User login
Autoimmune Hemolytic Anemia: Treatment of Common Types
Autoimmune hemolytic anemia (AIHA) is mediated by antibodies, and in most cases immunoglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C. AIHA caused by a drug reaction is rare, with an estimated annual incidence of 1:1,000,000 for severe drug-related AIHA.1 This article reviews the management of the more common types of AIHA, with a focus on warm, cold, and drug-induced AIHA; the evaluation and diagnosis of AIHA is reviewed in a separate article.
Warm Autoimmune Hemolytic Anemia
In AIHA, hemolysis is mediated by antibodies that bind to the surface of red blood cells. AIHA in which IgG antibodies are the offending antibodies is referred to as warm AIHA. “Warm” refers to the fact that the antibody binds best at body temperature (37°C). In warm AIHA, testing will show IgG molecules attached to the surface of the red cells, with 50% of patients also showing C3. Between 50% and 90% of AIHA cases are due to warm antibodies.2,3 The incidence of warm AIHA varies by series but is approximately 1 case per 100,000 patients per year; this form of hemolysis affects women more frequently than men.4,5
Therapeutic Options
First Line
Steroids. The goal of therapy in warm AIHA can be hard to define. However, most would agree that a hematocrit above 30% (or higher to prevent symptoms) with a minimal increase in the reticulocyte count—reflective of a significantly slowed hemolytic process—is a reasonable goal. Initial management of warm AIHA is prednisone at a standard dose of 1 mg/kg daily (Table 1).6,7 Patients should be also started on proton-pump inhibitors to prevent ulcers. It can take up to 3 weeks for patients to respond to prednisone therapy. Once the patient’s hematocrit is above 30%, the prednisone is slowly tapered. Although approximately 80% of patients will respond to steroids, only 30% can be fully tapered off steroids. For patients who can be maintained on a daily steroid dose of 10 mg or less, steroids may be the most reasonable long-term therapy. In addition, because active hemolysis leads to an increased demand for folic acid, patients with warm AIHA are often prescribed folic acid 1 mg daily to prevent megaloblastic anemia due to folic acid deficiency.

Rituximab. Increasingly, rituximab (anti-CD20) therapy is added to the initial steroids. Two clinical trials showed both increased long-term and short-term responses with the use of rituximab.8,9 An important consideration is that most patients respond gradually to rituximab over weeks, so a rapid response should not be expected. Most studies have used the traditional dosing of 375 mg/m2 weekly for 4 weeks. These responses appear to be durable, but as in immune thrombocytopenia (ITP), repeat treatment with rituximab is effective.
The major side effects of rituximab are infusion reactions, which are often worse with the first dose. These reactions can be controlled with antihistamines, steroids, and, for severe rigors, meperidine. Rarely, patients can develop neutropenia (approximately 1:500) that appears to be autoimmune in nature. Infections appear to be only minimally increased with the use of rituximab.10 One group at risk is chronic carriers of hepatitis B virus, who may experience a reactivation of the virus that can be fatal. Thus, patients being considered for rituximab need to be screened for hepatitis B virus carrier state.11 Patients receiving rituximab are at very slight risk for progressive multifocal leukoencephalopathy, which is more common in patients with cancer and in heavily immunosuppressed patients. The overall risk is unknown but is less than 1:50,000.
Second Line
Splenectomy. For patients who cannot be weaned from steroids or in whom steroid therapy fails, there is no standard therapy. Currently, the 2 main choices are splenectomy or rituximab (anti-CD20) therapy if the patient did not receive it first line. Splenectomy is the classic therapy for warm AIHA. Reported response rates in the literature range from 50% to 80%, with 50% to 60% remaining in remission.12-16 Timing of the procedure is a balance between allowing time for the steroids to work and the risk of toxicity of steroids. In a patient who has low presurgical risk and has either refractory disease or cannot be weaned from high doses of steroids, splenectomy should be done sooner. Splenectomy can be delayed or other therapy tried first in patients who require lower doses of steroids or have medical risk factors for surgery. Most splenectomies are performed via laparoscopy. The small incisions allow for quicker healing, and the laparoscopic approach provides better visualization of the abdomen to find and remove accessory spleens. When splenectomy is performed by experienced surgeons, the mortality rate is low (< 0.5%).17
The most concerning complication of splenectomy is overwhelming post-splenectomy infection (OPSI).18 In adults, the spleen appears to play a minimal role in immunity except for protecting against certain encap-sulated organisms. Splenectomized patients infected with an encapsulated organism (eg, Pneumococcus) will develop overwhelming sepsis within hours. These patients will often have disseminated intravascular coagulation and will rapidly progress to purpura fulminans. Approximately 40% to 50% of patients will die of sepsis even when the infection is detected early. The overall lifetime risk of sepsis may be as high as 1:500. The organism that most commonly causes sepsis in splenectomized patients is Streptococcus pneumoniae, reported in over 50% of cases. Neisseria meningitidis and Haemophilus influenzae have also been implicated in many cases.19 Overwhelming sepsis after dog bites has been reported due to Capnocytophaga canimorsus infections. Patients are also at increased risk of developing severe malaria and severe babesiosis.18
Patients who have undergone splenectomy need to be warned about the risk of OPSI and instructed to report to the emergency department readily if they develop a fever greater than 101°F (38.3°C) or shaking chills. Once in the emergency department, blood cultures should be obtained rapidly and the patient started on antibiotic coverage with vancomycin and ceftriaxone (or levofloxacin if allergic to beta-lactams).20 For patients in remote areas, some physicians will prescribe prophylactic antibiotics to take while they are traveling to a health care provider or even recommend a “standby” antibiotic dose to take while traveling to health care.5 This usually consists of amoxicillin or a macrolide for penicillin-allergic patients.
Patients in whom splenectomy is being planned or considered should be vaccinated for pneumococcal, meningococcal, and influenza infections (Table 2).18 If there is a plan to treat with rituximab, patients should first be vaccinated since they will not be able to mount an immune response after being treated with rituximab.

Third Line
The therapeutic options for patients who do not respond to either splenectomy or rituximab are much less certain.5,6 Although intravenous immune globulin is a standard therapy for ITP, response rates are low in warm AIHA.17 Numerous therapies have been reported in small series, but no clear approach has emerged. Options include azathioprine, cyclophosphamide, mycophenolate, cyclosporine, danazol, and alemtuzumab. Our approach has been to use mycophenolate for patients requiring high doses of steroids or transfusions. Patients who respond to lower doses of steroids may be good candidates for danazol to help wean them off steroids.
Treatment of Warm AIHA with Associated Diseases
Warm AIHA can complicate several diseases. Patients with systemic lupus erythematosus (SLE) can develop warm AIHA as part of their disease complex. The initial treatment approach is the same, but data suggest that splenectomy may not be as effective.13,17 Also, many SLE patients have complex medical conditions, making surgeries riskier. For SLE patients who are refractory or cannot be weaned from steroids, rituximab may be the better choice. Babesiosis, particularly in asplenic patients, has been associated with the development of AIHA.21,22
Of the malignances associated with AIHA, chronic lymphocytic leukemia (CLL) has the strongest association.4,23 Series show that 5% to 10% of patients with CLL will have warm AIHA. AIHA can appear concurrent with CLL or develop during the course of the disease. The introduction of purine analogs such as fludarabine led to a dramatic increase in the incidence of warm AIHA in treated patients.24 It is speculated that these powerful agents reduce the number and effectiveness of T cells that hold in check the autoantibody response, leading to warm AIHA.25 However, when these purine analogs are used in com-bination with agents such as cyclophosphamide or rituximab (with their immunosuppressive effects), the rates of warm AIHA have been lower.23
The approach to patients with CLL and warm AIHA depends on the state of their CLL.23 For patients who have low-stage CLL that does not need treatment, the standard approach to warm AIHA should be steroids, splenectomy, and rituximab.24 For patients with higher-stage CLL, the treatment for the leukemia will often provide therapy for the warm AIHA. The combination of rituximab-cyclophosphamide-dexamethasone has been reported to be effective for both the AIHA and CLL components.26 The use of ibrutinib has also been reported to be effective.27
A rare but important variant of warm AIHA is Evans syndrome.28,29 This is the combination of AIHA and ITP. Approximately 1% to 3% of AIHA cases are the Evans variant. The ITP can precede, be concurrent with, or develop after the AIHA. The diagnosis of Evans syndrome should raise concern for underlying disorders. In young adults, immunodeficiency disorders such as autoimmune lymphoproliferative disease (ALPS) need to be considered. In older patients, Evans syndrome is often associated with T cell lymphomas. The sparse literature on Evans syndrome suggests that it can be more refractory to standard therapy, with response rates to splenectomy in the 50% range.28,30 In patients with lymphoma, antineoplastic therapy is crucial. There is increasing data showing that mycophenolate or sirolimus may be effective for patients with ALPS in whom splenectomy or rituximab therapy is unsuccessful.31
Warm AIHA with IgA or IgM Antibodies
In rare patients with warm AIHA, IgA or IgM is the implicated antibody. The literature suggests that patients with IgA AIHA may have more severe hemolysis.32 Patients with IgM AIHA often have a severe course with a fatal outcome.33 In such cases, the patient’s plasma may show spontaneous hemolysis and agglutination. The DAT may not be strongly positive or may show C3 reactivity only. The clinical clues are C3 reactivity with no cold agglutinins and severe hemolysis, sometimes with an intravascular component. Treatment is the same as for warm AIHA, including the use of rituximab.34
Cold Autoimmune Hemolytic Anemia
In cold AIHA, the hemolysis is mediated by IgM antibodies directed against red cells.35 As discussed earlier, the term “cold” refers to the fact that the antibody binds maximally at temperatures below 37°C. The most efficient temperature for binding is called the “thermal amplitude,” and, in theory, the higher the thermal amplitude, the more virulent the antibody. An antibody titer can be calculated at each reaction temperature from 4°C to 37°C by serial dilutions of the patient’s serum prior to incubating with reagent red cells. Rarely, the IgM can fix complement rapidly, leading to intravascular hemolysis. In most patients, complement is fixed through C3, and the C3-coated red cells are taken up by macrophages in the mononuclear phagocyte system, primarily in the liver.3
The DAT in patients with cold AIHA will show cells coated with C3. The blood smear will often show ag-glutination of the blood, and if the blood cools before being analyzed, the agglutination will interfere with the analysis. Titers of cold agglutinin can range from 1:1000 to over 1 million. The IgM autoantibodies are most often directed against the I/i antigens on the red blood cell membrane, with 90% against I antigen.35 The I antigen specificity is typical with primary cold agglutinin disease and after Mycoplasma infection. The i antigen specificity is most typical of Epstein-Barr virus and cytomegalovirus infections in children. In young patients, cold AIHA often occurs following an infection, including viral and Mycoplasma infections, and the course is self-limited.35,36 The hemolysis usually starts 2 to 3 weeks after the illness and will last for 4 to 6 weeks. In older patients, the etiology in over 90% of cases is a B-cell lymphoproliferative disorder, usually with monoclonal kappa B-cells.37 The most common disorders are marginal zone lymphoma, small lymphocytic lymphoma, and lymphoplasmacytic lymphoma.3
Therapeutic Options
It is important to diagnose cold AIHA because the standard therapy for warm AIHA (steroids) is ineffective in cold AIHA. Because C3-coated red cells are taken up primarily in the liver, removing the spleen is also an ineffective therapy. Simple measures to help with cold AIHA should be employed.37 Patients should be kept in a warm environment and should try to avoid the cold. If transfusions are needed, they should be infused via blood warmers to prevent hemolysis. In rare patients with severe hemolysis, therapeutic plasma exchange (TPE) can be considered.38 Given that the culprit antibody is IgM—mostly intravascular—use of TPE may slow the hemolysis to give time for other therapies to take effect.
Treatment of cold AIHA remains difficult (Table 3). Because most patients with primary AIHA have underlying lymphoproliferative diseases, chlorambucil has been used in the past to treat cold AIHA. However, responses were rare and the drug could worsen the anemia.38 Currently, the drug of choice is rituximab. Response is seen in 45% to 75% of patients, but is almost always a partial response and retreatment is often necessary.17,37,39 As with other autoimmune hematologic diseases, there can be a delay in response that ranges from 2 weeks to 4 months (median time, 1.5 months).37 Given the lack of robust response (complete and durable) with rituximab, the Berentsen group has explored adding bendamustine to rituximab.40 In a prospective trial, 71% of patients responded with a 40% complete response rate. Therapy was well tolerated, with only 29% of patients needing dose reduction Although more toxic, this combination can be considered in patients with aggressive disease. A small study of the use of bortezomib showed a good response in one-third of patients.41 There is increasing use of the C5 complement inhibitor eculizumab to halt the hemolysis, but further study of this agent is also required.36,42,43 Blockers of C1s complement, which block hemolysis by preventing complement activation, are currently being studied in clinical trials.44

Since most patients with cold AIHA are older, a frequent issue that must be considered is cardiac surgery. The concern is that the hypothermia involved with most heart bypass procedures will lead to agglutination and hemolysis. The development of normothermic bypass has expanded treatment options. A recommended approach in patients who have known cold agglutinins is to measure the thermal amplitude of the antibody preoperatively. If the thermal amplitude is above 18°C, normothermic bypass should be done, if feasible.45 If not feasible, preoperative TPE should be considered.
Paroxysmal Cold Hemoglobinuria
A unique cold AIHA is paroxysmal cold hemoglobinuria (PCH).3,46 This cold hemolytic syndrome most often occurs in children following a viral infection, but in the past it complicated any stage of syphilis.47 The mediating antibody in PCH is an IgG antibody directed against the P antigen on the red blood cell. This antibody binds best at temperatures below 37°C, fixing complement at cold temperatures, but then activates the complement cascade at body temperature.48 Because this antibody can fix complement, hemolysis can be rapid and severe, leading to extreme anemia. The DAT is often weakly positive but can be negative. The diagnostic test for PCH is the Donath–Landsteiner test. This complex test is performed by incubating 3 blood samples, 1 at 0° to 4°C, another at 37°C, and a third at 0° to 4°C and then at 37°C. The diagnosis of PCH is made if only the third tube shows hemolysis.35 PCH can persist for 1 to 3 months but is almost always self-limiting. In severe case, steroids may be of benefit.
Drug-Induced Hemolytic Anemia
AIHA caused by a drug reaction is rare, with a lower incidence than drug-related ITP. The rate of severe drug-related AIHA is estimated at 1:1,000,000, but less severe cases may be missed.1 Most patients will have a positive DAT without signs of hemolysis, but in rare cases patients will have relentless hemolysis resulting in death.
Mechanisms
Multiple mechanisms for drug-induced immune hemolysis have been proposed, including drug-absorption (hapten-induced) and immune complex mechanisms.1,49 The hapten mechanism is most often associated with the use of high-dose penicillin.50 High doses of penicillin or similar drugs such as piperacillin lead to incorporation of the drug into the red cell membrane by binding to proteins. Patients will manifest a positive DAT with IgG antibody but not complement.51 The patient’s plasma will be reactive only with penicillin-coated red cells and not with normal cells. As mentioned, very few patients will have hemolysis, and if they have hemolysis, it will resolve in a few days after discontinuation of the offending drug.52
Binding of a drug-antibody complex to the red cell membrane may cause hemolysis via the immune com-plex mechanism.53 Again, most often the patient will have just a positive DAT, but rarely patients will have life-threatening hemolysis upon exposure or reexposure to the drug. The onset of hemolysis is rapid, with signs of acute illness and intravascular hemolysis. The paradigm drug is quinine, but many other drugs have been implicated. Testing shows a positive Coombs test with anti-complement but not anti-IgG.50 This pattern is due to the effectiveness of the tertiary complex at fixing complement. The patient’s plasma reacts with red cells only when the drug is added.
A form of immune complex hemolysis associated with both disseminated intravascular coagulation (DIC) and brisk hemolysis has been recognized. Patients who receive certain second- and third-generation cephalosporins (especially cefotetan and ceftriaxone.53,54) have developed this syndrome.50,55-59 The clinical symptoms start 7 to 10 days after the drug is administered; often the patient has only received the antibiotic for surgical prophylaxis. Immune hemolysis with acute hematocrit drop, hypotension, and DIC ensues. The patients are often believed to have sepsis and are often reexposed to the cephalosporin, resulting in worsening of the clinical status. The outcome is often fatal due to massive hemolysis and thrombosis.56,60,62
Finally, 8% to 36% of patients taking methyldopa will develop a positive DAT after 6 months of therapy, with less than 1% showing hemolysis.52,63 The hemolysis in these patients is indistinguishable from warm AIHA, consistent with the notion that methyldopa induces an AIHA. The hemolysis often resolves rapidly after the methyldopa is stopped, but the Coombs test may remain positive for months.63 This type of drug-induced hemolytic anemia has been reported with levodopa, procainamide, and chlorpromazine, but fludarabine is the most common cause currently. This form of AIHA is now being seen with increased use of checkpoint inhibitors.64
Diagnosis
In many patients, the first clue to the presence of drug AIHA is the finding of a positive DAT. Rarely, patients will have severe hemolysis, but in many patients the hemolytic process is mild and may be wrongly assumed to be part of the underlying illness. Finding the offending drug can be a challenge, unless a patient has recently started a new drug; in a hospitalized patient on multiple agents, identifying the problem drug is difficult. Patients recently started on “suspect drugs,” especially the most common agents cefotetan, ceftriaxone, and piperacillin, should have these agents stopped (Table 4).1,49,65 Specialty laboratories such as the Blood Center of Wisconsin or the Los Angeles Red Cross can perform in vitro studies of drug interactions that can confirm the clinical diagnosis of drug-induced AIHA.

Treatment
Therapy for patients with positive DAT without signs of hemolysis is uncertain. If the drug is essential, then the patient can be observed. If the patient has hemolysis, the drug needs to be stopped and the patient observed for signs of end-organ damage. It is doubtful that steroids or other autoimmune-directed therapy is effective. For patients with the DIC-hemolysis syndrome, there are anecdotal reports that TPE may be helpful.1
Summary
AIHA can range from an abnormal laboratory test (positive DAT and signs of hemolysis) to an acute, life-threatening illness. Treatment is guided by the laboratory work-up and evaluation of the patient’s clinical status. While rituximab is promising for many patients, the lack of robust clinical trials hinders the treatment of patients who fail standard therapies.
1. Garratty G. Immune hemolytic anemia associated with drug therapy. Blood Rev. 2010;24:143-150.
2. Ness PM. How do I encourage clinicians to transfuse mismatched blood to patients with autoimmune hemolytic anemia in urgent situations? Transfusion. 2006;46:1859-1862.
3. Berentsen S. Cold agglutinin disease. Hematology Am Soc Hematol Educ Program. 2016;2016:226-231.
4. Liebman HA, Weitz IC. Autoimmune hemolytic anemia. Med Clin North Am. 2017;101:351-359.
5. Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24:195–210.
6. Kyrle PA, Rosendaal FR, Eichinger S. Risk assessment for re¬current venous thrombosis. Lancet. 2010;376:2032-2039.
7. Go RS, Winters JL, Kay NE. How I treat autoimmune hemolytic anemia. Blood. 2017;129:2971-2979.
8. Birgens H, Frederiksen H, Hasselbalch HC, et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013;163:393-399
9. Michel M, Terriou L, Roudot-Thoraval F, et al. A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm auto-immune hemolytic anemia in adults (the RAIHA study). Am J Hematol. 2017;92:23-27.
10. Gea-Banacloche JC. Rituximab-associated infections. Semin Hematol. 2010;47:187-198.
11. Loomba R, Liang TJ. Hepatitis B reactivation associated with immune suppressive and biological modifier therapies: current concepts, management strategies, and future directions. Gastroenterology. 2017;152:1297-1309.
12. Coon WW. Splenectomy in the treatment of hemolytic anemia. Arch Surg. 1985;120:625-628.
13. Akpek G, McAneny D, Weintraub L. Comparative response to splenectomy in coombs-positive autoimmune hemolytic anemia with or without associated disease. Am J Hematol. 1999;61:98-102.
14. Patel NY, Chilsen AM, Mathiason MA, et al. Outcomes and complications after splenectomy for hematologic disorders. Am J Surg. 2012;204:1014-1020.
15. Crowther M, Chan YL, Garbett IK, et al. Evidence-based focused review of the treatment of idiopathic warm immune hemolytic anemia in adults. Blood. 2011;118:4036-4040.
16. Giudice V, Rosamilio R, Ferrara I, et al. Efficacy and safety of splenectomy in adult autoimmune hemolytic anemia. Open Med (Wars). 2016;11:374-380.
17. Lechner K, Jager U. How I treat autoimmune hemolytic anemias in adults. Blood. 2010;116:1831-1838.
18. Rodeghiero F, Ruggeri M. Short- and long-term risks of splenectomy for benign haematological disorders: should we revisit the indications? Br J Haematol. 2012;158:16-29.
19. Ahmed N, Bialowas C, Kuo YH, Zawodniak L. Impact of preinjury anticoagulation in patients with traumatic brain injury. South Med J. 2009;102:476-480.
20. Morgan TL, Tomich EB. Overwhelming post-splenectomy infection (OPSI): a case report and review of the literature. J Emerg Med. 2012;43:758-763.
21. Woolley AE, Montgomery MW, Savage WJ, et al. Post-babesiosis warm autoimmune hemolytic anemia. N Engl J Med. 2017;376:939-946.
22. Shatzel JJ, Donohoe K, Chu NQ, et al. Profound autoimmune hemolysis and Evans syndrome in two asplenic patients with babesiosis. Transfusion. 2015;55:661-665.
23. Hodgson K, Ferrer G, Montserrat E, Moreno C. Chronic lymphocytic leukemia and autoimmunity: a systematic review. Haematologica. 2011;96:752-761.
24. Hamblin TJ. Autoimmune complications of chronic lymphocytic leukemia. Semin Oncol. 2006;33:230-239.
25. Tertian G, Cartron J, Bayle C, et al. Fatal intravascular au¬toimmune hemolytic anemia after fludarabine treatment for chronic lymphocytic leukemia. Hematol Cell Ther. 1996;38:359-360.
26. Rossignol J, Michallet AS, Oberic L, et al. Rituximab-cyclophosphamide-dexamethasone combination in the management of autoimmune cytopenias associated with chronic lymphocytic leukemia. Leukemia. 2011;25:473-478.
27. Hampel PJ, Larson MC, Kabat B, et al. Autoimmune cytopenias in patients with chronic lymphocytic leukaemia treated with ibrutinib in routine clinical practice at an academic medical centre. Br J Haematol. 2018;183:421-427.
28. Michel M, Chanet V, Dechartres A, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood. 2009;114:3167-3172.
29. Jaime-Pérez JC, Aguilar-Calderón PE, Salazar-Cavazos L, Gómez-Almaguer D. Evans syndrome: clinical perspectives, biological insights and treatment modalities. J Blood Med. 2018;9:171-184.
30. Dhingra KK, Jain D, Mandal S, et al. Evans syndrome: a study of six cases with review of literature. Hematology. 2008;13:356-360.
31. Bride KL, Vincent T, Smith-Whitley K, et al. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial. Blood. 2016;127:17-28.
32. Sokol RJ, Booker DJ, Stamps R, et al. IgA red cell au¬toantibodies and autoimmune hemolysis. Transfusion. 1997;37:175-181.
33. Garratty G, Arndt P, Domen R, et al. Severe autoimmune hemolytic anemia associated with IgM warm autoantibodies directed against determinants on or associated with glycophorin A. Vox Sang. 1997;72:124-130.
34. Wakim M, Shah A, Arndt PA, et al. Successful anti-CD20 monoclonal antibody treatment of severe autoimmune hemolytic anemia due to warm reactive IgM autoantibody in a child with common variable immunodeficiency. Am J Hematol. 2004;76:152-155.
35. Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
36. Berentsen S, Tjonnfjord GE. Diagnosis and treatment of cold agglutinin mediated autoimmune hemolytic anemia. Blood Rev. 2012;26:107-115.
37. Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011;153:309-317.
38. King KE, Ness PM. Treatment of autoimmune hemolytic anemia. Semin Hematol. 2005;42:131136.
39. Barcellini W, Zaja F, Zaninoni A, et al. Low-dose rituximab in adult patients with idiopathic autoimmune hemolytic anemia: clinical efficacy and biologic studies. Blood. 2012;119:3691-3697.
40. Berentsen S, Randen U, Oksman M, et al. Bendamustine plus rituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood. 2017;130:537-541.
41. Rossi G, Gramegna D, Paoloni F, et al. Short course of bortezomib in anemic patients with relapsed cold agglutinin disease: a phase 2 prospective GIMEMA study. Blood. 2018;132:547-550.
42. Makishima K, Obara N, Ishitsuka K, et al. High efficacy of eculizumab treatment for fulminant hemolytic anemia in primary cold agglutinin disease. Ann Hematol. 2019;98:1031-1032.
43. Roth A, Huttmann A, Rother RP, et al. Long-term efficacy of the complement inhibitor eculizumab in cold agglutinin disease. Blood. 2009;113:38853886.
44. Jäger U, D'Sa S, Schörgenhofer C, et al. Inhibition of complement C1s improves severe hemolytic anemia in cold agglutinin disease: a first-in-human trial. Blood. 2019;133:893-901.
45. Agarwal SK, Ghosh PK, Gupta D. Cardiac surgery and cold-reactive proteins. Ann Thorac Surg. 1995;60:1143-1150.
46. Shanbhag S, Spivak J. Paroxysmal cold hemoglobinuria. Hematol Oncol Clin North Am. 2015;29:473-478.
47. Kumar ND, Sethi S, Pandhi RK. Paroxysmal cold haemoglobinuria in syphilis patients. Genitourin Med. 1993;69:76.
48. Zantek ND, Koepsell SA, Tharp DR Jr, Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. 2012;87:707-709.
49. Pierce A, Nester T. Pathology consultation on drug-induced hemolytic anemia. Am J Clin Pathol. 2011;136:7-12.
50. Garratty G. Immune cytopenia associated with antibiotics. Transfusion Med Rev. 1993;7:255-267.
51. Petz LD, Mueller-Eckhardt C. Drug-induced immune hemolytic anemia. Transfusion. 1992;32:202-204.
52. Packman CH, Leddy JP. Drug-related immune hemolytic anemia. In: Beutler E, Lichtman MA, Coller BS, Kipps TJ, eds. William’s Hematology. 5th ed. New York: McGraw-Hill; 1995:691-704.
53. Garratty G. Drug-induced immune hemolytic anemia. Hematology Am Soc Hematol Educ Program. 2009;73-79.
54. Leicht HB, Weinig E, Mayer B, et al. Ceftriaxone-induced hemolytic anemia with severe renal failure: a case report and review of literature. BMC Pharmacol Toxicol. 2018;19:67.
55. Chenoweth CE, Judd WJ, Steiner EA, Kauffman CA. Cefotetan-induced immune hemolytic anemia. Clin Infect Dis. 1992;15:863-865.
56. Garratty G, Nance S, Lloyd M, Domen R. Fatal im¬mune hemolytic anemia due to cefotetan. Transfusion. 1992;32:269-271.
57. Endoh T, Yagihashi A, Sasaki M, Watanabe N. Ceftizoxime-induced hemolysis due to immune complexes: case report and determination of the epitope responsible for immune complex-mediated hemolysis. Transfusion. 1999;39:306-309.
58. Arndt PA, Leger RM, Garratty G. Serology of antibodies to second- and third-generation cephalosporins associated with immune hemolytic anemia and/or positive direct antiglobulin tests. Transfusion. 1999;39:1239-1246.
59. Martin ME, Laber DA. Cefotetan-induced hemolytic anemia after perioperative prophylaxis. Am J Hematol. 2006;81:186-188.
60. Bernini JC, Mustafa MM, Sutor LJ, Buchanan GR. Fatal hemolysis induced by ceftriaxone in a child with sickle cell anemia. J Pediatr. 1995;126(5 Pt 1):813-815.
61. Borgna-Pignatti C, Bezzi TM, Reverberi R. Fatal ceftriaxone-induced hemolysis in a child with acquired immunodeficiency syndrome. Pediatr Infect Dis J. 1995;14:1116-1117.
62. Lascari AD, Amyot K. Fatal hemolysis caused by ceftriaxone [see comments]. J Pediatr. 1995;126(5 Pt 1):816-817.
63. Petz LD. Drug-induced autoimmune hemolytic anemia. Transfusion Med Rev. 1993;7:242-254.
64. Leaf RK, Ferreri C, Rangachari D, et al. Clinical and laboratory features of autoimmune hemolytic anemia associated with immune checkpoint inhibitors. Am J Hematol. 2019;94:563-574.
65. DeLoughery T. Drug induced immune hematological disease. Allerg Immunol Clin. 1998;18:829-841.
Autoimmune hemolytic anemia (AIHA) is mediated by antibodies, and in most cases immunoglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C. AIHA caused by a drug reaction is rare, with an estimated annual incidence of 1:1,000,000 for severe drug-related AIHA.1 This article reviews the management of the more common types of AIHA, with a focus on warm, cold, and drug-induced AIHA; the evaluation and diagnosis of AIHA is reviewed in a separate article.
Warm Autoimmune Hemolytic Anemia
In AIHA, hemolysis is mediated by antibodies that bind to the surface of red blood cells. AIHA in which IgG antibodies are the offending antibodies is referred to as warm AIHA. “Warm” refers to the fact that the antibody binds best at body temperature (37°C). In warm AIHA, testing will show IgG molecules attached to the surface of the red cells, with 50% of patients also showing C3. Between 50% and 90% of AIHA cases are due to warm antibodies.2,3 The incidence of warm AIHA varies by series but is approximately 1 case per 100,000 patients per year; this form of hemolysis affects women more frequently than men.4,5
Therapeutic Options
First Line
Steroids. The goal of therapy in warm AIHA can be hard to define. However, most would agree that a hematocrit above 30% (or higher to prevent symptoms) with a minimal increase in the reticulocyte count—reflective of a significantly slowed hemolytic process—is a reasonable goal. Initial management of warm AIHA is prednisone at a standard dose of 1 mg/kg daily (Table 1).6,7 Patients should be also started on proton-pump inhibitors to prevent ulcers. It can take up to 3 weeks for patients to respond to prednisone therapy. Once the patient’s hematocrit is above 30%, the prednisone is slowly tapered. Although approximately 80% of patients will respond to steroids, only 30% can be fully tapered off steroids. For patients who can be maintained on a daily steroid dose of 10 mg or less, steroids may be the most reasonable long-term therapy. In addition, because active hemolysis leads to an increased demand for folic acid, patients with warm AIHA are often prescribed folic acid 1 mg daily to prevent megaloblastic anemia due to folic acid deficiency.
Rituximab. Increasingly, rituximab (anti-CD20) therapy is added to the initial steroids. Two clinical trials showed both increased long-term and short-term responses with the use of rituximab.8,9 An important consideration is that most patients respond gradually to rituximab over weeks, so a rapid response should not be expected. Most studies have used the traditional dosing of 375 mg/m2 weekly for 4 weeks. These responses appear to be durable, but as in immune thrombocytopenia (ITP), repeat treatment with rituximab is effective.
The major side effects of rituximab are infusion reactions, which are often worse with the first dose. These reactions can be controlled with antihistamines, steroids, and, for severe rigors, meperidine. Rarely, patients can develop neutropenia (approximately 1:500) that appears to be autoimmune in nature. Infections appear to be only minimally increased with the use of rituximab.10 One group at risk is chronic carriers of hepatitis B virus, who may experience a reactivation of the virus that can be fatal. Thus, patients being considered for rituximab need to be screened for hepatitis B virus carrier state.11 Patients receiving rituximab are at very slight risk for progressive multifocal leukoencephalopathy, which is more common in patients with cancer and in heavily immunosuppressed patients. The overall risk is unknown but is less than 1:50,000.
Second Line
Splenectomy. For patients who cannot be weaned from steroids or in whom steroid therapy fails, there is no standard therapy. Currently, the 2 main choices are splenectomy or rituximab (anti-CD20) therapy if the patient did not receive it first line. Splenectomy is the classic therapy for warm AIHA. Reported response rates in the literature range from 50% to 80%, with 50% to 60% remaining in remission.12-16 Timing of the procedure is a balance between allowing time for the steroids to work and the risk of toxicity of steroids. In a patient who has low presurgical risk and has either refractory disease or cannot be weaned from high doses of steroids, splenectomy should be done sooner. Splenectomy can be delayed or other therapy tried first in patients who require lower doses of steroids or have medical risk factors for surgery. Most splenectomies are performed via laparoscopy. The small incisions allow for quicker healing, and the laparoscopic approach provides better visualization of the abdomen to find and remove accessory spleens. When splenectomy is performed by experienced surgeons, the mortality rate is low (< 0.5%).17
The most concerning complication of splenectomy is overwhelming post-splenectomy infection (OPSI).18 In adults, the spleen appears to play a minimal role in immunity except for protecting against certain encap-sulated organisms. Splenectomized patients infected with an encapsulated organism (eg, Pneumococcus) will develop overwhelming sepsis within hours. These patients will often have disseminated intravascular coagulation and will rapidly progress to purpura fulminans. Approximately 40% to 50% of patients will die of sepsis even when the infection is detected early. The overall lifetime risk of sepsis may be as high as 1:500. The organism that most commonly causes sepsis in splenectomized patients is Streptococcus pneumoniae, reported in over 50% of cases. Neisseria meningitidis and Haemophilus influenzae have also been implicated in many cases.19 Overwhelming sepsis after dog bites has been reported due to Capnocytophaga canimorsus infections. Patients are also at increased risk of developing severe malaria and severe babesiosis.18
Patients who have undergone splenectomy need to be warned about the risk of OPSI and instructed to report to the emergency department readily if they develop a fever greater than 101°F (38.3°C) or shaking chills. Once in the emergency department, blood cultures should be obtained rapidly and the patient started on antibiotic coverage with vancomycin and ceftriaxone (or levofloxacin if allergic to beta-lactams).20 For patients in remote areas, some physicians will prescribe prophylactic antibiotics to take while they are traveling to a health care provider or even recommend a “standby” antibiotic dose to take while traveling to health care.5 This usually consists of amoxicillin or a macrolide for penicillin-allergic patients.
Patients in whom splenectomy is being planned or considered should be vaccinated for pneumococcal, meningococcal, and influenza infections (Table 2).18 If there is a plan to treat with rituximab, patients should first be vaccinated since they will not be able to mount an immune response after being treated with rituximab.
Third Line
The therapeutic options for patients who do not respond to either splenectomy or rituximab are much less certain.5,6 Although intravenous immune globulin is a standard therapy for ITP, response rates are low in warm AIHA.17 Numerous therapies have been reported in small series, but no clear approach has emerged. Options include azathioprine, cyclophosphamide, mycophenolate, cyclosporine, danazol, and alemtuzumab. Our approach has been to use mycophenolate for patients requiring high doses of steroids or transfusions. Patients who respond to lower doses of steroids may be good candidates for danazol to help wean them off steroids.
Treatment of Warm AIHA with Associated Diseases
Warm AIHA can complicate several diseases. Patients with systemic lupus erythematosus (SLE) can develop warm AIHA as part of their disease complex. The initial treatment approach is the same, but data suggest that splenectomy may not be as effective.13,17 Also, many SLE patients have complex medical conditions, making surgeries riskier. For SLE patients who are refractory or cannot be weaned from steroids, rituximab may be the better choice. Babesiosis, particularly in asplenic patients, has been associated with the development of AIHA.21,22
Of the malignances associated with AIHA, chronic lymphocytic leukemia (CLL) has the strongest association.4,23 Series show that 5% to 10% of patients with CLL will have warm AIHA. AIHA can appear concurrent with CLL or develop during the course of the disease. The introduction of purine analogs such as fludarabine led to a dramatic increase in the incidence of warm AIHA in treated patients.24 It is speculated that these powerful agents reduce the number and effectiveness of T cells that hold in check the autoantibody response, leading to warm AIHA.25 However, when these purine analogs are used in com-bination with agents such as cyclophosphamide or rituximab (with their immunosuppressive effects), the rates of warm AIHA have been lower.23
The approach to patients with CLL and warm AIHA depends on the state of their CLL.23 For patients who have low-stage CLL that does not need treatment, the standard approach to warm AIHA should be steroids, splenectomy, and rituximab.24 For patients with higher-stage CLL, the treatment for the leukemia will often provide therapy for the warm AIHA. The combination of rituximab-cyclophosphamide-dexamethasone has been reported to be effective for both the AIHA and CLL components.26 The use of ibrutinib has also been reported to be effective.27
A rare but important variant of warm AIHA is Evans syndrome.28,29 This is the combination of AIHA and ITP. Approximately 1% to 3% of AIHA cases are the Evans variant. The ITP can precede, be concurrent with, or develop after the AIHA. The diagnosis of Evans syndrome should raise concern for underlying disorders. In young adults, immunodeficiency disorders such as autoimmune lymphoproliferative disease (ALPS) need to be considered. In older patients, Evans syndrome is often associated with T cell lymphomas. The sparse literature on Evans syndrome suggests that it can be more refractory to standard therapy, with response rates to splenectomy in the 50% range.28,30 In patients with lymphoma, antineoplastic therapy is crucial. There is increasing data showing that mycophenolate or sirolimus may be effective for patients with ALPS in whom splenectomy or rituximab therapy is unsuccessful.31
Warm AIHA with IgA or IgM Antibodies
In rare patients with warm AIHA, IgA or IgM is the implicated antibody. The literature suggests that patients with IgA AIHA may have more severe hemolysis.32 Patients with IgM AIHA often have a severe course with a fatal outcome.33 In such cases, the patient’s plasma may show spontaneous hemolysis and agglutination. The DAT may not be strongly positive or may show C3 reactivity only. The clinical clues are C3 reactivity with no cold agglutinins and severe hemolysis, sometimes with an intravascular component. Treatment is the same as for warm AIHA, including the use of rituximab.34
Cold Autoimmune Hemolytic Anemia
In cold AIHA, the hemolysis is mediated by IgM antibodies directed against red cells.35 As discussed earlier, the term “cold” refers to the fact that the antibody binds maximally at temperatures below 37°C. The most efficient temperature for binding is called the “thermal amplitude,” and, in theory, the higher the thermal amplitude, the more virulent the antibody. An antibody titer can be calculated at each reaction temperature from 4°C to 37°C by serial dilutions of the patient’s serum prior to incubating with reagent red cells. Rarely, the IgM can fix complement rapidly, leading to intravascular hemolysis. In most patients, complement is fixed through C3, and the C3-coated red cells are taken up by macrophages in the mononuclear phagocyte system, primarily in the liver.3
The DAT in patients with cold AIHA will show cells coated with C3. The blood smear will often show ag-glutination of the blood, and if the blood cools before being analyzed, the agglutination will interfere with the analysis. Titers of cold agglutinin can range from 1:1000 to over 1 million. The IgM autoantibodies are most often directed against the I/i antigens on the red blood cell membrane, with 90% against I antigen.35 The I antigen specificity is typical with primary cold agglutinin disease and after Mycoplasma infection. The i antigen specificity is most typical of Epstein-Barr virus and cytomegalovirus infections in children. In young patients, cold AIHA often occurs following an infection, including viral and Mycoplasma infections, and the course is self-limited.35,36 The hemolysis usually starts 2 to 3 weeks after the illness and will last for 4 to 6 weeks. In older patients, the etiology in over 90% of cases is a B-cell lymphoproliferative disorder, usually with monoclonal kappa B-cells.37 The most common disorders are marginal zone lymphoma, small lymphocytic lymphoma, and lymphoplasmacytic lymphoma.3
Therapeutic Options
It is important to diagnose cold AIHA because the standard therapy for warm AIHA (steroids) is ineffective in cold AIHA. Because C3-coated red cells are taken up primarily in the liver, removing the spleen is also an ineffective therapy. Simple measures to help with cold AIHA should be employed.37 Patients should be kept in a warm environment and should try to avoid the cold. If transfusions are needed, they should be infused via blood warmers to prevent hemolysis. In rare patients with severe hemolysis, therapeutic plasma exchange (TPE) can be considered.38 Given that the culprit antibody is IgM—mostly intravascular—use of TPE may slow the hemolysis to give time for other therapies to take effect.
Treatment of cold AIHA remains difficult (Table 3). Because most patients with primary AIHA have underlying lymphoproliferative diseases, chlorambucil has been used in the past to treat cold AIHA. However, responses were rare and the drug could worsen the anemia.38 Currently, the drug of choice is rituximab. Response is seen in 45% to 75% of patients, but is almost always a partial response and retreatment is often necessary.17,37,39 As with other autoimmune hematologic diseases, there can be a delay in response that ranges from 2 weeks to 4 months (median time, 1.5 months).37 Given the lack of robust response (complete and durable) with rituximab, the Berentsen group has explored adding bendamustine to rituximab.40 In a prospective trial, 71% of patients responded with a 40% complete response rate. Therapy was well tolerated, with only 29% of patients needing dose reduction Although more toxic, this combination can be considered in patients with aggressive disease. A small study of the use of bortezomib showed a good response in one-third of patients.41 There is increasing use of the C5 complement inhibitor eculizumab to halt the hemolysis, but further study of this agent is also required.36,42,43 Blockers of C1s complement, which block hemolysis by preventing complement activation, are currently being studied in clinical trials.44
Since most patients with cold AIHA are older, a frequent issue that must be considered is cardiac surgery. The concern is that the hypothermia involved with most heart bypass procedures will lead to agglutination and hemolysis. The development of normothermic bypass has expanded treatment options. A recommended approach in patients who have known cold agglutinins is to measure the thermal amplitude of the antibody preoperatively. If the thermal amplitude is above 18°C, normothermic bypass should be done, if feasible.45 If not feasible, preoperative TPE should be considered.
Paroxysmal Cold Hemoglobinuria
A unique cold AIHA is paroxysmal cold hemoglobinuria (PCH).3,46 This cold hemolytic syndrome most often occurs in children following a viral infection, but in the past it complicated any stage of syphilis.47 The mediating antibody in PCH is an IgG antibody directed against the P antigen on the red blood cell. This antibody binds best at temperatures below 37°C, fixing complement at cold temperatures, but then activates the complement cascade at body temperature.48 Because this antibody can fix complement, hemolysis can be rapid and severe, leading to extreme anemia. The DAT is often weakly positive but can be negative. The diagnostic test for PCH is the Donath–Landsteiner test. This complex test is performed by incubating 3 blood samples, 1 at 0° to 4°C, another at 37°C, and a third at 0° to 4°C and then at 37°C. The diagnosis of PCH is made if only the third tube shows hemolysis.35 PCH can persist for 1 to 3 months but is almost always self-limiting. In severe case, steroids may be of benefit.
Drug-Induced Hemolytic Anemia
AIHA caused by a drug reaction is rare, with a lower incidence than drug-related ITP. The rate of severe drug-related AIHA is estimated at 1:1,000,000, but less severe cases may be missed.1 Most patients will have a positive DAT without signs of hemolysis, but in rare cases patients will have relentless hemolysis resulting in death.
Mechanisms
Multiple mechanisms for drug-induced immune hemolysis have been proposed, including drug-absorption (hapten-induced) and immune complex mechanisms.1,49 The hapten mechanism is most often associated with the use of high-dose penicillin.50 High doses of penicillin or similar drugs such as piperacillin lead to incorporation of the drug into the red cell membrane by binding to proteins. Patients will manifest a positive DAT with IgG antibody but not complement.51 The patient’s plasma will be reactive only with penicillin-coated red cells and not with normal cells. As mentioned, very few patients will have hemolysis, and if they have hemolysis, it will resolve in a few days after discontinuation of the offending drug.52
Binding of a drug-antibody complex to the red cell membrane may cause hemolysis via the immune com-plex mechanism.53 Again, most often the patient will have just a positive DAT, but rarely patients will have life-threatening hemolysis upon exposure or reexposure to the drug. The onset of hemolysis is rapid, with signs of acute illness and intravascular hemolysis. The paradigm drug is quinine, but many other drugs have been implicated. Testing shows a positive Coombs test with anti-complement but not anti-IgG.50 This pattern is due to the effectiveness of the tertiary complex at fixing complement. The patient’s plasma reacts with red cells only when the drug is added.
A form of immune complex hemolysis associated with both disseminated intravascular coagulation (DIC) and brisk hemolysis has been recognized. Patients who receive certain second- and third-generation cephalosporins (especially cefotetan and ceftriaxone.53,54) have developed this syndrome.50,55-59 The clinical symptoms start 7 to 10 days after the drug is administered; often the patient has only received the antibiotic for surgical prophylaxis. Immune hemolysis with acute hematocrit drop, hypotension, and DIC ensues. The patients are often believed to have sepsis and are often reexposed to the cephalosporin, resulting in worsening of the clinical status. The outcome is often fatal due to massive hemolysis and thrombosis.56,60,62
Finally, 8% to 36% of patients taking methyldopa will develop a positive DAT after 6 months of therapy, with less than 1% showing hemolysis.52,63 The hemolysis in these patients is indistinguishable from warm AIHA, consistent with the notion that methyldopa induces an AIHA. The hemolysis often resolves rapidly after the methyldopa is stopped, but the Coombs test may remain positive for months.63 This type of drug-induced hemolytic anemia has been reported with levodopa, procainamide, and chlorpromazine, but fludarabine is the most common cause currently. This form of AIHA is now being seen with increased use of checkpoint inhibitors.64
Diagnosis
In many patients, the first clue to the presence of drug AIHA is the finding of a positive DAT. Rarely, patients will have severe hemolysis, but in many patients the hemolytic process is mild and may be wrongly assumed to be part of the underlying illness. Finding the offending drug can be a challenge, unless a patient has recently started a new drug; in a hospitalized patient on multiple agents, identifying the problem drug is difficult. Patients recently started on “suspect drugs,” especially the most common agents cefotetan, ceftriaxone, and piperacillin, should have these agents stopped (Table 4).1,49,65 Specialty laboratories such as the Blood Center of Wisconsin or the Los Angeles Red Cross can perform in vitro studies of drug interactions that can confirm the clinical diagnosis of drug-induced AIHA.
Treatment
Therapy for patients with positive DAT without signs of hemolysis is uncertain. If the drug is essential, then the patient can be observed. If the patient has hemolysis, the drug needs to be stopped and the patient observed for signs of end-organ damage. It is doubtful that steroids or other autoimmune-directed therapy is effective. For patients with the DIC-hemolysis syndrome, there are anecdotal reports that TPE may be helpful.1
Summary
AIHA can range from an abnormal laboratory test (positive DAT and signs of hemolysis) to an acute, life-threatening illness. Treatment is guided by the laboratory work-up and evaluation of the patient’s clinical status. While rituximab is promising for many patients, the lack of robust clinical trials hinders the treatment of patients who fail standard therapies.
Autoimmune hemolytic anemia (AIHA) is mediated by antibodies, and in most cases immunoglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C. AIHA caused by a drug reaction is rare, with an estimated annual incidence of 1:1,000,000 for severe drug-related AIHA.1 This article reviews the management of the more common types of AIHA, with a focus on warm, cold, and drug-induced AIHA; the evaluation and diagnosis of AIHA is reviewed in a separate article.
Warm Autoimmune Hemolytic Anemia
In AIHA, hemolysis is mediated by antibodies that bind to the surface of red blood cells. AIHA in which IgG antibodies are the offending antibodies is referred to as warm AIHA. “Warm” refers to the fact that the antibody binds best at body temperature (37°C). In warm AIHA, testing will show IgG molecules attached to the surface of the red cells, with 50% of patients also showing C3. Between 50% and 90% of AIHA cases are due to warm antibodies.2,3 The incidence of warm AIHA varies by series but is approximately 1 case per 100,000 patients per year; this form of hemolysis affects women more frequently than men.4,5
Therapeutic Options
First Line
Steroids. The goal of therapy in warm AIHA can be hard to define. However, most would agree that a hematocrit above 30% (or higher to prevent symptoms) with a minimal increase in the reticulocyte count—reflective of a significantly slowed hemolytic process—is a reasonable goal. Initial management of warm AIHA is prednisone at a standard dose of 1 mg/kg daily (Table 1).6,7 Patients should be also started on proton-pump inhibitors to prevent ulcers. It can take up to 3 weeks for patients to respond to prednisone therapy. Once the patient’s hematocrit is above 30%, the prednisone is slowly tapered. Although approximately 80% of patients will respond to steroids, only 30% can be fully tapered off steroids. For patients who can be maintained on a daily steroid dose of 10 mg or less, steroids may be the most reasonable long-term therapy. In addition, because active hemolysis leads to an increased demand for folic acid, patients with warm AIHA are often prescribed folic acid 1 mg daily to prevent megaloblastic anemia due to folic acid deficiency.
Rituximab. Increasingly, rituximab (anti-CD20) therapy is added to the initial steroids. Two clinical trials showed both increased long-term and short-term responses with the use of rituximab.8,9 An important consideration is that most patients respond gradually to rituximab over weeks, so a rapid response should not be expected. Most studies have used the traditional dosing of 375 mg/m2 weekly for 4 weeks. These responses appear to be durable, but as in immune thrombocytopenia (ITP), repeat treatment with rituximab is effective.
The major side effects of rituximab are infusion reactions, which are often worse with the first dose. These reactions can be controlled with antihistamines, steroids, and, for severe rigors, meperidine. Rarely, patients can develop neutropenia (approximately 1:500) that appears to be autoimmune in nature. Infections appear to be only minimally increased with the use of rituximab.10 One group at risk is chronic carriers of hepatitis B virus, who may experience a reactivation of the virus that can be fatal. Thus, patients being considered for rituximab need to be screened for hepatitis B virus carrier state.11 Patients receiving rituximab are at very slight risk for progressive multifocal leukoencephalopathy, which is more common in patients with cancer and in heavily immunosuppressed patients. The overall risk is unknown but is less than 1:50,000.
Second Line
Splenectomy. For patients who cannot be weaned from steroids or in whom steroid therapy fails, there is no standard therapy. Currently, the 2 main choices are splenectomy or rituximab (anti-CD20) therapy if the patient did not receive it first line. Splenectomy is the classic therapy for warm AIHA. Reported response rates in the literature range from 50% to 80%, with 50% to 60% remaining in remission.12-16 Timing of the procedure is a balance between allowing time for the steroids to work and the risk of toxicity of steroids. In a patient who has low presurgical risk and has either refractory disease or cannot be weaned from high doses of steroids, splenectomy should be done sooner. Splenectomy can be delayed or other therapy tried first in patients who require lower doses of steroids or have medical risk factors for surgery. Most splenectomies are performed via laparoscopy. The small incisions allow for quicker healing, and the laparoscopic approach provides better visualization of the abdomen to find and remove accessory spleens. When splenectomy is performed by experienced surgeons, the mortality rate is low (< 0.5%).17
The most concerning complication of splenectomy is overwhelming post-splenectomy infection (OPSI).18 In adults, the spleen appears to play a minimal role in immunity except for protecting against certain encap-sulated organisms. Splenectomized patients infected with an encapsulated organism (eg, Pneumococcus) will develop overwhelming sepsis within hours. These patients will often have disseminated intravascular coagulation and will rapidly progress to purpura fulminans. Approximately 40% to 50% of patients will die of sepsis even when the infection is detected early. The overall lifetime risk of sepsis may be as high as 1:500. The organism that most commonly causes sepsis in splenectomized patients is Streptococcus pneumoniae, reported in over 50% of cases. Neisseria meningitidis and Haemophilus influenzae have also been implicated in many cases.19 Overwhelming sepsis after dog bites has been reported due to Capnocytophaga canimorsus infections. Patients are also at increased risk of developing severe malaria and severe babesiosis.18
Patients who have undergone splenectomy need to be warned about the risk of OPSI and instructed to report to the emergency department readily if they develop a fever greater than 101°F (38.3°C) or shaking chills. Once in the emergency department, blood cultures should be obtained rapidly and the patient started on antibiotic coverage with vancomycin and ceftriaxone (or levofloxacin if allergic to beta-lactams).20 For patients in remote areas, some physicians will prescribe prophylactic antibiotics to take while they are traveling to a health care provider or even recommend a “standby” antibiotic dose to take while traveling to health care.5 This usually consists of amoxicillin or a macrolide for penicillin-allergic patients.
Patients in whom splenectomy is being planned or considered should be vaccinated for pneumococcal, meningococcal, and influenza infections (Table 2).18 If there is a plan to treat with rituximab, patients should first be vaccinated since they will not be able to mount an immune response after being treated with rituximab.
Third Line
The therapeutic options for patients who do not respond to either splenectomy or rituximab are much less certain.5,6 Although intravenous immune globulin is a standard therapy for ITP, response rates are low in warm AIHA.17 Numerous therapies have been reported in small series, but no clear approach has emerged. Options include azathioprine, cyclophosphamide, mycophenolate, cyclosporine, danazol, and alemtuzumab. Our approach has been to use mycophenolate for patients requiring high doses of steroids or transfusions. Patients who respond to lower doses of steroids may be good candidates for danazol to help wean them off steroids.
Treatment of Warm AIHA with Associated Diseases
Warm AIHA can complicate several diseases. Patients with systemic lupus erythematosus (SLE) can develop warm AIHA as part of their disease complex. The initial treatment approach is the same, but data suggest that splenectomy may not be as effective.13,17 Also, many SLE patients have complex medical conditions, making surgeries riskier. For SLE patients who are refractory or cannot be weaned from steroids, rituximab may be the better choice. Babesiosis, particularly in asplenic patients, has been associated with the development of AIHA.21,22
Of the malignances associated with AIHA, chronic lymphocytic leukemia (CLL) has the strongest association.4,23 Series show that 5% to 10% of patients with CLL will have warm AIHA. AIHA can appear concurrent with CLL or develop during the course of the disease. The introduction of purine analogs such as fludarabine led to a dramatic increase in the incidence of warm AIHA in treated patients.24 It is speculated that these powerful agents reduce the number and effectiveness of T cells that hold in check the autoantibody response, leading to warm AIHA.25 However, when these purine analogs are used in com-bination with agents such as cyclophosphamide or rituximab (with their immunosuppressive effects), the rates of warm AIHA have been lower.23
The approach to patients with CLL and warm AIHA depends on the state of their CLL.23 For patients who have low-stage CLL that does not need treatment, the standard approach to warm AIHA should be steroids, splenectomy, and rituximab.24 For patients with higher-stage CLL, the treatment for the leukemia will often provide therapy for the warm AIHA. The combination of rituximab-cyclophosphamide-dexamethasone has been reported to be effective for both the AIHA and CLL components.26 The use of ibrutinib has also been reported to be effective.27
A rare but important variant of warm AIHA is Evans syndrome.28,29 This is the combination of AIHA and ITP. Approximately 1% to 3% of AIHA cases are the Evans variant. The ITP can precede, be concurrent with, or develop after the AIHA. The diagnosis of Evans syndrome should raise concern for underlying disorders. In young adults, immunodeficiency disorders such as autoimmune lymphoproliferative disease (ALPS) need to be considered. In older patients, Evans syndrome is often associated with T cell lymphomas. The sparse literature on Evans syndrome suggests that it can be more refractory to standard therapy, with response rates to splenectomy in the 50% range.28,30 In patients with lymphoma, antineoplastic therapy is crucial. There is increasing data showing that mycophenolate or sirolimus may be effective for patients with ALPS in whom splenectomy or rituximab therapy is unsuccessful.31
Warm AIHA with IgA or IgM Antibodies
In rare patients with warm AIHA, IgA or IgM is the implicated antibody. The literature suggests that patients with IgA AIHA may have more severe hemolysis.32 Patients with IgM AIHA often have a severe course with a fatal outcome.33 In such cases, the patient’s plasma may show spontaneous hemolysis and agglutination. The DAT may not be strongly positive or may show C3 reactivity only. The clinical clues are C3 reactivity with no cold agglutinins and severe hemolysis, sometimes with an intravascular component. Treatment is the same as for warm AIHA, including the use of rituximab.34
Cold Autoimmune Hemolytic Anemia
In cold AIHA, the hemolysis is mediated by IgM antibodies directed against red cells.35 As discussed earlier, the term “cold” refers to the fact that the antibody binds maximally at temperatures below 37°C. The most efficient temperature for binding is called the “thermal amplitude,” and, in theory, the higher the thermal amplitude, the more virulent the antibody. An antibody titer can be calculated at each reaction temperature from 4°C to 37°C by serial dilutions of the patient’s serum prior to incubating with reagent red cells. Rarely, the IgM can fix complement rapidly, leading to intravascular hemolysis. In most patients, complement is fixed through C3, and the C3-coated red cells are taken up by macrophages in the mononuclear phagocyte system, primarily in the liver.3
The DAT in patients with cold AIHA will show cells coated with C3. The blood smear will often show ag-glutination of the blood, and if the blood cools before being analyzed, the agglutination will interfere with the analysis. Titers of cold agglutinin can range from 1:1000 to over 1 million. The IgM autoantibodies are most often directed against the I/i antigens on the red blood cell membrane, with 90% against I antigen.35 The I antigen specificity is typical with primary cold agglutinin disease and after Mycoplasma infection. The i antigen specificity is most typical of Epstein-Barr virus and cytomegalovirus infections in children. In young patients, cold AIHA often occurs following an infection, including viral and Mycoplasma infections, and the course is self-limited.35,36 The hemolysis usually starts 2 to 3 weeks after the illness and will last for 4 to 6 weeks. In older patients, the etiology in over 90% of cases is a B-cell lymphoproliferative disorder, usually with monoclonal kappa B-cells.37 The most common disorders are marginal zone lymphoma, small lymphocytic lymphoma, and lymphoplasmacytic lymphoma.3
Therapeutic Options
It is important to diagnose cold AIHA because the standard therapy for warm AIHA (steroids) is ineffective in cold AIHA. Because C3-coated red cells are taken up primarily in the liver, removing the spleen is also an ineffective therapy. Simple measures to help with cold AIHA should be employed.37 Patients should be kept in a warm environment and should try to avoid the cold. If transfusions are needed, they should be infused via blood warmers to prevent hemolysis. In rare patients with severe hemolysis, therapeutic plasma exchange (TPE) can be considered.38 Given that the culprit antibody is IgM—mostly intravascular—use of TPE may slow the hemolysis to give time for other therapies to take effect.
Treatment of cold AIHA remains difficult (Table 3). Because most patients with primary AIHA have underlying lymphoproliferative diseases, chlorambucil has been used in the past to treat cold AIHA. However, responses were rare and the drug could worsen the anemia.38 Currently, the drug of choice is rituximab. Response is seen in 45% to 75% of patients, but is almost always a partial response and retreatment is often necessary.17,37,39 As with other autoimmune hematologic diseases, there can be a delay in response that ranges from 2 weeks to 4 months (median time, 1.5 months).37 Given the lack of robust response (complete and durable) with rituximab, the Berentsen group has explored adding bendamustine to rituximab.40 In a prospective trial, 71% of patients responded with a 40% complete response rate. Therapy was well tolerated, with only 29% of patients needing dose reduction Although more toxic, this combination can be considered in patients with aggressive disease. A small study of the use of bortezomib showed a good response in one-third of patients.41 There is increasing use of the C5 complement inhibitor eculizumab to halt the hemolysis, but further study of this agent is also required.36,42,43 Blockers of C1s complement, which block hemolysis by preventing complement activation, are currently being studied in clinical trials.44
Since most patients with cold AIHA are older, a frequent issue that must be considered is cardiac surgery. The concern is that the hypothermia involved with most heart bypass procedures will lead to agglutination and hemolysis. The development of normothermic bypass has expanded treatment options. A recommended approach in patients who have known cold agglutinins is to measure the thermal amplitude of the antibody preoperatively. If the thermal amplitude is above 18°C, normothermic bypass should be done, if feasible.45 If not feasible, preoperative TPE should be considered.
Paroxysmal Cold Hemoglobinuria
A unique cold AIHA is paroxysmal cold hemoglobinuria (PCH).3,46 This cold hemolytic syndrome most often occurs in children following a viral infection, but in the past it complicated any stage of syphilis.47 The mediating antibody in PCH is an IgG antibody directed against the P antigen on the red blood cell. This antibody binds best at temperatures below 37°C, fixing complement at cold temperatures, but then activates the complement cascade at body temperature.48 Because this antibody can fix complement, hemolysis can be rapid and severe, leading to extreme anemia. The DAT is often weakly positive but can be negative. The diagnostic test for PCH is the Donath–Landsteiner test. This complex test is performed by incubating 3 blood samples, 1 at 0° to 4°C, another at 37°C, and a third at 0° to 4°C and then at 37°C. The diagnosis of PCH is made if only the third tube shows hemolysis.35 PCH can persist for 1 to 3 months but is almost always self-limiting. In severe case, steroids may be of benefit.
Drug-Induced Hemolytic Anemia
AIHA caused by a drug reaction is rare, with a lower incidence than drug-related ITP. The rate of severe drug-related AIHA is estimated at 1:1,000,000, but less severe cases may be missed.1 Most patients will have a positive DAT without signs of hemolysis, but in rare cases patients will have relentless hemolysis resulting in death.
Mechanisms
Multiple mechanisms for drug-induced immune hemolysis have been proposed, including drug-absorption (hapten-induced) and immune complex mechanisms.1,49 The hapten mechanism is most often associated with the use of high-dose penicillin.50 High doses of penicillin or similar drugs such as piperacillin lead to incorporation of the drug into the red cell membrane by binding to proteins. Patients will manifest a positive DAT with IgG antibody but not complement.51 The patient’s plasma will be reactive only with penicillin-coated red cells and not with normal cells. As mentioned, very few patients will have hemolysis, and if they have hemolysis, it will resolve in a few days after discontinuation of the offending drug.52
Binding of a drug-antibody complex to the red cell membrane may cause hemolysis via the immune com-plex mechanism.53 Again, most often the patient will have just a positive DAT, but rarely patients will have life-threatening hemolysis upon exposure or reexposure to the drug. The onset of hemolysis is rapid, with signs of acute illness and intravascular hemolysis. The paradigm drug is quinine, but many other drugs have been implicated. Testing shows a positive Coombs test with anti-complement but not anti-IgG.50 This pattern is due to the effectiveness of the tertiary complex at fixing complement. The patient’s plasma reacts with red cells only when the drug is added.
A form of immune complex hemolysis associated with both disseminated intravascular coagulation (DIC) and brisk hemolysis has been recognized. Patients who receive certain second- and third-generation cephalosporins (especially cefotetan and ceftriaxone.53,54) have developed this syndrome.50,55-59 The clinical symptoms start 7 to 10 days after the drug is administered; often the patient has only received the antibiotic for surgical prophylaxis. Immune hemolysis with acute hematocrit drop, hypotension, and DIC ensues. The patients are often believed to have sepsis and are often reexposed to the cephalosporin, resulting in worsening of the clinical status. The outcome is often fatal due to massive hemolysis and thrombosis.56,60,62
Finally, 8% to 36% of patients taking methyldopa will develop a positive DAT after 6 months of therapy, with less than 1% showing hemolysis.52,63 The hemolysis in these patients is indistinguishable from warm AIHA, consistent with the notion that methyldopa induces an AIHA. The hemolysis often resolves rapidly after the methyldopa is stopped, but the Coombs test may remain positive for months.63 This type of drug-induced hemolytic anemia has been reported with levodopa, procainamide, and chlorpromazine, but fludarabine is the most common cause currently. This form of AIHA is now being seen with increased use of checkpoint inhibitors.64
Diagnosis
In many patients, the first clue to the presence of drug AIHA is the finding of a positive DAT. Rarely, patients will have severe hemolysis, but in many patients the hemolytic process is mild and may be wrongly assumed to be part of the underlying illness. Finding the offending drug can be a challenge, unless a patient has recently started a new drug; in a hospitalized patient on multiple agents, identifying the problem drug is difficult. Patients recently started on “suspect drugs,” especially the most common agents cefotetan, ceftriaxone, and piperacillin, should have these agents stopped (Table 4).1,49,65 Specialty laboratories such as the Blood Center of Wisconsin or the Los Angeles Red Cross can perform in vitro studies of drug interactions that can confirm the clinical diagnosis of drug-induced AIHA.
Treatment
Therapy for patients with positive DAT without signs of hemolysis is uncertain. If the drug is essential, then the patient can be observed. If the patient has hemolysis, the drug needs to be stopped and the patient observed for signs of end-organ damage. It is doubtful that steroids or other autoimmune-directed therapy is effective. For patients with the DIC-hemolysis syndrome, there are anecdotal reports that TPE may be helpful.1
Summary
AIHA can range from an abnormal laboratory test (positive DAT and signs of hemolysis) to an acute, life-threatening illness. Treatment is guided by the laboratory work-up and evaluation of the patient’s clinical status. While rituximab is promising for many patients, the lack of robust clinical trials hinders the treatment of patients who fail standard therapies.
1. Garratty G. Immune hemolytic anemia associated with drug therapy. Blood Rev. 2010;24:143-150.
2. Ness PM. How do I encourage clinicians to transfuse mismatched blood to patients with autoimmune hemolytic anemia in urgent situations? Transfusion. 2006;46:1859-1862.
3. Berentsen S. Cold agglutinin disease. Hematology Am Soc Hematol Educ Program. 2016;2016:226-231.
4. Liebman HA, Weitz IC. Autoimmune hemolytic anemia. Med Clin North Am. 2017;101:351-359.
5. Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24:195–210.
6. Kyrle PA, Rosendaal FR, Eichinger S. Risk assessment for re¬current venous thrombosis. Lancet. 2010;376:2032-2039.
7. Go RS, Winters JL, Kay NE. How I treat autoimmune hemolytic anemia. Blood. 2017;129:2971-2979.
8. Birgens H, Frederiksen H, Hasselbalch HC, et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013;163:393-399
9. Michel M, Terriou L, Roudot-Thoraval F, et al. A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm auto-immune hemolytic anemia in adults (the RAIHA study). Am J Hematol. 2017;92:23-27.
10. Gea-Banacloche JC. Rituximab-associated infections. Semin Hematol. 2010;47:187-198.
11. Loomba R, Liang TJ. Hepatitis B reactivation associated with immune suppressive and biological modifier therapies: current concepts, management strategies, and future directions. Gastroenterology. 2017;152:1297-1309.
12. Coon WW. Splenectomy in the treatment of hemolytic anemia. Arch Surg. 1985;120:625-628.
13. Akpek G, McAneny D, Weintraub L. Comparative response to splenectomy in coombs-positive autoimmune hemolytic anemia with or without associated disease. Am J Hematol. 1999;61:98-102.
14. Patel NY, Chilsen AM, Mathiason MA, et al. Outcomes and complications after splenectomy for hematologic disorders. Am J Surg. 2012;204:1014-1020.
15. Crowther M, Chan YL, Garbett IK, et al. Evidence-based focused review of the treatment of idiopathic warm immune hemolytic anemia in adults. Blood. 2011;118:4036-4040.
16. Giudice V, Rosamilio R, Ferrara I, et al. Efficacy and safety of splenectomy in adult autoimmune hemolytic anemia. Open Med (Wars). 2016;11:374-380.
17. Lechner K, Jager U. How I treat autoimmune hemolytic anemias in adults. Blood. 2010;116:1831-1838.
18. Rodeghiero F, Ruggeri M. Short- and long-term risks of splenectomy for benign haematological disorders: should we revisit the indications? Br J Haematol. 2012;158:16-29.
19. Ahmed N, Bialowas C, Kuo YH, Zawodniak L. Impact of preinjury anticoagulation in patients with traumatic brain injury. South Med J. 2009;102:476-480.
20. Morgan TL, Tomich EB. Overwhelming post-splenectomy infection (OPSI): a case report and review of the literature. J Emerg Med. 2012;43:758-763.
21. Woolley AE, Montgomery MW, Savage WJ, et al. Post-babesiosis warm autoimmune hemolytic anemia. N Engl J Med. 2017;376:939-946.
22. Shatzel JJ, Donohoe K, Chu NQ, et al. Profound autoimmune hemolysis and Evans syndrome in two asplenic patients with babesiosis. Transfusion. 2015;55:661-665.
23. Hodgson K, Ferrer G, Montserrat E, Moreno C. Chronic lymphocytic leukemia and autoimmunity: a systematic review. Haematologica. 2011;96:752-761.
24. Hamblin TJ. Autoimmune complications of chronic lymphocytic leukemia. Semin Oncol. 2006;33:230-239.
25. Tertian G, Cartron J, Bayle C, et al. Fatal intravascular au¬toimmune hemolytic anemia after fludarabine treatment for chronic lymphocytic leukemia. Hematol Cell Ther. 1996;38:359-360.
26. Rossignol J, Michallet AS, Oberic L, et al. Rituximab-cyclophosphamide-dexamethasone combination in the management of autoimmune cytopenias associated with chronic lymphocytic leukemia. Leukemia. 2011;25:473-478.
27. Hampel PJ, Larson MC, Kabat B, et al. Autoimmune cytopenias in patients with chronic lymphocytic leukaemia treated with ibrutinib in routine clinical practice at an academic medical centre. Br J Haematol. 2018;183:421-427.
28. Michel M, Chanet V, Dechartres A, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood. 2009;114:3167-3172.
29. Jaime-Pérez JC, Aguilar-Calderón PE, Salazar-Cavazos L, Gómez-Almaguer D. Evans syndrome: clinical perspectives, biological insights and treatment modalities. J Blood Med. 2018;9:171-184.
30. Dhingra KK, Jain D, Mandal S, et al. Evans syndrome: a study of six cases with review of literature. Hematology. 2008;13:356-360.
31. Bride KL, Vincent T, Smith-Whitley K, et al. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial. Blood. 2016;127:17-28.
32. Sokol RJ, Booker DJ, Stamps R, et al. IgA red cell au¬toantibodies and autoimmune hemolysis. Transfusion. 1997;37:175-181.
33. Garratty G, Arndt P, Domen R, et al. Severe autoimmune hemolytic anemia associated with IgM warm autoantibodies directed against determinants on or associated with glycophorin A. Vox Sang. 1997;72:124-130.
34. Wakim M, Shah A, Arndt PA, et al. Successful anti-CD20 monoclonal antibody treatment of severe autoimmune hemolytic anemia due to warm reactive IgM autoantibody in a child with common variable immunodeficiency. Am J Hematol. 2004;76:152-155.
35. Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
36. Berentsen S, Tjonnfjord GE. Diagnosis and treatment of cold agglutinin mediated autoimmune hemolytic anemia. Blood Rev. 2012;26:107-115.
37. Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011;153:309-317.
38. King KE, Ness PM. Treatment of autoimmune hemolytic anemia. Semin Hematol. 2005;42:131136.
39. Barcellini W, Zaja F, Zaninoni A, et al. Low-dose rituximab in adult patients with idiopathic autoimmune hemolytic anemia: clinical efficacy and biologic studies. Blood. 2012;119:3691-3697.
40. Berentsen S, Randen U, Oksman M, et al. Bendamustine plus rituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood. 2017;130:537-541.
41. Rossi G, Gramegna D, Paoloni F, et al. Short course of bortezomib in anemic patients with relapsed cold agglutinin disease: a phase 2 prospective GIMEMA study. Blood. 2018;132:547-550.
42. Makishima K, Obara N, Ishitsuka K, et al. High efficacy of eculizumab treatment for fulminant hemolytic anemia in primary cold agglutinin disease. Ann Hematol. 2019;98:1031-1032.
43. Roth A, Huttmann A, Rother RP, et al. Long-term efficacy of the complement inhibitor eculizumab in cold agglutinin disease. Blood. 2009;113:38853886.
44. Jäger U, D'Sa S, Schörgenhofer C, et al. Inhibition of complement C1s improves severe hemolytic anemia in cold agglutinin disease: a first-in-human trial. Blood. 2019;133:893-901.
45. Agarwal SK, Ghosh PK, Gupta D. Cardiac surgery and cold-reactive proteins. Ann Thorac Surg. 1995;60:1143-1150.
46. Shanbhag S, Spivak J. Paroxysmal cold hemoglobinuria. Hematol Oncol Clin North Am. 2015;29:473-478.
47. Kumar ND, Sethi S, Pandhi RK. Paroxysmal cold haemoglobinuria in syphilis patients. Genitourin Med. 1993;69:76.
48. Zantek ND, Koepsell SA, Tharp DR Jr, Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. 2012;87:707-709.
49. Pierce A, Nester T. Pathology consultation on drug-induced hemolytic anemia. Am J Clin Pathol. 2011;136:7-12.
50. Garratty G. Immune cytopenia associated with antibiotics. Transfusion Med Rev. 1993;7:255-267.
51. Petz LD, Mueller-Eckhardt C. Drug-induced immune hemolytic anemia. Transfusion. 1992;32:202-204.
52. Packman CH, Leddy JP. Drug-related immune hemolytic anemia. In: Beutler E, Lichtman MA, Coller BS, Kipps TJ, eds. William’s Hematology. 5th ed. New York: McGraw-Hill; 1995:691-704.
53. Garratty G. Drug-induced immune hemolytic anemia. Hematology Am Soc Hematol Educ Program. 2009;73-79.
54. Leicht HB, Weinig E, Mayer B, et al. Ceftriaxone-induced hemolytic anemia with severe renal failure: a case report and review of literature. BMC Pharmacol Toxicol. 2018;19:67.
55. Chenoweth CE, Judd WJ, Steiner EA, Kauffman CA. Cefotetan-induced immune hemolytic anemia. Clin Infect Dis. 1992;15:863-865.
56. Garratty G, Nance S, Lloyd M, Domen R. Fatal im¬mune hemolytic anemia due to cefotetan. Transfusion. 1992;32:269-271.
57. Endoh T, Yagihashi A, Sasaki M, Watanabe N. Ceftizoxime-induced hemolysis due to immune complexes: case report and determination of the epitope responsible for immune complex-mediated hemolysis. Transfusion. 1999;39:306-309.
58. Arndt PA, Leger RM, Garratty G. Serology of antibodies to second- and third-generation cephalosporins associated with immune hemolytic anemia and/or positive direct antiglobulin tests. Transfusion. 1999;39:1239-1246.
59. Martin ME, Laber DA. Cefotetan-induced hemolytic anemia after perioperative prophylaxis. Am J Hematol. 2006;81:186-188.
60. Bernini JC, Mustafa MM, Sutor LJ, Buchanan GR. Fatal hemolysis induced by ceftriaxone in a child with sickle cell anemia. J Pediatr. 1995;126(5 Pt 1):813-815.
61. Borgna-Pignatti C, Bezzi TM, Reverberi R. Fatal ceftriaxone-induced hemolysis in a child with acquired immunodeficiency syndrome. Pediatr Infect Dis J. 1995;14:1116-1117.
62. Lascari AD, Amyot K. Fatal hemolysis caused by ceftriaxone [see comments]. J Pediatr. 1995;126(5 Pt 1):816-817.
63. Petz LD. Drug-induced autoimmune hemolytic anemia. Transfusion Med Rev. 1993;7:242-254.
64. Leaf RK, Ferreri C, Rangachari D, et al. Clinical and laboratory features of autoimmune hemolytic anemia associated with immune checkpoint inhibitors. Am J Hematol. 2019;94:563-574.
65. DeLoughery T. Drug induced immune hematological disease. Allerg Immunol Clin. 1998;18:829-841.
1. Garratty G. Immune hemolytic anemia associated with drug therapy. Blood Rev. 2010;24:143-150.
2. Ness PM. How do I encourage clinicians to transfuse mismatched blood to patients with autoimmune hemolytic anemia in urgent situations? Transfusion. 2006;46:1859-1862.
3. Berentsen S. Cold agglutinin disease. Hematology Am Soc Hematol Educ Program. 2016;2016:226-231.
4. Liebman HA, Weitz IC. Autoimmune hemolytic anemia. Med Clin North Am. 2017;101:351-359.
5. Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24:195–210.
6. Kyrle PA, Rosendaal FR, Eichinger S. Risk assessment for re¬current venous thrombosis. Lancet. 2010;376:2032-2039.
7. Go RS, Winters JL, Kay NE. How I treat autoimmune hemolytic anemia. Blood. 2017;129:2971-2979.
8. Birgens H, Frederiksen H, Hasselbalch HC, et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013;163:393-399
9. Michel M, Terriou L, Roudot-Thoraval F, et al. A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm auto-immune hemolytic anemia in adults (the RAIHA study). Am J Hematol. 2017;92:23-27.
10. Gea-Banacloche JC. Rituximab-associated infections. Semin Hematol. 2010;47:187-198.
11. Loomba R, Liang TJ. Hepatitis B reactivation associated with immune suppressive and biological modifier therapies: current concepts, management strategies, and future directions. Gastroenterology. 2017;152:1297-1309.
12. Coon WW. Splenectomy in the treatment of hemolytic anemia. Arch Surg. 1985;120:625-628.
13. Akpek G, McAneny D, Weintraub L. Comparative response to splenectomy in coombs-positive autoimmune hemolytic anemia with or without associated disease. Am J Hematol. 1999;61:98-102.
14. Patel NY, Chilsen AM, Mathiason MA, et al. Outcomes and complications after splenectomy for hematologic disorders. Am J Surg. 2012;204:1014-1020.
15. Crowther M, Chan YL, Garbett IK, et al. Evidence-based focused review of the treatment of idiopathic warm immune hemolytic anemia in adults. Blood. 2011;118:4036-4040.
16. Giudice V, Rosamilio R, Ferrara I, et al. Efficacy and safety of splenectomy in adult autoimmune hemolytic anemia. Open Med (Wars). 2016;11:374-380.
17. Lechner K, Jager U. How I treat autoimmune hemolytic anemias in adults. Blood. 2010;116:1831-1838.
18. Rodeghiero F, Ruggeri M. Short- and long-term risks of splenectomy for benign haematological disorders: should we revisit the indications? Br J Haematol. 2012;158:16-29.
19. Ahmed N, Bialowas C, Kuo YH, Zawodniak L. Impact of preinjury anticoagulation in patients with traumatic brain injury. South Med J. 2009;102:476-480.
20. Morgan TL, Tomich EB. Overwhelming post-splenectomy infection (OPSI): a case report and review of the literature. J Emerg Med. 2012;43:758-763.
21. Woolley AE, Montgomery MW, Savage WJ, et al. Post-babesiosis warm autoimmune hemolytic anemia. N Engl J Med. 2017;376:939-946.
22. Shatzel JJ, Donohoe K, Chu NQ, et al. Profound autoimmune hemolysis and Evans syndrome in two asplenic patients with babesiosis. Transfusion. 2015;55:661-665.
23. Hodgson K, Ferrer G, Montserrat E, Moreno C. Chronic lymphocytic leukemia and autoimmunity: a systematic review. Haematologica. 2011;96:752-761.
24. Hamblin TJ. Autoimmune complications of chronic lymphocytic leukemia. Semin Oncol. 2006;33:230-239.
25. Tertian G, Cartron J, Bayle C, et al. Fatal intravascular au¬toimmune hemolytic anemia after fludarabine treatment for chronic lymphocytic leukemia. Hematol Cell Ther. 1996;38:359-360.
26. Rossignol J, Michallet AS, Oberic L, et al. Rituximab-cyclophosphamide-dexamethasone combination in the management of autoimmune cytopenias associated with chronic lymphocytic leukemia. Leukemia. 2011;25:473-478.
27. Hampel PJ, Larson MC, Kabat B, et al. Autoimmune cytopenias in patients with chronic lymphocytic leukaemia treated with ibrutinib in routine clinical practice at an academic medical centre. Br J Haematol. 2018;183:421-427.
28. Michel M, Chanet V, Dechartres A, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood. 2009;114:3167-3172.
29. Jaime-Pérez JC, Aguilar-Calderón PE, Salazar-Cavazos L, Gómez-Almaguer D. Evans syndrome: clinical perspectives, biological insights and treatment modalities. J Blood Med. 2018;9:171-184.
30. Dhingra KK, Jain D, Mandal S, et al. Evans syndrome: a study of six cases with review of literature. Hematology. 2008;13:356-360.
31. Bride KL, Vincent T, Smith-Whitley K, et al. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial. Blood. 2016;127:17-28.
32. Sokol RJ, Booker DJ, Stamps R, et al. IgA red cell au¬toantibodies and autoimmune hemolysis. Transfusion. 1997;37:175-181.
33. Garratty G, Arndt P, Domen R, et al. Severe autoimmune hemolytic anemia associated with IgM warm autoantibodies directed against determinants on or associated with glycophorin A. Vox Sang. 1997;72:124-130.
34. Wakim M, Shah A, Arndt PA, et al. Successful anti-CD20 monoclonal antibody treatment of severe autoimmune hemolytic anemia due to warm reactive IgM autoantibody in a child with common variable immunodeficiency. Am J Hematol. 2004;76:152-155.
35. Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
36. Berentsen S, Tjonnfjord GE. Diagnosis and treatment of cold agglutinin mediated autoimmune hemolytic anemia. Blood Rev. 2012;26:107-115.
37. Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011;153:309-317.
38. King KE, Ness PM. Treatment of autoimmune hemolytic anemia. Semin Hematol. 2005;42:131136.
39. Barcellini W, Zaja F, Zaninoni A, et al. Low-dose rituximab in adult patients with idiopathic autoimmune hemolytic anemia: clinical efficacy and biologic studies. Blood. 2012;119:3691-3697.
40. Berentsen S, Randen U, Oksman M, et al. Bendamustine plus rituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood. 2017;130:537-541.
41. Rossi G, Gramegna D, Paoloni F, et al. Short course of bortezomib in anemic patients with relapsed cold agglutinin disease: a phase 2 prospective GIMEMA study. Blood. 2018;132:547-550.
42. Makishima K, Obara N, Ishitsuka K, et al. High efficacy of eculizumab treatment for fulminant hemolytic anemia in primary cold agglutinin disease. Ann Hematol. 2019;98:1031-1032.
43. Roth A, Huttmann A, Rother RP, et al. Long-term efficacy of the complement inhibitor eculizumab in cold agglutinin disease. Blood. 2009;113:38853886.
44. Jäger U, D'Sa S, Schörgenhofer C, et al. Inhibition of complement C1s improves severe hemolytic anemia in cold agglutinin disease: a first-in-human trial. Blood. 2019;133:893-901.
45. Agarwal SK, Ghosh PK, Gupta D. Cardiac surgery and cold-reactive proteins. Ann Thorac Surg. 1995;60:1143-1150.
46. Shanbhag S, Spivak J. Paroxysmal cold hemoglobinuria. Hematol Oncol Clin North Am. 2015;29:473-478.
47. Kumar ND, Sethi S, Pandhi RK. Paroxysmal cold haemoglobinuria in syphilis patients. Genitourin Med. 1993;69:76.
48. Zantek ND, Koepsell SA, Tharp DR Jr, Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. 2012;87:707-709.
49. Pierce A, Nester T. Pathology consultation on drug-induced hemolytic anemia. Am J Clin Pathol. 2011;136:7-12.
50. Garratty G. Immune cytopenia associated with antibiotics. Transfusion Med Rev. 1993;7:255-267.
51. Petz LD, Mueller-Eckhardt C. Drug-induced immune hemolytic anemia. Transfusion. 1992;32:202-204.
52. Packman CH, Leddy JP. Drug-related immune hemolytic anemia. In: Beutler E, Lichtman MA, Coller BS, Kipps TJ, eds. William’s Hematology. 5th ed. New York: McGraw-Hill; 1995:691-704.
53. Garratty G. Drug-induced immune hemolytic anemia. Hematology Am Soc Hematol Educ Program. 2009;73-79.
54. Leicht HB, Weinig E, Mayer B, et al. Ceftriaxone-induced hemolytic anemia with severe renal failure: a case report and review of literature. BMC Pharmacol Toxicol. 2018;19:67.
55. Chenoweth CE, Judd WJ, Steiner EA, Kauffman CA. Cefotetan-induced immune hemolytic anemia. Clin Infect Dis. 1992;15:863-865.
56. Garratty G, Nance S, Lloyd M, Domen R. Fatal im¬mune hemolytic anemia due to cefotetan. Transfusion. 1992;32:269-271.
57. Endoh T, Yagihashi A, Sasaki M, Watanabe N. Ceftizoxime-induced hemolysis due to immune complexes: case report and determination of the epitope responsible for immune complex-mediated hemolysis. Transfusion. 1999;39:306-309.
58. Arndt PA, Leger RM, Garratty G. Serology of antibodies to second- and third-generation cephalosporins associated with immune hemolytic anemia and/or positive direct antiglobulin tests. Transfusion. 1999;39:1239-1246.
59. Martin ME, Laber DA. Cefotetan-induced hemolytic anemia after perioperative prophylaxis. Am J Hematol. 2006;81:186-188.
60. Bernini JC, Mustafa MM, Sutor LJ, Buchanan GR. Fatal hemolysis induced by ceftriaxone in a child with sickle cell anemia. J Pediatr. 1995;126(5 Pt 1):813-815.
61. Borgna-Pignatti C, Bezzi TM, Reverberi R. Fatal ceftriaxone-induced hemolysis in a child with acquired immunodeficiency syndrome. Pediatr Infect Dis J. 1995;14:1116-1117.
62. Lascari AD, Amyot K. Fatal hemolysis caused by ceftriaxone [see comments]. J Pediatr. 1995;126(5 Pt 1):816-817.
63. Petz LD. Drug-induced autoimmune hemolytic anemia. Transfusion Med Rev. 1993;7:242-254.
64. Leaf RK, Ferreri C, Rangachari D, et al. Clinical and laboratory features of autoimmune hemolytic anemia associated with immune checkpoint inhibitors. Am J Hematol. 2019;94:563-574.
65. DeLoughery T. Drug induced immune hematological disease. Allerg Immunol Clin. 1998;18:829-841.
Autoimmune Hemolytic Anemia: Evaluation and Diagnosis
The autoimmune hemolytic anemias (AIHA) are rare but important hematologic diseases. They can range in severity from mildly symptomatic illness to a rapidly fatal syndrome. The incidence of AIHA is estimated to be between 0.6 and 3 cases per 100,000 persons.1,2 AIHA is mediated by antibodies, and in the majority of cases immunoglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C (Table 1). This article series reviews the most common types of AIHA, with an overview of evaluation and diagnosis presented in this article and management of warm, cold, and drug-induced AIHA reviewed in a separate article.

Pathogenesis
In most cases, the ultimate etiology of AIHA is unknown. In warm AIHA, the target epitopes in most cases are Rh proteins.2 What leads the immune system to target these proteins is unidentified, but one theory is that an initial immune response to a foreign antigen starts to cross-react with the Rh proteins and the immune system fails to suppress this autoreactive response, leading to hemolysis. In IgG-mediated (warm) hemolysis, the red cells become coated with IgG molecules, which mark the cells for uptake and destruction by splenic macrophages.3 In "cold" AIHA, IgM molecules fix complement to the surface of red blood cells. Rarely, this can lead to activation of the full complement cascade, resulting in red cell lysis, but more often it is stopped at the C3 stage, leading to C3-coated red cells which are then taken up by hepatic macrophages.4
Suspecting the Diagnosis
In many patients, it is the symptoms and signs of anemia that lead to suspicion of hemolysis. Older patients often present earlier in the course of the disease due to lack of tolerance of anemia, especially if there is a sudden drop in the red blood cell count. Dark, cola-colored urine resulting from the presence of free hemoglobin may be noted by some patients. Patients with rapid-onset hemolysis may note lumbar back pain, and those with cold agglutinins often note symptoms related to agglutination of red cells in the peripheral circulation, such as the development of acrocyanosis in cold weather.5 In rare cases, patients will have abdominal pain when eating cold food due to ischemia related to agglutination of red cells in the viscera. Some patients with cold agglutinins can have an exacerbation of their hemolysis with cold exposure.
Unlike patients with immune thrombocytopenia, those with AIHA may have mild splenomegaly on exam. The presence of enlarged lymph nodes or massive splenomegaly should raise concern about concomitant lymphoma or chronic lymphocytic leukemia.
Making the Diagnosis
The 2 key steps in diagnosis are (1) demonstrating hemolysis and (2) demonstrating the autoimmune component.
Laboratory Evaluation for Hemolysis
Hemolysis is proven by finding evidence of both red cell breakdown and the compensatory increase in red cell production this stimulates (Table 2). The following sections discuss the laboratory tests that are performed to investigate hemolysis.

Lactate Dehydrogenase
When red cells undergo hemolysis, they release their contents, which are mostly comprised of hemoglobin but also include lactate dehydrogenase (LDH), an enzyme found in high concentration in red cells. Most patients with hemolysis will have an elevated LDH level, making this a sensitive test. However, because many other processes, including liver disease and pneumonia, also raise the serum LDH level, this finding is not specific for hemolysis.
Serum Bilirubin
Hemoglobin is salvaged by haptoglobin, and the heme moiety is broken down first to bilirubin and then to urobilinogen, which is excreted in the urine.2 Bilirubin produced from the breakdown of heme is not conjugated, but rather is delivered to the liver, where it is conjugated and excreted into the bile. In hemolysis, the concentration of unconjugated bilirubin (indirect bilirubin) is increased, while in liver disease the level of conjugated bilirubin (direct bilirubin) is increased. However, if the patient has concomitant liver disease with an increased direct bilirubin level, the serum bilirubin test is not reliable.
Serum Haptoglobin
Haptoglobin binds free serum hemoglobin and is taken up by the liver. Haptoglobin usually falls to very low levels in hemolysis. A confounder is that haptoglobin is an acute phase reactant and can rise with systemic disease or inflammation. However, patients with advanced liver disease will have low haptoglobin levels due to lack of synthesis, and up to 2% of the population may congenitally lack haptoglobin.1
Serum Hemoglobin
If the hemolysis is very rapid, the amount of free hemoglobin released will overwhelm the binding capacity of haptoglobin and lead to free hemoglobin in the plasma. This can be crudely quantified by examining the plasma color. Even minute amounts of free hemoglobin will turn the plasma pink. In fulminant hemolysis, the plasma will be cola-colored.
Reticulocyte Count
In most patients with hemolysis, the destruction of red cells is accompanied by an increase in the reticulocyte count. Reticulocytes are red cells that still contain RNA and are a marker of red cells that are approximately 24 hours old or less. Traditionally, reticulocytes were measured manually by staining the blood smear with vital blue and counting the percentage of cells that absorb the stain; this percentage needs to be adjusted for the hematocrit. Usually a percentage above 1.5% is considered indicative of an elevated reticulocyte count. Recently, automated complete blood count machines have taken advantage of the fact that reticulocytes will absorb certain stains; these machines can directly measure the reticulocyte count via flow cytometry, which results in an “absolute” reticulocyte count. The reticulocyte count obtained using this method does not have to be corrected for hematocrit, and levels of approximately 90,000/μL are considered raised. However, the reticulocyte count can also be raised in blood loss or in patients who have other causes of anemia (eg, iron deficiency) under treatment. In addition, as many as 25% of patients with AIHA will never have raised counts for various reasons, such as nutritional deficiency, autoimmune destruction of red cell precursors, or lack of erythropoietin.
Blood Smear
The blood smear provides vital information. The hallmark laboratory parameter of AIHA is spherocytes seen on the blood smear. In AIHA, antibodies and/or complement attach to the red cells, and when the antibodies or complement are taken up by macrophages in the spleen some of the red blood cell mem-brane is removed as well, decreasing the surface area of the cell. As the surface area of the red cell decreases with each pass through the spleen, the cell's shape changes from a biconcave disk to a sphere before the cell is destroyed, reflecting the fact that a sphere has the smallest surface area for a given volume. The vast majority of patients with AIHA will have spherocytes on the blood smear. However, spherocytes are not specific to AIHA, as they can be seen in hereditary spherocytosis, Wilson’s disease, clostridial sepsis, and severe burns.
Patients with cold agglutinins will often have red cell agglutination on the blood smear. In addition, patients with AIHA will often have a raised mean corpuscular volume (MCV) for 2 reasons. In patients with brisk reticulocytosis, the MCV will be raised due to the large size of the reticulocyte. In patients with cold agglutinin disease, the MCV may be falsely raised due to clumping of the red blood cells.
Urinary Hemosiderin
When hemoglobin is excreted by the kidney, the iron is deposited in the tubules. When the tubule cells are sloughed off, they appear in the urine. The urine can be stained for iron, and a positive result is another sign of hemolysis. Hemosiderinuria is a later sign of hemolysis, as it takes 1 week for iron-laden tubule cells to be excreted in sufficient quantities to be detected in the urine.
Urinary Hemoglobin
One other sign of hemolysis is the presence of hemoglobin in the urine. A quick way to demonstrate hemoglobinuria is to check the urine with a dipstick followed by a microscopic exam. In hemolysis, the dipstick will detect “blood,” while the microscopic exam will be negative for red cells.
Laboratory Evaluation for Autoimmune Component
The autoimmune component is shown by demonstrating the presence of either IgG molecules or complement on the surface of red blood cells.4,6 This can be done by performing the direct antiglobulin test (DAT) or Coombs test. IgG bound to red cells will not agglutinate them, but if IgM that is directed against IgG or C3 is added, the red cells will agglutinate, proving that there is IgG and/or C3 on the red cell membrane. The finding of a positive DAT in the setting of a hemolytic anemia is diagnostic of AIHA. Beware of individuals with concomitant weak positive DAT and other causes of hemolysis. The strength of the DAT result and the degree of hemolysis must match in order to conclude that the hemolysis is immune-mediated.
There are several pitfalls to the DAT. One is that a positive DAT is found in 1:1000 patients in the normal population and in up to several percent of ill patients, especially those with elevated gamma globulin, such as patients with liver disease or HIV infection.6 Administration of intravenous immunoglobulin (IVIG) can also create a positive DAT. Conversely, patients can have AIHA with a negative DAT.7-9 For some patients, the number of IgG molecules bound to the red cell is below the detection limit of the DAT reagents. Other patients can have IgA or “warm” IgM as the cause of the AIHA.10 Specialty laboratories can test for these possibilities. The diagnosis of DAT-negative AIHA should be made with caution, and other causes of hemolysis, such as hereditary spherocytosis or paroxysmal hematuria, should be excluded.
Transfusion Therapy
Performing transfusions can be very difficult in patients with AIHA.2 The presence of the autoantibody can interfere with typing of the blood and almost always interferes with the crossmatch, since this final step consists of mixing the patient’s serum or plasma with donor red cells. In most patients with AIHA, the autoantibodies will react with any donor cells, rendering a negative crossmatch impossible. Without the crossmatch, the concern is that underlying alloantibodies can be missed. Studies indicate that 15% to 32% of patients will have underlying alloantibodies, which can lead to transfusion reactions.2 However, there are 2 considerations that may mitigate these concerns.11,12 First, patients who have never been transfused or pregnant will rarely have alloantibodies. Second, a patient who has been transfused in the remote past may have an anamnestic antibody response but not an immediate hemolytic reaction.
The transfusion service can take several steps to identify alloantibodies. Occasionally, if the autoantibody is weakly reacting when the patient’s serum is tested against a panel of reagent red cells, the alloantibodies can be identified by their stronger reactions as compared with the weakly reactive autoantibody. The most common technique for identifying alloantibodies is the autoadsorption technique.4,13 This involves incu-bating the patient’s red cells with the patient’s serum to adsorb the autoantibody. After a period of incubation, the cells are pelleted and the serum is collected as the supernatant. The adsorbed serum may be incubated with another sample of the patient’s cells for a second adsorption if the initial agglutination reactions of the patient’s serum with the reagent cells were strong. After 1 to 3 adsorptions, the adsorbed serum is tested with a red cell panel in order to check for “leftover” alloantibodies.
When a patient is first suspected of having AIHA, a generous sample of blood should be given to the transfusion service to allow for adequate testing. Many centers will test the blood not only for blood groups ABO and D but also perform full Rh typing plus check for Kidd, Duffy and Kell status.14 Increasingly, this is performed by direct genetic sequencing for the appropriate genotypes. This can allow transfusion of phenotypically matched red blood cells to lessen the risk of alloantibody formation.
One difficult issue is timing of transfusion. Clinicians are often hesitant to transfuse patients with AIHA due to fear of reactions, but in patients with severe anemia, especially elderly patients or those with heart disease, transfusion can be lifesaving. Since in some cases it may take hours to screen for alloantibodies, it is often preferable to transfuse patients with severe anemia and observe carefully for reaction.
1. Liebman HA, Weitz IC. Autoimmune hemolytic anemia. Med Clin North Am. 2017;101:351-359.
2. Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24:195–210.
3. Seve P, Philippe P, Dufour JF, et al. Autoimmune hemolytic anemia: classification and therapeutic approaches. Expert Rev Hematol. 2008;1:189-204.
4. Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
5. Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011;153:309-317.
6. Zantek ND, Koepsell SA, Tharp DR Jr, Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. 2012;87:707-709.
7. Michel M. Classification and therapeutic approaches in autoimmune hemolytic anemia: an update. Expert Rev Hematol. 2011;4:607-618.
8. Garratty G. Immune hemolytic anemia associated with negative routine serology. Semin Hematol. 2005;42:156-164.
9. Sachs UJ, Roder L, Santoso S, Bein G. Does a negative direct antiglobulin test exclude warm autoimmune haemolytic anaemia? A prospective study of 504 cases. Br J Haematol. 2006;132:655-656.
10. Sokol RJ, Booker DJ, Stamps R, et al. IgA red cell autoantibodies and autoimmune hemolysis. Transfusion. 1997;37:175-181.
11. Petz LD. “Least incompatible” units for transfusion in autoimmune hemolytic anemia: should we eliminate this meaningless term? A commentary for clinicians and transfusion medicine professionals. Transfusion. 2003;43:1503-1507.
12. Blackall DP. How do I approach patients with warm-reactive autoantibodies? Transfusion. 2011;51:14-17.
13. Winkelmayer WC, Liu J, Setoguchi S, Choudhry NK. Effectiveness and safety of warfarin initiation in older hemodialysis patients with incident atrial fibrillation. Clin J Am Soc Nephrol. 2011;6:2662-2668.
14. Ness PM. How do I encourage clinicians to transfuse mismatched blood to patients with autoimmune hemolytic anemia in urgent situations? Transfusion. 2006;46:1859-1862.
The autoimmune hemolytic anemias (AIHA) are rare but important hematologic diseases. They can range in severity from mildly symptomatic illness to a rapidly fatal syndrome. The incidence of AIHA is estimated to be between 0.6 and 3 cases per 100,000 persons.1,2 AIHA is mediated by antibodies, and in the majority of cases immunoglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C (Table 1). This article series reviews the most common types of AIHA, with an overview of evaluation and diagnosis presented in this article and management of warm, cold, and drug-induced AIHA reviewed in a separate article.
Pathogenesis
In most cases, the ultimate etiology of AIHA is unknown. In warm AIHA, the target epitopes in most cases are Rh proteins.2 What leads the immune system to target these proteins is unidentified, but one theory is that an initial immune response to a foreign antigen starts to cross-react with the Rh proteins and the immune system fails to suppress this autoreactive response, leading to hemolysis. In IgG-mediated (warm) hemolysis, the red cells become coated with IgG molecules, which mark the cells for uptake and destruction by splenic macrophages.3 In "cold" AIHA, IgM molecules fix complement to the surface of red blood cells. Rarely, this can lead to activation of the full complement cascade, resulting in red cell lysis, but more often it is stopped at the C3 stage, leading to C3-coated red cells which are then taken up by hepatic macrophages.4
Suspecting the Diagnosis
In many patients, it is the symptoms and signs of anemia that lead to suspicion of hemolysis. Older patients often present earlier in the course of the disease due to lack of tolerance of anemia, especially if there is a sudden drop in the red blood cell count. Dark, cola-colored urine resulting from the presence of free hemoglobin may be noted by some patients. Patients with rapid-onset hemolysis may note lumbar back pain, and those with cold agglutinins often note symptoms related to agglutination of red cells in the peripheral circulation, such as the development of acrocyanosis in cold weather.5 In rare cases, patients will have abdominal pain when eating cold food due to ischemia related to agglutination of red cells in the viscera. Some patients with cold agglutinins can have an exacerbation of their hemolysis with cold exposure.
Unlike patients with immune thrombocytopenia, those with AIHA may have mild splenomegaly on exam. The presence of enlarged lymph nodes or massive splenomegaly should raise concern about concomitant lymphoma or chronic lymphocytic leukemia.
Making the Diagnosis
The 2 key steps in diagnosis are (1) demonstrating hemolysis and (2) demonstrating the autoimmune component.
Laboratory Evaluation for Hemolysis
Hemolysis is proven by finding evidence of both red cell breakdown and the compensatory increase in red cell production this stimulates (Table 2). The following sections discuss the laboratory tests that are performed to investigate hemolysis.
Lactate Dehydrogenase
When red cells undergo hemolysis, they release their contents, which are mostly comprised of hemoglobin but also include lactate dehydrogenase (LDH), an enzyme found in high concentration in red cells. Most patients with hemolysis will have an elevated LDH level, making this a sensitive test. However, because many other processes, including liver disease and pneumonia, also raise the serum LDH level, this finding is not specific for hemolysis.
Serum Bilirubin
Hemoglobin is salvaged by haptoglobin, and the heme moiety is broken down first to bilirubin and then to urobilinogen, which is excreted in the urine.2 Bilirubin produced from the breakdown of heme is not conjugated, but rather is delivered to the liver, where it is conjugated and excreted into the bile. In hemolysis, the concentration of unconjugated bilirubin (indirect bilirubin) is increased, while in liver disease the level of conjugated bilirubin (direct bilirubin) is increased. However, if the patient has concomitant liver disease with an increased direct bilirubin level, the serum bilirubin test is not reliable.
Serum Haptoglobin
Haptoglobin binds free serum hemoglobin and is taken up by the liver. Haptoglobin usually falls to very low levels in hemolysis. A confounder is that haptoglobin is an acute phase reactant and can rise with systemic disease or inflammation. However, patients with advanced liver disease will have low haptoglobin levels due to lack of synthesis, and up to 2% of the population may congenitally lack haptoglobin.1
Serum Hemoglobin
If the hemolysis is very rapid, the amount of free hemoglobin released will overwhelm the binding capacity of haptoglobin and lead to free hemoglobin in the plasma. This can be crudely quantified by examining the plasma color. Even minute amounts of free hemoglobin will turn the plasma pink. In fulminant hemolysis, the plasma will be cola-colored.
Reticulocyte Count
In most patients with hemolysis, the destruction of red cells is accompanied by an increase in the reticulocyte count. Reticulocytes are red cells that still contain RNA and are a marker of red cells that are approximately 24 hours old or less. Traditionally, reticulocytes were measured manually by staining the blood smear with vital blue and counting the percentage of cells that absorb the stain; this percentage needs to be adjusted for the hematocrit. Usually a percentage above 1.5% is considered indicative of an elevated reticulocyte count. Recently, automated complete blood count machines have taken advantage of the fact that reticulocytes will absorb certain stains; these machines can directly measure the reticulocyte count via flow cytometry, which results in an “absolute” reticulocyte count. The reticulocyte count obtained using this method does not have to be corrected for hematocrit, and levels of approximately 90,000/μL are considered raised. However, the reticulocyte count can also be raised in blood loss or in patients who have other causes of anemia (eg, iron deficiency) under treatment. In addition, as many as 25% of patients with AIHA will never have raised counts for various reasons, such as nutritional deficiency, autoimmune destruction of red cell precursors, or lack of erythropoietin.
Blood Smear
The blood smear provides vital information. The hallmark laboratory parameter of AIHA is spherocytes seen on the blood smear. In AIHA, antibodies and/or complement attach to the red cells, and when the antibodies or complement are taken up by macrophages in the spleen some of the red blood cell mem-brane is removed as well, decreasing the surface area of the cell. As the surface area of the red cell decreases with each pass through the spleen, the cell's shape changes from a biconcave disk to a sphere before the cell is destroyed, reflecting the fact that a sphere has the smallest surface area for a given volume. The vast majority of patients with AIHA will have spherocytes on the blood smear. However, spherocytes are not specific to AIHA, as they can be seen in hereditary spherocytosis, Wilson’s disease, clostridial sepsis, and severe burns.
Patients with cold agglutinins will often have red cell agglutination on the blood smear. In addition, patients with AIHA will often have a raised mean corpuscular volume (MCV) for 2 reasons. In patients with brisk reticulocytosis, the MCV will be raised due to the large size of the reticulocyte. In patients with cold agglutinin disease, the MCV may be falsely raised due to clumping of the red blood cells.
Urinary Hemosiderin
When hemoglobin is excreted by the kidney, the iron is deposited in the tubules. When the tubule cells are sloughed off, they appear in the urine. The urine can be stained for iron, and a positive result is another sign of hemolysis. Hemosiderinuria is a later sign of hemolysis, as it takes 1 week for iron-laden tubule cells to be excreted in sufficient quantities to be detected in the urine.
Urinary Hemoglobin
One other sign of hemolysis is the presence of hemoglobin in the urine. A quick way to demonstrate hemoglobinuria is to check the urine with a dipstick followed by a microscopic exam. In hemolysis, the dipstick will detect “blood,” while the microscopic exam will be negative for red cells.
Laboratory Evaluation for Autoimmune Component
The autoimmune component is shown by demonstrating the presence of either IgG molecules or complement on the surface of red blood cells.4,6 This can be done by performing the direct antiglobulin test (DAT) or Coombs test. IgG bound to red cells will not agglutinate them, but if IgM that is directed against IgG or C3 is added, the red cells will agglutinate, proving that there is IgG and/or C3 on the red cell membrane. The finding of a positive DAT in the setting of a hemolytic anemia is diagnostic of AIHA. Beware of individuals with concomitant weak positive DAT and other causes of hemolysis. The strength of the DAT result and the degree of hemolysis must match in order to conclude that the hemolysis is immune-mediated.
There are several pitfalls to the DAT. One is that a positive DAT is found in 1:1000 patients in the normal population and in up to several percent of ill patients, especially those with elevated gamma globulin, such as patients with liver disease or HIV infection.6 Administration of intravenous immunoglobulin (IVIG) can also create a positive DAT. Conversely, patients can have AIHA with a negative DAT.7-9 For some patients, the number of IgG molecules bound to the red cell is below the detection limit of the DAT reagents. Other patients can have IgA or “warm” IgM as the cause of the AIHA.10 Specialty laboratories can test for these possibilities. The diagnosis of DAT-negative AIHA should be made with caution, and other causes of hemolysis, such as hereditary spherocytosis or paroxysmal hematuria, should be excluded.
Transfusion Therapy
Performing transfusions can be very difficult in patients with AIHA.2 The presence of the autoantibody can interfere with typing of the blood and almost always interferes with the crossmatch, since this final step consists of mixing the patient’s serum or plasma with donor red cells. In most patients with AIHA, the autoantibodies will react with any donor cells, rendering a negative crossmatch impossible. Without the crossmatch, the concern is that underlying alloantibodies can be missed. Studies indicate that 15% to 32% of patients will have underlying alloantibodies, which can lead to transfusion reactions.2 However, there are 2 considerations that may mitigate these concerns.11,12 First, patients who have never been transfused or pregnant will rarely have alloantibodies. Second, a patient who has been transfused in the remote past may have an anamnestic antibody response but not an immediate hemolytic reaction.
The transfusion service can take several steps to identify alloantibodies. Occasionally, if the autoantibody is weakly reacting when the patient’s serum is tested against a panel of reagent red cells, the alloantibodies can be identified by their stronger reactions as compared with the weakly reactive autoantibody. The most common technique for identifying alloantibodies is the autoadsorption technique.4,13 This involves incu-bating the patient’s red cells with the patient’s serum to adsorb the autoantibody. After a period of incubation, the cells are pelleted and the serum is collected as the supernatant. The adsorbed serum may be incubated with another sample of the patient’s cells for a second adsorption if the initial agglutination reactions of the patient’s serum with the reagent cells were strong. After 1 to 3 adsorptions, the adsorbed serum is tested with a red cell panel in order to check for “leftover” alloantibodies.
When a patient is first suspected of having AIHA, a generous sample of blood should be given to the transfusion service to allow for adequate testing. Many centers will test the blood not only for blood groups ABO and D but also perform full Rh typing plus check for Kidd, Duffy and Kell status.14 Increasingly, this is performed by direct genetic sequencing for the appropriate genotypes. This can allow transfusion of phenotypically matched red blood cells to lessen the risk of alloantibody formation.
One difficult issue is timing of transfusion. Clinicians are often hesitant to transfuse patients with AIHA due to fear of reactions, but in patients with severe anemia, especially elderly patients or those with heart disease, transfusion can be lifesaving. Since in some cases it may take hours to screen for alloantibodies, it is often preferable to transfuse patients with severe anemia and observe carefully for reaction.
The autoimmune hemolytic anemias (AIHA) are rare but important hematologic diseases. They can range in severity from mildly symptomatic illness to a rapidly fatal syndrome. The incidence of AIHA is estimated to be between 0.6 and 3 cases per 100,000 persons.1,2 AIHA is mediated by antibodies, and in the majority of cases immunoglobulin (Ig) G is the mediating antibody. This type of AIHA is referred to as "warm" AIHA because IgG antibodies bind best at body temperature. "Cold" AIHA is mediated by IgM antibodies, which bind maximally at temperatures below 37°C (Table 1). This article series reviews the most common types of AIHA, with an overview of evaluation and diagnosis presented in this article and management of warm, cold, and drug-induced AIHA reviewed in a separate article.
Pathogenesis
In most cases, the ultimate etiology of AIHA is unknown. In warm AIHA, the target epitopes in most cases are Rh proteins.2 What leads the immune system to target these proteins is unidentified, but one theory is that an initial immune response to a foreign antigen starts to cross-react with the Rh proteins and the immune system fails to suppress this autoreactive response, leading to hemolysis. In IgG-mediated (warm) hemolysis, the red cells become coated with IgG molecules, which mark the cells for uptake and destruction by splenic macrophages.3 In "cold" AIHA, IgM molecules fix complement to the surface of red blood cells. Rarely, this can lead to activation of the full complement cascade, resulting in red cell lysis, but more often it is stopped at the C3 stage, leading to C3-coated red cells which are then taken up by hepatic macrophages.4
Suspecting the Diagnosis
In many patients, it is the symptoms and signs of anemia that lead to suspicion of hemolysis. Older patients often present earlier in the course of the disease due to lack of tolerance of anemia, especially if there is a sudden drop in the red blood cell count. Dark, cola-colored urine resulting from the presence of free hemoglobin may be noted by some patients. Patients with rapid-onset hemolysis may note lumbar back pain, and those with cold agglutinins often note symptoms related to agglutination of red cells in the peripheral circulation, such as the development of acrocyanosis in cold weather.5 In rare cases, patients will have abdominal pain when eating cold food due to ischemia related to agglutination of red cells in the viscera. Some patients with cold agglutinins can have an exacerbation of their hemolysis with cold exposure.
Unlike patients with immune thrombocytopenia, those with AIHA may have mild splenomegaly on exam. The presence of enlarged lymph nodes or massive splenomegaly should raise concern about concomitant lymphoma or chronic lymphocytic leukemia.
Making the Diagnosis
The 2 key steps in diagnosis are (1) demonstrating hemolysis and (2) demonstrating the autoimmune component.
Laboratory Evaluation for Hemolysis
Hemolysis is proven by finding evidence of both red cell breakdown and the compensatory increase in red cell production this stimulates (Table 2). The following sections discuss the laboratory tests that are performed to investigate hemolysis.
Lactate Dehydrogenase
When red cells undergo hemolysis, they release their contents, which are mostly comprised of hemoglobin but also include lactate dehydrogenase (LDH), an enzyme found in high concentration in red cells. Most patients with hemolysis will have an elevated LDH level, making this a sensitive test. However, because many other processes, including liver disease and pneumonia, also raise the serum LDH level, this finding is not specific for hemolysis.
Serum Bilirubin
Hemoglobin is salvaged by haptoglobin, and the heme moiety is broken down first to bilirubin and then to urobilinogen, which is excreted in the urine.2 Bilirubin produced from the breakdown of heme is not conjugated, but rather is delivered to the liver, where it is conjugated and excreted into the bile. In hemolysis, the concentration of unconjugated bilirubin (indirect bilirubin) is increased, while in liver disease the level of conjugated bilirubin (direct bilirubin) is increased. However, if the patient has concomitant liver disease with an increased direct bilirubin level, the serum bilirubin test is not reliable.
Serum Haptoglobin
Haptoglobin binds free serum hemoglobin and is taken up by the liver. Haptoglobin usually falls to very low levels in hemolysis. A confounder is that haptoglobin is an acute phase reactant and can rise with systemic disease or inflammation. However, patients with advanced liver disease will have low haptoglobin levels due to lack of synthesis, and up to 2% of the population may congenitally lack haptoglobin.1
Serum Hemoglobin
If the hemolysis is very rapid, the amount of free hemoglobin released will overwhelm the binding capacity of haptoglobin and lead to free hemoglobin in the plasma. This can be crudely quantified by examining the plasma color. Even minute amounts of free hemoglobin will turn the plasma pink. In fulminant hemolysis, the plasma will be cola-colored.
Reticulocyte Count
In most patients with hemolysis, the destruction of red cells is accompanied by an increase in the reticulocyte count. Reticulocytes are red cells that still contain RNA and are a marker of red cells that are approximately 24 hours old or less. Traditionally, reticulocytes were measured manually by staining the blood smear with vital blue and counting the percentage of cells that absorb the stain; this percentage needs to be adjusted for the hematocrit. Usually a percentage above 1.5% is considered indicative of an elevated reticulocyte count. Recently, automated complete blood count machines have taken advantage of the fact that reticulocytes will absorb certain stains; these machines can directly measure the reticulocyte count via flow cytometry, which results in an “absolute” reticulocyte count. The reticulocyte count obtained using this method does not have to be corrected for hematocrit, and levels of approximately 90,000/μL are considered raised. However, the reticulocyte count can also be raised in blood loss or in patients who have other causes of anemia (eg, iron deficiency) under treatment. In addition, as many as 25% of patients with AIHA will never have raised counts for various reasons, such as nutritional deficiency, autoimmune destruction of red cell precursors, or lack of erythropoietin.
Blood Smear
The blood smear provides vital information. The hallmark laboratory parameter of AIHA is spherocytes seen on the blood smear. In AIHA, antibodies and/or complement attach to the red cells, and when the antibodies or complement are taken up by macrophages in the spleen some of the red blood cell mem-brane is removed as well, decreasing the surface area of the cell. As the surface area of the red cell decreases with each pass through the spleen, the cell's shape changes from a biconcave disk to a sphere before the cell is destroyed, reflecting the fact that a sphere has the smallest surface area for a given volume. The vast majority of patients with AIHA will have spherocytes on the blood smear. However, spherocytes are not specific to AIHA, as they can be seen in hereditary spherocytosis, Wilson’s disease, clostridial sepsis, and severe burns.
Patients with cold agglutinins will often have red cell agglutination on the blood smear. In addition, patients with AIHA will often have a raised mean corpuscular volume (MCV) for 2 reasons. In patients with brisk reticulocytosis, the MCV will be raised due to the large size of the reticulocyte. In patients with cold agglutinin disease, the MCV may be falsely raised due to clumping of the red blood cells.
Urinary Hemosiderin
When hemoglobin is excreted by the kidney, the iron is deposited in the tubules. When the tubule cells are sloughed off, they appear in the urine. The urine can be stained for iron, and a positive result is another sign of hemolysis. Hemosiderinuria is a later sign of hemolysis, as it takes 1 week for iron-laden tubule cells to be excreted in sufficient quantities to be detected in the urine.
Urinary Hemoglobin
One other sign of hemolysis is the presence of hemoglobin in the urine. A quick way to demonstrate hemoglobinuria is to check the urine with a dipstick followed by a microscopic exam. In hemolysis, the dipstick will detect “blood,” while the microscopic exam will be negative for red cells.
Laboratory Evaluation for Autoimmune Component
The autoimmune component is shown by demonstrating the presence of either IgG molecules or complement on the surface of red blood cells.4,6 This can be done by performing the direct antiglobulin test (DAT) or Coombs test. IgG bound to red cells will not agglutinate them, but if IgM that is directed against IgG or C3 is added, the red cells will agglutinate, proving that there is IgG and/or C3 on the red cell membrane. The finding of a positive DAT in the setting of a hemolytic anemia is diagnostic of AIHA. Beware of individuals with concomitant weak positive DAT and other causes of hemolysis. The strength of the DAT result and the degree of hemolysis must match in order to conclude that the hemolysis is immune-mediated.
There are several pitfalls to the DAT. One is that a positive DAT is found in 1:1000 patients in the normal population and in up to several percent of ill patients, especially those with elevated gamma globulin, such as patients with liver disease or HIV infection.6 Administration of intravenous immunoglobulin (IVIG) can also create a positive DAT. Conversely, patients can have AIHA with a negative DAT.7-9 For some patients, the number of IgG molecules bound to the red cell is below the detection limit of the DAT reagents. Other patients can have IgA or “warm” IgM as the cause of the AIHA.10 Specialty laboratories can test for these possibilities. The diagnosis of DAT-negative AIHA should be made with caution, and other causes of hemolysis, such as hereditary spherocytosis or paroxysmal hematuria, should be excluded.
Transfusion Therapy
Performing transfusions can be very difficult in patients with AIHA.2 The presence of the autoantibody can interfere with typing of the blood and almost always interferes with the crossmatch, since this final step consists of mixing the patient’s serum or plasma with donor red cells. In most patients with AIHA, the autoantibodies will react with any donor cells, rendering a negative crossmatch impossible. Without the crossmatch, the concern is that underlying alloantibodies can be missed. Studies indicate that 15% to 32% of patients will have underlying alloantibodies, which can lead to transfusion reactions.2 However, there are 2 considerations that may mitigate these concerns.11,12 First, patients who have never been transfused or pregnant will rarely have alloantibodies. Second, a patient who has been transfused in the remote past may have an anamnestic antibody response but not an immediate hemolytic reaction.
The transfusion service can take several steps to identify alloantibodies. Occasionally, if the autoantibody is weakly reacting when the patient’s serum is tested against a panel of reagent red cells, the alloantibodies can be identified by their stronger reactions as compared with the weakly reactive autoantibody. The most common technique for identifying alloantibodies is the autoadsorption technique.4,13 This involves incu-bating the patient’s red cells with the patient’s serum to adsorb the autoantibody. After a period of incubation, the cells are pelleted and the serum is collected as the supernatant. The adsorbed serum may be incubated with another sample of the patient’s cells for a second adsorption if the initial agglutination reactions of the patient’s serum with the reagent cells were strong. After 1 to 3 adsorptions, the adsorbed serum is tested with a red cell panel in order to check for “leftover” alloantibodies.
When a patient is first suspected of having AIHA, a generous sample of blood should be given to the transfusion service to allow for adequate testing. Many centers will test the blood not only for blood groups ABO and D but also perform full Rh typing plus check for Kidd, Duffy and Kell status.14 Increasingly, this is performed by direct genetic sequencing for the appropriate genotypes. This can allow transfusion of phenotypically matched red blood cells to lessen the risk of alloantibody formation.
One difficult issue is timing of transfusion. Clinicians are often hesitant to transfuse patients with AIHA due to fear of reactions, but in patients with severe anemia, especially elderly patients or those with heart disease, transfusion can be lifesaving. Since in some cases it may take hours to screen for alloantibodies, it is often preferable to transfuse patients with severe anemia and observe carefully for reaction.
1. Liebman HA, Weitz IC. Autoimmune hemolytic anemia. Med Clin North Am. 2017;101:351-359.
2. Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24:195–210.
3. Seve P, Philippe P, Dufour JF, et al. Autoimmune hemolytic anemia: classification and therapeutic approaches. Expert Rev Hematol. 2008;1:189-204.
4. Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
5. Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011;153:309-317.
6. Zantek ND, Koepsell SA, Tharp DR Jr, Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. 2012;87:707-709.
7. Michel M. Classification and therapeutic approaches in autoimmune hemolytic anemia: an update. Expert Rev Hematol. 2011;4:607-618.
8. Garratty G. Immune hemolytic anemia associated with negative routine serology. Semin Hematol. 2005;42:156-164.
9. Sachs UJ, Roder L, Santoso S, Bein G. Does a negative direct antiglobulin test exclude warm autoimmune haemolytic anaemia? A prospective study of 504 cases. Br J Haematol. 2006;132:655-656.
10. Sokol RJ, Booker DJ, Stamps R, et al. IgA red cell autoantibodies and autoimmune hemolysis. Transfusion. 1997;37:175-181.
11. Petz LD. “Least incompatible” units for transfusion in autoimmune hemolytic anemia: should we eliminate this meaningless term? A commentary for clinicians and transfusion medicine professionals. Transfusion. 2003;43:1503-1507.
12. Blackall DP. How do I approach patients with warm-reactive autoantibodies? Transfusion. 2011;51:14-17.
13. Winkelmayer WC, Liu J, Setoguchi S, Choudhry NK. Effectiveness and safety of warfarin initiation in older hemodialysis patients with incident atrial fibrillation. Clin J Am Soc Nephrol. 2011;6:2662-2668.
14. Ness PM. How do I encourage clinicians to transfuse mismatched blood to patients with autoimmune hemolytic anemia in urgent situations? Transfusion. 2006;46:1859-1862.
1. Liebman HA, Weitz IC. Autoimmune hemolytic anemia. Med Clin North Am. 2017;101:351-359.
2. Barros MM, Blajchman MA, Bordin JO. Warm autoimmune hemolytic anemia: recent progress in understanding the immunobiology and the treatment. Transfus Med Rev. 2010;24:195–210.
3. Seve P, Philippe P, Dufour JF, et al. Autoimmune hemolytic anemia: classification and therapeutic approaches. Expert Rev Hematol. 2008;1:189-204.
4. Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
5. Berentsen S. How I manage cold agglutinin disease. Br J Haematol. 2011;153:309-317.
6. Zantek ND, Koepsell SA, Tharp DR Jr, Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. 2012;87:707-709.
7. Michel M. Classification and therapeutic approaches in autoimmune hemolytic anemia: an update. Expert Rev Hematol. 2011;4:607-618.
8. Garratty G. Immune hemolytic anemia associated with negative routine serology. Semin Hematol. 2005;42:156-164.
9. Sachs UJ, Roder L, Santoso S, Bein G. Does a negative direct antiglobulin test exclude warm autoimmune haemolytic anaemia? A prospective study of 504 cases. Br J Haematol. 2006;132:655-656.
10. Sokol RJ, Booker DJ, Stamps R, et al. IgA red cell autoantibodies and autoimmune hemolysis. Transfusion. 1997;37:175-181.
11. Petz LD. “Least incompatible” units for transfusion in autoimmune hemolytic anemia: should we eliminate this meaningless term? A commentary for clinicians and transfusion medicine professionals. Transfusion. 2003;43:1503-1507.
12. Blackall DP. How do I approach patients with warm-reactive autoantibodies? Transfusion. 2011;51:14-17.
13. Winkelmayer WC, Liu J, Setoguchi S, Choudhry NK. Effectiveness and safety of warfarin initiation in older hemodialysis patients with incident atrial fibrillation. Clin J Am Soc Nephrol. 2011;6:2662-2668.
14. Ness PM. How do I encourage clinicians to transfuse mismatched blood to patients with autoimmune hemolytic anemia in urgent situations? Transfusion. 2006;46:1859-1862.
Gastrointestinal Stromal Tumors: Management of Localized Disease
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumor of the gastrointestinal (GI) tract and arise from the interstitial cells of Cajal of the myenteric plexus. These tumors are rare, with about 1 case per 100,000 persons diagnosed in the United States annually, but may be incidentally discovered in up to 1 in 5 autopsy specimens of older adults.1,2 Epidemiologic risk factors include increasing age, with a peak incidence between age 60 and 65 years, male gender, black race, and non-Hispanic white ethnicity. Germline predisposition can also increase the risk of developing GISTs; molecular drivers of GIST include gain-of-function mutations in the KIT proto-oncogene and platelet-derived growth factor receptor α (PDGFRA) gene, which both encode structurally similar tyrosine kinase receptors; germline mutations of succinate dehydrogenase (SDH) subunit genes; and mutations associated with neurofibromatosis type 1.
GISTs most commonly involve the stomach, followed by the small intestine, but can arise anywhere within the GI tract (esophagus, colon, rectum, and anus). They can also develop outside the GI tract, arising from the mesentery, omentum, and retroperitoneum. The majority of cases are localized or locoregional, whereas about 20% are metastatic at presentation.1 GISTs can occur in children, adolescents, and young adults. Pediatric GISTs represent a distinct subset marked by female predominance and gastric origin, are often multifocal, can sometimes have lymph node involvement, and typically lack mutations in the KIT and PDGFRA genes.
This review is the first of 2 articles focusing on the diagnosis and management of GISTs. Here, we review the evaluation and diagnosis of GISTs along with management of localized disease. Management of advanced disease is reviewed in a separate article.
Case Presentation
A 64-year-old African American man with progressive iron deficiency and abdominal discomfort undergoes upper and lower endoscopy and is found to have a bulging mass within his abdominal cavity. He undergoes a computed tomography (CT) evaluation of the chest, abdomen, and pelvis with contrast, which reveals the presence of a 10-cm gastric mass, with no other lesions identified. He undergoes surgical resection of the mass and presents for review of his pathology and to discuss his treatment plan.
What histopathologic features are consistent with GIST?
What factors are used for risk stratification and to predict likelihood of recurrence?
Clinical Presentation and Diagnosis
Most patients present with symptoms of overt or occult GI bleeding or abdominal discomfort, but a significant proportion of GISTs are discovered incidentally. Lymph node involvement is not typical, except for GISTs occurring in children and/or with rare syndromes. Most syndromic GISTs are multifocal and multicentric. After surgical resection, GISTs usually recur or metastasize within the abdominal cavity, including the omentum, peritoneum, or liver. These tumors rarely spread to the lungs, brain, or bones; when tumor spread does occur, it tends to be in heavily pre-treated patients with advanced disease who have been on multiple lines of therapy for a long duration of time.
The diagnosis usually can be made by histopathology. Specimens can be obtained by endoscopic ultrasound (EUS)– or CT-guided methods, the latter of which carries a very small risk of contamination from percutaneous biopsy. In terms of morphology, GISTs can be spindle cell, epithelioid, or mixed neoplasms. Epithelioid tumors are more commonly seen in the stomach and are often PDGFRA-mutated or SDH-deficient. The differential diagnosis includes other soft-tissue GI wall tumors such as leiomyosarcomas/leiomyomas, germ cell tumors, lymphomas, fibromatosis, and neuroendocrine and neurogenic tumors. A unique feature of GISTs that differentiates them from leiomyomas is near universal expression of CD117 by immunohistochemistry (IHC); this characteristic has allowed pathologists and providers to accurately distinguish true GISTs from other GI mesenchymal tumors.3 Recently, DOG1 (discovered on GIST1) immunoreactivity has been found to be helpful in identifying patients with CD117-negative GISTs. Initially identified through gene expression analysis of GISTs, DOG1 IHC can identify the common mutant c-Kit-driven CD117-positive GISTs as well as the rare CD117-negative GISTs, which are often driven by mutated PDGFRA.4 Importantly, IHC for KIT and DOG1 are not surrogates for mutational status, nor are they predictive of tyrosine kinase inhibitor (TKI) sensitivity. If IHC of a tumor specimen is CD117- and DOG1-negative, the specimen can be sent for KIT and PDGFRA mutational analysis to confirm the diagnosis. If analysis reveals that these genes are wild-type, then IHC staining for SDH B (SDHB) should follow to assess for an SDH-deficient GIST (negative staining).
Risk Stratification for Recurrence
The clinical behavior of GISTs can be variable. Some are indolent, while others behave more aggressively, with a greater malignant potential and a higher propensity to recur and metastasize. Clinical and pathologic features can provide important prognostic information that allows providers to risk-stratify patients. Various institutions have assessed prognostic variables for GISTs. In 2001, the National Institutes of Health (NIH) held a GIST workshop that proposed an approach to estimating metastatic risk based on tumor size and mitotic index (NIH or Fletcher criteria).5 Joensuu et al later proposed a modification of the NIH risk classification to include tumor location and tumor rupture (modified NIH criteria or Joensuu criteria).6-8 Similarly, the Armed Forces Institute of Pathology (AFIP) identified tumor site as a prognostic factor, with gastric GISTs having the best prognosis (AFIP-Miettinen criteria).9-11 Tabular schemes were designed which stratified patients into discrete groups with ranges for mitotic rate and tumor size. Nomograms for ease of use were then constructed utilizing a bimodal mitotic rate and included tumor site and size.12 Finally, contour maps were developed, which have the advantage of evaluating mitotic rate and tumor size as continuous nonlinear variables and also include tumor site and rupture (associated with a high risk of peritoneal metastasis) separately, further improving risk assessment. These contour maps have been validated against pooled data from 10 series (2560 patients).13 High-risk features identified from these studies include tumor location, size, mitotic rate and tumor rupture and are now used for deciding on the use of adjuvant imatinib and as requirements to enter clinical trials assessing adjuvant therapy for resected GISTs.
Case Continued
The patient’s operative and pathology reports indicate that the tumor is a spindle cell neoplasm of the stomach that is positive for CD117, DOG1, and CD34 and negative for smooth muscle actin and S-100, consistent with a diagnosis of GIST. Resection margins are negative. There are 10 mitoses per 50 high-power fields (HPF). Per the operative report, there was no intraoperative or intraperitoneal tumor rupture. Thus, while his GIST was gastric, which generally has a more favorable prognosis, the tumor harbors high-risk features based on its size and mitotic index.
What further testing should be requested?
Molecular Alterations
It is recommended that a mutational analysis be performed as part of the diagnostic work-up of all GISTs.14 Mutational analysis can provide prognostic and predictive information for sensitivity to imatinib and should be considered standard of care. It may also be useful for confirming a GIST diagnosis, or, if negative, lead to further evaluation with an IHC stain for SDHB. The c-Kit receptor is a member of the tyrosine kinase family and, through direct interactions with stem cell factor (SCF), can upregulate the PI3K/AKT/mTOR, Ras/Raf/MEK/ERK, and JAK-STAT pathways, resulting in transcription and translation of genes that enhance cell growth and survival.15 The cell of origin of GISTs, the interstitial cells of Cajal, are dependent on the SCF–c-Kit interaction for development.16 Likewise, the large majority of GISTs (about 70%) are driven by upregulation and constitutive activation of c-Kit, which is normally autoinhibited. About 80% of KIT mutations involve exon 11; these GISTs are most often associated with a gastric location and are associated with a favorable recurrence-free survival (RFS) rate with surgery alone.17KIT exon 9 mutations are much less common, encompassing only about 10% of GIST KIT mutations, and GISTs with these mutations are more likely to arise from the small bowel.17
About 8% of GISTs harbor gain-of-function PDGFRA driver mutations rendering constitutively active PDGFRA.18PDGFRA mutations are mutually exclusive from KIT mutations, and PDGFRA-mutated tumors most often occur in the stomach. PDGFRA mutations generally are associated with a lower mitotic rate and gastric location. Identification of the PDGFRA D842V mutation on exon 18, which is the most common, is important, as it is associated with imatinib resistance, and these patients should not be offered imatinib.19
Several other mutations associated with GISTs outside of the KIT and PDGFRA spectrum have been identified. About 10% of GISTs are wildtype for KIT and PDGFRA, and not all KIT/PDGFRA-wildtype GISTs are imatinib-sensitive and/or respond to other TKIs.18 These tumors may harbor aberrations in SDH and NF1, or less commonly, BRAF V600E, FGFR, and NTRK.20,21 SDH subunits B, C and D play a role in the Krebs cycle and electron transport chain. Germline mutations in these SDH subunits can result in the Carney-Stratakis syndrome characterized by the dyad of multifocal GISTs and multicentric paragangliomas.22 This syndrome is most likely to manifest in the pediatric or young adult population. In contradistinction is the Carney triad, which is associated with acquired loss of function of the SDHC gene due to promoter hypermethylation. This syndrome classically occurs in young women and is characterized by an indolent-behaving triad of multicentric GISTs, non-adrenal paragangliomas, and pulmonary chondromas.23 Like PDGFRA D842V–mutated GISTs, SDH-deficient and NF1-associated GISTs are considered imatinib resistant, and these patients should not be offered imatinib therapy.14
Case Continued
The patient’s GIST is found to harbor a KIT exon 11 single codon deletion. He appears anxious and asks to have everything done to prevent his GIST from coming back and to improve his lifespan.
What are the next steps in the management of this patient?
Management
A multidisciplinary team approach to the management of all GISTs is essential and includes input from radiology, gastroenterology, pathology, medical and surgical oncology, nuclear medicine, and nursing.
Surgical Resection
Small esophagogastric and duodenal GISTs ≤ 2 cm can be asymptomatic and managed with serial endoscopic surveillance, typically every 6 to 12 months, with biopsies if the tumors increase in size. GISTs larger than 2 cm require surgical resection, with resection of the full pseudocapsule and an R0 resection, if possible, since larger GISTs carry a higher risk of growth and recurrence. If an R0 resection would lead to significant morbidity or functional sequelae, an R1 may suffice. Rectal GISTs are an exception, where microscopic margins have been shown to be associated with an increased risk of local failure.24 It is important to explore the abdomen thoroughly for peritoneal, rectovaginal, and vesicular implants and metastasis to the liver. A formal lymph node dissection is not necessary because lymph nodes are rarely involved and should only be removed when clinically suspicious. Tumor rupture must be avoided. A laparoscopic approach should only be considered for smaller tumors, since there is a risk of tumor rupture with larger tumors.14
When is adjuvant imatinib indicated?
Adjuvant Imatinib
Among patients with local or locally advanced GISTs, the risk of death from recurrence with surgery alone can be high, with a historical 5-year overall survival (OS) of about 35%.25 As a result, multiple studies have assessed the benefit of adjuvant imatinib, which is now considered standard of care for patients with imatinib-sensitive, high-risk GISTs. In addition to inhibiting BCR-ABL, imatinib mesylate inhibits multiple other receptor tyrosine kinases, including PDGFR, SCF and c-Kit. As a result, imatinib has demonstrated in vitro inhibition of cell proliferation and apoptosis and clinical activity against GISTs expressing CD117.26 Importantly, adjuvant imatinib should only be offered to patients with imatinib-sensitive mutations, such as KIT exon 11 and KIT exon 9 mutations. Adjuvant imatinib should not be offered to patients with imatinib-insensitive mutations such as PDGFR 842V, NF1, or BRAF-related or SDH-deficient GISTs.
The ACOSOG Z9000 was the first study of adjuvant imatinib in patients with resected GISTs.25 This was a single-arm, phase 2 study involving 106 patients with surgically resected GISTs deemed high-risk for recurrence, defined as size > 10 cm, tumor rupture, or up to 4 peritoneal implants. Patients were treated with imatinib 400 mg daily for 1 year. The primary and secondary endpoints were OS and RFS, respectively. Long-term follow-up of this study demonstrated 1-, 3-, and 5-year OS of 99%, 97%, and 83%, and 1-, 3-, and 5-year RFS of 96%, 60%, and 40%, which compared favorably with historical controls. In a multivariable analysis, increasing tumor size, small bowel location, KIT exon 9 mutation, high mitotic rate, and older age were independent risk factors for a poor RFS.25 It is important to note that the benefit of adjuvant imatinib waned after discontinuation of therapy, creating a rationale to study adjuvant imatinib for longer periods of time.
As a result of the promising phase 2 data, ACOSOG opened a phase 3 randomized trial (Z9001) comparing 1 year of adjuvant imatinib to placebo among patients with surgically resected GISTs that were > 3 cm in size and that stained positive for CD117 on pathology. The trial accrued 713 patients and was stopped early at a planned interim analysis, which revealed a 1-year RFS of 98% for imatinib versus 83% for placebo (hazard ratio [HR], 0.35; P < 0.001). The 1-year OS did not differ between the 2 arms (92.2% vs 99.7%; HR, 0.66; P = 0.47).27 When comparing the 2 arms, imatinib was associated with a higher RFS among patients with a KIT exon 11 deletion, but not among patients with other KIT mutation types, PDGFRA mutations, or who were KIT/PDGFRA wildtype.28 Imatinib was granted approval by the US Food and Drug Administration (FDA) for the adjuvant treatment of high-risk GISTs based on the results of the ACOSOG Z9001 trial.
The EORTC 62024 study was a randomized placebo-controlled trial assessing the benefit of 2 years of adjuvant imatinib.29 Patients had to be considered intermediate or high risk per the 2002 NIH consensus classification to be eligible. The trial enrolled 918 patients. The 5-year OS rate, the original primary endpoint, did not differ between the 2 groups (100% vs 99%). The 3-year and 5-year RFS rates, secondary endpoints, were significantly longer among patients treated with imatinib (84% vs 66% and 69% vs 63%, respectively). Again, it was noted that the benefit of imatinib waned over time after treatment discontinuation.
The Scandinavian Sarcoma Group (SSG XVIII) trial was a prospective randomized phase 3 trial that compared 3 years versus 1 year of adjuvant imatinib.30 Patients had to be enrolled within 12 weeks of the postoperative period and had to have GISTs that were CD117-positive and with a high estimated risk of recurrence, per the modified NIH consensus criteria (size > 10 cm, > 10 mitoses per 50 HPF, diameter > 5 cm with mitotic count > 5, or tumor rupture before or at surgery). Three years of adjuvant imatinib was associated with a 54% reduction in the hazard for recurrence at 5 years (65.6% vs 47.9%; HR, 0.46; P < 0.001) and a 55% reduction in the hazard for death at 5 years (OS 92% vs 81.7%; HR, 0.45; P = 0.02). Based on the results of this study, the FDA granted approval for the use of 3 years of adjuvant imatinib in patients with high-risk resected GISTs.
The observation that a longer duration of adjuvant imatinib was associated with superior RFS and OS led to studies to further explore longer durations of adjuvant imatinib. The PERSIST-5 (Postresection Evaluation of Recurrence-free Survival for Gastrointestinal Stromal Tumors With 5 Years of Adjuvant Imatinib) was a multicenter, single-arm, phase 2 prospective study of adjuvant imatinib with a primary endpoint of RFS after 5 years.31 Patients had to have an intermediate or high risk of recurrence, which included GISTs at any site > 2 cm with > 5 mitoses per 50 HPF or nongastric GISTs that were ≥ 5 cm. With 91 patients enrolled, the estimated 5-year RFS was 90% and the OS was 95%. Of note, about half of the patients stopped treatment early due to a variety of reasons, including patient choice or adverse events. Importantly, there were no recurrences in patients with imatinib-sensitive mutations while on therapy. We know that in patients at high risk of relapse, adjuvant imatinib delays recurrence and improves survival, but whether any patients are cured, or their survival curves are just shifted to the right, is unknown. Only longer follow-up of existing studies, and the results of newer trials utilizing longer durations of adjuvant treatment, will help to determine the real value of adjuvant therapy for GIST patients.32 Based on this study, it would be reasonable to discuss a longer duration of imatinib with patients deemed to be at very high risk of recurrence and who are tolerating therapy well. We are awaiting the data from the randomized phase 3 Scandinavian Sarcoma Group XII trial comparing 5 years versus 3 years of adjuvant imatinib therapy, and from the French ImadGIST trial of adjuvant imatinib for 3 versus 6 years. A summary of the aforementioned key adjuvant trials is shown in the Table.

When imatinib is commenced, careful monitoring for treatment toxicities and drug interactions should ensue in order to improve compliance. Dose density should be maintained if possible, as retrospective studies suggest suboptimal plasma levels are associated with a worse outcome.33
When should neoadjuvant imatinib be considered?
Neoadjuvant Imatinib
Neoadjuvant imatinib should be considered for patients requiring total gastrectomy, esophagectomy, or abdominoperineal resection of the rectum in order to reduce tumor size, limit subsequent surgical morbidity, mitigate tumor bleeding and rupture, and aid with organ preservation. Patients with rectal GISTs that may otherwise warrant an abdominoperineal resection should be offered a trial of imatinib in the neoadjuvant setting. There is no evidence for the use of any other TKI aside from imatinib in the neoadjuvant or adjuvant setting. With neoadjuvant imatinib, it is difficult to accurately assess the mitotic rate in the resected tumor specimen.
The RTOG 0132/ACRIN 6665 trial was a prospective phase 2 study evaluating the efficacy of imatinib 600 mg daily in the perioperative setting.34 The trial enrolled 50 patients, 30 with primary GISTs (group A) and 22 with recurrent metastatic GISTs (group B). Based on data from the metastatic setting revealing a time to treatment response of about 2.5 months, patients were treated with 8 to 12 weeks of preoperative imatinib followed by 2 years of adjuvant imatinib. Imatinib was stopped 24 hours preoperatively and resumed as soon as possible postoperatively. In group A, 7% of patients achieved a partial response (PR), 83% achieved stable disease, and 2-year progression-free survival (PFS) and OS were 83% and 93%, respectively. In group B, 4.5% of patients achieved a PR, 91% achieved stable disease, and 4.5% experienced progressive disease in the preoperative period; the 2-year PFS and OS were 77% and 91%, respectively. The results of this trial demonstrated the feasibility of using perioperative imatinib with minimal effects on surgical outcomes and set the rationale to use neoadjuvant imatinib in select patients with borderline resectable or rectal GISTs. Another EORTC pooled analysis from 10 sarcoma centers revealed that after a median of 10 months of neoadjuvant imatinib, 83.2% of patients achieved an R0 resection and only 1% progressed during treatment.35 After a median follow-up of 46 months, the 5-year disease-free survival and OS were 65% and 87%, respectively.
Mutational testing should be performed beforehand to ensure the tumor is imatinib-sensitive. If a KIT exon 9 mutation is identified, then 400 mg twice daily should be considered (given the benefit seen with 800 mg imatinib for advanced GIST patients), although there are no studies to confirm this practice. Neoadjuvant imatinib is recommended for a total of 6 to 12 months to ensure maximal tumor debulking, but with very close monitoring and surgical input for disease resistance and growth.14 Imatinib should be stopped 1 to 2 days preoperatively and resumed once the patient has recovered from surgery for a total of 3 years (pre-/postoperatively combined). Neoadjuvant therapy has been shown to be safe and effective, but there have been no randomized trials to assess survival.
What is appropriate surveillance for resected GISTs?
Surveillance
There have been no randomized studies to guide the management of surveillance after surgical resection and adjuvant therapy. There is no known optimal follow-up schedule, but several have been proposed.13,36 Among high-risk patients, it is suggested to image every 3 to 6 months during adjuvant therapy, followed by every 3 months for 2 years after discontinuing therapy, then every 6 months for another 3 years and annually thereafter for an additional 5 years. High-risk patients usually relapse within 1 to 3 years after finishing adjuvant therapy, while low-risk patients can relapse later given that their disease can be slower growing. It has been recommended that low-risk patients undergo imaging every 6 months for 5 years, with follow-up individualized thereafter. Very-low-risk patients may not require more than annual imaging. Because most relapses occur within the peritoneum or liver, imaging should encompass the abdomen and pelvis. Surveillance imaging usually consists of CT scans of the abdomen and pelvis. MRI scans can be utilized for patients at lower risk or who are out several years in order to avoid excess radiation exposure. MRI is also specifically helpful for rectal and esophageal lesions. Chest CT or chest radiograph and bone scan are not routinely required for follow-up.
Case Conclusion
The patient receives adjuvant imatinib and experiences grade 2 myalgias, periorbital edema, and macrocytic anemia, which result in imatinib discontinuation after 3 years of treatment. He is seen every 3 to 6 months and a contrast CT abdomen and pelvis is obtained every 6 months for 5 years. During this 5-year follow-up period, he does not have any clinical or radiographic evidence of disease recurrence.
Further follow-up of this patient is presented in the second article in this 2-part review of management of GISTs.
Key Points
- GISTs are the most common mesenchymal neoplasms of the GI tract and can occasionally occur in extragastrointestinal locations as well.
- GISTs encompass a heterogeneous family of tumor subsets with different natural histories, mutations, and TKI responsiveness.
- Surgery is the mainstay of treatment for localized GISTs, with cure rates greater than 50%.
- For very small (< 2 cm) esophagogastric GISTs, endoscopic ultrasound evaluation and follow-up is recommended.
- For tumors ≥ 2 cm, biopsy and excision is the standard approach.
- For localized GISTs, complete surgical resection (R0) is standard treatment, with no lymphadenectomy for clinically negative lymph nodes.
- Mutational analysis should be considered standard of practice. It can be helpful for confirming the diagnosis and can be predictive and prognostic in determining specific TKI therapy and dose.
- Adjuvant imatinib at a dose of 400 mg for 3 years is standard of care for GISTs that are at high risk of relapse and are imatinib-sensitive, and it is the only TKI approved for adjuvant therapy. Patients with PDGFRA D842V, NF1, BRAF or SDH-deficient GISTs should not receive adjuvant imatinib therapy.
- Neoadjuvant therapy can be utilized for sites where extensive resection would lead to significant morbidity. It should be given for 6 to 12 months, but patients need to be monitored closely for tumor growth.
1. Ma GL, Murphy JD, Martinez ME et al. Epidemiology of gastrointestinal stromal tumors in the era of histology codes: results of a population-based study. Cancer Epidemiol Biomarkers Prev. 2015;24:298-302.
2. Agaimy A, Wunsch PH, Hofstaedter F, et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol. 2007;31:113-120.
3. Miettinen M, Sobin LH, Sarlomo-Rikala M. Immunohistochemical spectrum of GISTs at different sites and their differential diagnosis with a reference to CD117 (KIT). Mod Pathol. 2000;13:1134-1142.
4. West RB, Corless CL, Chen X, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutational status. Am J Pathol. 2004;165:107-113.
5. Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Int J Surg Pathol. 2002;10:81-89.
6. Joensuu H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol. 2008;39:1411-1419.
7. Hohenberger P, Ronellenfitsch U, Oladeji O, et al. Pattern of recurrence in patients with ruptured primary gastrointestinal stromal tumor. Br J Surg. 2010;97:1854-1859.
8. Holmenbakk T, Bjerkehagen B, Boye K, et al. Definition and clinical significance of tumor rupture in gastrointestinal stromal tumours of the small intestine. Br J Surg. 2016;103:684-691.
9. Emory TS, Sobin LH, Lukes L, et al. Prognosis of gastrointestinal smooth-muscle (stromal) tumors: dependence on anatomic site. Am J Surg Pathol. 1999;23:82-87.
10. Miettinen M, Makhlouf H, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol. 2006;30:477-489.
11. Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol. 2005;29:52-68.
12. Gold JS, Gonen M, Gutierrez A, et al. Development and validation of a prognostic nomogram for recurrence-free survival after complete surgical resection of localized primary gastrointestinal stromal tumour: a retrospective analysis. Lancet Oncol. 2009;10:1045-1052.
13. Joensuu H, Vehtari A, Rihimaki J et al. Risk of recurrence of gastrointestinal stromal tumor after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13:265-274.
14. Casali PG, Abecassis N, Bauer S, et al. Gastrointestinal stromal tumours: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow up. Ann Oncol. 2018;29(Supplement_4): iv267.
15. Jing L, Yan-Ling W, Bing-Jia C, et al. The c-kit receptor-mediated signal transduction and tumor-related diseases. Int J Biol Sci. 2013;9:435-443.
16. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577-580.
17. Joensuu H, Rutkowski P, Nishida T, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015;33:634-642.
18. Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22:3813-3825.
19. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342-4349.
20. Huss S, Pasternack H, Ihle MA, et al. Clinicopathological and molecular features of a large cohort of gastrointestinal stromal tumors (GISTs) and review of the literature: BRAF mutations in KIT/PDGFRA wild-type GISTs are rare events. Hum Pathol. 2017;62:206-214.
21. Shi E, Chmielecki J, Tang CM, et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J Transl Med. 2016;14:339.
22. Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet. 2002;108:132-139.
23. Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc. 1999;74:543-552.
24. Jakob J, Mussi C, Ronellenfitsch U, et al. Gastrointestinal stromal tumor of the rectum: results of surgical and multimodality therapy in the era of imatinib. Ann Surg Oncol. 2013;20:586-592.
25. DeMatteo RP, Ballman KV, Antonescu CR, et al. Long-term results of adjuvant imatinib mesylate in localized, high-risk, primary gastrointestinal stromal tumor (GIST): ACOSOG Z9000 (Alliance) intergroup phase 2 trial. Ann Surg. 2013;258:422-429.
26. Gleevac (imatinib) [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2016.
27. DeMatteo RP, Ballman KV, Antonescu CR, et al. Placebo-controlled randomized trial of adjuvant imatinib mesylate following the resection of localized, primary gastrointestinal stromal tumor (GIST). Lancet. 2009;373:1097-1104.
28. Corless CL, Ballman KV, Antonescu CR, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32:1563-1570.
29. Casali PG, Le Cesne A, Poveda Velasco A, et al. Imatinib failure-free survival (IFS) in patients with localized gastrointestinal stromal tumors (GIST) treated with adjuvant imatinib (IM): the EORTC/AGITG/FSG/GEIS/ISG randomized controlled phase III trial. J Clin Oncol. 2013;31. Abstract 10500.
30. Joensuu H, Eriksson M, Sundby HK, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307:1265-1272.
31. Raut CP, Espat NJ, Maki RG, et al. Efficacy and tolerability of 5-year adjuvant imatinib treatment for patients with resected intermediate- or high-risk primary gastrointestinal stromal tumor: The PERSIST-5 Clinical Trial. JAMA Oncol. 2018: e184060.
32. Benjamin RS, Casali PG. Adjuvant imatinib for GI stromal tumors: when and for how long? J Clin Oncol. 2016;34:215-218.
33. Demetri GD, Wang Y, Wehrle E, et al. Imatinib plasma levels are correlated with clinical benefit in patients with unresectable/metastatic gastrointestinal stromal tumors. J Clin Oncol. 2009;27:3141-3147.
34. Eisenberg BL, Harris J, Blanke CD, et al. Phase II trial of neoadjuvant/adjuvant imatinib mesylate (IM) for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumor (GIST): early results of RTOG 0132/ACRIN 6665. J Surg Oncol. 2009;99:42-47.
35. Rutkowski P, Gronchi A, Hohenberger P, et al. Neoadjuvant imatinib in locally advanced gastrointestinal stromal tumors (GIST): the EORTC STBSG experience. Ann Surg Oncol. 2013;20:2937-2943.
36. Joensuu H, Martin-Broto J, Nishida T, et al. Follow-up strategies for patients with gastrointestinal stromal tumour treated with or without adjuvant imatinib after surgery. Eur J Cancer. 2015;51:1611-1617.
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumor of the gastrointestinal (GI) tract and arise from the interstitial cells of Cajal of the myenteric plexus. These tumors are rare, with about 1 case per 100,000 persons diagnosed in the United States annually, but may be incidentally discovered in up to 1 in 5 autopsy specimens of older adults.1,2 Epidemiologic risk factors include increasing age, with a peak incidence between age 60 and 65 years, male gender, black race, and non-Hispanic white ethnicity. Germline predisposition can also increase the risk of developing GISTs; molecular drivers of GIST include gain-of-function mutations in the KIT proto-oncogene and platelet-derived growth factor receptor α (PDGFRA) gene, which both encode structurally similar tyrosine kinase receptors; germline mutations of succinate dehydrogenase (SDH) subunit genes; and mutations associated with neurofibromatosis type 1.
GISTs most commonly involve the stomach, followed by the small intestine, but can arise anywhere within the GI tract (esophagus, colon, rectum, and anus). They can also develop outside the GI tract, arising from the mesentery, omentum, and retroperitoneum. The majority of cases are localized or locoregional, whereas about 20% are metastatic at presentation.1 GISTs can occur in children, adolescents, and young adults. Pediatric GISTs represent a distinct subset marked by female predominance and gastric origin, are often multifocal, can sometimes have lymph node involvement, and typically lack mutations in the KIT and PDGFRA genes.
This review is the first of 2 articles focusing on the diagnosis and management of GISTs. Here, we review the evaluation and diagnosis of GISTs along with management of localized disease. Management of advanced disease is reviewed in a separate article.
Case Presentation
A 64-year-old African American man with progressive iron deficiency and abdominal discomfort undergoes upper and lower endoscopy and is found to have a bulging mass within his abdominal cavity. He undergoes a computed tomography (CT) evaluation of the chest, abdomen, and pelvis with contrast, which reveals the presence of a 10-cm gastric mass, with no other lesions identified. He undergoes surgical resection of the mass and presents for review of his pathology and to discuss his treatment plan.
What histopathologic features are consistent with GIST?
What factors are used for risk stratification and to predict likelihood of recurrence?
Clinical Presentation and Diagnosis
Most patients present with symptoms of overt or occult GI bleeding or abdominal discomfort, but a significant proportion of GISTs are discovered incidentally. Lymph node involvement is not typical, except for GISTs occurring in children and/or with rare syndromes. Most syndromic GISTs are multifocal and multicentric. After surgical resection, GISTs usually recur or metastasize within the abdominal cavity, including the omentum, peritoneum, or liver. These tumors rarely spread to the lungs, brain, or bones; when tumor spread does occur, it tends to be in heavily pre-treated patients with advanced disease who have been on multiple lines of therapy for a long duration of time.
The diagnosis usually can be made by histopathology. Specimens can be obtained by endoscopic ultrasound (EUS)– or CT-guided methods, the latter of which carries a very small risk of contamination from percutaneous biopsy. In terms of morphology, GISTs can be spindle cell, epithelioid, or mixed neoplasms. Epithelioid tumors are more commonly seen in the stomach and are often PDGFRA-mutated or SDH-deficient. The differential diagnosis includes other soft-tissue GI wall tumors such as leiomyosarcomas/leiomyomas, germ cell tumors, lymphomas, fibromatosis, and neuroendocrine and neurogenic tumors. A unique feature of GISTs that differentiates them from leiomyomas is near universal expression of CD117 by immunohistochemistry (IHC); this characteristic has allowed pathologists and providers to accurately distinguish true GISTs from other GI mesenchymal tumors.3 Recently, DOG1 (discovered on GIST1) immunoreactivity has been found to be helpful in identifying patients with CD117-negative GISTs. Initially identified through gene expression analysis of GISTs, DOG1 IHC can identify the common mutant c-Kit-driven CD117-positive GISTs as well as the rare CD117-negative GISTs, which are often driven by mutated PDGFRA.4 Importantly, IHC for KIT and DOG1 are not surrogates for mutational status, nor are they predictive of tyrosine kinase inhibitor (TKI) sensitivity. If IHC of a tumor specimen is CD117- and DOG1-negative, the specimen can be sent for KIT and PDGFRA mutational analysis to confirm the diagnosis. If analysis reveals that these genes are wild-type, then IHC staining for SDH B (SDHB) should follow to assess for an SDH-deficient GIST (negative staining).
Risk Stratification for Recurrence
The clinical behavior of GISTs can be variable. Some are indolent, while others behave more aggressively, with a greater malignant potential and a higher propensity to recur and metastasize. Clinical and pathologic features can provide important prognostic information that allows providers to risk-stratify patients. Various institutions have assessed prognostic variables for GISTs. In 2001, the National Institutes of Health (NIH) held a GIST workshop that proposed an approach to estimating metastatic risk based on tumor size and mitotic index (NIH or Fletcher criteria).5 Joensuu et al later proposed a modification of the NIH risk classification to include tumor location and tumor rupture (modified NIH criteria or Joensuu criteria).6-8 Similarly, the Armed Forces Institute of Pathology (AFIP) identified tumor site as a prognostic factor, with gastric GISTs having the best prognosis (AFIP-Miettinen criteria).9-11 Tabular schemes were designed which stratified patients into discrete groups with ranges for mitotic rate and tumor size. Nomograms for ease of use were then constructed utilizing a bimodal mitotic rate and included tumor site and size.12 Finally, contour maps were developed, which have the advantage of evaluating mitotic rate and tumor size as continuous nonlinear variables and also include tumor site and rupture (associated with a high risk of peritoneal metastasis) separately, further improving risk assessment. These contour maps have been validated against pooled data from 10 series (2560 patients).13 High-risk features identified from these studies include tumor location, size, mitotic rate and tumor rupture and are now used for deciding on the use of adjuvant imatinib and as requirements to enter clinical trials assessing adjuvant therapy for resected GISTs.
Case Continued
The patient’s operative and pathology reports indicate that the tumor is a spindle cell neoplasm of the stomach that is positive for CD117, DOG1, and CD34 and negative for smooth muscle actin and S-100, consistent with a diagnosis of GIST. Resection margins are negative. There are 10 mitoses per 50 high-power fields (HPF). Per the operative report, there was no intraoperative or intraperitoneal tumor rupture. Thus, while his GIST was gastric, which generally has a more favorable prognosis, the tumor harbors high-risk features based on its size and mitotic index.
What further testing should be requested?
Molecular Alterations
It is recommended that a mutational analysis be performed as part of the diagnostic work-up of all GISTs.14 Mutational analysis can provide prognostic and predictive information for sensitivity to imatinib and should be considered standard of care. It may also be useful for confirming a GIST diagnosis, or, if negative, lead to further evaluation with an IHC stain for SDHB. The c-Kit receptor is a member of the tyrosine kinase family and, through direct interactions with stem cell factor (SCF), can upregulate the PI3K/AKT/mTOR, Ras/Raf/MEK/ERK, and JAK-STAT pathways, resulting in transcription and translation of genes that enhance cell growth and survival.15 The cell of origin of GISTs, the interstitial cells of Cajal, are dependent on the SCF–c-Kit interaction for development.16 Likewise, the large majority of GISTs (about 70%) are driven by upregulation and constitutive activation of c-Kit, which is normally autoinhibited. About 80% of KIT mutations involve exon 11; these GISTs are most often associated with a gastric location and are associated with a favorable recurrence-free survival (RFS) rate with surgery alone.17KIT exon 9 mutations are much less common, encompassing only about 10% of GIST KIT mutations, and GISTs with these mutations are more likely to arise from the small bowel.17
About 8% of GISTs harbor gain-of-function PDGFRA driver mutations rendering constitutively active PDGFRA.18PDGFRA mutations are mutually exclusive from KIT mutations, and PDGFRA-mutated tumors most often occur in the stomach. PDGFRA mutations generally are associated with a lower mitotic rate and gastric location. Identification of the PDGFRA D842V mutation on exon 18, which is the most common, is important, as it is associated with imatinib resistance, and these patients should not be offered imatinib.19
Several other mutations associated with GISTs outside of the KIT and PDGFRA spectrum have been identified. About 10% of GISTs are wildtype for KIT and PDGFRA, and not all KIT/PDGFRA-wildtype GISTs are imatinib-sensitive and/or respond to other TKIs.18 These tumors may harbor aberrations in SDH and NF1, or less commonly, BRAF V600E, FGFR, and NTRK.20,21 SDH subunits B, C and D play a role in the Krebs cycle and electron transport chain. Germline mutations in these SDH subunits can result in the Carney-Stratakis syndrome characterized by the dyad of multifocal GISTs and multicentric paragangliomas.22 This syndrome is most likely to manifest in the pediatric or young adult population. In contradistinction is the Carney triad, which is associated with acquired loss of function of the SDHC gene due to promoter hypermethylation. This syndrome classically occurs in young women and is characterized by an indolent-behaving triad of multicentric GISTs, non-adrenal paragangliomas, and pulmonary chondromas.23 Like PDGFRA D842V–mutated GISTs, SDH-deficient and NF1-associated GISTs are considered imatinib resistant, and these patients should not be offered imatinib therapy.14
Case Continued
The patient’s GIST is found to harbor a KIT exon 11 single codon deletion. He appears anxious and asks to have everything done to prevent his GIST from coming back and to improve his lifespan.
What are the next steps in the management of this patient?
Management
A multidisciplinary team approach to the management of all GISTs is essential and includes input from radiology, gastroenterology, pathology, medical and surgical oncology, nuclear medicine, and nursing.
Surgical Resection
Small esophagogastric and duodenal GISTs ≤ 2 cm can be asymptomatic and managed with serial endoscopic surveillance, typically every 6 to 12 months, with biopsies if the tumors increase in size. GISTs larger than 2 cm require surgical resection, with resection of the full pseudocapsule and an R0 resection, if possible, since larger GISTs carry a higher risk of growth and recurrence. If an R0 resection would lead to significant morbidity or functional sequelae, an R1 may suffice. Rectal GISTs are an exception, where microscopic margins have been shown to be associated with an increased risk of local failure.24 It is important to explore the abdomen thoroughly for peritoneal, rectovaginal, and vesicular implants and metastasis to the liver. A formal lymph node dissection is not necessary because lymph nodes are rarely involved and should only be removed when clinically suspicious. Tumor rupture must be avoided. A laparoscopic approach should only be considered for smaller tumors, since there is a risk of tumor rupture with larger tumors.14
When is adjuvant imatinib indicated?
Adjuvant Imatinib
Among patients with local or locally advanced GISTs, the risk of death from recurrence with surgery alone can be high, with a historical 5-year overall survival (OS) of about 35%.25 As a result, multiple studies have assessed the benefit of adjuvant imatinib, which is now considered standard of care for patients with imatinib-sensitive, high-risk GISTs. In addition to inhibiting BCR-ABL, imatinib mesylate inhibits multiple other receptor tyrosine kinases, including PDGFR, SCF and c-Kit. As a result, imatinib has demonstrated in vitro inhibition of cell proliferation and apoptosis and clinical activity against GISTs expressing CD117.26 Importantly, adjuvant imatinib should only be offered to patients with imatinib-sensitive mutations, such as KIT exon 11 and KIT exon 9 mutations. Adjuvant imatinib should not be offered to patients with imatinib-insensitive mutations such as PDGFR 842V, NF1, or BRAF-related or SDH-deficient GISTs.
The ACOSOG Z9000 was the first study of adjuvant imatinib in patients with resected GISTs.25 This was a single-arm, phase 2 study involving 106 patients with surgically resected GISTs deemed high-risk for recurrence, defined as size > 10 cm, tumor rupture, or up to 4 peritoneal implants. Patients were treated with imatinib 400 mg daily for 1 year. The primary and secondary endpoints were OS and RFS, respectively. Long-term follow-up of this study demonstrated 1-, 3-, and 5-year OS of 99%, 97%, and 83%, and 1-, 3-, and 5-year RFS of 96%, 60%, and 40%, which compared favorably with historical controls. In a multivariable analysis, increasing tumor size, small bowel location, KIT exon 9 mutation, high mitotic rate, and older age were independent risk factors for a poor RFS.25 It is important to note that the benefit of adjuvant imatinib waned after discontinuation of therapy, creating a rationale to study adjuvant imatinib for longer periods of time.
As a result of the promising phase 2 data, ACOSOG opened a phase 3 randomized trial (Z9001) comparing 1 year of adjuvant imatinib to placebo among patients with surgically resected GISTs that were > 3 cm in size and that stained positive for CD117 on pathology. The trial accrued 713 patients and was stopped early at a planned interim analysis, which revealed a 1-year RFS of 98% for imatinib versus 83% for placebo (hazard ratio [HR], 0.35; P < 0.001). The 1-year OS did not differ between the 2 arms (92.2% vs 99.7%; HR, 0.66; P = 0.47).27 When comparing the 2 arms, imatinib was associated with a higher RFS among patients with a KIT exon 11 deletion, but not among patients with other KIT mutation types, PDGFRA mutations, or who were KIT/PDGFRA wildtype.28 Imatinib was granted approval by the US Food and Drug Administration (FDA) for the adjuvant treatment of high-risk GISTs based on the results of the ACOSOG Z9001 trial.
The EORTC 62024 study was a randomized placebo-controlled trial assessing the benefit of 2 years of adjuvant imatinib.29 Patients had to be considered intermediate or high risk per the 2002 NIH consensus classification to be eligible. The trial enrolled 918 patients. The 5-year OS rate, the original primary endpoint, did not differ between the 2 groups (100% vs 99%). The 3-year and 5-year RFS rates, secondary endpoints, were significantly longer among patients treated with imatinib (84% vs 66% and 69% vs 63%, respectively). Again, it was noted that the benefit of imatinib waned over time after treatment discontinuation.
The Scandinavian Sarcoma Group (SSG XVIII) trial was a prospective randomized phase 3 trial that compared 3 years versus 1 year of adjuvant imatinib.30 Patients had to be enrolled within 12 weeks of the postoperative period and had to have GISTs that were CD117-positive and with a high estimated risk of recurrence, per the modified NIH consensus criteria (size > 10 cm, > 10 mitoses per 50 HPF, diameter > 5 cm with mitotic count > 5, or tumor rupture before or at surgery). Three years of adjuvant imatinib was associated with a 54% reduction in the hazard for recurrence at 5 years (65.6% vs 47.9%; HR, 0.46; P < 0.001) and a 55% reduction in the hazard for death at 5 years (OS 92% vs 81.7%; HR, 0.45; P = 0.02). Based on the results of this study, the FDA granted approval for the use of 3 years of adjuvant imatinib in patients with high-risk resected GISTs.
The observation that a longer duration of adjuvant imatinib was associated with superior RFS and OS led to studies to further explore longer durations of adjuvant imatinib. The PERSIST-5 (Postresection Evaluation of Recurrence-free Survival for Gastrointestinal Stromal Tumors With 5 Years of Adjuvant Imatinib) was a multicenter, single-arm, phase 2 prospective study of adjuvant imatinib with a primary endpoint of RFS after 5 years.31 Patients had to have an intermediate or high risk of recurrence, which included GISTs at any site > 2 cm with > 5 mitoses per 50 HPF or nongastric GISTs that were ≥ 5 cm. With 91 patients enrolled, the estimated 5-year RFS was 90% and the OS was 95%. Of note, about half of the patients stopped treatment early due to a variety of reasons, including patient choice or adverse events. Importantly, there were no recurrences in patients with imatinib-sensitive mutations while on therapy. We know that in patients at high risk of relapse, adjuvant imatinib delays recurrence and improves survival, but whether any patients are cured, or their survival curves are just shifted to the right, is unknown. Only longer follow-up of existing studies, and the results of newer trials utilizing longer durations of adjuvant treatment, will help to determine the real value of adjuvant therapy for GIST patients.32 Based on this study, it would be reasonable to discuss a longer duration of imatinib with patients deemed to be at very high risk of recurrence and who are tolerating therapy well. We are awaiting the data from the randomized phase 3 Scandinavian Sarcoma Group XII trial comparing 5 years versus 3 years of adjuvant imatinib therapy, and from the French ImadGIST trial of adjuvant imatinib for 3 versus 6 years. A summary of the aforementioned key adjuvant trials is shown in the Table.
When imatinib is commenced, careful monitoring for treatment toxicities and drug interactions should ensue in order to improve compliance. Dose density should be maintained if possible, as retrospective studies suggest suboptimal plasma levels are associated with a worse outcome.33
When should neoadjuvant imatinib be considered?
Neoadjuvant Imatinib
Neoadjuvant imatinib should be considered for patients requiring total gastrectomy, esophagectomy, or abdominoperineal resection of the rectum in order to reduce tumor size, limit subsequent surgical morbidity, mitigate tumor bleeding and rupture, and aid with organ preservation. Patients with rectal GISTs that may otherwise warrant an abdominoperineal resection should be offered a trial of imatinib in the neoadjuvant setting. There is no evidence for the use of any other TKI aside from imatinib in the neoadjuvant or adjuvant setting. With neoadjuvant imatinib, it is difficult to accurately assess the mitotic rate in the resected tumor specimen.
The RTOG 0132/ACRIN 6665 trial was a prospective phase 2 study evaluating the efficacy of imatinib 600 mg daily in the perioperative setting.34 The trial enrolled 50 patients, 30 with primary GISTs (group A) and 22 with recurrent metastatic GISTs (group B). Based on data from the metastatic setting revealing a time to treatment response of about 2.5 months, patients were treated with 8 to 12 weeks of preoperative imatinib followed by 2 years of adjuvant imatinib. Imatinib was stopped 24 hours preoperatively and resumed as soon as possible postoperatively. In group A, 7% of patients achieved a partial response (PR), 83% achieved stable disease, and 2-year progression-free survival (PFS) and OS were 83% and 93%, respectively. In group B, 4.5% of patients achieved a PR, 91% achieved stable disease, and 4.5% experienced progressive disease in the preoperative period; the 2-year PFS and OS were 77% and 91%, respectively. The results of this trial demonstrated the feasibility of using perioperative imatinib with minimal effects on surgical outcomes and set the rationale to use neoadjuvant imatinib in select patients with borderline resectable or rectal GISTs. Another EORTC pooled analysis from 10 sarcoma centers revealed that after a median of 10 months of neoadjuvant imatinib, 83.2% of patients achieved an R0 resection and only 1% progressed during treatment.35 After a median follow-up of 46 months, the 5-year disease-free survival and OS were 65% and 87%, respectively.
Mutational testing should be performed beforehand to ensure the tumor is imatinib-sensitive. If a KIT exon 9 mutation is identified, then 400 mg twice daily should be considered (given the benefit seen with 800 mg imatinib for advanced GIST patients), although there are no studies to confirm this practice. Neoadjuvant imatinib is recommended for a total of 6 to 12 months to ensure maximal tumor debulking, but with very close monitoring and surgical input for disease resistance and growth.14 Imatinib should be stopped 1 to 2 days preoperatively and resumed once the patient has recovered from surgery for a total of 3 years (pre-/postoperatively combined). Neoadjuvant therapy has been shown to be safe and effective, but there have been no randomized trials to assess survival.
What is appropriate surveillance for resected GISTs?
Surveillance
There have been no randomized studies to guide the management of surveillance after surgical resection and adjuvant therapy. There is no known optimal follow-up schedule, but several have been proposed.13,36 Among high-risk patients, it is suggested to image every 3 to 6 months during adjuvant therapy, followed by every 3 months for 2 years after discontinuing therapy, then every 6 months for another 3 years and annually thereafter for an additional 5 years. High-risk patients usually relapse within 1 to 3 years after finishing adjuvant therapy, while low-risk patients can relapse later given that their disease can be slower growing. It has been recommended that low-risk patients undergo imaging every 6 months for 5 years, with follow-up individualized thereafter. Very-low-risk patients may not require more than annual imaging. Because most relapses occur within the peritoneum or liver, imaging should encompass the abdomen and pelvis. Surveillance imaging usually consists of CT scans of the abdomen and pelvis. MRI scans can be utilized for patients at lower risk or who are out several years in order to avoid excess radiation exposure. MRI is also specifically helpful for rectal and esophageal lesions. Chest CT or chest radiograph and bone scan are not routinely required for follow-up.
Case Conclusion
The patient receives adjuvant imatinib and experiences grade 2 myalgias, periorbital edema, and macrocytic anemia, which result in imatinib discontinuation after 3 years of treatment. He is seen every 3 to 6 months and a contrast CT abdomen and pelvis is obtained every 6 months for 5 years. During this 5-year follow-up period, he does not have any clinical or radiographic evidence of disease recurrence.
Further follow-up of this patient is presented in the second article in this 2-part review of management of GISTs.
Key Points
- GISTs are the most common mesenchymal neoplasms of the GI tract and can occasionally occur in extragastrointestinal locations as well.
- GISTs encompass a heterogeneous family of tumor subsets with different natural histories, mutations, and TKI responsiveness.
- Surgery is the mainstay of treatment for localized GISTs, with cure rates greater than 50%.
- For very small (< 2 cm) esophagogastric GISTs, endoscopic ultrasound evaluation and follow-up is recommended.
- For tumors ≥ 2 cm, biopsy and excision is the standard approach.
- For localized GISTs, complete surgical resection (R0) is standard treatment, with no lymphadenectomy for clinically negative lymph nodes.
- Mutational analysis should be considered standard of practice. It can be helpful for confirming the diagnosis and can be predictive and prognostic in determining specific TKI therapy and dose.
- Adjuvant imatinib at a dose of 400 mg for 3 years is standard of care for GISTs that are at high risk of relapse and are imatinib-sensitive, and it is the only TKI approved for adjuvant therapy. Patients with PDGFRA D842V, NF1, BRAF or SDH-deficient GISTs should not receive adjuvant imatinib therapy.
- Neoadjuvant therapy can be utilized for sites where extensive resection would lead to significant morbidity. It should be given for 6 to 12 months, but patients need to be monitored closely for tumor growth.
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumor of the gastrointestinal (GI) tract and arise from the interstitial cells of Cajal of the myenteric plexus. These tumors are rare, with about 1 case per 100,000 persons diagnosed in the United States annually, but may be incidentally discovered in up to 1 in 5 autopsy specimens of older adults.1,2 Epidemiologic risk factors include increasing age, with a peak incidence between age 60 and 65 years, male gender, black race, and non-Hispanic white ethnicity. Germline predisposition can also increase the risk of developing GISTs; molecular drivers of GIST include gain-of-function mutations in the KIT proto-oncogene and platelet-derived growth factor receptor α (PDGFRA) gene, which both encode structurally similar tyrosine kinase receptors; germline mutations of succinate dehydrogenase (SDH) subunit genes; and mutations associated with neurofibromatosis type 1.
GISTs most commonly involve the stomach, followed by the small intestine, but can arise anywhere within the GI tract (esophagus, colon, rectum, and anus). They can also develop outside the GI tract, arising from the mesentery, omentum, and retroperitoneum. The majority of cases are localized or locoregional, whereas about 20% are metastatic at presentation.1 GISTs can occur in children, adolescents, and young adults. Pediatric GISTs represent a distinct subset marked by female predominance and gastric origin, are often multifocal, can sometimes have lymph node involvement, and typically lack mutations in the KIT and PDGFRA genes.
This review is the first of 2 articles focusing on the diagnosis and management of GISTs. Here, we review the evaluation and diagnosis of GISTs along with management of localized disease. Management of advanced disease is reviewed in a separate article.
Case Presentation
A 64-year-old African American man with progressive iron deficiency and abdominal discomfort undergoes upper and lower endoscopy and is found to have a bulging mass within his abdominal cavity. He undergoes a computed tomography (CT) evaluation of the chest, abdomen, and pelvis with contrast, which reveals the presence of a 10-cm gastric mass, with no other lesions identified. He undergoes surgical resection of the mass and presents for review of his pathology and to discuss his treatment plan.
What histopathologic features are consistent with GIST?
What factors are used for risk stratification and to predict likelihood of recurrence?
Clinical Presentation and Diagnosis
Most patients present with symptoms of overt or occult GI bleeding or abdominal discomfort, but a significant proportion of GISTs are discovered incidentally. Lymph node involvement is not typical, except for GISTs occurring in children and/or with rare syndromes. Most syndromic GISTs are multifocal and multicentric. After surgical resection, GISTs usually recur or metastasize within the abdominal cavity, including the omentum, peritoneum, or liver. These tumors rarely spread to the lungs, brain, or bones; when tumor spread does occur, it tends to be in heavily pre-treated patients with advanced disease who have been on multiple lines of therapy for a long duration of time.
The diagnosis usually can be made by histopathology. Specimens can be obtained by endoscopic ultrasound (EUS)– or CT-guided methods, the latter of which carries a very small risk of contamination from percutaneous biopsy. In terms of morphology, GISTs can be spindle cell, epithelioid, or mixed neoplasms. Epithelioid tumors are more commonly seen in the stomach and are often PDGFRA-mutated or SDH-deficient. The differential diagnosis includes other soft-tissue GI wall tumors such as leiomyosarcomas/leiomyomas, germ cell tumors, lymphomas, fibromatosis, and neuroendocrine and neurogenic tumors. A unique feature of GISTs that differentiates them from leiomyomas is near universal expression of CD117 by immunohistochemistry (IHC); this characteristic has allowed pathologists and providers to accurately distinguish true GISTs from other GI mesenchymal tumors.3 Recently, DOG1 (discovered on GIST1) immunoreactivity has been found to be helpful in identifying patients with CD117-negative GISTs. Initially identified through gene expression analysis of GISTs, DOG1 IHC can identify the common mutant c-Kit-driven CD117-positive GISTs as well as the rare CD117-negative GISTs, which are often driven by mutated PDGFRA.4 Importantly, IHC for KIT and DOG1 are not surrogates for mutational status, nor are they predictive of tyrosine kinase inhibitor (TKI) sensitivity. If IHC of a tumor specimen is CD117- and DOG1-negative, the specimen can be sent for KIT and PDGFRA mutational analysis to confirm the diagnosis. If analysis reveals that these genes are wild-type, then IHC staining for SDH B (SDHB) should follow to assess for an SDH-deficient GIST (negative staining).
Risk Stratification for Recurrence
The clinical behavior of GISTs can be variable. Some are indolent, while others behave more aggressively, with a greater malignant potential and a higher propensity to recur and metastasize. Clinical and pathologic features can provide important prognostic information that allows providers to risk-stratify patients. Various institutions have assessed prognostic variables for GISTs. In 2001, the National Institutes of Health (NIH) held a GIST workshop that proposed an approach to estimating metastatic risk based on tumor size and mitotic index (NIH or Fletcher criteria).5 Joensuu et al later proposed a modification of the NIH risk classification to include tumor location and tumor rupture (modified NIH criteria or Joensuu criteria).6-8 Similarly, the Armed Forces Institute of Pathology (AFIP) identified tumor site as a prognostic factor, with gastric GISTs having the best prognosis (AFIP-Miettinen criteria).9-11 Tabular schemes were designed which stratified patients into discrete groups with ranges for mitotic rate and tumor size. Nomograms for ease of use were then constructed utilizing a bimodal mitotic rate and included tumor site and size.12 Finally, contour maps were developed, which have the advantage of evaluating mitotic rate and tumor size as continuous nonlinear variables and also include tumor site and rupture (associated with a high risk of peritoneal metastasis) separately, further improving risk assessment. These contour maps have been validated against pooled data from 10 series (2560 patients).13 High-risk features identified from these studies include tumor location, size, mitotic rate and tumor rupture and are now used for deciding on the use of adjuvant imatinib and as requirements to enter clinical trials assessing adjuvant therapy for resected GISTs.
Case Continued
The patient’s operative and pathology reports indicate that the tumor is a spindle cell neoplasm of the stomach that is positive for CD117, DOG1, and CD34 and negative for smooth muscle actin and S-100, consistent with a diagnosis of GIST. Resection margins are negative. There are 10 mitoses per 50 high-power fields (HPF). Per the operative report, there was no intraoperative or intraperitoneal tumor rupture. Thus, while his GIST was gastric, which generally has a more favorable prognosis, the tumor harbors high-risk features based on its size and mitotic index.
What further testing should be requested?
Molecular Alterations
It is recommended that a mutational analysis be performed as part of the diagnostic work-up of all GISTs.14 Mutational analysis can provide prognostic and predictive information for sensitivity to imatinib and should be considered standard of care. It may also be useful for confirming a GIST diagnosis, or, if negative, lead to further evaluation with an IHC stain for SDHB. The c-Kit receptor is a member of the tyrosine kinase family and, through direct interactions with stem cell factor (SCF), can upregulate the PI3K/AKT/mTOR, Ras/Raf/MEK/ERK, and JAK-STAT pathways, resulting in transcription and translation of genes that enhance cell growth and survival.15 The cell of origin of GISTs, the interstitial cells of Cajal, are dependent on the SCF–c-Kit interaction for development.16 Likewise, the large majority of GISTs (about 70%) are driven by upregulation and constitutive activation of c-Kit, which is normally autoinhibited. About 80% of KIT mutations involve exon 11; these GISTs are most often associated with a gastric location and are associated with a favorable recurrence-free survival (RFS) rate with surgery alone.17KIT exon 9 mutations are much less common, encompassing only about 10% of GIST KIT mutations, and GISTs with these mutations are more likely to arise from the small bowel.17
About 8% of GISTs harbor gain-of-function PDGFRA driver mutations rendering constitutively active PDGFRA.18PDGFRA mutations are mutually exclusive from KIT mutations, and PDGFRA-mutated tumors most often occur in the stomach. PDGFRA mutations generally are associated with a lower mitotic rate and gastric location. Identification of the PDGFRA D842V mutation on exon 18, which is the most common, is important, as it is associated with imatinib resistance, and these patients should not be offered imatinib.19
Several other mutations associated with GISTs outside of the KIT and PDGFRA spectrum have been identified. About 10% of GISTs are wildtype for KIT and PDGFRA, and not all KIT/PDGFRA-wildtype GISTs are imatinib-sensitive and/or respond to other TKIs.18 These tumors may harbor aberrations in SDH and NF1, or less commonly, BRAF V600E, FGFR, and NTRK.20,21 SDH subunits B, C and D play a role in the Krebs cycle and electron transport chain. Germline mutations in these SDH subunits can result in the Carney-Stratakis syndrome characterized by the dyad of multifocal GISTs and multicentric paragangliomas.22 This syndrome is most likely to manifest in the pediatric or young adult population. In contradistinction is the Carney triad, which is associated with acquired loss of function of the SDHC gene due to promoter hypermethylation. This syndrome classically occurs in young women and is characterized by an indolent-behaving triad of multicentric GISTs, non-adrenal paragangliomas, and pulmonary chondromas.23 Like PDGFRA D842V–mutated GISTs, SDH-deficient and NF1-associated GISTs are considered imatinib resistant, and these patients should not be offered imatinib therapy.14
Case Continued
The patient’s GIST is found to harbor a KIT exon 11 single codon deletion. He appears anxious and asks to have everything done to prevent his GIST from coming back and to improve his lifespan.
What are the next steps in the management of this patient?
Management
A multidisciplinary team approach to the management of all GISTs is essential and includes input from radiology, gastroenterology, pathology, medical and surgical oncology, nuclear medicine, and nursing.
Surgical Resection
Small esophagogastric and duodenal GISTs ≤ 2 cm can be asymptomatic and managed with serial endoscopic surveillance, typically every 6 to 12 months, with biopsies if the tumors increase in size. GISTs larger than 2 cm require surgical resection, with resection of the full pseudocapsule and an R0 resection, if possible, since larger GISTs carry a higher risk of growth and recurrence. If an R0 resection would lead to significant morbidity or functional sequelae, an R1 may suffice. Rectal GISTs are an exception, where microscopic margins have been shown to be associated with an increased risk of local failure.24 It is important to explore the abdomen thoroughly for peritoneal, rectovaginal, and vesicular implants and metastasis to the liver. A formal lymph node dissection is not necessary because lymph nodes are rarely involved and should only be removed when clinically suspicious. Tumor rupture must be avoided. A laparoscopic approach should only be considered for smaller tumors, since there is a risk of tumor rupture with larger tumors.14
When is adjuvant imatinib indicated?
Adjuvant Imatinib
Among patients with local or locally advanced GISTs, the risk of death from recurrence with surgery alone can be high, with a historical 5-year overall survival (OS) of about 35%.25 As a result, multiple studies have assessed the benefit of adjuvant imatinib, which is now considered standard of care for patients with imatinib-sensitive, high-risk GISTs. In addition to inhibiting BCR-ABL, imatinib mesylate inhibits multiple other receptor tyrosine kinases, including PDGFR, SCF and c-Kit. As a result, imatinib has demonstrated in vitro inhibition of cell proliferation and apoptosis and clinical activity against GISTs expressing CD117.26 Importantly, adjuvant imatinib should only be offered to patients with imatinib-sensitive mutations, such as KIT exon 11 and KIT exon 9 mutations. Adjuvant imatinib should not be offered to patients with imatinib-insensitive mutations such as PDGFR 842V, NF1, or BRAF-related or SDH-deficient GISTs.
The ACOSOG Z9000 was the first study of adjuvant imatinib in patients with resected GISTs.25 This was a single-arm, phase 2 study involving 106 patients with surgically resected GISTs deemed high-risk for recurrence, defined as size > 10 cm, tumor rupture, or up to 4 peritoneal implants. Patients were treated with imatinib 400 mg daily for 1 year. The primary and secondary endpoints were OS and RFS, respectively. Long-term follow-up of this study demonstrated 1-, 3-, and 5-year OS of 99%, 97%, and 83%, and 1-, 3-, and 5-year RFS of 96%, 60%, and 40%, which compared favorably with historical controls. In a multivariable analysis, increasing tumor size, small bowel location, KIT exon 9 mutation, high mitotic rate, and older age were independent risk factors for a poor RFS.25 It is important to note that the benefit of adjuvant imatinib waned after discontinuation of therapy, creating a rationale to study adjuvant imatinib for longer periods of time.
As a result of the promising phase 2 data, ACOSOG opened a phase 3 randomized trial (Z9001) comparing 1 year of adjuvant imatinib to placebo among patients with surgically resected GISTs that were > 3 cm in size and that stained positive for CD117 on pathology. The trial accrued 713 patients and was stopped early at a planned interim analysis, which revealed a 1-year RFS of 98% for imatinib versus 83% for placebo (hazard ratio [HR], 0.35; P < 0.001). The 1-year OS did not differ between the 2 arms (92.2% vs 99.7%; HR, 0.66; P = 0.47).27 When comparing the 2 arms, imatinib was associated with a higher RFS among patients with a KIT exon 11 deletion, but not among patients with other KIT mutation types, PDGFRA mutations, or who were KIT/PDGFRA wildtype.28 Imatinib was granted approval by the US Food and Drug Administration (FDA) for the adjuvant treatment of high-risk GISTs based on the results of the ACOSOG Z9001 trial.
The EORTC 62024 study was a randomized placebo-controlled trial assessing the benefit of 2 years of adjuvant imatinib.29 Patients had to be considered intermediate or high risk per the 2002 NIH consensus classification to be eligible. The trial enrolled 918 patients. The 5-year OS rate, the original primary endpoint, did not differ between the 2 groups (100% vs 99%). The 3-year and 5-year RFS rates, secondary endpoints, were significantly longer among patients treated with imatinib (84% vs 66% and 69% vs 63%, respectively). Again, it was noted that the benefit of imatinib waned over time after treatment discontinuation.
The Scandinavian Sarcoma Group (SSG XVIII) trial was a prospective randomized phase 3 trial that compared 3 years versus 1 year of adjuvant imatinib.30 Patients had to be enrolled within 12 weeks of the postoperative period and had to have GISTs that were CD117-positive and with a high estimated risk of recurrence, per the modified NIH consensus criteria (size > 10 cm, > 10 mitoses per 50 HPF, diameter > 5 cm with mitotic count > 5, or tumor rupture before or at surgery). Three years of adjuvant imatinib was associated with a 54% reduction in the hazard for recurrence at 5 years (65.6% vs 47.9%; HR, 0.46; P < 0.001) and a 55% reduction in the hazard for death at 5 years (OS 92% vs 81.7%; HR, 0.45; P = 0.02). Based on the results of this study, the FDA granted approval for the use of 3 years of adjuvant imatinib in patients with high-risk resected GISTs.
The observation that a longer duration of adjuvant imatinib was associated with superior RFS and OS led to studies to further explore longer durations of adjuvant imatinib. The PERSIST-5 (Postresection Evaluation of Recurrence-free Survival for Gastrointestinal Stromal Tumors With 5 Years of Adjuvant Imatinib) was a multicenter, single-arm, phase 2 prospective study of adjuvant imatinib with a primary endpoint of RFS after 5 years.31 Patients had to have an intermediate or high risk of recurrence, which included GISTs at any site > 2 cm with > 5 mitoses per 50 HPF or nongastric GISTs that were ≥ 5 cm. With 91 patients enrolled, the estimated 5-year RFS was 90% and the OS was 95%. Of note, about half of the patients stopped treatment early due to a variety of reasons, including patient choice or adverse events. Importantly, there were no recurrences in patients with imatinib-sensitive mutations while on therapy. We know that in patients at high risk of relapse, adjuvant imatinib delays recurrence and improves survival, but whether any patients are cured, or their survival curves are just shifted to the right, is unknown. Only longer follow-up of existing studies, and the results of newer trials utilizing longer durations of adjuvant treatment, will help to determine the real value of adjuvant therapy for GIST patients.32 Based on this study, it would be reasonable to discuss a longer duration of imatinib with patients deemed to be at very high risk of recurrence and who are tolerating therapy well. We are awaiting the data from the randomized phase 3 Scandinavian Sarcoma Group XII trial comparing 5 years versus 3 years of adjuvant imatinib therapy, and from the French ImadGIST trial of adjuvant imatinib for 3 versus 6 years. A summary of the aforementioned key adjuvant trials is shown in the Table.
When imatinib is commenced, careful monitoring for treatment toxicities and drug interactions should ensue in order to improve compliance. Dose density should be maintained if possible, as retrospective studies suggest suboptimal plasma levels are associated with a worse outcome.33
When should neoadjuvant imatinib be considered?
Neoadjuvant Imatinib
Neoadjuvant imatinib should be considered for patients requiring total gastrectomy, esophagectomy, or abdominoperineal resection of the rectum in order to reduce tumor size, limit subsequent surgical morbidity, mitigate tumor bleeding and rupture, and aid with organ preservation. Patients with rectal GISTs that may otherwise warrant an abdominoperineal resection should be offered a trial of imatinib in the neoadjuvant setting. There is no evidence for the use of any other TKI aside from imatinib in the neoadjuvant or adjuvant setting. With neoadjuvant imatinib, it is difficult to accurately assess the mitotic rate in the resected tumor specimen.
The RTOG 0132/ACRIN 6665 trial was a prospective phase 2 study evaluating the efficacy of imatinib 600 mg daily in the perioperative setting.34 The trial enrolled 50 patients, 30 with primary GISTs (group A) and 22 with recurrent metastatic GISTs (group B). Based on data from the metastatic setting revealing a time to treatment response of about 2.5 months, patients were treated with 8 to 12 weeks of preoperative imatinib followed by 2 years of adjuvant imatinib. Imatinib was stopped 24 hours preoperatively and resumed as soon as possible postoperatively. In group A, 7% of patients achieved a partial response (PR), 83% achieved stable disease, and 2-year progression-free survival (PFS) and OS were 83% and 93%, respectively. In group B, 4.5% of patients achieved a PR, 91% achieved stable disease, and 4.5% experienced progressive disease in the preoperative period; the 2-year PFS and OS were 77% and 91%, respectively. The results of this trial demonstrated the feasibility of using perioperative imatinib with minimal effects on surgical outcomes and set the rationale to use neoadjuvant imatinib in select patients with borderline resectable or rectal GISTs. Another EORTC pooled analysis from 10 sarcoma centers revealed that after a median of 10 months of neoadjuvant imatinib, 83.2% of patients achieved an R0 resection and only 1% progressed during treatment.35 After a median follow-up of 46 months, the 5-year disease-free survival and OS were 65% and 87%, respectively.
Mutational testing should be performed beforehand to ensure the tumor is imatinib-sensitive. If a KIT exon 9 mutation is identified, then 400 mg twice daily should be considered (given the benefit seen with 800 mg imatinib for advanced GIST patients), although there are no studies to confirm this practice. Neoadjuvant imatinib is recommended for a total of 6 to 12 months to ensure maximal tumor debulking, but with very close monitoring and surgical input for disease resistance and growth.14 Imatinib should be stopped 1 to 2 days preoperatively and resumed once the patient has recovered from surgery for a total of 3 years (pre-/postoperatively combined). Neoadjuvant therapy has been shown to be safe and effective, but there have been no randomized trials to assess survival.
What is appropriate surveillance for resected GISTs?
Surveillance
There have been no randomized studies to guide the management of surveillance after surgical resection and adjuvant therapy. There is no known optimal follow-up schedule, but several have been proposed.13,36 Among high-risk patients, it is suggested to image every 3 to 6 months during adjuvant therapy, followed by every 3 months for 2 years after discontinuing therapy, then every 6 months for another 3 years and annually thereafter for an additional 5 years. High-risk patients usually relapse within 1 to 3 years after finishing adjuvant therapy, while low-risk patients can relapse later given that their disease can be slower growing. It has been recommended that low-risk patients undergo imaging every 6 months for 5 years, with follow-up individualized thereafter. Very-low-risk patients may not require more than annual imaging. Because most relapses occur within the peritoneum or liver, imaging should encompass the abdomen and pelvis. Surveillance imaging usually consists of CT scans of the abdomen and pelvis. MRI scans can be utilized for patients at lower risk or who are out several years in order to avoid excess radiation exposure. MRI is also specifically helpful for rectal and esophageal lesions. Chest CT or chest radiograph and bone scan are not routinely required for follow-up.
Case Conclusion
The patient receives adjuvant imatinib and experiences grade 2 myalgias, periorbital edema, and macrocytic anemia, which result in imatinib discontinuation after 3 years of treatment. He is seen every 3 to 6 months and a contrast CT abdomen and pelvis is obtained every 6 months for 5 years. During this 5-year follow-up period, he does not have any clinical or radiographic evidence of disease recurrence.
Further follow-up of this patient is presented in the second article in this 2-part review of management of GISTs.
Key Points
- GISTs are the most common mesenchymal neoplasms of the GI tract and can occasionally occur in extragastrointestinal locations as well.
- GISTs encompass a heterogeneous family of tumor subsets with different natural histories, mutations, and TKI responsiveness.
- Surgery is the mainstay of treatment for localized GISTs, with cure rates greater than 50%.
- For very small (< 2 cm) esophagogastric GISTs, endoscopic ultrasound evaluation and follow-up is recommended.
- For tumors ≥ 2 cm, biopsy and excision is the standard approach.
- For localized GISTs, complete surgical resection (R0) is standard treatment, with no lymphadenectomy for clinically negative lymph nodes.
- Mutational analysis should be considered standard of practice. It can be helpful for confirming the diagnosis and can be predictive and prognostic in determining specific TKI therapy and dose.
- Adjuvant imatinib at a dose of 400 mg for 3 years is standard of care for GISTs that are at high risk of relapse and are imatinib-sensitive, and it is the only TKI approved for adjuvant therapy. Patients with PDGFRA D842V, NF1, BRAF or SDH-deficient GISTs should not receive adjuvant imatinib therapy.
- Neoadjuvant therapy can be utilized for sites where extensive resection would lead to significant morbidity. It should be given for 6 to 12 months, but patients need to be monitored closely for tumor growth.
1. Ma GL, Murphy JD, Martinez ME et al. Epidemiology of gastrointestinal stromal tumors in the era of histology codes: results of a population-based study. Cancer Epidemiol Biomarkers Prev. 2015;24:298-302.
2. Agaimy A, Wunsch PH, Hofstaedter F, et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol. 2007;31:113-120.
3. Miettinen M, Sobin LH, Sarlomo-Rikala M. Immunohistochemical spectrum of GISTs at different sites and their differential diagnosis with a reference to CD117 (KIT). Mod Pathol. 2000;13:1134-1142.
4. West RB, Corless CL, Chen X, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutational status. Am J Pathol. 2004;165:107-113.
5. Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Int J Surg Pathol. 2002;10:81-89.
6. Joensuu H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol. 2008;39:1411-1419.
7. Hohenberger P, Ronellenfitsch U, Oladeji O, et al. Pattern of recurrence in patients with ruptured primary gastrointestinal stromal tumor. Br J Surg. 2010;97:1854-1859.
8. Holmenbakk T, Bjerkehagen B, Boye K, et al. Definition and clinical significance of tumor rupture in gastrointestinal stromal tumours of the small intestine. Br J Surg. 2016;103:684-691.
9. Emory TS, Sobin LH, Lukes L, et al. Prognosis of gastrointestinal smooth-muscle (stromal) tumors: dependence on anatomic site. Am J Surg Pathol. 1999;23:82-87.
10. Miettinen M, Makhlouf H, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol. 2006;30:477-489.
11. Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol. 2005;29:52-68.
12. Gold JS, Gonen M, Gutierrez A, et al. Development and validation of a prognostic nomogram for recurrence-free survival after complete surgical resection of localized primary gastrointestinal stromal tumour: a retrospective analysis. Lancet Oncol. 2009;10:1045-1052.
13. Joensuu H, Vehtari A, Rihimaki J et al. Risk of recurrence of gastrointestinal stromal tumor after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13:265-274.
14. Casali PG, Abecassis N, Bauer S, et al. Gastrointestinal stromal tumours: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow up. Ann Oncol. 2018;29(Supplement_4): iv267.
15. Jing L, Yan-Ling W, Bing-Jia C, et al. The c-kit receptor-mediated signal transduction and tumor-related diseases. Int J Biol Sci. 2013;9:435-443.
16. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577-580.
17. Joensuu H, Rutkowski P, Nishida T, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015;33:634-642.
18. Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22:3813-3825.
19. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342-4349.
20. Huss S, Pasternack H, Ihle MA, et al. Clinicopathological and molecular features of a large cohort of gastrointestinal stromal tumors (GISTs) and review of the literature: BRAF mutations in KIT/PDGFRA wild-type GISTs are rare events. Hum Pathol. 2017;62:206-214.
21. Shi E, Chmielecki J, Tang CM, et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J Transl Med. 2016;14:339.
22. Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet. 2002;108:132-139.
23. Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc. 1999;74:543-552.
24. Jakob J, Mussi C, Ronellenfitsch U, et al. Gastrointestinal stromal tumor of the rectum: results of surgical and multimodality therapy in the era of imatinib. Ann Surg Oncol. 2013;20:586-592.
25. DeMatteo RP, Ballman KV, Antonescu CR, et al. Long-term results of adjuvant imatinib mesylate in localized, high-risk, primary gastrointestinal stromal tumor (GIST): ACOSOG Z9000 (Alliance) intergroup phase 2 trial. Ann Surg. 2013;258:422-429.
26. Gleevac (imatinib) [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2016.
27. DeMatteo RP, Ballman KV, Antonescu CR, et al. Placebo-controlled randomized trial of adjuvant imatinib mesylate following the resection of localized, primary gastrointestinal stromal tumor (GIST). Lancet. 2009;373:1097-1104.
28. Corless CL, Ballman KV, Antonescu CR, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32:1563-1570.
29. Casali PG, Le Cesne A, Poveda Velasco A, et al. Imatinib failure-free survival (IFS) in patients with localized gastrointestinal stromal tumors (GIST) treated with adjuvant imatinib (IM): the EORTC/AGITG/FSG/GEIS/ISG randomized controlled phase III trial. J Clin Oncol. 2013;31. Abstract 10500.
30. Joensuu H, Eriksson M, Sundby HK, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307:1265-1272.
31. Raut CP, Espat NJ, Maki RG, et al. Efficacy and tolerability of 5-year adjuvant imatinib treatment for patients with resected intermediate- or high-risk primary gastrointestinal stromal tumor: The PERSIST-5 Clinical Trial. JAMA Oncol. 2018: e184060.
32. Benjamin RS, Casali PG. Adjuvant imatinib for GI stromal tumors: when and for how long? J Clin Oncol. 2016;34:215-218.
33. Demetri GD, Wang Y, Wehrle E, et al. Imatinib plasma levels are correlated with clinical benefit in patients with unresectable/metastatic gastrointestinal stromal tumors. J Clin Oncol. 2009;27:3141-3147.
34. Eisenberg BL, Harris J, Blanke CD, et al. Phase II trial of neoadjuvant/adjuvant imatinib mesylate (IM) for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumor (GIST): early results of RTOG 0132/ACRIN 6665. J Surg Oncol. 2009;99:42-47.
35. Rutkowski P, Gronchi A, Hohenberger P, et al. Neoadjuvant imatinib in locally advanced gastrointestinal stromal tumors (GIST): the EORTC STBSG experience. Ann Surg Oncol. 2013;20:2937-2943.
36. Joensuu H, Martin-Broto J, Nishida T, et al. Follow-up strategies for patients with gastrointestinal stromal tumour treated with or without adjuvant imatinib after surgery. Eur J Cancer. 2015;51:1611-1617.
1. Ma GL, Murphy JD, Martinez ME et al. Epidemiology of gastrointestinal stromal tumors in the era of histology codes: results of a population-based study. Cancer Epidemiol Biomarkers Prev. 2015;24:298-302.
2. Agaimy A, Wunsch PH, Hofstaedter F, et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol. 2007;31:113-120.
3. Miettinen M, Sobin LH, Sarlomo-Rikala M. Immunohistochemical spectrum of GISTs at different sites and their differential diagnosis with a reference to CD117 (KIT). Mod Pathol. 2000;13:1134-1142.
4. West RB, Corless CL, Chen X, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutational status. Am J Pathol. 2004;165:107-113.
5. Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Int J Surg Pathol. 2002;10:81-89.
6. Joensuu H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol. 2008;39:1411-1419.
7. Hohenberger P, Ronellenfitsch U, Oladeji O, et al. Pattern of recurrence in patients with ruptured primary gastrointestinal stromal tumor. Br J Surg. 2010;97:1854-1859.
8. Holmenbakk T, Bjerkehagen B, Boye K, et al. Definition and clinical significance of tumor rupture in gastrointestinal stromal tumours of the small intestine. Br J Surg. 2016;103:684-691.
9. Emory TS, Sobin LH, Lukes L, et al. Prognosis of gastrointestinal smooth-muscle (stromal) tumors: dependence on anatomic site. Am J Surg Pathol. 1999;23:82-87.
10. Miettinen M, Makhlouf H, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol. 2006;30:477-489.
11. Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol. 2005;29:52-68.
12. Gold JS, Gonen M, Gutierrez A, et al. Development and validation of a prognostic nomogram for recurrence-free survival after complete surgical resection of localized primary gastrointestinal stromal tumour: a retrospective analysis. Lancet Oncol. 2009;10:1045-1052.
13. Joensuu H, Vehtari A, Rihimaki J et al. Risk of recurrence of gastrointestinal stromal tumor after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13:265-274.
14. Casali PG, Abecassis N, Bauer S, et al. Gastrointestinal stromal tumours: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow up. Ann Oncol. 2018;29(Supplement_4): iv267.
15. Jing L, Yan-Ling W, Bing-Jia C, et al. The c-kit receptor-mediated signal transduction and tumor-related diseases. Int J Biol Sci. 2013;9:435-443.
16. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577-580.
17. Joensuu H, Rutkowski P, Nishida T, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015;33:634-642.
18. Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22:3813-3825.
19. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342-4349.
20. Huss S, Pasternack H, Ihle MA, et al. Clinicopathological and molecular features of a large cohort of gastrointestinal stromal tumors (GISTs) and review of the literature: BRAF mutations in KIT/PDGFRA wild-type GISTs are rare events. Hum Pathol. 2017;62:206-214.
21. Shi E, Chmielecki J, Tang CM, et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J Transl Med. 2016;14:339.
22. Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet. 2002;108:132-139.
23. Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc. 1999;74:543-552.
24. Jakob J, Mussi C, Ronellenfitsch U, et al. Gastrointestinal stromal tumor of the rectum: results of surgical and multimodality therapy in the era of imatinib. Ann Surg Oncol. 2013;20:586-592.
25. DeMatteo RP, Ballman KV, Antonescu CR, et al. Long-term results of adjuvant imatinib mesylate in localized, high-risk, primary gastrointestinal stromal tumor (GIST): ACOSOG Z9000 (Alliance) intergroup phase 2 trial. Ann Surg. 2013;258:422-429.
26. Gleevac (imatinib) [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2016.
27. DeMatteo RP, Ballman KV, Antonescu CR, et al. Placebo-controlled randomized trial of adjuvant imatinib mesylate following the resection of localized, primary gastrointestinal stromal tumor (GIST). Lancet. 2009;373:1097-1104.
28. Corless CL, Ballman KV, Antonescu CR, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32:1563-1570.
29. Casali PG, Le Cesne A, Poveda Velasco A, et al. Imatinib failure-free survival (IFS) in patients with localized gastrointestinal stromal tumors (GIST) treated with adjuvant imatinib (IM): the EORTC/AGITG/FSG/GEIS/ISG randomized controlled phase III trial. J Clin Oncol. 2013;31. Abstract 10500.
30. Joensuu H, Eriksson M, Sundby HK, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307:1265-1272.
31. Raut CP, Espat NJ, Maki RG, et al. Efficacy and tolerability of 5-year adjuvant imatinib treatment for patients with resected intermediate- or high-risk primary gastrointestinal stromal tumor: The PERSIST-5 Clinical Trial. JAMA Oncol. 2018: e184060.
32. Benjamin RS, Casali PG. Adjuvant imatinib for GI stromal tumors: when and for how long? J Clin Oncol. 2016;34:215-218.
33. Demetri GD, Wang Y, Wehrle E, et al. Imatinib plasma levels are correlated with clinical benefit in patients with unresectable/metastatic gastrointestinal stromal tumors. J Clin Oncol. 2009;27:3141-3147.
34. Eisenberg BL, Harris J, Blanke CD, et al. Phase II trial of neoadjuvant/adjuvant imatinib mesylate (IM) for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumor (GIST): early results of RTOG 0132/ACRIN 6665. J Surg Oncol. 2009;99:42-47.
35. Rutkowski P, Gronchi A, Hohenberger P, et al. Neoadjuvant imatinib in locally advanced gastrointestinal stromal tumors (GIST): the EORTC STBSG experience. Ann Surg Oncol. 2013;20:2937-2943.
36. Joensuu H, Martin-Broto J, Nishida T, et al. Follow-up strategies for patients with gastrointestinal stromal tumour treated with or without adjuvant imatinib after surgery. Eur J Cancer. 2015;51:1611-1617.
Gastrointestinal Stromal Tumors: Management of Advanced Disease
Most advanced gastrointestinal stromal tumors (GISTs) are due to a recurrence of localized disease, with only a small minority presenting with metastatic disease.1 Compared with chemotherapy, tyrosine kinase inhibitors (TKIs) have significantly improved the natural history of the disease, with median overall survival (OS) increasing from less than 1 year to about 5 years and approximately 1 in 5 patients achieving long-term survival.2 In addition, newer drugs in development and in clinical trials appear promising and have the potential to improve outcomes even further. This article reviews current evidence on options for treating metastatic or recurrent GISTs and GISTs that have progressed following initial therapy. The evaluation and diagnosis of GIST along with management of localized disease are reviewed in a separate article.
Case Presentation
A 64-year-old African American man underwent surgical resection of a 10-cm gastric mass, which pathology reported was positive for CD117, DOG1, and CD34 and negative for smooth muscle actin and S-100, consistent with a diagnosis of GIST. There were 10 mitoses per 50 HPF, and there was no intraoperative or intraperitoneal tumor rupture. The patient was treated with adjuvant imatinib, which was discontinued after 3 years due to grade 2 myalgias, periorbital edema, and macrocytic anemia. Surveillance included office visits every 3 to 6 months and a contrast CT abdomen and pelvis every 6 months. For the past 5 years, he has not had any clinical or radiographic evidence of disease recurrence. New imaging reveals multiple liver metastases and peritoneal implants. He feels fatigued and has lost about 10 lb since his last visit. He is 5 years out from his initial diagnosis and 2 years out from last receiving imatinib. His original tumor harbored a KIT exon 11 deletion.
What treatment should you recommend now?
Imatinib for Advanced GISTs
Before the first report of the efficacy of imatinib for metastatic GISTs in 2002, patients with advanced unresectable or metastatic GISTs were routinely treated with doxorubicin-based chemotherapy regimens, which were largely ineffective, with response rates (RRs) of around 5% and a median overall survival (OS) of less than 1 year.3,4 In 2002 a landmark phase 2 study revealed imatinib’s significant efficacy profile in advanced or metastatic GISTs, resulting in its approval by the US Food and Drug Administration (FDA).5 In this study, 147 patients with CD117-positive GISTs were randomly assigned to receive daily imatinib 400 mg or 600 mg for up to 36 months. The RRs were similar between the 2 groups (68.5% vs 67.6%), with a median time to response of 12 weeks and median duration of response of 118 days. Results of this study were much more favorable when compared to doxorubicin, rendering imatinib the new standard of care for advanced GISTs. A long-term follow-up of this study after a median of 63 months confirmed near identical RRs, progression-free survival (PFS), and median survival of 57 months among the 2 groups.6
Imatinib Daily Dosing
Although 400 mg of daily imatinib proved to be efficacious, it was unclear if a dose-response relationship existed for imatinib. An EORTC phase 2 study demonstrated a benefit of using a higher dose of imatinib at 400 mg twice daily, producing a RR of 71% (4% complete , 67% partial) and 1-year PFS of 73%, which appeared favorable compared with once-daily dosing and set the framework for larger phase 3 studies.7 Two phase 3 studies compared imatinib 400 mg once daily versus twice daily (until disease progression or unacceptable toxicity) among patients with CD117-positive advanced or metastatic GISTs. These studies were eventually combined into a meta-analysis (metaGIST) to compare RR, PFS and OS between the treatment groups. Both studies allowed cross-over to the 800 mg dose for patients who progressed on 400 mg daily.
The first study, conducted jointly by the EORTC, Italian Sarcoma Group, and Australasian Gastro-Intestinal Trials Group (EU-AUS),8 randomly assigned 946 patients to 400 mg once daily or twice daily. There were no differences in response rates between the groups, but the twice-daily group had a predicted 18% reduction in the hazard for progression compared with the once-daily group (estimated HR, 0.82; P = 0.026), which came at the expense of greater toxicities warranting dose reductions (60%) and treatment interruptions (64%). Cross-over to high-dose imatinib was feasible and safe, producing a partial response in 2%, stable disease in 27%, and a median PFS of 81 days. The second study was an intergroup study conducted jointly by SWOG, CALGB, NCI-C, and ECOG (S0033, US-CDN), with a nearly identical study design as the EU-AUS trial.9 The trial enrolled 746 patients. After a median follow up of 4.5 years, the median PFS and OS were not statistically different (18 vs 20 months and 55 vs 51 months, respectively). There were also no differences in response rates. One third of patients initially placed on the once-daily arm who crossed over after progression achieved a treatment response or stable disease.
The combined EU-AUS and US-CDN analysis (metaGIST) included 1640 patients with a median age of 60 years and 58% of whom were men; 818 and 822 patients were assigned to the 400 mg and 800 mg total daily doses, respectively.10 The median follow-up was 37.5 months. There were no differences in OS (49 vs 48.7 months), median PFS (18.9 vs 23.2 months), or overall response rates (51.4% vs 53.9%). Patients who had crossed over (n = 347) to the 800 mg total daily dose arm had a 7.7-month average PFS while on the higher daily dose. An analysis was performed on 377 patients in the EU-AUS trial assessing the impact of mutational status on clinical outcomes among imatinib-treated patients. KIT exon 9 activating mutations were found to be a significant independent prognostic factor for death when compared with KIT exon 11 mutations. However, the adverse prognostic value of KIT exon 9 mutations was partially overcome with higher doses of imatinib, as those who received 800 mg total had a significantly better PFS, with a 61% relative risk reduction, than those who received 400 mg. Altogether, it was concluded that imatinib 400 mg once daily should be the standard-of-care first-line treatment for advanced or metastatic GISTs, unless a KIT exon 9 mutation is present, in which case imatinib 800 mg should be considered, if 400 mg is well tolerated. In addition, patients treated with frontline imatinib at 400 mg once daily, if tolerated well, should be considered for imatinib 800 mg upon progression of disease.
Despite there being problems with secondary resistance, significant progress has occurred in the treatment of metastatic disease over a short period of time. Prior to 2000, median OS for patients with metastatic GISTs was 9 months. With the introduction of imatinib and other TKIs, the median OS has increased to 5 years, with an estimated 10-year OS rate of approximately 20%.2
Imatinib Interruption
Since at this point, imatinib was a well-established standard of care for advanced GISTs, it was questioned whether imatinib therapy could be interrupted. At this time, treatment interruption in a stop-and-go fashion was deemed feasible in other metastatic solid tumors such as colorectal cancer (OPTIMOX1).11 The BFR French trial showed that stopping imatinib therapy in patients who had a response or stable disease after 1, 3, or 5 years was generally followed by relatively rapid tumor progression (approximately 50% of patients within 6 months), even when tumors were previously removed.12 Therefore, it is recommended that treatment in the metastatic setting should be continued indefinitely, unless there is disease progression. Hence, unlike with colorectal cancer or chronic myelogenous leukemia, as of now there is no role for imatinib interruption in metastatic GISTs.
Case Continued
The patient is started on imatinib 400 mg daily, and overall he tolerates therapy well. Interval CT imaging reveals a treatment response. Two years later, imaging reveals an increase in the tumor size and density with a new nodule present within a preexisting mass. There are no clinical trials in the area.
What defines tumor progression?
Disease Progression
When GISTs are responding to treatment, on imaging the tumors can become more cystic and less dense but with an increase in size. In addition, tumor progression may not always be associated with increased size—increased density of the tumor or a nodule within a mass that may indicate progression. If CT imaging is equivocal for progression, positron emission tomography (PET) can play a role in identifying true progression. It is critically important that tumor size and density are carefully assessed when performing interval imaging. Of note, radiofrequency ablation, cryotherapy, or chemoembolization can be used for symptomatic liver metastases or oligometastatic disease. When evaluating for progression, one needs to ask patients about compliance (ie, maintaining dose intensity related to side effects of therapy as well as the financial burden of treatment—copay toxicity).
What are mechanisms of secondary imatinib resistance?
Imatinib resistance can be subtle in patients with GISTs, manifesting with new nodular, enhancing foci enclosed within a preexisting mass (resistant clonal nodule), or can be clinically or radiographically overt.13 Imatinib resistance occurs through multiple mechanisms including acquisition of secondary activating KIT mutations in the intracellular ATP-binding domain (exons 13 and 14) and the activation loop (exons 17 and 18).14
What are the treatment options for this patient?
Second-line Therapy
Sunitinib malate is a multitargeted TKI that not only targets c-Kit and PDGFRA, but also has anti-angiogenic activity through inhibition of vascular endothelial growth factor receptors (VEGFR). Sunitinib gained FDA approval for the second-line treatment of advanced GISTs based on an international double-blind trial that randomized 312 patients with imatinib-resistant metastatic GISTs in a 2:1 fashion to receive sunitinib 50 mg daily for 4 weeks on and 2 weeks off or placebo.15,16 The trial was unblinded early at the planned interim analysis, which revealed a marked benefit, producing a 66% reduction in the hazard risk of progression (27.3 vs 6.4 weeks, HR, 0.33; P < 0.001). The most common treatment-related adverse events were fatigue, diarrhea, skin discoloration, nausea, and hand-foot syndrome. Another open-label phase 2 study assessed a continuous dosing schema of sunitinib 37.5 mg daily, which has been shown to be effective with less toxicity.17 Among the 60 patients enrolled, the primary endpoint of clinical benefit rate at 24 weeks was reached in 53%, which consisted of 13% partial responses and 40% stable disease. Most toxicities were grade 1 or 2 and easily manageable through standard interventions. This has been recommended as an alternative to the initial scheduled regimen.18 Part of sunitinib’s success is its activity against GISTs harboring secondary KIT exon 13 and 14 mutations, and possibly its anti-angiogenic activity.19 Sunitinib is particularly efficacious among GISTs harboring KIT exon 9 mutations.
Third-line Therapy
Patients who have progressed on prior imatinib and sunitinib can receive third-line regorafenib, a multi-TKI that differs chemically from sorafenib by a fluorouracil group (fluoro-sorafenib). FDA approval of regorafenib was based on the phase 3 GRID (GIST Regorafenib In progressive Disease) multicenter international trial.20 This trial randomly assigned 199 patients in a 2:1 fashion to receive regorafenib 160 mg daily for 21 days out of 28-day cycles plus best supportive care (BSC) versus placebo plus BSC. Cross-over was allowed. Regorafenib significantly reduced the hazard risk of progression by 73% compared with placebo (4.8 vs 0.9 months; HR, 0.27; P < 0.001). There was no difference in OS, which may be because of cross-over (median OS, 17.4 months in both arms). As a result, regorafenib is now considered standard third-line treatment for patients with metastatic GISTs. It has a less favorable toxicity profile than imatinib, with hand-foot syndrome, transaminitis, hypertension and fatigue being the most common treatment toxicities. In order to avoid noncompliance, it is recommended to start at 80 mg and carefully titrate upwards to the 160 mg dose.
A list of landmark studies for advanced GISTs is provided in Table 1.

A summary of FDA-approved drugs for treating GISTs is provided in Table 2.

Clinical Trials
Clinical trial enrollment should be considered for all patients with advanced or unresectable GISTs throughout their treatment continuum. Owing to significant advances in genomic profiling through next-generation sequencing, multiple driver mutations have recently been identified, and targeted therapies are being explored in clinical trials.21 For example, the neurotrophic receptor tyrosine kinase (NTRK) gene appears to be mutated in a small number of advanced GISTs, and these can respond to the highly selective TRK inhibitor larotrectinib.22 Additionally, ongoing studies are assessing immunotherapies for sporadic GISTs and treatment for familial GISTs (Table 3). Some notable studies include those assessing the efficacy of agents that target KIT and PDGFR secondary mutations, including avapritinib (BLU-285) and DCC-2618, MEK inhibitors, and the multi-kinase inhibitor crenolanib for GISTs harboring the imatinib-resistant PDGFRA D842V mutation. There are also studies utilizing checkpoint inhibitors alone or in combination with imatinib.

Case Conclusion
Given the patient’s progression on imatinib, he is started on second-line sunitinib malate. He experiences grade 1 fatigue and hand-foot syndrome, which are managed supportively. After he has been on sunitinib for approximately 8 months, his disease progresses. He subsequently undergoes genomic profiling of his tumor and starts BLU-285 on a clinical trial.
Key Points
- For advanced and metastatic disease, TKIs have substantially improved the prognosis of KIT mutated GISTs, with 3 FDA-approved drugs: imatinib, sunitinib, and regorafenib. Imatinib 400 mg is the standard-of-care frontline therapy for locally advanced, unresectable, or metastatic imatinib-sensitive GISTs. If a patient has a KIT exon 9 mutation and 400 mg is well-tolerated, increasing to 800 mg is recommended. Imatinib should be continued indefinitely unless there is intolerance, a specific patient request for interruption, or progression of disease.
- When there is progression of disease in a patient with a sensitive mutation on 400 mg of imatinib, the dose can be increased to 800 mg.
- For patients who are imatinib-intolerant or have progression, standard second line is sunitinib.
- For patients who further progress or are sunitinib-intolerant, regorafenib is the standard third-line treatment.
- There needs to be close attention to side effects, drug and food interactions, and patient copay costs in order to maintain patient compliance while on TKI therapy.
- There are still major limitations in the systemic treatment of GISTs marked by their inherent genetic heterogeneity and secondary resistance. Continued translational and clinical research is needed in order to improve treatment for patients who develop secondary resistance or who have less common primary resistant mutations. Patients are encouraged to participate in clinical trials of new therapies.
Summary
GISTs are the most common mesenchymal tumors of the GI tract. They comprise an expanding landscape of tumors that are heterogenous in terms of natural history, mutations, and response to systemic treatments. The mainstay of treatment for localized GISTs is surgical resection followed by at least 3-years of adjuvant imatinib for patients with high-risk features who are imatinib-sensitive. Patients with GISTs harboring resistance mutations such as PDGFRA D842V or with SDH-deficient or NF1-associated GISTs should not receive adjuvant imatinib. Patients with more advanced GISTs and/or in difficult to resect sites harboring a sensitive mutation can be considered for neoadjuvant imatinib. Those with metastatic GISTs can receive first-, second-, and third-line imatinib, sunitinib, or regorafenib, respectively. Clinical trial enrollment should be encouraged for patients whose GISTs harbor primary imatinib-resistant mutations, and those with advanced or unresectable GISTs with secondary resistance.
1. Ma GL, Murphy JD, Martinez ME et al. Epidemiology of gastrointestinal stromal tumors in the era of histology codes: results of a population-based study. Cancer Epidemiol Biomarkers Prev. 2015;24:298-302.
2. Heinrich MC, Rankin C, Blanke CD, et al. Correlation of long-term results of imatinib in advanced gastrointestinal stromal tumors with next-generation sequencing results: analysis of phase 3 SWOG Intergroup Trial S0033. JAMA Oncol. 2017;3:944-952.
3. DeMatteo RP, Lewis JJ, Leung D, et al. Two hundred gastrointestinal stromal tumors recurrence patterns and prognostic factors for survival. Ann Surg. 2000;231:51-58.
4. Goss GA, Merriam P, Manola J, et al. Clinical and pathological characteristics of gastrointestinal stromal tumors (GIST). Prog Proc Am Soc Clin Oncol. 2000;19:599a.
5. Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002; 347:472-480.
6. Blanke CD, Demetri GD, von Mehren M, et al. Long-term results from a randomized phase ii trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620-625.
7. Verweij J, van Oosterom A, Blay JY, et al. Imatinib mesylate (STI-571 Glivec, Gleevac) is an active agent for gastrointestinal stromal tumours, but does not yield responses in other soft-tissue sarcomas that are unselected for a molecular target. Results from an EORTC Soft Tissue and Bone Sarcoma Group phase II study. Eur J Cancer. 2003;39:2006-2011.
8. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomized trial. Lancet. 2004;364:1127-1134.
9. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26:626-632.
10. Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST). Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol. 2010;28:1247-1253.
11. Tournigand C, Cervantes A, Figer A, et al. OPTIMOX1: a randomized study of FOLFOX4 or FOLFOX7 with oxaliplatin in a stop-and-Go fashion in advanced colorectal cancer –a GERCOR study. J Clin Oncol. 2006;24:394-400.
12. Blay JV, Cesne AL, Ray-Coquard I, et al. Prospective multicentric randomized phase iii study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: The French Sarcoma Group. J Clin Oncol. 2007;25:1107-1113.
13. Desai J, Shankar S, Heinrich MC, et al. Clonal evolution of resistance to imatinib in patients with metastatic gastrointestinal stromal tumors. Clin Cancer Res. 2007;13(18 Pt 1): 5398-5405.
14. Gramza AW, Corless CL, Heinrich MC. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin Cancer Res. 2009;15:7510-7518.
15. Sutent (sunitinib malate) [package insert]. New York, NY: Pfizer Labs; 2017.
16. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomized controlled trial. Lancet. 2006;368:1329-1338.
17. George S, Blay JY, Casali PG, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer. 2009;45:1959-1968.
18. Brennan MF, Antonescu CR, Maki RG. Management of Soft Tissue Sarcomas. Switzerland: Springer International Publishing; 2013.
19. Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumors. J Clin Oncol. 2008;26:5352-5359.
20. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295-302.
21. Wilky BA, Villalobos VM. Emerging role for precision therapy through next-generation sequencing for sarcomas. JCO Precision Oncology. 2018;2:1-4.
22. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in trk fusion-positive cancers in adults and children. N Engl J Med. 2018;378:731-739.
Most advanced gastrointestinal stromal tumors (GISTs) are due to a recurrence of localized disease, with only a small minority presenting with metastatic disease.1 Compared with chemotherapy, tyrosine kinase inhibitors (TKIs) have significantly improved the natural history of the disease, with median overall survival (OS) increasing from less than 1 year to about 5 years and approximately 1 in 5 patients achieving long-term survival.2 In addition, newer drugs in development and in clinical trials appear promising and have the potential to improve outcomes even further. This article reviews current evidence on options for treating metastatic or recurrent GISTs and GISTs that have progressed following initial therapy. The evaluation and diagnosis of GIST along with management of localized disease are reviewed in a separate article.
Case Presentation
A 64-year-old African American man underwent surgical resection of a 10-cm gastric mass, which pathology reported was positive for CD117, DOG1, and CD34 and negative for smooth muscle actin and S-100, consistent with a diagnosis of GIST. There were 10 mitoses per 50 HPF, and there was no intraoperative or intraperitoneal tumor rupture. The patient was treated with adjuvant imatinib, which was discontinued after 3 years due to grade 2 myalgias, periorbital edema, and macrocytic anemia. Surveillance included office visits every 3 to 6 months and a contrast CT abdomen and pelvis every 6 months. For the past 5 years, he has not had any clinical or radiographic evidence of disease recurrence. New imaging reveals multiple liver metastases and peritoneal implants. He feels fatigued and has lost about 10 lb since his last visit. He is 5 years out from his initial diagnosis and 2 years out from last receiving imatinib. His original tumor harbored a KIT exon 11 deletion.
What treatment should you recommend now?
Imatinib for Advanced GISTs
Before the first report of the efficacy of imatinib for metastatic GISTs in 2002, patients with advanced unresectable or metastatic GISTs were routinely treated with doxorubicin-based chemotherapy regimens, which were largely ineffective, with response rates (RRs) of around 5% and a median overall survival (OS) of less than 1 year.3,4 In 2002 a landmark phase 2 study revealed imatinib’s significant efficacy profile in advanced or metastatic GISTs, resulting in its approval by the US Food and Drug Administration (FDA).5 In this study, 147 patients with CD117-positive GISTs were randomly assigned to receive daily imatinib 400 mg or 600 mg for up to 36 months. The RRs were similar between the 2 groups (68.5% vs 67.6%), with a median time to response of 12 weeks and median duration of response of 118 days. Results of this study were much more favorable when compared to doxorubicin, rendering imatinib the new standard of care for advanced GISTs. A long-term follow-up of this study after a median of 63 months confirmed near identical RRs, progression-free survival (PFS), and median survival of 57 months among the 2 groups.6
Imatinib Daily Dosing
Although 400 mg of daily imatinib proved to be efficacious, it was unclear if a dose-response relationship existed for imatinib. An EORTC phase 2 study demonstrated a benefit of using a higher dose of imatinib at 400 mg twice daily, producing a RR of 71% (4% complete , 67% partial) and 1-year PFS of 73%, which appeared favorable compared with once-daily dosing and set the framework for larger phase 3 studies.7 Two phase 3 studies compared imatinib 400 mg once daily versus twice daily (until disease progression or unacceptable toxicity) among patients with CD117-positive advanced or metastatic GISTs. These studies were eventually combined into a meta-analysis (metaGIST) to compare RR, PFS and OS between the treatment groups. Both studies allowed cross-over to the 800 mg dose for patients who progressed on 400 mg daily.
The first study, conducted jointly by the EORTC, Italian Sarcoma Group, and Australasian Gastro-Intestinal Trials Group (EU-AUS),8 randomly assigned 946 patients to 400 mg once daily or twice daily. There were no differences in response rates between the groups, but the twice-daily group had a predicted 18% reduction in the hazard for progression compared with the once-daily group (estimated HR, 0.82; P = 0.026), which came at the expense of greater toxicities warranting dose reductions (60%) and treatment interruptions (64%). Cross-over to high-dose imatinib was feasible and safe, producing a partial response in 2%, stable disease in 27%, and a median PFS of 81 days. The second study was an intergroup study conducted jointly by SWOG, CALGB, NCI-C, and ECOG (S0033, US-CDN), with a nearly identical study design as the EU-AUS trial.9 The trial enrolled 746 patients. After a median follow up of 4.5 years, the median PFS and OS were not statistically different (18 vs 20 months and 55 vs 51 months, respectively). There were also no differences in response rates. One third of patients initially placed on the once-daily arm who crossed over after progression achieved a treatment response or stable disease.
The combined EU-AUS and US-CDN analysis (metaGIST) included 1640 patients with a median age of 60 years and 58% of whom were men; 818 and 822 patients were assigned to the 400 mg and 800 mg total daily doses, respectively.10 The median follow-up was 37.5 months. There were no differences in OS (49 vs 48.7 months), median PFS (18.9 vs 23.2 months), or overall response rates (51.4% vs 53.9%). Patients who had crossed over (n = 347) to the 800 mg total daily dose arm had a 7.7-month average PFS while on the higher daily dose. An analysis was performed on 377 patients in the EU-AUS trial assessing the impact of mutational status on clinical outcomes among imatinib-treated patients. KIT exon 9 activating mutations were found to be a significant independent prognostic factor for death when compared with KIT exon 11 mutations. However, the adverse prognostic value of KIT exon 9 mutations was partially overcome with higher doses of imatinib, as those who received 800 mg total had a significantly better PFS, with a 61% relative risk reduction, than those who received 400 mg. Altogether, it was concluded that imatinib 400 mg once daily should be the standard-of-care first-line treatment for advanced or metastatic GISTs, unless a KIT exon 9 mutation is present, in which case imatinib 800 mg should be considered, if 400 mg is well tolerated. In addition, patients treated with frontline imatinib at 400 mg once daily, if tolerated well, should be considered for imatinib 800 mg upon progression of disease.
Despite there being problems with secondary resistance, significant progress has occurred in the treatment of metastatic disease over a short period of time. Prior to 2000, median OS for patients with metastatic GISTs was 9 months. With the introduction of imatinib and other TKIs, the median OS has increased to 5 years, with an estimated 10-year OS rate of approximately 20%.2
Imatinib Interruption
Since at this point, imatinib was a well-established standard of care for advanced GISTs, it was questioned whether imatinib therapy could be interrupted. At this time, treatment interruption in a stop-and-go fashion was deemed feasible in other metastatic solid tumors such as colorectal cancer (OPTIMOX1).11 The BFR French trial showed that stopping imatinib therapy in patients who had a response or stable disease after 1, 3, or 5 years was generally followed by relatively rapid tumor progression (approximately 50% of patients within 6 months), even when tumors were previously removed.12 Therefore, it is recommended that treatment in the metastatic setting should be continued indefinitely, unless there is disease progression. Hence, unlike with colorectal cancer or chronic myelogenous leukemia, as of now there is no role for imatinib interruption in metastatic GISTs.
Case Continued
The patient is started on imatinib 400 mg daily, and overall he tolerates therapy well. Interval CT imaging reveals a treatment response. Two years later, imaging reveals an increase in the tumor size and density with a new nodule present within a preexisting mass. There are no clinical trials in the area.
What defines tumor progression?
Disease Progression
When GISTs are responding to treatment, on imaging the tumors can become more cystic and less dense but with an increase in size. In addition, tumor progression may not always be associated with increased size—increased density of the tumor or a nodule within a mass that may indicate progression. If CT imaging is equivocal for progression, positron emission tomography (PET) can play a role in identifying true progression. It is critically important that tumor size and density are carefully assessed when performing interval imaging. Of note, radiofrequency ablation, cryotherapy, or chemoembolization can be used for symptomatic liver metastases or oligometastatic disease. When evaluating for progression, one needs to ask patients about compliance (ie, maintaining dose intensity related to side effects of therapy as well as the financial burden of treatment—copay toxicity).
What are mechanisms of secondary imatinib resistance?
Imatinib resistance can be subtle in patients with GISTs, manifesting with new nodular, enhancing foci enclosed within a preexisting mass (resistant clonal nodule), or can be clinically or radiographically overt.13 Imatinib resistance occurs through multiple mechanisms including acquisition of secondary activating KIT mutations in the intracellular ATP-binding domain (exons 13 and 14) and the activation loop (exons 17 and 18).14
What are the treatment options for this patient?
Second-line Therapy
Sunitinib malate is a multitargeted TKI that not only targets c-Kit and PDGFRA, but also has anti-angiogenic activity through inhibition of vascular endothelial growth factor receptors (VEGFR). Sunitinib gained FDA approval for the second-line treatment of advanced GISTs based on an international double-blind trial that randomized 312 patients with imatinib-resistant metastatic GISTs in a 2:1 fashion to receive sunitinib 50 mg daily for 4 weeks on and 2 weeks off or placebo.15,16 The trial was unblinded early at the planned interim analysis, which revealed a marked benefit, producing a 66% reduction in the hazard risk of progression (27.3 vs 6.4 weeks, HR, 0.33; P < 0.001). The most common treatment-related adverse events were fatigue, diarrhea, skin discoloration, nausea, and hand-foot syndrome. Another open-label phase 2 study assessed a continuous dosing schema of sunitinib 37.5 mg daily, which has been shown to be effective with less toxicity.17 Among the 60 patients enrolled, the primary endpoint of clinical benefit rate at 24 weeks was reached in 53%, which consisted of 13% partial responses and 40% stable disease. Most toxicities were grade 1 or 2 and easily manageable through standard interventions. This has been recommended as an alternative to the initial scheduled regimen.18 Part of sunitinib’s success is its activity against GISTs harboring secondary KIT exon 13 and 14 mutations, and possibly its anti-angiogenic activity.19 Sunitinib is particularly efficacious among GISTs harboring KIT exon 9 mutations.
Third-line Therapy
Patients who have progressed on prior imatinib and sunitinib can receive third-line regorafenib, a multi-TKI that differs chemically from sorafenib by a fluorouracil group (fluoro-sorafenib). FDA approval of regorafenib was based on the phase 3 GRID (GIST Regorafenib In progressive Disease) multicenter international trial.20 This trial randomly assigned 199 patients in a 2:1 fashion to receive regorafenib 160 mg daily for 21 days out of 28-day cycles plus best supportive care (BSC) versus placebo plus BSC. Cross-over was allowed. Regorafenib significantly reduced the hazard risk of progression by 73% compared with placebo (4.8 vs 0.9 months; HR, 0.27; P < 0.001). There was no difference in OS, which may be because of cross-over (median OS, 17.4 months in both arms). As a result, regorafenib is now considered standard third-line treatment for patients with metastatic GISTs. It has a less favorable toxicity profile than imatinib, with hand-foot syndrome, transaminitis, hypertension and fatigue being the most common treatment toxicities. In order to avoid noncompliance, it is recommended to start at 80 mg and carefully titrate upwards to the 160 mg dose.
A list of landmark studies for advanced GISTs is provided in Table 1.
A summary of FDA-approved drugs for treating GISTs is provided in Table 2.
Clinical Trials
Clinical trial enrollment should be considered for all patients with advanced or unresectable GISTs throughout their treatment continuum. Owing to significant advances in genomic profiling through next-generation sequencing, multiple driver mutations have recently been identified, and targeted therapies are being explored in clinical trials.21 For example, the neurotrophic receptor tyrosine kinase (NTRK) gene appears to be mutated in a small number of advanced GISTs, and these can respond to the highly selective TRK inhibitor larotrectinib.22 Additionally, ongoing studies are assessing immunotherapies for sporadic GISTs and treatment for familial GISTs (Table 3). Some notable studies include those assessing the efficacy of agents that target KIT and PDGFR secondary mutations, including avapritinib (BLU-285) and DCC-2618, MEK inhibitors, and the multi-kinase inhibitor crenolanib for GISTs harboring the imatinib-resistant PDGFRA D842V mutation. There are also studies utilizing checkpoint inhibitors alone or in combination with imatinib.
Case Conclusion
Given the patient’s progression on imatinib, he is started on second-line sunitinib malate. He experiences grade 1 fatigue and hand-foot syndrome, which are managed supportively. After he has been on sunitinib for approximately 8 months, his disease progresses. He subsequently undergoes genomic profiling of his tumor and starts BLU-285 on a clinical trial.
Key Points
- For advanced and metastatic disease, TKIs have substantially improved the prognosis of KIT mutated GISTs, with 3 FDA-approved drugs: imatinib, sunitinib, and regorafenib. Imatinib 400 mg is the standard-of-care frontline therapy for locally advanced, unresectable, or metastatic imatinib-sensitive GISTs. If a patient has a KIT exon 9 mutation and 400 mg is well-tolerated, increasing to 800 mg is recommended. Imatinib should be continued indefinitely unless there is intolerance, a specific patient request for interruption, or progression of disease.
- When there is progression of disease in a patient with a sensitive mutation on 400 mg of imatinib, the dose can be increased to 800 mg.
- For patients who are imatinib-intolerant or have progression, standard second line is sunitinib.
- For patients who further progress or are sunitinib-intolerant, regorafenib is the standard third-line treatment.
- There needs to be close attention to side effects, drug and food interactions, and patient copay costs in order to maintain patient compliance while on TKI therapy.
- There are still major limitations in the systemic treatment of GISTs marked by their inherent genetic heterogeneity and secondary resistance. Continued translational and clinical research is needed in order to improve treatment for patients who develop secondary resistance or who have less common primary resistant mutations. Patients are encouraged to participate in clinical trials of new therapies.
Summary
GISTs are the most common mesenchymal tumors of the GI tract. They comprise an expanding landscape of tumors that are heterogenous in terms of natural history, mutations, and response to systemic treatments. The mainstay of treatment for localized GISTs is surgical resection followed by at least 3-years of adjuvant imatinib for patients with high-risk features who are imatinib-sensitive. Patients with GISTs harboring resistance mutations such as PDGFRA D842V or with SDH-deficient or NF1-associated GISTs should not receive adjuvant imatinib. Patients with more advanced GISTs and/or in difficult to resect sites harboring a sensitive mutation can be considered for neoadjuvant imatinib. Those with metastatic GISTs can receive first-, second-, and third-line imatinib, sunitinib, or regorafenib, respectively. Clinical trial enrollment should be encouraged for patients whose GISTs harbor primary imatinib-resistant mutations, and those with advanced or unresectable GISTs with secondary resistance.
Most advanced gastrointestinal stromal tumors (GISTs) are due to a recurrence of localized disease, with only a small minority presenting with metastatic disease.1 Compared with chemotherapy, tyrosine kinase inhibitors (TKIs) have significantly improved the natural history of the disease, with median overall survival (OS) increasing from less than 1 year to about 5 years and approximately 1 in 5 patients achieving long-term survival.2 In addition, newer drugs in development and in clinical trials appear promising and have the potential to improve outcomes even further. This article reviews current evidence on options for treating metastatic or recurrent GISTs and GISTs that have progressed following initial therapy. The evaluation and diagnosis of GIST along with management of localized disease are reviewed in a separate article.
Case Presentation
A 64-year-old African American man underwent surgical resection of a 10-cm gastric mass, which pathology reported was positive for CD117, DOG1, and CD34 and negative for smooth muscle actin and S-100, consistent with a diagnosis of GIST. There were 10 mitoses per 50 HPF, and there was no intraoperative or intraperitoneal tumor rupture. The patient was treated with adjuvant imatinib, which was discontinued after 3 years due to grade 2 myalgias, periorbital edema, and macrocytic anemia. Surveillance included office visits every 3 to 6 months and a contrast CT abdomen and pelvis every 6 months. For the past 5 years, he has not had any clinical or radiographic evidence of disease recurrence. New imaging reveals multiple liver metastases and peritoneal implants. He feels fatigued and has lost about 10 lb since his last visit. He is 5 years out from his initial diagnosis and 2 years out from last receiving imatinib. His original tumor harbored a KIT exon 11 deletion.
What treatment should you recommend now?
Imatinib for Advanced GISTs
Before the first report of the efficacy of imatinib for metastatic GISTs in 2002, patients with advanced unresectable or metastatic GISTs were routinely treated with doxorubicin-based chemotherapy regimens, which were largely ineffective, with response rates (RRs) of around 5% and a median overall survival (OS) of less than 1 year.3,4 In 2002 a landmark phase 2 study revealed imatinib’s significant efficacy profile in advanced or metastatic GISTs, resulting in its approval by the US Food and Drug Administration (FDA).5 In this study, 147 patients with CD117-positive GISTs were randomly assigned to receive daily imatinib 400 mg or 600 mg for up to 36 months. The RRs were similar between the 2 groups (68.5% vs 67.6%), with a median time to response of 12 weeks and median duration of response of 118 days. Results of this study were much more favorable when compared to doxorubicin, rendering imatinib the new standard of care for advanced GISTs. A long-term follow-up of this study after a median of 63 months confirmed near identical RRs, progression-free survival (PFS), and median survival of 57 months among the 2 groups.6
Imatinib Daily Dosing
Although 400 mg of daily imatinib proved to be efficacious, it was unclear if a dose-response relationship existed for imatinib. An EORTC phase 2 study demonstrated a benefit of using a higher dose of imatinib at 400 mg twice daily, producing a RR of 71% (4% complete , 67% partial) and 1-year PFS of 73%, which appeared favorable compared with once-daily dosing and set the framework for larger phase 3 studies.7 Two phase 3 studies compared imatinib 400 mg once daily versus twice daily (until disease progression or unacceptable toxicity) among patients with CD117-positive advanced or metastatic GISTs. These studies were eventually combined into a meta-analysis (metaGIST) to compare RR, PFS and OS between the treatment groups. Both studies allowed cross-over to the 800 mg dose for patients who progressed on 400 mg daily.
The first study, conducted jointly by the EORTC, Italian Sarcoma Group, and Australasian Gastro-Intestinal Trials Group (EU-AUS),8 randomly assigned 946 patients to 400 mg once daily or twice daily. There were no differences in response rates between the groups, but the twice-daily group had a predicted 18% reduction in the hazard for progression compared with the once-daily group (estimated HR, 0.82; P = 0.026), which came at the expense of greater toxicities warranting dose reductions (60%) and treatment interruptions (64%). Cross-over to high-dose imatinib was feasible and safe, producing a partial response in 2%, stable disease in 27%, and a median PFS of 81 days. The second study was an intergroup study conducted jointly by SWOG, CALGB, NCI-C, and ECOG (S0033, US-CDN), with a nearly identical study design as the EU-AUS trial.9 The trial enrolled 746 patients. After a median follow up of 4.5 years, the median PFS and OS were not statistically different (18 vs 20 months and 55 vs 51 months, respectively). There were also no differences in response rates. One third of patients initially placed on the once-daily arm who crossed over after progression achieved a treatment response or stable disease.
The combined EU-AUS and US-CDN analysis (metaGIST) included 1640 patients with a median age of 60 years and 58% of whom were men; 818 and 822 patients were assigned to the 400 mg and 800 mg total daily doses, respectively.10 The median follow-up was 37.5 months. There were no differences in OS (49 vs 48.7 months), median PFS (18.9 vs 23.2 months), or overall response rates (51.4% vs 53.9%). Patients who had crossed over (n = 347) to the 800 mg total daily dose arm had a 7.7-month average PFS while on the higher daily dose. An analysis was performed on 377 patients in the EU-AUS trial assessing the impact of mutational status on clinical outcomes among imatinib-treated patients. KIT exon 9 activating mutations were found to be a significant independent prognostic factor for death when compared with KIT exon 11 mutations. However, the adverse prognostic value of KIT exon 9 mutations was partially overcome with higher doses of imatinib, as those who received 800 mg total had a significantly better PFS, with a 61% relative risk reduction, than those who received 400 mg. Altogether, it was concluded that imatinib 400 mg once daily should be the standard-of-care first-line treatment for advanced or metastatic GISTs, unless a KIT exon 9 mutation is present, in which case imatinib 800 mg should be considered, if 400 mg is well tolerated. In addition, patients treated with frontline imatinib at 400 mg once daily, if tolerated well, should be considered for imatinib 800 mg upon progression of disease.
Despite there being problems with secondary resistance, significant progress has occurred in the treatment of metastatic disease over a short period of time. Prior to 2000, median OS for patients with metastatic GISTs was 9 months. With the introduction of imatinib and other TKIs, the median OS has increased to 5 years, with an estimated 10-year OS rate of approximately 20%.2
Imatinib Interruption
Since at this point, imatinib was a well-established standard of care for advanced GISTs, it was questioned whether imatinib therapy could be interrupted. At this time, treatment interruption in a stop-and-go fashion was deemed feasible in other metastatic solid tumors such as colorectal cancer (OPTIMOX1).11 The BFR French trial showed that stopping imatinib therapy in patients who had a response or stable disease after 1, 3, or 5 years was generally followed by relatively rapid tumor progression (approximately 50% of patients within 6 months), even when tumors were previously removed.12 Therefore, it is recommended that treatment in the metastatic setting should be continued indefinitely, unless there is disease progression. Hence, unlike with colorectal cancer or chronic myelogenous leukemia, as of now there is no role for imatinib interruption in metastatic GISTs.
Case Continued
The patient is started on imatinib 400 mg daily, and overall he tolerates therapy well. Interval CT imaging reveals a treatment response. Two years later, imaging reveals an increase in the tumor size and density with a new nodule present within a preexisting mass. There are no clinical trials in the area.
What defines tumor progression?
Disease Progression
When GISTs are responding to treatment, on imaging the tumors can become more cystic and less dense but with an increase in size. In addition, tumor progression may not always be associated with increased size—increased density of the tumor or a nodule within a mass that may indicate progression. If CT imaging is equivocal for progression, positron emission tomography (PET) can play a role in identifying true progression. It is critically important that tumor size and density are carefully assessed when performing interval imaging. Of note, radiofrequency ablation, cryotherapy, or chemoembolization can be used for symptomatic liver metastases or oligometastatic disease. When evaluating for progression, one needs to ask patients about compliance (ie, maintaining dose intensity related to side effects of therapy as well as the financial burden of treatment—copay toxicity).
What are mechanisms of secondary imatinib resistance?
Imatinib resistance can be subtle in patients with GISTs, manifesting with new nodular, enhancing foci enclosed within a preexisting mass (resistant clonal nodule), or can be clinically or radiographically overt.13 Imatinib resistance occurs through multiple mechanisms including acquisition of secondary activating KIT mutations in the intracellular ATP-binding domain (exons 13 and 14) and the activation loop (exons 17 and 18).14
What are the treatment options for this patient?
Second-line Therapy
Sunitinib malate is a multitargeted TKI that not only targets c-Kit and PDGFRA, but also has anti-angiogenic activity through inhibition of vascular endothelial growth factor receptors (VEGFR). Sunitinib gained FDA approval for the second-line treatment of advanced GISTs based on an international double-blind trial that randomized 312 patients with imatinib-resistant metastatic GISTs in a 2:1 fashion to receive sunitinib 50 mg daily for 4 weeks on and 2 weeks off or placebo.15,16 The trial was unblinded early at the planned interim analysis, which revealed a marked benefit, producing a 66% reduction in the hazard risk of progression (27.3 vs 6.4 weeks, HR, 0.33; P < 0.001). The most common treatment-related adverse events were fatigue, diarrhea, skin discoloration, nausea, and hand-foot syndrome. Another open-label phase 2 study assessed a continuous dosing schema of sunitinib 37.5 mg daily, which has been shown to be effective with less toxicity.17 Among the 60 patients enrolled, the primary endpoint of clinical benefit rate at 24 weeks was reached in 53%, which consisted of 13% partial responses and 40% stable disease. Most toxicities were grade 1 or 2 and easily manageable through standard interventions. This has been recommended as an alternative to the initial scheduled regimen.18 Part of sunitinib’s success is its activity against GISTs harboring secondary KIT exon 13 and 14 mutations, and possibly its anti-angiogenic activity.19 Sunitinib is particularly efficacious among GISTs harboring KIT exon 9 mutations.
Third-line Therapy
Patients who have progressed on prior imatinib and sunitinib can receive third-line regorafenib, a multi-TKI that differs chemically from sorafenib by a fluorouracil group (fluoro-sorafenib). FDA approval of regorafenib was based on the phase 3 GRID (GIST Regorafenib In progressive Disease) multicenter international trial.20 This trial randomly assigned 199 patients in a 2:1 fashion to receive regorafenib 160 mg daily for 21 days out of 28-day cycles plus best supportive care (BSC) versus placebo plus BSC. Cross-over was allowed. Regorafenib significantly reduced the hazard risk of progression by 73% compared with placebo (4.8 vs 0.9 months; HR, 0.27; P < 0.001). There was no difference in OS, which may be because of cross-over (median OS, 17.4 months in both arms). As a result, regorafenib is now considered standard third-line treatment for patients with metastatic GISTs. It has a less favorable toxicity profile than imatinib, with hand-foot syndrome, transaminitis, hypertension and fatigue being the most common treatment toxicities. In order to avoid noncompliance, it is recommended to start at 80 mg and carefully titrate upwards to the 160 mg dose.
A list of landmark studies for advanced GISTs is provided in Table 1.
A summary of FDA-approved drugs for treating GISTs is provided in Table 2.
Clinical Trials
Clinical trial enrollment should be considered for all patients with advanced or unresectable GISTs throughout their treatment continuum. Owing to significant advances in genomic profiling through next-generation sequencing, multiple driver mutations have recently been identified, and targeted therapies are being explored in clinical trials.21 For example, the neurotrophic receptor tyrosine kinase (NTRK) gene appears to be mutated in a small number of advanced GISTs, and these can respond to the highly selective TRK inhibitor larotrectinib.22 Additionally, ongoing studies are assessing immunotherapies for sporadic GISTs and treatment for familial GISTs (Table 3). Some notable studies include those assessing the efficacy of agents that target KIT and PDGFR secondary mutations, including avapritinib (BLU-285) and DCC-2618, MEK inhibitors, and the multi-kinase inhibitor crenolanib for GISTs harboring the imatinib-resistant PDGFRA D842V mutation. There are also studies utilizing checkpoint inhibitors alone or in combination with imatinib.
Case Conclusion
Given the patient’s progression on imatinib, he is started on second-line sunitinib malate. He experiences grade 1 fatigue and hand-foot syndrome, which are managed supportively. After he has been on sunitinib for approximately 8 months, his disease progresses. He subsequently undergoes genomic profiling of his tumor and starts BLU-285 on a clinical trial.
Key Points
- For advanced and metastatic disease, TKIs have substantially improved the prognosis of KIT mutated GISTs, with 3 FDA-approved drugs: imatinib, sunitinib, and regorafenib. Imatinib 400 mg is the standard-of-care frontline therapy for locally advanced, unresectable, or metastatic imatinib-sensitive GISTs. If a patient has a KIT exon 9 mutation and 400 mg is well-tolerated, increasing to 800 mg is recommended. Imatinib should be continued indefinitely unless there is intolerance, a specific patient request for interruption, or progression of disease.
- When there is progression of disease in a patient with a sensitive mutation on 400 mg of imatinib, the dose can be increased to 800 mg.
- For patients who are imatinib-intolerant or have progression, standard second line is sunitinib.
- For patients who further progress or are sunitinib-intolerant, regorafenib is the standard third-line treatment.
- There needs to be close attention to side effects, drug and food interactions, and patient copay costs in order to maintain patient compliance while on TKI therapy.
- There are still major limitations in the systemic treatment of GISTs marked by their inherent genetic heterogeneity and secondary resistance. Continued translational and clinical research is needed in order to improve treatment for patients who develop secondary resistance or who have less common primary resistant mutations. Patients are encouraged to participate in clinical trials of new therapies.
Summary
GISTs are the most common mesenchymal tumors of the GI tract. They comprise an expanding landscape of tumors that are heterogenous in terms of natural history, mutations, and response to systemic treatments. The mainstay of treatment for localized GISTs is surgical resection followed by at least 3-years of adjuvant imatinib for patients with high-risk features who are imatinib-sensitive. Patients with GISTs harboring resistance mutations such as PDGFRA D842V or with SDH-deficient or NF1-associated GISTs should not receive adjuvant imatinib. Patients with more advanced GISTs and/or in difficult to resect sites harboring a sensitive mutation can be considered for neoadjuvant imatinib. Those with metastatic GISTs can receive first-, second-, and third-line imatinib, sunitinib, or regorafenib, respectively. Clinical trial enrollment should be encouraged for patients whose GISTs harbor primary imatinib-resistant mutations, and those with advanced or unresectable GISTs with secondary resistance.
1. Ma GL, Murphy JD, Martinez ME et al. Epidemiology of gastrointestinal stromal tumors in the era of histology codes: results of a population-based study. Cancer Epidemiol Biomarkers Prev. 2015;24:298-302.
2. Heinrich MC, Rankin C, Blanke CD, et al. Correlation of long-term results of imatinib in advanced gastrointestinal stromal tumors with next-generation sequencing results: analysis of phase 3 SWOG Intergroup Trial S0033. JAMA Oncol. 2017;3:944-952.
3. DeMatteo RP, Lewis JJ, Leung D, et al. Two hundred gastrointestinal stromal tumors recurrence patterns and prognostic factors for survival. Ann Surg. 2000;231:51-58.
4. Goss GA, Merriam P, Manola J, et al. Clinical and pathological characteristics of gastrointestinal stromal tumors (GIST). Prog Proc Am Soc Clin Oncol. 2000;19:599a.
5. Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002; 347:472-480.
6. Blanke CD, Demetri GD, von Mehren M, et al. Long-term results from a randomized phase ii trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620-625.
7. Verweij J, van Oosterom A, Blay JY, et al. Imatinib mesylate (STI-571 Glivec, Gleevac) is an active agent for gastrointestinal stromal tumours, but does not yield responses in other soft-tissue sarcomas that are unselected for a molecular target. Results from an EORTC Soft Tissue and Bone Sarcoma Group phase II study. Eur J Cancer. 2003;39:2006-2011.
8. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomized trial. Lancet. 2004;364:1127-1134.
9. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26:626-632.
10. Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST). Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol. 2010;28:1247-1253.
11. Tournigand C, Cervantes A, Figer A, et al. OPTIMOX1: a randomized study of FOLFOX4 or FOLFOX7 with oxaliplatin in a stop-and-Go fashion in advanced colorectal cancer –a GERCOR study. J Clin Oncol. 2006;24:394-400.
12. Blay JV, Cesne AL, Ray-Coquard I, et al. Prospective multicentric randomized phase iii study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: The French Sarcoma Group. J Clin Oncol. 2007;25:1107-1113.
13. Desai J, Shankar S, Heinrich MC, et al. Clonal evolution of resistance to imatinib in patients with metastatic gastrointestinal stromal tumors. Clin Cancer Res. 2007;13(18 Pt 1): 5398-5405.
14. Gramza AW, Corless CL, Heinrich MC. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin Cancer Res. 2009;15:7510-7518.
15. Sutent (sunitinib malate) [package insert]. New York, NY: Pfizer Labs; 2017.
16. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomized controlled trial. Lancet. 2006;368:1329-1338.
17. George S, Blay JY, Casali PG, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer. 2009;45:1959-1968.
18. Brennan MF, Antonescu CR, Maki RG. Management of Soft Tissue Sarcomas. Switzerland: Springer International Publishing; 2013.
19. Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumors. J Clin Oncol. 2008;26:5352-5359.
20. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295-302.
21. Wilky BA, Villalobos VM. Emerging role for precision therapy through next-generation sequencing for sarcomas. JCO Precision Oncology. 2018;2:1-4.
22. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in trk fusion-positive cancers in adults and children. N Engl J Med. 2018;378:731-739.
1. Ma GL, Murphy JD, Martinez ME et al. Epidemiology of gastrointestinal stromal tumors in the era of histology codes: results of a population-based study. Cancer Epidemiol Biomarkers Prev. 2015;24:298-302.
2. Heinrich MC, Rankin C, Blanke CD, et al. Correlation of long-term results of imatinib in advanced gastrointestinal stromal tumors with next-generation sequencing results: analysis of phase 3 SWOG Intergroup Trial S0033. JAMA Oncol. 2017;3:944-952.
3. DeMatteo RP, Lewis JJ, Leung D, et al. Two hundred gastrointestinal stromal tumors recurrence patterns and prognostic factors for survival. Ann Surg. 2000;231:51-58.
4. Goss GA, Merriam P, Manola J, et al. Clinical and pathological characteristics of gastrointestinal stromal tumors (GIST). Prog Proc Am Soc Clin Oncol. 2000;19:599a.
5. Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002; 347:472-480.
6. Blanke CD, Demetri GD, von Mehren M, et al. Long-term results from a randomized phase ii trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620-625.
7. Verweij J, van Oosterom A, Blay JY, et al. Imatinib mesylate (STI-571 Glivec, Gleevac) is an active agent for gastrointestinal stromal tumours, but does not yield responses in other soft-tissue sarcomas that are unselected for a molecular target. Results from an EORTC Soft Tissue and Bone Sarcoma Group phase II study. Eur J Cancer. 2003;39:2006-2011.
8. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomized trial. Lancet. 2004;364:1127-1134.
9. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26:626-632.
10. Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST). Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol. 2010;28:1247-1253.
11. Tournigand C, Cervantes A, Figer A, et al. OPTIMOX1: a randomized study of FOLFOX4 or FOLFOX7 with oxaliplatin in a stop-and-Go fashion in advanced colorectal cancer –a GERCOR study. J Clin Oncol. 2006;24:394-400.
12. Blay JV, Cesne AL, Ray-Coquard I, et al. Prospective multicentric randomized phase iii study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: The French Sarcoma Group. J Clin Oncol. 2007;25:1107-1113.
13. Desai J, Shankar S, Heinrich MC, et al. Clonal evolution of resistance to imatinib in patients with metastatic gastrointestinal stromal tumors. Clin Cancer Res. 2007;13(18 Pt 1): 5398-5405.
14. Gramza AW, Corless CL, Heinrich MC. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin Cancer Res. 2009;15:7510-7518.
15. Sutent (sunitinib malate) [package insert]. New York, NY: Pfizer Labs; 2017.
16. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomized controlled trial. Lancet. 2006;368:1329-1338.
17. George S, Blay JY, Casali PG, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer. 2009;45:1959-1968.
18. Brennan MF, Antonescu CR, Maki RG. Management of Soft Tissue Sarcomas. Switzerland: Springer International Publishing; 2013.
19. Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumors. J Clin Oncol. 2008;26:5352-5359.
20. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295-302.
21. Wilky BA, Villalobos VM. Emerging role for precision therapy through next-generation sequencing for sarcomas. JCO Precision Oncology. 2018;2:1-4.
22. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in trk fusion-positive cancers in adults and children. N Engl J Med. 2018;378:731-739.
Testicular Cancer: Diagnosis and Treatment
Malignant testicular neoplasms can arise from either the germ cells or sex-cord stromal cells, with the former comprising approximately 95% of all testicular cancers (Table 1). Germ cell tumors may contain a single histology or a mix of multiple histologies. For clinical decision making, testicular tumors are categorized as either pure seminoma (no nonseminomatous elements present) or nonseminomatous germ cell tumors (NSGCT). The prevalence of seminoma and NSGCT is roughly equal. If a testicular tumor contains both seminomatous and nonseminomatous components, it is called a mixed germ cell tumor. Because of similarities in biological behavior, the approach to treatment of mixed germ cell tumors is similar to that for NSGCT.

The key points to remember for testicular cancer are:
- With early diagnosis and aggressive multidisciplinary therapy, the overwhelming majority of patients can be cured;
- Specialized care is often critical and affects outcomes; and
- Survivorship, or post-treatment care, is very important for these patients, as they often have lifespan of several decades and a unique set of short- and long-term treatment-related complications.
Developmental Biology and Genetics
The developmental biology of germ cells and germ cell neoplasms is beyond the scope of this review, and interested readers are recommended to refer to pertinent articles on the topic.1,2 A characteristic genetic marker of all germ cell tumors is an isochromosome of the short arm of chromosome 12, i(12p). This is present in testicular tumors regardless of histologic subtype as well as in carcinoma-in-situ. In germ cell tumors without i(12p) karyotype, excess 12p genetic material consisting of repetitive segments has been found, suggesting that this is an early and potentially critical change in oncogenesis.3 Several recent studies have revealed a diverse genomic landscape in testicular cancers, including KIT, KRAS and NRAS mutations in addition to a hyperdiploid karyotype.4,5
Evaluation and Diagnosis
Case Presentation
A 23-year-old Caucasian man presents to a primary care clinic for a pre-employment history and physical exam. He reports testicular pain on the sexually transmitted infections screening questionnaire. On examination, the physician finds a firm, mobile, minimally-tender, 1.5-cm mass in the inferior aspect of left testicle. No contralateral testicular mass or inguinal lymphadenopathy is noted, and a detailed physical exam is otherwise unremarkable. The physician immediately orders an ultrasound of the testicles, which shows a 1.5-cm hypoechoic mass in the inferior aspect of the left testicle, with an unremarkable contralateral testicle. After discussion of the results, the patient is referred a urologic oncologist with expertise in testicular cancer for further care.
The urologic oncologist orders a computed tomography (CT) abdomen and pelvis with and without contrast, which shows a 1.8-cm pathologic-appearing retroperitoneal lymph node at the level of the left renal vein. Chest radiograph with anteroposterior and lateral views is unremarkable. Tumor markers are as follows: beta human chorionic gonadotropin (beta-HCG) 8 mIU/mL (normal range, 0–4 mIU/mL), alpha-fetoprotein (AFP) 2 ng/mL (normal range, 0–8.5 ng/mL), and lactate dehydrogenase (LDH) 195 U/L (normal range, 119–213 U/L).
What is the approach to the initial workup and diagnosis of testicular cancer?
Clinical Presentation and Physical Exam
The majority of testicular cancers are diagnosed on work-up of a nodule or painless swelling of one testicle, usually noted incidentally by the patient. Approximately 30% to 40% of patients complain of a dull ache or heavy sensation in the lower abdomen, perianal area, or scrotum, while acute pain is the presenting symptom in 10%.3
In approximately 10% of patients, the presenting symptom is a result of distant metastatic involvement, such as cough and dyspnea on exertion (pulmonary or mediastinal metastasis), intractable bone pain (skeletal metastasis), intractable back/flank pain, presence of psoas sign or unexplained lower extremity deep vein thrombosis (bulky retroperitoneal metastasis), or central nervous system symptoms (vertebral, spinal or brain metastasis). Constitutional symptoms (unexplained weight loss, anorexia, fatigue) often accompany these symptoms.3
Rarely (5% or less), testicular cancer may present with systemic endocrine symptoms or paraneoplastic symptoms. Gynecomastia is the most common in this category, occurring in approximately 2% of germ cell tumors and more commonly (20%–30%) in Leydig cell tumors of testis.6 Classically, these patients are either 6- to 10-year-old boys with precocious puberty or young men (mid 20s-mid 30s) with a combination of testicular mass, gynecomastia, loss of libido, and impotence. Workup typically reveals increased beta-HCG levels in blood.
Anti-Ma2-antibody-associated limbic encephalitis is the most common (and still quite rare) paraneoplastic complication associated with testicular germ cell tumors. The Ma2 antigen is selectively expressed in the neuronal nucleoli of normal brain tissue and the testicular tumor of the patient. Importantly, in a subset of these patients, the treatment of testicular cancer may result in improvement of symptoms of encephalitis.7
The first step in the diagnosis of testicular neoplasm is a physical exam. This should include a bimanual examination of the scrotal contents, starting with the normal contralateral testis. Normal testicle has a homogeneous texture and consistency, is freely movable, and is separable from the epididymis. Any firm, hard, or fixed mass within the substance of the tunica albuginea should be considered suspicious until proven otherwise. Spread to the epididymis or spermatic cord occurs in 10% to 15% of patients and examination should include these structures as well.3 A comprehensive system-wise examination for features of metastatic spread as discussed above should then be performed. If the patient has cryptorchidism, ultrasound is a mandatory part of the diagnostic workup.
If clinical evaluation suggests a possibility of testicular cancer, the patient must be counseled to undergo an expedited diagnostic workup and specialist evaluation, as a prompt diagnosis and treatment is key to not only improving the likelihood of cure, but also minimizing the treatments needed to achieve it.
Role of Imaging
Scrotal Ultrasound
Scrotal ultrasound is the first imaging modality used in the diagnostic workup of patient with suspected testicular cancer. Bilateral scrotal ultrasound can detect lesions as small as 1 to 2 mm in diameter and help differentiate intratesticular lesions from extrinsic masses. A cystic mass on ultrasound is unlikely to be malignant. Seminomas appear as well-defined hypoechoic lesions without cystic areas, while NSGCTs are typically inhomogeneous with calcifications, cystic areas, and indistinct margins. However, this distinction is not always apparent or reliable. Ultrasound alone is also insufficient for tumor staging.8 For these reasons, a radical inguinal orchiectomy must be pursued for accurate determination of histology and local stage.
If testicular ultrasound shows a suspicious intratesticular mass, the following workup is typically done:
- Measurement of serum tumor markers (beta-HCG, AFP and LDH);
- CT abdomen and pelvis with and without contrast;
- Chest radiograph anteroposterior and lateral views, or CT chest with and without contrast if clinically indicated;
- Any additional focal imaging based on symptoms (eg, magnetic resonance imaging [MRI] scan with and without contrast to evaluate the brain if the patient has CNS symptoms).
CT Scan
CT scan is the preferred imaging modality for staging of testicular cancers, specifically for evaluation of the retroperitoneum, as it is the predominant site for metastases.9 CT scan should encompass the abdomen and pelvis, and contrast-enhanced sequences should be obtained unless medically contraindicated. CT scan of the chest (if not initially done) is compulsory should a CT of abdomen and pelvis and/or a chest radiograph show abnormal findings.
The sensitivity and specificity of CT scans for detection of nodal metastases can vary significantly based on the cutoff. For example, in a series of 70 patients using a cutoff of 10 mm, the sensitivity and specificity of CT scans for patients undergoing retroperitoneal lymph node dissection were 37% and 100%, respectively.10 In the same study, a cutoff of 4 mm increased the sensitivity to 93% and decreased the specificity to 58%. The current general consensus for this cutoff value is 8 to 10 mm measured in the short axis in the transverse (axial) plane.
Approximately 20% of men with clinical stage I testicular cancer (ie, those with non-enlarged retroperitoneal lymph nodes) who do not undergo any adjuvant therapy will have disease relapse in the retroperitoneum, suggesting that they had occult micrometastases that were missed on the initial CT scans.11,12
MRI/Radionuclide Bone Scan/PET Scan
Abdominal or pelvic MRI, whole-body radionuclide bone scan, and positron emission tomography (PET) scans are almost never needed as part of the initial staging workup for testicular cancers due to several limitations, including a high false-negative rate, specifically for the PET scans, and lack of any additional value compared with CT and testicular ultrasound alone.9,13,14 If necessary, these should only be ordered after a multidisciplinary oncology consultation to prevent unnecessary delays in treatment, inappropriate changes to treatment, and unnecessary increases in cost of care.
Tumor Markers, Biopsy, and Staging
What is the role of tumor markers in the management of testicular cancers?
Serum AFP, beta-hCG, and LDH have a well-established role as tumor markers in testicular cancer. The alpha subunit of hCG is shared between multiple pituitary hormones and hence does not serve as a specific marker for testicular cancer. Serum levels of AFP and/or beta-hCG are elevated in approximately 80% percent of men with NSGCTs, even in absence of metastatic spread. On the other hand, serum beta-hCG is elevated in less than 20% and AFP is not elevated in pure seminomas.3
Tumor markers by themselves are not sufficiently sensitive or specific for the diagnosis of testicular cancer, in general, or to differentiate among its subtypes. Despite this limitation, marked elevations in these markers are rarely due to causes other than germ cell tumor. For example, serum beta-hCG concentrations greater than 10,000 mIU/mL occur only in germ cell tumors, trophoblastic differentiation of a primary lung or gastric cancer, gestational trophoblastic disease, or pregnancy. Serum AFP concentrations greater than 10,000 ng/mL occur almost exclusively in germ cell tumors and hepatocellular carcinoma.15
The pattern of marker elevation may play an important role in management of testicular cancer patients. For example, in our practice, several patients have had discordant serum tumor markers and pathology results (eg, elevated AFP with pure seminoma on orchiectomy). One of these patients was treated with adjuvant retroperitoneal lymph node dissection, which confirmed that he had a NSGCT with a seminoma, choriocarcinoma, and teratoma on pathology evaluation of retroperitoneal lymph nodes.
Serum tumor markers have 2 additional critical roles—(1) in the American Joint Committee on Cancer (AJCC)/Union for International Cancer Control (UICC) staging16 and International Germ Cell Cancer Collaboration Group (IGCCCG) risk stratification of testicular cancer,17 and (2) in post-treatment disease monitoring.
Is a testicular biopsy necessary for diagnosis?
A testicular biopsy is almost never pursued to confirm the diagnosis of testicular cancer. There is a concern that percutaneous testicular biopsy, which is associated with scrotal skin violation, can adversely affect outcomes due to tumor seeding of scrotal sac or metastatic spread into the inguinal nodes via scrotal skin lymphatics.
Tissue diagnosis is made by radical orchiectomy in a majority of cases. Rarely in our practice, we obtain a biopsy of metastatic lesion for a tissue diagnosis. This is only done in cases where chemotherapy must be started urgently to prevent worsening of complications from metastatic spread. This decision should be made only after a multidisciplinary consultation with urologic and medical oncology teams.
How is testicular cancer staged?
Both seminomatous and nonseminomatous germ cell tumors of the testis are staged using the AJCC/UICC staging system, which incorporates assessments of the primary tumor (T), lymph nodes (N), and distant metastases (M) and serum tumor marker values (S). Details of this staging system are beyond the scope of this review and further information can be obtained through the AJCC website (www.cancerstaging.org). This TNMS staging enables a prognostic assessment and helps with the therapeutic approach.
For patients with advanced germ cell tumors, a risk group classification developed by the IGCCCG is used to classify patients into good-risk, intermediate-risk, and poor-risk category (Table 2). This classification has been extensively validated for the past 2 decades, provides important prognostic information, and helps inform therapy decisions.

Treatment
Case 1 Continued
Based on the patient’s imaging and biomarker results, the patient undergoes a left radical inguinal orchiectomy. The physician’s operative note mentions that the left testicle was delivered without violation of scrotal integrity. A pathology report shows pure spermatocytic seminoma (unifocal, 1.4 cm size) with negative margins and no evidence of lymphovascular invasion. No lymph nodes are identified in the resection specimen. Post-orchiectomy markers are “negative,” meaning within normal range. After discussions with medical and radiation oncology physicians, the patient opts to pursue active surveillance.
Surgery alone followed by active surveillance is an appropriate option for this patient, as the likelihood of recurrence is low and most recurrences can be subsequently salvaged using treatment options detailed below.
What are the therapeutic options for testicular cancer?
An overview of management for most testicular cancers is presented in Table 3. Note that the actual treatments are significantly more complex and need a comprehensive multidisciplinary consultation (urologic, medical and radiation oncology) at centers with specialized testicular cancer teams, if possible.

Fertility Preservation
All patients initiating treatment for testicular cancer must be offered options for fertility preservation and consultation with a reproductive health team, if available. At the time of diagnosis, approximately 50% patients have some degree of impairment in spermatogenesis, but with effective fertility preservation, successful pregnancy can occur for as many as 30% to 60% of patients.18,19
Orchiectomy
Radical inguinal orchiectomy with high ligation of the spermatic cord at the level of the internal ring is the procedure of choice for suspected testicular cancer. The goal is to provide a definitive tissue diagnosis and local tumor control with minimal morbidity. It can be performed under general, regional, or local anesthesia. Depending on the complexity and surgical expertise, it can be done in an inpatient or outpatient setting. During the procedure, the testicle is delivered from the scrotum through an incision in the inguinal region and then resected. A testicular prosthesis is usually inserted, with resultant excellent cosmetic and patient satisfaction outcomes.20
Testicular sparing surgery (TSS) has been explored as an alternative to radical orchiectomy but is not considered a standard-of-care option at this time. Small studies have shown evidence for comparable short-term oncologic outcomes in a very select group of patients, generally with solitary tumors < 2 cm in size and solitary testicle. If this is being considered as an option, we recommended obtaining a consultation from a urologist at a high-volume center. For a majority of patients, the value of a TSS is diminished due to excellent anatomic/cosmetic outcomes with a testicular prosthesis implanted during the radical orchiectomy, and resumption of sexual functions by the unaffected contralateral testicle.
Retroperitoneal Lymph Node Dissection
As discussed, conventional cross-sectional imaging has a high false-negative rate for detection of retroperitoneal involvement. General indications for RPLND in various stages and histologies of testicular cancer germ cell tumors are outlined in Table 3. Seminoma tends to most commonly metastasize to retroperitoneum, but RPLND for seminoma is generally reserved for a very small subset of these patients. Patterns of metastases of NSGCT (except choriocarcinoma) are considered to be well-defined. In a series of patients with stage II NSGCTs, left-sided tumors metastasized to the pre- and para-aortic nodes in 88% and 86% of cases, respectively (drainage basin of left testicular vein); and right-sided tumors involved the interaortocaval nodes in 93% of patients.3 Inguinal and pelvic nodal metastases may rarely be seen and should not be used to rule out the diagnosis of testicular cancer.
Choriocarcinoma is an exception to this pattern of retroperitoneal spread, as it tends to have a higher likelihood of hematogenous metastases to distant organs. Compared with NSGCTs, pure seminomas are either localized to the testis (80% of all cases) or limited to the retroperitoneum (an additional 15% of all cases) at presentation.3
Depending on the case and expertise of the surgical team, robotic or open RPLND can be performed.21 Regardless of the approach used, RPLND remains a technically challenging surgery. The retroperitoneal “landing zone” lymph nodes lie in close proximity to, and are often densely adherent to, the abdominal great vessels. Complication rates vary widely in the reported literature, but can be as high as 50%.21-23 As detailed in Table 2, the number and size of involved retroperitoneal lymph nodes have prognostic importance.
In summary, RPLND is considered to be a viable option for a subset of early-stage NSGCT (T1-3, N0-2, M0) and for those with advanced seminoma, NSGCT, or mixed germ cell tumors with post-chemotherapy residual disease.
Systemic Chemotherapy
Except for the single-agent carboplatin, most chemotherapy regimens used to treat testicular cancer are combinations of 2 or more chemotherapy agents. For this review, we will focus on the 3 most commonly used regimens: bleomycin, etoposide, and cisplatin (BEP), etoposide and cisplatin (EP), and etoposide, ifosfamide, and cisplatin (VIP).
The core principles of testicular cancer chemotherapy are:
- Minimize dose interruptions, delays, or reductions, as these adversely affect outcomes without clearly improving side effect profile;
- Do not substitute carboplatin for cisplatin in combination regimens because carboplatin-containing combination regimens have been shown to result in significantly poorer outcomes in multiple trials of adults with germ cell tumors;24-27 and
- Give myeloid growth factor support, if necessary.
BEP
The standard BEP regimen comprises a 21-day cycle with bleomycin 30 units on days 1, 8, and 15; etoposide 100 mg/m2 on days 1 to 5; and cisplatin 20 mg/m2 on days 1 to 5. Number of cycles varies based on histology and stage (Table 3). A strong justification to maintain treatment intensity comes from the Australian and New Zealand Germ Cell Trial Group trial. In this study, 166 men were randomly assigned to treatment using 3 cycles of standard BEP or 4 cycles of a modified BEP regimen (bleomycin 30 units day 1; etoposide 120 mg/m2 days 1 to 3; cisplatin 100 mg/m2 day 1) every 21 days. This trial was stopped at interim analyses because the modified BEP arm was inferior to the standard BEP arm. With a median follow-up of 8.5 years, 8-year overall survival was 92% with standard BEP and 83% with modified BEP (P = 0.037).28
Bleomycin used in the BEP regimen has been associated with uncommon but potentially fatal pulmonary toxicity that tends to present as interstitial pneumonitis, which may ultimately progress to fibrosis or bronchiolitis obliterans with organizing pneumonia.29 This has led to evaluation of EP as an alternative to BEP.
EP
The standard EP regimen consists of a 21-day cycle with etoposide 100 mg/m2 on days 1 to 5, and cisplatin 20 mg/m2 on days 1 to 5. Due to conflicting data from multiple randomized trials, there is considerable debate in the field regarding whether 4 cycles of EP are equivalent to 3 cycles of BEP.30,31 The benefit of the EP regimen is that it avoids the higher rates of pulmonary, cutaneous, and neurologic toxicities associated bleomycin, but it does result in the patient receiving an up to 33% higher cumulative dose of cisplatin and etoposide due to the extra cycle of treatment. This has important implications in terms of tolerability and side effects, including delayed toxicities such as second malignancies, which increase with a higher cumulative dose of these agents (etoposide in particular).
VIP
The standard VIP regimen consists of a 21-day cycle with etoposide 75 mg/m2 on days 1 to 5; cisplatin 20 mg/m2 on days 1 to 5; ifosfamide 1200 mg/m2 on days 1 to 5; and mesna 120 mg/m2 IV push on day 1 followed by 1200 mg/m2 on days 1 to 5. For patients with intermediate- or poor-risk disease, 4 cycles of VIP has demonstrated comparable efficacy but higher rates of hematologic toxicities compared with 4 cycles of BEP.32-34 It remains an option for upfront treatment of patients who are not good candidates for a bleomycin-based regimen, and for patients who need salvage chemotherapy.
Adverse Effects of Chemotherapy
Acute and late chemotherapy toxicities vary significantly between regimens depending on the chemotherapy drugs used. Bleomycin-induced pneumonitis may masquerade as a “pneumonia,” which can lead to a delay in diagnosis or institution of treatment, as well as institution of an incorrect treatment (for example, there is a concern that bleomycin toxicity can be precipitated or worsened by a high fraction of inspired oxygen). Chemotherapy-associated neutropenia tends to occur a few days (7–10 days) after initiation of chemotherapy, and neutrophil counts recover without intervention in most patients after an additional 7 to 10 days. Myeloid growth factor support (eg, filgrastim, pegfilgrastim) can be given to patients either prophylactically (if they had an episode of febrile or prolonged neutropenia with the preceding cycle) or secondarily if they present with neutropenia (an absolute neutrophil count ≤ 500 cells/µL) with fever or active infection. Such interventions tend to shorten the duration of neutropenia but does not affect overall survival. Patients with asymptomatic neutropenia do not benefit from growth factor use.35
Stem Cell Transplant
Autologous stem cell transplant (SCT) is the preferred type of SCT for patients with testicular cancer and involves delivery of high doses of chemotherapy followed by infusion of patient-derived myeloid stem cells. While the details of this treatment are outside the scope of this review, decades of experience has shown that this is an effective curative option for a subset of patients with poor prognosis, such as those with platinum-refractory or relapsed disease.36
Clinical Trials
Due to excellent clinical outcomes with front-line therapy, as described, and the relatively low incidence of testicular and other germ cell tumors, clinical trial options for patients with testicular cancer are limited. The TIGER trial is an ongoing international, randomized, phase 3 trial comparing conventional TIP (paclitaxel, ifosfamide, and cisplatin) chemotherapy with high-dose chemotherapy with SCT as the first salvage treatment for relapsed/refractory germ cell tumors (NCT02375204). It is enrolling at multiple centers in the United States and results are expected in 2022. At least 2 ongoing trials are evaluating the role of immunotherapy in patients with relapsed/refractory germ cell tumors (NCT03081923 and NCT03726281). Cluster of differentiation antigen-30 (CD30) has emerged as a potential target of interest in germ cell tumors, and brentuximab vedotin, an anti-CD30 monoclonal antibody, is undergoing evaluation in a phase 2 trial of CD-30–expressing germ cell tumors (NCT01851200). This trial has completed enrollment and results are expected to be available in late 2019 or early 2020.
When possible, patients with relapsed/refractory germ cell tumors should be referred to centers of excellence with access to either testicular/germ-cell tumor specific clinical trials or phase 1 clinical trials.
Radiation Therapy
Adjuvant radiation to the retroperitoneum has a role in the management of stage I and IIA seminomas (Table 3). In a randomized noninferiority trial of radiation therapy versus single-dose carboplatin in stage I seminoma patients, 5-year recurrence-free survival was comparable at approximately 95% in either arm.37,38 In a retrospective database review of 2437 patients receiving either radiation therapy or multi-agent chemotherapy for stage II seminoma, the 5-year survival exceeded 90% in both treatment groups.39 Typically, a total of 30 to 36 Gy of radiation is delivered to para-aortic and ipsilateral external iliac lymph nodes (“dog-leg” field), followed by an optional boost to the involved nodal areas.40 Radiation is associated with acute side effects such as fatigue, gastrointestinal effects, myelosuppression as well as late side effects such as second cancers in the irradiated field (eg, sarcoma, bladder cancer).
Evaluation of Treatment Response
Monitoring of treatment response is fairly straightforward for patients with testicular cancer. Our practice is the following:
- Measure tumor markers on day 1 of each chemotherapy cycle and 3 to 4 weeks after completion of treatment.
- CT of the chest, abdomen, and pelvis with intravenous contrast prior to chemotherapy and upon completion of chemotherapy. Interim imaging is only needed for a small subset of patients with additional clinical indications (eg, new symptoms, lack of improvement in existing symptoms).
- For patients with stage II/III seminoma who have a residual mass ≥ 3 cm on post-treatment CT scan, a PET-CT scan is indicated 6 to 8 weeks after the completion of chemotherapy to determine the need for further treatment.
Active Surveillance
Because testicular cancer has high cure rates even when patients have disease relapse after primary therapy, and additional therapies have significant short- and long-term side effects in these generally young patients, active surveillance is a critical option used in the management of testicular cancer.41
Patients must be counseled that active surveillance is a form of treatment itself in that it involves close clinical and radiographic monitoring. Because there is a risk of disease relapse, patients opting to undergo active surveillance must fully understand the risks of disease recurrence and be willing to abide by the recommended follow-up schedule.
Surveillance is necessary for a minimum of 5 years and possibly 10 years following orchiectomy, and most relapses tend to occur within the first 2 years. Late relapses such as skeletal metastatic disease from seminoma have been reported to occur more than 15 years after orchiectomy, but are generally rare and unpredictable.
The general guidelines for active surveillance are as follows:
For patients with seminoma, history and physical exam and tumor marker assessment should be performed every 3 to 6 months for the first year, then every 6 to 12 months in years 2 and 3, and then annually. CT of the abdomen and pelvis should be done at 3, 6, and 12 months, every 6 to 12 months in years 2 and 3, and then every 12 to 24 months in years 4 and 5. A chest radiograph is performed only if clinically indicated, as the likelihood of distant metastatic recurrence is low.
For patients with nonseminoma, history and physical exam and tumor markers assessment should be performed every 2 to 3 months for first 2 years, every 4 to 6 months in years 3 and 4, and then annually. CT of the abdomen and pelvis should be obtained every 4 to 6 months in year 1, gradually decreasing to annually in year 3 or 4. Chest radiograph is indicated at 4 and 12 months and annually thereafter for stage IA disease. For those with stage IB disease, chest radiograph is indicated every 2 months during the first year and then gradually decreasing to annually beginning year 5.
These recommendations are expected to change over time, and treating physicians are recommended to exercise discretion and consider the patient and tumor characteristics to develop the optimal surveillance plan.
Conclusion
Testicular cancer is the most common cancer afflicting young men. Prompt diagnostic workup initiated in a primary care or hospital setting followed by a referral to a multidisciplinary team of urologists, medical oncologists, and radiation oncologists enables cure in a majority of patients. For patients with stage I seminoma, a radical inguinal orchiectomy followed by active surveillance may offer the best long-term outcome with minimal side effects. For patients with relapsed/refractory testicular cancers, clinical trial participation is strongly encouraged. Patients with a history of testicular cancer benefit from robust survivorship care tailored to their prior therapies. This can be safely delivered through their primary care providers in collaboration with the multidisciplinary oncology team.
1. van der Zwan YG, Biermann K, Wolffenbuttel KP, et al. Gonadal maldevelopment as risk factor for germ cell cancer: towards a clinical decision model. Eur Urol. 2015; 67:692–701.
2. Pierce JL, Frazier AL, Amatruda JF. Pediatric germ cell tumors: a developmental perspective. Adv Urol. 2018 Feb 4;2018.
3. Bosl GJ, Motzer RJ. Testicular germ-cell cancer. N Engl J Med. 1997;337:242-253.
4. Pyle LC, Nathanson KL. Genetic changes associated with testicular cancer susceptibility. Semin Oncol. 2016;43:575-581.
5. Shen H, Shih J, Hollern DP, et al. Integrated molecular characterization of testicular germ cell tumors. Cell Rep. 2018;23:3392-3406.
6. Barry M, Rao A, Lauer R. Sex cord-stromal tumors of the testis. In: Pagliaro L, ed. Rare Genitourinary Tumors. Cham: Springer International Publishing; 2016: 231-251.
7. Dalmau J, Graus F, Villarejo A, et al. Clinical analysis of anti-Ma2-associated encephalitis. Brain J Neurol. 2004;127:1831-1844.
8. Coursey Moreno C, Small WC, Camacho JC, et al. Testicular tumors: what radiologists need to know—differential diagnosis, staging, and management. RadioGraphics. 2015;35:400-415.
9. Kreydin EI, Barrisford GW, Feldman AS, Preston MA. Testicular cancer: what the radiologist needs to know. Am J Roentgenol. 2013;200:1215-1225.
10. Hilton S, Herr HW, Teitcher JB, et al. CT detection of retroperitoneal lymph node metastases in patients with clinical stage I testicular nonseminomatous germ cell cancer: assessment of size and distribution criteria. Am J Roentgenol. 1997;169:521-525.
11. Thompson PI, Nixon J, Harvey VJ. Disease relapse in patients with stage I nonseminomatous germ cell tumor of the testis on active surveillance. J Clin Oncol. 1988;6:1597-1603.
12. Nicolai N, Pizzocaro G. A surveillance study of clinical stage I nonseminomatous germ cell tumors of the testis: 10-year followup. J Urol. 1995;154:1045-1049.
13. Kok HK, Leong S, Torreggiani WC. Is magnetic resonance imaging comparable with computed tomography in the diagnosis of retroperitoneal metastasis in patients with testicular cancer? Can Assoc Radiol J. 2014;65:196-198.
14. Hale GR, Teplitsky S, Truong H, et al. Lymph node imaging in testicular cancer. Transl Androl Urol. 2018;7:864-874.
15. Honecker F, Aparicio J, Berney D, et al. ESMO Consensus Conference on testicular germ cell cancer: diagnosis, treatment and follow-up. Ann Oncol. 2018;29:1658-1686.
16. Paner GP, Stadler WM, Hansel DE, et al. Updates in the Eighth Edition of the Tumor-Node-Metastasis Staging Classification for Urologic Cancers. Eur Urol. 2018;73:560-569.
17. International Germ Cell Cancer Collaborative Group. International Germ Cell Consensus Classification: a prognostic factor-based staging system for metastatic germ cell cancers. International Germ Cell Cancer Collaborative Group. J Clin Oncol. 1997;15:594-603.
18. Lopategui DM, Ibrahim E, Aballa TC, et al. Effect of a formal oncofertility program on fertility preservation rates-first year experience. Transl Androl Urol. 2018;7:S271-S275.
19. Moody JA, Ahmed K, Horsfield C, et al. Fertility preservation in testicular cancer - predictors of spermatogenesis. BJU Int. 2018;122:236-242.
20. Dieckmann KP, Anheuser P, Schmidt S, et al. Testicular prostheses in patients with testicular cancer - acceptance rate and patient satisfaction. BMC Urol. 2015;15:16.
21. Schwen ZR, Gupta M, Pierorazio PM. A review of outcomes and technique for the robotic-assisted laparoscopic retroperitoneal lymph node dissection for testicular cancer. Adv Urol. 2018;2146080.
22. Singh P, Yadav S, Mahapatra S, Seth A. Outcomes following retroperitoneal lymph node dissection in postchemotherapy residual masses in advanced testicular germ cell tumors. Indian J Urol. 2016;32:40-44.
23. Heidenreich A, Thüer D, Polyakov S. Postchemotherapy retroperitoneal lymph node dissection in advanced germ cell tumours of the testis. Eur Urol. 2008;53:260-272.
24. Bajorin DF, Sarosdy MF, Pfister DG, et al. Randomized trial of etoposide and cisplatin versus etoposide and carboplatin in patients with good-risk germ cell tumors: a multiinstitutional study. J Clin Oncol. 1993;11:598-606.
25. Bokemeyer C, Köhrmann O, Tischler J, et al. A randomized trial of cisplatin, etoposide and bleomycin (PEB) versus carboplatin, etoposide and bleomycin (CEB) for patients with “good-risk” metastatic non-seminomatous germ cell tumors. Ann Oncol. 1996;7:1015-1021.
26. Horwich A, Sleijfer DT, Fosså SD, et al. Randomized trial of bleomycin, etoposide, and cisplatin compared with bleomycin, etoposide, and carboplatin in good-prognosis metastatic nonseminomatous germ cell cancer: a Multiinstitutional Medical Research Council/European Organization for Research and Treatment of Cancer Trial. J Clin Oncol. 1997;15:1844-1852.
27. Shaikh F, Nathan PC, Hale J, et al. Is there a role for carboplatin in the treatment of malignant germ cell tumors? A systematic review of adult and pediatric trials. Pediatr Blood Cancer. 2013;60:587-592.
28. Grimison PS, Stockler MR, Thomson DB, et al. Comparison of two standard chemotherapy regimens for good-prognosis germ cell tumors: updated analysis of a randomized trial. J Natl Cancer Inst. 2010;102:1253-1262.
29. Reinert T, da Rocha Baldotto CS, Nunes FAP, de Souza Scheliga AA. Bleomycin-induced lung injury. J Cancer Res. 2013;480608.
30. Jones RH, Vasey PA. Part II: Testicular cancer—management of advanced disease. Lancet Oncol. 2003;4:738-747.
31. Jankilevich G. BEP versus EP for treatment of metastatic germ-cell tumours. Lancet Oncol. 2004;5, 146.
32. Nichols CR, Catalano PJ, Crawford ED, et al. Randomized comparison of cisplatin and etoposide and either bleomycin or ifosfamide in treatment of advanced disseminated germ cell tumors: an Eastern Cooperative Oncology Group, Southwest Oncology Group, and Cancer and Leukemia Group B Study. J Clin Oncol. 1998;16:12871293.
33. Hinton S, Catalano PJ, Einhorn LH, et al. Cisplatin, etoposide and either bleomycin or ifosfamide in the treatment of disseminated germ cell tumors: final analysis of an intergroup trial. Cancer. 2003;97: 1869-1875.
34. de Wit R, Stoter G, Sleijfer DT, et al. Four cycles of BEP vs four cycles of VIP in patients with intermediate-prognosis metastatic testicular non-seminoma: a randomized study of the EORTC Genitourinary Tract Cancer Cooperative Group. European Organization for Research and Treatment of Cancer. Br J Cancer. 1998;78:828-832.
35. Mhaskar R, Clark OA, Lyman G, et al. Colony-stimulating factors for chemotherapy-induced febrile neutropenia. Cochrane Database Syst. Rev. 2014;CD003039.
36. Adra N, Abonour R, Althouse SK, et al. High-dose chemotherapy and autologous peripheral-blood stem-cell transplantation for relapsed metastatic germ cell tumors: The Indiana University experience. J Clin Oncol. 2017;35:1096-1102.
37. Oliver RT, Mason MD, Mead GM, et al. Radiotherapy versus single-dose carboplatin in adjuvant treatment of stage I seminoma: a randomised trial. Lancet. 2005;366:293-300.
38. Oliver RT, Mead GM, Rustin GJ, et al. Randomized trial of carboplatin versus radiotherapy for stage I seminoma: mature results on relapse and contralateral testis cancer rates in MRC TE19/EORTC 30982 study (ISRCTN27163214). J Clin Oncol. 2011;29:957-962.
39. Glaser SM, Vargo JA, Balasubramani GK, Beriwal S. Stage II testicular seminoma: patterns of care and survival by treatment strategy. Clin Oncol. 2016;28:513-521.
40. Boujelbene N, Cosinschi A, Boujelbene N, et al. Pure seminoma: A review and update. Radiat Oncol. 2011;6:90.
41. Nichols CR, Roth B, Albers P, et al. Active surveillance is the preferred approach to clinical stage I testicular cancer. J Clin Oncol. 2013;31;3490-3493.
Malignant testicular neoplasms can arise from either the germ cells or sex-cord stromal cells, with the former comprising approximately 95% of all testicular cancers (Table 1). Germ cell tumors may contain a single histology or a mix of multiple histologies. For clinical decision making, testicular tumors are categorized as either pure seminoma (no nonseminomatous elements present) or nonseminomatous germ cell tumors (NSGCT). The prevalence of seminoma and NSGCT is roughly equal. If a testicular tumor contains both seminomatous and nonseminomatous components, it is called a mixed germ cell tumor. Because of similarities in biological behavior, the approach to treatment of mixed germ cell tumors is similar to that for NSGCT.
The key points to remember for testicular cancer are:
- With early diagnosis and aggressive multidisciplinary therapy, the overwhelming majority of patients can be cured;
- Specialized care is often critical and affects outcomes; and
- Survivorship, or post-treatment care, is very important for these patients, as they often have lifespan of several decades and a unique set of short- and long-term treatment-related complications.
Developmental Biology and Genetics
The developmental biology of germ cells and germ cell neoplasms is beyond the scope of this review, and interested readers are recommended to refer to pertinent articles on the topic.1,2 A characteristic genetic marker of all germ cell tumors is an isochromosome of the short arm of chromosome 12, i(12p). This is present in testicular tumors regardless of histologic subtype as well as in carcinoma-in-situ. In germ cell tumors without i(12p) karyotype, excess 12p genetic material consisting of repetitive segments has been found, suggesting that this is an early and potentially critical change in oncogenesis.3 Several recent studies have revealed a diverse genomic landscape in testicular cancers, including KIT, KRAS and NRAS mutations in addition to a hyperdiploid karyotype.4,5
Evaluation and Diagnosis
Case Presentation
A 23-year-old Caucasian man presents to a primary care clinic for a pre-employment history and physical exam. He reports testicular pain on the sexually transmitted infections screening questionnaire. On examination, the physician finds a firm, mobile, minimally-tender, 1.5-cm mass in the inferior aspect of left testicle. No contralateral testicular mass or inguinal lymphadenopathy is noted, and a detailed physical exam is otherwise unremarkable. The physician immediately orders an ultrasound of the testicles, which shows a 1.5-cm hypoechoic mass in the inferior aspect of the left testicle, with an unremarkable contralateral testicle. After discussion of the results, the patient is referred a urologic oncologist with expertise in testicular cancer for further care.
The urologic oncologist orders a computed tomography (CT) abdomen and pelvis with and without contrast, which shows a 1.8-cm pathologic-appearing retroperitoneal lymph node at the level of the left renal vein. Chest radiograph with anteroposterior and lateral views is unremarkable. Tumor markers are as follows: beta human chorionic gonadotropin (beta-HCG) 8 mIU/mL (normal range, 0–4 mIU/mL), alpha-fetoprotein (AFP) 2 ng/mL (normal range, 0–8.5 ng/mL), and lactate dehydrogenase (LDH) 195 U/L (normal range, 119–213 U/L).
What is the approach to the initial workup and diagnosis of testicular cancer?
Clinical Presentation and Physical Exam
The majority of testicular cancers are diagnosed on work-up of a nodule or painless swelling of one testicle, usually noted incidentally by the patient. Approximately 30% to 40% of patients complain of a dull ache or heavy sensation in the lower abdomen, perianal area, or scrotum, while acute pain is the presenting symptom in 10%.3
In approximately 10% of patients, the presenting symptom is a result of distant metastatic involvement, such as cough and dyspnea on exertion (pulmonary or mediastinal metastasis), intractable bone pain (skeletal metastasis), intractable back/flank pain, presence of psoas sign or unexplained lower extremity deep vein thrombosis (bulky retroperitoneal metastasis), or central nervous system symptoms (vertebral, spinal or brain metastasis). Constitutional symptoms (unexplained weight loss, anorexia, fatigue) often accompany these symptoms.3
Rarely (5% or less), testicular cancer may present with systemic endocrine symptoms or paraneoplastic symptoms. Gynecomastia is the most common in this category, occurring in approximately 2% of germ cell tumors and more commonly (20%–30%) in Leydig cell tumors of testis.6 Classically, these patients are either 6- to 10-year-old boys with precocious puberty or young men (mid 20s-mid 30s) with a combination of testicular mass, gynecomastia, loss of libido, and impotence. Workup typically reveals increased beta-HCG levels in blood.
Anti-Ma2-antibody-associated limbic encephalitis is the most common (and still quite rare) paraneoplastic complication associated with testicular germ cell tumors. The Ma2 antigen is selectively expressed in the neuronal nucleoli of normal brain tissue and the testicular tumor of the patient. Importantly, in a subset of these patients, the treatment of testicular cancer may result in improvement of symptoms of encephalitis.7
The first step in the diagnosis of testicular neoplasm is a physical exam. This should include a bimanual examination of the scrotal contents, starting with the normal contralateral testis. Normal testicle has a homogeneous texture and consistency, is freely movable, and is separable from the epididymis. Any firm, hard, or fixed mass within the substance of the tunica albuginea should be considered suspicious until proven otherwise. Spread to the epididymis or spermatic cord occurs in 10% to 15% of patients and examination should include these structures as well.3 A comprehensive system-wise examination for features of metastatic spread as discussed above should then be performed. If the patient has cryptorchidism, ultrasound is a mandatory part of the diagnostic workup.
If clinical evaluation suggests a possibility of testicular cancer, the patient must be counseled to undergo an expedited diagnostic workup and specialist evaluation, as a prompt diagnosis and treatment is key to not only improving the likelihood of cure, but also minimizing the treatments needed to achieve it.
Role of Imaging
Scrotal Ultrasound
Scrotal ultrasound is the first imaging modality used in the diagnostic workup of patient with suspected testicular cancer. Bilateral scrotal ultrasound can detect lesions as small as 1 to 2 mm in diameter and help differentiate intratesticular lesions from extrinsic masses. A cystic mass on ultrasound is unlikely to be malignant. Seminomas appear as well-defined hypoechoic lesions without cystic areas, while NSGCTs are typically inhomogeneous with calcifications, cystic areas, and indistinct margins. However, this distinction is not always apparent or reliable. Ultrasound alone is also insufficient for tumor staging.8 For these reasons, a radical inguinal orchiectomy must be pursued for accurate determination of histology and local stage.
If testicular ultrasound shows a suspicious intratesticular mass, the following workup is typically done:
- Measurement of serum tumor markers (beta-HCG, AFP and LDH);
- CT abdomen and pelvis with and without contrast;
- Chest radiograph anteroposterior and lateral views, or CT chest with and without contrast if clinically indicated;
- Any additional focal imaging based on symptoms (eg, magnetic resonance imaging [MRI] scan with and without contrast to evaluate the brain if the patient has CNS symptoms).
CT Scan
CT scan is the preferred imaging modality for staging of testicular cancers, specifically for evaluation of the retroperitoneum, as it is the predominant site for metastases.9 CT scan should encompass the abdomen and pelvis, and contrast-enhanced sequences should be obtained unless medically contraindicated. CT scan of the chest (if not initially done) is compulsory should a CT of abdomen and pelvis and/or a chest radiograph show abnormal findings.
The sensitivity and specificity of CT scans for detection of nodal metastases can vary significantly based on the cutoff. For example, in a series of 70 patients using a cutoff of 10 mm, the sensitivity and specificity of CT scans for patients undergoing retroperitoneal lymph node dissection were 37% and 100%, respectively.10 In the same study, a cutoff of 4 mm increased the sensitivity to 93% and decreased the specificity to 58%. The current general consensus for this cutoff value is 8 to 10 mm measured in the short axis in the transverse (axial) plane.
Approximately 20% of men with clinical stage I testicular cancer (ie, those with non-enlarged retroperitoneal lymph nodes) who do not undergo any adjuvant therapy will have disease relapse in the retroperitoneum, suggesting that they had occult micrometastases that were missed on the initial CT scans.11,12
MRI/Radionuclide Bone Scan/PET Scan
Abdominal or pelvic MRI, whole-body radionuclide bone scan, and positron emission tomography (PET) scans are almost never needed as part of the initial staging workup for testicular cancers due to several limitations, including a high false-negative rate, specifically for the PET scans, and lack of any additional value compared with CT and testicular ultrasound alone.9,13,14 If necessary, these should only be ordered after a multidisciplinary oncology consultation to prevent unnecessary delays in treatment, inappropriate changes to treatment, and unnecessary increases in cost of care.
Tumor Markers, Biopsy, and Staging
What is the role of tumor markers in the management of testicular cancers?
Serum AFP, beta-hCG, and LDH have a well-established role as tumor markers in testicular cancer. The alpha subunit of hCG is shared between multiple pituitary hormones and hence does not serve as a specific marker for testicular cancer. Serum levels of AFP and/or beta-hCG are elevated in approximately 80% percent of men with NSGCTs, even in absence of metastatic spread. On the other hand, serum beta-hCG is elevated in less than 20% and AFP is not elevated in pure seminomas.3
Tumor markers by themselves are not sufficiently sensitive or specific for the diagnosis of testicular cancer, in general, or to differentiate among its subtypes. Despite this limitation, marked elevations in these markers are rarely due to causes other than germ cell tumor. For example, serum beta-hCG concentrations greater than 10,000 mIU/mL occur only in germ cell tumors, trophoblastic differentiation of a primary lung or gastric cancer, gestational trophoblastic disease, or pregnancy. Serum AFP concentrations greater than 10,000 ng/mL occur almost exclusively in germ cell tumors and hepatocellular carcinoma.15
The pattern of marker elevation may play an important role in management of testicular cancer patients. For example, in our practice, several patients have had discordant serum tumor markers and pathology results (eg, elevated AFP with pure seminoma on orchiectomy). One of these patients was treated with adjuvant retroperitoneal lymph node dissection, which confirmed that he had a NSGCT with a seminoma, choriocarcinoma, and teratoma on pathology evaluation of retroperitoneal lymph nodes.
Serum tumor markers have 2 additional critical roles—(1) in the American Joint Committee on Cancer (AJCC)/Union for International Cancer Control (UICC) staging16 and International Germ Cell Cancer Collaboration Group (IGCCCG) risk stratification of testicular cancer,17 and (2) in post-treatment disease monitoring.
Is a testicular biopsy necessary for diagnosis?
A testicular biopsy is almost never pursued to confirm the diagnosis of testicular cancer. There is a concern that percutaneous testicular biopsy, which is associated with scrotal skin violation, can adversely affect outcomes due to tumor seeding of scrotal sac or metastatic spread into the inguinal nodes via scrotal skin lymphatics.
Tissue diagnosis is made by radical orchiectomy in a majority of cases. Rarely in our practice, we obtain a biopsy of metastatic lesion for a tissue diagnosis. This is only done in cases where chemotherapy must be started urgently to prevent worsening of complications from metastatic spread. This decision should be made only after a multidisciplinary consultation with urologic and medical oncology teams.
How is testicular cancer staged?
Both seminomatous and nonseminomatous germ cell tumors of the testis are staged using the AJCC/UICC staging system, which incorporates assessments of the primary tumor (T), lymph nodes (N), and distant metastases (M) and serum tumor marker values (S). Details of this staging system are beyond the scope of this review and further information can be obtained through the AJCC website (www.cancerstaging.org). This TNMS staging enables a prognostic assessment and helps with the therapeutic approach.
For patients with advanced germ cell tumors, a risk group classification developed by the IGCCCG is used to classify patients into good-risk, intermediate-risk, and poor-risk category (Table 2). This classification has been extensively validated for the past 2 decades, provides important prognostic information, and helps inform therapy decisions.
Treatment
Case 1 Continued
Based on the patient’s imaging and biomarker results, the patient undergoes a left radical inguinal orchiectomy. The physician’s operative note mentions that the left testicle was delivered without violation of scrotal integrity. A pathology report shows pure spermatocytic seminoma (unifocal, 1.4 cm size) with negative margins and no evidence of lymphovascular invasion. No lymph nodes are identified in the resection specimen. Post-orchiectomy markers are “negative,” meaning within normal range. After discussions with medical and radiation oncology physicians, the patient opts to pursue active surveillance.
Surgery alone followed by active surveillance is an appropriate option for this patient, as the likelihood of recurrence is low and most recurrences can be subsequently salvaged using treatment options detailed below.
What are the therapeutic options for testicular cancer?
An overview of management for most testicular cancers is presented in Table 3. Note that the actual treatments are significantly more complex and need a comprehensive multidisciplinary consultation (urologic, medical and radiation oncology) at centers with specialized testicular cancer teams, if possible.
Fertility Preservation
All patients initiating treatment for testicular cancer must be offered options for fertility preservation and consultation with a reproductive health team, if available. At the time of diagnosis, approximately 50% patients have some degree of impairment in spermatogenesis, but with effective fertility preservation, successful pregnancy can occur for as many as 30% to 60% of patients.18,19
Orchiectomy
Radical inguinal orchiectomy with high ligation of the spermatic cord at the level of the internal ring is the procedure of choice for suspected testicular cancer. The goal is to provide a definitive tissue diagnosis and local tumor control with minimal morbidity. It can be performed under general, regional, or local anesthesia. Depending on the complexity and surgical expertise, it can be done in an inpatient or outpatient setting. During the procedure, the testicle is delivered from the scrotum through an incision in the inguinal region and then resected. A testicular prosthesis is usually inserted, with resultant excellent cosmetic and patient satisfaction outcomes.20
Testicular sparing surgery (TSS) has been explored as an alternative to radical orchiectomy but is not considered a standard-of-care option at this time. Small studies have shown evidence for comparable short-term oncologic outcomes in a very select group of patients, generally with solitary tumors < 2 cm in size and solitary testicle. If this is being considered as an option, we recommended obtaining a consultation from a urologist at a high-volume center. For a majority of patients, the value of a TSS is diminished due to excellent anatomic/cosmetic outcomes with a testicular prosthesis implanted during the radical orchiectomy, and resumption of sexual functions by the unaffected contralateral testicle.
Retroperitoneal Lymph Node Dissection
As discussed, conventional cross-sectional imaging has a high false-negative rate for detection of retroperitoneal involvement. General indications for RPLND in various stages and histologies of testicular cancer germ cell tumors are outlined in Table 3. Seminoma tends to most commonly metastasize to retroperitoneum, but RPLND for seminoma is generally reserved for a very small subset of these patients. Patterns of metastases of NSGCT (except choriocarcinoma) are considered to be well-defined. In a series of patients with stage II NSGCTs, left-sided tumors metastasized to the pre- and para-aortic nodes in 88% and 86% of cases, respectively (drainage basin of left testicular vein); and right-sided tumors involved the interaortocaval nodes in 93% of patients.3 Inguinal and pelvic nodal metastases may rarely be seen and should not be used to rule out the diagnosis of testicular cancer.
Choriocarcinoma is an exception to this pattern of retroperitoneal spread, as it tends to have a higher likelihood of hematogenous metastases to distant organs. Compared with NSGCTs, pure seminomas are either localized to the testis (80% of all cases) or limited to the retroperitoneum (an additional 15% of all cases) at presentation.3
Depending on the case and expertise of the surgical team, robotic or open RPLND can be performed.21 Regardless of the approach used, RPLND remains a technically challenging surgery. The retroperitoneal “landing zone” lymph nodes lie in close proximity to, and are often densely adherent to, the abdominal great vessels. Complication rates vary widely in the reported literature, but can be as high as 50%.21-23 As detailed in Table 2, the number and size of involved retroperitoneal lymph nodes have prognostic importance.
In summary, RPLND is considered to be a viable option for a subset of early-stage NSGCT (T1-3, N0-2, M0) and for those with advanced seminoma, NSGCT, or mixed germ cell tumors with post-chemotherapy residual disease.
Systemic Chemotherapy
Except for the single-agent carboplatin, most chemotherapy regimens used to treat testicular cancer are combinations of 2 or more chemotherapy agents. For this review, we will focus on the 3 most commonly used regimens: bleomycin, etoposide, and cisplatin (BEP), etoposide and cisplatin (EP), and etoposide, ifosfamide, and cisplatin (VIP).
The core principles of testicular cancer chemotherapy are:
- Minimize dose interruptions, delays, or reductions, as these adversely affect outcomes without clearly improving side effect profile;
- Do not substitute carboplatin for cisplatin in combination regimens because carboplatin-containing combination regimens have been shown to result in significantly poorer outcomes in multiple trials of adults with germ cell tumors;24-27 and
- Give myeloid growth factor support, if necessary.
BEP
The standard BEP regimen comprises a 21-day cycle with bleomycin 30 units on days 1, 8, and 15; etoposide 100 mg/m2 on days 1 to 5; and cisplatin 20 mg/m2 on days 1 to 5. Number of cycles varies based on histology and stage (Table 3). A strong justification to maintain treatment intensity comes from the Australian and New Zealand Germ Cell Trial Group trial. In this study, 166 men were randomly assigned to treatment using 3 cycles of standard BEP or 4 cycles of a modified BEP regimen (bleomycin 30 units day 1; etoposide 120 mg/m2 days 1 to 3; cisplatin 100 mg/m2 day 1) every 21 days. This trial was stopped at interim analyses because the modified BEP arm was inferior to the standard BEP arm. With a median follow-up of 8.5 years, 8-year overall survival was 92% with standard BEP and 83% with modified BEP (P = 0.037).28
Bleomycin used in the BEP regimen has been associated with uncommon but potentially fatal pulmonary toxicity that tends to present as interstitial pneumonitis, which may ultimately progress to fibrosis or bronchiolitis obliterans with organizing pneumonia.29 This has led to evaluation of EP as an alternative to BEP.
EP
The standard EP regimen consists of a 21-day cycle with etoposide 100 mg/m2 on days 1 to 5, and cisplatin 20 mg/m2 on days 1 to 5. Due to conflicting data from multiple randomized trials, there is considerable debate in the field regarding whether 4 cycles of EP are equivalent to 3 cycles of BEP.30,31 The benefit of the EP regimen is that it avoids the higher rates of pulmonary, cutaneous, and neurologic toxicities associated bleomycin, but it does result in the patient receiving an up to 33% higher cumulative dose of cisplatin and etoposide due to the extra cycle of treatment. This has important implications in terms of tolerability and side effects, including delayed toxicities such as second malignancies, which increase with a higher cumulative dose of these agents (etoposide in particular).
VIP
The standard VIP regimen consists of a 21-day cycle with etoposide 75 mg/m2 on days 1 to 5; cisplatin 20 mg/m2 on days 1 to 5; ifosfamide 1200 mg/m2 on days 1 to 5; and mesna 120 mg/m2 IV push on day 1 followed by 1200 mg/m2 on days 1 to 5. For patients with intermediate- or poor-risk disease, 4 cycles of VIP has demonstrated comparable efficacy but higher rates of hematologic toxicities compared with 4 cycles of BEP.32-34 It remains an option for upfront treatment of patients who are not good candidates for a bleomycin-based regimen, and for patients who need salvage chemotherapy.
Adverse Effects of Chemotherapy
Acute and late chemotherapy toxicities vary significantly between regimens depending on the chemotherapy drugs used. Bleomycin-induced pneumonitis may masquerade as a “pneumonia,” which can lead to a delay in diagnosis or institution of treatment, as well as institution of an incorrect treatment (for example, there is a concern that bleomycin toxicity can be precipitated or worsened by a high fraction of inspired oxygen). Chemotherapy-associated neutropenia tends to occur a few days (7–10 days) after initiation of chemotherapy, and neutrophil counts recover without intervention in most patients after an additional 7 to 10 days. Myeloid growth factor support (eg, filgrastim, pegfilgrastim) can be given to patients either prophylactically (if they had an episode of febrile or prolonged neutropenia with the preceding cycle) or secondarily if they present with neutropenia (an absolute neutrophil count ≤ 500 cells/µL) with fever or active infection. Such interventions tend to shorten the duration of neutropenia but does not affect overall survival. Patients with asymptomatic neutropenia do not benefit from growth factor use.35
Stem Cell Transplant
Autologous stem cell transplant (SCT) is the preferred type of SCT for patients with testicular cancer and involves delivery of high doses of chemotherapy followed by infusion of patient-derived myeloid stem cells. While the details of this treatment are outside the scope of this review, decades of experience has shown that this is an effective curative option for a subset of patients with poor prognosis, such as those with platinum-refractory or relapsed disease.36
Clinical Trials
Due to excellent clinical outcomes with front-line therapy, as described, and the relatively low incidence of testicular and other germ cell tumors, clinical trial options for patients with testicular cancer are limited. The TIGER trial is an ongoing international, randomized, phase 3 trial comparing conventional TIP (paclitaxel, ifosfamide, and cisplatin) chemotherapy with high-dose chemotherapy with SCT as the first salvage treatment for relapsed/refractory germ cell tumors (NCT02375204). It is enrolling at multiple centers in the United States and results are expected in 2022. At least 2 ongoing trials are evaluating the role of immunotherapy in patients with relapsed/refractory germ cell tumors (NCT03081923 and NCT03726281). Cluster of differentiation antigen-30 (CD30) has emerged as a potential target of interest in germ cell tumors, and brentuximab vedotin, an anti-CD30 monoclonal antibody, is undergoing evaluation in a phase 2 trial of CD-30–expressing germ cell tumors (NCT01851200). This trial has completed enrollment and results are expected to be available in late 2019 or early 2020.
When possible, patients with relapsed/refractory germ cell tumors should be referred to centers of excellence with access to either testicular/germ-cell tumor specific clinical trials or phase 1 clinical trials.
Radiation Therapy
Adjuvant radiation to the retroperitoneum has a role in the management of stage I and IIA seminomas (Table 3). In a randomized noninferiority trial of radiation therapy versus single-dose carboplatin in stage I seminoma patients, 5-year recurrence-free survival was comparable at approximately 95% in either arm.37,38 In a retrospective database review of 2437 patients receiving either radiation therapy or multi-agent chemotherapy for stage II seminoma, the 5-year survival exceeded 90% in both treatment groups.39 Typically, a total of 30 to 36 Gy of radiation is delivered to para-aortic and ipsilateral external iliac lymph nodes (“dog-leg” field), followed by an optional boost to the involved nodal areas.40 Radiation is associated with acute side effects such as fatigue, gastrointestinal effects, myelosuppression as well as late side effects such as second cancers in the irradiated field (eg, sarcoma, bladder cancer).
Evaluation of Treatment Response
Monitoring of treatment response is fairly straightforward for patients with testicular cancer. Our practice is the following:
- Measure tumor markers on day 1 of each chemotherapy cycle and 3 to 4 weeks after completion of treatment.
- CT of the chest, abdomen, and pelvis with intravenous contrast prior to chemotherapy and upon completion of chemotherapy. Interim imaging is only needed for a small subset of patients with additional clinical indications (eg, new symptoms, lack of improvement in existing symptoms).
- For patients with stage II/III seminoma who have a residual mass ≥ 3 cm on post-treatment CT scan, a PET-CT scan is indicated 6 to 8 weeks after the completion of chemotherapy to determine the need for further treatment.
Active Surveillance
Because testicular cancer has high cure rates even when patients have disease relapse after primary therapy, and additional therapies have significant short- and long-term side effects in these generally young patients, active surveillance is a critical option used in the management of testicular cancer.41
Patients must be counseled that active surveillance is a form of treatment itself in that it involves close clinical and radiographic monitoring. Because there is a risk of disease relapse, patients opting to undergo active surveillance must fully understand the risks of disease recurrence and be willing to abide by the recommended follow-up schedule.
Surveillance is necessary for a minimum of 5 years and possibly 10 years following orchiectomy, and most relapses tend to occur within the first 2 years. Late relapses such as skeletal metastatic disease from seminoma have been reported to occur more than 15 years after orchiectomy, but are generally rare and unpredictable.
The general guidelines for active surveillance are as follows:
For patients with seminoma, history and physical exam and tumor marker assessment should be performed every 3 to 6 months for the first year, then every 6 to 12 months in years 2 and 3, and then annually. CT of the abdomen and pelvis should be done at 3, 6, and 12 months, every 6 to 12 months in years 2 and 3, and then every 12 to 24 months in years 4 and 5. A chest radiograph is performed only if clinically indicated, as the likelihood of distant metastatic recurrence is low.
For patients with nonseminoma, history and physical exam and tumor markers assessment should be performed every 2 to 3 months for first 2 years, every 4 to 6 months in years 3 and 4, and then annually. CT of the abdomen and pelvis should be obtained every 4 to 6 months in year 1, gradually decreasing to annually in year 3 or 4. Chest radiograph is indicated at 4 and 12 months and annually thereafter for stage IA disease. For those with stage IB disease, chest radiograph is indicated every 2 months during the first year and then gradually decreasing to annually beginning year 5.
These recommendations are expected to change over time, and treating physicians are recommended to exercise discretion and consider the patient and tumor characteristics to develop the optimal surveillance plan.
Conclusion
Testicular cancer is the most common cancer afflicting young men. Prompt diagnostic workup initiated in a primary care or hospital setting followed by a referral to a multidisciplinary team of urologists, medical oncologists, and radiation oncologists enables cure in a majority of patients. For patients with stage I seminoma, a radical inguinal orchiectomy followed by active surveillance may offer the best long-term outcome with minimal side effects. For patients with relapsed/refractory testicular cancers, clinical trial participation is strongly encouraged. Patients with a history of testicular cancer benefit from robust survivorship care tailored to their prior therapies. This can be safely delivered through their primary care providers in collaboration with the multidisciplinary oncology team.
Malignant testicular neoplasms can arise from either the germ cells or sex-cord stromal cells, with the former comprising approximately 95% of all testicular cancers (Table 1). Germ cell tumors may contain a single histology or a mix of multiple histologies. For clinical decision making, testicular tumors are categorized as either pure seminoma (no nonseminomatous elements present) or nonseminomatous germ cell tumors (NSGCT). The prevalence of seminoma and NSGCT is roughly equal. If a testicular tumor contains both seminomatous and nonseminomatous components, it is called a mixed germ cell tumor. Because of similarities in biological behavior, the approach to treatment of mixed germ cell tumors is similar to that for NSGCT.
The key points to remember for testicular cancer are:
- With early diagnosis and aggressive multidisciplinary therapy, the overwhelming majority of patients can be cured;
- Specialized care is often critical and affects outcomes; and
- Survivorship, or post-treatment care, is very important for these patients, as they often have lifespan of several decades and a unique set of short- and long-term treatment-related complications.
Developmental Biology and Genetics
The developmental biology of germ cells and germ cell neoplasms is beyond the scope of this review, and interested readers are recommended to refer to pertinent articles on the topic.1,2 A characteristic genetic marker of all germ cell tumors is an isochromosome of the short arm of chromosome 12, i(12p). This is present in testicular tumors regardless of histologic subtype as well as in carcinoma-in-situ. In germ cell tumors without i(12p) karyotype, excess 12p genetic material consisting of repetitive segments has been found, suggesting that this is an early and potentially critical change in oncogenesis.3 Several recent studies have revealed a diverse genomic landscape in testicular cancers, including KIT, KRAS and NRAS mutations in addition to a hyperdiploid karyotype.4,5
Evaluation and Diagnosis
Case Presentation
A 23-year-old Caucasian man presents to a primary care clinic for a pre-employment history and physical exam. He reports testicular pain on the sexually transmitted infections screening questionnaire. On examination, the physician finds a firm, mobile, minimally-tender, 1.5-cm mass in the inferior aspect of left testicle. No contralateral testicular mass or inguinal lymphadenopathy is noted, and a detailed physical exam is otherwise unremarkable. The physician immediately orders an ultrasound of the testicles, which shows a 1.5-cm hypoechoic mass in the inferior aspect of the left testicle, with an unremarkable contralateral testicle. After discussion of the results, the patient is referred a urologic oncologist with expertise in testicular cancer for further care.
The urologic oncologist orders a computed tomography (CT) abdomen and pelvis with and without contrast, which shows a 1.8-cm pathologic-appearing retroperitoneal lymph node at the level of the left renal vein. Chest radiograph with anteroposterior and lateral views is unremarkable. Tumor markers are as follows: beta human chorionic gonadotropin (beta-HCG) 8 mIU/mL (normal range, 0–4 mIU/mL), alpha-fetoprotein (AFP) 2 ng/mL (normal range, 0–8.5 ng/mL), and lactate dehydrogenase (LDH) 195 U/L (normal range, 119–213 U/L).
What is the approach to the initial workup and diagnosis of testicular cancer?
Clinical Presentation and Physical Exam
The majority of testicular cancers are diagnosed on work-up of a nodule or painless swelling of one testicle, usually noted incidentally by the patient. Approximately 30% to 40% of patients complain of a dull ache or heavy sensation in the lower abdomen, perianal area, or scrotum, while acute pain is the presenting symptom in 10%.3
In approximately 10% of patients, the presenting symptom is a result of distant metastatic involvement, such as cough and dyspnea on exertion (pulmonary or mediastinal metastasis), intractable bone pain (skeletal metastasis), intractable back/flank pain, presence of psoas sign or unexplained lower extremity deep vein thrombosis (bulky retroperitoneal metastasis), or central nervous system symptoms (vertebral, spinal or brain metastasis). Constitutional symptoms (unexplained weight loss, anorexia, fatigue) often accompany these symptoms.3
Rarely (5% or less), testicular cancer may present with systemic endocrine symptoms or paraneoplastic symptoms. Gynecomastia is the most common in this category, occurring in approximately 2% of germ cell tumors and more commonly (20%–30%) in Leydig cell tumors of testis.6 Classically, these patients are either 6- to 10-year-old boys with precocious puberty or young men (mid 20s-mid 30s) with a combination of testicular mass, gynecomastia, loss of libido, and impotence. Workup typically reveals increased beta-HCG levels in blood.
Anti-Ma2-antibody-associated limbic encephalitis is the most common (and still quite rare) paraneoplastic complication associated with testicular germ cell tumors. The Ma2 antigen is selectively expressed in the neuronal nucleoli of normal brain tissue and the testicular tumor of the patient. Importantly, in a subset of these patients, the treatment of testicular cancer may result in improvement of symptoms of encephalitis.7
The first step in the diagnosis of testicular neoplasm is a physical exam. This should include a bimanual examination of the scrotal contents, starting with the normal contralateral testis. Normal testicle has a homogeneous texture and consistency, is freely movable, and is separable from the epididymis. Any firm, hard, or fixed mass within the substance of the tunica albuginea should be considered suspicious until proven otherwise. Spread to the epididymis or spermatic cord occurs in 10% to 15% of patients and examination should include these structures as well.3 A comprehensive system-wise examination for features of metastatic spread as discussed above should then be performed. If the patient has cryptorchidism, ultrasound is a mandatory part of the diagnostic workup.
If clinical evaluation suggests a possibility of testicular cancer, the patient must be counseled to undergo an expedited diagnostic workup and specialist evaluation, as a prompt diagnosis and treatment is key to not only improving the likelihood of cure, but also minimizing the treatments needed to achieve it.
Role of Imaging
Scrotal Ultrasound
Scrotal ultrasound is the first imaging modality used in the diagnostic workup of patient with suspected testicular cancer. Bilateral scrotal ultrasound can detect lesions as small as 1 to 2 mm in diameter and help differentiate intratesticular lesions from extrinsic masses. A cystic mass on ultrasound is unlikely to be malignant. Seminomas appear as well-defined hypoechoic lesions without cystic areas, while NSGCTs are typically inhomogeneous with calcifications, cystic areas, and indistinct margins. However, this distinction is not always apparent or reliable. Ultrasound alone is also insufficient for tumor staging.8 For these reasons, a radical inguinal orchiectomy must be pursued for accurate determination of histology and local stage.
If testicular ultrasound shows a suspicious intratesticular mass, the following workup is typically done:
- Measurement of serum tumor markers (beta-HCG, AFP and LDH);
- CT abdomen and pelvis with and without contrast;
- Chest radiograph anteroposterior and lateral views, or CT chest with and without contrast if clinically indicated;
- Any additional focal imaging based on symptoms (eg, magnetic resonance imaging [MRI] scan with and without contrast to evaluate the brain if the patient has CNS symptoms).
CT Scan
CT scan is the preferred imaging modality for staging of testicular cancers, specifically for evaluation of the retroperitoneum, as it is the predominant site for metastases.9 CT scan should encompass the abdomen and pelvis, and contrast-enhanced sequences should be obtained unless medically contraindicated. CT scan of the chest (if not initially done) is compulsory should a CT of abdomen and pelvis and/or a chest radiograph show abnormal findings.
The sensitivity and specificity of CT scans for detection of nodal metastases can vary significantly based on the cutoff. For example, in a series of 70 patients using a cutoff of 10 mm, the sensitivity and specificity of CT scans for patients undergoing retroperitoneal lymph node dissection were 37% and 100%, respectively.10 In the same study, a cutoff of 4 mm increased the sensitivity to 93% and decreased the specificity to 58%. The current general consensus for this cutoff value is 8 to 10 mm measured in the short axis in the transverse (axial) plane.
Approximately 20% of men with clinical stage I testicular cancer (ie, those with non-enlarged retroperitoneal lymph nodes) who do not undergo any adjuvant therapy will have disease relapse in the retroperitoneum, suggesting that they had occult micrometastases that were missed on the initial CT scans.11,12
MRI/Radionuclide Bone Scan/PET Scan
Abdominal or pelvic MRI, whole-body radionuclide bone scan, and positron emission tomography (PET) scans are almost never needed as part of the initial staging workup for testicular cancers due to several limitations, including a high false-negative rate, specifically for the PET scans, and lack of any additional value compared with CT and testicular ultrasound alone.9,13,14 If necessary, these should only be ordered after a multidisciplinary oncology consultation to prevent unnecessary delays in treatment, inappropriate changes to treatment, and unnecessary increases in cost of care.
Tumor Markers, Biopsy, and Staging
What is the role of tumor markers in the management of testicular cancers?
Serum AFP, beta-hCG, and LDH have a well-established role as tumor markers in testicular cancer. The alpha subunit of hCG is shared between multiple pituitary hormones and hence does not serve as a specific marker for testicular cancer. Serum levels of AFP and/or beta-hCG are elevated in approximately 80% percent of men with NSGCTs, even in absence of metastatic spread. On the other hand, serum beta-hCG is elevated in less than 20% and AFP is not elevated in pure seminomas.3
Tumor markers by themselves are not sufficiently sensitive or specific for the diagnosis of testicular cancer, in general, or to differentiate among its subtypes. Despite this limitation, marked elevations in these markers are rarely due to causes other than germ cell tumor. For example, serum beta-hCG concentrations greater than 10,000 mIU/mL occur only in germ cell tumors, trophoblastic differentiation of a primary lung or gastric cancer, gestational trophoblastic disease, or pregnancy. Serum AFP concentrations greater than 10,000 ng/mL occur almost exclusively in germ cell tumors and hepatocellular carcinoma.15
The pattern of marker elevation may play an important role in management of testicular cancer patients. For example, in our practice, several patients have had discordant serum tumor markers and pathology results (eg, elevated AFP with pure seminoma on orchiectomy). One of these patients was treated with adjuvant retroperitoneal lymph node dissection, which confirmed that he had a NSGCT with a seminoma, choriocarcinoma, and teratoma on pathology evaluation of retroperitoneal lymph nodes.
Serum tumor markers have 2 additional critical roles—(1) in the American Joint Committee on Cancer (AJCC)/Union for International Cancer Control (UICC) staging16 and International Germ Cell Cancer Collaboration Group (IGCCCG) risk stratification of testicular cancer,17 and (2) in post-treatment disease monitoring.
Is a testicular biopsy necessary for diagnosis?
A testicular biopsy is almost never pursued to confirm the diagnosis of testicular cancer. There is a concern that percutaneous testicular biopsy, which is associated with scrotal skin violation, can adversely affect outcomes due to tumor seeding of scrotal sac or metastatic spread into the inguinal nodes via scrotal skin lymphatics.
Tissue diagnosis is made by radical orchiectomy in a majority of cases. Rarely in our practice, we obtain a biopsy of metastatic lesion for a tissue diagnosis. This is only done in cases where chemotherapy must be started urgently to prevent worsening of complications from metastatic spread. This decision should be made only after a multidisciplinary consultation with urologic and medical oncology teams.
How is testicular cancer staged?
Both seminomatous and nonseminomatous germ cell tumors of the testis are staged using the AJCC/UICC staging system, which incorporates assessments of the primary tumor (T), lymph nodes (N), and distant metastases (M) and serum tumor marker values (S). Details of this staging system are beyond the scope of this review and further information can be obtained through the AJCC website (www.cancerstaging.org). This TNMS staging enables a prognostic assessment and helps with the therapeutic approach.
For patients with advanced germ cell tumors, a risk group classification developed by the IGCCCG is used to classify patients into good-risk, intermediate-risk, and poor-risk category (Table 2). This classification has been extensively validated for the past 2 decades, provides important prognostic information, and helps inform therapy decisions.
Treatment
Case 1 Continued
Based on the patient’s imaging and biomarker results, the patient undergoes a left radical inguinal orchiectomy. The physician’s operative note mentions that the left testicle was delivered without violation of scrotal integrity. A pathology report shows pure spermatocytic seminoma (unifocal, 1.4 cm size) with negative margins and no evidence of lymphovascular invasion. No lymph nodes are identified in the resection specimen. Post-orchiectomy markers are “negative,” meaning within normal range. After discussions with medical and radiation oncology physicians, the patient opts to pursue active surveillance.
Surgery alone followed by active surveillance is an appropriate option for this patient, as the likelihood of recurrence is low and most recurrences can be subsequently salvaged using treatment options detailed below.
What are the therapeutic options for testicular cancer?
An overview of management for most testicular cancers is presented in Table 3. Note that the actual treatments are significantly more complex and need a comprehensive multidisciplinary consultation (urologic, medical and radiation oncology) at centers with specialized testicular cancer teams, if possible.
Fertility Preservation
All patients initiating treatment for testicular cancer must be offered options for fertility preservation and consultation with a reproductive health team, if available. At the time of diagnosis, approximately 50% patients have some degree of impairment in spermatogenesis, but with effective fertility preservation, successful pregnancy can occur for as many as 30% to 60% of patients.18,19
Orchiectomy
Radical inguinal orchiectomy with high ligation of the spermatic cord at the level of the internal ring is the procedure of choice for suspected testicular cancer. The goal is to provide a definitive tissue diagnosis and local tumor control with minimal morbidity. It can be performed under general, regional, or local anesthesia. Depending on the complexity and surgical expertise, it can be done in an inpatient or outpatient setting. During the procedure, the testicle is delivered from the scrotum through an incision in the inguinal region and then resected. A testicular prosthesis is usually inserted, with resultant excellent cosmetic and patient satisfaction outcomes.20
Testicular sparing surgery (TSS) has been explored as an alternative to radical orchiectomy but is not considered a standard-of-care option at this time. Small studies have shown evidence for comparable short-term oncologic outcomes in a very select group of patients, generally with solitary tumors < 2 cm in size and solitary testicle. If this is being considered as an option, we recommended obtaining a consultation from a urologist at a high-volume center. For a majority of patients, the value of a TSS is diminished due to excellent anatomic/cosmetic outcomes with a testicular prosthesis implanted during the radical orchiectomy, and resumption of sexual functions by the unaffected contralateral testicle.
Retroperitoneal Lymph Node Dissection
As discussed, conventional cross-sectional imaging has a high false-negative rate for detection of retroperitoneal involvement. General indications for RPLND in various stages and histologies of testicular cancer germ cell tumors are outlined in Table 3. Seminoma tends to most commonly metastasize to retroperitoneum, but RPLND for seminoma is generally reserved for a very small subset of these patients. Patterns of metastases of NSGCT (except choriocarcinoma) are considered to be well-defined. In a series of patients with stage II NSGCTs, left-sided tumors metastasized to the pre- and para-aortic nodes in 88% and 86% of cases, respectively (drainage basin of left testicular vein); and right-sided tumors involved the interaortocaval nodes in 93% of patients.3 Inguinal and pelvic nodal metastases may rarely be seen and should not be used to rule out the diagnosis of testicular cancer.
Choriocarcinoma is an exception to this pattern of retroperitoneal spread, as it tends to have a higher likelihood of hematogenous metastases to distant organs. Compared with NSGCTs, pure seminomas are either localized to the testis (80% of all cases) or limited to the retroperitoneum (an additional 15% of all cases) at presentation.3
Depending on the case and expertise of the surgical team, robotic or open RPLND can be performed.21 Regardless of the approach used, RPLND remains a technically challenging surgery. The retroperitoneal “landing zone” lymph nodes lie in close proximity to, and are often densely adherent to, the abdominal great vessels. Complication rates vary widely in the reported literature, but can be as high as 50%.21-23 As detailed in Table 2, the number and size of involved retroperitoneal lymph nodes have prognostic importance.
In summary, RPLND is considered to be a viable option for a subset of early-stage NSGCT (T1-3, N0-2, M0) and for those with advanced seminoma, NSGCT, or mixed germ cell tumors with post-chemotherapy residual disease.
Systemic Chemotherapy
Except for the single-agent carboplatin, most chemotherapy regimens used to treat testicular cancer are combinations of 2 or more chemotherapy agents. For this review, we will focus on the 3 most commonly used regimens: bleomycin, etoposide, and cisplatin (BEP), etoposide and cisplatin (EP), and etoposide, ifosfamide, and cisplatin (VIP).
The core principles of testicular cancer chemotherapy are:
- Minimize dose interruptions, delays, or reductions, as these adversely affect outcomes without clearly improving side effect profile;
- Do not substitute carboplatin for cisplatin in combination regimens because carboplatin-containing combination regimens have been shown to result in significantly poorer outcomes in multiple trials of adults with germ cell tumors;24-27 and
- Give myeloid growth factor support, if necessary.
BEP
The standard BEP regimen comprises a 21-day cycle with bleomycin 30 units on days 1, 8, and 15; etoposide 100 mg/m2 on days 1 to 5; and cisplatin 20 mg/m2 on days 1 to 5. Number of cycles varies based on histology and stage (Table 3). A strong justification to maintain treatment intensity comes from the Australian and New Zealand Germ Cell Trial Group trial. In this study, 166 men were randomly assigned to treatment using 3 cycles of standard BEP or 4 cycles of a modified BEP regimen (bleomycin 30 units day 1; etoposide 120 mg/m2 days 1 to 3; cisplatin 100 mg/m2 day 1) every 21 days. This trial was stopped at interim analyses because the modified BEP arm was inferior to the standard BEP arm. With a median follow-up of 8.5 years, 8-year overall survival was 92% with standard BEP and 83% with modified BEP (P = 0.037).28
Bleomycin used in the BEP regimen has been associated with uncommon but potentially fatal pulmonary toxicity that tends to present as interstitial pneumonitis, which may ultimately progress to fibrosis or bronchiolitis obliterans with organizing pneumonia.29 This has led to evaluation of EP as an alternative to BEP.
EP
The standard EP regimen consists of a 21-day cycle with etoposide 100 mg/m2 on days 1 to 5, and cisplatin 20 mg/m2 on days 1 to 5. Due to conflicting data from multiple randomized trials, there is considerable debate in the field regarding whether 4 cycles of EP are equivalent to 3 cycles of BEP.30,31 The benefit of the EP regimen is that it avoids the higher rates of pulmonary, cutaneous, and neurologic toxicities associated bleomycin, but it does result in the patient receiving an up to 33% higher cumulative dose of cisplatin and etoposide due to the extra cycle of treatment. This has important implications in terms of tolerability and side effects, including delayed toxicities such as second malignancies, which increase with a higher cumulative dose of these agents (etoposide in particular).
VIP
The standard VIP regimen consists of a 21-day cycle with etoposide 75 mg/m2 on days 1 to 5; cisplatin 20 mg/m2 on days 1 to 5; ifosfamide 1200 mg/m2 on days 1 to 5; and mesna 120 mg/m2 IV push on day 1 followed by 1200 mg/m2 on days 1 to 5. For patients with intermediate- or poor-risk disease, 4 cycles of VIP has demonstrated comparable efficacy but higher rates of hematologic toxicities compared with 4 cycles of BEP.32-34 It remains an option for upfront treatment of patients who are not good candidates for a bleomycin-based regimen, and for patients who need salvage chemotherapy.
Adverse Effects of Chemotherapy
Acute and late chemotherapy toxicities vary significantly between regimens depending on the chemotherapy drugs used. Bleomycin-induced pneumonitis may masquerade as a “pneumonia,” which can lead to a delay in diagnosis or institution of treatment, as well as institution of an incorrect treatment (for example, there is a concern that bleomycin toxicity can be precipitated or worsened by a high fraction of inspired oxygen). Chemotherapy-associated neutropenia tends to occur a few days (7–10 days) after initiation of chemotherapy, and neutrophil counts recover without intervention in most patients after an additional 7 to 10 days. Myeloid growth factor support (eg, filgrastim, pegfilgrastim) can be given to patients either prophylactically (if they had an episode of febrile or prolonged neutropenia with the preceding cycle) or secondarily if they present with neutropenia (an absolute neutrophil count ≤ 500 cells/µL) with fever or active infection. Such interventions tend to shorten the duration of neutropenia but does not affect overall survival. Patients with asymptomatic neutropenia do not benefit from growth factor use.35
Stem Cell Transplant
Autologous stem cell transplant (SCT) is the preferred type of SCT for patients with testicular cancer and involves delivery of high doses of chemotherapy followed by infusion of patient-derived myeloid stem cells. While the details of this treatment are outside the scope of this review, decades of experience has shown that this is an effective curative option for a subset of patients with poor prognosis, such as those with platinum-refractory or relapsed disease.36
Clinical Trials
Due to excellent clinical outcomes with front-line therapy, as described, and the relatively low incidence of testicular and other germ cell tumors, clinical trial options for patients with testicular cancer are limited. The TIGER trial is an ongoing international, randomized, phase 3 trial comparing conventional TIP (paclitaxel, ifosfamide, and cisplatin) chemotherapy with high-dose chemotherapy with SCT as the first salvage treatment for relapsed/refractory germ cell tumors (NCT02375204). It is enrolling at multiple centers in the United States and results are expected in 2022. At least 2 ongoing trials are evaluating the role of immunotherapy in patients with relapsed/refractory germ cell tumors (NCT03081923 and NCT03726281). Cluster of differentiation antigen-30 (CD30) has emerged as a potential target of interest in germ cell tumors, and brentuximab vedotin, an anti-CD30 monoclonal antibody, is undergoing evaluation in a phase 2 trial of CD-30–expressing germ cell tumors (NCT01851200). This trial has completed enrollment and results are expected to be available in late 2019 or early 2020.
When possible, patients with relapsed/refractory germ cell tumors should be referred to centers of excellence with access to either testicular/germ-cell tumor specific clinical trials or phase 1 clinical trials.
Radiation Therapy
Adjuvant radiation to the retroperitoneum has a role in the management of stage I and IIA seminomas (Table 3). In a randomized noninferiority trial of radiation therapy versus single-dose carboplatin in stage I seminoma patients, 5-year recurrence-free survival was comparable at approximately 95% in either arm.37,38 In a retrospective database review of 2437 patients receiving either radiation therapy or multi-agent chemotherapy for stage II seminoma, the 5-year survival exceeded 90% in both treatment groups.39 Typically, a total of 30 to 36 Gy of radiation is delivered to para-aortic and ipsilateral external iliac lymph nodes (“dog-leg” field), followed by an optional boost to the involved nodal areas.40 Radiation is associated with acute side effects such as fatigue, gastrointestinal effects, myelosuppression as well as late side effects such as second cancers in the irradiated field (eg, sarcoma, bladder cancer).
Evaluation of Treatment Response
Monitoring of treatment response is fairly straightforward for patients with testicular cancer. Our practice is the following:
- Measure tumor markers on day 1 of each chemotherapy cycle and 3 to 4 weeks after completion of treatment.
- CT of the chest, abdomen, and pelvis with intravenous contrast prior to chemotherapy and upon completion of chemotherapy. Interim imaging is only needed for a small subset of patients with additional clinical indications (eg, new symptoms, lack of improvement in existing symptoms).
- For patients with stage II/III seminoma who have a residual mass ≥ 3 cm on post-treatment CT scan, a PET-CT scan is indicated 6 to 8 weeks after the completion of chemotherapy to determine the need for further treatment.
Active Surveillance
Because testicular cancer has high cure rates even when patients have disease relapse after primary therapy, and additional therapies have significant short- and long-term side effects in these generally young patients, active surveillance is a critical option used in the management of testicular cancer.41
Patients must be counseled that active surveillance is a form of treatment itself in that it involves close clinical and radiographic monitoring. Because there is a risk of disease relapse, patients opting to undergo active surveillance must fully understand the risks of disease recurrence and be willing to abide by the recommended follow-up schedule.
Surveillance is necessary for a minimum of 5 years and possibly 10 years following orchiectomy, and most relapses tend to occur within the first 2 years. Late relapses such as skeletal metastatic disease from seminoma have been reported to occur more than 15 years after orchiectomy, but are generally rare and unpredictable.
The general guidelines for active surveillance are as follows:
For patients with seminoma, history and physical exam and tumor marker assessment should be performed every 3 to 6 months for the first year, then every 6 to 12 months in years 2 and 3, and then annually. CT of the abdomen and pelvis should be done at 3, 6, and 12 months, every 6 to 12 months in years 2 and 3, and then every 12 to 24 months in years 4 and 5. A chest radiograph is performed only if clinically indicated, as the likelihood of distant metastatic recurrence is low.
For patients with nonseminoma, history and physical exam and tumor markers assessment should be performed every 2 to 3 months for first 2 years, every 4 to 6 months in years 3 and 4, and then annually. CT of the abdomen and pelvis should be obtained every 4 to 6 months in year 1, gradually decreasing to annually in year 3 or 4. Chest radiograph is indicated at 4 and 12 months and annually thereafter for stage IA disease. For those with stage IB disease, chest radiograph is indicated every 2 months during the first year and then gradually decreasing to annually beginning year 5.
These recommendations are expected to change over time, and treating physicians are recommended to exercise discretion and consider the patient and tumor characteristics to develop the optimal surveillance plan.
Conclusion
Testicular cancer is the most common cancer afflicting young men. Prompt diagnostic workup initiated in a primary care or hospital setting followed by a referral to a multidisciplinary team of urologists, medical oncologists, and radiation oncologists enables cure in a majority of patients. For patients with stage I seminoma, a radical inguinal orchiectomy followed by active surveillance may offer the best long-term outcome with minimal side effects. For patients with relapsed/refractory testicular cancers, clinical trial participation is strongly encouraged. Patients with a history of testicular cancer benefit from robust survivorship care tailored to their prior therapies. This can be safely delivered through their primary care providers in collaboration with the multidisciplinary oncology team.
1. van der Zwan YG, Biermann K, Wolffenbuttel KP, et al. Gonadal maldevelopment as risk factor for germ cell cancer: towards a clinical decision model. Eur Urol. 2015; 67:692–701.
2. Pierce JL, Frazier AL, Amatruda JF. Pediatric germ cell tumors: a developmental perspective. Adv Urol. 2018 Feb 4;2018.
3. Bosl GJ, Motzer RJ. Testicular germ-cell cancer. N Engl J Med. 1997;337:242-253.
4. Pyle LC, Nathanson KL. Genetic changes associated with testicular cancer susceptibility. Semin Oncol. 2016;43:575-581.
5. Shen H, Shih J, Hollern DP, et al. Integrated molecular characterization of testicular germ cell tumors. Cell Rep. 2018;23:3392-3406.
6. Barry M, Rao A, Lauer R. Sex cord-stromal tumors of the testis. In: Pagliaro L, ed. Rare Genitourinary Tumors. Cham: Springer International Publishing; 2016: 231-251.
7. Dalmau J, Graus F, Villarejo A, et al. Clinical analysis of anti-Ma2-associated encephalitis. Brain J Neurol. 2004;127:1831-1844.
8. Coursey Moreno C, Small WC, Camacho JC, et al. Testicular tumors: what radiologists need to know—differential diagnosis, staging, and management. RadioGraphics. 2015;35:400-415.
9. Kreydin EI, Barrisford GW, Feldman AS, Preston MA. Testicular cancer: what the radiologist needs to know. Am J Roentgenol. 2013;200:1215-1225.
10. Hilton S, Herr HW, Teitcher JB, et al. CT detection of retroperitoneal lymph node metastases in patients with clinical stage I testicular nonseminomatous germ cell cancer: assessment of size and distribution criteria. Am J Roentgenol. 1997;169:521-525.
11. Thompson PI, Nixon J, Harvey VJ. Disease relapse in patients with stage I nonseminomatous germ cell tumor of the testis on active surveillance. J Clin Oncol. 1988;6:1597-1603.
12. Nicolai N, Pizzocaro G. A surveillance study of clinical stage I nonseminomatous germ cell tumors of the testis: 10-year followup. J Urol. 1995;154:1045-1049.
13. Kok HK, Leong S, Torreggiani WC. Is magnetic resonance imaging comparable with computed tomography in the diagnosis of retroperitoneal metastasis in patients with testicular cancer? Can Assoc Radiol J. 2014;65:196-198.
14. Hale GR, Teplitsky S, Truong H, et al. Lymph node imaging in testicular cancer. Transl Androl Urol. 2018;7:864-874.
15. Honecker F, Aparicio J, Berney D, et al. ESMO Consensus Conference on testicular germ cell cancer: diagnosis, treatment and follow-up. Ann Oncol. 2018;29:1658-1686.
16. Paner GP, Stadler WM, Hansel DE, et al. Updates in the Eighth Edition of the Tumor-Node-Metastasis Staging Classification for Urologic Cancers. Eur Urol. 2018;73:560-569.
17. International Germ Cell Cancer Collaborative Group. International Germ Cell Consensus Classification: a prognostic factor-based staging system for metastatic germ cell cancers. International Germ Cell Cancer Collaborative Group. J Clin Oncol. 1997;15:594-603.
18. Lopategui DM, Ibrahim E, Aballa TC, et al. Effect of a formal oncofertility program on fertility preservation rates-first year experience. Transl Androl Urol. 2018;7:S271-S275.
19. Moody JA, Ahmed K, Horsfield C, et al. Fertility preservation in testicular cancer - predictors of spermatogenesis. BJU Int. 2018;122:236-242.
20. Dieckmann KP, Anheuser P, Schmidt S, et al. Testicular prostheses in patients with testicular cancer - acceptance rate and patient satisfaction. BMC Urol. 2015;15:16.
21. Schwen ZR, Gupta M, Pierorazio PM. A review of outcomes and technique for the robotic-assisted laparoscopic retroperitoneal lymph node dissection for testicular cancer. Adv Urol. 2018;2146080.
22. Singh P, Yadav S, Mahapatra S, Seth A. Outcomes following retroperitoneal lymph node dissection in postchemotherapy residual masses in advanced testicular germ cell tumors. Indian J Urol. 2016;32:40-44.
23. Heidenreich A, Thüer D, Polyakov S. Postchemotherapy retroperitoneal lymph node dissection in advanced germ cell tumours of the testis. Eur Urol. 2008;53:260-272.
24. Bajorin DF, Sarosdy MF, Pfister DG, et al. Randomized trial of etoposide and cisplatin versus etoposide and carboplatin in patients with good-risk germ cell tumors: a multiinstitutional study. J Clin Oncol. 1993;11:598-606.
25. Bokemeyer C, Köhrmann O, Tischler J, et al. A randomized trial of cisplatin, etoposide and bleomycin (PEB) versus carboplatin, etoposide and bleomycin (CEB) for patients with “good-risk” metastatic non-seminomatous germ cell tumors. Ann Oncol. 1996;7:1015-1021.
26. Horwich A, Sleijfer DT, Fosså SD, et al. Randomized trial of bleomycin, etoposide, and cisplatin compared with bleomycin, etoposide, and carboplatin in good-prognosis metastatic nonseminomatous germ cell cancer: a Multiinstitutional Medical Research Council/European Organization for Research and Treatment of Cancer Trial. J Clin Oncol. 1997;15:1844-1852.
27. Shaikh F, Nathan PC, Hale J, et al. Is there a role for carboplatin in the treatment of malignant germ cell tumors? A systematic review of adult and pediatric trials. Pediatr Blood Cancer. 2013;60:587-592.
28. Grimison PS, Stockler MR, Thomson DB, et al. Comparison of two standard chemotherapy regimens for good-prognosis germ cell tumors: updated analysis of a randomized trial. J Natl Cancer Inst. 2010;102:1253-1262.
29. Reinert T, da Rocha Baldotto CS, Nunes FAP, de Souza Scheliga AA. Bleomycin-induced lung injury. J Cancer Res. 2013;480608.
30. Jones RH, Vasey PA. Part II: Testicular cancer—management of advanced disease. Lancet Oncol. 2003;4:738-747.
31. Jankilevich G. BEP versus EP for treatment of metastatic germ-cell tumours. Lancet Oncol. 2004;5, 146.
32. Nichols CR, Catalano PJ, Crawford ED, et al. Randomized comparison of cisplatin and etoposide and either bleomycin or ifosfamide in treatment of advanced disseminated germ cell tumors: an Eastern Cooperative Oncology Group, Southwest Oncology Group, and Cancer and Leukemia Group B Study. J Clin Oncol. 1998;16:12871293.
33. Hinton S, Catalano PJ, Einhorn LH, et al. Cisplatin, etoposide and either bleomycin or ifosfamide in the treatment of disseminated germ cell tumors: final analysis of an intergroup trial. Cancer. 2003;97: 1869-1875.
34. de Wit R, Stoter G, Sleijfer DT, et al. Four cycles of BEP vs four cycles of VIP in patients with intermediate-prognosis metastatic testicular non-seminoma: a randomized study of the EORTC Genitourinary Tract Cancer Cooperative Group. European Organization for Research and Treatment of Cancer. Br J Cancer. 1998;78:828-832.
35. Mhaskar R, Clark OA, Lyman G, et al. Colony-stimulating factors for chemotherapy-induced febrile neutropenia. Cochrane Database Syst. Rev. 2014;CD003039.
36. Adra N, Abonour R, Althouse SK, et al. High-dose chemotherapy and autologous peripheral-blood stem-cell transplantation for relapsed metastatic germ cell tumors: The Indiana University experience. J Clin Oncol. 2017;35:1096-1102.
37. Oliver RT, Mason MD, Mead GM, et al. Radiotherapy versus single-dose carboplatin in adjuvant treatment of stage I seminoma: a randomised trial. Lancet. 2005;366:293-300.
38. Oliver RT, Mead GM, Rustin GJ, et al. Randomized trial of carboplatin versus radiotherapy for stage I seminoma: mature results on relapse and contralateral testis cancer rates in MRC TE19/EORTC 30982 study (ISRCTN27163214). J Clin Oncol. 2011;29:957-962.
39. Glaser SM, Vargo JA, Balasubramani GK, Beriwal S. Stage II testicular seminoma: patterns of care and survival by treatment strategy. Clin Oncol. 2016;28:513-521.
40. Boujelbene N, Cosinschi A, Boujelbene N, et al. Pure seminoma: A review and update. Radiat Oncol. 2011;6:90.
41. Nichols CR, Roth B, Albers P, et al. Active surveillance is the preferred approach to clinical stage I testicular cancer. J Clin Oncol. 2013;31;3490-3493.
1. van der Zwan YG, Biermann K, Wolffenbuttel KP, et al. Gonadal maldevelopment as risk factor for germ cell cancer: towards a clinical decision model. Eur Urol. 2015; 67:692–701.
2. Pierce JL, Frazier AL, Amatruda JF. Pediatric germ cell tumors: a developmental perspective. Adv Urol. 2018 Feb 4;2018.
3. Bosl GJ, Motzer RJ. Testicular germ-cell cancer. N Engl J Med. 1997;337:242-253.
4. Pyle LC, Nathanson KL. Genetic changes associated with testicular cancer susceptibility. Semin Oncol. 2016;43:575-581.
5. Shen H, Shih J, Hollern DP, et al. Integrated molecular characterization of testicular germ cell tumors. Cell Rep. 2018;23:3392-3406.
6. Barry M, Rao A, Lauer R. Sex cord-stromal tumors of the testis. In: Pagliaro L, ed. Rare Genitourinary Tumors. Cham: Springer International Publishing; 2016: 231-251.
7. Dalmau J, Graus F, Villarejo A, et al. Clinical analysis of anti-Ma2-associated encephalitis. Brain J Neurol. 2004;127:1831-1844.
8. Coursey Moreno C, Small WC, Camacho JC, et al. Testicular tumors: what radiologists need to know—differential diagnosis, staging, and management. RadioGraphics. 2015;35:400-415.
9. Kreydin EI, Barrisford GW, Feldman AS, Preston MA. Testicular cancer: what the radiologist needs to know. Am J Roentgenol. 2013;200:1215-1225.
10. Hilton S, Herr HW, Teitcher JB, et al. CT detection of retroperitoneal lymph node metastases in patients with clinical stage I testicular nonseminomatous germ cell cancer: assessment of size and distribution criteria. Am J Roentgenol. 1997;169:521-525.
11. Thompson PI, Nixon J, Harvey VJ. Disease relapse in patients with stage I nonseminomatous germ cell tumor of the testis on active surveillance. J Clin Oncol. 1988;6:1597-1603.
12. Nicolai N, Pizzocaro G. A surveillance study of clinical stage I nonseminomatous germ cell tumors of the testis: 10-year followup. J Urol. 1995;154:1045-1049.
13. Kok HK, Leong S, Torreggiani WC. Is magnetic resonance imaging comparable with computed tomography in the diagnosis of retroperitoneal metastasis in patients with testicular cancer? Can Assoc Radiol J. 2014;65:196-198.
14. Hale GR, Teplitsky S, Truong H, et al. Lymph node imaging in testicular cancer. Transl Androl Urol. 2018;7:864-874.
15. Honecker F, Aparicio J, Berney D, et al. ESMO Consensus Conference on testicular germ cell cancer: diagnosis, treatment and follow-up. Ann Oncol. 2018;29:1658-1686.
16. Paner GP, Stadler WM, Hansel DE, et al. Updates in the Eighth Edition of the Tumor-Node-Metastasis Staging Classification for Urologic Cancers. Eur Urol. 2018;73:560-569.
17. International Germ Cell Cancer Collaborative Group. International Germ Cell Consensus Classification: a prognostic factor-based staging system for metastatic germ cell cancers. International Germ Cell Cancer Collaborative Group. J Clin Oncol. 1997;15:594-603.
18. Lopategui DM, Ibrahim E, Aballa TC, et al. Effect of a formal oncofertility program on fertility preservation rates-first year experience. Transl Androl Urol. 2018;7:S271-S275.
19. Moody JA, Ahmed K, Horsfield C, et al. Fertility preservation in testicular cancer - predictors of spermatogenesis. BJU Int. 2018;122:236-242.
20. Dieckmann KP, Anheuser P, Schmidt S, et al. Testicular prostheses in patients with testicular cancer - acceptance rate and patient satisfaction. BMC Urol. 2015;15:16.
21. Schwen ZR, Gupta M, Pierorazio PM. A review of outcomes and technique for the robotic-assisted laparoscopic retroperitoneal lymph node dissection for testicular cancer. Adv Urol. 2018;2146080.
22. Singh P, Yadav S, Mahapatra S, Seth A. Outcomes following retroperitoneal lymph node dissection in postchemotherapy residual masses in advanced testicular germ cell tumors. Indian J Urol. 2016;32:40-44.
23. Heidenreich A, Thüer D, Polyakov S. Postchemotherapy retroperitoneal lymph node dissection in advanced germ cell tumours of the testis. Eur Urol. 2008;53:260-272.
24. Bajorin DF, Sarosdy MF, Pfister DG, et al. Randomized trial of etoposide and cisplatin versus etoposide and carboplatin in patients with good-risk germ cell tumors: a multiinstitutional study. J Clin Oncol. 1993;11:598-606.
25. Bokemeyer C, Köhrmann O, Tischler J, et al. A randomized trial of cisplatin, etoposide and bleomycin (PEB) versus carboplatin, etoposide and bleomycin (CEB) for patients with “good-risk” metastatic non-seminomatous germ cell tumors. Ann Oncol. 1996;7:1015-1021.
26. Horwich A, Sleijfer DT, Fosså SD, et al. Randomized trial of bleomycin, etoposide, and cisplatin compared with bleomycin, etoposide, and carboplatin in good-prognosis metastatic nonseminomatous germ cell cancer: a Multiinstitutional Medical Research Council/European Organization for Research and Treatment of Cancer Trial. J Clin Oncol. 1997;15:1844-1852.
27. Shaikh F, Nathan PC, Hale J, et al. Is there a role for carboplatin in the treatment of malignant germ cell tumors? A systematic review of adult and pediatric trials. Pediatr Blood Cancer. 2013;60:587-592.
28. Grimison PS, Stockler MR, Thomson DB, et al. Comparison of two standard chemotherapy regimens for good-prognosis germ cell tumors: updated analysis of a randomized trial. J Natl Cancer Inst. 2010;102:1253-1262.
29. Reinert T, da Rocha Baldotto CS, Nunes FAP, de Souza Scheliga AA. Bleomycin-induced lung injury. J Cancer Res. 2013;480608.
30. Jones RH, Vasey PA. Part II: Testicular cancer—management of advanced disease. Lancet Oncol. 2003;4:738-747.
31. Jankilevich G. BEP versus EP for treatment of metastatic germ-cell tumours. Lancet Oncol. 2004;5, 146.
32. Nichols CR, Catalano PJ, Crawford ED, et al. Randomized comparison of cisplatin and etoposide and either bleomycin or ifosfamide in treatment of advanced disseminated germ cell tumors: an Eastern Cooperative Oncology Group, Southwest Oncology Group, and Cancer and Leukemia Group B Study. J Clin Oncol. 1998;16:12871293.
33. Hinton S, Catalano PJ, Einhorn LH, et al. Cisplatin, etoposide and either bleomycin or ifosfamide in the treatment of disseminated germ cell tumors: final analysis of an intergroup trial. Cancer. 2003;97: 1869-1875.
34. de Wit R, Stoter G, Sleijfer DT, et al. Four cycles of BEP vs four cycles of VIP in patients with intermediate-prognosis metastatic testicular non-seminoma: a randomized study of the EORTC Genitourinary Tract Cancer Cooperative Group. European Organization for Research and Treatment of Cancer. Br J Cancer. 1998;78:828-832.
35. Mhaskar R, Clark OA, Lyman G, et al. Colony-stimulating factors for chemotherapy-induced febrile neutropenia. Cochrane Database Syst. Rev. 2014;CD003039.
36. Adra N, Abonour R, Althouse SK, et al. High-dose chemotherapy and autologous peripheral-blood stem-cell transplantation for relapsed metastatic germ cell tumors: The Indiana University experience. J Clin Oncol. 2017;35:1096-1102.
37. Oliver RT, Mason MD, Mead GM, et al. Radiotherapy versus single-dose carboplatin in adjuvant treatment of stage I seminoma: a randomised trial. Lancet. 2005;366:293-300.
38. Oliver RT, Mead GM, Rustin GJ, et al. Randomized trial of carboplatin versus radiotherapy for stage I seminoma: mature results on relapse and contralateral testis cancer rates in MRC TE19/EORTC 30982 study (ISRCTN27163214). J Clin Oncol. 2011;29:957-962.
39. Glaser SM, Vargo JA, Balasubramani GK, Beriwal S. Stage II testicular seminoma: patterns of care and survival by treatment strategy. Clin Oncol. 2016;28:513-521.
40. Boujelbene N, Cosinschi A, Boujelbene N, et al. Pure seminoma: A review and update. Radiat Oncol. 2011;6:90.
41. Nichols CR, Roth B, Albers P, et al. Active surveillance is the preferred approach to clinical stage I testicular cancer. J Clin Oncol. 2013;31;3490-3493.
Advanced Melanoma: Treatment After Progression on First-line Therapy
The past decade has brought rapid advancements in treatment with immune checkpoint inhibitors and molecularly targeted agents, which have significantly improved objective response rates (ORRs), progression-free survival (PFS), and overall survival (OS) for patients with metastatic melanoma. This article reviews current evidence for immune checkpoint blockade and molecularly targeted agents in the treatment of metastatic melanoma after progression on first-line therapy. The selection of first-line therapy for metastatic melanoma is reviewed in a separate article.
Case Presentation
A 62-year-old man was diagnosed with stage IIA melanoma after undergoing wide local excision of a right scalp lesion (final staging was consistent with pT3aN0M0). After 3.5 years of follow-up, he developed symptoms of vertigo, diplopia, and recurrent falls prompting medical attention. Magnetic resonance imaging (MRI) brain revealed multiple supratentorial and infratentorial lesions concerning for intracranial metastases and computed tomography (CT) chest/abdomen/pelvis revealed a right lower lobe pulmonary mass with right hilar and subcarinal lymphadenopathy. He was treated with intravenous dexamethasone and further evaluation with an endobronchial ultrasound-guided fine-needle aspiration of the right lower lobe mass revealed metastatic melanoma. The patient underwent whole brain radiation therapy for symptomatic relief prior to initiating systemic therapy. Testing showed the melanoma was positive for a BRAF V600K mutation. He was started on combination molecularly targeted therapy with dabrafenib and trametinib. He initially did well, with a partial response noted by resolution of symptoms and decreased size of his intracranial metastases and decreased size of the right lower lobe mass.
After 3 months of therapy, surveillance PET-CT notes increasing size and FDG avidity of the right lower lobe mass. MRI brain reveals resolution of several previously noted metastases, but with interval development of a new left frontal lobe mass concerning for progressive disease.
What is the general approach to treatment of metastatic melanoma after progression on first-line therapy?
Based on the current evidence, there is no definitive algorithm for the treatment of metastatic melanoma after progression on first-line therapy. Enrollment in clinical trials is encouraged to further elucidate the best sequencing of treatment. The current practice is to typically switch class of agents after progression on front-line therapy to either immunotherapy that has not yet been tried or to molecularly targeted therapy in patients harboring a BRAF V600 mutation. After further progression of disease, retreatment with a previously received agent is possible, and this may be combined with investigational therapies.
Immune Checkpoint Inhibitors in Progressive Disease
The 2 major populations of patients to consider are those with BRAF wild-type melanomas who progress on first-line immunotherapy and those with BRAF V600 mutation–positive melanoma who progress on molecularly targeted therapy with BRAF and MEK inhibitors. There is relatively limited data on the efficacy of immune checkpoint inhibition after progression on anti-programmed cell death 1 (PD-1) monotherapy. A small retrospective study of patients who progressed on anti-PD-1 monotherapy were treated with ipilimumab, with a 10% ORR and another 8% having stable disease for more than 6 months; however, 35% of patients experienced grade 3 to 5 immune-related adverse events.1 The only prospective data supports the efficacy of anti-PD-1 therapy after progression on ipilimumab, as supported by the CheckMate 037 trial (nivolumab versus chemotherapy)2 and KEYNOTE-002 trial (pembrolizumab versus chemotherapy)3,4; however, this is no longer applicable as ipilimumab is no longer given in the first-line setting and has been replaced by anti-PD-1 monotherapy or combination immunotherapy.
Another interesting facet of PD-1 monotherapy is the idea of treatment beyond progression. The concept of pseudoprogression—whereby patients receiving PD-1 inhibitors initially meet Response Evaluation Criteria in Solid Tumors (RECIST) criteria for progression, but then later go on to demonstrate significant decreases in tumor burden on subsequent imaging studies—has been described in melanoma patients receiving such immunotherapies. It is thought that pseudoprogression occurs due to either an initial delay in anti-tumor response to the immunotherapy or from the measured target lesion appearing larger due to surrounding immune/inflammatory infiltrate. In an analysis of individual patient data pooled from 8 multicenter clinical trials, 19% of patients were treated beyond initially documented RECIST progression and had subsequent imaging to evaluate the tumor burden; in these patients, the same target lesion later met RECIST criteria for response, with a greater than 30% reduction in tumor size. Furthermore, of the evaluable cohort, the median OS in patients who did receive treatment beyond progression was 24.4 months compared to 11.2 months in those who did not receive treatment beyond progression.5 While further randomized studies are warranted to characterize the potential benefit, the existing data suggests that selected patients who are doing well clinically despite evidence of radiographic progressive disease may benefit from continued treatment with PD-1 inhibitors.
Combination immunotherapy with both PD-1 and CTLA-4 blockade has been studied retrospectively in the second-line setting. A retrospective analysis of patients who had progressive disease on PD-1 inhibitor monotherapy compared the outcomes of patients who received just ipilimumab to those of patients who received both ipilimumab and nivolumab. The ORR (16% ipilimumab vs 21% combination group) and 1-year OS (54% vs 55%) were similar in both groups,6 and this demonstrated significantly less efficacy with combination therapy when compared to use in the first-line setting, albeit in a separate prospective trial.7 A multicenter, retrospective study by Tétu and colleagues compared outcomes with ipilimumab plus nivolumab across 3 groups that included previously untreated patients, patients who had progressed on single-agent immunotherapy, and patients who had progressed on prior molecularly targeted therapy.8 Despite clearly inferior efficacy in previously treated patients, the results support combination immunotherapy as a viable treatment option in the second-line setting. Outcomes are reported in Table 1 below. Of note, there is an ongoing phase 2 trial to assess the use of combined PD-1 and CTLA-4 inhibitors versus CTLA-4 inhibition alone after progression on first-line PD-1 inhibitor monotherapy (NCT03033576).

For patients with BRAF V600–mutation positive melanoma who progress on front-line molecularly targeted therapy, immune checkpoint inhibitor therapy with either anti-PD-1 monotherapy or combination anti-PD-1 and ipilimumab should be considered. The KEYNOTE-006 trial that demonstrated superiority of pembrolizumab compared to ipilimumab included patients who had received up to 1 prior systemic therapy that was not a PD-1 or CTLA-4 inhibitor, and subgroup analysis demonstrated efficacy with pembrolizumab in patients who had received prior treatment with a BRAF inhibitor.9 The retrospective analysis by Tétu et al (Table 1) noted efficacy of combination nivolumab and ipilimumab in patients treated with prior molecularly targeted therapy, as evidenced by an ORR of 35% and median OS of 16.5 months.8
A retrospective trial by Ackerman et al analyzed ORR, median PFS, and median OS from the time of commencement of BRAF inhibitor therapy (with or without a MEK inhibitor), and the comparison was made between those who received ipilimumab before or after molecularly targeted therapy. While ipilimumab is no longer the first-line immunotherapy agent used in advanced melanoma, the study did highlight some important concepts. First, ORRs to BRAF inhibitors were similar between the 2 treatment groups. The conclusions of the analysis were that there was no significant difference in median PFS or OS in regard to which therapy was given first, but median OS after BRAF inhibitors were discontinued was very short and patients had poor responses to ipilimumab after stopping a BRAF inhibitor. This highlights the concern that patients who have progressive disease on molecularly targeted therapy often have a poor performance status and undergo too rapid of a clinical decline to derive benefit from immunotherapy, which can often take weeks to months to take effect.10
A more recent retrospective study by Johnson et al compared efficacy outcomes in patients who received single-agent anti-PD-1 therapy prior to molecularly targeted therapy (BRAF inhibitor with or without MEK inhibitor) to those who received molecularly targeted therapy prior to anti-PD-1 therapy. The difference in median OS was not statistically significant (27.5 months with PD-1 inhibitor first vs 40.3 months with molecularly targeted therapy first). Both treatments demonstrated second-line efficacy, but outcomes were inferior to those reported when either type of therapy was used in the first-line setting. Interestingly, patients who were maintained on molecularly targeted therapy for more than 6 months prior to progression demonstrated an improved ORR to subsequent anti-PD-1 therapy (34% vs 15%).11
Molecularly Targeted Therapy in Progressive Disease
When melanoma patients with a BRAF V600 mutation are treated initially with immunotherapy and demonstrate progressive disease, molecularly targeted therapy with combined BRAF and MEK inhibition should be considered for second-line therapy. While there are no dedicated prospective trial results with BRAF/MEK inhibitors after progression on immune checkpoint inhibitors, for practical purposes, it may be reasonable to extrapolate outcomes from the currently available first-line studies.12-16 An ongoing study (NCT02224781) in which patients are randomized to receive ipilimumab/nivolumab followed by dabrafenib/trametinib at progression versus the reverse order is designed to help answer the question of optimal sequencing and timing of therapy. Johnson et al’s retrospective analysis of patients receiving single-agent anti-PD-1 therapy prior to molecularly targeted therapy compared to the reverse order concluded that there was no statistically significant difference in median OS.11 Ackerman et al’s retrospective study of patients who had received ipilimumab before or after molecularly targeted therapy noted similar response rates to molecularly targeted therapy in each treatment group.10
The issue of re-treatment with a BRAF/MEK inhibitor in a patient already progressing on targeted therapy is a more challenging situation, and currently available data suggests there is limited benefit. However, select patients may be considered for this approach. The combination of dabrafenib/trametinib demonstrated an ORR of approximately 15% in a cohort of patients who progressed on single-agent BRAF inhibitor therapy, with a suggestion that those patients who had previously derived benefit for more than 6 months may have a more favorable outcome.17
Based on the hypothesis that acquired resistance to BRAF/MEK inhibition may be reversible if the selective pressure of the medication is held for a period of time, a phase 2 trial analyzed outcomes with retreatment. The study included patients with BRAF V600–mutant melanoma who had progressed on prior BRAF inhibition (with or without MEK inhibitor) and required that they had been off of therapy for at least 12 weeks. Of the 25 patients who received dabrafenib plus trametinib as retreatment, 32% demonstrated a partial response and 40% had stable disease.18 While further studies are warranted, retreatment with molecularly targeted therapy may be a viable option, especially in light of the multiple approved BRAF and MEK inhibitor combinations.
Another concept that has been studied is treatment beyond disease progression with molecularly targeted therapy. In a retrospective analysis of patients who had progressed on a single-agent BRAF inhibitor, 39% of those patients were continued on the same BRAF inhibitor and compared to patients who received no subsequent therapy or changed to an alternative systemic therapy. In the multivariable analysis adjusting for other prognostic factors, continued treatment with the BRAF inhibitor was associated with prolonged OS.19
Case Conclusion
The patient is started on second-line therapy with nivolumab and ipilimumab and demonstrates a partial response. One year later he continues to feel well with decreased size of the intracranial and right lower lobe lesions, and without any interval development of new areas of metastatic disease.
Special Considerations
Intralesional Therapies
Talimogene laherparepvec (T-VEC) is a genetically modified herpesvirus-1 oncolytic virus that is injected into melanoma skin lesions and leads to the expression of granulocyte-macrophage colony-stimulating factor. While T-VEC is currently approved for local treatment of unresectable cutaneous, subcutaneous, or nodal recurrences,20 it has also been investigated in combination with other therapies for patients with advanced disease. In patients with previously treated melanoma, T-VEC plus ipilimumab demonstrated superior ORR to ipilimumab alone (39% vs 18%), and the tumor response was not limited to the injected lesions. The observation of systemic response suggests synergy between T-VEC and immune checkpoint blockade in enhancing the anti-tumor immune response.21 The phase 1b MASTERKEY-265 trial combining pembrolizumab and T-VEC led to an ORR of 62% and CR of 33%.22 A phase 3 trial comparing pembrolizumab plus T-VEC to pembrolizumab alone is ongoing (NCT02263508).
Melanoma Brain Metastases
The presence of brain metastases is a common event in patients with metastatic melanoma, and often confers a poor prognosis.23 The approach to the management of brain metastases should be multidisciplinary among medical oncology, neurosurgery, and radiation oncology providers, as treatment algorithms continue to rapidly evolve. Historically, there has been little prospective clinical trial data regarding optimal systemic therapy, and local therapies such as surgery or stereotactic radiation have long been the mainstay of therapy for intracranial disease.24 However, recent data with both immunotherapy and molecularly targeted therapy has demonstrated efficacy with intracranial metastases.
A recent trial of combined nivolumab and ipilimumab as frontline therapy in patients with asymptomatic melanoma brain metastases demonstrated a complete response rate of 26% and partial response rate of 30% in patients with a median follow-up of 14 months.25 In a separate study, ipilimumab plus nivolumab demonstrated better intracranial ORR when compared to nivolumab alone in asymptomatic, previously untreated patients. Outcomes were better in patients presenting with asymptomatic versus symptomatic brain metastases.26 Collectively, these results suggest that systemic immunotherapy alone may be adequate for patients with asymptomatic, previously untreated brain metastases.
For molecularly targeted therapy in patients with BRAF mutations and brain metastases, the BREAK-MB trial demonstrated that an intracranial response was attainable with dabrafenib regardless of whether the patient had previously received local therapy in the form of surgery or radiation.27 The COMBI-MB trial enhanced the preexisting data by testing the intracranial efficacy of dabrafenib plus trametinib in 4 different cohorts of patients, further supporting that systemic molecularly targeted therapy can provide significant intracranial activity in patients with both symptomatic and asymptomatic brain lesions and regardless of prior local therapy (Table 2).28

Conclusion
The treatment of advanced melanoma has been drastically improved over the past decade by the development and study of immune checkpoint inhibitors and molecularly targeted agents. There is still much to learn regarding the optimal combination and sequencing of therapies. Many of these trials are ongoing and will provide additional evidence to guide treatment decisions moving forward.
1. Bowyer S, Prithviraj P, Lorigan P, et al. Efficacy and toxicity of treatment with the anti-CTLA-4 antibody ipilimumab in patients with metastatic melanoma after prior anti-PD-1 therapy. Br J Cancer. 2016;114:1084-1089.
2. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-384.
3. Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908-918.
4. Hamid O, Puzanov I, Dummer R, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer. 2017;86:37-45.
5. Beaver JA, Hazarika M, Mulkey F, et al. Patients with melanoma treated with an anti-PD-1 antibody beyond RECIST progression: a US Food and Drug Administration pooled analysis. Lancet Oncol. 2018;19:229-239.
6. Zimmer L, Apuri S, Eroglu Z, et al. Ipilimumab alone or in combination with nivolumab after progression on anti-PD-1 therapy in advanced melanoma. Eur J Cancer. 2017;75:47-55.
7. Larkin J, Chiarion-Sileni V, Gonazalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34.
8. Tétu P, Mangana J, Dummer R, et al. Benefit of the nivolumab and ipilimumab combination in pretreated advanced melanoma. Eur J Cancer. 2018;93:147-149.
9. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2522-2532.
10. Ackerman A, Klein O, McDermott D, et al. Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer. 2014;120:1695-1701.
11. Johnson DB, Pectasides E, Feld E, et al. Sequencing treatment in BRAFV600 mutant melanoma: anti-pd-1 before and after BRAF inhibition. J Immunother. 2017;40:31-35.
12. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicenter, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2015;386:444-451.
13. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28:1631-1639.
14. Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248-1260.
15. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicenter, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:603-615.
16. Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19:1315-1327.
17. Johnson DB, Flaherty KT, Weber, JS et al. Combined BRAF (Dabrafenib) and MEK inhibition (Trametinib) in patients with BRAFV600-mutant melanoma experiencing progression with single-agent BRAF inhibitor. J Clin Oncol. 2014;32:3697-3704.
18. Schreuer M, Jansen Y, Planken S, et al. Combination of dabrafenib plus trametinib for BRAF and MEK inhibitor pretreated patients with advanced BRAFV600-mutant melanoma: an open-label, single arm, dual-centre, phase 2 clinical trial. Lancet Oncol. 2017;18:464-472.
19. Chan MM, Haydu LE, Azer MW, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer. 2014;120:3142-3153.
20. Imlygic (talimogene laherparepvec) suspension for intralesional injection [package insert]. Thousand Oaks, CA: BioVex; 2015.
21. Chesney J, Puzanov I, Collichio F, et al. Randomized, open-label phase ii study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. 2018;36:1658-1667.
22. Ribas A, Dummer R, Puzanov I, et al. Oncolytic virotherapy promotes intratumoral t cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2018;174:1031-1032.
23. Sampson JH, Carter Jr. JH, Friedman AH, Seigler HF. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg. 1998;88:11-20.
24. Yamamoto M, Serizawa T, Shuto T, et al. Stereotactic radiosurgery for patients with multiple brain metastases (JLGK0901): a multi-institutional prospective observational study. Lancet Oncol. 2014;15:387-395.
25. Tawbi HA, Forsyth PA, Hamid O, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med. 2018;379:722-730.
26. Long GV, Atkinson V, La S, et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicenter randomised phase 2 study. Lancet Oncol. 2018;19:672-681.
27. Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicenter, open-label, phase 2 trial. Lancet Oncol. 2012;13:1087-1095.
28. Davies MA, Saiag P, Robert C, et al. Dabrafenib plus trametinib in patients with BRAFV600-mutant melanoma brain metastases (COMBI-MB): a multicenter, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017;18:863-873.
The past decade has brought rapid advancements in treatment with immune checkpoint inhibitors and molecularly targeted agents, which have significantly improved objective response rates (ORRs), progression-free survival (PFS), and overall survival (OS) for patients with metastatic melanoma. This article reviews current evidence for immune checkpoint blockade and molecularly targeted agents in the treatment of metastatic melanoma after progression on first-line therapy. The selection of first-line therapy for metastatic melanoma is reviewed in a separate article.
Case Presentation
A 62-year-old man was diagnosed with stage IIA melanoma after undergoing wide local excision of a right scalp lesion (final staging was consistent with pT3aN0M0). After 3.5 years of follow-up, he developed symptoms of vertigo, diplopia, and recurrent falls prompting medical attention. Magnetic resonance imaging (MRI) brain revealed multiple supratentorial and infratentorial lesions concerning for intracranial metastases and computed tomography (CT) chest/abdomen/pelvis revealed a right lower lobe pulmonary mass with right hilar and subcarinal lymphadenopathy. He was treated with intravenous dexamethasone and further evaluation with an endobronchial ultrasound-guided fine-needle aspiration of the right lower lobe mass revealed metastatic melanoma. The patient underwent whole brain radiation therapy for symptomatic relief prior to initiating systemic therapy. Testing showed the melanoma was positive for a BRAF V600K mutation. He was started on combination molecularly targeted therapy with dabrafenib and trametinib. He initially did well, with a partial response noted by resolution of symptoms and decreased size of his intracranial metastases and decreased size of the right lower lobe mass.
After 3 months of therapy, surveillance PET-CT notes increasing size and FDG avidity of the right lower lobe mass. MRI brain reveals resolution of several previously noted metastases, but with interval development of a new left frontal lobe mass concerning for progressive disease.
What is the general approach to treatment of metastatic melanoma after progression on first-line therapy?
Based on the current evidence, there is no definitive algorithm for the treatment of metastatic melanoma after progression on first-line therapy. Enrollment in clinical trials is encouraged to further elucidate the best sequencing of treatment. The current practice is to typically switch class of agents after progression on front-line therapy to either immunotherapy that has not yet been tried or to molecularly targeted therapy in patients harboring a BRAF V600 mutation. After further progression of disease, retreatment with a previously received agent is possible, and this may be combined with investigational therapies.
Immune Checkpoint Inhibitors in Progressive Disease
The 2 major populations of patients to consider are those with BRAF wild-type melanomas who progress on first-line immunotherapy and those with BRAF V600 mutation–positive melanoma who progress on molecularly targeted therapy with BRAF and MEK inhibitors. There is relatively limited data on the efficacy of immune checkpoint inhibition after progression on anti-programmed cell death 1 (PD-1) monotherapy. A small retrospective study of patients who progressed on anti-PD-1 monotherapy were treated with ipilimumab, with a 10% ORR and another 8% having stable disease for more than 6 months; however, 35% of patients experienced grade 3 to 5 immune-related adverse events.1 The only prospective data supports the efficacy of anti-PD-1 therapy after progression on ipilimumab, as supported by the CheckMate 037 trial (nivolumab versus chemotherapy)2 and KEYNOTE-002 trial (pembrolizumab versus chemotherapy)3,4; however, this is no longer applicable as ipilimumab is no longer given in the first-line setting and has been replaced by anti-PD-1 monotherapy or combination immunotherapy.
Another interesting facet of PD-1 monotherapy is the idea of treatment beyond progression. The concept of pseudoprogression—whereby patients receiving PD-1 inhibitors initially meet Response Evaluation Criteria in Solid Tumors (RECIST) criteria for progression, but then later go on to demonstrate significant decreases in tumor burden on subsequent imaging studies—has been described in melanoma patients receiving such immunotherapies. It is thought that pseudoprogression occurs due to either an initial delay in anti-tumor response to the immunotherapy or from the measured target lesion appearing larger due to surrounding immune/inflammatory infiltrate. In an analysis of individual patient data pooled from 8 multicenter clinical trials, 19% of patients were treated beyond initially documented RECIST progression and had subsequent imaging to evaluate the tumor burden; in these patients, the same target lesion later met RECIST criteria for response, with a greater than 30% reduction in tumor size. Furthermore, of the evaluable cohort, the median OS in patients who did receive treatment beyond progression was 24.4 months compared to 11.2 months in those who did not receive treatment beyond progression.5 While further randomized studies are warranted to characterize the potential benefit, the existing data suggests that selected patients who are doing well clinically despite evidence of radiographic progressive disease may benefit from continued treatment with PD-1 inhibitors.
Combination immunotherapy with both PD-1 and CTLA-4 blockade has been studied retrospectively in the second-line setting. A retrospective analysis of patients who had progressive disease on PD-1 inhibitor monotherapy compared the outcomes of patients who received just ipilimumab to those of patients who received both ipilimumab and nivolumab. The ORR (16% ipilimumab vs 21% combination group) and 1-year OS (54% vs 55%) were similar in both groups,6 and this demonstrated significantly less efficacy with combination therapy when compared to use in the first-line setting, albeit in a separate prospective trial.7 A multicenter, retrospective study by Tétu and colleagues compared outcomes with ipilimumab plus nivolumab across 3 groups that included previously untreated patients, patients who had progressed on single-agent immunotherapy, and patients who had progressed on prior molecularly targeted therapy.8 Despite clearly inferior efficacy in previously treated patients, the results support combination immunotherapy as a viable treatment option in the second-line setting. Outcomes are reported in Table 1 below. Of note, there is an ongoing phase 2 trial to assess the use of combined PD-1 and CTLA-4 inhibitors versus CTLA-4 inhibition alone after progression on first-line PD-1 inhibitor monotherapy (NCT03033576).
For patients with BRAF V600–mutation positive melanoma who progress on front-line molecularly targeted therapy, immune checkpoint inhibitor therapy with either anti-PD-1 monotherapy or combination anti-PD-1 and ipilimumab should be considered. The KEYNOTE-006 trial that demonstrated superiority of pembrolizumab compared to ipilimumab included patients who had received up to 1 prior systemic therapy that was not a PD-1 or CTLA-4 inhibitor, and subgroup analysis demonstrated efficacy with pembrolizumab in patients who had received prior treatment with a BRAF inhibitor.9 The retrospective analysis by Tétu et al (Table 1) noted efficacy of combination nivolumab and ipilimumab in patients treated with prior molecularly targeted therapy, as evidenced by an ORR of 35% and median OS of 16.5 months.8
A retrospective trial by Ackerman et al analyzed ORR, median PFS, and median OS from the time of commencement of BRAF inhibitor therapy (with or without a MEK inhibitor), and the comparison was made between those who received ipilimumab before or after molecularly targeted therapy. While ipilimumab is no longer the first-line immunotherapy agent used in advanced melanoma, the study did highlight some important concepts. First, ORRs to BRAF inhibitors were similar between the 2 treatment groups. The conclusions of the analysis were that there was no significant difference in median PFS or OS in regard to which therapy was given first, but median OS after BRAF inhibitors were discontinued was very short and patients had poor responses to ipilimumab after stopping a BRAF inhibitor. This highlights the concern that patients who have progressive disease on molecularly targeted therapy often have a poor performance status and undergo too rapid of a clinical decline to derive benefit from immunotherapy, which can often take weeks to months to take effect.10
A more recent retrospective study by Johnson et al compared efficacy outcomes in patients who received single-agent anti-PD-1 therapy prior to molecularly targeted therapy (BRAF inhibitor with or without MEK inhibitor) to those who received molecularly targeted therapy prior to anti-PD-1 therapy. The difference in median OS was not statistically significant (27.5 months with PD-1 inhibitor first vs 40.3 months with molecularly targeted therapy first). Both treatments demonstrated second-line efficacy, but outcomes were inferior to those reported when either type of therapy was used in the first-line setting. Interestingly, patients who were maintained on molecularly targeted therapy for more than 6 months prior to progression demonstrated an improved ORR to subsequent anti-PD-1 therapy (34% vs 15%).11
Molecularly Targeted Therapy in Progressive Disease
When melanoma patients with a BRAF V600 mutation are treated initially with immunotherapy and demonstrate progressive disease, molecularly targeted therapy with combined BRAF and MEK inhibition should be considered for second-line therapy. While there are no dedicated prospective trial results with BRAF/MEK inhibitors after progression on immune checkpoint inhibitors, for practical purposes, it may be reasonable to extrapolate outcomes from the currently available first-line studies.12-16 An ongoing study (NCT02224781) in which patients are randomized to receive ipilimumab/nivolumab followed by dabrafenib/trametinib at progression versus the reverse order is designed to help answer the question of optimal sequencing and timing of therapy. Johnson et al’s retrospective analysis of patients receiving single-agent anti-PD-1 therapy prior to molecularly targeted therapy compared to the reverse order concluded that there was no statistically significant difference in median OS.11 Ackerman et al’s retrospective study of patients who had received ipilimumab before or after molecularly targeted therapy noted similar response rates to molecularly targeted therapy in each treatment group.10
The issue of re-treatment with a BRAF/MEK inhibitor in a patient already progressing on targeted therapy is a more challenging situation, and currently available data suggests there is limited benefit. However, select patients may be considered for this approach. The combination of dabrafenib/trametinib demonstrated an ORR of approximately 15% in a cohort of patients who progressed on single-agent BRAF inhibitor therapy, with a suggestion that those patients who had previously derived benefit for more than 6 months may have a more favorable outcome.17
Based on the hypothesis that acquired resistance to BRAF/MEK inhibition may be reversible if the selective pressure of the medication is held for a period of time, a phase 2 trial analyzed outcomes with retreatment. The study included patients with BRAF V600–mutant melanoma who had progressed on prior BRAF inhibition (with or without MEK inhibitor) and required that they had been off of therapy for at least 12 weeks. Of the 25 patients who received dabrafenib plus trametinib as retreatment, 32% demonstrated a partial response and 40% had stable disease.18 While further studies are warranted, retreatment with molecularly targeted therapy may be a viable option, especially in light of the multiple approved BRAF and MEK inhibitor combinations.
Another concept that has been studied is treatment beyond disease progression with molecularly targeted therapy. In a retrospective analysis of patients who had progressed on a single-agent BRAF inhibitor, 39% of those patients were continued on the same BRAF inhibitor and compared to patients who received no subsequent therapy or changed to an alternative systemic therapy. In the multivariable analysis adjusting for other prognostic factors, continued treatment with the BRAF inhibitor was associated with prolonged OS.19
Case Conclusion
The patient is started on second-line therapy with nivolumab and ipilimumab and demonstrates a partial response. One year later he continues to feel well with decreased size of the intracranial and right lower lobe lesions, and without any interval development of new areas of metastatic disease.
Special Considerations
Intralesional Therapies
Talimogene laherparepvec (T-VEC) is a genetically modified herpesvirus-1 oncolytic virus that is injected into melanoma skin lesions and leads to the expression of granulocyte-macrophage colony-stimulating factor. While T-VEC is currently approved for local treatment of unresectable cutaneous, subcutaneous, or nodal recurrences,20 it has also been investigated in combination with other therapies for patients with advanced disease. In patients with previously treated melanoma, T-VEC plus ipilimumab demonstrated superior ORR to ipilimumab alone (39% vs 18%), and the tumor response was not limited to the injected lesions. The observation of systemic response suggests synergy between T-VEC and immune checkpoint blockade in enhancing the anti-tumor immune response.21 The phase 1b MASTERKEY-265 trial combining pembrolizumab and T-VEC led to an ORR of 62% and CR of 33%.22 A phase 3 trial comparing pembrolizumab plus T-VEC to pembrolizumab alone is ongoing (NCT02263508).
Melanoma Brain Metastases
The presence of brain metastases is a common event in patients with metastatic melanoma, and often confers a poor prognosis.23 The approach to the management of brain metastases should be multidisciplinary among medical oncology, neurosurgery, and radiation oncology providers, as treatment algorithms continue to rapidly evolve. Historically, there has been little prospective clinical trial data regarding optimal systemic therapy, and local therapies such as surgery or stereotactic radiation have long been the mainstay of therapy for intracranial disease.24 However, recent data with both immunotherapy and molecularly targeted therapy has demonstrated efficacy with intracranial metastases.
A recent trial of combined nivolumab and ipilimumab as frontline therapy in patients with asymptomatic melanoma brain metastases demonstrated a complete response rate of 26% and partial response rate of 30% in patients with a median follow-up of 14 months.25 In a separate study, ipilimumab plus nivolumab demonstrated better intracranial ORR when compared to nivolumab alone in asymptomatic, previously untreated patients. Outcomes were better in patients presenting with asymptomatic versus symptomatic brain metastases.26 Collectively, these results suggest that systemic immunotherapy alone may be adequate for patients with asymptomatic, previously untreated brain metastases.
For molecularly targeted therapy in patients with BRAF mutations and brain metastases, the BREAK-MB trial demonstrated that an intracranial response was attainable with dabrafenib regardless of whether the patient had previously received local therapy in the form of surgery or radiation.27 The COMBI-MB trial enhanced the preexisting data by testing the intracranial efficacy of dabrafenib plus trametinib in 4 different cohorts of patients, further supporting that systemic molecularly targeted therapy can provide significant intracranial activity in patients with both symptomatic and asymptomatic brain lesions and regardless of prior local therapy (Table 2).28
Conclusion
The treatment of advanced melanoma has been drastically improved over the past decade by the development and study of immune checkpoint inhibitors and molecularly targeted agents. There is still much to learn regarding the optimal combination and sequencing of therapies. Many of these trials are ongoing and will provide additional evidence to guide treatment decisions moving forward.
The past decade has brought rapid advancements in treatment with immune checkpoint inhibitors and molecularly targeted agents, which have significantly improved objective response rates (ORRs), progression-free survival (PFS), and overall survival (OS) for patients with metastatic melanoma. This article reviews current evidence for immune checkpoint blockade and molecularly targeted agents in the treatment of metastatic melanoma after progression on first-line therapy. The selection of first-line therapy for metastatic melanoma is reviewed in a separate article.
Case Presentation
A 62-year-old man was diagnosed with stage IIA melanoma after undergoing wide local excision of a right scalp lesion (final staging was consistent with pT3aN0M0). After 3.5 years of follow-up, he developed symptoms of vertigo, diplopia, and recurrent falls prompting medical attention. Magnetic resonance imaging (MRI) brain revealed multiple supratentorial and infratentorial lesions concerning for intracranial metastases and computed tomography (CT) chest/abdomen/pelvis revealed a right lower lobe pulmonary mass with right hilar and subcarinal lymphadenopathy. He was treated with intravenous dexamethasone and further evaluation with an endobronchial ultrasound-guided fine-needle aspiration of the right lower lobe mass revealed metastatic melanoma. The patient underwent whole brain radiation therapy for symptomatic relief prior to initiating systemic therapy. Testing showed the melanoma was positive for a BRAF V600K mutation. He was started on combination molecularly targeted therapy with dabrafenib and trametinib. He initially did well, with a partial response noted by resolution of symptoms and decreased size of his intracranial metastases and decreased size of the right lower lobe mass.
After 3 months of therapy, surveillance PET-CT notes increasing size and FDG avidity of the right lower lobe mass. MRI brain reveals resolution of several previously noted metastases, but with interval development of a new left frontal lobe mass concerning for progressive disease.
What is the general approach to treatment of metastatic melanoma after progression on first-line therapy?
Based on the current evidence, there is no definitive algorithm for the treatment of metastatic melanoma after progression on first-line therapy. Enrollment in clinical trials is encouraged to further elucidate the best sequencing of treatment. The current practice is to typically switch class of agents after progression on front-line therapy to either immunotherapy that has not yet been tried or to molecularly targeted therapy in patients harboring a BRAF V600 mutation. After further progression of disease, retreatment with a previously received agent is possible, and this may be combined with investigational therapies.
Immune Checkpoint Inhibitors in Progressive Disease
The 2 major populations of patients to consider are those with BRAF wild-type melanomas who progress on first-line immunotherapy and those with BRAF V600 mutation–positive melanoma who progress on molecularly targeted therapy with BRAF and MEK inhibitors. There is relatively limited data on the efficacy of immune checkpoint inhibition after progression on anti-programmed cell death 1 (PD-1) monotherapy. A small retrospective study of patients who progressed on anti-PD-1 monotherapy were treated with ipilimumab, with a 10% ORR and another 8% having stable disease for more than 6 months; however, 35% of patients experienced grade 3 to 5 immune-related adverse events.1 The only prospective data supports the efficacy of anti-PD-1 therapy after progression on ipilimumab, as supported by the CheckMate 037 trial (nivolumab versus chemotherapy)2 and KEYNOTE-002 trial (pembrolizumab versus chemotherapy)3,4; however, this is no longer applicable as ipilimumab is no longer given in the first-line setting and has been replaced by anti-PD-1 monotherapy or combination immunotherapy.
Another interesting facet of PD-1 monotherapy is the idea of treatment beyond progression. The concept of pseudoprogression—whereby patients receiving PD-1 inhibitors initially meet Response Evaluation Criteria in Solid Tumors (RECIST) criteria for progression, but then later go on to demonstrate significant decreases in tumor burden on subsequent imaging studies—has been described in melanoma patients receiving such immunotherapies. It is thought that pseudoprogression occurs due to either an initial delay in anti-tumor response to the immunotherapy or from the measured target lesion appearing larger due to surrounding immune/inflammatory infiltrate. In an analysis of individual patient data pooled from 8 multicenter clinical trials, 19% of patients were treated beyond initially documented RECIST progression and had subsequent imaging to evaluate the tumor burden; in these patients, the same target lesion later met RECIST criteria for response, with a greater than 30% reduction in tumor size. Furthermore, of the evaluable cohort, the median OS in patients who did receive treatment beyond progression was 24.4 months compared to 11.2 months in those who did not receive treatment beyond progression.5 While further randomized studies are warranted to characterize the potential benefit, the existing data suggests that selected patients who are doing well clinically despite evidence of radiographic progressive disease may benefit from continued treatment with PD-1 inhibitors.
Combination immunotherapy with both PD-1 and CTLA-4 blockade has been studied retrospectively in the second-line setting. A retrospective analysis of patients who had progressive disease on PD-1 inhibitor monotherapy compared the outcomes of patients who received just ipilimumab to those of patients who received both ipilimumab and nivolumab. The ORR (16% ipilimumab vs 21% combination group) and 1-year OS (54% vs 55%) were similar in both groups,6 and this demonstrated significantly less efficacy with combination therapy when compared to use in the first-line setting, albeit in a separate prospective trial.7 A multicenter, retrospective study by Tétu and colleagues compared outcomes with ipilimumab plus nivolumab across 3 groups that included previously untreated patients, patients who had progressed on single-agent immunotherapy, and patients who had progressed on prior molecularly targeted therapy.8 Despite clearly inferior efficacy in previously treated patients, the results support combination immunotherapy as a viable treatment option in the second-line setting. Outcomes are reported in Table 1 below. Of note, there is an ongoing phase 2 trial to assess the use of combined PD-1 and CTLA-4 inhibitors versus CTLA-4 inhibition alone after progression on first-line PD-1 inhibitor monotherapy (NCT03033576).
For patients with BRAF V600–mutation positive melanoma who progress on front-line molecularly targeted therapy, immune checkpoint inhibitor therapy with either anti-PD-1 monotherapy or combination anti-PD-1 and ipilimumab should be considered. The KEYNOTE-006 trial that demonstrated superiority of pembrolizumab compared to ipilimumab included patients who had received up to 1 prior systemic therapy that was not a PD-1 or CTLA-4 inhibitor, and subgroup analysis demonstrated efficacy with pembrolizumab in patients who had received prior treatment with a BRAF inhibitor.9 The retrospective analysis by Tétu et al (Table 1) noted efficacy of combination nivolumab and ipilimumab in patients treated with prior molecularly targeted therapy, as evidenced by an ORR of 35% and median OS of 16.5 months.8
A retrospective trial by Ackerman et al analyzed ORR, median PFS, and median OS from the time of commencement of BRAF inhibitor therapy (with or without a MEK inhibitor), and the comparison was made between those who received ipilimumab before or after molecularly targeted therapy. While ipilimumab is no longer the first-line immunotherapy agent used in advanced melanoma, the study did highlight some important concepts. First, ORRs to BRAF inhibitors were similar between the 2 treatment groups. The conclusions of the analysis were that there was no significant difference in median PFS or OS in regard to which therapy was given first, but median OS after BRAF inhibitors were discontinued was very short and patients had poor responses to ipilimumab after stopping a BRAF inhibitor. This highlights the concern that patients who have progressive disease on molecularly targeted therapy often have a poor performance status and undergo too rapid of a clinical decline to derive benefit from immunotherapy, which can often take weeks to months to take effect.10
A more recent retrospective study by Johnson et al compared efficacy outcomes in patients who received single-agent anti-PD-1 therapy prior to molecularly targeted therapy (BRAF inhibitor with or without MEK inhibitor) to those who received molecularly targeted therapy prior to anti-PD-1 therapy. The difference in median OS was not statistically significant (27.5 months with PD-1 inhibitor first vs 40.3 months with molecularly targeted therapy first). Both treatments demonstrated second-line efficacy, but outcomes were inferior to those reported when either type of therapy was used in the first-line setting. Interestingly, patients who were maintained on molecularly targeted therapy for more than 6 months prior to progression demonstrated an improved ORR to subsequent anti-PD-1 therapy (34% vs 15%).11
Molecularly Targeted Therapy in Progressive Disease
When melanoma patients with a BRAF V600 mutation are treated initially with immunotherapy and demonstrate progressive disease, molecularly targeted therapy with combined BRAF and MEK inhibition should be considered for second-line therapy. While there are no dedicated prospective trial results with BRAF/MEK inhibitors after progression on immune checkpoint inhibitors, for practical purposes, it may be reasonable to extrapolate outcomes from the currently available first-line studies.12-16 An ongoing study (NCT02224781) in which patients are randomized to receive ipilimumab/nivolumab followed by dabrafenib/trametinib at progression versus the reverse order is designed to help answer the question of optimal sequencing and timing of therapy. Johnson et al’s retrospective analysis of patients receiving single-agent anti-PD-1 therapy prior to molecularly targeted therapy compared to the reverse order concluded that there was no statistically significant difference in median OS.11 Ackerman et al’s retrospective study of patients who had received ipilimumab before or after molecularly targeted therapy noted similar response rates to molecularly targeted therapy in each treatment group.10
The issue of re-treatment with a BRAF/MEK inhibitor in a patient already progressing on targeted therapy is a more challenging situation, and currently available data suggests there is limited benefit. However, select patients may be considered for this approach. The combination of dabrafenib/trametinib demonstrated an ORR of approximately 15% in a cohort of patients who progressed on single-agent BRAF inhibitor therapy, with a suggestion that those patients who had previously derived benefit for more than 6 months may have a more favorable outcome.17
Based on the hypothesis that acquired resistance to BRAF/MEK inhibition may be reversible if the selective pressure of the medication is held for a period of time, a phase 2 trial analyzed outcomes with retreatment. The study included patients with BRAF V600–mutant melanoma who had progressed on prior BRAF inhibition (with or without MEK inhibitor) and required that they had been off of therapy for at least 12 weeks. Of the 25 patients who received dabrafenib plus trametinib as retreatment, 32% demonstrated a partial response and 40% had stable disease.18 While further studies are warranted, retreatment with molecularly targeted therapy may be a viable option, especially in light of the multiple approved BRAF and MEK inhibitor combinations.
Another concept that has been studied is treatment beyond disease progression with molecularly targeted therapy. In a retrospective analysis of patients who had progressed on a single-agent BRAF inhibitor, 39% of those patients were continued on the same BRAF inhibitor and compared to patients who received no subsequent therapy or changed to an alternative systemic therapy. In the multivariable analysis adjusting for other prognostic factors, continued treatment with the BRAF inhibitor was associated with prolonged OS.19
Case Conclusion
The patient is started on second-line therapy with nivolumab and ipilimumab and demonstrates a partial response. One year later he continues to feel well with decreased size of the intracranial and right lower lobe lesions, and without any interval development of new areas of metastatic disease.
Special Considerations
Intralesional Therapies
Talimogene laherparepvec (T-VEC) is a genetically modified herpesvirus-1 oncolytic virus that is injected into melanoma skin lesions and leads to the expression of granulocyte-macrophage colony-stimulating factor. While T-VEC is currently approved for local treatment of unresectable cutaneous, subcutaneous, or nodal recurrences,20 it has also been investigated in combination with other therapies for patients with advanced disease. In patients with previously treated melanoma, T-VEC plus ipilimumab demonstrated superior ORR to ipilimumab alone (39% vs 18%), and the tumor response was not limited to the injected lesions. The observation of systemic response suggests synergy between T-VEC and immune checkpoint blockade in enhancing the anti-tumor immune response.21 The phase 1b MASTERKEY-265 trial combining pembrolizumab and T-VEC led to an ORR of 62% and CR of 33%.22 A phase 3 trial comparing pembrolizumab plus T-VEC to pembrolizumab alone is ongoing (NCT02263508).
Melanoma Brain Metastases
The presence of brain metastases is a common event in patients with metastatic melanoma, and often confers a poor prognosis.23 The approach to the management of brain metastases should be multidisciplinary among medical oncology, neurosurgery, and radiation oncology providers, as treatment algorithms continue to rapidly evolve. Historically, there has been little prospective clinical trial data regarding optimal systemic therapy, and local therapies such as surgery or stereotactic radiation have long been the mainstay of therapy for intracranial disease.24 However, recent data with both immunotherapy and molecularly targeted therapy has demonstrated efficacy with intracranial metastases.
A recent trial of combined nivolumab and ipilimumab as frontline therapy in patients with asymptomatic melanoma brain metastases demonstrated a complete response rate of 26% and partial response rate of 30% in patients with a median follow-up of 14 months.25 In a separate study, ipilimumab plus nivolumab demonstrated better intracranial ORR when compared to nivolumab alone in asymptomatic, previously untreated patients. Outcomes were better in patients presenting with asymptomatic versus symptomatic brain metastases.26 Collectively, these results suggest that systemic immunotherapy alone may be adequate for patients with asymptomatic, previously untreated brain metastases.
For molecularly targeted therapy in patients with BRAF mutations and brain metastases, the BREAK-MB trial demonstrated that an intracranial response was attainable with dabrafenib regardless of whether the patient had previously received local therapy in the form of surgery or radiation.27 The COMBI-MB trial enhanced the preexisting data by testing the intracranial efficacy of dabrafenib plus trametinib in 4 different cohorts of patients, further supporting that systemic molecularly targeted therapy can provide significant intracranial activity in patients with both symptomatic and asymptomatic brain lesions and regardless of prior local therapy (Table 2).28
Conclusion
The treatment of advanced melanoma has been drastically improved over the past decade by the development and study of immune checkpoint inhibitors and molecularly targeted agents. There is still much to learn regarding the optimal combination and sequencing of therapies. Many of these trials are ongoing and will provide additional evidence to guide treatment decisions moving forward.
1. Bowyer S, Prithviraj P, Lorigan P, et al. Efficacy and toxicity of treatment with the anti-CTLA-4 antibody ipilimumab in patients with metastatic melanoma after prior anti-PD-1 therapy. Br J Cancer. 2016;114:1084-1089.
2. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-384.
3. Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908-918.
4. Hamid O, Puzanov I, Dummer R, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer. 2017;86:37-45.
5. Beaver JA, Hazarika M, Mulkey F, et al. Patients with melanoma treated with an anti-PD-1 antibody beyond RECIST progression: a US Food and Drug Administration pooled analysis. Lancet Oncol. 2018;19:229-239.
6. Zimmer L, Apuri S, Eroglu Z, et al. Ipilimumab alone or in combination with nivolumab after progression on anti-PD-1 therapy in advanced melanoma. Eur J Cancer. 2017;75:47-55.
7. Larkin J, Chiarion-Sileni V, Gonazalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34.
8. Tétu P, Mangana J, Dummer R, et al. Benefit of the nivolumab and ipilimumab combination in pretreated advanced melanoma. Eur J Cancer. 2018;93:147-149.
9. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2522-2532.
10. Ackerman A, Klein O, McDermott D, et al. Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer. 2014;120:1695-1701.
11. Johnson DB, Pectasides E, Feld E, et al. Sequencing treatment in BRAFV600 mutant melanoma: anti-pd-1 before and after BRAF inhibition. J Immunother. 2017;40:31-35.
12. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicenter, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2015;386:444-451.
13. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28:1631-1639.
14. Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248-1260.
15. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicenter, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:603-615.
16. Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19:1315-1327.
17. Johnson DB, Flaherty KT, Weber, JS et al. Combined BRAF (Dabrafenib) and MEK inhibition (Trametinib) in patients with BRAFV600-mutant melanoma experiencing progression with single-agent BRAF inhibitor. J Clin Oncol. 2014;32:3697-3704.
18. Schreuer M, Jansen Y, Planken S, et al. Combination of dabrafenib plus trametinib for BRAF and MEK inhibitor pretreated patients with advanced BRAFV600-mutant melanoma: an open-label, single arm, dual-centre, phase 2 clinical trial. Lancet Oncol. 2017;18:464-472.
19. Chan MM, Haydu LE, Azer MW, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer. 2014;120:3142-3153.
20. Imlygic (talimogene laherparepvec) suspension for intralesional injection [package insert]. Thousand Oaks, CA: BioVex; 2015.
21. Chesney J, Puzanov I, Collichio F, et al. Randomized, open-label phase ii study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. 2018;36:1658-1667.
22. Ribas A, Dummer R, Puzanov I, et al. Oncolytic virotherapy promotes intratumoral t cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2018;174:1031-1032.
23. Sampson JH, Carter Jr. JH, Friedman AH, Seigler HF. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg. 1998;88:11-20.
24. Yamamoto M, Serizawa T, Shuto T, et al. Stereotactic radiosurgery for patients with multiple brain metastases (JLGK0901): a multi-institutional prospective observational study. Lancet Oncol. 2014;15:387-395.
25. Tawbi HA, Forsyth PA, Hamid O, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med. 2018;379:722-730.
26. Long GV, Atkinson V, La S, et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicenter randomised phase 2 study. Lancet Oncol. 2018;19:672-681.
27. Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicenter, open-label, phase 2 trial. Lancet Oncol. 2012;13:1087-1095.
28. Davies MA, Saiag P, Robert C, et al. Dabrafenib plus trametinib in patients with BRAFV600-mutant melanoma brain metastases (COMBI-MB): a multicenter, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017;18:863-873.
1. Bowyer S, Prithviraj P, Lorigan P, et al. Efficacy and toxicity of treatment with the anti-CTLA-4 antibody ipilimumab in patients with metastatic melanoma after prior anti-PD-1 therapy. Br J Cancer. 2016;114:1084-1089.
2. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-384.
3. Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908-918.
4. Hamid O, Puzanov I, Dummer R, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer. 2017;86:37-45.
5. Beaver JA, Hazarika M, Mulkey F, et al. Patients with melanoma treated with an anti-PD-1 antibody beyond RECIST progression: a US Food and Drug Administration pooled analysis. Lancet Oncol. 2018;19:229-239.
6. Zimmer L, Apuri S, Eroglu Z, et al. Ipilimumab alone or in combination with nivolumab after progression on anti-PD-1 therapy in advanced melanoma. Eur J Cancer. 2017;75:47-55.
7. Larkin J, Chiarion-Sileni V, Gonazalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34.
8. Tétu P, Mangana J, Dummer R, et al. Benefit of the nivolumab and ipilimumab combination in pretreated advanced melanoma. Eur J Cancer. 2018;93:147-149.
9. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2522-2532.
10. Ackerman A, Klein O, McDermott D, et al. Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer. 2014;120:1695-1701.
11. Johnson DB, Pectasides E, Feld E, et al. Sequencing treatment in BRAFV600 mutant melanoma: anti-pd-1 before and after BRAF inhibition. J Immunother. 2017;40:31-35.
12. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicenter, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2015;386:444-451.
13. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28:1631-1639.
14. Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248-1260.
15. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicenter, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:603-615.
16. Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19:1315-1327.
17. Johnson DB, Flaherty KT, Weber, JS et al. Combined BRAF (Dabrafenib) and MEK inhibition (Trametinib) in patients with BRAFV600-mutant melanoma experiencing progression with single-agent BRAF inhibitor. J Clin Oncol. 2014;32:3697-3704.
18. Schreuer M, Jansen Y, Planken S, et al. Combination of dabrafenib plus trametinib for BRAF and MEK inhibitor pretreated patients with advanced BRAFV600-mutant melanoma: an open-label, single arm, dual-centre, phase 2 clinical trial. Lancet Oncol. 2017;18:464-472.
19. Chan MM, Haydu LE, Azer MW, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer. 2014;120:3142-3153.
20. Imlygic (talimogene laherparepvec) suspension for intralesional injection [package insert]. Thousand Oaks, CA: BioVex; 2015.
21. Chesney J, Puzanov I, Collichio F, et al. Randomized, open-label phase ii study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. 2018;36:1658-1667.
22. Ribas A, Dummer R, Puzanov I, et al. Oncolytic virotherapy promotes intratumoral t cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2018;174:1031-1032.
23. Sampson JH, Carter Jr. JH, Friedman AH, Seigler HF. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg. 1998;88:11-20.
24. Yamamoto M, Serizawa T, Shuto T, et al. Stereotactic radiosurgery for patients with multiple brain metastases (JLGK0901): a multi-institutional prospective observational study. Lancet Oncol. 2014;15:387-395.
25. Tawbi HA, Forsyth PA, Hamid O, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med. 2018;379:722-730.
26. Long GV, Atkinson V, La S, et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicenter randomised phase 2 study. Lancet Oncol. 2018;19:672-681.
27. Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicenter, open-label, phase 2 trial. Lancet Oncol. 2012;13:1087-1095.
28. Davies MA, Saiag P, Robert C, et al. Dabrafenib plus trametinib in patients with BRAFV600-mutant melanoma brain metastases (COMBI-MB): a multicenter, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017;18:863-873.
Advanced Melanoma: First-line Therapy
Malignant melanoma is the most serious form of primary skin cancer and one of the only malignancies in which the incidence rate has been rising. It is estimated that in 2018 there were 91,270 newly diagnosed cases and 9320 deaths from advanced melanoma in the United States. Melanoma is the fifth most common cancer type in males and the sixth most common in females. Despite rising incidence rates, improvement in the treatment of advanced melanoma has resulted in declining death rates over the past decade.1 Although most melanoma is diagnosed at an early stage and can be cured with surgical excision, the prognosis for metastatic melanoma had been historically poor prior to recent advancements in treatment. Conventional chemotherapy treatment with dacarbazine or temozolomide resulted in response rates ranging from 7.5% to 12.1%, but without much impact on median overall survival (OS), with reported OS ranging from 6.4 to 7.8 months. Combination approaches with interferon alfa-2B and low-dose interleukin-2 resulted in improved response rates compared with traditional chemotherapy, but again without survival benefit.2
Immunotherapy in the form of high-dose interleukin-2 emerged as the first therapy to alter the natural history of advanced melanoma, with both improved response rates (objective response rate [ORR], 16%) and median OS (2 months), with some patients achieving durable responses lasting more than 30 months. However, significant systemic toxicity limited its application to carefully selected patients.3 The past decade has brought rapid advancements in treatment with immune checkpoint inhibitors and molecularly targeted agents, which have significantly improved ORRs, progression-free survival (PFS), and OS for patients with metastatic melanoma.4-8
This review is the first of 2 articles focusing on the treatment and sequencing of therapies in advanced melanoma. Here, we review the selection of first-line therapy for metastatic melanoma. Current evidence for immune checkpoint blockade and molecularly targeted agents in the treatment of metastatic melanoma after progression on first-line therapy is discussed in a separate article.
Pathogenesis
The incidence of melanoma is strongly associated with ultraviolet light–mediated DNA damage related to sun exposure. Specifically, melanoma is associated to a greater degree with intense intermittent sun exposure and sunburn, but not associated with higher occupational exposure.9 Ultraviolet radiation can induce DNA damage by a number of mechanisms, and deficient DNA repair leads to somatic mutations that drive the progression from normal melanocyte to melanoma.10
The most commonly identified genetic mutations in cutaneous melanomas are alterations in the mitogen-activated protein kinase (MAPK) pathway. Typically, an extracellular growth factor causes dimerization of the growth factor receptor, which activates the intracellular RAS GTPase protein. Subsequently BRAF is phosphorylated within the kinase domain, which leads to downstream activation of the MEK and ERK kinases through phosphorylation. Activated ERK leads to phosphorylation of various cytoplasmic and nuclear targets, and the downstream effects of these changes promote cellular proliferation. While activation of this pathway usually requires phosphorylation of BRAF by RAS, mutations placing an acidic amino acid near the kinase domain mimics phosphorylation and leads to constitutive activation of the BRAF serine/threonine kinase in the absence of upstream signaling from extracellular growth factors mediated through RAS.11 One study of tumor samples of 71 patients with cutaneous melanoma detected NRAS mutations in 30% and BRAF mutations in 59% of all tumors tested. Of the BRAF mutation–positive tumors, 88% harbored the Val599Glu mutation, now commonly referred to as the BRAF V600E mutation. The same study demonstrated that the vast majority of BRAF mutations were seen in the primary tumor and were preserved when metastases were analyzed. Additionally, both NRAS and BRAF mutations were detected in the radial growth phase of the melanoma tumor. These findings indicate that alterations in the MAPK pathway occur early in the pathogenesis of advanced melanoma.11 Another group demonstrated that 66% of malignant melanoma tumor samples harbored BRAF mutations, of which 80% were specifically the V600E mutation. In vitro assays showed that the BRAF V600E–mutated kinase had greater than 10-fold kinase activity compared to wild-type BRAF, and that this kinase enhanced cellular proliferation even when upstream NRAS signaling was inhibited.12
The Cancer Genome Atlas Network performed a large analysis of tumor samples from 331 different melanoma patients and studied variations at the DNA, RNA, and protein levels. The study established a framework of 4 notable genomic subtypes, including mutant BRAF (52%), mutant RAS (28%), mutant NF1 (14%), and triple wild-type (6%). Additionally, mRNA transcriptomic analysis of overexpressed genes identified 3 different subclasses, which were labeled as “immune,” “keratin,” and “MITF-low.” The immune subclass was characterized by increased expression of proteins found in immune cells, immune signaling molecules, immune checkpoint proteins, cytokines, and chemokines, and correlated with increased lymphocyte invasion within the tumor. Interestingly, in the post-accession survival analysis, the “immune” transcriptomic subclass was statistically correlated with an improved prognosis.13 Having an understanding of the molecular pathogenesis of advanced melanoma helps to create a framework for understanding the mechanisms of current standard of care therapies for the disease.
Case Presentation
A 62-year-old Caucasian man with a history of well-controlled type 2 diabetes mellitus and hypertension is being followed by his dermatologist for surveillance of melanocytic nevi. On follow-up he is noted to have an asymmetrical melanocytic lesion over the right scalp with irregular borders and variegated color. He is asymptomatic and the remainder of physical examination is unremarkable, as he has no other concerning skin lesions and no cervical, axillary, or inguinal lymphadenopathy.
How is melanoma diagnosed?
Detailed discussion about diagnosis and staging will be deferred in this review of treatment of advanced melanoma. In brief, melanoma is best diagnosed by excisional biopsy and histopathology. Staging of melanoma is done according to the American Joint Committee on Cancer’s (AJCC) Cancer Staging Manual, 8th edition, using a TNM staging system that incorporates tumor thickness (Breslow depth); ulceration; number of involved regional lymph nodes; presence of in-transit, satellite, and/or microsatellite metastases; distant metastases; and serum lactate dehydrogenase level.14
Case Continued
The patient undergoes a wide excisional biopsy of the right scalp lesion, which is consistent with malignant melanoma. Pathology demonstrates a Breslow depth of 2.6 mm, 2 mitotic figures/mm2, and no evidence of ulceration. He subsequently undergoes wide local excision with 0/3 sentinel lymph nodes positive for malignancy. His final staging is consistent with pT3aN0M0, stage IIA melanoma.
He is seen in follow-up with medical oncology for the next 3.5 years without any evidence of disease recurrence. He then develops symptoms of vertigo, diplopia, and recurrent falls, prompting medical attention. Magnetic resonance imaging (MRI) brain reveals multiple supratentorial and infratentorial lesions concerning for intracranial metastases. Further imaging with computed tomography (CT) chest/abdomen/pelvis reveals a right lower lobe pulmonary mass with right hilar and subcarinal lymphadenopathy. He is admitted for treatment with intravenous dexamethasone and further evaluation with endobronchial ultrasound-guided fine-needle aspiration of the right lower lobe mass, which reveals metastatic melanoma. Given the extent of his intracranial metastases, he is treated with whole brain radiation therapy for symptomatic relief prior to initiating systemic therapy.
What is the general approach to first-line treatment for metastatic melanoma?
The past decade has brought an abundance of data supporting the use of immunotherapy with immune checkpoint inhibitors or molecularly targeted therapy with combined BRAF/MEK inhibitors in the first-line setting.4-8 After the diagnosis of metastatic melanoma has been made, molecular testing is recommended to determine the BRAF status of the tumor. Immunotherapy is the clear choice for first-line therapy in the absence of an activating BRAF V600 mutation. When a BRAF V600 mutation is present, current evidence supports the use of either immunotherapy or molecularly targeted therapy as first-line therapy.
To date, there have been no prospective clinical trials comparing the sequencing of immunotherapy and molecularly targeted therapy in the first-line setting. An ongoing clinical trial (NCT02224781) is comparing dabrafenib and trametinib followed by ipilimumab and nivolumab at time of progression to ipilimumab and nivolumab followed by dabrafenib and trametinib in patients with newly diagnosed stage III/IV BRAF V600 mutation–positive melanoma. The primary outcome measure is 2-year OS. Until completion of that trial, current practice regarding which type of therapy to use in the first-line setting is based on a number of factors including clinical characteristics and provider preferences.
Data suggest that immunotherapies can produce durable responses, especially after treatment completion or discontinuation, albeit at the expense of taking a longer time to achieve clinical benefit and the risk of potentially serious immune-related adverse effects. This idea of a durable, off-treatment response is highlighted by a study that followed 105 patients who had achieved a complete response (CR) and found that 24-month disease-free survival from the time of CR was 90.9% in all patients and 89.9% in the 67 patients who had discontinued pembrolizumab after attaining CR.15 BRAF/MEK inhibition has the potential for rapid clinical responses, though concerns exist about the development of resistance to therapy. The following sections explore the evidence supporting the use of these therapies.
Immunotherapy with Immune Checkpoint Inhibitors
Immunotherapy via immune checkpoint blockade has revolutionized the treatment of many solid tumors over the past decade. The promise of immunotherapy revolves around the potential for achieving a dynamic and durable systemic response against cancer by augmenting the antitumor effects of the immune system. T-cells are central to mounting a systemic antitumor response, and, in addition to antigen recognition, their function depends heavily on fine tuning between co-stimulatory and co-inhibitory signaling. The cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) expressed on T-cells was the first discovered co-inhibitory receptor of T-cell activation.16 Later, it was discovered that the programmed cell death 1 receptor (PD-1), expressed on T-cells, and its ligands PD-L1 and PD-L2, expressed on antigen presenting cells, tumor cells, or other cells in the tumor microenvironment, also served as a potent negative regulator of T-cell function.17
Together, these 2 signaling pathways help to maintain peripheral immune tolerance, whereby autoreactive T-cells that have escaped from the thymus are silenced to prevent autoimmunity. However, these pathways can also be utilized by cancer cells to escape immune surveillance. Monoclonal antibodies that inhibit the aforementioned co-inhibitory signaling pathways, and thus augment the immune response, have proven to be an effective anticancer therapy capable of producing profound and durable responses in certain malignancies.16,17
Ipilimumab
Ipilimumab is a monoclonal antibody that inhibits the function of the CTLA-4 co-inhibitory immune checkpoint. In a phase 3 randomized controlled trial of 676 patients with previously treated metastatic melanoma, ipilimumab at a dose of 3 mg/kg every 3 weeks for 4 cycles, with or without a gp100 peptide vaccine, resulted in an improved median OS of 10.0 and 10.1 months, respectively, compared to 6.4 months in those receiving the peptide vaccine alone, meeting the primary endpoint.4 Subsequently, a phase 3 trial of 502 patients with untreated metastatic melanoma compared ipilimumab at a dose of 10 mg/kg every 3 weeks for 4 cycles plus dacarbazine to dacarbazine plus placebo and found a significant increase in median OS (11.2 months vs 9.1 months), with no additive benefit of chemotherapy. There was a higher reported rate of grade 3 or 4 adverse events in this trial with ipilimumab dosed at 10 mg/kg, which was felt to be dose-related.18 These trials were the first to show improved OS with any systemic therapy in metastatic melanoma and led to US Food and Drug Administration approval of ipilimumab for this indication in 2011.
PD-1 Inhibitor Monotherapy
The PD-1 inhibitors nivolumab and pembrolizumab were initially approved for metastatic melanoma after progression on ipilimumab. In the phase 1 trial of patients with previously treated metastatic melanoma, nivolumab therapy resulted in an ORR of 28%.19 The subsequent phase 2 trial conducted in pretreated patients, including patients who had progressed on ipilimumab, confirmed a similar ORR of 31%, as well as a median PFS of 3.7 months and a median OS of 16.8 months. The estimated response duration in patients who did achieve a response to therapy was 2 years.20 A phase 3 trial (CheckMate 037) comparing nivolumab (n = 120) to investigator’s choice chemotherapy (n = 47) in those with melanoma refractory to ipilimumab demonstrated that nivolumab was superior for the primary endpoint of ORR (31.7% vs 10.6%), had less toxicity (5% rate of grade 3 or 4 adverse events versus 9%), and increased median duration of response (32 months vs 13 months).21
The phase 1 trial (KEYNOTE-001) testing the efficacy of pembrolizumab demonstrated an ORR of 33% in the total population of patients treated and an ORR of 45% in those who were treatment-naive. Additionally, the median OS was 23 months for the total population and 31 months for treatment-naive patients, with only 14% of patients experiencing a grade 3 or 4 adverse event.22 The KEYNOTE-002 phase 2 trial compared 2 different pembrolizumab doses (2 mg/kg and 10 mg/kg every 3 weeks) to investigator’s choice chemotherapy (paclitaxel plus carboplatin, paclitaxel, carboplatin, dacarbazine, or oral temozolomide) in 540 patients with advanced melanoma with documented progression on ipilimumab with or without prior progression on molecularly targeted therapy if positive for a BRAF V600 mutation. The final analysis demonstrated significantly improved ORR with pembrolizumab (22% at 2 mg/kg vs 26% at 10 mg/kg vs 4% chemotherapy) and significantly improved 24-month PFS (16% vs 22% vs 0.6%, respectively). There was a nonstatistically significant improvement in median OS (13.4 months vs 14.7 months vs 10 months), although 55% of the patients initially assigned to the chemotherapy arm crossed over and received pembrolizumab after documentation of progressive disease.23,24
Because PD-1 inhibition improved efficacy with less toxicity than chemotherapy when studied in progressive disease, subsequent studies focused on PD-1 inhibition in the frontline setting. CheckMate 066 was a phase 3 trial comparing nivolumab to dacarbazine as first-line therapy for 418 patients with untreated metastatic melanoma who did not have a BRAF mutation. For the primary end point of 1-year OS, nivolumab was superior to dacarbazine (72.9% vs 42.1%; hazard ratio [HR], 0.42; P < 0.001). Treatment with nivolumab also resulted in superior ORR (40% vs 14%) and PFS (5.1 months vs 2.2 months). Additionally, nivolumab therapy had a lower rate of grade 3 or 4 toxicity compared to dacarbazine (11.7% vs 17.6%).25
The KEYNOTE-006 trial compared 2 separate dosing schedules of pembrolizumab (10 mg/kg every 2 weeks versus every 3 weeks) to ipilimumab (3 mg/kg every 3 weeks for 4 cycles) in a 1:1:1 ratio in 834 patients with metastatic melanoma who had received up to 1 prior systemic therapy, but no prior CTLA-4 or PD-1 inhibitors. The first published data reported statistically significant outcomes for the co-primary end points of 6-month PFS (47.3% for pembrolizumab every 2 weeks vs 46.4% for pembrolizumab every 3 weeks vs 26.5% for ipilimumab; HR, 0.58 for both pembrolizumab groups compared to ipilimumab; P < 0.001) and 12-month OS (74.1% vs 68.4% vs 58.2%) with pembrolizumab compared to ipilimumab. Compared to ipilimumab, pembrolizumab every 2 weeks had a hazard ratio of 0.63 (P = 0.0005) and pembrolizumab every 3 weeks had a hazard ratio of 0.69 (P = 0.0036). The pembrolizumab groups was also had lower rates of grade 3 to 5 toxicity (13.3% vs 10.1% vs 19.9%).5 Updated outcomes demonstrated improved ORR compared to the first analysis (37% vs 36% vs 13%), and improved OS (median OS, not reached for the pembrolizumab groups vs 16.0 months for the ipilimumab group; HR, 0.68, P = 0.0009 for pembrolizumab every 2 weeks versus HR 0.68, P = 0.0008 for pembrolizumab every 3 weeks).26 In addition, 24-month OS was 55% in both pembrolizumab groups compared to 43% in the ipilimumab group. Grade 3 or 4 toxicity occurred less frequently with pembrolizumab (17% vs 17% vs 20%).
Further analysis from the KEYNOTE-006 trial data demonstrated improved ORR, PFS, and OS with pembrolizumab compared to ipilimumab in tumors positive for PD-L1 expression. For PD-L1-negative tumors, response rate was higher, and PFS and OS rates were similar with pembrolizumab compared to ipilimumab. Given that pembrolizumab was associated with similar survival outcomes in PD-L1-negative tumors and with less toxicity than ipilimumab, the superiority of PD-L1 inhibitors over ipilimumab was further supported, regardless of tumor PD-L1 status.27
In sum, PD-1 inhibition should be considered the first-line immunotherapy in advanced melanoma, either alone or in combination with ipilimumab, as discussed in the following section. There is no longer a role for ipilimumab monotherapy in the first-line setting, based on evidence from direct comparison to single-agent PD-1 inhibition in clinical trials that demonstrated superior efficacy and less serious toxicity with PD-1 inhibitors.5,26 The finding that ORR and OS outcomes with single-agent PD-1 inhibitors are higher in treatment-naive patients compared to those receiving prior therapies also supports this approach.22
Combination CTLA-4 and PD-1 Therapy
Despite the potential for durable responses, the majority of patients fail to respond to single-agent PD-1 therapy. Given that preclinical data had suggested the potential for synergy between dual inhibition of CTLA-4 and PD-1, clinical trials were designed to test this approach. The first randomized phase 2 trial that established superior efficacy with combination therapy was the CheckMate 069 trial comparing nivolumab plus ipilimumab to ipilimumab monotherapy. Combination therapy resulted in increased ORR (59% vs 11%), median PFS (not reached vs 3.0 months), 2-year PFS (51.3% vs 12.0%), and 2-year OS (63.8% vs 53.6%).28 Similarly, a phase 1b trial of pembrolizumab plus reduced-dose ipilimumab demonstrated an ORR of 61%, with a 1-year PFS of 69% and 1-year OS of 89%.29
The landmark phase 3 CheckMate 067 trial analyzed efficacy outcomes for 3 different treatment regimens including nivolumab plus ipilimumab, nivolumab monotherapy, and ipilimumab monotherapy in previously untreated patients with unresectable stage III or IV melanoma. The trial was powered to compare survival outcomes for both the combination therapy arm against ipilimumab and the nivolumab monotherapy arm against ipilimumab, but not to compare combination therapy to nivolumab monotherapy. The initial analysis demonstrated a median PFS of 11.5 months with combination therapy versus 6.9 months with nivolumab and 2.9 months with ipilimumab, as well as an ORR of 58% versus 44% and 19%, respectively (Table 1).6 The updated 3-year survival outcomes from CheckMate 067 were notable for superior median OS with combination therapy (not reached in combination vs 37.6 months for nivolumab vs 19.9 months ipilimumab), improved 3-year OS (58% vs 52% vs 34%), and improved 3-year PFS (39% vs 32% vs 10%).7 In the reported 4-year survival outcomes, median OS was not reached in the combination therapy group, and was 36.9 months in the nivolumab monotherapy group and 19.9 months in the ipilimumab monotherapy group. Rates of grade 3 or 4 adverse events were significantly higher in the combination therapy group, at 59% compared to 22% with nivolumab monotherapy and 28% with ipilimumab alone.30 The 3- and 4-year OS outcomes (58% and 54%, respectively) with combination therapy were the highest seen in any phase 3 trial for treatment of advanced melanoma, supporting its use as the best approved first-line therapy in those who can tolerate the potential toxicity of combination therapy7,30 The conclusions from this landmark trial were that both combination therapy and nivolumab monotherapy resulted in statistically significant improvement in OS compared to ipilimumab.

Toxicity Associated with Immune Checkpoint Inhibitors
While immune checkpoint inhibitors have revolutionized the treatment of many solid tumor malignancies, this new class of cancer therapy has brought about a new type of toxicity for clinicians to be aware of, termed immune-related adverse events (irAEs). As immune checkpoint inhibitors amplify the immune response against malignancy, they also increase the likelihood that autoreactive T-cells persist and proliferate within the circulation. Therefore, these therapies can result in almost any type of autoimmune side effect. The most commonly reported irAEs in large clinical trials studying CTLA-4 and PD-1 inhibitors include rash/pruritus, diarrhea/colitis, hepatitis, endocrinopathies (thyroiditis, hypophysitis, adrenalitis), and pneumonitis. Other more rare toxicities include pancreatitis, autoimmune hematologic toxicities, cardiac toxicity (myocarditis, heart failure), and neurologic toxicities (neuropathies, myasthenia gravis-like syndrome, Guillain-Barré syndrome). It has been observed that PD-1 inhibitors have a lower incidence of irAEs than CTLA-4 inhibitors, and that the combined use of PD-1 and CTLA-4 inhibitors is associated with a greater incidence of irAEs compared to monotherapy with either agent.31 Toxicities associated with ipilimumab have been noted to be dose dependent.18 Generally, these toxicities are treated with immunosuppression in the form of glucocorticoids and are often reversible.31 There are several published guidelines that include algorithms for the management of irAEs by organizations such as the National Comprehensive Cancer Network.32
For example, previously untreated patients treated with ipilimumab plus dacarbazine as compared to dacarbazine plus placebo had greater grade 3 or 4 adverse events (56.3% vs 27.5%), and 77.7% of patients experiencing an irAE of any grade.18 In the CheckMate 066 trial comparing frontline nivolumab to dacarbazine, nivolumab had a lower rate of grade 3 or 4 toxicity (11.7% vs 17.6%) and irAEs were relatively infrequent, with diarrhea and elevated alanine aminotransferase level each being the most prominent irAE (affecting 1.0% of patients).25 In the KEYNOTE-006 trial, irAEs seen in more than 1% of patients treated with pembrolizumab included colitis, hepatitis, hypothyroidism, and hyperthyroidism, whereas those occurring in more than 1% of patients treated with ipilimumab included colitis and hypophysitis. Overall, there were lower rates of grade 3 to 5 toxicity with the 2 pembrolizumab doses compared to ipilimumab (13.3% pembrolizumab every 2 weeks vs 10.1% pembrolizumab every 3 weeks vs 19.9% ipilimumab).5 In the CheckMate 067 trial comparing nivolumab plus ipilimumab, nivolumab monotherapy, and ipilimumab monotherapy, rates of treatment-related adverse events of any grade were higher in the combination group (96% combination vs 86% nivolumab vs 86% ipilimumab), as were rates of grade 3 or 4 adverse events (59% vs 21% vs 28%, respectively). The irAE profile was similar to that demonstrated in prior studies: rash/pruritus were the most common, and diarrhea/colitis, elevated aminotransferases, and endocrinopathies were among the more common irAEs.7
Alternative dosing strategies have been investigated in an effort to preserve efficacy and minimize toxicity. A phase 1b trial of pembrolizumab plus reduced-dose ipilimumab demonstrated an ORR of 61%, with a 1-year PFS of 69% and a 1-year OS of 89%. This combination led to 45% of patients having a grade 3 or 4 adverse event, 60% having irAEs of any grade, and only 27% having grade 3 or 4 irAEs.29 The CheckMate 067 trial studied the combination of nivolumab 1 mg/kg plus ipilimumab 3 mg/kg.6 The CheckMate 511 trial compared different combination dosing strategies (nivolumab 3 mg/kg + ipilimumab 1 mg/kg versus nivolumab 1 mg/kg + ipilimumab 3 mg/kg) to assess for safety benefit. In the results published in abstract form, the reduced ipilimumab dose (nivolumab 3 mg/kg + ipilimumab 1 mg/kg arm) resulted in significantly decreased grade 3 to 5 adverse events (33.9% vs 48.3%) without significant differences in ORR, PFS, or OS.33
The question about the efficacy of checkpoint inhibitors in patients who discontinue treatment due to irAEs has been raised, as one hypothesis suggests that such toxicities may also indicate that the antitumor immune response has been activated. In a retrospective pooled analysis of phase 2 and 3 trials where patients received combination therapy with ipilimumab and nivolumab and discontinued therapy during the induction phase due to irAEs, outcomes did not appear to be inferior. Median PFS was 8.4 months in those who discontinued therapy compared to 10.8 months in those who continued therapy, but this did not reach statistical significance. Median OS had not been reached in either group and ORR was actually higher in those who discontinued due to adverse events (58.3% vs 50.2%). While this retrospective analysis needs to be validated, it does suggest that patients likely derive antitumor benefit from immunotherapy even if they have to discontinue therapy due to irAEs. Of note, patients in this analysis were not trialed on nivolumab monotherapy after receiving immunosuppressive treatment for toxicity related to combination therapy, which has since been deemed a reasonable treatment option.34
Molecularly Targeted Therapy for Metastatic Melanoma
As previously mentioned, the MAPK pathway is frequently altered in metastatic melanoma and thus serves as a target for therapy. Mutations in BRAF can cause constitutive activation of the protein’s kinase function, which subsequently phosphorylates/activates MEK in the absence of extracellular growth signals and causes increased cellular proliferation. For the roughly half of patients diagnosed with metastatic melanoma who harbor a BRAF V600 mutation, molecularly targeted therapy with BRAF/MEK inhibitors has emerged as a standard of care treatment option. As such, all patients with advanced disease should be tested for BRAF mutations.
After early phase 1 studies of the BRAF inhibitor vemurafenib demonstrated successful inhibition of mutated BRAF,35 subsequent studies confirmed the benefit of BRAF targeted therapy. In the phase 3 randomized controlled BRIM-3 trial comparing vemurafenib with dacarbazine for treatment of 675 patients with previously untreated metastatic melanoma positive for a BRAF V600E mutation, the vemurafenib group had superior ORR and 6-month OS during the first analysis.36 In a subsequent analysis, median PFS and median OS were also superior with vemurafenib compared to dacarbazine, as vemurafenib had a median OS of 13.6 months compared to 9.7 months with dacarbazine (HR, 0.70; P = 0.0008).37 Dabrafenib was the next BRAF inhibitor to demonstrate clinical efficacy with superior PFS compared to dacarbazine.38
Despite tumor shrinkage in the majority of patients, the development of resistance to therapy was an issue early on. The development of acquired resistance emerged as a heterogeneous process, though many of the identified resistance mechanisms involved reactivation of the MAPK pathway.39 A phase 3 trial of 322 patients with metastatic melanoma comparing the MEK inhibitor trametinib as monotherapy against chemotherapy demonstrated a modest improvement in both median PFS and OS.40 As a result, subsequent efforts focused on a strategy of concurrent MEK inhibition as a means to overcome resistance to molecularly targeted monotherapy
At least 4 large phase 3 randomized controlled trials of combination therapy with BRAF plus MEK inhibitors showed an improved ORR, PFS, and OS when compared to BRAF inhibition alone. The COMBI-d trial comparing dabrafenib plus trametinib versus dabrafenib alone was the first to demonstrate the superiority of combined BRAF/MEK inhibition and made combination therapy the current standard of care for patients with metastatic melanoma and a BRAF V600 mutation. In the final analysis of this trial, 3-year PFS was 22% with combination therapy compared to 12% with dabrafenib alone, and 3-year OS was 44% compared to 32%.8,41,42 A second trial with the combination of dabrafenib and trametinib (COMBI-V) also demonstrated superior efficacy when compared to single-agent vemurafenib without increased toxicity.43 Subsequently, the combination of vemurafenib with MEK inhibitor cobimetinib demonstrated superiority compared to vemurafenib alone,44 followed by the newest combination encorafenib (BRAF inhibitor) and binimetinib (MEK inhibitor) proving superior to either vemurafenib or encorafenib alone.45,46
It is important to note that there have been no studies directly comparing the efficacy of the 3 approved BRAF/MEK inhibitor combinations, but the 3 different regimens have some differences in their toxicity profiles (Table 2). Of note, single-agent BRAF inhibition was associated with increased cutaneous toxicity, including secondary squamous cell carcinoma and keratoacanthoma,47 which was demonstrated to be driven by paradoxical activation of the MAPK pathway.48 The concerning cutaneous toxicities such as squamous cell carcinoma were substantially reduced by combination BRAF/MEK inhibitor therapy.47 Collectively, the higher efficacy along with manageable toxicity profile established combination BRAF/MEK inhibition as the preferred regimen for patients with BRAF-mutated metastatic melanoma who are being considered for molecularly targeted therapy. BRAF inhibitor monotherapy should only be used when there is a specific concern regarding the use of a MEK inhibitor in certain clinical circumstances.

Other driver mutations associated with metastatic melanoma such as NRAS-mutated tumors have proven more difficult to effectively treat with molecularly targeted therapy, with one study showing that the MEK inhibitor binimetinib resulted in a modest improvement in ORR and median PFS without OS benefit compared to dacarbazine.49 Several phase 2 trials involving metastatic melanoma harboring a c-Kit alteration have demonstrated some efficacy with the tyrosine kinase inhibitor imatinib. The largest phase 2 trial of 43 patients treated with imatinib resulted in a 53.5% disease control rate (23.3% partial response and 30.2% stable disease), with 9 of the 10 patients who achieved partial response having a mutation in either exon 11 or 13. Median PFS was 3.5 months and 1-year OS was 51.0%.50
Case Conclusion
Prior to initiation of systemic therapy, the patient’s melanoma is tested and is found to be positive for a BRAF V600K mutation. At his follow-up appointment, the patient continues to endorse generalized weakness, fatigue, issues with balance, and residual pulmonary symptoms after being treated for post-obstructive pneumonia. Given his current symptoms and extent of metastatic disease, immunotherapy is deferred and he is started on combination molecularly targeted therapy with dabrafenib and trametinib. He initially does well, with a partial response noted by resolution of symptoms and decreased size of his intracranial metastases and decreased size of the right lower lobe mass. Further follow-up of this patient is presented in the second article in this 2-part review of advanced melanoma.
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2018. CA Cancer J Clin. 2018;68:7-30.
2. Ives NJ, Stowe RL, Lorigan P, Wheatley K. Chemotherapy compared with biochemotherapy for the treatment of metastatic melanoma: a meta-analysis of 18 trials involving 2621 patients. J Clin Oncol. 2007;25:5426-34.
3. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105-16.
4. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-23.
5. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2522-2532.
6. Larkin J, Chiarion-Sileni V, Gonazalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34.
7. Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377:1345-1356.
8. Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
9. Elwood JM, Jopson J. Melanoma and sun exposure: an overview of published studies. Int J Cancer. 1997;73:198-203.
10. Gilchrest BA, Eller MS, Geller AC, Yaar M. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 199;340:1341-1348.
11. Omholt K, Platz A, Kanter L, et al. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9:6483-8.
12. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-54.
13. Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681-96.
14. Gershenwald JE, Scolyer RA, Hess KR, et al. Melanoma staging: evidence-based changes in the American Joint Committee on Cancer Eighth Edition Cancer Staging Manual. CA Cancer J Clin. 2017;67:472-492.
15. Robert C, Ribas A, Hamid O, et al. Durable complete response after discontinuation of pembrolizumab in patients with metastatic melanoma. J Clin Oncol. 2018;36:1668-1674.
16. Salama AKS, Hodi FS. Cytotoxic T-lymphocyte-associated antigen-4. Clin Cancer Res. 2011;17:4622-8.
17. Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375:1767-1778.
18. Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517-2526.
19. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-2454.
20. Topalian S, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020-30.
21. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-84.
22. Ribas A, Hamid O, Daud A, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. 2016;315:1600-1609.
23. Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908-18.
24. Hamid O, Puzanov I, Dummer R, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer. 2017;86:37-45.
25. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-330.
26. Schachter J, Ribas A, Long GV, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicenter, randomised, open-label phase 3 study (KEYNOTE-006). Lancet Oncol. 2017;390:1853-1862.
27. Carlino MS, Long GV, Schadendorf D, et al. Outcomes by line of therapy and programmed death ligand 1 expression in patients with advanced melanoma treated with pembrolizumab or ipilimumab in KEYNOTE-006. A randomised clinical trial. Eur J Cancer. 2018;101:236-243.
28. Hodi FS, Chesney J, Pavlick AC, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2016;17:1558-1568.
29. Long GV, Atkinson V, Cebon JS, et al. Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): an open-label, phase 1b trial. Lancet Oncol. 2017;18:1202-10.
30. Hodi FS, Chiarion-Sileni V, Gonzalez R, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018;19:1480-1492.
31. Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the immune-related adverse effects of immune checkpoint inhibitors: a review. JAMA Oncol. 2016;2:1346-1353.
32. National Comprehensive Cancer Network. Management of immunotherapy-related toxicities (version 2.2019). www.nccn.org/professionals/physician_gls/pdf/immunotherapy.pdf. Accessed April 8, 2019.
33. Lebbé C, Meyer N, Mortier L, et al. Initial results from a phase IIIb/IV study evaluating two dosing regimens of nivolumab (NIVO) in combination with ipilimumab (IPI) in patients with advanced melanoma (CheckMate 511) [Abstract LBA47]. Ann Oncol. 2018;29:mdy424.057.
34. Schadendorf D, Wolchok JD, Hodi FS, et al. Efficacy and safety outcomes in patients with advanced melanoma who discontinued treatment with nivolumab and ipilimumab because of adverse events: a pooled analysis of randomized phase ii and iii trials. J Clin Oncol. 2017;35:3807-3814.
35. Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
36. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
37. McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): extended follow up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
38. Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicenter, open-label, phase 3 randomised controlled trial. Lancet Oncol. 2012;380:358-365.
39. Rizos H, Menzies AM, Pupo GM, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20:1965-1977.
40. Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107-114.
41. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicenter, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2015;386:444-451.
42. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28:1631-1639.
43. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
44. Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248-260.
45. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicenter, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:603-615.
46. Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19:1315-1327.
47. Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA. Dermatol 2015;151:1103-1109.
48. Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
49. Dummer R, Schadendorf D, Ascierto P, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18:435-445.
50. Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29:2904-2909.
Malignant melanoma is the most serious form of primary skin cancer and one of the only malignancies in which the incidence rate has been rising. It is estimated that in 2018 there were 91,270 newly diagnosed cases and 9320 deaths from advanced melanoma in the United States. Melanoma is the fifth most common cancer type in males and the sixth most common in females. Despite rising incidence rates, improvement in the treatment of advanced melanoma has resulted in declining death rates over the past decade.1 Although most melanoma is diagnosed at an early stage and can be cured with surgical excision, the prognosis for metastatic melanoma had been historically poor prior to recent advancements in treatment. Conventional chemotherapy treatment with dacarbazine or temozolomide resulted in response rates ranging from 7.5% to 12.1%, but without much impact on median overall survival (OS), with reported OS ranging from 6.4 to 7.8 months. Combination approaches with interferon alfa-2B and low-dose interleukin-2 resulted in improved response rates compared with traditional chemotherapy, but again without survival benefit.2
Immunotherapy in the form of high-dose interleukin-2 emerged as the first therapy to alter the natural history of advanced melanoma, with both improved response rates (objective response rate [ORR], 16%) and median OS (2 months), with some patients achieving durable responses lasting more than 30 months. However, significant systemic toxicity limited its application to carefully selected patients.3 The past decade has brought rapid advancements in treatment with immune checkpoint inhibitors and molecularly targeted agents, which have significantly improved ORRs, progression-free survival (PFS), and OS for patients with metastatic melanoma.4-8
This review is the first of 2 articles focusing on the treatment and sequencing of therapies in advanced melanoma. Here, we review the selection of first-line therapy for metastatic melanoma. Current evidence for immune checkpoint blockade and molecularly targeted agents in the treatment of metastatic melanoma after progression on first-line therapy is discussed in a separate article.
Pathogenesis
The incidence of melanoma is strongly associated with ultraviolet light–mediated DNA damage related to sun exposure. Specifically, melanoma is associated to a greater degree with intense intermittent sun exposure and sunburn, but not associated with higher occupational exposure.9 Ultraviolet radiation can induce DNA damage by a number of mechanisms, and deficient DNA repair leads to somatic mutations that drive the progression from normal melanocyte to melanoma.10
The most commonly identified genetic mutations in cutaneous melanomas are alterations in the mitogen-activated protein kinase (MAPK) pathway. Typically, an extracellular growth factor causes dimerization of the growth factor receptor, which activates the intracellular RAS GTPase protein. Subsequently BRAF is phosphorylated within the kinase domain, which leads to downstream activation of the MEK and ERK kinases through phosphorylation. Activated ERK leads to phosphorylation of various cytoplasmic and nuclear targets, and the downstream effects of these changes promote cellular proliferation. While activation of this pathway usually requires phosphorylation of BRAF by RAS, mutations placing an acidic amino acid near the kinase domain mimics phosphorylation and leads to constitutive activation of the BRAF serine/threonine kinase in the absence of upstream signaling from extracellular growth factors mediated through RAS.11 One study of tumor samples of 71 patients with cutaneous melanoma detected NRAS mutations in 30% and BRAF mutations in 59% of all tumors tested. Of the BRAF mutation–positive tumors, 88% harbored the Val599Glu mutation, now commonly referred to as the BRAF V600E mutation. The same study demonstrated that the vast majority of BRAF mutations were seen in the primary tumor and were preserved when metastases were analyzed. Additionally, both NRAS and BRAF mutations were detected in the radial growth phase of the melanoma tumor. These findings indicate that alterations in the MAPK pathway occur early in the pathogenesis of advanced melanoma.11 Another group demonstrated that 66% of malignant melanoma tumor samples harbored BRAF mutations, of which 80% were specifically the V600E mutation. In vitro assays showed that the BRAF V600E–mutated kinase had greater than 10-fold kinase activity compared to wild-type BRAF, and that this kinase enhanced cellular proliferation even when upstream NRAS signaling was inhibited.12
The Cancer Genome Atlas Network performed a large analysis of tumor samples from 331 different melanoma patients and studied variations at the DNA, RNA, and protein levels. The study established a framework of 4 notable genomic subtypes, including mutant BRAF (52%), mutant RAS (28%), mutant NF1 (14%), and triple wild-type (6%). Additionally, mRNA transcriptomic analysis of overexpressed genes identified 3 different subclasses, which were labeled as “immune,” “keratin,” and “MITF-low.” The immune subclass was characterized by increased expression of proteins found in immune cells, immune signaling molecules, immune checkpoint proteins, cytokines, and chemokines, and correlated with increased lymphocyte invasion within the tumor. Interestingly, in the post-accession survival analysis, the “immune” transcriptomic subclass was statistically correlated with an improved prognosis.13 Having an understanding of the molecular pathogenesis of advanced melanoma helps to create a framework for understanding the mechanisms of current standard of care therapies for the disease.
Case Presentation
A 62-year-old Caucasian man with a history of well-controlled type 2 diabetes mellitus and hypertension is being followed by his dermatologist for surveillance of melanocytic nevi. On follow-up he is noted to have an asymmetrical melanocytic lesion over the right scalp with irregular borders and variegated color. He is asymptomatic and the remainder of physical examination is unremarkable, as he has no other concerning skin lesions and no cervical, axillary, or inguinal lymphadenopathy.
How is melanoma diagnosed?
Detailed discussion about diagnosis and staging will be deferred in this review of treatment of advanced melanoma. In brief, melanoma is best diagnosed by excisional biopsy and histopathology. Staging of melanoma is done according to the American Joint Committee on Cancer’s (AJCC) Cancer Staging Manual, 8th edition, using a TNM staging system that incorporates tumor thickness (Breslow depth); ulceration; number of involved regional lymph nodes; presence of in-transit, satellite, and/or microsatellite metastases; distant metastases; and serum lactate dehydrogenase level.14
Case Continued
The patient undergoes a wide excisional biopsy of the right scalp lesion, which is consistent with malignant melanoma. Pathology demonstrates a Breslow depth of 2.6 mm, 2 mitotic figures/mm2, and no evidence of ulceration. He subsequently undergoes wide local excision with 0/3 sentinel lymph nodes positive for malignancy. His final staging is consistent with pT3aN0M0, stage IIA melanoma.
He is seen in follow-up with medical oncology for the next 3.5 years without any evidence of disease recurrence. He then develops symptoms of vertigo, diplopia, and recurrent falls, prompting medical attention. Magnetic resonance imaging (MRI) brain reveals multiple supratentorial and infratentorial lesions concerning for intracranial metastases. Further imaging with computed tomography (CT) chest/abdomen/pelvis reveals a right lower lobe pulmonary mass with right hilar and subcarinal lymphadenopathy. He is admitted for treatment with intravenous dexamethasone and further evaluation with endobronchial ultrasound-guided fine-needle aspiration of the right lower lobe mass, which reveals metastatic melanoma. Given the extent of his intracranial metastases, he is treated with whole brain radiation therapy for symptomatic relief prior to initiating systemic therapy.
What is the general approach to first-line treatment for metastatic melanoma?
The past decade has brought an abundance of data supporting the use of immunotherapy with immune checkpoint inhibitors or molecularly targeted therapy with combined BRAF/MEK inhibitors in the first-line setting.4-8 After the diagnosis of metastatic melanoma has been made, molecular testing is recommended to determine the BRAF status of the tumor. Immunotherapy is the clear choice for first-line therapy in the absence of an activating BRAF V600 mutation. When a BRAF V600 mutation is present, current evidence supports the use of either immunotherapy or molecularly targeted therapy as first-line therapy.
To date, there have been no prospective clinical trials comparing the sequencing of immunotherapy and molecularly targeted therapy in the first-line setting. An ongoing clinical trial (NCT02224781) is comparing dabrafenib and trametinib followed by ipilimumab and nivolumab at time of progression to ipilimumab and nivolumab followed by dabrafenib and trametinib in patients with newly diagnosed stage III/IV BRAF V600 mutation–positive melanoma. The primary outcome measure is 2-year OS. Until completion of that trial, current practice regarding which type of therapy to use in the first-line setting is based on a number of factors including clinical characteristics and provider preferences.
Data suggest that immunotherapies can produce durable responses, especially after treatment completion or discontinuation, albeit at the expense of taking a longer time to achieve clinical benefit and the risk of potentially serious immune-related adverse effects. This idea of a durable, off-treatment response is highlighted by a study that followed 105 patients who had achieved a complete response (CR) and found that 24-month disease-free survival from the time of CR was 90.9% in all patients and 89.9% in the 67 patients who had discontinued pembrolizumab after attaining CR.15 BRAF/MEK inhibition has the potential for rapid clinical responses, though concerns exist about the development of resistance to therapy. The following sections explore the evidence supporting the use of these therapies.
Immunotherapy with Immune Checkpoint Inhibitors
Immunotherapy via immune checkpoint blockade has revolutionized the treatment of many solid tumors over the past decade. The promise of immunotherapy revolves around the potential for achieving a dynamic and durable systemic response against cancer by augmenting the antitumor effects of the immune system. T-cells are central to mounting a systemic antitumor response, and, in addition to antigen recognition, their function depends heavily on fine tuning between co-stimulatory and co-inhibitory signaling. The cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) expressed on T-cells was the first discovered co-inhibitory receptor of T-cell activation.16 Later, it was discovered that the programmed cell death 1 receptor (PD-1), expressed on T-cells, and its ligands PD-L1 and PD-L2, expressed on antigen presenting cells, tumor cells, or other cells in the tumor microenvironment, also served as a potent negative regulator of T-cell function.17
Together, these 2 signaling pathways help to maintain peripheral immune tolerance, whereby autoreactive T-cells that have escaped from the thymus are silenced to prevent autoimmunity. However, these pathways can also be utilized by cancer cells to escape immune surveillance. Monoclonal antibodies that inhibit the aforementioned co-inhibitory signaling pathways, and thus augment the immune response, have proven to be an effective anticancer therapy capable of producing profound and durable responses in certain malignancies.16,17
Ipilimumab
Ipilimumab is a monoclonal antibody that inhibits the function of the CTLA-4 co-inhibitory immune checkpoint. In a phase 3 randomized controlled trial of 676 patients with previously treated metastatic melanoma, ipilimumab at a dose of 3 mg/kg every 3 weeks for 4 cycles, with or without a gp100 peptide vaccine, resulted in an improved median OS of 10.0 and 10.1 months, respectively, compared to 6.4 months in those receiving the peptide vaccine alone, meeting the primary endpoint.4 Subsequently, a phase 3 trial of 502 patients with untreated metastatic melanoma compared ipilimumab at a dose of 10 mg/kg every 3 weeks for 4 cycles plus dacarbazine to dacarbazine plus placebo and found a significant increase in median OS (11.2 months vs 9.1 months), with no additive benefit of chemotherapy. There was a higher reported rate of grade 3 or 4 adverse events in this trial with ipilimumab dosed at 10 mg/kg, which was felt to be dose-related.18 These trials were the first to show improved OS with any systemic therapy in metastatic melanoma and led to US Food and Drug Administration approval of ipilimumab for this indication in 2011.
PD-1 Inhibitor Monotherapy
The PD-1 inhibitors nivolumab and pembrolizumab were initially approved for metastatic melanoma after progression on ipilimumab. In the phase 1 trial of patients with previously treated metastatic melanoma, nivolumab therapy resulted in an ORR of 28%.19 The subsequent phase 2 trial conducted in pretreated patients, including patients who had progressed on ipilimumab, confirmed a similar ORR of 31%, as well as a median PFS of 3.7 months and a median OS of 16.8 months. The estimated response duration in patients who did achieve a response to therapy was 2 years.20 A phase 3 trial (CheckMate 037) comparing nivolumab (n = 120) to investigator’s choice chemotherapy (n = 47) in those with melanoma refractory to ipilimumab demonstrated that nivolumab was superior for the primary endpoint of ORR (31.7% vs 10.6%), had less toxicity (5% rate of grade 3 or 4 adverse events versus 9%), and increased median duration of response (32 months vs 13 months).21
The phase 1 trial (KEYNOTE-001) testing the efficacy of pembrolizumab demonstrated an ORR of 33% in the total population of patients treated and an ORR of 45% in those who were treatment-naive. Additionally, the median OS was 23 months for the total population and 31 months for treatment-naive patients, with only 14% of patients experiencing a grade 3 or 4 adverse event.22 The KEYNOTE-002 phase 2 trial compared 2 different pembrolizumab doses (2 mg/kg and 10 mg/kg every 3 weeks) to investigator’s choice chemotherapy (paclitaxel plus carboplatin, paclitaxel, carboplatin, dacarbazine, or oral temozolomide) in 540 patients with advanced melanoma with documented progression on ipilimumab with or without prior progression on molecularly targeted therapy if positive for a BRAF V600 mutation. The final analysis demonstrated significantly improved ORR with pembrolizumab (22% at 2 mg/kg vs 26% at 10 mg/kg vs 4% chemotherapy) and significantly improved 24-month PFS (16% vs 22% vs 0.6%, respectively). There was a nonstatistically significant improvement in median OS (13.4 months vs 14.7 months vs 10 months), although 55% of the patients initially assigned to the chemotherapy arm crossed over and received pembrolizumab after documentation of progressive disease.23,24
Because PD-1 inhibition improved efficacy with less toxicity than chemotherapy when studied in progressive disease, subsequent studies focused on PD-1 inhibition in the frontline setting. CheckMate 066 was a phase 3 trial comparing nivolumab to dacarbazine as first-line therapy for 418 patients with untreated metastatic melanoma who did not have a BRAF mutation. For the primary end point of 1-year OS, nivolumab was superior to dacarbazine (72.9% vs 42.1%; hazard ratio [HR], 0.42; P < 0.001). Treatment with nivolumab also resulted in superior ORR (40% vs 14%) and PFS (5.1 months vs 2.2 months). Additionally, nivolumab therapy had a lower rate of grade 3 or 4 toxicity compared to dacarbazine (11.7% vs 17.6%).25
The KEYNOTE-006 trial compared 2 separate dosing schedules of pembrolizumab (10 mg/kg every 2 weeks versus every 3 weeks) to ipilimumab (3 mg/kg every 3 weeks for 4 cycles) in a 1:1:1 ratio in 834 patients with metastatic melanoma who had received up to 1 prior systemic therapy, but no prior CTLA-4 or PD-1 inhibitors. The first published data reported statistically significant outcomes for the co-primary end points of 6-month PFS (47.3% for pembrolizumab every 2 weeks vs 46.4% for pembrolizumab every 3 weeks vs 26.5% for ipilimumab; HR, 0.58 for both pembrolizumab groups compared to ipilimumab; P < 0.001) and 12-month OS (74.1% vs 68.4% vs 58.2%) with pembrolizumab compared to ipilimumab. Compared to ipilimumab, pembrolizumab every 2 weeks had a hazard ratio of 0.63 (P = 0.0005) and pembrolizumab every 3 weeks had a hazard ratio of 0.69 (P = 0.0036). The pembrolizumab groups was also had lower rates of grade 3 to 5 toxicity (13.3% vs 10.1% vs 19.9%).5 Updated outcomes demonstrated improved ORR compared to the first analysis (37% vs 36% vs 13%), and improved OS (median OS, not reached for the pembrolizumab groups vs 16.0 months for the ipilimumab group; HR, 0.68, P = 0.0009 for pembrolizumab every 2 weeks versus HR 0.68, P = 0.0008 for pembrolizumab every 3 weeks).26 In addition, 24-month OS was 55% in both pembrolizumab groups compared to 43% in the ipilimumab group. Grade 3 or 4 toxicity occurred less frequently with pembrolizumab (17% vs 17% vs 20%).
Further analysis from the KEYNOTE-006 trial data demonstrated improved ORR, PFS, and OS with pembrolizumab compared to ipilimumab in tumors positive for PD-L1 expression. For PD-L1-negative tumors, response rate was higher, and PFS and OS rates were similar with pembrolizumab compared to ipilimumab. Given that pembrolizumab was associated with similar survival outcomes in PD-L1-negative tumors and with less toxicity than ipilimumab, the superiority of PD-L1 inhibitors over ipilimumab was further supported, regardless of tumor PD-L1 status.27
In sum, PD-1 inhibition should be considered the first-line immunotherapy in advanced melanoma, either alone or in combination with ipilimumab, as discussed in the following section. There is no longer a role for ipilimumab monotherapy in the first-line setting, based on evidence from direct comparison to single-agent PD-1 inhibition in clinical trials that demonstrated superior efficacy and less serious toxicity with PD-1 inhibitors.5,26 The finding that ORR and OS outcomes with single-agent PD-1 inhibitors are higher in treatment-naive patients compared to those receiving prior therapies also supports this approach.22
Combination CTLA-4 and PD-1 Therapy
Despite the potential for durable responses, the majority of patients fail to respond to single-agent PD-1 therapy. Given that preclinical data had suggested the potential for synergy between dual inhibition of CTLA-4 and PD-1, clinical trials were designed to test this approach. The first randomized phase 2 trial that established superior efficacy with combination therapy was the CheckMate 069 trial comparing nivolumab plus ipilimumab to ipilimumab monotherapy. Combination therapy resulted in increased ORR (59% vs 11%), median PFS (not reached vs 3.0 months), 2-year PFS (51.3% vs 12.0%), and 2-year OS (63.8% vs 53.6%).28 Similarly, a phase 1b trial of pembrolizumab plus reduced-dose ipilimumab demonstrated an ORR of 61%, with a 1-year PFS of 69% and 1-year OS of 89%.29
The landmark phase 3 CheckMate 067 trial analyzed efficacy outcomes for 3 different treatment regimens including nivolumab plus ipilimumab, nivolumab monotherapy, and ipilimumab monotherapy in previously untreated patients with unresectable stage III or IV melanoma. The trial was powered to compare survival outcomes for both the combination therapy arm against ipilimumab and the nivolumab monotherapy arm against ipilimumab, but not to compare combination therapy to nivolumab monotherapy. The initial analysis demonstrated a median PFS of 11.5 months with combination therapy versus 6.9 months with nivolumab and 2.9 months with ipilimumab, as well as an ORR of 58% versus 44% and 19%, respectively (Table 1).6 The updated 3-year survival outcomes from CheckMate 067 were notable for superior median OS with combination therapy (not reached in combination vs 37.6 months for nivolumab vs 19.9 months ipilimumab), improved 3-year OS (58% vs 52% vs 34%), and improved 3-year PFS (39% vs 32% vs 10%).7 In the reported 4-year survival outcomes, median OS was not reached in the combination therapy group, and was 36.9 months in the nivolumab monotherapy group and 19.9 months in the ipilimumab monotherapy group. Rates of grade 3 or 4 adverse events were significantly higher in the combination therapy group, at 59% compared to 22% with nivolumab monotherapy and 28% with ipilimumab alone.30 The 3- and 4-year OS outcomes (58% and 54%, respectively) with combination therapy were the highest seen in any phase 3 trial for treatment of advanced melanoma, supporting its use as the best approved first-line therapy in those who can tolerate the potential toxicity of combination therapy7,30 The conclusions from this landmark trial were that both combination therapy and nivolumab monotherapy resulted in statistically significant improvement in OS compared to ipilimumab.
Toxicity Associated with Immune Checkpoint Inhibitors
While immune checkpoint inhibitors have revolutionized the treatment of many solid tumor malignancies, this new class of cancer therapy has brought about a new type of toxicity for clinicians to be aware of, termed immune-related adverse events (irAEs). As immune checkpoint inhibitors amplify the immune response against malignancy, they also increase the likelihood that autoreactive T-cells persist and proliferate within the circulation. Therefore, these therapies can result in almost any type of autoimmune side effect. The most commonly reported irAEs in large clinical trials studying CTLA-4 and PD-1 inhibitors include rash/pruritus, diarrhea/colitis, hepatitis, endocrinopathies (thyroiditis, hypophysitis, adrenalitis), and pneumonitis. Other more rare toxicities include pancreatitis, autoimmune hematologic toxicities, cardiac toxicity (myocarditis, heart failure), and neurologic toxicities (neuropathies, myasthenia gravis-like syndrome, Guillain-Barré syndrome). It has been observed that PD-1 inhibitors have a lower incidence of irAEs than CTLA-4 inhibitors, and that the combined use of PD-1 and CTLA-4 inhibitors is associated with a greater incidence of irAEs compared to monotherapy with either agent.31 Toxicities associated with ipilimumab have been noted to be dose dependent.18 Generally, these toxicities are treated with immunosuppression in the form of glucocorticoids and are often reversible.31 There are several published guidelines that include algorithms for the management of irAEs by organizations such as the National Comprehensive Cancer Network.32
For example, previously untreated patients treated with ipilimumab plus dacarbazine as compared to dacarbazine plus placebo had greater grade 3 or 4 adverse events (56.3% vs 27.5%), and 77.7% of patients experiencing an irAE of any grade.18 In the CheckMate 066 trial comparing frontline nivolumab to dacarbazine, nivolumab had a lower rate of grade 3 or 4 toxicity (11.7% vs 17.6%) and irAEs were relatively infrequent, with diarrhea and elevated alanine aminotransferase level each being the most prominent irAE (affecting 1.0% of patients).25 In the KEYNOTE-006 trial, irAEs seen in more than 1% of patients treated with pembrolizumab included colitis, hepatitis, hypothyroidism, and hyperthyroidism, whereas those occurring in more than 1% of patients treated with ipilimumab included colitis and hypophysitis. Overall, there were lower rates of grade 3 to 5 toxicity with the 2 pembrolizumab doses compared to ipilimumab (13.3% pembrolizumab every 2 weeks vs 10.1% pembrolizumab every 3 weeks vs 19.9% ipilimumab).5 In the CheckMate 067 trial comparing nivolumab plus ipilimumab, nivolumab monotherapy, and ipilimumab monotherapy, rates of treatment-related adverse events of any grade were higher in the combination group (96% combination vs 86% nivolumab vs 86% ipilimumab), as were rates of grade 3 or 4 adverse events (59% vs 21% vs 28%, respectively). The irAE profile was similar to that demonstrated in prior studies: rash/pruritus were the most common, and diarrhea/colitis, elevated aminotransferases, and endocrinopathies were among the more common irAEs.7
Alternative dosing strategies have been investigated in an effort to preserve efficacy and minimize toxicity. A phase 1b trial of pembrolizumab plus reduced-dose ipilimumab demonstrated an ORR of 61%, with a 1-year PFS of 69% and a 1-year OS of 89%. This combination led to 45% of patients having a grade 3 or 4 adverse event, 60% having irAEs of any grade, and only 27% having grade 3 or 4 irAEs.29 The CheckMate 067 trial studied the combination of nivolumab 1 mg/kg plus ipilimumab 3 mg/kg.6 The CheckMate 511 trial compared different combination dosing strategies (nivolumab 3 mg/kg + ipilimumab 1 mg/kg versus nivolumab 1 mg/kg + ipilimumab 3 mg/kg) to assess for safety benefit. In the results published in abstract form, the reduced ipilimumab dose (nivolumab 3 mg/kg + ipilimumab 1 mg/kg arm) resulted in significantly decreased grade 3 to 5 adverse events (33.9% vs 48.3%) without significant differences in ORR, PFS, or OS.33
The question about the efficacy of checkpoint inhibitors in patients who discontinue treatment due to irAEs has been raised, as one hypothesis suggests that such toxicities may also indicate that the antitumor immune response has been activated. In a retrospective pooled analysis of phase 2 and 3 trials where patients received combination therapy with ipilimumab and nivolumab and discontinued therapy during the induction phase due to irAEs, outcomes did not appear to be inferior. Median PFS was 8.4 months in those who discontinued therapy compared to 10.8 months in those who continued therapy, but this did not reach statistical significance. Median OS had not been reached in either group and ORR was actually higher in those who discontinued due to adverse events (58.3% vs 50.2%). While this retrospective analysis needs to be validated, it does suggest that patients likely derive antitumor benefit from immunotherapy even if they have to discontinue therapy due to irAEs. Of note, patients in this analysis were not trialed on nivolumab monotherapy after receiving immunosuppressive treatment for toxicity related to combination therapy, which has since been deemed a reasonable treatment option.34
Molecularly Targeted Therapy for Metastatic Melanoma
As previously mentioned, the MAPK pathway is frequently altered in metastatic melanoma and thus serves as a target for therapy. Mutations in BRAF can cause constitutive activation of the protein’s kinase function, which subsequently phosphorylates/activates MEK in the absence of extracellular growth signals and causes increased cellular proliferation. For the roughly half of patients diagnosed with metastatic melanoma who harbor a BRAF V600 mutation, molecularly targeted therapy with BRAF/MEK inhibitors has emerged as a standard of care treatment option. As such, all patients with advanced disease should be tested for BRAF mutations.
After early phase 1 studies of the BRAF inhibitor vemurafenib demonstrated successful inhibition of mutated BRAF,35 subsequent studies confirmed the benefit of BRAF targeted therapy. In the phase 3 randomized controlled BRIM-3 trial comparing vemurafenib with dacarbazine for treatment of 675 patients with previously untreated metastatic melanoma positive for a BRAF V600E mutation, the vemurafenib group had superior ORR and 6-month OS during the first analysis.36 In a subsequent analysis, median PFS and median OS were also superior with vemurafenib compared to dacarbazine, as vemurafenib had a median OS of 13.6 months compared to 9.7 months with dacarbazine (HR, 0.70; P = 0.0008).37 Dabrafenib was the next BRAF inhibitor to demonstrate clinical efficacy with superior PFS compared to dacarbazine.38
Despite tumor shrinkage in the majority of patients, the development of resistance to therapy was an issue early on. The development of acquired resistance emerged as a heterogeneous process, though many of the identified resistance mechanisms involved reactivation of the MAPK pathway.39 A phase 3 trial of 322 patients with metastatic melanoma comparing the MEK inhibitor trametinib as monotherapy against chemotherapy demonstrated a modest improvement in both median PFS and OS.40 As a result, subsequent efforts focused on a strategy of concurrent MEK inhibition as a means to overcome resistance to molecularly targeted monotherapy
At least 4 large phase 3 randomized controlled trials of combination therapy with BRAF plus MEK inhibitors showed an improved ORR, PFS, and OS when compared to BRAF inhibition alone. The COMBI-d trial comparing dabrafenib plus trametinib versus dabrafenib alone was the first to demonstrate the superiority of combined BRAF/MEK inhibition and made combination therapy the current standard of care for patients with metastatic melanoma and a BRAF V600 mutation. In the final analysis of this trial, 3-year PFS was 22% with combination therapy compared to 12% with dabrafenib alone, and 3-year OS was 44% compared to 32%.8,41,42 A second trial with the combination of dabrafenib and trametinib (COMBI-V) also demonstrated superior efficacy when compared to single-agent vemurafenib without increased toxicity.43 Subsequently, the combination of vemurafenib with MEK inhibitor cobimetinib demonstrated superiority compared to vemurafenib alone,44 followed by the newest combination encorafenib (BRAF inhibitor) and binimetinib (MEK inhibitor) proving superior to either vemurafenib or encorafenib alone.45,46
It is important to note that there have been no studies directly comparing the efficacy of the 3 approved BRAF/MEK inhibitor combinations, but the 3 different regimens have some differences in their toxicity profiles (Table 2). Of note, single-agent BRAF inhibition was associated with increased cutaneous toxicity, including secondary squamous cell carcinoma and keratoacanthoma,47 which was demonstrated to be driven by paradoxical activation of the MAPK pathway.48 The concerning cutaneous toxicities such as squamous cell carcinoma were substantially reduced by combination BRAF/MEK inhibitor therapy.47 Collectively, the higher efficacy along with manageable toxicity profile established combination BRAF/MEK inhibition as the preferred regimen for patients with BRAF-mutated metastatic melanoma who are being considered for molecularly targeted therapy. BRAF inhibitor monotherapy should only be used when there is a specific concern regarding the use of a MEK inhibitor in certain clinical circumstances.
Other driver mutations associated with metastatic melanoma such as NRAS-mutated tumors have proven more difficult to effectively treat with molecularly targeted therapy, with one study showing that the MEK inhibitor binimetinib resulted in a modest improvement in ORR and median PFS without OS benefit compared to dacarbazine.49 Several phase 2 trials involving metastatic melanoma harboring a c-Kit alteration have demonstrated some efficacy with the tyrosine kinase inhibitor imatinib. The largest phase 2 trial of 43 patients treated with imatinib resulted in a 53.5% disease control rate (23.3% partial response and 30.2% stable disease), with 9 of the 10 patients who achieved partial response having a mutation in either exon 11 or 13. Median PFS was 3.5 months and 1-year OS was 51.0%.50
Case Conclusion
Prior to initiation of systemic therapy, the patient’s melanoma is tested and is found to be positive for a BRAF V600K mutation. At his follow-up appointment, the patient continues to endorse generalized weakness, fatigue, issues with balance, and residual pulmonary symptoms after being treated for post-obstructive pneumonia. Given his current symptoms and extent of metastatic disease, immunotherapy is deferred and he is started on combination molecularly targeted therapy with dabrafenib and trametinib. He initially does well, with a partial response noted by resolution of symptoms and decreased size of his intracranial metastases and decreased size of the right lower lobe mass. Further follow-up of this patient is presented in the second article in this 2-part review of advanced melanoma.
Malignant melanoma is the most serious form of primary skin cancer and one of the only malignancies in which the incidence rate has been rising. It is estimated that in 2018 there were 91,270 newly diagnosed cases and 9320 deaths from advanced melanoma in the United States. Melanoma is the fifth most common cancer type in males and the sixth most common in females. Despite rising incidence rates, improvement in the treatment of advanced melanoma has resulted in declining death rates over the past decade.1 Although most melanoma is diagnosed at an early stage and can be cured with surgical excision, the prognosis for metastatic melanoma had been historically poor prior to recent advancements in treatment. Conventional chemotherapy treatment with dacarbazine or temozolomide resulted in response rates ranging from 7.5% to 12.1%, but without much impact on median overall survival (OS), with reported OS ranging from 6.4 to 7.8 months. Combination approaches with interferon alfa-2B and low-dose interleukin-2 resulted in improved response rates compared with traditional chemotherapy, but again without survival benefit.2
Immunotherapy in the form of high-dose interleukin-2 emerged as the first therapy to alter the natural history of advanced melanoma, with both improved response rates (objective response rate [ORR], 16%) and median OS (2 months), with some patients achieving durable responses lasting more than 30 months. However, significant systemic toxicity limited its application to carefully selected patients.3 The past decade has brought rapid advancements in treatment with immune checkpoint inhibitors and molecularly targeted agents, which have significantly improved ORRs, progression-free survival (PFS), and OS for patients with metastatic melanoma.4-8
This review is the first of 2 articles focusing on the treatment and sequencing of therapies in advanced melanoma. Here, we review the selection of first-line therapy for metastatic melanoma. Current evidence for immune checkpoint blockade and molecularly targeted agents in the treatment of metastatic melanoma after progression on first-line therapy is discussed in a separate article.
Pathogenesis
The incidence of melanoma is strongly associated with ultraviolet light–mediated DNA damage related to sun exposure. Specifically, melanoma is associated to a greater degree with intense intermittent sun exposure and sunburn, but not associated with higher occupational exposure.9 Ultraviolet radiation can induce DNA damage by a number of mechanisms, and deficient DNA repair leads to somatic mutations that drive the progression from normal melanocyte to melanoma.10
The most commonly identified genetic mutations in cutaneous melanomas are alterations in the mitogen-activated protein kinase (MAPK) pathway. Typically, an extracellular growth factor causes dimerization of the growth factor receptor, which activates the intracellular RAS GTPase protein. Subsequently BRAF is phosphorylated within the kinase domain, which leads to downstream activation of the MEK and ERK kinases through phosphorylation. Activated ERK leads to phosphorylation of various cytoplasmic and nuclear targets, and the downstream effects of these changes promote cellular proliferation. While activation of this pathway usually requires phosphorylation of BRAF by RAS, mutations placing an acidic amino acid near the kinase domain mimics phosphorylation and leads to constitutive activation of the BRAF serine/threonine kinase in the absence of upstream signaling from extracellular growth factors mediated through RAS.11 One study of tumor samples of 71 patients with cutaneous melanoma detected NRAS mutations in 30% and BRAF mutations in 59% of all tumors tested. Of the BRAF mutation–positive tumors, 88% harbored the Val599Glu mutation, now commonly referred to as the BRAF V600E mutation. The same study demonstrated that the vast majority of BRAF mutations were seen in the primary tumor and were preserved when metastases were analyzed. Additionally, both NRAS and BRAF mutations were detected in the radial growth phase of the melanoma tumor. These findings indicate that alterations in the MAPK pathway occur early in the pathogenesis of advanced melanoma.11 Another group demonstrated that 66% of malignant melanoma tumor samples harbored BRAF mutations, of which 80% were specifically the V600E mutation. In vitro assays showed that the BRAF V600E–mutated kinase had greater than 10-fold kinase activity compared to wild-type BRAF, and that this kinase enhanced cellular proliferation even when upstream NRAS signaling was inhibited.12
The Cancer Genome Atlas Network performed a large analysis of tumor samples from 331 different melanoma patients and studied variations at the DNA, RNA, and protein levels. The study established a framework of 4 notable genomic subtypes, including mutant BRAF (52%), mutant RAS (28%), mutant NF1 (14%), and triple wild-type (6%). Additionally, mRNA transcriptomic analysis of overexpressed genes identified 3 different subclasses, which were labeled as “immune,” “keratin,” and “MITF-low.” The immune subclass was characterized by increased expression of proteins found in immune cells, immune signaling molecules, immune checkpoint proteins, cytokines, and chemokines, and correlated with increased lymphocyte invasion within the tumor. Interestingly, in the post-accession survival analysis, the “immune” transcriptomic subclass was statistically correlated with an improved prognosis.13 Having an understanding of the molecular pathogenesis of advanced melanoma helps to create a framework for understanding the mechanisms of current standard of care therapies for the disease.
Case Presentation
A 62-year-old Caucasian man with a history of well-controlled type 2 diabetes mellitus and hypertension is being followed by his dermatologist for surveillance of melanocytic nevi. On follow-up he is noted to have an asymmetrical melanocytic lesion over the right scalp with irregular borders and variegated color. He is asymptomatic and the remainder of physical examination is unremarkable, as he has no other concerning skin lesions and no cervical, axillary, or inguinal lymphadenopathy.
How is melanoma diagnosed?
Detailed discussion about diagnosis and staging will be deferred in this review of treatment of advanced melanoma. In brief, melanoma is best diagnosed by excisional biopsy and histopathology. Staging of melanoma is done according to the American Joint Committee on Cancer’s (AJCC) Cancer Staging Manual, 8th edition, using a TNM staging system that incorporates tumor thickness (Breslow depth); ulceration; number of involved regional lymph nodes; presence of in-transit, satellite, and/or microsatellite metastases; distant metastases; and serum lactate dehydrogenase level.14
Case Continued
The patient undergoes a wide excisional biopsy of the right scalp lesion, which is consistent with malignant melanoma. Pathology demonstrates a Breslow depth of 2.6 mm, 2 mitotic figures/mm2, and no evidence of ulceration. He subsequently undergoes wide local excision with 0/3 sentinel lymph nodes positive for malignancy. His final staging is consistent with pT3aN0M0, stage IIA melanoma.
He is seen in follow-up with medical oncology for the next 3.5 years without any evidence of disease recurrence. He then develops symptoms of vertigo, diplopia, and recurrent falls, prompting medical attention. Magnetic resonance imaging (MRI) brain reveals multiple supratentorial and infratentorial lesions concerning for intracranial metastases. Further imaging with computed tomography (CT) chest/abdomen/pelvis reveals a right lower lobe pulmonary mass with right hilar and subcarinal lymphadenopathy. He is admitted for treatment with intravenous dexamethasone and further evaluation with endobronchial ultrasound-guided fine-needle aspiration of the right lower lobe mass, which reveals metastatic melanoma. Given the extent of his intracranial metastases, he is treated with whole brain radiation therapy for symptomatic relief prior to initiating systemic therapy.
What is the general approach to first-line treatment for metastatic melanoma?
The past decade has brought an abundance of data supporting the use of immunotherapy with immune checkpoint inhibitors or molecularly targeted therapy with combined BRAF/MEK inhibitors in the first-line setting.4-8 After the diagnosis of metastatic melanoma has been made, molecular testing is recommended to determine the BRAF status of the tumor. Immunotherapy is the clear choice for first-line therapy in the absence of an activating BRAF V600 mutation. When a BRAF V600 mutation is present, current evidence supports the use of either immunotherapy or molecularly targeted therapy as first-line therapy.
To date, there have been no prospective clinical trials comparing the sequencing of immunotherapy and molecularly targeted therapy in the first-line setting. An ongoing clinical trial (NCT02224781) is comparing dabrafenib and trametinib followed by ipilimumab and nivolumab at time of progression to ipilimumab and nivolumab followed by dabrafenib and trametinib in patients with newly diagnosed stage III/IV BRAF V600 mutation–positive melanoma. The primary outcome measure is 2-year OS. Until completion of that trial, current practice regarding which type of therapy to use in the first-line setting is based on a number of factors including clinical characteristics and provider preferences.
Data suggest that immunotherapies can produce durable responses, especially after treatment completion or discontinuation, albeit at the expense of taking a longer time to achieve clinical benefit and the risk of potentially serious immune-related adverse effects. This idea of a durable, off-treatment response is highlighted by a study that followed 105 patients who had achieved a complete response (CR) and found that 24-month disease-free survival from the time of CR was 90.9% in all patients and 89.9% in the 67 patients who had discontinued pembrolizumab after attaining CR.15 BRAF/MEK inhibition has the potential for rapid clinical responses, though concerns exist about the development of resistance to therapy. The following sections explore the evidence supporting the use of these therapies.
Immunotherapy with Immune Checkpoint Inhibitors
Immunotherapy via immune checkpoint blockade has revolutionized the treatment of many solid tumors over the past decade. The promise of immunotherapy revolves around the potential for achieving a dynamic and durable systemic response against cancer by augmenting the antitumor effects of the immune system. T-cells are central to mounting a systemic antitumor response, and, in addition to antigen recognition, their function depends heavily on fine tuning between co-stimulatory and co-inhibitory signaling. The cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) expressed on T-cells was the first discovered co-inhibitory receptor of T-cell activation.16 Later, it was discovered that the programmed cell death 1 receptor (PD-1), expressed on T-cells, and its ligands PD-L1 and PD-L2, expressed on antigen presenting cells, tumor cells, or other cells in the tumor microenvironment, also served as a potent negative regulator of T-cell function.17
Together, these 2 signaling pathways help to maintain peripheral immune tolerance, whereby autoreactive T-cells that have escaped from the thymus are silenced to prevent autoimmunity. However, these pathways can also be utilized by cancer cells to escape immune surveillance. Monoclonal antibodies that inhibit the aforementioned co-inhibitory signaling pathways, and thus augment the immune response, have proven to be an effective anticancer therapy capable of producing profound and durable responses in certain malignancies.16,17
Ipilimumab
Ipilimumab is a monoclonal antibody that inhibits the function of the CTLA-4 co-inhibitory immune checkpoint. In a phase 3 randomized controlled trial of 676 patients with previously treated metastatic melanoma, ipilimumab at a dose of 3 mg/kg every 3 weeks for 4 cycles, with or without a gp100 peptide vaccine, resulted in an improved median OS of 10.0 and 10.1 months, respectively, compared to 6.4 months in those receiving the peptide vaccine alone, meeting the primary endpoint.4 Subsequently, a phase 3 trial of 502 patients with untreated metastatic melanoma compared ipilimumab at a dose of 10 mg/kg every 3 weeks for 4 cycles plus dacarbazine to dacarbazine plus placebo and found a significant increase in median OS (11.2 months vs 9.1 months), with no additive benefit of chemotherapy. There was a higher reported rate of grade 3 or 4 adverse events in this trial with ipilimumab dosed at 10 mg/kg, which was felt to be dose-related.18 These trials were the first to show improved OS with any systemic therapy in metastatic melanoma and led to US Food and Drug Administration approval of ipilimumab for this indication in 2011.
PD-1 Inhibitor Monotherapy
The PD-1 inhibitors nivolumab and pembrolizumab were initially approved for metastatic melanoma after progression on ipilimumab. In the phase 1 trial of patients with previously treated metastatic melanoma, nivolumab therapy resulted in an ORR of 28%.19 The subsequent phase 2 trial conducted in pretreated patients, including patients who had progressed on ipilimumab, confirmed a similar ORR of 31%, as well as a median PFS of 3.7 months and a median OS of 16.8 months. The estimated response duration in patients who did achieve a response to therapy was 2 years.20 A phase 3 trial (CheckMate 037) comparing nivolumab (n = 120) to investigator’s choice chemotherapy (n = 47) in those with melanoma refractory to ipilimumab demonstrated that nivolumab was superior for the primary endpoint of ORR (31.7% vs 10.6%), had less toxicity (5% rate of grade 3 or 4 adverse events versus 9%), and increased median duration of response (32 months vs 13 months).21
The phase 1 trial (KEYNOTE-001) testing the efficacy of pembrolizumab demonstrated an ORR of 33% in the total population of patients treated and an ORR of 45% in those who were treatment-naive. Additionally, the median OS was 23 months for the total population and 31 months for treatment-naive patients, with only 14% of patients experiencing a grade 3 or 4 adverse event.22 The KEYNOTE-002 phase 2 trial compared 2 different pembrolizumab doses (2 mg/kg and 10 mg/kg every 3 weeks) to investigator’s choice chemotherapy (paclitaxel plus carboplatin, paclitaxel, carboplatin, dacarbazine, or oral temozolomide) in 540 patients with advanced melanoma with documented progression on ipilimumab with or without prior progression on molecularly targeted therapy if positive for a BRAF V600 mutation. The final analysis demonstrated significantly improved ORR with pembrolizumab (22% at 2 mg/kg vs 26% at 10 mg/kg vs 4% chemotherapy) and significantly improved 24-month PFS (16% vs 22% vs 0.6%, respectively). There was a nonstatistically significant improvement in median OS (13.4 months vs 14.7 months vs 10 months), although 55% of the patients initially assigned to the chemotherapy arm crossed over and received pembrolizumab after documentation of progressive disease.23,24
Because PD-1 inhibition improved efficacy with less toxicity than chemotherapy when studied in progressive disease, subsequent studies focused on PD-1 inhibition in the frontline setting. CheckMate 066 was a phase 3 trial comparing nivolumab to dacarbazine as first-line therapy for 418 patients with untreated metastatic melanoma who did not have a BRAF mutation. For the primary end point of 1-year OS, nivolumab was superior to dacarbazine (72.9% vs 42.1%; hazard ratio [HR], 0.42; P < 0.001). Treatment with nivolumab also resulted in superior ORR (40% vs 14%) and PFS (5.1 months vs 2.2 months). Additionally, nivolumab therapy had a lower rate of grade 3 or 4 toxicity compared to dacarbazine (11.7% vs 17.6%).25
The KEYNOTE-006 trial compared 2 separate dosing schedules of pembrolizumab (10 mg/kg every 2 weeks versus every 3 weeks) to ipilimumab (3 mg/kg every 3 weeks for 4 cycles) in a 1:1:1 ratio in 834 patients with metastatic melanoma who had received up to 1 prior systemic therapy, but no prior CTLA-4 or PD-1 inhibitors. The first published data reported statistically significant outcomes for the co-primary end points of 6-month PFS (47.3% for pembrolizumab every 2 weeks vs 46.4% for pembrolizumab every 3 weeks vs 26.5% for ipilimumab; HR, 0.58 for both pembrolizumab groups compared to ipilimumab; P < 0.001) and 12-month OS (74.1% vs 68.4% vs 58.2%) with pembrolizumab compared to ipilimumab. Compared to ipilimumab, pembrolizumab every 2 weeks had a hazard ratio of 0.63 (P = 0.0005) and pembrolizumab every 3 weeks had a hazard ratio of 0.69 (P = 0.0036). The pembrolizumab groups was also had lower rates of grade 3 to 5 toxicity (13.3% vs 10.1% vs 19.9%).5 Updated outcomes demonstrated improved ORR compared to the first analysis (37% vs 36% vs 13%), and improved OS (median OS, not reached for the pembrolizumab groups vs 16.0 months for the ipilimumab group; HR, 0.68, P = 0.0009 for pembrolizumab every 2 weeks versus HR 0.68, P = 0.0008 for pembrolizumab every 3 weeks).26 In addition, 24-month OS was 55% in both pembrolizumab groups compared to 43% in the ipilimumab group. Grade 3 or 4 toxicity occurred less frequently with pembrolizumab (17% vs 17% vs 20%).
Further analysis from the KEYNOTE-006 trial data demonstrated improved ORR, PFS, and OS with pembrolizumab compared to ipilimumab in tumors positive for PD-L1 expression. For PD-L1-negative tumors, response rate was higher, and PFS and OS rates were similar with pembrolizumab compared to ipilimumab. Given that pembrolizumab was associated with similar survival outcomes in PD-L1-negative tumors and with less toxicity than ipilimumab, the superiority of PD-L1 inhibitors over ipilimumab was further supported, regardless of tumor PD-L1 status.27
In sum, PD-1 inhibition should be considered the first-line immunotherapy in advanced melanoma, either alone or in combination with ipilimumab, as discussed in the following section. There is no longer a role for ipilimumab monotherapy in the first-line setting, based on evidence from direct comparison to single-agent PD-1 inhibition in clinical trials that demonstrated superior efficacy and less serious toxicity with PD-1 inhibitors.5,26 The finding that ORR and OS outcomes with single-agent PD-1 inhibitors are higher in treatment-naive patients compared to those receiving prior therapies also supports this approach.22
Combination CTLA-4 and PD-1 Therapy
Despite the potential for durable responses, the majority of patients fail to respond to single-agent PD-1 therapy. Given that preclinical data had suggested the potential for synergy between dual inhibition of CTLA-4 and PD-1, clinical trials were designed to test this approach. The first randomized phase 2 trial that established superior efficacy with combination therapy was the CheckMate 069 trial comparing nivolumab plus ipilimumab to ipilimumab monotherapy. Combination therapy resulted in increased ORR (59% vs 11%), median PFS (not reached vs 3.0 months), 2-year PFS (51.3% vs 12.0%), and 2-year OS (63.8% vs 53.6%).28 Similarly, a phase 1b trial of pembrolizumab plus reduced-dose ipilimumab demonstrated an ORR of 61%, with a 1-year PFS of 69% and 1-year OS of 89%.29
The landmark phase 3 CheckMate 067 trial analyzed efficacy outcomes for 3 different treatment regimens including nivolumab plus ipilimumab, nivolumab monotherapy, and ipilimumab monotherapy in previously untreated patients with unresectable stage III or IV melanoma. The trial was powered to compare survival outcomes for both the combination therapy arm against ipilimumab and the nivolumab monotherapy arm against ipilimumab, but not to compare combination therapy to nivolumab monotherapy. The initial analysis demonstrated a median PFS of 11.5 months with combination therapy versus 6.9 months with nivolumab and 2.9 months with ipilimumab, as well as an ORR of 58% versus 44% and 19%, respectively (Table 1).6 The updated 3-year survival outcomes from CheckMate 067 were notable for superior median OS with combination therapy (not reached in combination vs 37.6 months for nivolumab vs 19.9 months ipilimumab), improved 3-year OS (58% vs 52% vs 34%), and improved 3-year PFS (39% vs 32% vs 10%).7 In the reported 4-year survival outcomes, median OS was not reached in the combination therapy group, and was 36.9 months in the nivolumab monotherapy group and 19.9 months in the ipilimumab monotherapy group. Rates of grade 3 or 4 adverse events were significantly higher in the combination therapy group, at 59% compared to 22% with nivolumab monotherapy and 28% with ipilimumab alone.30 The 3- and 4-year OS outcomes (58% and 54%, respectively) with combination therapy were the highest seen in any phase 3 trial for treatment of advanced melanoma, supporting its use as the best approved first-line therapy in those who can tolerate the potential toxicity of combination therapy7,30 The conclusions from this landmark trial were that both combination therapy and nivolumab monotherapy resulted in statistically significant improvement in OS compared to ipilimumab.
Toxicity Associated with Immune Checkpoint Inhibitors
While immune checkpoint inhibitors have revolutionized the treatment of many solid tumor malignancies, this new class of cancer therapy has brought about a new type of toxicity for clinicians to be aware of, termed immune-related adverse events (irAEs). As immune checkpoint inhibitors amplify the immune response against malignancy, they also increase the likelihood that autoreactive T-cells persist and proliferate within the circulation. Therefore, these therapies can result in almost any type of autoimmune side effect. The most commonly reported irAEs in large clinical trials studying CTLA-4 and PD-1 inhibitors include rash/pruritus, diarrhea/colitis, hepatitis, endocrinopathies (thyroiditis, hypophysitis, adrenalitis), and pneumonitis. Other more rare toxicities include pancreatitis, autoimmune hematologic toxicities, cardiac toxicity (myocarditis, heart failure), and neurologic toxicities (neuropathies, myasthenia gravis-like syndrome, Guillain-Barré syndrome). It has been observed that PD-1 inhibitors have a lower incidence of irAEs than CTLA-4 inhibitors, and that the combined use of PD-1 and CTLA-4 inhibitors is associated with a greater incidence of irAEs compared to monotherapy with either agent.31 Toxicities associated with ipilimumab have been noted to be dose dependent.18 Generally, these toxicities are treated with immunosuppression in the form of glucocorticoids and are often reversible.31 There are several published guidelines that include algorithms for the management of irAEs by organizations such as the National Comprehensive Cancer Network.32
For example, previously untreated patients treated with ipilimumab plus dacarbazine as compared to dacarbazine plus placebo had greater grade 3 or 4 adverse events (56.3% vs 27.5%), and 77.7% of patients experiencing an irAE of any grade.18 In the CheckMate 066 trial comparing frontline nivolumab to dacarbazine, nivolumab had a lower rate of grade 3 or 4 toxicity (11.7% vs 17.6%) and irAEs were relatively infrequent, with diarrhea and elevated alanine aminotransferase level each being the most prominent irAE (affecting 1.0% of patients).25 In the KEYNOTE-006 trial, irAEs seen in more than 1% of patients treated with pembrolizumab included colitis, hepatitis, hypothyroidism, and hyperthyroidism, whereas those occurring in more than 1% of patients treated with ipilimumab included colitis and hypophysitis. Overall, there were lower rates of grade 3 to 5 toxicity with the 2 pembrolizumab doses compared to ipilimumab (13.3% pembrolizumab every 2 weeks vs 10.1% pembrolizumab every 3 weeks vs 19.9% ipilimumab).5 In the CheckMate 067 trial comparing nivolumab plus ipilimumab, nivolumab monotherapy, and ipilimumab monotherapy, rates of treatment-related adverse events of any grade were higher in the combination group (96% combination vs 86% nivolumab vs 86% ipilimumab), as were rates of grade 3 or 4 adverse events (59% vs 21% vs 28%, respectively). The irAE profile was similar to that demonstrated in prior studies: rash/pruritus were the most common, and diarrhea/colitis, elevated aminotransferases, and endocrinopathies were among the more common irAEs.7
Alternative dosing strategies have been investigated in an effort to preserve efficacy and minimize toxicity. A phase 1b trial of pembrolizumab plus reduced-dose ipilimumab demonstrated an ORR of 61%, with a 1-year PFS of 69% and a 1-year OS of 89%. This combination led to 45% of patients having a grade 3 or 4 adverse event, 60% having irAEs of any grade, and only 27% having grade 3 or 4 irAEs.29 The CheckMate 067 trial studied the combination of nivolumab 1 mg/kg plus ipilimumab 3 mg/kg.6 The CheckMate 511 trial compared different combination dosing strategies (nivolumab 3 mg/kg + ipilimumab 1 mg/kg versus nivolumab 1 mg/kg + ipilimumab 3 mg/kg) to assess for safety benefit. In the results published in abstract form, the reduced ipilimumab dose (nivolumab 3 mg/kg + ipilimumab 1 mg/kg arm) resulted in significantly decreased grade 3 to 5 adverse events (33.9% vs 48.3%) without significant differences in ORR, PFS, or OS.33
The question about the efficacy of checkpoint inhibitors in patients who discontinue treatment due to irAEs has been raised, as one hypothesis suggests that such toxicities may also indicate that the antitumor immune response has been activated. In a retrospective pooled analysis of phase 2 and 3 trials where patients received combination therapy with ipilimumab and nivolumab and discontinued therapy during the induction phase due to irAEs, outcomes did not appear to be inferior. Median PFS was 8.4 months in those who discontinued therapy compared to 10.8 months in those who continued therapy, but this did not reach statistical significance. Median OS had not been reached in either group and ORR was actually higher in those who discontinued due to adverse events (58.3% vs 50.2%). While this retrospective analysis needs to be validated, it does suggest that patients likely derive antitumor benefit from immunotherapy even if they have to discontinue therapy due to irAEs. Of note, patients in this analysis were not trialed on nivolumab monotherapy after receiving immunosuppressive treatment for toxicity related to combination therapy, which has since been deemed a reasonable treatment option.34
Molecularly Targeted Therapy for Metastatic Melanoma
As previously mentioned, the MAPK pathway is frequently altered in metastatic melanoma and thus serves as a target for therapy. Mutations in BRAF can cause constitutive activation of the protein’s kinase function, which subsequently phosphorylates/activates MEK in the absence of extracellular growth signals and causes increased cellular proliferation. For the roughly half of patients diagnosed with metastatic melanoma who harbor a BRAF V600 mutation, molecularly targeted therapy with BRAF/MEK inhibitors has emerged as a standard of care treatment option. As such, all patients with advanced disease should be tested for BRAF mutations.
After early phase 1 studies of the BRAF inhibitor vemurafenib demonstrated successful inhibition of mutated BRAF,35 subsequent studies confirmed the benefit of BRAF targeted therapy. In the phase 3 randomized controlled BRIM-3 trial comparing vemurafenib with dacarbazine for treatment of 675 patients with previously untreated metastatic melanoma positive for a BRAF V600E mutation, the vemurafenib group had superior ORR and 6-month OS during the first analysis.36 In a subsequent analysis, median PFS and median OS were also superior with vemurafenib compared to dacarbazine, as vemurafenib had a median OS of 13.6 months compared to 9.7 months with dacarbazine (HR, 0.70; P = 0.0008).37 Dabrafenib was the next BRAF inhibitor to demonstrate clinical efficacy with superior PFS compared to dacarbazine.38
Despite tumor shrinkage in the majority of patients, the development of resistance to therapy was an issue early on. The development of acquired resistance emerged as a heterogeneous process, though many of the identified resistance mechanisms involved reactivation of the MAPK pathway.39 A phase 3 trial of 322 patients with metastatic melanoma comparing the MEK inhibitor trametinib as monotherapy against chemotherapy demonstrated a modest improvement in both median PFS and OS.40 As a result, subsequent efforts focused on a strategy of concurrent MEK inhibition as a means to overcome resistance to molecularly targeted monotherapy
At least 4 large phase 3 randomized controlled trials of combination therapy with BRAF plus MEK inhibitors showed an improved ORR, PFS, and OS when compared to BRAF inhibition alone. The COMBI-d trial comparing dabrafenib plus trametinib versus dabrafenib alone was the first to demonstrate the superiority of combined BRAF/MEK inhibition and made combination therapy the current standard of care for patients with metastatic melanoma and a BRAF V600 mutation. In the final analysis of this trial, 3-year PFS was 22% with combination therapy compared to 12% with dabrafenib alone, and 3-year OS was 44% compared to 32%.8,41,42 A second trial with the combination of dabrafenib and trametinib (COMBI-V) also demonstrated superior efficacy when compared to single-agent vemurafenib without increased toxicity.43 Subsequently, the combination of vemurafenib with MEK inhibitor cobimetinib demonstrated superiority compared to vemurafenib alone,44 followed by the newest combination encorafenib (BRAF inhibitor) and binimetinib (MEK inhibitor) proving superior to either vemurafenib or encorafenib alone.45,46
It is important to note that there have been no studies directly comparing the efficacy of the 3 approved BRAF/MEK inhibitor combinations, but the 3 different regimens have some differences in their toxicity profiles (Table 2). Of note, single-agent BRAF inhibition was associated with increased cutaneous toxicity, including secondary squamous cell carcinoma and keratoacanthoma,47 which was demonstrated to be driven by paradoxical activation of the MAPK pathway.48 The concerning cutaneous toxicities such as squamous cell carcinoma were substantially reduced by combination BRAF/MEK inhibitor therapy.47 Collectively, the higher efficacy along with manageable toxicity profile established combination BRAF/MEK inhibition as the preferred regimen for patients with BRAF-mutated metastatic melanoma who are being considered for molecularly targeted therapy. BRAF inhibitor monotherapy should only be used when there is a specific concern regarding the use of a MEK inhibitor in certain clinical circumstances.
Other driver mutations associated with metastatic melanoma such as NRAS-mutated tumors have proven more difficult to effectively treat with molecularly targeted therapy, with one study showing that the MEK inhibitor binimetinib resulted in a modest improvement in ORR and median PFS without OS benefit compared to dacarbazine.49 Several phase 2 trials involving metastatic melanoma harboring a c-Kit alteration have demonstrated some efficacy with the tyrosine kinase inhibitor imatinib. The largest phase 2 trial of 43 patients treated with imatinib resulted in a 53.5% disease control rate (23.3% partial response and 30.2% stable disease), with 9 of the 10 patients who achieved partial response having a mutation in either exon 11 or 13. Median PFS was 3.5 months and 1-year OS was 51.0%.50
Case Conclusion
Prior to initiation of systemic therapy, the patient’s melanoma is tested and is found to be positive for a BRAF V600K mutation. At his follow-up appointment, the patient continues to endorse generalized weakness, fatigue, issues with balance, and residual pulmonary symptoms after being treated for post-obstructive pneumonia. Given his current symptoms and extent of metastatic disease, immunotherapy is deferred and he is started on combination molecularly targeted therapy with dabrafenib and trametinib. He initially does well, with a partial response noted by resolution of symptoms and decreased size of his intracranial metastases and decreased size of the right lower lobe mass. Further follow-up of this patient is presented in the second article in this 2-part review of advanced melanoma.
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2018. CA Cancer J Clin. 2018;68:7-30.
2. Ives NJ, Stowe RL, Lorigan P, Wheatley K. Chemotherapy compared with biochemotherapy for the treatment of metastatic melanoma: a meta-analysis of 18 trials involving 2621 patients. J Clin Oncol. 2007;25:5426-34.
3. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105-16.
4. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-23.
5. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2522-2532.
6. Larkin J, Chiarion-Sileni V, Gonazalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34.
7. Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377:1345-1356.
8. Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
9. Elwood JM, Jopson J. Melanoma and sun exposure: an overview of published studies. Int J Cancer. 1997;73:198-203.
10. Gilchrest BA, Eller MS, Geller AC, Yaar M. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 199;340:1341-1348.
11. Omholt K, Platz A, Kanter L, et al. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9:6483-8.
12. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-54.
13. Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681-96.
14. Gershenwald JE, Scolyer RA, Hess KR, et al. Melanoma staging: evidence-based changes in the American Joint Committee on Cancer Eighth Edition Cancer Staging Manual. CA Cancer J Clin. 2017;67:472-492.
15. Robert C, Ribas A, Hamid O, et al. Durable complete response after discontinuation of pembrolizumab in patients with metastatic melanoma. J Clin Oncol. 2018;36:1668-1674.
16. Salama AKS, Hodi FS. Cytotoxic T-lymphocyte-associated antigen-4. Clin Cancer Res. 2011;17:4622-8.
17. Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375:1767-1778.
18. Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517-2526.
19. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-2454.
20. Topalian S, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020-30.
21. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-84.
22. Ribas A, Hamid O, Daud A, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. 2016;315:1600-1609.
23. Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908-18.
24. Hamid O, Puzanov I, Dummer R, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer. 2017;86:37-45.
25. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-330.
26. Schachter J, Ribas A, Long GV, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicenter, randomised, open-label phase 3 study (KEYNOTE-006). Lancet Oncol. 2017;390:1853-1862.
27. Carlino MS, Long GV, Schadendorf D, et al. Outcomes by line of therapy and programmed death ligand 1 expression in patients with advanced melanoma treated with pembrolizumab or ipilimumab in KEYNOTE-006. A randomised clinical trial. Eur J Cancer. 2018;101:236-243.
28. Hodi FS, Chesney J, Pavlick AC, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2016;17:1558-1568.
29. Long GV, Atkinson V, Cebon JS, et al. Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): an open-label, phase 1b trial. Lancet Oncol. 2017;18:1202-10.
30. Hodi FS, Chiarion-Sileni V, Gonzalez R, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018;19:1480-1492.
31. Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the immune-related adverse effects of immune checkpoint inhibitors: a review. JAMA Oncol. 2016;2:1346-1353.
32. National Comprehensive Cancer Network. Management of immunotherapy-related toxicities (version 2.2019). www.nccn.org/professionals/physician_gls/pdf/immunotherapy.pdf. Accessed April 8, 2019.
33. Lebbé C, Meyer N, Mortier L, et al. Initial results from a phase IIIb/IV study evaluating two dosing regimens of nivolumab (NIVO) in combination with ipilimumab (IPI) in patients with advanced melanoma (CheckMate 511) [Abstract LBA47]. Ann Oncol. 2018;29:mdy424.057.
34. Schadendorf D, Wolchok JD, Hodi FS, et al. Efficacy and safety outcomes in patients with advanced melanoma who discontinued treatment with nivolumab and ipilimumab because of adverse events: a pooled analysis of randomized phase ii and iii trials. J Clin Oncol. 2017;35:3807-3814.
35. Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
36. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
37. McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): extended follow up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
38. Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicenter, open-label, phase 3 randomised controlled trial. Lancet Oncol. 2012;380:358-365.
39. Rizos H, Menzies AM, Pupo GM, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20:1965-1977.
40. Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107-114.
41. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicenter, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2015;386:444-451.
42. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28:1631-1639.
43. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
44. Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248-260.
45. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicenter, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:603-615.
46. Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19:1315-1327.
47. Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA. Dermatol 2015;151:1103-1109.
48. Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
49. Dummer R, Schadendorf D, Ascierto P, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18:435-445.
50. Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29:2904-2909.
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2018. CA Cancer J Clin. 2018;68:7-30.
2. Ives NJ, Stowe RL, Lorigan P, Wheatley K. Chemotherapy compared with biochemotherapy for the treatment of metastatic melanoma: a meta-analysis of 18 trials involving 2621 patients. J Clin Oncol. 2007;25:5426-34.
3. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105-16.
4. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-23.
5. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2522-2532.
6. Larkin J, Chiarion-Sileni V, Gonazalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34.
7. Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377:1345-1356.
8. Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
9. Elwood JM, Jopson J. Melanoma and sun exposure: an overview of published studies. Int J Cancer. 1997;73:198-203.
10. Gilchrest BA, Eller MS, Geller AC, Yaar M. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 199;340:1341-1348.
11. Omholt K, Platz A, Kanter L, et al. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9:6483-8.
12. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-54.
13. Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681-96.
14. Gershenwald JE, Scolyer RA, Hess KR, et al. Melanoma staging: evidence-based changes in the American Joint Committee on Cancer Eighth Edition Cancer Staging Manual. CA Cancer J Clin. 2017;67:472-492.
15. Robert C, Ribas A, Hamid O, et al. Durable complete response after discontinuation of pembrolizumab in patients with metastatic melanoma. J Clin Oncol. 2018;36:1668-1674.
16. Salama AKS, Hodi FS. Cytotoxic T-lymphocyte-associated antigen-4. Clin Cancer Res. 2011;17:4622-8.
17. Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375:1767-1778.
18. Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517-2526.
19. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-2454.
20. Topalian S, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020-30.
21. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-84.
22. Ribas A, Hamid O, Daud A, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. 2016;315:1600-1609.
23. Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908-18.
24. Hamid O, Puzanov I, Dummer R, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer. 2017;86:37-45.
25. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-330.
26. Schachter J, Ribas A, Long GV, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicenter, randomised, open-label phase 3 study (KEYNOTE-006). Lancet Oncol. 2017;390:1853-1862.
27. Carlino MS, Long GV, Schadendorf D, et al. Outcomes by line of therapy and programmed death ligand 1 expression in patients with advanced melanoma treated with pembrolizumab or ipilimumab in KEYNOTE-006. A randomised clinical trial. Eur J Cancer. 2018;101:236-243.
28. Hodi FS, Chesney J, Pavlick AC, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2016;17:1558-1568.
29. Long GV, Atkinson V, Cebon JS, et al. Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): an open-label, phase 1b trial. Lancet Oncol. 2017;18:1202-10.
30. Hodi FS, Chiarion-Sileni V, Gonzalez R, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018;19:1480-1492.
31. Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the immune-related adverse effects of immune checkpoint inhibitors: a review. JAMA Oncol. 2016;2:1346-1353.
32. National Comprehensive Cancer Network. Management of immunotherapy-related toxicities (version 2.2019). www.nccn.org/professionals/physician_gls/pdf/immunotherapy.pdf. Accessed April 8, 2019.
33. Lebbé C, Meyer N, Mortier L, et al. Initial results from a phase IIIb/IV study evaluating two dosing regimens of nivolumab (NIVO) in combination with ipilimumab (IPI) in patients with advanced melanoma (CheckMate 511) [Abstract LBA47]. Ann Oncol. 2018;29:mdy424.057.
34. Schadendorf D, Wolchok JD, Hodi FS, et al. Efficacy and safety outcomes in patients with advanced melanoma who discontinued treatment with nivolumab and ipilimumab because of adverse events: a pooled analysis of randomized phase ii and iii trials. J Clin Oncol. 2017;35:3807-3814.
35. Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
36. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
37. McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): extended follow up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
38. Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicenter, open-label, phase 3 randomised controlled trial. Lancet Oncol. 2012;380:358-365.
39. Rizos H, Menzies AM, Pupo GM, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20:1965-1977.
40. Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107-114.
41. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicenter, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2015;386:444-451.
42. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28:1631-1639.
43. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
44. Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248-260.
45. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicenter, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:603-615.
46. Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19:1315-1327.
47. Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA. Dermatol 2015;151:1103-1109.
48. Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
49. Dummer R, Schadendorf D, Ascierto P, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18:435-445.
50. Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29:2904-2909.
Management of Late Pulmonary Complications After Hematopoietic Stem Cell Transplantation
Hematopoietic stem cell transplantation (HSCT) is increasingly being used to treat hematologic malignancies as well as nonmalignant diseases and solid tumors. Over the past 2 decades overall survival following transplant and transplant-related mortality have improved.1 With this increased survival, there is a need to focus on late complications after transplantation. Pulmonary complications are a common but sometimes underrecognized cause of late morbidity and mortality in HSCT patients. This article, the second of 2 articles on post-HSCT pulmonary complications, reviews late-onset complications, with a focus on the evaluation and treatment of bronchiolitis obliterans syndrome (BOS), one of the most common and serious late pulmonary complications in HSCT patients. The first article reviewed the management of early-onset pulmonary complications and included a basic overview of stem cell transplantation, discussion of factors associated with pulmonary complications, and a review of methods for assessing pretransplant risk for pulmonary complications in patients undergoing HSCT.2
Case Presentation
A 40-year-old white woman with a history of acute myeloid leukemia status post peripheral blood stem cell transplant presents with dyspnea on exertion, which she states started about 1 month ago and now is limiting her with even 1 flight of stairs. She also complains of mild dry cough and a 4- to 5-lb weight loss over the past 1 to 2 months. She has an occasional runny nose, but denies gastroesophageal reflux, fevers, chills, or night sweats. She has a history of matched related sibling donor transplant with busulfan and cyclophosphamide conditioning 1 year prior to presentation. She has had significant graft-versus-host disease (GVHD), affecting the liver, gastrointestinal tract, skin, and eyes.
On physical examination, heart rate is 110 beats/min, respiratory rate is 16 breaths/min, blood pressure is 92/58 mm Hg, and the patient is afebrile. Eye exam reveals scleral injection, mouth shows dry mucous membranes with a few white plaques, and the skin has chronic changes with a rash over both arms. Cardiac exam reveals tachycardia but regular rhythm and there are no murmurs, rubs, or gallops. Lungs are clear bilaterally and abdomen shows no organomegaly.
Laboratory exam shows a white blood cell count of 7800 cells/μL, hemoglobin level of 12.4 g/dL, and platelet count of 186 × 103/μL. Liver enzymes are mildly elevated. Chest radiograph shows clear lung fields bilaterally.
- What is the differential in this patient with dyspnea 1 year after transplantation?
Late pulmonary complications are generally accepted as those occurring more than 100 days post transplant. This period of time is characterized by chronic GVHD and impaired cellular and humoral immunity. Results of longitudinal studies of infections in adult HSCT patients suggest that special attention should be paid to allogeneic HSCT recipients for post-engraftment infectious pulmonary complications.3 Encapsulated bacteria such as Haemophilus influenzae and Streptococcus pneumoniae are the most frequent bacterial organisms causing late infectious pulmonary complications. Nontuberculous mycobacteria and Nocardia should also be considered. Depending upon geographic location, social and occupational risk factors, and prevalence, tuberculosis should also enter the differential.
There are many noninfectious late-onset pulmonary complications after HSCT. Unfortunately, the literature has divided pulmonary complications after HSCT using a range of criteria and classifications based upon timing, predominant pulmonary function test (PFT) findings, and etiology. These include early versus late, obstructive versus restrictive, and infectious versus noninfectious, which makes a comprehensive literature review of late pulmonary complications difficult. The most common noninfectious late-onset complications are bronchiolitis obliterans, cryptogenic organizing pneumonia (previously referred to as bronchiolitis obliterans organizing pneumonia, or BOOP), and interstitial pneumonia. Other rarely reported complications include eosinophilic pneumonia, pulmonary alveolar proteinosis, air leak syndrome, and pulmonary hypertension.
Case Continued
Because the patient does not have symptoms of infection, PFTs are obtained. Pretransplant PFTs and current PFTs are shown in Table 1. 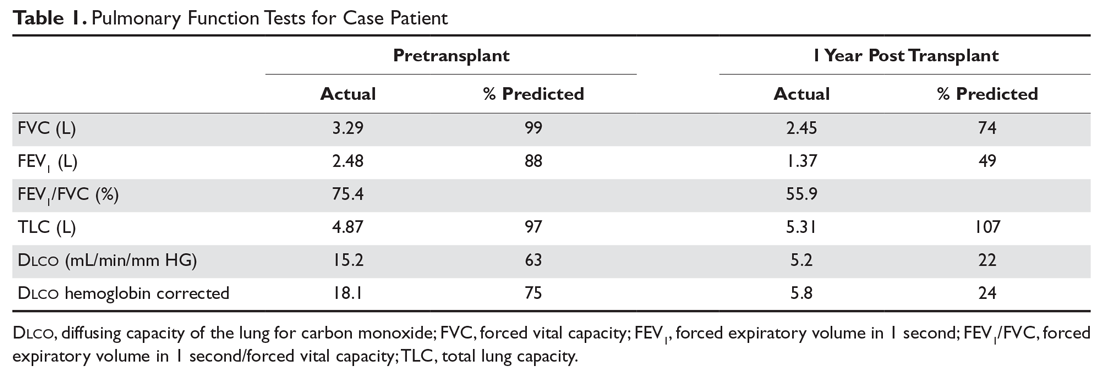
- What is the diagnosis in this case?
Bronchiolitis Obliterans
BOS is one of the most common and most serious late-onset pulmonary diseases after allogeneic transplantation. It is considered the pulmonary form of chronic GVHD. BOS was first described in 1982 in patients with chronic GVHD after bone marrow transplantation.4 Many differing definitions of bronchiolitis obliterans have been described in the literature. A recent review of the topic cites 10 different published sets of criteria for the diagnosis of bronchiolitis obliterans.5 Traditionally, bronchiolitis obliterans was thought to occur in 2% to 8% of patients undergoing allogeneic HSCT, but these findings were from older studies that used a diagnosis based on very specific pathology findings. When more liberal diagnostic criteria are used, the incidence may be as high as 26% of allogeneic HSCT patients.6
Bronchiolitis obliterans is a progressive lung disease characterized by narrowing of the terminal airways and obliteration of the terminal bronchi. Pathology may show constrictive bronchiolitis but can also show lymphocytic bronchiolitis, which may be associated with a better outcome.7 As noted, bronchiolitis obliterans has traditionally been considered a pathologic diagnosis. Current diagnostic criteria have evolved based upon the difficulty in obtaining this diagnosis through transbronchial biopsy given the patchy nature of the disease.8 The gold standard of open lung biopsy is seldom pursued in the post-HSCT population as the procedure continues to carry a worrisome risk-benefit profile.
The 2005 National Institutes of Health (NIH) consensus development project on criteria for clinical trials in chronic GVHD developed a clinical strategy for diagnosing BOS using the following criteria: absence of active infection, decreased forced expiratory volume in 1 second (FEV1) < 75%, FEV1/forced vital capacity (FVC) ratio of < 70%, and evidence of air trapping on high-resolution computed tomography (HRCT) or PFTs (residual volume > 120%). These diagnostic criteria were applied to a small series of patients with clinically identified bronchiolitis obliterans or biopsy-proven bronchiolitis obliterans. Only 18% of these patients met the requirements for the NIH consensus definition.5 A 2011 study that applied the NIH criteria found an overall prevalence of 5.5% among all transplant recipients but a prevalence of 14% in patients with GVHD.9 In 2014, the NIH consensus development group updated their recommendations. The new criteria for diagnosis of BOS require the presence of airflow obstruction (FEV1/FVC < 70% or 5th percentile of predicted), FEV1 < 75% predicted with a ≥ 10% decline in fewer than 2 years, absence of infection, and presence of air trapping (by expiratory computed tomography [CT] scan or PFT with residual volume >120% predicted) (Table 2). 
Some issues must be considered when determining airflow obstruction. The 2005 NIH working group recommends using Crapo as the reference set,11 but the National Health and Nutrition Examination Survey (NHANES) III reference values are the preferred reference set at this time12 and should be used in the United States. A recent article showed that the NHANES values were superior to older reference sets (however, they did not use Crapo as the comparison), although this study used the lower limit of normal as compared with the fixed 70% ratio.13 The 2014 NIH consensus group does not recommend a specific reference set and recognizes an FEV1/FVC ratio of 70% or less than the lower limit of normal as the cutoff value for airflow obstruction.10
Another issue in PFT interpretation is the finding of a decrease in FEV1 and FVC and normal total lung capacity, which is termed a nonspecific pattern. This pattern has been reported to occur in 9% of all PFTs and usually is associated with obstructive lung disease or obesity.14 A 2013 study described the nonspecific pattern as a BOS subgroup occurring in up to 31% of bronchiolitis obliterans patients.15
- What are the radiographic findings of BOS?
Chest radiograph is often normal in BOS. As discussed, air trapping can be documented using HRCT, according to the NIH clinical definition of bronchiolitis obliterans.16 A study that explored findings and trends seen on HRCT in HSCT patients with BOS found that the syndrome in these patients is characterized by central airway dilatation.17 Expiratory airway trapping on HRCT is the main finding, and this is best demonstrated on HRCT during inspiratory and expiratory phases.18 Other findings are bronchial wall thickening, parenchymal hypoattenuation, bronchiectasis, and centrilobular nodules.19
Galbán and colleagues developed a new technique called parametric response mapping that uses CT scanners to quantify normal parenchyma, functional small airway disease, emphysema, and parenchymal disease as relative lung volumes.20 This technique can detect airflow obstruction and small airway disease and was found to be a good method for detecting BOS after HSCT. In their study of parametric response mapping, the authors found that functional small airway disease affecting 28% or more of the total lung was highly indicative of bronchiolitis obliterans.20
- What therapies are used to treat BOS?
Traditionally, BOS has been treated with systemic immunosuppression. The recommended treatment had been systemic steroids at approximately 1 mg/ kg. However, it is increasingly recognized that BOS responds poorly to systemic steroids, and systemic steroids may actually be harmful and associated with increased mortality.15,21 The chronic GVHD recommendations from 2005 recommend ancillary therapy with inhaled corticosteroids and pulmonary rehabilitation.11 The updated 2011 German consensus statement lays out a clear management strategy for mild and moderate-severe disease with monitoring recommendations.22 The 2014 NIH chronic GVHD working group recommends fluticasone, azithromycin, and montelukast (ie, the FAM protocol) for treating BOS.23 FAM therapy in BOS may help lower the systemic steroid dose.24,25 Montelukast is not considered a treatment mainstay for BOS after lung transplant, but there is a study showing possible benefit in chronic GVHD.26 An evaluation of the natural history of a cohort of BOS patients treated with FAM therapy showed a rapid decline of FEV1 in the 6 months prior to diagnosis and treatment of BOS and subsequent stabilization following diagnosis and treatment.27 The benefit of high-dose inhaled corticosteroids or the combination of inhaled corticosteroids and long-acting beta-agonists has been demonstrated in small studies, which showed that these agents stabilized FEV1 and avoided the untoward side effects of systemic corticosteroids.28–30
Macrolide antibiotics have been explored as a treatment for BOS post HSCT because pilot studies suggested that azithromycin improved or stabilized FEV1 in patients with BOS after lung transplant or HSCT.31–33 Other studies of azithromycin have not shown benefit in the HSCT population after 3 months of therapy.34 A recent meta-analysis could neither support or refute the benefit of azithromycin for BOS after HSCT.35 In the lung transplant population, a study showed that patients who were started on azithromycin after transplant and continued on it 3 times a week had improved FEV1; these patients also had a reduced rate of BOS and improved overall and BOS-free survival 2 years after transplant.36 However, these benefits of azithromycin have not been observed in patients after HSCT. In fact, the ALLOZITHRO trial was stopped early because prophylactic azithromycin started at the time of the conditioning regimen with HSCT was associated with increased hematologic disease relapse, a decrease in airflow-decline-free survival, and reduced 2-year survival.30
Azithromycin is believed to exert an effect by its anti-inflammatory properties and perhaps by decreasing lung neutrophilia (it may be most beneficial in the subset of patients with high neutrophilia on bronchoalveolar lavage [BAL]).30 Adverse effects of chronic azithromycin include QT prolongation, cardiac arrhythmia, hearing loss, and antibiotic-resistant organism colonization.37,38
Other therapies include pulmonary rehabilitation, which may improve health-related quality of life and 6-minute walk distance,39 extracorporeal photopheresis,40 immunosuppression with calcineurin inhibitors or mycophenolate mofetil,21,41 and lung transplantation.42–44 A study with imatinib for the treatment of lung disease in steroid-refractory GVHD has shown promising results, but further validation with larger clinical trials is required.45
Case Continued
The patient is diagnosed with BOS and is treated for several months with prednisone 40 mg/day weaned over 3 months. She is started on inhaled corticosteroids, a proton pump inhibitor, and azithromycin 3 times per week, but she has a progressive decline in FEV1. She starts pulmonary rehabilitation but continues to functionally decline. Over the next year she develops bilateral pneumothoraces and bilateral cavitary nodules (Figure 1).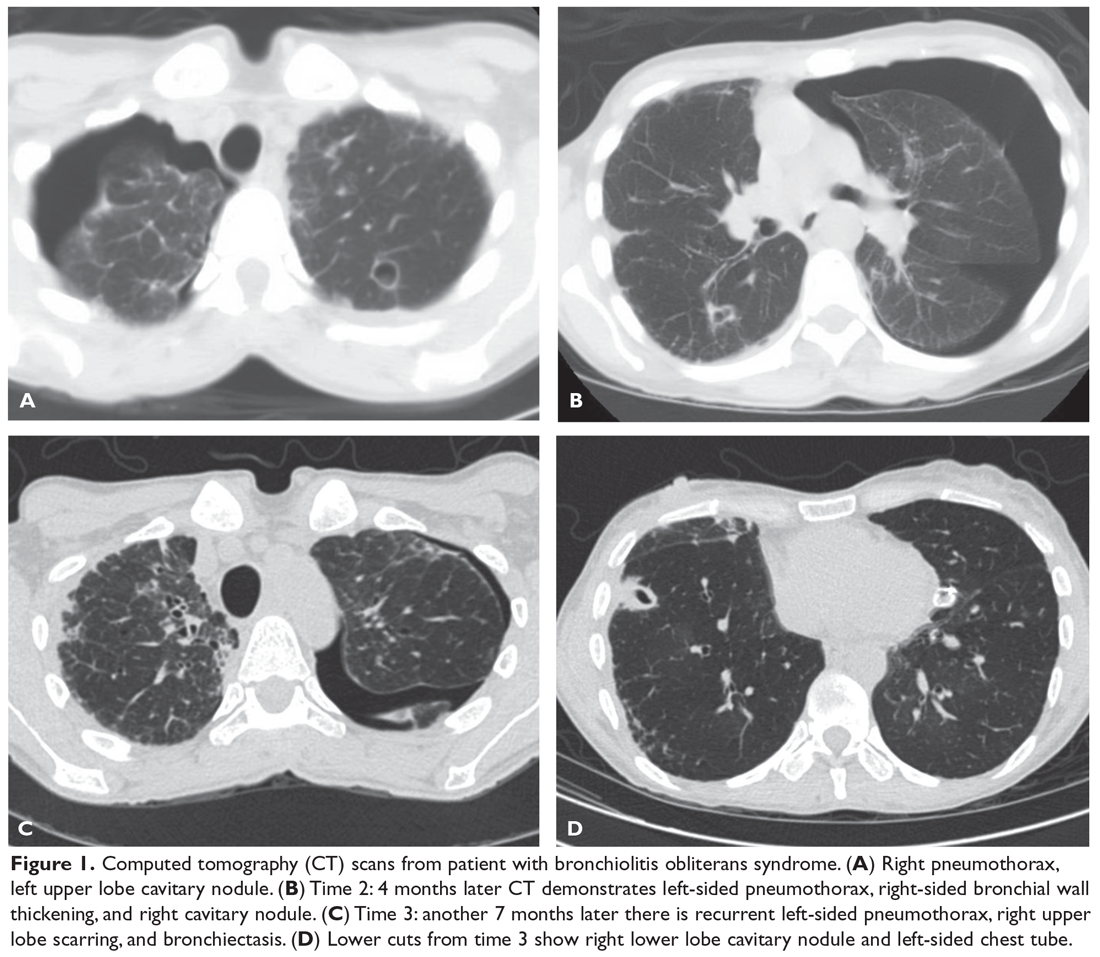
- What is causing this decline and the radiographic abnormalities?
Spontaneous air leak syndrome has been described in a little more than 1% of patients undergoing HSCT and has included pneumothorax and mediastinal and subcutaneous emphysema.46 It appears that air leak syndrome is more likely to occur in patients with chronic GVHD.47 The association between chronic GVHD and air leak syndrome could explain this patient’s recurrent pneumothoraces. The recurrent cavitary nodules are suspicious for infectious etiologies such as nontuberculous mycobacteria, tuberculosis, and fungal infections.
Case Continued
During an episode of pneumothorax, the patient undergoes chest tube placement, pleurodesis, and lung biopsy. Pathology reveals bronchiolitis obliterans as well as organizing pneumonia (Figure 2). No organisms are seen on acid-fast bacilli or GMS stains.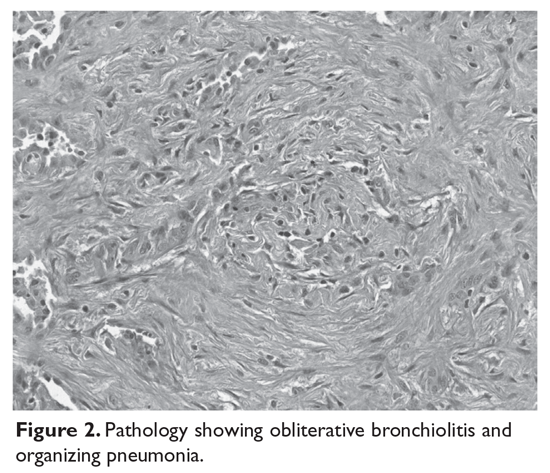
- What are the other late-onset noninfectious pulmonary complications?
Definitions of other late noninfectious pulmonary complications following HSCT are shown in Table 3.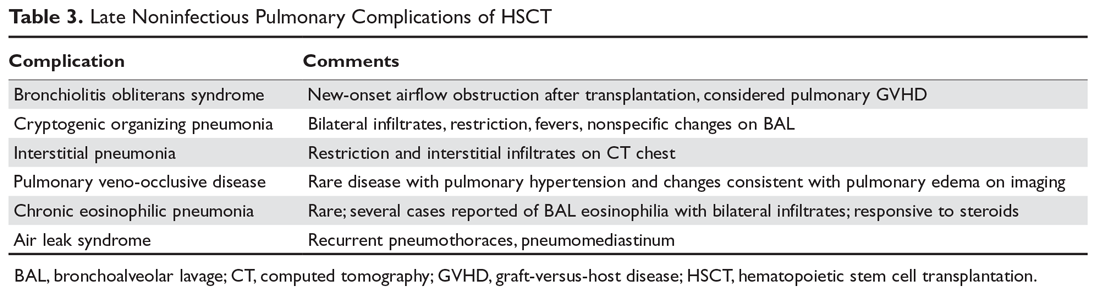
Interstitial pneumonias may represent COP or may be idiopathic pneumonia syndrome with a later onset or a nonspecific interstitial pneumonia. This syndrome is poorly defined, with a number of differing definitions of the syndrome published in the literature.50–55
A rare pulmonary complication after HSCT is pulmonary veno-occlusive disease (PVOD). Pulmonary hypertension has been reported after HSCT,56 but PVOD is a subset of pulmonary hypertension. It is associated with pleural effusions and volume overload on chest radiography.57,58 It may present early or late after transplant and is poorly understood.
Besides obstructive and restrictive PFT abnormalities, changes in small airway function59 after transplant and loss in diffusing capacity of the lungs for carbon monoxide (D
Case Conclusion
The patient’s lung function continues to worsen, but no infectious etiologies are discovered. Ultimately, she dies of respiratory failure caused by progressive bronchiolitis obliterans.
Conclusion
Late pulmonary complications occur frequently in patients who have undergone HSCT. These complications can be classified as infectious versus noninfectious etiologies. Late-onset complications are more common in allogeneic transplantations because they are associated with chronic GVHD. These complications can be manifestations of pulmonary GHVD or can be infectious complications associated with prolonged immunosuppression. Appropriate monitoring for the development of BOS is essential. Early and aggressive treatment of respiratory infections and diagnostic bronchoscopy with BAL can help elucidate most infectious causes. Still, diagnostic challenges remain and multiple causes of respiratory deterioration can be present concurrently in the post-HSCT patient. Steroid therapy remains the mainstay treatment for most noninfectious pulmonary complications and should be strongly considered once infection is effectively ruled out.
1. Remberger M, Ackefors M, Berglund S, et al. Improved survival after allogeneic hematopoietic stem cell transplantation in recent years. A single-center study. Biol Blood Marrow Transplant 2011;17:1688–97.
2. Wood KL, Esguerra VG. Management of late pulmonary complications after hematopoietic stem cell transplantation. Hosp Phys Hematology-Oncology Board Review Manual 2018;13(1):36–48.
3. Ninin E, Milpied N, Moreau P, et al. Longitudinal study of bacterial, viral, and fungal infections in adult recipients of bone marrow transplants. Clin Infect Dis 2001;33:41–7.
4. Roca J, Granena A, Rodriguez-Roisin R, et al. Fatal airway disease in an adult with chronic graft-versus-host disease. Thorax 1982;37:77–8.
5. Williams KM, Chien JW, Gladwin MT, Pavletic SZ. Bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. JAMA 2009;302:306–14.
6. Chien JW, Martin PJ, Gooley TA, et al. Airflow obstruction after myeloablative allogeneic hematopoietic stem cell transplantation. Am J Respir Crit Care Med 2003;168:208–14.
7. Holbro A, Lehmann T, Girsberger S, et al. Lung histology predicts outcome of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:973–80.
8. Chamberlain D, Maurer J, Chaparro C, Idolor L. Evaluation of transbronchial lung biopsy specimens in the diagnosis of bronchiolitis obliterans after lung transplantation. J Heart Lung Transplant 1994;13:963–71.
9. Au BK, Au MA, Chien JW. Bronchiolitis obliterans syndrome epidemiology after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 2011;17:1072–8.
10. Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant 2015;21:389–401.
11. Couriel D, Carpenter PA, Cutler C, et al. Ancillary therapy and supportive care of chronic graft-versus-host disease: national institutes of health consensus development project on criteria for clinical trials in chronic Graft-versus-host disease: V. Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 2006;12:375–96.
12. Pellegrino R, Viegi G, Brusasco V, et al. Interpretative strategies for lung function tests. Eur Respir J 2005;26:948–68.
13. Williams KM, Hnatiuk O, Mitchell SA, et al. NHANES III equations enhance early detection and mortality prediction of bronchiolitis obliterans syndrome after hematopoietic SCT. Bone Marrow Transplant 2014;49:561–6.
14. Hyatt RE, Cowl CT, Bjoraker JA, Scanlon PD. Conditions associated with an abnormal nonspecific pattern of pulmonary function tests. Chest 2009;135:419–24.
15. Bergeron A, Godet C, Chevret S, et al. Bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: phenotypes and prognosis. Bone Marrow Transplant 2013;48:819–24.
16. Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant 2005;11:945–56.
17. Gazourian L, Coronata AM, Rogers AJ, et al. Airway dilation in bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. Respir Med 2013;107:276–83.
18. Gunn ML, Godwin JD, Kanne JP, et al. High-resolution CT findings of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. J Thorac Imaging 2008;23:244–50.
19. Sargent MA, Cairns RA, Murdoch MJ, et al. Obstructive lung disease in children after allogeneic bone marrow transplantation: evaluation with high-resolution CT. AJR Am J Roentgenol 1995;164:693–6.
20. Galban CJ, Boes JL, Bule M, et al. Parametric response mapping as an indicator of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2014;20:1592–8.
21. Meyer KC, Raghu G, Verleden GM, et al. An international ISHLT/ATS/ERS clinical practice guideline: diagnosis and management of bronchiolitis obliterans syndrome. Eur Respir J 2014;44:1479–1503.
22. Hildebrandt GC, Fazekas T, Lawitschka A, et al. Diagnosis and treatment of pulmonary chronic GVHD: report from the consensus conference on clinical practice in chronic GVHD. Bone Marrow Transplant 2011;46:1283–95.
23. Carpenter PA, Kitko CL, Elad S, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: V. The 2014 Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 2015;21:1167–87.
24. Norman BC, Jacobsohn DA, Williams KM, et al. Fluticasone, azithromycin and montelukast therapy in reducing corticosteroid exposure in bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: a case series of eight patients. Bone Marrow Transplant 2011;46:1369–73.
25. Williams KM, Cheng GS, Pusic I, et al. Fluticasone, azithromycin, and montelukast treatment for new-onset bronchiolitis obliterans syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transplant 2016;22:710–6.
26. Or R, Gesundheit B, Resnick I, et al. Sparing effect by montelukast treatment for chronic graft versus host disease: a pilot study. Transplantation 2007;83:577–81.
27. Cheng GS, Storer B, Chien JW, et al. Lung function trajectory in bronchiolitis obliterans syndrome after allogeneic hematopoietic cell transplant. Ann Am Thorac Soc 2016;13:1932–9.
28. Bergeron A, Belle A, Chevret S, et al. Combined inhaled steroids and bronchodilatators in obstructive airway disease after allogeneic stem cell transplantation. Bone Marrow Transplant 2007;39:547–53.
29. Bashoura L, Gupta S, Jain A, et al. Inhaled corticosteroids stabilize constrictive bronchiolitis after hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;41:63–7.
30. Bergeron A, Chevret S, Granata A, et al. Effect of azithromycin on airflow decline-free survival after allogeneic hematopoietic stem cell transplant: the ALLOZITHRO randomized clinical trial. JAMA 2017;318:557–66.
31. Gerhardt SG, McDyer JF, Girgis RE, et al. Maintenance azithromycin therapy for bronchiolitis obliterans syndrome: results of a pilot study. Am J Respir Crit Care Med 2003;168:121–5.
32. Khalid M, Al Saghir A, Saleemi S, et al. Azithromycin in bronchiolitis obliterans complicating bone marrow transplantation: a preliminary study. Eur Respir J 2005;25:490–3.
33. Maimon N, Lipton JH, Chan CK, Marras TK. Macrolides in the treatment of bronchiolitis obliterans in allograft recipients. Bone Marrow Transplant 2009;44:69–73.
34. Lam DC, Lam B, Wong MK, et al. Effects of azithromycin in bronchiolitis obliterans syndrome after hematopoietic SCT--a randomized double-blinded placebo-controlled study. Bone Marrow Transplant 2011;46:1551–6.
35. Yadav H, Peters SG, Keogh KA, et al. Azithromycin for the treatment of obliterative bronchiolitis after hematopoietic stem cell transplantation: a systematic review and meta-analysis. Biol Blood Marrow Transplant 2016;22:2264–9.
36. Vos R, Vanaudenaerde BM, Verleden SE, et al. A randomised controlled trial of azithromycin to prevent chronic rejection after lung transplantation. Eur Respir J 2011;37:164–72.
37. Svanstrom H, Pasternak B, Hviid A. Use of azithromycin and death from cardiovascular causes. N Engl J Med 2013;368:1704–12.
38. Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011;365:689–98.
39. Tran J, Norder EE, Diaz PT, et al. Pulmonary rehabilitation for bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2012;18:1250–4.
40. Lucid CE, Savani BN, Engelhardt BG, et al. Extracorporeal photopheresis in patients with refractory bronchiolitis obliterans developing after allo-SCT. Bone Marrow Transplant 2011;46:426–9.
41. Hostettler KE, Halter JP, Gerull S, et al. Calcineurin inhibitors in bronchiolitis obliterans syndrome following stem cell transplantation. Eur Respir J 2014;43:221–32.
42. Holm AM, Riise GC, Brinch L, et al. Lung transplantation for bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation: unresolved questions. Transplantation 2013;96:e21–22.
43. Cheng GS, Edelman JD, Madtes DK, et al. Outcomes of lung transplantation after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2014;20:1169–75.
44. Okumura H, Ohtake S, Ontachi Y, et al. Living-donor lobar lung transplantation for broncho-bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation: does bronchiolitis obliterans recur in transplanted lungs? Int J Hematol 2007;86:369–73.
45. Olivieri A, Cimminiello M, Corradini P, et al. Long-term outcome and prospective validation of NIH response criteria in 39 patients receiving imatinib for steroid-refractory chronic GVHD. Blood 2013;122:4111–8.
46. Rahmanian S, Wood KL. Bronchiolitis obliterans and the risk of pneumothorax after transbronchial biopsy. Respiratory Medicine CME 2010;3:87–9.
47. Sakai R, Kanamori H, Nakaseko C, et al. Air-leak syndrome following allo-SCT in adult patients: report from the Kanto Study Group for Cell Therapy in Japan. Bone Marrow Transplant 2011;46:379–84.
48. Visscher DW, Myers JL. Histologic spectrum of idiopathic interstitial pneumonias. Proc Am Thorac Soc 2006;3:322–9.
49. Cordier JF. Cryptogenic organising pneumonia. Eur Respir J 2006;28:422–46.
50. Nishio N, Yagasaki H, Takahashi Y, et al. Late-onset non-infectious pulmonary complications following allogeneic hematopoietic stem cell transplantation in children. Bone Marrow Transplant 2009;44:303–8.
51. Ueda K, Watadani T, Maeda E, et al. Outcome and treatment of late-onset noninfectious pulmonary complications after allogeneic haematopoietic SCT. Bone Marrow Transplant 2010;45:1719–27.
52. Schlemmer F, Chevret S, Lorillon G, et al. Late-onset noninfectious interstitial lung disease after allogeneic hematopoietic stem cell transplantation. Respir Med 2014;108:1525–33.
53. Palmas A, Tefferi A, Myers JL, et al. Late-onset noninfectious pulmonary complications after allogeneic bone marrow transplantation. Br J Haematol 1998;100:680–7.
54. Sakaida E, Nakaseko C, Harima A, et al. Late-onset noninfectious pulmonary complications after allogeneic stem cell transplantation are significantly associated with chronic graft-versus-host disease and with the graft-versus-leukemia effect. Blood 2003;102:4236–42.
55. Solh M, Arat M, Cao Q, et al. Late-onset noninfectious pulmonary complications in adult allogeneic hematopoietic cell transplant recipients. Transplantation 2011;91:798–803.
56. Dandoy CE, Hirsch R, Chima R, et al. Pulmonary hypertension after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:1546–56.
57. Bunte MC, Patnaik MM, Pritzker MR, Burns LJ. Pulmonary veno-occlusive disease following hematopoietic stem cell transplantation: a rare model of endothelial dysfunction. Bone Marrow Transplant 2008;41:677–86.
58. Troussard X, Bernaudin JF, Cordonnier C, et al. Pulmonary veno-occlusive disease after bone marrow transplantation. Thorax 1984;39:956–7.
59. Lahzami S, Schoeffel RE, Pechey V, et al. Small airways function declines after allogeneic haematopoietic stem cell transplantation. Eur Respir J 2011;38:1180–8.
60. Jain NA, Pophali PA, Klotz JK, et al. Repair of impaired pulmonary function is possible in very-long-term allogeneic stem cell transplantation survivors. Biol Blood Marrow Transplant 2014;20:209–13.
61. Barisione G, Bacigalupo A, Crimi E, et al. Changes in lung volumes and airway responsiveness following haematopoietic stem cell transplantation. Eur Respir J 2008;32:1576–82.
62. Kovalszki A, Schumaker GL, Klein A, et al. Reduced respiratory and skeletal muscle strength in survivors of sibling or unrelated donor hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;41:965–9.
63. Mathiesen S, Uhlving HH, Buchvald F, et al. Aerobic exercise capacity at long-term follow-up after paediatric allogeneic haematopoietic SCT. Bone Marrow Transplant 2014;49:1393–9.
Hematopoietic stem cell transplantation (HSCT) is increasingly being used to treat hematologic malignancies as well as nonmalignant diseases and solid tumors. Over the past 2 decades overall survival following transplant and transplant-related mortality have improved.1 With this increased survival, there is a need to focus on late complications after transplantation. Pulmonary complications are a common but sometimes underrecognized cause of late morbidity and mortality in HSCT patients. This article, the second of 2 articles on post-HSCT pulmonary complications, reviews late-onset complications, with a focus on the evaluation and treatment of bronchiolitis obliterans syndrome (BOS), one of the most common and serious late pulmonary complications in HSCT patients. The first article reviewed the management of early-onset pulmonary complications and included a basic overview of stem cell transplantation, discussion of factors associated with pulmonary complications, and a review of methods for assessing pretransplant risk for pulmonary complications in patients undergoing HSCT.2
Case Presentation
A 40-year-old white woman with a history of acute myeloid leukemia status post peripheral blood stem cell transplant presents with dyspnea on exertion, which she states started about 1 month ago and now is limiting her with even 1 flight of stairs. She also complains of mild dry cough and a 4- to 5-lb weight loss over the past 1 to 2 months. She has an occasional runny nose, but denies gastroesophageal reflux, fevers, chills, or night sweats. She has a history of matched related sibling donor transplant with busulfan and cyclophosphamide conditioning 1 year prior to presentation. She has had significant graft-versus-host disease (GVHD), affecting the liver, gastrointestinal tract, skin, and eyes.
On physical examination, heart rate is 110 beats/min, respiratory rate is 16 breaths/min, blood pressure is 92/58 mm Hg, and the patient is afebrile. Eye exam reveals scleral injection, mouth shows dry mucous membranes with a few white plaques, and the skin has chronic changes with a rash over both arms. Cardiac exam reveals tachycardia but regular rhythm and there are no murmurs, rubs, or gallops. Lungs are clear bilaterally and abdomen shows no organomegaly.
Laboratory exam shows a white blood cell count of 7800 cells/μL, hemoglobin level of 12.4 g/dL, and platelet count of 186 × 103/μL. Liver enzymes are mildly elevated. Chest radiograph shows clear lung fields bilaterally.
- What is the differential in this patient with dyspnea 1 year after transplantation?
Late pulmonary complications are generally accepted as those occurring more than 100 days post transplant. This period of time is characterized by chronic GVHD and impaired cellular and humoral immunity. Results of longitudinal studies of infections in adult HSCT patients suggest that special attention should be paid to allogeneic HSCT recipients for post-engraftment infectious pulmonary complications.3 Encapsulated bacteria such as Haemophilus influenzae and Streptococcus pneumoniae are the most frequent bacterial organisms causing late infectious pulmonary complications. Nontuberculous mycobacteria and Nocardia should also be considered. Depending upon geographic location, social and occupational risk factors, and prevalence, tuberculosis should also enter the differential.
There are many noninfectious late-onset pulmonary complications after HSCT. Unfortunately, the literature has divided pulmonary complications after HSCT using a range of criteria and classifications based upon timing, predominant pulmonary function test (PFT) findings, and etiology. These include early versus late, obstructive versus restrictive, and infectious versus noninfectious, which makes a comprehensive literature review of late pulmonary complications difficult. The most common noninfectious late-onset complications are bronchiolitis obliterans, cryptogenic organizing pneumonia (previously referred to as bronchiolitis obliterans organizing pneumonia, or BOOP), and interstitial pneumonia. Other rarely reported complications include eosinophilic pneumonia, pulmonary alveolar proteinosis, air leak syndrome, and pulmonary hypertension.
Case Continued
Because the patient does not have symptoms of infection, PFTs are obtained. Pretransplant PFTs and current PFTs are shown in Table 1.
- What is the diagnosis in this case?
Bronchiolitis Obliterans
BOS is one of the most common and most serious late-onset pulmonary diseases after allogeneic transplantation. It is considered the pulmonary form of chronic GVHD. BOS was first described in 1982 in patients with chronic GVHD after bone marrow transplantation.4 Many differing definitions of bronchiolitis obliterans have been described in the literature. A recent review of the topic cites 10 different published sets of criteria for the diagnosis of bronchiolitis obliterans.5 Traditionally, bronchiolitis obliterans was thought to occur in 2% to 8% of patients undergoing allogeneic HSCT, but these findings were from older studies that used a diagnosis based on very specific pathology findings. When more liberal diagnostic criteria are used, the incidence may be as high as 26% of allogeneic HSCT patients.6
Bronchiolitis obliterans is a progressive lung disease characterized by narrowing of the terminal airways and obliteration of the terminal bronchi. Pathology may show constrictive bronchiolitis but can also show lymphocytic bronchiolitis, which may be associated with a better outcome.7 As noted, bronchiolitis obliterans has traditionally been considered a pathologic diagnosis. Current diagnostic criteria have evolved based upon the difficulty in obtaining this diagnosis through transbronchial biopsy given the patchy nature of the disease.8 The gold standard of open lung biopsy is seldom pursued in the post-HSCT population as the procedure continues to carry a worrisome risk-benefit profile.
The 2005 National Institutes of Health (NIH) consensus development project on criteria for clinical trials in chronic GVHD developed a clinical strategy for diagnosing BOS using the following criteria: absence of active infection, decreased forced expiratory volume in 1 second (FEV1) < 75%, FEV1/forced vital capacity (FVC) ratio of < 70%, and evidence of air trapping on high-resolution computed tomography (HRCT) or PFTs (residual volume > 120%). These diagnostic criteria were applied to a small series of patients with clinically identified bronchiolitis obliterans or biopsy-proven bronchiolitis obliterans. Only 18% of these patients met the requirements for the NIH consensus definition.5 A 2011 study that applied the NIH criteria found an overall prevalence of 5.5% among all transplant recipients but a prevalence of 14% in patients with GVHD.9 In 2014, the NIH consensus development group updated their recommendations. The new criteria for diagnosis of BOS require the presence of airflow obstruction (FEV1/FVC < 70% or 5th percentile of predicted), FEV1 < 75% predicted with a ≥ 10% decline in fewer than 2 years, absence of infection, and presence of air trapping (by expiratory computed tomography [CT] scan or PFT with residual volume >120% predicted) (Table 2).
Some issues must be considered when determining airflow obstruction. The 2005 NIH working group recommends using Crapo as the reference set,11 but the National Health and Nutrition Examination Survey (NHANES) III reference values are the preferred reference set at this time12 and should be used in the United States. A recent article showed that the NHANES values were superior to older reference sets (however, they did not use Crapo as the comparison), although this study used the lower limit of normal as compared with the fixed 70% ratio.13 The 2014 NIH consensus group does not recommend a specific reference set and recognizes an FEV1/FVC ratio of 70% or less than the lower limit of normal as the cutoff value for airflow obstruction.10
Another issue in PFT interpretation is the finding of a decrease in FEV1 and FVC and normal total lung capacity, which is termed a nonspecific pattern. This pattern has been reported to occur in 9% of all PFTs and usually is associated with obstructive lung disease or obesity.14 A 2013 study described the nonspecific pattern as a BOS subgroup occurring in up to 31% of bronchiolitis obliterans patients.15
- What are the radiographic findings of BOS?
Chest radiograph is often normal in BOS. As discussed, air trapping can be documented using HRCT, according to the NIH clinical definition of bronchiolitis obliterans.16 A study that explored findings and trends seen on HRCT in HSCT patients with BOS found that the syndrome in these patients is characterized by central airway dilatation.17 Expiratory airway trapping on HRCT is the main finding, and this is best demonstrated on HRCT during inspiratory and expiratory phases.18 Other findings are bronchial wall thickening, parenchymal hypoattenuation, bronchiectasis, and centrilobular nodules.19
Galbán and colleagues developed a new technique called parametric response mapping that uses CT scanners to quantify normal parenchyma, functional small airway disease, emphysema, and parenchymal disease as relative lung volumes.20 This technique can detect airflow obstruction and small airway disease and was found to be a good method for detecting BOS after HSCT. In their study of parametric response mapping, the authors found that functional small airway disease affecting 28% or more of the total lung was highly indicative of bronchiolitis obliterans.20
- What therapies are used to treat BOS?
Traditionally, BOS has been treated with systemic immunosuppression. The recommended treatment had been systemic steroids at approximately 1 mg/ kg. However, it is increasingly recognized that BOS responds poorly to systemic steroids, and systemic steroids may actually be harmful and associated with increased mortality.15,21 The chronic GVHD recommendations from 2005 recommend ancillary therapy with inhaled corticosteroids and pulmonary rehabilitation.11 The updated 2011 German consensus statement lays out a clear management strategy for mild and moderate-severe disease with monitoring recommendations.22 The 2014 NIH chronic GVHD working group recommends fluticasone, azithromycin, and montelukast (ie, the FAM protocol) for treating BOS.23 FAM therapy in BOS may help lower the systemic steroid dose.24,25 Montelukast is not considered a treatment mainstay for BOS after lung transplant, but there is a study showing possible benefit in chronic GVHD.26 An evaluation of the natural history of a cohort of BOS patients treated with FAM therapy showed a rapid decline of FEV1 in the 6 months prior to diagnosis and treatment of BOS and subsequent stabilization following diagnosis and treatment.27 The benefit of high-dose inhaled corticosteroids or the combination of inhaled corticosteroids and long-acting beta-agonists has been demonstrated in small studies, which showed that these agents stabilized FEV1 and avoided the untoward side effects of systemic corticosteroids.28–30
Macrolide antibiotics have been explored as a treatment for BOS post HSCT because pilot studies suggested that azithromycin improved or stabilized FEV1 in patients with BOS after lung transplant or HSCT.31–33 Other studies of azithromycin have not shown benefit in the HSCT population after 3 months of therapy.34 A recent meta-analysis could neither support or refute the benefit of azithromycin for BOS after HSCT.35 In the lung transplant population, a study showed that patients who were started on azithromycin after transplant and continued on it 3 times a week had improved FEV1; these patients also had a reduced rate of BOS and improved overall and BOS-free survival 2 years after transplant.36 However, these benefits of azithromycin have not been observed in patients after HSCT. In fact, the ALLOZITHRO trial was stopped early because prophylactic azithromycin started at the time of the conditioning regimen with HSCT was associated with increased hematologic disease relapse, a decrease in airflow-decline-free survival, and reduced 2-year survival.30
Azithromycin is believed to exert an effect by its anti-inflammatory properties and perhaps by decreasing lung neutrophilia (it may be most beneficial in the subset of patients with high neutrophilia on bronchoalveolar lavage [BAL]).30 Adverse effects of chronic azithromycin include QT prolongation, cardiac arrhythmia, hearing loss, and antibiotic-resistant organism colonization.37,38
Other therapies include pulmonary rehabilitation, which may improve health-related quality of life and 6-minute walk distance,39 extracorporeal photopheresis,40 immunosuppression with calcineurin inhibitors or mycophenolate mofetil,21,41 and lung transplantation.42–44 A study with imatinib for the treatment of lung disease in steroid-refractory GVHD has shown promising results, but further validation with larger clinical trials is required.45
Case Continued
The patient is diagnosed with BOS and is treated for several months with prednisone 40 mg/day weaned over 3 months. She is started on inhaled corticosteroids, a proton pump inhibitor, and azithromycin 3 times per week, but she has a progressive decline in FEV1. She starts pulmonary rehabilitation but continues to functionally decline. Over the next year she develops bilateral pneumothoraces and bilateral cavitary nodules (Figure 1).
- What is causing this decline and the radiographic abnormalities?
Spontaneous air leak syndrome has been described in a little more than 1% of patients undergoing HSCT and has included pneumothorax and mediastinal and subcutaneous emphysema.46 It appears that air leak syndrome is more likely to occur in patients with chronic GVHD.47 The association between chronic GVHD and air leak syndrome could explain this patient’s recurrent pneumothoraces. The recurrent cavitary nodules are suspicious for infectious etiologies such as nontuberculous mycobacteria, tuberculosis, and fungal infections.
Case Continued
During an episode of pneumothorax, the patient undergoes chest tube placement, pleurodesis, and lung biopsy. Pathology reveals bronchiolitis obliterans as well as organizing pneumonia (Figure 2). No organisms are seen on acid-fast bacilli or GMS stains.
- What are the other late-onset noninfectious pulmonary complications?
Definitions of other late noninfectious pulmonary complications following HSCT are shown in Table 3.
Interstitial pneumonias may represent COP or may be idiopathic pneumonia syndrome with a later onset or a nonspecific interstitial pneumonia. This syndrome is poorly defined, with a number of differing definitions of the syndrome published in the literature.50–55
A rare pulmonary complication after HSCT is pulmonary veno-occlusive disease (PVOD). Pulmonary hypertension has been reported after HSCT,56 but PVOD is a subset of pulmonary hypertension. It is associated with pleural effusions and volume overload on chest radiography.57,58 It may present early or late after transplant and is poorly understood.
Besides obstructive and restrictive PFT abnormalities, changes in small airway function59 after transplant and loss in diffusing capacity of the lungs for carbon monoxide (D
Case Conclusion
The patient’s lung function continues to worsen, but no infectious etiologies are discovered. Ultimately, she dies of respiratory failure caused by progressive bronchiolitis obliterans.
Conclusion
Late pulmonary complications occur frequently in patients who have undergone HSCT. These complications can be classified as infectious versus noninfectious etiologies. Late-onset complications are more common in allogeneic transplantations because they are associated with chronic GVHD. These complications can be manifestations of pulmonary GHVD or can be infectious complications associated with prolonged immunosuppression. Appropriate monitoring for the development of BOS is essential. Early and aggressive treatment of respiratory infections and diagnostic bronchoscopy with BAL can help elucidate most infectious causes. Still, diagnostic challenges remain and multiple causes of respiratory deterioration can be present concurrently in the post-HSCT patient. Steroid therapy remains the mainstay treatment for most noninfectious pulmonary complications and should be strongly considered once infection is effectively ruled out.
Hematopoietic stem cell transplantation (HSCT) is increasingly being used to treat hematologic malignancies as well as nonmalignant diseases and solid tumors. Over the past 2 decades overall survival following transplant and transplant-related mortality have improved.1 With this increased survival, there is a need to focus on late complications after transplantation. Pulmonary complications are a common but sometimes underrecognized cause of late morbidity and mortality in HSCT patients. This article, the second of 2 articles on post-HSCT pulmonary complications, reviews late-onset complications, with a focus on the evaluation and treatment of bronchiolitis obliterans syndrome (BOS), one of the most common and serious late pulmonary complications in HSCT patients. The first article reviewed the management of early-onset pulmonary complications and included a basic overview of stem cell transplantation, discussion of factors associated with pulmonary complications, and a review of methods for assessing pretransplant risk for pulmonary complications in patients undergoing HSCT.2
Case Presentation
A 40-year-old white woman with a history of acute myeloid leukemia status post peripheral blood stem cell transplant presents with dyspnea on exertion, which she states started about 1 month ago and now is limiting her with even 1 flight of stairs. She also complains of mild dry cough and a 4- to 5-lb weight loss over the past 1 to 2 months. She has an occasional runny nose, but denies gastroesophageal reflux, fevers, chills, or night sweats. She has a history of matched related sibling donor transplant with busulfan and cyclophosphamide conditioning 1 year prior to presentation. She has had significant graft-versus-host disease (GVHD), affecting the liver, gastrointestinal tract, skin, and eyes.
On physical examination, heart rate is 110 beats/min, respiratory rate is 16 breaths/min, blood pressure is 92/58 mm Hg, and the patient is afebrile. Eye exam reveals scleral injection, mouth shows dry mucous membranes with a few white plaques, and the skin has chronic changes with a rash over both arms. Cardiac exam reveals tachycardia but regular rhythm and there are no murmurs, rubs, or gallops. Lungs are clear bilaterally and abdomen shows no organomegaly.
Laboratory exam shows a white blood cell count of 7800 cells/μL, hemoglobin level of 12.4 g/dL, and platelet count of 186 × 103/μL. Liver enzymes are mildly elevated. Chest radiograph shows clear lung fields bilaterally.
- What is the differential in this patient with dyspnea 1 year after transplantation?
Late pulmonary complications are generally accepted as those occurring more than 100 days post transplant. This period of time is characterized by chronic GVHD and impaired cellular and humoral immunity. Results of longitudinal studies of infections in adult HSCT patients suggest that special attention should be paid to allogeneic HSCT recipients for post-engraftment infectious pulmonary complications.3 Encapsulated bacteria such as Haemophilus influenzae and Streptococcus pneumoniae are the most frequent bacterial organisms causing late infectious pulmonary complications. Nontuberculous mycobacteria and Nocardia should also be considered. Depending upon geographic location, social and occupational risk factors, and prevalence, tuberculosis should also enter the differential.
There are many noninfectious late-onset pulmonary complications after HSCT. Unfortunately, the literature has divided pulmonary complications after HSCT using a range of criteria and classifications based upon timing, predominant pulmonary function test (PFT) findings, and etiology. These include early versus late, obstructive versus restrictive, and infectious versus noninfectious, which makes a comprehensive literature review of late pulmonary complications difficult. The most common noninfectious late-onset complications are bronchiolitis obliterans, cryptogenic organizing pneumonia (previously referred to as bronchiolitis obliterans organizing pneumonia, or BOOP), and interstitial pneumonia. Other rarely reported complications include eosinophilic pneumonia, pulmonary alveolar proteinosis, air leak syndrome, and pulmonary hypertension.
Case Continued
Because the patient does not have symptoms of infection, PFTs are obtained. Pretransplant PFTs and current PFTs are shown in Table 1.
- What is the diagnosis in this case?
Bronchiolitis Obliterans
BOS is one of the most common and most serious late-onset pulmonary diseases after allogeneic transplantation. It is considered the pulmonary form of chronic GVHD. BOS was first described in 1982 in patients with chronic GVHD after bone marrow transplantation.4 Many differing definitions of bronchiolitis obliterans have been described in the literature. A recent review of the topic cites 10 different published sets of criteria for the diagnosis of bronchiolitis obliterans.5 Traditionally, bronchiolitis obliterans was thought to occur in 2% to 8% of patients undergoing allogeneic HSCT, but these findings were from older studies that used a diagnosis based on very specific pathology findings. When more liberal diagnostic criteria are used, the incidence may be as high as 26% of allogeneic HSCT patients.6
Bronchiolitis obliterans is a progressive lung disease characterized by narrowing of the terminal airways and obliteration of the terminal bronchi. Pathology may show constrictive bronchiolitis but can also show lymphocytic bronchiolitis, which may be associated with a better outcome.7 As noted, bronchiolitis obliterans has traditionally been considered a pathologic diagnosis. Current diagnostic criteria have evolved based upon the difficulty in obtaining this diagnosis through transbronchial biopsy given the patchy nature of the disease.8 The gold standard of open lung biopsy is seldom pursued in the post-HSCT population as the procedure continues to carry a worrisome risk-benefit profile.
The 2005 National Institutes of Health (NIH) consensus development project on criteria for clinical trials in chronic GVHD developed a clinical strategy for diagnosing BOS using the following criteria: absence of active infection, decreased forced expiratory volume in 1 second (FEV1) < 75%, FEV1/forced vital capacity (FVC) ratio of < 70%, and evidence of air trapping on high-resolution computed tomography (HRCT) or PFTs (residual volume > 120%). These diagnostic criteria were applied to a small series of patients with clinically identified bronchiolitis obliterans or biopsy-proven bronchiolitis obliterans. Only 18% of these patients met the requirements for the NIH consensus definition.5 A 2011 study that applied the NIH criteria found an overall prevalence of 5.5% among all transplant recipients but a prevalence of 14% in patients with GVHD.9 In 2014, the NIH consensus development group updated their recommendations. The new criteria for diagnosis of BOS require the presence of airflow obstruction (FEV1/FVC < 70% or 5th percentile of predicted), FEV1 < 75% predicted with a ≥ 10% decline in fewer than 2 years, absence of infection, and presence of air trapping (by expiratory computed tomography [CT] scan or PFT with residual volume >120% predicted) (Table 2).
Some issues must be considered when determining airflow obstruction. The 2005 NIH working group recommends using Crapo as the reference set,11 but the National Health and Nutrition Examination Survey (NHANES) III reference values are the preferred reference set at this time12 and should be used in the United States. A recent article showed that the NHANES values were superior to older reference sets (however, they did not use Crapo as the comparison), although this study used the lower limit of normal as compared with the fixed 70% ratio.13 The 2014 NIH consensus group does not recommend a specific reference set and recognizes an FEV1/FVC ratio of 70% or less than the lower limit of normal as the cutoff value for airflow obstruction.10
Another issue in PFT interpretation is the finding of a decrease in FEV1 and FVC and normal total lung capacity, which is termed a nonspecific pattern. This pattern has been reported to occur in 9% of all PFTs and usually is associated with obstructive lung disease or obesity.14 A 2013 study described the nonspecific pattern as a BOS subgroup occurring in up to 31% of bronchiolitis obliterans patients.15
- What are the radiographic findings of BOS?
Chest radiograph is often normal in BOS. As discussed, air trapping can be documented using HRCT, according to the NIH clinical definition of bronchiolitis obliterans.16 A study that explored findings and trends seen on HRCT in HSCT patients with BOS found that the syndrome in these patients is characterized by central airway dilatation.17 Expiratory airway trapping on HRCT is the main finding, and this is best demonstrated on HRCT during inspiratory and expiratory phases.18 Other findings are bronchial wall thickening, parenchymal hypoattenuation, bronchiectasis, and centrilobular nodules.19
Galbán and colleagues developed a new technique called parametric response mapping that uses CT scanners to quantify normal parenchyma, functional small airway disease, emphysema, and parenchymal disease as relative lung volumes.20 This technique can detect airflow obstruction and small airway disease and was found to be a good method for detecting BOS after HSCT. In their study of parametric response mapping, the authors found that functional small airway disease affecting 28% or more of the total lung was highly indicative of bronchiolitis obliterans.20
- What therapies are used to treat BOS?
Traditionally, BOS has been treated with systemic immunosuppression. The recommended treatment had been systemic steroids at approximately 1 mg/ kg. However, it is increasingly recognized that BOS responds poorly to systemic steroids, and systemic steroids may actually be harmful and associated with increased mortality.15,21 The chronic GVHD recommendations from 2005 recommend ancillary therapy with inhaled corticosteroids and pulmonary rehabilitation.11 The updated 2011 German consensus statement lays out a clear management strategy for mild and moderate-severe disease with monitoring recommendations.22 The 2014 NIH chronic GVHD working group recommends fluticasone, azithromycin, and montelukast (ie, the FAM protocol) for treating BOS.23 FAM therapy in BOS may help lower the systemic steroid dose.24,25 Montelukast is not considered a treatment mainstay for BOS after lung transplant, but there is a study showing possible benefit in chronic GVHD.26 An evaluation of the natural history of a cohort of BOS patients treated with FAM therapy showed a rapid decline of FEV1 in the 6 months prior to diagnosis and treatment of BOS and subsequent stabilization following diagnosis and treatment.27 The benefit of high-dose inhaled corticosteroids or the combination of inhaled corticosteroids and long-acting beta-agonists has been demonstrated in small studies, which showed that these agents stabilized FEV1 and avoided the untoward side effects of systemic corticosteroids.28–30
Macrolide antibiotics have been explored as a treatment for BOS post HSCT because pilot studies suggested that azithromycin improved or stabilized FEV1 in patients with BOS after lung transplant or HSCT.31–33 Other studies of azithromycin have not shown benefit in the HSCT population after 3 months of therapy.34 A recent meta-analysis could neither support or refute the benefit of azithromycin for BOS after HSCT.35 In the lung transplant population, a study showed that patients who were started on azithromycin after transplant and continued on it 3 times a week had improved FEV1; these patients also had a reduced rate of BOS and improved overall and BOS-free survival 2 years after transplant.36 However, these benefits of azithromycin have not been observed in patients after HSCT. In fact, the ALLOZITHRO trial was stopped early because prophylactic azithromycin started at the time of the conditioning regimen with HSCT was associated with increased hematologic disease relapse, a decrease in airflow-decline-free survival, and reduced 2-year survival.30
Azithromycin is believed to exert an effect by its anti-inflammatory properties and perhaps by decreasing lung neutrophilia (it may be most beneficial in the subset of patients with high neutrophilia on bronchoalveolar lavage [BAL]).30 Adverse effects of chronic azithromycin include QT prolongation, cardiac arrhythmia, hearing loss, and antibiotic-resistant organism colonization.37,38
Other therapies include pulmonary rehabilitation, which may improve health-related quality of life and 6-minute walk distance,39 extracorporeal photopheresis,40 immunosuppression with calcineurin inhibitors or mycophenolate mofetil,21,41 and lung transplantation.42–44 A study with imatinib for the treatment of lung disease in steroid-refractory GVHD has shown promising results, but further validation with larger clinical trials is required.45
Case Continued
The patient is diagnosed with BOS and is treated for several months with prednisone 40 mg/day weaned over 3 months. She is started on inhaled corticosteroids, a proton pump inhibitor, and azithromycin 3 times per week, but she has a progressive decline in FEV1. She starts pulmonary rehabilitation but continues to functionally decline. Over the next year she develops bilateral pneumothoraces and bilateral cavitary nodules (Figure 1).
- What is causing this decline and the radiographic abnormalities?
Spontaneous air leak syndrome has been described in a little more than 1% of patients undergoing HSCT and has included pneumothorax and mediastinal and subcutaneous emphysema.46 It appears that air leak syndrome is more likely to occur in patients with chronic GVHD.47 The association between chronic GVHD and air leak syndrome could explain this patient’s recurrent pneumothoraces. The recurrent cavitary nodules are suspicious for infectious etiologies such as nontuberculous mycobacteria, tuberculosis, and fungal infections.
Case Continued
During an episode of pneumothorax, the patient undergoes chest tube placement, pleurodesis, and lung biopsy. Pathology reveals bronchiolitis obliterans as well as organizing pneumonia (Figure 2). No organisms are seen on acid-fast bacilli or GMS stains.
- What are the other late-onset noninfectious pulmonary complications?
Definitions of other late noninfectious pulmonary complications following HSCT are shown in Table 3.
Interstitial pneumonias may represent COP or may be idiopathic pneumonia syndrome with a later onset or a nonspecific interstitial pneumonia. This syndrome is poorly defined, with a number of differing definitions of the syndrome published in the literature.50–55
A rare pulmonary complication after HSCT is pulmonary veno-occlusive disease (PVOD). Pulmonary hypertension has been reported after HSCT,56 but PVOD is a subset of pulmonary hypertension. It is associated with pleural effusions and volume overload on chest radiography.57,58 It may present early or late after transplant and is poorly understood.
Besides obstructive and restrictive PFT abnormalities, changes in small airway function59 after transplant and loss in diffusing capacity of the lungs for carbon monoxide (D
Case Conclusion
The patient’s lung function continues to worsen, but no infectious etiologies are discovered. Ultimately, she dies of respiratory failure caused by progressive bronchiolitis obliterans.
Conclusion
Late pulmonary complications occur frequently in patients who have undergone HSCT. These complications can be classified as infectious versus noninfectious etiologies. Late-onset complications are more common in allogeneic transplantations because they are associated with chronic GVHD. These complications can be manifestations of pulmonary GHVD or can be infectious complications associated with prolonged immunosuppression. Appropriate monitoring for the development of BOS is essential. Early and aggressive treatment of respiratory infections and diagnostic bronchoscopy with BAL can help elucidate most infectious causes. Still, diagnostic challenges remain and multiple causes of respiratory deterioration can be present concurrently in the post-HSCT patient. Steroid therapy remains the mainstay treatment for most noninfectious pulmonary complications and should be strongly considered once infection is effectively ruled out.
1. Remberger M, Ackefors M, Berglund S, et al. Improved survival after allogeneic hematopoietic stem cell transplantation in recent years. A single-center study. Biol Blood Marrow Transplant 2011;17:1688–97.
2. Wood KL, Esguerra VG. Management of late pulmonary complications after hematopoietic stem cell transplantation. Hosp Phys Hematology-Oncology Board Review Manual 2018;13(1):36–48.
3. Ninin E, Milpied N, Moreau P, et al. Longitudinal study of bacterial, viral, and fungal infections in adult recipients of bone marrow transplants. Clin Infect Dis 2001;33:41–7.
4. Roca J, Granena A, Rodriguez-Roisin R, et al. Fatal airway disease in an adult with chronic graft-versus-host disease. Thorax 1982;37:77–8.
5. Williams KM, Chien JW, Gladwin MT, Pavletic SZ. Bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. JAMA 2009;302:306–14.
6. Chien JW, Martin PJ, Gooley TA, et al. Airflow obstruction after myeloablative allogeneic hematopoietic stem cell transplantation. Am J Respir Crit Care Med 2003;168:208–14.
7. Holbro A, Lehmann T, Girsberger S, et al. Lung histology predicts outcome of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:973–80.
8. Chamberlain D, Maurer J, Chaparro C, Idolor L. Evaluation of transbronchial lung biopsy specimens in the diagnosis of bronchiolitis obliterans after lung transplantation. J Heart Lung Transplant 1994;13:963–71.
9. Au BK, Au MA, Chien JW. Bronchiolitis obliterans syndrome epidemiology after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 2011;17:1072–8.
10. Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant 2015;21:389–401.
11. Couriel D, Carpenter PA, Cutler C, et al. Ancillary therapy and supportive care of chronic graft-versus-host disease: national institutes of health consensus development project on criteria for clinical trials in chronic Graft-versus-host disease: V. Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 2006;12:375–96.
12. Pellegrino R, Viegi G, Brusasco V, et al. Interpretative strategies for lung function tests. Eur Respir J 2005;26:948–68.
13. Williams KM, Hnatiuk O, Mitchell SA, et al. NHANES III equations enhance early detection and mortality prediction of bronchiolitis obliterans syndrome after hematopoietic SCT. Bone Marrow Transplant 2014;49:561–6.
14. Hyatt RE, Cowl CT, Bjoraker JA, Scanlon PD. Conditions associated with an abnormal nonspecific pattern of pulmonary function tests. Chest 2009;135:419–24.
15. Bergeron A, Godet C, Chevret S, et al. Bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: phenotypes and prognosis. Bone Marrow Transplant 2013;48:819–24.
16. Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant 2005;11:945–56.
17. Gazourian L, Coronata AM, Rogers AJ, et al. Airway dilation in bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. Respir Med 2013;107:276–83.
18. Gunn ML, Godwin JD, Kanne JP, et al. High-resolution CT findings of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. J Thorac Imaging 2008;23:244–50.
19. Sargent MA, Cairns RA, Murdoch MJ, et al. Obstructive lung disease in children after allogeneic bone marrow transplantation: evaluation with high-resolution CT. AJR Am J Roentgenol 1995;164:693–6.
20. Galban CJ, Boes JL, Bule M, et al. Parametric response mapping as an indicator of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2014;20:1592–8.
21. Meyer KC, Raghu G, Verleden GM, et al. An international ISHLT/ATS/ERS clinical practice guideline: diagnosis and management of bronchiolitis obliterans syndrome. Eur Respir J 2014;44:1479–1503.
22. Hildebrandt GC, Fazekas T, Lawitschka A, et al. Diagnosis and treatment of pulmonary chronic GVHD: report from the consensus conference on clinical practice in chronic GVHD. Bone Marrow Transplant 2011;46:1283–95.
23. Carpenter PA, Kitko CL, Elad S, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: V. The 2014 Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 2015;21:1167–87.
24. Norman BC, Jacobsohn DA, Williams KM, et al. Fluticasone, azithromycin and montelukast therapy in reducing corticosteroid exposure in bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: a case series of eight patients. Bone Marrow Transplant 2011;46:1369–73.
25. Williams KM, Cheng GS, Pusic I, et al. Fluticasone, azithromycin, and montelukast treatment for new-onset bronchiolitis obliterans syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transplant 2016;22:710–6.
26. Or R, Gesundheit B, Resnick I, et al. Sparing effect by montelukast treatment for chronic graft versus host disease: a pilot study. Transplantation 2007;83:577–81.
27. Cheng GS, Storer B, Chien JW, et al. Lung function trajectory in bronchiolitis obliterans syndrome after allogeneic hematopoietic cell transplant. Ann Am Thorac Soc 2016;13:1932–9.
28. Bergeron A, Belle A, Chevret S, et al. Combined inhaled steroids and bronchodilatators in obstructive airway disease after allogeneic stem cell transplantation. Bone Marrow Transplant 2007;39:547–53.
29. Bashoura L, Gupta S, Jain A, et al. Inhaled corticosteroids stabilize constrictive bronchiolitis after hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;41:63–7.
30. Bergeron A, Chevret S, Granata A, et al. Effect of azithromycin on airflow decline-free survival after allogeneic hematopoietic stem cell transplant: the ALLOZITHRO randomized clinical trial. JAMA 2017;318:557–66.
31. Gerhardt SG, McDyer JF, Girgis RE, et al. Maintenance azithromycin therapy for bronchiolitis obliterans syndrome: results of a pilot study. Am J Respir Crit Care Med 2003;168:121–5.
32. Khalid M, Al Saghir A, Saleemi S, et al. Azithromycin in bronchiolitis obliterans complicating bone marrow transplantation: a preliminary study. Eur Respir J 2005;25:490–3.
33. Maimon N, Lipton JH, Chan CK, Marras TK. Macrolides in the treatment of bronchiolitis obliterans in allograft recipients. Bone Marrow Transplant 2009;44:69–73.
34. Lam DC, Lam B, Wong MK, et al. Effects of azithromycin in bronchiolitis obliterans syndrome after hematopoietic SCT--a randomized double-blinded placebo-controlled study. Bone Marrow Transplant 2011;46:1551–6.
35. Yadav H, Peters SG, Keogh KA, et al. Azithromycin for the treatment of obliterative bronchiolitis after hematopoietic stem cell transplantation: a systematic review and meta-analysis. Biol Blood Marrow Transplant 2016;22:2264–9.
36. Vos R, Vanaudenaerde BM, Verleden SE, et al. A randomised controlled trial of azithromycin to prevent chronic rejection after lung transplantation. Eur Respir J 2011;37:164–72.
37. Svanstrom H, Pasternak B, Hviid A. Use of azithromycin and death from cardiovascular causes. N Engl J Med 2013;368:1704–12.
38. Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011;365:689–98.
39. Tran J, Norder EE, Diaz PT, et al. Pulmonary rehabilitation for bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2012;18:1250–4.
40. Lucid CE, Savani BN, Engelhardt BG, et al. Extracorporeal photopheresis in patients with refractory bronchiolitis obliterans developing after allo-SCT. Bone Marrow Transplant 2011;46:426–9.
41. Hostettler KE, Halter JP, Gerull S, et al. Calcineurin inhibitors in bronchiolitis obliterans syndrome following stem cell transplantation. Eur Respir J 2014;43:221–32.
42. Holm AM, Riise GC, Brinch L, et al. Lung transplantation for bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation: unresolved questions. Transplantation 2013;96:e21–22.
43. Cheng GS, Edelman JD, Madtes DK, et al. Outcomes of lung transplantation after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2014;20:1169–75.
44. Okumura H, Ohtake S, Ontachi Y, et al. Living-donor lobar lung transplantation for broncho-bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation: does bronchiolitis obliterans recur in transplanted lungs? Int J Hematol 2007;86:369–73.
45. Olivieri A, Cimminiello M, Corradini P, et al. Long-term outcome and prospective validation of NIH response criteria in 39 patients receiving imatinib for steroid-refractory chronic GVHD. Blood 2013;122:4111–8.
46. Rahmanian S, Wood KL. Bronchiolitis obliterans and the risk of pneumothorax after transbronchial biopsy. Respiratory Medicine CME 2010;3:87–9.
47. Sakai R, Kanamori H, Nakaseko C, et al. Air-leak syndrome following allo-SCT in adult patients: report from the Kanto Study Group for Cell Therapy in Japan. Bone Marrow Transplant 2011;46:379–84.
48. Visscher DW, Myers JL. Histologic spectrum of idiopathic interstitial pneumonias. Proc Am Thorac Soc 2006;3:322–9.
49. Cordier JF. Cryptogenic organising pneumonia. Eur Respir J 2006;28:422–46.
50. Nishio N, Yagasaki H, Takahashi Y, et al. Late-onset non-infectious pulmonary complications following allogeneic hematopoietic stem cell transplantation in children. Bone Marrow Transplant 2009;44:303–8.
51. Ueda K, Watadani T, Maeda E, et al. Outcome and treatment of late-onset noninfectious pulmonary complications after allogeneic haematopoietic SCT. Bone Marrow Transplant 2010;45:1719–27.
52. Schlemmer F, Chevret S, Lorillon G, et al. Late-onset noninfectious interstitial lung disease after allogeneic hematopoietic stem cell transplantation. Respir Med 2014;108:1525–33.
53. Palmas A, Tefferi A, Myers JL, et al. Late-onset noninfectious pulmonary complications after allogeneic bone marrow transplantation. Br J Haematol 1998;100:680–7.
54. Sakaida E, Nakaseko C, Harima A, et al. Late-onset noninfectious pulmonary complications after allogeneic stem cell transplantation are significantly associated with chronic graft-versus-host disease and with the graft-versus-leukemia effect. Blood 2003;102:4236–42.
55. Solh M, Arat M, Cao Q, et al. Late-onset noninfectious pulmonary complications in adult allogeneic hematopoietic cell transplant recipients. Transplantation 2011;91:798–803.
56. Dandoy CE, Hirsch R, Chima R, et al. Pulmonary hypertension after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:1546–56.
57. Bunte MC, Patnaik MM, Pritzker MR, Burns LJ. Pulmonary veno-occlusive disease following hematopoietic stem cell transplantation: a rare model of endothelial dysfunction. Bone Marrow Transplant 2008;41:677–86.
58. Troussard X, Bernaudin JF, Cordonnier C, et al. Pulmonary veno-occlusive disease after bone marrow transplantation. Thorax 1984;39:956–7.
59. Lahzami S, Schoeffel RE, Pechey V, et al. Small airways function declines after allogeneic haematopoietic stem cell transplantation. Eur Respir J 2011;38:1180–8.
60. Jain NA, Pophali PA, Klotz JK, et al. Repair of impaired pulmonary function is possible in very-long-term allogeneic stem cell transplantation survivors. Biol Blood Marrow Transplant 2014;20:209–13.
61. Barisione G, Bacigalupo A, Crimi E, et al. Changes in lung volumes and airway responsiveness following haematopoietic stem cell transplantation. Eur Respir J 2008;32:1576–82.
62. Kovalszki A, Schumaker GL, Klein A, et al. Reduced respiratory and skeletal muscle strength in survivors of sibling or unrelated donor hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;41:965–9.
63. Mathiesen S, Uhlving HH, Buchvald F, et al. Aerobic exercise capacity at long-term follow-up after paediatric allogeneic haematopoietic SCT. Bone Marrow Transplant 2014;49:1393–9.
1. Remberger M, Ackefors M, Berglund S, et al. Improved survival after allogeneic hematopoietic stem cell transplantation in recent years. A single-center study. Biol Blood Marrow Transplant 2011;17:1688–97.
2. Wood KL, Esguerra VG. Management of late pulmonary complications after hematopoietic stem cell transplantation. Hosp Phys Hematology-Oncology Board Review Manual 2018;13(1):36–48.
3. Ninin E, Milpied N, Moreau P, et al. Longitudinal study of bacterial, viral, and fungal infections in adult recipients of bone marrow transplants. Clin Infect Dis 2001;33:41–7.
4. Roca J, Granena A, Rodriguez-Roisin R, et al. Fatal airway disease in an adult with chronic graft-versus-host disease. Thorax 1982;37:77–8.
5. Williams KM, Chien JW, Gladwin MT, Pavletic SZ. Bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. JAMA 2009;302:306–14.
6. Chien JW, Martin PJ, Gooley TA, et al. Airflow obstruction after myeloablative allogeneic hematopoietic stem cell transplantation. Am J Respir Crit Care Med 2003;168:208–14.
7. Holbro A, Lehmann T, Girsberger S, et al. Lung histology predicts outcome of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:973–80.
8. Chamberlain D, Maurer J, Chaparro C, Idolor L. Evaluation of transbronchial lung biopsy specimens in the diagnosis of bronchiolitis obliterans after lung transplantation. J Heart Lung Transplant 1994;13:963–71.
9. Au BK, Au MA, Chien JW. Bronchiolitis obliterans syndrome epidemiology after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 2011;17:1072–8.
10. Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant 2015;21:389–401.
11. Couriel D, Carpenter PA, Cutler C, et al. Ancillary therapy and supportive care of chronic graft-versus-host disease: national institutes of health consensus development project on criteria for clinical trials in chronic Graft-versus-host disease: V. Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 2006;12:375–96.
12. Pellegrino R, Viegi G, Brusasco V, et al. Interpretative strategies for lung function tests. Eur Respir J 2005;26:948–68.
13. Williams KM, Hnatiuk O, Mitchell SA, et al. NHANES III equations enhance early detection and mortality prediction of bronchiolitis obliterans syndrome after hematopoietic SCT. Bone Marrow Transplant 2014;49:561–6.
14. Hyatt RE, Cowl CT, Bjoraker JA, Scanlon PD. Conditions associated with an abnormal nonspecific pattern of pulmonary function tests. Chest 2009;135:419–24.
15. Bergeron A, Godet C, Chevret S, et al. Bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: phenotypes and prognosis. Bone Marrow Transplant 2013;48:819–24.
16. Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant 2005;11:945–56.
17. Gazourian L, Coronata AM, Rogers AJ, et al. Airway dilation in bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. Respir Med 2013;107:276–83.
18. Gunn ML, Godwin JD, Kanne JP, et al. High-resolution CT findings of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. J Thorac Imaging 2008;23:244–50.
19. Sargent MA, Cairns RA, Murdoch MJ, et al. Obstructive lung disease in children after allogeneic bone marrow transplantation: evaluation with high-resolution CT. AJR Am J Roentgenol 1995;164:693–6.
20. Galban CJ, Boes JL, Bule M, et al. Parametric response mapping as an indicator of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2014;20:1592–8.
21. Meyer KC, Raghu G, Verleden GM, et al. An international ISHLT/ATS/ERS clinical practice guideline: diagnosis and management of bronchiolitis obliterans syndrome. Eur Respir J 2014;44:1479–1503.
22. Hildebrandt GC, Fazekas T, Lawitschka A, et al. Diagnosis and treatment of pulmonary chronic GVHD: report from the consensus conference on clinical practice in chronic GVHD. Bone Marrow Transplant 2011;46:1283–95.
23. Carpenter PA, Kitko CL, Elad S, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: V. The 2014 Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 2015;21:1167–87.
24. Norman BC, Jacobsohn DA, Williams KM, et al. Fluticasone, azithromycin and montelukast therapy in reducing corticosteroid exposure in bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: a case series of eight patients. Bone Marrow Transplant 2011;46:1369–73.
25. Williams KM, Cheng GS, Pusic I, et al. Fluticasone, azithromycin, and montelukast treatment for new-onset bronchiolitis obliterans syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transplant 2016;22:710–6.
26. Or R, Gesundheit B, Resnick I, et al. Sparing effect by montelukast treatment for chronic graft versus host disease: a pilot study. Transplantation 2007;83:577–81.
27. Cheng GS, Storer B, Chien JW, et al. Lung function trajectory in bronchiolitis obliterans syndrome after allogeneic hematopoietic cell transplant. Ann Am Thorac Soc 2016;13:1932–9.
28. Bergeron A, Belle A, Chevret S, et al. Combined inhaled steroids and bronchodilatators in obstructive airway disease after allogeneic stem cell transplantation. Bone Marrow Transplant 2007;39:547–53.
29. Bashoura L, Gupta S, Jain A, et al. Inhaled corticosteroids stabilize constrictive bronchiolitis after hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;41:63–7.
30. Bergeron A, Chevret S, Granata A, et al. Effect of azithromycin on airflow decline-free survival after allogeneic hematopoietic stem cell transplant: the ALLOZITHRO randomized clinical trial. JAMA 2017;318:557–66.
31. Gerhardt SG, McDyer JF, Girgis RE, et al. Maintenance azithromycin therapy for bronchiolitis obliterans syndrome: results of a pilot study. Am J Respir Crit Care Med 2003;168:121–5.
32. Khalid M, Al Saghir A, Saleemi S, et al. Azithromycin in bronchiolitis obliterans complicating bone marrow transplantation: a preliminary study. Eur Respir J 2005;25:490–3.
33. Maimon N, Lipton JH, Chan CK, Marras TK. Macrolides in the treatment of bronchiolitis obliterans in allograft recipients. Bone Marrow Transplant 2009;44:69–73.
34. Lam DC, Lam B, Wong MK, et al. Effects of azithromycin in bronchiolitis obliterans syndrome after hematopoietic SCT--a randomized double-blinded placebo-controlled study. Bone Marrow Transplant 2011;46:1551–6.
35. Yadav H, Peters SG, Keogh KA, et al. Azithromycin for the treatment of obliterative bronchiolitis after hematopoietic stem cell transplantation: a systematic review and meta-analysis. Biol Blood Marrow Transplant 2016;22:2264–9.
36. Vos R, Vanaudenaerde BM, Verleden SE, et al. A randomised controlled trial of azithromycin to prevent chronic rejection after lung transplantation. Eur Respir J 2011;37:164–72.
37. Svanstrom H, Pasternak B, Hviid A. Use of azithromycin and death from cardiovascular causes. N Engl J Med 2013;368:1704–12.
38. Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011;365:689–98.
39. Tran J, Norder EE, Diaz PT, et al. Pulmonary rehabilitation for bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2012;18:1250–4.
40. Lucid CE, Savani BN, Engelhardt BG, et al. Extracorporeal photopheresis in patients with refractory bronchiolitis obliterans developing after allo-SCT. Bone Marrow Transplant 2011;46:426–9.
41. Hostettler KE, Halter JP, Gerull S, et al. Calcineurin inhibitors in bronchiolitis obliterans syndrome following stem cell transplantation. Eur Respir J 2014;43:221–32.
42. Holm AM, Riise GC, Brinch L, et al. Lung transplantation for bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation: unresolved questions. Transplantation 2013;96:e21–22.
43. Cheng GS, Edelman JD, Madtes DK, et al. Outcomes of lung transplantation after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2014;20:1169–75.
44. Okumura H, Ohtake S, Ontachi Y, et al. Living-donor lobar lung transplantation for broncho-bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation: does bronchiolitis obliterans recur in transplanted lungs? Int J Hematol 2007;86:369–73.
45. Olivieri A, Cimminiello M, Corradini P, et al. Long-term outcome and prospective validation of NIH response criteria in 39 patients receiving imatinib for steroid-refractory chronic GVHD. Blood 2013;122:4111–8.
46. Rahmanian S, Wood KL. Bronchiolitis obliterans and the risk of pneumothorax after transbronchial biopsy. Respiratory Medicine CME 2010;3:87–9.
47. Sakai R, Kanamori H, Nakaseko C, et al. Air-leak syndrome following allo-SCT in adult patients: report from the Kanto Study Group for Cell Therapy in Japan. Bone Marrow Transplant 2011;46:379–84.
48. Visscher DW, Myers JL. Histologic spectrum of idiopathic interstitial pneumonias. Proc Am Thorac Soc 2006;3:322–9.
49. Cordier JF. Cryptogenic organising pneumonia. Eur Respir J 2006;28:422–46.
50. Nishio N, Yagasaki H, Takahashi Y, et al. Late-onset non-infectious pulmonary complications following allogeneic hematopoietic stem cell transplantation in children. Bone Marrow Transplant 2009;44:303–8.
51. Ueda K, Watadani T, Maeda E, et al. Outcome and treatment of late-onset noninfectious pulmonary complications after allogeneic haematopoietic SCT. Bone Marrow Transplant 2010;45:1719–27.
52. Schlemmer F, Chevret S, Lorillon G, et al. Late-onset noninfectious interstitial lung disease after allogeneic hematopoietic stem cell transplantation. Respir Med 2014;108:1525–33.
53. Palmas A, Tefferi A, Myers JL, et al. Late-onset noninfectious pulmonary complications after allogeneic bone marrow transplantation. Br J Haematol 1998;100:680–7.
54. Sakaida E, Nakaseko C, Harima A, et al. Late-onset noninfectious pulmonary complications after allogeneic stem cell transplantation are significantly associated with chronic graft-versus-host disease and with the graft-versus-leukemia effect. Blood 2003;102:4236–42.
55. Solh M, Arat M, Cao Q, et al. Late-onset noninfectious pulmonary complications in adult allogeneic hematopoietic cell transplant recipients. Transplantation 2011;91:798–803.
56. Dandoy CE, Hirsch R, Chima R, et al. Pulmonary hypertension after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:1546–56.
57. Bunte MC, Patnaik MM, Pritzker MR, Burns LJ. Pulmonary veno-occlusive disease following hematopoietic stem cell transplantation: a rare model of endothelial dysfunction. Bone Marrow Transplant 2008;41:677–86.
58. Troussard X, Bernaudin JF, Cordonnier C, et al. Pulmonary veno-occlusive disease after bone marrow transplantation. Thorax 1984;39:956–7.
59. Lahzami S, Schoeffel RE, Pechey V, et al. Small airways function declines after allogeneic haematopoietic stem cell transplantation. Eur Respir J 2011;38:1180–8.
60. Jain NA, Pophali PA, Klotz JK, et al. Repair of impaired pulmonary function is possible in very-long-term allogeneic stem cell transplantation survivors. Biol Blood Marrow Transplant 2014;20:209–13.
61. Barisione G, Bacigalupo A, Crimi E, et al. Changes in lung volumes and airway responsiveness following haematopoietic stem cell transplantation. Eur Respir J 2008;32:1576–82.
62. Kovalszki A, Schumaker GL, Klein A, et al. Reduced respiratory and skeletal muscle strength in survivors of sibling or unrelated donor hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;41:965–9.
63. Mathiesen S, Uhlving HH, Buchvald F, et al. Aerobic exercise capacity at long-term follow-up after paediatric allogeneic haematopoietic SCT. Bone Marrow Transplant 2014;49:1393–9.
Management of Early Pulmonary Complications After Hematopoietic Stem Cell Transplantation
Hematopoietic stem cell transplantation (HSCT) is widely used in the economically developed world to treat a variety of hematologic malignancies as well as nonmalignant diseases and solid tumors. An estimated 17,900 HSCTs were performed in 2011, and survival rates continue to increase.1 Pulmonary complications post HSCT are common, with rates ranging from 40% to 60%, and are associated with increased morbidity and mortality.2
Clinical diagnosis of pulmonary complications in the HSCT population has been aided by a previously well-defined chronology of the most common diseases.3 Historically, early pulmonary complications were defined as pulmonary complications occurring within 100 days of HSCT (corresponding to the acute graft-versus-host disease [GVHD] period). Late pulmonary complications are those that occur thereafter. This timeline, however, is now more variable given the increasing indications for HSCT, the use of reduced-intensity conditioning strategies, and varied individual immune reconstitution. This article discusses the management of early post-HSCT pulmonary complications; late post-HSCT pulmonary complications will be discussed in a separate follow-up article.
Transplant Basics
The development of pulmonary complications is affected by many factors associated with the transplant. Autologous transplantation involves the collection of a patient’s own stem cells, appropriate storage and processing, and re-implantation after induction therapy. During induction therapy, the patient undergoes high-dose chemotherapy or radiation therapy that ablates the bone marrow. The stem cells are then transfused back into the patient to repopulate the bone marrow. Allogeneic transplants involve the collection of stem cells from a donor. Donors are matched as closely as possible to the recipient’s histocompatibility antigen (HLA) haplotypes to prevent graft failure and rejection. The donor can be related or unrelated to the recipient. If there is not a possibility of a related match (from a sibling), then a national search is undertaken to look for a match through the National Marrow Donor Program. There are fewer transplant reactions and occurrences of GVHD if the major HLAs of the donor and recipient match. Table 1 reviews basic definitions pertaining to HSCT.
How the cells for transplantation are obtained is also an important factor in the rate of complications. There are 3 main sources: peripheral blood, bone marrow, and umbilical cord. Peripheral stem cell harvesting involves exposing the donor to granulocyte-colony stimulating factor (gCSF), which increases peripheral circulation of stem cells. These cells are then collected and infused into the recipient after the recipient has completed an induction regimen involving chemotherapy and/or radiation, depending on the protocol. This procedure is called peripheral blood stem cell transplant (PBSCT). Stem cells can also be directly harvested from bone marrow cells, which are collected from repeated aspiration of bone marrow from the posterior iliac crest.4 This technique is most common in children, whereas in adults peripheral blood stem cells are the most common source. Overall mortality does not differ based on the source of the stem cells. It is postulated that GVHD may be more common in patients undergoing PBSCT, but the graft failure rate may be lower.5
The third option is umbilical cord blood (UCB) as the source of stem cells. This involves the collection of umbilical cord blood that is prepared and frozen after birth. It has a smaller volume of cells, and although fewer cells are needed when using UCB, 2 separate donors may be required for a single adult recipient. The engraftment of the stem cells is slower and infections in the post-transplant period are more common. Prior reports indicate GVHD rates may be lower.4 While the use of UCB is not common in adults, the incidence has doubled over the past decade, increasing from 3% to 6%.
The conditioning regimen can influence pulmonary complications. Traditionally, an ablative transplant involves high-dose chemotherapy or radiation to eradicate the recipient’s bone marrow. This regimen can lead to many complications, especially in the immediate post-transplant period. In the past 10 years, there has been increasing interest in non-myeloablative, or reduced-intensity, conditioning transplants.6 These “mini transplants” involve smaller doses of chemotherapy or radiation, which do not totally eradicate the bone marrow; after the transplant a degree of chimerism develops where the donor and recipient stem cells coexist. The medications in the preparative regimen also should be considered because they can affect pulmonary complications after transplant. Certain chemotherapeutic agents such as carmustine, bleomycin, and many others can lead to acute and chronic presentations of pulmonary diseases such as hypersensitivity pneumonitis, pulmonary fibrosis, acute respiratory distress syndrome, and abnormal pulmonary function testing.
After the HSCT, GVHD can develop in more than 50% of allogeneic recipients.3 The incidence of GVHD has been reported to be increasing over the past 12 years.It is divided into acute GVHD (which traditionally happens in the first 100 days after transplant) and chronic GVHD (after day 100). This calendar-day–based system has been augmented based on a 2006 National Institutes of Health working group report emphasizing the importance of organ-specific features of chronic GVHD in the clinical presentation of GVHD.7 Histologic changes in chronic organ GVHD tend to include more fibrotic features, whereas in acute GVHD more inflammatory changes are seen. The NIH working group report also stressed the importance of obtaining a biopsy specimen for histopathologic review and interdisciplinary collaboration to arrive at a consensus diagnosis, and noted the limitations of using histologic changes as the sole determinant of a “gold standard” diagnosis.7 GVHD can directly predispose patients to pulmonary GVHD and indirectly predispose them to infectious complications because the mainstay of therapy for GVHD is increased immunosuppression.
Pretransplant Evaluation
Case Patient 1
A 56-year-old man is diagnosed with acute myeloid leukemia (AML) after presenting with signs and symptoms consistent with pancytopenia. He has a past medical history of chronic sinus congestion, arthritis, depression, chronic pain, and carpal tunnel surgery. He is employed as an oilfield worker and has a 40-pack-year smoking history, but he recently cut back to half a pack per day. He is being evaluated for allogeneic transplant with his brother as the donor and the planned conditioning regimen is total body irradiation (TBI), thiotepa, cyclophosphamide, and antithymocyte globulin with T-cell depletion. Routine pretransplant pulmonary function testing (PFT) reveals a restrictive pattern and he is sent for pretransplant pulmonary evaluation.
Physical exam reveals a chronically ill appearing man. He is afebrile, the respiratory rate is 16 breaths/min, blood pressure is 145/88 mm Hg, heart rate is 92 beats/min, and oxygen saturation is 95%. He is in no distress. Auscultation of the chest reveals slightly diminished breath sounds bilaterally but is clear and without wheezes, rhonchi, or rales. Heart exam shows regular rate and rhythm without murmurs, rubs, or gallops. Extremities reveal no edema or rashes. Otherwise, the remainder of the exam is normal. The patient’s PFT results are shown in Table 2. 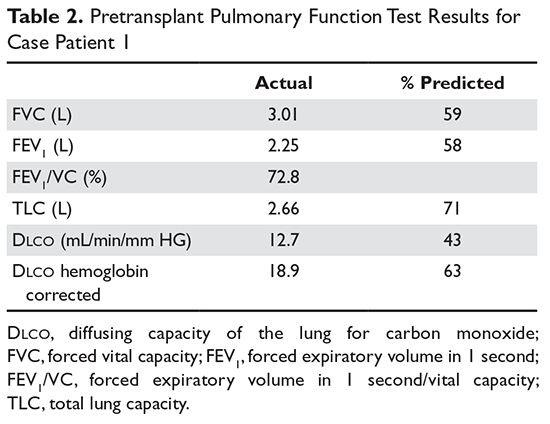
- What aspects of this patient’s history put him at risk for pulmonary complications after transplantation?
Risk Factors for Pulmonary Complications
Predicting who is at risk for pulmonary complications is difficult. Complications are generally divided into infectious and noninfectious categories. Regardless of category, allogeneic HSCT recipients are at increased risk compared with autologous recipients, but even in autologous transplants, more than 25% of patients will develop pulmonary complications in the first year.8 Prior to transplant, patients undergo full PFT. Early on, many studies attempted to show relationships between various factors and post-transplant pulmonary complications. Factors that were implicated were forced expiratory volume in 1 second (FEV1), diffusing capacity of the lung for carbon monoxide (D
Another sometimes overlooked risk before transplantation is restrictive lung disease. One study showed a twofold increase in respiratory failure and mortality if there was pretransplant restriction based on TLC < 80%.16
An interesting study by one group in pretransplant evaluation found decreased muscle strength by maximal inspiratory muscle strength (PImax), maximal expiratory muscle strength (PEmax), dominant hand grip strength, and 6-minute walk test (6MWT) distance prior to allogeneic transplant, but did not find a relationship between these variables and mortality.17 While this study had a small sample size, these findings likely deserve continued investigation.18
- What methods are used to calculate risk for complications?
Risk Scoring Systems
Several pretransplantation risk scores have been developed. In a study that looked at more than 2500 allogeneic transplants, Parimon et al showed that risk of mortality and respiratory failure could be estimated prior to transplant using a scoring system—the Lung Function Score (LFS)—that combines the FEV1 and D
The Pretransplantation Assessment of Mortality score, initially developed in 2006, predicts mortality within the first 2 years after HSCT based on 8 clinical factors: disease risk, age at transplant, donor type, conditioning regimen, and markers of organ function (percentage of predicted FEV1, percentage of predicted D
- What other preoperative testing or interventions should be considered in this patient?
Since there is a high risk of infectious complications after transplant, the question of whether pretransplantation patients should undergo screening imaging may arise. There is no evidence that routine chest computed tomography (CT) reduces the risk of infectious complications after transplantation.26 An area that may be insufficiently addressed in the pretransplantation evaluation is smoking cessation counseling.27 Studies have shown an elevated risk of mortality in smokers.28-30 Others have found a higher incidence of respiratory failure but not an increased mortality.31 Overall, with the good rates of smoking cessation that can be accomplished, smokers should be counseled to quit before transplantation.
In summary, patients should undergo full PFTs prior to transplantation to help stratify risk for pulmonary complications and mortality and to establish a clinical baseline. The LFS (using FEV1 and D
Case Patient 1 Conclusion
The patient undergoes transplantation due to his lack of other treatment options. Evaluation prior to transplant, however, shows that he is at high risk for pulmonary complications. He has a LFS of 7 prior to transplant (using the D
Early Infectious Pulmonary Complications
Case Patient 2
A 27-year-old man with a medical history significant for AML and allogeneic HSCT presents with cough productive of a small amount of clear to white sputum, dyspnea on exertion, and fevers for 1 week. He also has mild nausea and a decrease in appetite. He underwent HSCT 2.5 months prior to admission, which was a matched unrelated bone marrow transplant with TBI and cyclophosphamide conditioning. His past medical history is significant only for exercise-induced asthma for which he takes a rescue inhaler infrequently prior to transplantation. His pretransplant PFTs showed normal spirometry with an FEV1 of 106% of predicted and D
Physical exam is notable for fever of 101.0°F, heart rate 80 beats/min, respiratory rate 16 breaths/ min, and blood pressure 142/78 mm Hg; an admission oxygen saturation is 93% on room air. Lungs show bibasilar crackles and the remainder of the exam is normal. Laboratory testing shows a white blood cell count of 2400 cells/μL, hemoglobin 7.6 g/dL, and platelet count 66 × 103/μL. Creatinine is 1.0 mg/dL. Chest radiograph shows ill-defined bilateral lower-lobe infiltrates. CT scans are shown in the Figure. 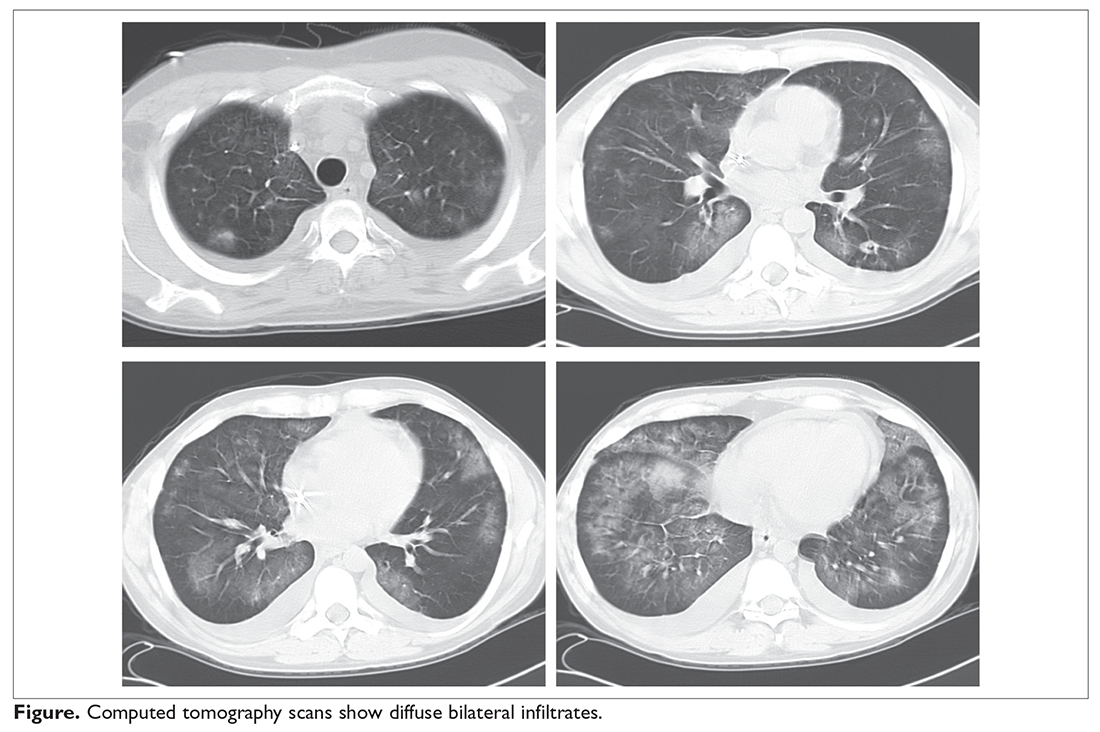
- For which infectious complications is this patient most at risk?
Pneumonia
A prospective trial in the HSCT population reported a pneumonia incidence rate of 68%, and pneumonia is more common in allogeneic HSCT with prolonged immunosuppressive therapy.32 Development of pneumonia within 100 days of transplant directly correlates with nonrelapsed mortality.33 Early detection is key, and bronchoscopy within the first 5 days of symptoms has been shown to change therapy in approximately 40% of cases but has not been shown to affect mortality.34 The clinical presentation of pneumonia in the HSCT population can be variable because of the presence of neutropenia and profound immunosuppression. Traditionally accepted diagnostic criteria of fevers, sputum production, and new infiltrates should be used with caution, and an appropriately high index of suspicion should be maintained. Progression to respiratory failure, regardless of causative organism of infection, portends a poor prognosis, with mortality rates estimated at 70% to 90%.35,36 Several transplant-specific factors may affect early infections. For instance, UCB transplants have been found to have a higher incidence of invasive aspergillosis and cytomegalovirus (CMV) infections but without higher mortality attributed to the infections.37
Bacterial Pneumonia
Bacterial pneumonia accounts for 20% to 50% of pneumonia cases in HSCT recipients.38 Gram-negative organisms, specifically Pseudomonas aeruginosa and Escherichia coli, were reported to be the most common pathologic bacteria in recent prospective trials, whereas previous retrospective trials showed that common community-acquired organisms were the most common cause of pneumonia in HSCT recipients.32,39 This underscores the importance of being aware of the clinical prevalence of microorganisms and local antibiograms, along with associated institutional susceptibility profiles. Initiation of immediate empiric broad-spectrum antibiotics is essential when bacterial pneumonia is suspected.
Viral Pneumonia
The prevalence of viral pneumonia in stem cell transplant recipients is estimated at 28%,32 with most cases being caused by community viral pathogens such as rhinovirus, respiratory syncytial virus (RSV), influenza A and B, and parainfluenza.39 The prevention, prophylaxis, and early treatment of viral pneumonias, specifically CMV infection, have decreased the mortality associated with early pneumonia after HSCT. Co-infection with bacterial organisms must be considered and has been associated with increased mortality in the intensive care unit setting.40
Supportive treatment with rhinovirus infection is sufficient as the disease is usually self-limited in immunocompromised patients. In contrast, infection with RSV in the lower respiratory tract is associated with increased mortality in prior reports, and recent studies suggest that further exploration of prophylaxis strategies is warranted.41 Treatment with ribavirin remains the backbone of therapy, but drug toxicity continues to limit its use. The addition of immunomodulators such as RSV immune globulin or palivizumab to ribavirin remains controversial, but a retrospective review suggests that early treatment may prevent progression to lower respiratory tract infection and lead to improved mortality.42 Infection with influenza A/B must be considered during influenza season. Treatment with oseltamivir may shorten the duration of disease when influenza A/B or parainfluenza are detected. Reactivation of latent herpes simplex virus during the pre-engraftment phase should also be considered. Treatment is similar to that in nonimmunocompromised hosts. When CMV pneumonia is suspected, careful history regarding compliance with prophylactic antivirals and CMV status of both the recipient and donor are key. A presumptive diagnosis can be made with the presence of appropriate clinical scenario, supportive radiographic images showing areas of ground-glass opacification or consolidation, and positive CMV polymerase chain reaction (PCR) assay. Visualization of inclusion bodies on lung biopsy tissue remains the gold standard for diagnosis. Treatment consists of CMV immunoglobulin and ganciclovir.
Fungal Pneumonia
Early fungal pneumonias have been associated with increased mortality in the HSCT population.43 Clinical suspicion should remain high and compliance with antifungal prophylaxis should be questioned thoroughly. Invasive aspergillosis (IA) remains the most common fungal infection. A bimodal distribution of onset of infection peaking on day 16 and again on day 96 has been described in the literature.44 Patients often present with classic pneumonia symptoms, but these may be accompanied by hemoptysis. Proven IA diagnosis requires visualization of fungal forms from biopsy or needle aspiration or a positive culture obtained in a sterile fashion.45 Most clinical data comes from experience with probable and possible diagnosis of IA. Bronchoalveolar lavage with testing with Aspergillus galactomannan assay has been shown to be clinically useful in establishing the clinical diagnosis in the HSCT population.46 Classic air-crescent findings on chest CT are helpful in establishing a possible diagnosis, but retrospective analysis reveals CT findings such as focal infiltrates and pulmonary nodular patterns are more common.47 First-line treatment with voriconazole has been shown to decrease short-term mortality attributable to IA but has not had an effect on long-term, all-cause mortality.48 Surgical resection is reserved for patients with refractory disease or patients presenting with massive hemoptysis.
Mucormycosis is an emerging disease with ever increasing prevalence in the HSCT population, reflecting the improved prophylaxis and treatment of IA. Initial clinical presentation is similar to IA, most commonly affecting the lung, although craniofacial involvement is classic for mucormycosis, especially in HSCT patients with diabetes.49Mucor infections can present with massive hemoptysis due to tissue invasion and disregard for tissue and fascial planes. Diagnosis of mucormycosis is associated with as much as a six-fold increase in risk for death. Diagnosis requires identification of the organism by examination or culture and biopsy is often necessary.50,51 Amphotericin B remains first-line therapy as mucormycosis is resistant to azole antifungals, with higher doses recommended for cerebral involvement.52
Candida pulmonary infections during the early HSCT period are becoming increasingly rare due to widespread use of fluconazole prophylaxis and early treatment of mucosal involvement during neutropenia. Endemic fungal infections such as blastomycosis, coccidioidomycosis, and histoplasmosis should be considered in patients inhabiting specific geographic areas or with recent travel to these areas.
- What test should be performed to evaluate for infectious causes of pneumonia?
Role of Flexible Fiberoptic Bronchoscopy
The utility of flexible fiberoptic bronchoscopy (FOB) in immune-compromised patients for the evaluation of pulmonary infiltrates is a frequently debated topic. Current studies suggest a diagnosis can be made in approximately 80% of cases in the immune-compromised population.32,53 Noninvasive testing such as urine and serum antigens, sputum cultures, Aspergillus galactomannan assays, viral nasal swabs, and PCR studies often lead to a diagnosis in appropriate clinical scenarios. Conservative management would dictate the use of noninvasive testing whenever possible, and randomized controlled trials have shown noninvasive testing to be noninferior to FOB in preventing need for mechanical ventilation, with no difference in overall mortality.54 FOB has been shown to be most useful in establishing a diagnosis when an infectious etiology is suspected.55 In multivariate analysis, a delay in the identification of the etiology of pulmonary infiltrate was associated with increased mortality.56 Additionally, early FOB was found to be superior to late FOB in revealing a diagnosis. 32,57 Despite its ability to detect the cause of pulmonary disease, direct antibiotic therapy, and possibly change therapy, FOB with diagnostic maneuvers has not been shown to affect mortality.58 In a large case series, FOB with bronchoalveolar lavage (BAL) revealed a diagnosis in approximately 30% to 50% of cases. The addition of transbronchial biopsy did not improve diagnostic utility.58 More recent studies have confirmed that the addition of transbronchial biopsy does not add to diagnostic yield and is associated with increased adverse events.59 The appropriate use of advanced techniques such as endobronchial ultrasound–guided transbronchial needle aspirations, endobronchial biopsy, and CT-guided navigational bronchoscopy has not been established and should be considered on a case-by-case basis. In summary, routine early BAL is the diagnostic test of choice, especially when infectious pulmonary complications are suspected.
Contraindications for FOB in this population mirror those in the general population. These include acute severe hypoxemic respiratory failure, myocardial ischemia or acute coronary syndrome within 2 weeks of procedure, severe thrombocytopenia, and inability to provide or obtain informed consent from patient or health care power of attorney. Coagulopathy and thrombocytopenia are common comorbid conditions in the HSCT population. A platelet count of < 20 × 103/µL has generally been used as a cut-off for routine FOB with BAL.60 Risks of the procedures should be discussed clearly with the patient, but simple FOB for airway evaluation and BAL is generally well tolerated even under these conditions.
Early Nonifectious Pulmonary Complications
Case Patient 2 Continued
Bronchoscopy with BAL performed the day after admission is unremarkable and stains and cultures are negative for viral, bacterial, and fungal organisms. The patient is initially started on broad-spectrum antibiotics, but his oxygenation continues to worsen to the point that he is placed on noninvasive positive pressure ventilation. He is started empirically on amphotericin B and eventually is intubated. VATS lung biopsy is ultimately performed and pathology is consistent with diffuse alveolar damage.
- Based on these biopsy findings, what is the diagnosis?
Based on the pathology consistent with diffuse alveolar damage, a diagnosis of idiopathic pneumonia syndrome (IPS) is made.
- What noninfectious pulmonary complications occur in the early post-transplant period?
The overall incidence of noninfectious pulmonary complications after HSCT is generally estimated at 20% to 30%.32 Acute pulmonary edema is a common very early noninfectious pulmonary complication and clinically the most straightforward to treat. Three distinct clinical syndromes—peri-engraftment respiratory distress syndrome (PERDS), diffuse alveolar hemorrhage (DAH), and IPS—comprise the remainder of the pertinent early noninfectious complications. Clinical presentation differs based upon the disease entity. Recent studies have evaluated the role of angiotensin-converting enzyme polymorphisms as a predictive marker for risk of developing early noninfectious pulmonary complications.61
Peri-Engraftment Respiratory Distress Syndrome
PERDS is a clinical syndrome comprising the cardinal features of erythematous rash and fever along with noncardiogenic pulmonary infiltrates and hypoxemia that occur in the peri-engraftment period, defined as recovery of absolute neutrophil count to > 500/μL on 2 consecutive days.62 PERDS occurs in the autologous HSCT population and may be a clinical correlate to early GVHD in the allogeneic HSCT population. It is hypothesized that the pathophysiology underlying PERDS is an autoimmune-related capillary leak caused by pro-inflammatory cytokine release.63 Treatment remains anecdotal and currently consists of supportive care and high-dose corticosteroids. Some have favored limiting the use of gCSF given its role in stimulating rapid white blood cell recovery.33 Prognosis is favorable, but progression to fulminant respiratory failure requiring mechanical ventilation portends a poor prognosis.
Diffuse Alveolar Hemorrhage
DAH is clinical syndrome consisting of diffuse alveolar infiltrates on pulmonary imaging combined with progressively bloodier return per aliquot during BAL in 3 different subsegments or more than 20% hemosiderin-laden macrophages on BAL fluid evaluation. Classically, DAH is defined in the absence of pulmonary infection or cardiac dysfunction. The pathophysiology is thought to be related to inflammation of pulmonary vasculature within the alveolar walls leading to alveolitis. Although no prospective trials exist, early use of high-dose corticosteroid therapy is thought to improve outcomes;64,65 a recent study, however, showed low-dose steroids may be associated with the lowest mortality.66 Mortality is directly linked to the presence of superimposed infection, need for mechanical ventilation, late onset, and development of multiorgan failure.67
Idiopathic Pneumonia Syndrome
IPS is a complex clinical syndrome whose pathology is felt to stem from a variety of possible lung insults such as direct myeloablative drug toxicity, occult pulmonary infection, or cytokine-driven inflammation. The ATS published an article further subcategorizing IPS as different clinical entities based upon whether the primary insult involves the vascular endothelium, interstitial tissue, and airway tissue, truly idiopathic, or unclassified.68 In clinical practice, IPS is defined as widespread alveolar injury in the absence of evidence of renal failure, heart failure, and excessive fluid resuscitation. In addition, negative testing for a variety of bacterial, viral, and fungal causes is also necessary.69 Clinical syndromes included within the IPS definition are ARDS, acute interstitial pneumonia, DAH, cryptogenic organizing pneumonia, and BOS.70 Risk factors for developing IPS include TBI, older age of recipient, acute GVHD, and underlying diagnosis of AML or myelodysplastic syndrome.12 In addition, it has been shown that risk for developing IPS is lower in patients undergoing allogeneic HSCT who receive non-myeloablative conditioning regimens.71 The pathologic finding in IPS is diffuse alveolar damage. A 2006 study in which investigators reviewed BAL samples from patients with IPS found that 3% of the patients had PCR evidence of human metapneumovirus infection, and a study in 2015 found PCR evidence of infection in 53% of BAL samples from patients diagnosed with IPS.72,73 This fuels the debate on whether IPS is truly an infection-driven process where the source of infection, pulmonary or otherwise, simply escapes detection. Various surfactant proteins, which play a role in decreasing surface tension within the alveolar interface and function as mediators within the innate immunity of the lung, have been studied in regard to development of IPS. Small retrospective studies have shown a trend toward lower pre-transplant serum protein surfactant D and the development of IPS.74
The diagnosis of IPS does not require pathologic diagnosis in most circumstances. The correct clinical findings in association with a negative infectious workup lead to a presumptive diagnosis of IPS. The extent of the infectious workup that must be completed to adequately rule out infection is often a difficult clinical question. Recent recommendations include BAL fluid evaluation for routine bacterial cultures, appropriate viral culture, and consideration of PCR testing to evaluate for Mycoplasma, Chlamydia, and Aspergillus antigens.75 Transbronchial biopsy continues to appear in recommendations, but is not routinely performed and should be completed as the patient’s clinical status permits.8,68 Table 3 reviews basic features of early noninfectious pulmonary complications. 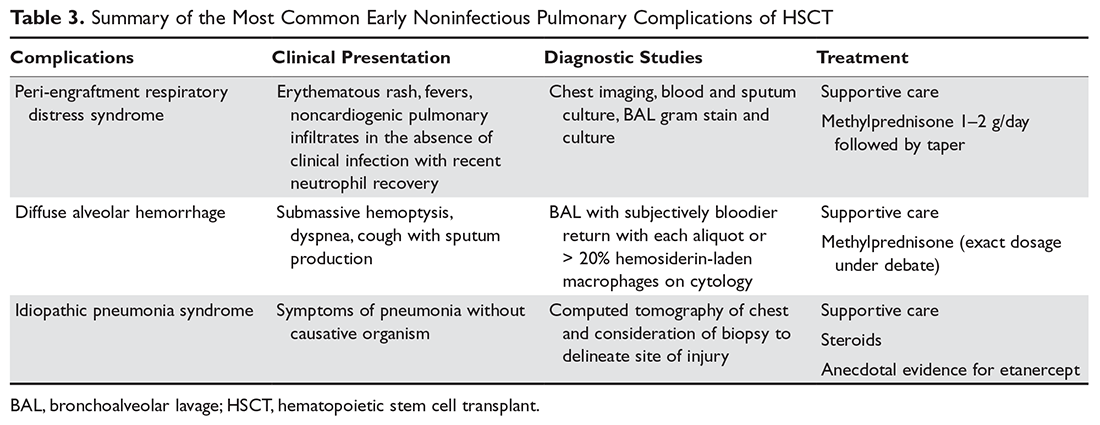
Treatment of IPS centers around moderate to high doses of corticosteroids. Based on IPS experimental modes, tumor necrosis factor (TNF)-α has been implicated as an important mediator. Unfortunately, several studies evaluating etanercept have produced conflicting results, and this agent’s clinical effects on morbidity and mortality remain in question.76
- What treatment should be offered to the patient with diffuse alveolar damage on biopsy?
Treatment consists of supportive care and empiric broad-spectrum antibiotics with consideration of high-dose corticosteroids. Based upon early studies in murine models implicating TNF, pilot studies were performed evaluating etanercept as a possible safe and effective addition to high-dose systemic corticosteroids.77 Although these results were promising, data from a truncated randomized control clinical trial failed to show improvement in patient response in the adult population.76 More recent data from the same author suggests that pediatric populations with IPS are, however, responsive to etanercept and high-dose corticosteroid therapy.78 When IPS develops as a late complication, treatment with high-dose corticosteroids (2 mg/kg/day) and etanercept (0.4 mg/kg twice weekly) has been shown to improve 2-year survival.79
Case Patient 2 Conclusion
The patient is started on steroids and makes a speedy recovery. He is successfully extubated 5 days later.
Conclusion
Careful pretransplant evaluation, including a full set of pulmonary function tests, can help predict a patient’s risk for pulmonary complications after transplant, allowing risk factor modification strategies to be implemented prior to transplant, including smoking cessation. It also helps identify patients at high risk for complications who will require closer monitoring after transplantation. Early posttransplant complications include infectious and noninfectious entities. Bacterial, viral, and fungal pneumonias are in the differential of infectious pneumonia, and bronchoscopy can be helpful in establishing a diagnosis. A common, important noninfectious cause of early pulmonary complications is IPS, which is treated with steroids and sometimes anti-TNF therapy.
1. Gratwohl A, Baldomero H, Aljurf M, et al. Hematopoietic stem cell transplantation: a global perspective. JAMA 2010;303:1617–24.
2. Kotloff RM, Ahya VN, Crawford SW. Pulmonary complications of solid organ and hematopoietic stem cell transplantation. Am J Respir Crit Care Med 2004;170:22–48.
4. Copelan EA. Hematopoietic stem-cell transplantation. N Engl J Med 2006;354:1813–26.
5. Anasetti C, Logan BR, Lee SJ, et al. Peripheral-blood stem cells versus bone marrow from unrelated donors. N Engl J Med 2012;367:1487–96.
6. Giralt S, Ballen K, Rizzo D, et al. Reduced-intensity conditioning regimen workshop: defining the dose spectrum. Report of a workshop convened by the center for international blood and marrow transplant research. Biol Blood Marrow Transplant 2009;15:367–9.
7. Shulman HM, Kleiner D, Lee SJ, et al. Histopathologic diagnosis of chronic graft-versus-host disease: National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: II. Pathology Working Group Report. Biol Blood Marrow Transplant 2006;12:31–47.
8. Afessa B, Abdulai RM, Kremers WK, et al. Risk factors and outcome of pulmonary complications after autologous hematopoietic stem cell transplant. Chest 2012;141:442–50.
9. Bolwell BJ. Are predictive factors clinically useful in bone marrow transplantation? Bone Marrow Transplant 2003;32:853–61.
10. Carlson K, Backlund L, Smedmyr B, et al. Pulmonary function and complications subsequent to autologous bone marrow transplantation. Bone Marrow Transplant 1994;14:805–11.
11. Clark JG, Schwartz DA, Flournoy N, et al. Risk factors for airflow obstruction in recipients of bone marrow transplants. Ann Intern Med 1987;107:648–56.
12. Crawford SW, Fisher L. Predictive value of pulmonary function tests before marrow transplantation. Chest 1992; 101:1257–64.
13. Ghalie R, Szidon JP, Thompson L, et al. Evaluation of pulmonary complications after bone marrow transplantation: the role of pretransplant pulmonary function tests. Bone Marrow Transplant 1992;10:359–65.
14. Ho VT, Weller E, Lee SJ, et al. Prognostic factors for early severe pulmonary complications after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2001;7:223–9.
15. Horak DA, Schmidt GM, Zaia JA, et al. Pretransplant pulmonary function predicts cytomegalovirus-associated interstitial pneumonia following bone marrow transplantation. Chest 1992;102:1484–90.
16. Ramirez-Sarmiento A, Orozco-Levi M, Walter EC, et al. Influence of pretransplantation restrictive lung disease on allogeneic hematopoietic cell transplantation outcomes. Biol Blood Marrow Transplant 2010;16:199–206.
17. White AC, Terrin N, Miller KB, Ryan HF. Impaired respiratory and skeletal muscle strength in patients prior to hematopoietic stem-cell transplantation. Chest 2005;128145–52.
18. Afessa B. Pretransplant pulmonary evaluation of the blood and marrow transplant recipient. Chest 2005;128:8–10.
19. Parimon T, Madtes DK, Au DH, et al. Pretransplant lung function, respiratory failure, and mortality after stem cell transplantation. Am J Respir Crit Care Med 2005;172:384–90.
20. Pavletic SZ, Martin P, Lee SJ, et al. Measuring therapeutic response in chronic graft-versus-host disease: National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: IV. Response Criteria Working Group report. Biol Blood Marrow Transplant 2006;12:252–66.
21. Parimon T, Au DH, Martin PJ, Chien JW. A risk score for mortality after allogeneic hematopoietic cell transplantation. Ann Intern Med 2006;144:407–14.
22. Au BK, Gooley TA, Armand P, et al. Reevaluation of the pretransplant assessment of mortality score after allogeneic hematopoietic transplantation. Biol Blood Marrow Transplant 2015;21:848–54.
23. Sorror ML, Maris MB, Storb R, et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood 2005;106:2912–9.
24. Chien JW, Sullivan KM. Carbon monoxide diffusion capacity: how low can you go for hematopoietic cell transplantation eligibility? Biol Blood Marrow Transplant 2009;15: 447–53.
25. Coffey DG, Pollyea DA, Myint H, et al. Adjusting DLCO for Hb and its effects on the Hematopoietic Cell Transplantation-specific Comorbidity Index. Bone Marrow Transplant 2013;48:1253–6.
26. Kasow KA, Krueger J, Srivastava DK, et al. Clinical utility of computed tomography screening of chest, abdomen, and sinuses before hematopoietic stem cell transplantation: the St. Jude experience. Biol Blood Marrow Transplant 2009;15:490–5.
27. Hamadani M, Craig M, Awan FT, Devine SM. How we approach patient evaluation for hematopoietic stem cell transplantation. Bone Marrow Transplant 2010;45: 1259–68.
28. Savani BN, Montero A, Wu C, et al. Prediction and prevention of transplant-related mortality from pulmonary causes after total body irradiation and allogeneic stem cell transplantation. Biol Blood Marrow Transplant 2005;11:223–30.
29. Ehlers SL, Gastineau DA, Patten CA, et al. The impact of smoking on outcomes among patients undergoing hematopoietic SCT for the treatment of acute leukemia. Bone Marrow Transplant 2011;46:285–90.
30. Marks DI, Ballen K, Logan BR, et al. The effect of smoking on allogeneic transplant outcomes. Biol Blood Marrow Transplant 2009;15:1277–87.
31. Tran BT, Halperin A, Chien JW. Cigarette smoking and outcomes after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2011;17:1004–11.
32. Lucena CM, Torres A, Rovira M, et al. Pulmonary complications in hematopoietic SCT: a prospective study. Bone Marrow Transplant 2014;49:1293–9.
33. Chi AK, Soubani AO, White AC, Miller KB. An update on pulmonary complications of hematopoietic stem cell transplantation. Chest 2013;144:1913–22.
34. Dunagan DP, Baker AM, Hurd DD, Haponik EF. Bronchoscopic evaluation of pulmonary infiltrates following bone marrow transplantation. Chest 1997;111:135–41.
35. Naeem N, Reed MD, Creger RJ, et al. Transfer of the hematopoietic stem cell transplant patient to the intensive care unit: does it really matter? Bone Marrow Transplant 2006;37:119–33.
36. Afessa B, Tefferi A, Hoagland HC, et al. Outcome of recipients of bone marrow transplants who require intensive care unit support. Mayo Clin Proc 1992;67:117–22.
37. Parody R, Martino R, de la Camara R, et al. Fungal and viral infections after allogeneic hematopoietic transplantation from unrelated donors in adults: improving outcomes over time. Bone Marrow Transplant 2015;50:274–81.
38. Orasch C, Weisser M, Mertz D, et al. Comparison of infectious complications during induction/consolidation chemotherapy versus allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant 2010;45:521–6.
39. Aguilar-Guisado M, Jimenez-Jambrina M, Espigado I, et al. Pneumonia in allogeneic stem cell transplantation recipients: a multicenter prospective study. Clin Transplant 2011;25:E629–38.
40. Palacios G, Hornig M, Cisterna D, et al. Streptococcus pneumoniae coinfection is correlated with the severity of H1N1 pandemic influenza. PLoS One 2009;4:e8540.
41. Hynicka LM, Ensor CR. Prophylaxis and treatment of respiratory syncytial virus in adult immunocompromised patients. Ann Pharmacother 2012;46:558–66.
42. Shah JN, Chemaly RF. Management of RSV infections in adult recipients of hematopoietic stem cell transplantation. Blood 2011;2755–63.
43. Marr KA, Bowden RA. Fungal infections in patients undergoing blood and marrow transplantation. Transpl Infect Dis 1999;1:237–46.
44. Wald A, Leisenring W, van Burik JA, Bowden RA. Epidemiology of Aspergillus infections in a large cohort of patients undergoing bone marrow transplantation. J Infect Dis 1997;175:1459–66.
45. Ascioglu S, Rex JH, de Pauw B, et al. Defining opportunistic invasive fungal infections in immunocompromised patients with cancer and hematopoietic stem cell transplants: an international consensus. Clin Infect Dis 2002;34:7–14.
46. Fisher CE, Stevens AM, Leisenring W, et al. Independent contribution of bronchoalveolar lavage and serum galactomannan in the diagnosis of invasive pulmonary aspergillosis. Transpl Infect Dis 2014;16:505–10.
47. Kojima R, Tateishi U, Kami M, et al. Chest computed tomography of late invasive aspergillosis after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2005;11:506–11.
48. Salmeron G, Porcher R, Bergeron A, et al. Persistent poor long-term prognosis of allogeneic hematopoietic stem cell transplant recipients surviving invasive aspergillosis. Haematologica 2012;97:1357–63.
49. McNulty JS. Rhinocerebral mucormycosis: predisposing factors. Laryngoscope 1982;92(10 Pt 1):1140.
50. Walsh TJ, Gamaletsou MN, McGinnis MR, et al. Early clinical and laboratory diagnosis of invasive pulmonary, extrapulmonary, and disseminated mucormycosis (zygomycosis). Clin Infect Dis 2012;54 Suppl 1:S55–60.
51. Klingspor L, Saaedi B, Ljungman P, Szakos A. Epidemiology and outcomes of patients with invasive mould infections: a retrospective observational study from a single centre (2005-2009). Mycoses 2015;58:470–7.
52. Danion F, Aguilar C, Catherinot E, et al. Mucormycosis: new developments in a persistently devastating infection. Semin Respir Crit Care Med 2015;36:692–70.
53. Rano A, Agusti C, Jimenez P, et al. Pulmonary infiltrates in non-HIV immunocompromised patients: a diagnostic approach using non-invasive and bronchoscopic procedures. Thorax 2001;56:379–87.
54. Azoulay E, Mokart D, Rabbat A, et al. Diagnostic bronchoscopy in hematology and oncology patients with acute respiratory failure: prospective multicenter data. Crit Care Med 2008;36:100–7.
55. Jain P, Sandur S, Meli Y, et al. Role of flexible bronchoscopy in immunocompromised patients with lung infiltrates. Chest 2004;125:712–22.
56. Rano A, Agusti C, Benito N, et al. Prognostic factors of non-HIV immunocompromised patients with pulmonary infiltrates. Chest 2002;122:253–61.
57. Shannon VR, Andersson BS, Lei X, et al. Utility of early versus late fiberoptic bronchoscopy in the evaluation of new pulmonary infiltrates following hematopoietic stem cell transplantation. Bone Marrow Transplant 2010;45:647–55.
58. Patel NR, Lee PS, Kim JH, et al. The influence of diagnostic bronchoscopy on clinical outcomes comparing adult autologous and allogeneic bone marrow transplant patients. Chest 2005;127:1388–96.
59. Chellapandian D, Lehrnbecher T, Phillips B, et al. Bronchoalveolar lavage and lung biopsy in patients with cancer and hematopoietic stem-cell transplantation recipients: a systematic review and meta-analysis. J Clin Oncol 2015;33:501–9.
60. Carr IM, Koegelenberg CF, von Groote-Bidlingmaier F, et al. Blood loss during flexible bronchoscopy: a prospective observational study. Respiration 2012;84:312–8.
61. Miyamoto M, Onizuka M, Machida S, et al. ACE deletion polymorphism is associated with a high risk of non-infectious pulmonary complications after stem cell transplantation. Int J Hematol 2014;99:175–83.
62. Capizzi SA, Kumar S, Huneke NE, et al. Peri-engraftment respiratory distress syndrome during autologous hematopoietic stem cell transplantation. Bone Marrow Transplant 2001;27:1299–303.
63. Spitzer TR. Engraftment syndrome following hematopoietic stem cell transplantation. Bone Marrow Transplant 2001;27:893–8.
64. Wanko SO, Broadwater G, Folz RJ, Chao NJ. Diffuse alveolar hemorrhage: retrospective review of clinical outcome in allogeneic transplant recipients treated with aminocaproic acid. Biol Blood Marrow Transplant 2006;12:949–53.
65. Metcalf JP, Rennard SI, Reed EC, et al. Corticosteroids as adjunctive therapy for diffuse alveolar hemorrhage associated with bone marrow transplantation. University of Nebraska Medical Center Bone Marrow Transplant Group. Am J Med 1994;96:327–34.
66. Rathi NK, Tanner AR, Dinh A, et al. Low-, medium- and high-dose steroids with or without aminocaproic acid in adult hematopoietic SCT patients with diffuse alveolar hemorrhage. Bone Marrow Transplant 2015;50:420–6.
67. Afessa B, Tefferi A, Litzow MR, Peters SG. Outcome of diffuse alveolar hemorrhage in hematopoietic stem cell transplant recipients. Am J Respir Crit Care Med 2002;166:1364–8.
68. Panoskaltsis-Mortari A, Griese M, Madtes DK, et al. An official American Thoracic Society research statement: noninfectious lung injury after hematopoietic stem cell transplantation: idiopathic pneumonia syndrome. Am J Respir Crit Care Med 2011;183:1262–79.
69. Clark JG, Hansen JA, Hertz MI, Pet al. NHLBI workshop summary. Idiopathic pneumonia syndrome after bone marrow transplantation. Am Rev Resp Dis 1993;147:1601–6.
70. Vande Vusse LK, Madtes DK. Early onset noninfectious pulmonary syndromes after hematopoietic cell transplantation. Clin Chest Med 2017;38:233–48.
71. Fukuda T, Hackman RC, Guthrie KA, et al. Risks and outcomes of idiopathic pneumonia syndrome after nonmyeloablative and conventional conditioning regimens for allogeneic hematopoietic stem cell transplantation. Blood 2003;102:2777–85.
72. Englund JA, Boeckh M, Kuypers J, et al. Brief communication: fatal human metapneumovirus infection in stem-cell transplant recipients. Ann Intern Med 2006;144:344–9.
73. Seo S, Renaud C, Kuypers JM, et al. Idiopathic pneumonia syndrome after hematopoietic cell transplantation: evidence of occult infectious etiologies. Blood 2015;125:3789–97.
74. Nakane T, Nakamae H, Kamoi H, et al. Prognostic value of serum surfactant protein D level prior to transplant for the development of bronchiolitis obliterans syndrome and idiopathic pneumonia syndrome following allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;42:43–9.
75. Gilbert CR, Lerner A, Baram M, Awsare BK. Utility of flexible bronchoscopy in the evaluation of pulmonary infiltrates in the hematopoietic stem cell transplant population—a single center fourteen year experience. Arch Bronconeumol 2013;49:189–95.
76. Yanik GA, Horowitz MM, Weisdorf DJ, et al. Randomized, double-blind, placebo-controlled trial of soluble tumor necrosis factor receptor: enbrel (etanercept) for the treatment of idiopathic pneumonia syndrome after allogeneic stem cell transplantation: blood and marrow transplant clinical trials network protocol. Biol Blood Marrow Transplant 2014;20:858–64.
77. Levine JE, Paczesny S, Mineishi S, et al. Etanercept plus methylprednisolone as initial therapy for acute graft-versus-host disease. Blood 2008;111:2470–5.
78. Yanik GA, Grupp SA, Pulsipher MA, et al. TNF-receptor inhibitor therapy for the treatment of children with idiopathic pneumonia syndrome. A joint Pediatric Blood and Marrow Transplant Consortium and Children’s Oncology Group Study (ASCT0521). Biol Blood Marrow Transplant 2015;21:67–73.
79. Thompson J, Yin Z, D’Souza A, et al. Etanercept and corticosteroid therapy for the treatment of late-onset idiopathic pneumonia syndrome. Biol Blood Marrow Transplant J 2017; 23:1955–60.
Hematopoietic stem cell transplantation (HSCT) is widely used in the economically developed world to treat a variety of hematologic malignancies as well as nonmalignant diseases and solid tumors. An estimated 17,900 HSCTs were performed in 2011, and survival rates continue to increase.1 Pulmonary complications post HSCT are common, with rates ranging from 40% to 60%, and are associated with increased morbidity and mortality.2
Clinical diagnosis of pulmonary complications in the HSCT population has been aided by a previously well-defined chronology of the most common diseases.3 Historically, early pulmonary complications were defined as pulmonary complications occurring within 100 days of HSCT (corresponding to the acute graft-versus-host disease [GVHD] period). Late pulmonary complications are those that occur thereafter. This timeline, however, is now more variable given the increasing indications for HSCT, the use of reduced-intensity conditioning strategies, and varied individual immune reconstitution. This article discusses the management of early post-HSCT pulmonary complications; late post-HSCT pulmonary complications will be discussed in a separate follow-up article.
Transplant Basics
The development of pulmonary complications is affected by many factors associated with the transplant. Autologous transplantation involves the collection of a patient’s own stem cells, appropriate storage and processing, and re-implantation after induction therapy. During induction therapy, the patient undergoes high-dose chemotherapy or radiation therapy that ablates the bone marrow. The stem cells are then transfused back into the patient to repopulate the bone marrow. Allogeneic transplants involve the collection of stem cells from a donor. Donors are matched as closely as possible to the recipient’s histocompatibility antigen (HLA) haplotypes to prevent graft failure and rejection. The donor can be related or unrelated to the recipient. If there is not a possibility of a related match (from a sibling), then a national search is undertaken to look for a match through the National Marrow Donor Program. There are fewer transplant reactions and occurrences of GVHD if the major HLAs of the donor and recipient match. Table 1 reviews basic definitions pertaining to HSCT.
How the cells for transplantation are obtained is also an important factor in the rate of complications. There are 3 main sources: peripheral blood, bone marrow, and umbilical cord. Peripheral stem cell harvesting involves exposing the donor to granulocyte-colony stimulating factor (gCSF), which increases peripheral circulation of stem cells. These cells are then collected and infused into the recipient after the recipient has completed an induction regimen involving chemotherapy and/or radiation, depending on the protocol. This procedure is called peripheral blood stem cell transplant (PBSCT). Stem cells can also be directly harvested from bone marrow cells, which are collected from repeated aspiration of bone marrow from the posterior iliac crest.4 This technique is most common in children, whereas in adults peripheral blood stem cells are the most common source. Overall mortality does not differ based on the source of the stem cells. It is postulated that GVHD may be more common in patients undergoing PBSCT, but the graft failure rate may be lower.5
The third option is umbilical cord blood (UCB) as the source of stem cells. This involves the collection of umbilical cord blood that is prepared and frozen after birth. It has a smaller volume of cells, and although fewer cells are needed when using UCB, 2 separate donors may be required for a single adult recipient. The engraftment of the stem cells is slower and infections in the post-transplant period are more common. Prior reports indicate GVHD rates may be lower.4 While the use of UCB is not common in adults, the incidence has doubled over the past decade, increasing from 3% to 6%.
The conditioning regimen can influence pulmonary complications. Traditionally, an ablative transplant involves high-dose chemotherapy or radiation to eradicate the recipient’s bone marrow. This regimen can lead to many complications, especially in the immediate post-transplant period. In the past 10 years, there has been increasing interest in non-myeloablative, or reduced-intensity, conditioning transplants.6 These “mini transplants” involve smaller doses of chemotherapy or radiation, which do not totally eradicate the bone marrow; after the transplant a degree of chimerism develops where the donor and recipient stem cells coexist. The medications in the preparative regimen also should be considered because they can affect pulmonary complications after transplant. Certain chemotherapeutic agents such as carmustine, bleomycin, and many others can lead to acute and chronic presentations of pulmonary diseases such as hypersensitivity pneumonitis, pulmonary fibrosis, acute respiratory distress syndrome, and abnormal pulmonary function testing.
After the HSCT, GVHD can develop in more than 50% of allogeneic recipients.3 The incidence of GVHD has been reported to be increasing over the past 12 years.It is divided into acute GVHD (which traditionally happens in the first 100 days after transplant) and chronic GVHD (after day 100). This calendar-day–based system has been augmented based on a 2006 National Institutes of Health working group report emphasizing the importance of organ-specific features of chronic GVHD in the clinical presentation of GVHD.7 Histologic changes in chronic organ GVHD tend to include more fibrotic features, whereas in acute GVHD more inflammatory changes are seen. The NIH working group report also stressed the importance of obtaining a biopsy specimen for histopathologic review and interdisciplinary collaboration to arrive at a consensus diagnosis, and noted the limitations of using histologic changes as the sole determinant of a “gold standard” diagnosis.7 GVHD can directly predispose patients to pulmonary GVHD and indirectly predispose them to infectious complications because the mainstay of therapy for GVHD is increased immunosuppression.
Pretransplant Evaluation
Case Patient 1
A 56-year-old man is diagnosed with acute myeloid leukemia (AML) after presenting with signs and symptoms consistent with pancytopenia. He has a past medical history of chronic sinus congestion, arthritis, depression, chronic pain, and carpal tunnel surgery. He is employed as an oilfield worker and has a 40-pack-year smoking history, but he recently cut back to half a pack per day. He is being evaluated for allogeneic transplant with his brother as the donor and the planned conditioning regimen is total body irradiation (TBI), thiotepa, cyclophosphamide, and antithymocyte globulin with T-cell depletion. Routine pretransplant pulmonary function testing (PFT) reveals a restrictive pattern and he is sent for pretransplant pulmonary evaluation.
Physical exam reveals a chronically ill appearing man. He is afebrile, the respiratory rate is 16 breaths/min, blood pressure is 145/88 mm Hg, heart rate is 92 beats/min, and oxygen saturation is 95%. He is in no distress. Auscultation of the chest reveals slightly diminished breath sounds bilaterally but is clear and without wheezes, rhonchi, or rales. Heart exam shows regular rate and rhythm without murmurs, rubs, or gallops. Extremities reveal no edema or rashes. Otherwise, the remainder of the exam is normal. The patient’s PFT results are shown in Table 2.
- What aspects of this patient’s history put him at risk for pulmonary complications after transplantation?
Risk Factors for Pulmonary Complications
Predicting who is at risk for pulmonary complications is difficult. Complications are generally divided into infectious and noninfectious categories. Regardless of category, allogeneic HSCT recipients are at increased risk compared with autologous recipients, but even in autologous transplants, more than 25% of patients will develop pulmonary complications in the first year.8 Prior to transplant, patients undergo full PFT. Early on, many studies attempted to show relationships between various factors and post-transplant pulmonary complications. Factors that were implicated were forced expiratory volume in 1 second (FEV1), diffusing capacity of the lung for carbon monoxide (D
Another sometimes overlooked risk before transplantation is restrictive lung disease. One study showed a twofold increase in respiratory failure and mortality if there was pretransplant restriction based on TLC < 80%.16
An interesting study by one group in pretransplant evaluation found decreased muscle strength by maximal inspiratory muscle strength (PImax), maximal expiratory muscle strength (PEmax), dominant hand grip strength, and 6-minute walk test (6MWT) distance prior to allogeneic transplant, but did not find a relationship between these variables and mortality.17 While this study had a small sample size, these findings likely deserve continued investigation.18
- What methods are used to calculate risk for complications?
Risk Scoring Systems
Several pretransplantation risk scores have been developed. In a study that looked at more than 2500 allogeneic transplants, Parimon et al showed that risk of mortality and respiratory failure could be estimated prior to transplant using a scoring system—the Lung Function Score (LFS)—that combines the FEV1 and D
The Pretransplantation Assessment of Mortality score, initially developed in 2006, predicts mortality within the first 2 years after HSCT based on 8 clinical factors: disease risk, age at transplant, donor type, conditioning regimen, and markers of organ function (percentage of predicted FEV1, percentage of predicted D
- What other preoperative testing or interventions should be considered in this patient?
Since there is a high risk of infectious complications after transplant, the question of whether pretransplantation patients should undergo screening imaging may arise. There is no evidence that routine chest computed tomography (CT) reduces the risk of infectious complications after transplantation.26 An area that may be insufficiently addressed in the pretransplantation evaluation is smoking cessation counseling.27 Studies have shown an elevated risk of mortality in smokers.28-30 Others have found a higher incidence of respiratory failure but not an increased mortality.31 Overall, with the good rates of smoking cessation that can be accomplished, smokers should be counseled to quit before transplantation.
In summary, patients should undergo full PFTs prior to transplantation to help stratify risk for pulmonary complications and mortality and to establish a clinical baseline. The LFS (using FEV1 and D
Case Patient 1 Conclusion
The patient undergoes transplantation due to his lack of other treatment options. Evaluation prior to transplant, however, shows that he is at high risk for pulmonary complications. He has a LFS of 7 prior to transplant (using the D
Early Infectious Pulmonary Complications
Case Patient 2
A 27-year-old man with a medical history significant for AML and allogeneic HSCT presents with cough productive of a small amount of clear to white sputum, dyspnea on exertion, and fevers for 1 week. He also has mild nausea and a decrease in appetite. He underwent HSCT 2.5 months prior to admission, which was a matched unrelated bone marrow transplant with TBI and cyclophosphamide conditioning. His past medical history is significant only for exercise-induced asthma for which he takes a rescue inhaler infrequently prior to transplantation. His pretransplant PFTs showed normal spirometry with an FEV1 of 106% of predicted and D
Physical exam is notable for fever of 101.0°F, heart rate 80 beats/min, respiratory rate 16 breaths/ min, and blood pressure 142/78 mm Hg; an admission oxygen saturation is 93% on room air. Lungs show bibasilar crackles and the remainder of the exam is normal. Laboratory testing shows a white blood cell count of 2400 cells/μL, hemoglobin 7.6 g/dL, and platelet count 66 × 103/μL. Creatinine is 1.0 mg/dL. Chest radiograph shows ill-defined bilateral lower-lobe infiltrates. CT scans are shown in the Figure.
- For which infectious complications is this patient most at risk?
Pneumonia
A prospective trial in the HSCT population reported a pneumonia incidence rate of 68%, and pneumonia is more common in allogeneic HSCT with prolonged immunosuppressive therapy.32 Development of pneumonia within 100 days of transplant directly correlates with nonrelapsed mortality.33 Early detection is key, and bronchoscopy within the first 5 days of symptoms has been shown to change therapy in approximately 40% of cases but has not been shown to affect mortality.34 The clinical presentation of pneumonia in the HSCT population can be variable because of the presence of neutropenia and profound immunosuppression. Traditionally accepted diagnostic criteria of fevers, sputum production, and new infiltrates should be used with caution, and an appropriately high index of suspicion should be maintained. Progression to respiratory failure, regardless of causative organism of infection, portends a poor prognosis, with mortality rates estimated at 70% to 90%.35,36 Several transplant-specific factors may affect early infections. For instance, UCB transplants have been found to have a higher incidence of invasive aspergillosis and cytomegalovirus (CMV) infections but without higher mortality attributed to the infections.37
Bacterial Pneumonia
Bacterial pneumonia accounts for 20% to 50% of pneumonia cases in HSCT recipients.38 Gram-negative organisms, specifically Pseudomonas aeruginosa and Escherichia coli, were reported to be the most common pathologic bacteria in recent prospective trials, whereas previous retrospective trials showed that common community-acquired organisms were the most common cause of pneumonia in HSCT recipients.32,39 This underscores the importance of being aware of the clinical prevalence of microorganisms and local antibiograms, along with associated institutional susceptibility profiles. Initiation of immediate empiric broad-spectrum antibiotics is essential when bacterial pneumonia is suspected.
Viral Pneumonia
The prevalence of viral pneumonia in stem cell transplant recipients is estimated at 28%,32 with most cases being caused by community viral pathogens such as rhinovirus, respiratory syncytial virus (RSV), influenza A and B, and parainfluenza.39 The prevention, prophylaxis, and early treatment of viral pneumonias, specifically CMV infection, have decreased the mortality associated with early pneumonia after HSCT. Co-infection with bacterial organisms must be considered and has been associated with increased mortality in the intensive care unit setting.40
Supportive treatment with rhinovirus infection is sufficient as the disease is usually self-limited in immunocompromised patients. In contrast, infection with RSV in the lower respiratory tract is associated with increased mortality in prior reports, and recent studies suggest that further exploration of prophylaxis strategies is warranted.41 Treatment with ribavirin remains the backbone of therapy, but drug toxicity continues to limit its use. The addition of immunomodulators such as RSV immune globulin or palivizumab to ribavirin remains controversial, but a retrospective review suggests that early treatment may prevent progression to lower respiratory tract infection and lead to improved mortality.42 Infection with influenza A/B must be considered during influenza season. Treatment with oseltamivir may shorten the duration of disease when influenza A/B or parainfluenza are detected. Reactivation of latent herpes simplex virus during the pre-engraftment phase should also be considered. Treatment is similar to that in nonimmunocompromised hosts. When CMV pneumonia is suspected, careful history regarding compliance with prophylactic antivirals and CMV status of both the recipient and donor are key. A presumptive diagnosis can be made with the presence of appropriate clinical scenario, supportive radiographic images showing areas of ground-glass opacification or consolidation, and positive CMV polymerase chain reaction (PCR) assay. Visualization of inclusion bodies on lung biopsy tissue remains the gold standard for diagnosis. Treatment consists of CMV immunoglobulin and ganciclovir.
Fungal Pneumonia
Early fungal pneumonias have been associated with increased mortality in the HSCT population.43 Clinical suspicion should remain high and compliance with antifungal prophylaxis should be questioned thoroughly. Invasive aspergillosis (IA) remains the most common fungal infection. A bimodal distribution of onset of infection peaking on day 16 and again on day 96 has been described in the literature.44 Patients often present with classic pneumonia symptoms, but these may be accompanied by hemoptysis. Proven IA diagnosis requires visualization of fungal forms from biopsy or needle aspiration or a positive culture obtained in a sterile fashion.45 Most clinical data comes from experience with probable and possible diagnosis of IA. Bronchoalveolar lavage with testing with Aspergillus galactomannan assay has been shown to be clinically useful in establishing the clinical diagnosis in the HSCT population.46 Classic air-crescent findings on chest CT are helpful in establishing a possible diagnosis, but retrospective analysis reveals CT findings such as focal infiltrates and pulmonary nodular patterns are more common.47 First-line treatment with voriconazole has been shown to decrease short-term mortality attributable to IA but has not had an effect on long-term, all-cause mortality.48 Surgical resection is reserved for patients with refractory disease or patients presenting with massive hemoptysis.
Mucormycosis is an emerging disease with ever increasing prevalence in the HSCT population, reflecting the improved prophylaxis and treatment of IA. Initial clinical presentation is similar to IA, most commonly affecting the lung, although craniofacial involvement is classic for mucormycosis, especially in HSCT patients with diabetes.49Mucor infections can present with massive hemoptysis due to tissue invasion and disregard for tissue and fascial planes. Diagnosis of mucormycosis is associated with as much as a six-fold increase in risk for death. Diagnosis requires identification of the organism by examination or culture and biopsy is often necessary.50,51 Amphotericin B remains first-line therapy as mucormycosis is resistant to azole antifungals, with higher doses recommended for cerebral involvement.52
Candida pulmonary infections during the early HSCT period are becoming increasingly rare due to widespread use of fluconazole prophylaxis and early treatment of mucosal involvement during neutropenia. Endemic fungal infections such as blastomycosis, coccidioidomycosis, and histoplasmosis should be considered in patients inhabiting specific geographic areas or with recent travel to these areas.
- What test should be performed to evaluate for infectious causes of pneumonia?
Role of Flexible Fiberoptic Bronchoscopy
The utility of flexible fiberoptic bronchoscopy (FOB) in immune-compromised patients for the evaluation of pulmonary infiltrates is a frequently debated topic. Current studies suggest a diagnosis can be made in approximately 80% of cases in the immune-compromised population.32,53 Noninvasive testing such as urine and serum antigens, sputum cultures, Aspergillus galactomannan assays, viral nasal swabs, and PCR studies often lead to a diagnosis in appropriate clinical scenarios. Conservative management would dictate the use of noninvasive testing whenever possible, and randomized controlled trials have shown noninvasive testing to be noninferior to FOB in preventing need for mechanical ventilation, with no difference in overall mortality.54 FOB has been shown to be most useful in establishing a diagnosis when an infectious etiology is suspected.55 In multivariate analysis, a delay in the identification of the etiology of pulmonary infiltrate was associated with increased mortality.56 Additionally, early FOB was found to be superior to late FOB in revealing a diagnosis. 32,57 Despite its ability to detect the cause of pulmonary disease, direct antibiotic therapy, and possibly change therapy, FOB with diagnostic maneuvers has not been shown to affect mortality.58 In a large case series, FOB with bronchoalveolar lavage (BAL) revealed a diagnosis in approximately 30% to 50% of cases. The addition of transbronchial biopsy did not improve diagnostic utility.58 More recent studies have confirmed that the addition of transbronchial biopsy does not add to diagnostic yield and is associated with increased adverse events.59 The appropriate use of advanced techniques such as endobronchial ultrasound–guided transbronchial needle aspirations, endobronchial biopsy, and CT-guided navigational bronchoscopy has not been established and should be considered on a case-by-case basis. In summary, routine early BAL is the diagnostic test of choice, especially when infectious pulmonary complications are suspected.
Contraindications for FOB in this population mirror those in the general population. These include acute severe hypoxemic respiratory failure, myocardial ischemia or acute coronary syndrome within 2 weeks of procedure, severe thrombocytopenia, and inability to provide or obtain informed consent from patient or health care power of attorney. Coagulopathy and thrombocytopenia are common comorbid conditions in the HSCT population. A platelet count of < 20 × 103/µL has generally been used as a cut-off for routine FOB with BAL.60 Risks of the procedures should be discussed clearly with the patient, but simple FOB for airway evaluation and BAL is generally well tolerated even under these conditions.
Early Nonifectious Pulmonary Complications
Case Patient 2 Continued
Bronchoscopy with BAL performed the day after admission is unremarkable and stains and cultures are negative for viral, bacterial, and fungal organisms. The patient is initially started on broad-spectrum antibiotics, but his oxygenation continues to worsen to the point that he is placed on noninvasive positive pressure ventilation. He is started empirically on amphotericin B and eventually is intubated. VATS lung biopsy is ultimately performed and pathology is consistent with diffuse alveolar damage.
- Based on these biopsy findings, what is the diagnosis?
Based on the pathology consistent with diffuse alveolar damage, a diagnosis of idiopathic pneumonia syndrome (IPS) is made.
- What noninfectious pulmonary complications occur in the early post-transplant period?
The overall incidence of noninfectious pulmonary complications after HSCT is generally estimated at 20% to 30%.32 Acute pulmonary edema is a common very early noninfectious pulmonary complication and clinically the most straightforward to treat. Three distinct clinical syndromes—peri-engraftment respiratory distress syndrome (PERDS), diffuse alveolar hemorrhage (DAH), and IPS—comprise the remainder of the pertinent early noninfectious complications. Clinical presentation differs based upon the disease entity. Recent studies have evaluated the role of angiotensin-converting enzyme polymorphisms as a predictive marker for risk of developing early noninfectious pulmonary complications.61
Peri-Engraftment Respiratory Distress Syndrome
PERDS is a clinical syndrome comprising the cardinal features of erythematous rash and fever along with noncardiogenic pulmonary infiltrates and hypoxemia that occur in the peri-engraftment period, defined as recovery of absolute neutrophil count to > 500/μL on 2 consecutive days.62 PERDS occurs in the autologous HSCT population and may be a clinical correlate to early GVHD in the allogeneic HSCT population. It is hypothesized that the pathophysiology underlying PERDS is an autoimmune-related capillary leak caused by pro-inflammatory cytokine release.63 Treatment remains anecdotal and currently consists of supportive care and high-dose corticosteroids. Some have favored limiting the use of gCSF given its role in stimulating rapid white blood cell recovery.33 Prognosis is favorable, but progression to fulminant respiratory failure requiring mechanical ventilation portends a poor prognosis.
Diffuse Alveolar Hemorrhage
DAH is clinical syndrome consisting of diffuse alveolar infiltrates on pulmonary imaging combined with progressively bloodier return per aliquot during BAL in 3 different subsegments or more than 20% hemosiderin-laden macrophages on BAL fluid evaluation. Classically, DAH is defined in the absence of pulmonary infection or cardiac dysfunction. The pathophysiology is thought to be related to inflammation of pulmonary vasculature within the alveolar walls leading to alveolitis. Although no prospective trials exist, early use of high-dose corticosteroid therapy is thought to improve outcomes;64,65 a recent study, however, showed low-dose steroids may be associated with the lowest mortality.66 Mortality is directly linked to the presence of superimposed infection, need for mechanical ventilation, late onset, and development of multiorgan failure.67
Idiopathic Pneumonia Syndrome
IPS is a complex clinical syndrome whose pathology is felt to stem from a variety of possible lung insults such as direct myeloablative drug toxicity, occult pulmonary infection, or cytokine-driven inflammation. The ATS published an article further subcategorizing IPS as different clinical entities based upon whether the primary insult involves the vascular endothelium, interstitial tissue, and airway tissue, truly idiopathic, or unclassified.68 In clinical practice, IPS is defined as widespread alveolar injury in the absence of evidence of renal failure, heart failure, and excessive fluid resuscitation. In addition, negative testing for a variety of bacterial, viral, and fungal causes is also necessary.69 Clinical syndromes included within the IPS definition are ARDS, acute interstitial pneumonia, DAH, cryptogenic organizing pneumonia, and BOS.70 Risk factors for developing IPS include TBI, older age of recipient, acute GVHD, and underlying diagnosis of AML or myelodysplastic syndrome.12 In addition, it has been shown that risk for developing IPS is lower in patients undergoing allogeneic HSCT who receive non-myeloablative conditioning regimens.71 The pathologic finding in IPS is diffuse alveolar damage. A 2006 study in which investigators reviewed BAL samples from patients with IPS found that 3% of the patients had PCR evidence of human metapneumovirus infection, and a study in 2015 found PCR evidence of infection in 53% of BAL samples from patients diagnosed with IPS.72,73 This fuels the debate on whether IPS is truly an infection-driven process where the source of infection, pulmonary or otherwise, simply escapes detection. Various surfactant proteins, which play a role in decreasing surface tension within the alveolar interface and function as mediators within the innate immunity of the lung, have been studied in regard to development of IPS. Small retrospective studies have shown a trend toward lower pre-transplant serum protein surfactant D and the development of IPS.74
The diagnosis of IPS does not require pathologic diagnosis in most circumstances. The correct clinical findings in association with a negative infectious workup lead to a presumptive diagnosis of IPS. The extent of the infectious workup that must be completed to adequately rule out infection is often a difficult clinical question. Recent recommendations include BAL fluid evaluation for routine bacterial cultures, appropriate viral culture, and consideration of PCR testing to evaluate for Mycoplasma, Chlamydia, and Aspergillus antigens.75 Transbronchial biopsy continues to appear in recommendations, but is not routinely performed and should be completed as the patient’s clinical status permits.8,68 Table 3 reviews basic features of early noninfectious pulmonary complications.
Treatment of IPS centers around moderate to high doses of corticosteroids. Based on IPS experimental modes, tumor necrosis factor (TNF)-α has been implicated as an important mediator. Unfortunately, several studies evaluating etanercept have produced conflicting results, and this agent’s clinical effects on morbidity and mortality remain in question.76
- What treatment should be offered to the patient with diffuse alveolar damage on biopsy?
Treatment consists of supportive care and empiric broad-spectrum antibiotics with consideration of high-dose corticosteroids. Based upon early studies in murine models implicating TNF, pilot studies were performed evaluating etanercept as a possible safe and effective addition to high-dose systemic corticosteroids.77 Although these results were promising, data from a truncated randomized control clinical trial failed to show improvement in patient response in the adult population.76 More recent data from the same author suggests that pediatric populations with IPS are, however, responsive to etanercept and high-dose corticosteroid therapy.78 When IPS develops as a late complication, treatment with high-dose corticosteroids (2 mg/kg/day) and etanercept (0.4 mg/kg twice weekly) has been shown to improve 2-year survival.79
Case Patient 2 Conclusion
The patient is started on steroids and makes a speedy recovery. He is successfully extubated 5 days later.
Conclusion
Careful pretransplant evaluation, including a full set of pulmonary function tests, can help predict a patient’s risk for pulmonary complications after transplant, allowing risk factor modification strategies to be implemented prior to transplant, including smoking cessation. It also helps identify patients at high risk for complications who will require closer monitoring after transplantation. Early posttransplant complications include infectious and noninfectious entities. Bacterial, viral, and fungal pneumonias are in the differential of infectious pneumonia, and bronchoscopy can be helpful in establishing a diagnosis. A common, important noninfectious cause of early pulmonary complications is IPS, which is treated with steroids and sometimes anti-TNF therapy.
Hematopoietic stem cell transplantation (HSCT) is widely used in the economically developed world to treat a variety of hematologic malignancies as well as nonmalignant diseases and solid tumors. An estimated 17,900 HSCTs were performed in 2011, and survival rates continue to increase.1 Pulmonary complications post HSCT are common, with rates ranging from 40% to 60%, and are associated with increased morbidity and mortality.2
Clinical diagnosis of pulmonary complications in the HSCT population has been aided by a previously well-defined chronology of the most common diseases.3 Historically, early pulmonary complications were defined as pulmonary complications occurring within 100 days of HSCT (corresponding to the acute graft-versus-host disease [GVHD] period). Late pulmonary complications are those that occur thereafter. This timeline, however, is now more variable given the increasing indications for HSCT, the use of reduced-intensity conditioning strategies, and varied individual immune reconstitution. This article discusses the management of early post-HSCT pulmonary complications; late post-HSCT pulmonary complications will be discussed in a separate follow-up article.
Transplant Basics
The development of pulmonary complications is affected by many factors associated with the transplant. Autologous transplantation involves the collection of a patient’s own stem cells, appropriate storage and processing, and re-implantation after induction therapy. During induction therapy, the patient undergoes high-dose chemotherapy or radiation therapy that ablates the bone marrow. The stem cells are then transfused back into the patient to repopulate the bone marrow. Allogeneic transplants involve the collection of stem cells from a donor. Donors are matched as closely as possible to the recipient’s histocompatibility antigen (HLA) haplotypes to prevent graft failure and rejection. The donor can be related or unrelated to the recipient. If there is not a possibility of a related match (from a sibling), then a national search is undertaken to look for a match through the National Marrow Donor Program. There are fewer transplant reactions and occurrences of GVHD if the major HLAs of the donor and recipient match. Table 1 reviews basic definitions pertaining to HSCT.
How the cells for transplantation are obtained is also an important factor in the rate of complications. There are 3 main sources: peripheral blood, bone marrow, and umbilical cord. Peripheral stem cell harvesting involves exposing the donor to granulocyte-colony stimulating factor (gCSF), which increases peripheral circulation of stem cells. These cells are then collected and infused into the recipient after the recipient has completed an induction regimen involving chemotherapy and/or radiation, depending on the protocol. This procedure is called peripheral blood stem cell transplant (PBSCT). Stem cells can also be directly harvested from bone marrow cells, which are collected from repeated aspiration of bone marrow from the posterior iliac crest.4 This technique is most common in children, whereas in adults peripheral blood stem cells are the most common source. Overall mortality does not differ based on the source of the stem cells. It is postulated that GVHD may be more common in patients undergoing PBSCT, but the graft failure rate may be lower.5
The third option is umbilical cord blood (UCB) as the source of stem cells. This involves the collection of umbilical cord blood that is prepared and frozen after birth. It has a smaller volume of cells, and although fewer cells are needed when using UCB, 2 separate donors may be required for a single adult recipient. The engraftment of the stem cells is slower and infections in the post-transplant period are more common. Prior reports indicate GVHD rates may be lower.4 While the use of UCB is not common in adults, the incidence has doubled over the past decade, increasing from 3% to 6%.
The conditioning regimen can influence pulmonary complications. Traditionally, an ablative transplant involves high-dose chemotherapy or radiation to eradicate the recipient’s bone marrow. This regimen can lead to many complications, especially in the immediate post-transplant period. In the past 10 years, there has been increasing interest in non-myeloablative, or reduced-intensity, conditioning transplants.6 These “mini transplants” involve smaller doses of chemotherapy or radiation, which do not totally eradicate the bone marrow; after the transplant a degree of chimerism develops where the donor and recipient stem cells coexist. The medications in the preparative regimen also should be considered because they can affect pulmonary complications after transplant. Certain chemotherapeutic agents such as carmustine, bleomycin, and many others can lead to acute and chronic presentations of pulmonary diseases such as hypersensitivity pneumonitis, pulmonary fibrosis, acute respiratory distress syndrome, and abnormal pulmonary function testing.
After the HSCT, GVHD can develop in more than 50% of allogeneic recipients.3 The incidence of GVHD has been reported to be increasing over the past 12 years.It is divided into acute GVHD (which traditionally happens in the first 100 days after transplant) and chronic GVHD (after day 100). This calendar-day–based system has been augmented based on a 2006 National Institutes of Health working group report emphasizing the importance of organ-specific features of chronic GVHD in the clinical presentation of GVHD.7 Histologic changes in chronic organ GVHD tend to include more fibrotic features, whereas in acute GVHD more inflammatory changes are seen. The NIH working group report also stressed the importance of obtaining a biopsy specimen for histopathologic review and interdisciplinary collaboration to arrive at a consensus diagnosis, and noted the limitations of using histologic changes as the sole determinant of a “gold standard” diagnosis.7 GVHD can directly predispose patients to pulmonary GVHD and indirectly predispose them to infectious complications because the mainstay of therapy for GVHD is increased immunosuppression.
Pretransplant Evaluation
Case Patient 1
A 56-year-old man is diagnosed with acute myeloid leukemia (AML) after presenting with signs and symptoms consistent with pancytopenia. He has a past medical history of chronic sinus congestion, arthritis, depression, chronic pain, and carpal tunnel surgery. He is employed as an oilfield worker and has a 40-pack-year smoking history, but he recently cut back to half a pack per day. He is being evaluated for allogeneic transplant with his brother as the donor and the planned conditioning regimen is total body irradiation (TBI), thiotepa, cyclophosphamide, and antithymocyte globulin with T-cell depletion. Routine pretransplant pulmonary function testing (PFT) reveals a restrictive pattern and he is sent for pretransplant pulmonary evaluation.
Physical exam reveals a chronically ill appearing man. He is afebrile, the respiratory rate is 16 breaths/min, blood pressure is 145/88 mm Hg, heart rate is 92 beats/min, and oxygen saturation is 95%. He is in no distress. Auscultation of the chest reveals slightly diminished breath sounds bilaterally but is clear and without wheezes, rhonchi, or rales. Heart exam shows regular rate and rhythm without murmurs, rubs, or gallops. Extremities reveal no edema or rashes. Otherwise, the remainder of the exam is normal. The patient’s PFT results are shown in Table 2.
- What aspects of this patient’s history put him at risk for pulmonary complications after transplantation?
Risk Factors for Pulmonary Complications
Predicting who is at risk for pulmonary complications is difficult. Complications are generally divided into infectious and noninfectious categories. Regardless of category, allogeneic HSCT recipients are at increased risk compared with autologous recipients, but even in autologous transplants, more than 25% of patients will develop pulmonary complications in the first year.8 Prior to transplant, patients undergo full PFT. Early on, many studies attempted to show relationships between various factors and post-transplant pulmonary complications. Factors that were implicated were forced expiratory volume in 1 second (FEV1), diffusing capacity of the lung for carbon monoxide (D
Another sometimes overlooked risk before transplantation is restrictive lung disease. One study showed a twofold increase in respiratory failure and mortality if there was pretransplant restriction based on TLC < 80%.16
An interesting study by one group in pretransplant evaluation found decreased muscle strength by maximal inspiratory muscle strength (PImax), maximal expiratory muscle strength (PEmax), dominant hand grip strength, and 6-minute walk test (6MWT) distance prior to allogeneic transplant, but did not find a relationship between these variables and mortality.17 While this study had a small sample size, these findings likely deserve continued investigation.18
- What methods are used to calculate risk for complications?
Risk Scoring Systems
Several pretransplantation risk scores have been developed. In a study that looked at more than 2500 allogeneic transplants, Parimon et al showed that risk of mortality and respiratory failure could be estimated prior to transplant using a scoring system—the Lung Function Score (LFS)—that combines the FEV1 and D
The Pretransplantation Assessment of Mortality score, initially developed in 2006, predicts mortality within the first 2 years after HSCT based on 8 clinical factors: disease risk, age at transplant, donor type, conditioning regimen, and markers of organ function (percentage of predicted FEV1, percentage of predicted D
- What other preoperative testing or interventions should be considered in this patient?
Since there is a high risk of infectious complications after transplant, the question of whether pretransplantation patients should undergo screening imaging may arise. There is no evidence that routine chest computed tomography (CT) reduces the risk of infectious complications after transplantation.26 An area that may be insufficiently addressed in the pretransplantation evaluation is smoking cessation counseling.27 Studies have shown an elevated risk of mortality in smokers.28-30 Others have found a higher incidence of respiratory failure but not an increased mortality.31 Overall, with the good rates of smoking cessation that can be accomplished, smokers should be counseled to quit before transplantation.
In summary, patients should undergo full PFTs prior to transplantation to help stratify risk for pulmonary complications and mortality and to establish a clinical baseline. The LFS (using FEV1 and D
Case Patient 1 Conclusion
The patient undergoes transplantation due to his lack of other treatment options. Evaluation prior to transplant, however, shows that he is at high risk for pulmonary complications. He has a LFS of 7 prior to transplant (using the D
Early Infectious Pulmonary Complications
Case Patient 2
A 27-year-old man with a medical history significant for AML and allogeneic HSCT presents with cough productive of a small amount of clear to white sputum, dyspnea on exertion, and fevers for 1 week. He also has mild nausea and a decrease in appetite. He underwent HSCT 2.5 months prior to admission, which was a matched unrelated bone marrow transplant with TBI and cyclophosphamide conditioning. His past medical history is significant only for exercise-induced asthma for which he takes a rescue inhaler infrequently prior to transplantation. His pretransplant PFTs showed normal spirometry with an FEV1 of 106% of predicted and D
Physical exam is notable for fever of 101.0°F, heart rate 80 beats/min, respiratory rate 16 breaths/ min, and blood pressure 142/78 mm Hg; an admission oxygen saturation is 93% on room air. Lungs show bibasilar crackles and the remainder of the exam is normal. Laboratory testing shows a white blood cell count of 2400 cells/μL, hemoglobin 7.6 g/dL, and platelet count 66 × 103/μL. Creatinine is 1.0 mg/dL. Chest radiograph shows ill-defined bilateral lower-lobe infiltrates. CT scans are shown in the Figure.
- For which infectious complications is this patient most at risk?
Pneumonia
A prospective trial in the HSCT population reported a pneumonia incidence rate of 68%, and pneumonia is more common in allogeneic HSCT with prolonged immunosuppressive therapy.32 Development of pneumonia within 100 days of transplant directly correlates with nonrelapsed mortality.33 Early detection is key, and bronchoscopy within the first 5 days of symptoms has been shown to change therapy in approximately 40% of cases but has not been shown to affect mortality.34 The clinical presentation of pneumonia in the HSCT population can be variable because of the presence of neutropenia and profound immunosuppression. Traditionally accepted diagnostic criteria of fevers, sputum production, and new infiltrates should be used with caution, and an appropriately high index of suspicion should be maintained. Progression to respiratory failure, regardless of causative organism of infection, portends a poor prognosis, with mortality rates estimated at 70% to 90%.35,36 Several transplant-specific factors may affect early infections. For instance, UCB transplants have been found to have a higher incidence of invasive aspergillosis and cytomegalovirus (CMV) infections but without higher mortality attributed to the infections.37
Bacterial Pneumonia
Bacterial pneumonia accounts for 20% to 50% of pneumonia cases in HSCT recipients.38 Gram-negative organisms, specifically Pseudomonas aeruginosa and Escherichia coli, were reported to be the most common pathologic bacteria in recent prospective trials, whereas previous retrospective trials showed that common community-acquired organisms were the most common cause of pneumonia in HSCT recipients.32,39 This underscores the importance of being aware of the clinical prevalence of microorganisms and local antibiograms, along with associated institutional susceptibility profiles. Initiation of immediate empiric broad-spectrum antibiotics is essential when bacterial pneumonia is suspected.
Viral Pneumonia
The prevalence of viral pneumonia in stem cell transplant recipients is estimated at 28%,32 with most cases being caused by community viral pathogens such as rhinovirus, respiratory syncytial virus (RSV), influenza A and B, and parainfluenza.39 The prevention, prophylaxis, and early treatment of viral pneumonias, specifically CMV infection, have decreased the mortality associated with early pneumonia after HSCT. Co-infection with bacterial organisms must be considered and has been associated with increased mortality in the intensive care unit setting.40
Supportive treatment with rhinovirus infection is sufficient as the disease is usually self-limited in immunocompromised patients. In contrast, infection with RSV in the lower respiratory tract is associated with increased mortality in prior reports, and recent studies suggest that further exploration of prophylaxis strategies is warranted.41 Treatment with ribavirin remains the backbone of therapy, but drug toxicity continues to limit its use. The addition of immunomodulators such as RSV immune globulin or palivizumab to ribavirin remains controversial, but a retrospective review suggests that early treatment may prevent progression to lower respiratory tract infection and lead to improved mortality.42 Infection with influenza A/B must be considered during influenza season. Treatment with oseltamivir may shorten the duration of disease when influenza A/B or parainfluenza are detected. Reactivation of latent herpes simplex virus during the pre-engraftment phase should also be considered. Treatment is similar to that in nonimmunocompromised hosts. When CMV pneumonia is suspected, careful history regarding compliance with prophylactic antivirals and CMV status of both the recipient and donor are key. A presumptive diagnosis can be made with the presence of appropriate clinical scenario, supportive radiographic images showing areas of ground-glass opacification or consolidation, and positive CMV polymerase chain reaction (PCR) assay. Visualization of inclusion bodies on lung biopsy tissue remains the gold standard for diagnosis. Treatment consists of CMV immunoglobulin and ganciclovir.
Fungal Pneumonia
Early fungal pneumonias have been associated with increased mortality in the HSCT population.43 Clinical suspicion should remain high and compliance with antifungal prophylaxis should be questioned thoroughly. Invasive aspergillosis (IA) remains the most common fungal infection. A bimodal distribution of onset of infection peaking on day 16 and again on day 96 has been described in the literature.44 Patients often present with classic pneumonia symptoms, but these may be accompanied by hemoptysis. Proven IA diagnosis requires visualization of fungal forms from biopsy or needle aspiration or a positive culture obtained in a sterile fashion.45 Most clinical data comes from experience with probable and possible diagnosis of IA. Bronchoalveolar lavage with testing with Aspergillus galactomannan assay has been shown to be clinically useful in establishing the clinical diagnosis in the HSCT population.46 Classic air-crescent findings on chest CT are helpful in establishing a possible diagnosis, but retrospective analysis reveals CT findings such as focal infiltrates and pulmonary nodular patterns are more common.47 First-line treatment with voriconazole has been shown to decrease short-term mortality attributable to IA but has not had an effect on long-term, all-cause mortality.48 Surgical resection is reserved for patients with refractory disease or patients presenting with massive hemoptysis.
Mucormycosis is an emerging disease with ever increasing prevalence in the HSCT population, reflecting the improved prophylaxis and treatment of IA. Initial clinical presentation is similar to IA, most commonly affecting the lung, although craniofacial involvement is classic for mucormycosis, especially in HSCT patients with diabetes.49Mucor infections can present with massive hemoptysis due to tissue invasion and disregard for tissue and fascial planes. Diagnosis of mucormycosis is associated with as much as a six-fold increase in risk for death. Diagnosis requires identification of the organism by examination or culture and biopsy is often necessary.50,51 Amphotericin B remains first-line therapy as mucormycosis is resistant to azole antifungals, with higher doses recommended for cerebral involvement.52
Candida pulmonary infections during the early HSCT period are becoming increasingly rare due to widespread use of fluconazole prophylaxis and early treatment of mucosal involvement during neutropenia. Endemic fungal infections such as blastomycosis, coccidioidomycosis, and histoplasmosis should be considered in patients inhabiting specific geographic areas or with recent travel to these areas.
- What test should be performed to evaluate for infectious causes of pneumonia?
Role of Flexible Fiberoptic Bronchoscopy
The utility of flexible fiberoptic bronchoscopy (FOB) in immune-compromised patients for the evaluation of pulmonary infiltrates is a frequently debated topic. Current studies suggest a diagnosis can be made in approximately 80% of cases in the immune-compromised population.32,53 Noninvasive testing such as urine and serum antigens, sputum cultures, Aspergillus galactomannan assays, viral nasal swabs, and PCR studies often lead to a diagnosis in appropriate clinical scenarios. Conservative management would dictate the use of noninvasive testing whenever possible, and randomized controlled trials have shown noninvasive testing to be noninferior to FOB in preventing need for mechanical ventilation, with no difference in overall mortality.54 FOB has been shown to be most useful in establishing a diagnosis when an infectious etiology is suspected.55 In multivariate analysis, a delay in the identification of the etiology of pulmonary infiltrate was associated with increased mortality.56 Additionally, early FOB was found to be superior to late FOB in revealing a diagnosis. 32,57 Despite its ability to detect the cause of pulmonary disease, direct antibiotic therapy, and possibly change therapy, FOB with diagnostic maneuvers has not been shown to affect mortality.58 In a large case series, FOB with bronchoalveolar lavage (BAL) revealed a diagnosis in approximately 30% to 50% of cases. The addition of transbronchial biopsy did not improve diagnostic utility.58 More recent studies have confirmed that the addition of transbronchial biopsy does not add to diagnostic yield and is associated with increased adverse events.59 The appropriate use of advanced techniques such as endobronchial ultrasound–guided transbronchial needle aspirations, endobronchial biopsy, and CT-guided navigational bronchoscopy has not been established and should be considered on a case-by-case basis. In summary, routine early BAL is the diagnostic test of choice, especially when infectious pulmonary complications are suspected.
Contraindications for FOB in this population mirror those in the general population. These include acute severe hypoxemic respiratory failure, myocardial ischemia or acute coronary syndrome within 2 weeks of procedure, severe thrombocytopenia, and inability to provide or obtain informed consent from patient or health care power of attorney. Coagulopathy and thrombocytopenia are common comorbid conditions in the HSCT population. A platelet count of < 20 × 103/µL has generally been used as a cut-off for routine FOB with BAL.60 Risks of the procedures should be discussed clearly with the patient, but simple FOB for airway evaluation and BAL is generally well tolerated even under these conditions.
Early Nonifectious Pulmonary Complications
Case Patient 2 Continued
Bronchoscopy with BAL performed the day after admission is unremarkable and stains and cultures are negative for viral, bacterial, and fungal organisms. The patient is initially started on broad-spectrum antibiotics, but his oxygenation continues to worsen to the point that he is placed on noninvasive positive pressure ventilation. He is started empirically on amphotericin B and eventually is intubated. VATS lung biopsy is ultimately performed and pathology is consistent with diffuse alveolar damage.
- Based on these biopsy findings, what is the diagnosis?
Based on the pathology consistent with diffuse alveolar damage, a diagnosis of idiopathic pneumonia syndrome (IPS) is made.
- What noninfectious pulmonary complications occur in the early post-transplant period?
The overall incidence of noninfectious pulmonary complications after HSCT is generally estimated at 20% to 30%.32 Acute pulmonary edema is a common very early noninfectious pulmonary complication and clinically the most straightforward to treat. Three distinct clinical syndromes—peri-engraftment respiratory distress syndrome (PERDS), diffuse alveolar hemorrhage (DAH), and IPS—comprise the remainder of the pertinent early noninfectious complications. Clinical presentation differs based upon the disease entity. Recent studies have evaluated the role of angiotensin-converting enzyme polymorphisms as a predictive marker for risk of developing early noninfectious pulmonary complications.61
Peri-Engraftment Respiratory Distress Syndrome
PERDS is a clinical syndrome comprising the cardinal features of erythematous rash and fever along with noncardiogenic pulmonary infiltrates and hypoxemia that occur in the peri-engraftment period, defined as recovery of absolute neutrophil count to > 500/μL on 2 consecutive days.62 PERDS occurs in the autologous HSCT population and may be a clinical correlate to early GVHD in the allogeneic HSCT population. It is hypothesized that the pathophysiology underlying PERDS is an autoimmune-related capillary leak caused by pro-inflammatory cytokine release.63 Treatment remains anecdotal and currently consists of supportive care and high-dose corticosteroids. Some have favored limiting the use of gCSF given its role in stimulating rapid white blood cell recovery.33 Prognosis is favorable, but progression to fulminant respiratory failure requiring mechanical ventilation portends a poor prognosis.
Diffuse Alveolar Hemorrhage
DAH is clinical syndrome consisting of diffuse alveolar infiltrates on pulmonary imaging combined with progressively bloodier return per aliquot during BAL in 3 different subsegments or more than 20% hemosiderin-laden macrophages on BAL fluid evaluation. Classically, DAH is defined in the absence of pulmonary infection or cardiac dysfunction. The pathophysiology is thought to be related to inflammation of pulmonary vasculature within the alveolar walls leading to alveolitis. Although no prospective trials exist, early use of high-dose corticosteroid therapy is thought to improve outcomes;64,65 a recent study, however, showed low-dose steroids may be associated with the lowest mortality.66 Mortality is directly linked to the presence of superimposed infection, need for mechanical ventilation, late onset, and development of multiorgan failure.67
Idiopathic Pneumonia Syndrome
IPS is a complex clinical syndrome whose pathology is felt to stem from a variety of possible lung insults such as direct myeloablative drug toxicity, occult pulmonary infection, or cytokine-driven inflammation. The ATS published an article further subcategorizing IPS as different clinical entities based upon whether the primary insult involves the vascular endothelium, interstitial tissue, and airway tissue, truly idiopathic, or unclassified.68 In clinical practice, IPS is defined as widespread alveolar injury in the absence of evidence of renal failure, heart failure, and excessive fluid resuscitation. In addition, negative testing for a variety of bacterial, viral, and fungal causes is also necessary.69 Clinical syndromes included within the IPS definition are ARDS, acute interstitial pneumonia, DAH, cryptogenic organizing pneumonia, and BOS.70 Risk factors for developing IPS include TBI, older age of recipient, acute GVHD, and underlying diagnosis of AML or myelodysplastic syndrome.12 In addition, it has been shown that risk for developing IPS is lower in patients undergoing allogeneic HSCT who receive non-myeloablative conditioning regimens.71 The pathologic finding in IPS is diffuse alveolar damage. A 2006 study in which investigators reviewed BAL samples from patients with IPS found that 3% of the patients had PCR evidence of human metapneumovirus infection, and a study in 2015 found PCR evidence of infection in 53% of BAL samples from patients diagnosed with IPS.72,73 This fuels the debate on whether IPS is truly an infection-driven process where the source of infection, pulmonary or otherwise, simply escapes detection. Various surfactant proteins, which play a role in decreasing surface tension within the alveolar interface and function as mediators within the innate immunity of the lung, have been studied in regard to development of IPS. Small retrospective studies have shown a trend toward lower pre-transplant serum protein surfactant D and the development of IPS.74
The diagnosis of IPS does not require pathologic diagnosis in most circumstances. The correct clinical findings in association with a negative infectious workup lead to a presumptive diagnosis of IPS. The extent of the infectious workup that must be completed to adequately rule out infection is often a difficult clinical question. Recent recommendations include BAL fluid evaluation for routine bacterial cultures, appropriate viral culture, and consideration of PCR testing to evaluate for Mycoplasma, Chlamydia, and Aspergillus antigens.75 Transbronchial biopsy continues to appear in recommendations, but is not routinely performed and should be completed as the patient’s clinical status permits.8,68 Table 3 reviews basic features of early noninfectious pulmonary complications.
Treatment of IPS centers around moderate to high doses of corticosteroids. Based on IPS experimental modes, tumor necrosis factor (TNF)-α has been implicated as an important mediator. Unfortunately, several studies evaluating etanercept have produced conflicting results, and this agent’s clinical effects on morbidity and mortality remain in question.76
- What treatment should be offered to the patient with diffuse alveolar damage on biopsy?
Treatment consists of supportive care and empiric broad-spectrum antibiotics with consideration of high-dose corticosteroids. Based upon early studies in murine models implicating TNF, pilot studies were performed evaluating etanercept as a possible safe and effective addition to high-dose systemic corticosteroids.77 Although these results were promising, data from a truncated randomized control clinical trial failed to show improvement in patient response in the adult population.76 More recent data from the same author suggests that pediatric populations with IPS are, however, responsive to etanercept and high-dose corticosteroid therapy.78 When IPS develops as a late complication, treatment with high-dose corticosteroids (2 mg/kg/day) and etanercept (0.4 mg/kg twice weekly) has been shown to improve 2-year survival.79
Case Patient 2 Conclusion
The patient is started on steroids and makes a speedy recovery. He is successfully extubated 5 days later.
Conclusion
Careful pretransplant evaluation, including a full set of pulmonary function tests, can help predict a patient’s risk for pulmonary complications after transplant, allowing risk factor modification strategies to be implemented prior to transplant, including smoking cessation. It also helps identify patients at high risk for complications who will require closer monitoring after transplantation. Early posttransplant complications include infectious and noninfectious entities. Bacterial, viral, and fungal pneumonias are in the differential of infectious pneumonia, and bronchoscopy can be helpful in establishing a diagnosis. A common, important noninfectious cause of early pulmonary complications is IPS, which is treated with steroids and sometimes anti-TNF therapy.
1. Gratwohl A, Baldomero H, Aljurf M, et al. Hematopoietic stem cell transplantation: a global perspective. JAMA 2010;303:1617–24.
2. Kotloff RM, Ahya VN, Crawford SW. Pulmonary complications of solid organ and hematopoietic stem cell transplantation. Am J Respir Crit Care Med 2004;170:22–48.
4. Copelan EA. Hematopoietic stem-cell transplantation. N Engl J Med 2006;354:1813–26.
5. Anasetti C, Logan BR, Lee SJ, et al. Peripheral-blood stem cells versus bone marrow from unrelated donors. N Engl J Med 2012;367:1487–96.
6. Giralt S, Ballen K, Rizzo D, et al. Reduced-intensity conditioning regimen workshop: defining the dose spectrum. Report of a workshop convened by the center for international blood and marrow transplant research. Biol Blood Marrow Transplant 2009;15:367–9.
7. Shulman HM, Kleiner D, Lee SJ, et al. Histopathologic diagnosis of chronic graft-versus-host disease: National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: II. Pathology Working Group Report. Biol Blood Marrow Transplant 2006;12:31–47.
8. Afessa B, Abdulai RM, Kremers WK, et al. Risk factors and outcome of pulmonary complications after autologous hematopoietic stem cell transplant. Chest 2012;141:442–50.
9. Bolwell BJ. Are predictive factors clinically useful in bone marrow transplantation? Bone Marrow Transplant 2003;32:853–61.
10. Carlson K, Backlund L, Smedmyr B, et al. Pulmonary function and complications subsequent to autologous bone marrow transplantation. Bone Marrow Transplant 1994;14:805–11.
11. Clark JG, Schwartz DA, Flournoy N, et al. Risk factors for airflow obstruction in recipients of bone marrow transplants. Ann Intern Med 1987;107:648–56.
12. Crawford SW, Fisher L. Predictive value of pulmonary function tests before marrow transplantation. Chest 1992; 101:1257–64.
13. Ghalie R, Szidon JP, Thompson L, et al. Evaluation of pulmonary complications after bone marrow transplantation: the role of pretransplant pulmonary function tests. Bone Marrow Transplant 1992;10:359–65.
14. Ho VT, Weller E, Lee SJ, et al. Prognostic factors for early severe pulmonary complications after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2001;7:223–9.
15. Horak DA, Schmidt GM, Zaia JA, et al. Pretransplant pulmonary function predicts cytomegalovirus-associated interstitial pneumonia following bone marrow transplantation. Chest 1992;102:1484–90.
16. Ramirez-Sarmiento A, Orozco-Levi M, Walter EC, et al. Influence of pretransplantation restrictive lung disease on allogeneic hematopoietic cell transplantation outcomes. Biol Blood Marrow Transplant 2010;16:199–206.
17. White AC, Terrin N, Miller KB, Ryan HF. Impaired respiratory and skeletal muscle strength in patients prior to hematopoietic stem-cell transplantation. Chest 2005;128145–52.
18. Afessa B. Pretransplant pulmonary evaluation of the blood and marrow transplant recipient. Chest 2005;128:8–10.
19. Parimon T, Madtes DK, Au DH, et al. Pretransplant lung function, respiratory failure, and mortality after stem cell transplantation. Am J Respir Crit Care Med 2005;172:384–90.
20. Pavletic SZ, Martin P, Lee SJ, et al. Measuring therapeutic response in chronic graft-versus-host disease: National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: IV. Response Criteria Working Group report. Biol Blood Marrow Transplant 2006;12:252–66.
21. Parimon T, Au DH, Martin PJ, Chien JW. A risk score for mortality after allogeneic hematopoietic cell transplantation. Ann Intern Med 2006;144:407–14.
22. Au BK, Gooley TA, Armand P, et al. Reevaluation of the pretransplant assessment of mortality score after allogeneic hematopoietic transplantation. Biol Blood Marrow Transplant 2015;21:848–54.
23. Sorror ML, Maris MB, Storb R, et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood 2005;106:2912–9.
24. Chien JW, Sullivan KM. Carbon monoxide diffusion capacity: how low can you go for hematopoietic cell transplantation eligibility? Biol Blood Marrow Transplant 2009;15: 447–53.
25. Coffey DG, Pollyea DA, Myint H, et al. Adjusting DLCO for Hb and its effects on the Hematopoietic Cell Transplantation-specific Comorbidity Index. Bone Marrow Transplant 2013;48:1253–6.
26. Kasow KA, Krueger J, Srivastava DK, et al. Clinical utility of computed tomography screening of chest, abdomen, and sinuses before hematopoietic stem cell transplantation: the St. Jude experience. Biol Blood Marrow Transplant 2009;15:490–5.
27. Hamadani M, Craig M, Awan FT, Devine SM. How we approach patient evaluation for hematopoietic stem cell transplantation. Bone Marrow Transplant 2010;45: 1259–68.
28. Savani BN, Montero A, Wu C, et al. Prediction and prevention of transplant-related mortality from pulmonary causes after total body irradiation and allogeneic stem cell transplantation. Biol Blood Marrow Transplant 2005;11:223–30.
29. Ehlers SL, Gastineau DA, Patten CA, et al. The impact of smoking on outcomes among patients undergoing hematopoietic SCT for the treatment of acute leukemia. Bone Marrow Transplant 2011;46:285–90.
30. Marks DI, Ballen K, Logan BR, et al. The effect of smoking on allogeneic transplant outcomes. Biol Blood Marrow Transplant 2009;15:1277–87.
31. Tran BT, Halperin A, Chien JW. Cigarette smoking and outcomes after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2011;17:1004–11.
32. Lucena CM, Torres A, Rovira M, et al. Pulmonary complications in hematopoietic SCT: a prospective study. Bone Marrow Transplant 2014;49:1293–9.
33. Chi AK, Soubani AO, White AC, Miller KB. An update on pulmonary complications of hematopoietic stem cell transplantation. Chest 2013;144:1913–22.
34. Dunagan DP, Baker AM, Hurd DD, Haponik EF. Bronchoscopic evaluation of pulmonary infiltrates following bone marrow transplantation. Chest 1997;111:135–41.
35. Naeem N, Reed MD, Creger RJ, et al. Transfer of the hematopoietic stem cell transplant patient to the intensive care unit: does it really matter? Bone Marrow Transplant 2006;37:119–33.
36. Afessa B, Tefferi A, Hoagland HC, et al. Outcome of recipients of bone marrow transplants who require intensive care unit support. Mayo Clin Proc 1992;67:117–22.
37. Parody R, Martino R, de la Camara R, et al. Fungal and viral infections after allogeneic hematopoietic transplantation from unrelated donors in adults: improving outcomes over time. Bone Marrow Transplant 2015;50:274–81.
38. Orasch C, Weisser M, Mertz D, et al. Comparison of infectious complications during induction/consolidation chemotherapy versus allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant 2010;45:521–6.
39. Aguilar-Guisado M, Jimenez-Jambrina M, Espigado I, et al. Pneumonia in allogeneic stem cell transplantation recipients: a multicenter prospective study. Clin Transplant 2011;25:E629–38.
40. Palacios G, Hornig M, Cisterna D, et al. Streptococcus pneumoniae coinfection is correlated with the severity of H1N1 pandemic influenza. PLoS One 2009;4:e8540.
41. Hynicka LM, Ensor CR. Prophylaxis and treatment of respiratory syncytial virus in adult immunocompromised patients. Ann Pharmacother 2012;46:558–66.
42. Shah JN, Chemaly RF. Management of RSV infections in adult recipients of hematopoietic stem cell transplantation. Blood 2011;2755–63.
43. Marr KA, Bowden RA. Fungal infections in patients undergoing blood and marrow transplantation. Transpl Infect Dis 1999;1:237–46.
44. Wald A, Leisenring W, van Burik JA, Bowden RA. Epidemiology of Aspergillus infections in a large cohort of patients undergoing bone marrow transplantation. J Infect Dis 1997;175:1459–66.
45. Ascioglu S, Rex JH, de Pauw B, et al. Defining opportunistic invasive fungal infections in immunocompromised patients with cancer and hematopoietic stem cell transplants: an international consensus. Clin Infect Dis 2002;34:7–14.
46. Fisher CE, Stevens AM, Leisenring W, et al. Independent contribution of bronchoalveolar lavage and serum galactomannan in the diagnosis of invasive pulmonary aspergillosis. Transpl Infect Dis 2014;16:505–10.
47. Kojima R, Tateishi U, Kami M, et al. Chest computed tomography of late invasive aspergillosis after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2005;11:506–11.
48. Salmeron G, Porcher R, Bergeron A, et al. Persistent poor long-term prognosis of allogeneic hematopoietic stem cell transplant recipients surviving invasive aspergillosis. Haematologica 2012;97:1357–63.
49. McNulty JS. Rhinocerebral mucormycosis: predisposing factors. Laryngoscope 1982;92(10 Pt 1):1140.
50. Walsh TJ, Gamaletsou MN, McGinnis MR, et al. Early clinical and laboratory diagnosis of invasive pulmonary, extrapulmonary, and disseminated mucormycosis (zygomycosis). Clin Infect Dis 2012;54 Suppl 1:S55–60.
51. Klingspor L, Saaedi B, Ljungman P, Szakos A. Epidemiology and outcomes of patients with invasive mould infections: a retrospective observational study from a single centre (2005-2009). Mycoses 2015;58:470–7.
52. Danion F, Aguilar C, Catherinot E, et al. Mucormycosis: new developments in a persistently devastating infection. Semin Respir Crit Care Med 2015;36:692–70.
53. Rano A, Agusti C, Jimenez P, et al. Pulmonary infiltrates in non-HIV immunocompromised patients: a diagnostic approach using non-invasive and bronchoscopic procedures. Thorax 2001;56:379–87.
54. Azoulay E, Mokart D, Rabbat A, et al. Diagnostic bronchoscopy in hematology and oncology patients with acute respiratory failure: prospective multicenter data. Crit Care Med 2008;36:100–7.
55. Jain P, Sandur S, Meli Y, et al. Role of flexible bronchoscopy in immunocompromised patients with lung infiltrates. Chest 2004;125:712–22.
56. Rano A, Agusti C, Benito N, et al. Prognostic factors of non-HIV immunocompromised patients with pulmonary infiltrates. Chest 2002;122:253–61.
57. Shannon VR, Andersson BS, Lei X, et al. Utility of early versus late fiberoptic bronchoscopy in the evaluation of new pulmonary infiltrates following hematopoietic stem cell transplantation. Bone Marrow Transplant 2010;45:647–55.
58. Patel NR, Lee PS, Kim JH, et al. The influence of diagnostic bronchoscopy on clinical outcomes comparing adult autologous and allogeneic bone marrow transplant patients. Chest 2005;127:1388–96.
59. Chellapandian D, Lehrnbecher T, Phillips B, et al. Bronchoalveolar lavage and lung biopsy in patients with cancer and hematopoietic stem-cell transplantation recipients: a systematic review and meta-analysis. J Clin Oncol 2015;33:501–9.
60. Carr IM, Koegelenberg CF, von Groote-Bidlingmaier F, et al. Blood loss during flexible bronchoscopy: a prospective observational study. Respiration 2012;84:312–8.
61. Miyamoto M, Onizuka M, Machida S, et al. ACE deletion polymorphism is associated with a high risk of non-infectious pulmonary complications after stem cell transplantation. Int J Hematol 2014;99:175–83.
62. Capizzi SA, Kumar S, Huneke NE, et al. Peri-engraftment respiratory distress syndrome during autologous hematopoietic stem cell transplantation. Bone Marrow Transplant 2001;27:1299–303.
63. Spitzer TR. Engraftment syndrome following hematopoietic stem cell transplantation. Bone Marrow Transplant 2001;27:893–8.
64. Wanko SO, Broadwater G, Folz RJ, Chao NJ. Diffuse alveolar hemorrhage: retrospective review of clinical outcome in allogeneic transplant recipients treated with aminocaproic acid. Biol Blood Marrow Transplant 2006;12:949–53.
65. Metcalf JP, Rennard SI, Reed EC, et al. Corticosteroids as adjunctive therapy for diffuse alveolar hemorrhage associated with bone marrow transplantation. University of Nebraska Medical Center Bone Marrow Transplant Group. Am J Med 1994;96:327–34.
66. Rathi NK, Tanner AR, Dinh A, et al. Low-, medium- and high-dose steroids with or without aminocaproic acid in adult hematopoietic SCT patients with diffuse alveolar hemorrhage. Bone Marrow Transplant 2015;50:420–6.
67. Afessa B, Tefferi A, Litzow MR, Peters SG. Outcome of diffuse alveolar hemorrhage in hematopoietic stem cell transplant recipients. Am J Respir Crit Care Med 2002;166:1364–8.
68. Panoskaltsis-Mortari A, Griese M, Madtes DK, et al. An official American Thoracic Society research statement: noninfectious lung injury after hematopoietic stem cell transplantation: idiopathic pneumonia syndrome. Am J Respir Crit Care Med 2011;183:1262–79.
69. Clark JG, Hansen JA, Hertz MI, Pet al. NHLBI workshop summary. Idiopathic pneumonia syndrome after bone marrow transplantation. Am Rev Resp Dis 1993;147:1601–6.
70. Vande Vusse LK, Madtes DK. Early onset noninfectious pulmonary syndromes after hematopoietic cell transplantation. Clin Chest Med 2017;38:233–48.
71. Fukuda T, Hackman RC, Guthrie KA, et al. Risks and outcomes of idiopathic pneumonia syndrome after nonmyeloablative and conventional conditioning regimens for allogeneic hematopoietic stem cell transplantation. Blood 2003;102:2777–85.
72. Englund JA, Boeckh M, Kuypers J, et al. Brief communication: fatal human metapneumovirus infection in stem-cell transplant recipients. Ann Intern Med 2006;144:344–9.
73. Seo S, Renaud C, Kuypers JM, et al. Idiopathic pneumonia syndrome after hematopoietic cell transplantation: evidence of occult infectious etiologies. Blood 2015;125:3789–97.
74. Nakane T, Nakamae H, Kamoi H, et al. Prognostic value of serum surfactant protein D level prior to transplant for the development of bronchiolitis obliterans syndrome and idiopathic pneumonia syndrome following allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;42:43–9.
75. Gilbert CR, Lerner A, Baram M, Awsare BK. Utility of flexible bronchoscopy in the evaluation of pulmonary infiltrates in the hematopoietic stem cell transplant population—a single center fourteen year experience. Arch Bronconeumol 2013;49:189–95.
76. Yanik GA, Horowitz MM, Weisdorf DJ, et al. Randomized, double-blind, placebo-controlled trial of soluble tumor necrosis factor receptor: enbrel (etanercept) for the treatment of idiopathic pneumonia syndrome after allogeneic stem cell transplantation: blood and marrow transplant clinical trials network protocol. Biol Blood Marrow Transplant 2014;20:858–64.
77. Levine JE, Paczesny S, Mineishi S, et al. Etanercept plus methylprednisolone as initial therapy for acute graft-versus-host disease. Blood 2008;111:2470–5.
78. Yanik GA, Grupp SA, Pulsipher MA, et al. TNF-receptor inhibitor therapy for the treatment of children with idiopathic pneumonia syndrome. A joint Pediatric Blood and Marrow Transplant Consortium and Children’s Oncology Group Study (ASCT0521). Biol Blood Marrow Transplant 2015;21:67–73.
79. Thompson J, Yin Z, D’Souza A, et al. Etanercept and corticosteroid therapy for the treatment of late-onset idiopathic pneumonia syndrome. Biol Blood Marrow Transplant J 2017; 23:1955–60.
1. Gratwohl A, Baldomero H, Aljurf M, et al. Hematopoietic stem cell transplantation: a global perspective. JAMA 2010;303:1617–24.
2. Kotloff RM, Ahya VN, Crawford SW. Pulmonary complications of solid organ and hematopoietic stem cell transplantation. Am J Respir Crit Care Med 2004;170:22–48.
4. Copelan EA. Hematopoietic stem-cell transplantation. N Engl J Med 2006;354:1813–26.
5. Anasetti C, Logan BR, Lee SJ, et al. Peripheral-blood stem cells versus bone marrow from unrelated donors. N Engl J Med 2012;367:1487–96.
6. Giralt S, Ballen K, Rizzo D, et al. Reduced-intensity conditioning regimen workshop: defining the dose spectrum. Report of a workshop convened by the center for international blood and marrow transplant research. Biol Blood Marrow Transplant 2009;15:367–9.
7. Shulman HM, Kleiner D, Lee SJ, et al. Histopathologic diagnosis of chronic graft-versus-host disease: National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: II. Pathology Working Group Report. Biol Blood Marrow Transplant 2006;12:31–47.
8. Afessa B, Abdulai RM, Kremers WK, et al. Risk factors and outcome of pulmonary complications after autologous hematopoietic stem cell transplant. Chest 2012;141:442–50.
9. Bolwell BJ. Are predictive factors clinically useful in bone marrow transplantation? Bone Marrow Transplant 2003;32:853–61.
10. Carlson K, Backlund L, Smedmyr B, et al. Pulmonary function and complications subsequent to autologous bone marrow transplantation. Bone Marrow Transplant 1994;14:805–11.
11. Clark JG, Schwartz DA, Flournoy N, et al. Risk factors for airflow obstruction in recipients of bone marrow transplants. Ann Intern Med 1987;107:648–56.
12. Crawford SW, Fisher L. Predictive value of pulmonary function tests before marrow transplantation. Chest 1992; 101:1257–64.
13. Ghalie R, Szidon JP, Thompson L, et al. Evaluation of pulmonary complications after bone marrow transplantation: the role of pretransplant pulmonary function tests. Bone Marrow Transplant 1992;10:359–65.
14. Ho VT, Weller E, Lee SJ, et al. Prognostic factors for early severe pulmonary complications after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2001;7:223–9.
15. Horak DA, Schmidt GM, Zaia JA, et al. Pretransplant pulmonary function predicts cytomegalovirus-associated interstitial pneumonia following bone marrow transplantation. Chest 1992;102:1484–90.
16. Ramirez-Sarmiento A, Orozco-Levi M, Walter EC, et al. Influence of pretransplantation restrictive lung disease on allogeneic hematopoietic cell transplantation outcomes. Biol Blood Marrow Transplant 2010;16:199–206.
17. White AC, Terrin N, Miller KB, Ryan HF. Impaired respiratory and skeletal muscle strength in patients prior to hematopoietic stem-cell transplantation. Chest 2005;128145–52.
18. Afessa B. Pretransplant pulmonary evaluation of the blood and marrow transplant recipient. Chest 2005;128:8–10.
19. Parimon T, Madtes DK, Au DH, et al. Pretransplant lung function, respiratory failure, and mortality after stem cell transplantation. Am J Respir Crit Care Med 2005;172:384–90.
20. Pavletic SZ, Martin P, Lee SJ, et al. Measuring therapeutic response in chronic graft-versus-host disease: National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: IV. Response Criteria Working Group report. Biol Blood Marrow Transplant 2006;12:252–66.
21. Parimon T, Au DH, Martin PJ, Chien JW. A risk score for mortality after allogeneic hematopoietic cell transplantation. Ann Intern Med 2006;144:407–14.
22. Au BK, Gooley TA, Armand P, et al. Reevaluation of the pretransplant assessment of mortality score after allogeneic hematopoietic transplantation. Biol Blood Marrow Transplant 2015;21:848–54.
23. Sorror ML, Maris MB, Storb R, et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood 2005;106:2912–9.
24. Chien JW, Sullivan KM. Carbon monoxide diffusion capacity: how low can you go for hematopoietic cell transplantation eligibility? Biol Blood Marrow Transplant 2009;15: 447–53.
25. Coffey DG, Pollyea DA, Myint H, et al. Adjusting DLCO for Hb and its effects on the Hematopoietic Cell Transplantation-specific Comorbidity Index. Bone Marrow Transplant 2013;48:1253–6.
26. Kasow KA, Krueger J, Srivastava DK, et al. Clinical utility of computed tomography screening of chest, abdomen, and sinuses before hematopoietic stem cell transplantation: the St. Jude experience. Biol Blood Marrow Transplant 2009;15:490–5.
27. Hamadani M, Craig M, Awan FT, Devine SM. How we approach patient evaluation for hematopoietic stem cell transplantation. Bone Marrow Transplant 2010;45: 1259–68.
28. Savani BN, Montero A, Wu C, et al. Prediction and prevention of transplant-related mortality from pulmonary causes after total body irradiation and allogeneic stem cell transplantation. Biol Blood Marrow Transplant 2005;11:223–30.
29. Ehlers SL, Gastineau DA, Patten CA, et al. The impact of smoking on outcomes among patients undergoing hematopoietic SCT for the treatment of acute leukemia. Bone Marrow Transplant 2011;46:285–90.
30. Marks DI, Ballen K, Logan BR, et al. The effect of smoking on allogeneic transplant outcomes. Biol Blood Marrow Transplant 2009;15:1277–87.
31. Tran BT, Halperin A, Chien JW. Cigarette smoking and outcomes after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2011;17:1004–11.
32. Lucena CM, Torres A, Rovira M, et al. Pulmonary complications in hematopoietic SCT: a prospective study. Bone Marrow Transplant 2014;49:1293–9.
33. Chi AK, Soubani AO, White AC, Miller KB. An update on pulmonary complications of hematopoietic stem cell transplantation. Chest 2013;144:1913–22.
34. Dunagan DP, Baker AM, Hurd DD, Haponik EF. Bronchoscopic evaluation of pulmonary infiltrates following bone marrow transplantation. Chest 1997;111:135–41.
35. Naeem N, Reed MD, Creger RJ, et al. Transfer of the hematopoietic stem cell transplant patient to the intensive care unit: does it really matter? Bone Marrow Transplant 2006;37:119–33.
36. Afessa B, Tefferi A, Hoagland HC, et al. Outcome of recipients of bone marrow transplants who require intensive care unit support. Mayo Clin Proc 1992;67:117–22.
37. Parody R, Martino R, de la Camara R, et al. Fungal and viral infections after allogeneic hematopoietic transplantation from unrelated donors in adults: improving outcomes over time. Bone Marrow Transplant 2015;50:274–81.
38. Orasch C, Weisser M, Mertz D, et al. Comparison of infectious complications during induction/consolidation chemotherapy versus allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant 2010;45:521–6.
39. Aguilar-Guisado M, Jimenez-Jambrina M, Espigado I, et al. Pneumonia in allogeneic stem cell transplantation recipients: a multicenter prospective study. Clin Transplant 2011;25:E629–38.
40. Palacios G, Hornig M, Cisterna D, et al. Streptococcus pneumoniae coinfection is correlated with the severity of H1N1 pandemic influenza. PLoS One 2009;4:e8540.
41. Hynicka LM, Ensor CR. Prophylaxis and treatment of respiratory syncytial virus in adult immunocompromised patients. Ann Pharmacother 2012;46:558–66.
42. Shah JN, Chemaly RF. Management of RSV infections in adult recipients of hematopoietic stem cell transplantation. Blood 2011;2755–63.
43. Marr KA, Bowden RA. Fungal infections in patients undergoing blood and marrow transplantation. Transpl Infect Dis 1999;1:237–46.
44. Wald A, Leisenring W, van Burik JA, Bowden RA. Epidemiology of Aspergillus infections in a large cohort of patients undergoing bone marrow transplantation. J Infect Dis 1997;175:1459–66.
45. Ascioglu S, Rex JH, de Pauw B, et al. Defining opportunistic invasive fungal infections in immunocompromised patients with cancer and hematopoietic stem cell transplants: an international consensus. Clin Infect Dis 2002;34:7–14.
46. Fisher CE, Stevens AM, Leisenring W, et al. Independent contribution of bronchoalveolar lavage and serum galactomannan in the diagnosis of invasive pulmonary aspergillosis. Transpl Infect Dis 2014;16:505–10.
47. Kojima R, Tateishi U, Kami M, et al. Chest computed tomography of late invasive aspergillosis after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2005;11:506–11.
48. Salmeron G, Porcher R, Bergeron A, et al. Persistent poor long-term prognosis of allogeneic hematopoietic stem cell transplant recipients surviving invasive aspergillosis. Haematologica 2012;97:1357–63.
49. McNulty JS. Rhinocerebral mucormycosis: predisposing factors. Laryngoscope 1982;92(10 Pt 1):1140.
50. Walsh TJ, Gamaletsou MN, McGinnis MR, et al. Early clinical and laboratory diagnosis of invasive pulmonary, extrapulmonary, and disseminated mucormycosis (zygomycosis). Clin Infect Dis 2012;54 Suppl 1:S55–60.
51. Klingspor L, Saaedi B, Ljungman P, Szakos A. Epidemiology and outcomes of patients with invasive mould infections: a retrospective observational study from a single centre (2005-2009). Mycoses 2015;58:470–7.
52. Danion F, Aguilar C, Catherinot E, et al. Mucormycosis: new developments in a persistently devastating infection. Semin Respir Crit Care Med 2015;36:692–70.
53. Rano A, Agusti C, Jimenez P, et al. Pulmonary infiltrates in non-HIV immunocompromised patients: a diagnostic approach using non-invasive and bronchoscopic procedures. Thorax 2001;56:379–87.
54. Azoulay E, Mokart D, Rabbat A, et al. Diagnostic bronchoscopy in hematology and oncology patients with acute respiratory failure: prospective multicenter data. Crit Care Med 2008;36:100–7.
55. Jain P, Sandur S, Meli Y, et al. Role of flexible bronchoscopy in immunocompromised patients with lung infiltrates. Chest 2004;125:712–22.
56. Rano A, Agusti C, Benito N, et al. Prognostic factors of non-HIV immunocompromised patients with pulmonary infiltrates. Chest 2002;122:253–61.
57. Shannon VR, Andersson BS, Lei X, et al. Utility of early versus late fiberoptic bronchoscopy in the evaluation of new pulmonary infiltrates following hematopoietic stem cell transplantation. Bone Marrow Transplant 2010;45:647–55.
58. Patel NR, Lee PS, Kim JH, et al. The influence of diagnostic bronchoscopy on clinical outcomes comparing adult autologous and allogeneic bone marrow transplant patients. Chest 2005;127:1388–96.
59. Chellapandian D, Lehrnbecher T, Phillips B, et al. Bronchoalveolar lavage and lung biopsy in patients with cancer and hematopoietic stem-cell transplantation recipients: a systematic review and meta-analysis. J Clin Oncol 2015;33:501–9.
60. Carr IM, Koegelenberg CF, von Groote-Bidlingmaier F, et al. Blood loss during flexible bronchoscopy: a prospective observational study. Respiration 2012;84:312–8.
61. Miyamoto M, Onizuka M, Machida S, et al. ACE deletion polymorphism is associated with a high risk of non-infectious pulmonary complications after stem cell transplantation. Int J Hematol 2014;99:175–83.
62. Capizzi SA, Kumar S, Huneke NE, et al. Peri-engraftment respiratory distress syndrome during autologous hematopoietic stem cell transplantation. Bone Marrow Transplant 2001;27:1299–303.
63. Spitzer TR. Engraftment syndrome following hematopoietic stem cell transplantation. Bone Marrow Transplant 2001;27:893–8.
64. Wanko SO, Broadwater G, Folz RJ, Chao NJ. Diffuse alveolar hemorrhage: retrospective review of clinical outcome in allogeneic transplant recipients treated with aminocaproic acid. Biol Blood Marrow Transplant 2006;12:949–53.
65. Metcalf JP, Rennard SI, Reed EC, et al. Corticosteroids as adjunctive therapy for diffuse alveolar hemorrhage associated with bone marrow transplantation. University of Nebraska Medical Center Bone Marrow Transplant Group. Am J Med 1994;96:327–34.
66. Rathi NK, Tanner AR, Dinh A, et al. Low-, medium- and high-dose steroids with or without aminocaproic acid in adult hematopoietic SCT patients with diffuse alveolar hemorrhage. Bone Marrow Transplant 2015;50:420–6.
67. Afessa B, Tefferi A, Litzow MR, Peters SG. Outcome of diffuse alveolar hemorrhage in hematopoietic stem cell transplant recipients. Am J Respir Crit Care Med 2002;166:1364–8.
68. Panoskaltsis-Mortari A, Griese M, Madtes DK, et al. An official American Thoracic Society research statement: noninfectious lung injury after hematopoietic stem cell transplantation: idiopathic pneumonia syndrome. Am J Respir Crit Care Med 2011;183:1262–79.
69. Clark JG, Hansen JA, Hertz MI, Pet al. NHLBI workshop summary. Idiopathic pneumonia syndrome after bone marrow transplantation. Am Rev Resp Dis 1993;147:1601–6.
70. Vande Vusse LK, Madtes DK. Early onset noninfectious pulmonary syndromes after hematopoietic cell transplantation. Clin Chest Med 2017;38:233–48.
71. Fukuda T, Hackman RC, Guthrie KA, et al. Risks and outcomes of idiopathic pneumonia syndrome after nonmyeloablative and conventional conditioning regimens for allogeneic hematopoietic stem cell transplantation. Blood 2003;102:2777–85.
72. Englund JA, Boeckh M, Kuypers J, et al. Brief communication: fatal human metapneumovirus infection in stem-cell transplant recipients. Ann Intern Med 2006;144:344–9.
73. Seo S, Renaud C, Kuypers JM, et al. Idiopathic pneumonia syndrome after hematopoietic cell transplantation: evidence of occult infectious etiologies. Blood 2015;125:3789–97.
74. Nakane T, Nakamae H, Kamoi H, et al. Prognostic value of serum surfactant protein D level prior to transplant for the development of bronchiolitis obliterans syndrome and idiopathic pneumonia syndrome following allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;42:43–9.
75. Gilbert CR, Lerner A, Baram M, Awsare BK. Utility of flexible bronchoscopy in the evaluation of pulmonary infiltrates in the hematopoietic stem cell transplant population—a single center fourteen year experience. Arch Bronconeumol 2013;49:189–95.
76. Yanik GA, Horowitz MM, Weisdorf DJ, et al. Randomized, double-blind, placebo-controlled trial of soluble tumor necrosis factor receptor: enbrel (etanercept) for the treatment of idiopathic pneumonia syndrome after allogeneic stem cell transplantation: blood and marrow transplant clinical trials network protocol. Biol Blood Marrow Transplant 2014;20:858–64.
77. Levine JE, Paczesny S, Mineishi S, et al. Etanercept plus methylprednisolone as initial therapy for acute graft-versus-host disease. Blood 2008;111:2470–5.
78. Yanik GA, Grupp SA, Pulsipher MA, et al. TNF-receptor inhibitor therapy for the treatment of children with idiopathic pneumonia syndrome. A joint Pediatric Blood and Marrow Transplant Consortium and Children’s Oncology Group Study (ASCT0521). Biol Blood Marrow Transplant 2015;21:67–73.
79. Thompson J, Yin Z, D’Souza A, et al. Etanercept and corticosteroid therapy for the treatment of late-onset idiopathic pneumonia syndrome. Biol Blood Marrow Transplant J 2017; 23:1955–60.
Management of Castration-Resistant Prostate Cancer
Prostate cancer is the most common malignancy in men, with an estimated 165,000 new prostate cancer diagnoses and 29,000 prostate cancer deaths occurring in the United States in 2018.1 Due to the widespread use of screening prostate-specific antigen (PSA), prostate cancer has been mainly diagnosed when the tumor is confined to the prostate. Despite definitive treatment of localized prostate cancer, some men develop systemic disease, either biochemical failure, as defined by rising PSA level, or metastatic disease.1 Several factors have been demonstrated to predict risk of relapse, including higher pretreatment PSA, higher Gleason score, and a greater anatomic extent of disease.2 In addition, the incidence of de novo metastatic prostate cancer was recently noted to be increasing. This may be due to changes in the United States Preventive Services Task Force prostate cancer screening guidelines in 2012, which recommended against screening for prostate cancer in men of any age. The updated 2018 guidelines recommend a discussion of the risks versus benefits of screening for prostate cancer for all men aged 55 to 69 years,recommend against screening for men older than 70 years, and do not have recommendations for high-risk subgroups.3
Androgen deprivation therapy (ADT) has been the cornerstone of therapy since 1941 for men with hormone-sensitive systemic disease, both in biochemically relapsed and metastatic disease.4,5 While more than 90% of patients respond to initial ADT, castration resistance is inevitable in some men.6,7 Prostate cancer will become castration-resistant typically after 18 to 24 months of ADT, with the majority of patients developing castration-resistant prostate cancer (CRPC) within 5 years of initiation of ADT.8
Pathogenesis
CRPC (previously called androgen independent prostate cancer) is defined as progression of disease despite serum total testosterone levels less than 50 ng/dL. CRPC is characterized by a rising PSA level and/or radiographic progression. One mechanism of castration resistance is genetic modification of the androgen receptor (AR), including increased expression of the wild-type AR.9 Alternatively, mutations of the steroid-binding domain may play a role in the development of castration resistance by allowing the AR to become activated by non-androgen steroid hormones or even paradoxically by antiandrogens. Studies suggest, however, that AR mutations may be seen in only 10% of prostate cancers that have developed castration resistance.10 The AR-V7 splice variant of the AR lacks an androgen binding site altogether, and may play an important role in castration resistance. In one study, the presence of this splice variant in circulating prostate cancer tumor cells predicted resistance to enzalutamide and abiraterone as well as poor outcomes.11 Intratumoral androgen synthesis also may play a role in the development of CRPC.12,13
CRPC can be broadly categorized into 2 categories, metastatic (mCRPC) and nonmetastatic (nmCRPC; Figure). The exact proportion of patients entering CRPC at a nonmetastatic stage (M0) is largely unknown.14 In one study of patients at the time of diagnosis of CRPC, ≥ 84% of patients were shown to have metastases.8 In this article, we review key aspects of management of CRPC, including selection of first- and second-line therapy, and briefly discuss upcoming clinical trials.
.")
Treatment of Nonmetastatic CRPC (M0 Disease)
Early identification of M0 CRPC is important because patients with nonmetastatic CRPC are at risk for metastasis, as demonstrated by Smith and colleagues.15 In this study that evaluated data from patients with nmCRPC in the placebo group (n = 331) of a randomized controlled trial, at 2 years 46% had developed ≥ 1 bone metastasis, 20% had died, and the median bone metastasis-free survival (MFS) was 25 months.15
Rapid PSA doubling time (PSADT) is linked to shorter time to metastasis in this group of patients. Patients with a PSADT of < 10 months had a risk for bone metastasis 12 times greater and a risk for death 4 times greater than patients with a PSADT of ≥ 10 months.16 Accordingly, observation should be reserved for those patients with a PSADT of ≥ 10 months.
Options for secondary hormonal therapy in those with a PSADT of ≤ 10 months include a first-generation antiandrogen (bicalutamide, flutamide, nilutamide), ketoconazole with hydrocortisone, and more recently second-generation antiandrogens (apalutamide or enzalutamide).
Bicalutamide competitively inhibits dihydrotestosterone and testosterone binding to the AR and is generally well-tolerated; it is given in conjunction with a GnRH agonist/castration.17 The use of other first-generation antiandrogens is limited mainly due to their toxicity profile. When compared to flutamide in a randomized, double-blinded control study, bicalutamide had significantly improved time to treatment failure.18 Due to promiscuous binding to AR, withdrawal of first-generation antiandrogen therapy has been associated with a biochemical response in a small proportion of patients, with response typically seen after 5 to 7 half-lives of the drug have elapsed.19
Although traditionally used as an antifungal agent, ketoconazole also inhibits androgen synthesis in the adrenal glands and acts as a direct cytotoxin to cancer cells.20 Ketoconazole (with hydrocortisone) has been considered as a treatment option, typically at the time of antiandrogen withdrawal. However, ketoconazole offers no survival benefit, and with the approval of abiraterone in M1 CRPC, its use has declined significantly.21 Additionally, ketoconazole poses a risk for severe hepatotoxicity and QT prolongation, and has significant interactions with numerous drugs, thereby limiting its use. Given the typically short duration of response to first-generation antiandrogens, the second-generation antiandrogens were developed and are associated with a significantly greater progression-free survival (PFS) in M0 CRPC.22,23
The second-generation antiandrogens enzalutamide and apalutamide not only competitively bind to the AR, inhibiting formation of the androgen/AR complex, but they also inhibit androgen/AR complex nuclear translocation and binding to nuclear DNA. In the PROSPER trial, enzalutamide significantly increased radiographic PFS and improved quality of life compared to placebo in chemotherapy-naive patients (Table 1).24 Apalutamide significantly increased MFS as well as PFS and time to PSA progression compared to placebo in the phase 3 SPARTAN trial.25 Apalutamide is generally well tolerated, with hypertension and rash being the most common severe adverse effects. Apalutamide also has less potential for central nervous system toxicities than enzalutamide. The recent approval of these agents is likely to change responses to subsequent treatments, especially in the metastatic setting.

Treatment of Metastatic CRPC (M1 Disease)
As with M0 CRPC, ADT should be continued in patients with mCRPC to maintain castration levels of testosterone while initiating additional treatments. Several drugs for the treatment of mCRPC have been approved by the US Food and Drug Administration (FDA) since 2010, including abiraterone with prednisone (or methylprednisolone), enzalutamide (but not apalutamide), radium-223, sipuleucel-T, and cabazitaxel (Table 2).

Given the availability of numerous treatment options for men with mCRPC, the sequencing of treatments should be based on careful consideration of the efficacy and adverse effect profiles of each drug as well as the anatomic and molecular characteristics of the cancer, comorbidities, and patient preference. If there is no evidence of visceral disease and the patient has an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0 or 1 with an estimated life expectancy of greater than 6 months and is minimally symptomatic, then treatment with either oral targeted agents or immunotherapy with sipuleucel-T is considered appropriate.
Sipuleucel-T is an autologous dendritic cell vaccination designed to enhance T-cell–mediated response to prostatic acid phosphatase (PAP). The treatments are prepared from leukapheresed host mononuclear cells that are then exposed to PAP fused to granulocyte-macrophage colony-stimulating factor. The activated dendritic cells are then infused back into the host once every 2 weeks for a total of 3 treatments. The main side effects of this treatment include chills, fever, and headache, but it is generally well-tolerated and has demonstrated a survival benefit.26
Both enzalutamide and abiraterone (abiraterone given with physiologic-dose steroid replacement) confer a survival benefit in chemotherapy-naive patients with M1 CRPC. Per the PREVAIL study, enzalutamide (when compared to placebo) offers a median improvement in overall survival (OS) by about 2 months and in radiographic PFS by about 14.6 months.24 Abiraterone blocks the synthesis of androgens via inhibition of CYP17 in the testes and adrenal glands. Abiraterone also confers an overall survival advantage for patients with M1 CRPC who are chemotherapy-naïve, with an estimated 25% decrease in the risk of death (hazard ratio, 0.75, P = 0.009) when compared to prednisone.27
In patients with symptomatic M1 CRPC who have visceral disease or rapidly progressive disease and who are candidates for chemotherapy, docetaxel is frequently used and is given concurrently with steroids. Docetaxel has been given for up to 10 cycles in clinical trials (assuming no progression of disease or dose-limiting toxicities were observed), and at least 6 cycles of treatment are recommended. When compared to mitoxantrone plus prednisone in the TAX 327 phase 3 trial, docetaxel plus prednisone offered a significant OS benefit of about 3 months (19.2 months versus 16.3 months).28 For patients who are not candidates for docetaxel (eg, due to preexisting peripheral neuropathy), cabazitaxel should be considered. OS is similar for mCRPC with docetaxel versus cabazitaxel when given in the first-line setting.29 Additionally, cabazitaxel dosed at 20 mg/m2 is noninferior to cabazitaxel dosed at 25 mg/m2, and the lower dose is associated with lower rates of peripheral neuropathy.30 Cabazitaxel should also be considered for mCRPC that has progressed following treatment with docetaxel, with improved OS and PFS when compared to treatment with mitoxantrone and prednisone in this setting, as shown in the TROPIC study.31 Mitoxantrone given with prednisone has been shown to improve quality of life, but it is associated with significant cardiac toxicity. Additionally, mitoxantrone does not improve disease-free survival or OS in chemotherapy-naive patients32 or in patients who have progressed on docetaxel, and therefore should not be given to patients prior to a taxane chemotherapy unless the patient absolutely cannot tolerate docetaxel or cabazitaxel.
Once a patient’s prostate cancer progresses following treatment with a taxane, the sequence in which to administer subsequent therapies should involve careful consideration of previous treatments and duration of response to each of these treatments. Both enzalutamide and abiraterone are FDA-approved for use following treatment with chemotherapy. Per the AFFIRM trial, heavily pre-treated patients (including those who have received docetaxel) have a median 5-month OS benefit with enzalutamide compared to placebo.33 Another study of M1 CRPC patients who had previously received docetaxel demonstrated an OS benefit with abiraterone (versus placebo),34 but this regimen has limited benefit in patients who have previously received both docetaxel and enzalutamide.35 A rechallenge with docetaxel therapy also can be considered if the patient’s disease responded to docetaxel in the metastatic hormone-sensitive setting.
If the patient’s metastases are limited to bone (ie, no visceral disease), then radiotherapy with radium-223 should be considered. Radium-223 is an alpha-emitting calcium-mimetic radioactive compound that tracks to bone to delay the onset of symptoms from bone metastases.36 Radium-223 also confers a median OS benefit of about 3 months.37 However, this treatment is often limited by preexisting cytopenias.
Diethylstilbestrol (1 mg daily) competes with androgens for AR binding and is also cytotoxic to androgen-sensitive and insensitive prostate cancer cells. While its efficacy is similar to bicalutamide in terms of PSA response rate and median response duration, diethylstilbestrol is associated with significantly more cardiovascular toxicity, including stroke, pulmonary embolism, and heart failure, and its use is therefore limited.38 The glucocorticoids—prednisone (5 mg orally twice daily), dexamethasone (0.5 mg daily), and hydrocortisone (40 mg daily)—inhibit pituitary synthesis of adrenocorticotropic hormone, resulting in decreased adrenal androgen synthesis. Data suggest that among the glucocorticoids, dexamethasone monotherapy may produce superior response rates compared to prednisone monotherapy.39 While the glucocorticoids do produce a PSA response, prolong time to disease progression, and can provide symptomatic relief (eg, from bone pain), they have not been shown to confer a survival benefit and therefore are not commonly used as monotherapy.
Future of CRPC Treatment
Patients with CRPC should be considered for clinical trials when available. These patients’ tumors should be assessed with next-generation sequencing for analysis of microsatellite instability (MSI) or mismatch repair (MMR) as well as the presence of other potentially targetable mutations, as this information may bring into consideration additional investigational as well as FDA-approved treatment options. As of May 2017, immunotherapy with pembrolizumab is approved for patients whose prostate cancer is deficient in MMR or has a high MSI burden based on a study of 12 solid tumor types (including prostate cancer) with deficient MMR.40 Additionally, for patients whose tumor has ≥ 1% programmed death ligand 1 (PD-L1) expression, pembrolizumab has a 17% overall response rate and confers stability of disease in 35%, with a median response duration of 13.5 months.41 Cabozantinib is a mesenchymal epithelial transition (MET) kinase and vascular endothelial growth factor receptor (VEGF-R) inhibitor. When used in heavily pre-treated patients with mCRPC, it showed a radiographic PFS benefit but no survival benefit over prednisone monotherapy.42 One study showed that for patients whose mCRPC had a homozygous deletion and/or a deleterious mutation in the homologous recombination repair genes BRCA1/2, ATM, and CHEK2 or the Fanconi anemia genes, the response rate to the poly ADP ribose polymerase (PARP) inhibitor olaparib was 88%, with a 100% response rate in those with BRCA2 mutations.43 Furthermore, mutations in these DNA repair genes predict increased sensitivity to platinum-based chemotherapy.
Supportive Care
Zoledronic acid or denosumab are FDA approved for men with CRPC and bone metastasis based on the ability of these agents to delay skeletal-related events, including pathologic fracture and spinal cord compression.46 Bisphosphonates, however, do not decrease the incidence of bone metastases.47 And while denosumab does delay the time to first bone metastasis in nmCRPC (particularly in patients with a PSADT of ≤ 6 months), it does not improve OS.48 Other supportive measures include exercise and nutrition. Moderate aerobic exercise for 150 minutes in addition to 2 or 3 strength training sessions per week is recommended by the American College of Sport Medicine to combat cancer-related fatigue.49 There are currently no dietary changes that are routinely recommended to improve the outcome of prostate cancer, but a study noted a shorter biochemical failure–free survival in men with prostate cancer who were obese and consumed a diet high in saturated fat.50
Conclusion
Prostate cancer affects more men in the United States than any other cancer. Once a patient is started on hormone therapy, in all likelihood their prostate cancer will become castration-resistant. Once prostate cancer has developed hormone resistance, there are a host of further treatment options available, including further hormone therapy, chemotherapy, immunotherapy, radiation therapy, bone-targeting agents, and clinical trials. Determining the appropriate sequence in which to use these therapies requires knowledge of the natural history of CRPC, the indications for changing therapies, the mechanism of action and adverse event profile of each treatment, and the optimal time to enroll in a clinical trial.
1. Pound CR, Partin AW, Epstein JI, Walsh PC. Prostate-specific antigen after anatomic radical retropubic prostatectomy. Patterns of recurrence and cancer control. Urol Clin North Am. 1997;24:395-406.
2. Caire AA, Sun L, Ode O, et al. Delayed prostate-specific antigen recurrence after radical prostatectomy: how to identify and what are their clinical outcomes? Urology. 2009;74:643-647.
3. US Preventive Services Task Force, Grossman DC, Curry SJ, Owens DK, et al. Screening for prostate cancer: US Preventive Services Task Force recommendation statement. JAMA. 2018;319:1901-1913.
4. Huggins C, Hodges CV. Studies on prostatic cancer. I: The effects of castration, of estrogen, and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941;1:293-297.
5. Huggins C, Stevens RE Jr, Hodges CV. Studies on prostatic cancer. II: The effects of castration on advanced carcinoma of the prostate gland. Arch Surg. 1941;43:209-223.
6. Pomerantz M, Kantoff P. Clinical progression to castration recurrent prostate cancer. In: Tindall DJ, James M, eds. Androgen Action in Prostate Cancer. New York: Springer; 2009:57-72.
7. Kirby M, Hirst C, Crawford ED. Characterising the castration-resistant prostate cancer population: a systematic review. Int J Clin Pract. 2011;65:1180-1192.
8. Sharifi N, Dahut WL, Steinberg SM, et al. A retrospective study of the time to clinical endpoints for advanced prostate cancer. BJU Int. 2005;96:985-989.
9. Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33-39.
10. Taplin ME, Rajeshkumar B, Halabi S, et al. Androgen receptor mutations in androgen-independent prostate cancer: Cancer and Leukemia Group B Study 9663. J Clin Oncol. 2003;21:2673-2678.
11. Antonarakis ES, Lu C, Luber B, et al. Clinical significance of androgen receptor splice variant-7 (AR-V7) mRNA detection in circulating tumor cells of men with metastatic castration-resistant prostate cancer treated with first- and second-line abiraterone and enzalutamide. J Clin Oncol. 2017;35:2149-2156.
12. Kahn B, Collazo J, Kyprianou N. Androgen receptor as a driver of therapeutic resistance in advanced prostate cancer. Int J Biol Sci. 2014;10:588-595.
13. Logothetis CJ, Gallick GE, Maity SN, et al. Molecular classification of prostate cancer progression: foundation for marker-driven treatment of prostate cancer. Cancer Discov. 2013;3:849-861
14. Tombal B. Non-metastatic CRPC and asymptomatic metastatic CRPC: which treatment for which patient? Ann Oncol. 2012;23(suppl 10):251-258
15. Smith MR, Cook R, Lee KA, et al. Disease and host characteristics as predictors of time to first bone metastasis and death in men with progressive castration-resistant non-metastatic prostate cancer. Cancer. 2011;117:2077-2085.
16. Metwalli AR, Rosner IL, Cullen J, et al. Elevated alkaline phosphatase velocity strongly predicts overall survival and the risk of bone metastases in castrate-resistant prostate cancer. Urol Oncol. 2014;32:761-768
17. Akaza H, Yamaguchi A, Matsuda T, et al. Superior anti-tumor efficacy of bicalutamide 80mg in combination with luteinizing hormone-releasing hormone (LHRH) against versus LHRH agonist monotherapy as first line treatment for advanced prostate cancer: Interim results of a randomized study in Japanese patients. J Clin Oncol. 2004;34:20-28
18. Schellhammer P, Patterson AL, Sharifi R, et al. A controlled trial of bicalutamide versus flutamide, each in combination with luteinizing hormone-releasing hormone analogue therapy, in patients with advanced prostate cancer. Urology. 1995;45(5):745-752.
19. Sartor AO, Tangen CM, Hussain MH, et al. Antiandrogen withdrawal in castrate-refractory prostate cancer: a Southwest Oncology Group Trial (SWOG 9426). Cancer. 2008;112:2393-2400.
20. Eichenberger T, Trachtenberg J, Toor P, et al. Ketoconazole: a possible direct cytotoxic effect on prostate carcinoma cells. J Urol. 1989;141:190-191.
21. Small EJ, Halabi S, Dawson NA, et al. Antiandrogen withdrawal alone or in combination with ketoconazole in androgen-independent prostate cancer patients: a phase III trial (CALGB 9583). J Clin Oncol. 2004;22:1025-1033.
22. Penson DF, Armstrong AJ, Concepcion R, et al. Enzalutamide versus bicalutamide in castration resistant prostate cancer: the STRIVE trial. J Clin Oncol. 2016;34:2098-2106.
23. Shore ND, Chowdhury S, Villers A, et al. Efficacy and safety of enzalutamide versus bicalutamide for patients with metastatic prostate cancer (TERRAIN): a randomized, double-blind, phase 2 study. Lancet Oncol. 2016;199:147-154.
24. Beer TM, Armstrong AJ, Rathkopf D, et al. Enzalutamide in men with chemotherapy-naïve castration-resistant prostate cancer: extended analysis of the phase 3 PREVAIL study. Eur Urol. 2017;71:151-154.
25. Smith MR, Saad F, Chowdhury S; SPARTAN Investigators, et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med. 2018;378:1408-1418.
26. Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411-422.
27. Ryan CJ, Smith MR, de Bono JS, et al. Randomized phase 3 trial of abiraterone acetate in men with metastatic castration-resistant prostate cancer and no prior chemotherapy. N Engl J Med. 2013;368:138-148
28. Berthold DR, Pond GR, Soban F, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: updated survival in the TAX 327 study. J Clin Oncol. 2008;26:242-245.
29. Oudard S, Fizazi K, Sengelov L, et al. Cabazitaxel versus docetaxel as first-line therapy for patients with metastatic castration-resistant prostate cancer: a randomized phase III trial—FIRSTANA. J Clin Oncol. 2017;35:3189-3197.
30. Eisenberger M, Hardy-Bessard A-C, Kim CS, et al. Phase III study comparing a reduced dose of cabazitaxel (20 mg/m2) and the currently approved dose (25 mg/m2) in postdocetaxel patients with metastatic castration-resistant prostate cancer—PROSELICA. J Clin Oncol. 2017;35:3198-3206.
31. de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376(9747):P1147-1154.
32. Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 2004;351:1502-1512.
33. Fizazi K, Scher HI, Miller K, et al. Effect of enzalutamide on time to first skeletal-related event, pain, and quality of life in men with castration-resistant prostate cancer: results from the randomised, phase 3 AFFIRM trial. Lancet Oncol. 2014;15:1147-1156.
34. de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995-2005.
35. Loriot Y, Bianchini D, Ileana E, et al. Antitumor activity of abiraterone acetate against metastatic castration-resistant prostate cancer progressing after docetaxel and enzalutamide. Ann Oncol. 2013;24:1807-1812.
36. Sartor O, Coleman R, Nilsson S, et al. Effect of radium-223 dichloride on symptomatic skeletal events in patients with castration-resistant prostate cancer and bone metastases: results from a phase 3, double blind, randomized trial. Lancet Oncol. 2014;15:738-746.
37. Parker C, Nilsson S, Heinrich D, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013; 369:213-223.
38. Turo R, Smolski M, Esler R, et al. Diethylstilboestrol for the treatment of prostate cancer: past, present and future. Scand J Urol. 2014;48:4-14.
39. Venkitaraman R, Lorente D, Murthy V. A randomized phase 2 trial of dexamethasone versus prednisolone in castration-resistant prostate cancer. Eur Urol. 2015 67:673-679.
40. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409-413.
41. Hansen AR, Massard C, Ott PA, et al. Pembrolizumab for advanced prostate adenocarcinoma: findings of the KEYNOTE-028 study. Ann Oncol. 2018;29:1807-1813.
42. Smith M, De Bono J, Sternberg C, et al. Phase III study of cabozantinib in previously treated metastatic castration-resistant prostate cancer: COMET-1. J Clin Oncol. 2016;34:3005-3013.
43. Mateo J, Carreira S, Sandhu S, et al. DNA-Repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697-1708.
44. Kentepozidis N, Soultati A, Giassas S, et al. Paclitaxel in combination with carboplatin as salvage treatment in patients with castration-resistant prostate cancer: a Hellenic oncology research group multicenter phase II study. Cancer Chemother Pharmacol. 2012;70:161-168.
45. Smith M, De Bono J, Sternberg C, et al. Phase III study of cabozantinib in previously treated metastatic castration-resistant prostate cancer: COMET-1. J Clin Oncol. 2016;34:3005-3013.
46. Saad F, Gleason DM, Murray R, et al. A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma. J Natl Cancer Inst. 2002;94:1458-1468.
47. Wirth M, Tammela T, Cicalese V, et al. Prevention of bone metastases in patients with high-risk nonmetastatic prostate cancer treated with zoledronic acid: efficacy and safety results of the Zometa European Study (ZEUS). Eur Urol. 2015;67:482-491.
48. Smith MR, Saad F, Coleman R, et al. Denosumab and bone-metastases-free survival in men with castration-resistant prostate cancer: results of a phase 3, randomized, placebo-controlled trial. Lancet. 2012;379(9810):39-46.
49. Schmitz KH, Courneya KS, Matthews C, et al. American College of Sports Medicine roundtable on exercise guidelines for cancer survivors. Med Sci Sports Exerc. 2010;42:1409-1426.
50. Strom SS, Yamamura Y, Flores-Sandoval FN, et al. Prostate cancer in Mexican-Americans: identification of risk factors. Prostate. 2008;68:563-570.
Prostate cancer is the most common malignancy in men, with an estimated 165,000 new prostate cancer diagnoses and 29,000 prostate cancer deaths occurring in the United States in 2018.1 Due to the widespread use of screening prostate-specific antigen (PSA), prostate cancer has been mainly diagnosed when the tumor is confined to the prostate. Despite definitive treatment of localized prostate cancer, some men develop systemic disease, either biochemical failure, as defined by rising PSA level, or metastatic disease.1 Several factors have been demonstrated to predict risk of relapse, including higher pretreatment PSA, higher Gleason score, and a greater anatomic extent of disease.2 In addition, the incidence of de novo metastatic prostate cancer was recently noted to be increasing. This may be due to changes in the United States Preventive Services Task Force prostate cancer screening guidelines in 2012, which recommended against screening for prostate cancer in men of any age. The updated 2018 guidelines recommend a discussion of the risks versus benefits of screening for prostate cancer for all men aged 55 to 69 years,recommend against screening for men older than 70 years, and do not have recommendations for high-risk subgroups.3
Androgen deprivation therapy (ADT) has been the cornerstone of therapy since 1941 for men with hormone-sensitive systemic disease, both in biochemically relapsed and metastatic disease.4,5 While more than 90% of patients respond to initial ADT, castration resistance is inevitable in some men.6,7 Prostate cancer will become castration-resistant typically after 18 to 24 months of ADT, with the majority of patients developing castration-resistant prostate cancer (CRPC) within 5 years of initiation of ADT.8
Pathogenesis
CRPC (previously called androgen independent prostate cancer) is defined as progression of disease despite serum total testosterone levels less than 50 ng/dL. CRPC is characterized by a rising PSA level and/or radiographic progression. One mechanism of castration resistance is genetic modification of the androgen receptor (AR), including increased expression of the wild-type AR.9 Alternatively, mutations of the steroid-binding domain may play a role in the development of castration resistance by allowing the AR to become activated by non-androgen steroid hormones or even paradoxically by antiandrogens. Studies suggest, however, that AR mutations may be seen in only 10% of prostate cancers that have developed castration resistance.10 The AR-V7 splice variant of the AR lacks an androgen binding site altogether, and may play an important role in castration resistance. In one study, the presence of this splice variant in circulating prostate cancer tumor cells predicted resistance to enzalutamide and abiraterone as well as poor outcomes.11 Intratumoral androgen synthesis also may play a role in the development of CRPC.12,13
CRPC can be broadly categorized into 2 categories, metastatic (mCRPC) and nonmetastatic (nmCRPC; Figure). The exact proportion of patients entering CRPC at a nonmetastatic stage (M0) is largely unknown.14 In one study of patients at the time of diagnosis of CRPC, ≥ 84% of patients were shown to have metastases.8 In this article, we review key aspects of management of CRPC, including selection of first- and second-line therapy, and briefly discuss upcoming clinical trials.
Treatment of Nonmetastatic CRPC (M0 Disease)
Early identification of M0 CRPC is important because patients with nonmetastatic CRPC are at risk for metastasis, as demonstrated by Smith and colleagues.15 In this study that evaluated data from patients with nmCRPC in the placebo group (n = 331) of a randomized controlled trial, at 2 years 46% had developed ≥ 1 bone metastasis, 20% had died, and the median bone metastasis-free survival (MFS) was 25 months.15
Rapid PSA doubling time (PSADT) is linked to shorter time to metastasis in this group of patients. Patients with a PSADT of < 10 months had a risk for bone metastasis 12 times greater and a risk for death 4 times greater than patients with a PSADT of ≥ 10 months.16 Accordingly, observation should be reserved for those patients with a PSADT of ≥ 10 months.
Options for secondary hormonal therapy in those with a PSADT of ≤ 10 months include a first-generation antiandrogen (bicalutamide, flutamide, nilutamide), ketoconazole with hydrocortisone, and more recently second-generation antiandrogens (apalutamide or enzalutamide).
Bicalutamide competitively inhibits dihydrotestosterone and testosterone binding to the AR and is generally well-tolerated; it is given in conjunction with a GnRH agonist/castration.17 The use of other first-generation antiandrogens is limited mainly due to their toxicity profile. When compared to flutamide in a randomized, double-blinded control study, bicalutamide had significantly improved time to treatment failure.18 Due to promiscuous binding to AR, withdrawal of first-generation antiandrogen therapy has been associated with a biochemical response in a small proportion of patients, with response typically seen after 5 to 7 half-lives of the drug have elapsed.19
Although traditionally used as an antifungal agent, ketoconazole also inhibits androgen synthesis in the adrenal glands and acts as a direct cytotoxin to cancer cells.20 Ketoconazole (with hydrocortisone) has been considered as a treatment option, typically at the time of antiandrogen withdrawal. However, ketoconazole offers no survival benefit, and with the approval of abiraterone in M1 CRPC, its use has declined significantly.21 Additionally, ketoconazole poses a risk for severe hepatotoxicity and QT prolongation, and has significant interactions with numerous drugs, thereby limiting its use. Given the typically short duration of response to first-generation antiandrogens, the second-generation antiandrogens were developed and are associated with a significantly greater progression-free survival (PFS) in M0 CRPC.22,23
The second-generation antiandrogens enzalutamide and apalutamide not only competitively bind to the AR, inhibiting formation of the androgen/AR complex, but they also inhibit androgen/AR complex nuclear translocation and binding to nuclear DNA. In the PROSPER trial, enzalutamide significantly increased radiographic PFS and improved quality of life compared to placebo in chemotherapy-naive patients (Table 1).24 Apalutamide significantly increased MFS as well as PFS and time to PSA progression compared to placebo in the phase 3 SPARTAN trial.25 Apalutamide is generally well tolerated, with hypertension and rash being the most common severe adverse effects. Apalutamide also has less potential for central nervous system toxicities than enzalutamide. The recent approval of these agents is likely to change responses to subsequent treatments, especially in the metastatic setting.
Treatment of Metastatic CRPC (M1 Disease)
As with M0 CRPC, ADT should be continued in patients with mCRPC to maintain castration levels of testosterone while initiating additional treatments. Several drugs for the treatment of mCRPC have been approved by the US Food and Drug Administration (FDA) since 2010, including abiraterone with prednisone (or methylprednisolone), enzalutamide (but not apalutamide), radium-223, sipuleucel-T, and cabazitaxel (Table 2).
Given the availability of numerous treatment options for men with mCRPC, the sequencing of treatments should be based on careful consideration of the efficacy and adverse effect profiles of each drug as well as the anatomic and molecular characteristics of the cancer, comorbidities, and patient preference. If there is no evidence of visceral disease and the patient has an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0 or 1 with an estimated life expectancy of greater than 6 months and is minimally symptomatic, then treatment with either oral targeted agents or immunotherapy with sipuleucel-T is considered appropriate.
Sipuleucel-T is an autologous dendritic cell vaccination designed to enhance T-cell–mediated response to prostatic acid phosphatase (PAP). The treatments are prepared from leukapheresed host mononuclear cells that are then exposed to PAP fused to granulocyte-macrophage colony-stimulating factor. The activated dendritic cells are then infused back into the host once every 2 weeks for a total of 3 treatments. The main side effects of this treatment include chills, fever, and headache, but it is generally well-tolerated and has demonstrated a survival benefit.26
Both enzalutamide and abiraterone (abiraterone given with physiologic-dose steroid replacement) confer a survival benefit in chemotherapy-naive patients with M1 CRPC. Per the PREVAIL study, enzalutamide (when compared to placebo) offers a median improvement in overall survival (OS) by about 2 months and in radiographic PFS by about 14.6 months.24 Abiraterone blocks the synthesis of androgens via inhibition of CYP17 in the testes and adrenal glands. Abiraterone also confers an overall survival advantage for patients with M1 CRPC who are chemotherapy-naïve, with an estimated 25% decrease in the risk of death (hazard ratio, 0.75, P = 0.009) when compared to prednisone.27
In patients with symptomatic M1 CRPC who have visceral disease or rapidly progressive disease and who are candidates for chemotherapy, docetaxel is frequently used and is given concurrently with steroids. Docetaxel has been given for up to 10 cycles in clinical trials (assuming no progression of disease or dose-limiting toxicities were observed), and at least 6 cycles of treatment are recommended. When compared to mitoxantrone plus prednisone in the TAX 327 phase 3 trial, docetaxel plus prednisone offered a significant OS benefit of about 3 months (19.2 months versus 16.3 months).28 For patients who are not candidates for docetaxel (eg, due to preexisting peripheral neuropathy), cabazitaxel should be considered. OS is similar for mCRPC with docetaxel versus cabazitaxel when given in the first-line setting.29 Additionally, cabazitaxel dosed at 20 mg/m2 is noninferior to cabazitaxel dosed at 25 mg/m2, and the lower dose is associated with lower rates of peripheral neuropathy.30 Cabazitaxel should also be considered for mCRPC that has progressed following treatment with docetaxel, with improved OS and PFS when compared to treatment with mitoxantrone and prednisone in this setting, as shown in the TROPIC study.31 Mitoxantrone given with prednisone has been shown to improve quality of life, but it is associated with significant cardiac toxicity. Additionally, mitoxantrone does not improve disease-free survival or OS in chemotherapy-naive patients32 or in patients who have progressed on docetaxel, and therefore should not be given to patients prior to a taxane chemotherapy unless the patient absolutely cannot tolerate docetaxel or cabazitaxel.
Once a patient’s prostate cancer progresses following treatment with a taxane, the sequence in which to administer subsequent therapies should involve careful consideration of previous treatments and duration of response to each of these treatments. Both enzalutamide and abiraterone are FDA-approved for use following treatment with chemotherapy. Per the AFFIRM trial, heavily pre-treated patients (including those who have received docetaxel) have a median 5-month OS benefit with enzalutamide compared to placebo.33 Another study of M1 CRPC patients who had previously received docetaxel demonstrated an OS benefit with abiraterone (versus placebo),34 but this regimen has limited benefit in patients who have previously received both docetaxel and enzalutamide.35 A rechallenge with docetaxel therapy also can be considered if the patient’s disease responded to docetaxel in the metastatic hormone-sensitive setting.
If the patient’s metastases are limited to bone (ie, no visceral disease), then radiotherapy with radium-223 should be considered. Radium-223 is an alpha-emitting calcium-mimetic radioactive compound that tracks to bone to delay the onset of symptoms from bone metastases.36 Radium-223 also confers a median OS benefit of about 3 months.37 However, this treatment is often limited by preexisting cytopenias.
Diethylstilbestrol (1 mg daily) competes with androgens for AR binding and is also cytotoxic to androgen-sensitive and insensitive prostate cancer cells. While its efficacy is similar to bicalutamide in terms of PSA response rate and median response duration, diethylstilbestrol is associated with significantly more cardiovascular toxicity, including stroke, pulmonary embolism, and heart failure, and its use is therefore limited.38 The glucocorticoids—prednisone (5 mg orally twice daily), dexamethasone (0.5 mg daily), and hydrocortisone (40 mg daily)—inhibit pituitary synthesis of adrenocorticotropic hormone, resulting in decreased adrenal androgen synthesis. Data suggest that among the glucocorticoids, dexamethasone monotherapy may produce superior response rates compared to prednisone monotherapy.39 While the glucocorticoids do produce a PSA response, prolong time to disease progression, and can provide symptomatic relief (eg, from bone pain), they have not been shown to confer a survival benefit and therefore are not commonly used as monotherapy.
Future of CRPC Treatment
Patients with CRPC should be considered for clinical trials when available. These patients’ tumors should be assessed with next-generation sequencing for analysis of microsatellite instability (MSI) or mismatch repair (MMR) as well as the presence of other potentially targetable mutations, as this information may bring into consideration additional investigational as well as FDA-approved treatment options. As of May 2017, immunotherapy with pembrolizumab is approved for patients whose prostate cancer is deficient in MMR or has a high MSI burden based on a study of 12 solid tumor types (including prostate cancer) with deficient MMR.40 Additionally, for patients whose tumor has ≥ 1% programmed death ligand 1 (PD-L1) expression, pembrolizumab has a 17% overall response rate and confers stability of disease in 35%, with a median response duration of 13.5 months.41 Cabozantinib is a mesenchymal epithelial transition (MET) kinase and vascular endothelial growth factor receptor (VEGF-R) inhibitor. When used in heavily pre-treated patients with mCRPC, it showed a radiographic PFS benefit but no survival benefit over prednisone monotherapy.42 One study showed that for patients whose mCRPC had a homozygous deletion and/or a deleterious mutation in the homologous recombination repair genes BRCA1/2, ATM, and CHEK2 or the Fanconi anemia genes, the response rate to the poly ADP ribose polymerase (PARP) inhibitor olaparib was 88%, with a 100% response rate in those with BRCA2 mutations.43 Furthermore, mutations in these DNA repair genes predict increased sensitivity to platinum-based chemotherapy.
Supportive Care
Zoledronic acid or denosumab are FDA approved for men with CRPC and bone metastasis based on the ability of these agents to delay skeletal-related events, including pathologic fracture and spinal cord compression.46 Bisphosphonates, however, do not decrease the incidence of bone metastases.47 And while denosumab does delay the time to first bone metastasis in nmCRPC (particularly in patients with a PSADT of ≤ 6 months), it does not improve OS.48 Other supportive measures include exercise and nutrition. Moderate aerobic exercise for 150 minutes in addition to 2 or 3 strength training sessions per week is recommended by the American College of Sport Medicine to combat cancer-related fatigue.49 There are currently no dietary changes that are routinely recommended to improve the outcome of prostate cancer, but a study noted a shorter biochemical failure–free survival in men with prostate cancer who were obese and consumed a diet high in saturated fat.50
Conclusion
Prostate cancer affects more men in the United States than any other cancer. Once a patient is started on hormone therapy, in all likelihood their prostate cancer will become castration-resistant. Once prostate cancer has developed hormone resistance, there are a host of further treatment options available, including further hormone therapy, chemotherapy, immunotherapy, radiation therapy, bone-targeting agents, and clinical trials. Determining the appropriate sequence in which to use these therapies requires knowledge of the natural history of CRPC, the indications for changing therapies, the mechanism of action and adverse event profile of each treatment, and the optimal time to enroll in a clinical trial.
Prostate cancer is the most common malignancy in men, with an estimated 165,000 new prostate cancer diagnoses and 29,000 prostate cancer deaths occurring in the United States in 2018.1 Due to the widespread use of screening prostate-specific antigen (PSA), prostate cancer has been mainly diagnosed when the tumor is confined to the prostate. Despite definitive treatment of localized prostate cancer, some men develop systemic disease, either biochemical failure, as defined by rising PSA level, or metastatic disease.1 Several factors have been demonstrated to predict risk of relapse, including higher pretreatment PSA, higher Gleason score, and a greater anatomic extent of disease.2 In addition, the incidence of de novo metastatic prostate cancer was recently noted to be increasing. This may be due to changes in the United States Preventive Services Task Force prostate cancer screening guidelines in 2012, which recommended against screening for prostate cancer in men of any age. The updated 2018 guidelines recommend a discussion of the risks versus benefits of screening for prostate cancer for all men aged 55 to 69 years,recommend against screening for men older than 70 years, and do not have recommendations for high-risk subgroups.3
Androgen deprivation therapy (ADT) has been the cornerstone of therapy since 1941 for men with hormone-sensitive systemic disease, both in biochemically relapsed and metastatic disease.4,5 While more than 90% of patients respond to initial ADT, castration resistance is inevitable in some men.6,7 Prostate cancer will become castration-resistant typically after 18 to 24 months of ADT, with the majority of patients developing castration-resistant prostate cancer (CRPC) within 5 years of initiation of ADT.8
Pathogenesis
CRPC (previously called androgen independent prostate cancer) is defined as progression of disease despite serum total testosterone levels less than 50 ng/dL. CRPC is characterized by a rising PSA level and/or radiographic progression. One mechanism of castration resistance is genetic modification of the androgen receptor (AR), including increased expression of the wild-type AR.9 Alternatively, mutations of the steroid-binding domain may play a role in the development of castration resistance by allowing the AR to become activated by non-androgen steroid hormones or even paradoxically by antiandrogens. Studies suggest, however, that AR mutations may be seen in only 10% of prostate cancers that have developed castration resistance.10 The AR-V7 splice variant of the AR lacks an androgen binding site altogether, and may play an important role in castration resistance. In one study, the presence of this splice variant in circulating prostate cancer tumor cells predicted resistance to enzalutamide and abiraterone as well as poor outcomes.11 Intratumoral androgen synthesis also may play a role in the development of CRPC.12,13
CRPC can be broadly categorized into 2 categories, metastatic (mCRPC) and nonmetastatic (nmCRPC; Figure). The exact proportion of patients entering CRPC at a nonmetastatic stage (M0) is largely unknown.14 In one study of patients at the time of diagnosis of CRPC, ≥ 84% of patients were shown to have metastases.8 In this article, we review key aspects of management of CRPC, including selection of first- and second-line therapy, and briefly discuss upcoming clinical trials.
Treatment of Nonmetastatic CRPC (M0 Disease)
Early identification of M0 CRPC is important because patients with nonmetastatic CRPC are at risk for metastasis, as demonstrated by Smith and colleagues.15 In this study that evaluated data from patients with nmCRPC in the placebo group (n = 331) of a randomized controlled trial, at 2 years 46% had developed ≥ 1 bone metastasis, 20% had died, and the median bone metastasis-free survival (MFS) was 25 months.15
Rapid PSA doubling time (PSADT) is linked to shorter time to metastasis in this group of patients. Patients with a PSADT of < 10 months had a risk for bone metastasis 12 times greater and a risk for death 4 times greater than patients with a PSADT of ≥ 10 months.16 Accordingly, observation should be reserved for those patients with a PSADT of ≥ 10 months.
Options for secondary hormonal therapy in those with a PSADT of ≤ 10 months include a first-generation antiandrogen (bicalutamide, flutamide, nilutamide), ketoconazole with hydrocortisone, and more recently second-generation antiandrogens (apalutamide or enzalutamide).
Bicalutamide competitively inhibits dihydrotestosterone and testosterone binding to the AR and is generally well-tolerated; it is given in conjunction with a GnRH agonist/castration.17 The use of other first-generation antiandrogens is limited mainly due to their toxicity profile. When compared to flutamide in a randomized, double-blinded control study, bicalutamide had significantly improved time to treatment failure.18 Due to promiscuous binding to AR, withdrawal of first-generation antiandrogen therapy has been associated with a biochemical response in a small proportion of patients, with response typically seen after 5 to 7 half-lives of the drug have elapsed.19
Although traditionally used as an antifungal agent, ketoconazole also inhibits androgen synthesis in the adrenal glands and acts as a direct cytotoxin to cancer cells.20 Ketoconazole (with hydrocortisone) has been considered as a treatment option, typically at the time of antiandrogen withdrawal. However, ketoconazole offers no survival benefit, and with the approval of abiraterone in M1 CRPC, its use has declined significantly.21 Additionally, ketoconazole poses a risk for severe hepatotoxicity and QT prolongation, and has significant interactions with numerous drugs, thereby limiting its use. Given the typically short duration of response to first-generation antiandrogens, the second-generation antiandrogens were developed and are associated with a significantly greater progression-free survival (PFS) in M0 CRPC.22,23
The second-generation antiandrogens enzalutamide and apalutamide not only competitively bind to the AR, inhibiting formation of the androgen/AR complex, but they also inhibit androgen/AR complex nuclear translocation and binding to nuclear DNA. In the PROSPER trial, enzalutamide significantly increased radiographic PFS and improved quality of life compared to placebo in chemotherapy-naive patients (Table 1).24 Apalutamide significantly increased MFS as well as PFS and time to PSA progression compared to placebo in the phase 3 SPARTAN trial.25 Apalutamide is generally well tolerated, with hypertension and rash being the most common severe adverse effects. Apalutamide also has less potential for central nervous system toxicities than enzalutamide. The recent approval of these agents is likely to change responses to subsequent treatments, especially in the metastatic setting.
Treatment of Metastatic CRPC (M1 Disease)
As with M0 CRPC, ADT should be continued in patients with mCRPC to maintain castration levels of testosterone while initiating additional treatments. Several drugs for the treatment of mCRPC have been approved by the US Food and Drug Administration (FDA) since 2010, including abiraterone with prednisone (or methylprednisolone), enzalutamide (but not apalutamide), radium-223, sipuleucel-T, and cabazitaxel (Table 2).
Given the availability of numerous treatment options for men with mCRPC, the sequencing of treatments should be based on careful consideration of the efficacy and adverse effect profiles of each drug as well as the anatomic and molecular characteristics of the cancer, comorbidities, and patient preference. If there is no evidence of visceral disease and the patient has an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0 or 1 with an estimated life expectancy of greater than 6 months and is minimally symptomatic, then treatment with either oral targeted agents or immunotherapy with sipuleucel-T is considered appropriate.
Sipuleucel-T is an autologous dendritic cell vaccination designed to enhance T-cell–mediated response to prostatic acid phosphatase (PAP). The treatments are prepared from leukapheresed host mononuclear cells that are then exposed to PAP fused to granulocyte-macrophage colony-stimulating factor. The activated dendritic cells are then infused back into the host once every 2 weeks for a total of 3 treatments. The main side effects of this treatment include chills, fever, and headache, but it is generally well-tolerated and has demonstrated a survival benefit.26
Both enzalutamide and abiraterone (abiraterone given with physiologic-dose steroid replacement) confer a survival benefit in chemotherapy-naive patients with M1 CRPC. Per the PREVAIL study, enzalutamide (when compared to placebo) offers a median improvement in overall survival (OS) by about 2 months and in radiographic PFS by about 14.6 months.24 Abiraterone blocks the synthesis of androgens via inhibition of CYP17 in the testes and adrenal glands. Abiraterone also confers an overall survival advantage for patients with M1 CRPC who are chemotherapy-naïve, with an estimated 25% decrease in the risk of death (hazard ratio, 0.75, P = 0.009) when compared to prednisone.27
In patients with symptomatic M1 CRPC who have visceral disease or rapidly progressive disease and who are candidates for chemotherapy, docetaxel is frequently used and is given concurrently with steroids. Docetaxel has been given for up to 10 cycles in clinical trials (assuming no progression of disease or dose-limiting toxicities were observed), and at least 6 cycles of treatment are recommended. When compared to mitoxantrone plus prednisone in the TAX 327 phase 3 trial, docetaxel plus prednisone offered a significant OS benefit of about 3 months (19.2 months versus 16.3 months).28 For patients who are not candidates for docetaxel (eg, due to preexisting peripheral neuropathy), cabazitaxel should be considered. OS is similar for mCRPC with docetaxel versus cabazitaxel when given in the first-line setting.29 Additionally, cabazitaxel dosed at 20 mg/m2 is noninferior to cabazitaxel dosed at 25 mg/m2, and the lower dose is associated with lower rates of peripheral neuropathy.30 Cabazitaxel should also be considered for mCRPC that has progressed following treatment with docetaxel, with improved OS and PFS when compared to treatment with mitoxantrone and prednisone in this setting, as shown in the TROPIC study.31 Mitoxantrone given with prednisone has been shown to improve quality of life, but it is associated with significant cardiac toxicity. Additionally, mitoxantrone does not improve disease-free survival or OS in chemotherapy-naive patients32 or in patients who have progressed on docetaxel, and therefore should not be given to patients prior to a taxane chemotherapy unless the patient absolutely cannot tolerate docetaxel or cabazitaxel.
Once a patient’s prostate cancer progresses following treatment with a taxane, the sequence in which to administer subsequent therapies should involve careful consideration of previous treatments and duration of response to each of these treatments. Both enzalutamide and abiraterone are FDA-approved for use following treatment with chemotherapy. Per the AFFIRM trial, heavily pre-treated patients (including those who have received docetaxel) have a median 5-month OS benefit with enzalutamide compared to placebo.33 Another study of M1 CRPC patients who had previously received docetaxel demonstrated an OS benefit with abiraterone (versus placebo),34 but this regimen has limited benefit in patients who have previously received both docetaxel and enzalutamide.35 A rechallenge with docetaxel therapy also can be considered if the patient’s disease responded to docetaxel in the metastatic hormone-sensitive setting.
If the patient’s metastases are limited to bone (ie, no visceral disease), then radiotherapy with radium-223 should be considered. Radium-223 is an alpha-emitting calcium-mimetic radioactive compound that tracks to bone to delay the onset of symptoms from bone metastases.36 Radium-223 also confers a median OS benefit of about 3 months.37 However, this treatment is often limited by preexisting cytopenias.
Diethylstilbestrol (1 mg daily) competes with androgens for AR binding and is also cytotoxic to androgen-sensitive and insensitive prostate cancer cells. While its efficacy is similar to bicalutamide in terms of PSA response rate and median response duration, diethylstilbestrol is associated with significantly more cardiovascular toxicity, including stroke, pulmonary embolism, and heart failure, and its use is therefore limited.38 The glucocorticoids—prednisone (5 mg orally twice daily), dexamethasone (0.5 mg daily), and hydrocortisone (40 mg daily)—inhibit pituitary synthesis of adrenocorticotropic hormone, resulting in decreased adrenal androgen synthesis. Data suggest that among the glucocorticoids, dexamethasone monotherapy may produce superior response rates compared to prednisone monotherapy.39 While the glucocorticoids do produce a PSA response, prolong time to disease progression, and can provide symptomatic relief (eg, from bone pain), they have not been shown to confer a survival benefit and therefore are not commonly used as monotherapy.
Future of CRPC Treatment
Patients with CRPC should be considered for clinical trials when available. These patients’ tumors should be assessed with next-generation sequencing for analysis of microsatellite instability (MSI) or mismatch repair (MMR) as well as the presence of other potentially targetable mutations, as this information may bring into consideration additional investigational as well as FDA-approved treatment options. As of May 2017, immunotherapy with pembrolizumab is approved for patients whose prostate cancer is deficient in MMR or has a high MSI burden based on a study of 12 solid tumor types (including prostate cancer) with deficient MMR.40 Additionally, for patients whose tumor has ≥ 1% programmed death ligand 1 (PD-L1) expression, pembrolizumab has a 17% overall response rate and confers stability of disease in 35%, with a median response duration of 13.5 months.41 Cabozantinib is a mesenchymal epithelial transition (MET) kinase and vascular endothelial growth factor receptor (VEGF-R) inhibitor. When used in heavily pre-treated patients with mCRPC, it showed a radiographic PFS benefit but no survival benefit over prednisone monotherapy.42 One study showed that for patients whose mCRPC had a homozygous deletion and/or a deleterious mutation in the homologous recombination repair genes BRCA1/2, ATM, and CHEK2 or the Fanconi anemia genes, the response rate to the poly ADP ribose polymerase (PARP) inhibitor olaparib was 88%, with a 100% response rate in those with BRCA2 mutations.43 Furthermore, mutations in these DNA repair genes predict increased sensitivity to platinum-based chemotherapy.
Supportive Care
Zoledronic acid or denosumab are FDA approved for men with CRPC and bone metastasis based on the ability of these agents to delay skeletal-related events, including pathologic fracture and spinal cord compression.46 Bisphosphonates, however, do not decrease the incidence of bone metastases.47 And while denosumab does delay the time to first bone metastasis in nmCRPC (particularly in patients with a PSADT of ≤ 6 months), it does not improve OS.48 Other supportive measures include exercise and nutrition. Moderate aerobic exercise for 150 minutes in addition to 2 or 3 strength training sessions per week is recommended by the American College of Sport Medicine to combat cancer-related fatigue.49 There are currently no dietary changes that are routinely recommended to improve the outcome of prostate cancer, but a study noted a shorter biochemical failure–free survival in men with prostate cancer who were obese and consumed a diet high in saturated fat.50
Conclusion
Prostate cancer affects more men in the United States than any other cancer. Once a patient is started on hormone therapy, in all likelihood their prostate cancer will become castration-resistant. Once prostate cancer has developed hormone resistance, there are a host of further treatment options available, including further hormone therapy, chemotherapy, immunotherapy, radiation therapy, bone-targeting agents, and clinical trials. Determining the appropriate sequence in which to use these therapies requires knowledge of the natural history of CRPC, the indications for changing therapies, the mechanism of action and adverse event profile of each treatment, and the optimal time to enroll in a clinical trial.
1. Pound CR, Partin AW, Epstein JI, Walsh PC. Prostate-specific antigen after anatomic radical retropubic prostatectomy. Patterns of recurrence and cancer control. Urol Clin North Am. 1997;24:395-406.
2. Caire AA, Sun L, Ode O, et al. Delayed prostate-specific antigen recurrence after radical prostatectomy: how to identify and what are their clinical outcomes? Urology. 2009;74:643-647.
3. US Preventive Services Task Force, Grossman DC, Curry SJ, Owens DK, et al. Screening for prostate cancer: US Preventive Services Task Force recommendation statement. JAMA. 2018;319:1901-1913.
4. Huggins C, Hodges CV. Studies on prostatic cancer. I: The effects of castration, of estrogen, and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941;1:293-297.
5. Huggins C, Stevens RE Jr, Hodges CV. Studies on prostatic cancer. II: The effects of castration on advanced carcinoma of the prostate gland. Arch Surg. 1941;43:209-223.
6. Pomerantz M, Kantoff P. Clinical progression to castration recurrent prostate cancer. In: Tindall DJ, James M, eds. Androgen Action in Prostate Cancer. New York: Springer; 2009:57-72.
7. Kirby M, Hirst C, Crawford ED. Characterising the castration-resistant prostate cancer population: a systematic review. Int J Clin Pract. 2011;65:1180-1192.
8. Sharifi N, Dahut WL, Steinberg SM, et al. A retrospective study of the time to clinical endpoints for advanced prostate cancer. BJU Int. 2005;96:985-989.
9. Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33-39.
10. Taplin ME, Rajeshkumar B, Halabi S, et al. Androgen receptor mutations in androgen-independent prostate cancer: Cancer and Leukemia Group B Study 9663. J Clin Oncol. 2003;21:2673-2678.
11. Antonarakis ES, Lu C, Luber B, et al. Clinical significance of androgen receptor splice variant-7 (AR-V7) mRNA detection in circulating tumor cells of men with metastatic castration-resistant prostate cancer treated with first- and second-line abiraterone and enzalutamide. J Clin Oncol. 2017;35:2149-2156.
12. Kahn B, Collazo J, Kyprianou N. Androgen receptor as a driver of therapeutic resistance in advanced prostate cancer. Int J Biol Sci. 2014;10:588-595.
13. Logothetis CJ, Gallick GE, Maity SN, et al. Molecular classification of prostate cancer progression: foundation for marker-driven treatment of prostate cancer. Cancer Discov. 2013;3:849-861
14. Tombal B. Non-metastatic CRPC and asymptomatic metastatic CRPC: which treatment for which patient? Ann Oncol. 2012;23(suppl 10):251-258
15. Smith MR, Cook R, Lee KA, et al. Disease and host characteristics as predictors of time to first bone metastasis and death in men with progressive castration-resistant non-metastatic prostate cancer. Cancer. 2011;117:2077-2085.
16. Metwalli AR, Rosner IL, Cullen J, et al. Elevated alkaline phosphatase velocity strongly predicts overall survival and the risk of bone metastases in castrate-resistant prostate cancer. Urol Oncol. 2014;32:761-768
17. Akaza H, Yamaguchi A, Matsuda T, et al. Superior anti-tumor efficacy of bicalutamide 80mg in combination with luteinizing hormone-releasing hormone (LHRH) against versus LHRH agonist monotherapy as first line treatment for advanced prostate cancer: Interim results of a randomized study in Japanese patients. J Clin Oncol. 2004;34:20-28
18. Schellhammer P, Patterson AL, Sharifi R, et al. A controlled trial of bicalutamide versus flutamide, each in combination with luteinizing hormone-releasing hormone analogue therapy, in patients with advanced prostate cancer. Urology. 1995;45(5):745-752.
19. Sartor AO, Tangen CM, Hussain MH, et al. Antiandrogen withdrawal in castrate-refractory prostate cancer: a Southwest Oncology Group Trial (SWOG 9426). Cancer. 2008;112:2393-2400.
20. Eichenberger T, Trachtenberg J, Toor P, et al. Ketoconazole: a possible direct cytotoxic effect on prostate carcinoma cells. J Urol. 1989;141:190-191.
21. Small EJ, Halabi S, Dawson NA, et al. Antiandrogen withdrawal alone or in combination with ketoconazole in androgen-independent prostate cancer patients: a phase III trial (CALGB 9583). J Clin Oncol. 2004;22:1025-1033.
22. Penson DF, Armstrong AJ, Concepcion R, et al. Enzalutamide versus bicalutamide in castration resistant prostate cancer: the STRIVE trial. J Clin Oncol. 2016;34:2098-2106.
23. Shore ND, Chowdhury S, Villers A, et al. Efficacy and safety of enzalutamide versus bicalutamide for patients with metastatic prostate cancer (TERRAIN): a randomized, double-blind, phase 2 study. Lancet Oncol. 2016;199:147-154.
24. Beer TM, Armstrong AJ, Rathkopf D, et al. Enzalutamide in men with chemotherapy-naïve castration-resistant prostate cancer: extended analysis of the phase 3 PREVAIL study. Eur Urol. 2017;71:151-154.
25. Smith MR, Saad F, Chowdhury S; SPARTAN Investigators, et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med. 2018;378:1408-1418.
26. Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411-422.
27. Ryan CJ, Smith MR, de Bono JS, et al. Randomized phase 3 trial of abiraterone acetate in men with metastatic castration-resistant prostate cancer and no prior chemotherapy. N Engl J Med. 2013;368:138-148
28. Berthold DR, Pond GR, Soban F, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: updated survival in the TAX 327 study. J Clin Oncol. 2008;26:242-245.
29. Oudard S, Fizazi K, Sengelov L, et al. Cabazitaxel versus docetaxel as first-line therapy for patients with metastatic castration-resistant prostate cancer: a randomized phase III trial—FIRSTANA. J Clin Oncol. 2017;35:3189-3197.
30. Eisenberger M, Hardy-Bessard A-C, Kim CS, et al. Phase III study comparing a reduced dose of cabazitaxel (20 mg/m2) and the currently approved dose (25 mg/m2) in postdocetaxel patients with metastatic castration-resistant prostate cancer—PROSELICA. J Clin Oncol. 2017;35:3198-3206.
31. de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376(9747):P1147-1154.
32. Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 2004;351:1502-1512.
33. Fizazi K, Scher HI, Miller K, et al. Effect of enzalutamide on time to first skeletal-related event, pain, and quality of life in men with castration-resistant prostate cancer: results from the randomised, phase 3 AFFIRM trial. Lancet Oncol. 2014;15:1147-1156.
34. de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995-2005.
35. Loriot Y, Bianchini D, Ileana E, et al. Antitumor activity of abiraterone acetate against metastatic castration-resistant prostate cancer progressing after docetaxel and enzalutamide. Ann Oncol. 2013;24:1807-1812.
36. Sartor O, Coleman R, Nilsson S, et al. Effect of radium-223 dichloride on symptomatic skeletal events in patients with castration-resistant prostate cancer and bone metastases: results from a phase 3, double blind, randomized trial. Lancet Oncol. 2014;15:738-746.
37. Parker C, Nilsson S, Heinrich D, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013; 369:213-223.
38. Turo R, Smolski M, Esler R, et al. Diethylstilboestrol for the treatment of prostate cancer: past, present and future. Scand J Urol. 2014;48:4-14.
39. Venkitaraman R, Lorente D, Murthy V. A randomized phase 2 trial of dexamethasone versus prednisolone in castration-resistant prostate cancer. Eur Urol. 2015 67:673-679.
40. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409-413.
41. Hansen AR, Massard C, Ott PA, et al. Pembrolizumab for advanced prostate adenocarcinoma: findings of the KEYNOTE-028 study. Ann Oncol. 2018;29:1807-1813.
42. Smith M, De Bono J, Sternberg C, et al. Phase III study of cabozantinib in previously treated metastatic castration-resistant prostate cancer: COMET-1. J Clin Oncol. 2016;34:3005-3013.
43. Mateo J, Carreira S, Sandhu S, et al. DNA-Repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697-1708.
44. Kentepozidis N, Soultati A, Giassas S, et al. Paclitaxel in combination with carboplatin as salvage treatment in patients with castration-resistant prostate cancer: a Hellenic oncology research group multicenter phase II study. Cancer Chemother Pharmacol. 2012;70:161-168.
45. Smith M, De Bono J, Sternberg C, et al. Phase III study of cabozantinib in previously treated metastatic castration-resistant prostate cancer: COMET-1. J Clin Oncol. 2016;34:3005-3013.
46. Saad F, Gleason DM, Murray R, et al. A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma. J Natl Cancer Inst. 2002;94:1458-1468.
47. Wirth M, Tammela T, Cicalese V, et al. Prevention of bone metastases in patients with high-risk nonmetastatic prostate cancer treated with zoledronic acid: efficacy and safety results of the Zometa European Study (ZEUS). Eur Urol. 2015;67:482-491.
48. Smith MR, Saad F, Coleman R, et al. Denosumab and bone-metastases-free survival in men with castration-resistant prostate cancer: results of a phase 3, randomized, placebo-controlled trial. Lancet. 2012;379(9810):39-46.
49. Schmitz KH, Courneya KS, Matthews C, et al. American College of Sports Medicine roundtable on exercise guidelines for cancer survivors. Med Sci Sports Exerc. 2010;42:1409-1426.
50. Strom SS, Yamamura Y, Flores-Sandoval FN, et al. Prostate cancer in Mexican-Americans: identification of risk factors. Prostate. 2008;68:563-570.
1. Pound CR, Partin AW, Epstein JI, Walsh PC. Prostate-specific antigen after anatomic radical retropubic prostatectomy. Patterns of recurrence and cancer control. Urol Clin North Am. 1997;24:395-406.
2. Caire AA, Sun L, Ode O, et al. Delayed prostate-specific antigen recurrence after radical prostatectomy: how to identify and what are their clinical outcomes? Urology. 2009;74:643-647.
3. US Preventive Services Task Force, Grossman DC, Curry SJ, Owens DK, et al. Screening for prostate cancer: US Preventive Services Task Force recommendation statement. JAMA. 2018;319:1901-1913.
4. Huggins C, Hodges CV. Studies on prostatic cancer. I: The effects of castration, of estrogen, and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941;1:293-297.
5. Huggins C, Stevens RE Jr, Hodges CV. Studies on prostatic cancer. II: The effects of castration on advanced carcinoma of the prostate gland. Arch Surg. 1941;43:209-223.
6. Pomerantz M, Kantoff P. Clinical progression to castration recurrent prostate cancer. In: Tindall DJ, James M, eds. Androgen Action in Prostate Cancer. New York: Springer; 2009:57-72.
7. Kirby M, Hirst C, Crawford ED. Characterising the castration-resistant prostate cancer population: a systematic review. Int J Clin Pract. 2011;65:1180-1192.
8. Sharifi N, Dahut WL, Steinberg SM, et al. A retrospective study of the time to clinical endpoints for advanced prostate cancer. BJU Int. 2005;96:985-989.
9. Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33-39.
10. Taplin ME, Rajeshkumar B, Halabi S, et al. Androgen receptor mutations in androgen-independent prostate cancer: Cancer and Leukemia Group B Study 9663. J Clin Oncol. 2003;21:2673-2678.
11. Antonarakis ES, Lu C, Luber B, et al. Clinical significance of androgen receptor splice variant-7 (AR-V7) mRNA detection in circulating tumor cells of men with metastatic castration-resistant prostate cancer treated with first- and second-line abiraterone and enzalutamide. J Clin Oncol. 2017;35:2149-2156.
12. Kahn B, Collazo J, Kyprianou N. Androgen receptor as a driver of therapeutic resistance in advanced prostate cancer. Int J Biol Sci. 2014;10:588-595.
13. Logothetis CJ, Gallick GE, Maity SN, et al. Molecular classification of prostate cancer progression: foundation for marker-driven treatment of prostate cancer. Cancer Discov. 2013;3:849-861
14. Tombal B. Non-metastatic CRPC and asymptomatic metastatic CRPC: which treatment for which patient? Ann Oncol. 2012;23(suppl 10):251-258
15. Smith MR, Cook R, Lee KA, et al. Disease and host characteristics as predictors of time to first bone metastasis and death in men with progressive castration-resistant non-metastatic prostate cancer. Cancer. 2011;117:2077-2085.
16. Metwalli AR, Rosner IL, Cullen J, et al. Elevated alkaline phosphatase velocity strongly predicts overall survival and the risk of bone metastases in castrate-resistant prostate cancer. Urol Oncol. 2014;32:761-768
17. Akaza H, Yamaguchi A, Matsuda T, et al. Superior anti-tumor efficacy of bicalutamide 80mg in combination with luteinizing hormone-releasing hormone (LHRH) against versus LHRH agonist monotherapy as first line treatment for advanced prostate cancer: Interim results of a randomized study in Japanese patients. J Clin Oncol. 2004;34:20-28
18. Schellhammer P, Patterson AL, Sharifi R, et al. A controlled trial of bicalutamide versus flutamide, each in combination with luteinizing hormone-releasing hormone analogue therapy, in patients with advanced prostate cancer. Urology. 1995;45(5):745-752.
19. Sartor AO, Tangen CM, Hussain MH, et al. Antiandrogen withdrawal in castrate-refractory prostate cancer: a Southwest Oncology Group Trial (SWOG 9426). Cancer. 2008;112:2393-2400.
20. Eichenberger T, Trachtenberg J, Toor P, et al. Ketoconazole: a possible direct cytotoxic effect on prostate carcinoma cells. J Urol. 1989;141:190-191.
21. Small EJ, Halabi S, Dawson NA, et al. Antiandrogen withdrawal alone or in combination with ketoconazole in androgen-independent prostate cancer patients: a phase III trial (CALGB 9583). J Clin Oncol. 2004;22:1025-1033.
22. Penson DF, Armstrong AJ, Concepcion R, et al. Enzalutamide versus bicalutamide in castration resistant prostate cancer: the STRIVE trial. J Clin Oncol. 2016;34:2098-2106.
23. Shore ND, Chowdhury S, Villers A, et al. Efficacy and safety of enzalutamide versus bicalutamide for patients with metastatic prostate cancer (TERRAIN): a randomized, double-blind, phase 2 study. Lancet Oncol. 2016;199:147-154.
24. Beer TM, Armstrong AJ, Rathkopf D, et al. Enzalutamide in men with chemotherapy-naïve castration-resistant prostate cancer: extended analysis of the phase 3 PREVAIL study. Eur Urol. 2017;71:151-154.
25. Smith MR, Saad F, Chowdhury S; SPARTAN Investigators, et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med. 2018;378:1408-1418.
26. Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411-422.
27. Ryan CJ, Smith MR, de Bono JS, et al. Randomized phase 3 trial of abiraterone acetate in men with metastatic castration-resistant prostate cancer and no prior chemotherapy. N Engl J Med. 2013;368:138-148
28. Berthold DR, Pond GR, Soban F, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: updated survival in the TAX 327 study. J Clin Oncol. 2008;26:242-245.
29. Oudard S, Fizazi K, Sengelov L, et al. Cabazitaxel versus docetaxel as first-line therapy for patients with metastatic castration-resistant prostate cancer: a randomized phase III trial—FIRSTANA. J Clin Oncol. 2017;35:3189-3197.
30. Eisenberger M, Hardy-Bessard A-C, Kim CS, et al. Phase III study comparing a reduced dose of cabazitaxel (20 mg/m2) and the currently approved dose (25 mg/m2) in postdocetaxel patients with metastatic castration-resistant prostate cancer—PROSELICA. J Clin Oncol. 2017;35:3198-3206.
31. de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376(9747):P1147-1154.
32. Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 2004;351:1502-1512.
33. Fizazi K, Scher HI, Miller K, et al. Effect of enzalutamide on time to first skeletal-related event, pain, and quality of life in men with castration-resistant prostate cancer: results from the randomised, phase 3 AFFIRM trial. Lancet Oncol. 2014;15:1147-1156.
34. de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995-2005.
35. Loriot Y, Bianchini D, Ileana E, et al. Antitumor activity of abiraterone acetate against metastatic castration-resistant prostate cancer progressing after docetaxel and enzalutamide. Ann Oncol. 2013;24:1807-1812.
36. Sartor O, Coleman R, Nilsson S, et al. Effect of radium-223 dichloride on symptomatic skeletal events in patients with castration-resistant prostate cancer and bone metastases: results from a phase 3, double blind, randomized trial. Lancet Oncol. 2014;15:738-746.
37. Parker C, Nilsson S, Heinrich D, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013; 369:213-223.
38. Turo R, Smolski M, Esler R, et al. Diethylstilboestrol for the treatment of prostate cancer: past, present and future. Scand J Urol. 2014;48:4-14.
39. Venkitaraman R, Lorente D, Murthy V. A randomized phase 2 trial of dexamethasone versus prednisolone in castration-resistant prostate cancer. Eur Urol. 2015 67:673-679.
40. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409-413.
41. Hansen AR, Massard C, Ott PA, et al. Pembrolizumab for advanced prostate adenocarcinoma: findings of the KEYNOTE-028 study. Ann Oncol. 2018;29:1807-1813.
42. Smith M, De Bono J, Sternberg C, et al. Phase III study of cabozantinib in previously treated metastatic castration-resistant prostate cancer: COMET-1. J Clin Oncol. 2016;34:3005-3013.
43. Mateo J, Carreira S, Sandhu S, et al. DNA-Repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697-1708.
44. Kentepozidis N, Soultati A, Giassas S, et al. Paclitaxel in combination with carboplatin as salvage treatment in patients with castration-resistant prostate cancer: a Hellenic oncology research group multicenter phase II study. Cancer Chemother Pharmacol. 2012;70:161-168.
45. Smith M, De Bono J, Sternberg C, et al. Phase III study of cabozantinib in previously treated metastatic castration-resistant prostate cancer: COMET-1. J Clin Oncol. 2016;34:3005-3013.
46. Saad F, Gleason DM, Murray R, et al. A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma. J Natl Cancer Inst. 2002;94:1458-1468.
47. Wirth M, Tammela T, Cicalese V, et al. Prevention of bone metastases in patients with high-risk nonmetastatic prostate cancer treated with zoledronic acid: efficacy and safety results of the Zometa European Study (ZEUS). Eur Urol. 2015;67:482-491.
48. Smith MR, Saad F, Coleman R, et al. Denosumab and bone-metastases-free survival in men with castration-resistant prostate cancer: results of a phase 3, randomized, placebo-controlled trial. Lancet. 2012;379(9810):39-46.
49. Schmitz KH, Courneya KS, Matthews C, et al. American College of Sports Medicine roundtable on exercise guidelines for cancer survivors. Med Sci Sports Exerc. 2010;42:1409-1426.
50. Strom SS, Yamamura Y, Flores-Sandoval FN, et al. Prostate cancer in Mexican-Americans: identification of risk factors. Prostate. 2008;68:563-570.