User login
Darkening and Eruptive Nevi During Treatment With Erlotinib
To the Editor:
Erlotinib is a small-molecule selective tyrosine kinase inhibitor that functions by blocking the intracellular portion of the epidermal growth factor receptor (EGFR)1,2; EGFR normally is expressed in the basal layer of the epidermis, sweat glands, and hair follicles, and is overexpressed in some cancers.1,3 Normal activation of EGFR leads to signal transduction through the mitogen-activated protein kinase (MAPK) signaling pathway, which stimulates cell survival and proliferation.4,5 Erlotinib-induced inhibition of EGFR prevents tyrosine kinase phosphorylation and aims to decrease cell proliferation in these tumors.
Erlotinib is indicated as once-daily oral monotherapy for the treatment of advanced-stage non–small cell lung cancer (NSCLCA) and in combination with gemcitabine for treatment of advanced-stage pancreatic cancer.1 A number of cutaneous side effects have been reported, including acneform eruption, xerosis, paronychia, and pruritus.6 Other tyrosine kinase inhibitors, which also decrease signal transduction through the MAPK pathway, have some overlapping side effects; among these are vemurafenib, a selective BRAF inhibitor, and sorafenib, a multikinase inhibitor.7,8
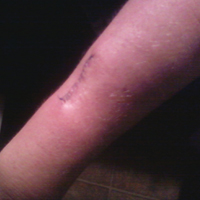
A 70-year-old man with NSCLCA presented with eruptive nevi and darkening of existing nevi 3 months after starting monotherapy with erlotinib. Physical examination demonstrated the simultaneous appearance of scattered acneform papules and pustules; diffuse xerosis; and numerous dark brown to black nevi on the trunk, arms, and legs. Compared to prior clinical photographs taken in our office, darkening of existing medium brown nevi was noted, and new nevi developed in areas where no prior nevi had been visible (Figure 1).
The patient’s medical history included 3 invasive melanomas, all of which were diagnosed at least 7 years prior to the initiation of erlotinib and were treated by surgical excision alone. Prior treatment of NSCLCA consisted of a left lower lobectomy followed by docetaxel, carboplatin, pegfilgrastim, dexamethasone, and pemetrexed. A thorough review of all of the patient’s medications revealed no associations with changes in nevi.
A review of the patient’s treatment timeline revealed that all other chemotherapeutic medications had been discontinued a minimum of 5 weeks before starting erlotinib. A complete cutaneous examination performed in our office after completion of these chemotherapeutic agents and prior to initiation of erlotinib was unremarkable for abnormally dark or eruptive nevi.
Since starting erlotinib treatment, the patient underwent 10 biopsies of clinically suspicious dark nevi performed by a dermatologist in our office. Two of these were diagnosed as melanoma in situ and one as an atypical nevus. A temporal association of the darkening and eruptive nevi with erlotinib treatment was established; however, because erlotinib was essential to his NSCLCA treatment, he continued erlotinib with frequent complete cutaneous examinations.
A number of cutaneous side effects have been described during treatment with erlotinib, the most common being acneform eruption.6 The incidence and severity of acneform eruptions have been positively correlated to survival in patients with NSCLCA.3,5,6 Other common side effects include xerosis, paronychia, and pruritus.1,5,6 Less common side effects include periungual pyogenic granulomas and hair growth abnormalities.1
Eruptive nevi previously were reported in a patient who was treated with erlotinib.1 Other tyrosine kinase inhibitors that also decrease signal transduction through the MAPK pathway, including sorafenib and vemurafenib, have been reported to cause eruptive nevi. There are 7 reports of eruptive nevi with sorafenib and 5 reports with vemurafenib.7-9 Development of nevi were noted within a few months of initiating treatment with these medications.7
A PubMed search of articles indexed for MEDLINE using the terms erlotinib and melanoma and erlotinib and nevi yielded no prior reports of darkening of existing nevi or the development of melanoma during treatment with erlotinib. However, vemurafenib has been reported to cause dysplastic nevi, melanomas, and darkening of existing nevi, in addition to eruptive nevi.8-10 The side effects of vemurafenib have been ascribed to a paradoxical upregulation of MAPK in BRAF wild-type cells. This effect has been well documented and demonstrated in vivo.8,10 Perhaps erlotinib has a similar potential to paradoxically upregulate the MAPK pathway, thus stimulating cellular proliferation and survival.
Another tyrosine kinase receptor, c-KIT, is found on the cell membrane of melanocytes along with EGFR.11,12 The c-KIT receptor also activates the MAPK pathway and is critical to the development, migration, and survival of melanocytes.11,13 Stimulation of the c-KIT tyrosine kinase receptor also can induce melanocyte proliferation and melanogenesis.11 The c-KIT receptor is encoded by the KIT gene (KIT proto-oncogene receptor tyrosine kinase). Mutations in this gene are associated with melanocytic disorders. Inherited KIT mutation leading to c-KIT receptor deficiency is associated with piebaldism. Acquired activating KIT mutations increasing c-KIT expression are associated with acral and mucosal melanomas as well as melanomas in chronically sun-damaged skin.13
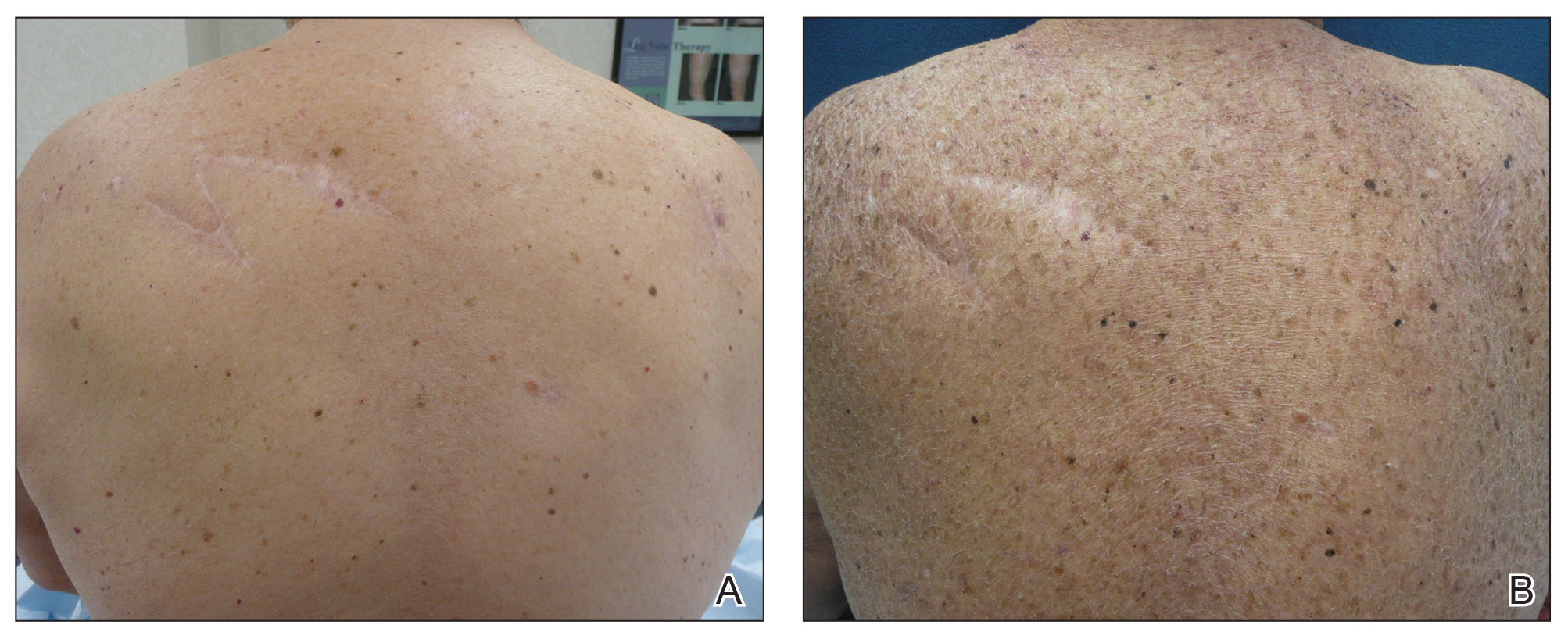
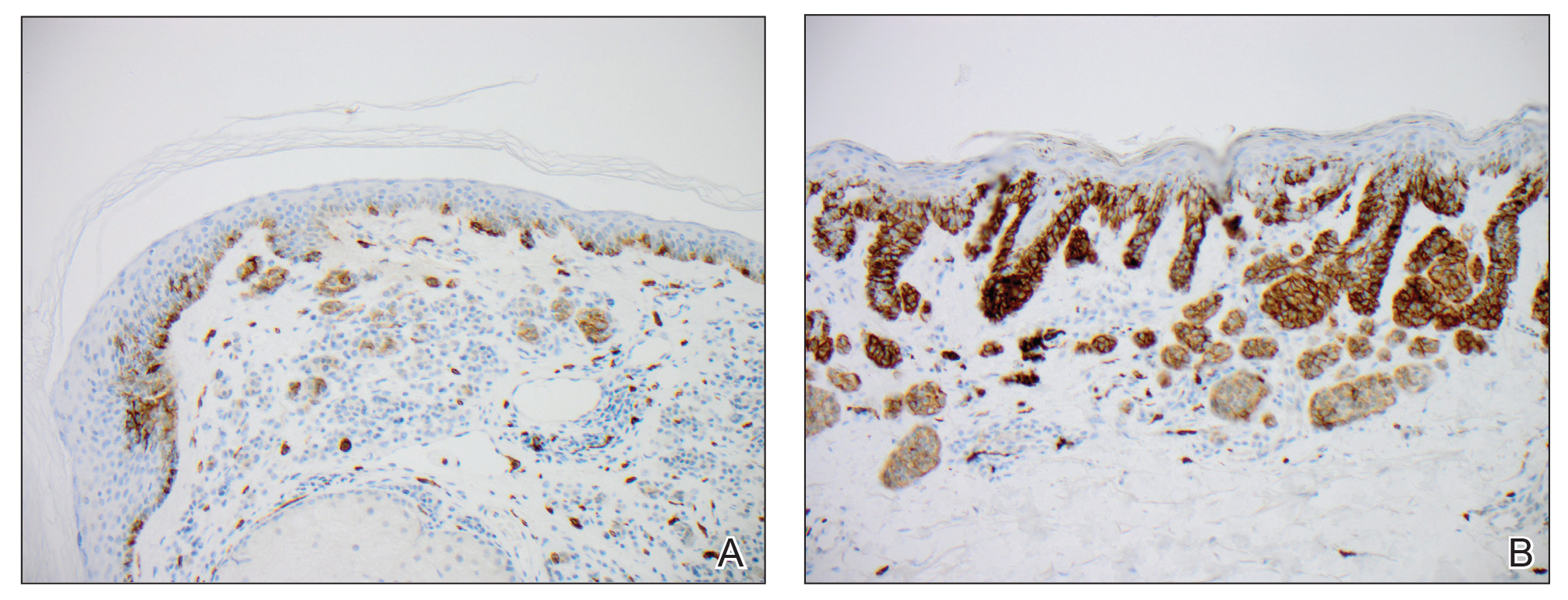
We hypothesized that erlotinib-induced inhibition of the MAPK pathway could lead to a reactive increase in expression of c-KIT and thus stimulate melanocyte proliferation and pigment production. Similar feedback upregulation of an MAPK pathway stimulating receptor during downstream MAPK inhibition has been demonstrated in colon adenocarcinoma; in this setting, BRAF inhibitors blocking the MAPK pathway leads to upregulation of EGFR.14 In our patient, c-KIT immunostaining revealed a mild to moderate increase in intensity (ie, the darkness of the staining) in nevi and melanomas during treatment with erlotinib compared to nevi biopsied before erlotinib treatment (Figure 2). The increased intensity of c-KIT immunostaining was further confirmed via semiquantitative digital image analysis. Using this method, a darkened nevus biopsied during treatment with erlotinib demonstrated 43.16% of cells (N=31,451) had very strong c-KIT staining, while a nevus biopsied before treatment with erlotinib demonstrated only 3.32% of cells (N=7507) with very strong c-KIT staining. Increased expression of c-KIT, possibly reactive to downstream inhibition the MAPK pathway from erlotinib, could be implicated in our case of eruptive nevi.
In summary, we report a rare case of darkening of existing nevi and development of melanoma in situ during treatment with erlotinib. The patient’s therapeutic timeline and concurrence of other well-documented side effects provided support for erlotinib as the causative agent in our patient. Additional support is provided through reports of other medications affecting the same pathway as erlotinib causing eruptive nevi, darkening of existing nevi, and melanoma in situ.7-10 Through c-KIT immunostaining, we demonstrated that increased expression of c-KIT might be responsible for the changes in nevi in our patient. We, therefore, suggest frequent full-body skin examinations in patients treated with erlotinib to monitor for the possible development of malignant melanomas.
- Santiago F, Goncalo M, Reis J, et al. Adverse cutaneous reactions to epidermal growth factor receptor inhibitors: a study of 14 patients. An Bras Dermatol 2011;86:483-490.
- Lubbe J, Masouye I, Dietrich P. Generalized xerotic dermatitis with neutrophilic spongiosis induced by erlotinib (Tarceva). Dermatology. 2008;216:247-249.
- Dessinioti C, Antoniou C, Katsambas A. Acneiform eruptions. Clin Dermatol. 2014;32:24-34.
- Herbst R, Fukuoka M, Baselga J. Gefitinib—a novel targeted approach to treating cancer. Nat Rev Cancer. 2004;4:979-987.
- Brodell L, Hepper D, Lind A, et al. Histopathology of acneiform eruptions in patients treated with epidermal growth factor receptor inhibitors. J Cutan Pathol. 2013;40:865-870.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol 2013;69:463-472.
- Uhlenhake E, Watson A, Aronson P. Sorafenib induced eruptive melanocytic lesions. Dermatol Online J. 2013;19:181-84.
- Chu E, Wanat K, Miller C, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol 2012;67:1265-1272.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Cohen P, Bedikian A, Kim K. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Longley B, Tyrrell L, Lu S, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312-314.
- Yun W, Bang S, Min K, et al. Epidermal growth factor and epidermal growth factor signaling attenuate laser-induced melanogenesis. Dermatol Surg. 2013;39:1903-1911.
- Swick J, Maize J. Molecular biology of melanoma. J Am Acad Dermatol. 2012;67:1049-1054.
- Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118-122.
To the Editor:
Erlotinib is a small-molecule selective tyrosine kinase inhibitor that functions by blocking the intracellular portion of the epidermal growth factor receptor (EGFR)1,2; EGFR normally is expressed in the basal layer of the epidermis, sweat glands, and hair follicles, and is overexpressed in some cancers.1,3 Normal activation of EGFR leads to signal transduction through the mitogen-activated protein kinase (MAPK) signaling pathway, which stimulates cell survival and proliferation.4,5 Erlotinib-induced inhibition of EGFR prevents tyrosine kinase phosphorylation and aims to decrease cell proliferation in these tumors.
Erlotinib is indicated as once-daily oral monotherapy for the treatment of advanced-stage non–small cell lung cancer (NSCLCA) and in combination with gemcitabine for treatment of advanced-stage pancreatic cancer.1 A number of cutaneous side effects have been reported, including acneform eruption, xerosis, paronychia, and pruritus.6 Other tyrosine kinase inhibitors, which also decrease signal transduction through the MAPK pathway, have some overlapping side effects; among these are vemurafenib, a selective BRAF inhibitor, and sorafenib, a multikinase inhibitor.7,8
A 70-year-old man with NSCLCA presented with eruptive nevi and darkening of existing nevi 3 months after starting monotherapy with erlotinib. Physical examination demonstrated the simultaneous appearance of scattered acneform papules and pustules; diffuse xerosis; and numerous dark brown to black nevi on the trunk, arms, and legs. Compared to prior clinical photographs taken in our office, darkening of existing medium brown nevi was noted, and new nevi developed in areas where no prior nevi had been visible (Figure 1).
The patient’s medical history included 3 invasive melanomas, all of which were diagnosed at least 7 years prior to the initiation of erlotinib and were treated by surgical excision alone. Prior treatment of NSCLCA consisted of a left lower lobectomy followed by docetaxel, carboplatin, pegfilgrastim, dexamethasone, and pemetrexed. A thorough review of all of the patient’s medications revealed no associations with changes in nevi.
A review of the patient’s treatment timeline revealed that all other chemotherapeutic medications had been discontinued a minimum of 5 weeks before starting erlotinib. A complete cutaneous examination performed in our office after completion of these chemotherapeutic agents and prior to initiation of erlotinib was unremarkable for abnormally dark or eruptive nevi.
Since starting erlotinib treatment, the patient underwent 10 biopsies of clinically suspicious dark nevi performed by a dermatologist in our office. Two of these were diagnosed as melanoma in situ and one as an atypical nevus. A temporal association of the darkening and eruptive nevi with erlotinib treatment was established; however, because erlotinib was essential to his NSCLCA treatment, he continued erlotinib with frequent complete cutaneous examinations.
A number of cutaneous side effects have been described during treatment with erlotinib, the most common being acneform eruption.6 The incidence and severity of acneform eruptions have been positively correlated to survival in patients with NSCLCA.3,5,6 Other common side effects include xerosis, paronychia, and pruritus.1,5,6 Less common side effects include periungual pyogenic granulomas and hair growth abnormalities.1
Eruptive nevi previously were reported in a patient who was treated with erlotinib.1 Other tyrosine kinase inhibitors that also decrease signal transduction through the MAPK pathway, including sorafenib and vemurafenib, have been reported to cause eruptive nevi. There are 7 reports of eruptive nevi with sorafenib and 5 reports with vemurafenib.7-9 Development of nevi were noted within a few months of initiating treatment with these medications.7
A PubMed search of articles indexed for MEDLINE using the terms erlotinib and melanoma and erlotinib and nevi yielded no prior reports of darkening of existing nevi or the development of melanoma during treatment with erlotinib. However, vemurafenib has been reported to cause dysplastic nevi, melanomas, and darkening of existing nevi, in addition to eruptive nevi.8-10 The side effects of vemurafenib have been ascribed to a paradoxical upregulation of MAPK in BRAF wild-type cells. This effect has been well documented and demonstrated in vivo.8,10 Perhaps erlotinib has a similar potential to paradoxically upregulate the MAPK pathway, thus stimulating cellular proliferation and survival.
Another tyrosine kinase receptor, c-KIT, is found on the cell membrane of melanocytes along with EGFR.11,12 The c-KIT receptor also activates the MAPK pathway and is critical to the development, migration, and survival of melanocytes.11,13 Stimulation of the c-KIT tyrosine kinase receptor also can induce melanocyte proliferation and melanogenesis.11 The c-KIT receptor is encoded by the KIT gene (KIT proto-oncogene receptor tyrosine kinase). Mutations in this gene are associated with melanocytic disorders. Inherited KIT mutation leading to c-KIT receptor deficiency is associated with piebaldism. Acquired activating KIT mutations increasing c-KIT expression are associated with acral and mucosal melanomas as well as melanomas in chronically sun-damaged skin.13
We hypothesized that erlotinib-induced inhibition of the MAPK pathway could lead to a reactive increase in expression of c-KIT and thus stimulate melanocyte proliferation and pigment production. Similar feedback upregulation of an MAPK pathway stimulating receptor during downstream MAPK inhibition has been demonstrated in colon adenocarcinoma; in this setting, BRAF inhibitors blocking the MAPK pathway leads to upregulation of EGFR.14 In our patient, c-KIT immunostaining revealed a mild to moderate increase in intensity (ie, the darkness of the staining) in nevi and melanomas during treatment with erlotinib compared to nevi biopsied before erlotinib treatment (Figure 2). The increased intensity of c-KIT immunostaining was further confirmed via semiquantitative digital image analysis. Using this method, a darkened nevus biopsied during treatment with erlotinib demonstrated 43.16% of cells (N=31,451) had very strong c-KIT staining, while a nevus biopsied before treatment with erlotinib demonstrated only 3.32% of cells (N=7507) with very strong c-KIT staining. Increased expression of c-KIT, possibly reactive to downstream inhibition the MAPK pathway from erlotinib, could be implicated in our case of eruptive nevi.
In summary, we report a rare case of darkening of existing nevi and development of melanoma in situ during treatment with erlotinib. The patient’s therapeutic timeline and concurrence of other well-documented side effects provided support for erlotinib as the causative agent in our patient. Additional support is provided through reports of other medications affecting the same pathway as erlotinib causing eruptive nevi, darkening of existing nevi, and melanoma in situ.7-10 Through c-KIT immunostaining, we demonstrated that increased expression of c-KIT might be responsible for the changes in nevi in our patient. We, therefore, suggest frequent full-body skin examinations in patients treated with erlotinib to monitor for the possible development of malignant melanomas.
To the Editor:
Erlotinib is a small-molecule selective tyrosine kinase inhibitor that functions by blocking the intracellular portion of the epidermal growth factor receptor (EGFR)1,2; EGFR normally is expressed in the basal layer of the epidermis, sweat glands, and hair follicles, and is overexpressed in some cancers.1,3 Normal activation of EGFR leads to signal transduction through the mitogen-activated protein kinase (MAPK) signaling pathway, which stimulates cell survival and proliferation.4,5 Erlotinib-induced inhibition of EGFR prevents tyrosine kinase phosphorylation and aims to decrease cell proliferation in these tumors.
Erlotinib is indicated as once-daily oral monotherapy for the treatment of advanced-stage non–small cell lung cancer (NSCLCA) and in combination with gemcitabine for treatment of advanced-stage pancreatic cancer.1 A number of cutaneous side effects have been reported, including acneform eruption, xerosis, paronychia, and pruritus.6 Other tyrosine kinase inhibitors, which also decrease signal transduction through the MAPK pathway, have some overlapping side effects; among these are vemurafenib, a selective BRAF inhibitor, and sorafenib, a multikinase inhibitor.7,8
A 70-year-old man with NSCLCA presented with eruptive nevi and darkening of existing nevi 3 months after starting monotherapy with erlotinib. Physical examination demonstrated the simultaneous appearance of scattered acneform papules and pustules; diffuse xerosis; and numerous dark brown to black nevi on the trunk, arms, and legs. Compared to prior clinical photographs taken in our office, darkening of existing medium brown nevi was noted, and new nevi developed in areas where no prior nevi had been visible (Figure 1).
The patient’s medical history included 3 invasive melanomas, all of which were diagnosed at least 7 years prior to the initiation of erlotinib and were treated by surgical excision alone. Prior treatment of NSCLCA consisted of a left lower lobectomy followed by docetaxel, carboplatin, pegfilgrastim, dexamethasone, and pemetrexed. A thorough review of all of the patient’s medications revealed no associations with changes in nevi.
A review of the patient’s treatment timeline revealed that all other chemotherapeutic medications had been discontinued a minimum of 5 weeks before starting erlotinib. A complete cutaneous examination performed in our office after completion of these chemotherapeutic agents and prior to initiation of erlotinib was unremarkable for abnormally dark or eruptive nevi.
Since starting erlotinib treatment, the patient underwent 10 biopsies of clinically suspicious dark nevi performed by a dermatologist in our office. Two of these were diagnosed as melanoma in situ and one as an atypical nevus. A temporal association of the darkening and eruptive nevi with erlotinib treatment was established; however, because erlotinib was essential to his NSCLCA treatment, he continued erlotinib with frequent complete cutaneous examinations.
A number of cutaneous side effects have been described during treatment with erlotinib, the most common being acneform eruption.6 The incidence and severity of acneform eruptions have been positively correlated to survival in patients with NSCLCA.3,5,6 Other common side effects include xerosis, paronychia, and pruritus.1,5,6 Less common side effects include periungual pyogenic granulomas and hair growth abnormalities.1
Eruptive nevi previously were reported in a patient who was treated with erlotinib.1 Other tyrosine kinase inhibitors that also decrease signal transduction through the MAPK pathway, including sorafenib and vemurafenib, have been reported to cause eruptive nevi. There are 7 reports of eruptive nevi with sorafenib and 5 reports with vemurafenib.7-9 Development of nevi were noted within a few months of initiating treatment with these medications.7
A PubMed search of articles indexed for MEDLINE using the terms erlotinib and melanoma and erlotinib and nevi yielded no prior reports of darkening of existing nevi or the development of melanoma during treatment with erlotinib. However, vemurafenib has been reported to cause dysplastic nevi, melanomas, and darkening of existing nevi, in addition to eruptive nevi.8-10 The side effects of vemurafenib have been ascribed to a paradoxical upregulation of MAPK in BRAF wild-type cells. This effect has been well documented and demonstrated in vivo.8,10 Perhaps erlotinib has a similar potential to paradoxically upregulate the MAPK pathway, thus stimulating cellular proliferation and survival.
Another tyrosine kinase receptor, c-KIT, is found on the cell membrane of melanocytes along with EGFR.11,12 The c-KIT receptor also activates the MAPK pathway and is critical to the development, migration, and survival of melanocytes.11,13 Stimulation of the c-KIT tyrosine kinase receptor also can induce melanocyte proliferation and melanogenesis.11 The c-KIT receptor is encoded by the KIT gene (KIT proto-oncogene receptor tyrosine kinase). Mutations in this gene are associated with melanocytic disorders. Inherited KIT mutation leading to c-KIT receptor deficiency is associated with piebaldism. Acquired activating KIT mutations increasing c-KIT expression are associated with acral and mucosal melanomas as well as melanomas in chronically sun-damaged skin.13
We hypothesized that erlotinib-induced inhibition of the MAPK pathway could lead to a reactive increase in expression of c-KIT and thus stimulate melanocyte proliferation and pigment production. Similar feedback upregulation of an MAPK pathway stimulating receptor during downstream MAPK inhibition has been demonstrated in colon adenocarcinoma; in this setting, BRAF inhibitors blocking the MAPK pathway leads to upregulation of EGFR.14 In our patient, c-KIT immunostaining revealed a mild to moderate increase in intensity (ie, the darkness of the staining) in nevi and melanomas during treatment with erlotinib compared to nevi biopsied before erlotinib treatment (Figure 2). The increased intensity of c-KIT immunostaining was further confirmed via semiquantitative digital image analysis. Using this method, a darkened nevus biopsied during treatment with erlotinib demonstrated 43.16% of cells (N=31,451) had very strong c-KIT staining, while a nevus biopsied before treatment with erlotinib demonstrated only 3.32% of cells (N=7507) with very strong c-KIT staining. Increased expression of c-KIT, possibly reactive to downstream inhibition the MAPK pathway from erlotinib, could be implicated in our case of eruptive nevi.
In summary, we report a rare case of darkening of existing nevi and development of melanoma in situ during treatment with erlotinib. The patient’s therapeutic timeline and concurrence of other well-documented side effects provided support for erlotinib as the causative agent in our patient. Additional support is provided through reports of other medications affecting the same pathway as erlotinib causing eruptive nevi, darkening of existing nevi, and melanoma in situ.7-10 Through c-KIT immunostaining, we demonstrated that increased expression of c-KIT might be responsible for the changes in nevi in our patient. We, therefore, suggest frequent full-body skin examinations in patients treated with erlotinib to monitor for the possible development of malignant melanomas.
- Santiago F, Goncalo M, Reis J, et al. Adverse cutaneous reactions to epidermal growth factor receptor inhibitors: a study of 14 patients. An Bras Dermatol 2011;86:483-490.
- Lubbe J, Masouye I, Dietrich P. Generalized xerotic dermatitis with neutrophilic spongiosis induced by erlotinib (Tarceva). Dermatology. 2008;216:247-249.
- Dessinioti C, Antoniou C, Katsambas A. Acneiform eruptions. Clin Dermatol. 2014;32:24-34.
- Herbst R, Fukuoka M, Baselga J. Gefitinib—a novel targeted approach to treating cancer. Nat Rev Cancer. 2004;4:979-987.
- Brodell L, Hepper D, Lind A, et al. Histopathology of acneiform eruptions in patients treated with epidermal growth factor receptor inhibitors. J Cutan Pathol. 2013;40:865-870.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol 2013;69:463-472.
- Uhlenhake E, Watson A, Aronson P. Sorafenib induced eruptive melanocytic lesions. Dermatol Online J. 2013;19:181-84.
- Chu E, Wanat K, Miller C, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol 2012;67:1265-1272.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Cohen P, Bedikian A, Kim K. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Longley B, Tyrrell L, Lu S, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312-314.
- Yun W, Bang S, Min K, et al. Epidermal growth factor and epidermal growth factor signaling attenuate laser-induced melanogenesis. Dermatol Surg. 2013;39:1903-1911.
- Swick J, Maize J. Molecular biology of melanoma. J Am Acad Dermatol. 2012;67:1049-1054.
- Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118-122.
- Santiago F, Goncalo M, Reis J, et al. Adverse cutaneous reactions to epidermal growth factor receptor inhibitors: a study of 14 patients. An Bras Dermatol 2011;86:483-490.
- Lubbe J, Masouye I, Dietrich P. Generalized xerotic dermatitis with neutrophilic spongiosis induced by erlotinib (Tarceva). Dermatology. 2008;216:247-249.
- Dessinioti C, Antoniou C, Katsambas A. Acneiform eruptions. Clin Dermatol. 2014;32:24-34.
- Herbst R, Fukuoka M, Baselga J. Gefitinib—a novel targeted approach to treating cancer. Nat Rev Cancer. 2004;4:979-987.
- Brodell L, Hepper D, Lind A, et al. Histopathology of acneiform eruptions in patients treated with epidermal growth factor receptor inhibitors. J Cutan Pathol. 2013;40:865-870.
- Kiyohara Y, Yamazaki N, Kishi A. Erlotinib-related skin toxicities: treatment strategies in patients with metastatic non-small cell lung cancer. J Am Acad Dermatol 2013;69:463-472.
- Uhlenhake E, Watson A, Aronson P. Sorafenib induced eruptive melanocytic lesions. Dermatol Online J. 2013;19:181-84.
- Chu E, Wanat K, Miller C, et al. Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol 2012;67:1265-1272.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Cohen P, Bedikian A, Kim K. Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol. 2013;6:27-37.
- Longley B, Tyrrell L, Lu S, et al. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312-314.
- Yun W, Bang S, Min K, et al. Epidermal growth factor and epidermal growth factor signaling attenuate laser-induced melanogenesis. Dermatol Surg. 2013;39:1903-1911.
- Swick J, Maize J. Molecular biology of melanoma. J Am Acad Dermatol. 2012;67:1049-1054.
- Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature. 2014;508:118-122.
Practice Points
- Cutaneous side effects of erlotinib include acneform eruption, xerosis, paronychia, and pruritus.
- Clinicians should monitor patients for darkening and/or eruptive nevi as well as melanoma during treatment with erlotinib.
Ill-Defined Macule on the Abdomen
The Diagnosis: Microvenular Hemangioma
Microvenular hemangioma is an acquired benign vascular neoplasm that was described by Hunt et al1 in 1991, though Bantel et al2 reported a similar entity termed micropapillary angioma in 1989. Microvenular hemangioma typically presents as a solitary, slowly enlarging, red to violaceous, asymptomatic papule, plaque, or nodule measuring 5 to 20 mm in diameter. It usually is located on the trunk, arms, or legs of young adults without any gender predilection. Microvenular hemangioma is rare.3 The etiology has not been elucidated, though a relationship with hormonal factors such as pregnancy or hormonal contraceptives has been described.2
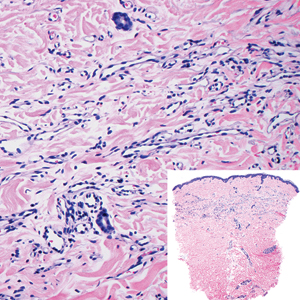
Histopathologically, microvenular hemangioma has a characteristic morphology. It is comprised of a well-circumscribed collection of thin-walled blood vessels with narrow lumens (quiz image).4 The blood vessels tend to infiltrate the superficial and deep dermis and are surrounded by a collagenous or desmoplastic stroma. The endothelial cells are normal in size without atypia, mitotic figures, or pleomorphism. A mild lymphoplasmacytic inflammatory infiltrate sometimes is present. Microvenular hemangioma expresses many vascular markers confirming its endothelial origin, including CD34, CD31, WT1, factor VIII-related antigen, and von Willebrand factor.3 Moreover, WT1 staining suggests the lesion is a vascular proliferative growth, as it usually is negative in vascular malformations due to errors of endothelial development.5 In addition, it lacks expression of podoplanin (D2-40), which also supports a vascular as opposed to a lymphatic origin.4
Cutaneous angiosarcoma is a rare and highly aggressive malignant neoplasm of the vascular endothelium with a predilection for the skin and superficial soft tissue. Clinical presentation is variable, as it can arise sporadically, commonly on the scalp and face of elderly patients, in areas of chronic radiation therapy, or in association with chronic lymphedema (Stewart-Treves syndrome).6 Sporadic neoplasms appear clinically as purpuric macules, plaques, or nodules and are more common in elderly men than women. They are aggressive tumors that tend to recur and metastasize despite aggressive therapy and therefore carry a poor prognosis.7 Histopathologically, well-differentiated tumors are characterized by irregular dissecting vessels lined with crowded inconspicuous endothelial cells (Figure 1). Cutaneous angiosarcoma is poorly circumscribed with marked cytologic atypia, and the vessels can take on a sinusoidal growth pattern.8
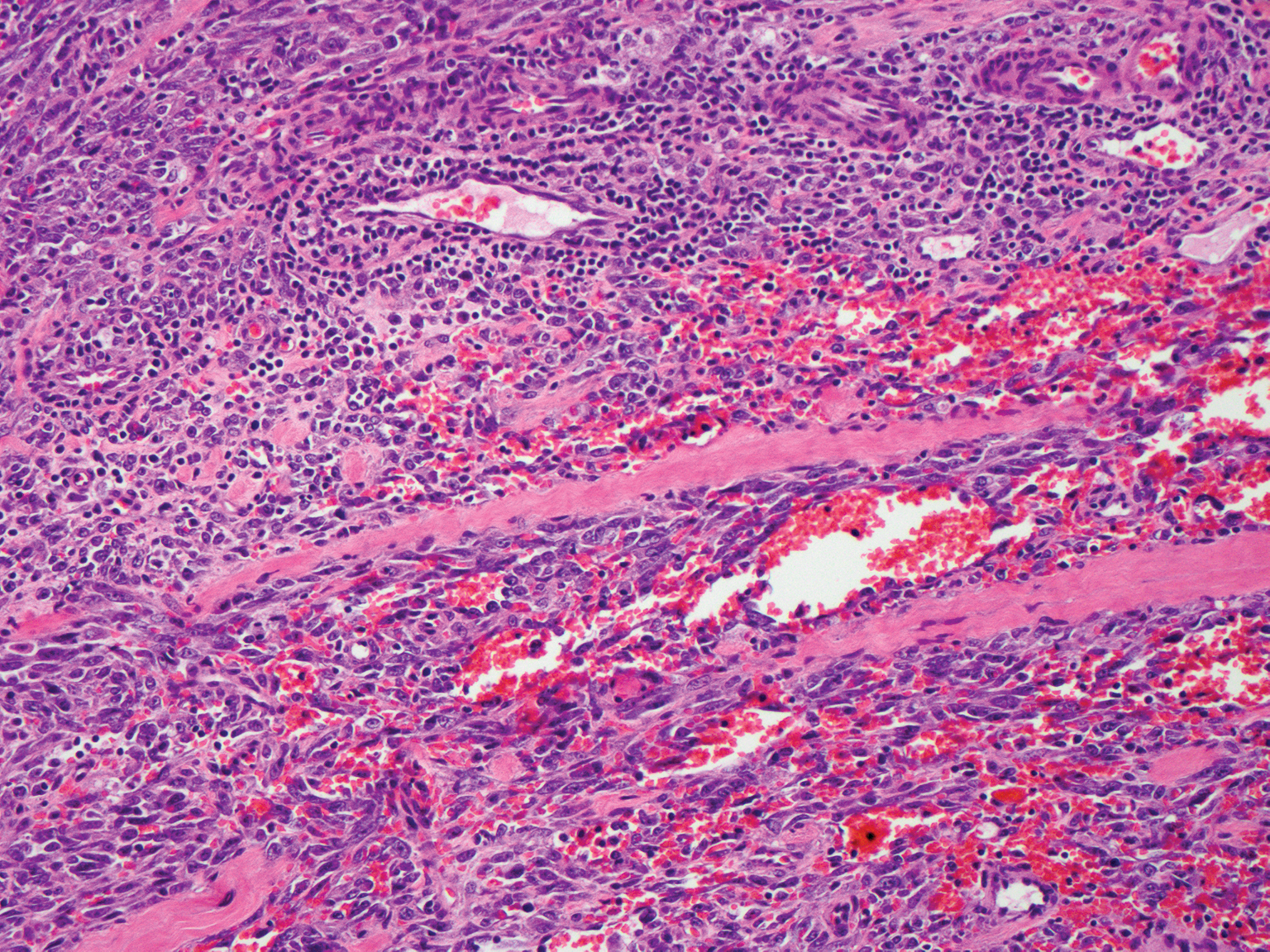
Kaposi sarcoma (KS) is a virally induced lymphoangioproliferative disease, with human herpesvirus 8 as the implicated agent. There are 4 principal clinical variants of KS: epidemic or AIDS-associated KS, endemic or African KS, KS due to iatrogenic immunosuppression, and Mediterranean or classic KS.9 Cutaneous lesions vary from pink patches to dark purple plaques or nodules that commonly occur on the lower legs10; however, the clinical appearance of KS varies depending on the clinical variant and stage. Histopathologically, early lesions of KS exhibit a superficial dermal proliferation of small angulated and jagged vessels that tend to separate into collagen bundles and are surrounded by a lymphoplasmacytic perivascular infiltrate. These native vascular structures often are surrounded by more ectatic neoplastic channels with plump endothelial cells, known as the promontory sign (Figure 2).11 With more advanced lesions, the proliferation of slitlike vessels becomes more cellular and extends deeper into the dermis and subcutis. Although the histopathologic features vary with the stage of the lesion, they do not notably vary between clinical subtypes.
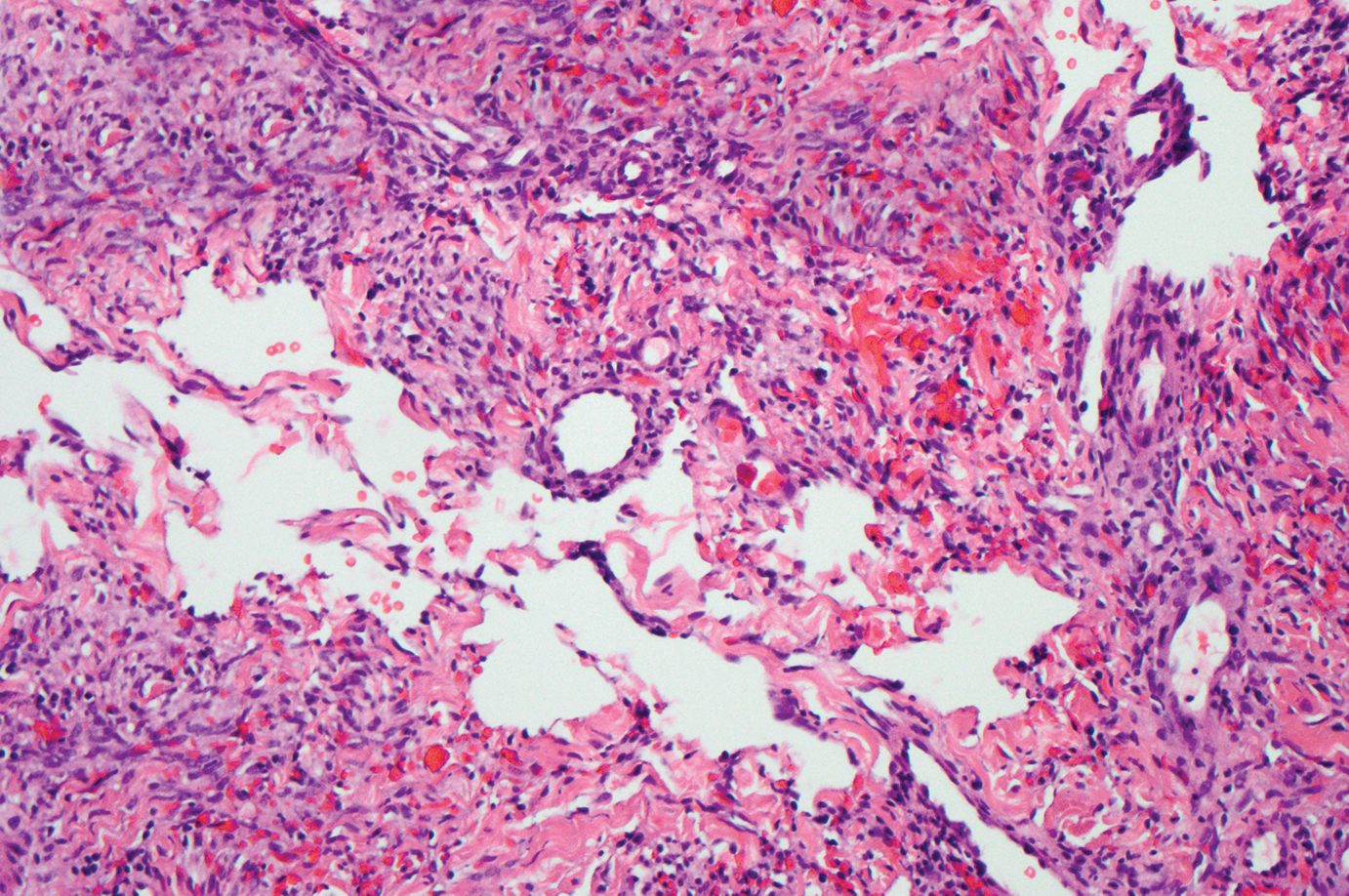
Targetoid hemosiderotic hemangioma, also known as hobnail hemangioma, is a small, benign, vascular tumor that usually affects the trunk, arms, and legs in young to middle-aged adults without a gender predilection. Clinically, it appears as a small, solitary, red to purple papule or macule that typically is surrounded by a pale thin area and a peripheral ecchymotic ring, creating a targetoid appearance, thus the term targetoid hemosiderotic hemangioma.12 Histopathologically, there is a prominent dermal vascular proliferation. In the papillary dermis, there are dilated superficial vessels lined with a single layer of endothelial cells characterized by a plump, hobnail-like appearance that protrude into the lumen (Figure 3). In the deeper dermis, the vascular spaces are angulated and slitlike and appear to dissect through collagen bundles. Hemosiderin, thrombi, extravasated erythrocytes, and a lymphocytic infiltrate also are often seen.13
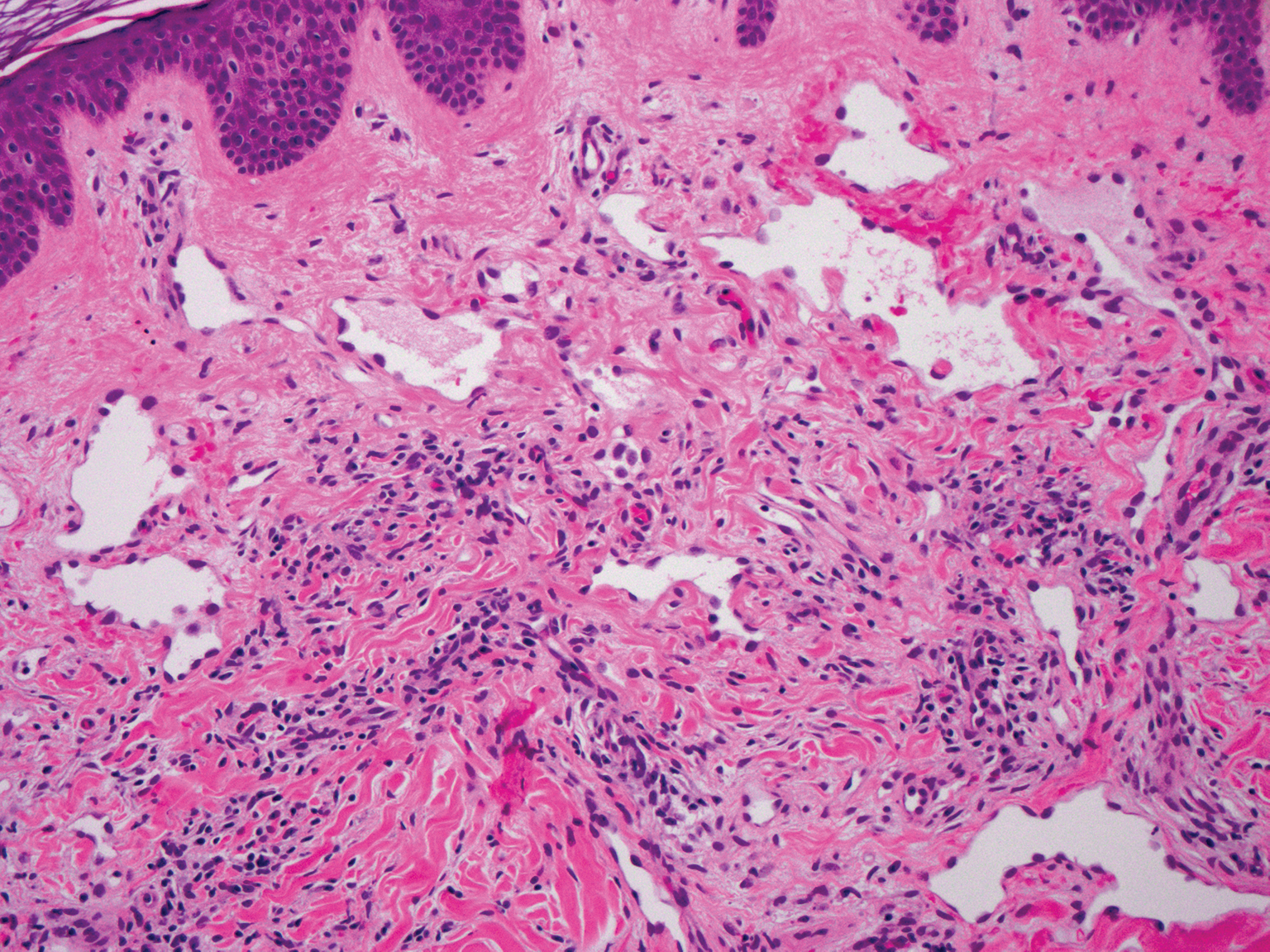
Tufted angioma is a rare benign vascular lesion that usually presents as an acquired lesion in children and young adults, though it may be congenital. It is commonly localized to the skin and subcutaneous tissues. Clinically, the lesions appear as red to purple patches and plaques that typically are located on the neck or trunk. More than 50% of cases present during the first year of life and slowly spread to involve large areas before stabilizing in size.14 Partial spontaneous regression may occur, but complete regression is rare.15 Lesions usually are asymptomatic but may be painful during periods of platelet trapping (Kasabach-Merritt phenomenon), which may develop in congenital cases. Tufted angioma is named for its characteristic histopathologic appearance, which consists of multiple discrete lobules or tufts of tightly packed capillaries in a cannonball-like appearance throughout the dermis and subcutis (Figure 4).14,15

- Hunt SJ, Santa Cruz DJ, Barr RJ. Microvenular hemangioma. J Cutan Pathol. 1991;18:235-240.
- Bantel E, Grosshans E, Ortonne JP. Understanding microcapillary angioma, observations in pregnant patients and in females treated with hormonal contraceptives [in German]. Z Hautkr. 1989;64:1071-1074.
- Mansur AT, Demirci GT, Ozbal Koc E, et al. An unusual lesion on the nose: microvenular hemangioma. Dermatol Pract Concept. 2018;8:7-11.
- Napekoski KM, Fernandez AP, Billings SD. Microvenular hemangioma: a clinicopathologic review of 13 cases. J Cutan Pathol. 2014;41:816-822.
- Trinidade F, Tellechea O, Torrelo A, et al. Wilms tumor 1 expression in vascular neoplasms and vascular malformations. Am J Dermatopathol. 2011;33:569-572.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.
- Morgan M, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Shon W, Billings SD. Cutaneous malignant vascular neoplasms. Clin Lab Med. 2017;37:633-646.
- Régnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol. 2013;68:313-331.
- Tappero JW, Conant MA, Wolfe SF, et al. Kaposi's sarcoma: epidemiology, pathogenesis, histology, clinical spectrum, staging criteria and therapy. J Am Acad Dermatol. 1993;28:371-395.
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Mentzel T, Partanen TA, Kutzner H. Hobnail hemangioma ("targetoid hemosiderotic hemangioma"): clinicopathologic and immunohistochemical analysis of 62 cases. J Cutan Pathol. 1999;26:279-286.
- Morales-Callaghan AM, Martinez-Garcia G, Aragoneses-Fraile H, et al. Targetoid hemosiderotic hemangioma: clinical and dermoscopical findings. J Eur Acad Dermatol Venereol. 2007;21:267-269.
- Kamath GH, Bhat RM, Kumar S. Tufted angioma. Int J Dermatol. 2005;44:1045-1047.
- Prasuna A, Rao P. A tufted angioma. Indian Dermatol Online J. 2015;6:266-268.
The Diagnosis: Microvenular Hemangioma
Microvenular hemangioma is an acquired benign vascular neoplasm that was described by Hunt et al1 in 1991, though Bantel et al2 reported a similar entity termed micropapillary angioma in 1989. Microvenular hemangioma typically presents as a solitary, slowly enlarging, red to violaceous, asymptomatic papule, plaque, or nodule measuring 5 to 20 mm in diameter. It usually is located on the trunk, arms, or legs of young adults without any gender predilection. Microvenular hemangioma is rare.3 The etiology has not been elucidated, though a relationship with hormonal factors such as pregnancy or hormonal contraceptives has been described.2
Histopathologically, microvenular hemangioma has a characteristic morphology. It is comprised of a well-circumscribed collection of thin-walled blood vessels with narrow lumens (quiz image).4 The blood vessels tend to infiltrate the superficial and deep dermis and are surrounded by a collagenous or desmoplastic stroma. The endothelial cells are normal in size without atypia, mitotic figures, or pleomorphism. A mild lymphoplasmacytic inflammatory infiltrate sometimes is present. Microvenular hemangioma expresses many vascular markers confirming its endothelial origin, including CD34, CD31, WT1, factor VIII-related antigen, and von Willebrand factor.3 Moreover, WT1 staining suggests the lesion is a vascular proliferative growth, as it usually is negative in vascular malformations due to errors of endothelial development.5 In addition, it lacks expression of podoplanin (D2-40), which also supports a vascular as opposed to a lymphatic origin.4
Cutaneous angiosarcoma is a rare and highly aggressive malignant neoplasm of the vascular endothelium with a predilection for the skin and superficial soft tissue. Clinical presentation is variable, as it can arise sporadically, commonly on the scalp and face of elderly patients, in areas of chronic radiation therapy, or in association with chronic lymphedema (Stewart-Treves syndrome).6 Sporadic neoplasms appear clinically as purpuric macules, plaques, or nodules and are more common in elderly men than women. They are aggressive tumors that tend to recur and metastasize despite aggressive therapy and therefore carry a poor prognosis.7 Histopathologically, well-differentiated tumors are characterized by irregular dissecting vessels lined with crowded inconspicuous endothelial cells (Figure 1). Cutaneous angiosarcoma is poorly circumscribed with marked cytologic atypia, and the vessels can take on a sinusoidal growth pattern.8
Kaposi sarcoma (KS) is a virally induced lymphoangioproliferative disease, with human herpesvirus 8 as the implicated agent. There are 4 principal clinical variants of KS: epidemic or AIDS-associated KS, endemic or African KS, KS due to iatrogenic immunosuppression, and Mediterranean or classic KS.9 Cutaneous lesions vary from pink patches to dark purple plaques or nodules that commonly occur on the lower legs10; however, the clinical appearance of KS varies depending on the clinical variant and stage. Histopathologically, early lesions of KS exhibit a superficial dermal proliferation of small angulated and jagged vessels that tend to separate into collagen bundles and are surrounded by a lymphoplasmacytic perivascular infiltrate. These native vascular structures often are surrounded by more ectatic neoplastic channels with plump endothelial cells, known as the promontory sign (Figure 2).11 With more advanced lesions, the proliferation of slitlike vessels becomes more cellular and extends deeper into the dermis and subcutis. Although the histopathologic features vary with the stage of the lesion, they do not notably vary between clinical subtypes.
Targetoid hemosiderotic hemangioma, also known as hobnail hemangioma, is a small, benign, vascular tumor that usually affects the trunk, arms, and legs in young to middle-aged adults without a gender predilection. Clinically, it appears as a small, solitary, red to purple papule or macule that typically is surrounded by a pale thin area and a peripheral ecchymotic ring, creating a targetoid appearance, thus the term targetoid hemosiderotic hemangioma.12 Histopathologically, there is a prominent dermal vascular proliferation. In the papillary dermis, there are dilated superficial vessels lined with a single layer of endothelial cells characterized by a plump, hobnail-like appearance that protrude into the lumen (Figure 3). In the deeper dermis, the vascular spaces are angulated and slitlike and appear to dissect through collagen bundles. Hemosiderin, thrombi, extravasated erythrocytes, and a lymphocytic infiltrate also are often seen.13
Tufted angioma is a rare benign vascular lesion that usually presents as an acquired lesion in children and young adults, though it may be congenital. It is commonly localized to the skin and subcutaneous tissues. Clinically, the lesions appear as red to purple patches and plaques that typically are located on the neck or trunk. More than 50% of cases present during the first year of life and slowly spread to involve large areas before stabilizing in size.14 Partial spontaneous regression may occur, but complete regression is rare.15 Lesions usually are asymptomatic but may be painful during periods of platelet trapping (Kasabach-Merritt phenomenon), which may develop in congenital cases. Tufted angioma is named for its characteristic histopathologic appearance, which consists of multiple discrete lobules or tufts of tightly packed capillaries in a cannonball-like appearance throughout the dermis and subcutis (Figure 4).14,15
The Diagnosis: Microvenular Hemangioma
Microvenular hemangioma is an acquired benign vascular neoplasm that was described by Hunt et al1 in 1991, though Bantel et al2 reported a similar entity termed micropapillary angioma in 1989. Microvenular hemangioma typically presents as a solitary, slowly enlarging, red to violaceous, asymptomatic papule, plaque, or nodule measuring 5 to 20 mm in diameter. It usually is located on the trunk, arms, or legs of young adults without any gender predilection. Microvenular hemangioma is rare.3 The etiology has not been elucidated, though a relationship with hormonal factors such as pregnancy or hormonal contraceptives has been described.2
Histopathologically, microvenular hemangioma has a characteristic morphology. It is comprised of a well-circumscribed collection of thin-walled blood vessels with narrow lumens (quiz image).4 The blood vessels tend to infiltrate the superficial and deep dermis and are surrounded by a collagenous or desmoplastic stroma. The endothelial cells are normal in size without atypia, mitotic figures, or pleomorphism. A mild lymphoplasmacytic inflammatory infiltrate sometimes is present. Microvenular hemangioma expresses many vascular markers confirming its endothelial origin, including CD34, CD31, WT1, factor VIII-related antigen, and von Willebrand factor.3 Moreover, WT1 staining suggests the lesion is a vascular proliferative growth, as it usually is negative in vascular malformations due to errors of endothelial development.5 In addition, it lacks expression of podoplanin (D2-40), which also supports a vascular as opposed to a lymphatic origin.4
Cutaneous angiosarcoma is a rare and highly aggressive malignant neoplasm of the vascular endothelium with a predilection for the skin and superficial soft tissue. Clinical presentation is variable, as it can arise sporadically, commonly on the scalp and face of elderly patients, in areas of chronic radiation therapy, or in association with chronic lymphedema (Stewart-Treves syndrome).6 Sporadic neoplasms appear clinically as purpuric macules, plaques, or nodules and are more common in elderly men than women. They are aggressive tumors that tend to recur and metastasize despite aggressive therapy and therefore carry a poor prognosis.7 Histopathologically, well-differentiated tumors are characterized by irregular dissecting vessels lined with crowded inconspicuous endothelial cells (Figure 1). Cutaneous angiosarcoma is poorly circumscribed with marked cytologic atypia, and the vessels can take on a sinusoidal growth pattern.8
Kaposi sarcoma (KS) is a virally induced lymphoangioproliferative disease, with human herpesvirus 8 as the implicated agent. There are 4 principal clinical variants of KS: epidemic or AIDS-associated KS, endemic or African KS, KS due to iatrogenic immunosuppression, and Mediterranean or classic KS.9 Cutaneous lesions vary from pink patches to dark purple plaques or nodules that commonly occur on the lower legs10; however, the clinical appearance of KS varies depending on the clinical variant and stage. Histopathologically, early lesions of KS exhibit a superficial dermal proliferation of small angulated and jagged vessels that tend to separate into collagen bundles and are surrounded by a lymphoplasmacytic perivascular infiltrate. These native vascular structures often are surrounded by more ectatic neoplastic channels with plump endothelial cells, known as the promontory sign (Figure 2).11 With more advanced lesions, the proliferation of slitlike vessels becomes more cellular and extends deeper into the dermis and subcutis. Although the histopathologic features vary with the stage of the lesion, they do not notably vary between clinical subtypes.
Targetoid hemosiderotic hemangioma, also known as hobnail hemangioma, is a small, benign, vascular tumor that usually affects the trunk, arms, and legs in young to middle-aged adults without a gender predilection. Clinically, it appears as a small, solitary, red to purple papule or macule that typically is surrounded by a pale thin area and a peripheral ecchymotic ring, creating a targetoid appearance, thus the term targetoid hemosiderotic hemangioma.12 Histopathologically, there is a prominent dermal vascular proliferation. In the papillary dermis, there are dilated superficial vessels lined with a single layer of endothelial cells characterized by a plump, hobnail-like appearance that protrude into the lumen (Figure 3). In the deeper dermis, the vascular spaces are angulated and slitlike and appear to dissect through collagen bundles. Hemosiderin, thrombi, extravasated erythrocytes, and a lymphocytic infiltrate also are often seen.13
Tufted angioma is a rare benign vascular lesion that usually presents as an acquired lesion in children and young adults, though it may be congenital. It is commonly localized to the skin and subcutaneous tissues. Clinically, the lesions appear as red to purple patches and plaques that typically are located on the neck or trunk. More than 50% of cases present during the first year of life and slowly spread to involve large areas before stabilizing in size.14 Partial spontaneous regression may occur, but complete regression is rare.15 Lesions usually are asymptomatic but may be painful during periods of platelet trapping (Kasabach-Merritt phenomenon), which may develop in congenital cases. Tufted angioma is named for its characteristic histopathologic appearance, which consists of multiple discrete lobules or tufts of tightly packed capillaries in a cannonball-like appearance throughout the dermis and subcutis (Figure 4).14,15
- Hunt SJ, Santa Cruz DJ, Barr RJ. Microvenular hemangioma. J Cutan Pathol. 1991;18:235-240.
- Bantel E, Grosshans E, Ortonne JP. Understanding microcapillary angioma, observations in pregnant patients and in females treated with hormonal contraceptives [in German]. Z Hautkr. 1989;64:1071-1074.
- Mansur AT, Demirci GT, Ozbal Koc E, et al. An unusual lesion on the nose: microvenular hemangioma. Dermatol Pract Concept. 2018;8:7-11.
- Napekoski KM, Fernandez AP, Billings SD. Microvenular hemangioma: a clinicopathologic review of 13 cases. J Cutan Pathol. 2014;41:816-822.
- Trinidade F, Tellechea O, Torrelo A, et al. Wilms tumor 1 expression in vascular neoplasms and vascular malformations. Am J Dermatopathol. 2011;33:569-572.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.
- Morgan M, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Shon W, Billings SD. Cutaneous malignant vascular neoplasms. Clin Lab Med. 2017;37:633-646.
- Régnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol. 2013;68:313-331.
- Tappero JW, Conant MA, Wolfe SF, et al. Kaposi's sarcoma: epidemiology, pathogenesis, histology, clinical spectrum, staging criteria and therapy. J Am Acad Dermatol. 1993;28:371-395.
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Mentzel T, Partanen TA, Kutzner H. Hobnail hemangioma ("targetoid hemosiderotic hemangioma"): clinicopathologic and immunohistochemical analysis of 62 cases. J Cutan Pathol. 1999;26:279-286.
- Morales-Callaghan AM, Martinez-Garcia G, Aragoneses-Fraile H, et al. Targetoid hemosiderotic hemangioma: clinical and dermoscopical findings. J Eur Acad Dermatol Venereol. 2007;21:267-269.
- Kamath GH, Bhat RM, Kumar S. Tufted angioma. Int J Dermatol. 2005;44:1045-1047.
- Prasuna A, Rao P. A tufted angioma. Indian Dermatol Online J. 2015;6:266-268.
- Hunt SJ, Santa Cruz DJ, Barr RJ. Microvenular hemangioma. J Cutan Pathol. 1991;18:235-240.
- Bantel E, Grosshans E, Ortonne JP. Understanding microcapillary angioma, observations in pregnant patients and in females treated with hormonal contraceptives [in German]. Z Hautkr. 1989;64:1071-1074.
- Mansur AT, Demirci GT, Ozbal Koc E, et al. An unusual lesion on the nose: microvenular hemangioma. Dermatol Pract Concept. 2018;8:7-11.
- Napekoski KM, Fernandez AP, Billings SD. Microvenular hemangioma: a clinicopathologic review of 13 cases. J Cutan Pathol. 2014;41:816-822.
- Trinidade F, Tellechea O, Torrelo A, et al. Wilms tumor 1 expression in vascular neoplasms and vascular malformations. Am J Dermatopathol. 2011;33:569-572.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.
- Morgan M, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Shon W, Billings SD. Cutaneous malignant vascular neoplasms. Clin Lab Med. 2017;37:633-646.
- Régnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol. 2013;68:313-331.
- Tappero JW, Conant MA, Wolfe SF, et al. Kaposi's sarcoma: epidemiology, pathogenesis, histology, clinical spectrum, staging criteria and therapy. J Am Acad Dermatol. 1993;28:371-395.
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Mentzel T, Partanen TA, Kutzner H. Hobnail hemangioma ("targetoid hemosiderotic hemangioma"): clinicopathologic and immunohistochemical analysis of 62 cases. J Cutan Pathol. 1999;26:279-286.
- Morales-Callaghan AM, Martinez-Garcia G, Aragoneses-Fraile H, et al. Targetoid hemosiderotic hemangioma: clinical and dermoscopical findings. J Eur Acad Dermatol Venereol. 2007;21:267-269.
- Kamath GH, Bhat RM, Kumar S. Tufted angioma. Int J Dermatol. 2005;44:1045-1047.
- Prasuna A, Rao P. A tufted angioma. Indian Dermatol Online J. 2015;6:266-268.
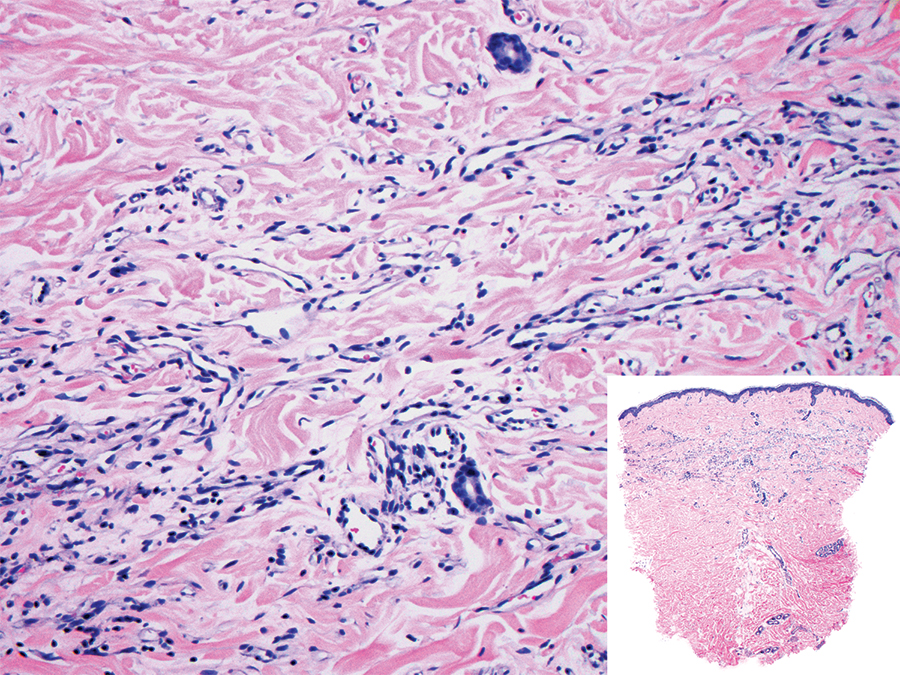
A 38-year-old woman presented with an asymptomatic lesion on the abdomen. On physical examination, there was a 5×2-mm, solitary, ill-defined pink macule on the right side of the abdomen. The patient denied recent change in size or color of the lesion, prior trauma, or a personal or family history of similar lesions. Due to the uncertain diagnostic appearance, a punch biopsy was performed.
Necrobiosis Lipoidica With Superimposed Pyoderma Vegetans
Case Report
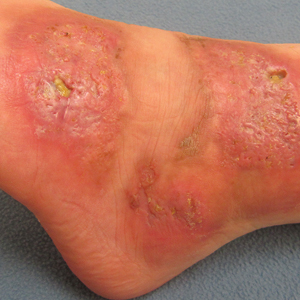
A 26-year-old woman with a medical history of newly diagnosed diabetes mellitus (DM), obesity, and asthma was evaluated as a hospital consultation with a vegetative plaque on the left lateral ankle of 13 months’ duration. The lesion first appeared as a red scaly rash that became purulent. The lesion had been treated with multiple rounds of topical antibiotics, oral antibiotics, topical antifungals, and corticosteroids without resolution. The patient denied pain or any decrease in ankle mobility. Review of systems was otherwise negative.
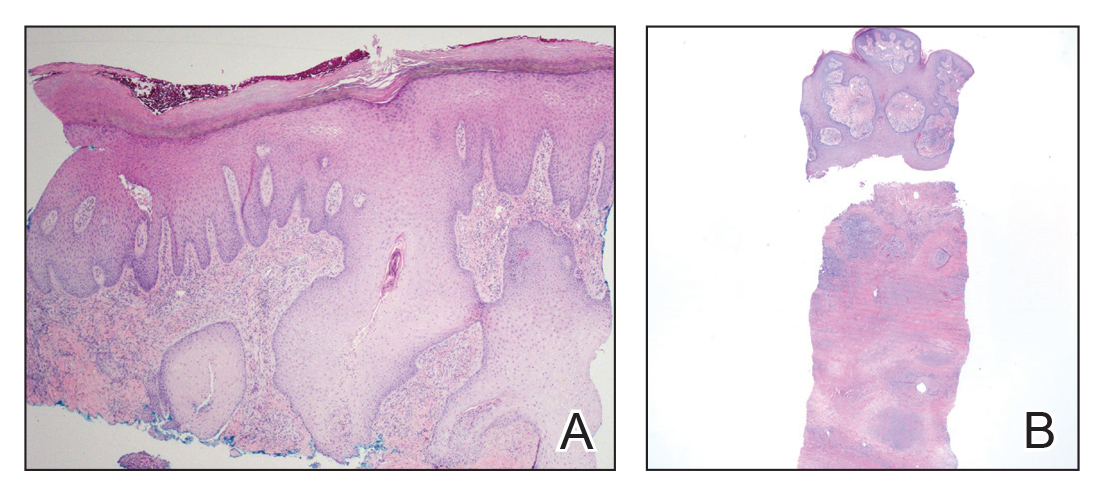
On physical examination, 3 large, pink, scaly, crusted plaques with surrounding erythema were observed (Figure 1A). On palpation, purulent drainage with a foul odor was noted in the area underlying the lesion. Initial punch biopsy demonstrated epidermal hyperplasia with neutrophil-rich sinus tracts consistent with pyoderma vegetans (PV)(Figure 2A). Tissue culture was positive for Staphylococcus aureus and Streptococcus anginosus. Cultures for both fungi and acid-fast bacilli were negative for growth.

The patient was treated with mupirocin ointment 2% and 3 months of cephalexin 250 mg twice daily, which cleared the purulent crust; however, serous drainage, ulceration, and erythema persisted. The patient needed an extended course of antibiotics, which had not been previously administered to clear the purulence. During this treatment regimen, the patient’s DM remained uncontrolled.
A second deeper punch biopsy revealed a layered granulomatous infiltrate with sclerosis throughout the dermis most consistent with necrobiosis lipoidica (NL)(Figure 2B). Direct immunofluorescence biopsy was negative. Once the PV was clear, betamethasone dipropionate ointment 0.05% was initiated to address the residual lesions (Figure 1B).
Physical examination combined with histopathologic findings and staphylococcal- and streptococcal-positive tissue cultures supported a diagnosis of NL with superimposed PV.
Comment
Necrobiosis lipoidica is a chronic granulomatous disease characterized by collagen degeneration, granulomatous formation, and endothelial wall thickening.1 The condition is most commonly seen in association with insulin-dependent DM, though it also has been described in other inflammatory conditions. A case of NL in monozygotic twins has been reported, suggesting a genetic component in nondiabetic patients with NL.2 Necrobiosis lipoidica affects females more often than males.
The pathogenesis of NL is not well understood but likely involves secondary microangiopathy because of glycoprotein deposition in vessel walls, leading to vascular thickening. Histopathology reveals palisading and necrobiotic granulomas comprising large confluent areas of necrobiosis throughout the dermis, giving a layered appearance.3
Clinically, NL presents with asymptomatic, well-circumscribed, violaceous papules and nodules that coalesce into plaques on the lower extremities, face, or trunk. The plaques have a central red-brown hue that progressively becomes more yellow and atrophic. The lesions can become eroded and ulcerated if left untreated.1
Clinical diagnosis of NL can be challenging due to the similar clinical findings of other granulomatous lesions, such as granuloma annulare and cutaneous sarcoidosis. As reported by Pellicano and colleagues,4 dermoscopy has proved to be an excellent tool for differentiating these granulomatous skin lesions. Necrobiosis lipoidica demonstrates elongated serpentine telangiectases overlying a white structureless background, whereas granuloma annulare reveals orange-red structureless peripheral borders.5
Treatment of NL is difficult; patients often are refractory. Tight control of blood glucose alone has not been proven to cure NL. The mainstay of treatment is topical and intralesional corticosteroids at the active borders of the lesions. Tumor necrosis factor α inhibitors have shown some success, though recurrence has been reported.6 Other treatments, such as topical tretinoin and topical tacrolimus, may be of some benefit for atrophic NL lesions. Studies also have shown that skin grafting can be of surgical benefit in ulcerative NL with a low rate of recurrence.6 Control and management of DM plus lifestyle modifications may play a role in decreasing the severity of NL.7 Topical psoralen plus UVA light therapy and other experimental treatments, such as antiplatelet medications,8 also have been utilized.
The case of NL presented here was complicated by a superimposed suppurative infection consistent with PV, a rare chronic bacterial infection of the skin that presents with vegetative plaques. Pyoderma vegetans is most commonly observed in patients with underlying immunosuppression, likely secondary to DM in this case. Pyoderma vegetans is most often caused by S aureus and β-hemolytic streptococci. The clinical presentation of PV reveals verrucous vegetative plaques with pustules and abscesses. The borders of the lesions may be elevated and have a granulomatous appearance, thus complicating clinical diagnosis. There often is foul-smelling, purulent discharge within the plaques.9
Histopathology reveals pseudoepitheliomatous hyperplasia with abscesses and sinus tracts. An acute or chronic granulomatous inflammatory infiltrate may be observed. Basophilic fungus like granules are not seen within specimens of PV, which helps differentiate the disease from botryomycosis.10
There is no standardized treatment of PV; topical and systemic antibiotics are mainstays.10 One reported case of PV responded well to acitretin.9 Our patient responded well to 3 months of oral antibiotic therapy, followed by topical corticosteroids.
1. Reid SD, Ladizinski B, Lee K, et al. Update on necrobiosis lipoidica: a review of etiology, diagnosis, and treatment options. J Am Acad Dermatol. 2013;69:783-791.
2. Shimanovich I, Erdmann H, Grabbe J, et al. Necrobiosis lipoidica in monozygotic twins. Arch Dermatol. 2008;144:119-120.
3. Ghazarian D, Al Habeeb A. Necrobiotic lesions of the skin: an approach and review of the literature. Diagn Histopathol. 2009;15:186-194.
4. Pellicano R, Caldarola G, Filabozzi P, et al. Dermoscopy of necrobiosis lipoidica and granuloma annulare. Dermatology. 2013;226:319-323.
5. Bakos RM, Cartell A, Bakos L. Dermatoscopy of early-onset necrobiosis lipoidica. J Am Acad Dermatol. 2012;66:143-144.
6. Feily A, Mehraban S. Treatment modalities of necrobiosis lipoidica: a concise systematic review. Dermatol Reports. 2015;7:5749.
7. Yigit S, Estrada E. Recurrent necrobiosis lipoidica diabeticorum associated with venous insufficiency in an adolescent with poorly controlled type 2 diabetes mellitus. J Pediatr. 2002;141:280-282.
8. Heng MC, Song MK, Heng MK. Healing of necrobiotic ulcers with antiplatelet therapy. Correlation with plasma thromboxane levels. Int J Dermatol. 1989;28:195-197.
9. Lee Y, Jung SW, Sim HS, et al. Blastomycosis-like pyoderma with good response to acitretin. Ann Dermatol. 2011;23:365-368.
10. Marschalko M, Preisz K, Harsing J, et al. Pyoderma vegetans. report on a case and review of data on pyoderma vegetans and cutaneous botryomycosis. Acta Dermatovenerol. 1995;95:55-59.
Case Report
A 26-year-old woman with a medical history of newly diagnosed diabetes mellitus (DM), obesity, and asthma was evaluated as a hospital consultation with a vegetative plaque on the left lateral ankle of 13 months’ duration. The lesion first appeared as a red scaly rash that became purulent. The lesion had been treated with multiple rounds of topical antibiotics, oral antibiotics, topical antifungals, and corticosteroids without resolution. The patient denied pain or any decrease in ankle mobility. Review of systems was otherwise negative.
On physical examination, 3 large, pink, scaly, crusted plaques with surrounding erythema were observed (Figure 1A). On palpation, purulent drainage with a foul odor was noted in the area underlying the lesion. Initial punch biopsy demonstrated epidermal hyperplasia with neutrophil-rich sinus tracts consistent with pyoderma vegetans (PV)(Figure 2A). Tissue culture was positive for Staphylococcus aureus and Streptococcus anginosus. Cultures for both fungi and acid-fast bacilli were negative for growth.
The patient was treated with mupirocin ointment 2% and 3 months of cephalexin 250 mg twice daily, which cleared the purulent crust; however, serous drainage, ulceration, and erythema persisted. The patient needed an extended course of antibiotics, which had not been previously administered to clear the purulence. During this treatment regimen, the patient’s DM remained uncontrolled.
A second deeper punch biopsy revealed a layered granulomatous infiltrate with sclerosis throughout the dermis most consistent with necrobiosis lipoidica (NL)(Figure 2B). Direct immunofluorescence biopsy was negative. Once the PV was clear, betamethasone dipropionate ointment 0.05% was initiated to address the residual lesions (Figure 1B).
Physical examination combined with histopathologic findings and staphylococcal- and streptococcal-positive tissue cultures supported a diagnosis of NL with superimposed PV.
Comment
Necrobiosis lipoidica is a chronic granulomatous disease characterized by collagen degeneration, granulomatous formation, and endothelial wall thickening.1 The condition is most commonly seen in association with insulin-dependent DM, though it also has been described in other inflammatory conditions. A case of NL in monozygotic twins has been reported, suggesting a genetic component in nondiabetic patients with NL.2 Necrobiosis lipoidica affects females more often than males.
The pathogenesis of NL is not well understood but likely involves secondary microangiopathy because of glycoprotein deposition in vessel walls, leading to vascular thickening. Histopathology reveals palisading and necrobiotic granulomas comprising large confluent areas of necrobiosis throughout the dermis, giving a layered appearance.3
Clinically, NL presents with asymptomatic, well-circumscribed, violaceous papules and nodules that coalesce into plaques on the lower extremities, face, or trunk. The plaques have a central red-brown hue that progressively becomes more yellow and atrophic. The lesions can become eroded and ulcerated if left untreated.1
Clinical diagnosis of NL can be challenging due to the similar clinical findings of other granulomatous lesions, such as granuloma annulare and cutaneous sarcoidosis. As reported by Pellicano and colleagues,4 dermoscopy has proved to be an excellent tool for differentiating these granulomatous skin lesions. Necrobiosis lipoidica demonstrates elongated serpentine telangiectases overlying a white structureless background, whereas granuloma annulare reveals orange-red structureless peripheral borders.5
Treatment of NL is difficult; patients often are refractory. Tight control of blood glucose alone has not been proven to cure NL. The mainstay of treatment is topical and intralesional corticosteroids at the active borders of the lesions. Tumor necrosis factor α inhibitors have shown some success, though recurrence has been reported.6 Other treatments, such as topical tretinoin and topical tacrolimus, may be of some benefit for atrophic NL lesions. Studies also have shown that skin grafting can be of surgical benefit in ulcerative NL with a low rate of recurrence.6 Control and management of DM plus lifestyle modifications may play a role in decreasing the severity of NL.7 Topical psoralen plus UVA light therapy and other experimental treatments, such as antiplatelet medications,8 also have been utilized.
The case of NL presented here was complicated by a superimposed suppurative infection consistent with PV, a rare chronic bacterial infection of the skin that presents with vegetative plaques. Pyoderma vegetans is most commonly observed in patients with underlying immunosuppression, likely secondary to DM in this case. Pyoderma vegetans is most often caused by S aureus and β-hemolytic streptococci. The clinical presentation of PV reveals verrucous vegetative plaques with pustules and abscesses. The borders of the lesions may be elevated and have a granulomatous appearance, thus complicating clinical diagnosis. There often is foul-smelling, purulent discharge within the plaques.9
Histopathology reveals pseudoepitheliomatous hyperplasia with abscesses and sinus tracts. An acute or chronic granulomatous inflammatory infiltrate may be observed. Basophilic fungus like granules are not seen within specimens of PV, which helps differentiate the disease from botryomycosis.10
There is no standardized treatment of PV; topical and systemic antibiotics are mainstays.10 One reported case of PV responded well to acitretin.9 Our patient responded well to 3 months of oral antibiotic therapy, followed by topical corticosteroids.
Case Report
A 26-year-old woman with a medical history of newly diagnosed diabetes mellitus (DM), obesity, and asthma was evaluated as a hospital consultation with a vegetative plaque on the left lateral ankle of 13 months’ duration. The lesion first appeared as a red scaly rash that became purulent. The lesion had been treated with multiple rounds of topical antibiotics, oral antibiotics, topical antifungals, and corticosteroids without resolution. The patient denied pain or any decrease in ankle mobility. Review of systems was otherwise negative.
On physical examination, 3 large, pink, scaly, crusted plaques with surrounding erythema were observed (Figure 1A). On palpation, purulent drainage with a foul odor was noted in the area underlying the lesion. Initial punch biopsy demonstrated epidermal hyperplasia with neutrophil-rich sinus tracts consistent with pyoderma vegetans (PV)(Figure 2A). Tissue culture was positive for Staphylococcus aureus and Streptococcus anginosus. Cultures for both fungi and acid-fast bacilli were negative for growth.
The patient was treated with mupirocin ointment 2% and 3 months of cephalexin 250 mg twice daily, which cleared the purulent crust; however, serous drainage, ulceration, and erythema persisted. The patient needed an extended course of antibiotics, which had not been previously administered to clear the purulence. During this treatment regimen, the patient’s DM remained uncontrolled.
A second deeper punch biopsy revealed a layered granulomatous infiltrate with sclerosis throughout the dermis most consistent with necrobiosis lipoidica (NL)(Figure 2B). Direct immunofluorescence biopsy was negative. Once the PV was clear, betamethasone dipropionate ointment 0.05% was initiated to address the residual lesions (Figure 1B).
Physical examination combined with histopathologic findings and staphylococcal- and streptococcal-positive tissue cultures supported a diagnosis of NL with superimposed PV.
Comment
Necrobiosis lipoidica is a chronic granulomatous disease characterized by collagen degeneration, granulomatous formation, and endothelial wall thickening.1 The condition is most commonly seen in association with insulin-dependent DM, though it also has been described in other inflammatory conditions. A case of NL in monozygotic twins has been reported, suggesting a genetic component in nondiabetic patients with NL.2 Necrobiosis lipoidica affects females more often than males.
The pathogenesis of NL is not well understood but likely involves secondary microangiopathy because of glycoprotein deposition in vessel walls, leading to vascular thickening. Histopathology reveals palisading and necrobiotic granulomas comprising large confluent areas of necrobiosis throughout the dermis, giving a layered appearance.3
Clinically, NL presents with asymptomatic, well-circumscribed, violaceous papules and nodules that coalesce into plaques on the lower extremities, face, or trunk. The plaques have a central red-brown hue that progressively becomes more yellow and atrophic. The lesions can become eroded and ulcerated if left untreated.1
Clinical diagnosis of NL can be challenging due to the similar clinical findings of other granulomatous lesions, such as granuloma annulare and cutaneous sarcoidosis. As reported by Pellicano and colleagues,4 dermoscopy has proved to be an excellent tool for differentiating these granulomatous skin lesions. Necrobiosis lipoidica demonstrates elongated serpentine telangiectases overlying a white structureless background, whereas granuloma annulare reveals orange-red structureless peripheral borders.5
Treatment of NL is difficult; patients often are refractory. Tight control of blood glucose alone has not been proven to cure NL. The mainstay of treatment is topical and intralesional corticosteroids at the active borders of the lesions. Tumor necrosis factor α inhibitors have shown some success, though recurrence has been reported.6 Other treatments, such as topical tretinoin and topical tacrolimus, may be of some benefit for atrophic NL lesions. Studies also have shown that skin grafting can be of surgical benefit in ulcerative NL with a low rate of recurrence.6 Control and management of DM plus lifestyle modifications may play a role in decreasing the severity of NL.7 Topical psoralen plus UVA light therapy and other experimental treatments, such as antiplatelet medications,8 also have been utilized.
The case of NL presented here was complicated by a superimposed suppurative infection consistent with PV, a rare chronic bacterial infection of the skin that presents with vegetative plaques. Pyoderma vegetans is most commonly observed in patients with underlying immunosuppression, likely secondary to DM in this case. Pyoderma vegetans is most often caused by S aureus and β-hemolytic streptococci. The clinical presentation of PV reveals verrucous vegetative plaques with pustules and abscesses. The borders of the lesions may be elevated and have a granulomatous appearance, thus complicating clinical diagnosis. There often is foul-smelling, purulent discharge within the plaques.9
Histopathology reveals pseudoepitheliomatous hyperplasia with abscesses and sinus tracts. An acute or chronic granulomatous inflammatory infiltrate may be observed. Basophilic fungus like granules are not seen within specimens of PV, which helps differentiate the disease from botryomycosis.10
There is no standardized treatment of PV; topical and systemic antibiotics are mainstays.10 One reported case of PV responded well to acitretin.9 Our patient responded well to 3 months of oral antibiotic therapy, followed by topical corticosteroids.
1. Reid SD, Ladizinski B, Lee K, et al. Update on necrobiosis lipoidica: a review of etiology, diagnosis, and treatment options. J Am Acad Dermatol. 2013;69:783-791.
2. Shimanovich I, Erdmann H, Grabbe J, et al. Necrobiosis lipoidica in monozygotic twins. Arch Dermatol. 2008;144:119-120.
3. Ghazarian D, Al Habeeb A. Necrobiotic lesions of the skin: an approach and review of the literature. Diagn Histopathol. 2009;15:186-194.
4. Pellicano R, Caldarola G, Filabozzi P, et al. Dermoscopy of necrobiosis lipoidica and granuloma annulare. Dermatology. 2013;226:319-323.
5. Bakos RM, Cartell A, Bakos L. Dermatoscopy of early-onset necrobiosis lipoidica. J Am Acad Dermatol. 2012;66:143-144.
6. Feily A, Mehraban S. Treatment modalities of necrobiosis lipoidica: a concise systematic review. Dermatol Reports. 2015;7:5749.
7. Yigit S, Estrada E. Recurrent necrobiosis lipoidica diabeticorum associated with venous insufficiency in an adolescent with poorly controlled type 2 diabetes mellitus. J Pediatr. 2002;141:280-282.
8. Heng MC, Song MK, Heng MK. Healing of necrobiotic ulcers with antiplatelet therapy. Correlation with plasma thromboxane levels. Int J Dermatol. 1989;28:195-197.
9. Lee Y, Jung SW, Sim HS, et al. Blastomycosis-like pyoderma with good response to acitretin. Ann Dermatol. 2011;23:365-368.
10. Marschalko M, Preisz K, Harsing J, et al. Pyoderma vegetans. report on a case and review of data on pyoderma vegetans and cutaneous botryomycosis. Acta Dermatovenerol. 1995;95:55-59.
1. Reid SD, Ladizinski B, Lee K, et al. Update on necrobiosis lipoidica: a review of etiology, diagnosis, and treatment options. J Am Acad Dermatol. 2013;69:783-791.
2. Shimanovich I, Erdmann H, Grabbe J, et al. Necrobiosis lipoidica in monozygotic twins. Arch Dermatol. 2008;144:119-120.
3. Ghazarian D, Al Habeeb A. Necrobiotic lesions of the skin: an approach and review of the literature. Diagn Histopathol. 2009;15:186-194.
4. Pellicano R, Caldarola G, Filabozzi P, et al. Dermoscopy of necrobiosis lipoidica and granuloma annulare. Dermatology. 2013;226:319-323.
5. Bakos RM, Cartell A, Bakos L. Dermatoscopy of early-onset necrobiosis lipoidica. J Am Acad Dermatol. 2012;66:143-144.
6. Feily A, Mehraban S. Treatment modalities of necrobiosis lipoidica: a concise systematic review. Dermatol Reports. 2015;7:5749.
7. Yigit S, Estrada E. Recurrent necrobiosis lipoidica diabeticorum associated with venous insufficiency in an adolescent with poorly controlled type 2 diabetes mellitus. J Pediatr. 2002;141:280-282.
8. Heng MC, Song MK, Heng MK. Healing of necrobiotic ulcers with antiplatelet therapy. Correlation with plasma thromboxane levels. Int J Dermatol. 1989;28:195-197.
9. Lee Y, Jung SW, Sim HS, et al. Blastomycosis-like pyoderma with good response to acitretin. Ann Dermatol. 2011;23:365-368.
10. Marschalko M, Preisz K, Harsing J, et al. Pyoderma vegetans. report on a case and review of data on pyoderma vegetans and cutaneous botryomycosis. Acta Dermatovenerol. 1995;95:55-59.
Practice Points
- Necrobiosis lipoidica (NL), a chronic granulomatous disease characterized by collagen degeneration, granulomatous formation, and endothelial-wall thickening, is most often seen in association with insulin-dependent diabetes mellitus (DM).
- Asymptomatic, well-circumscribed, violaceous papules and nodules coalesce into plaques on the lower extremities, face, or trunk in NL.
- Treatment mainstay is topical and intralesional corticosteroids at active borders of lesions. Other treatments used with some success include tumor necrosis factor 11α inhibitors, topical tretinoin, topical tacrolimus, and skin grafting. Control and management of DM can be helpful.
Muckle-Wells Syndrome in the Setting of Basal Cell Nevus Syndrome
Muckle-Wells syndrome (MWS) was first described in 1962 and is part of a broad category of hereditary periodic fever syndromes that include the autoinflammatory syndromes and the cryopyrin-associated periodic syndromes (CAPSs). Unlike autoimmune diseases, autoinflammatory syndromes are not associated with antigen-specific T-cell responses or high titers of autoantibodies but are related to disorders of the innate immune system. Basal cell nevus syndrome (BCNS), or Gorlin syndrome, is a rare genodermatosis inherited in an autosomal-dominant fashion that is characterized by a broad range of anomalies. Most notable is the early and strong predisposition to develop several to hundreds of basal cell carcinomas (BCCs). Classic clinical features of MWS and a thorough history and physical examination can assist in the diagnosis of this rare entity.
Case Report
A 35-year-old woman with a history of BCNS, which had been diagnosed at 24 years of age based on the presence of more than 2 BCCs and a family history of BCNS in her mother, presented with intermittent pruritic urticaria on the chest and back, episodic fevers, associated joint pain and swelling that worsened several hours after exercise, headache, conjunctivitis, blurred vision, and severe debilitating fatigue that had been present since childhood. The symptoms had progressively worsened with age and symptom-free intervals became shorter. She was diagnosed by her rheumatologist with biopsy-proven MWS and a positive NLRP3 (NLR family pyrin domain containing 3) gene mutation at 29 years of age. She was treated unsuccessfully with prednisone and antihistamines and entered a trial with anakinra. She showed improvement for 2 weeks but developed severe swelling and erythema at the injection sites at week 3, along with large leathery patches on the legs and difficulty ambulating.
The patient subsequently underwent excision of her BCCs and reported each site became erythematous, edematous, warm, and painful 6 hours after excision, which lasted for hours to days (Figures 1–3). After the first excision on the right forearm, she was seen in the emergency department, started on intravenous antibiotics and prednisone, and kept overnight in the hospital. She was discharged the following day and the edema in the right forearm subsided over several days. Bacterial culture and laboratory evaluation for infection were negative after the first excision on the right forearm. Because of the symptoms she experienced following this excision, she was referred to the plastic surgery department for excision followed by postoperative monitoring in the hospital. The patient continued to undergo excisions for BCCs and developed more severe symptoms including erythema, edema, warmth, and tenderness at the surrounding sites. Once again, the excision sites were cultured and laboratory work to rule out infection was ordered with a negative result. After several excisions and subsequent clinical findings, the patients’ symptoms were deemed consistent with MWS and not a result of infectious etiology. A diagnosis of MWS and BCNS with exacerbation of MWS with surgical procedures was made.

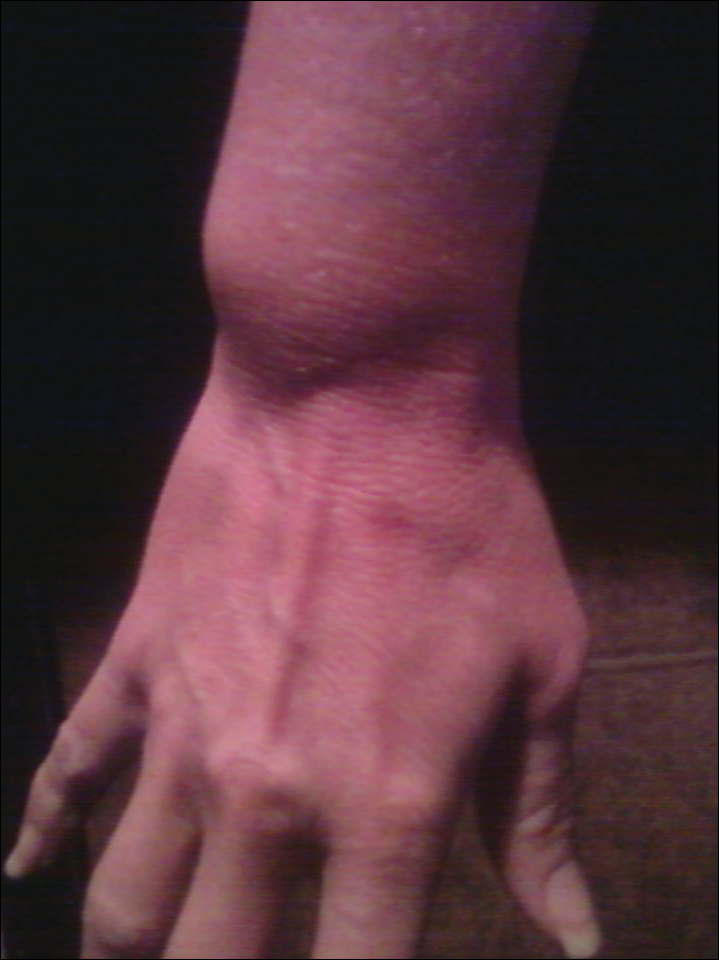
The patient has continued therapy with rilonacept for MWS, which is managed by her rheumatologist. She has tolerated rilonacept without adverse effects and has experienced a reduction in symptoms that has enhanced her quality of life and allows for further treatment of her BCNS. Her dermatologist (J.W.L.) has been treating her BCCs with vismodegib, but treatment has been sporadic due to muscle cramping after 7 days of therapy. She reported subjective improvement to her dermatologist and has tried alternating 7 days on and 7 days off vismodegib. The muscle cramping still has limited her treatment with this regimen, and she is currently on a trial of 3 days on, 4 days off per week.
Comment
Classification and Clinical Presentation
The hereditary periodic fever syndromes include the autoinflammatory syndromes and the CAPSs. The autoinflammatory syndromes include familial Mediterranean fever, hyperimmunoglobulinemia D with periodic fever syndrome, and tumor necrosis factor receptor–associated periodic syndrome. The CAPSs are similar but distinct and include familial cold autoinflammatory syndrome, neonatal-onset multisystem inflammatory disease (also known as chronic infantile neurologic cutaneous and articular syndrome, or cutaneous articular syndrome) and MWS.1,2
Cryopyrin-associated periodic syndromes are rare inherited diseases that result from mutations in the NLRP3 gene. There is a gain-of-function mutation on the NLRP3 gene located on the long arm of chromosome 1 at position 44, which codes for cryopyrin. An NLRP3 gene mutation causes cryopyrin to become hyperactive, leading to the formation of an inflammasome, which is a group of cryopyrin molecules. Inflammasomes, along with other proteins, activate caspase 1 to produce excess IL-1β, leading to persistent inflammatory symptoms.3 IL-1β is one of the key mediators of the body’s response to microbial invasion, inflammation, immunologic reactions, and tissue injury. It affects a large range of cells and organs. Although IL-1β production is critical for the control of pathogenic infections, excessive cytokine production is harmful to the host and can even be fatal.3,4
Cryopyrin-associated periodic syndromes encompass a disease continuum. The 3 distinct entities share many overlapping features as well as unique and distinguishing characteristics. Familial cold autoinflammatory syndrome is the mildest phenotype and is inherited in an autosomal-dominant fashion. It is characterized by a chronic urticarial eruption that starts early in infancy or childhood. The distribution of the cutaneous eruption is widespread and favors the arms and legs over the face and trunk. A low-grade fever often is seen along with musculoskeletal concerns of arthralgia and pain. Other commonly reported symptoms include conjunctivitis, myalgia, fatigue, and headache. Neurologic symptoms can include headaches. Symptoms usually begin 1 to 2 hours after cold exposure and last less than 24 hours.5-8
Neonatal-onset multisystem inflammatory disease is the most severe phenotype and occurs sporadically. Continuous symptoms and flares are characteristic and the length of the flare can vary from minutes to days. The cutaneous eruption favors the face, trunk, arms, and legs, and varies in intensity, beginning in infancy or childhood. Fever may be intermittent, mild, or absent. Rheumatologic manifestations include arthralgia and swelling, with approximately one-third of patients experiencing severe disabling arthropathy that causes gross joint deformity. Ocular findings include conjunctivitis, uveitis, papilledema, and even blindness. Neurologic sequelae include headaches, sensorineural hearing loss, and aseptic meningitis. Amyloidosis has been seen as a late complication.5,8
Muckle-Wells syndrome is a rare hereditary inflammatory disorder. It has no ethnic predisposition and is mostly inherited in an autosomal-dominant fashion. Classically, the condition is characterized by recurrent urticaria beginning at birth with intermittent episodic fever and malaise. The eruption has a predilection for the face, trunk, arms, and legs, which is similar to neonatal-onset multisystem inflammatory disease. Associated myalgia and arthralgia are common as well as ocular findings of conjunctivitis and episcleritis. Neurologic manifestations include headache and progressive sensorineural hearing loss in 60% to 70% of patients.6 Abdominal pain may be seen along with rare serositis in MWS but is rare in the other CAPSs. Amyloidosis caused by chronic inflammation is the most serious complication of MWS and is seen in approximately one-third of patients, manifesting as proteinuria followed by renal impairment. Symptoms of MWS may occur daily but vary individually, are broad in intensity and duration, and can last 1 to 2 days before resolving spontaneously. The symptoms can result from metabolic stressors including cold, stress, and exercise, as well as microbial pathogens. Leukocytosis and increased acute-phase reactants are observed during episodes of inflammation.4,6,8
Histopathology
Mild phenotypic variability exists between individuals, and many of the symptoms overlap in CAPSs. Although CAPSs display several distinguishing clinical characteristics, interestingly they share the same histopathological features regardless of the syndrome. The typical histopathological finding is a dermal neutrophilic infiltrate that tends to be perivascular and also may be perieccrine. Vasodilation and dermal edema also may be seen. These histopathological findings contrast with the typical lymphocytic and eosinophilic infiltrate seen in classic urticaria. Similar histopathologic findings have been seen in other neutrophilic urticarial dermatoses such as Schnitzler syndrome.4,6
Differential
The differential diagnoses for CAPSs include Schnitzler syndrome, cold urticaria, systemic-onset juvenile idiopathic arthritis/adult-onset Still disease, and deficiency in IL-1ra. It is important to consider these differential diagnoses for management and treatment options.
Management
The discovery of the NLRP3 gene mutation as well as an understanding of IL-1 biology has led to targeted therapy for these syndromes. Cryopyrin-associated periodic syndromes are mediated by IL-1β with an in vivo rate 5 times higher than in healthy patients.4 The blockade of IL-1β results in complete resolution of symptoms.
In the last several years, anakinra, rilonacept, and canakinumab have shown efficacy in targeting IL-1β as receptor antagonists. Anakinra is a short-acting recombinant IL-1ra with a half-life of 4 to 6 hours. This short half-life requires daily injections and the most common adverse events included injection-site reaction and upper respiratory tract infection.2,4 Rilonacept is a dimeric fusion protein that contains binding regions for the type 1 receptor and the IL-1 receptor accessory protein and is fused to the fragment, crystallizable (Fc) portion of human IgG1. Rilonacept is long acting with a circulating half-life of 8.6 days and offers patients ease of dosing with weekly subcutaneous injections. Rilonacept generally is well tolerated, with the most frequent adverse effects being injection-site reaction, upper respiratory tract infection, headache, arthralgia, and diarrhea.2,7
The newest of the treatments for patients with CAPS is canakinumab. It is a fully human IL-1β monoclonal antibody that is specific for IL-1β and not other members of the IL-1 family. It has a mean half-life of 26 days and is dosed subcutaneously once every 8 weeks. The most common adverse effects include nasopharyngitis, rhinitis, nausea, diarrhea, and vertigo.4 In one study, most patients did not report injection-site reactions.7 Studies also are underway on VX-765, a caspace-1 targeted therapy that acts upstream in the IL-1β pathway. Treatment with anakinra, rilonacept, and canakinumab generally offers rapid and sustained remission in the majority of MWS patients and helps prevent the development of systemic amyloidosis and lessens the potential for end organ damage.2,7
MWS and BCNS
Our patient had an unusual presentation of MWS complicated by BCNS, another rare autosomal-dominant inherited genodermatosis. In an extensive review of PubMed articles indexed for MEDLINE using the search terms Muckle-Wells syndrome and basal cell nevus syndrome, no association was identified between MWS and BCNS. Basal cell nevus syndrome is linked to PTCH1 (patched 1) gene mutation with an incidence of 1:150,000 in the United States and Europe and is characterized by a broad range of anomalies including skeletal abnormalities, ectopic calcification, odontogenic keratocysts, facial dysmorphism with macrocephaly, palmoplantar pits, and numerous tumors. Most notable is the early and strong predisposition to develop several to hundreds of BCCs.9
Conclusion
Muckle-Wells syndrome may go undiagnosed for many years or may be misdiagnosed as refractory urticaria, as in our patient. It is important to include periodic fever syndromes in the differential diagnosis of refractory urticaria with episodic fever to diagnose these cases of MWS earlier.
- Kagami S, Saeki H, Kuwano Y, et al. A probable case of Muckle-Wells syndrome. J Dermatol. 2006;2:118-121.
- Kanazawa N, Furukawa F. Autoinflammatory syndromes with a dermatological perspective. J Dermatol. 2007;34:601-618.
- Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561-574.
- Mueller SM, Itin P, Haeusermann P. Muckle-Wells syndrome effectively treated with canakinumab: is the recommended dosing schedule mandatory? Dermatology. 2011;223:113-118.
- Neven B, Prieur A, Quartier dit Maire P. Cryopyrinopathies: update on pathogenesis and treatment. Nat Clin Pract Rheumatol. 2008;4:481-489.
- Newell L, August S, Foria V, et al. Lifelong urticaria and multiple unexplained systemic symptoms. Clin Exp Dermatol. 2011;36:431-433.
- Yu JR, Kieron KS. Cryopyrin-associated periodic syndrome: an update on diagnosis and treatment response. Curr Allergy Asthma Rep. 2011;11:12-20.
- Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. 2nd ed. Barcelona, Spain: Mosby Elsevier; 2008. 9. Göppner D, Leverkus M. Basal cell carcinoma: from the molecular understanding of the pathogenesis to targeted therapy of progressive disease. J Skin Cancer. 2011;2011:650258.
Muckle-Wells syndrome (MWS) was first described in 1962 and is part of a broad category of hereditary periodic fever syndromes that include the autoinflammatory syndromes and the cryopyrin-associated periodic syndromes (CAPSs). Unlike autoimmune diseases, autoinflammatory syndromes are not associated with antigen-specific T-cell responses or high titers of autoantibodies but are related to disorders of the innate immune system. Basal cell nevus syndrome (BCNS), or Gorlin syndrome, is a rare genodermatosis inherited in an autosomal-dominant fashion that is characterized by a broad range of anomalies. Most notable is the early and strong predisposition to develop several to hundreds of basal cell carcinomas (BCCs). Classic clinical features of MWS and a thorough history and physical examination can assist in the diagnosis of this rare entity.
Case Report
A 35-year-old woman with a history of BCNS, which had been diagnosed at 24 years of age based on the presence of more than 2 BCCs and a family history of BCNS in her mother, presented with intermittent pruritic urticaria on the chest and back, episodic fevers, associated joint pain and swelling that worsened several hours after exercise, headache, conjunctivitis, blurred vision, and severe debilitating fatigue that had been present since childhood. The symptoms had progressively worsened with age and symptom-free intervals became shorter. She was diagnosed by her rheumatologist with biopsy-proven MWS and a positive NLRP3 (NLR family pyrin domain containing 3) gene mutation at 29 years of age. She was treated unsuccessfully with prednisone and antihistamines and entered a trial with anakinra. She showed improvement for 2 weeks but developed severe swelling and erythema at the injection sites at week 3, along with large leathery patches on the legs and difficulty ambulating.
The patient subsequently underwent excision of her BCCs and reported each site became erythematous, edematous, warm, and painful 6 hours after excision, which lasted for hours to days (Figures 1–3). After the first excision on the right forearm, she was seen in the emergency department, started on intravenous antibiotics and prednisone, and kept overnight in the hospital. She was discharged the following day and the edema in the right forearm subsided over several days. Bacterial culture and laboratory evaluation for infection were negative after the first excision on the right forearm. Because of the symptoms she experienced following this excision, she was referred to the plastic surgery department for excision followed by postoperative monitoring in the hospital. The patient continued to undergo excisions for BCCs and developed more severe symptoms including erythema, edema, warmth, and tenderness at the surrounding sites. Once again, the excision sites were cultured and laboratory work to rule out infection was ordered with a negative result. After several excisions and subsequent clinical findings, the patients’ symptoms were deemed consistent with MWS and not a result of infectious etiology. A diagnosis of MWS and BCNS with exacerbation of MWS with surgical procedures was made.
The patient has continued therapy with rilonacept for MWS, which is managed by her rheumatologist. She has tolerated rilonacept without adverse effects and has experienced a reduction in symptoms that has enhanced her quality of life and allows for further treatment of her BCNS. Her dermatologist (J.W.L.) has been treating her BCCs with vismodegib, but treatment has been sporadic due to muscle cramping after 7 days of therapy. She reported subjective improvement to her dermatologist and has tried alternating 7 days on and 7 days off vismodegib. The muscle cramping still has limited her treatment with this regimen, and she is currently on a trial of 3 days on, 4 days off per week.
Comment
Classification and Clinical Presentation
The hereditary periodic fever syndromes include the autoinflammatory syndromes and the CAPSs. The autoinflammatory syndromes include familial Mediterranean fever, hyperimmunoglobulinemia D with periodic fever syndrome, and tumor necrosis factor receptor–associated periodic syndrome. The CAPSs are similar but distinct and include familial cold autoinflammatory syndrome, neonatal-onset multisystem inflammatory disease (also known as chronic infantile neurologic cutaneous and articular syndrome, or cutaneous articular syndrome) and MWS.1,2
Cryopyrin-associated periodic syndromes are rare inherited diseases that result from mutations in the NLRP3 gene. There is a gain-of-function mutation on the NLRP3 gene located on the long arm of chromosome 1 at position 44, which codes for cryopyrin. An NLRP3 gene mutation causes cryopyrin to become hyperactive, leading to the formation of an inflammasome, which is a group of cryopyrin molecules. Inflammasomes, along with other proteins, activate caspase 1 to produce excess IL-1β, leading to persistent inflammatory symptoms.3 IL-1β is one of the key mediators of the body’s response to microbial invasion, inflammation, immunologic reactions, and tissue injury. It affects a large range of cells and organs. Although IL-1β production is critical for the control of pathogenic infections, excessive cytokine production is harmful to the host and can even be fatal.3,4
Cryopyrin-associated periodic syndromes encompass a disease continuum. The 3 distinct entities share many overlapping features as well as unique and distinguishing characteristics. Familial cold autoinflammatory syndrome is the mildest phenotype and is inherited in an autosomal-dominant fashion. It is characterized by a chronic urticarial eruption that starts early in infancy or childhood. The distribution of the cutaneous eruption is widespread and favors the arms and legs over the face and trunk. A low-grade fever often is seen along with musculoskeletal concerns of arthralgia and pain. Other commonly reported symptoms include conjunctivitis, myalgia, fatigue, and headache. Neurologic symptoms can include headaches. Symptoms usually begin 1 to 2 hours after cold exposure and last less than 24 hours.5-8
Neonatal-onset multisystem inflammatory disease is the most severe phenotype and occurs sporadically. Continuous symptoms and flares are characteristic and the length of the flare can vary from minutes to days. The cutaneous eruption favors the face, trunk, arms, and legs, and varies in intensity, beginning in infancy or childhood. Fever may be intermittent, mild, or absent. Rheumatologic manifestations include arthralgia and swelling, with approximately one-third of patients experiencing severe disabling arthropathy that causes gross joint deformity. Ocular findings include conjunctivitis, uveitis, papilledema, and even blindness. Neurologic sequelae include headaches, sensorineural hearing loss, and aseptic meningitis. Amyloidosis has been seen as a late complication.5,8
Muckle-Wells syndrome is a rare hereditary inflammatory disorder. It has no ethnic predisposition and is mostly inherited in an autosomal-dominant fashion. Classically, the condition is characterized by recurrent urticaria beginning at birth with intermittent episodic fever and malaise. The eruption has a predilection for the face, trunk, arms, and legs, which is similar to neonatal-onset multisystem inflammatory disease. Associated myalgia and arthralgia are common as well as ocular findings of conjunctivitis and episcleritis. Neurologic manifestations include headache and progressive sensorineural hearing loss in 60% to 70% of patients.6 Abdominal pain may be seen along with rare serositis in MWS but is rare in the other CAPSs. Amyloidosis caused by chronic inflammation is the most serious complication of MWS and is seen in approximately one-third of patients, manifesting as proteinuria followed by renal impairment. Symptoms of MWS may occur daily but vary individually, are broad in intensity and duration, and can last 1 to 2 days before resolving spontaneously. The symptoms can result from metabolic stressors including cold, stress, and exercise, as well as microbial pathogens. Leukocytosis and increased acute-phase reactants are observed during episodes of inflammation.4,6,8
Histopathology
Mild phenotypic variability exists between individuals, and many of the symptoms overlap in CAPSs. Although CAPSs display several distinguishing clinical characteristics, interestingly they share the same histopathological features regardless of the syndrome. The typical histopathological finding is a dermal neutrophilic infiltrate that tends to be perivascular and also may be perieccrine. Vasodilation and dermal edema also may be seen. These histopathological findings contrast with the typical lymphocytic and eosinophilic infiltrate seen in classic urticaria. Similar histopathologic findings have been seen in other neutrophilic urticarial dermatoses such as Schnitzler syndrome.4,6
Differential
The differential diagnoses for CAPSs include Schnitzler syndrome, cold urticaria, systemic-onset juvenile idiopathic arthritis/adult-onset Still disease, and deficiency in IL-1ra. It is important to consider these differential diagnoses for management and treatment options.
Management
The discovery of the NLRP3 gene mutation as well as an understanding of IL-1 biology has led to targeted therapy for these syndromes. Cryopyrin-associated periodic syndromes are mediated by IL-1β with an in vivo rate 5 times higher than in healthy patients.4 The blockade of IL-1β results in complete resolution of symptoms.
In the last several years, anakinra, rilonacept, and canakinumab have shown efficacy in targeting IL-1β as receptor antagonists. Anakinra is a short-acting recombinant IL-1ra with a half-life of 4 to 6 hours. This short half-life requires daily injections and the most common adverse events included injection-site reaction and upper respiratory tract infection.2,4 Rilonacept is a dimeric fusion protein that contains binding regions for the type 1 receptor and the IL-1 receptor accessory protein and is fused to the fragment, crystallizable (Fc) portion of human IgG1. Rilonacept is long acting with a circulating half-life of 8.6 days and offers patients ease of dosing with weekly subcutaneous injections. Rilonacept generally is well tolerated, with the most frequent adverse effects being injection-site reaction, upper respiratory tract infection, headache, arthralgia, and diarrhea.2,7
The newest of the treatments for patients with CAPS is canakinumab. It is a fully human IL-1β monoclonal antibody that is specific for IL-1β and not other members of the IL-1 family. It has a mean half-life of 26 days and is dosed subcutaneously once every 8 weeks. The most common adverse effects include nasopharyngitis, rhinitis, nausea, diarrhea, and vertigo.4 In one study, most patients did not report injection-site reactions.7 Studies also are underway on VX-765, a caspace-1 targeted therapy that acts upstream in the IL-1β pathway. Treatment with anakinra, rilonacept, and canakinumab generally offers rapid and sustained remission in the majority of MWS patients and helps prevent the development of systemic amyloidosis and lessens the potential for end organ damage.2,7
MWS and BCNS
Our patient had an unusual presentation of MWS complicated by BCNS, another rare autosomal-dominant inherited genodermatosis. In an extensive review of PubMed articles indexed for MEDLINE using the search terms Muckle-Wells syndrome and basal cell nevus syndrome, no association was identified between MWS and BCNS. Basal cell nevus syndrome is linked to PTCH1 (patched 1) gene mutation with an incidence of 1:150,000 in the United States and Europe and is characterized by a broad range of anomalies including skeletal abnormalities, ectopic calcification, odontogenic keratocysts, facial dysmorphism with macrocephaly, palmoplantar pits, and numerous tumors. Most notable is the early and strong predisposition to develop several to hundreds of BCCs.9
Conclusion
Muckle-Wells syndrome may go undiagnosed for many years or may be misdiagnosed as refractory urticaria, as in our patient. It is important to include periodic fever syndromes in the differential diagnosis of refractory urticaria with episodic fever to diagnose these cases of MWS earlier.
Muckle-Wells syndrome (MWS) was first described in 1962 and is part of a broad category of hereditary periodic fever syndromes that include the autoinflammatory syndromes and the cryopyrin-associated periodic syndromes (CAPSs). Unlike autoimmune diseases, autoinflammatory syndromes are not associated with antigen-specific T-cell responses or high titers of autoantibodies but are related to disorders of the innate immune system. Basal cell nevus syndrome (BCNS), or Gorlin syndrome, is a rare genodermatosis inherited in an autosomal-dominant fashion that is characterized by a broad range of anomalies. Most notable is the early and strong predisposition to develop several to hundreds of basal cell carcinomas (BCCs). Classic clinical features of MWS and a thorough history and physical examination can assist in the diagnosis of this rare entity.
Case Report
A 35-year-old woman with a history of BCNS, which had been diagnosed at 24 years of age based on the presence of more than 2 BCCs and a family history of BCNS in her mother, presented with intermittent pruritic urticaria on the chest and back, episodic fevers, associated joint pain and swelling that worsened several hours after exercise, headache, conjunctivitis, blurred vision, and severe debilitating fatigue that had been present since childhood. The symptoms had progressively worsened with age and symptom-free intervals became shorter. She was diagnosed by her rheumatologist with biopsy-proven MWS and a positive NLRP3 (NLR family pyrin domain containing 3) gene mutation at 29 years of age. She was treated unsuccessfully with prednisone and antihistamines and entered a trial with anakinra. She showed improvement for 2 weeks but developed severe swelling and erythema at the injection sites at week 3, along with large leathery patches on the legs and difficulty ambulating.
The patient subsequently underwent excision of her BCCs and reported each site became erythematous, edematous, warm, and painful 6 hours after excision, which lasted for hours to days (Figures 1–3). After the first excision on the right forearm, she was seen in the emergency department, started on intravenous antibiotics and prednisone, and kept overnight in the hospital. She was discharged the following day and the edema in the right forearm subsided over several days. Bacterial culture and laboratory evaluation for infection were negative after the first excision on the right forearm. Because of the symptoms she experienced following this excision, she was referred to the plastic surgery department for excision followed by postoperative monitoring in the hospital. The patient continued to undergo excisions for BCCs and developed more severe symptoms including erythema, edema, warmth, and tenderness at the surrounding sites. Once again, the excision sites were cultured and laboratory work to rule out infection was ordered with a negative result. After several excisions and subsequent clinical findings, the patients’ symptoms were deemed consistent with MWS and not a result of infectious etiology. A diagnosis of MWS and BCNS with exacerbation of MWS with surgical procedures was made.
The patient has continued therapy with rilonacept for MWS, which is managed by her rheumatologist. She has tolerated rilonacept without adverse effects and has experienced a reduction in symptoms that has enhanced her quality of life and allows for further treatment of her BCNS. Her dermatologist (J.W.L.) has been treating her BCCs with vismodegib, but treatment has been sporadic due to muscle cramping after 7 days of therapy. She reported subjective improvement to her dermatologist and has tried alternating 7 days on and 7 days off vismodegib. The muscle cramping still has limited her treatment with this regimen, and she is currently on a trial of 3 days on, 4 days off per week.
Comment
Classification and Clinical Presentation
The hereditary periodic fever syndromes include the autoinflammatory syndromes and the CAPSs. The autoinflammatory syndromes include familial Mediterranean fever, hyperimmunoglobulinemia D with periodic fever syndrome, and tumor necrosis factor receptor–associated periodic syndrome. The CAPSs are similar but distinct and include familial cold autoinflammatory syndrome, neonatal-onset multisystem inflammatory disease (also known as chronic infantile neurologic cutaneous and articular syndrome, or cutaneous articular syndrome) and MWS.1,2
Cryopyrin-associated periodic syndromes are rare inherited diseases that result from mutations in the NLRP3 gene. There is a gain-of-function mutation on the NLRP3 gene located on the long arm of chromosome 1 at position 44, which codes for cryopyrin. An NLRP3 gene mutation causes cryopyrin to become hyperactive, leading to the formation of an inflammasome, which is a group of cryopyrin molecules. Inflammasomes, along with other proteins, activate caspase 1 to produce excess IL-1β, leading to persistent inflammatory symptoms.3 IL-1β is one of the key mediators of the body’s response to microbial invasion, inflammation, immunologic reactions, and tissue injury. It affects a large range of cells and organs. Although IL-1β production is critical for the control of pathogenic infections, excessive cytokine production is harmful to the host and can even be fatal.3,4
Cryopyrin-associated periodic syndromes encompass a disease continuum. The 3 distinct entities share many overlapping features as well as unique and distinguishing characteristics. Familial cold autoinflammatory syndrome is the mildest phenotype and is inherited in an autosomal-dominant fashion. It is characterized by a chronic urticarial eruption that starts early in infancy or childhood. The distribution of the cutaneous eruption is widespread and favors the arms and legs over the face and trunk. A low-grade fever often is seen along with musculoskeletal concerns of arthralgia and pain. Other commonly reported symptoms include conjunctivitis, myalgia, fatigue, and headache. Neurologic symptoms can include headaches. Symptoms usually begin 1 to 2 hours after cold exposure and last less than 24 hours.5-8
Neonatal-onset multisystem inflammatory disease is the most severe phenotype and occurs sporadically. Continuous symptoms and flares are characteristic and the length of the flare can vary from minutes to days. The cutaneous eruption favors the face, trunk, arms, and legs, and varies in intensity, beginning in infancy or childhood. Fever may be intermittent, mild, or absent. Rheumatologic manifestations include arthralgia and swelling, with approximately one-third of patients experiencing severe disabling arthropathy that causes gross joint deformity. Ocular findings include conjunctivitis, uveitis, papilledema, and even blindness. Neurologic sequelae include headaches, sensorineural hearing loss, and aseptic meningitis. Amyloidosis has been seen as a late complication.5,8
Muckle-Wells syndrome is a rare hereditary inflammatory disorder. It has no ethnic predisposition and is mostly inherited in an autosomal-dominant fashion. Classically, the condition is characterized by recurrent urticaria beginning at birth with intermittent episodic fever and malaise. The eruption has a predilection for the face, trunk, arms, and legs, which is similar to neonatal-onset multisystem inflammatory disease. Associated myalgia and arthralgia are common as well as ocular findings of conjunctivitis and episcleritis. Neurologic manifestations include headache and progressive sensorineural hearing loss in 60% to 70% of patients.6 Abdominal pain may be seen along with rare serositis in MWS but is rare in the other CAPSs. Amyloidosis caused by chronic inflammation is the most serious complication of MWS and is seen in approximately one-third of patients, manifesting as proteinuria followed by renal impairment. Symptoms of MWS may occur daily but vary individually, are broad in intensity and duration, and can last 1 to 2 days before resolving spontaneously. The symptoms can result from metabolic stressors including cold, stress, and exercise, as well as microbial pathogens. Leukocytosis and increased acute-phase reactants are observed during episodes of inflammation.4,6,8
Histopathology
Mild phenotypic variability exists between individuals, and many of the symptoms overlap in CAPSs. Although CAPSs display several distinguishing clinical characteristics, interestingly they share the same histopathological features regardless of the syndrome. The typical histopathological finding is a dermal neutrophilic infiltrate that tends to be perivascular and also may be perieccrine. Vasodilation and dermal edema also may be seen. These histopathological findings contrast with the typical lymphocytic and eosinophilic infiltrate seen in classic urticaria. Similar histopathologic findings have been seen in other neutrophilic urticarial dermatoses such as Schnitzler syndrome.4,6
Differential
The differential diagnoses for CAPSs include Schnitzler syndrome, cold urticaria, systemic-onset juvenile idiopathic arthritis/adult-onset Still disease, and deficiency in IL-1ra. It is important to consider these differential diagnoses for management and treatment options.
Management
The discovery of the NLRP3 gene mutation as well as an understanding of IL-1 biology has led to targeted therapy for these syndromes. Cryopyrin-associated periodic syndromes are mediated by IL-1β with an in vivo rate 5 times higher than in healthy patients.4 The blockade of IL-1β results in complete resolution of symptoms.
In the last several years, anakinra, rilonacept, and canakinumab have shown efficacy in targeting IL-1β as receptor antagonists. Anakinra is a short-acting recombinant IL-1ra with a half-life of 4 to 6 hours. This short half-life requires daily injections and the most common adverse events included injection-site reaction and upper respiratory tract infection.2,4 Rilonacept is a dimeric fusion protein that contains binding regions for the type 1 receptor and the IL-1 receptor accessory protein and is fused to the fragment, crystallizable (Fc) portion of human IgG1. Rilonacept is long acting with a circulating half-life of 8.6 days and offers patients ease of dosing with weekly subcutaneous injections. Rilonacept generally is well tolerated, with the most frequent adverse effects being injection-site reaction, upper respiratory tract infection, headache, arthralgia, and diarrhea.2,7
The newest of the treatments for patients with CAPS is canakinumab. It is a fully human IL-1β monoclonal antibody that is specific for IL-1β and not other members of the IL-1 family. It has a mean half-life of 26 days and is dosed subcutaneously once every 8 weeks. The most common adverse effects include nasopharyngitis, rhinitis, nausea, diarrhea, and vertigo.4 In one study, most patients did not report injection-site reactions.7 Studies also are underway on VX-765, a caspace-1 targeted therapy that acts upstream in the IL-1β pathway. Treatment with anakinra, rilonacept, and canakinumab generally offers rapid and sustained remission in the majority of MWS patients and helps prevent the development of systemic amyloidosis and lessens the potential for end organ damage.2,7
MWS and BCNS
Our patient had an unusual presentation of MWS complicated by BCNS, another rare autosomal-dominant inherited genodermatosis. In an extensive review of PubMed articles indexed for MEDLINE using the search terms Muckle-Wells syndrome and basal cell nevus syndrome, no association was identified between MWS and BCNS. Basal cell nevus syndrome is linked to PTCH1 (patched 1) gene mutation with an incidence of 1:150,000 in the United States and Europe and is characterized by a broad range of anomalies including skeletal abnormalities, ectopic calcification, odontogenic keratocysts, facial dysmorphism with macrocephaly, palmoplantar pits, and numerous tumors. Most notable is the early and strong predisposition to develop several to hundreds of BCCs.9
Conclusion
Muckle-Wells syndrome may go undiagnosed for many years or may be misdiagnosed as refractory urticaria, as in our patient. It is important to include periodic fever syndromes in the differential diagnosis of refractory urticaria with episodic fever to diagnose these cases of MWS earlier.
- Kagami S, Saeki H, Kuwano Y, et al. A probable case of Muckle-Wells syndrome. J Dermatol. 2006;2:118-121.
- Kanazawa N, Furukawa F. Autoinflammatory syndromes with a dermatological perspective. J Dermatol. 2007;34:601-618.
- Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561-574.
- Mueller SM, Itin P, Haeusermann P. Muckle-Wells syndrome effectively treated with canakinumab: is the recommended dosing schedule mandatory? Dermatology. 2011;223:113-118.
- Neven B, Prieur A, Quartier dit Maire P. Cryopyrinopathies: update on pathogenesis and treatment. Nat Clin Pract Rheumatol. 2008;4:481-489.
- Newell L, August S, Foria V, et al. Lifelong urticaria and multiple unexplained systemic symptoms. Clin Exp Dermatol. 2011;36:431-433.
- Yu JR, Kieron KS. Cryopyrin-associated periodic syndrome: an update on diagnosis and treatment response. Curr Allergy Asthma Rep. 2011;11:12-20.
- Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. 2nd ed. Barcelona, Spain: Mosby Elsevier; 2008. 9. Göppner D, Leverkus M. Basal cell carcinoma: from the molecular understanding of the pathogenesis to targeted therapy of progressive disease. J Skin Cancer. 2011;2011:650258.
- Kagami S, Saeki H, Kuwano Y, et al. A probable case of Muckle-Wells syndrome. J Dermatol. 2006;2:118-121.
- Kanazawa N, Furukawa F. Autoinflammatory syndromes with a dermatological perspective. J Dermatol. 2007;34:601-618.
- Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561-574.
- Mueller SM, Itin P, Haeusermann P. Muckle-Wells syndrome effectively treated with canakinumab: is the recommended dosing schedule mandatory? Dermatology. 2011;223:113-118.
- Neven B, Prieur A, Quartier dit Maire P. Cryopyrinopathies: update on pathogenesis and treatment. Nat Clin Pract Rheumatol. 2008;4:481-489.
- Newell L, August S, Foria V, et al. Lifelong urticaria and multiple unexplained systemic symptoms. Clin Exp Dermatol. 2011;36:431-433.
- Yu JR, Kieron KS. Cryopyrin-associated periodic syndrome: an update on diagnosis and treatment response. Curr Allergy Asthma Rep. 2011;11:12-20.
- Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. 2nd ed. Barcelona, Spain: Mosby Elsevier; 2008. 9. Göppner D, Leverkus M. Basal cell carcinoma: from the molecular understanding of the pathogenesis to targeted therapy of progressive disease. J Skin Cancer. 2011;2011:650258.
Practice Points
- An urticarial rash occurring in childhood with symptoms of fever, joint pain, and swelling along with visual symptoms should prompt consideration of a cryopyrin-associated periodic syndrome.
- Histopathology shows a dermal neutrophilic infiltrate that tends to be perivascular and also may be perieccrine. This atypical urticaria contrasts with the typical lymphocytic and eosinophilic infiltrate seen in classic urticaria.