User login
Research and Reviews for the Practicing Oncologist
Trastuzumab-dkst approval adds to the biosimilar cancer drug market
The human epidermal growth factor receptor-2 (HER2)-targeting monoclonal antibody trastuzumab-dkst, was approved by the US Food and Drug Administration in 2017 for the treatment of patients with HER2-positive breast or metastatic gastric or gastroesophageal junction adenocarcinoma.1 Trastuzumab-dkst, marketed as Ogviri by Mylan NV and Biocon Ltd, is a copy, known as a biosimilar, of Genentech’s trastuzumab (Herceptin), which has been approved in the US since 1998. Genentech’s patent on trastuzumab expires in 2018
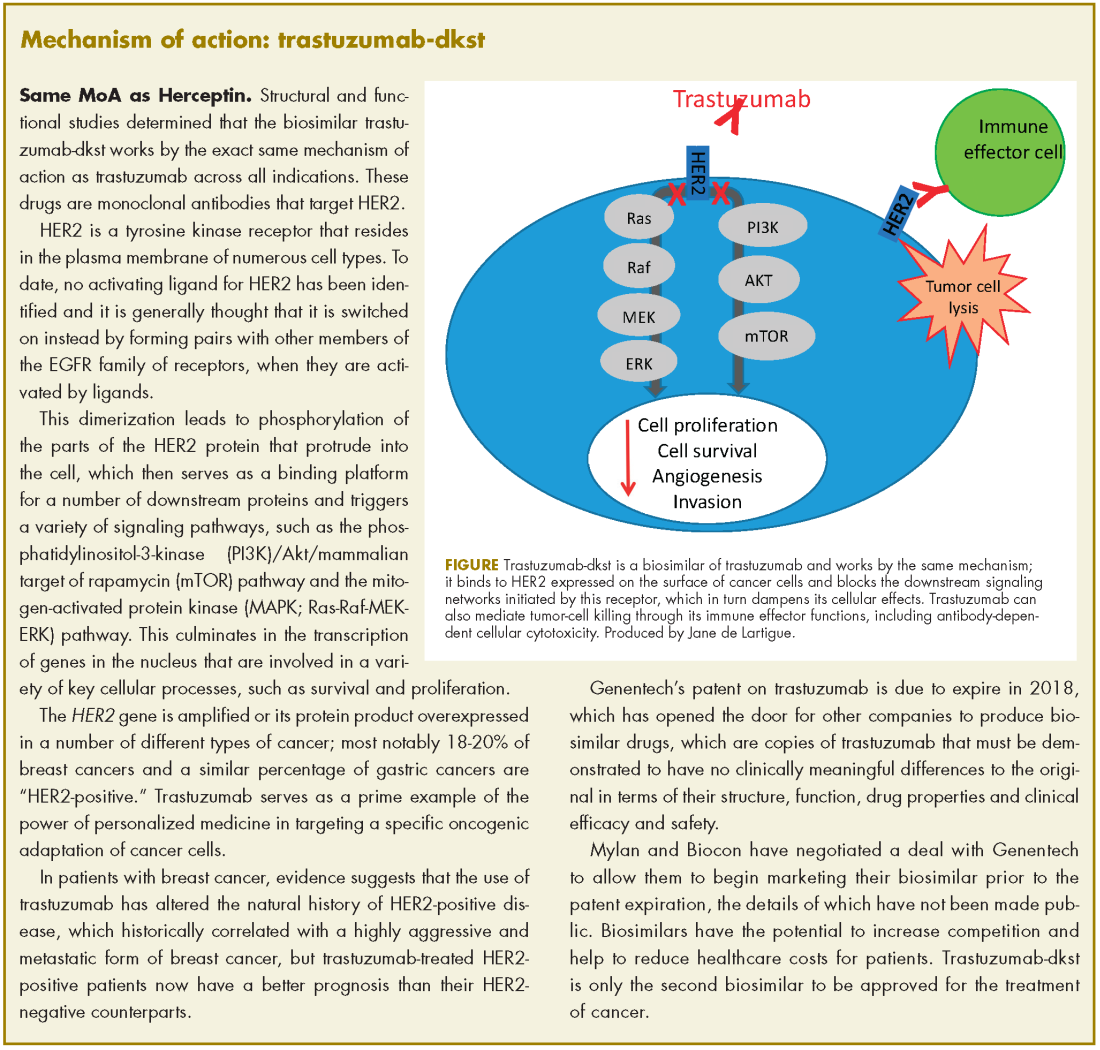
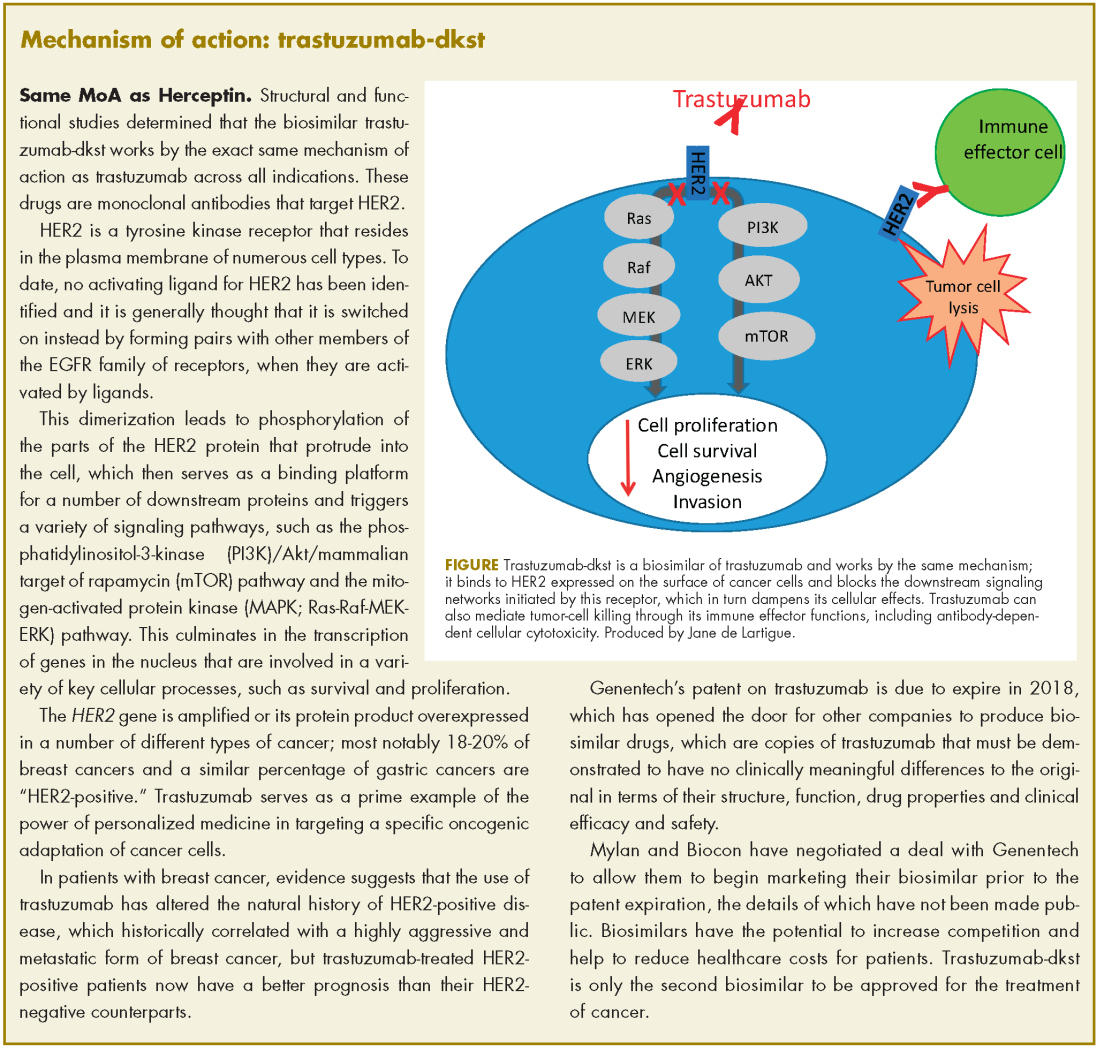
Approval was based on a comparison of the 2 drugs, which demonstrated that there were no clinically meaningful differences between the biosimilar and the reference product (trastuzumab) in terms of structure and function, pharmacokinetics (PKs), pharmacodynamics, and clinical efficacy and safety.
In structural and functional studies, trastuzumab-dkst was shown to have an identical amino acid sequence and a highly similar 3-dimensional structure, as well as equivalency in an inhibition of proliferation assay, a HER2-binding assay, and an antibody-dependent cellular cytotoxicity assay, compared with trastuzumab.
Two nonclinical animal studies were performed in cynomolgus monkeys; a single-dose comparative PK study and a 4-week, repeat-dose toxicity study. That was further supported by data from a single-dose, randomized, double-blind, comparative 3-way PK study (MYL-HER-1002) in which 120 healthy men were given an 8 mg/kg infusion of trastuzumab-dkst, US-approved trastuzumab, or European Union (EU)-approved trastuzumab.
The key clinical study was the phase 3 HERiTAge trial, a 2-part, multicenter, double-blind, randomized, parallel group trial that was performed in patients with HER2-positive metastatic breast cancer who had not been previously treated with either chemotherapy or trastuzumab in the metastatic setting.2
Eligible patients included males or females with measurably HER2-positive disease (as defined by HER2 overexpression determined by immunohistochemistry performed by a central laboratory), no exposure to chemotherapy or trastuzumab in the metastatic setting, an Eastern Cooperative Oncology Group Performance Status of 0 or 2, left ventricular ejection fraction (LVEF) within institutional range of normal, and who had completed adjuvant trastuzumab therapy at least 1 year before.
Patients with central nervous system metastases had to have stable disease after treatment, and hormonal agents were required to be discontinued before the start of the study. Patients with a history of unstable angina, heart failure, myocardial infarction less than 1 year from randomization, other clinically significant cardiac disease, grade 2 or higher peripheral neuropathy, a history of any other cancer within 4 years before screening, or any significant medical illness that increased treatment risk or impeded evaluation, were excluded from the study.
Patients were randomly assigned 1:1 to receive trastuzumab-dkst or trastuzumab, both in combination with paclitaxel or docetaxel, at a loading dose of 8 mg/kg, followed by a maintenance dose of 6 mg/kg, every 3 weeks for a minimum of 7 cycles in part 1 of the study. Patients who had stable disease or better were enrolled in part 2 and continued treatment until disease progression or unacceptable toxicity.
The primary endpoint was overall response rate (ORR) and, after 24 weeks, the ORR was 69.6% in the trastuzumab-dkst arm, compared with 64% in the trastuzumab arm, with a ratio of ORR of 1.09. Progression-free survival was also nearly identical in the 2 groups and median overall survival had not been reached in either arm.
The safety of the biosimilar and reference product were also highly similar. Serious adverse events occurred in 39.3%, compared with 37% of patients, respectively, with neutropenia the most frequently reported in both arms. Overall, treatment-emergent AEs occurred in 96.8%, compared with 94.7% of patients, respectively, with the majority of events mild or moderate in severity in both groups. This study also confirmed the low immunogenicity of the 2 drug products.
The prescribing information details the recommended doses of trastuzumab-dkst for each approved indication and warnings and precautions for cardiomyopathy, infusion reactions, pulmonary toxicity, exacerbation of chemotherapy-induced neutropenia and embryofetal toxicity.3
Patients should undergo thorough cardiac assessments, including baseline LVEF measurement immediately before starting therapy, every 3 months during therapy, and upon completion of therapy. Patients who complete adjuvant therapy should have cardiac assessments every 6 months for at least 2 years. Treatment should be withheld for ≥16% absolute decrease in LVEF from pre-treatment values or an LVEF value below institutional limits of normal and ≥10% absolute decrease in LVEF from pre-treatment values. When treatment is withheld for significant LVEF cardiac dysfunction, patients should undergo cardiac assessment at 4-week intervals.
To combat infusion reactions, infusion should be interrupted in all patients experiencing dyspnea or clinically significant hypotension and medical therapy administered. Patients should be evaluated and monitored carefully until signs and symptoms resolve and permanent discontinuation considered in patients with severe reactions. Patients should be warned of the potential for fetal harm with trastuzumab-dkst and of the need for effective contraceptive use during and for 6 months after treatment
1. FDA approves first biosimilar for the treatment of certain breast and stomach cancers. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm587378.htm. December 1, 2017. Accessed January 31, 2018.
2. Rugo HS, Barve A, Waller CF, et al. Effect of a proposed trastuzumab biosimilar compared with trastuzumab on overall response rate in patients with ERBB2 (HER2)-positive metastatic breast cancer: a randomized clinical trial. JAMA. 2017;317(1):37-47.
3. Ogviri (trastuzumab-dkst) injection, for intravenous use. Prescribing information. Mylan, GMBH. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761074s000lbl.pdf. December, 2017. Accessed July 31, 2015.
The human epidermal growth factor receptor-2 (HER2)-targeting monoclonal antibody trastuzumab-dkst, was approved by the US Food and Drug Administration in 2017 for the treatment of patients with HER2-positive breast or metastatic gastric or gastroesophageal junction adenocarcinoma.1 Trastuzumab-dkst, marketed as Ogviri by Mylan NV and Biocon Ltd, is a copy, known as a biosimilar, of Genentech’s trastuzumab (Herceptin), which has been approved in the US since 1998. Genentech’s patent on trastuzumab expires in 2018
Approval was based on a comparison of the 2 drugs, which demonstrated that there were no clinically meaningful differences between the biosimilar and the reference product (trastuzumab) in terms of structure and function, pharmacokinetics (PKs), pharmacodynamics, and clinical efficacy and safety.
In structural and functional studies, trastuzumab-dkst was shown to have an identical amino acid sequence and a highly similar 3-dimensional structure, as well as equivalency in an inhibition of proliferation assay, a HER2-binding assay, and an antibody-dependent cellular cytotoxicity assay, compared with trastuzumab.
Two nonclinical animal studies were performed in cynomolgus monkeys; a single-dose comparative PK study and a 4-week, repeat-dose toxicity study. That was further supported by data from a single-dose, randomized, double-blind, comparative 3-way PK study (MYL-HER-1002) in which 120 healthy men were given an 8 mg/kg infusion of trastuzumab-dkst, US-approved trastuzumab, or European Union (EU)-approved trastuzumab.
The key clinical study was the phase 3 HERiTAge trial, a 2-part, multicenter, double-blind, randomized, parallel group trial that was performed in patients with HER2-positive metastatic breast cancer who had not been previously treated with either chemotherapy or trastuzumab in the metastatic setting.2
Eligible patients included males or females with measurably HER2-positive disease (as defined by HER2 overexpression determined by immunohistochemistry performed by a central laboratory), no exposure to chemotherapy or trastuzumab in the metastatic setting, an Eastern Cooperative Oncology Group Performance Status of 0 or 2, left ventricular ejection fraction (LVEF) within institutional range of normal, and who had completed adjuvant trastuzumab therapy at least 1 year before.
Patients with central nervous system metastases had to have stable disease after treatment, and hormonal agents were required to be discontinued before the start of the study. Patients with a history of unstable angina, heart failure, myocardial infarction less than 1 year from randomization, other clinically significant cardiac disease, grade 2 or higher peripheral neuropathy, a history of any other cancer within 4 years before screening, or any significant medical illness that increased treatment risk or impeded evaluation, were excluded from the study.
Patients were randomly assigned 1:1 to receive trastuzumab-dkst or trastuzumab, both in combination with paclitaxel or docetaxel, at a loading dose of 8 mg/kg, followed by a maintenance dose of 6 mg/kg, every 3 weeks for a minimum of 7 cycles in part 1 of the study. Patients who had stable disease or better were enrolled in part 2 and continued treatment until disease progression or unacceptable toxicity.
The primary endpoint was overall response rate (ORR) and, after 24 weeks, the ORR was 69.6% in the trastuzumab-dkst arm, compared with 64% in the trastuzumab arm, with a ratio of ORR of 1.09. Progression-free survival was also nearly identical in the 2 groups and median overall survival had not been reached in either arm.
The safety of the biosimilar and reference product were also highly similar. Serious adverse events occurred in 39.3%, compared with 37% of patients, respectively, with neutropenia the most frequently reported in both arms. Overall, treatment-emergent AEs occurred in 96.8%, compared with 94.7% of patients, respectively, with the majority of events mild or moderate in severity in both groups. This study also confirmed the low immunogenicity of the 2 drug products.
The prescribing information details the recommended doses of trastuzumab-dkst for each approved indication and warnings and precautions for cardiomyopathy, infusion reactions, pulmonary toxicity, exacerbation of chemotherapy-induced neutropenia and embryofetal toxicity.3
Patients should undergo thorough cardiac assessments, including baseline LVEF measurement immediately before starting therapy, every 3 months during therapy, and upon completion of therapy. Patients who complete adjuvant therapy should have cardiac assessments every 6 months for at least 2 years. Treatment should be withheld for ≥16% absolute decrease in LVEF from pre-treatment values or an LVEF value below institutional limits of normal and ≥10% absolute decrease in LVEF from pre-treatment values. When treatment is withheld for significant LVEF cardiac dysfunction, patients should undergo cardiac assessment at 4-week intervals.
To combat infusion reactions, infusion should be interrupted in all patients experiencing dyspnea or clinically significant hypotension and medical therapy administered. Patients should be evaluated and monitored carefully until signs and symptoms resolve and permanent discontinuation considered in patients with severe reactions. Patients should be warned of the potential for fetal harm with trastuzumab-dkst and of the need for effective contraceptive use during and for 6 months after treatment
The human epidermal growth factor receptor-2 (HER2)-targeting monoclonal antibody trastuzumab-dkst, was approved by the US Food and Drug Administration in 2017 for the treatment of patients with HER2-positive breast or metastatic gastric or gastroesophageal junction adenocarcinoma.1 Trastuzumab-dkst, marketed as Ogviri by Mylan NV and Biocon Ltd, is a copy, known as a biosimilar, of Genentech’s trastuzumab (Herceptin), which has been approved in the US since 1998. Genentech’s patent on trastuzumab expires in 2018
Approval was based on a comparison of the 2 drugs, which demonstrated that there were no clinically meaningful differences between the biosimilar and the reference product (trastuzumab) in terms of structure and function, pharmacokinetics (PKs), pharmacodynamics, and clinical efficacy and safety.
In structural and functional studies, trastuzumab-dkst was shown to have an identical amino acid sequence and a highly similar 3-dimensional structure, as well as equivalency in an inhibition of proliferation assay, a HER2-binding assay, and an antibody-dependent cellular cytotoxicity assay, compared with trastuzumab.
Two nonclinical animal studies were performed in cynomolgus monkeys; a single-dose comparative PK study and a 4-week, repeat-dose toxicity study. That was further supported by data from a single-dose, randomized, double-blind, comparative 3-way PK study (MYL-HER-1002) in which 120 healthy men were given an 8 mg/kg infusion of trastuzumab-dkst, US-approved trastuzumab, or European Union (EU)-approved trastuzumab.
The key clinical study was the phase 3 HERiTAge trial, a 2-part, multicenter, double-blind, randomized, parallel group trial that was performed in patients with HER2-positive metastatic breast cancer who had not been previously treated with either chemotherapy or trastuzumab in the metastatic setting.2
Eligible patients included males or females with measurably HER2-positive disease (as defined by HER2 overexpression determined by immunohistochemistry performed by a central laboratory), no exposure to chemotherapy or trastuzumab in the metastatic setting, an Eastern Cooperative Oncology Group Performance Status of 0 or 2, left ventricular ejection fraction (LVEF) within institutional range of normal, and who had completed adjuvant trastuzumab therapy at least 1 year before.
Patients with central nervous system metastases had to have stable disease after treatment, and hormonal agents were required to be discontinued before the start of the study. Patients with a history of unstable angina, heart failure, myocardial infarction less than 1 year from randomization, other clinically significant cardiac disease, grade 2 or higher peripheral neuropathy, a history of any other cancer within 4 years before screening, or any significant medical illness that increased treatment risk or impeded evaluation, were excluded from the study.
Patients were randomly assigned 1:1 to receive trastuzumab-dkst or trastuzumab, both in combination with paclitaxel or docetaxel, at a loading dose of 8 mg/kg, followed by a maintenance dose of 6 mg/kg, every 3 weeks for a minimum of 7 cycles in part 1 of the study. Patients who had stable disease or better were enrolled in part 2 and continued treatment until disease progression or unacceptable toxicity.
The primary endpoint was overall response rate (ORR) and, after 24 weeks, the ORR was 69.6% in the trastuzumab-dkst arm, compared with 64% in the trastuzumab arm, with a ratio of ORR of 1.09. Progression-free survival was also nearly identical in the 2 groups and median overall survival had not been reached in either arm.
The safety of the biosimilar and reference product were also highly similar. Serious adverse events occurred in 39.3%, compared with 37% of patients, respectively, with neutropenia the most frequently reported in both arms. Overall, treatment-emergent AEs occurred in 96.8%, compared with 94.7% of patients, respectively, with the majority of events mild or moderate in severity in both groups. This study also confirmed the low immunogenicity of the 2 drug products.
The prescribing information details the recommended doses of trastuzumab-dkst for each approved indication and warnings and precautions for cardiomyopathy, infusion reactions, pulmonary toxicity, exacerbation of chemotherapy-induced neutropenia and embryofetal toxicity.3
Patients should undergo thorough cardiac assessments, including baseline LVEF measurement immediately before starting therapy, every 3 months during therapy, and upon completion of therapy. Patients who complete adjuvant therapy should have cardiac assessments every 6 months for at least 2 years. Treatment should be withheld for ≥16% absolute decrease in LVEF from pre-treatment values or an LVEF value below institutional limits of normal and ≥10% absolute decrease in LVEF from pre-treatment values. When treatment is withheld for significant LVEF cardiac dysfunction, patients should undergo cardiac assessment at 4-week intervals.
To combat infusion reactions, infusion should be interrupted in all patients experiencing dyspnea or clinically significant hypotension and medical therapy administered. Patients should be evaluated and monitored carefully until signs and symptoms resolve and permanent discontinuation considered in patients with severe reactions. Patients should be warned of the potential for fetal harm with trastuzumab-dkst and of the need for effective contraceptive use during and for 6 months after treatment
1. FDA approves first biosimilar for the treatment of certain breast and stomach cancers. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm587378.htm. December 1, 2017. Accessed January 31, 2018.
2. Rugo HS, Barve A, Waller CF, et al. Effect of a proposed trastuzumab biosimilar compared with trastuzumab on overall response rate in patients with ERBB2 (HER2)-positive metastatic breast cancer: a randomized clinical trial. JAMA. 2017;317(1):37-47.
3. Ogviri (trastuzumab-dkst) injection, for intravenous use. Prescribing information. Mylan, GMBH. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761074s000lbl.pdf. December, 2017. Accessed July 31, 2015.
1. FDA approves first biosimilar for the treatment of certain breast and stomach cancers. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm587378.htm. December 1, 2017. Accessed January 31, 2018.
2. Rugo HS, Barve A, Waller CF, et al. Effect of a proposed trastuzumab biosimilar compared with trastuzumab on overall response rate in patients with ERBB2 (HER2)-positive metastatic breast cancer: a randomized clinical trial. JAMA. 2017;317(1):37-47.
3. Ogviri (trastuzumab-dkst) injection, for intravenous use. Prescribing information. Mylan, GMBH. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761074s000lbl.pdf. December, 2017. Accessed July 31, 2015.
Biosimilars: same ol’ – but with a suffix, and cheaper
Biosimilars have arrived, and chances are that you’re already prescribing them. Last September, the US Food and Drug Administration (FDA) approved the first cancer-specific biosimilar, bevacizumab-awwb, for multiple cancer types (p. e60);1 and in November, it approved trastuzumab-dkst for HER2-positive breast and gastrointestinal cancers (p. e63).1 Briefly, biosimilars are biologic products that show comparable quality, efficacy, and safety to an existing, approved biologic known as the reference product.
Small-molecule drugs such as aspirin are easy to replicate identically, whereas biosimilars are large, complex proteins that are manufactured in nature’s factory, a micro-organism or biologic cell.2 The manufacturing process must be nearly identical to that for the reference product, so that only insignificant/nonclinically significant impurities occur in the final product. The protein-amino acid sequence is key and must therefore be identical. The 2010 Biologics Price Competition and Innovation Act established an abbreviated pathway for the FDA to consider and approve biosimilars, and 5 years later, the bone marrow stimulant filgrastim-sndz became the first biosimilar approved for use in the United States.3 The development of biosimilars is not inexpensive. The law and the FDA approval system require preclinical and phase 1 testing, and a robust phase 3 trial against the reference product to demonstrate that safety and efficacy are statistically not different and that any chemical differences between the biosimilar and reference product are clinically and safety or immunogenically insignificant. When those criteria have been met, and the biosimilar approved, the clinical and cost benefits to patients could be significant. In general, the cost of a biosimilar is about 20% to 30% lower than that of the reference product.
Biosimilarity does not yet allow interchangeability. Small-molecule generics under FDA regulations are interchangeable in the drug store and the hospital without the prescriber or patient being aware. That is not yet the case with biosimilars, but their lower prices could have a notable impact on overall cost of care. In 2013, 7 of the top 8 best-selling drugs in the global market were biologics.4 Three of the top 8 – rituximab, trastuzumab, and bevacizumab – were used to treat cancer, and 1 (pegfilgrastim) was for therapy-related neutropenia. Their total cost was US$27 billion. Biosimilars of those therapies could significantly lower that amount.
Nabhan and colleagues interviewed 510 US-based community oncologists about their understanding of biosimilars. They found that only 29% of respondents said they prescribed filgrastim-sndz for supportive care by personal choice, but upward of 73% said they would prescribe biosimilars for the active anticancer therapies, trastuzumab and bevacizumab. There’s no question that biosimilars are here to stay. The requirements to make them have been well worked out. Their safety and efficacy therefore can be assured, and their lower prices promise cost savings for patients and society as a whole.
1. Bosserman L. Cancer care in 2017: the promise of more cures with the challenges of an unstable health care system. https://www. mdedge.com/jcso/article/154559/cancer-care-2017-promise-morecures- challenges-unstable-health-care-system. December 15, 2017. Accessed April 23, 2018.
2. Biosimilar and interchangeable products. FDA website. https://www. fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsare DevelopedandApproved/ApprovalApplications/Therapeutic BiologicApplications/Biosimilars/ucm580419.htm#biological. Last updated October 23, 2017. Accessed April 25, 2018.
3. de Lartigue J. Filgrastim-sndz debuts as the first biosimilar approved in United States. https://www.mdedge.com/jcso/article/105177/patientsurvivor- care/filgrastim-sndz-debuts-first-biosimilar-approved-united. Published December 2015. Accessed April 23, 2018.
4 . The Dish. Biologics still on top in best selling drugs of 2013. http:// cellculturedish.com/2014/03/top-ten-biologics-2013-us-pharmaceutical- sales-2/. March 13, 2014. Accessed April 26, 2018.
5. Nabhan C, Jeune-Smith Y, Valley A, Feinberg BA. Community Oncologists’ Perception and Acceptance of Biosimilars in Oncology. https://www.journalofclinicalpathways.com/article/communityoncologists- perception-and-acceptance-biosimilars-oncology. Published March 2018. Accessed April 24, 2018.
Biosimilars have arrived, and chances are that you’re already prescribing them. Last September, the US Food and Drug Administration (FDA) approved the first cancer-specific biosimilar, bevacizumab-awwb, for multiple cancer types (p. e60);1 and in November, it approved trastuzumab-dkst for HER2-positive breast and gastrointestinal cancers (p. e63).1 Briefly, biosimilars are biologic products that show comparable quality, efficacy, and safety to an existing, approved biologic known as the reference product.
Small-molecule drugs such as aspirin are easy to replicate identically, whereas biosimilars are large, complex proteins that are manufactured in nature’s factory, a micro-organism or biologic cell.2 The manufacturing process must be nearly identical to that for the reference product, so that only insignificant/nonclinically significant impurities occur in the final product. The protein-amino acid sequence is key and must therefore be identical. The 2010 Biologics Price Competition and Innovation Act established an abbreviated pathway for the FDA to consider and approve biosimilars, and 5 years later, the bone marrow stimulant filgrastim-sndz became the first biosimilar approved for use in the United States.3 The development of biosimilars is not inexpensive. The law and the FDA approval system require preclinical and phase 1 testing, and a robust phase 3 trial against the reference product to demonstrate that safety and efficacy are statistically not different and that any chemical differences between the biosimilar and reference product are clinically and safety or immunogenically insignificant. When those criteria have been met, and the biosimilar approved, the clinical and cost benefits to patients could be significant. In general, the cost of a biosimilar is about 20% to 30% lower than that of the reference product.
Biosimilarity does not yet allow interchangeability. Small-molecule generics under FDA regulations are interchangeable in the drug store and the hospital without the prescriber or patient being aware. That is not yet the case with biosimilars, but their lower prices could have a notable impact on overall cost of care. In 2013, 7 of the top 8 best-selling drugs in the global market were biologics.4 Three of the top 8 – rituximab, trastuzumab, and bevacizumab – were used to treat cancer, and 1 (pegfilgrastim) was for therapy-related neutropenia. Their total cost was US$27 billion. Biosimilars of those therapies could significantly lower that amount.
Nabhan and colleagues interviewed 510 US-based community oncologists about their understanding of biosimilars. They found that only 29% of respondents said they prescribed filgrastim-sndz for supportive care by personal choice, but upward of 73% said they would prescribe biosimilars for the active anticancer therapies, trastuzumab and bevacizumab. There’s no question that biosimilars are here to stay. The requirements to make them have been well worked out. Their safety and efficacy therefore can be assured, and their lower prices promise cost savings for patients and society as a whole.
Biosimilars have arrived, and chances are that you’re already prescribing them. Last September, the US Food and Drug Administration (FDA) approved the first cancer-specific biosimilar, bevacizumab-awwb, for multiple cancer types (p. e60);1 and in November, it approved trastuzumab-dkst for HER2-positive breast and gastrointestinal cancers (p. e63).1 Briefly, biosimilars are biologic products that show comparable quality, efficacy, and safety to an existing, approved biologic known as the reference product.
Small-molecule drugs such as aspirin are easy to replicate identically, whereas biosimilars are large, complex proteins that are manufactured in nature’s factory, a micro-organism or biologic cell.2 The manufacturing process must be nearly identical to that for the reference product, so that only insignificant/nonclinically significant impurities occur in the final product. The protein-amino acid sequence is key and must therefore be identical. The 2010 Biologics Price Competition and Innovation Act established an abbreviated pathway for the FDA to consider and approve biosimilars, and 5 years later, the bone marrow stimulant filgrastim-sndz became the first biosimilar approved for use in the United States.3 The development of biosimilars is not inexpensive. The law and the FDA approval system require preclinical and phase 1 testing, and a robust phase 3 trial against the reference product to demonstrate that safety and efficacy are statistically not different and that any chemical differences between the biosimilar and reference product are clinically and safety or immunogenically insignificant. When those criteria have been met, and the biosimilar approved, the clinical and cost benefits to patients could be significant. In general, the cost of a biosimilar is about 20% to 30% lower than that of the reference product.
Biosimilarity does not yet allow interchangeability. Small-molecule generics under FDA regulations are interchangeable in the drug store and the hospital without the prescriber or patient being aware. That is not yet the case with biosimilars, but their lower prices could have a notable impact on overall cost of care. In 2013, 7 of the top 8 best-selling drugs in the global market were biologics.4 Three of the top 8 – rituximab, trastuzumab, and bevacizumab – were used to treat cancer, and 1 (pegfilgrastim) was for therapy-related neutropenia. Their total cost was US$27 billion. Biosimilars of those therapies could significantly lower that amount.
Nabhan and colleagues interviewed 510 US-based community oncologists about their understanding of biosimilars. They found that only 29% of respondents said they prescribed filgrastim-sndz for supportive care by personal choice, but upward of 73% said they would prescribe biosimilars for the active anticancer therapies, trastuzumab and bevacizumab. There’s no question that biosimilars are here to stay. The requirements to make them have been well worked out. Their safety and efficacy therefore can be assured, and their lower prices promise cost savings for patients and society as a whole.
1. Bosserman L. Cancer care in 2017: the promise of more cures with the challenges of an unstable health care system. https://www. mdedge.com/jcso/article/154559/cancer-care-2017-promise-morecures- challenges-unstable-health-care-system. December 15, 2017. Accessed April 23, 2018.
2. Biosimilar and interchangeable products. FDA website. https://www. fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsare DevelopedandApproved/ApprovalApplications/Therapeutic BiologicApplications/Biosimilars/ucm580419.htm#biological. Last updated October 23, 2017. Accessed April 25, 2018.
3. de Lartigue J. Filgrastim-sndz debuts as the first biosimilar approved in United States. https://www.mdedge.com/jcso/article/105177/patientsurvivor- care/filgrastim-sndz-debuts-first-biosimilar-approved-united. Published December 2015. Accessed April 23, 2018.
4 . The Dish. Biologics still on top in best selling drugs of 2013. http:// cellculturedish.com/2014/03/top-ten-biologics-2013-us-pharmaceutical- sales-2/. March 13, 2014. Accessed April 26, 2018.
5. Nabhan C, Jeune-Smith Y, Valley A, Feinberg BA. Community Oncologists’ Perception and Acceptance of Biosimilars in Oncology. https://www.journalofclinicalpathways.com/article/communityoncologists- perception-and-acceptance-biosimilars-oncology. Published March 2018. Accessed April 24, 2018.
1. Bosserman L. Cancer care in 2017: the promise of more cures with the challenges of an unstable health care system. https://www. mdedge.com/jcso/article/154559/cancer-care-2017-promise-morecures- challenges-unstable-health-care-system. December 15, 2017. Accessed April 23, 2018.
2. Biosimilar and interchangeable products. FDA website. https://www. fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsare DevelopedandApproved/ApprovalApplications/Therapeutic BiologicApplications/Biosimilars/ucm580419.htm#biological. Last updated October 23, 2017. Accessed April 25, 2018.
3. de Lartigue J. Filgrastim-sndz debuts as the first biosimilar approved in United States. https://www.mdedge.com/jcso/article/105177/patientsurvivor- care/filgrastim-sndz-debuts-first-biosimilar-approved-united. Published December 2015. Accessed April 23, 2018.
4 . The Dish. Biologics still on top in best selling drugs of 2013. http:// cellculturedish.com/2014/03/top-ten-biologics-2013-us-pharmaceutical- sales-2/. March 13, 2014. Accessed April 26, 2018.
5. Nabhan C, Jeune-Smith Y, Valley A, Feinberg BA. Community Oncologists’ Perception and Acceptance of Biosimilars in Oncology. https://www.journalofclinicalpathways.com/article/communityoncologists- perception-and-acceptance-biosimilars-oncology. Published March 2018. Accessed April 24, 2018.
Isolated ocular metastases from lung cancer
Non–small cell lung cancer constitutes 80%-85% of lung cancers, and 40% of NSCLC are adenocarcinoma. It is rare to find intraocular metastasis from lung cancer. In this article, we present the case of a patient who presented with complaints of diminished vision redness of the eye and was found to have intra-ocular metastases from lung cancer.
Case presentation and summary
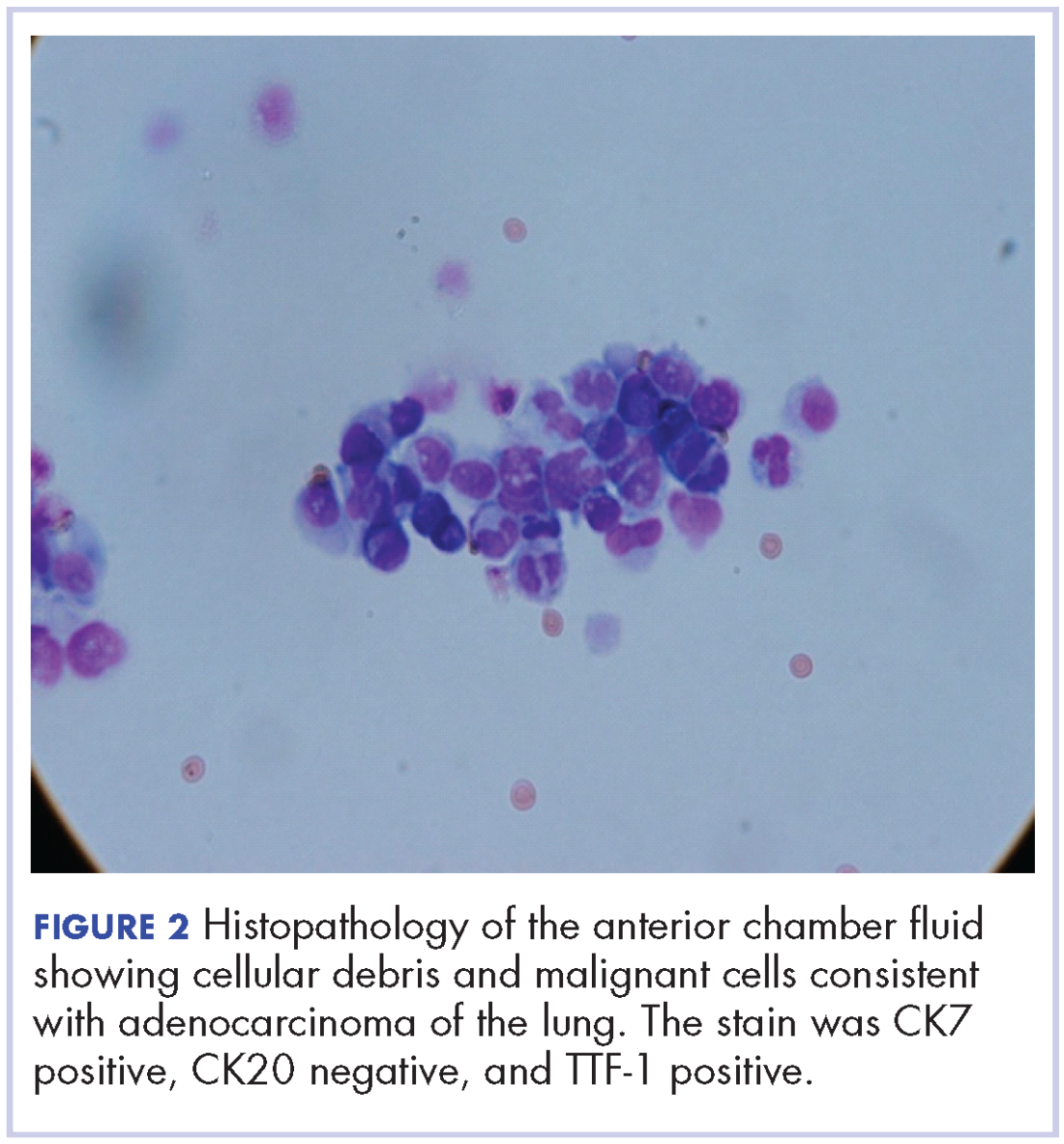
A 60-year-old man with a 40-pack per year history of smoking presented to multiple ophthalmologists with complaints of decreased vision and redness of the left eye. He was eventually evaluated by an ophthalmologist who performed a biopsy of the anterior chamber of the eye. Histologic findings were consistent with adenocarcinoma of lung primary (Figures 1 and 2).
After the diagnosis, a chest X-ray showed that the patient had a left lower lung mass. The results of his physical exam were all within normal limits, with the exception of decreased visual acuity in the left eye. The results of his laboratory studies, including complete blood count and serum chemistries, were also within normal limits. Imaging studies – including a computed-tomography (CT) scan of the chest, abdomen, and pelvis and a full-body positron-emission tomography–CT scan – showed a hypermetabolic left lower lobe mass 4.5 cm and right lower paratracheal lymph node metastasis 2 cm with a small focus of increased uptake alone the medial aspect of the left globe (Figures 3 and 4).
An MRI orbit was performed in an attempt to better characterize the left eye mass, but no optic lesion was identified. A biopsy of the left lower lung mass was consistent with non–small-cell lung cancer (NSCLC). Aside from the isolated left eye metastases, the patient did not have evidence of other distant metastatic involvement.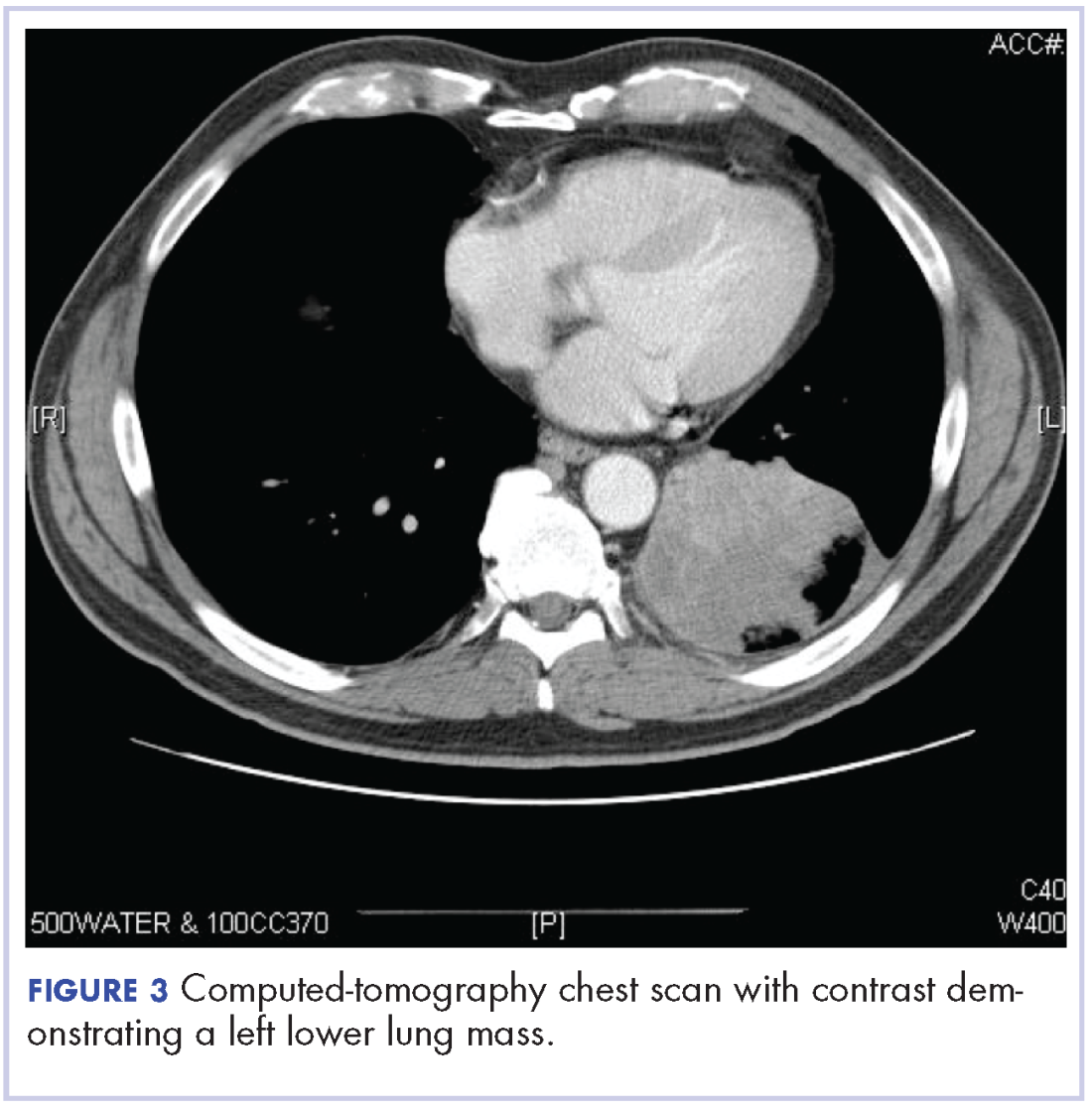

He was started on palliative chemotherapy on a clinical trial and received intravenous carboplatin AUC 6, pemetrexed 500 mg/m2, and bevacizumab 15 mg/kg every 3 weeks. He received 1 dose intraocular bevacizumab injection before initiation of systemic chemotherapy as he was symptomatic from the intraocular metastases. Within 2 weeks after intravitreal bevacizumab was administered, the patient had subjective improvement in vision. Mutational analysis to identify if the patient would benefit from targeted therapy showed no presence of EGFR mutation and ALK gene rearrangement, and that the patient was K-RAS mutant.
After treatment initiation, interval imaging studies (a computed-tomography scan of the chest, abdomen, pelvis; and magnetic-resonance imaging of the brain) after 3 cycles showed no evidence of disease progression, and after 4 cycles of chemotherapy with these drugs, the patient was started on maintenance chemotherapy with bevacizumab 15 mg/kg and pemetrexed 500 mg/m2.
Discussion
Choroidal metastasis is the most common site of intraocular tumor. In an autopsy study of 230 patients with carcinoma, 12% of cases demonstrated histologic evidence of ocular metastasis.1 A retrospective series of patients with malignant involvement of the eye, 66% of patients had a known history of primary cancer and in 34% of patients the ocular tumor was the first sign of cancer.2 The most common cancers that were found to have ocular metastasis were lung and breast cancer.2 Adenocarcinoma was the most common histologic type of lung cancer to result in ocular metastases and was seen in 41% of patients.3
Decreased or blurred vision with redness as the primary complaint of NSCLC is rare. Only a few case reports are available. Abundo and colleagues reported that 0.7%-12% of patients with lung cancer develop ocular metastases.4 Therefore, routine ophthalmologic screening for ocular metastases in patients with cancer has not been pursued in asymptomatic patients.5 Ophthalmological evaluation is recommended in symptomatic patients.
Metastatic involvement of two or more other organs was found to be a risk factor for development of choroidal metastasis in patients with lung cancer though in our patient no evidence of other organ involvement was found.5 The most common site of metastases in patients with NSCLC with ocular metastases was found to be the liver. Choroidal metastases was reported to be the sixth common site of metastases in patients with lung cancer.5
Treatment of ocular manifestations has been generally confined to surgical resection or radiation therapy, but advances in chemotherapy and development of novel targeted agents have shown promising results.7 Median life expectancy after a diagnosis of uveal metastases was reported to be 12 months in a retrospective study, which is similar to the reported median survival in metastatic NSCLC.8
Our patient was enrolled in a clinical trial and was treated with a regimen of carboplatin, paclitaxel, and bevacizumab. On presentation, he had significant impairment of vision with pain. He was treated with intravitreal bevacizumab yielding improvement in his visual symptoms. Bevacizumab is a vascular endothelial growth factor receptor monoclonal antibody approved for use in patients with metastatic lung cancer. Other pathways that have been reported in development of lung cancer involve the ALK gene translocation, and EGFR and K-RAS mutations, and targeted therapy has shown good results in cancer patients with these molecular defects. Randomized clinical trials in patients with advanced NSCLC and an EGFR mutation have shown significant improvement in overall survival with the use of erlotinib, a tyrosine kinase inhibitor targeting the epidermal growth factor receptor.9 Similarly, crizotinib has shown promising results in patients with metastatic NSCLC who have ELM-ALK rearrangement.10 As our patient’s tumor did not have either of these mutations, he was initiated on chemotherapy with bevacizumab. The presence of a K-RAS mutation in this patient further supported the use of front-line chemotherapy given that it may confer resistance against agents that target the EGFR pathway.
In our review of the literature, we found cases of patients with ocular metastases who responded well to therapy with targeted agents (Table).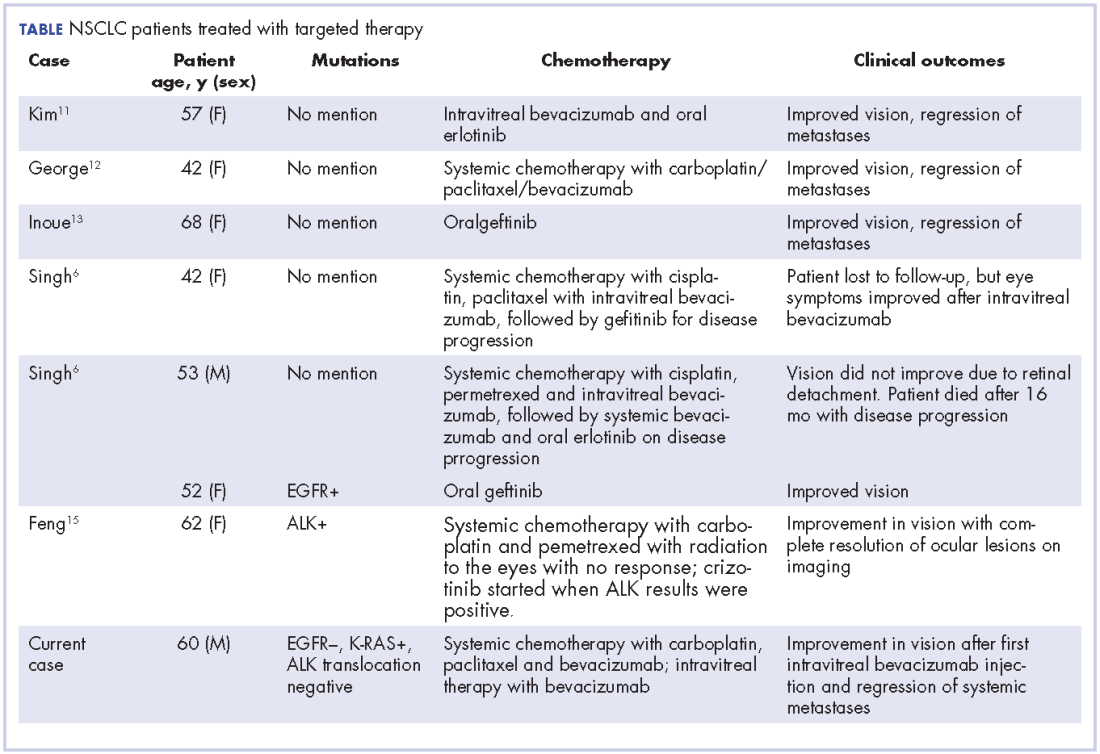
Singh and colleagues did a systematic review of 55 cases of patients with lung cancer and choroidal metastases and found that the type of therapy depended on when the diagnosis had been made in relation to the advent of targeted therapy: cases diagnosed before targeted therapy had received radiation therapy or enucleation.6 As far as we could ascertain, there have been no randomized studies evaluating the impact of various targeted therapies or systemic chemotherapy on ocular metastases, although case reports have documented improvement in vision and regression of metastases with such therapy.
Conclusion
The goal of therapy in metastatic lung cancer is palliation of symptoms and improvement in patient quality of life with prolongation in overall survival. The newer targeted chemotherapeutic agents assist in achieving these goals and may decrease the morbidity associated from radiation or surgery with improvement in vision and regression of ocular metastatic lesions. Targeted therapies should be considered in the treatment of patients with ocular metastases from NSCLC.
1. Bloch RS, Gartner S. The incidence of ocular metastatic carcinoma. Arch Ophthalmol-Chic. 1971;85(6):673-675.
2. Shields CL, Shields JA, Gross NE, Schwartz GP, Lally SE. Survey of 520 eyes with uveal metastases. Ophthalmology. 1997;104(8):1265-1276.
3. Kreusel KM, Bechrakis NE, Wiegel T, Krause L, Foerster MH. Incidence and clinical characteristics of symptomatic choroidal metastasis from lung cancer. Acta Ophthalmol. 2008;86(5):515-519.
4. Abundo RE, Orenic CJ, Anderson SF, Townsend JC. Choroidal metastases resulting from carcinoma of the lung. J Am Optom Assoc. 1997;68(2):95-108.
5. Kreusel KM, Wiegel T, Stange M, Bornfeld N, Hinkelbein W, Foerster MH. Choroidal metastasis in disseminated lung cancer: frequency and risk factors. Am J Ophthalmol. 2002;134(3):445-447.
6. Singh N, Kulkarni P, Aggarwal AN, et al. Choroidal metastasis as a presenting manifestation of lung cancer: a report of 3 cases and systematic review of the literature. Medicine (Baltimore). 2012;91(4):179-194.
7. Chen CJ, McCoy AN, Brahmer J, Handa JT. Emerging treatments for choroidal metastases. Surv Ophthalmol. 2011;56(6):511-521.
8. Shah SU, Mashayekhi A, Shields CL, et al. Uveal metastasis from lung cancer: clinical features, treatment, and outcome in 194 patients. Ophthalmology. 2014;121(1):352-357.
9. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123-132.
10. Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385-2394.
11. Kim SW, Kim MJ, Huh K, Oh J. Complete regression of choroidal metastasis secondary to non-small-cell lung cancer with intravitreal bevacizumab and oral erlotinib combination therapy. Ophthalmologica. 2009;223(6):411-413.
12. George B, Wirostko WJ, Connor TB, Choong NW. Complete and durable response of choroid metastasis from non-small cell lung cancer with systemic bevacizumab and chemotherapy. J Thorac Oncol. 2009;4(5):661-662.
13. Inoue M, Watanabe Y, Yamane S, et al. Choroidal metastasis with adenocarcinoma of the lung treated with gefitinib. Eur J Ophthalmol. 2010;20(5):963-965.
14. Shimomura I, Tada Y, Miura G, et al. Choroidal metastasis of non-small cell lung cancer that responded to gefitinib. https://www.hindawi.com/journals/criopm/2013/213124/. Published 2013. Accessed May 4, 2017.
15. Feng Y, Singh AD, Lanigan C, Tubbs RR, Ma PC. Choroidal metastases responsive to crizotinib therapy in a lung adenocarcinoma patient with ALK 2p23 fusion identified by ALK immunohistochemistry. J Thorac Oncol. 2013;8(12):e109-111.
Non–small cell lung cancer constitutes 80%-85% of lung cancers, and 40% of NSCLC are adenocarcinoma. It is rare to find intraocular metastasis from lung cancer. In this article, we present the case of a patient who presented with complaints of diminished vision redness of the eye and was found to have intra-ocular metastases from lung cancer.
Case presentation and summary
A 60-year-old man with a 40-pack per year history of smoking presented to multiple ophthalmologists with complaints of decreased vision and redness of the left eye. He was eventually evaluated by an ophthalmologist who performed a biopsy of the anterior chamber of the eye. Histologic findings were consistent with adenocarcinoma of lung primary (Figures 1 and 2).
After the diagnosis, a chest X-ray showed that the patient had a left lower lung mass. The results of his physical exam were all within normal limits, with the exception of decreased visual acuity in the left eye. The results of his laboratory studies, including complete blood count and serum chemistries, were also within normal limits. Imaging studies – including a computed-tomography (CT) scan of the chest, abdomen, and pelvis and a full-body positron-emission tomography–CT scan – showed a hypermetabolic left lower lobe mass 4.5 cm and right lower paratracheal lymph node metastasis 2 cm with a small focus of increased uptake alone the medial aspect of the left globe (Figures 3 and 4).
An MRI orbit was performed in an attempt to better characterize the left eye mass, but no optic lesion was identified. A biopsy of the left lower lung mass was consistent with non–small-cell lung cancer (NSCLC). Aside from the isolated left eye metastases, the patient did not have evidence of other distant metastatic involvement.
He was started on palliative chemotherapy on a clinical trial and received intravenous carboplatin AUC 6, pemetrexed 500 mg/m2, and bevacizumab 15 mg/kg every 3 weeks. He received 1 dose intraocular bevacizumab injection before initiation of systemic chemotherapy as he was symptomatic from the intraocular metastases. Within 2 weeks after intravitreal bevacizumab was administered, the patient had subjective improvement in vision. Mutational analysis to identify if the patient would benefit from targeted therapy showed no presence of EGFR mutation and ALK gene rearrangement, and that the patient was K-RAS mutant.
After treatment initiation, interval imaging studies (a computed-tomography scan of the chest, abdomen, pelvis; and magnetic-resonance imaging of the brain) after 3 cycles showed no evidence of disease progression, and after 4 cycles of chemotherapy with these drugs, the patient was started on maintenance chemotherapy with bevacizumab 15 mg/kg and pemetrexed 500 mg/m2.
Discussion
Choroidal metastasis is the most common site of intraocular tumor. In an autopsy study of 230 patients with carcinoma, 12% of cases demonstrated histologic evidence of ocular metastasis.1 A retrospective series of patients with malignant involvement of the eye, 66% of patients had a known history of primary cancer and in 34% of patients the ocular tumor was the first sign of cancer.2 The most common cancers that were found to have ocular metastasis were lung and breast cancer.2 Adenocarcinoma was the most common histologic type of lung cancer to result in ocular metastases and was seen in 41% of patients.3
Decreased or blurred vision with redness as the primary complaint of NSCLC is rare. Only a few case reports are available. Abundo and colleagues reported that 0.7%-12% of patients with lung cancer develop ocular metastases.4 Therefore, routine ophthalmologic screening for ocular metastases in patients with cancer has not been pursued in asymptomatic patients.5 Ophthalmological evaluation is recommended in symptomatic patients.
Metastatic involvement of two or more other organs was found to be a risk factor for development of choroidal metastasis in patients with lung cancer though in our patient no evidence of other organ involvement was found.5 The most common site of metastases in patients with NSCLC with ocular metastases was found to be the liver. Choroidal metastases was reported to be the sixth common site of metastases in patients with lung cancer.5
Treatment of ocular manifestations has been generally confined to surgical resection or radiation therapy, but advances in chemotherapy and development of novel targeted agents have shown promising results.7 Median life expectancy after a diagnosis of uveal metastases was reported to be 12 months in a retrospective study, which is similar to the reported median survival in metastatic NSCLC.8
Our patient was enrolled in a clinical trial and was treated with a regimen of carboplatin, paclitaxel, and bevacizumab. On presentation, he had significant impairment of vision with pain. He was treated with intravitreal bevacizumab yielding improvement in his visual symptoms. Bevacizumab is a vascular endothelial growth factor receptor monoclonal antibody approved for use in patients with metastatic lung cancer. Other pathways that have been reported in development of lung cancer involve the ALK gene translocation, and EGFR and K-RAS mutations, and targeted therapy has shown good results in cancer patients with these molecular defects. Randomized clinical trials in patients with advanced NSCLC and an EGFR mutation have shown significant improvement in overall survival with the use of erlotinib, a tyrosine kinase inhibitor targeting the epidermal growth factor receptor.9 Similarly, crizotinib has shown promising results in patients with metastatic NSCLC who have ELM-ALK rearrangement.10 As our patient’s tumor did not have either of these mutations, he was initiated on chemotherapy with bevacizumab. The presence of a K-RAS mutation in this patient further supported the use of front-line chemotherapy given that it may confer resistance against agents that target the EGFR pathway.
In our review of the literature, we found cases of patients with ocular metastases who responded well to therapy with targeted agents (Table).
Singh and colleagues did a systematic review of 55 cases of patients with lung cancer and choroidal metastases and found that the type of therapy depended on when the diagnosis had been made in relation to the advent of targeted therapy: cases diagnosed before targeted therapy had received radiation therapy or enucleation.6 As far as we could ascertain, there have been no randomized studies evaluating the impact of various targeted therapies or systemic chemotherapy on ocular metastases, although case reports have documented improvement in vision and regression of metastases with such therapy.
Conclusion
The goal of therapy in metastatic lung cancer is palliation of symptoms and improvement in patient quality of life with prolongation in overall survival. The newer targeted chemotherapeutic agents assist in achieving these goals and may decrease the morbidity associated from radiation or surgery with improvement in vision and regression of ocular metastatic lesions. Targeted therapies should be considered in the treatment of patients with ocular metastases from NSCLC.
Non–small cell lung cancer constitutes 80%-85% of lung cancers, and 40% of NSCLC are adenocarcinoma. It is rare to find intraocular metastasis from lung cancer. In this article, we present the case of a patient who presented with complaints of diminished vision redness of the eye and was found to have intra-ocular metastases from lung cancer.
Case presentation and summary
A 60-year-old man with a 40-pack per year history of smoking presented to multiple ophthalmologists with complaints of decreased vision and redness of the left eye. He was eventually evaluated by an ophthalmologist who performed a biopsy of the anterior chamber of the eye. Histologic findings were consistent with adenocarcinoma of lung primary (Figures 1 and 2).
After the diagnosis, a chest X-ray showed that the patient had a left lower lung mass. The results of his physical exam were all within normal limits, with the exception of decreased visual acuity in the left eye. The results of his laboratory studies, including complete blood count and serum chemistries, were also within normal limits. Imaging studies – including a computed-tomography (CT) scan of the chest, abdomen, and pelvis and a full-body positron-emission tomography–CT scan – showed a hypermetabolic left lower lobe mass 4.5 cm and right lower paratracheal lymph node metastasis 2 cm with a small focus of increased uptake alone the medial aspect of the left globe (Figures 3 and 4).
An MRI orbit was performed in an attempt to better characterize the left eye mass, but no optic lesion was identified. A biopsy of the left lower lung mass was consistent with non–small-cell lung cancer (NSCLC). Aside from the isolated left eye metastases, the patient did not have evidence of other distant metastatic involvement.
He was started on palliative chemotherapy on a clinical trial and received intravenous carboplatin AUC 6, pemetrexed 500 mg/m2, and bevacizumab 15 mg/kg every 3 weeks. He received 1 dose intraocular bevacizumab injection before initiation of systemic chemotherapy as he was symptomatic from the intraocular metastases. Within 2 weeks after intravitreal bevacizumab was administered, the patient had subjective improvement in vision. Mutational analysis to identify if the patient would benefit from targeted therapy showed no presence of EGFR mutation and ALK gene rearrangement, and that the patient was K-RAS mutant.
After treatment initiation, interval imaging studies (a computed-tomography scan of the chest, abdomen, pelvis; and magnetic-resonance imaging of the brain) after 3 cycles showed no evidence of disease progression, and after 4 cycles of chemotherapy with these drugs, the patient was started on maintenance chemotherapy with bevacizumab 15 mg/kg and pemetrexed 500 mg/m2.
Discussion
Choroidal metastasis is the most common site of intraocular tumor. In an autopsy study of 230 patients with carcinoma, 12% of cases demonstrated histologic evidence of ocular metastasis.1 A retrospective series of patients with malignant involvement of the eye, 66% of patients had a known history of primary cancer and in 34% of patients the ocular tumor was the first sign of cancer.2 The most common cancers that were found to have ocular metastasis were lung and breast cancer.2 Adenocarcinoma was the most common histologic type of lung cancer to result in ocular metastases and was seen in 41% of patients.3
Decreased or blurred vision with redness as the primary complaint of NSCLC is rare. Only a few case reports are available. Abundo and colleagues reported that 0.7%-12% of patients with lung cancer develop ocular metastases.4 Therefore, routine ophthalmologic screening for ocular metastases in patients with cancer has not been pursued in asymptomatic patients.5 Ophthalmological evaluation is recommended in symptomatic patients.
Metastatic involvement of two or more other organs was found to be a risk factor for development of choroidal metastasis in patients with lung cancer though in our patient no evidence of other organ involvement was found.5 The most common site of metastases in patients with NSCLC with ocular metastases was found to be the liver. Choroidal metastases was reported to be the sixth common site of metastases in patients with lung cancer.5
Treatment of ocular manifestations has been generally confined to surgical resection or radiation therapy, but advances in chemotherapy and development of novel targeted agents have shown promising results.7 Median life expectancy after a diagnosis of uveal metastases was reported to be 12 months in a retrospective study, which is similar to the reported median survival in metastatic NSCLC.8
Our patient was enrolled in a clinical trial and was treated with a regimen of carboplatin, paclitaxel, and bevacizumab. On presentation, he had significant impairment of vision with pain. He was treated with intravitreal bevacizumab yielding improvement in his visual symptoms. Bevacizumab is a vascular endothelial growth factor receptor monoclonal antibody approved for use in patients with metastatic lung cancer. Other pathways that have been reported in development of lung cancer involve the ALK gene translocation, and EGFR and K-RAS mutations, and targeted therapy has shown good results in cancer patients with these molecular defects. Randomized clinical trials in patients with advanced NSCLC and an EGFR mutation have shown significant improvement in overall survival with the use of erlotinib, a tyrosine kinase inhibitor targeting the epidermal growth factor receptor.9 Similarly, crizotinib has shown promising results in patients with metastatic NSCLC who have ELM-ALK rearrangement.10 As our patient’s tumor did not have either of these mutations, he was initiated on chemotherapy with bevacizumab. The presence of a K-RAS mutation in this patient further supported the use of front-line chemotherapy given that it may confer resistance against agents that target the EGFR pathway.
In our review of the literature, we found cases of patients with ocular metastases who responded well to therapy with targeted agents (Table).
Singh and colleagues did a systematic review of 55 cases of patients with lung cancer and choroidal metastases and found that the type of therapy depended on when the diagnosis had been made in relation to the advent of targeted therapy: cases diagnosed before targeted therapy had received radiation therapy or enucleation.6 As far as we could ascertain, there have been no randomized studies evaluating the impact of various targeted therapies or systemic chemotherapy on ocular metastases, although case reports have documented improvement in vision and regression of metastases with such therapy.
Conclusion
The goal of therapy in metastatic lung cancer is palliation of symptoms and improvement in patient quality of life with prolongation in overall survival. The newer targeted chemotherapeutic agents assist in achieving these goals and may decrease the morbidity associated from radiation or surgery with improvement in vision and regression of ocular metastatic lesions. Targeted therapies should be considered in the treatment of patients with ocular metastases from NSCLC.
1. Bloch RS, Gartner S. The incidence of ocular metastatic carcinoma. Arch Ophthalmol-Chic. 1971;85(6):673-675.
2. Shields CL, Shields JA, Gross NE, Schwartz GP, Lally SE. Survey of 520 eyes with uveal metastases. Ophthalmology. 1997;104(8):1265-1276.
3. Kreusel KM, Bechrakis NE, Wiegel T, Krause L, Foerster MH. Incidence and clinical characteristics of symptomatic choroidal metastasis from lung cancer. Acta Ophthalmol. 2008;86(5):515-519.
4. Abundo RE, Orenic CJ, Anderson SF, Townsend JC. Choroidal metastases resulting from carcinoma of the lung. J Am Optom Assoc. 1997;68(2):95-108.
5. Kreusel KM, Wiegel T, Stange M, Bornfeld N, Hinkelbein W, Foerster MH. Choroidal metastasis in disseminated lung cancer: frequency and risk factors. Am J Ophthalmol. 2002;134(3):445-447.
6. Singh N, Kulkarni P, Aggarwal AN, et al. Choroidal metastasis as a presenting manifestation of lung cancer: a report of 3 cases and systematic review of the literature. Medicine (Baltimore). 2012;91(4):179-194.
7. Chen CJ, McCoy AN, Brahmer J, Handa JT. Emerging treatments for choroidal metastases. Surv Ophthalmol. 2011;56(6):511-521.
8. Shah SU, Mashayekhi A, Shields CL, et al. Uveal metastasis from lung cancer: clinical features, treatment, and outcome in 194 patients. Ophthalmology. 2014;121(1):352-357.
9. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123-132.
10. Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385-2394.
11. Kim SW, Kim MJ, Huh K, Oh J. Complete regression of choroidal metastasis secondary to non-small-cell lung cancer with intravitreal bevacizumab and oral erlotinib combination therapy. Ophthalmologica. 2009;223(6):411-413.
12. George B, Wirostko WJ, Connor TB, Choong NW. Complete and durable response of choroid metastasis from non-small cell lung cancer with systemic bevacizumab and chemotherapy. J Thorac Oncol. 2009;4(5):661-662.
13. Inoue M, Watanabe Y, Yamane S, et al. Choroidal metastasis with adenocarcinoma of the lung treated with gefitinib. Eur J Ophthalmol. 2010;20(5):963-965.
14. Shimomura I, Tada Y, Miura G, et al. Choroidal metastasis of non-small cell lung cancer that responded to gefitinib. https://www.hindawi.com/journals/criopm/2013/213124/. Published 2013. Accessed May 4, 2017.
15. Feng Y, Singh AD, Lanigan C, Tubbs RR, Ma PC. Choroidal metastases responsive to crizotinib therapy in a lung adenocarcinoma patient with ALK 2p23 fusion identified by ALK immunohistochemistry. J Thorac Oncol. 2013;8(12):e109-111.
1. Bloch RS, Gartner S. The incidence of ocular metastatic carcinoma. Arch Ophthalmol-Chic. 1971;85(6):673-675.
2. Shields CL, Shields JA, Gross NE, Schwartz GP, Lally SE. Survey of 520 eyes with uveal metastases. Ophthalmology. 1997;104(8):1265-1276.
3. Kreusel KM, Bechrakis NE, Wiegel T, Krause L, Foerster MH. Incidence and clinical characteristics of symptomatic choroidal metastasis from lung cancer. Acta Ophthalmol. 2008;86(5):515-519.
4. Abundo RE, Orenic CJ, Anderson SF, Townsend JC. Choroidal metastases resulting from carcinoma of the lung. J Am Optom Assoc. 1997;68(2):95-108.
5. Kreusel KM, Wiegel T, Stange M, Bornfeld N, Hinkelbein W, Foerster MH. Choroidal metastasis in disseminated lung cancer: frequency and risk factors. Am J Ophthalmol. 2002;134(3):445-447.
6. Singh N, Kulkarni P, Aggarwal AN, et al. Choroidal metastasis as a presenting manifestation of lung cancer: a report of 3 cases and systematic review of the literature. Medicine (Baltimore). 2012;91(4):179-194.
7. Chen CJ, McCoy AN, Brahmer J, Handa JT. Emerging treatments for choroidal metastases. Surv Ophthalmol. 2011;56(6):511-521.
8. Shah SU, Mashayekhi A, Shields CL, et al. Uveal metastasis from lung cancer: clinical features, treatment, and outcome in 194 patients. Ophthalmology. 2014;121(1):352-357.
9. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123-132.
10. Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385-2394.
11. Kim SW, Kim MJ, Huh K, Oh J. Complete regression of choroidal metastasis secondary to non-small-cell lung cancer with intravitreal bevacizumab and oral erlotinib combination therapy. Ophthalmologica. 2009;223(6):411-413.
12. George B, Wirostko WJ, Connor TB, Choong NW. Complete and durable response of choroid metastasis from non-small cell lung cancer with systemic bevacizumab and chemotherapy. J Thorac Oncol. 2009;4(5):661-662.
13. Inoue M, Watanabe Y, Yamane S, et al. Choroidal metastasis with adenocarcinoma of the lung treated with gefitinib. Eur J Ophthalmol. 2010;20(5):963-965.
14. Shimomura I, Tada Y, Miura G, et al. Choroidal metastasis of non-small cell lung cancer that responded to gefitinib. https://www.hindawi.com/journals/criopm/2013/213124/. Published 2013. Accessed May 4, 2017.
15. Feng Y, Singh AD, Lanigan C, Tubbs RR, Ma PC. Choroidal metastases responsive to crizotinib therapy in a lung adenocarcinoma patient with ALK 2p23 fusion identified by ALK immunohistochemistry. J Thorac Oncol. 2013;8(12):e109-111.
Resolution of refractory pruritus with aprepitant in a patient with microcystic adnexal carcinoma
Substance P is an important neurotransmitter implicated in itch pathways.1 After binding to its receptor, neurokinin-1 (NK-1), substance P induces release of factors including histamine, which may cause pruritus.2 Recent literature has reported successful use of aprepitant, an NK-1 antagonist that has been approved by the US Food and Drug Administration for the treatment of chemotherapy-induced nausea and vomiting, for treatment of pruritus. We report here the case of a patient with microcystic adnexal carcinoma (MAC) who presented with refractory pruritus and who had rapid and complete resolution of itch after administration of aprepitant.
Case presentation and summary
A 73-year-old man presented with a 12-year history of a small nodule on his philtrum, which had been increasing in size. He subsequently developed upper-lip numbness and nasal induration. He complained of 2.5 months of severe, debilitating, full-body pruritus. His symptoms were refractory to treatment with prednisone, gabapentin, doxycycline, doxepin, antihistamines, and topical steroids. At the time of consultation, he was being treated with hydroxyzine and topical pramocaine lotion with minimal relief.
At initial dermatologic evaluation, his tumor involved the lower two-thirds of the nose and entire upper cutaneous lip. There was a 4-mm rolled ulcer on the nasal tip and a 1-cm exophytic, smooth nodule on the left upper lip with palpable 4-cm submandibular adenopathy (Figure). Skin examination otherwise revealed linear excoriations on the upper back with no additional primary lesions. The nodule was biopsied, and the patient was diagnosed with MAC with gross nodal involvement. Laboratory findings including serum chemistries, blood urea nitrogen, complete blood cell count, thyroid, and liver function were normal. Positron emission tomography-computed tomography (PET-CT) imaging was negative for distant metastases.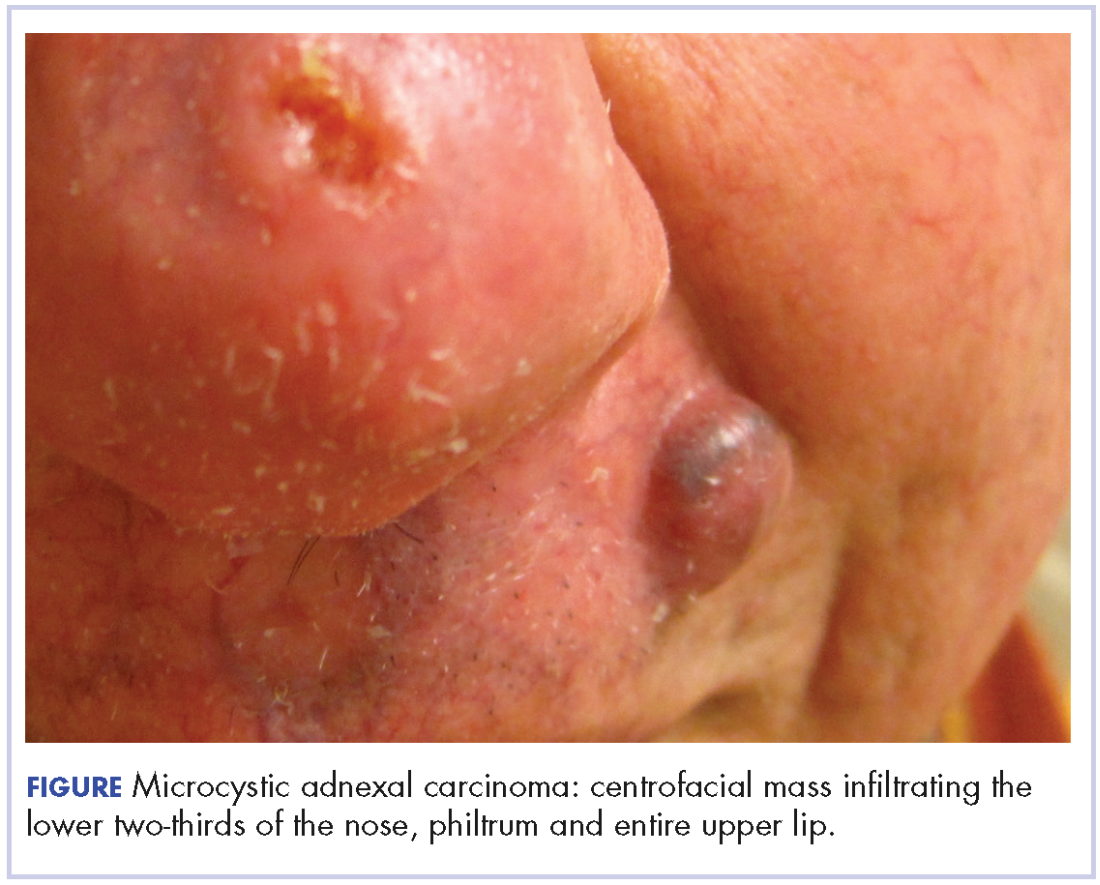
Treatment was initiated with oral aprepitant – 125 mg on day 1, 80 mg on day 2, and 80 mg on day 3 –with concomitant weekly carboplatin (AUC 1.5) and paclitaxel (30 mg/m2) as well as radiation. Within hours after the first dose of aprepitant, the patient reported a notable cessation in his pruritus. He reported that after 5 hours, his skin “finally turned off” and over the hour that followed, he had complete resolution of symptoms. He completed chemoradiation with a significant disease response. Despite persistent MAC confined to the philtrum, he has been followed for over 2 years without recurrence of itch.
Discussion
MAC is an uncommon cutaneous malignancy of sweat and eccrine gland differentiation. In all, 700 cases of MAC have been described in the literature; a 2008 review estimated the incidence of metastasis at around 2.1%.3 Though metastasis is exceedingly rare, the tumor is locally aggressive and there are reports of invasion into the muscle, perichondrium, periosteum, bone marrow, as well as perineural spaces and vascular adventitia.4
The clinical presentation of MAC includes smooth, flesh-colored or yellow papules, nodules, or plaques.3 Patients often present with numbness, paresthesia, and burning in the area of involvement because of neural infiltration with tumor. Despite the rarity of MAC, pruritus has been reported as a presenting symptom in 1 other case in the literature.4 Our case represents the first report of MAC presenting with a grossly enlarging centrofacial mass, lymph node involvement, and severe full-body pruritus. Our patient responded completely, and within hours, to treatment with aprepitant after experiencing months of failure with conventional antipruritus treatments and without recurrence in symptoms in more than 2 years of follow-up.
Aprepitant blocks the binding of substance P to its receptor NK-1 and has been approved as an anti-emetic for chemotherapy patients. Substance P has been shown to be important in both nausea and itch pathways. The largest prospective study to date on aprepitant for the indication of pruritus in 45 patients with metastatic solid tumors demonstrated a 91% response rate, defined by >50% reduction in pruritus intensity, and 13% recurrence rate that occurred at a median of 7 weeks after initial treatment.5 Aprepitant treatment has been used with success for pruritus associated with both malignant and nonmalignant conditions in at least 74 patients,6 among whom the malignant conditions included cutaneous T-cell lymphoma, Hodgkin lymphoma, and metastatic solid tumors.5-7 Aprepitant has also been used for erlotinib- and nivolumab-induced pruritus in non–small cell lung cancer, which suggests a possible future role for aprepitant in the treatment of pruritus secondary to novel cancer therapies, perhaps including immune checkpoint inhibitors.8-10
However, despite those reports, and likely owing to the multifactorial nature of pruritus, aprepitant is not unviversally effective. Mechanisms of malignancy-associated itch are yet to be elucidated, and optimal patient selection for aprepitant use needs to be determined. However, our patient’s notable response supports the increasing evidence that substance P is a key mediator of pruritus and that disruption of binding to its receptor may result in significant improvement in symptoms in certain patients. It remains to be seen whether the cell type or the tendency toward neural invasion plays a role. Large, randomized studies are needed to guide patient selection and confirm the findings reported here and in the literature, with careful documentation of and close attention paid to timing of pruritus relief and improvement in patient quality of life. Aprepitant might be an important therapeutic tool for refractory, malignancy-associated pruritus, in which patient quality of life is especially critical.
Acknowledgments
This work was presented at the Multinational Association of Supportive Care and Cancer Meeting, in Miami Florida, June 26-28, 2014. The authors are indebted to Saajar Jadeja for his assistance preparing the manuscript.
1. Wallengren J. Neuroanatomy and neurophysiology of itch. Dermatol Ther. 2005;18(4):292-303.
2. Kulka M, Sheen CH, Tancowny BP, Grammer LC, Schleimer RP. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology. 2008;123(3):398-410.
3. Wetter R, Goldstein GD. Microcystic adnexal carcinoma: a diagnostic and therapeutic challenge. Dermatol Ther. 2008;21(6):452-458.
4. Adamson T. Microcystic adnexal carcinoma. Dermatol Nurs. 2004;16(4):365.
5. Santini D, Vincenzi B, Guida FM, et al. Aprepitant for management of severe pruritus related to biological cancer treatments: a pilot study. Lancet Oncol. 2012;13(10):1020-1024.
6. Song JS, Tawa M, Chau NG, Kupper TS, LeBoeuf NR. Aprepitant for refractory cutaneous T-cell lymphoma-associated pruritus: 4 cases and a review of the literature. BMC Cancer. 2017;17.
7. Villafranca JJA, Siles MG, Casanova M, Goitia BT, Domínguez AR. Paraneoplastic pruritus presenting with Hodgkin’s lymphoma: a case report. J Med Case Reports. 2014;8:300.
8. Ito J, Fujimoto D, Nakamura A, et al. Aprepitant for refractory nivolumab-induced pruritus. Lung Cancer Amst Neth. 2017;109:58-61.
9. Levêque D. Aprepitant for erlotinib-induced pruritus. N Engl J Med. 2010;363(17):1680-1681; author reply 1681.
10. Gerber PA, Buhren BA, Homey B. More on aprepitant for erlotinib-induced pruritus. N Engl J Med. 2011;364(5):486-487.
Substance P is an important neurotransmitter implicated in itch pathways.1 After binding to its receptor, neurokinin-1 (NK-1), substance P induces release of factors including histamine, which may cause pruritus.2 Recent literature has reported successful use of aprepitant, an NK-1 antagonist that has been approved by the US Food and Drug Administration for the treatment of chemotherapy-induced nausea and vomiting, for treatment of pruritus. We report here the case of a patient with microcystic adnexal carcinoma (MAC) who presented with refractory pruritus and who had rapid and complete resolution of itch after administration of aprepitant.
Case presentation and summary
A 73-year-old man presented with a 12-year history of a small nodule on his philtrum, which had been increasing in size. He subsequently developed upper-lip numbness and nasal induration. He complained of 2.5 months of severe, debilitating, full-body pruritus. His symptoms were refractory to treatment with prednisone, gabapentin, doxycycline, doxepin, antihistamines, and topical steroids. At the time of consultation, he was being treated with hydroxyzine and topical pramocaine lotion with minimal relief.
At initial dermatologic evaluation, his tumor involved the lower two-thirds of the nose and entire upper cutaneous lip. There was a 4-mm rolled ulcer on the nasal tip and a 1-cm exophytic, smooth nodule on the left upper lip with palpable 4-cm submandibular adenopathy (Figure). Skin examination otherwise revealed linear excoriations on the upper back with no additional primary lesions. The nodule was biopsied, and the patient was diagnosed with MAC with gross nodal involvement. Laboratory findings including serum chemistries, blood urea nitrogen, complete blood cell count, thyroid, and liver function were normal. Positron emission tomography-computed tomography (PET-CT) imaging was negative for distant metastases.
Treatment was initiated with oral aprepitant – 125 mg on day 1, 80 mg on day 2, and 80 mg on day 3 –with concomitant weekly carboplatin (AUC 1.5) and paclitaxel (30 mg/m2) as well as radiation. Within hours after the first dose of aprepitant, the patient reported a notable cessation in his pruritus. He reported that after 5 hours, his skin “finally turned off” and over the hour that followed, he had complete resolution of symptoms. He completed chemoradiation with a significant disease response. Despite persistent MAC confined to the philtrum, he has been followed for over 2 years without recurrence of itch.
Discussion
MAC is an uncommon cutaneous malignancy of sweat and eccrine gland differentiation. In all, 700 cases of MAC have been described in the literature; a 2008 review estimated the incidence of metastasis at around 2.1%.3 Though metastasis is exceedingly rare, the tumor is locally aggressive and there are reports of invasion into the muscle, perichondrium, periosteum, bone marrow, as well as perineural spaces and vascular adventitia.4
The clinical presentation of MAC includes smooth, flesh-colored or yellow papules, nodules, or plaques.3 Patients often present with numbness, paresthesia, and burning in the area of involvement because of neural infiltration with tumor. Despite the rarity of MAC, pruritus has been reported as a presenting symptom in 1 other case in the literature.4 Our case represents the first report of MAC presenting with a grossly enlarging centrofacial mass, lymph node involvement, and severe full-body pruritus. Our patient responded completely, and within hours, to treatment with aprepitant after experiencing months of failure with conventional antipruritus treatments and without recurrence in symptoms in more than 2 years of follow-up.
Aprepitant blocks the binding of substance P to its receptor NK-1 and has been approved as an anti-emetic for chemotherapy patients. Substance P has been shown to be important in both nausea and itch pathways. The largest prospective study to date on aprepitant for the indication of pruritus in 45 patients with metastatic solid tumors demonstrated a 91% response rate, defined by >50% reduction in pruritus intensity, and 13% recurrence rate that occurred at a median of 7 weeks after initial treatment.5 Aprepitant treatment has been used with success for pruritus associated with both malignant and nonmalignant conditions in at least 74 patients,6 among whom the malignant conditions included cutaneous T-cell lymphoma, Hodgkin lymphoma, and metastatic solid tumors.5-7 Aprepitant has also been used for erlotinib- and nivolumab-induced pruritus in non–small cell lung cancer, which suggests a possible future role for aprepitant in the treatment of pruritus secondary to novel cancer therapies, perhaps including immune checkpoint inhibitors.8-10
However, despite those reports, and likely owing to the multifactorial nature of pruritus, aprepitant is not unviversally effective. Mechanisms of malignancy-associated itch are yet to be elucidated, and optimal patient selection for aprepitant use needs to be determined. However, our patient’s notable response supports the increasing evidence that substance P is a key mediator of pruritus and that disruption of binding to its receptor may result in significant improvement in symptoms in certain patients. It remains to be seen whether the cell type or the tendency toward neural invasion plays a role. Large, randomized studies are needed to guide patient selection and confirm the findings reported here and in the literature, with careful documentation of and close attention paid to timing of pruritus relief and improvement in patient quality of life. Aprepitant might be an important therapeutic tool for refractory, malignancy-associated pruritus, in which patient quality of life is especially critical.
Acknowledgments
This work was presented at the Multinational Association of Supportive Care and Cancer Meeting, in Miami Florida, June 26-28, 2014. The authors are indebted to Saajar Jadeja for his assistance preparing the manuscript.
Substance P is an important neurotransmitter implicated in itch pathways.1 After binding to its receptor, neurokinin-1 (NK-1), substance P induces release of factors including histamine, which may cause pruritus.2 Recent literature has reported successful use of aprepitant, an NK-1 antagonist that has been approved by the US Food and Drug Administration for the treatment of chemotherapy-induced nausea and vomiting, for treatment of pruritus. We report here the case of a patient with microcystic adnexal carcinoma (MAC) who presented with refractory pruritus and who had rapid and complete resolution of itch after administration of aprepitant.
Case presentation and summary
A 73-year-old man presented with a 12-year history of a small nodule on his philtrum, which had been increasing in size. He subsequently developed upper-lip numbness and nasal induration. He complained of 2.5 months of severe, debilitating, full-body pruritus. His symptoms were refractory to treatment with prednisone, gabapentin, doxycycline, doxepin, antihistamines, and topical steroids. At the time of consultation, he was being treated with hydroxyzine and topical pramocaine lotion with minimal relief.
At initial dermatologic evaluation, his tumor involved the lower two-thirds of the nose and entire upper cutaneous lip. There was a 4-mm rolled ulcer on the nasal tip and a 1-cm exophytic, smooth nodule on the left upper lip with palpable 4-cm submandibular adenopathy (Figure). Skin examination otherwise revealed linear excoriations on the upper back with no additional primary lesions. The nodule was biopsied, and the patient was diagnosed with MAC with gross nodal involvement. Laboratory findings including serum chemistries, blood urea nitrogen, complete blood cell count, thyroid, and liver function were normal. Positron emission tomography-computed tomography (PET-CT) imaging was negative for distant metastases.
Treatment was initiated with oral aprepitant – 125 mg on day 1, 80 mg on day 2, and 80 mg on day 3 –with concomitant weekly carboplatin (AUC 1.5) and paclitaxel (30 mg/m2) as well as radiation. Within hours after the first dose of aprepitant, the patient reported a notable cessation in his pruritus. He reported that after 5 hours, his skin “finally turned off” and over the hour that followed, he had complete resolution of symptoms. He completed chemoradiation with a significant disease response. Despite persistent MAC confined to the philtrum, he has been followed for over 2 years without recurrence of itch.
Discussion
MAC is an uncommon cutaneous malignancy of sweat and eccrine gland differentiation. In all, 700 cases of MAC have been described in the literature; a 2008 review estimated the incidence of metastasis at around 2.1%.3 Though metastasis is exceedingly rare, the tumor is locally aggressive and there are reports of invasion into the muscle, perichondrium, periosteum, bone marrow, as well as perineural spaces and vascular adventitia.4
The clinical presentation of MAC includes smooth, flesh-colored or yellow papules, nodules, or plaques.3 Patients often present with numbness, paresthesia, and burning in the area of involvement because of neural infiltration with tumor. Despite the rarity of MAC, pruritus has been reported as a presenting symptom in 1 other case in the literature.4 Our case represents the first report of MAC presenting with a grossly enlarging centrofacial mass, lymph node involvement, and severe full-body pruritus. Our patient responded completely, and within hours, to treatment with aprepitant after experiencing months of failure with conventional antipruritus treatments and without recurrence in symptoms in more than 2 years of follow-up.
Aprepitant blocks the binding of substance P to its receptor NK-1 and has been approved as an anti-emetic for chemotherapy patients. Substance P has been shown to be important in both nausea and itch pathways. The largest prospective study to date on aprepitant for the indication of pruritus in 45 patients with metastatic solid tumors demonstrated a 91% response rate, defined by >50% reduction in pruritus intensity, and 13% recurrence rate that occurred at a median of 7 weeks after initial treatment.5 Aprepitant treatment has been used with success for pruritus associated with both malignant and nonmalignant conditions in at least 74 patients,6 among whom the malignant conditions included cutaneous T-cell lymphoma, Hodgkin lymphoma, and metastatic solid tumors.5-7 Aprepitant has also been used for erlotinib- and nivolumab-induced pruritus in non–small cell lung cancer, which suggests a possible future role for aprepitant in the treatment of pruritus secondary to novel cancer therapies, perhaps including immune checkpoint inhibitors.8-10
However, despite those reports, and likely owing to the multifactorial nature of pruritus, aprepitant is not unviversally effective. Mechanisms of malignancy-associated itch are yet to be elucidated, and optimal patient selection for aprepitant use needs to be determined. However, our patient’s notable response supports the increasing evidence that substance P is a key mediator of pruritus and that disruption of binding to its receptor may result in significant improvement in symptoms in certain patients. It remains to be seen whether the cell type or the tendency toward neural invasion plays a role. Large, randomized studies are needed to guide patient selection and confirm the findings reported here and in the literature, with careful documentation of and close attention paid to timing of pruritus relief and improvement in patient quality of life. Aprepitant might be an important therapeutic tool for refractory, malignancy-associated pruritus, in which patient quality of life is especially critical.
Acknowledgments
This work was presented at the Multinational Association of Supportive Care and Cancer Meeting, in Miami Florida, June 26-28, 2014. The authors are indebted to Saajar Jadeja for his assistance preparing the manuscript.
1. Wallengren J. Neuroanatomy and neurophysiology of itch. Dermatol Ther. 2005;18(4):292-303.
2. Kulka M, Sheen CH, Tancowny BP, Grammer LC, Schleimer RP. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology. 2008;123(3):398-410.
3. Wetter R, Goldstein GD. Microcystic adnexal carcinoma: a diagnostic and therapeutic challenge. Dermatol Ther. 2008;21(6):452-458.
4. Adamson T. Microcystic adnexal carcinoma. Dermatol Nurs. 2004;16(4):365.
5. Santini D, Vincenzi B, Guida FM, et al. Aprepitant for management of severe pruritus related to biological cancer treatments: a pilot study. Lancet Oncol. 2012;13(10):1020-1024.
6. Song JS, Tawa M, Chau NG, Kupper TS, LeBoeuf NR. Aprepitant for refractory cutaneous T-cell lymphoma-associated pruritus: 4 cases and a review of the literature. BMC Cancer. 2017;17.
7. Villafranca JJA, Siles MG, Casanova M, Goitia BT, Domínguez AR. Paraneoplastic pruritus presenting with Hodgkin’s lymphoma: a case report. J Med Case Reports. 2014;8:300.
8. Ito J, Fujimoto D, Nakamura A, et al. Aprepitant for refractory nivolumab-induced pruritus. Lung Cancer Amst Neth. 2017;109:58-61.
9. Levêque D. Aprepitant for erlotinib-induced pruritus. N Engl J Med. 2010;363(17):1680-1681; author reply 1681.
10. Gerber PA, Buhren BA, Homey B. More on aprepitant for erlotinib-induced pruritus. N Engl J Med. 2011;364(5):486-487.
1. Wallengren J. Neuroanatomy and neurophysiology of itch. Dermatol Ther. 2005;18(4):292-303.
2. Kulka M, Sheen CH, Tancowny BP, Grammer LC, Schleimer RP. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology. 2008;123(3):398-410.
3. Wetter R, Goldstein GD. Microcystic adnexal carcinoma: a diagnostic and therapeutic challenge. Dermatol Ther. 2008;21(6):452-458.
4. Adamson T. Microcystic adnexal carcinoma. Dermatol Nurs. 2004;16(4):365.
5. Santini D, Vincenzi B, Guida FM, et al. Aprepitant for management of severe pruritus related to biological cancer treatments: a pilot study. Lancet Oncol. 2012;13(10):1020-1024.
6. Song JS, Tawa M, Chau NG, Kupper TS, LeBoeuf NR. Aprepitant for refractory cutaneous T-cell lymphoma-associated pruritus: 4 cases and a review of the literature. BMC Cancer. 2017;17.
7. Villafranca JJA, Siles MG, Casanova M, Goitia BT, Domínguez AR. Paraneoplastic pruritus presenting with Hodgkin’s lymphoma: a case report. J Med Case Reports. 2014;8:300.
8. Ito J, Fujimoto D, Nakamura A, et al. Aprepitant for refractory nivolumab-induced pruritus. Lung Cancer Amst Neth. 2017;109:58-61.
9. Levêque D. Aprepitant for erlotinib-induced pruritus. N Engl J Med. 2010;363(17):1680-1681; author reply 1681.
10. Gerber PA, Buhren BA, Homey B. More on aprepitant for erlotinib-induced pruritus. N Engl J Med. 2011;364(5):486-487.
Rare paraneoplastic dermatomyositis secondary to high-grade bladder cancer
The clinical presentation of bladder cancer typically presents with hematuria; changes in voiding habits such as urgency, frequency, and pain; or less commonly, obstructive symptoms. Rarely does bladder cancer first present as part of a paraneoplastic syndrome with an inflammatory myopathy. Inflammatory myopathies such as dermatomyositis have been known to be associated with malignancy, however, in a meta-analysis by Yang and colleagues of 449 patients with dermatomyositis and malignancy there were only 8 cases reported of bladder cancer.1 Herein, we report a paraneoplastic dermatomyositis in the setting of a bladder cancer.
Case presentation and summary
A 65-year-old man with a medical history of hypertension and alcohol use presented to the emergency department with worsening pain, stiffness in the neck, shoulders, and inability to lift his arms above his shoulders. During the physical exam, an erythematous purple rash was noted over his chest, neck, and arms. Upon further evaluation, his creatine phosphokinase was 3,500 U/L (reference range 52-336 U/L) suggesting muscle breakdown and possible inflammatory myopathy. A biopsy of the left deltoid and quadriceps muscles was performed and yielded a diagnosis of dermatomyositis. He was treated with prednisone 60 mg daily for his inflammatory myopathy. The patient also reported an unintentional weight loss of 20 lbs. and increasing weakness and inability to swallow, which caused aspiration events without developing pneumonia.
The patient’s symptoms worsened while he was on steroids, and we became concerned about the possibility of a primary malignancy, which led to further work-up. The results of a computed-tomography (CT) scan of the abdomen and pelvis showed right-sided hydronephrosis and hydrourteter with an irregular, soft-tissue density mass of 4.7 x 3.2 x 4.2 cm along the posterior wall of the bladder (Figure 1).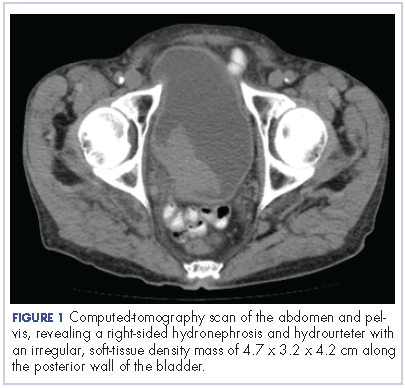
A cystoscopy was performed with transurethral resection of a bladder tumor that was more than 8 cm in diameter. Because the mass was not fully resectable, only 25% of the tumor burden was removed. The pathology report revealed an invasive, high-grade urothelial cell carcinoma (Figure 2, see PDF). Further imaging ruled out metastatic spread. The patient was continued on steroids. He was not a candidate for neoadjuvant chemotherapy because of his comorbidities and cisplatin ineligibility owing to his significant bilateral hearing deficiencies. Members of a multidisciplinary tumor board decided to move forward with definitive surgery. The patient underwent a robotic-assisted laparoscoptic cystoprostatectomy with bilateral pelvic lymph node dissection and open ileal conduit urinary diversion. Staging of tumor was determined as pT3b N1 (1/30) M0, LVI+. After the surgery, the patient had resolution of his rash and significant improvement in his muscle weakness with the ability to raise his arms over his head and climb stairs. Adjuvant chemotherapy was not given since he was cisplatin ineligible as a result of his hearing loss. Active surveillance was preferred.
Four months after his cystoprostatectomy, he experienced new-onset hip pain and further imaging, including a bone scan, was performed. It showed metastatic disease in the ischium and iliac crest (Figure 3).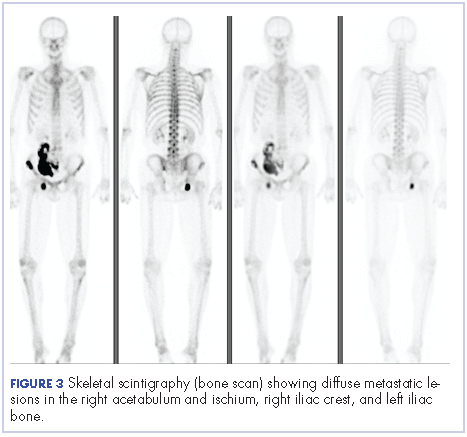
The patient decided to forgo any palliative chemotherapy and to have palliative radiation for pain and enroll in hospice. He died nine months after the initial diagnosis of urothelial cell carcinoma.
Discussion
Dermatomyositis is one of the inflammatory myopathies with a clinical presentation of proximal muscle weakness and characteristic skin findings of Gottron papules and heliotrope eruption. The most common subgroups of inflammatory myopathies are dermatomyositis, polymyositis, necrotizing autoimmune myopathy, and inclusion body myopathy. The pathogenesis of inflammatory myopathies is not well understood; however, some theories have been described, including: type 1 interferon signaling causing myofiber injury and antibody-complement mediated processes causing ischemia resulting in myofiber injury. 2,3 The diagnoses of inflammatory myopathies may be suggested based on history, physical examination findings, laboratory values showing muscle injury (creatine kinase, aldolase, ALT, AST, LDH), myositis-specific antibodies (antisynthetase autoantibodies), electromyogram, and magnetic-resonance imaging. However, muscle biopsy remains the gold standard.4
The initial treatment of inflammatory myopathies begins with glucocorticoid therapy at 0.5-1.0 mg/kg. This regimen may be titrated down over 6 weeks to a level adequate to control symptoms. Even while on glucocorticoid therapy, this patient’s symptoms continued, along with the development of dysphagia. Dysphagia is another notable symptom of dermatomyositis that may result in aspiration pneumonia with fatal outcomes.5,6,7 Not only did this patient initially respond poorly to corticosteroids, but the unintentional weight loss was another alarming feature prompting further evaluation. That led to the diagnosis of urothelial cell carcinoma, which was causing the paraneoplastic syndrome.
A paraneoplastic syndrome is a collection of symptoms that are observed in organ systems separate from the primary disease. This process is mostly caused by an autoimmune response to the tumor and nervous system.8 Inflammatory myopathies, such as dermatomyositis, have been shown to be associated with a variety of malignancies as part of a paraneoplastic syndrome. The most common cancers associated with dermatomyositis are ovarian, lung, pancreatic, stomach, colorectal, and non-Hodgkin lymphoma.9 Although an association between dermatomyositis and bladder cancer has been established, very few cases have been reported in the literature. In the Yang meta-analysis, the relative risk of malignancy for patients with dermatomyositis was 5.5%, and of the 449 patients with dermatomyositis who had malignancy, only 8 cases of bladder cancer were reported.1
After a patient has been diagnosed with an inflammatory myopathy, there should be further evaluation for an underling malignancy causing a paraneoplastic process. The risk of these patients having a malignancy overall is 4.5 times higher than patients without dermatomyositis.1 Definite screening recommendations have not been established, but screening should be based on patient’s age, gender, and clinical scenario. The European Federation of Neurological Societies formed a task force to focus on malignancy screening of paraneoplastic neurological syndromes and included dermatomyositis as one of the signs.10 Patients should have a CT scan of the chest, abdomen, and pelvis. Women should have a mammogram and a pelvis ultrasound. Men younger than 50 years should consider testes ultrasound, and patients older than 50 years should undergo usual colonoscopy screening.
The risk of malignancy is highest in the first year after diagnosis, but may extend to 5 years after the diagnosis, so repeat screening should be performed 3-6 months after diagnosis, followed with biannual testing for 4 years. If a malignancy is present, then treatment should be tailored to the neoplasm to improve symptoms of myositis; however, response is generally worse than it would be with dermatomyositis in the absence of malignancy. In the present case with bladder cancer, therapies may include platinum-based-chemotherapy, resection, and radiation. Dermatomyositis as a result of a bladder cancer paraneoplastic syndrome is associated with a poor prognosis as demonstrated in the case of this patient and others reported in the literature.11
Even though dermatomyositis is usually a chronic disease process, 87% of patients respond initially to corticosteroid treatment.12 Therefore, treatment should be escalated with an agent such as azathioprine or methotrexate, or, like in this case, an underlying malignancy should be suspected. This case emphasizes the importance of screening patients appropriately for malignancy in patients with an inflammatory myopathy and reveals the poor prognosis associated with this disease.
1. Yang Z, Lin F, Qin B, Liang Y, Zhong R. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42(2):282-291.
2. Greenberg, SA. Dermatomyositis and type 1 interferons. Curr Rheumatol Rep. 2010;12(3):198-203.
3. Dalakas, MC, Hohlfeld, R. Polymyositis and dermatomyositis. Lancet. 2003;362(9388):971-982.
4. Malik A, Hayat G, Kalia JS, Guzman MA. Idiopathic inflammatory myopathies: clinical approach and management. Front Neurol. 2016;7:64.
5. Sabio JM, Vargas-Hitos JA, Jiménez-Alonso J. Paraneoplastic dermatomyositis associated with bladder cancer. Lupus. 2006;15(9):619-620.
6. Mallon E, Osborne G, Dinneen M, Lane RJ, Glaser M, Bunker CB. Dermatomyositis in association with transitional cell carcinoma of the bladder. Clin Exp Dermatol. 1999;24(2):94-96.
7. Hafejee A, Coulson IH. Dysphagia in dermatomyositis secondary to bladder cancer: rapid response to combined immunoglobulin and methylprednisolone. Clin Exp Dermatol. 2005;30(1):93-94.
8. Dalmau J, Gultekin HS, Posner JB. Paraneoplastic neurologic syndromes: pathogenesis and physiopathology. Brain Pathol. 1999;9(2):275-284.
9. Hill CL, Zhang Y, Sigureirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357(9250):96-100.
10. Titulaer, MJ, Soffietti R, Dalmau J, et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS Task Force. Eur J Neurol. 2011;18(1):19-e3.
11. Xu R, Zhong Z, Jiang H, Zhang L, Zhao X. A rare paraneoplastic dermatomyositis in bladder cancer with fatal outcome. Urol J. 2013;10(1):815-817.
12. Troyanov Y, Targoff IN, Tremblay JL, Goulet JR, Raymond Y, Senecal JL. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore), 2005;84(4):231-249.
The clinical presentation of bladder cancer typically presents with hematuria; changes in voiding habits such as urgency, frequency, and pain; or less commonly, obstructive symptoms. Rarely does bladder cancer first present as part of a paraneoplastic syndrome with an inflammatory myopathy. Inflammatory myopathies such as dermatomyositis have been known to be associated with malignancy, however, in a meta-analysis by Yang and colleagues of 449 patients with dermatomyositis and malignancy there were only 8 cases reported of bladder cancer.1 Herein, we report a paraneoplastic dermatomyositis in the setting of a bladder cancer.
Case presentation and summary
A 65-year-old man with a medical history of hypertension and alcohol use presented to the emergency department with worsening pain, stiffness in the neck, shoulders, and inability to lift his arms above his shoulders. During the physical exam, an erythematous purple rash was noted over his chest, neck, and arms. Upon further evaluation, his creatine phosphokinase was 3,500 U/L (reference range 52-336 U/L) suggesting muscle breakdown and possible inflammatory myopathy. A biopsy of the left deltoid and quadriceps muscles was performed and yielded a diagnosis of dermatomyositis. He was treated with prednisone 60 mg daily for his inflammatory myopathy. The patient also reported an unintentional weight loss of 20 lbs. and increasing weakness and inability to swallow, which caused aspiration events without developing pneumonia.
The patient’s symptoms worsened while he was on steroids, and we became concerned about the possibility of a primary malignancy, which led to further work-up. The results of a computed-tomography (CT) scan of the abdomen and pelvis showed right-sided hydronephrosis and hydrourteter with an irregular, soft-tissue density mass of 4.7 x 3.2 x 4.2 cm along the posterior wall of the bladder (Figure 1).
A cystoscopy was performed with transurethral resection of a bladder tumor that was more than 8 cm in diameter. Because the mass was not fully resectable, only 25% of the tumor burden was removed. The pathology report revealed an invasive, high-grade urothelial cell carcinoma (Figure 2, see PDF). Further imaging ruled out metastatic spread. The patient was continued on steroids. He was not a candidate for neoadjuvant chemotherapy because of his comorbidities and cisplatin ineligibility owing to his significant bilateral hearing deficiencies. Members of a multidisciplinary tumor board decided to move forward with definitive surgery. The patient underwent a robotic-assisted laparoscoptic cystoprostatectomy with bilateral pelvic lymph node dissection and open ileal conduit urinary diversion. Staging of tumor was determined as pT3b N1 (1/30) M0, LVI+. After the surgery, the patient had resolution of his rash and significant improvement in his muscle weakness with the ability to raise his arms over his head and climb stairs. Adjuvant chemotherapy was not given since he was cisplatin ineligible as a result of his hearing loss. Active surveillance was preferred.
Four months after his cystoprostatectomy, he experienced new-onset hip pain and further imaging, including a bone scan, was performed. It showed metastatic disease in the ischium and iliac crest (Figure 3).
The patient decided to forgo any palliative chemotherapy and to have palliative radiation for pain and enroll in hospice. He died nine months after the initial diagnosis of urothelial cell carcinoma.
Discussion
Dermatomyositis is one of the inflammatory myopathies with a clinical presentation of proximal muscle weakness and characteristic skin findings of Gottron papules and heliotrope eruption. The most common subgroups of inflammatory myopathies are dermatomyositis, polymyositis, necrotizing autoimmune myopathy, and inclusion body myopathy. The pathogenesis of inflammatory myopathies is not well understood; however, some theories have been described, including: type 1 interferon signaling causing myofiber injury and antibody-complement mediated processes causing ischemia resulting in myofiber injury. 2,3 The diagnoses of inflammatory myopathies may be suggested based on history, physical examination findings, laboratory values showing muscle injury (creatine kinase, aldolase, ALT, AST, LDH), myositis-specific antibodies (antisynthetase autoantibodies), electromyogram, and magnetic-resonance imaging. However, muscle biopsy remains the gold standard.4
The initial treatment of inflammatory myopathies begins with glucocorticoid therapy at 0.5-1.0 mg/kg. This regimen may be titrated down over 6 weeks to a level adequate to control symptoms. Even while on glucocorticoid therapy, this patient’s symptoms continued, along with the development of dysphagia. Dysphagia is another notable symptom of dermatomyositis that may result in aspiration pneumonia with fatal outcomes.5,6,7 Not only did this patient initially respond poorly to corticosteroids, but the unintentional weight loss was another alarming feature prompting further evaluation. That led to the diagnosis of urothelial cell carcinoma, which was causing the paraneoplastic syndrome.
A paraneoplastic syndrome is a collection of symptoms that are observed in organ systems separate from the primary disease. This process is mostly caused by an autoimmune response to the tumor and nervous system.8 Inflammatory myopathies, such as dermatomyositis, have been shown to be associated with a variety of malignancies as part of a paraneoplastic syndrome. The most common cancers associated with dermatomyositis are ovarian, lung, pancreatic, stomach, colorectal, and non-Hodgkin lymphoma.9 Although an association between dermatomyositis and bladder cancer has been established, very few cases have been reported in the literature. In the Yang meta-analysis, the relative risk of malignancy for patients with dermatomyositis was 5.5%, and of the 449 patients with dermatomyositis who had malignancy, only 8 cases of bladder cancer were reported.1
After a patient has been diagnosed with an inflammatory myopathy, there should be further evaluation for an underling malignancy causing a paraneoplastic process. The risk of these patients having a malignancy overall is 4.5 times higher than patients without dermatomyositis.1 Definite screening recommendations have not been established, but screening should be based on patient’s age, gender, and clinical scenario. The European Federation of Neurological Societies formed a task force to focus on malignancy screening of paraneoplastic neurological syndromes and included dermatomyositis as one of the signs.10 Patients should have a CT scan of the chest, abdomen, and pelvis. Women should have a mammogram and a pelvis ultrasound. Men younger than 50 years should consider testes ultrasound, and patients older than 50 years should undergo usual colonoscopy screening.
The risk of malignancy is highest in the first year after diagnosis, but may extend to 5 years after the diagnosis, so repeat screening should be performed 3-6 months after diagnosis, followed with biannual testing for 4 years. If a malignancy is present, then treatment should be tailored to the neoplasm to improve symptoms of myositis; however, response is generally worse than it would be with dermatomyositis in the absence of malignancy. In the present case with bladder cancer, therapies may include platinum-based-chemotherapy, resection, and radiation. Dermatomyositis as a result of a bladder cancer paraneoplastic syndrome is associated with a poor prognosis as demonstrated in the case of this patient and others reported in the literature.11
Even though dermatomyositis is usually a chronic disease process, 87% of patients respond initially to corticosteroid treatment.12 Therefore, treatment should be escalated with an agent such as azathioprine or methotrexate, or, like in this case, an underlying malignancy should be suspected. This case emphasizes the importance of screening patients appropriately for malignancy in patients with an inflammatory myopathy and reveals the poor prognosis associated with this disease.
The clinical presentation of bladder cancer typically presents with hematuria; changes in voiding habits such as urgency, frequency, and pain; or less commonly, obstructive symptoms. Rarely does bladder cancer first present as part of a paraneoplastic syndrome with an inflammatory myopathy. Inflammatory myopathies such as dermatomyositis have been known to be associated with malignancy, however, in a meta-analysis by Yang and colleagues of 449 patients with dermatomyositis and malignancy there were only 8 cases reported of bladder cancer.1 Herein, we report a paraneoplastic dermatomyositis in the setting of a bladder cancer.
Case presentation and summary
A 65-year-old man with a medical history of hypertension and alcohol use presented to the emergency department with worsening pain, stiffness in the neck, shoulders, and inability to lift his arms above his shoulders. During the physical exam, an erythematous purple rash was noted over his chest, neck, and arms. Upon further evaluation, his creatine phosphokinase was 3,500 U/L (reference range 52-336 U/L) suggesting muscle breakdown and possible inflammatory myopathy. A biopsy of the left deltoid and quadriceps muscles was performed and yielded a diagnosis of dermatomyositis. He was treated with prednisone 60 mg daily for his inflammatory myopathy. The patient also reported an unintentional weight loss of 20 lbs. and increasing weakness and inability to swallow, which caused aspiration events without developing pneumonia.
The patient’s symptoms worsened while he was on steroids, and we became concerned about the possibility of a primary malignancy, which led to further work-up. The results of a computed-tomography (CT) scan of the abdomen and pelvis showed right-sided hydronephrosis and hydrourteter with an irregular, soft-tissue density mass of 4.7 x 3.2 x 4.2 cm along the posterior wall of the bladder (Figure 1).
A cystoscopy was performed with transurethral resection of a bladder tumor that was more than 8 cm in diameter. Because the mass was not fully resectable, only 25% of the tumor burden was removed. The pathology report revealed an invasive, high-grade urothelial cell carcinoma (Figure 2, see PDF). Further imaging ruled out metastatic spread. The patient was continued on steroids. He was not a candidate for neoadjuvant chemotherapy because of his comorbidities and cisplatin ineligibility owing to his significant bilateral hearing deficiencies. Members of a multidisciplinary tumor board decided to move forward with definitive surgery. The patient underwent a robotic-assisted laparoscoptic cystoprostatectomy with bilateral pelvic lymph node dissection and open ileal conduit urinary diversion. Staging of tumor was determined as pT3b N1 (1/30) M0, LVI+. After the surgery, the patient had resolution of his rash and significant improvement in his muscle weakness with the ability to raise his arms over his head and climb stairs. Adjuvant chemotherapy was not given since he was cisplatin ineligible as a result of his hearing loss. Active surveillance was preferred.
Four months after his cystoprostatectomy, he experienced new-onset hip pain and further imaging, including a bone scan, was performed. It showed metastatic disease in the ischium and iliac crest (Figure 3).
The patient decided to forgo any palliative chemotherapy and to have palliative radiation for pain and enroll in hospice. He died nine months after the initial diagnosis of urothelial cell carcinoma.
Discussion
Dermatomyositis is one of the inflammatory myopathies with a clinical presentation of proximal muscle weakness and characteristic skin findings of Gottron papules and heliotrope eruption. The most common subgroups of inflammatory myopathies are dermatomyositis, polymyositis, necrotizing autoimmune myopathy, and inclusion body myopathy. The pathogenesis of inflammatory myopathies is not well understood; however, some theories have been described, including: type 1 interferon signaling causing myofiber injury and antibody-complement mediated processes causing ischemia resulting in myofiber injury. 2,3 The diagnoses of inflammatory myopathies may be suggested based on history, physical examination findings, laboratory values showing muscle injury (creatine kinase, aldolase, ALT, AST, LDH), myositis-specific antibodies (antisynthetase autoantibodies), electromyogram, and magnetic-resonance imaging. However, muscle biopsy remains the gold standard.4
The initial treatment of inflammatory myopathies begins with glucocorticoid therapy at 0.5-1.0 mg/kg. This regimen may be titrated down over 6 weeks to a level adequate to control symptoms. Even while on glucocorticoid therapy, this patient’s symptoms continued, along with the development of dysphagia. Dysphagia is another notable symptom of dermatomyositis that may result in aspiration pneumonia with fatal outcomes.5,6,7 Not only did this patient initially respond poorly to corticosteroids, but the unintentional weight loss was another alarming feature prompting further evaluation. That led to the diagnosis of urothelial cell carcinoma, which was causing the paraneoplastic syndrome.
A paraneoplastic syndrome is a collection of symptoms that are observed in organ systems separate from the primary disease. This process is mostly caused by an autoimmune response to the tumor and nervous system.8 Inflammatory myopathies, such as dermatomyositis, have been shown to be associated with a variety of malignancies as part of a paraneoplastic syndrome. The most common cancers associated with dermatomyositis are ovarian, lung, pancreatic, stomach, colorectal, and non-Hodgkin lymphoma.9 Although an association between dermatomyositis and bladder cancer has been established, very few cases have been reported in the literature. In the Yang meta-analysis, the relative risk of malignancy for patients with dermatomyositis was 5.5%, and of the 449 patients with dermatomyositis who had malignancy, only 8 cases of bladder cancer were reported.1
After a patient has been diagnosed with an inflammatory myopathy, there should be further evaluation for an underling malignancy causing a paraneoplastic process. The risk of these patients having a malignancy overall is 4.5 times higher than patients without dermatomyositis.1 Definite screening recommendations have not been established, but screening should be based on patient’s age, gender, and clinical scenario. The European Federation of Neurological Societies formed a task force to focus on malignancy screening of paraneoplastic neurological syndromes and included dermatomyositis as one of the signs.10 Patients should have a CT scan of the chest, abdomen, and pelvis. Women should have a mammogram and a pelvis ultrasound. Men younger than 50 years should consider testes ultrasound, and patients older than 50 years should undergo usual colonoscopy screening.
The risk of malignancy is highest in the first year after diagnosis, but may extend to 5 years after the diagnosis, so repeat screening should be performed 3-6 months after diagnosis, followed with biannual testing for 4 years. If a malignancy is present, then treatment should be tailored to the neoplasm to improve symptoms of myositis; however, response is generally worse than it would be with dermatomyositis in the absence of malignancy. In the present case with bladder cancer, therapies may include platinum-based-chemotherapy, resection, and radiation. Dermatomyositis as a result of a bladder cancer paraneoplastic syndrome is associated with a poor prognosis as demonstrated in the case of this patient and others reported in the literature.11
Even though dermatomyositis is usually a chronic disease process, 87% of patients respond initially to corticosteroid treatment.12 Therefore, treatment should be escalated with an agent such as azathioprine or methotrexate, or, like in this case, an underlying malignancy should be suspected. This case emphasizes the importance of screening patients appropriately for malignancy in patients with an inflammatory myopathy and reveals the poor prognosis associated with this disease.
1. Yang Z, Lin F, Qin B, Liang Y, Zhong R. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42(2):282-291.
2. Greenberg, SA. Dermatomyositis and type 1 interferons. Curr Rheumatol Rep. 2010;12(3):198-203.
3. Dalakas, MC, Hohlfeld, R. Polymyositis and dermatomyositis. Lancet. 2003;362(9388):971-982.
4. Malik A, Hayat G, Kalia JS, Guzman MA. Idiopathic inflammatory myopathies: clinical approach and management. Front Neurol. 2016;7:64.
5. Sabio JM, Vargas-Hitos JA, Jiménez-Alonso J. Paraneoplastic dermatomyositis associated with bladder cancer. Lupus. 2006;15(9):619-620.
6. Mallon E, Osborne G, Dinneen M, Lane RJ, Glaser M, Bunker CB. Dermatomyositis in association with transitional cell carcinoma of the bladder. Clin Exp Dermatol. 1999;24(2):94-96.
7. Hafejee A, Coulson IH. Dysphagia in dermatomyositis secondary to bladder cancer: rapid response to combined immunoglobulin and methylprednisolone. Clin Exp Dermatol. 2005;30(1):93-94.
8. Dalmau J, Gultekin HS, Posner JB. Paraneoplastic neurologic syndromes: pathogenesis and physiopathology. Brain Pathol. 1999;9(2):275-284.
9. Hill CL, Zhang Y, Sigureirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357(9250):96-100.
10. Titulaer, MJ, Soffietti R, Dalmau J, et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS Task Force. Eur J Neurol. 2011;18(1):19-e3.
11. Xu R, Zhong Z, Jiang H, Zhang L, Zhao X. A rare paraneoplastic dermatomyositis in bladder cancer with fatal outcome. Urol J. 2013;10(1):815-817.
12. Troyanov Y, Targoff IN, Tremblay JL, Goulet JR, Raymond Y, Senecal JL. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore), 2005;84(4):231-249.
1. Yang Z, Lin F, Qin B, Liang Y, Zhong R. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42(2):282-291.
2. Greenberg, SA. Dermatomyositis and type 1 interferons. Curr Rheumatol Rep. 2010;12(3):198-203.
3. Dalakas, MC, Hohlfeld, R. Polymyositis and dermatomyositis. Lancet. 2003;362(9388):971-982.
4. Malik A, Hayat G, Kalia JS, Guzman MA. Idiopathic inflammatory myopathies: clinical approach and management. Front Neurol. 2016;7:64.
5. Sabio JM, Vargas-Hitos JA, Jiménez-Alonso J. Paraneoplastic dermatomyositis associated with bladder cancer. Lupus. 2006;15(9):619-620.
6. Mallon E, Osborne G, Dinneen M, Lane RJ, Glaser M, Bunker CB. Dermatomyositis in association with transitional cell carcinoma of the bladder. Clin Exp Dermatol. 1999;24(2):94-96.
7. Hafejee A, Coulson IH. Dysphagia in dermatomyositis secondary to bladder cancer: rapid response to combined immunoglobulin and methylprednisolone. Clin Exp Dermatol. 2005;30(1):93-94.
8. Dalmau J, Gultekin HS, Posner JB. Paraneoplastic neurologic syndromes: pathogenesis and physiopathology. Brain Pathol. 1999;9(2):275-284.
9. Hill CL, Zhang Y, Sigureirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357(9250):96-100.
10. Titulaer, MJ, Soffietti R, Dalmau J, et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS Task Force. Eur J Neurol. 2011;18(1):19-e3.
11. Xu R, Zhong Z, Jiang H, Zhang L, Zhao X. A rare paraneoplastic dermatomyositis in bladder cancer with fatal outcome. Urol J. 2013;10(1):815-817.
12. Troyanov Y, Targoff IN, Tremblay JL, Goulet JR, Raymond Y, Senecal JL. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore), 2005;84(4):231-249.
Myelodysplastic syndromes: etiologies, evaluation, and therapy
In this interview, Dr David Henry, MD, the Editor-in-Chief of JCSO, and David Steensma, MD, of the Dana-Farber Cancer Institute in Boston, talk about myelodysplasic syndromes, from diagnosis, evaluation, and etiologies, to therapy options and molecular sequencing.
Listen to the podcast below, or click on the PDF icon at the top of this introduction to read a transcript of the interview.
In this interview, Dr David Henry, MD, the Editor-in-Chief of JCSO, and David Steensma, MD, of the Dana-Farber Cancer Institute in Boston, talk about myelodysplasic syndromes, from diagnosis, evaluation, and etiologies, to therapy options and molecular sequencing.
Listen to the podcast below, or click on the PDF icon at the top of this introduction to read a transcript of the interview.
In this interview, Dr David Henry, MD, the Editor-in-Chief of JCSO, and David Steensma, MD, of the Dana-Farber Cancer Institute in Boston, talk about myelodysplasic syndromes, from diagnosis, evaluation, and etiologies, to therapy options and molecular sequencing.
Listen to the podcast below, or click on the PDF icon at the top of this introduction to read a transcript of the interview.
Gastrointestinal cancers: new standards of care from landmark trials
In this interview, Dr David Henry, the Editor-in-Chief of JCSO, and Dr Daniel Haller, of the Abramson Cancer Center, University of Pennsylvania Perelman School of Medicine in Philadelphia, discuss the findings from recent groundbreaking studies in gastrointestinal cancers.
Listen to the podcast below.
In this interview, Dr David Henry, the Editor-in-Chief of JCSO, and Dr Daniel Haller, of the Abramson Cancer Center, University of Pennsylvania Perelman School of Medicine in Philadelphia, discuss the findings from recent groundbreaking studies in gastrointestinal cancers.
Listen to the podcast below.
In this interview, Dr David Henry, the Editor-in-Chief of JCSO, and Dr Daniel Haller, of the Abramson Cancer Center, University of Pennsylvania Perelman School of Medicine in Philadelphia, discuss the findings from recent groundbreaking studies in gastrointestinal cancers.
Listen to the podcast below.
Analgesic management in radiation oncology for painful bone metastases
Bone metastases are a common cause of pain in patients with advanced cancer, with about three-quarters of patients with bone metastases experiencing pain as the dominant symptom.1 Inadequately treated cancer pain impairs patient quality of life, and is associated with higher rates of depression, anxiety, and fatigue. Palliative radiotherapy (RT) is effective in alleviating pain from bone metastases.4 Local field external beam radiotherapy can provide some pain relief at the site of treated metastasis in 80%-90% of cases, with complete pain relief in 50%-60% of cases.5,6 However, maximal pain relief from RT is delayed, in some cases taking days to up to multiple weeks to attain.7,8 Therefore, optimal management of bone metastases pain may require the use of analgesics until RT takes adequate effect.
National Comprehensive Cancer Network (NCCN) Guidelines for Adult Cancer Pain (v. 2.2015) recommend that pain intensity rating (PIR; range, 0-10, where 0 denotes no pain and 10, worst pain imaginable) be used to quantify pain for all symptomatic patients. These guidelines also recommend the pain medication regimen be assessed for all symptomatic patients. For patients with moderate or severe pain (PIR of ≥4), NCCN guidelines recommend that analgesic regimen be intervened upon by alteration of the analgesic regimen (initiating, rotating, or titrating analgesic) or consideration of referral to pain/symptom management specialty.
Previous findings have demonstrated inadequate analgesic management for cancer pain,2,9 including within the radiation oncology (RO) clinic, suggesting that patients seen in consultation for palliative RT may experience uncontrolled pain for days to weeks before the onset of relief from RT. Possible reasons for inadequate acute pain intervention in the RO clinic may be provider discomfort with analgesic management and infrequent formal integration of palliative care within RO.10
Limited single-institution data from the few institutions with dedicated palliative RO services have suggested that these services improve the quality of palliative care delivery, as demonstrated by providers perceptions’ of the clinical impact of a dedicated service11 and the implementation of expedited palliative RT delivery for acute cancer pain.12,13 To our knowledge, the impact of a dedicated palliative RO service on analgesic management for cancer pain has not been assessed.
Here, we report how often patients with symptomatic bone metastases had assessments of existing analgesic regimens and interventions at RO consultation at 2 cancer centers. Center 1 had implemented a dedicated palliative RO service in 2011, consisting of rotating attending physicians and residents as well as dedicated palliative care trained nurse practitioners and a fellow, with the service structured around daily rounds,11 whereas Center 2 had not yet implemented a dedicated service. Using data from both centers, we assessed the impact of a palliative RO service on analgesic assessment and management in patients with bone metastases.
Methods
We searched our institutional databases for patients seen in RO consultation for bone metastases using ICD-9 code 198.5, and retrospectively reviewed consultation notes for those patients during June-July 2008, January-February 2010, January-February 2013, and June-July 2014. Those time periods were chosen as evenly spaced representative samples before and after implementation of a dedicated palliative RO service in 2011 at Center 1. Center 2 did not implement a dedicated palliative RO service in these time periods.
Within consultation notes, we recorded the following data from the History of the Present Illness section: symptoms from bone metastases (symptomatic was defined as any pain present); PIR (range, 0-10); and whether or not the preconsultation analgesic regimen was reported for symptomatic patients (including analgesic type, dosing, effectiveness, and adherence).
Documentation of the analgesic regimen in the history section of the notes was considered the proxy for analgesic regimen assessment at time of RO consultation. Analgesics within the Medications list, which were autopopulated in the consultation note by the electronic medical record, were recorded.
Whether or not pain was addressed with initiation or titration of analgesics for patients with a PIR of ≥4 was recorded from the Assessment and Plan portion of the notes, and that metric was considered the proxy for pain intervention. In addition, the case was coded as having had pain intervention if there was documentation of the patient declining recommended analgesic intervention, or the patient had been referred to a symptom management service for intervention (eg, referral to a specialty palliative care clinic), or there was recommendation for the patient to discuss uncontrolled pain with the original prescriber. A PIR of 4 was chosen as the threshold for analgesic intervention because at that level, NCCN guidelines for cancer pain state that the analgesic regimen should be titrated, whereas for a PIR of 3 or less, the guidelines recommend only consideration of titrating the analgesic. Only patients with a documented PIR were included in the pain intervention analysis.
Frequencies of analgesic assessment and analgesic intervention were compared using t tests (Wizard Pro, v1.8.5; Evan Miller, Chicago IL).
Results
A total of 271 patients with RO consultation notes were identified at the 2 centers within the 4 time periods (Table 1).
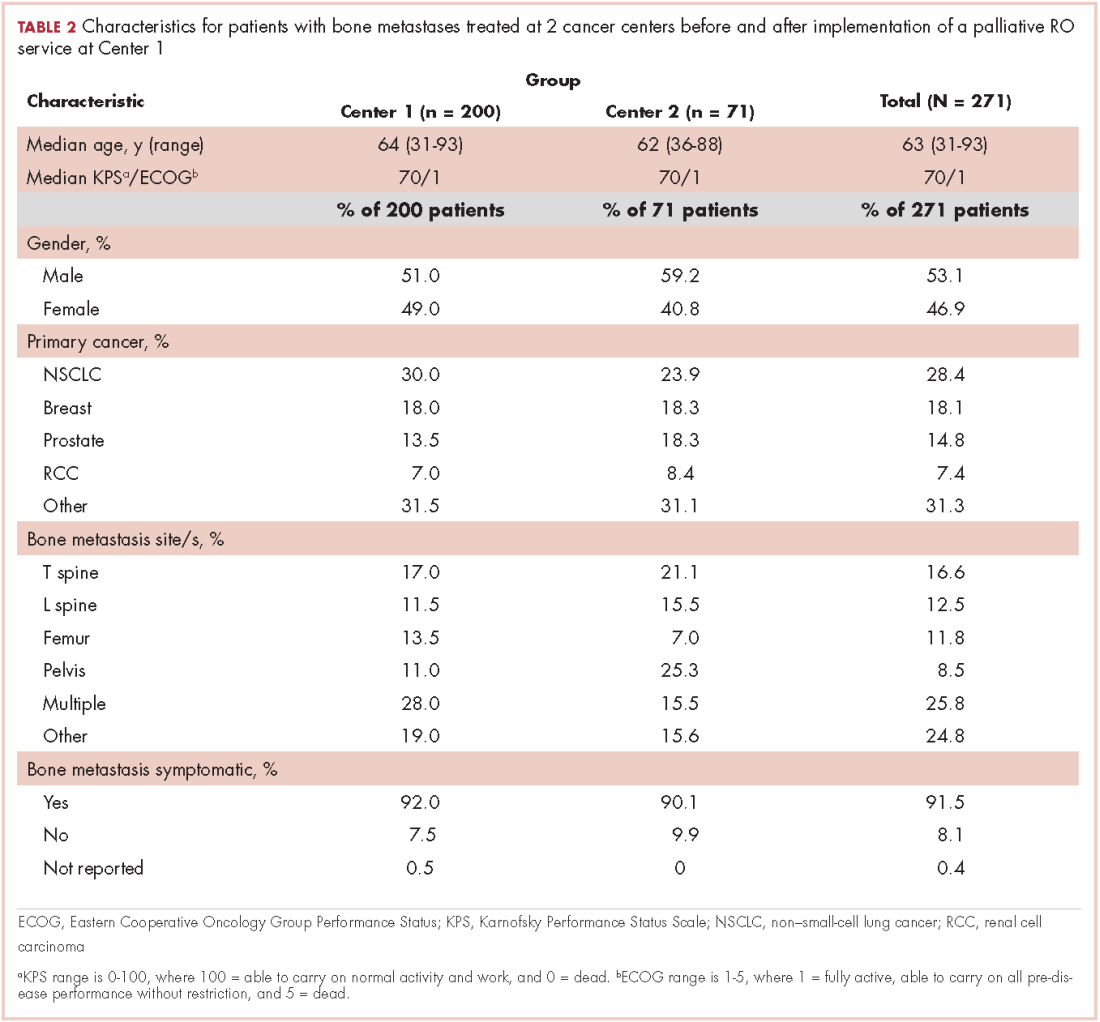
Among symptomatic patients, any component of the preconsultation analgesic regimen (including analgesic type, dosing, pain response, and adherence) was documented for 37.9% of the entire cohort at RO consultation (Table 3). At Centers 1 and 2, the frequencies of analgesic regimen assessment were documented for 41.3% and 28.1%, respectively (P = .06). Among symptomatic patients, 81.5% had an opioid or nonopioid analgesic listed in the Medications section in the electronic medical record at time of consultation.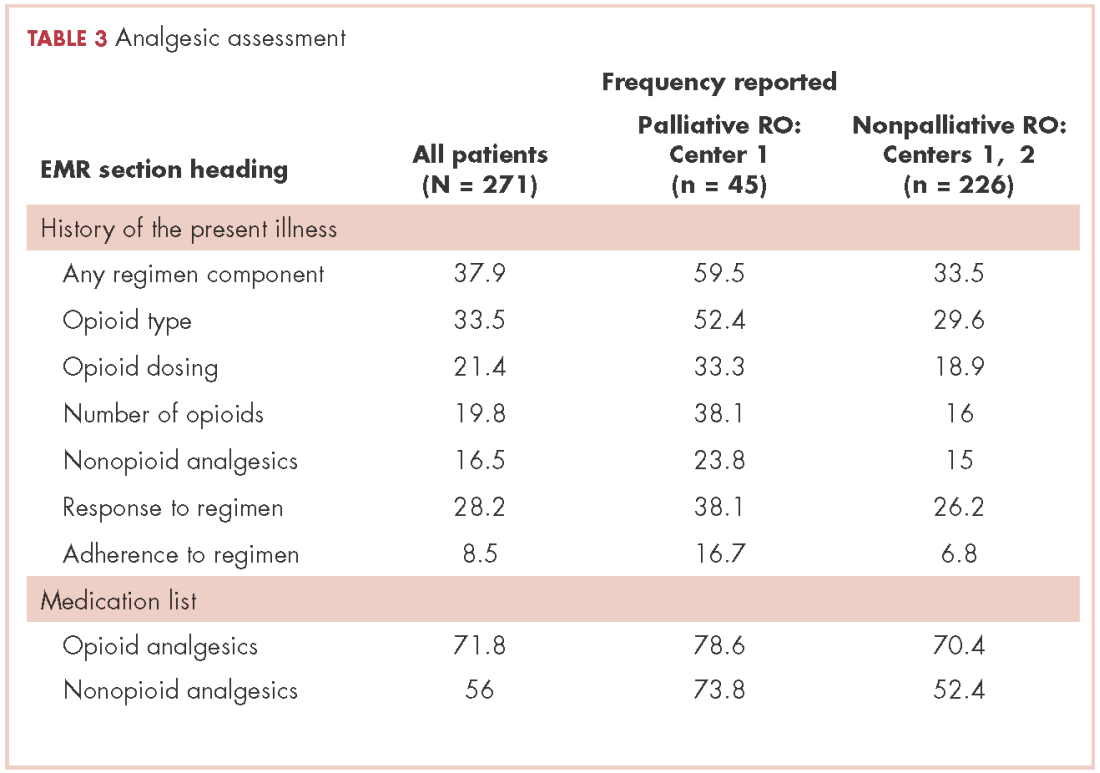
Patients seen on the dedicated palliative RO service at Center 1 had an analgesic assessment documentation rate of 59.5%, whereas the patients not seen on a palliative RO service (ie, patients seen on a nonpalliative RO service at Center 1 plus all patients at Center 2) had an assessment documentation rate of 33.5% (P = .002; Figure 1). There was no significant difference between rates of analgesic regimen assessment between patients seen at Center 2 and patients seen within nondedicated palliative RO services at Center 1 (28.1% vs 35.9%, respectively; P = .27).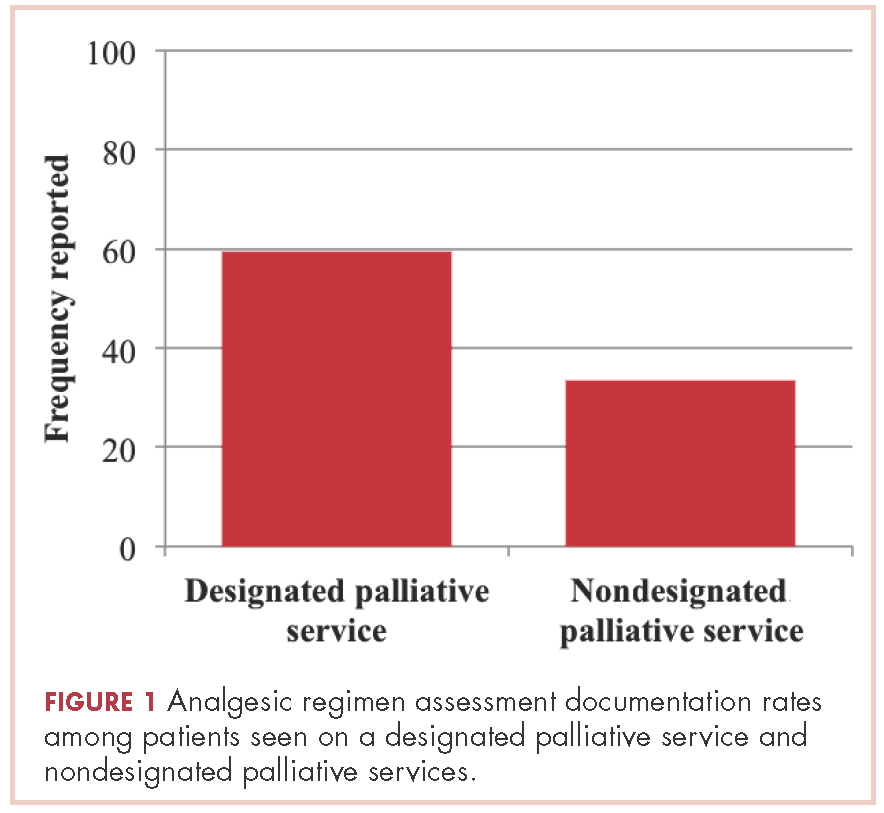
In patients seen at Center 1 only, those seen on the palliative RO service had a higher documentation rate of analgesic assessment compared with those seen by other services after implementation of the dedicated service (59.5% vs 38%, respectively; P = .018). Time period (after versus before 2011) was not significantly associated with the rate of documentation of analgesic assessment at either Center 1 (after vs before 2011: 44.4% vs 31%, P = .23) or Center 2 (31.4% vs 24.1%, P = .60).
Among patients with a PIR of ≥4, analgesic intervention was reported for 17.2% of patients within the entire cohort (20.8% at Center 1 and 0% at Center 2, P = .05). Among those with a PIR of ≥4, documentation of analgesic assessment noted in the History of the Present Illness section was associated with increased documentation of an analgesic intervention in the Assessment and Plan section (25% vs 7.3%; odds ratio [OR], 4.22; 95% confidence interval [CI], 1.1-16.0; P = .03).
Patients seen on the dedicated palliative RO service at Center 1 had a documented analgesic intervention rate of 31.6%, whereas the patients not seen on a palliative RO service (ie, those seen on a nonpalliative RO service at Center 1 plus all patients at Center 2) had a documented analgesic intervention rate of 9.2% (P = .01; Figure 2). There was no statistically significant difference between rates of documentation of an analgesic regimen intervention between patients seen at Center 2 and patients seen within nondedicated palliative RO services at Center 1 (0% vs 17.2%, respectively; P = .07).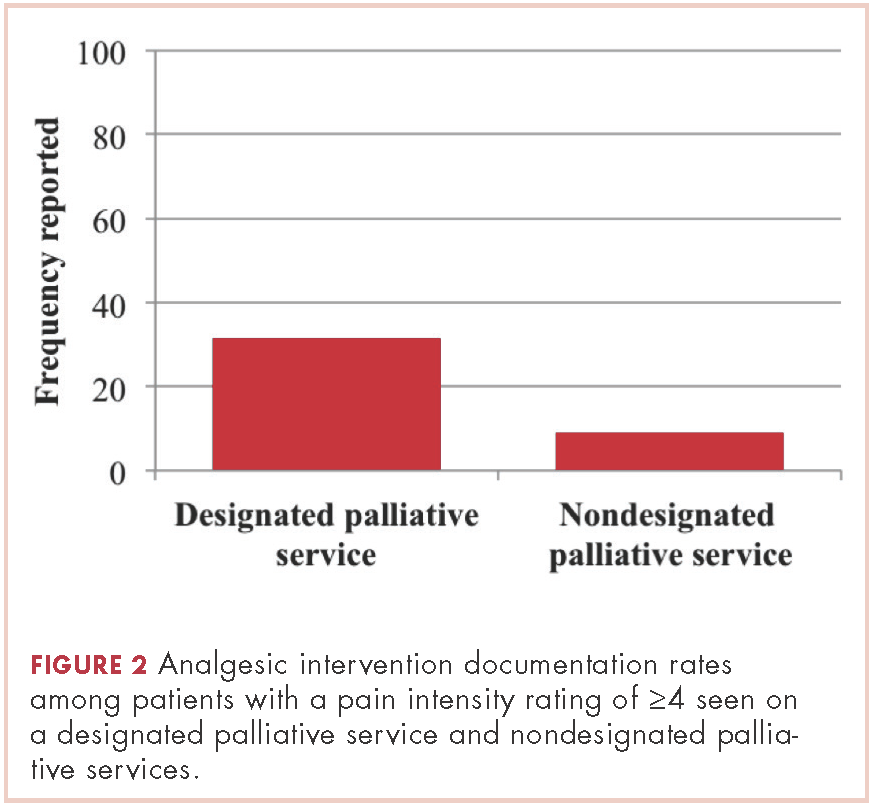
Looking at only patients seen at Center 1, patients with a PIR of ≥4 seen on the dedicated palliative RO service had a nearly significant higher rate of documented analgesic interventions in the time period after implementation of the dedicate service (31.6% if seen on the dedicated service vs 12% if seen on a nondedicated service, P = .06).
Discussion
Multiple studies demonstrate the undertreatment of cancer pain in the outpatient setting.2,9,14,15 At 2 cancer centers, we found that about half of patients who present for consideration of palliative RT for bone metastases had a PIR of ≥4, yet only 17% of them had documentation of analgesic intervention as recommended by NCCN guidelines for cancer pain. Underlying this low rate of appropriate intervention may be the assumption of rapid pain relief by RT. However, RT often does not begin at time of consultation,16 and maximal pain relief may take days to weeks after commencement of RT.17 It is estimated that a quarter of all patients with cancer develop bone metastases during the course of their disease,12 and most of those patients suffer from pain. Thus, inherent delay in pain relief before, during, and after RT results in significant morbidity for the cancer patient population if adequate analgesic management is not provided.
The low rate of appropriate analgesic intervention at the time of RO consultation may also be related to the low incidence of proper analgesic assessment. In our cohort, 80% of symptomatic patients had an opioid or nonopioid analgesic listed in their medications within the electronic medical record at time of consultation, but only 38% had the analgesic regimen and/or its effectiveness described in the History of the Present Illness section of the record. Inattentiveness to analgesic type, dosing, and effectiveness during consultation may result in any inadequacies of the analgesic regimen going unnoticed. Consistent with this notion, we found that the rate of appropriate intervention for patients with a PIR of ≥4 was higher among patients who had analgesic regimen reported in the consultation note. Thus, interventions to implement routine review and documentation of the analgesic regimen, for example within the electronic medical record, may be one way to improve pain management.
Another possible reason for low rates of acute pain management within the RO clinic is low provider confidence in regard to analgesic management. In a recent national survey, 96% of radiation oncologists stated they were at least moderately confident with assessment of pain, yet only 77% were at least moderately confident with titrating opioids, and just 56% were at least moderately confident with rotating opioids.10 Educational interventions that improve providers’ facility with analgesic management may increase the frequency of pain management in the RO clinic.
Patients seen on the dedicated palliative RO service had significantly higher rates of documented analgesic regimen assessment and appropriate intervention during RO consultation, compared with patients seen at Center 2 and those not seen on the dedicated palliative RO service at Center 1. The improvements we observed in analgesic assessment and intervention at Center 1 for patients seen on the palliative RO service are likely owing to involvement of palliative RO and not to secular trends, because there were not similar improvements for patients at Center 1 who were not seen by the palliative RO service and those at Center 2, where there was no service.
At Center 1, the dedicated palliative RO service was created to provide specialized care to patients with metastatic disease undergoing palliative radiation. Within its structure, topics within palliative RO, such as technical aspects of palliative RT, symptom management, and communication are taught and reinforced in a case-based approach. Such palliative care awareness, integration, and education within RO achieved by the palliative RO service likely contribute to the improved rates of analgesic management we found in our study. We do note that rate of analgesic intervention in the palliative RO cohort, though higher than in the nonpalliative RO group, was still low, with only a third of patients receiving proper analgesic management. These findings highlight the importance of continued effort in increasing providers’ awareness of the need to assess pain and raise comfort with analgesic initiation and titration and of having dedicated palliative care clinicians embedded within the RO setting.
Since the data for this study was acquired, Center 2 has implemented a short palliative RO didactic course for residents, which improved their comfort levels in assessing analgesic effectiveness and intervening for uncontrolled pain.18 The impact of this intervention on clinical care will need to be evaluated, but the improved provider comfort levels may translate into better-quality care.
Limitations
An important limitation of this retrospective study is the reliance on the documentation provided in the consultation note for determining frequencies of analgesic regimen assessment and intervention. The actual rates of analgesic management that occurred in clinic may have been higher than reported in the documentation. However, such discrepancy in documentation of analgesic management would also be an area for quality improvement. Inadequate documentation limits the ability for proper follow-up of cancer pain as recommended by a joint guidance statement from the American Society of Clinical Oncology and the American Academy of Hospice and Palliative Medicine.19,20 The results of our study may also partly reflect a positive impact in documentation of analgesic management by a dedicated palliative RO service.
Given the multi-institutional nature of this study, it may be that general practice differences confound the impact of the dedicated palliative RO service at Center 1. However, with excluding Center 2, the dedicated service was still strongly associated with a higher rate of analgesic assessment within Center 1 and was almost significantly associated with appropriate analgesic intervention within Center 1.
We used a PIR of ≥4 as a threshold for appropriate analgesic regimen intervention because it is what is recommended by the NCCN guidelines. However, close attention should be paid to the impact that any amount of pain has on an individual patient. The functional, spiritual, and existential impact of pain is unique to each patient’s experience, and optimal symptom management should take those elements into account.
Conclusion
In conclusion, this study indicates that advanced cancer patient pain assessment and intervention according to NCCN cancer pain management guidelines is not common in the RO setting, and it is an area that should be targeted for quality improvement because of the positive implications for patient well-being. Pain assessment and intervention were greater in the setting of a dedicated structure for palliative care within RO, suggesting that the integration of palliative care within RO is a promising means of improving quality of pain management.
This work was presented at the 2016 ASCO Palliative Care in Oncology Symposium (September 9-10, 2016), where this work received a Conquer Cancer Foundation Merit Award.
1. Amichetti M, Orrù P, Madeddu A, et al. Comparative evaluation of two hypofractionated radiotherapy regimens for painful bone metastases. Tumori. 2004;90(1):91-95.
2. Vuong S, Pulenzas N, DeAngelis C, et al. Inadequate pain management in cancer patients attending an outpatient palliative radiotherapy clinic. Support Care Cancer. 2016;24(2):887-892.
3. Portenoy RK, Payne D, Jacobsen P. Breakthrough pain: characteristics and impact in patients with cancer pain. Pain. 1999;81(1-2):129-134.
4. Sze WM, Shelley M, Held I, Mason M. Palliation of metastatic bone pain: single fraction versus multifraction radiotherapy - a systematic review of the randomised trials. Sze WM, ed. Cochrane Database Syst Rev. 2004;(2):CD004721-CD004721.
5. Ratanatharathorn V, Powers WE, Moss WT, Perez CA. Bone metastasis: review and critical analysis of random allocation trials of local field treatment. Int J Radiat Oncol Biol Phys. 1999;44(1):1-18.
6. Kirou-Mauro A, Hird A, Wong J, et al. Is response to radiotherapy in patients related to the severity of pretreatment pain? Int J Radiat Oncol Biol Phys. 2008;71(4):1208-1212.
7. Frassica DA. General principles of external beam radiation therapy for skeletal metastases. Clin Orthop Relat Res. 2003;(415 Suppl):S158-S164.
8. McDonald R, Ding K, Brundage M, et al. Effect of radiotherapy on painful bone metastases: a secondary analysis of the NCIC Clinical Trials Group Symptom Control Trial SC.23. JAMA Oncol. 2017 Jul 1;3(7):953-959.
9. Greco MT, Roberto A, Corli O, et al. Quality of cancer pain management: an update of a systematic review of undertreatment of patients with cancer. J Clin Oncol. 2014;32(36):4149-4154.
10. Wei RL, Mattes MD, Yu J, et al. Attitudes of radiation oncologists toward palliative and supportive care in the united states: report on national membership survey by the American Society for Radiation Oncology (ASTRO). Pract Radiat Oncol. 2017;7(2):113-119.
11. Tseng YD, Krishnan MS, Jones JA, et al. Supportive and palliative radiation oncology service: impact of a dedicated service on palliative cancer care. Pract Radiat Oncol. 2014;4(4):247-253.
12. Fairchild A, Pituskin E, Rose B, et al. The rapid access palliative radiotherapy program: blueprint for initiation of a one-stop multidisciplinary bone metastases clinic. Support Care Cancer. 2009;17(2):163-170.
13. de Sa E, Sinclair E, Mitera G, et al. Continued success of the rapid response radiotherapy program: a review of 2004-2008. Support Care Cancer. 2009;17(7):757-762.
14. Deandrea S, Montanari M, Moja L, Apolone G. Prevalence of undertreatment in cancer pain. A review of published literature. Ann Oncol. 2008;19(12):1985-1991.
15. Mitera G, Zeiadin N, Kirou-Mauro A, et al. Retrospective assessment of cancer pain management in an outpatient palliative radiotherapy clinic using the Pain Management Index. J Pain Symptom Manage. 2010;39(2):259-267.
16. Danjoux C, Chow E, Drossos A, et al. An innovative rapid response radiotherapy program to reduce waiting time for palliative radiotherapy. Support Care Cancer. 2006;14(1):38-43.
17. Feyer PC, Steingraeber M. Radiotherapy of bone metastasis in breast cancer patients – current approaches. Breast Care (Basel). 2012;7(2):108-112.
18. Garcia MA, Braunstein SE, Anderson WG. Palliative Care Didactic Course for Radiation Oncology Residents. Int J Radiat Oncol Biol Phys. 2017;97(5):884-885.
19. Ferrell BR, Temel JS, Temin S, et al. Integration of palliative care into standard oncology care: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol. 2017;35(1):96-112.
20. Bickel KE, McNiff K, Buss MK, et al. Defining high-quality palliative care in oncology practice: an American Society of Clinical Oncology/American Academy of Hospice and Palliative Medicine guidance statement. J Oncol Pract. 2016;12(9):e828-e838.
Bone metastases are a common cause of pain in patients with advanced cancer, with about three-quarters of patients with bone metastases experiencing pain as the dominant symptom.1 Inadequately treated cancer pain impairs patient quality of life, and is associated with higher rates of depression, anxiety, and fatigue. Palliative radiotherapy (RT) is effective in alleviating pain from bone metastases.4 Local field external beam radiotherapy can provide some pain relief at the site of treated metastasis in 80%-90% of cases, with complete pain relief in 50%-60% of cases.5,6 However, maximal pain relief from RT is delayed, in some cases taking days to up to multiple weeks to attain.7,8 Therefore, optimal management of bone metastases pain may require the use of analgesics until RT takes adequate effect.
National Comprehensive Cancer Network (NCCN) Guidelines for Adult Cancer Pain (v. 2.2015) recommend that pain intensity rating (PIR; range, 0-10, where 0 denotes no pain and 10, worst pain imaginable) be used to quantify pain for all symptomatic patients. These guidelines also recommend the pain medication regimen be assessed for all symptomatic patients. For patients with moderate or severe pain (PIR of ≥4), NCCN guidelines recommend that analgesic regimen be intervened upon by alteration of the analgesic regimen (initiating, rotating, or titrating analgesic) or consideration of referral to pain/symptom management specialty.
Previous findings have demonstrated inadequate analgesic management for cancer pain,2,9 including within the radiation oncology (RO) clinic, suggesting that patients seen in consultation for palliative RT may experience uncontrolled pain for days to weeks before the onset of relief from RT. Possible reasons for inadequate acute pain intervention in the RO clinic may be provider discomfort with analgesic management and infrequent formal integration of palliative care within RO.10
Limited single-institution data from the few institutions with dedicated palliative RO services have suggested that these services improve the quality of palliative care delivery, as demonstrated by providers perceptions’ of the clinical impact of a dedicated service11 and the implementation of expedited palliative RT delivery for acute cancer pain.12,13 To our knowledge, the impact of a dedicated palliative RO service on analgesic management for cancer pain has not been assessed.
Here, we report how often patients with symptomatic bone metastases had assessments of existing analgesic regimens and interventions at RO consultation at 2 cancer centers. Center 1 had implemented a dedicated palliative RO service in 2011, consisting of rotating attending physicians and residents as well as dedicated palliative care trained nurse practitioners and a fellow, with the service structured around daily rounds,11 whereas Center 2 had not yet implemented a dedicated service. Using data from both centers, we assessed the impact of a palliative RO service on analgesic assessment and management in patients with bone metastases.
Methods
We searched our institutional databases for patients seen in RO consultation for bone metastases using ICD-9 code 198.5, and retrospectively reviewed consultation notes for those patients during June-July 2008, January-February 2010, January-February 2013, and June-July 2014. Those time periods were chosen as evenly spaced representative samples before and after implementation of a dedicated palliative RO service in 2011 at Center 1. Center 2 did not implement a dedicated palliative RO service in these time periods.
Within consultation notes, we recorded the following data from the History of the Present Illness section: symptoms from bone metastases (symptomatic was defined as any pain present); PIR (range, 0-10); and whether or not the preconsultation analgesic regimen was reported for symptomatic patients (including analgesic type, dosing, effectiveness, and adherence).
Documentation of the analgesic regimen in the history section of the notes was considered the proxy for analgesic regimen assessment at time of RO consultation. Analgesics within the Medications list, which were autopopulated in the consultation note by the electronic medical record, were recorded.
Whether or not pain was addressed with initiation or titration of analgesics for patients with a PIR of ≥4 was recorded from the Assessment and Plan portion of the notes, and that metric was considered the proxy for pain intervention. In addition, the case was coded as having had pain intervention if there was documentation of the patient declining recommended analgesic intervention, or the patient had been referred to a symptom management service for intervention (eg, referral to a specialty palliative care clinic), or there was recommendation for the patient to discuss uncontrolled pain with the original prescriber. A PIR of 4 was chosen as the threshold for analgesic intervention because at that level, NCCN guidelines for cancer pain state that the analgesic regimen should be titrated, whereas for a PIR of 3 or less, the guidelines recommend only consideration of titrating the analgesic. Only patients with a documented PIR were included in the pain intervention analysis.
Frequencies of analgesic assessment and analgesic intervention were compared using t tests (Wizard Pro, v1.8.5; Evan Miller, Chicago IL).
Results
A total of 271 patients with RO consultation notes were identified at the 2 centers within the 4 time periods (Table 1).
Among symptomatic patients, any component of the preconsultation analgesic regimen (including analgesic type, dosing, pain response, and adherence) was documented for 37.9% of the entire cohort at RO consultation (Table 3). At Centers 1 and 2, the frequencies of analgesic regimen assessment were documented for 41.3% and 28.1%, respectively (P = .06). Among symptomatic patients, 81.5% had an opioid or nonopioid analgesic listed in the Medications section in the electronic medical record at time of consultation.
Patients seen on the dedicated palliative RO service at Center 1 had an analgesic assessment documentation rate of 59.5%, whereas the patients not seen on a palliative RO service (ie, patients seen on a nonpalliative RO service at Center 1 plus all patients at Center 2) had an assessment documentation rate of 33.5% (P = .002; Figure 1). There was no significant difference between rates of analgesic regimen assessment between patients seen at Center 2 and patients seen within nondedicated palliative RO services at Center 1 (28.1% vs 35.9%, respectively; P = .27).
In patients seen at Center 1 only, those seen on the palliative RO service had a higher documentation rate of analgesic assessment compared with those seen by other services after implementation of the dedicated service (59.5% vs 38%, respectively; P = .018). Time period (after versus before 2011) was not significantly associated with the rate of documentation of analgesic assessment at either Center 1 (after vs before 2011: 44.4% vs 31%, P = .23) or Center 2 (31.4% vs 24.1%, P = .60).
Among patients with a PIR of ≥4, analgesic intervention was reported for 17.2% of patients within the entire cohort (20.8% at Center 1 and 0% at Center 2, P = .05). Among those with a PIR of ≥4, documentation of analgesic assessment noted in the History of the Present Illness section was associated with increased documentation of an analgesic intervention in the Assessment and Plan section (25% vs 7.3%; odds ratio [OR], 4.22; 95% confidence interval [CI], 1.1-16.0; P = .03).
Patients seen on the dedicated palliative RO service at Center 1 had a documented analgesic intervention rate of 31.6%, whereas the patients not seen on a palliative RO service (ie, those seen on a nonpalliative RO service at Center 1 plus all patients at Center 2) had a documented analgesic intervention rate of 9.2% (P = .01; Figure 2). There was no statistically significant difference between rates of documentation of an analgesic regimen intervention between patients seen at Center 2 and patients seen within nondedicated palliative RO services at Center 1 (0% vs 17.2%, respectively; P = .07).
Looking at only patients seen at Center 1, patients with a PIR of ≥4 seen on the dedicated palliative RO service had a nearly significant higher rate of documented analgesic interventions in the time period after implementation of the dedicate service (31.6% if seen on the dedicated service vs 12% if seen on a nondedicated service, P = .06).
Discussion
Multiple studies demonstrate the undertreatment of cancer pain in the outpatient setting.2,9,14,15 At 2 cancer centers, we found that about half of patients who present for consideration of palliative RT for bone metastases had a PIR of ≥4, yet only 17% of them had documentation of analgesic intervention as recommended by NCCN guidelines for cancer pain. Underlying this low rate of appropriate intervention may be the assumption of rapid pain relief by RT. However, RT often does not begin at time of consultation,16 and maximal pain relief may take days to weeks after commencement of RT.17 It is estimated that a quarter of all patients with cancer develop bone metastases during the course of their disease,12 and most of those patients suffer from pain. Thus, inherent delay in pain relief before, during, and after RT results in significant morbidity for the cancer patient population if adequate analgesic management is not provided.
The low rate of appropriate analgesic intervention at the time of RO consultation may also be related to the low incidence of proper analgesic assessment. In our cohort, 80% of symptomatic patients had an opioid or nonopioid analgesic listed in their medications within the electronic medical record at time of consultation, but only 38% had the analgesic regimen and/or its effectiveness described in the History of the Present Illness section of the record. Inattentiveness to analgesic type, dosing, and effectiveness during consultation may result in any inadequacies of the analgesic regimen going unnoticed. Consistent with this notion, we found that the rate of appropriate intervention for patients with a PIR of ≥4 was higher among patients who had analgesic regimen reported in the consultation note. Thus, interventions to implement routine review and documentation of the analgesic regimen, for example within the electronic medical record, may be one way to improve pain management.
Another possible reason for low rates of acute pain management within the RO clinic is low provider confidence in regard to analgesic management. In a recent national survey, 96% of radiation oncologists stated they were at least moderately confident with assessment of pain, yet only 77% were at least moderately confident with titrating opioids, and just 56% were at least moderately confident with rotating opioids.10 Educational interventions that improve providers’ facility with analgesic management may increase the frequency of pain management in the RO clinic.
Patients seen on the dedicated palliative RO service had significantly higher rates of documented analgesic regimen assessment and appropriate intervention during RO consultation, compared with patients seen at Center 2 and those not seen on the dedicated palliative RO service at Center 1. The improvements we observed in analgesic assessment and intervention at Center 1 for patients seen on the palliative RO service are likely owing to involvement of palliative RO and not to secular trends, because there were not similar improvements for patients at Center 1 who were not seen by the palliative RO service and those at Center 2, where there was no service.
At Center 1, the dedicated palliative RO service was created to provide specialized care to patients with metastatic disease undergoing palliative radiation. Within its structure, topics within palliative RO, such as technical aspects of palliative RT, symptom management, and communication are taught and reinforced in a case-based approach. Such palliative care awareness, integration, and education within RO achieved by the palliative RO service likely contribute to the improved rates of analgesic management we found in our study. We do note that rate of analgesic intervention in the palliative RO cohort, though higher than in the nonpalliative RO group, was still low, with only a third of patients receiving proper analgesic management. These findings highlight the importance of continued effort in increasing providers’ awareness of the need to assess pain and raise comfort with analgesic initiation and titration and of having dedicated palliative care clinicians embedded within the RO setting.
Since the data for this study was acquired, Center 2 has implemented a short palliative RO didactic course for residents, which improved their comfort levels in assessing analgesic effectiveness and intervening for uncontrolled pain.18 The impact of this intervention on clinical care will need to be evaluated, but the improved provider comfort levels may translate into better-quality care.
Limitations
An important limitation of this retrospective study is the reliance on the documentation provided in the consultation note for determining frequencies of analgesic regimen assessment and intervention. The actual rates of analgesic management that occurred in clinic may have been higher than reported in the documentation. However, such discrepancy in documentation of analgesic management would also be an area for quality improvement. Inadequate documentation limits the ability for proper follow-up of cancer pain as recommended by a joint guidance statement from the American Society of Clinical Oncology and the American Academy of Hospice and Palliative Medicine.19,20 The results of our study may also partly reflect a positive impact in documentation of analgesic management by a dedicated palliative RO service.
Given the multi-institutional nature of this study, it may be that general practice differences confound the impact of the dedicated palliative RO service at Center 1. However, with excluding Center 2, the dedicated service was still strongly associated with a higher rate of analgesic assessment within Center 1 and was almost significantly associated with appropriate analgesic intervention within Center 1.
We used a PIR of ≥4 as a threshold for appropriate analgesic regimen intervention because it is what is recommended by the NCCN guidelines. However, close attention should be paid to the impact that any amount of pain has on an individual patient. The functional, spiritual, and existential impact of pain is unique to each patient’s experience, and optimal symptom management should take those elements into account.
Conclusion
In conclusion, this study indicates that advanced cancer patient pain assessment and intervention according to NCCN cancer pain management guidelines is not common in the RO setting, and it is an area that should be targeted for quality improvement because of the positive implications for patient well-being. Pain assessment and intervention were greater in the setting of a dedicated structure for palliative care within RO, suggesting that the integration of palliative care within RO is a promising means of improving quality of pain management.
This work was presented at the 2016 ASCO Palliative Care in Oncology Symposium (September 9-10, 2016), where this work received a Conquer Cancer Foundation Merit Award.
Bone metastases are a common cause of pain in patients with advanced cancer, with about three-quarters of patients with bone metastases experiencing pain as the dominant symptom.1 Inadequately treated cancer pain impairs patient quality of life, and is associated with higher rates of depression, anxiety, and fatigue. Palliative radiotherapy (RT) is effective in alleviating pain from bone metastases.4 Local field external beam radiotherapy can provide some pain relief at the site of treated metastasis in 80%-90% of cases, with complete pain relief in 50%-60% of cases.5,6 However, maximal pain relief from RT is delayed, in some cases taking days to up to multiple weeks to attain.7,8 Therefore, optimal management of bone metastases pain may require the use of analgesics until RT takes adequate effect.
National Comprehensive Cancer Network (NCCN) Guidelines for Adult Cancer Pain (v. 2.2015) recommend that pain intensity rating (PIR; range, 0-10, where 0 denotes no pain and 10, worst pain imaginable) be used to quantify pain for all symptomatic patients. These guidelines also recommend the pain medication regimen be assessed for all symptomatic patients. For patients with moderate or severe pain (PIR of ≥4), NCCN guidelines recommend that analgesic regimen be intervened upon by alteration of the analgesic regimen (initiating, rotating, or titrating analgesic) or consideration of referral to pain/symptom management specialty.
Previous findings have demonstrated inadequate analgesic management for cancer pain,2,9 including within the radiation oncology (RO) clinic, suggesting that patients seen in consultation for palliative RT may experience uncontrolled pain for days to weeks before the onset of relief from RT. Possible reasons for inadequate acute pain intervention in the RO clinic may be provider discomfort with analgesic management and infrequent formal integration of palliative care within RO.10
Limited single-institution data from the few institutions with dedicated palliative RO services have suggested that these services improve the quality of palliative care delivery, as demonstrated by providers perceptions’ of the clinical impact of a dedicated service11 and the implementation of expedited palliative RT delivery for acute cancer pain.12,13 To our knowledge, the impact of a dedicated palliative RO service on analgesic management for cancer pain has not been assessed.
Here, we report how often patients with symptomatic bone metastases had assessments of existing analgesic regimens and interventions at RO consultation at 2 cancer centers. Center 1 had implemented a dedicated palliative RO service in 2011, consisting of rotating attending physicians and residents as well as dedicated palliative care trained nurse practitioners and a fellow, with the service structured around daily rounds,11 whereas Center 2 had not yet implemented a dedicated service. Using data from both centers, we assessed the impact of a palliative RO service on analgesic assessment and management in patients with bone metastases.
Methods
We searched our institutional databases for patients seen in RO consultation for bone metastases using ICD-9 code 198.5, and retrospectively reviewed consultation notes for those patients during June-July 2008, January-February 2010, January-February 2013, and June-July 2014. Those time periods were chosen as evenly spaced representative samples before and after implementation of a dedicated palliative RO service in 2011 at Center 1. Center 2 did not implement a dedicated palliative RO service in these time periods.
Within consultation notes, we recorded the following data from the History of the Present Illness section: symptoms from bone metastases (symptomatic was defined as any pain present); PIR (range, 0-10); and whether or not the preconsultation analgesic regimen was reported for symptomatic patients (including analgesic type, dosing, effectiveness, and adherence).
Documentation of the analgesic regimen in the history section of the notes was considered the proxy for analgesic regimen assessment at time of RO consultation. Analgesics within the Medications list, which were autopopulated in the consultation note by the electronic medical record, were recorded.
Whether or not pain was addressed with initiation or titration of analgesics for patients with a PIR of ≥4 was recorded from the Assessment and Plan portion of the notes, and that metric was considered the proxy for pain intervention. In addition, the case was coded as having had pain intervention if there was documentation of the patient declining recommended analgesic intervention, or the patient had been referred to a symptom management service for intervention (eg, referral to a specialty palliative care clinic), or there was recommendation for the patient to discuss uncontrolled pain with the original prescriber. A PIR of 4 was chosen as the threshold for analgesic intervention because at that level, NCCN guidelines for cancer pain state that the analgesic regimen should be titrated, whereas for a PIR of 3 or less, the guidelines recommend only consideration of titrating the analgesic. Only patients with a documented PIR were included in the pain intervention analysis.
Frequencies of analgesic assessment and analgesic intervention were compared using t tests (Wizard Pro, v1.8.5; Evan Miller, Chicago IL).
Results
A total of 271 patients with RO consultation notes were identified at the 2 centers within the 4 time periods (Table 1).
Among symptomatic patients, any component of the preconsultation analgesic regimen (including analgesic type, dosing, pain response, and adherence) was documented for 37.9% of the entire cohort at RO consultation (Table 3). At Centers 1 and 2, the frequencies of analgesic regimen assessment were documented for 41.3% and 28.1%, respectively (P = .06). Among symptomatic patients, 81.5% had an opioid or nonopioid analgesic listed in the Medications section in the electronic medical record at time of consultation.
Patients seen on the dedicated palliative RO service at Center 1 had an analgesic assessment documentation rate of 59.5%, whereas the patients not seen on a palliative RO service (ie, patients seen on a nonpalliative RO service at Center 1 plus all patients at Center 2) had an assessment documentation rate of 33.5% (P = .002; Figure 1). There was no significant difference between rates of analgesic regimen assessment between patients seen at Center 2 and patients seen within nondedicated palliative RO services at Center 1 (28.1% vs 35.9%, respectively; P = .27).
In patients seen at Center 1 only, those seen on the palliative RO service had a higher documentation rate of analgesic assessment compared with those seen by other services after implementation of the dedicated service (59.5% vs 38%, respectively; P = .018). Time period (after versus before 2011) was not significantly associated with the rate of documentation of analgesic assessment at either Center 1 (after vs before 2011: 44.4% vs 31%, P = .23) or Center 2 (31.4% vs 24.1%, P = .60).
Among patients with a PIR of ≥4, analgesic intervention was reported for 17.2% of patients within the entire cohort (20.8% at Center 1 and 0% at Center 2, P = .05). Among those with a PIR of ≥4, documentation of analgesic assessment noted in the History of the Present Illness section was associated with increased documentation of an analgesic intervention in the Assessment and Plan section (25% vs 7.3%; odds ratio [OR], 4.22; 95% confidence interval [CI], 1.1-16.0; P = .03).
Patients seen on the dedicated palliative RO service at Center 1 had a documented analgesic intervention rate of 31.6%, whereas the patients not seen on a palliative RO service (ie, those seen on a nonpalliative RO service at Center 1 plus all patients at Center 2) had a documented analgesic intervention rate of 9.2% (P = .01; Figure 2). There was no statistically significant difference between rates of documentation of an analgesic regimen intervention between patients seen at Center 2 and patients seen within nondedicated palliative RO services at Center 1 (0% vs 17.2%, respectively; P = .07).
Looking at only patients seen at Center 1, patients with a PIR of ≥4 seen on the dedicated palliative RO service had a nearly significant higher rate of documented analgesic interventions in the time period after implementation of the dedicate service (31.6% if seen on the dedicated service vs 12% if seen on a nondedicated service, P = .06).
Discussion
Multiple studies demonstrate the undertreatment of cancer pain in the outpatient setting.2,9,14,15 At 2 cancer centers, we found that about half of patients who present for consideration of palliative RT for bone metastases had a PIR of ≥4, yet only 17% of them had documentation of analgesic intervention as recommended by NCCN guidelines for cancer pain. Underlying this low rate of appropriate intervention may be the assumption of rapid pain relief by RT. However, RT often does not begin at time of consultation,16 and maximal pain relief may take days to weeks after commencement of RT.17 It is estimated that a quarter of all patients with cancer develop bone metastases during the course of their disease,12 and most of those patients suffer from pain. Thus, inherent delay in pain relief before, during, and after RT results in significant morbidity for the cancer patient population if adequate analgesic management is not provided.
The low rate of appropriate analgesic intervention at the time of RO consultation may also be related to the low incidence of proper analgesic assessment. In our cohort, 80% of symptomatic patients had an opioid or nonopioid analgesic listed in their medications within the electronic medical record at time of consultation, but only 38% had the analgesic regimen and/or its effectiveness described in the History of the Present Illness section of the record. Inattentiveness to analgesic type, dosing, and effectiveness during consultation may result in any inadequacies of the analgesic regimen going unnoticed. Consistent with this notion, we found that the rate of appropriate intervention for patients with a PIR of ≥4 was higher among patients who had analgesic regimen reported in the consultation note. Thus, interventions to implement routine review and documentation of the analgesic regimen, for example within the electronic medical record, may be one way to improve pain management.
Another possible reason for low rates of acute pain management within the RO clinic is low provider confidence in regard to analgesic management. In a recent national survey, 96% of radiation oncologists stated they were at least moderately confident with assessment of pain, yet only 77% were at least moderately confident with titrating opioids, and just 56% were at least moderately confident with rotating opioids.10 Educational interventions that improve providers’ facility with analgesic management may increase the frequency of pain management in the RO clinic.
Patients seen on the dedicated palliative RO service had significantly higher rates of documented analgesic regimen assessment and appropriate intervention during RO consultation, compared with patients seen at Center 2 and those not seen on the dedicated palliative RO service at Center 1. The improvements we observed in analgesic assessment and intervention at Center 1 for patients seen on the palliative RO service are likely owing to involvement of palliative RO and not to secular trends, because there were not similar improvements for patients at Center 1 who were not seen by the palliative RO service and those at Center 2, where there was no service.
At Center 1, the dedicated palliative RO service was created to provide specialized care to patients with metastatic disease undergoing palliative radiation. Within its structure, topics within palliative RO, such as technical aspects of palliative RT, symptom management, and communication are taught and reinforced in a case-based approach. Such palliative care awareness, integration, and education within RO achieved by the palliative RO service likely contribute to the improved rates of analgesic management we found in our study. We do note that rate of analgesic intervention in the palliative RO cohort, though higher than in the nonpalliative RO group, was still low, with only a third of patients receiving proper analgesic management. These findings highlight the importance of continued effort in increasing providers’ awareness of the need to assess pain and raise comfort with analgesic initiation and titration and of having dedicated palliative care clinicians embedded within the RO setting.
Since the data for this study was acquired, Center 2 has implemented a short palliative RO didactic course for residents, which improved their comfort levels in assessing analgesic effectiveness and intervening for uncontrolled pain.18 The impact of this intervention on clinical care will need to be evaluated, but the improved provider comfort levels may translate into better-quality care.
Limitations
An important limitation of this retrospective study is the reliance on the documentation provided in the consultation note for determining frequencies of analgesic regimen assessment and intervention. The actual rates of analgesic management that occurred in clinic may have been higher than reported in the documentation. However, such discrepancy in documentation of analgesic management would also be an area for quality improvement. Inadequate documentation limits the ability for proper follow-up of cancer pain as recommended by a joint guidance statement from the American Society of Clinical Oncology and the American Academy of Hospice and Palliative Medicine.19,20 The results of our study may also partly reflect a positive impact in documentation of analgesic management by a dedicated palliative RO service.
Given the multi-institutional nature of this study, it may be that general practice differences confound the impact of the dedicated palliative RO service at Center 1. However, with excluding Center 2, the dedicated service was still strongly associated with a higher rate of analgesic assessment within Center 1 and was almost significantly associated with appropriate analgesic intervention within Center 1.
We used a PIR of ≥4 as a threshold for appropriate analgesic regimen intervention because it is what is recommended by the NCCN guidelines. However, close attention should be paid to the impact that any amount of pain has on an individual patient. The functional, spiritual, and existential impact of pain is unique to each patient’s experience, and optimal symptom management should take those elements into account.
Conclusion
In conclusion, this study indicates that advanced cancer patient pain assessment and intervention according to NCCN cancer pain management guidelines is not common in the RO setting, and it is an area that should be targeted for quality improvement because of the positive implications for patient well-being. Pain assessment and intervention were greater in the setting of a dedicated structure for palliative care within RO, suggesting that the integration of palliative care within RO is a promising means of improving quality of pain management.
This work was presented at the 2016 ASCO Palliative Care in Oncology Symposium (September 9-10, 2016), where this work received a Conquer Cancer Foundation Merit Award.
1. Amichetti M, Orrù P, Madeddu A, et al. Comparative evaluation of two hypofractionated radiotherapy regimens for painful bone metastases. Tumori. 2004;90(1):91-95.
2. Vuong S, Pulenzas N, DeAngelis C, et al. Inadequate pain management in cancer patients attending an outpatient palliative radiotherapy clinic. Support Care Cancer. 2016;24(2):887-892.
3. Portenoy RK, Payne D, Jacobsen P. Breakthrough pain: characteristics and impact in patients with cancer pain. Pain. 1999;81(1-2):129-134.
4. Sze WM, Shelley M, Held I, Mason M. Palliation of metastatic bone pain: single fraction versus multifraction radiotherapy - a systematic review of the randomised trials. Sze WM, ed. Cochrane Database Syst Rev. 2004;(2):CD004721-CD004721.
5. Ratanatharathorn V, Powers WE, Moss WT, Perez CA. Bone metastasis: review and critical analysis of random allocation trials of local field treatment. Int J Radiat Oncol Biol Phys. 1999;44(1):1-18.
6. Kirou-Mauro A, Hird A, Wong J, et al. Is response to radiotherapy in patients related to the severity of pretreatment pain? Int J Radiat Oncol Biol Phys. 2008;71(4):1208-1212.
7. Frassica DA. General principles of external beam radiation therapy for skeletal metastases. Clin Orthop Relat Res. 2003;(415 Suppl):S158-S164.
8. McDonald R, Ding K, Brundage M, et al. Effect of radiotherapy on painful bone metastases: a secondary analysis of the NCIC Clinical Trials Group Symptom Control Trial SC.23. JAMA Oncol. 2017 Jul 1;3(7):953-959.
9. Greco MT, Roberto A, Corli O, et al. Quality of cancer pain management: an update of a systematic review of undertreatment of patients with cancer. J Clin Oncol. 2014;32(36):4149-4154.
10. Wei RL, Mattes MD, Yu J, et al. Attitudes of radiation oncologists toward palliative and supportive care in the united states: report on national membership survey by the American Society for Radiation Oncology (ASTRO). Pract Radiat Oncol. 2017;7(2):113-119.
11. Tseng YD, Krishnan MS, Jones JA, et al. Supportive and palliative radiation oncology service: impact of a dedicated service on palliative cancer care. Pract Radiat Oncol. 2014;4(4):247-253.
12. Fairchild A, Pituskin E, Rose B, et al. The rapid access palliative radiotherapy program: blueprint for initiation of a one-stop multidisciplinary bone metastases clinic. Support Care Cancer. 2009;17(2):163-170.
13. de Sa E, Sinclair E, Mitera G, et al. Continued success of the rapid response radiotherapy program: a review of 2004-2008. Support Care Cancer. 2009;17(7):757-762.
14. Deandrea S, Montanari M, Moja L, Apolone G. Prevalence of undertreatment in cancer pain. A review of published literature. Ann Oncol. 2008;19(12):1985-1991.
15. Mitera G, Zeiadin N, Kirou-Mauro A, et al. Retrospective assessment of cancer pain management in an outpatient palliative radiotherapy clinic using the Pain Management Index. J Pain Symptom Manage. 2010;39(2):259-267.
16. Danjoux C, Chow E, Drossos A, et al. An innovative rapid response radiotherapy program to reduce waiting time for palliative radiotherapy. Support Care Cancer. 2006;14(1):38-43.
17. Feyer PC, Steingraeber M. Radiotherapy of bone metastasis in breast cancer patients – current approaches. Breast Care (Basel). 2012;7(2):108-112.
18. Garcia MA, Braunstein SE, Anderson WG. Palliative Care Didactic Course for Radiation Oncology Residents. Int J Radiat Oncol Biol Phys. 2017;97(5):884-885.
19. Ferrell BR, Temel JS, Temin S, et al. Integration of palliative care into standard oncology care: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol. 2017;35(1):96-112.
20. Bickel KE, McNiff K, Buss MK, et al. Defining high-quality palliative care in oncology practice: an American Society of Clinical Oncology/American Academy of Hospice and Palliative Medicine guidance statement. J Oncol Pract. 2016;12(9):e828-e838.
1. Amichetti M, Orrù P, Madeddu A, et al. Comparative evaluation of two hypofractionated radiotherapy regimens for painful bone metastases. Tumori. 2004;90(1):91-95.
2. Vuong S, Pulenzas N, DeAngelis C, et al. Inadequate pain management in cancer patients attending an outpatient palliative radiotherapy clinic. Support Care Cancer. 2016;24(2):887-892.
3. Portenoy RK, Payne D, Jacobsen P. Breakthrough pain: characteristics and impact in patients with cancer pain. Pain. 1999;81(1-2):129-134.
4. Sze WM, Shelley M, Held I, Mason M. Palliation of metastatic bone pain: single fraction versus multifraction radiotherapy - a systematic review of the randomised trials. Sze WM, ed. Cochrane Database Syst Rev. 2004;(2):CD004721-CD004721.
5. Ratanatharathorn V, Powers WE, Moss WT, Perez CA. Bone metastasis: review and critical analysis of random allocation trials of local field treatment. Int J Radiat Oncol Biol Phys. 1999;44(1):1-18.
6. Kirou-Mauro A, Hird A, Wong J, et al. Is response to radiotherapy in patients related to the severity of pretreatment pain? Int J Radiat Oncol Biol Phys. 2008;71(4):1208-1212.
7. Frassica DA. General principles of external beam radiation therapy for skeletal metastases. Clin Orthop Relat Res. 2003;(415 Suppl):S158-S164.
8. McDonald R, Ding K, Brundage M, et al. Effect of radiotherapy on painful bone metastases: a secondary analysis of the NCIC Clinical Trials Group Symptom Control Trial SC.23. JAMA Oncol. 2017 Jul 1;3(7):953-959.
9. Greco MT, Roberto A, Corli O, et al. Quality of cancer pain management: an update of a systematic review of undertreatment of patients with cancer. J Clin Oncol. 2014;32(36):4149-4154.
10. Wei RL, Mattes MD, Yu J, et al. Attitudes of radiation oncologists toward palliative and supportive care in the united states: report on national membership survey by the American Society for Radiation Oncology (ASTRO). Pract Radiat Oncol. 2017;7(2):113-119.
11. Tseng YD, Krishnan MS, Jones JA, et al. Supportive and palliative radiation oncology service: impact of a dedicated service on palliative cancer care. Pract Radiat Oncol. 2014;4(4):247-253.
12. Fairchild A, Pituskin E, Rose B, et al. The rapid access palliative radiotherapy program: blueprint for initiation of a one-stop multidisciplinary bone metastases clinic. Support Care Cancer. 2009;17(2):163-170.
13. de Sa E, Sinclair E, Mitera G, et al. Continued success of the rapid response radiotherapy program: a review of 2004-2008. Support Care Cancer. 2009;17(7):757-762.
14. Deandrea S, Montanari M, Moja L, Apolone G. Prevalence of undertreatment in cancer pain. A review of published literature. Ann Oncol. 2008;19(12):1985-1991.
15. Mitera G, Zeiadin N, Kirou-Mauro A, et al. Retrospective assessment of cancer pain management in an outpatient palliative radiotherapy clinic using the Pain Management Index. J Pain Symptom Manage. 2010;39(2):259-267.
16. Danjoux C, Chow E, Drossos A, et al. An innovative rapid response radiotherapy program to reduce waiting time for palliative radiotherapy. Support Care Cancer. 2006;14(1):38-43.
17. Feyer PC, Steingraeber M. Radiotherapy of bone metastasis in breast cancer patients – current approaches. Breast Care (Basel). 2012;7(2):108-112.
18. Garcia MA, Braunstein SE, Anderson WG. Palliative Care Didactic Course for Radiation Oncology Residents. Int J Radiat Oncol Biol Phys. 2017;97(5):884-885.
19. Ferrell BR, Temel JS, Temin S, et al. Integration of palliative care into standard oncology care: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol. 2017;35(1):96-112.
20. Bickel KE, McNiff K, Buss MK, et al. Defining high-quality palliative care in oncology practice: an American Society of Clinical Oncology/American Academy of Hospice and Palliative Medicine guidance statement. J Oncol Pract. 2016;12(9):e828-e838.
Gastrointestinal cancers: new standards of care from landmark trials
DR HENRY I am Dr David Henry, the Editor-in-Chief of 
DR HALLER The IDEA collaboration was the brainchild of the late Dan Sargent, a biostatistician who was at the Mayo Clinic. It was his idea, since 6 international groups were all testing the same question of 3 months for oxaliplatin to 6 months of oxaliplatin, to combine the data in an individual patient database – which is the best way to do it – so there were these six trials that were all completed.
Three of them were individually reported at ASCO this year, and then the totality was presented at the plenary session – the first time in 12 years that a gastrointestinal (GI) cancer trial made the plenary session. The whole point, obviously, is neuropathy. With 6 months of FOLFOX or XELOX, about 13% or more patients will develop grade 3 neuropathy, even if people stop short of the full-cycle length, and that is a big deal for the 50,000 patients or so who get adjuvant therapy. At the plenary session, the data were presented and the next day three individual trials were presented and discussed by Jeff Meyerhardt (of Dana-Farber Cancer Institute, Boston).
There were 6 different trials: a few included rectum, some included stage II, some used CAPOX and FOLFOX-4 or 6. The only trial that used only FOLFOX was the Cancer and Leukemia Group B (CALGB) trial in the United States (US). There was a lot of heterogeneity, but when Dan was around, I asked him whether that was a problem, and he said on the contrary, was a better thing because it allowed for real-life practice.
The primary endpoint of the study was to look for noninferiority of 3 months versus 6 months of treatment. The noninferiority margin was at a hazard ratio of 1.12, so they were willing to barter down a few percentage points from benefit. If you looked at the primary disease-free survival analysis, the hazard ratio was 1.07, which was an absolute difference of 0.9%, favoring 3 months of therapy. But because the hazard ratio crossed the 1.12 boundary, it was considered inconclusive and not proven.
If you looked at the regimens, CAPOX outperformed FOLFOX. That’s a regimen we don’t do much in the US. We tend to use more FOLFOX, but CAPOX looked better. What they then did was look at the different subsets of patients, and the subsets that it was obviously as good in was the group that had T1-3N1 disease, where 3 months of therapy was clearly just as good as 6 months of therapy, with only a 3% risk of grade 3 neuropathy.
DR HENRY That would be one to three nodes?
DR HALLER Exactly. That’s about 50% of patients. In the T4N2 patients, neither regimen did very well and the 3-year disease-free survival was in the range of 50%, which is clearly unacceptable. Jeff discussed two things. Why could CAPOX be better? If you do the math, when you do CAPOX, you get more oxaliplatin during the first few months of therapy, because it’s 130 mg every 3 weeks, rather than 85 mg every 2 weeks. His conclusion was, “for my next patient who has T4N2 disease, I’ll offer 6 months of FOLFOX.” The study that really need
He discussed the two new trials. One is a study called ARGO, which is being done by the National Surgical Adjuvant Breast and Bowel Project, where people get standard adjuvant chemotherapy, and they’re then randomized to either 24 months of regorafenib 120 mg per day or a placebo. This is an attempt to recreate the transient benefit from bevacizumab in the NSABP C-08 trial. It’s accruing slowly because regorafenib has some toxicity associated with it, but it probably will be completed. Will it continue the benefit as seen in the 12 months of bevacizumab and C-08? We’ll see.
The other, more interesting study is being done in the cooperative groups looking at FOLFOX plus atezolizumab, one of the checkpoint inhibitors. The difficulty here is that only 15% of people with stage III disease have microsatellite instability (MSI)-high tumors, but it’s certainly compelling. This is a straight up comparison. It’s 6 months of FOLFOX in the control arm, or 6 months of FOLFOX plus atezolizumab concurrently for 6 months, and then an additional 6 months of atezolizumab. These are both very fascinating ideas.
DR HENRY To go back to one of your original points, this 3 versus 6 months: the neuropathy is significantly less in those getting the 3 months?
DR HALLER It went to 3%.
DR HENRY We all see that is very bothersome to patients. Before we leave colorectal, I must ask about the right-sided versus left-sided colorectal cancer that we hear a lot about now. Could you comment on how right-sided is worse than left-sided, and do we understand why?
DR HALLER There are two things to consider. If you look back even to simple trials of 5-FU or biochemical modulated 5-FU from 20 years ago, there were clear differences showing worse prognosis in patients with right-sided tumors, so that’s one point to be made. It’s been consistently seen but never acted upon. Then, the explanation for it, possibly, is that the right colon and left colon are two biologically different organs – and they are. Embryologically, the right colon comes from the midgut and the left colon comes from the hindgut, and there were several presentations at ASCO and at prior meetings showing that when you look at different mutations, they differ between the right and left colons. The right-sided tumors are more MSI-high and more BRAF-mutated, left-sided mutations less so.
Then, people started analyzing many of the very large colon cancer trials, including the US trial CALB/SWOG C80405 and the FIRE-3 trials in Europe, where backbone chemotherapy of FOLFIRI or FOLFOX was given with either cetuximab or bevacizumab in RAS wild-type patients. For one study, C80405, they saw that for cetuximab, on the right side, the median survival was 16.7 months and on the left side, it’s 36 months – a 20-month difference. In fact, if you look at the totality of the data, 16.7 almost looks like cetuximab is harming them, as if you were giving it to a RAS-mutated patient, but they were not. They were all RAS wild-type.
For bevacizumab, the right side was 24 months; the left side was 31.4 months. If you look at the left, cetuximab was 36 months and bevacizumab was 31.4 months, so it appears left-sided tumors should get more cetuximab than they are now getting in the US with a 5-month difference, but that decrement is much different on the right, where there’s an 8-month benefit for bevacizumab compared with cetuximab. There is a very good review by Dirk Arnold, who looked at a totality of 6 studies to really examine this more carefully.2
The National Comprehensive Cancer Network has chimed in on this, and is suggesting that for the 25% of people who have right-sided tumors, epidermal growth factor receptor (EGFR) agents not be considered in first-line therapy. NCCN did not go as far to say that EGFR agents should be given on the left side. As I said, the differences are much more impressive in the right, so this is a real sea change for people to consider which side of the tumor affects outcome.
Deb Schrag (Dana-Farber Cancer Institute) presented data at last year’s ASCO not only for stage IV disease showing the same thing, but also stage III disease where there are also right-versus-left differences in terms of recurrence, with a hazard ratio on the right side of about 1.4 compared with the left-sided tumors. Maybe it should be true that 3 months is especially good if you’re treating left-sided tumors, and maybe the right-sided tumor needs to be also calculated with the factors we just talked about. These are two big changes in an area in which we literally haven’t made any change since FOLFOX was introduced a decade ago.
DR HENRY That’s really fascinating, and if not practice changing, then practice challenging. Staying with the mutations idea, in my patients, I’m checking the RAS family and the BRAF mutation, where I’ve learned that’s a particularly bad mutation. I wonder if you might comment on the Kopetz trial, which took a cohort of BRAF mutants and treated them (Figure 2).3 How did that turn out?
DR HALLER It turned out well. We’re turning colon cancer into non–small cell lung cancer in that we’re getting small groups of patients who now have very dedicated care. The backstory here is that there was some thought that you should be treating mutations, not tumor sites. Drugs such as vemurafenib, for example, which is a BRAF inhibitor, worked well in melanoma for the same mutation that’s in colon cancer, V600E. But when vemurafenib was used in the BRAF-mutant patients – these are 10% of the population – median survivorship was one-third that of the rest of the patients, so roughly 12 months. People looked like they were doing worse when vemurafenib was used. They had no benefit.
Scott Kopetz at MD Anderson (Houston, Texas) is a very good bench-to-bed-and-back sort of doc. He looked at this in cell lines and found that when you give a BRAF inhibitor, you upregulate EGFR so you add an EGFR inhibitor. He did a phase 1 and 1B study, and then in the co-operative groups, a study was done – a randomized phase 2 trial for people who had the BRAF-V600E mutation failing first-line therapy, and then went on to receive either irinotecan single agent or irinotecan plus cetuximab or a triple arm of irinotecan, cetuximab, and vemurafenib. There was a crossover, and so the primary endpoint was progression-free survival. It accrued rapidly.
Again, small study, about 100 patients, but for the double-agent arm, or cetuximab–irinotecan, the median survivorship was 2 months. It was 4.4 months for the combination, so more than double. The response rate quadrupled from 4% to 16%, and the people who had disease control tripled, from 22% to 67%. Many of these patients had bulky disease, BRAF mutations. They need response, so this is a very important endpoint.
Overall survival was not different, in part because it was a crossover, and the crossover patients did pretty well. This is going to move more toward first-line therapy, because we don’t talk about fourth- and fifth-line therapies, TAS-102 or regorafenib. These patients don’t make it to even third line. We’re chipping away at what we think is a very homogenous group of peoples’ metastatic disease. They’re obviously not.
DR HENRY In the BRAF-mutant patient, the vemurafenib might drive them toward EGFR, and then the cetuximab could come in and handle that diversion of the pathway. Fascinating.
DR HALLER The preferred regimen in first-line therapy for a BRAF mutant might be FOLFIRI, cetuximab, and vemurafenib, especially on the left side.
DR HENRY Certainly makes sense. We’ll continue the theme at ASCO of “new standard of care.” Let’s move to gastroesophageal junction. There was a so-called FLOT (5-FU, leucovorin, oxaliplatin, Taxotere) presentation in the neoadjuvant/adjuvant setting, 4 cycles preoperatively and 4 cycles postoperatively. Could you comment on that study?
DR HALLER Gastric cancer for metastatic disease has a very large buffet of treatment regimens, and some just become entrenched, like the ECF regimen with epirubicin (epirubicin, cisplatin, 5-FU), where most people don’t exactly know what the contribution of that drug is, and so some people use EOX (epirubicin, oxaliplatin, capecitabine), some people use FOLFOX, some people use FOLFIRI. It gets a little bit confusing as to whether you use taxanes, platinums, or 5-FU or capecitabine.
The Germans came up with a regimen called FLOT – it’s sort of like FOLFOX with Taxotere attached. They did a very large study comparing it with ECF or ECX (epirubicin, cisplatin, capecitabine; Figure 3).4 The overall endpoint with over 700 patients was survival. This is an adjuvant regimen. Only 37% of people got ECF or ECX postoperatively, and 50% of the FLOT patients got the regimens postoperatively.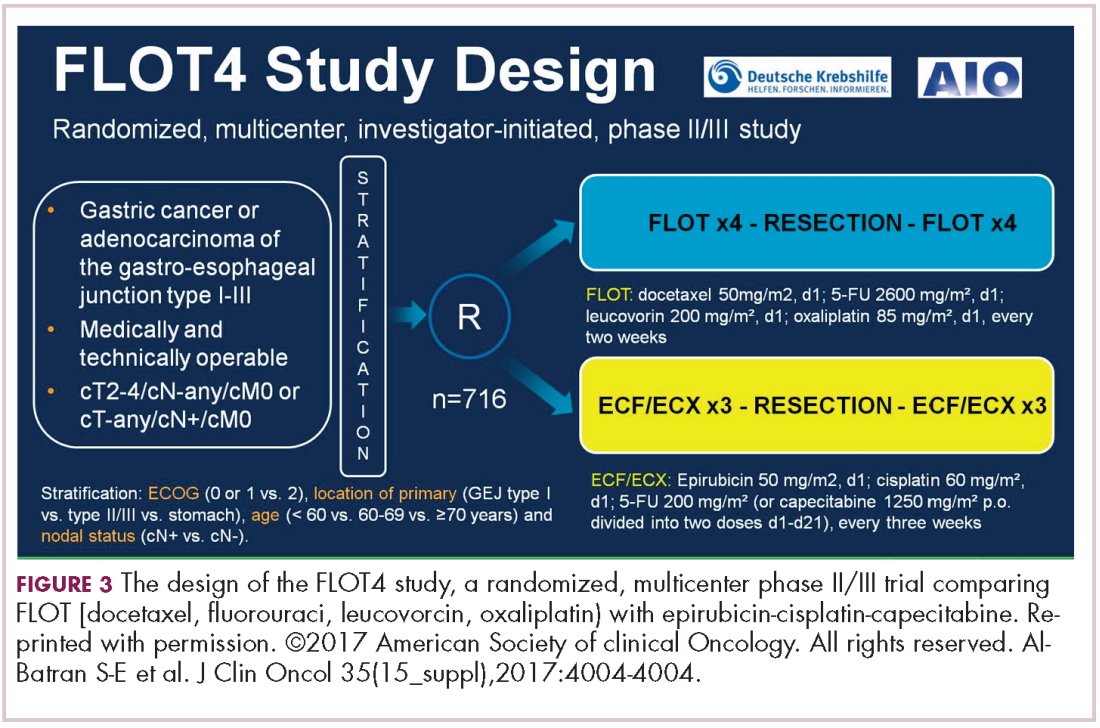
One of the reasons FLOT might be more beneficial is that more people were given postoperative treatment, and it’s one reason why many adjuvant regimens are being moved completely preoperatively, because so few people get the planned treatment. The FLOT regimen improved overall survival with a P value of .0112 and a hazard ratio of 0.77. The difference was 35 months versus 50 months. With the uncertainty as to what epirubicin actually does and the fact that it’s been around for a while and that fewer people receive postoperative treatment, with that 15-month benefit, if you’re using chemotherapy alone, and there’s no radiotherapy component for true gastric cancer, this is a new standard of care.
DR HENRY I struggle with this in my patients as well. This concept of getting more therapy preoperatively to those who can’t get it postoperatively certainly resonates with most of us in practice.
DR HALLER If I were redesigning the trial, I would probably say just give 4-6 cycles of treatment, and give it all preoperatively. In rectal cancer, there’s the total neoadjuvant approach, where it’s being tested in people who get all their chemotherapy first, then chemoradiotherapy, then surgery, and you’re done.
DR HENRY Yes, right. Thank you for mentioning that. Staying with the gastric GE junction, you couldn’t get away from ASCO this year without hearing about the checkpoint inhibitor immunotherapies in this population. In the CHECKMATE-142 trial with nivolumab versus placebo, response rates were good, especially in the MSI-high (microsatellite instability). Could you comment on that study?
DR HALLER We already know that in May and July 2017, pembrolizumab and nivolumab were both approved for any MSI-high solid tumor based on phase 2 data only, and based on response. That’s the first time we’ve seen that happen. It’s remarkable. For nivolumab, the approval was based on 53 patients with MSI-high metastatic colon cancer. So these were people who failed standard therapy and got nivolumab by standard infusion every 2 weeks. The overall response rate was almost 30% in this population, which is typically quite resistant to any treatment, so one expects much lower response rates with anything in that setting – chemotherapy, TAS-102, regorafenib, et cetera (Table).5
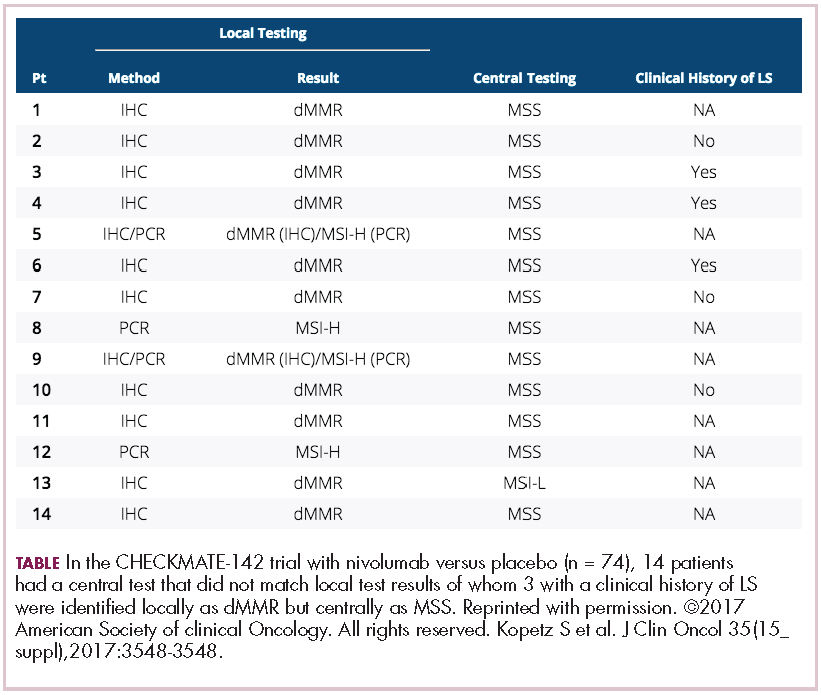
More importantly, as we’re seeing with Jimmy Carter with checkpoint treatment (for melanoma that had metastasized to the brain), responses lasted for more than 6 months in about two-thirds of patients, even a complete response, so this is just off the wall. I mean, this is not what you would expect with almost any other treatment. The data are the same for atezolizumab and for pembrolizumab. What seems to be true is that in the GI tumors and colon cancer, MSI-high seems more important than expression of PD-1 or PD-L1 (programmed cell death protein-1 or programmed cell death protein-ligand 1).
In different tumor sites, PD-1 or PD-L1 measurement may be important, but in these tumors, and in colorectal cancer, it looks as if MSI-high is the preferred measurement. Recently ASCO, together with the American Society for Clinical Pathology, College of American Pathologists, and Association for Molecular Pathology, came out with guidelines on what you should measure in colorectal cancer specimens. Obviously, one is extended RAS. They say you should get BRAF for prognosis, but it may also be a prognostic factor that leads you to treat, which ultimately makes it a predictive factor, so the data from Kopetz might suggest that will move up to something you also must measure. If patients have the BRAF mutation, it’s important they know that it’s a poor prognostic sign. But if they come in with literature saying they might live 36 months when their actual outcome is about a third of that, you need to frame your discussion in that regard and make sure they understand it.
The guidelines also suggested getting MSI-high, and certainly prognostically in early-stage disease, but now it’s going to be a predictive factor, so in the month in which these recommendations are made, two of them are already out of date. They also didn’t include human epidermal growth factor receptor 2 (HER2), and what we’ve heard from the HERACLES (HER2 Amplification for Colorectal Cancer Enhanced Stratification) trial is that for those patients who got the trastuzumab and pertuzumab combination – and this is another 5% of patients – almost the same data was seen as in the MSI-high patients with checkpoint inhibitors. That is double-digit response rates and durable responses. As I said, we’re very much nearing in colorectal cancer what’s now being done in non-small cell lung cancer.
DR HENRY Indeed. Could you comment on the BILCAP study and adjuvant capecitabine for biliary tract cancer?
DR HALLER There are large meta-analyses looking at adjuvant therapy for biliary tract cancers typically from fairly small, fairly old studies that all suggest that in certain stages of resected biliary tumors, either bile duct or gall bladder, adjuvant treatment works, and typically either chemotherapy and radiotherapy, or chemotherapy alone, but not radiotherapy alone.
Capecitabine has been used for metastatic disease for years, mostly by default, and because most GI tumors have some response to fluoropyridines. But we’re finally able now to do large trials in biliary tumors, so this trial was a very large study with almost 450 patients from the United Kingdom over an 8-year period. About 20% were gallbladder, so the R0 surgery was about 60%, R1 at about 40% (Figure 4).6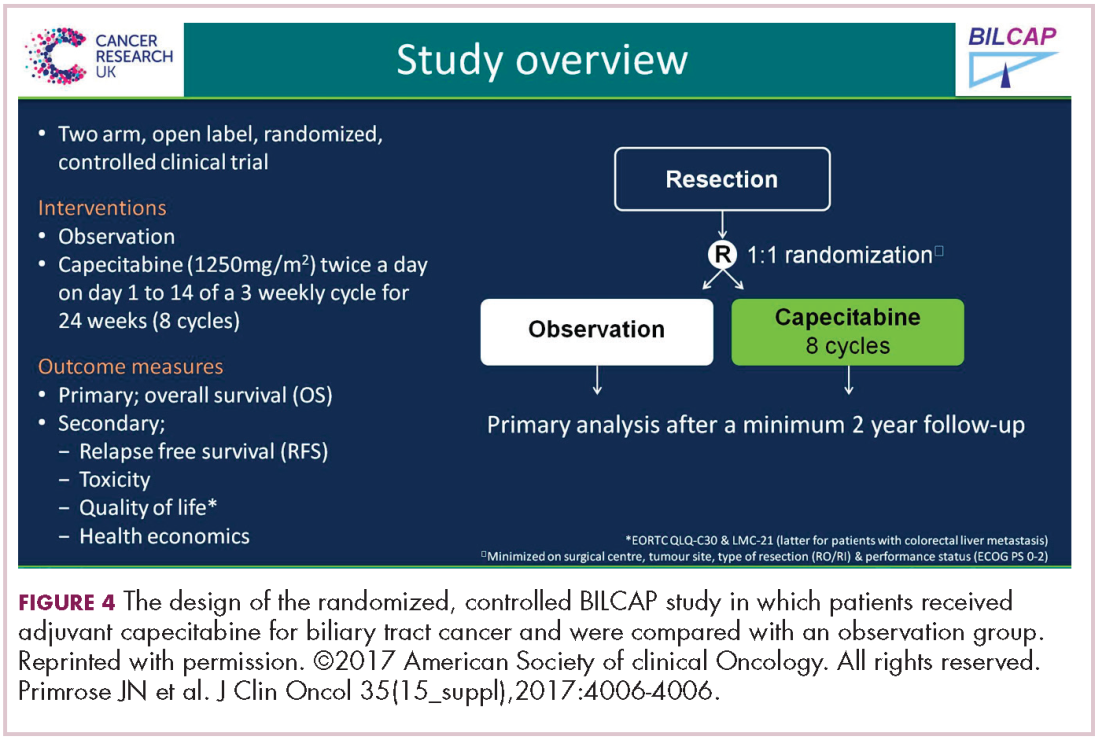
The endpoint of the study was survival advantage, and when they did the protocol analysis, the survival for the treated population was 53 months and for the observation arm, 36 months, so that was a hazard ratio of 0.75, which is acceptable in an adjuvant study. It’s simple drug to give, and usually tolerable, so this will represent a new standard of care. Of course, in the advanced disease setting, the gemcitabine–cisplatin combination is the standard of care for metastatic disease. It’s a little more toxic combination, but we know that’s standard. There’s an ongoing study in Europe called the ACTICCA-1 trial, and this is gemcitabine–cisplatin for 6 months versus not capecitabine, but a control arm. My guess is if the capecitabine study was positive, that this also will be a positive trial, because gemcitabine–cisplatin is probably more active. Then, we’ll have 2 standards, and I don’t think anyone is going to compare capecitabine with gemcitabine–cisplatin.
What you’ll have are two regimens for two different populations of patients. Perhaps for the elderly and people who have renal problems, capecitabine alone will give them benefit, and then you’ll have gemcitabine–cisplatin, which may be just a more toxic regimen, but also more effective for the younger, healthier people with fewer comorbidities.
DR HENRY Great data and a small population, but a population in need. That moves us on to pancreatic cancer, and I don’t know if this is happening nationwide, but in my practice, I’m seeing more. These patients tend to present beyond surgery, so they have metastatic or advanced pancreatic cancer. Any comment on where you think this field is going?
DR HALLER We were a bit bereft of new pancreatic cancer studies at ASCO this year. We’re certainly looking more at neoadjuvant therapy for pancreatic cancer, primarily because of ease of administration and the increased ability to tolerate treatments in the preoperative setting. There aren’t many people that get downstaged, but some are. Unfortunately, even in the MSI-high pancreas, which is a small subset, they don’t seem to get as big a bang out of the checkpoint inhibitors as in other tumor sites, so I’m afraid I didn’t come home with much new about this subset of patients.
DR HENRY We’ve covered a nice group of studies and practice-changing new standard-of-care comments from ASCO and other studies. Thank Dr Dan Haller for being with us and commenting. This podcast and discussion are brought to you from
1. Andre T, Bonnetain F, Mineur L, et al. Oxaliplatin-based chemotherapy for patients with stage III colon cancer: disease free survival results of the three versus six months adjuvant IDEA France trial. Abstract presented at: 2017 American Society of Clinical Oncology Annual Meeting; June 2-6, 2017; Chicago, IL. Abstract 3500.
2. Arnold D, Lueza B, Douillard JY, et al. Prognostic and predictive value of primary tumour side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann Oncol. 2017;28(8):1713-1729.
3. Kopetz S, McDonough SL, Lenz H-J, et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG S1406). Abstract presented at: 2017 American Society of Clinical Oncology Annual Meeting; June 2-6, 2017; Chicago, IL. Abstract 3505.
4. Al-Batran S-E, Homann N, Schmalenberg H, et al. Perioperative chemotherapy with docetaxel, oxaliplatin. Abstract presented at: 2017 American Society of Clinical Oncology Annu, and fluorouracil/leucovorin (FLOT) versus epirubicin, cisplatin, and fluorouracil or capecitabine (ECF/ECX) for resectable gastric or gastroesophageal junction (GEJ) adenocarcinoma (FLOT4-AIO): a multicenter, randomized phase 3 trial. Abstract presented at: 2017 American Society of Clinical Oncology Annual Meeting; June 2-6, 2017; Chicago, IL. Abstract 4004.
5. Kopetz S, Lonardi S, McDermott RS, et al. Concordance of DNA mismatch repair deficient (dMMR)/microsatellite instability (MSI) assessment by local and central testing in patients with metastatic CRC (mCRC) receiving nivolumab (nivo) in Checkmate 142 study. Abstract presented at: 2017 American Society of Clinical Oncology Annual Meeting; June 2-6, 2017; Chicago, IL. Abstract 3548.
6. Primrose JN, Fox R, Palmer DH, et al. Adjuvant capecitabine for biliary tract cancer: the BILCAP randomized study. Abstract presented at: 2017 American Society of Clinical Oncology Annual Meeting; June 2-6, 2017; Chicago, IL. Abstract 4006.
DR HENRY I am Dr David Henry, the Editor-in-Chief of
DR HALLER The IDEA collaboration was the brainchild of the late Dan Sargent, a biostatistician who was at the Mayo Clinic. It was his idea, since 6 international groups were all testing the same question of 3 months for oxaliplatin to 6 months of oxaliplatin, to combine the data in an individual patient database – which is the best way to do it – so there were these six trials that were all completed.
Three of them were individually reported at ASCO this year, and then the totality was presented at the plenary session – the first time in 12 years that a gastrointestinal (GI) cancer trial made the plenary session. The whole point, obviously, is neuropathy. With 6 months of FOLFOX or XELOX, about 13% or more patients will develop grade 3 neuropathy, even if people stop short of the full-cycle length, and that is a big deal for the 50,000 patients or so who get adjuvant therapy. At the plenary session, the data were presented and the next day three individual trials were presented and discussed by Jeff Meyerhardt (of Dana-Farber Cancer Institute, Boston).
There were 6 different trials: a few included rectum, some included stage II, some used CAPOX and FOLFOX-4 or 6. The only trial that used only FOLFOX was the Cancer and Leukemia Group B (CALGB) trial in the United States (US). There was a lot of heterogeneity, but when Dan was around, I asked him whether that was a problem, and he said on the contrary, was a better thing because it allowed for real-life practice.
The primary endpoint of the study was to look for noninferiority of 3 months versus 6 months of treatment. The noninferiority margin was at a hazard ratio of 1.12, so they were willing to barter down a few percentage points from benefit. If you looked at the primary disease-free survival analysis, the hazard ratio was 1.07, which was an absolute difference of 0.9%, favoring 3 months of therapy. But because the hazard ratio crossed the 1.12 boundary, it was considered inconclusive and not proven.
If you looked at the regimens, CAPOX outperformed FOLFOX. That’s a regimen we don’t do much in the US. We tend to use more FOLFOX, but CAPOX looked better. What they then did was look at the different subsets of patients, and the subsets that it was obviously as good in was the group that had T1-3N1 disease, where 3 months of therapy was clearly just as good as 6 months of therapy, with only a 3% risk of grade 3 neuropathy.
DR HENRY That would be one to three nodes?
DR HALLER Exactly. That’s about 50% of patients. In the T4N2 patients, neither regimen did very well and the 3-year disease-free survival was in the range of 50%, which is clearly unacceptable. Jeff discussed two things. Why could CAPOX be better? If you do the math, when you do CAPOX, you get more oxaliplatin during the first few months of therapy, because it’s 130 mg every 3 weeks, rather than 85 mg every 2 weeks. His conclusion was, “for my next patient who has T4N2 disease, I’ll offer 6 months of FOLFOX.” The study that really need
He discussed the two new trials. One is a study called ARGO, which is being done by the National Surgical Adjuvant Breast and Bowel Project, where people get standard adjuvant chemotherapy, and they’re then randomized to either 24 months of regorafenib 120 mg per day or a placebo. This is an attempt to recreate the transient benefit from bevacizumab in the NSABP C-08 trial. It’s accruing slowly because regorafenib has some toxicity associated with it, but it probably will be completed. Will it continue the benefit as seen in the 12 months of bevacizumab and C-08? We’ll see.
The other, more interesting study is being done in the cooperative groups looking at FOLFOX plus atezolizumab, one of the checkpoint inhibitors. The difficulty here is that only 15% of people with stage III disease have microsatellite instability (MSI)-high tumors, but it’s certainly compelling. This is a straight up comparison. It’s 6 months of FOLFOX in the control arm, or 6 months of FOLFOX plus atezolizumab concurrently for 6 months, and then an additional 6 months of atezolizumab. These are both very fascinating ideas.
DR HENRY To go back to one of your original points, this 3 versus 6 months: the neuropathy is significantly less in those getting the 3 months?
DR HALLER It went to 3%.
DR HENRY We all see that is very bothersome to patients. Before we leave colorectal, I must ask about the right-sided versus left-sided colorectal cancer that we hear a lot about now. Could you comment on how right-sided is worse than left-sided, and do we understand why?
DR HALLER There are two things to consider. If you look back even to simple trials of 5-FU or biochemical modulated 5-FU from 20 years ago, there were clear differences showing worse prognosis in patients with right-sided tumors, so that’s one point to be made. It’s been consistently seen but never acted upon. Then, the explanation for it, possibly, is that the right colon and left colon are two biologically different organs – and they are. Embryologically, the right colon comes from the midgut and the left colon comes from the hindgut, and there were several presentations at ASCO and at prior meetings showing that when you look at different mutations, they differ between the right and left colons. The right-sided tumors are more MSI-high and more BRAF-mutated, left-sided mutations less so.
Then, people started analyzing many of the very large colon cancer trials, including the US trial CALB/SWOG C80405 and the FIRE-3 trials in Europe, where backbone chemotherapy of FOLFIRI or FOLFOX was given with either cetuximab or bevacizumab in RAS wild-type patients. For one study, C80405, they saw that for cetuximab, on the right side, the median survival was 16.7 months and on the left side, it’s 36 months – a 20-month difference. In fact, if you look at the totality of the data, 16.7 almost looks like cetuximab is harming them, as if you were giving it to a RAS-mutated patient, but they were not. They were all RAS wild-type.
For bevacizumab, the right side was 24 months; the left side was 31.4 months. If you look at the left, cetuximab was 36 months and bevacizumab was 31.4 months, so it appears left-sided tumors should get more cetuximab than they are now getting in the US with a 5-month difference, but that decrement is much different on the right, where there’s an 8-month benefit for bevacizumab compared with cetuximab. There is a very good review by Dirk Arnold, who looked at a totality of 6 studies to really examine this more carefully.2
The National Comprehensive Cancer Network has chimed in on this, and is suggesting that for the 25% of people who have right-sided tumors, epidermal growth factor receptor (EGFR) agents not be considered in first-line therapy. NCCN did not go as far to say that EGFR agents should be given on the left side. As I said, the differences are much more impressive in the right, so this is a real sea change for people to consider which side of the tumor affects outcome.
Deb Schrag (Dana-Farber Cancer Institute) presented data at last year’s ASCO not only for stage IV disease showing the same thing, but also stage III disease where there are also right-versus-left differences in terms of recurrence, with a hazard ratio on the right side of about 1.4 compared with the left-sided tumors. Maybe it should be true that 3 months is especially good if you’re treating left-sided tumors, and maybe the right-sided tumor needs to be also calculated with the factors we just talked about. These are two big changes in an area in which we literally haven’t made any change since FOLFOX was introduced a decade ago.
DR HENRY That’s really fascinating, and if not practice changing, then practice challenging. Staying with the mutations idea, in my patients, I’m checking the RAS family and the BRAF mutation, where I’ve learned that’s a particularly bad mutation. I wonder if you might comment on the Kopetz trial, which took a cohort of BRAF mutants and treated them (Figure 2).3 How did that turn out?
DR HALLER It turned out well. We’re turning colon cancer into non–small cell lung cancer in that we’re getting small groups of patients who now have very dedicated care. The backstory here is that there was some thought that you should be treating mutations, not tumor sites. Drugs such as vemurafenib, for example, which is a BRAF inhibitor, worked well in melanoma for the same mutation that’s in colon cancer, V600E. But when vemurafenib was used in the BRAF-mutant patients – these are 10% of the population – median survivorship was one-third that of the rest of the patients, so roughly 12 months. People looked like they were doing worse when vemurafenib was used. They had no benefit.
Scott Kopetz at MD Anderson (Houston, Texas) is a very good bench-to-bed-and-back sort of doc. He looked at this in cell lines and found that when you give a BRAF inhibitor, you upregulate EGFR so you add an EGFR inhibitor. He did a phase 1 and 1B study, and then in the co-operative groups, a study was done – a randomized phase 2 trial for people who had the BRAF-V600E mutation failing first-line therapy, and then went on to receive either irinotecan single agent or irinotecan plus cetuximab or a triple arm of irinotecan, cetuximab, and vemurafenib. There was a crossover, and so the primary endpoint was progression-free survival. It accrued rapidly.
Again, small study, about 100 patients, but for the double-agent arm, or cetuximab–irinotecan, the median survivorship was 2 months. It was 4.4 months for the combination, so more than double. The response rate quadrupled from 4% to 16%, and the people who had disease control tripled, from 22% to 67%. Many of these patients had bulky disease, BRAF mutations. They need response, so this is a very important endpoint.
Overall survival was not different, in part because it was a crossover, and the crossover patients did pretty well. This is going to move more toward first-line therapy, because we don’t talk about fourth- and fifth-line therapies, TAS-102 or regorafenib. These patients don’t make it to even third line. We’re chipping away at what we think is a very homogenous group of peoples’ metastatic disease. They’re obviously not.
DR HENRY In the BRAF-mutant patient, the vemurafenib might drive them toward EGFR, and then the cetuximab could come in and handle that diversion of the pathway. Fascinating.
DR HALLER The preferred regimen in first-line therapy for a BRAF mutant might be FOLFIRI, cetuximab, and vemurafenib, especially on the left side.
DR HENRY Certainly makes sense. We’ll continue the theme at ASCO of “new standard of care.” Let’s move to gastroesophageal junction. There was a so-called FLOT (5-FU, leucovorin, oxaliplatin, Taxotere) presentation in the neoadjuvant/adjuvant setting, 4 cycles preoperatively and 4 cycles postoperatively. Could you comment on that study?
DR HALLER Gastric cancer for metastatic disease has a very large buffet of treatment regimens, and some just become entrenched, like the ECF regimen with epirubicin (epirubicin, cisplatin, 5-FU), where most people don’t exactly know what the contribution of that drug is, and so some people use EOX (epirubicin, oxaliplatin, capecitabine), some people use FOLFOX, some people use FOLFIRI. It gets a little bit confusing as to whether you use taxanes, platinums, or 5-FU or capecitabine.
The Germans came up with a regimen called FLOT – it’s sort of like FOLFOX with Taxotere attached. They did a very large study comparing it with ECF or ECX (epirubicin, cisplatin, capecitabine; Figure 3).4 The overall endpoint with over 700 patients was survival. This is an adjuvant regimen. Only 37% of people got ECF or ECX postoperatively, and 50% of the FLOT patients got the regimens postoperatively.
One of the reasons FLOT might be more beneficial is that more people were given postoperative treatment, and it’s one reason why many adjuvant regimens are being moved completely preoperatively, because so few people get the planned treatment. The FLOT regimen improved overall survival with a P value of .0112 and a hazard ratio of 0.77. The difference was 35 months versus 50 months. With the uncertainty as to what epirubicin actually does and the fact that it’s been around for a while and that fewer people receive postoperative treatment, with that 15-month benefit, if you’re using chemotherapy alone, and there’s no radiotherapy component for true gastric cancer, this is a new standard of care.
DR HENRY I struggle with this in my patients as well. This concept of getting more therapy preoperatively to those who can’t get it postoperatively certainly resonates with most of us in practice.
DR HALLER If I were redesigning the trial, I would probably say just give 4-6 cycles of treatment, and give it all preoperatively. In rectal cancer, there’s the total neoadjuvant approach, where it’s being tested in people who get all their chemotherapy first, then chemoradiotherapy, then surgery, and you’re done.
DR HENRY Yes, right. Thank you for mentioning that. Staying with the gastric GE junction, you couldn’t get away from ASCO this year without hearing about the checkpoint inhibitor immunotherapies in this population. In the CHECKMATE-142 trial with nivolumab versus placebo, response rates were good, especially in the MSI-high (microsatellite instability). Could you comment on that study?
DR HALLER We already know that in May and July 2017, pembrolizumab and nivolumab were both approved for any MSI-high solid tumor based on phase 2 data only, and based on response. That’s the first time we’ve seen that happen. It’s remarkable. For nivolumab, the approval was based on 53 patients with MSI-high metastatic colon cancer. So these were people who failed standard therapy and got nivolumab by standard infusion every 2 weeks. The overall response rate was almost 30% in this population, which is typically quite resistant to any treatment, so one expects much lower response rates with anything in that setting – chemotherapy, TAS-102, regorafenib, et cetera (Table).5
More importantly, as we’re seeing with Jimmy Carter with checkpoint treatment (for melanoma that had metastasized to the brain), responses lasted for more than 6 months in about two-thirds of patients, even a complete response, so this is just off the wall. I mean, this is not what you would expect with almost any other treatment. The data are the same for atezolizumab and for pembrolizumab. What seems to be true is that in the GI tumors and colon cancer, MSI-high seems more important than expression of PD-1 or PD-L1 (programmed cell death protein-1 or programmed cell death protein-ligand 1).
In different tumor sites, PD-1 or PD-L1 measurement may be important, but in these tumors, and in colorectal cancer, it looks as if MSI-high is the preferred measurement. Recently ASCO, together with the American Society for Clinical Pathology, College of American Pathologists, and Association for Molecular Pathology, came out with guidelines on what you should measure in colorectal cancer specimens. Obviously, one is extended RAS. They say you should get BRAF for prognosis, but it may also be a prognostic factor that leads you to treat, which ultimately makes it a predictive factor, so the data from Kopetz might suggest that will move up to something you also must measure. If patients have the BRAF mutation, it’s important they know that it’s a poor prognostic sign. But if they come in with literature saying they might live 36 months when their actual outcome is about a third of that, you need to frame your discussion in that regard and make sure they understand it.
The guidelines also suggested getting MSI-high, and certainly prognostically in early-stage disease, but now it’s going to be a predictive factor, so in the month in which these recommendations are made, two of them are already out of date. They also didn’t include human epidermal growth factor receptor 2 (HER2), and what we’ve heard from the HERACLES (HER2 Amplification for Colorectal Cancer Enhanced Stratification) trial is that for those patients who got the trastuzumab and pertuzumab combination – and this is another 5% of patients – almost the same data was seen as in the MSI-high patients with checkpoint inhibitors. That is double-digit response rates and durable responses. As I said, we’re very much nearing in colorectal cancer what’s now being done in non-small cell lung cancer.
DR HENRY Indeed. Could you comment on the BILCAP study and adjuvant capecitabine for biliary tract cancer?
DR HALLER There are large meta-analyses looking at adjuvant therapy for biliary tract cancers typically from fairly small, fairly old studies that all suggest that in certain stages of resected biliary tumors, either bile duct or gall bladder, adjuvant treatment works, and typically either chemotherapy and radiotherapy, or chemotherapy alone, but not radiotherapy alone.
Capecitabine has been used for metastatic disease for years, mostly by default, and because most GI tumors have some response to fluoropyridines. But we’re finally able now to do large trials in biliary tumors, so this trial was a very large study with almost 450 patients from the United Kingdom over an 8-year period. About 20% were gallbladder, so the R0 surgery was about 60%, R1 at about 40% (Figure 4).6
The endpoint of the study was survival advantage, and when they did the protocol analysis, the survival for the treated population was 53 months and for the observation arm, 36 months, so that was a hazard ratio of 0.75, which is acceptable in an adjuvant study. It’s simple drug to give, and usually tolerable, so this will represent a new standard of care. Of course, in the advanced disease setting, the gemcitabine–cisplatin combination is the standard of care for metastatic disease. It’s a little more toxic combination, but we know that’s standard. There’s an ongoing study in Europe called the ACTICCA-1 trial, and this is gemcitabine–cisplatin for 6 months versus not capecitabine, but a control arm. My guess is if the capecitabine study was positive, that this also will be a positive trial, because gemcitabine–cisplatin is probably more active. Then, we’ll have 2 standards, and I don’t think anyone is going to compare capecitabine with gemcitabine–cisplatin.
What you’ll have are two regimens for two different populations of patients. Perhaps for the elderly and people who have renal problems, capecitabine alone will give them benefit, and then you’ll have gemcitabine–cisplatin, which may be just a more toxic regimen, but also more effective for the younger, healthier people with fewer comorbidities.
DR HENRY Great data and a small population, but a population in need. That moves us on to pancreatic cancer, and I don’t know if this is happening nationwide, but in my practice, I’m seeing more. These patients tend to present beyond surgery, so they have metastatic or advanced pancreatic cancer. Any comment on where you think this field is going?
DR HALLER We were a bit bereft of new pancreatic cancer studies at ASCO this year. We’re certainly looking more at neoadjuvant therapy for pancreatic cancer, primarily because of ease of administration and the increased ability to tolerate treatments in the preoperative setting. There aren’t many people that get downstaged, but some are. Unfortunately, even in the MSI-high pancreas, which is a small subset, they don’t seem to get as big a bang out of the checkpoint inhibitors as in other tumor sites, so I’m afraid I didn’t come home with much new about this subset of patients.
DR HENRY We’ve covered a nice group of studies and practice-changing new standard-of-care comments from ASCO and other studies. Thank Dr Dan Haller for being with us and commenting. This podcast and discussion are brought to you from
DR HENRY I am Dr David Henry, the Editor-in-Chief of
DR HALLER The IDEA collaboration was the brainchild of the late Dan Sargent, a biostatistician who was at the Mayo Clinic. It was his idea, since 6 international groups were all testing the same question of 3 months for oxaliplatin to 6 months of oxaliplatin, to combine the data in an individual patient database – which is the best way to do it – so there were these six trials that were all completed.
Three of them were individually reported at ASCO this year, and then the totality was presented at the plenary session – the first time in 12 years that a gastrointestinal (GI) cancer trial made the plenary session. The whole point, obviously, is neuropathy. With 6 months of FOLFOX or XELOX, about 13% or more patients will develop grade 3 neuropathy, even if people stop short of the full-cycle length, and that is a big deal for the 50,000 patients or so who get adjuvant therapy. At the plenary session, the data were presented and the next day three individual trials were presented and discussed by Jeff Meyerhardt (of Dana-Farber Cancer Institute, Boston).
There were 6 different trials: a few included rectum, some included stage II, some used CAPOX and FOLFOX-4 or 6. The only trial that used only FOLFOX was the Cancer and Leukemia Group B (CALGB) trial in the United States (US). There was a lot of heterogeneity, but when Dan was around, I asked him whether that was a problem, and he said on the contrary, was a better thing because it allowed for real-life practice.
The primary endpoint of the study was to look for noninferiority of 3 months versus 6 months of treatment. The noninferiority margin was at a hazard ratio of 1.12, so they were willing to barter down a few percentage points from benefit. If you looked at the primary disease-free survival analysis, the hazard ratio was 1.07, which was an absolute difference of 0.9%, favoring 3 months of therapy. But because the hazard ratio crossed the 1.12 boundary, it was considered inconclusive and not proven.
If you looked at the regimens, CAPOX outperformed FOLFOX. That’s a regimen we don’t do much in the US. We tend to use more FOLFOX, but CAPOX looked better. What they then did was look at the different subsets of patients, and the subsets that it was obviously as good in was the group that had T1-3N1 disease, where 3 months of therapy was clearly just as good as 6 months of therapy, with only a 3% risk of grade 3 neuropathy.
DR HENRY That would be one to three nodes?
DR HALLER Exactly. That’s about 50% of patients. In the T4N2 patients, neither regimen did very well and the 3-year disease-free survival was in the range of 50%, which is clearly unacceptable. Jeff discussed two things. Why could CAPOX be better? If you do the math, when you do CAPOX, you get more oxaliplatin during the first few months of therapy, because it’s 130 mg every 3 weeks, rather than 85 mg every 2 weeks. His conclusion was, “for my next patient who has T4N2 disease, I’ll offer 6 months of FOLFOX.” The study that really need
He discussed the two new trials. One is a study called ARGO, which is being done by the National Surgical Adjuvant Breast and Bowel Project, where people get standard adjuvant chemotherapy, and they’re then randomized to either 24 months of regorafenib 120 mg per day or a placebo. This is an attempt to recreate the transient benefit from bevacizumab in the NSABP C-08 trial. It’s accruing slowly because regorafenib has some toxicity associated with it, but it probably will be completed. Will it continue the benefit as seen in the 12 months of bevacizumab and C-08? We’ll see.
The other, more interesting study is being done in the cooperative groups looking at FOLFOX plus atezolizumab, one of the checkpoint inhibitors. The difficulty here is that only 15% of people with stage III disease have microsatellite instability (MSI)-high tumors, but it’s certainly compelling. This is a straight up comparison. It’s 6 months of FOLFOX in the control arm, or 6 months of FOLFOX plus atezolizumab concurrently for 6 months, and then an additional 6 months of atezolizumab. These are both very fascinating ideas.
DR HENRY To go back to one of your original points, this 3 versus 6 months: the neuropathy is significantly less in those getting the 3 months?
DR HALLER It went to 3%.
DR HENRY We all see that is very bothersome to patients. Before we leave colorectal, I must ask about the right-sided versus left-sided colorectal cancer that we hear a lot about now. Could you comment on how right-sided is worse than left-sided, and do we understand why?
DR HALLER There are two things to consider. If you look back even to simple trials of 5-FU or biochemical modulated 5-FU from 20 years ago, there were clear differences showing worse prognosis in patients with right-sided tumors, so that’s one point to be made. It’s been consistently seen but never acted upon. Then, the explanation for it, possibly, is that the right colon and left colon are two biologically different organs – and they are. Embryologically, the right colon comes from the midgut and the left colon comes from the hindgut, and there were several presentations at ASCO and at prior meetings showing that when you look at different mutations, they differ between the right and left colons. The right-sided tumors are more MSI-high and more BRAF-mutated, left-sided mutations less so.
Then, people started analyzing many of the very large colon cancer trials, including the US trial CALB/SWOG C80405 and the FIRE-3 trials in Europe, where backbone chemotherapy of FOLFIRI or FOLFOX was given with either cetuximab or bevacizumab in RAS wild-type patients. For one study, C80405, they saw that for cetuximab, on the right side, the median survival was 16.7 months and on the left side, it’s 36 months – a 20-month difference. In fact, if you look at the totality of the data, 16.7 almost looks like cetuximab is harming them, as if you were giving it to a RAS-mutated patient, but they were not. They were all RAS wild-type.
For bevacizumab, the right side was 24 months; the left side was 31.4 months. If you look at the left, cetuximab was 36 months and bevacizumab was 31.4 months, so it appears left-sided tumors should get more cetuximab than they are now getting in the US with a 5-month difference, but that decrement is much different on the right, where there’s an 8-month benefit for bevacizumab compared with cetuximab. There is a very good review by Dirk Arnold, who looked at a totality of 6 studies to really examine this more carefully.2
The National Comprehensive Cancer Network has chimed in on this, and is suggesting that for the 25% of people who have right-sided tumors, epidermal growth factor receptor (EGFR) agents not be considered in first-line therapy. NCCN did not go as far to say that EGFR agents should be given on the left side. As I said, the differences are much more impressive in the right, so this is a real sea change for people to consider which side of the tumor affects outcome.
Deb Schrag (Dana-Farber Cancer Institute) presented data at last year’s ASCO not only for stage IV disease showing the same thing, but also stage III disease where there are also right-versus-left differences in terms of recurrence, with a hazard ratio on the right side of about 1.4 compared with the left-sided tumors. Maybe it should be true that 3 months is especially good if you’re treating left-sided tumors, and maybe the right-sided tumor needs to be also calculated with the factors we just talked about. These are two big changes in an area in which we literally haven’t made any change since FOLFOX was introduced a decade ago.
DR HENRY That’s really fascinating, and if not practice changing, then practice challenging. Staying with the mutations idea, in my patients, I’m checking the RAS family and the BRAF mutation, where I’ve learned that’s a particularly bad mutation. I wonder if you might comment on the Kopetz trial, which took a cohort of BRAF mutants and treated them (Figure 2).3 How did that turn out?
DR HALLER It turned out well. We’re turning colon cancer into non–small cell lung cancer in that we’re getting small groups of patients who now have very dedicated care. The backstory here is that there was some thought that you should be treating mutations, not tumor sites. Drugs such as vemurafenib, for example, which is a BRAF inhibitor, worked well in melanoma for the same mutation that’s in colon cancer, V600E. But when vemurafenib was used in the BRAF-mutant patients – these are 10% of the population – median survivorship was one-third that of the rest of the patients, so roughly 12 months. People looked like they were doing worse when vemurafenib was used. They had no benefit.
Scott Kopetz at MD Anderson (Houston, Texas) is a very good bench-to-bed-and-back sort of doc. He looked at this in cell lines and found that when you give a BRAF inhibitor, you upregulate EGFR so you add an EGFR inhibitor. He did a phase 1 and 1B study, and then in the co-operative groups, a study was done – a randomized phase 2 trial for people who had the BRAF-V600E mutation failing first-line therapy, and then went on to receive either irinotecan single agent or irinotecan plus cetuximab or a triple arm of irinotecan, cetuximab, and vemurafenib. There was a crossover, and so the primary endpoint was progression-free survival. It accrued rapidly.
Again, small study, about 100 patients, but for the double-agent arm, or cetuximab–irinotecan, the median survivorship was 2 months. It was 4.4 months for the combination, so more than double. The response rate quadrupled from 4% to 16%, and the people who had disease control tripled, from 22% to 67%. Many of these patients had bulky disease, BRAF mutations. They need response, so this is a very important endpoint.
Overall survival was not different, in part because it was a crossover, and the crossover patients did pretty well. This is going to move more toward first-line therapy, because we don’t talk about fourth- and fifth-line therapies, TAS-102 or regorafenib. These patients don’t make it to even third line. We’re chipping away at what we think is a very homogenous group of peoples’ metastatic disease. They’re obviously not.
DR HENRY In the BRAF-mutant patient, the vemurafenib might drive them toward EGFR, and then the cetuximab could come in and handle that diversion of the pathway. Fascinating.
DR HALLER The preferred regimen in first-line therapy for a BRAF mutant might be FOLFIRI, cetuximab, and vemurafenib, especially on the left side.
DR HENRY Certainly makes sense. We’ll continue the theme at ASCO of “new standard of care.” Let’s move to gastroesophageal junction. There was a so-called FLOT (5-FU, leucovorin, oxaliplatin, Taxotere) presentation in the neoadjuvant/adjuvant setting, 4 cycles preoperatively and 4 cycles postoperatively. Could you comment on that study?
DR HALLER Gastric cancer for metastatic disease has a very large buffet of treatment regimens, and some just become entrenched, like the ECF regimen with epirubicin (epirubicin, cisplatin, 5-FU), where most people don’t exactly know what the contribution of that drug is, and so some people use EOX (epirubicin, oxaliplatin, capecitabine), some people use FOLFOX, some people use FOLFIRI. It gets a little bit confusing as to whether you use taxanes, platinums, or 5-FU or capecitabine.
The Germans came up with a regimen called FLOT – it’s sort of like FOLFOX with Taxotere attached. They did a very large study comparing it with ECF or ECX (epirubicin, cisplatin, capecitabine; Figure 3).4 The overall endpoint with over 700 patients was survival. This is an adjuvant regimen. Only 37% of people got ECF or ECX postoperatively, and 50% of the FLOT patients got the regimens postoperatively.
One of the reasons FLOT might be more beneficial is that more people were given postoperative treatment, and it’s one reason why many adjuvant regimens are being moved completely preoperatively, because so few people get the planned treatment. The FLOT regimen improved overall survival with a P value of .0112 and a hazard ratio of 0.77. The difference was 35 months versus 50 months. With the uncertainty as to what epirubicin actually does and the fact that it’s been around for a while and that fewer people receive postoperative treatment, with that 15-month benefit, if you’re using chemotherapy alone, and there’s no radiotherapy component for true gastric cancer, this is a new standard of care.
DR HENRY I struggle with this in my patients as well. This concept of getting more therapy preoperatively to those who can’t get it postoperatively certainly resonates with most of us in practice.
DR HALLER If I were redesigning the trial, I would probably say just give 4-6 cycles of treatment, and give it all preoperatively. In rectal cancer, there’s the total neoadjuvant approach, where it’s being tested in people who get all their chemotherapy first, then chemoradiotherapy, then surgery, and you’re done.
DR HENRY Yes, right. Thank you for mentioning that. Staying with the gastric GE junction, you couldn’t get away from ASCO this year without hearing about the checkpoint inhibitor immunotherapies in this population. In the CHECKMATE-142 trial with nivolumab versus placebo, response rates were good, especially in the MSI-high (microsatellite instability). Could you comment on that study?
DR HALLER We already know that in May and July 2017, pembrolizumab and nivolumab were both approved for any MSI-high solid tumor based on phase 2 data only, and based on response. That’s the first time we’ve seen that happen. It’s remarkable. For nivolumab, the approval was based on 53 patients with MSI-high metastatic colon cancer. So these were people who failed standard therapy and got nivolumab by standard infusion every 2 weeks. The overall response rate was almost 30% in this population, which is typically quite resistant to any treatment, so one expects much lower response rates with anything in that setting – chemotherapy, TAS-102, regorafenib, et cetera (Table).5
More importantly, as we’re seeing with Jimmy Carter with checkpoint treatment (for melanoma that had metastasized to the brain), responses lasted for more than 6 months in about two-thirds of patients, even a complete response, so this is just off the wall. I mean, this is not what you would expect with almost any other treatment. The data are the same for atezolizumab and for pembrolizumab. What seems to be true is that in the GI tumors and colon cancer, MSI-high seems more important than expression of PD-1 or PD-L1 (programmed cell death protein-1 or programmed cell death protein-ligand 1).
In different tumor sites, PD-1 or PD-L1 measurement may be important, but in these tumors, and in colorectal cancer, it looks as if MSI-high is the preferred measurement. Recently ASCO, together with the American Society for Clinical Pathology, College of American Pathologists, and Association for Molecular Pathology, came out with guidelines on what you should measure in colorectal cancer specimens. Obviously, one is extended RAS. They say you should get BRAF for prognosis, but it may also be a prognostic factor that leads you to treat, which ultimately makes it a predictive factor, so the data from Kopetz might suggest that will move up to something you also must measure. If patients have the BRAF mutation, it’s important they know that it’s a poor prognostic sign. But if they come in with literature saying they might live 36 months when their actual outcome is about a third of that, you need to frame your discussion in that regard and make sure they understand it.
The guidelines also suggested getting MSI-high, and certainly prognostically in early-stage disease, but now it’s going to be a predictive factor, so in the month in which these recommendations are made, two of them are already out of date. They also didn’t include human epidermal growth factor receptor 2 (HER2), and what we’ve heard from the HERACLES (HER2 Amplification for Colorectal Cancer Enhanced Stratification) trial is that for those patients who got the trastuzumab and pertuzumab combination – and this is another 5% of patients – almost the same data was seen as in the MSI-high patients with checkpoint inhibitors. That is double-digit response rates and durable responses. As I said, we’re very much nearing in colorectal cancer what’s now being done in non-small cell lung cancer.
DR HENRY Indeed. Could you comment on the BILCAP study and adjuvant capecitabine for biliary tract cancer?
DR HALLER There are large meta-analyses looking at adjuvant therapy for biliary tract cancers typically from fairly small, fairly old studies that all suggest that in certain stages of resected biliary tumors, either bile duct or gall bladder, adjuvant treatment works, and typically either chemotherapy and radiotherapy, or chemotherapy alone, but not radiotherapy alone.
Capecitabine has been used for metastatic disease for years, mostly by default, and because most GI tumors have some response to fluoropyridines. But we’re finally able now to do large trials in biliary tumors, so this trial was a very large study with almost 450 patients from the United Kingdom over an 8-year period. About 20% were gallbladder, so the R0 surgery was about 60%, R1 at about 40% (Figure 4).6
The endpoint of the study was survival advantage, and when they did the protocol analysis, the survival for the treated population was 53 months and for the observation arm, 36 months, so that was a hazard ratio of 0.75, which is acceptable in an adjuvant study. It’s simple drug to give, and usually tolerable, so this will represent a new standard of care. Of course, in the advanced disease setting, the gemcitabine–cisplatin combination is the standard of care for metastatic disease. It’s a little more toxic combination, but we know that’s standard. There’s an ongoing study in Europe called the ACTICCA-1 trial, and this is gemcitabine–cisplatin for 6 months versus not capecitabine, but a control arm. My guess is if the capecitabine study was positive, that this also will be a positive trial, because gemcitabine–cisplatin is probably more active. Then, we’ll have 2 standards, and I don’t think anyone is going to compare capecitabine with gemcitabine–cisplatin.
What you’ll have are two regimens for two different populations of patients. Perhaps for the elderly and people who have renal problems, capecitabine alone will give them benefit, and then you’ll have gemcitabine–cisplatin, which may be just a more toxic regimen, but also more effective for the younger, healthier people with fewer comorbidities.
DR HENRY Great data and a small population, but a population in need. That moves us on to pancreatic cancer, and I don’t know if this is happening nationwide, but in my practice, I’m seeing more. These patients tend to present beyond surgery, so they have metastatic or advanced pancreatic cancer. Any comment on where you think this field is going?
DR HALLER We were a bit bereft of new pancreatic cancer studies at ASCO this year. We’re certainly looking more at neoadjuvant therapy for pancreatic cancer, primarily because of ease of administration and the increased ability to tolerate treatments in the preoperative setting. There aren’t many people that get downstaged, but some are. Unfortunately, even in the MSI-high pancreas, which is a small subset, they don’t seem to get as big a bang out of the checkpoint inhibitors as in other tumor sites, so I’m afraid I didn’t come home with much new about this subset of patients.
DR HENRY We’ve covered a nice group of studies and practice-changing new standard-of-care comments from ASCO and other studies. Thank Dr Dan Haller for being with us and commenting. This podcast and discussion are brought to you from
1. Andre T, Bonnetain F, Mineur L, et al. Oxaliplatin-based chemotherapy for patients with stage III colon cancer: disease free survival results of the three versus six months adjuvant IDEA France trial. Abstract presented at: 2017 American Society of Clinical Oncology Annual Meeting; June 2-6, 2017; Chicago, IL. Abstract 3500.
2. Arnold D, Lueza B, Douillard JY, et al. Prognostic and predictive value of primary tumour side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann Oncol. 2017;28(8):1713-1729.
3. Kopetz S, McDonough SL, Lenz H-J, et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG S1406). Abstract presented at: 2017 American Society of Clinical Oncology Annual Meeting; June 2-6, 2017; Chicago, IL. Abstract 3505.
4. Al-Batran S-E, Homann N, Schmalenberg H, et al. Perioperative chemotherapy with docetaxel, oxaliplatin. Abstract presented at: 2017 American Society of Clinical Oncology Annu, and fluorouracil/leucovorin (FLOT) versus epirubicin, cisplatin, and fluorouracil or capecitabine (ECF/ECX) for resectable gastric or gastroesophageal junction (GEJ) adenocarcinoma (FLOT4-AIO): a multicenter, randomized phase 3 trial. Abstract presented at: 2017 American Society of Clinical Oncology Annual Meeting; June 2-6, 2017; Chicago, IL. Abstract 4004.
5. Kopetz S, Lonardi S, McDermott RS, et al. Concordance of DNA mismatch repair deficient (dMMR)/microsatellite instability (MSI) assessment by local and central testing in patients with metastatic CRC (mCRC) receiving nivolumab (nivo) in Checkmate 142 study. Abstract presented at: 2017 American Society of Clinical Oncology Annual Meeting; June 2-6, 2017; Chicago, IL. Abstract 3548.
6. Primrose JN, Fox R, Palmer DH, et al. Adjuvant capecitabine for biliary tract cancer: the BILCAP randomized study. Abstract presented at: 2017 American Society of Clinical Oncology Annual Meeting; June 2-6, 2017; Chicago, IL. Abstract 4006.
1. Andre T, Bonnetain F, Mineur L, et al. Oxaliplatin-based chemotherapy for patients with stage III colon cancer: disease free survival results of the three versus six months adjuvant IDEA France trial. Abstract presented at: 2017 American Society of Clinical Oncology Annual Meeting; June 2-6, 2017; Chicago, IL. Abstract 3500.
2. Arnold D, Lueza B, Douillard JY, et al. Prognostic and predictive value of primary tumour side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann Oncol. 2017;28(8):1713-1729.
3. Kopetz S, McDonough SL, Lenz H-J, et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG S1406). Abstract presented at: 2017 American Society of Clinical Oncology Annual Meeting; June 2-6, 2017; Chicago, IL. Abstract 3505.
4. Al-Batran S-E, Homann N, Schmalenberg H, et al. Perioperative chemotherapy with docetaxel, oxaliplatin. Abstract presented at: 2017 American Society of Clinical Oncology Annu, and fluorouracil/leucovorin (FLOT) versus epirubicin, cisplatin, and fluorouracil or capecitabine (ECF/ECX) for resectable gastric or gastroesophageal junction (GEJ) adenocarcinoma (FLOT4-AIO): a multicenter, randomized phase 3 trial. Abstract presented at: 2017 American Society of Clinical Oncology Annual Meeting; June 2-6, 2017; Chicago, IL. Abstract 4004.
5. Kopetz S, Lonardi S, McDermott RS, et al. Concordance of DNA mismatch repair deficient (dMMR)/microsatellite instability (MSI) assessment by local and central testing in patients with metastatic CRC (mCRC) receiving nivolumab (nivo) in Checkmate 142 study. Abstract presented at: 2017 American Society of Clinical Oncology Annual Meeting; June 2-6, 2017; Chicago, IL. Abstract 3548.
6. Primrose JN, Fox R, Palmer DH, et al. Adjuvant capecitabine for biliary tract cancer: the BILCAP randomized study. Abstract presented at: 2017 American Society of Clinical Oncology Annual Meeting; June 2-6, 2017; Chicago, IL. Abstract 4006.