Nevus of Ota, originally referred to as nevus fusco-caeruleus ophthalmomaxillaris, initially was described in 1939 by Ota and Tanino.1 It is a dermal melanocytic hamartoma arising from incomplete migration of neural crest melanocytes to the epidermis during embryogenesis, resulting in nesting of subtle bands of dendritic melanocytes in the upper dermis. More common in Asians, Native Americans, and females, this hyperpigmented dermatosis most often is unilaterally distributed along the ophthalmic (V1) and maxillary (V2) branches of the trigeminal nerve.2 In some patients, nevus of Ota also is associated with ocular, orbital, and leptomeningeal melanocytosis. Approximately 15% of nevi of Ota have an activating guanine nucleotide-binding protein G(q) subunit alpha (GNAQ) or G protein subunit alpha 11 (GNAQ) mutation; 85% of uveal melanomas harbor one of these mutations.3 Although uncommon, neoplastic transformation with extension or metastasis to the brain has been reported in patients with nevus of Ota.4
We report the case of a 29-year-old woman with a long-standing history of nevus of Ota who presented acutely with an intracranial melanoma as an extension of a primary uveal melanoma.
Case Report
A 29-year-old woman with a history of a nevus of Ota involving the left inner canthus, eyelids, sclera, and superior malar cheek that had been present since birth presented to the emergency department with an acute onset of severe headache, blurred vision, and vomiting. Computed tomography (CT) and magnetic resonance imaging of the brain revealed a hemorrhagic mass in the left frontal lobe. Subsequent frontal craniotomy and resection revealed an intracranial melanoma.
Two weeks following surgery, the patient underwent magnetic resonance imaging and combined positron emission tomography and CT scans that demonstrated a fluorodeoxyglucose-avid left retro-orbital mass. Histopathology of a biopsy from the left retro-orbital mass that had been obtained intraoperatively demonstrated a pigmented, spindled to epithelioid neoplasm with areas of marked atypia and a high mitotic rate that was compatible with malignant melanoma (Figure 1). Intracranial biopsies were sent for genetic study and were found to harbor GNAQ (Q209P) and BRCA1-associated protein 1 (BAP1)(p.P324fs*11) mutations.
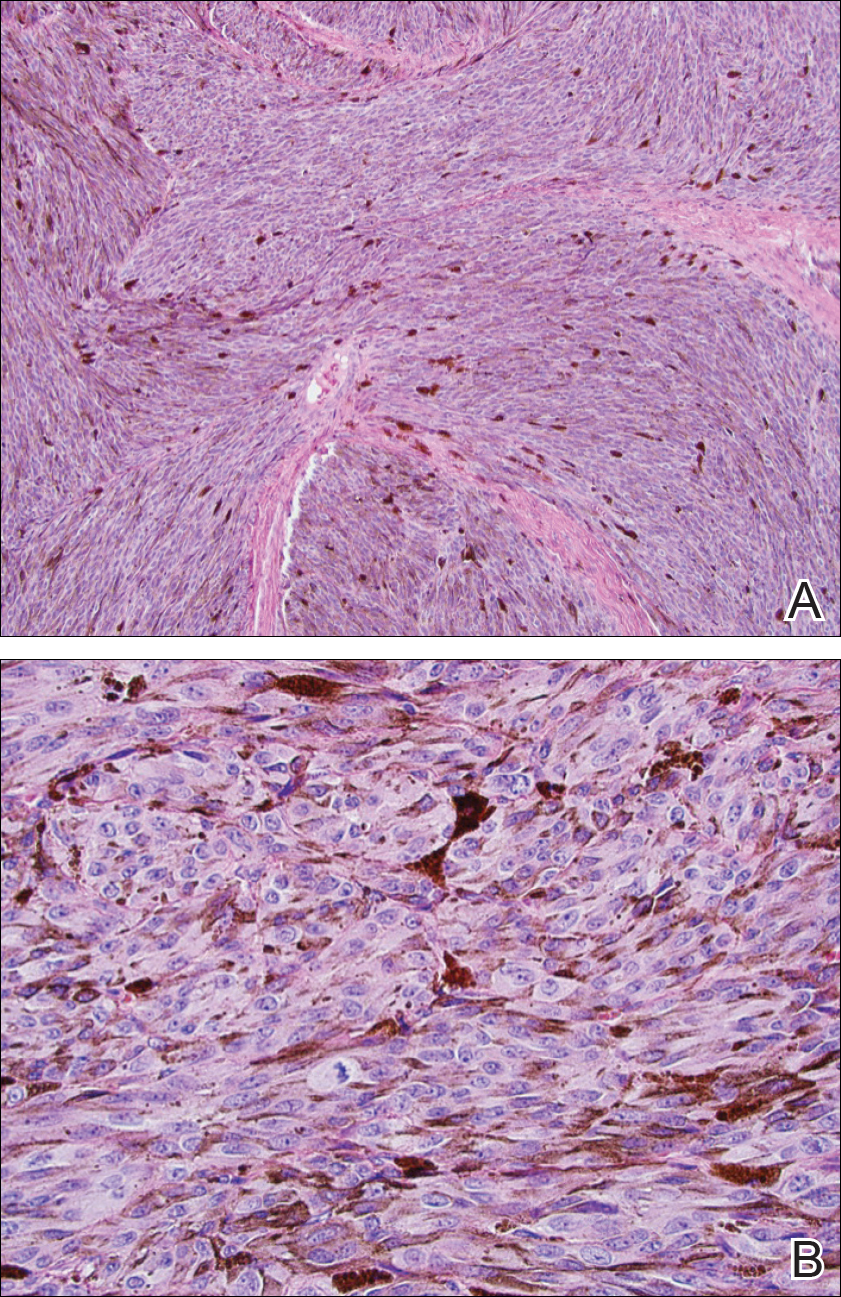
Figure 1. Histopathology of the intracranial biopsy. On low-power view, fascicles of atypical, pigmented, spindled to epithelioid melanocytes were noted (A)(H&E, original magnification ×10). A higher-power view revealed increased mitotic activity (B)(H&E, original magnification ×40). Findings were consistent with malignant melanoma.
The patient was referred to dermatology by neurosurgery for evaluation of a suspected primary cutaneous melanoma. Biopsies of 2 blue papules that had appeared over the last 2 years within the nevus of Ota on the left medial canthus and left malar cheek (Figure 2) revealed cellular blue nevi (Figure 3). No primary cutaneous melanoma was identified. Based on the genetic profile described above and the presence of GNAQ and BAP1 mutations, the patient was referred to ophthalmology. Inferotemporal darkening of the choroid, most likely consistent with a primary uveal melanoma, was discovered. The intracranial melanoma was thought to have arisen from the primary uveal melanoma.
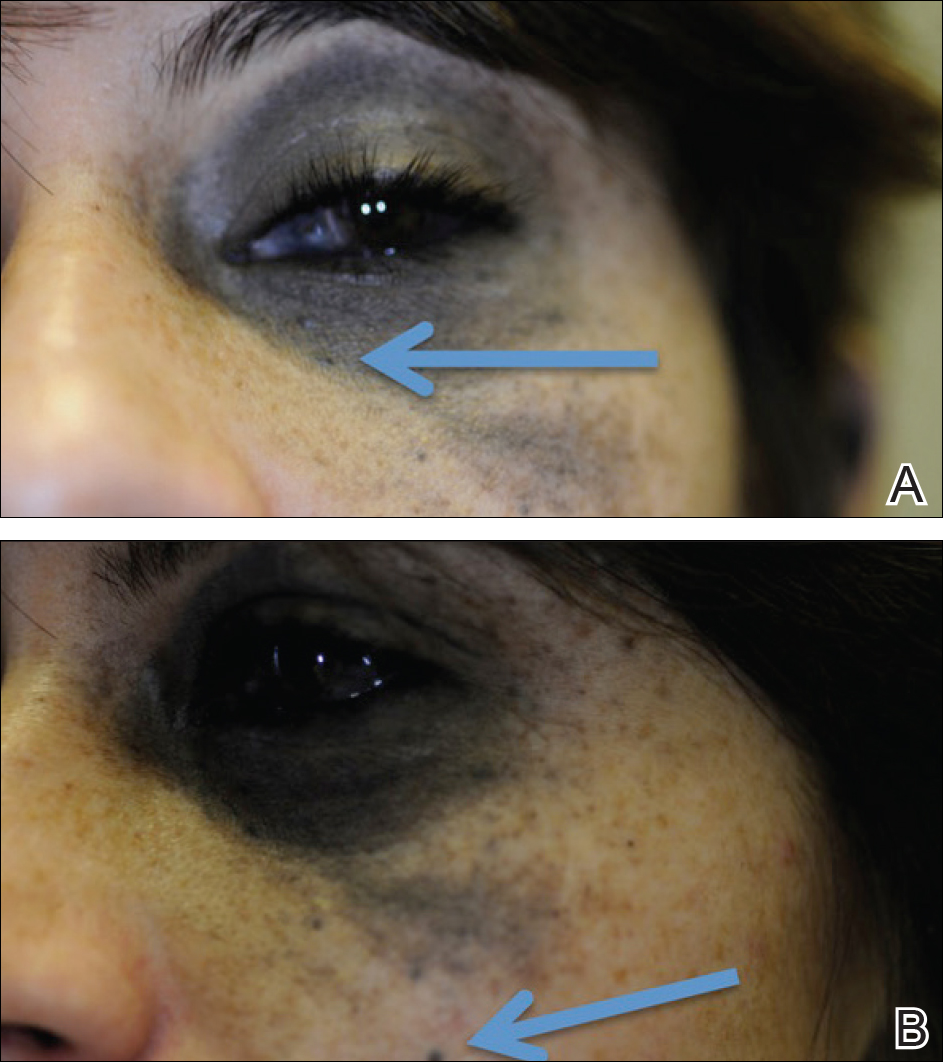
Figure 2. Nevus of ota extending from the left medial canthus (A), encompassing the sclera and the malar cheek (B), containing the 2 papules that were biopsied (arrows).

Figure 3. Punch biopsies of the left ear (A) and malar cheek (B) demonstrated bland, spindled, melanocytic proliferations with melanophages, consistent with cellular blue nevi (H&E, original magnifications ×10 and ×40).
The patient entered a clinical trial at an outside institution several weeks after initial presentation to our institution for treatment with a mitogen-activated protein kinase MEK1 inhibitor as well as radiation therapy. The patient was lost to follow-up.