Cutaneous amyloidosis encompasses a variety of clinical presentations. Primary localized cutaneous amyloidosis comprises lichen amyloidosis, macular amyloidosis, and nodular amyloidosis.1 Macular and lichen amyloidosis result from keratin deposits, while nodular amyloidosis results from cutaneous infiltration of plasma cells.2 Primary systemic amyloidosis is due to a plasma cell dyscrasia, particularly multiple myeloma, while secondary systemic amyloidosis occurs in the setting of restrictive cardiomyopathy, congestive heart failure, renal dysfunction, or chronic inflammation, as seen with rheumatoid arthritis, tuberculosis, and various autoinflammatory disorders.2 Plasma cell proliferative disorders are associated with various skin disorders, which may result from aggregated misfolded monoclonal immunoglobulins, indicating light chain–related systemic amyloidosis. Mucocutaneous lesions can occur in 30% to 40% of cases of primary systemic amyloidosis and may present as purpura, ecchymoses, waxy thickening, plaques, subcutaneous nodules, and/or bullae.3,4 When blistering is present, the differential diagnosis is broad and includes autoimmune bullous disease, drug eruptions, enoxaparin-induced bullous hemorrhagic dermatosis, deposition diseases, allergic contact dermatitis, bullous cellulitis, bullous bite reactions, neutrophilic dermatosis, and bullous lichen sclerosus.5 Herein, we present a case of a woman with a bullous skin eruption who eventually was diagnosed with bullous amyloidosis subsequent to a diagnosis of multiple myeloma.
Case Report
A 70-year-old woman presented to our dermatology clinic for evaluation of well-demarcated, hemorrhagic, flaccid vesicles and focal erosions with a rim of erythema on the distal forearms and hands. A shave biopsy from the right forearm showed cell-poor subepidermal vesicular dermatitis. Enzyme-linked immunosorbent assays for bullous pemphigoid antigens 1 and 2 as well as urinary porphyrins were negative. Direct immunofluorescence showed granular IgM at the basement membrane zone around vessels and cytoid bodies. At this time, a preliminary diagnosis of pseudoporphyria was suspected, though no classic medications (eg, nonsteroidal anti-inflammatory drugs, furosemide, antibiotics) or exogenous trigger factors (eg, UV light exposure, dialysis) were temporally related. Three months later, the patient presented with a large hemorrhagic bulla on the distal left forearm (Figure 1) and healing erosions on the dorsal fingers and upper back. Clobetasol ointment was initiated, as an autoimmune bullous dermatosis was suspected.

FIGURE 1. Large hemorrhagic bulla on the distal left forearm later confirmed to be bullous amyloidosis.
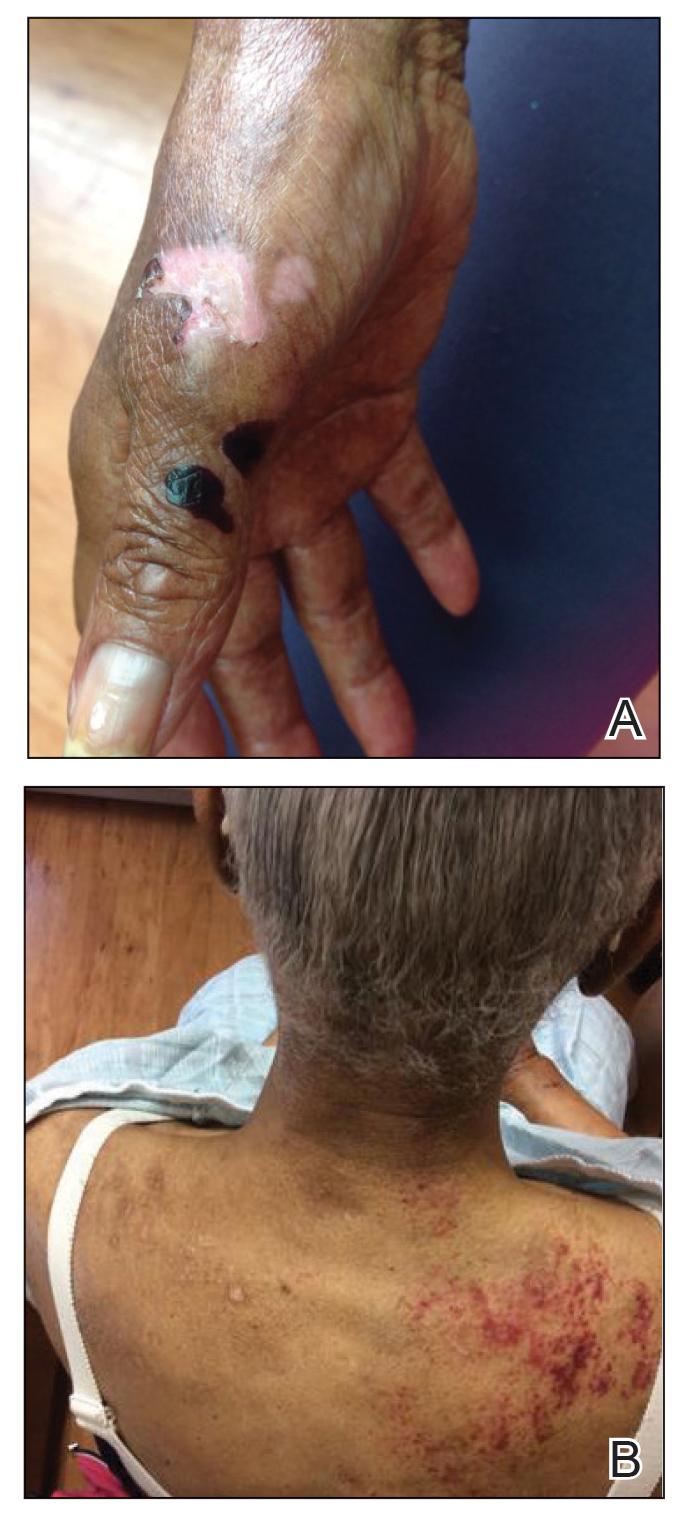
Approximately 1 year after she was first seen in our outpatient clinic, the patient was hospitalized for induction of chemotherapy—cyclophosphamide, bortezomib, and dexamethasone—for a new diagnosis of stage III multiple myeloma. A workup for back pain revealed multiple compression fractures and a plasma cell neoplasm with elevated λ light chains, which was confirmed with a bone marrow biopsy. During an inpatient dermatology consultation, we noted the development of intraoral hemorrhagic vesicles and worsening generalization of the hemorrhagic bullae, with healing erosions and intact hemorrhagic bullae on the dorsal hands, fingers (Figure 2), and upper back.
A repeat biopsy displayed bullous amyloidosis. Histopathologic examination revealed an ulcerated subepidermal blister with fibrin deposition at the ulcer base. A periadnexal, scant, eosinophilic deposition with extravasated red blood cells was appreciated. Amorphous eosinophilic deposits were found within the detached fragment of the epidermis and inflammatory infiltrate. A Congo red stain highlighted these areas with a salmon pink–colored material. Congo red staining showed a moderate amount of pale, apple green, birefringent deposit within these areas on polarized light examination.
A few months later, the patient was re-admitted, and the amount of skin detachment prompted the primary team to ask for another consultation. Although the extensive skin sloughing resembled toxic epidermal necrolysis, a repeat biopsy confirmed bullous amyloidosis.
Comment
Amyloidosis Histopathology—Amyloidoses represent a wide array of disorders with deposition of β-pleated sheets or amyloid fibrils, often with cutaneous manifestations.2,3 Primary systemic amyloidosis has been associated with underlying dyscrasia or multiple myeloma.6 In such cases, the skin lesions of multiple myeloma may result from a collection of misfolded monoclonal immunoglobulins or their fragments, as in light chain–related systemic amyloidosis.3 Histopathologically, both systemic and cutaneous amyloidosis appear similar and display deposition of amorphous, eosinophilic, fissured amyloid material in the dermis. Congo red stains the material orange-red and will display a characteristic apple green birefringence under polarized light.4 Although bullous amyloid lesions are rare, the cutaneous forms of these lesions can be an important sign of plasma cell dyscrasia.7
Presentation of Bullous Amyloidosis—Bullous manifestations rarely have been noted in the primary cutaneous forms of amyloidosis.5,8,9 Importantly, cutaneous blistering more often is linked to systemic forms of amyloidosis with multiorgan involvement, including primary systemic and myeloma-associated amyloidosis.5,10 However, patients with localized bullous cutaneous amyloidosis without systemic involvement also have been seen.10,11 Bullae may occur at any time, with contents that frequently are hemorrhagic due to capillary fragility.12,13 Bullous manifestations raise the differential diagnoses of bullous pemphigoid, epidermolysis bullosa acquisita, linear IgA disease, porphyria cutanea tarda, pseudoporphyria, bullous drug eruption, bullous eruption of renal dialysis, or bullous lupus erythematosus.5,13-17
In our patient, the acral distribution of bullae, presence of hemorrhage, chronicity of symptoms, and negative enzyme-linked immunosorbent assay initially suggested a diagnosis of pseudoporphyria. However, the presence of intraoral hemorrhagic vesicles and subsequent confirmatory pathology aided in differentiating bullous amyloidosis from pseudoporphyria. Nodular localized primary cutaneous amyloidosis, a rare form of skin-restricted amyloidoses, can coexist with bullous lesions. Of note, reported cases of nodular localized primary cutaneous amyloidosis did not result in development of multiple myeloma.5,10
Bullae are located either subepidermally or intradermally, and bullous lesions of cutaneous amyloidosis typically demonstrate subepidermal or superficial intradermal clefting on light microscopy.5,10,12 Histopathology of bullous amyloidosis shows intradermal or subepidermal blister formation and amorphous eosinophilic material showing apple green birefringence with Congo red staining deposited in the dermis and/or around the adipocytes and blood vessel walls.12,18-20 In prior cases, direct immunofluorescence of bullous amyloidosis revealed absent immunoglobulin (IgG, IgA, IgM) or complement (C3 and C9) deposits in the basement membrane zone or dermis.13,21,22 In these cases, electron microscopy was useful in diagnosis, as it showed the presence of amyloid deposits.21,22
Cause of Bullae—Various mechanisms are thought to trigger the blister formation in amyloidosis. Bullae created from trauma or friction often present as tense painful blisters that commonly are hemorrhagic.10,23 Amyloid deposits in the walls of blood vessels and the affinity of dermal amyloid in blood vessel walls to surrounding collagen likely leads to increased fragility of capillaries and the dermal matrix, hemorrhagic tendency, and infrapapillary blisters, thus creating hemorrhagic bullous eruptions.24,25 Specifically, close proximity of immunoglobulin-derived amyloid oligomers to epidermal keratinocytes may be toxic and therefore could trigger subepidermal bullous change.5 Additionally, alteration in the physicochemical properties of the amyloidal protein might explain bullous eruption.9 Trauma or rubbing of the hands and feet may precipitate the acral blister formation in bullous amyloidosis.5,11
Due to deposition of these amyloid fibrils, skin bleeding in these patients is called amyloid or pinch purpura. Vessel wall fragility and damage by amyloid are the principal causes of periorbital and gastrointestinal tract bleeding.26 Destruction of the lamina densa and widening of the intercellular space between keratinocytes by amyloid globules induce skin fragility.11
Although uncommon, various cases of bullous amyloidosis have been reported in the literature. Multiple myeloma patients represent the majority of those reported to have bullous amyloidosis.6,7,13,24,27-30 Plasmacytoma-associated bullous amyloid purpura and paraproteinemia also have been noted.25 Multiple myeloma with secondary AL amyloidosis has been seen with amyloid purpura and atraumatic ecchymoses of the face, highlighting the hemorrhage noted in these patients.26
Management of Amyloidosis—Various treatment options have been attempted for primary cutaneous amyloidosis, including oral retinoids, corticosteroids, cyclophosphamide, cyclosporine, amitriptyline, colchicine, cepharanthin, tacrolimus, dimethyl sulfoxide, vitamin D3 analogs, capsaicin, menthol, hydrocolloid dressings, surgical modalities, laser treatment, and phototherapy.1 There is no clear consensus for therapeutic modalities except for treating the underlying plasma cell dyscrasia in primary systemic amyloidosis.
Conclusion
We report the case of a patient displaying signs of pseudoporphyria that ultimately proved to be bullous amyloidosis, or what we termed pseudopseudoporphyria. Bullous amyloidosis should be considered in the differential diagnoses of hemorrhagic bullous skin eruptions. Particular attention should be given to a systemic workup for multiple myeloma when hemorrhagic vesicles/bullae are chronic and coexist with purpura, angina bullosa hemorrhagica, fatigue/weight loss, and/or macroglossia.