User login
Of Mice and Men
Fever in an elderly man is a nonspecific finding, occurring most commonly with infections but also with certain malignancies, rheumatologic disorders, and drug exposures. The complaint of rigors with diaphoresis makes an infection most likely. The acuity of his illness makes infections with more chronic presentations such as tuberculosis or actinomycosis less likely. The presence of frontal headache might suggest a sinus or brain source, but headache also occurs in generalized infections such as pneumonia, bacteremia from any cause, malaria, rickettsial infections, viral illnesses, and others. Additional history should include detailed inquiry into travel, vocational, and avocational exposures.
The patient's difficulty standing implies the development of lower extremity weakness and infections associated with neurological syndromes. His leg weakness may be related to early Guillain‐Barre syndrome, which is associated most commonly with Campylobacter jejuni, but also other bacteria and viruses such as Haemophilus influenza, Mycoplasma pneumonia, Influenza virus, Cytomegalovirus and hepatitis E. Other viral infections associated with pure motor deficits include echovirus, coxsackie virus, enterovirus, and West Nile virus (WNV). The paralytic syndrome associated with enteroviruses is more common in children, whereas the neuroinvasive variant of WNV more often affects the elderly and can be associated with encephalitis as well as a flaccid paralysis. Although acute paralytic shellfish poisoning could account for both his weakness and his acute gastrointestinal syndrome, this diagnosis is unlikely because the symptoms often have a prominent sensory component, and there is usually the history of recent ingestion of the suspect bivalves. Like all adults presenting for medical care, he should be screened for human immunodeficiency virus (HIV) infection; if testing is positive, the differential diagnosis for his current illness broadens significantly. Finally, he may have a spinal cord disorder or infection such as an epidural abscess, or transverse myelitis, which would present with lower extremity weakness and fever. It would be helpful to know the time of year of his illness, exposure to mosquito bites, his neurological exam findings, and results of blood and stool cultures. If the patient had signs of meningitis or encephalitis, cerebral spinal fluid analysis would be helpful. If his neurological exam was suggestive of cord involvement, it would be helpful to know the results of magnetic resonance imaging of the spinal cord.
The patient's past medical history includes relatively common problems for a 73‐year‐old man and does not substantially influence the differential diagnosis of his current illness. His travel history to Uganda a year previously may be relevant, because malaria (Plasmodium vivax) could present with fever and weakness. Less commonly, African trypanosomiasis (Trypanosoma brucei gambiense) can, in the late phase, present with fever and malaise, but also typically includes symptoms of encephalitis, including depressed mental status, confusion, ataxia, and possibly personality changes. His travel to Zurich should not impose any particular infection risk, unless he was hiking in the mountains around Zurich, where he could have contracted tick‐borne encephalitis; however, his travel more than 6 months prior to presentation makes this unlikely. Lyme disease due to Borrelia burgdorferi is also a potential exposure in the Swiss mountains, and can present with fever in the acute phase, as well as arthritis with chronic disease, but should not cause fever, rigor, diaphoresis, and headache many months later. Summering in Cape Cod puts him at risk for babesiosis, but an incubation period of 5 months is too long. Keeping chickens places him at risk for Salmonella exposure and typhoid fever. Ingesting raw oysters carries a risk for shellfish poisoning and Vibrio infections, but the incubation period (1 month) again seems too long to cause his current symptoms.
Notable physical findings are an ill‐appearing man with injected sclera and a high fever but normal blood pressure and heart rate. He also demonstrates proximal lower extremity weakness manifested by difficulty rising from a chair and a slow gait with short strides and deliberate (possibly on‐block) turning. His neurological exam is most consistent with Parkinsonian symptoms that have been described in patients with severe influenza A, which would explain all of his other symptoms as well. Pulse‐temperature dissociation is classically described with typhoid fever but usually occurs later in the disease course, and could be masked by the patient's metoprolol. Typhoid fever can also be associated with neurological symptoms including meningitis and movement disorders.
The patient has a remarkable bandemia, suggesting a bacterial infection, as well as a slight reduction in hematocrit and platelet count. Additionally, his labs revealed a mild transaminitis, but with significantly elevated alkaline phosphatase and GGT, and microscopic hematuria. His ferritin is significantly elevated, which may simply represent an acute phase reactant. Infections associated with hepatitis, cytopenias, and hematuria include sepsis with disseminated intravascular coagulation, previously mentioned malaria, leptospirosis, dengue, ehrlichiosis, and rickettsial diseases, but he has no special risks for these infections, and other aspects of his illness (Parkinsonian features, bandemia) do not fit. His lung findings with hematuria might suggest a pulmonary/renal syndrome, but, once again, other features of his illness are not typical of these syndromes. Salmonella (typhoid fever) or influenza, now complicated by an early bacterial pneumonia, are viable possibilities.
The patient's ongoing clinical course is notable for a nontoxic (non‐SIRS) appearance but continued high‐grade fever with blood and urine cultures that are sterile. This argues against a common bacteremia with sepsis, and for either relapsing malaria (P vivax), influenza with a Parkinsonian‐like illness, typhoid fever, leptospirosis, dengue, or a rickettsial infection. Mycoplasma pneumonia is also possible given the atypical chest x‐ray appearance, slightly low hematocrit with elevated bilirubin, and neurological symptoms that may represent ataxia.
The subsequent negative laboratory tests listed are helpful in likely excluding many of the diagnoses suggested such as malaria, Babesia, common bacteremias, viral hepatitis, HIV, and WNV. Furthermore, the new history of mouse exposure brings to the forefront rodent‐associated infections, specifically exposure to mouse urine, a vehicle for leptospirosis. The patient's hepatitis, anemia, thrombocytopenia, scleral injection, along with the rest of his symptoms in the context of exposure to mouse urine makes leptospirosis the likely diagnosis. A negative Leptospira antibody early in his illness does not rule out the disease, and a convalescent titer should be obtained to confirm the diagnosis.
COMMENTARY
This case describes an elderly man who presented with a fever of unknown origin (FUO), and was eventually diagnosed with leptospirosis. FUO presents slightly differently in elderly patients, as elderly patients are less likely to mount a high fever, and when they do, the etiology is more likely to indicate a serious bacterial or viral infection. Additionally, an etiology for FUO in the elderly is found in over 70% of presenting cases, compared to 51% in patients under the age of 65 years.[1] A detailed, comprehensive social, travel, and exposure history and physical examination remains the cornerstone of elucidating the diagnosis for FUO. The exposure to mouse urine in this case was an unusual and a helpful piece of the history to further focus the differential diagnosis.
Leptospirosis is an emerging bacterial zoonosis, and causes both endemic and epidemic severe multisystem disease. The Leptospira spirochete is maintained in nature through a chronic renal infection in mammalian reservoir hosts, such as mice,[2, 4] and is transmitted through direct or aerosolized contact with infected urine or tissue. After a mean incubation period of 10 days, a variety of clinical manifestations may be seen. In this case, the patient's clinical presentation revealed many classic symptoms of leptospirosis, including fevers, rigors, headache, lower extremity myalgias, nausea, vomiting, and diarrhea; however, these symptoms are nonspecific. The presence of a conjunctival suffusion in leptospirosis infection had a specificity of 98% in a high‐incidence cohort of febrile patients in Sri Lanka,[3] and was an important diagnostic clue in this case. Leptospirosis is a self‐limited illness in most patients, with an initial septicemic, febrile phase followed by an immune phase. A more severe presentation may be seen in the immune phase of the illness, which includes renal and hepatic dysfunction (known as Weil's disease), as well as cardiac, pulmonary, and central nervous system abnormalities. With a 14% case fatality rate, the risk of death has been shown to be higher in patients over 40 years old, with altered mental status and multiorgan failure.[4]
The early diagnosis of leptospirosis relies heavily on physical exam findings and epidemiologic history. In this case, the patient's laboratory abnormalities, including immature granulocytes, thrombocytopenia, hyponatremia, hypokalemia, mild hepatitis, and pyuria with granular casts are all reported with leptospirosis infection2; however, independently, these laboratory findings are nonspecific. Patients may not have a detectable antibody levels in the acute phase of the disease. In this case, given the strong clinical suspicion based on the findings of conjunctival suffusion and exposure to mouse urine history, the lack of a more plausible alternate diagnosis, and known delay in antibody positivity, the patient was treated empirically with doxycycline for presumed leptospirosis.[5] Forthcoming novel diagnostic strategies such as next‐generation DNA sequencing techniques may provide real‐time diagnosis of this zoonotic infection, thus decreasing the window period between empirical antimicrobial coverage and diagnostic confirmation.[6]
Leptospirosis is prevalent in tropical climates and has been associated with impoverished communities.[7] Urban slums, with poor sanitation and high rodent density, are an ideal environment for leptospirosis. The reported risk of infection in a Brazilian slum was as high as 3% per year.[8] Additionally, rodent sightings, as well as the presence of chickens, were risk factors for leptospirosis transmission in urban slums.[9] Correspondingly in this case, we hypothesize that the patient's interest in urban farming, specifically the chickens he kept, likely attracted the mice infected with leptospirosis. Urban chicken farming is becoming increasingly popular in the United States,[10] and may be a developing risk factor for human leptospirosis infection. Leptospirosis is one of many emerging zoonoses, such as avian influenza, tick‐borne illness, and ebola, resulting from changing human ecology. Thus, when considering infectious etiologies, clinicians should ask patients about vocational and avocational exposures, including new trends such as urban farming, which may expose them to previously underappreciated illnesses.
TEACHING POINTS
- Elderly patients with a FUO are more likely to be diagnosed with an underlying serious bacterial or viral infection when compared to a younger cohort of FUO patients.
- The diagnosis of leptospirosis may initially be based on clinical suspicion in patients with classic features and exposures, noting the high specificity of conjunctival suffusion, and initial titers may be nondiagnostic; therefore, empiric treatment should be considered when clinical suspicion is high.
- Increased interest in urban chicken farming in the United States, with associated higher rodent density, may represent a newly recognized risk factor for human leptospirosis infection.
Disclosures
The authors report no conflicts of interest.
- , , . Fever of unknown origin in older persons. Infect Dis Clin North Am. 2007;21(4):937–945.
- . Leptospirosis. Clin Microbiol Rev. 2011;14(2):296–326.
- , , , et al. Leptospirosis as frequent cause of acute febrile illness in southern Sri Lanka. Emerg Infect Dis. 2011;17(9):1678–1684.
- , , , et al. Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases. Philadelphia, PA: Elsevier; 2010.
- , . Antibiotics for leptospirosis. The Cochrane Database Syst Rev. 2012;2:CD008264.
- , , , et al. Actionable diagnosis of neuroleptospirosis by next‐generation sequencing. N Engl J Med. 2014;370(25):2408–2417.
- , , , . Cases distribution of leptospirosis in City of Manaus, State of Amazonas, Brazil, 2000–2010. Rev Soc Bras Med Trop. 2012;45(6):713–716.
- , , , et al. Prospective study of leptospirosis transmission in an urban slum community: role of poor environment in repeated exposures to the leptospira agent. PLoS Negl Trop Dis. 2014;8(5):e2927.
- , , , et al. Impact of environment and social gradient on leptospira infection in urban slums. PLoS Negl Trop Dis. 2008;2(4):e228.
- Urban chicken ownership in four U.S. cities. United States Department of Agriculture website. Available at: http://www.aphis.usda.gov/animal_health/nahms/poultry/downloads/poultry10/Poultry10_dr_Urban_Chicken_Four.pdf. Published April 2013. Accessed June 9, 2015.
Fever in an elderly man is a nonspecific finding, occurring most commonly with infections but also with certain malignancies, rheumatologic disorders, and drug exposures. The complaint of rigors with diaphoresis makes an infection most likely. The acuity of his illness makes infections with more chronic presentations such as tuberculosis or actinomycosis less likely. The presence of frontal headache might suggest a sinus or brain source, but headache also occurs in generalized infections such as pneumonia, bacteremia from any cause, malaria, rickettsial infections, viral illnesses, and others. Additional history should include detailed inquiry into travel, vocational, and avocational exposures.
The patient's difficulty standing implies the development of lower extremity weakness and infections associated with neurological syndromes. His leg weakness may be related to early Guillain‐Barre syndrome, which is associated most commonly with Campylobacter jejuni, but also other bacteria and viruses such as Haemophilus influenza, Mycoplasma pneumonia, Influenza virus, Cytomegalovirus and hepatitis E. Other viral infections associated with pure motor deficits include echovirus, coxsackie virus, enterovirus, and West Nile virus (WNV). The paralytic syndrome associated with enteroviruses is more common in children, whereas the neuroinvasive variant of WNV more often affects the elderly and can be associated with encephalitis as well as a flaccid paralysis. Although acute paralytic shellfish poisoning could account for both his weakness and his acute gastrointestinal syndrome, this diagnosis is unlikely because the symptoms often have a prominent sensory component, and there is usually the history of recent ingestion of the suspect bivalves. Like all adults presenting for medical care, he should be screened for human immunodeficiency virus (HIV) infection; if testing is positive, the differential diagnosis for his current illness broadens significantly. Finally, he may have a spinal cord disorder or infection such as an epidural abscess, or transverse myelitis, which would present with lower extremity weakness and fever. It would be helpful to know the time of year of his illness, exposure to mosquito bites, his neurological exam findings, and results of blood and stool cultures. If the patient had signs of meningitis or encephalitis, cerebral spinal fluid analysis would be helpful. If his neurological exam was suggestive of cord involvement, it would be helpful to know the results of magnetic resonance imaging of the spinal cord.
The patient's past medical history includes relatively common problems for a 73‐year‐old man and does not substantially influence the differential diagnosis of his current illness. His travel history to Uganda a year previously may be relevant, because malaria (Plasmodium vivax) could present with fever and weakness. Less commonly, African trypanosomiasis (Trypanosoma brucei gambiense) can, in the late phase, present with fever and malaise, but also typically includes symptoms of encephalitis, including depressed mental status, confusion, ataxia, and possibly personality changes. His travel to Zurich should not impose any particular infection risk, unless he was hiking in the mountains around Zurich, where he could have contracted tick‐borne encephalitis; however, his travel more than 6 months prior to presentation makes this unlikely. Lyme disease due to Borrelia burgdorferi is also a potential exposure in the Swiss mountains, and can present with fever in the acute phase, as well as arthritis with chronic disease, but should not cause fever, rigor, diaphoresis, and headache many months later. Summering in Cape Cod puts him at risk for babesiosis, but an incubation period of 5 months is too long. Keeping chickens places him at risk for Salmonella exposure and typhoid fever. Ingesting raw oysters carries a risk for shellfish poisoning and Vibrio infections, but the incubation period (1 month) again seems too long to cause his current symptoms.
Notable physical findings are an ill‐appearing man with injected sclera and a high fever but normal blood pressure and heart rate. He also demonstrates proximal lower extremity weakness manifested by difficulty rising from a chair and a slow gait with short strides and deliberate (possibly on‐block) turning. His neurological exam is most consistent with Parkinsonian symptoms that have been described in patients with severe influenza A, which would explain all of his other symptoms as well. Pulse‐temperature dissociation is classically described with typhoid fever but usually occurs later in the disease course, and could be masked by the patient's metoprolol. Typhoid fever can also be associated with neurological symptoms including meningitis and movement disorders.
The patient has a remarkable bandemia, suggesting a bacterial infection, as well as a slight reduction in hematocrit and platelet count. Additionally, his labs revealed a mild transaminitis, but with significantly elevated alkaline phosphatase and GGT, and microscopic hematuria. His ferritin is significantly elevated, which may simply represent an acute phase reactant. Infections associated with hepatitis, cytopenias, and hematuria include sepsis with disseminated intravascular coagulation, previously mentioned malaria, leptospirosis, dengue, ehrlichiosis, and rickettsial diseases, but he has no special risks for these infections, and other aspects of his illness (Parkinsonian features, bandemia) do not fit. His lung findings with hematuria might suggest a pulmonary/renal syndrome, but, once again, other features of his illness are not typical of these syndromes. Salmonella (typhoid fever) or influenza, now complicated by an early bacterial pneumonia, are viable possibilities.
The patient's ongoing clinical course is notable for a nontoxic (non‐SIRS) appearance but continued high‐grade fever with blood and urine cultures that are sterile. This argues against a common bacteremia with sepsis, and for either relapsing malaria (P vivax), influenza with a Parkinsonian‐like illness, typhoid fever, leptospirosis, dengue, or a rickettsial infection. Mycoplasma pneumonia is also possible given the atypical chest x‐ray appearance, slightly low hematocrit with elevated bilirubin, and neurological symptoms that may represent ataxia.
The subsequent negative laboratory tests listed are helpful in likely excluding many of the diagnoses suggested such as malaria, Babesia, common bacteremias, viral hepatitis, HIV, and WNV. Furthermore, the new history of mouse exposure brings to the forefront rodent‐associated infections, specifically exposure to mouse urine, a vehicle for leptospirosis. The patient's hepatitis, anemia, thrombocytopenia, scleral injection, along with the rest of his symptoms in the context of exposure to mouse urine makes leptospirosis the likely diagnosis. A negative Leptospira antibody early in his illness does not rule out the disease, and a convalescent titer should be obtained to confirm the diagnosis.
COMMENTARY
This case describes an elderly man who presented with a fever of unknown origin (FUO), and was eventually diagnosed with leptospirosis. FUO presents slightly differently in elderly patients, as elderly patients are less likely to mount a high fever, and when they do, the etiology is more likely to indicate a serious bacterial or viral infection. Additionally, an etiology for FUO in the elderly is found in over 70% of presenting cases, compared to 51% in patients under the age of 65 years.[1] A detailed, comprehensive social, travel, and exposure history and physical examination remains the cornerstone of elucidating the diagnosis for FUO. The exposure to mouse urine in this case was an unusual and a helpful piece of the history to further focus the differential diagnosis.
Leptospirosis is an emerging bacterial zoonosis, and causes both endemic and epidemic severe multisystem disease. The Leptospira spirochete is maintained in nature through a chronic renal infection in mammalian reservoir hosts, such as mice,[2, 4] and is transmitted through direct or aerosolized contact with infected urine or tissue. After a mean incubation period of 10 days, a variety of clinical manifestations may be seen. In this case, the patient's clinical presentation revealed many classic symptoms of leptospirosis, including fevers, rigors, headache, lower extremity myalgias, nausea, vomiting, and diarrhea; however, these symptoms are nonspecific. The presence of a conjunctival suffusion in leptospirosis infection had a specificity of 98% in a high‐incidence cohort of febrile patients in Sri Lanka,[3] and was an important diagnostic clue in this case. Leptospirosis is a self‐limited illness in most patients, with an initial septicemic, febrile phase followed by an immune phase. A more severe presentation may be seen in the immune phase of the illness, which includes renal and hepatic dysfunction (known as Weil's disease), as well as cardiac, pulmonary, and central nervous system abnormalities. With a 14% case fatality rate, the risk of death has been shown to be higher in patients over 40 years old, with altered mental status and multiorgan failure.[4]
The early diagnosis of leptospirosis relies heavily on physical exam findings and epidemiologic history. In this case, the patient's laboratory abnormalities, including immature granulocytes, thrombocytopenia, hyponatremia, hypokalemia, mild hepatitis, and pyuria with granular casts are all reported with leptospirosis infection2; however, independently, these laboratory findings are nonspecific. Patients may not have a detectable antibody levels in the acute phase of the disease. In this case, given the strong clinical suspicion based on the findings of conjunctival suffusion and exposure to mouse urine history, the lack of a more plausible alternate diagnosis, and known delay in antibody positivity, the patient was treated empirically with doxycycline for presumed leptospirosis.[5] Forthcoming novel diagnostic strategies such as next‐generation DNA sequencing techniques may provide real‐time diagnosis of this zoonotic infection, thus decreasing the window period between empirical antimicrobial coverage and diagnostic confirmation.[6]
Leptospirosis is prevalent in tropical climates and has been associated with impoverished communities.[7] Urban slums, with poor sanitation and high rodent density, are an ideal environment for leptospirosis. The reported risk of infection in a Brazilian slum was as high as 3% per year.[8] Additionally, rodent sightings, as well as the presence of chickens, were risk factors for leptospirosis transmission in urban slums.[9] Correspondingly in this case, we hypothesize that the patient's interest in urban farming, specifically the chickens he kept, likely attracted the mice infected with leptospirosis. Urban chicken farming is becoming increasingly popular in the United States,[10] and may be a developing risk factor for human leptospirosis infection. Leptospirosis is one of many emerging zoonoses, such as avian influenza, tick‐borne illness, and ebola, resulting from changing human ecology. Thus, when considering infectious etiologies, clinicians should ask patients about vocational and avocational exposures, including new trends such as urban farming, which may expose them to previously underappreciated illnesses.
TEACHING POINTS
- Elderly patients with a FUO are more likely to be diagnosed with an underlying serious bacterial or viral infection when compared to a younger cohort of FUO patients.
- The diagnosis of leptospirosis may initially be based on clinical suspicion in patients with classic features and exposures, noting the high specificity of conjunctival suffusion, and initial titers may be nondiagnostic; therefore, empiric treatment should be considered when clinical suspicion is high.
- Increased interest in urban chicken farming in the United States, with associated higher rodent density, may represent a newly recognized risk factor for human leptospirosis infection.
Disclosures
The authors report no conflicts of interest.
Fever in an elderly man is a nonspecific finding, occurring most commonly with infections but also with certain malignancies, rheumatologic disorders, and drug exposures. The complaint of rigors with diaphoresis makes an infection most likely. The acuity of his illness makes infections with more chronic presentations such as tuberculosis or actinomycosis less likely. The presence of frontal headache might suggest a sinus or brain source, but headache also occurs in generalized infections such as pneumonia, bacteremia from any cause, malaria, rickettsial infections, viral illnesses, and others. Additional history should include detailed inquiry into travel, vocational, and avocational exposures.
The patient's difficulty standing implies the development of lower extremity weakness and infections associated with neurological syndromes. His leg weakness may be related to early Guillain‐Barre syndrome, which is associated most commonly with Campylobacter jejuni, but also other bacteria and viruses such as Haemophilus influenza, Mycoplasma pneumonia, Influenza virus, Cytomegalovirus and hepatitis E. Other viral infections associated with pure motor deficits include echovirus, coxsackie virus, enterovirus, and West Nile virus (WNV). The paralytic syndrome associated with enteroviruses is more common in children, whereas the neuroinvasive variant of WNV more often affects the elderly and can be associated with encephalitis as well as a flaccid paralysis. Although acute paralytic shellfish poisoning could account for both his weakness and his acute gastrointestinal syndrome, this diagnosis is unlikely because the symptoms often have a prominent sensory component, and there is usually the history of recent ingestion of the suspect bivalves. Like all adults presenting for medical care, he should be screened for human immunodeficiency virus (HIV) infection; if testing is positive, the differential diagnosis for his current illness broadens significantly. Finally, he may have a spinal cord disorder or infection such as an epidural abscess, or transverse myelitis, which would present with lower extremity weakness and fever. It would be helpful to know the time of year of his illness, exposure to mosquito bites, his neurological exam findings, and results of blood and stool cultures. If the patient had signs of meningitis or encephalitis, cerebral spinal fluid analysis would be helpful. If his neurological exam was suggestive of cord involvement, it would be helpful to know the results of magnetic resonance imaging of the spinal cord.
The patient's past medical history includes relatively common problems for a 73‐year‐old man and does not substantially influence the differential diagnosis of his current illness. His travel history to Uganda a year previously may be relevant, because malaria (Plasmodium vivax) could present with fever and weakness. Less commonly, African trypanosomiasis (Trypanosoma brucei gambiense) can, in the late phase, present with fever and malaise, but also typically includes symptoms of encephalitis, including depressed mental status, confusion, ataxia, and possibly personality changes. His travel to Zurich should not impose any particular infection risk, unless he was hiking in the mountains around Zurich, where he could have contracted tick‐borne encephalitis; however, his travel more than 6 months prior to presentation makes this unlikely. Lyme disease due to Borrelia burgdorferi is also a potential exposure in the Swiss mountains, and can present with fever in the acute phase, as well as arthritis with chronic disease, but should not cause fever, rigor, diaphoresis, and headache many months later. Summering in Cape Cod puts him at risk for babesiosis, but an incubation period of 5 months is too long. Keeping chickens places him at risk for Salmonella exposure and typhoid fever. Ingesting raw oysters carries a risk for shellfish poisoning and Vibrio infections, but the incubation period (1 month) again seems too long to cause his current symptoms.
Notable physical findings are an ill‐appearing man with injected sclera and a high fever but normal blood pressure and heart rate. He also demonstrates proximal lower extremity weakness manifested by difficulty rising from a chair and a slow gait with short strides and deliberate (possibly on‐block) turning. His neurological exam is most consistent with Parkinsonian symptoms that have been described in patients with severe influenza A, which would explain all of his other symptoms as well. Pulse‐temperature dissociation is classically described with typhoid fever but usually occurs later in the disease course, and could be masked by the patient's metoprolol. Typhoid fever can also be associated with neurological symptoms including meningitis and movement disorders.
The patient has a remarkable bandemia, suggesting a bacterial infection, as well as a slight reduction in hematocrit and platelet count. Additionally, his labs revealed a mild transaminitis, but with significantly elevated alkaline phosphatase and GGT, and microscopic hematuria. His ferritin is significantly elevated, which may simply represent an acute phase reactant. Infections associated with hepatitis, cytopenias, and hematuria include sepsis with disseminated intravascular coagulation, previously mentioned malaria, leptospirosis, dengue, ehrlichiosis, and rickettsial diseases, but he has no special risks for these infections, and other aspects of his illness (Parkinsonian features, bandemia) do not fit. His lung findings with hematuria might suggest a pulmonary/renal syndrome, but, once again, other features of his illness are not typical of these syndromes. Salmonella (typhoid fever) or influenza, now complicated by an early bacterial pneumonia, are viable possibilities.
The patient's ongoing clinical course is notable for a nontoxic (non‐SIRS) appearance but continued high‐grade fever with blood and urine cultures that are sterile. This argues against a common bacteremia with sepsis, and for either relapsing malaria (P vivax), influenza with a Parkinsonian‐like illness, typhoid fever, leptospirosis, dengue, or a rickettsial infection. Mycoplasma pneumonia is also possible given the atypical chest x‐ray appearance, slightly low hematocrit with elevated bilirubin, and neurological symptoms that may represent ataxia.
The subsequent negative laboratory tests listed are helpful in likely excluding many of the diagnoses suggested such as malaria, Babesia, common bacteremias, viral hepatitis, HIV, and WNV. Furthermore, the new history of mouse exposure brings to the forefront rodent‐associated infections, specifically exposure to mouse urine, a vehicle for leptospirosis. The patient's hepatitis, anemia, thrombocytopenia, scleral injection, along with the rest of his symptoms in the context of exposure to mouse urine makes leptospirosis the likely diagnosis. A negative Leptospira antibody early in his illness does not rule out the disease, and a convalescent titer should be obtained to confirm the diagnosis.
COMMENTARY
This case describes an elderly man who presented with a fever of unknown origin (FUO), and was eventually diagnosed with leptospirosis. FUO presents slightly differently in elderly patients, as elderly patients are less likely to mount a high fever, and when they do, the etiology is more likely to indicate a serious bacterial or viral infection. Additionally, an etiology for FUO in the elderly is found in over 70% of presenting cases, compared to 51% in patients under the age of 65 years.[1] A detailed, comprehensive social, travel, and exposure history and physical examination remains the cornerstone of elucidating the diagnosis for FUO. The exposure to mouse urine in this case was an unusual and a helpful piece of the history to further focus the differential diagnosis.
Leptospirosis is an emerging bacterial zoonosis, and causes both endemic and epidemic severe multisystem disease. The Leptospira spirochete is maintained in nature through a chronic renal infection in mammalian reservoir hosts, such as mice,[2, 4] and is transmitted through direct or aerosolized contact with infected urine or tissue. After a mean incubation period of 10 days, a variety of clinical manifestations may be seen. In this case, the patient's clinical presentation revealed many classic symptoms of leptospirosis, including fevers, rigors, headache, lower extremity myalgias, nausea, vomiting, and diarrhea; however, these symptoms are nonspecific. The presence of a conjunctival suffusion in leptospirosis infection had a specificity of 98% in a high‐incidence cohort of febrile patients in Sri Lanka,[3] and was an important diagnostic clue in this case. Leptospirosis is a self‐limited illness in most patients, with an initial septicemic, febrile phase followed by an immune phase. A more severe presentation may be seen in the immune phase of the illness, which includes renal and hepatic dysfunction (known as Weil's disease), as well as cardiac, pulmonary, and central nervous system abnormalities. With a 14% case fatality rate, the risk of death has been shown to be higher in patients over 40 years old, with altered mental status and multiorgan failure.[4]
The early diagnosis of leptospirosis relies heavily on physical exam findings and epidemiologic history. In this case, the patient's laboratory abnormalities, including immature granulocytes, thrombocytopenia, hyponatremia, hypokalemia, mild hepatitis, and pyuria with granular casts are all reported with leptospirosis infection2; however, independently, these laboratory findings are nonspecific. Patients may not have a detectable antibody levels in the acute phase of the disease. In this case, given the strong clinical suspicion based on the findings of conjunctival suffusion and exposure to mouse urine history, the lack of a more plausible alternate diagnosis, and known delay in antibody positivity, the patient was treated empirically with doxycycline for presumed leptospirosis.[5] Forthcoming novel diagnostic strategies such as next‐generation DNA sequencing techniques may provide real‐time diagnosis of this zoonotic infection, thus decreasing the window period between empirical antimicrobial coverage and diagnostic confirmation.[6]
Leptospirosis is prevalent in tropical climates and has been associated with impoverished communities.[7] Urban slums, with poor sanitation and high rodent density, are an ideal environment for leptospirosis. The reported risk of infection in a Brazilian slum was as high as 3% per year.[8] Additionally, rodent sightings, as well as the presence of chickens, were risk factors for leptospirosis transmission in urban slums.[9] Correspondingly in this case, we hypothesize that the patient's interest in urban farming, specifically the chickens he kept, likely attracted the mice infected with leptospirosis. Urban chicken farming is becoming increasingly popular in the United States,[10] and may be a developing risk factor for human leptospirosis infection. Leptospirosis is one of many emerging zoonoses, such as avian influenza, tick‐borne illness, and ebola, resulting from changing human ecology. Thus, when considering infectious etiologies, clinicians should ask patients about vocational and avocational exposures, including new trends such as urban farming, which may expose them to previously underappreciated illnesses.
TEACHING POINTS
- Elderly patients with a FUO are more likely to be diagnosed with an underlying serious bacterial or viral infection when compared to a younger cohort of FUO patients.
- The diagnosis of leptospirosis may initially be based on clinical suspicion in patients with classic features and exposures, noting the high specificity of conjunctival suffusion, and initial titers may be nondiagnostic; therefore, empiric treatment should be considered when clinical suspicion is high.
- Increased interest in urban chicken farming in the United States, with associated higher rodent density, may represent a newly recognized risk factor for human leptospirosis infection.
Disclosures
The authors report no conflicts of interest.
- , , . Fever of unknown origin in older persons. Infect Dis Clin North Am. 2007;21(4):937–945.
- . Leptospirosis. Clin Microbiol Rev. 2011;14(2):296–326.
- , , , et al. Leptospirosis as frequent cause of acute febrile illness in southern Sri Lanka. Emerg Infect Dis. 2011;17(9):1678–1684.
- , , , et al. Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases. Philadelphia, PA: Elsevier; 2010.
- , . Antibiotics for leptospirosis. The Cochrane Database Syst Rev. 2012;2:CD008264.
- , , , et al. Actionable diagnosis of neuroleptospirosis by next‐generation sequencing. N Engl J Med. 2014;370(25):2408–2417.
- , , , . Cases distribution of leptospirosis in City of Manaus, State of Amazonas, Brazil, 2000–2010. Rev Soc Bras Med Trop. 2012;45(6):713–716.
- , , , et al. Prospective study of leptospirosis transmission in an urban slum community: role of poor environment in repeated exposures to the leptospira agent. PLoS Negl Trop Dis. 2014;8(5):e2927.
- , , , et al. Impact of environment and social gradient on leptospira infection in urban slums. PLoS Negl Trop Dis. 2008;2(4):e228.
- Urban chicken ownership in four U.S. cities. United States Department of Agriculture website. Available at: http://www.aphis.usda.gov/animal_health/nahms/poultry/downloads/poultry10/Poultry10_dr_Urban_Chicken_Four.pdf. Published April 2013. Accessed June 9, 2015.
- , , . Fever of unknown origin in older persons. Infect Dis Clin North Am. 2007;21(4):937–945.
- . Leptospirosis. Clin Microbiol Rev. 2011;14(2):296–326.
- , , , et al. Leptospirosis as frequent cause of acute febrile illness in southern Sri Lanka. Emerg Infect Dis. 2011;17(9):1678–1684.
- , , , et al. Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases. Philadelphia, PA: Elsevier; 2010.
- , . Antibiotics for leptospirosis. The Cochrane Database Syst Rev. 2012;2:CD008264.
- , , , et al. Actionable diagnosis of neuroleptospirosis by next‐generation sequencing. N Engl J Med. 2014;370(25):2408–2417.
- , , , . Cases distribution of leptospirosis in City of Manaus, State of Amazonas, Brazil, 2000–2010. Rev Soc Bras Med Trop. 2012;45(6):713–716.
- , , , et al. Prospective study of leptospirosis transmission in an urban slum community: role of poor environment in repeated exposures to the leptospira agent. PLoS Negl Trop Dis. 2014;8(5):e2927.
- , , , et al. Impact of environment and social gradient on leptospira infection in urban slums. PLoS Negl Trop Dis. 2008;2(4):e228.
- Urban chicken ownership in four U.S. cities. United States Department of Agriculture website. Available at: http://www.aphis.usda.gov/animal_health/nahms/poultry/downloads/poultry10/Poultry10_dr_Urban_Chicken_Four.pdf. Published April 2013. Accessed June 9, 2015.
More Than a Mnemonic
A 51‐year‐old man presented to the emergency department after 1 day of progressive dyspnea and increasing confusion.
Acute dyspnea most commonly stems from a cardiac or pulmonary disorder such as heart failure, acute coronary syndrome, pneumonia, pulmonary embolism, or exacerbations of asthma or chronic obstructive pulmonary disease. Less frequent cardiopulmonary considerations include pericardial or pleural effusion, pneumothorax, aspiration, and upper airway obstruction. Dyspnea might also be the initial manifestation of profound anemia or metabolic acidosis.
The presence of confusion suggests either a severe presentation of any of the aforementioned possibilities (with confusion resulting from hypoxia, hypercapnia, or hypotension); a multiorgan illness such as sepsis, malignancy, thromboembolic disease, vasculitis, thyroid dysfunction, or toxic ingestion; or a metabolic derangement related to the underlying cause of dyspnea (for example, hypercalcemia or hyponatremia associated with lung cancer).
Twelve hours prior to presentation, he started to have visual hallucinations. He denied fever, chills, cough, chest discomfort, palpitations, weight gain, headache, neck pain, or weakness.
Visual hallucinations could result from a toxic‐metabolic encephalopathy, such as drug overdose or withdrawal, liver or kidney failure, or hypoxia. A structural brain abnormality may also manifest with visual hallucination. Acute onset at age 51 and the absence of auditory hallucinations argue against a neurodegenerative illness and a primary psychiatric disturbance, respectively.
Episodic hallucinations would support the possibility of seizures, monocular hallucinations would point to a retinal or ocular problem, and a description of yellow‐green hue would suggest a side effect of digoxin.
His past medical history was remarkable for diet‐controlled type 2 diabetes mellitus, hypertension, hyperlipidemia, and chronic low back pain. His medications included metoprolol tartrate 25 mg twice daily, omeprazole 40 mg daily, baclofen 15 mg twice daily, oxycodone 30 mg 3 times daily, and hydrocodone 10 mg/acetaminophen 325 mg, 2 tablets 3 times daily as needed for back pain. He was a smoker with a 30 pack‐year history. He had a history of alcohol and cocaine use, but denied any recent substance use. He had no known history of obstructive pulmonary disease.
The patient takes 3 medications well known to cause confusion and hallucinations (oxycodone, hydrocodone, and baclofen), especially when they accumulate due to excessive ingestion or impaired clearance. Although these medications may suppress ventilatory drive, dyspnea would not be a common presenting complaint. He has risk factors for ischemic heart disease and cardiomyopathy, and his smoking history raises the possibility of malignancy.
On exam, the patient's temperature was 94.4C, heart rate 128 beats per minute, respiration rate 28 breaths per minute, blood pressure 155/63 mm Hg, and oxygen saturation 100% while breathing ambient air. The patient was cachectic and appeared in moderate respiratory distress. His pupils were equal and reactive to light, and extraocular movements were intact. He did not have scleral icterus, or cervical or clavicular lymphadenopathy. His oropharynx was negative for erythema, edema, or exudate. His cardiovascular exam revealed a regular tachycardia without rubs or diastolic gallops. There was a 2/6 systolic murmur heard best at left sternal border, without radiation. He did not have jugular venous distention. His pulmonary exam was notable for tachypnea but with normal vesicular breath sounds throughout. He did not have stridor, wheezing, rhonchi, or rales. His abdomen had normal bowel tones and was soft without tenderness, distention, or organomegaly. His extremities were warm, revealed normal pulses, and no edema was present. His joints were cool to palpation, without effusion. On neurologic exam, he was oriented to person and place and able to answer yes/no questions, but unable to provide detailed history. His speech was fluent. His motor exam was without focal deficits. His skin was without any notable lesions.
The constellation of findings does not point to a specific toxidrome. The finding of warm extremities in a hypothermic patient suggests heat loss due to inappropriate peripheral vasodilation. In the absence of vasodilators or features of aortic insufficiency, sepsis becomes a leading consideration. Infection could result in hypothermia and altered sensorium, and accompanying lactic acidosis could trigger tachypnea.
Shortly after admission, he became more somnolent and developed progressive respiratory distress, requiring intubation. Arterial blood gas revealed a pH of 6.93, PaCO2<20 mm Hg, PaO2 127 mm Hg, and HCO3<5 mEq/L. Other laboratory results included a lactate of 4.1 mmol/L, blood urea nitrogen 49 mg/dL, creatinine 2.3 mg/dL (0.8 at 1 month prior), sodium level of 143 mmol/L, chloride of 106 mmol/L, and bicarbonate level of <5 mg/dL. His aspartate aminotransferase was 34 IU/L, alanine transaminase was 28 IU/L, total bilirubin was 0.6 mg/dL, International Normalized Ratio was 1.3. A complete blood count revealed a white blood cell count of 23,000/L, hemoglobin of 10.6 g/dL, and platelet count of 454,000/L. A urinalysis was unremarkable. Cultures of blood, urine, and sputum were collected. Head computed tomography was negative.
This patient has a combined anion gap and nongap metabolic acidosis, as well as respiratory alkalosis. Although his acute kidney failure could produce these 2 types of metabolic acidosis, the modest elevation of the serum creatinine is not commensurate with such profound acidosis. Similarly, sepsis without hypotension or more striking elevation in lactate levels would not account for the entirety of the acidosis. Severe diabetic ketoacidosis can result in profound metabolic acidosis, and marked hyperglycemia or hyperosmolarity could result in somnolence; however, his diabetes has been controlled without medication and there is no obvious precipitant for an episode of ketoacidosis.
Remaining causes of anion gap acidosis include ingestion of methanol, ethylene glycol, ethanol, or salicylates. A careful history of ingestions and medications from witnesses including any prehospital personnel might suggest a source of intoxication. Absent this information, the hypothermia favors an ingestion of an alcohol over salicylates, and the lack of urine crystals and the presence of prominent visual hallucinations would point more toward methanol poisoning than ethylene glycol. A serum osmolarity measurement would allow determination of the osmolar gap, which would be elevated in the setting of methanol or ethylene glycol poisoning. If he were this ill from ethanol, I would have expected to see evidence of hepatotoxicity.
I would administer sodium bicarbonate to reverse the acidosis and to promote renal clearance of salicylates, methanol, ethylene glycol, and their metabolites. Orogastric decontamination with activated charcoal should be considered. If the osmolar gap is elevated, I would also administer intravenous fomepizole to attempt to reverse methanol or ethylene glycol poisoning. I would not delay treatment while waiting for these serum levels to return.
Initial serologic toxicology performed in the emergency department revealed negative ethanol, salicylates, and ketones. His osmolar gap was 13 mOsm/kg. His acetaminophen level was 69 g/mL (normal <120 g/mL). A creatinine phosphokinase was 84 IU/L and myoglobin was 93 ng/mL. His subsequent serum toxicology screen was negative for methanol, ethylene glycol, isopropranol, and hippuric acid. Urine toxicology was positive for opiates, but negative for amphetamine, benzodiazepine, cannabinoid, and cocaine.
Serum and urine ketone assays typically involve the nitroprusside reaction and detect acetoacetate, but not ‐hydroxybutyrate, and can lead to negative test results early in diabetic or alcoholic ketoacidosis. However, the normal ethanol level argues against alcoholic ketoacidosis. Rare causes of elevated anion gap acidosis include toluene toxicity, acetaminophen poisoning, and ingestion of other alcohols. Toluene is metabolized to hippuric acid, and acetaminophen toxicity and associated glutathione depletion can lead to 5‐oxoproline accumulation, producing an anion gap. Patients who abuse alcohol are at risk for acetaminophen toxicity even at doses considered normal. However, this degree of encephalopathy would be unusual for acetaminophen toxicity unless liver failure had developed or unless there was another ingestion that might alter sensorium. Furthermore, the elevated osmolar gap is not a feature of acetaminophen poisoning. I would monitor liver enzyme tests and consider a serum ammonia level, but would not attribute the entire picture to acetaminophen.
The combination of elevated anion gap with an elevated osmolar gap narrows the diagnostic possibilities. Ingestion of several alcohols (ethanol, methanol, ethylene glycol, diethylene glycol) or toluene could produce these abnormalities. Of note, the osmolar gap is typically most markedly elevated early in methanol and ethylene glycol ingestions, and then as the parent compound is metabolized, the osmolar gap closes and the accumulation of metabolites produces the anion gap. Hallucinations are more common with methanol and toluene, and renal failure is more typical of ethylene glycol or toluene. The lack of oxalate crystalluria does not exclude ethylene glycol poisoning. Unfortunately, urine testing for oxalate crystals or fluorescein examination are neither sensitive nor specific enough to diagnosis ethylene glycol toxicity reliably. In most hospitals, assays used for serum testing for alcohols are insensitive, and require confirmation with gas chromatography performed at a specialty lab.
Additional history might reveal the likely culprit or culprits. Inhalant abuse including huffing would point to toluene or organic acid exposure. Solvent ingestion (eg, antifreeze, brake fluid) would suggest methanol or ethylene glycol. Absent this history, I remain suspicious for poisoning with methanol or ethylene glycol and would consider empiric treatment after urgent consultation with a medical toxicologist. A careful ophthalmologic exam might demonstrate characteristic features of methanol poisoning. Serum samples should be sent to a regional lab for analysis for alcohols and organic acids.
He was admitted to the intensive care unit, and empiric antibiotics started. He was empirically started on N‐acetylcysteine and sodium bicarbonate drips. However, his acidemia persisted and he required hemodialysis, which was initiated 12 hours after initial presentation. His acidemia and mental status quickly improved after hemodialysis. He was extubated on hospital day 2 and no longer required hemodialysis.
The differential diagnosis at this point consists of 3 main possibilities: ingestion of methanol, ethylene glycol, or inhalant abuse such as from toluene. The normal hippuric acid level points away from toluene, whereas serum levels can be misleading in the alcohol poisonings. Other discriminating features to consider include exposure history and unique clinical aspects. In this patient, an exposure history is lacking, but 4 clinical features stand out: visual hallucinations, acute kidney injury, mild lactic acidosis, and rapid improvement with hemodialysis. Both ethylene glycol and methanol toxicity may produce a mild lactic acidosis by increasing hepatic metabolism of pyruvate to lactate, and both are rapidly cleared by dialysis. Although it is tempting to place methanol at the top of the list of possibilities due to the report of visual hallucinations, the subjective visual complaints without objective exam corollaries (loss of visual acuity, abnormal pupillary reflexes, or optic disc hyperemia) are nonspecific and might be provoked by alcohol or an inhalant. Furthermore, the acute renal failure is much more typical of ethylene glycol, and thus I would consider ethylene glycol as being the more likely of the ingestions. Coingestion of multiple alcohols is a possibility, but it would be statistically less likely. Confirmation of ethylene glycol poisoning would consist of further insight into his exposures and measurement of levels using gas chromatography.
A urine sample from his emergency department presentation was sent to an outside lab for organic acid levels. Based on high clinical suspicion for 5‐oxoprolinemia (pyroglutamic acidemia) the patient was counseled to avoid any acetaminophen. His primary care provider was informed of this and acetaminophen was added as an adverse drug reaction. The patient left against medical advice soon after extubation. Following discharge, his 5‐oxoproline (pyroglutamic acid) level returned markedly elevated at greater than 10,000 mmol/mol creatinine (200 times the upper limit of normal).
Elevations in 5‐oxoproline levels in this patient most likely stem from glutathione depletion related to chronic acetaminophen use. Alcohol use and malnutrition may have heightened this patient's susceptibility. Despite the common occurrence of acetaminophen use in alcohol abusers or the malnourished, the rarity of severe 5‐oxoproline toxicity suggests unknown factors may be present in predisposed individuals, or under‐recognition. Although acetaminophen‐induced hepatotoxicity may occur along with 5‐oxoprolinemia, this does not always occur.
Several features led me away from this syndrome. First, its rarity lowered my pretest probability. Second, the lack of exposure history and details about the serum assays, specifically whether the measurements were confirmed by gas chromatography, reduced my confidence in eliminating more common ingestions. Third, several aspects proved to be less useful discriminating features: the mild elevation in osmolar gap, renal failure, and hallucinations, which in retrospect proved to be nonspecific.
The patient admitted that he had a longstanding use of acetaminophen in addition to using his girlfriend's acetaminophen‐hydrocodone. He had significant weight loss of over 50 pounds over the previous year, which he attributed to poor appetite. On further chart review, he had been admitted 3 times with a similar clinical presentation and recovered quickly with intensive and supportive care, with no etiology found at those times. He had 2 subsequent hospital admissions for altered mental status and respiratory failure, and his final hospitalization resulted in cardiac arrest and death.
DISCUSSION
5‐Oxoprolinemia is a rare, but potentially lethal cause of severe anion gap metabolic acidosis.[1, 2] The mechanism is thought to be impairment of glutathione metabolism, in the context of other predisposing factors. This can be a congenital error of metabolism, or can be acquired and exacerbated by acetaminophen use. Ingestion of acetaminophen leads to glutathione depletion, which in turn may precipitate accumulation of pyroglutamic acid and subsequent anion gap metabolic acidosis (Figure 1). Additional risk factors that may predispose patients to this condition include malnutrition, renal insufficiency, concurrent infection, and female gender.[1, 2, 3]
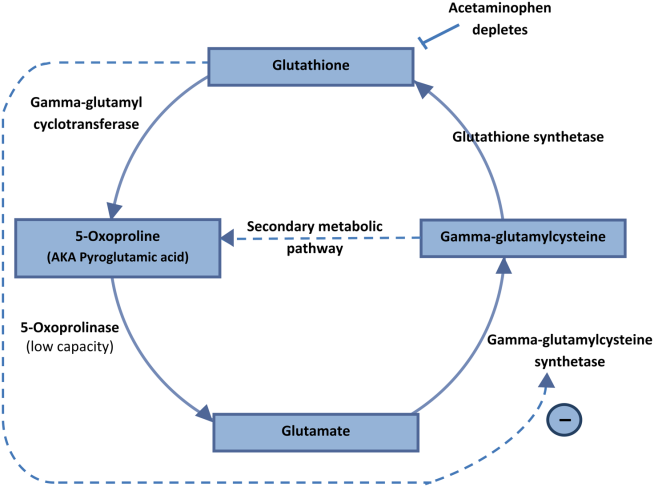
The diagnosis of 5‐oxoprolinemia is made via urine or serum organic acid analysis, testing routinely performed in pediatric populations when screening for congenital metabolic disorders. The pathophysiology suggests that obtaining a urine sample early in presentation, when acidosis is greatest, would lead to the highest 5‐oxoproline levels and best chance for diagnosis. Case patients have had normal levels prior to and in convalescent phases after the acute episode.[4] Given the long turnaround time for lab testing, presumptive diagnosis and treatment may be necessary.
Treatment of 5‐oxoprolinemia is primarily supportive, aimed at the metabolic acidosis. Fluid resuscitation and bicarbonate therapy are reasonable temporizing measures. Hemodialysis can clear 5‐oxoproline and may be indicated in severe acidosis.[5] Furthermore, the proposed pathophysiology suggests that administration of N‐acetylcysteine (NAC) may help to address the underlying process, but there are no trials to support a specific dosing regimen. However, given the fulminant presentation and common competing concern for acetaminophen toxicity, it is reasonable to initiate NAC aimed at treatment for possible acetaminophen overdose. Prevention of recurrence includes avoidance of acetaminophen, and counseling the patient to avoid acetaminophen in prescription combination medications and over‐the‐counter preparations.
Recent regulatory changes regarding acetaminophen/opioid combinations may reduce the incidence of 5‐oxoprolinemia. The US Food and Drug Administration has taken action to reduce adverse effects from acetaminophen exposure by limiting the amount of acetaminophen in opioid combination pills from 500 mg to a maximum of 325 mg per pill. This is aimed at preventing hepatotoxicity from ingestion of higher‐than‐recommended doses. However, clinicians should remember that 5‐oxoprolinemia can result from ingestion of acetaminophen at therapeutic levels.
Given its rare incidence, low clinical suspicion, and transient nature of confirmatory testing, it is likely this remains an underdiagnosed syndrome. In the case discussed, subsequent chart review demonstrated 5 previous admissions in multiple hospitals for severe transient anion gap acidosis. The likelihood that 5‐oxoprolinemia was missed in each of these cases supports a lack of awareness of this syndrome. In this patient, the discussant appropriately identified the possibility of 5‐oxoproline toxicity, but felt ethylene glycol ingestion was more likely. As this case underscores, a cornerstone in the management of suspected ingestions is empiric treatment for the most likely etiologies. Here, treatment for acetaminophen overdose and for methanol or ethylene glycol were warranted, and fortunately also addressed the rarer possibility of 5‐oxoproline toxicity.
The mnemonic MUDPILES is commonly used to identify possible causes of life‐threatening anion gap metabolic acidosis, as such heuristics have benefits in rapidly generating a differential diagnosis to guide initial evaluation. Given the fact that the traditional letter P (paraldehyde) in MUDPILES is no longer clinically utilized, some authors have suggested replacing this with pyroglutamic acid (a synonym of 5‐oxoproline). Such a change may help providers who have ruled out other causes of a high anion gap metabolic acidosis, facilitating diagnosis of this life‐threatening syndrome. In any case, clinicians must be mindful that simple memory aids may mislead clinicians, and a complete differential diagnosis may require more than a mnemonic.
TEACHING POINTS
- Acetaminophen use, even at therapeutic levels, can lead to 5‐oxoprolinemia, a potentially lethal anion gap metabolic acidosis.
- 5‐oxoprolinemia is likely related to glutathione depletion, worsened by acetaminophen, malnutrition, renal insufficiency, female gender, and infection. This implies theoretical benefit from administration of NAC for glutathione repletion.
- Mnemonics can be useful, but have limitations by way of oversimplification. This case suggests that changing the letter P in MUDPILES from paraldehyde to pyroglutamic acid could reduce underdiagnosis.
Disclosure: Nothing to report.
- , , , , . 5‐oxoprolinemia causing elevated anion gap metabolic acidosis in the setting of acetaminophen use. J Emerg Med. 2012;43(1):54–57.
- , , , . What is the clinical significance of 5‐oxoproline (pyroglutamic acid) in high anion gap metabolic acidosis following paracetamol (acetaminophen) exposure? Clin Toxicol (Phila). 2013;51(9):817–827.
- , , , , . Increased anion gap metabolic acidosis as a result of 5‐oxoproline (pyroglutamic acid): a role for acetaminophen. Clin J Am Soc Nephrol. 2006;1(3):441–447.
- , , , et al. Recurrent high anion gap metabolic acidosis secondary to 5‐oxoproline (pyroglutamic acid). Am J Kidney Dis. 2005;46(1):e4–e10.
- , , . Profound metabolic acidosis from pyroglutamic acidemia: an underappreciated cause of high anion gap metabolic acidosis. CJEM. 2010;12(5):449–452.
A 51‐year‐old man presented to the emergency department after 1 day of progressive dyspnea and increasing confusion.
Acute dyspnea most commonly stems from a cardiac or pulmonary disorder such as heart failure, acute coronary syndrome, pneumonia, pulmonary embolism, or exacerbations of asthma or chronic obstructive pulmonary disease. Less frequent cardiopulmonary considerations include pericardial or pleural effusion, pneumothorax, aspiration, and upper airway obstruction. Dyspnea might also be the initial manifestation of profound anemia or metabolic acidosis.
The presence of confusion suggests either a severe presentation of any of the aforementioned possibilities (with confusion resulting from hypoxia, hypercapnia, or hypotension); a multiorgan illness such as sepsis, malignancy, thromboembolic disease, vasculitis, thyroid dysfunction, or toxic ingestion; or a metabolic derangement related to the underlying cause of dyspnea (for example, hypercalcemia or hyponatremia associated with lung cancer).
Twelve hours prior to presentation, he started to have visual hallucinations. He denied fever, chills, cough, chest discomfort, palpitations, weight gain, headache, neck pain, or weakness.
Visual hallucinations could result from a toxic‐metabolic encephalopathy, such as drug overdose or withdrawal, liver or kidney failure, or hypoxia. A structural brain abnormality may also manifest with visual hallucination. Acute onset at age 51 and the absence of auditory hallucinations argue against a neurodegenerative illness and a primary psychiatric disturbance, respectively.
Episodic hallucinations would support the possibility of seizures, monocular hallucinations would point to a retinal or ocular problem, and a description of yellow‐green hue would suggest a side effect of digoxin.
His past medical history was remarkable for diet‐controlled type 2 diabetes mellitus, hypertension, hyperlipidemia, and chronic low back pain. His medications included metoprolol tartrate 25 mg twice daily, omeprazole 40 mg daily, baclofen 15 mg twice daily, oxycodone 30 mg 3 times daily, and hydrocodone 10 mg/acetaminophen 325 mg, 2 tablets 3 times daily as needed for back pain. He was a smoker with a 30 pack‐year history. He had a history of alcohol and cocaine use, but denied any recent substance use. He had no known history of obstructive pulmonary disease.
The patient takes 3 medications well known to cause confusion and hallucinations (oxycodone, hydrocodone, and baclofen), especially when they accumulate due to excessive ingestion or impaired clearance. Although these medications may suppress ventilatory drive, dyspnea would not be a common presenting complaint. He has risk factors for ischemic heart disease and cardiomyopathy, and his smoking history raises the possibility of malignancy.
On exam, the patient's temperature was 94.4C, heart rate 128 beats per minute, respiration rate 28 breaths per minute, blood pressure 155/63 mm Hg, and oxygen saturation 100% while breathing ambient air. The patient was cachectic and appeared in moderate respiratory distress. His pupils were equal and reactive to light, and extraocular movements were intact. He did not have scleral icterus, or cervical or clavicular lymphadenopathy. His oropharynx was negative for erythema, edema, or exudate. His cardiovascular exam revealed a regular tachycardia without rubs or diastolic gallops. There was a 2/6 systolic murmur heard best at left sternal border, without radiation. He did not have jugular venous distention. His pulmonary exam was notable for tachypnea but with normal vesicular breath sounds throughout. He did not have stridor, wheezing, rhonchi, or rales. His abdomen had normal bowel tones and was soft without tenderness, distention, or organomegaly. His extremities were warm, revealed normal pulses, and no edema was present. His joints were cool to palpation, without effusion. On neurologic exam, he was oriented to person and place and able to answer yes/no questions, but unable to provide detailed history. His speech was fluent. His motor exam was without focal deficits. His skin was without any notable lesions.
The constellation of findings does not point to a specific toxidrome. The finding of warm extremities in a hypothermic patient suggests heat loss due to inappropriate peripheral vasodilation. In the absence of vasodilators or features of aortic insufficiency, sepsis becomes a leading consideration. Infection could result in hypothermia and altered sensorium, and accompanying lactic acidosis could trigger tachypnea.
Shortly after admission, he became more somnolent and developed progressive respiratory distress, requiring intubation. Arterial blood gas revealed a pH of 6.93, PaCO2<20 mm Hg, PaO2 127 mm Hg, and HCO3<5 mEq/L. Other laboratory results included a lactate of 4.1 mmol/L, blood urea nitrogen 49 mg/dL, creatinine 2.3 mg/dL (0.8 at 1 month prior), sodium level of 143 mmol/L, chloride of 106 mmol/L, and bicarbonate level of <5 mg/dL. His aspartate aminotransferase was 34 IU/L, alanine transaminase was 28 IU/L, total bilirubin was 0.6 mg/dL, International Normalized Ratio was 1.3. A complete blood count revealed a white blood cell count of 23,000/L, hemoglobin of 10.6 g/dL, and platelet count of 454,000/L. A urinalysis was unremarkable. Cultures of blood, urine, and sputum were collected. Head computed tomography was negative.
This patient has a combined anion gap and nongap metabolic acidosis, as well as respiratory alkalosis. Although his acute kidney failure could produce these 2 types of metabolic acidosis, the modest elevation of the serum creatinine is not commensurate with such profound acidosis. Similarly, sepsis without hypotension or more striking elevation in lactate levels would not account for the entirety of the acidosis. Severe diabetic ketoacidosis can result in profound metabolic acidosis, and marked hyperglycemia or hyperosmolarity could result in somnolence; however, his diabetes has been controlled without medication and there is no obvious precipitant for an episode of ketoacidosis.
Remaining causes of anion gap acidosis include ingestion of methanol, ethylene glycol, ethanol, or salicylates. A careful history of ingestions and medications from witnesses including any prehospital personnel might suggest a source of intoxication. Absent this information, the hypothermia favors an ingestion of an alcohol over salicylates, and the lack of urine crystals and the presence of prominent visual hallucinations would point more toward methanol poisoning than ethylene glycol. A serum osmolarity measurement would allow determination of the osmolar gap, which would be elevated in the setting of methanol or ethylene glycol poisoning. If he were this ill from ethanol, I would have expected to see evidence of hepatotoxicity.
I would administer sodium bicarbonate to reverse the acidosis and to promote renal clearance of salicylates, methanol, ethylene glycol, and their metabolites. Orogastric decontamination with activated charcoal should be considered. If the osmolar gap is elevated, I would also administer intravenous fomepizole to attempt to reverse methanol or ethylene glycol poisoning. I would not delay treatment while waiting for these serum levels to return.
Initial serologic toxicology performed in the emergency department revealed negative ethanol, salicylates, and ketones. His osmolar gap was 13 mOsm/kg. His acetaminophen level was 69 g/mL (normal <120 g/mL). A creatinine phosphokinase was 84 IU/L and myoglobin was 93 ng/mL. His subsequent serum toxicology screen was negative for methanol, ethylene glycol, isopropranol, and hippuric acid. Urine toxicology was positive for opiates, but negative for amphetamine, benzodiazepine, cannabinoid, and cocaine.
Serum and urine ketone assays typically involve the nitroprusside reaction and detect acetoacetate, but not ‐hydroxybutyrate, and can lead to negative test results early in diabetic or alcoholic ketoacidosis. However, the normal ethanol level argues against alcoholic ketoacidosis. Rare causes of elevated anion gap acidosis include toluene toxicity, acetaminophen poisoning, and ingestion of other alcohols. Toluene is metabolized to hippuric acid, and acetaminophen toxicity and associated glutathione depletion can lead to 5‐oxoproline accumulation, producing an anion gap. Patients who abuse alcohol are at risk for acetaminophen toxicity even at doses considered normal. However, this degree of encephalopathy would be unusual for acetaminophen toxicity unless liver failure had developed or unless there was another ingestion that might alter sensorium. Furthermore, the elevated osmolar gap is not a feature of acetaminophen poisoning. I would monitor liver enzyme tests and consider a serum ammonia level, but would not attribute the entire picture to acetaminophen.
The combination of elevated anion gap with an elevated osmolar gap narrows the diagnostic possibilities. Ingestion of several alcohols (ethanol, methanol, ethylene glycol, diethylene glycol) or toluene could produce these abnormalities. Of note, the osmolar gap is typically most markedly elevated early in methanol and ethylene glycol ingestions, and then as the parent compound is metabolized, the osmolar gap closes and the accumulation of metabolites produces the anion gap. Hallucinations are more common with methanol and toluene, and renal failure is more typical of ethylene glycol or toluene. The lack of oxalate crystalluria does not exclude ethylene glycol poisoning. Unfortunately, urine testing for oxalate crystals or fluorescein examination are neither sensitive nor specific enough to diagnosis ethylene glycol toxicity reliably. In most hospitals, assays used for serum testing for alcohols are insensitive, and require confirmation with gas chromatography performed at a specialty lab.
Additional history might reveal the likely culprit or culprits. Inhalant abuse including huffing would point to toluene or organic acid exposure. Solvent ingestion (eg, antifreeze, brake fluid) would suggest methanol or ethylene glycol. Absent this history, I remain suspicious for poisoning with methanol or ethylene glycol and would consider empiric treatment after urgent consultation with a medical toxicologist. A careful ophthalmologic exam might demonstrate characteristic features of methanol poisoning. Serum samples should be sent to a regional lab for analysis for alcohols and organic acids.
He was admitted to the intensive care unit, and empiric antibiotics started. He was empirically started on N‐acetylcysteine and sodium bicarbonate drips. However, his acidemia persisted and he required hemodialysis, which was initiated 12 hours after initial presentation. His acidemia and mental status quickly improved after hemodialysis. He was extubated on hospital day 2 and no longer required hemodialysis.
The differential diagnosis at this point consists of 3 main possibilities: ingestion of methanol, ethylene glycol, or inhalant abuse such as from toluene. The normal hippuric acid level points away from toluene, whereas serum levels can be misleading in the alcohol poisonings. Other discriminating features to consider include exposure history and unique clinical aspects. In this patient, an exposure history is lacking, but 4 clinical features stand out: visual hallucinations, acute kidney injury, mild lactic acidosis, and rapid improvement with hemodialysis. Both ethylene glycol and methanol toxicity may produce a mild lactic acidosis by increasing hepatic metabolism of pyruvate to lactate, and both are rapidly cleared by dialysis. Although it is tempting to place methanol at the top of the list of possibilities due to the report of visual hallucinations, the subjective visual complaints without objective exam corollaries (loss of visual acuity, abnormal pupillary reflexes, or optic disc hyperemia) are nonspecific and might be provoked by alcohol or an inhalant. Furthermore, the acute renal failure is much more typical of ethylene glycol, and thus I would consider ethylene glycol as being the more likely of the ingestions. Coingestion of multiple alcohols is a possibility, but it would be statistically less likely. Confirmation of ethylene glycol poisoning would consist of further insight into his exposures and measurement of levels using gas chromatography.
A urine sample from his emergency department presentation was sent to an outside lab for organic acid levels. Based on high clinical suspicion for 5‐oxoprolinemia (pyroglutamic acidemia) the patient was counseled to avoid any acetaminophen. His primary care provider was informed of this and acetaminophen was added as an adverse drug reaction. The patient left against medical advice soon after extubation. Following discharge, his 5‐oxoproline (pyroglutamic acid) level returned markedly elevated at greater than 10,000 mmol/mol creatinine (200 times the upper limit of normal).
Elevations in 5‐oxoproline levels in this patient most likely stem from glutathione depletion related to chronic acetaminophen use. Alcohol use and malnutrition may have heightened this patient's susceptibility. Despite the common occurrence of acetaminophen use in alcohol abusers or the malnourished, the rarity of severe 5‐oxoproline toxicity suggests unknown factors may be present in predisposed individuals, or under‐recognition. Although acetaminophen‐induced hepatotoxicity may occur along with 5‐oxoprolinemia, this does not always occur.
Several features led me away from this syndrome. First, its rarity lowered my pretest probability. Second, the lack of exposure history and details about the serum assays, specifically whether the measurements were confirmed by gas chromatography, reduced my confidence in eliminating more common ingestions. Third, several aspects proved to be less useful discriminating features: the mild elevation in osmolar gap, renal failure, and hallucinations, which in retrospect proved to be nonspecific.
The patient admitted that he had a longstanding use of acetaminophen in addition to using his girlfriend's acetaminophen‐hydrocodone. He had significant weight loss of over 50 pounds over the previous year, which he attributed to poor appetite. On further chart review, he had been admitted 3 times with a similar clinical presentation and recovered quickly with intensive and supportive care, with no etiology found at those times. He had 2 subsequent hospital admissions for altered mental status and respiratory failure, and his final hospitalization resulted in cardiac arrest and death.
DISCUSSION
5‐Oxoprolinemia is a rare, but potentially lethal cause of severe anion gap metabolic acidosis.[1, 2] The mechanism is thought to be impairment of glutathione metabolism, in the context of other predisposing factors. This can be a congenital error of metabolism, or can be acquired and exacerbated by acetaminophen use. Ingestion of acetaminophen leads to glutathione depletion, which in turn may precipitate accumulation of pyroglutamic acid and subsequent anion gap metabolic acidosis (Figure 1). Additional risk factors that may predispose patients to this condition include malnutrition, renal insufficiency, concurrent infection, and female gender.[1, 2, 3]
The diagnosis of 5‐oxoprolinemia is made via urine or serum organic acid analysis, testing routinely performed in pediatric populations when screening for congenital metabolic disorders. The pathophysiology suggests that obtaining a urine sample early in presentation, when acidosis is greatest, would lead to the highest 5‐oxoproline levels and best chance for diagnosis. Case patients have had normal levels prior to and in convalescent phases after the acute episode.[4] Given the long turnaround time for lab testing, presumptive diagnosis and treatment may be necessary.
Treatment of 5‐oxoprolinemia is primarily supportive, aimed at the metabolic acidosis. Fluid resuscitation and bicarbonate therapy are reasonable temporizing measures. Hemodialysis can clear 5‐oxoproline and may be indicated in severe acidosis.[5] Furthermore, the proposed pathophysiology suggests that administration of N‐acetylcysteine (NAC) may help to address the underlying process, but there are no trials to support a specific dosing regimen. However, given the fulminant presentation and common competing concern for acetaminophen toxicity, it is reasonable to initiate NAC aimed at treatment for possible acetaminophen overdose. Prevention of recurrence includes avoidance of acetaminophen, and counseling the patient to avoid acetaminophen in prescription combination medications and over‐the‐counter preparations.
Recent regulatory changes regarding acetaminophen/opioid combinations may reduce the incidence of 5‐oxoprolinemia. The US Food and Drug Administration has taken action to reduce adverse effects from acetaminophen exposure by limiting the amount of acetaminophen in opioid combination pills from 500 mg to a maximum of 325 mg per pill. This is aimed at preventing hepatotoxicity from ingestion of higher‐than‐recommended doses. However, clinicians should remember that 5‐oxoprolinemia can result from ingestion of acetaminophen at therapeutic levels.
Given its rare incidence, low clinical suspicion, and transient nature of confirmatory testing, it is likely this remains an underdiagnosed syndrome. In the case discussed, subsequent chart review demonstrated 5 previous admissions in multiple hospitals for severe transient anion gap acidosis. The likelihood that 5‐oxoprolinemia was missed in each of these cases supports a lack of awareness of this syndrome. In this patient, the discussant appropriately identified the possibility of 5‐oxoproline toxicity, but felt ethylene glycol ingestion was more likely. As this case underscores, a cornerstone in the management of suspected ingestions is empiric treatment for the most likely etiologies. Here, treatment for acetaminophen overdose and for methanol or ethylene glycol were warranted, and fortunately also addressed the rarer possibility of 5‐oxoproline toxicity.
The mnemonic MUDPILES is commonly used to identify possible causes of life‐threatening anion gap metabolic acidosis, as such heuristics have benefits in rapidly generating a differential diagnosis to guide initial evaluation. Given the fact that the traditional letter P (paraldehyde) in MUDPILES is no longer clinically utilized, some authors have suggested replacing this with pyroglutamic acid (a synonym of 5‐oxoproline). Such a change may help providers who have ruled out other causes of a high anion gap metabolic acidosis, facilitating diagnosis of this life‐threatening syndrome. In any case, clinicians must be mindful that simple memory aids may mislead clinicians, and a complete differential diagnosis may require more than a mnemonic.
TEACHING POINTS
- Acetaminophen use, even at therapeutic levels, can lead to 5‐oxoprolinemia, a potentially lethal anion gap metabolic acidosis.
- 5‐oxoprolinemia is likely related to glutathione depletion, worsened by acetaminophen, malnutrition, renal insufficiency, female gender, and infection. This implies theoretical benefit from administration of NAC for glutathione repletion.
- Mnemonics can be useful, but have limitations by way of oversimplification. This case suggests that changing the letter P in MUDPILES from paraldehyde to pyroglutamic acid could reduce underdiagnosis.
Disclosure: Nothing to report.
A 51‐year‐old man presented to the emergency department after 1 day of progressive dyspnea and increasing confusion.
Acute dyspnea most commonly stems from a cardiac or pulmonary disorder such as heart failure, acute coronary syndrome, pneumonia, pulmonary embolism, or exacerbations of asthma or chronic obstructive pulmonary disease. Less frequent cardiopulmonary considerations include pericardial or pleural effusion, pneumothorax, aspiration, and upper airway obstruction. Dyspnea might also be the initial manifestation of profound anemia or metabolic acidosis.
The presence of confusion suggests either a severe presentation of any of the aforementioned possibilities (with confusion resulting from hypoxia, hypercapnia, or hypotension); a multiorgan illness such as sepsis, malignancy, thromboembolic disease, vasculitis, thyroid dysfunction, or toxic ingestion; or a metabolic derangement related to the underlying cause of dyspnea (for example, hypercalcemia or hyponatremia associated with lung cancer).
Twelve hours prior to presentation, he started to have visual hallucinations. He denied fever, chills, cough, chest discomfort, palpitations, weight gain, headache, neck pain, or weakness.
Visual hallucinations could result from a toxic‐metabolic encephalopathy, such as drug overdose or withdrawal, liver or kidney failure, or hypoxia. A structural brain abnormality may also manifest with visual hallucination. Acute onset at age 51 and the absence of auditory hallucinations argue against a neurodegenerative illness and a primary psychiatric disturbance, respectively.
Episodic hallucinations would support the possibility of seizures, monocular hallucinations would point to a retinal or ocular problem, and a description of yellow‐green hue would suggest a side effect of digoxin.
His past medical history was remarkable for diet‐controlled type 2 diabetes mellitus, hypertension, hyperlipidemia, and chronic low back pain. His medications included metoprolol tartrate 25 mg twice daily, omeprazole 40 mg daily, baclofen 15 mg twice daily, oxycodone 30 mg 3 times daily, and hydrocodone 10 mg/acetaminophen 325 mg, 2 tablets 3 times daily as needed for back pain. He was a smoker with a 30 pack‐year history. He had a history of alcohol and cocaine use, but denied any recent substance use. He had no known history of obstructive pulmonary disease.
The patient takes 3 medications well known to cause confusion and hallucinations (oxycodone, hydrocodone, and baclofen), especially when they accumulate due to excessive ingestion or impaired clearance. Although these medications may suppress ventilatory drive, dyspnea would not be a common presenting complaint. He has risk factors for ischemic heart disease and cardiomyopathy, and his smoking history raises the possibility of malignancy.
On exam, the patient's temperature was 94.4C, heart rate 128 beats per minute, respiration rate 28 breaths per minute, blood pressure 155/63 mm Hg, and oxygen saturation 100% while breathing ambient air. The patient was cachectic and appeared in moderate respiratory distress. His pupils were equal and reactive to light, and extraocular movements were intact. He did not have scleral icterus, or cervical or clavicular lymphadenopathy. His oropharynx was negative for erythema, edema, or exudate. His cardiovascular exam revealed a regular tachycardia without rubs or diastolic gallops. There was a 2/6 systolic murmur heard best at left sternal border, without radiation. He did not have jugular venous distention. His pulmonary exam was notable for tachypnea but with normal vesicular breath sounds throughout. He did not have stridor, wheezing, rhonchi, or rales. His abdomen had normal bowel tones and was soft without tenderness, distention, or organomegaly. His extremities were warm, revealed normal pulses, and no edema was present. His joints were cool to palpation, without effusion. On neurologic exam, he was oriented to person and place and able to answer yes/no questions, but unable to provide detailed history. His speech was fluent. His motor exam was without focal deficits. His skin was without any notable lesions.
The constellation of findings does not point to a specific toxidrome. The finding of warm extremities in a hypothermic patient suggests heat loss due to inappropriate peripheral vasodilation. In the absence of vasodilators or features of aortic insufficiency, sepsis becomes a leading consideration. Infection could result in hypothermia and altered sensorium, and accompanying lactic acidosis could trigger tachypnea.
Shortly after admission, he became more somnolent and developed progressive respiratory distress, requiring intubation. Arterial blood gas revealed a pH of 6.93, PaCO2<20 mm Hg, PaO2 127 mm Hg, and HCO3<5 mEq/L. Other laboratory results included a lactate of 4.1 mmol/L, blood urea nitrogen 49 mg/dL, creatinine 2.3 mg/dL (0.8 at 1 month prior), sodium level of 143 mmol/L, chloride of 106 mmol/L, and bicarbonate level of <5 mg/dL. His aspartate aminotransferase was 34 IU/L, alanine transaminase was 28 IU/L, total bilirubin was 0.6 mg/dL, International Normalized Ratio was 1.3. A complete blood count revealed a white blood cell count of 23,000/L, hemoglobin of 10.6 g/dL, and platelet count of 454,000/L. A urinalysis was unremarkable. Cultures of blood, urine, and sputum were collected. Head computed tomography was negative.
This patient has a combined anion gap and nongap metabolic acidosis, as well as respiratory alkalosis. Although his acute kidney failure could produce these 2 types of metabolic acidosis, the modest elevation of the serum creatinine is not commensurate with such profound acidosis. Similarly, sepsis without hypotension or more striking elevation in lactate levels would not account for the entirety of the acidosis. Severe diabetic ketoacidosis can result in profound metabolic acidosis, and marked hyperglycemia or hyperosmolarity could result in somnolence; however, his diabetes has been controlled without medication and there is no obvious precipitant for an episode of ketoacidosis.
Remaining causes of anion gap acidosis include ingestion of methanol, ethylene glycol, ethanol, or salicylates. A careful history of ingestions and medications from witnesses including any prehospital personnel might suggest a source of intoxication. Absent this information, the hypothermia favors an ingestion of an alcohol over salicylates, and the lack of urine crystals and the presence of prominent visual hallucinations would point more toward methanol poisoning than ethylene glycol. A serum osmolarity measurement would allow determination of the osmolar gap, which would be elevated in the setting of methanol or ethylene glycol poisoning. If he were this ill from ethanol, I would have expected to see evidence of hepatotoxicity.
I would administer sodium bicarbonate to reverse the acidosis and to promote renal clearance of salicylates, methanol, ethylene glycol, and their metabolites. Orogastric decontamination with activated charcoal should be considered. If the osmolar gap is elevated, I would also administer intravenous fomepizole to attempt to reverse methanol or ethylene glycol poisoning. I would not delay treatment while waiting for these serum levels to return.
Initial serologic toxicology performed in the emergency department revealed negative ethanol, salicylates, and ketones. His osmolar gap was 13 mOsm/kg. His acetaminophen level was 69 g/mL (normal <120 g/mL). A creatinine phosphokinase was 84 IU/L and myoglobin was 93 ng/mL. His subsequent serum toxicology screen was negative for methanol, ethylene glycol, isopropranol, and hippuric acid. Urine toxicology was positive for opiates, but negative for amphetamine, benzodiazepine, cannabinoid, and cocaine.
Serum and urine ketone assays typically involve the nitroprusside reaction and detect acetoacetate, but not ‐hydroxybutyrate, and can lead to negative test results early in diabetic or alcoholic ketoacidosis. However, the normal ethanol level argues against alcoholic ketoacidosis. Rare causes of elevated anion gap acidosis include toluene toxicity, acetaminophen poisoning, and ingestion of other alcohols. Toluene is metabolized to hippuric acid, and acetaminophen toxicity and associated glutathione depletion can lead to 5‐oxoproline accumulation, producing an anion gap. Patients who abuse alcohol are at risk for acetaminophen toxicity even at doses considered normal. However, this degree of encephalopathy would be unusual for acetaminophen toxicity unless liver failure had developed or unless there was another ingestion that might alter sensorium. Furthermore, the elevated osmolar gap is not a feature of acetaminophen poisoning. I would monitor liver enzyme tests and consider a serum ammonia level, but would not attribute the entire picture to acetaminophen.
The combination of elevated anion gap with an elevated osmolar gap narrows the diagnostic possibilities. Ingestion of several alcohols (ethanol, methanol, ethylene glycol, diethylene glycol) or toluene could produce these abnormalities. Of note, the osmolar gap is typically most markedly elevated early in methanol and ethylene glycol ingestions, and then as the parent compound is metabolized, the osmolar gap closes and the accumulation of metabolites produces the anion gap. Hallucinations are more common with methanol and toluene, and renal failure is more typical of ethylene glycol or toluene. The lack of oxalate crystalluria does not exclude ethylene glycol poisoning. Unfortunately, urine testing for oxalate crystals or fluorescein examination are neither sensitive nor specific enough to diagnosis ethylene glycol toxicity reliably. In most hospitals, assays used for serum testing for alcohols are insensitive, and require confirmation with gas chromatography performed at a specialty lab.
Additional history might reveal the likely culprit or culprits. Inhalant abuse including huffing would point to toluene or organic acid exposure. Solvent ingestion (eg, antifreeze, brake fluid) would suggest methanol or ethylene glycol. Absent this history, I remain suspicious for poisoning with methanol or ethylene glycol and would consider empiric treatment after urgent consultation with a medical toxicologist. A careful ophthalmologic exam might demonstrate characteristic features of methanol poisoning. Serum samples should be sent to a regional lab for analysis for alcohols and organic acids.
He was admitted to the intensive care unit, and empiric antibiotics started. He was empirically started on N‐acetylcysteine and sodium bicarbonate drips. However, his acidemia persisted and he required hemodialysis, which was initiated 12 hours after initial presentation. His acidemia and mental status quickly improved after hemodialysis. He was extubated on hospital day 2 and no longer required hemodialysis.
The differential diagnosis at this point consists of 3 main possibilities: ingestion of methanol, ethylene glycol, or inhalant abuse such as from toluene. The normal hippuric acid level points away from toluene, whereas serum levels can be misleading in the alcohol poisonings. Other discriminating features to consider include exposure history and unique clinical aspects. In this patient, an exposure history is lacking, but 4 clinical features stand out: visual hallucinations, acute kidney injury, mild lactic acidosis, and rapid improvement with hemodialysis. Both ethylene glycol and methanol toxicity may produce a mild lactic acidosis by increasing hepatic metabolism of pyruvate to lactate, and both are rapidly cleared by dialysis. Although it is tempting to place methanol at the top of the list of possibilities due to the report of visual hallucinations, the subjective visual complaints without objective exam corollaries (loss of visual acuity, abnormal pupillary reflexes, or optic disc hyperemia) are nonspecific and might be provoked by alcohol or an inhalant. Furthermore, the acute renal failure is much more typical of ethylene glycol, and thus I would consider ethylene glycol as being the more likely of the ingestions. Coingestion of multiple alcohols is a possibility, but it would be statistically less likely. Confirmation of ethylene glycol poisoning would consist of further insight into his exposures and measurement of levels using gas chromatography.
A urine sample from his emergency department presentation was sent to an outside lab for organic acid levels. Based on high clinical suspicion for 5‐oxoprolinemia (pyroglutamic acidemia) the patient was counseled to avoid any acetaminophen. His primary care provider was informed of this and acetaminophen was added as an adverse drug reaction. The patient left against medical advice soon after extubation. Following discharge, his 5‐oxoproline (pyroglutamic acid) level returned markedly elevated at greater than 10,000 mmol/mol creatinine (200 times the upper limit of normal).
Elevations in 5‐oxoproline levels in this patient most likely stem from glutathione depletion related to chronic acetaminophen use. Alcohol use and malnutrition may have heightened this patient's susceptibility. Despite the common occurrence of acetaminophen use in alcohol abusers or the malnourished, the rarity of severe 5‐oxoproline toxicity suggests unknown factors may be present in predisposed individuals, or under‐recognition. Although acetaminophen‐induced hepatotoxicity may occur along with 5‐oxoprolinemia, this does not always occur.
Several features led me away from this syndrome. First, its rarity lowered my pretest probability. Second, the lack of exposure history and details about the serum assays, specifically whether the measurements were confirmed by gas chromatography, reduced my confidence in eliminating more common ingestions. Third, several aspects proved to be less useful discriminating features: the mild elevation in osmolar gap, renal failure, and hallucinations, which in retrospect proved to be nonspecific.
The patient admitted that he had a longstanding use of acetaminophen in addition to using his girlfriend's acetaminophen‐hydrocodone. He had significant weight loss of over 50 pounds over the previous year, which he attributed to poor appetite. On further chart review, he had been admitted 3 times with a similar clinical presentation and recovered quickly with intensive and supportive care, with no etiology found at those times. He had 2 subsequent hospital admissions for altered mental status and respiratory failure, and his final hospitalization resulted in cardiac arrest and death.
DISCUSSION
5‐Oxoprolinemia is a rare, but potentially lethal cause of severe anion gap metabolic acidosis.[1, 2] The mechanism is thought to be impairment of glutathione metabolism, in the context of other predisposing factors. This can be a congenital error of metabolism, or can be acquired and exacerbated by acetaminophen use. Ingestion of acetaminophen leads to glutathione depletion, which in turn may precipitate accumulation of pyroglutamic acid and subsequent anion gap metabolic acidosis (Figure 1). Additional risk factors that may predispose patients to this condition include malnutrition, renal insufficiency, concurrent infection, and female gender.[1, 2, 3]
The diagnosis of 5‐oxoprolinemia is made via urine or serum organic acid analysis, testing routinely performed in pediatric populations when screening for congenital metabolic disorders. The pathophysiology suggests that obtaining a urine sample early in presentation, when acidosis is greatest, would lead to the highest 5‐oxoproline levels and best chance for diagnosis. Case patients have had normal levels prior to and in convalescent phases after the acute episode.[4] Given the long turnaround time for lab testing, presumptive diagnosis and treatment may be necessary.
Treatment of 5‐oxoprolinemia is primarily supportive, aimed at the metabolic acidosis. Fluid resuscitation and bicarbonate therapy are reasonable temporizing measures. Hemodialysis can clear 5‐oxoproline and may be indicated in severe acidosis.[5] Furthermore, the proposed pathophysiology suggests that administration of N‐acetylcysteine (NAC) may help to address the underlying process, but there are no trials to support a specific dosing regimen. However, given the fulminant presentation and common competing concern for acetaminophen toxicity, it is reasonable to initiate NAC aimed at treatment for possible acetaminophen overdose. Prevention of recurrence includes avoidance of acetaminophen, and counseling the patient to avoid acetaminophen in prescription combination medications and over‐the‐counter preparations.
Recent regulatory changes regarding acetaminophen/opioid combinations may reduce the incidence of 5‐oxoprolinemia. The US Food and Drug Administration has taken action to reduce adverse effects from acetaminophen exposure by limiting the amount of acetaminophen in opioid combination pills from 500 mg to a maximum of 325 mg per pill. This is aimed at preventing hepatotoxicity from ingestion of higher‐than‐recommended doses. However, clinicians should remember that 5‐oxoprolinemia can result from ingestion of acetaminophen at therapeutic levels.
Given its rare incidence, low clinical suspicion, and transient nature of confirmatory testing, it is likely this remains an underdiagnosed syndrome. In the case discussed, subsequent chart review demonstrated 5 previous admissions in multiple hospitals for severe transient anion gap acidosis. The likelihood that 5‐oxoprolinemia was missed in each of these cases supports a lack of awareness of this syndrome. In this patient, the discussant appropriately identified the possibility of 5‐oxoproline toxicity, but felt ethylene glycol ingestion was more likely. As this case underscores, a cornerstone in the management of suspected ingestions is empiric treatment for the most likely etiologies. Here, treatment for acetaminophen overdose and for methanol or ethylene glycol were warranted, and fortunately also addressed the rarer possibility of 5‐oxoproline toxicity.
The mnemonic MUDPILES is commonly used to identify possible causes of life‐threatening anion gap metabolic acidosis, as such heuristics have benefits in rapidly generating a differential diagnosis to guide initial evaluation. Given the fact that the traditional letter P (paraldehyde) in MUDPILES is no longer clinically utilized, some authors have suggested replacing this with pyroglutamic acid (a synonym of 5‐oxoproline). Such a change may help providers who have ruled out other causes of a high anion gap metabolic acidosis, facilitating diagnosis of this life‐threatening syndrome. In any case, clinicians must be mindful that simple memory aids may mislead clinicians, and a complete differential diagnosis may require more than a mnemonic.
TEACHING POINTS
- Acetaminophen use, even at therapeutic levels, can lead to 5‐oxoprolinemia, a potentially lethal anion gap metabolic acidosis.
- 5‐oxoprolinemia is likely related to glutathione depletion, worsened by acetaminophen, malnutrition, renal insufficiency, female gender, and infection. This implies theoretical benefit from administration of NAC for glutathione repletion.
- Mnemonics can be useful, but have limitations by way of oversimplification. This case suggests that changing the letter P in MUDPILES from paraldehyde to pyroglutamic acid could reduce underdiagnosis.
Disclosure: Nothing to report.
- , , , , . 5‐oxoprolinemia causing elevated anion gap metabolic acidosis in the setting of acetaminophen use. J Emerg Med. 2012;43(1):54–57.
- , , , . What is the clinical significance of 5‐oxoproline (pyroglutamic acid) in high anion gap metabolic acidosis following paracetamol (acetaminophen) exposure? Clin Toxicol (Phila). 2013;51(9):817–827.
- , , , , . Increased anion gap metabolic acidosis as a result of 5‐oxoproline (pyroglutamic acid): a role for acetaminophen. Clin J Am Soc Nephrol. 2006;1(3):441–447.
- , , , et al. Recurrent high anion gap metabolic acidosis secondary to 5‐oxoproline (pyroglutamic acid). Am J Kidney Dis. 2005;46(1):e4–e10.
- , , . Profound metabolic acidosis from pyroglutamic acidemia: an underappreciated cause of high anion gap metabolic acidosis. CJEM. 2010;12(5):449–452.
- , , , , . 5‐oxoprolinemia causing elevated anion gap metabolic acidosis in the setting of acetaminophen use. J Emerg Med. 2012;43(1):54–57.
- , , , . What is the clinical significance of 5‐oxoproline (pyroglutamic acid) in high anion gap metabolic acidosis following paracetamol (acetaminophen) exposure? Clin Toxicol (Phila). 2013;51(9):817–827.
- , , , , . Increased anion gap metabolic acidosis as a result of 5‐oxoproline (pyroglutamic acid): a role for acetaminophen. Clin J Am Soc Nephrol. 2006;1(3):441–447.
- , , , et al. Recurrent high anion gap metabolic acidosis secondary to 5‐oxoproline (pyroglutamic acid). Am J Kidney Dis. 2005;46(1):e4–e10.
- , , . Profound metabolic acidosis from pyroglutamic acidemia: an underappreciated cause of high anion gap metabolic acidosis. CJEM. 2010;12(5):449–452.
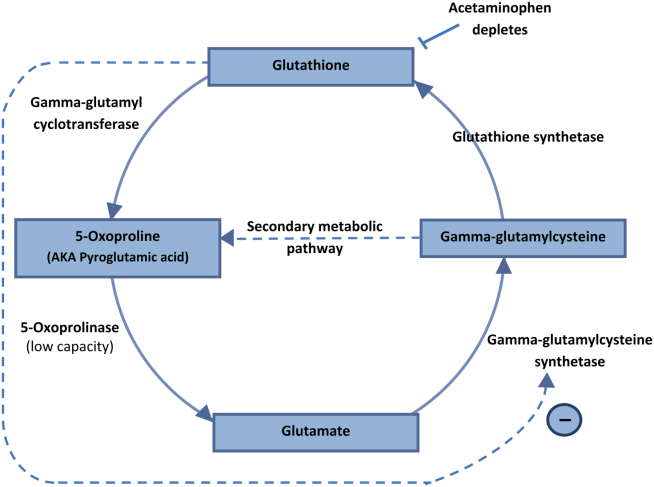
Stranger than Fiction
A 65‐year‐old man suffered a myocardial infarction (MI) while traveling in Thailand. After 7 days of recovery, the patient departed for his home in the United States. He developed substernal, nonexertional, inspiratory chest pain and shortness of breath during his return flight and presented directly to an emergency room after arrival.
Initially, the evaluation should focus on life‐threatening diagnoses and not be distracted by the travel history. The immediate diagnostic concerns are active cardiac ischemia, complications of MI, and pulmonary embolus. Other cardiac causes of dyspnea include ischemic mitral regurgitation, postinfarction pericarditis with or without pericardial effusion, and heart failure. Mechanical complications of infarction, such as left ventricular free wall rupture or rupture of the interventricular septum, can occur in this time frame and are associated with significant morbidity. Pneumothorax may be precipitated by air travel, especially in patients with underlying lung disease. The immobilization associated with long airline flights is a risk factor for thromboembolic disease, which is classically associated with pleuritic chest pain. Inspiratory chest pain is also associated with inflammatory processes involving the pericardium or pleura. If pneumonia, pericarditis, or pleural effusion is present, details of his travel history will become more important in his evaluation.
The patient elaborated that he spent 10 days in Thailand. On the third day of his trip he developed severe chest pain while hiking toward a waterfall in a rural northern district. He was transferred to a large private hospital, where he received a stent in the proximal left anterior descending coronary artery 4 hours after symptom onset. At discharge he was prescribed ticagrelor 90 mg twice daily and daily doses of losartan 50 mg, furosemide 20 mg, spironolactone 12.5 mg, aspirin 81 mg, ivabradine 2.5 mg, and pravastatin 40 mg. He had also been taking doxycycline for malaria prophylaxis since departing the United States.
His past medical history was notable for hypertension and hyperlipidemia. The patient was a lifelong nonsmoker, did not use illicit substances, and consumed no more than 2 alcoholic beverages per day. He denied cough, fevers, chills, diaphoresis, weight loss, recent upper respiratory infection, abdominal pain, hematuria, and nausea. However, he reported exertional dyspnea following his MI and nonbloody diarrhea that occurred a few days prior to his return flight and resolved without intervention.
The remainder of his past medical history confirms that he received appropriate post‐MI care, but does not substantially alter the high priority concerns in his differential diagnosis. Diarrhea may occur in up to 50% of international travelers, and is especially common when returning from Southeast Asia or the Indian subcontinent. Disease processes that may explain diarrhea and subsequent dyspnea include intestinal infections that spread to the lung (eg, ascariasis and Loeffler syndrome), infection that precipitates neuromuscular weakness (eg, Campylobacter and Guillain‐Barr syndrome), or infection that precipitates heart failure (eg, coxsackievirus, myocarditis).
On admission, his temperature was 36.2C, heart rate 91 beats per minute, blood pressure 135/81 mm Hg, respiratory rate 16 breaths per minute, and oxygen saturation 98% on room air. Cardiac exam revealed a regular rhythm without rubs, murmurs, or diastolic gallops. He had no jugular venous distention, and no lower extremity edema. His distal pulses were equal and palpable throughout. Pulmonary exam was notable for decreased breath sounds at both bases without wheezing, rhonchi, or crackles noted. He had no rashes, joint effusions, or jaundice. Abdominal and neurologic examinations were unremarkable.
Diminished breath sounds may suggest atelectasis or pleural effusion; the latter could account for the patient's inspiratory chest pain. A chest radiograph is essential to evaluate this finding further. The physical examination is not suggestive of decompensated heart failure; measurement of serum brain natriuretic peptide level would further exclude that diagnosis.
Laboratory evaluation revealed a leukocytosis of 16,000/L, with 76% polymorphonuclear cells and 12% lymphocytes without eosinophils or band forms; a hematocrit of 38%; and a platelet count of 363,000/L. The patient had a creatinine of 1.6 mg/dL, potassium of 2.7 mEq/L, and a troponin‐I of 1.0 ng/mL (normal <0.40 ng/mL), with the remainder of the routine serum chemistries within normal limits. An electrocardiogram (ECG) showed QS complexes in the anteroseptal leads, and a chest radiograph showed bibasilar consolidations and a left pleural effusion. A ventilation‐perfusion scan of the chest was performed to evaluate for pulmonary embolism, and was interpreted as low probability. Transthoracic echocardiography demonstrated severe left ventricular systolic dysfunction with anterior wall akinesis, and an aneurysmal left ventricle with an apical thrombus. No significant valvular pathology or other structural defects were noted.
The ECG and echocardiogram confirm the history of a large anteroseptal infarction with severe left ventricular dysfunction. Serial troponin testing would be reasonable. However, the absence of any acute ischemic ECG changes, typical angina symptoms, and a relatively normal troponin level all suggest his chest pain does not represent active ischemia. His low abnormal troponin‐I is consistent with slow resolution after a large ischemic event in the recent past, and his anterior wall akinesis is consistent with prior infarction in the territory of his culprit left anterior descending coronary artery.
Although acute cardiac conditions appear less likely, the brisk leukocytosis in a returned traveler prompts consideration of infection. His lung consolidations could represent either new or resolving pneumonia. The complete absence of cough and fever is unusual for pneumonia, yet clinical findings are not as sensitive as chest radiograph for this diagnosis. At this point, typical organisms as well as uncommon pathogens associated with diarrhea or his travel history should be included in the differential.
After 24 hours, the patient was discharged on warfarin to treat the apical thrombus and moxifloxacin for a presumed community‐acquired pneumonia. Eight days after discharge, the patient visited his primary care physician with improving, but not resolved, shortness of breath and pleuritic pain despite completing the 7‐day course of moxifloxacin. A chest radiograph showed a large posterior left basal pleural fluid collection, increased from previous.
In the setting of a recent infection, the symptoms and radiographic findings suggest a complicated parapneumonic effusion or empyema. Failure to drain a previously seeded fluid collection leaves bacterial pathogens susceptible to moxifloxacin on the differential, including Streptococcus pneumoniae, Staphylococcus aureus, Legionella species, and other enterobacteriaciae (eg, Klebsiella pneumoniae).
The indolent course should also prompt consideration of more unusual pathogens, including roundworms (such as Ascaris) or lung flukes (Paragonimus), either of which can cause a lung infection without traditional pneumonia symptoms. Tuberculosis tends to present months (or years) after exposure. Older adults may manifest primary pulmonary tuberculosis with lower lobe infiltrates, consistent with this presentation. However, moxifloxacin is quite active against tuberculosis, and although single drug therapy would not be expected to cure the patient, it would be surprising for him to progress this quickly on moxifloxacin.
In northern Thailand, Burkholderia pseudomallei is a common cause of bacteremic pneumonia. The organism often has high‐level resistance to fluoroquinolones, and may present in a more insidious fashion than other causes of community‐acquired pneumonia. Although infection with B pseudomallei (melioidosis) can occasionally mimic apical pulmonary tuberculosis and may present after a prolonged latent period, it most commonly manifests as an acute pneumonia.
The patient was prescribed 10 days of amoxicillin‐clavulanic acid and clindamycin, and decubitus films were ordered to assess the effusion. These films, obtained 5 days later, showed a persistent pleural effusion. Subsequent ultrasound demonstrated loculated fluid, but a thoracentesis was not performed at that time due to the patient's therapeutic international normalized ratio and dual antiplatelet therapy.
The loculation further suggests a complicated parapneumonic effusion or empyema. Clindamycin adds very little to amoxicillin‐clavulanate as far as coverage of oral anaerobes or common pneumonia pathogens and may add to the risk of antibiotic side effects. A susceptible organism might not clear because of failure to drain this collection; if undertreated bacterial infection is suspected, tube thoracentesis is the established standard of care. However, the protracted course of illness makes untreated pyogenic bacterial infections unlikely.
At this point, the top 2 diagnostic considerations are Paragonimus westermani and B pseudomallei. P westermani is initially ingested, usually from an undercooked freshwater crustacean. Infected patients may experience a brief diarrheal illness, as this patient reported. However, infected patients typically have a brisk peripheral eosinophilia.
Melioidosis is thus the leading concern. Amoxicillin‐clavulanate is active against many strains of B pseudomallei, so the failure of the patient to worsen could be seen as a partial treatment and supports this diagnosis. However, as prolonged therapy is necessary for complete eradication of B pseudomallei, a definitive, culture‐based diagnosis should be established before committing the patient to months of antibiotics.
After completing 10 days of clindamycin and amoxicillin‐clavulanate, the patient reported improvement of his pleuritic pain, and repeat physical exam suggested interval decrease in the size of the effusion. Two days later, the patient began experiencing dysuria that persisted despite 3 days of nitrofurantoin.
Melioidosis can also involve the genitourinary tract. Hematogenous spread of B pseudomallei can seed a number of visceral organs including the bladder, joints, and bones. Men with suspected urinary infection should be evaluated for the possibility of prostatitis, an infection with considerable morbidity that requires extended therapy. This gentleman should have a prostate exam, and blood and urine cultures should be collected if prostatitis is suspected. Empiric antibiotics are not recommended without culture in a patient with complicated urinary tract infection.
Prostate exam was unremarkable. A urine culture grew a gram‐negative rod identified as B pseudomallei. Because B pseudomallei can cause fulminant sepsis, the infectious disease consultant requested that he return for admission, further evaluation, and initiation of intravenous antibiotics. Computed tomography (CT) of the chest, abdomen, and pelvis revealed multiple pulmonary nodules, a persistent left pleural effusion, and a rim‐enhancing hypodensity in the prostate consistent with abscess (Figure 1). Blood and pleural fluid cultures were negative.
Initial treatment for a patient with severe or metastatic B pseudomallei infection requires high‐dose intravenous antibiotic therapy. Ceftazidime, imipenem, and meropenem are the best studied agents for this purpose. Surgical drainage should be considered for the abscess. Following the completion of intensive intravenous therapy, relapse rates are high unless a longer‐term, consolidation therapy is pursued. Trimethoprim‐sulfamethoxazole is the recommended agent.
The patient was treated with high‐dose ceftazidime for 2 weeks, followed by 6 months of high‐dose oral trimethoprim‐sulfamethoxazole. His symptoms resolved, and 7 months after presentation, he continued to feel well.
DISCUSSION
Melioidosis refers to any infection caused by B pseudomallei, a gram‐negative bacillus found in soil and water, most commonly in Southeast Asia and Australia.[1] It is an important cause of pneumonia in endemic regions; in Thailand, the incidence is as high as 12 cases per 100,000 people, and it is the third leading infectious cause of death, following human immunodeficiency virus and tuberculosis.[2] However, it occurs only as an imported infection in the United States and remains an unfamiliar infection for many US medical practitioners. Melioidosis should be considered in patients returning from endemic regions presenting with sepsis, pneumonia, urinary symptoms, or abscesses.
B pseudomallei can be transmitted to humans through exposure to contaminated soil or water via ingestion, inhalation, or percutaneous inoculation.[1] Outbreaks typically occur during the rainy season and after typhoons.[1, 3] Presumably, this patient's exposure to B pseudomallei occurred while hiking and wading in freshwater lakes and waterfalls. Although hospital‐acquired melioidosis has not been reported, and isolation precautions are not necessary, rare cases of disease acquired via laboratory exposure have been reported among US healthcare workers. Clinicians suspecting melioidosis should alert the receiving laboratory.[4]
The treatment course for melioidosis is lengthy and should involve consultation with an infectious disease specialist. B pseudomallei is known to be resistant to penicillin, first‐ and second‐generation cephalosporins, and moxifloxacin. The standard treatment includes 10 to 14 days of intravenous ceftazidime, meropenem, or imipenem, and then trimethoprim‐sulfamethoxazole for 3 to 6 months.[1] Treatment should be guided by culture susceptibility data when available. There are reports of B pseudomallei having different resistance patterns within the same host; clinicians should culture all drained fluid collections and tailor antibiotics to the most resistant strain recovered.[5, 6] Although melioidosis is a life‐threatening infection, previously healthy patients have an excellent prognosis assuming prompt diagnosis and treatment are provided.[3]
After excluding common causes of chest pain, the discussant identified the need to definitively establish a microbiologic diagnosis by obtaining pleural fluid. Although common clinical scenarios can often be treated with guideline‐supported empiric antibiotics, the use of serial courses of empiric antibiotics should be carefully questioned and is generally discouraged. Specific data to prove or disprove the presence of infection should be obtained before exposing a patient to the risks of multiple drugs or prolonged antibiotic therapy, as well as the risks of delayed (or missed) diagnosis. Unfortunately, a complete evaluation was delayed by clinical contraindications to diagnostic thoracentesis, and a definitive diagnosis was reached only after development of more widespread symptoms.
This patient's protean presentation is not surprising given his ultimate diagnosis. B pseudomallei has been termed the great mimicker, as disease presentation and organ involvement can vary from an indolent localized infection to acute severe sepsis.[7] Pneumonia and genitourinary infections are the most common manifestations, although skin infections, bacteremia, septic arthritis, and neurologic disease are also possible.[1, 3] In addition, melioidosis may develop after a lengthy incubation. In a case series, confirmed incubation periods ranged from 1 to 21 days (mean, 9 days); however, cases of chronic (>2 months) infection, mimicking tuberculosis, are estimated to occur in about 12% of cases.[4] B pseudomallei is also capable of causing reactivation disease, similar to tuberculosis. It was referred to as the Vietnamese time bomb when US Vietnam War veterans, exposed to the disease when helicopters aerosolized the bacteria in the soil, developed the disease only after their return to the United States.[8] Fortunately, only a tiny fraction of the quarter‐million soldiers with serologically confirmed exposure to the bacteria ultimately developed disease.
In The Adventure of the Dying Detective, Sherlock Holmes fakes a serious illness characterized by shortness of breath and weakness to trick an adversary into confessing to murder. The abrupt, crippling infection mimicked by Holmes is thought by some to be melioidosis.[9, 10] Conan Doyle's story was published in 1913, a year after melioidosis was first reported in the medical literature, and the exotic, protean infection may well have sparked Doyle's imagination. However, this patient's case of melioidosis proved stranger than fiction in its untimely concomitant development with an MI. Cracking our case required imagination and nimble thinking to avoid a number of cognitive pitfalls. The patient's recent MI anchored reasoning at his initial presentation, and the initial diagnosis of community‐acquired pneumonia raised the danger of premature closure. Reaching the correct diagnosis required an open mind, a detailed travel history, and firm microbiologic evidence. Hospitalists need not be expert in the health risks of travel to specific foreign destinations, but investigating those risks can hasten proper diagnosis and treatment.
TEACHING POINTS
- Melioidosis should be considered in patients returning from endemic regions who present with sepsis, pneumonia, urinary symptoms, or an abscess.
- For patients with a loculated parapneumonic effusion, tube thoracentesis for culture and drainage is the standard of care for diagnosis and treatment.
- Culture identification and antibiotic sensitivities are critical for management of B pseudomallei, because prolonged antibiotic treatment is needed.
Disclosure
Nothing to report.
- , , . Melioidosis. N Engl J Med. 2012;367(11):1035–1044.
- , , , et al. Increasing incidence of human melioidosis in Northeast Thailand. Am J Trop Med Hyg. 2010;82(6):1113–1117.
- , , . The epidemiology and clinical spectrum of melioidosis: 540 cases from the 20 year Darwin prospective study. PLoS Negl Trop Dis. 2010;4(11):e900.
- , , , et al. Management of accidental laboratory exposure to Burkholderia pseudomallei and B. mallei. Emerg Infect Dis. 2008;14(7):e2.
- , , . Variations in ceftazidime and amoxicillin‐clavulanate susceptibilities within a clonal infection of Burkholderia pseudomallei. J Clin Microbiol. 2009;47(5):1556–1558.
- , , , et al. Within‐host evolution of Burkholderia pseudomallei in four cases of acute melioidosis. PLoS Pathog. 2010;6(1):e1000725.
- , , , , . Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat Rev Microbiol. 2006;4(4):272–282.
- , . Melioidosis. In: Dembeck ZF, ed. Medical Aspects of Biological Warfare. 2nd ed. Washington, DC: Office of the Surgeon General; 2007:146–166.
- . Sherlock Holmes and a biological weapon. J R Soc Med. 2002;95(2):101–103.
- . Sherlock Holmes and tropical medicine: a centennial appraisal. Am J Trop Med Hyg. 1994;50:99–101.
A 65‐year‐old man suffered a myocardial infarction (MI) while traveling in Thailand. After 7 days of recovery, the patient departed for his home in the United States. He developed substernal, nonexertional, inspiratory chest pain and shortness of breath during his return flight and presented directly to an emergency room after arrival.
Initially, the evaluation should focus on life‐threatening diagnoses and not be distracted by the travel history. The immediate diagnostic concerns are active cardiac ischemia, complications of MI, and pulmonary embolus. Other cardiac causes of dyspnea include ischemic mitral regurgitation, postinfarction pericarditis with or without pericardial effusion, and heart failure. Mechanical complications of infarction, such as left ventricular free wall rupture or rupture of the interventricular septum, can occur in this time frame and are associated with significant morbidity. Pneumothorax may be precipitated by air travel, especially in patients with underlying lung disease. The immobilization associated with long airline flights is a risk factor for thromboembolic disease, which is classically associated with pleuritic chest pain. Inspiratory chest pain is also associated with inflammatory processes involving the pericardium or pleura. If pneumonia, pericarditis, or pleural effusion is present, details of his travel history will become more important in his evaluation.
The patient elaborated that he spent 10 days in Thailand. On the third day of his trip he developed severe chest pain while hiking toward a waterfall in a rural northern district. He was transferred to a large private hospital, where he received a stent in the proximal left anterior descending coronary artery 4 hours after symptom onset. At discharge he was prescribed ticagrelor 90 mg twice daily and daily doses of losartan 50 mg, furosemide 20 mg, spironolactone 12.5 mg, aspirin 81 mg, ivabradine 2.5 mg, and pravastatin 40 mg. He had also been taking doxycycline for malaria prophylaxis since departing the United States.
His past medical history was notable for hypertension and hyperlipidemia. The patient was a lifelong nonsmoker, did not use illicit substances, and consumed no more than 2 alcoholic beverages per day. He denied cough, fevers, chills, diaphoresis, weight loss, recent upper respiratory infection, abdominal pain, hematuria, and nausea. However, he reported exertional dyspnea following his MI and nonbloody diarrhea that occurred a few days prior to his return flight and resolved without intervention.
The remainder of his past medical history confirms that he received appropriate post‐MI care, but does not substantially alter the high priority concerns in his differential diagnosis. Diarrhea may occur in up to 50% of international travelers, and is especially common when returning from Southeast Asia or the Indian subcontinent. Disease processes that may explain diarrhea and subsequent dyspnea include intestinal infections that spread to the lung (eg, ascariasis and Loeffler syndrome), infection that precipitates neuromuscular weakness (eg, Campylobacter and Guillain‐Barr syndrome), or infection that precipitates heart failure (eg, coxsackievirus, myocarditis).
On admission, his temperature was 36.2C, heart rate 91 beats per minute, blood pressure 135/81 mm Hg, respiratory rate 16 breaths per minute, and oxygen saturation 98% on room air. Cardiac exam revealed a regular rhythm without rubs, murmurs, or diastolic gallops. He had no jugular venous distention, and no lower extremity edema. His distal pulses were equal and palpable throughout. Pulmonary exam was notable for decreased breath sounds at both bases without wheezing, rhonchi, or crackles noted. He had no rashes, joint effusions, or jaundice. Abdominal and neurologic examinations were unremarkable.
Diminished breath sounds may suggest atelectasis or pleural effusion; the latter could account for the patient's inspiratory chest pain. A chest radiograph is essential to evaluate this finding further. The physical examination is not suggestive of decompensated heart failure; measurement of serum brain natriuretic peptide level would further exclude that diagnosis.
Laboratory evaluation revealed a leukocytosis of 16,000/L, with 76% polymorphonuclear cells and 12% lymphocytes without eosinophils or band forms; a hematocrit of 38%; and a platelet count of 363,000/L. The patient had a creatinine of 1.6 mg/dL, potassium of 2.7 mEq/L, and a troponin‐I of 1.0 ng/mL (normal <0.40 ng/mL), with the remainder of the routine serum chemistries within normal limits. An electrocardiogram (ECG) showed QS complexes in the anteroseptal leads, and a chest radiograph showed bibasilar consolidations and a left pleural effusion. A ventilation‐perfusion scan of the chest was performed to evaluate for pulmonary embolism, and was interpreted as low probability. Transthoracic echocardiography demonstrated severe left ventricular systolic dysfunction with anterior wall akinesis, and an aneurysmal left ventricle with an apical thrombus. No significant valvular pathology or other structural defects were noted.
The ECG and echocardiogram confirm the history of a large anteroseptal infarction with severe left ventricular dysfunction. Serial troponin testing would be reasonable. However, the absence of any acute ischemic ECG changes, typical angina symptoms, and a relatively normal troponin level all suggest his chest pain does not represent active ischemia. His low abnormal troponin‐I is consistent with slow resolution after a large ischemic event in the recent past, and his anterior wall akinesis is consistent with prior infarction in the territory of his culprit left anterior descending coronary artery.
Although acute cardiac conditions appear less likely, the brisk leukocytosis in a returned traveler prompts consideration of infection. His lung consolidations could represent either new or resolving pneumonia. The complete absence of cough and fever is unusual for pneumonia, yet clinical findings are not as sensitive as chest radiograph for this diagnosis. At this point, typical organisms as well as uncommon pathogens associated with diarrhea or his travel history should be included in the differential.
After 24 hours, the patient was discharged on warfarin to treat the apical thrombus and moxifloxacin for a presumed community‐acquired pneumonia. Eight days after discharge, the patient visited his primary care physician with improving, but not resolved, shortness of breath and pleuritic pain despite completing the 7‐day course of moxifloxacin. A chest radiograph showed a large posterior left basal pleural fluid collection, increased from previous.
In the setting of a recent infection, the symptoms and radiographic findings suggest a complicated parapneumonic effusion or empyema. Failure to drain a previously seeded fluid collection leaves bacterial pathogens susceptible to moxifloxacin on the differential, including Streptococcus pneumoniae, Staphylococcus aureus, Legionella species, and other enterobacteriaciae (eg, Klebsiella pneumoniae).
The indolent course should also prompt consideration of more unusual pathogens, including roundworms (such as Ascaris) or lung flukes (Paragonimus), either of which can cause a lung infection without traditional pneumonia symptoms. Tuberculosis tends to present months (or years) after exposure. Older adults may manifest primary pulmonary tuberculosis with lower lobe infiltrates, consistent with this presentation. However, moxifloxacin is quite active against tuberculosis, and although single drug therapy would not be expected to cure the patient, it would be surprising for him to progress this quickly on moxifloxacin.
In northern Thailand, Burkholderia pseudomallei is a common cause of bacteremic pneumonia. The organism often has high‐level resistance to fluoroquinolones, and may present in a more insidious fashion than other causes of community‐acquired pneumonia. Although infection with B pseudomallei (melioidosis) can occasionally mimic apical pulmonary tuberculosis and may present after a prolonged latent period, it most commonly manifests as an acute pneumonia.
The patient was prescribed 10 days of amoxicillin‐clavulanic acid and clindamycin, and decubitus films were ordered to assess the effusion. These films, obtained 5 days later, showed a persistent pleural effusion. Subsequent ultrasound demonstrated loculated fluid, but a thoracentesis was not performed at that time due to the patient's therapeutic international normalized ratio and dual antiplatelet therapy.
The loculation further suggests a complicated parapneumonic effusion or empyema. Clindamycin adds very little to amoxicillin‐clavulanate as far as coverage of oral anaerobes or common pneumonia pathogens and may add to the risk of antibiotic side effects. A susceptible organism might not clear because of failure to drain this collection; if undertreated bacterial infection is suspected, tube thoracentesis is the established standard of care. However, the protracted course of illness makes untreated pyogenic bacterial infections unlikely.
At this point, the top 2 diagnostic considerations are Paragonimus westermani and B pseudomallei. P westermani is initially ingested, usually from an undercooked freshwater crustacean. Infected patients may experience a brief diarrheal illness, as this patient reported. However, infected patients typically have a brisk peripheral eosinophilia.
Melioidosis is thus the leading concern. Amoxicillin‐clavulanate is active against many strains of B pseudomallei, so the failure of the patient to worsen could be seen as a partial treatment and supports this diagnosis. However, as prolonged therapy is necessary for complete eradication of B pseudomallei, a definitive, culture‐based diagnosis should be established before committing the patient to months of antibiotics.
After completing 10 days of clindamycin and amoxicillin‐clavulanate, the patient reported improvement of his pleuritic pain, and repeat physical exam suggested interval decrease in the size of the effusion. Two days later, the patient began experiencing dysuria that persisted despite 3 days of nitrofurantoin.
Melioidosis can also involve the genitourinary tract. Hematogenous spread of B pseudomallei can seed a number of visceral organs including the bladder, joints, and bones. Men with suspected urinary infection should be evaluated for the possibility of prostatitis, an infection with considerable morbidity that requires extended therapy. This gentleman should have a prostate exam, and blood and urine cultures should be collected if prostatitis is suspected. Empiric antibiotics are not recommended without culture in a patient with complicated urinary tract infection.
Prostate exam was unremarkable. A urine culture grew a gram‐negative rod identified as B pseudomallei. Because B pseudomallei can cause fulminant sepsis, the infectious disease consultant requested that he return for admission, further evaluation, and initiation of intravenous antibiotics. Computed tomography (CT) of the chest, abdomen, and pelvis revealed multiple pulmonary nodules, a persistent left pleural effusion, and a rim‐enhancing hypodensity in the prostate consistent with abscess (Figure 1). Blood and pleural fluid cultures were negative.
Initial treatment for a patient with severe or metastatic B pseudomallei infection requires high‐dose intravenous antibiotic therapy. Ceftazidime, imipenem, and meropenem are the best studied agents for this purpose. Surgical drainage should be considered for the abscess. Following the completion of intensive intravenous therapy, relapse rates are high unless a longer‐term, consolidation therapy is pursued. Trimethoprim‐sulfamethoxazole is the recommended agent.
The patient was treated with high‐dose ceftazidime for 2 weeks, followed by 6 months of high‐dose oral trimethoprim‐sulfamethoxazole. His symptoms resolved, and 7 months after presentation, he continued to feel well.
DISCUSSION
Melioidosis refers to any infection caused by B pseudomallei, a gram‐negative bacillus found in soil and water, most commonly in Southeast Asia and Australia.[1] It is an important cause of pneumonia in endemic regions; in Thailand, the incidence is as high as 12 cases per 100,000 people, and it is the third leading infectious cause of death, following human immunodeficiency virus and tuberculosis.[2] However, it occurs only as an imported infection in the United States and remains an unfamiliar infection for many US medical practitioners. Melioidosis should be considered in patients returning from endemic regions presenting with sepsis, pneumonia, urinary symptoms, or abscesses.
B pseudomallei can be transmitted to humans through exposure to contaminated soil or water via ingestion, inhalation, or percutaneous inoculation.[1] Outbreaks typically occur during the rainy season and after typhoons.[1, 3] Presumably, this patient's exposure to B pseudomallei occurred while hiking and wading in freshwater lakes and waterfalls. Although hospital‐acquired melioidosis has not been reported, and isolation precautions are not necessary, rare cases of disease acquired via laboratory exposure have been reported among US healthcare workers. Clinicians suspecting melioidosis should alert the receiving laboratory.[4]
The treatment course for melioidosis is lengthy and should involve consultation with an infectious disease specialist. B pseudomallei is known to be resistant to penicillin, first‐ and second‐generation cephalosporins, and moxifloxacin. The standard treatment includes 10 to 14 days of intravenous ceftazidime, meropenem, or imipenem, and then trimethoprim‐sulfamethoxazole for 3 to 6 months.[1] Treatment should be guided by culture susceptibility data when available. There are reports of B pseudomallei having different resistance patterns within the same host; clinicians should culture all drained fluid collections and tailor antibiotics to the most resistant strain recovered.[5, 6] Although melioidosis is a life‐threatening infection, previously healthy patients have an excellent prognosis assuming prompt diagnosis and treatment are provided.[3]
After excluding common causes of chest pain, the discussant identified the need to definitively establish a microbiologic diagnosis by obtaining pleural fluid. Although common clinical scenarios can often be treated with guideline‐supported empiric antibiotics, the use of serial courses of empiric antibiotics should be carefully questioned and is generally discouraged. Specific data to prove or disprove the presence of infection should be obtained before exposing a patient to the risks of multiple drugs or prolonged antibiotic therapy, as well as the risks of delayed (or missed) diagnosis. Unfortunately, a complete evaluation was delayed by clinical contraindications to diagnostic thoracentesis, and a definitive diagnosis was reached only after development of more widespread symptoms.
This patient's protean presentation is not surprising given his ultimate diagnosis. B pseudomallei has been termed the great mimicker, as disease presentation and organ involvement can vary from an indolent localized infection to acute severe sepsis.[7] Pneumonia and genitourinary infections are the most common manifestations, although skin infections, bacteremia, septic arthritis, and neurologic disease are also possible.[1, 3] In addition, melioidosis may develop after a lengthy incubation. In a case series, confirmed incubation periods ranged from 1 to 21 days (mean, 9 days); however, cases of chronic (>2 months) infection, mimicking tuberculosis, are estimated to occur in about 12% of cases.[4] B pseudomallei is also capable of causing reactivation disease, similar to tuberculosis. It was referred to as the Vietnamese time bomb when US Vietnam War veterans, exposed to the disease when helicopters aerosolized the bacteria in the soil, developed the disease only after their return to the United States.[8] Fortunately, only a tiny fraction of the quarter‐million soldiers with serologically confirmed exposure to the bacteria ultimately developed disease.
In The Adventure of the Dying Detective, Sherlock Holmes fakes a serious illness characterized by shortness of breath and weakness to trick an adversary into confessing to murder. The abrupt, crippling infection mimicked by Holmes is thought by some to be melioidosis.[9, 10] Conan Doyle's story was published in 1913, a year after melioidosis was first reported in the medical literature, and the exotic, protean infection may well have sparked Doyle's imagination. However, this patient's case of melioidosis proved stranger than fiction in its untimely concomitant development with an MI. Cracking our case required imagination and nimble thinking to avoid a number of cognitive pitfalls. The patient's recent MI anchored reasoning at his initial presentation, and the initial diagnosis of community‐acquired pneumonia raised the danger of premature closure. Reaching the correct diagnosis required an open mind, a detailed travel history, and firm microbiologic evidence. Hospitalists need not be expert in the health risks of travel to specific foreign destinations, but investigating those risks can hasten proper diagnosis and treatment.
TEACHING POINTS
- Melioidosis should be considered in patients returning from endemic regions who present with sepsis, pneumonia, urinary symptoms, or an abscess.
- For patients with a loculated parapneumonic effusion, tube thoracentesis for culture and drainage is the standard of care for diagnosis and treatment.
- Culture identification and antibiotic sensitivities are critical for management of B pseudomallei, because prolonged antibiotic treatment is needed.
Disclosure
Nothing to report.
A 65‐year‐old man suffered a myocardial infarction (MI) while traveling in Thailand. After 7 days of recovery, the patient departed for his home in the United States. He developed substernal, nonexertional, inspiratory chest pain and shortness of breath during his return flight and presented directly to an emergency room after arrival.
Initially, the evaluation should focus on life‐threatening diagnoses and not be distracted by the travel history. The immediate diagnostic concerns are active cardiac ischemia, complications of MI, and pulmonary embolus. Other cardiac causes of dyspnea include ischemic mitral regurgitation, postinfarction pericarditis with or without pericardial effusion, and heart failure. Mechanical complications of infarction, such as left ventricular free wall rupture or rupture of the interventricular septum, can occur in this time frame and are associated with significant morbidity. Pneumothorax may be precipitated by air travel, especially in patients with underlying lung disease. The immobilization associated with long airline flights is a risk factor for thromboembolic disease, which is classically associated with pleuritic chest pain. Inspiratory chest pain is also associated with inflammatory processes involving the pericardium or pleura. If pneumonia, pericarditis, or pleural effusion is present, details of his travel history will become more important in his evaluation.
The patient elaborated that he spent 10 days in Thailand. On the third day of his trip he developed severe chest pain while hiking toward a waterfall in a rural northern district. He was transferred to a large private hospital, where he received a stent in the proximal left anterior descending coronary artery 4 hours after symptom onset. At discharge he was prescribed ticagrelor 90 mg twice daily and daily doses of losartan 50 mg, furosemide 20 mg, spironolactone 12.5 mg, aspirin 81 mg, ivabradine 2.5 mg, and pravastatin 40 mg. He had also been taking doxycycline for malaria prophylaxis since departing the United States.
His past medical history was notable for hypertension and hyperlipidemia. The patient was a lifelong nonsmoker, did not use illicit substances, and consumed no more than 2 alcoholic beverages per day. He denied cough, fevers, chills, diaphoresis, weight loss, recent upper respiratory infection, abdominal pain, hematuria, and nausea. However, he reported exertional dyspnea following his MI and nonbloody diarrhea that occurred a few days prior to his return flight and resolved without intervention.
The remainder of his past medical history confirms that he received appropriate post‐MI care, but does not substantially alter the high priority concerns in his differential diagnosis. Diarrhea may occur in up to 50% of international travelers, and is especially common when returning from Southeast Asia or the Indian subcontinent. Disease processes that may explain diarrhea and subsequent dyspnea include intestinal infections that spread to the lung (eg, ascariasis and Loeffler syndrome), infection that precipitates neuromuscular weakness (eg, Campylobacter and Guillain‐Barr syndrome), or infection that precipitates heart failure (eg, coxsackievirus, myocarditis).
On admission, his temperature was 36.2C, heart rate 91 beats per minute, blood pressure 135/81 mm Hg, respiratory rate 16 breaths per minute, and oxygen saturation 98% on room air. Cardiac exam revealed a regular rhythm without rubs, murmurs, or diastolic gallops. He had no jugular venous distention, and no lower extremity edema. His distal pulses were equal and palpable throughout. Pulmonary exam was notable for decreased breath sounds at both bases without wheezing, rhonchi, or crackles noted. He had no rashes, joint effusions, or jaundice. Abdominal and neurologic examinations were unremarkable.
Diminished breath sounds may suggest atelectasis or pleural effusion; the latter could account for the patient's inspiratory chest pain. A chest radiograph is essential to evaluate this finding further. The physical examination is not suggestive of decompensated heart failure; measurement of serum brain natriuretic peptide level would further exclude that diagnosis.
Laboratory evaluation revealed a leukocytosis of 16,000/L, with 76% polymorphonuclear cells and 12% lymphocytes without eosinophils or band forms; a hematocrit of 38%; and a platelet count of 363,000/L. The patient had a creatinine of 1.6 mg/dL, potassium of 2.7 mEq/L, and a troponin‐I of 1.0 ng/mL (normal <0.40 ng/mL), with the remainder of the routine serum chemistries within normal limits. An electrocardiogram (ECG) showed QS complexes in the anteroseptal leads, and a chest radiograph showed bibasilar consolidations and a left pleural effusion. A ventilation‐perfusion scan of the chest was performed to evaluate for pulmonary embolism, and was interpreted as low probability. Transthoracic echocardiography demonstrated severe left ventricular systolic dysfunction with anterior wall akinesis, and an aneurysmal left ventricle with an apical thrombus. No significant valvular pathology or other structural defects were noted.
The ECG and echocardiogram confirm the history of a large anteroseptal infarction with severe left ventricular dysfunction. Serial troponin testing would be reasonable. However, the absence of any acute ischemic ECG changes, typical angina symptoms, and a relatively normal troponin level all suggest his chest pain does not represent active ischemia. His low abnormal troponin‐I is consistent with slow resolution after a large ischemic event in the recent past, and his anterior wall akinesis is consistent with prior infarction in the territory of his culprit left anterior descending coronary artery.
Although acute cardiac conditions appear less likely, the brisk leukocytosis in a returned traveler prompts consideration of infection. His lung consolidations could represent either new or resolving pneumonia. The complete absence of cough and fever is unusual for pneumonia, yet clinical findings are not as sensitive as chest radiograph for this diagnosis. At this point, typical organisms as well as uncommon pathogens associated with diarrhea or his travel history should be included in the differential.
After 24 hours, the patient was discharged on warfarin to treat the apical thrombus and moxifloxacin for a presumed community‐acquired pneumonia. Eight days after discharge, the patient visited his primary care physician with improving, but not resolved, shortness of breath and pleuritic pain despite completing the 7‐day course of moxifloxacin. A chest radiograph showed a large posterior left basal pleural fluid collection, increased from previous.
In the setting of a recent infection, the symptoms and radiographic findings suggest a complicated parapneumonic effusion or empyema. Failure to drain a previously seeded fluid collection leaves bacterial pathogens susceptible to moxifloxacin on the differential, including Streptococcus pneumoniae, Staphylococcus aureus, Legionella species, and other enterobacteriaciae (eg, Klebsiella pneumoniae).
The indolent course should also prompt consideration of more unusual pathogens, including roundworms (such as Ascaris) or lung flukes (Paragonimus), either of which can cause a lung infection without traditional pneumonia symptoms. Tuberculosis tends to present months (or years) after exposure. Older adults may manifest primary pulmonary tuberculosis with lower lobe infiltrates, consistent with this presentation. However, moxifloxacin is quite active against tuberculosis, and although single drug therapy would not be expected to cure the patient, it would be surprising for him to progress this quickly on moxifloxacin.
In northern Thailand, Burkholderia pseudomallei is a common cause of bacteremic pneumonia. The organism often has high‐level resistance to fluoroquinolones, and may present in a more insidious fashion than other causes of community‐acquired pneumonia. Although infection with B pseudomallei (melioidosis) can occasionally mimic apical pulmonary tuberculosis and may present after a prolonged latent period, it most commonly manifests as an acute pneumonia.
The patient was prescribed 10 days of amoxicillin‐clavulanic acid and clindamycin, and decubitus films were ordered to assess the effusion. These films, obtained 5 days later, showed a persistent pleural effusion. Subsequent ultrasound demonstrated loculated fluid, but a thoracentesis was not performed at that time due to the patient's therapeutic international normalized ratio and dual antiplatelet therapy.
The loculation further suggests a complicated parapneumonic effusion or empyema. Clindamycin adds very little to amoxicillin‐clavulanate as far as coverage of oral anaerobes or common pneumonia pathogens and may add to the risk of antibiotic side effects. A susceptible organism might not clear because of failure to drain this collection; if undertreated bacterial infection is suspected, tube thoracentesis is the established standard of care. However, the protracted course of illness makes untreated pyogenic bacterial infections unlikely.
At this point, the top 2 diagnostic considerations are Paragonimus westermani and B pseudomallei. P westermani is initially ingested, usually from an undercooked freshwater crustacean. Infected patients may experience a brief diarrheal illness, as this patient reported. However, infected patients typically have a brisk peripheral eosinophilia.
Melioidosis is thus the leading concern. Amoxicillin‐clavulanate is active against many strains of B pseudomallei, so the failure of the patient to worsen could be seen as a partial treatment and supports this diagnosis. However, as prolonged therapy is necessary for complete eradication of B pseudomallei, a definitive, culture‐based diagnosis should be established before committing the patient to months of antibiotics.
After completing 10 days of clindamycin and amoxicillin‐clavulanate, the patient reported improvement of his pleuritic pain, and repeat physical exam suggested interval decrease in the size of the effusion. Two days later, the patient began experiencing dysuria that persisted despite 3 days of nitrofurantoin.
Melioidosis can also involve the genitourinary tract. Hematogenous spread of B pseudomallei can seed a number of visceral organs including the bladder, joints, and bones. Men with suspected urinary infection should be evaluated for the possibility of prostatitis, an infection with considerable morbidity that requires extended therapy. This gentleman should have a prostate exam, and blood and urine cultures should be collected if prostatitis is suspected. Empiric antibiotics are not recommended without culture in a patient with complicated urinary tract infection.
Prostate exam was unremarkable. A urine culture grew a gram‐negative rod identified as B pseudomallei. Because B pseudomallei can cause fulminant sepsis, the infectious disease consultant requested that he return for admission, further evaluation, and initiation of intravenous antibiotics. Computed tomography (CT) of the chest, abdomen, and pelvis revealed multiple pulmonary nodules, a persistent left pleural effusion, and a rim‐enhancing hypodensity in the prostate consistent with abscess (Figure 1). Blood and pleural fluid cultures were negative.
Initial treatment for a patient with severe or metastatic B pseudomallei infection requires high‐dose intravenous antibiotic therapy. Ceftazidime, imipenem, and meropenem are the best studied agents for this purpose. Surgical drainage should be considered for the abscess. Following the completion of intensive intravenous therapy, relapse rates are high unless a longer‐term, consolidation therapy is pursued. Trimethoprim‐sulfamethoxazole is the recommended agent.
The patient was treated with high‐dose ceftazidime for 2 weeks, followed by 6 months of high‐dose oral trimethoprim‐sulfamethoxazole. His symptoms resolved, and 7 months after presentation, he continued to feel well.
DISCUSSION
Melioidosis refers to any infection caused by B pseudomallei, a gram‐negative bacillus found in soil and water, most commonly in Southeast Asia and Australia.[1] It is an important cause of pneumonia in endemic regions; in Thailand, the incidence is as high as 12 cases per 100,000 people, and it is the third leading infectious cause of death, following human immunodeficiency virus and tuberculosis.[2] However, it occurs only as an imported infection in the United States and remains an unfamiliar infection for many US medical practitioners. Melioidosis should be considered in patients returning from endemic regions presenting with sepsis, pneumonia, urinary symptoms, or abscesses.
B pseudomallei can be transmitted to humans through exposure to contaminated soil or water via ingestion, inhalation, or percutaneous inoculation.[1] Outbreaks typically occur during the rainy season and after typhoons.[1, 3] Presumably, this patient's exposure to B pseudomallei occurred while hiking and wading in freshwater lakes and waterfalls. Although hospital‐acquired melioidosis has not been reported, and isolation precautions are not necessary, rare cases of disease acquired via laboratory exposure have been reported among US healthcare workers. Clinicians suspecting melioidosis should alert the receiving laboratory.[4]
The treatment course for melioidosis is lengthy and should involve consultation with an infectious disease specialist. B pseudomallei is known to be resistant to penicillin, first‐ and second‐generation cephalosporins, and moxifloxacin. The standard treatment includes 10 to 14 days of intravenous ceftazidime, meropenem, or imipenem, and then trimethoprim‐sulfamethoxazole for 3 to 6 months.[1] Treatment should be guided by culture susceptibility data when available. There are reports of B pseudomallei having different resistance patterns within the same host; clinicians should culture all drained fluid collections and tailor antibiotics to the most resistant strain recovered.[5, 6] Although melioidosis is a life‐threatening infection, previously healthy patients have an excellent prognosis assuming prompt diagnosis and treatment are provided.[3]
After excluding common causes of chest pain, the discussant identified the need to definitively establish a microbiologic diagnosis by obtaining pleural fluid. Although common clinical scenarios can often be treated with guideline‐supported empiric antibiotics, the use of serial courses of empiric antibiotics should be carefully questioned and is generally discouraged. Specific data to prove or disprove the presence of infection should be obtained before exposing a patient to the risks of multiple drugs or prolonged antibiotic therapy, as well as the risks of delayed (or missed) diagnosis. Unfortunately, a complete evaluation was delayed by clinical contraindications to diagnostic thoracentesis, and a definitive diagnosis was reached only after development of more widespread symptoms.
This patient's protean presentation is not surprising given his ultimate diagnosis. B pseudomallei has been termed the great mimicker, as disease presentation and organ involvement can vary from an indolent localized infection to acute severe sepsis.[7] Pneumonia and genitourinary infections are the most common manifestations, although skin infections, bacteremia, septic arthritis, and neurologic disease are also possible.[1, 3] In addition, melioidosis may develop after a lengthy incubation. In a case series, confirmed incubation periods ranged from 1 to 21 days (mean, 9 days); however, cases of chronic (>2 months) infection, mimicking tuberculosis, are estimated to occur in about 12% of cases.[4] B pseudomallei is also capable of causing reactivation disease, similar to tuberculosis. It was referred to as the Vietnamese time bomb when US Vietnam War veterans, exposed to the disease when helicopters aerosolized the bacteria in the soil, developed the disease only after their return to the United States.[8] Fortunately, only a tiny fraction of the quarter‐million soldiers with serologically confirmed exposure to the bacteria ultimately developed disease.
In The Adventure of the Dying Detective, Sherlock Holmes fakes a serious illness characterized by shortness of breath and weakness to trick an adversary into confessing to murder. The abrupt, crippling infection mimicked by Holmes is thought by some to be melioidosis.[9, 10] Conan Doyle's story was published in 1913, a year after melioidosis was first reported in the medical literature, and the exotic, protean infection may well have sparked Doyle's imagination. However, this patient's case of melioidosis proved stranger than fiction in its untimely concomitant development with an MI. Cracking our case required imagination and nimble thinking to avoid a number of cognitive pitfalls. The patient's recent MI anchored reasoning at his initial presentation, and the initial diagnosis of community‐acquired pneumonia raised the danger of premature closure. Reaching the correct diagnosis required an open mind, a detailed travel history, and firm microbiologic evidence. Hospitalists need not be expert in the health risks of travel to specific foreign destinations, but investigating those risks can hasten proper diagnosis and treatment.
TEACHING POINTS
- Melioidosis should be considered in patients returning from endemic regions who present with sepsis, pneumonia, urinary symptoms, or an abscess.
- For patients with a loculated parapneumonic effusion, tube thoracentesis for culture and drainage is the standard of care for diagnosis and treatment.
- Culture identification and antibiotic sensitivities are critical for management of B pseudomallei, because prolonged antibiotic treatment is needed.
Disclosure
Nothing to report.
- , , . Melioidosis. N Engl J Med. 2012;367(11):1035–1044.
- , , , et al. Increasing incidence of human melioidosis in Northeast Thailand. Am J Trop Med Hyg. 2010;82(6):1113–1117.
- , , . The epidemiology and clinical spectrum of melioidosis: 540 cases from the 20 year Darwin prospective study. PLoS Negl Trop Dis. 2010;4(11):e900.
- , , , et al. Management of accidental laboratory exposure to Burkholderia pseudomallei and B. mallei. Emerg Infect Dis. 2008;14(7):e2.
- , , . Variations in ceftazidime and amoxicillin‐clavulanate susceptibilities within a clonal infection of Burkholderia pseudomallei. J Clin Microbiol. 2009;47(5):1556–1558.
- , , , et al. Within‐host evolution of Burkholderia pseudomallei in four cases of acute melioidosis. PLoS Pathog. 2010;6(1):e1000725.
- , , , , . Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat Rev Microbiol. 2006;4(4):272–282.
- , . Melioidosis. In: Dembeck ZF, ed. Medical Aspects of Biological Warfare. 2nd ed. Washington, DC: Office of the Surgeon General; 2007:146–166.
- . Sherlock Holmes and a biological weapon. J R Soc Med. 2002;95(2):101–103.
- . Sherlock Holmes and tropical medicine: a centennial appraisal. Am J Trop Med Hyg. 1994;50:99–101.
- , , . Melioidosis. N Engl J Med. 2012;367(11):1035–1044.
- , , , et al. Increasing incidence of human melioidosis in Northeast Thailand. Am J Trop Med Hyg. 2010;82(6):1113–1117.
- , , . The epidemiology and clinical spectrum of melioidosis: 540 cases from the 20 year Darwin prospective study. PLoS Negl Trop Dis. 2010;4(11):e900.
- , , , et al. Management of accidental laboratory exposure to Burkholderia pseudomallei and B. mallei. Emerg Infect Dis. 2008;14(7):e2.
- , , . Variations in ceftazidime and amoxicillin‐clavulanate susceptibilities within a clonal infection of Burkholderia pseudomallei. J Clin Microbiol. 2009;47(5):1556–1558.
- , , , et al. Within‐host evolution of Burkholderia pseudomallei in four cases of acute melioidosis. PLoS Pathog. 2010;6(1):e1000725.
- , , , , . Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat Rev Microbiol. 2006;4(4):272–282.
- , . Melioidosis. In: Dembeck ZF, ed. Medical Aspects of Biological Warfare. 2nd ed. Washington, DC: Office of the Surgeon General; 2007:146–166.
- . Sherlock Holmes and a biological weapon. J R Soc Med. 2002;95(2):101–103.
- . Sherlock Holmes and tropical medicine: a centennial appraisal. Am J Trop Med Hyg. 1994;50:99–101.