User login
Acrokeratoelastoidosis and Knuckle Pads Coexisting in a Child
Case Report
An 11-year-old boy presented with atraumatic thickening of the skin on the bilateral distal and proximal interphalangeal joints of 1 year’s duration. The patient also noted small bumps of unknown duration across the bilateral palms and soles with prominence on the lateral aspects. The patient previously used over-the-counter topical wart removal treatment and topical salicylic acid with minimal improvement. The patient reported no pertinent medical or surgical history, although there was a family history of Alport syndrome, predominantly in male relatives. The patient’s father and paternal grandfather were noted to have similar lesions on the palms.
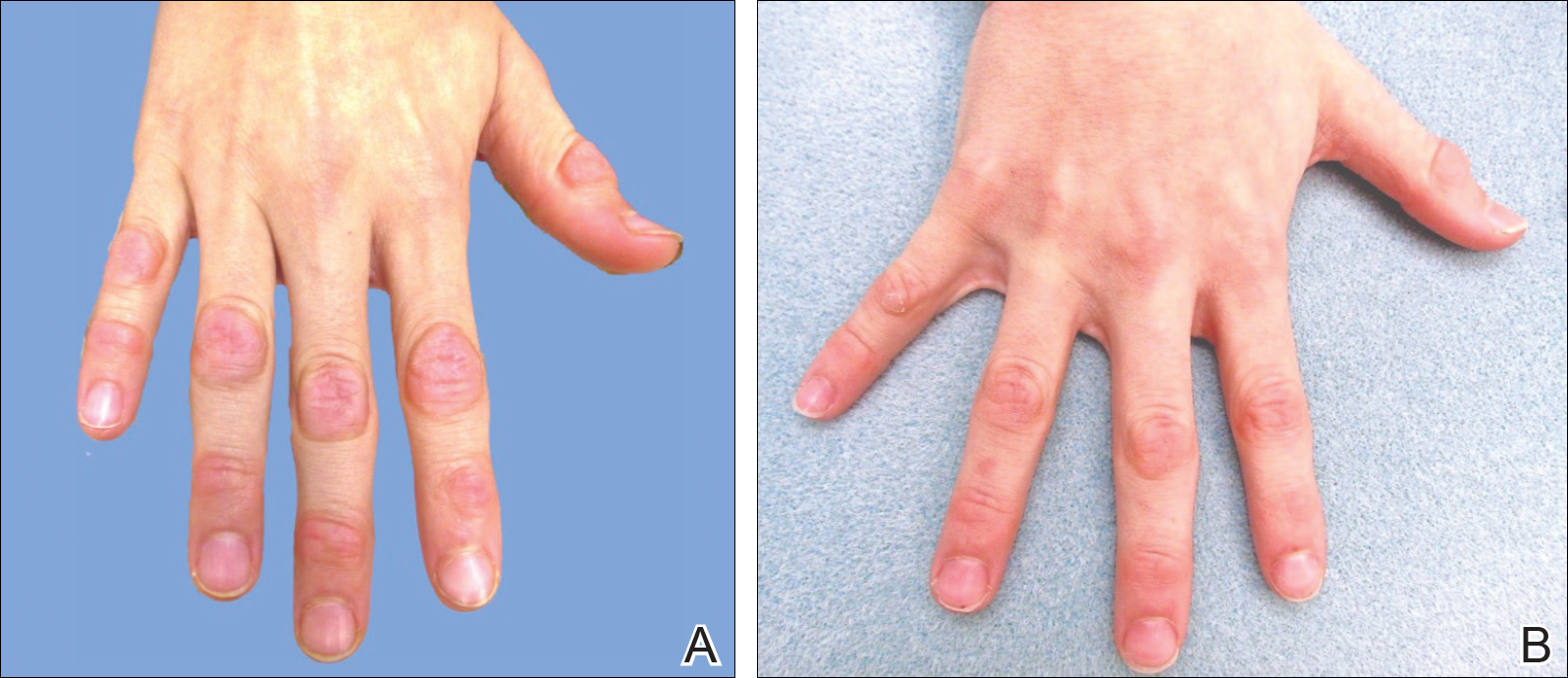
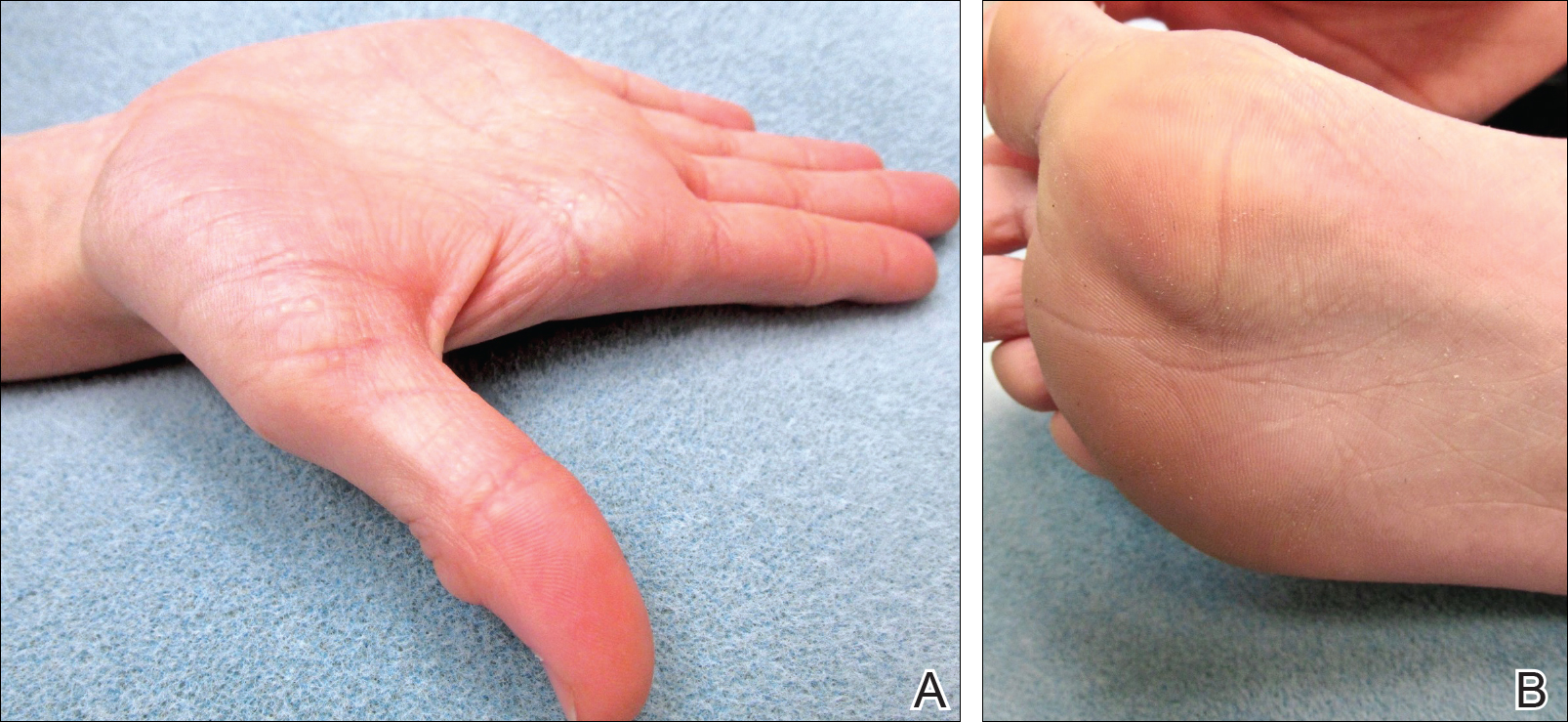
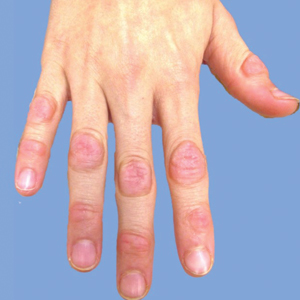
On physical examination, multiple pink to flesh-colored hyperkeratotic plaques were noted over the proximal and distal interphalangeal joints of the bilateral hands (Figure 1A). Upon close inspection, there were small flesh-colored and slightly translucent papules in a linear distribution on the palmar surfaces of the hands (Figure 2A) with predominance on the thenar and hypothenar eminences. The flexural creases of the bilateral wrists also revealed linear flesh-colored papules. The same small flesh-colored and translucent papules also were noted on the plantar surfaces of the bilateral feet (Figure 2B).
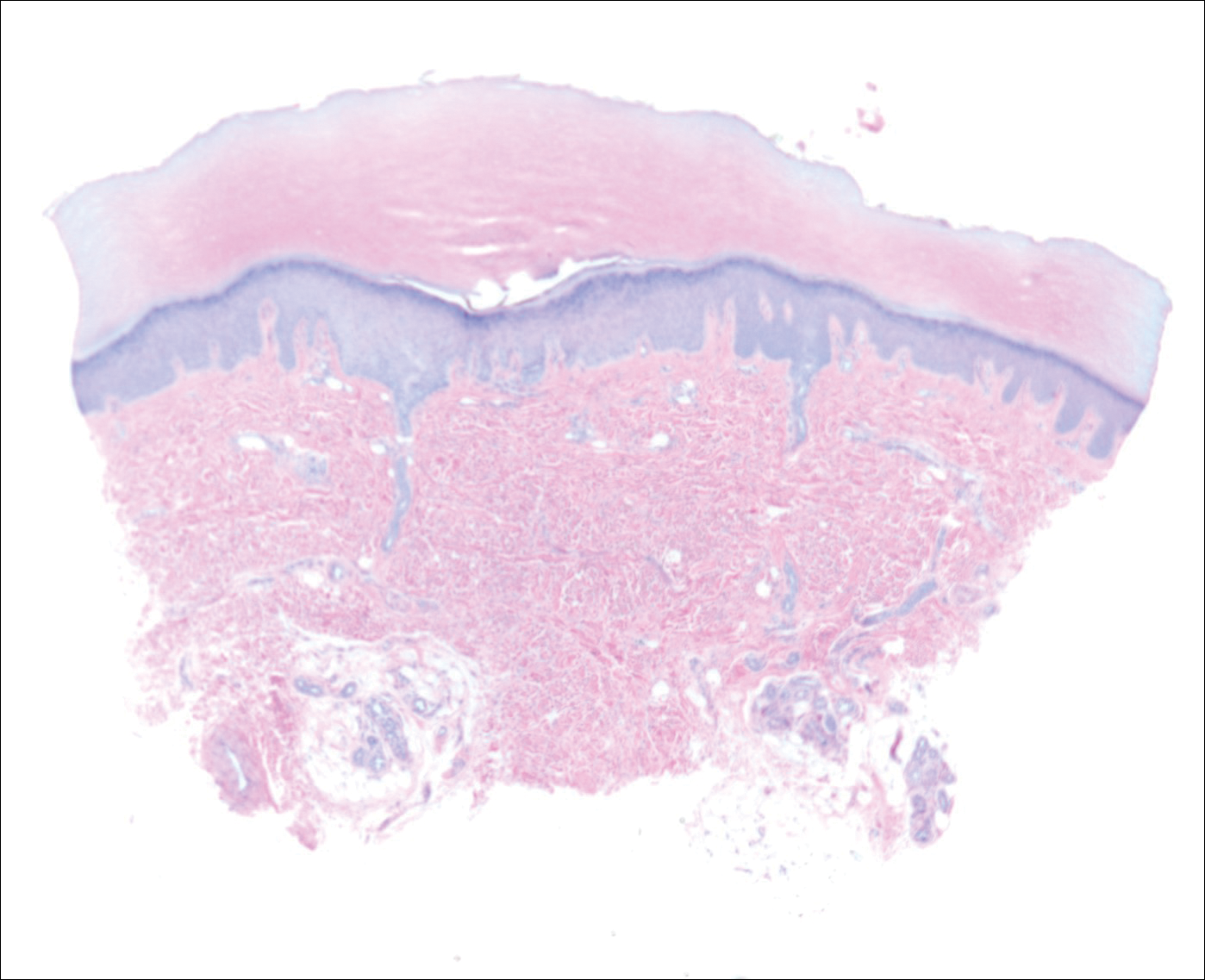
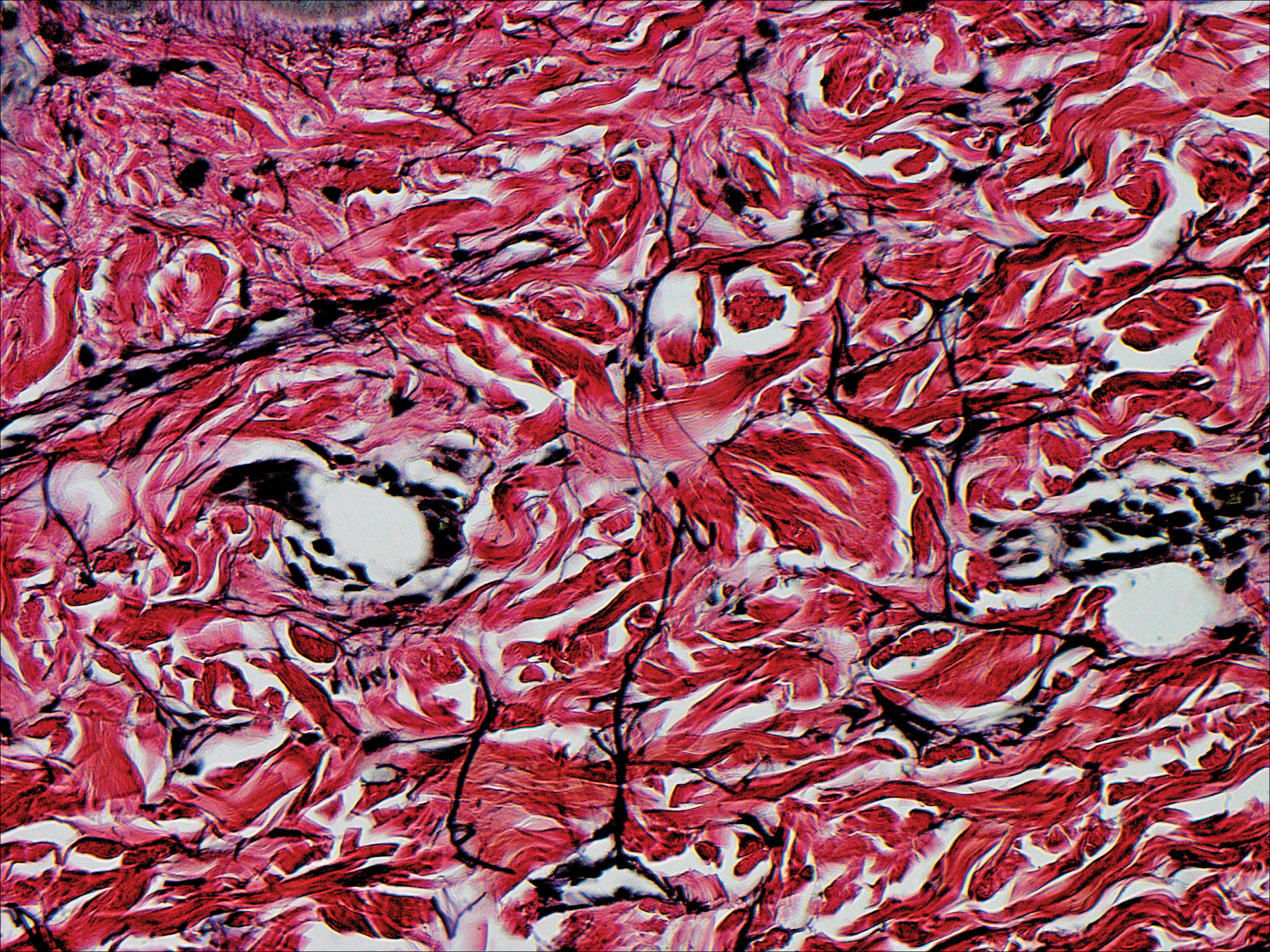
A biopsy was obtained from one of the small translucent papules on the left palm. Hematoxylin and eosin–stained sections revealed elevated compact orthokeratosis with an underlying central epidermal dell (Figure 3). A diagnosis of marginal papular keratoderma was made and further elastin staining was completed. Elastin stains showed marked thinning of the elastin fibers throughout the reticular dermis. Many elastin fibers in the reticular dermis demonstrated a fine arborizing pattern that normally is only evident in the papillary dermis (Figure 4). Acrokeratoelastoidosis (AKE) was diagnosed histopathologically, and knuckle pads were diagnosed clinically.
Because the patient was asymptomatic, he did not want treatment of AKE. He had marked improvement of the knuckle pads after 1 month with daily application of urea cream 10% (Figure 1B), and intermittent use was required for maintenance.
Comment
Etiology
Acrokeratoelastoidosis was first described in 1953 and is considered a type of palmoplantar marginal papular keratoderma.1 There is overlap within the marginal papular keratodermas that makes precise diagnosis difficult within this group. The marginal papular keratodermas on the palms and soles are a group of disorders that include AKE, focal acral hyperkeratosis (FAH), mosaic acral keratosis, degenerative collagenous plaques on the hands, and digital papular calcific elastosis. These diseases are similar in clinical and histopathological features; some argue these diseases are the same entity.2
Acrokeratoelastoidosis has been hypothesized to originate from altered elastic fiber synthesis from fibroblasts.3 Because AKE is rare, most cases of common knuckle pads do not coexist with AKE; therefore, it is unknown if the underlying etiology remains the same for both entities. Unlike AKE, knuckle pads are often associated with Dupuytren contractures, repetitive trauma, or friction to the area.1,2
Presentation
Acrokeratoelastoidosis is a rare disease with onset in childhood or young adulthood. Childhood cases are inherited in an autosomal-dominant fashion.1 Adulthood onset suggests a sporadic form of inheritance. Acrokeratoelastoidosis has no gender or racial predilection.4 It presents over the thenar and hypothenar eminences, as well as the lateral digits, calcaneal tendon, and dorsal digits.1 Most often, AKE occurs symmetrically along the border separating the ventral and dorsal aspects on the palms and soles. These lesions present as small, firm, translucent papules that align linearly on the ventral-dorsal palmoplantar junction in a pattern resembling paving stones.1 Coalescence of papules into plaques has been reported. Extension of lesions to the dorsal and palmar surfaces can occur. Small circumscribed callosities may develop over the metacarpophalangeal and interphalangeal joints resembling knuckle pads.2
Histopathology
Histopathologically, AKE is distinguished by elastorrhexis—thinning, fragmenting, and rarefaction of elastin fibers—in the epidermis and reticular dermis layers.3 Acrokeratoelastoidosis also presents with orthokeratosis overlying a cuplike epithelial depression and possible epithelial acanthosis.2,5 Many cases exhibit hypergranulosis at the base of the epidermal dell. Dense basophilic granules may be seen in the peripheral cytoplasm of fibroblast cells coming from the hypothesized defect in elastin secretion.1,3,4
Differential Diagnosis
The main differential diagnosis of AKE is FAH. Clinically and histopathologically they appear identical; both diseases have cuplike epidermal depressions with overlying orthohyperkeratosis and prominent hypergranulosis.5 The elastin stains, Verhoeff-van Gieson or acid orcein stain, are imperative for distinguishing these two diseases. Although AKE demonstrates elastorrhexis and reduced elastic fibers, FAH reveals no alteration of elastic fibers. It has been suggested that FAH is a clinical variant of AKE and should be titled AKE without elastorrhexis.1
Treatment
Acrokeratoelastoidosis is asymptomatic except for mild palmoplantar hyperhidrosis and typically does not require treatment4; however, the condition can be of cosmetic concern for patients. Lesions can be treated topically with keratolytics such as tretinoin and salicylic acid. A wide variety of systemic treatments including methotrexate, prednisolone, dapsone, and acitretin have been reported with variable clinical response.2-4 Copresenting knuckle pads can be treated with urea cream, salicylic acid cream, or intralesional corticosteroids.1
- Erkek E, Koçak M, Bozdog˘an O, et al. Focal acral hyperkeratosis: a rare cutaneous disorder within the spectrum are Costa acrokeratoelastoidosis. Pediatr Dermatol. 2004;21:128-130.
- Abulafia J, Vignale R. Degenerative collagenous plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
- Nelson-Adesokan P, Mallory SB, Leonardi CL, et al. Acrokeratoelastoidosis of Costa. Int J Dermatol. 1995;34:431-433.
- Shbaklo Z, Jamaleddine NF, Kibbi AG, et al. Acrokeratoelastoidosis. Int J Dermatol. 1990;29:333-336.
- Ming M. Papules overlying finger joints—diagnosis. Arch Dermatol. 2006;142:235-240.
Case Report
An 11-year-old boy presented with atraumatic thickening of the skin on the bilateral distal and proximal interphalangeal joints of 1 year’s duration. The patient also noted small bumps of unknown duration across the bilateral palms and soles with prominence on the lateral aspects. The patient previously used over-the-counter topical wart removal treatment and topical salicylic acid with minimal improvement. The patient reported no pertinent medical or surgical history, although there was a family history of Alport syndrome, predominantly in male relatives. The patient’s father and paternal grandfather were noted to have similar lesions on the palms.
On physical examination, multiple pink to flesh-colored hyperkeratotic plaques were noted over the proximal and distal interphalangeal joints of the bilateral hands (Figure 1A). Upon close inspection, there were small flesh-colored and slightly translucent papules in a linear distribution on the palmar surfaces of the hands (Figure 2A) with predominance on the thenar and hypothenar eminences. The flexural creases of the bilateral wrists also revealed linear flesh-colored papules. The same small flesh-colored and translucent papules also were noted on the plantar surfaces of the bilateral feet (Figure 2B).
A biopsy was obtained from one of the small translucent papules on the left palm. Hematoxylin and eosin–stained sections revealed elevated compact orthokeratosis with an underlying central epidermal dell (Figure 3). A diagnosis of marginal papular keratoderma was made and further elastin staining was completed. Elastin stains showed marked thinning of the elastin fibers throughout the reticular dermis. Many elastin fibers in the reticular dermis demonstrated a fine arborizing pattern that normally is only evident in the papillary dermis (Figure 4). Acrokeratoelastoidosis (AKE) was diagnosed histopathologically, and knuckle pads were diagnosed clinically.
Because the patient was asymptomatic, he did not want treatment of AKE. He had marked improvement of the knuckle pads after 1 month with daily application of urea cream 10% (Figure 1B), and intermittent use was required for maintenance.
Comment
Etiology
Acrokeratoelastoidosis was first described in 1953 and is considered a type of palmoplantar marginal papular keratoderma.1 There is overlap within the marginal papular keratodermas that makes precise diagnosis difficult within this group. The marginal papular keratodermas on the palms and soles are a group of disorders that include AKE, focal acral hyperkeratosis (FAH), mosaic acral keratosis, degenerative collagenous plaques on the hands, and digital papular calcific elastosis. These diseases are similar in clinical and histopathological features; some argue these diseases are the same entity.2
Acrokeratoelastoidosis has been hypothesized to originate from altered elastic fiber synthesis from fibroblasts.3 Because AKE is rare, most cases of common knuckle pads do not coexist with AKE; therefore, it is unknown if the underlying etiology remains the same for both entities. Unlike AKE, knuckle pads are often associated with Dupuytren contractures, repetitive trauma, or friction to the area.1,2
Presentation
Acrokeratoelastoidosis is a rare disease with onset in childhood or young adulthood. Childhood cases are inherited in an autosomal-dominant fashion.1 Adulthood onset suggests a sporadic form of inheritance. Acrokeratoelastoidosis has no gender or racial predilection.4 It presents over the thenar and hypothenar eminences, as well as the lateral digits, calcaneal tendon, and dorsal digits.1 Most often, AKE occurs symmetrically along the border separating the ventral and dorsal aspects on the palms and soles. These lesions present as small, firm, translucent papules that align linearly on the ventral-dorsal palmoplantar junction in a pattern resembling paving stones.1 Coalescence of papules into plaques has been reported. Extension of lesions to the dorsal and palmar surfaces can occur. Small circumscribed callosities may develop over the metacarpophalangeal and interphalangeal joints resembling knuckle pads.2
Histopathology
Histopathologically, AKE is distinguished by elastorrhexis—thinning, fragmenting, and rarefaction of elastin fibers—in the epidermis and reticular dermis layers.3 Acrokeratoelastoidosis also presents with orthokeratosis overlying a cuplike epithelial depression and possible epithelial acanthosis.2,5 Many cases exhibit hypergranulosis at the base of the epidermal dell. Dense basophilic granules may be seen in the peripheral cytoplasm of fibroblast cells coming from the hypothesized defect in elastin secretion.1,3,4
Differential Diagnosis
The main differential diagnosis of AKE is FAH. Clinically and histopathologically they appear identical; both diseases have cuplike epidermal depressions with overlying orthohyperkeratosis and prominent hypergranulosis.5 The elastin stains, Verhoeff-van Gieson or acid orcein stain, are imperative for distinguishing these two diseases. Although AKE demonstrates elastorrhexis and reduced elastic fibers, FAH reveals no alteration of elastic fibers. It has been suggested that FAH is a clinical variant of AKE and should be titled AKE without elastorrhexis.1
Treatment
Acrokeratoelastoidosis is asymptomatic except for mild palmoplantar hyperhidrosis and typically does not require treatment4; however, the condition can be of cosmetic concern for patients. Lesions can be treated topically with keratolytics such as tretinoin and salicylic acid. A wide variety of systemic treatments including methotrexate, prednisolone, dapsone, and acitretin have been reported with variable clinical response.2-4 Copresenting knuckle pads can be treated with urea cream, salicylic acid cream, or intralesional corticosteroids.1
Case Report
An 11-year-old boy presented with atraumatic thickening of the skin on the bilateral distal and proximal interphalangeal joints of 1 year’s duration. The patient also noted small bumps of unknown duration across the bilateral palms and soles with prominence on the lateral aspects. The patient previously used over-the-counter topical wart removal treatment and topical salicylic acid with minimal improvement. The patient reported no pertinent medical or surgical history, although there was a family history of Alport syndrome, predominantly in male relatives. The patient’s father and paternal grandfather were noted to have similar lesions on the palms.
On physical examination, multiple pink to flesh-colored hyperkeratotic plaques were noted over the proximal and distal interphalangeal joints of the bilateral hands (Figure 1A). Upon close inspection, there were small flesh-colored and slightly translucent papules in a linear distribution on the palmar surfaces of the hands (Figure 2A) with predominance on the thenar and hypothenar eminences. The flexural creases of the bilateral wrists also revealed linear flesh-colored papules. The same small flesh-colored and translucent papules also were noted on the plantar surfaces of the bilateral feet (Figure 2B).
A biopsy was obtained from one of the small translucent papules on the left palm. Hematoxylin and eosin–stained sections revealed elevated compact orthokeratosis with an underlying central epidermal dell (Figure 3). A diagnosis of marginal papular keratoderma was made and further elastin staining was completed. Elastin stains showed marked thinning of the elastin fibers throughout the reticular dermis. Many elastin fibers in the reticular dermis demonstrated a fine arborizing pattern that normally is only evident in the papillary dermis (Figure 4). Acrokeratoelastoidosis (AKE) was diagnosed histopathologically, and knuckle pads were diagnosed clinically.
Because the patient was asymptomatic, he did not want treatment of AKE. He had marked improvement of the knuckle pads after 1 month with daily application of urea cream 10% (Figure 1B), and intermittent use was required for maintenance.
Comment
Etiology
Acrokeratoelastoidosis was first described in 1953 and is considered a type of palmoplantar marginal papular keratoderma.1 There is overlap within the marginal papular keratodermas that makes precise diagnosis difficult within this group. The marginal papular keratodermas on the palms and soles are a group of disorders that include AKE, focal acral hyperkeratosis (FAH), mosaic acral keratosis, degenerative collagenous plaques on the hands, and digital papular calcific elastosis. These diseases are similar in clinical and histopathological features; some argue these diseases are the same entity.2
Acrokeratoelastoidosis has been hypothesized to originate from altered elastic fiber synthesis from fibroblasts.3 Because AKE is rare, most cases of common knuckle pads do not coexist with AKE; therefore, it is unknown if the underlying etiology remains the same for both entities. Unlike AKE, knuckle pads are often associated with Dupuytren contractures, repetitive trauma, or friction to the area.1,2
Presentation
Acrokeratoelastoidosis is a rare disease with onset in childhood or young adulthood. Childhood cases are inherited in an autosomal-dominant fashion.1 Adulthood onset suggests a sporadic form of inheritance. Acrokeratoelastoidosis has no gender or racial predilection.4 It presents over the thenar and hypothenar eminences, as well as the lateral digits, calcaneal tendon, and dorsal digits.1 Most often, AKE occurs symmetrically along the border separating the ventral and dorsal aspects on the palms and soles. These lesions present as small, firm, translucent papules that align linearly on the ventral-dorsal palmoplantar junction in a pattern resembling paving stones.1 Coalescence of papules into plaques has been reported. Extension of lesions to the dorsal and palmar surfaces can occur. Small circumscribed callosities may develop over the metacarpophalangeal and interphalangeal joints resembling knuckle pads.2
Histopathology
Histopathologically, AKE is distinguished by elastorrhexis—thinning, fragmenting, and rarefaction of elastin fibers—in the epidermis and reticular dermis layers.3 Acrokeratoelastoidosis also presents with orthokeratosis overlying a cuplike epithelial depression and possible epithelial acanthosis.2,5 Many cases exhibit hypergranulosis at the base of the epidermal dell. Dense basophilic granules may be seen in the peripheral cytoplasm of fibroblast cells coming from the hypothesized defect in elastin secretion.1,3,4
Differential Diagnosis
The main differential diagnosis of AKE is FAH. Clinically and histopathologically they appear identical; both diseases have cuplike epidermal depressions with overlying orthohyperkeratosis and prominent hypergranulosis.5 The elastin stains, Verhoeff-van Gieson or acid orcein stain, are imperative for distinguishing these two diseases. Although AKE demonstrates elastorrhexis and reduced elastic fibers, FAH reveals no alteration of elastic fibers. It has been suggested that FAH is a clinical variant of AKE and should be titled AKE without elastorrhexis.1
Treatment
Acrokeratoelastoidosis is asymptomatic except for mild palmoplantar hyperhidrosis and typically does not require treatment4; however, the condition can be of cosmetic concern for patients. Lesions can be treated topically with keratolytics such as tretinoin and salicylic acid. A wide variety of systemic treatments including methotrexate, prednisolone, dapsone, and acitretin have been reported with variable clinical response.2-4 Copresenting knuckle pads can be treated with urea cream, salicylic acid cream, or intralesional corticosteroids.1
- Erkek E, Koçak M, Bozdog˘an O, et al. Focal acral hyperkeratosis: a rare cutaneous disorder within the spectrum are Costa acrokeratoelastoidosis. Pediatr Dermatol. 2004;21:128-130.
- Abulafia J, Vignale R. Degenerative collagenous plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
- Nelson-Adesokan P, Mallory SB, Leonardi CL, et al. Acrokeratoelastoidosis of Costa. Int J Dermatol. 1995;34:431-433.
- Shbaklo Z, Jamaleddine NF, Kibbi AG, et al. Acrokeratoelastoidosis. Int J Dermatol. 1990;29:333-336.
- Ming M. Papules overlying finger joints—diagnosis. Arch Dermatol. 2006;142:235-240.
- Erkek E, Koçak M, Bozdog˘an O, et al. Focal acral hyperkeratosis: a rare cutaneous disorder within the spectrum are Costa acrokeratoelastoidosis. Pediatr Dermatol. 2004;21:128-130.
- Abulafia J, Vignale R. Degenerative collagenous plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
- Nelson-Adesokan P, Mallory SB, Leonardi CL, et al. Acrokeratoelastoidosis of Costa. Int J Dermatol. 1995;34:431-433.
- Shbaklo Z, Jamaleddine NF, Kibbi AG, et al. Acrokeratoelastoidosis. Int J Dermatol. 1990;29:333-336.
- Ming M. Papules overlying finger joints—diagnosis. Arch Dermatol. 2006;142:235-240.
Practice Points
- Acrokeratoelastoidosis presents as small, firm, translucent, linear papules on the ventral-dorsal palmoplantar junction.
- Acrokeratoelastoidosis does not require treatment but can be treated topically with keratolytics such as tretinoin and salicylic acid.
- Knuckle pads may respond to urea cream, salicylic acid cream, or intralesional corticosteroids.
Muckle-Wells Syndrome in the Setting of Basal Cell Nevus Syndrome
Muckle-Wells syndrome (MWS) was first described in 1962 and is part of a broad category of hereditary periodic fever syndromes that include the autoinflammatory syndromes and the cryopyrin-associated periodic syndromes (CAPSs). Unlike autoimmune diseases, autoinflammatory syndromes are not associated with antigen-specific T-cell responses or high titers of autoantibodies but are related to disorders of the innate immune system. Basal cell nevus syndrome (BCNS), or Gorlin syndrome, is a rare genodermatosis inherited in an autosomal-dominant fashion that is characterized by a broad range of anomalies. Most notable is the early and strong predisposition to develop several to hundreds of basal cell carcinomas (BCCs). Classic clinical features of MWS and a thorough history and physical examination can assist in the diagnosis of this rare entity.
Case Report
A 35-year-old woman with a history of BCNS, which had been diagnosed at 24 years of age based on the presence of more than 2 BCCs and a family history of BCNS in her mother, presented with intermittent pruritic urticaria on the chest and back, episodic fevers, associated joint pain and swelling that worsened several hours after exercise, headache, conjunctivitis, blurred vision, and severe debilitating fatigue that had been present since childhood. The symptoms had progressively worsened with age and symptom-free intervals became shorter. She was diagnosed by her rheumatologist with biopsy-proven MWS and a positive NLRP3 (NLR family pyrin domain containing 3) gene mutation at 29 years of age. She was treated unsuccessfully with prednisone and antihistamines and entered a trial with anakinra. She showed improvement for 2 weeks but developed severe swelling and erythema at the injection sites at week 3, along with large leathery patches on the legs and difficulty ambulating.
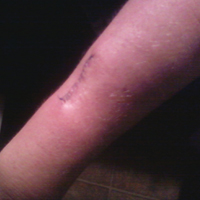
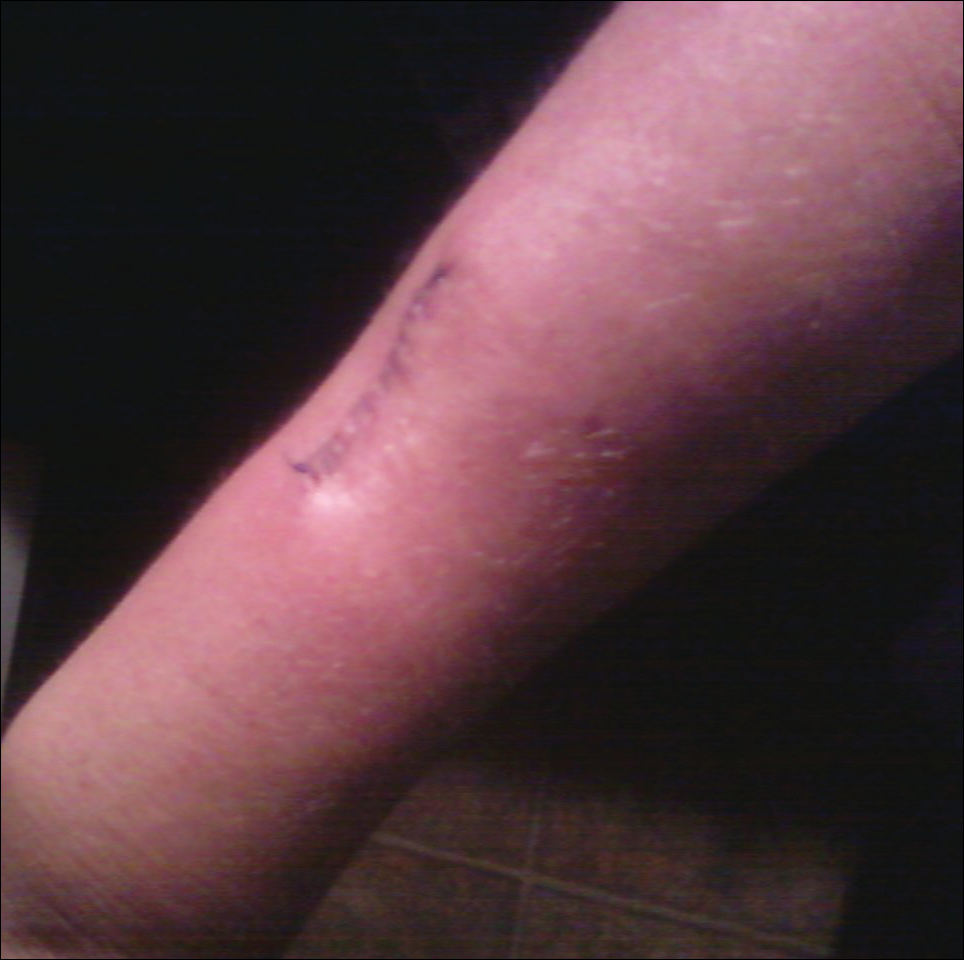
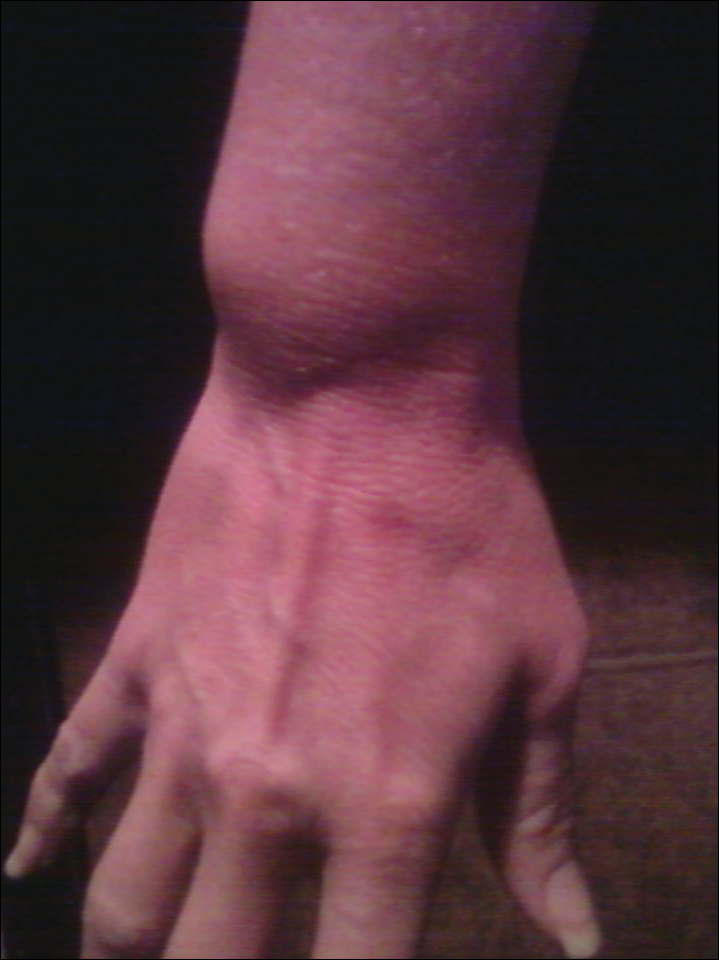
The patient subsequently underwent excision of her BCCs and reported each site became erythematous, edematous, warm, and painful 6 hours after excision, which lasted for hours to days (Figures 1–3). After the first excision on the right forearm, she was seen in the emergency department, started on intravenous antibiotics and prednisone, and kept overnight in the hospital. She was discharged the following day and the edema in the right forearm subsided over several days. Bacterial culture and laboratory evaluation for infection were negative after the first excision on the right forearm. Because of the symptoms she experienced following this excision, she was referred to the plastic surgery department for excision followed by postoperative monitoring in the hospital. The patient continued to undergo excisions for BCCs and developed more severe symptoms including erythema, edema, warmth, and tenderness at the surrounding sites. Once again, the excision sites were cultured and laboratory work to rule out infection was ordered with a negative result. After several excisions and subsequent clinical findings, the patients’ symptoms were deemed consistent with MWS and not a result of infectious etiology. A diagnosis of MWS and BCNS with exacerbation of MWS with surgical procedures was made.

The patient has continued therapy with rilonacept for MWS, which is managed by her rheumatologist. She has tolerated rilonacept without adverse effects and has experienced a reduction in symptoms that has enhanced her quality of life and allows for further treatment of her BCNS. Her dermatologist (J.W.L.) has been treating her BCCs with vismodegib, but treatment has been sporadic due to muscle cramping after 7 days of therapy. She reported subjective improvement to her dermatologist and has tried alternating 7 days on and 7 days off vismodegib. The muscle cramping still has limited her treatment with this regimen, and she is currently on a trial of 3 days on, 4 days off per week.
Comment
Classification and Clinical Presentation
The hereditary periodic fever syndromes include the autoinflammatory syndromes and the CAPSs. The autoinflammatory syndromes include familial Mediterranean fever, hyperimmunoglobulinemia D with periodic fever syndrome, and tumor necrosis factor receptor–associated periodic syndrome. The CAPSs are similar but distinct and include familial cold autoinflammatory syndrome, neonatal-onset multisystem inflammatory disease (also known as chronic infantile neurologic cutaneous and articular syndrome, or cutaneous articular syndrome) and MWS.1,2
Cryopyrin-associated periodic syndromes are rare inherited diseases that result from mutations in the NLRP3 gene. There is a gain-of-function mutation on the NLRP3 gene located on the long arm of chromosome 1 at position 44, which codes for cryopyrin. An NLRP3 gene mutation causes cryopyrin to become hyperactive, leading to the formation of an inflammasome, which is a group of cryopyrin molecules. Inflammasomes, along with other proteins, activate caspase 1 to produce excess IL-1β, leading to persistent inflammatory symptoms.3 IL-1β is one of the key mediators of the body’s response to microbial invasion, inflammation, immunologic reactions, and tissue injury. It affects a large range of cells and organs. Although IL-1β production is critical for the control of pathogenic infections, excessive cytokine production is harmful to the host and can even be fatal.3,4
Cryopyrin-associated periodic syndromes encompass a disease continuum. The 3 distinct entities share many overlapping features as well as unique and distinguishing characteristics. Familial cold autoinflammatory syndrome is the mildest phenotype and is inherited in an autosomal-dominant fashion. It is characterized by a chronic urticarial eruption that starts early in infancy or childhood. The distribution of the cutaneous eruption is widespread and favors the arms and legs over the face and trunk. A low-grade fever often is seen along with musculoskeletal concerns of arthralgia and pain. Other commonly reported symptoms include conjunctivitis, myalgia, fatigue, and headache. Neurologic symptoms can include headaches. Symptoms usually begin 1 to 2 hours after cold exposure and last less than 24 hours.5-8
Neonatal-onset multisystem inflammatory disease is the most severe phenotype and occurs sporadically. Continuous symptoms and flares are characteristic and the length of the flare can vary from minutes to days. The cutaneous eruption favors the face, trunk, arms, and legs, and varies in intensity, beginning in infancy or childhood. Fever may be intermittent, mild, or absent. Rheumatologic manifestations include arthralgia and swelling, with approximately one-third of patients experiencing severe disabling arthropathy that causes gross joint deformity. Ocular findings include conjunctivitis, uveitis, papilledema, and even blindness. Neurologic sequelae include headaches, sensorineural hearing loss, and aseptic meningitis. Amyloidosis has been seen as a late complication.5,8
Muckle-Wells syndrome is a rare hereditary inflammatory disorder. It has no ethnic predisposition and is mostly inherited in an autosomal-dominant fashion. Classically, the condition is characterized by recurrent urticaria beginning at birth with intermittent episodic fever and malaise. The eruption has a predilection for the face, trunk, arms, and legs, which is similar to neonatal-onset multisystem inflammatory disease. Associated myalgia and arthralgia are common as well as ocular findings of conjunctivitis and episcleritis. Neurologic manifestations include headache and progressive sensorineural hearing loss in 60% to 70% of patients.6 Abdominal pain may be seen along with rare serositis in MWS but is rare in the other CAPSs. Amyloidosis caused by chronic inflammation is the most serious complication of MWS and is seen in approximately one-third of patients, manifesting as proteinuria followed by renal impairment. Symptoms of MWS may occur daily but vary individually, are broad in intensity and duration, and can last 1 to 2 days before resolving spontaneously. The symptoms can result from metabolic stressors including cold, stress, and exercise, as well as microbial pathogens. Leukocytosis and increased acute-phase reactants are observed during episodes of inflammation.4,6,8
Histopathology
Mild phenotypic variability exists between individuals, and many of the symptoms overlap in CAPSs. Although CAPSs display several distinguishing clinical characteristics, interestingly they share the same histopathological features regardless of the syndrome. The typical histopathological finding is a dermal neutrophilic infiltrate that tends to be perivascular and also may be perieccrine. Vasodilation and dermal edema also may be seen. These histopathological findings contrast with the typical lymphocytic and eosinophilic infiltrate seen in classic urticaria. Similar histopathologic findings have been seen in other neutrophilic urticarial dermatoses such as Schnitzler syndrome.4,6
Differential
The differential diagnoses for CAPSs include Schnitzler syndrome, cold urticaria, systemic-onset juvenile idiopathic arthritis/adult-onset Still disease, and deficiency in IL-1ra. It is important to consider these differential diagnoses for management and treatment options.
Management
The discovery of the NLRP3 gene mutation as well as an understanding of IL-1 biology has led to targeted therapy for these syndromes. Cryopyrin-associated periodic syndromes are mediated by IL-1β with an in vivo rate 5 times higher than in healthy patients.4 The blockade of IL-1β results in complete resolution of symptoms.
In the last several years, anakinra, rilonacept, and canakinumab have shown efficacy in targeting IL-1β as receptor antagonists. Anakinra is a short-acting recombinant IL-1ra with a half-life of 4 to 6 hours. This short half-life requires daily injections and the most common adverse events included injection-site reaction and upper respiratory tract infection.2,4 Rilonacept is a dimeric fusion protein that contains binding regions for the type 1 receptor and the IL-1 receptor accessory protein and is fused to the fragment, crystallizable (Fc) portion of human IgG1. Rilonacept is long acting with a circulating half-life of 8.6 days and offers patients ease of dosing with weekly subcutaneous injections. Rilonacept generally is well tolerated, with the most frequent adverse effects being injection-site reaction, upper respiratory tract infection, headache, arthralgia, and diarrhea.2,7
The newest of the treatments for patients with CAPS is canakinumab. It is a fully human IL-1β monoclonal antibody that is specific for IL-1β and not other members of the IL-1 family. It has a mean half-life of 26 days and is dosed subcutaneously once every 8 weeks. The most common adverse effects include nasopharyngitis, rhinitis, nausea, diarrhea, and vertigo.4 In one study, most patients did not report injection-site reactions.7 Studies also are underway on VX-765, a caspace-1 targeted therapy that acts upstream in the IL-1β pathway. Treatment with anakinra, rilonacept, and canakinumab generally offers rapid and sustained remission in the majority of MWS patients and helps prevent the development of systemic amyloidosis and lessens the potential for end organ damage.2,7
MWS and BCNS
Our patient had an unusual presentation of MWS complicated by BCNS, another rare autosomal-dominant inherited genodermatosis. In an extensive review of PubMed articles indexed for MEDLINE using the search terms Muckle-Wells syndrome and basal cell nevus syndrome, no association was identified between MWS and BCNS. Basal cell nevus syndrome is linked to PTCH1 (patched 1) gene mutation with an incidence of 1:150,000 in the United States and Europe and is characterized by a broad range of anomalies including skeletal abnormalities, ectopic calcification, odontogenic keratocysts, facial dysmorphism with macrocephaly, palmoplantar pits, and numerous tumors. Most notable is the early and strong predisposition to develop several to hundreds of BCCs.9
Conclusion
Muckle-Wells syndrome may go undiagnosed for many years or may be misdiagnosed as refractory urticaria, as in our patient. It is important to include periodic fever syndromes in the differential diagnosis of refractory urticaria with episodic fever to diagnose these cases of MWS earlier.
- Kagami S, Saeki H, Kuwano Y, et al. A probable case of Muckle-Wells syndrome. J Dermatol. 2006;2:118-121.
- Kanazawa N, Furukawa F. Autoinflammatory syndromes with a dermatological perspective. J Dermatol. 2007;34:601-618.
- Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561-574.
- Mueller SM, Itin P, Haeusermann P. Muckle-Wells syndrome effectively treated with canakinumab: is the recommended dosing schedule mandatory? Dermatology. 2011;223:113-118.
- Neven B, Prieur A, Quartier dit Maire P. Cryopyrinopathies: update on pathogenesis and treatment. Nat Clin Pract Rheumatol. 2008;4:481-489.
- Newell L, August S, Foria V, et al. Lifelong urticaria and multiple unexplained systemic symptoms. Clin Exp Dermatol. 2011;36:431-433.
- Yu JR, Kieron KS. Cryopyrin-associated periodic syndrome: an update on diagnosis and treatment response. Curr Allergy Asthma Rep. 2011;11:12-20.
- Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. 2nd ed. Barcelona, Spain: Mosby Elsevier; 2008. 9. Göppner D, Leverkus M. Basal cell carcinoma: from the molecular understanding of the pathogenesis to targeted therapy of progressive disease. J Skin Cancer. 2011;2011:650258.
Muckle-Wells syndrome (MWS) was first described in 1962 and is part of a broad category of hereditary periodic fever syndromes that include the autoinflammatory syndromes and the cryopyrin-associated periodic syndromes (CAPSs). Unlike autoimmune diseases, autoinflammatory syndromes are not associated with antigen-specific T-cell responses or high titers of autoantibodies but are related to disorders of the innate immune system. Basal cell nevus syndrome (BCNS), or Gorlin syndrome, is a rare genodermatosis inherited in an autosomal-dominant fashion that is characterized by a broad range of anomalies. Most notable is the early and strong predisposition to develop several to hundreds of basal cell carcinomas (BCCs). Classic clinical features of MWS and a thorough history and physical examination can assist in the diagnosis of this rare entity.
Case Report
A 35-year-old woman with a history of BCNS, which had been diagnosed at 24 years of age based on the presence of more than 2 BCCs and a family history of BCNS in her mother, presented with intermittent pruritic urticaria on the chest and back, episodic fevers, associated joint pain and swelling that worsened several hours after exercise, headache, conjunctivitis, blurred vision, and severe debilitating fatigue that had been present since childhood. The symptoms had progressively worsened with age and symptom-free intervals became shorter. She was diagnosed by her rheumatologist with biopsy-proven MWS and a positive NLRP3 (NLR family pyrin domain containing 3) gene mutation at 29 years of age. She was treated unsuccessfully with prednisone and antihistamines and entered a trial with anakinra. She showed improvement for 2 weeks but developed severe swelling and erythema at the injection sites at week 3, along with large leathery patches on the legs and difficulty ambulating.
The patient subsequently underwent excision of her BCCs and reported each site became erythematous, edematous, warm, and painful 6 hours after excision, which lasted for hours to days (Figures 1–3). After the first excision on the right forearm, she was seen in the emergency department, started on intravenous antibiotics and prednisone, and kept overnight in the hospital. She was discharged the following day and the edema in the right forearm subsided over several days. Bacterial culture and laboratory evaluation for infection were negative after the first excision on the right forearm. Because of the symptoms she experienced following this excision, she was referred to the plastic surgery department for excision followed by postoperative monitoring in the hospital. The patient continued to undergo excisions for BCCs and developed more severe symptoms including erythema, edema, warmth, and tenderness at the surrounding sites. Once again, the excision sites were cultured and laboratory work to rule out infection was ordered with a negative result. After several excisions and subsequent clinical findings, the patients’ symptoms were deemed consistent with MWS and not a result of infectious etiology. A diagnosis of MWS and BCNS with exacerbation of MWS with surgical procedures was made.
The patient has continued therapy with rilonacept for MWS, which is managed by her rheumatologist. She has tolerated rilonacept without adverse effects and has experienced a reduction in symptoms that has enhanced her quality of life and allows for further treatment of her BCNS. Her dermatologist (J.W.L.) has been treating her BCCs with vismodegib, but treatment has been sporadic due to muscle cramping after 7 days of therapy. She reported subjective improvement to her dermatologist and has tried alternating 7 days on and 7 days off vismodegib. The muscle cramping still has limited her treatment with this regimen, and she is currently on a trial of 3 days on, 4 days off per week.
Comment
Classification and Clinical Presentation
The hereditary periodic fever syndromes include the autoinflammatory syndromes and the CAPSs. The autoinflammatory syndromes include familial Mediterranean fever, hyperimmunoglobulinemia D with periodic fever syndrome, and tumor necrosis factor receptor–associated periodic syndrome. The CAPSs are similar but distinct and include familial cold autoinflammatory syndrome, neonatal-onset multisystem inflammatory disease (also known as chronic infantile neurologic cutaneous and articular syndrome, or cutaneous articular syndrome) and MWS.1,2
Cryopyrin-associated periodic syndromes are rare inherited diseases that result from mutations in the NLRP3 gene. There is a gain-of-function mutation on the NLRP3 gene located on the long arm of chromosome 1 at position 44, which codes for cryopyrin. An NLRP3 gene mutation causes cryopyrin to become hyperactive, leading to the formation of an inflammasome, which is a group of cryopyrin molecules. Inflammasomes, along with other proteins, activate caspase 1 to produce excess IL-1β, leading to persistent inflammatory symptoms.3 IL-1β is one of the key mediators of the body’s response to microbial invasion, inflammation, immunologic reactions, and tissue injury. It affects a large range of cells and organs. Although IL-1β production is critical for the control of pathogenic infections, excessive cytokine production is harmful to the host and can even be fatal.3,4
Cryopyrin-associated periodic syndromes encompass a disease continuum. The 3 distinct entities share many overlapping features as well as unique and distinguishing characteristics. Familial cold autoinflammatory syndrome is the mildest phenotype and is inherited in an autosomal-dominant fashion. It is characterized by a chronic urticarial eruption that starts early in infancy or childhood. The distribution of the cutaneous eruption is widespread and favors the arms and legs over the face and trunk. A low-grade fever often is seen along with musculoskeletal concerns of arthralgia and pain. Other commonly reported symptoms include conjunctivitis, myalgia, fatigue, and headache. Neurologic symptoms can include headaches. Symptoms usually begin 1 to 2 hours after cold exposure and last less than 24 hours.5-8
Neonatal-onset multisystem inflammatory disease is the most severe phenotype and occurs sporadically. Continuous symptoms and flares are characteristic and the length of the flare can vary from minutes to days. The cutaneous eruption favors the face, trunk, arms, and legs, and varies in intensity, beginning in infancy or childhood. Fever may be intermittent, mild, or absent. Rheumatologic manifestations include arthralgia and swelling, with approximately one-third of patients experiencing severe disabling arthropathy that causes gross joint deformity. Ocular findings include conjunctivitis, uveitis, papilledema, and even blindness. Neurologic sequelae include headaches, sensorineural hearing loss, and aseptic meningitis. Amyloidosis has been seen as a late complication.5,8
Muckle-Wells syndrome is a rare hereditary inflammatory disorder. It has no ethnic predisposition and is mostly inherited in an autosomal-dominant fashion. Classically, the condition is characterized by recurrent urticaria beginning at birth with intermittent episodic fever and malaise. The eruption has a predilection for the face, trunk, arms, and legs, which is similar to neonatal-onset multisystem inflammatory disease. Associated myalgia and arthralgia are common as well as ocular findings of conjunctivitis and episcleritis. Neurologic manifestations include headache and progressive sensorineural hearing loss in 60% to 70% of patients.6 Abdominal pain may be seen along with rare serositis in MWS but is rare in the other CAPSs. Amyloidosis caused by chronic inflammation is the most serious complication of MWS and is seen in approximately one-third of patients, manifesting as proteinuria followed by renal impairment. Symptoms of MWS may occur daily but vary individually, are broad in intensity and duration, and can last 1 to 2 days before resolving spontaneously. The symptoms can result from metabolic stressors including cold, stress, and exercise, as well as microbial pathogens. Leukocytosis and increased acute-phase reactants are observed during episodes of inflammation.4,6,8
Histopathology
Mild phenotypic variability exists between individuals, and many of the symptoms overlap in CAPSs. Although CAPSs display several distinguishing clinical characteristics, interestingly they share the same histopathological features regardless of the syndrome. The typical histopathological finding is a dermal neutrophilic infiltrate that tends to be perivascular and also may be perieccrine. Vasodilation and dermal edema also may be seen. These histopathological findings contrast with the typical lymphocytic and eosinophilic infiltrate seen in classic urticaria. Similar histopathologic findings have been seen in other neutrophilic urticarial dermatoses such as Schnitzler syndrome.4,6
Differential
The differential diagnoses for CAPSs include Schnitzler syndrome, cold urticaria, systemic-onset juvenile idiopathic arthritis/adult-onset Still disease, and deficiency in IL-1ra. It is important to consider these differential diagnoses for management and treatment options.
Management
The discovery of the NLRP3 gene mutation as well as an understanding of IL-1 biology has led to targeted therapy for these syndromes. Cryopyrin-associated periodic syndromes are mediated by IL-1β with an in vivo rate 5 times higher than in healthy patients.4 The blockade of IL-1β results in complete resolution of symptoms.
In the last several years, anakinra, rilonacept, and canakinumab have shown efficacy in targeting IL-1β as receptor antagonists. Anakinra is a short-acting recombinant IL-1ra with a half-life of 4 to 6 hours. This short half-life requires daily injections and the most common adverse events included injection-site reaction and upper respiratory tract infection.2,4 Rilonacept is a dimeric fusion protein that contains binding regions for the type 1 receptor and the IL-1 receptor accessory protein and is fused to the fragment, crystallizable (Fc) portion of human IgG1. Rilonacept is long acting with a circulating half-life of 8.6 days and offers patients ease of dosing with weekly subcutaneous injections. Rilonacept generally is well tolerated, with the most frequent adverse effects being injection-site reaction, upper respiratory tract infection, headache, arthralgia, and diarrhea.2,7
The newest of the treatments for patients with CAPS is canakinumab. It is a fully human IL-1β monoclonal antibody that is specific for IL-1β and not other members of the IL-1 family. It has a mean half-life of 26 days and is dosed subcutaneously once every 8 weeks. The most common adverse effects include nasopharyngitis, rhinitis, nausea, diarrhea, and vertigo.4 In one study, most patients did not report injection-site reactions.7 Studies also are underway on VX-765, a caspace-1 targeted therapy that acts upstream in the IL-1β pathway. Treatment with anakinra, rilonacept, and canakinumab generally offers rapid and sustained remission in the majority of MWS patients and helps prevent the development of systemic amyloidosis and lessens the potential for end organ damage.2,7
MWS and BCNS
Our patient had an unusual presentation of MWS complicated by BCNS, another rare autosomal-dominant inherited genodermatosis. In an extensive review of PubMed articles indexed for MEDLINE using the search terms Muckle-Wells syndrome and basal cell nevus syndrome, no association was identified between MWS and BCNS. Basal cell nevus syndrome is linked to PTCH1 (patched 1) gene mutation with an incidence of 1:150,000 in the United States and Europe and is characterized by a broad range of anomalies including skeletal abnormalities, ectopic calcification, odontogenic keratocysts, facial dysmorphism with macrocephaly, palmoplantar pits, and numerous tumors. Most notable is the early and strong predisposition to develop several to hundreds of BCCs.9
Conclusion
Muckle-Wells syndrome may go undiagnosed for many years or may be misdiagnosed as refractory urticaria, as in our patient. It is important to include periodic fever syndromes in the differential diagnosis of refractory urticaria with episodic fever to diagnose these cases of MWS earlier.
Muckle-Wells syndrome (MWS) was first described in 1962 and is part of a broad category of hereditary periodic fever syndromes that include the autoinflammatory syndromes and the cryopyrin-associated periodic syndromes (CAPSs). Unlike autoimmune diseases, autoinflammatory syndromes are not associated with antigen-specific T-cell responses or high titers of autoantibodies but are related to disorders of the innate immune system. Basal cell nevus syndrome (BCNS), or Gorlin syndrome, is a rare genodermatosis inherited in an autosomal-dominant fashion that is characterized by a broad range of anomalies. Most notable is the early and strong predisposition to develop several to hundreds of basal cell carcinomas (BCCs). Classic clinical features of MWS and a thorough history and physical examination can assist in the diagnosis of this rare entity.
Case Report
A 35-year-old woman with a history of BCNS, which had been diagnosed at 24 years of age based on the presence of more than 2 BCCs and a family history of BCNS in her mother, presented with intermittent pruritic urticaria on the chest and back, episodic fevers, associated joint pain and swelling that worsened several hours after exercise, headache, conjunctivitis, blurred vision, and severe debilitating fatigue that had been present since childhood. The symptoms had progressively worsened with age and symptom-free intervals became shorter. She was diagnosed by her rheumatologist with biopsy-proven MWS and a positive NLRP3 (NLR family pyrin domain containing 3) gene mutation at 29 years of age. She was treated unsuccessfully with prednisone and antihistamines and entered a trial with anakinra. She showed improvement for 2 weeks but developed severe swelling and erythema at the injection sites at week 3, along with large leathery patches on the legs and difficulty ambulating.
The patient subsequently underwent excision of her BCCs and reported each site became erythematous, edematous, warm, and painful 6 hours after excision, which lasted for hours to days (Figures 1–3). After the first excision on the right forearm, she was seen in the emergency department, started on intravenous antibiotics and prednisone, and kept overnight in the hospital. She was discharged the following day and the edema in the right forearm subsided over several days. Bacterial culture and laboratory evaluation for infection were negative after the first excision on the right forearm. Because of the symptoms she experienced following this excision, she was referred to the plastic surgery department for excision followed by postoperative monitoring in the hospital. The patient continued to undergo excisions for BCCs and developed more severe symptoms including erythema, edema, warmth, and tenderness at the surrounding sites. Once again, the excision sites were cultured and laboratory work to rule out infection was ordered with a negative result. After several excisions and subsequent clinical findings, the patients’ symptoms were deemed consistent with MWS and not a result of infectious etiology. A diagnosis of MWS and BCNS with exacerbation of MWS with surgical procedures was made.
The patient has continued therapy with rilonacept for MWS, which is managed by her rheumatologist. She has tolerated rilonacept without adverse effects and has experienced a reduction in symptoms that has enhanced her quality of life and allows for further treatment of her BCNS. Her dermatologist (J.W.L.) has been treating her BCCs with vismodegib, but treatment has been sporadic due to muscle cramping after 7 days of therapy. She reported subjective improvement to her dermatologist and has tried alternating 7 days on and 7 days off vismodegib. The muscle cramping still has limited her treatment with this regimen, and she is currently on a trial of 3 days on, 4 days off per week.
Comment
Classification and Clinical Presentation
The hereditary periodic fever syndromes include the autoinflammatory syndromes and the CAPSs. The autoinflammatory syndromes include familial Mediterranean fever, hyperimmunoglobulinemia D with periodic fever syndrome, and tumor necrosis factor receptor–associated periodic syndrome. The CAPSs are similar but distinct and include familial cold autoinflammatory syndrome, neonatal-onset multisystem inflammatory disease (also known as chronic infantile neurologic cutaneous and articular syndrome, or cutaneous articular syndrome) and MWS.1,2
Cryopyrin-associated periodic syndromes are rare inherited diseases that result from mutations in the NLRP3 gene. There is a gain-of-function mutation on the NLRP3 gene located on the long arm of chromosome 1 at position 44, which codes for cryopyrin. An NLRP3 gene mutation causes cryopyrin to become hyperactive, leading to the formation of an inflammasome, which is a group of cryopyrin molecules. Inflammasomes, along with other proteins, activate caspase 1 to produce excess IL-1β, leading to persistent inflammatory symptoms.3 IL-1β is one of the key mediators of the body’s response to microbial invasion, inflammation, immunologic reactions, and tissue injury. It affects a large range of cells and organs. Although IL-1β production is critical for the control of pathogenic infections, excessive cytokine production is harmful to the host and can even be fatal.3,4
Cryopyrin-associated periodic syndromes encompass a disease continuum. The 3 distinct entities share many overlapping features as well as unique and distinguishing characteristics. Familial cold autoinflammatory syndrome is the mildest phenotype and is inherited in an autosomal-dominant fashion. It is characterized by a chronic urticarial eruption that starts early in infancy or childhood. The distribution of the cutaneous eruption is widespread and favors the arms and legs over the face and trunk. A low-grade fever often is seen along with musculoskeletal concerns of arthralgia and pain. Other commonly reported symptoms include conjunctivitis, myalgia, fatigue, and headache. Neurologic symptoms can include headaches. Symptoms usually begin 1 to 2 hours after cold exposure and last less than 24 hours.5-8
Neonatal-onset multisystem inflammatory disease is the most severe phenotype and occurs sporadically. Continuous symptoms and flares are characteristic and the length of the flare can vary from minutes to days. The cutaneous eruption favors the face, trunk, arms, and legs, and varies in intensity, beginning in infancy or childhood. Fever may be intermittent, mild, or absent. Rheumatologic manifestations include arthralgia and swelling, with approximately one-third of patients experiencing severe disabling arthropathy that causes gross joint deformity. Ocular findings include conjunctivitis, uveitis, papilledema, and even blindness. Neurologic sequelae include headaches, sensorineural hearing loss, and aseptic meningitis. Amyloidosis has been seen as a late complication.5,8
Muckle-Wells syndrome is a rare hereditary inflammatory disorder. It has no ethnic predisposition and is mostly inherited in an autosomal-dominant fashion. Classically, the condition is characterized by recurrent urticaria beginning at birth with intermittent episodic fever and malaise. The eruption has a predilection for the face, trunk, arms, and legs, which is similar to neonatal-onset multisystem inflammatory disease. Associated myalgia and arthralgia are common as well as ocular findings of conjunctivitis and episcleritis. Neurologic manifestations include headache and progressive sensorineural hearing loss in 60% to 70% of patients.6 Abdominal pain may be seen along with rare serositis in MWS but is rare in the other CAPSs. Amyloidosis caused by chronic inflammation is the most serious complication of MWS and is seen in approximately one-third of patients, manifesting as proteinuria followed by renal impairment. Symptoms of MWS may occur daily but vary individually, are broad in intensity and duration, and can last 1 to 2 days before resolving spontaneously. The symptoms can result from metabolic stressors including cold, stress, and exercise, as well as microbial pathogens. Leukocytosis and increased acute-phase reactants are observed during episodes of inflammation.4,6,8
Histopathology
Mild phenotypic variability exists between individuals, and many of the symptoms overlap in CAPSs. Although CAPSs display several distinguishing clinical characteristics, interestingly they share the same histopathological features regardless of the syndrome. The typical histopathological finding is a dermal neutrophilic infiltrate that tends to be perivascular and also may be perieccrine. Vasodilation and dermal edema also may be seen. These histopathological findings contrast with the typical lymphocytic and eosinophilic infiltrate seen in classic urticaria. Similar histopathologic findings have been seen in other neutrophilic urticarial dermatoses such as Schnitzler syndrome.4,6
Differential
The differential diagnoses for CAPSs include Schnitzler syndrome, cold urticaria, systemic-onset juvenile idiopathic arthritis/adult-onset Still disease, and deficiency in IL-1ra. It is important to consider these differential diagnoses for management and treatment options.
Management
The discovery of the NLRP3 gene mutation as well as an understanding of IL-1 biology has led to targeted therapy for these syndromes. Cryopyrin-associated periodic syndromes are mediated by IL-1β with an in vivo rate 5 times higher than in healthy patients.4 The blockade of IL-1β results in complete resolution of symptoms.
In the last several years, anakinra, rilonacept, and canakinumab have shown efficacy in targeting IL-1β as receptor antagonists. Anakinra is a short-acting recombinant IL-1ra with a half-life of 4 to 6 hours. This short half-life requires daily injections and the most common adverse events included injection-site reaction and upper respiratory tract infection.2,4 Rilonacept is a dimeric fusion protein that contains binding regions for the type 1 receptor and the IL-1 receptor accessory protein and is fused to the fragment, crystallizable (Fc) portion of human IgG1. Rilonacept is long acting with a circulating half-life of 8.6 days and offers patients ease of dosing with weekly subcutaneous injections. Rilonacept generally is well tolerated, with the most frequent adverse effects being injection-site reaction, upper respiratory tract infection, headache, arthralgia, and diarrhea.2,7
The newest of the treatments for patients with CAPS is canakinumab. It is a fully human IL-1β monoclonal antibody that is specific for IL-1β and not other members of the IL-1 family. It has a mean half-life of 26 days and is dosed subcutaneously once every 8 weeks. The most common adverse effects include nasopharyngitis, rhinitis, nausea, diarrhea, and vertigo.4 In one study, most patients did not report injection-site reactions.7 Studies also are underway on VX-765, a caspace-1 targeted therapy that acts upstream in the IL-1β pathway. Treatment with anakinra, rilonacept, and canakinumab generally offers rapid and sustained remission in the majority of MWS patients and helps prevent the development of systemic amyloidosis and lessens the potential for end organ damage.2,7
MWS and BCNS
Our patient had an unusual presentation of MWS complicated by BCNS, another rare autosomal-dominant inherited genodermatosis. In an extensive review of PubMed articles indexed for MEDLINE using the search terms Muckle-Wells syndrome and basal cell nevus syndrome, no association was identified between MWS and BCNS. Basal cell nevus syndrome is linked to PTCH1 (patched 1) gene mutation with an incidence of 1:150,000 in the United States and Europe and is characterized by a broad range of anomalies including skeletal abnormalities, ectopic calcification, odontogenic keratocysts, facial dysmorphism with macrocephaly, palmoplantar pits, and numerous tumors. Most notable is the early and strong predisposition to develop several to hundreds of BCCs.9
Conclusion
Muckle-Wells syndrome may go undiagnosed for many years or may be misdiagnosed as refractory urticaria, as in our patient. It is important to include periodic fever syndromes in the differential diagnosis of refractory urticaria with episodic fever to diagnose these cases of MWS earlier.
- Kagami S, Saeki H, Kuwano Y, et al. A probable case of Muckle-Wells syndrome. J Dermatol. 2006;2:118-121.
- Kanazawa N, Furukawa F. Autoinflammatory syndromes with a dermatological perspective. J Dermatol. 2007;34:601-618.
- Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561-574.
- Mueller SM, Itin P, Haeusermann P. Muckle-Wells syndrome effectively treated with canakinumab: is the recommended dosing schedule mandatory? Dermatology. 2011;223:113-118.
- Neven B, Prieur A, Quartier dit Maire P. Cryopyrinopathies: update on pathogenesis and treatment. Nat Clin Pract Rheumatol. 2008;4:481-489.
- Newell L, August S, Foria V, et al. Lifelong urticaria and multiple unexplained systemic symptoms. Clin Exp Dermatol. 2011;36:431-433.
- Yu JR, Kieron KS. Cryopyrin-associated periodic syndrome: an update on diagnosis and treatment response. Curr Allergy Asthma Rep. 2011;11:12-20.
- Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. 2nd ed. Barcelona, Spain: Mosby Elsevier; 2008. 9. Göppner D, Leverkus M. Basal cell carcinoma: from the molecular understanding of the pathogenesis to targeted therapy of progressive disease. J Skin Cancer. 2011;2011:650258.
- Kagami S, Saeki H, Kuwano Y, et al. A probable case of Muckle-Wells syndrome. J Dermatol. 2006;2:118-121.
- Kanazawa N, Furukawa F. Autoinflammatory syndromes with a dermatological perspective. J Dermatol. 2007;34:601-618.
- Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561-574.
- Mueller SM, Itin P, Haeusermann P. Muckle-Wells syndrome effectively treated with canakinumab: is the recommended dosing schedule mandatory? Dermatology. 2011;223:113-118.
- Neven B, Prieur A, Quartier dit Maire P. Cryopyrinopathies: update on pathogenesis and treatment. Nat Clin Pract Rheumatol. 2008;4:481-489.
- Newell L, August S, Foria V, et al. Lifelong urticaria and multiple unexplained systemic symptoms. Clin Exp Dermatol. 2011;36:431-433.
- Yu JR, Kieron KS. Cryopyrin-associated periodic syndrome: an update on diagnosis and treatment response. Curr Allergy Asthma Rep. 2011;11:12-20.
- Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. 2nd ed. Barcelona, Spain: Mosby Elsevier; 2008. 9. Göppner D, Leverkus M. Basal cell carcinoma: from the molecular understanding of the pathogenesis to targeted therapy of progressive disease. J Skin Cancer. 2011;2011:650258.
Practice Points
- An urticarial rash occurring in childhood with symptoms of fever, joint pain, and swelling along with visual symptoms should prompt consideration of a cryopyrin-associated periodic syndrome.
- Histopathology shows a dermal neutrophilic infiltrate that tends to be perivascular and also may be perieccrine. This atypical urticaria contrasts with the typical lymphocytic and eosinophilic infiltrate seen in classic urticaria.