User login
Hand-foot-and-mouth Disease Caused by Coxsackievirus A6 on the Rise
Hand-foot-and-mouth disease (HFMD) is a viral illness caused by several enteroviruses, most commonly coxsackievirus A16 (CVA16) and enterovirus 71 (EV71). The disease is generally seen in children younger than 5 years, characterized by lesions of the oral mucosa, palms, and soles, usually lasting 7 to 10 days. Other coxsackie type A viruses, including CVA6, CVA9, and CVA10, also are associated with HFMD.1-5 Although CVA16 has traditionally been the primary strain causing HFMD, CVA6 has become a major cause of HFMD outbreaks in the United States and worldwide in recent years.6-12 Interestingly, CVA6 also has been found to be associated with adult HFMD, which has increased in incidence. The CVA6 strain was first identified in association with the disease during HFMD outbreaks in Finland and Singapore in 2008,13,14 with similar strains detected in subsequent outbreaks in Taiwan, Japan, Spain, France, China, India, and the United States.12,15-25 Most cases took place in warmer months, with one winter outbreak in Massachusetts in 2012.24
Herein, we review the incidence of CVA6, as well as its atypical presentation, diagnosis, and treatment to aid dermatologists. Given the increasing incidence of HFMD caused by CVA6 and its often atypical presentation, it is important for dermatologists to be aware of this increasingly notable disease state and its viral cause.
Incidence of CVA6
Coxsackievirus A6 has been identified as the cause of many reported outbreaks of HFMD since it was first identified in 2008, and it is known to cause both pediatric and adult outbreaks.7-12 It may even be surpassing other strains in frequency in certain areas. In Tianjin, China, for example, EV71 and CVA16 were the most common serotypes causing HFMD from 2008 to 2012; however, in 2013, CVA6 was the most prevalent strain.26
The incidence of CVA6 also has been increasing in other areas.28
In 2015, an outbreak of HFMD took place at Lackland Air Force Base in Texas during a basic military training. Eight cases were confirmed and 45 cases were suspected. The rate of infection was 0.4% (50/12,270) among trainees and 0.3% (2/602) among instructors.7 Eight of 12 nasopharyngeal swabs tested positive for EV by way of local real-time reverse transcription–polymerase chain reaction (RT-PCR). Four nasopharyngeal swabs were sent to the CDC for evaluation and all were positive for CVA6.7
Presentation
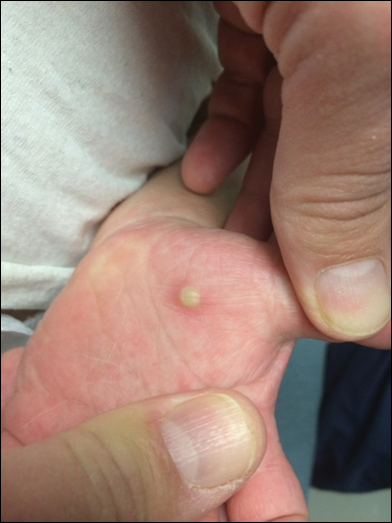
Because the prevalence of CVA6 has increased, it is important to be able to identify the presentation of HFMD caused by this strain. Coxsackievirus A6 has been found to affect a broader demographic and cause more severe cases of HFMD with its unique constellation of findings compared to other known strains. Patients present with flulike symptoms; higher fever than present in typical HFMD; and a longer duration of disease, typically lasting 2 weeks. Patients also may present with more severe skin disease compared to classic HFMD, not only including vesicles but also large bullae, erosions, and ulcers on the dorsal and plantar feet (Figure 1).
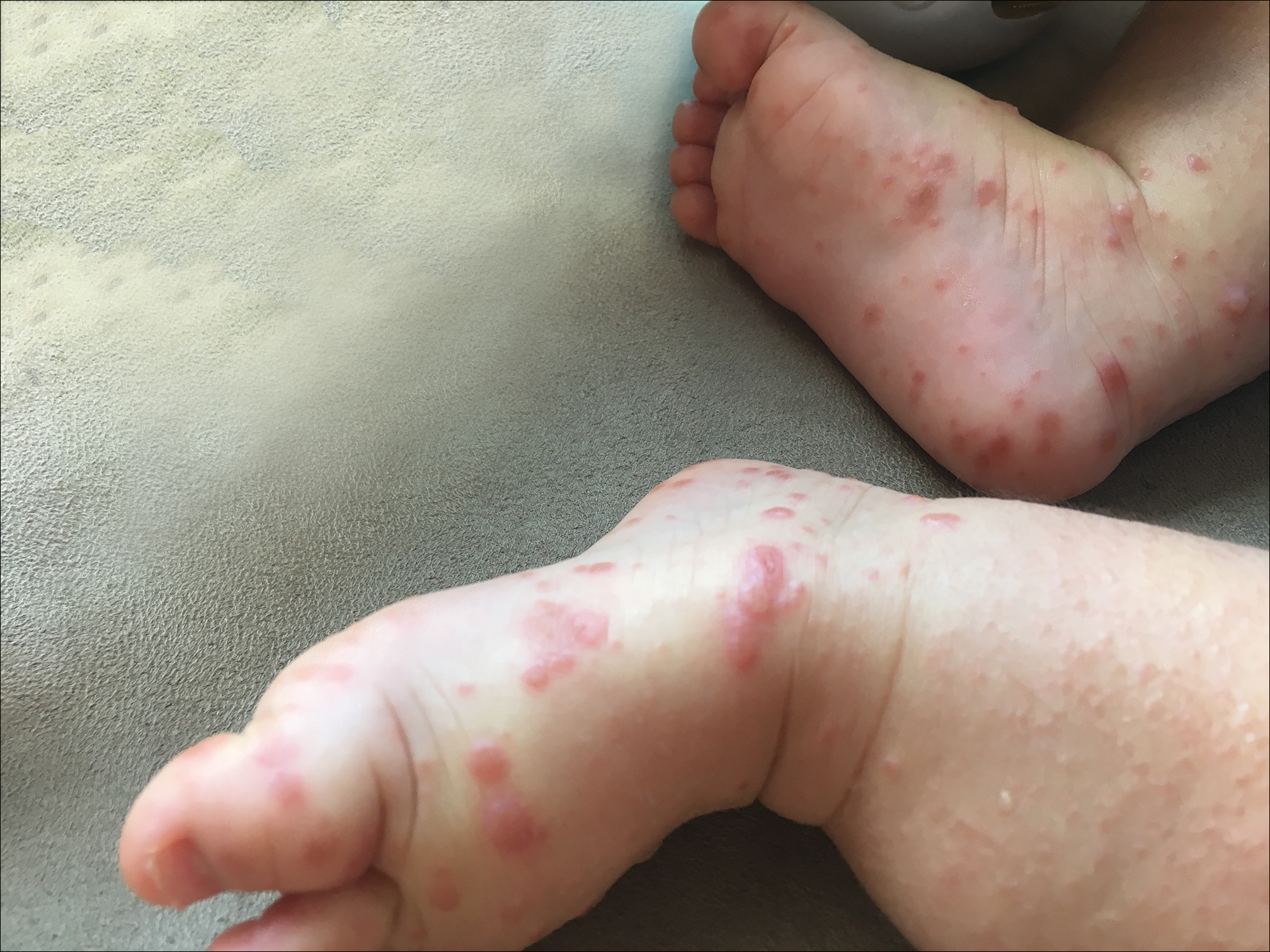
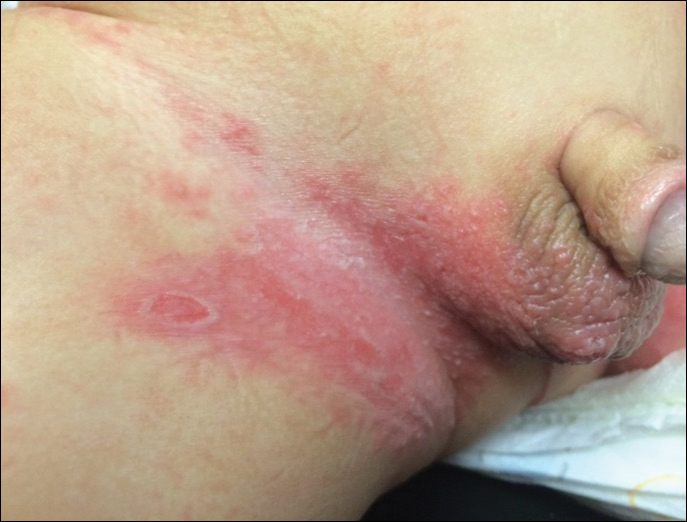
In patients with atopic dermatitis, CVA6 also shows a predilection to appear in areas of skin disease, such as the flexural regions of the arms and legs, and is referred to as eczema coxsackium.24,38,39 It can mimic eczema herpeticum or varicella superinfection, which are important considerations to include in the differential diagnosis. Additionally, CVA6-induced lesions often show up in previously irritated or traumatized areas such as sunburns, fungal infections, and diaper dermatitis in children. Lesions have been described to sometimes mimic Gianotti-Crosti syndrome, with involvement of the extensor surfaces, buttocks, and cheeks, and sparing of the trunk.24
Clinical Diagnosis
Because HFMD is uncommon and atypical in adults, skin biopsies may be used in the initial workup and evaluation of patients. It is important to understand the histologic features associated with HFMD, including spongiosis with exocytosis of neutrophils as well as keratinocyte necrosis and pallor with associated shadow cells.6 In one series, the most extensively involved areas of keratinocyte necrosis were the stratum granulosum and upper half of the stratum spinosum.40 In the dermis, vascular involvement may be present on a spectrum with the extravasation of red blood cells and leukocytoclasis or true leukocytoclastic vasculitis.6,40 Vesicular lesions show severe dermal edema and inflammatory infiltrate.6,41 CD3+ and CD8+ lymphocytes predominate. Cytotoxic T lymphocytes are present and express granzyme B and granulysin, both important mediators of apoptosis in virally infected keratinocytes.6
Adult HFMD primarily is a clinical diagnosis, and histopathologic analysis can be a useful tool in certain cases. Coxsackievirus A6 does not grow well on culture and is not detected by standard serologic testing laboratories, necessitating the use of quantitative RT-PCR analysis.41,42 In one study, culture was able to detect only 14% to 16% of samples that tested positive by quantitative RT-PCR.43 This form of PCR identifies viral subtype through amplification of enterovirus viral protein 1 capsid gene sequence.24 Unfortunately, this testing often is not offered in most readily available laboratories and often necessitates being sent out to more well-equipped laboratories.2,24
Treatment
Hand-foot-and-mouth disease is a self-limited illness and requires only supportive care with a focus on hydration and pain management. Lesions heal without scarring but may leave notable postinflammatory pigment alteration that may last months to years, depending on extent of disease and skin type. Secondarily infected individuals should be treated with appropriate antibiotics or antivirals depending on the infectious agent. Hand hygiene is of great importance, and hospitalized patients should be put on strict contact precautions. It also is important to isolate patients from vulnerable individuals, especially pregnant women, as coxsackievirus has been linked to intrauterine infections and loss of pregnancy.24
Genetic Analysis
Genetic studies of the virus have suggested that nonstructural genes may be playing an interesting role in clinical phenotypes and outcomes of CVA6 infection.44 These genetic studies also are being implemented into the understanding of the virus’ evolution as well as the construction of vaccinations.27,44
Conclusion
With the increasing prevalence of CVA6-associated HFMD, it is important to understand the clinical presentation and histologic findings associated with this atypical presentation of the disease as well as the changing epidemiology of the viral strains causing HFMD.
- Galen WK. Cutaneous manifestations of enterovirus infections. In: Tyring SK, ed. Mucocutaneous Manifestations of Viral Diseases. New York, NY: Marcel Dekker; 2002:455-467.
- Ramirez-Fort M, Downing C, Doan H, et al. Coxsackievirus A6 associated hand, foot and mouth disease in adults: clinical presentation and review of the literature. J Clin Virol. 2014;60:381-386.
- Khetsuriani N, Lamonte-Fowlkes A, Oberst S, et al. Enterovirus surveillance—United States, 1970-2005. MMWR Surveill Summ. 2006;55:1-20.
- Yang F, Zhang T, Hu Y, et al. Survey of enterovirus infections from hand, foot and mouth disease outbreak in China, 2009. Virol J. 2011;8:508.
- Ho M, Chen ER, Hsu KH, et al. An epidemic of enterovirus 71 infection in Taiwan. Taiwan Enterovirus Epidemic Working Group. N Engl J Med. 1999;341:929-935.
- Second J, Velter C, Calès S, et al. Clinicopathologic analysis of atypical hand, foot, and mouth disease in adult patients. J Am Acad Dermatol. 2016;76:722-729.
- Banta J, Lenz B, Pawlak M, et al. Notes from the field: outbreak of hand, foot, and mouth disease caused by coxsackievirus A6 among basic military trainees—Texas, 2015. MMWR Morb Mortal Wkly Rep. 2016;65.26:678-680.
- Bian L, Wang Y, Yao X, et al. Coxsackievirus A6: a new emerging pathogen causing hand, foot and mouth disease outbreaks worldwide. Expert Rev Anti Infect Ther. 2015;13:1061-1071.
- Buttery VW, Kenyon C, Grunewald S, et al. Notes from the field: atypical presentations of hand, foot, and mouth disease caused by coxsackievirus A6—Minnesota, 2014. MMWR Morb Mortal Wkly Rep. 2015;64:805.
- Puenpa J, Chieochansin T, Linsuwanon P, et al. Hand, foot, and mouth disease caused by coxsackievirus A6, Thailand, 2012. Emerg Infect Dis. 2013;19:641-643.
- Flett K, Youngster I, Huang J, et al. Hand, foot, and mouth disease caused by coxsackievirus A6. Emerg Infect Dis. 2012;18:1702-1704.
- Centers for Disease Control and Prevention. Notes from the field: severe hand, foot, and mouth disease associated with coxsackievirus A6—Alabama, Connecticut, California, and Nevada, November 2011-February 2012. MMWR Morb Mortal Wkly Rep. 2012;61:213-214.
- Blomqvist S, Klemola P, Kaijalainen S, et al. Co-circulation of coxsackieviruses A6 and A10 in hand, foot and mouth disease outbreak in Finland. J Clin Virol. 2010;48:49-54.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Zeng H, Lu J, Zheng H, et al. The epidemiological study of coxsackievirus A6 revealing hand, foot and mouth disease epidemic patterns in Guandong, China. Sci Rep. 2015;5:10550.
- Mirand A, Henquell C, Archimbaud C, et al. Outbreak of hand, foot and mouth disease/herpangina associated with coxsackievirus A6 andA10 infections in 2010, France: a large citywide, prospective observational study. Clin Microbiol Infect. 2012;18:E110-E118.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Fujimoto T, Iizuka S, Enomoto M, et al. Hand, foot, and mouth disease caused by coxsackievirus A6, Japan, 2011. Emerg Infect Dis. 2012;18:337-339.
- Bracho MA, Gonzalez-Candelas F, Valero A, et al. Enterovirus co-infections and onychomadesis after hand, foot, and mouth disease, Spain, 2008. Emerg Infect Dis. 2011;17:2223-2231.
- Gopalkrishna V, Patil PR, Patil GP, et al. Circulation of multiple enterovirus serotypes causing hand, foot and mouth disease in India. J Med Microbiol. 2012;61:420-425.
- Lo SH, Huang YC, Huang CG, et al. Clinical and epidemiologic features of coxsackievirus A6 infection in children in northern Taiwan between 2004 and 2009. J Microbiol Immunol Infect. 2011;44:252-257.
- Lu QB, Zhang XA, Wo Y, et al. Circulation of coxsackievirus A10 and A6 in hand-foot-mouth disease in China, 2009-2011. PLoS One. 2012;7:E52073.
- Wu Y, Yeo A, Phoon MC, et al. The largest outbreak of hand; foot and mouth disease in Singapore in 2008: the role of enterovirus 71 and coxsackievirus A strains. Int J Infect Dis. 2010;14:E1076-E1081.
- Ventarola D, Bordone L, Silverberg N. Update on hand-foot-and-mouth disease. Clin Dermatol. 2015;33:340-346.
- Li Y, Chang Z, Wu P, et al. Emerging enteroviruses causing hand, foot and mouth disease, China. 2010-2016. Emerg Infect Dis. 2018;24:1902-1906.
- Tan X, Li L, Zhang B, et al. Molecular epidemiology of coxsackievirus A6 associated with outbreaks of hand, foot, and mouth disease in Tianjin, China, in 2013. Arch Virol. 2015;160:1097-1104.
- Li Y, Bao H, Zhang X, et al. Epidemiological and genetic analysis concerning the non-enterovirus 71 and non-coxsackievirus A16 causative agents related to hand, foot and mouth disease in Anyang City, Henan Province, China, from 2011 to 2015. J Med Virol. 2017;89:1749-1758.
- Guan H, Wang J, Wang C, et al. Etiology of multiple non-EV71 and non-CVA16 enteroviruses associated with hand, foot, and mouth disease in Jinan, China, 2009-2013. PLoS One. 2015;10:E0142733.
- Cabrerizo M, Tarrago´ D, Muñoz-Almagro C, et al. Mollecular epidemiology of enterovirus 71, coxsackievirus A16 and A6 associated with hand, foot and mouth disease in Spain. Clin Microbiol Infect. 2014;20:O150-O156.
- Lønnberg A, Elberling J, Fischer T, et al. Two cases of hand, foot, and mouth disease involving the scalp. Acta Derm Venereol. 2013;93:467-468.
- Lott JP, Liu K, Landry ML, et al. Atypical hand-foot-and-mouth disease associated with coxsackievirus A6 infection. J Am Acad Dermatol. 2013;69:736-741.
- Kaminska K, Martinetti G, Lucchini R, et al. Coxsackievirus A6 and hand, foot and mouth disease: three case reports of familial child-to-immunocompetent adult transmission and a literature review. Case Rep Dermatol. 2013;5:203-209.
- Shin JU, Oh SH, Lee JH. A case of hand-foot-mouth disease in an immunocompetent adult. Ann Dermatol. 2010;22:216-218.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Feder HM, Bennett N, Modlin JF. Atypical hand, foot, and mouth disease: a vesiculobullous eruption caused by coxsackie virus A6. Lancet Infect Dis. 2014;14:83-86.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Kim M, Kim B, Byun S, et al. Beau’s lines and onychomadesis after hand-foot-mouth disease. Clin Pediatr Dermatol. 2015;1:1.
- Mathes EF, Oza V, Frieden IJ, et al. “Eczema coxsackium” and unusual cutaneous findings in an enterovirus outbreak. Pediatrics. 2013;132:E149-E157.
- Lynch M, Sears A, Cookson H, et al. Disseminated coxsackievirus A6 affecting children with atopic dermatitis. Clin Exp Dermatol. 2015;40:525-528.
- Laga A, Shroba S, Hanna J. Atypical hand, foot and mouth disease in adults associated with coxsackievirus A6: a clinicopathologic study. J Cutan Pathol. 2016;43:940-945.
- Schmidt NJ, Ho HH, Lennette EH. Propagation and isolation of group A coxsackieviruses in RD cells. J Clin Microbiol. 1975;2:183-185.
- Oberste MS, Penaranda S, Rogers SL, et al. Comparative evaluation of Taqman real-time PCR and semi-nested VP1 PCR for detection of enteroviruses in clinical specimens. J Clin Virol. 2010;49:73-74.
- Lee MK, Chan PK, Ho II, et al. Enterovirus infection among patients admitted to hospital in Hong Kong in 2010: epidemiology, clinical characteristics, and importance of molecular diagnosis. J Med Virol. 2013;85:1811-1817.
- Yee PTI, Laa Poh C. Impact of genetic changes, pathogenicity and antigenicity on enterovirus A71 vaccine development. Virology. 2017;506:121-129.
Hand-foot-and-mouth disease (HFMD) is a viral illness caused by several enteroviruses, most commonly coxsackievirus A16 (CVA16) and enterovirus 71 (EV71). The disease is generally seen in children younger than 5 years, characterized by lesions of the oral mucosa, palms, and soles, usually lasting 7 to 10 days. Other coxsackie type A viruses, including CVA6, CVA9, and CVA10, also are associated with HFMD.1-5 Although CVA16 has traditionally been the primary strain causing HFMD, CVA6 has become a major cause of HFMD outbreaks in the United States and worldwide in recent years.6-12 Interestingly, CVA6 also has been found to be associated with adult HFMD, which has increased in incidence. The CVA6 strain was first identified in association with the disease during HFMD outbreaks in Finland and Singapore in 2008,13,14 with similar strains detected in subsequent outbreaks in Taiwan, Japan, Spain, France, China, India, and the United States.12,15-25 Most cases took place in warmer months, with one winter outbreak in Massachusetts in 2012.24
Herein, we review the incidence of CVA6, as well as its atypical presentation, diagnosis, and treatment to aid dermatologists. Given the increasing incidence of HFMD caused by CVA6 and its often atypical presentation, it is important for dermatologists to be aware of this increasingly notable disease state and its viral cause.
Incidence of CVA6
Coxsackievirus A6 has been identified as the cause of many reported outbreaks of HFMD since it was first identified in 2008, and it is known to cause both pediatric and adult outbreaks.7-12 It may even be surpassing other strains in frequency in certain areas. In Tianjin, China, for example, EV71 and CVA16 were the most common serotypes causing HFMD from 2008 to 2012; however, in 2013, CVA6 was the most prevalent strain.26
The incidence of CVA6 also has been increasing in other areas.28
In 2015, an outbreak of HFMD took place at Lackland Air Force Base in Texas during a basic military training. Eight cases were confirmed and 45 cases were suspected. The rate of infection was 0.4% (50/12,270) among trainees and 0.3% (2/602) among instructors.7 Eight of 12 nasopharyngeal swabs tested positive for EV by way of local real-time reverse transcription–polymerase chain reaction (RT-PCR). Four nasopharyngeal swabs were sent to the CDC for evaluation and all were positive for CVA6.7
Presentation
Because the prevalence of CVA6 has increased, it is important to be able to identify the presentation of HFMD caused by this strain. Coxsackievirus A6 has been found to affect a broader demographic and cause more severe cases of HFMD with its unique constellation of findings compared to other known strains. Patients present with flulike symptoms; higher fever than present in typical HFMD; and a longer duration of disease, typically lasting 2 weeks. Patients also may present with more severe skin disease compared to classic HFMD, not only including vesicles but also large bullae, erosions, and ulcers on the dorsal and plantar feet (Figure 1).
In patients with atopic dermatitis, CVA6 also shows a predilection to appear in areas of skin disease, such as the flexural regions of the arms and legs, and is referred to as eczema coxsackium.24,38,39 It can mimic eczema herpeticum or varicella superinfection, which are important considerations to include in the differential diagnosis. Additionally, CVA6-induced lesions often show up in previously irritated or traumatized areas such as sunburns, fungal infections, and diaper dermatitis in children. Lesions have been described to sometimes mimic Gianotti-Crosti syndrome, with involvement of the extensor surfaces, buttocks, and cheeks, and sparing of the trunk.24
Clinical Diagnosis
Because HFMD is uncommon and atypical in adults, skin biopsies may be used in the initial workup and evaluation of patients. It is important to understand the histologic features associated with HFMD, including spongiosis with exocytosis of neutrophils as well as keratinocyte necrosis and pallor with associated shadow cells.6 In one series, the most extensively involved areas of keratinocyte necrosis were the stratum granulosum and upper half of the stratum spinosum.40 In the dermis, vascular involvement may be present on a spectrum with the extravasation of red blood cells and leukocytoclasis or true leukocytoclastic vasculitis.6,40 Vesicular lesions show severe dermal edema and inflammatory infiltrate.6,41 CD3+ and CD8+ lymphocytes predominate. Cytotoxic T lymphocytes are present and express granzyme B and granulysin, both important mediators of apoptosis in virally infected keratinocytes.6
Adult HFMD primarily is a clinical diagnosis, and histopathologic analysis can be a useful tool in certain cases. Coxsackievirus A6 does not grow well on culture and is not detected by standard serologic testing laboratories, necessitating the use of quantitative RT-PCR analysis.41,42 In one study, culture was able to detect only 14% to 16% of samples that tested positive by quantitative RT-PCR.43 This form of PCR identifies viral subtype through amplification of enterovirus viral protein 1 capsid gene sequence.24 Unfortunately, this testing often is not offered in most readily available laboratories and often necessitates being sent out to more well-equipped laboratories.2,24
Treatment
Hand-foot-and-mouth disease is a self-limited illness and requires only supportive care with a focus on hydration and pain management. Lesions heal without scarring but may leave notable postinflammatory pigment alteration that may last months to years, depending on extent of disease and skin type. Secondarily infected individuals should be treated with appropriate antibiotics or antivirals depending on the infectious agent. Hand hygiene is of great importance, and hospitalized patients should be put on strict contact precautions. It also is important to isolate patients from vulnerable individuals, especially pregnant women, as coxsackievirus has been linked to intrauterine infections and loss of pregnancy.24
Genetic Analysis
Genetic studies of the virus have suggested that nonstructural genes may be playing an interesting role in clinical phenotypes and outcomes of CVA6 infection.44 These genetic studies also are being implemented into the understanding of the virus’ evolution as well as the construction of vaccinations.27,44
Conclusion
With the increasing prevalence of CVA6-associated HFMD, it is important to understand the clinical presentation and histologic findings associated with this atypical presentation of the disease as well as the changing epidemiology of the viral strains causing HFMD.
Hand-foot-and-mouth disease (HFMD) is a viral illness caused by several enteroviruses, most commonly coxsackievirus A16 (CVA16) and enterovirus 71 (EV71). The disease is generally seen in children younger than 5 years, characterized by lesions of the oral mucosa, palms, and soles, usually lasting 7 to 10 days. Other coxsackie type A viruses, including CVA6, CVA9, and CVA10, also are associated with HFMD.1-5 Although CVA16 has traditionally been the primary strain causing HFMD, CVA6 has become a major cause of HFMD outbreaks in the United States and worldwide in recent years.6-12 Interestingly, CVA6 also has been found to be associated with adult HFMD, which has increased in incidence. The CVA6 strain was first identified in association with the disease during HFMD outbreaks in Finland and Singapore in 2008,13,14 with similar strains detected in subsequent outbreaks in Taiwan, Japan, Spain, France, China, India, and the United States.12,15-25 Most cases took place in warmer months, with one winter outbreak in Massachusetts in 2012.24
Herein, we review the incidence of CVA6, as well as its atypical presentation, diagnosis, and treatment to aid dermatologists. Given the increasing incidence of HFMD caused by CVA6 and its often atypical presentation, it is important for dermatologists to be aware of this increasingly notable disease state and its viral cause.
Incidence of CVA6
Coxsackievirus A6 has been identified as the cause of many reported outbreaks of HFMD since it was first identified in 2008, and it is known to cause both pediatric and adult outbreaks.7-12 It may even be surpassing other strains in frequency in certain areas. In Tianjin, China, for example, EV71 and CVA16 were the most common serotypes causing HFMD from 2008 to 2012; however, in 2013, CVA6 was the most prevalent strain.26
The incidence of CVA6 also has been increasing in other areas.28
In 2015, an outbreak of HFMD took place at Lackland Air Force Base in Texas during a basic military training. Eight cases were confirmed and 45 cases were suspected. The rate of infection was 0.4% (50/12,270) among trainees and 0.3% (2/602) among instructors.7 Eight of 12 nasopharyngeal swabs tested positive for EV by way of local real-time reverse transcription–polymerase chain reaction (RT-PCR). Four nasopharyngeal swabs were sent to the CDC for evaluation and all were positive for CVA6.7
Presentation
Because the prevalence of CVA6 has increased, it is important to be able to identify the presentation of HFMD caused by this strain. Coxsackievirus A6 has been found to affect a broader demographic and cause more severe cases of HFMD with its unique constellation of findings compared to other known strains. Patients present with flulike symptoms; higher fever than present in typical HFMD; and a longer duration of disease, typically lasting 2 weeks. Patients also may present with more severe skin disease compared to classic HFMD, not only including vesicles but also large bullae, erosions, and ulcers on the dorsal and plantar feet (Figure 1).
In patients with atopic dermatitis, CVA6 also shows a predilection to appear in areas of skin disease, such as the flexural regions of the arms and legs, and is referred to as eczema coxsackium.24,38,39 It can mimic eczema herpeticum or varicella superinfection, which are important considerations to include in the differential diagnosis. Additionally, CVA6-induced lesions often show up in previously irritated or traumatized areas such as sunburns, fungal infections, and diaper dermatitis in children. Lesions have been described to sometimes mimic Gianotti-Crosti syndrome, with involvement of the extensor surfaces, buttocks, and cheeks, and sparing of the trunk.24
Clinical Diagnosis
Because HFMD is uncommon and atypical in adults, skin biopsies may be used in the initial workup and evaluation of patients. It is important to understand the histologic features associated with HFMD, including spongiosis with exocytosis of neutrophils as well as keratinocyte necrosis and pallor with associated shadow cells.6 In one series, the most extensively involved areas of keratinocyte necrosis were the stratum granulosum and upper half of the stratum spinosum.40 In the dermis, vascular involvement may be present on a spectrum with the extravasation of red blood cells and leukocytoclasis or true leukocytoclastic vasculitis.6,40 Vesicular lesions show severe dermal edema and inflammatory infiltrate.6,41 CD3+ and CD8+ lymphocytes predominate. Cytotoxic T lymphocytes are present and express granzyme B and granulysin, both important mediators of apoptosis in virally infected keratinocytes.6
Adult HFMD primarily is a clinical diagnosis, and histopathologic analysis can be a useful tool in certain cases. Coxsackievirus A6 does not grow well on culture and is not detected by standard serologic testing laboratories, necessitating the use of quantitative RT-PCR analysis.41,42 In one study, culture was able to detect only 14% to 16% of samples that tested positive by quantitative RT-PCR.43 This form of PCR identifies viral subtype through amplification of enterovirus viral protein 1 capsid gene sequence.24 Unfortunately, this testing often is not offered in most readily available laboratories and often necessitates being sent out to more well-equipped laboratories.2,24
Treatment
Hand-foot-and-mouth disease is a self-limited illness and requires only supportive care with a focus on hydration and pain management. Lesions heal without scarring but may leave notable postinflammatory pigment alteration that may last months to years, depending on extent of disease and skin type. Secondarily infected individuals should be treated with appropriate antibiotics or antivirals depending on the infectious agent. Hand hygiene is of great importance, and hospitalized patients should be put on strict contact precautions. It also is important to isolate patients from vulnerable individuals, especially pregnant women, as coxsackievirus has been linked to intrauterine infections and loss of pregnancy.24
Genetic Analysis
Genetic studies of the virus have suggested that nonstructural genes may be playing an interesting role in clinical phenotypes and outcomes of CVA6 infection.44 These genetic studies also are being implemented into the understanding of the virus’ evolution as well as the construction of vaccinations.27,44
Conclusion
With the increasing prevalence of CVA6-associated HFMD, it is important to understand the clinical presentation and histologic findings associated with this atypical presentation of the disease as well as the changing epidemiology of the viral strains causing HFMD.
- Galen WK. Cutaneous manifestations of enterovirus infections. In: Tyring SK, ed. Mucocutaneous Manifestations of Viral Diseases. New York, NY: Marcel Dekker; 2002:455-467.
- Ramirez-Fort M, Downing C, Doan H, et al. Coxsackievirus A6 associated hand, foot and mouth disease in adults: clinical presentation and review of the literature. J Clin Virol. 2014;60:381-386.
- Khetsuriani N, Lamonte-Fowlkes A, Oberst S, et al. Enterovirus surveillance—United States, 1970-2005. MMWR Surveill Summ. 2006;55:1-20.
- Yang F, Zhang T, Hu Y, et al. Survey of enterovirus infections from hand, foot and mouth disease outbreak in China, 2009. Virol J. 2011;8:508.
- Ho M, Chen ER, Hsu KH, et al. An epidemic of enterovirus 71 infection in Taiwan. Taiwan Enterovirus Epidemic Working Group. N Engl J Med. 1999;341:929-935.
- Second J, Velter C, Calès S, et al. Clinicopathologic analysis of atypical hand, foot, and mouth disease in adult patients. J Am Acad Dermatol. 2016;76:722-729.
- Banta J, Lenz B, Pawlak M, et al. Notes from the field: outbreak of hand, foot, and mouth disease caused by coxsackievirus A6 among basic military trainees—Texas, 2015. MMWR Morb Mortal Wkly Rep. 2016;65.26:678-680.
- Bian L, Wang Y, Yao X, et al. Coxsackievirus A6: a new emerging pathogen causing hand, foot and mouth disease outbreaks worldwide. Expert Rev Anti Infect Ther. 2015;13:1061-1071.
- Buttery VW, Kenyon C, Grunewald S, et al. Notes from the field: atypical presentations of hand, foot, and mouth disease caused by coxsackievirus A6—Minnesota, 2014. MMWR Morb Mortal Wkly Rep. 2015;64:805.
- Puenpa J, Chieochansin T, Linsuwanon P, et al. Hand, foot, and mouth disease caused by coxsackievirus A6, Thailand, 2012. Emerg Infect Dis. 2013;19:641-643.
- Flett K, Youngster I, Huang J, et al. Hand, foot, and mouth disease caused by coxsackievirus A6. Emerg Infect Dis. 2012;18:1702-1704.
- Centers for Disease Control and Prevention. Notes from the field: severe hand, foot, and mouth disease associated with coxsackievirus A6—Alabama, Connecticut, California, and Nevada, November 2011-February 2012. MMWR Morb Mortal Wkly Rep. 2012;61:213-214.
- Blomqvist S, Klemola P, Kaijalainen S, et al. Co-circulation of coxsackieviruses A6 and A10 in hand, foot and mouth disease outbreak in Finland. J Clin Virol. 2010;48:49-54.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Zeng H, Lu J, Zheng H, et al. The epidemiological study of coxsackievirus A6 revealing hand, foot and mouth disease epidemic patterns in Guandong, China. Sci Rep. 2015;5:10550.
- Mirand A, Henquell C, Archimbaud C, et al. Outbreak of hand, foot and mouth disease/herpangina associated with coxsackievirus A6 andA10 infections in 2010, France: a large citywide, prospective observational study. Clin Microbiol Infect. 2012;18:E110-E118.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Fujimoto T, Iizuka S, Enomoto M, et al. Hand, foot, and mouth disease caused by coxsackievirus A6, Japan, 2011. Emerg Infect Dis. 2012;18:337-339.
- Bracho MA, Gonzalez-Candelas F, Valero A, et al. Enterovirus co-infections and onychomadesis after hand, foot, and mouth disease, Spain, 2008. Emerg Infect Dis. 2011;17:2223-2231.
- Gopalkrishna V, Patil PR, Patil GP, et al. Circulation of multiple enterovirus serotypes causing hand, foot and mouth disease in India. J Med Microbiol. 2012;61:420-425.
- Lo SH, Huang YC, Huang CG, et al. Clinical and epidemiologic features of coxsackievirus A6 infection in children in northern Taiwan between 2004 and 2009. J Microbiol Immunol Infect. 2011;44:252-257.
- Lu QB, Zhang XA, Wo Y, et al. Circulation of coxsackievirus A10 and A6 in hand-foot-mouth disease in China, 2009-2011. PLoS One. 2012;7:E52073.
- Wu Y, Yeo A, Phoon MC, et al. The largest outbreak of hand; foot and mouth disease in Singapore in 2008: the role of enterovirus 71 and coxsackievirus A strains. Int J Infect Dis. 2010;14:E1076-E1081.
- Ventarola D, Bordone L, Silverberg N. Update on hand-foot-and-mouth disease. Clin Dermatol. 2015;33:340-346.
- Li Y, Chang Z, Wu P, et al. Emerging enteroviruses causing hand, foot and mouth disease, China. 2010-2016. Emerg Infect Dis. 2018;24:1902-1906.
- Tan X, Li L, Zhang B, et al. Molecular epidemiology of coxsackievirus A6 associated with outbreaks of hand, foot, and mouth disease in Tianjin, China, in 2013. Arch Virol. 2015;160:1097-1104.
- Li Y, Bao H, Zhang X, et al. Epidemiological and genetic analysis concerning the non-enterovirus 71 and non-coxsackievirus A16 causative agents related to hand, foot and mouth disease in Anyang City, Henan Province, China, from 2011 to 2015. J Med Virol. 2017;89:1749-1758.
- Guan H, Wang J, Wang C, et al. Etiology of multiple non-EV71 and non-CVA16 enteroviruses associated with hand, foot, and mouth disease in Jinan, China, 2009-2013. PLoS One. 2015;10:E0142733.
- Cabrerizo M, Tarrago´ D, Muñoz-Almagro C, et al. Mollecular epidemiology of enterovirus 71, coxsackievirus A16 and A6 associated with hand, foot and mouth disease in Spain. Clin Microbiol Infect. 2014;20:O150-O156.
- Lønnberg A, Elberling J, Fischer T, et al. Two cases of hand, foot, and mouth disease involving the scalp. Acta Derm Venereol. 2013;93:467-468.
- Lott JP, Liu K, Landry ML, et al. Atypical hand-foot-and-mouth disease associated with coxsackievirus A6 infection. J Am Acad Dermatol. 2013;69:736-741.
- Kaminska K, Martinetti G, Lucchini R, et al. Coxsackievirus A6 and hand, foot and mouth disease: three case reports of familial child-to-immunocompetent adult transmission and a literature review. Case Rep Dermatol. 2013;5:203-209.
- Shin JU, Oh SH, Lee JH. A case of hand-foot-mouth disease in an immunocompetent adult. Ann Dermatol. 2010;22:216-218.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Feder HM, Bennett N, Modlin JF. Atypical hand, foot, and mouth disease: a vesiculobullous eruption caused by coxsackie virus A6. Lancet Infect Dis. 2014;14:83-86.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Kim M, Kim B, Byun S, et al. Beau’s lines and onychomadesis after hand-foot-mouth disease. Clin Pediatr Dermatol. 2015;1:1.
- Mathes EF, Oza V, Frieden IJ, et al. “Eczema coxsackium” and unusual cutaneous findings in an enterovirus outbreak. Pediatrics. 2013;132:E149-E157.
- Lynch M, Sears A, Cookson H, et al. Disseminated coxsackievirus A6 affecting children with atopic dermatitis. Clin Exp Dermatol. 2015;40:525-528.
- Laga A, Shroba S, Hanna J. Atypical hand, foot and mouth disease in adults associated with coxsackievirus A6: a clinicopathologic study. J Cutan Pathol. 2016;43:940-945.
- Schmidt NJ, Ho HH, Lennette EH. Propagation and isolation of group A coxsackieviruses in RD cells. J Clin Microbiol. 1975;2:183-185.
- Oberste MS, Penaranda S, Rogers SL, et al. Comparative evaluation of Taqman real-time PCR and semi-nested VP1 PCR for detection of enteroviruses in clinical specimens. J Clin Virol. 2010;49:73-74.
- Lee MK, Chan PK, Ho II, et al. Enterovirus infection among patients admitted to hospital in Hong Kong in 2010: epidemiology, clinical characteristics, and importance of molecular diagnosis. J Med Virol. 2013;85:1811-1817.
- Yee PTI, Laa Poh C. Impact of genetic changes, pathogenicity and antigenicity on enterovirus A71 vaccine development. Virology. 2017;506:121-129.
- Galen WK. Cutaneous manifestations of enterovirus infections. In: Tyring SK, ed. Mucocutaneous Manifestations of Viral Diseases. New York, NY: Marcel Dekker; 2002:455-467.
- Ramirez-Fort M, Downing C, Doan H, et al. Coxsackievirus A6 associated hand, foot and mouth disease in adults: clinical presentation and review of the literature. J Clin Virol. 2014;60:381-386.
- Khetsuriani N, Lamonte-Fowlkes A, Oberst S, et al. Enterovirus surveillance—United States, 1970-2005. MMWR Surveill Summ. 2006;55:1-20.
- Yang F, Zhang T, Hu Y, et al. Survey of enterovirus infections from hand, foot and mouth disease outbreak in China, 2009. Virol J. 2011;8:508.
- Ho M, Chen ER, Hsu KH, et al. An epidemic of enterovirus 71 infection in Taiwan. Taiwan Enterovirus Epidemic Working Group. N Engl J Med. 1999;341:929-935.
- Second J, Velter C, Calès S, et al. Clinicopathologic analysis of atypical hand, foot, and mouth disease in adult patients. J Am Acad Dermatol. 2016;76:722-729.
- Banta J, Lenz B, Pawlak M, et al. Notes from the field: outbreak of hand, foot, and mouth disease caused by coxsackievirus A6 among basic military trainees—Texas, 2015. MMWR Morb Mortal Wkly Rep. 2016;65.26:678-680.
- Bian L, Wang Y, Yao X, et al. Coxsackievirus A6: a new emerging pathogen causing hand, foot and mouth disease outbreaks worldwide. Expert Rev Anti Infect Ther. 2015;13:1061-1071.
- Buttery VW, Kenyon C, Grunewald S, et al. Notes from the field: atypical presentations of hand, foot, and mouth disease caused by coxsackievirus A6—Minnesota, 2014. MMWR Morb Mortal Wkly Rep. 2015;64:805.
- Puenpa J, Chieochansin T, Linsuwanon P, et al. Hand, foot, and mouth disease caused by coxsackievirus A6, Thailand, 2012. Emerg Infect Dis. 2013;19:641-643.
- Flett K, Youngster I, Huang J, et al. Hand, foot, and mouth disease caused by coxsackievirus A6. Emerg Infect Dis. 2012;18:1702-1704.
- Centers for Disease Control and Prevention. Notes from the field: severe hand, foot, and mouth disease associated with coxsackievirus A6—Alabama, Connecticut, California, and Nevada, November 2011-February 2012. MMWR Morb Mortal Wkly Rep. 2012;61:213-214.
- Blomqvist S, Klemola P, Kaijalainen S, et al. Co-circulation of coxsackieviruses A6 and A10 in hand, foot and mouth disease outbreak in Finland. J Clin Virol. 2010;48:49-54.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Zeng H, Lu J, Zheng H, et al. The epidemiological study of coxsackievirus A6 revealing hand, foot and mouth disease epidemic patterns in Guandong, China. Sci Rep. 2015;5:10550.
- Mirand A, Henquell C, Archimbaud C, et al. Outbreak of hand, foot and mouth disease/herpangina associated with coxsackievirus A6 andA10 infections in 2010, France: a large citywide, prospective observational study. Clin Microbiol Infect. 2012;18:E110-E118.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Fujimoto T, Iizuka S, Enomoto M, et al. Hand, foot, and mouth disease caused by coxsackievirus A6, Japan, 2011. Emerg Infect Dis. 2012;18:337-339.
- Bracho MA, Gonzalez-Candelas F, Valero A, et al. Enterovirus co-infections and onychomadesis after hand, foot, and mouth disease, Spain, 2008. Emerg Infect Dis. 2011;17:2223-2231.
- Gopalkrishna V, Patil PR, Patil GP, et al. Circulation of multiple enterovirus serotypes causing hand, foot and mouth disease in India. J Med Microbiol. 2012;61:420-425.
- Lo SH, Huang YC, Huang CG, et al. Clinical and epidemiologic features of coxsackievirus A6 infection in children in northern Taiwan between 2004 and 2009. J Microbiol Immunol Infect. 2011;44:252-257.
- Lu QB, Zhang XA, Wo Y, et al. Circulation of coxsackievirus A10 and A6 in hand-foot-mouth disease in China, 2009-2011. PLoS One. 2012;7:E52073.
- Wu Y, Yeo A, Phoon MC, et al. The largest outbreak of hand; foot and mouth disease in Singapore in 2008: the role of enterovirus 71 and coxsackievirus A strains. Int J Infect Dis. 2010;14:E1076-E1081.
- Ventarola D, Bordone L, Silverberg N. Update on hand-foot-and-mouth disease. Clin Dermatol. 2015;33:340-346.
- Li Y, Chang Z, Wu P, et al. Emerging enteroviruses causing hand, foot and mouth disease, China. 2010-2016. Emerg Infect Dis. 2018;24:1902-1906.
- Tan X, Li L, Zhang B, et al. Molecular epidemiology of coxsackievirus A6 associated with outbreaks of hand, foot, and mouth disease in Tianjin, China, in 2013. Arch Virol. 2015;160:1097-1104.
- Li Y, Bao H, Zhang X, et al. Epidemiological and genetic analysis concerning the non-enterovirus 71 and non-coxsackievirus A16 causative agents related to hand, foot and mouth disease in Anyang City, Henan Province, China, from 2011 to 2015. J Med Virol. 2017;89:1749-1758.
- Guan H, Wang J, Wang C, et al. Etiology of multiple non-EV71 and non-CVA16 enteroviruses associated with hand, foot, and mouth disease in Jinan, China, 2009-2013. PLoS One. 2015;10:E0142733.
- Cabrerizo M, Tarrago´ D, Muñoz-Almagro C, et al. Mollecular epidemiology of enterovirus 71, coxsackievirus A16 and A6 associated with hand, foot and mouth disease in Spain. Clin Microbiol Infect. 2014;20:O150-O156.
- Lønnberg A, Elberling J, Fischer T, et al. Two cases of hand, foot, and mouth disease involving the scalp. Acta Derm Venereol. 2013;93:467-468.
- Lott JP, Liu K, Landry ML, et al. Atypical hand-foot-and-mouth disease associated with coxsackievirus A6 infection. J Am Acad Dermatol. 2013;69:736-741.
- Kaminska K, Martinetti G, Lucchini R, et al. Coxsackievirus A6 and hand, foot and mouth disease: three case reports of familial child-to-immunocompetent adult transmission and a literature review. Case Rep Dermatol. 2013;5:203-209.
- Shin JU, Oh SH, Lee JH. A case of hand-foot-mouth disease in an immunocompetent adult. Ann Dermatol. 2010;22:216-218.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Feder HM, Bennett N, Modlin JF. Atypical hand, foot, and mouth disease: a vesiculobullous eruption caused by coxsackie virus A6. Lancet Infect Dis. 2014;14:83-86.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Kim M, Kim B, Byun S, et al. Beau’s lines and onychomadesis after hand-foot-mouth disease. Clin Pediatr Dermatol. 2015;1:1.
- Mathes EF, Oza V, Frieden IJ, et al. “Eczema coxsackium” and unusual cutaneous findings in an enterovirus outbreak. Pediatrics. 2013;132:E149-E157.
- Lynch M, Sears A, Cookson H, et al. Disseminated coxsackievirus A6 affecting children with atopic dermatitis. Clin Exp Dermatol. 2015;40:525-528.
- Laga A, Shroba S, Hanna J. Atypical hand, foot and mouth disease in adults associated with coxsackievirus A6: a clinicopathologic study. J Cutan Pathol. 2016;43:940-945.
- Schmidt NJ, Ho HH, Lennette EH. Propagation and isolation of group A coxsackieviruses in RD cells. J Clin Microbiol. 1975;2:183-185.
- Oberste MS, Penaranda S, Rogers SL, et al. Comparative evaluation of Taqman real-time PCR and semi-nested VP1 PCR for detection of enteroviruses in clinical specimens. J Clin Virol. 2010;49:73-74.
- Lee MK, Chan PK, Ho II, et al. Enterovirus infection among patients admitted to hospital in Hong Kong in 2010: epidemiology, clinical characteristics, and importance of molecular diagnosis. J Med Virol. 2013;85:1811-1817.
- Yee PTI, Laa Poh C. Impact of genetic changes, pathogenicity and antigenicity on enterovirus A71 vaccine development. Virology. 2017;506:121-129.
Practice Points
- Coxsackievirus A6 is an increasingly more common cause of hand-foot-and-mouth disease (HFMD), often with atypical presentation, more severe disease, and association with HFMD in adults.
- Coxsackievirus A6 has become a major cause of HFMD outbreak in the United States and worldwide.
Patient-Reported Outcomes of Azelaic Acid Foam 15% for Patients With Papulopustular Rosacea: Secondary Efficacy Results From a Randomized, Controlled, Double-blind, Phase 3 Trial
Rosacea is a chronic inflammatory disorder that may negatively impact patients’ quality of life (QOL).1,2 Papulopustular rosacea (PPR) is characterized by centrofacial inflammatory lesions and erythema as well as burning and stinging secondary to skin barrier dysfunction.3-5 Increasing rosacea severity is associated with greater rates of anxiety and depression and lower QOL6 as well as low self-esteem and feelings of embarrassment.7,8 Accordingly, assessing patient perceptions of rosacea treatments is necessary for understanding its impact on patient health.6,9
The Rosacea International Expert Group has emphasized the need to incorporate patient assessments of disease severity and QOL when developing therapeutic strategies for rosacea.7 Ease of use, sensory experience, and patient preference also are important dimensions in the evaluation of topical medications, as attributes of specific formulations may affect usability, adherence, and efficacy.10,11
An azelaic acid (AzA) 15% foam formulation, which was approved by the US Food and Drug Administration in 2015, was developed to deliver AzA in a vehicle designed to improve treatment experience in patients with mild to moderate PPR.12 Results from a clinical trial demonstrated superiority of AzA foam to vehicle foam for primary end points that included therapeutic success rate and change in inflammatory lesion count.13,14 Secondary end points assessed in the current analysis included patient perception of product usability, efficacy, and effect on QOL. These patient-reported outcome (PRO) results are reported here.
Methods
Study Design
The design of this phase 3 multicenter, randomized, double-blind, vehicle-controlled, parallel-group clinical trial was described in more detail in an earlier report.13 This study was approved by all appropriate institutional review boards. Eligible participants were 18 years and older with moderate or severe PPR, 12 to 50 inflammatory lesions, and persistent erythema with or without telangiectasia. Exclusion criteria included known nonresponse to AzA, current or prior use (within 6 weeks of randomization) of noninvestigational products to treat rosacea, and presence of other dermatoses that could interfere with rosacea evaluation.
Participants were randomized into the AzA foam or vehicle group (1:1 ratio). The study medication (0.5 g) or vehicle foam was applied twice daily to the entire face until the end of treatment (EoT) at 12 weeks. Efficacy and safety parameters were evaluated at baseline and at 4, 8, and 12 weeks of treatment, and at a follow-up visit 4 weeks after EoT (week 16).
Results for the coprimary efficacy end points—therapeutic success rate according to investigator global assessment and nominal change in inflammatory lesion count—were previously reported,13 as well as secondary efficacy outcomes including change in inflammatory lesion count, therapeutic response rate, and change in erythema rating.14
Patient-Reported Secondary Efficacy Outcomes
The secondary PRO end points were patient-reported global assessment of treatment response (rated as excellent, good, fair, none, or worse), global assessment of tolerability (rated as excellent, good, acceptable despite minor irritation, less acceptable due to continuous irritation, not acceptable, or no opinion), and opinion on cosmetic acceptability and practicability of product use in areas adjacent to the hairline (rated as very good, good, satisfactory, poor, or no opinion).
Additionally, QOL was measured by 3 validated standardized PRO tools, including the Rosacea Quality of Life Index (RosaQOL),15 the EuroQOL 5-dimension 5-level questionnaire (EQ-5D-5L),16 and the Dermatology Life Quality Index (DLQI). The RosaQOL is a rosacea-specific instrument assessing 3 constructs: (1) symptom, (2) emotion, and (3) function. The EQ-5D-5L questionnaire measures overall health status and comprises 5 constructs: (1) mobility, (2) self-care, (3) usual activities, (4) pain/discomfort, and (5) anxiety/depression. The DLQI is a general, dermatology-oriented instrument categorized into 6 constructs: (1) symptoms and feelings, (2) daily activities, (3) leisure, (4) work and school, (5) personal relationships, and (6) treatment.
Statistical Analyses
Patient-reported outcomes were analyzed in an exploratory manner and evaluated at EoT relative to baseline. Self-reported global assessment of treatment response and change in RosaQOL, EQ-5D-5L, and DLQI scores between AzA foam and vehicle foam groups were evaluated using the Wilcoxon rank sum test. Categorical change in the number of participants achieving an increase of 5 or more points in overall DLQI score was evaluated using a χ2 test.
Safety
Safety was analyzed for all randomized patients who were dispensed any study medication. All analyses were performed using SAS version 9.2.
Results
Of the 961 participants included in the study, 483 were randomized to receive AzA foam and 478 were randomized to receive vehicle foam. The mean age was 51.5 years, and the majority of participants were female (73.0%) and white (95.5%)(Table). At baseline, 834 (86.8%) participants had moderate PPR and 127 (13.2%) had severe PPR. The mean inflammatory lesion count (SD) was 21.4 (8.9). No significant differences in baseline characteristics were observed between treatment groups.

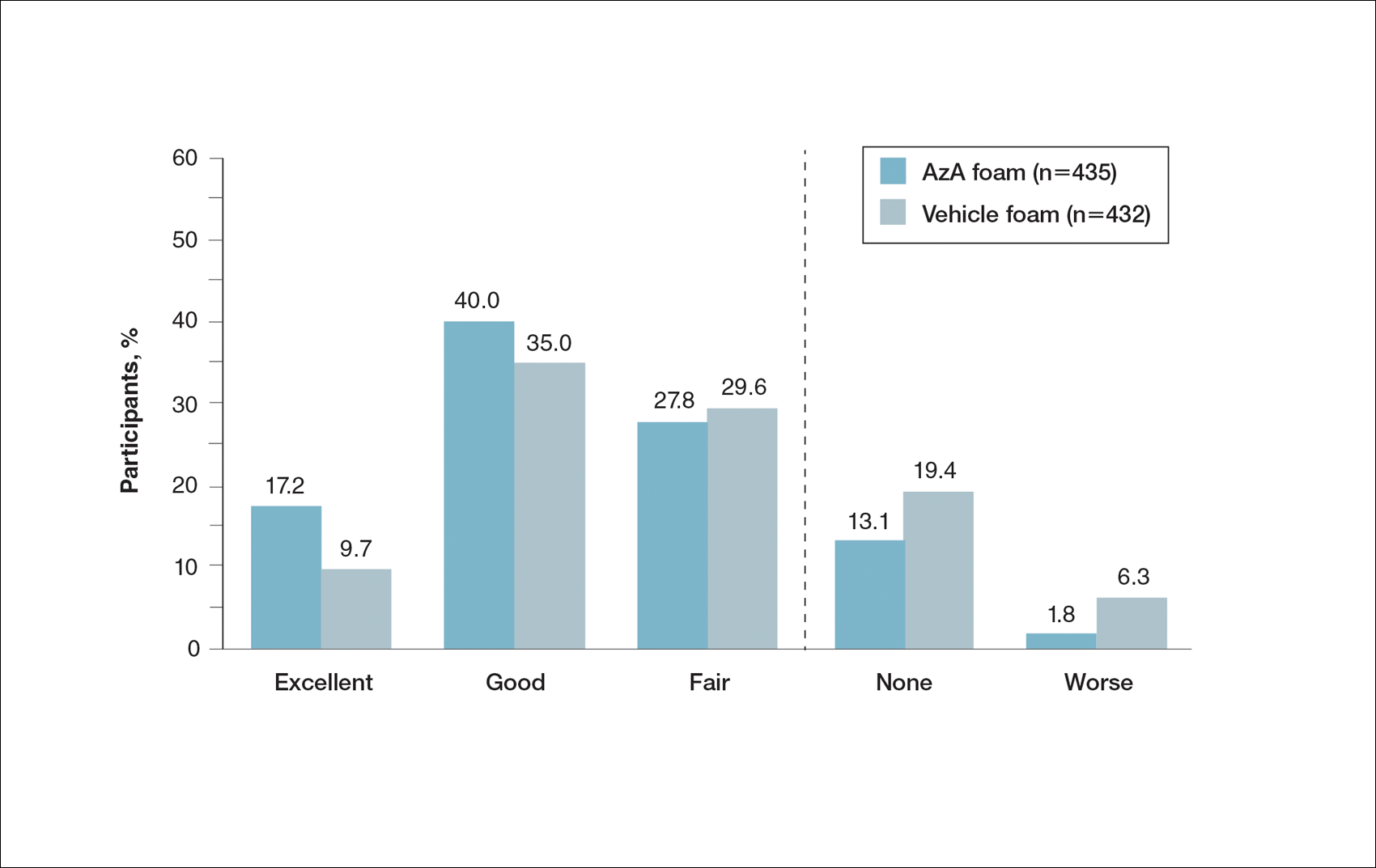
Patient-reported global assessment of treatment response differed between treatment groups at EoT (P<.001)(Figure 1). Higher ratings of treatment response were reported among the AzA foam group (excellent, 17.2%; good, 40.0%) versus vehicle foam (excellent, 9.7%; good, 35.0%). The number of participants reporting no treatment response was 13.1% in the AzA foam group, with 1.8% reporting worsening of their condition, while 19.4% of participants in the vehicle foam group reported no response, with 6.3% reporting worsening of their condition (Figure 1).
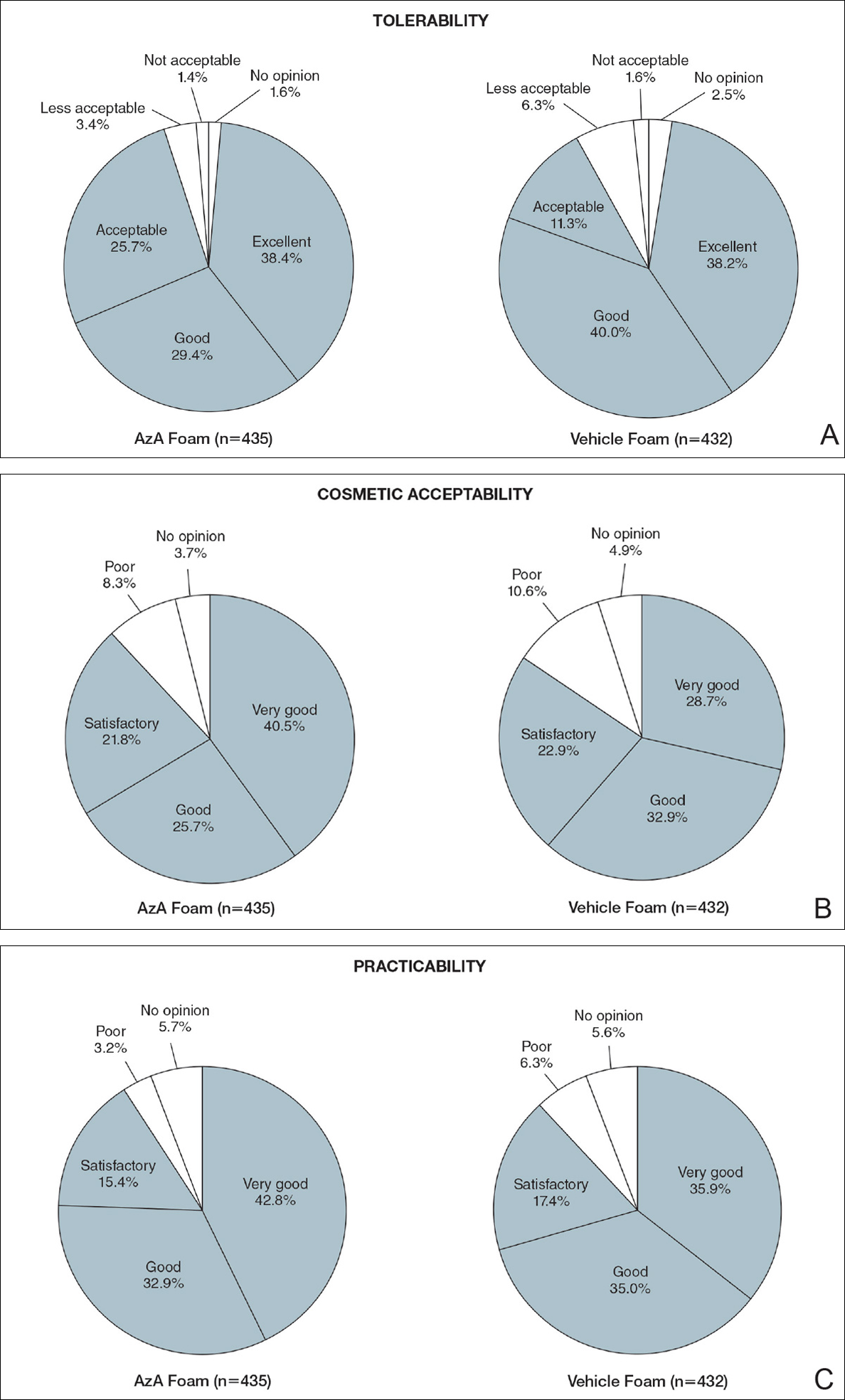
Tolerability was rated excellent or good in 67.8% of the AzA foam group versus 78.2% of the vehicle foam group (Figure 2A). Approximately 38.4% of the AzA foam group versus 38.2% of the vehicle foam group rated treatment tolerability as excellent, while 93.5% of the AzA foam group rated tolerability as acceptable, good, or excellent compared with 89.5% of the vehicle foam group. Only 1.4% of participants in the AzA foam group indicated that treatment was not acceptable due to irritation. In addition, a greater proportion of the AzA foam group reported cosmetic acceptability as very good versus the vehicle foam group (40.5% vs 28.7%)(Figure 2B), with two-thirds reporting cosmetic acceptability as very good or good. Practicability of product use in areas adjacent to the hairline was rated very good by substantial proportions of both the AzA foam and vehicle foam groups (42.8% vs 35.9%)(Figure 2C).
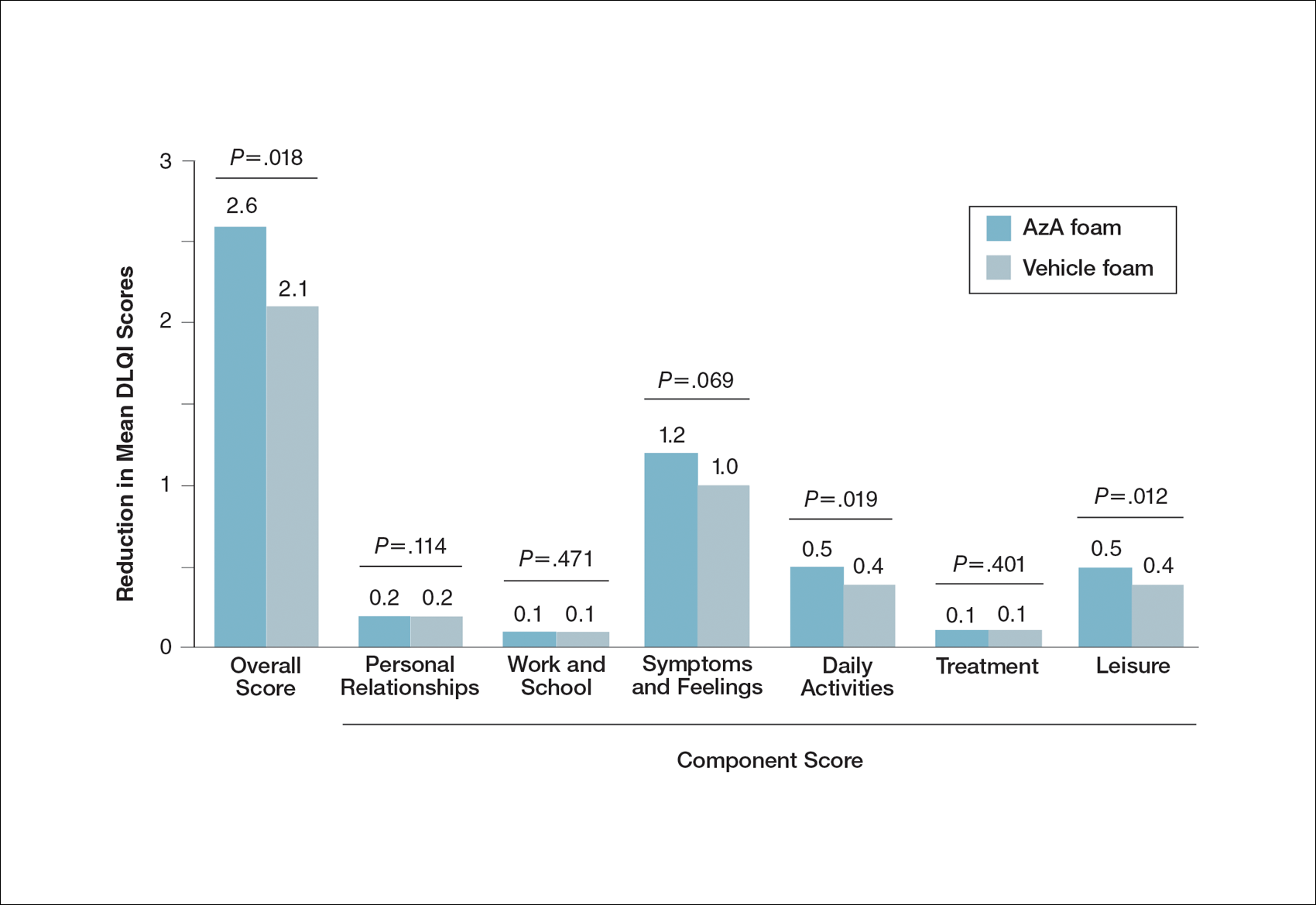
At baseline, average disease burden was moderate according to mean overall DLQI scores (SD) for the AzA foam (5.4 [4.8]) and vehicle foam (5.4 [4.9]) groups. Mean overall DLQI scores improved at EoT, with greater improvement occurring in the AzA foam group (2.6 vs 2.1; P=.018)(Figure 3). A larger proportion of participants in the AzA foam group versus the vehicle foam group also achieved a 5-point or more improvement in overall DLQI score (24.6% vs 19.0%; P=.047). Changes in specific DLQI subscore components were either balanced or in favor of the AzA foam group, including daily activities (0.5 vs 0.4; P=.019), symptoms and feelings (1.2 vs 1.0; P=.069), and leisure (0.5 vs 0.4; P=.012). Specific DLQI items with differences in scores between treatment groups from baseline included the following questions: Over the last week, how embarrassed or self-conscious have you been because of your skin? (P<.001); Over the last week, how much has your skin interfered with you going shopping or looking after your home or garden? (P=.005); Over the last week, how much has your skin affected any social or leisure activities? (P=.040); Over the last week, how much has your skin created problems with your partner or any of your close friends or relatives? (P=.001). Differences between treatment groups favored the AzA foam group for each of these items.
Participants in the AzA foam and vehicle foam groups also showed improvement in RosaQOL scores at EoT (6.8 vs 6.4; P=.67), while EQ-5D-5L scores changed minimally from baseline (0.006 vs 0.007; P=.50).
Safety
The incidence of drug-related adverse events (AEs) was greater in the AzA foam group versus the vehicle foam group (7.7% vs 4.8%). Drug-related AEs occurring in 1% of the AzA foam group were application-site pain including tenderness, stinging, and burning (3.5% for AzA foam vs 1.3% for vehicle foam); application-site pruritus (1.4% vs 0.4%); and application-site dryness (1.0% vs 0.6%). One drug-related AE of severe intensity—application-site dermatitis—occurred in the vehicle foam group; all other drug-related AEs were mild or moderate.14 More detailed safety results are described in a previous report.13
Comment
The PRO outcome data reported here are consistent with previously reported statistically significant improvements in investigator-assessed primary end points for the treatment of PPR with AzA foam.13,14 The data demonstrate that AzA foam benefits both clinical and patient-oriented dimensions of rosacea disease burden and suggest an association between positive treatment response and improved QOL.
Specifically, patient evaluation of treatment response to AzA foam was highly favorable, with 57.2% reporting excellent or good response and 85.1% reporting positive response overall. Recognizing the relapsing-remitting course of PPR, only 1.8% of the AzA foam group experienced worsening of disease at EoT.
The DLQI and RosaQOL instruments revealed notable improvements in QOL from baseline for both treatment groups. Although no significant differences in RosaQOL scores were observed between groups at EoT, significant differences in DLQI scores were detected. Almost one-quarter of participants in the AzA foam group achieved at least a 5-point improvement in DLQI score, exceeding the 4-point threshold for clinically meaningful change.17 Although little change in EQ-5D-5L scores was observed at EoT for both groups with no between-group differences, this finding is not unexpected, as this instrument assesses QOL dimensions such as loss of function, mobility, and ability to wash or dress, which are unlikely to be compromised in most rosacea patients.
Our results also underscore the importance of vehicle in the treatment of compromised skin. Studies of topical treatments for other dermatoses suggest that vehicle properties may reduce disease severity and improve QOL independent of active ingredients.10,18 For example, ease of application, minimal residue, and less time spent in application may explain the superiority of foam to other vehicles in the treatment of psoriasis.18 Our data demonstrating high cosmetic favorability of AzA foam are consistent with these prior observations. Increased tolerability of foam formulations also may affect response to treatment, in part by supporting adherence.18 Most participants receiving AzA foam described tolerability as excellent or good, and the discontinuation rate was low (1.2% of participants in the AzA foam group left the study due to AEs) in the setting of near-complete dosage administration (97% of expected doses applied).13
Conclusion
These results indicate that use of AzA foam as well as its novel vehicle results in high patient satisfaction and improved QOL. Although additional research is necessary to further delineate the relationship between PROs and other measures of clinical efficacy, our data demonstrate a positive treatment experience as perceived by patients that parallels the clinical efficacy of AzA foam for the treatment of PPR.13,14
Acknowledgment
Editorial support through inVentiv Medical Communications (New York, New York) was provided by Bayer Pharmaceuticals.
- Cardwell LA, Farhangian ME, Alinia H, et al. Psychological disorders associated with rosacea: analysis of unscripted comments. J Dermatol Surg. 2015;19:99-103.
- Moustafa F, Lewallen RS, Feldman SR. The psychological impact of rosacea and the influence of current management options. J Am Acad Dermatol. 2014;71:973-980.
- Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the Classification and Staging of Rosacea. J Am Acad Dermatol. 2002;46:584-587.
- Yamasaki K, Gallo RL. The molecular pathology of rosacea. J Dermatol Sci. 2009;55:77-81.
- Del Rosso JQ. Advances in understanding and managing rosacea: part 1: connecting the dots between pathophysiological mechanisms and common clinical features of rosacea with emphasis on vascular changes and facial erythema. J Clin Aesthet Dermatol. 2012;5:16-25.
- Bohm D, Schwanitz P, Stock Gissendanner S, et al. Symptom severity and psychological sequelae in rosacea: results of a survey. Psychol Health Med. 2014;19:586-591.
- Elewski BE, Draelos Z, Dreno B, et al. Rosacea—global diversity and optimized outcome: proposed international consensus from the Rosacea International Expert Group. J Eur Acad Dermatol Venereol. 2011;25:188-200.
- Dirschka T, Micali G, Papadopoulos L, et al. Perceptions on the psychological impact of facial erythema associated with rosacea: results of international survey [published online May 29, 2015]. Dermatol Ther (Heidelb). 2015;5:117-127.
- Abram K, Silm H, Maaroos HI, et al. Subjective disease perception and symptoms of depression in relation to healthcare-seeking behaviour in patients with rosacea. Acta Derm Venereol. 2009;89:488-491.
- Stein L. Clinical studies of a new vehicle formulation for topical corticosteroids in the treatment of psoriasis. J Am Acad Dermatol. 2005;53(1, suppl 1):S39-S49.
- Yentzer BA, Camacho FT, Young T, et al. Good adherence and early efficacy using desonide hydrogel for atopic dermatitis: results from a program addressing patient compliance. J Drugs Dermatol. 2010;9:324-329.
- Finacea (azelaic acid) foam 15% [package insert]. Whippany, NJ: Bayer Pharmaceuticals; 2015.
- Draelos ZD, Elewski BE, Harper JC, et al. A phase 3 randomized, double-blind, vehicle-controlled trial of azelaic acid foam 15% in the treatment of papulopustular rosacea. Cutis. 2015;96:54-61.
- Solomon JA, Tyring S, Staedtler G, et al. Investigator-reported efficacy of azelaic acid foam 15% in patients with papulopustular rosacea: secondary efficacy outcomes from a randomized, controlled, double-blind, phase 3 trial. Cutis. 2016;98:187-194.
- Nicholson K, Abramova L, Chren MM, et al. A pilot quality-of-life instrument for acne rosacea. J Am Acad Dermatol. 2007;57:213-221.
- Herdman M, Gudex C, Lloyd A, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. 2011;20:1727-1736.
- Basra MK, Salek MS, Camilleri L, et al. Determining the minimal clinically important difference and responsiveness of the Dermatology Life Quality Index (DLQI): further data. Dermatology. 2015;230:27-33.
- Bergstrom KG, Arambula K, Kimball AB. Medication formulation affects quality of life: a randomized single-blind study of clobetasol propionate foam 0.05% compared with a combined program of clobetasol cream 0.05% and solution 0.05% for the treatment of psoriasis. Cutis. 2003;72:407-411.
Rosacea is a chronic inflammatory disorder that may negatively impact patients’ quality of life (QOL).1,2 Papulopustular rosacea (PPR) is characterized by centrofacial inflammatory lesions and erythema as well as burning and stinging secondary to skin barrier dysfunction.3-5 Increasing rosacea severity is associated with greater rates of anxiety and depression and lower QOL6 as well as low self-esteem and feelings of embarrassment.7,8 Accordingly, assessing patient perceptions of rosacea treatments is necessary for understanding its impact on patient health.6,9
The Rosacea International Expert Group has emphasized the need to incorporate patient assessments of disease severity and QOL when developing therapeutic strategies for rosacea.7 Ease of use, sensory experience, and patient preference also are important dimensions in the evaluation of topical medications, as attributes of specific formulations may affect usability, adherence, and efficacy.10,11
An azelaic acid (AzA) 15% foam formulation, which was approved by the US Food and Drug Administration in 2015, was developed to deliver AzA in a vehicle designed to improve treatment experience in patients with mild to moderate PPR.12 Results from a clinical trial demonstrated superiority of AzA foam to vehicle foam for primary end points that included therapeutic success rate and change in inflammatory lesion count.13,14 Secondary end points assessed in the current analysis included patient perception of product usability, efficacy, and effect on QOL. These patient-reported outcome (PRO) results are reported here.
Methods
Study Design
The design of this phase 3 multicenter, randomized, double-blind, vehicle-controlled, parallel-group clinical trial was described in more detail in an earlier report.13 This study was approved by all appropriate institutional review boards. Eligible participants were 18 years and older with moderate or severe PPR, 12 to 50 inflammatory lesions, and persistent erythema with or without telangiectasia. Exclusion criteria included known nonresponse to AzA, current or prior use (within 6 weeks of randomization) of noninvestigational products to treat rosacea, and presence of other dermatoses that could interfere with rosacea evaluation.
Participants were randomized into the AzA foam or vehicle group (1:1 ratio). The study medication (0.5 g) or vehicle foam was applied twice daily to the entire face until the end of treatment (EoT) at 12 weeks. Efficacy and safety parameters were evaluated at baseline and at 4, 8, and 12 weeks of treatment, and at a follow-up visit 4 weeks after EoT (week 16).
Results for the coprimary efficacy end points—therapeutic success rate according to investigator global assessment and nominal change in inflammatory lesion count—were previously reported,13 as well as secondary efficacy outcomes including change in inflammatory lesion count, therapeutic response rate, and change in erythema rating.14
Patient-Reported Secondary Efficacy Outcomes
The secondary PRO end points were patient-reported global assessment of treatment response (rated as excellent, good, fair, none, or worse), global assessment of tolerability (rated as excellent, good, acceptable despite minor irritation, less acceptable due to continuous irritation, not acceptable, or no opinion), and opinion on cosmetic acceptability and practicability of product use in areas adjacent to the hairline (rated as very good, good, satisfactory, poor, or no opinion).
Additionally, QOL was measured by 3 validated standardized PRO tools, including the Rosacea Quality of Life Index (RosaQOL),15 the EuroQOL 5-dimension 5-level questionnaire (EQ-5D-5L),16 and the Dermatology Life Quality Index (DLQI). The RosaQOL is a rosacea-specific instrument assessing 3 constructs: (1) symptom, (2) emotion, and (3) function. The EQ-5D-5L questionnaire measures overall health status and comprises 5 constructs: (1) mobility, (2) self-care, (3) usual activities, (4) pain/discomfort, and (5) anxiety/depression. The DLQI is a general, dermatology-oriented instrument categorized into 6 constructs: (1) symptoms and feelings, (2) daily activities, (3) leisure, (4) work and school, (5) personal relationships, and (6) treatment.
Statistical Analyses
Patient-reported outcomes were analyzed in an exploratory manner and evaluated at EoT relative to baseline. Self-reported global assessment of treatment response and change in RosaQOL, EQ-5D-5L, and DLQI scores between AzA foam and vehicle foam groups were evaluated using the Wilcoxon rank sum test. Categorical change in the number of participants achieving an increase of 5 or more points in overall DLQI score was evaluated using a χ2 test.
Safety
Safety was analyzed for all randomized patients who were dispensed any study medication. All analyses were performed using SAS version 9.2.
Results
Of the 961 participants included in the study, 483 were randomized to receive AzA foam and 478 were randomized to receive vehicle foam. The mean age was 51.5 years, and the majority of participants were female (73.0%) and white (95.5%)(Table). At baseline, 834 (86.8%) participants had moderate PPR and 127 (13.2%) had severe PPR. The mean inflammatory lesion count (SD) was 21.4 (8.9). No significant differences in baseline characteristics were observed between treatment groups.
Patient-reported global assessment of treatment response differed between treatment groups at EoT (P<.001)(Figure 1). Higher ratings of treatment response were reported among the AzA foam group (excellent, 17.2%; good, 40.0%) versus vehicle foam (excellent, 9.7%; good, 35.0%). The number of participants reporting no treatment response was 13.1% in the AzA foam group, with 1.8% reporting worsening of their condition, while 19.4% of participants in the vehicle foam group reported no response, with 6.3% reporting worsening of their condition (Figure 1).
Tolerability was rated excellent or good in 67.8% of the AzA foam group versus 78.2% of the vehicle foam group (Figure 2A). Approximately 38.4% of the AzA foam group versus 38.2% of the vehicle foam group rated treatment tolerability as excellent, while 93.5% of the AzA foam group rated tolerability as acceptable, good, or excellent compared with 89.5% of the vehicle foam group. Only 1.4% of participants in the AzA foam group indicated that treatment was not acceptable due to irritation. In addition, a greater proportion of the AzA foam group reported cosmetic acceptability as very good versus the vehicle foam group (40.5% vs 28.7%)(Figure 2B), with two-thirds reporting cosmetic acceptability as very good or good. Practicability of product use in areas adjacent to the hairline was rated very good by substantial proportions of both the AzA foam and vehicle foam groups (42.8% vs 35.9%)(Figure 2C).
At baseline, average disease burden was moderate according to mean overall DLQI scores (SD) for the AzA foam (5.4 [4.8]) and vehicle foam (5.4 [4.9]) groups. Mean overall DLQI scores improved at EoT, with greater improvement occurring in the AzA foam group (2.6 vs 2.1; P=.018)(Figure 3). A larger proportion of participants in the AzA foam group versus the vehicle foam group also achieved a 5-point or more improvement in overall DLQI score (24.6% vs 19.0%; P=.047). Changes in specific DLQI subscore components were either balanced or in favor of the AzA foam group, including daily activities (0.5 vs 0.4; P=.019), symptoms and feelings (1.2 vs 1.0; P=.069), and leisure (0.5 vs 0.4; P=.012). Specific DLQI items with differences in scores between treatment groups from baseline included the following questions: Over the last week, how embarrassed or self-conscious have you been because of your skin? (P<.001); Over the last week, how much has your skin interfered with you going shopping or looking after your home or garden? (P=.005); Over the last week, how much has your skin affected any social or leisure activities? (P=.040); Over the last week, how much has your skin created problems with your partner or any of your close friends or relatives? (P=.001). Differences between treatment groups favored the AzA foam group for each of these items.
Participants in the AzA foam and vehicle foam groups also showed improvement in RosaQOL scores at EoT (6.8 vs 6.4; P=.67), while EQ-5D-5L scores changed minimally from baseline (0.006 vs 0.007; P=.50).
Safety
The incidence of drug-related adverse events (AEs) was greater in the AzA foam group versus the vehicle foam group (7.7% vs 4.8%). Drug-related AEs occurring in 1% of the AzA foam group were application-site pain including tenderness, stinging, and burning (3.5% for AzA foam vs 1.3% for vehicle foam); application-site pruritus (1.4% vs 0.4%); and application-site dryness (1.0% vs 0.6%). One drug-related AE of severe intensity—application-site dermatitis—occurred in the vehicle foam group; all other drug-related AEs were mild or moderate.14 More detailed safety results are described in a previous report.13
Comment
The PRO outcome data reported here are consistent with previously reported statistically significant improvements in investigator-assessed primary end points for the treatment of PPR with AzA foam.13,14 The data demonstrate that AzA foam benefits both clinical and patient-oriented dimensions of rosacea disease burden and suggest an association between positive treatment response and improved QOL.
Specifically, patient evaluation of treatment response to AzA foam was highly favorable, with 57.2% reporting excellent or good response and 85.1% reporting positive response overall. Recognizing the relapsing-remitting course of PPR, only 1.8% of the AzA foam group experienced worsening of disease at EoT.
The DLQI and RosaQOL instruments revealed notable improvements in QOL from baseline for both treatment groups. Although no significant differences in RosaQOL scores were observed between groups at EoT, significant differences in DLQI scores were detected. Almost one-quarter of participants in the AzA foam group achieved at least a 5-point improvement in DLQI score, exceeding the 4-point threshold for clinically meaningful change.17 Although little change in EQ-5D-5L scores was observed at EoT for both groups with no between-group differences, this finding is not unexpected, as this instrument assesses QOL dimensions such as loss of function, mobility, and ability to wash or dress, which are unlikely to be compromised in most rosacea patients.
Our results also underscore the importance of vehicle in the treatment of compromised skin. Studies of topical treatments for other dermatoses suggest that vehicle properties may reduce disease severity and improve QOL independent of active ingredients.10,18 For example, ease of application, minimal residue, and less time spent in application may explain the superiority of foam to other vehicles in the treatment of psoriasis.18 Our data demonstrating high cosmetic favorability of AzA foam are consistent with these prior observations. Increased tolerability of foam formulations also may affect response to treatment, in part by supporting adherence.18 Most participants receiving AzA foam described tolerability as excellent or good, and the discontinuation rate was low (1.2% of participants in the AzA foam group left the study due to AEs) in the setting of near-complete dosage administration (97% of expected doses applied).13
Conclusion
These results indicate that use of AzA foam as well as its novel vehicle results in high patient satisfaction and improved QOL. Although additional research is necessary to further delineate the relationship between PROs and other measures of clinical efficacy, our data demonstrate a positive treatment experience as perceived by patients that parallels the clinical efficacy of AzA foam for the treatment of PPR.13,14
Acknowledgment
Editorial support through inVentiv Medical Communications (New York, New York) was provided by Bayer Pharmaceuticals.
Rosacea is a chronic inflammatory disorder that may negatively impact patients’ quality of life (QOL).1,2 Papulopustular rosacea (PPR) is characterized by centrofacial inflammatory lesions and erythema as well as burning and stinging secondary to skin barrier dysfunction.3-5 Increasing rosacea severity is associated with greater rates of anxiety and depression and lower QOL6 as well as low self-esteem and feelings of embarrassment.7,8 Accordingly, assessing patient perceptions of rosacea treatments is necessary for understanding its impact on patient health.6,9
The Rosacea International Expert Group has emphasized the need to incorporate patient assessments of disease severity and QOL when developing therapeutic strategies for rosacea.7 Ease of use, sensory experience, and patient preference also are important dimensions in the evaluation of topical medications, as attributes of specific formulations may affect usability, adherence, and efficacy.10,11
An azelaic acid (AzA) 15% foam formulation, which was approved by the US Food and Drug Administration in 2015, was developed to deliver AzA in a vehicle designed to improve treatment experience in patients with mild to moderate PPR.12 Results from a clinical trial demonstrated superiority of AzA foam to vehicle foam for primary end points that included therapeutic success rate and change in inflammatory lesion count.13,14 Secondary end points assessed in the current analysis included patient perception of product usability, efficacy, and effect on QOL. These patient-reported outcome (PRO) results are reported here.
Methods
Study Design
The design of this phase 3 multicenter, randomized, double-blind, vehicle-controlled, parallel-group clinical trial was described in more detail in an earlier report.13 This study was approved by all appropriate institutional review boards. Eligible participants were 18 years and older with moderate or severe PPR, 12 to 50 inflammatory lesions, and persistent erythema with or without telangiectasia. Exclusion criteria included known nonresponse to AzA, current or prior use (within 6 weeks of randomization) of noninvestigational products to treat rosacea, and presence of other dermatoses that could interfere with rosacea evaluation.
Participants were randomized into the AzA foam or vehicle group (1:1 ratio). The study medication (0.5 g) or vehicle foam was applied twice daily to the entire face until the end of treatment (EoT) at 12 weeks. Efficacy and safety parameters were evaluated at baseline and at 4, 8, and 12 weeks of treatment, and at a follow-up visit 4 weeks after EoT (week 16).
Results for the coprimary efficacy end points—therapeutic success rate according to investigator global assessment and nominal change in inflammatory lesion count—were previously reported,13 as well as secondary efficacy outcomes including change in inflammatory lesion count, therapeutic response rate, and change in erythema rating.14
Patient-Reported Secondary Efficacy Outcomes
The secondary PRO end points were patient-reported global assessment of treatment response (rated as excellent, good, fair, none, or worse), global assessment of tolerability (rated as excellent, good, acceptable despite minor irritation, less acceptable due to continuous irritation, not acceptable, or no opinion), and opinion on cosmetic acceptability and practicability of product use in areas adjacent to the hairline (rated as very good, good, satisfactory, poor, or no opinion).
Additionally, QOL was measured by 3 validated standardized PRO tools, including the Rosacea Quality of Life Index (RosaQOL),15 the EuroQOL 5-dimension 5-level questionnaire (EQ-5D-5L),16 and the Dermatology Life Quality Index (DLQI). The RosaQOL is a rosacea-specific instrument assessing 3 constructs: (1) symptom, (2) emotion, and (3) function. The EQ-5D-5L questionnaire measures overall health status and comprises 5 constructs: (1) mobility, (2) self-care, (3) usual activities, (4) pain/discomfort, and (5) anxiety/depression. The DLQI is a general, dermatology-oriented instrument categorized into 6 constructs: (1) symptoms and feelings, (2) daily activities, (3) leisure, (4) work and school, (5) personal relationships, and (6) treatment.
Statistical Analyses
Patient-reported outcomes were analyzed in an exploratory manner and evaluated at EoT relative to baseline. Self-reported global assessment of treatment response and change in RosaQOL, EQ-5D-5L, and DLQI scores between AzA foam and vehicle foam groups were evaluated using the Wilcoxon rank sum test. Categorical change in the number of participants achieving an increase of 5 or more points in overall DLQI score was evaluated using a χ2 test.
Safety
Safety was analyzed for all randomized patients who were dispensed any study medication. All analyses were performed using SAS version 9.2.
Results
Of the 961 participants included in the study, 483 were randomized to receive AzA foam and 478 were randomized to receive vehicle foam. The mean age was 51.5 years, and the majority of participants were female (73.0%) and white (95.5%)(Table). At baseline, 834 (86.8%) participants had moderate PPR and 127 (13.2%) had severe PPR. The mean inflammatory lesion count (SD) was 21.4 (8.9). No significant differences in baseline characteristics were observed between treatment groups.
Patient-reported global assessment of treatment response differed between treatment groups at EoT (P<.001)(Figure 1). Higher ratings of treatment response were reported among the AzA foam group (excellent, 17.2%; good, 40.0%) versus vehicle foam (excellent, 9.7%; good, 35.0%). The number of participants reporting no treatment response was 13.1% in the AzA foam group, with 1.8% reporting worsening of their condition, while 19.4% of participants in the vehicle foam group reported no response, with 6.3% reporting worsening of their condition (Figure 1).
Tolerability was rated excellent or good in 67.8% of the AzA foam group versus 78.2% of the vehicle foam group (Figure 2A). Approximately 38.4% of the AzA foam group versus 38.2% of the vehicle foam group rated treatment tolerability as excellent, while 93.5% of the AzA foam group rated tolerability as acceptable, good, or excellent compared with 89.5% of the vehicle foam group. Only 1.4% of participants in the AzA foam group indicated that treatment was not acceptable due to irritation. In addition, a greater proportion of the AzA foam group reported cosmetic acceptability as very good versus the vehicle foam group (40.5% vs 28.7%)(Figure 2B), with two-thirds reporting cosmetic acceptability as very good or good. Practicability of product use in areas adjacent to the hairline was rated very good by substantial proportions of both the AzA foam and vehicle foam groups (42.8% vs 35.9%)(Figure 2C).
At baseline, average disease burden was moderate according to mean overall DLQI scores (SD) for the AzA foam (5.4 [4.8]) and vehicle foam (5.4 [4.9]) groups. Mean overall DLQI scores improved at EoT, with greater improvement occurring in the AzA foam group (2.6 vs 2.1; P=.018)(Figure 3). A larger proportion of participants in the AzA foam group versus the vehicle foam group also achieved a 5-point or more improvement in overall DLQI score (24.6% vs 19.0%; P=.047). Changes in specific DLQI subscore components were either balanced or in favor of the AzA foam group, including daily activities (0.5 vs 0.4; P=.019), symptoms and feelings (1.2 vs 1.0; P=.069), and leisure (0.5 vs 0.4; P=.012). Specific DLQI items with differences in scores between treatment groups from baseline included the following questions: Over the last week, how embarrassed or self-conscious have you been because of your skin? (P<.001); Over the last week, how much has your skin interfered with you going shopping or looking after your home or garden? (P=.005); Over the last week, how much has your skin affected any social or leisure activities? (P=.040); Over the last week, how much has your skin created problems with your partner or any of your close friends or relatives? (P=.001). Differences between treatment groups favored the AzA foam group for each of these items.
Participants in the AzA foam and vehicle foam groups also showed improvement in RosaQOL scores at EoT (6.8 vs 6.4; P=.67), while EQ-5D-5L scores changed minimally from baseline (0.006 vs 0.007; P=.50).
Safety
The incidence of drug-related adverse events (AEs) was greater in the AzA foam group versus the vehicle foam group (7.7% vs 4.8%). Drug-related AEs occurring in 1% of the AzA foam group were application-site pain including tenderness, stinging, and burning (3.5% for AzA foam vs 1.3% for vehicle foam); application-site pruritus (1.4% vs 0.4%); and application-site dryness (1.0% vs 0.6%). One drug-related AE of severe intensity—application-site dermatitis—occurred in the vehicle foam group; all other drug-related AEs were mild or moderate.14 More detailed safety results are described in a previous report.13
Comment
The PRO outcome data reported here are consistent with previously reported statistically significant improvements in investigator-assessed primary end points for the treatment of PPR with AzA foam.13,14 The data demonstrate that AzA foam benefits both clinical and patient-oriented dimensions of rosacea disease burden and suggest an association between positive treatment response and improved QOL.
Specifically, patient evaluation of treatment response to AzA foam was highly favorable, with 57.2% reporting excellent or good response and 85.1% reporting positive response overall. Recognizing the relapsing-remitting course of PPR, only 1.8% of the AzA foam group experienced worsening of disease at EoT.
The DLQI and RosaQOL instruments revealed notable improvements in QOL from baseline for both treatment groups. Although no significant differences in RosaQOL scores were observed between groups at EoT, significant differences in DLQI scores were detected. Almost one-quarter of participants in the AzA foam group achieved at least a 5-point improvement in DLQI score, exceeding the 4-point threshold for clinically meaningful change.17 Although little change in EQ-5D-5L scores was observed at EoT for both groups with no between-group differences, this finding is not unexpected, as this instrument assesses QOL dimensions such as loss of function, mobility, and ability to wash or dress, which are unlikely to be compromised in most rosacea patients.
Our results also underscore the importance of vehicle in the treatment of compromised skin. Studies of topical treatments for other dermatoses suggest that vehicle properties may reduce disease severity and improve QOL independent of active ingredients.10,18 For example, ease of application, minimal residue, and less time spent in application may explain the superiority of foam to other vehicles in the treatment of psoriasis.18 Our data demonstrating high cosmetic favorability of AzA foam are consistent with these prior observations. Increased tolerability of foam formulations also may affect response to treatment, in part by supporting adherence.18 Most participants receiving AzA foam described tolerability as excellent or good, and the discontinuation rate was low (1.2% of participants in the AzA foam group left the study due to AEs) in the setting of near-complete dosage administration (97% of expected doses applied).13
Conclusion
These results indicate that use of AzA foam as well as its novel vehicle results in high patient satisfaction and improved QOL. Although additional research is necessary to further delineate the relationship between PROs and other measures of clinical efficacy, our data demonstrate a positive treatment experience as perceived by patients that parallels the clinical efficacy of AzA foam for the treatment of PPR.13,14
Acknowledgment
Editorial support through inVentiv Medical Communications (New York, New York) was provided by Bayer Pharmaceuticals.
- Cardwell LA, Farhangian ME, Alinia H, et al. Psychological disorders associated with rosacea: analysis of unscripted comments. J Dermatol Surg. 2015;19:99-103.
- Moustafa F, Lewallen RS, Feldman SR. The psychological impact of rosacea and the influence of current management options. J Am Acad Dermatol. 2014;71:973-980.
- Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the Classification and Staging of Rosacea. J Am Acad Dermatol. 2002;46:584-587.
- Yamasaki K, Gallo RL. The molecular pathology of rosacea. J Dermatol Sci. 2009;55:77-81.
- Del Rosso JQ. Advances in understanding and managing rosacea: part 1: connecting the dots between pathophysiological mechanisms and common clinical features of rosacea with emphasis on vascular changes and facial erythema. J Clin Aesthet Dermatol. 2012;5:16-25.
- Bohm D, Schwanitz P, Stock Gissendanner S, et al. Symptom severity and psychological sequelae in rosacea: results of a survey. Psychol Health Med. 2014;19:586-591.
- Elewski BE, Draelos Z, Dreno B, et al. Rosacea—global diversity and optimized outcome: proposed international consensus from the Rosacea International Expert Group. J Eur Acad Dermatol Venereol. 2011;25:188-200.
- Dirschka T, Micali G, Papadopoulos L, et al. Perceptions on the psychological impact of facial erythema associated with rosacea: results of international survey [published online May 29, 2015]. Dermatol Ther (Heidelb). 2015;5:117-127.
- Abram K, Silm H, Maaroos HI, et al. Subjective disease perception and symptoms of depression in relation to healthcare-seeking behaviour in patients with rosacea. Acta Derm Venereol. 2009;89:488-491.
- Stein L. Clinical studies of a new vehicle formulation for topical corticosteroids in the treatment of psoriasis. J Am Acad Dermatol. 2005;53(1, suppl 1):S39-S49.
- Yentzer BA, Camacho FT, Young T, et al. Good adherence and early efficacy using desonide hydrogel for atopic dermatitis: results from a program addressing patient compliance. J Drugs Dermatol. 2010;9:324-329.
- Finacea (azelaic acid) foam 15% [package insert]. Whippany, NJ: Bayer Pharmaceuticals; 2015.
- Draelos ZD, Elewski BE, Harper JC, et al. A phase 3 randomized, double-blind, vehicle-controlled trial of azelaic acid foam 15% in the treatment of papulopustular rosacea. Cutis. 2015;96:54-61.
- Solomon JA, Tyring S, Staedtler G, et al. Investigator-reported efficacy of azelaic acid foam 15% in patients with papulopustular rosacea: secondary efficacy outcomes from a randomized, controlled, double-blind, phase 3 trial. Cutis. 2016;98:187-194.
- Nicholson K, Abramova L, Chren MM, et al. A pilot quality-of-life instrument for acne rosacea. J Am Acad Dermatol. 2007;57:213-221.
- Herdman M, Gudex C, Lloyd A, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. 2011;20:1727-1736.
- Basra MK, Salek MS, Camilleri L, et al. Determining the minimal clinically important difference and responsiveness of the Dermatology Life Quality Index (DLQI): further data. Dermatology. 2015;230:27-33.
- Bergstrom KG, Arambula K, Kimball AB. Medication formulation affects quality of life: a randomized single-blind study of clobetasol propionate foam 0.05% compared with a combined program of clobetasol cream 0.05% and solution 0.05% for the treatment of psoriasis. Cutis. 2003;72:407-411.
- Cardwell LA, Farhangian ME, Alinia H, et al. Psychological disorders associated with rosacea: analysis of unscripted comments. J Dermatol Surg. 2015;19:99-103.
- Moustafa F, Lewallen RS, Feldman SR. The psychological impact of rosacea and the influence of current management options. J Am Acad Dermatol. 2014;71:973-980.
- Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the Classification and Staging of Rosacea. J Am Acad Dermatol. 2002;46:584-587.
- Yamasaki K, Gallo RL. The molecular pathology of rosacea. J Dermatol Sci. 2009;55:77-81.
- Del Rosso JQ. Advances in understanding and managing rosacea: part 1: connecting the dots between pathophysiological mechanisms and common clinical features of rosacea with emphasis on vascular changes and facial erythema. J Clin Aesthet Dermatol. 2012;5:16-25.
- Bohm D, Schwanitz P, Stock Gissendanner S, et al. Symptom severity and psychological sequelae in rosacea: results of a survey. Psychol Health Med. 2014;19:586-591.
- Elewski BE, Draelos Z, Dreno B, et al. Rosacea—global diversity and optimized outcome: proposed international consensus from the Rosacea International Expert Group. J Eur Acad Dermatol Venereol. 2011;25:188-200.
- Dirschka T, Micali G, Papadopoulos L, et al. Perceptions on the psychological impact of facial erythema associated with rosacea: results of international survey [published online May 29, 2015]. Dermatol Ther (Heidelb). 2015;5:117-127.
- Abram K, Silm H, Maaroos HI, et al. Subjective disease perception and symptoms of depression in relation to healthcare-seeking behaviour in patients with rosacea. Acta Derm Venereol. 2009;89:488-491.
- Stein L. Clinical studies of a new vehicle formulation for topical corticosteroids in the treatment of psoriasis. J Am Acad Dermatol. 2005;53(1, suppl 1):S39-S49.
- Yentzer BA, Camacho FT, Young T, et al. Good adherence and early efficacy using desonide hydrogel for atopic dermatitis: results from a program addressing patient compliance. J Drugs Dermatol. 2010;9:324-329.
- Finacea (azelaic acid) foam 15% [package insert]. Whippany, NJ: Bayer Pharmaceuticals; 2015.
- Draelos ZD, Elewski BE, Harper JC, et al. A phase 3 randomized, double-blind, vehicle-controlled trial of azelaic acid foam 15% in the treatment of papulopustular rosacea. Cutis. 2015;96:54-61.
- Solomon JA, Tyring S, Staedtler G, et al. Investigator-reported efficacy of azelaic acid foam 15% in patients with papulopustular rosacea: secondary efficacy outcomes from a randomized, controlled, double-blind, phase 3 trial. Cutis. 2016;98:187-194.
- Nicholson K, Abramova L, Chren MM, et al. A pilot quality-of-life instrument for acne rosacea. J Am Acad Dermatol. 2007;57:213-221.
- Herdman M, Gudex C, Lloyd A, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. 2011;20:1727-1736.
- Basra MK, Salek MS, Camilleri L, et al. Determining the minimal clinically important difference and responsiveness of the Dermatology Life Quality Index (DLQI): further data. Dermatology. 2015;230:27-33.
- Bergstrom KG, Arambula K, Kimball AB. Medication formulation affects quality of life: a randomized single-blind study of clobetasol propionate foam 0.05% compared with a combined program of clobetasol cream 0.05% and solution 0.05% for the treatment of psoriasis. Cutis. 2003;72:407-411.
Practice Points
- Patient perceptions of treatment are an important consideration in developing topical therapeutic strategies for papulopustular rosacea.
- A novel hydrophilic foam formulation of azelaic acid (AzA) provided substantial benefits in patient-reported measures of treatment response and quality of life.
- Patients reported high levels of satisfaction with the usability, tolerability, and practicability of AzA foam.
- The positive treatment experience described by patients parallels investigator-reported measures of clinical efficacy reported elsewhere.
Investigator-Reported Efficacy of Azelaic Acid Foam 15% in Patients With Papulopustular Rosacea: Secondary Efficacy Outcomes From a Randomized, Controlled, Double-blind, Phase 3 Trial
Papulopustular rosacea (PPR) is characterized by centrofacial papules, pustules, erythema, and occasionally telangiectasia.1,2 A myriad of factors, including genetic predisposition3 and environmental triggers,4 have been associated with dysregulated inflammatory responses,5 contributing to the disease pathogenesis and symptoms. Inflammation associated with PPR may decrease skin barrier function, increase transepidermal water loss, and reduce stratum corneum hydration,6,7 resulting in heightened skin sensitivity, pain, burning, and/or stinging.5,8
Azelaic acid (AzA), which historically has only been available in gel or cream formulations, is well established for the treatment of rosacea9; however, these formulations have been associated with application-site adverse events (AEs)(eg, burning, erythema, irritation), limited cosmetic acceptability, and reduced compliance or efficacy.10
For select skin conditions, active agents delivered in foam vehicles may offer superior tolerability with improved outcomes.11 An AzA foam 15% formulation was approved for the treatment of mild to moderate PPR. Primary outcomes from a phase 3 trial demonstrated the efficacy and safety of AzA foam in improving inflammatory lesion counts (ILCs) and disease severity in participants with PPR. The trial also evaluated additional secondary end points, including the effect of AzA foam on erythema, inflammatory lesions, treatment response, and other manifestations of PPR.12 The current study evaluated investigator-reported efficacy outcomes for these secondary end points for AzA foam 15% versus vehicle foam.
Methods
Study Design
This phase 3 multicenter, randomized, double-blind, vehicle-controlled, parallel-group clinical trial was conducted from September 2012 to January 2014 at 48 US study centers comparing the efficacy of AzA foam versus vehicle foam in patients with PPR. Eligible participants were 18 years and older with PPR rated as moderate or severe according to investigator global assessment (IGA), plus 12 to 50 inflammatory lesions and persistent erythema with or without telangiectasia. Exclusion criteria included known nonresponse to AzA, current or prior use (within 6 weeks of randomization) of noninvestigational products to treat rosacea, and presence of other dermatoses that could interfere with rosacea evaluation.
Participants were randomized into the AzA foam or vehicle group (1:1 ratio). The study medication was applied in 0.5-g doses twice daily until the end of treatment (EoT) at 12 weeks. Efficacy and safety parameters were evaluated at baseline and at 4, 8, and 12 weeks of treatment, and at a follow-up visit 4 weeks after EoT (week 16).
Results for the coprimary efficacy end points—therapeutic success rate according to IGA and nominal change in ILC—were previously reported.12
Investigator-Reported Secondary Efficacy Outcomes
The secondary efficacy end points were grouped change in erythema rating, grouped change in telangiectasia rating, grouped change in IGA score, therapeutic response rate according to IGA, percentage change in ILC from baseline, and facial skin color rating at EoT.
Grouped change for all secondary end points was measured as improved, no change, or worsened relative to baseline. For grouped change in erythema and telangiectasia ratings, a participant was considered improved if the rating at the postbaseline visit was lower than the baseline rating, no change if the postbaseline and baseline ratings were identical, and worsened if the postbaseline rating was higher than at baseline. For grouped change in IGA score, a participant was considered improved if a responder showed at least a 1-step improvement postbaseline compared to baseline, no change if postbaseline and baseline ratings were identical, and worsened if the postbaseline rating was higher than at baseline.
For the therapeutic response rate, a participant was considered a treatment responder if the IGA score improved from baseline and resulted in clear, minimal, or mild disease severity at EoT.
Safety
Adverse events also were assessed.
Statistical Analyses
Secondary efficacy and safety end points were assessed for all randomized participants who were dispensed the study medication. Missing data were imputed using last observation carried forward.
For the percentage change in ILC from baseline, therapeutic response rate, and grouped change in erythema rating, confirmatory analyses were conducted in a hierarchical manner (in the order listed), with testing stopped as soon as a null hypothesis of superior treatment effect could not be rejected. Analyses without significance level were exploratory. The Cochran-Mantel-Haenszel van Elteren test stratified by study center was used for grouped change in erythema rating (1-tailed, 2.5%) and IGA score (2-tailed, 5%); Wilcoxon rank sum tests also were performed. Percentage change in ILC from baseline was evaluated using the Student t test and F test of analysis of covariance (1-tailed, 2.5%). Therapeutic response rate was evaluated using the Cochran-Mantel-Haenszel van Elteren test stratified by study center and the Pearson χ2 test. Facial skin color and grouped change in telangiectasia rating were evaluated using the Wilcoxon rank sum test.
Adverse events beginning or worsening after the first dose of the study drug were considered treatment emergent and were coded using the Medical Dictionary for Regulatory Activities (MedDRA) Version 16.1. Statistical analyses were performed using SAS software version 9.2.
Results
Study Participants
The study included 961 total participants; 483 were randomized to the AzA foam group and 478 to the vehicle group (Figure 1). Overall, 803 participants completed follow-up; however, week 16 results for the efficacy outcomes include data for 4 additional patients (2 per study arm) who did not formally meet all requirements for follow-up completion. The mean age was 51.5 years, and the majority of the participants were white and female (Table 1). Most participants (86.8%) had moderate PPR at baseline, with the remaining rated as having severe disease (13.2%). The majority (76.4%) had more than 14 inflammatory lesions with moderate (76.4%) or severe (15.1%) erythema at baseline.
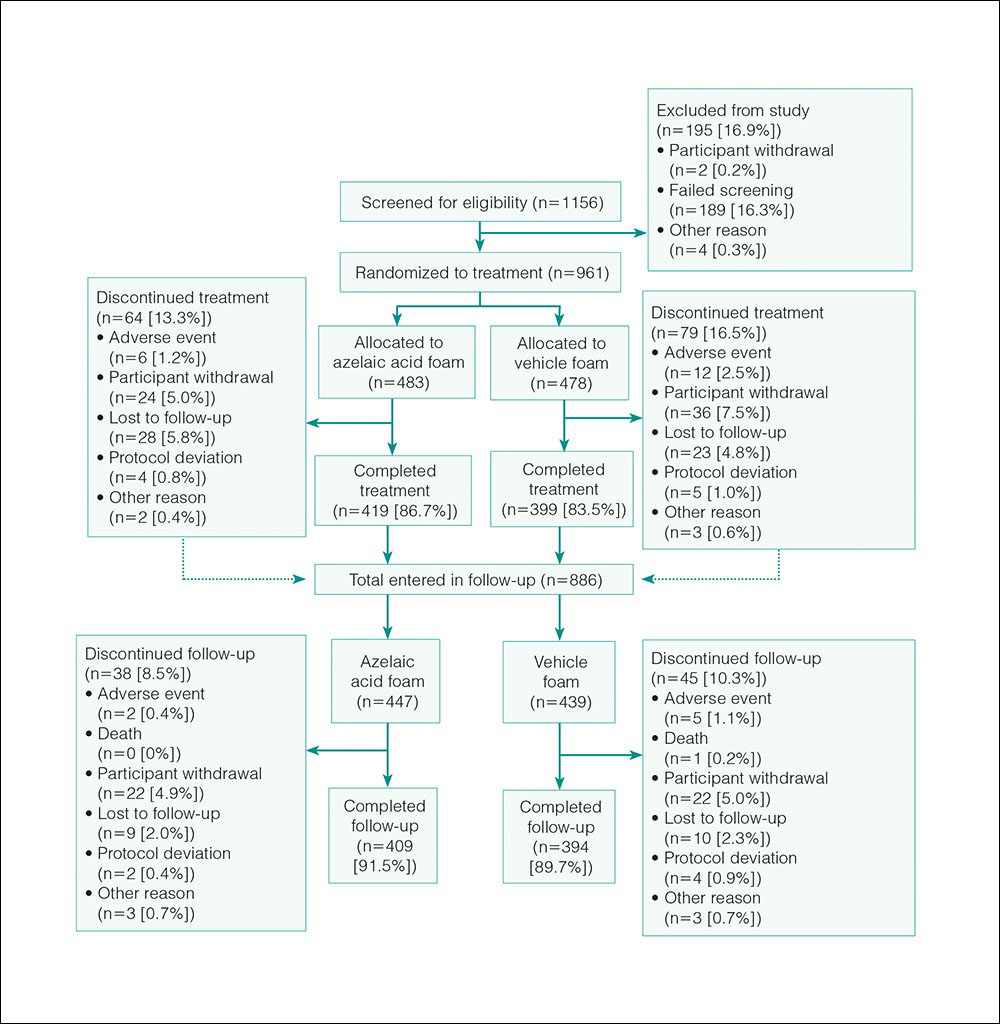
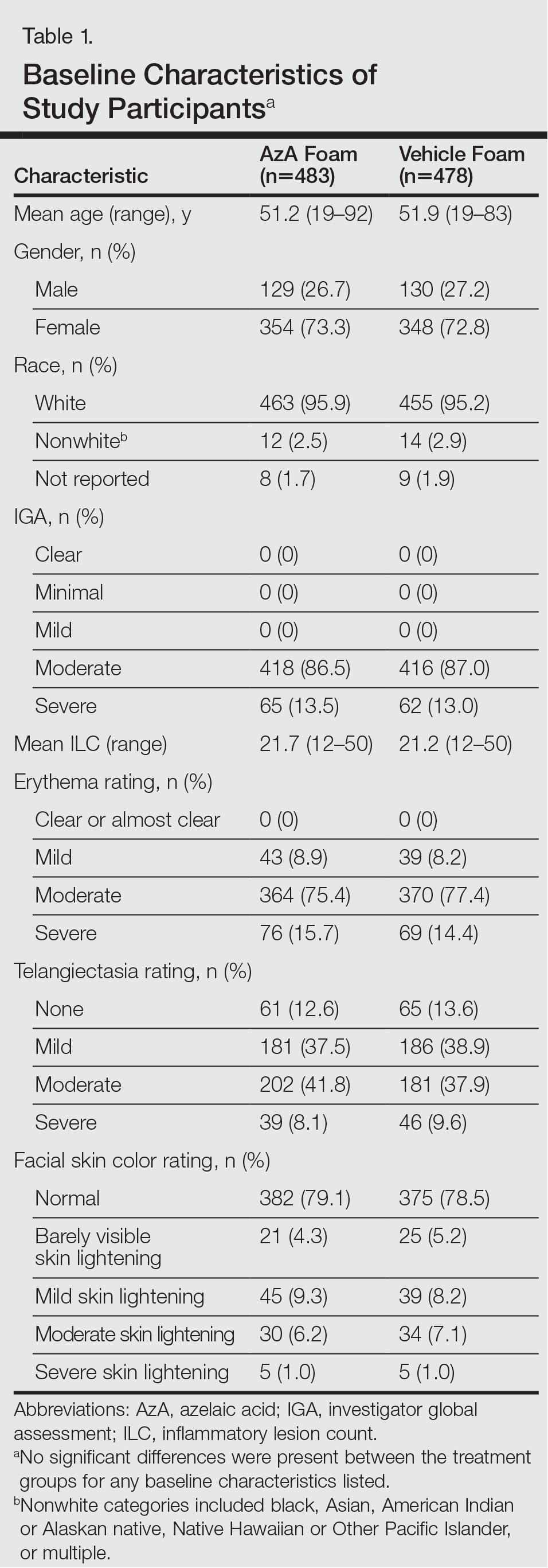
Efficacy
Significantly more participants in the AzA group than in the vehicle group showed an improved erythema rating at EoT (61.5% vs 51.3%; P<.001)(Figure 2), with more participants in the AzA group showing improvement at weeks 4 (P=.022) and 8 (P=.002).
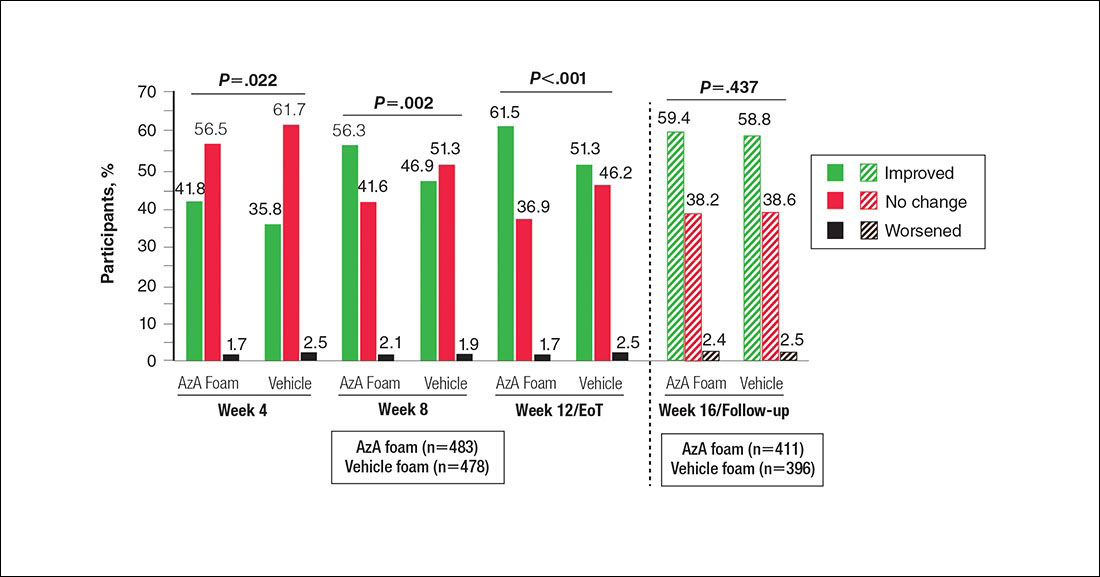
A significantly greater mean percentage reduction in ILC from baseline to EoT was observed in the AzA group versus the vehicle group (61.6% vs 50.8%; P<.001)(Figure 3), and between-group differences were observed at week 4 (P<.001), week 8 (P=.003), and week 16 (end of study/follow-up)(P=.002).
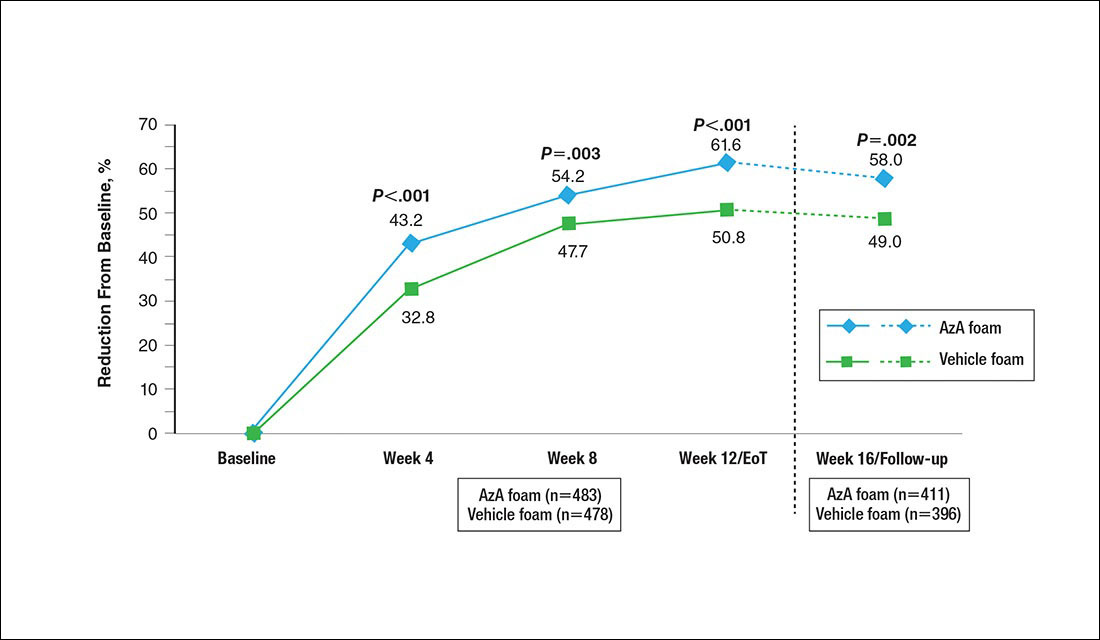
A significantly higher proportion of participants treated with AzA foam versus vehicle were considered responders at week 12/EoT (66.3% vs 54.4%; P<.001)(Figure 4). Differences in responder rate also were observed at week 4 (P=.026) and week 8 (P=.026).
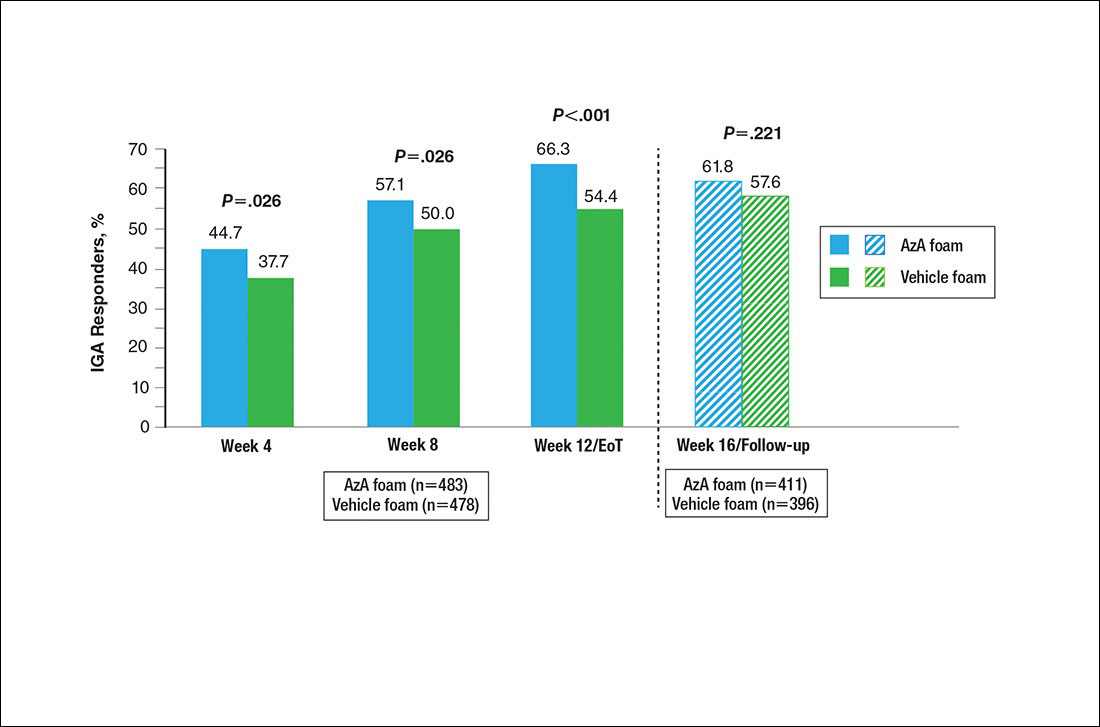
No study drug was administered between week 12/EoT and week 16/follow-up; last observation carried forward was not applied to week 16/follow-up analysis. AzA indicates azelaic acid; IGA, investigator global assessment.
Differences in grouped change in IGA score were observed between groups at every evaluation during the treatment phase (Figure 5). Specifically, IGA score was improved at week 12/EoT relative to baseline in 71.2% of participants in the AzA group versus 58.8% in the vehicle group (P<.001).
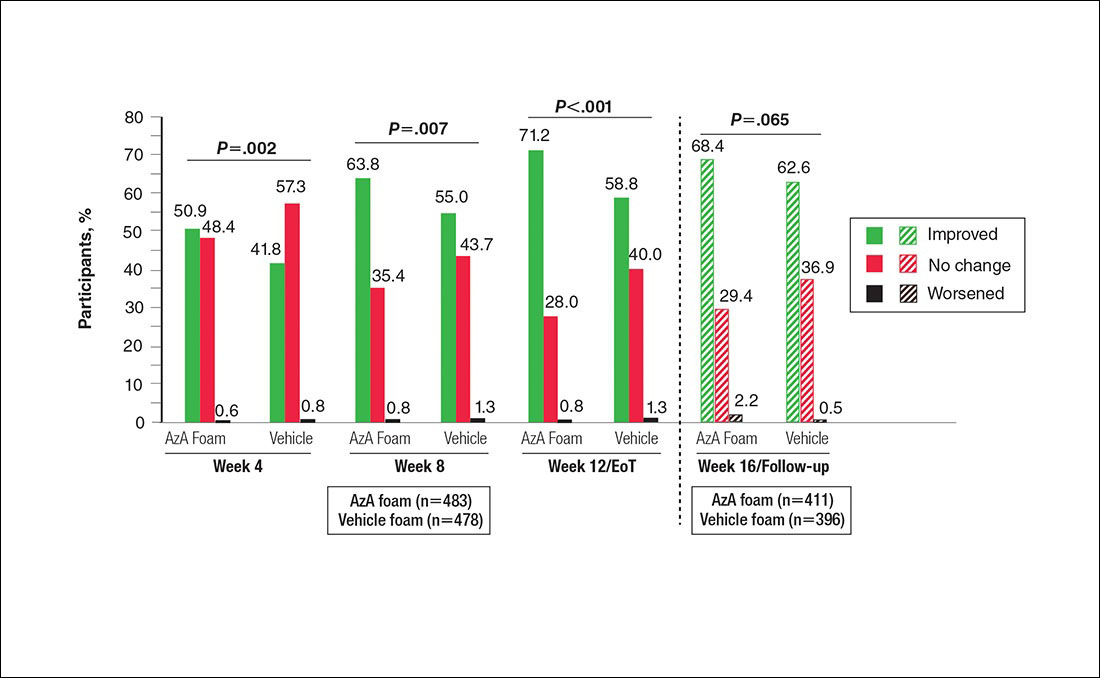
For grouped change in telangiectasia rating at EoT, the majority of participants in both treatment groups showed no change (Table 2). Regarding facial skin color, the majority of participants in both the AzA and vehicle treatment groups (80.1% and 78.7%, respectively) showed normal skin color compared to nontreated skin EoT; no between-group differences were detected for facial skin color rating (P=.315, Wilcoxon rank sum test).
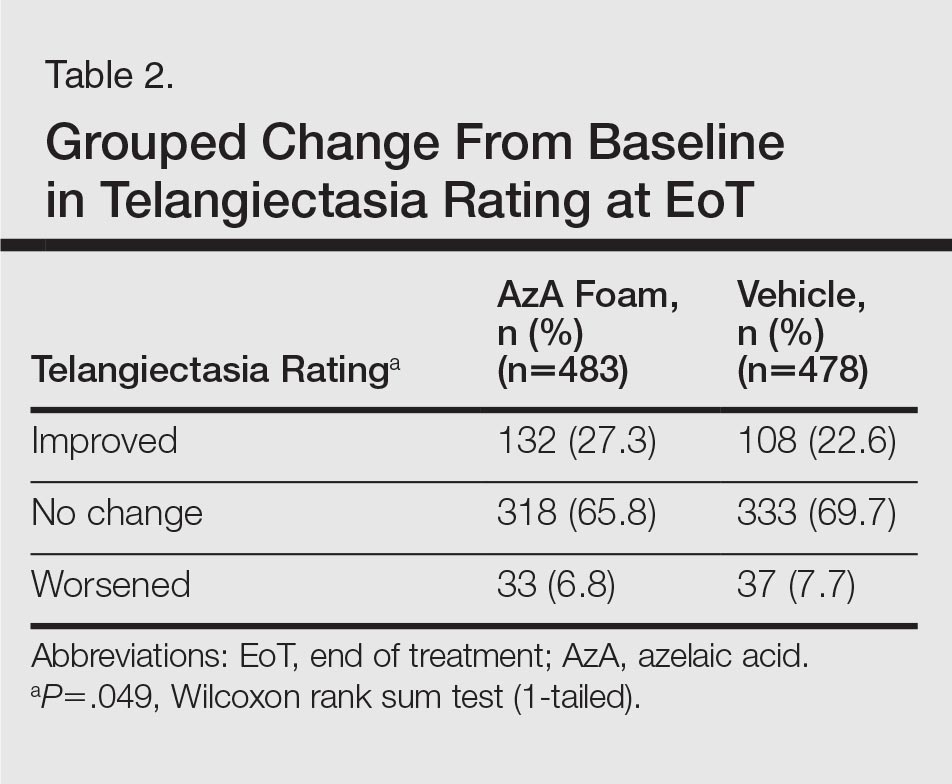
Safety
The incidence of drug-related AEs was greater in the AzA group than the vehicle group (7.7% vs 4.8%)(Table 3). Drug-related AEs occurring in at least 1% of the AzA group were pain at application site (eg, tenderness, stinging, burning)(AzA group, 3.5%; vehicle group, 1.3%), application-site pruritus (1.4% vs 0.4%), and application-site dryness (1.0% vs 0.6%). A single drug-related AE of severe intensity (ie, application-site dermatitis) was observed in the vehicle group; all other drug-related AEs were mild or moderate. The incidence of withdrawals due to AEs was lower in the AzA group than the vehicle group (1.2% vs 2.5%). This AE profile correlated with a treatment compliance (the percentage of expected doses that were actually administered) of 97.0% in the AzA group and 95.9% in the vehicle group. One participant in the vehicle group died due to head trauma unrelated to administration of the study drug.
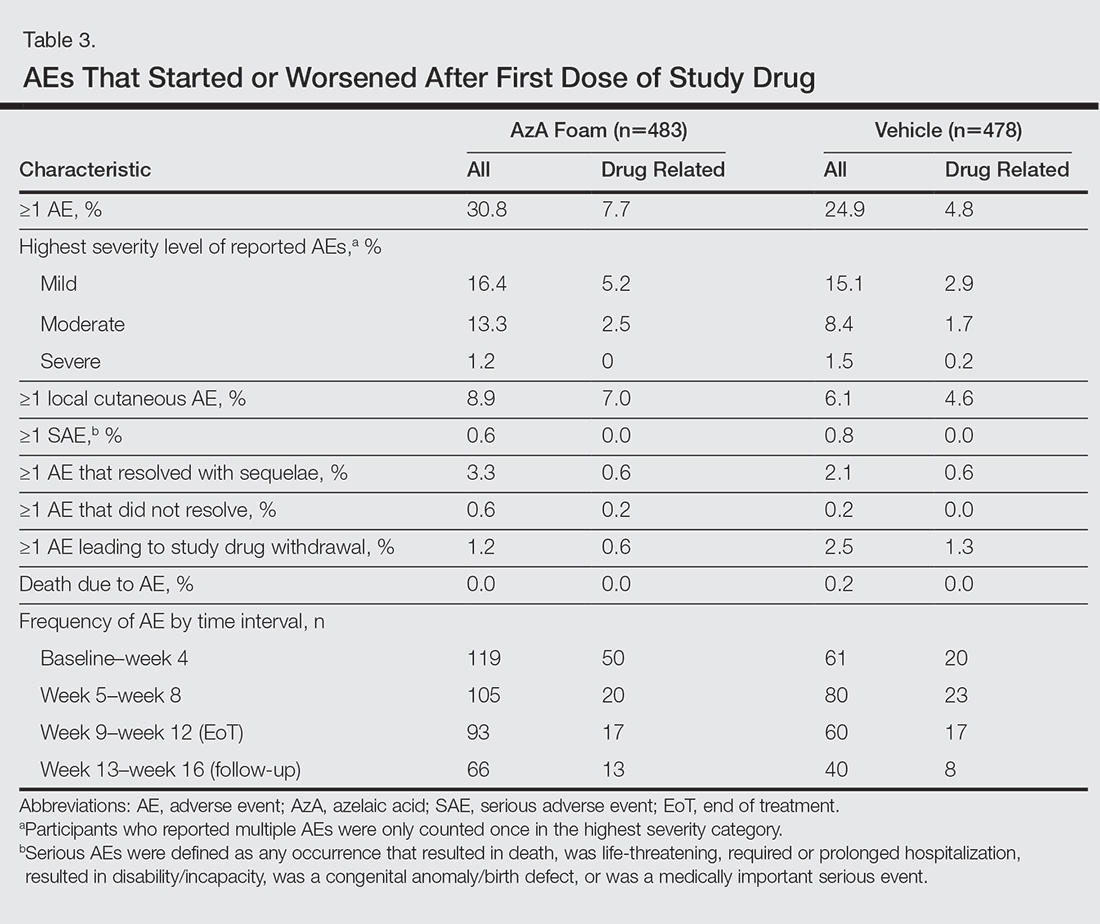
Comment
The results of this study further support the efficacy of AzA foam for the treatment of PPR. The percentage reduction in ILC was consistent with nominal decreases in ILC, a coprimary efficacy end point of this study.12 Almost two-thirds of participants treated with AzA foam achieved a therapeutic response, indicating that many participants who did not strictly achieve the primary outcome of therapeutic success nevertheless attained notable reductions in disease severity. The number of participants who showed any improvement on the IGA scale increased throughout the course of treatment (63.8% AzA foam vs 55.0% vehicle at week 8) up to EoT (71.2% vs 58.8%)(Figure 5). In addition, the number of participants showing any improvement at week 8 (63.8% AzA foam vs 55.0% vehicle)(Figure 5) was comparable to the number of participants achieving therapeutic response at week 12/EoT (66.3% vs 54.4%)(Figure 4). These data suggest that increasing time of treatment increases the likelihood of achieving better results.
Erythema also appeared to respond to AzA foam, with 10.2% more participants in the AzA group demonstrating improvement at week 12/EoT compared to vehicle. The difference in grouped change in erythema rating also was statistically significant and favored AzA foam, sustained up to 4 weeks after EoT.
The outcomes for percentage change in ILC, therapeutic response rate, and grouped change in erythema rating consequently led to the rejection of all 3 null hypotheses in hierarchical confirmatory analyses, underscoring the benefits of AzA foam treatment.
The therapeutic effects of AzA foam were apparent at the first postbaseline evaluation and persisted throughout treatment. Differences favoring AzA foam were observed at every on-treatment evaluation for grouped change in erythema rating, percentage change in ILC, therapeutic response rate, and grouped change in IGA score. Symptoms showed minimal resurgence after treatment cessation, and there were no signs of disease flare-up within the 4 weeks of observational follow-up. In addition, the percentage reduction in ILC remained higher in the AzA foam group during follow-up.
These results also show that AzA foam was well tolerated with a low incidence of discontinuation because of drug-related AEs. No serious drug-related AEs were reported for this study or in the preceding phase 2 trial.12,13 Although not directly evaluated, the low incidence of cutaneous AEs suggests that AzA foam may be better tolerated than prior formulations of AzA14,15 and correlates with high compliance observed during the study.12 Azelaic acid foam appeared to have minimal to no effect on skin color, with more than 88% of participants reporting barely visible or no skin lightening.
Interestingly, the vehicle foam showed appreciable efficacy independent of AzA. Improvements in erythema were recorded in approximately half of the vehicle group at week 12/EoT. A similar proportion attained a therapeutic response, and ILC was reduced by 50.8% at week 12/EoT. Comparable results also were evident in the vehicle group for the primary end points of this study.12 Vehicles in dermatologic trials frequently exert effects on diseased skin16,17 via a skin care regimen effect (eg, moisturization and other vehicle-related effects that may improve skin barrier integrity and function) and thus should not be regarded as placebo controls. The mechanism underlying this efficacy may be due to the impact of vehicle composition on skin barrier integrity and transepidermal water loss.18 The hydrophilic emulsion or other constituents of AzA foam (eg, fatty alcohols) may play a role.
A notable strength of our study is detailed clinical characterization using carefully chosen parameters and preplanned analyses that complement the primary end points. As the latter are often driven by regulatory requirements, opportunities to characterize other outcomes of interest to clinicians may be missed. The additional analyses reported here hopefully will aid dermatologists in both assessing the role of AzA foam in the treatment armamentarium for PPR and counseling patients.
Because participants with lighter skin pigmentation dominated our study population, the impact of AzA foam among patients with darker skin complexions is unknown. Although AzA is unlikely to cause hypopigmentation in normal undiseased skin, patients should be monitored for early signs of hypopigmentation.19,20 Our data also do not allow assessment of the differential effect, if any, of AzA foam on erythema of different etiologies in PPR, as corresponding information was not collected in the trial.
Conclusion
Azelaic acid foam 15% combines a well-established treatment of PPR with new vehicle technology to deliver effective therapy across multiple disease dimensions. In addition, the vehicle foam appears to demonstrate inherent therapeutic properties independent of AzA. The availability of this novel, efficacious, and well-tolerated option for PPR has the potential to improve patient care, reduce disease burden, and minimize unnecessary costs through increased tolerability and compliance.21
Acknowledgment
Editorial support through inVentiv Medical Communications (New York, New York) was provided by Bayer Pharmaceuticals.
- Tan J, Berg M. Rosacea: current state of epidemiology. J Am Acad Dermatol. 2013;69(6, suppl 1):S27-S35.
- Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the classification and staging of rosacea. J Am Acad Dermatol. 2002;46:584-587.
- Chang AL, Raber I, Xu J, et al. Assessment of the genetic basis of rosacea by genome-wide association study. J Invest Dermatol. 2015;135:1548-1555.
- Abram K, Silm H, Maaroos HI, et al. Risk factors associated with rosacea. J Eur Acad Dermatol Venereol. 2010;24:565-571.
- Yamasaki K, Di Nardo A, Bardan A, et al. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat Med. 2007;13:975-980.
- Yamasaki K, Kanada K, Macleod DT, et al. TLR2 expression is increased in rosacea and stimulates enhanced serine protease production by keratinocytes. J Invest Dermatol. 2011;131:688-697.
- Darlenski R, Kazandjieva J, Tsankov N, et al. Acute irritant threshold correlates with barrier function, skin hydration and contact hypersensitivity in atopic dermatitis and rosacea. Exp Dermatol. 2013;22:752-753.
- Del Rosso JQ, Levin J. The clinical relevance of maintaining the functional integrity of the stratum corneum in both healthy and disease-affected skin. J Clin Aesthet Dermatol. 2011;4:22-42.
- van Zuuren EJ, Kramer SF, Carter BR, et al. Effective and evidence-based management strategies for rosacea: summary of a Cochrane systematic review. Br J Dermatol. 2011;165:760-781.
- Tan X, Feldman SR, Chang J, et al. Topical drug delivery systems in dermatology: a review of patient adherence issues. Expert Opin Drug Deliv. 2012;9:1263-1271.
- Stein L. Clinical studies of a new vehicle formulation for topical corticosteroids in the treatment of psoriasis. J Am Acad Dermatol. 2005;53(1, suppl 1):S39-S49.
- Draelos ZD, Elewski BE, Harper JC, et al. A phase 3 randomized, double-blind, vehicle-controlled trial of azelaic acid foam 15% in the treatment of papulopustular rosacea. Cutis. 2015;96:54-61.
- Draelos ZD, Elewski B, Staedtler G, et al. Azelaic acid foam 15% in the treatment of papulopustular rosacea: a randomized, double-blind, vehicle-controlled study. Cutis. 2013;92:306-317.
- Finacea gel [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2016.
- Elewski BE, Fleischer AB Jr, Pariser DM. A comparison of 15% azelaic acid gel and 0.75% metronidazole gel in the topical treatment of papulopustular rosacea: results of a randomized trial. Arch Dermatol. 2003;139:1444-1450.
- Daniels R, Knie U. Galenics of dermal products—vehicles, properties and drug release. J Dtsch Dermatol Ges. 2007;5:367-383.
- Shamsudin N, Fleischer AB Jr. Vehicle or placebo? Investigators use incorrect terminology in randomized controlled trials half of the time: a systematic review of randomized controlled trials published in three major dermatology journals. J Drugs Dermatol. 2010;9:1221-1226.
- Del Rosso JQ, Thiboutot D, Gallo R, et al. Consensus recommendations from the American Acne & Rosacea Society on the management of rosacea, part 2: a status report on topical agents. Cutis. 2013;92:277-284.
- Finacea foam [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2015.
- Solano F, Briganti S, Picardo M, et al. Hypopigmenting agents: an updated review on biological, chemical and clinical aspects. Pigment Cell Res. 2006;19:550-571.
- Hammarstrom B, Wessling A, Nilsson JL. Pharmaceutical care for patients with skin diseases: a campaign year at Swedish pharmacies. J Clin Pharm Ther. 1995;20:327-334.
Papulopustular rosacea (PPR) is characterized by centrofacial papules, pustules, erythema, and occasionally telangiectasia.1,2 A myriad of factors, including genetic predisposition3 and environmental triggers,4 have been associated with dysregulated inflammatory responses,5 contributing to the disease pathogenesis and symptoms. Inflammation associated with PPR may decrease skin barrier function, increase transepidermal water loss, and reduce stratum corneum hydration,6,7 resulting in heightened skin sensitivity, pain, burning, and/or stinging.5,8
Azelaic acid (AzA), which historically has only been available in gel or cream formulations, is well established for the treatment of rosacea9; however, these formulations have been associated with application-site adverse events (AEs)(eg, burning, erythema, irritation), limited cosmetic acceptability, and reduced compliance or efficacy.10
For select skin conditions, active agents delivered in foam vehicles may offer superior tolerability with improved outcomes.11 An AzA foam 15% formulation was approved for the treatment of mild to moderate PPR. Primary outcomes from a phase 3 trial demonstrated the efficacy and safety of AzA foam in improving inflammatory lesion counts (ILCs) and disease severity in participants with PPR. The trial also evaluated additional secondary end points, including the effect of AzA foam on erythema, inflammatory lesions, treatment response, and other manifestations of PPR.12 The current study evaluated investigator-reported efficacy outcomes for these secondary end points for AzA foam 15% versus vehicle foam.
Methods
Study Design
This phase 3 multicenter, randomized, double-blind, vehicle-controlled, parallel-group clinical trial was conducted from September 2012 to January 2014 at 48 US study centers comparing the efficacy of AzA foam versus vehicle foam in patients with PPR. Eligible participants were 18 years and older with PPR rated as moderate or severe according to investigator global assessment (IGA), plus 12 to 50 inflammatory lesions and persistent erythema with or without telangiectasia. Exclusion criteria included known nonresponse to AzA, current or prior use (within 6 weeks of randomization) of noninvestigational products to treat rosacea, and presence of other dermatoses that could interfere with rosacea evaluation.
Participants were randomized into the AzA foam or vehicle group (1:1 ratio). The study medication was applied in 0.5-g doses twice daily until the end of treatment (EoT) at 12 weeks. Efficacy and safety parameters were evaluated at baseline and at 4, 8, and 12 weeks of treatment, and at a follow-up visit 4 weeks after EoT (week 16).
Results for the coprimary efficacy end points—therapeutic success rate according to IGA and nominal change in ILC—were previously reported.12
Investigator-Reported Secondary Efficacy Outcomes
The secondary efficacy end points were grouped change in erythema rating, grouped change in telangiectasia rating, grouped change in IGA score, therapeutic response rate according to IGA, percentage change in ILC from baseline, and facial skin color rating at EoT.
Grouped change for all secondary end points was measured as improved, no change, or worsened relative to baseline. For grouped change in erythema and telangiectasia ratings, a participant was considered improved if the rating at the postbaseline visit was lower than the baseline rating, no change if the postbaseline and baseline ratings were identical, and worsened if the postbaseline rating was higher than at baseline. For grouped change in IGA score, a participant was considered improved if a responder showed at least a 1-step improvement postbaseline compared to baseline, no change if postbaseline and baseline ratings were identical, and worsened if the postbaseline rating was higher than at baseline.
For the therapeutic response rate, a participant was considered a treatment responder if the IGA score improved from baseline and resulted in clear, minimal, or mild disease severity at EoT.
Safety
Adverse events also were assessed.
Statistical Analyses
Secondary efficacy and safety end points were assessed for all randomized participants who were dispensed the study medication. Missing data were imputed using last observation carried forward.
For the percentage change in ILC from baseline, therapeutic response rate, and grouped change in erythema rating, confirmatory analyses were conducted in a hierarchical manner (in the order listed), with testing stopped as soon as a null hypothesis of superior treatment effect could not be rejected. Analyses without significance level were exploratory. The Cochran-Mantel-Haenszel van Elteren test stratified by study center was used for grouped change in erythema rating (1-tailed, 2.5%) and IGA score (2-tailed, 5%); Wilcoxon rank sum tests also were performed. Percentage change in ILC from baseline was evaluated using the Student t test and F test of analysis of covariance (1-tailed, 2.5%). Therapeutic response rate was evaluated using the Cochran-Mantel-Haenszel van Elteren test stratified by study center and the Pearson χ2 test. Facial skin color and grouped change in telangiectasia rating were evaluated using the Wilcoxon rank sum test.
Adverse events beginning or worsening after the first dose of the study drug were considered treatment emergent and were coded using the Medical Dictionary for Regulatory Activities (MedDRA) Version 16.1. Statistical analyses were performed using SAS software version 9.2.
Results
Study Participants
The study included 961 total participants; 483 were randomized to the AzA foam group and 478 to the vehicle group (Figure 1). Overall, 803 participants completed follow-up; however, week 16 results for the efficacy outcomes include data for 4 additional patients (2 per study arm) who did not formally meet all requirements for follow-up completion. The mean age was 51.5 years, and the majority of the participants were white and female (Table 1). Most participants (86.8%) had moderate PPR at baseline, with the remaining rated as having severe disease (13.2%). The majority (76.4%) had more than 14 inflammatory lesions with moderate (76.4%) or severe (15.1%) erythema at baseline.
Efficacy
Significantly more participants in the AzA group than in the vehicle group showed an improved erythema rating at EoT (61.5% vs 51.3%; P<.001)(Figure 2), with more participants in the AzA group showing improvement at weeks 4 (P=.022) and 8 (P=.002).
A significantly greater mean percentage reduction in ILC from baseline to EoT was observed in the AzA group versus the vehicle group (61.6% vs 50.8%; P<.001)(Figure 3), and between-group differences were observed at week 4 (P<.001), week 8 (P=.003), and week 16 (end of study/follow-up)(P=.002).
A significantly higher proportion of participants treated with AzA foam versus vehicle were considered responders at week 12/EoT (66.3% vs 54.4%; P<.001)(Figure 4). Differences in responder rate also were observed at week 4 (P=.026) and week 8 (P=.026).
No study drug was administered between week 12/EoT and week 16/follow-up; last observation carried forward was not applied to week 16/follow-up analysis. AzA indicates azelaic acid; IGA, investigator global assessment.
Differences in grouped change in IGA score were observed between groups at every evaluation during the treatment phase (Figure 5). Specifically, IGA score was improved at week 12/EoT relative to baseline in 71.2% of participants in the AzA group versus 58.8% in the vehicle group (P<.001).
For grouped change in telangiectasia rating at EoT, the majority of participants in both treatment groups showed no change (Table 2). Regarding facial skin color, the majority of participants in both the AzA and vehicle treatment groups (80.1% and 78.7%, respectively) showed normal skin color compared to nontreated skin EoT; no between-group differences were detected for facial skin color rating (P=.315, Wilcoxon rank sum test).
Safety
The incidence of drug-related AEs was greater in the AzA group than the vehicle group (7.7% vs 4.8%)(Table 3). Drug-related AEs occurring in at least 1% of the AzA group were pain at application site (eg, tenderness, stinging, burning)(AzA group, 3.5%; vehicle group, 1.3%), application-site pruritus (1.4% vs 0.4%), and application-site dryness (1.0% vs 0.6%). A single drug-related AE of severe intensity (ie, application-site dermatitis) was observed in the vehicle group; all other drug-related AEs were mild or moderate. The incidence of withdrawals due to AEs was lower in the AzA group than the vehicle group (1.2% vs 2.5%). This AE profile correlated with a treatment compliance (the percentage of expected doses that were actually administered) of 97.0% in the AzA group and 95.9% in the vehicle group. One participant in the vehicle group died due to head trauma unrelated to administration of the study drug.
Comment
The results of this study further support the efficacy of AzA foam for the treatment of PPR. The percentage reduction in ILC was consistent with nominal decreases in ILC, a coprimary efficacy end point of this study.12 Almost two-thirds of participants treated with AzA foam achieved a therapeutic response, indicating that many participants who did not strictly achieve the primary outcome of therapeutic success nevertheless attained notable reductions in disease severity. The number of participants who showed any improvement on the IGA scale increased throughout the course of treatment (63.8% AzA foam vs 55.0% vehicle at week 8) up to EoT (71.2% vs 58.8%)(Figure 5). In addition, the number of participants showing any improvement at week 8 (63.8% AzA foam vs 55.0% vehicle)(Figure 5) was comparable to the number of participants achieving therapeutic response at week 12/EoT (66.3% vs 54.4%)(Figure 4). These data suggest that increasing time of treatment increases the likelihood of achieving better results.
Erythema also appeared to respond to AzA foam, with 10.2% more participants in the AzA group demonstrating improvement at week 12/EoT compared to vehicle. The difference in grouped change in erythema rating also was statistically significant and favored AzA foam, sustained up to 4 weeks after EoT.
The outcomes for percentage change in ILC, therapeutic response rate, and grouped change in erythema rating consequently led to the rejection of all 3 null hypotheses in hierarchical confirmatory analyses, underscoring the benefits of AzA foam treatment.
The therapeutic effects of AzA foam were apparent at the first postbaseline evaluation and persisted throughout treatment. Differences favoring AzA foam were observed at every on-treatment evaluation for grouped change in erythema rating, percentage change in ILC, therapeutic response rate, and grouped change in IGA score. Symptoms showed minimal resurgence after treatment cessation, and there were no signs of disease flare-up within the 4 weeks of observational follow-up. In addition, the percentage reduction in ILC remained higher in the AzA foam group during follow-up.
These results also show that AzA foam was well tolerated with a low incidence of discontinuation because of drug-related AEs. No serious drug-related AEs were reported for this study or in the preceding phase 2 trial.12,13 Although not directly evaluated, the low incidence of cutaneous AEs suggests that AzA foam may be better tolerated than prior formulations of AzA14,15 and correlates with high compliance observed during the study.12 Azelaic acid foam appeared to have minimal to no effect on skin color, with more than 88% of participants reporting barely visible or no skin lightening.
Interestingly, the vehicle foam showed appreciable efficacy independent of AzA. Improvements in erythema were recorded in approximately half of the vehicle group at week 12/EoT. A similar proportion attained a therapeutic response, and ILC was reduced by 50.8% at week 12/EoT. Comparable results also were evident in the vehicle group for the primary end points of this study.12 Vehicles in dermatologic trials frequently exert effects on diseased skin16,17 via a skin care regimen effect (eg, moisturization and other vehicle-related effects that may improve skin barrier integrity and function) and thus should not be regarded as placebo controls. The mechanism underlying this efficacy may be due to the impact of vehicle composition on skin barrier integrity and transepidermal water loss.18 The hydrophilic emulsion or other constituents of AzA foam (eg, fatty alcohols) may play a role.
A notable strength of our study is detailed clinical characterization using carefully chosen parameters and preplanned analyses that complement the primary end points. As the latter are often driven by regulatory requirements, opportunities to characterize other outcomes of interest to clinicians may be missed. The additional analyses reported here hopefully will aid dermatologists in both assessing the role of AzA foam in the treatment armamentarium for PPR and counseling patients.
Because participants with lighter skin pigmentation dominated our study population, the impact of AzA foam among patients with darker skin complexions is unknown. Although AzA is unlikely to cause hypopigmentation in normal undiseased skin, patients should be monitored for early signs of hypopigmentation.19,20 Our data also do not allow assessment of the differential effect, if any, of AzA foam on erythema of different etiologies in PPR, as corresponding information was not collected in the trial.
Conclusion
Azelaic acid foam 15% combines a well-established treatment of PPR with new vehicle technology to deliver effective therapy across multiple disease dimensions. In addition, the vehicle foam appears to demonstrate inherent therapeutic properties independent of AzA. The availability of this novel, efficacious, and well-tolerated option for PPR has the potential to improve patient care, reduce disease burden, and minimize unnecessary costs through increased tolerability and compliance.21
Acknowledgment
Editorial support through inVentiv Medical Communications (New York, New York) was provided by Bayer Pharmaceuticals.
Papulopustular rosacea (PPR) is characterized by centrofacial papules, pustules, erythema, and occasionally telangiectasia.1,2 A myriad of factors, including genetic predisposition3 and environmental triggers,4 have been associated with dysregulated inflammatory responses,5 contributing to the disease pathogenesis and symptoms. Inflammation associated with PPR may decrease skin barrier function, increase transepidermal water loss, and reduce stratum corneum hydration,6,7 resulting in heightened skin sensitivity, pain, burning, and/or stinging.5,8
Azelaic acid (AzA), which historically has only been available in gel or cream formulations, is well established for the treatment of rosacea9; however, these formulations have been associated with application-site adverse events (AEs)(eg, burning, erythema, irritation), limited cosmetic acceptability, and reduced compliance or efficacy.10
For select skin conditions, active agents delivered in foam vehicles may offer superior tolerability with improved outcomes.11 An AzA foam 15% formulation was approved for the treatment of mild to moderate PPR. Primary outcomes from a phase 3 trial demonstrated the efficacy and safety of AzA foam in improving inflammatory lesion counts (ILCs) and disease severity in participants with PPR. The trial also evaluated additional secondary end points, including the effect of AzA foam on erythema, inflammatory lesions, treatment response, and other manifestations of PPR.12 The current study evaluated investigator-reported efficacy outcomes for these secondary end points for AzA foam 15% versus vehicle foam.
Methods
Study Design
This phase 3 multicenter, randomized, double-blind, vehicle-controlled, parallel-group clinical trial was conducted from September 2012 to January 2014 at 48 US study centers comparing the efficacy of AzA foam versus vehicle foam in patients with PPR. Eligible participants were 18 years and older with PPR rated as moderate or severe according to investigator global assessment (IGA), plus 12 to 50 inflammatory lesions and persistent erythema with or without telangiectasia. Exclusion criteria included known nonresponse to AzA, current or prior use (within 6 weeks of randomization) of noninvestigational products to treat rosacea, and presence of other dermatoses that could interfere with rosacea evaluation.
Participants were randomized into the AzA foam or vehicle group (1:1 ratio). The study medication was applied in 0.5-g doses twice daily until the end of treatment (EoT) at 12 weeks. Efficacy and safety parameters were evaluated at baseline and at 4, 8, and 12 weeks of treatment, and at a follow-up visit 4 weeks after EoT (week 16).
Results for the coprimary efficacy end points—therapeutic success rate according to IGA and nominal change in ILC—were previously reported.12
Investigator-Reported Secondary Efficacy Outcomes
The secondary efficacy end points were grouped change in erythema rating, grouped change in telangiectasia rating, grouped change in IGA score, therapeutic response rate according to IGA, percentage change in ILC from baseline, and facial skin color rating at EoT.
Grouped change for all secondary end points was measured as improved, no change, or worsened relative to baseline. For grouped change in erythema and telangiectasia ratings, a participant was considered improved if the rating at the postbaseline visit was lower than the baseline rating, no change if the postbaseline and baseline ratings were identical, and worsened if the postbaseline rating was higher than at baseline. For grouped change in IGA score, a participant was considered improved if a responder showed at least a 1-step improvement postbaseline compared to baseline, no change if postbaseline and baseline ratings were identical, and worsened if the postbaseline rating was higher than at baseline.
For the therapeutic response rate, a participant was considered a treatment responder if the IGA score improved from baseline and resulted in clear, minimal, or mild disease severity at EoT.
Safety
Adverse events also were assessed.
Statistical Analyses
Secondary efficacy and safety end points were assessed for all randomized participants who were dispensed the study medication. Missing data were imputed using last observation carried forward.
For the percentage change in ILC from baseline, therapeutic response rate, and grouped change in erythema rating, confirmatory analyses were conducted in a hierarchical manner (in the order listed), with testing stopped as soon as a null hypothesis of superior treatment effect could not be rejected. Analyses without significance level were exploratory. The Cochran-Mantel-Haenszel van Elteren test stratified by study center was used for grouped change in erythema rating (1-tailed, 2.5%) and IGA score (2-tailed, 5%); Wilcoxon rank sum tests also were performed. Percentage change in ILC from baseline was evaluated using the Student t test and F test of analysis of covariance (1-tailed, 2.5%). Therapeutic response rate was evaluated using the Cochran-Mantel-Haenszel van Elteren test stratified by study center and the Pearson χ2 test. Facial skin color and grouped change in telangiectasia rating were evaluated using the Wilcoxon rank sum test.
Adverse events beginning or worsening after the first dose of the study drug were considered treatment emergent and were coded using the Medical Dictionary for Regulatory Activities (MedDRA) Version 16.1. Statistical analyses were performed using SAS software version 9.2.
Results
Study Participants
The study included 961 total participants; 483 were randomized to the AzA foam group and 478 to the vehicle group (Figure 1). Overall, 803 participants completed follow-up; however, week 16 results for the efficacy outcomes include data for 4 additional patients (2 per study arm) who did not formally meet all requirements for follow-up completion. The mean age was 51.5 years, and the majority of the participants were white and female (Table 1). Most participants (86.8%) had moderate PPR at baseline, with the remaining rated as having severe disease (13.2%). The majority (76.4%) had more than 14 inflammatory lesions with moderate (76.4%) or severe (15.1%) erythema at baseline.
Efficacy
Significantly more participants in the AzA group than in the vehicle group showed an improved erythema rating at EoT (61.5% vs 51.3%; P<.001)(Figure 2), with more participants in the AzA group showing improvement at weeks 4 (P=.022) and 8 (P=.002).
A significantly greater mean percentage reduction in ILC from baseline to EoT was observed in the AzA group versus the vehicle group (61.6% vs 50.8%; P<.001)(Figure 3), and between-group differences were observed at week 4 (P<.001), week 8 (P=.003), and week 16 (end of study/follow-up)(P=.002).
A significantly higher proportion of participants treated with AzA foam versus vehicle were considered responders at week 12/EoT (66.3% vs 54.4%; P<.001)(Figure 4). Differences in responder rate also were observed at week 4 (P=.026) and week 8 (P=.026).
No study drug was administered between week 12/EoT and week 16/follow-up; last observation carried forward was not applied to week 16/follow-up analysis. AzA indicates azelaic acid; IGA, investigator global assessment.
Differences in grouped change in IGA score were observed between groups at every evaluation during the treatment phase (Figure 5). Specifically, IGA score was improved at week 12/EoT relative to baseline in 71.2% of participants in the AzA group versus 58.8% in the vehicle group (P<.001).
For grouped change in telangiectasia rating at EoT, the majority of participants in both treatment groups showed no change (Table 2). Regarding facial skin color, the majority of participants in both the AzA and vehicle treatment groups (80.1% and 78.7%, respectively) showed normal skin color compared to nontreated skin EoT; no between-group differences were detected for facial skin color rating (P=.315, Wilcoxon rank sum test).
Safety
The incidence of drug-related AEs was greater in the AzA group than the vehicle group (7.7% vs 4.8%)(Table 3). Drug-related AEs occurring in at least 1% of the AzA group were pain at application site (eg, tenderness, stinging, burning)(AzA group, 3.5%; vehicle group, 1.3%), application-site pruritus (1.4% vs 0.4%), and application-site dryness (1.0% vs 0.6%). A single drug-related AE of severe intensity (ie, application-site dermatitis) was observed in the vehicle group; all other drug-related AEs were mild or moderate. The incidence of withdrawals due to AEs was lower in the AzA group than the vehicle group (1.2% vs 2.5%). This AE profile correlated with a treatment compliance (the percentage of expected doses that were actually administered) of 97.0% in the AzA group and 95.9% in the vehicle group. One participant in the vehicle group died due to head trauma unrelated to administration of the study drug.
Comment
The results of this study further support the efficacy of AzA foam for the treatment of PPR. The percentage reduction in ILC was consistent with nominal decreases in ILC, a coprimary efficacy end point of this study.12 Almost two-thirds of participants treated with AzA foam achieved a therapeutic response, indicating that many participants who did not strictly achieve the primary outcome of therapeutic success nevertheless attained notable reductions in disease severity. The number of participants who showed any improvement on the IGA scale increased throughout the course of treatment (63.8% AzA foam vs 55.0% vehicle at week 8) up to EoT (71.2% vs 58.8%)(Figure 5). In addition, the number of participants showing any improvement at week 8 (63.8% AzA foam vs 55.0% vehicle)(Figure 5) was comparable to the number of participants achieving therapeutic response at week 12/EoT (66.3% vs 54.4%)(Figure 4). These data suggest that increasing time of treatment increases the likelihood of achieving better results.
Erythema also appeared to respond to AzA foam, with 10.2% more participants in the AzA group demonstrating improvement at week 12/EoT compared to vehicle. The difference in grouped change in erythema rating also was statistically significant and favored AzA foam, sustained up to 4 weeks after EoT.
The outcomes for percentage change in ILC, therapeutic response rate, and grouped change in erythema rating consequently led to the rejection of all 3 null hypotheses in hierarchical confirmatory analyses, underscoring the benefits of AzA foam treatment.
The therapeutic effects of AzA foam were apparent at the first postbaseline evaluation and persisted throughout treatment. Differences favoring AzA foam were observed at every on-treatment evaluation for grouped change in erythema rating, percentage change in ILC, therapeutic response rate, and grouped change in IGA score. Symptoms showed minimal resurgence after treatment cessation, and there were no signs of disease flare-up within the 4 weeks of observational follow-up. In addition, the percentage reduction in ILC remained higher in the AzA foam group during follow-up.
These results also show that AzA foam was well tolerated with a low incidence of discontinuation because of drug-related AEs. No serious drug-related AEs were reported for this study or in the preceding phase 2 trial.12,13 Although not directly evaluated, the low incidence of cutaneous AEs suggests that AzA foam may be better tolerated than prior formulations of AzA14,15 and correlates with high compliance observed during the study.12 Azelaic acid foam appeared to have minimal to no effect on skin color, with more than 88% of participants reporting barely visible or no skin lightening.
Interestingly, the vehicle foam showed appreciable efficacy independent of AzA. Improvements in erythema were recorded in approximately half of the vehicle group at week 12/EoT. A similar proportion attained a therapeutic response, and ILC was reduced by 50.8% at week 12/EoT. Comparable results also were evident in the vehicle group for the primary end points of this study.12 Vehicles in dermatologic trials frequently exert effects on diseased skin16,17 via a skin care regimen effect (eg, moisturization and other vehicle-related effects that may improve skin barrier integrity and function) and thus should not be regarded as placebo controls. The mechanism underlying this efficacy may be due to the impact of vehicle composition on skin barrier integrity and transepidermal water loss.18 The hydrophilic emulsion or other constituents of AzA foam (eg, fatty alcohols) may play a role.
A notable strength of our study is detailed clinical characterization using carefully chosen parameters and preplanned analyses that complement the primary end points. As the latter are often driven by regulatory requirements, opportunities to characterize other outcomes of interest to clinicians may be missed. The additional analyses reported here hopefully will aid dermatologists in both assessing the role of AzA foam in the treatment armamentarium for PPR and counseling patients.
Because participants with lighter skin pigmentation dominated our study population, the impact of AzA foam among patients with darker skin complexions is unknown. Although AzA is unlikely to cause hypopigmentation in normal undiseased skin, patients should be monitored for early signs of hypopigmentation.19,20 Our data also do not allow assessment of the differential effect, if any, of AzA foam on erythema of different etiologies in PPR, as corresponding information was not collected in the trial.
Conclusion
Azelaic acid foam 15% combines a well-established treatment of PPR with new vehicle technology to deliver effective therapy across multiple disease dimensions. In addition, the vehicle foam appears to demonstrate inherent therapeutic properties independent of AzA. The availability of this novel, efficacious, and well-tolerated option for PPR has the potential to improve patient care, reduce disease burden, and minimize unnecessary costs through increased tolerability and compliance.21
Acknowledgment
Editorial support through inVentiv Medical Communications (New York, New York) was provided by Bayer Pharmaceuticals.
- Tan J, Berg M. Rosacea: current state of epidemiology. J Am Acad Dermatol. 2013;69(6, suppl 1):S27-S35.
- Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the classification and staging of rosacea. J Am Acad Dermatol. 2002;46:584-587.
- Chang AL, Raber I, Xu J, et al. Assessment of the genetic basis of rosacea by genome-wide association study. J Invest Dermatol. 2015;135:1548-1555.
- Abram K, Silm H, Maaroos HI, et al. Risk factors associated with rosacea. J Eur Acad Dermatol Venereol. 2010;24:565-571.
- Yamasaki K, Di Nardo A, Bardan A, et al. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat Med. 2007;13:975-980.
- Yamasaki K, Kanada K, Macleod DT, et al. TLR2 expression is increased in rosacea and stimulates enhanced serine protease production by keratinocytes. J Invest Dermatol. 2011;131:688-697.
- Darlenski R, Kazandjieva J, Tsankov N, et al. Acute irritant threshold correlates with barrier function, skin hydration and contact hypersensitivity in atopic dermatitis and rosacea. Exp Dermatol. 2013;22:752-753.
- Del Rosso JQ, Levin J. The clinical relevance of maintaining the functional integrity of the stratum corneum in both healthy and disease-affected skin. J Clin Aesthet Dermatol. 2011;4:22-42.
- van Zuuren EJ, Kramer SF, Carter BR, et al. Effective and evidence-based management strategies for rosacea: summary of a Cochrane systematic review. Br J Dermatol. 2011;165:760-781.
- Tan X, Feldman SR, Chang J, et al. Topical drug delivery systems in dermatology: a review of patient adherence issues. Expert Opin Drug Deliv. 2012;9:1263-1271.
- Stein L. Clinical studies of a new vehicle formulation for topical corticosteroids in the treatment of psoriasis. J Am Acad Dermatol. 2005;53(1, suppl 1):S39-S49.
- Draelos ZD, Elewski BE, Harper JC, et al. A phase 3 randomized, double-blind, vehicle-controlled trial of azelaic acid foam 15% in the treatment of papulopustular rosacea. Cutis. 2015;96:54-61.
- Draelos ZD, Elewski B, Staedtler G, et al. Azelaic acid foam 15% in the treatment of papulopustular rosacea: a randomized, double-blind, vehicle-controlled study. Cutis. 2013;92:306-317.
- Finacea gel [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2016.
- Elewski BE, Fleischer AB Jr, Pariser DM. A comparison of 15% azelaic acid gel and 0.75% metronidazole gel in the topical treatment of papulopustular rosacea: results of a randomized trial. Arch Dermatol. 2003;139:1444-1450.
- Daniels R, Knie U. Galenics of dermal products—vehicles, properties and drug release. J Dtsch Dermatol Ges. 2007;5:367-383.
- Shamsudin N, Fleischer AB Jr. Vehicle or placebo? Investigators use incorrect terminology in randomized controlled trials half of the time: a systematic review of randomized controlled trials published in three major dermatology journals. J Drugs Dermatol. 2010;9:1221-1226.
- Del Rosso JQ, Thiboutot D, Gallo R, et al. Consensus recommendations from the American Acne & Rosacea Society on the management of rosacea, part 2: a status report on topical agents. Cutis. 2013;92:277-284.
- Finacea foam [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2015.
- Solano F, Briganti S, Picardo M, et al. Hypopigmenting agents: an updated review on biological, chemical and clinical aspects. Pigment Cell Res. 2006;19:550-571.
- Hammarstrom B, Wessling A, Nilsson JL. Pharmaceutical care for patients with skin diseases: a campaign year at Swedish pharmacies. J Clin Pharm Ther. 1995;20:327-334.
- Tan J, Berg M. Rosacea: current state of epidemiology. J Am Acad Dermatol. 2013;69(6, suppl 1):S27-S35.
- Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the classification and staging of rosacea. J Am Acad Dermatol. 2002;46:584-587.
- Chang AL, Raber I, Xu J, et al. Assessment of the genetic basis of rosacea by genome-wide association study. J Invest Dermatol. 2015;135:1548-1555.
- Abram K, Silm H, Maaroos HI, et al. Risk factors associated with rosacea. J Eur Acad Dermatol Venereol. 2010;24:565-571.
- Yamasaki K, Di Nardo A, Bardan A, et al. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat Med. 2007;13:975-980.
- Yamasaki K, Kanada K, Macleod DT, et al. TLR2 expression is increased in rosacea and stimulates enhanced serine protease production by keratinocytes. J Invest Dermatol. 2011;131:688-697.
- Darlenski R, Kazandjieva J, Tsankov N, et al. Acute irritant threshold correlates with barrier function, skin hydration and contact hypersensitivity in atopic dermatitis and rosacea. Exp Dermatol. 2013;22:752-753.
- Del Rosso JQ, Levin J. The clinical relevance of maintaining the functional integrity of the stratum corneum in both healthy and disease-affected skin. J Clin Aesthet Dermatol. 2011;4:22-42.
- van Zuuren EJ, Kramer SF, Carter BR, et al. Effective and evidence-based management strategies for rosacea: summary of a Cochrane systematic review. Br J Dermatol. 2011;165:760-781.
- Tan X, Feldman SR, Chang J, et al. Topical drug delivery systems in dermatology: a review of patient adherence issues. Expert Opin Drug Deliv. 2012;9:1263-1271.
- Stein L. Clinical studies of a new vehicle formulation for topical corticosteroids in the treatment of psoriasis. J Am Acad Dermatol. 2005;53(1, suppl 1):S39-S49.
- Draelos ZD, Elewski BE, Harper JC, et al. A phase 3 randomized, double-blind, vehicle-controlled trial of azelaic acid foam 15% in the treatment of papulopustular rosacea. Cutis. 2015;96:54-61.
- Draelos ZD, Elewski B, Staedtler G, et al. Azelaic acid foam 15% in the treatment of papulopustular rosacea: a randomized, double-blind, vehicle-controlled study. Cutis. 2013;92:306-317.
- Finacea gel [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2016.
- Elewski BE, Fleischer AB Jr, Pariser DM. A comparison of 15% azelaic acid gel and 0.75% metronidazole gel in the topical treatment of papulopustular rosacea: results of a randomized trial. Arch Dermatol. 2003;139:1444-1450.
- Daniels R, Knie U. Galenics of dermal products—vehicles, properties and drug release. J Dtsch Dermatol Ges. 2007;5:367-383.
- Shamsudin N, Fleischer AB Jr. Vehicle or placebo? Investigators use incorrect terminology in randomized controlled trials half of the time: a systematic review of randomized controlled trials published in three major dermatology journals. J Drugs Dermatol. 2010;9:1221-1226.
- Del Rosso JQ, Thiboutot D, Gallo R, et al. Consensus recommendations from the American Acne & Rosacea Society on the management of rosacea, part 2: a status report on topical agents. Cutis. 2013;92:277-284.
- Finacea foam [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2015.
- Solano F, Briganti S, Picardo M, et al. Hypopigmenting agents: an updated review on biological, chemical and clinical aspects. Pigment Cell Res. 2006;19:550-571.
- Hammarstrom B, Wessling A, Nilsson JL. Pharmaceutical care for patients with skin diseases: a campaign year at Swedish pharmacies. J Clin Pharm Ther. 1995;20:327-334.
Practice Points
- Papulopustular rosacea (PPR) is a common chronic inflammatory dermatosis.
- A novel hydrophilic foam formulation of azelaic acid (AzA) was approved for the treatment of PPR.
- In addition to effectively treating papules and pustules, AzA foam also may reduce rosacea-associated erythema.
- The unique AzA foam vehicle may improve epidermal barrier integrity and function, thereby offering patients a distinct topical approach to rosacea management.
Short-course therapy for recurrent genital herpes and herpes labialis
- Consider giving patients an oral antiviral (OAV) medication to self-administer when HSV prodromal symptoms occur.
- Patient-initiated, short-course, high-dose OAV treatment of recurrent HSV outbreaks may be as effective as the traditional, longer-course regimens.
Hit early, hit hard. That expression arose during the evolution of treatment for human immunodeficiency virus (HIV).1 While this approach has not lived up to expectations for HIV treatment, it may have found its place in the treatment of recurrent herpes simplex virus (HSV) infections.
Our review focuses on episodic treatment of acute recurrent HSV outbreaks for immunocompetent persons. We do not discuss suppressive therapy, which may be indicated for frequent or severe recurrences (6 or more per year) in immunocompetent persons, for immunocompromised patients, or as an adjunctive measure to reduce genital herpes transmission.2
As we will describe in detail, the efficacy of the new short-course therapy is, at minimum, comparable to that seen with the older, longer-course trials of topical and oral antiviral therapy. In one head-to-head comparison, Leone et al compared a short-course regimen (3 days) of valacyclovir with 5 days of treatment; they found no difference in results.3 If the efficacy of short-course treatment is the same as that of longer courses, the increased convenience and expected improvement in patient adherence with these new regimens argue strongly in their favor. (See Scope of the problem.)
The strategy: Take advantage of a brief therapeutic window
The innate and acquired immune responses of chronically infected, immunocompetent persons rapidly limit cutaneous viral replication, thereby truncating the duration of recurrent HSV outbreaks.13,14 In both recurrent herpes labialis and genital herpes, HSV viral titers peak in the first 24 hours following lesion onset (FIGURE 1A).13-15
Herpes labialis lesion size and pain are also greatest in the first 24 hours.13,16 Most herpes labialis lesions progress from the vesicle stage to the ulcer/soft crust stage within 48 hours, with a hard crust forming by day 2 or 3 (FIGURE 1B).17
With genital lesions, crust formation depends on whether the skin area is dry (3–4 days) or moist (8–9 days).14
The likely events are a burst of virus replication in the first 24 hours of outbreak that lyses basal keratinocytes in a discreet area of epidermis innervated by the infected neuron(s), followed by a vigorous immune response that curtails the infection and creates, in part, the clinical disease (erythema, swelling, vesiculation, and ulceration). The subsequent elements of the illness, which are the majority of the lesion course, are related to wound healing
Recognizing the window. Given the brief period of viral replication and the rapid evolution of lesions, the therapeutic window for treating HSV outbreaks with antiviral drugs is both early and short, making it problematic to effectively treat HSV recurrences. Patients often have mature lesions by the time they consult a physician, rendering subsequent anti-viral treatment less effective.18 However, before lesions appear, many patients experience prodromal symptoms such as pain, burning, or itching.13,18 These symptoms can be a prompt to start treatment early, thereby taking advantage of the transient therapeutic window.
If a patient is able to self-administer therapy when prodromal symptoms occur, there may be a greater benefit to treatment. Giving patients drugs for self-administration is therefore an important strategy in managing HSV recurrences.
Traditionally, patient-initiated episodic therapy for recurrent genital herpes and herpes labialis has involved multiple daily doses of topical or oral antiviral agents for 4 to 5 days.19-26 Studies of the pathogenesis of HSV recurrences, however, indicate—as said earlier—that the period of virus replication is early and brief, such that a shorter duration of treatment might be more appropriate and equally effective. Other recent clinical studies have indicated that patient-initiated, short-course, high-dose OAV treatment of recurrent HSV infections may be as effective as the traditional therapies.3,27-30 In the section that follows, we examine and compare the results of these trials. (See The agents and how they work.)
FIGURE 1A
Lesion HSV-1 titer peaks within 24 hours of onset of herpes labialis lesions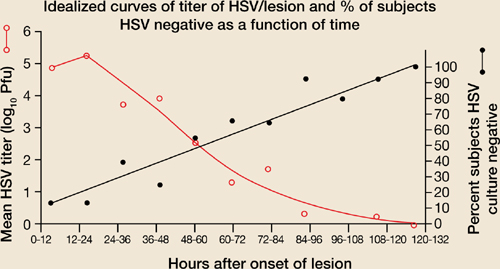
Source: Krueger et al, J Clin Epidemiol Derm 1978.15 Reproduced with permission from GG Krueger.
FIGURE 1B
Hard crust formation occurs by 48 to 72 hours of onset of herpes labialis lesions
Source: Spruance, Sem Dermatol 1992.17 With permission from Elsevier.
Clinical trials: Short-course, high-dose, patient-initiated episodic OAV therapy for recurrent genital herpes
Three-day vs 5-day valacyclovir therapy. The efficacy of 3-day treatment with oral valacyclovir was compared with that of 5-day treatment in immunocompetent adults with a history of ≥4 episodes of recurrent genital herpes and confirmed HSV infection.3 Eight hundred participants were randomized to receive 500 mg twice daily valacyclovir for 3 days (and placebo for the remaining 2 days) or 500 mg twice daily for 5 days, and were required to self-administer therapy no later than 24 hours after the onset of symptoms.
Herpes simplex virus (HSV) type 1 (HSV-1) or type 2 (HSV-2) results in periodic, recurrent outbreaks of skin lesions after first infection. Herpes labialis (fever blisters or cold sores) is usually caused by HSV-1, while genital herpes is usually caused by HSV-2.4 HSV-2 lesions of the lips have been reported, and the incidence of genital herpes caused by HSV-1 is on the rise in the developed world, likely because of increased oral-genital sexual behavior.5,6 Patients with HSV-1 genital herpes typically have fewer recurrences than those with HSV-2 genital infection.7
The prevalence of HSV-1 and HSV-2 infection varies according to age, geography, gender, and population subgroup, such as people who exhibit high-risk sexual behavior.8 approximately 45% of americans are infected with HSV-1 by adolescence,8 and approximately 22% of all american adults are infected with HSV-2.9 The global prevalence of HSV is even greater: as many as 60% to 90% of older adults worldwide are seropositive for HSV-1, and as many as 30% are seropositive for HSV-2. HSV-2 seropositivity is more prevalent among women than men.8 overall, the burden of recurrent genital herpes outbreaks can have a profound, negative impact on patient quality of life.10,11 The psychological impact of recurrent herpes labialis has not been thoroughly investigated, but an undefined burden is thought to exist, particularly in young patients with frequent or severe recurrences.12
The primary endpoint was time to lesion healing (defined as the number of days from initiation of therapy to lesion reepithelialization). Secondary endpoints were pain duration, episode duration (defined as time from initiation of therapy to resolution of all symptoms) and percentage of patients with aborted lesions.
The 3-day valacyclovir treatment exhibited similar time to lesion healing, length of episode, and percentage of patients with aborted lesions as the 5-day treatment (TABLE 1), suggesting equal efficacy. Duration of pain was also similar (data not shown). Adverse events were similar for both treatment groups, with the most common being headache (10%), nausea (4%), and diarrhea (4%, 5-day treatment vs 2%, 3-day treatment).
Placebo-controlled trial of 2-day acyclovir therapy. Wald and coworkers examined the effect of a shorter treatment regimen of acyclovir (2 days) on recurrent genital herpes.28 Eighty-four immunocompetent HSV-2–infected patients with a history of ≥3 recurrences in the previous 12 months were randomized to receive either 2 days of 800 mg 3 times daily acyclovir or matching placebo. Patients were asked to take their medication no later than 12 hours after the first sign or symptom of an episode.
Efficacy endpoints were time to lesion healing, episode duration, and percentage of patients with aborted lesions. Short-course acyclovir therapy was shown to decrease time to healing (P=.001) and episode duration (P<.001) by 2 days compared with placebo (TABLE 1). Short-course acyclovir therapy also increased the percentage of patients with aborted lesions compared with placebo (27% vs 11%; P=.029 (TABLE 1). Adverse events were not recorded in this analysis.
Placebo-controlled trial of single-day famciclovir therapy. Aoki and colleagues29 performed a randomized, double-blind, patient-initiated, placebo-controlled trial to assess the efficacy and safety of patient-initiated, single-day famciclovir 1000 mg twice daily in immunocompetent adults with recurrent genital herpes. The 329 patients in the study were instructed to self-initiate therapy within 6 hours of the onset of prodromal symptoms or genital herpes lesions, and were asked to return to the clinic no later than 24 hours after initiation of therapy. Patients were followed until their lesions healed or for up to 14 days.
The primary endpoint was time to lesion healing of nonaborted lesions. Secondary endpoints were time to healing of all lesions (aborted and nonaborted), time to resolution of pain and other symptoms, and the percentage of patients who did not progress to a full outbreak.
Single-day treatment with famciclovir shortened the time to healing of nonaborted genital herpes lesions by approximately 2 days (P<.001), and the time to healing of all lesions by 1.5 days (P<.001) compared with placebo, and increased the percentage of patients who did not progress to a full outbreak (23% vs 13%;P=.003) (TABLE 1). Famciclovir also reduced the time to resolution of all symptoms by approximately 2 days (P<.001) (data not shown).
Adverse events were mild to moderate; the most common in the famciclovir and placebo groups, respectively, were headache (13.5% vs 5.4%), nausea (2.5% vs 3.6%), and diarrhea (4.9% vs 1.2%).
TABLE 1
Short-course, patient-initiated OAV therapy is effective for treating episodic genital herpes
| DRUG | TREATMENT DURATION | TREATMENT DOSE | CONTROL | MEDIAN TIME (DAYS) TO LESION HEALING (TREATMENT VS CONTROL) | MEDIAN EP ISODE DURATION (DAYS) (TREATMENT VS CONTROL) | PATIENTS WITH ABORTED EPISODES (%) (TREATMENT VS CONTROL) |
|---|---|---|---|---|---|---|
| Valacyclovir3 | 3 days | 500 mg 2×daily | valacyclovir 500 mg 2×/day for 5 days | 4.4 vs 4.7 (P=NS) | 4.3 vs 4.4 (P=NS) | 25 vs 27 (P=NS) |
| Acyclovir28 | 2 days | 800 mg 3×daily | Placebo | 4.0 vs 6.0 (P=.001) | 4.0 vs 6.0 (P=.001) | 27 vs 11 (P=.029) |
| Famciclovir29 | 1 day | 1000 mg 2×daily | Placebo | 4.3 vs 6.1 (P<.001) | 3.5 vs 5.0 (P<.001) | 23 vs 13 (P=.003) |
| Lesion healing time measures the duration of a subset of severe or classical herpetic outbreaks, characterized by the formation of vesicles, ulcers, or crusts (also papules in some studies28,29). The endpoint is lesion reepithelialization/loss of crust. Episodes where there were only prodromal symptoms, erythema, and/or papule formation (or only symptoms and/or erythema in some studies28,29) were considered “aborted” or prevented lesions. The occurrence of these favorable episode outcomes is described as a percentage of all episodes. Episode duration, sometimes called healing time of all lesions or time to return to normal skin, is the time to resolution of all episodes, regardless of lesion severity. The definition of normal skin varies among the different studies. | ||||||
| NS=not significant. | ||||||
Short-course, high-dose, patient-initiated episodic OAV therapy for recurrent herpes labialis
Placebo-controlled trial of single-day and 2-day valacyclovir therapy. Spruance and coworkers studied the efficacy of single-day and 2-day valacyclovir treatments in comparison with placebo for an episode of herpes labialis.27 Two identical studies were performed on individuals who were at least 12 years old, had a clinical history of recurrent cold sores, and had experienced ≥3 episodes in the preceding year. Participants in both studies (study 1, N=1524; study 2, N=1627) were required to self-administer 2 g valacyclovir twice daily for 1 day (valacyclovir 1 day), 2 g valacyclovir twice daily for 1 day followed by 1 g twice daily for 1 day (valacyclovir 2 days), or matching placebo at the earliest onset of prodromal symptoms and before the appearance of lesions. Patients were asked to return to the clinic within 24 hours of initiation of therapy.
The primary endpoint in study 1 was clinician-observed duration of all herpes labialis lesions and the secondary endpoint was the percentage of subjects who had herpes labialis lesions that did not progress beyond the papule stage. In study 2, the endpoints were reversed: the primary endpoint was the percentage of patients with lesions that did not progress and the secondary endpoint was the duration of lesions. Other efficacy endpoints were time to healing of vesicular (classical) lesions and duration of pain and discomfort.
Both studies demonstrated that single-day valacyclovir treatment significantly decreased lesion healing time and the duration of herpes labialis episodes by 0.5 to 1.0 days compared with placebo (TABLE 2). A statistically significant decrease in the duration of pain and other symptoms was also seen with single-day valacyclovir compared with placebo (data not shown). In both studies, a higher percentage of patients in the valacyclovir group did not progress to full outbreak compared with placebo, but these differences were not statistically significant. The results with 2 days of valacyclovir treatment were similar. Adverse events were similar between the treatment groups and the placebo group.
Placebo-controlled trial of single-dose and single-day famciclovir therapy. Spruance and coworkers assessed patient-initiated famciclovir 1500 mg (single-dose) and 750 mg twice daily (single-day) in immunocompetent adults with recurrent cold sores.30 Subjects (N=1376) were at least 18 years of age and had experienced ≥3 episodes of cold sores over the previous 12 months. Subjects were instructed to administer 1500 mg (single-dose), 750 mg twice daily (single-day), or matching placebo within 1 hour of the onset of prodromal symptoms and before the onset of lesions, and were asked to return to the clinic within 24 hours of initiating medication.
Topical antiviral drug formulations were the first treatments approved for recurrent HSV-1 and HSV-2 outbreaks, but these were only marginally efficacious.19-21,31 orally-administered antiviral agents appear to be more effective, possibly because of better delivery of the drug to the site of infection. Three oral antiviral agents (OAVs) are currently approved for the treatment of recurrent genital herpes: acyclovir, an acyclic nucleoside analog; valacyclovir, the prodrug of acyclovir; and famciclovir, the prodrug of penciclovir, another acyclic nucleoside analog. one OAV (valacyclovir) is currently approved for the treatment of herpes labialis in immunocompetent patients.27 The prodrugs of acyclovir and penciclovir, valacyclovir and famciclovir, respectively, were synthesized to provide high oral bioavailability and thus permit less frequent administration and potentially greater efficacy compared to the parent compounds.
Following oral administration, valacyclovir and famciclovir undergo first-pass metabolism to acyclovir and penciclovir, respectively.4,32 acyclovir and penciclovir are selectively phosphorylated by the viral thymidine kinase of infected cells and then converted to the active triphosphate by cellular enzymes. The triphosphate forms (which have different half-lives depending upon the compound)33 inhibit viral DNA polymerase and interfere with DNA chain extension,34 thereby halting viral DNA synthesis. The drugs cannot prevent the death of a cell once it is infected, but they can reduce, in a dose-dependent manner, the quantity of virions produced by an infected cell. The mechanism of action of HSV-selective antiviral drugs suggests that the most logical strategy for episodic treatment is to maximally inhibit HSV replication using high doses.18,35
The primary endpoint was time to healing of primary vesicular lesions. Secondary endpoints included time to healing of all vesicular lesions (primary and secondary [secondary lesions are defined as lesions that developed in addition to and on 1 or more days after primary lesions and that were located at least 1 cm from primary lesions]), time to return to normal skin for all lesions (defined as loss of crust, swelling, and dry flaking), duration of lesion tenderness and pain, and proportion of patients with aborted lesions.
There was a statistically significant decrease in time to healing of primary vesicular lesions by approximately 2 days with both single-dose and single-day famciclovir compared with placebo, with no significant difference between the 2 famciclovir regimens in time to healing of primary vesicular lesions (TABLE 2). There was also a statistically significant decrease in the time to healing of all lesions (primary and secondary) by approximately 2 days with both famciclovir treatments compared with placebo, with no significant differences seen in healing between the famciclovir arms (data not shown).
However, only single-dose famciclovir had a statistically significant decrease in the duration of lesion tenderness and pain and the time to return to normal skin compared with placebo (data not shown). No difference was noted between the famciclovir arms in the percentage of patients with aborted lesions compared with placebo. Adverse events in both famciclovir groups were similar to those in the placebo group.
TABLE 2
Short-course, patient-initiated OAV therapy is effective against recurrent herpes labialis
| DRUG | TREATMENT DURATION | TREATMENT DOSE | COMPARATOR REGIMEN | CONTROL | MEDIAN TIME (DAYS) TO LESION HEALING (TREATMENT VS COMPARATOR VS CONTROL)* | MEDIAN EP ISODE DURATION (DAYS) (TREATMENT VS COMPARATOR VS CONTROL)* | PATIENTS WITH ABORTED LESIONS (%)(TREATMENT VS COMPARATOR VS CONTROL)† |
|---|---|---|---|---|---|---|---|
| Valacyclovir27 | 1 day | 2000 mg 2×daily | valacyclovir 2000 mg 2×daily×1 day 1000 mg 2×daily for a 2nd day | Placebo | study 1 4.3 vs 4.3 vs 5.1 study 2 4.8 vs 4.6 vs 5.4 | study 1 4.0 vs 4.5 vs 5.0 study 2 2 5.0 vs 5.0 vs 5.5 | study 1 44 vs 46 vs 38 study 2 43 vs 43 vs 35 |
| Famciclovir30 1 dose | 1 Does | 1500 mg | famciclovir 750 mg 2x daily for 1 day | Placebo | 4.4 vs 4.0 vs 6.2 | 4.5 vs 5.7 vs 7.0 | 33 vs 29 vs 34 |
| Lesion healing time measures the duration of a subset of severe or classical herpetic outbreaks, characterized by the formation of vesicles, ulcers, or crusts (also papules in some studies28,29). The endpoint is lesion reepithelialization/loss of crust. Episodes where there were only prodromal symptoms, erythema, and/or papule formation (or only symptoms and/or erythema in some studies28,29) were considered “aborted” or prevented lesions. The occurrence of these favorable episode outcomes is described as a percentage of all episodes. Episode duration, sometimes called healing time of all lesions or time to return to normal skin, is the time to resolution of all episodes, regardless of lesion severity. The definition of normal skin varies among the different studies. | |||||||
| *All of the healing time and episode duration values for the active treatment arms in both studies differed statistically significantly from placebo, except for famciclovir 750 mg twice daily for 1 day. | |||||||
| †None of the frequencies of aborted lesions in the active treatment arms in either study differed statistically significantly from placebo. | |||||||
CORRESPONDENCE
Spotswood Spruance MD Professor of Medicine, Division of Infectious Diseases, University of Utah School of Medicine, Room 4B319, 30 North 1900 East, Salt Lake City, UT 84132-2405. E-mail:woody.spruance@hsc.utah.edu
1. Ho D. Time to hit HIV, early and hard. N Engl J Med 1995;333:450-451.
2. Corey L, Wald A, Patel R, et al. Once-daily valacyclovir to reduce the risk of transmission of genital herpes. N Engl J Med 2004;350:11-20.
3. Leone PA, Trottier S, Miller JM. Valacyclovir for episodic treatment of genital herpes: a shorter 3-day treatment course compared with 5-day treatment. Clin Infect Dis 2002;34:958-962.
4. Whitley RJ, Kimberlin DW, Roizman B. Herpes simplex viruses. Clin Infect Dis 1998;26:541-555.
5. Wald A, Ericsson M, Krantz E, Selkes S, Corey L. Oral shedding of herpes simplex virus type 2. Sex Transm Infect 2004;80:272-276.
6. Mertz GJ, Rosenthal Sl, Stanberry LR. Editorial response: Is herpes simplex virus type 1 (HSV-1) now more common than HSV-2 in first episodes of genital herpes? Sex Transm Dis 2003;30:801-802.
7. Lafferty WE, Coombs RW, Benedetti J, Critchlow C, Corey L. Recurrences after oral and genital herpes simplex virus infection. N Engl J Med 1987;316:1444-1449.
8. Smith JS, Robinson RJ. Age-specific prevalence of infection with herpes simplex virus types 2 and 1: a global review. J Infect Dis 2002;186(suppl 1):S3-S28.
9. Fleming DT, McQuillan GM, Johnson RE, et al. Herpes simplex virus type 2 in the united States, 1976 to 1994. N Engl J Med 1997;337:1105-1111.
10. Bierman SM. A retrospective study of 375 patients with genital herpes simplex infections seen between 1973 and 1980. Cutis 1983;31:548-565.
11. Drob S, Loemer M, Lifshutz H. Genital herpes: the psychological consequences. Br J Med Psychol 1985;58:307-315.
12. Spruance SL, Kriesel JD. Treatment of herpes simplex labialis. Herpes 2002;9:64-69.
13. Spruance SL, overall JC, Jr, Kern ER, Krueger GG, Pliam V, Miller W. The natural history of recurrent herpes simplex labialis. N Engl J Med 1977;297:69-75.
14. Brown ZA, Kern ER, Spruance SL, Overall JC, Jr. Clinical and virologic course of herpes simplex genitalis. West J Med 1979;130:414-421.
15. Krueger GG, Spruance SL, Overall JC, Jr. Herpes simplex labialis: a review of pathogenesis and therapy. J Clin Epidemiol Derm 1978;1:19-37.
16. Spruance SL, Wenerstrom G. Pathogenesis of herpes simplex labialis: IV. Maturation of lesions during within 8 hours after onset and implications for antiviral treatment. Oral Surg Oral Med Oral Path 1984;58:667-671.
17. Spruance SL. The natural history of recurrent oral-facial herpes simplex virus infection. Sem Dermatol 1992;11:200-206.
18. Spruance SL. Herpes simplex labialis. In: Clinical Management of Herpes Viruses. Sacks SL, Straus SE, Whitley RJ, Griffiths PD, eds. amsterdam, Netherlands: IOS Press; 1995.
19. Spruance SL, Nett R, Marbury T, Wolff R, Johnson J, Spaulding T. Acyclovir cream for treatment of herpes simplex labialis: results of two randomized, double-blind, vehicle-controlled, multicenter clinical trials. Antimicrob Agents Chemother 2002;46:2238-2243.
20. Spruance SL, Rea TL, Thoming C, Tucker R, Saltzman R, Boon R. Penciclovir cream for the treatment of herpes simplex labialis. JAMA 1997;277:1374-1379.
21. Raborn GW, Martel AY, lassonde M, et al. Worldwide Topical Penciclovir Collaborative Study Group. Effective treatment of herpes simplex labialis with penciclovir cream: combined results of two trials. J Am Dent Assoc 2002;133:303-309.
22. Reichman RC, Badger GJ, Mertz GJ, et al. Treatment of recurrent genital herpes simplex infections with oral acyclovir: a controlled trial. JAMA 1984;251:2103-2107.
23. Sacks SL, Aoki FY, Diaz-Mitoma F, Sellors J, Shafran SD. Canadian Famciclovir Study Group. Patient-initiated, twice-daily oral famciclovir for early recurrent genital herpes: a randomized, double-blind multicenter trial. JAMA 1996;276:44-49.
24. Tyring SK, Douglas JM, Jr, Corey L, Spruance SL, Esmann J. The valaciclovir International Study Group. A randomized, placebo-controlled comparison of oral valcyclovir and acyclovir in immunocompetent patients with recurrent genital herpes infections. Arch Dermatol 1998;134:185-191.
25. Spruance S, Stewart JCB, Rowe NH, McKeough MB, Wenerstrom G, Freeman DJ. Treatment of herpes simplex labialis with oral acyclovir. J Infect Dis 1990;161:185-190.
26. Spruance SL, Tyring SK, DeGregorio B, Miller C, Beutner K; valaciclovir HSV Study Group. A large-scale, placebo-controlled, dose-ranging trial of peroral valaciclovir for episodic treatment of recurrent herpes genitalis. Arch Intern Med 1996;156:1729-1735.
27. Spruance SL, Jones TM, Blatter MM, et al. High-dose, short-duration, early valacyclovir therapy for episodic treatment of cold sores: results of two randomized, placebo-controlled, multicenter studies. Antimicrob Agents Chemother 2003;47:1072-1080.
28. Wald A, Carrell D, Remington M, Kexel E, Zeh J, Corey L. Two-day regimen of acyclovir for treatment of recurrent genital herpes simplex virus type 2 infection. Clin Infect Dis 2002;34:944-948.
29. Aoki FY, Tyring S, Dias-Mitoma F, Gross G, Gao J, Hamed K. Single-day patient initiated famciclovir therapy for recurrent genital herpes: a randomized double-blind, placebo-controlled trial. Clin Infect Dis 2006;42:8-13.
30. Spruance S, Bodsworth N, Resnick H, et al. Single-dose, patient-initiated famciclovir: a randomized, double-blind, placebo-controlled trial for episodic treatment of herpes labialis. J Am Acad Dermatol 2006;55:47-53.
31. Reichman RC, Badger GJ, Guinan ME, et al. Topically administered acyclovir in the treatment of recurrent herpes simplex genitalis: a controlled trial. J Infect Dis 1983;147:336-340.
32. Gill KS, Wood MJ. The clinical pharmacokinetics of famciclovir. Clin Pharmacokinet 1996;31:1-8.
33. Earnshaw DL, Bacon TH, Darlison SJ, Edmonds K, Perkins RM, Vere Hodge RA. Mode of antiviral action of penciclovir in MRC-5 cells infected with herpes simplex virus type 1 (HSV-1), HSV-2, and varicella-zoster virus. Antimicrob Agents Chemother 1992;36:2747-2757.
34. Vere Hodge RA, Perkins RM. Mode of action of 9-(4-hydroxy-3-hydroxymethylbut-1-yl)guanine (Brl 39123) against herpes simplex virus in MrC-5 cells. Antimicrob Agents Chemother 1989;33:223-229.
35. Spruance SL, Freeman DJ. Topical treatment of cutaneous herpes simplex virus infections. Antivir Res 1990;14:305-321.
- Consider giving patients an oral antiviral (OAV) medication to self-administer when HSV prodromal symptoms occur.
- Patient-initiated, short-course, high-dose OAV treatment of recurrent HSV outbreaks may be as effective as the traditional, longer-course regimens.
Hit early, hit hard. That expression arose during the evolution of treatment for human immunodeficiency virus (HIV).1 While this approach has not lived up to expectations for HIV treatment, it may have found its place in the treatment of recurrent herpes simplex virus (HSV) infections.
Our review focuses on episodic treatment of acute recurrent HSV outbreaks for immunocompetent persons. We do not discuss suppressive therapy, which may be indicated for frequent or severe recurrences (6 or more per year) in immunocompetent persons, for immunocompromised patients, or as an adjunctive measure to reduce genital herpes transmission.2
As we will describe in detail, the efficacy of the new short-course therapy is, at minimum, comparable to that seen with the older, longer-course trials of topical and oral antiviral therapy. In one head-to-head comparison, Leone et al compared a short-course regimen (3 days) of valacyclovir with 5 days of treatment; they found no difference in results.3 If the efficacy of short-course treatment is the same as that of longer courses, the increased convenience and expected improvement in patient adherence with these new regimens argue strongly in their favor. (See Scope of the problem.)
The strategy: Take advantage of a brief therapeutic window
The innate and acquired immune responses of chronically infected, immunocompetent persons rapidly limit cutaneous viral replication, thereby truncating the duration of recurrent HSV outbreaks.13,14 In both recurrent herpes labialis and genital herpes, HSV viral titers peak in the first 24 hours following lesion onset (FIGURE 1A).13-15
Herpes labialis lesion size and pain are also greatest in the first 24 hours.13,16 Most herpes labialis lesions progress from the vesicle stage to the ulcer/soft crust stage within 48 hours, with a hard crust forming by day 2 or 3 (FIGURE 1B).17
With genital lesions, crust formation depends on whether the skin area is dry (3–4 days) or moist (8–9 days).14
The likely events are a burst of virus replication in the first 24 hours of outbreak that lyses basal keratinocytes in a discreet area of epidermis innervated by the infected neuron(s), followed by a vigorous immune response that curtails the infection and creates, in part, the clinical disease (erythema, swelling, vesiculation, and ulceration). The subsequent elements of the illness, which are the majority of the lesion course, are related to wound healing
Recognizing the window. Given the brief period of viral replication and the rapid evolution of lesions, the therapeutic window for treating HSV outbreaks with antiviral drugs is both early and short, making it problematic to effectively treat HSV recurrences. Patients often have mature lesions by the time they consult a physician, rendering subsequent anti-viral treatment less effective.18 However, before lesions appear, many patients experience prodromal symptoms such as pain, burning, or itching.13,18 These symptoms can be a prompt to start treatment early, thereby taking advantage of the transient therapeutic window.
If a patient is able to self-administer therapy when prodromal symptoms occur, there may be a greater benefit to treatment. Giving patients drugs for self-administration is therefore an important strategy in managing HSV recurrences.
Traditionally, patient-initiated episodic therapy for recurrent genital herpes and herpes labialis has involved multiple daily doses of topical or oral antiviral agents for 4 to 5 days.19-26 Studies of the pathogenesis of HSV recurrences, however, indicate—as said earlier—that the period of virus replication is early and brief, such that a shorter duration of treatment might be more appropriate and equally effective. Other recent clinical studies have indicated that patient-initiated, short-course, high-dose OAV treatment of recurrent HSV infections may be as effective as the traditional therapies.3,27-30 In the section that follows, we examine and compare the results of these trials. (See The agents and how they work.)
FIGURE 1A
Lesion HSV-1 titer peaks within 24 hours of onset of herpes labialis lesions
Source: Krueger et al, J Clin Epidemiol Derm 1978.15 Reproduced with permission from GG Krueger.
FIGURE 1B
Hard crust formation occurs by 48 to 72 hours of onset of herpes labialis lesions
Source: Spruance, Sem Dermatol 1992.17 With permission from Elsevier.
Clinical trials: Short-course, high-dose, patient-initiated episodic OAV therapy for recurrent genital herpes
Three-day vs 5-day valacyclovir therapy. The efficacy of 3-day treatment with oral valacyclovir was compared with that of 5-day treatment in immunocompetent adults with a history of ≥4 episodes of recurrent genital herpes and confirmed HSV infection.3 Eight hundred participants were randomized to receive 500 mg twice daily valacyclovir for 3 days (and placebo for the remaining 2 days) or 500 mg twice daily for 5 days, and were required to self-administer therapy no later than 24 hours after the onset of symptoms.
Herpes simplex virus (HSV) type 1 (HSV-1) or type 2 (HSV-2) results in periodic, recurrent outbreaks of skin lesions after first infection. Herpes labialis (fever blisters or cold sores) is usually caused by HSV-1, while genital herpes is usually caused by HSV-2.4 HSV-2 lesions of the lips have been reported, and the incidence of genital herpes caused by HSV-1 is on the rise in the developed world, likely because of increased oral-genital sexual behavior.5,6 Patients with HSV-1 genital herpes typically have fewer recurrences than those with HSV-2 genital infection.7
The prevalence of HSV-1 and HSV-2 infection varies according to age, geography, gender, and population subgroup, such as people who exhibit high-risk sexual behavior.8 approximately 45% of americans are infected with HSV-1 by adolescence,8 and approximately 22% of all american adults are infected with HSV-2.9 The global prevalence of HSV is even greater: as many as 60% to 90% of older adults worldwide are seropositive for HSV-1, and as many as 30% are seropositive for HSV-2. HSV-2 seropositivity is more prevalent among women than men.8 overall, the burden of recurrent genital herpes outbreaks can have a profound, negative impact on patient quality of life.10,11 The psychological impact of recurrent herpes labialis has not been thoroughly investigated, but an undefined burden is thought to exist, particularly in young patients with frequent or severe recurrences.12
The primary endpoint was time to lesion healing (defined as the number of days from initiation of therapy to lesion reepithelialization). Secondary endpoints were pain duration, episode duration (defined as time from initiation of therapy to resolution of all symptoms) and percentage of patients with aborted lesions.
The 3-day valacyclovir treatment exhibited similar time to lesion healing, length of episode, and percentage of patients with aborted lesions as the 5-day treatment (TABLE 1), suggesting equal efficacy. Duration of pain was also similar (data not shown). Adverse events were similar for both treatment groups, with the most common being headache (10%), nausea (4%), and diarrhea (4%, 5-day treatment vs 2%, 3-day treatment).
Placebo-controlled trial of 2-day acyclovir therapy. Wald and coworkers examined the effect of a shorter treatment regimen of acyclovir (2 days) on recurrent genital herpes.28 Eighty-four immunocompetent HSV-2–infected patients with a history of ≥3 recurrences in the previous 12 months were randomized to receive either 2 days of 800 mg 3 times daily acyclovir or matching placebo. Patients were asked to take their medication no later than 12 hours after the first sign or symptom of an episode.
Efficacy endpoints were time to lesion healing, episode duration, and percentage of patients with aborted lesions. Short-course acyclovir therapy was shown to decrease time to healing (P=.001) and episode duration (P<.001) by 2 days compared with placebo (TABLE 1). Short-course acyclovir therapy also increased the percentage of patients with aborted lesions compared with placebo (27% vs 11%; P=.029 (TABLE 1). Adverse events were not recorded in this analysis.
Placebo-controlled trial of single-day famciclovir therapy. Aoki and colleagues29 performed a randomized, double-blind, patient-initiated, placebo-controlled trial to assess the efficacy and safety of patient-initiated, single-day famciclovir 1000 mg twice daily in immunocompetent adults with recurrent genital herpes. The 329 patients in the study were instructed to self-initiate therapy within 6 hours of the onset of prodromal symptoms or genital herpes lesions, and were asked to return to the clinic no later than 24 hours after initiation of therapy. Patients were followed until their lesions healed or for up to 14 days.
The primary endpoint was time to lesion healing of nonaborted lesions. Secondary endpoints were time to healing of all lesions (aborted and nonaborted), time to resolution of pain and other symptoms, and the percentage of patients who did not progress to a full outbreak.
Single-day treatment with famciclovir shortened the time to healing of nonaborted genital herpes lesions by approximately 2 days (P<.001), and the time to healing of all lesions by 1.5 days (P<.001) compared with placebo, and increased the percentage of patients who did not progress to a full outbreak (23% vs 13%;P=.003) (TABLE 1). Famciclovir also reduced the time to resolution of all symptoms by approximately 2 days (P<.001) (data not shown).
Adverse events were mild to moderate; the most common in the famciclovir and placebo groups, respectively, were headache (13.5% vs 5.4%), nausea (2.5% vs 3.6%), and diarrhea (4.9% vs 1.2%).
TABLE 1
Short-course, patient-initiated OAV therapy is effective for treating episodic genital herpes
| DRUG | TREATMENT DURATION | TREATMENT DOSE | CONTROL | MEDIAN TIME (DAYS) TO LESION HEALING (TREATMENT VS CONTROL) | MEDIAN EP ISODE DURATION (DAYS) (TREATMENT VS CONTROL) | PATIENTS WITH ABORTED EPISODES (%) (TREATMENT VS CONTROL) |
|---|---|---|---|---|---|---|
| Valacyclovir3 | 3 days | 500 mg 2×daily | valacyclovir 500 mg 2×/day for 5 days | 4.4 vs 4.7 (P=NS) | 4.3 vs 4.4 (P=NS) | 25 vs 27 (P=NS) |
| Acyclovir28 | 2 days | 800 mg 3×daily | Placebo | 4.0 vs 6.0 (P=.001) | 4.0 vs 6.0 (P=.001) | 27 vs 11 (P=.029) |
| Famciclovir29 | 1 day | 1000 mg 2×daily | Placebo | 4.3 vs 6.1 (P<.001) | 3.5 vs 5.0 (P<.001) | 23 vs 13 (P=.003) |
| Lesion healing time measures the duration of a subset of severe or classical herpetic outbreaks, characterized by the formation of vesicles, ulcers, or crusts (also papules in some studies28,29). The endpoint is lesion reepithelialization/loss of crust. Episodes where there were only prodromal symptoms, erythema, and/or papule formation (or only symptoms and/or erythema in some studies28,29) were considered “aborted” or prevented lesions. The occurrence of these favorable episode outcomes is described as a percentage of all episodes. Episode duration, sometimes called healing time of all lesions or time to return to normal skin, is the time to resolution of all episodes, regardless of lesion severity. The definition of normal skin varies among the different studies. | ||||||
| NS=not significant. | ||||||
Short-course, high-dose, patient-initiated episodic OAV therapy for recurrent herpes labialis
Placebo-controlled trial of single-day and 2-day valacyclovir therapy. Spruance and coworkers studied the efficacy of single-day and 2-day valacyclovir treatments in comparison with placebo for an episode of herpes labialis.27 Two identical studies were performed on individuals who were at least 12 years old, had a clinical history of recurrent cold sores, and had experienced ≥3 episodes in the preceding year. Participants in both studies (study 1, N=1524; study 2, N=1627) were required to self-administer 2 g valacyclovir twice daily for 1 day (valacyclovir 1 day), 2 g valacyclovir twice daily for 1 day followed by 1 g twice daily for 1 day (valacyclovir 2 days), or matching placebo at the earliest onset of prodromal symptoms and before the appearance of lesions. Patients were asked to return to the clinic within 24 hours of initiation of therapy.
The primary endpoint in study 1 was clinician-observed duration of all herpes labialis lesions and the secondary endpoint was the percentage of subjects who had herpes labialis lesions that did not progress beyond the papule stage. In study 2, the endpoints were reversed: the primary endpoint was the percentage of patients with lesions that did not progress and the secondary endpoint was the duration of lesions. Other efficacy endpoints were time to healing of vesicular (classical) lesions and duration of pain and discomfort.
Both studies demonstrated that single-day valacyclovir treatment significantly decreased lesion healing time and the duration of herpes labialis episodes by 0.5 to 1.0 days compared with placebo (TABLE 2). A statistically significant decrease in the duration of pain and other symptoms was also seen with single-day valacyclovir compared with placebo (data not shown). In both studies, a higher percentage of patients in the valacyclovir group did not progress to full outbreak compared with placebo, but these differences were not statistically significant. The results with 2 days of valacyclovir treatment were similar. Adverse events were similar between the treatment groups and the placebo group.
Placebo-controlled trial of single-dose and single-day famciclovir therapy. Spruance and coworkers assessed patient-initiated famciclovir 1500 mg (single-dose) and 750 mg twice daily (single-day) in immunocompetent adults with recurrent cold sores.30 Subjects (N=1376) were at least 18 years of age and had experienced ≥3 episodes of cold sores over the previous 12 months. Subjects were instructed to administer 1500 mg (single-dose), 750 mg twice daily (single-day), or matching placebo within 1 hour of the onset of prodromal symptoms and before the onset of lesions, and were asked to return to the clinic within 24 hours of initiating medication.
Topical antiviral drug formulations were the first treatments approved for recurrent HSV-1 and HSV-2 outbreaks, but these were only marginally efficacious.19-21,31 orally-administered antiviral agents appear to be more effective, possibly because of better delivery of the drug to the site of infection. Three oral antiviral agents (OAVs) are currently approved for the treatment of recurrent genital herpes: acyclovir, an acyclic nucleoside analog; valacyclovir, the prodrug of acyclovir; and famciclovir, the prodrug of penciclovir, another acyclic nucleoside analog. one OAV (valacyclovir) is currently approved for the treatment of herpes labialis in immunocompetent patients.27 The prodrugs of acyclovir and penciclovir, valacyclovir and famciclovir, respectively, were synthesized to provide high oral bioavailability and thus permit less frequent administration and potentially greater efficacy compared to the parent compounds.
Following oral administration, valacyclovir and famciclovir undergo first-pass metabolism to acyclovir and penciclovir, respectively.4,32 acyclovir and penciclovir are selectively phosphorylated by the viral thymidine kinase of infected cells and then converted to the active triphosphate by cellular enzymes. The triphosphate forms (which have different half-lives depending upon the compound)33 inhibit viral DNA polymerase and interfere with DNA chain extension,34 thereby halting viral DNA synthesis. The drugs cannot prevent the death of a cell once it is infected, but they can reduce, in a dose-dependent manner, the quantity of virions produced by an infected cell. The mechanism of action of HSV-selective antiviral drugs suggests that the most logical strategy for episodic treatment is to maximally inhibit HSV replication using high doses.18,35
The primary endpoint was time to healing of primary vesicular lesions. Secondary endpoints included time to healing of all vesicular lesions (primary and secondary [secondary lesions are defined as lesions that developed in addition to and on 1 or more days after primary lesions and that were located at least 1 cm from primary lesions]), time to return to normal skin for all lesions (defined as loss of crust, swelling, and dry flaking), duration of lesion tenderness and pain, and proportion of patients with aborted lesions.
There was a statistically significant decrease in time to healing of primary vesicular lesions by approximately 2 days with both single-dose and single-day famciclovir compared with placebo, with no significant difference between the 2 famciclovir regimens in time to healing of primary vesicular lesions (TABLE 2). There was also a statistically significant decrease in the time to healing of all lesions (primary and secondary) by approximately 2 days with both famciclovir treatments compared with placebo, with no significant differences seen in healing between the famciclovir arms (data not shown).
However, only single-dose famciclovir had a statistically significant decrease in the duration of lesion tenderness and pain and the time to return to normal skin compared with placebo (data not shown). No difference was noted between the famciclovir arms in the percentage of patients with aborted lesions compared with placebo. Adverse events in both famciclovir groups were similar to those in the placebo group.
TABLE 2
Short-course, patient-initiated OAV therapy is effective against recurrent herpes labialis
| DRUG | TREATMENT DURATION | TREATMENT DOSE | COMPARATOR REGIMEN | CONTROL | MEDIAN TIME (DAYS) TO LESION HEALING (TREATMENT VS COMPARATOR VS CONTROL)* | MEDIAN EP ISODE DURATION (DAYS) (TREATMENT VS COMPARATOR VS CONTROL)* | PATIENTS WITH ABORTED LESIONS (%)(TREATMENT VS COMPARATOR VS CONTROL)† |
|---|---|---|---|---|---|---|---|
| Valacyclovir27 | 1 day | 2000 mg 2×daily | valacyclovir 2000 mg 2×daily×1 day 1000 mg 2×daily for a 2nd day | Placebo | study 1 4.3 vs 4.3 vs 5.1 study 2 4.8 vs 4.6 vs 5.4 | study 1 4.0 vs 4.5 vs 5.0 study 2 2 5.0 vs 5.0 vs 5.5 | study 1 44 vs 46 vs 38 study 2 43 vs 43 vs 35 |
| Famciclovir30 1 dose | 1 Does | 1500 mg | famciclovir 750 mg 2x daily for 1 day | Placebo | 4.4 vs 4.0 vs 6.2 | 4.5 vs 5.7 vs 7.0 | 33 vs 29 vs 34 |
| Lesion healing time measures the duration of a subset of severe or classical herpetic outbreaks, characterized by the formation of vesicles, ulcers, or crusts (also papules in some studies28,29). The endpoint is lesion reepithelialization/loss of crust. Episodes where there were only prodromal symptoms, erythema, and/or papule formation (or only symptoms and/or erythema in some studies28,29) were considered “aborted” or prevented lesions. The occurrence of these favorable episode outcomes is described as a percentage of all episodes. Episode duration, sometimes called healing time of all lesions or time to return to normal skin, is the time to resolution of all episodes, regardless of lesion severity. The definition of normal skin varies among the different studies. | |||||||
| *All of the healing time and episode duration values for the active treatment arms in both studies differed statistically significantly from placebo, except for famciclovir 750 mg twice daily for 1 day. | |||||||
| †None of the frequencies of aborted lesions in the active treatment arms in either study differed statistically significantly from placebo. | |||||||
CORRESPONDENCE
Spotswood Spruance MD Professor of Medicine, Division of Infectious Diseases, University of Utah School of Medicine, Room 4B319, 30 North 1900 East, Salt Lake City, UT 84132-2405. E-mail:woody.spruance@hsc.utah.edu
- Consider giving patients an oral antiviral (OAV) medication to self-administer when HSV prodromal symptoms occur.
- Patient-initiated, short-course, high-dose OAV treatment of recurrent HSV outbreaks may be as effective as the traditional, longer-course regimens.
Hit early, hit hard. That expression arose during the evolution of treatment for human immunodeficiency virus (HIV).1 While this approach has not lived up to expectations for HIV treatment, it may have found its place in the treatment of recurrent herpes simplex virus (HSV) infections.
Our review focuses on episodic treatment of acute recurrent HSV outbreaks for immunocompetent persons. We do not discuss suppressive therapy, which may be indicated for frequent or severe recurrences (6 or more per year) in immunocompetent persons, for immunocompromised patients, or as an adjunctive measure to reduce genital herpes transmission.2
As we will describe in detail, the efficacy of the new short-course therapy is, at minimum, comparable to that seen with the older, longer-course trials of topical and oral antiviral therapy. In one head-to-head comparison, Leone et al compared a short-course regimen (3 days) of valacyclovir with 5 days of treatment; they found no difference in results.3 If the efficacy of short-course treatment is the same as that of longer courses, the increased convenience and expected improvement in patient adherence with these new regimens argue strongly in their favor. (See Scope of the problem.)
The strategy: Take advantage of a brief therapeutic window
The innate and acquired immune responses of chronically infected, immunocompetent persons rapidly limit cutaneous viral replication, thereby truncating the duration of recurrent HSV outbreaks.13,14 In both recurrent herpes labialis and genital herpes, HSV viral titers peak in the first 24 hours following lesion onset (FIGURE 1A).13-15
Herpes labialis lesion size and pain are also greatest in the first 24 hours.13,16 Most herpes labialis lesions progress from the vesicle stage to the ulcer/soft crust stage within 48 hours, with a hard crust forming by day 2 or 3 (FIGURE 1B).17
With genital lesions, crust formation depends on whether the skin area is dry (3–4 days) or moist (8–9 days).14
The likely events are a burst of virus replication in the first 24 hours of outbreak that lyses basal keratinocytes in a discreet area of epidermis innervated by the infected neuron(s), followed by a vigorous immune response that curtails the infection and creates, in part, the clinical disease (erythema, swelling, vesiculation, and ulceration). The subsequent elements of the illness, which are the majority of the lesion course, are related to wound healing
Recognizing the window. Given the brief period of viral replication and the rapid evolution of lesions, the therapeutic window for treating HSV outbreaks with antiviral drugs is both early and short, making it problematic to effectively treat HSV recurrences. Patients often have mature lesions by the time they consult a physician, rendering subsequent anti-viral treatment less effective.18 However, before lesions appear, many patients experience prodromal symptoms such as pain, burning, or itching.13,18 These symptoms can be a prompt to start treatment early, thereby taking advantage of the transient therapeutic window.
If a patient is able to self-administer therapy when prodromal symptoms occur, there may be a greater benefit to treatment. Giving patients drugs for self-administration is therefore an important strategy in managing HSV recurrences.
Traditionally, patient-initiated episodic therapy for recurrent genital herpes and herpes labialis has involved multiple daily doses of topical or oral antiviral agents for 4 to 5 days.19-26 Studies of the pathogenesis of HSV recurrences, however, indicate—as said earlier—that the period of virus replication is early and brief, such that a shorter duration of treatment might be more appropriate and equally effective. Other recent clinical studies have indicated that patient-initiated, short-course, high-dose OAV treatment of recurrent HSV infections may be as effective as the traditional therapies.3,27-30 In the section that follows, we examine and compare the results of these trials. (See The agents and how they work.)
FIGURE 1A
Lesion HSV-1 titer peaks within 24 hours of onset of herpes labialis lesions
Source: Krueger et al, J Clin Epidemiol Derm 1978.15 Reproduced with permission from GG Krueger.
FIGURE 1B
Hard crust formation occurs by 48 to 72 hours of onset of herpes labialis lesions
Source: Spruance, Sem Dermatol 1992.17 With permission from Elsevier.
Clinical trials: Short-course, high-dose, patient-initiated episodic OAV therapy for recurrent genital herpes
Three-day vs 5-day valacyclovir therapy. The efficacy of 3-day treatment with oral valacyclovir was compared with that of 5-day treatment in immunocompetent adults with a history of ≥4 episodes of recurrent genital herpes and confirmed HSV infection.3 Eight hundred participants were randomized to receive 500 mg twice daily valacyclovir for 3 days (and placebo for the remaining 2 days) or 500 mg twice daily for 5 days, and were required to self-administer therapy no later than 24 hours after the onset of symptoms.
Herpes simplex virus (HSV) type 1 (HSV-1) or type 2 (HSV-2) results in periodic, recurrent outbreaks of skin lesions after first infection. Herpes labialis (fever blisters or cold sores) is usually caused by HSV-1, while genital herpes is usually caused by HSV-2.4 HSV-2 lesions of the lips have been reported, and the incidence of genital herpes caused by HSV-1 is on the rise in the developed world, likely because of increased oral-genital sexual behavior.5,6 Patients with HSV-1 genital herpes typically have fewer recurrences than those with HSV-2 genital infection.7
The prevalence of HSV-1 and HSV-2 infection varies according to age, geography, gender, and population subgroup, such as people who exhibit high-risk sexual behavior.8 approximately 45% of americans are infected with HSV-1 by adolescence,8 and approximately 22% of all american adults are infected with HSV-2.9 The global prevalence of HSV is even greater: as many as 60% to 90% of older adults worldwide are seropositive for HSV-1, and as many as 30% are seropositive for HSV-2. HSV-2 seropositivity is more prevalent among women than men.8 overall, the burden of recurrent genital herpes outbreaks can have a profound, negative impact on patient quality of life.10,11 The psychological impact of recurrent herpes labialis has not been thoroughly investigated, but an undefined burden is thought to exist, particularly in young patients with frequent or severe recurrences.12
The primary endpoint was time to lesion healing (defined as the number of days from initiation of therapy to lesion reepithelialization). Secondary endpoints were pain duration, episode duration (defined as time from initiation of therapy to resolution of all symptoms) and percentage of patients with aborted lesions.
The 3-day valacyclovir treatment exhibited similar time to lesion healing, length of episode, and percentage of patients with aborted lesions as the 5-day treatment (TABLE 1), suggesting equal efficacy. Duration of pain was also similar (data not shown). Adverse events were similar for both treatment groups, with the most common being headache (10%), nausea (4%), and diarrhea (4%, 5-day treatment vs 2%, 3-day treatment).
Placebo-controlled trial of 2-day acyclovir therapy. Wald and coworkers examined the effect of a shorter treatment regimen of acyclovir (2 days) on recurrent genital herpes.28 Eighty-four immunocompetent HSV-2–infected patients with a history of ≥3 recurrences in the previous 12 months were randomized to receive either 2 days of 800 mg 3 times daily acyclovir or matching placebo. Patients were asked to take their medication no later than 12 hours after the first sign or symptom of an episode.
Efficacy endpoints were time to lesion healing, episode duration, and percentage of patients with aborted lesions. Short-course acyclovir therapy was shown to decrease time to healing (P=.001) and episode duration (P<.001) by 2 days compared with placebo (TABLE 1). Short-course acyclovir therapy also increased the percentage of patients with aborted lesions compared with placebo (27% vs 11%; P=.029 (TABLE 1). Adverse events were not recorded in this analysis.
Placebo-controlled trial of single-day famciclovir therapy. Aoki and colleagues29 performed a randomized, double-blind, patient-initiated, placebo-controlled trial to assess the efficacy and safety of patient-initiated, single-day famciclovir 1000 mg twice daily in immunocompetent adults with recurrent genital herpes. The 329 patients in the study were instructed to self-initiate therapy within 6 hours of the onset of prodromal symptoms or genital herpes lesions, and were asked to return to the clinic no later than 24 hours after initiation of therapy. Patients were followed until their lesions healed or for up to 14 days.
The primary endpoint was time to lesion healing of nonaborted lesions. Secondary endpoints were time to healing of all lesions (aborted and nonaborted), time to resolution of pain and other symptoms, and the percentage of patients who did not progress to a full outbreak.
Single-day treatment with famciclovir shortened the time to healing of nonaborted genital herpes lesions by approximately 2 days (P<.001), and the time to healing of all lesions by 1.5 days (P<.001) compared with placebo, and increased the percentage of patients who did not progress to a full outbreak (23% vs 13%;P=.003) (TABLE 1). Famciclovir also reduced the time to resolution of all symptoms by approximately 2 days (P<.001) (data not shown).
Adverse events were mild to moderate; the most common in the famciclovir and placebo groups, respectively, were headache (13.5% vs 5.4%), nausea (2.5% vs 3.6%), and diarrhea (4.9% vs 1.2%).
TABLE 1
Short-course, patient-initiated OAV therapy is effective for treating episodic genital herpes
| DRUG | TREATMENT DURATION | TREATMENT DOSE | CONTROL | MEDIAN TIME (DAYS) TO LESION HEALING (TREATMENT VS CONTROL) | MEDIAN EP ISODE DURATION (DAYS) (TREATMENT VS CONTROL) | PATIENTS WITH ABORTED EPISODES (%) (TREATMENT VS CONTROL) |
|---|---|---|---|---|---|---|
| Valacyclovir3 | 3 days | 500 mg 2×daily | valacyclovir 500 mg 2×/day for 5 days | 4.4 vs 4.7 (P=NS) | 4.3 vs 4.4 (P=NS) | 25 vs 27 (P=NS) |
| Acyclovir28 | 2 days | 800 mg 3×daily | Placebo | 4.0 vs 6.0 (P=.001) | 4.0 vs 6.0 (P=.001) | 27 vs 11 (P=.029) |
| Famciclovir29 | 1 day | 1000 mg 2×daily | Placebo | 4.3 vs 6.1 (P<.001) | 3.5 vs 5.0 (P<.001) | 23 vs 13 (P=.003) |
| Lesion healing time measures the duration of a subset of severe or classical herpetic outbreaks, characterized by the formation of vesicles, ulcers, or crusts (also papules in some studies28,29). The endpoint is lesion reepithelialization/loss of crust. Episodes where there were only prodromal symptoms, erythema, and/or papule formation (or only symptoms and/or erythema in some studies28,29) were considered “aborted” or prevented lesions. The occurrence of these favorable episode outcomes is described as a percentage of all episodes. Episode duration, sometimes called healing time of all lesions or time to return to normal skin, is the time to resolution of all episodes, regardless of lesion severity. The definition of normal skin varies among the different studies. | ||||||
| NS=not significant. | ||||||
Short-course, high-dose, patient-initiated episodic OAV therapy for recurrent herpes labialis
Placebo-controlled trial of single-day and 2-day valacyclovir therapy. Spruance and coworkers studied the efficacy of single-day and 2-day valacyclovir treatments in comparison with placebo for an episode of herpes labialis.27 Two identical studies were performed on individuals who were at least 12 years old, had a clinical history of recurrent cold sores, and had experienced ≥3 episodes in the preceding year. Participants in both studies (study 1, N=1524; study 2, N=1627) were required to self-administer 2 g valacyclovir twice daily for 1 day (valacyclovir 1 day), 2 g valacyclovir twice daily for 1 day followed by 1 g twice daily for 1 day (valacyclovir 2 days), or matching placebo at the earliest onset of prodromal symptoms and before the appearance of lesions. Patients were asked to return to the clinic within 24 hours of initiation of therapy.
The primary endpoint in study 1 was clinician-observed duration of all herpes labialis lesions and the secondary endpoint was the percentage of subjects who had herpes labialis lesions that did not progress beyond the papule stage. In study 2, the endpoints were reversed: the primary endpoint was the percentage of patients with lesions that did not progress and the secondary endpoint was the duration of lesions. Other efficacy endpoints were time to healing of vesicular (classical) lesions and duration of pain and discomfort.
Both studies demonstrated that single-day valacyclovir treatment significantly decreased lesion healing time and the duration of herpes labialis episodes by 0.5 to 1.0 days compared with placebo (TABLE 2). A statistically significant decrease in the duration of pain and other symptoms was also seen with single-day valacyclovir compared with placebo (data not shown). In both studies, a higher percentage of patients in the valacyclovir group did not progress to full outbreak compared with placebo, but these differences were not statistically significant. The results with 2 days of valacyclovir treatment were similar. Adverse events were similar between the treatment groups and the placebo group.
Placebo-controlled trial of single-dose and single-day famciclovir therapy. Spruance and coworkers assessed patient-initiated famciclovir 1500 mg (single-dose) and 750 mg twice daily (single-day) in immunocompetent adults with recurrent cold sores.30 Subjects (N=1376) were at least 18 years of age and had experienced ≥3 episodes of cold sores over the previous 12 months. Subjects were instructed to administer 1500 mg (single-dose), 750 mg twice daily (single-day), or matching placebo within 1 hour of the onset of prodromal symptoms and before the onset of lesions, and were asked to return to the clinic within 24 hours of initiating medication.
Topical antiviral drug formulations were the first treatments approved for recurrent HSV-1 and HSV-2 outbreaks, but these were only marginally efficacious.19-21,31 orally-administered antiviral agents appear to be more effective, possibly because of better delivery of the drug to the site of infection. Three oral antiviral agents (OAVs) are currently approved for the treatment of recurrent genital herpes: acyclovir, an acyclic nucleoside analog; valacyclovir, the prodrug of acyclovir; and famciclovir, the prodrug of penciclovir, another acyclic nucleoside analog. one OAV (valacyclovir) is currently approved for the treatment of herpes labialis in immunocompetent patients.27 The prodrugs of acyclovir and penciclovir, valacyclovir and famciclovir, respectively, were synthesized to provide high oral bioavailability and thus permit less frequent administration and potentially greater efficacy compared to the parent compounds.
Following oral administration, valacyclovir and famciclovir undergo first-pass metabolism to acyclovir and penciclovir, respectively.4,32 acyclovir and penciclovir are selectively phosphorylated by the viral thymidine kinase of infected cells and then converted to the active triphosphate by cellular enzymes. The triphosphate forms (which have different half-lives depending upon the compound)33 inhibit viral DNA polymerase and interfere with DNA chain extension,34 thereby halting viral DNA synthesis. The drugs cannot prevent the death of a cell once it is infected, but they can reduce, in a dose-dependent manner, the quantity of virions produced by an infected cell. The mechanism of action of HSV-selective antiviral drugs suggests that the most logical strategy for episodic treatment is to maximally inhibit HSV replication using high doses.18,35
The primary endpoint was time to healing of primary vesicular lesions. Secondary endpoints included time to healing of all vesicular lesions (primary and secondary [secondary lesions are defined as lesions that developed in addition to and on 1 or more days after primary lesions and that were located at least 1 cm from primary lesions]), time to return to normal skin for all lesions (defined as loss of crust, swelling, and dry flaking), duration of lesion tenderness and pain, and proportion of patients with aborted lesions.
There was a statistically significant decrease in time to healing of primary vesicular lesions by approximately 2 days with both single-dose and single-day famciclovir compared with placebo, with no significant difference between the 2 famciclovir regimens in time to healing of primary vesicular lesions (TABLE 2). There was also a statistically significant decrease in the time to healing of all lesions (primary and secondary) by approximately 2 days with both famciclovir treatments compared with placebo, with no significant differences seen in healing between the famciclovir arms (data not shown).
However, only single-dose famciclovir had a statistically significant decrease in the duration of lesion tenderness and pain and the time to return to normal skin compared with placebo (data not shown). No difference was noted between the famciclovir arms in the percentage of patients with aborted lesions compared with placebo. Adverse events in both famciclovir groups were similar to those in the placebo group.
TABLE 2
Short-course, patient-initiated OAV therapy is effective against recurrent herpes labialis
| DRUG | TREATMENT DURATION | TREATMENT DOSE | COMPARATOR REGIMEN | CONTROL | MEDIAN TIME (DAYS) TO LESION HEALING (TREATMENT VS COMPARATOR VS CONTROL)* | MEDIAN EP ISODE DURATION (DAYS) (TREATMENT VS COMPARATOR VS CONTROL)* | PATIENTS WITH ABORTED LESIONS (%)(TREATMENT VS COMPARATOR VS CONTROL)† |
|---|---|---|---|---|---|---|---|
| Valacyclovir27 | 1 day | 2000 mg 2×daily | valacyclovir 2000 mg 2×daily×1 day 1000 mg 2×daily for a 2nd day | Placebo | study 1 4.3 vs 4.3 vs 5.1 study 2 4.8 vs 4.6 vs 5.4 | study 1 4.0 vs 4.5 vs 5.0 study 2 2 5.0 vs 5.0 vs 5.5 | study 1 44 vs 46 vs 38 study 2 43 vs 43 vs 35 |
| Famciclovir30 1 dose | 1 Does | 1500 mg | famciclovir 750 mg 2x daily for 1 day | Placebo | 4.4 vs 4.0 vs 6.2 | 4.5 vs 5.7 vs 7.0 | 33 vs 29 vs 34 |
| Lesion healing time measures the duration of a subset of severe or classical herpetic outbreaks, characterized by the formation of vesicles, ulcers, or crusts (also papules in some studies28,29). The endpoint is lesion reepithelialization/loss of crust. Episodes where there were only prodromal symptoms, erythema, and/or papule formation (or only symptoms and/or erythema in some studies28,29) were considered “aborted” or prevented lesions. The occurrence of these favorable episode outcomes is described as a percentage of all episodes. Episode duration, sometimes called healing time of all lesions or time to return to normal skin, is the time to resolution of all episodes, regardless of lesion severity. The definition of normal skin varies among the different studies. | |||||||
| *All of the healing time and episode duration values for the active treatment arms in both studies differed statistically significantly from placebo, except for famciclovir 750 mg twice daily for 1 day. | |||||||
| †None of the frequencies of aborted lesions in the active treatment arms in either study differed statistically significantly from placebo. | |||||||
CORRESPONDENCE
Spotswood Spruance MD Professor of Medicine, Division of Infectious Diseases, University of Utah School of Medicine, Room 4B319, 30 North 1900 East, Salt Lake City, UT 84132-2405. E-mail:woody.spruance@hsc.utah.edu
1. Ho D. Time to hit HIV, early and hard. N Engl J Med 1995;333:450-451.
2. Corey L, Wald A, Patel R, et al. Once-daily valacyclovir to reduce the risk of transmission of genital herpes. N Engl J Med 2004;350:11-20.
3. Leone PA, Trottier S, Miller JM. Valacyclovir for episodic treatment of genital herpes: a shorter 3-day treatment course compared with 5-day treatment. Clin Infect Dis 2002;34:958-962.
4. Whitley RJ, Kimberlin DW, Roizman B. Herpes simplex viruses. Clin Infect Dis 1998;26:541-555.
5. Wald A, Ericsson M, Krantz E, Selkes S, Corey L. Oral shedding of herpes simplex virus type 2. Sex Transm Infect 2004;80:272-276.
6. Mertz GJ, Rosenthal Sl, Stanberry LR. Editorial response: Is herpes simplex virus type 1 (HSV-1) now more common than HSV-2 in first episodes of genital herpes? Sex Transm Dis 2003;30:801-802.
7. Lafferty WE, Coombs RW, Benedetti J, Critchlow C, Corey L. Recurrences after oral and genital herpes simplex virus infection. N Engl J Med 1987;316:1444-1449.
8. Smith JS, Robinson RJ. Age-specific prevalence of infection with herpes simplex virus types 2 and 1: a global review. J Infect Dis 2002;186(suppl 1):S3-S28.
9. Fleming DT, McQuillan GM, Johnson RE, et al. Herpes simplex virus type 2 in the united States, 1976 to 1994. N Engl J Med 1997;337:1105-1111.
10. Bierman SM. A retrospective study of 375 patients with genital herpes simplex infections seen between 1973 and 1980. Cutis 1983;31:548-565.
11. Drob S, Loemer M, Lifshutz H. Genital herpes: the psychological consequences. Br J Med Psychol 1985;58:307-315.
12. Spruance SL, Kriesel JD. Treatment of herpes simplex labialis. Herpes 2002;9:64-69.
13. Spruance SL, overall JC, Jr, Kern ER, Krueger GG, Pliam V, Miller W. The natural history of recurrent herpes simplex labialis. N Engl J Med 1977;297:69-75.
14. Brown ZA, Kern ER, Spruance SL, Overall JC, Jr. Clinical and virologic course of herpes simplex genitalis. West J Med 1979;130:414-421.
15. Krueger GG, Spruance SL, Overall JC, Jr. Herpes simplex labialis: a review of pathogenesis and therapy. J Clin Epidemiol Derm 1978;1:19-37.
16. Spruance SL, Wenerstrom G. Pathogenesis of herpes simplex labialis: IV. Maturation of lesions during within 8 hours after onset and implications for antiviral treatment. Oral Surg Oral Med Oral Path 1984;58:667-671.
17. Spruance SL. The natural history of recurrent oral-facial herpes simplex virus infection. Sem Dermatol 1992;11:200-206.
18. Spruance SL. Herpes simplex labialis. In: Clinical Management of Herpes Viruses. Sacks SL, Straus SE, Whitley RJ, Griffiths PD, eds. amsterdam, Netherlands: IOS Press; 1995.
19. Spruance SL, Nett R, Marbury T, Wolff R, Johnson J, Spaulding T. Acyclovir cream for treatment of herpes simplex labialis: results of two randomized, double-blind, vehicle-controlled, multicenter clinical trials. Antimicrob Agents Chemother 2002;46:2238-2243.
20. Spruance SL, Rea TL, Thoming C, Tucker R, Saltzman R, Boon R. Penciclovir cream for the treatment of herpes simplex labialis. JAMA 1997;277:1374-1379.
21. Raborn GW, Martel AY, lassonde M, et al. Worldwide Topical Penciclovir Collaborative Study Group. Effective treatment of herpes simplex labialis with penciclovir cream: combined results of two trials. J Am Dent Assoc 2002;133:303-309.
22. Reichman RC, Badger GJ, Mertz GJ, et al. Treatment of recurrent genital herpes simplex infections with oral acyclovir: a controlled trial. JAMA 1984;251:2103-2107.
23. Sacks SL, Aoki FY, Diaz-Mitoma F, Sellors J, Shafran SD. Canadian Famciclovir Study Group. Patient-initiated, twice-daily oral famciclovir for early recurrent genital herpes: a randomized, double-blind multicenter trial. JAMA 1996;276:44-49.
24. Tyring SK, Douglas JM, Jr, Corey L, Spruance SL, Esmann J. The valaciclovir International Study Group. A randomized, placebo-controlled comparison of oral valcyclovir and acyclovir in immunocompetent patients with recurrent genital herpes infections. Arch Dermatol 1998;134:185-191.
25. Spruance S, Stewart JCB, Rowe NH, McKeough MB, Wenerstrom G, Freeman DJ. Treatment of herpes simplex labialis with oral acyclovir. J Infect Dis 1990;161:185-190.
26. Spruance SL, Tyring SK, DeGregorio B, Miller C, Beutner K; valaciclovir HSV Study Group. A large-scale, placebo-controlled, dose-ranging trial of peroral valaciclovir for episodic treatment of recurrent herpes genitalis. Arch Intern Med 1996;156:1729-1735.
27. Spruance SL, Jones TM, Blatter MM, et al. High-dose, short-duration, early valacyclovir therapy for episodic treatment of cold sores: results of two randomized, placebo-controlled, multicenter studies. Antimicrob Agents Chemother 2003;47:1072-1080.
28. Wald A, Carrell D, Remington M, Kexel E, Zeh J, Corey L. Two-day regimen of acyclovir for treatment of recurrent genital herpes simplex virus type 2 infection. Clin Infect Dis 2002;34:944-948.
29. Aoki FY, Tyring S, Dias-Mitoma F, Gross G, Gao J, Hamed K. Single-day patient initiated famciclovir therapy for recurrent genital herpes: a randomized double-blind, placebo-controlled trial. Clin Infect Dis 2006;42:8-13.
30. Spruance S, Bodsworth N, Resnick H, et al. Single-dose, patient-initiated famciclovir: a randomized, double-blind, placebo-controlled trial for episodic treatment of herpes labialis. J Am Acad Dermatol 2006;55:47-53.
31. Reichman RC, Badger GJ, Guinan ME, et al. Topically administered acyclovir in the treatment of recurrent herpes simplex genitalis: a controlled trial. J Infect Dis 1983;147:336-340.
32. Gill KS, Wood MJ. The clinical pharmacokinetics of famciclovir. Clin Pharmacokinet 1996;31:1-8.
33. Earnshaw DL, Bacon TH, Darlison SJ, Edmonds K, Perkins RM, Vere Hodge RA. Mode of antiviral action of penciclovir in MRC-5 cells infected with herpes simplex virus type 1 (HSV-1), HSV-2, and varicella-zoster virus. Antimicrob Agents Chemother 1992;36:2747-2757.
34. Vere Hodge RA, Perkins RM. Mode of action of 9-(4-hydroxy-3-hydroxymethylbut-1-yl)guanine (Brl 39123) against herpes simplex virus in MrC-5 cells. Antimicrob Agents Chemother 1989;33:223-229.
35. Spruance SL, Freeman DJ. Topical treatment of cutaneous herpes simplex virus infections. Antivir Res 1990;14:305-321.
1. Ho D. Time to hit HIV, early and hard. N Engl J Med 1995;333:450-451.
2. Corey L, Wald A, Patel R, et al. Once-daily valacyclovir to reduce the risk of transmission of genital herpes. N Engl J Med 2004;350:11-20.
3. Leone PA, Trottier S, Miller JM. Valacyclovir for episodic treatment of genital herpes: a shorter 3-day treatment course compared with 5-day treatment. Clin Infect Dis 2002;34:958-962.
4. Whitley RJ, Kimberlin DW, Roizman B. Herpes simplex viruses. Clin Infect Dis 1998;26:541-555.
5. Wald A, Ericsson M, Krantz E, Selkes S, Corey L. Oral shedding of herpes simplex virus type 2. Sex Transm Infect 2004;80:272-276.
6. Mertz GJ, Rosenthal Sl, Stanberry LR. Editorial response: Is herpes simplex virus type 1 (HSV-1) now more common than HSV-2 in first episodes of genital herpes? Sex Transm Dis 2003;30:801-802.
7. Lafferty WE, Coombs RW, Benedetti J, Critchlow C, Corey L. Recurrences after oral and genital herpes simplex virus infection. N Engl J Med 1987;316:1444-1449.
8. Smith JS, Robinson RJ. Age-specific prevalence of infection with herpes simplex virus types 2 and 1: a global review. J Infect Dis 2002;186(suppl 1):S3-S28.
9. Fleming DT, McQuillan GM, Johnson RE, et al. Herpes simplex virus type 2 in the united States, 1976 to 1994. N Engl J Med 1997;337:1105-1111.
10. Bierman SM. A retrospective study of 375 patients with genital herpes simplex infections seen between 1973 and 1980. Cutis 1983;31:548-565.
11. Drob S, Loemer M, Lifshutz H. Genital herpes: the psychological consequences. Br J Med Psychol 1985;58:307-315.
12. Spruance SL, Kriesel JD. Treatment of herpes simplex labialis. Herpes 2002;9:64-69.
13. Spruance SL, overall JC, Jr, Kern ER, Krueger GG, Pliam V, Miller W. The natural history of recurrent herpes simplex labialis. N Engl J Med 1977;297:69-75.
14. Brown ZA, Kern ER, Spruance SL, Overall JC, Jr. Clinical and virologic course of herpes simplex genitalis. West J Med 1979;130:414-421.
15. Krueger GG, Spruance SL, Overall JC, Jr. Herpes simplex labialis: a review of pathogenesis and therapy. J Clin Epidemiol Derm 1978;1:19-37.
16. Spruance SL, Wenerstrom G. Pathogenesis of herpes simplex labialis: IV. Maturation of lesions during within 8 hours after onset and implications for antiviral treatment. Oral Surg Oral Med Oral Path 1984;58:667-671.
17. Spruance SL. The natural history of recurrent oral-facial herpes simplex virus infection. Sem Dermatol 1992;11:200-206.
18. Spruance SL. Herpes simplex labialis. In: Clinical Management of Herpes Viruses. Sacks SL, Straus SE, Whitley RJ, Griffiths PD, eds. amsterdam, Netherlands: IOS Press; 1995.
19. Spruance SL, Nett R, Marbury T, Wolff R, Johnson J, Spaulding T. Acyclovir cream for treatment of herpes simplex labialis: results of two randomized, double-blind, vehicle-controlled, multicenter clinical trials. Antimicrob Agents Chemother 2002;46:2238-2243.
20. Spruance SL, Rea TL, Thoming C, Tucker R, Saltzman R, Boon R. Penciclovir cream for the treatment of herpes simplex labialis. JAMA 1997;277:1374-1379.
21. Raborn GW, Martel AY, lassonde M, et al. Worldwide Topical Penciclovir Collaborative Study Group. Effective treatment of herpes simplex labialis with penciclovir cream: combined results of two trials. J Am Dent Assoc 2002;133:303-309.
22. Reichman RC, Badger GJ, Mertz GJ, et al. Treatment of recurrent genital herpes simplex infections with oral acyclovir: a controlled trial. JAMA 1984;251:2103-2107.
23. Sacks SL, Aoki FY, Diaz-Mitoma F, Sellors J, Shafran SD. Canadian Famciclovir Study Group. Patient-initiated, twice-daily oral famciclovir for early recurrent genital herpes: a randomized, double-blind multicenter trial. JAMA 1996;276:44-49.
24. Tyring SK, Douglas JM, Jr, Corey L, Spruance SL, Esmann J. The valaciclovir International Study Group. A randomized, placebo-controlled comparison of oral valcyclovir and acyclovir in immunocompetent patients with recurrent genital herpes infections. Arch Dermatol 1998;134:185-191.
25. Spruance S, Stewart JCB, Rowe NH, McKeough MB, Wenerstrom G, Freeman DJ. Treatment of herpes simplex labialis with oral acyclovir. J Infect Dis 1990;161:185-190.
26. Spruance SL, Tyring SK, DeGregorio B, Miller C, Beutner K; valaciclovir HSV Study Group. A large-scale, placebo-controlled, dose-ranging trial of peroral valaciclovir for episodic treatment of recurrent herpes genitalis. Arch Intern Med 1996;156:1729-1735.
27. Spruance SL, Jones TM, Blatter MM, et al. High-dose, short-duration, early valacyclovir therapy for episodic treatment of cold sores: results of two randomized, placebo-controlled, multicenter studies. Antimicrob Agents Chemother 2003;47:1072-1080.
28. Wald A, Carrell D, Remington M, Kexel E, Zeh J, Corey L. Two-day regimen of acyclovir for treatment of recurrent genital herpes simplex virus type 2 infection. Clin Infect Dis 2002;34:944-948.
29. Aoki FY, Tyring S, Dias-Mitoma F, Gross G, Gao J, Hamed K. Single-day patient initiated famciclovir therapy for recurrent genital herpes: a randomized double-blind, placebo-controlled trial. Clin Infect Dis 2006;42:8-13.
30. Spruance S, Bodsworth N, Resnick H, et al. Single-dose, patient-initiated famciclovir: a randomized, double-blind, placebo-controlled trial for episodic treatment of herpes labialis. J Am Acad Dermatol 2006;55:47-53.
31. Reichman RC, Badger GJ, Guinan ME, et al. Topically administered acyclovir in the treatment of recurrent herpes simplex genitalis: a controlled trial. J Infect Dis 1983;147:336-340.
32. Gill KS, Wood MJ. The clinical pharmacokinetics of famciclovir. Clin Pharmacokinet 1996;31:1-8.
33. Earnshaw DL, Bacon TH, Darlison SJ, Edmonds K, Perkins RM, Vere Hodge RA. Mode of antiviral action of penciclovir in MRC-5 cells infected with herpes simplex virus type 1 (HSV-1), HSV-2, and varicella-zoster virus. Antimicrob Agents Chemother 1992;36:2747-2757.
34. Vere Hodge RA, Perkins RM. Mode of action of 9-(4-hydroxy-3-hydroxymethylbut-1-yl)guanine (Brl 39123) against herpes simplex virus in MrC-5 cells. Antimicrob Agents Chemother 1989;33:223-229.
35. Spruance SL, Freeman DJ. Topical treatment of cutaneous herpes simplex virus infections. Antivir Res 1990;14:305-321.