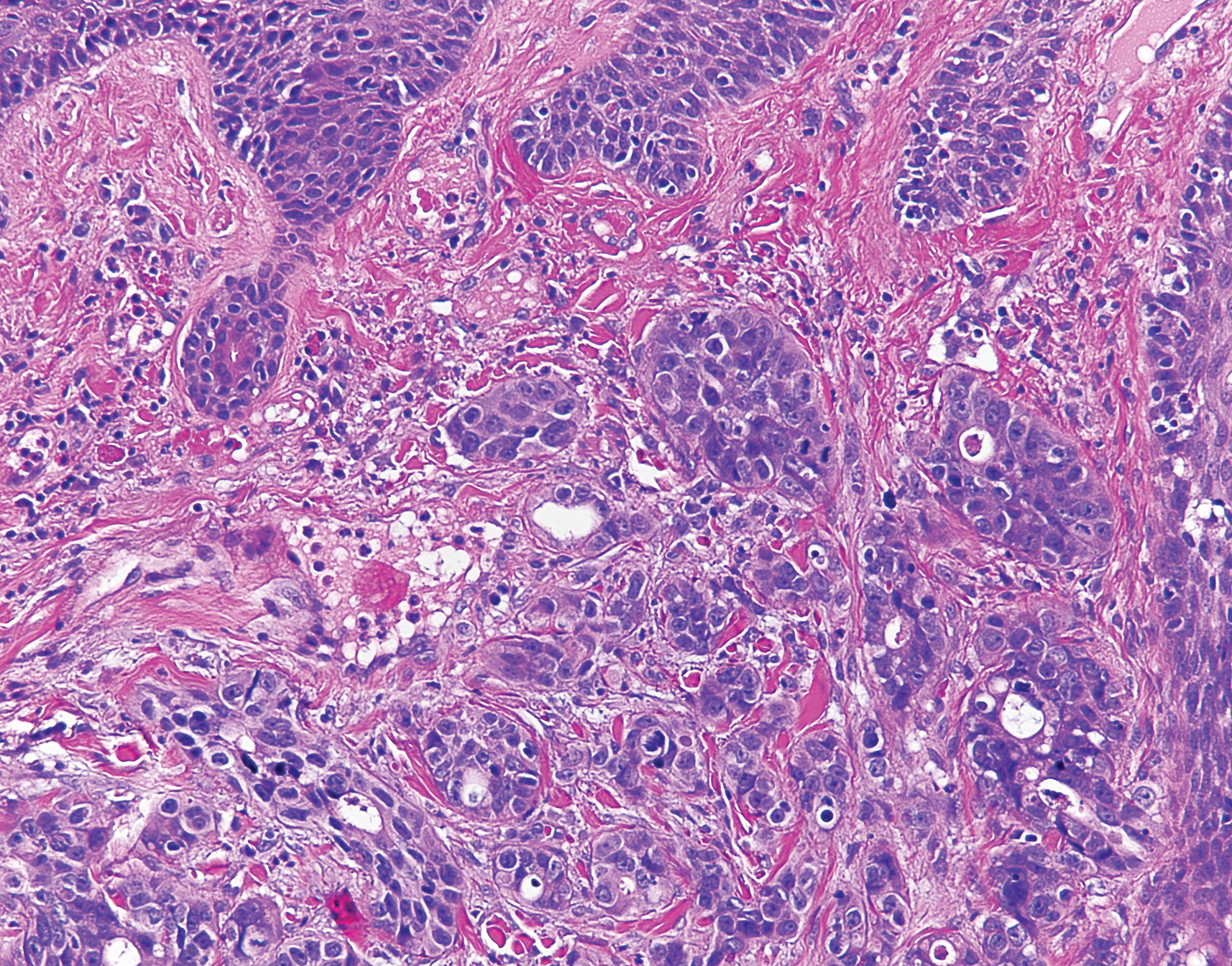
Esophageal carcinoma is the eighth most common cause of death related to cancer worldwide. Adenocarcinoma is the most prevalent histologic type of esophageal carcinoma, with an incidence as high as 5.69 per 100,000 individuals in the United States.1 Internal malignancies that lead to cutaneous metastases are not uncommon; however, the literature is limited on cutaneous scalp metastases from esophageal cancer. Cutaneous metastases secondary to internal malignancies present in less than 10% of overall cases; tend to derive from the breasts, lungs, and large bowel; and usually present in the sixth to seventh decades of life.2 Further, roughly 1% of all skin metastases originate from the esophagus.3 When there are cutaneous metastases to the scalp, they often arise from breast carcinomas and renal cell carcinomas.4,5 Rarely does esophageal cancer spread to the scalp.2,6-9 When cutaneous metastases originate from the esophagus, multiple cancers such as squamous cell carcinomas, mucoepidermoid carcinomas, small cell carcinomas, and adenocarcinomas can be the etiology of origin.10 Metastases originating from esophageal carcinomas frequently are diagnosed in the abdominal lymph nodes (45%), liver (35%), lungs (20%), cervical/supraclavicular lymph nodes (18%), bones (9%), adrenals (5%), peritoneum (2%), brain (2%), stomach (1%), pancreas (1%), pleura (1%), skin/body wall (1%), pericardium (1%), and spleen (1%).3 Additionally, multiple cutaneous scalp metastases from esophageal adenocarcinoma have been reported,7,9 as were seen in our case.
The clinical appearance of cutaneous scalp metastases has been described as inflammatory papules, indurated plaques, or nodules,2 which is consistent with our case, though the spectrum of presentation is admittedly broad. Histopathology of lesions characteristically shows prominent intraluminal necrotic cellular debris, which is common for adenocarcinomas of the gastrointestinal tract.7 However, utilizing immunohistochemical stains to detect specific antigens within tumor cells allows for better specificity of the tumor origin. More specifically, cytokeratin 7 and cytokeratin 20 are stained in esophageal metaplasia, such as Barrett esophagus, rather than in intestinal metaplasia inside the stomach.2,11 Therefore, discerning the location of the adenocarcinoma proves fruitful when using cytokeratin 7 and cytokeratin 20. Although CDX2 is an additional marker that can be used for gastrointestinal adenocarcinomas with decent sensitivity and specificity, it can still be expressed in mucinous ovarian carcinomas and urinary bladder adenocarcinomas.12 In our patient, the strong reactivity of cytokeratin 7 in addition to the characteristic morphology in both presenting biopsies was sufficient to make the diagnosis of cutaneous metastasis of esophageal adenocarcinoma to the scalp.
Our case highlights multiple cutaneous metastases of the scalp from a primary esophageal adenocarcinoma. Although cutaneous scalp metastasis of esophageal adenocarcinoma is rare, it is essential to provide a full-body skin examination, including the scalp, in patients with a history of esophageal cancer and to biopsy any suspicious nodules or plaques. The 1-year survival rate after diagnosis of esophageal carcinoma is less than 50%, and the 5-year survival rate is less than 10%.13 Identifying cutaneous metastasis of an esophageal adenocarcinoma can either change the staging of the cancer (if it was the first distant metastasis noted) or indicate an insufficient response to treatment in a patient with known metastatic disease, prompting a potential change in treatment.7
This case illustrates a rare site of metastasis of a fairly common cancer and highlights the histopathology and accompanying immunohistochemical stains that can be useful in diagnosis as well as the spectrum of its clinical presentation.
- Melhado R, Alderson D, Tucker O. The changing face of esophageal cancer. Cancers (Basel). 2010;2:1379-1404.
- Park JM, Kim DS, Oh SH, et al. A case of esophageal adenocarcinoma metastasized to the scalp [published online May 31, 2009]. Ann Dermatol. 2009;21:164-167.
- Quint LE, Hepburn LM, Francis IR, et al. Incidence and distribution of distant metastases from newly diagnosed esophageal carcinoma. Cancer. 1995;76:1120.
- Dobson C, Tagor V, Myint A, et al. Telangiectatic metastatic breast carcinoma in face and scalp mimicking cutaneous angiosarcoma. J Am Acad Dermatol. 2003;48:635-636.
- Riter H, Ghobrial I. Renal cell carcinoma with acrometastasis and scalp metastasis. Mayo Clin Proc. 2004;79:76.
- Roh EK, Nord R, Jukic DM. Scalp metastasis from esophageal adenocarcinoma. Cutis. 2006;77:106.
- Doumit G, Abouhassan W, Piliang M, et al. Scalp metastasis from esophageal adenocarcinoma: comparative histopathology dictates surgical approach. Ann Plast Surg. 2011;71:60-62.
- Roy AD, Sherparpa M, Prasad PR, et al. Scalp metastasis of gastro-esophageal junction adenocarcinoma: a rare occurrence. 2014;8:159-160.
- Stein R, Spencer J. Painful cutaneous metastases from esophageal carcinoma. Cutis. 2002;70:230.
- Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33(2 pt 1):161-182.
- Ormsby AH, Goldblum JR, Rice TW, et al. Cytokeratin subsets can reliably distinguish Barrett’s esophagus from intestinal metaplasia of the stomach. Hum Pathol. 1999;30:288-294.
- Werling RW, Yaziji H, Bacchi CE, et al. CDX2, a highly sensitive and specific marker of adenocarcinomas of intestinal origin: an immunohistochemical survey of 476 primary and metastatic carcinomas. Am J Surg Pathol. 2003;27:303-310.
- Smith KJ, Williams J, Skelton H. Metastatic adenocarcinoma of the esophagus to the skin: new patterns of tumor recurrence and alternate treatments for palliation. J Cutan Pathol. 2001;28:425-431.
To the Editor:
A 59-year-old man presented with a lesion on the right frontal scalp of 4 months’ duration and a lesion on the left frontal scalp of 1 month’s duration. Both lesions were tender, bleeding, nonhealing, and growing in size. The patient reported no improvement with the use of triple antibiotic ointment. He denied any associated symptoms or trauma to the affected areas. He had a history of stage IV esophageal adenocarcinoma that initially had been surgically removed 6 years prior but metastasized to the lungs and bone. The patient subsequently underwent treatment with FOLFOX (folinic acid, fluorouracil, oxaliplatin), trastuzumab, and radiation therapy.
Physical examination revealed a hyperkeratotic pink nodule with a central erosion and crust on the right frontal scalp measuring 1.5×2 cm in diameter (Figure 1A). The left frontal scalp lesion was a smooth pearly papule measuring 5×5 mm in diameter (Figure 1B). The differential diagnosis included basal cell carcinoma, squamous cell carcinoma, and cutaneous metastases from esophageal adenocarcinoma. Shave biopsies were taken of both scalp lesions.
Histologic examination of both scalp lesions demonstrated a dermal gland-forming neoplasm with an infiltrative distribution that was comprised of irregular cribriform glands containing cellular debris (Figure 2). The cells of interest were enlarged and contained pleomorphic crowded nuclei that formed aberrant mitotic division figures. Both biopsies were positive for cytokeratin 7 and negative for cytokeratin 20 and CDX2. The final diagnosis for both scalp lesions was poorly differentiated adenocarcinoma, which was most suggestive of cutaneous metastases of the patient’s known esophageal adenocarcinoma. Given further metastasis, the patient was ultimately switched to ramucirumab and paclitaxel per oncology.
Esophageal carcinoma is the eighth most common cause of death related to cancer worldwide. Adenocarcinoma is the most prevalent histologic type of esophageal carcinoma, with an incidence as high as 5.69 per 100,000 individuals in the United States.1 Internal malignancies that lead to cutaneous metastases are not uncommon; however, the literature is limited on cutaneous scalp metastases from esophageal cancer. Cutaneous metastases secondary to internal malignancies present in less than 10% of overall cases; tend to derive from the breasts, lungs, and large bowel; and usually present in the sixth to seventh decades of life.2 Further, roughly 1% of all skin metastases originate from the esophagus.3 When there are cutaneous metastases to the scalp, they often arise from breast carcinomas and renal cell carcinomas.4,5 Rarely does esophageal cancer spread to the scalp.2,6-9 When cutaneous metastases originate from the esophagus, multiple cancers such as squamous cell carcinomas, mucoepidermoid carcinomas, small cell carcinomas, and adenocarcinomas can be the etiology of origin.10 Metastases originating from esophageal carcinomas frequently are diagnosed in the abdominal lymph nodes (45%), liver (35%), lungs (20%), cervical/supraclavicular lymph nodes (18%), bones (9%), adrenals (5%), peritoneum (2%), brain (2%), stomach (1%), pancreas (1%), pleura (1%), skin/body wall (1%), pericardium (1%), and spleen (1%).3 Additionally, multiple cutaneous scalp metastases from esophageal adenocarcinoma have been reported,7,9 as were seen in our case.
The clinical appearance of cutaneous scalp metastases has been described as inflammatory papules, indurated plaques, or nodules,2 which is consistent with our case, though the spectrum of presentation is admittedly broad. Histopathology of lesions characteristically shows prominent intraluminal necrotic cellular debris, which is common for adenocarcinomas of the gastrointestinal tract.7 However, utilizing immunohistochemical stains to detect specific antigens within tumor cells allows for better specificity of the tumor origin. More specifically, cytokeratin 7 and cytokeratin 20 are stained in esophageal metaplasia, such as Barrett esophagus, rather than in intestinal metaplasia inside the stomach.2,11 Therefore, discerning the location of the adenocarcinoma proves fruitful when using cytokeratin 7 and cytokeratin 20. Although CDX2 is an additional marker that can be used for gastrointestinal adenocarcinomas with decent sensitivity and specificity, it can still be expressed in mucinous ovarian carcinomas and urinary bladder adenocarcinomas.12 In our patient, the strong reactivity of cytokeratin 7 in addition to the characteristic morphology in both presenting biopsies was sufficient to make the diagnosis of cutaneous metastasis of esophageal adenocarcinoma to the scalp.
Our case highlights multiple cutaneous metastases of the scalp from a primary esophageal adenocarcinoma. Although cutaneous scalp metastasis of esophageal adenocarcinoma is rare, it is essential to provide a full-body skin examination, including the scalp, in patients with a history of esophageal cancer and to biopsy any suspicious nodules or plaques. The 1-year survival rate after diagnosis of esophageal carcinoma is less than 50%, and the 5-year survival rate is less than 10%.13 Identifying cutaneous metastasis of an esophageal adenocarcinoma can either change the staging of the cancer (if it was the first distant metastasis noted) or indicate an insufficient response to treatment in a patient with known metastatic disease, prompting a potential change in treatment.7
This case illustrates a rare site of metastasis of a fairly common cancer and highlights the histopathology and accompanying immunohistochemical stains that can be useful in diagnosis as well as the spectrum of its clinical presentation.
To the Editor:
A 59-year-old man presented with a lesion on the right frontal scalp of 4 months’ duration and a lesion on the left frontal scalp of 1 month’s duration. Both lesions were tender, bleeding, nonhealing, and growing in size. The patient reported no improvement with the use of triple antibiotic ointment. He denied any associated symptoms or trauma to the affected areas. He had a history of stage IV esophageal adenocarcinoma that initially had been surgically removed 6 years prior but metastasized to the lungs and bone. The patient subsequently underwent treatment with FOLFOX (folinic acid, fluorouracil, oxaliplatin), trastuzumab, and radiation therapy.
Physical examination revealed a hyperkeratotic pink nodule with a central erosion and crust on the right frontal scalp measuring 1.5×2 cm in diameter (Figure 1A). The left frontal scalp lesion was a smooth pearly papule measuring 5×5 mm in diameter (Figure 1B). The differential diagnosis included basal cell carcinoma, squamous cell carcinoma, and cutaneous metastases from esophageal adenocarcinoma. Shave biopsies were taken of both scalp lesions.
Histologic examination of both scalp lesions demonstrated a dermal gland-forming neoplasm with an infiltrative distribution that was comprised of irregular cribriform glands containing cellular debris (Figure 2). The cells of interest were enlarged and contained pleomorphic crowded nuclei that formed aberrant mitotic division figures. Both biopsies were positive for cytokeratin 7 and negative for cytokeratin 20 and CDX2. The final diagnosis for both scalp lesions was poorly differentiated adenocarcinoma, which was most suggestive of cutaneous metastases of the patient’s known esophageal adenocarcinoma. Given further metastasis, the patient was ultimately switched to ramucirumab and paclitaxel per oncology.
Esophageal carcinoma is the eighth most common cause of death related to cancer worldwide. Adenocarcinoma is the most prevalent histologic type of esophageal carcinoma, with an incidence as high as 5.69 per 100,000 individuals in the United States.1 Internal malignancies that lead to cutaneous metastases are not uncommon; however, the literature is limited on cutaneous scalp metastases from esophageal cancer. Cutaneous metastases secondary to internal malignancies present in less than 10% of overall cases; tend to derive from the breasts, lungs, and large bowel; and usually present in the sixth to seventh decades of life.2 Further, roughly 1% of all skin metastases originate from the esophagus.3 When there are cutaneous metastases to the scalp, they often arise from breast carcinomas and renal cell carcinomas.4,5 Rarely does esophageal cancer spread to the scalp.2,6-9 When cutaneous metastases originate from the esophagus, multiple cancers such as squamous cell carcinomas, mucoepidermoid carcinomas, small cell carcinomas, and adenocarcinomas can be the etiology of origin.10 Metastases originating from esophageal carcinomas frequently are diagnosed in the abdominal lymph nodes (45%), liver (35%), lungs (20%), cervical/supraclavicular lymph nodes (18%), bones (9%), adrenals (5%), peritoneum (2%), brain (2%), stomach (1%), pancreas (1%), pleura (1%), skin/body wall (1%), pericardium (1%), and spleen (1%).3 Additionally, multiple cutaneous scalp metastases from esophageal adenocarcinoma have been reported,7,9 as were seen in our case.
The clinical appearance of cutaneous scalp metastases has been described as inflammatory papules, indurated plaques, or nodules,2 which is consistent with our case, though the spectrum of presentation is admittedly broad. Histopathology of lesions characteristically shows prominent intraluminal necrotic cellular debris, which is common for adenocarcinomas of the gastrointestinal tract.7 However, utilizing immunohistochemical stains to detect specific antigens within tumor cells allows for better specificity of the tumor origin. More specifically, cytokeratin 7 and cytokeratin 20 are stained in esophageal metaplasia, such as Barrett esophagus, rather than in intestinal metaplasia inside the stomach.2,11 Therefore, discerning the location of the adenocarcinoma proves fruitful when using cytokeratin 7 and cytokeratin 20. Although CDX2 is an additional marker that can be used for gastrointestinal adenocarcinomas with decent sensitivity and specificity, it can still be expressed in mucinous ovarian carcinomas and urinary bladder adenocarcinomas.12 In our patient, the strong reactivity of cytokeratin 7 in addition to the characteristic morphology in both presenting biopsies was sufficient to make the diagnosis of cutaneous metastasis of esophageal adenocarcinoma to the scalp.
Our case highlights multiple cutaneous metastases of the scalp from a primary esophageal adenocarcinoma. Although cutaneous scalp metastasis of esophageal adenocarcinoma is rare, it is essential to provide a full-body skin examination, including the scalp, in patients with a history of esophageal cancer and to biopsy any suspicious nodules or plaques. The 1-year survival rate after diagnosis of esophageal carcinoma is less than 50%, and the 5-year survival rate is less than 10%.13 Identifying cutaneous metastasis of an esophageal adenocarcinoma can either change the staging of the cancer (if it was the first distant metastasis noted) or indicate an insufficient response to treatment in a patient with known metastatic disease, prompting a potential change in treatment.7
This case illustrates a rare site of metastasis of a fairly common cancer and highlights the histopathology and accompanying immunohistochemical stains that can be useful in diagnosis as well as the spectrum of its clinical presentation.
- Melhado R, Alderson D, Tucker O. The changing face of esophageal cancer. Cancers (Basel). 2010;2:1379-1404.
- Park JM, Kim DS, Oh SH, et al. A case of esophageal adenocarcinoma metastasized to the scalp [published online May 31, 2009]. Ann Dermatol. 2009;21:164-167.
- Quint LE, Hepburn LM, Francis IR, et al. Incidence and distribution of distant metastases from newly diagnosed esophageal carcinoma. Cancer. 1995;76:1120.
- Dobson C, Tagor V, Myint A, et al. Telangiectatic metastatic breast carcinoma in face and scalp mimicking cutaneous angiosarcoma. J Am Acad Dermatol. 2003;48:635-636.
- Riter H, Ghobrial I. Renal cell carcinoma with acrometastasis and scalp metastasis. Mayo Clin Proc. 2004;79:76.
- Roh EK, Nord R, Jukic DM. Scalp metastasis from esophageal adenocarcinoma. Cutis. 2006;77:106.
- Doumit G, Abouhassan W, Piliang M, et al. Scalp metastasis from esophageal adenocarcinoma: comparative histopathology dictates surgical approach. Ann Plast Surg. 2011;71:60-62.
- Roy AD, Sherparpa M, Prasad PR, et al. Scalp metastasis of gastro-esophageal junction adenocarcinoma: a rare occurrence. 2014;8:159-160.
- Stein R, Spencer J. Painful cutaneous metastases from esophageal carcinoma. Cutis. 2002;70:230.
- Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33(2 pt 1):161-182.
- Ormsby AH, Goldblum JR, Rice TW, et al. Cytokeratin subsets can reliably distinguish Barrett’s esophagus from intestinal metaplasia of the stomach. Hum Pathol. 1999;30:288-294.
- Werling RW, Yaziji H, Bacchi CE, et al. CDX2, a highly sensitive and specific marker of adenocarcinomas of intestinal origin: an immunohistochemical survey of 476 primary and metastatic carcinomas. Am J Surg Pathol. 2003;27:303-310.
- Smith KJ, Williams J, Skelton H. Metastatic adenocarcinoma of the esophagus to the skin: new patterns of tumor recurrence and alternate treatments for palliation. J Cutan Pathol. 2001;28:425-431.
- Melhado R, Alderson D, Tucker O. The changing face of esophageal cancer. Cancers (Basel). 2010;2:1379-1404.
- Park JM, Kim DS, Oh SH, et al. A case of esophageal adenocarcinoma metastasized to the scalp [published online May 31, 2009]. Ann Dermatol. 2009;21:164-167.
- Quint LE, Hepburn LM, Francis IR, et al. Incidence and distribution of distant metastases from newly diagnosed esophageal carcinoma. Cancer. 1995;76:1120.
- Dobson C, Tagor V, Myint A, et al. Telangiectatic metastatic breast carcinoma in face and scalp mimicking cutaneous angiosarcoma. J Am Acad Dermatol. 2003;48:635-636.
- Riter H, Ghobrial I. Renal cell carcinoma with acrometastasis and scalp metastasis. Mayo Clin Proc. 2004;79:76.
- Roh EK, Nord R, Jukic DM. Scalp metastasis from esophageal adenocarcinoma. Cutis. 2006;77:106.
- Doumit G, Abouhassan W, Piliang M, et al. Scalp metastasis from esophageal adenocarcinoma: comparative histopathology dictates surgical approach. Ann Plast Surg. 2011;71:60-62.
- Roy AD, Sherparpa M, Prasad PR, et al. Scalp metastasis of gastro-esophageal junction adenocarcinoma: a rare occurrence. 2014;8:159-160.
- Stein R, Spencer J. Painful cutaneous metastases from esophageal carcinoma. Cutis. 2002;70:230.
- Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33(2 pt 1):161-182.
- Ormsby AH, Goldblum JR, Rice TW, et al. Cytokeratin subsets can reliably distinguish Barrett’s esophagus from intestinal metaplasia of the stomach. Hum Pathol. 1999;30:288-294.
- Werling RW, Yaziji H, Bacchi CE, et al. CDX2, a highly sensitive and specific marker of adenocarcinomas of intestinal origin: an immunohistochemical survey of 476 primary and metastatic carcinomas. Am J Surg Pathol. 2003;27:303-310.
- Smith KJ, Williams J, Skelton H. Metastatic adenocarcinoma of the esophagus to the skin: new patterns of tumor recurrence and alternate treatments for palliation. J Cutan Pathol. 2001;28:425-431.
Practice Points
- In the setting of underlying esophageal adenocarcinoma, metastatic spread to the scalp should be considered in the differential diagnosis for any suspicious scalp lesions.
- Coupling histopathology with immunohistochemical stains may aid in the diagnosis for cutaneous metastasis of esophageal adenocarcinoma.
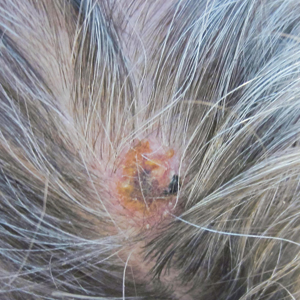
Telangiectatic Patch on the Neck
The Diagnosis: Unilateral Nevoid Telangiectasia
Unilateral nevoid telangiectasia (UNT) is an uncommon, or perhaps underreported, cutaneous condition involving telangiectatic patches in a unilateral dermatomal or blaschkoid pattern.1 The condition has been described as either congenital or acquired. Congenital UNT is thought to be a result of somatic mosaicism, whereby a mutation during embryogenesis leads to a distinct population of cells expressing the vascular malformation.1 Congenital UNT has been associated with Becker nevus, which also is thought to be a result of somatic mosaicism, further providing evidence for this theory, though it is unclear whether this finding is incidental.2 The acquired form often is associated with fluctuation of hormones, such as in pregnancy or with oral contraceptive initiation, as well as with hepatic disease as seen in our patient. However, there are many cases of acquired UNT with no implicated underlying disease, alcohol abuse, or hormonal changes, which calls into question if UNT is definitively an estrogen-related condition.3 One study demonstrated an increased level of estrogen and progesterone receptors in affected skin, which may have led to expression of the cutaneous changes at that site.4 More research is needed to elucidate this point, as other studies have not reproduced similar findings.
Congenital UNT occurs more commonly in males, whereas the acquired variant is seen more frequently in females. The third and fourth cervical dermatomes most often are involved.5 Most lesions persist without spontaneous resolution. Treatment options are limited and include pulsed dye laser treatment and makeup application to cover the telangiectatic patches. The main side effect seen with pulsed dye laser treatment is reversible pigmentary changes, with 1 report of textural skin change.6
A biopsy was deemed unnecessary for the clinical diagnosis in our patient because there was a clear explanation for the physical examination findings due to long-standing underlying liver disease. When biopsied, UNT characteristically demonstrates dilated dermal capillaries.5 Our patient elected not to pursue laser therapy but expressed interest in using makeup to camouflage the lesion.
The differential diagnosis includes acquired nevus flammeus, which typically is present on the face and often appears following mechanical or thermal trauma. Angioma serpiginosum most often occurs on the buttocks and legs as small red papules or puncta coalescing into a serpiginous linear arrangement. It often appears in childhood. Angiosarcoma is an aggressive malignancy that often occurs on the head and neck in elderly patients. It is associated with areas of long-standing lymphedema and often appears as a bruiselike lesion. Rosacea typically is not fixed in its clinical appearance and presents as transitory flushing of the head and neck with or without a history of acneform eruptions on the face. It typically is not unilateral.
- Wilkin JK. Unilateral dermatomal superficial telangiectasia. Arch Dermatol. 1984;120:579-580.
- Karakaş M, Durdu M, Sönmezoğlu S, et al. Unilateral nevoid telangiectasia. J Dermatol. 2004;31:109-112.
- Taskapan O, Harmanyeri Y, Sener O, et al. Acquired unilateral nevoid telangiectasia syndrome. Acta Derm Venereol. 1997;77:62-63.
- Uhlin SR, McCarty KS Jr. Unilateral nevoid telangiectatic syndrome: the role of estrogen and progesterone receptors. Arch Dermatol. 1983;119:226-228.
- Derrow AE, Adams BB, Timani S, et al. Acquired unilateral nevoid telangiectasia in a 51-year-old female. Int J Dermatol. 2008;47:1331-1333.
- Sharma VK, Khandpur S. Unilateral nevoid telangiectasia--response to pulsed dye laser. Int J Dermatol. 2006;45:960-964.
The Diagnosis: Unilateral Nevoid Telangiectasia
Unilateral nevoid telangiectasia (UNT) is an uncommon, or perhaps underreported, cutaneous condition involving telangiectatic patches in a unilateral dermatomal or blaschkoid pattern.1 The condition has been described as either congenital or acquired. Congenital UNT is thought to be a result of somatic mosaicism, whereby a mutation during embryogenesis leads to a distinct population of cells expressing the vascular malformation.1 Congenital UNT has been associated with Becker nevus, which also is thought to be a result of somatic mosaicism, further providing evidence for this theory, though it is unclear whether this finding is incidental.2 The acquired form often is associated with fluctuation of hormones, such as in pregnancy or with oral contraceptive initiation, as well as with hepatic disease as seen in our patient. However, there are many cases of acquired UNT with no implicated underlying disease, alcohol abuse, or hormonal changes, which calls into question if UNT is definitively an estrogen-related condition.3 One study demonstrated an increased level of estrogen and progesterone receptors in affected skin, which may have led to expression of the cutaneous changes at that site.4 More research is needed to elucidate this point, as other studies have not reproduced similar findings.
Congenital UNT occurs more commonly in males, whereas the acquired variant is seen more frequently in females. The third and fourth cervical dermatomes most often are involved.5 Most lesions persist without spontaneous resolution. Treatment options are limited and include pulsed dye laser treatment and makeup application to cover the telangiectatic patches. The main side effect seen with pulsed dye laser treatment is reversible pigmentary changes, with 1 report of textural skin change.6
A biopsy was deemed unnecessary for the clinical diagnosis in our patient because there was a clear explanation for the physical examination findings due to long-standing underlying liver disease. When biopsied, UNT characteristically demonstrates dilated dermal capillaries.5 Our patient elected not to pursue laser therapy but expressed interest in using makeup to camouflage the lesion.
The differential diagnosis includes acquired nevus flammeus, which typically is present on the face and often appears following mechanical or thermal trauma. Angioma serpiginosum most often occurs on the buttocks and legs as small red papules or puncta coalescing into a serpiginous linear arrangement. It often appears in childhood. Angiosarcoma is an aggressive malignancy that often occurs on the head and neck in elderly patients. It is associated with areas of long-standing lymphedema and often appears as a bruiselike lesion. Rosacea typically is not fixed in its clinical appearance and presents as transitory flushing of the head and neck with or without a history of acneform eruptions on the face. It typically is not unilateral.
The Diagnosis: Unilateral Nevoid Telangiectasia
Unilateral nevoid telangiectasia (UNT) is an uncommon, or perhaps underreported, cutaneous condition involving telangiectatic patches in a unilateral dermatomal or blaschkoid pattern.1 The condition has been described as either congenital or acquired. Congenital UNT is thought to be a result of somatic mosaicism, whereby a mutation during embryogenesis leads to a distinct population of cells expressing the vascular malformation.1 Congenital UNT has been associated with Becker nevus, which also is thought to be a result of somatic mosaicism, further providing evidence for this theory, though it is unclear whether this finding is incidental.2 The acquired form often is associated with fluctuation of hormones, such as in pregnancy or with oral contraceptive initiation, as well as with hepatic disease as seen in our patient. However, there are many cases of acquired UNT with no implicated underlying disease, alcohol abuse, or hormonal changes, which calls into question if UNT is definitively an estrogen-related condition.3 One study demonstrated an increased level of estrogen and progesterone receptors in affected skin, which may have led to expression of the cutaneous changes at that site.4 More research is needed to elucidate this point, as other studies have not reproduced similar findings.
Congenital UNT occurs more commonly in males, whereas the acquired variant is seen more frequently in females. The third and fourth cervical dermatomes most often are involved.5 Most lesions persist without spontaneous resolution. Treatment options are limited and include pulsed dye laser treatment and makeup application to cover the telangiectatic patches. The main side effect seen with pulsed dye laser treatment is reversible pigmentary changes, with 1 report of textural skin change.6
A biopsy was deemed unnecessary for the clinical diagnosis in our patient because there was a clear explanation for the physical examination findings due to long-standing underlying liver disease. When biopsied, UNT characteristically demonstrates dilated dermal capillaries.5 Our patient elected not to pursue laser therapy but expressed interest in using makeup to camouflage the lesion.
The differential diagnosis includes acquired nevus flammeus, which typically is present on the face and often appears following mechanical or thermal trauma. Angioma serpiginosum most often occurs on the buttocks and legs as small red papules or puncta coalescing into a serpiginous linear arrangement. It often appears in childhood. Angiosarcoma is an aggressive malignancy that often occurs on the head and neck in elderly patients. It is associated with areas of long-standing lymphedema and often appears as a bruiselike lesion. Rosacea typically is not fixed in its clinical appearance and presents as transitory flushing of the head and neck with or without a history of acneform eruptions on the face. It typically is not unilateral.
- Wilkin JK. Unilateral dermatomal superficial telangiectasia. Arch Dermatol. 1984;120:579-580.
- Karakaş M, Durdu M, Sönmezoğlu S, et al. Unilateral nevoid telangiectasia. J Dermatol. 2004;31:109-112.
- Taskapan O, Harmanyeri Y, Sener O, et al. Acquired unilateral nevoid telangiectasia syndrome. Acta Derm Venereol. 1997;77:62-63.
- Uhlin SR, McCarty KS Jr. Unilateral nevoid telangiectatic syndrome: the role of estrogen and progesterone receptors. Arch Dermatol. 1983;119:226-228.
- Derrow AE, Adams BB, Timani S, et al. Acquired unilateral nevoid telangiectasia in a 51-year-old female. Int J Dermatol. 2008;47:1331-1333.
- Sharma VK, Khandpur S. Unilateral nevoid telangiectasia--response to pulsed dye laser. Int J Dermatol. 2006;45:960-964.
- Wilkin JK. Unilateral dermatomal superficial telangiectasia. Arch Dermatol. 1984;120:579-580.
- Karakaş M, Durdu M, Sönmezoğlu S, et al. Unilateral nevoid telangiectasia. J Dermatol. 2004;31:109-112.
- Taskapan O, Harmanyeri Y, Sener O, et al. Acquired unilateral nevoid telangiectasia syndrome. Acta Derm Venereol. 1997;77:62-63.
- Uhlin SR, McCarty KS Jr. Unilateral nevoid telangiectatic syndrome: the role of estrogen and progesterone receptors. Arch Dermatol. 1983;119:226-228.
- Derrow AE, Adams BB, Timani S, et al. Acquired unilateral nevoid telangiectasia in a 51-year-old female. Int J Dermatol. 2008;47:1331-1333.
- Sharma VK, Khandpur S. Unilateral nevoid telangiectasia--response to pulsed dye laser. Int J Dermatol. 2006;45:960-964.
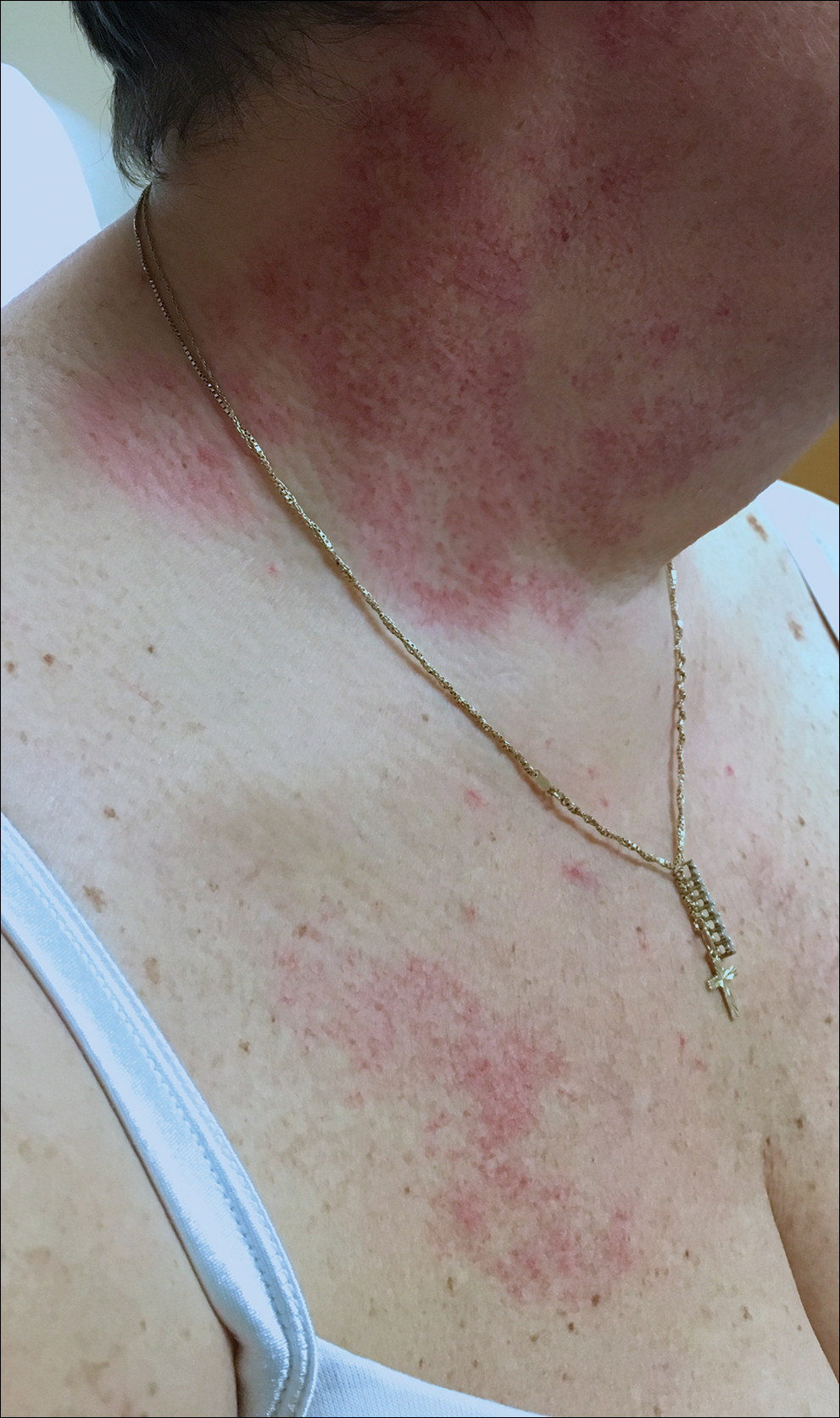
A 55-year-old woman presented to our clinic for a total-body skin examination and was noted to have a completely blanchable telangiectatic patch on the right side of the neck extending down onto the chest and breast. The patient reported that it had been present for 15 years and had slowly expanded in size. The lesion was asymptomatic. Pertinent medical history included cryptogenic cirrhosis of the liver, and she was undergoing a workup for a liver transplant.
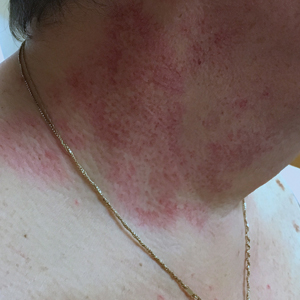
Irregular Yellow-Brown Plaques on the Trunk and Thighs
The Diagnosis: Necrobiotic Xanthogranuloma
A 4-mm punch biopsy was performed for routine stain with hematoxylin and eosin. The differential diagnosis included sarcoidosis, necrobiosis lipoidica, xanthoma disseminatum, and multicentric reticulohistiocytosis. Histopathologic examination demonstrated a dermal infiltrate of foamy histiocytes and neutrophils (Figure). There were surrounding areas of degenerated collagen containing numerous cholesterol clefts. After clinical pathologic correlation, a diagnosis of necrobiotic xanthogranuloma (NXG) was elucidated.
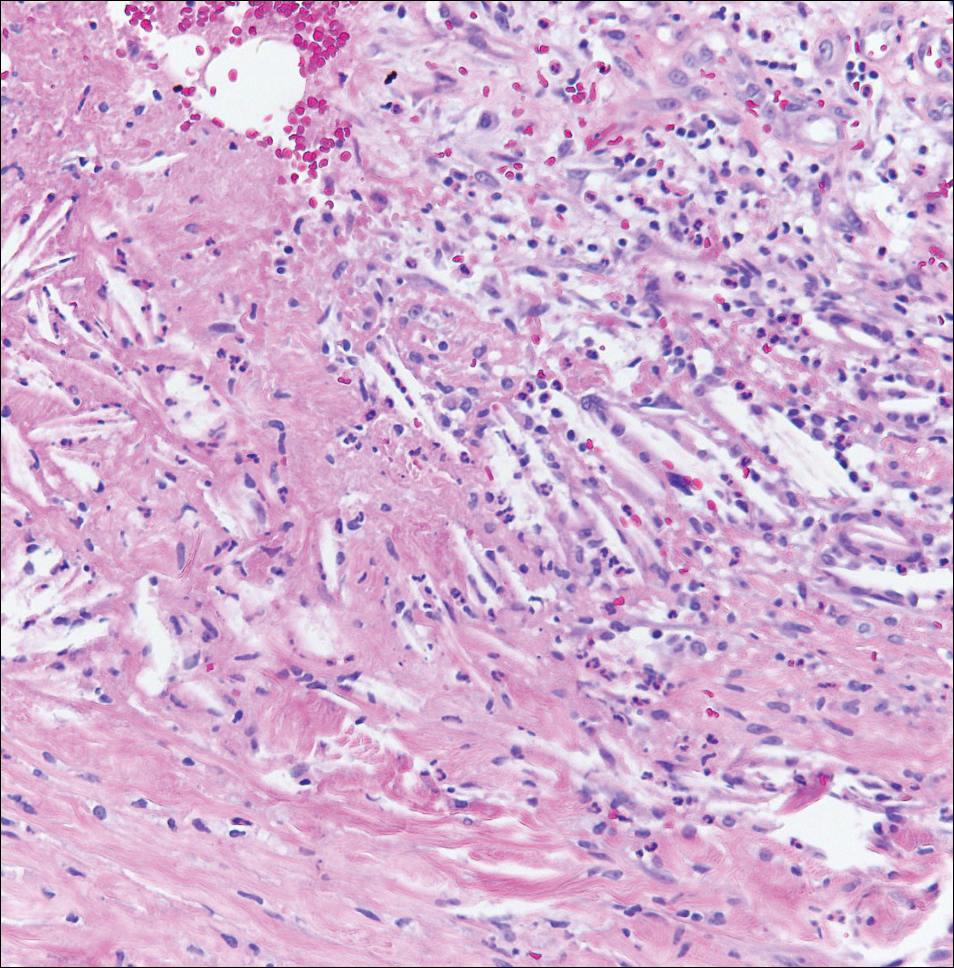
The patient was referred to general surgery for elective excision of 1 or more of the lesions. Excision of an abdominal lesion was performed without complication. After several months, a new lesion reformed within the excisional scar that also was consistent with NXG. At further dermatologic visits, a trial of intralesional corticosteroids was attempted to the largest lesions with modest improvement. In addition, follow-up with hematology and oncology was recommended for routine surveillance of the known blood dyscrasia.
Necrobiotic xanthogranuloma is a multisystem non-Langerhans cell histiocytic disease. Clinically, NXG is characterized by infiltrative plaques and ulcerative nodules. Lesions may appear red, brown, or yellow with associated atrophy and telangiectasia.1 Koch et al2 described a predilection for granuloma formation within preexisting scars. Periorbital location is the most common cutaneous site of involvement of NXG, seen in 80% of cases, but the trunk and extremities also may be involved.1,3 Approximately half of those with periocular involvement experience ocular symptoms including prop- tosis, blepharoptosis, and restricted eye movements.4 The onset of NXG most commonly is seen in middle age.
Characteristic systemic associations have been reported in the setting of NXG. More than 20% of patients may exhibit hepatomegaly. Hematologic abnormalities, hyperlipidemia, and cryoglobulinemia also may be seen.1 In addition, a monoclonal gammopathy of uncertain significance is found in more than 80% of NXG cases. The IgG κ light chain is most commonly identified.2 A foreign body reaction is incited by the immunoglobulin-lipid complex, which is thought to contribute to the formation of cutaneous lesions. There may be associated plasma cell dyscrasia such as multiple myeloma or B-cell lymphoma in approximately 13% of cases.2 Evaluation for underlying plasma cell dyscrasia or lymphoproliferative disorder should be performed regularly with serum protein electrophoresis or immunofixation electrophoresis, and in some cases full-body imaging with computed tomography or magnetic resonance imaging may be warranted.1
Treatment of NXG often is unsuccessful. Surgical excision, systemic immunosuppressive agents, electron beam radiation, and destructive therapies such as cryotherapy may be trialed, often with little success.1 Cutaneous regression has been reported with combination treatment of high-dose dexamethasone and high-dose lenalidomide.5
- Efebera Y, Blanchard E, Allam C, et al. Complete response to thalidomide and dexamethasone in a patient with necrobiotic xanthogranuloma associated with monoclonal gammopathy: a case report and review of the literature. Clin Lymphoma Myeloma Leuk. 2011;11:298-302.
- Koch PS, Goerdt S, Géraud C. Erythematous papules, plaques, and nodular lesions on the trunk and within preexisting scars. JAMA Dermatol. 2013;149:1103-1104.
- Kerstetter J, Wang J. Adult orbital xanthogranulomatous disease: a review with emphasis on etiology, systemic associations, diagnostic tools, and treatment. Dermatol Clin. 2015;33:457-463.
- Spicknall KE, Mehregan DA. Necrobiotic xanthogranuloma. Int J Dermatol. 2009;48:1-10.
- Dholaria BR, Cappel M, Roy V. Necrobiotic xanthogranuloma associated with monoclonal gammopathy: successful treatment with lenalidomide and dexamethasone [published online Jan 27, 2016]. Ann Hematol. 2016;95:671-672.
The Diagnosis: Necrobiotic Xanthogranuloma
A 4-mm punch biopsy was performed for routine stain with hematoxylin and eosin. The differential diagnosis included sarcoidosis, necrobiosis lipoidica, xanthoma disseminatum, and multicentric reticulohistiocytosis. Histopathologic examination demonstrated a dermal infiltrate of foamy histiocytes and neutrophils (Figure). There were surrounding areas of degenerated collagen containing numerous cholesterol clefts. After clinical pathologic correlation, a diagnosis of necrobiotic xanthogranuloma (NXG) was elucidated.
The patient was referred to general surgery for elective excision of 1 or more of the lesions. Excision of an abdominal lesion was performed without complication. After several months, a new lesion reformed within the excisional scar that also was consistent with NXG. At further dermatologic visits, a trial of intralesional corticosteroids was attempted to the largest lesions with modest improvement. In addition, follow-up with hematology and oncology was recommended for routine surveillance of the known blood dyscrasia.
Necrobiotic xanthogranuloma is a multisystem non-Langerhans cell histiocytic disease. Clinically, NXG is characterized by infiltrative plaques and ulcerative nodules. Lesions may appear red, brown, or yellow with associated atrophy and telangiectasia.1 Koch et al2 described a predilection for granuloma formation within preexisting scars. Periorbital location is the most common cutaneous site of involvement of NXG, seen in 80% of cases, but the trunk and extremities also may be involved.1,3 Approximately half of those with periocular involvement experience ocular symptoms including prop- tosis, blepharoptosis, and restricted eye movements.4 The onset of NXG most commonly is seen in middle age.
Characteristic systemic associations have been reported in the setting of NXG. More than 20% of patients may exhibit hepatomegaly. Hematologic abnormalities, hyperlipidemia, and cryoglobulinemia also may be seen.1 In addition, a monoclonal gammopathy of uncertain significance is found in more than 80% of NXG cases. The IgG κ light chain is most commonly identified.2 A foreign body reaction is incited by the immunoglobulin-lipid complex, which is thought to contribute to the formation of cutaneous lesions. There may be associated plasma cell dyscrasia such as multiple myeloma or B-cell lymphoma in approximately 13% of cases.2 Evaluation for underlying plasma cell dyscrasia or lymphoproliferative disorder should be performed regularly with serum protein electrophoresis or immunofixation electrophoresis, and in some cases full-body imaging with computed tomography or magnetic resonance imaging may be warranted.1
Treatment of NXG often is unsuccessful. Surgical excision, systemic immunosuppressive agents, electron beam radiation, and destructive therapies such as cryotherapy may be trialed, often with little success.1 Cutaneous regression has been reported with combination treatment of high-dose dexamethasone and high-dose lenalidomide.5
The Diagnosis: Necrobiotic Xanthogranuloma
A 4-mm punch biopsy was performed for routine stain with hematoxylin and eosin. The differential diagnosis included sarcoidosis, necrobiosis lipoidica, xanthoma disseminatum, and multicentric reticulohistiocytosis. Histopathologic examination demonstrated a dermal infiltrate of foamy histiocytes and neutrophils (Figure). There were surrounding areas of degenerated collagen containing numerous cholesterol clefts. After clinical pathologic correlation, a diagnosis of necrobiotic xanthogranuloma (NXG) was elucidated.
The patient was referred to general surgery for elective excision of 1 or more of the lesions. Excision of an abdominal lesion was performed without complication. After several months, a new lesion reformed within the excisional scar that also was consistent with NXG. At further dermatologic visits, a trial of intralesional corticosteroids was attempted to the largest lesions with modest improvement. In addition, follow-up with hematology and oncology was recommended for routine surveillance of the known blood dyscrasia.
Necrobiotic xanthogranuloma is a multisystem non-Langerhans cell histiocytic disease. Clinically, NXG is characterized by infiltrative plaques and ulcerative nodules. Lesions may appear red, brown, or yellow with associated atrophy and telangiectasia.1 Koch et al2 described a predilection for granuloma formation within preexisting scars. Periorbital location is the most common cutaneous site of involvement of NXG, seen in 80% of cases, but the trunk and extremities also may be involved.1,3 Approximately half of those with periocular involvement experience ocular symptoms including prop- tosis, blepharoptosis, and restricted eye movements.4 The onset of NXG most commonly is seen in middle age.
Characteristic systemic associations have been reported in the setting of NXG. More than 20% of patients may exhibit hepatomegaly. Hematologic abnormalities, hyperlipidemia, and cryoglobulinemia also may be seen.1 In addition, a monoclonal gammopathy of uncertain significance is found in more than 80% of NXG cases. The IgG κ light chain is most commonly identified.2 A foreign body reaction is incited by the immunoglobulin-lipid complex, which is thought to contribute to the formation of cutaneous lesions. There may be associated plasma cell dyscrasia such as multiple myeloma or B-cell lymphoma in approximately 13% of cases.2 Evaluation for underlying plasma cell dyscrasia or lymphoproliferative disorder should be performed regularly with serum protein electrophoresis or immunofixation electrophoresis, and in some cases full-body imaging with computed tomography or magnetic resonance imaging may be warranted.1
Treatment of NXG often is unsuccessful. Surgical excision, systemic immunosuppressive agents, electron beam radiation, and destructive therapies such as cryotherapy may be trialed, often with little success.1 Cutaneous regression has been reported with combination treatment of high-dose dexamethasone and high-dose lenalidomide.5
- Efebera Y, Blanchard E, Allam C, et al. Complete response to thalidomide and dexamethasone in a patient with necrobiotic xanthogranuloma associated with monoclonal gammopathy: a case report and review of the literature. Clin Lymphoma Myeloma Leuk. 2011;11:298-302.
- Koch PS, Goerdt S, Géraud C. Erythematous papules, plaques, and nodular lesions on the trunk and within preexisting scars. JAMA Dermatol. 2013;149:1103-1104.
- Kerstetter J, Wang J. Adult orbital xanthogranulomatous disease: a review with emphasis on etiology, systemic associations, diagnostic tools, and treatment. Dermatol Clin. 2015;33:457-463.
- Spicknall KE, Mehregan DA. Necrobiotic xanthogranuloma. Int J Dermatol. 2009;48:1-10.
- Dholaria BR, Cappel M, Roy V. Necrobiotic xanthogranuloma associated with monoclonal gammopathy: successful treatment with lenalidomide and dexamethasone [published online Jan 27, 2016]. Ann Hematol. 2016;95:671-672.
- Efebera Y, Blanchard E, Allam C, et al. Complete response to thalidomide and dexamethasone in a patient with necrobiotic xanthogranuloma associated with monoclonal gammopathy: a case report and review of the literature. Clin Lymphoma Myeloma Leuk. 2011;11:298-302.
- Koch PS, Goerdt S, Géraud C. Erythematous papules, plaques, and nodular lesions on the trunk and within preexisting scars. JAMA Dermatol. 2013;149:1103-1104.
- Kerstetter J, Wang J. Adult orbital xanthogranulomatous disease: a review with emphasis on etiology, systemic associations, diagnostic tools, and treatment. Dermatol Clin. 2015;33:457-463.
- Spicknall KE, Mehregan DA. Necrobiotic xanthogranuloma. Int J Dermatol. 2009;48:1-10.
- Dholaria BR, Cappel M, Roy V. Necrobiotic xanthogranuloma associated with monoclonal gammopathy: successful treatment with lenalidomide and dexamethasone [published online Jan 27, 2016]. Ann Hematol. 2016;95:671-672.
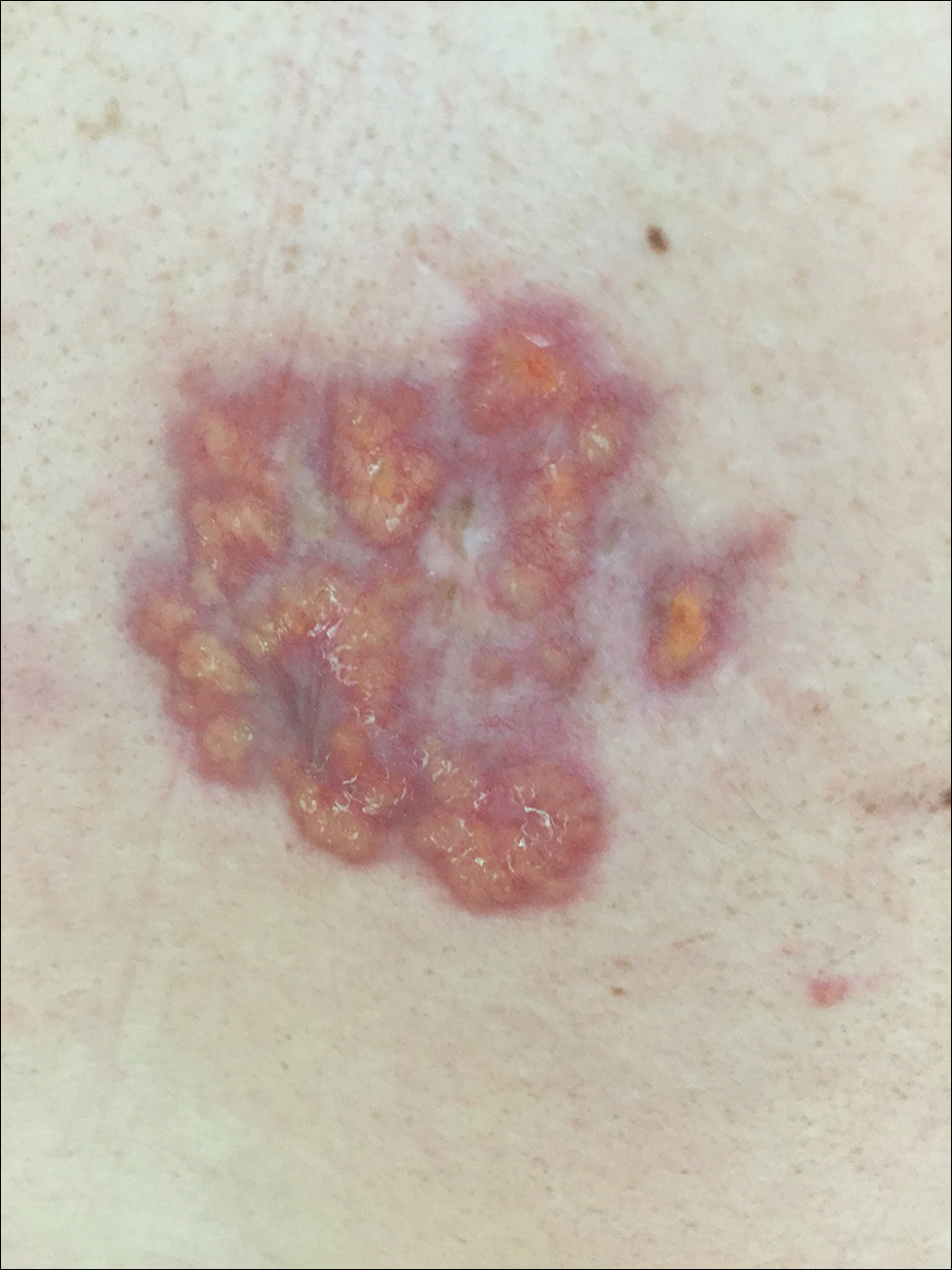
A 40-year-old man presented with tender lesions on the back, abdomen, and thighs of 10 years' duration. His medical history was remarkable for follicular lymphoma treated with chemotherapy and a monoclonal gammopathy of uncertain significance diagnosed 5 years after the onset of skin symptoms. Physical examination revealed numerous irregularly shaped, yellow plaques on the back, abdomen, and thighs with overlying telangiectasia. A single lesion was noted to extend from a scar.
Recalcitrant Solitary Erythematous Scaly Patch on the Foot
The Diagnosis: Pagetoid Reticulosis
Histopathologic examination demonstrated a dense infiltrate and psoriasiform pattern epidermal hyperplasia (Figure, A). There was conspicuous epidermotropism of moderately enlarged, hyperchromatic lymphocytes. Intraepidermal lymphocytes were slightly larger, darker, and more convoluted than those in the subjacent dermis (Figure, B). These cells exhibited CD3+ T-cell differentiation with an abnormal CD4-CD7-CD8- phenotype (Figure, C). The histopathologic finding of atypical epidermotropic T-cell infiltrate was compatible with a rare variant of mycosis fungoides known as pagetoid reticulosis (PR). After discussing the diagnosis and treatment options, the patient elected to begin with a conservative approach to therapy. We prescribed fluocinonide ointment 0.05% twice daily under occlusion. At 1 month follow-up, the patient experienced marked improvement of the erythema and scaling of the lesion.
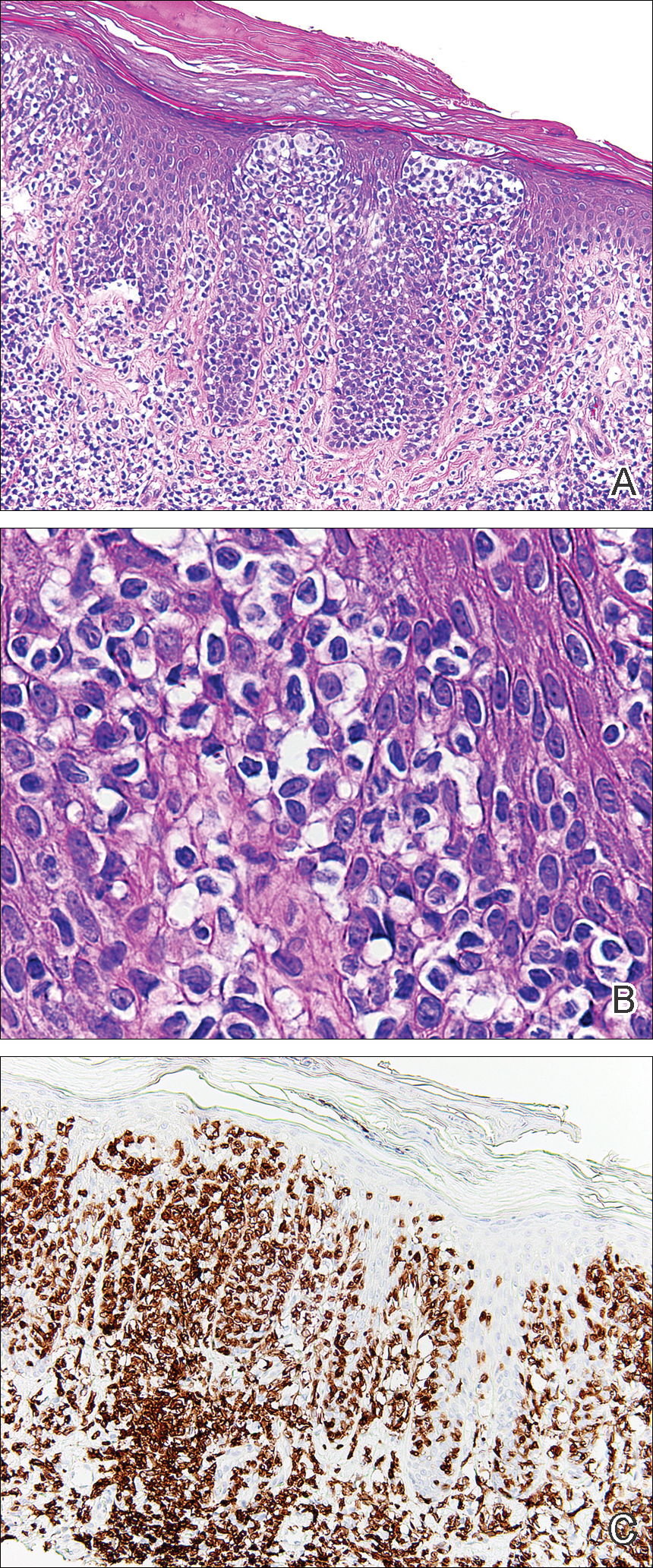
Pagetoid reticulosis is a primary cutaneous T-cell lymphoma that has been categorized as an indolent localized variant of mycosis fungoides. This rare skin disorder was originally described by Woringer and Kolopp in 19391 and was further renamed in 1973 by Braun-Falco et al.2 At that time the term pagetoid reticulosis was introduced due to similarities in histopathologic findings seen in Paget disease of the nipple. Two variants of the disease have been described since then: the localized type and the disseminated type. The localized type, also known as Woringer-Kolopp disease (WKD), typically presents as a persistent, sharply localized, scaly patch that slowly expands over several years. The lesion is classically located on the extensor surface of the hand or foot and often is asymptomatic. Due to the benign presentation, WKD can easily be confused with much more common diseases, such as psoriasis or fungal infections, resulting in a substantial delay in the diagnosis. The patient will often report a medical history notable for frequent office visits and numerous failed therapies. Even though it is exceedingly uncommon, these findings should prompt the practitioner to add WKD to their differential. The disseminated type of PR (also known as Ketron-Goodman disease) is characterized by diffuse cutaneous involvement, carries a much more progressive course, and often leads to a poor outcome.3 The histopathologic features of WKD and Ketron-Goodman disease are identical, and the 2 types are distinguished on clinical grounds alone.
Histopathologic features of PR are unique and often distinct in comparison to mycosis fungoides. Pagetoid reticulosis often is described as epidermal hyperplasia with parakeratosis, prominent acanthosis, and excessive epidermotropism of atypical lymphocytes scattered throughout the epidermis.3 The distinct pattern of epidermotropism seen in PR is the characteristic finding. Review of immunocytochemistry from reported cases has shown that CD marker expression of neoplastic T cells in PR can be variable in nature.4 Although it is known that immunophenotyping can be useful in diagnosing and distinguishing PR from other types of primary cutaneous T-cell lymphoma, the clinical significance of the observed phenotypic variation remains a mystery. As of now, it appears to be prognostically irrelevant.5
There are numerous therapeutic options available for PR. Depending on the size and extent of the disease, surgical excision and radiotherapy may be an option and are the most effective.6 For patients who are not good candidates or opt out of these options, there are various pharmacotherapies that also have proven to work. Traditional therapies include topical corticosteroids, corticosteroid injections, and phototherapy. However, more recent trials with retinoids, such as alitretinoin or bexarotene, appear to offer a promising therapeutic approach.7
Pagetoid reticulosis is a true malignant lymphoma of T-cell lineage, but it typically carries an excellent prognosis. Rare cases have been reported to progress to disseminated lymphoma.8 Therefore, long-term follow-up for a patient diagnosed with PR is recommended.
- Woringer FR, Kolopp P. Lésion érythémato-squameuse polycyclique de l'avant-bras évoluantdepuis 6 ans chez un garçonnet de 13 ans. Ann Dermatol Venereol. 1939;10:945-948.
- Braun-Falco O, Marghescu S, Wolff HH. Pagetoid reticulosis--Woringer-Kolopp's disease [in German]. Hautarzt. 1973;24:11-21.
- Haghighi B, Smoller BR, Leboit PE, et al. Pagetoid reticulosis (Woringer-Kolopp disease): an immunophenotypic, molecular, and clinicopathologic study. Mod Pathol. 2000;13:502-510.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Mourtzinos N, Puri PK, Wang G, et al. CD4/CD8 double negative pagetoid reticulosis: a case report and literature review. J Cutan Pathol. 2010;37:491-496.
- Lee J, Viakhireva N, Cesca C, et al. Clinicopathologic features and treatment outcomes in Woringer-Kolopp disease. J Am Acad Dermatol. 2008;59:706-712.
- Schmitz L, Bierhoff E, Dirschka T. Alitretinoin: an effective treatment option for pagetoid reticulosis. J Dtsch Dermatol Ges. 2013;11:1194-1195.
- Ioannides G, Engel MF, Rywlin AM. Woringer-Kolopp disease (pagetoid reticulosis). Am J Dermatopathol. 1983;5:153-158.
The Diagnosis: Pagetoid Reticulosis
Histopathologic examination demonstrated a dense infiltrate and psoriasiform pattern epidermal hyperplasia (Figure, A). There was conspicuous epidermotropism of moderately enlarged, hyperchromatic lymphocytes. Intraepidermal lymphocytes were slightly larger, darker, and more convoluted than those in the subjacent dermis (Figure, B). These cells exhibited CD3+ T-cell differentiation with an abnormal CD4-CD7-CD8- phenotype (Figure, C). The histopathologic finding of atypical epidermotropic T-cell infiltrate was compatible with a rare variant of mycosis fungoides known as pagetoid reticulosis (PR). After discussing the diagnosis and treatment options, the patient elected to begin with a conservative approach to therapy. We prescribed fluocinonide ointment 0.05% twice daily under occlusion. At 1 month follow-up, the patient experienced marked improvement of the erythema and scaling of the lesion.
Pagetoid reticulosis is a primary cutaneous T-cell lymphoma that has been categorized as an indolent localized variant of mycosis fungoides. This rare skin disorder was originally described by Woringer and Kolopp in 19391 and was further renamed in 1973 by Braun-Falco et al.2 At that time the term pagetoid reticulosis was introduced due to similarities in histopathologic findings seen in Paget disease of the nipple. Two variants of the disease have been described since then: the localized type and the disseminated type. The localized type, also known as Woringer-Kolopp disease (WKD), typically presents as a persistent, sharply localized, scaly patch that slowly expands over several years. The lesion is classically located on the extensor surface of the hand or foot and often is asymptomatic. Due to the benign presentation, WKD can easily be confused with much more common diseases, such as psoriasis or fungal infections, resulting in a substantial delay in the diagnosis. The patient will often report a medical history notable for frequent office visits and numerous failed therapies. Even though it is exceedingly uncommon, these findings should prompt the practitioner to add WKD to their differential. The disseminated type of PR (also known as Ketron-Goodman disease) is characterized by diffuse cutaneous involvement, carries a much more progressive course, and often leads to a poor outcome.3 The histopathologic features of WKD and Ketron-Goodman disease are identical, and the 2 types are distinguished on clinical grounds alone.
Histopathologic features of PR are unique and often distinct in comparison to mycosis fungoides. Pagetoid reticulosis often is described as epidermal hyperplasia with parakeratosis, prominent acanthosis, and excessive epidermotropism of atypical lymphocytes scattered throughout the epidermis.3 The distinct pattern of epidermotropism seen in PR is the characteristic finding. Review of immunocytochemistry from reported cases has shown that CD marker expression of neoplastic T cells in PR can be variable in nature.4 Although it is known that immunophenotyping can be useful in diagnosing and distinguishing PR from other types of primary cutaneous T-cell lymphoma, the clinical significance of the observed phenotypic variation remains a mystery. As of now, it appears to be prognostically irrelevant.5
There are numerous therapeutic options available for PR. Depending on the size and extent of the disease, surgical excision and radiotherapy may be an option and are the most effective.6 For patients who are not good candidates or opt out of these options, there are various pharmacotherapies that also have proven to work. Traditional therapies include topical corticosteroids, corticosteroid injections, and phototherapy. However, more recent trials with retinoids, such as alitretinoin or bexarotene, appear to offer a promising therapeutic approach.7
Pagetoid reticulosis is a true malignant lymphoma of T-cell lineage, but it typically carries an excellent prognosis. Rare cases have been reported to progress to disseminated lymphoma.8 Therefore, long-term follow-up for a patient diagnosed with PR is recommended.
The Diagnosis: Pagetoid Reticulosis
Histopathologic examination demonstrated a dense infiltrate and psoriasiform pattern epidermal hyperplasia (Figure, A). There was conspicuous epidermotropism of moderately enlarged, hyperchromatic lymphocytes. Intraepidermal lymphocytes were slightly larger, darker, and more convoluted than those in the subjacent dermis (Figure, B). These cells exhibited CD3+ T-cell differentiation with an abnormal CD4-CD7-CD8- phenotype (Figure, C). The histopathologic finding of atypical epidermotropic T-cell infiltrate was compatible with a rare variant of mycosis fungoides known as pagetoid reticulosis (PR). After discussing the diagnosis and treatment options, the patient elected to begin with a conservative approach to therapy. We prescribed fluocinonide ointment 0.05% twice daily under occlusion. At 1 month follow-up, the patient experienced marked improvement of the erythema and scaling of the lesion.
Pagetoid reticulosis is a primary cutaneous T-cell lymphoma that has been categorized as an indolent localized variant of mycosis fungoides. This rare skin disorder was originally described by Woringer and Kolopp in 19391 and was further renamed in 1973 by Braun-Falco et al.2 At that time the term pagetoid reticulosis was introduced due to similarities in histopathologic findings seen in Paget disease of the nipple. Two variants of the disease have been described since then: the localized type and the disseminated type. The localized type, also known as Woringer-Kolopp disease (WKD), typically presents as a persistent, sharply localized, scaly patch that slowly expands over several years. The lesion is classically located on the extensor surface of the hand or foot and often is asymptomatic. Due to the benign presentation, WKD can easily be confused with much more common diseases, such as psoriasis or fungal infections, resulting in a substantial delay in the diagnosis. The patient will often report a medical history notable for frequent office visits and numerous failed therapies. Even though it is exceedingly uncommon, these findings should prompt the practitioner to add WKD to their differential. The disseminated type of PR (also known as Ketron-Goodman disease) is characterized by diffuse cutaneous involvement, carries a much more progressive course, and often leads to a poor outcome.3 The histopathologic features of WKD and Ketron-Goodman disease are identical, and the 2 types are distinguished on clinical grounds alone.
Histopathologic features of PR are unique and often distinct in comparison to mycosis fungoides. Pagetoid reticulosis often is described as epidermal hyperplasia with parakeratosis, prominent acanthosis, and excessive epidermotropism of atypical lymphocytes scattered throughout the epidermis.3 The distinct pattern of epidermotropism seen in PR is the characteristic finding. Review of immunocytochemistry from reported cases has shown that CD marker expression of neoplastic T cells in PR can be variable in nature.4 Although it is known that immunophenotyping can be useful in diagnosing and distinguishing PR from other types of primary cutaneous T-cell lymphoma, the clinical significance of the observed phenotypic variation remains a mystery. As of now, it appears to be prognostically irrelevant.5
There are numerous therapeutic options available for PR. Depending on the size and extent of the disease, surgical excision and radiotherapy may be an option and are the most effective.6 For patients who are not good candidates or opt out of these options, there are various pharmacotherapies that also have proven to work. Traditional therapies include topical corticosteroids, corticosteroid injections, and phototherapy. However, more recent trials with retinoids, such as alitretinoin or bexarotene, appear to offer a promising therapeutic approach.7
Pagetoid reticulosis is a true malignant lymphoma of T-cell lineage, but it typically carries an excellent prognosis. Rare cases have been reported to progress to disseminated lymphoma.8 Therefore, long-term follow-up for a patient diagnosed with PR is recommended.
- Woringer FR, Kolopp P. Lésion érythémato-squameuse polycyclique de l'avant-bras évoluantdepuis 6 ans chez un garçonnet de 13 ans. Ann Dermatol Venereol. 1939;10:945-948.
- Braun-Falco O, Marghescu S, Wolff HH. Pagetoid reticulosis--Woringer-Kolopp's disease [in German]. Hautarzt. 1973;24:11-21.
- Haghighi B, Smoller BR, Leboit PE, et al. Pagetoid reticulosis (Woringer-Kolopp disease): an immunophenotypic, molecular, and clinicopathologic study. Mod Pathol. 2000;13:502-510.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Mourtzinos N, Puri PK, Wang G, et al. CD4/CD8 double negative pagetoid reticulosis: a case report and literature review. J Cutan Pathol. 2010;37:491-496.
- Lee J, Viakhireva N, Cesca C, et al. Clinicopathologic features and treatment outcomes in Woringer-Kolopp disease. J Am Acad Dermatol. 2008;59:706-712.
- Schmitz L, Bierhoff E, Dirschka T. Alitretinoin: an effective treatment option for pagetoid reticulosis. J Dtsch Dermatol Ges. 2013;11:1194-1195.
- Ioannides G, Engel MF, Rywlin AM. Woringer-Kolopp disease (pagetoid reticulosis). Am J Dermatopathol. 1983;5:153-158.
- Woringer FR, Kolopp P. Lésion érythémato-squameuse polycyclique de l'avant-bras évoluantdepuis 6 ans chez un garçonnet de 13 ans. Ann Dermatol Venereol. 1939;10:945-948.
- Braun-Falco O, Marghescu S, Wolff HH. Pagetoid reticulosis--Woringer-Kolopp's disease [in German]. Hautarzt. 1973;24:11-21.
- Haghighi B, Smoller BR, Leboit PE, et al. Pagetoid reticulosis (Woringer-Kolopp disease): an immunophenotypic, molecular, and clinicopathologic study. Mod Pathol. 2000;13:502-510.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Mourtzinos N, Puri PK, Wang G, et al. CD4/CD8 double negative pagetoid reticulosis: a case report and literature review. J Cutan Pathol. 2010;37:491-496.
- Lee J, Viakhireva N, Cesca C, et al. Clinicopathologic features and treatment outcomes in Woringer-Kolopp disease. J Am Acad Dermatol. 2008;59:706-712.
- Schmitz L, Bierhoff E, Dirschka T. Alitretinoin: an effective treatment option for pagetoid reticulosis. J Dtsch Dermatol Ges. 2013;11:1194-1195.
- Ioannides G, Engel MF, Rywlin AM. Woringer-Kolopp disease (pagetoid reticulosis). Am J Dermatopathol. 1983;5:153-158.
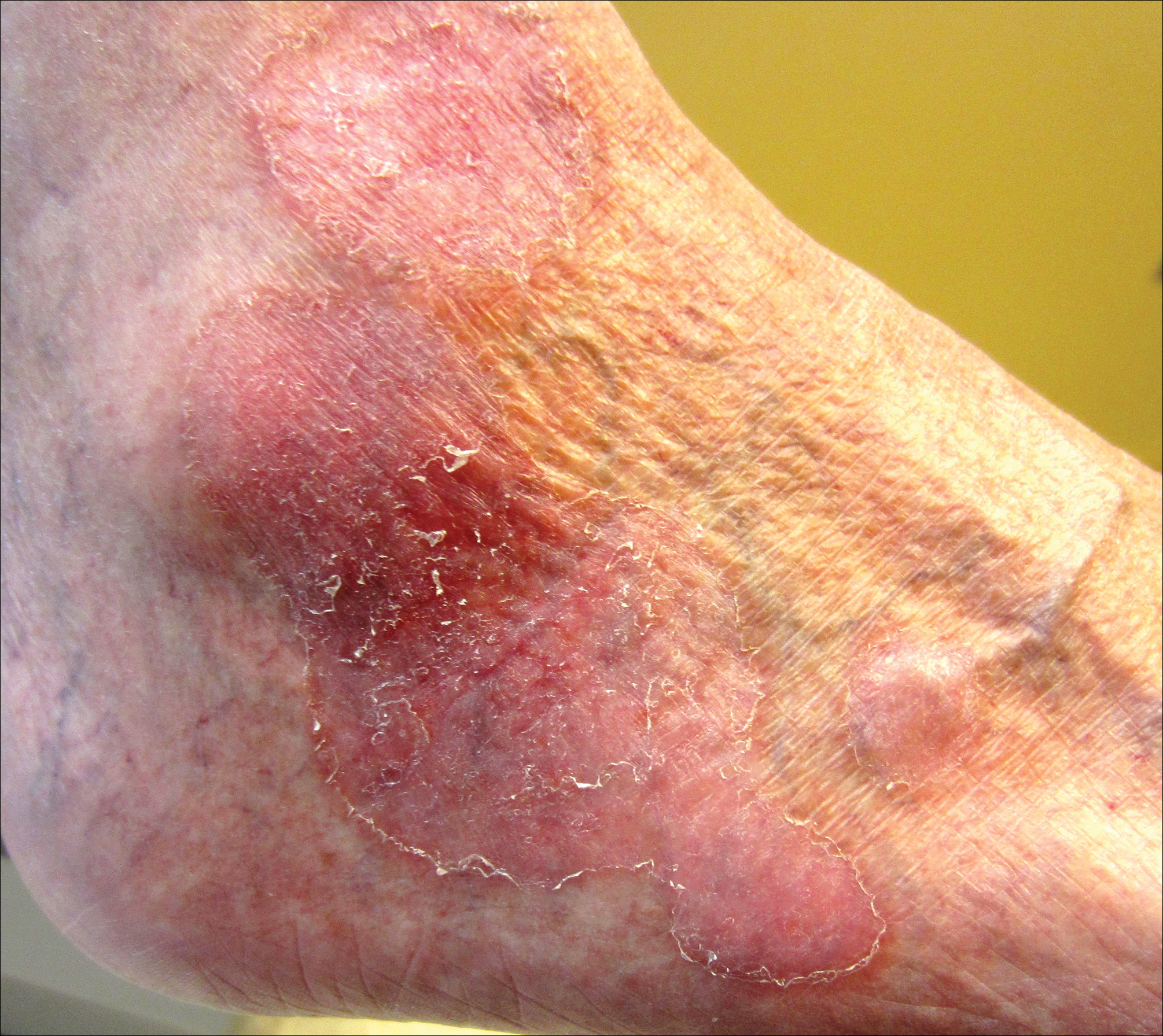
An 80-year-old man with a history of malignant melanoma and squamous cell carcinoma presented to the dermatology clinic with a chronic rash of 20 years' duration on the right ankle that extended to the instep of the right foot. His medical history was notable for hypertension and hyperlipidemia. Family history was unremarkable. The patient described the rash as red and scaly but denied associated pain or pruritus. Over the last 2 to 3 years he had tried treating the affected area with petroleum jelly, topical and oral antifungals, and mild topical steroids with minimal improvement. Complete review of systems was performed and was negative other than some mild constipation. Physical examination revealed an erythematous scaly patch on the dorsal aspect of the right ankle. Potassium hydroxide preparation and fungal culture swab yielded negative results, and a shave biopsy was performed.