User login
How to spot heritable breast cancer: A primary care physician’s guide
PATIENT 1: A PERSONAL AND FAMILY HISTORY OF BREAST CANCER
A 55-year-old Ashkenazi Jewish woman presents to your clinic for her annual physical. She reports that she had been diagnosed with breast cancer 10 years ago and that it had been treated with lumpectomy. You recall that Ashkenazi Jewish ethnicity and a diagnosis of breast cancer before age 50 are red flags for a hereditary cancer syndrome, and you ask about her family history of cancer. She reports that her mother was diagnosed with breast cancer in her 60s. The patient wants to know if her daughter should start breast cancer screening.
What do you do next?
Facing increasing demands and a plethora of information to be discussed in a short time, primary care physicians may find it challenging to inform patients about the possibility of a hereditary cancer syndrome, to assess the risk, to organize genetic testing if appropriate, and to counsel patients about their management options. As our knowledge of the genetics of breast cancer continues to expand, this information will become more detailed and complex.
Nevertheless, primary care physicians can help identify patients who may have a syndrome of inherited cancer predisposition or whose family history raises concern for familial breast cancer. Patients in both groups may be candidates for genetic risk assessment, for special management options for women at high risk, or for both.
This article provides an overview of inherited conditions associated with higher breast cancer risk, and guidelines to help physicians recognize patients in their own practice for whom a genetics referral may be appropriate.
BREAST CANCER IS COMPLEX AND HETEROGENEOUS
Breast cancer is the second-leading cause of cancer deaths in women. According to the American Cancer Society, an estimated 234,340 new cases of breast cancer are expected to be diagnosed in women in the United States in 2013, and about 2,240 new cases are expected in men; 39,620 women and 410 men are expected to die of it.1

Breast cancer is a complex and heterogeneous disease, influenced by many factors, of which female sex and increasing age are the most significant. Modifiable risk factors include obesity, use of combined hormone replacement therapy, and physical inactivity. Other risk factors include dense breast tissue, having had a breast biopsy in the past, the finding of atypical hyperplasia on biopsy, a history of high-dose chest radiation, and reproductive factors that include early menarche, late menopause, nulliparity, and birth of first child after age 30.
After female sex and age, family history of the disease is the most significant risk factor for breast cancer.2 If a woman has a first-degree relative (mother, sister, daughter) with breast cancer, her risk is 1.8 times higher, and if she has a second-degree relative (aunt, grandmother) with breast cancer, her risk is 1.3 times higher.3
Hereditary cancer predisposition syndromes account for 5% to 10% of cases of breast cancer. These are caused by a germline mutation in a highly penetrant gene that considerably increases the risk of malignancies of the breast and other tissues. These conditions are inherited in an autosomal-dominant fashion, with age of onset tending to be significantly—several decades—younger than the median age of onset in the general population. The most common of these is hereditary breast and ovarian cancer syndrome, caused by germline mutations of the BRCA1 or BRCA2 gene.
Familial breast cancers account for 15% to 20% of cases. Here, the women who develop breast cancer have multiple family members who are also affected but without an obvious inheritance pattern, and the age of onset is similar to that in the general population.4
Sporadic forms of breast cancer account for the remaining 70% to 80% of cases. Their development can be attributed mainly to nonhereditary causes, such as the environmental and personal risk factors listed above. In general, sporadic forms of breast cancer occur at older ages, with no particular inheritance pattern and with frequency of occurrence in a family comparable to that in the general population.
IS A GENETICS CONSULTATION NEEDED?

In the case described above, the primary care physician gathered basic information about the patient’s cancer-related personal and family history. Asking a few key questions (Table 1)5,6 can help physicians understand two important things: whether a more detailed assessment of genetic risk and counseling by a genetics professional are indicated, and whether the patient would benefit from additional cancer screening and prevention.

Table 2 summarizes the National Comprehensive Cancer Network’s recommendations for cancer genetics consultation.5 These red flags for a hereditary breast cancer syndrome can help primary care providers identify patients for whom a cancer genetics referral is appropriate. Of note: the maternal and paternal family histories are equally important.
Because our patient was diagnosed with breast cancer before age 50 and is of Ashkenazi Jewish ethnicity, she meets these criteria and warrants a cancer genetics consultation.
What is a cancer-focused genetic counseling session?
The tenets of genetic counseling, described previously in this series,7 are relevant to hereditary cancer syndromes. Cancer risk assessment and genetic counseling constitute the process of identifying and counseling individuals at risk of familial or hereditary cancer.8
As in other genetic counseling scenarios, a detailed pedigree (family tree) is taken, and this information, along with the patient’s personal medical history, allows a genetics specialist to determine if the presentation is most suggestive of sporadic, familial, or hereditary cancer.
A common misconception among patients is that there is a single genetic test for hereditary breast cancer, when in fact many highly penetrant predisposition genes have been linked to heightened risk (see below). The syndromes summarized in Table 35,9–18 are part of the differential diagnosis for every patient presenting with a personal or family history of breast cancer, and the detailed information from the personal and family history, ascertained during the assessment, ensures the right syndrome is explored within a family.
Cancer-focused genetic counseling may also help a patient or family process the psychological and emotional responses that can occur when cancer risk is discussed: eg, fear of cancer and death; guilt a parent may feel for passing on a genetic predisposition; and survivor guilt experienced by family members who test negative.
Genetic counselors are trained to recognize patients who may benefit from additional counseling. Not all patients pursuing cancer-focused genetic testing need a thorough evaluation by a psychologist, unlike those with adult-onset neurodegenerative conditions such as Huntington disease. Rather, the genetic counselor discusses the psychological implications of cancer-focused genetic testing and can refer the patient to a psychologist, therapist, social worker, or others if he or she feels the patient may benefit.8
Some patients come to a genetic counseling session with concerns about whether their insurance will pay for testing, and about whether they will face discrimination because of the testing results. In most situations, genetic testing is deemed medically necessary and is covered by the patient’s insurance. When testing is necessary, genetic counselors are skilled at preauthorizing it and writing letters of medical necessity. They are also familiar with laws and regulations that protect patients, such as the Genetic Information Nondiscrimination Act, which protects patients from insurance and employment discrimination.
Because a cancer-focused genetic counseling session typically lasts 1 hour, the counselor has enough time to address these and any other concerns that might prevent a patient who is otherwise interested in genetic testing from pursuing it.
HOW CAN GENETIC TESTING HELP?
Genetic testing for hereditary cancer syndromes can have personal benefit for the patient and at-risk family members.
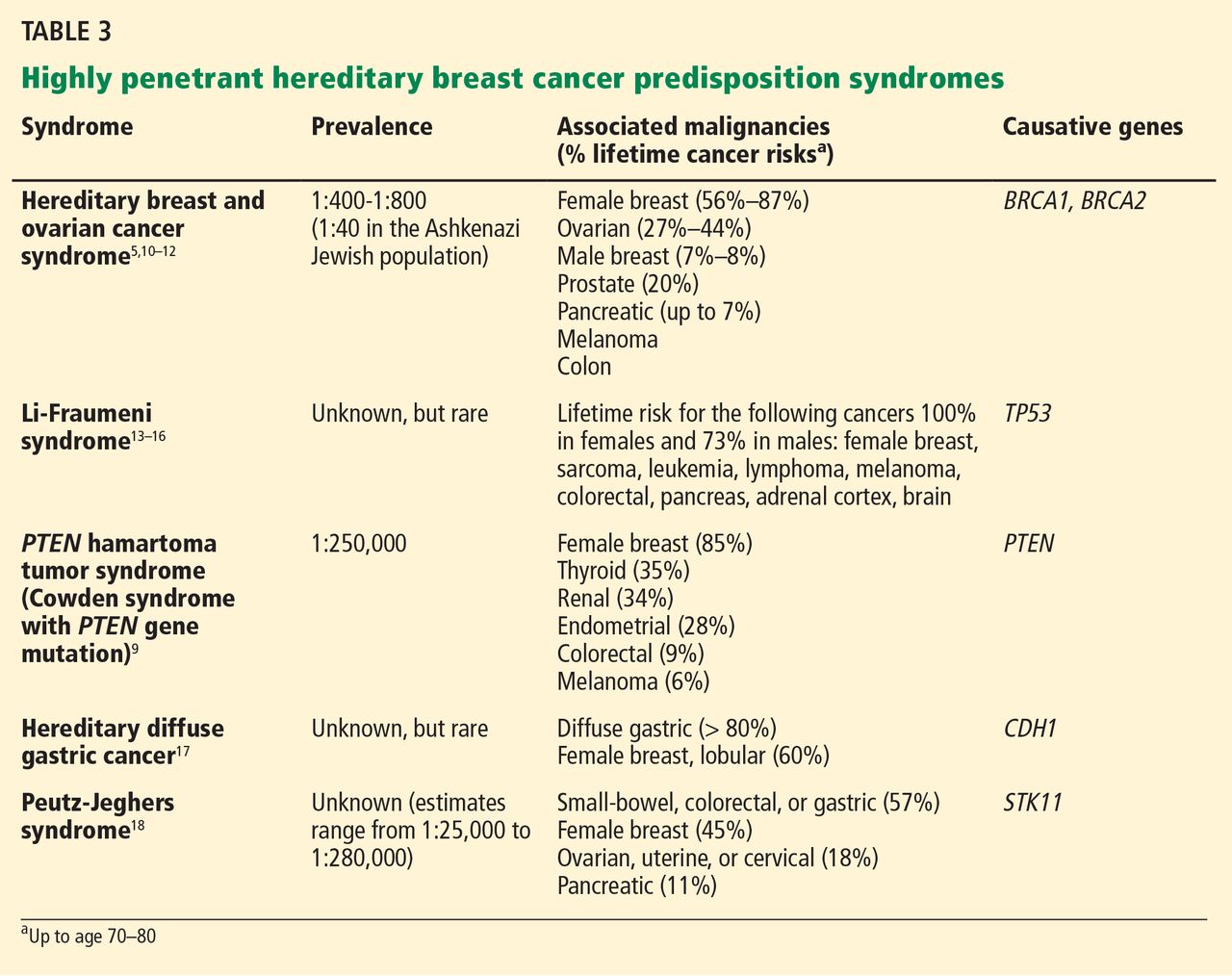
Note that the syndromes in Table 3 all increase the risk of more than one type of cancer. Patients with these syndromes frequently receive care from multiple subspecialists to mitigate those risks. Guidelines exist for each of these syndromes and, if followed, may prevent the morbidity and possibly death from the genotype-specific cancers that would otherwise be in the patient’s future. For patients found to have a hereditary cancer syndrome, medical management options include more-frequent cancer screening or surveillance, prophylactic surgery, and preventive medical treatment, which will be reviewed in a future article in this series.
Identifying the specific mutation in one family member allows at-risk relatives, both female and male, to then take advantage of predictive testing, with genetic counseling. If they test positive for the risk-increasing mutation, they too can take advantage of the management options for people at high risk. If they test negative, they can continue to undergo the same screening as recommended for the general population. Also, they may be relieved to know that their cancer risk is no greater than that in the general population.
The American Society of Clinical Oncology9 recommends genetic counseling and testing when all of the following are true:
- There is a personal or family history suggesting genetic cancer susceptibility
- The test can be adequately interpreted
- The results will aid in the diagnosis or influence the medical or surgical management of the patient or family at hereditary risk of cancer.
Professional society guidelines also recommend that genetic testing be done only with genetic counseling before and after.5,6,8 The National Society of Genetic Counselors provides a list of clinical genetic counselors, organized by geographical area, at www.nsgc.org.
PATIENT 1 RECEIVES GENETIC TESTING AND COUNSELING
Let’s return to the Ashkenazi Jewish patient who has a personal and family history of breast cancer, whom you referred for cancer genetics consultation and who attends this appointment. A detailed personal and family history is gathered, and a brief physical examination is done, which reveals that the patient has macrocephaly and a history of multiple uterine fibroids.
The genetic differential diagnosis for your patient includes hereditary breast and ovarian cancer syndrome (resulting from mutations in the BRCA1 and BRCA2 genes) and Cowden syndrome (from mutations in the PTEN gene) (TABLE 3). The counselor uses BRCAPRO, a statistical risk-assessment tool that estimates a patient’s risk of harboring a BRCA1 or BRCA2 mutation based on ethnicity and personal and family history of cancer, and finds her risk to be 31%. In view of this risk, genetic testing for BRCA1 and BRCA2 is offered after a detailed discussion of the genetic differential diagnosis, the implications of a positive vs a negative test result, the possibility of finding gene changes (variants) of unknown significance, and the implications of the test results for family members.
Your patient elects to pursue BRCA1 and BRCA2 genetic testing and the results are negative—no mutations in either gene are found. PTEN testing is recommended next, which your patient elects to undergo. A mutation in the PTEN gene is found, indicating that she has Cowden syndrome. This result and its implications are discussed in a posttest genetic counseling session.
Cowden syndrome is an autosomal-dominant condition that carries a heightened risk of benign and malignant neoplasms, including a lifetime risk of breast cancer of up to 85%, with the average age at diagnosis in the 40s. Mutations in the PTEN gene also predispose to other cancer types, including nonmedullary thyroid, uterine, renal, and colorectal cancers, as well as melanoma.9 Multiple benign skin lesions and gastrointestinal polyposis are common.20
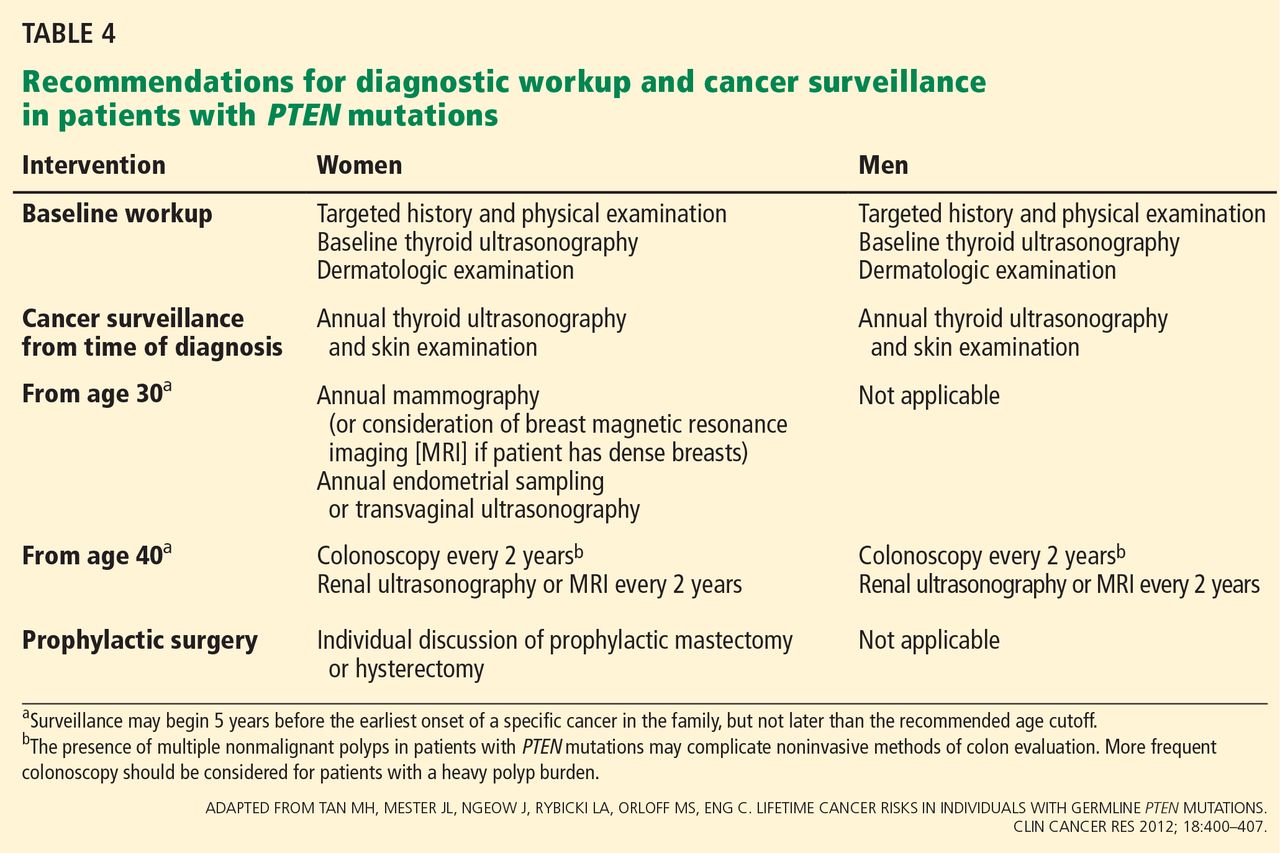
During the appointment, medical management options for patients with PTEN mutations are presented (Table 4).9 Given that your patient’s breast cancer was initially treated with lumpectomy, her remaining breast tissue is at risk of a second malignancy. She has never undergone thyroid imaging, colonoscopy, or kidney imaging. She reports that lately she has had occasional abnormal uterine bleeding and pain, which she believes are caused by her uterine fibroids. Given these symptoms and in light of her PTEN mutation, hysterectomy may be presented to her as an option. The genetics team sends a detailed clinical note directly to the primary care physician so they can coordinate and “quarterback” the patient’s care.
Like many patients, your patient is very concerned about how this information may affect her daughter. She first expresses some guilt at having to tell her daughter that she may have “given” her a risk of cancer. However, during the course of the genetic counseling session, she accepts that she could not have prevented her daughter from possibly inheriting this mutation, and understands that sharing this information will enable her daughter to pursue testing to help her understand her own risks.
When a known mutation exists in the family, as is the case with your patient, predictive testing only for that mutation gives a 100% accurate result. During a separate genetic counseling appointment, the patient’s daughter opts to proceed with testing and is found to be negative for her mother’s PTEN mutation.
WHAT HAPPENS WHEN GENETIC TESTING IS NOT INDICATED?
Cancer genetic risk assessment and counseling provides benefits even when genetic testing is not indicated. In some situations genetic testing is not warranted, but referral for heightened surveillance for breast cancer is deemed necessary. Patients who have a personal or family history of cancer can still gain from a detailed assessment of their personal and family history and may come away relieved after learning that they or their family members are not at high risk of developing cancer. Such patients or families may be classified as demonstrating either familial or sporadic breast cancer diagnoses.
Familial breast cancer
Familial breast cancers, believed to account for 15% to 20% of all cases of breast cancer, share features with hereditary breast cancer syndromes.4 In affected families, the frequency of breast cancer is higher than in the general population (multiple family members may be affected), and the age of onset tends to be close to that in the general population.
Members of a family with familial breast cancer who have not yet developed the disease may be at increased risk of it. Several risk-assessment tools (the Gail, Tyrer-Cuzick, Claus, and other models)21–25 use personal and family history to estimate breast cancer risk.
Depending on the assessed risk, additional options for screening and surveillance are available. The American Cancer Society recommends magnetic resonance imaging (MRI) in addition to annual mammography for women whose lifetime risk of breast cancer is greater than 20%. They also recommend that women at moderately increased risk (ie, 15%–20% lifetime risk) talk to their doctor about the benefits and limitations of adding MRI screening to yearly mammography.1
Sporadic breast cancer
Sporadic forms of breast cancer account for 70% to 80% of cases of breast cancer. Sporadic breast cancers are thought to have mainly nonhereditary causes, with environment and personal risk factors playing a large role.
Women with apparently sporadic breast cancers are diagnosed at or beyond the average age at diagnosis in the general population and do not have a family history that suggests either a hereditary cancer syndrome or familial breast cancer. If they undergo a cancer risk assessment, they may be relieved to learn that other women in their family do not have a high probability of being affected, and that they themselves do not appear to be at increased risk of other malignancies.
PATIENT 2: NEGATIVE TEST RESULTS ARE SOMETIMES ‘UNINFORMATIVE’
A healthy 35-year-old woman is referred for a genetics consultation by her gynecologist because her mother developed breast cancer at age 40 and died of the disease. A detailed personal and family history and risk assessment are done. After pretest genetic counseling, testing for BRCA1 and BRCA2 mutations (hereditary breast and ovarian cancer syndrome) is ordered, and the patient’s test results are negative. Risk assessment determines that no other hereditary cancer syndrome is likely. Therefore, no other genetic testing is offered at this time.
Genetic testing is most informative when performed first on the family member at highest risk of having a mutation. For families with breast cancer, this is typically the person with cancer diagnosed at the earliest age.
Unfortunately, sometimes these family members cannot be tested because they are deceased or otherwise unavailable. In such situations, it is acceptable to offer testing to a close, unaffected relative, such as your patient. Pretest genetic counseling in these circumstances is key, highlighting the fact that negative (normal) results would be uninformative. In your case, we cannot know whether the patient’s mother would have tested positive for a BRCA1 or BRCA2 mutation and your patient is a “true negative,” or whether her mother would have tested negative as well.
In unaffected patients with uninformative genetic testing results, medical management is based on the patient’s personal risk factors and family history of cancer. For your patient, statistical risk modeling tools (the Gail, Claus, Couch, and Tyrer-Cuzick models) determine that her risk of developing breast cancer is 22% to 28.5%, qualifying her for MRI along with yearly mammography per the American Cancer Society guidelines previously discussed.
KNOWLEDGE CONTINUES TO EXPAND
Major advances in the understanding of breast cancer susceptibility were made in the last decade through genetic linkage mapping in families that have an overabundance of members with breast cancer.26–28 Additionally, as more information is acquired, other genes predisposing to cancer or modifying cancer risk may be identified and additional knowledge gained.
With the advent of gene-panel-based testing and exome sequencing, we will incidentally discover mutations that predispose to cancer in patients in whom we were not looking for these mutations. With improving technology and value-based health care delivery, providers must continue to embrace multidisciplinary care, and genetics will become central in guiding medical management. In the event of an incidental finding suggesting susceptibility to heritable cancer, a consult to genetic counseling is recommended.
Many studies of the genetics of breast cancer are now focusing on known hereditary breast cancer syndromes and on possibilities for risk reduction, lifestyle modification, and identification of genetic variations that may increase or decrease cancer risk for an individual patient. The Center for Personalized Genetic Healthcare at Cleveland Clinic is collaborating in one such study. Titled “Risk Factor Analysis of Hereditary Breast and Ovarian Cancer Syndrome,” it is an international study led by a leading breast cancer researcher, Dr. Steven Narod from the Women’s College Research Institute in Toronto, ON. This study is focusing on women with a BRCA1 or BRCA2 mutation and their personal cancer risk factors, lifestyle choices, and overall development of cancer. This research group and others are also focusing on identifying genetic “modifiers” of cancer risk in these high-risk women.29
For patients who do not have a hereditary cancer syndrome, research is further exploring novel genes and their relation to breast cancer risk. One such study in our laboratory has found that several genes once thought only to cause an increased risk of hereditary paraganglioma may also predispose to breast and thyroid cancer.29,31 Additional research in this area is under way to clarify these risks.
GOOD SCIENCE, BAD MEDICINE?
Other research studies have identified a number of genes currently thought to be “moderately penetrant” for breast cancer risk, meaning that they may confer a risk of breast cancer slightly greater than that in the general population, but in some instances the risk has not been proven to be high enough to alter a patient’s management.32,33
Although a few clinical laboratories currently offer testing for these kinds of genes, the clinical utility of this testing is questionable. Before offering testing on a clinical basis, we need clear, consistent data on the types of cancers associated with these genes and on the lifetime percentage risk of acquiring these cancers. Currently, it is difficult to understand whether a variant in a moderately penetrant gene is the true explanation behind a patient’s breast cancer diagnosis. If such a variant is identified and family members pursue testing for it, should those family members who test negative be considered to have the same risk of cancer as the general population? And should family members testing positive be offered prophylactic surgical options?
Without more data these questions cannot be answered, and until such data are gathered, we believe that testing for moderately penetrant genes should not be performed outside of a research study. The Center for Personalized Genetic Healthcare in Cleveland Clinic’s Genomic Medicine Institute can assist in educating and coordinating patients’ enrollment in such research studies.
PUTTING IT ALL TOGETHER
Primary care physicians are the first-line providers to individuals and families, many of whom have a personal or family history of breast cancer. Identifying patients at risk of breast cancer and hereditary cancer syndromes can be challenging in this era of shortened appointment times and patients with complex medical histories.
Reviewing an individual’s personal and cancer family history is a necessary first step in considering appropriate medical management recommendations for cancer screening and prevention, the cornerstone of personalized health care. Patients with hereditary breast cancer syndromes and those with familial breast cancer can benefit from high-risk breast cancer surveillance.
Cancer genetics risk assessment ensures that the correct genetic testing is offered to the most appropriate patients, with personalized interpretation of results and provision of future management recommendations based on the individual patient’s personal and family history. Genetic counselors empower patients to make educated and informed decisions about genetic testing, cancer screening, and prevention.
As health care continues to focus more on prevention in this new era of genomic medicine and value-based delivery of health care, genetic counselors will serve as powerful allies to physicians.34
Acknowledgments: We would like to thank Dr. Colleen Clayton and Dr. Lynn Pattimakiel of the Medicine Institute, Cleveland Clinic, for their critical review of and thoughtful feedback on this manuscript.
- American Cancer Society. Breast cancer: detailed guide( 2013). http://www.cancer.org/Cancer/BreastCancer/DetailedGuide/index. Accessed November 12, 2013.
- McTiernan A, Gilligan MA, Redmond C. Assessing individual risk for breast cancer: risky business. J Clin Epidemiol 1997; 50:547–556.
- Teerlink CC, Albright FS, Lins L, Cannon-Albright LA. A comprehensive survey of cancer risks in extended families. Genet Med 2012; 14:107–114.
- National Comprehensive Cancer Network (NCCN). NCCN clinical practice guidelines in oncology. Breast cancer risk reduction (version 1.2013). http://www.nccn.org. Accessed November 21, 2013.
- National Comprehensive Cancer Network (NCCN). NCCN clinical practice guidelines in oncology. Genetic/familial high risk assessment: breast and ovarian (version 4.2013). http://www.nccn.org. Accessed November 21, 2013.
- National Comprehensive Cancer Network (NCCN). NCCN clinical practice guidelines in oncology. Breast cancer screening and diagnosis (version 2.2013). http://www.nccn.org. Accessed November 21, 2013.
- Mester JL, Schreiber AH, Moran RT. Genetic counselors: your partners in clinical practice. Cleve Clin J Med 2012; 79:560–568.
- Trepanier A, Ahrens M, McKinnon W, et al; National Society of Genetic Counselors. Genetic cancer risk assessment and counseling: recommendations of the National Society of Genetic Counselors. J Genet Couns 2004; 13:83–114.
- Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 2012; 18:400–407.
- Ford D, Easton DF, Stratton M, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998; 62:676–689.
- Liede A, Karlan BY, Narod SA. Cancer risks for male carriers of germline mutations in BRCA1 or BRCA2: a review of the literature. J Clin Oncol 2004; 22:735–742.
- Struewing JP, Hartge P, Wacholder S, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med 1997; 336:1401–1408.
- Birch JM, Hartley AL, Tricker KJ, et al. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res 1994; 54:1298–1304.
- Chompret A, Brugières L, Ronsin M, et al. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer 2000; 82:1932–1937.
- Gonzalez KD, Noltner KA, Buzin CH, et al. Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol 2009; 27:1250–1256.
- Varley JM. Germline TP53 mutations and Li-Fraumeni syndrome. Hum Mutat 2003; 21:313–320.
- Fitzgerald RC, Hardwick R, Huntsman D, et al; International Gastric Cancer Linkage Consortium. Hereditary diffuse gastric cancer: updated consensus guidelines for clinical management and directions for future research. J Med Genet 2010; 47:436–444.
- Hearle N, Schumacher V, Menko FH, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res 2006; 12:3209–3215.
- American Society of Clinical Oncology. American Society of Clinical Oncology policy statement update: genetic testing for cancer susceptibility. J Clin Oncol 2003; 21:2397–2406.
- Mester J, Eng C. When overgrowth bumps into cancer: the PTEN-opathies. Am J Med Genet C Semin Med Genet 2013; 163:114–121.
- Claus EB, Risch N, Thompson WD. Autosomal dominant inheritance of early-onset breast cancer. Implications for risk prediction. Cancer 1994; 73:643–651.
- Couch FJ, DeShano ML, Blackwood MA, et al. BRCA1 mutations in women attending clinics that evaluate the risk of breast cancer. N Engl J Med 1997; 336:1409–1415.
- Tyrer J, Duffy SW, Cuzick J. A breast cancer prediction model incorporating familial and personal risk factors. Stat Med 2004; 23:1111–1130.
- Gail MH, Anderson WF, Garcia-Closas M, Sherman ME. Absolute risk models for subtypes of breast cancer. J Natl Cancer Inst 2007; 99:1657–1659.
- Gail MH, Brinton LA, Byar DP, et al. Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst 1989; 81:1879–1886.
- Kent P, O’Donoghue JM, O’Hanlon DM, Kerin MJ, Maher DJ, Given HF. Linkage analysis and the susceptibility gene (BRCA-1) in familial breast cancer. Eur J Surg Oncol 1995; 21:240–241.
- Easton DF, Bishop DT, Ford D, Crockford GP. Genetic linkage analysis in familial breast and ovarian cancer: results from 214 families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1993; 52:678–701.
- Ormiston W. Hereditary breast cancer. Eur J Cancer Care (Engl) 1996; 5:13–20.
- Couch FJ, Wang X, McGuffog L, et al. Genome-wide association study in BRCA1 mutation carriers identifies novel loci associated with breast and ovarian cancer risk. PLoS Genet 2013; 9:e1003212.
- Bennett KL, Mester J, Eng C. Germline epigenetic regulation of KILLIN in Cowden and Cowden-like syndrome. JAMA 2010; 304:2724–2731.
- Ni Y, He X, Chen J, et al. Germline SDHx variants modify breast and thyroid cancer risks in Cowden and Cowden-like syndrome via FAD/NAD-dependent destabilization of p53. Hum Mol Genet 2012; 21:300–310.
- Casadei S, Norquist BM, Walsh T, et al. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res 2011; 71:2222–2229.
- Walsh T, Lee MK, Casadei S, et al. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci U S A 2010; 107:12629–12633.
- Eng C. Molecular genetics to genomic medicine: at the heart of value-based delivery of healthcare. Mol Genet Genom Med 2013; 1:4–6.
PATIENT 1: A PERSONAL AND FAMILY HISTORY OF BREAST CANCER
A 55-year-old Ashkenazi Jewish woman presents to your clinic for her annual physical. She reports that she had been diagnosed with breast cancer 10 years ago and that it had been treated with lumpectomy. You recall that Ashkenazi Jewish ethnicity and a diagnosis of breast cancer before age 50 are red flags for a hereditary cancer syndrome, and you ask about her family history of cancer. She reports that her mother was diagnosed with breast cancer in her 60s. The patient wants to know if her daughter should start breast cancer screening.
What do you do next?
Facing increasing demands and a plethora of information to be discussed in a short time, primary care physicians may find it challenging to inform patients about the possibility of a hereditary cancer syndrome, to assess the risk, to organize genetic testing if appropriate, and to counsel patients about their management options. As our knowledge of the genetics of breast cancer continues to expand, this information will become more detailed and complex.
Nevertheless, primary care physicians can help identify patients who may have a syndrome of inherited cancer predisposition or whose family history raises concern for familial breast cancer. Patients in both groups may be candidates for genetic risk assessment, for special management options for women at high risk, or for both.
This article provides an overview of inherited conditions associated with higher breast cancer risk, and guidelines to help physicians recognize patients in their own practice for whom a genetics referral may be appropriate.
BREAST CANCER IS COMPLEX AND HETEROGENEOUS
Breast cancer is the second-leading cause of cancer deaths in women. According to the American Cancer Society, an estimated 234,340 new cases of breast cancer are expected to be diagnosed in women in the United States in 2013, and about 2,240 new cases are expected in men; 39,620 women and 410 men are expected to die of it.1
Breast cancer is a complex and heterogeneous disease, influenced by many factors, of which female sex and increasing age are the most significant. Modifiable risk factors include obesity, use of combined hormone replacement therapy, and physical inactivity. Other risk factors include dense breast tissue, having had a breast biopsy in the past, the finding of atypical hyperplasia on biopsy, a history of high-dose chest radiation, and reproductive factors that include early menarche, late menopause, nulliparity, and birth of first child after age 30.
After female sex and age, family history of the disease is the most significant risk factor for breast cancer.2 If a woman has a first-degree relative (mother, sister, daughter) with breast cancer, her risk is 1.8 times higher, and if she has a second-degree relative (aunt, grandmother) with breast cancer, her risk is 1.3 times higher.3
Hereditary cancer predisposition syndromes account for 5% to 10% of cases of breast cancer. These are caused by a germline mutation in a highly penetrant gene that considerably increases the risk of malignancies of the breast and other tissues. These conditions are inherited in an autosomal-dominant fashion, with age of onset tending to be significantly—several decades—younger than the median age of onset in the general population. The most common of these is hereditary breast and ovarian cancer syndrome, caused by germline mutations of the BRCA1 or BRCA2 gene.
Familial breast cancers account for 15% to 20% of cases. Here, the women who develop breast cancer have multiple family members who are also affected but without an obvious inheritance pattern, and the age of onset is similar to that in the general population.4
Sporadic forms of breast cancer account for the remaining 70% to 80% of cases. Their development can be attributed mainly to nonhereditary causes, such as the environmental and personal risk factors listed above. In general, sporadic forms of breast cancer occur at older ages, with no particular inheritance pattern and with frequency of occurrence in a family comparable to that in the general population.
IS A GENETICS CONSULTATION NEEDED?
In the case described above, the primary care physician gathered basic information about the patient’s cancer-related personal and family history. Asking a few key questions (Table 1)5,6 can help physicians understand two important things: whether a more detailed assessment of genetic risk and counseling by a genetics professional are indicated, and whether the patient would benefit from additional cancer screening and prevention.
Table 2 summarizes the National Comprehensive Cancer Network’s recommendations for cancer genetics consultation.5 These red flags for a hereditary breast cancer syndrome can help primary care providers identify patients for whom a cancer genetics referral is appropriate. Of note: the maternal and paternal family histories are equally important.
Because our patient was diagnosed with breast cancer before age 50 and is of Ashkenazi Jewish ethnicity, she meets these criteria and warrants a cancer genetics consultation.
What is a cancer-focused genetic counseling session?
The tenets of genetic counseling, described previously in this series,7 are relevant to hereditary cancer syndromes. Cancer risk assessment and genetic counseling constitute the process of identifying and counseling individuals at risk of familial or hereditary cancer.8
As in other genetic counseling scenarios, a detailed pedigree (family tree) is taken, and this information, along with the patient’s personal medical history, allows a genetics specialist to determine if the presentation is most suggestive of sporadic, familial, or hereditary cancer.
A common misconception among patients is that there is a single genetic test for hereditary breast cancer, when in fact many highly penetrant predisposition genes have been linked to heightened risk (see below). The syndromes summarized in Table 35,9–18 are part of the differential diagnosis for every patient presenting with a personal or family history of breast cancer, and the detailed information from the personal and family history, ascertained during the assessment, ensures the right syndrome is explored within a family.
Cancer-focused genetic counseling may also help a patient or family process the psychological and emotional responses that can occur when cancer risk is discussed: eg, fear of cancer and death; guilt a parent may feel for passing on a genetic predisposition; and survivor guilt experienced by family members who test negative.
Genetic counselors are trained to recognize patients who may benefit from additional counseling. Not all patients pursuing cancer-focused genetic testing need a thorough evaluation by a psychologist, unlike those with adult-onset neurodegenerative conditions such as Huntington disease. Rather, the genetic counselor discusses the psychological implications of cancer-focused genetic testing and can refer the patient to a psychologist, therapist, social worker, or others if he or she feels the patient may benefit.8
Some patients come to a genetic counseling session with concerns about whether their insurance will pay for testing, and about whether they will face discrimination because of the testing results. In most situations, genetic testing is deemed medically necessary and is covered by the patient’s insurance. When testing is necessary, genetic counselors are skilled at preauthorizing it and writing letters of medical necessity. They are also familiar with laws and regulations that protect patients, such as the Genetic Information Nondiscrimination Act, which protects patients from insurance and employment discrimination.
Because a cancer-focused genetic counseling session typically lasts 1 hour, the counselor has enough time to address these and any other concerns that might prevent a patient who is otherwise interested in genetic testing from pursuing it.
HOW CAN GENETIC TESTING HELP?
Genetic testing for hereditary cancer syndromes can have personal benefit for the patient and at-risk family members.
Note that the syndromes in Table 3 all increase the risk of more than one type of cancer. Patients with these syndromes frequently receive care from multiple subspecialists to mitigate those risks. Guidelines exist for each of these syndromes and, if followed, may prevent the morbidity and possibly death from the genotype-specific cancers that would otherwise be in the patient’s future. For patients found to have a hereditary cancer syndrome, medical management options include more-frequent cancer screening or surveillance, prophylactic surgery, and preventive medical treatment, which will be reviewed in a future article in this series.
Identifying the specific mutation in one family member allows at-risk relatives, both female and male, to then take advantage of predictive testing, with genetic counseling. If they test positive for the risk-increasing mutation, they too can take advantage of the management options for people at high risk. If they test negative, they can continue to undergo the same screening as recommended for the general population. Also, they may be relieved to know that their cancer risk is no greater than that in the general population.
The American Society of Clinical Oncology9 recommends genetic counseling and testing when all of the following are true:
- There is a personal or family history suggesting genetic cancer susceptibility
- The test can be adequately interpreted
- The results will aid in the diagnosis or influence the medical or surgical management of the patient or family at hereditary risk of cancer.
Professional society guidelines also recommend that genetic testing be done only with genetic counseling before and after.5,6,8 The National Society of Genetic Counselors provides a list of clinical genetic counselors, organized by geographical area, at www.nsgc.org.
PATIENT 1 RECEIVES GENETIC TESTING AND COUNSELING
Let’s return to the Ashkenazi Jewish patient who has a personal and family history of breast cancer, whom you referred for cancer genetics consultation and who attends this appointment. A detailed personal and family history is gathered, and a brief physical examination is done, which reveals that the patient has macrocephaly and a history of multiple uterine fibroids.
The genetic differential diagnosis for your patient includes hereditary breast and ovarian cancer syndrome (resulting from mutations in the BRCA1 and BRCA2 genes) and Cowden syndrome (from mutations in the PTEN gene) (TABLE 3). The counselor uses BRCAPRO, a statistical risk-assessment tool that estimates a patient’s risk of harboring a BRCA1 or BRCA2 mutation based on ethnicity and personal and family history of cancer, and finds her risk to be 31%. In view of this risk, genetic testing for BRCA1 and BRCA2 is offered after a detailed discussion of the genetic differential diagnosis, the implications of a positive vs a negative test result, the possibility of finding gene changes (variants) of unknown significance, and the implications of the test results for family members.
Your patient elects to pursue BRCA1 and BRCA2 genetic testing and the results are negative—no mutations in either gene are found. PTEN testing is recommended next, which your patient elects to undergo. A mutation in the PTEN gene is found, indicating that she has Cowden syndrome. This result and its implications are discussed in a posttest genetic counseling session.
Cowden syndrome is an autosomal-dominant condition that carries a heightened risk of benign and malignant neoplasms, including a lifetime risk of breast cancer of up to 85%, with the average age at diagnosis in the 40s. Mutations in the PTEN gene also predispose to other cancer types, including nonmedullary thyroid, uterine, renal, and colorectal cancers, as well as melanoma.9 Multiple benign skin lesions and gastrointestinal polyposis are common.20
During the appointment, medical management options for patients with PTEN mutations are presented (Table 4).9 Given that your patient’s breast cancer was initially treated with lumpectomy, her remaining breast tissue is at risk of a second malignancy. She has never undergone thyroid imaging, colonoscopy, or kidney imaging. She reports that lately she has had occasional abnormal uterine bleeding and pain, which she believes are caused by her uterine fibroids. Given these symptoms and in light of her PTEN mutation, hysterectomy may be presented to her as an option. The genetics team sends a detailed clinical note directly to the primary care physician so they can coordinate and “quarterback” the patient’s care.
Like many patients, your patient is very concerned about how this information may affect her daughter. She first expresses some guilt at having to tell her daughter that she may have “given” her a risk of cancer. However, during the course of the genetic counseling session, she accepts that she could not have prevented her daughter from possibly inheriting this mutation, and understands that sharing this information will enable her daughter to pursue testing to help her understand her own risks.
When a known mutation exists in the family, as is the case with your patient, predictive testing only for that mutation gives a 100% accurate result. During a separate genetic counseling appointment, the patient’s daughter opts to proceed with testing and is found to be negative for her mother’s PTEN mutation.
WHAT HAPPENS WHEN GENETIC TESTING IS NOT INDICATED?
Cancer genetic risk assessment and counseling provides benefits even when genetic testing is not indicated. In some situations genetic testing is not warranted, but referral for heightened surveillance for breast cancer is deemed necessary. Patients who have a personal or family history of cancer can still gain from a detailed assessment of their personal and family history and may come away relieved after learning that they or their family members are not at high risk of developing cancer. Such patients or families may be classified as demonstrating either familial or sporadic breast cancer diagnoses.
Familial breast cancer
Familial breast cancers, believed to account for 15% to 20% of all cases of breast cancer, share features with hereditary breast cancer syndromes.4 In affected families, the frequency of breast cancer is higher than in the general population (multiple family members may be affected), and the age of onset tends to be close to that in the general population.
Members of a family with familial breast cancer who have not yet developed the disease may be at increased risk of it. Several risk-assessment tools (the Gail, Tyrer-Cuzick, Claus, and other models)21–25 use personal and family history to estimate breast cancer risk.
Depending on the assessed risk, additional options for screening and surveillance are available. The American Cancer Society recommends magnetic resonance imaging (MRI) in addition to annual mammography for women whose lifetime risk of breast cancer is greater than 20%. They also recommend that women at moderately increased risk (ie, 15%–20% lifetime risk) talk to their doctor about the benefits and limitations of adding MRI screening to yearly mammography.1
Sporadic breast cancer
Sporadic forms of breast cancer account for 70% to 80% of cases of breast cancer. Sporadic breast cancers are thought to have mainly nonhereditary causes, with environment and personal risk factors playing a large role.
Women with apparently sporadic breast cancers are diagnosed at or beyond the average age at diagnosis in the general population and do not have a family history that suggests either a hereditary cancer syndrome or familial breast cancer. If they undergo a cancer risk assessment, they may be relieved to learn that other women in their family do not have a high probability of being affected, and that they themselves do not appear to be at increased risk of other malignancies.
PATIENT 2: NEGATIVE TEST RESULTS ARE SOMETIMES ‘UNINFORMATIVE’
A healthy 35-year-old woman is referred for a genetics consultation by her gynecologist because her mother developed breast cancer at age 40 and died of the disease. A detailed personal and family history and risk assessment are done. After pretest genetic counseling, testing for BRCA1 and BRCA2 mutations (hereditary breast and ovarian cancer syndrome) is ordered, and the patient’s test results are negative. Risk assessment determines that no other hereditary cancer syndrome is likely. Therefore, no other genetic testing is offered at this time.
Genetic testing is most informative when performed first on the family member at highest risk of having a mutation. For families with breast cancer, this is typically the person with cancer diagnosed at the earliest age.
Unfortunately, sometimes these family members cannot be tested because they are deceased or otherwise unavailable. In such situations, it is acceptable to offer testing to a close, unaffected relative, such as your patient. Pretest genetic counseling in these circumstances is key, highlighting the fact that negative (normal) results would be uninformative. In your case, we cannot know whether the patient’s mother would have tested positive for a BRCA1 or BRCA2 mutation and your patient is a “true negative,” or whether her mother would have tested negative as well.
In unaffected patients with uninformative genetic testing results, medical management is based on the patient’s personal risk factors and family history of cancer. For your patient, statistical risk modeling tools (the Gail, Claus, Couch, and Tyrer-Cuzick models) determine that her risk of developing breast cancer is 22% to 28.5%, qualifying her for MRI along with yearly mammography per the American Cancer Society guidelines previously discussed.
KNOWLEDGE CONTINUES TO EXPAND
Major advances in the understanding of breast cancer susceptibility were made in the last decade through genetic linkage mapping in families that have an overabundance of members with breast cancer.26–28 Additionally, as more information is acquired, other genes predisposing to cancer or modifying cancer risk may be identified and additional knowledge gained.
With the advent of gene-panel-based testing and exome sequencing, we will incidentally discover mutations that predispose to cancer in patients in whom we were not looking for these mutations. With improving technology and value-based health care delivery, providers must continue to embrace multidisciplinary care, and genetics will become central in guiding medical management. In the event of an incidental finding suggesting susceptibility to heritable cancer, a consult to genetic counseling is recommended.
Many studies of the genetics of breast cancer are now focusing on known hereditary breast cancer syndromes and on possibilities for risk reduction, lifestyle modification, and identification of genetic variations that may increase or decrease cancer risk for an individual patient. The Center for Personalized Genetic Healthcare at Cleveland Clinic is collaborating in one such study. Titled “Risk Factor Analysis of Hereditary Breast and Ovarian Cancer Syndrome,” it is an international study led by a leading breast cancer researcher, Dr. Steven Narod from the Women’s College Research Institute in Toronto, ON. This study is focusing on women with a BRCA1 or BRCA2 mutation and their personal cancer risk factors, lifestyle choices, and overall development of cancer. This research group and others are also focusing on identifying genetic “modifiers” of cancer risk in these high-risk women.29
For patients who do not have a hereditary cancer syndrome, research is further exploring novel genes and their relation to breast cancer risk. One such study in our laboratory has found that several genes once thought only to cause an increased risk of hereditary paraganglioma may also predispose to breast and thyroid cancer.29,31 Additional research in this area is under way to clarify these risks.
GOOD SCIENCE, BAD MEDICINE?
Other research studies have identified a number of genes currently thought to be “moderately penetrant” for breast cancer risk, meaning that they may confer a risk of breast cancer slightly greater than that in the general population, but in some instances the risk has not been proven to be high enough to alter a patient’s management.32,33
Although a few clinical laboratories currently offer testing for these kinds of genes, the clinical utility of this testing is questionable. Before offering testing on a clinical basis, we need clear, consistent data on the types of cancers associated with these genes and on the lifetime percentage risk of acquiring these cancers. Currently, it is difficult to understand whether a variant in a moderately penetrant gene is the true explanation behind a patient’s breast cancer diagnosis. If such a variant is identified and family members pursue testing for it, should those family members who test negative be considered to have the same risk of cancer as the general population? And should family members testing positive be offered prophylactic surgical options?
Without more data these questions cannot be answered, and until such data are gathered, we believe that testing for moderately penetrant genes should not be performed outside of a research study. The Center for Personalized Genetic Healthcare in Cleveland Clinic’s Genomic Medicine Institute can assist in educating and coordinating patients’ enrollment in such research studies.
PUTTING IT ALL TOGETHER
Primary care physicians are the first-line providers to individuals and families, many of whom have a personal or family history of breast cancer. Identifying patients at risk of breast cancer and hereditary cancer syndromes can be challenging in this era of shortened appointment times and patients with complex medical histories.
Reviewing an individual’s personal and cancer family history is a necessary first step in considering appropriate medical management recommendations for cancer screening and prevention, the cornerstone of personalized health care. Patients with hereditary breast cancer syndromes and those with familial breast cancer can benefit from high-risk breast cancer surveillance.
Cancer genetics risk assessment ensures that the correct genetic testing is offered to the most appropriate patients, with personalized interpretation of results and provision of future management recommendations based on the individual patient’s personal and family history. Genetic counselors empower patients to make educated and informed decisions about genetic testing, cancer screening, and prevention.
As health care continues to focus more on prevention in this new era of genomic medicine and value-based delivery of health care, genetic counselors will serve as powerful allies to physicians.34
Acknowledgments: We would like to thank Dr. Colleen Clayton and Dr. Lynn Pattimakiel of the Medicine Institute, Cleveland Clinic, for their critical review of and thoughtful feedback on this manuscript.
PATIENT 1: A PERSONAL AND FAMILY HISTORY OF BREAST CANCER
A 55-year-old Ashkenazi Jewish woman presents to your clinic for her annual physical. She reports that she had been diagnosed with breast cancer 10 years ago and that it had been treated with lumpectomy. You recall that Ashkenazi Jewish ethnicity and a diagnosis of breast cancer before age 50 are red flags for a hereditary cancer syndrome, and you ask about her family history of cancer. She reports that her mother was diagnosed with breast cancer in her 60s. The patient wants to know if her daughter should start breast cancer screening.
What do you do next?
Facing increasing demands and a plethora of information to be discussed in a short time, primary care physicians may find it challenging to inform patients about the possibility of a hereditary cancer syndrome, to assess the risk, to organize genetic testing if appropriate, and to counsel patients about their management options. As our knowledge of the genetics of breast cancer continues to expand, this information will become more detailed and complex.
Nevertheless, primary care physicians can help identify patients who may have a syndrome of inherited cancer predisposition or whose family history raises concern for familial breast cancer. Patients in both groups may be candidates for genetic risk assessment, for special management options for women at high risk, or for both.
This article provides an overview of inherited conditions associated with higher breast cancer risk, and guidelines to help physicians recognize patients in their own practice for whom a genetics referral may be appropriate.
BREAST CANCER IS COMPLEX AND HETEROGENEOUS
Breast cancer is the second-leading cause of cancer deaths in women. According to the American Cancer Society, an estimated 234,340 new cases of breast cancer are expected to be diagnosed in women in the United States in 2013, and about 2,240 new cases are expected in men; 39,620 women and 410 men are expected to die of it.1
Breast cancer is a complex and heterogeneous disease, influenced by many factors, of which female sex and increasing age are the most significant. Modifiable risk factors include obesity, use of combined hormone replacement therapy, and physical inactivity. Other risk factors include dense breast tissue, having had a breast biopsy in the past, the finding of atypical hyperplasia on biopsy, a history of high-dose chest radiation, and reproductive factors that include early menarche, late menopause, nulliparity, and birth of first child after age 30.
After female sex and age, family history of the disease is the most significant risk factor for breast cancer.2 If a woman has a first-degree relative (mother, sister, daughter) with breast cancer, her risk is 1.8 times higher, and if she has a second-degree relative (aunt, grandmother) with breast cancer, her risk is 1.3 times higher.3
Hereditary cancer predisposition syndromes account for 5% to 10% of cases of breast cancer. These are caused by a germline mutation in a highly penetrant gene that considerably increases the risk of malignancies of the breast and other tissues. These conditions are inherited in an autosomal-dominant fashion, with age of onset tending to be significantly—several decades—younger than the median age of onset in the general population. The most common of these is hereditary breast and ovarian cancer syndrome, caused by germline mutations of the BRCA1 or BRCA2 gene.
Familial breast cancers account for 15% to 20% of cases. Here, the women who develop breast cancer have multiple family members who are also affected but without an obvious inheritance pattern, and the age of onset is similar to that in the general population.4
Sporadic forms of breast cancer account for the remaining 70% to 80% of cases. Their development can be attributed mainly to nonhereditary causes, such as the environmental and personal risk factors listed above. In general, sporadic forms of breast cancer occur at older ages, with no particular inheritance pattern and with frequency of occurrence in a family comparable to that in the general population.
IS A GENETICS CONSULTATION NEEDED?
In the case described above, the primary care physician gathered basic information about the patient’s cancer-related personal and family history. Asking a few key questions (Table 1)5,6 can help physicians understand two important things: whether a more detailed assessment of genetic risk and counseling by a genetics professional are indicated, and whether the patient would benefit from additional cancer screening and prevention.
Table 2 summarizes the National Comprehensive Cancer Network’s recommendations for cancer genetics consultation.5 These red flags for a hereditary breast cancer syndrome can help primary care providers identify patients for whom a cancer genetics referral is appropriate. Of note: the maternal and paternal family histories are equally important.
Because our patient was diagnosed with breast cancer before age 50 and is of Ashkenazi Jewish ethnicity, she meets these criteria and warrants a cancer genetics consultation.
What is a cancer-focused genetic counseling session?
The tenets of genetic counseling, described previously in this series,7 are relevant to hereditary cancer syndromes. Cancer risk assessment and genetic counseling constitute the process of identifying and counseling individuals at risk of familial or hereditary cancer.8
As in other genetic counseling scenarios, a detailed pedigree (family tree) is taken, and this information, along with the patient’s personal medical history, allows a genetics specialist to determine if the presentation is most suggestive of sporadic, familial, or hereditary cancer.
A common misconception among patients is that there is a single genetic test for hereditary breast cancer, when in fact many highly penetrant predisposition genes have been linked to heightened risk (see below). The syndromes summarized in Table 35,9–18 are part of the differential diagnosis for every patient presenting with a personal or family history of breast cancer, and the detailed information from the personal and family history, ascertained during the assessment, ensures the right syndrome is explored within a family.
Cancer-focused genetic counseling may also help a patient or family process the psychological and emotional responses that can occur when cancer risk is discussed: eg, fear of cancer and death; guilt a parent may feel for passing on a genetic predisposition; and survivor guilt experienced by family members who test negative.
Genetic counselors are trained to recognize patients who may benefit from additional counseling. Not all patients pursuing cancer-focused genetic testing need a thorough evaluation by a psychologist, unlike those with adult-onset neurodegenerative conditions such as Huntington disease. Rather, the genetic counselor discusses the psychological implications of cancer-focused genetic testing and can refer the patient to a psychologist, therapist, social worker, or others if he or she feels the patient may benefit.8
Some patients come to a genetic counseling session with concerns about whether their insurance will pay for testing, and about whether they will face discrimination because of the testing results. In most situations, genetic testing is deemed medically necessary and is covered by the patient’s insurance. When testing is necessary, genetic counselors are skilled at preauthorizing it and writing letters of medical necessity. They are also familiar with laws and regulations that protect patients, such as the Genetic Information Nondiscrimination Act, which protects patients from insurance and employment discrimination.
Because a cancer-focused genetic counseling session typically lasts 1 hour, the counselor has enough time to address these and any other concerns that might prevent a patient who is otherwise interested in genetic testing from pursuing it.
HOW CAN GENETIC TESTING HELP?
Genetic testing for hereditary cancer syndromes can have personal benefit for the patient and at-risk family members.
Note that the syndromes in Table 3 all increase the risk of more than one type of cancer. Patients with these syndromes frequently receive care from multiple subspecialists to mitigate those risks. Guidelines exist for each of these syndromes and, if followed, may prevent the morbidity and possibly death from the genotype-specific cancers that would otherwise be in the patient’s future. For patients found to have a hereditary cancer syndrome, medical management options include more-frequent cancer screening or surveillance, prophylactic surgery, and preventive medical treatment, which will be reviewed in a future article in this series.
Identifying the specific mutation in one family member allows at-risk relatives, both female and male, to then take advantage of predictive testing, with genetic counseling. If they test positive for the risk-increasing mutation, they too can take advantage of the management options for people at high risk. If they test negative, they can continue to undergo the same screening as recommended for the general population. Also, they may be relieved to know that their cancer risk is no greater than that in the general population.
The American Society of Clinical Oncology9 recommends genetic counseling and testing when all of the following are true:
- There is a personal or family history suggesting genetic cancer susceptibility
- The test can be adequately interpreted
- The results will aid in the diagnosis or influence the medical or surgical management of the patient or family at hereditary risk of cancer.
Professional society guidelines also recommend that genetic testing be done only with genetic counseling before and after.5,6,8 The National Society of Genetic Counselors provides a list of clinical genetic counselors, organized by geographical area, at www.nsgc.org.
PATIENT 1 RECEIVES GENETIC TESTING AND COUNSELING
Let’s return to the Ashkenazi Jewish patient who has a personal and family history of breast cancer, whom you referred for cancer genetics consultation and who attends this appointment. A detailed personal and family history is gathered, and a brief physical examination is done, which reveals that the patient has macrocephaly and a history of multiple uterine fibroids.
The genetic differential diagnosis for your patient includes hereditary breast and ovarian cancer syndrome (resulting from mutations in the BRCA1 and BRCA2 genes) and Cowden syndrome (from mutations in the PTEN gene) (TABLE 3). The counselor uses BRCAPRO, a statistical risk-assessment tool that estimates a patient’s risk of harboring a BRCA1 or BRCA2 mutation based on ethnicity and personal and family history of cancer, and finds her risk to be 31%. In view of this risk, genetic testing for BRCA1 and BRCA2 is offered after a detailed discussion of the genetic differential diagnosis, the implications of a positive vs a negative test result, the possibility of finding gene changes (variants) of unknown significance, and the implications of the test results for family members.
Your patient elects to pursue BRCA1 and BRCA2 genetic testing and the results are negative—no mutations in either gene are found. PTEN testing is recommended next, which your patient elects to undergo. A mutation in the PTEN gene is found, indicating that she has Cowden syndrome. This result and its implications are discussed in a posttest genetic counseling session.
Cowden syndrome is an autosomal-dominant condition that carries a heightened risk of benign and malignant neoplasms, including a lifetime risk of breast cancer of up to 85%, with the average age at diagnosis in the 40s. Mutations in the PTEN gene also predispose to other cancer types, including nonmedullary thyroid, uterine, renal, and colorectal cancers, as well as melanoma.9 Multiple benign skin lesions and gastrointestinal polyposis are common.20
During the appointment, medical management options for patients with PTEN mutations are presented (Table 4).9 Given that your patient’s breast cancer was initially treated with lumpectomy, her remaining breast tissue is at risk of a second malignancy. She has never undergone thyroid imaging, colonoscopy, or kidney imaging. She reports that lately she has had occasional abnormal uterine bleeding and pain, which she believes are caused by her uterine fibroids. Given these symptoms and in light of her PTEN mutation, hysterectomy may be presented to her as an option. The genetics team sends a detailed clinical note directly to the primary care physician so they can coordinate and “quarterback” the patient’s care.
Like many patients, your patient is very concerned about how this information may affect her daughter. She first expresses some guilt at having to tell her daughter that she may have “given” her a risk of cancer. However, during the course of the genetic counseling session, she accepts that she could not have prevented her daughter from possibly inheriting this mutation, and understands that sharing this information will enable her daughter to pursue testing to help her understand her own risks.
When a known mutation exists in the family, as is the case with your patient, predictive testing only for that mutation gives a 100% accurate result. During a separate genetic counseling appointment, the patient’s daughter opts to proceed with testing and is found to be negative for her mother’s PTEN mutation.
WHAT HAPPENS WHEN GENETIC TESTING IS NOT INDICATED?
Cancer genetic risk assessment and counseling provides benefits even when genetic testing is not indicated. In some situations genetic testing is not warranted, but referral for heightened surveillance for breast cancer is deemed necessary. Patients who have a personal or family history of cancer can still gain from a detailed assessment of their personal and family history and may come away relieved after learning that they or their family members are not at high risk of developing cancer. Such patients or families may be classified as demonstrating either familial or sporadic breast cancer diagnoses.
Familial breast cancer
Familial breast cancers, believed to account for 15% to 20% of all cases of breast cancer, share features with hereditary breast cancer syndromes.4 In affected families, the frequency of breast cancer is higher than in the general population (multiple family members may be affected), and the age of onset tends to be close to that in the general population.
Members of a family with familial breast cancer who have not yet developed the disease may be at increased risk of it. Several risk-assessment tools (the Gail, Tyrer-Cuzick, Claus, and other models)21–25 use personal and family history to estimate breast cancer risk.
Depending on the assessed risk, additional options for screening and surveillance are available. The American Cancer Society recommends magnetic resonance imaging (MRI) in addition to annual mammography for women whose lifetime risk of breast cancer is greater than 20%. They also recommend that women at moderately increased risk (ie, 15%–20% lifetime risk) talk to their doctor about the benefits and limitations of adding MRI screening to yearly mammography.1
Sporadic breast cancer
Sporadic forms of breast cancer account for 70% to 80% of cases of breast cancer. Sporadic breast cancers are thought to have mainly nonhereditary causes, with environment and personal risk factors playing a large role.
Women with apparently sporadic breast cancers are diagnosed at or beyond the average age at diagnosis in the general population and do not have a family history that suggests either a hereditary cancer syndrome or familial breast cancer. If they undergo a cancer risk assessment, they may be relieved to learn that other women in their family do not have a high probability of being affected, and that they themselves do not appear to be at increased risk of other malignancies.
PATIENT 2: NEGATIVE TEST RESULTS ARE SOMETIMES ‘UNINFORMATIVE’
A healthy 35-year-old woman is referred for a genetics consultation by her gynecologist because her mother developed breast cancer at age 40 and died of the disease. A detailed personal and family history and risk assessment are done. After pretest genetic counseling, testing for BRCA1 and BRCA2 mutations (hereditary breast and ovarian cancer syndrome) is ordered, and the patient’s test results are negative. Risk assessment determines that no other hereditary cancer syndrome is likely. Therefore, no other genetic testing is offered at this time.
Genetic testing is most informative when performed first on the family member at highest risk of having a mutation. For families with breast cancer, this is typically the person with cancer diagnosed at the earliest age.
Unfortunately, sometimes these family members cannot be tested because they are deceased or otherwise unavailable. In such situations, it is acceptable to offer testing to a close, unaffected relative, such as your patient. Pretest genetic counseling in these circumstances is key, highlighting the fact that negative (normal) results would be uninformative. In your case, we cannot know whether the patient’s mother would have tested positive for a BRCA1 or BRCA2 mutation and your patient is a “true negative,” or whether her mother would have tested negative as well.
In unaffected patients with uninformative genetic testing results, medical management is based on the patient’s personal risk factors and family history of cancer. For your patient, statistical risk modeling tools (the Gail, Claus, Couch, and Tyrer-Cuzick models) determine that her risk of developing breast cancer is 22% to 28.5%, qualifying her for MRI along with yearly mammography per the American Cancer Society guidelines previously discussed.
KNOWLEDGE CONTINUES TO EXPAND
Major advances in the understanding of breast cancer susceptibility were made in the last decade through genetic linkage mapping in families that have an overabundance of members with breast cancer.26–28 Additionally, as more information is acquired, other genes predisposing to cancer or modifying cancer risk may be identified and additional knowledge gained.
With the advent of gene-panel-based testing and exome sequencing, we will incidentally discover mutations that predispose to cancer in patients in whom we were not looking for these mutations. With improving technology and value-based health care delivery, providers must continue to embrace multidisciplinary care, and genetics will become central in guiding medical management. In the event of an incidental finding suggesting susceptibility to heritable cancer, a consult to genetic counseling is recommended.
Many studies of the genetics of breast cancer are now focusing on known hereditary breast cancer syndromes and on possibilities for risk reduction, lifestyle modification, and identification of genetic variations that may increase or decrease cancer risk for an individual patient. The Center for Personalized Genetic Healthcare at Cleveland Clinic is collaborating in one such study. Titled “Risk Factor Analysis of Hereditary Breast and Ovarian Cancer Syndrome,” it is an international study led by a leading breast cancer researcher, Dr. Steven Narod from the Women’s College Research Institute in Toronto, ON. This study is focusing on women with a BRCA1 or BRCA2 mutation and their personal cancer risk factors, lifestyle choices, and overall development of cancer. This research group and others are also focusing on identifying genetic “modifiers” of cancer risk in these high-risk women.29
For patients who do not have a hereditary cancer syndrome, research is further exploring novel genes and their relation to breast cancer risk. One such study in our laboratory has found that several genes once thought only to cause an increased risk of hereditary paraganglioma may also predispose to breast and thyroid cancer.29,31 Additional research in this area is under way to clarify these risks.
GOOD SCIENCE, BAD MEDICINE?
Other research studies have identified a number of genes currently thought to be “moderately penetrant” for breast cancer risk, meaning that they may confer a risk of breast cancer slightly greater than that in the general population, but in some instances the risk has not been proven to be high enough to alter a patient’s management.32,33
Although a few clinical laboratories currently offer testing for these kinds of genes, the clinical utility of this testing is questionable. Before offering testing on a clinical basis, we need clear, consistent data on the types of cancers associated with these genes and on the lifetime percentage risk of acquiring these cancers. Currently, it is difficult to understand whether a variant in a moderately penetrant gene is the true explanation behind a patient’s breast cancer diagnosis. If such a variant is identified and family members pursue testing for it, should those family members who test negative be considered to have the same risk of cancer as the general population? And should family members testing positive be offered prophylactic surgical options?
Without more data these questions cannot be answered, and until such data are gathered, we believe that testing for moderately penetrant genes should not be performed outside of a research study. The Center for Personalized Genetic Healthcare in Cleveland Clinic’s Genomic Medicine Institute can assist in educating and coordinating patients’ enrollment in such research studies.
PUTTING IT ALL TOGETHER
Primary care physicians are the first-line providers to individuals and families, many of whom have a personal or family history of breast cancer. Identifying patients at risk of breast cancer and hereditary cancer syndromes can be challenging in this era of shortened appointment times and patients with complex medical histories.
Reviewing an individual’s personal and cancer family history is a necessary first step in considering appropriate medical management recommendations for cancer screening and prevention, the cornerstone of personalized health care. Patients with hereditary breast cancer syndromes and those with familial breast cancer can benefit from high-risk breast cancer surveillance.
Cancer genetics risk assessment ensures that the correct genetic testing is offered to the most appropriate patients, with personalized interpretation of results and provision of future management recommendations based on the individual patient’s personal and family history. Genetic counselors empower patients to make educated and informed decisions about genetic testing, cancer screening, and prevention.
As health care continues to focus more on prevention in this new era of genomic medicine and value-based delivery of health care, genetic counselors will serve as powerful allies to physicians.34
Acknowledgments: We would like to thank Dr. Colleen Clayton and Dr. Lynn Pattimakiel of the Medicine Institute, Cleveland Clinic, for their critical review of and thoughtful feedback on this manuscript.
- American Cancer Society. Breast cancer: detailed guide( 2013). http://www.cancer.org/Cancer/BreastCancer/DetailedGuide/index. Accessed November 12, 2013.
- McTiernan A, Gilligan MA, Redmond C. Assessing individual risk for breast cancer: risky business. J Clin Epidemiol 1997; 50:547–556.
- Teerlink CC, Albright FS, Lins L, Cannon-Albright LA. A comprehensive survey of cancer risks in extended families. Genet Med 2012; 14:107–114.
- National Comprehensive Cancer Network (NCCN). NCCN clinical practice guidelines in oncology. Breast cancer risk reduction (version 1.2013). http://www.nccn.org. Accessed November 21, 2013.
- National Comprehensive Cancer Network (NCCN). NCCN clinical practice guidelines in oncology. Genetic/familial high risk assessment: breast and ovarian (version 4.2013). http://www.nccn.org. Accessed November 21, 2013.
- National Comprehensive Cancer Network (NCCN). NCCN clinical practice guidelines in oncology. Breast cancer screening and diagnosis (version 2.2013). http://www.nccn.org. Accessed November 21, 2013.
- Mester JL, Schreiber AH, Moran RT. Genetic counselors: your partners in clinical practice. Cleve Clin J Med 2012; 79:560–568.
- Trepanier A, Ahrens M, McKinnon W, et al; National Society of Genetic Counselors. Genetic cancer risk assessment and counseling: recommendations of the National Society of Genetic Counselors. J Genet Couns 2004; 13:83–114.
- Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 2012; 18:400–407.
- Ford D, Easton DF, Stratton M, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998; 62:676–689.
- Liede A, Karlan BY, Narod SA. Cancer risks for male carriers of germline mutations in BRCA1 or BRCA2: a review of the literature. J Clin Oncol 2004; 22:735–742.
- Struewing JP, Hartge P, Wacholder S, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med 1997; 336:1401–1408.
- Birch JM, Hartley AL, Tricker KJ, et al. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res 1994; 54:1298–1304.
- Chompret A, Brugières L, Ronsin M, et al. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer 2000; 82:1932–1937.
- Gonzalez KD, Noltner KA, Buzin CH, et al. Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol 2009; 27:1250–1256.
- Varley JM. Germline TP53 mutations and Li-Fraumeni syndrome. Hum Mutat 2003; 21:313–320.
- Fitzgerald RC, Hardwick R, Huntsman D, et al; International Gastric Cancer Linkage Consortium. Hereditary diffuse gastric cancer: updated consensus guidelines for clinical management and directions for future research. J Med Genet 2010; 47:436–444.
- Hearle N, Schumacher V, Menko FH, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res 2006; 12:3209–3215.
- American Society of Clinical Oncology. American Society of Clinical Oncology policy statement update: genetic testing for cancer susceptibility. J Clin Oncol 2003; 21:2397–2406.
- Mester J, Eng C. When overgrowth bumps into cancer: the PTEN-opathies. Am J Med Genet C Semin Med Genet 2013; 163:114–121.
- Claus EB, Risch N, Thompson WD. Autosomal dominant inheritance of early-onset breast cancer. Implications for risk prediction. Cancer 1994; 73:643–651.
- Couch FJ, DeShano ML, Blackwood MA, et al. BRCA1 mutations in women attending clinics that evaluate the risk of breast cancer. N Engl J Med 1997; 336:1409–1415.
- Tyrer J, Duffy SW, Cuzick J. A breast cancer prediction model incorporating familial and personal risk factors. Stat Med 2004; 23:1111–1130.
- Gail MH, Anderson WF, Garcia-Closas M, Sherman ME. Absolute risk models for subtypes of breast cancer. J Natl Cancer Inst 2007; 99:1657–1659.
- Gail MH, Brinton LA, Byar DP, et al. Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst 1989; 81:1879–1886.
- Kent P, O’Donoghue JM, O’Hanlon DM, Kerin MJ, Maher DJ, Given HF. Linkage analysis and the susceptibility gene (BRCA-1) in familial breast cancer. Eur J Surg Oncol 1995; 21:240–241.
- Easton DF, Bishop DT, Ford D, Crockford GP. Genetic linkage analysis in familial breast and ovarian cancer: results from 214 families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1993; 52:678–701.
- Ormiston W. Hereditary breast cancer. Eur J Cancer Care (Engl) 1996; 5:13–20.
- Couch FJ, Wang X, McGuffog L, et al. Genome-wide association study in BRCA1 mutation carriers identifies novel loci associated with breast and ovarian cancer risk. PLoS Genet 2013; 9:e1003212.
- Bennett KL, Mester J, Eng C. Germline epigenetic regulation of KILLIN in Cowden and Cowden-like syndrome. JAMA 2010; 304:2724–2731.
- Ni Y, He X, Chen J, et al. Germline SDHx variants modify breast and thyroid cancer risks in Cowden and Cowden-like syndrome via FAD/NAD-dependent destabilization of p53. Hum Mol Genet 2012; 21:300–310.
- Casadei S, Norquist BM, Walsh T, et al. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res 2011; 71:2222–2229.
- Walsh T, Lee MK, Casadei S, et al. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci U S A 2010; 107:12629–12633.
- Eng C. Molecular genetics to genomic medicine: at the heart of value-based delivery of healthcare. Mol Genet Genom Med 2013; 1:4–6.
- American Cancer Society. Breast cancer: detailed guide( 2013). http://www.cancer.org/Cancer/BreastCancer/DetailedGuide/index. Accessed November 12, 2013.
- McTiernan A, Gilligan MA, Redmond C. Assessing individual risk for breast cancer: risky business. J Clin Epidemiol 1997; 50:547–556.
- Teerlink CC, Albright FS, Lins L, Cannon-Albright LA. A comprehensive survey of cancer risks in extended families. Genet Med 2012; 14:107–114.
- National Comprehensive Cancer Network (NCCN). NCCN clinical practice guidelines in oncology. Breast cancer risk reduction (version 1.2013). http://www.nccn.org. Accessed November 21, 2013.
- National Comprehensive Cancer Network (NCCN). NCCN clinical practice guidelines in oncology. Genetic/familial high risk assessment: breast and ovarian (version 4.2013). http://www.nccn.org. Accessed November 21, 2013.
- National Comprehensive Cancer Network (NCCN). NCCN clinical practice guidelines in oncology. Breast cancer screening and diagnosis (version 2.2013). http://www.nccn.org. Accessed November 21, 2013.
- Mester JL, Schreiber AH, Moran RT. Genetic counselors: your partners in clinical practice. Cleve Clin J Med 2012; 79:560–568.
- Trepanier A, Ahrens M, McKinnon W, et al; National Society of Genetic Counselors. Genetic cancer risk assessment and counseling: recommendations of the National Society of Genetic Counselors. J Genet Couns 2004; 13:83–114.
- Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 2012; 18:400–407.
- Ford D, Easton DF, Stratton M, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998; 62:676–689.
- Liede A, Karlan BY, Narod SA. Cancer risks for male carriers of germline mutations in BRCA1 or BRCA2: a review of the literature. J Clin Oncol 2004; 22:735–742.
- Struewing JP, Hartge P, Wacholder S, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med 1997; 336:1401–1408.
- Birch JM, Hartley AL, Tricker KJ, et al. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res 1994; 54:1298–1304.
- Chompret A, Brugières L, Ronsin M, et al. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer 2000; 82:1932–1937.
- Gonzalez KD, Noltner KA, Buzin CH, et al. Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol 2009; 27:1250–1256.
- Varley JM. Germline TP53 mutations and Li-Fraumeni syndrome. Hum Mutat 2003; 21:313–320.
- Fitzgerald RC, Hardwick R, Huntsman D, et al; International Gastric Cancer Linkage Consortium. Hereditary diffuse gastric cancer: updated consensus guidelines for clinical management and directions for future research. J Med Genet 2010; 47:436–444.
- Hearle N, Schumacher V, Menko FH, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res 2006; 12:3209–3215.
- American Society of Clinical Oncology. American Society of Clinical Oncology policy statement update: genetic testing for cancer susceptibility. J Clin Oncol 2003; 21:2397–2406.
- Mester J, Eng C. When overgrowth bumps into cancer: the PTEN-opathies. Am J Med Genet C Semin Med Genet 2013; 163:114–121.
- Claus EB, Risch N, Thompson WD. Autosomal dominant inheritance of early-onset breast cancer. Implications for risk prediction. Cancer 1994; 73:643–651.
- Couch FJ, DeShano ML, Blackwood MA, et al. BRCA1 mutations in women attending clinics that evaluate the risk of breast cancer. N Engl J Med 1997; 336:1409–1415.
- Tyrer J, Duffy SW, Cuzick J. A breast cancer prediction model incorporating familial and personal risk factors. Stat Med 2004; 23:1111–1130.
- Gail MH, Anderson WF, Garcia-Closas M, Sherman ME. Absolute risk models for subtypes of breast cancer. J Natl Cancer Inst 2007; 99:1657–1659.
- Gail MH, Brinton LA, Byar DP, et al. Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst 1989; 81:1879–1886.
- Kent P, O’Donoghue JM, O’Hanlon DM, Kerin MJ, Maher DJ, Given HF. Linkage analysis and the susceptibility gene (BRCA-1) in familial breast cancer. Eur J Surg Oncol 1995; 21:240–241.
- Easton DF, Bishop DT, Ford D, Crockford GP. Genetic linkage analysis in familial breast and ovarian cancer: results from 214 families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1993; 52:678–701.
- Ormiston W. Hereditary breast cancer. Eur J Cancer Care (Engl) 1996; 5:13–20.
- Couch FJ, Wang X, McGuffog L, et al. Genome-wide association study in BRCA1 mutation carriers identifies novel loci associated with breast and ovarian cancer risk. PLoS Genet 2013; 9:e1003212.
- Bennett KL, Mester J, Eng C. Germline epigenetic regulation of KILLIN in Cowden and Cowden-like syndrome. JAMA 2010; 304:2724–2731.
- Ni Y, He X, Chen J, et al. Germline SDHx variants modify breast and thyroid cancer risks in Cowden and Cowden-like syndrome via FAD/NAD-dependent destabilization of p53. Hum Mol Genet 2012; 21:300–310.
- Casadei S, Norquist BM, Walsh T, et al. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res 2011; 71:2222–2229.
- Walsh T, Lee MK, Casadei S, et al. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci U S A 2010; 107:12629–12633.
- Eng C. Molecular genetics to genomic medicine: at the heart of value-based delivery of healthcare. Mol Genet Genom Med 2013; 1:4–6.
KEY POINTS
- Primary care physicians play a critical role in identifying patients at risk of inherited health problems.
- Hereditary cancers are important to detect because the age of onset is early, multiple primary cancers can develop, and cancer predisposition may be inherited.
- Hereditary syndromes account for only a minority of cases of breast cancer, but women who bear the responsible mutations have an extremely high risk.
- Patients with hereditary breast cancer syndromes and those with familial breast cancer can benefit from heightened surveillance for breast cancer.
- Cancer genetics risk assessment ensures that the correct genetic testing is offered to the most appropriate patients, with personalized interpretation of results and provision of future management recommendations based on the individual patient’s personal and family history.
Clinical applications of pharmacogenetics: Present and near future
“Change is the only constant.”
—Heraclitus (c 535–475 bce)
With the cost of health care rising and money to pay for it shrinking, there has never been a greater need to reduce waste.
Ineffective treatments and adverse drug effects account for much preventable morbidity and expense. New treatments, touted as more potent, are often introduced as replacements for traditional ones that are still effective in many patients, adding to costs and the potential for harm. For the pharmaceutical industry, the search for new “blockbuster” drugs seems to have hit a wall, at least in cardiovascular medicine.1 Advances often come at the cost of adverse effects, such as bleeding with triple antiplatelet therapy and diabetes with potent statin drugs.
The path to maximizing benefit and reducing harm now appears to lie in stratifying populations and appreciating patient individuality in response to treatment. For many decades we have known that patients vary widely in their response to drugs, owing to personal factors such as body surface area, age, environment, and genetics. And indeed, we treat our patients as individuals, for example by tailoring aminoglycoside dose to weight and renal function.
However, clinical trials typically give us an idea of the benefits only to the average patient. While subgroup analyses identify groups that may benefit more or less from treatment, the additional information they provide is not easily integrated into the clinical model of prescribing, in which one size fits all.
THE PROMISE OF PHARMACOGENETICS
The emerging field of pharmacogenetics promises to give clinicians the tools to make informed treatment decisions based on predictive genetic testing. This genetic testing aims to match treatment to an individual’s genetic profile. This often involves analyzing single-nucleotide polymorphisms in genes for enzymes that metabolize drugs, such as the cytochrome P450 enzymes, to predict efficacy or an adverse event with treatment.
Pharmacogenetics is playing an increasing role in clinical trials, particularly in the early stages of drug development, by helping to reduce the number of patients needed, prove efficacy, and identify subgroups in which alternative treatment can be targeted. At another level, a molecular understanding of disease is leading to truly targeted treatments based on genomics.
Over recent years, genetic testing has been increasingly used in clinical practice, thanks to a convergence of factors such as rapid, low-cost tests, a growing evidence base, and emerging interest among doctors and payers.
An advantage to using genetic testing as opposed to other types of laboratory testing, such as measuring the concentration of the drug in the blood during treatment, is that genetic tests can predict the response to treatment before the treatment is started. Moreover, with therapeutic drug monitoring after treatment has begun, there are sometimes no detectable measures of toxicity. For example, both carbamazepine and the antiviral drug abacavir can—fortunately only rarely—cause Stevens-Johnson syndrome. But before genetic markers were discovered, there was no method of estimating this risk apart from taking a family history.2,3 Considering the numbers of people involved, it was not feasible until recently to suggest genetic screening for patients starting on these drugs. However, the cost of genotyping and gene sequencing has been falling at a rate inversely faster than Moore’s law (an approximate annual doubling in computer power), and population genomics is becoming a reality.4
The US Food and Drug Administration (FDA) recognizes the current and future value of pharmacogenetics in drug safety and development. A number of approved pharmacogenetic biomarkers are listed on the FDA website (Table 1). Black box warnings have been mandated for a number of drugs on the basis of observational evidence.
The FDA also promotes rapid approval for novel drugs with pharmacogenetic “companion diagnostics.” A recent example of this was the approval of ivacaftor for cystic fibrosis patients who have the G551D mutation.5 Here, a molecular understanding of the condition led to the development of a targeted treatment. Although the cost of developing this drug was high, the path is now paved for similar advances. Oncologists are familiar with these advances with the emergence of new molecularly targeted treatments, eg, BRAF inhibitors in metastatic melanoma, imatinib in chronic myeloid leukemia, and gefitinib in non-small-cell lung cancer.
PHARMACOGENETICS IN CARDIOVASCULAR MEDICINE
Cardiovascular medicine also stands to benefit from rapid advances in pharmacogenetics.
While no treatment has been developed that targets the molecular basis of cardiovascular disease, a number of genomic biomarkers have emerged that identify patients at risk of adverse reactions or treatment failure. These include genetic tests to predict the maintenance dose and risk of bleeding with warfarin,6 the likelihood of myopathy and myositis with simvastatin,7 and the risk of recurrent thrombotic events with clopidogrel.8–10
Using pharmacogenetics in prescribing warfarin and its alternatives
The pharmacogenetics of warfarin has been extensively researched, but genotyping before prescribing this drug is not yet widely done.
In 2007, the FDA updated the labeling of warfarin to include information about the influence of two genes, VKORC1 and CYP2C9, on a patient’s response to this drug. In 2010 this was updated to add that testing for these genes could be used to predict the maintenance dose of the drug. Difficulties with algorithms used to integrate this into clinical practice have hindered adoption of this testing.
With the advent of new anticoagulants such as dabigatran, rivaroxaban, and apixaban, many have expected warfarin and its pharmacogenetics to become obsolete. However, the new agents cost considerably more. Further, they may not offer a very great advantage over warfarin: in the Randomized Evaluation of Long-term Anticoagulant Therapy (RE-LY) trial, the absolute risk reduction in intracranial hemorrhage with dabigatran vs warfarin was small.11 Therefore, dabigatran is probably not cost-effective in populations at low risk of bleeding.12 A cost-effectiveness analysis comparing warfarin with dabigatran in patients with uncomplicated atrial fibrillation has suggested that dabigatran is, however, cost-effective in patients at moderate risk.12
In the RE-LY trial, the international normalized ratios (INRs) of the patients in the warfarin group were in the therapeutic range only 64% of the time. The advantages of dabigatran over warfarin become less pronounced as warfarin control is tightened.13 Of note, pharmacogenetics and home monitoring of the INR have both been shown to lead to tighter control of the INR, with greater time within the therapeutic range.14,15
Moreover, genetic testing can help us reduce the number of bleeding events in patients taking warfarin.16 Patients who carry the CYP2C9*2 or CYP2C9*3 polymorphism metabolize S-warfarin slower and therefore have a threefold higher risk of hemorrhage after starting warfarin.17 We could speculate that patients carrying these variants may be better served by the newer anticoagulants, though this has not been tested in any clinical trial.
It is also worth appreciating that the conditions requiring anticoagulation, such as atrial fibrillation, also have a strong genetic basis. Variants in chromosomes 4q25, 1q21, and 16q22 have all been associated with atrial fibrillation.18 The risk of atrial fibrillation is five to six times higher in carriers of multiple variants within all of these loci.19 Genetic variants at 4q25 have been associated with the response to specific antiarrhythmic drug treatments,20 response to pulmonary vein isolation, 21,22 and direct-current cardioversion.23 One can imagine a future in which patients with palpitations, carrying multiple gene risk variants, will choose prolonged monitoring at home to confirm a diagnosis. They would then be provided with a personalized best management strategy, using their personal preferences, clinical data, and genetic profile to make a treatment decision.
Using pharmacogenetics in prescribing clopidogrel and its alternatives
The pharmacogenetics of clopidogrel is of particular interest, as it has the potential of establishing a rational basis for using newer antiplatelet drugs such as ticagrelor and prasugrel, which are considerably more expensive than generic clopidogrel.
Most of the people who do not respond to clopidogrel carry the common cytochrome P450 2C19 variants CYP2C19*2 or CYP2C19*3.9 These variants are present in particularly high frequency in Asians and African Americans, who often do not feature in large randomized trials.
Newer antiplatelet agents have failed to demonstrate consistent superiority to clopidogrel without a tradeoff of more bleeding. However, in the Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel-Thrombolysis in Myocardial Infarction 39 (TRITON TIMI-38),24,25 patients with the *2 variant receiving prasugrel had lower cardiovascular event rates than *2 carriers receiving clopidogrel.
Similarly, the patients who benefit the most from ticagrelor are carriers of the 2C19 nonresponder variants. In a large study, clopidogrel responders who did not carry either 2C19 nonresponder genetic variants or ABCB1 variants had cardiovascular outcomes similar to those of patients receiving ticagrelor.26
Clinicians have been cautious in prescribing potent antiplatelet agents to all patients because of the risk of bleeding. One could assume that by reserving newer agents for clopidogrel nonresponders, the bleeding risk could be minimized and overall benefit could be preserved with this strategy.
Cost may also be contained. The cost-effectiveness of such an approach with prasugrel has been tested with computer modeling and appears favorable.27 On the other hand, a similar yet limited analysis did not find genotype-driven use of ticagrelor to be cost-effective.28 This was mostly due to fewer deaths in patients receiving ticagrelor. However, the cost estimate for genotype-guided therapy was overestimated, as heterozygotes in the model were treated with ticagrelor instead of a high dose of clopidogrel.
It now appears that heterozygotes, ie, patients with one copy of the nonresponder variant, can achieve similar platelet inhibition with clopidogrel 225 mg daily as noncarriers on 75 mg daily.29 Since genotype-guided antiplatelet therapy has not been tested in a randomized outcomes trial, this tailored strategy has not been widely accepted.
THE FUTURE
The barriers to adoption of pharmacogenetics are considerable. Clinicians need to be educated about it, reimbursement needs to be worked out, and the pharmaceutical industry needs to get behind it. Nevertheless, the future of pharmacogenetics is extremely promising.
Research networks are forming to support the use of pharmacogenetics in clinical practice. The Pharmgkb (www.pharmgkb.org) database serves as a hub for educating clinicians and researchers as well as curating data for reference. Vanderbilt University is piloting the BioVu project, in which DNA and genotype data on patients are being stored and matched to the electronic clinical records.30 These projects not only provide clinically useful information on the current state of the art of pharmacogenetics, they also aid in disseminating new information about genotype-phenotype relationships.
Analytical software that uses “natural language processing” is being applied to clinician-generated notes to derive new observations and associations between genetic variants and clinical phenotypes. Integrating this information in real-time decision-support modules in the electronic health record provides a feedback loop for a rapid assimilation of new knowledge. Similar innovative decision-support modules are being established by Cleveland Clinic’s Center for Personalized Healthcare.31
The rise of ‘omic’ sciences
Pharmacogenetics and pharmacogenomics are part of a larger set of “omic” sciences. The suffix “-omics” implies a larger, more holistic view and is being applied to a number of fields—for example, the study of proteins (proteomics) and the study of metabolites (metabolomics). Profiling proteins and metabolites delivers a deluge of information on a patient that can be clustered, using pattern-recognition software, into population subgroups. Patterns of multiple proteins or metabolites are extracted from this spectral data to identify disease or response to treatment (pharmacometabolomics).
Metabolomics has been shown to predict the response to statins,32 diagnose myocardial infarction,33 and reclassify cardiovascular risk status.34 In addition, whereas traditional laboratory chemistry is reductionist, using single biomarkers for single-disease diagnosis, omic technologies hold the potential to reveal information on a number of possible health or disease states. The identification of “healthy” profiles using these technologies can potentially provide positive feedback to patients undertaking lifestyle changes and treatment.
The instrument costs for proteomic and metabolomic profiling are relatively high. However, the ongoing running costs are minimal, estimated at as low as less than $13 per test, as there are no expensive reagents.33 High-volume testing therefore becomes very cost-effective.
Although omic science appears futuristic, proteomics and metabolomics are already used in many clinical laboratories to rapidly identify bacteria. These methods have already revolutionized the way laboratories identify microbes, since they are automated, reduce workload, and give very fast results.
The cost of genetic testing is falling
Critics of pharmacogenetics claim that the predictive value of genetic testing is poor, that evidence is lacking, and that the cost is too high. In all new technologies, the first iteration is coarse, but performance improves with use. The first major barrier is adoption. Projects like BioVu are establishing the infrastructure for a feedback loop to iteratively improve upon the status quo and provide the evidence base clinicians demand.
The cost of genetic testing is falling rapidly, with whole-genome sequencing and annotation now costing less than $5,000. The cost of a pharmacogenetic test can be as low as $100 using low-cost nanotechnology, and the test needs to be performed only once in a patient’s lifetime.27
As other related molecular technologies such as proteomics and metabolomics become available and are integrated with genomics, the predictive ability of this science will improve.
AWAY FROM ONE-SIZE-FITS-ALL MEDICINE
Over the last decade there has been a trend away from “one size fits all” to customized “markets of one” in everything from consumer products to education to medicine. Mass customizing, also known as personalization, has been embraced by the internet community as a means to increase efficiency and reduce cost. This occurs by eliminating waste in redundant work or production of ineffective products.
Personalization on the Internet has been enabled through the use of informatics, mathematics, and supercomputing. The same tools that have personalized the delivery of consumer products are also being applied to the field of pharmacogenetics. Applied in an evidence-based fashion, these new technologies should profoundly improve patient care now and in the future.
- Topol EJ. Past the wall in cardiovascular R&D. Nat Rev Drug Discov 2009; 8:259.
- Mallal S, Phillips E, Carosi G, et al; PREDICT-1 Study Team. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med 2008; 358:568–579.
- Hung SI, Chung WH, Jee SH, et al. Genetic susceptibility to carbamazepine-induced cutaneous adverse drug reactions. Pharmacogenet Genomics 2006; 16:297–306.
- Phimister EG, Feero WG, Guttmacher AE. Realizing genomic medicine. N Engl J Med 2012; 366:757–759.
- Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA 2009; 106:18825–18830.
- International Warfarin Pharmacogenetics Consortium; Klein TE, Altman RB, Eriksson N, et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med 2009; 360:753–764.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Simon T, Verstuyft C, Mary-Krause M, et al; French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360:363–375.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med 2009; 360:354–362
- Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 2009; 373:309–317.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Shah SV, Gage BF. Cost-effectiveness of dabigatran for stroke prophylaxis in atrial fibrillation. Circulation 2011; 123:2562–2570.
- Wallentin L, Yusuf S, Ezekowitz MD, et al; RE-LY investigators. Efficacy and safety of dabigatran compared with warfarin at different levels of international normalised ratio control for stroke prevention in atrial fibrillation: an analysis of the RE-LY trial. Lancet 2010; 376:975–983.
- Anderson JL, Horne BD, Stevens SM, et al; Couma-Gen Investigators. Randomized trial of genotype-guided versus standard warfarin dosing in patients initiating oral anticoagulation. Circulation 2007; 116:2563–2570.
- Matchar DB, Jacobson A, Dolor R, et al; THINRS Executive Committee and Site Investigators. Effect of home testing of international normalized ratio on clinical events. N Engl J Med 2010; 363:1608–1620.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study). J Am Coll Cardiol 2010; 55:2804–2812.
- Sanderson S, Emery J, Higgins J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: a HuGEnet systematic review and meta-analysis. Genet Med 2005; 7:97–104.
- Ellinor PT, Lunetta KL, Albert CM, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet 2012; 44:670–675.
- Lubitz SA, Sinner MF, Lunetta KL, et al. Independent susceptibility markers for atrial fibrillation on chromosome 4q25. Circulation 2010; 122:976–984.
- Parvez B, Vaglio J, Rowan S, et al. Symptomatic response to antiarrhythmic drug therapy is modulated by a common single nucleotide polymorphism in atrial fibrillation. J Am Coll Cardiol 2012; 60:539–545.
- Husser D, Adams V, Piorkowski C, Hindricks G, Bollmann A. Chromosome 4q25 variants and atrial fibrillation recurrence after catheter ablation. J Am Coll Cardiol 2010; 55:747–753.
- Benjamin Shoemaker M, Muhammad R, Parvez B, et al. Common atrial fibrillation risk alleles at 4q25 predict recurrence after catheter-based atrial fibrillation ablation.” Heart Rhythm 2012; Nov 23.pii: S1547-5271(12)01340–9. 10.1016/j.hrthm.2012.11.012. [Epub ahead of print]
- Parvez B, Benjamin Shoemaker M, Muhammad R, et al. Common genetic polymorphism at 4q25 locus predicts atrial fibrillation recurrence after successful cardioversion. Heart Rhythm 2013 Feb 18.pii: S1547-5271(13)00161–6. 10.1016/j.hrthm.2013.02.018. [Epub ahead of print]
- Mega JL, Close SL, Wiviott SD, et al. Genetic variants in ABCB1 and CYP2C19 and cardiovascular outcomes after treatment with clopidogrel and prasugrel in the TRITON-TIMI 38 trial: a pharmacogenetic analysis. Lancet 2010; 376:1312–1319.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome P450 genetic polymorphisms and the response to prasugrel: relationship to pharmacokinetic, pharmacodynamic, and clinical outcomes. Circulation 2009; 119:2553–2560.
- Wallentin L, James S, Storey RF, et al; PLATO investigators. Effect of CYP2C19 and ABCB1 single nucleotide polymorphisms on outcomes of treatment with ticagrelor versus clopidogrel for acute coronary syndromes: a genetic substudy of the PLATO trial. Lancet 2010; 376:1320–1328.
- Guzauskas GF, Hughes DA, Bradley SM, Veenstra DL. A risk-benefit assessment of prasugrel, clopidogrel, and genotype-guided therapy in patients undergoing percutaneous coronary intervention. Clin Pharmacol Ther 2012; 91:829–837.
- Crespin DJ, Federspiel JJ, Biddle AK, Jonas DE, Rossi JS. Ticagrelor versus genotype-driven antiplatelet therapy for secondary prevention after acute coronary syndrome: a cost-effectiveness analysis. Value Health 2011; 14:483–491.
- Mega JL, Hochholzer W, Frelinger AL, et al. Dosing clopidogrel based on CYP2C19 genotype and the effect on platelet reactivity in patients with stable cardiovascular disease. JAMA 2011; 306:2221–2228.
- Xu H, Jiang M, Oetjens M, et al. Facilitating pharmacogenetic studies using electronic health records and natural-language processing: a case study of warfarin. J Am Med Inform Assoc 2011; 18:387–391.
- Teng K, Eng C, Hess CA, et al. Building an innovative model for personalized healthcare. Cleve Clin J Med 2012; 79( suppl 1):S1–S9.
- Kaddurah-Daouk R, Baillie RA, Zhu H, et al. Enteric microbiome metabolites correlate with response to simvastatin treatment. PLoS One 2011; 6:e25482.
- Bodi V, Sanchis J, Morales JM, et al. Metabolomic profile of human myocardial ischemia by nuclear magnetic resonance spectroscopy of peripheral blood serum: a translational study based on transient coronary occlusion models. J Am Coll Cardiol 2012; 59:1629–1641.
- Shah SH, Sun JL, Stevens RD, et al. Baseline metabolomic profiles predict cardiovascular events in patients at risk for coronary artery disease. Am Heart J 2012; 163:844–850.e1.
“Change is the only constant.”
—Heraclitus (c 535–475 bce)
With the cost of health care rising and money to pay for it shrinking, there has never been a greater need to reduce waste.
Ineffective treatments and adverse drug effects account for much preventable morbidity and expense. New treatments, touted as more potent, are often introduced as replacements for traditional ones that are still effective in many patients, adding to costs and the potential for harm. For the pharmaceutical industry, the search for new “blockbuster” drugs seems to have hit a wall, at least in cardiovascular medicine.1 Advances often come at the cost of adverse effects, such as bleeding with triple antiplatelet therapy and diabetes with potent statin drugs.
The path to maximizing benefit and reducing harm now appears to lie in stratifying populations and appreciating patient individuality in response to treatment. For many decades we have known that patients vary widely in their response to drugs, owing to personal factors such as body surface area, age, environment, and genetics. And indeed, we treat our patients as individuals, for example by tailoring aminoglycoside dose to weight and renal function.
However, clinical trials typically give us an idea of the benefits only to the average patient. While subgroup analyses identify groups that may benefit more or less from treatment, the additional information they provide is not easily integrated into the clinical model of prescribing, in which one size fits all.
THE PROMISE OF PHARMACOGENETICS
The emerging field of pharmacogenetics promises to give clinicians the tools to make informed treatment decisions based on predictive genetic testing. This genetic testing aims to match treatment to an individual’s genetic profile. This often involves analyzing single-nucleotide polymorphisms in genes for enzymes that metabolize drugs, such as the cytochrome P450 enzymes, to predict efficacy or an adverse event with treatment.
Pharmacogenetics is playing an increasing role in clinical trials, particularly in the early stages of drug development, by helping to reduce the number of patients needed, prove efficacy, and identify subgroups in which alternative treatment can be targeted. At another level, a molecular understanding of disease is leading to truly targeted treatments based on genomics.
Over recent years, genetic testing has been increasingly used in clinical practice, thanks to a convergence of factors such as rapid, low-cost tests, a growing evidence base, and emerging interest among doctors and payers.
An advantage to using genetic testing as opposed to other types of laboratory testing, such as measuring the concentration of the drug in the blood during treatment, is that genetic tests can predict the response to treatment before the treatment is started. Moreover, with therapeutic drug monitoring after treatment has begun, there are sometimes no detectable measures of toxicity. For example, both carbamazepine and the antiviral drug abacavir can—fortunately only rarely—cause Stevens-Johnson syndrome. But before genetic markers were discovered, there was no method of estimating this risk apart from taking a family history.2,3 Considering the numbers of people involved, it was not feasible until recently to suggest genetic screening for patients starting on these drugs. However, the cost of genotyping and gene sequencing has been falling at a rate inversely faster than Moore’s law (an approximate annual doubling in computer power), and population genomics is becoming a reality.4
The US Food and Drug Administration (FDA) recognizes the current and future value of pharmacogenetics in drug safety and development. A number of approved pharmacogenetic biomarkers are listed on the FDA website (Table 1). Black box warnings have been mandated for a number of drugs on the basis of observational evidence.
The FDA also promotes rapid approval for novel drugs with pharmacogenetic “companion diagnostics.” A recent example of this was the approval of ivacaftor for cystic fibrosis patients who have the G551D mutation.5 Here, a molecular understanding of the condition led to the development of a targeted treatment. Although the cost of developing this drug was high, the path is now paved for similar advances. Oncologists are familiar with these advances with the emergence of new molecularly targeted treatments, eg, BRAF inhibitors in metastatic melanoma, imatinib in chronic myeloid leukemia, and gefitinib in non-small-cell lung cancer.
PHARMACOGENETICS IN CARDIOVASCULAR MEDICINE
Cardiovascular medicine also stands to benefit from rapid advances in pharmacogenetics.
While no treatment has been developed that targets the molecular basis of cardiovascular disease, a number of genomic biomarkers have emerged that identify patients at risk of adverse reactions or treatment failure. These include genetic tests to predict the maintenance dose and risk of bleeding with warfarin,6 the likelihood of myopathy and myositis with simvastatin,7 and the risk of recurrent thrombotic events with clopidogrel.8–10
Using pharmacogenetics in prescribing warfarin and its alternatives
The pharmacogenetics of warfarin has been extensively researched, but genotyping before prescribing this drug is not yet widely done.
In 2007, the FDA updated the labeling of warfarin to include information about the influence of two genes, VKORC1 and CYP2C9, on a patient’s response to this drug. In 2010 this was updated to add that testing for these genes could be used to predict the maintenance dose of the drug. Difficulties with algorithms used to integrate this into clinical practice have hindered adoption of this testing.
With the advent of new anticoagulants such as dabigatran, rivaroxaban, and apixaban, many have expected warfarin and its pharmacogenetics to become obsolete. However, the new agents cost considerably more. Further, they may not offer a very great advantage over warfarin: in the Randomized Evaluation of Long-term Anticoagulant Therapy (RE-LY) trial, the absolute risk reduction in intracranial hemorrhage with dabigatran vs warfarin was small.11 Therefore, dabigatran is probably not cost-effective in populations at low risk of bleeding.12 A cost-effectiveness analysis comparing warfarin with dabigatran in patients with uncomplicated atrial fibrillation has suggested that dabigatran is, however, cost-effective in patients at moderate risk.12
In the RE-LY trial, the international normalized ratios (INRs) of the patients in the warfarin group were in the therapeutic range only 64% of the time. The advantages of dabigatran over warfarin become less pronounced as warfarin control is tightened.13 Of note, pharmacogenetics and home monitoring of the INR have both been shown to lead to tighter control of the INR, with greater time within the therapeutic range.14,15
Moreover, genetic testing can help us reduce the number of bleeding events in patients taking warfarin.16 Patients who carry the CYP2C9*2 or CYP2C9*3 polymorphism metabolize S-warfarin slower and therefore have a threefold higher risk of hemorrhage after starting warfarin.17 We could speculate that patients carrying these variants may be better served by the newer anticoagulants, though this has not been tested in any clinical trial.
It is also worth appreciating that the conditions requiring anticoagulation, such as atrial fibrillation, also have a strong genetic basis. Variants in chromosomes 4q25, 1q21, and 16q22 have all been associated with atrial fibrillation.18 The risk of atrial fibrillation is five to six times higher in carriers of multiple variants within all of these loci.19 Genetic variants at 4q25 have been associated with the response to specific antiarrhythmic drug treatments,20 response to pulmonary vein isolation, 21,22 and direct-current cardioversion.23 One can imagine a future in which patients with palpitations, carrying multiple gene risk variants, will choose prolonged monitoring at home to confirm a diagnosis. They would then be provided with a personalized best management strategy, using their personal preferences, clinical data, and genetic profile to make a treatment decision.
Using pharmacogenetics in prescribing clopidogrel and its alternatives
The pharmacogenetics of clopidogrel is of particular interest, as it has the potential of establishing a rational basis for using newer antiplatelet drugs such as ticagrelor and prasugrel, which are considerably more expensive than generic clopidogrel.
Most of the people who do not respond to clopidogrel carry the common cytochrome P450 2C19 variants CYP2C19*2 or CYP2C19*3.9 These variants are present in particularly high frequency in Asians and African Americans, who often do not feature in large randomized trials.
Newer antiplatelet agents have failed to demonstrate consistent superiority to clopidogrel without a tradeoff of more bleeding. However, in the Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel-Thrombolysis in Myocardial Infarction 39 (TRITON TIMI-38),24,25 patients with the *2 variant receiving prasugrel had lower cardiovascular event rates than *2 carriers receiving clopidogrel.
Similarly, the patients who benefit the most from ticagrelor are carriers of the 2C19 nonresponder variants. In a large study, clopidogrel responders who did not carry either 2C19 nonresponder genetic variants or ABCB1 variants had cardiovascular outcomes similar to those of patients receiving ticagrelor.26
Clinicians have been cautious in prescribing potent antiplatelet agents to all patients because of the risk of bleeding. One could assume that by reserving newer agents for clopidogrel nonresponders, the bleeding risk could be minimized and overall benefit could be preserved with this strategy.
Cost may also be contained. The cost-effectiveness of such an approach with prasugrel has been tested with computer modeling and appears favorable.27 On the other hand, a similar yet limited analysis did not find genotype-driven use of ticagrelor to be cost-effective.28 This was mostly due to fewer deaths in patients receiving ticagrelor. However, the cost estimate for genotype-guided therapy was overestimated, as heterozygotes in the model were treated with ticagrelor instead of a high dose of clopidogrel.
It now appears that heterozygotes, ie, patients with one copy of the nonresponder variant, can achieve similar platelet inhibition with clopidogrel 225 mg daily as noncarriers on 75 mg daily.29 Since genotype-guided antiplatelet therapy has not been tested in a randomized outcomes trial, this tailored strategy has not been widely accepted.
THE FUTURE
The barriers to adoption of pharmacogenetics are considerable. Clinicians need to be educated about it, reimbursement needs to be worked out, and the pharmaceutical industry needs to get behind it. Nevertheless, the future of pharmacogenetics is extremely promising.
Research networks are forming to support the use of pharmacogenetics in clinical practice. The Pharmgkb (www.pharmgkb.org) database serves as a hub for educating clinicians and researchers as well as curating data for reference. Vanderbilt University is piloting the BioVu project, in which DNA and genotype data on patients are being stored and matched to the electronic clinical records.30 These projects not only provide clinically useful information on the current state of the art of pharmacogenetics, they also aid in disseminating new information about genotype-phenotype relationships.
Analytical software that uses “natural language processing” is being applied to clinician-generated notes to derive new observations and associations between genetic variants and clinical phenotypes. Integrating this information in real-time decision-support modules in the electronic health record provides a feedback loop for a rapid assimilation of new knowledge. Similar innovative decision-support modules are being established by Cleveland Clinic’s Center for Personalized Healthcare.31
The rise of ‘omic’ sciences
Pharmacogenetics and pharmacogenomics are part of a larger set of “omic” sciences. The suffix “-omics” implies a larger, more holistic view and is being applied to a number of fields—for example, the study of proteins (proteomics) and the study of metabolites (metabolomics). Profiling proteins and metabolites delivers a deluge of information on a patient that can be clustered, using pattern-recognition software, into population subgroups. Patterns of multiple proteins or metabolites are extracted from this spectral data to identify disease or response to treatment (pharmacometabolomics).
Metabolomics has been shown to predict the response to statins,32 diagnose myocardial infarction,33 and reclassify cardiovascular risk status.34 In addition, whereas traditional laboratory chemistry is reductionist, using single biomarkers for single-disease diagnosis, omic technologies hold the potential to reveal information on a number of possible health or disease states. The identification of “healthy” profiles using these technologies can potentially provide positive feedback to patients undertaking lifestyle changes and treatment.
The instrument costs for proteomic and metabolomic profiling are relatively high. However, the ongoing running costs are minimal, estimated at as low as less than $13 per test, as there are no expensive reagents.33 High-volume testing therefore becomes very cost-effective.
Although omic science appears futuristic, proteomics and metabolomics are already used in many clinical laboratories to rapidly identify bacteria. These methods have already revolutionized the way laboratories identify microbes, since they are automated, reduce workload, and give very fast results.
The cost of genetic testing is falling
Critics of pharmacogenetics claim that the predictive value of genetic testing is poor, that evidence is lacking, and that the cost is too high. In all new technologies, the first iteration is coarse, but performance improves with use. The first major barrier is adoption. Projects like BioVu are establishing the infrastructure for a feedback loop to iteratively improve upon the status quo and provide the evidence base clinicians demand.
The cost of genetic testing is falling rapidly, with whole-genome sequencing and annotation now costing less than $5,000. The cost of a pharmacogenetic test can be as low as $100 using low-cost nanotechnology, and the test needs to be performed only once in a patient’s lifetime.27
As other related molecular technologies such as proteomics and metabolomics become available and are integrated with genomics, the predictive ability of this science will improve.
AWAY FROM ONE-SIZE-FITS-ALL MEDICINE
Over the last decade there has been a trend away from “one size fits all” to customized “markets of one” in everything from consumer products to education to medicine. Mass customizing, also known as personalization, has been embraced by the internet community as a means to increase efficiency and reduce cost. This occurs by eliminating waste in redundant work or production of ineffective products.
Personalization on the Internet has been enabled through the use of informatics, mathematics, and supercomputing. The same tools that have personalized the delivery of consumer products are also being applied to the field of pharmacogenetics. Applied in an evidence-based fashion, these new technologies should profoundly improve patient care now and in the future.
“Change is the only constant.”
—Heraclitus (c 535–475 bce)
With the cost of health care rising and money to pay for it shrinking, there has never been a greater need to reduce waste.
Ineffective treatments and adverse drug effects account for much preventable morbidity and expense. New treatments, touted as more potent, are often introduced as replacements for traditional ones that are still effective in many patients, adding to costs and the potential for harm. For the pharmaceutical industry, the search for new “blockbuster” drugs seems to have hit a wall, at least in cardiovascular medicine.1 Advances often come at the cost of adverse effects, such as bleeding with triple antiplatelet therapy and diabetes with potent statin drugs.
The path to maximizing benefit and reducing harm now appears to lie in stratifying populations and appreciating patient individuality in response to treatment. For many decades we have known that patients vary widely in their response to drugs, owing to personal factors such as body surface area, age, environment, and genetics. And indeed, we treat our patients as individuals, for example by tailoring aminoglycoside dose to weight and renal function.
However, clinical trials typically give us an idea of the benefits only to the average patient. While subgroup analyses identify groups that may benefit more or less from treatment, the additional information they provide is not easily integrated into the clinical model of prescribing, in which one size fits all.
THE PROMISE OF PHARMACOGENETICS
The emerging field of pharmacogenetics promises to give clinicians the tools to make informed treatment decisions based on predictive genetic testing. This genetic testing aims to match treatment to an individual’s genetic profile. This often involves analyzing single-nucleotide polymorphisms in genes for enzymes that metabolize drugs, such as the cytochrome P450 enzymes, to predict efficacy or an adverse event with treatment.
Pharmacogenetics is playing an increasing role in clinical trials, particularly in the early stages of drug development, by helping to reduce the number of patients needed, prove efficacy, and identify subgroups in which alternative treatment can be targeted. At another level, a molecular understanding of disease is leading to truly targeted treatments based on genomics.
Over recent years, genetic testing has been increasingly used in clinical practice, thanks to a convergence of factors such as rapid, low-cost tests, a growing evidence base, and emerging interest among doctors and payers.
An advantage to using genetic testing as opposed to other types of laboratory testing, such as measuring the concentration of the drug in the blood during treatment, is that genetic tests can predict the response to treatment before the treatment is started. Moreover, with therapeutic drug monitoring after treatment has begun, there are sometimes no detectable measures of toxicity. For example, both carbamazepine and the antiviral drug abacavir can—fortunately only rarely—cause Stevens-Johnson syndrome. But before genetic markers were discovered, there was no method of estimating this risk apart from taking a family history.2,3 Considering the numbers of people involved, it was not feasible until recently to suggest genetic screening for patients starting on these drugs. However, the cost of genotyping and gene sequencing has been falling at a rate inversely faster than Moore’s law (an approximate annual doubling in computer power), and population genomics is becoming a reality.4
The US Food and Drug Administration (FDA) recognizes the current and future value of pharmacogenetics in drug safety and development. A number of approved pharmacogenetic biomarkers are listed on the FDA website (Table 1). Black box warnings have been mandated for a number of drugs on the basis of observational evidence.
The FDA also promotes rapid approval for novel drugs with pharmacogenetic “companion diagnostics.” A recent example of this was the approval of ivacaftor for cystic fibrosis patients who have the G551D mutation.5 Here, a molecular understanding of the condition led to the development of a targeted treatment. Although the cost of developing this drug was high, the path is now paved for similar advances. Oncologists are familiar with these advances with the emergence of new molecularly targeted treatments, eg, BRAF inhibitors in metastatic melanoma, imatinib in chronic myeloid leukemia, and gefitinib in non-small-cell lung cancer.
PHARMACOGENETICS IN CARDIOVASCULAR MEDICINE
Cardiovascular medicine also stands to benefit from rapid advances in pharmacogenetics.
While no treatment has been developed that targets the molecular basis of cardiovascular disease, a number of genomic biomarkers have emerged that identify patients at risk of adverse reactions or treatment failure. These include genetic tests to predict the maintenance dose and risk of bleeding with warfarin,6 the likelihood of myopathy and myositis with simvastatin,7 and the risk of recurrent thrombotic events with clopidogrel.8–10
Using pharmacogenetics in prescribing warfarin and its alternatives
The pharmacogenetics of warfarin has been extensively researched, but genotyping before prescribing this drug is not yet widely done.
In 2007, the FDA updated the labeling of warfarin to include information about the influence of two genes, VKORC1 and CYP2C9, on a patient’s response to this drug. In 2010 this was updated to add that testing for these genes could be used to predict the maintenance dose of the drug. Difficulties with algorithms used to integrate this into clinical practice have hindered adoption of this testing.
With the advent of new anticoagulants such as dabigatran, rivaroxaban, and apixaban, many have expected warfarin and its pharmacogenetics to become obsolete. However, the new agents cost considerably more. Further, they may not offer a very great advantage over warfarin: in the Randomized Evaluation of Long-term Anticoagulant Therapy (RE-LY) trial, the absolute risk reduction in intracranial hemorrhage with dabigatran vs warfarin was small.11 Therefore, dabigatran is probably not cost-effective in populations at low risk of bleeding.12 A cost-effectiveness analysis comparing warfarin with dabigatran in patients with uncomplicated atrial fibrillation has suggested that dabigatran is, however, cost-effective in patients at moderate risk.12
In the RE-LY trial, the international normalized ratios (INRs) of the patients in the warfarin group were in the therapeutic range only 64% of the time. The advantages of dabigatran over warfarin become less pronounced as warfarin control is tightened.13 Of note, pharmacogenetics and home monitoring of the INR have both been shown to lead to tighter control of the INR, with greater time within the therapeutic range.14,15
Moreover, genetic testing can help us reduce the number of bleeding events in patients taking warfarin.16 Patients who carry the CYP2C9*2 or CYP2C9*3 polymorphism metabolize S-warfarin slower and therefore have a threefold higher risk of hemorrhage after starting warfarin.17 We could speculate that patients carrying these variants may be better served by the newer anticoagulants, though this has not been tested in any clinical trial.
It is also worth appreciating that the conditions requiring anticoagulation, such as atrial fibrillation, also have a strong genetic basis. Variants in chromosomes 4q25, 1q21, and 16q22 have all been associated with atrial fibrillation.18 The risk of atrial fibrillation is five to six times higher in carriers of multiple variants within all of these loci.19 Genetic variants at 4q25 have been associated with the response to specific antiarrhythmic drug treatments,20 response to pulmonary vein isolation, 21,22 and direct-current cardioversion.23 One can imagine a future in which patients with palpitations, carrying multiple gene risk variants, will choose prolonged monitoring at home to confirm a diagnosis. They would then be provided with a personalized best management strategy, using their personal preferences, clinical data, and genetic profile to make a treatment decision.
Using pharmacogenetics in prescribing clopidogrel and its alternatives
The pharmacogenetics of clopidogrel is of particular interest, as it has the potential of establishing a rational basis for using newer antiplatelet drugs such as ticagrelor and prasugrel, which are considerably more expensive than generic clopidogrel.
Most of the people who do not respond to clopidogrel carry the common cytochrome P450 2C19 variants CYP2C19*2 or CYP2C19*3.9 These variants are present in particularly high frequency in Asians and African Americans, who often do not feature in large randomized trials.
Newer antiplatelet agents have failed to demonstrate consistent superiority to clopidogrel without a tradeoff of more bleeding. However, in the Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition With Prasugrel-Thrombolysis in Myocardial Infarction 39 (TRITON TIMI-38),24,25 patients with the *2 variant receiving prasugrel had lower cardiovascular event rates than *2 carriers receiving clopidogrel.
Similarly, the patients who benefit the most from ticagrelor are carriers of the 2C19 nonresponder variants. In a large study, clopidogrel responders who did not carry either 2C19 nonresponder genetic variants or ABCB1 variants had cardiovascular outcomes similar to those of patients receiving ticagrelor.26
Clinicians have been cautious in prescribing potent antiplatelet agents to all patients because of the risk of bleeding. One could assume that by reserving newer agents for clopidogrel nonresponders, the bleeding risk could be minimized and overall benefit could be preserved with this strategy.
Cost may also be contained. The cost-effectiveness of such an approach with prasugrel has been tested with computer modeling and appears favorable.27 On the other hand, a similar yet limited analysis did not find genotype-driven use of ticagrelor to be cost-effective.28 This was mostly due to fewer deaths in patients receiving ticagrelor. However, the cost estimate for genotype-guided therapy was overestimated, as heterozygotes in the model were treated with ticagrelor instead of a high dose of clopidogrel.
It now appears that heterozygotes, ie, patients with one copy of the nonresponder variant, can achieve similar platelet inhibition with clopidogrel 225 mg daily as noncarriers on 75 mg daily.29 Since genotype-guided antiplatelet therapy has not been tested in a randomized outcomes trial, this tailored strategy has not been widely accepted.
THE FUTURE
The barriers to adoption of pharmacogenetics are considerable. Clinicians need to be educated about it, reimbursement needs to be worked out, and the pharmaceutical industry needs to get behind it. Nevertheless, the future of pharmacogenetics is extremely promising.
Research networks are forming to support the use of pharmacogenetics in clinical practice. The Pharmgkb (www.pharmgkb.org) database serves as a hub for educating clinicians and researchers as well as curating data for reference. Vanderbilt University is piloting the BioVu project, in which DNA and genotype data on patients are being stored and matched to the electronic clinical records.30 These projects not only provide clinically useful information on the current state of the art of pharmacogenetics, they also aid in disseminating new information about genotype-phenotype relationships.
Analytical software that uses “natural language processing” is being applied to clinician-generated notes to derive new observations and associations between genetic variants and clinical phenotypes. Integrating this information in real-time decision-support modules in the electronic health record provides a feedback loop for a rapid assimilation of new knowledge. Similar innovative decision-support modules are being established by Cleveland Clinic’s Center for Personalized Healthcare.31
The rise of ‘omic’ sciences
Pharmacogenetics and pharmacogenomics are part of a larger set of “omic” sciences. The suffix “-omics” implies a larger, more holistic view and is being applied to a number of fields—for example, the study of proteins (proteomics) and the study of metabolites (metabolomics). Profiling proteins and metabolites delivers a deluge of information on a patient that can be clustered, using pattern-recognition software, into population subgroups. Patterns of multiple proteins or metabolites are extracted from this spectral data to identify disease or response to treatment (pharmacometabolomics).
Metabolomics has been shown to predict the response to statins,32 diagnose myocardial infarction,33 and reclassify cardiovascular risk status.34 In addition, whereas traditional laboratory chemistry is reductionist, using single biomarkers for single-disease diagnosis, omic technologies hold the potential to reveal information on a number of possible health or disease states. The identification of “healthy” profiles using these technologies can potentially provide positive feedback to patients undertaking lifestyle changes and treatment.
The instrument costs for proteomic and metabolomic profiling are relatively high. However, the ongoing running costs are minimal, estimated at as low as less than $13 per test, as there are no expensive reagents.33 High-volume testing therefore becomes very cost-effective.
Although omic science appears futuristic, proteomics and metabolomics are already used in many clinical laboratories to rapidly identify bacteria. These methods have already revolutionized the way laboratories identify microbes, since they are automated, reduce workload, and give very fast results.
The cost of genetic testing is falling
Critics of pharmacogenetics claim that the predictive value of genetic testing is poor, that evidence is lacking, and that the cost is too high. In all new technologies, the first iteration is coarse, but performance improves with use. The first major barrier is adoption. Projects like BioVu are establishing the infrastructure for a feedback loop to iteratively improve upon the status quo and provide the evidence base clinicians demand.
The cost of genetic testing is falling rapidly, with whole-genome sequencing and annotation now costing less than $5,000. The cost of a pharmacogenetic test can be as low as $100 using low-cost nanotechnology, and the test needs to be performed only once in a patient’s lifetime.27
As other related molecular technologies such as proteomics and metabolomics become available and are integrated with genomics, the predictive ability of this science will improve.
AWAY FROM ONE-SIZE-FITS-ALL MEDICINE
Over the last decade there has been a trend away from “one size fits all” to customized “markets of one” in everything from consumer products to education to medicine. Mass customizing, also known as personalization, has been embraced by the internet community as a means to increase efficiency and reduce cost. This occurs by eliminating waste in redundant work or production of ineffective products.
Personalization on the Internet has been enabled through the use of informatics, mathematics, and supercomputing. The same tools that have personalized the delivery of consumer products are also being applied to the field of pharmacogenetics. Applied in an evidence-based fashion, these new technologies should profoundly improve patient care now and in the future.
- Topol EJ. Past the wall in cardiovascular R&D. Nat Rev Drug Discov 2009; 8:259.
- Mallal S, Phillips E, Carosi G, et al; PREDICT-1 Study Team. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med 2008; 358:568–579.
- Hung SI, Chung WH, Jee SH, et al. Genetic susceptibility to carbamazepine-induced cutaneous adverse drug reactions. Pharmacogenet Genomics 2006; 16:297–306.
- Phimister EG, Feero WG, Guttmacher AE. Realizing genomic medicine. N Engl J Med 2012; 366:757–759.
- Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA 2009; 106:18825–18830.
- International Warfarin Pharmacogenetics Consortium; Klein TE, Altman RB, Eriksson N, et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med 2009; 360:753–764.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Simon T, Verstuyft C, Mary-Krause M, et al; French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360:363–375.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med 2009; 360:354–362
- Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 2009; 373:309–317.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Shah SV, Gage BF. Cost-effectiveness of dabigatran for stroke prophylaxis in atrial fibrillation. Circulation 2011; 123:2562–2570.
- Wallentin L, Yusuf S, Ezekowitz MD, et al; RE-LY investigators. Efficacy and safety of dabigatran compared with warfarin at different levels of international normalised ratio control for stroke prevention in atrial fibrillation: an analysis of the RE-LY trial. Lancet 2010; 376:975–983.
- Anderson JL, Horne BD, Stevens SM, et al; Couma-Gen Investigators. Randomized trial of genotype-guided versus standard warfarin dosing in patients initiating oral anticoagulation. Circulation 2007; 116:2563–2570.
- Matchar DB, Jacobson A, Dolor R, et al; THINRS Executive Committee and Site Investigators. Effect of home testing of international normalized ratio on clinical events. N Engl J Med 2010; 363:1608–1620.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study). J Am Coll Cardiol 2010; 55:2804–2812.
- Sanderson S, Emery J, Higgins J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: a HuGEnet systematic review and meta-analysis. Genet Med 2005; 7:97–104.
- Ellinor PT, Lunetta KL, Albert CM, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet 2012; 44:670–675.
- Lubitz SA, Sinner MF, Lunetta KL, et al. Independent susceptibility markers for atrial fibrillation on chromosome 4q25. Circulation 2010; 122:976–984.
- Parvez B, Vaglio J, Rowan S, et al. Symptomatic response to antiarrhythmic drug therapy is modulated by a common single nucleotide polymorphism in atrial fibrillation. J Am Coll Cardiol 2012; 60:539–545.
- Husser D, Adams V, Piorkowski C, Hindricks G, Bollmann A. Chromosome 4q25 variants and atrial fibrillation recurrence after catheter ablation. J Am Coll Cardiol 2010; 55:747–753.
- Benjamin Shoemaker M, Muhammad R, Parvez B, et al. Common atrial fibrillation risk alleles at 4q25 predict recurrence after catheter-based atrial fibrillation ablation.” Heart Rhythm 2012; Nov 23.pii: S1547-5271(12)01340–9. 10.1016/j.hrthm.2012.11.012. [Epub ahead of print]
- Parvez B, Benjamin Shoemaker M, Muhammad R, et al. Common genetic polymorphism at 4q25 locus predicts atrial fibrillation recurrence after successful cardioversion. Heart Rhythm 2013 Feb 18.pii: S1547-5271(13)00161–6. 10.1016/j.hrthm.2013.02.018. [Epub ahead of print]
- Mega JL, Close SL, Wiviott SD, et al. Genetic variants in ABCB1 and CYP2C19 and cardiovascular outcomes after treatment with clopidogrel and prasugrel in the TRITON-TIMI 38 trial: a pharmacogenetic analysis. Lancet 2010; 376:1312–1319.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome P450 genetic polymorphisms and the response to prasugrel: relationship to pharmacokinetic, pharmacodynamic, and clinical outcomes. Circulation 2009; 119:2553–2560.
- Wallentin L, James S, Storey RF, et al; PLATO investigators. Effect of CYP2C19 and ABCB1 single nucleotide polymorphisms on outcomes of treatment with ticagrelor versus clopidogrel for acute coronary syndromes: a genetic substudy of the PLATO trial. Lancet 2010; 376:1320–1328.
- Guzauskas GF, Hughes DA, Bradley SM, Veenstra DL. A risk-benefit assessment of prasugrel, clopidogrel, and genotype-guided therapy in patients undergoing percutaneous coronary intervention. Clin Pharmacol Ther 2012; 91:829–837.
- Crespin DJ, Federspiel JJ, Biddle AK, Jonas DE, Rossi JS. Ticagrelor versus genotype-driven antiplatelet therapy for secondary prevention after acute coronary syndrome: a cost-effectiveness analysis. Value Health 2011; 14:483–491.
- Mega JL, Hochholzer W, Frelinger AL, et al. Dosing clopidogrel based on CYP2C19 genotype and the effect on platelet reactivity in patients with stable cardiovascular disease. JAMA 2011; 306:2221–2228.
- Xu H, Jiang M, Oetjens M, et al. Facilitating pharmacogenetic studies using electronic health records and natural-language processing: a case study of warfarin. J Am Med Inform Assoc 2011; 18:387–391.
- Teng K, Eng C, Hess CA, et al. Building an innovative model for personalized healthcare. Cleve Clin J Med 2012; 79( suppl 1):S1–S9.
- Kaddurah-Daouk R, Baillie RA, Zhu H, et al. Enteric microbiome metabolites correlate with response to simvastatin treatment. PLoS One 2011; 6:e25482.
- Bodi V, Sanchis J, Morales JM, et al. Metabolomic profile of human myocardial ischemia by nuclear magnetic resonance spectroscopy of peripheral blood serum: a translational study based on transient coronary occlusion models. J Am Coll Cardiol 2012; 59:1629–1641.
- Shah SH, Sun JL, Stevens RD, et al. Baseline metabolomic profiles predict cardiovascular events in patients at risk for coronary artery disease. Am Heart J 2012; 163:844–850.e1.
- Topol EJ. Past the wall in cardiovascular R&D. Nat Rev Drug Discov 2009; 8:259.
- Mallal S, Phillips E, Carosi G, et al; PREDICT-1 Study Team. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med 2008; 358:568–579.
- Hung SI, Chung WH, Jee SH, et al. Genetic susceptibility to carbamazepine-induced cutaneous adverse drug reactions. Pharmacogenet Genomics 2006; 16:297–306.
- Phimister EG, Feero WG, Guttmacher AE. Realizing genomic medicine. N Engl J Med 2012; 366:757–759.
- Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA 2009; 106:18825–18830.
- International Warfarin Pharmacogenetics Consortium; Klein TE, Altman RB, Eriksson N, et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med 2009; 360:753–764.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Simon T, Verstuyft C, Mary-Krause M, et al; French Registry of Acute ST-Elevation and Non-ST-Elevation Myocardial Infarction (FAST-MI) Investigators. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 2009; 360:363–375.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med 2009; 360:354–362
- Collet JP, Hulot JS, Pena A, et al. Cytochrome P450 2C19 polymorphism in young patients treated with clopidogrel after myocardial infarction: a cohort study. Lancet 2009; 373:309–317.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Shah SV, Gage BF. Cost-effectiveness of dabigatran for stroke prophylaxis in atrial fibrillation. Circulation 2011; 123:2562–2570.
- Wallentin L, Yusuf S, Ezekowitz MD, et al; RE-LY investigators. Efficacy and safety of dabigatran compared with warfarin at different levels of international normalised ratio control for stroke prevention in atrial fibrillation: an analysis of the RE-LY trial. Lancet 2010; 376:975–983.
- Anderson JL, Horne BD, Stevens SM, et al; Couma-Gen Investigators. Randomized trial of genotype-guided versus standard warfarin dosing in patients initiating oral anticoagulation. Circulation 2007; 116:2563–2570.
- Matchar DB, Jacobson A, Dolor R, et al; THINRS Executive Committee and Site Investigators. Effect of home testing of international normalized ratio on clinical events. N Engl J Med 2010; 363:1608–1620.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study). J Am Coll Cardiol 2010; 55:2804–2812.
- Sanderson S, Emery J, Higgins J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: a HuGEnet systematic review and meta-analysis. Genet Med 2005; 7:97–104.
- Ellinor PT, Lunetta KL, Albert CM, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet 2012; 44:670–675.
- Lubitz SA, Sinner MF, Lunetta KL, et al. Independent susceptibility markers for atrial fibrillation on chromosome 4q25. Circulation 2010; 122:976–984.
- Parvez B, Vaglio J, Rowan S, et al. Symptomatic response to antiarrhythmic drug therapy is modulated by a common single nucleotide polymorphism in atrial fibrillation. J Am Coll Cardiol 2012; 60:539–545.
- Husser D, Adams V, Piorkowski C, Hindricks G, Bollmann A. Chromosome 4q25 variants and atrial fibrillation recurrence after catheter ablation. J Am Coll Cardiol 2010; 55:747–753.
- Benjamin Shoemaker M, Muhammad R, Parvez B, et al. Common atrial fibrillation risk alleles at 4q25 predict recurrence after catheter-based atrial fibrillation ablation.” Heart Rhythm 2012; Nov 23.pii: S1547-5271(12)01340–9. 10.1016/j.hrthm.2012.11.012. [Epub ahead of print]
- Parvez B, Benjamin Shoemaker M, Muhammad R, et al. Common genetic polymorphism at 4q25 locus predicts atrial fibrillation recurrence after successful cardioversion. Heart Rhythm 2013 Feb 18.pii: S1547-5271(13)00161–6. 10.1016/j.hrthm.2013.02.018. [Epub ahead of print]
- Mega JL, Close SL, Wiviott SD, et al. Genetic variants in ABCB1 and CYP2C19 and cardiovascular outcomes after treatment with clopidogrel and prasugrel in the TRITON-TIMI 38 trial: a pharmacogenetic analysis. Lancet 2010; 376:1312–1319.
- Mega JL, Close SL, Wiviott SD, et al. Cytochrome P450 genetic polymorphisms and the response to prasugrel: relationship to pharmacokinetic, pharmacodynamic, and clinical outcomes. Circulation 2009; 119:2553–2560.
- Wallentin L, James S, Storey RF, et al; PLATO investigators. Effect of CYP2C19 and ABCB1 single nucleotide polymorphisms on outcomes of treatment with ticagrelor versus clopidogrel for acute coronary syndromes: a genetic substudy of the PLATO trial. Lancet 2010; 376:1320–1328.
- Guzauskas GF, Hughes DA, Bradley SM, Veenstra DL. A risk-benefit assessment of prasugrel, clopidogrel, and genotype-guided therapy in patients undergoing percutaneous coronary intervention. Clin Pharmacol Ther 2012; 91:829–837.
- Crespin DJ, Federspiel JJ, Biddle AK, Jonas DE, Rossi JS. Ticagrelor versus genotype-driven antiplatelet therapy for secondary prevention after acute coronary syndrome: a cost-effectiveness analysis. Value Health 2011; 14:483–491.
- Mega JL, Hochholzer W, Frelinger AL, et al. Dosing clopidogrel based on CYP2C19 genotype and the effect on platelet reactivity in patients with stable cardiovascular disease. JAMA 2011; 306:2221–2228.
- Xu H, Jiang M, Oetjens M, et al. Facilitating pharmacogenetic studies using electronic health records and natural-language processing: a case study of warfarin. J Am Med Inform Assoc 2011; 18:387–391.
- Teng K, Eng C, Hess CA, et al. Building an innovative model for personalized healthcare. Cleve Clin J Med 2012; 79( suppl 1):S1–S9.
- Kaddurah-Daouk R, Baillie RA, Zhu H, et al. Enteric microbiome metabolites correlate with response to simvastatin treatment. PLoS One 2011; 6:e25482.
- Bodi V, Sanchis J, Morales JM, et al. Metabolomic profile of human myocardial ischemia by nuclear magnetic resonance spectroscopy of peripheral blood serum: a translational study based on transient coronary occlusion models. J Am Coll Cardiol 2012; 59:1629–1641.
- Shah SH, Sun JL, Stevens RD, et al. Baseline metabolomic profiles predict cardiovascular events in patients at risk for coronary artery disease. Am Heart J 2012; 163:844–850.e1.
Should we use pharmacogenetic testing when prescribing warfarin?
The answer is not clear. There is evidence in favor of pharmacogenetic testing, but not yet enough to strongly recommend it. However, we do believe that physicians should consider it when starting patients on warfarin therapy.
WARFARIN HAS A NARROW THERAPEUTIC WINDOW
Although newer drugs are available, warfarin is still the most commonly used oral anticoagulant for preventing and treating thromboembolism.1 It is highly effective but has a narrow therapeutic window and wide interindividual variability in dosage requirements, which poses challenges to achieving adequate anticoagulation.1–3 Inappropriate dosing contributes to a high rate of bleeding events and emergency room visits.4
Warfarin is monitored using the prothrombin time. Because the prothrombin time varies depending on the assay used, the standardized value called the international normalized ratio (INR) is more commonly used.
Clinical factors such as age, body size, and drug interactions affect warfarin dosage requirements and are important to consider,5 even though they account for only 15% to 20% of the variability in warfarin dose.6
Genetic factors also affect warfarin dosage requirements. The combination of genetic and clinical factors accounts for up to 47% of the dose variability.7
GENES THAT AFFECT WARFARIN
Several genes are known to influence warfarin’s pharmacokinetics and pharmacodynamics. Of these, the two most clinically relevant and well studied are CYP2C9 (which codes for cytochrome P450 2C9) and VKORC1 (which codes for vitamin K epoxide reductase).7 These genes are polymorphic, with some variants producing less-active enzymes that allow warfarin to be more active. Therefore, patients who carry these variants need lower doses of this drug (see below).
CYP2C9 variants
The CYP2C9 gene has several variants. Of these, CYP2C9*2 and CYP2C9*3 are associated with the lowest enzyme activity.
Patients with either of these variants require significantly lower warfarin doses to reach therapeutic levels than those with the wild-type gene (ie, CYP2C9*1). CYP2C9*2 reduces warfarin clearance by 40%, and the CYP2C9*3 variant reduces it by 75%.7 Having a *2 or *3 allele increases the risk of bleeding during warfarin therapy and the time needed to achieve a stable dose.8 Other variants associated with lower warfarin dose requirements are *5, *6, and *11.
The prevalence of these variants is significantly higher in people of European ancestry (roughly one-third) than in Asian people and African Americans,7 although no one has recommended not testing in these low-prevalence populations. Limdi et al9 reported that by including the *5, *6, and *11 variants in genetic testing (in addition to *2 and *3), they could identify more African Americans (9.7%) who carried at least one of these abnormal variants than reported previously. Differences among ethnic groups need to be taken into account when interpreting pharmacogenetic studies.
VKORC1 variants
Patients also need lower doses of warfarin if they carry the VKORC1 −1639G>A variant, and they spend more time with an INR above the therapeutic range and have higher overall INR values. However, having this variant does not appear to increase the risk of bleeding.
The −1639G>A variant is the most common variant of VKORC1. Rarer ones have also been described, but most commercially available tests do not detect them.
Racial differences exist in the prevalence rates of the various VKORC1 polymorphisms, with the most sensitive (low-dose) genotype predominating in Asians and the least sensitive (high-dose) genotype predominating in African Americans. Over 50% of people of European ancestry carry the intermediate-sensitivity genotype (typical dose).7
CURRENT RECOMMENDATIONS FOR OR AGAINST TESTING
FDA labeling
In 2007, the US Food and Drug Administration (FDA) required that the warfarin package insert carry information about initial dosing based on CYP2C9 and VKORC1 testing. This recommendation was revised in 2010 to include a table to help clinicians select an initial warfarin dose if CYP2C9 and VKORC1 genotype information is available. However, the FDA does not require pharmacogenetic testing, leaving the decision to the discretion of the clinician.7
American College of Chest Physicians
The American College of Chest Physicians recommends against routine pharmacogenetic testing (grade 1B) because of a lack of evidence that it improves clinical end points or that it is cost-effective.5
WHAT EVIDENCE SUPORTS GENETIC TESTING TO GUIDE WARFARIN THERAPY?
To date, no large randomized, controlled trial has been published that looked at clinical outcomes with warfarin dosing based on pharmacogenetic testing. However, several smaller studies have suggested it is beneficial.
One trial found that when dosing was informed by pharmacogenetic testing, patients had significantly more time in the therapeutic range, a lower percentage of INRs greater than 4 or less than 1.5, and fewer serious adverse events (death, myocardial infarction, stroke, thromboembolism, and clinically significant bleeding events).10 Patients whose dosage was determined using pharmacogenetic algorithms as opposed to traditional clinical algorithms maintained a therapeutic INR more consistently.11
In addition, compared with historical controls, patients whose physician used pharmacogenetic testing to guide warfarin dosing had a rate of hospitalization 31% lower and a rate of hospitalization specifically for bleeding or thromboembolism 28% lower during 6 months of follow-up.12,13
Several studies have attempted to assess the cost-effectiveness and utility of pharmacogenetic testing in warfarin therapy. As yet, the results have been inconclusive.14 Larger prospective trials are under way and are estimated to be completed in late 2013.15 These include:
- COAG (Clarification of Optimal Anticoagulation Through Genetics)
- GIFT (Genetics Informatics Trial of Warfarin to Prevent Venous Thrombosis)
- EU-PACT (European Pharmacogenetics of Anticoagulant Therapy-Warfarin).
We hope these studies will provide greater clarity on the clinical utility and cost-effectiveness of pharmacogenetic testing to guide warfarin dosing.
HOW SHOULD GENETIC INFORMATION BE USED TO GUIDE OR ALTER THERAPY?
Algorithms are available for estimating initial and maintenance warfarin doses based on genetic information (CYP2C9 and VKORC1), race or ethnicity, age, sex, body mass index, smoking status, and other medications taken. In addition, models incorporating the INR on day 4 and days 6 to 11 have been developed for dose refinement.15 The algorithms explain 30% to 60% of the variability of the data, with lower values for African Americans.7
A well-developed dosing model that includes traditional clinical factors and patient genetic status is publicly available online at www.warfarindosing.org.4
CPIC: A leader in applied pharmacogenetics
In late 2009, PharmGKB joined forces with the Pharmacogenomics Research Network of the National Institutes of Health to form the Clinical Pharmacogenetics Implementation Consortium (CPIC). This organization issues guidelines that are written by expert clinicians and scientists and then are peer-reviewed, published in leading journals, and simultaneously posted to the PharmGKB website along with supplemental information and updates.
CPIC’s goal is to review the current evidence and to address barriers to the adoption of pharmacogenetic testing into clinical practice. Its guidelines do not advise when or which pharmacogenetic tests should be ordered. Rather, they provide guidance on interpreting and applying such testing, should the test results be available.7
CPIC has guidelines on CYP2C9 and VKORC1 genotypes and warfarin dosing.8 If a patient’s genetic information is available, CPIC strongly recommends the use of pharmacogenetic algorithm-based dosing. If such an algorithm is not accessible, use of a genotype dosing table is recommended.8
Monitoring is still needed
Many factors can affect an individual’s response to warfarin above and beyond the above-noted clinical and genetic traits. These include diet, concomitant medications (both prescription and over-the-counter and herbal), and disease state. There may also be additional genetic polymorphisms not yet identified in various racial and ethnic groups that may affect dosing requirements. And as with all medications, patient compliance and dosing errors have a large potential to affect individual response. Therefore, clinicians should still be diligent about clinical monitoring.15
Most useful for initial dose
As with most pharmacogenetic information, the greatest benefit can be achieved when this information is used to guide the initial dose, although there is also some effect noted when this information is known and acted upon into the 2nd week of treatment.8
Patients on long-term warfarin treatment with stable doses and those unable to achieve stable dosing because of variable adherence or dietary vitamin K intake are less likely to benefit from genetic testing.
There are no published guidelines on the utility of pharmacogenetic testing if a patient is already on a stable dose of warfarin or has a known historical stable dose. There are also no published guidelines on changing the frequency of monitoring based on known genotype.
In children, the data are sparse at this time regarding the utility of pharmacogenetically informed dosing.
HOW DOES ONE ORDER TESTING, AND WHAT IS THE COST?
The FDA has approved four warfarin pharmacogenetic test kits. To be used in clinical decision-making, these tests must be done in a laboratory certified by the Clinical Laboratory Improvement Amendments (CLIA) program.
Testing typically costs a few hundred dollars and may take days for results to be returned if not available on site.15 At Cleveland Clinic, CYP2C9 and VKORC1 testing can be run in-house at a cost of about $700. Generally, many third-party payers do not reimburse for testing without a prior-approval process.
TO TEST OR NOT TO TEST
Pharmacogenetic testing is available and may help optimize warfarin dosing early in treatment, as well as help maintain therapeutic INRs more consistently. There is preliminary evidence that using this information to guide dosing improves clinical outcomes. Several large trials are under way to address additional questions of clinical utility, with results expected in the next year. There are also readily available decision-support tools to guide therapeutic dosing, and when pharmacogenetic test results are available, utilization of a warfarin dosing algorithm is recommended.
The largest barrier remaining appears to be cost (relative to perceived benefit), and until larger trials of clinical utility and cost-effectiveness are completed and analyzed, hurdles exist to obtaining coverage for such testing.
If it is readily available (and can be paid for by insurance companies or out-of-pocket) and test results can be obtained within 24 to 48 hours or before prescribing, pharmacogenetic testing can be a valuable tool to guide and manage warfarin dosing. Particularly for patients who want to be as proactive as possible, warfarin pharmacogenetic testing offers the ability to participate in this decision-making and to potentially reduce their risk of adverse drug events. And in view of the evidence and FDA recommendations, we propose that the discussion with our patients is not whether we should consider pharmacogenetic testing, but that we have considered pharmacogenetic testing, and why we have decided for or against it.
- Jacobs LG. Warfarin pharmacology, clinical management, and evaluation of hemorrhagic risk for the elderly. Clin Geriatr Med 2006; 22:17–32,vii–viii.
- Rieder MJ, Reiner AP, Gage BF, et al. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N Engl J Med 2005; 352:2285–2293.
- Higashi MK, Veenstra DL, Kondo LM, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 2002; 287:1690–1698.
- Shehab N, Sperling LS, Kegler SR, Budnitz DS. National estimates of emergency department visits for hemorrhage-related adverse events from clopidogrel plus aspirin and from warfarin. Arch Intern Med 2010; 170:1926–1933.
- Holbrook A, Schulman S, Witt DM, et al; American College of Chest Physicians. Evidence-based management of anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e152S–e184S.
- Gage BF, Eby C, Johnson JA, et al. Use of pharmacogenetic and clinical factors to predict the therapeutic dose of warfarin. Clin Pharmacol Ther 2008; 84:326–331.
- Cavallari LH, Shin J, Perera MA. Role of pharmacogenomics in the management of traditional and novel oral anticoagulants. Pharmacotherapy 2011; 31:1192–1207.
- Johnson JA, Gong L, Whirl-Carillo M, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin Pharmacol Ther 2011; 90:625–629.
- Limdi NA, McGwin G, Goldstein JA, et al. Influence of CYP2C9 and VKORC1 1173C/T genotype on the risk of hemorrhagic complications in African-American and European-American patients on warfarin. Clin Pharmacol Ther 2008; 83:312–321.
- Anderson JL, Horne BD, Stevens SM, et al. A randomized and clinical effectiveness trial comparing two pharmacogenetic algorithms and standard care for individualizing warfarin dosing (CoumaGen-II). Circulation 2012; 125:1997–2005.
- Yip VL, Pirmohamed M. Expanding role of pharmacogenomics in the management of cardiovascular disorders. Am J Cardiovasc Drugs 2013; 12 Apr; Epub ahead of print.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates: results from the MM-WES (Medco-Mayo Warfarin Effectiveness Study). J Am Coll Cardiol 2010; 55:2804–2812.
- Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med 2011; 364:1144–1153.
- Kitzmiller JP, Groen DK, Phelps MA, Sadee W. Pharmacogenomic testing: relevance in medical practice: why drugs work in some patients but not in others. Cleve Clin J Med 2011; 78:243–257.
- Carlquist JF, Anderson JL. Using pharmacogenetics in real time to guide warfarin initiation: a clinician update. Circulation 2011; 124:2554–2559.
The answer is not clear. There is evidence in favor of pharmacogenetic testing, but not yet enough to strongly recommend it. However, we do believe that physicians should consider it when starting patients on warfarin therapy.
WARFARIN HAS A NARROW THERAPEUTIC WINDOW
Although newer drugs are available, warfarin is still the most commonly used oral anticoagulant for preventing and treating thromboembolism.1 It is highly effective but has a narrow therapeutic window and wide interindividual variability in dosage requirements, which poses challenges to achieving adequate anticoagulation.1–3 Inappropriate dosing contributes to a high rate of bleeding events and emergency room visits.4
Warfarin is monitored using the prothrombin time. Because the prothrombin time varies depending on the assay used, the standardized value called the international normalized ratio (INR) is more commonly used.
Clinical factors such as age, body size, and drug interactions affect warfarin dosage requirements and are important to consider,5 even though they account for only 15% to 20% of the variability in warfarin dose.6
Genetic factors also affect warfarin dosage requirements. The combination of genetic and clinical factors accounts for up to 47% of the dose variability.7
GENES THAT AFFECT WARFARIN
Several genes are known to influence warfarin’s pharmacokinetics and pharmacodynamics. Of these, the two most clinically relevant and well studied are CYP2C9 (which codes for cytochrome P450 2C9) and VKORC1 (which codes for vitamin K epoxide reductase).7 These genes are polymorphic, with some variants producing less-active enzymes that allow warfarin to be more active. Therefore, patients who carry these variants need lower doses of this drug (see below).
CYP2C9 variants
The CYP2C9 gene has several variants. Of these, CYP2C9*2 and CYP2C9*3 are associated with the lowest enzyme activity.
Patients with either of these variants require significantly lower warfarin doses to reach therapeutic levels than those with the wild-type gene (ie, CYP2C9*1). CYP2C9*2 reduces warfarin clearance by 40%, and the CYP2C9*3 variant reduces it by 75%.7 Having a *2 or *3 allele increases the risk of bleeding during warfarin therapy and the time needed to achieve a stable dose.8 Other variants associated with lower warfarin dose requirements are *5, *6, and *11.
The prevalence of these variants is significantly higher in people of European ancestry (roughly one-third) than in Asian people and African Americans,7 although no one has recommended not testing in these low-prevalence populations. Limdi et al9 reported that by including the *5, *6, and *11 variants in genetic testing (in addition to *2 and *3), they could identify more African Americans (9.7%) who carried at least one of these abnormal variants than reported previously. Differences among ethnic groups need to be taken into account when interpreting pharmacogenetic studies.
VKORC1 variants
Patients also need lower doses of warfarin if they carry the VKORC1 −1639G>A variant, and they spend more time with an INR above the therapeutic range and have higher overall INR values. However, having this variant does not appear to increase the risk of bleeding.
The −1639G>A variant is the most common variant of VKORC1. Rarer ones have also been described, but most commercially available tests do not detect them.
Racial differences exist in the prevalence rates of the various VKORC1 polymorphisms, with the most sensitive (low-dose) genotype predominating in Asians and the least sensitive (high-dose) genotype predominating in African Americans. Over 50% of people of European ancestry carry the intermediate-sensitivity genotype (typical dose).7
CURRENT RECOMMENDATIONS FOR OR AGAINST TESTING
FDA labeling
In 2007, the US Food and Drug Administration (FDA) required that the warfarin package insert carry information about initial dosing based on CYP2C9 and VKORC1 testing. This recommendation was revised in 2010 to include a table to help clinicians select an initial warfarin dose if CYP2C9 and VKORC1 genotype information is available. However, the FDA does not require pharmacogenetic testing, leaving the decision to the discretion of the clinician.7
American College of Chest Physicians
The American College of Chest Physicians recommends against routine pharmacogenetic testing (grade 1B) because of a lack of evidence that it improves clinical end points or that it is cost-effective.5
WHAT EVIDENCE SUPORTS GENETIC TESTING TO GUIDE WARFARIN THERAPY?
To date, no large randomized, controlled trial has been published that looked at clinical outcomes with warfarin dosing based on pharmacogenetic testing. However, several smaller studies have suggested it is beneficial.
One trial found that when dosing was informed by pharmacogenetic testing, patients had significantly more time in the therapeutic range, a lower percentage of INRs greater than 4 or less than 1.5, and fewer serious adverse events (death, myocardial infarction, stroke, thromboembolism, and clinically significant bleeding events).10 Patients whose dosage was determined using pharmacogenetic algorithms as opposed to traditional clinical algorithms maintained a therapeutic INR more consistently.11
In addition, compared with historical controls, patients whose physician used pharmacogenetic testing to guide warfarin dosing had a rate of hospitalization 31% lower and a rate of hospitalization specifically for bleeding or thromboembolism 28% lower during 6 months of follow-up.12,13
Several studies have attempted to assess the cost-effectiveness and utility of pharmacogenetic testing in warfarin therapy. As yet, the results have been inconclusive.14 Larger prospective trials are under way and are estimated to be completed in late 2013.15 These include:
- COAG (Clarification of Optimal Anticoagulation Through Genetics)
- GIFT (Genetics Informatics Trial of Warfarin to Prevent Venous Thrombosis)
- EU-PACT (European Pharmacogenetics of Anticoagulant Therapy-Warfarin).
We hope these studies will provide greater clarity on the clinical utility and cost-effectiveness of pharmacogenetic testing to guide warfarin dosing.
HOW SHOULD GENETIC INFORMATION BE USED TO GUIDE OR ALTER THERAPY?
Algorithms are available for estimating initial and maintenance warfarin doses based on genetic information (CYP2C9 and VKORC1), race or ethnicity, age, sex, body mass index, smoking status, and other medications taken. In addition, models incorporating the INR on day 4 and days 6 to 11 have been developed for dose refinement.15 The algorithms explain 30% to 60% of the variability of the data, with lower values for African Americans.7
A well-developed dosing model that includes traditional clinical factors and patient genetic status is publicly available online at www.warfarindosing.org.4
CPIC: A leader in applied pharmacogenetics
In late 2009, PharmGKB joined forces with the Pharmacogenomics Research Network of the National Institutes of Health to form the Clinical Pharmacogenetics Implementation Consortium (CPIC). This organization issues guidelines that are written by expert clinicians and scientists and then are peer-reviewed, published in leading journals, and simultaneously posted to the PharmGKB website along with supplemental information and updates.
CPIC’s goal is to review the current evidence and to address barriers to the adoption of pharmacogenetic testing into clinical practice. Its guidelines do not advise when or which pharmacogenetic tests should be ordered. Rather, they provide guidance on interpreting and applying such testing, should the test results be available.7
CPIC has guidelines on CYP2C9 and VKORC1 genotypes and warfarin dosing.8 If a patient’s genetic information is available, CPIC strongly recommends the use of pharmacogenetic algorithm-based dosing. If such an algorithm is not accessible, use of a genotype dosing table is recommended.8
Monitoring is still needed
Many factors can affect an individual’s response to warfarin above and beyond the above-noted clinical and genetic traits. These include diet, concomitant medications (both prescription and over-the-counter and herbal), and disease state. There may also be additional genetic polymorphisms not yet identified in various racial and ethnic groups that may affect dosing requirements. And as with all medications, patient compliance and dosing errors have a large potential to affect individual response. Therefore, clinicians should still be diligent about clinical monitoring.15
Most useful for initial dose
As with most pharmacogenetic information, the greatest benefit can be achieved when this information is used to guide the initial dose, although there is also some effect noted when this information is known and acted upon into the 2nd week of treatment.8
Patients on long-term warfarin treatment with stable doses and those unable to achieve stable dosing because of variable adherence or dietary vitamin K intake are less likely to benefit from genetic testing.
There are no published guidelines on the utility of pharmacogenetic testing if a patient is already on a stable dose of warfarin or has a known historical stable dose. There are also no published guidelines on changing the frequency of monitoring based on known genotype.
In children, the data are sparse at this time regarding the utility of pharmacogenetically informed dosing.
HOW DOES ONE ORDER TESTING, AND WHAT IS THE COST?
The FDA has approved four warfarin pharmacogenetic test kits. To be used in clinical decision-making, these tests must be done in a laboratory certified by the Clinical Laboratory Improvement Amendments (CLIA) program.
Testing typically costs a few hundred dollars and may take days for results to be returned if not available on site.15 At Cleveland Clinic, CYP2C9 and VKORC1 testing can be run in-house at a cost of about $700. Generally, many third-party payers do not reimburse for testing without a prior-approval process.
TO TEST OR NOT TO TEST
Pharmacogenetic testing is available and may help optimize warfarin dosing early in treatment, as well as help maintain therapeutic INRs more consistently. There is preliminary evidence that using this information to guide dosing improves clinical outcomes. Several large trials are under way to address additional questions of clinical utility, with results expected in the next year. There are also readily available decision-support tools to guide therapeutic dosing, and when pharmacogenetic test results are available, utilization of a warfarin dosing algorithm is recommended.
The largest barrier remaining appears to be cost (relative to perceived benefit), and until larger trials of clinical utility and cost-effectiveness are completed and analyzed, hurdles exist to obtaining coverage for such testing.
If it is readily available (and can be paid for by insurance companies or out-of-pocket) and test results can be obtained within 24 to 48 hours or before prescribing, pharmacogenetic testing can be a valuable tool to guide and manage warfarin dosing. Particularly for patients who want to be as proactive as possible, warfarin pharmacogenetic testing offers the ability to participate in this decision-making and to potentially reduce their risk of adverse drug events. And in view of the evidence and FDA recommendations, we propose that the discussion with our patients is not whether we should consider pharmacogenetic testing, but that we have considered pharmacogenetic testing, and why we have decided for or against it.
The answer is not clear. There is evidence in favor of pharmacogenetic testing, but not yet enough to strongly recommend it. However, we do believe that physicians should consider it when starting patients on warfarin therapy.
WARFARIN HAS A NARROW THERAPEUTIC WINDOW
Although newer drugs are available, warfarin is still the most commonly used oral anticoagulant for preventing and treating thromboembolism.1 It is highly effective but has a narrow therapeutic window and wide interindividual variability in dosage requirements, which poses challenges to achieving adequate anticoagulation.1–3 Inappropriate dosing contributes to a high rate of bleeding events and emergency room visits.4
Warfarin is monitored using the prothrombin time. Because the prothrombin time varies depending on the assay used, the standardized value called the international normalized ratio (INR) is more commonly used.
Clinical factors such as age, body size, and drug interactions affect warfarin dosage requirements and are important to consider,5 even though they account for only 15% to 20% of the variability in warfarin dose.6
Genetic factors also affect warfarin dosage requirements. The combination of genetic and clinical factors accounts for up to 47% of the dose variability.7
GENES THAT AFFECT WARFARIN
Several genes are known to influence warfarin’s pharmacokinetics and pharmacodynamics. Of these, the two most clinically relevant and well studied are CYP2C9 (which codes for cytochrome P450 2C9) and VKORC1 (which codes for vitamin K epoxide reductase).7 These genes are polymorphic, with some variants producing less-active enzymes that allow warfarin to be more active. Therefore, patients who carry these variants need lower doses of this drug (see below).
CYP2C9 variants
The CYP2C9 gene has several variants. Of these, CYP2C9*2 and CYP2C9*3 are associated with the lowest enzyme activity.
Patients with either of these variants require significantly lower warfarin doses to reach therapeutic levels than those with the wild-type gene (ie, CYP2C9*1). CYP2C9*2 reduces warfarin clearance by 40%, and the CYP2C9*3 variant reduces it by 75%.7 Having a *2 or *3 allele increases the risk of bleeding during warfarin therapy and the time needed to achieve a stable dose.8 Other variants associated with lower warfarin dose requirements are *5, *6, and *11.
The prevalence of these variants is significantly higher in people of European ancestry (roughly one-third) than in Asian people and African Americans,7 although no one has recommended not testing in these low-prevalence populations. Limdi et al9 reported that by including the *5, *6, and *11 variants in genetic testing (in addition to *2 and *3), they could identify more African Americans (9.7%) who carried at least one of these abnormal variants than reported previously. Differences among ethnic groups need to be taken into account when interpreting pharmacogenetic studies.
VKORC1 variants
Patients also need lower doses of warfarin if they carry the VKORC1 −1639G>A variant, and they spend more time with an INR above the therapeutic range and have higher overall INR values. However, having this variant does not appear to increase the risk of bleeding.
The −1639G>A variant is the most common variant of VKORC1. Rarer ones have also been described, but most commercially available tests do not detect them.
Racial differences exist in the prevalence rates of the various VKORC1 polymorphisms, with the most sensitive (low-dose) genotype predominating in Asians and the least sensitive (high-dose) genotype predominating in African Americans. Over 50% of people of European ancestry carry the intermediate-sensitivity genotype (typical dose).7
CURRENT RECOMMENDATIONS FOR OR AGAINST TESTING
FDA labeling
In 2007, the US Food and Drug Administration (FDA) required that the warfarin package insert carry information about initial dosing based on CYP2C9 and VKORC1 testing. This recommendation was revised in 2010 to include a table to help clinicians select an initial warfarin dose if CYP2C9 and VKORC1 genotype information is available. However, the FDA does not require pharmacogenetic testing, leaving the decision to the discretion of the clinician.7
American College of Chest Physicians
The American College of Chest Physicians recommends against routine pharmacogenetic testing (grade 1B) because of a lack of evidence that it improves clinical end points or that it is cost-effective.5
WHAT EVIDENCE SUPORTS GENETIC TESTING TO GUIDE WARFARIN THERAPY?
To date, no large randomized, controlled trial has been published that looked at clinical outcomes with warfarin dosing based on pharmacogenetic testing. However, several smaller studies have suggested it is beneficial.
One trial found that when dosing was informed by pharmacogenetic testing, patients had significantly more time in the therapeutic range, a lower percentage of INRs greater than 4 or less than 1.5, and fewer serious adverse events (death, myocardial infarction, stroke, thromboembolism, and clinically significant bleeding events).10 Patients whose dosage was determined using pharmacogenetic algorithms as opposed to traditional clinical algorithms maintained a therapeutic INR more consistently.11
In addition, compared with historical controls, patients whose physician used pharmacogenetic testing to guide warfarin dosing had a rate of hospitalization 31% lower and a rate of hospitalization specifically for bleeding or thromboembolism 28% lower during 6 months of follow-up.12,13
Several studies have attempted to assess the cost-effectiveness and utility of pharmacogenetic testing in warfarin therapy. As yet, the results have been inconclusive.14 Larger prospective trials are under way and are estimated to be completed in late 2013.15 These include:
- COAG (Clarification of Optimal Anticoagulation Through Genetics)
- GIFT (Genetics Informatics Trial of Warfarin to Prevent Venous Thrombosis)
- EU-PACT (European Pharmacogenetics of Anticoagulant Therapy-Warfarin).
We hope these studies will provide greater clarity on the clinical utility and cost-effectiveness of pharmacogenetic testing to guide warfarin dosing.
HOW SHOULD GENETIC INFORMATION BE USED TO GUIDE OR ALTER THERAPY?
Algorithms are available for estimating initial and maintenance warfarin doses based on genetic information (CYP2C9 and VKORC1), race or ethnicity, age, sex, body mass index, smoking status, and other medications taken. In addition, models incorporating the INR on day 4 and days 6 to 11 have been developed for dose refinement.15 The algorithms explain 30% to 60% of the variability of the data, with lower values for African Americans.7
A well-developed dosing model that includes traditional clinical factors and patient genetic status is publicly available online at www.warfarindosing.org.4
CPIC: A leader in applied pharmacogenetics
In late 2009, PharmGKB joined forces with the Pharmacogenomics Research Network of the National Institutes of Health to form the Clinical Pharmacogenetics Implementation Consortium (CPIC). This organization issues guidelines that are written by expert clinicians and scientists and then are peer-reviewed, published in leading journals, and simultaneously posted to the PharmGKB website along with supplemental information and updates.
CPIC’s goal is to review the current evidence and to address barriers to the adoption of pharmacogenetic testing into clinical practice. Its guidelines do not advise when or which pharmacogenetic tests should be ordered. Rather, they provide guidance on interpreting and applying such testing, should the test results be available.7
CPIC has guidelines on CYP2C9 and VKORC1 genotypes and warfarin dosing.8 If a patient’s genetic information is available, CPIC strongly recommends the use of pharmacogenetic algorithm-based dosing. If such an algorithm is not accessible, use of a genotype dosing table is recommended.8
Monitoring is still needed
Many factors can affect an individual’s response to warfarin above and beyond the above-noted clinical and genetic traits. These include diet, concomitant medications (both prescription and over-the-counter and herbal), and disease state. There may also be additional genetic polymorphisms not yet identified in various racial and ethnic groups that may affect dosing requirements. And as with all medications, patient compliance and dosing errors have a large potential to affect individual response. Therefore, clinicians should still be diligent about clinical monitoring.15
Most useful for initial dose
As with most pharmacogenetic information, the greatest benefit can be achieved when this information is used to guide the initial dose, although there is also some effect noted when this information is known and acted upon into the 2nd week of treatment.8
Patients on long-term warfarin treatment with stable doses and those unable to achieve stable dosing because of variable adherence or dietary vitamin K intake are less likely to benefit from genetic testing.
There are no published guidelines on the utility of pharmacogenetic testing if a patient is already on a stable dose of warfarin or has a known historical stable dose. There are also no published guidelines on changing the frequency of monitoring based on known genotype.
In children, the data are sparse at this time regarding the utility of pharmacogenetically informed dosing.
HOW DOES ONE ORDER TESTING, AND WHAT IS THE COST?
The FDA has approved four warfarin pharmacogenetic test kits. To be used in clinical decision-making, these tests must be done in a laboratory certified by the Clinical Laboratory Improvement Amendments (CLIA) program.
Testing typically costs a few hundred dollars and may take days for results to be returned if not available on site.15 At Cleveland Clinic, CYP2C9 and VKORC1 testing can be run in-house at a cost of about $700. Generally, many third-party payers do not reimburse for testing without a prior-approval process.
TO TEST OR NOT TO TEST
Pharmacogenetic testing is available and may help optimize warfarin dosing early in treatment, as well as help maintain therapeutic INRs more consistently. There is preliminary evidence that using this information to guide dosing improves clinical outcomes. Several large trials are under way to address additional questions of clinical utility, with results expected in the next year. There are also readily available decision-support tools to guide therapeutic dosing, and when pharmacogenetic test results are available, utilization of a warfarin dosing algorithm is recommended.
The largest barrier remaining appears to be cost (relative to perceived benefit), and until larger trials of clinical utility and cost-effectiveness are completed and analyzed, hurdles exist to obtaining coverage for such testing.
If it is readily available (and can be paid for by insurance companies or out-of-pocket) and test results can be obtained within 24 to 48 hours or before prescribing, pharmacogenetic testing can be a valuable tool to guide and manage warfarin dosing. Particularly for patients who want to be as proactive as possible, warfarin pharmacogenetic testing offers the ability to participate in this decision-making and to potentially reduce their risk of adverse drug events. And in view of the evidence and FDA recommendations, we propose that the discussion with our patients is not whether we should consider pharmacogenetic testing, but that we have considered pharmacogenetic testing, and why we have decided for or against it.
- Jacobs LG. Warfarin pharmacology, clinical management, and evaluation of hemorrhagic risk for the elderly. Clin Geriatr Med 2006; 22:17–32,vii–viii.
- Rieder MJ, Reiner AP, Gage BF, et al. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N Engl J Med 2005; 352:2285–2293.
- Higashi MK, Veenstra DL, Kondo LM, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 2002; 287:1690–1698.
- Shehab N, Sperling LS, Kegler SR, Budnitz DS. National estimates of emergency department visits for hemorrhage-related adverse events from clopidogrel plus aspirin and from warfarin. Arch Intern Med 2010; 170:1926–1933.
- Holbrook A, Schulman S, Witt DM, et al; American College of Chest Physicians. Evidence-based management of anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e152S–e184S.
- Gage BF, Eby C, Johnson JA, et al. Use of pharmacogenetic and clinical factors to predict the therapeutic dose of warfarin. Clin Pharmacol Ther 2008; 84:326–331.
- Cavallari LH, Shin J, Perera MA. Role of pharmacogenomics in the management of traditional and novel oral anticoagulants. Pharmacotherapy 2011; 31:1192–1207.
- Johnson JA, Gong L, Whirl-Carillo M, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin Pharmacol Ther 2011; 90:625–629.
- Limdi NA, McGwin G, Goldstein JA, et al. Influence of CYP2C9 and VKORC1 1173C/T genotype on the risk of hemorrhagic complications in African-American and European-American patients on warfarin. Clin Pharmacol Ther 2008; 83:312–321.
- Anderson JL, Horne BD, Stevens SM, et al. A randomized and clinical effectiveness trial comparing two pharmacogenetic algorithms and standard care for individualizing warfarin dosing (CoumaGen-II). Circulation 2012; 125:1997–2005.
- Yip VL, Pirmohamed M. Expanding role of pharmacogenomics in the management of cardiovascular disorders. Am J Cardiovasc Drugs 2013; 12 Apr; Epub ahead of print.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates: results from the MM-WES (Medco-Mayo Warfarin Effectiveness Study). J Am Coll Cardiol 2010; 55:2804–2812.
- Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med 2011; 364:1144–1153.
- Kitzmiller JP, Groen DK, Phelps MA, Sadee W. Pharmacogenomic testing: relevance in medical practice: why drugs work in some patients but not in others. Cleve Clin J Med 2011; 78:243–257.
- Carlquist JF, Anderson JL. Using pharmacogenetics in real time to guide warfarin initiation: a clinician update. Circulation 2011; 124:2554–2559.
- Jacobs LG. Warfarin pharmacology, clinical management, and evaluation of hemorrhagic risk for the elderly. Clin Geriatr Med 2006; 22:17–32,vii–viii.
- Rieder MJ, Reiner AP, Gage BF, et al. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N Engl J Med 2005; 352:2285–2293.
- Higashi MK, Veenstra DL, Kondo LM, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 2002; 287:1690–1698.
- Shehab N, Sperling LS, Kegler SR, Budnitz DS. National estimates of emergency department visits for hemorrhage-related adverse events from clopidogrel plus aspirin and from warfarin. Arch Intern Med 2010; 170:1926–1933.
- Holbrook A, Schulman S, Witt DM, et al; American College of Chest Physicians. Evidence-based management of anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e152S–e184S.
- Gage BF, Eby C, Johnson JA, et al. Use of pharmacogenetic and clinical factors to predict the therapeutic dose of warfarin. Clin Pharmacol Ther 2008; 84:326–331.
- Cavallari LH, Shin J, Perera MA. Role of pharmacogenomics in the management of traditional and novel oral anticoagulants. Pharmacotherapy 2011; 31:1192–1207.
- Johnson JA, Gong L, Whirl-Carillo M, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin Pharmacol Ther 2011; 90:625–629.
- Limdi NA, McGwin G, Goldstein JA, et al. Influence of CYP2C9 and VKORC1 1173C/T genotype on the risk of hemorrhagic complications in African-American and European-American patients on warfarin. Clin Pharmacol Ther 2008; 83:312–321.
- Anderson JL, Horne BD, Stevens SM, et al. A randomized and clinical effectiveness trial comparing two pharmacogenetic algorithms and standard care for individualizing warfarin dosing (CoumaGen-II). Circulation 2012; 125:1997–2005.
- Yip VL, Pirmohamed M. Expanding role of pharmacogenomics in the management of cardiovascular disorders. Am J Cardiovasc Drugs 2013; 12 Apr; Epub ahead of print.
- Epstein RS, Moyer TP, Aubert RE, et al. Warfarin genotyping reduces hospitalization rates: results from the MM-WES (Medco-Mayo Warfarin Effectiveness Study). J Am Coll Cardiol 2010; 55:2804–2812.
- Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med 2011; 364:1144–1153.
- Kitzmiller JP, Groen DK, Phelps MA, Sadee W. Pharmacogenomic testing: relevance in medical practice: why drugs work in some patients but not in others. Cleve Clin J Med 2011; 78:243–257.
- Carlquist JF, Anderson JL. Using pharmacogenetics in real time to guide warfarin initiation: a clinician update. Circulation 2011; 124:2554–2559.
A 67-year old man with an abdominal aortic aneurysm
A 67-year-old man presented for evaluation of an abdominal aortic aneurysm, noted 1 month previously after his primary care physician ordered screening ultrasonography as part of a routine annual physical examination. The man was experiencing no symptoms.
He had type 2 diabetes mellitus, chronic obstructive pulmonary disease, hypertension, and hyperlipidemia. He smoked two packs of cigarettes a day. He had never had surgery. His current medications included diltiazem, fenofibrate, niacin, and aspirin; because he had chronic obstructive pulmonary disease, he was not on a beta-blocker.
His father had died suddenly at the age of 77; his death was attributed to a cardiac cause, but no formal autopsy was performed. Neither the patient’s siblings nor his children were screened for aneurysms.
On physical examination, he was comfortable and in no acute distress. His blood pressure was 156/71 mm Hg, pulse 60, temperature 36.1°C (97.0°F), and body mass index 30.15 kg/m2, which is in the obese range.
He had no jugular venous distention, no carotid bruits, and no lymphadenopathy. The cardiac examination was unremarkable, with regular rate, normal sinus rhythm, and no murmurs. On pulmonary examination, inspiratory and expiratory wheezes were noted in all lung fields.
His abdomen was obese but not tender to palpation. The aneurysm was not palpable. His pedal pulses were normal. The remainder of the examination was normal.
WHO SHOULD BE SCREENED?
1. For which of the following groups does the United States Preventive Services Task Force (USPSTF) strongly recommend screening for abdominal aortic aneurysms?
- Men and women over age 65
- Men and women who have ever smoked and are over age 65
- Men over age 75 and men over age 65 who smoke
- Men age 65 to 75 who have ever smoked
In 2005, the USPSTF recommended one-time screening ultrasonography for all men age 65 to 75 who have ever smoked. On the basis of evidence available at the time, it made no recommendation for men age 65 to 75 who have never smoked, and it recommended against screening women.1
ANEURYSMS ARE COMMON, OFTEN ASYMPTOMATIC, UNTIL THEY RUPTURE
Abdominal aortic aneurysms are relatively common in older adults, with a prevalence of 1.4% in the US population age 50 to 84 years.2 In four randomized controlled trials of aneurysm screening in Europe and Australia, the prevalence of any aneurysm (not just abdominal aortic aneurysms) in men was 6% (95% confidence interval 5–6).3–6
Fewer studies are available on the prevalence in women. One study found a prevalence of 0.7% in 10,012 US women, compared with 3.9% in men.7
In a recent report of the aneurysm screening program in the United Kingdom, the incidence of aneurysms had decreased from historically reported estimates.8,9
In the year 2000, abdominal aortic aneurysms caused 15,000 deaths in the United States and were the 10th leading cause of death in white men age 65 to 74.10 The actual number of deaths may be larger, since some people may die suddenly of an aneurysm with no evaluation for attributable cause.11
Aortic aneurysms are often asymptomatic until they rupture, making them difficult to detect without a focused screening program. The goal of treatment is to avoid spontaneous rupture and death. When aneurysms rupture, the estimated death rate is 80%.6
EVIDENCE IN FAVOR OF SCREENING
Ultrasonography is nearly 100% sensitive and specific in detecting abdominal aortic aneurysms in patients without symptoms.12 In comparison, abdominal palpation is 68% sensitive and 75% specific.13
The larger the aneurysm, the higher the risk of rupture.14–16 The annual risk of rupture is:
- 0.5% with aneurysms smaller than 4.0 cm
- 1.0% with aneurysms 4.0–4.9 cm
- 11% with aneurysms 5.0–5.9 cm
- 26% with aneurysms 6.0–6.9 cm.
Several large randomized controlled trials in men over age 65 evaluated the effect of screening programs for abdominal aortic aneurysms on the rate of deaths from this cause.3–6,17 A meta-analysis of these trials found a relative risk of 0.60 in favor of screening—ie, men over age 65 who were screened had a 40% lower risk of dying of an abdominal aortic aneurysm than men who were not screened.18 In long-term follow-up, the rate continued to be about 50% lower with screening than without.19,20 The absolute reduction in risk of death was 0.13%.21
Absolute risk reduction and number needed to screen
2. If screening offers an absolute risk reduction in the death rate of 0.13%, how many need to be screened to prevent one death?
- 769
- 856
- 1,300
- 13,000
The number of patients that need to be screened to prevent one death is called the number needed to screen.22 It is calculated as 1 divided by the absolute risk reduction. Therefore, in screening for abdominal aortic aneurysms, the number needed to screen is 1/0.0013, or 769. Recall that these numbers are from men over age 65, with no upper limit in age. If we consider only men age 65 to 75, the absolute risk reduction is 0.16%, which corresponds to a number needed to screen of 625.
To put this in perspective, the number of people who need to be screened using fecal occult blood testing to prevent one death from colon cancer is 808, and the number of women who need to undergo mammography to prevent one breast cancer death is 1,887.21,22
Criteria for a good screening test
3. Which of the following is not one of the World Health Organization’s guiding principles for adopting a screening test?
- The disease must be common, or it must have grave consequences if it is not detected
- The disease must be detectable in a latent or early stage
- A screening test must exist that is acceptable to patients
- A treatment must exist that affects the natural history of the disease and its prognosis
- The cost of screening must be reasonable
- The screening test must have high sensitivity and specificity
In 1968, the World Health Organization published guidelines that continue to be used to determine the acceptability of screening tests.23 These principles state that for a screening test to be acceptable, the disease must be highly prevalent or result in grave consequences if not detected. The disease must have a latent or early stage in which it can be detected, and treatment must be available at that stage that affects the natural history and prognosis of the illness. The test must also be acceptable to patients physically, and the cost of it should be balanced in relation to possible expenditure on medical care as a whole.
As discussed previously, abdominal aortic aneurysms are common, and the consequences of rupture are grave. If the condition is detected early, treatment is available that can be lifesaving. Additionally, abdominal ultrasonography is noninvasive and inexpensive (costing roughly a few hundred dollars).24 Therefore, all of the World Health Organization criteria are satisfied. Improved outcomes with newer endovascular techniques for repair23 will likely also improve the value of screening.
Although high sensitivity and specificity are not required to satisfy the criteria, abdominal ultrasonography is nearly 100% sensitive and specific for detecting abdominal aortic aneurysms in patients without symptoms.12
Given the prevalence of the disease, by one estimate, if current USPSTF guidelines are followed (ie, if we screen only men age 65 to 75 who have ever smoked), for every 20 men we screen, we would detect one abdominal aortic aneurysm, and we would detect 29.5% of all of these aneurysms.2 If we screen all patients age 50 to 84, 74 people would need to be screened to detect one abdominal aortic aneurysm, but a much greater percentage of all of these aneurysms would be detected.
SHOULD OTHER GROUPS BE SCREENED?
4. The patient has a 40-year-old daughter who has hypertension and a 20-pack-year history of smoking. Should she be screened for an abdominal aortic aneurysm?
- Yes
- No
The 2005 USPSTF report recommends onetime ultrasonographic screening for all men age 65 to 75 who have ever smoked.1
The American Heart Association made a similar recommendation in 2005 in conjunction with the Society for Vascular Surgery, the American Association of Vascular Surgery, the Society for Vascular Medicine and Biology, and others.25 However, these groups also support screening men age 60 and older who are siblings or children of patients with abdominal aortic aneurysms, using physical examination and abdominal ultrasonography.
Both of the guidelines exclude women (who account for 41% of all deaths from this disease by one estimate) and nonsmokers (who account for 22%).2
The USPSTF makes no recommendation about nonsmokers, but it specifically recommends against screening women, stating that women have a low prevalence of large abdominal aortic aneurysms and that few women die of this disease. Therefore, according to the USPSTF, the risks of early treatment in women—including morbidity and death with surgical treatment and associated psychological harms—are not worth the benefits.1
However, a study of 3.1 million Americans found that women who have multiple cardiovascular risk factors such as smoking, hypertension, hyperlipidemia, and a family history of abdominal aortic aneurysm are at as great or greater risk of abdominal aortic aneurysm as men who fit the USPSTF criteria.2 Additionally, a positive family history of abdominal aortic aneurysm was among the strongest predictors of a diagnosis of abdominal aortic aneurysm on screening.2
Since 2005, newer guidelines have been released that broaden the recommendations for who should be screened. The Society for Vascular Surgery12 recommends screening:
- All men age 65 and older
- Men age 55 and older and women age 65 and older who have a family history of abdominal aortic aneurysm
- Women age 65 and older who have ever smoked.
A recent Swedish study demonstrated that the prevalence of abdominal aortic aneurysms in siblings of patients known to have this condition is significantly higher than in the general population; of the siblings who were screened, 11% had an abdominal aortic aneurysm, as did 17% of brothers and 6% of sisters.26
Nevertheless, broadened screening remains controversial, and more investigations of family history-based screening are ongoing.
WHEN DOES AN ABDOMINAL AORTIC ANEURYSM NEED SURGERY ?
Our patient was diagnosed with an infrarenal abdominal aortic aneurysm 6.5 cm in diameter and with bilateral common iliac artery aneurysms measuring 3.8 cm on the left and 5.2 cm on the right.
Computed tomography (CT) was done for preoperative planning (Figures 1 and 2), as it can define the aneurysm better for surgical intervention. Ultrasonography, while nearly 99% sensitive and specific for finding abdominal aortic aneurysms,12 does not provide the view of the abdominal anatomy that may be needed in surgical planning. The patient was seen by a vascular surgeon, and appropriate preoperative testing was done; the results showed that his risk during an open surgical procedure would be slightly above average.
The decision that needed to be made in this case was whether the patient should undergo surgery (either open or endovascular) or only medical intervention. In two randomized controlled trials comparing immediate intervention vs ongoing surveillance, the best threshold for surgical intervention was an aneurysm larger than 5.5 cm.27–29 Both trials found no benefit in terms of survival with surgical repair of aneurysms 4.0 to 5.4 cm: there was no long-term difference in the rate of survival in patients who underwent early surgical intervention compared with surveillance until the aneurysm was larger than 5.5 cm.
But this was with open surgery. What about endovascular repair? More recent studies that evaluated endovascular repair of small aneurysms (4.0–5.0 cm) found no improvement in end points, including time to aneurysm rupture and rate of aneurysm-related death, compared with surveillance.30,31
Treat risk factors
Medical therapy currently focuses on reducing risk factors for aneurysm growth and rupture, including hypertension, hyperlipidemia, and smoking, but research is focusing on angiotensin-converting enzyme inhibitors and experimental agents such as metalloproteinase inhibitors.32,33
Smoking is a major risk factor in the development, growth, and rupture of abdominal aortic aneurysms,34 and the 2005 joint guidelines of the American College of Cardiology and the American Heart Association (ACC/AHA) recommend that everyone with an abdominal aortic aneurysm or a family history of it be advised to stop smoking.25 This is especially important in light of data that show a higher risk of abdominal aortic aneurysm with a higher volume of smoking (total pack-years) and a decrease in risk with time since quitting.2
Medical management also includes treating other associated cardiovascular risk factors, including hypertension and dyslipidemia. The ACC/AHA guidelines recommend that patients with abdominal aortic aneurysms be treated similarly to patients with atherosclerotic disease or a coronary artery disease equivalent, including giving them a statin and an antiplatelet drug such as aspirin.
The ACC/AHA guidelines also recommend that patients who are managed medically and have an aneurysm of 3.0 to 4.0 cm undergo ultrasonographic monitoring every 2 to 3 years, and those with an aneurysm of 4.0 to 5.4 cm undergo monitoring with ultrasonography or CT every 6 to 12 months.25
5. Which of the following is the treatment of choice for our patient’s high blood pressure?
- Propranolol
- Lisinopril
- Hydralazine
- Hydrochlorothiazide
The recommended agents for blood pressure control in this patient population are betablockers, such as propranolol. In a small study of patients with infrarenal aortic aneurysms, beta-blockers reduced the mean expansion rate from 0.68 cm/year to 0.36 cm/year, although larger trials have not yet confirmed this benefit.35,36 The 2005 ACC/AHA guidelines recommend beta-blockers for patients who are being managed medically.25 Other antihypertensive drugs can be added to achieve optimal blood pressure control after the addition of a beta-blocker.
Open vs endovascular repair
If a patient has an abdominal aortic aneurysm larger than 5.5 cm or if the benefits of surgery are determined to outweigh the risks, a surgical plan should be developed. Patients should be evaluated for surgical risk factors, and this should guide the choice of surgical approach—ie, open repair or endovascular repair.
Compared with open repair, endovascular repair has been increasing in popularity. It has a lower rate of complications, including a significantly lower rate of perioperative death, even though patients who undergo endovascular repair are on average significantly older than those who undergo open repair.37–39
Endovascular repair is performed with open or percutaneous access of the common femoral artery. An endograft, which is packed into an introductory sheath, is introduced into the aorta and expands upon unsheathing. It is positioned to “land” in sealing zones of normal-caliber aorta, where it seals to exclude the aneurysm from circulatory flow (Figure 3).
This is different from the open approach in that it avoids the large incision and aortic cross-clamping necessary in open repair. In open repair, a large incision is made in the patient’s abdomen and the aorta is cross-clamped to stop blood flow. The aneurysm is then incised and a graft is sutured into place to protect the vessel wall from stress (Figure 4).
CASE CONCLUDED
Our patient elected to undergo endovascular repair of his aneurysm with a bifurcated graft (Figure 3). He was able to walk the day after his procedure, and he was sent home that same day. According to the guidelines of the Society for Vascular Surgery,40 he will have surveillance CT angiography at 1 and 12 months to detect “endoleak” or aneurysm enlargement. If these are not seen, he will then undergo routine surveillance with abdominal duplex ultrasonography.
- US Preventive Services Task Force. Screening for abdominal aortic aneurysm: recommendation statement. Ann Intern Med 2005; 142:198–202.
- Kent KC, Zwolak RM, Egorova NN, et al. Analysis of risk factors for abdominal aortic aneurysm in a cohort of more than 3 million individuals. J Vasc Surg 2010; 52:539–548.
- Lindholt JS, Juul S, Fasting H, Henneberg EW. Screening for abdominal aortic aneurysms: single centre randomised controlled trial. BMJ 2005; 330:750.
- Ashton HA, Buxton MJ, Day NE, et al; Multicentre Aneurysm Screening Study Group. The Multicentre Aneurysm Screening Study (MASS) into the effect of abdominal aortic aneurysm screening on mortality in men: a randomised controlled trial. Lancet 2002; 360:1531–1539.
- Norman PE, Jamrozik K, Lawrence-Brown MM, et al. Population based randomised controlled trial on impact of screening on mortality from abdominal aortic aneurysm. BMJ 2004; 329:1259.
- Vardulaki KA, Walker NM, Couto E, et al. Late results concerning feasibility and compliance from a randomized trial of ultrasonographic screening for abdominal aortic aneurysm. Br J Surg 2002; 89:861–864.
- Derubertis BG, Trocciola SM, Ryer EJ, et al. Abdominal aortic aneurysm in women: prevalence, risk factors, and implications for screening. J Vasc Surg 2007; 46:630–635.
- Sandiford P, Mosquera D, Bramley D. Trends in incidence and mortality from abdominal aortic aneurysm in New Zealand. Br J Surg 2011; 98:645–651.
- Anjum A, Powell JT. Is the incidence of abdominal aortic aneurysm declining in the 21st century? Mortality and hospital admissions for England & Wales and Scotland. Eur J Vasc Endovasc Surg 2012; 43:161–166.
- Anderson RN. Deaths: leading causes for 2000. Natl Vital Stat Rep 2002; 50:1–85.
- Kent KC, Zwolak RM, Jaff MR, et al; Society for Vascular Surgery; American Association of Vascular Surgery; Society for Vascular Medicine and Biology. Screening for abdominal aortic aneurysm: a consensus statement. J Vasc Surg 2004; 39:267–269.
- Chaikof EL, Brewster DC, Dalman RL, et al; Society for Vascular Surgery. The care of patients with an abdominal aortic aneurysm: the Society for Vascular Surgery practice guidelines. J Vasc Surg 2009; 50(suppl 4):S2–S49.
- Fink HA, Lederle FA, Roth CS, Bowles CA, Nelson DB, Haas MA. The accuracy of physical examination to detect abdominal aortic aneurysm. Arch Intern Med 2000; 160:833–836.
- Reed WW, Hallett JW, Damiano MA, Ballard DJ. Learning from the last ultrasound. A population-based study of patients with abdominal aortic aneurysm. Arch Intern Med 1997; 157:2064–2068.
- Bernstein EF, Dilley RB, Goldberger LE, Gosink BB, Leopold GR. Growth rates of small abdominal aortic aneurysms. Surgery 1976; 80:765–773.
- Cronenwett JL, Sargent SK, Wall MH, et al. Variables that affect the expansion rate and outcome of small abdominal aortic aneurysms. J Vasc Surg 1990; 11:260–268.
- Scott RA, Bridgewater SG, Ashton HA. Randomized clinical trial of screening for abdominal aortic aneurysm in women. Br J Surg 2002; 89:283–285.
- Fleming C, Whitlock EP, Beil TL, Lederle FA. Screening for abdominal aortic aneurysm: a best-evidence systematic review for the US Preventive Services Task Force. Ann Intern Med 2005; 142:203–211.
- Lindholt JS, Sørensen J, Søgaard R, Henneberg EW. Long-term benefit and cost-effectiveness analysis of screening for abdominal aortic aneurysms from a randomized controlled trial. Br J Surg 2010; 97:826–834.
- Thompson SG, Ashton HA, Gao L, Scott RA; Multicentre Aneurysm Screening Study Group. Screening men for abdominal aortic aneurysm: 10 year mortality and cost effectiveness results from the randomised Multicentre Aneurysm Screening Study. BMJ 2009; 338:b2307.
- Mastracci TM, Cina CS. Regarding Screening for abdominal aortic aneurysm reduces both aneurysm-related and all-cause mortality (letter). J Vasc Surg 2007; 46:1312.
- Rembold CM. Number needed to screen: development of a statistic for disease screening. BMJ 1998; 317:307–312.
- Wilson JMG, Jungner G. Principles and practice of screening for disease. World Health Organization. Public Health Papers #34.
- Lee TY, Korn P, Heller JA, et al. The cost-effectiveness of a “quickscreen” program for abdominal aortic aneurysms. Surgery 2002; 132:399–407.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al; American Association for Vascular Surgery; Society for Vascular Surgery; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine and Biology; Society of Interventional Radiology; ACC/AHA Task Force on Practice Guidelines Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease; American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; Vascular Disease Foundation. ACC/AHA 2005 Practice Guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation 2006; 113:e463–e654.
- Linné A, Lindström D, Hultgren R. High prevalence of abdominal aortic aneurysms in brothers and sisters of patients despite a low prevalence in the population. J Vasc Surg 2012; 56:305–310.
- The UK Small Aneurysm Trial Participants. Mortality results for randomised controlled trial of early elective surgery or ultrasonographic surveillance for small abdominal aortic aneurysms. Lancet 1998; 352:1649–1655.
- Lederle FA, Johnson GR, Wilson SE, et al. Prevalence and associations of abdominal aortic aneurysm detected through screening. Aneurysm Detection and Management (ADAM) Veterans Affairs Cooperative Study Group. Ann Intern Med 1997; 126:441–449.
- Brewster DC, Cronenwett JL, Hallett JW, Johnston KW, Krupski WC, Matsumura JS; Joint Council of the American Association for Vascular Surgery and Society for Vascular Surgery. Guidelines for the treatment of abdominal aortic aneurysms. Report of a subcommittee of the Joint Council of the American Association for Vascular Surgery and Society for Vascular Surgery. J Vasc Surg 2003; 37:1106–117.
- Ouriel K, Clair DG, Kent KC, Zarins CK; Positive Impact of Endovascular Options for treating Aneurysms Early (PIVOTAL) Investigators. Endovascular repair compared with surveillance for patients with small abdominal aortic aneurysms. J Vasc Surg 2010; 51:1081–1087.
- De Rango P, Verzini F, Parlani G; Comparison of surveillance vs Aortic Endografting for Small Aneurysm Repair (CAESAR) Investigators. Quality of life in patients with small abdominal aortic aneurysm: the effect of early endovascular repair versus surveillance in the CAESAR trial. Eur J Vasc Endovasc Surg 2011; 41:324–331.
- Antoniou GA, Lazarides MK, Patera S, et al. Assessment of insertion/deletion polymorphism of the angiotensin-converting enzyme gene in abdominal aortic aneurysm and inguinal hernia. Vascular 2012; Epub ahead of print.
- Ogata T, Shibamura H, Tromp G, et al. Genetic analysis of polymorphisms in biologically relevant candidate genes in patients with abdominal aortic aneurysms. J Vasc Surg 2005; 41:1036–1042.
- Powell JT, Greenhalgh RM. Clinical practice. Small abdominal aortic aneurysms. N Engl J Med 2003; 348:1895–1901.
- Gadowski GR, Pilcher DB, Ricci MA. Abdominal aortic aneurysm expansion rate: effect of size and beta-adrenergic blockade. J Vasc Surg 1994; 19:727–731.
- Propanolol Aneurysm Trial Investigators. Propranolol for small abdominal aortic aneurysms: results of a randomized trial. J Vasc Surg 2002; 35:72–79.
- Jackson RS, Chang DC, Freischlag JA. Comparison of long-term survival after open vs endovascular repair of intact abdominal aortic aneurysm among Medicare beneficiaries. JAMA 2012; 307:1621–1628.
- Dillavou ED, Muluk SC, Makaroun MS. Improving aneurysm-related outcomes: nationwide benefits of endovascular repair. J Vasc Surg 2006; 43:446–451.
- Giles KA, Pomposelli F, Hamdan A, Wyers M, Jhaveri A, Schermerhorn ML. Decrease in total aneurysm-related deaths in the era of endovascular aneurysm repair. J Vasc Surg 2009; 49:543–550.
- Chaikof EL, Blankensteijn JD, Harris PL, et al; Ad Hoc Committee for Standardized Reporting Practices in Vascular Surgery of The Society for Vascular Surgery/American Association for Vascular Surgery. Reporting standards for endovascular aortic aneurysm repair. J Vasc Surg 2002; 35:1048–1060.
A 67-year-old man presented for evaluation of an abdominal aortic aneurysm, noted 1 month previously after his primary care physician ordered screening ultrasonography as part of a routine annual physical examination. The man was experiencing no symptoms.
He had type 2 diabetes mellitus, chronic obstructive pulmonary disease, hypertension, and hyperlipidemia. He smoked two packs of cigarettes a day. He had never had surgery. His current medications included diltiazem, fenofibrate, niacin, and aspirin; because he had chronic obstructive pulmonary disease, he was not on a beta-blocker.
His father had died suddenly at the age of 77; his death was attributed to a cardiac cause, but no formal autopsy was performed. Neither the patient’s siblings nor his children were screened for aneurysms.
On physical examination, he was comfortable and in no acute distress. His blood pressure was 156/71 mm Hg, pulse 60, temperature 36.1°C (97.0°F), and body mass index 30.15 kg/m2, which is in the obese range.
He had no jugular venous distention, no carotid bruits, and no lymphadenopathy. The cardiac examination was unremarkable, with regular rate, normal sinus rhythm, and no murmurs. On pulmonary examination, inspiratory and expiratory wheezes were noted in all lung fields.
His abdomen was obese but not tender to palpation. The aneurysm was not palpable. His pedal pulses were normal. The remainder of the examination was normal.
WHO SHOULD BE SCREENED?
1. For which of the following groups does the United States Preventive Services Task Force (USPSTF) strongly recommend screening for abdominal aortic aneurysms?
- Men and women over age 65
- Men and women who have ever smoked and are over age 65
- Men over age 75 and men over age 65 who smoke
- Men age 65 to 75 who have ever smoked
In 2005, the USPSTF recommended one-time screening ultrasonography for all men age 65 to 75 who have ever smoked. On the basis of evidence available at the time, it made no recommendation for men age 65 to 75 who have never smoked, and it recommended against screening women.1
ANEURYSMS ARE COMMON, OFTEN ASYMPTOMATIC, UNTIL THEY RUPTURE
Abdominal aortic aneurysms are relatively common in older adults, with a prevalence of 1.4% in the US population age 50 to 84 years.2 In four randomized controlled trials of aneurysm screening in Europe and Australia, the prevalence of any aneurysm (not just abdominal aortic aneurysms) in men was 6% (95% confidence interval 5–6).3–6
Fewer studies are available on the prevalence in women. One study found a prevalence of 0.7% in 10,012 US women, compared with 3.9% in men.7
In a recent report of the aneurysm screening program in the United Kingdom, the incidence of aneurysms had decreased from historically reported estimates.8,9
In the year 2000, abdominal aortic aneurysms caused 15,000 deaths in the United States and were the 10th leading cause of death in white men age 65 to 74.10 The actual number of deaths may be larger, since some people may die suddenly of an aneurysm with no evaluation for attributable cause.11
Aortic aneurysms are often asymptomatic until they rupture, making them difficult to detect without a focused screening program. The goal of treatment is to avoid spontaneous rupture and death. When aneurysms rupture, the estimated death rate is 80%.6
EVIDENCE IN FAVOR OF SCREENING
Ultrasonography is nearly 100% sensitive and specific in detecting abdominal aortic aneurysms in patients without symptoms.12 In comparison, abdominal palpation is 68% sensitive and 75% specific.13
The larger the aneurysm, the higher the risk of rupture.14–16 The annual risk of rupture is:
- 0.5% with aneurysms smaller than 4.0 cm
- 1.0% with aneurysms 4.0–4.9 cm
- 11% with aneurysms 5.0–5.9 cm
- 26% with aneurysms 6.0–6.9 cm.
Several large randomized controlled trials in men over age 65 evaluated the effect of screening programs for abdominal aortic aneurysms on the rate of deaths from this cause.3–6,17 A meta-analysis of these trials found a relative risk of 0.60 in favor of screening—ie, men over age 65 who were screened had a 40% lower risk of dying of an abdominal aortic aneurysm than men who were not screened.18 In long-term follow-up, the rate continued to be about 50% lower with screening than without.19,20 The absolute reduction in risk of death was 0.13%.21
Absolute risk reduction and number needed to screen
2. If screening offers an absolute risk reduction in the death rate of 0.13%, how many need to be screened to prevent one death?
- 769
- 856
- 1,300
- 13,000
The number of patients that need to be screened to prevent one death is called the number needed to screen.22 It is calculated as 1 divided by the absolute risk reduction. Therefore, in screening for abdominal aortic aneurysms, the number needed to screen is 1/0.0013, or 769. Recall that these numbers are from men over age 65, with no upper limit in age. If we consider only men age 65 to 75, the absolute risk reduction is 0.16%, which corresponds to a number needed to screen of 625.
To put this in perspective, the number of people who need to be screened using fecal occult blood testing to prevent one death from colon cancer is 808, and the number of women who need to undergo mammography to prevent one breast cancer death is 1,887.21,22
Criteria for a good screening test
3. Which of the following is not one of the World Health Organization’s guiding principles for adopting a screening test?
- The disease must be common, or it must have grave consequences if it is not detected
- The disease must be detectable in a latent or early stage
- A screening test must exist that is acceptable to patients
- A treatment must exist that affects the natural history of the disease and its prognosis
- The cost of screening must be reasonable
- The screening test must have high sensitivity and specificity
In 1968, the World Health Organization published guidelines that continue to be used to determine the acceptability of screening tests.23 These principles state that for a screening test to be acceptable, the disease must be highly prevalent or result in grave consequences if not detected. The disease must have a latent or early stage in which it can be detected, and treatment must be available at that stage that affects the natural history and prognosis of the illness. The test must also be acceptable to patients physically, and the cost of it should be balanced in relation to possible expenditure on medical care as a whole.
As discussed previously, abdominal aortic aneurysms are common, and the consequences of rupture are grave. If the condition is detected early, treatment is available that can be lifesaving. Additionally, abdominal ultrasonography is noninvasive and inexpensive (costing roughly a few hundred dollars).24 Therefore, all of the World Health Organization criteria are satisfied. Improved outcomes with newer endovascular techniques for repair23 will likely also improve the value of screening.
Although high sensitivity and specificity are not required to satisfy the criteria, abdominal ultrasonography is nearly 100% sensitive and specific for detecting abdominal aortic aneurysms in patients without symptoms.12
Given the prevalence of the disease, by one estimate, if current USPSTF guidelines are followed (ie, if we screen only men age 65 to 75 who have ever smoked), for every 20 men we screen, we would detect one abdominal aortic aneurysm, and we would detect 29.5% of all of these aneurysms.2 If we screen all patients age 50 to 84, 74 people would need to be screened to detect one abdominal aortic aneurysm, but a much greater percentage of all of these aneurysms would be detected.
SHOULD OTHER GROUPS BE SCREENED?
4. The patient has a 40-year-old daughter who has hypertension and a 20-pack-year history of smoking. Should she be screened for an abdominal aortic aneurysm?
- Yes
- No
The 2005 USPSTF report recommends onetime ultrasonographic screening for all men age 65 to 75 who have ever smoked.1
The American Heart Association made a similar recommendation in 2005 in conjunction with the Society for Vascular Surgery, the American Association of Vascular Surgery, the Society for Vascular Medicine and Biology, and others.25 However, these groups also support screening men age 60 and older who are siblings or children of patients with abdominal aortic aneurysms, using physical examination and abdominal ultrasonography.
Both of the guidelines exclude women (who account for 41% of all deaths from this disease by one estimate) and nonsmokers (who account for 22%).2
The USPSTF makes no recommendation about nonsmokers, but it specifically recommends against screening women, stating that women have a low prevalence of large abdominal aortic aneurysms and that few women die of this disease. Therefore, according to the USPSTF, the risks of early treatment in women—including morbidity and death with surgical treatment and associated psychological harms—are not worth the benefits.1
However, a study of 3.1 million Americans found that women who have multiple cardiovascular risk factors such as smoking, hypertension, hyperlipidemia, and a family history of abdominal aortic aneurysm are at as great or greater risk of abdominal aortic aneurysm as men who fit the USPSTF criteria.2 Additionally, a positive family history of abdominal aortic aneurysm was among the strongest predictors of a diagnosis of abdominal aortic aneurysm on screening.2
Since 2005, newer guidelines have been released that broaden the recommendations for who should be screened. The Society for Vascular Surgery12 recommends screening:
- All men age 65 and older
- Men age 55 and older and women age 65 and older who have a family history of abdominal aortic aneurysm
- Women age 65 and older who have ever smoked.
A recent Swedish study demonstrated that the prevalence of abdominal aortic aneurysms in siblings of patients known to have this condition is significantly higher than in the general population; of the siblings who were screened, 11% had an abdominal aortic aneurysm, as did 17% of brothers and 6% of sisters.26
Nevertheless, broadened screening remains controversial, and more investigations of family history-based screening are ongoing.
WHEN DOES AN ABDOMINAL AORTIC ANEURYSM NEED SURGERY ?
Our patient was diagnosed with an infrarenal abdominal aortic aneurysm 6.5 cm in diameter and with bilateral common iliac artery aneurysms measuring 3.8 cm on the left and 5.2 cm on the right.
Computed tomography (CT) was done for preoperative planning (Figures 1 and 2), as it can define the aneurysm better for surgical intervention. Ultrasonography, while nearly 99% sensitive and specific for finding abdominal aortic aneurysms,12 does not provide the view of the abdominal anatomy that may be needed in surgical planning. The patient was seen by a vascular surgeon, and appropriate preoperative testing was done; the results showed that his risk during an open surgical procedure would be slightly above average.
The decision that needed to be made in this case was whether the patient should undergo surgery (either open or endovascular) or only medical intervention. In two randomized controlled trials comparing immediate intervention vs ongoing surveillance, the best threshold for surgical intervention was an aneurysm larger than 5.5 cm.27–29 Both trials found no benefit in terms of survival with surgical repair of aneurysms 4.0 to 5.4 cm: there was no long-term difference in the rate of survival in patients who underwent early surgical intervention compared with surveillance until the aneurysm was larger than 5.5 cm.
But this was with open surgery. What about endovascular repair? More recent studies that evaluated endovascular repair of small aneurysms (4.0–5.0 cm) found no improvement in end points, including time to aneurysm rupture and rate of aneurysm-related death, compared with surveillance.30,31
Treat risk factors
Medical therapy currently focuses on reducing risk factors for aneurysm growth and rupture, including hypertension, hyperlipidemia, and smoking, but research is focusing on angiotensin-converting enzyme inhibitors and experimental agents such as metalloproteinase inhibitors.32,33
Smoking is a major risk factor in the development, growth, and rupture of abdominal aortic aneurysms,34 and the 2005 joint guidelines of the American College of Cardiology and the American Heart Association (ACC/AHA) recommend that everyone with an abdominal aortic aneurysm or a family history of it be advised to stop smoking.25 This is especially important in light of data that show a higher risk of abdominal aortic aneurysm with a higher volume of smoking (total pack-years) and a decrease in risk with time since quitting.2
Medical management also includes treating other associated cardiovascular risk factors, including hypertension and dyslipidemia. The ACC/AHA guidelines recommend that patients with abdominal aortic aneurysms be treated similarly to patients with atherosclerotic disease or a coronary artery disease equivalent, including giving them a statin and an antiplatelet drug such as aspirin.
The ACC/AHA guidelines also recommend that patients who are managed medically and have an aneurysm of 3.0 to 4.0 cm undergo ultrasonographic monitoring every 2 to 3 years, and those with an aneurysm of 4.0 to 5.4 cm undergo monitoring with ultrasonography or CT every 6 to 12 months.25
5. Which of the following is the treatment of choice for our patient’s high blood pressure?
- Propranolol
- Lisinopril
- Hydralazine
- Hydrochlorothiazide
The recommended agents for blood pressure control in this patient population are betablockers, such as propranolol. In a small study of patients with infrarenal aortic aneurysms, beta-blockers reduced the mean expansion rate from 0.68 cm/year to 0.36 cm/year, although larger trials have not yet confirmed this benefit.35,36 The 2005 ACC/AHA guidelines recommend beta-blockers for patients who are being managed medically.25 Other antihypertensive drugs can be added to achieve optimal blood pressure control after the addition of a beta-blocker.
Open vs endovascular repair
If a patient has an abdominal aortic aneurysm larger than 5.5 cm or if the benefits of surgery are determined to outweigh the risks, a surgical plan should be developed. Patients should be evaluated for surgical risk factors, and this should guide the choice of surgical approach—ie, open repair or endovascular repair.
Compared with open repair, endovascular repair has been increasing in popularity. It has a lower rate of complications, including a significantly lower rate of perioperative death, even though patients who undergo endovascular repair are on average significantly older than those who undergo open repair.37–39
Endovascular repair is performed with open or percutaneous access of the common femoral artery. An endograft, which is packed into an introductory sheath, is introduced into the aorta and expands upon unsheathing. It is positioned to “land” in sealing zones of normal-caliber aorta, where it seals to exclude the aneurysm from circulatory flow (Figure 3).
This is different from the open approach in that it avoids the large incision and aortic cross-clamping necessary in open repair. In open repair, a large incision is made in the patient’s abdomen and the aorta is cross-clamped to stop blood flow. The aneurysm is then incised and a graft is sutured into place to protect the vessel wall from stress (Figure 4).
CASE CONCLUDED
Our patient elected to undergo endovascular repair of his aneurysm with a bifurcated graft (Figure 3). He was able to walk the day after his procedure, and he was sent home that same day. According to the guidelines of the Society for Vascular Surgery,40 he will have surveillance CT angiography at 1 and 12 months to detect “endoleak” or aneurysm enlargement. If these are not seen, he will then undergo routine surveillance with abdominal duplex ultrasonography.
A 67-year-old man presented for evaluation of an abdominal aortic aneurysm, noted 1 month previously after his primary care physician ordered screening ultrasonography as part of a routine annual physical examination. The man was experiencing no symptoms.
He had type 2 diabetes mellitus, chronic obstructive pulmonary disease, hypertension, and hyperlipidemia. He smoked two packs of cigarettes a day. He had never had surgery. His current medications included diltiazem, fenofibrate, niacin, and aspirin; because he had chronic obstructive pulmonary disease, he was not on a beta-blocker.
His father had died suddenly at the age of 77; his death was attributed to a cardiac cause, but no formal autopsy was performed. Neither the patient’s siblings nor his children were screened for aneurysms.
On physical examination, he was comfortable and in no acute distress. His blood pressure was 156/71 mm Hg, pulse 60, temperature 36.1°C (97.0°F), and body mass index 30.15 kg/m2, which is in the obese range.
He had no jugular venous distention, no carotid bruits, and no lymphadenopathy. The cardiac examination was unremarkable, with regular rate, normal sinus rhythm, and no murmurs. On pulmonary examination, inspiratory and expiratory wheezes were noted in all lung fields.
His abdomen was obese but not tender to palpation. The aneurysm was not palpable. His pedal pulses were normal. The remainder of the examination was normal.
WHO SHOULD BE SCREENED?
1. For which of the following groups does the United States Preventive Services Task Force (USPSTF) strongly recommend screening for abdominal aortic aneurysms?
- Men and women over age 65
- Men and women who have ever smoked and are over age 65
- Men over age 75 and men over age 65 who smoke
- Men age 65 to 75 who have ever smoked
In 2005, the USPSTF recommended one-time screening ultrasonography for all men age 65 to 75 who have ever smoked. On the basis of evidence available at the time, it made no recommendation for men age 65 to 75 who have never smoked, and it recommended against screening women.1
ANEURYSMS ARE COMMON, OFTEN ASYMPTOMATIC, UNTIL THEY RUPTURE
Abdominal aortic aneurysms are relatively common in older adults, with a prevalence of 1.4% in the US population age 50 to 84 years.2 In four randomized controlled trials of aneurysm screening in Europe and Australia, the prevalence of any aneurysm (not just abdominal aortic aneurysms) in men was 6% (95% confidence interval 5–6).3–6
Fewer studies are available on the prevalence in women. One study found a prevalence of 0.7% in 10,012 US women, compared with 3.9% in men.7
In a recent report of the aneurysm screening program in the United Kingdom, the incidence of aneurysms had decreased from historically reported estimates.8,9
In the year 2000, abdominal aortic aneurysms caused 15,000 deaths in the United States and were the 10th leading cause of death in white men age 65 to 74.10 The actual number of deaths may be larger, since some people may die suddenly of an aneurysm with no evaluation for attributable cause.11
Aortic aneurysms are often asymptomatic until they rupture, making them difficult to detect without a focused screening program. The goal of treatment is to avoid spontaneous rupture and death. When aneurysms rupture, the estimated death rate is 80%.6
EVIDENCE IN FAVOR OF SCREENING
Ultrasonography is nearly 100% sensitive and specific in detecting abdominal aortic aneurysms in patients without symptoms.12 In comparison, abdominal palpation is 68% sensitive and 75% specific.13
The larger the aneurysm, the higher the risk of rupture.14–16 The annual risk of rupture is:
- 0.5% with aneurysms smaller than 4.0 cm
- 1.0% with aneurysms 4.0–4.9 cm
- 11% with aneurysms 5.0–5.9 cm
- 26% with aneurysms 6.0–6.9 cm.
Several large randomized controlled trials in men over age 65 evaluated the effect of screening programs for abdominal aortic aneurysms on the rate of deaths from this cause.3–6,17 A meta-analysis of these trials found a relative risk of 0.60 in favor of screening—ie, men over age 65 who were screened had a 40% lower risk of dying of an abdominal aortic aneurysm than men who were not screened.18 In long-term follow-up, the rate continued to be about 50% lower with screening than without.19,20 The absolute reduction in risk of death was 0.13%.21
Absolute risk reduction and number needed to screen
2. If screening offers an absolute risk reduction in the death rate of 0.13%, how many need to be screened to prevent one death?
- 769
- 856
- 1,300
- 13,000
The number of patients that need to be screened to prevent one death is called the number needed to screen.22 It is calculated as 1 divided by the absolute risk reduction. Therefore, in screening for abdominal aortic aneurysms, the number needed to screen is 1/0.0013, or 769. Recall that these numbers are from men over age 65, with no upper limit in age. If we consider only men age 65 to 75, the absolute risk reduction is 0.16%, which corresponds to a number needed to screen of 625.
To put this in perspective, the number of people who need to be screened using fecal occult blood testing to prevent one death from colon cancer is 808, and the number of women who need to undergo mammography to prevent one breast cancer death is 1,887.21,22
Criteria for a good screening test
3. Which of the following is not one of the World Health Organization’s guiding principles for adopting a screening test?
- The disease must be common, or it must have grave consequences if it is not detected
- The disease must be detectable in a latent or early stage
- A screening test must exist that is acceptable to patients
- A treatment must exist that affects the natural history of the disease and its prognosis
- The cost of screening must be reasonable
- The screening test must have high sensitivity and specificity
In 1968, the World Health Organization published guidelines that continue to be used to determine the acceptability of screening tests.23 These principles state that for a screening test to be acceptable, the disease must be highly prevalent or result in grave consequences if not detected. The disease must have a latent or early stage in which it can be detected, and treatment must be available at that stage that affects the natural history and prognosis of the illness. The test must also be acceptable to patients physically, and the cost of it should be balanced in relation to possible expenditure on medical care as a whole.
As discussed previously, abdominal aortic aneurysms are common, and the consequences of rupture are grave. If the condition is detected early, treatment is available that can be lifesaving. Additionally, abdominal ultrasonography is noninvasive and inexpensive (costing roughly a few hundred dollars).24 Therefore, all of the World Health Organization criteria are satisfied. Improved outcomes with newer endovascular techniques for repair23 will likely also improve the value of screening.
Although high sensitivity and specificity are not required to satisfy the criteria, abdominal ultrasonography is nearly 100% sensitive and specific for detecting abdominal aortic aneurysms in patients without symptoms.12
Given the prevalence of the disease, by one estimate, if current USPSTF guidelines are followed (ie, if we screen only men age 65 to 75 who have ever smoked), for every 20 men we screen, we would detect one abdominal aortic aneurysm, and we would detect 29.5% of all of these aneurysms.2 If we screen all patients age 50 to 84, 74 people would need to be screened to detect one abdominal aortic aneurysm, but a much greater percentage of all of these aneurysms would be detected.
SHOULD OTHER GROUPS BE SCREENED?
4. The patient has a 40-year-old daughter who has hypertension and a 20-pack-year history of smoking. Should she be screened for an abdominal aortic aneurysm?
- Yes
- No
The 2005 USPSTF report recommends onetime ultrasonographic screening for all men age 65 to 75 who have ever smoked.1
The American Heart Association made a similar recommendation in 2005 in conjunction with the Society for Vascular Surgery, the American Association of Vascular Surgery, the Society for Vascular Medicine and Biology, and others.25 However, these groups also support screening men age 60 and older who are siblings or children of patients with abdominal aortic aneurysms, using physical examination and abdominal ultrasonography.
Both of the guidelines exclude women (who account for 41% of all deaths from this disease by one estimate) and nonsmokers (who account for 22%).2
The USPSTF makes no recommendation about nonsmokers, but it specifically recommends against screening women, stating that women have a low prevalence of large abdominal aortic aneurysms and that few women die of this disease. Therefore, according to the USPSTF, the risks of early treatment in women—including morbidity and death with surgical treatment and associated psychological harms—are not worth the benefits.1
However, a study of 3.1 million Americans found that women who have multiple cardiovascular risk factors such as smoking, hypertension, hyperlipidemia, and a family history of abdominal aortic aneurysm are at as great or greater risk of abdominal aortic aneurysm as men who fit the USPSTF criteria.2 Additionally, a positive family history of abdominal aortic aneurysm was among the strongest predictors of a diagnosis of abdominal aortic aneurysm on screening.2
Since 2005, newer guidelines have been released that broaden the recommendations for who should be screened. The Society for Vascular Surgery12 recommends screening:
- All men age 65 and older
- Men age 55 and older and women age 65 and older who have a family history of abdominal aortic aneurysm
- Women age 65 and older who have ever smoked.
A recent Swedish study demonstrated that the prevalence of abdominal aortic aneurysms in siblings of patients known to have this condition is significantly higher than in the general population; of the siblings who were screened, 11% had an abdominal aortic aneurysm, as did 17% of brothers and 6% of sisters.26
Nevertheless, broadened screening remains controversial, and more investigations of family history-based screening are ongoing.
WHEN DOES AN ABDOMINAL AORTIC ANEURYSM NEED SURGERY ?
Our patient was diagnosed with an infrarenal abdominal aortic aneurysm 6.5 cm in diameter and with bilateral common iliac artery aneurysms measuring 3.8 cm on the left and 5.2 cm on the right.
Computed tomography (CT) was done for preoperative planning (Figures 1 and 2), as it can define the aneurysm better for surgical intervention. Ultrasonography, while nearly 99% sensitive and specific for finding abdominal aortic aneurysms,12 does not provide the view of the abdominal anatomy that may be needed in surgical planning. The patient was seen by a vascular surgeon, and appropriate preoperative testing was done; the results showed that his risk during an open surgical procedure would be slightly above average.
The decision that needed to be made in this case was whether the patient should undergo surgery (either open or endovascular) or only medical intervention. In two randomized controlled trials comparing immediate intervention vs ongoing surveillance, the best threshold for surgical intervention was an aneurysm larger than 5.5 cm.27–29 Both trials found no benefit in terms of survival with surgical repair of aneurysms 4.0 to 5.4 cm: there was no long-term difference in the rate of survival in patients who underwent early surgical intervention compared with surveillance until the aneurysm was larger than 5.5 cm.
But this was with open surgery. What about endovascular repair? More recent studies that evaluated endovascular repair of small aneurysms (4.0–5.0 cm) found no improvement in end points, including time to aneurysm rupture and rate of aneurysm-related death, compared with surveillance.30,31
Treat risk factors
Medical therapy currently focuses on reducing risk factors for aneurysm growth and rupture, including hypertension, hyperlipidemia, and smoking, but research is focusing on angiotensin-converting enzyme inhibitors and experimental agents such as metalloproteinase inhibitors.32,33
Smoking is a major risk factor in the development, growth, and rupture of abdominal aortic aneurysms,34 and the 2005 joint guidelines of the American College of Cardiology and the American Heart Association (ACC/AHA) recommend that everyone with an abdominal aortic aneurysm or a family history of it be advised to stop smoking.25 This is especially important in light of data that show a higher risk of abdominal aortic aneurysm with a higher volume of smoking (total pack-years) and a decrease in risk with time since quitting.2
Medical management also includes treating other associated cardiovascular risk factors, including hypertension and dyslipidemia. The ACC/AHA guidelines recommend that patients with abdominal aortic aneurysms be treated similarly to patients with atherosclerotic disease or a coronary artery disease equivalent, including giving them a statin and an antiplatelet drug such as aspirin.
The ACC/AHA guidelines also recommend that patients who are managed medically and have an aneurysm of 3.0 to 4.0 cm undergo ultrasonographic monitoring every 2 to 3 years, and those with an aneurysm of 4.0 to 5.4 cm undergo monitoring with ultrasonography or CT every 6 to 12 months.25
5. Which of the following is the treatment of choice for our patient’s high blood pressure?
- Propranolol
- Lisinopril
- Hydralazine
- Hydrochlorothiazide
The recommended agents for blood pressure control in this patient population are betablockers, such as propranolol. In a small study of patients with infrarenal aortic aneurysms, beta-blockers reduced the mean expansion rate from 0.68 cm/year to 0.36 cm/year, although larger trials have not yet confirmed this benefit.35,36 The 2005 ACC/AHA guidelines recommend beta-blockers for patients who are being managed medically.25 Other antihypertensive drugs can be added to achieve optimal blood pressure control after the addition of a beta-blocker.
Open vs endovascular repair
If a patient has an abdominal aortic aneurysm larger than 5.5 cm or if the benefits of surgery are determined to outweigh the risks, a surgical plan should be developed. Patients should be evaluated for surgical risk factors, and this should guide the choice of surgical approach—ie, open repair or endovascular repair.
Compared with open repair, endovascular repair has been increasing in popularity. It has a lower rate of complications, including a significantly lower rate of perioperative death, even though patients who undergo endovascular repair are on average significantly older than those who undergo open repair.37–39
Endovascular repair is performed with open or percutaneous access of the common femoral artery. An endograft, which is packed into an introductory sheath, is introduced into the aorta and expands upon unsheathing. It is positioned to “land” in sealing zones of normal-caliber aorta, where it seals to exclude the aneurysm from circulatory flow (Figure 3).
This is different from the open approach in that it avoids the large incision and aortic cross-clamping necessary in open repair. In open repair, a large incision is made in the patient’s abdomen and the aorta is cross-clamped to stop blood flow. The aneurysm is then incised and a graft is sutured into place to protect the vessel wall from stress (Figure 4).
CASE CONCLUDED
Our patient elected to undergo endovascular repair of his aneurysm with a bifurcated graft (Figure 3). He was able to walk the day after his procedure, and he was sent home that same day. According to the guidelines of the Society for Vascular Surgery,40 he will have surveillance CT angiography at 1 and 12 months to detect “endoleak” or aneurysm enlargement. If these are not seen, he will then undergo routine surveillance with abdominal duplex ultrasonography.
- US Preventive Services Task Force. Screening for abdominal aortic aneurysm: recommendation statement. Ann Intern Med 2005; 142:198–202.
- Kent KC, Zwolak RM, Egorova NN, et al. Analysis of risk factors for abdominal aortic aneurysm in a cohort of more than 3 million individuals. J Vasc Surg 2010; 52:539–548.
- Lindholt JS, Juul S, Fasting H, Henneberg EW. Screening for abdominal aortic aneurysms: single centre randomised controlled trial. BMJ 2005; 330:750.
- Ashton HA, Buxton MJ, Day NE, et al; Multicentre Aneurysm Screening Study Group. The Multicentre Aneurysm Screening Study (MASS) into the effect of abdominal aortic aneurysm screening on mortality in men: a randomised controlled trial. Lancet 2002; 360:1531–1539.
- Norman PE, Jamrozik K, Lawrence-Brown MM, et al. Population based randomised controlled trial on impact of screening on mortality from abdominal aortic aneurysm. BMJ 2004; 329:1259.
- Vardulaki KA, Walker NM, Couto E, et al. Late results concerning feasibility and compliance from a randomized trial of ultrasonographic screening for abdominal aortic aneurysm. Br J Surg 2002; 89:861–864.
- Derubertis BG, Trocciola SM, Ryer EJ, et al. Abdominal aortic aneurysm in women: prevalence, risk factors, and implications for screening. J Vasc Surg 2007; 46:630–635.
- Sandiford P, Mosquera D, Bramley D. Trends in incidence and mortality from abdominal aortic aneurysm in New Zealand. Br J Surg 2011; 98:645–651.
- Anjum A, Powell JT. Is the incidence of abdominal aortic aneurysm declining in the 21st century? Mortality and hospital admissions for England & Wales and Scotland. Eur J Vasc Endovasc Surg 2012; 43:161–166.
- Anderson RN. Deaths: leading causes for 2000. Natl Vital Stat Rep 2002; 50:1–85.
- Kent KC, Zwolak RM, Jaff MR, et al; Society for Vascular Surgery; American Association of Vascular Surgery; Society for Vascular Medicine and Biology. Screening for abdominal aortic aneurysm: a consensus statement. J Vasc Surg 2004; 39:267–269.
- Chaikof EL, Brewster DC, Dalman RL, et al; Society for Vascular Surgery. The care of patients with an abdominal aortic aneurysm: the Society for Vascular Surgery practice guidelines. J Vasc Surg 2009; 50(suppl 4):S2–S49.
- Fink HA, Lederle FA, Roth CS, Bowles CA, Nelson DB, Haas MA. The accuracy of physical examination to detect abdominal aortic aneurysm. Arch Intern Med 2000; 160:833–836.
- Reed WW, Hallett JW, Damiano MA, Ballard DJ. Learning from the last ultrasound. A population-based study of patients with abdominal aortic aneurysm. Arch Intern Med 1997; 157:2064–2068.
- Bernstein EF, Dilley RB, Goldberger LE, Gosink BB, Leopold GR. Growth rates of small abdominal aortic aneurysms. Surgery 1976; 80:765–773.
- Cronenwett JL, Sargent SK, Wall MH, et al. Variables that affect the expansion rate and outcome of small abdominal aortic aneurysms. J Vasc Surg 1990; 11:260–268.
- Scott RA, Bridgewater SG, Ashton HA. Randomized clinical trial of screening for abdominal aortic aneurysm in women. Br J Surg 2002; 89:283–285.
- Fleming C, Whitlock EP, Beil TL, Lederle FA. Screening for abdominal aortic aneurysm: a best-evidence systematic review for the US Preventive Services Task Force. Ann Intern Med 2005; 142:203–211.
- Lindholt JS, Sørensen J, Søgaard R, Henneberg EW. Long-term benefit and cost-effectiveness analysis of screening for abdominal aortic aneurysms from a randomized controlled trial. Br J Surg 2010; 97:826–834.
- Thompson SG, Ashton HA, Gao L, Scott RA; Multicentre Aneurysm Screening Study Group. Screening men for abdominal aortic aneurysm: 10 year mortality and cost effectiveness results from the randomised Multicentre Aneurysm Screening Study. BMJ 2009; 338:b2307.
- Mastracci TM, Cina CS. Regarding Screening for abdominal aortic aneurysm reduces both aneurysm-related and all-cause mortality (letter). J Vasc Surg 2007; 46:1312.
- Rembold CM. Number needed to screen: development of a statistic for disease screening. BMJ 1998; 317:307–312.
- Wilson JMG, Jungner G. Principles and practice of screening for disease. World Health Organization. Public Health Papers #34.
- Lee TY, Korn P, Heller JA, et al. The cost-effectiveness of a “quickscreen” program for abdominal aortic aneurysms. Surgery 2002; 132:399–407.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al; American Association for Vascular Surgery; Society for Vascular Surgery; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine and Biology; Society of Interventional Radiology; ACC/AHA Task Force on Practice Guidelines Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease; American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; Vascular Disease Foundation. ACC/AHA 2005 Practice Guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation 2006; 113:e463–e654.
- Linné A, Lindström D, Hultgren R. High prevalence of abdominal aortic aneurysms in brothers and sisters of patients despite a low prevalence in the population. J Vasc Surg 2012; 56:305–310.
- The UK Small Aneurysm Trial Participants. Mortality results for randomised controlled trial of early elective surgery or ultrasonographic surveillance for small abdominal aortic aneurysms. Lancet 1998; 352:1649–1655.
- Lederle FA, Johnson GR, Wilson SE, et al. Prevalence and associations of abdominal aortic aneurysm detected through screening. Aneurysm Detection and Management (ADAM) Veterans Affairs Cooperative Study Group. Ann Intern Med 1997; 126:441–449.
- Brewster DC, Cronenwett JL, Hallett JW, Johnston KW, Krupski WC, Matsumura JS; Joint Council of the American Association for Vascular Surgery and Society for Vascular Surgery. Guidelines for the treatment of abdominal aortic aneurysms. Report of a subcommittee of the Joint Council of the American Association for Vascular Surgery and Society for Vascular Surgery. J Vasc Surg 2003; 37:1106–117.
- Ouriel K, Clair DG, Kent KC, Zarins CK; Positive Impact of Endovascular Options for treating Aneurysms Early (PIVOTAL) Investigators. Endovascular repair compared with surveillance for patients with small abdominal aortic aneurysms. J Vasc Surg 2010; 51:1081–1087.
- De Rango P, Verzini F, Parlani G; Comparison of surveillance vs Aortic Endografting for Small Aneurysm Repair (CAESAR) Investigators. Quality of life in patients with small abdominal aortic aneurysm: the effect of early endovascular repair versus surveillance in the CAESAR trial. Eur J Vasc Endovasc Surg 2011; 41:324–331.
- Antoniou GA, Lazarides MK, Patera S, et al. Assessment of insertion/deletion polymorphism of the angiotensin-converting enzyme gene in abdominal aortic aneurysm and inguinal hernia. Vascular 2012; Epub ahead of print.
- Ogata T, Shibamura H, Tromp G, et al. Genetic analysis of polymorphisms in biologically relevant candidate genes in patients with abdominal aortic aneurysms. J Vasc Surg 2005; 41:1036–1042.
- Powell JT, Greenhalgh RM. Clinical practice. Small abdominal aortic aneurysms. N Engl J Med 2003; 348:1895–1901.
- Gadowski GR, Pilcher DB, Ricci MA. Abdominal aortic aneurysm expansion rate: effect of size and beta-adrenergic blockade. J Vasc Surg 1994; 19:727–731.
- Propanolol Aneurysm Trial Investigators. Propranolol for small abdominal aortic aneurysms: results of a randomized trial. J Vasc Surg 2002; 35:72–79.
- Jackson RS, Chang DC, Freischlag JA. Comparison of long-term survival after open vs endovascular repair of intact abdominal aortic aneurysm among Medicare beneficiaries. JAMA 2012; 307:1621–1628.
- Dillavou ED, Muluk SC, Makaroun MS. Improving aneurysm-related outcomes: nationwide benefits of endovascular repair. J Vasc Surg 2006; 43:446–451.
- Giles KA, Pomposelli F, Hamdan A, Wyers M, Jhaveri A, Schermerhorn ML. Decrease in total aneurysm-related deaths in the era of endovascular aneurysm repair. J Vasc Surg 2009; 49:543–550.
- Chaikof EL, Blankensteijn JD, Harris PL, et al; Ad Hoc Committee for Standardized Reporting Practices in Vascular Surgery of The Society for Vascular Surgery/American Association for Vascular Surgery. Reporting standards for endovascular aortic aneurysm repair. J Vasc Surg 2002; 35:1048–1060.
- US Preventive Services Task Force. Screening for abdominal aortic aneurysm: recommendation statement. Ann Intern Med 2005; 142:198–202.
- Kent KC, Zwolak RM, Egorova NN, et al. Analysis of risk factors for abdominal aortic aneurysm in a cohort of more than 3 million individuals. J Vasc Surg 2010; 52:539–548.
- Lindholt JS, Juul S, Fasting H, Henneberg EW. Screening for abdominal aortic aneurysms: single centre randomised controlled trial. BMJ 2005; 330:750.
- Ashton HA, Buxton MJ, Day NE, et al; Multicentre Aneurysm Screening Study Group. The Multicentre Aneurysm Screening Study (MASS) into the effect of abdominal aortic aneurysm screening on mortality in men: a randomised controlled trial. Lancet 2002; 360:1531–1539.
- Norman PE, Jamrozik K, Lawrence-Brown MM, et al. Population based randomised controlled trial on impact of screening on mortality from abdominal aortic aneurysm. BMJ 2004; 329:1259.
- Vardulaki KA, Walker NM, Couto E, et al. Late results concerning feasibility and compliance from a randomized trial of ultrasonographic screening for abdominal aortic aneurysm. Br J Surg 2002; 89:861–864.
- Derubertis BG, Trocciola SM, Ryer EJ, et al. Abdominal aortic aneurysm in women: prevalence, risk factors, and implications for screening. J Vasc Surg 2007; 46:630–635.
- Sandiford P, Mosquera D, Bramley D. Trends in incidence and mortality from abdominal aortic aneurysm in New Zealand. Br J Surg 2011; 98:645–651.
- Anjum A, Powell JT. Is the incidence of abdominal aortic aneurysm declining in the 21st century? Mortality and hospital admissions for England & Wales and Scotland. Eur J Vasc Endovasc Surg 2012; 43:161–166.
- Anderson RN. Deaths: leading causes for 2000. Natl Vital Stat Rep 2002; 50:1–85.
- Kent KC, Zwolak RM, Jaff MR, et al; Society for Vascular Surgery; American Association of Vascular Surgery; Society for Vascular Medicine and Biology. Screening for abdominal aortic aneurysm: a consensus statement. J Vasc Surg 2004; 39:267–269.
- Chaikof EL, Brewster DC, Dalman RL, et al; Society for Vascular Surgery. The care of patients with an abdominal aortic aneurysm: the Society for Vascular Surgery practice guidelines. J Vasc Surg 2009; 50(suppl 4):S2–S49.
- Fink HA, Lederle FA, Roth CS, Bowles CA, Nelson DB, Haas MA. The accuracy of physical examination to detect abdominal aortic aneurysm. Arch Intern Med 2000; 160:833–836.
- Reed WW, Hallett JW, Damiano MA, Ballard DJ. Learning from the last ultrasound. A population-based study of patients with abdominal aortic aneurysm. Arch Intern Med 1997; 157:2064–2068.
- Bernstein EF, Dilley RB, Goldberger LE, Gosink BB, Leopold GR. Growth rates of small abdominal aortic aneurysms. Surgery 1976; 80:765–773.
- Cronenwett JL, Sargent SK, Wall MH, et al. Variables that affect the expansion rate and outcome of small abdominal aortic aneurysms. J Vasc Surg 1990; 11:260–268.
- Scott RA, Bridgewater SG, Ashton HA. Randomized clinical trial of screening for abdominal aortic aneurysm in women. Br J Surg 2002; 89:283–285.
- Fleming C, Whitlock EP, Beil TL, Lederle FA. Screening for abdominal aortic aneurysm: a best-evidence systematic review for the US Preventive Services Task Force. Ann Intern Med 2005; 142:203–211.
- Lindholt JS, Sørensen J, Søgaard R, Henneberg EW. Long-term benefit and cost-effectiveness analysis of screening for abdominal aortic aneurysms from a randomized controlled trial. Br J Surg 2010; 97:826–834.
- Thompson SG, Ashton HA, Gao L, Scott RA; Multicentre Aneurysm Screening Study Group. Screening men for abdominal aortic aneurysm: 10 year mortality and cost effectiveness results from the randomised Multicentre Aneurysm Screening Study. BMJ 2009; 338:b2307.
- Mastracci TM, Cina CS. Regarding Screening for abdominal aortic aneurysm reduces both aneurysm-related and all-cause mortality (letter). J Vasc Surg 2007; 46:1312.
- Rembold CM. Number needed to screen: development of a statistic for disease screening. BMJ 1998; 317:307–312.
- Wilson JMG, Jungner G. Principles and practice of screening for disease. World Health Organization. Public Health Papers #34.
- Lee TY, Korn P, Heller JA, et al. The cost-effectiveness of a “quickscreen” program for abdominal aortic aneurysms. Surgery 2002; 132:399–407.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al; American Association for Vascular Surgery; Society for Vascular Surgery; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine and Biology; Society of Interventional Radiology; ACC/AHA Task Force on Practice Guidelines Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease; American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; Vascular Disease Foundation. ACC/AHA 2005 Practice Guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation 2006; 113:e463–e654.
- Linné A, Lindström D, Hultgren R. High prevalence of abdominal aortic aneurysms in brothers and sisters of patients despite a low prevalence in the population. J Vasc Surg 2012; 56:305–310.
- The UK Small Aneurysm Trial Participants. Mortality results for randomised controlled trial of early elective surgery or ultrasonographic surveillance for small abdominal aortic aneurysms. Lancet 1998; 352:1649–1655.
- Lederle FA, Johnson GR, Wilson SE, et al. Prevalence and associations of abdominal aortic aneurysm detected through screening. Aneurysm Detection and Management (ADAM) Veterans Affairs Cooperative Study Group. Ann Intern Med 1997; 126:441–449.
- Brewster DC, Cronenwett JL, Hallett JW, Johnston KW, Krupski WC, Matsumura JS; Joint Council of the American Association for Vascular Surgery and Society for Vascular Surgery. Guidelines for the treatment of abdominal aortic aneurysms. Report of a subcommittee of the Joint Council of the American Association for Vascular Surgery and Society for Vascular Surgery. J Vasc Surg 2003; 37:1106–117.
- Ouriel K, Clair DG, Kent KC, Zarins CK; Positive Impact of Endovascular Options for treating Aneurysms Early (PIVOTAL) Investigators. Endovascular repair compared with surveillance for patients with small abdominal aortic aneurysms. J Vasc Surg 2010; 51:1081–1087.
- De Rango P, Verzini F, Parlani G; Comparison of surveillance vs Aortic Endografting for Small Aneurysm Repair (CAESAR) Investigators. Quality of life in patients with small abdominal aortic aneurysm: the effect of early endovascular repair versus surveillance in the CAESAR trial. Eur J Vasc Endovasc Surg 2011; 41:324–331.
- Antoniou GA, Lazarides MK, Patera S, et al. Assessment of insertion/deletion polymorphism of the angiotensin-converting enzyme gene in abdominal aortic aneurysm and inguinal hernia. Vascular 2012; Epub ahead of print.
- Ogata T, Shibamura H, Tromp G, et al. Genetic analysis of polymorphisms in biologically relevant candidate genes in patients with abdominal aortic aneurysms. J Vasc Surg 2005; 41:1036–1042.
- Powell JT, Greenhalgh RM. Clinical practice. Small abdominal aortic aneurysms. N Engl J Med 2003; 348:1895–1901.
- Gadowski GR, Pilcher DB, Ricci MA. Abdominal aortic aneurysm expansion rate: effect of size and beta-adrenergic blockade. J Vasc Surg 1994; 19:727–731.
- Propanolol Aneurysm Trial Investigators. Propranolol for small abdominal aortic aneurysms: results of a randomized trial. J Vasc Surg 2002; 35:72–79.
- Jackson RS, Chang DC, Freischlag JA. Comparison of long-term survival after open vs endovascular repair of intact abdominal aortic aneurysm among Medicare beneficiaries. JAMA 2012; 307:1621–1628.
- Dillavou ED, Muluk SC, Makaroun MS. Improving aneurysm-related outcomes: nationwide benefits of endovascular repair. J Vasc Surg 2006; 43:446–451.
- Giles KA, Pomposelli F, Hamdan A, Wyers M, Jhaveri A, Schermerhorn ML. Decrease in total aneurysm-related deaths in the era of endovascular aneurysm repair. J Vasc Surg 2009; 49:543–550.
- Chaikof EL, Blankensteijn JD, Harris PL, et al; Ad Hoc Committee for Standardized Reporting Practices in Vascular Surgery of The Society for Vascular Surgery/American Association for Vascular Surgery. Reporting standards for endovascular aortic aneurysm repair. J Vasc Surg 2002; 35:1048–1060.