User login
Bringing you the latest case reports, original research, clinical trial reviews, perspectives, patient resources, and more.

Sorafenib extends PFS for refractory desmoid tumors
For patients with progressive, refractory, or symptomatic desmoid tumors – also known as aggressive fibromatosis – treatment with daily sorafenib (Nexavar) was associated with durable responses and a significant improvement in progression-free survival.
After a median follow-up of 27.2 months, the 2-year progression-free survival (PFS) rate for patients randomly assigned to receive 400 mg sorafenib daily was 81%, compared with 36% for patients assigned to placebo (P less than .001), reported Mrinal M. Gounder, MD, from Memorial Sloan Kettering Cancer Center in New York City, and his colleagues.
“Other agents that are used to treat these tumors include anthracyclines [e.g., pegylated liposomal doxorubicin], vinca alkaloids, and pazopanib. On the basis of the predictable toxic-effects profile and substantial progression-free survival advantage conferred by sorafenib, the drug has antitumor activity as first-line therapy or as subsequent therapy for desmoid tumors,” they wrote in the New England Journal of Medicine.
There is no accepted standard of care for the systemic treatment for desmoid tumors, with options ranging from hormonal blockade, cytotoxic chemotherapy, and targeted agents such as tyrosine kinase inhibitors (TKIs).
Based on a retrospective study showing that the multitargeting oral TKI sorafenib was associated with a 25% response rate and acceptable safety in patients with desmoid tumors, the investigators initiated a phase 3, randomized trial to evaluate the efficacy and safety of sorafenib in this population.
They enrolled 87 patients aged 18 years or older with a histologically documented desmoid tumor that showed clinical and radiographic progression of at least 10% in maximum unidimensional measurement within the last 6 months, symptomatic disease, or recurrent or primary disease that was either inoperable or deemed to require extensive surgery.
The patients were randomized in double-blinded fashion on a 2:1 basis to receive either sorafenib 400 mg daily or placebo until progression. Crossover to sorafenib was allowed for patients assigned to placebo who experienced disease progressions.
As noted before, investigator-assessed PFS, the primary endpoint, clearly favored sorafenib.
Objective response rates before crossover were 33% in the sorafenib arm, consisting of 1 complete and 15 partial responses, and 20% in the placebo arm, consisting of 7 partial responses. The respective median times to objective response were 9.6 months versus 13.3 months. The earliest response, defined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, occurred at 2.2 months in the sorafenib arm versus 8.8 months in the placebo arm.
The authors also performed an exploratory analysis looking at MRI as a measure of response evaluation and found that “changes in T2-weighted signal intensity and volumetric measurements may be better measures of treatment effect than RECIST. This is particularly evident when the best response according to RECIST is stable disease.”
The most frequently reported adverse events among patients treated with sorafenib were grade 1 or 2 rash in 73%, fatigue in 67%, hypertension in 55%, and diarrhea in 51%. The most frequent treatment-emergent adverse events in the placebo group were rash of any kind in 42% and palmar-plantar erythrodysesthesia syndrome in 22%.
The investigators acknowledged that the mechanism of action of sorafenib in desmoid tumors is unknown, but noted that they are looking for clues in 25 sets of paired biopsy samples.
The study was supported by grants from the National Cancer Institute, Bayer, Memorial Sloan Kettering Cancer Center, the American Society of Clinical Oncology, Desmoid Tumor Research Foundation, and an Orphan Products Clinical Trials Grant from the Food and Drug Administration. Dr. Gounder reported fees for advisory board activities/consulting for Bayer, Epizyme, Karyopharm Therapeutics, Daiichi Sankyo, TRACON Pharmaceuticals, and Amgen, and travel expenses from Epizyme.
SOURCE: Gounder MM et al. N Engl J Med. 2018 Dec 19. doi: 10.1056/NEJMoa1805052.
For patients with progressive, refractory, or symptomatic desmoid tumors – also known as aggressive fibromatosis – treatment with daily sorafenib (Nexavar) was associated with durable responses and a significant improvement in progression-free survival.
After a median follow-up of 27.2 months, the 2-year progression-free survival (PFS) rate for patients randomly assigned to receive 400 mg sorafenib daily was 81%, compared with 36% for patients assigned to placebo (P less than .001), reported Mrinal M. Gounder, MD, from Memorial Sloan Kettering Cancer Center in New York City, and his colleagues.
“Other agents that are used to treat these tumors include anthracyclines [e.g., pegylated liposomal doxorubicin], vinca alkaloids, and pazopanib. On the basis of the predictable toxic-effects profile and substantial progression-free survival advantage conferred by sorafenib, the drug has antitumor activity as first-line therapy or as subsequent therapy for desmoid tumors,” they wrote in the New England Journal of Medicine.
There is no accepted standard of care for the systemic treatment for desmoid tumors, with options ranging from hormonal blockade, cytotoxic chemotherapy, and targeted agents such as tyrosine kinase inhibitors (TKIs).
Based on a retrospective study showing that the multitargeting oral TKI sorafenib was associated with a 25% response rate and acceptable safety in patients with desmoid tumors, the investigators initiated a phase 3, randomized trial to evaluate the efficacy and safety of sorafenib in this population.
They enrolled 87 patients aged 18 years or older with a histologically documented desmoid tumor that showed clinical and radiographic progression of at least 10% in maximum unidimensional measurement within the last 6 months, symptomatic disease, or recurrent or primary disease that was either inoperable or deemed to require extensive surgery.
The patients were randomized in double-blinded fashion on a 2:1 basis to receive either sorafenib 400 mg daily or placebo until progression. Crossover to sorafenib was allowed for patients assigned to placebo who experienced disease progressions.
As noted before, investigator-assessed PFS, the primary endpoint, clearly favored sorafenib.
Objective response rates before crossover were 33% in the sorafenib arm, consisting of 1 complete and 15 partial responses, and 20% in the placebo arm, consisting of 7 partial responses. The respective median times to objective response were 9.6 months versus 13.3 months. The earliest response, defined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, occurred at 2.2 months in the sorafenib arm versus 8.8 months in the placebo arm.
The authors also performed an exploratory analysis looking at MRI as a measure of response evaluation and found that “changes in T2-weighted signal intensity and volumetric measurements may be better measures of treatment effect than RECIST. This is particularly evident when the best response according to RECIST is stable disease.”
The most frequently reported adverse events among patients treated with sorafenib were grade 1 or 2 rash in 73%, fatigue in 67%, hypertension in 55%, and diarrhea in 51%. The most frequent treatment-emergent adverse events in the placebo group were rash of any kind in 42% and palmar-plantar erythrodysesthesia syndrome in 22%.
The investigators acknowledged that the mechanism of action of sorafenib in desmoid tumors is unknown, but noted that they are looking for clues in 25 sets of paired biopsy samples.
The study was supported by grants from the National Cancer Institute, Bayer, Memorial Sloan Kettering Cancer Center, the American Society of Clinical Oncology, Desmoid Tumor Research Foundation, and an Orphan Products Clinical Trials Grant from the Food and Drug Administration. Dr. Gounder reported fees for advisory board activities/consulting for Bayer, Epizyme, Karyopharm Therapeutics, Daiichi Sankyo, TRACON Pharmaceuticals, and Amgen, and travel expenses from Epizyme.
SOURCE: Gounder MM et al. N Engl J Med. 2018 Dec 19. doi: 10.1056/NEJMoa1805052.
For patients with progressive, refractory, or symptomatic desmoid tumors – also known as aggressive fibromatosis – treatment with daily sorafenib (Nexavar) was associated with durable responses and a significant improvement in progression-free survival.
After a median follow-up of 27.2 months, the 2-year progression-free survival (PFS) rate for patients randomly assigned to receive 400 mg sorafenib daily was 81%, compared with 36% for patients assigned to placebo (P less than .001), reported Mrinal M. Gounder, MD, from Memorial Sloan Kettering Cancer Center in New York City, and his colleagues.
“Other agents that are used to treat these tumors include anthracyclines [e.g., pegylated liposomal doxorubicin], vinca alkaloids, and pazopanib. On the basis of the predictable toxic-effects profile and substantial progression-free survival advantage conferred by sorafenib, the drug has antitumor activity as first-line therapy or as subsequent therapy for desmoid tumors,” they wrote in the New England Journal of Medicine.
There is no accepted standard of care for the systemic treatment for desmoid tumors, with options ranging from hormonal blockade, cytotoxic chemotherapy, and targeted agents such as tyrosine kinase inhibitors (TKIs).
Based on a retrospective study showing that the multitargeting oral TKI sorafenib was associated with a 25% response rate and acceptable safety in patients with desmoid tumors, the investigators initiated a phase 3, randomized trial to evaluate the efficacy and safety of sorafenib in this population.
They enrolled 87 patients aged 18 years or older with a histologically documented desmoid tumor that showed clinical and radiographic progression of at least 10% in maximum unidimensional measurement within the last 6 months, symptomatic disease, or recurrent or primary disease that was either inoperable or deemed to require extensive surgery.
The patients were randomized in double-blinded fashion on a 2:1 basis to receive either sorafenib 400 mg daily or placebo until progression. Crossover to sorafenib was allowed for patients assigned to placebo who experienced disease progressions.
As noted before, investigator-assessed PFS, the primary endpoint, clearly favored sorafenib.
Objective response rates before crossover were 33% in the sorafenib arm, consisting of 1 complete and 15 partial responses, and 20% in the placebo arm, consisting of 7 partial responses. The respective median times to objective response were 9.6 months versus 13.3 months. The earliest response, defined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, occurred at 2.2 months in the sorafenib arm versus 8.8 months in the placebo arm.
The authors also performed an exploratory analysis looking at MRI as a measure of response evaluation and found that “changes in T2-weighted signal intensity and volumetric measurements may be better measures of treatment effect than RECIST. This is particularly evident when the best response according to RECIST is stable disease.”
The most frequently reported adverse events among patients treated with sorafenib were grade 1 or 2 rash in 73%, fatigue in 67%, hypertension in 55%, and diarrhea in 51%. The most frequent treatment-emergent adverse events in the placebo group were rash of any kind in 42% and palmar-plantar erythrodysesthesia syndrome in 22%.
The investigators acknowledged that the mechanism of action of sorafenib in desmoid tumors is unknown, but noted that they are looking for clues in 25 sets of paired biopsy samples.
The study was supported by grants from the National Cancer Institute, Bayer, Memorial Sloan Kettering Cancer Center, the American Society of Clinical Oncology, Desmoid Tumor Research Foundation, and an Orphan Products Clinical Trials Grant from the Food and Drug Administration. Dr. Gounder reported fees for advisory board activities/consulting for Bayer, Epizyme, Karyopharm Therapeutics, Daiichi Sankyo, TRACON Pharmaceuticals, and Amgen, and travel expenses from Epizyme.
SOURCE: Gounder MM et al. N Engl J Med. 2018 Dec 19. doi: 10.1056/NEJMoa1805052.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: There is no accepted standard of systemic therapy for recurrent, refractory, or symptomatic desmoid tumors.
Major finding: Median progression-free survival with sorafenib after a median follow-up of 27.2 months was 81% versus 36% for placebo.
Study details: A double-blind, phase 3 trial with 2:1 randomization of sorafenib to placebo in 87 patients.
Disclosures: The study was supported by grants from the National Cancer Institute, Bayer, Memorial Sloan Kettering Cancer Center, the American Society of Clinical Oncology, Desmoid Tumor Research Foundation, and an Orphan Products Clinical Trials Grant from the Food and Drug Administration. Dr. Gounder reported fees for advisory board activities/consulting for Bayer, Epizyme, Karyopharm Therapeutics, Daiichi Sankyo, TRACON Pharmaceuticals, and Amgen, and travel expenses from Epizyme.
Source: Gounder MM et al. N Engl J Med. 2018 Dec 19. doi: 10.1056/NEJMoa1805052.
Cardiac failure due to left atrial angiosarcoma
Abstract
Primary heart sarcomas are rare and represent 20% of all primary cardiac tumors. Symptoms depend on which chambers and cardiac structures are involved. Angiosarcoma is one of the most common and the most aggressive types of primary heart sarcomas. Typically, these tumors are found in the right atrium, however, cardiac angiosarcomas may involve any part of the heart. Most of these tumors are diagnosed in advanced stages and the patient prognosis is poor. Most tumors are diagnosed using echocardiography. Computed tomography (CT) and magnetic resonance imaging (MRI) provide useful information on tumor size and location for planning surgery, which is the only treatment shown to increase survival. We present the case of a 69-year-old woman who presented to the emergency department with hypotension, dyspnea and progressive shortness of breath. After adequate resuscitation, a cardiac mass was identified and surgery was successfully performed. Pathology confirmed a grade 2 primary heart angiosarcoma. Following surgery, the patient was admitted to the intensive care unit and later died secondary to multi-organ system failure.
Introduction
Primary heart angiosarcoma is an aggressive and usually fatal cardiac neoplasm (1). Angiosarcomas can originate at any location in the heart (2, 3), but these tumors typically reside in the right atrium and frequently cause nonspecific symptoms such as dyspnea, cough, heart failure, and arrhythmias. (2) Surgery followed by chemotherapy is the typical approach to these tumors. (4)
We present the case of a 69-year-old woman who presented to the emergency department with hypotension and severe dyspnea.
Case Report
The patient was a 69-year-old woman with a medical history of diabetes. A week before seeking care in the emergency department, she experienced a general feeling of unwellness, dyspnea, and mild respiratory distress. She reported these symptoms had become more and more severe in the last 24 hours and were accompanied by acute chest pain and progressive shortness of breath.
On clinical examination, the patient was hypotensive, had tachypnea and tachycardia, and was hypoxic. Cardiac auscultation detected a systolic murmur in the apex, and auscultation of the lungs revealed crackles and rales, especially at the bases of the lungs. The remainder of her clinical examination was unremarkable. She had sinus tachycardia on an electrocardiogram. A chest X-ray showed a left atrial enlargement along with some patchy opacities in the middle and lower zones of the lungs, along with Kerley B lines suggestive of pulmonary edema.
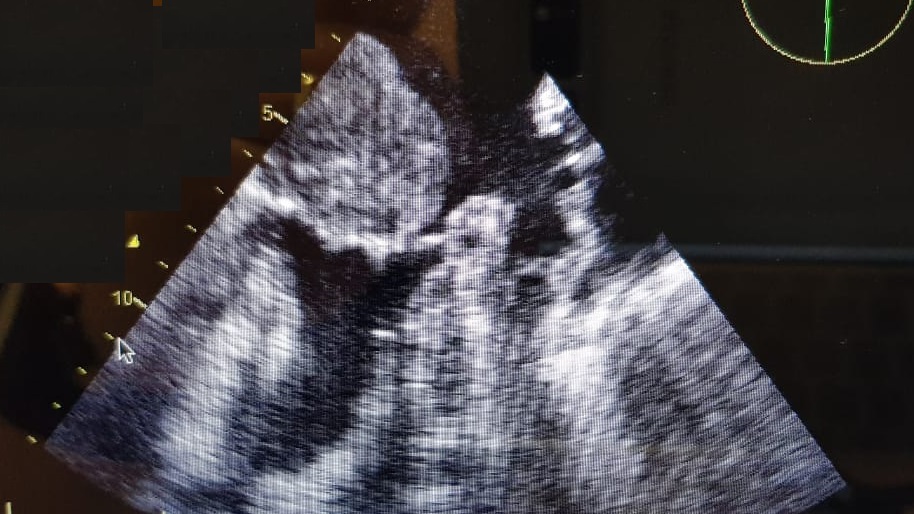
With these findings, and after adequate resuscitation, a contrast-enhanced computed tomography (CT) scan detected a filling defect in the left atrium suggestive of a large intra-cardiac mass with a thick and hyper-enhanced interatrial septum. Bilateral pleural effusions also were evident, (Figure 1A) hence an echocardiogram was requested and it confirmed the presence of a 30 x 29 x 40 mm lobulated highly mobile mass in the left atrium. 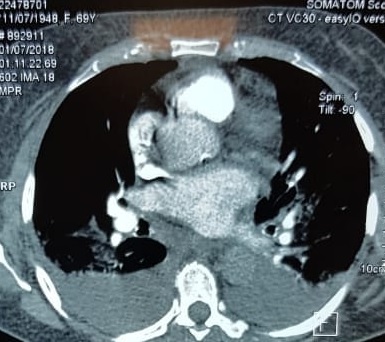
After a cardiothoracic consultation, cardiac magnetic resonance imaging (MRI) was performed. The findings showed the presence of a 58 x 45 x 6 mm well-circumscribed hyperemic mass on the anterior leaflet of the mitral valve and a second 10 x 10 x 6 mm smaller mass firmly adhered to the posterior leaflet of the mitral valve. 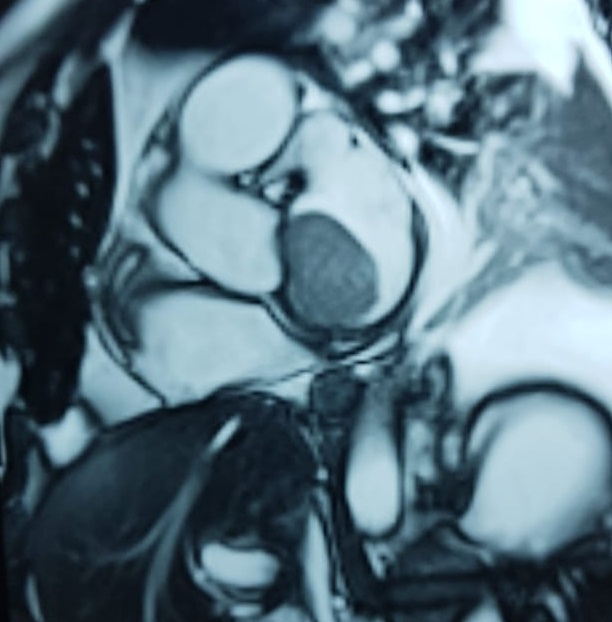
The patient, who was hypotensive and hypoxic, was admitted to the hospital for surgical treatment.
Following sternotomy and cardiopulmonary bypass, a right atriotomy was performed using a trans-septal approach. The large left atrial mass was firmly adhered to the endocardium at the level of the anterior leaflet of the mitral valve and the interatrial septum. The mass had a grey and whitish appearance with some bluish necrotic patches, (Figure 1B, 2B, 3B). 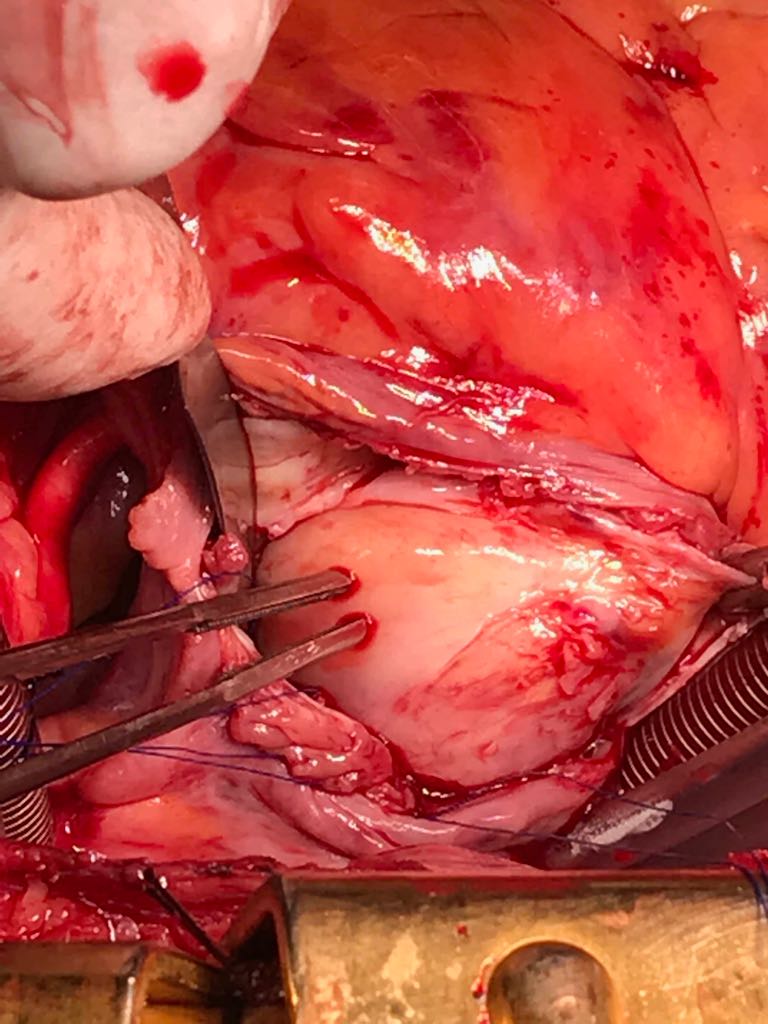
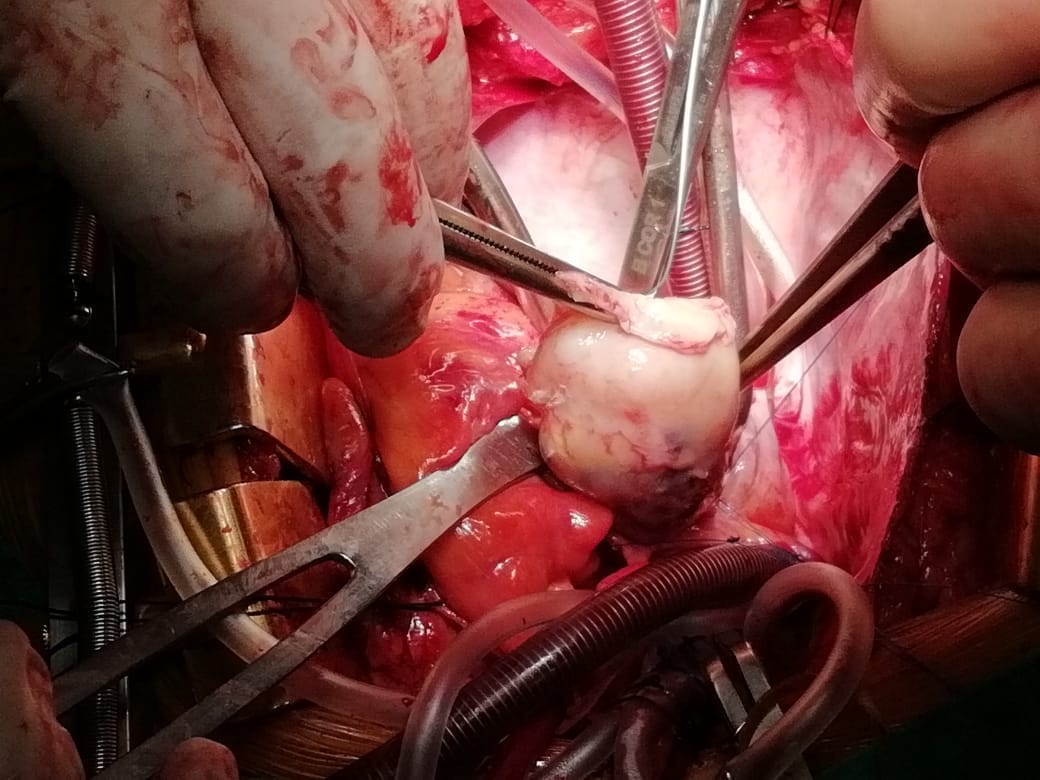
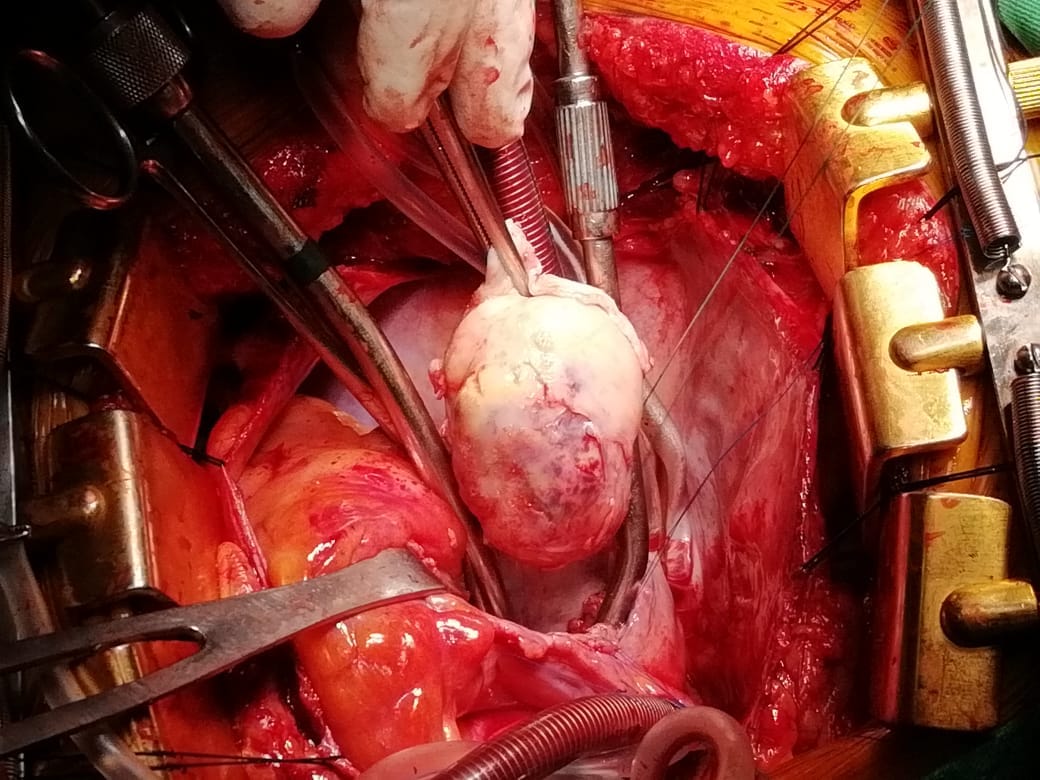
The patient had a complicated postoperative course in the Intensive Care Unit (ICU) and needed inotropic support and vasoactive agents. A postop echocardiogram indicated appropriate left ventricle systolic function, nonetheless, the patient persisted in a hypotensive status that caused refractory shock and ultimately provoked severe organ dysfunction that led to the patient’s death.
Discussion
Primary heart sarcomas are extremely rare malignant neoplasms derived from mesenchymal cells, (1) with an incidence ranging from 0.001% to 0.28% at autopsy. 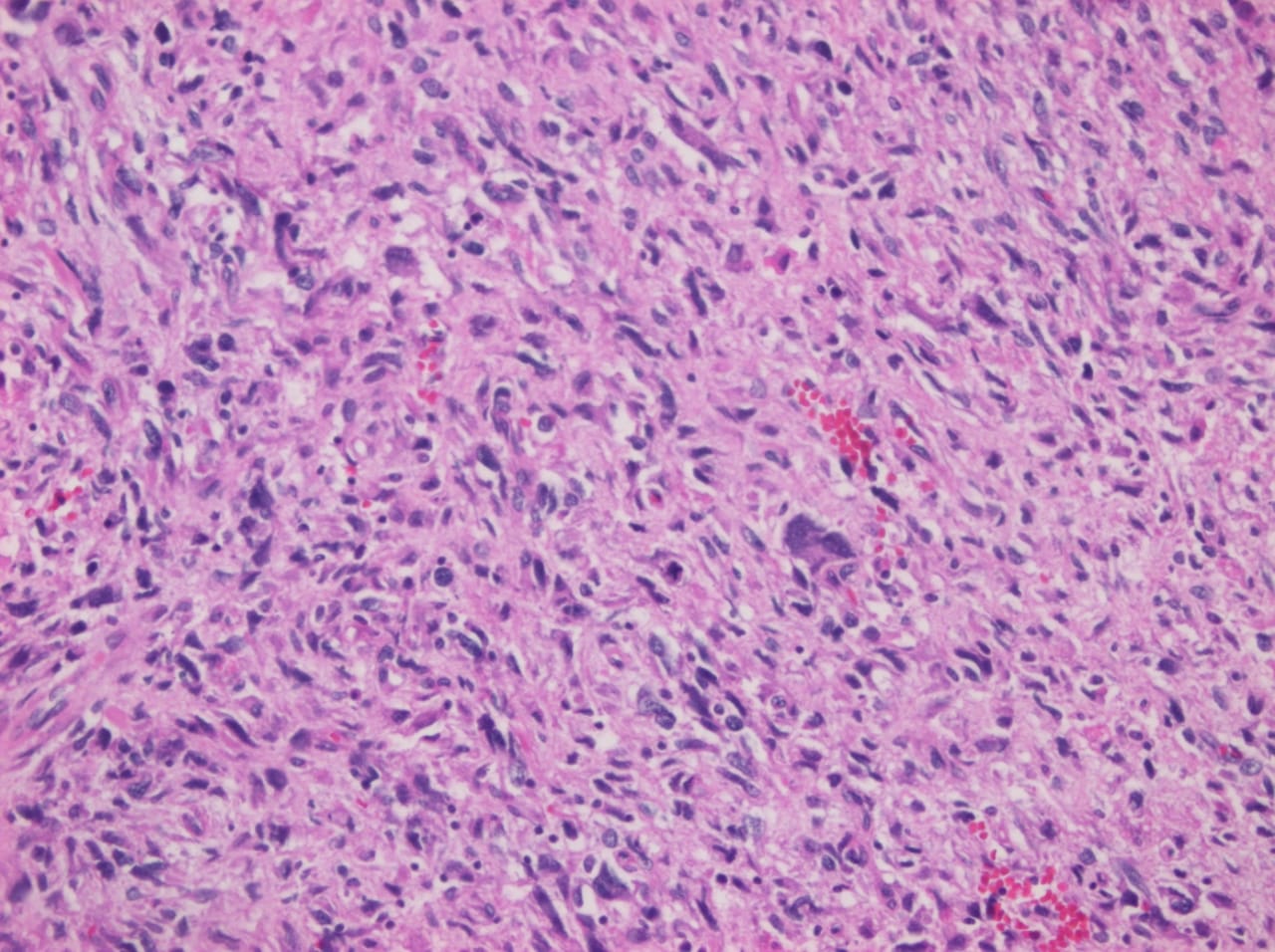
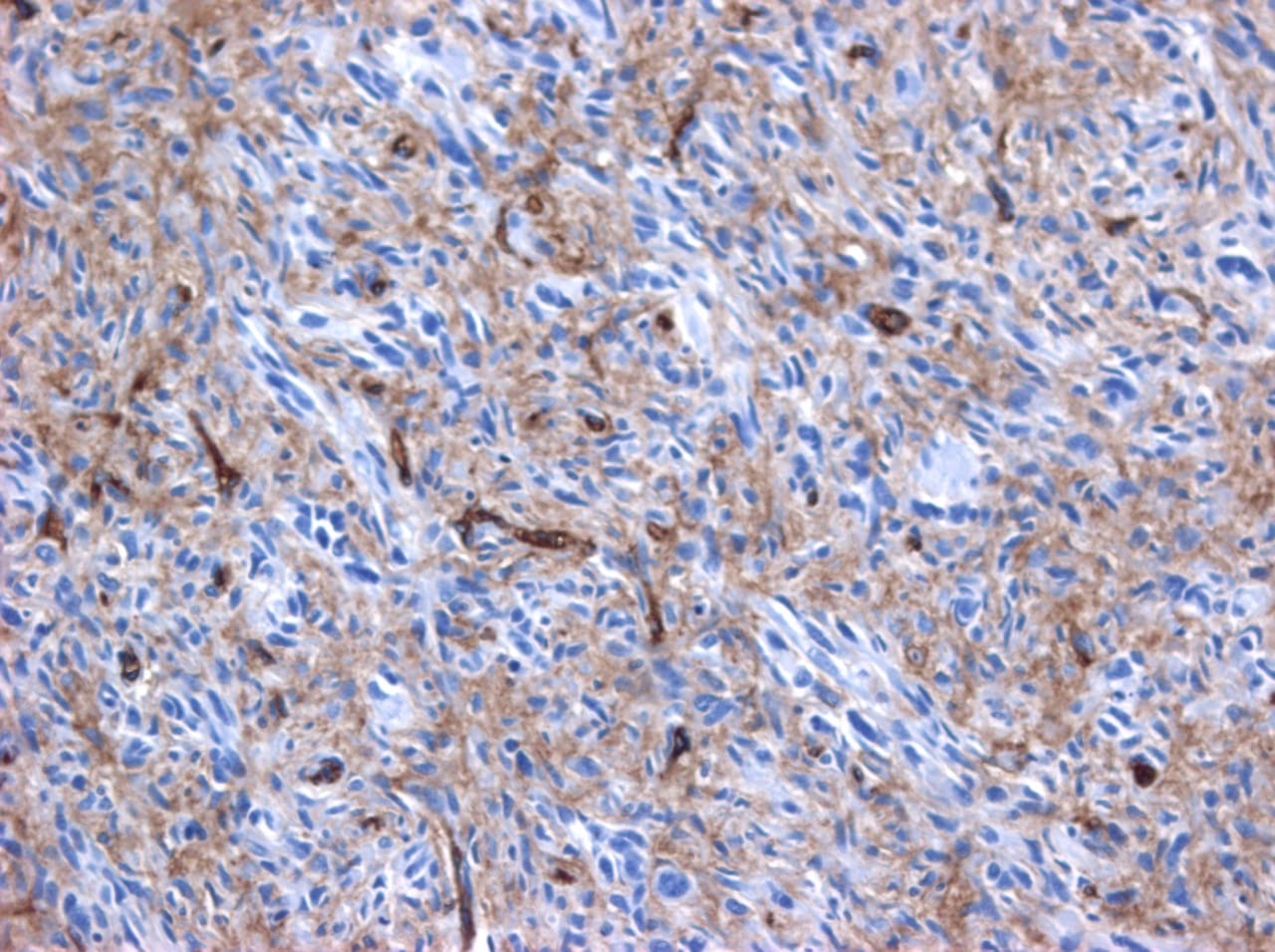
Cardiac angiosarcomas (CA) account for one-third of all primary heart sarcomas (4) and usually develop as gray-brown masses with hemorrhagic patches in the right atrium of male patients. The tumors are filled with vascular channels and their cells are positive for CD34 and factor VIII. (5) Left-sided cardiac angiosarcoma can cause heart failure early in the disease process, but the tumors tend to be more circumscribed, less infiltrative, and associated with better overall survival. (6, 7) Most patients are asymptomatic early in their disease, (2) making the diagnosis even more difficult and worsening its already poor prognosis. (1) The preference of cardiac angiosarcomas for the right heart often leads to a presentation with right-sided congestive heart failure. (2) At later stages, symptoms depend on the structures compromised and range from mild dyspnea on exertion to cardiogenic shock. (8) Cardiac angiosarcomas tend to have a notable intracavitary element, and in some cases may intermittently compromise a cardiac valve, thereby simulating a stenosis or regurgitation. (2, 7)
Our patient presented with acute cardiac failure, pulmonary edema and severe valve dysfunction due to a mass in the left atrium. The tumor had a vascular supply and showed positivity for CD34.
Most patients with cardiac angiosarcoma have metastases, typically to the lung, at diagnosis. (1) Several decades ago, cardiac angiosarcoma was mainly diagnosed postmortem. (1) Now, it can be suspected when cardiomegaly or pleural effusions are seen on chest x-rays (8). Echocardiography is the most useful diagnostic tool, (2) however, CT and MRI can provide useful information on tumor size, invasion and localization. (2, 9) This imaging combination generally provides an excellent anatomic description for preoperative planning. (1, 9)
In our patient, progressive dyspnea was the main symptom and after a prompt evaluation an intracardiac mass was identified as the cause of severe cardiac dysfunction. Because of this finding and the clinical condition of the patient, surgery was planned.
Complete resection of the tumor is the treatment of choice, and is the only therapy currently seen to influence survival. (8) But because of the highly aggressive behavior and a high incidence of systemic metastases with cardiac angiosarcomas, a complete surgical resection is often hampered. (1) Cardiac angiosarcoma carries a grim prognosis as these tumors are universally fatal with a mean survival time of several months after initial presentation even after successful surgery. (2) Chemotherapy is recommended after surgery, even when clear surgical margins are obtained because of the high probability of missed microscopic disease. (1, 2)
High clinical suspicion together with an appropriate history, a thorough physical examination, and precise complementary tests are vital for timely diagnosis and proper treatment.
Authors and Affiliations
Santiago A. Endara: Department of General Surgery, Division of Cardiothoracic Surgery, Hospital Metropolitano, Quito, Ecuador, MD
Gerardo A. Dávalos: Department of General Surgery, Division of Cardiothoracic Surgery, Hospital Metropolitano, Quito, Ecuador, MD
Patricia M. Pontón: Hospital Metropolitano, Quito, Ecuador. Department of Internal Medicine Division of Pathology, MD
Gabriel A. Molina: Pontificia Universidad Católica del Ecuador (PUCE), Quito, Ecuador. PGY4 General Surgery Resident, MD
Daniel L. Mogrovejo: Pontificia Universidad Católica del Ecuador (PUCE), Quito, Ecuador. PGY1 General Surgery Resident, MD
Corresponding Author Info:
Santiago A. Endara, Hospital Metropolitano, Av. Mariana de Jesus Oe 7/47 y Conclina, Edificio Diagnostico 2000 tercer piso 3/3, Quito, Ecuador, + 593 9 98416157
Email: drsantiagoendara@gmail.com
1. Orlandi A, Ferlosio A, Roselli M, Chiariello L, Spagnoli L. Cardiac Sarcomas: An Update. Journal of Thoracic Oncology. 2010;5(9):1483-1489.
2. Brandt R, Arnold R, Bohle R, Dill T, Hamm C. Cardiac angiosarcoma: case report and review of the literature. Zeitschrift für Kardiologie. 2005;94(12):824-828.
3. Kurian K, Weisshaar D, Parekh H, Berry G, Reitz B. Primary cardiac angiosarcoma: case report and review of the literature. Cardiovascular Pathology. 2006;15(2):110-112.
4. Habibi R, Faramarzi N, Altamirano A, Dadkhah S. A Patient Presenting with Cardiac Tamponade and the Challenges of Finding Its Cause: A Cardiac Angiosarcoma. Case Reports in Cardiology. 2018;2018:1-3.
5. Leduc C, Jenkins S, Sukov W, Rustin J, Maleszewski J. Cardiac angiosarcoma: histopathologic, immunohistochemical, and cytogenetic analysis of 10 cases. Human Pathology. 2017;60:199-207.
6. Ramlawi B, Leja M, Abu Saleh W, Al Jabbari O, Benjamin R, Ravi V et al. Surgical Treatment of Primary Cardiac Sarcomas: Review of a Single-Institution Experience. The Annals of Thoracic Surgery. 2016;101(2):698-702.
7.Engelen M. Primary left atrial angiosarcoma mimicking severe mitral valve stenosis. Heart. 2005;91(4):e27-e27.
8. Chenier M, Johnson D, Ohman M, Pavlisko E. Cardiac angiosarcoma presenting as progressive dyspnea on exertion. Journal of Cardiovascular Medicine. 2011;12(12):904-907.
9. Lindsey J, Stacey R. Cardiac magnetic resonance in cardiac angiosarcoma. Echocardiography. 2017;34(7):1077-1081.
Abstract
Primary heart sarcomas are rare and represent 20% of all primary cardiac tumors. Symptoms depend on which chambers and cardiac structures are involved. Angiosarcoma is one of the most common and the most aggressive types of primary heart sarcomas. Typically, these tumors are found in the right atrium, however, cardiac angiosarcomas may involve any part of the heart. Most of these tumors are diagnosed in advanced stages and the patient prognosis is poor. Most tumors are diagnosed using echocardiography. Computed tomography (CT) and magnetic resonance imaging (MRI) provide useful information on tumor size and location for planning surgery, which is the only treatment shown to increase survival. We present the case of a 69-year-old woman who presented to the emergency department with hypotension, dyspnea and progressive shortness of breath. After adequate resuscitation, a cardiac mass was identified and surgery was successfully performed. Pathology confirmed a grade 2 primary heart angiosarcoma. Following surgery, the patient was admitted to the intensive care unit and later died secondary to multi-organ system failure.
Introduction
Primary heart angiosarcoma is an aggressive and usually fatal cardiac neoplasm (1). Angiosarcomas can originate at any location in the heart (2, 3), but these tumors typically reside in the right atrium and frequently cause nonspecific symptoms such as dyspnea, cough, heart failure, and arrhythmias. (2) Surgery followed by chemotherapy is the typical approach to these tumors. (4)
We present the case of a 69-year-old woman who presented to the emergency department with hypotension and severe dyspnea.
Case Report
The patient was a 69-year-old woman with a medical history of diabetes. A week before seeking care in the emergency department, she experienced a general feeling of unwellness, dyspnea, and mild respiratory distress. She reported these symptoms had become more and more severe in the last 24 hours and were accompanied by acute chest pain and progressive shortness of breath.
On clinical examination, the patient was hypotensive, had tachypnea and tachycardia, and was hypoxic. Cardiac auscultation detected a systolic murmur in the apex, and auscultation of the lungs revealed crackles and rales, especially at the bases of the lungs. The remainder of her clinical examination was unremarkable. She had sinus tachycardia on an electrocardiogram. A chest X-ray showed a left atrial enlargement along with some patchy opacities in the middle and lower zones of the lungs, along with Kerley B lines suggestive of pulmonary edema.
With these findings, and after adequate resuscitation, a contrast-enhanced computed tomography (CT) scan detected a filling defect in the left atrium suggestive of a large intra-cardiac mass with a thick and hyper-enhanced interatrial septum. Bilateral pleural effusions also were evident, (Figure 1A) hence an echocardiogram was requested and it confirmed the presence of a 30 x 29 x 40 mm lobulated highly mobile mass in the left atrium.
After a cardiothoracic consultation, cardiac magnetic resonance imaging (MRI) was performed. The findings showed the presence of a 58 x 45 x 6 mm well-circumscribed hyperemic mass on the anterior leaflet of the mitral valve and a second 10 x 10 x 6 mm smaller mass firmly adhered to the posterior leaflet of the mitral valve.
The patient, who was hypotensive and hypoxic, was admitted to the hospital for surgical treatment.
Following sternotomy and cardiopulmonary bypass, a right atriotomy was performed using a trans-septal approach. The large left atrial mass was firmly adhered to the endocardium at the level of the anterior leaflet of the mitral valve and the interatrial septum. The mass had a grey and whitish appearance with some bluish necrotic patches, (Figure 1B, 2B, 3B).
The patient had a complicated postoperative course in the Intensive Care Unit (ICU) and needed inotropic support and vasoactive agents. A postop echocardiogram indicated appropriate left ventricle systolic function, nonetheless, the patient persisted in a hypotensive status that caused refractory shock and ultimately provoked severe organ dysfunction that led to the patient’s death.
Discussion
Primary heart sarcomas are extremely rare malignant neoplasms derived from mesenchymal cells, (1) with an incidence ranging from 0.001% to 0.28% at autopsy.
Cardiac angiosarcomas (CA) account for one-third of all primary heart sarcomas (4) and usually develop as gray-brown masses with hemorrhagic patches in the right atrium of male patients. The tumors are filled with vascular channels and their cells are positive for CD34 and factor VIII. (5) Left-sided cardiac angiosarcoma can cause heart failure early in the disease process, but the tumors tend to be more circumscribed, less infiltrative, and associated with better overall survival. (6, 7) Most patients are asymptomatic early in their disease, (2) making the diagnosis even more difficult and worsening its already poor prognosis. (1) The preference of cardiac angiosarcomas for the right heart often leads to a presentation with right-sided congestive heart failure. (2) At later stages, symptoms depend on the structures compromised and range from mild dyspnea on exertion to cardiogenic shock. (8) Cardiac angiosarcomas tend to have a notable intracavitary element, and in some cases may intermittently compromise a cardiac valve, thereby simulating a stenosis or regurgitation. (2, 7)
Our patient presented with acute cardiac failure, pulmonary edema and severe valve dysfunction due to a mass in the left atrium. The tumor had a vascular supply and showed positivity for CD34.
Most patients with cardiac angiosarcoma have metastases, typically to the lung, at diagnosis. (1) Several decades ago, cardiac angiosarcoma was mainly diagnosed postmortem. (1) Now, it can be suspected when cardiomegaly or pleural effusions are seen on chest x-rays (8). Echocardiography is the most useful diagnostic tool, (2) however, CT and MRI can provide useful information on tumor size, invasion and localization. (2, 9) This imaging combination generally provides an excellent anatomic description for preoperative planning. (1, 9)
In our patient, progressive dyspnea was the main symptom and after a prompt evaluation an intracardiac mass was identified as the cause of severe cardiac dysfunction. Because of this finding and the clinical condition of the patient, surgery was planned.
Complete resection of the tumor is the treatment of choice, and is the only therapy currently seen to influence survival. (8) But because of the highly aggressive behavior and a high incidence of systemic metastases with cardiac angiosarcomas, a complete surgical resection is often hampered. (1) Cardiac angiosarcoma carries a grim prognosis as these tumors are universally fatal with a mean survival time of several months after initial presentation even after successful surgery. (2) Chemotherapy is recommended after surgery, even when clear surgical margins are obtained because of the high probability of missed microscopic disease. (1, 2)
High clinical suspicion together with an appropriate history, a thorough physical examination, and precise complementary tests are vital for timely diagnosis and proper treatment.
Authors and Affiliations
Santiago A. Endara: Department of General Surgery, Division of Cardiothoracic Surgery, Hospital Metropolitano, Quito, Ecuador, MD
Gerardo A. Dávalos: Department of General Surgery, Division of Cardiothoracic Surgery, Hospital Metropolitano, Quito, Ecuador, MD
Patricia M. Pontón: Hospital Metropolitano, Quito, Ecuador. Department of Internal Medicine Division of Pathology, MD
Gabriel A. Molina: Pontificia Universidad Católica del Ecuador (PUCE), Quito, Ecuador. PGY4 General Surgery Resident, MD
Daniel L. Mogrovejo: Pontificia Universidad Católica del Ecuador (PUCE), Quito, Ecuador. PGY1 General Surgery Resident, MD
Corresponding Author Info:
Santiago A. Endara, Hospital Metropolitano, Av. Mariana de Jesus Oe 7/47 y Conclina, Edificio Diagnostico 2000 tercer piso 3/3, Quito, Ecuador, + 593 9 98416157
Email: drsantiagoendara@gmail.com
Abstract
Primary heart sarcomas are rare and represent 20% of all primary cardiac tumors. Symptoms depend on which chambers and cardiac structures are involved. Angiosarcoma is one of the most common and the most aggressive types of primary heart sarcomas. Typically, these tumors are found in the right atrium, however, cardiac angiosarcomas may involve any part of the heart. Most of these tumors are diagnosed in advanced stages and the patient prognosis is poor. Most tumors are diagnosed using echocardiography. Computed tomography (CT) and magnetic resonance imaging (MRI) provide useful information on tumor size and location for planning surgery, which is the only treatment shown to increase survival. We present the case of a 69-year-old woman who presented to the emergency department with hypotension, dyspnea and progressive shortness of breath. After adequate resuscitation, a cardiac mass was identified and surgery was successfully performed. Pathology confirmed a grade 2 primary heart angiosarcoma. Following surgery, the patient was admitted to the intensive care unit and later died secondary to multi-organ system failure.
Introduction
Primary heart angiosarcoma is an aggressive and usually fatal cardiac neoplasm (1). Angiosarcomas can originate at any location in the heart (2, 3), but these tumors typically reside in the right atrium and frequently cause nonspecific symptoms such as dyspnea, cough, heart failure, and arrhythmias. (2) Surgery followed by chemotherapy is the typical approach to these tumors. (4)
We present the case of a 69-year-old woman who presented to the emergency department with hypotension and severe dyspnea.
Case Report
The patient was a 69-year-old woman with a medical history of diabetes. A week before seeking care in the emergency department, she experienced a general feeling of unwellness, dyspnea, and mild respiratory distress. She reported these symptoms had become more and more severe in the last 24 hours and were accompanied by acute chest pain and progressive shortness of breath.
On clinical examination, the patient was hypotensive, had tachypnea and tachycardia, and was hypoxic. Cardiac auscultation detected a systolic murmur in the apex, and auscultation of the lungs revealed crackles and rales, especially at the bases of the lungs. The remainder of her clinical examination was unremarkable. She had sinus tachycardia on an electrocardiogram. A chest X-ray showed a left atrial enlargement along with some patchy opacities in the middle and lower zones of the lungs, along with Kerley B lines suggestive of pulmonary edema.
With these findings, and after adequate resuscitation, a contrast-enhanced computed tomography (CT) scan detected a filling defect in the left atrium suggestive of a large intra-cardiac mass with a thick and hyper-enhanced interatrial septum. Bilateral pleural effusions also were evident, (Figure 1A) hence an echocardiogram was requested and it confirmed the presence of a 30 x 29 x 40 mm lobulated highly mobile mass in the left atrium.
After a cardiothoracic consultation, cardiac magnetic resonance imaging (MRI) was performed. The findings showed the presence of a 58 x 45 x 6 mm well-circumscribed hyperemic mass on the anterior leaflet of the mitral valve and a second 10 x 10 x 6 mm smaller mass firmly adhered to the posterior leaflet of the mitral valve.
The patient, who was hypotensive and hypoxic, was admitted to the hospital for surgical treatment.
Following sternotomy and cardiopulmonary bypass, a right atriotomy was performed using a trans-septal approach. The large left atrial mass was firmly adhered to the endocardium at the level of the anterior leaflet of the mitral valve and the interatrial septum. The mass had a grey and whitish appearance with some bluish necrotic patches, (Figure 1B, 2B, 3B).
The patient had a complicated postoperative course in the Intensive Care Unit (ICU) and needed inotropic support and vasoactive agents. A postop echocardiogram indicated appropriate left ventricle systolic function, nonetheless, the patient persisted in a hypotensive status that caused refractory shock and ultimately provoked severe organ dysfunction that led to the patient’s death.
Discussion
Primary heart sarcomas are extremely rare malignant neoplasms derived from mesenchymal cells, (1) with an incidence ranging from 0.001% to 0.28% at autopsy.
Cardiac angiosarcomas (CA) account for one-third of all primary heart sarcomas (4) and usually develop as gray-brown masses with hemorrhagic patches in the right atrium of male patients. The tumors are filled with vascular channels and their cells are positive for CD34 and factor VIII. (5) Left-sided cardiac angiosarcoma can cause heart failure early in the disease process, but the tumors tend to be more circumscribed, less infiltrative, and associated with better overall survival. (6, 7) Most patients are asymptomatic early in their disease, (2) making the diagnosis even more difficult and worsening its already poor prognosis. (1) The preference of cardiac angiosarcomas for the right heart often leads to a presentation with right-sided congestive heart failure. (2) At later stages, symptoms depend on the structures compromised and range from mild dyspnea on exertion to cardiogenic shock. (8) Cardiac angiosarcomas tend to have a notable intracavitary element, and in some cases may intermittently compromise a cardiac valve, thereby simulating a stenosis or regurgitation. (2, 7)
Our patient presented with acute cardiac failure, pulmonary edema and severe valve dysfunction due to a mass in the left atrium. The tumor had a vascular supply and showed positivity for CD34.
Most patients with cardiac angiosarcoma have metastases, typically to the lung, at diagnosis. (1) Several decades ago, cardiac angiosarcoma was mainly diagnosed postmortem. (1) Now, it can be suspected when cardiomegaly or pleural effusions are seen on chest x-rays (8). Echocardiography is the most useful diagnostic tool, (2) however, CT and MRI can provide useful information on tumor size, invasion and localization. (2, 9) This imaging combination generally provides an excellent anatomic description for preoperative planning. (1, 9)
In our patient, progressive dyspnea was the main symptom and after a prompt evaluation an intracardiac mass was identified as the cause of severe cardiac dysfunction. Because of this finding and the clinical condition of the patient, surgery was planned.
Complete resection of the tumor is the treatment of choice, and is the only therapy currently seen to influence survival. (8) But because of the highly aggressive behavior and a high incidence of systemic metastases with cardiac angiosarcomas, a complete surgical resection is often hampered. (1) Cardiac angiosarcoma carries a grim prognosis as these tumors are universally fatal with a mean survival time of several months after initial presentation even after successful surgery. (2) Chemotherapy is recommended after surgery, even when clear surgical margins are obtained because of the high probability of missed microscopic disease. (1, 2)
High clinical suspicion together with an appropriate history, a thorough physical examination, and precise complementary tests are vital for timely diagnosis and proper treatment.
Authors and Affiliations
Santiago A. Endara: Department of General Surgery, Division of Cardiothoracic Surgery, Hospital Metropolitano, Quito, Ecuador, MD
Gerardo A. Dávalos: Department of General Surgery, Division of Cardiothoracic Surgery, Hospital Metropolitano, Quito, Ecuador, MD
Patricia M. Pontón: Hospital Metropolitano, Quito, Ecuador. Department of Internal Medicine Division of Pathology, MD
Gabriel A. Molina: Pontificia Universidad Católica del Ecuador (PUCE), Quito, Ecuador. PGY4 General Surgery Resident, MD
Daniel L. Mogrovejo: Pontificia Universidad Católica del Ecuador (PUCE), Quito, Ecuador. PGY1 General Surgery Resident, MD
Corresponding Author Info:
Santiago A. Endara, Hospital Metropolitano, Av. Mariana de Jesus Oe 7/47 y Conclina, Edificio Diagnostico 2000 tercer piso 3/3, Quito, Ecuador, + 593 9 98416157
Email: drsantiagoendara@gmail.com
1. Orlandi A, Ferlosio A, Roselli M, Chiariello L, Spagnoli L. Cardiac Sarcomas: An Update. Journal of Thoracic Oncology. 2010;5(9):1483-1489.
2. Brandt R, Arnold R, Bohle R, Dill T, Hamm C. Cardiac angiosarcoma: case report and review of the literature. Zeitschrift für Kardiologie. 2005;94(12):824-828.
3. Kurian K, Weisshaar D, Parekh H, Berry G, Reitz B. Primary cardiac angiosarcoma: case report and review of the literature. Cardiovascular Pathology. 2006;15(2):110-112.
4. Habibi R, Faramarzi N, Altamirano A, Dadkhah S. A Patient Presenting with Cardiac Tamponade and the Challenges of Finding Its Cause: A Cardiac Angiosarcoma. Case Reports in Cardiology. 2018;2018:1-3.
5. Leduc C, Jenkins S, Sukov W, Rustin J, Maleszewski J. Cardiac angiosarcoma: histopathologic, immunohistochemical, and cytogenetic analysis of 10 cases. Human Pathology. 2017;60:199-207.
6. Ramlawi B, Leja M, Abu Saleh W, Al Jabbari O, Benjamin R, Ravi V et al. Surgical Treatment of Primary Cardiac Sarcomas: Review of a Single-Institution Experience. The Annals of Thoracic Surgery. 2016;101(2):698-702.
7.Engelen M. Primary left atrial angiosarcoma mimicking severe mitral valve stenosis. Heart. 2005;91(4):e27-e27.
8. Chenier M, Johnson D, Ohman M, Pavlisko E. Cardiac angiosarcoma presenting as progressive dyspnea on exertion. Journal of Cardiovascular Medicine. 2011;12(12):904-907.
9. Lindsey J, Stacey R. Cardiac magnetic resonance in cardiac angiosarcoma. Echocardiography. 2017;34(7):1077-1081.
1. Orlandi A, Ferlosio A, Roselli M, Chiariello L, Spagnoli L. Cardiac Sarcomas: An Update. Journal of Thoracic Oncology. 2010;5(9):1483-1489.
2. Brandt R, Arnold R, Bohle R, Dill T, Hamm C. Cardiac angiosarcoma: case report and review of the literature. Zeitschrift für Kardiologie. 2005;94(12):824-828.
3. Kurian K, Weisshaar D, Parekh H, Berry G, Reitz B. Primary cardiac angiosarcoma: case report and review of the literature. Cardiovascular Pathology. 2006;15(2):110-112.
4. Habibi R, Faramarzi N, Altamirano A, Dadkhah S. A Patient Presenting with Cardiac Tamponade and the Challenges of Finding Its Cause: A Cardiac Angiosarcoma. Case Reports in Cardiology. 2018;2018:1-3.
5. Leduc C, Jenkins S, Sukov W, Rustin J, Maleszewski J. Cardiac angiosarcoma: histopathologic, immunohistochemical, and cytogenetic analysis of 10 cases. Human Pathology. 2017;60:199-207.
6. Ramlawi B, Leja M, Abu Saleh W, Al Jabbari O, Benjamin R, Ravi V et al. Surgical Treatment of Primary Cardiac Sarcomas: Review of a Single-Institution Experience. The Annals of Thoracic Surgery. 2016;101(2):698-702.
7.Engelen M. Primary left atrial angiosarcoma mimicking severe mitral valve stenosis. Heart. 2005;91(4):e27-e27.
8. Chenier M, Johnson D, Ohman M, Pavlisko E. Cardiac angiosarcoma presenting as progressive dyspnea on exertion. Journal of Cardiovascular Medicine. 2011;12(12):904-907.
9. Lindsey J, Stacey R. Cardiac magnetic resonance in cardiac angiosarcoma. Echocardiography. 2017;34(7):1077-1081.
Soft Tissue Sarcoma Chemotherapy
Predicting response to chemotherapy
The prognostic nomogram called Sarculator was used effectively to define a high-risk subgroup of patients likely to benefit from adjuvant chemotherapy, Sandro Pasquali, MD, of the Fondazione IRCCS Istituto Nazionale dei Tumori, Milano, Italy and his colleagues reported at the meeting.
Perioperative chemotherapy was shown to afford no survival advantage over observation in the EORTC 62931 (European Organization for Research and Treatment of Cancer—62931) study of adjuvant doxorubicin plus ifosfamide (Lancet Oncol 2012;13:1045-54). However, subsequent analyses of that data attributed this finding to variations in treatment schedules and the inclusion of low-risk tumors, which may have diluted the effect of chemotherapy, the researchers said in their abstract.
Further, a recent interim report of the ISG-1001 trial showed a survival benefit for patients who received neoadjuvant epirubicin plus ifosfamide therapy for localized high-risk soft-tissue sarcoma of the extremities or trunk wall (Lancet Oncol 2017;18:812-822).
The researchers performed a retrospective analysis of individual data for 290 patients with extremity and trunk wall soft-tissue sarcomas in the EORTC-STBSG 62931 study. The Sarculator was used to calculate 10-year predicted probability of overall survival (pr-OS) for each patient.
Patients were grouped in two categories of predicted overall survival: high predicted survival (over 60%) and low predicted overall survival (60% or less). Overall survival and disease-free survival were calculated at 8 years, the study’s median follow-up.
The 8-year probability of overall survival and disease-free survival was 58% [95% confidence interval (CI): 52–63%] and 51% (95% CI: 46–57%), respectively. In the 290 patients with extremity and trunk wall soft tissue sarcomas, adjuvant chemotherapy was not associated with an overall survival benefit [Hazard ratio (HR) = 0.91, 95%CI 0.63–1.31]. The Sarcolator Nomogram detected 80 patients who were at greater risk of death compared to the 210 patients with higher predicted overall survival. The risk of death was significantly lower with adjuvant chemotherapy in the group with low predicted survival based on the Sarculator Nomogram (HR=0.50, 95%CI 0.30-0.90). Consistently, the risk of recurrence was significantly lower when adjuvant chemotherapy was used in the group with predicted overall survival of less than 60% (HR = 0.49, 95%CI 0.28-0.85) while this difference was not observed in patients with high predicted overall survival (HR = 0.95, 95%CI 0.62-1.44).
Doxorubicin plus dacarbazine deserve evaluation in prospective trials in leiomyosarcoma
Doxorubicin plus dacarbazine appeared to best the outcomes seen with doxorubicin plus ifosfamide and with doxorubicin alone in terms of overall response rate and progression free survival as first-line treatment in patients with advanced leiomyosarcomas, based on a retrospective analysis presented by Lorenzo D’Ambrosio, MD, of the Unitversity of Torino, Italy, and his associates.
As patients in the trial were not randomized to therapy, the researchers used a logistic regression model that accounted for histology, site of primary, age, gender, performance status, tumor extent, and tumor grade. Patients were then matched across the different groups by their propensity scores.The 303 patients, 216 of them women, were enrolled from 18 EORTC STBSG (European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group) sites. Doxorubicin plus dacarbazine was given to 117 patients (39%), doxorubicin plus ifosfamide was given to 71 (23%), and doxorubicin alone was given to 115 (38%). There were no significant differences among the regimens in terms of dose reductions of more than 10%, delays of greater than 72 hours, or granulocyte-colony stimulating factor use.
In the whole population, unadjusted median progression free survival was 9.4 months (95% CI 6.1-9.7 months) for those given doxorubicin plus dacarbazine, 6.8 months (4.5-9.5 months) for those given doxorubicin plus ifosfamide), and 5.4 months (3.8-6.8 months) for those given doxorubicin alone. The respective overall response rates for the three regimens were 36.8%, 21.5%, and 25.9%. When using propensity scores to adjust for lack of randomization, progression free survival was significantly longer with doxorubicin plus dacarbazine [median 9.2 months (95%CI 5.2-9.7 months) than with doxorubicin [median 4.8 months (2.3-6.0 months); HR 0.72 (0.52-0.99)]. The difference was not significant when compared to doxorubicin plus ifosfamide [8.2 months (5.2-10.1 months), HR 1.01 (0.68-1.50)]. Progression free survival did not differ significantly between doxorubicin plus ifosfamide, and doxorubicin [HR 0.71 (0.48-1.06)]. In the same matched population, overall response rates were 30.9%, 19.5%, and 25.6% for doxorubicin plus dacarbazine, doxorubicin plus ifosfamide, and doxorubicin, respectively.
Overall survival comparisons were weakened by a shorter median follow-up in the doxorubicin plus dacarbazine groups (32 months) compared to the doxorubicin plus ifosfamide group (50 months) and the doxorubicin group (46 months). With this limit, patients in the doxorubicin plus dacarbazine arm had longer overall survival [median 36.8 (27.9-47.2) months] when compared to both doxorubicin plus ifosfamide [21.9 (16.7-33.4), HR 0.65 (0.40-1.06); and doxorubicin arms 30.3 (21.0-36.3) months, HR 0.66 (0.43-0.99).
Subsequent treatments were well balanced across arms. None of the selected factors for multivariate analysis (age, sex, ECOG performance status, histotype, site of primary tumor, tumor grade, and tumor extent) significantly affected the progression free survival and overall survival associated with the treatments.
Olaratumab in combination with doxorubicin plus ifosfamide
Olaratumab at 15 mg/kg has been shown to be safe in combination with doxorubicin plus ifosfamide in a Phase 1b study (NCT03283696), reported Sebastian Bauer, MD, of the West German Cancer Center, University of Duisburg-Essen, Essen, Germany, and his colleagues.
Given that 8 of 10 evaluable patients have completed the drug-limiting toxicity period without drug-limiting toxicities at the 15 mg/kg dose level of olaratumab, the study has proceeded to the next cohort. In those patients, an olaratumab loading dose of 20 mg/kg will be evaluated in cycle 1, followed by 15 mg/kg of olaratumab in subsequent cycles with the same doses of doxorubicin plus ifosfamide, the researchers wrote in their abstract.
The phase 1 trial enrolled 16 patients with advanced or metastatic soft tissue sarcomas and no prior lines of systemic therapy and ECOG performance status 0-1. Adequate follow up data was available for 10 patients.
Olaratumab, (Lartruvo), which binds platelet-derived growth factor receptor alpha (PDGFRα), was given at 15 mg/kg in combination with doxorubicin (75 mg/m2 on days 1-3) and ifosfamide (10 g/m2 on days 1-4) followed by mandatory granulocyte-colony-stimulating factor therapy in cycles 1-6 on a 21-day cycle. Doxorubicin was allowed to be administered by continuous infusion or bolus administration and with cardiac protection. Mesna dosing was at least 60% of the ifosfamide dose.
Two of the 10 patients had dose-limiting toxicities; one had Grade 4 febrile neutropenia and the other had Grade 3 febrile neutropenia and Grade 3 mucositis. Common related adverse events occurring in over 30% of patients included fatigue, anemia, neutropenia, thrombocytopenia, constipation, and nausea. One patient discontinued study treatment due to progressive disease, and all others were on study treatment as of data cutoff. Among 7 patients evaluated for tumor response assessment, 3 patients had a partial response according to RECIST and 3 further patients had stabilized disease as best overall response for a disease control rate of 86%.
Anthracycline-based regimen excels in FIGO-1 uterine leiomyosarcoma
Future trials to assess the efficacy of adjuvant chemotherapy in uterine leiomyosarcoma should incorporate anthracyclines, according to Roberta Sanfilippo, MD, of Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues.
Disease-free survival was extended in patients with uterine leiomyosarcomas treated with anthracycline-based regimens as compared to gemcitabine and docetaxel, based on a retrospective analysis reported at the meeting by Dr. Sanfilippo.
They reviewed all patients with FIGO stage I uterine leiomyosarcomas who underwent hysterectomy with or without oophorectomy and were treated with adjuvant chemotherapy with either anthracycline-based or gemcitabine-based chemotherapy at two Italian centers.
Of 145 patients, 97 were treated with an anthracycline-based regimen and 48 with gemcitabine and docetaxel. The median number of cycles of anthracycline based regimen received was 4 (range 2-6) and with gemcitabine and docetaxel was 5 (range 3-7). Disease free survival was 31 months in patients treated with anthracycline-based chemotherapy and 19 months in patients treated with gemcitabine and docetaxel.
Trabectedin and low-dose radiotherapy
Trabectedin concurrent with low-dose radiotherapy is being examined as an option for patients with pulmonary metastatic soft tissue sarcoma (NCT02275286).
In a phase 1 study, long-lasting dimensional responses were seen in 71% of the irradiated lesions showed. Based on those results, trabectedin (Yondelis) at 1.5 mg/m 2 will be the recommended dose for phase 2, according to Javier Martín-Broto, MD, of the Institute of Biomedicine Research (IBIS)-University Hospital Virgen del Rocio/CSIC/University of Seville, Spain, and his colleagues.
For the study, trabectedin was given along with radiotherapy (30 Gy) in 10 fractions (3 Gy/fraction). Three dose levels of trabectedin were administered: -1 (1.1 mg/m 2), 1 (1.3 mg/m 2) and 2 (1.5 mg/m 2). Dose-limiting toxicity was defined as grade 3 or greater events excluding grade 3/4 neutropenia lasting less than 5 days, grade 3 transaminitis if it did not lead to trabectedin delay, and grade 3/4 nausea/vomiting due to inadequate prophylaxis.
Ten of the 18 patients enrolled had synovial sarcoma; 3 had undifferentiated pleomorphic sarcomas and the other patients had either myxoid liposarcoma, dedifferentiated liposarcoma, G3 not otherwise specified sarcoma, leiomyosarcoma, and malignant peripheral nerve sheath tumor.
Patients received a median of 1 prior line of chemotherapy (range: 0-3). Twelve patients received trabectedin at dose level 1 and 6 patients at dose level 2. Grade 3/4 adverse events were neutropenia, seen in 8 patients; alanine aminotransferase (ALT) elevation, seen in 2 patients; gamma-glutamyl transferase (GGT) elevation, seen in 2 patients; anemia, seen in 2 patients; febrile neutropenia, seen in 1 patient; and pneumonitis, seen in 1 patient.
There were two dose-limiting toxicities: transient grade 4 ALT elevation at the level 1 dose and grade 4 neutropenia for more than 5 days at the level 2 dose.
Based on central radiological review of 17 evaluable patients, 2 patients achieved complete response, 3 had partial responses, 6 had stable disease, and 6 had progressive disease. The local review reported complete responses in 2 patients, partial responses in 5, stable disease in 4, and progressive disease in 6.
On the irradiated lesions, 4 had complete responses, 8 had partial responses, 4 had stable disease, and 1 had progressive disease. With a median follow-up of 18 months, median progression-free survival was 2.83 months (95%CI: 2.3-3.3 months). Thirteen patients have died, with a median overall survival of 8.77 months (95%CI: 3.6-13.9) and a 12-month overall survival rate of 48%.
Predicting response to chemotherapy
The prognostic nomogram called Sarculator was used effectively to define a high-risk subgroup of patients likely to benefit from adjuvant chemotherapy, Sandro Pasquali, MD, of the Fondazione IRCCS Istituto Nazionale dei Tumori, Milano, Italy and his colleagues reported at the meeting.
Perioperative chemotherapy was shown to afford no survival advantage over observation in the EORTC 62931 (European Organization for Research and Treatment of Cancer—62931) study of adjuvant doxorubicin plus ifosfamide (Lancet Oncol 2012;13:1045-54). However, subsequent analyses of that data attributed this finding to variations in treatment schedules and the inclusion of low-risk tumors, which may have diluted the effect of chemotherapy, the researchers said in their abstract.
Further, a recent interim report of the ISG-1001 trial showed a survival benefit for patients who received neoadjuvant epirubicin plus ifosfamide therapy for localized high-risk soft-tissue sarcoma of the extremities or trunk wall (Lancet Oncol 2017;18:812-822).
The researchers performed a retrospective analysis of individual data for 290 patients with extremity and trunk wall soft-tissue sarcomas in the EORTC-STBSG 62931 study. The Sarculator was used to calculate 10-year predicted probability of overall survival (pr-OS) for each patient.
Patients were grouped in two categories of predicted overall survival: high predicted survival (over 60%) and low predicted overall survival (60% or less). Overall survival and disease-free survival were calculated at 8 years, the study’s median follow-up.
The 8-year probability of overall survival and disease-free survival was 58% [95% confidence interval (CI): 52–63%] and 51% (95% CI: 46–57%), respectively. In the 290 patients with extremity and trunk wall soft tissue sarcomas, adjuvant chemotherapy was not associated with an overall survival benefit [Hazard ratio (HR) = 0.91, 95%CI 0.63–1.31]. The Sarcolator Nomogram detected 80 patients who were at greater risk of death compared to the 210 patients with higher predicted overall survival. The risk of death was significantly lower with adjuvant chemotherapy in the group with low predicted survival based on the Sarculator Nomogram (HR=0.50, 95%CI 0.30-0.90). Consistently, the risk of recurrence was significantly lower when adjuvant chemotherapy was used in the group with predicted overall survival of less than 60% (HR = 0.49, 95%CI 0.28-0.85) while this difference was not observed in patients with high predicted overall survival (HR = 0.95, 95%CI 0.62-1.44).
Doxorubicin plus dacarbazine deserve evaluation in prospective trials in leiomyosarcoma
Doxorubicin plus dacarbazine appeared to best the outcomes seen with doxorubicin plus ifosfamide and with doxorubicin alone in terms of overall response rate and progression free survival as first-line treatment in patients with advanced leiomyosarcomas, based on a retrospective analysis presented by Lorenzo D’Ambrosio, MD, of the Unitversity of Torino, Italy, and his associates.
As patients in the trial were not randomized to therapy, the researchers used a logistic regression model that accounted for histology, site of primary, age, gender, performance status, tumor extent, and tumor grade. Patients were then matched across the different groups by their propensity scores.The 303 patients, 216 of them women, were enrolled from 18 EORTC STBSG (European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group) sites. Doxorubicin plus dacarbazine was given to 117 patients (39%), doxorubicin plus ifosfamide was given to 71 (23%), and doxorubicin alone was given to 115 (38%). There were no significant differences among the regimens in terms of dose reductions of more than 10%, delays of greater than 72 hours, or granulocyte-colony stimulating factor use.
In the whole population, unadjusted median progression free survival was 9.4 months (95% CI 6.1-9.7 months) for those given doxorubicin plus dacarbazine, 6.8 months (4.5-9.5 months) for those given doxorubicin plus ifosfamide), and 5.4 months (3.8-6.8 months) for those given doxorubicin alone. The respective overall response rates for the three regimens were 36.8%, 21.5%, and 25.9%. When using propensity scores to adjust for lack of randomization, progression free survival was significantly longer with doxorubicin plus dacarbazine [median 9.2 months (95%CI 5.2-9.7 months) than with doxorubicin [median 4.8 months (2.3-6.0 months); HR 0.72 (0.52-0.99)]. The difference was not significant when compared to doxorubicin plus ifosfamide [8.2 months (5.2-10.1 months), HR 1.01 (0.68-1.50)]. Progression free survival did not differ significantly between doxorubicin plus ifosfamide, and doxorubicin [HR 0.71 (0.48-1.06)]. In the same matched population, overall response rates were 30.9%, 19.5%, and 25.6% for doxorubicin plus dacarbazine, doxorubicin plus ifosfamide, and doxorubicin, respectively.
Overall survival comparisons were weakened by a shorter median follow-up in the doxorubicin plus dacarbazine groups (32 months) compared to the doxorubicin plus ifosfamide group (50 months) and the doxorubicin group (46 months). With this limit, patients in the doxorubicin plus dacarbazine arm had longer overall survival [median 36.8 (27.9-47.2) months] when compared to both doxorubicin plus ifosfamide [21.9 (16.7-33.4), HR 0.65 (0.40-1.06); and doxorubicin arms 30.3 (21.0-36.3) months, HR 0.66 (0.43-0.99).
Subsequent treatments were well balanced across arms. None of the selected factors for multivariate analysis (age, sex, ECOG performance status, histotype, site of primary tumor, tumor grade, and tumor extent) significantly affected the progression free survival and overall survival associated with the treatments.
Olaratumab in combination with doxorubicin plus ifosfamide
Olaratumab at 15 mg/kg has been shown to be safe in combination with doxorubicin plus ifosfamide in a Phase 1b study (NCT03283696), reported Sebastian Bauer, MD, of the West German Cancer Center, University of Duisburg-Essen, Essen, Germany, and his colleagues.
Given that 8 of 10 evaluable patients have completed the drug-limiting toxicity period without drug-limiting toxicities at the 15 mg/kg dose level of olaratumab, the study has proceeded to the next cohort. In those patients, an olaratumab loading dose of 20 mg/kg will be evaluated in cycle 1, followed by 15 mg/kg of olaratumab in subsequent cycles with the same doses of doxorubicin plus ifosfamide, the researchers wrote in their abstract.
The phase 1 trial enrolled 16 patients with advanced or metastatic soft tissue sarcomas and no prior lines of systemic therapy and ECOG performance status 0-1. Adequate follow up data was available for 10 patients.
Olaratumab, (Lartruvo), which binds platelet-derived growth factor receptor alpha (PDGFRα), was given at 15 mg/kg in combination with doxorubicin (75 mg/m2 on days 1-3) and ifosfamide (10 g/m2 on days 1-4) followed by mandatory granulocyte-colony-stimulating factor therapy in cycles 1-6 on a 21-day cycle. Doxorubicin was allowed to be administered by continuous infusion or bolus administration and with cardiac protection. Mesna dosing was at least 60% of the ifosfamide dose.
Two of the 10 patients had dose-limiting toxicities; one had Grade 4 febrile neutropenia and the other had Grade 3 febrile neutropenia and Grade 3 mucositis. Common related adverse events occurring in over 30% of patients included fatigue, anemia, neutropenia, thrombocytopenia, constipation, and nausea. One patient discontinued study treatment due to progressive disease, and all others were on study treatment as of data cutoff. Among 7 patients evaluated for tumor response assessment, 3 patients had a partial response according to RECIST and 3 further patients had stabilized disease as best overall response for a disease control rate of 86%.
Anthracycline-based regimen excels in FIGO-1 uterine leiomyosarcoma
Future trials to assess the efficacy of adjuvant chemotherapy in uterine leiomyosarcoma should incorporate anthracyclines, according to Roberta Sanfilippo, MD, of Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues.
Disease-free survival was extended in patients with uterine leiomyosarcomas treated with anthracycline-based regimens as compared to gemcitabine and docetaxel, based on a retrospective analysis reported at the meeting by Dr. Sanfilippo.
They reviewed all patients with FIGO stage I uterine leiomyosarcomas who underwent hysterectomy with or without oophorectomy and were treated with adjuvant chemotherapy with either anthracycline-based or gemcitabine-based chemotherapy at two Italian centers.
Of 145 patients, 97 were treated with an anthracycline-based regimen and 48 with gemcitabine and docetaxel. The median number of cycles of anthracycline based regimen received was 4 (range 2-6) and with gemcitabine and docetaxel was 5 (range 3-7). Disease free survival was 31 months in patients treated with anthracycline-based chemotherapy and 19 months in patients treated with gemcitabine and docetaxel.
Trabectedin and low-dose radiotherapy
Trabectedin concurrent with low-dose radiotherapy is being examined as an option for patients with pulmonary metastatic soft tissue sarcoma (NCT02275286).
In a phase 1 study, long-lasting dimensional responses were seen in 71% of the irradiated lesions showed. Based on those results, trabectedin (Yondelis) at 1.5 mg/m 2 will be the recommended dose for phase 2, according to Javier Martín-Broto, MD, of the Institute of Biomedicine Research (IBIS)-University Hospital Virgen del Rocio/CSIC/University of Seville, Spain, and his colleagues.
For the study, trabectedin was given along with radiotherapy (30 Gy) in 10 fractions (3 Gy/fraction). Three dose levels of trabectedin were administered: -1 (1.1 mg/m 2), 1 (1.3 mg/m 2) and 2 (1.5 mg/m 2). Dose-limiting toxicity was defined as grade 3 or greater events excluding grade 3/4 neutropenia lasting less than 5 days, grade 3 transaminitis if it did not lead to trabectedin delay, and grade 3/4 nausea/vomiting due to inadequate prophylaxis.
Ten of the 18 patients enrolled had synovial sarcoma; 3 had undifferentiated pleomorphic sarcomas and the other patients had either myxoid liposarcoma, dedifferentiated liposarcoma, G3 not otherwise specified sarcoma, leiomyosarcoma, and malignant peripheral nerve sheath tumor.
Patients received a median of 1 prior line of chemotherapy (range: 0-3). Twelve patients received trabectedin at dose level 1 and 6 patients at dose level 2. Grade 3/4 adverse events were neutropenia, seen in 8 patients; alanine aminotransferase (ALT) elevation, seen in 2 patients; gamma-glutamyl transferase (GGT) elevation, seen in 2 patients; anemia, seen in 2 patients; febrile neutropenia, seen in 1 patient; and pneumonitis, seen in 1 patient.
There were two dose-limiting toxicities: transient grade 4 ALT elevation at the level 1 dose and grade 4 neutropenia for more than 5 days at the level 2 dose.
Based on central radiological review of 17 evaluable patients, 2 patients achieved complete response, 3 had partial responses, 6 had stable disease, and 6 had progressive disease. The local review reported complete responses in 2 patients, partial responses in 5, stable disease in 4, and progressive disease in 6.
On the irradiated lesions, 4 had complete responses, 8 had partial responses, 4 had stable disease, and 1 had progressive disease. With a median follow-up of 18 months, median progression-free survival was 2.83 months (95%CI: 2.3-3.3 months). Thirteen patients have died, with a median overall survival of 8.77 months (95%CI: 3.6-13.9) and a 12-month overall survival rate of 48%.
Predicting response to chemotherapy
The prognostic nomogram called Sarculator was used effectively to define a high-risk subgroup of patients likely to benefit from adjuvant chemotherapy, Sandro Pasquali, MD, of the Fondazione IRCCS Istituto Nazionale dei Tumori, Milano, Italy and his colleagues reported at the meeting.
Perioperative chemotherapy was shown to afford no survival advantage over observation in the EORTC 62931 (European Organization for Research and Treatment of Cancer—62931) study of adjuvant doxorubicin plus ifosfamide (Lancet Oncol 2012;13:1045-54). However, subsequent analyses of that data attributed this finding to variations in treatment schedules and the inclusion of low-risk tumors, which may have diluted the effect of chemotherapy, the researchers said in their abstract.
Further, a recent interim report of the ISG-1001 trial showed a survival benefit for patients who received neoadjuvant epirubicin plus ifosfamide therapy for localized high-risk soft-tissue sarcoma of the extremities or trunk wall (Lancet Oncol 2017;18:812-822).
The researchers performed a retrospective analysis of individual data for 290 patients with extremity and trunk wall soft-tissue sarcomas in the EORTC-STBSG 62931 study. The Sarculator was used to calculate 10-year predicted probability of overall survival (pr-OS) for each patient.
Patients were grouped in two categories of predicted overall survival: high predicted survival (over 60%) and low predicted overall survival (60% or less). Overall survival and disease-free survival were calculated at 8 years, the study’s median follow-up.
The 8-year probability of overall survival and disease-free survival was 58% [95% confidence interval (CI): 52–63%] and 51% (95% CI: 46–57%), respectively. In the 290 patients with extremity and trunk wall soft tissue sarcomas, adjuvant chemotherapy was not associated with an overall survival benefit [Hazard ratio (HR) = 0.91, 95%CI 0.63–1.31]. The Sarcolator Nomogram detected 80 patients who were at greater risk of death compared to the 210 patients with higher predicted overall survival. The risk of death was significantly lower with adjuvant chemotherapy in the group with low predicted survival based on the Sarculator Nomogram (HR=0.50, 95%CI 0.30-0.90). Consistently, the risk of recurrence was significantly lower when adjuvant chemotherapy was used in the group with predicted overall survival of less than 60% (HR = 0.49, 95%CI 0.28-0.85) while this difference was not observed in patients with high predicted overall survival (HR = 0.95, 95%CI 0.62-1.44).
Doxorubicin plus dacarbazine deserve evaluation in prospective trials in leiomyosarcoma
Doxorubicin plus dacarbazine appeared to best the outcomes seen with doxorubicin plus ifosfamide and with doxorubicin alone in terms of overall response rate and progression free survival as first-line treatment in patients with advanced leiomyosarcomas, based on a retrospective analysis presented by Lorenzo D’Ambrosio, MD, of the Unitversity of Torino, Italy, and his associates.
As patients in the trial were not randomized to therapy, the researchers used a logistic regression model that accounted for histology, site of primary, age, gender, performance status, tumor extent, and tumor grade. Patients were then matched across the different groups by their propensity scores.The 303 patients, 216 of them women, were enrolled from 18 EORTC STBSG (European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group) sites. Doxorubicin plus dacarbazine was given to 117 patients (39%), doxorubicin plus ifosfamide was given to 71 (23%), and doxorubicin alone was given to 115 (38%). There were no significant differences among the regimens in terms of dose reductions of more than 10%, delays of greater than 72 hours, or granulocyte-colony stimulating factor use.
In the whole population, unadjusted median progression free survival was 9.4 months (95% CI 6.1-9.7 months) for those given doxorubicin plus dacarbazine, 6.8 months (4.5-9.5 months) for those given doxorubicin plus ifosfamide), and 5.4 months (3.8-6.8 months) for those given doxorubicin alone. The respective overall response rates for the three regimens were 36.8%, 21.5%, and 25.9%. When using propensity scores to adjust for lack of randomization, progression free survival was significantly longer with doxorubicin plus dacarbazine [median 9.2 months (95%CI 5.2-9.7 months) than with doxorubicin [median 4.8 months (2.3-6.0 months); HR 0.72 (0.52-0.99)]. The difference was not significant when compared to doxorubicin plus ifosfamide [8.2 months (5.2-10.1 months), HR 1.01 (0.68-1.50)]. Progression free survival did not differ significantly between doxorubicin plus ifosfamide, and doxorubicin [HR 0.71 (0.48-1.06)]. In the same matched population, overall response rates were 30.9%, 19.5%, and 25.6% for doxorubicin plus dacarbazine, doxorubicin plus ifosfamide, and doxorubicin, respectively.
Overall survival comparisons were weakened by a shorter median follow-up in the doxorubicin plus dacarbazine groups (32 months) compared to the doxorubicin plus ifosfamide group (50 months) and the doxorubicin group (46 months). With this limit, patients in the doxorubicin plus dacarbazine arm had longer overall survival [median 36.8 (27.9-47.2) months] when compared to both doxorubicin plus ifosfamide [21.9 (16.7-33.4), HR 0.65 (0.40-1.06); and doxorubicin arms 30.3 (21.0-36.3) months, HR 0.66 (0.43-0.99).
Subsequent treatments were well balanced across arms. None of the selected factors for multivariate analysis (age, sex, ECOG performance status, histotype, site of primary tumor, tumor grade, and tumor extent) significantly affected the progression free survival and overall survival associated with the treatments.
Olaratumab in combination with doxorubicin plus ifosfamide
Olaratumab at 15 mg/kg has been shown to be safe in combination with doxorubicin plus ifosfamide in a Phase 1b study (NCT03283696), reported Sebastian Bauer, MD, of the West German Cancer Center, University of Duisburg-Essen, Essen, Germany, and his colleagues.
Given that 8 of 10 evaluable patients have completed the drug-limiting toxicity period without drug-limiting toxicities at the 15 mg/kg dose level of olaratumab, the study has proceeded to the next cohort. In those patients, an olaratumab loading dose of 20 mg/kg will be evaluated in cycle 1, followed by 15 mg/kg of olaratumab in subsequent cycles with the same doses of doxorubicin plus ifosfamide, the researchers wrote in their abstract.
The phase 1 trial enrolled 16 patients with advanced or metastatic soft tissue sarcomas and no prior lines of systemic therapy and ECOG performance status 0-1. Adequate follow up data was available for 10 patients.
Olaratumab, (Lartruvo), which binds platelet-derived growth factor receptor alpha (PDGFRα), was given at 15 mg/kg in combination with doxorubicin (75 mg/m2 on days 1-3) and ifosfamide (10 g/m2 on days 1-4) followed by mandatory granulocyte-colony-stimulating factor therapy in cycles 1-6 on a 21-day cycle. Doxorubicin was allowed to be administered by continuous infusion or bolus administration and with cardiac protection. Mesna dosing was at least 60% of the ifosfamide dose.
Two of the 10 patients had dose-limiting toxicities; one had Grade 4 febrile neutropenia and the other had Grade 3 febrile neutropenia and Grade 3 mucositis. Common related adverse events occurring in over 30% of patients included fatigue, anemia, neutropenia, thrombocytopenia, constipation, and nausea. One patient discontinued study treatment due to progressive disease, and all others were on study treatment as of data cutoff. Among 7 patients evaluated for tumor response assessment, 3 patients had a partial response according to RECIST and 3 further patients had stabilized disease as best overall response for a disease control rate of 86%.
Anthracycline-based regimen excels in FIGO-1 uterine leiomyosarcoma
Future trials to assess the efficacy of adjuvant chemotherapy in uterine leiomyosarcoma should incorporate anthracyclines, according to Roberta Sanfilippo, MD, of Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues.
Disease-free survival was extended in patients with uterine leiomyosarcomas treated with anthracycline-based regimens as compared to gemcitabine and docetaxel, based on a retrospective analysis reported at the meeting by Dr. Sanfilippo.
They reviewed all patients with FIGO stage I uterine leiomyosarcomas who underwent hysterectomy with or without oophorectomy and were treated with adjuvant chemotherapy with either anthracycline-based or gemcitabine-based chemotherapy at two Italian centers.
Of 145 patients, 97 were treated with an anthracycline-based regimen and 48 with gemcitabine and docetaxel. The median number of cycles of anthracycline based regimen received was 4 (range 2-6) and with gemcitabine and docetaxel was 5 (range 3-7). Disease free survival was 31 months in patients treated with anthracycline-based chemotherapy and 19 months in patients treated with gemcitabine and docetaxel.
Trabectedin and low-dose radiotherapy
Trabectedin concurrent with low-dose radiotherapy is being examined as an option for patients with pulmonary metastatic soft tissue sarcoma (NCT02275286).
In a phase 1 study, long-lasting dimensional responses were seen in 71% of the irradiated lesions showed. Based on those results, trabectedin (Yondelis) at 1.5 mg/m 2 will be the recommended dose for phase 2, according to Javier Martín-Broto, MD, of the Institute of Biomedicine Research (IBIS)-University Hospital Virgen del Rocio/CSIC/University of Seville, Spain, and his colleagues.
For the study, trabectedin was given along with radiotherapy (30 Gy) in 10 fractions (3 Gy/fraction). Three dose levels of trabectedin were administered: -1 (1.1 mg/m 2), 1 (1.3 mg/m 2) and 2 (1.5 mg/m 2). Dose-limiting toxicity was defined as grade 3 or greater events excluding grade 3/4 neutropenia lasting less than 5 days, grade 3 transaminitis if it did not lead to trabectedin delay, and grade 3/4 nausea/vomiting due to inadequate prophylaxis.
Ten of the 18 patients enrolled had synovial sarcoma; 3 had undifferentiated pleomorphic sarcomas and the other patients had either myxoid liposarcoma, dedifferentiated liposarcoma, G3 not otherwise specified sarcoma, leiomyosarcoma, and malignant peripheral nerve sheath tumor.
Patients received a median of 1 prior line of chemotherapy (range: 0-3). Twelve patients received trabectedin at dose level 1 and 6 patients at dose level 2. Grade 3/4 adverse events were neutropenia, seen in 8 patients; alanine aminotransferase (ALT) elevation, seen in 2 patients; gamma-glutamyl transferase (GGT) elevation, seen in 2 patients; anemia, seen in 2 patients; febrile neutropenia, seen in 1 patient; and pneumonitis, seen in 1 patient.
There were two dose-limiting toxicities: transient grade 4 ALT elevation at the level 1 dose and grade 4 neutropenia for more than 5 days at the level 2 dose.
Based on central radiological review of 17 evaluable patients, 2 patients achieved complete response, 3 had partial responses, 6 had stable disease, and 6 had progressive disease. The local review reported complete responses in 2 patients, partial responses in 5, stable disease in 4, and progressive disease in 6.
On the irradiated lesions, 4 had complete responses, 8 had partial responses, 4 had stable disease, and 1 had progressive disease. With a median follow-up of 18 months, median progression-free survival was 2.83 months (95%CI: 2.3-3.3 months). Thirteen patients have died, with a median overall survival of 8.77 months (95%CI: 3.6-13.9) and a 12-month overall survival rate of 48%.
Reports from the annual meeting of The Connective Tissue Oncology Society held in Rome, November 14-17, 2018 Sarcoma of the Year: Intimal Sarcoma
This year’s annual meeting of The Connective Tissue Oncology Society brought new insights on intimal sarcoma. Four studies in a featured session at the meeting examined both current and novel treatments for this rare and aggressive cancer, and emphasized the need for new therapies.
Anthracycline-based regimens as preferred first-line therapies
Anthracycline-based regimens were the preferred first-line therapies used in 83 adults with intimal sarcomas in a retrospective study of data from the World Sarcoma Network, reported by Anna Maria Frezza, MD, of the, Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues. The researchers described the experience with anthracycline-based regimens as well as gemcitabine-based regimens and pazopanib among MDM2-positive patients with intimal sarcomas treated at 16 sarcoma reference centers in Europe, the United States, and Japan. Their findings speak to the need for new active drugs, which they said should target the MDM2 and CDK4 overexpression seen in patients with this rare sarcoma.Of the 83 patients studied, nearly all (76 patients) initially received an anthracycline-based regimen. Gemcitabine-based regimens were used in 29 patients and pazopanib in 10 patients; 20 of the 39 patients received more than one treatment.
Anthracycline-based regimens were associated with a 12-month progression-free survival rate of 38% in 76 patients with intimal sarcomas. All of the 76 patients received anthracycline regimens as their initial systemic therapy; 27 were treated for localized disease with a curative intent and the remaining 49 had advanced disease. The researchers also noted that anthracycline regimens were safely used in 22 patients with cardiac intimal sarcomas, as none of them died of cardiotoxicity.
Based on RECIST 1.1 measures, the overall response rate was 37% in 57 evaluable patients: 3 patients had a complete response, 18 had a partial response, 27 had stable disease, and 9 had progressive disease. For those with localized disease, the median time to progression was 14 months, and overall survival time was 51. For patients with advanced disease, the median time to progression was 8 months and overall survival was 22 months.
Outcomes were less favorable when patients were treated with gemcitabine regimens or pazopanib. In most of these cases, however, patients were either on their second (gemcitabine) or third (pazopanib) lines of therapy.
In the gemcitabine group, 2 patients were treated for localized disease with curative intent and 27 for advanced disease. Of 28 evaluable patients, best response was partial remission in 3, stable disease in 8, and progressive disease in 17. In the 27 patients with advanced disease, the median progression free survival time was 3 months and overall survival was 13 months.
All 10 patients in the pazopanib group had advanced disease and had undergone a median of two prior lines of therapy. One patient had a partial remission, 3 had stable disease, and 6 had progressive disease. The median progression free survival was 4 months and median overall survival was 12 months.
Rarest of the rare: Primary malignant sarcoma of the heart
Luke Smith, of the School of Clinical Medicine, University of Cambridge, U.K., detailed the experience of 28 patients diagnosed with sarcomas of the heart or great vessels at the university’s Royal Papworth Hospital and Addenbrooke’s Hospital between 2000 and 2018.
Based on this retrospective review, surgery offers the best chance for long-term survival for these patients, who would otherwise experience progressive heart failure and die. Adjuvant chemotherapy and radiation therapy might be able to extend their survival and improve symptomatic relief, he said, but these outcomes have not been prospectively studied.
Typically, the patients in this series, 20 with pulmonary artery sarcoma and 8 with cardiac sarcoma, presented with symptoms mimicking heart failure, pulmonary hypertension, or thromboembolic disease. Nearly all, 24 patients reported breathlessness. Eight patients had chest pain or tightness, 6 had cough, 6 had peripheral edema, 6 had constitutional symptoms, 3 had hemoptysis, and 1 had a TIA. Only 1 patient had a seriously impaired left ventricular ejection fraction of less than 30%. LVEF was normal at 55% or more in 16 patients, and moderately impaired at 30% or more in 10 patients.
Median overall survival was 17 months. The 19 patients who underwent surgical resection of their primary tumor survived much longer than the 10 patients who did not--median overall survival of 20 months vs. 9 months--but this finding may simply reflect more advanced disease in patients with inoperable disease. There were 3 perioperative deaths among the 19 patients who underwent surgery: 14 with pulmonary artery sarcomas had pulmonary endarterectomy and 4 with cardiac sarcomas underwent resection or maximal debulking of their tumors.
Based on the retrospective study, adjuvant chemotherapy and radiation were safe and may lead to better outcomes for these patients. Active chemotherapy regimens in the palliative setting included paclitaxel (angiosarcoma) and anthracycline ± ifosfamide.
Nine patients received post-surgical chemotherapy, and after completion five also had radiotherapy. The 3 cardiac sarcoma patients who had surgical resection with curative intent were treated with adjuvant ifosfamide-based chemotherapy (with close monitoring of fluid balance), and showed no evidence of disease on last follow-ups. One patient received post-operative paclitaxel following maximal debulking of a cardiac angiosarcoma.
Post-surgical anthracycline with and without ifosfamide were used in patients with pulmonary artery sarcomas with no clinical cardiotoxicity. Although the median overall survival for patients who received post-operative chemo- and radio-therapy was 28 months and the median overall survival with surgery alone was 9 months, the difference was not statistically significant.
In the palliative setting, partial responses were observed with paclitaxel and anthracycline (including liposomal doxorubicin) in patients with cardiac angiosarcoma. For pulmonary artery intimal sarcomas, partial responses were achieved with anthracycline with and without ifosfamide. Radiotherapy provided good local control.
The longest surviving pulmonary artery sarcoma patient, at 103 months, had pulmonary artery endarterectomy, followed by adjuvant epirubicin and radiotherapy. She developed lung metastases 7 years later and was treated with radiofrequency ablation. The longest surviving cardiac sarcoma patient, at 24 months, remains disease free. He had surgery to resect a high-grade undifferentiated sarcoma with involved margins, followed by adjuvant ifosfamide and radiotherapy to the right atrium.
Therapeutically exploitable genetic aberrations in intimal sarcomas
Imatinib and olaratumab might prove to be therapeutic approaches for some patients with intimal sarcomas, based on a retrospective evaluation of genetic aberrations in 11 patients with intimal sarcomas, Jason Roszik, PhD, MBA, reported at the meeting.
Dr. Roszik and his colleagues at the University of Texas MD Anderson Cancer Center, Houston, analyzed information on 11 patients with intimal sarcomas in the American Association for Cancer Research (AACR) project, Genomics Evidence Neoplasia Information Exchange (GENIE). Sampling was taken from the primary tumor in 8 patients and from the metastatic site in the other 3.
MDM2 amplifications were seen in 8 of 10 patients with available copy number alterations. Amplifications in the CDK pathway were present in 5, PDGFRA gain was seen in 4, and CDKN2A copy number loss was present in 3. Mutations that could be targeted with drugs included ALK, ATM/ATR, PTCH1 and PDGFRB, he said.
Unique genomic rearrangement events included PDE4DIP-NOTCH2 and MRPS30-ARID2 fusions. Co-occurring alterations included a NOTCH2 copy number gain in the PDE4DIP-NOTCH2 fusion tumor, and PDGFRB mutations in both fusion-positive cases.
The researchers also drew on the published findings of whole-exome sequencing and array-comparative genomic hybridization from an autopsy case of cardiac intimal sarcoma (Virchows Arch. 2017 Sep;471(3):423-428). That study identified concurrent PDGFRA amplification and PDGFRB mutation.
The researchers additionally examined clinical trial enrollments and could find no patient with intimal sarcoma among 406 sarcoma enrolled patients. Intimal sarcomas were not eligible for any clinical trial given the location of the tumors in major blood vessels.
“The somatic mutations and DNA copy number alterations in the PDGFR pathway relevant to the pathogenesis and potential targeted therapy of cardiac intimal sarcoma may be targeted by imatinib or olaratumab. Inclusion of such rare tumors in targeted therapy basket trials with a waiver for inclusion criteria is warranted,” Dr. Roszik and his colleagues concluded in the abstract of their presentation.
The promise of combination therapy
The “largest experience using multimodality therapy with proton based local therapy” for sarcomas involving the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries was reported by Yen-Lin E. Chen, MD, and her colleagues at Massachusetts General Hospital, Boston.
They examined an institutional sarcoma data repository of 13,950 patients and found 37 patients with sarcomas arising from the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries. These included 9 with unclassified pleomorphic sarcoma/malignant fibrous histiocytoma, 8 with angiosarcoma, 4 with spindle cell sarcoma, 4 with sarcoma not otherwise specified, 3 with leiomyosarcoma, 2 with osteosarcoma, 2 with Ewing sarcoma, and 1 each with chondrosarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, synovial sarcoma, and intimal sarcoma.
Two-thirds of the patients had induction chemotherapy with or without maintenance therapy. Adriamycin, ifosfamide, and taxol therapies were most common. Two-thirds received proton based radiotherapy. Of the 23 patients who underwent resection, 11 were R2 (macroscopic positive margins), 3 were R1 (microscopic positive margins), and 9 were R0 (clear margins).
The 1-year overall survival rate was 64%, which fell to 37% at 3 years and to 28% at 5 years. Median survival was 28 months, twice that typically seen in the literature, Dr. Chen said.
For patients receiving proton based radiotherapy to a median dose of 64.8 GyRBE (range 63-72 GyRBE, 3 with additional intraoperative electrons), local failure free survivals were 80%, 64%, and 52% at 1, 3, and 5 years, respectively. For patients who did not receive radiotherapy, local failure free survival rates were 13%, 10%, 10%, respectively.
Overall, the 1, 3, and 5 year metastatic free survival rates were 25%, 14%, and 14%.
Survival rate was significantly better for patients with tumors smaller than 5 cm ( P =0.036), those over 40 years old ( P =0.028), those able to have surgery ( P =0.011), and those with non-angiosarcoma histologies ( P = 0.002).
This year’s annual meeting of The Connective Tissue Oncology Society brought new insights on intimal sarcoma. Four studies in a featured session at the meeting examined both current and novel treatments for this rare and aggressive cancer, and emphasized the need for new therapies.
Anthracycline-based regimens as preferred first-line therapies
Anthracycline-based regimens were the preferred first-line therapies used in 83 adults with intimal sarcomas in a retrospective study of data from the World Sarcoma Network, reported by Anna Maria Frezza, MD, of the, Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues. The researchers described the experience with anthracycline-based regimens as well as gemcitabine-based regimens and pazopanib among MDM2-positive patients with intimal sarcomas treated at 16 sarcoma reference centers in Europe, the United States, and Japan. Their findings speak to the need for new active drugs, which they said should target the MDM2 and CDK4 overexpression seen in patients with this rare sarcoma.Of the 83 patients studied, nearly all (76 patients) initially received an anthracycline-based regimen. Gemcitabine-based regimens were used in 29 patients and pazopanib in 10 patients; 20 of the 39 patients received more than one treatment.
Anthracycline-based regimens were associated with a 12-month progression-free survival rate of 38% in 76 patients with intimal sarcomas. All of the 76 patients received anthracycline regimens as their initial systemic therapy; 27 were treated for localized disease with a curative intent and the remaining 49 had advanced disease. The researchers also noted that anthracycline regimens were safely used in 22 patients with cardiac intimal sarcomas, as none of them died of cardiotoxicity.
Based on RECIST 1.1 measures, the overall response rate was 37% in 57 evaluable patients: 3 patients had a complete response, 18 had a partial response, 27 had stable disease, and 9 had progressive disease. For those with localized disease, the median time to progression was 14 months, and overall survival time was 51. For patients with advanced disease, the median time to progression was 8 months and overall survival was 22 months.
Outcomes were less favorable when patients were treated with gemcitabine regimens or pazopanib. In most of these cases, however, patients were either on their second (gemcitabine) or third (pazopanib) lines of therapy.
In the gemcitabine group, 2 patients were treated for localized disease with curative intent and 27 for advanced disease. Of 28 evaluable patients, best response was partial remission in 3, stable disease in 8, and progressive disease in 17. In the 27 patients with advanced disease, the median progression free survival time was 3 months and overall survival was 13 months.
All 10 patients in the pazopanib group had advanced disease and had undergone a median of two prior lines of therapy. One patient had a partial remission, 3 had stable disease, and 6 had progressive disease. The median progression free survival was 4 months and median overall survival was 12 months.
Rarest of the rare: Primary malignant sarcoma of the heart
Luke Smith, of the School of Clinical Medicine, University of Cambridge, U.K., detailed the experience of 28 patients diagnosed with sarcomas of the heart or great vessels at the university’s Royal Papworth Hospital and Addenbrooke’s Hospital between 2000 and 2018.
Based on this retrospective review, surgery offers the best chance for long-term survival for these patients, who would otherwise experience progressive heart failure and die. Adjuvant chemotherapy and radiation therapy might be able to extend their survival and improve symptomatic relief, he said, but these outcomes have not been prospectively studied.
Typically, the patients in this series, 20 with pulmonary artery sarcoma and 8 with cardiac sarcoma, presented with symptoms mimicking heart failure, pulmonary hypertension, or thromboembolic disease. Nearly all, 24 patients reported breathlessness. Eight patients had chest pain or tightness, 6 had cough, 6 had peripheral edema, 6 had constitutional symptoms, 3 had hemoptysis, and 1 had a TIA. Only 1 patient had a seriously impaired left ventricular ejection fraction of less than 30%. LVEF was normal at 55% or more in 16 patients, and moderately impaired at 30% or more in 10 patients.
Median overall survival was 17 months. The 19 patients who underwent surgical resection of their primary tumor survived much longer than the 10 patients who did not--median overall survival of 20 months vs. 9 months--but this finding may simply reflect more advanced disease in patients with inoperable disease. There were 3 perioperative deaths among the 19 patients who underwent surgery: 14 with pulmonary artery sarcomas had pulmonary endarterectomy and 4 with cardiac sarcomas underwent resection or maximal debulking of their tumors.
Based on the retrospective study, adjuvant chemotherapy and radiation were safe and may lead to better outcomes for these patients. Active chemotherapy regimens in the palliative setting included paclitaxel (angiosarcoma) and anthracycline ± ifosfamide.
Nine patients received post-surgical chemotherapy, and after completion five also had radiotherapy. The 3 cardiac sarcoma patients who had surgical resection with curative intent were treated with adjuvant ifosfamide-based chemotherapy (with close monitoring of fluid balance), and showed no evidence of disease on last follow-ups. One patient received post-operative paclitaxel following maximal debulking of a cardiac angiosarcoma.
Post-surgical anthracycline with and without ifosfamide were used in patients with pulmonary artery sarcomas with no clinical cardiotoxicity. Although the median overall survival for patients who received post-operative chemo- and radio-therapy was 28 months and the median overall survival with surgery alone was 9 months, the difference was not statistically significant.
In the palliative setting, partial responses were observed with paclitaxel and anthracycline (including liposomal doxorubicin) in patients with cardiac angiosarcoma. For pulmonary artery intimal sarcomas, partial responses were achieved with anthracycline with and without ifosfamide. Radiotherapy provided good local control.
The longest surviving pulmonary artery sarcoma patient, at 103 months, had pulmonary artery endarterectomy, followed by adjuvant epirubicin and radiotherapy. She developed lung metastases 7 years later and was treated with radiofrequency ablation. The longest surviving cardiac sarcoma patient, at 24 months, remains disease free. He had surgery to resect a high-grade undifferentiated sarcoma with involved margins, followed by adjuvant ifosfamide and radiotherapy to the right atrium.
Therapeutically exploitable genetic aberrations in intimal sarcomas
Imatinib and olaratumab might prove to be therapeutic approaches for some patients with intimal sarcomas, based on a retrospective evaluation of genetic aberrations in 11 patients with intimal sarcomas, Jason Roszik, PhD, MBA, reported at the meeting.
Dr. Roszik and his colleagues at the University of Texas MD Anderson Cancer Center, Houston, analyzed information on 11 patients with intimal sarcomas in the American Association for Cancer Research (AACR) project, Genomics Evidence Neoplasia Information Exchange (GENIE). Sampling was taken from the primary tumor in 8 patients and from the metastatic site in the other 3.
MDM2 amplifications were seen in 8 of 10 patients with available copy number alterations. Amplifications in the CDK pathway were present in 5, PDGFRA gain was seen in 4, and CDKN2A copy number loss was present in 3. Mutations that could be targeted with drugs included ALK, ATM/ATR, PTCH1 and PDGFRB, he said.
Unique genomic rearrangement events included PDE4DIP-NOTCH2 and MRPS30-ARID2 fusions. Co-occurring alterations included a NOTCH2 copy number gain in the PDE4DIP-NOTCH2 fusion tumor, and PDGFRB mutations in both fusion-positive cases.
The researchers also drew on the published findings of whole-exome sequencing and array-comparative genomic hybridization from an autopsy case of cardiac intimal sarcoma (Virchows Arch. 2017 Sep;471(3):423-428). That study identified concurrent PDGFRA amplification and PDGFRB mutation.
The researchers additionally examined clinical trial enrollments and could find no patient with intimal sarcoma among 406 sarcoma enrolled patients. Intimal sarcomas were not eligible for any clinical trial given the location of the tumors in major blood vessels.
“The somatic mutations and DNA copy number alterations in the PDGFR pathway relevant to the pathogenesis and potential targeted therapy of cardiac intimal sarcoma may be targeted by imatinib or olaratumab. Inclusion of such rare tumors in targeted therapy basket trials with a waiver for inclusion criteria is warranted,” Dr. Roszik and his colleagues concluded in the abstract of their presentation.
The promise of combination therapy
The “largest experience using multimodality therapy with proton based local therapy” for sarcomas involving the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries was reported by Yen-Lin E. Chen, MD, and her colleagues at Massachusetts General Hospital, Boston.
They examined an institutional sarcoma data repository of 13,950 patients and found 37 patients with sarcomas arising from the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries. These included 9 with unclassified pleomorphic sarcoma/malignant fibrous histiocytoma, 8 with angiosarcoma, 4 with spindle cell sarcoma, 4 with sarcoma not otherwise specified, 3 with leiomyosarcoma, 2 with osteosarcoma, 2 with Ewing sarcoma, and 1 each with chondrosarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, synovial sarcoma, and intimal sarcoma.
Two-thirds of the patients had induction chemotherapy with or without maintenance therapy. Adriamycin, ifosfamide, and taxol therapies were most common. Two-thirds received proton based radiotherapy. Of the 23 patients who underwent resection, 11 were R2 (macroscopic positive margins), 3 were R1 (microscopic positive margins), and 9 were R0 (clear margins).
The 1-year overall survival rate was 64%, which fell to 37% at 3 years and to 28% at 5 years. Median survival was 28 months, twice that typically seen in the literature, Dr. Chen said.
For patients receiving proton based radiotherapy to a median dose of 64.8 GyRBE (range 63-72 GyRBE, 3 with additional intraoperative electrons), local failure free survivals were 80%, 64%, and 52% at 1, 3, and 5 years, respectively. For patients who did not receive radiotherapy, local failure free survival rates were 13%, 10%, 10%, respectively.
Overall, the 1, 3, and 5 year metastatic free survival rates were 25%, 14%, and 14%.
Survival rate was significantly better for patients with tumors smaller than 5 cm ( P =0.036), those over 40 years old ( P =0.028), those able to have surgery ( P =0.011), and those with non-angiosarcoma histologies ( P = 0.002).
This year’s annual meeting of The Connective Tissue Oncology Society brought new insights on intimal sarcoma. Four studies in a featured session at the meeting examined both current and novel treatments for this rare and aggressive cancer, and emphasized the need for new therapies.
Anthracycline-based regimens as preferred first-line therapies
Anthracycline-based regimens were the preferred first-line therapies used in 83 adults with intimal sarcomas in a retrospective study of data from the World Sarcoma Network, reported by Anna Maria Frezza, MD, of the, Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues. The researchers described the experience with anthracycline-based regimens as well as gemcitabine-based regimens and pazopanib among MDM2-positive patients with intimal sarcomas treated at 16 sarcoma reference centers in Europe, the United States, and Japan. Their findings speak to the need for new active drugs, which they said should target the MDM2 and CDK4 overexpression seen in patients with this rare sarcoma.Of the 83 patients studied, nearly all (76 patients) initially received an anthracycline-based regimen. Gemcitabine-based regimens were used in 29 patients and pazopanib in 10 patients; 20 of the 39 patients received more than one treatment.
Anthracycline-based regimens were associated with a 12-month progression-free survival rate of 38% in 76 patients with intimal sarcomas. All of the 76 patients received anthracycline regimens as their initial systemic therapy; 27 were treated for localized disease with a curative intent and the remaining 49 had advanced disease. The researchers also noted that anthracycline regimens were safely used in 22 patients with cardiac intimal sarcomas, as none of them died of cardiotoxicity.
Based on RECIST 1.1 measures, the overall response rate was 37% in 57 evaluable patients: 3 patients had a complete response, 18 had a partial response, 27 had stable disease, and 9 had progressive disease. For those with localized disease, the median time to progression was 14 months, and overall survival time was 51. For patients with advanced disease, the median time to progression was 8 months and overall survival was 22 months.
Outcomes were less favorable when patients were treated with gemcitabine regimens or pazopanib. In most of these cases, however, patients were either on their second (gemcitabine) or third (pazopanib) lines of therapy.
In the gemcitabine group, 2 patients were treated for localized disease with curative intent and 27 for advanced disease. Of 28 evaluable patients, best response was partial remission in 3, stable disease in 8, and progressive disease in 17. In the 27 patients with advanced disease, the median progression free survival time was 3 months and overall survival was 13 months.
All 10 patients in the pazopanib group had advanced disease and had undergone a median of two prior lines of therapy. One patient had a partial remission, 3 had stable disease, and 6 had progressive disease. The median progression free survival was 4 months and median overall survival was 12 months.
Rarest of the rare: Primary malignant sarcoma of the heart
Luke Smith, of the School of Clinical Medicine, University of Cambridge, U.K., detailed the experience of 28 patients diagnosed with sarcomas of the heart or great vessels at the university’s Royal Papworth Hospital and Addenbrooke’s Hospital between 2000 and 2018.
Based on this retrospective review, surgery offers the best chance for long-term survival for these patients, who would otherwise experience progressive heart failure and die. Adjuvant chemotherapy and radiation therapy might be able to extend their survival and improve symptomatic relief, he said, but these outcomes have not been prospectively studied.
Typically, the patients in this series, 20 with pulmonary artery sarcoma and 8 with cardiac sarcoma, presented with symptoms mimicking heart failure, pulmonary hypertension, or thromboembolic disease. Nearly all, 24 patients reported breathlessness. Eight patients had chest pain or tightness, 6 had cough, 6 had peripheral edema, 6 had constitutional symptoms, 3 had hemoptysis, and 1 had a TIA. Only 1 patient had a seriously impaired left ventricular ejection fraction of less than 30%. LVEF was normal at 55% or more in 16 patients, and moderately impaired at 30% or more in 10 patients.
Median overall survival was 17 months. The 19 patients who underwent surgical resection of their primary tumor survived much longer than the 10 patients who did not--median overall survival of 20 months vs. 9 months--but this finding may simply reflect more advanced disease in patients with inoperable disease. There were 3 perioperative deaths among the 19 patients who underwent surgery: 14 with pulmonary artery sarcomas had pulmonary endarterectomy and 4 with cardiac sarcomas underwent resection or maximal debulking of their tumors.
Based on the retrospective study, adjuvant chemotherapy and radiation were safe and may lead to better outcomes for these patients. Active chemotherapy regimens in the palliative setting included paclitaxel (angiosarcoma) and anthracycline ± ifosfamide.
Nine patients received post-surgical chemotherapy, and after completion five also had radiotherapy. The 3 cardiac sarcoma patients who had surgical resection with curative intent were treated with adjuvant ifosfamide-based chemotherapy (with close monitoring of fluid balance), and showed no evidence of disease on last follow-ups. One patient received post-operative paclitaxel following maximal debulking of a cardiac angiosarcoma.
Post-surgical anthracycline with and without ifosfamide were used in patients with pulmonary artery sarcomas with no clinical cardiotoxicity. Although the median overall survival for patients who received post-operative chemo- and radio-therapy was 28 months and the median overall survival with surgery alone was 9 months, the difference was not statistically significant.
In the palliative setting, partial responses were observed with paclitaxel and anthracycline (including liposomal doxorubicin) in patients with cardiac angiosarcoma. For pulmonary artery intimal sarcomas, partial responses were achieved with anthracycline with and without ifosfamide. Radiotherapy provided good local control.
The longest surviving pulmonary artery sarcoma patient, at 103 months, had pulmonary artery endarterectomy, followed by adjuvant epirubicin and radiotherapy. She developed lung metastases 7 years later and was treated with radiofrequency ablation. The longest surviving cardiac sarcoma patient, at 24 months, remains disease free. He had surgery to resect a high-grade undifferentiated sarcoma with involved margins, followed by adjuvant ifosfamide and radiotherapy to the right atrium.
Therapeutically exploitable genetic aberrations in intimal sarcomas
Imatinib and olaratumab might prove to be therapeutic approaches for some patients with intimal sarcomas, based on a retrospective evaluation of genetic aberrations in 11 patients with intimal sarcomas, Jason Roszik, PhD, MBA, reported at the meeting.
Dr. Roszik and his colleagues at the University of Texas MD Anderson Cancer Center, Houston, analyzed information on 11 patients with intimal sarcomas in the American Association for Cancer Research (AACR) project, Genomics Evidence Neoplasia Information Exchange (GENIE). Sampling was taken from the primary tumor in 8 patients and from the metastatic site in the other 3.
MDM2 amplifications were seen in 8 of 10 patients with available copy number alterations. Amplifications in the CDK pathway were present in 5, PDGFRA gain was seen in 4, and CDKN2A copy number loss was present in 3. Mutations that could be targeted with drugs included ALK, ATM/ATR, PTCH1 and PDGFRB, he said.
Unique genomic rearrangement events included PDE4DIP-NOTCH2 and MRPS30-ARID2 fusions. Co-occurring alterations included a NOTCH2 copy number gain in the PDE4DIP-NOTCH2 fusion tumor, and PDGFRB mutations in both fusion-positive cases.
The researchers also drew on the published findings of whole-exome sequencing and array-comparative genomic hybridization from an autopsy case of cardiac intimal sarcoma (Virchows Arch. 2017 Sep;471(3):423-428). That study identified concurrent PDGFRA amplification and PDGFRB mutation.
The researchers additionally examined clinical trial enrollments and could find no patient with intimal sarcoma among 406 sarcoma enrolled patients. Intimal sarcomas were not eligible for any clinical trial given the location of the tumors in major blood vessels.
“The somatic mutations and DNA copy number alterations in the PDGFR pathway relevant to the pathogenesis and potential targeted therapy of cardiac intimal sarcoma may be targeted by imatinib or olaratumab. Inclusion of such rare tumors in targeted therapy basket trials with a waiver for inclusion criteria is warranted,” Dr. Roszik and his colleagues concluded in the abstract of their presentation.
The promise of combination therapy
The “largest experience using multimodality therapy with proton based local therapy” for sarcomas involving the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries was reported by Yen-Lin E. Chen, MD, and her colleagues at Massachusetts General Hospital, Boston.
They examined an institutional sarcoma data repository of 13,950 patients and found 37 patients with sarcomas arising from the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries. These included 9 with unclassified pleomorphic sarcoma/malignant fibrous histiocytoma, 8 with angiosarcoma, 4 with spindle cell sarcoma, 4 with sarcoma not otherwise specified, 3 with leiomyosarcoma, 2 with osteosarcoma, 2 with Ewing sarcoma, and 1 each with chondrosarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, synovial sarcoma, and intimal sarcoma.
Two-thirds of the patients had induction chemotherapy with or without maintenance therapy. Adriamycin, ifosfamide, and taxol therapies were most common. Two-thirds received proton based radiotherapy. Of the 23 patients who underwent resection, 11 were R2 (macroscopic positive margins), 3 were R1 (microscopic positive margins), and 9 were R0 (clear margins).
The 1-year overall survival rate was 64%, which fell to 37% at 3 years and to 28% at 5 years. Median survival was 28 months, twice that typically seen in the literature, Dr. Chen said.
For patients receiving proton based radiotherapy to a median dose of 64.8 GyRBE (range 63-72 GyRBE, 3 with additional intraoperative electrons), local failure free survivals were 80%, 64%, and 52% at 1, 3, and 5 years, respectively. For patients who did not receive radiotherapy, local failure free survival rates were 13%, 10%, 10%, respectively.
Overall, the 1, 3, and 5 year metastatic free survival rates were 25%, 14%, and 14%.
Survival rate was significantly better for patients with tumors smaller than 5 cm ( P =0.036), those over 40 years old ( P =0.028), those able to have surgery ( P =0.011), and those with non-angiosarcoma histologies ( P = 0.002).
FDA approves larotrectinib for cancers with NTRK gene fusions
for adult and pediatric patients with solid tumors that harbor a genetic aberration known as an neurotrophic receptor tyrosine kinase (NTRK) fusion.
Specifically, the oral inhibitor of tropomyosin receptor kinase is approved for patients with solid tumors that have a NTRK gene fusion without a known acquired resistance mutation and have metastatic disease, are likely to experience severe morbidity from surgical resection, have no satisfactory alternative treatments, or have cancer that has progressed following treatment, the FDA said in a press release.
NTRK fusions are found in both children and adults with dozens of different cancer types. The genetic aberration tends to be rare in common cancers and nearly universal in certain uncommon cancers.
Approval of larotrectinib (Vitrakvi) was based on overall response rate and response duration in the first 55 patients with unresectable or metastatic solid tumors harboring an NTRK gene fusion enrolled across three multicenter, open-label, single-arm clinical trials. The ORR was 75% (95% confidence interval, 61%-85%), including 22% complete responses and 53% partial responses. At the time of database lock, median duration of response had not been reached, the FDA said.
The most common tumors were salivary gland tumors (22%), soft tissue sarcomas (20%), infantile fibrosarcomas (13%), and thyroid cancers (9%). Identification of positive NTRK gene fusion status was prospectively determined in local laboratories using next-generation sequencing or fluorescence in situ hybridization.
Results of the three trials, a phase 1 trial among 8 adult patients (LOXO-TRK-14001), a phase 1/2 trial among 12 pediatric patients (SCOUT), and a phase 2 basket trial among 35 adult and adolescent patients (NAVIGATE), were presented at the annual meeting of the American Society of Clinical Oncology in 2017.
The safety of larotrectinib was evaluated in 176 patients enrolled across the three clinical trials, including 44 pediatric patients. The most common adverse reactions with larotrectinib were fatigue, nausea, dizziness, vomiting, increased AST, cough, increased ALT, constipation, and diarrhea, the FDA said.
The recommended larotrectinib doses are 100 mg orally twice daily for adults and 100 mg/m2 orally twice daily (maximum of 100 mg per dose) for pediatric patients.
The approval of larotrectinib follows the approval of pembrolizumab for certain solid tumors with the MSI-H biomarker, as the first approval for the treatment of cancer based on a biomarker rather than the particular body part or organ system affected by the tumor.
The FDA granted the accelerated approval of Vitrakvi to Loxo Oncology and Bayer.
for adult and pediatric patients with solid tumors that harbor a genetic aberration known as an neurotrophic receptor tyrosine kinase (NTRK) fusion.
Specifically, the oral inhibitor of tropomyosin receptor kinase is approved for patients with solid tumors that have a NTRK gene fusion without a known acquired resistance mutation and have metastatic disease, are likely to experience severe morbidity from surgical resection, have no satisfactory alternative treatments, or have cancer that has progressed following treatment, the FDA said in a press release.
NTRK fusions are found in both children and adults with dozens of different cancer types. The genetic aberration tends to be rare in common cancers and nearly universal in certain uncommon cancers.
Approval of larotrectinib (Vitrakvi) was based on overall response rate and response duration in the first 55 patients with unresectable or metastatic solid tumors harboring an NTRK gene fusion enrolled across three multicenter, open-label, single-arm clinical trials. The ORR was 75% (95% confidence interval, 61%-85%), including 22% complete responses and 53% partial responses. At the time of database lock, median duration of response had not been reached, the FDA said.
The most common tumors were salivary gland tumors (22%), soft tissue sarcomas (20%), infantile fibrosarcomas (13%), and thyroid cancers (9%). Identification of positive NTRK gene fusion status was prospectively determined in local laboratories using next-generation sequencing or fluorescence in situ hybridization.
Results of the three trials, a phase 1 trial among 8 adult patients (LOXO-TRK-14001), a phase 1/2 trial among 12 pediatric patients (SCOUT), and a phase 2 basket trial among 35 adult and adolescent patients (NAVIGATE), were presented at the annual meeting of the American Society of Clinical Oncology in 2017.
The safety of larotrectinib was evaluated in 176 patients enrolled across the three clinical trials, including 44 pediatric patients. The most common adverse reactions with larotrectinib were fatigue, nausea, dizziness, vomiting, increased AST, cough, increased ALT, constipation, and diarrhea, the FDA said.
The recommended larotrectinib doses are 100 mg orally twice daily for adults and 100 mg/m2 orally twice daily (maximum of 100 mg per dose) for pediatric patients.
The approval of larotrectinib follows the approval of pembrolizumab for certain solid tumors with the MSI-H biomarker, as the first approval for the treatment of cancer based on a biomarker rather than the particular body part or organ system affected by the tumor.
The FDA granted the accelerated approval of Vitrakvi to Loxo Oncology and Bayer.
for adult and pediatric patients with solid tumors that harbor a genetic aberration known as an neurotrophic receptor tyrosine kinase (NTRK) fusion.
Specifically, the oral inhibitor of tropomyosin receptor kinase is approved for patients with solid tumors that have a NTRK gene fusion without a known acquired resistance mutation and have metastatic disease, are likely to experience severe morbidity from surgical resection, have no satisfactory alternative treatments, or have cancer that has progressed following treatment, the FDA said in a press release.
NTRK fusions are found in both children and adults with dozens of different cancer types. The genetic aberration tends to be rare in common cancers and nearly universal in certain uncommon cancers.
Approval of larotrectinib (Vitrakvi) was based on overall response rate and response duration in the first 55 patients with unresectable or metastatic solid tumors harboring an NTRK gene fusion enrolled across three multicenter, open-label, single-arm clinical trials. The ORR was 75% (95% confidence interval, 61%-85%), including 22% complete responses and 53% partial responses. At the time of database lock, median duration of response had not been reached, the FDA said.
The most common tumors were salivary gland tumors (22%), soft tissue sarcomas (20%), infantile fibrosarcomas (13%), and thyroid cancers (9%). Identification of positive NTRK gene fusion status was prospectively determined in local laboratories using next-generation sequencing or fluorescence in situ hybridization.
Results of the three trials, a phase 1 trial among 8 adult patients (LOXO-TRK-14001), a phase 1/2 trial among 12 pediatric patients (SCOUT), and a phase 2 basket trial among 35 adult and adolescent patients (NAVIGATE), were presented at the annual meeting of the American Society of Clinical Oncology in 2017.
The safety of larotrectinib was evaluated in 176 patients enrolled across the three clinical trials, including 44 pediatric patients. The most common adverse reactions with larotrectinib were fatigue, nausea, dizziness, vomiting, increased AST, cough, increased ALT, constipation, and diarrhea, the FDA said.
The recommended larotrectinib doses are 100 mg orally twice daily for adults and 100 mg/m2 orally twice daily (maximum of 100 mg per dose) for pediatric patients.
The approval of larotrectinib follows the approval of pembrolizumab for certain solid tumors with the MSI-H biomarker, as the first approval for the treatment of cancer based on a biomarker rather than the particular body part or organ system affected by the tumor.
The FDA granted the accelerated approval of Vitrakvi to Loxo Oncology and Bayer.

Primary renal synovial sarcoma – a diagnostic dilemma
Soft tissue sarcomas are rare mesenchymal tumors that comprise 1% of all malignancies. Synovial sarcoma accounts for 5% to 10% of adult soft tissue sarcomas and usually occurs in close association with joint capsules, tendon sheaths, and bursa in the extremities of young and middle-aged adults.1 Synovial sarcomas have been reported in other unusual sites, including the head and neck, thoracic and abdominal wall, retroperitoneum, bone, pleura, and visceral organs such as the lung, prostate, or kidney.2 Primary renal synovial sarcoma is an extremely rare tumor accounting for <2% of all malignant renal tumors.3 To the best of our knowledge, fewer than 50 cases of primary renal synovial sarcoma have been described in the English literature.4 It presents as a diagnostic dilemma because of the dearth of specific clinical and imaging findings and is often confused with benign and malignant tumors. The differential diagnosis includes angiomyolipoma, renal cell carcinoma with sarcomatoid differentiation, metastatic sarcoma, hemangiopericytoma, malignant solitary fibrous tumor, Wilms tumor, and malignant peripheral nerve sheath tumor. Hence, a combination of histomorphologic, immunohistochemical, cytogenetic, and molecular studies that show a unique chromosomal translocation t(X;18) (p11;q11) is imperative in the diagnosis of primary renal synovial sarcoma.4 In the present report, we present the case of a 38-year-old man who was diagnosed with primary renal synovial sarcoma.
Case presentation and summary
A 38-year-old man with a medical history of gastroesophageal reflux disease and Barrett’s esophagus presented to our hospital for the first time with persistent and progressive right-sided flank and abdominal pain that was aggravated after a minor trauma to the back. There was no associated hematuria or dysuria.
Of note is that he had experienced intermittent flank pain for 2 years before this transfer. He had initially been diagnosed at his local hospital close to his home by ultrasound with an angiomyolipoma of 2 × 3 cm arising from the upper pole of his right kidney, which remained stable on repeat sonograms. About 22 months after his initial presentation at his local hospital, the flank pain increased, and a computed-tomographic (CT) scan revealed a perinephric hematoma that was thought to originate from a ruptured angiomyolipoma. He subsequently underwent embolization, but his symptoms recurred soon after. He presented again to his local hospital where CT imaging revealed a significant increase in the size of the retroperitoneal mass, and findings were suggestive of a hematoma. Subsequent angiogram did not reveal active extravasation, so a biopsy was performed.
Before confirmatory pathologic evaluation could be completed, the patient presented to his local hospital again in excruciating pain. A CT scan of his abdomen and pelvis demonstrated a massive subacute on chronic hematoma in the right retroperitoneum measuring 22 × 19 × 18 cm, with calcifications originating from an upper pole right renal neoplasm. The right kidney was displaced antero-inferiorly, and the inferior vena cava was displaced anteriorly and to the left. The preliminary pathology returned with findings suggestive of sarcoma (Figures 1 and 2).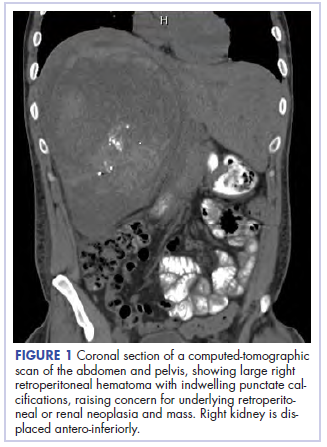
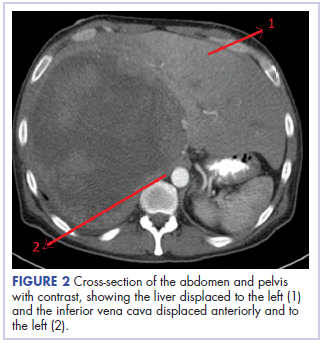
The patient was then transferred to our institution, where he was evaluated by medical and surgical oncology. A CT scan of the chest and magnetic-resonance imaging (MRI) of the brain did not reveal metastatic disease. He underwent exploratory laparotomy that involved the resection of a 22-cm retroperitoneal mass, right nephrectomy, right adrenalectomy, partial right hepatectomy, and a full thickness resection of the right postero-inferior diaphragm followed by mesh repair because of involvement by the tumor.
In its entirety, the specimen was a mass of 26 × 24 × 14 cm. It was sectioned to show extensively necrotic and hemorrhagic variegated white to tan-red parenchyma (Figure 3). Histology revealed a poorly differentiated malignant neoplasm composed of round cells with scant amphophilic cytoplasm arranged in solid, variably sized nests separated by prominent thin-walled branching vascular channels (Figure 4). The mitotic rate was high. It was determined to be a histologically ungraded sarcoma according to the French Federation of Comprehensive Cancer Centers system of grading soft tissue sarcomas; the margins were indeterminate. Immunohistochemistry was positive for EMA, TLE1, and negative for AE1/AE3, S100, STAT6, and Nkx2.2. Molecular pathology fluorescent in situ hybridization (FISH) analysis demonstrated positivity for SS18 gene rearrangement (SS18-SSX1 fusion).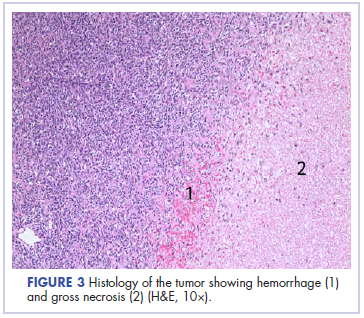
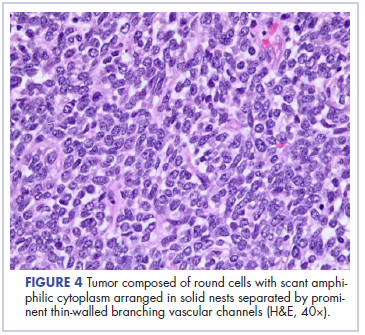
After recovering from surgery, the patient received adjuvant chemotherapy with doxorubicin and ifosfamide. It has been almost 16 months since we first saw this patient. He was started on doxorubicin 20 mg/m2 on days 1 to 4, ifosfamide 2,500 mg on days 1 to 4, and mesna 800 mg on days 1 to 4, for a total of 6 cycles. He did well for the first 5 months, after which he developed disease recurrence in the postoperative nephrectomy bed (a biopsy showed it to be recurrent synovial sarcoma) as well as pulmonary nodules, for which he was started on trabectedin 1.5 mg/m2 every 3 weeks. Two months later, a CT scan showed an increase in the size of his retroperitoneal mass, and the treatment was changed to pazopanib 400 mg daily orally, on which he remained at the time of publication.
Discussion
Synovial sarcoma is the fourth most common type of soft tissue sarcoma, accounting for 2.5% to 10.5% of all primary soft tissue malignancies worldwide. It occurs most frequently in adolescents and young adults, with most patients presenting between the ages of 15 and 40 years. Median age of presentation is 36 years. Despite the nomenclature, synovial sarcoma does not arise in intra-articular locations but typically occurs in proximity to joints in the extremities. Synovial sarcomas are less commonly described in other sites, including the head and neck, mediastinum, intraperitoneum, retroperitoneum, lung, pleura, and kidney.4,5 Renal synovial sarcoma was first described in a published article by Argani and colleagues in 2000.5
Adult renal mesenchymal tumors are classified into benign and malignant tumors on the basis of the histologic features and clinicobiologic behavior.6,7 The benign esenchymal renal tumors include angiomyolipoma, leiomyoma, hemangioma, lymphangioma, juxtaglomerular cell tumor, renomedullary interstitial cell tumor (medullary fibroma), lipoma, solitary fibrous tumor, and schwannoma. Malignant renal tumors of mesenchymal origin include leiomyosarcoma, rhabdomyosarcoma, angiosarcoma, osteosarcoma, fibrosarcoma, malignant fibrous histiocytoma, solitary fibrous tumor, and synovial sarcoma.
Most of these tumor types cause the same nonspecific symptoms in patients – abdominal pain, flank pain, abdominal fullness, a palpable mass, and hematuria – although they can be clinically silent. The average duration of symptoms in synovial sarcoma is 2 to 4 years.8 The long duration of symptoms and initial slow growth of synovial sarcomas may give a false impression of a benign process.
A preoperative radiological diagnosis of primary renal synovial sarcoma may be suspected by analyzing the tumor’s growth patterns on CT scans.9 Renal synovial sarcomas often appear as large, well-defined soft tissue masses that can extend into the renal pelvis or into the perinephric region.9 A CT scan may identify soft tissue calcifications, especially subtle ones in areas where the tumor anatomy is complex. A CT scan may also reveal areas of hemorrhage, necrosis, or cyst formation within the tumor, and can easily confirm bone involvement. Intravenous contrast may help in differentiating the mass from adjacent muscle and neurovascular complex.9,10 On MRI, renal synovial sarcomas are often described as nonspecific heterogeneous masses, although they may also exhibit heterogeneous enhancement of hemorrhagic areas, calcifications, and air-fluid levels (known as “triple sign”) as well as septae. The triple sign may be identified as areas of low, intermediate, and high signal intensity, correlating with areas of hemorrhage, calcification, and air-fluid level.9,10 Signal intensity is about equal to that of skeletal muscle on T1-weighted MRI and higher than that of subcutaneous fat on T2-weighted MRI.
In the present case, the tumor was initially misdiagnosed as an angiomyolipoma, the most common benign tumor of the kidney. Angiomyolipomas are usually solid triphasic tumors arising from the renal cortex and are composed of 3 major elements: dysmorphic blood vessels, smooth muscle components, and adipose tissue. When angiomyolipomas are large enough, they are readily recognized by the identification of macroscopic fat within the tumor, either by CT scan or MRI.11 When they are small, they may be difficult to distinguish from a small cyst on CT because of volume averaging.
On pathology, synovial sarcoma has dual epithelial and mesenchymal differentiation. They are frequently multi-lobulated, and areas of necrosis, hemorrhage, and cyst formation are also common. There are 3 main histologic subtypes of synovial sarcoma: biphasic (20%-30%), monophasic (50%-60%), and poorly differentiated (15%-25%). Poorly differentiated synovial sarcomas are generally epithelioid in morphology, have high mitotic activity (usually 10-20 mitoses/10 high-power field; range is <5 for well differentiated, low-grade tumors), and can be confused with round cell tumors such as Ewing sarcoma. Poorly differentiated synovial sarcomas are high-grade tumors.
Immunohistochemical studies can confirm the pathological diagnosis. Synovial sarcomas usually stain positive for Bcl2, CD99/Mic2, CD56, Vim, and focally for EMA but negatively for desmin, actin, WT1, S-100, CD34, and CD31.5 Currently, the gold standard for diagnosis and hallmark for synovial sarcomas are the t (X;18) translocation and SYT-SSX gene fusion products (SYT-SSX1 in 67% and SYT-SSX2 in 33% of cases). These can be detected either by FISH or reverse-transcription polymerase chain reaction. This genetic alteration is identified in more than 90% of synovial sarcomas and is highly specific.
The role of SYT-SSX gene fusion in the pathogenesis of synovial sarcoma is an active area of investigation. The fusion of SYT with SSX translates into a fusion protein that binds to the transcription activator SMARCA4 that is involved in chromatin remodeling, thus displacing both the wildtype SYT and the tumor suppressor gene SMARCB1. The modified protein complex then binds at several super-enhancer loci, unlocking suppressed genes such as Sox2, which is known to be necessary for synovial sarcoma proliferation. Alterations in SMARCB1 are involved in several cancer types, implicating this event as a driver of these malignancies.12 This results in a global alteration in chromatin remodeling that needs to be better understood to design targeted therapies.
The clinical course of synovial sarcoma, regardless of the tissue of origin, is typically poor. Multiple clinical and pathologic factors, including tumor size, location, patient age, and presence of poorly differentiated areas, are thought to have prognostic significance. A tumor size of more than 5 cm at presentation has the greatest impact on prognosis, with studies showing 5-year survival rates of 64% for patients with tumors smaller than 5 cm and 26% for patients with masses greater than 5 cm.13,14 High-grade synovial sarcoma is favored in tumors that have cystic components, hemorrhage, and fluid levels and the triple sign.
Patients with tumors in the extremities have a more favorable prognosis than those with lesions in the head and neck area or axially, a feature that likely reflects better surgical control available for extremity lesions. Patient age of less than 15 to 20 years is also associated with a better long-term prognosis.15,16 Varela-Duran and Enzinger17 reported that the presence of extensive calcifications suggests improved long-term survival, with 5-year survival rates of 82% and decreased rates of local recurrence (32%) and metastatic disease (29%). The poorly differentiated subtype is associated with a worsened prognosis, with a 5-year survival rate of 20% through 30%.18,19 Other pathologic factors associated with worsened prognosis include presence of rhabdoid cells, extensive tumor necrosis, high nuclear grade, p53 mutations, and high mitotic rate (>10 mitoses/10 high-power field). More recently, the gene fusion type SYT-SSX2 (more common in monophasic lesions) has been associated with an improved prognosis, compared with that for SYT-SSX1, and an 89% metastasis-free survival.20
Although there are no guidelines for the treatment of primary renal synovial sarcoma because of the limited number of cases reported, surgery is considered the first choice. Adjuvant chemotherapy with an anthracycline (doxorubicin or epirubicin) combined with ifosfamide has been the most frequently used regimen in published cases, especially in those in which patients have poor prognostic factors as mentioned above.
Overall, the 5-year survival rate ranges from 36% to 76%.14 The clinical course of synovial sarcoma is characterized by a high rate of local recurrence (30%-50%) and metastatic disease (41%). Most metastases occur within the first 2 to 5 years after treatment cessation. Metastases are present in 16% to 25% of patients at their initial presentation, with the most frequent metastatic site being the lung, followed by the lymph nodes (4%-18%) and bone (8%-11%).
Conclusion
Primary renal synovial sarcoma is extremely rare, and preoperative diagnosis is difficult in the absence of specific clinical or imaging findings. A high index of suspicion combined with pathologic, immunohistochemical, cytogenetic, and molecular studies is essential for accurate diagnosis and subsequent treatment planning. The differential diagnosis of renal synovial sarcoma can be extensive, and our experience with this patient illustrates the diagnostic dilemma associated with renal synovial sarcoma.
1. Majumder A, Dey S, Khandakar B, Medda S, Chandra Paul P. Primary renal synovial sarcoma: a rare tumor with an atypical presentation. Arch Iran Med. 2014;17(10):726-728.
2. Fetsch JF, Meis JM. Synovial sarcoma of the abdominal wall. Cancer. 1993;72(2):469 477.
3. Wang Z, Zhong Z, Zhu L, et al. Primary synovial sarcoma of the kidney: a case report. Oncol Lett. 2015;10(6):3542-3544.
4. Abbas M, Dämmrich ME, Braubach P, et al. Synovial sarcoma of the kidney in a young patient with a review of the literature. Rare tumors. 2014;6(2):5393
5. Argani P, Faria PA, Epstein JI, et al. Primary renal synovial sarcoma: molecular and morphologic delineation of an entity previously included among embryonal sarcomas of the kidney. Am J Surg Pathol. 2000;24(8):1087-1096.
6. Eble JN, Sauter G, Epstein JI, Sesterhenn IA, eds. World Health Organization classification of tumours: pathology and genetics of tumours of the urinary system and male genital organs. Lyon, France: IARC; 2004.
7. Tamboli P, Ro JY, Amin MB, Ligato S, Ayala AG. Benign tumors and tumor-like lesions of the adult kidney. Part II: benign mesenchymal and mixed neoplasms, and tumor-like lesions. Adv Anat Pathol. 2000;7(1):47-66.
8. Weiss SW, Goldblum JR. Malignant soft tissue tumors of uncertain type. In: Weiss SW, Goldblum JR, eds. Enzinger and Weiss’s soft tissue tumors. 4th ed. St. Louis, MO: Mosby, 2001; 1483-1565.
9. Lacovelli R, Altavilla A, Ciardi A, et al. Clinical and pathological features of primary renal synovial sarcoma: analysis of 64 cases from 11 years of medical literature. BJU Int. 2012;110(10):1449-1454.
10. Alhazzani AR, El-Sharkawy MS, Hassan H. Primary retroperitoneal synovial sarcoma in CT and MRI. Urol Ann. 2010;2(1):39-41.
11. Katabathina VS, Vikram R, Nagar AM, Tamboli P, Menias CO, Prasad SR. Mesenchymal neoplasms of the kidney in adults: imaging spectrum with radiologic-pathologic correlation. Radiographics. 2010;30(6):1525-1540.
12. Sápi Z, Papp G, Szendrői M, et al. Epigenetic regulation of SMARCB1 by miR-206, -381 and -671- 5p is evident in a variety of SMARCB1 immunonegative soft tissue sarcomas, while miR-765 appears specific for epithelioid sarcoma. A miRNA study of 223 soft tissue sarcomas. Genes Chromosomes Cancer. 2016;55(10):786-802.
13. Ferrari A, Gronchi A, Casanova M, et al. Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer. 2004;101(3):627-634.
14. Rangheard AS, Vanel D, Viala J, Schwaab G, Casiraghi O, Sigal R. Synovial sarcomas of the head and neck: CT and MR imaging findings of eight patients. Am J Neuroradiol. 2001;22(5):851-857.
15. Oda Y, Hashimoto H, Tsuneyoshi M, Takeshita S. Survival in synovial sarcoma: a multivariate study of prognostic factors with special emphasis on the comparison between early death and long-term survival. Am J Surg Pathol. 1993;17(1):35-44.
16. Raney RB. Synovial sarcoma in young people: background, prognostic factors and therapeutic questions. J Pediatr Hematol Oncol. 2005;27(4):207-211.
17. Varela-Duran J, Enzinger FM. Calcifying synovial sarcoma. Cancer. 1982;50(2):345-352.
18. Cagle LA, Mirra JM, Storm FK, Roe DJ, Eilber FR. Histologic features relating to prognosis in synovial sarcoma. Cancer. 1987;59(10):1810-1814.
19. Skytting B, Meis-Kindblom JM, Larsson O, et al. Synovial sarcoma – identification of favorable and unfavorable histologic types: a Scandinavian sarcoma group study of 104 cases. Acta Orthop Scand. 1999:70(6):543-554.
20. Murphey MD, Gibson MS, Jennings BT, Crespo-Rodríguez AM, Fanburg-Smith J, Gajewski DA. Imaging of synovial sarcoma with radiologic-pathologic correlation. Radiographics. 2006;26(5):1543-1565.
Soft tissue sarcomas are rare mesenchymal tumors that comprise 1% of all malignancies. Synovial sarcoma accounts for 5% to 10% of adult soft tissue sarcomas and usually occurs in close association with joint capsules, tendon sheaths, and bursa in the extremities of young and middle-aged adults.1 Synovial sarcomas have been reported in other unusual sites, including the head and neck, thoracic and abdominal wall, retroperitoneum, bone, pleura, and visceral organs such as the lung, prostate, or kidney.2 Primary renal synovial sarcoma is an extremely rare tumor accounting for <2% of all malignant renal tumors.3 To the best of our knowledge, fewer than 50 cases of primary renal synovial sarcoma have been described in the English literature.4 It presents as a diagnostic dilemma because of the dearth of specific clinical and imaging findings and is often confused with benign and malignant tumors. The differential diagnosis includes angiomyolipoma, renal cell carcinoma with sarcomatoid differentiation, metastatic sarcoma, hemangiopericytoma, malignant solitary fibrous tumor, Wilms tumor, and malignant peripheral nerve sheath tumor. Hence, a combination of histomorphologic, immunohistochemical, cytogenetic, and molecular studies that show a unique chromosomal translocation t(X;18) (p11;q11) is imperative in the diagnosis of primary renal synovial sarcoma.4 In the present report, we present the case of a 38-year-old man who was diagnosed with primary renal synovial sarcoma.
Case presentation and summary
A 38-year-old man with a medical history of gastroesophageal reflux disease and Barrett’s esophagus presented to our hospital for the first time with persistent and progressive right-sided flank and abdominal pain that was aggravated after a minor trauma to the back. There was no associated hematuria or dysuria.
Of note is that he had experienced intermittent flank pain for 2 years before this transfer. He had initially been diagnosed at his local hospital close to his home by ultrasound with an angiomyolipoma of 2 × 3 cm arising from the upper pole of his right kidney, which remained stable on repeat sonograms. About 22 months after his initial presentation at his local hospital, the flank pain increased, and a computed-tomographic (CT) scan revealed a perinephric hematoma that was thought to originate from a ruptured angiomyolipoma. He subsequently underwent embolization, but his symptoms recurred soon after. He presented again to his local hospital where CT imaging revealed a significant increase in the size of the retroperitoneal mass, and findings were suggestive of a hematoma. Subsequent angiogram did not reveal active extravasation, so a biopsy was performed.
Before confirmatory pathologic evaluation could be completed, the patient presented to his local hospital again in excruciating pain. A CT scan of his abdomen and pelvis demonstrated a massive subacute on chronic hematoma in the right retroperitoneum measuring 22 × 19 × 18 cm, with calcifications originating from an upper pole right renal neoplasm. The right kidney was displaced antero-inferiorly, and the inferior vena cava was displaced anteriorly and to the left. The preliminary pathology returned with findings suggestive of sarcoma (Figures 1 and 2).
The patient was then transferred to our institution, where he was evaluated by medical and surgical oncology. A CT scan of the chest and magnetic-resonance imaging (MRI) of the brain did not reveal metastatic disease. He underwent exploratory laparotomy that involved the resection of a 22-cm retroperitoneal mass, right nephrectomy, right adrenalectomy, partial right hepatectomy, and a full thickness resection of the right postero-inferior diaphragm followed by mesh repair because of involvement by the tumor.
In its entirety, the specimen was a mass of 26 × 24 × 14 cm. It was sectioned to show extensively necrotic and hemorrhagic variegated white to tan-red parenchyma (Figure 3). Histology revealed a poorly differentiated malignant neoplasm composed of round cells with scant amphophilic cytoplasm arranged in solid, variably sized nests separated by prominent thin-walled branching vascular channels (Figure 4). The mitotic rate was high. It was determined to be a histologically ungraded sarcoma according to the French Federation of Comprehensive Cancer Centers system of grading soft tissue sarcomas; the margins were indeterminate. Immunohistochemistry was positive for EMA, TLE1, and negative for AE1/AE3, S100, STAT6, and Nkx2.2. Molecular pathology fluorescent in situ hybridization (FISH) analysis demonstrated positivity for SS18 gene rearrangement (SS18-SSX1 fusion).
After recovering from surgery, the patient received adjuvant chemotherapy with doxorubicin and ifosfamide. It has been almost 16 months since we first saw this patient. He was started on doxorubicin 20 mg/m2 on days 1 to 4, ifosfamide 2,500 mg on days 1 to 4, and mesna 800 mg on days 1 to 4, for a total of 6 cycles. He did well for the first 5 months, after which he developed disease recurrence in the postoperative nephrectomy bed (a biopsy showed it to be recurrent synovial sarcoma) as well as pulmonary nodules, for which he was started on trabectedin 1.5 mg/m2 every 3 weeks. Two months later, a CT scan showed an increase in the size of his retroperitoneal mass, and the treatment was changed to pazopanib 400 mg daily orally, on which he remained at the time of publication.
Discussion
Synovial sarcoma is the fourth most common type of soft tissue sarcoma, accounting for 2.5% to 10.5% of all primary soft tissue malignancies worldwide. It occurs most frequently in adolescents and young adults, with most patients presenting between the ages of 15 and 40 years. Median age of presentation is 36 years. Despite the nomenclature, synovial sarcoma does not arise in intra-articular locations but typically occurs in proximity to joints in the extremities. Synovial sarcomas are less commonly described in other sites, including the head and neck, mediastinum, intraperitoneum, retroperitoneum, lung, pleura, and kidney.4,5 Renal synovial sarcoma was first described in a published article by Argani and colleagues in 2000.5
Adult renal mesenchymal tumors are classified into benign and malignant tumors on the basis of the histologic features and clinicobiologic behavior.6,7 The benign esenchymal renal tumors include angiomyolipoma, leiomyoma, hemangioma, lymphangioma, juxtaglomerular cell tumor, renomedullary interstitial cell tumor (medullary fibroma), lipoma, solitary fibrous tumor, and schwannoma. Malignant renal tumors of mesenchymal origin include leiomyosarcoma, rhabdomyosarcoma, angiosarcoma, osteosarcoma, fibrosarcoma, malignant fibrous histiocytoma, solitary fibrous tumor, and synovial sarcoma.
Most of these tumor types cause the same nonspecific symptoms in patients – abdominal pain, flank pain, abdominal fullness, a palpable mass, and hematuria – although they can be clinically silent. The average duration of symptoms in synovial sarcoma is 2 to 4 years.8 The long duration of symptoms and initial slow growth of synovial sarcomas may give a false impression of a benign process.
A preoperative radiological diagnosis of primary renal synovial sarcoma may be suspected by analyzing the tumor’s growth patterns on CT scans.9 Renal synovial sarcomas often appear as large, well-defined soft tissue masses that can extend into the renal pelvis or into the perinephric region.9 A CT scan may identify soft tissue calcifications, especially subtle ones in areas where the tumor anatomy is complex. A CT scan may also reveal areas of hemorrhage, necrosis, or cyst formation within the tumor, and can easily confirm bone involvement. Intravenous contrast may help in differentiating the mass from adjacent muscle and neurovascular complex.9,10 On MRI, renal synovial sarcomas are often described as nonspecific heterogeneous masses, although they may also exhibit heterogeneous enhancement of hemorrhagic areas, calcifications, and air-fluid levels (known as “triple sign”) as well as septae. The triple sign may be identified as areas of low, intermediate, and high signal intensity, correlating with areas of hemorrhage, calcification, and air-fluid level.9,10 Signal intensity is about equal to that of skeletal muscle on T1-weighted MRI and higher than that of subcutaneous fat on T2-weighted MRI.
In the present case, the tumor was initially misdiagnosed as an angiomyolipoma, the most common benign tumor of the kidney. Angiomyolipomas are usually solid triphasic tumors arising from the renal cortex and are composed of 3 major elements: dysmorphic blood vessels, smooth muscle components, and adipose tissue. When angiomyolipomas are large enough, they are readily recognized by the identification of macroscopic fat within the tumor, either by CT scan or MRI.11 When they are small, they may be difficult to distinguish from a small cyst on CT because of volume averaging.
On pathology, synovial sarcoma has dual epithelial and mesenchymal differentiation. They are frequently multi-lobulated, and areas of necrosis, hemorrhage, and cyst formation are also common. There are 3 main histologic subtypes of synovial sarcoma: biphasic (20%-30%), monophasic (50%-60%), and poorly differentiated (15%-25%). Poorly differentiated synovial sarcomas are generally epithelioid in morphology, have high mitotic activity (usually 10-20 mitoses/10 high-power field; range is <5 for well differentiated, low-grade tumors), and can be confused with round cell tumors such as Ewing sarcoma. Poorly differentiated synovial sarcomas are high-grade tumors.
Immunohistochemical studies can confirm the pathological diagnosis. Synovial sarcomas usually stain positive for Bcl2, CD99/Mic2, CD56, Vim, and focally for EMA but negatively for desmin, actin, WT1, S-100, CD34, and CD31.5 Currently, the gold standard for diagnosis and hallmark for synovial sarcomas are the t (X;18) translocation and SYT-SSX gene fusion products (SYT-SSX1 in 67% and SYT-SSX2 in 33% of cases). These can be detected either by FISH or reverse-transcription polymerase chain reaction. This genetic alteration is identified in more than 90% of synovial sarcomas and is highly specific.
The role of SYT-SSX gene fusion in the pathogenesis of synovial sarcoma is an active area of investigation. The fusion of SYT with SSX translates into a fusion protein that binds to the transcription activator SMARCA4 that is involved in chromatin remodeling, thus displacing both the wildtype SYT and the tumor suppressor gene SMARCB1. The modified protein complex then binds at several super-enhancer loci, unlocking suppressed genes such as Sox2, which is known to be necessary for synovial sarcoma proliferation. Alterations in SMARCB1 are involved in several cancer types, implicating this event as a driver of these malignancies.12 This results in a global alteration in chromatin remodeling that needs to be better understood to design targeted therapies.
The clinical course of synovial sarcoma, regardless of the tissue of origin, is typically poor. Multiple clinical and pathologic factors, including tumor size, location, patient age, and presence of poorly differentiated areas, are thought to have prognostic significance. A tumor size of more than 5 cm at presentation has the greatest impact on prognosis, with studies showing 5-year survival rates of 64% for patients with tumors smaller than 5 cm and 26% for patients with masses greater than 5 cm.13,14 High-grade synovial sarcoma is favored in tumors that have cystic components, hemorrhage, and fluid levels and the triple sign.
Patients with tumors in the extremities have a more favorable prognosis than those with lesions in the head and neck area or axially, a feature that likely reflects better surgical control available for extremity lesions. Patient age of less than 15 to 20 years is also associated with a better long-term prognosis.15,16 Varela-Duran and Enzinger17 reported that the presence of extensive calcifications suggests improved long-term survival, with 5-year survival rates of 82% and decreased rates of local recurrence (32%) and metastatic disease (29%). The poorly differentiated subtype is associated with a worsened prognosis, with a 5-year survival rate of 20% through 30%.18,19 Other pathologic factors associated with worsened prognosis include presence of rhabdoid cells, extensive tumor necrosis, high nuclear grade, p53 mutations, and high mitotic rate (>10 mitoses/10 high-power field). More recently, the gene fusion type SYT-SSX2 (more common in monophasic lesions) has been associated with an improved prognosis, compared with that for SYT-SSX1, and an 89% metastasis-free survival.20
Although there are no guidelines for the treatment of primary renal synovial sarcoma because of the limited number of cases reported, surgery is considered the first choice. Adjuvant chemotherapy with an anthracycline (doxorubicin or epirubicin) combined with ifosfamide has been the most frequently used regimen in published cases, especially in those in which patients have poor prognostic factors as mentioned above.
Overall, the 5-year survival rate ranges from 36% to 76%.14 The clinical course of synovial sarcoma is characterized by a high rate of local recurrence (30%-50%) and metastatic disease (41%). Most metastases occur within the first 2 to 5 years after treatment cessation. Metastases are present in 16% to 25% of patients at their initial presentation, with the most frequent metastatic site being the lung, followed by the lymph nodes (4%-18%) and bone (8%-11%).
Conclusion
Primary renal synovial sarcoma is extremely rare, and preoperative diagnosis is difficult in the absence of specific clinical or imaging findings. A high index of suspicion combined with pathologic, immunohistochemical, cytogenetic, and molecular studies is essential for accurate diagnosis and subsequent treatment planning. The differential diagnosis of renal synovial sarcoma can be extensive, and our experience with this patient illustrates the diagnostic dilemma associated with renal synovial sarcoma.
Soft tissue sarcomas are rare mesenchymal tumors that comprise 1% of all malignancies. Synovial sarcoma accounts for 5% to 10% of adult soft tissue sarcomas and usually occurs in close association with joint capsules, tendon sheaths, and bursa in the extremities of young and middle-aged adults.1 Synovial sarcomas have been reported in other unusual sites, including the head and neck, thoracic and abdominal wall, retroperitoneum, bone, pleura, and visceral organs such as the lung, prostate, or kidney.2 Primary renal synovial sarcoma is an extremely rare tumor accounting for <2% of all malignant renal tumors.3 To the best of our knowledge, fewer than 50 cases of primary renal synovial sarcoma have been described in the English literature.4 It presents as a diagnostic dilemma because of the dearth of specific clinical and imaging findings and is often confused with benign and malignant tumors. The differential diagnosis includes angiomyolipoma, renal cell carcinoma with sarcomatoid differentiation, metastatic sarcoma, hemangiopericytoma, malignant solitary fibrous tumor, Wilms tumor, and malignant peripheral nerve sheath tumor. Hence, a combination of histomorphologic, immunohistochemical, cytogenetic, and molecular studies that show a unique chromosomal translocation t(X;18) (p11;q11) is imperative in the diagnosis of primary renal synovial sarcoma.4 In the present report, we present the case of a 38-year-old man who was diagnosed with primary renal synovial sarcoma.
Case presentation and summary
A 38-year-old man with a medical history of gastroesophageal reflux disease and Barrett’s esophagus presented to our hospital for the first time with persistent and progressive right-sided flank and abdominal pain that was aggravated after a minor trauma to the back. There was no associated hematuria or dysuria.
Of note is that he had experienced intermittent flank pain for 2 years before this transfer. He had initially been diagnosed at his local hospital close to his home by ultrasound with an angiomyolipoma of 2 × 3 cm arising from the upper pole of his right kidney, which remained stable on repeat sonograms. About 22 months after his initial presentation at his local hospital, the flank pain increased, and a computed-tomographic (CT) scan revealed a perinephric hematoma that was thought to originate from a ruptured angiomyolipoma. He subsequently underwent embolization, but his symptoms recurred soon after. He presented again to his local hospital where CT imaging revealed a significant increase in the size of the retroperitoneal mass, and findings were suggestive of a hematoma. Subsequent angiogram did not reveal active extravasation, so a biopsy was performed.
Before confirmatory pathologic evaluation could be completed, the patient presented to his local hospital again in excruciating pain. A CT scan of his abdomen and pelvis demonstrated a massive subacute on chronic hematoma in the right retroperitoneum measuring 22 × 19 × 18 cm, with calcifications originating from an upper pole right renal neoplasm. The right kidney was displaced antero-inferiorly, and the inferior vena cava was displaced anteriorly and to the left. The preliminary pathology returned with findings suggestive of sarcoma (Figures 1 and 2).
The patient was then transferred to our institution, where he was evaluated by medical and surgical oncology. A CT scan of the chest and magnetic-resonance imaging (MRI) of the brain did not reveal metastatic disease. He underwent exploratory laparotomy that involved the resection of a 22-cm retroperitoneal mass, right nephrectomy, right adrenalectomy, partial right hepatectomy, and a full thickness resection of the right postero-inferior diaphragm followed by mesh repair because of involvement by the tumor.
In its entirety, the specimen was a mass of 26 × 24 × 14 cm. It was sectioned to show extensively necrotic and hemorrhagic variegated white to tan-red parenchyma (Figure 3). Histology revealed a poorly differentiated malignant neoplasm composed of round cells with scant amphophilic cytoplasm arranged in solid, variably sized nests separated by prominent thin-walled branching vascular channels (Figure 4). The mitotic rate was high. It was determined to be a histologically ungraded sarcoma according to the French Federation of Comprehensive Cancer Centers system of grading soft tissue sarcomas; the margins were indeterminate. Immunohistochemistry was positive for EMA, TLE1, and negative for AE1/AE3, S100, STAT6, and Nkx2.2. Molecular pathology fluorescent in situ hybridization (FISH) analysis demonstrated positivity for SS18 gene rearrangement (SS18-SSX1 fusion).
After recovering from surgery, the patient received adjuvant chemotherapy with doxorubicin and ifosfamide. It has been almost 16 months since we first saw this patient. He was started on doxorubicin 20 mg/m2 on days 1 to 4, ifosfamide 2,500 mg on days 1 to 4, and mesna 800 mg on days 1 to 4, for a total of 6 cycles. He did well for the first 5 months, after which he developed disease recurrence in the postoperative nephrectomy bed (a biopsy showed it to be recurrent synovial sarcoma) as well as pulmonary nodules, for which he was started on trabectedin 1.5 mg/m2 every 3 weeks. Two months later, a CT scan showed an increase in the size of his retroperitoneal mass, and the treatment was changed to pazopanib 400 mg daily orally, on which he remained at the time of publication.
Discussion
Synovial sarcoma is the fourth most common type of soft tissue sarcoma, accounting for 2.5% to 10.5% of all primary soft tissue malignancies worldwide. It occurs most frequently in adolescents and young adults, with most patients presenting between the ages of 15 and 40 years. Median age of presentation is 36 years. Despite the nomenclature, synovial sarcoma does not arise in intra-articular locations but typically occurs in proximity to joints in the extremities. Synovial sarcomas are less commonly described in other sites, including the head and neck, mediastinum, intraperitoneum, retroperitoneum, lung, pleura, and kidney.4,5 Renal synovial sarcoma was first described in a published article by Argani and colleagues in 2000.5
Adult renal mesenchymal tumors are classified into benign and malignant tumors on the basis of the histologic features and clinicobiologic behavior.6,7 The benign esenchymal renal tumors include angiomyolipoma, leiomyoma, hemangioma, lymphangioma, juxtaglomerular cell tumor, renomedullary interstitial cell tumor (medullary fibroma), lipoma, solitary fibrous tumor, and schwannoma. Malignant renal tumors of mesenchymal origin include leiomyosarcoma, rhabdomyosarcoma, angiosarcoma, osteosarcoma, fibrosarcoma, malignant fibrous histiocytoma, solitary fibrous tumor, and synovial sarcoma.
Most of these tumor types cause the same nonspecific symptoms in patients – abdominal pain, flank pain, abdominal fullness, a palpable mass, and hematuria – although they can be clinically silent. The average duration of symptoms in synovial sarcoma is 2 to 4 years.8 The long duration of symptoms and initial slow growth of synovial sarcomas may give a false impression of a benign process.
A preoperative radiological diagnosis of primary renal synovial sarcoma may be suspected by analyzing the tumor’s growth patterns on CT scans.9 Renal synovial sarcomas often appear as large, well-defined soft tissue masses that can extend into the renal pelvis or into the perinephric region.9 A CT scan may identify soft tissue calcifications, especially subtle ones in areas where the tumor anatomy is complex. A CT scan may also reveal areas of hemorrhage, necrosis, or cyst formation within the tumor, and can easily confirm bone involvement. Intravenous contrast may help in differentiating the mass from adjacent muscle and neurovascular complex.9,10 On MRI, renal synovial sarcomas are often described as nonspecific heterogeneous masses, although they may also exhibit heterogeneous enhancement of hemorrhagic areas, calcifications, and air-fluid levels (known as “triple sign”) as well as septae. The triple sign may be identified as areas of low, intermediate, and high signal intensity, correlating with areas of hemorrhage, calcification, and air-fluid level.9,10 Signal intensity is about equal to that of skeletal muscle on T1-weighted MRI and higher than that of subcutaneous fat on T2-weighted MRI.
In the present case, the tumor was initially misdiagnosed as an angiomyolipoma, the most common benign tumor of the kidney. Angiomyolipomas are usually solid triphasic tumors arising from the renal cortex and are composed of 3 major elements: dysmorphic blood vessels, smooth muscle components, and adipose tissue. When angiomyolipomas are large enough, they are readily recognized by the identification of macroscopic fat within the tumor, either by CT scan or MRI.11 When they are small, they may be difficult to distinguish from a small cyst on CT because of volume averaging.
On pathology, synovial sarcoma has dual epithelial and mesenchymal differentiation. They are frequently multi-lobulated, and areas of necrosis, hemorrhage, and cyst formation are also common. There are 3 main histologic subtypes of synovial sarcoma: biphasic (20%-30%), monophasic (50%-60%), and poorly differentiated (15%-25%). Poorly differentiated synovial sarcomas are generally epithelioid in morphology, have high mitotic activity (usually 10-20 mitoses/10 high-power field; range is <5 for well differentiated, low-grade tumors), and can be confused with round cell tumors such as Ewing sarcoma. Poorly differentiated synovial sarcomas are high-grade tumors.
Immunohistochemical studies can confirm the pathological diagnosis. Synovial sarcomas usually stain positive for Bcl2, CD99/Mic2, CD56, Vim, and focally for EMA but negatively for desmin, actin, WT1, S-100, CD34, and CD31.5 Currently, the gold standard for diagnosis and hallmark for synovial sarcomas are the t (X;18) translocation and SYT-SSX gene fusion products (SYT-SSX1 in 67% and SYT-SSX2 in 33% of cases). These can be detected either by FISH or reverse-transcription polymerase chain reaction. This genetic alteration is identified in more than 90% of synovial sarcomas and is highly specific.
The role of SYT-SSX gene fusion in the pathogenesis of synovial sarcoma is an active area of investigation. The fusion of SYT with SSX translates into a fusion protein that binds to the transcription activator SMARCA4 that is involved in chromatin remodeling, thus displacing both the wildtype SYT and the tumor suppressor gene SMARCB1. The modified protein complex then binds at several super-enhancer loci, unlocking suppressed genes such as Sox2, which is known to be necessary for synovial sarcoma proliferation. Alterations in SMARCB1 are involved in several cancer types, implicating this event as a driver of these malignancies.12 This results in a global alteration in chromatin remodeling that needs to be better understood to design targeted therapies.
The clinical course of synovial sarcoma, regardless of the tissue of origin, is typically poor. Multiple clinical and pathologic factors, including tumor size, location, patient age, and presence of poorly differentiated areas, are thought to have prognostic significance. A tumor size of more than 5 cm at presentation has the greatest impact on prognosis, with studies showing 5-year survival rates of 64% for patients with tumors smaller than 5 cm and 26% for patients with masses greater than 5 cm.13,14 High-grade synovial sarcoma is favored in tumors that have cystic components, hemorrhage, and fluid levels and the triple sign.
Patients with tumors in the extremities have a more favorable prognosis than those with lesions in the head and neck area or axially, a feature that likely reflects better surgical control available for extremity lesions. Patient age of less than 15 to 20 years is also associated with a better long-term prognosis.15,16 Varela-Duran and Enzinger17 reported that the presence of extensive calcifications suggests improved long-term survival, with 5-year survival rates of 82% and decreased rates of local recurrence (32%) and metastatic disease (29%). The poorly differentiated subtype is associated with a worsened prognosis, with a 5-year survival rate of 20% through 30%.18,19 Other pathologic factors associated with worsened prognosis include presence of rhabdoid cells, extensive tumor necrosis, high nuclear grade, p53 mutations, and high mitotic rate (>10 mitoses/10 high-power field). More recently, the gene fusion type SYT-SSX2 (more common in monophasic lesions) has been associated with an improved prognosis, compared with that for SYT-SSX1, and an 89% metastasis-free survival.20
Although there are no guidelines for the treatment of primary renal synovial sarcoma because of the limited number of cases reported, surgery is considered the first choice. Adjuvant chemotherapy with an anthracycline (doxorubicin or epirubicin) combined with ifosfamide has been the most frequently used regimen in published cases, especially in those in which patients have poor prognostic factors as mentioned above.
Overall, the 5-year survival rate ranges from 36% to 76%.14 The clinical course of synovial sarcoma is characterized by a high rate of local recurrence (30%-50%) and metastatic disease (41%). Most metastases occur within the first 2 to 5 years after treatment cessation. Metastases are present in 16% to 25% of patients at their initial presentation, with the most frequent metastatic site being the lung, followed by the lymph nodes (4%-18%) and bone (8%-11%).
Conclusion
Primary renal synovial sarcoma is extremely rare, and preoperative diagnosis is difficult in the absence of specific clinical or imaging findings. A high index of suspicion combined with pathologic, immunohistochemical, cytogenetic, and molecular studies is essential for accurate diagnosis and subsequent treatment planning. The differential diagnosis of renal synovial sarcoma can be extensive, and our experience with this patient illustrates the diagnostic dilemma associated with renal synovial sarcoma.
1. Majumder A, Dey S, Khandakar B, Medda S, Chandra Paul P. Primary renal synovial sarcoma: a rare tumor with an atypical presentation. Arch Iran Med. 2014;17(10):726-728.
2. Fetsch JF, Meis JM. Synovial sarcoma of the abdominal wall. Cancer. 1993;72(2):469 477.
3. Wang Z, Zhong Z, Zhu L, et al. Primary synovial sarcoma of the kidney: a case report. Oncol Lett. 2015;10(6):3542-3544.
4. Abbas M, Dämmrich ME, Braubach P, et al. Synovial sarcoma of the kidney in a young patient with a review of the literature. Rare tumors. 2014;6(2):5393
5. Argani P, Faria PA, Epstein JI, et al. Primary renal synovial sarcoma: molecular and morphologic delineation of an entity previously included among embryonal sarcomas of the kidney. Am J Surg Pathol. 2000;24(8):1087-1096.
6. Eble JN, Sauter G, Epstein JI, Sesterhenn IA, eds. World Health Organization classification of tumours: pathology and genetics of tumours of the urinary system and male genital organs. Lyon, France: IARC; 2004.
7. Tamboli P, Ro JY, Amin MB, Ligato S, Ayala AG. Benign tumors and tumor-like lesions of the adult kidney. Part II: benign mesenchymal and mixed neoplasms, and tumor-like lesions. Adv Anat Pathol. 2000;7(1):47-66.
8. Weiss SW, Goldblum JR. Malignant soft tissue tumors of uncertain type. In: Weiss SW, Goldblum JR, eds. Enzinger and Weiss’s soft tissue tumors. 4th ed. St. Louis, MO: Mosby, 2001; 1483-1565.
9. Lacovelli R, Altavilla A, Ciardi A, et al. Clinical and pathological features of primary renal synovial sarcoma: analysis of 64 cases from 11 years of medical literature. BJU Int. 2012;110(10):1449-1454.
10. Alhazzani AR, El-Sharkawy MS, Hassan H. Primary retroperitoneal synovial sarcoma in CT and MRI. Urol Ann. 2010;2(1):39-41.
11. Katabathina VS, Vikram R, Nagar AM, Tamboli P, Menias CO, Prasad SR. Mesenchymal neoplasms of the kidney in adults: imaging spectrum with radiologic-pathologic correlation. Radiographics. 2010;30(6):1525-1540.
12. Sápi Z, Papp G, Szendrői M, et al. Epigenetic regulation of SMARCB1 by miR-206, -381 and -671- 5p is evident in a variety of SMARCB1 immunonegative soft tissue sarcomas, while miR-765 appears specific for epithelioid sarcoma. A miRNA study of 223 soft tissue sarcomas. Genes Chromosomes Cancer. 2016;55(10):786-802.
13. Ferrari A, Gronchi A, Casanova M, et al. Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer. 2004;101(3):627-634.
14. Rangheard AS, Vanel D, Viala J, Schwaab G, Casiraghi O, Sigal R. Synovial sarcomas of the head and neck: CT and MR imaging findings of eight patients. Am J Neuroradiol. 2001;22(5):851-857.
15. Oda Y, Hashimoto H, Tsuneyoshi M, Takeshita S. Survival in synovial sarcoma: a multivariate study of prognostic factors with special emphasis on the comparison between early death and long-term survival. Am J Surg Pathol. 1993;17(1):35-44.
16. Raney RB. Synovial sarcoma in young people: background, prognostic factors and therapeutic questions. J Pediatr Hematol Oncol. 2005;27(4):207-211.
17. Varela-Duran J, Enzinger FM. Calcifying synovial sarcoma. Cancer. 1982;50(2):345-352.
18. Cagle LA, Mirra JM, Storm FK, Roe DJ, Eilber FR. Histologic features relating to prognosis in synovial sarcoma. Cancer. 1987;59(10):1810-1814.
19. Skytting B, Meis-Kindblom JM, Larsson O, et al. Synovial sarcoma – identification of favorable and unfavorable histologic types: a Scandinavian sarcoma group study of 104 cases. Acta Orthop Scand. 1999:70(6):543-554.
20. Murphey MD, Gibson MS, Jennings BT, Crespo-Rodríguez AM, Fanburg-Smith J, Gajewski DA. Imaging of synovial sarcoma with radiologic-pathologic correlation. Radiographics. 2006;26(5):1543-1565.
1. Majumder A, Dey S, Khandakar B, Medda S, Chandra Paul P. Primary renal synovial sarcoma: a rare tumor with an atypical presentation. Arch Iran Med. 2014;17(10):726-728.
2. Fetsch JF, Meis JM. Synovial sarcoma of the abdominal wall. Cancer. 1993;72(2):469 477.
3. Wang Z, Zhong Z, Zhu L, et al. Primary synovial sarcoma of the kidney: a case report. Oncol Lett. 2015;10(6):3542-3544.
4. Abbas M, Dämmrich ME, Braubach P, et al. Synovial sarcoma of the kidney in a young patient with a review of the literature. Rare tumors. 2014;6(2):5393
5. Argani P, Faria PA, Epstein JI, et al. Primary renal synovial sarcoma: molecular and morphologic delineation of an entity previously included among embryonal sarcomas of the kidney. Am J Surg Pathol. 2000;24(8):1087-1096.
6. Eble JN, Sauter G, Epstein JI, Sesterhenn IA, eds. World Health Organization classification of tumours: pathology and genetics of tumours of the urinary system and male genital organs. Lyon, France: IARC; 2004.
7. Tamboli P, Ro JY, Amin MB, Ligato S, Ayala AG. Benign tumors and tumor-like lesions of the adult kidney. Part II: benign mesenchymal and mixed neoplasms, and tumor-like lesions. Adv Anat Pathol. 2000;7(1):47-66.
8. Weiss SW, Goldblum JR. Malignant soft tissue tumors of uncertain type. In: Weiss SW, Goldblum JR, eds. Enzinger and Weiss’s soft tissue tumors. 4th ed. St. Louis, MO: Mosby, 2001; 1483-1565.
9. Lacovelli R, Altavilla A, Ciardi A, et al. Clinical and pathological features of primary renal synovial sarcoma: analysis of 64 cases from 11 years of medical literature. BJU Int. 2012;110(10):1449-1454.
10. Alhazzani AR, El-Sharkawy MS, Hassan H. Primary retroperitoneal synovial sarcoma in CT and MRI. Urol Ann. 2010;2(1):39-41.
11. Katabathina VS, Vikram R, Nagar AM, Tamboli P, Menias CO, Prasad SR. Mesenchymal neoplasms of the kidney in adults: imaging spectrum with radiologic-pathologic correlation. Radiographics. 2010;30(6):1525-1540.
12. Sápi Z, Papp G, Szendrői M, et al. Epigenetic regulation of SMARCB1 by miR-206, -381 and -671- 5p is evident in a variety of SMARCB1 immunonegative soft tissue sarcomas, while miR-765 appears specific for epithelioid sarcoma. A miRNA study of 223 soft tissue sarcomas. Genes Chromosomes Cancer. 2016;55(10):786-802.
13. Ferrari A, Gronchi A, Casanova M, et al. Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer. 2004;101(3):627-634.
14. Rangheard AS, Vanel D, Viala J, Schwaab G, Casiraghi O, Sigal R. Synovial sarcomas of the head and neck: CT and MR imaging findings of eight patients. Am J Neuroradiol. 2001;22(5):851-857.
15. Oda Y, Hashimoto H, Tsuneyoshi M, Takeshita S. Survival in synovial sarcoma: a multivariate study of prognostic factors with special emphasis on the comparison between early death and long-term survival. Am J Surg Pathol. 1993;17(1):35-44.
16. Raney RB. Synovial sarcoma in young people: background, prognostic factors and therapeutic questions. J Pediatr Hematol Oncol. 2005;27(4):207-211.
17. Varela-Duran J, Enzinger FM. Calcifying synovial sarcoma. Cancer. 1982;50(2):345-352.
18. Cagle LA, Mirra JM, Storm FK, Roe DJ, Eilber FR. Histologic features relating to prognosis in synovial sarcoma. Cancer. 1987;59(10):1810-1814.
19. Skytting B, Meis-Kindblom JM, Larsson O, et al. Synovial sarcoma – identification of favorable and unfavorable histologic types: a Scandinavian sarcoma group study of 104 cases. Acta Orthop Scand. 1999:70(6):543-554.
20. Murphey MD, Gibson MS, Jennings BT, Crespo-Rodríguez AM, Fanburg-Smith J, Gajewski DA. Imaging of synovial sarcoma with radiologic-pathologic correlation. Radiographics. 2006;26(5):1543-1565.
TKIs and immunotherapy hold promise for alveolar soft part sarcoma
Alveolar soft part sarcoma (ASPS) has often proven to be resistant to conventional doxorubicin-based chemotherapy, but tyrosine kinase inhibitors (TKIs) and immune checkpoint inhibitors (ICIs) may provide new treatment strategies for this rare type of sarcoma, according to a literature review.
A rare, translocation-driven sarcoma of the soft tissues, ASPS often affects young adults and is characterized by indolent behavior and early metastasis. Despite its resistance to chemotherapy, studies indicate that survival is often prolonged in patients with metastatic disease. The literature has shown 5-year survival rates at about 60%, and this percentage has remained fairly consistent for the past 3 decades.
Luca Paoluzzi, MD, of New York University, and Robert G. Maki, MD, PhD, of Hofstra University, Hempstead, N.Y., reviewed the literature from 1952 to March 2018, in order to gain a better understanding of ASPS and the opportunities “for the translation of such knowledge into clinical practice,” they wrote in JAMA.
From a therapeutic standpoint, ASPS is characterized by sensitivity to vascular endothelial growth factor receptor–predominant TKIs, compared with other soft tissue sarcomas (STS), and recent data have emphasized that it is responsive to new immunotherapy regimens including ICIs. Pazopanib is currently the only agent that has received regulatory approval for use in STS refractory to other treatments and it appears to have consistent activity in metastatic ASPS. Management of ASPS generally also involves surgical resection and/or systemic treatment for metastatic disease. Conventional agents such as anthracycline-based chemotherapy have demonstrated a poor response rate lower than 10%, and while a complete resection may be curative, metastases are common and can occur years after resection of the primary tumor.
Conversely, ICIs “represent a promising area of drug development in ASPS; the data to date are limited but encouraging,” wrote Dr. Paoluzzi and Dr. Maki.
They pointed to one study that included 50 patients with sarcoma with 14 different subtypes of STS who were enrolled in immunotherapy trials conducted at the University of Texas MD Anderson Cancer Center, Houston. There were two pretreated patients with ASPS (two to four prior lines) in the cohort who received antiprogrammed death-ligand 1–based therapy, and achieved a partial response bordering on a complete response that lasted 8 and 12 months. An additional two patients achieved stable disease.
Another paper, presented at the 2017 Connective Tissue Oncology Society annual meeting, presented preliminary data from a phase 2 study that showed four of nine evaluable patients with ASPS treated with the TKI axitinib, combined with pembrolizumab, achieved a partial response. Three others had stable disease.
“Pathway-driven basket trials facilitate the enrollment of patients with such uncommon cancers and should provide valuable information regarding a second type of immune responsiveness to ICIs, one that is not a function of high tumor mutational burden,” the authors concluded.
No outside funding sources were reported. Dr. Maki reported receiving consultant fees from numerous sources and research support to New York University from Immune Design, Immunocore, Eli Lilly, Presage Biosciences, TRACON Pharmaceuticals, SARC, Regeneron, and Genentech. No other conflicts were reported.
SOURCE: doi: 10.1001/jamaoncol.2018.4490.
Alveolar soft part sarcoma (ASPS) has often proven to be resistant to conventional doxorubicin-based chemotherapy, but tyrosine kinase inhibitors (TKIs) and immune checkpoint inhibitors (ICIs) may provide new treatment strategies for this rare type of sarcoma, according to a literature review.
A rare, translocation-driven sarcoma of the soft tissues, ASPS often affects young adults and is characterized by indolent behavior and early metastasis. Despite its resistance to chemotherapy, studies indicate that survival is often prolonged in patients with metastatic disease. The literature has shown 5-year survival rates at about 60%, and this percentage has remained fairly consistent for the past 3 decades.
Luca Paoluzzi, MD, of New York University, and Robert G. Maki, MD, PhD, of Hofstra University, Hempstead, N.Y., reviewed the literature from 1952 to March 2018, in order to gain a better understanding of ASPS and the opportunities “for the translation of such knowledge into clinical practice,” they wrote in JAMA.
From a therapeutic standpoint, ASPS is characterized by sensitivity to vascular endothelial growth factor receptor–predominant TKIs, compared with other soft tissue sarcomas (STS), and recent data have emphasized that it is responsive to new immunotherapy regimens including ICIs. Pazopanib is currently the only agent that has received regulatory approval for use in STS refractory to other treatments and it appears to have consistent activity in metastatic ASPS. Management of ASPS generally also involves surgical resection and/or systemic treatment for metastatic disease. Conventional agents such as anthracycline-based chemotherapy have demonstrated a poor response rate lower than 10%, and while a complete resection may be curative, metastases are common and can occur years after resection of the primary tumor.
Conversely, ICIs “represent a promising area of drug development in ASPS; the data to date are limited but encouraging,” wrote Dr. Paoluzzi and Dr. Maki.
They pointed to one study that included 50 patients with sarcoma with 14 different subtypes of STS who were enrolled in immunotherapy trials conducted at the University of Texas MD Anderson Cancer Center, Houston. There were two pretreated patients with ASPS (two to four prior lines) in the cohort who received antiprogrammed death-ligand 1–based therapy, and achieved a partial response bordering on a complete response that lasted 8 and 12 months. An additional two patients achieved stable disease.
Another paper, presented at the 2017 Connective Tissue Oncology Society annual meeting, presented preliminary data from a phase 2 study that showed four of nine evaluable patients with ASPS treated with the TKI axitinib, combined with pembrolizumab, achieved a partial response. Three others had stable disease.
“Pathway-driven basket trials facilitate the enrollment of patients with such uncommon cancers and should provide valuable information regarding a second type of immune responsiveness to ICIs, one that is not a function of high tumor mutational burden,” the authors concluded.
No outside funding sources were reported. Dr. Maki reported receiving consultant fees from numerous sources and research support to New York University from Immune Design, Immunocore, Eli Lilly, Presage Biosciences, TRACON Pharmaceuticals, SARC, Regeneron, and Genentech. No other conflicts were reported.
SOURCE: doi: 10.1001/jamaoncol.2018.4490.
Alveolar soft part sarcoma (ASPS) has often proven to be resistant to conventional doxorubicin-based chemotherapy, but tyrosine kinase inhibitors (TKIs) and immune checkpoint inhibitors (ICIs) may provide new treatment strategies for this rare type of sarcoma, according to a literature review.
A rare, translocation-driven sarcoma of the soft tissues, ASPS often affects young adults and is characterized by indolent behavior and early metastasis. Despite its resistance to chemotherapy, studies indicate that survival is often prolonged in patients with metastatic disease. The literature has shown 5-year survival rates at about 60%, and this percentage has remained fairly consistent for the past 3 decades.
Luca Paoluzzi, MD, of New York University, and Robert G. Maki, MD, PhD, of Hofstra University, Hempstead, N.Y., reviewed the literature from 1952 to March 2018, in order to gain a better understanding of ASPS and the opportunities “for the translation of such knowledge into clinical practice,” they wrote in JAMA.
From a therapeutic standpoint, ASPS is characterized by sensitivity to vascular endothelial growth factor receptor–predominant TKIs, compared with other soft tissue sarcomas (STS), and recent data have emphasized that it is responsive to new immunotherapy regimens including ICIs. Pazopanib is currently the only agent that has received regulatory approval for use in STS refractory to other treatments and it appears to have consistent activity in metastatic ASPS. Management of ASPS generally also involves surgical resection and/or systemic treatment for metastatic disease. Conventional agents such as anthracycline-based chemotherapy have demonstrated a poor response rate lower than 10%, and while a complete resection may be curative, metastases are common and can occur years after resection of the primary tumor.
Conversely, ICIs “represent a promising area of drug development in ASPS; the data to date are limited but encouraging,” wrote Dr. Paoluzzi and Dr. Maki.
They pointed to one study that included 50 patients with sarcoma with 14 different subtypes of STS who were enrolled in immunotherapy trials conducted at the University of Texas MD Anderson Cancer Center, Houston. There were two pretreated patients with ASPS (two to four prior lines) in the cohort who received antiprogrammed death-ligand 1–based therapy, and achieved a partial response bordering on a complete response that lasted 8 and 12 months. An additional two patients achieved stable disease.
Another paper, presented at the 2017 Connective Tissue Oncology Society annual meeting, presented preliminary data from a phase 2 study that showed four of nine evaluable patients with ASPS treated with the TKI axitinib, combined with pembrolizumab, achieved a partial response. Three others had stable disease.
“Pathway-driven basket trials facilitate the enrollment of patients with such uncommon cancers and should provide valuable information regarding a second type of immune responsiveness to ICIs, one that is not a function of high tumor mutational burden,” the authors concluded.
No outside funding sources were reported. Dr. Maki reported receiving consultant fees from numerous sources and research support to New York University from Immune Design, Immunocore, Eli Lilly, Presage Biosciences, TRACON Pharmaceuticals, SARC, Regeneron, and Genentech. No other conflicts were reported.
SOURCE: doi: 10.1001/jamaoncol.2018.4490.
FROM JAMA
Key clinical point: Alveolar soft part sarcoma has often proven to be resistant to conventional doxorubicin-based chemotherapy, tyrosine kinase inhibitors and immune checkpoint inhibitors may provide new treatment strategies.
Major finding: In one study of sarcoma patients enrolled in immunotherapy trials, two pretreated patients with alveolar soft part sarcoma (two to four prior lines) who received antiprogrammed death-ligand 1–based therapy achieved partial responses, bordering on a complete response, that lasted 8 and 12 months.
Study details: A review of literature concerning treatment for alveolar soft part sarcoma.
Disclosures: No outside funding sources were reported. Dr. Maki reported receiving consultant fees from numerous sources and research support to New York University from Immune Design, Immunocore, Eli Lilly, Presage Biosciences, TRACON Pharmaceuticals, SARC, Regeneron, and Genentech. No other conflicts were reported.
Source: