The development of biologic therapies has led to some of the most significant advances in the treatment of cancer, but these drugs are also very expensive. As patents for the biologics begin to expire, the development of biosimilars has the potential to dramatically cut therapy costs thereby making the therapies more readily accessible to patients. Here, we discuss biosimilar development and the challenges that need to be overcome to create a robust market.
Biosimilar, not generic
Biologic therapies are derived from living organisms and include the targeted monoclonal antibodies (mAbs) and cell-based therapies that have revolutionized the treatment of certain cancer types. Yet, their greater complexity makes them more difficult to manufacture, store, and administer, making them a costly therapeutic option that ultimately drives up health care costs. According to a 2011 drug expenditure analysis, biologic therapies accounted for more than half of the total expenditure on anticancer drugs in the US health care system.1,2
Generally, when drug patents expire, other companies can develop their own identical generic versions to increase competition in the marketplace and drive down costs. However, the paradigm for generic development cannot be applied to biologic therapies because the way in which they are manufactured makes it impossible to generate an identical copy.
Instead, the Biologics Price Competition and Innovation Act, a provision of the Patient Protection and Affordable Care Act, has allowed for submission of an application for “licensure of a biologic product based on its similarity to a licensed biologic product”.3
These “biosimilars” have been positioned as game-changers in oncology, with the potential to reduce costs and improve access to biologic therapies. With the patents on several blockbuster cancer biologics already expired or due to expire by 2020, an increasing number of biosimilars are being developed.4
Totality of evidence
Biosimilars require more rigorous testing than generics, but they don’t require the same type of scientific data that the original biologic products, termed “reference products,” did. Therefore, they are governed by legislation unique to them and approved by different regulatory pathways. The US Food and Drug Administration (FDA) has established a unique shortened regulatory pathway for their approval, known as the 351(k) pathway. So whereas the pathway for reference products is geared toward demonstrating patient benefit, biosimilars are required instead to show equivalence to the reference product.5
Biosimilars are produced through reverse engineering the reference product. Then, through a stepwise process, to generate what the FDA calls a “totality of evidence,” biosimilar manufacturers must demonstrate structural and functional similarities (through comparative quality studies) and comparable pharmacokinetics and pharmacodynamics (through comparative nonclinical and clinical studies) to the reference product. Final approval is based on 1 or more comparative clinical studies performed in the most sensitive patient population(s) (Figure 1).6
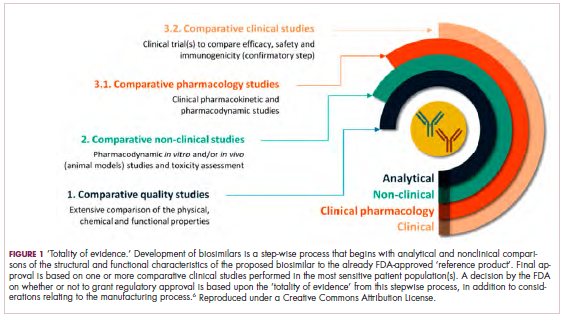
The primary endpoint of biosimilar clinical trials is chosen to detect clinically relevant differences and may not be the same as that used in pivotal trials of the reference product. Endpoints such as progression-free survival (PFS) and overall survival (OS) may not be feasible or sensitive enough to demonstrate biosimilarity.
Clinical trials of biosimilars should also be carried out in the most sensitive patient population, so that any potential differences can be attributed to the drug and not the patient population itself. If the reference product is approved across several different indications and there is sufficient scientific evidence to allow it, including the demonstration that the mechanism of action of the drug is the same across all indications, the FDA can extend the approval of the biosimilar to all of these indications without the need for individual clinical trials through a process known as extrapolation.
Biosimilar manufacturers must also provide evidence of the composition of their formulation and of quality control in their manufacturing processes, to ensure that biosimilarity can be maintained from batch to batch. As with the reference product, even small changes in the manufacturing process can have serious ramifications for clinical efficacy and safety.7,8
A flurry of approvals
The first biosimilar approvals in oncology in the United States came in the supportive care niche (Table 1). Filgrastim-sndz (Zarxio), approved in March 2015, is a biosimilar of the granulocyte-macrophage colony stimulating factor (G-CSF) analog filgrastim (Neupogen). Owing to its mechanism of action in stimulating the production of neutrophils in the bone marrow, filgrastim is used to help reduce the risk or severity of neutropenia in patients undergoing myelosuppressive chemotherapy regimens.
Filgrastim-sndz was approved for use across all 5 indications for which the reference product is approved, based on the totality of evidence, which included results from the key phase 3 PIONEER study.9 Market entry was initially delayed by lawsuits filed by Amgen, the maker of the reference product, but the biosimilar was subsequently cleared by the US Court of Appeals for the Federal Circuit. The wholesale acquisition cost (WAC) for a 300µg syringe is $324.30 for filgrastim and $275.66 figrastim-sndz, representing a 15% reduction on the reference product.10
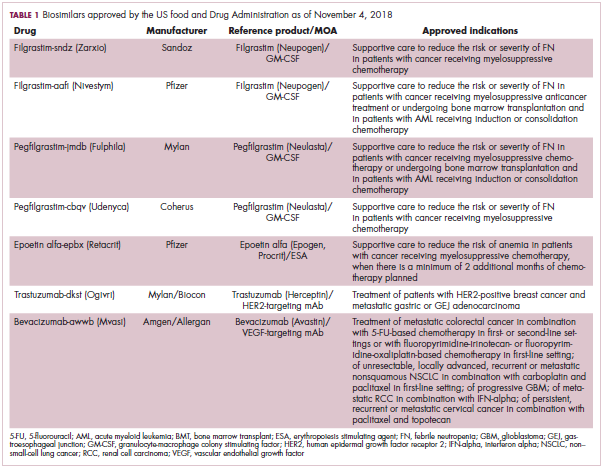
In 2018, the FDA approved a second filgrastim biosimilar, filgrastim-aafi (Nivestym),11 in addition to 2 biosimilars of the pegylated form of filgrastim, pegfilgrastim-jmdb (Fulphila)12 and pegfilgrastim-cbqv (Udenyca)13 – these forms of filgrastim have been modified by the addition of polyethylene glycol polymer chains that help to increase circulation time.
Approval for the 2 pegfilgrastm biosimilars was originally delayed by complete response letters (CRLs) from the FDA. For pegfilgrastim-jmdb, the CRL was reported to be related to a pending update of the Biologic’s License Application (BLA) to include information regarding facility requalification activities that had been taken after the addition of plant modifications. The CRL for pegfilgrastim-cbqv requested that the company provide additional manufacturing information and reanalyze a subset of samples with a revised immunogenicity assay.
Once the CRL concerns were addressed, regulatory approval was awarded and Mylan recently confirmed that pegfilgrastim-jmdb has been launched in the US marketplace at a WAC that reflects a 33% discount over the reference product.14
Approval data for filgrastim-aafi and pegfilgrastim-cbqv have not yet been published, however the respective manufacturers reported that approval was based on totality of evidence demonstrating a high degree of similarity to the reference products. Filgrastim-aafi was approved for all of the indications of the reference product and launched in the US on October 1, 2018 at a 30% discounted WAC.15
Epoetin alfa-epbx (Retacrit), a biosimilar of epoetin alfa, was also approved in 2018. It is a recombinant analog of erythropoietin (EPO), which stimulates the production of blood cells and has proved useful for the treatment of anemia, including in cancer patients receiving myelosuppressive chemotherapy. Approval of the biosimilar followed earlier receipt of a CRL from the FDA citing concerns relating to the manufacturing facility, which the company addressed. Pfizer has said that it expects to launch the biosimilar this year (2018), but a WAC has not been disclosed.16The FDA also recently approved the first biosimilars for the treatment of cancer. Trastuzumab-dkst (Ogivri) and bevacizumab-awwb (Mvasi) were approved in the second half of 2017 for the same indications as their respective reference products, which are mAbs directed at the human epidermal growth factor receptor 2 (HER2) and vascular endothelial growth factor, respectively.17,18
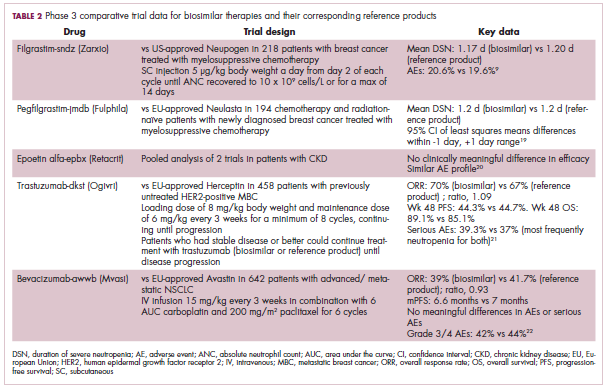
Approval data for bevacizumab-awwb included a comparative clinical trial in patients with advanced/metastatic non–small-cell lung cancer (NSCLC), which was considered the most sensitive patient population. The BLA for trastuzumab-dkst included data from the phase 3 comparative HERiTAge clinical trial, in which the biosimilar was compared with the reference product, both in combination with docetaxel or paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Neither biosimilar has been launched on the US market yet because the patents for their reference products do not expire until 2019, so it is not clear what the price discount will be for these drugs (Table 2).9,19-22
Biosimilars in development
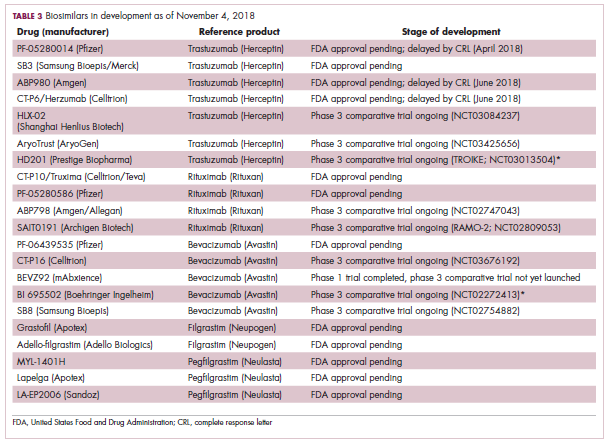
While numerous other biosimilars of filgrastim and pegfilgrastim are in development, the major focus has been on the development of more biosimilars to treat cancer (Table 3). BLAs have been submitted for 4 biosimilars of trastuzumab and 1 bevacizumab biosimilar. Approval for several of the trastuzumab biosimilars has been delayed by CRLs from the FDA, mostly regarding issues with the manufacturing process or facility. Several other trastuzumab and bevacizumab biosimilars are in late-stage clinical trials.
The results of several phase 3 comparative clinical trials were recently published or reported at annual conferences. Pfizer’s PF-05280014 was compared with the European Union (EU)–approved trastuzumab, both in combination with paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Data reported at the European Society for Medical Oncology congress in 2017 demonstrated equivalence between the reference product and biosimilar in overall response rate (ORR).23
Another recently published trial compared this biosimilar to EU-trastuzumab, both in combination with carboplatin and docetaxel, as neoadjuvant treatment for patients with resectable HER2-positive breast cancer. Among 226 patients randomized to receive 8 mg/kg in cycle 1 and 6 mg/kg thereafter of the biosimilar or reference product, every 3 weeks for 6 cycles, the pathologic complete response (pCR) rates were 47% and 50%, respectively.24
The results of a phase 3 study comparing Samsung Bioepis/Merck’s joint offering SB3 were recently published. A total of 875 patients were randomized 1:1 to receive SB3 or reference trastuzumab in combination with chemotherapy (4 cycles docetaxel followed by 4 cycles 5-fluorouracil/epirubicin/cyclophosphamide) prior to surgery, followed by 10 cycles of adjuvant SB3 or trastuzumab reference. Rates of event-free survival (EFS) were comparable between the 2 groups at 12 months (93.7% vs 96.1%, respectively).25
Amgen’s ABP980 was evaluated in the phase 3 LILAC trial, which measured the effect of the biosimilar on pCR in women with HER2-positive early breast cancer compared with reference trastuzumab. After 4 cycles of run-in anthracycline-based chemotherapy, ABP980 or reference trastuzumab were administered in combination with paclitaxel. This was followed by surgery and then ABP980 or reference trastuzumab in the adjuvant setting for up to 1 year, with the option to continue on the same drug as the neoadjuvant setting or to switch to the other. Among 696 assessable patients, the pCR rates were 48% and 42%, respectively.26
Most advanced in clinical testing among the upcoming bevacizumab biosimilars is Pfizer’s PF-06439535, for which the results of a phase 3 comparative trial were presented at the 2018 annual meeting of the American Society for Clinical Oncology. PF-06439535 was compared with the EU-approved bevacizumab, both in combination with paclitaxel and carboplatin, as first-line therapy for patients with advanced non-squamous NSCLC. Among 719 patients, the primary endpoint of ORR was 45.3% and 44.6%, respectively.27
Biosimilars of a third blockbuster cancer drug, the CD20-targeting mAb rituximab (Rituxan) are also in development and FDA approval is pending for 2. The patent for Rituxan expired in 2016, so these drugs could hit the market as soon as they are approved.
In a race to the finish for the first US-approved rituximab biosimilar, Celltrion-Teva’s CT-P10 (Truxima) seems most likely to come first; the Oncologic Drugs Advisory Committee voted unanimously in October 2018 to recommend its approval. Phase 3 comparative data were recently published; patients with newly diagnosed advanced-stage follicular lymphoma were randomized to receive intravenous infusions of 375 mg/m2 CT-P10 or reference rituximab, both in combination with cyclophosphamide, vincristine, and prednisone, on day 1 of 8 21-day cycles. The ORRs were identical (92.6%) for both drugs, pharmacokinetics data also suggested bioequivalence, and the incidence of AEs was also comparable (83% vs 80%).28
Biosimilars of the epidermal growth factor receptor (EGFR)-targeting mAb cetuximab are also listed in the pipeline for several biosimilar developers, but there is no indication of their developmental status as yet and no clinical trials are ongoing in the US.
Sorrento is developing STI-001, a cetuximab biosimilar, and reported that a phase 3 trial had been completed. Instead of a comparison with the reference product, however, the trial compared STI-001 in combination with irinotecan with irinotecan alone. They reported significantly higher ORR, PFS, and OS with the biosimilar compared with irinotecan alone, and a significant increase over historical data with the reference product, as well as fewer side effects and immunogenicity, which they attribute to its manufacture in a different cell line. However, no data has been published and no trials are ongoing in the United States, so the status of its development remains unclear.29
Challenges to a robust market
It is an exciting time for biosimilars, with many approvals and drugs being brought to market in the US in the past several years and more poised to follow suit as patents expire. Yet many challenges remain around the growth of a robust biosimilars market.
Several surveys conducted in recent years have demonstrated suboptimal knowledge of all aspects of biosimilars and highlighted the need for evidence-based education across specialties.30,31 In response, the FDA recently announced that it was launching an educational campaign to further understanding of biosimilars, including naming conventions (Figure 2).32,33 Numerous other medical professional societies have produced or are in the process of producing biosimilar guidelines.

Educational outreach by the FDA forms part of their 4-step plan to aid biosimilar development, which also aims to improve the efficiency of biosimilar development and approval, to provide regulatory clarity for manufacturers, to facilitate public understanding and acceptance, and to support a competitive marketplace.
Among the most critical educational gaps is confusion over the issue of interchangeability. Once approved by the FDA, generic drugs are considered interchangeable with the brand name drug and can be substituted at the pharmacy level without referring to the prescribing physician. This is not the case for biosimilars; owing to their more complex nature, biosimilars require a separate designation for interchangeability and none of those approved so far have been given this designation by the FDA.
There has been some confusion about what will be required to demonstrate interchangeability, and the FDA recently produced draft guidance, saying that essentially it should be proven that switching out the reference product for a biosimilar does not increase risk in terms of diminished efficacy or safety. Several companies are beginning to incorporate a switching component into their clinical trials of biosimilars.
Continued postmarketing and real-world studies will also be particularly important for biosimilars to increase confidence in prescribing them by demonstrating their continued efficacy and safety in the long-term. Several real-world studies are now ongoing, including the MONITOR-GCSF trial of filgrastim biosimilars.
Another major barrier to the development of a thriving biosimilars market that achieves the goals of reduced costs and increased access is the financial burden of their development. They are vastly more costly to develop and produce than generics. Added to litigation costs, this can limit their ability to compete in terms of price, which has been reflected in the lower-than-anticipated cost savings with some approved biosimilars thus far.
Experts have suggested that there might be much to learn from the European market, where biosimilars have been available for more than a decade and over time have reached even higher-than-expected savings. With high financial stakes and an increasingly important role in the treatment of cancer, the need to iron out the kinks is more pressing than ever.7,8,34,35