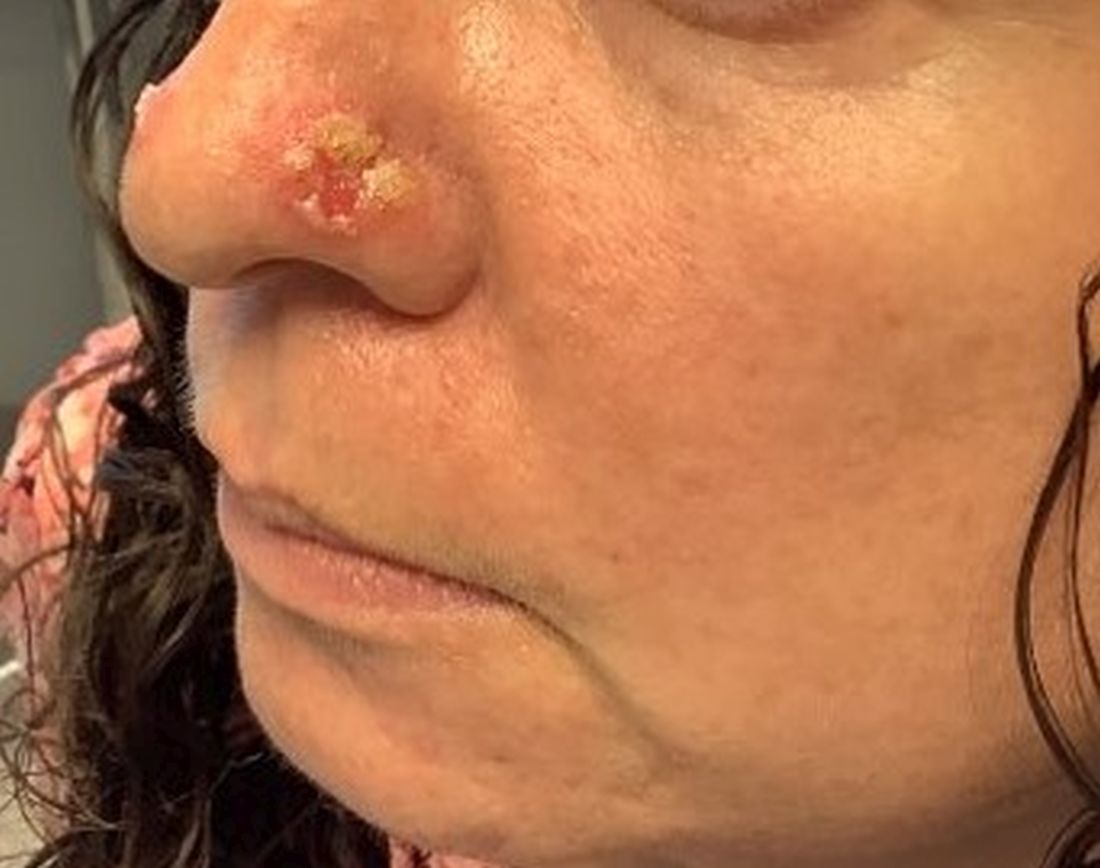
Cultures for bacteria, varicella zoster virus, herpes simplex virus, and mpox virus were all negative. A biopsy revealed suprabasilar acantholysis with follicular involvement in association with blister formation and inflammation. Direct immunofluorescence was positive for suprabasilar IgG and C3 deposition, consistent with pemphigus vulgaris (PV).
. There is likely a genetic predisposition. Medications that may induce pemphigus include penicillamine, nifedipine, or captopril.
Clinically, flaccid blistering lesions are present that may be cutaneous and/or mucosal. Bullae can progress to erosions and crusting, which then heal with pigment alteration but not scarring. The most commonly affected sites are the mouth, intertriginous areas, face, and neck. Mucosal lesions may involve the lips, esophagus, conjunctiva, and genitals.

Dr. Donna Bilu Martin
Biopsy for histology and direct immunofluorescence is important in distinguishing between PV and other blistering disorders. Up to 75% of patients with active disease also have a positive indirect immunofluorescence with circulating IgG.
Treatment is generally immunosuppressive. Systemic therapy usually begins with prednisone and then is transitioned to a steroid-sparing agent such as mycophenolate mofetil. Other steroid-sparing agents include azathioprine, methotrexate, cyclophosphamide, and intravenous immunoglobulin. Secondary infections are possible and should be treated. Topical therapies aimed at reducing pain, especially in mucosal lesions, can be beneficial.
This case and the photos are from Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.