User login
Bullous Pemphigoid Masquerading as a Prosthesis Allergy
To the Editor:
Bullous pemphigoid (BP) is an autoimmune bullous dermatosis characterized by tense subepidermal blisters. It primarily affects older individuals who typically report pruritus in the affected area. Subepidermal blisters are caused by a humoral and cellular autoimmune attack directed against 2 BP antigens—BP180 and BP230—which are 2 critical components of the hemidesmosome whose primary function is to anchor the epidermis to the underlying dermis. Although tense bullae typically prompt immediate consideration of BP in the differential diagnosis, early disease often is characterized by urticarial plaques that require a high degree of suspicion to make the appropriate diagnosis. Locus minoris resistentiae is a term used to describe the phenomenon of skin disease occurring at the point of least resistance.1
A 79-year-old woman with type 2 diabetes mellitus, peptic ulcer disease, and hypertension was referred to the dermatology clinic due to concern for allergic contact dermatitis limited to the area of and adjacent to a well-healed surgical wound. History and examination revealed that the patient had sustained a left femoral neck fracture 10 months prior to presentation that required closed reduction and surgical pinning. The surgical site healed well postoperatively; however, 7 months after surgery, she began to develop edema and erythema within and immediately adjacent to the surgical scar. She subsequently developed areas of superficial erosion within the erythema and was evaluated by her surgeon who was concerned for suture granuloma. Superficial wound debridement of the area was performed without improvement. Approximately 9 months after surgery, the patient developed bullae along the old surgical site, which raised concern for an allergic reaction to the implanted screws. Orthopedics elected to remove the hardware but also sent intraoperative tissue for pathologic examination, which revealed subepidermal bullae containing eosinophils and neutrophils, most consistent with a bullous drug eruption. During the ensuing weeks after hardware removal, the plaque spread along the old surgical wound, and several bullous lesions began to appear. The patient’s primary care physician became concerned for allergic contact dermatitis, possibly to the surgical scrub employed during hardware removal. He prescribed triamcinolone ointment 0.1% and referred the patient to dermatology.
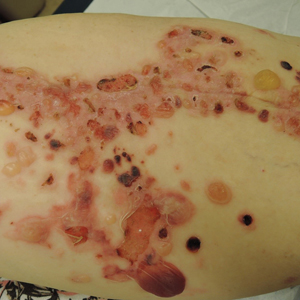
Upon presentation to dermatology, the patient noted stinging pain and intense pruritus of the affected area. Examination revealed a pink edematous plaque distributed along a well-healed surgical wound (Figure). Numerous fluid-filled tense bullae were superimposed on this plaque as well as areas of superficial erosion with serum crust. An expanded examination revealed similar smaller lesions on the upper arms, inner thighs, and lateral breasts. A 4-mm punch biopsy of lesional and perilesional skin was sent for hematoxylin and eosin staining and direct immunofluorescence, which demonstrated a subepidermal bullous dermatosis with a predominance of neutrophilic inflammation as well as a band of linear IgG deposition at the dermal-epidermal junction. The patient was diagnosed with BP exhibiting a locus minoris resistentiae phenomenon within the surgical site. She was started on prednisone 1 mg/kg daily and doxycycline 100 mg twice daily and demonstrated rapid improvement.

Although the tense bullae seen in well-developed BP are fairly characteristic, the prodromal phase of this disease can present with urticarial plaques that are nonspecific. This progression is well described, but our case demonstrates the difficulty of considering BP when a patient presents with an urticarial plaque. As lesions progress to the bullous phase, they may be inappropriately diagnosed as allergic contact dermatitis, an error that may lead to unnecessary interventions (eg, removal of an implicated prosthesis). This case is a reminder that not all cutaneous eruptions in and around postsurgical scars are allergic in nature.
This case also depicts BP appearing in the locus minoris resistentiae, a well-healed surgical wound in our patient. Although many diseases have been shown to exhibit this type of isomorphic response, this phenomenon may pose diagnostic and management conundrums. Locus minoris resistentiae has been reported in many different diseases, both cutaneous and otherwise, but there likely are distinct disease- and case-specific mechanisms via which this occurs. Local phenomena reported to trigger BP include contact dermatitis, vaccination, radiation therapy, phototherapy, infection, and surgery.2 We suspect that the mechanism of locus minoris resistentiae in our patient was disruption of the architecture of the dermal-epidermal basement membrane zone due to surgical trauma. Disruption of this architecture may have resulted in exposure of previously occult antigens, recognition by T cells, T-cell stimulation of autoantibody production by B cells, binding of autoantibodies to BP180, complement deposition, recruitment of inflammatory cells, release of proteinases, and degradation of BP180 and extracellular matrix proteins.2
- Lo Schiavo A, Ruocco E, Russo T, et al. Locus minoris resistentiae: an old but still valid way of thinking in medicine. Clin Dermatol. 2014;32:553-556.
- Lo Schiavo A, Ruocco E, Brancaccio G, et al. Bullous pemphigoid: etiology, pathogenesis, and inducing factors: facts and controversies. Clin Dermatol. 2013;31:391-399.
To the Editor:
Bullous pemphigoid (BP) is an autoimmune bullous dermatosis characterized by tense subepidermal blisters. It primarily affects older individuals who typically report pruritus in the affected area. Subepidermal blisters are caused by a humoral and cellular autoimmune attack directed against 2 BP antigens—BP180 and BP230—which are 2 critical components of the hemidesmosome whose primary function is to anchor the epidermis to the underlying dermis. Although tense bullae typically prompt immediate consideration of BP in the differential diagnosis, early disease often is characterized by urticarial plaques that require a high degree of suspicion to make the appropriate diagnosis. Locus minoris resistentiae is a term used to describe the phenomenon of skin disease occurring at the point of least resistance.1
A 79-year-old woman with type 2 diabetes mellitus, peptic ulcer disease, and hypertension was referred to the dermatology clinic due to concern for allergic contact dermatitis limited to the area of and adjacent to a well-healed surgical wound. History and examination revealed that the patient had sustained a left femoral neck fracture 10 months prior to presentation that required closed reduction and surgical pinning. The surgical site healed well postoperatively; however, 7 months after surgery, she began to develop edema and erythema within and immediately adjacent to the surgical scar. She subsequently developed areas of superficial erosion within the erythema and was evaluated by her surgeon who was concerned for suture granuloma. Superficial wound debridement of the area was performed without improvement. Approximately 9 months after surgery, the patient developed bullae along the old surgical site, which raised concern for an allergic reaction to the implanted screws. Orthopedics elected to remove the hardware but also sent intraoperative tissue for pathologic examination, which revealed subepidermal bullae containing eosinophils and neutrophils, most consistent with a bullous drug eruption. During the ensuing weeks after hardware removal, the plaque spread along the old surgical wound, and several bullous lesions began to appear. The patient’s primary care physician became concerned for allergic contact dermatitis, possibly to the surgical scrub employed during hardware removal. He prescribed triamcinolone ointment 0.1% and referred the patient to dermatology.
Upon presentation to dermatology, the patient noted stinging pain and intense pruritus of the affected area. Examination revealed a pink edematous plaque distributed along a well-healed surgical wound (Figure). Numerous fluid-filled tense bullae were superimposed on this plaque as well as areas of superficial erosion with serum crust. An expanded examination revealed similar smaller lesions on the upper arms, inner thighs, and lateral breasts. A 4-mm punch biopsy of lesional and perilesional skin was sent for hematoxylin and eosin staining and direct immunofluorescence, which demonstrated a subepidermal bullous dermatosis with a predominance of neutrophilic inflammation as well as a band of linear IgG deposition at the dermal-epidermal junction. The patient was diagnosed with BP exhibiting a locus minoris resistentiae phenomenon within the surgical site. She was started on prednisone 1 mg/kg daily and doxycycline 100 mg twice daily and demonstrated rapid improvement.
Although the tense bullae seen in well-developed BP are fairly characteristic, the prodromal phase of this disease can present with urticarial plaques that are nonspecific. This progression is well described, but our case demonstrates the difficulty of considering BP when a patient presents with an urticarial plaque. As lesions progress to the bullous phase, they may be inappropriately diagnosed as allergic contact dermatitis, an error that may lead to unnecessary interventions (eg, removal of an implicated prosthesis). This case is a reminder that not all cutaneous eruptions in and around postsurgical scars are allergic in nature.
This case also depicts BP appearing in the locus minoris resistentiae, a well-healed surgical wound in our patient. Although many diseases have been shown to exhibit this type of isomorphic response, this phenomenon may pose diagnostic and management conundrums. Locus minoris resistentiae has been reported in many different diseases, both cutaneous and otherwise, but there likely are distinct disease- and case-specific mechanisms via which this occurs. Local phenomena reported to trigger BP include contact dermatitis, vaccination, radiation therapy, phototherapy, infection, and surgery.2 We suspect that the mechanism of locus minoris resistentiae in our patient was disruption of the architecture of the dermal-epidermal basement membrane zone due to surgical trauma. Disruption of this architecture may have resulted in exposure of previously occult antigens, recognition by T cells, T-cell stimulation of autoantibody production by B cells, binding of autoantibodies to BP180, complement deposition, recruitment of inflammatory cells, release of proteinases, and degradation of BP180 and extracellular matrix proteins.2
To the Editor:
Bullous pemphigoid (BP) is an autoimmune bullous dermatosis characterized by tense subepidermal blisters. It primarily affects older individuals who typically report pruritus in the affected area. Subepidermal blisters are caused by a humoral and cellular autoimmune attack directed against 2 BP antigens—BP180 and BP230—which are 2 critical components of the hemidesmosome whose primary function is to anchor the epidermis to the underlying dermis. Although tense bullae typically prompt immediate consideration of BP in the differential diagnosis, early disease often is characterized by urticarial plaques that require a high degree of suspicion to make the appropriate diagnosis. Locus minoris resistentiae is a term used to describe the phenomenon of skin disease occurring at the point of least resistance.1
A 79-year-old woman with type 2 diabetes mellitus, peptic ulcer disease, and hypertension was referred to the dermatology clinic due to concern for allergic contact dermatitis limited to the area of and adjacent to a well-healed surgical wound. History and examination revealed that the patient had sustained a left femoral neck fracture 10 months prior to presentation that required closed reduction and surgical pinning. The surgical site healed well postoperatively; however, 7 months after surgery, she began to develop edema and erythema within and immediately adjacent to the surgical scar. She subsequently developed areas of superficial erosion within the erythema and was evaluated by her surgeon who was concerned for suture granuloma. Superficial wound debridement of the area was performed without improvement. Approximately 9 months after surgery, the patient developed bullae along the old surgical site, which raised concern for an allergic reaction to the implanted screws. Orthopedics elected to remove the hardware but also sent intraoperative tissue for pathologic examination, which revealed subepidermal bullae containing eosinophils and neutrophils, most consistent with a bullous drug eruption. During the ensuing weeks after hardware removal, the plaque spread along the old surgical wound, and several bullous lesions began to appear. The patient’s primary care physician became concerned for allergic contact dermatitis, possibly to the surgical scrub employed during hardware removal. He prescribed triamcinolone ointment 0.1% and referred the patient to dermatology.
Upon presentation to dermatology, the patient noted stinging pain and intense pruritus of the affected area. Examination revealed a pink edematous plaque distributed along a well-healed surgical wound (Figure). Numerous fluid-filled tense bullae were superimposed on this plaque as well as areas of superficial erosion with serum crust. An expanded examination revealed similar smaller lesions on the upper arms, inner thighs, and lateral breasts. A 4-mm punch biopsy of lesional and perilesional skin was sent for hematoxylin and eosin staining and direct immunofluorescence, which demonstrated a subepidermal bullous dermatosis with a predominance of neutrophilic inflammation as well as a band of linear IgG deposition at the dermal-epidermal junction. The patient was diagnosed with BP exhibiting a locus minoris resistentiae phenomenon within the surgical site. She was started on prednisone 1 mg/kg daily and doxycycline 100 mg twice daily and demonstrated rapid improvement.
Although the tense bullae seen in well-developed BP are fairly characteristic, the prodromal phase of this disease can present with urticarial plaques that are nonspecific. This progression is well described, but our case demonstrates the difficulty of considering BP when a patient presents with an urticarial plaque. As lesions progress to the bullous phase, they may be inappropriately diagnosed as allergic contact dermatitis, an error that may lead to unnecessary interventions (eg, removal of an implicated prosthesis). This case is a reminder that not all cutaneous eruptions in and around postsurgical scars are allergic in nature.
This case also depicts BP appearing in the locus minoris resistentiae, a well-healed surgical wound in our patient. Although many diseases have been shown to exhibit this type of isomorphic response, this phenomenon may pose diagnostic and management conundrums. Locus minoris resistentiae has been reported in many different diseases, both cutaneous and otherwise, but there likely are distinct disease- and case-specific mechanisms via which this occurs. Local phenomena reported to trigger BP include contact dermatitis, vaccination, radiation therapy, phototherapy, infection, and surgery.2 We suspect that the mechanism of locus minoris resistentiae in our patient was disruption of the architecture of the dermal-epidermal basement membrane zone due to surgical trauma. Disruption of this architecture may have resulted in exposure of previously occult antigens, recognition by T cells, T-cell stimulation of autoantibody production by B cells, binding of autoantibodies to BP180, complement deposition, recruitment of inflammatory cells, release of proteinases, and degradation of BP180 and extracellular matrix proteins.2
- Lo Schiavo A, Ruocco E, Russo T, et al. Locus minoris resistentiae: an old but still valid way of thinking in medicine. Clin Dermatol. 2014;32:553-556.
- Lo Schiavo A, Ruocco E, Brancaccio G, et al. Bullous pemphigoid: etiology, pathogenesis, and inducing factors: facts and controversies. Clin Dermatol. 2013;31:391-399.
- Lo Schiavo A, Ruocco E, Russo T, et al. Locus minoris resistentiae: an old but still valid way of thinking in medicine. Clin Dermatol. 2014;32:553-556.
- Lo Schiavo A, Ruocco E, Brancaccio G, et al. Bullous pemphigoid: etiology, pathogenesis, and inducing factors: facts and controversies. Clin Dermatol. 2013;31:391-399.
Practice Points
- Bullous pemphigoid frequently presents with urticarial plaques without classic tense blisters in the early phase of disease.
- The phenomenon of locus minoris resistentiae can lead to the presentation of bullous pemphigoid in locations traumatized by surgery.
- Bullous pemphigoid can present as urticarial plaques at surgery sites mimicking allergic contact dermatitis or reaction to surgical sutures or hardware.
Granulomatous Facial Dermatoses
Cutaneous granulomatous diseases encompass many entities that are skin-limited or systemic. The prototypical cutaneous granuloma is a painless, rounded, well-defined, red-pink or flesh-colored papule1 and is smooth, owing to minimal epidermal involvement. Examples of conditions that present with such lesions include granulomatous periorificial dermatitis (GPD), granulomatous rosacea (GR), lupus miliaris disseminatus faciei (LMDF), and papular sarcoidosis. These entities commonly are seen on the face and can be a source of distress to patients when they are extensive. Several reports have raised the possibility that these conditions lie on a spectrum.2-4 We present 2 cases of patients with facial papular granulomas, discuss potential causes of the lesions, review historical aspects from the literature, and highlight the challenges that these lesions can pose to the clinician.
Case Reports
Patient 1—A 10-year-old Ethiopian girl with a history of atopic dermatitis presented with a facial rash of 4 months’ duration. Her pediatrician initially treated the rash as pityriasis alba and prescribed hydrocortisone cream. Two months into treatment, the patient developed an otherwise asymptomatic, unilateral, papular dermatosis on the right cheek. She subsequently was switched to treatment with benzoyl peroxide and topical clindamycin, which she had been using for 2 months with no improvement at the time of the current presentation. The lesions then spread bilaterally and periorally.
At the current presentation, physical examination demonstrated fine, diffuse, follicular-based, flesh-colored papules over both cheeks, the right side of the nose, and the perioral region (Figure 1). A biopsy of a papular lesion from the right cheek revealed well-formed, noncaseating granulomas in the superficial and mid dermis with an associated lymphocytic infiltrate (Figure 2). No organisms were identified on acid-fast, Fite, or periodic acid–Schiff staining. A tuberculin skin test was negative. A chest radiograph showed small calcified hilar lymph nodes bilaterally. Pulmonary function tests were unremarkable. Calcium and angiotensin-converting enzyme levels were normal.
The patient denied any fever, chills, hemoptysis, cough, dyspnea, lymphadenopathy, scleral or conjunctival pain or erythema, visual disturbances, or arthralgias. Hydroxychloroquine 200 mg twice daily was started with minimal improvement after 5 months. Methotrexate 20 mg once weekly was then added. Topical fluocinonide 0.05% also was started at this time, as the patient had required several prednisone tapers over the past 3 months for symptomatic relief. The lesions improved minimally after 5 more months of treatment, at which time she had developed inflammatory papules, pustules, and open comedones in the same areas as well as the glabella.
Repeat biopsy of a papular lesion demonstrated noncaseating granulomas and an associated chronic lymphocytic infiltrate in a follicular and perifollicular distribution (Figure 3). Biopsy of a pustule demonstrated acute Demodex folliculitis. Fluocinonide was stopped, and anti-mite therapy with ivermectin, permethrin cream 5%, and selenium sulfide lotion 2.5% was started, with good response from the pustular lesions.
The patient continued taking methotrexate 20 mg once weekly during this time, with improvement in the papular lesions. She discontinued methotrexate after 12 months with complete resolution. At follow-up 12 months after stopping the methotrexate (roughly 2 years after initial presentation), she showed sustained resolution, with small pitted scars on both cheeks and the nasal tip.
Patient 2—A 33-year-old Ethiopian woman presented with a facial rash of 15 years’ duration. The lesions had been accumulating slowly and were asymptomatic. Physical examination revealed multiple follicular-based, flesh-colored, and erythematous papules on the cheeks, chin, perioral area, and forehead (Figure 4). There were no pustules or telangiectasias. Treatment with tretinoin cream 0.05% for 6 months offered minimal relief.
Biopsy of a papule from the left mandible showed superficial vascular telangiectasias, noncaseating granulomas comprising epithelioid histiocytes and lymphocytes in the superficial dermis, and a perifollicular lymphocytic infiltrate (Figure 5). No organisms were identified on Fite or Gomori methenamine silver staining.
Comment
The first step in differentiating cutaneous granulomatous lesions should be to distinguish infectious from noninfectious causes.1 Noninfectious cutaneous granulomas can appear nearly anywhere; however, certain processes have a predilection for the face, including GPD, GR, LMDF, and papular sarcoidosis.5-7 These conditions generally present with papular granulomas with features as described above.
Granulomatous Periorificial Dermatitis—In 1970, Gianotti and colleagues8 briefly described the first possible cases of GPD in 5 children. The eruption comprised numerous yellow, dome-shaped papules in a mostly perioral distribution. Tuberculin and the Kveim tests were nonreactive; histopathology was described as sarcoid-type and not necessarily follicular or perifollicular.8 In 1974, Marten et al9 described 22 Afro-Caribbean children with flesh-colored, papular eruptions on the face that did not show histologic granulomatous changes but were morphologically similar to the reports by Gianotti et al.8 By 1989, Frieden and colleagues10 described this facial eruption as “granulomatous perioral dermatitis in children”. Additionally, the investigators observed granulomatous infiltrates in a perifollicular distribution and suggested follicular disruption as a possible cause. It was clear from the case discussions that these eruptions were not uncommonly diagnosed as papular sarcoidosis.10 The following year, Williams et al11 reported 5 cases of similar papular eruptions in 5 Afro-Caribbean children, coining the term facial Afro-Caribbean eruption.11 Knautz and Lesher12 referred to this entity as “childhood GPD” in 1996 to avoid limiting the diagnosis to Afro-Caribbean patients and to a perioral distribution; this is the most popular current terminology.12 Since then, reports of extrafacial involvement and disease in adults have been published.13,14
Granulomatous periorificial dermatitis often is seen in the perinasal, periocular, and perioral regions of the face.2 It is associated with topical steroid exposure.5 Histologically, noncaseating granulomas around the upper half of undisrupted hair follicles with a lymphocytic infiltrate are typical.13 Treatment should begin with cessation of any topical steroids; first-line agents are oral tetracycline or macrolide antibiotics.5 These agents can be used alone or in combination with topical erythromycin, metronidazole, or sulfur-based lotions.13 Rarely, GPD presents extrafacially.13 Even so, it usually resolves within 2 weeks to 6 months, especially with therapy; scarring is unusual.5,13,15
Granulomatous Rosacea—A report in the early 20th century described patients with tuberculoid granulomas resembling papular rosacea; the initial belief was that this finding represented a rosacealike tuberculid eruption.5 However, this belief was questioned by Snapp,16 among others, who demonstrated near universal lack of reactivity to tuberculin among 20 of these patients in 1949; more recent evidence has substantiated these findings.17 Still, Snapp16 postulated that these rosacealike granulomatous lesions were distinct from classic rosacea because they lacked vascular symptoms and pustules and were recalcitrant to rosacea treatment modalities.
In 1970, Mullanax and colleagues18 introduced the term granulomatous rosacea, reiterating that this entity was not tuberculous. They documented papulopustular lesions as well as telangiectasias, raising the possibility that GR does overlap with acne rosacea. More recent studies have established the current theory that GR is a histologic variant of acne rosacea because, in addition to typical granulomatous papules, its microscopic features can be seen across subtypes of acne rosacea.19,20
Various causes have been proposed for GR. Demodex mites have been reported in association with GR for nearly 30 years.19,20 In the past 10 years, molecular studies have started to define the role of metalloproteinases, UV radiation, and cutaneous peptides in the pathogenesis of acne rosacea and GR.21,22
Granulomatous rosacea typically is seen in middle-aged women.20,23 Hallmarks of rosacea, such as facial erythema, flushing, telangiectasias, pustules, and rhinophyma, are not always present in GR.5,20,23 Lesions usually are distributed around the central face, although extension to the cheeks, total facial involvement, and extrafacial lesions are possible.5,20 Histologically, perifollicular and follicular-based noncaseating granulomas with dilatation of the dermal papillary vasculature are seen.17,23 As a whole, rosacea is comparatively uncommon in dark-skinned patients; when it does occur, GR is a frequent presentation.24
First-line treatment for GR is tetracycline antibiotics.5 Unresponsive cases have been treated—largely anecdotally—with topical modalities (eg, metronidazole, steroids, immunomodulators), systemic agents (eg, dapsone, erythromycin, isotretinoin), and other therapies.5 Granulomatous rosacea tends to have a chronic course.5,23
Lupus Miliaris Disseminatus Faciei—Classic LMDF demonstrates caseating perifollicular granulomas histologically.6,17,25 Lesions tend to appear on the central face, particularly the eyelids, and can be seen extrafacially.3,6,25,26 Although LMDF originally was categorized as a tuberculid eruption, this no longer is thought to be the case.27 It is now regarded by some as a variant of GR25; however, LMDF responds poorly to tetracyclines, is more common in males, and lacks rosacealike vascular abnormalities, leading some to question this association.3,6,17 In the past 20 years, some have proposed renaming LMDF to better reflect its clinical course and to consider it independent of tuberculosis and GR.28 It usually resolves spontaneously after 1 to 3 years, leaving pitted scars.3,6
Papular Sarcoidosis—The first potential documented case of sarcoidosis was by Hutchinson29 in 1869 in a patient seen in London. The author labeled purple plaques on the index patient’s legs and hands as “livid papillary psoriasis.” In 1889, Besnier30 described a patient with violaceous swellings on the nose, ears, and fingers, which he called “lupus pernio”; his contemporary, Tenneson,31 published a case of lupus pernio and described its histologic profile as comprising epithelioid cells and giant cells. It was not until 1899 that the term sarkoid was used to describe these cutaneous lesions by Boeck,32 who thought they were reminiscent of sarcoma. In 1915, Kuznitsky and Bittorf33 described a patient with cutaneous lesions histologically consistent with Boeck’s sarkoid but additionally with hilar lymphadenopathy and pulmonary infiltrates. Around 1916 or 1917, Schaumann34 described patients with cutaneous lesions and additionally with involvement of pulmonary, osseous, hepatosplenic, and tonsillar tissue. These reports are among the first to recognize the multisystemic nature of sarcoidosis. The first possible case of childhood sarcoidosis might have been reported by Osler35 in the United States in 1898.
In the past century or so, an ongoing effort by researchers has focused on identifying etiologic triggers for sarcoidosis. Microbial agents have been considered in this role, with Mycobacterium and Propionibacterium organisms the most intensively studied; the possibility that foreign material contributes to the formation of granulomas also has been raised.36 Current models of the pathogenesis of sarcoidosis involve an interplay between the immune system in genetically predisposed patients and an infection that leads to a hyperimmune type 1 T–helper cell response that clears the infection but not antigens generated by the microbes and the acute host response, including proteins such as serum amyloid A and vimentin.36,37 These antigens aggregate and serve as a nidus for granuloma formation and maintenance long after infection has resolved.
Cutaneous lesions of sarcoidosis include macules, papules, plaques, and lupus pernio, as well as lesions arising within scars or tattoos, with many less common presentations.7,38 Papular sarcoidosis is common on the face but also can involve the extremities.4,7 Strictly, at least 2 organ systems must be involved to diagnose sarcoidosis, but this is debatable.4,7 Among 41 patients with cutaneous sarcoidosis, 24 (58.5%) had systemic disease; cutaneous lesions were the presenting sign in 87.5% (21/24) of patients.38 Histologic analysis, regardless of the lesion, usually shows noncaseating so-called “naked” granulomas, which have minimal lymphocytic infiltrate associated with the epithelioid histiocytes.38,39 Perifollicular granulomas are possible but unusual.40
Treatment depends on the extent of cutaneous and systemic involvement. Pharmacotherapeutic modalities include topical steroids, immunomodulators, and retinoids; systemic immunomodulators and immunosuppressants; and biologic agents.7 Isolated cutaneous sarcoidosis, particularly the papular variant, usually is associated with acute disease lasting less than 2 years, with resolution of skin lesions.7,38 That said, a recent report suggested that cutaneous sarcoidosis can progress to multisystemic disease as long as 7 years after the initial diagnosis.41
Clinical and Histologic Overlap—Despite this categorization of noninfectious facial granulomatous conditions, each has some clinical and histologic overlap with the others, which must be considered when encountering a granulomatous facial dermatosis. Both GPD and GR tend to present with lesions near the eyes, mouth, and nose, although GR can extend to lateral aspects of the face, below the mandible, and the forehead and has different demographic features.15,20,23 Granulomas in both GPD and GR generally are noncaseating and form in a follicular or perifollicular distribution within the dermis.2,15,23 Lupus miliaris disseminatus faciei and GR share a similar facial distribution in some cases.17,20 Even papular cutaneous sarcoidosis has masqueraded as GR clinically and histologically.4
Diagnostic and Treatment Difficulty—Our cases illustrate the range of difficulty in evaluating and managing patients with facial papular granulomas. On one hand, our adult patient’s clinical and histologic findings were highly consistent with GR; on the other hand, our younger patient had clinicopathologic features of both sarcoidosis and GPD at varying times. Both conditions are more common in dark-skinned patients.11,42
Juvenile sarcoidosis is comparatively rare, with a reported annual incidence of 0.22 to 0.27 for every 100,000 children younger than 15 years; however, juvenile sarcoidosis commonly presents around 8 to 15 years of age.43
It is unusual for sarcoid granulomas to be isolated to the skin, much less to the face.4,7,43,44 Patient 1 initially presented in this manner and lacked convincing laboratory or radiographic evidence of systemic sarcoidosis. Bilateral hilar calcifications in sarcoidosis are more typical among adults after 5 to 20 years; there were no signs or symptoms of active infection that could account for the pulmonary and cutaneous lesions.45
The presence of perifollicular granulomas with associated lymphocytic infiltrates on repeat biopsy, coupled with the use of topical steroids, made it difficult to rule out a contribution by GPD to her clinical course. That her lesions resolved with pitted scarring while she was taking methotrexate and after topical steroids had been stopped could be the result of successful management or spontaneous resolution of her dermatosis; both papular sarcoidosis and GPD tend to have a self-limited course.7,13
Conclusion
We present 2 cases of papular facial granulomas in patients with similar skin types who had different clinical courses. Evaluation of such lesions remains challenging given the similarity between specific entities that present in this manner. Certainly, it is reasonable to consider a spectrum upon which all of these conditions fall, in light of the findings of these cases and those reported previously.
- Beretta-Piccoli BT, Mainetti C, Peeters M-A, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146. doi:10.1007/s12016-017-8666-8
- Lucas CR, Korman NJ, Gilliam AC. Granulomatous periorificial dermatitis: a variant of granulomatous rosacea in children? J Cutan Med Surg. 2009;13:115-118. doi:10.2310/7750.2008.07088
- van de Scheur MR, van der Waal RIF, Starink TM. Lupus miliaris disseminatus faciei: a distinctive rosacea-like syndrome and not a granulomatous form of rosacea. Dermatology. 2003;206:120-123. doi:10.1159/000068457
- Simonart T, Lowy M, Rasquin F, et al. Overlap of sarcoidosis and rosacea. Dermatology. 1997;194:416-418. doi:10.1159/000246165
- Lee GL, Zirwas MJ. Granulomatous rosacea and periorificial dermatitis: controversies and review of management. Dermatol Clin. 2015;33:447-455. doi:10.1016/j.det.2015.03.009
- Michaels JD, Cook-Norris RH, Lehman JS, et al. Adult with papular eruption of the central aspect of the face. J Am Acad Dermatol. 2014;71:410-412. doi:10.1016/j.jaad.2012.06.039
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med. 2015;38:685-702. doi:10.1016/j.ccm.2015.08.010
- Gianotti F, Ermacora E, Benelli MG, et al. Particulière dermatite peri-orale infantile. observations sur 5 cas. Bull Soc Fr Dermatol Syphiligr. 1970;77:341.
- Marten RH, Presbury DG, Adamson JE, et al. An unusual papular and acneiform facial eruption in the negro child. Br J Dermatol. 1974;91:435-438. doi:10.1111/j.1365-2133.1974.tb13083.x
- Frieden IJ, Prose NS, Fletcher V, et al. Granulomatous perioral dermatitis in children. Arch Dermatol. 1989;125:369-373.
- Williams HC, Ashworth J, Pembroke AC, et al. FACE—facial Afro-Caribbean childhood eruption. Clin Exp Dermatol. 1990;15:163-166. doi:10.1111/j.1365-2230.1990.tb02063.x
- Knautz MA, Lesher JL Jr. Childhood granulomatous periorificial dermatitis. Pediatr Dermatol. 1996;13:131-134. doi:10.1111/j.1525-1470.1996.tb01419.x
- Urbatsch AJ, Frieden I, Williams ML, et al. Extrafacial and generalized granulomatous periorificial dermatitis. Arch Dermatol. 2002;138:1354-1358. doi:10.1001/archderm.138.10.1354
- Vincenzi C, Parente G, Tosti A. Perioral granulomatous dermatitis: two cases treated with clarithromycin. J Dermatol Treat. 2000;11:57-61.
- Kim YJ, Shin JW, Lee JS, et al. Childhood granulomatous periorificial dermatitis. Ann Dermatol. 2011;23:386-388. doi:10.5021/ad.2011.23.3.386
- Snapp RH. Lewandowsky’s rosacea-like eruption; a clinical study. J Invest Dermatol. 1949;13:175-190. doi:10.1038/jid.1949.86
- Chougule A, Chatterjee D, Sethi S, et al. Granulomatous rosacea versus lupus miliaris disseminatus faciei—2 faces of facial granulomatous disorder: a clinicohistological and molecular study. Am J Dermatopathol. 2018;40:819-823. doi:10.1097/DAD.0000000000001243
- Mullanax MG, Kierland RR. Granulomatous rosacea. Arch Dermatol. 1970;101:206-211.
- Sánchez JL, Berlingeri-Ramos AC, Dueño DV. Granulomatous rosacea. Am J Dermatopathol. 2008;30:6-9. doi:10.1097/DAD.0b013e31815bc191
- Helm KF, Menz J, Gibson LE, et al. A clinical and histopathologic study of granulomatous rosacea. J Am Acad Dermatol. 1991;25:1038-1043. doi:10.1016/0190-9622(91)70304-k
- Kanada KN, Nakatsuji T, Gallo RL. Doxycycline indirectly inhibits proteolytic activation of tryptic kallikrein-related peptidases and activation of cathelicidin. J Invest Dermatol. 2012;132:1435-1442. doi:10.1038/jid.2012.14
- Jang YH, Sim JH, Kang HY, et al. Immunohistochemical expression of matrix metalloproteinases in the granulomatous rosacea compared with the non-granulomatous rosacea. J Eur Acad Dermatol Venereol. 2011;25:544-548. doi:10.1111/j.1468-3083.2010.03825.x
- Khokhar O, Khachemoune A. A case of granulomatous rosacea: sorting granulomatous rosacea from other granulomatous diseases that affect the face. Dermatol Online J. 2004;10:6.
- Rosen T, Stone MS. Acne rosacea in blacks. J Am Acad Dermatol. 1987;17:70-73. doi:10.1016/s0190-9622(87)70173-x
- Adams AK, Davis JL, Davis MDP, et al. What is your diagnosis? granulomatous rosacea (lupus miliaris disseminatus faciei, acne agminata). Cutis. 2008;82:103-112.
- Shitara A. Lupus miliaris disseminatus faciei. Int J Dermatol. 1984;23:542-544. doi:10.1111/j.1365-4362.1984.tb04206.x
- Hodak E, Trattner A, Feuerman H, et al. Lupus miliaris disseminatus faciei—the DNA of Mycobacterium tuberculosis is not detectable in active lesions by polymerase chain reaction. Br J Dermatol. 1997;137:614-619. doi: 10.1111/j.1365-2133.1997.tb03797.x
- Skowron F, Causeret AS, Pabion C, et al. F.I.GU.R.E.: facial idiopathic granulomas with regressive evolution. Dermatology. 2000;201:287-289. doi:10.1159/000051539
- Hutchinson J. Case of livid papillary psoriasis. In: London J, Churchill A, eds. Illustrations of Clinical Surgery. J&A Churchill; 1877:42-43.
- Besnier E. Lupus pernio of the face [in French]. Ann Dermatol Syphiligr (Paris). 1889;10:33-36.
- Tenneson H. Lupus pernio. Ann Dermatol Syphiligr (Paris). 1889;10:333-336.
- Boeck C. Multiple benign sarkoid of the skin [in Norwegian]. Norsk Mag Laegevidensk. 1899;14:1321-1334.
- Kuznitsky E, Bittorf A. Sarkoid mit beteiligung innerer organe. Münch Med Wochenschr. 1915;62:1349-1353.
- Schaumann J. Etude sur le lupus pernio et ses rapports avec les sarcoides et la tuberculose. Ann Dermatol Syphiligr. 1916-1917;6:357-373.
- Osler W. On chronic symmetrical enlargement of the salivary and lacrimal glands. Am J Med Sci. 1898;115:27-30.
- Chen ES, Moller DR. Etiologies of sarcoidosis. Clin Rev Allergy Immunol. 2015;49:6-18. doi:10.1007/s12016-015-8481-z
- Eberhardt C, Thillai M, Parker R, et al. Proteomic analysis of Kveim reagent identifies targets of cellular immunity in sarcoidosis. PLoS One. 2017;12:e0170285. doi:10.1371/journal.pone.0170285
- Esteves TC, Aparicio G, Ferrer B, et al. Prognostic value of skin lesions in sarcoidosis: clinical and histopathological clues. Eur J Dermatol. 2015;25:556-562. doi:10.1684/ejd.2015.2666
- Cardoso JC, Cravo M, Reis JP, et al. Cutaneous sarcoidosis: a histopathological study. J Eur Acad Dermatol Venereol. 2009;23:678-682. doi:10.1111/j.1468-3083.2009.03153.x
- Mangas C, Fernández-Figueras M-T, Fité E, et al. Clinical spectrum and histological analysis of 32 cases of specific cutaneous sarcoidosis. J Cutan Pathol. 2006;33:772-777. doi:10.1111/j.1600-0560.2006.00563.x
- García-Colmenero L, Sánchez-Schmidt JM, Barranco C, et al. The natural history of cutaneous sarcoidosis. clinical spectrum and histological analysis of 40 cases. Int J Dermatol. 2019;58:178-184. doi: 10.1111/ijd.14218
- Shetty AK, Gedalia A. Childhood sarcoidosis: a rare but fascinating disorder. Pediatr Rheumatol Online J. 2008;6:16. doi:10.1186/1546-0096-6-16
- Milman N, Hoffmann AL, Byg KE. Sarcoidosis in children. epidemiology in Danes, clinical features, diagnosis, treatment and prognosis. Acta Paediatr. 1998;87:871-878. doi:10.1080/08035259875001366244. A, H, Yapıcı I. Isolated cutaneous sarcoidosis. Arch Bronconeumol. 2016;52:220.
- Scadding JG. The late stages of pulmonary sarcoidosis. Postgrad Med J. 1970;46:530-536. doi:10.1136/pgmj.46.538.530
Cutaneous granulomatous diseases encompass many entities that are skin-limited or systemic. The prototypical cutaneous granuloma is a painless, rounded, well-defined, red-pink or flesh-colored papule1 and is smooth, owing to minimal epidermal involvement. Examples of conditions that present with such lesions include granulomatous periorificial dermatitis (GPD), granulomatous rosacea (GR), lupus miliaris disseminatus faciei (LMDF), and papular sarcoidosis. These entities commonly are seen on the face and can be a source of distress to patients when they are extensive. Several reports have raised the possibility that these conditions lie on a spectrum.2-4 We present 2 cases of patients with facial papular granulomas, discuss potential causes of the lesions, review historical aspects from the literature, and highlight the challenges that these lesions can pose to the clinician.
Case Reports
Patient 1—A 10-year-old Ethiopian girl with a history of atopic dermatitis presented with a facial rash of 4 months’ duration. Her pediatrician initially treated the rash as pityriasis alba and prescribed hydrocortisone cream. Two months into treatment, the patient developed an otherwise asymptomatic, unilateral, papular dermatosis on the right cheek. She subsequently was switched to treatment with benzoyl peroxide and topical clindamycin, which she had been using for 2 months with no improvement at the time of the current presentation. The lesions then spread bilaterally and periorally.
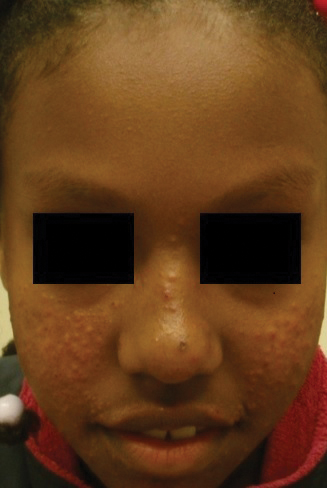
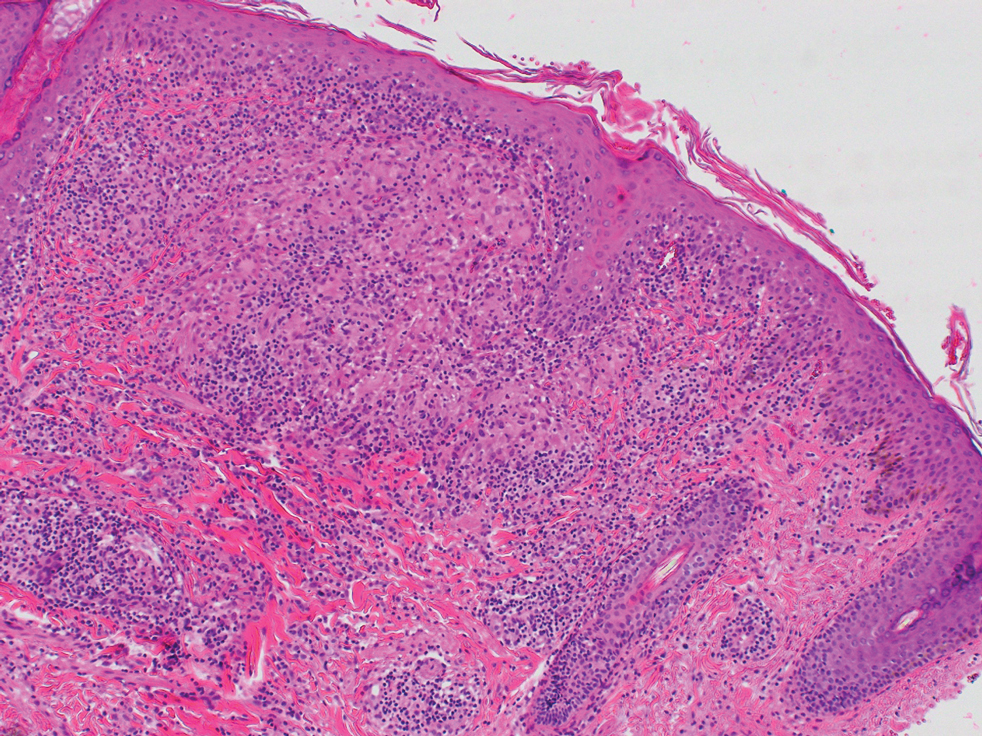
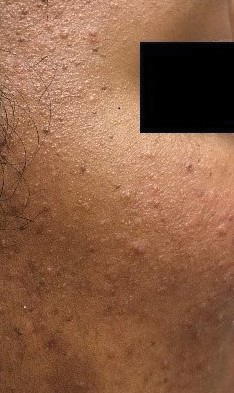
At the current presentation, physical examination demonstrated fine, diffuse, follicular-based, flesh-colored papules over both cheeks, the right side of the nose, and the perioral region (Figure 1). A biopsy of a papular lesion from the right cheek revealed well-formed, noncaseating granulomas in the superficial and mid dermis with an associated lymphocytic infiltrate (Figure 2). No organisms were identified on acid-fast, Fite, or periodic acid–Schiff staining. A tuberculin skin test was negative. A chest radiograph showed small calcified hilar lymph nodes bilaterally. Pulmonary function tests were unremarkable. Calcium and angiotensin-converting enzyme levels were normal.
The patient denied any fever, chills, hemoptysis, cough, dyspnea, lymphadenopathy, scleral or conjunctival pain or erythema, visual disturbances, or arthralgias. Hydroxychloroquine 200 mg twice daily was started with minimal improvement after 5 months. Methotrexate 20 mg once weekly was then added. Topical fluocinonide 0.05% also was started at this time, as the patient had required several prednisone tapers over the past 3 months for symptomatic relief. The lesions improved minimally after 5 more months of treatment, at which time she had developed inflammatory papules, pustules, and open comedones in the same areas as well as the glabella.
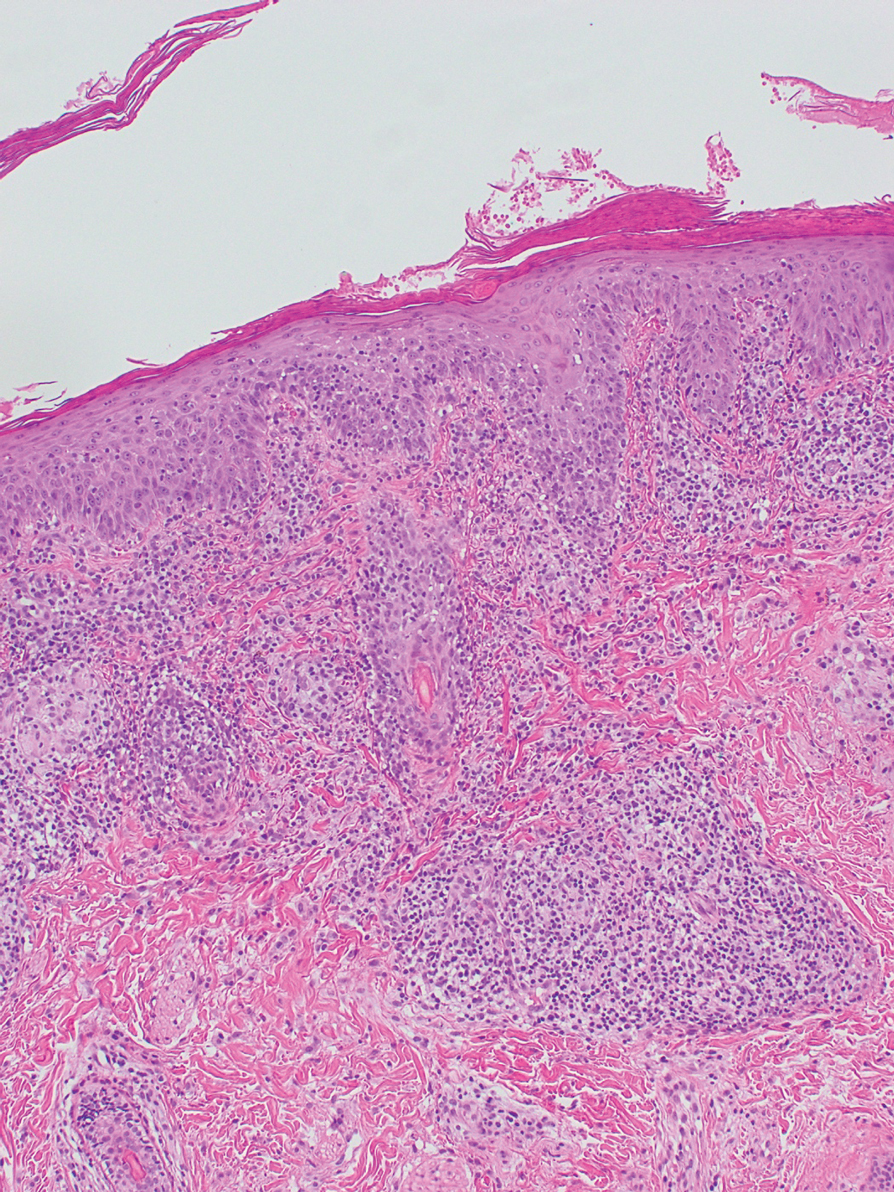
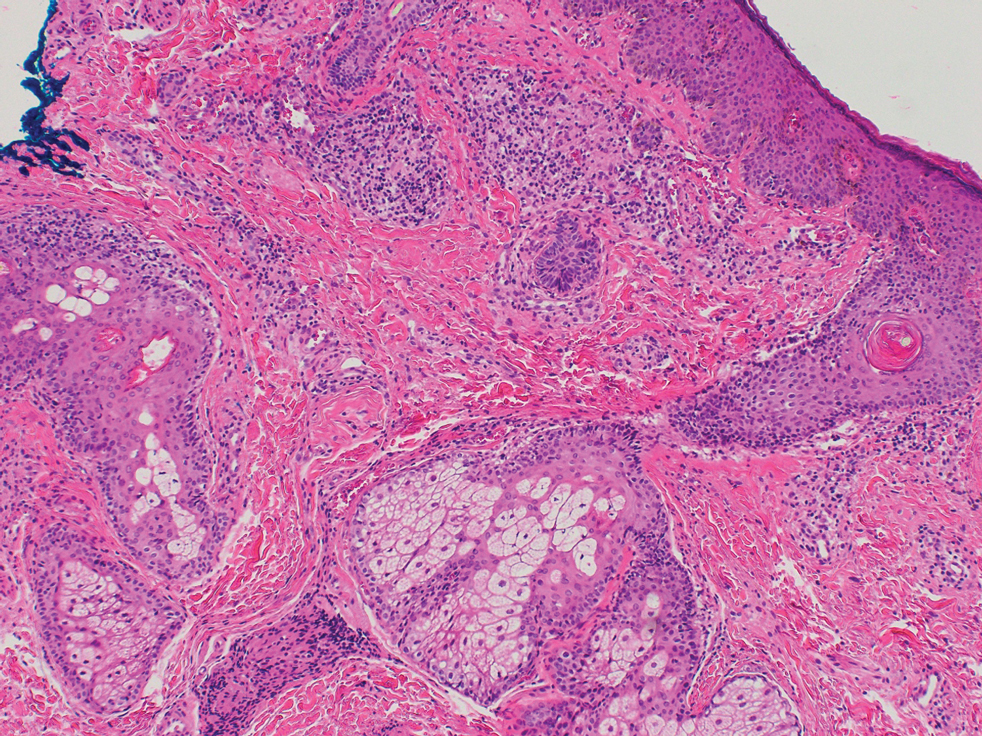
Repeat biopsy of a papular lesion demonstrated noncaseating granulomas and an associated chronic lymphocytic infiltrate in a follicular and perifollicular distribution (Figure 3). Biopsy of a pustule demonstrated acute Demodex folliculitis. Fluocinonide was stopped, and anti-mite therapy with ivermectin, permethrin cream 5%, and selenium sulfide lotion 2.5% was started, with good response from the pustular lesions.
The patient continued taking methotrexate 20 mg once weekly during this time, with improvement in the papular lesions. She discontinued methotrexate after 12 months with complete resolution. At follow-up 12 months after stopping the methotrexate (roughly 2 years after initial presentation), she showed sustained resolution, with small pitted scars on both cheeks and the nasal tip.
Patient 2—A 33-year-old Ethiopian woman presented with a facial rash of 15 years’ duration. The lesions had been accumulating slowly and were asymptomatic. Physical examination revealed multiple follicular-based, flesh-colored, and erythematous papules on the cheeks, chin, perioral area, and forehead (Figure 4). There were no pustules or telangiectasias. Treatment with tretinoin cream 0.05% for 6 months offered minimal relief.
Biopsy of a papule from the left mandible showed superficial vascular telangiectasias, noncaseating granulomas comprising epithelioid histiocytes and lymphocytes in the superficial dermis, and a perifollicular lymphocytic infiltrate (Figure 5). No organisms were identified on Fite or Gomori methenamine silver staining.
Comment
The first step in differentiating cutaneous granulomatous lesions should be to distinguish infectious from noninfectious causes.1 Noninfectious cutaneous granulomas can appear nearly anywhere; however, certain processes have a predilection for the face, including GPD, GR, LMDF, and papular sarcoidosis.5-7 These conditions generally present with papular granulomas with features as described above.
Granulomatous Periorificial Dermatitis—In 1970, Gianotti and colleagues8 briefly described the first possible cases of GPD in 5 children. The eruption comprised numerous yellow, dome-shaped papules in a mostly perioral distribution. Tuberculin and the Kveim tests were nonreactive; histopathology was described as sarcoid-type and not necessarily follicular or perifollicular.8 In 1974, Marten et al9 described 22 Afro-Caribbean children with flesh-colored, papular eruptions on the face that did not show histologic granulomatous changes but were morphologically similar to the reports by Gianotti et al.8 By 1989, Frieden and colleagues10 described this facial eruption as “granulomatous perioral dermatitis in children”. Additionally, the investigators observed granulomatous infiltrates in a perifollicular distribution and suggested follicular disruption as a possible cause. It was clear from the case discussions that these eruptions were not uncommonly diagnosed as papular sarcoidosis.10 The following year, Williams et al11 reported 5 cases of similar papular eruptions in 5 Afro-Caribbean children, coining the term facial Afro-Caribbean eruption.11 Knautz and Lesher12 referred to this entity as “childhood GPD” in 1996 to avoid limiting the diagnosis to Afro-Caribbean patients and to a perioral distribution; this is the most popular current terminology.12 Since then, reports of extrafacial involvement and disease in adults have been published.13,14
Granulomatous periorificial dermatitis often is seen in the perinasal, periocular, and perioral regions of the face.2 It is associated with topical steroid exposure.5 Histologically, noncaseating granulomas around the upper half of undisrupted hair follicles with a lymphocytic infiltrate are typical.13 Treatment should begin with cessation of any topical steroids; first-line agents are oral tetracycline or macrolide antibiotics.5 These agents can be used alone or in combination with topical erythromycin, metronidazole, or sulfur-based lotions.13 Rarely, GPD presents extrafacially.13 Even so, it usually resolves within 2 weeks to 6 months, especially with therapy; scarring is unusual.5,13,15
Granulomatous Rosacea—A report in the early 20th century described patients with tuberculoid granulomas resembling papular rosacea; the initial belief was that this finding represented a rosacealike tuberculid eruption.5 However, this belief was questioned by Snapp,16 among others, who demonstrated near universal lack of reactivity to tuberculin among 20 of these patients in 1949; more recent evidence has substantiated these findings.17 Still, Snapp16 postulated that these rosacealike granulomatous lesions were distinct from classic rosacea because they lacked vascular symptoms and pustules and were recalcitrant to rosacea treatment modalities.
In 1970, Mullanax and colleagues18 introduced the term granulomatous rosacea, reiterating that this entity was not tuberculous. They documented papulopustular lesions as well as telangiectasias, raising the possibility that GR does overlap with acne rosacea. More recent studies have established the current theory that GR is a histologic variant of acne rosacea because, in addition to typical granulomatous papules, its microscopic features can be seen across subtypes of acne rosacea.19,20
Various causes have been proposed for GR. Demodex mites have been reported in association with GR for nearly 30 years.19,20 In the past 10 years, molecular studies have started to define the role of metalloproteinases, UV radiation, and cutaneous peptides in the pathogenesis of acne rosacea and GR.21,22
Granulomatous rosacea typically is seen in middle-aged women.20,23 Hallmarks of rosacea, such as facial erythema, flushing, telangiectasias, pustules, and rhinophyma, are not always present in GR.5,20,23 Lesions usually are distributed around the central face, although extension to the cheeks, total facial involvement, and extrafacial lesions are possible.5,20 Histologically, perifollicular and follicular-based noncaseating granulomas with dilatation of the dermal papillary vasculature are seen.17,23 As a whole, rosacea is comparatively uncommon in dark-skinned patients; when it does occur, GR is a frequent presentation.24
First-line treatment for GR is tetracycline antibiotics.5 Unresponsive cases have been treated—largely anecdotally—with topical modalities (eg, metronidazole, steroids, immunomodulators), systemic agents (eg, dapsone, erythromycin, isotretinoin), and other therapies.5 Granulomatous rosacea tends to have a chronic course.5,23
Lupus Miliaris Disseminatus Faciei—Classic LMDF demonstrates caseating perifollicular granulomas histologically.6,17,25 Lesions tend to appear on the central face, particularly the eyelids, and can be seen extrafacially.3,6,25,26 Although LMDF originally was categorized as a tuberculid eruption, this no longer is thought to be the case.27 It is now regarded by some as a variant of GR25; however, LMDF responds poorly to tetracyclines, is more common in males, and lacks rosacealike vascular abnormalities, leading some to question this association.3,6,17 In the past 20 years, some have proposed renaming LMDF to better reflect its clinical course and to consider it independent of tuberculosis and GR.28 It usually resolves spontaneously after 1 to 3 years, leaving pitted scars.3,6
Papular Sarcoidosis—The first potential documented case of sarcoidosis was by Hutchinson29 in 1869 in a patient seen in London. The author labeled purple plaques on the index patient’s legs and hands as “livid papillary psoriasis.” In 1889, Besnier30 described a patient with violaceous swellings on the nose, ears, and fingers, which he called “lupus pernio”; his contemporary, Tenneson,31 published a case of lupus pernio and described its histologic profile as comprising epithelioid cells and giant cells. It was not until 1899 that the term sarkoid was used to describe these cutaneous lesions by Boeck,32 who thought they were reminiscent of sarcoma. In 1915, Kuznitsky and Bittorf33 described a patient with cutaneous lesions histologically consistent with Boeck’s sarkoid but additionally with hilar lymphadenopathy and pulmonary infiltrates. Around 1916 or 1917, Schaumann34 described patients with cutaneous lesions and additionally with involvement of pulmonary, osseous, hepatosplenic, and tonsillar tissue. These reports are among the first to recognize the multisystemic nature of sarcoidosis. The first possible case of childhood sarcoidosis might have been reported by Osler35 in the United States in 1898.
In the past century or so, an ongoing effort by researchers has focused on identifying etiologic triggers for sarcoidosis. Microbial agents have been considered in this role, with Mycobacterium and Propionibacterium organisms the most intensively studied; the possibility that foreign material contributes to the formation of granulomas also has been raised.36 Current models of the pathogenesis of sarcoidosis involve an interplay between the immune system in genetically predisposed patients and an infection that leads to a hyperimmune type 1 T–helper cell response that clears the infection but not antigens generated by the microbes and the acute host response, including proteins such as serum amyloid A and vimentin.36,37 These antigens aggregate and serve as a nidus for granuloma formation and maintenance long after infection has resolved.
Cutaneous lesions of sarcoidosis include macules, papules, plaques, and lupus pernio, as well as lesions arising within scars or tattoos, with many less common presentations.7,38 Papular sarcoidosis is common on the face but also can involve the extremities.4,7 Strictly, at least 2 organ systems must be involved to diagnose sarcoidosis, but this is debatable.4,7 Among 41 patients with cutaneous sarcoidosis, 24 (58.5%) had systemic disease; cutaneous lesions were the presenting sign in 87.5% (21/24) of patients.38 Histologic analysis, regardless of the lesion, usually shows noncaseating so-called “naked” granulomas, which have minimal lymphocytic infiltrate associated with the epithelioid histiocytes.38,39 Perifollicular granulomas are possible but unusual.40
Treatment depends on the extent of cutaneous and systemic involvement. Pharmacotherapeutic modalities include topical steroids, immunomodulators, and retinoids; systemic immunomodulators and immunosuppressants; and biologic agents.7 Isolated cutaneous sarcoidosis, particularly the papular variant, usually is associated with acute disease lasting less than 2 years, with resolution of skin lesions.7,38 That said, a recent report suggested that cutaneous sarcoidosis can progress to multisystemic disease as long as 7 years after the initial diagnosis.41
Clinical and Histologic Overlap—Despite this categorization of noninfectious facial granulomatous conditions, each has some clinical and histologic overlap with the others, which must be considered when encountering a granulomatous facial dermatosis. Both GPD and GR tend to present with lesions near the eyes, mouth, and nose, although GR can extend to lateral aspects of the face, below the mandible, and the forehead and has different demographic features.15,20,23 Granulomas in both GPD and GR generally are noncaseating and form in a follicular or perifollicular distribution within the dermis.2,15,23 Lupus miliaris disseminatus faciei and GR share a similar facial distribution in some cases.17,20 Even papular cutaneous sarcoidosis has masqueraded as GR clinically and histologically.4
Diagnostic and Treatment Difficulty—Our cases illustrate the range of difficulty in evaluating and managing patients with facial papular granulomas. On one hand, our adult patient’s clinical and histologic findings were highly consistent with GR; on the other hand, our younger patient had clinicopathologic features of both sarcoidosis and GPD at varying times. Both conditions are more common in dark-skinned patients.11,42
Juvenile sarcoidosis is comparatively rare, with a reported annual incidence of 0.22 to 0.27 for every 100,000 children younger than 15 years; however, juvenile sarcoidosis commonly presents around 8 to 15 years of age.43
It is unusual for sarcoid granulomas to be isolated to the skin, much less to the face.4,7,43,44 Patient 1 initially presented in this manner and lacked convincing laboratory or radiographic evidence of systemic sarcoidosis. Bilateral hilar calcifications in sarcoidosis are more typical among adults after 5 to 20 years; there were no signs or symptoms of active infection that could account for the pulmonary and cutaneous lesions.45
The presence of perifollicular granulomas with associated lymphocytic infiltrates on repeat biopsy, coupled with the use of topical steroids, made it difficult to rule out a contribution by GPD to her clinical course. That her lesions resolved with pitted scarring while she was taking methotrexate and after topical steroids had been stopped could be the result of successful management or spontaneous resolution of her dermatosis; both papular sarcoidosis and GPD tend to have a self-limited course.7,13
Conclusion
We present 2 cases of papular facial granulomas in patients with similar skin types who had different clinical courses. Evaluation of such lesions remains challenging given the similarity between specific entities that present in this manner. Certainly, it is reasonable to consider a spectrum upon which all of these conditions fall, in light of the findings of these cases and those reported previously.
Cutaneous granulomatous diseases encompass many entities that are skin-limited or systemic. The prototypical cutaneous granuloma is a painless, rounded, well-defined, red-pink or flesh-colored papule1 and is smooth, owing to minimal epidermal involvement. Examples of conditions that present with such lesions include granulomatous periorificial dermatitis (GPD), granulomatous rosacea (GR), lupus miliaris disseminatus faciei (LMDF), and papular sarcoidosis. These entities commonly are seen on the face and can be a source of distress to patients when they are extensive. Several reports have raised the possibility that these conditions lie on a spectrum.2-4 We present 2 cases of patients with facial papular granulomas, discuss potential causes of the lesions, review historical aspects from the literature, and highlight the challenges that these lesions can pose to the clinician.
Case Reports
Patient 1—A 10-year-old Ethiopian girl with a history of atopic dermatitis presented with a facial rash of 4 months’ duration. Her pediatrician initially treated the rash as pityriasis alba and prescribed hydrocortisone cream. Two months into treatment, the patient developed an otherwise asymptomatic, unilateral, papular dermatosis on the right cheek. She subsequently was switched to treatment with benzoyl peroxide and topical clindamycin, which she had been using for 2 months with no improvement at the time of the current presentation. The lesions then spread bilaterally and periorally.
At the current presentation, physical examination demonstrated fine, diffuse, follicular-based, flesh-colored papules over both cheeks, the right side of the nose, and the perioral region (Figure 1). A biopsy of a papular lesion from the right cheek revealed well-formed, noncaseating granulomas in the superficial and mid dermis with an associated lymphocytic infiltrate (Figure 2). No organisms were identified on acid-fast, Fite, or periodic acid–Schiff staining. A tuberculin skin test was negative. A chest radiograph showed small calcified hilar lymph nodes bilaterally. Pulmonary function tests were unremarkable. Calcium and angiotensin-converting enzyme levels were normal.
The patient denied any fever, chills, hemoptysis, cough, dyspnea, lymphadenopathy, scleral or conjunctival pain or erythema, visual disturbances, or arthralgias. Hydroxychloroquine 200 mg twice daily was started with minimal improvement after 5 months. Methotrexate 20 mg once weekly was then added. Topical fluocinonide 0.05% also was started at this time, as the patient had required several prednisone tapers over the past 3 months for symptomatic relief. The lesions improved minimally after 5 more months of treatment, at which time she had developed inflammatory papules, pustules, and open comedones in the same areas as well as the glabella.
Repeat biopsy of a papular lesion demonstrated noncaseating granulomas and an associated chronic lymphocytic infiltrate in a follicular and perifollicular distribution (Figure 3). Biopsy of a pustule demonstrated acute Demodex folliculitis. Fluocinonide was stopped, and anti-mite therapy with ivermectin, permethrin cream 5%, and selenium sulfide lotion 2.5% was started, with good response from the pustular lesions.
The patient continued taking methotrexate 20 mg once weekly during this time, with improvement in the papular lesions. She discontinued methotrexate after 12 months with complete resolution. At follow-up 12 months after stopping the methotrexate (roughly 2 years after initial presentation), she showed sustained resolution, with small pitted scars on both cheeks and the nasal tip.
Patient 2—A 33-year-old Ethiopian woman presented with a facial rash of 15 years’ duration. The lesions had been accumulating slowly and were asymptomatic. Physical examination revealed multiple follicular-based, flesh-colored, and erythematous papules on the cheeks, chin, perioral area, and forehead (Figure 4). There were no pustules or telangiectasias. Treatment with tretinoin cream 0.05% for 6 months offered minimal relief.
Biopsy of a papule from the left mandible showed superficial vascular telangiectasias, noncaseating granulomas comprising epithelioid histiocytes and lymphocytes in the superficial dermis, and a perifollicular lymphocytic infiltrate (Figure 5). No organisms were identified on Fite or Gomori methenamine silver staining.
Comment
The first step in differentiating cutaneous granulomatous lesions should be to distinguish infectious from noninfectious causes.1 Noninfectious cutaneous granulomas can appear nearly anywhere; however, certain processes have a predilection for the face, including GPD, GR, LMDF, and papular sarcoidosis.5-7 These conditions generally present with papular granulomas with features as described above.
Granulomatous Periorificial Dermatitis—In 1970, Gianotti and colleagues8 briefly described the first possible cases of GPD in 5 children. The eruption comprised numerous yellow, dome-shaped papules in a mostly perioral distribution. Tuberculin and the Kveim tests were nonreactive; histopathology was described as sarcoid-type and not necessarily follicular or perifollicular.8 In 1974, Marten et al9 described 22 Afro-Caribbean children with flesh-colored, papular eruptions on the face that did not show histologic granulomatous changes but were morphologically similar to the reports by Gianotti et al.8 By 1989, Frieden and colleagues10 described this facial eruption as “granulomatous perioral dermatitis in children”. Additionally, the investigators observed granulomatous infiltrates in a perifollicular distribution and suggested follicular disruption as a possible cause. It was clear from the case discussions that these eruptions were not uncommonly diagnosed as papular sarcoidosis.10 The following year, Williams et al11 reported 5 cases of similar papular eruptions in 5 Afro-Caribbean children, coining the term facial Afro-Caribbean eruption.11 Knautz and Lesher12 referred to this entity as “childhood GPD” in 1996 to avoid limiting the diagnosis to Afro-Caribbean patients and to a perioral distribution; this is the most popular current terminology.12 Since then, reports of extrafacial involvement and disease in adults have been published.13,14
Granulomatous periorificial dermatitis often is seen in the perinasal, periocular, and perioral regions of the face.2 It is associated with topical steroid exposure.5 Histologically, noncaseating granulomas around the upper half of undisrupted hair follicles with a lymphocytic infiltrate are typical.13 Treatment should begin with cessation of any topical steroids; first-line agents are oral tetracycline or macrolide antibiotics.5 These agents can be used alone or in combination with topical erythromycin, metronidazole, or sulfur-based lotions.13 Rarely, GPD presents extrafacially.13 Even so, it usually resolves within 2 weeks to 6 months, especially with therapy; scarring is unusual.5,13,15
Granulomatous Rosacea—A report in the early 20th century described patients with tuberculoid granulomas resembling papular rosacea; the initial belief was that this finding represented a rosacealike tuberculid eruption.5 However, this belief was questioned by Snapp,16 among others, who demonstrated near universal lack of reactivity to tuberculin among 20 of these patients in 1949; more recent evidence has substantiated these findings.17 Still, Snapp16 postulated that these rosacealike granulomatous lesions were distinct from classic rosacea because they lacked vascular symptoms and pustules and were recalcitrant to rosacea treatment modalities.
In 1970, Mullanax and colleagues18 introduced the term granulomatous rosacea, reiterating that this entity was not tuberculous. They documented papulopustular lesions as well as telangiectasias, raising the possibility that GR does overlap with acne rosacea. More recent studies have established the current theory that GR is a histologic variant of acne rosacea because, in addition to typical granulomatous papules, its microscopic features can be seen across subtypes of acne rosacea.19,20
Various causes have been proposed for GR. Demodex mites have been reported in association with GR for nearly 30 years.19,20 In the past 10 years, molecular studies have started to define the role of metalloproteinases, UV radiation, and cutaneous peptides in the pathogenesis of acne rosacea and GR.21,22
Granulomatous rosacea typically is seen in middle-aged women.20,23 Hallmarks of rosacea, such as facial erythema, flushing, telangiectasias, pustules, and rhinophyma, are not always present in GR.5,20,23 Lesions usually are distributed around the central face, although extension to the cheeks, total facial involvement, and extrafacial lesions are possible.5,20 Histologically, perifollicular and follicular-based noncaseating granulomas with dilatation of the dermal papillary vasculature are seen.17,23 As a whole, rosacea is comparatively uncommon in dark-skinned patients; when it does occur, GR is a frequent presentation.24
First-line treatment for GR is tetracycline antibiotics.5 Unresponsive cases have been treated—largely anecdotally—with topical modalities (eg, metronidazole, steroids, immunomodulators), systemic agents (eg, dapsone, erythromycin, isotretinoin), and other therapies.5 Granulomatous rosacea tends to have a chronic course.5,23
Lupus Miliaris Disseminatus Faciei—Classic LMDF demonstrates caseating perifollicular granulomas histologically.6,17,25 Lesions tend to appear on the central face, particularly the eyelids, and can be seen extrafacially.3,6,25,26 Although LMDF originally was categorized as a tuberculid eruption, this no longer is thought to be the case.27 It is now regarded by some as a variant of GR25; however, LMDF responds poorly to tetracyclines, is more common in males, and lacks rosacealike vascular abnormalities, leading some to question this association.3,6,17 In the past 20 years, some have proposed renaming LMDF to better reflect its clinical course and to consider it independent of tuberculosis and GR.28 It usually resolves spontaneously after 1 to 3 years, leaving pitted scars.3,6
Papular Sarcoidosis—The first potential documented case of sarcoidosis was by Hutchinson29 in 1869 in a patient seen in London. The author labeled purple plaques on the index patient’s legs and hands as “livid papillary psoriasis.” In 1889, Besnier30 described a patient with violaceous swellings on the nose, ears, and fingers, which he called “lupus pernio”; his contemporary, Tenneson,31 published a case of lupus pernio and described its histologic profile as comprising epithelioid cells and giant cells. It was not until 1899 that the term sarkoid was used to describe these cutaneous lesions by Boeck,32 who thought they were reminiscent of sarcoma. In 1915, Kuznitsky and Bittorf33 described a patient with cutaneous lesions histologically consistent with Boeck’s sarkoid but additionally with hilar lymphadenopathy and pulmonary infiltrates. Around 1916 or 1917, Schaumann34 described patients with cutaneous lesions and additionally with involvement of pulmonary, osseous, hepatosplenic, and tonsillar tissue. These reports are among the first to recognize the multisystemic nature of sarcoidosis. The first possible case of childhood sarcoidosis might have been reported by Osler35 in the United States in 1898.
In the past century or so, an ongoing effort by researchers has focused on identifying etiologic triggers for sarcoidosis. Microbial agents have been considered in this role, with Mycobacterium and Propionibacterium organisms the most intensively studied; the possibility that foreign material contributes to the formation of granulomas also has been raised.36 Current models of the pathogenesis of sarcoidosis involve an interplay between the immune system in genetically predisposed patients and an infection that leads to a hyperimmune type 1 T–helper cell response that clears the infection but not antigens generated by the microbes and the acute host response, including proteins such as serum amyloid A and vimentin.36,37 These antigens aggregate and serve as a nidus for granuloma formation and maintenance long after infection has resolved.
Cutaneous lesions of sarcoidosis include macules, papules, plaques, and lupus pernio, as well as lesions arising within scars or tattoos, with many less common presentations.7,38 Papular sarcoidosis is common on the face but also can involve the extremities.4,7 Strictly, at least 2 organ systems must be involved to diagnose sarcoidosis, but this is debatable.4,7 Among 41 patients with cutaneous sarcoidosis, 24 (58.5%) had systemic disease; cutaneous lesions were the presenting sign in 87.5% (21/24) of patients.38 Histologic analysis, regardless of the lesion, usually shows noncaseating so-called “naked” granulomas, which have minimal lymphocytic infiltrate associated with the epithelioid histiocytes.38,39 Perifollicular granulomas are possible but unusual.40
Treatment depends on the extent of cutaneous and systemic involvement. Pharmacotherapeutic modalities include topical steroids, immunomodulators, and retinoids; systemic immunomodulators and immunosuppressants; and biologic agents.7 Isolated cutaneous sarcoidosis, particularly the papular variant, usually is associated with acute disease lasting less than 2 years, with resolution of skin lesions.7,38 That said, a recent report suggested that cutaneous sarcoidosis can progress to multisystemic disease as long as 7 years after the initial diagnosis.41
Clinical and Histologic Overlap—Despite this categorization of noninfectious facial granulomatous conditions, each has some clinical and histologic overlap with the others, which must be considered when encountering a granulomatous facial dermatosis. Both GPD and GR tend to present with lesions near the eyes, mouth, and nose, although GR can extend to lateral aspects of the face, below the mandible, and the forehead and has different demographic features.15,20,23 Granulomas in both GPD and GR generally are noncaseating and form in a follicular or perifollicular distribution within the dermis.2,15,23 Lupus miliaris disseminatus faciei and GR share a similar facial distribution in some cases.17,20 Even papular cutaneous sarcoidosis has masqueraded as GR clinically and histologically.4
Diagnostic and Treatment Difficulty—Our cases illustrate the range of difficulty in evaluating and managing patients with facial papular granulomas. On one hand, our adult patient’s clinical and histologic findings were highly consistent with GR; on the other hand, our younger patient had clinicopathologic features of both sarcoidosis and GPD at varying times. Both conditions are more common in dark-skinned patients.11,42
Juvenile sarcoidosis is comparatively rare, with a reported annual incidence of 0.22 to 0.27 for every 100,000 children younger than 15 years; however, juvenile sarcoidosis commonly presents around 8 to 15 years of age.43
It is unusual for sarcoid granulomas to be isolated to the skin, much less to the face.4,7,43,44 Patient 1 initially presented in this manner and lacked convincing laboratory or radiographic evidence of systemic sarcoidosis. Bilateral hilar calcifications in sarcoidosis are more typical among adults after 5 to 20 years; there were no signs or symptoms of active infection that could account for the pulmonary and cutaneous lesions.45
The presence of perifollicular granulomas with associated lymphocytic infiltrates on repeat biopsy, coupled with the use of topical steroids, made it difficult to rule out a contribution by GPD to her clinical course. That her lesions resolved with pitted scarring while she was taking methotrexate and after topical steroids had been stopped could be the result of successful management or spontaneous resolution of her dermatosis; both papular sarcoidosis and GPD tend to have a self-limited course.7,13
Conclusion
We present 2 cases of papular facial granulomas in patients with similar skin types who had different clinical courses. Evaluation of such lesions remains challenging given the similarity between specific entities that present in this manner. Certainly, it is reasonable to consider a spectrum upon which all of these conditions fall, in light of the findings of these cases and those reported previously.
- Beretta-Piccoli BT, Mainetti C, Peeters M-A, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146. doi:10.1007/s12016-017-8666-8
- Lucas CR, Korman NJ, Gilliam AC. Granulomatous periorificial dermatitis: a variant of granulomatous rosacea in children? J Cutan Med Surg. 2009;13:115-118. doi:10.2310/7750.2008.07088
- van de Scheur MR, van der Waal RIF, Starink TM. Lupus miliaris disseminatus faciei: a distinctive rosacea-like syndrome and not a granulomatous form of rosacea. Dermatology. 2003;206:120-123. doi:10.1159/000068457
- Simonart T, Lowy M, Rasquin F, et al. Overlap of sarcoidosis and rosacea. Dermatology. 1997;194:416-418. doi:10.1159/000246165
- Lee GL, Zirwas MJ. Granulomatous rosacea and periorificial dermatitis: controversies and review of management. Dermatol Clin. 2015;33:447-455. doi:10.1016/j.det.2015.03.009
- Michaels JD, Cook-Norris RH, Lehman JS, et al. Adult with papular eruption of the central aspect of the face. J Am Acad Dermatol. 2014;71:410-412. doi:10.1016/j.jaad.2012.06.039
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med. 2015;38:685-702. doi:10.1016/j.ccm.2015.08.010
- Gianotti F, Ermacora E, Benelli MG, et al. Particulière dermatite peri-orale infantile. observations sur 5 cas. Bull Soc Fr Dermatol Syphiligr. 1970;77:341.
- Marten RH, Presbury DG, Adamson JE, et al. An unusual papular and acneiform facial eruption in the negro child. Br J Dermatol. 1974;91:435-438. doi:10.1111/j.1365-2133.1974.tb13083.x
- Frieden IJ, Prose NS, Fletcher V, et al. Granulomatous perioral dermatitis in children. Arch Dermatol. 1989;125:369-373.
- Williams HC, Ashworth J, Pembroke AC, et al. FACE—facial Afro-Caribbean childhood eruption. Clin Exp Dermatol. 1990;15:163-166. doi:10.1111/j.1365-2230.1990.tb02063.x
- Knautz MA, Lesher JL Jr. Childhood granulomatous periorificial dermatitis. Pediatr Dermatol. 1996;13:131-134. doi:10.1111/j.1525-1470.1996.tb01419.x
- Urbatsch AJ, Frieden I, Williams ML, et al. Extrafacial and generalized granulomatous periorificial dermatitis. Arch Dermatol. 2002;138:1354-1358. doi:10.1001/archderm.138.10.1354
- Vincenzi C, Parente G, Tosti A. Perioral granulomatous dermatitis: two cases treated with clarithromycin. J Dermatol Treat. 2000;11:57-61.
- Kim YJ, Shin JW, Lee JS, et al. Childhood granulomatous periorificial dermatitis. Ann Dermatol. 2011;23:386-388. doi:10.5021/ad.2011.23.3.386
- Snapp RH. Lewandowsky’s rosacea-like eruption; a clinical study. J Invest Dermatol. 1949;13:175-190. doi:10.1038/jid.1949.86
- Chougule A, Chatterjee D, Sethi S, et al. Granulomatous rosacea versus lupus miliaris disseminatus faciei—2 faces of facial granulomatous disorder: a clinicohistological and molecular study. Am J Dermatopathol. 2018;40:819-823. doi:10.1097/DAD.0000000000001243
- Mullanax MG, Kierland RR. Granulomatous rosacea. Arch Dermatol. 1970;101:206-211.
- Sánchez JL, Berlingeri-Ramos AC, Dueño DV. Granulomatous rosacea. Am J Dermatopathol. 2008;30:6-9. doi:10.1097/DAD.0b013e31815bc191
- Helm KF, Menz J, Gibson LE, et al. A clinical and histopathologic study of granulomatous rosacea. J Am Acad Dermatol. 1991;25:1038-1043. doi:10.1016/0190-9622(91)70304-k
- Kanada KN, Nakatsuji T, Gallo RL. Doxycycline indirectly inhibits proteolytic activation of tryptic kallikrein-related peptidases and activation of cathelicidin. J Invest Dermatol. 2012;132:1435-1442. doi:10.1038/jid.2012.14
- Jang YH, Sim JH, Kang HY, et al. Immunohistochemical expression of matrix metalloproteinases in the granulomatous rosacea compared with the non-granulomatous rosacea. J Eur Acad Dermatol Venereol. 2011;25:544-548. doi:10.1111/j.1468-3083.2010.03825.x
- Khokhar O, Khachemoune A. A case of granulomatous rosacea: sorting granulomatous rosacea from other granulomatous diseases that affect the face. Dermatol Online J. 2004;10:6.
- Rosen T, Stone MS. Acne rosacea in blacks. J Am Acad Dermatol. 1987;17:70-73. doi:10.1016/s0190-9622(87)70173-x
- Adams AK, Davis JL, Davis MDP, et al. What is your diagnosis? granulomatous rosacea (lupus miliaris disseminatus faciei, acne agminata). Cutis. 2008;82:103-112.
- Shitara A. Lupus miliaris disseminatus faciei. Int J Dermatol. 1984;23:542-544. doi:10.1111/j.1365-4362.1984.tb04206.x
- Hodak E, Trattner A, Feuerman H, et al. Lupus miliaris disseminatus faciei—the DNA of Mycobacterium tuberculosis is not detectable in active lesions by polymerase chain reaction. Br J Dermatol. 1997;137:614-619. doi: 10.1111/j.1365-2133.1997.tb03797.x
- Skowron F, Causeret AS, Pabion C, et al. F.I.GU.R.E.: facial idiopathic granulomas with regressive evolution. Dermatology. 2000;201:287-289. doi:10.1159/000051539
- Hutchinson J. Case of livid papillary psoriasis. In: London J, Churchill A, eds. Illustrations of Clinical Surgery. J&A Churchill; 1877:42-43.
- Besnier E. Lupus pernio of the face [in French]. Ann Dermatol Syphiligr (Paris). 1889;10:33-36.
- Tenneson H. Lupus pernio. Ann Dermatol Syphiligr (Paris). 1889;10:333-336.
- Boeck C. Multiple benign sarkoid of the skin [in Norwegian]. Norsk Mag Laegevidensk. 1899;14:1321-1334.
- Kuznitsky E, Bittorf A. Sarkoid mit beteiligung innerer organe. Münch Med Wochenschr. 1915;62:1349-1353.
- Schaumann J. Etude sur le lupus pernio et ses rapports avec les sarcoides et la tuberculose. Ann Dermatol Syphiligr. 1916-1917;6:357-373.
- Osler W. On chronic symmetrical enlargement of the salivary and lacrimal glands. Am J Med Sci. 1898;115:27-30.
- Chen ES, Moller DR. Etiologies of sarcoidosis. Clin Rev Allergy Immunol. 2015;49:6-18. doi:10.1007/s12016-015-8481-z
- Eberhardt C, Thillai M, Parker R, et al. Proteomic analysis of Kveim reagent identifies targets of cellular immunity in sarcoidosis. PLoS One. 2017;12:e0170285. doi:10.1371/journal.pone.0170285
- Esteves TC, Aparicio G, Ferrer B, et al. Prognostic value of skin lesions in sarcoidosis: clinical and histopathological clues. Eur J Dermatol. 2015;25:556-562. doi:10.1684/ejd.2015.2666
- Cardoso JC, Cravo M, Reis JP, et al. Cutaneous sarcoidosis: a histopathological study. J Eur Acad Dermatol Venereol. 2009;23:678-682. doi:10.1111/j.1468-3083.2009.03153.x
- Mangas C, Fernández-Figueras M-T, Fité E, et al. Clinical spectrum and histological analysis of 32 cases of specific cutaneous sarcoidosis. J Cutan Pathol. 2006;33:772-777. doi:10.1111/j.1600-0560.2006.00563.x
- García-Colmenero L, Sánchez-Schmidt JM, Barranco C, et al. The natural history of cutaneous sarcoidosis. clinical spectrum and histological analysis of 40 cases. Int J Dermatol. 2019;58:178-184. doi: 10.1111/ijd.14218
- Shetty AK, Gedalia A. Childhood sarcoidosis: a rare but fascinating disorder. Pediatr Rheumatol Online J. 2008;6:16. doi:10.1186/1546-0096-6-16
- Milman N, Hoffmann AL, Byg KE. Sarcoidosis in children. epidemiology in Danes, clinical features, diagnosis, treatment and prognosis. Acta Paediatr. 1998;87:871-878. doi:10.1080/08035259875001366244. A, H, Yapıcı I. Isolated cutaneous sarcoidosis. Arch Bronconeumol. 2016;52:220.
- Scadding JG. The late stages of pulmonary sarcoidosis. Postgrad Med J. 1970;46:530-536. doi:10.1136/pgmj.46.538.530
- Beretta-Piccoli BT, Mainetti C, Peeters M-A, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146. doi:10.1007/s12016-017-8666-8
- Lucas CR, Korman NJ, Gilliam AC. Granulomatous periorificial dermatitis: a variant of granulomatous rosacea in children? J Cutan Med Surg. 2009;13:115-118. doi:10.2310/7750.2008.07088
- van de Scheur MR, van der Waal RIF, Starink TM. Lupus miliaris disseminatus faciei: a distinctive rosacea-like syndrome and not a granulomatous form of rosacea. Dermatology. 2003;206:120-123. doi:10.1159/000068457
- Simonart T, Lowy M, Rasquin F, et al. Overlap of sarcoidosis and rosacea. Dermatology. 1997;194:416-418. doi:10.1159/000246165
- Lee GL, Zirwas MJ. Granulomatous rosacea and periorificial dermatitis: controversies and review of management. Dermatol Clin. 2015;33:447-455. doi:10.1016/j.det.2015.03.009
- Michaels JD, Cook-Norris RH, Lehman JS, et al. Adult with papular eruption of the central aspect of the face. J Am Acad Dermatol. 2014;71:410-412. doi:10.1016/j.jaad.2012.06.039
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med. 2015;38:685-702. doi:10.1016/j.ccm.2015.08.010
- Gianotti F, Ermacora E, Benelli MG, et al. Particulière dermatite peri-orale infantile. observations sur 5 cas. Bull Soc Fr Dermatol Syphiligr. 1970;77:341.
- Marten RH, Presbury DG, Adamson JE, et al. An unusual papular and acneiform facial eruption in the negro child. Br J Dermatol. 1974;91:435-438. doi:10.1111/j.1365-2133.1974.tb13083.x
- Frieden IJ, Prose NS, Fletcher V, et al. Granulomatous perioral dermatitis in children. Arch Dermatol. 1989;125:369-373.
- Williams HC, Ashworth J, Pembroke AC, et al. FACE—facial Afro-Caribbean childhood eruption. Clin Exp Dermatol. 1990;15:163-166. doi:10.1111/j.1365-2230.1990.tb02063.x
- Knautz MA, Lesher JL Jr. Childhood granulomatous periorificial dermatitis. Pediatr Dermatol. 1996;13:131-134. doi:10.1111/j.1525-1470.1996.tb01419.x
- Urbatsch AJ, Frieden I, Williams ML, et al. Extrafacial and generalized granulomatous periorificial dermatitis. Arch Dermatol. 2002;138:1354-1358. doi:10.1001/archderm.138.10.1354
- Vincenzi C, Parente G, Tosti A. Perioral granulomatous dermatitis: two cases treated with clarithromycin. J Dermatol Treat. 2000;11:57-61.
- Kim YJ, Shin JW, Lee JS, et al. Childhood granulomatous periorificial dermatitis. Ann Dermatol. 2011;23:386-388. doi:10.5021/ad.2011.23.3.386
- Snapp RH. Lewandowsky’s rosacea-like eruption; a clinical study. J Invest Dermatol. 1949;13:175-190. doi:10.1038/jid.1949.86
- Chougule A, Chatterjee D, Sethi S, et al. Granulomatous rosacea versus lupus miliaris disseminatus faciei—2 faces of facial granulomatous disorder: a clinicohistological and molecular study. Am J Dermatopathol. 2018;40:819-823. doi:10.1097/DAD.0000000000001243
- Mullanax MG, Kierland RR. Granulomatous rosacea. Arch Dermatol. 1970;101:206-211.
- Sánchez JL, Berlingeri-Ramos AC, Dueño DV. Granulomatous rosacea. Am J Dermatopathol. 2008;30:6-9. doi:10.1097/DAD.0b013e31815bc191
- Helm KF, Menz J, Gibson LE, et al. A clinical and histopathologic study of granulomatous rosacea. J Am Acad Dermatol. 1991;25:1038-1043. doi:10.1016/0190-9622(91)70304-k
- Kanada KN, Nakatsuji T, Gallo RL. Doxycycline indirectly inhibits proteolytic activation of tryptic kallikrein-related peptidases and activation of cathelicidin. J Invest Dermatol. 2012;132:1435-1442. doi:10.1038/jid.2012.14
- Jang YH, Sim JH, Kang HY, et al. Immunohistochemical expression of matrix metalloproteinases in the granulomatous rosacea compared with the non-granulomatous rosacea. J Eur Acad Dermatol Venereol. 2011;25:544-548. doi:10.1111/j.1468-3083.2010.03825.x
- Khokhar O, Khachemoune A. A case of granulomatous rosacea: sorting granulomatous rosacea from other granulomatous diseases that affect the face. Dermatol Online J. 2004;10:6.
- Rosen T, Stone MS. Acne rosacea in blacks. J Am Acad Dermatol. 1987;17:70-73. doi:10.1016/s0190-9622(87)70173-x
- Adams AK, Davis JL, Davis MDP, et al. What is your diagnosis? granulomatous rosacea (lupus miliaris disseminatus faciei, acne agminata). Cutis. 2008;82:103-112.
- Shitara A. Lupus miliaris disseminatus faciei. Int J Dermatol. 1984;23:542-544. doi:10.1111/j.1365-4362.1984.tb04206.x
- Hodak E, Trattner A, Feuerman H, et al. Lupus miliaris disseminatus faciei—the DNA of Mycobacterium tuberculosis is not detectable in active lesions by polymerase chain reaction. Br J Dermatol. 1997;137:614-619. doi: 10.1111/j.1365-2133.1997.tb03797.x
- Skowron F, Causeret AS, Pabion C, et al. F.I.GU.R.E.: facial idiopathic granulomas with regressive evolution. Dermatology. 2000;201:287-289. doi:10.1159/000051539
- Hutchinson J. Case of livid papillary psoriasis. In: London J, Churchill A, eds. Illustrations of Clinical Surgery. J&A Churchill; 1877:42-43.
- Besnier E. Lupus pernio of the face [in French]. Ann Dermatol Syphiligr (Paris). 1889;10:33-36.
- Tenneson H. Lupus pernio. Ann Dermatol Syphiligr (Paris). 1889;10:333-336.
- Boeck C. Multiple benign sarkoid of the skin [in Norwegian]. Norsk Mag Laegevidensk. 1899;14:1321-1334.
- Kuznitsky E, Bittorf A. Sarkoid mit beteiligung innerer organe. Münch Med Wochenschr. 1915;62:1349-1353.
- Schaumann J. Etude sur le lupus pernio et ses rapports avec les sarcoides et la tuberculose. Ann Dermatol Syphiligr. 1916-1917;6:357-373.
- Osler W. On chronic symmetrical enlargement of the salivary and lacrimal glands. Am J Med Sci. 1898;115:27-30.
- Chen ES, Moller DR. Etiologies of sarcoidosis. Clin Rev Allergy Immunol. 2015;49:6-18. doi:10.1007/s12016-015-8481-z
- Eberhardt C, Thillai M, Parker R, et al. Proteomic analysis of Kveim reagent identifies targets of cellular immunity in sarcoidosis. PLoS One. 2017;12:e0170285. doi:10.1371/journal.pone.0170285
- Esteves TC, Aparicio G, Ferrer B, et al. Prognostic value of skin lesions in sarcoidosis: clinical and histopathological clues. Eur J Dermatol. 2015;25:556-562. doi:10.1684/ejd.2015.2666
- Cardoso JC, Cravo M, Reis JP, et al. Cutaneous sarcoidosis: a histopathological study. J Eur Acad Dermatol Venereol. 2009;23:678-682. doi:10.1111/j.1468-3083.2009.03153.x
- Mangas C, Fernández-Figueras M-T, Fité E, et al. Clinical spectrum and histological analysis of 32 cases of specific cutaneous sarcoidosis. J Cutan Pathol. 2006;33:772-777. doi:10.1111/j.1600-0560.2006.00563.x
- García-Colmenero L, Sánchez-Schmidt JM, Barranco C, et al. The natural history of cutaneous sarcoidosis. clinical spectrum and histological analysis of 40 cases. Int J Dermatol. 2019;58:178-184. doi: 10.1111/ijd.14218
- Shetty AK, Gedalia A. Childhood sarcoidosis: a rare but fascinating disorder. Pediatr Rheumatol Online J. 2008;6:16. doi:10.1186/1546-0096-6-16
- Milman N, Hoffmann AL, Byg KE. Sarcoidosis in children. epidemiology in Danes, clinical features, diagnosis, treatment and prognosis. Acta Paediatr. 1998;87:871-878. doi:10.1080/08035259875001366244. A, H, Yapıcı I. Isolated cutaneous sarcoidosis. Arch Bronconeumol. 2016;52:220.
- Scadding JG. The late stages of pulmonary sarcoidosis. Postgrad Med J. 1970;46:530-536. doi:10.1136/pgmj.46.538.530
Practice Points
- Dermatologists should be aware that noninfectious granulomatous dermatosis of the face can be caused by granulomatous periorificial dermatitis, granulomatous rosacea, lupus miliaris disseminatus faciei, and papular sarcoidosis.
- These conditions lie on a spectrum, suggested by their historical description and clinical and histological features.
- Because their clinical courses can vary considerably from patient to patient, a thorough effort should be made to differentiate these conditions.
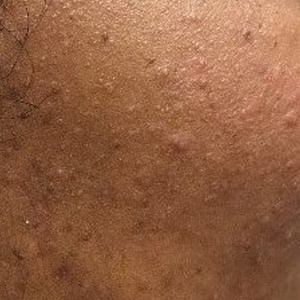
Unilateral Papules on the Face
The Diagnosis: Mosaic Tuberous Sclerosis
A punch biopsy of the facial lesion was stained with hematoxylin and eosin, which demonstrated spindled and stellate fibroblasts with dilated blood vessels (Figure), consistent with an angiofibroma. Given the clinical presentation and histologic findings, there was concern for a diagnosis of tuberous sclerosis (TSC). The patient was referred for genetic workup but tested negative for mutations of the TSC genes in the blood. Because the patient had only unilateral facial lesions, a possible cortical tuber, no other symptoms, and tested negative for TSC gene mutations, mosaic TSC was considered a likely diagnosis. Her facial lesions were treated with pulsed dye vascular laser therapy.
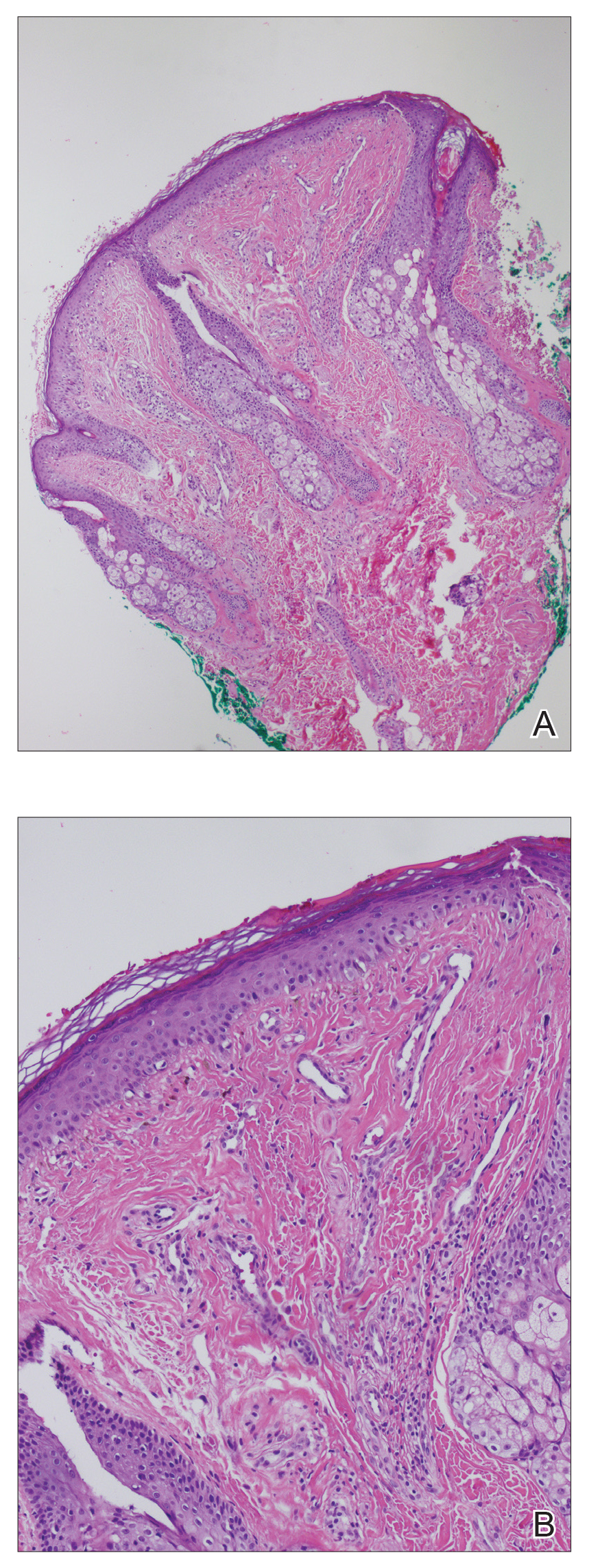
Tuberous sclerosis is an autosomal-dominant neurocutaneous disorder caused by inactivation of the genes TSC1 (encoding hamartin) and TSC2 (encoding tuberin). Mutation results in overactivation of the downstream mTOR (mammalian target of rapamycin) pathway, resulting in abnormal cellular proliferation and hamartomas. These benign tumors can be found in nearly every organ, most often in the central nervous system and skin, and they provide for a highly variable presentation of the disease.1
Tuberous sclerosis affects 1 in 6000 to 10,000 live births and has a prevalence of 1 in 20,000 individuals. Of these individuals, 75% carry sporadic mutations, and 75% to 90% eventually test positive for a TSC gene mutation.2 Genetic mosaicism has been reported in 28% of cases affected by large deletions1 and as few as 1% of cases involving small mutations.3
The dermatologic manifestation of mosaic TSC most often includes unilateral angiofibromas, whereas in nonmosaic cases, angiofibromas cover both cheeks, the forehead, and the eyelids. The other skin lesions of TSC--shagreen patches, forehead plaques, hypomelanotic macules, and ungual fibromas--are seen less frequently.4-6 Additionally, neurologic disease in mosaic patients is notably milder, with 57% of mosaic patients found to have epilepsy compared to 91% of nonmosaic patients.7 Our patient had both unilateral facial angiofibromas and a cortical lesion suspicious for a tuber, prompting a suspected diagnosis of mosaic TSC.
The methods of diagnosis outlined by the International Tuberous Sclerosis Complex Consensus Group pose a challenge in diagnosing mosaic TSC. The clinical criteria require 2 major (eg, multiple angiofibromas, angiomyolipomas, a shagreen patch) and 1 minor feature (eg, dental enamel pit, renal cyst).2 However, case reports detailing unilateral facial angiofibromas have described patients with isolated dermatologic findings.5,6 Further, it has been demonstrated that genetic studies in mosaic TSC can be unreliable depending on the tissue sampled.8 Thus, for patients who have mosaic TSC, establishing a definitive diagnosis is not always possible and may rely solely on the clinical picture.
Considering the differential diagnosis, benign cephalic histiocytosis usually would present with small red-brown macules and papules symmetrically located on the head and neck. The lesions occur at a younger age, usually in the first year or two of life. Fibrofolliculomas present as multiple whitish, slightly larger papules found on the central face. They are a marker for Birt-Hogg-Dubé syndrome, which also is associated with spontaneous pneumothorax.
Agminated means clustering or grouping of lesions. Agminated melanocytic nevi and agminated Spitz nevi are clusters of nevi. These lesions can vary in size and color. They may be elevated or flat. Melanocytic nevi usually are tan-brown or black. Spitz nevi may be pink or pigmented brown or black. To definitively differentiate between these 2 diagnoses and this patient's diagnosis of angiofibroma, a biopsy is needed.
- Curatolo P, Moavero R, Roberto D, et al. Genotype/phenotype correlations in tuberous sclerosis complex. Semin Pediatr Neurol. 2015;22:259-273.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Kwiatkowski DJ, Whittemore VH, Thiele EA. Tuberous Sclerosis Complex: Genes, Clinical Features and Therapeutics. Weinham, Germany: Wiley-Blackwell; 2011.
- Alshaiji JM, Spock CR, Connelly EA, et al. Facial angiofibromas in a mosaic pattern tuberous sclerosis: a case report. Dermatol Online J. 2012;18:8.
- Gutte R, Khopkar U. Unilateral multiple facial angiofibromas: a case report with brief review of literature. Indian J Dermatol. 2013;58:159.
- Silvestre JF, Bañuls J, Ramón R, et al. Unilateral multiple facial angiofibromas: a mosaic form of TSC. J Am Acad Dermatol. 2000;43(1, pt 1):127-129.
- Kozlowski P, Roberts P, Dabora S, et al. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet. 2007;121:389-400.
- Kwiatkowska J, Wigowska-Sowinska J, Napierala D, et al. Mosaicism in TSC as a potential cause of the failure of molecular diagnosis. N Engl J Med. 1999;340:703-707.
The Diagnosis: Mosaic Tuberous Sclerosis
A punch biopsy of the facial lesion was stained with hematoxylin and eosin, which demonstrated spindled and stellate fibroblasts with dilated blood vessels (Figure), consistent with an angiofibroma. Given the clinical presentation and histologic findings, there was concern for a diagnosis of tuberous sclerosis (TSC). The patient was referred for genetic workup but tested negative for mutations of the TSC genes in the blood. Because the patient had only unilateral facial lesions, a possible cortical tuber, no other symptoms, and tested negative for TSC gene mutations, mosaic TSC was considered a likely diagnosis. Her facial lesions were treated with pulsed dye vascular laser therapy.
Tuberous sclerosis is an autosomal-dominant neurocutaneous disorder caused by inactivation of the genes TSC1 (encoding hamartin) and TSC2 (encoding tuberin). Mutation results in overactivation of the downstream mTOR (mammalian target of rapamycin) pathway, resulting in abnormal cellular proliferation and hamartomas. These benign tumors can be found in nearly every organ, most often in the central nervous system and skin, and they provide for a highly variable presentation of the disease.1
Tuberous sclerosis affects 1 in 6000 to 10,000 live births and has a prevalence of 1 in 20,000 individuals. Of these individuals, 75% carry sporadic mutations, and 75% to 90% eventually test positive for a TSC gene mutation.2 Genetic mosaicism has been reported in 28% of cases affected by large deletions1 and as few as 1% of cases involving small mutations.3
The dermatologic manifestation of mosaic TSC most often includes unilateral angiofibromas, whereas in nonmosaic cases, angiofibromas cover both cheeks, the forehead, and the eyelids. The other skin lesions of TSC--shagreen patches, forehead plaques, hypomelanotic macules, and ungual fibromas--are seen less frequently.4-6 Additionally, neurologic disease in mosaic patients is notably milder, with 57% of mosaic patients found to have epilepsy compared to 91% of nonmosaic patients.7 Our patient had both unilateral facial angiofibromas and a cortical lesion suspicious for a tuber, prompting a suspected diagnosis of mosaic TSC.
The methods of diagnosis outlined by the International Tuberous Sclerosis Complex Consensus Group pose a challenge in diagnosing mosaic TSC. The clinical criteria require 2 major (eg, multiple angiofibromas, angiomyolipomas, a shagreen patch) and 1 minor feature (eg, dental enamel pit, renal cyst).2 However, case reports detailing unilateral facial angiofibromas have described patients with isolated dermatologic findings.5,6 Further, it has been demonstrated that genetic studies in mosaic TSC can be unreliable depending on the tissue sampled.8 Thus, for patients who have mosaic TSC, establishing a definitive diagnosis is not always possible and may rely solely on the clinical picture.
Considering the differential diagnosis, benign cephalic histiocytosis usually would present with small red-brown macules and papules symmetrically located on the head and neck. The lesions occur at a younger age, usually in the first year or two of life. Fibrofolliculomas present as multiple whitish, slightly larger papules found on the central face. They are a marker for Birt-Hogg-Dubé syndrome, which also is associated with spontaneous pneumothorax.
Agminated means clustering or grouping of lesions. Agminated melanocytic nevi and agminated Spitz nevi are clusters of nevi. These lesions can vary in size and color. They may be elevated or flat. Melanocytic nevi usually are tan-brown or black. Spitz nevi may be pink or pigmented brown or black. To definitively differentiate between these 2 diagnoses and this patient's diagnosis of angiofibroma, a biopsy is needed.
The Diagnosis: Mosaic Tuberous Sclerosis
A punch biopsy of the facial lesion was stained with hematoxylin and eosin, which demonstrated spindled and stellate fibroblasts with dilated blood vessels (Figure), consistent with an angiofibroma. Given the clinical presentation and histologic findings, there was concern for a diagnosis of tuberous sclerosis (TSC). The patient was referred for genetic workup but tested negative for mutations of the TSC genes in the blood. Because the patient had only unilateral facial lesions, a possible cortical tuber, no other symptoms, and tested negative for TSC gene mutations, mosaic TSC was considered a likely diagnosis. Her facial lesions were treated with pulsed dye vascular laser therapy.
Tuberous sclerosis is an autosomal-dominant neurocutaneous disorder caused by inactivation of the genes TSC1 (encoding hamartin) and TSC2 (encoding tuberin). Mutation results in overactivation of the downstream mTOR (mammalian target of rapamycin) pathway, resulting in abnormal cellular proliferation and hamartomas. These benign tumors can be found in nearly every organ, most often in the central nervous system and skin, and they provide for a highly variable presentation of the disease.1
Tuberous sclerosis affects 1 in 6000 to 10,000 live births and has a prevalence of 1 in 20,000 individuals. Of these individuals, 75% carry sporadic mutations, and 75% to 90% eventually test positive for a TSC gene mutation.2 Genetic mosaicism has been reported in 28% of cases affected by large deletions1 and as few as 1% of cases involving small mutations.3
The dermatologic manifestation of mosaic TSC most often includes unilateral angiofibromas, whereas in nonmosaic cases, angiofibromas cover both cheeks, the forehead, and the eyelids. The other skin lesions of TSC--shagreen patches, forehead plaques, hypomelanotic macules, and ungual fibromas--are seen less frequently.4-6 Additionally, neurologic disease in mosaic patients is notably milder, with 57% of mosaic patients found to have epilepsy compared to 91% of nonmosaic patients.7 Our patient had both unilateral facial angiofibromas and a cortical lesion suspicious for a tuber, prompting a suspected diagnosis of mosaic TSC.
The methods of diagnosis outlined by the International Tuberous Sclerosis Complex Consensus Group pose a challenge in diagnosing mosaic TSC. The clinical criteria require 2 major (eg, multiple angiofibromas, angiomyolipomas, a shagreen patch) and 1 minor feature (eg, dental enamel pit, renal cyst).2 However, case reports detailing unilateral facial angiofibromas have described patients with isolated dermatologic findings.5,6 Further, it has been demonstrated that genetic studies in mosaic TSC can be unreliable depending on the tissue sampled.8 Thus, for patients who have mosaic TSC, establishing a definitive diagnosis is not always possible and may rely solely on the clinical picture.
Considering the differential diagnosis, benign cephalic histiocytosis usually would present with small red-brown macules and papules symmetrically located on the head and neck. The lesions occur at a younger age, usually in the first year or two of life. Fibrofolliculomas present as multiple whitish, slightly larger papules found on the central face. They are a marker for Birt-Hogg-Dubé syndrome, which also is associated with spontaneous pneumothorax.
Agminated means clustering or grouping of lesions. Agminated melanocytic nevi and agminated Spitz nevi are clusters of nevi. These lesions can vary in size and color. They may be elevated or flat. Melanocytic nevi usually are tan-brown or black. Spitz nevi may be pink or pigmented brown or black. To definitively differentiate between these 2 diagnoses and this patient's diagnosis of angiofibroma, a biopsy is needed.
- Curatolo P, Moavero R, Roberto D, et al. Genotype/phenotype correlations in tuberous sclerosis complex. Semin Pediatr Neurol. 2015;22:259-273.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Kwiatkowski DJ, Whittemore VH, Thiele EA. Tuberous Sclerosis Complex: Genes, Clinical Features and Therapeutics. Weinham, Germany: Wiley-Blackwell; 2011.
- Alshaiji JM, Spock CR, Connelly EA, et al. Facial angiofibromas in a mosaic pattern tuberous sclerosis: a case report. Dermatol Online J. 2012;18:8.
- Gutte R, Khopkar U. Unilateral multiple facial angiofibromas: a case report with brief review of literature. Indian J Dermatol. 2013;58:159.
- Silvestre JF, Bañuls J, Ramón R, et al. Unilateral multiple facial angiofibromas: a mosaic form of TSC. J Am Acad Dermatol. 2000;43(1, pt 1):127-129.
- Kozlowski P, Roberts P, Dabora S, et al. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet. 2007;121:389-400.
- Kwiatkowska J, Wigowska-Sowinska J, Napierala D, et al. Mosaicism in TSC as a potential cause of the failure of molecular diagnosis. N Engl J Med. 1999;340:703-707.
- Curatolo P, Moavero R, Roberto D, et al. Genotype/phenotype correlations in tuberous sclerosis complex. Semin Pediatr Neurol. 2015;22:259-273.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Kwiatkowski DJ, Whittemore VH, Thiele EA. Tuberous Sclerosis Complex: Genes, Clinical Features and Therapeutics. Weinham, Germany: Wiley-Blackwell; 2011.
- Alshaiji JM, Spock CR, Connelly EA, et al. Facial angiofibromas in a mosaic pattern tuberous sclerosis: a case report. Dermatol Online J. 2012;18:8.
- Gutte R, Khopkar U. Unilateral multiple facial angiofibromas: a case report with brief review of literature. Indian J Dermatol. 2013;58:159.
- Silvestre JF, Bañuls J, Ramón R, et al. Unilateral multiple facial angiofibromas: a mosaic form of TSC. J Am Acad Dermatol. 2000;43(1, pt 1):127-129.
- Kozlowski P, Roberts P, Dabora S, et al. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet. 2007;121:389-400.
- Kwiatkowska J, Wigowska-Sowinska J, Napierala D, et al. Mosaicism in TSC as a potential cause of the failure of molecular diagnosis. N Engl J Med. 1999;340:703-707.
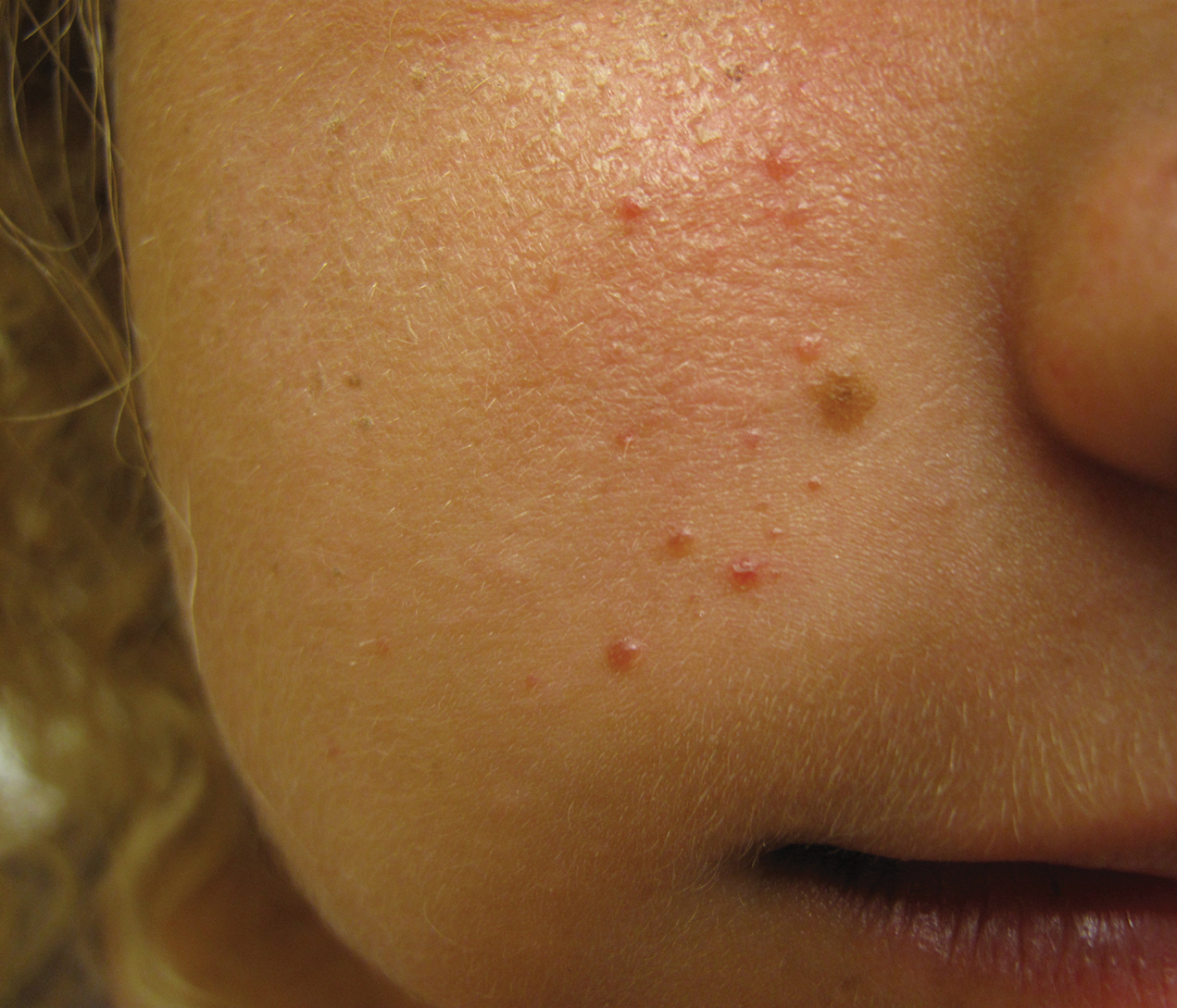
An 18-year-old woman presented with a progressive appearance of firm, red-brown, asymptomatic, 1- to 3-mm, dome-shaped papules on the right cheek that developed over the course of 2 years. She had 10 lesions that covered a 2.2 ×4-cm area on the right medial cheek. No similar-appearing lesions were detectable on a full-body skin examination, and no periungual tumors, café au lait macules, or shagreen patches were noted. A full-body skin examination using a Wood lamp revealed 1 small hypopigmented macule on the right second finger. The patient had a history of treatment-refractory migraines; magnetic resonance imaging 5 years prior to the current presentation revealed a nonspecific lesion in the left parietal gyrus. There was no personal or family history of seizures, cognitive delay, kidney disease, or ocular disease. Punch biopsy of a facial lesion was performed for histopathologic correlation.
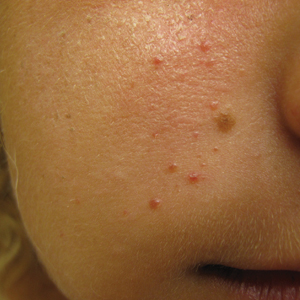
Larval Tick Infestation Causing an Eruption of Pruritic Papules and Pustules
Case Reports
Patient 1
A 65-year-old woman presented to the dermatology clinic in July with a pruritic rash of 2 days’ duration that started on the back and spread diffusely. The patient gardened regularly. Physical examination showed inflammatory papules and pustules on the back (Figure 1), as well as the groin, breasts, and ears. There was a punctate black dot in the center of some papules, and dermoscopy revealed ticks (Figure 2). Removal and microscopic examination confirmed larval-stage lone star ticks (Figure 3). The patient was prescribed topical steroids for pruritus as well as oral doxycycline for prophylaxis against tick-borne illnesses.
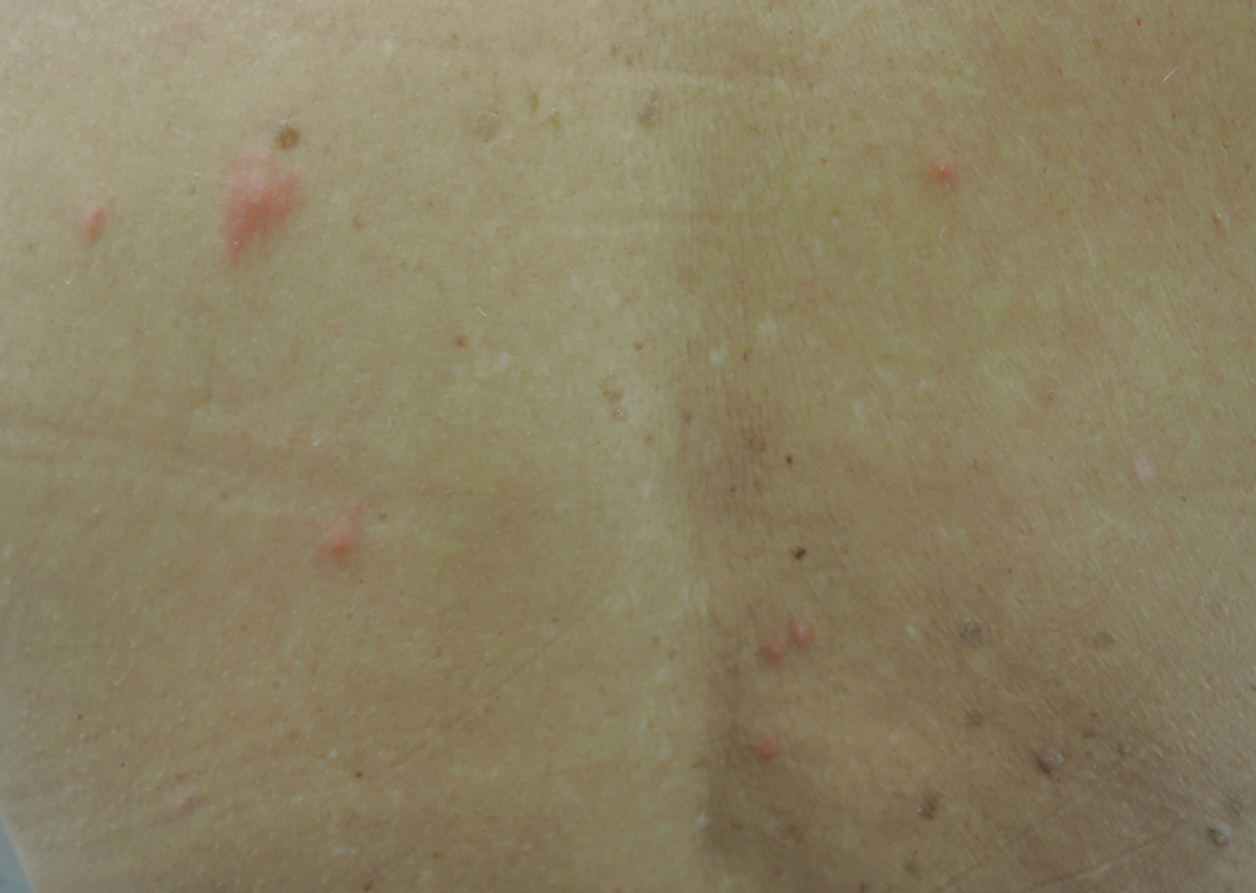


Patient 2
A 54-year-old man presented to the same clinic in July with pruritic lesions on the back, legs, ankles, and scrotum of 3 days’ duration that first appeared 24 hours after performing yardwork. Physical examination revealed diffusely distributed papules, pustules, and vesicles on the back (Figure 4). Some papules featured a punctate black dot in the center (similar to patient 1), and dermoscopy again revealed ticks. Removal and microscopic examination confirmed larval-stage ticks. The patient was treated with topical steroids and oral antihistamines for pruritus as well as prophylactic oral doxycycline.
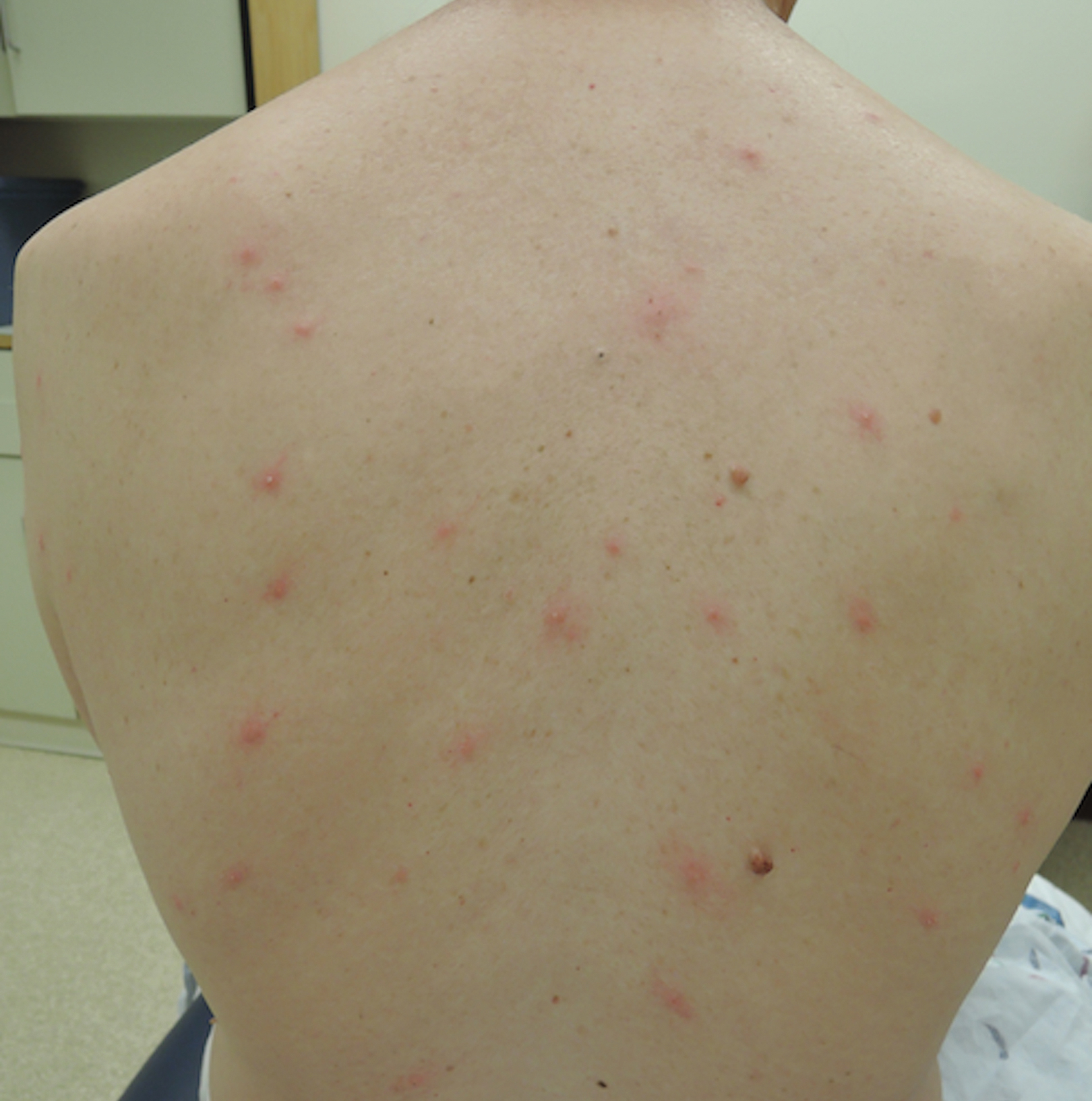
Comment
Ticks are well-known human parasites, representing the second most common vector of human infectious disease.1 Ticks have 3 motile stages: larva (or “seed”), nymph, and adult. They can bite humans during all stages. Larval ticks, distinguished by having 6 legs rather than 8 legs in nymphs and adults, can attack in droves and cause an infestation that presents as diffuse, pruritic, erythematous papules and pustules.2-4 The first report of larval tick infestation in humans may have been in 1728 by William Byrd who described finding ticks on the skin that were too small to see without a microscope.5
Identification
The ticks in both of our cases were lone star ticks (Amblyomma americanum). The larval stage of A americanum is a proven cause of cutaneous reaction.6,7 A PubMed search of articles indexed for MEDLINE as well as a Google Scholar search using the terms tick, seed tick, or tick bite in combination with rash, eruption, infestation, papule, pustule, or pruritic revealed 6 reported cases of larval tick infestation in the literature (including our case); 5 were caused by A americanum and 1 by Ixodes dammini (now known as Ixodes scapularis); all occurred in July or August.3,7-10 This time frame is consistent with the general tick life cycle across species: Adults feed from April to June, then lay eggs that hatch into larval ticks within 4 to 6 weeks. After hatching, larval ticks climb grass and weeds awaiting a passing host.4
Diagnosis
Larval tick infestation remains a frequently misdiagnosed etiology of diffuse pruritic papules and pustules, especially in urban settings where physicians are less likely to be familiar with this type of manifestation.3,9-11 Larval ticks are submillimeter in size and difficult to appreciate with the naked eye, contributing to misdiagnosis. A punctate black dot may sometimes be seen in papules; however, dermoscopy is critical for accurate diagnosis, as hemorrhagic crust is a frequent misdiagnosis.
Management
In addition to symptomatic therapy, both of our patients received doxycycline as antibiotic prophylaxis for tick-borne illnesses given that a high number of ticks had been attached for more than 2 days.12,13 Antibiotic prophylaxis for tick-borne illness is controversial. The exception is Lyme disease transmitted by nymphal or adult I scapularis when specific conditions are met: the bite must have occurred in an endemic area, doxycycline cannot be contraindicated, estimated duration of attachment is at least 36 hours, and prophylaxis must be started within 72 hours of tick removal.13 There are no official recommendations for the A americanum species or for larval-stage ticks of any species. Larval-stage ticks acting as vectors for disease transmission is not well documented in recent literature, and there currently is limited evidence supporting prophylactic antibiotics for larval tick bites. The presence of spotted fever rickettsioses has been reported (with the exception of Rickettsia rickettsii and Ehrlichia chaffeensis) in larval A americanum ticks, suggesting a theoretical possibility that they could act as disease vectors.3,8,11,14-17 At a minimum, both prompt tick removal and close patient follow-up is warranted.
Conclusion
Human infestation with larval ticks is a common occurrence but can present a diagnostic challenge to an unfamiliar physician. We encourage consideration of larval tick infestation as the etiology of multiple or diffuse pruritic papules with a history of outdoor exposure.
- Sonenshine DE. Biology of Ticks. New York, NY: Oxford University; 1991.
- Alexander JOD. The effects of tick bites. In: Alexander JOD. Arthropods and Human Skin. London, England: Springer London; 1984:363-382.
- Duckworth PF Jr, Hayden GF, Reed CN. Human infestation by Amblyomma americanum larvae (“seed ticks”). South Med J. 1985;78:751-753.
- Parola P, Raoult D. Ticks and tickborne bacterial diseases in humans: an emerging infectious threat. Clin Infect Dis. 2001;32:897-928.
- Cropley TG. William Byrd on ticks, 1728. Arch Dermatol. 2009;145:187.
- Goddard J. A ten-year study of tick biting in Mississippi: implications for human disease transmission. J Agromedicine. 2002;8:25-32.
- Goddard J, Portugal JS. Cutaneous lesions due to bites by larval Amblyomma americanum ticks. JAMA Dermatol. 2015;151:1373-1375.
- Fibeger EA, Erickson QL, Weintraub BD, et al. Larval tick infestation: a case report and review of tick-borne disease. Cutis. 2008;82:38-46.
- Jones BE. Human ‘seed tick’ infestation: Amblyomma americanum larvae. Arch Dermatol. 1981;117:812-814.
- Fisher EJ, Mo J, Lucky AW. Multiple pruritic papules from lone star tick larvae bites. Arch Dermatol. 2006;142:491-494.
- Culp JS. Seed ticks. Am Fam Physician. 1987;36:121-123.
- Perea AE, Hinckley AF, Mead PS. Tick bite prophylaxis: results from a 2012 survey of healthcare providers. Zoonoses Public Health. 2015;62:388-392.
- Tick bites/prevention. Centers for Disease Control and Prevention website. https://www.cdc.gov/ticks/tickbornediseases/tick-bites-prevention.html. Revised January 10, 2019. Accessed September 17, 2019.
- Moncayo AC, Cohen SB, Fritzen CM, et al. Absence of Rickettsia rickettsii and occurrence of other spotted fever group rickettsiae in ticks from Tennessee. Am J Trop Med Hyg. 2010;83:653-657.
- Castellaw AH, Showers J, Goddard J, et al. Detection of vector-borne agents in lone star ticks, Amblyomma americanum (Acari: Ixodidae), from Mississippi. J Med Entomol. 2010;47:473-476.
- Stromdahl EY, Vince MA, Billingsley PM, et al. Rickettsia amblyommii infecting Amblyomma americanum larvae. Vector Borne Zoonotic Dis. 2008;8:15-24.
- Long SW, Zhang X, Zhang J, et al. Evaluation of transovarial transmission and transmissibility of Ehrlichia chaffeensis (Rickettsiales: Anaplasmataceae) in Amblyomma americanum (Acari: Ixodidae). J Med Entomol. 2003;40:1000-1004.
Case Reports
Patient 1
A 65-year-old woman presented to the dermatology clinic in July with a pruritic rash of 2 days’ duration that started on the back and spread diffusely. The patient gardened regularly. Physical examination showed inflammatory papules and pustules on the back (Figure 1), as well as the groin, breasts, and ears. There was a punctate black dot in the center of some papules, and dermoscopy revealed ticks (Figure 2). Removal and microscopic examination confirmed larval-stage lone star ticks (Figure 3). The patient was prescribed topical steroids for pruritus as well as oral doxycycline for prophylaxis against tick-borne illnesses.
Patient 2
A 54-year-old man presented to the same clinic in July with pruritic lesions on the back, legs, ankles, and scrotum of 3 days’ duration that first appeared 24 hours after performing yardwork. Physical examination revealed diffusely distributed papules, pustules, and vesicles on the back (Figure 4). Some papules featured a punctate black dot in the center (similar to patient 1), and dermoscopy again revealed ticks. Removal and microscopic examination confirmed larval-stage ticks. The patient was treated with topical steroids and oral antihistamines for pruritus as well as prophylactic oral doxycycline.
Comment
Ticks are well-known human parasites, representing the second most common vector of human infectious disease.1 Ticks have 3 motile stages: larva (or “seed”), nymph, and adult. They can bite humans during all stages. Larval ticks, distinguished by having 6 legs rather than 8 legs in nymphs and adults, can attack in droves and cause an infestation that presents as diffuse, pruritic, erythematous papules and pustules.2-4 The first report of larval tick infestation in humans may have been in 1728 by William Byrd who described finding ticks on the skin that were too small to see without a microscope.5
Identification
The ticks in both of our cases were lone star ticks (Amblyomma americanum). The larval stage of A americanum is a proven cause of cutaneous reaction.6,7 A PubMed search of articles indexed for MEDLINE as well as a Google Scholar search using the terms tick, seed tick, or tick bite in combination with rash, eruption, infestation, papule, pustule, or pruritic revealed 6 reported cases of larval tick infestation in the literature (including our case); 5 were caused by A americanum and 1 by Ixodes dammini (now known as Ixodes scapularis); all occurred in July or August.3,7-10 This time frame is consistent with the general tick life cycle across species: Adults feed from April to June, then lay eggs that hatch into larval ticks within 4 to 6 weeks. After hatching, larval ticks climb grass and weeds awaiting a passing host.4
Diagnosis
Larval tick infestation remains a frequently misdiagnosed etiology of diffuse pruritic papules and pustules, especially in urban settings where physicians are less likely to be familiar with this type of manifestation.3,9-11 Larval ticks are submillimeter in size and difficult to appreciate with the naked eye, contributing to misdiagnosis. A punctate black dot may sometimes be seen in papules; however, dermoscopy is critical for accurate diagnosis, as hemorrhagic crust is a frequent misdiagnosis.
Management
In addition to symptomatic therapy, both of our patients received doxycycline as antibiotic prophylaxis for tick-borne illnesses given that a high number of ticks had been attached for more than 2 days.12,13 Antibiotic prophylaxis for tick-borne illness is controversial. The exception is Lyme disease transmitted by nymphal or adult I scapularis when specific conditions are met: the bite must have occurred in an endemic area, doxycycline cannot be contraindicated, estimated duration of attachment is at least 36 hours, and prophylaxis must be started within 72 hours of tick removal.13 There are no official recommendations for the A americanum species or for larval-stage ticks of any species. Larval-stage ticks acting as vectors for disease transmission is not well documented in recent literature, and there currently is limited evidence supporting prophylactic antibiotics for larval tick bites. The presence of spotted fever rickettsioses has been reported (with the exception of Rickettsia rickettsii and Ehrlichia chaffeensis) in larval A americanum ticks, suggesting a theoretical possibility that they could act as disease vectors.3,8,11,14-17 At a minimum, both prompt tick removal and close patient follow-up is warranted.
Conclusion
Human infestation with larval ticks is a common occurrence but can present a diagnostic challenge to an unfamiliar physician. We encourage consideration of larval tick infestation as the etiology of multiple or diffuse pruritic papules with a history of outdoor exposure.
Case Reports
Patient 1
A 65-year-old woman presented to the dermatology clinic in July with a pruritic rash of 2 days’ duration that started on the back and spread diffusely. The patient gardened regularly. Physical examination showed inflammatory papules and pustules on the back (Figure 1), as well as the groin, breasts, and ears. There was a punctate black dot in the center of some papules, and dermoscopy revealed ticks (Figure 2). Removal and microscopic examination confirmed larval-stage lone star ticks (Figure 3). The patient was prescribed topical steroids for pruritus as well as oral doxycycline for prophylaxis against tick-borne illnesses.
Patient 2
A 54-year-old man presented to the same clinic in July with pruritic lesions on the back, legs, ankles, and scrotum of 3 days’ duration that first appeared 24 hours after performing yardwork. Physical examination revealed diffusely distributed papules, pustules, and vesicles on the back (Figure 4). Some papules featured a punctate black dot in the center (similar to patient 1), and dermoscopy again revealed ticks. Removal and microscopic examination confirmed larval-stage ticks. The patient was treated with topical steroids and oral antihistamines for pruritus as well as prophylactic oral doxycycline.
Comment
Ticks are well-known human parasites, representing the second most common vector of human infectious disease.1 Ticks have 3 motile stages: larva (or “seed”), nymph, and adult. They can bite humans during all stages. Larval ticks, distinguished by having 6 legs rather than 8 legs in nymphs and adults, can attack in droves and cause an infestation that presents as diffuse, pruritic, erythematous papules and pustules.2-4 The first report of larval tick infestation in humans may have been in 1728 by William Byrd who described finding ticks on the skin that were too small to see without a microscope.5
Identification
The ticks in both of our cases were lone star ticks (Amblyomma americanum). The larval stage of A americanum is a proven cause of cutaneous reaction.6,7 A PubMed search of articles indexed for MEDLINE as well as a Google Scholar search using the terms tick, seed tick, or tick bite in combination with rash, eruption, infestation, papule, pustule, or pruritic revealed 6 reported cases of larval tick infestation in the literature (including our case); 5 were caused by A americanum and 1 by Ixodes dammini (now known as Ixodes scapularis); all occurred in July or August.3,7-10 This time frame is consistent with the general tick life cycle across species: Adults feed from April to June, then lay eggs that hatch into larval ticks within 4 to 6 weeks. After hatching, larval ticks climb grass and weeds awaiting a passing host.4
Diagnosis
Larval tick infestation remains a frequently misdiagnosed etiology of diffuse pruritic papules and pustules, especially in urban settings where physicians are less likely to be familiar with this type of manifestation.3,9-11 Larval ticks are submillimeter in size and difficult to appreciate with the naked eye, contributing to misdiagnosis. A punctate black dot may sometimes be seen in papules; however, dermoscopy is critical for accurate diagnosis, as hemorrhagic crust is a frequent misdiagnosis.
Management
In addition to symptomatic therapy, both of our patients received doxycycline as antibiotic prophylaxis for tick-borne illnesses given that a high number of ticks had been attached for more than 2 days.12,13 Antibiotic prophylaxis for tick-borne illness is controversial. The exception is Lyme disease transmitted by nymphal or adult I scapularis when specific conditions are met: the bite must have occurred in an endemic area, doxycycline cannot be contraindicated, estimated duration of attachment is at least 36 hours, and prophylaxis must be started within 72 hours of tick removal.13 There are no official recommendations for the A americanum species or for larval-stage ticks of any species. Larval-stage ticks acting as vectors for disease transmission is not well documented in recent literature, and there currently is limited evidence supporting prophylactic antibiotics for larval tick bites. The presence of spotted fever rickettsioses has been reported (with the exception of Rickettsia rickettsii and Ehrlichia chaffeensis) in larval A americanum ticks, suggesting a theoretical possibility that they could act as disease vectors.3,8,11,14-17 At a minimum, both prompt tick removal and close patient follow-up is warranted.
Conclusion
Human infestation with larval ticks is a common occurrence but can present a diagnostic challenge to an unfamiliar physician. We encourage consideration of larval tick infestation as the etiology of multiple or diffuse pruritic papules with a history of outdoor exposure.
- Sonenshine DE. Biology of Ticks. New York, NY: Oxford University; 1991.
- Alexander JOD. The effects of tick bites. In: Alexander JOD. Arthropods and Human Skin. London, England: Springer London; 1984:363-382.
- Duckworth PF Jr, Hayden GF, Reed CN. Human infestation by Amblyomma americanum larvae (“seed ticks”). South Med J. 1985;78:751-753.
- Parola P, Raoult D. Ticks and tickborne bacterial diseases in humans: an emerging infectious threat. Clin Infect Dis. 2001;32:897-928.
- Cropley TG. William Byrd on ticks, 1728. Arch Dermatol. 2009;145:187.
- Goddard J. A ten-year study of tick biting in Mississippi: implications for human disease transmission. J Agromedicine. 2002;8:25-32.
- Goddard J, Portugal JS. Cutaneous lesions due to bites by larval Amblyomma americanum ticks. JAMA Dermatol. 2015;151:1373-1375.
- Fibeger EA, Erickson QL, Weintraub BD, et al. Larval tick infestation: a case report and review of tick-borne disease. Cutis. 2008;82:38-46.
- Jones BE. Human ‘seed tick’ infestation: Amblyomma americanum larvae. Arch Dermatol. 1981;117:812-814.
- Fisher EJ, Mo J, Lucky AW. Multiple pruritic papules from lone star tick larvae bites. Arch Dermatol. 2006;142:491-494.
- Culp JS. Seed ticks. Am Fam Physician. 1987;36:121-123.
- Perea AE, Hinckley AF, Mead PS. Tick bite prophylaxis: results from a 2012 survey of healthcare providers. Zoonoses Public Health. 2015;62:388-392.
- Tick bites/prevention. Centers for Disease Control and Prevention website. https://www.cdc.gov/ticks/tickbornediseases/tick-bites-prevention.html. Revised January 10, 2019. Accessed September 17, 2019.
- Moncayo AC, Cohen SB, Fritzen CM, et al. Absence of Rickettsia rickettsii and occurrence of other spotted fever group rickettsiae in ticks from Tennessee. Am J Trop Med Hyg. 2010;83:653-657.
- Castellaw AH, Showers J, Goddard J, et al. Detection of vector-borne agents in lone star ticks, Amblyomma americanum (Acari: Ixodidae), from Mississippi. J Med Entomol. 2010;47:473-476.
- Stromdahl EY, Vince MA, Billingsley PM, et al. Rickettsia amblyommii infecting Amblyomma americanum larvae. Vector Borne Zoonotic Dis. 2008;8:15-24.
- Long SW, Zhang X, Zhang J, et al. Evaluation of transovarial transmission and transmissibility of Ehrlichia chaffeensis (Rickettsiales: Anaplasmataceae) in Amblyomma americanum (Acari: Ixodidae). J Med Entomol. 2003;40:1000-1004.
- Sonenshine DE. Biology of Ticks. New York, NY: Oxford University; 1991.
- Alexander JOD. The effects of tick bites. In: Alexander JOD. Arthropods and Human Skin. London, England: Springer London; 1984:363-382.
- Duckworth PF Jr, Hayden GF, Reed CN. Human infestation by Amblyomma americanum larvae (“seed ticks”). South Med J. 1985;78:751-753.
- Parola P, Raoult D. Ticks and tickborne bacterial diseases in humans: an emerging infectious threat. Clin Infect Dis. 2001;32:897-928.
- Cropley TG. William Byrd on ticks, 1728. Arch Dermatol. 2009;145:187.
- Goddard J. A ten-year study of tick biting in Mississippi: implications for human disease transmission. J Agromedicine. 2002;8:25-32.
- Goddard J, Portugal JS. Cutaneous lesions due to bites by larval Amblyomma americanum ticks. JAMA Dermatol. 2015;151:1373-1375.
- Fibeger EA, Erickson QL, Weintraub BD, et al. Larval tick infestation: a case report and review of tick-borne disease. Cutis. 2008;82:38-46.
- Jones BE. Human ‘seed tick’ infestation: Amblyomma americanum larvae. Arch Dermatol. 1981;117:812-814.
- Fisher EJ, Mo J, Lucky AW. Multiple pruritic papules from lone star tick larvae bites. Arch Dermatol. 2006;142:491-494.
- Culp JS. Seed ticks. Am Fam Physician. 1987;36:121-123.
- Perea AE, Hinckley AF, Mead PS. Tick bite prophylaxis: results from a 2012 survey of healthcare providers. Zoonoses Public Health. 2015;62:388-392.
- Tick bites/prevention. Centers for Disease Control and Prevention website. https://www.cdc.gov/ticks/tickbornediseases/tick-bites-prevention.html. Revised January 10, 2019. Accessed September 17, 2019.
- Moncayo AC, Cohen SB, Fritzen CM, et al. Absence of Rickettsia rickettsii and occurrence of other spotted fever group rickettsiae in ticks from Tennessee. Am J Trop Med Hyg. 2010;83:653-657.
- Castellaw AH, Showers J, Goddard J, et al. Detection of vector-borne agents in lone star ticks, Amblyomma americanum (Acari: Ixodidae), from Mississippi. J Med Entomol. 2010;47:473-476.
- Stromdahl EY, Vince MA, Billingsley PM, et al. Rickettsia amblyommii infecting Amblyomma americanum larvae. Vector Borne Zoonotic Dis. 2008;8:15-24.
- Long SW, Zhang X, Zhang J, et al. Evaluation of transovarial transmission and transmissibility of Ehrlichia chaffeensis (Rickettsiales: Anaplasmataceae) in Amblyomma americanum (Acari: Ixodidae). J Med Entomol. 2003;40:1000-1004.
Practice Points
- Larval (“seed”) ticks can attack in droves, causing a widespread rash consisting of pruritic erythematous papules and pustules.
- Tiny black dots can be seen in some papules, which are the seed ticks themselves. Careful dermoscopic examination is critical to avoid easy misdiagnosis as hemorrhagic crust.
- We encourage providers to include larval tick infestation in the differential for eruptive pruritic papules and pustules with a history of outdoor exposure, especially during the summer months.

Nevus of Ota Associated With a Primary Uveal Melanoma and Intracranial Melanoma Metastasis
Nevus of Ota, originally referred to as nevus fusco-caeruleus ophthalmomaxillaris, initially was described in 1939 by Ota and Tanino.1 It is a dermal melanocytic hamartoma arising from incomplete migration of neural crest melanocytes to the epidermis during embryogenesis, resulting in nesting of subtle bands of dendritic melanocytes in the upper dermis. More common in Asians, Native Americans, and females, this hyperpigmented dermatosis most often is unilaterally distributed along the ophthalmic (V1) and maxillary (V2) branches of the trigeminal nerve.2 In some patients, nevus of Ota also is associated with ocular, orbital, and leptomeningeal melanocytosis. Approximately 15% of nevi of Ota have an activating guanine nucleotide-binding protein G(q) subunit alpha (GNAQ) or G protein subunit alpha 11 (GNAQ) mutation; 85% of uveal melanomas harbor one of these mutations.3 Although uncommon, neoplastic transformation with extension or metastasis to the brain has been reported in patients with nevus of Ota.4
We report the case of a 29-year-old woman with a long-standing history of nevus of Ota who presented acutely with an intracranial melanoma as an extension of a primary uveal melanoma.
Case Report
A 29-year-old woman with a history of a nevus of Ota involving the left inner canthus, eyelids, sclera, and superior malar cheek that had been present since birth presented to the emergency department with an acute onset of severe headache, blurred vision, and vomiting. Computed tomography (CT) and magnetic resonance imaging of the brain revealed a hemorrhagic mass in the left frontal lobe. Subsequent frontal craniotomy and resection revealed an intracranial melanoma.
Two weeks following surgery, the patient underwent magnetic resonance imaging and combined positron emission tomography and CT scans that demonstrated a fluorodeoxyglucose-avid left retro-orbital mass. Histopathology of a biopsy from the left retro-orbital mass that had been obtained intraoperatively demonstrated a pigmented, spindled to epithelioid neoplasm with areas of marked atypia and a high mitotic rate that was compatible with malignant melanoma (Figure 1). Intracranial biopsies were sent for genetic study and were found to harbor GNAQ (Q209P) and BRCA1-associated protein 1 (BAP1)(p.P324fs*11) mutations.
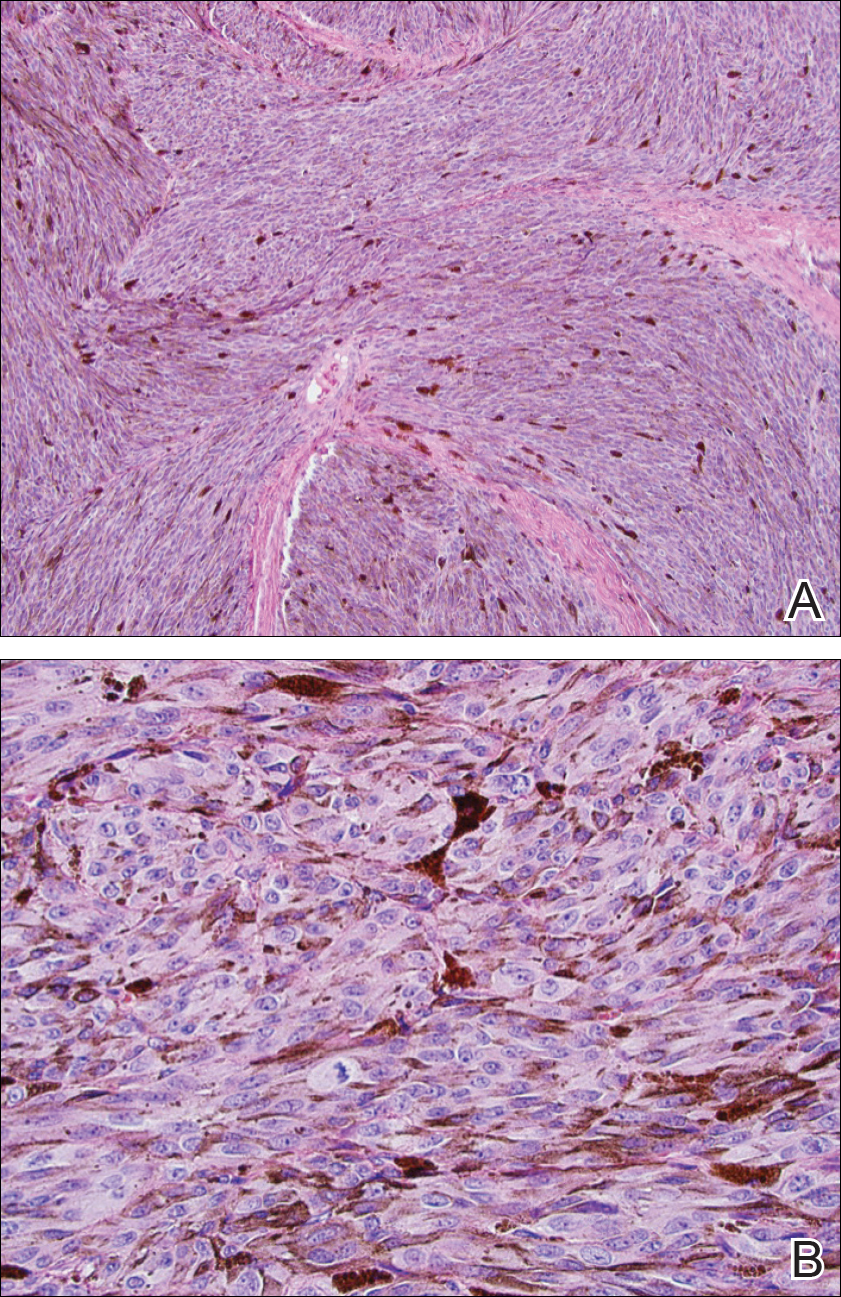
The patient was referred to dermatology by neurosurgery for evaluation of a suspected primary cutaneous melanoma.

The patient entered a clinical trial at an outside institution several weeks after initial presentation to our institution for treatment with a mitogen-activated protein kinase MEK1 inhibitor as well as radiation therapy. The patient was lost to follow-up.
Comment
It has been demonstrated that homozygous loss of BAP1, located on the chromosome 3p21.1 locus, allows for progression to metastatic disease in uveal melanoma. The BAP1 gene codes for ubiquitin carboxyl-terminal hydrolase 7, which is involved in the removal of ubiquitin from proteins. This enzyme binds to BRCA1 (BRCA1, DNA repair associated) via the RING (Really Interesting New Gene) finger domain and acts as a tumor suppressor.5 Biallelic BAP1 mutations allow the transition to malignancy in concert with other mutations, such as GNAQ. Identification of a BAP1 mutation may serve as a valuable diagnostic and future therapeutic target in uveal melanoma.
Currently, there are no drugs that directly target mutated GNA11 and GNAQ proteins. Because aberrant GNA11 and GNAQ proteins activate MEK1, several MEK1 inhibitors are being tested with the hope of achieving indirect suppression of GNA11/GNAQ.6
We present a rare case of BAP1 and GNAQ mutations in intracranial melanoma associated with nevus of Ota. Although the uveal melanoma was not confirmed on histopathology, the clear mention of foci within the eye by ophthalmology, positron emission tomography–CT scan showing a fluorodeoxyglucose-avid left retro-orbital mass, and genetic studies of the intracranial biopsies were highly suggestive of a primary uveal melanoma.
Our case highlights the importance of ongoing ocular screening in patients with nevus of Ota, noting the possibility of malignant transformation. Furthermore, patients with nevus of Ota with ocular involvement may benefit from testing of BAP1 protein expression by immunohistochemistry.7 Identification of BAP1 and GNAQ mutations in patients with nevus of Ota place them at markedly higher risk for malignant melanoma. Therefore, dermatologic evaluation of patients with nevus of Ota should include a thorough review of the patient’s history and skin examination as well as referral for ophthalmologic evaluation.
- Ota M, Tanino H. A variety of nevus, frequently encountered in Japan, nevus fusco-caeruleus ophthalmomaxillaris and its relationship to pigmentary changes in the eye. Tokyo Med J. 1939;63:1243-1244.
- Swann PG, Kwong E. The naevus of Ota. Clin Exp Optom. 2010;93:264-267.
- Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma [published online November 17, 2010]. N Engl J Med. 2010;363:2191-2199.
- Nitta K, Kashima T, Mayuzumi H, et al. Animal-type malignancy melanoma associated with nevus of Ota in the orbit of a Japanese woman: a case report. Melanoma Res. 2014;24:286-289.
- Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas [published online November 4, 2010]. Science. 2010;330:1410-1413.
- Chen X, Wu Q, Tan L, et al. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene. 2014;33:4724-4734.
- Kalirai H, Dodson A, Faqir S, et al. Lack of BAP1 protein expression in uveal melanoma is associated with increased metastatic risk and has utility in routine prognostic testing [published online July 24, 2010]. Br J Cancer. 2014;111:1373-1380.
Nevus of Ota, originally referred to as nevus fusco-caeruleus ophthalmomaxillaris, initially was described in 1939 by Ota and Tanino.1 It is a dermal melanocytic hamartoma arising from incomplete migration of neural crest melanocytes to the epidermis during embryogenesis, resulting in nesting of subtle bands of dendritic melanocytes in the upper dermis. More common in Asians, Native Americans, and females, this hyperpigmented dermatosis most often is unilaterally distributed along the ophthalmic (V1) and maxillary (V2) branches of the trigeminal nerve.2 In some patients, nevus of Ota also is associated with ocular, orbital, and leptomeningeal melanocytosis. Approximately 15% of nevi of Ota have an activating guanine nucleotide-binding protein G(q) subunit alpha (GNAQ) or G protein subunit alpha 11 (GNAQ) mutation; 85% of uveal melanomas harbor one of these mutations.3 Although uncommon, neoplastic transformation with extension or metastasis to the brain has been reported in patients with nevus of Ota.4
We report the case of a 29-year-old woman with a long-standing history of nevus of Ota who presented acutely with an intracranial melanoma as an extension of a primary uveal melanoma.
Case Report
A 29-year-old woman with a history of a nevus of Ota involving the left inner canthus, eyelids, sclera, and superior malar cheek that had been present since birth presented to the emergency department with an acute onset of severe headache, blurred vision, and vomiting. Computed tomography (CT) and magnetic resonance imaging of the brain revealed a hemorrhagic mass in the left frontal lobe. Subsequent frontal craniotomy and resection revealed an intracranial melanoma.
Two weeks following surgery, the patient underwent magnetic resonance imaging and combined positron emission tomography and CT scans that demonstrated a fluorodeoxyglucose-avid left retro-orbital mass. Histopathology of a biopsy from the left retro-orbital mass that had been obtained intraoperatively demonstrated a pigmented, spindled to epithelioid neoplasm with areas of marked atypia and a high mitotic rate that was compatible with malignant melanoma (Figure 1). Intracranial biopsies were sent for genetic study and were found to harbor GNAQ (Q209P) and BRCA1-associated protein 1 (BAP1)(p.P324fs*11) mutations.
The patient was referred to dermatology by neurosurgery for evaluation of a suspected primary cutaneous melanoma.
The patient entered a clinical trial at an outside institution several weeks after initial presentation to our institution for treatment with a mitogen-activated protein kinase MEK1 inhibitor as well as radiation therapy. The patient was lost to follow-up.
Comment
It has been demonstrated that homozygous loss of BAP1, located on the chromosome 3p21.1 locus, allows for progression to metastatic disease in uveal melanoma. The BAP1 gene codes for ubiquitin carboxyl-terminal hydrolase 7, which is involved in the removal of ubiquitin from proteins. This enzyme binds to BRCA1 (BRCA1, DNA repair associated) via the RING (Really Interesting New Gene) finger domain and acts as a tumor suppressor.5 Biallelic BAP1 mutations allow the transition to malignancy in concert with other mutations, such as GNAQ. Identification of a BAP1 mutation may serve as a valuable diagnostic and future therapeutic target in uveal melanoma.
Currently, there are no drugs that directly target mutated GNA11 and GNAQ proteins. Because aberrant GNA11 and GNAQ proteins activate MEK1, several MEK1 inhibitors are being tested with the hope of achieving indirect suppression of GNA11/GNAQ.6
We present a rare case of BAP1 and GNAQ mutations in intracranial melanoma associated with nevus of Ota. Although the uveal melanoma was not confirmed on histopathology, the clear mention of foci within the eye by ophthalmology, positron emission tomography–CT scan showing a fluorodeoxyglucose-avid left retro-orbital mass, and genetic studies of the intracranial biopsies were highly suggestive of a primary uveal melanoma.
Our case highlights the importance of ongoing ocular screening in patients with nevus of Ota, noting the possibility of malignant transformation. Furthermore, patients with nevus of Ota with ocular involvement may benefit from testing of BAP1 protein expression by immunohistochemistry.7 Identification of BAP1 and GNAQ mutations in patients with nevus of Ota place them at markedly higher risk for malignant melanoma. Therefore, dermatologic evaluation of patients with nevus of Ota should include a thorough review of the patient’s history and skin examination as well as referral for ophthalmologic evaluation.
Nevus of Ota, originally referred to as nevus fusco-caeruleus ophthalmomaxillaris, initially was described in 1939 by Ota and Tanino.1 It is a dermal melanocytic hamartoma arising from incomplete migration of neural crest melanocytes to the epidermis during embryogenesis, resulting in nesting of subtle bands of dendritic melanocytes in the upper dermis. More common in Asians, Native Americans, and females, this hyperpigmented dermatosis most often is unilaterally distributed along the ophthalmic (V1) and maxillary (V2) branches of the trigeminal nerve.2 In some patients, nevus of Ota also is associated with ocular, orbital, and leptomeningeal melanocytosis. Approximately 15% of nevi of Ota have an activating guanine nucleotide-binding protein G(q) subunit alpha (GNAQ) or G protein subunit alpha 11 (GNAQ) mutation; 85% of uveal melanomas harbor one of these mutations.3 Although uncommon, neoplastic transformation with extension or metastasis to the brain has been reported in patients with nevus of Ota.4
We report the case of a 29-year-old woman with a long-standing history of nevus of Ota who presented acutely with an intracranial melanoma as an extension of a primary uveal melanoma.
Case Report
A 29-year-old woman with a history of a nevus of Ota involving the left inner canthus, eyelids, sclera, and superior malar cheek that had been present since birth presented to the emergency department with an acute onset of severe headache, blurred vision, and vomiting. Computed tomography (CT) and magnetic resonance imaging of the brain revealed a hemorrhagic mass in the left frontal lobe. Subsequent frontal craniotomy and resection revealed an intracranial melanoma.
Two weeks following surgery, the patient underwent magnetic resonance imaging and combined positron emission tomography and CT scans that demonstrated a fluorodeoxyglucose-avid left retro-orbital mass. Histopathology of a biopsy from the left retro-orbital mass that had been obtained intraoperatively demonstrated a pigmented, spindled to epithelioid neoplasm with areas of marked atypia and a high mitotic rate that was compatible with malignant melanoma (Figure 1). Intracranial biopsies were sent for genetic study and were found to harbor GNAQ (Q209P) and BRCA1-associated protein 1 (BAP1)(p.P324fs*11) mutations.
The patient was referred to dermatology by neurosurgery for evaluation of a suspected primary cutaneous melanoma.
The patient entered a clinical trial at an outside institution several weeks after initial presentation to our institution for treatment with a mitogen-activated protein kinase MEK1 inhibitor as well as radiation therapy. The patient was lost to follow-up.
Comment
It has been demonstrated that homozygous loss of BAP1, located on the chromosome 3p21.1 locus, allows for progression to metastatic disease in uveal melanoma. The BAP1 gene codes for ubiquitin carboxyl-terminal hydrolase 7, which is involved in the removal of ubiquitin from proteins. This enzyme binds to BRCA1 (BRCA1, DNA repair associated) via the RING (Really Interesting New Gene) finger domain and acts as a tumor suppressor.5 Biallelic BAP1 mutations allow the transition to malignancy in concert with other mutations, such as GNAQ. Identification of a BAP1 mutation may serve as a valuable diagnostic and future therapeutic target in uveal melanoma.
Currently, there are no drugs that directly target mutated GNA11 and GNAQ proteins. Because aberrant GNA11 and GNAQ proteins activate MEK1, several MEK1 inhibitors are being tested with the hope of achieving indirect suppression of GNA11/GNAQ.6
We present a rare case of BAP1 and GNAQ mutations in intracranial melanoma associated with nevus of Ota. Although the uveal melanoma was not confirmed on histopathology, the clear mention of foci within the eye by ophthalmology, positron emission tomography–CT scan showing a fluorodeoxyglucose-avid left retro-orbital mass, and genetic studies of the intracranial biopsies were highly suggestive of a primary uveal melanoma.
Our case highlights the importance of ongoing ocular screening in patients with nevus of Ota, noting the possibility of malignant transformation. Furthermore, patients with nevus of Ota with ocular involvement may benefit from testing of BAP1 protein expression by immunohistochemistry.7 Identification of BAP1 and GNAQ mutations in patients with nevus of Ota place them at markedly higher risk for malignant melanoma. Therefore, dermatologic evaluation of patients with nevus of Ota should include a thorough review of the patient’s history and skin examination as well as referral for ophthalmologic evaluation.
- Ota M, Tanino H. A variety of nevus, frequently encountered in Japan, nevus fusco-caeruleus ophthalmomaxillaris and its relationship to pigmentary changes in the eye. Tokyo Med J. 1939;63:1243-1244.
- Swann PG, Kwong E. The naevus of Ota. Clin Exp Optom. 2010;93:264-267.
- Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma [published online November 17, 2010]. N Engl J Med. 2010;363:2191-2199.
- Nitta K, Kashima T, Mayuzumi H, et al. Animal-type malignancy melanoma associated with nevus of Ota in the orbit of a Japanese woman: a case report. Melanoma Res. 2014;24:286-289.
- Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas [published online November 4, 2010]. Science. 2010;330:1410-1413.
- Chen X, Wu Q, Tan L, et al. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene. 2014;33:4724-4734.
- Kalirai H, Dodson A, Faqir S, et al. Lack of BAP1 protein expression in uveal melanoma is associated with increased metastatic risk and has utility in routine prognostic testing [published online July 24, 2010]. Br J Cancer. 2014;111:1373-1380.
- Ota M, Tanino H. A variety of nevus, frequently encountered in Japan, nevus fusco-caeruleus ophthalmomaxillaris and its relationship to pigmentary changes in the eye. Tokyo Med J. 1939;63:1243-1244.
- Swann PG, Kwong E. The naevus of Ota. Clin Exp Optom. 2010;93:264-267.
- Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma [published online November 17, 2010]. N Engl J Med. 2010;363:2191-2199.
- Nitta K, Kashima T, Mayuzumi H, et al. Animal-type malignancy melanoma associated with nevus of Ota in the orbit of a Japanese woman: a case report. Melanoma Res. 2014;24:286-289.
- Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas [published online November 4, 2010]. Science. 2010;330:1410-1413.
- Chen X, Wu Q, Tan L, et al. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene. 2014;33:4724-4734.
- Kalirai H, Dodson A, Faqir S, et al. Lack of BAP1 protein expression in uveal melanoma is associated with increased metastatic risk and has utility in routine prognostic testing [published online July 24, 2010]. Br J Cancer. 2014;111:1373-1380.
Practice Points
- Nevus of Ota is a hyperpigmented dermatosis that typically is distributed along the ophthalmic (V1) and maxillary (V2) branches of the trigeminal nerve.
- GNAQ and BAP1 mutations in patients with nevus of Ota confer a greater risk for malignant melanoma and metastatic progression.
- Ongoing ophthalmologic screening is paramount in patients with nevus of Ota and may prevent devastating sequelae.
Levonorgestrel-Releasing Intrauterine System Causes a Lichenoid Drug Eruption
To the Editor:
Numerous drugs have been implicated as possible causes of lichenoid drug eruptions (LDEs). We describe a case of an LDE secondary to placement of a levonorgestrel-releasing intrauterine system (IUS).
A 28-year-old woman presented with an extensive pruritic rash of 2 months’ duration. She reported that it began on the wrists; progressed inward to involve the trunk; and then became generalized over the trunk, back, wrists, and legs. A levonorgestrel-releasing IUS had been placed 6 weeks prior to the onset of the rash. She was otherwise healthy and took loratadine and pseudoephedrine on occasion for environmental allergies. On examination there were violaceous, lichenified, flat-topped, polygonal papules scattered over the arms, legs, and trunk (Figure 1). Some papules demonstrated a Köbner phenomenon. No Wickham striae or mucosal involvement was noted. Rapid plasma reagin and hepatitis panel were negative. The patient was treated empirically with fluocinonide ointment 0.05% twice daily.
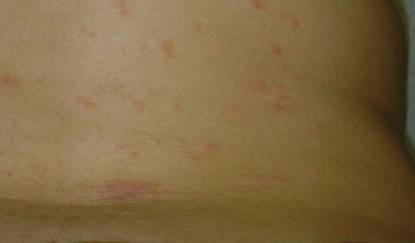
|

A shave biopsy was taken at the initial visit prior to steroid treatment. Histology revealed a classic lichenoid reaction pattern (Figure 2) and irregular acanthosis lying above the dense bandlike infiltrate of lymphocytes with liquefaction degeneration of the basal layer, rare Civatte bodies in the epidermis, and melanophages in the dermis.
At 5-week follow-up, the patient showed some improvement but not complete control of the lesions with topical steroids. Because the patient was on no other regular medications, we recommended a 3-month trial removal of the IUS. The patient decided to have the IUS removed and noted complete clearance of the skin lesions within 1 month. Challenge with oral or intradermal levonorgestrel was not conducted after clearance of the rash, which is a weakness in this report. Accordingly, the possibility that this patient’s condition was caused by idiopathic lichen planus, which may resolve spontaneously, cannot be ruled out. However, because the patient noted substantial improvement following removal of the device and remained symptom free 2 years after removal, we concluded that the cutaneous lesions were secondary to an LDE in response to the IUS.
It should be noted that as-needed use of pseudoephedrine and loratadine continued during this 2-year follow-up period and again the patient experienced no return of symptoms, which is particularly important because both of these agents have been associated with drug eruption patterns akin to lichenoid tissue reaction/interface dermatitis patterns. Pseudoephedrine is particularly notorious for causing nonpigmenting fixed drug eruptions such as those that heal without hyperpigmentation, while antihistamines such as loratadine have been associated with lichenoid and subacute lupus erythematosus–pattern drug reactions.1,2
Lichenoid drug reactions fall into the category of lymphocyte-rich lichenoid tissue reaction/interface dermatitis skin disorders.3 There are currently 202 different drugs reported to cause lichen planus or lichenoid eruptions as collected in Litt’s Drug Eruption & Reaction Database.4 Some of the more common causes of an LDE include angiotensin-converting enzyme inhibitors, antimalarials, calcium channel blockers, gold salts, and nonsteroidal anti-inflammatory drugs.3,4 Lichenoid eruptions typically are attributed to oral hormonal contraceptives only.5,6 An eruption in response to intrauterine levonorgestrel treatment is rare. One case report of a lichenoid eruption in response to a copper IUS was hypothesized to be due to presence of nickel salts as a manufacturing contaminant; however, the manufacturer denied the presence of the contaminant.7
The manufacturer’s information for health care professionals prescribing levonorgestrel-releasing IUS describes rashes as an adverse reaction present in less than 5% of individuals.8 Levonorgestrel-releasing IUS consists of a polyethylene frame compounded with barium sulfate, 52 mg of levonorgestrel, silicone (polydimethylsiloxane), and a monofilament brown polyethylene removal thread. The device initially releases 20 μg levonorgestrel daily, with a stable levonorgestrel plasma level of 150 to 200 pg/mL reached after the first few weeks following insertion of the device.8 Levonorgestrel is an agonist at the progesterone and androgen receptors.9 In clinical trials, levonorgestrel was implicated as the cause of increased acne, hair loss, and hirsutism as cutaneous side effects from use of levonorgestrel implants.10 However, to our knowledge, none of the other components of the levonorgestrel-releasing IUS have previously been reported to cause lichen planus or LDE.
The levonorgestrel-releasing IUS has been implicated as the cause of biopsy-proven Sweet disease,11 exacerbation of preexisting seborrheic dermatitis,12 rosacea,13 and autoimmune progesterone dermatitis.14 The skin findings in these cases resolved after removal of the IUS and appropriate treatment.
Identification of the causative drug can be difficult in LDE, as timing of the eruption can vary. The latent period has been reported to range from a few months to 1 to 2 years.15 Additionally, the clinical picture is often complicated in patients with a history of different drug dosages or multiple medications. When present, the histologic features of parakeratosis and eosinophils can be clues that a lichen planus–like eruption is drug related rather than idiopathic. However, the absence of these features does not rule out a medication or environmental trigger. In this case, the time-event relationship likely indicates that the eruption was related to the levonorgestrel-releasing IUS and not triggered by other medications or not idiopathic in nature. Lichenoid drug eruptions can resolve within a few weeks or up to 2 years after drug cessation and can occasionally be complicated by partial or complete resolution and recurrence even when the drug has not been discontinued.16,17 Lichenoid drug eruptions or idiopathic lichen planus generally are treated with topical immunomodulators or corticosteroids.3
Based on the time-event relationship, morphology, distribution, and histopathologic findings, we conclude that our patient developed LDE in response to the placement of a levonorgestrel-releasing IUS. Clinicians should be aware of the possibility of LDE occurring as a rare adverse effect of these devices.
1. Shelley WB, Shelley ED. Nonpigmenting fixed drug eruption as a distinctive reaction pattern: examples caused by sensitivity to pseudoephedrine hydrochloride and tetrahydrozoline. J Am Acad Dermatol. 1987;17:403-407.
2. Crowson AN, Magro CM. Lichenoid and subacute cutaneous lupus erythematosus-like dermatitis associated with antihistamine therapy. J Cutan Pathol. 1999;26:95-99.
3. Sontheimer RD. Lichenoid tissue reaction/interface dermatitis: clinical and histological perspectives [published online ahead of print February 26, 2009]. J Invest Dermatol. 2009;129:1088-1099.
4. Litt’s Drug Eruption & Reaction Database. Boca Raton, FL: Taylor & Francis Group; 2015. http://www.drugeruptiondata.com/searchresults/index/reaction_type/id/1/char/L. Accessed June 11, 2015.
5. Coskey RJ. Eruptions due to oral contraceptives. Arch Dermatol. 1977;113:333-334.
6. Thomas P, Dalle E, Revillon B, et al. Cutaneous effects in hormonal contraception [in French]. NPN Med. 1985;5:19-24.
7. Lombardi P, Campolmi P, Sertoli A. Lichenoid dermatitis caused by nickel salts? Contact Dermatitis. 1983;9:520-521.
8. Mirena [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2014.
9. Lemus AE, Vilchis F, Damsky R, et al. Mechanism of action of levonorgestrel: in vitro metabolism and specific interactions with steroid receptors in target organs. J Steroid Biochem Mol Biol. 1992;41:881-890.
10. Brache V, Faundes A, Alvarex F, et al. Nonmenstrual adverse events during use of implantable contraceptives for women: data from clinical trials. Contraception. 2002;65:63-74.
11. Hamill M, Bowling J, Vega-Lopez F. Sweet’s syndrome and a Mirena intrauterine system. J Fam Plann Reprod Health Care. 2004;30:115-116.
12. Karri K, Mowbray D, Adams S, et al. Severe seborrhoeic dermatitis: side-effect of the Mirena intra-uterine system. Eur J Contracept Reprod Health Care. 2006;11:53-54.
13. Choudry K, Humphreys F, Menage J. Rosacea in association with the progesterone-releasing intrauterine contraceptive device. Clin Exp Dermatol. 2001;26:102.
14. Pereira A, Coker A. Hypersensitivity to Mirena—a rare complication. J Obstet Gynaecol. 2003;23:81.
15. Halevy S, Shai A. Lichenoid drug eruptions. J Am Acad Dermatol. 1993;29(2, pt 1):249-255.
16. Seehafer JR, Rogers RS 3rd, Fleming CR, et al. Lichen planus-like lesions caused by penicillamine in primary biliary cirrhosis. Arch Dermatol. 1981;117:140-142.
17. Anderson TE. Lichen planus following quinidine therapy. Br J Dermatol. 1967;79:500.
To the Editor:
Numerous drugs have been implicated as possible causes of lichenoid drug eruptions (LDEs). We describe a case of an LDE secondary to placement of a levonorgestrel-releasing intrauterine system (IUS).
A 28-year-old woman presented with an extensive pruritic rash of 2 months’ duration. She reported that it began on the wrists; progressed inward to involve the trunk; and then became generalized over the trunk, back, wrists, and legs. A levonorgestrel-releasing IUS had been placed 6 weeks prior to the onset of the rash. She was otherwise healthy and took loratadine and pseudoephedrine on occasion for environmental allergies. On examination there were violaceous, lichenified, flat-topped, polygonal papules scattered over the arms, legs, and trunk (Figure 1). Some papules demonstrated a Köbner phenomenon. No Wickham striae or mucosal involvement was noted. Rapid plasma reagin and hepatitis panel were negative. The patient was treated empirically with fluocinonide ointment 0.05% twice daily.
|
A shave biopsy was taken at the initial visit prior to steroid treatment. Histology revealed a classic lichenoid reaction pattern (Figure 2) and irregular acanthosis lying above the dense bandlike infiltrate of lymphocytes with liquefaction degeneration of the basal layer, rare Civatte bodies in the epidermis, and melanophages in the dermis.
At 5-week follow-up, the patient showed some improvement but not complete control of the lesions with topical steroids. Because the patient was on no other regular medications, we recommended a 3-month trial removal of the IUS. The patient decided to have the IUS removed and noted complete clearance of the skin lesions within 1 month. Challenge with oral or intradermal levonorgestrel was not conducted after clearance of the rash, which is a weakness in this report. Accordingly, the possibility that this patient’s condition was caused by idiopathic lichen planus, which may resolve spontaneously, cannot be ruled out. However, because the patient noted substantial improvement following removal of the device and remained symptom free 2 years after removal, we concluded that the cutaneous lesions were secondary to an LDE in response to the IUS.
It should be noted that as-needed use of pseudoephedrine and loratadine continued during this 2-year follow-up period and again the patient experienced no return of symptoms, which is particularly important because both of these agents have been associated with drug eruption patterns akin to lichenoid tissue reaction/interface dermatitis patterns. Pseudoephedrine is particularly notorious for causing nonpigmenting fixed drug eruptions such as those that heal without hyperpigmentation, while antihistamines such as loratadine have been associated with lichenoid and subacute lupus erythematosus–pattern drug reactions.1,2
Lichenoid drug reactions fall into the category of lymphocyte-rich lichenoid tissue reaction/interface dermatitis skin disorders.3 There are currently 202 different drugs reported to cause lichen planus or lichenoid eruptions as collected in Litt’s Drug Eruption & Reaction Database.4 Some of the more common causes of an LDE include angiotensin-converting enzyme inhibitors, antimalarials, calcium channel blockers, gold salts, and nonsteroidal anti-inflammatory drugs.3,4 Lichenoid eruptions typically are attributed to oral hormonal contraceptives only.5,6 An eruption in response to intrauterine levonorgestrel treatment is rare. One case report of a lichenoid eruption in response to a copper IUS was hypothesized to be due to presence of nickel salts as a manufacturing contaminant; however, the manufacturer denied the presence of the contaminant.7
The manufacturer’s information for health care professionals prescribing levonorgestrel-releasing IUS describes rashes as an adverse reaction present in less than 5% of individuals.8 Levonorgestrel-releasing IUS consists of a polyethylene frame compounded with barium sulfate, 52 mg of levonorgestrel, silicone (polydimethylsiloxane), and a monofilament brown polyethylene removal thread. The device initially releases 20 μg levonorgestrel daily, with a stable levonorgestrel plasma level of 150 to 200 pg/mL reached after the first few weeks following insertion of the device.8 Levonorgestrel is an agonist at the progesterone and androgen receptors.9 In clinical trials, levonorgestrel was implicated as the cause of increased acne, hair loss, and hirsutism as cutaneous side effects from use of levonorgestrel implants.10 However, to our knowledge, none of the other components of the levonorgestrel-releasing IUS have previously been reported to cause lichen planus or LDE.
The levonorgestrel-releasing IUS has been implicated as the cause of biopsy-proven Sweet disease,11 exacerbation of preexisting seborrheic dermatitis,12 rosacea,13 and autoimmune progesterone dermatitis.14 The skin findings in these cases resolved after removal of the IUS and appropriate treatment.
Identification of the causative drug can be difficult in LDE, as timing of the eruption can vary. The latent period has been reported to range from a few months to 1 to 2 years.15 Additionally, the clinical picture is often complicated in patients with a history of different drug dosages or multiple medications. When present, the histologic features of parakeratosis and eosinophils can be clues that a lichen planus–like eruption is drug related rather than idiopathic. However, the absence of these features does not rule out a medication or environmental trigger. In this case, the time-event relationship likely indicates that the eruption was related to the levonorgestrel-releasing IUS and not triggered by other medications or not idiopathic in nature. Lichenoid drug eruptions can resolve within a few weeks or up to 2 years after drug cessation and can occasionally be complicated by partial or complete resolution and recurrence even when the drug has not been discontinued.16,17 Lichenoid drug eruptions or idiopathic lichen planus generally are treated with topical immunomodulators or corticosteroids.3
Based on the time-event relationship, morphology, distribution, and histopathologic findings, we conclude that our patient developed LDE in response to the placement of a levonorgestrel-releasing IUS. Clinicians should be aware of the possibility of LDE occurring as a rare adverse effect of these devices.
To the Editor:
Numerous drugs have been implicated as possible causes of lichenoid drug eruptions (LDEs). We describe a case of an LDE secondary to placement of a levonorgestrel-releasing intrauterine system (IUS).
A 28-year-old woman presented with an extensive pruritic rash of 2 months’ duration. She reported that it began on the wrists; progressed inward to involve the trunk; and then became generalized over the trunk, back, wrists, and legs. A levonorgestrel-releasing IUS had been placed 6 weeks prior to the onset of the rash. She was otherwise healthy and took loratadine and pseudoephedrine on occasion for environmental allergies. On examination there were violaceous, lichenified, flat-topped, polygonal papules scattered over the arms, legs, and trunk (Figure 1). Some papules demonstrated a Köbner phenomenon. No Wickham striae or mucosal involvement was noted. Rapid plasma reagin and hepatitis panel were negative. The patient was treated empirically with fluocinonide ointment 0.05% twice daily.
|
A shave biopsy was taken at the initial visit prior to steroid treatment. Histology revealed a classic lichenoid reaction pattern (Figure 2) and irregular acanthosis lying above the dense bandlike infiltrate of lymphocytes with liquefaction degeneration of the basal layer, rare Civatte bodies in the epidermis, and melanophages in the dermis.
At 5-week follow-up, the patient showed some improvement but not complete control of the lesions with topical steroids. Because the patient was on no other regular medications, we recommended a 3-month trial removal of the IUS. The patient decided to have the IUS removed and noted complete clearance of the skin lesions within 1 month. Challenge with oral or intradermal levonorgestrel was not conducted after clearance of the rash, which is a weakness in this report. Accordingly, the possibility that this patient’s condition was caused by idiopathic lichen planus, which may resolve spontaneously, cannot be ruled out. However, because the patient noted substantial improvement following removal of the device and remained symptom free 2 years after removal, we concluded that the cutaneous lesions were secondary to an LDE in response to the IUS.
It should be noted that as-needed use of pseudoephedrine and loratadine continued during this 2-year follow-up period and again the patient experienced no return of symptoms, which is particularly important because both of these agents have been associated with drug eruption patterns akin to lichenoid tissue reaction/interface dermatitis patterns. Pseudoephedrine is particularly notorious for causing nonpigmenting fixed drug eruptions such as those that heal without hyperpigmentation, while antihistamines such as loratadine have been associated with lichenoid and subacute lupus erythematosus–pattern drug reactions.1,2
Lichenoid drug reactions fall into the category of lymphocyte-rich lichenoid tissue reaction/interface dermatitis skin disorders.3 There are currently 202 different drugs reported to cause lichen planus or lichenoid eruptions as collected in Litt’s Drug Eruption & Reaction Database.4 Some of the more common causes of an LDE include angiotensin-converting enzyme inhibitors, antimalarials, calcium channel blockers, gold salts, and nonsteroidal anti-inflammatory drugs.3,4 Lichenoid eruptions typically are attributed to oral hormonal contraceptives only.5,6 An eruption in response to intrauterine levonorgestrel treatment is rare. One case report of a lichenoid eruption in response to a copper IUS was hypothesized to be due to presence of nickel salts as a manufacturing contaminant; however, the manufacturer denied the presence of the contaminant.7
The manufacturer’s information for health care professionals prescribing levonorgestrel-releasing IUS describes rashes as an adverse reaction present in less than 5% of individuals.8 Levonorgestrel-releasing IUS consists of a polyethylene frame compounded with barium sulfate, 52 mg of levonorgestrel, silicone (polydimethylsiloxane), and a monofilament brown polyethylene removal thread. The device initially releases 20 μg levonorgestrel daily, with a stable levonorgestrel plasma level of 150 to 200 pg/mL reached after the first few weeks following insertion of the device.8 Levonorgestrel is an agonist at the progesterone and androgen receptors.9 In clinical trials, levonorgestrel was implicated as the cause of increased acne, hair loss, and hirsutism as cutaneous side effects from use of levonorgestrel implants.10 However, to our knowledge, none of the other components of the levonorgestrel-releasing IUS have previously been reported to cause lichen planus or LDE.
The levonorgestrel-releasing IUS has been implicated as the cause of biopsy-proven Sweet disease,11 exacerbation of preexisting seborrheic dermatitis,12 rosacea,13 and autoimmune progesterone dermatitis.14 The skin findings in these cases resolved after removal of the IUS and appropriate treatment.
Identification of the causative drug can be difficult in LDE, as timing of the eruption can vary. The latent period has been reported to range from a few months to 1 to 2 years.15 Additionally, the clinical picture is often complicated in patients with a history of different drug dosages or multiple medications. When present, the histologic features of parakeratosis and eosinophils can be clues that a lichen planus–like eruption is drug related rather than idiopathic. However, the absence of these features does not rule out a medication or environmental trigger. In this case, the time-event relationship likely indicates that the eruption was related to the levonorgestrel-releasing IUS and not triggered by other medications or not idiopathic in nature. Lichenoid drug eruptions can resolve within a few weeks or up to 2 years after drug cessation and can occasionally be complicated by partial or complete resolution and recurrence even when the drug has not been discontinued.16,17 Lichenoid drug eruptions or idiopathic lichen planus generally are treated with topical immunomodulators or corticosteroids.3
Based on the time-event relationship, morphology, distribution, and histopathologic findings, we conclude that our patient developed LDE in response to the placement of a levonorgestrel-releasing IUS. Clinicians should be aware of the possibility of LDE occurring as a rare adverse effect of these devices.
1. Shelley WB, Shelley ED. Nonpigmenting fixed drug eruption as a distinctive reaction pattern: examples caused by sensitivity to pseudoephedrine hydrochloride and tetrahydrozoline. J Am Acad Dermatol. 1987;17:403-407.
2. Crowson AN, Magro CM. Lichenoid and subacute cutaneous lupus erythematosus-like dermatitis associated with antihistamine therapy. J Cutan Pathol. 1999;26:95-99.
3. Sontheimer RD. Lichenoid tissue reaction/interface dermatitis: clinical and histological perspectives [published online ahead of print February 26, 2009]. J Invest Dermatol. 2009;129:1088-1099.
4. Litt’s Drug Eruption & Reaction Database. Boca Raton, FL: Taylor & Francis Group; 2015. http://www.drugeruptiondata.com/searchresults/index/reaction_type/id/1/char/L. Accessed June 11, 2015.
5. Coskey RJ. Eruptions due to oral contraceptives. Arch Dermatol. 1977;113:333-334.
6. Thomas P, Dalle E, Revillon B, et al. Cutaneous effects in hormonal contraception [in French]. NPN Med. 1985;5:19-24.
7. Lombardi P, Campolmi P, Sertoli A. Lichenoid dermatitis caused by nickel salts? Contact Dermatitis. 1983;9:520-521.
8. Mirena [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2014.
9. Lemus AE, Vilchis F, Damsky R, et al. Mechanism of action of levonorgestrel: in vitro metabolism and specific interactions with steroid receptors in target organs. J Steroid Biochem Mol Biol. 1992;41:881-890.
10. Brache V, Faundes A, Alvarex F, et al. Nonmenstrual adverse events during use of implantable contraceptives for women: data from clinical trials. Contraception. 2002;65:63-74.
11. Hamill M, Bowling J, Vega-Lopez F. Sweet’s syndrome and a Mirena intrauterine system. J Fam Plann Reprod Health Care. 2004;30:115-116.
12. Karri K, Mowbray D, Adams S, et al. Severe seborrhoeic dermatitis: side-effect of the Mirena intra-uterine system. Eur J Contracept Reprod Health Care. 2006;11:53-54.
13. Choudry K, Humphreys F, Menage J. Rosacea in association with the progesterone-releasing intrauterine contraceptive device. Clin Exp Dermatol. 2001;26:102.
14. Pereira A, Coker A. Hypersensitivity to Mirena—a rare complication. J Obstet Gynaecol. 2003;23:81.
15. Halevy S, Shai A. Lichenoid drug eruptions. J Am Acad Dermatol. 1993;29(2, pt 1):249-255.
16. Seehafer JR, Rogers RS 3rd, Fleming CR, et al. Lichen planus-like lesions caused by penicillamine in primary biliary cirrhosis. Arch Dermatol. 1981;117:140-142.
17. Anderson TE. Lichen planus following quinidine therapy. Br J Dermatol. 1967;79:500.
1. Shelley WB, Shelley ED. Nonpigmenting fixed drug eruption as a distinctive reaction pattern: examples caused by sensitivity to pseudoephedrine hydrochloride and tetrahydrozoline. J Am Acad Dermatol. 1987;17:403-407.
2. Crowson AN, Magro CM. Lichenoid and subacute cutaneous lupus erythematosus-like dermatitis associated with antihistamine therapy. J Cutan Pathol. 1999;26:95-99.
3. Sontheimer RD. Lichenoid tissue reaction/interface dermatitis: clinical and histological perspectives [published online ahead of print February 26, 2009]. J Invest Dermatol. 2009;129:1088-1099.
4. Litt’s Drug Eruption & Reaction Database. Boca Raton, FL: Taylor & Francis Group; 2015. http://www.drugeruptiondata.com/searchresults/index/reaction_type/id/1/char/L. Accessed June 11, 2015.
5. Coskey RJ. Eruptions due to oral contraceptives. Arch Dermatol. 1977;113:333-334.
6. Thomas P, Dalle E, Revillon B, et al. Cutaneous effects in hormonal contraception [in French]. NPN Med. 1985;5:19-24.
7. Lombardi P, Campolmi P, Sertoli A. Lichenoid dermatitis caused by nickel salts? Contact Dermatitis. 1983;9:520-521.
8. Mirena [package insert]. Whippany, NJ: Bayer HealthCare Pharmaceuticals Inc; 2014.
9. Lemus AE, Vilchis F, Damsky R, et al. Mechanism of action of levonorgestrel: in vitro metabolism and specific interactions with steroid receptors in target organs. J Steroid Biochem Mol Biol. 1992;41:881-890.
10. Brache V, Faundes A, Alvarex F, et al. Nonmenstrual adverse events during use of implantable contraceptives for women: data from clinical trials. Contraception. 2002;65:63-74.
11. Hamill M, Bowling J, Vega-Lopez F. Sweet’s syndrome and a Mirena intrauterine system. J Fam Plann Reprod Health Care. 2004;30:115-116.
12. Karri K, Mowbray D, Adams S, et al. Severe seborrhoeic dermatitis: side-effect of the Mirena intra-uterine system. Eur J Contracept Reprod Health Care. 2006;11:53-54.
13. Choudry K, Humphreys F, Menage J. Rosacea in association with the progesterone-releasing intrauterine contraceptive device. Clin Exp Dermatol. 2001;26:102.
14. Pereira A, Coker A. Hypersensitivity to Mirena—a rare complication. J Obstet Gynaecol. 2003;23:81.
15. Halevy S, Shai A. Lichenoid drug eruptions. J Am Acad Dermatol. 1993;29(2, pt 1):249-255.
16. Seehafer JR, Rogers RS 3rd, Fleming CR, et al. Lichen planus-like lesions caused by penicillamine in primary biliary cirrhosis. Arch Dermatol. 1981;117:140-142.
17. Anderson TE. Lichen planus following quinidine therapy. Br J Dermatol. 1967;79:500.
