User login
The urgent need to diagnose Sanfilippo syndrome at an early age
Sanfilippo syndrome is a rare inherited neurodegenerative metabolic disorder for which there are no approved therapies. Symptoms of the more severe subtypes typically begin within the first years of life, rapidly producing serious and progressive physical and cognitive deficits. The underlying pathophysiology is targetable, but the delay in diagnosis of this as well as other lysosomal storage disorders (LSDs) is slowing progress toward effective therapies.
“Lack of awareness and the delays to diagnosis have been a real challenge for us. There is reason for cautious optimism about treatments now in or approaching clinical studies, but to evaluate efficacy on cognitive outcomes we need to enroll more children at a very young age, before loss of milestones,” according to Cara O’Neill, MD, a co-founder and chief science officer of Cure Sanfilippo Foundation.
Epidemiology and description
Sanfilippo syndrome, like the more than 50 other LSDs, is caused by a gene mutation that leads to an enzyme deficiency in the lysosome.1 In the case of Sanfilippo syndrome, also known as mucopolysaccharidosis (MPS III), there are hundreds of mutations that can lead to Sanfilippo by altering the function of one of the four genes essential to degradation of heparan sulfate.2 Lysosomal accumulation of heparan sulfate drives a broad spectrum of progressive and largely irreversible symptoms that typically begin with somatic manifestations, such as bowel dysfunction and recurrent ear and upper respiratory infections.

Impairment of the central nervous system (CNS) usually occurs early in life, halting physical and mental development. As it progresses, accumulation of heparan sulfate in a variety of cells leads to a cascade of abnormal cellular signaling and dysfunction. Disruption of these processes, which are critical for normal neurodevelopment, result in loss of the developmental skills already gained and eventually loss of brain tissue.3 Although life expectancy has improved with supportive care, survival into adulthood is typically limited to milder forms.4
Over the past several years, progress in this and other LSDs has yielded therapeutic targets, including those involving gene repair and enzyme replacement. Already approved for use in some LSDs, these therapies have also shown promise in the experimental setting for Sanfilippo syndrome, leading to several completed clinical trials.5
So far, none of these treatments has advanced beyond clinical trials in Sanfilippo syndrome, but there have been favorable changes in the markers of disease, suggesting that better methods of treatment delivery and/or more sensitive tools to measure clinical change might lead to evidence of disease attenuation. However, the promise of treatment in all cases has been to prevent, slow, or halt progression, not to reverse it. This point is important, because it indicates that degree of benefit will depend on enrolling patients early in life. Even if effective therapies are identified, few patients will benefit without strategies to accelerate diagnosis.
In fact, “one study6 reported that the average age of diagnosis for Sanfilippo syndrome has not improved over the past 30 years,” according to Dr. O’Neill. She indicated that this has been frustrating, given the availability of clinical trials on which progress is dependent. There is no widely accepted protocol for who and when to test for Sanfilippo syndrome or other LSDs, but Dr. O’Neill’s organization is among those advocating for strategies to detect these diseases earlier, including screening at birth.
Almost by definition, the clinical diagnosis of rare diseases poses a challenge. With nonspecific symptoms and a broad range of potential diagnoses, diseases with a low incidence are not the first ones that are typically considered. In the case of Sanfilippo syndrome, published studies indicate incidence rates at or below 1 per 70,000 live births.7 However, the incidence rates have been highly variable not only by geographical regions but even across neighboring countries where genetic risk would be expected to be similar.
In Europe, for example, epidemiologic studies suggest the lifetime risk of MPS IIIA is approximately two times greater in Germany and the Netherlands relative to France and Sweden.7 It is possible that the methodology for identifying cases might be a more important factor than differences in genetic risk to explain this variability. Many experts, including Dr. O’Neill, believe that prevalence figures for Sanfilippo syndrome are typically underestimates because of the frequency with which LSDs are attributed to other pathology.
“For these types of rare disorders, a clinician might only see a single case over a career, and the symptoms can vary in presentation and severity with many alternatives to consider in the differential diagnosis,” Dr. O’Neill explained. She cited case reports in which symptoms of Sanfilippo syndrome after a period of initial normal development has been initially attributed to autism, which is a comorbid feature of the disease, idiopathic developmental delay, or other nonprogressive disorders until further clinical deterioration leads to additional testing. The implication is that LSDs must be considered far earlier despite their rarity.
For the least common of the four clinical subtypes, MPS IIIC and MPS IIID, the median ages of diagnosis have ranged from 4.5 to 19 years of age.7 This is likely a reflection of a slower progression and a later onset of clinical manifestations.
For the more rapidly progressing and typically more severe subtypes, MPS IIIA and MPS IIIB, the diagnosis is typically made earlier. In one review of epidemiologic studies in different countries, the earliest reported median age at diagnosis was 2.5 years,7 a point at which significant disease progression is likely to have already occurred. If the promise of treatments in development is prevention of disease progression, disability in many patients might be substantial if the time to diagnosis is not reduced.
Screening and testing
Independent of the potential to enroll children in clinical trials, early diagnosis also advances the opportunities for supportive care to lessen the burden of the disease on patients and families. Perhaps even more important, early diagnosis is vital to family planning. Since the American pediatrician Sylvester Sanfilippo, MD, first described this syndrome in 1963,7 the genetic profile and many of the features of the disease have become well characterized.8

“One reason to emphasize the importance of early diagnosis is the heritability of this disorder. With prompt diagnosis, genetic counseling can be offered to families to provide them with critical information for future family planning and for cascade testing of other potentially affected siblings,” Dr. O’Neill reported. The inheritance pattern of Sanfilippo syndrome is autosomal recessive.3 In families with an affected child, the risk for any subsequent child to have the same disorder is 25%. The chance of a sibling to be unaffected and not a carrier is also 25%. There is a 50% chance of a sibling to be a carrier but asymptomatic. Of priorities, spreading awareness has been a critical mission of the Cure Sanfilippo Foundation since it was founded 8 years ago, according to Glenn O’Neill, the president. He and his wife, Dr. O’Neill, who is a pediatrician, founded the organization after their own child’s diagnosis of Sanfilippo syndrome. Creating awareness is fundamental to the mission of attracting funds for research, but support to patients and their families as well as early enrollment in clinical trials are among other initiatives being pursued by the foundation to improve care and prognosis.
These strategies include some novel ideas, including an algorithm based on artificial intelligence (AI) that can accelerate suspicion of Sanfilippo syndrome in advance of laboratory or genetic testing, according to Dr. O’Neill. She reported that the facial phenotype, which is observed in a high proportion of but not in all Sanfilippo patients, includes coarse facial features such as puffiness around the eyes, heavy eyebrows, full lips, and macrocephaly.9 Interpretation of photos for AI-based analysis is enhanced when combined with other clinical symptoms.
“The Foundation was involved in honing such a tool by submitting the photos that were used to teach the AI to recognize the Sanfilippo syndrome phenotype,” Dr. O’Neill said. The AI-based tool (Face2Gene.com) is available from FDNA, a company that has been involved in analyzing complex phenotypic and genomic information to guide diagnosis and therapeutic strategies for an array of diseases, not just Sanfilippo syndrome.
The preferred method for diagnosis is biochemical or genetic testing. Of these, urine testing for elevated levels of heparan sulfate glycosaminoglycans (GAG) can be useful for screening, although false-negative tests occur. Analysis of the blood can be performed to detect abnormal levels or activity of the enzymes that break down this GAG. In addition, genetic testing can be performed on blood, fibroblast, buccal swab, or saliva samples. Genetic testing of the blood is the most frequently performed.
For the four MPS III subtypes – MPS IIIA, IIIB, IIIC, and IIID – the presence of two pathogenic mutations in the SGSH (17q25.3), NAGLU (17q21.2), HGSNAT (8p11.21), and GNS (12q14.3) genes, respectively, are likely diagnostic, but enzymatic testing or GAG analysis should be performed to confirm disease status, according to Dr. O’Neill, who said that global consensus based clinical care guidelines led by the Foundation were recently accepted for publication and also include a section on the approach to diagnosis.
While laboratory testing is sensitive, urinary excretion of GAG can be variable, with the potential for ambiguous results. Typically, biochemical and genetic testing provide more reliable results for the diagnosis. They can be readily performed in utero or at the time of birth. In addition, gene panels can permit the diagnosis of multiple types of LSDs, not just Sanfilippo, making screening a cost-effective strategy to consider multiple diseases with overlapping symptoms when an LSD is suspected. Dr. O’Neill said clinical guidelines recommend confirmation of enzyme deficiency or evidence of GAG substrate accumulation as confirmatory tests when genetic testing is positive.
“Ultimately, our goal is to promote universal screening at birth for these serious genetic disorders affecting children,” Dr. O’Neill said.
“We are in a catch-22 when it comes to newborn screening. Currently our federal system requires there be an available treatment before recommending routine screening for a disease. However, it is extremely difficult to power trials with patients who are most likely to show benefit in a trial setting without that very early diagnosis. Universal newborn screening would pave the way for accelerated drug development for children,” she added.
In the meantime, Dr. O’Neill suggests that clinicians should employ a low threshold of suspicion to pursue diagnostic studies of LSDs in infants and children with developmental delays or otherwise unexplained progressive disorders.
Importantly, clinicians can now act quickly on their suspicions and order testing without concern for delays or denial by insurers through a special program, according to Dr. O’Neill. Free genetic testing, offered by the Invitae Corporation, evaluates a panel of 58 genes associated with lysosomal disorders, permitting detection of Sanfilippo syndrome and other LSDs, according to Dr. O’Neill. The Invitae testing is typically performed on 3 mL of whole blood delivered to a central testing facility.
“Results can be obtained within a few weeks or sooner. This can seem like a long wait for families, but it is much more efficient than ordering tests sequentially,” Dr. O’Neill said.
Diagnosis: Signs and symptoms
Despite the differences in progression of the MPS III subtypes, the clinical characteristics are more similar than different. In all patients, prenatal and infant development are typically normal. The initial signs of disease can be found in the newborn, such as neonatal tachypnea, through the early infancy period, such as macrocephaly. However, these are not commonly recognized until about age 1 or soon after in those with MPS IIIA and IIIB.3 Speech delay is the first developmental delay seen in most patients. In those with MPS IIIC, initial symptoms are typically detected at age 3 or later and progress more slowly.10,11 The same is likely to be true of MPS IIID, although this subtype is less well characterized than the other three.7
Although many organs can be involved, degeneration of the CNS is regarded as the most characteristic.3 In aggressive disease, this includes slower acquisition of and failure to meet developmental milestones with progressive intellectual disability, while behavioral difficulties are a more common initial compliant in children with milder disease.13,14 These behavioral changes include hyperactivity, inattention, autistic behaviors, worsening safety awareness, and in some cases aggressive behavior that can be destructive. Sleep disturbances are common.15Because of variability inherent in descriptions of relatively small numbers of patients, the characterization of each of the MPS III subgroups is based on a limited number of small studies, but most patients demonstrate behavior disorders, have coarse facial features, and develop speech delay, according to a survey conducted of published studies.7 Collectively, abnormal behavior was identified as an early symptom in 77% of those with MPS IIIA, 69% of those with IIIB, and 77% of those with IIIC.
For MPS IIIA, loss of speech was observed at a median age of 3.8 years and loss of walking ability at 10.4 years. The median survival has been reported to range between 13 and 18 years. In children with MPS IIIB, the median age of speech loss was reported to about the same age, while loss of walking ability occurred at 11 years. In one study of MPS IIIB, 24% of patients had developed dementia by age 6 years, and the reported median survival has ranged between 17 and 19 years. For MPS IIIC, the onset of clinical symptoms has been observed at a median age of 3.5 years with evidence of cognitive loss observed in 33% of children by the age of 6 years. The median survival has ranged from 19 to 34 years in three studies tracing the natural history of this MPS III subtype.
The differential diagnosis reasonably includes other types of mucopolysaccharidosis disorders with cognitive impairment, including Hurler, Hunter, or Sly syndromes, other neurodevelopmental disorders, and inborn errors of metabolism. The heterogeneity of the features makes definitive laboratory or genetic testing, rather than the effort to differentiate clinical features, appropriate for a definitive diagnosis.
Once the diagnosis is made, other examinations for the common complications of Sanfilippo syndrome are appropriate. Abdominal imaging is appropriate for detecting complications in the gastrointestinal tract, including hepatomegaly, which has been reported in more than half of patients with MPS IIIA and IIIB and in 39% of patients with IIIC.7 In patients with breathing concerns at night and/or sleep disturbance, polysomnography can be useful for identifying sleep apnea and nocturnal seizure activity. In children suspected of seizures, EEG is appropriate. In one study, 66% of patients with MPS IIIA developed seizure activity.16 This has been less commonly reported in MPS IIIB and IIIC, ranging from 8% to 13%.15
Formal hearing evaluation is indicated for any child with speech delays. Hearing loss typically develops after the newborn period in Sanfilippo and may affect peak language acquisition if not treated, according to Dr. O’Neill.
Radiographic studies for dysostosis multiplex or other skeletal abnormalities are also appropriate based on clinical presentation.
Treatment: Present and future
In the absence of treatments to improve the prognosis of Sanfilippo syndrome, current management is based on supportive care and managing organ-specific complications. However, several strategies have proven viable in experimental models and led to clinical trials. None of these therapies has reached approval yet, but several have been associated with attenuation of biomarkers of MPS III disease activity.
Of nearly 30 Sanfilippo clinical trials conducted over the past 20 years, at least 9 have now been completed.5 In addition to studying gene therapy and enzyme replacement therapy, these trials have included stem cell transplantation and substrate reduction therapy, for which the goal is to reduce synthesis of the heparan sulfate GAG to prevent accumulation.5 Of this latter approach, promising initial results with genistein, an isoflavone that breaks down heparan sulfate, reached a phase 3 evaluation.18 Although heparan sulfate levels in the CNS were non-significantly reduced over the course of the trial, the reduction was not sufficient to attenuate cognitive decline.
In other LSDs, several forms of enzyme replacement therapy are now approved. In Fabry disease, for example, recombinant alpha-galactosidase A has now been used for more than 15 years.19 Clinical benefit has not yet been demonstrated in patients with Sanfilippo syndrome because of the difficulty of delivering these therapies past the blood-brain barrier. Several strategies have been pursued. For example, intrathecal delivery of recombinant heparan-N-sulfatase reduced CNS levels of GAG heparan sulfate in one phase 2B study, but it approached but fell short of the statistical significance for the primary endpoint of predefined cognitive stabilization.20 The signal of activity and generally acceptable tolerability has encouraged further study, including an ongoing study with promising interim results of intracerebroventricular enzyme replacement in MPS IIIB, according to Dr. O’Neill.
Acceptable safety and promising activity on disease biomarkers have also been seen with gene therapy in clinical trials. In one study that showed attenuation of brain atrophy, there was moderate improvement in behavior and sleep in three of the four patients enrolled.21 Other studies using various strategies for gene delivery have also produced signals of activity against the underlying pathology, generating persistent interest in ongoing and planned clinical studies with this form of treatment.22Unmodified hematopoietic stem cell transplantation (HSCT), an approach that has demonstrated efficacy when delivered early in the course of other LSDs, such as Hurler syndrome,23 has not yet been associated with significant activity in clinical studies of MPS III, including those that initiated treatment prior to the onset of neurological symptoms.24 However, promising early results have been reported in a study of gene-modified HSCT, which overexpresses the MPS IIIA enzyme.
“The clinical trial landscape fluctuates quite a bit, so I always encourage clinicians and families to check back often for updates. Patient organizations can also be helpful for understanding the most up-to-date and emerging trial options,” Dr. O’Neill reported.
Although it is expected that the greatest benefit would be derived from treatments initiated before or very early after the onset of symptoms, based on the limited potential for reversing cognitive loss, Dr. O’Neill said that she and others are also striving to offer treatments for individuals now living with Sanfilippo syndrome.
“We have to be willing to test treatments that are symptomatic in nature. To that aim, the Cure Sanfilippo Foundation has sponsored a study of a CNS-penetrating anti-inflammatory agent in advanced-disease patients more than 4 years of age,” Dr. O’Neill said. This group of patients typically been ineligible for clinical trials in the past. Dr. O’Neill hopes to change this orientation.
“It is important to highlight that all patients deserve our efforts to improve their quality of life and alleviate suffering, regardless of how old they are or how progressed in the disease they happen to be,” she said.
However, whether the goal is enrollment before or early in disease or later in disease progression, the challenge of enrolling sufficient numbers of patients to confirm clinical activity has been and continues to be a hurdle to progress.
“Clinical studies in Sanfilippo enroll relatively small numbers of patients, often 20 or less,” said Dr. O’Neill, explaining one of the reasons why her organization has been so active in raising awareness and funding such studies. For patients and families, the Cure Sanfilippo Foundation can offer a variety of guidance and support, but information about opportunities for clinical trial participation is a key resource they provide for families and their physicians.
Conclusion
For most children with Sanfilippo syndrome, life expectancy is limited. However, the characterization of the genetic causes and the biochemistry of the subtypes has led to several viable therapeutic approaches under development. There has been progress in delivery of therapeutic enzymes to the CNS, and there is substantial optimism that more progress is coming. One issue for treatment development, is the last of a clear regulatory pathway addressing important biomarkers of pathology, such as heparan sulfate burden. Developing treatments that address this issue or impaired enzyme activity levels have promise for preventing progression, particularly if started in infancy. However, the effort to draw awareness to this disease is the first step toward accelerating the time to an early diagnosis and subsequent opportunities to enroll in clinical trials.
References
1. Sun A. Lysosomal storage disease overview. Ann Transl Med. 2018 Dec;6(24):476. doi: 10.21037/atm.2018.11.39.
2. Andrade F et al. Sanfilippo syndrome: Overall review. Pediatr Int. 2015 Jun;57(3):331-8. doi: 10.1111/ped.12636.
3. Fedele AO. Sanfilippo syndrome: Causes, consequences, and treatments. Appl Clin Genet. 2015 Nov 25;8:269-81. doi: 10.2147/TACG.S57672.
4. Lavery C et al. Mortality in patients with Sanfilippo syndrome. Orphanet J Rare Dis. 2017 Oct 23;12(1):168. doi: 10.1186/s13023-017-0717-y.
5. Pearse Y et al. A cure for Sanfilippo syndrome? A summary of current therapeutic approaches and their promise. Med Res Arch. 2020 Feb 1;8(2). doi: 10.18103/mra.v8i2.2045.
6. Kuiper GA et al. Failure to shorten the diagnostic delay in two ultrao-rphan diseases (mucopolysaccharidosis types I and III): potential causes and implication. Orphanet J Rare Dis. 2018;13:2. Doi: 10.1186/s13023-017-0733-y.
7. Zelei T et al. Epidemiology of Sanfilippo syndrome: Results of a systematic literature review. Orphanet J Rare Dis. 2018 Apr 10;13(1):53. doi: 10.1186/s13023-018-0796-4.
8. Wagner VF, Northrup H. Mucopolysaccaharidosis type III. Gene Reviews. 2019 Sep 19. University of Washington, Seattle. https://www.ncbi.nlm.nih.gov/books/NBK546574/8.
9. O’Neill C et al. Natural history of facial features observed in Sanfilippo syndrome (MPS IIIB) using a next generation phenotyping tool. Mol Genet Metab. 2019 Feb;126:S112.
10. Ruijter GJ et al. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in the Netherlands. Mol Genet Metab. 2008 Feb;93(2):104-11. doi: 10.1016/j.ymgme.2007.09.011.
11. Valstar MJ et al. Mucopolysaccharidosis type IIID: 12 new patients and 15 novel mutations. Hum Mutat. 2010 May;31(5):E1348-60. doi: 10.1002/humu.21234.
12. Nijmeijer SCM. The attenuated end of phenotypic spectrum in MPS III: from late-onset stable cognitive impairment to non-neuronopathic phenotype. Orphanet J Rare Dis. 2019;14:249. Doi10.1186/s13023-019-1232-0.
13. Nidiffer FD, Kelly TE. Developmental and degenerative patterns associated with cognitive, behavioural and motor difficulties in the Sanfilippo syndrome: An epidemiological study. J Ment Defic Res. 1983 Sep;27 (Pt 3):185-203. doi: 10.1111/j.1365-2788.1983.tb00291.x.
14. Bax MC, Colville GA. Behaviour in mucopolysaccharide disorders. Arch Dis Child. 1995 Jul;73(1):77-81. doi: 10.1136/adc.73.1.77.
15. Fraser J et al. Sleep disturbance in mucopolysaccharidosis type III (Sanfilippo syndrome): A survey of managing clinicians. Clin Genet. 2002 Nov;62(5):418-21. doi: 10.1034/j.1399-0004.2002.620512.x.
16. Valstar MJ et al. Mucopolysaccharidosis type IIIA: Clinical spectrum and genotype-phenotype correlations. Ann Neurol. 2010 Dec;68(6):876-87. doi: 10.1002/ana.22092.
17. Heron B et al. Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am J Med Genet A. 2011 Jan;155A(1):58-68. doi: 10.1002/ajmg.a.33779.
18. Delgadillo V et al. Genistein supplementation in patients affected by Sanfilippo disease. J Inherit Metab Dis. 2011 Oct;34(5):1039-44. doi: 10.1007/s10545-011-9342-4.
19. van der Veen SJ et al. Developments in the treatment of Fabry disease. J Inherit Metab Dis. 2020 Sep;43(5):908-21. doi: 10.1002/jimd.12228.
20. Wijburg FA et al. Intrathecal heparan-N-sulfatase in patients with Sanfilippo syndrome type A: A phase IIb randomized trial. Mol Genet Metab. 2019 Feb;126(2):121-30. doi: 10.1016/j.ymgme.2018.10.006.
21. Tardieu M et al. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: Results of a phase I/II trial. Hum Gene Ther. 2014 Jun;25(6):506-16. doi: 10.1089/hum.2013.238.
22. Marco S et al. In vivo gene therapy for mucopolysaccharidosis type III (Sanfilippo syndrome): A new treatment horizon. Hum Gene Ther. 2019 Oct;30(10):1211-1121. doi: 10.1089/hum.2019.217.
23. Taylor M et al. Hematopoietic stem cell transplantation for mucopolysaccharidoses: Past, present, and future. Biol Blood Marrow Transplant. 2019 Jul;25(7):e226-e246. doi: 10.1016/j.bbmt.2019.02.012.
24. Sivakumur P, Wraith JE. Bone marrow transplantation in mucopolysaccharidosis type IIIA: A comparison of an early treated patient with his untreated sibling. J Inherit Metab Dis. 1999 Oct;22(7):849-50. doi: 10.1023/a:1005526628598.
Sanfilippo syndrome is a rare inherited neurodegenerative metabolic disorder for which there are no approved therapies. Symptoms of the more severe subtypes typically begin within the first years of life, rapidly producing serious and progressive physical and cognitive deficits. The underlying pathophysiology is targetable, but the delay in diagnosis of this as well as other lysosomal storage disorders (LSDs) is slowing progress toward effective therapies.
“Lack of awareness and the delays to diagnosis have been a real challenge for us. There is reason for cautious optimism about treatments now in or approaching clinical studies, but to evaluate efficacy on cognitive outcomes we need to enroll more children at a very young age, before loss of milestones,” according to Cara O’Neill, MD, a co-founder and chief science officer of Cure Sanfilippo Foundation.
Epidemiology and description
Sanfilippo syndrome, like the more than 50 other LSDs, is caused by a gene mutation that leads to an enzyme deficiency in the lysosome.1 In the case of Sanfilippo syndrome, also known as mucopolysaccharidosis (MPS III), there are hundreds of mutations that can lead to Sanfilippo by altering the function of one of the four genes essential to degradation of heparan sulfate.2 Lysosomal accumulation of heparan sulfate drives a broad spectrum of progressive and largely irreversible symptoms that typically begin with somatic manifestations, such as bowel dysfunction and recurrent ear and upper respiratory infections.
Impairment of the central nervous system (CNS) usually occurs early in life, halting physical and mental development. As it progresses, accumulation of heparan sulfate in a variety of cells leads to a cascade of abnormal cellular signaling and dysfunction. Disruption of these processes, which are critical for normal neurodevelopment, result in loss of the developmental skills already gained and eventually loss of brain tissue.3 Although life expectancy has improved with supportive care, survival into adulthood is typically limited to milder forms.4
Over the past several years, progress in this and other LSDs has yielded therapeutic targets, including those involving gene repair and enzyme replacement. Already approved for use in some LSDs, these therapies have also shown promise in the experimental setting for Sanfilippo syndrome, leading to several completed clinical trials.5
So far, none of these treatments has advanced beyond clinical trials in Sanfilippo syndrome, but there have been favorable changes in the markers of disease, suggesting that better methods of treatment delivery and/or more sensitive tools to measure clinical change might lead to evidence of disease attenuation. However, the promise of treatment in all cases has been to prevent, slow, or halt progression, not to reverse it. This point is important, because it indicates that degree of benefit will depend on enrolling patients early in life. Even if effective therapies are identified, few patients will benefit without strategies to accelerate diagnosis.
In fact, “one study6 reported that the average age of diagnosis for Sanfilippo syndrome has not improved over the past 30 years,” according to Dr. O’Neill. She indicated that this has been frustrating, given the availability of clinical trials on which progress is dependent. There is no widely accepted protocol for who and when to test for Sanfilippo syndrome or other LSDs, but Dr. O’Neill’s organization is among those advocating for strategies to detect these diseases earlier, including screening at birth.
Almost by definition, the clinical diagnosis of rare diseases poses a challenge. With nonspecific symptoms and a broad range of potential diagnoses, diseases with a low incidence are not the first ones that are typically considered. In the case of Sanfilippo syndrome, published studies indicate incidence rates at or below 1 per 70,000 live births.7 However, the incidence rates have been highly variable not only by geographical regions but even across neighboring countries where genetic risk would be expected to be similar.
In Europe, for example, epidemiologic studies suggest the lifetime risk of MPS IIIA is approximately two times greater in Germany and the Netherlands relative to France and Sweden.7 It is possible that the methodology for identifying cases might be a more important factor than differences in genetic risk to explain this variability. Many experts, including Dr. O’Neill, believe that prevalence figures for Sanfilippo syndrome are typically underestimates because of the frequency with which LSDs are attributed to other pathology.
“For these types of rare disorders, a clinician might only see a single case over a career, and the symptoms can vary in presentation and severity with many alternatives to consider in the differential diagnosis,” Dr. O’Neill explained. She cited case reports in which symptoms of Sanfilippo syndrome after a period of initial normal development has been initially attributed to autism, which is a comorbid feature of the disease, idiopathic developmental delay, or other nonprogressive disorders until further clinical deterioration leads to additional testing. The implication is that LSDs must be considered far earlier despite their rarity.
For the least common of the four clinical subtypes, MPS IIIC and MPS IIID, the median ages of diagnosis have ranged from 4.5 to 19 years of age.7 This is likely a reflection of a slower progression and a later onset of clinical manifestations.
For the more rapidly progressing and typically more severe subtypes, MPS IIIA and MPS IIIB, the diagnosis is typically made earlier. In one review of epidemiologic studies in different countries, the earliest reported median age at diagnosis was 2.5 years,7 a point at which significant disease progression is likely to have already occurred. If the promise of treatments in development is prevention of disease progression, disability in many patients might be substantial if the time to diagnosis is not reduced.
Screening and testing
Independent of the potential to enroll children in clinical trials, early diagnosis also advances the opportunities for supportive care to lessen the burden of the disease on patients and families. Perhaps even more important, early diagnosis is vital to family planning. Since the American pediatrician Sylvester Sanfilippo, MD, first described this syndrome in 1963,7 the genetic profile and many of the features of the disease have become well characterized.8
“One reason to emphasize the importance of early diagnosis is the heritability of this disorder. With prompt diagnosis, genetic counseling can be offered to families to provide them with critical information for future family planning and for cascade testing of other potentially affected siblings,” Dr. O’Neill reported. The inheritance pattern of Sanfilippo syndrome is autosomal recessive.3 In families with an affected child, the risk for any subsequent child to have the same disorder is 25%. The chance of a sibling to be unaffected and not a carrier is also 25%. There is a 50% chance of a sibling to be a carrier but asymptomatic. Of priorities, spreading awareness has been a critical mission of the Cure Sanfilippo Foundation since it was founded 8 years ago, according to Glenn O’Neill, the president. He and his wife, Dr. O’Neill, who is a pediatrician, founded the organization after their own child’s diagnosis of Sanfilippo syndrome. Creating awareness is fundamental to the mission of attracting funds for research, but support to patients and their families as well as early enrollment in clinical trials are among other initiatives being pursued by the foundation to improve care and prognosis.
These strategies include some novel ideas, including an algorithm based on artificial intelligence (AI) that can accelerate suspicion of Sanfilippo syndrome in advance of laboratory or genetic testing, according to Dr. O’Neill. She reported that the facial phenotype, which is observed in a high proportion of but not in all Sanfilippo patients, includes coarse facial features such as puffiness around the eyes, heavy eyebrows, full lips, and macrocephaly.9 Interpretation of photos for AI-based analysis is enhanced when combined with other clinical symptoms.
“The Foundation was involved in honing such a tool by submitting the photos that were used to teach the AI to recognize the Sanfilippo syndrome phenotype,” Dr. O’Neill said. The AI-based tool (Face2Gene.com) is available from FDNA, a company that has been involved in analyzing complex phenotypic and genomic information to guide diagnosis and therapeutic strategies for an array of diseases, not just Sanfilippo syndrome.
The preferred method for diagnosis is biochemical or genetic testing. Of these, urine testing for elevated levels of heparan sulfate glycosaminoglycans (GAG) can be useful for screening, although false-negative tests occur. Analysis of the blood can be performed to detect abnormal levels or activity of the enzymes that break down this GAG. In addition, genetic testing can be performed on blood, fibroblast, buccal swab, or saliva samples. Genetic testing of the blood is the most frequently performed.
For the four MPS III subtypes – MPS IIIA, IIIB, IIIC, and IIID – the presence of two pathogenic mutations in the SGSH (17q25.3), NAGLU (17q21.2), HGSNAT (8p11.21), and GNS (12q14.3) genes, respectively, are likely diagnostic, but enzymatic testing or GAG analysis should be performed to confirm disease status, according to Dr. O’Neill, who said that global consensus based clinical care guidelines led by the Foundation were recently accepted for publication and also include a section on the approach to diagnosis.
While laboratory testing is sensitive, urinary excretion of GAG can be variable, with the potential for ambiguous results. Typically, biochemical and genetic testing provide more reliable results for the diagnosis. They can be readily performed in utero or at the time of birth. In addition, gene panels can permit the diagnosis of multiple types of LSDs, not just Sanfilippo, making screening a cost-effective strategy to consider multiple diseases with overlapping symptoms when an LSD is suspected. Dr. O’Neill said clinical guidelines recommend confirmation of enzyme deficiency or evidence of GAG substrate accumulation as confirmatory tests when genetic testing is positive.
“Ultimately, our goal is to promote universal screening at birth for these serious genetic disorders affecting children,” Dr. O’Neill said.
“We are in a catch-22 when it comes to newborn screening. Currently our federal system requires there be an available treatment before recommending routine screening for a disease. However, it is extremely difficult to power trials with patients who are most likely to show benefit in a trial setting without that very early diagnosis. Universal newborn screening would pave the way for accelerated drug development for children,” she added.
In the meantime, Dr. O’Neill suggests that clinicians should employ a low threshold of suspicion to pursue diagnostic studies of LSDs in infants and children with developmental delays or otherwise unexplained progressive disorders.
Importantly, clinicians can now act quickly on their suspicions and order testing without concern for delays or denial by insurers through a special program, according to Dr. O’Neill. Free genetic testing, offered by the Invitae Corporation, evaluates a panel of 58 genes associated with lysosomal disorders, permitting detection of Sanfilippo syndrome and other LSDs, according to Dr. O’Neill. The Invitae testing is typically performed on 3 mL of whole blood delivered to a central testing facility.
“Results can be obtained within a few weeks or sooner. This can seem like a long wait for families, but it is much more efficient than ordering tests sequentially,” Dr. O’Neill said.
Diagnosis: Signs and symptoms
Despite the differences in progression of the MPS III subtypes, the clinical characteristics are more similar than different. In all patients, prenatal and infant development are typically normal. The initial signs of disease can be found in the newborn, such as neonatal tachypnea, through the early infancy period, such as macrocephaly. However, these are not commonly recognized until about age 1 or soon after in those with MPS IIIA and IIIB.3 Speech delay is the first developmental delay seen in most patients. In those with MPS IIIC, initial symptoms are typically detected at age 3 or later and progress more slowly.10,11 The same is likely to be true of MPS IIID, although this subtype is less well characterized than the other three.7
Although many organs can be involved, degeneration of the CNS is regarded as the most characteristic.3 In aggressive disease, this includes slower acquisition of and failure to meet developmental milestones with progressive intellectual disability, while behavioral difficulties are a more common initial compliant in children with milder disease.13,14 These behavioral changes include hyperactivity, inattention, autistic behaviors, worsening safety awareness, and in some cases aggressive behavior that can be destructive. Sleep disturbances are common.15Because of variability inherent in descriptions of relatively small numbers of patients, the characterization of each of the MPS III subgroups is based on a limited number of small studies, but most patients demonstrate behavior disorders, have coarse facial features, and develop speech delay, according to a survey conducted of published studies.7 Collectively, abnormal behavior was identified as an early symptom in 77% of those with MPS IIIA, 69% of those with IIIB, and 77% of those with IIIC.
For MPS IIIA, loss of speech was observed at a median age of 3.8 years and loss of walking ability at 10.4 years. The median survival has been reported to range between 13 and 18 years. In children with MPS IIIB, the median age of speech loss was reported to about the same age, while loss of walking ability occurred at 11 years. In one study of MPS IIIB, 24% of patients had developed dementia by age 6 years, and the reported median survival has ranged between 17 and 19 years. For MPS IIIC, the onset of clinical symptoms has been observed at a median age of 3.5 years with evidence of cognitive loss observed in 33% of children by the age of 6 years. The median survival has ranged from 19 to 34 years in three studies tracing the natural history of this MPS III subtype.
The differential diagnosis reasonably includes other types of mucopolysaccharidosis disorders with cognitive impairment, including Hurler, Hunter, or Sly syndromes, other neurodevelopmental disorders, and inborn errors of metabolism. The heterogeneity of the features makes definitive laboratory or genetic testing, rather than the effort to differentiate clinical features, appropriate for a definitive diagnosis.
Once the diagnosis is made, other examinations for the common complications of Sanfilippo syndrome are appropriate. Abdominal imaging is appropriate for detecting complications in the gastrointestinal tract, including hepatomegaly, which has been reported in more than half of patients with MPS IIIA and IIIB and in 39% of patients with IIIC.7 In patients with breathing concerns at night and/or sleep disturbance, polysomnography can be useful for identifying sleep apnea and nocturnal seizure activity. In children suspected of seizures, EEG is appropriate. In one study, 66% of patients with MPS IIIA developed seizure activity.16 This has been less commonly reported in MPS IIIB and IIIC, ranging from 8% to 13%.15
Formal hearing evaluation is indicated for any child with speech delays. Hearing loss typically develops after the newborn period in Sanfilippo and may affect peak language acquisition if not treated, according to Dr. O’Neill.
Radiographic studies for dysostosis multiplex or other skeletal abnormalities are also appropriate based on clinical presentation.
Treatment: Present and future
In the absence of treatments to improve the prognosis of Sanfilippo syndrome, current management is based on supportive care and managing organ-specific complications. However, several strategies have proven viable in experimental models and led to clinical trials. None of these therapies has reached approval yet, but several have been associated with attenuation of biomarkers of MPS III disease activity.
Of nearly 30 Sanfilippo clinical trials conducted over the past 20 years, at least 9 have now been completed.5 In addition to studying gene therapy and enzyme replacement therapy, these trials have included stem cell transplantation and substrate reduction therapy, for which the goal is to reduce synthesis of the heparan sulfate GAG to prevent accumulation.5 Of this latter approach, promising initial results with genistein, an isoflavone that breaks down heparan sulfate, reached a phase 3 evaluation.18 Although heparan sulfate levels in the CNS were non-significantly reduced over the course of the trial, the reduction was not sufficient to attenuate cognitive decline.
In other LSDs, several forms of enzyme replacement therapy are now approved. In Fabry disease, for example, recombinant alpha-galactosidase A has now been used for more than 15 years.19 Clinical benefit has not yet been demonstrated in patients with Sanfilippo syndrome because of the difficulty of delivering these therapies past the blood-brain barrier. Several strategies have been pursued. For example, intrathecal delivery of recombinant heparan-N-sulfatase reduced CNS levels of GAG heparan sulfate in one phase 2B study, but it approached but fell short of the statistical significance for the primary endpoint of predefined cognitive stabilization.20 The signal of activity and generally acceptable tolerability has encouraged further study, including an ongoing study with promising interim results of intracerebroventricular enzyme replacement in MPS IIIB, according to Dr. O’Neill.
Acceptable safety and promising activity on disease biomarkers have also been seen with gene therapy in clinical trials. In one study that showed attenuation of brain atrophy, there was moderate improvement in behavior and sleep in three of the four patients enrolled.21 Other studies using various strategies for gene delivery have also produced signals of activity against the underlying pathology, generating persistent interest in ongoing and planned clinical studies with this form of treatment.22Unmodified hematopoietic stem cell transplantation (HSCT), an approach that has demonstrated efficacy when delivered early in the course of other LSDs, such as Hurler syndrome,23 has not yet been associated with significant activity in clinical studies of MPS III, including those that initiated treatment prior to the onset of neurological symptoms.24 However, promising early results have been reported in a study of gene-modified HSCT, which overexpresses the MPS IIIA enzyme.
“The clinical trial landscape fluctuates quite a bit, so I always encourage clinicians and families to check back often for updates. Patient organizations can also be helpful for understanding the most up-to-date and emerging trial options,” Dr. O’Neill reported.
Although it is expected that the greatest benefit would be derived from treatments initiated before or very early after the onset of symptoms, based on the limited potential for reversing cognitive loss, Dr. O’Neill said that she and others are also striving to offer treatments for individuals now living with Sanfilippo syndrome.
“We have to be willing to test treatments that are symptomatic in nature. To that aim, the Cure Sanfilippo Foundation has sponsored a study of a CNS-penetrating anti-inflammatory agent in advanced-disease patients more than 4 years of age,” Dr. O’Neill said. This group of patients typically been ineligible for clinical trials in the past. Dr. O’Neill hopes to change this orientation.
“It is important to highlight that all patients deserve our efforts to improve their quality of life and alleviate suffering, regardless of how old they are or how progressed in the disease they happen to be,” she said.
However, whether the goal is enrollment before or early in disease or later in disease progression, the challenge of enrolling sufficient numbers of patients to confirm clinical activity has been and continues to be a hurdle to progress.
“Clinical studies in Sanfilippo enroll relatively small numbers of patients, often 20 or less,” said Dr. O’Neill, explaining one of the reasons why her organization has been so active in raising awareness and funding such studies. For patients and families, the Cure Sanfilippo Foundation can offer a variety of guidance and support, but information about opportunities for clinical trial participation is a key resource they provide for families and their physicians.
Conclusion
For most children with Sanfilippo syndrome, life expectancy is limited. However, the characterization of the genetic causes and the biochemistry of the subtypes has led to several viable therapeutic approaches under development. There has been progress in delivery of therapeutic enzymes to the CNS, and there is substantial optimism that more progress is coming. One issue for treatment development, is the last of a clear regulatory pathway addressing important biomarkers of pathology, such as heparan sulfate burden. Developing treatments that address this issue or impaired enzyme activity levels have promise for preventing progression, particularly if started in infancy. However, the effort to draw awareness to this disease is the first step toward accelerating the time to an early diagnosis and subsequent opportunities to enroll in clinical trials.
References
1. Sun A. Lysosomal storage disease overview. Ann Transl Med. 2018 Dec;6(24):476. doi: 10.21037/atm.2018.11.39.
2. Andrade F et al. Sanfilippo syndrome: Overall review. Pediatr Int. 2015 Jun;57(3):331-8. doi: 10.1111/ped.12636.
3. Fedele AO. Sanfilippo syndrome: Causes, consequences, and treatments. Appl Clin Genet. 2015 Nov 25;8:269-81. doi: 10.2147/TACG.S57672.
4. Lavery C et al. Mortality in patients with Sanfilippo syndrome. Orphanet J Rare Dis. 2017 Oct 23;12(1):168. doi: 10.1186/s13023-017-0717-y.
5. Pearse Y et al. A cure for Sanfilippo syndrome? A summary of current therapeutic approaches and their promise. Med Res Arch. 2020 Feb 1;8(2). doi: 10.18103/mra.v8i2.2045.
6. Kuiper GA et al. Failure to shorten the diagnostic delay in two ultrao-rphan diseases (mucopolysaccharidosis types I and III): potential causes and implication. Orphanet J Rare Dis. 2018;13:2. Doi: 10.1186/s13023-017-0733-y.
7. Zelei T et al. Epidemiology of Sanfilippo syndrome: Results of a systematic literature review. Orphanet J Rare Dis. 2018 Apr 10;13(1):53. doi: 10.1186/s13023-018-0796-4.
8. Wagner VF, Northrup H. Mucopolysaccaharidosis type III. Gene Reviews. 2019 Sep 19. University of Washington, Seattle. https://www.ncbi.nlm.nih.gov/books/NBK546574/8.
9. O’Neill C et al. Natural history of facial features observed in Sanfilippo syndrome (MPS IIIB) using a next generation phenotyping tool. Mol Genet Metab. 2019 Feb;126:S112.
10. Ruijter GJ et al. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in the Netherlands. Mol Genet Metab. 2008 Feb;93(2):104-11. doi: 10.1016/j.ymgme.2007.09.011.
11. Valstar MJ et al. Mucopolysaccharidosis type IIID: 12 new patients and 15 novel mutations. Hum Mutat. 2010 May;31(5):E1348-60. doi: 10.1002/humu.21234.
12. Nijmeijer SCM. The attenuated end of phenotypic spectrum in MPS III: from late-onset stable cognitive impairment to non-neuronopathic phenotype. Orphanet J Rare Dis. 2019;14:249. Doi10.1186/s13023-019-1232-0.
13. Nidiffer FD, Kelly TE. Developmental and degenerative patterns associated with cognitive, behavioural and motor difficulties in the Sanfilippo syndrome: An epidemiological study. J Ment Defic Res. 1983 Sep;27 (Pt 3):185-203. doi: 10.1111/j.1365-2788.1983.tb00291.x.
14. Bax MC, Colville GA. Behaviour in mucopolysaccharide disorders. Arch Dis Child. 1995 Jul;73(1):77-81. doi: 10.1136/adc.73.1.77.
15. Fraser J et al. Sleep disturbance in mucopolysaccharidosis type III (Sanfilippo syndrome): A survey of managing clinicians. Clin Genet. 2002 Nov;62(5):418-21. doi: 10.1034/j.1399-0004.2002.620512.x.
16. Valstar MJ et al. Mucopolysaccharidosis type IIIA: Clinical spectrum and genotype-phenotype correlations. Ann Neurol. 2010 Dec;68(6):876-87. doi: 10.1002/ana.22092.
17. Heron B et al. Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am J Med Genet A. 2011 Jan;155A(1):58-68. doi: 10.1002/ajmg.a.33779.
18. Delgadillo V et al. Genistein supplementation in patients affected by Sanfilippo disease. J Inherit Metab Dis. 2011 Oct;34(5):1039-44. doi: 10.1007/s10545-011-9342-4.
19. van der Veen SJ et al. Developments in the treatment of Fabry disease. J Inherit Metab Dis. 2020 Sep;43(5):908-21. doi: 10.1002/jimd.12228.
20. Wijburg FA et al. Intrathecal heparan-N-sulfatase in patients with Sanfilippo syndrome type A: A phase IIb randomized trial. Mol Genet Metab. 2019 Feb;126(2):121-30. doi: 10.1016/j.ymgme.2018.10.006.
21. Tardieu M et al. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: Results of a phase I/II trial. Hum Gene Ther. 2014 Jun;25(6):506-16. doi: 10.1089/hum.2013.238.
22. Marco S et al. In vivo gene therapy for mucopolysaccharidosis type III (Sanfilippo syndrome): A new treatment horizon. Hum Gene Ther. 2019 Oct;30(10):1211-1121. doi: 10.1089/hum.2019.217.
23. Taylor M et al. Hematopoietic stem cell transplantation for mucopolysaccharidoses: Past, present, and future. Biol Blood Marrow Transplant. 2019 Jul;25(7):e226-e246. doi: 10.1016/j.bbmt.2019.02.012.
24. Sivakumur P, Wraith JE. Bone marrow transplantation in mucopolysaccharidosis type IIIA: A comparison of an early treated patient with his untreated sibling. J Inherit Metab Dis. 1999 Oct;22(7):849-50. doi: 10.1023/a:1005526628598.
Sanfilippo syndrome is a rare inherited neurodegenerative metabolic disorder for which there are no approved therapies. Symptoms of the more severe subtypes typically begin within the first years of life, rapidly producing serious and progressive physical and cognitive deficits. The underlying pathophysiology is targetable, but the delay in diagnosis of this as well as other lysosomal storage disorders (LSDs) is slowing progress toward effective therapies.
“Lack of awareness and the delays to diagnosis have been a real challenge for us. There is reason for cautious optimism about treatments now in or approaching clinical studies, but to evaluate efficacy on cognitive outcomes we need to enroll more children at a very young age, before loss of milestones,” according to Cara O’Neill, MD, a co-founder and chief science officer of Cure Sanfilippo Foundation.
Epidemiology and description
Sanfilippo syndrome, like the more than 50 other LSDs, is caused by a gene mutation that leads to an enzyme deficiency in the lysosome.1 In the case of Sanfilippo syndrome, also known as mucopolysaccharidosis (MPS III), there are hundreds of mutations that can lead to Sanfilippo by altering the function of one of the four genes essential to degradation of heparan sulfate.2 Lysosomal accumulation of heparan sulfate drives a broad spectrum of progressive and largely irreversible symptoms that typically begin with somatic manifestations, such as bowel dysfunction and recurrent ear and upper respiratory infections.
Impairment of the central nervous system (CNS) usually occurs early in life, halting physical and mental development. As it progresses, accumulation of heparan sulfate in a variety of cells leads to a cascade of abnormal cellular signaling and dysfunction. Disruption of these processes, which are critical for normal neurodevelopment, result in loss of the developmental skills already gained and eventually loss of brain tissue.3 Although life expectancy has improved with supportive care, survival into adulthood is typically limited to milder forms.4
Over the past several years, progress in this and other LSDs has yielded therapeutic targets, including those involving gene repair and enzyme replacement. Already approved for use in some LSDs, these therapies have also shown promise in the experimental setting for Sanfilippo syndrome, leading to several completed clinical trials.5
So far, none of these treatments has advanced beyond clinical trials in Sanfilippo syndrome, but there have been favorable changes in the markers of disease, suggesting that better methods of treatment delivery and/or more sensitive tools to measure clinical change might lead to evidence of disease attenuation. However, the promise of treatment in all cases has been to prevent, slow, or halt progression, not to reverse it. This point is important, because it indicates that degree of benefit will depend on enrolling patients early in life. Even if effective therapies are identified, few patients will benefit without strategies to accelerate diagnosis.
In fact, “one study6 reported that the average age of diagnosis for Sanfilippo syndrome has not improved over the past 30 years,” according to Dr. O’Neill. She indicated that this has been frustrating, given the availability of clinical trials on which progress is dependent. There is no widely accepted protocol for who and when to test for Sanfilippo syndrome or other LSDs, but Dr. O’Neill’s organization is among those advocating for strategies to detect these diseases earlier, including screening at birth.
Almost by definition, the clinical diagnosis of rare diseases poses a challenge. With nonspecific symptoms and a broad range of potential diagnoses, diseases with a low incidence are not the first ones that are typically considered. In the case of Sanfilippo syndrome, published studies indicate incidence rates at or below 1 per 70,000 live births.7 However, the incidence rates have been highly variable not only by geographical regions but even across neighboring countries where genetic risk would be expected to be similar.
In Europe, for example, epidemiologic studies suggest the lifetime risk of MPS IIIA is approximately two times greater in Germany and the Netherlands relative to France and Sweden.7 It is possible that the methodology for identifying cases might be a more important factor than differences in genetic risk to explain this variability. Many experts, including Dr. O’Neill, believe that prevalence figures for Sanfilippo syndrome are typically underestimates because of the frequency with which LSDs are attributed to other pathology.
“For these types of rare disorders, a clinician might only see a single case over a career, and the symptoms can vary in presentation and severity with many alternatives to consider in the differential diagnosis,” Dr. O’Neill explained. She cited case reports in which symptoms of Sanfilippo syndrome after a period of initial normal development has been initially attributed to autism, which is a comorbid feature of the disease, idiopathic developmental delay, or other nonprogressive disorders until further clinical deterioration leads to additional testing. The implication is that LSDs must be considered far earlier despite their rarity.
For the least common of the four clinical subtypes, MPS IIIC and MPS IIID, the median ages of diagnosis have ranged from 4.5 to 19 years of age.7 This is likely a reflection of a slower progression and a later onset of clinical manifestations.
For the more rapidly progressing and typically more severe subtypes, MPS IIIA and MPS IIIB, the diagnosis is typically made earlier. In one review of epidemiologic studies in different countries, the earliest reported median age at diagnosis was 2.5 years,7 a point at which significant disease progression is likely to have already occurred. If the promise of treatments in development is prevention of disease progression, disability in many patients might be substantial if the time to diagnosis is not reduced.
Screening and testing
Independent of the potential to enroll children in clinical trials, early diagnosis also advances the opportunities for supportive care to lessen the burden of the disease on patients and families. Perhaps even more important, early diagnosis is vital to family planning. Since the American pediatrician Sylvester Sanfilippo, MD, first described this syndrome in 1963,7 the genetic profile and many of the features of the disease have become well characterized.8
“One reason to emphasize the importance of early diagnosis is the heritability of this disorder. With prompt diagnosis, genetic counseling can be offered to families to provide them with critical information for future family planning and for cascade testing of other potentially affected siblings,” Dr. O’Neill reported. The inheritance pattern of Sanfilippo syndrome is autosomal recessive.3 In families with an affected child, the risk for any subsequent child to have the same disorder is 25%. The chance of a sibling to be unaffected and not a carrier is also 25%. There is a 50% chance of a sibling to be a carrier but asymptomatic. Of priorities, spreading awareness has been a critical mission of the Cure Sanfilippo Foundation since it was founded 8 years ago, according to Glenn O’Neill, the president. He and his wife, Dr. O’Neill, who is a pediatrician, founded the organization after their own child’s diagnosis of Sanfilippo syndrome. Creating awareness is fundamental to the mission of attracting funds for research, but support to patients and their families as well as early enrollment in clinical trials are among other initiatives being pursued by the foundation to improve care and prognosis.
These strategies include some novel ideas, including an algorithm based on artificial intelligence (AI) that can accelerate suspicion of Sanfilippo syndrome in advance of laboratory or genetic testing, according to Dr. O’Neill. She reported that the facial phenotype, which is observed in a high proportion of but not in all Sanfilippo patients, includes coarse facial features such as puffiness around the eyes, heavy eyebrows, full lips, and macrocephaly.9 Interpretation of photos for AI-based analysis is enhanced when combined with other clinical symptoms.
“The Foundation was involved in honing such a tool by submitting the photos that were used to teach the AI to recognize the Sanfilippo syndrome phenotype,” Dr. O’Neill said. The AI-based tool (Face2Gene.com) is available from FDNA, a company that has been involved in analyzing complex phenotypic and genomic information to guide diagnosis and therapeutic strategies for an array of diseases, not just Sanfilippo syndrome.
The preferred method for diagnosis is biochemical or genetic testing. Of these, urine testing for elevated levels of heparan sulfate glycosaminoglycans (GAG) can be useful for screening, although false-negative tests occur. Analysis of the blood can be performed to detect abnormal levels or activity of the enzymes that break down this GAG. In addition, genetic testing can be performed on blood, fibroblast, buccal swab, or saliva samples. Genetic testing of the blood is the most frequently performed.
For the four MPS III subtypes – MPS IIIA, IIIB, IIIC, and IIID – the presence of two pathogenic mutations in the SGSH (17q25.3), NAGLU (17q21.2), HGSNAT (8p11.21), and GNS (12q14.3) genes, respectively, are likely diagnostic, but enzymatic testing or GAG analysis should be performed to confirm disease status, according to Dr. O’Neill, who said that global consensus based clinical care guidelines led by the Foundation were recently accepted for publication and also include a section on the approach to diagnosis.
While laboratory testing is sensitive, urinary excretion of GAG can be variable, with the potential for ambiguous results. Typically, biochemical and genetic testing provide more reliable results for the diagnosis. They can be readily performed in utero or at the time of birth. In addition, gene panels can permit the diagnosis of multiple types of LSDs, not just Sanfilippo, making screening a cost-effective strategy to consider multiple diseases with overlapping symptoms when an LSD is suspected. Dr. O’Neill said clinical guidelines recommend confirmation of enzyme deficiency or evidence of GAG substrate accumulation as confirmatory tests when genetic testing is positive.
“Ultimately, our goal is to promote universal screening at birth for these serious genetic disorders affecting children,” Dr. O’Neill said.
“We are in a catch-22 when it comes to newborn screening. Currently our federal system requires there be an available treatment before recommending routine screening for a disease. However, it is extremely difficult to power trials with patients who are most likely to show benefit in a trial setting without that very early diagnosis. Universal newborn screening would pave the way for accelerated drug development for children,” she added.
In the meantime, Dr. O’Neill suggests that clinicians should employ a low threshold of suspicion to pursue diagnostic studies of LSDs in infants and children with developmental delays or otherwise unexplained progressive disorders.
Importantly, clinicians can now act quickly on their suspicions and order testing without concern for delays or denial by insurers through a special program, according to Dr. O’Neill. Free genetic testing, offered by the Invitae Corporation, evaluates a panel of 58 genes associated with lysosomal disorders, permitting detection of Sanfilippo syndrome and other LSDs, according to Dr. O’Neill. The Invitae testing is typically performed on 3 mL of whole blood delivered to a central testing facility.
“Results can be obtained within a few weeks or sooner. This can seem like a long wait for families, but it is much more efficient than ordering tests sequentially,” Dr. O’Neill said.
Diagnosis: Signs and symptoms
Despite the differences in progression of the MPS III subtypes, the clinical characteristics are more similar than different. In all patients, prenatal and infant development are typically normal. The initial signs of disease can be found in the newborn, such as neonatal tachypnea, through the early infancy period, such as macrocephaly. However, these are not commonly recognized until about age 1 or soon after in those with MPS IIIA and IIIB.3 Speech delay is the first developmental delay seen in most patients. In those with MPS IIIC, initial symptoms are typically detected at age 3 or later and progress more slowly.10,11 The same is likely to be true of MPS IIID, although this subtype is less well characterized than the other three.7
Although many organs can be involved, degeneration of the CNS is regarded as the most characteristic.3 In aggressive disease, this includes slower acquisition of and failure to meet developmental milestones with progressive intellectual disability, while behavioral difficulties are a more common initial compliant in children with milder disease.13,14 These behavioral changes include hyperactivity, inattention, autistic behaviors, worsening safety awareness, and in some cases aggressive behavior that can be destructive. Sleep disturbances are common.15Because of variability inherent in descriptions of relatively small numbers of patients, the characterization of each of the MPS III subgroups is based on a limited number of small studies, but most patients demonstrate behavior disorders, have coarse facial features, and develop speech delay, according to a survey conducted of published studies.7 Collectively, abnormal behavior was identified as an early symptom in 77% of those with MPS IIIA, 69% of those with IIIB, and 77% of those with IIIC.
For MPS IIIA, loss of speech was observed at a median age of 3.8 years and loss of walking ability at 10.4 years. The median survival has been reported to range between 13 and 18 years. In children with MPS IIIB, the median age of speech loss was reported to about the same age, while loss of walking ability occurred at 11 years. In one study of MPS IIIB, 24% of patients had developed dementia by age 6 years, and the reported median survival has ranged between 17 and 19 years. For MPS IIIC, the onset of clinical symptoms has been observed at a median age of 3.5 years with evidence of cognitive loss observed in 33% of children by the age of 6 years. The median survival has ranged from 19 to 34 years in three studies tracing the natural history of this MPS III subtype.
The differential diagnosis reasonably includes other types of mucopolysaccharidosis disorders with cognitive impairment, including Hurler, Hunter, or Sly syndromes, other neurodevelopmental disorders, and inborn errors of metabolism. The heterogeneity of the features makes definitive laboratory or genetic testing, rather than the effort to differentiate clinical features, appropriate for a definitive diagnosis.
Once the diagnosis is made, other examinations for the common complications of Sanfilippo syndrome are appropriate. Abdominal imaging is appropriate for detecting complications in the gastrointestinal tract, including hepatomegaly, which has been reported in more than half of patients with MPS IIIA and IIIB and in 39% of patients with IIIC.7 In patients with breathing concerns at night and/or sleep disturbance, polysomnography can be useful for identifying sleep apnea and nocturnal seizure activity. In children suspected of seizures, EEG is appropriate. In one study, 66% of patients with MPS IIIA developed seizure activity.16 This has been less commonly reported in MPS IIIB and IIIC, ranging from 8% to 13%.15
Formal hearing evaluation is indicated for any child with speech delays. Hearing loss typically develops after the newborn period in Sanfilippo and may affect peak language acquisition if not treated, according to Dr. O’Neill.
Radiographic studies for dysostosis multiplex or other skeletal abnormalities are also appropriate based on clinical presentation.
Treatment: Present and future
In the absence of treatments to improve the prognosis of Sanfilippo syndrome, current management is based on supportive care and managing organ-specific complications. However, several strategies have proven viable in experimental models and led to clinical trials. None of these therapies has reached approval yet, but several have been associated with attenuation of biomarkers of MPS III disease activity.
Of nearly 30 Sanfilippo clinical trials conducted over the past 20 years, at least 9 have now been completed.5 In addition to studying gene therapy and enzyme replacement therapy, these trials have included stem cell transplantation and substrate reduction therapy, for which the goal is to reduce synthesis of the heparan sulfate GAG to prevent accumulation.5 Of this latter approach, promising initial results with genistein, an isoflavone that breaks down heparan sulfate, reached a phase 3 evaluation.18 Although heparan sulfate levels in the CNS were non-significantly reduced over the course of the trial, the reduction was not sufficient to attenuate cognitive decline.
In other LSDs, several forms of enzyme replacement therapy are now approved. In Fabry disease, for example, recombinant alpha-galactosidase A has now been used for more than 15 years.19 Clinical benefit has not yet been demonstrated in patients with Sanfilippo syndrome because of the difficulty of delivering these therapies past the blood-brain barrier. Several strategies have been pursued. For example, intrathecal delivery of recombinant heparan-N-sulfatase reduced CNS levels of GAG heparan sulfate in one phase 2B study, but it approached but fell short of the statistical significance for the primary endpoint of predefined cognitive stabilization.20 The signal of activity and generally acceptable tolerability has encouraged further study, including an ongoing study with promising interim results of intracerebroventricular enzyme replacement in MPS IIIB, according to Dr. O’Neill.
Acceptable safety and promising activity on disease biomarkers have also been seen with gene therapy in clinical trials. In one study that showed attenuation of brain atrophy, there was moderate improvement in behavior and sleep in three of the four patients enrolled.21 Other studies using various strategies for gene delivery have also produced signals of activity against the underlying pathology, generating persistent interest in ongoing and planned clinical studies with this form of treatment.22Unmodified hematopoietic stem cell transplantation (HSCT), an approach that has demonstrated efficacy when delivered early in the course of other LSDs, such as Hurler syndrome,23 has not yet been associated with significant activity in clinical studies of MPS III, including those that initiated treatment prior to the onset of neurological symptoms.24 However, promising early results have been reported in a study of gene-modified HSCT, which overexpresses the MPS IIIA enzyme.
“The clinical trial landscape fluctuates quite a bit, so I always encourage clinicians and families to check back often for updates. Patient organizations can also be helpful for understanding the most up-to-date and emerging trial options,” Dr. O’Neill reported.
Although it is expected that the greatest benefit would be derived from treatments initiated before or very early after the onset of symptoms, based on the limited potential for reversing cognitive loss, Dr. O’Neill said that she and others are also striving to offer treatments for individuals now living with Sanfilippo syndrome.
“We have to be willing to test treatments that are symptomatic in nature. To that aim, the Cure Sanfilippo Foundation has sponsored a study of a CNS-penetrating anti-inflammatory agent in advanced-disease patients more than 4 years of age,” Dr. O’Neill said. This group of patients typically been ineligible for clinical trials in the past. Dr. O’Neill hopes to change this orientation.
“It is important to highlight that all patients deserve our efforts to improve their quality of life and alleviate suffering, regardless of how old they are or how progressed in the disease they happen to be,” she said.
However, whether the goal is enrollment before or early in disease or later in disease progression, the challenge of enrolling sufficient numbers of patients to confirm clinical activity has been and continues to be a hurdle to progress.
“Clinical studies in Sanfilippo enroll relatively small numbers of patients, often 20 or less,” said Dr. O’Neill, explaining one of the reasons why her organization has been so active in raising awareness and funding such studies. For patients and families, the Cure Sanfilippo Foundation can offer a variety of guidance and support, but information about opportunities for clinical trial participation is a key resource they provide for families and their physicians.
Conclusion
For most children with Sanfilippo syndrome, life expectancy is limited. However, the characterization of the genetic causes and the biochemistry of the subtypes has led to several viable therapeutic approaches under development. There has been progress in delivery of therapeutic enzymes to the CNS, and there is substantial optimism that more progress is coming. One issue for treatment development, is the last of a clear regulatory pathway addressing important biomarkers of pathology, such as heparan sulfate burden. Developing treatments that address this issue or impaired enzyme activity levels have promise for preventing progression, particularly if started in infancy. However, the effort to draw awareness to this disease is the first step toward accelerating the time to an early diagnosis and subsequent opportunities to enroll in clinical trials.
References
1. Sun A. Lysosomal storage disease overview. Ann Transl Med. 2018 Dec;6(24):476. doi: 10.21037/atm.2018.11.39.
2. Andrade F et al. Sanfilippo syndrome: Overall review. Pediatr Int. 2015 Jun;57(3):331-8. doi: 10.1111/ped.12636.
3. Fedele AO. Sanfilippo syndrome: Causes, consequences, and treatments. Appl Clin Genet. 2015 Nov 25;8:269-81. doi: 10.2147/TACG.S57672.
4. Lavery C et al. Mortality in patients with Sanfilippo syndrome. Orphanet J Rare Dis. 2017 Oct 23;12(1):168. doi: 10.1186/s13023-017-0717-y.
5. Pearse Y et al. A cure for Sanfilippo syndrome? A summary of current therapeutic approaches and their promise. Med Res Arch. 2020 Feb 1;8(2). doi: 10.18103/mra.v8i2.2045.
6. Kuiper GA et al. Failure to shorten the diagnostic delay in two ultrao-rphan diseases (mucopolysaccharidosis types I and III): potential causes and implication. Orphanet J Rare Dis. 2018;13:2. Doi: 10.1186/s13023-017-0733-y.
7. Zelei T et al. Epidemiology of Sanfilippo syndrome: Results of a systematic literature review. Orphanet J Rare Dis. 2018 Apr 10;13(1):53. doi: 10.1186/s13023-018-0796-4.
8. Wagner VF, Northrup H. Mucopolysaccaharidosis type III. Gene Reviews. 2019 Sep 19. University of Washington, Seattle. https://www.ncbi.nlm.nih.gov/books/NBK546574/8.
9. O’Neill C et al. Natural history of facial features observed in Sanfilippo syndrome (MPS IIIB) using a next generation phenotyping tool. Mol Genet Metab. 2019 Feb;126:S112.
10. Ruijter GJ et al. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in the Netherlands. Mol Genet Metab. 2008 Feb;93(2):104-11. doi: 10.1016/j.ymgme.2007.09.011.
11. Valstar MJ et al. Mucopolysaccharidosis type IIID: 12 new patients and 15 novel mutations. Hum Mutat. 2010 May;31(5):E1348-60. doi: 10.1002/humu.21234.
12. Nijmeijer SCM. The attenuated end of phenotypic spectrum in MPS III: from late-onset stable cognitive impairment to non-neuronopathic phenotype. Orphanet J Rare Dis. 2019;14:249. Doi10.1186/s13023-019-1232-0.
13. Nidiffer FD, Kelly TE. Developmental and degenerative patterns associated with cognitive, behavioural and motor difficulties in the Sanfilippo syndrome: An epidemiological study. J Ment Defic Res. 1983 Sep;27 (Pt 3):185-203. doi: 10.1111/j.1365-2788.1983.tb00291.x.
14. Bax MC, Colville GA. Behaviour in mucopolysaccharide disorders. Arch Dis Child. 1995 Jul;73(1):77-81. doi: 10.1136/adc.73.1.77.
15. Fraser J et al. Sleep disturbance in mucopolysaccharidosis type III (Sanfilippo syndrome): A survey of managing clinicians. Clin Genet. 2002 Nov;62(5):418-21. doi: 10.1034/j.1399-0004.2002.620512.x.
16. Valstar MJ et al. Mucopolysaccharidosis type IIIA: Clinical spectrum and genotype-phenotype correlations. Ann Neurol. 2010 Dec;68(6):876-87. doi: 10.1002/ana.22092.
17. Heron B et al. Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am J Med Genet A. 2011 Jan;155A(1):58-68. doi: 10.1002/ajmg.a.33779.
18. Delgadillo V et al. Genistein supplementation in patients affected by Sanfilippo disease. J Inherit Metab Dis. 2011 Oct;34(5):1039-44. doi: 10.1007/s10545-011-9342-4.
19. van der Veen SJ et al. Developments in the treatment of Fabry disease. J Inherit Metab Dis. 2020 Sep;43(5):908-21. doi: 10.1002/jimd.12228.
20. Wijburg FA et al. Intrathecal heparan-N-sulfatase in patients with Sanfilippo syndrome type A: A phase IIb randomized trial. Mol Genet Metab. 2019 Feb;126(2):121-30. doi: 10.1016/j.ymgme.2018.10.006.
21. Tardieu M et al. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: Results of a phase I/II trial. Hum Gene Ther. 2014 Jun;25(6):506-16. doi: 10.1089/hum.2013.238.
22. Marco S et al. In vivo gene therapy for mucopolysaccharidosis type III (Sanfilippo syndrome): A new treatment horizon. Hum Gene Ther. 2019 Oct;30(10):1211-1121. doi: 10.1089/hum.2019.217.
23. Taylor M et al. Hematopoietic stem cell transplantation for mucopolysaccharidoses: Past, present, and future. Biol Blood Marrow Transplant. 2019 Jul;25(7):e226-e246. doi: 10.1016/j.bbmt.2019.02.012.
24. Sivakumur P, Wraith JE. Bone marrow transplantation in mucopolysaccharidosis type IIIA: A comparison of an early treated patient with his untreated sibling. J Inherit Metab Dis. 1999 Oct;22(7):849-50. doi: 10.1023/a:1005526628598.

Novel gene-based therapies for neuromuscular diseases
Neuromuscular diseases (NMDs) are a broad classification of heterogeneous groups of disorders characterized by progressive muscle weakness resulting from muscle or nerve dysfunction.1 Diagnosis is based on symptoms and a full medical history, as well as on muscle and imaging tests (including electromyography, nerve-conduction studies, magnetic resonance imaging, muscle biopsy, and blood tests) to confirm or rule out specific NMDs.2 Early diagnosis of NMDs can be difficult because symptoms overlap with those of many other diseases.
Although individually, NMDs are rare, collectively, they affect approximately 250,000 people in the United States. Disease types vary in regard to cause, symptoms, prevalence, age of onset, progression, and severity. Functional impairment from any NMD can lead to lifelong morbidities and shortened life expectancy.1,3
Treatment options for NMDs are limited; most target symptoms, not disease progression. Although there is a need for safe and effective gene-based therapies for NMDs, there are challenges to developing and delivering such treatments that have impeded clinical success. These include a lack of understanding about disease pathology and drug targets, limited animal model systems, and few reliable biomarkers that are predictive of therapeutic success.4,5
Notwithstanding that challenges remain, our understanding of gene expression in NMDs has greatly advanced in the past few decades. This progress has translated into promising results in the gene-therapy field – thereby setting the stage for therapeutic approaches that use novel gene-delivery and gene-manipulation tools.6 These novel approaches include nonviral strategies, such as antisense oligonucleotides (ASOs), and viral-based strategies, such as adeno-associated virus (AAV)-mediated gene silencing and AAV-mediated gene delivery.
In this article, we highlight advancements in the clinical development of gene-based therapies for NMDs. We focus on amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), and Duchenne muscular dystrophy (DMD) because of recent clinical successes in developing such therapies.1,6,7 We also catalog completed and ongoing clinical trials for ALS, SMA, and DMD (Tables 1-3).
Amyotrophic lateral sclerosis
ALS is caused by progressive degeneration of upper- and lower-motor neurons, which eventually leads to respiratory failure and death 3 to 5 years after disease onset.7-9 There are two subtypes: Familial ALS (10% of cases) and sporadic ALS (90% of cases). Commonly mutated ALS-associated genes6,8 are:
- Superoxide dismutase type 1 (SOD1).
- Chromosome 9 open reading frame 72 (C9orf72).
- Transactive response DNA-binding protein 43 (TARDBP).
- Fused in sarcoma (FUS).
SOD1-targeted therapy is being studied, with early evidence of clinical success. Mutations in SOD1 account for 10% to 20% of familial ALS cases and 1% to 2% of sporadic ALS cases.6,10 10 Mutations in C9orf72 account for 25 to 40% of familial ALS cases and 7% of sporadic ALS cases.8,9,11 Mutations in TARDBP account for 3% of familial ALS cases and 2% of sporadic cases.12 Mutations in FUS account for 4% of familial ALS cases and 1% of sporadic cases. Overall, these mutant proteins can trigger neurotoxicity, thus inducing motor-neuron death.6,10
Treatment of ALS
Two treatments for ALS are Food and Drug Administration approved: riluzole (Rilutek), approved in 1995, and edaravone (Radicava), approved in 2017.

Riluzole is an oral anti-excitotoxic glutamate antagonist.11 Approval of riluzole was based on the results of two studies that demonstrated a 2- to 3-month survival benefit.10,14 For patients who have difficulty swallowing, an oral suspension (Tiglutik, approved in 2018) and an oral film (Exservan, approved in 2019) are available.
Edaravone is a free-radical scavenger that decreases oxidative stress and is administered intravenously (IV).9,13,14 Findings from clinical trials suggest functional improvement or slower decline in function for some patients.
Although these two agents demonstrate modest therapeutic benefit, neither reverses progression of disease.10,14
Gene-based therapy for ALS
Many non-viral strategies, including antisense oligonucleotide (ASO), monoclonal antibodies, reverse transcriptase inhibitors, and HGF gene replacement therapy are used as therapeutic approaches to SOD1, C9orf72, and FUS gene mutations in ALS patients, and are being evaluated in clinical studies14,15 (Table 113-17).
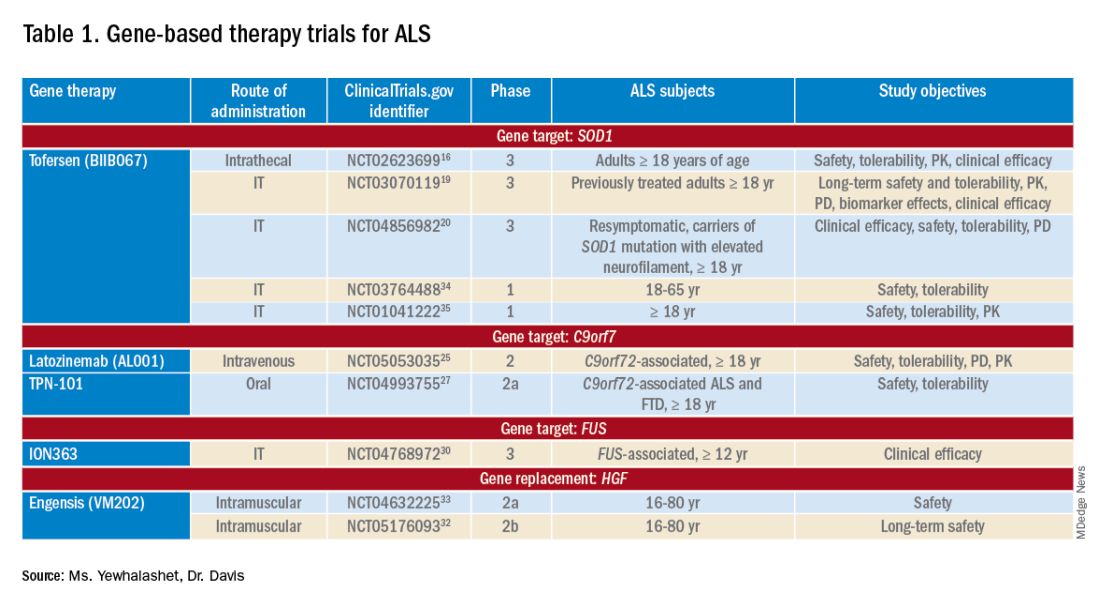
Tofersen, also known as BIIB067, is an investigational ASO, administered by intrathecal (IT) injection, that binds to SOD1 mRNA, thus reducing its protein levels.16 Tofersen was evaluated in the VALOR phase 3 study (ClinicalTrials.gov Identifier: NCT02623699), a three-part randomized, double-blind, placebo-controlled trial: single ascending dose (Part A), multiple ascending dose (B), and fixed dose (C).10 In Parts A and B, 48 participants received five IT injections of tofersen or placebo over 12 weeks and were followed for an additional 12 weeks. Reduction in SOD1 protein production and neurofilament level in cerebrospinal fluid (CSF) (a potential biomarker of motor-neuron degeneration) was observed, which determined the fixed-dose for Part C.16,17
Part C examined the efficacy, safety and tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of tofersen, compared with placebo, in adults with ALS who had a confirmed SOD1 mutation.17 A total of 108 participants were enrolled; 60 were identified as “faster-progressing”; 48, as “slower-progressing.”18 The primary endpoint of Part C was change from baseline to Week 28 on the Revised ALS Functional Rating Scale (ALSFRS-R) total score. (ALSFRS-R measures overall clinical effect; the score ranges from 0 [no function] to 4 [full function].17)
Tofersen failed to meet the primary efficacy outcome because statistically significant findings were lacking in the faster-progressing population, as measured by joint-rank analysis (difference of 1.2 on the ALSFRS-R score; P = .97). However, trends favoring tofersen were observed across key secondary clinical outcome measures18:
- Change from baseline in CSF SOD1 protein concentration.17 Percent reduction in the total SOD1 protein level was much higher in the tofersen-treated group than in the control group (38% more than controls in the faster-progressing population; 26% more than controls in the slower-progressing population).18
- Change from baseline in neurofilament light-chain concentration in plasma.17,18 Percent reduction in the level of neurofilament light chain was also observed to be higher in the tofersen-treated group than in the control group (67% more than controls in the faster-progressing population and 48% more than controls in the slower-progressing population).18
Because of these encouraging results, VALOR participants were moved to the ongoing open-label extension trial of tofersen (ClinicalTri-als.gov Identifier: NCT03070119), in which both groups were treated with the active agent.
These data suggest that early tofersen treatment might slow decline in faster-progressing patients and stabilize clinical function in slower-progressing patients.18,19 Overall, most adverse events (AEs) in the trial among patients receiving active treatment were of mild or moderate severity, and were largely consistent with either disease progression or lumbar puncture–related complications.18
Because data from VALOR suggested potential benefit from tofersen, the ATLAS trial (ClinicalTrials.gov Identifier: NCT04856982) is investigating the clinical value of presymptomatic treatment and the optimal timing of initiation of therapy.20,21 ATLAS is a phase 3, randomized, placebo-controlled trial that examines the clinical efficacy, safety, and tolerability of tofersen in presymptomatic adult carriers of SOD1 mutation who have an elevated neurofilament light-chain concentration.21 ATLAS will also evaluate the efficacy of tofersen when initiated before, rather than after, ALS manifests clinically. Enrollment is still open for this trial.20,21
Latozinemab, also known as AL001, is a first-in-class monoclonal antibody, administered by IV infusion, that elevates levels of progranulin, a key regulator of the immune activity and lysosomal function in the brain.22,23 Latozinemab limits progranulin endocytosis and degradation by sortilin inhibition.22 Progranulin gene mutations can reduce progranulin expression (by 50 to 70 percent reduction), which may cause neuro-degeneration due to abnormal accumulation of TAR-DNA-binding protein 43 (TDP-43) in the brain cells.22,24 TDP-43 pathology has also been shown to be associated with C9orf72 mutations.23 Although the mechanism is not fully understood, the role of progranulin deficiency in TDP-43 pathology is believed to be associated with neurodegenerative diseases like ALS.11,23,24,43 Previous animal models of chronic neurodegenera-tion have demonstrated how increased progranulin levels can be protective against TDP-43 pathology, increasing neuronal development and survival, thus potentially slowing disease progression.23,24,43 Currently, latozinemab is being investigated in a randomized, double-blind, placebo-controlled, multicenter phase 2 trial (ClinicalTrials.gov Identifier: NCT05053035). Approximately, 45 C90rf72-associated ALS participants (≥ 18 years of age) will receive latozinemab or placebo infusions every 4 weeks (for 24 weeks). Study endpoints include safety, tolerability, PK, PD, as well as plasma, and CSF progranulin levels.25 In previous studies, latozinemab demonstrated encouraging results in frontotemporal dementia (FTD) patients who carry a progranulin mutation. Because FTD was revealed to have significant genetic overlap with ALS, there is disease-modifying potential for latozinemab in ALS patients.23,24
TPN-101 is a nucleoside analog reverse transcriptase inhibitor, administered orally, that was originally developed for human immunodeficiency virus (HIV) treatment. However, due to recent findings suggesting retrotransposon activity contributing to neurodegeneration in TDP-43 mediated diseases, including ALS and FTD, TNP-101 is being repurposed.26 The safety and tolerability of TNP-101 are currently being evaluated in C9orf72-associated ALS and FTD patients (≥ 18 years of age). The study is a randomized, double-blind, placebo-controlled paral-lel-group phase 2a trial (ClinicalTrials.gov Identifier: NCT04993755) The study includes a screening period of 6 weeks, double-blind treatment period of 24 weeks, an open-label treatment period of 24 weeks, and 4 weeks of the post-treatment follow-up visit. Study endpoints include the incidence and severity of spontaneously reported treatment-emergent adverse events (TEAEs) associated with TNP-101 and placebo for a to-tal of 48 weeks.27
ION363 is an investigational ASO, administered by IT injection, that selectively targets one of the FUS mutations (p.P525L), which is responsible for earlier disease onset and rapid ALS progression.28,29 The clinical efficacy of ION363, specifically in clinical function and survival is being assessed in FUS-associated ALS patients (≥ 12 years of age). This randomized phase 3 study (ClinicalTrials.gov Identifier: NCT04768972) includes two parts; part 1 will consist of participants receiving a multi-dose regimen (1 dose every 4-12 weeks) of ION363 or placebo for 61 weeks followed by an open-label extension treatment period in part 2, which will consist of participants receiving ION363 (every 12 weeks) for 85 weeks. The primary endpoint of the study is the change from baseline to day 505 in functional impairment, using ALS Functional Rating Scale-Revised (ALSFRS-R). This measures functional disease severity, specifically in bulbar function, gross motor skills, fine motor skills, and respiratory. The score for all 12 questions can range from 0 (no function) to 4 (full function) with a total possible score of 48.30
Engensis, also known as VM202, is a non-viral gene therapy, administered by intramuscular (IM) injection, that uses a plasmid to deliver the hepatocyte growth factor (HGF) gene to promote HGF protein production. The HGF protein plays a role in angiogenesis, the previous of muscle atrophy, and the promotion of neuronal survival and growth. Based on preclinical studies, increasing HGF protein production has been shown to reduce neurodegeneration, thus potentially halting or slowing ALS progression.31 Currently, the safety of engensis is being evaluated in ALS patients (18-80 years of age) in the REViVALS phase 2a (ClinicalTrials.gov Identifier: NCT04632225)/2b (ClinicalTrial.gov Identifier: NCT05176093).32,33 The ReViVALS trial is a double-blind, randomized, placebo-controlled, multi-center study. The phase 2a study endpoints include the incidence of TEAEs, treatment-emergent serious adverse events (TESAEs), injection site reactions, and clinically significant labor-atory values post-treatment (engensis vs placebo group) for 180 days.33 A phase 2b study will evaluate the long-term safety of engensis for an additional 6 months. Study endpoints include the incidence of AEs, changes from baseline in ALSFRS-R scores to evaluate improvement in muscle function, changes from baseline in quality of life using the ALS patient assessment questionnaire, time to all-cause mortality compared to placebo, etc.32
Spinal muscular atrophy
SMA is a hereditary lower motor-neuron disease caused (in 95% of cases) by deletions or, less commonly, by mutations of the survival motor neuron 1 (SMN1) gene on chromosome 5q13 that encodes the SMN protein.6 Reduction in expression of the SMN protein causes motor neurons to degenerate.36-38 Because of a large inverted duplication in chromosome 5q, two variants of SMN (SMN1 and SMN2) exist on each allele. The paralog gene, SMN2, also produces the SMN protein – although at a lower level (10% to 20% of total SMN protein production) than SMN1 does.
A single nucleotide substitution in SMN2 alters splicing and suppresses transcription of exon 7, resulting in a shortened mRNA strand that yields a truncated SMN protein product.6,37,39 SMA is classified based on age of onset and maximum motor abilities achieved, ranging from the most severe (Type 0) to mildest (Type 4) disease.36,40 Because SMA patients lack functional SMN1 (due to polymorphisms), disease severity is determined by copy numbers of SMN2.6,39
Gene-based therapy for SMA
Three FDA-approved SMN treatments demonstrate clinically meaningful benefit in SMA: SMN2-targeting nusinersen [Spinraza] and risdiplam [Evrysdi], and SMN1-targeting onasemnogene abeparvovec-xioi [Zolgensma]38 Additional approaches to SMA treatment are through SMN-independent therapies, which target muscle and nerve function. Research has strongly suggested that combined SMA therapies, specifically approved SMN-targeted and investigational SMN-independent treatments, such as GYM329 (also known as RO7204239) may be the best strategy to treat all ages, stages, and types of SMA.41 (Table 226-41).
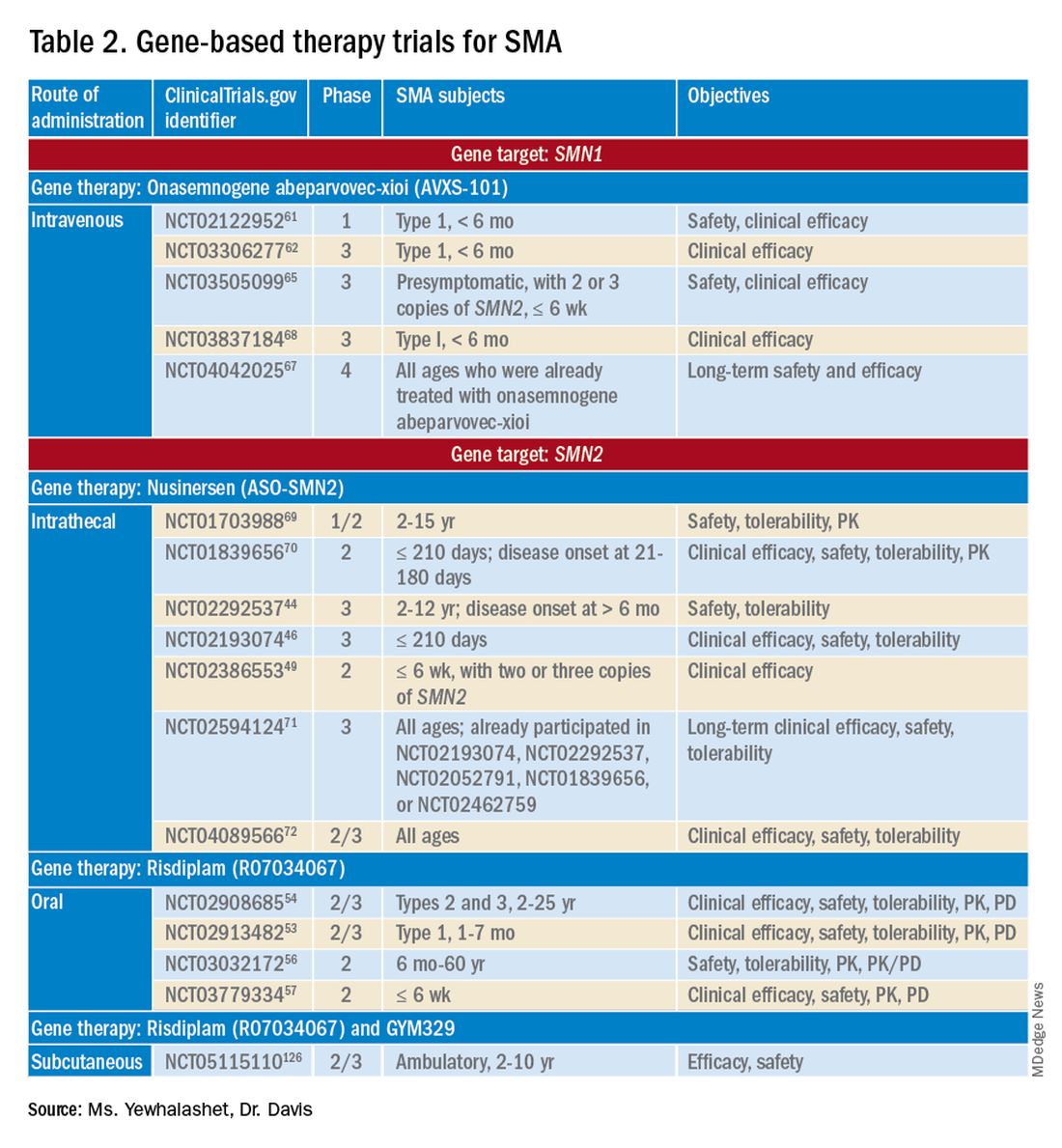
Agents that modulate SMN2. Nusinersen, approved by the FDA in 2016, was the first treatment indicated for all SMA types in pediatric and adult patients.42 The agent is an ASO that targets exon 7 of SMN2, thus stabilizing transcription. Inclusion of exon 7 increases SMN protein production, improving motor function.6,38 Nusinersen is a lifelong treatment that requires IT administration every 4 months because it cannot cross the blood-brain barrier.38,43
Pivotal clinical studies that led to approval of nusinersen include CHERISH (ClinicalTrial.gov Identifier: NCT02292537) and ENDEAR (ClinicalTrial.gov Identifier: NCT02193074) studies.
CHERISH was a phase 3, randomized, double-blind, sham procedure–controlled trial that examined the clinical efficacy and safety of nusinersen in 126 participants with later-onset SMA (2-12 years of age). The primary endpoint was the change from baseline using the Hammersmith Functional Motor Scale Expanded (HFMSE) at 15 months. HFMSE looks at 33 activities to assess improvement in motor function. The study met the primary efficacy outcome, demonstrating statistically significant (P = .0000001) improvement in overall motor function. The nusinersen group showed a 3.9-point increase in the HFMSE score from baseline, which indicates improvement, compared with a 1.0-point decline from baseline in the control group.46,47
ENDEAR was also a randomized, double-blind, sham procedure–controlled phase 3 trial, which investigated the efficacy and safety of nusinersen in 121 participants with early-onset SMA Type 1 (≤ 210 days of age). Coprimary endpoints were:
- Percentage of motor milestones responders, as determined using Section 2 of the Hammersmith Infant Neurological Examination–Part 2.
- Event-free survival (that is, avoidance of combined endpoint of death or permanent ventilation).
ENDEAR met the first primary efficacy outcome, demonstrating statistically significant (P < .0001) improvement in motor milestones (head control, rolling, independent sitting, and standing). By 13 months of age, approximately 51% of nusinersen-treated participants showed improvement, compared with none in the control group.46,47
The second primary endpoint was also met, with a statistically significant (P = .005) 47% decrease in mortality or permanent ventilation use.46-48
The NURTURE (ClinicalTrial.gov Identifier: NCT02386553) study is also investigating the efficacy and safety of nusinersen. An ongoing, open-label, supportive phase 2 trial, NURTURE is evaluating the efficacy and safety of multiple doses of nusinersen in 25 presymptomatic SMA patients (≤ 6 weeks of age). The primary endpoint of this study is time to death or respiratory intervention.49 Interim results demonstrate that 100% of presymptomatic infants are functioning without respiratory intervention after median follow-up of 2.9 years.46-48
Although nusinersen has been shown to be generally safe in clinical studies, development of lumbar puncture–related complications, as well as the need for sedation during IT administration, might affect treatment tolerability in some patients.39
Risdiplam was approved by the FDA in 2020 as the first orally administered small-molecule treatment of SMA (for patients ≤ 2 months of age).52 Risdiplam is a SMN2 splicing modifier, binding to the 5’ splice site of intron 7 and exonic splicing enhancer 2 in exon 7 of SMN2 pre-mRNA. This alternative splicing increases efficiency in SMN2 gene transcription, thus increasing SMN protein production in motor-neuron cells.36 An important advantage of risdiplam is the convenience of oral administration: A large percentage of SMA patients (that is, those with Type 2 disease) have severe scoliosis, which can further complicate therapy or deter patients from using a treatment that is administered through the IT route.40
FDA approval of risdiplam was based on clinical data from two pivotal studies, FIREFISH (ClinicalTrial.gov Identifier: NCT02913482) and SUNFISH (ClinicalTrial.gov Identifier: NCT02908685).53-54
FIREFISH is an open-label, phase 2/3 ongoing trial in infants (1-7 months of age) with SMA Type 1. The study comprises two parts; Part 1 determined the dose of risdiplam used in Part 2, which assessed the efficacy and safety of risdiplam for 24 months. The primary endpoint was the percentage of infants sitting without support for 5 seconds after 12 months of treatment using the gross motor scale of the Bayley Scales of Infant and Toddler Development–Third Edition. A statistically significant (P < .0001) therapeutic benefit was observed in motor milestones. Approximately 29% of infants achieved the motor milestone of independent sitting for 5 seconds, which had not been observed in the natural history of SMA.53-55
SUNFISH is an ongoing randomized, double-blind, placebo-controlled trial of risdiplam in adult and pediatric patients with SMA Types 2 and 3 (2-25 years old). This phase 2/3 study comprises two parts: Part 1 determined the dose (for 12 weeks) to be used for confirmatory Part 2 (for 12 to 24 months). The primary endpoint was the change from baseline on the 32-item Motor Function Measure at 12 months. The study met its primary endpoint, demonstrating statistically significant (P = .0156) improvement in motor function scores, with a 1.36-point increase in the risdiplam group, compared with a 0.19-point decrease in the control group.54,55
Ongoing risdiplam clinical trials also include JEWELFISH (ClinicalTrial.gov Identifier: NCT03032172) and RAINBOW (ClinicalTrial.gov Identifier: NCT03779334).56-57 JEWELFISH is an open-label, phase 2 trial assessing the safety of risdiplam in patients (6 months to 60 years old) who received prior treatment. The study has completed recruitment; results are pending.56 RAINBOW is an ongoing, open-label, single-arm, phase 2 trial, evaluating the clinical efficacy and safety of risdiplam in SMA-presymptomatic newborns (≤ 6 weeks old). The study is open for enrollment.57 Overall, interim results for JEWELFISH and RAINBOW appear promising.
In addition, combined SMA therapies, specifically risdiplam and GYM329 are currently being investigated to address the underlying cause and symptoms of SMA concurrently.58 GYM329, is an investigational anti-myostatin antibody, selectively binding preforms of myostatin - pro-myostatin and latent myostatin, thus improving muscle mass and strength for SMA patients.59 The safety and efficacy of GYM329 in combination with risdiplam is currently being investigated in 180 ambulant participants with SMA (2-10 years of age) in the MANATEE (ClinicalTrial.gov Identifier: NCT05115110) phase 2/3 trial. The MANATEE study is a two-part, seamless, randomized, placebo-controlled, double-blind trial. Part 1 will assess the safety of the combination treatment in approximately 36 participants; participants will receive both GYM329 (every 4 weeks) by subcutaneous (SC) injection into the abdomen and risdiplam (once per day) for 24 weeks followed by a 72-week open-label treatment period. 54,58 The outcome measures include the incidence of AEs, percentage change from baseline in the contractile area of skeletal muscle (in dominant thigh and calf), change from baseline in RHS total score, and incidence of change from baseline in serum concentration (total myostatin, free latent myostatin, and mature myostatin) etc.54 Part 2 will be conducted on 144 participants, specifically assessing the efficacy and safety of the optimal dose of GYM329 selected from Part 1 (combined with risdiplam) for 72 weeks. Once the treatment period is completed in either part, participants can partake in a 2-year open-label extension period.54,58 Other outcome measures include change from baseline in lean muscle mass (assessed by full body dual-energy X- ray absorptiometry (DXA) scan), in time taken to walk/run 10 meters (measured by RHS), in time taken to rise from the floor (measured by RHS), etc.54 Overall, this combination treatment has the potential to further improve SMA patient outcomes and will be further investigated in other patient populations (including non-ambulant patients and a broader age range) in the future.58
An agent that alters SMN1 expression. Onasemnogene abeparvovec-xioi, FDA approved in 2019, was the first gene-replacement therapy indicated for treating SMA in children ≤ 2 years old.60 Treatment utilizes an AAV vector type 9 (AAV9) to deliver a functional copy of SMN1 into target motor-neuron cells, thus increasing SMN protein production and improving motor function. This AAV serotype is ideal because it crosses the blood-brain barrier. Treatment is administered as a one-time IV fusion.38,39,43
FDA approval was based on the STR1VE (ClinicalTrial.gov Identifier: NCT03306277) phase 3 study and START (ClinicalTrial.gov Identifier: NCT02122952) phase 1 study.61,62 START was the first trial to investigate the safety and efficacy of onasemnogene abeparvovec-xioi in SMA Type 1 infants (< 6 months old). Results demonstrated remarkable clinical benefit, including 100% permanent ventilation-free survival and a 92% (11 of 12 patients) rate of improvement in motor function. Improvement in development milestones was also observed: 92% (11 of 12 patients) could sit without support for 5 seconds and 75% (9 of 12) could sit without support for 30 seconds.14,61,63
The efficacy of onasemnogene abeparvovec-xioi seen in STR1VE was consistent with what was observed in START. STRIVE, a phase 3 open-label, single-dose trial, examined treatment efficacy and safety in 22 symptomatic infants (< 6 months old) with SMA Type 1 (one or two SMN2 copies). The primary endpoint was 30 seconds of independent sitting and event-free survival. Patients were followed for as long as 18 months. Treatment showed statistically significant (P < .0001) improvement in motor milestone development and event-free survival, which had not been observed in SMA Type 1 historically. Approximately 59% (13 of 22 patients) could sit independently for 30 seconds at 18 months of age. At 14 months of age, 91% (20 of 22 patients) were alive and achieved independence from ventilatory support.34,35,53
Although many clinical studies suggest that onasemnogene abeparvovec-xioi can slow disease progression, the benefits and risks of long-term effects are still unknown. A 15-year observational study is investigating the long-term therapeutic effects and potential complications of onasemnogene abeparvovec-xioi. Participants in START were invited to enroll in this long-term follow-up study (ClinicalTrial.gov Identifier: NCT04042025).66-67
Duchenne muscular dystrophy
DMD is the most common muscular dystrophy of childhood. With an X-linked pattern of inheritance, DMD is seen mostly in young males (1 in every 3,500 male births).38,39,73 DMD is caused by mutation of the dystrophin encoding gene, or DMD, on the X chromosome. Deletion of one or more exons of DMD prevents production of the dystrophin protein, which leads to muscle degeneration.38,39,43 Common DMD deletion hotspots are exon 51 (20% of cases), exon 53 (13% of cases), exon 44 (11% of cases), and exon 45 (12% of cases).74 Nonsense mutations, which account for another 10% of DMD cases, occur when premature termination codons are found in the DMD gene. Those mutations yield truncated dystrophin protein products.39,66
Therapy for DMD
There are many therapeutic options for DMD, including deflazacort (Emflaza), FDA approved in 2017, which has been shown to reduce inflammation and immune system activity in DMD patients (≥ 5 years old). Deflazacort is a corticosteroid prodrug; its active metabolite acts on the glucocorticoid receptor to exert anti-inflammatory and immunosuppressive effects. Studies have shown that muscle strength scores over 6-12 months and average time to loss of ambulation numerically favored deflazacort over placebo.74,75
Gene-based therapy for DMD
Mutation-specific therapeutic approaches, such as exon skipping and nonsense suppression, have shown promise for the treatment of DMD (Table 358-79):
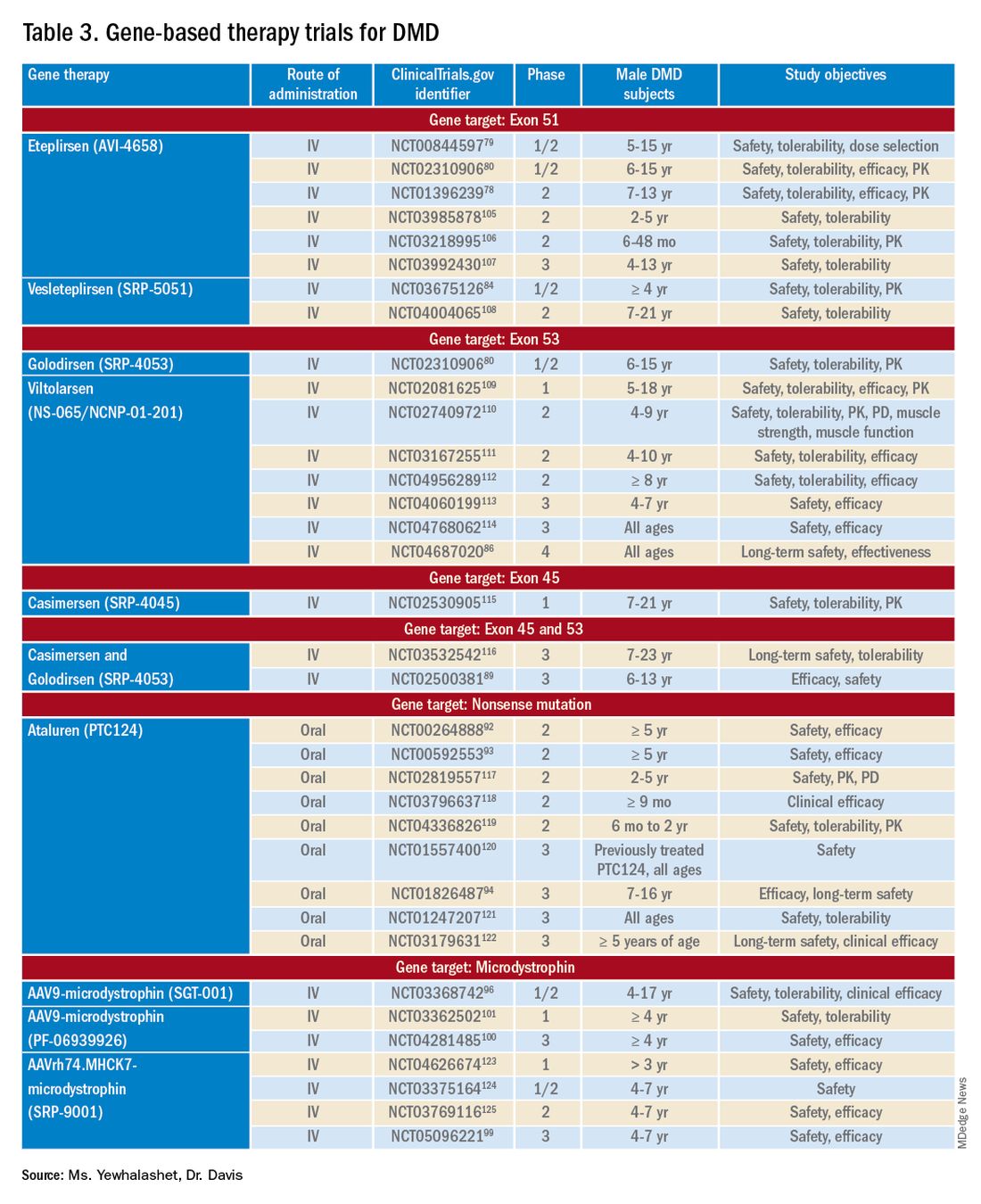
- ASO-mediated exon skipping allows one or more exons to be omitted from the mutated DMD mRNA.74,75 Effective FDA-approved ASOs include golodirsen [Vyondys 53], viltolarsen [Viltepso], and casimersen [Amondys 45].74
- An example of therapeutic suppression of nonsense mutations is ataluren [Translarna], an investigational agent that can promote premature termination codon read-through in DMD patients.66
Another potential treatment approach is through the use of AAV gene transfer to treat DMD. However, because DMD is too large for the AAV vector (packaging size, 5.0 kb), microdystrophin genes (3.5-4 kb, are used as an alternative to fit into a single AAV vector.39,76
Exon skipping targeting exon 51. Eteplirsen, approved in 2016, is indicated for the treatment of DMD patients with the confirmed DMD gene mutation that is amenable to exon 51 skipping. Eteplirsen binds to exon 51 of dystrophin pre-mRNA, causing it to be skipped, thus, restoring the reading frame in patients with DMD gene mutation amenable to exon 51 skipping. This exclusion promotes dystrophin production. Though the dystrophin protein is still functional, it is shortened.38,77 Treatment is administered IV, once a week (over 35-60 minutes). Eteplirsen’s accelerated approval was based on 3 clinical studies (ClinicalTrial.gov Identifier: NCT01396239, NCT01540409, and NCT00844597.) 78-81 The data demonstrated an increased expression of dystrophin in skeletal muscles in some DMD patients treated with eteplirsen. Though the clinical benefit of eteplirsen (including improved motor function) was not established, it was concluded by the FDA that the data were reasonably likely to predict clinical benefit. Continued approval for this indication may depend on the verification of a clinical benefit in confirmatory trials. Ongoing clinical trials include (ClinicalTrial.gov Identifier: NCT03992430 (MIS51ON), NCT03218995, and NCT03218995).77,81,82
Vesleteplirsen, is an investigational agent that is designed for DMD patients who are amendable to exon 51 skip-ping. The mechanism of action of vesleteplirsen appears to be similar to that of eteplirsen.83 The ongoing MOMENTUM (ClinicalTrial.gov Identifier: NCT04004065) phase 2 trial is assessing the safety and tolerability of vesleteplirsen at multiple-ascending dose levels (administered via IV infusion) in 60 participants (7-21 years of age). The study consists of two parts; participants receive escalating dose levels of vesleteplirsen (every 4 weeks) for 72 weeks during part A and participants receive the selected doses from part A (every 4 weeks) for 2 years during part B. Study endpoints include the number of AEs (up to 75 weeks) and the change from baseline to week 28 in dystrophin protein level. 84 Serious AEs of reversible hypomagnesemia were observed in part B, and as a result, the study protocol was amended to include magnesium supplementation and monitoring of magnesium levels.83
Exon skipping targeting exon 53. Golodirsen, FDA approved in 2019, is indicated for the treatment of DMD in patients who have a confirmed DMD mutation that is amenable to exon 53 skipping. The mechanism of action is similar to eteplirsen, however, golodirsen is designed to bind to exon 53.38,39 Treatment is administered by IV infusion over 35-60 minutes.
Approval of golodirsen was based primarily on a two-part, phase 1/2 clinical trial (ClinicalTrial.gov Identifier: NCT02310906). Part 1 was a randomized, placebo-controlled, dose-titration study that assessed multiple-dose efficacy in 12 DMD male patients, 6 to 15 years old, with deletions that were amenable to exon 53 skipping.
Part 2 was an open-label trial in 12 DMD patients from Part 1 of the trial plus 13 newly enrolled male DMD patients who were also amenable to exon 53 skipping and who had not already received treatment. Primary endpoints were change from baseline in total distance walked during the 6-minute walk test at Week 144 and dystrophin protein levels (measured by western blot testing) at Week 48. A statistically significant increase in the mean dystrophin level was observed, from a baseline 0.10% mean dystrophin level to a 1.02% mean dystrophin level after 48 weeks of treatment (P < .001). Common reported adverse events associated with golodirsen were headache, fever, abdominal pain, rash, and dermatitis. Renal toxicity was observed in preclinical studies of golodirsen but not in clinical studies.80,85
Viltolarsen, approved in 2020, is also indicated for the treatment of DMD in patients with deletions amenable to exon 53 skipping. The mechanism of action and administration (IV infusion over 60 minutes) are similar to that of golodirsen.
Approval of viltolarsen was based on two phase 2 clinical trials (ClinicalTrial.gov Identifier: NCT02740972 and NCT03167255) in a total of 32 patients. NCT02740972 was a randomized, double-blind, placebo-controlled, dose-finding study that evaluated the clinical efficacy of viltolarsen in 16 male DMD patients (4-9 years old) for 24 weeks.
NCT03167255 was an open-label study that evaluated the safety and tolerability of viltolarsen in DMD male patients (5-18 years old) for 192 weeks. The efficacy endpoint was the change in dystrophin production from baseline after 24 weeks of treatment. A statistically significant increase in the mean dystrophin level was observed, from a 0.6% mean dystrophin level at baseline to a 5.9% mean dystrophin level at Week 25 (P = .01). The most common adverse events observed were upper respiratory tract infection, cough, fever, and injection-site reaction.86-87
Exon skipping targeting exon 45. Casimersen was approved in 2021 for the treatment of DMD in patients with deletions amenable to exon 45 skipping.88 Treatment is administered by IV infusion over 30-60 minutes. Approval was based on an increase in dystrophin production in skeletal muscle in treated patients. Clinical benefit was reported in interim results from the ESSENCE (ClinicalTrial.gov Identifier: NCT02500381) study, an ongoing double-blind, placebo-controlled phase 3 trial that is evaluating the efficacy of casimersen, compared with placebo, in male participants (6-13 years old) for 48 weeks. Efficacy is based on the change from baseline dystrophin intensity level, determined by immunohistochemistry, at Week 48.
Interim results from ESSENCE show a statistically significant increase in dystrophin production in the casimersen group, from a 0.9% mean dystrophin level at baseline to a 1.7% mean dystrophin level at Week 48 (P = .004); in the control group, a 0.54% mean dystrophin level at baseline increased to a 0.76% mean dystrophin level at Week 48 (P = .09). Common adverse events have included respiratory tract infection, headache, arthralgia, fever, and oropharyngeal pain. Renal toxicity was observed in preclinical data but not in clinical studies.60,84
Targeting nonsense mutations. Ataluren is an investigational, orally administered nonsense mutation suppression therapy (through the read-through of stop codons).37 Early clinical evidence supporting the use of ataluren in DMD was seen in an open-label, dose-ranging, phase 2a study (ClinicalTrial.gov Identifier: NCT00264888) in male DMD patients (≥ 5 years old) caused by nonsense mutation. The study demonstrated a modest (61% ) increase in dystrophin expression in 23 of 38 patients after 28 days of treatment.37,91,92
However, a phase 2b randomized, double-blind, placebo-controlled trial (ClinicalTrial.gov Identifier: NCT00592553) and a subsequent confirmatory ACT DMD phase 3 study (ClinicalTrial.gov Identifier: NCT01826487) did not meet their primary endpoint of improvement in ambulation after 48 weeks as measured by the 6-minute walk test.37,93,94 In ACT DMD, approximately 74% of the ataluren group did not experience disease progression, compared with 56% of the control group (P = 0386), measured by a change in the 6-minute walk test, which assessed ambulatory decline.37,95
Based on limited data showing that ataluren is effective and well tolerated, the European Medicines Agency has given conditional approval for clinical use of the drug in Europe. However, ataluren was rejected by the FDA as a candidate therapy for DMD in the United States.22 Late-stage clinical studies of ataluren are ongoing in the United States.
AAV gene transfer with microdystrophin. Limitations on traditional gene-replacement therapy prompted exploration of gene-editing strategies for treating DMD, including using AAV-based vectors to transfer microdystrophin, an engineered version of DMD, into target muscles.43 The microdystrophin gene is designed to produce a functional, truncated form of dystrophin, thus improving muscular function.
There are 3 ongoing investigational microdystrophin gene therapies that are in clinical development (ClinicalTrial.gov Identifier: NCT03368742 (IGNITE DMD), NCT04281485 (CIFFREO), and NCT05096221 (EMBARK)).38,82
IGNITE DMD is a phase 1/2 randomized, controlled, single-ascending dose trial evaluating the safety and efficacy of a SGT-001, single IV infusion of AAV9 vector containing a microdystrophin construct in DMD patients (4-17 years old) for 12 months. At the conclusion of the trial, treatment and control groups will be followed for 5 years. The primary efficacy endpoint is the change from baseline in microdystrophin protein production in muscle-biopsy material, using western blot testing.96 Long-term interim data on biopsy findings from three patients demonstrated clinical evidence of durable microdystrophin protein expression after 2 years of treatment.96,97
The CIFFREO trial will assess the safety and efficacy of the PF-06939926 microdystrophin gene therapy, an investigational AAV9 containing microdystrophin, in approximately 99 ambulatory DMD patients (4-7 years of age). The study is a randomized, double-blind, placebo-controlled, multicenter phase 3 trial. The primary efficacy end-point is the change from baseline in the North Star Ambulatory Assessment (NSAA), which measures gross motor function. This will be assessed at 52 weeks; all study participants will be followed for a total of 5 years post-treatment.98,99,100 Due to unexpected patient death (in a non-ambulatory cohort) in the phase 1b (in a non-ambulatory cohort) in the phase 1b (ClinicalTrial.gov Identifier: (NCT03362502) trial, microdystrophin gene therapy was immediately placed on clinical hold.101,102 The amended study protocol required that all participants undergo one week of in-hospital observation after receiving treatment.102
The EMBARK study is a global, randomized, double-blind, placebo-controlled, phase 3 trial that is evaluating the safety and efficacy of SRP-9001, which is a rAAVrh74.MHCK7.microdystrophin gene therapy. The AAV vector (rAAVrh74) contains the microdystrophin construct, driven by the skeletal and cardiac muscle–specific promoter, MHCK7.98,99 In the EMBARK study, approximately 120 participants with DMD (4-7 years of age) will be enrolled. The primary efficacy endpoint includes the change from baseline to week 52 in the NSAA total score.99 Based on SRP-9001, data demonstrating consistent statistically significant functional improvements in NSAA total scores and timed function tests (after one-year post- treatment) in DMD patients from previous studies and an integrated analysis from multiple studies (ClinicalTrial.gov Identifier: NCT03375164, NCT03769116, and NCT04626674), the ongoing EMBARK has great promise.103,104
Challenges ahead, but advancements realized
Novel gene-based therapies show significant potential for transforming the treatment of NMDs. The complex pathologies of NMDs have been a huge challenge to disease management in an area once considered unremediable by gene-based therapy. However, advancements in precision medicine – specifically, gene-delivery systems (for example, AAV9 and AAVrh74 vectors) combined with gene modification strategies (ASOs and AAV-mediated silencing) – have the potential to, first, revolutionize standards of care for sporadic and inherited NMDs and, second, significantly reduce disease burden.6
What will be determined to be the “best” therapeutic approach will, likely, vary from NMD to NMD; further investigation is required to determine which agents offer optimal clinical efficacy and safety profiles.43 Furthermore, the key to therapeutic success will continue to be early detection and diagnosis – first, by better understanding disease pathology and drug targets and, second, by validation of reliable biomarkers that are predictive of therapeutic benefit.4,5
To sum up, development challenges remain, but therapeutic approaches to ALS, SMA, and DMD that utilize novel gene-delivery and gene-manipulation tools show great promise.
Ms. Yewhalashet is a student in the masters of business and science program, with a concentration in healthcare economics, at Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences, Claremont, Calif. Dr. Davis is professor of practice in clinical and regulatory affairs, Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences.
References
1. Aitken M et al. Understanding neuromuscular disease care. IQVIA [Internet]. Oct 30, 2018. Accessed Mar 1, 2022. https://www.iqvia.com/insights/the-iqvia-institute/reports/understanding-neuromuscular-disease-care.
2. National Institute of Neurological Disorders and Stroke. Neurological diagnostic tests and procedures fact sheet. Updated Nov 15, 2021. Ac-cessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Neurological-Diagnostic-Tests-and-Procedures-Fact.
3. Deenen JCW et al. The epidemiology of neuromuscular disorders: A comprehensive overview of the literature. J Neuromuscul Dis. 2015;2(1):73-85.
4. Cavazzoni P. The path forward: Advancing treatments and cures for neurodegenerative diseases. U.S. Food and Drug Administration. Jul 29, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/congressional-testimony/path-forward-advancing-treatments-and-cures-neurodegenerative-diseases-07292021.
5. Martier R, Konstantinova P. Gene therapy for neurodegenerative diseases: Slowing down the ticking clock. Front Neurosci. 2020 Sep 18;14:580179. doi: 10.3389/fnins.2020.580179.
6. Sun J, Roy S. Gene-based therapies for neurodegenerative diseases. Nat Neurosci. 2021 Mar;24(3):297-311. doi:10.1038/s41593-020-00778-1.
7. Amado DA, Davidson BL. Gene therapy for ALS: A review. Mol Ther. 2021 Dec 1;29(12):3345-58. doi:10.1016/j.ymthe.2021.04.008.
8. Yun Y, Ha Y. CRISPR/Cas9-mediated gene correction to understand ALS. Int J Mol Sci. 2020;21(11):3801. doi:10.3390/ijms21113801.
9. National Institute of Neurological Disorders and Stroke. Amyotrophic lateral sclerosis (ALS) fact sheet. Updated Nov 15, 2021. Accessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Amyotrophic-Lateral-Sclerosis-ALS-Fact-Sheet.
10. Cappella M et al. Gene therapy for ALS – A perspective. Int J Mol Sci. 2019;20(18):4388. doi:10.3390/ijms20184388.
11. Abramzon YA, Fratta P, Traynor BJ, Chia R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front Neurosci. 2020;14. Accessed August 18, 2022. https://www.frontiersin.org/articles/10.3389/fnins.2020.00042
12. Giannini M, Bayona-Feliu A, Sproviero D, Barroso SI, Cereda C, Aguilera A. TDP-43 mutations link Amyotrophic Lateral Sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLOS Genet. 2020;16(12):e1009260. doi:10.1371/journal.pgen.1009260
13. FDA-approved drugs for treating ALS. The ALS Association [Internet]. Accessed Mar 1, 2022. http://www.als.org/navigating-als/living-with-als/fda-approved-drugs.
14. Jensen TL et al. Current and future prospects for gene therapy for rare genetic diseases affecting the brain and spinal cord. Front Mol Neurosci. 2021 Oct 6;14:695937. doi:10.3389/fnmol.2021.695937.
15. ALS Gene Targeted Therapies. The ALS Association. Accessed August 22, 2022. https://www.als.org/understanding-als/who-gets-als/genetic-testing/als-gene-targeted-therapies
16. Tofersen for ALS clears phase 1/2 trial, now in phase 3. Advances in Motion. Massachusetts General Hospital [Internet]. Sep 30, 2020. Accessed Mar 1, 2022. https://advances.massgeneral.org/neuro/journal.aspx?id=1699.17. Biogen. A study to evaluate the efficacy, safety, tol-erability, pharmacokinetics, and pharmacodynamics of BIIB067 administered to adult subjects with amyotrophic lateral sclerosis and confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT02623699. Updated Jul 25, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT02623699.
18. Biogen. Biogen announces topline results from the tofersen phase 3 study and its open-label Extension in SOD1-ALS. Press release. Oct 17, 2021. Accessed Mar 1, 2022. https://investors.biogen.com/news-releases/news-release-details/biogen-announces-topline-results-tofersen-phase-3-study-and-its.
19. Biogen. An extension study to assess the long-term safety, tolerability, pharmacokinetics, and effect on disease progression of BIIB067 ad-ministered to previously treated adults with amyotrophic lateral sclerosis caused by superoxide dismutase 1 mutation. ClinicalTrials.gov Identi-fier: NCT03070119. Updated Sep 10, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT03070119.
20. MS MW. #AANAM – ATLAS Trial to Assess Tofersen in Presymptomatic SOD1 ALS. Accessed February 19, 2022. https://alsnewstoday.com/news-posts/2021/04/23/aanam-atlas-clinical-trial- tofersen-presymptomatic-sod1-als-patients/
21.Biogen. A phase 3 randomized, placebo-controlled trial with a longitudinal natural history run-in and open-label extension to evaluate BIIB067 initiated in clinically presymptomatic adults with a confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT04856982. Updated Feb 18, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04856982.
22. Latozinemab | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/latozinemab
23. Alector Presents AL001 (latozinemab) Data from the FTD-C9orf72 Cohort of the INFRONT-2 Phase 2 Clinical Trial | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releas-es/news-release-details/alector-presents-al001-latozinemab-data-ftd-c9orf72-cohort/
24. Alector Announces First Participant Dosed in Phase 2 Study Evaluating AL001 in Amyotrophic Lateral Sclerosis (ALS) | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releases/news-release-details/lector-announces-first-participant-dosed-phase-2-study-0/ 25. A Phase 2 Study to Evaluate AL001 in C9orf72-Associated ALS - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT05053035
26.TPN-101 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/tpn- 101
27. Transposon Therapeutics, Inc. A Phase 2a Study of TPN-101 in Patients With Amyotrophic Lateral Sclerosis (ALS) and/or Frontotemporal Dementia (FTD) Associated With Hexanucleotide Repeat Expansion in the C9orf72 Gene (C9ORF72 ALS/FTD). clinicaltrials.gov; 2022. Ac-cessed August 17, 2022. https://clinicaltrials.gov/ct2/show/NCT04993755
28. Kerk SY, Bai Y, Smith J, et al. Homozygous ALS-linked FUS P525L mutations cell- autonomously perturb transcriptome profile and chem-oreceptor signaling in human iPSC microglia. Stem Cell Rep. 2022;17(3):678-692. doi:10.1016/j.stemcr.2022.01.004
29. ION363 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/ion363 30. Ionis Pharmaceuticals, Inc. A Phase 1-3 Study to Evaluate the Efficacy, Safety, Pharmacokinetics and Pharmacodynamics of Intrathecally Administered ION363 in Amyo-trophic Lateral Sclerosis Patients With Fused in Sarcoma Mutations (FUS-ALS). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04768972
31. PhD LF. Engensis (VM202) - ALS News Today. Accessed August 19, 2022. https://alsnewstoday.com/vm202/
32. Helixmith Co., Ltd. A 6-Month Extension Study Following Protocol VMALS-002-2 (A Phase 2a, Double-Blind, Randomized, Place-bo-Controlled, Multicenter Study to Assess the Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05176093 33. Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT04632225
34. Biogen. A phase 1, safety, tolerability, and distribution study of a microdose of radiolabeled BIIB067 co-administered with BIIB067 to healthy adults. ClinicalTrials.gov Identifier: NCT03764488. Updated Jul 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03764488.
35. Ionis Pharmaceuticals Inc. A phase 1, double-blind, placebo-controlled, dose-escalation study of the safety, tolerability, and pharmacokinet-ics of ISIS 333611 administered intrathecally to patients with familial amyotrophic lateral sclerosis due to superoxide dismutase 1 gene muta-tions. ClinicalTrials.gov Identifier: NCT01041222. Updated Apr 13, 2012. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01041222.
36. Messina S, Sframeli M. New treatments in spinal muscular atrophy: Positive results and new challenges. J Clin Med. 2020;9(7):2222. doi:10.3390/jcm9072222.
37. Scoto M et al. Genetic therapies for inherited neuromuscular disorders. Lancet Child Adolesc Health. 2018 Aug;2(8):600-9. doi:10.1016/S2352-4642(18)30140-8.
38. Abreu NJ, Waldrop MA. Overview of gene therapy in spinal muscular atrophy and Duchenne muscular dystrophy. Pediatr Pulmonol. 2021 Apr;56(4):710-20. doi:10.1002/ppul.25055.
39. Brandsema J, Cappa R. Genetically targeted therapies for inherited neuromuscular disorders. Practical Neurology [Internet]. Jul/Aug 2021:69-73. Accessed Mar 1, 2022. https://practicalneurology.com/articles/2021-july-aug/genetically-targeted-therapies-for-inherited-neuromuscular-disorders/pdf.
40. Ojala KS et al. In search of a cure: The development of therapeutics to alter the progression of spinal muscular atrophy. Brain Sci. 2021;11(2):194. doi:10.3390/brainsci11020194.
41. McCall S. Cure SMA Releases Updated Drug Pipeline. Cure SMA. Published December 13, 2021. Accessed August 21, 2022. https://www.curesma.org/cure-sma-releases-updated-drug-pipeline- 2021/ 42. FDA approves first drug for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Dec 23, 2016. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy.43. Kirschner J. Postnatal gene therapy for neuromuscular diseases – Opportunities and limitations. J Perinat Med. 2021 Sep;49(8):1011-5. doi:10.1515/jpm-2021-0435.
43. Terryn J, Verfaillie CM, Van Damme P. Tweaking Progranulin Expression: Therapeutic Avenues and Opportunities. Front Mol Neurosci. 2021;14. Accessed September 4, 2022. https://www.frontiersin.org/articles/10.3389/fnmol.2021.71303144.
44. Biogen. A phase 3, randomized, double-blind, sham-procedure controlled study to assess the clinical efficacy and safety of ISIS 396443 administered intrathecally in patients with later-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02292537. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/study/NCT02292537.
45. Why Spinraza/later-onset studies. SPINRAZA® (nusinersen) [Internet]. Accessed Mar 1, 2022. www.spinraza.com/en_us/home/why-spinraza/later-onset-studies.html#scroll-tabs.
46. Biogen. A Phase 3, Randomized, Double-Blind, Sham-Procedure Controlled Study to Assess the Clinical Efficacy and Safety of ISIS 396443 Administered Intrathecally in Patients With Infantile- Onset Spinal Muscular Atrophy. clinicaltrials.gov; 2021. Accessed February 10, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02193074
47. Early-onset SMA (Type 1) | SPINRAZA® (nusinersen). Accessed Mar 1, 2022. https://www.spinraza-hcp.com/en_us/home/why-spinraza/about-spinraza.html.
48. Finkel RS et al; ENDEAR Study Group. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723-32. doi: 10.1056/NEJMoa1702752.
49. Biogen. An open-label study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to subjects with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02386553. Updated Nov 18, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02386553.
50. De Vivo DC et al; NURTURE Study Group. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: In-terim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul Disord. 2019 Nov;29(11):842-56. doi:10.1016/j.nmd.2019.09.007.
51. Why Spinraza/presymptomatic study. SPINRAZA® (nusinersen) [Internet]. Accessed Feb 22, 2022. www.spinraza.com/en_us/home/why-spinraza/presymptomatic-study.html#scroll-tabs.
52. FDA approves oral treatment for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Aug 7, 2020. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy.
53. Hoffmann-La Roche. A two-part seamless, open-label, multicenter study to investigate the safety, tolerability, pharmacokinetics, pharmaco-dynamics and efficacy of risdiplam (RO7034067) in infants with type 1 spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02913482. Updated Jan 21, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02913482.
54. Hoffmann-La Roche. A two-part seamless, multi-center randomized, placebo-controlled, double-blind study to investigate the safety, tolera-bility, pharmacokinetics, pharmacodynamics and efficacy of risdiplam (RO7034067) in type 2 and 3 spinal muscular atrophy patients. Clinical-Trials.gov Identifier: NCT02908685. Updated Dec 28, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02908685.
55. Genentech. Genentech’s risdiplam shows significant improvement in survival and motor milestones in infants with type 1 spinal muscular atrophy (SMA). Press release. Apr 27, 2020. Accessed Mar 1, 2022. http://www.gene.com/media/press-releases/14847/2020-04-27/genentechs-risdiplam-shows-significant-i
56. Hoffmann-La Roche. An open-label study to investigate the safety, tolerability, and pharmacokinetics/pharmacodynamics of risdiplam (RO7034067) in adult and pediatric patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03032172. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03032172.
57. Hoffmann-La Roche. An open-label study of risdiplam in infants with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03779334. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03779334.
58. McCall S. Update on Genentech/Roche Initiation of MANATEE Clinical Study. Cure SMA. Published October 20, 2021. Accessed August 20, 2022. https://www.curesma.org/update-on- genentech-roche-initiation-of-manatee-clinical-study/
59. Abati E, Manini A, Comi GP, Corti S. Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases. Cell Mol Life Sci. 2022;79(7):374. doi:10.1007/s00018-022-04408-w
60. FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality. U.S. Food and Drug Administration. News release. May 24, 2019. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease.
61. Novartis Gene Therapies. Phase I gene transfer clinical trial for spinal muscular atrophy type 1 delivering AVXS-101. ClinicalTrials.gov Identifier: NCT02122952. Updated Jun 14, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02122952.
62. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT03306277. Updated Jun 14, 2021. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT03306277.
63. Mendell JR et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713-22. doi:10.1056/NEJMoa1706198.
64. Symptomatic study results. ZOLGENSMA [Internet]. Updated Nov 2021. Accessed Mar 1, 2022. Error! Hyperlink reference not valid..
65. Novartis Gene Therapies. A global study of a single, one-time dose of AVXS-101 delivered to infants with genetically diagnosed and pre-symptomatic spinal muscular atrophy with multiple copies of SMN2. ClinicalTrials.gov Identifier: NCT03505099. Updated Jan 1, 2022. Ac-cessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03505099.
66. Chiu W et al. Current genetics and potential gene-targeting therapeutics for neuromuscular diseases. Int J Mol Sci. 2020 Dec;21(24):9589. doi:10.3390/ijms21249589.
67. Novartis Gene Therapies. A long-term follow-up study of patients in the clinical trials for spinal muscular atrophy receiving AVXS-101. Clini-calTrials.gov Identifier: NCT04042025. Updated Jun 9, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04042025.
68. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT0383718. Up-dated Jan 11, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03837184.
69. Biogen. An open-label, dose escalation study to assess the safety, tolerability and dose-range finding of multiple doses of ISIS 396443 de-livered intrathecally to patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01703988. Updated Apr 13, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01703988.
70. Biogen. A study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to patients with infantile-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01839656. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01839656.
71. Biogen. An open-label extension study for patients with spinal muscular atrophy who previously participated in investigational studies of ISIS 396443. ClinicalTrials.gov Identifier: NCT02594124. Updated Nov 15, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02594124.
72. Biogen. Escalating dose and randomized, controlled study of nusinersen (BIIB058) in participants with spinal muscular atrophy. ClinicalTri-als.gov Identifier: NCT04089566. Updated Feb 24, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04089566.
73. National Center for Advancing Translational Sciences. Duchenne muscular dystrophy. Genetic and Rare Diseases Information Center. Up-dated Nov 2, 2020. Accessed Mar 1, 2022. https://rarediseases.info.nih.gov/diseases/6291/duchenne-muscular-dystrophy.
74. Matsuo M. Antisense oligonucleotide-mediated exon-skipping therapies: Precision medicine spreading from Duchenne muscular dystrophy. JMA J. 2021 Jul 15;4(3):232-40. doi:10.31662/jmaj.2021-0019.
75. FDA approves drug to treat Duchenne muscular dystrophy. U.S. Food and Drug Administration. News release. Feb 9, 2017. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-drug-treat-duchenne-muscular-dystrophy.74.
76. Duan D. Dystrophin gene replacement and gene repair therapy for Duchenne muscular dystrophy in 2016: An interview. Hum Gene Ther Clin Dev. 2016 Mar;27(1):9-18. doi:10.1089/humc.2016.001.
77. EXONDYS 51®. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/drug-development-pipeline/exondys-51/
78. Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Placebo-Controlled, Multiple Dose Efficacy, Safety, Tolerability and Pharmacoki-netics Study of AVI-4658(Eteplirsen),in the Treatment of Ambulant Subjects With Duchenne Muscular Dystrophy. clinicaltrials.gov; 2020. Ac-cessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT01396239
79. Sarepta Therapeutics, Inc. Clinical Study to Assess the Safety Fo AVI-4658 in Subjects With Duchenne Muscular Dystrophy Due to a Frame-Shift Mutation Amenable to Correction by Skipping Exon 51. clinicaltrials.gov; 2015. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/study/NCT00844597
80. Sarepta Therapeutics, Inc. A 2-part, randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study (Part 1) followed by an open-label efficacy and safety evaluation (Part 2) of SRP-4053 in patients with Duchenne muscular dystrophy amenable to exon 53 skipping. ClinicalTrials.gov Identifier: NCT02310906. Updated Oct 19, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02310906.
81. Commissioner O of the. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. FDA. Published March 24, 2020. Accessed August 21, 2022. hDuchenne Muscular Dystrophy Amenable to Exon 51-Skipping Treatment. clinicaltrials.gov; 2022. Accessed Au-gust 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04004065
109. National Center of Neurology and Psychiatry, Japan. Exploratory study of NS-065/NCNP-01 in Duchenne muscular dystrophy. ClinicalTri-als.gov Identifier: NCT02081625; Updated Feb 26, 2020. Accessed Mar 2, 2022. https://clinicaltrialsttps://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-drug-duchenne-muscular- dys-trophy
82. Duchenne Drug Development Pipeline. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/duchenne-drug-development-pipeline/
83. Sarepta Therapeutics Provides Update on SRP-5051 for the Treatment of Duchenne Muscular Dystrophy | Sarepta Therapeutics, Inc. Ac-cessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics- pro-vides-update-srp-5051-treatment-duchenne
84. Sarepta Therapeutics, Inc. An Open-Label Extension Study for Patients With Duchenne Muscular Dystrophy Who Participated in Studies of SRP-5051. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03675126
85. VYONDYS 53. Prescribing information. Sarepta Therapeutics Inc.; 2019. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211970s000lbl.pdf.
86. NS Pharma Inc. Long-term use of viltolarsen in boys with Duchenne muscular dystrophy in clinical practice (VILT-502). ClinicalTrials.gov Identifier: NCT04687020. Updated Nov 22, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04687020.
87. VILTEPSO. Prescribing information. NS Pharma; 2020. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212154s000lbl.pdf.
88. FDA approves targeted treatment for rare Duchenne muscular dystrophy mutation. U.S. Food and Drug Administration. News release. Feb 25, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation-0.
89. Sarepta Therapeutics Inc. A double-blind, placebo-controlled, multi-center study with an open-label extension to evaluate the efficacy and safety of SRP-4045 and SRP-4053 in patients with Duchenne muscular dystrophy. Clinicaltrials.gov Identifier: NCT02500381. Updated Aug 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02500381.
90. AMONDYS 45. Prescribing information. Sarepta Therapeutics Inc.; 2021. Accessed Feb 22, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213026lbl.pdf.
91. Finkel RS et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dys-trophy. PLoS ONE. 2013;8(12):e81302. doi:10.1371/journal.pone.0081302.
92. PTC Therapeutics. A phase 2 study of PTC124 as an oral treatment for nonsense-mutation-mediated Duchenne muscular dystrophy. Clini-calTrials.gov Identifier: NCT00264888. Updated Jan 14, 2009. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00264888.
93. PTC Therapeutics. A phase 2B efficacy and safety study of PTC124 in subjects with nonsense-mutation-mediated Duchenne and Becker muscular dystrophy. ClinicalTrials.gov Identifier: NCT00592553. Updated Apr 7, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00592553.
94. PTC Therapeutics. A phase 3 efficacy and safety study of ataluren in patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01826487. Updated Aug 4, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01826487.
95. Bushby K et al; PTC124-GD-007-DMD Study Group. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014 Oct;50(4):477-87. doi:10.1002/mus.24332.
96. Solid Biosciences LLC. A randomized, controlled, open-label, single-ascending dose, phase I/II study to investigate the safety and tolerabil-ity, and efficacy of intravenous SGT-001 in male adolescents and children with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03368742. Updated Aug 24, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03368742.
97. Solid Biosciences reports 1.5-year data from patients in the ongoing IGNITE DMD phase I/II clinical trial of SGT-001. Press release. Solid Biosciences. Sep 27, 2021. Accessed Mar 2, 2022. http://www.solidbio.com/about/media/press-releases/solid-biosciences-reports-1-5-year-data-from-patients-in-the-ongoing-ignite-dmd-phase-i-ii-clinical-trial-of-sgt-001.
98. Potter RA et al. Dose-escalation study of systemically delivered rAAVrh74.MHCK7.microdystrophin in the mdx mouse model of Duchenne muscular dystrophy. Hum Gene Ther. 2021 Apr;32(7-8):375-89. doi:10.1089/hum.2019.255.
99. Sarepta Therapeutics, Inc. A Phase 3 Multinational, Randomized, Double-Blind, Placebo- Controlled Systemic Gene Delivery Study to Evaluate the Safety and Efficacy of SRP-9001 in Patients With Duchenne Muscular Dystrophy (EMBARK). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05096221
100. Pfizer. A PHASE 3, MULTICENTER, RANDOMIZED, DOUBLE-BLIND, PLACEBO CONTROLLED STUDY TO EVALUATE THE SAFETY AND EFFICACY OF PF 06939926 FOR THE TREATMENT OF DUCHENNE MUSCULAR DYSTROPHY. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04281485
101. Pfizer. A phase 1B multicenter open-label, single ascending dose study to evaluate the safety and tolerability of PF-06939926 in ambula-tory and non-ambulatory subjects with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03362502. Updated Mar 2, 2022. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03362502.
102. MS MW. Phase 3 CIFFREO DMD Gene Therapy Trial Slated to Begin in June in US. Accessed August 21, 2022. https://musculardystrophynews.com/news/phase-3-trial-of-pfizers-gene-therapy- expected-to-open-in-us-in-june/
103. SRP-9001. Parent Project Muscular Dystrophy. Accessed August 22, 2022. https://www.parentprojectmd.org/drug-development-pipeline/srp-9001-micro-dystrophin-gene- transfer/
104. Sarepta Therapeutics’ Investigational Gene Therapy SRP-9001 for Duchenne Muscular Dystrophy Demonstrates Significant Functional Improvements Across Multiple Studies | Sarepta Therapeutics, Inc. Accessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release- details/sarepta-therapeutics-investigational-gene-therapy-srp-9001
105. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Efficacy Study of Eteplirsen in Patients With Duchenne Muscular Dys-trophy Who Have Completed Study 4658-102.clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03985878
106. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Pharmacokinetics Study of Eteplirsen in Young Patients With Duchenne Mus-cular Dystrophy Amenable to Exon 51 Skipping. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03218995
107.Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Dose Finding and Comparison Study of the Safety and Efficacy of a High Dose of Eteplirsen, Preceded by an Open-Label Dose Escalation, in Patients With Duchenne Muscular Dystrophy With Deletion Mutations Amenable to Exon 51 Skipping. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03992430
108. Sarepta Therapeutics, Inc. A Phase 2, Two-Part, Multiple-Ascending-Dose Study of SRP-5051 for Dose Determination, Then Dose Ex-pansion, in Patients With .gov/ct2/show/NCT02081625.
110. NS Pharma Inc. A phase II, dose finding study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT02740972. Updated Dec 7, 2021. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02740972.
111. NS Pharma Inc. A phase II, open-label, extension study to assess the safety and efficacy of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT03167255. Updated Nov 24, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03167255.
112. NS Pharma Inc. A phase 2 open label study to assess the safety, tolerability, and efficacy of viltolarsen in ambulant and non-ambulant boys with Duchenne muscular dystrophy (DMD) compared with natural history controls. ClinicalTrials.gov Identifier: NCT04956289. Updated Feb 1, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04956289.
113. NS Pharma Inc. A phase 3 randomized, double-blind, placebo-controlled, multi-center study to assess the efficacy and safety of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04060199. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04060199.
114. NS Pharma Inc. A phase 3, multi-center, open-label extension study to assess the safety and efficacy of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04768062. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04768062.
115. Sarepta Therapeutics Inc. A randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study followed by an open-label safety and efficacy evaluation of SRP-4045 in advanced-stage patients with Duchenne muscular dystrophy amena-ble to exon 45 skipping. ClinicalTrials.gov Identifier: NCT02530905. Updated May 17, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02530905.
116. Sarepta Therapeutics Inc. Long-term, open-label extension study for patients with Duchenne muscular dystrophy enrolled in clinical trials evaluating casimersen or golodirsen. ClinicalTrials.gov Identifier: NCT03532542. Updated Dec 20, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03532542.
117. PTC Therapeutics. A phase 2 study of the safety, pharmacokinetics, and pharmacodynamics of ataluren (PTC124®) in patients aged ≥2 to <5 years old with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT02819557. Updated Aug 28, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02819557.
118. PTC Therapeutics. Phase 2, non-interventional, clinical study to assess dystrophin levels in subjects with nonsense mutation Duchenne muscular dystrophy who have been treated with ataluren for ≥ 9 months. ClinicalTrials.gov Identifier: NCT03796637. Updated Apr 10, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03796637.
119. PTC Therapeutics. An Open-Label Study Evaluating the Safety and Pharmacokinetics of Ataluren in Children From ≥6 Months to <2 Years of Age With Nonsense Mutation Duchenne Muscular Dystrophy. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04336826 120. PTC Therapeutics. An open-label study for previously treated ataluren (PTC124®) pa-tients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01557400. Updated Nov 25, 2020. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT01557400.
121. PTC Therapeutics. An open-label, safety study for ataluren (PTC124) patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01247207. Updated Feb 16, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT01247207.
122. PTC Therapeutics. A phase 3, randomized, double-blind, placebo-controlled efficacy and safety study of ataluren in patients with non-sense mutation Duchenne muscular dystrophy and open-label extension. ClinicalTrials.gov Identifier: NCT03179631. Updated Feb 8, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03179631.
123. Sarepta Therapeutics, Inc. An Open-Label, Systemic Gene Delivery Study Using Commercial Process Material to Evaluate the Safety of and Expression From SRP-9001 in Subjects With Duchenne Muscular Dystrophy (ENDEAVOR). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04626674
124. Sarepta Therapeutics, Inc. Systemic Gene Delivery Phase I/IIa Clinical Trial for Duchenne Muscular Dystrophy Using RAA-Vrh74.MHCK7.Micro-Dystrophin (MicroDys-IV-001). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03375164
125. Sarepta Therapeutics Inc. A multicenter, randomized, double-blind, placebo-controlled trial for Duchenne muscular dystrophy using SRP-9001. ClinicalTrials.gov Identifier: NCT03769116. Updated Dec 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03769116.
126. Hoffmann-La Roche. A Two-Part, Seamless, Multi-Center, Randomized, Placebo-Controlled, Double-Blind Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of RO7204239 in Combination With Risdiplam (RO7034067) in Ambulant Pa-tients With Spinal Muscular Atrophy. clinicaltrials.gov; 2022. Accessed September 1, 2022. https://clinicaltrials.gov/ct2/show/NCT05115110
Neuromuscular diseases (NMDs) are a broad classification of heterogeneous groups of disorders characterized by progressive muscle weakness resulting from muscle or nerve dysfunction.1 Diagnosis is based on symptoms and a full medical history, as well as on muscle and imaging tests (including electromyography, nerve-conduction studies, magnetic resonance imaging, muscle biopsy, and blood tests) to confirm or rule out specific NMDs.2 Early diagnosis of NMDs can be difficult because symptoms overlap with those of many other diseases.
Although individually, NMDs are rare, collectively, they affect approximately 250,000 people in the United States. Disease types vary in regard to cause, symptoms, prevalence, age of onset, progression, and severity. Functional impairment from any NMD can lead to lifelong morbidities and shortened life expectancy.1,3
Treatment options for NMDs are limited; most target symptoms, not disease progression. Although there is a need for safe and effective gene-based therapies for NMDs, there are challenges to developing and delivering such treatments that have impeded clinical success. These include a lack of understanding about disease pathology and drug targets, limited animal model systems, and few reliable biomarkers that are predictive of therapeutic success.4,5
Notwithstanding that challenges remain, our understanding of gene expression in NMDs has greatly advanced in the past few decades. This progress has translated into promising results in the gene-therapy field – thereby setting the stage for therapeutic approaches that use novel gene-delivery and gene-manipulation tools.6 These novel approaches include nonviral strategies, such as antisense oligonucleotides (ASOs), and viral-based strategies, such as adeno-associated virus (AAV)-mediated gene silencing and AAV-mediated gene delivery.
In this article, we highlight advancements in the clinical development of gene-based therapies for NMDs. We focus on amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), and Duchenne muscular dystrophy (DMD) because of recent clinical successes in developing such therapies.1,6,7 We also catalog completed and ongoing clinical trials for ALS, SMA, and DMD (Tables 1-3).
Amyotrophic lateral sclerosis
ALS is caused by progressive degeneration of upper- and lower-motor neurons, which eventually leads to respiratory failure and death 3 to 5 years after disease onset.7-9 There are two subtypes: Familial ALS (10% of cases) and sporadic ALS (90% of cases). Commonly mutated ALS-associated genes6,8 are:
- Superoxide dismutase type 1 (SOD1).
- Chromosome 9 open reading frame 72 (C9orf72).
- Transactive response DNA-binding protein 43 (TARDBP).
- Fused in sarcoma (FUS).
SOD1-targeted therapy is being studied, with early evidence of clinical success. Mutations in SOD1 account for 10% to 20% of familial ALS cases and 1% to 2% of sporadic ALS cases.6,10 10 Mutations in C9orf72 account for 25 to 40% of familial ALS cases and 7% of sporadic ALS cases.8,9,11 Mutations in TARDBP account for 3% of familial ALS cases and 2% of sporadic cases.12 Mutations in FUS account for 4% of familial ALS cases and 1% of sporadic cases. Overall, these mutant proteins can trigger neurotoxicity, thus inducing motor-neuron death.6,10
Treatment of ALS
Two treatments for ALS are Food and Drug Administration approved: riluzole (Rilutek), approved in 1995, and edaravone (Radicava), approved in 2017.
Riluzole is an oral anti-excitotoxic glutamate antagonist.11 Approval of riluzole was based on the results of two studies that demonstrated a 2- to 3-month survival benefit.10,14 For patients who have difficulty swallowing, an oral suspension (Tiglutik, approved in 2018) and an oral film (Exservan, approved in 2019) are available.
Edaravone is a free-radical scavenger that decreases oxidative stress and is administered intravenously (IV).9,13,14 Findings from clinical trials suggest functional improvement or slower decline in function for some patients.
Although these two agents demonstrate modest therapeutic benefit, neither reverses progression of disease.10,14
Gene-based therapy for ALS
Many non-viral strategies, including antisense oligonucleotide (ASO), monoclonal antibodies, reverse transcriptase inhibitors, and HGF gene replacement therapy are used as therapeutic approaches to SOD1, C9orf72, and FUS gene mutations in ALS patients, and are being evaluated in clinical studies14,15 (Table 113-17).
Tofersen, also known as BIIB067, is an investigational ASO, administered by intrathecal (IT) injection, that binds to SOD1 mRNA, thus reducing its protein levels.16 Tofersen was evaluated in the VALOR phase 3 study (ClinicalTrials.gov Identifier: NCT02623699), a three-part randomized, double-blind, placebo-controlled trial: single ascending dose (Part A), multiple ascending dose (B), and fixed dose (C).10 In Parts A and B, 48 participants received five IT injections of tofersen or placebo over 12 weeks and were followed for an additional 12 weeks. Reduction in SOD1 protein production and neurofilament level in cerebrospinal fluid (CSF) (a potential biomarker of motor-neuron degeneration) was observed, which determined the fixed-dose for Part C.16,17
Part C examined the efficacy, safety and tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of tofersen, compared with placebo, in adults with ALS who had a confirmed SOD1 mutation.17 A total of 108 participants were enrolled; 60 were identified as “faster-progressing”; 48, as “slower-progressing.”18 The primary endpoint of Part C was change from baseline to Week 28 on the Revised ALS Functional Rating Scale (ALSFRS-R) total score. (ALSFRS-R measures overall clinical effect; the score ranges from 0 [no function] to 4 [full function].17)
Tofersen failed to meet the primary efficacy outcome because statistically significant findings were lacking in the faster-progressing population, as measured by joint-rank analysis (difference of 1.2 on the ALSFRS-R score; P = .97). However, trends favoring tofersen were observed across key secondary clinical outcome measures18:
- Change from baseline in CSF SOD1 protein concentration.17 Percent reduction in the total SOD1 protein level was much higher in the tofersen-treated group than in the control group (38% more than controls in the faster-progressing population; 26% more than controls in the slower-progressing population).18
- Change from baseline in neurofilament light-chain concentration in plasma.17,18 Percent reduction in the level of neurofilament light chain was also observed to be higher in the tofersen-treated group than in the control group (67% more than controls in the faster-progressing population and 48% more than controls in the slower-progressing population).18
Because of these encouraging results, VALOR participants were moved to the ongoing open-label extension trial of tofersen (ClinicalTri-als.gov Identifier: NCT03070119), in which both groups were treated with the active agent.
These data suggest that early tofersen treatment might slow decline in faster-progressing patients and stabilize clinical function in slower-progressing patients.18,19 Overall, most adverse events (AEs) in the trial among patients receiving active treatment were of mild or moderate severity, and were largely consistent with either disease progression or lumbar puncture–related complications.18
Because data from VALOR suggested potential benefit from tofersen, the ATLAS trial (ClinicalTrials.gov Identifier: NCT04856982) is investigating the clinical value of presymptomatic treatment and the optimal timing of initiation of therapy.20,21 ATLAS is a phase 3, randomized, placebo-controlled trial that examines the clinical efficacy, safety, and tolerability of tofersen in presymptomatic adult carriers of SOD1 mutation who have an elevated neurofilament light-chain concentration.21 ATLAS will also evaluate the efficacy of tofersen when initiated before, rather than after, ALS manifests clinically. Enrollment is still open for this trial.20,21
Latozinemab, also known as AL001, is a first-in-class monoclonal antibody, administered by IV infusion, that elevates levels of progranulin, a key regulator of the immune activity and lysosomal function in the brain.22,23 Latozinemab limits progranulin endocytosis and degradation by sortilin inhibition.22 Progranulin gene mutations can reduce progranulin expression (by 50 to 70 percent reduction), which may cause neuro-degeneration due to abnormal accumulation of TAR-DNA-binding protein 43 (TDP-43) in the brain cells.22,24 TDP-43 pathology has also been shown to be associated with C9orf72 mutations.23 Although the mechanism is not fully understood, the role of progranulin deficiency in TDP-43 pathology is believed to be associated with neurodegenerative diseases like ALS.11,23,24,43 Previous animal models of chronic neurodegenera-tion have demonstrated how increased progranulin levels can be protective against TDP-43 pathology, increasing neuronal development and survival, thus potentially slowing disease progression.23,24,43 Currently, latozinemab is being investigated in a randomized, double-blind, placebo-controlled, multicenter phase 2 trial (ClinicalTrials.gov Identifier: NCT05053035). Approximately, 45 C90rf72-associated ALS participants (≥ 18 years of age) will receive latozinemab or placebo infusions every 4 weeks (for 24 weeks). Study endpoints include safety, tolerability, PK, PD, as well as plasma, and CSF progranulin levels.25 In previous studies, latozinemab demonstrated encouraging results in frontotemporal dementia (FTD) patients who carry a progranulin mutation. Because FTD was revealed to have significant genetic overlap with ALS, there is disease-modifying potential for latozinemab in ALS patients.23,24
TPN-101 is a nucleoside analog reverse transcriptase inhibitor, administered orally, that was originally developed for human immunodeficiency virus (HIV) treatment. However, due to recent findings suggesting retrotransposon activity contributing to neurodegeneration in TDP-43 mediated diseases, including ALS and FTD, TNP-101 is being repurposed.26 The safety and tolerability of TNP-101 are currently being evaluated in C9orf72-associated ALS and FTD patients (≥ 18 years of age). The study is a randomized, double-blind, placebo-controlled paral-lel-group phase 2a trial (ClinicalTrials.gov Identifier: NCT04993755) The study includes a screening period of 6 weeks, double-blind treatment period of 24 weeks, an open-label treatment period of 24 weeks, and 4 weeks of the post-treatment follow-up visit. Study endpoints include the incidence and severity of spontaneously reported treatment-emergent adverse events (TEAEs) associated with TNP-101 and placebo for a to-tal of 48 weeks.27
ION363 is an investigational ASO, administered by IT injection, that selectively targets one of the FUS mutations (p.P525L), which is responsible for earlier disease onset and rapid ALS progression.28,29 The clinical efficacy of ION363, specifically in clinical function and survival is being assessed in FUS-associated ALS patients (≥ 12 years of age). This randomized phase 3 study (ClinicalTrials.gov Identifier: NCT04768972) includes two parts; part 1 will consist of participants receiving a multi-dose regimen (1 dose every 4-12 weeks) of ION363 or placebo for 61 weeks followed by an open-label extension treatment period in part 2, which will consist of participants receiving ION363 (every 12 weeks) for 85 weeks. The primary endpoint of the study is the change from baseline to day 505 in functional impairment, using ALS Functional Rating Scale-Revised (ALSFRS-R). This measures functional disease severity, specifically in bulbar function, gross motor skills, fine motor skills, and respiratory. The score for all 12 questions can range from 0 (no function) to 4 (full function) with a total possible score of 48.30
Engensis, also known as VM202, is a non-viral gene therapy, administered by intramuscular (IM) injection, that uses a plasmid to deliver the hepatocyte growth factor (HGF) gene to promote HGF protein production. The HGF protein plays a role in angiogenesis, the previous of muscle atrophy, and the promotion of neuronal survival and growth. Based on preclinical studies, increasing HGF protein production has been shown to reduce neurodegeneration, thus potentially halting or slowing ALS progression.31 Currently, the safety of engensis is being evaluated in ALS patients (18-80 years of age) in the REViVALS phase 2a (ClinicalTrials.gov Identifier: NCT04632225)/2b (ClinicalTrial.gov Identifier: NCT05176093).32,33 The ReViVALS trial is a double-blind, randomized, placebo-controlled, multi-center study. The phase 2a study endpoints include the incidence of TEAEs, treatment-emergent serious adverse events (TESAEs), injection site reactions, and clinically significant labor-atory values post-treatment (engensis vs placebo group) for 180 days.33 A phase 2b study will evaluate the long-term safety of engensis for an additional 6 months. Study endpoints include the incidence of AEs, changes from baseline in ALSFRS-R scores to evaluate improvement in muscle function, changes from baseline in quality of life using the ALS patient assessment questionnaire, time to all-cause mortality compared to placebo, etc.32
Spinal muscular atrophy
SMA is a hereditary lower motor-neuron disease caused (in 95% of cases) by deletions or, less commonly, by mutations of the survival motor neuron 1 (SMN1) gene on chromosome 5q13 that encodes the SMN protein.6 Reduction in expression of the SMN protein causes motor neurons to degenerate.36-38 Because of a large inverted duplication in chromosome 5q, two variants of SMN (SMN1 and SMN2) exist on each allele. The paralog gene, SMN2, also produces the SMN protein – although at a lower level (10% to 20% of total SMN protein production) than SMN1 does.
A single nucleotide substitution in SMN2 alters splicing and suppresses transcription of exon 7, resulting in a shortened mRNA strand that yields a truncated SMN protein product.6,37,39 SMA is classified based on age of onset and maximum motor abilities achieved, ranging from the most severe (Type 0) to mildest (Type 4) disease.36,40 Because SMA patients lack functional SMN1 (due to polymorphisms), disease severity is determined by copy numbers of SMN2.6,39
Gene-based therapy for SMA
Three FDA-approved SMN treatments demonstrate clinically meaningful benefit in SMA: SMN2-targeting nusinersen [Spinraza] and risdiplam [Evrysdi], and SMN1-targeting onasemnogene abeparvovec-xioi [Zolgensma]38 Additional approaches to SMA treatment are through SMN-independent therapies, which target muscle and nerve function. Research has strongly suggested that combined SMA therapies, specifically approved SMN-targeted and investigational SMN-independent treatments, such as GYM329 (also known as RO7204239) may be the best strategy to treat all ages, stages, and types of SMA.41 (Table 226-41).
Agents that modulate SMN2. Nusinersen, approved by the FDA in 2016, was the first treatment indicated for all SMA types in pediatric and adult patients.42 The agent is an ASO that targets exon 7 of SMN2, thus stabilizing transcription. Inclusion of exon 7 increases SMN protein production, improving motor function.6,38 Nusinersen is a lifelong treatment that requires IT administration every 4 months because it cannot cross the blood-brain barrier.38,43
Pivotal clinical studies that led to approval of nusinersen include CHERISH (ClinicalTrial.gov Identifier: NCT02292537) and ENDEAR (ClinicalTrial.gov Identifier: NCT02193074) studies.
CHERISH was a phase 3, randomized, double-blind, sham procedure–controlled trial that examined the clinical efficacy and safety of nusinersen in 126 participants with later-onset SMA (2-12 years of age). The primary endpoint was the change from baseline using the Hammersmith Functional Motor Scale Expanded (HFMSE) at 15 months. HFMSE looks at 33 activities to assess improvement in motor function. The study met the primary efficacy outcome, demonstrating statistically significant (P = .0000001) improvement in overall motor function. The nusinersen group showed a 3.9-point increase in the HFMSE score from baseline, which indicates improvement, compared with a 1.0-point decline from baseline in the control group.46,47
ENDEAR was also a randomized, double-blind, sham procedure–controlled phase 3 trial, which investigated the efficacy and safety of nusinersen in 121 participants with early-onset SMA Type 1 (≤ 210 days of age). Coprimary endpoints were:
- Percentage of motor milestones responders, as determined using Section 2 of the Hammersmith Infant Neurological Examination–Part 2.
- Event-free survival (that is, avoidance of combined endpoint of death or permanent ventilation).
ENDEAR met the first primary efficacy outcome, demonstrating statistically significant (P < .0001) improvement in motor milestones (head control, rolling, independent sitting, and standing). By 13 months of age, approximately 51% of nusinersen-treated participants showed improvement, compared with none in the control group.46,47
The second primary endpoint was also met, with a statistically significant (P = .005) 47% decrease in mortality or permanent ventilation use.46-48
The NURTURE (ClinicalTrial.gov Identifier: NCT02386553) study is also investigating the efficacy and safety of nusinersen. An ongoing, open-label, supportive phase 2 trial, NURTURE is evaluating the efficacy and safety of multiple doses of nusinersen in 25 presymptomatic SMA patients (≤ 6 weeks of age). The primary endpoint of this study is time to death or respiratory intervention.49 Interim results demonstrate that 100% of presymptomatic infants are functioning without respiratory intervention after median follow-up of 2.9 years.46-48
Although nusinersen has been shown to be generally safe in clinical studies, development of lumbar puncture–related complications, as well as the need for sedation during IT administration, might affect treatment tolerability in some patients.39
Risdiplam was approved by the FDA in 2020 as the first orally administered small-molecule treatment of SMA (for patients ≤ 2 months of age).52 Risdiplam is a SMN2 splicing modifier, binding to the 5’ splice site of intron 7 and exonic splicing enhancer 2 in exon 7 of SMN2 pre-mRNA. This alternative splicing increases efficiency in SMN2 gene transcription, thus increasing SMN protein production in motor-neuron cells.36 An important advantage of risdiplam is the convenience of oral administration: A large percentage of SMA patients (that is, those with Type 2 disease) have severe scoliosis, which can further complicate therapy or deter patients from using a treatment that is administered through the IT route.40
FDA approval of risdiplam was based on clinical data from two pivotal studies, FIREFISH (ClinicalTrial.gov Identifier: NCT02913482) and SUNFISH (ClinicalTrial.gov Identifier: NCT02908685).53-54
FIREFISH is an open-label, phase 2/3 ongoing trial in infants (1-7 months of age) with SMA Type 1. The study comprises two parts; Part 1 determined the dose of risdiplam used in Part 2, which assessed the efficacy and safety of risdiplam for 24 months. The primary endpoint was the percentage of infants sitting without support for 5 seconds after 12 months of treatment using the gross motor scale of the Bayley Scales of Infant and Toddler Development–Third Edition. A statistically significant (P < .0001) therapeutic benefit was observed in motor milestones. Approximately 29% of infants achieved the motor milestone of independent sitting for 5 seconds, which had not been observed in the natural history of SMA.53-55
SUNFISH is an ongoing randomized, double-blind, placebo-controlled trial of risdiplam in adult and pediatric patients with SMA Types 2 and 3 (2-25 years old). This phase 2/3 study comprises two parts: Part 1 determined the dose (for 12 weeks) to be used for confirmatory Part 2 (for 12 to 24 months). The primary endpoint was the change from baseline on the 32-item Motor Function Measure at 12 months. The study met its primary endpoint, demonstrating statistically significant (P = .0156) improvement in motor function scores, with a 1.36-point increase in the risdiplam group, compared with a 0.19-point decrease in the control group.54,55
Ongoing risdiplam clinical trials also include JEWELFISH (ClinicalTrial.gov Identifier: NCT03032172) and RAINBOW (ClinicalTrial.gov Identifier: NCT03779334).56-57 JEWELFISH is an open-label, phase 2 trial assessing the safety of risdiplam in patients (6 months to 60 years old) who received prior treatment. The study has completed recruitment; results are pending.56 RAINBOW is an ongoing, open-label, single-arm, phase 2 trial, evaluating the clinical efficacy and safety of risdiplam in SMA-presymptomatic newborns (≤ 6 weeks old). The study is open for enrollment.57 Overall, interim results for JEWELFISH and RAINBOW appear promising.
In addition, combined SMA therapies, specifically risdiplam and GYM329 are currently being investigated to address the underlying cause and symptoms of SMA concurrently.58 GYM329, is an investigational anti-myostatin antibody, selectively binding preforms of myostatin - pro-myostatin and latent myostatin, thus improving muscle mass and strength for SMA patients.59 The safety and efficacy of GYM329 in combination with risdiplam is currently being investigated in 180 ambulant participants with SMA (2-10 years of age) in the MANATEE (ClinicalTrial.gov Identifier: NCT05115110) phase 2/3 trial. The MANATEE study is a two-part, seamless, randomized, placebo-controlled, double-blind trial. Part 1 will assess the safety of the combination treatment in approximately 36 participants; participants will receive both GYM329 (every 4 weeks) by subcutaneous (SC) injection into the abdomen and risdiplam (once per day) for 24 weeks followed by a 72-week open-label treatment period. 54,58 The outcome measures include the incidence of AEs, percentage change from baseline in the contractile area of skeletal muscle (in dominant thigh and calf), change from baseline in RHS total score, and incidence of change from baseline in serum concentration (total myostatin, free latent myostatin, and mature myostatin) etc.54 Part 2 will be conducted on 144 participants, specifically assessing the efficacy and safety of the optimal dose of GYM329 selected from Part 1 (combined with risdiplam) for 72 weeks. Once the treatment period is completed in either part, participants can partake in a 2-year open-label extension period.54,58 Other outcome measures include change from baseline in lean muscle mass (assessed by full body dual-energy X- ray absorptiometry (DXA) scan), in time taken to walk/run 10 meters (measured by RHS), in time taken to rise from the floor (measured by RHS), etc.54 Overall, this combination treatment has the potential to further improve SMA patient outcomes and will be further investigated in other patient populations (including non-ambulant patients and a broader age range) in the future.58
An agent that alters SMN1 expression. Onasemnogene abeparvovec-xioi, FDA approved in 2019, was the first gene-replacement therapy indicated for treating SMA in children ≤ 2 years old.60 Treatment utilizes an AAV vector type 9 (AAV9) to deliver a functional copy of SMN1 into target motor-neuron cells, thus increasing SMN protein production and improving motor function. This AAV serotype is ideal because it crosses the blood-brain barrier. Treatment is administered as a one-time IV fusion.38,39,43
FDA approval was based on the STR1VE (ClinicalTrial.gov Identifier: NCT03306277) phase 3 study and START (ClinicalTrial.gov Identifier: NCT02122952) phase 1 study.61,62 START was the first trial to investigate the safety and efficacy of onasemnogene abeparvovec-xioi in SMA Type 1 infants (< 6 months old). Results demonstrated remarkable clinical benefit, including 100% permanent ventilation-free survival and a 92% (11 of 12 patients) rate of improvement in motor function. Improvement in development milestones was also observed: 92% (11 of 12 patients) could sit without support for 5 seconds and 75% (9 of 12) could sit without support for 30 seconds.14,61,63
The efficacy of onasemnogene abeparvovec-xioi seen in STR1VE was consistent with what was observed in START. STRIVE, a phase 3 open-label, single-dose trial, examined treatment efficacy and safety in 22 symptomatic infants (< 6 months old) with SMA Type 1 (one or two SMN2 copies). The primary endpoint was 30 seconds of independent sitting and event-free survival. Patients were followed for as long as 18 months. Treatment showed statistically significant (P < .0001) improvement in motor milestone development and event-free survival, which had not been observed in SMA Type 1 historically. Approximately 59% (13 of 22 patients) could sit independently for 30 seconds at 18 months of age. At 14 months of age, 91% (20 of 22 patients) were alive and achieved independence from ventilatory support.34,35,53
Although many clinical studies suggest that onasemnogene abeparvovec-xioi can slow disease progression, the benefits and risks of long-term effects are still unknown. A 15-year observational study is investigating the long-term therapeutic effects and potential complications of onasemnogene abeparvovec-xioi. Participants in START were invited to enroll in this long-term follow-up study (ClinicalTrial.gov Identifier: NCT04042025).66-67
Duchenne muscular dystrophy
DMD is the most common muscular dystrophy of childhood. With an X-linked pattern of inheritance, DMD is seen mostly in young males (1 in every 3,500 male births).38,39,73 DMD is caused by mutation of the dystrophin encoding gene, or DMD, on the X chromosome. Deletion of one or more exons of DMD prevents production of the dystrophin protein, which leads to muscle degeneration.38,39,43 Common DMD deletion hotspots are exon 51 (20% of cases), exon 53 (13% of cases), exon 44 (11% of cases), and exon 45 (12% of cases).74 Nonsense mutations, which account for another 10% of DMD cases, occur when premature termination codons are found in the DMD gene. Those mutations yield truncated dystrophin protein products.39,66
Therapy for DMD
There are many therapeutic options for DMD, including deflazacort (Emflaza), FDA approved in 2017, which has been shown to reduce inflammation and immune system activity in DMD patients (≥ 5 years old). Deflazacort is a corticosteroid prodrug; its active metabolite acts on the glucocorticoid receptor to exert anti-inflammatory and immunosuppressive effects. Studies have shown that muscle strength scores over 6-12 months and average time to loss of ambulation numerically favored deflazacort over placebo.74,75
Gene-based therapy for DMD
Mutation-specific therapeutic approaches, such as exon skipping and nonsense suppression, have shown promise for the treatment of DMD (Table 358-79):
- ASO-mediated exon skipping allows one or more exons to be omitted from the mutated DMD mRNA.74,75 Effective FDA-approved ASOs include golodirsen [Vyondys 53], viltolarsen [Viltepso], and casimersen [Amondys 45].74
- An example of therapeutic suppression of nonsense mutations is ataluren [Translarna], an investigational agent that can promote premature termination codon read-through in DMD patients.66
Another potential treatment approach is through the use of AAV gene transfer to treat DMD. However, because DMD is too large for the AAV vector (packaging size, 5.0 kb), microdystrophin genes (3.5-4 kb, are used as an alternative to fit into a single AAV vector.39,76
Exon skipping targeting exon 51. Eteplirsen, approved in 2016, is indicated for the treatment of DMD patients with the confirmed DMD gene mutation that is amenable to exon 51 skipping. Eteplirsen binds to exon 51 of dystrophin pre-mRNA, causing it to be skipped, thus, restoring the reading frame in patients with DMD gene mutation amenable to exon 51 skipping. This exclusion promotes dystrophin production. Though the dystrophin protein is still functional, it is shortened.38,77 Treatment is administered IV, once a week (over 35-60 minutes). Eteplirsen’s accelerated approval was based on 3 clinical studies (ClinicalTrial.gov Identifier: NCT01396239, NCT01540409, and NCT00844597.) 78-81 The data demonstrated an increased expression of dystrophin in skeletal muscles in some DMD patients treated with eteplirsen. Though the clinical benefit of eteplirsen (including improved motor function) was not established, it was concluded by the FDA that the data were reasonably likely to predict clinical benefit. Continued approval for this indication may depend on the verification of a clinical benefit in confirmatory trials. Ongoing clinical trials include (ClinicalTrial.gov Identifier: NCT03992430 (MIS51ON), NCT03218995, and NCT03218995).77,81,82
Vesleteplirsen, is an investigational agent that is designed for DMD patients who are amendable to exon 51 skip-ping. The mechanism of action of vesleteplirsen appears to be similar to that of eteplirsen.83 The ongoing MOMENTUM (ClinicalTrial.gov Identifier: NCT04004065) phase 2 trial is assessing the safety and tolerability of vesleteplirsen at multiple-ascending dose levels (administered via IV infusion) in 60 participants (7-21 years of age). The study consists of two parts; participants receive escalating dose levels of vesleteplirsen (every 4 weeks) for 72 weeks during part A and participants receive the selected doses from part A (every 4 weeks) for 2 years during part B. Study endpoints include the number of AEs (up to 75 weeks) and the change from baseline to week 28 in dystrophin protein level. 84 Serious AEs of reversible hypomagnesemia were observed in part B, and as a result, the study protocol was amended to include magnesium supplementation and monitoring of magnesium levels.83
Exon skipping targeting exon 53. Golodirsen, FDA approved in 2019, is indicated for the treatment of DMD in patients who have a confirmed DMD mutation that is amenable to exon 53 skipping. The mechanism of action is similar to eteplirsen, however, golodirsen is designed to bind to exon 53.38,39 Treatment is administered by IV infusion over 35-60 minutes.
Approval of golodirsen was based primarily on a two-part, phase 1/2 clinical trial (ClinicalTrial.gov Identifier: NCT02310906). Part 1 was a randomized, placebo-controlled, dose-titration study that assessed multiple-dose efficacy in 12 DMD male patients, 6 to 15 years old, with deletions that were amenable to exon 53 skipping.
Part 2 was an open-label trial in 12 DMD patients from Part 1 of the trial plus 13 newly enrolled male DMD patients who were also amenable to exon 53 skipping and who had not already received treatment. Primary endpoints were change from baseline in total distance walked during the 6-minute walk test at Week 144 and dystrophin protein levels (measured by western blot testing) at Week 48. A statistically significant increase in the mean dystrophin level was observed, from a baseline 0.10% mean dystrophin level to a 1.02% mean dystrophin level after 48 weeks of treatment (P < .001). Common reported adverse events associated with golodirsen were headache, fever, abdominal pain, rash, and dermatitis. Renal toxicity was observed in preclinical studies of golodirsen but not in clinical studies.80,85
Viltolarsen, approved in 2020, is also indicated for the treatment of DMD in patients with deletions amenable to exon 53 skipping. The mechanism of action and administration (IV infusion over 60 minutes) are similar to that of golodirsen.
Approval of viltolarsen was based on two phase 2 clinical trials (ClinicalTrial.gov Identifier: NCT02740972 and NCT03167255) in a total of 32 patients. NCT02740972 was a randomized, double-blind, placebo-controlled, dose-finding study that evaluated the clinical efficacy of viltolarsen in 16 male DMD patients (4-9 years old) for 24 weeks.
NCT03167255 was an open-label study that evaluated the safety and tolerability of viltolarsen in DMD male patients (5-18 years old) for 192 weeks. The efficacy endpoint was the change in dystrophin production from baseline after 24 weeks of treatment. A statistically significant increase in the mean dystrophin level was observed, from a 0.6% mean dystrophin level at baseline to a 5.9% mean dystrophin level at Week 25 (P = .01). The most common adverse events observed were upper respiratory tract infection, cough, fever, and injection-site reaction.86-87
Exon skipping targeting exon 45. Casimersen was approved in 2021 for the treatment of DMD in patients with deletions amenable to exon 45 skipping.88 Treatment is administered by IV infusion over 30-60 minutes. Approval was based on an increase in dystrophin production in skeletal muscle in treated patients. Clinical benefit was reported in interim results from the ESSENCE (ClinicalTrial.gov Identifier: NCT02500381) study, an ongoing double-blind, placebo-controlled phase 3 trial that is evaluating the efficacy of casimersen, compared with placebo, in male participants (6-13 years old) for 48 weeks. Efficacy is based on the change from baseline dystrophin intensity level, determined by immunohistochemistry, at Week 48.
Interim results from ESSENCE show a statistically significant increase in dystrophin production in the casimersen group, from a 0.9% mean dystrophin level at baseline to a 1.7% mean dystrophin level at Week 48 (P = .004); in the control group, a 0.54% mean dystrophin level at baseline increased to a 0.76% mean dystrophin level at Week 48 (P = .09). Common adverse events have included respiratory tract infection, headache, arthralgia, fever, and oropharyngeal pain. Renal toxicity was observed in preclinical data but not in clinical studies.60,84
Targeting nonsense mutations. Ataluren is an investigational, orally administered nonsense mutation suppression therapy (through the read-through of stop codons).37 Early clinical evidence supporting the use of ataluren in DMD was seen in an open-label, dose-ranging, phase 2a study (ClinicalTrial.gov Identifier: NCT00264888) in male DMD patients (≥ 5 years old) caused by nonsense mutation. The study demonstrated a modest (61% ) increase in dystrophin expression in 23 of 38 patients after 28 days of treatment.37,91,92
However, a phase 2b randomized, double-blind, placebo-controlled trial (ClinicalTrial.gov Identifier: NCT00592553) and a subsequent confirmatory ACT DMD phase 3 study (ClinicalTrial.gov Identifier: NCT01826487) did not meet their primary endpoint of improvement in ambulation after 48 weeks as measured by the 6-minute walk test.37,93,94 In ACT DMD, approximately 74% of the ataluren group did not experience disease progression, compared with 56% of the control group (P = 0386), measured by a change in the 6-minute walk test, which assessed ambulatory decline.37,95
Based on limited data showing that ataluren is effective and well tolerated, the European Medicines Agency has given conditional approval for clinical use of the drug in Europe. However, ataluren was rejected by the FDA as a candidate therapy for DMD in the United States.22 Late-stage clinical studies of ataluren are ongoing in the United States.
AAV gene transfer with microdystrophin. Limitations on traditional gene-replacement therapy prompted exploration of gene-editing strategies for treating DMD, including using AAV-based vectors to transfer microdystrophin, an engineered version of DMD, into target muscles.43 The microdystrophin gene is designed to produce a functional, truncated form of dystrophin, thus improving muscular function.
There are 3 ongoing investigational microdystrophin gene therapies that are in clinical development (ClinicalTrial.gov Identifier: NCT03368742 (IGNITE DMD), NCT04281485 (CIFFREO), and NCT05096221 (EMBARK)).38,82
IGNITE DMD is a phase 1/2 randomized, controlled, single-ascending dose trial evaluating the safety and efficacy of a SGT-001, single IV infusion of AAV9 vector containing a microdystrophin construct in DMD patients (4-17 years old) for 12 months. At the conclusion of the trial, treatment and control groups will be followed for 5 years. The primary efficacy endpoint is the change from baseline in microdystrophin protein production in muscle-biopsy material, using western blot testing.96 Long-term interim data on biopsy findings from three patients demonstrated clinical evidence of durable microdystrophin protein expression after 2 years of treatment.96,97
The CIFFREO trial will assess the safety and efficacy of the PF-06939926 microdystrophin gene therapy, an investigational AAV9 containing microdystrophin, in approximately 99 ambulatory DMD patients (4-7 years of age). The study is a randomized, double-blind, placebo-controlled, multicenter phase 3 trial. The primary efficacy end-point is the change from baseline in the North Star Ambulatory Assessment (NSAA), which measures gross motor function. This will be assessed at 52 weeks; all study participants will be followed for a total of 5 years post-treatment.98,99,100 Due to unexpected patient death (in a non-ambulatory cohort) in the phase 1b (in a non-ambulatory cohort) in the phase 1b (ClinicalTrial.gov Identifier: (NCT03362502) trial, microdystrophin gene therapy was immediately placed on clinical hold.101,102 The amended study protocol required that all participants undergo one week of in-hospital observation after receiving treatment.102
The EMBARK study is a global, randomized, double-blind, placebo-controlled, phase 3 trial that is evaluating the safety and efficacy of SRP-9001, which is a rAAVrh74.MHCK7.microdystrophin gene therapy. The AAV vector (rAAVrh74) contains the microdystrophin construct, driven by the skeletal and cardiac muscle–specific promoter, MHCK7.98,99 In the EMBARK study, approximately 120 participants with DMD (4-7 years of age) will be enrolled. The primary efficacy endpoint includes the change from baseline to week 52 in the NSAA total score.99 Based on SRP-9001, data demonstrating consistent statistically significant functional improvements in NSAA total scores and timed function tests (after one-year post- treatment) in DMD patients from previous studies and an integrated analysis from multiple studies (ClinicalTrial.gov Identifier: NCT03375164, NCT03769116, and NCT04626674), the ongoing EMBARK has great promise.103,104
Challenges ahead, but advancements realized
Novel gene-based therapies show significant potential for transforming the treatment of NMDs. The complex pathologies of NMDs have been a huge challenge to disease management in an area once considered unremediable by gene-based therapy. However, advancements in precision medicine – specifically, gene-delivery systems (for example, AAV9 and AAVrh74 vectors) combined with gene modification strategies (ASOs and AAV-mediated silencing) – have the potential to, first, revolutionize standards of care for sporadic and inherited NMDs and, second, significantly reduce disease burden.6
What will be determined to be the “best” therapeutic approach will, likely, vary from NMD to NMD; further investigation is required to determine which agents offer optimal clinical efficacy and safety profiles.43 Furthermore, the key to therapeutic success will continue to be early detection and diagnosis – first, by better understanding disease pathology and drug targets and, second, by validation of reliable biomarkers that are predictive of therapeutic benefit.4,5
To sum up, development challenges remain, but therapeutic approaches to ALS, SMA, and DMD that utilize novel gene-delivery and gene-manipulation tools show great promise.
Ms. Yewhalashet is a student in the masters of business and science program, with a concentration in healthcare economics, at Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences, Claremont, Calif. Dr. Davis is professor of practice in clinical and regulatory affairs, Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences.
References
1. Aitken M et al. Understanding neuromuscular disease care. IQVIA [Internet]. Oct 30, 2018. Accessed Mar 1, 2022. https://www.iqvia.com/insights/the-iqvia-institute/reports/understanding-neuromuscular-disease-care.
2. National Institute of Neurological Disorders and Stroke. Neurological diagnostic tests and procedures fact sheet. Updated Nov 15, 2021. Ac-cessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Neurological-Diagnostic-Tests-and-Procedures-Fact.
3. Deenen JCW et al. The epidemiology of neuromuscular disorders: A comprehensive overview of the literature. J Neuromuscul Dis. 2015;2(1):73-85.
4. Cavazzoni P. The path forward: Advancing treatments and cures for neurodegenerative diseases. U.S. Food and Drug Administration. Jul 29, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/congressional-testimony/path-forward-advancing-treatments-and-cures-neurodegenerative-diseases-07292021.
5. Martier R, Konstantinova P. Gene therapy for neurodegenerative diseases: Slowing down the ticking clock. Front Neurosci. 2020 Sep 18;14:580179. doi: 10.3389/fnins.2020.580179.
6. Sun J, Roy S. Gene-based therapies for neurodegenerative diseases. Nat Neurosci. 2021 Mar;24(3):297-311. doi:10.1038/s41593-020-00778-1.
7. Amado DA, Davidson BL. Gene therapy for ALS: A review. Mol Ther. 2021 Dec 1;29(12):3345-58. doi:10.1016/j.ymthe.2021.04.008.
8. Yun Y, Ha Y. CRISPR/Cas9-mediated gene correction to understand ALS. Int J Mol Sci. 2020;21(11):3801. doi:10.3390/ijms21113801.
9. National Institute of Neurological Disorders and Stroke. Amyotrophic lateral sclerosis (ALS) fact sheet. Updated Nov 15, 2021. Accessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Amyotrophic-Lateral-Sclerosis-ALS-Fact-Sheet.
10. Cappella M et al. Gene therapy for ALS – A perspective. Int J Mol Sci. 2019;20(18):4388. doi:10.3390/ijms20184388.
11. Abramzon YA, Fratta P, Traynor BJ, Chia R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front Neurosci. 2020;14. Accessed August 18, 2022. https://www.frontiersin.org/articles/10.3389/fnins.2020.00042
12. Giannini M, Bayona-Feliu A, Sproviero D, Barroso SI, Cereda C, Aguilera A. TDP-43 mutations link Amyotrophic Lateral Sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLOS Genet. 2020;16(12):e1009260. doi:10.1371/journal.pgen.1009260
13. FDA-approved drugs for treating ALS. The ALS Association [Internet]. Accessed Mar 1, 2022. http://www.als.org/navigating-als/living-with-als/fda-approved-drugs.
14. Jensen TL et al. Current and future prospects for gene therapy for rare genetic diseases affecting the brain and spinal cord. Front Mol Neurosci. 2021 Oct 6;14:695937. doi:10.3389/fnmol.2021.695937.
15. ALS Gene Targeted Therapies. The ALS Association. Accessed August 22, 2022. https://www.als.org/understanding-als/who-gets-als/genetic-testing/als-gene-targeted-therapies
16. Tofersen for ALS clears phase 1/2 trial, now in phase 3. Advances in Motion. Massachusetts General Hospital [Internet]. Sep 30, 2020. Accessed Mar 1, 2022. https://advances.massgeneral.org/neuro/journal.aspx?id=1699.17. Biogen. A study to evaluate the efficacy, safety, tol-erability, pharmacokinetics, and pharmacodynamics of BIIB067 administered to adult subjects with amyotrophic lateral sclerosis and confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT02623699. Updated Jul 25, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT02623699.
18. Biogen. Biogen announces topline results from the tofersen phase 3 study and its open-label Extension in SOD1-ALS. Press release. Oct 17, 2021. Accessed Mar 1, 2022. https://investors.biogen.com/news-releases/news-release-details/biogen-announces-topline-results-tofersen-phase-3-study-and-its.
19. Biogen. An extension study to assess the long-term safety, tolerability, pharmacokinetics, and effect on disease progression of BIIB067 ad-ministered to previously treated adults with amyotrophic lateral sclerosis caused by superoxide dismutase 1 mutation. ClinicalTrials.gov Identi-fier: NCT03070119. Updated Sep 10, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT03070119.
20. MS MW. #AANAM – ATLAS Trial to Assess Tofersen in Presymptomatic SOD1 ALS. Accessed February 19, 2022. https://alsnewstoday.com/news-posts/2021/04/23/aanam-atlas-clinical-trial- tofersen-presymptomatic-sod1-als-patients/
21.Biogen. A phase 3 randomized, placebo-controlled trial with a longitudinal natural history run-in and open-label extension to evaluate BIIB067 initiated in clinically presymptomatic adults with a confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT04856982. Updated Feb 18, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04856982.
22. Latozinemab | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/latozinemab
23. Alector Presents AL001 (latozinemab) Data from the FTD-C9orf72 Cohort of the INFRONT-2 Phase 2 Clinical Trial | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releas-es/news-release-details/alector-presents-al001-latozinemab-data-ftd-c9orf72-cohort/
24. Alector Announces First Participant Dosed in Phase 2 Study Evaluating AL001 in Amyotrophic Lateral Sclerosis (ALS) | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releases/news-release-details/lector-announces-first-participant-dosed-phase-2-study-0/ 25. A Phase 2 Study to Evaluate AL001 in C9orf72-Associated ALS - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT05053035
26.TPN-101 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/tpn- 101
27. Transposon Therapeutics, Inc. A Phase 2a Study of TPN-101 in Patients With Amyotrophic Lateral Sclerosis (ALS) and/or Frontotemporal Dementia (FTD) Associated With Hexanucleotide Repeat Expansion in the C9orf72 Gene (C9ORF72 ALS/FTD). clinicaltrials.gov; 2022. Ac-cessed August 17, 2022. https://clinicaltrials.gov/ct2/show/NCT04993755
28. Kerk SY, Bai Y, Smith J, et al. Homozygous ALS-linked FUS P525L mutations cell- autonomously perturb transcriptome profile and chem-oreceptor signaling in human iPSC microglia. Stem Cell Rep. 2022;17(3):678-692. doi:10.1016/j.stemcr.2022.01.004
29. ION363 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/ion363 30. Ionis Pharmaceuticals, Inc. A Phase 1-3 Study to Evaluate the Efficacy, Safety, Pharmacokinetics and Pharmacodynamics of Intrathecally Administered ION363 in Amyo-trophic Lateral Sclerosis Patients With Fused in Sarcoma Mutations (FUS-ALS). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04768972
31. PhD LF. Engensis (VM202) - ALS News Today. Accessed August 19, 2022. https://alsnewstoday.com/vm202/
32. Helixmith Co., Ltd. A 6-Month Extension Study Following Protocol VMALS-002-2 (A Phase 2a, Double-Blind, Randomized, Place-bo-Controlled, Multicenter Study to Assess the Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05176093 33. Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT04632225
34. Biogen. A phase 1, safety, tolerability, and distribution study of a microdose of radiolabeled BIIB067 co-administered with BIIB067 to healthy adults. ClinicalTrials.gov Identifier: NCT03764488. Updated Jul 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03764488.
35. Ionis Pharmaceuticals Inc. A phase 1, double-blind, placebo-controlled, dose-escalation study of the safety, tolerability, and pharmacokinet-ics of ISIS 333611 administered intrathecally to patients with familial amyotrophic lateral sclerosis due to superoxide dismutase 1 gene muta-tions. ClinicalTrials.gov Identifier: NCT01041222. Updated Apr 13, 2012. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01041222.
36. Messina S, Sframeli M. New treatments in spinal muscular atrophy: Positive results and new challenges. J Clin Med. 2020;9(7):2222. doi:10.3390/jcm9072222.
37. Scoto M et al. Genetic therapies for inherited neuromuscular disorders. Lancet Child Adolesc Health. 2018 Aug;2(8):600-9. doi:10.1016/S2352-4642(18)30140-8.
38. Abreu NJ, Waldrop MA. Overview of gene therapy in spinal muscular atrophy and Duchenne muscular dystrophy. Pediatr Pulmonol. 2021 Apr;56(4):710-20. doi:10.1002/ppul.25055.
39. Brandsema J, Cappa R. Genetically targeted therapies for inherited neuromuscular disorders. Practical Neurology [Internet]. Jul/Aug 2021:69-73. Accessed Mar 1, 2022. https://practicalneurology.com/articles/2021-july-aug/genetically-targeted-therapies-for-inherited-neuromuscular-disorders/pdf.
40. Ojala KS et al. In search of a cure: The development of therapeutics to alter the progression of spinal muscular atrophy. Brain Sci. 2021;11(2):194. doi:10.3390/brainsci11020194.
41. McCall S. Cure SMA Releases Updated Drug Pipeline. Cure SMA. Published December 13, 2021. Accessed August 21, 2022. https://www.curesma.org/cure-sma-releases-updated-drug-pipeline- 2021/ 42. FDA approves first drug for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Dec 23, 2016. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy.43. Kirschner J. Postnatal gene therapy for neuromuscular diseases – Opportunities and limitations. J Perinat Med. 2021 Sep;49(8):1011-5. doi:10.1515/jpm-2021-0435.
43. Terryn J, Verfaillie CM, Van Damme P. Tweaking Progranulin Expression: Therapeutic Avenues and Opportunities. Front Mol Neurosci. 2021;14. Accessed September 4, 2022. https://www.frontiersin.org/articles/10.3389/fnmol.2021.71303144.
44. Biogen. A phase 3, randomized, double-blind, sham-procedure controlled study to assess the clinical efficacy and safety of ISIS 396443 administered intrathecally in patients with later-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02292537. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/study/NCT02292537.
45. Why Spinraza/later-onset studies. SPINRAZA® (nusinersen) [Internet]. Accessed Mar 1, 2022. www.spinraza.com/en_us/home/why-spinraza/later-onset-studies.html#scroll-tabs.
46. Biogen. A Phase 3, Randomized, Double-Blind, Sham-Procedure Controlled Study to Assess the Clinical Efficacy and Safety of ISIS 396443 Administered Intrathecally in Patients With Infantile- Onset Spinal Muscular Atrophy. clinicaltrials.gov; 2021. Accessed February 10, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02193074
47. Early-onset SMA (Type 1) | SPINRAZA® (nusinersen). Accessed Mar 1, 2022. https://www.spinraza-hcp.com/en_us/home/why-spinraza/about-spinraza.html.
48. Finkel RS et al; ENDEAR Study Group. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723-32. doi: 10.1056/NEJMoa1702752.
49. Biogen. An open-label study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to subjects with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02386553. Updated Nov 18, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02386553.
50. De Vivo DC et al; NURTURE Study Group. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: In-terim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul Disord. 2019 Nov;29(11):842-56. doi:10.1016/j.nmd.2019.09.007.
51. Why Spinraza/presymptomatic study. SPINRAZA® (nusinersen) [Internet]. Accessed Feb 22, 2022. www.spinraza.com/en_us/home/why-spinraza/presymptomatic-study.html#scroll-tabs.
52. FDA approves oral treatment for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Aug 7, 2020. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy.
53. Hoffmann-La Roche. A two-part seamless, open-label, multicenter study to investigate the safety, tolerability, pharmacokinetics, pharmaco-dynamics and efficacy of risdiplam (RO7034067) in infants with type 1 spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02913482. Updated Jan 21, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02913482.
54. Hoffmann-La Roche. A two-part seamless, multi-center randomized, placebo-controlled, double-blind study to investigate the safety, tolera-bility, pharmacokinetics, pharmacodynamics and efficacy of risdiplam (RO7034067) in type 2 and 3 spinal muscular atrophy patients. Clinical-Trials.gov Identifier: NCT02908685. Updated Dec 28, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02908685.
55. Genentech. Genentech’s risdiplam shows significant improvement in survival and motor milestones in infants with type 1 spinal muscular atrophy (SMA). Press release. Apr 27, 2020. Accessed Mar 1, 2022. http://www.gene.com/media/press-releases/14847/2020-04-27/genentechs-risdiplam-shows-significant-i
56. Hoffmann-La Roche. An open-label study to investigate the safety, tolerability, and pharmacokinetics/pharmacodynamics of risdiplam (RO7034067) in adult and pediatric patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03032172. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03032172.
57. Hoffmann-La Roche. An open-label study of risdiplam in infants with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03779334. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03779334.
58. McCall S. Update on Genentech/Roche Initiation of MANATEE Clinical Study. Cure SMA. Published October 20, 2021. Accessed August 20, 2022. https://www.curesma.org/update-on- genentech-roche-initiation-of-manatee-clinical-study/
59. Abati E, Manini A, Comi GP, Corti S. Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases. Cell Mol Life Sci. 2022;79(7):374. doi:10.1007/s00018-022-04408-w
60. FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality. U.S. Food and Drug Administration. News release. May 24, 2019. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease.
61. Novartis Gene Therapies. Phase I gene transfer clinical trial for spinal muscular atrophy type 1 delivering AVXS-101. ClinicalTrials.gov Identifier: NCT02122952. Updated Jun 14, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02122952.
62. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT03306277. Updated Jun 14, 2021. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT03306277.
63. Mendell JR et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713-22. doi:10.1056/NEJMoa1706198.
64. Symptomatic study results. ZOLGENSMA [Internet]. Updated Nov 2021. Accessed Mar 1, 2022. Error! Hyperlink reference not valid..
65. Novartis Gene Therapies. A global study of a single, one-time dose of AVXS-101 delivered to infants with genetically diagnosed and pre-symptomatic spinal muscular atrophy with multiple copies of SMN2. ClinicalTrials.gov Identifier: NCT03505099. Updated Jan 1, 2022. Ac-cessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03505099.
66. Chiu W et al. Current genetics and potential gene-targeting therapeutics for neuromuscular diseases. Int J Mol Sci. 2020 Dec;21(24):9589. doi:10.3390/ijms21249589.
67. Novartis Gene Therapies. A long-term follow-up study of patients in the clinical trials for spinal muscular atrophy receiving AVXS-101. Clini-calTrials.gov Identifier: NCT04042025. Updated Jun 9, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04042025.
68. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT0383718. Up-dated Jan 11, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03837184.
69. Biogen. An open-label, dose escalation study to assess the safety, tolerability and dose-range finding of multiple doses of ISIS 396443 de-livered intrathecally to patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01703988. Updated Apr 13, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01703988.
70. Biogen. A study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to patients with infantile-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01839656. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01839656.
71. Biogen. An open-label extension study for patients with spinal muscular atrophy who previously participated in investigational studies of ISIS 396443. ClinicalTrials.gov Identifier: NCT02594124. Updated Nov 15, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02594124.
72. Biogen. Escalating dose and randomized, controlled study of nusinersen (BIIB058) in participants with spinal muscular atrophy. ClinicalTri-als.gov Identifier: NCT04089566. Updated Feb 24, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04089566.
73. National Center for Advancing Translational Sciences. Duchenne muscular dystrophy. Genetic and Rare Diseases Information Center. Up-dated Nov 2, 2020. Accessed Mar 1, 2022. https://rarediseases.info.nih.gov/diseases/6291/duchenne-muscular-dystrophy.
74. Matsuo M. Antisense oligonucleotide-mediated exon-skipping therapies: Precision medicine spreading from Duchenne muscular dystrophy. JMA J. 2021 Jul 15;4(3):232-40. doi:10.31662/jmaj.2021-0019.
75. FDA approves drug to treat Duchenne muscular dystrophy. U.S. Food and Drug Administration. News release. Feb 9, 2017. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-drug-treat-duchenne-muscular-dystrophy.74.
76. Duan D. Dystrophin gene replacement and gene repair therapy for Duchenne muscular dystrophy in 2016: An interview. Hum Gene Ther Clin Dev. 2016 Mar;27(1):9-18. doi:10.1089/humc.2016.001.
77. EXONDYS 51®. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/drug-development-pipeline/exondys-51/
78. Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Placebo-Controlled, Multiple Dose Efficacy, Safety, Tolerability and Pharmacoki-netics Study of AVI-4658(Eteplirsen),in the Treatment of Ambulant Subjects With Duchenne Muscular Dystrophy. clinicaltrials.gov; 2020. Ac-cessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT01396239
79. Sarepta Therapeutics, Inc. Clinical Study to Assess the Safety Fo AVI-4658 in Subjects With Duchenne Muscular Dystrophy Due to a Frame-Shift Mutation Amenable to Correction by Skipping Exon 51. clinicaltrials.gov; 2015. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/study/NCT00844597
80. Sarepta Therapeutics, Inc. A 2-part, randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study (Part 1) followed by an open-label efficacy and safety evaluation (Part 2) of SRP-4053 in patients with Duchenne muscular dystrophy amenable to exon 53 skipping. ClinicalTrials.gov Identifier: NCT02310906. Updated Oct 19, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02310906.
81. Commissioner O of the. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. FDA. Published March 24, 2020. Accessed August 21, 2022. hDuchenne Muscular Dystrophy Amenable to Exon 51-Skipping Treatment. clinicaltrials.gov; 2022. Accessed Au-gust 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04004065
109. National Center of Neurology and Psychiatry, Japan. Exploratory study of NS-065/NCNP-01 in Duchenne muscular dystrophy. ClinicalTri-als.gov Identifier: NCT02081625; Updated Feb 26, 2020. Accessed Mar 2, 2022. https://clinicaltrialsttps://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-drug-duchenne-muscular- dys-trophy
82. Duchenne Drug Development Pipeline. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/duchenne-drug-development-pipeline/
83. Sarepta Therapeutics Provides Update on SRP-5051 for the Treatment of Duchenne Muscular Dystrophy | Sarepta Therapeutics, Inc. Ac-cessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics- pro-vides-update-srp-5051-treatment-duchenne
84. Sarepta Therapeutics, Inc. An Open-Label Extension Study for Patients With Duchenne Muscular Dystrophy Who Participated in Studies of SRP-5051. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03675126
85. VYONDYS 53. Prescribing information. Sarepta Therapeutics Inc.; 2019. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211970s000lbl.pdf.
86. NS Pharma Inc. Long-term use of viltolarsen in boys with Duchenne muscular dystrophy in clinical practice (VILT-502). ClinicalTrials.gov Identifier: NCT04687020. Updated Nov 22, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04687020.
87. VILTEPSO. Prescribing information. NS Pharma; 2020. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212154s000lbl.pdf.
88. FDA approves targeted treatment for rare Duchenne muscular dystrophy mutation. U.S. Food and Drug Administration. News release. Feb 25, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation-0.
89. Sarepta Therapeutics Inc. A double-blind, placebo-controlled, multi-center study with an open-label extension to evaluate the efficacy and safety of SRP-4045 and SRP-4053 in patients with Duchenne muscular dystrophy. Clinicaltrials.gov Identifier: NCT02500381. Updated Aug 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02500381.
90. AMONDYS 45. Prescribing information. Sarepta Therapeutics Inc.; 2021. Accessed Feb 22, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213026lbl.pdf.
91. Finkel RS et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dys-trophy. PLoS ONE. 2013;8(12):e81302. doi:10.1371/journal.pone.0081302.
92. PTC Therapeutics. A phase 2 study of PTC124 as an oral treatment for nonsense-mutation-mediated Duchenne muscular dystrophy. Clini-calTrials.gov Identifier: NCT00264888. Updated Jan 14, 2009. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00264888.
93. PTC Therapeutics. A phase 2B efficacy and safety study of PTC124 in subjects with nonsense-mutation-mediated Duchenne and Becker muscular dystrophy. ClinicalTrials.gov Identifier: NCT00592553. Updated Apr 7, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00592553.
94. PTC Therapeutics. A phase 3 efficacy and safety study of ataluren in patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01826487. Updated Aug 4, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01826487.
95. Bushby K et al; PTC124-GD-007-DMD Study Group. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014 Oct;50(4):477-87. doi:10.1002/mus.24332.
96. Solid Biosciences LLC. A randomized, controlled, open-label, single-ascending dose, phase I/II study to investigate the safety and tolerabil-ity, and efficacy of intravenous SGT-001 in male adolescents and children with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03368742. Updated Aug 24, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03368742.
97. Solid Biosciences reports 1.5-year data from patients in the ongoing IGNITE DMD phase I/II clinical trial of SGT-001. Press release. Solid Biosciences. Sep 27, 2021. Accessed Mar 2, 2022. http://www.solidbio.com/about/media/press-releases/solid-biosciences-reports-1-5-year-data-from-patients-in-the-ongoing-ignite-dmd-phase-i-ii-clinical-trial-of-sgt-001.
98. Potter RA et al. Dose-escalation study of systemically delivered rAAVrh74.MHCK7.microdystrophin in the mdx mouse model of Duchenne muscular dystrophy. Hum Gene Ther. 2021 Apr;32(7-8):375-89. doi:10.1089/hum.2019.255.
99. Sarepta Therapeutics, Inc. A Phase 3 Multinational, Randomized, Double-Blind, Placebo- Controlled Systemic Gene Delivery Study to Evaluate the Safety and Efficacy of SRP-9001 in Patients With Duchenne Muscular Dystrophy (EMBARK). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05096221
100. Pfizer. A PHASE 3, MULTICENTER, RANDOMIZED, DOUBLE-BLIND, PLACEBO CONTROLLED STUDY TO EVALUATE THE SAFETY AND EFFICACY OF PF 06939926 FOR THE TREATMENT OF DUCHENNE MUSCULAR DYSTROPHY. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04281485
101. Pfizer. A phase 1B multicenter open-label, single ascending dose study to evaluate the safety and tolerability of PF-06939926 in ambula-tory and non-ambulatory subjects with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03362502. Updated Mar 2, 2022. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03362502.
102. MS MW. Phase 3 CIFFREO DMD Gene Therapy Trial Slated to Begin in June in US. Accessed August 21, 2022. https://musculardystrophynews.com/news/phase-3-trial-of-pfizers-gene-therapy- expected-to-open-in-us-in-june/
103. SRP-9001. Parent Project Muscular Dystrophy. Accessed August 22, 2022. https://www.parentprojectmd.org/drug-development-pipeline/srp-9001-micro-dystrophin-gene- transfer/
104. Sarepta Therapeutics’ Investigational Gene Therapy SRP-9001 for Duchenne Muscular Dystrophy Demonstrates Significant Functional Improvements Across Multiple Studies | Sarepta Therapeutics, Inc. Accessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release- details/sarepta-therapeutics-investigational-gene-therapy-srp-9001
105. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Efficacy Study of Eteplirsen in Patients With Duchenne Muscular Dys-trophy Who Have Completed Study 4658-102.clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03985878
106. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Pharmacokinetics Study of Eteplirsen in Young Patients With Duchenne Mus-cular Dystrophy Amenable to Exon 51 Skipping. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03218995
107.Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Dose Finding and Comparison Study of the Safety and Efficacy of a High Dose of Eteplirsen, Preceded by an Open-Label Dose Escalation, in Patients With Duchenne Muscular Dystrophy With Deletion Mutations Amenable to Exon 51 Skipping. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03992430
108. Sarepta Therapeutics, Inc. A Phase 2, Two-Part, Multiple-Ascending-Dose Study of SRP-5051 for Dose Determination, Then Dose Ex-pansion, in Patients With .gov/ct2/show/NCT02081625.
110. NS Pharma Inc. A phase II, dose finding study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT02740972. Updated Dec 7, 2021. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02740972.
111. NS Pharma Inc. A phase II, open-label, extension study to assess the safety and efficacy of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT03167255. Updated Nov 24, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03167255.
112. NS Pharma Inc. A phase 2 open label study to assess the safety, tolerability, and efficacy of viltolarsen in ambulant and non-ambulant boys with Duchenne muscular dystrophy (DMD) compared with natural history controls. ClinicalTrials.gov Identifier: NCT04956289. Updated Feb 1, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04956289.
113. NS Pharma Inc. A phase 3 randomized, double-blind, placebo-controlled, multi-center study to assess the efficacy and safety of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04060199. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04060199.
114. NS Pharma Inc. A phase 3, multi-center, open-label extension study to assess the safety and efficacy of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04768062. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04768062.
115. Sarepta Therapeutics Inc. A randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study followed by an open-label safety and efficacy evaluation of SRP-4045 in advanced-stage patients with Duchenne muscular dystrophy amena-ble to exon 45 skipping. ClinicalTrials.gov Identifier: NCT02530905. Updated May 17, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02530905.
116. Sarepta Therapeutics Inc. Long-term, open-label extension study for patients with Duchenne muscular dystrophy enrolled in clinical trials evaluating casimersen or golodirsen. ClinicalTrials.gov Identifier: NCT03532542. Updated Dec 20, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03532542.
117. PTC Therapeutics. A phase 2 study of the safety, pharmacokinetics, and pharmacodynamics of ataluren (PTC124®) in patients aged ≥2 to <5 years old with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT02819557. Updated Aug 28, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02819557.
118. PTC Therapeutics. Phase 2, non-interventional, clinical study to assess dystrophin levels in subjects with nonsense mutation Duchenne muscular dystrophy who have been treated with ataluren for ≥ 9 months. ClinicalTrials.gov Identifier: NCT03796637. Updated Apr 10, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03796637.
119. PTC Therapeutics. An Open-Label Study Evaluating the Safety and Pharmacokinetics of Ataluren in Children From ≥6 Months to <2 Years of Age With Nonsense Mutation Duchenne Muscular Dystrophy. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04336826 120. PTC Therapeutics. An open-label study for previously treated ataluren (PTC124®) pa-tients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01557400. Updated Nov 25, 2020. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT01557400.
121. PTC Therapeutics. An open-label, safety study for ataluren (PTC124) patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01247207. Updated Feb 16, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT01247207.
122. PTC Therapeutics. A phase 3, randomized, double-blind, placebo-controlled efficacy and safety study of ataluren in patients with non-sense mutation Duchenne muscular dystrophy and open-label extension. ClinicalTrials.gov Identifier: NCT03179631. Updated Feb 8, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03179631.
123. Sarepta Therapeutics, Inc. An Open-Label, Systemic Gene Delivery Study Using Commercial Process Material to Evaluate the Safety of and Expression From SRP-9001 in Subjects With Duchenne Muscular Dystrophy (ENDEAVOR). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04626674
124. Sarepta Therapeutics, Inc. Systemic Gene Delivery Phase I/IIa Clinical Trial for Duchenne Muscular Dystrophy Using RAA-Vrh74.MHCK7.Micro-Dystrophin (MicroDys-IV-001). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03375164
125. Sarepta Therapeutics Inc. A multicenter, randomized, double-blind, placebo-controlled trial for Duchenne muscular dystrophy using SRP-9001. ClinicalTrials.gov Identifier: NCT03769116. Updated Dec 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03769116.
126. Hoffmann-La Roche. A Two-Part, Seamless, Multi-Center, Randomized, Placebo-Controlled, Double-Blind Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of RO7204239 in Combination With Risdiplam (RO7034067) in Ambulant Pa-tients With Spinal Muscular Atrophy. clinicaltrials.gov; 2022. Accessed September 1, 2022. https://clinicaltrials.gov/ct2/show/NCT05115110
Neuromuscular diseases (NMDs) are a broad classification of heterogeneous groups of disorders characterized by progressive muscle weakness resulting from muscle or nerve dysfunction.1 Diagnosis is based on symptoms and a full medical history, as well as on muscle and imaging tests (including electromyography, nerve-conduction studies, magnetic resonance imaging, muscle biopsy, and blood tests) to confirm or rule out specific NMDs.2 Early diagnosis of NMDs can be difficult because symptoms overlap with those of many other diseases.
Although individually, NMDs are rare, collectively, they affect approximately 250,000 people in the United States. Disease types vary in regard to cause, symptoms, prevalence, age of onset, progression, and severity. Functional impairment from any NMD can lead to lifelong morbidities and shortened life expectancy.1,3
Treatment options for NMDs are limited; most target symptoms, not disease progression. Although there is a need for safe and effective gene-based therapies for NMDs, there are challenges to developing and delivering such treatments that have impeded clinical success. These include a lack of understanding about disease pathology and drug targets, limited animal model systems, and few reliable biomarkers that are predictive of therapeutic success.4,5
Notwithstanding that challenges remain, our understanding of gene expression in NMDs has greatly advanced in the past few decades. This progress has translated into promising results in the gene-therapy field – thereby setting the stage for therapeutic approaches that use novel gene-delivery and gene-manipulation tools.6 These novel approaches include nonviral strategies, such as antisense oligonucleotides (ASOs), and viral-based strategies, such as adeno-associated virus (AAV)-mediated gene silencing and AAV-mediated gene delivery.
In this article, we highlight advancements in the clinical development of gene-based therapies for NMDs. We focus on amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), and Duchenne muscular dystrophy (DMD) because of recent clinical successes in developing such therapies.1,6,7 We also catalog completed and ongoing clinical trials for ALS, SMA, and DMD (Tables 1-3).
Amyotrophic lateral sclerosis
ALS is caused by progressive degeneration of upper- and lower-motor neurons, which eventually leads to respiratory failure and death 3 to 5 years after disease onset.7-9 There are two subtypes: Familial ALS (10% of cases) and sporadic ALS (90% of cases). Commonly mutated ALS-associated genes6,8 are:
- Superoxide dismutase type 1 (SOD1).
- Chromosome 9 open reading frame 72 (C9orf72).
- Transactive response DNA-binding protein 43 (TARDBP).
- Fused in sarcoma (FUS).
SOD1-targeted therapy is being studied, with early evidence of clinical success. Mutations in SOD1 account for 10% to 20% of familial ALS cases and 1% to 2% of sporadic ALS cases.6,10 10 Mutations in C9orf72 account for 25 to 40% of familial ALS cases and 7% of sporadic ALS cases.8,9,11 Mutations in TARDBP account for 3% of familial ALS cases and 2% of sporadic cases.12 Mutations in FUS account for 4% of familial ALS cases and 1% of sporadic cases. Overall, these mutant proteins can trigger neurotoxicity, thus inducing motor-neuron death.6,10
Treatment of ALS
Two treatments for ALS are Food and Drug Administration approved: riluzole (Rilutek), approved in 1995, and edaravone (Radicava), approved in 2017.
Riluzole is an oral anti-excitotoxic glutamate antagonist.11 Approval of riluzole was based on the results of two studies that demonstrated a 2- to 3-month survival benefit.10,14 For patients who have difficulty swallowing, an oral suspension (Tiglutik, approved in 2018) and an oral film (Exservan, approved in 2019) are available.
Edaravone is a free-radical scavenger that decreases oxidative stress and is administered intravenously (IV).9,13,14 Findings from clinical trials suggest functional improvement or slower decline in function for some patients.
Although these two agents demonstrate modest therapeutic benefit, neither reverses progression of disease.10,14
Gene-based therapy for ALS
Many non-viral strategies, including antisense oligonucleotide (ASO), monoclonal antibodies, reverse transcriptase inhibitors, and HGF gene replacement therapy are used as therapeutic approaches to SOD1, C9orf72, and FUS gene mutations in ALS patients, and are being evaluated in clinical studies14,15 (Table 113-17).
Tofersen, also known as BIIB067, is an investigational ASO, administered by intrathecal (IT) injection, that binds to SOD1 mRNA, thus reducing its protein levels.16 Tofersen was evaluated in the VALOR phase 3 study (ClinicalTrials.gov Identifier: NCT02623699), a three-part randomized, double-blind, placebo-controlled trial: single ascending dose (Part A), multiple ascending dose (B), and fixed dose (C).10 In Parts A and B, 48 participants received five IT injections of tofersen or placebo over 12 weeks and were followed for an additional 12 weeks. Reduction in SOD1 protein production and neurofilament level in cerebrospinal fluid (CSF) (a potential biomarker of motor-neuron degeneration) was observed, which determined the fixed-dose for Part C.16,17
Part C examined the efficacy, safety and tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of tofersen, compared with placebo, in adults with ALS who had a confirmed SOD1 mutation.17 A total of 108 participants were enrolled; 60 were identified as “faster-progressing”; 48, as “slower-progressing.”18 The primary endpoint of Part C was change from baseline to Week 28 on the Revised ALS Functional Rating Scale (ALSFRS-R) total score. (ALSFRS-R measures overall clinical effect; the score ranges from 0 [no function] to 4 [full function].17)
Tofersen failed to meet the primary efficacy outcome because statistically significant findings were lacking in the faster-progressing population, as measured by joint-rank analysis (difference of 1.2 on the ALSFRS-R score; P = .97). However, trends favoring tofersen were observed across key secondary clinical outcome measures18:
- Change from baseline in CSF SOD1 protein concentration.17 Percent reduction in the total SOD1 protein level was much higher in the tofersen-treated group than in the control group (38% more than controls in the faster-progressing population; 26% more than controls in the slower-progressing population).18
- Change from baseline in neurofilament light-chain concentration in plasma.17,18 Percent reduction in the level of neurofilament light chain was also observed to be higher in the tofersen-treated group than in the control group (67% more than controls in the faster-progressing population and 48% more than controls in the slower-progressing population).18
Because of these encouraging results, VALOR participants were moved to the ongoing open-label extension trial of tofersen (ClinicalTri-als.gov Identifier: NCT03070119), in which both groups were treated with the active agent.
These data suggest that early tofersen treatment might slow decline in faster-progressing patients and stabilize clinical function in slower-progressing patients.18,19 Overall, most adverse events (AEs) in the trial among patients receiving active treatment were of mild or moderate severity, and were largely consistent with either disease progression or lumbar puncture–related complications.18
Because data from VALOR suggested potential benefit from tofersen, the ATLAS trial (ClinicalTrials.gov Identifier: NCT04856982) is investigating the clinical value of presymptomatic treatment and the optimal timing of initiation of therapy.20,21 ATLAS is a phase 3, randomized, placebo-controlled trial that examines the clinical efficacy, safety, and tolerability of tofersen in presymptomatic adult carriers of SOD1 mutation who have an elevated neurofilament light-chain concentration.21 ATLAS will also evaluate the efficacy of tofersen when initiated before, rather than after, ALS manifests clinically. Enrollment is still open for this trial.20,21
Latozinemab, also known as AL001, is a first-in-class monoclonal antibody, administered by IV infusion, that elevates levels of progranulin, a key regulator of the immune activity and lysosomal function in the brain.22,23 Latozinemab limits progranulin endocytosis and degradation by sortilin inhibition.22 Progranulin gene mutations can reduce progranulin expression (by 50 to 70 percent reduction), which may cause neuro-degeneration due to abnormal accumulation of TAR-DNA-binding protein 43 (TDP-43) in the brain cells.22,24 TDP-43 pathology has also been shown to be associated with C9orf72 mutations.23 Although the mechanism is not fully understood, the role of progranulin deficiency in TDP-43 pathology is believed to be associated with neurodegenerative diseases like ALS.11,23,24,43 Previous animal models of chronic neurodegenera-tion have demonstrated how increased progranulin levels can be protective against TDP-43 pathology, increasing neuronal development and survival, thus potentially slowing disease progression.23,24,43 Currently, latozinemab is being investigated in a randomized, double-blind, placebo-controlled, multicenter phase 2 trial (ClinicalTrials.gov Identifier: NCT05053035). Approximately, 45 C90rf72-associated ALS participants (≥ 18 years of age) will receive latozinemab or placebo infusions every 4 weeks (for 24 weeks). Study endpoints include safety, tolerability, PK, PD, as well as plasma, and CSF progranulin levels.25 In previous studies, latozinemab demonstrated encouraging results in frontotemporal dementia (FTD) patients who carry a progranulin mutation. Because FTD was revealed to have significant genetic overlap with ALS, there is disease-modifying potential for latozinemab in ALS patients.23,24
TPN-101 is a nucleoside analog reverse transcriptase inhibitor, administered orally, that was originally developed for human immunodeficiency virus (HIV) treatment. However, due to recent findings suggesting retrotransposon activity contributing to neurodegeneration in TDP-43 mediated diseases, including ALS and FTD, TNP-101 is being repurposed.26 The safety and tolerability of TNP-101 are currently being evaluated in C9orf72-associated ALS and FTD patients (≥ 18 years of age). The study is a randomized, double-blind, placebo-controlled paral-lel-group phase 2a trial (ClinicalTrials.gov Identifier: NCT04993755) The study includes a screening period of 6 weeks, double-blind treatment period of 24 weeks, an open-label treatment period of 24 weeks, and 4 weeks of the post-treatment follow-up visit. Study endpoints include the incidence and severity of spontaneously reported treatment-emergent adverse events (TEAEs) associated with TNP-101 and placebo for a to-tal of 48 weeks.27
ION363 is an investigational ASO, administered by IT injection, that selectively targets one of the FUS mutations (p.P525L), which is responsible for earlier disease onset and rapid ALS progression.28,29 The clinical efficacy of ION363, specifically in clinical function and survival is being assessed in FUS-associated ALS patients (≥ 12 years of age). This randomized phase 3 study (ClinicalTrials.gov Identifier: NCT04768972) includes two parts; part 1 will consist of participants receiving a multi-dose regimen (1 dose every 4-12 weeks) of ION363 or placebo for 61 weeks followed by an open-label extension treatment period in part 2, which will consist of participants receiving ION363 (every 12 weeks) for 85 weeks. The primary endpoint of the study is the change from baseline to day 505 in functional impairment, using ALS Functional Rating Scale-Revised (ALSFRS-R). This measures functional disease severity, specifically in bulbar function, gross motor skills, fine motor skills, and respiratory. The score for all 12 questions can range from 0 (no function) to 4 (full function) with a total possible score of 48.30
Engensis, also known as VM202, is a non-viral gene therapy, administered by intramuscular (IM) injection, that uses a plasmid to deliver the hepatocyte growth factor (HGF) gene to promote HGF protein production. The HGF protein plays a role in angiogenesis, the previous of muscle atrophy, and the promotion of neuronal survival and growth. Based on preclinical studies, increasing HGF protein production has been shown to reduce neurodegeneration, thus potentially halting or slowing ALS progression.31 Currently, the safety of engensis is being evaluated in ALS patients (18-80 years of age) in the REViVALS phase 2a (ClinicalTrials.gov Identifier: NCT04632225)/2b (ClinicalTrial.gov Identifier: NCT05176093).32,33 The ReViVALS trial is a double-blind, randomized, placebo-controlled, multi-center study. The phase 2a study endpoints include the incidence of TEAEs, treatment-emergent serious adverse events (TESAEs), injection site reactions, and clinically significant labor-atory values post-treatment (engensis vs placebo group) for 180 days.33 A phase 2b study will evaluate the long-term safety of engensis for an additional 6 months. Study endpoints include the incidence of AEs, changes from baseline in ALSFRS-R scores to evaluate improvement in muscle function, changes from baseline in quality of life using the ALS patient assessment questionnaire, time to all-cause mortality compared to placebo, etc.32
Spinal muscular atrophy
SMA is a hereditary lower motor-neuron disease caused (in 95% of cases) by deletions or, less commonly, by mutations of the survival motor neuron 1 (SMN1) gene on chromosome 5q13 that encodes the SMN protein.6 Reduction in expression of the SMN protein causes motor neurons to degenerate.36-38 Because of a large inverted duplication in chromosome 5q, two variants of SMN (SMN1 and SMN2) exist on each allele. The paralog gene, SMN2, also produces the SMN protein – although at a lower level (10% to 20% of total SMN protein production) than SMN1 does.
A single nucleotide substitution in SMN2 alters splicing and suppresses transcription of exon 7, resulting in a shortened mRNA strand that yields a truncated SMN protein product.6,37,39 SMA is classified based on age of onset and maximum motor abilities achieved, ranging from the most severe (Type 0) to mildest (Type 4) disease.36,40 Because SMA patients lack functional SMN1 (due to polymorphisms), disease severity is determined by copy numbers of SMN2.6,39
Gene-based therapy for SMA
Three FDA-approved SMN treatments demonstrate clinically meaningful benefit in SMA: SMN2-targeting nusinersen [Spinraza] and risdiplam [Evrysdi], and SMN1-targeting onasemnogene abeparvovec-xioi [Zolgensma]38 Additional approaches to SMA treatment are through SMN-independent therapies, which target muscle and nerve function. Research has strongly suggested that combined SMA therapies, specifically approved SMN-targeted and investigational SMN-independent treatments, such as GYM329 (also known as RO7204239) may be the best strategy to treat all ages, stages, and types of SMA.41 (Table 226-41).
Agents that modulate SMN2. Nusinersen, approved by the FDA in 2016, was the first treatment indicated for all SMA types in pediatric and adult patients.42 The agent is an ASO that targets exon 7 of SMN2, thus stabilizing transcription. Inclusion of exon 7 increases SMN protein production, improving motor function.6,38 Nusinersen is a lifelong treatment that requires IT administration every 4 months because it cannot cross the blood-brain barrier.38,43
Pivotal clinical studies that led to approval of nusinersen include CHERISH (ClinicalTrial.gov Identifier: NCT02292537) and ENDEAR (ClinicalTrial.gov Identifier: NCT02193074) studies.
CHERISH was a phase 3, randomized, double-blind, sham procedure–controlled trial that examined the clinical efficacy and safety of nusinersen in 126 participants with later-onset SMA (2-12 years of age). The primary endpoint was the change from baseline using the Hammersmith Functional Motor Scale Expanded (HFMSE) at 15 months. HFMSE looks at 33 activities to assess improvement in motor function. The study met the primary efficacy outcome, demonstrating statistically significant (P = .0000001) improvement in overall motor function. The nusinersen group showed a 3.9-point increase in the HFMSE score from baseline, which indicates improvement, compared with a 1.0-point decline from baseline in the control group.46,47
ENDEAR was also a randomized, double-blind, sham procedure–controlled phase 3 trial, which investigated the efficacy and safety of nusinersen in 121 participants with early-onset SMA Type 1 (≤ 210 days of age). Coprimary endpoints were:
- Percentage of motor milestones responders, as determined using Section 2 of the Hammersmith Infant Neurological Examination–Part 2.
- Event-free survival (that is, avoidance of combined endpoint of death or permanent ventilation).
ENDEAR met the first primary efficacy outcome, demonstrating statistically significant (P < .0001) improvement in motor milestones (head control, rolling, independent sitting, and standing). By 13 months of age, approximately 51% of nusinersen-treated participants showed improvement, compared with none in the control group.46,47
The second primary endpoint was also met, with a statistically significant (P = .005) 47% decrease in mortality or permanent ventilation use.46-48
The NURTURE (ClinicalTrial.gov Identifier: NCT02386553) study is also investigating the efficacy and safety of nusinersen. An ongoing, open-label, supportive phase 2 trial, NURTURE is evaluating the efficacy and safety of multiple doses of nusinersen in 25 presymptomatic SMA patients (≤ 6 weeks of age). The primary endpoint of this study is time to death or respiratory intervention.49 Interim results demonstrate that 100% of presymptomatic infants are functioning without respiratory intervention after median follow-up of 2.9 years.46-48
Although nusinersen has been shown to be generally safe in clinical studies, development of lumbar puncture–related complications, as well as the need for sedation during IT administration, might affect treatment tolerability in some patients.39
Risdiplam was approved by the FDA in 2020 as the first orally administered small-molecule treatment of SMA (for patients ≤ 2 months of age).52 Risdiplam is a SMN2 splicing modifier, binding to the 5’ splice site of intron 7 and exonic splicing enhancer 2 in exon 7 of SMN2 pre-mRNA. This alternative splicing increases efficiency in SMN2 gene transcription, thus increasing SMN protein production in motor-neuron cells.36 An important advantage of risdiplam is the convenience of oral administration: A large percentage of SMA patients (that is, those with Type 2 disease) have severe scoliosis, which can further complicate therapy or deter patients from using a treatment that is administered through the IT route.40
FDA approval of risdiplam was based on clinical data from two pivotal studies, FIREFISH (ClinicalTrial.gov Identifier: NCT02913482) and SUNFISH (ClinicalTrial.gov Identifier: NCT02908685).53-54
FIREFISH is an open-label, phase 2/3 ongoing trial in infants (1-7 months of age) with SMA Type 1. The study comprises two parts; Part 1 determined the dose of risdiplam used in Part 2, which assessed the efficacy and safety of risdiplam for 24 months. The primary endpoint was the percentage of infants sitting without support for 5 seconds after 12 months of treatment using the gross motor scale of the Bayley Scales of Infant and Toddler Development–Third Edition. A statistically significant (P < .0001) therapeutic benefit was observed in motor milestones. Approximately 29% of infants achieved the motor milestone of independent sitting for 5 seconds, which had not been observed in the natural history of SMA.53-55
SUNFISH is an ongoing randomized, double-blind, placebo-controlled trial of risdiplam in adult and pediatric patients with SMA Types 2 and 3 (2-25 years old). This phase 2/3 study comprises two parts: Part 1 determined the dose (for 12 weeks) to be used for confirmatory Part 2 (for 12 to 24 months). The primary endpoint was the change from baseline on the 32-item Motor Function Measure at 12 months. The study met its primary endpoint, demonstrating statistically significant (P = .0156) improvement in motor function scores, with a 1.36-point increase in the risdiplam group, compared with a 0.19-point decrease in the control group.54,55
Ongoing risdiplam clinical trials also include JEWELFISH (ClinicalTrial.gov Identifier: NCT03032172) and RAINBOW (ClinicalTrial.gov Identifier: NCT03779334).56-57 JEWELFISH is an open-label, phase 2 trial assessing the safety of risdiplam in patients (6 months to 60 years old) who received prior treatment. The study has completed recruitment; results are pending.56 RAINBOW is an ongoing, open-label, single-arm, phase 2 trial, evaluating the clinical efficacy and safety of risdiplam in SMA-presymptomatic newborns (≤ 6 weeks old). The study is open for enrollment.57 Overall, interim results for JEWELFISH and RAINBOW appear promising.
In addition, combined SMA therapies, specifically risdiplam and GYM329 are currently being investigated to address the underlying cause and symptoms of SMA concurrently.58 GYM329, is an investigational anti-myostatin antibody, selectively binding preforms of myostatin - pro-myostatin and latent myostatin, thus improving muscle mass and strength for SMA patients.59 The safety and efficacy of GYM329 in combination with risdiplam is currently being investigated in 180 ambulant participants with SMA (2-10 years of age) in the MANATEE (ClinicalTrial.gov Identifier: NCT05115110) phase 2/3 trial. The MANATEE study is a two-part, seamless, randomized, placebo-controlled, double-blind trial. Part 1 will assess the safety of the combination treatment in approximately 36 participants; participants will receive both GYM329 (every 4 weeks) by subcutaneous (SC) injection into the abdomen and risdiplam (once per day) for 24 weeks followed by a 72-week open-label treatment period. 54,58 The outcome measures include the incidence of AEs, percentage change from baseline in the contractile area of skeletal muscle (in dominant thigh and calf), change from baseline in RHS total score, and incidence of change from baseline in serum concentration (total myostatin, free latent myostatin, and mature myostatin) etc.54 Part 2 will be conducted on 144 participants, specifically assessing the efficacy and safety of the optimal dose of GYM329 selected from Part 1 (combined with risdiplam) for 72 weeks. Once the treatment period is completed in either part, participants can partake in a 2-year open-label extension period.54,58 Other outcome measures include change from baseline in lean muscle mass (assessed by full body dual-energy X- ray absorptiometry (DXA) scan), in time taken to walk/run 10 meters (measured by RHS), in time taken to rise from the floor (measured by RHS), etc.54 Overall, this combination treatment has the potential to further improve SMA patient outcomes and will be further investigated in other patient populations (including non-ambulant patients and a broader age range) in the future.58
An agent that alters SMN1 expression. Onasemnogene abeparvovec-xioi, FDA approved in 2019, was the first gene-replacement therapy indicated for treating SMA in children ≤ 2 years old.60 Treatment utilizes an AAV vector type 9 (AAV9) to deliver a functional copy of SMN1 into target motor-neuron cells, thus increasing SMN protein production and improving motor function. This AAV serotype is ideal because it crosses the blood-brain barrier. Treatment is administered as a one-time IV fusion.38,39,43
FDA approval was based on the STR1VE (ClinicalTrial.gov Identifier: NCT03306277) phase 3 study and START (ClinicalTrial.gov Identifier: NCT02122952) phase 1 study.61,62 START was the first trial to investigate the safety and efficacy of onasemnogene abeparvovec-xioi in SMA Type 1 infants (< 6 months old). Results demonstrated remarkable clinical benefit, including 100% permanent ventilation-free survival and a 92% (11 of 12 patients) rate of improvement in motor function. Improvement in development milestones was also observed: 92% (11 of 12 patients) could sit without support for 5 seconds and 75% (9 of 12) could sit without support for 30 seconds.14,61,63
The efficacy of onasemnogene abeparvovec-xioi seen in STR1VE was consistent with what was observed in START. STRIVE, a phase 3 open-label, single-dose trial, examined treatment efficacy and safety in 22 symptomatic infants (< 6 months old) with SMA Type 1 (one or two SMN2 copies). The primary endpoint was 30 seconds of independent sitting and event-free survival. Patients were followed for as long as 18 months. Treatment showed statistically significant (P < .0001) improvement in motor milestone development and event-free survival, which had not been observed in SMA Type 1 historically. Approximately 59% (13 of 22 patients) could sit independently for 30 seconds at 18 months of age. At 14 months of age, 91% (20 of 22 patients) were alive and achieved independence from ventilatory support.34,35,53
Although many clinical studies suggest that onasemnogene abeparvovec-xioi can slow disease progression, the benefits and risks of long-term effects are still unknown. A 15-year observational study is investigating the long-term therapeutic effects and potential complications of onasemnogene abeparvovec-xioi. Participants in START were invited to enroll in this long-term follow-up study (ClinicalTrial.gov Identifier: NCT04042025).66-67
Duchenne muscular dystrophy
DMD is the most common muscular dystrophy of childhood. With an X-linked pattern of inheritance, DMD is seen mostly in young males (1 in every 3,500 male births).38,39,73 DMD is caused by mutation of the dystrophin encoding gene, or DMD, on the X chromosome. Deletion of one or more exons of DMD prevents production of the dystrophin protein, which leads to muscle degeneration.38,39,43 Common DMD deletion hotspots are exon 51 (20% of cases), exon 53 (13% of cases), exon 44 (11% of cases), and exon 45 (12% of cases).74 Nonsense mutations, which account for another 10% of DMD cases, occur when premature termination codons are found in the DMD gene. Those mutations yield truncated dystrophin protein products.39,66
Therapy for DMD
There are many therapeutic options for DMD, including deflazacort (Emflaza), FDA approved in 2017, which has been shown to reduce inflammation and immune system activity in DMD patients (≥ 5 years old). Deflazacort is a corticosteroid prodrug; its active metabolite acts on the glucocorticoid receptor to exert anti-inflammatory and immunosuppressive effects. Studies have shown that muscle strength scores over 6-12 months and average time to loss of ambulation numerically favored deflazacort over placebo.74,75
Gene-based therapy for DMD
Mutation-specific therapeutic approaches, such as exon skipping and nonsense suppression, have shown promise for the treatment of DMD (Table 358-79):
- ASO-mediated exon skipping allows one or more exons to be omitted from the mutated DMD mRNA.74,75 Effective FDA-approved ASOs include golodirsen [Vyondys 53], viltolarsen [Viltepso], and casimersen [Amondys 45].74
- An example of therapeutic suppression of nonsense mutations is ataluren [Translarna], an investigational agent that can promote premature termination codon read-through in DMD patients.66
Another potential treatment approach is through the use of AAV gene transfer to treat DMD. However, because DMD is too large for the AAV vector (packaging size, 5.0 kb), microdystrophin genes (3.5-4 kb, are used as an alternative to fit into a single AAV vector.39,76
Exon skipping targeting exon 51. Eteplirsen, approved in 2016, is indicated for the treatment of DMD patients with the confirmed DMD gene mutation that is amenable to exon 51 skipping. Eteplirsen binds to exon 51 of dystrophin pre-mRNA, causing it to be skipped, thus, restoring the reading frame in patients with DMD gene mutation amenable to exon 51 skipping. This exclusion promotes dystrophin production. Though the dystrophin protein is still functional, it is shortened.38,77 Treatment is administered IV, once a week (over 35-60 minutes). Eteplirsen’s accelerated approval was based on 3 clinical studies (ClinicalTrial.gov Identifier: NCT01396239, NCT01540409, and NCT00844597.) 78-81 The data demonstrated an increased expression of dystrophin in skeletal muscles in some DMD patients treated with eteplirsen. Though the clinical benefit of eteplirsen (including improved motor function) was not established, it was concluded by the FDA that the data were reasonably likely to predict clinical benefit. Continued approval for this indication may depend on the verification of a clinical benefit in confirmatory trials. Ongoing clinical trials include (ClinicalTrial.gov Identifier: NCT03992430 (MIS51ON), NCT03218995, and NCT03218995).77,81,82
Vesleteplirsen, is an investigational agent that is designed for DMD patients who are amendable to exon 51 skip-ping. The mechanism of action of vesleteplirsen appears to be similar to that of eteplirsen.83 The ongoing MOMENTUM (ClinicalTrial.gov Identifier: NCT04004065) phase 2 trial is assessing the safety and tolerability of vesleteplirsen at multiple-ascending dose levels (administered via IV infusion) in 60 participants (7-21 years of age). The study consists of two parts; participants receive escalating dose levels of vesleteplirsen (every 4 weeks) for 72 weeks during part A and participants receive the selected doses from part A (every 4 weeks) for 2 years during part B. Study endpoints include the number of AEs (up to 75 weeks) and the change from baseline to week 28 in dystrophin protein level. 84 Serious AEs of reversible hypomagnesemia were observed in part B, and as a result, the study protocol was amended to include magnesium supplementation and monitoring of magnesium levels.83
Exon skipping targeting exon 53. Golodirsen, FDA approved in 2019, is indicated for the treatment of DMD in patients who have a confirmed DMD mutation that is amenable to exon 53 skipping. The mechanism of action is similar to eteplirsen, however, golodirsen is designed to bind to exon 53.38,39 Treatment is administered by IV infusion over 35-60 minutes.
Approval of golodirsen was based primarily on a two-part, phase 1/2 clinical trial (ClinicalTrial.gov Identifier: NCT02310906). Part 1 was a randomized, placebo-controlled, dose-titration study that assessed multiple-dose efficacy in 12 DMD male patients, 6 to 15 years old, with deletions that were amenable to exon 53 skipping.
Part 2 was an open-label trial in 12 DMD patients from Part 1 of the trial plus 13 newly enrolled male DMD patients who were also amenable to exon 53 skipping and who had not already received treatment. Primary endpoints were change from baseline in total distance walked during the 6-minute walk test at Week 144 and dystrophin protein levels (measured by western blot testing) at Week 48. A statistically significant increase in the mean dystrophin level was observed, from a baseline 0.10% mean dystrophin level to a 1.02% mean dystrophin level after 48 weeks of treatment (P < .001). Common reported adverse events associated with golodirsen were headache, fever, abdominal pain, rash, and dermatitis. Renal toxicity was observed in preclinical studies of golodirsen but not in clinical studies.80,85
Viltolarsen, approved in 2020, is also indicated for the treatment of DMD in patients with deletions amenable to exon 53 skipping. The mechanism of action and administration (IV infusion over 60 minutes) are similar to that of golodirsen.
Approval of viltolarsen was based on two phase 2 clinical trials (ClinicalTrial.gov Identifier: NCT02740972 and NCT03167255) in a total of 32 patients. NCT02740972 was a randomized, double-blind, placebo-controlled, dose-finding study that evaluated the clinical efficacy of viltolarsen in 16 male DMD patients (4-9 years old) for 24 weeks.
NCT03167255 was an open-label study that evaluated the safety and tolerability of viltolarsen in DMD male patients (5-18 years old) for 192 weeks. The efficacy endpoint was the change in dystrophin production from baseline after 24 weeks of treatment. A statistically significant increase in the mean dystrophin level was observed, from a 0.6% mean dystrophin level at baseline to a 5.9% mean dystrophin level at Week 25 (P = .01). The most common adverse events observed were upper respiratory tract infection, cough, fever, and injection-site reaction.86-87
Exon skipping targeting exon 45. Casimersen was approved in 2021 for the treatment of DMD in patients with deletions amenable to exon 45 skipping.88 Treatment is administered by IV infusion over 30-60 minutes. Approval was based on an increase in dystrophin production in skeletal muscle in treated patients. Clinical benefit was reported in interim results from the ESSENCE (ClinicalTrial.gov Identifier: NCT02500381) study, an ongoing double-blind, placebo-controlled phase 3 trial that is evaluating the efficacy of casimersen, compared with placebo, in male participants (6-13 years old) for 48 weeks. Efficacy is based on the change from baseline dystrophin intensity level, determined by immunohistochemistry, at Week 48.
Interim results from ESSENCE show a statistically significant increase in dystrophin production in the casimersen group, from a 0.9% mean dystrophin level at baseline to a 1.7% mean dystrophin level at Week 48 (P = .004); in the control group, a 0.54% mean dystrophin level at baseline increased to a 0.76% mean dystrophin level at Week 48 (P = .09). Common adverse events have included respiratory tract infection, headache, arthralgia, fever, and oropharyngeal pain. Renal toxicity was observed in preclinical data but not in clinical studies.60,84
Targeting nonsense mutations. Ataluren is an investigational, orally administered nonsense mutation suppression therapy (through the read-through of stop codons).37 Early clinical evidence supporting the use of ataluren in DMD was seen in an open-label, dose-ranging, phase 2a study (ClinicalTrial.gov Identifier: NCT00264888) in male DMD patients (≥ 5 years old) caused by nonsense mutation. The study demonstrated a modest (61% ) increase in dystrophin expression in 23 of 38 patients after 28 days of treatment.37,91,92
However, a phase 2b randomized, double-blind, placebo-controlled trial (ClinicalTrial.gov Identifier: NCT00592553) and a subsequent confirmatory ACT DMD phase 3 study (ClinicalTrial.gov Identifier: NCT01826487) did not meet their primary endpoint of improvement in ambulation after 48 weeks as measured by the 6-minute walk test.37,93,94 In ACT DMD, approximately 74% of the ataluren group did not experience disease progression, compared with 56% of the control group (P = 0386), measured by a change in the 6-minute walk test, which assessed ambulatory decline.37,95
Based on limited data showing that ataluren is effective and well tolerated, the European Medicines Agency has given conditional approval for clinical use of the drug in Europe. However, ataluren was rejected by the FDA as a candidate therapy for DMD in the United States.22 Late-stage clinical studies of ataluren are ongoing in the United States.
AAV gene transfer with microdystrophin. Limitations on traditional gene-replacement therapy prompted exploration of gene-editing strategies for treating DMD, including using AAV-based vectors to transfer microdystrophin, an engineered version of DMD, into target muscles.43 The microdystrophin gene is designed to produce a functional, truncated form of dystrophin, thus improving muscular function.
There are 3 ongoing investigational microdystrophin gene therapies that are in clinical development (ClinicalTrial.gov Identifier: NCT03368742 (IGNITE DMD), NCT04281485 (CIFFREO), and NCT05096221 (EMBARK)).38,82
IGNITE DMD is a phase 1/2 randomized, controlled, single-ascending dose trial evaluating the safety and efficacy of a SGT-001, single IV infusion of AAV9 vector containing a microdystrophin construct in DMD patients (4-17 years old) for 12 months. At the conclusion of the trial, treatment and control groups will be followed for 5 years. The primary efficacy endpoint is the change from baseline in microdystrophin protein production in muscle-biopsy material, using western blot testing.96 Long-term interim data on biopsy findings from three patients demonstrated clinical evidence of durable microdystrophin protein expression after 2 years of treatment.96,97
The CIFFREO trial will assess the safety and efficacy of the PF-06939926 microdystrophin gene therapy, an investigational AAV9 containing microdystrophin, in approximately 99 ambulatory DMD patients (4-7 years of age). The study is a randomized, double-blind, placebo-controlled, multicenter phase 3 trial. The primary efficacy end-point is the change from baseline in the North Star Ambulatory Assessment (NSAA), which measures gross motor function. This will be assessed at 52 weeks; all study participants will be followed for a total of 5 years post-treatment.98,99,100 Due to unexpected patient death (in a non-ambulatory cohort) in the phase 1b (in a non-ambulatory cohort) in the phase 1b (ClinicalTrial.gov Identifier: (NCT03362502) trial, microdystrophin gene therapy was immediately placed on clinical hold.101,102 The amended study protocol required that all participants undergo one week of in-hospital observation after receiving treatment.102
The EMBARK study is a global, randomized, double-blind, placebo-controlled, phase 3 trial that is evaluating the safety and efficacy of SRP-9001, which is a rAAVrh74.MHCK7.microdystrophin gene therapy. The AAV vector (rAAVrh74) contains the microdystrophin construct, driven by the skeletal and cardiac muscle–specific promoter, MHCK7.98,99 In the EMBARK study, approximately 120 participants with DMD (4-7 years of age) will be enrolled. The primary efficacy endpoint includes the change from baseline to week 52 in the NSAA total score.99 Based on SRP-9001, data demonstrating consistent statistically significant functional improvements in NSAA total scores and timed function tests (after one-year post- treatment) in DMD patients from previous studies and an integrated analysis from multiple studies (ClinicalTrial.gov Identifier: NCT03375164, NCT03769116, and NCT04626674), the ongoing EMBARK has great promise.103,104
Challenges ahead, but advancements realized
Novel gene-based therapies show significant potential for transforming the treatment of NMDs. The complex pathologies of NMDs have been a huge challenge to disease management in an area once considered unremediable by gene-based therapy. However, advancements in precision medicine – specifically, gene-delivery systems (for example, AAV9 and AAVrh74 vectors) combined with gene modification strategies (ASOs and AAV-mediated silencing) – have the potential to, first, revolutionize standards of care for sporadic and inherited NMDs and, second, significantly reduce disease burden.6
What will be determined to be the “best” therapeutic approach will, likely, vary from NMD to NMD; further investigation is required to determine which agents offer optimal clinical efficacy and safety profiles.43 Furthermore, the key to therapeutic success will continue to be early detection and diagnosis – first, by better understanding disease pathology and drug targets and, second, by validation of reliable biomarkers that are predictive of therapeutic benefit.4,5
To sum up, development challenges remain, but therapeutic approaches to ALS, SMA, and DMD that utilize novel gene-delivery and gene-manipulation tools show great promise.
Ms. Yewhalashet is a student in the masters of business and science program, with a concentration in healthcare economics, at Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences, Claremont, Calif. Dr. Davis is professor of practice in clinical and regulatory affairs, Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences.
References
1. Aitken M et al. Understanding neuromuscular disease care. IQVIA [Internet]. Oct 30, 2018. Accessed Mar 1, 2022. https://www.iqvia.com/insights/the-iqvia-institute/reports/understanding-neuromuscular-disease-care.
2. National Institute of Neurological Disorders and Stroke. Neurological diagnostic tests and procedures fact sheet. Updated Nov 15, 2021. Ac-cessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Neurological-Diagnostic-Tests-and-Procedures-Fact.
3. Deenen JCW et al. The epidemiology of neuromuscular disorders: A comprehensive overview of the literature. J Neuromuscul Dis. 2015;2(1):73-85.
4. Cavazzoni P. The path forward: Advancing treatments and cures for neurodegenerative diseases. U.S. Food and Drug Administration. Jul 29, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/congressional-testimony/path-forward-advancing-treatments-and-cures-neurodegenerative-diseases-07292021.
5. Martier R, Konstantinova P. Gene therapy for neurodegenerative diseases: Slowing down the ticking clock. Front Neurosci. 2020 Sep 18;14:580179. doi: 10.3389/fnins.2020.580179.
6. Sun J, Roy S. Gene-based therapies for neurodegenerative diseases. Nat Neurosci. 2021 Mar;24(3):297-311. doi:10.1038/s41593-020-00778-1.
7. Amado DA, Davidson BL. Gene therapy for ALS: A review. Mol Ther. 2021 Dec 1;29(12):3345-58. doi:10.1016/j.ymthe.2021.04.008.
8. Yun Y, Ha Y. CRISPR/Cas9-mediated gene correction to understand ALS. Int J Mol Sci. 2020;21(11):3801. doi:10.3390/ijms21113801.
9. National Institute of Neurological Disorders and Stroke. Amyotrophic lateral sclerosis (ALS) fact sheet. Updated Nov 15, 2021. Accessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Amyotrophic-Lateral-Sclerosis-ALS-Fact-Sheet.
10. Cappella M et al. Gene therapy for ALS – A perspective. Int J Mol Sci. 2019;20(18):4388. doi:10.3390/ijms20184388.
11. Abramzon YA, Fratta P, Traynor BJ, Chia R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front Neurosci. 2020;14. Accessed August 18, 2022. https://www.frontiersin.org/articles/10.3389/fnins.2020.00042
12. Giannini M, Bayona-Feliu A, Sproviero D, Barroso SI, Cereda C, Aguilera A. TDP-43 mutations link Amyotrophic Lateral Sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLOS Genet. 2020;16(12):e1009260. doi:10.1371/journal.pgen.1009260
13. FDA-approved drugs for treating ALS. The ALS Association [Internet]. Accessed Mar 1, 2022. http://www.als.org/navigating-als/living-with-als/fda-approved-drugs.
14. Jensen TL et al. Current and future prospects for gene therapy for rare genetic diseases affecting the brain and spinal cord. Front Mol Neurosci. 2021 Oct 6;14:695937. doi:10.3389/fnmol.2021.695937.
15. ALS Gene Targeted Therapies. The ALS Association. Accessed August 22, 2022. https://www.als.org/understanding-als/who-gets-als/genetic-testing/als-gene-targeted-therapies
16. Tofersen for ALS clears phase 1/2 trial, now in phase 3. Advances in Motion. Massachusetts General Hospital [Internet]. Sep 30, 2020. Accessed Mar 1, 2022. https://advances.massgeneral.org/neuro/journal.aspx?id=1699.17. Biogen. A study to evaluate the efficacy, safety, tol-erability, pharmacokinetics, and pharmacodynamics of BIIB067 administered to adult subjects with amyotrophic lateral sclerosis and confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT02623699. Updated Jul 25, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT02623699.
18. Biogen. Biogen announces topline results from the tofersen phase 3 study and its open-label Extension in SOD1-ALS. Press release. Oct 17, 2021. Accessed Mar 1, 2022. https://investors.biogen.com/news-releases/news-release-details/biogen-announces-topline-results-tofersen-phase-3-study-and-its.
19. Biogen. An extension study to assess the long-term safety, tolerability, pharmacokinetics, and effect on disease progression of BIIB067 ad-ministered to previously treated adults with amyotrophic lateral sclerosis caused by superoxide dismutase 1 mutation. ClinicalTrials.gov Identi-fier: NCT03070119. Updated Sep 10, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT03070119.
20. MS MW. #AANAM – ATLAS Trial to Assess Tofersen in Presymptomatic SOD1 ALS. Accessed February 19, 2022. https://alsnewstoday.com/news-posts/2021/04/23/aanam-atlas-clinical-trial- tofersen-presymptomatic-sod1-als-patients/
21.Biogen. A phase 3 randomized, placebo-controlled trial with a longitudinal natural history run-in and open-label extension to evaluate BIIB067 initiated in clinically presymptomatic adults with a confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT04856982. Updated Feb 18, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04856982.
22. Latozinemab | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/latozinemab
23. Alector Presents AL001 (latozinemab) Data from the FTD-C9orf72 Cohort of the INFRONT-2 Phase 2 Clinical Trial | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releas-es/news-release-details/alector-presents-al001-latozinemab-data-ftd-c9orf72-cohort/
24. Alector Announces First Participant Dosed in Phase 2 Study Evaluating AL001 in Amyotrophic Lateral Sclerosis (ALS) | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releases/news-release-details/lector-announces-first-participant-dosed-phase-2-study-0/ 25. A Phase 2 Study to Evaluate AL001 in C9orf72-Associated ALS - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT05053035
26.TPN-101 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/tpn- 101
27. Transposon Therapeutics, Inc. A Phase 2a Study of TPN-101 in Patients With Amyotrophic Lateral Sclerosis (ALS) and/or Frontotemporal Dementia (FTD) Associated With Hexanucleotide Repeat Expansion in the C9orf72 Gene (C9ORF72 ALS/FTD). clinicaltrials.gov; 2022. Ac-cessed August 17, 2022. https://clinicaltrials.gov/ct2/show/NCT04993755
28. Kerk SY, Bai Y, Smith J, et al. Homozygous ALS-linked FUS P525L mutations cell- autonomously perturb transcriptome profile and chem-oreceptor signaling in human iPSC microglia. Stem Cell Rep. 2022;17(3):678-692. doi:10.1016/j.stemcr.2022.01.004
29. ION363 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/ion363 30. Ionis Pharmaceuticals, Inc. A Phase 1-3 Study to Evaluate the Efficacy, Safety, Pharmacokinetics and Pharmacodynamics of Intrathecally Administered ION363 in Amyo-trophic Lateral Sclerosis Patients With Fused in Sarcoma Mutations (FUS-ALS). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04768972
31. PhD LF. Engensis (VM202) - ALS News Today. Accessed August 19, 2022. https://alsnewstoday.com/vm202/
32. Helixmith Co., Ltd. A 6-Month Extension Study Following Protocol VMALS-002-2 (A Phase 2a, Double-Blind, Randomized, Place-bo-Controlled, Multicenter Study to Assess the Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05176093 33. Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT04632225
34. Biogen. A phase 1, safety, tolerability, and distribution study of a microdose of radiolabeled BIIB067 co-administered with BIIB067 to healthy adults. ClinicalTrials.gov Identifier: NCT03764488. Updated Jul 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03764488.
35. Ionis Pharmaceuticals Inc. A phase 1, double-blind, placebo-controlled, dose-escalation study of the safety, tolerability, and pharmacokinet-ics of ISIS 333611 administered intrathecally to patients with familial amyotrophic lateral sclerosis due to superoxide dismutase 1 gene muta-tions. ClinicalTrials.gov Identifier: NCT01041222. Updated Apr 13, 2012. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01041222.
36. Messina S, Sframeli M. New treatments in spinal muscular atrophy: Positive results and new challenges. J Clin Med. 2020;9(7):2222. doi:10.3390/jcm9072222.
37. Scoto M et al. Genetic therapies for inherited neuromuscular disorders. Lancet Child Adolesc Health. 2018 Aug;2(8):600-9. doi:10.1016/S2352-4642(18)30140-8.
38. Abreu NJ, Waldrop MA. Overview of gene therapy in spinal muscular atrophy and Duchenne muscular dystrophy. Pediatr Pulmonol. 2021 Apr;56(4):710-20. doi:10.1002/ppul.25055.
39. Brandsema J, Cappa R. Genetically targeted therapies for inherited neuromuscular disorders. Practical Neurology [Internet]. Jul/Aug 2021:69-73. Accessed Mar 1, 2022. https://practicalneurology.com/articles/2021-july-aug/genetically-targeted-therapies-for-inherited-neuromuscular-disorders/pdf.
40. Ojala KS et al. In search of a cure: The development of therapeutics to alter the progression of spinal muscular atrophy. Brain Sci. 2021;11(2):194. doi:10.3390/brainsci11020194.
41. McCall S. Cure SMA Releases Updated Drug Pipeline. Cure SMA. Published December 13, 2021. Accessed August 21, 2022. https://www.curesma.org/cure-sma-releases-updated-drug-pipeline- 2021/ 42. FDA approves first drug for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Dec 23, 2016. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy.43. Kirschner J. Postnatal gene therapy for neuromuscular diseases – Opportunities and limitations. J Perinat Med. 2021 Sep;49(8):1011-5. doi:10.1515/jpm-2021-0435.
43. Terryn J, Verfaillie CM, Van Damme P. Tweaking Progranulin Expression: Therapeutic Avenues and Opportunities. Front Mol Neurosci. 2021;14. Accessed September 4, 2022. https://www.frontiersin.org/articles/10.3389/fnmol.2021.71303144.
44. Biogen. A phase 3, randomized, double-blind, sham-procedure controlled study to assess the clinical efficacy and safety of ISIS 396443 administered intrathecally in patients with later-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02292537. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/study/NCT02292537.
45. Why Spinraza/later-onset studies. SPINRAZA® (nusinersen) [Internet]. Accessed Mar 1, 2022. www.spinraza.com/en_us/home/why-spinraza/later-onset-studies.html#scroll-tabs.
46. Biogen. A Phase 3, Randomized, Double-Blind, Sham-Procedure Controlled Study to Assess the Clinical Efficacy and Safety of ISIS 396443 Administered Intrathecally in Patients With Infantile- Onset Spinal Muscular Atrophy. clinicaltrials.gov; 2021. Accessed February 10, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02193074
47. Early-onset SMA (Type 1) | SPINRAZA® (nusinersen). Accessed Mar 1, 2022. https://www.spinraza-hcp.com/en_us/home/why-spinraza/about-spinraza.html.
48. Finkel RS et al; ENDEAR Study Group. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723-32. doi: 10.1056/NEJMoa1702752.
49. Biogen. An open-label study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to subjects with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02386553. Updated Nov 18, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02386553.
50. De Vivo DC et al; NURTURE Study Group. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: In-terim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul Disord. 2019 Nov;29(11):842-56. doi:10.1016/j.nmd.2019.09.007.
51. Why Spinraza/presymptomatic study. SPINRAZA® (nusinersen) [Internet]. Accessed Feb 22, 2022. www.spinraza.com/en_us/home/why-spinraza/presymptomatic-study.html#scroll-tabs.
52. FDA approves oral treatment for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Aug 7, 2020. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy.
53. Hoffmann-La Roche. A two-part seamless, open-label, multicenter study to investigate the safety, tolerability, pharmacokinetics, pharmaco-dynamics and efficacy of risdiplam (RO7034067) in infants with type 1 spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02913482. Updated Jan 21, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02913482.
54. Hoffmann-La Roche. A two-part seamless, multi-center randomized, placebo-controlled, double-blind study to investigate the safety, tolera-bility, pharmacokinetics, pharmacodynamics and efficacy of risdiplam (RO7034067) in type 2 and 3 spinal muscular atrophy patients. Clinical-Trials.gov Identifier: NCT02908685. Updated Dec 28, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02908685.
55. Genentech. Genentech’s risdiplam shows significant improvement in survival and motor milestones in infants with type 1 spinal muscular atrophy (SMA). Press release. Apr 27, 2020. Accessed Mar 1, 2022. http://www.gene.com/media/press-releases/14847/2020-04-27/genentechs-risdiplam-shows-significant-i
56. Hoffmann-La Roche. An open-label study to investigate the safety, tolerability, and pharmacokinetics/pharmacodynamics of risdiplam (RO7034067) in adult and pediatric patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03032172. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03032172.
57. Hoffmann-La Roche. An open-label study of risdiplam in infants with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03779334. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03779334.
58. McCall S. Update on Genentech/Roche Initiation of MANATEE Clinical Study. Cure SMA. Published October 20, 2021. Accessed August 20, 2022. https://www.curesma.org/update-on- genentech-roche-initiation-of-manatee-clinical-study/
59. Abati E, Manini A, Comi GP, Corti S. Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases. Cell Mol Life Sci. 2022;79(7):374. doi:10.1007/s00018-022-04408-w
60. FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality. U.S. Food and Drug Administration. News release. May 24, 2019. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease.
61. Novartis Gene Therapies. Phase I gene transfer clinical trial for spinal muscular atrophy type 1 delivering AVXS-101. ClinicalTrials.gov Identifier: NCT02122952. Updated Jun 14, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02122952.
62. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT03306277. Updated Jun 14, 2021. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT03306277.
63. Mendell JR et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713-22. doi:10.1056/NEJMoa1706198.
64. Symptomatic study results. ZOLGENSMA [Internet]. Updated Nov 2021. Accessed Mar 1, 2022. Error! Hyperlink reference not valid..
65. Novartis Gene Therapies. A global study of a single, one-time dose of AVXS-101 delivered to infants with genetically diagnosed and pre-symptomatic spinal muscular atrophy with multiple copies of SMN2. ClinicalTrials.gov Identifier: NCT03505099. Updated Jan 1, 2022. Ac-cessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03505099.
66. Chiu W et al. Current genetics and potential gene-targeting therapeutics for neuromuscular diseases. Int J Mol Sci. 2020 Dec;21(24):9589. doi:10.3390/ijms21249589.
67. Novartis Gene Therapies. A long-term follow-up study of patients in the clinical trials for spinal muscular atrophy receiving AVXS-101. Clini-calTrials.gov Identifier: NCT04042025. Updated Jun 9, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04042025.
68. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT0383718. Up-dated Jan 11, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03837184.
69. Biogen. An open-label, dose escalation study to assess the safety, tolerability and dose-range finding of multiple doses of ISIS 396443 de-livered intrathecally to patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01703988. Updated Apr 13, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01703988.
70. Biogen. A study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to patients with infantile-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01839656. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01839656.
71. Biogen. An open-label extension study for patients with spinal muscular atrophy who previously participated in investigational studies of ISIS 396443. ClinicalTrials.gov Identifier: NCT02594124. Updated Nov 15, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02594124.
72. Biogen. Escalating dose and randomized, controlled study of nusinersen (BIIB058) in participants with spinal muscular atrophy. ClinicalTri-als.gov Identifier: NCT04089566. Updated Feb 24, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04089566.
73. National Center for Advancing Translational Sciences. Duchenne muscular dystrophy. Genetic and Rare Diseases Information Center. Up-dated Nov 2, 2020. Accessed Mar 1, 2022. https://rarediseases.info.nih.gov/diseases/6291/duchenne-muscular-dystrophy.
74. Matsuo M. Antisense oligonucleotide-mediated exon-skipping therapies: Precision medicine spreading from Duchenne muscular dystrophy. JMA J. 2021 Jul 15;4(3):232-40. doi:10.31662/jmaj.2021-0019.
75. FDA approves drug to treat Duchenne muscular dystrophy. U.S. Food and Drug Administration. News release. Feb 9, 2017. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-drug-treat-duchenne-muscular-dystrophy.74.
76. Duan D. Dystrophin gene replacement and gene repair therapy for Duchenne muscular dystrophy in 2016: An interview. Hum Gene Ther Clin Dev. 2016 Mar;27(1):9-18. doi:10.1089/humc.2016.001.
77. EXONDYS 51®. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/drug-development-pipeline/exondys-51/
78. Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Placebo-Controlled, Multiple Dose Efficacy, Safety, Tolerability and Pharmacoki-netics Study of AVI-4658(Eteplirsen),in the Treatment of Ambulant Subjects With Duchenne Muscular Dystrophy. clinicaltrials.gov; 2020. Ac-cessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT01396239
79. Sarepta Therapeutics, Inc. Clinical Study to Assess the Safety Fo AVI-4658 in Subjects With Duchenne Muscular Dystrophy Due to a Frame-Shift Mutation Amenable to Correction by Skipping Exon 51. clinicaltrials.gov; 2015. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/study/NCT00844597
80. Sarepta Therapeutics, Inc. A 2-part, randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study (Part 1) followed by an open-label efficacy and safety evaluation (Part 2) of SRP-4053 in patients with Duchenne muscular dystrophy amenable to exon 53 skipping. ClinicalTrials.gov Identifier: NCT02310906. Updated Oct 19, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02310906.
81. Commissioner O of the. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. FDA. Published March 24, 2020. Accessed August 21, 2022. hDuchenne Muscular Dystrophy Amenable to Exon 51-Skipping Treatment. clinicaltrials.gov; 2022. Accessed Au-gust 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04004065
109. National Center of Neurology and Psychiatry, Japan. Exploratory study of NS-065/NCNP-01 in Duchenne muscular dystrophy. ClinicalTri-als.gov Identifier: NCT02081625; Updated Feb 26, 2020. Accessed Mar 2, 2022. https://clinicaltrialsttps://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-drug-duchenne-muscular- dys-trophy
82. Duchenne Drug Development Pipeline. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/duchenne-drug-development-pipeline/
83. Sarepta Therapeutics Provides Update on SRP-5051 for the Treatment of Duchenne Muscular Dystrophy | Sarepta Therapeutics, Inc. Ac-cessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics- pro-vides-update-srp-5051-treatment-duchenne
84. Sarepta Therapeutics, Inc. An Open-Label Extension Study for Patients With Duchenne Muscular Dystrophy Who Participated in Studies of SRP-5051. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03675126
85. VYONDYS 53. Prescribing information. Sarepta Therapeutics Inc.; 2019. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211970s000lbl.pdf.
86. NS Pharma Inc. Long-term use of viltolarsen in boys with Duchenne muscular dystrophy in clinical practice (VILT-502). ClinicalTrials.gov Identifier: NCT04687020. Updated Nov 22, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04687020.
87. VILTEPSO. Prescribing information. NS Pharma; 2020. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212154s000lbl.pdf.
88. FDA approves targeted treatment for rare Duchenne muscular dystrophy mutation. U.S. Food and Drug Administration. News release. Feb 25, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation-0.
89. Sarepta Therapeutics Inc. A double-blind, placebo-controlled, multi-center study with an open-label extension to evaluate the efficacy and safety of SRP-4045 and SRP-4053 in patients with Duchenne muscular dystrophy. Clinicaltrials.gov Identifier: NCT02500381. Updated Aug 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02500381.
90. AMONDYS 45. Prescribing information. Sarepta Therapeutics Inc.; 2021. Accessed Feb 22, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213026lbl.pdf.
91. Finkel RS et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dys-trophy. PLoS ONE. 2013;8(12):e81302. doi:10.1371/journal.pone.0081302.
92. PTC Therapeutics. A phase 2 study of PTC124 as an oral treatment for nonsense-mutation-mediated Duchenne muscular dystrophy. Clini-calTrials.gov Identifier: NCT00264888. Updated Jan 14, 2009. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00264888.
93. PTC Therapeutics. A phase 2B efficacy and safety study of PTC124 in subjects with nonsense-mutation-mediated Duchenne and Becker muscular dystrophy. ClinicalTrials.gov Identifier: NCT00592553. Updated Apr 7, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00592553.
94. PTC Therapeutics. A phase 3 efficacy and safety study of ataluren in patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01826487. Updated Aug 4, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01826487.
95. Bushby K et al; PTC124-GD-007-DMD Study Group. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014 Oct;50(4):477-87. doi:10.1002/mus.24332.
96. Solid Biosciences LLC. A randomized, controlled, open-label, single-ascending dose, phase I/II study to investigate the safety and tolerabil-ity, and efficacy of intravenous SGT-001 in male adolescents and children with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03368742. Updated Aug 24, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03368742.
97. Solid Biosciences reports 1.5-year data from patients in the ongoing IGNITE DMD phase I/II clinical trial of SGT-001. Press release. Solid Biosciences. Sep 27, 2021. Accessed Mar 2, 2022. http://www.solidbio.com/about/media/press-releases/solid-biosciences-reports-1-5-year-data-from-patients-in-the-ongoing-ignite-dmd-phase-i-ii-clinical-trial-of-sgt-001.
98. Potter RA et al. Dose-escalation study of systemically delivered rAAVrh74.MHCK7.microdystrophin in the mdx mouse model of Duchenne muscular dystrophy. Hum Gene Ther. 2021 Apr;32(7-8):375-89. doi:10.1089/hum.2019.255.
99. Sarepta Therapeutics, Inc. A Phase 3 Multinational, Randomized, Double-Blind, Placebo- Controlled Systemic Gene Delivery Study to Evaluate the Safety and Efficacy of SRP-9001 in Patients With Duchenne Muscular Dystrophy (EMBARK). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05096221
100. Pfizer. A PHASE 3, MULTICENTER, RANDOMIZED, DOUBLE-BLIND, PLACEBO CONTROLLED STUDY TO EVALUATE THE SAFETY AND EFFICACY OF PF 06939926 FOR THE TREATMENT OF DUCHENNE MUSCULAR DYSTROPHY. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04281485
101. Pfizer. A phase 1B multicenter open-label, single ascending dose study to evaluate the safety and tolerability of PF-06939926 in ambula-tory and non-ambulatory subjects with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03362502. Updated Mar 2, 2022. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03362502.
102. MS MW. Phase 3 CIFFREO DMD Gene Therapy Trial Slated to Begin in June in US. Accessed August 21, 2022. https://musculardystrophynews.com/news/phase-3-trial-of-pfizers-gene-therapy- expected-to-open-in-us-in-june/
103. SRP-9001. Parent Project Muscular Dystrophy. Accessed August 22, 2022. https://www.parentprojectmd.org/drug-development-pipeline/srp-9001-micro-dystrophin-gene- transfer/
104. Sarepta Therapeutics’ Investigational Gene Therapy SRP-9001 for Duchenne Muscular Dystrophy Demonstrates Significant Functional Improvements Across Multiple Studies | Sarepta Therapeutics, Inc. Accessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release- details/sarepta-therapeutics-investigational-gene-therapy-srp-9001
105. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Efficacy Study of Eteplirsen in Patients With Duchenne Muscular Dys-trophy Who Have Completed Study 4658-102.clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03985878
106. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Pharmacokinetics Study of Eteplirsen in Young Patients With Duchenne Mus-cular Dystrophy Amenable to Exon 51 Skipping. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03218995
107.Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Dose Finding and Comparison Study of the Safety and Efficacy of a High Dose of Eteplirsen, Preceded by an Open-Label Dose Escalation, in Patients With Duchenne Muscular Dystrophy With Deletion Mutations Amenable to Exon 51 Skipping. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03992430
108. Sarepta Therapeutics, Inc. A Phase 2, Two-Part, Multiple-Ascending-Dose Study of SRP-5051 for Dose Determination, Then Dose Ex-pansion, in Patients With .gov/ct2/show/NCT02081625.
110. NS Pharma Inc. A phase II, dose finding study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT02740972. Updated Dec 7, 2021. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02740972.
111. NS Pharma Inc. A phase II, open-label, extension study to assess the safety and efficacy of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT03167255. Updated Nov 24, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03167255.
112. NS Pharma Inc. A phase 2 open label study to assess the safety, tolerability, and efficacy of viltolarsen in ambulant and non-ambulant boys with Duchenne muscular dystrophy (DMD) compared with natural history controls. ClinicalTrials.gov Identifier: NCT04956289. Updated Feb 1, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04956289.
113. NS Pharma Inc. A phase 3 randomized, double-blind, placebo-controlled, multi-center study to assess the efficacy and safety of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04060199. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04060199.
114. NS Pharma Inc. A phase 3, multi-center, open-label extension study to assess the safety and efficacy of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04768062. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04768062.
115. Sarepta Therapeutics Inc. A randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study followed by an open-label safety and efficacy evaluation of SRP-4045 in advanced-stage patients with Duchenne muscular dystrophy amena-ble to exon 45 skipping. ClinicalTrials.gov Identifier: NCT02530905. Updated May 17, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02530905.
116. Sarepta Therapeutics Inc. Long-term, open-label extension study for patients with Duchenne muscular dystrophy enrolled in clinical trials evaluating casimersen or golodirsen. ClinicalTrials.gov Identifier: NCT03532542. Updated Dec 20, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03532542.
117. PTC Therapeutics. A phase 2 study of the safety, pharmacokinetics, and pharmacodynamics of ataluren (PTC124®) in patients aged ≥2 to <5 years old with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT02819557. Updated Aug 28, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02819557.
118. PTC Therapeutics. Phase 2, non-interventional, clinical study to assess dystrophin levels in subjects with nonsense mutation Duchenne muscular dystrophy who have been treated with ataluren for ≥ 9 months. ClinicalTrials.gov Identifier: NCT03796637. Updated Apr 10, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03796637.
119. PTC Therapeutics. An Open-Label Study Evaluating the Safety and Pharmacokinetics of Ataluren in Children From ≥6 Months to <2 Years of Age With Nonsense Mutation Duchenne Muscular Dystrophy. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04336826 120. PTC Therapeutics. An open-label study for previously treated ataluren (PTC124®) pa-tients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01557400. Updated Nov 25, 2020. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT01557400.
121. PTC Therapeutics. An open-label, safety study for ataluren (PTC124) patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01247207. Updated Feb 16, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT01247207.
122. PTC Therapeutics. A phase 3, randomized, double-blind, placebo-controlled efficacy and safety study of ataluren in patients with non-sense mutation Duchenne muscular dystrophy and open-label extension. ClinicalTrials.gov Identifier: NCT03179631. Updated Feb 8, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03179631.
123. Sarepta Therapeutics, Inc. An Open-Label, Systemic Gene Delivery Study Using Commercial Process Material to Evaluate the Safety of and Expression From SRP-9001 in Subjects With Duchenne Muscular Dystrophy (ENDEAVOR). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04626674
124. Sarepta Therapeutics, Inc. Systemic Gene Delivery Phase I/IIa Clinical Trial for Duchenne Muscular Dystrophy Using RAA-Vrh74.MHCK7.Micro-Dystrophin (MicroDys-IV-001). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03375164
125. Sarepta Therapeutics Inc. A multicenter, randomized, double-blind, placebo-controlled trial for Duchenne muscular dystrophy using SRP-9001. ClinicalTrials.gov Identifier: NCT03769116. Updated Dec 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03769116.
126. Hoffmann-La Roche. A Two-Part, Seamless, Multi-Center, Randomized, Placebo-Controlled, Double-Blind Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of RO7204239 in Combination With Risdiplam (RO7034067) in Ambulant Pa-tients With Spinal Muscular Atrophy. clinicaltrials.gov; 2022. Accessed September 1, 2022. https://clinicaltrials.gov/ct2/show/NCT05115110
Staying alert for patients with narcolepsy
Almost half of Americans report feeling daytime sleepiness on at least 3 days per week. For most patients, this sleepiness results from insufficient nighttime sleep. But a minority of these patients have narcolepsy, a chronic neurologic disorder that impairs the brain’s control of sleep-wake cycles. This disorder often goes undiagnosed, but neurologists can make a significant difference by learning how to recognize and treat it.

What is narcolepsy?
Narcolepsy is characterized by excessive daytime sleepiness (EDS) and sudden attacks of sleep. Patients have difficulty staying awake for long periods of time, and the disorder can make performing daily tasks difficult. Problems with concentration and alertness are common.
Narcolepsy is considered to have two subtypes. Patients with narcolepsy type 1 also have cataplexy, a sudden loss of muscle tone. Attacks of cataplexy are triggered by strong, usually positive, emotions. These attacks have manifestations ranging from slurred speech to complete weakness of most muscles. Patients with narcolepsy type 2, however, do not have cataplexy.
Dysregulation of rapid eye movement (REM) sleep, which is when most dreaming occurs, is another symptom of narcolepsy. The transition to REM sleep is quicker in patients with narcolepsy and usually occurs within 15 minutes of sleep onset. A related symptom is sleep paralysis, an inability to move while falling asleep or waking up. This symptom resembles a state that normally occurs during REM sleep.

Hallucinations also are common in patients with narcolepsy and can be especially vivid. Hypnagogic hallucinations occur during the transition to sleep, and hypnopompic hallucinations arise while the patient is waking up. Patients may think they see a stranger in their bedroom, and children sometimes report seeing animals.
Although it is easy for patients with narcolepsy to fall asleep at night, they often have disrupted sleep. Patients have frequent, brief arousals throughout the night that may become disturbing. Dream content often is affected in narcolepsy, too. Patients have described lucid dreams of flying or out-of-body experiences. After such intense dreams, patients often feel that their sleep has not been restful.
Criteria and diagnosis
To receive a diagnosis of narcolepsy type 1, a patient must have EDS that persists for at least 3 months and at least one of the following two features: cataplexy and objective evidence of quick sleep onset and early start of REM sleep or low cerebrospinal fluid (CSF) levels (that is, less than 110 pg/mL) of hypocretin. Hypocretin, also known as orexin, is a neuropeptide that regulates wakefulness and arousal.

Patients must meet five criteria to receive a diagnosis of narcolepsy type 2. They must have EDS that persists for at least 3 months. They must have test results that show quick sleep onset and early start of REM sleep. They must have no cataplexy. Their CSF levels of hypocretin must be normal or unknown. Finally, they must have no other conditions that provide a better explanation for their symptoms and test results.
“The diagnosis of narcolepsy is made primarily by history on the clinical features of the disorder,” said Michael J. Thorpy, MB, ChB, professor of neurology at Albert Einstein College of Medicine and director of the Sleep–Wake Disorders Center at Montefiore Medical Center in New York. When narcolepsy is suspected, testing is required to confirm the diagnosis. The patient should undergo all-night polysomnographic (PSG) testing, followed by a daytime multiple sleep latency test (MSLT). Measurement of CSF hypocretin can be diagnostic but is performed mainly in the research setting and is not common in the clinical setting, said Dr. Thorpy.
Patients with narcolepsy typically fall asleep in an average of less than 8 minutes during the nap opportunities of the MSLT. They also have at least two sleep-onset REM periods. “A new change in the diagnostic classification is that a sleep-onset REM period on the preceding night’s PSG can count as one of the two sleep-onset REM periods required for diagnosis,” said Dr. Thorpy.
“In the case of type 1 narcolepsy, the history is usually pretty clear, and the MSLT is usually positive, in the sense that it is consistent with a narcolepsy pattern,” said Thomas E. Scammell, MD, professor of neurology at Harvard Medical School and Beth Israel Deaconess Medical Center in Boston. “The PSG is also important, because other factors that disrupt the patient’s nighttime sleep (such as obstructive sleep apnea and periodic limb movements) must be ruled out, especially in type 2 narcolepsy,” said Dr. Scammell.
Early sleep onset, late diagnosis
Diagnostic delay is a common problem for patients with narcolepsy. Although the median age of onset is 16 years, a patient typically does not receive the appropriate diagnosis until adulthood. “It takes, on average, somewhere between 8 and 12 years for a patient to get a diagnosis of narcolepsy,” said Dr. Thorpy. Growing awareness and an increase in the number of sleep disorder centers have reduced but not eliminated the diagnostic delay.
Children with narcolepsy are often misdiagnosed. “One of the most common misdiagnoses in childhood is ADHD, because sleepiness in children differs from that in adults,” said Dr. Thorpy. Sleepy children often become hyperactive and display increased impulsivity, he explained. Stimulants prescribed for ADHD tend to mask the symptoms of narcolepsy and delay the correct diagnosis. Mood disorders, behavioral disorders, and psychogenic disorders are other common misdiagnoses for children with narcolepsy.
But when it comes to adults, sometimes patients themselves contribute to the diagnostic delay. EDS is “such a pervasive feeling that I think a lot of people just don’t make much of it,” said Dr. Scammell. The symptom is easily ascribed to insufficient sleep or a difficult work schedule. “It may take them months to get to see a doctor,” said Dr. Scammell.
Behavioral treatments
Nonpharmacologic treatments are one component of care for patients with narcolepsy. Patients must maintain a regular sleep-wake schedule and ensure that they are in bed for no less than 8 hours per night, said Dr. Thorpy. Taking no more than two daytime naps of less than 20 minutes each can help relieve some of the sleepiness, he added.
In addition to ensuring an adequate amount of sleep, it is important to promote good quality sleep, said Dr. Scammell. To do this, clinicians should address any conditions such as sleep apnea that disrupt patients’ sleep, he added.
Patients also tend to avoid situations that are likely to entail the emotional stimuli that could precipitate cataplexy. Some avoid laughter or try to suppress their emotions. “That’s not good,” said Kiran Maski, MD, MPH, assistant professor of neurology at Harvard Medical School and neurologist and sleep physician at Boston Children’s Hospital. “We worry that that might be a risk factor for depression or social isolation.” Cognitive-behavioral therapy can help patients with narcolepsy gradually increase their comfort with and exposure to social situations.
Although behavioral treatments are helpful, they are not sufficient to control all the symptoms of narcolepsy. Most patients require pharmacologic treatments, which are the most effective treatments for narcolepsy, said Dr. Thorpy.
Pharmacologic treatments
Previously, neurologists relied on the stimulants methylphenidate and amphetamine, which primarily treated patients’ EDS. But the field is moving away from these drugs because of their tendency to induce side effects and their potential for abuse, said Dr. Thorpy. In this context, modafinil and armodafinil became the mainstay for promoting alertness in patients with narcolepsy.
In recent years, newer medications have emerged that have slightly greater efficacy and better safety profiles than modafinil and armodafinil. Solriamfetol (Sunosi, Jazz Pharmaceuticals), for example, is effective for EDS but does not affect cataplexy. Pitolisant (Wakix, Harmony Biosciences), on the other hand, effectively treats EDS and cataplexy.
Sodium oxybate (Xyrem, Jazz Pharmaceuticals) is the only medication that treats all the symptoms of narcolepsy, said Dr. Thorpy. “That treats the sleepiness, the cataplexy, and the disturbed nocturnal sleep,” he added. Sodium oxybate also appears to reduce sleep paralysis, hallucinations, and disturbed dreams.
A potential concern about sodium oxybate, which has been used since approximately 2000, is its high sodium load. A new formulation called low-sodium oxybate (Xywav, Jazz Pharmaceuticals) “has a slightly better safety profile, particularly in people who have cardiovascular or renal disease,” said Dr. Thorpy. “This is tending to take over the role of regular sodium oxybate.”
Many clinicians who treat patients with narcolepsy develop their own approaches, but the choice of treatment generally depends on the patient’s symptoms, said Dr. Scammell. Modafinil is a good first choice for patients with mild to moderate sleepiness, he added. Pitolisant is another good choice for these patients but is more expensive. Both drugs are well tolerated.
Clinicians can consider solriamfetol and amphetamine for patients with moderate to severe sleepiness. “I generally consider the oxybates to be a second line,” said Dr. Scammell. Although these drugs may be the most effective, and they do help patients a great deal, they have a higher prevalence of side effects and are more expensive, he added. “If we can get good results with something gentle and simple like modafinil, that would be great.”
“There are differences of opinion as to what the first-line treatments are,” said Dr. Thorpy. Some patients prefer to use the traditional stimulants as first-line treatments, but others prefer to avoid them because of their adverse effects. They favor the newer, and unfortunately more expensive, medications instead. But there is no consensus among clinicians about which of the newer medications to use. “There’s no standard treatment, and it’s very hard to develop an algorithm that is acceptable to most physicians treating patients with narcolepsy,” said Dr. Thorpy. Treatment response varies, as well. Some patients respond extremely well to treatment, but clinical trials indicate that even optimal therapy helps patients achieve about 70% of the normal level of alertness. “If they’re sedentary, sitting in a boring meeting or at the computer, they can still fall asleep, even with our current medications,” said Dr. Scammell.
“The hardest symptom of all to treat is the EDS,” agreed Dr. Thorpy. Most patients cannot be treated with one medication alone, and polypharmacy tends to be necessary, he added. Typically, this means the addition of another medication to the regimen to maximize alertness. For other patients, cataplexy is difficult to control, and adding an anticataplectic medication is appropriate. Still, most patients can control their cataplexy with one drug, either oxybate or pitolisant, said Dr. Thorpy.
Investigational treatments
Researchers are trying to develop new medicines with greater potency, and several medications are under investigation. Early studies have shown that reboxetine, an antidepressant medication that affects dopamine and norepinephrine activity, is an effective treatment for EDS and cataplexy. Ongoing phase 3 studies are examining reboxetine for EDS. Another drug known as FT-218 is a once-nightly formulation of sodium oxybate, unlike the twice-nightly formulations of the drug that currently are available. In a phase 3 trial, the drug was associated with significant improvements in wakefulness and reductions in attacks of cataplexy. Avadel, which is developing the drug, submitted it to the U.S. Food and Drug Administration for approval in 2021, but the agency has not yet made a decision about it.
Researchers and patients alike have high hopes for medications that activate the orexin receptors. Orexin stimulates the wake-promoting neurons in the brain. Narcolepsy, and particularly narcolepsy type 1, is characterized by a loss of hypocretin cells in the central nervous system. The loss of these cells promotes sleepiness and disturbed REM sleep. To counteract this loss of cells, several companies are investigating new orexin agonists.
One such medication is TAK-994, which was developed by Takeda. The drug showed great promise for treating EDS and cataplexy, said Dr. Thorpy. But when phase 3 studies suggested that TAK-994 was associated with hepatotoxicity, the company terminated the studies. Nevertheless, other orexin agonists, including Takeda’s TAK-861, are under investigation.
“If we can restore orexin signaling, it could be like giving insulin to type 1 diabetics,” said Dr. Scammell. This class of medications could provide substantial improvements in sleepiness and other symptoms, he added. “I think when orexin agonists become available, it’s going to be quite transformative.” But these drugs are still in early development and will not be available in clinical practice for several years.
Common psychological comorbidities
Certain comorbidities are prevalent among patients with narcolepsy, and psychiatric disorders tend to be the most common. These comorbidities may complicate the management of narcolepsy. Nevertheless, they often are significant enough to require management in their own right, said Dr. Thorpy.
Depression is likely twice as common among patients with narcolepsy than among the general population, said Dr. Scammell. “Whether this is an actual neurobiologic feature of the disease, or whether it is just a reaction to having a challenging disorder isn’t entirely clear,” he added. “But it doesn’t get the attention or treatment that it deserves.”
Partnering with a psychologist or psychiatrist is important because many treatments can exacerbate mood disorders, said Dr. Maski. In general, stimulants, for example, can worsen depression and anxiety and are associated with increased suicide risk. “We oftentimes are using high-dose stimulants in patients, so mood has to be really carefully monitored and managed,” Dr. Maski added.
Cases of depression and suicidal ideation were reported in clinical trials of sodium oxybate. Although these serious adverse events were rare, patients must be monitored very closely even on treatments specifically approved for narcolepsy, said Dr. Maski. Mood disturbances are reported less frequently with modafinil and pitolisant than with stimulants, she noted.
Many times, patients need to take an antidepressant medication, but these drugs could affect the medicines administered for narcolepsy, said Dr. Thorpy. Pitolisant, in particular, may be adversely affected by current antidepressant medications. The only remedies are to change from pitolisant to another narcolepsy medication or to use an antidepressant that does not have histamine 1 receptor antagonism or affect the QTc interval.
Anxiety also is prevalent among patients with narcolepsy, and it can be worsened by traditional stimulants. These drugs also can increase the likelihood of irritability or obsessive-compulsive tendencies. “Traditional stimulants would be best avoided in these patients who have significant anxiety,” said Dr. Thorpy.
The social burden of narcolepsy
The burden of narcolepsy extends beyond psychiatric comorbidities into the social sphere. “Patients with narcolepsy do have greater difficulties in terms of social and interpersonal relationships,” said Dr. Thorpy. The disorder reduces patients’ quality of life, and educational difficulties and job loss are common in this population. “It’s a lifelong, incurable disorder, and these patients suffer an immense burden throughout their life because of the sleepiness that … affects their cognitive abilities,” said Dr. Thorpy.
“There’s an increased reporting of what probably amounts to social isolation,” said Dr. Maski. Patients often report that they must prioritize activities or events because they do not have the energy or alertness to participate in all of them. For instance, adolescents with narcolepsy frequently say that they must forgo after-school extracurricular activities because they need to prioritize studying and getting enough sleep. “Those priorities take away from their normal social life and events that they would like to participate in,” said Dr. Maski.
Another problem is that patients have the impression that others do not understand their condition. They are afraid that they will be perceived as lazy, uninterested, or unmotivated if they fall asleep. “Sometimes they withdraw from social events because they don’t want to be perceived in such a way,” said Dr. Maski. She and her colleagues encourage patients to participate in selected after-school events and to engage in social activities they find meaningful to maintain social networks.
An unpublished study of more than 300 patients with narcolepsy examined the effect of the disorder on patients’ social lives. At the end of the day, many patients “crash and burn,” said Dr. Scammell. Consequently, they do not have as much energy for social activities.
This lack of energy affects patients’ social relationships. The study suggests that patients with narcolepsy do not have as many friends as the general population does. Nevertheless, the frequency of close relationships and marriage was similar between patients with narcolepsy and the general population. “What people are doing is putting their energy into these close relationships, rather than having lots of friends and socializing a lot,” said Dr. Scammell. “I found that heartening, that people were doing their best and developed those close relationships,” which are vitally important for many reasons, he added.
The study, which has been submitted for publication, also asked patients about their sex lives. Many patients reported having had cataplexy during sex, and others reported that their medications caused problems with their sex lives. “Their doctors never ask about these things, and many patients actually would like their doctor to ask about them more,” said Dr. Scammell.
In addition, narcolepsy significantly affects a patient’s ability to drive. Patients with narcolepsy have a three- to fourfold increased risk of car accidents, said Dr. Scammell. This increased risk likely results from patients’ EDS.
But as important as this issue is for patients’ lives, there is no consensus on how to counsel patients about driving, said Dr. Maski. “For instance, it is not really clear if there is value in doing a maintenance of wakefulness test before allowing patients with narcolepsy to drive,” she said. The test is not validated in children or adolescents, which raises questions about how to advise beginning drivers with narcolepsy. “It’s not really clear that passing your maintenance of wakefulness test increases your safety behind the wheel,” said Dr. Maski.
“It’s the rare person with narcolepsy who can easily and safely do a 2-hour drive by themselves,” said Dr. Scammell. Patients must determine what their own limits are, and it is important for clinicians to discuss reasonable limits honestly with their patients. “I almost never would push to have somebody’s license taken away,” said Dr. Scammell. “But there are patients who only can drive around town for short errands, and if it’s anything more than half an hour, they start getting drowsy.”
There is a need for a public awareness campaign about narcolepsy, Dr. Scammell added. Such a campaign was carried out in Italy several years ago, and it included cartoons and TV segments. “It got a lot of people’s attention, and there was a real spike in new and correct diagnoses of narcolepsy,” said Dr. Scammell. But such a broad campaign is expensive, while narcolepsy is rare, and it might not be feasible to reach out to the general population. “But I certainly think it’s worth targeting doctors who are likely to see patients with sleepiness: neurologists, psychiatrists and psychologists, and primary care doctors,” said Dr. Scammell.
Almost half of Americans report feeling daytime sleepiness on at least 3 days per week. For most patients, this sleepiness results from insufficient nighttime sleep. But a minority of these patients have narcolepsy, a chronic neurologic disorder that impairs the brain’s control of sleep-wake cycles. This disorder often goes undiagnosed, but neurologists can make a significant difference by learning how to recognize and treat it.
What is narcolepsy?
Narcolepsy is characterized by excessive daytime sleepiness (EDS) and sudden attacks of sleep. Patients have difficulty staying awake for long periods of time, and the disorder can make performing daily tasks difficult. Problems with concentration and alertness are common.
Narcolepsy is considered to have two subtypes. Patients with narcolepsy type 1 also have cataplexy, a sudden loss of muscle tone. Attacks of cataplexy are triggered by strong, usually positive, emotions. These attacks have manifestations ranging from slurred speech to complete weakness of most muscles. Patients with narcolepsy type 2, however, do not have cataplexy.
Dysregulation of rapid eye movement (REM) sleep, which is when most dreaming occurs, is another symptom of narcolepsy. The transition to REM sleep is quicker in patients with narcolepsy and usually occurs within 15 minutes of sleep onset. A related symptom is sleep paralysis, an inability to move while falling asleep or waking up. This symptom resembles a state that normally occurs during REM sleep.
Hallucinations also are common in patients with narcolepsy and can be especially vivid. Hypnagogic hallucinations occur during the transition to sleep, and hypnopompic hallucinations arise while the patient is waking up. Patients may think they see a stranger in their bedroom, and children sometimes report seeing animals.
Although it is easy for patients with narcolepsy to fall asleep at night, they often have disrupted sleep. Patients have frequent, brief arousals throughout the night that may become disturbing. Dream content often is affected in narcolepsy, too. Patients have described lucid dreams of flying or out-of-body experiences. After such intense dreams, patients often feel that their sleep has not been restful.
Criteria and diagnosis
To receive a diagnosis of narcolepsy type 1, a patient must have EDS that persists for at least 3 months and at least one of the following two features: cataplexy and objective evidence of quick sleep onset and early start of REM sleep or low cerebrospinal fluid (CSF) levels (that is, less than 110 pg/mL) of hypocretin. Hypocretin, also known as orexin, is a neuropeptide that regulates wakefulness and arousal.
Patients must meet five criteria to receive a diagnosis of narcolepsy type 2. They must have EDS that persists for at least 3 months. They must have test results that show quick sleep onset and early start of REM sleep. They must have no cataplexy. Their CSF levels of hypocretin must be normal or unknown. Finally, they must have no other conditions that provide a better explanation for their symptoms and test results.
“The diagnosis of narcolepsy is made primarily by history on the clinical features of the disorder,” said Michael J. Thorpy, MB, ChB, professor of neurology at Albert Einstein College of Medicine and director of the Sleep–Wake Disorders Center at Montefiore Medical Center in New York. When narcolepsy is suspected, testing is required to confirm the diagnosis. The patient should undergo all-night polysomnographic (PSG) testing, followed by a daytime multiple sleep latency test (MSLT). Measurement of CSF hypocretin can be diagnostic but is performed mainly in the research setting and is not common in the clinical setting, said Dr. Thorpy.
Patients with narcolepsy typically fall asleep in an average of less than 8 minutes during the nap opportunities of the MSLT. They also have at least two sleep-onset REM periods. “A new change in the diagnostic classification is that a sleep-onset REM period on the preceding night’s PSG can count as one of the two sleep-onset REM periods required for diagnosis,” said Dr. Thorpy.
“In the case of type 1 narcolepsy, the history is usually pretty clear, and the MSLT is usually positive, in the sense that it is consistent with a narcolepsy pattern,” said Thomas E. Scammell, MD, professor of neurology at Harvard Medical School and Beth Israel Deaconess Medical Center in Boston. “The PSG is also important, because other factors that disrupt the patient’s nighttime sleep (such as obstructive sleep apnea and periodic limb movements) must be ruled out, especially in type 2 narcolepsy,” said Dr. Scammell.
Early sleep onset, late diagnosis
Diagnostic delay is a common problem for patients with narcolepsy. Although the median age of onset is 16 years, a patient typically does not receive the appropriate diagnosis until adulthood. “It takes, on average, somewhere between 8 and 12 years for a patient to get a diagnosis of narcolepsy,” said Dr. Thorpy. Growing awareness and an increase in the number of sleep disorder centers have reduced but not eliminated the diagnostic delay.
Children with narcolepsy are often misdiagnosed. “One of the most common misdiagnoses in childhood is ADHD, because sleepiness in children differs from that in adults,” said Dr. Thorpy. Sleepy children often become hyperactive and display increased impulsivity, he explained. Stimulants prescribed for ADHD tend to mask the symptoms of narcolepsy and delay the correct diagnosis. Mood disorders, behavioral disorders, and psychogenic disorders are other common misdiagnoses for children with narcolepsy.
But when it comes to adults, sometimes patients themselves contribute to the diagnostic delay. EDS is “such a pervasive feeling that I think a lot of people just don’t make much of it,” said Dr. Scammell. The symptom is easily ascribed to insufficient sleep or a difficult work schedule. “It may take them months to get to see a doctor,” said Dr. Scammell.
Behavioral treatments
Nonpharmacologic treatments are one component of care for patients with narcolepsy. Patients must maintain a regular sleep-wake schedule and ensure that they are in bed for no less than 8 hours per night, said Dr. Thorpy. Taking no more than two daytime naps of less than 20 minutes each can help relieve some of the sleepiness, he added.
In addition to ensuring an adequate amount of sleep, it is important to promote good quality sleep, said Dr. Scammell. To do this, clinicians should address any conditions such as sleep apnea that disrupt patients’ sleep, he added.
Patients also tend to avoid situations that are likely to entail the emotional stimuli that could precipitate cataplexy. Some avoid laughter or try to suppress their emotions. “That’s not good,” said Kiran Maski, MD, MPH, assistant professor of neurology at Harvard Medical School and neurologist and sleep physician at Boston Children’s Hospital. “We worry that that might be a risk factor for depression or social isolation.” Cognitive-behavioral therapy can help patients with narcolepsy gradually increase their comfort with and exposure to social situations.
Although behavioral treatments are helpful, they are not sufficient to control all the symptoms of narcolepsy. Most patients require pharmacologic treatments, which are the most effective treatments for narcolepsy, said Dr. Thorpy.
Pharmacologic treatments
Previously, neurologists relied on the stimulants methylphenidate and amphetamine, which primarily treated patients’ EDS. But the field is moving away from these drugs because of their tendency to induce side effects and their potential for abuse, said Dr. Thorpy. In this context, modafinil and armodafinil became the mainstay for promoting alertness in patients with narcolepsy.
In recent years, newer medications have emerged that have slightly greater efficacy and better safety profiles than modafinil and armodafinil. Solriamfetol (Sunosi, Jazz Pharmaceuticals), for example, is effective for EDS but does not affect cataplexy. Pitolisant (Wakix, Harmony Biosciences), on the other hand, effectively treats EDS and cataplexy.
Sodium oxybate (Xyrem, Jazz Pharmaceuticals) is the only medication that treats all the symptoms of narcolepsy, said Dr. Thorpy. “That treats the sleepiness, the cataplexy, and the disturbed nocturnal sleep,” he added. Sodium oxybate also appears to reduce sleep paralysis, hallucinations, and disturbed dreams.
A potential concern about sodium oxybate, which has been used since approximately 2000, is its high sodium load. A new formulation called low-sodium oxybate (Xywav, Jazz Pharmaceuticals) “has a slightly better safety profile, particularly in people who have cardiovascular or renal disease,” said Dr. Thorpy. “This is tending to take over the role of regular sodium oxybate.”
Many clinicians who treat patients with narcolepsy develop their own approaches, but the choice of treatment generally depends on the patient’s symptoms, said Dr. Scammell. Modafinil is a good first choice for patients with mild to moderate sleepiness, he added. Pitolisant is another good choice for these patients but is more expensive. Both drugs are well tolerated.
Clinicians can consider solriamfetol and amphetamine for patients with moderate to severe sleepiness. “I generally consider the oxybates to be a second line,” said Dr. Scammell. Although these drugs may be the most effective, and they do help patients a great deal, they have a higher prevalence of side effects and are more expensive, he added. “If we can get good results with something gentle and simple like modafinil, that would be great.”
“There are differences of opinion as to what the first-line treatments are,” said Dr. Thorpy. Some patients prefer to use the traditional stimulants as first-line treatments, but others prefer to avoid them because of their adverse effects. They favor the newer, and unfortunately more expensive, medications instead. But there is no consensus among clinicians about which of the newer medications to use. “There’s no standard treatment, and it’s very hard to develop an algorithm that is acceptable to most physicians treating patients with narcolepsy,” said Dr. Thorpy. Treatment response varies, as well. Some patients respond extremely well to treatment, but clinical trials indicate that even optimal therapy helps patients achieve about 70% of the normal level of alertness. “If they’re sedentary, sitting in a boring meeting or at the computer, they can still fall asleep, even with our current medications,” said Dr. Scammell.
“The hardest symptom of all to treat is the EDS,” agreed Dr. Thorpy. Most patients cannot be treated with one medication alone, and polypharmacy tends to be necessary, he added. Typically, this means the addition of another medication to the regimen to maximize alertness. For other patients, cataplexy is difficult to control, and adding an anticataplectic medication is appropriate. Still, most patients can control their cataplexy with one drug, either oxybate or pitolisant, said Dr. Thorpy.
Investigational treatments
Researchers are trying to develop new medicines with greater potency, and several medications are under investigation. Early studies have shown that reboxetine, an antidepressant medication that affects dopamine and norepinephrine activity, is an effective treatment for EDS and cataplexy. Ongoing phase 3 studies are examining reboxetine for EDS. Another drug known as FT-218 is a once-nightly formulation of sodium oxybate, unlike the twice-nightly formulations of the drug that currently are available. In a phase 3 trial, the drug was associated with significant improvements in wakefulness and reductions in attacks of cataplexy. Avadel, which is developing the drug, submitted it to the U.S. Food and Drug Administration for approval in 2021, but the agency has not yet made a decision about it.
Researchers and patients alike have high hopes for medications that activate the orexin receptors. Orexin stimulates the wake-promoting neurons in the brain. Narcolepsy, and particularly narcolepsy type 1, is characterized by a loss of hypocretin cells in the central nervous system. The loss of these cells promotes sleepiness and disturbed REM sleep. To counteract this loss of cells, several companies are investigating new orexin agonists.
One such medication is TAK-994, which was developed by Takeda. The drug showed great promise for treating EDS and cataplexy, said Dr. Thorpy. But when phase 3 studies suggested that TAK-994 was associated with hepatotoxicity, the company terminated the studies. Nevertheless, other orexin agonists, including Takeda’s TAK-861, are under investigation.
“If we can restore orexin signaling, it could be like giving insulin to type 1 diabetics,” said Dr. Scammell. This class of medications could provide substantial improvements in sleepiness and other symptoms, he added. “I think when orexin agonists become available, it’s going to be quite transformative.” But these drugs are still in early development and will not be available in clinical practice for several years.
Common psychological comorbidities
Certain comorbidities are prevalent among patients with narcolepsy, and psychiatric disorders tend to be the most common. These comorbidities may complicate the management of narcolepsy. Nevertheless, they often are significant enough to require management in their own right, said Dr. Thorpy.
Depression is likely twice as common among patients with narcolepsy than among the general population, said Dr. Scammell. “Whether this is an actual neurobiologic feature of the disease, or whether it is just a reaction to having a challenging disorder isn’t entirely clear,” he added. “But it doesn’t get the attention or treatment that it deserves.”
Partnering with a psychologist or psychiatrist is important because many treatments can exacerbate mood disorders, said Dr. Maski. In general, stimulants, for example, can worsen depression and anxiety and are associated with increased suicide risk. “We oftentimes are using high-dose stimulants in patients, so mood has to be really carefully monitored and managed,” Dr. Maski added.
Cases of depression and suicidal ideation were reported in clinical trials of sodium oxybate. Although these serious adverse events were rare, patients must be monitored very closely even on treatments specifically approved for narcolepsy, said Dr. Maski. Mood disturbances are reported less frequently with modafinil and pitolisant than with stimulants, she noted.
Many times, patients need to take an antidepressant medication, but these drugs could affect the medicines administered for narcolepsy, said Dr. Thorpy. Pitolisant, in particular, may be adversely affected by current antidepressant medications. The only remedies are to change from pitolisant to another narcolepsy medication or to use an antidepressant that does not have histamine 1 receptor antagonism or affect the QTc interval.
Anxiety also is prevalent among patients with narcolepsy, and it can be worsened by traditional stimulants. These drugs also can increase the likelihood of irritability or obsessive-compulsive tendencies. “Traditional stimulants would be best avoided in these patients who have significant anxiety,” said Dr. Thorpy.
The social burden of narcolepsy
The burden of narcolepsy extends beyond psychiatric comorbidities into the social sphere. “Patients with narcolepsy do have greater difficulties in terms of social and interpersonal relationships,” said Dr. Thorpy. The disorder reduces patients’ quality of life, and educational difficulties and job loss are common in this population. “It’s a lifelong, incurable disorder, and these patients suffer an immense burden throughout their life because of the sleepiness that … affects their cognitive abilities,” said Dr. Thorpy.
“There’s an increased reporting of what probably amounts to social isolation,” said Dr. Maski. Patients often report that they must prioritize activities or events because they do not have the energy or alertness to participate in all of them. For instance, adolescents with narcolepsy frequently say that they must forgo after-school extracurricular activities because they need to prioritize studying and getting enough sleep. “Those priorities take away from their normal social life and events that they would like to participate in,” said Dr. Maski.
Another problem is that patients have the impression that others do not understand their condition. They are afraid that they will be perceived as lazy, uninterested, or unmotivated if they fall asleep. “Sometimes they withdraw from social events because they don’t want to be perceived in such a way,” said Dr. Maski. She and her colleagues encourage patients to participate in selected after-school events and to engage in social activities they find meaningful to maintain social networks.
An unpublished study of more than 300 patients with narcolepsy examined the effect of the disorder on patients’ social lives. At the end of the day, many patients “crash and burn,” said Dr. Scammell. Consequently, they do not have as much energy for social activities.
This lack of energy affects patients’ social relationships. The study suggests that patients with narcolepsy do not have as many friends as the general population does. Nevertheless, the frequency of close relationships and marriage was similar between patients with narcolepsy and the general population. “What people are doing is putting their energy into these close relationships, rather than having lots of friends and socializing a lot,” said Dr. Scammell. “I found that heartening, that people were doing their best and developed those close relationships,” which are vitally important for many reasons, he added.
The study, which has been submitted for publication, also asked patients about their sex lives. Many patients reported having had cataplexy during sex, and others reported that their medications caused problems with their sex lives. “Their doctors never ask about these things, and many patients actually would like their doctor to ask about them more,” said Dr. Scammell.
In addition, narcolepsy significantly affects a patient’s ability to drive. Patients with narcolepsy have a three- to fourfold increased risk of car accidents, said Dr. Scammell. This increased risk likely results from patients’ EDS.
But as important as this issue is for patients’ lives, there is no consensus on how to counsel patients about driving, said Dr. Maski. “For instance, it is not really clear if there is value in doing a maintenance of wakefulness test before allowing patients with narcolepsy to drive,” she said. The test is not validated in children or adolescents, which raises questions about how to advise beginning drivers with narcolepsy. “It’s not really clear that passing your maintenance of wakefulness test increases your safety behind the wheel,” said Dr. Maski.
“It’s the rare person with narcolepsy who can easily and safely do a 2-hour drive by themselves,” said Dr. Scammell. Patients must determine what their own limits are, and it is important for clinicians to discuss reasonable limits honestly with their patients. “I almost never would push to have somebody’s license taken away,” said Dr. Scammell. “But there are patients who only can drive around town for short errands, and if it’s anything more than half an hour, they start getting drowsy.”
There is a need for a public awareness campaign about narcolepsy, Dr. Scammell added. Such a campaign was carried out in Italy several years ago, and it included cartoons and TV segments. “It got a lot of people’s attention, and there was a real spike in new and correct diagnoses of narcolepsy,” said Dr. Scammell. But such a broad campaign is expensive, while narcolepsy is rare, and it might not be feasible to reach out to the general population. “But I certainly think it’s worth targeting doctors who are likely to see patients with sleepiness: neurologists, psychiatrists and psychologists, and primary care doctors,” said Dr. Scammell.
Almost half of Americans report feeling daytime sleepiness on at least 3 days per week. For most patients, this sleepiness results from insufficient nighttime sleep. But a minority of these patients have narcolepsy, a chronic neurologic disorder that impairs the brain’s control of sleep-wake cycles. This disorder often goes undiagnosed, but neurologists can make a significant difference by learning how to recognize and treat it.
What is narcolepsy?
Narcolepsy is characterized by excessive daytime sleepiness (EDS) and sudden attacks of sleep. Patients have difficulty staying awake for long periods of time, and the disorder can make performing daily tasks difficult. Problems with concentration and alertness are common.
Narcolepsy is considered to have two subtypes. Patients with narcolepsy type 1 also have cataplexy, a sudden loss of muscle tone. Attacks of cataplexy are triggered by strong, usually positive, emotions. These attacks have manifestations ranging from slurred speech to complete weakness of most muscles. Patients with narcolepsy type 2, however, do not have cataplexy.
Dysregulation of rapid eye movement (REM) sleep, which is when most dreaming occurs, is another symptom of narcolepsy. The transition to REM sleep is quicker in patients with narcolepsy and usually occurs within 15 minutes of sleep onset. A related symptom is sleep paralysis, an inability to move while falling asleep or waking up. This symptom resembles a state that normally occurs during REM sleep.
Hallucinations also are common in patients with narcolepsy and can be especially vivid. Hypnagogic hallucinations occur during the transition to sleep, and hypnopompic hallucinations arise while the patient is waking up. Patients may think they see a stranger in their bedroom, and children sometimes report seeing animals.
Although it is easy for patients with narcolepsy to fall asleep at night, they often have disrupted sleep. Patients have frequent, brief arousals throughout the night that may become disturbing. Dream content often is affected in narcolepsy, too. Patients have described lucid dreams of flying or out-of-body experiences. After such intense dreams, patients often feel that their sleep has not been restful.
Criteria and diagnosis
To receive a diagnosis of narcolepsy type 1, a patient must have EDS that persists for at least 3 months and at least one of the following two features: cataplexy and objective evidence of quick sleep onset and early start of REM sleep or low cerebrospinal fluid (CSF) levels (that is, less than 110 pg/mL) of hypocretin. Hypocretin, also known as orexin, is a neuropeptide that regulates wakefulness and arousal.
Patients must meet five criteria to receive a diagnosis of narcolepsy type 2. They must have EDS that persists for at least 3 months. They must have test results that show quick sleep onset and early start of REM sleep. They must have no cataplexy. Their CSF levels of hypocretin must be normal or unknown. Finally, they must have no other conditions that provide a better explanation for their symptoms and test results.
“The diagnosis of narcolepsy is made primarily by history on the clinical features of the disorder,” said Michael J. Thorpy, MB, ChB, professor of neurology at Albert Einstein College of Medicine and director of the Sleep–Wake Disorders Center at Montefiore Medical Center in New York. When narcolepsy is suspected, testing is required to confirm the diagnosis. The patient should undergo all-night polysomnographic (PSG) testing, followed by a daytime multiple sleep latency test (MSLT). Measurement of CSF hypocretin can be diagnostic but is performed mainly in the research setting and is not common in the clinical setting, said Dr. Thorpy.
Patients with narcolepsy typically fall asleep in an average of less than 8 minutes during the nap opportunities of the MSLT. They also have at least two sleep-onset REM periods. “A new change in the diagnostic classification is that a sleep-onset REM period on the preceding night’s PSG can count as one of the two sleep-onset REM periods required for diagnosis,” said Dr. Thorpy.
“In the case of type 1 narcolepsy, the history is usually pretty clear, and the MSLT is usually positive, in the sense that it is consistent with a narcolepsy pattern,” said Thomas E. Scammell, MD, professor of neurology at Harvard Medical School and Beth Israel Deaconess Medical Center in Boston. “The PSG is also important, because other factors that disrupt the patient’s nighttime sleep (such as obstructive sleep apnea and periodic limb movements) must be ruled out, especially in type 2 narcolepsy,” said Dr. Scammell.
Early sleep onset, late diagnosis
Diagnostic delay is a common problem for patients with narcolepsy. Although the median age of onset is 16 years, a patient typically does not receive the appropriate diagnosis until adulthood. “It takes, on average, somewhere between 8 and 12 years for a patient to get a diagnosis of narcolepsy,” said Dr. Thorpy. Growing awareness and an increase in the number of sleep disorder centers have reduced but not eliminated the diagnostic delay.
Children with narcolepsy are often misdiagnosed. “One of the most common misdiagnoses in childhood is ADHD, because sleepiness in children differs from that in adults,” said Dr. Thorpy. Sleepy children often become hyperactive and display increased impulsivity, he explained. Stimulants prescribed for ADHD tend to mask the symptoms of narcolepsy and delay the correct diagnosis. Mood disorders, behavioral disorders, and psychogenic disorders are other common misdiagnoses for children with narcolepsy.
But when it comes to adults, sometimes patients themselves contribute to the diagnostic delay. EDS is “such a pervasive feeling that I think a lot of people just don’t make much of it,” said Dr. Scammell. The symptom is easily ascribed to insufficient sleep or a difficult work schedule. “It may take them months to get to see a doctor,” said Dr. Scammell.
Behavioral treatments
Nonpharmacologic treatments are one component of care for patients with narcolepsy. Patients must maintain a regular sleep-wake schedule and ensure that they are in bed for no less than 8 hours per night, said Dr. Thorpy. Taking no more than two daytime naps of less than 20 minutes each can help relieve some of the sleepiness, he added.
In addition to ensuring an adequate amount of sleep, it is important to promote good quality sleep, said Dr. Scammell. To do this, clinicians should address any conditions such as sleep apnea that disrupt patients’ sleep, he added.
Patients also tend to avoid situations that are likely to entail the emotional stimuli that could precipitate cataplexy. Some avoid laughter or try to suppress their emotions. “That’s not good,” said Kiran Maski, MD, MPH, assistant professor of neurology at Harvard Medical School and neurologist and sleep physician at Boston Children’s Hospital. “We worry that that might be a risk factor for depression or social isolation.” Cognitive-behavioral therapy can help patients with narcolepsy gradually increase their comfort with and exposure to social situations.
Although behavioral treatments are helpful, they are not sufficient to control all the symptoms of narcolepsy. Most patients require pharmacologic treatments, which are the most effective treatments for narcolepsy, said Dr. Thorpy.
Pharmacologic treatments
Previously, neurologists relied on the stimulants methylphenidate and amphetamine, which primarily treated patients’ EDS. But the field is moving away from these drugs because of their tendency to induce side effects and their potential for abuse, said Dr. Thorpy. In this context, modafinil and armodafinil became the mainstay for promoting alertness in patients with narcolepsy.
In recent years, newer medications have emerged that have slightly greater efficacy and better safety profiles than modafinil and armodafinil. Solriamfetol (Sunosi, Jazz Pharmaceuticals), for example, is effective for EDS but does not affect cataplexy. Pitolisant (Wakix, Harmony Biosciences), on the other hand, effectively treats EDS and cataplexy.
Sodium oxybate (Xyrem, Jazz Pharmaceuticals) is the only medication that treats all the symptoms of narcolepsy, said Dr. Thorpy. “That treats the sleepiness, the cataplexy, and the disturbed nocturnal sleep,” he added. Sodium oxybate also appears to reduce sleep paralysis, hallucinations, and disturbed dreams.
A potential concern about sodium oxybate, which has been used since approximately 2000, is its high sodium load. A new formulation called low-sodium oxybate (Xywav, Jazz Pharmaceuticals) “has a slightly better safety profile, particularly in people who have cardiovascular or renal disease,” said Dr. Thorpy. “This is tending to take over the role of regular sodium oxybate.”
Many clinicians who treat patients with narcolepsy develop their own approaches, but the choice of treatment generally depends on the patient’s symptoms, said Dr. Scammell. Modafinil is a good first choice for patients with mild to moderate sleepiness, he added. Pitolisant is another good choice for these patients but is more expensive. Both drugs are well tolerated.
Clinicians can consider solriamfetol and amphetamine for patients with moderate to severe sleepiness. “I generally consider the oxybates to be a second line,” said Dr. Scammell. Although these drugs may be the most effective, and they do help patients a great deal, they have a higher prevalence of side effects and are more expensive, he added. “If we can get good results with something gentle and simple like modafinil, that would be great.”
“There are differences of opinion as to what the first-line treatments are,” said Dr. Thorpy. Some patients prefer to use the traditional stimulants as first-line treatments, but others prefer to avoid them because of their adverse effects. They favor the newer, and unfortunately more expensive, medications instead. But there is no consensus among clinicians about which of the newer medications to use. “There’s no standard treatment, and it’s very hard to develop an algorithm that is acceptable to most physicians treating patients with narcolepsy,” said Dr. Thorpy. Treatment response varies, as well. Some patients respond extremely well to treatment, but clinical trials indicate that even optimal therapy helps patients achieve about 70% of the normal level of alertness. “If they’re sedentary, sitting in a boring meeting or at the computer, they can still fall asleep, even with our current medications,” said Dr. Scammell.
“The hardest symptom of all to treat is the EDS,” agreed Dr. Thorpy. Most patients cannot be treated with one medication alone, and polypharmacy tends to be necessary, he added. Typically, this means the addition of another medication to the regimen to maximize alertness. For other patients, cataplexy is difficult to control, and adding an anticataplectic medication is appropriate. Still, most patients can control their cataplexy with one drug, either oxybate or pitolisant, said Dr. Thorpy.
Investigational treatments
Researchers are trying to develop new medicines with greater potency, and several medications are under investigation. Early studies have shown that reboxetine, an antidepressant medication that affects dopamine and norepinephrine activity, is an effective treatment for EDS and cataplexy. Ongoing phase 3 studies are examining reboxetine for EDS. Another drug known as FT-218 is a once-nightly formulation of sodium oxybate, unlike the twice-nightly formulations of the drug that currently are available. In a phase 3 trial, the drug was associated with significant improvements in wakefulness and reductions in attacks of cataplexy. Avadel, which is developing the drug, submitted it to the U.S. Food and Drug Administration for approval in 2021, but the agency has not yet made a decision about it.
Researchers and patients alike have high hopes for medications that activate the orexin receptors. Orexin stimulates the wake-promoting neurons in the brain. Narcolepsy, and particularly narcolepsy type 1, is characterized by a loss of hypocretin cells in the central nervous system. The loss of these cells promotes sleepiness and disturbed REM sleep. To counteract this loss of cells, several companies are investigating new orexin agonists.
One such medication is TAK-994, which was developed by Takeda. The drug showed great promise for treating EDS and cataplexy, said Dr. Thorpy. But when phase 3 studies suggested that TAK-994 was associated with hepatotoxicity, the company terminated the studies. Nevertheless, other orexin agonists, including Takeda’s TAK-861, are under investigation.
“If we can restore orexin signaling, it could be like giving insulin to type 1 diabetics,” said Dr. Scammell. This class of medications could provide substantial improvements in sleepiness and other symptoms, he added. “I think when orexin agonists become available, it’s going to be quite transformative.” But these drugs are still in early development and will not be available in clinical practice for several years.
Common psychological comorbidities
Certain comorbidities are prevalent among patients with narcolepsy, and psychiatric disorders tend to be the most common. These comorbidities may complicate the management of narcolepsy. Nevertheless, they often are significant enough to require management in their own right, said Dr. Thorpy.
Depression is likely twice as common among patients with narcolepsy than among the general population, said Dr. Scammell. “Whether this is an actual neurobiologic feature of the disease, or whether it is just a reaction to having a challenging disorder isn’t entirely clear,” he added. “But it doesn’t get the attention or treatment that it deserves.”
Partnering with a psychologist or psychiatrist is important because many treatments can exacerbate mood disorders, said Dr. Maski. In general, stimulants, for example, can worsen depression and anxiety and are associated with increased suicide risk. “We oftentimes are using high-dose stimulants in patients, so mood has to be really carefully monitored and managed,” Dr. Maski added.
Cases of depression and suicidal ideation were reported in clinical trials of sodium oxybate. Although these serious adverse events were rare, patients must be monitored very closely even on treatments specifically approved for narcolepsy, said Dr. Maski. Mood disturbances are reported less frequently with modafinil and pitolisant than with stimulants, she noted.
Many times, patients need to take an antidepressant medication, but these drugs could affect the medicines administered for narcolepsy, said Dr. Thorpy. Pitolisant, in particular, may be adversely affected by current antidepressant medications. The only remedies are to change from pitolisant to another narcolepsy medication or to use an antidepressant that does not have histamine 1 receptor antagonism or affect the QTc interval.
Anxiety also is prevalent among patients with narcolepsy, and it can be worsened by traditional stimulants. These drugs also can increase the likelihood of irritability or obsessive-compulsive tendencies. “Traditional stimulants would be best avoided in these patients who have significant anxiety,” said Dr. Thorpy.
The social burden of narcolepsy
The burden of narcolepsy extends beyond psychiatric comorbidities into the social sphere. “Patients with narcolepsy do have greater difficulties in terms of social and interpersonal relationships,” said Dr. Thorpy. The disorder reduces patients’ quality of life, and educational difficulties and job loss are common in this population. “It’s a lifelong, incurable disorder, and these patients suffer an immense burden throughout their life because of the sleepiness that … affects their cognitive abilities,” said Dr. Thorpy.
“There’s an increased reporting of what probably amounts to social isolation,” said Dr. Maski. Patients often report that they must prioritize activities or events because they do not have the energy or alertness to participate in all of them. For instance, adolescents with narcolepsy frequently say that they must forgo after-school extracurricular activities because they need to prioritize studying and getting enough sleep. “Those priorities take away from their normal social life and events that they would like to participate in,” said Dr. Maski.
Another problem is that patients have the impression that others do not understand their condition. They are afraid that they will be perceived as lazy, uninterested, or unmotivated if they fall asleep. “Sometimes they withdraw from social events because they don’t want to be perceived in such a way,” said Dr. Maski. She and her colleagues encourage patients to participate in selected after-school events and to engage in social activities they find meaningful to maintain social networks.
An unpublished study of more than 300 patients with narcolepsy examined the effect of the disorder on patients’ social lives. At the end of the day, many patients “crash and burn,” said Dr. Scammell. Consequently, they do not have as much energy for social activities.
This lack of energy affects patients’ social relationships. The study suggests that patients with narcolepsy do not have as many friends as the general population does. Nevertheless, the frequency of close relationships and marriage was similar between patients with narcolepsy and the general population. “What people are doing is putting their energy into these close relationships, rather than having lots of friends and socializing a lot,” said Dr. Scammell. “I found that heartening, that people were doing their best and developed those close relationships,” which are vitally important for many reasons, he added.
The study, which has been submitted for publication, also asked patients about their sex lives. Many patients reported having had cataplexy during sex, and others reported that their medications caused problems with their sex lives. “Their doctors never ask about these things, and many patients actually would like their doctor to ask about them more,” said Dr. Scammell.
In addition, narcolepsy significantly affects a patient’s ability to drive. Patients with narcolepsy have a three- to fourfold increased risk of car accidents, said Dr. Scammell. This increased risk likely results from patients’ EDS.
But as important as this issue is for patients’ lives, there is no consensus on how to counsel patients about driving, said Dr. Maski. “For instance, it is not really clear if there is value in doing a maintenance of wakefulness test before allowing patients with narcolepsy to drive,” she said. The test is not validated in children or adolescents, which raises questions about how to advise beginning drivers with narcolepsy. “It’s not really clear that passing your maintenance of wakefulness test increases your safety behind the wheel,” said Dr. Maski.
“It’s the rare person with narcolepsy who can easily and safely do a 2-hour drive by themselves,” said Dr. Scammell. Patients must determine what their own limits are, and it is important for clinicians to discuss reasonable limits honestly with their patients. “I almost never would push to have somebody’s license taken away,” said Dr. Scammell. “But there are patients who only can drive around town for short errands, and if it’s anything more than half an hour, they start getting drowsy.”
There is a need for a public awareness campaign about narcolepsy, Dr. Scammell added. Such a campaign was carried out in Italy several years ago, and it included cartoons and TV segments. “It got a lot of people’s attention, and there was a real spike in new and correct diagnoses of narcolepsy,” said Dr. Scammell. But such a broad campaign is expensive, while narcolepsy is rare, and it might not be feasible to reach out to the general population. “But I certainly think it’s worth targeting doctors who are likely to see patients with sleepiness: neurologists, psychiatrists and psychologists, and primary care doctors,” said Dr. Scammell.
NORD Rare Disease Centers of Excellence: A new network seeks to break down barriers in rare disease care
In November 2021, the National Organization for Rare Disorders (NORD) announced that it had designated 31 institutions across the United States as “NORD Rare Disease Centers of Excellence.” More than just a stamp of approval, the new NORD network aims to change the way rare diseases are diagnosed and treated, creating more efficient pathways for collaboration among physicians, while helping patients get better care closer to home.

To understand better how the nascent network can benefit patients and clinicians, Neurology Reviews/MDedge Neurology spoke with Ed Neilan, MD, PhD, NORD’s chief scientific and medical officer. Dr. Neilan, a pediatrician and geneticist, is a former president of the medical staff at Boston Children’s Hospital and also served as head of global medical affairs for rare neurology at Sanofi Genzyme.
How did NORD choose its 31 centers?
We were looking for places that had both broad capabilities and deep expertise, where it was reasonable to expect that a patient with almost any condition could go and, without too many missteps or delays, get the right diagnosis or the right treatment. We also sought sites that were educating the next generation of rare disease specialists across departments. The sites had to be involved in research, because that moves the field forward, and sometimes it’s the only way to get a really impactful treatment for the 95% of rare diseases that don’t have an FDA-approved treatment. NORD sent a letter inviting different centers to apply, along with an application that had 120 questions. Most of the questions sought information about what kinds of expertise or services were available on-site, so that patients don’t have to go elsewhere to get, let’s say, a brain MRI scan or to see an immunologist. We wanted each site to be a place where you could go for almost any problem, at any age, and expect that while you’re being seen, and receiving treatment, it can also contribute to the education of the next generation of rare disease specialists and to research.
Several of the members of the network comprise more than one institution: They’re a children’s hospital combined with another facility.
Children’s hospitals, which are highly specialized and able to care for rare things in children, couldn’t apply by themselves. They had to apply in partnership with a center that could provide adult care as patients got older; otherwise, their care model would be incomplete. We’ve had some small victories already just by asking these questions and outlining this sort of approach. At one institution in the Great Plains, the director told us that he had been trying for years to get permission to hire someone who could make appointments across three different hospitals – a children’s hospital and two adult hospitals. He’d wanted to ensure that patients with rare and genetic diseases were seen in the appropriate places, and thanks to the NORD designation, he finally can. Now, regardless of age, the same office staff can handle the arrangements, and the patient will be scheduled in the right place.
You make clear that these are different from disease-specific centers of excellence – you specifically chose the 31 centers for their breadth of expertise. There’s no way to represent all 7,000 rare diseases equally, and disease-specific centers of excellence, which already exist for hemophilia, muscular dystrophy, cystic fibrosis, and some other conditions, have a very important role. We’re not aiming to compete with any other existing resources. What we are seeking to do is to fill the unmet need of, “What if there are no such designations for the disease that you’re concerned about?” Our goal was to find places that could help with unanswered questions, whether diagnostic questions or treatment questions. To identify places where a patient could reasonably expect to go and have a deeper dive – maybe an interdisciplinary deep dive.
The delay to diagnosis can be years in rare diseases. How can the network help speed up diagnoses?
With all these experts on different diseases, we hope to develop some better diagnostic algorithms within the network. Another thing we can do is to share resources. With 31 sites, everybody’s seeing patients with unknown diagnoses. Everyone is seeing patients for whom they would maybe like to get a whole genome done, or a whole exome done, but they are often encountering stiff resistance from insurance companies.
Meanwhile some sites, but not all 31, have multimillion-dollar grants to do sequencing and other kinds of advanced diagnostic tests to solve unknown cases. And there are people at those sites who say, “We need more samples. Can you get us samples from the other sites?”
One of the main things we aim to do is share information, including information about available diagnostic resources. We want all 31 sites to know which sites have funding and programs that enable them to study samples for other sites. We also want to know what criteria they’re putting on it. Someone might say: “I’ve got a grant to sequence genomes for people with unexplained seizures. Send me all your unexplained seizures.” Somebody else might have a grant for unexplained GI diseases. So, we want to put on our intranet a resource for the 31 sites, kind of a cookbook for – when if you can’t get it paid for by insurance, but you really think you need a particular special test – who might be able to do it for you within the network.
This would seem to benefit research across sites as well.
Yes, but we also want to share clinical advice and expertise for direct patient benefit. So, it doesn’t always have to fulfill the goals of a specific research project. For example, we might be able to create an undiagnosed patient quality improvement database across all 31 sites that could compliantly let Drs. X and Y know that they’re each seeing a patient with the same rare thing.
But let’s say you want to move the field forward by discovering a new disease. Rare genetic diseases are now being discovered at the rate of about 250 a year, so about 5 per week across the world. With two or three unrelated patients who have the same disease and a whole exome sequence, you can potentially discover a disease. Maybe you’ve found one unique patient with a genetic variant of possible significance, but you can’t be 100% sure, and you may not be able to convince your colleagues, or journal editors, until you find other cases. You need those two or three ultrarare patients. Within this network, a lot of sites want to share information about their ultrarare patients and be able to put together additional instances of the same thing, to prove that it is a real disease, to learn more about it and how to diagnose, manage, and treat it.
Part of the idea with a nationwide network is that patients aren’t going to have to move around among these centers of excellence, is that correct? They’re going to be seen at the closest ones, and it’s the expertise that is mobile.
Yes, that’s right. While we can’t eliminate the need for travel, what we are trying to do is increase the sharing of expertise, to improve results for patients while limiting the need for traveling very long distances. As a geneticist I’ve been on both the requesting and the receiving end of consultations with doctors at other sites, sometimes very far away, especially for ultrarare conditions for which any one physician’s experience is limited. We all try to honor these sorts of requests, but insurance doesn’t reimburse it and so hospitals don’t give doctors much credit for it.
We want to ultimately find ways to incentivize this type of collaboration. Hopefully we can get agreements with insurance companies to allow intersite consultations within our network, recognizing that they don’t want to pay for the patient to be seen out of state, but you also want the patient to get the best possible medical advice. This might require legislative changes in the long run. But what we can do more readily is create a culture within this network of mutual consultation and sharing of clinical experience. Outside of such a network, the idea of “cold calling” somebody, whom you may never have met, and asking them for help and free advice is a little bit of a bar, right? We want to lower that bar.
Can patients get telemedicine consults with physicians across the network?
NORD supports having telemedicine options for everybody regardless of diagnosis, rare or not, and we support legislation that would continue access and reimbursement for telemedicine post pandemic. I hope we can get that, or at least preserve telemedicine for rare diseases, for which there are often not enough, or sometimes not any, expert providers in the same state. Ultimately, we want patients to be able to get the expert assessments and advice they need. For rare diseases, that sometimes means battling back and forth with an insurance provider, seeking permission to see an expert clinician a thousand miles away. By sharing medical expertise, and through telemedicine when that’s allowed, we hope to reduce the need for that. But the telemedicine environment is still evolving and somewhat uncertain.
How will the network’s physician collaborations take place?
One of the important things NORD is providing to the network is an information technology setup and intranet across the 31 sites. That intranet is where center staff will go to access the network’s internal resources, including live and recorded case conferences. In those case conferences you can present a case you haven’t been able to solve. Experts you may have only heard of by reputation will now be streamed to your computer as part of the nationwide network. It benefits the patient because you get additional expert opinions, but it also benefits the physicians because we have this collegial space for discussion and learning. We’ll be linked by frequent meetings – some in person, most virtual – a common culture, and a common intranet.
On the intranet, we will also have a growing set of useful databases, links, and documents that are available to all members. These will be progressively updated with help from experts at the centers, so that clinicians can more directly learn from each other, instead of separately reinventing the wheel. The way things usually work, when you see a patient with an ultrarare condition that you’re not that familiar with, is that you tell them what little you can, then schedule them to come back in a few weeks. In the meantime, usually in your off time, you spend hours searching PubMed and other sources and you try to piece things together, to figure out what’s known that might help your patient. But imagine that this has already been figured out by someone else in the network. You can see on the network a list of articles the other expert read and found helpful in addressing this problem. And you then reach out directly to that other expert.
In recent months you’ve had one-on-one meetings with all 31 directors at the sites, and after that you convened 11 working groups. What are you trying to achieve?
Once the sites were chosen, we aimed to talk quickly and honestly about what everyone needed, what everyone saw as the biggest problems to tackle in rare diseases. Two things were very rewarding about those phone calls: one, all the centers were very enthusiastic, and two, they pretty much all agreed on what the key unmet needs are for rare disease patients and the practitioners trying to help them. So, we empaneled working groups of expert volunteers enthusiastic to work on each of those problems. These groups collectively comprise more than 200 volunteers – faculty, staff, and trainees – from the different sites nationwide. Each group is working on a key unmet need in rare diseases, and each group will be given its own space on our file-sharing platform, where they can share information and co-develop new ideas and documents. When something they produce is good enough to start to be a practice resource, such as a draft treatment guideline that the working group now wants to try in the real world, but it’s not yet ready to be published, they can share it and have it tested by all 31 sites through the dedicated intranet we are building for the network.
Jennie Smith is a freelance journalist specializing in medicine and health.
In November 2021, the National Organization for Rare Disorders (NORD) announced that it had designated 31 institutions across the United States as “NORD Rare Disease Centers of Excellence.” More than just a stamp of approval, the new NORD network aims to change the way rare diseases are diagnosed and treated, creating more efficient pathways for collaboration among physicians, while helping patients get better care closer to home.
To understand better how the nascent network can benefit patients and clinicians, Neurology Reviews/MDedge Neurology spoke with Ed Neilan, MD, PhD, NORD’s chief scientific and medical officer. Dr. Neilan, a pediatrician and geneticist, is a former president of the medical staff at Boston Children’s Hospital and also served as head of global medical affairs for rare neurology at Sanofi Genzyme.
How did NORD choose its 31 centers?
We were looking for places that had both broad capabilities and deep expertise, where it was reasonable to expect that a patient with almost any condition could go and, without too many missteps or delays, get the right diagnosis or the right treatment. We also sought sites that were educating the next generation of rare disease specialists across departments. The sites had to be involved in research, because that moves the field forward, and sometimes it’s the only way to get a really impactful treatment for the 95% of rare diseases that don’t have an FDA-approved treatment. NORD sent a letter inviting different centers to apply, along with an application that had 120 questions. Most of the questions sought information about what kinds of expertise or services were available on-site, so that patients don’t have to go elsewhere to get, let’s say, a brain MRI scan or to see an immunologist. We wanted each site to be a place where you could go for almost any problem, at any age, and expect that while you’re being seen, and receiving treatment, it can also contribute to the education of the next generation of rare disease specialists and to research.
Several of the members of the network comprise more than one institution: They’re a children’s hospital combined with another facility.
Children’s hospitals, which are highly specialized and able to care for rare things in children, couldn’t apply by themselves. They had to apply in partnership with a center that could provide adult care as patients got older; otherwise, their care model would be incomplete. We’ve had some small victories already just by asking these questions and outlining this sort of approach. At one institution in the Great Plains, the director told us that he had been trying for years to get permission to hire someone who could make appointments across three different hospitals – a children’s hospital and two adult hospitals. He’d wanted to ensure that patients with rare and genetic diseases were seen in the appropriate places, and thanks to the NORD designation, he finally can. Now, regardless of age, the same office staff can handle the arrangements, and the patient will be scheduled in the right place.
You make clear that these are different from disease-specific centers of excellence – you specifically chose the 31 centers for their breadth of expertise. There’s no way to represent all 7,000 rare diseases equally, and disease-specific centers of excellence, which already exist for hemophilia, muscular dystrophy, cystic fibrosis, and some other conditions, have a very important role. We’re not aiming to compete with any other existing resources. What we are seeking to do is to fill the unmet need of, “What if there are no such designations for the disease that you’re concerned about?” Our goal was to find places that could help with unanswered questions, whether diagnostic questions or treatment questions. To identify places where a patient could reasonably expect to go and have a deeper dive – maybe an interdisciplinary deep dive.
The delay to diagnosis can be years in rare diseases. How can the network help speed up diagnoses?
With all these experts on different diseases, we hope to develop some better diagnostic algorithms within the network. Another thing we can do is to share resources. With 31 sites, everybody’s seeing patients with unknown diagnoses. Everyone is seeing patients for whom they would maybe like to get a whole genome done, or a whole exome done, but they are often encountering stiff resistance from insurance companies.
Meanwhile some sites, but not all 31, have multimillion-dollar grants to do sequencing and other kinds of advanced diagnostic tests to solve unknown cases. And there are people at those sites who say, “We need more samples. Can you get us samples from the other sites?”
One of the main things we aim to do is share information, including information about available diagnostic resources. We want all 31 sites to know which sites have funding and programs that enable them to study samples for other sites. We also want to know what criteria they’re putting on it. Someone might say: “I’ve got a grant to sequence genomes for people with unexplained seizures. Send me all your unexplained seizures.” Somebody else might have a grant for unexplained GI diseases. So, we want to put on our intranet a resource for the 31 sites, kind of a cookbook for – when if you can’t get it paid for by insurance, but you really think you need a particular special test – who might be able to do it for you within the network.
This would seem to benefit research across sites as well.
Yes, but we also want to share clinical advice and expertise for direct patient benefit. So, it doesn’t always have to fulfill the goals of a specific research project. For example, we might be able to create an undiagnosed patient quality improvement database across all 31 sites that could compliantly let Drs. X and Y know that they’re each seeing a patient with the same rare thing.
But let’s say you want to move the field forward by discovering a new disease. Rare genetic diseases are now being discovered at the rate of about 250 a year, so about 5 per week across the world. With two or three unrelated patients who have the same disease and a whole exome sequence, you can potentially discover a disease. Maybe you’ve found one unique patient with a genetic variant of possible significance, but you can’t be 100% sure, and you may not be able to convince your colleagues, or journal editors, until you find other cases. You need those two or three ultrarare patients. Within this network, a lot of sites want to share information about their ultrarare patients and be able to put together additional instances of the same thing, to prove that it is a real disease, to learn more about it and how to diagnose, manage, and treat it.
Part of the idea with a nationwide network is that patients aren’t going to have to move around among these centers of excellence, is that correct? They’re going to be seen at the closest ones, and it’s the expertise that is mobile.
Yes, that’s right. While we can’t eliminate the need for travel, what we are trying to do is increase the sharing of expertise, to improve results for patients while limiting the need for traveling very long distances. As a geneticist I’ve been on both the requesting and the receiving end of consultations with doctors at other sites, sometimes very far away, especially for ultrarare conditions for which any one physician’s experience is limited. We all try to honor these sorts of requests, but insurance doesn’t reimburse it and so hospitals don’t give doctors much credit for it.
We want to ultimately find ways to incentivize this type of collaboration. Hopefully we can get agreements with insurance companies to allow intersite consultations within our network, recognizing that they don’t want to pay for the patient to be seen out of state, but you also want the patient to get the best possible medical advice. This might require legislative changes in the long run. But what we can do more readily is create a culture within this network of mutual consultation and sharing of clinical experience. Outside of such a network, the idea of “cold calling” somebody, whom you may never have met, and asking them for help and free advice is a little bit of a bar, right? We want to lower that bar.
Can patients get telemedicine consults with physicians across the network?
NORD supports having telemedicine options for everybody regardless of diagnosis, rare or not, and we support legislation that would continue access and reimbursement for telemedicine post pandemic. I hope we can get that, or at least preserve telemedicine for rare diseases, for which there are often not enough, or sometimes not any, expert providers in the same state. Ultimately, we want patients to be able to get the expert assessments and advice they need. For rare diseases, that sometimes means battling back and forth with an insurance provider, seeking permission to see an expert clinician a thousand miles away. By sharing medical expertise, and through telemedicine when that’s allowed, we hope to reduce the need for that. But the telemedicine environment is still evolving and somewhat uncertain.
How will the network’s physician collaborations take place?
One of the important things NORD is providing to the network is an information technology setup and intranet across the 31 sites. That intranet is where center staff will go to access the network’s internal resources, including live and recorded case conferences. In those case conferences you can present a case you haven’t been able to solve. Experts you may have only heard of by reputation will now be streamed to your computer as part of the nationwide network. It benefits the patient because you get additional expert opinions, but it also benefits the physicians because we have this collegial space for discussion and learning. We’ll be linked by frequent meetings – some in person, most virtual – a common culture, and a common intranet.
On the intranet, we will also have a growing set of useful databases, links, and documents that are available to all members. These will be progressively updated with help from experts at the centers, so that clinicians can more directly learn from each other, instead of separately reinventing the wheel. The way things usually work, when you see a patient with an ultrarare condition that you’re not that familiar with, is that you tell them what little you can, then schedule them to come back in a few weeks. In the meantime, usually in your off time, you spend hours searching PubMed and other sources and you try to piece things together, to figure out what’s known that might help your patient. But imagine that this has already been figured out by someone else in the network. You can see on the network a list of articles the other expert read and found helpful in addressing this problem. And you then reach out directly to that other expert.
In recent months you’ve had one-on-one meetings with all 31 directors at the sites, and after that you convened 11 working groups. What are you trying to achieve?
Once the sites were chosen, we aimed to talk quickly and honestly about what everyone needed, what everyone saw as the biggest problems to tackle in rare diseases. Two things were very rewarding about those phone calls: one, all the centers were very enthusiastic, and two, they pretty much all agreed on what the key unmet needs are for rare disease patients and the practitioners trying to help them. So, we empaneled working groups of expert volunteers enthusiastic to work on each of those problems. These groups collectively comprise more than 200 volunteers – faculty, staff, and trainees – from the different sites nationwide. Each group is working on a key unmet need in rare diseases, and each group will be given its own space on our file-sharing platform, where they can share information and co-develop new ideas and documents. When something they produce is good enough to start to be a practice resource, such as a draft treatment guideline that the working group now wants to try in the real world, but it’s not yet ready to be published, they can share it and have it tested by all 31 sites through the dedicated intranet we are building for the network.
Jennie Smith is a freelance journalist specializing in medicine and health.
In November 2021, the National Organization for Rare Disorders (NORD) announced that it had designated 31 institutions across the United States as “NORD Rare Disease Centers of Excellence.” More than just a stamp of approval, the new NORD network aims to change the way rare diseases are diagnosed and treated, creating more efficient pathways for collaboration among physicians, while helping patients get better care closer to home.
To understand better how the nascent network can benefit patients and clinicians, Neurology Reviews/MDedge Neurology spoke with Ed Neilan, MD, PhD, NORD’s chief scientific and medical officer. Dr. Neilan, a pediatrician and geneticist, is a former president of the medical staff at Boston Children’s Hospital and also served as head of global medical affairs for rare neurology at Sanofi Genzyme.
How did NORD choose its 31 centers?
We were looking for places that had both broad capabilities and deep expertise, where it was reasonable to expect that a patient with almost any condition could go and, without too many missteps or delays, get the right diagnosis or the right treatment. We also sought sites that were educating the next generation of rare disease specialists across departments. The sites had to be involved in research, because that moves the field forward, and sometimes it’s the only way to get a really impactful treatment for the 95% of rare diseases that don’t have an FDA-approved treatment. NORD sent a letter inviting different centers to apply, along with an application that had 120 questions. Most of the questions sought information about what kinds of expertise or services were available on-site, so that patients don’t have to go elsewhere to get, let’s say, a brain MRI scan or to see an immunologist. We wanted each site to be a place where you could go for almost any problem, at any age, and expect that while you’re being seen, and receiving treatment, it can also contribute to the education of the next generation of rare disease specialists and to research.
Several of the members of the network comprise more than one institution: They’re a children’s hospital combined with another facility.
Children’s hospitals, which are highly specialized and able to care for rare things in children, couldn’t apply by themselves. They had to apply in partnership with a center that could provide adult care as patients got older; otherwise, their care model would be incomplete. We’ve had some small victories already just by asking these questions and outlining this sort of approach. At one institution in the Great Plains, the director told us that he had been trying for years to get permission to hire someone who could make appointments across three different hospitals – a children’s hospital and two adult hospitals. He’d wanted to ensure that patients with rare and genetic diseases were seen in the appropriate places, and thanks to the NORD designation, he finally can. Now, regardless of age, the same office staff can handle the arrangements, and the patient will be scheduled in the right place.
You make clear that these are different from disease-specific centers of excellence – you specifically chose the 31 centers for their breadth of expertise. There’s no way to represent all 7,000 rare diseases equally, and disease-specific centers of excellence, which already exist for hemophilia, muscular dystrophy, cystic fibrosis, and some other conditions, have a very important role. We’re not aiming to compete with any other existing resources. What we are seeking to do is to fill the unmet need of, “What if there are no such designations for the disease that you’re concerned about?” Our goal was to find places that could help with unanswered questions, whether diagnostic questions or treatment questions. To identify places where a patient could reasonably expect to go and have a deeper dive – maybe an interdisciplinary deep dive.
The delay to diagnosis can be years in rare diseases. How can the network help speed up diagnoses?
With all these experts on different diseases, we hope to develop some better diagnostic algorithms within the network. Another thing we can do is to share resources. With 31 sites, everybody’s seeing patients with unknown diagnoses. Everyone is seeing patients for whom they would maybe like to get a whole genome done, or a whole exome done, but they are often encountering stiff resistance from insurance companies.
Meanwhile some sites, but not all 31, have multimillion-dollar grants to do sequencing and other kinds of advanced diagnostic tests to solve unknown cases. And there are people at those sites who say, “We need more samples. Can you get us samples from the other sites?”
One of the main things we aim to do is share information, including information about available diagnostic resources. We want all 31 sites to know which sites have funding and programs that enable them to study samples for other sites. We also want to know what criteria they’re putting on it. Someone might say: “I’ve got a grant to sequence genomes for people with unexplained seizures. Send me all your unexplained seizures.” Somebody else might have a grant for unexplained GI diseases. So, we want to put on our intranet a resource for the 31 sites, kind of a cookbook for – when if you can’t get it paid for by insurance, but you really think you need a particular special test – who might be able to do it for you within the network.
This would seem to benefit research across sites as well.
Yes, but we also want to share clinical advice and expertise for direct patient benefit. So, it doesn’t always have to fulfill the goals of a specific research project. For example, we might be able to create an undiagnosed patient quality improvement database across all 31 sites that could compliantly let Drs. X and Y know that they’re each seeing a patient with the same rare thing.
But let’s say you want to move the field forward by discovering a new disease. Rare genetic diseases are now being discovered at the rate of about 250 a year, so about 5 per week across the world. With two or three unrelated patients who have the same disease and a whole exome sequence, you can potentially discover a disease. Maybe you’ve found one unique patient with a genetic variant of possible significance, but you can’t be 100% sure, and you may not be able to convince your colleagues, or journal editors, until you find other cases. You need those two or three ultrarare patients. Within this network, a lot of sites want to share information about their ultrarare patients and be able to put together additional instances of the same thing, to prove that it is a real disease, to learn more about it and how to diagnose, manage, and treat it.
Part of the idea with a nationwide network is that patients aren’t going to have to move around among these centers of excellence, is that correct? They’re going to be seen at the closest ones, and it’s the expertise that is mobile.
Yes, that’s right. While we can’t eliminate the need for travel, what we are trying to do is increase the sharing of expertise, to improve results for patients while limiting the need for traveling very long distances. As a geneticist I’ve been on both the requesting and the receiving end of consultations with doctors at other sites, sometimes very far away, especially for ultrarare conditions for which any one physician’s experience is limited. We all try to honor these sorts of requests, but insurance doesn’t reimburse it and so hospitals don’t give doctors much credit for it.
We want to ultimately find ways to incentivize this type of collaboration. Hopefully we can get agreements with insurance companies to allow intersite consultations within our network, recognizing that they don’t want to pay for the patient to be seen out of state, but you also want the patient to get the best possible medical advice. This might require legislative changes in the long run. But what we can do more readily is create a culture within this network of mutual consultation and sharing of clinical experience. Outside of such a network, the idea of “cold calling” somebody, whom you may never have met, and asking them for help and free advice is a little bit of a bar, right? We want to lower that bar.
Can patients get telemedicine consults with physicians across the network?
NORD supports having telemedicine options for everybody regardless of diagnosis, rare or not, and we support legislation that would continue access and reimbursement for telemedicine post pandemic. I hope we can get that, or at least preserve telemedicine for rare diseases, for which there are often not enough, or sometimes not any, expert providers in the same state. Ultimately, we want patients to be able to get the expert assessments and advice they need. For rare diseases, that sometimes means battling back and forth with an insurance provider, seeking permission to see an expert clinician a thousand miles away. By sharing medical expertise, and through telemedicine when that’s allowed, we hope to reduce the need for that. But the telemedicine environment is still evolving and somewhat uncertain.
How will the network’s physician collaborations take place?
One of the important things NORD is providing to the network is an information technology setup and intranet across the 31 sites. That intranet is where center staff will go to access the network’s internal resources, including live and recorded case conferences. In those case conferences you can present a case you haven’t been able to solve. Experts you may have only heard of by reputation will now be streamed to your computer as part of the nationwide network. It benefits the patient because you get additional expert opinions, but it also benefits the physicians because we have this collegial space for discussion and learning. We’ll be linked by frequent meetings – some in person, most virtual – a common culture, and a common intranet.
On the intranet, we will also have a growing set of useful databases, links, and documents that are available to all members. These will be progressively updated with help from experts at the centers, so that clinicians can more directly learn from each other, instead of separately reinventing the wheel. The way things usually work, when you see a patient with an ultrarare condition that you’re not that familiar with, is that you tell them what little you can, then schedule them to come back in a few weeks. In the meantime, usually in your off time, you spend hours searching PubMed and other sources and you try to piece things together, to figure out what’s known that might help your patient. But imagine that this has already been figured out by someone else in the network. You can see on the network a list of articles the other expert read and found helpful in addressing this problem. And you then reach out directly to that other expert.
In recent months you’ve had one-on-one meetings with all 31 directors at the sites, and after that you convened 11 working groups. What are you trying to achieve?
Once the sites were chosen, we aimed to talk quickly and honestly about what everyone needed, what everyone saw as the biggest problems to tackle in rare diseases. Two things were very rewarding about those phone calls: one, all the centers were very enthusiastic, and two, they pretty much all agreed on what the key unmet needs are for rare disease patients and the practitioners trying to help them. So, we empaneled working groups of expert volunteers enthusiastic to work on each of those problems. These groups collectively comprise more than 200 volunteers – faculty, staff, and trainees – from the different sites nationwide. Each group is working on a key unmet need in rare diseases, and each group will be given its own space on our file-sharing platform, where they can share information and co-develop new ideas and documents. When something they produce is good enough to start to be a practice resource, such as a draft treatment guideline that the working group now wants to try in the real world, but it’s not yet ready to be published, they can share it and have it tested by all 31 sites through the dedicated intranet we are building for the network.
Jennie Smith is a freelance journalist specializing in medicine and health.
The broad and challenging – but promising – landscape of peripheral neuropathy
Peripheral neuropathy is becoming an increasing focal point for clinicians when treating patients because of the plethora of causes to which the disorder has been attributed. Characterized by damage to the peripheral nervous system, peripheral neuropathy causes sharp, burning pain; numbness of the extremities that can travel proximally; muscle weakness; and an overall diminished quality of life. Rather than being a self-developing disease, peripheral neuropathy has mostly been identified as a symptom of causative disorders and therapeutic agents – making prevention and treatment extremely important for patients and providers.

The etiology of peripheral neuropathy has been studied thoroughly over the past 2 decades. In this review, we summarize the landscape of peripheral neuropathy, including the more common causative entities; diagnostic tools that can potentially be employed to identify the disorder; and treatments that are in use or being tested to prevent, slow, or reverse the effects of peripheral neuropathy.
DIABETIC PERIPHERAL NEUROPATHY
The most common cause of peripheral neuropathy is diabetes mellitus. Diabetic peripheral neuropathy (DPN) is a symmetrical, length-dependent neuropathy that affects more than 50% of type I and type II diabetes patients.1 Not only is DPN an initiating factor of foot ulcers and nontraumatic lower-limb amputation, but it also leads to a severely lower quality of life, financial burden, and increased risk of death after major surgical procedures.2

Once DPN has progressed significantly, its effects are irreversible; there are no agents capable of reversing or halting DPN past initial stages of disease.3 It is important to detect and treat DPN early on, as it has a favorable prognosis and most DPN-related amputations are preventable.
Diagnosis
Nerve-conduction studies are the preferred diagnostic tool for DPN; however, these studies are costly and difficult to conduct in a clinical setting.2 Currently, such diagnostic tools as the 10-g monofilament and tuning fork are more commonly utilized to detect loss of protective foot sensation to decrease the risk of foot ulceration.2 In addition, other common aspects of diagnosing DPN include assessment of symptoms in the patient’s hands or feet and patient-reported symptoms.

Several diagnostic devices are in experimental stages and have shown potential for utilization in clinical settings.
DPNCheck is a handheld device, with a turnaround time of 3 minutes, that measures sural nerve conduction velocity, which can identify DPN early in asymptomatic cases; and amplitude of sensory-nerve action potentials, which decrease with the degeneration of axons, a clinical characteristic of DPN. In a study of patients with diabetes (n = 162 [type 1, n = 80; type 2, n = 82]) and healthy controls (n = 80), a comparative analysis of DPNCheck and reference techniques showed a strong linear relationship between between clinical neuropathy scores and LDIFLARE (r = 0.64-0.84; P < 0.03), which suggests that the device might be viable in clinical settings.4 LDIFLARE is a method developed to assess axon reflex to detect neuropathy in type 2 diabetes.4
Neuropad, a 10-minute test, measures foot plantar-surface sweat production, indicated by a cobalt compound color change on the device. The test is advantageous because it is highly sensitive – 73% more sensitive than DPNCheck – and does not rely on patient response or require operator training.5 A study of Neuropad showed that a drier foot and, therefore, increased risk of foot ulceration correlated with greater abnormal readings on the device, which might indicate onset of more severe DPN in the future.6
Sudoscan measures sudomotor function in 3 minutes through an electrochemical reaction between stimulated sweat glands and electrodes.2 A study performed in China in patients with type 2 diabetes (n = 394) showed that electrical conductance in the feet is associated with increasing risk and severity of symptoms of DPN in asymptomatic patients (r = 0.98 [95% confidence interval, 0.962-0.993]; P < .01) and might serve as a biomarker of DPN.7
Although these three techniques present favorable data, each is a nerve conduction study that can access only small-fiber nerves. Additional testing is required for larger-fiber nerves that are also affected by DPN.2 Also, some of the studies of these devices have high heterogeneity and a small sample size. Further research utilizing these three methods should include larger sample sizes to appropriately assess any clinically significant patient outcomes.
Corneal confocal microscopy (CCM), another potential technique for DPN screening, is a noninvasive ophthalmic device for assessing corneal small-fiber nerves. A study of patients with diabetes or obesity or both (n = 35) showed high reproducibility of corneal-nerve pathology identification using CCM.8 A larger-scale study showed that CCM can detect a reduction in corneal-nerve parameters in DPN patients, as well as in patients who have yet to develop DPN – thus demonstrating the technique’s ability to detect both early subclinical and established DPN.9 Once CCM is approved as a point-of-care device, it might provide a reliable, sensitive screening method for DPN as an early-intervention tool.
Therapeutic options
The three principal types of treatment for DPN are tricyclic antidepressants, anticonvulsants, and selective serotonin-norepinephrine reuptake inhibitors (SSNRIs). Only three medications are Food and Drug Administration (FDA) approved for the treatment of DPN: pregabalin, duloxetine, and the recently approved capsaicin patch. Some opioid analgesics, including extended-release tapentadol, are FDA approved for DPN-associated neuropathic pain; however, evidence of their efficacy is questionable, and they present a risk of addiction.10 Here, we focus on potential treatments for DPN and DPN-associated neuropathic pain.
Cinacalcet. Several potential treatments have been studied for alleviating DPN symptoms after progression. Cinacalcet is a calcimimetic agent that activates the adenosine monophosphate-activated protein kinase–endothelial nitric oxide synthase pathway, which mediates DPN development. The drug has shown evidence of improving sensorimotor function and restoring nerve function in human Schwann cells expressed in diabetes-induced mice.11 In these animal models, cinacalcet improved tactile response when interventional mice were compared with a control group (P < .01).11 Further research is necessary to determine similar efficacy in human subjects.
Traditional Chinese medicine. Recent studies have focused on traditional Chinese medicine and practice, such as acupuncture and moxibustion, for DPN.
Moxibustion is the technique of burning moxa floss (a plant also known as mugwort) on different points on the body, which is thought to alleviate disease. In a study performed on rats, moxibustion increased nerve velocity (P < .05) and preserved sciatic-nerve ultrastructure.12 Research on the use of moxibustion is preliminary. A meta-analysis of available data found that all clinical studies took place in China, and results were therefore subject to high heterogeneity and small sample size.13 Previously, a lack of high-quality data prevented moxibustion from being considered a potential treatment.3 The technique has demonstrated potential benefit, but larger-scale and more rigorous studies must be utilized to verify its clinical efficacy.
Quercetin. This common dietary flavonoid is in development. In rat models with induced DPN, treatment produced significant neuroprotective effects, such as rescued mechanical withdrawal threshold, lowered nerve densities (P = .0378), and rescued lowered levels of reactive O2 species (P < .0001), which contribute to neurotoxicity in many peripheral neuropathies.14 Another study of the anti-inflammatory effects of quercetin in rat models found significant lowering of inflammatory factors, including proteins encoded by toll-like receptor 4 and MyD88, and protein transcription factor nuclear factor kappa B (P < .001), which can be beneficial in the treatment of DPN.15 Future testing in human subjects might reveal similarly positive effects.
Vitamin B. A systematic review examined the therapeutic effects of vitamin B supplementation on DPN. Through a meta-analysis on 14 studies (N = 997), it was revealed that statistically significant improvements in pain and electrophysiological sensory outcomes were observed after vitamin B supplementation. However, the majority of the studies included in the analysis utilized combination therapies with different vitamins (such as vitamin D) and other vitamin B types. Furthermore, deficiencies in B vitamins – especially folic acid and vitamin B12 – have been observed in diabetic patients, and may be the potential cause of DPN in them. The validity of the studies and their findings are weakened by this observation. Therefore, the clinical efficacy of individual B vitamin supplements must be evaluated in long-term, larger scale future studies that exclude those with B vitamin deficiency and DPN to minimize potential error.71
CHEMOTHERAPY-INDUCED PERIPHERAL NEUROPATHY
Another cause of peripheral neuropathy has been directly linked to particular chemotherapeutic agents. Platinum-based agents have been widely accepted as an ideal solution for slowing tumor progression; however, it has been established that platinum adducts within DNA are the cause of neuronal degeneration – specifically in dorsal-root ganglion neurons of the peripheral nervous system. In a 2010 meta-analysis in the United States, the prevalence of chemotherapy-induced peripheral neuropathy (CIPN) was observed to range from 65% to 75%, depending on the platinum-based agent.16 This problem is often dose-limiting and can lead to cessation of treatment, causing patients physical and financial harm. CIPN can be acute or chronic, and symptoms affect motor, sensory, and autonomic function, which can lead to reduced quality of life.17
Diagnostic tools and strategies
A variety of avenues can be taken to assess whether a patient has CIPN. Because peripheral neuropathy is often subjective, it has been recommended that clinicians use patient-reported outcome measures in this setting, in the form of a questionnaire.
Common toxicity criteria. The most conventional measure of CIPN is the National Cancer Institute’s Common Toxicity Criteria, which grades severity of adverse effects on a scale of 1 to 5 and has been found to be statistically valid.18 This questionnaire assesses a patient’s neuropathic pain score and sensory deficits, and can detect other potential adverse findings, such as neutropenia.
Total neuropathy score. This commonly used questionnaire measures subjective autonomic, sensory, and motor symptoms on a scale of 0 to 4 for each item, with the individual item scores then summed. A score > 5 indicates CIPN.19 The tested validity of this measure shows that it has an inter-rater reliability of 0.966 and an intra-rater reliability of 0.986.19
Other questionnaires. The Neuropathy Screening Questionnaire, Treatment-Induced Neuropathy Assessment Scale, and Chemotherapy-Induced Peripheral Neuropathy Assessment Tool have been identified as means of understanding what a patient experiences following neurotoxic chemotherapy.18
Pain caused by CIPN can also be assessed with one of several general scales, such as the Neuropathic Pain Scale for Chemotherapy-Induced Neuropathy (NPS-CIN), which identifies a patient’s level of pain on a scale from 0 to 4 on six items: intensity, unpleasantness, sharpness, depth, numbness, and tingling. This scale has been found to be reliable.18
Other scales that can be used are the Neuropathic Pain Symptom Inventory, Patient-Reported Outcomes Measurement Information System: Pain Quality Neuro, and Leeds Assessment of Neuropathic Symptoms and Signs.18
Other diagnostic tests. Tests to determine a chemotherapy patient’s functional ability regarding their extremities include postural stability tests, the Timed Up and Go (TUG) test, the Fullerton Advance Balance (FAB) Scale, the 6-minute walk test, and the grooved pegboard test.
Nerve conduction studies have been identified as useful tools to assess the physiologic function of fibers, but are costly and used most often in research settings.18 Quantitative sensory testing and the Bumps test are used to assess threshold capacities for varying sensations. Nerve-imaging tools, such as high-resolution ultrasonography, magnetic resonance neurography, and positron emission and computed tomography, have been found to be successful in identifying nerve damage.18
Additionally, the accumulation of mitochondrial DNA (mtDNA) in the blood has been identified as a potential biomarker for CIPN following animal trials on rats.69 Researchers conducted a double-blind trial where healthy rats were given doses of paclitaxel, oxaliplatin, and bortezomib and compared to vehicle-treated rats. Researchers found that there was a correlation between the onset of CIPN and levels of mtDNA, with 1-2-fold increases of mtDNA found in paclitaxel and oxaliplatin treated patients (P < 0.01).69 Dysfunctional mitochondria can cause an increase in the activity of reactive oxygen species which results in damage to mtDNA; and abnormal bioenergetics, which may lead to irregular ATP production and result in cellular damage.
Navitoclax. The antineoplastic agent cisplatin is used to treat a variety of cancers, including ovarian, lung, head and neck, testicular, and bladder.20 Using single-cell RNA sequencing of dorsal-root ganglion cells in mouse models that have been given human equivalent doses of cisplatin to induce peripheral neuropathy, a study identified that the drug was upregulating the cyclin-dependent kinase inhibitor 1A gene (CDKN1A) and leading to overproduction of its product, the p21 protein.21 This is due to a cellular response to DNA damage that causes the dorsal-root ganglion sensory neuron to change into a senescence-like state to survive. Subsequently, accumulation of senescent sensory neurons correlates with induction of neuropathic pain and peripheral neuropathy. It has been established, in mouse models, that removing senescent cells has the potential to reduce or reverse peripheral neuropathy associated with cisplatin treatment.21
A study induced irreversible CIPN using cisplatin on mice that were subsequently treated with antineoplastic agent navitoclax (n = 5) or vehicle (n = 10). Using navitoclax, a broad-spectrum senolytic agent, the study examined the dorsal-root ganglia of the mice and found that CIPN was reversed following clearance of senescent cells, with baseline mechanical thresholds able to be reestablished without difference, compared with the control group (P = .7734).22 The investigators found that clearance of senescent cells using navitoclax proved a promising avenue toward mitigating CIPN. More studies should be completed to validate this treatment as an effective preventive.
NGF Monoclonal Antibody (Tanezumab). Tanezumab has been identified as a potential analgesic for CIPN having observed success during animal trials. This monoclonal antibody targets the NGF-TrkA pathway in a dose-dependent manner which results in a reduction of neuronal sensitivity and subsequently neuropathic pain (P < 0.05).70 NGF is a peripheral pain mediator that has functional properties relating to inflammation and neuropathy. Therefore, by targeting this protein and inhibiting its activation, patients could potentially see a dramatic improvement in their quality of life following a CIPN diagnosis. This potential analgesic was observed to be successful for a variety of chemotherapeutic agents including cisplatin, vincristine, and paclitaxel.70
SASP inhibitors. A second possible approach to neutralizing senescent cells would be by inhibiting the senescence-associated secretory phenotype (SASP). This could be accomplished through the use of nuclear factor kappa B inhibitors, mammalian target of rapamycin (mTOR) inhibitors, bromodomain and extra-terminal (BET) inhibitors, and inhibitors of secretory factors, such as interleukin (IL)-6 and tumor necrosis factor (TNF) alpha.23 Rapamycin, an mTOR inhibitor that is already used in clinical settings, has been found to reduce the inflammatory effects of senescent cells, expanding the lifespan of mice.24 JQ1, OTX015, and ARV825 are BET inhibitors that have been found to block bromodomain-containing protein 4, thus inducing senescent cell death.25 IL-6 inhibitors (for example, tocilizumab) and TNF alpha inhibitors (for example, adalimumab) are already used clinically and can mitigate the effects of SASP.23,26 However, further studies are needed to examine potential adverse effects of this type of therapy.
Mitigation of oxaliplatin adverse effects. This platinum-based chemotherapeutic agent associated with peripheral neuropathy is primarily used to treat colorectal cancer and digestive-tract malignancies.27 Oxaliplatin-induced peripheral neuropathy (OIPN) can be acute or chronic, and causes neuropathic pain, autonomic nerve dysfunction, and hypersensitivity to cold, which lead to abnormal nervous system effects, such as peripheral paresthesia.
These symptoms derive from oxaliplatin’s effects on a variety of cellular mechanisms, and differ in chronic and acute OIPN. Acute OIPN includes abnormal changes to sodium, potassium, calcium, and transient receptor potential channels, which lead to dysregulation and dysfunction in peripheral neurons; glia activation associated with dysregulation of pain modulation, by reducing thresholds; and upregulation of the octamer-binding transcription factor (OCT) protein.
Chronic OIPN has been associated with damage to nuclear DNA by platinum adducts, mitochondrial dysfunction (due to oxidative stress), and neuroinflammation caused by glia activation and gut microbiota.28
With increased understanding regarding cellular mechanisms affected in OIPN, treatment options are being established to prevent or reduce its effects. A treatment being tested for the treatment of OIPN is the serotonin and norepinephrine reuptake inhibitor (SSNRI) antidepressant duloxetine.29 In a clinical trial of 40 patients with gastrointestinal cancer, duloxetine was found to reduce cold sensitivity (P = .001), tingling or discomfort of hands (P < .002) and feet (P = .017), and peripheral neuropathic pain (P = .001), and was found to prevent paresthesia (P = .025).29 The SNRI antidepressant venlafaxine has also shown that it can alleviate neuropathic pain and motor neuropathy in clinical trials.30
Antioxidant agents, such as amifostine and calmangafodipir, have also been identified as possible preventive measures against OIPN. Amifostine prevents neuronal hyperactivation and nitrosative stress, while calmangafodipir modulates reactive O2 species, regulates ion channels, and protects axons and the myelin sheath.31,32
Treatments such as riluzole, lidocaine, and pregabalin have all shown promise in reducing the effects of OIPN by their action on potassium, sodium, and calcium channels, respectively.28 A study conducted on mice (n = 565) with OIPN found that riluzole effectively mitigated motor and sensory deficits associated with the use of oxaliplatin.33
TREK-1 and TRAAK, potassium channels that are important for thermal and motor sensitivity, and that act as silencing mechanisms to excitatory stimuli, were shown to degenerate following oxaliplatin treatment, leading to hypersensitivity. Riluzole performs its therapeutic function by activating TREK-1 and TRAAK channels and blocking excessive accumulation of glutamate. Following riluzole treatment, mice were observed to show a significant reduction in sensorimotor deficits. Interestingly, riluzole also aided in reducing depression associated with oxaliplatin (P < .01).33 However, more studies are necessary to ensure the safety and efficacy of riluzole in humans.
Pyridoxine, pyridostigmine for vincristine-induced peripheral neuropathy. Vinca alkaloids have also been identified as chemotherapeutic agents that induce peripheral neuropathy. One such agent, vincristine, which is used primarily to treat leukemia and brain cancer, has been observed to cause peripheral neuropathy, including motor, autonomic, and sensory symptoms, such as abnormal gait, mechanical allodynia, paresthesia, ptosis, and obstipation, and altered perception of stimuli.34,35 These symptoms are caused primarily by the ability of vincristine to activate neuroinflammatory mechanisms in dorsal-root ganglia. This is caused by activation of nucleotide-binding oligomerization domain 3 (NLRP3)-dependent release of IL-1b and subsequent cleavage of gasdermin D and caspase-1 in macrophages (observed in mouse models). Vincristine activates the NLRP3 signaling cascade that results in production of proinflammatory cytokines, thus inducing symptoms of peripheral neuropathy.36
Pyridoxine and pyridostigmine have been introduced as potential treatments for vincristine-induced peripheral neuropathy. Following a clinical trial of pediatric acute lymphoblastic leukemia patients, a study of 23 patients with vincristine-induced peripheral neuropathy found statistical validity for using pyridoxine and pyridostigmine because the drugs improved the neuropathy score (P < .001).37 However, more research is needed before implementing their use in point-of-care settings.
AUTOIMMUNE PERIPHERAL NEUROPATHY
Autoimmune peripheral neuropathies (APNs) occur when the immune system targets peripheral nervous system and its various cells. Although there is a wide range of conditions in this category of peripheral neuropathy, the two most common types – Guillain-Barré syndrome (GBS) and chronic inflammatory demyelinating polyneuropathy (CIDP) – have been targeted for clinical research.
Guillain-Barré syndrome: Diagnostic tools and strategies
Guillain-Barré syndrome encompasses a variety of acute inflammatory polyneuropathies, including axonal motor, sensory, and autonomic neuropathies and Miller Fisher syndrome (MFS).38 In particular, the anti-GQ1b ganglioside antibody is considered archetypical in APNs because it is detected in MFS patients and not found in normal and disease-control samples, which makes it a good clinical marker.39
It is difficult to distinguish GBS from CIDP because the time frame of onset of maximum deficit of neuropathy – 4 weeks – can overlap with subacute CIDP symptoms.40 Current diagnosis is based on elevated levels of cerebrospinal fluid (CSF) proteins, which can increase fourfold 6 weeks into the early phase of disease, and nerve conduction studies.40 However, electrodiagnostic readings and CSF protein levels are normal in 30% to 50% of patients in the first week after onset of disease and must be repeated in weeks that follow.41 A major disadvantage in the workup of suspected GBS is that the syndrome can be confirmed only several weeks after onset of symptoms.
Ultrasonography. A potential new diagnostic tool is serial peripheral nerve ultrasonographic (US) imaging. A pilot study of GBS patients (n = 16) showed that US can detect enlarged nerve cross-sections in median, ulnar, and sural nerves in the first 3 weeks of disease. Imaging performance was consistent with that of nerve conduction studies, and was advantageous because US is easier to perform and for patients to undergo.42
Spinal inflammation. Another study hints at the importance of spinal-root inflammation as an early indicator of disease, especially when nerve conduction study readings are normal.43 Further research is needed to demonstrate the clinical efficacy of this diagnostic method in larger population groups.
Guillain-Barré syndrome: Therapeutic options
The standard of care for GBS in the United States is intravenous immunoglobulin (IVIG) therapy and plasmapheresis, but there is no FDA-approved treatment.44 Although the two treatments have been shown to be equally effective in early stages of disease, early relapses can occur with both. One study found that 20% of patients who underwent plasmapheresis relapsed.40 Because nearly 50% of GBS patients do not respond to IVIG or plasmapheresis, the need is urgent for new therapies to decrease the risk of permanent disability.45
Antibody therapy. Recent developments include the use of monoclonal antibodies against GBS. ANX005 is an immunoglobulin G4 recombinant antibody that inhibits complement component 1q (C1q). Activation of this protein triggers the classical complement cascade, a natural part of the innate immune system that is nonetheless inappropriately activated in some autoimmune diseases, leading to neurodegeneration as a consequence of tissue damage.
ANX005 was found to have high-binding affinity to C1q in human, rat, cynomolgus monkey, and dog sera in nonclinical trials, and demonstrated low cross-reactivity despite being a plasma protein present throughout human tissue. Furthermore, studies show that ANX005 can deplete C1q completely in the CSF of monkeys.46 Phase 1b clinical trials in Bangladesh with GBS patients (n = 23) 18 to 58 years of age against a placebo group (n = 8) indicate that treatment is well tolerated. Drug-related serious adverse events were lacking and subjects’ GBS-Disability Score improved compared with placebo controls at week 1 (r2 = 0.48; P < .0001) and week 8, when an improvement of three or more in the score was observed.40
ANX005 is entering phase 2 trials, which are expected to be completed in 2023.47
Eculizumab. This promising treatment is a monoclonal antibody against C5 convertase, an enzyme that catalyzes formation of C5b-9, a membrane attack complex in nerve membranes. Studies in mouse models showed that treatment could significantly improve symptoms of terminal motor neuropathy and completely block formation of membrane attack complexes.48 Rats in this study were paralyzed by anti-GQ1b antibodies to emulate GBS pathogenesis.
A double-blind, placebo-controlled phase 2 clinical trial in Japan enrolled 34 patients (23 assigned to receive eculizumab; 11, to placebo); all were 18 years old or older and could not walk independently (3-5 on the GBS functional grading scale). Results showed that:
- Sixteen percent more patients receiving eculizumab treatment (n = 14; 42-78 years) than in the placebo group (n = 5; 20-73 years) could walk independently after 4 weeks.
- Fifty-six percent more patients in the functional group (n = 17; 52-90 years) than in the placebo group (n = 2; 20-52 years) could run after 6 months.49 While it is noted that the first portion of the trial failed to meet the predefined significance level, its long-term effects are observed to have therapeutic potential.
Eculizumab is in phase 3 clinical trials with primary data to be released in October 2022.50
Alemtuzumab, which inhibits the CD52 gene, was found to alleviate symptoms and restore strength in a rapidly deteriorating patient with MFS and chronic lymphocytic leukemia. By week 4 of treatment, anti-GQ1B antibodies were eliminated. However, the cause of this patient’s MFS is unclear; recovery might have been the result of multiple factors.51
IgG inhibition. Additional ongoing studies include therapies geared toward the neonatal Fc receptor as a potential clinical target for IgG inhibition.52
Chronic inflammatory demyelinating polyneuropathy (CIDP): Diagnostic tools and strategies
CIDP is the most common chronic APN and shares many similarities with GBS but differs in its responsiveness to corticosteroids, prognosis, and more. Lack of consensus on diagnostic criteria for CIDP has led to reliance on nerve conduction studies and clinical findings for making the diagnosis.53
Guidelines. European Federation of Neurological Societies/Peripheral Nerve Society guidelines have high sensitivity (81%) and specificity (96%) and are utilized as diagnostic criteria for CIDP; however, a survey found that these criteria may be underutilized in clinical practice – which might contribute to a high misdiagnosis rate.54 Furthermore, although current diagnostic methods are dependent on CSF proteins, this disease is lacking a diagnostic biomarker, leading to easy overdiagnosis and unnecessary immunotherapy.55
Electrodiagnostic testing, which is often used, is limited because it cannot evaluate small-fiber nerves, cannot access the CNS adequately, and does not provide a specific diagnosis.56
Sphingomyelin in CSF. Recently, a study in Italy explored the potential of CSF sphingomyelin as a biomarker for CIDP and for GBS. Findings reveal that sphingomyelin levels can be used to diagnose more than 80% of APN cases in the clinical setting. Different levels were identified in GBS, acute inflammatory demyelinating polyneuropathy, and typical and atypical CIDP patients. Additionally, sphingomyelin showed potential to diagnose the correct stage of disease. An increase in sphingomyelin in relapsing CIDP patients was noted, compared with what was seen in controls and stable CIDP patients.57 Larger-scale studies are needed to further test the efficacy of this method.
Chronic inflammatory demyelinating polyneuropathy: Therapeutic options
First-line therapy for CIDP comprises prednisone, 60-100 mg/d, plasmapheresis, and IVIG, all of which have proved effective. Some patients respond better to one treatment than to others40; some have subpar response to all these treatments and are categorized as having refractory CIDP.45
Although there are no newly approved treatments for CIDP, several show promise in ongoing clinical trials.
Rituximab is an anti-CD20 monoclonal antibody being studied in two phase 2 clinical trials of efficacy for refractory CIDP with IgG4 autoantibodies, after showing potential efficacy.58,59
Efgartigimod is an Fc fragment that blocks the neonatal Fc receptor, prevents lysosome degradation of IgGs, and thus allows them to be “recycled.”60 These autoantibodies are crucial in disease pathology because lowering their concentration provides effective therapy.61 Phase 1 trials showed that repeated doses of efgartigimod reduced IgG levels in healthy volunteers by 50%. Repeated dosing lowered IgG levels, on average by 75% in serum, which was an effect that was sustained for an 8-week period.62 Phase 2 trials are recruiting, with a projected primary completion in 2023.
INFECTION-INDUCED PERIPHERAL NEUROPATHY
Infections have been identified as a primary cause of peripheral neuropathy. Infection-induced peripheral neuropathy has been associated with Lyme disease, Epstein-Barr and human immunodeficiency virus (HIV) infection, shingles, hepatitis B and C, diphtheria, leprosy, and rabies.63 Extensive research on peripheral neuropathy has not been completed for most of the diseases, highlighting an unmet need for patients who experience this sequela of infection.
HIV is a well-documented viral cause of peripheral neuropathy. The most common symptom is distal sensory polyneuropathy, which affects more than 50% of patients with HIV.64 The incidence of distal sensory polyneuropathy in HIV has been correlated with the use of antiretroviral therapy – specifically, tenofovir disoproxil fumarate – and with certain proteins secreted by the virus.65 Symptoms include loss of sensory properties, neuropathic pain, and allodynia.66
Diagnostic tools and strategies
Nerve conduction studies have primarily been used to diagnose HIV-induced peripheral neuropathy, as well as electrophysiological testing and noninvasive CCM. These assays can detect changes or abnormalities in large- and small-fiber nerves in HIV infection patients.66
Therapeutic options
Studies in mouse models have illustrated how the Tat protein correlates with induction of motor and sensory distal symmetric polyneuropathy. Expression of Tat can lead to mitochondrial disruption, resulting in degeneration of sensory dorsal root ganglia and subsequent neuropathic pain.67
Pirenzepine. Studies on mice have identified a potential treatment for HIV infection-induced peripheral neuropathy with pirenzepine, targeting the muscarinic subtype-1 receptor. Pirenzepine activates a molecular pathway that promotes neurite growth and mitochondrial function. Researchers found that, following treatment with pirenzepine (n = 6), there was marked reduction in mitochondrial degeneration and HIV-induced distal sensory neuropathy.66 This outcome was due to the ability of pirenzepine to block the effects of Tat protein expression, leading to reversal of its neurodegenerative effects.
Exercise combined with analgesics has also been identified as a potential treatment for alleviating distal sensory polyneuropathy in HIV infection–induced peripheral neuropathy. In a 12-week study, researchers instructed subjects who were receiving a combination of HIV treatments, including tenofovir, lamivudine, and efavirenz, to perform aerobic and resistance exercises. This regimen was intended to improve peripheral nerve-conduction velocity and increase the density of nerve fibers and neurogenic branching.
The study identified baseline pain scores and divided participants into three groups: aerobic exercise (n = 45), resistance exercise (n = 44), and controls (n = 47), for whom the average level of pain was 2 on an ascending scale of 1 to 10. There was significant reduction in pain score in the experimental groups by the end of the study, as well as an increased sensory profile.64 This study has elucidated a pain management therapy for HIV-induced peripheral neuropathy that can prove beneficial for patients.
CRYPTOGENIC SENSORY POLYNEUROPATHY
Also known as idiopathic neuropathy or small-fiber sensory peripheral neuropathy, cryptogenic sensory polyneuropathy (CSPN) affects one-third of patients with peripheral neuropathy, in whom (despite extensive testing) no known cause of their condition is revealed.
Diagnostic tools and strategies
Applicable clinical and laboratory tests of any potential known underlying causes of neuropathy, including diabetes, hereditary disorders, and autoimmune disease, must be performed to rule out those causes and suggest an idiopathic cause.68
Therapeutic options
There are no FDA-approved treatments for CSPN, as most treatments are geared toward neuropathic pain management, rehabilitation, and supportive care.68 Due to a lack of research and data regarding these types of peripheral neuropathies, various studies suggest different first-line therapies. For example, anticonvulsants (pregabalin, gabapentin), antidepressants (duloxetine), and opioid-like compounds (tramadol) are all threapy options to treat DPN.3
Adequate data are lacking to support the efficacy of immunosuppressive therapy in CSPN.
Summing up
The combination of an understanding of a widening range of underlying diseases, advancements in cancer therapies, and the rising prevalence of diabetes have all led to an increasing incidence of peripheral neuropathy. Coupled with the fact that one-third of patients with peripheral neuropathy experience idiopathic neuropathy, this indicates that extensive studies must be undertaken to identify mitigation and prevention strategies for peripheral neuropathy. To summarize the landscape of treatment for peripheral neuropathy:
Diabetic peripheral neuropathy. Treatment for DPN comprises three FDA-approved products: pregabalin, duloxetine, and a higher (8%)-strength capsaicin patch.3 Pain-management therapies also exist to reduce diabetes-induced neuropathic pain, including gabapentin, amitriptyline, and extended-release tapentadol.10
Chemotherapy-induced peripheral neuropathy has yet to be effectively treated in humans; however, many trials are being completed in animals with promising results. Treatment for CIPN has been identified using senolytic agents, such as navitoclax,22 and through inhibition of SASP by a variety of agents, including ARV825, tocilizumab, and adalimumab.23-26
Oxaliplatin-induced peripheral neuropathy. Research has identified a potential preventive agent in duloxetine, with human trials already showing efficacy and safety.29 Animal models have shown progress studying antioxidant agents, such as amifostine31 and calmangafodipir,32 which target ion channels. In a similar mechanism of action, riluzole has been observed to reduce motor and sensory deficits and depression resulting from treatment with oxaliplatin.
Vincristine-induced peripheral neuropathy. Progress has been seen in treating vincristine-induced peripheral neuropathy with pyridoxine and pyridostigmine, which have improved neuropathy scores in trial subjects;37 more studies must be completed before these agents can be established as effective therapy.
Autoimmune PN. There are no FDA-approved drugs to mitigate the peripheral neuropathy induced by GBS and CIDP; however, studies are being conducted to resolve this impediment. Potential treatments, such as ANX005, a recombinant antibody, and eculizumab, a monoclonal antibody, have both shown efficacy in human trials and provide a potential path toward treatment against peripheral neuropathy caused by GBS.47,50 CIDP is currently treated using prednisone, plasmapheresis, and IVIG.40 Clinical trials are studying the efficacy of rituximab and efgartigimod for CIDP.58-60
Infection-induced peripheral neuropathy. Although many infections can induce peripheral neuropathy, HIV is most well documented and therefore was singled out for discussion in this article. Pirenzepine has been shown to promote neurite growth and reduce mitochondrial degeneration – both of which factors are associated with reduction of neuropathic pain.66 Exercise and analgesics have also been found to mitigate the effects of HIV-induced distal sensory neuropathy, with pain scores being reduced.61
Cryptogenic sensory polyneuropathy. Research has yet to identify a causative agent of, or subsequent potential therapy for, CSPN. Increased knowledge about this neuropathy will, it is hoped, bring patients closer to a cure – beyond current pain mitigation strategies with anticonvulsants, antidepressants, and opioid-like compounds.3
Ms. Lee is a first-year master of science candidate in applied life sciences, with an emphasis on infectious diseases, and Mr. Kosacki is a first-year master of science candidate in applied life sciences, with an emphasis on translational research, both at Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences, Claremont, Calif. Dr. Bhandari is professor of clinical sciences and Dr. Tran is professor of clinical sciences, Keck Graduate Institute School of Pharmacy and Health Sciences.
References
1. Barrell K, Smith AG. Peripheral neuropathy. Med Clin North Am. 2019 Mar;103(2):383-97. doi: 10.1016/j.mcna.2018.10.006.
2. Selvarajah D et al. Diabetic peripheral neuropathy: Advances in diagnosis and strategies for screening and early intervention. Lancet Diabetes Endocrinol. 2019 Dec;7(12):938-48. doi: 10.1016/S2213-8587(19)30081-6.
3. Snyder MJ et al. Treating painful diabetic peripheral neuropathy: An update. Am Fam Physician. 2016 Aug;94(3):227-334.
4. Sharma S et al. Assessment of diabetic neuropathy using a point-of-care nerve conduction device shows significant associations with the LDIFLARE method and clinical neuropathy scoring. J Diabetes Sci Technol. 2014 Jan;9(1):123-31. doi: 10.1177/1932296814551044.
5. Zografou I et al. Validation of Neuropad in the assessment of peripheral diabetic neuropathy in patients with diabetes mellitus versus the Michigan Neuropathy Screening Instrument, 10g monofilament application and biothesiometer measurement. Curr Vasc Pharmacol. 2020;18(5):517-22. doi: 10.2174/1570161117666190723155324.
6. Tentolouris N et al. Moisture status of the skin of the feet assessed by the visual test Neuropad correlates with foot ulceration in diabetes. Diabetes Care. 2010;33(5):1112-4. doi: 10.2337/dc09-2027.
7. Mao F et al. Sudoscan is an effective screening method for asymptomatic diabetic neuropathy in Chinese type 2 diabetes mellitus patients. J Diabetes Investig. 2017 May;8(3):363-8. doi: 10.1111/jdi.12575.
8. Kalteniece A et al. Corneal confocal microscopy is a rapid reproducible ophthalmic technique for quantifying corneal nerve abnormalities. PLoS One. 2017 Aug;12(8):e0183040. doi: 10.1371/journal.pone.0183040.
9. Gad H et al. Corneal confocal microscopy for the diagnosis of diabetic peripheral neuropathy: A systematic review and meta-analysis. J Diabetes Investig. 2022 Jan;13(1):134-47. doi: 10.1111/jdi.13643.
10. Pop-Busui R et al. Diabetic neuropathy: A position statement by the American Diabetes Association. Diabetes Care. 2017;40(1):136-54. doi: 10.2337/dc16-2042.
11. Chung YC et al. Calcimimetic restores diabetic peripheral neuropathy by ameliorating apoptosis and improving autophagy. Cell Death Dis. 2018 Nov;9(12):1163. doi: 10.1038/s41419-018-1192-7.
12. Li J et al. Therapeutic effects of moxibustion simultaneously targeting Nrf2 and NF-kB in diabetic peripheral neuropathy. Appl Biochem Biotechnol. 2019 Dec;189(4):1167-82. doi: 10.1007/s12010-019-03052-8.
13. Tan Y et al. Moxibustion for the treatment of diabetic peripheral neuropathy: A systematic review and meta-analysis following PRISMA guidelines. Medicine (Baltimore). 2020 Sep 26;99(39):e22286. doi: 10.1097/MD.0000000000022286.
14. Xie J et al. Protective effect of quercetin on streptozotocin-induced diabetic peripheral neuropathy rats through modulating gut microbiota and reactive oxygen species level. Biomed Pharmacother. 2020 Jul;127:110147. doi: 10.1016/j.biopha.2020.110147.
15. Zhao B et al. Quercetin reduces inflammation in a rat model of diabetic peripheral neuropathy by regulating the TLR4/MyD88/NF-kappa B signalling pathway. Eur J Pharmacol. 2021 Dec;912:174607. doi: 10.1016/j.ejphar.2021.174607.
16. McWhinney SR et al. Platinum neurotoxicity pharmacogenetics. Mol Cancer Ther. 2009;8(1):10-6. doi: 10.1158/1535-7163.MCT-08-0840.
17. Addington J, Freimer M. Chemotherapy-induced peripheral neuropathy: An update on the current understanding. F1000Res. 2016 Jun 22;5:F1000 Faculty Rev-1466. doi: 10.12688/f1000research.8053.1.
18. Lustberg M, Loprinzi C, eds. “Diagnosis, Management and Emerging Strategies for Chemotherapy-Induced Neuropathy: A MASCC Book.” Springer International Publishing; 2021.
19. Cornblath DR et al. Total neuropathy score: Validation and reliability study. Neurology. 1999 Nov;53(8):1660-4. doi: 10.1212/wnl.53.8.1660.
20. Aldossary SA. Review on pharmacology of cisplatin: Clinical use, toxicity and mechanism of resistance of cisplatin. Biomedical and Pharmacology Journal. 2019;12(1):7-15. http://dx.doi.org/10.13005/bpj/1608.
21. Calls A et al. Cisplatin-induced peripheral neuropathy is associated with neuronal senescence-like response. Neuro Oncol. 2021 Jan;23(1):88-99. doi: 10.1093/neuonc/noaa151.
22. Acklin S et al. Depletion of senescent-like neuronal cells alleviates cisplatin-induced peripheral neuropathy in mice. Sci Rep. 2020 Aug;10(1):14170. doi: 10.1038/s41598-020-71042-6.
23. Watanabe S et al. Impact of senescence‐associated secretory phenotype and its potential as a therapeutic target for senescence‐associated diseases. Cancer Sci. 2017 Apr;108(4):563-9. doi: 10.1111/cas.13184.
24. Harrison DE et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009 Jul 16;460(7253):392-5. doi: 10.1038/nature08221.
25. Wakita M et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat Commun. 2020;11(1):1935. doi: 10.1038/s41467-020-15719-6.
26. Prattichizzo F et al. Anti-TNF-alpha treatment modulates SASP and SASP-related microRNAs in endothelial cells and in circulating angiogenic cells. Oncotarget. 2016 Mar 15;7(11):11945-58. doi: 10.18632/oncotarget.7858.
27. Kang L et al. Oxaliplatin-induced peripheral neuropathy: Clinical features, mechanisms, prevention and treatment. J Neurol. 2021 Sep;268(9):3269-82. doi: 10.1007/s00415-020-09942-w.
28. Yang Y et al. Targeting strategies for oxaliplatin-induced peripheral neuropathy: Clinical syndrome, molecular basis, and drug development. J Exp Clin Cancer Res. 2021 Oct 22;40(1):331. doi: 10.1186/s13046-021-02141-z.
29. Rokhsareh S et al. Evaluating the effects of duloxetine on prophylaxis of oxaliplatin-induced peripheral neuropathy in patients with gastrointestinal cancer: A randomized double-blind placebo controlled clinical trial. J Oncol Pharm Pract. 2021 Nov 5;10781552211052646. doi: 10.1177/10781552211052646.
30. Farshchian N et al. Comparative study of the effects of venlafaxine and duloxetine on chemotherapy-induced peripheral neuropathy. Cancer Chemother Pharmacol. 2018 Nov;82(5):787-93. doi: 10.1007/s00280-018-3664-y.
31. Pereira AF et al. Amifostine protects from the peripheral sensory neuropathy induced by oxaliplatin in mice. Braz J Med Biol Res. 2020 Sep 18;53(11):e10263. doi: 10.1590/1414-431X202010263.
32. Glimelius B et al. Persistent prevention of oxaliplatin-induced peripheral neuropathy using calmangafodipir (PledOx®): A placebo-controlled randomised phase II study (PLIANT). Acta Oncol. 2018 Mar;57(3):393-402. doi: 10.1080/0284186X.2017.1398836.
33. Poupon L et al. Targeting the TREK-1 potassium channel via riluzole to eliminate the neuropathic and depressive-like effects of oxaliplatin. Neuropharmacology. 2018 Sep 15;140:43-61. doi: 10.1016/j.neuropharm.2018.07.026.
34. Mora J et al. Next generation ligand binding assays – Review of emerging technologies’ capabilities to enhance throughput and multiplexing. AAPS J. 2014 Nov;16(6):1175-84. doi: 10.1208/s12248-014-9660-1.
35. Starobova H, Vetter I. Pathophysiology of chemotherapy-induced peripheral neuropathy. Front Mol Neurosci. 2017 May 31;10:174. doi: 10.3389/fnmol.2017.00174.
36. Starobova H et al. Vincristine-induced peripheral neuropathy is driven by canonical NLRP3 activation and IL-1-beta release. J Exp Med. 2021;218(5):e20201452. doi: 10.1084/jem.20201452.
37. Aydin Köker S et al. Effect of pyridoxine plus pyridostigmine treatment on vincristine-induced peripheral neuropathy in pediatric patients with acute lymphoblastic leukemia: A single-center experience. Neurol Sci. 2021 Sep;42(9):3681-6. doi: 10.1007/s10072-020-04970-w.
38. Bourque PR et al. Autoimmune peripheral neuropathies. Clin Chim Acta. 2015 Sep 20;449:37-42. doi: 10.1016/j.cca.2015.02.039.
39. Paparounas K. Anti-GQ1b ganglioside antibody in peripheral nervous system disorders: Pathophysiologic role and clinical relevance. Arch Neurol. 2004 Jul;61(7):1013-6. doi: 10.1001/archneur.61.7.1013.
40. Dalakas MC. Autoimmune peripheral neuropathies, in Rich RR et al., eds., “Clinical Immunology.” 5th ed, (Amsterdam: Elsevier, 2019, pp. 903-915.e1). doi: 10.1016/B978-0-7020-6896-6.00067-3
41. Leonhard SE et al. Diagnosis and management of Guillain-Barré syndrome in ten steps. Nat Rev Neurol. 2019;15(11):671-83. doi: 10.1038/s41582-019-0250-9.
42. Razali SNO et al. Serial peripheral nerve ultrasound in Guillain–Barré syndrome. Clin Neurophysiol. 2016 Nov;127(2):1652-6. doi: 10.1016/j.clinph.2015.06.030.
43. Gallardo E et al. Spinal nerve involvement in early Guillain-Barré syndrome: A clinico-electrophysiological, ultrasonographic and pathological study. Clin Neurophysiol. 2015 Apr;126(4):810-9. doi: 10.1016/j.clinph.2014.06.051.
44. Islam Z et al. Inhibition of C1q, initiator of the classical complement cascade, by ANX005 for the treatment of Guillain–Barré syndrome: Results from a phase 1b study (763). Neurology. 2020 Apr;94(15 Suppl):763.
45. Hughes R et al.; . Oral fingolimod for chronic inflammatory demyelinating polyradiculoneuropathy (FORCIDP Trial): A double-blind, multicentre, randomised controlled trial. Lancet Neurol. 2018 Aug;17(8):689-98. doi: 10.1016/S1474-4422(18)30202-3.
46. Lansita JA et al. Nonclinical development of ANX005: A humanized anti-C1q antibody for treatment of autoimmune and neurodegenerative diseases. Int J Toxicol. 2017 Nov/Dec;36(6):449-62. doi: 10.1177/1091581817740873.
47. Annexon Inc. A randomized, double-blind, placebo-controlled phase 2/3 study to evaluate the efficacy, safety, pharmacokinetics, and pharmacodynamics of ANX005 in subjects with Guillain–Barré syndrome. ClinicalTrials.gov Identifier: NCT04701164. Updated Jan 8, 2021. Accessed Feb 23, 2022. https://clinicaltrials.gov/ct2/show/NCT04701164.
48. Halstead SK et al. Eculizumab prevents anti-ganglioside antibody-mediated neuropathy in a murine model. Brain. 2008 May;131(Pt 5):1197-1208. doi: 10.1093/brain/awm316.
49. Misawa S et al. Safety and efficacy of eculizumab in Guillain-Barré syndrome: A multicentre, double-blind, randomised phase 2 trial. Lancet Neurol. 2018 Jun;17(6):519-29. doi: 10.1016/S1474-4422(18)30114-5.
50. Alexion Pharmaceuticals. A phase 3, prospective, multicenter, double blind, randomized, placebo-controlled study to evaluate the efficacy and safety of eculizumab in patients with Guillain–Barré syndrome (GBS). ClinicalTrials.gov Identifier: NCT04752566. Updated Feb 18, 2022. Accessed Feb 23, 2022. https://clinicaltrials.gov/ct2/show/NCT04752566.
51. Tzachanis D et al. Successful treatment of refractory Guillain–Barré syndrome with alemtuzumab in a patient with chronic lymphocytic leukemia. Acta Haematol. 2014 Aug;132(2):240-3. doi: 10.1159/000358292.
52. Satkowiak K, Smith AG. Guillain-Barré syndrome, in Roos KL, ed. “Emergency Neurology.” (Springer, Cham, 2021, pp. 225-50). Accessed Feb 23, 2022. https://doi.org/10.1007/978-3-030-75778-6_12.
53. Gogia B et al. Chronic inflammatory demyelinating polyradiculoneuropathy, in “StatPearls [Internet].” (Treasure Island (Fla.): StatPearls Publishing; 2022 Jan). Updated Nov 22, 2021. Accessed Feb 23, 2022. www.ncbi.nlm.nih.gov/books/NBK563249.
54. Allen JA et al. Challenges in the diagnosis of chronic inflammatory demyelinating polyneuropathy. Brain Behav. 2018 Feb;8(3):e00932. doi: 10.1002/brb3.932.
55. Stino AM et al. Chronic inflammatory demyelinating polyradiculoneuropathy-diagnostic pitfalls and treatment approach. Muscle Nerve. 2021 Feb;63(2):157-69. doi: 10.1002/mus.27046.
56. Ginsberg MR et al. Using and interpreting electrodiagnostic tests. Cleve Clin J Med. 2020 Nov 2;87(11):671-82. doi: 10.3949/ccjm.87a.19154.
57. Capodivento G et al. CSF sphingomyelin: A new biomarker of demyelination in the diagnosis and management of CIDP and GBS. J Neurol Neurosurg Psychiatry. 2021;92(3):303-10. doi: 10.1136/jnnp-2020-324445.
58. Shimizu S et al. Efficacy and safety of rituximab in refractory CIDP with or without IgG4 autoantibodies (RECIPE): Protocol for a double-blind, randomized, placebo-controlled clinical trial. JMIR Res Protoc. 2020 Jan 4;9(4):e17117. doi: 10.2196/17117.
59. Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group. Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barré syndrome. Lancet. 1997;349(9047):225-30.
60. Zuercher AW et al. Next-generation Fc receptor–targeting biologics for autoimmune diseases. Autoimmun Rev. 2019 Oct;18(10):102366. doi: 10.1016/j.autrev.2019.102366.
61. Sesarman A et al. The neonatal Fc receptor as therapeutic target in IgG-mediated autoimmune diseases. Cell Mol Life Sci. 2010 Aug;67(15):2533-50. doi: 10.1007/s00018-010-0318-6.
62. Ulrichts P et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J Clin Invest. 2018 Oct;128(10):4372-86. doi: 10.1172/JCI97911.
63. Peripheral neuropathy [symptoms and causes]. Mayo Clinic [Internet]. Accessed Feb 23, 2022. http://www.mayoclinic.org/diseases-conditions/peripheral-neuropathy/symptoms-causes/syc-20352061.
64. Maharaj SS, Yakasai AM. Does a rehabilitation program of aerobic and progressive resisted exercises influence HIV-induced distal neuropathic pain? Am J Phys Med Rehabil. 2018 May;97(5):364-9. doi: 10.1097/PHM.0000000000000866.
65. Fields JA et al. Tenofovir disoproxil fumarate induces peripheral neuropathy and alters inflammation and mitochondrial biogenesis in the brains of mice. Sci Rep. 2019 Nov 20;9(1):17158. doi: 10.1038/s41598-019-53466-x.
66. Han MM et al. Prevention of HIV-1 TAT protein-induced peripheral neuropathy and mitochondrial disruption by the antimuscarinic pirenzepine. Front Neurol. 2021 Jun 15;12:663373. doi: 10.3389/fneur.2021.663373.
67. Rozzi SJ et al. Human immunodeficiency virus Tat impairs mitochondrial fission in neurons. Cell Death Discov. 2018;4:8. doi: 10.1038/s41420-017-0013-6.
68. Pasnoor M et al. Cryptogenic sensory polyneuropathy. Neurol Clin. 2013 May;31(2):463-76. doi: 10.1016/j.ncl.2013.01.008.
Peripheral neuropathy is becoming an increasing focal point for clinicians when treating patients because of the plethora of causes to which the disorder has been attributed. Characterized by damage to the peripheral nervous system, peripheral neuropathy causes sharp, burning pain; numbness of the extremities that can travel proximally; muscle weakness; and an overall diminished quality of life. Rather than being a self-developing disease, peripheral neuropathy has mostly been identified as a symptom of causative disorders and therapeutic agents – making prevention and treatment extremely important for patients and providers.
The etiology of peripheral neuropathy has been studied thoroughly over the past 2 decades. In this review, we summarize the landscape of peripheral neuropathy, including the more common causative entities; diagnostic tools that can potentially be employed to identify the disorder; and treatments that are in use or being tested to prevent, slow, or reverse the effects of peripheral neuropathy.
DIABETIC PERIPHERAL NEUROPATHY
The most common cause of peripheral neuropathy is diabetes mellitus. Diabetic peripheral neuropathy (DPN) is a symmetrical, length-dependent neuropathy that affects more than 50% of type I and type II diabetes patients.1 Not only is DPN an initiating factor of foot ulcers and nontraumatic lower-limb amputation, but it also leads to a severely lower quality of life, financial burden, and increased risk of death after major surgical procedures.2
Once DPN has progressed significantly, its effects are irreversible; there are no agents capable of reversing or halting DPN past initial stages of disease.3 It is important to detect and treat DPN early on, as it has a favorable prognosis and most DPN-related amputations are preventable.
Diagnosis
Nerve-conduction studies are the preferred diagnostic tool for DPN; however, these studies are costly and difficult to conduct in a clinical setting.2 Currently, such diagnostic tools as the 10-g monofilament and tuning fork are more commonly utilized to detect loss of protective foot sensation to decrease the risk of foot ulceration.2 In addition, other common aspects of diagnosing DPN include assessment of symptoms in the patient’s hands or feet and patient-reported symptoms.
Several diagnostic devices are in experimental stages and have shown potential for utilization in clinical settings.
DPNCheck is a handheld device, with a turnaround time of 3 minutes, that measures sural nerve conduction velocity, which can identify DPN early in asymptomatic cases; and amplitude of sensory-nerve action potentials, which decrease with the degeneration of axons, a clinical characteristic of DPN. In a study of patients with diabetes (n = 162 [type 1, n = 80; type 2, n = 82]) and healthy controls (n = 80), a comparative analysis of DPNCheck and reference techniques showed a strong linear relationship between between clinical neuropathy scores and LDIFLARE (r = 0.64-0.84; P < 0.03), which suggests that the device might be viable in clinical settings.4 LDIFLARE is a method developed to assess axon reflex to detect neuropathy in type 2 diabetes.4
Neuropad, a 10-minute test, measures foot plantar-surface sweat production, indicated by a cobalt compound color change on the device. The test is advantageous because it is highly sensitive – 73% more sensitive than DPNCheck – and does not rely on patient response or require operator training.5 A study of Neuropad showed that a drier foot and, therefore, increased risk of foot ulceration correlated with greater abnormal readings on the device, which might indicate onset of more severe DPN in the future.6
Sudoscan measures sudomotor function in 3 minutes through an electrochemical reaction between stimulated sweat glands and electrodes.2 A study performed in China in patients with type 2 diabetes (n = 394) showed that electrical conductance in the feet is associated with increasing risk and severity of symptoms of DPN in asymptomatic patients (r = 0.98 [95% confidence interval, 0.962-0.993]; P < .01) and might serve as a biomarker of DPN.7
Although these three techniques present favorable data, each is a nerve conduction study that can access only small-fiber nerves. Additional testing is required for larger-fiber nerves that are also affected by DPN.2 Also, some of the studies of these devices have high heterogeneity and a small sample size. Further research utilizing these three methods should include larger sample sizes to appropriately assess any clinically significant patient outcomes.
Corneal confocal microscopy (CCM), another potential technique for DPN screening, is a noninvasive ophthalmic device for assessing corneal small-fiber nerves. A study of patients with diabetes or obesity or both (n = 35) showed high reproducibility of corneal-nerve pathology identification using CCM.8 A larger-scale study showed that CCM can detect a reduction in corneal-nerve parameters in DPN patients, as well as in patients who have yet to develop DPN – thus demonstrating the technique’s ability to detect both early subclinical and established DPN.9 Once CCM is approved as a point-of-care device, it might provide a reliable, sensitive screening method for DPN as an early-intervention tool.
Therapeutic options
The three principal types of treatment for DPN are tricyclic antidepressants, anticonvulsants, and selective serotonin-norepinephrine reuptake inhibitors (SSNRIs). Only three medications are Food and Drug Administration (FDA) approved for the treatment of DPN: pregabalin, duloxetine, and the recently approved capsaicin patch. Some opioid analgesics, including extended-release tapentadol, are FDA approved for DPN-associated neuropathic pain; however, evidence of their efficacy is questionable, and they present a risk of addiction.10 Here, we focus on potential treatments for DPN and DPN-associated neuropathic pain.
Cinacalcet. Several potential treatments have been studied for alleviating DPN symptoms after progression. Cinacalcet is a calcimimetic agent that activates the adenosine monophosphate-activated protein kinase–endothelial nitric oxide synthase pathway, which mediates DPN development. The drug has shown evidence of improving sensorimotor function and restoring nerve function in human Schwann cells expressed in diabetes-induced mice.11 In these animal models, cinacalcet improved tactile response when interventional mice were compared with a control group (P < .01).11 Further research is necessary to determine similar efficacy in human subjects.
Traditional Chinese medicine. Recent studies have focused on traditional Chinese medicine and practice, such as acupuncture and moxibustion, for DPN.
Moxibustion is the technique of burning moxa floss (a plant also known as mugwort) on different points on the body, which is thought to alleviate disease. In a study performed on rats, moxibustion increased nerve velocity (P < .05) and preserved sciatic-nerve ultrastructure.12 Research on the use of moxibustion is preliminary. A meta-analysis of available data found that all clinical studies took place in China, and results were therefore subject to high heterogeneity and small sample size.13 Previously, a lack of high-quality data prevented moxibustion from being considered a potential treatment.3 The technique has demonstrated potential benefit, but larger-scale and more rigorous studies must be utilized to verify its clinical efficacy.
Quercetin. This common dietary flavonoid is in development. In rat models with induced DPN, treatment produced significant neuroprotective effects, such as rescued mechanical withdrawal threshold, lowered nerve densities (P = .0378), and rescued lowered levels of reactive O2 species (P < .0001), which contribute to neurotoxicity in many peripheral neuropathies.14 Another study of the anti-inflammatory effects of quercetin in rat models found significant lowering of inflammatory factors, including proteins encoded by toll-like receptor 4 and MyD88, and protein transcription factor nuclear factor kappa B (P < .001), which can be beneficial in the treatment of DPN.15 Future testing in human subjects might reveal similarly positive effects.
Vitamin B. A systematic review examined the therapeutic effects of vitamin B supplementation on DPN. Through a meta-analysis on 14 studies (N = 997), it was revealed that statistically significant improvements in pain and electrophysiological sensory outcomes were observed after vitamin B supplementation. However, the majority of the studies included in the analysis utilized combination therapies with different vitamins (such as vitamin D) and other vitamin B types. Furthermore, deficiencies in B vitamins – especially folic acid and vitamin B12 – have been observed in diabetic patients, and may be the potential cause of DPN in them. The validity of the studies and their findings are weakened by this observation. Therefore, the clinical efficacy of individual B vitamin supplements must be evaluated in long-term, larger scale future studies that exclude those with B vitamin deficiency and DPN to minimize potential error.71
CHEMOTHERAPY-INDUCED PERIPHERAL NEUROPATHY
Another cause of peripheral neuropathy has been directly linked to particular chemotherapeutic agents. Platinum-based agents have been widely accepted as an ideal solution for slowing tumor progression; however, it has been established that platinum adducts within DNA are the cause of neuronal degeneration – specifically in dorsal-root ganglion neurons of the peripheral nervous system. In a 2010 meta-analysis in the United States, the prevalence of chemotherapy-induced peripheral neuropathy (CIPN) was observed to range from 65% to 75%, depending on the platinum-based agent.16 This problem is often dose-limiting and can lead to cessation of treatment, causing patients physical and financial harm. CIPN can be acute or chronic, and symptoms affect motor, sensory, and autonomic function, which can lead to reduced quality of life.17
Diagnostic tools and strategies
A variety of avenues can be taken to assess whether a patient has CIPN. Because peripheral neuropathy is often subjective, it has been recommended that clinicians use patient-reported outcome measures in this setting, in the form of a questionnaire.
Common toxicity criteria. The most conventional measure of CIPN is the National Cancer Institute’s Common Toxicity Criteria, which grades severity of adverse effects on a scale of 1 to 5 and has been found to be statistically valid.18 This questionnaire assesses a patient’s neuropathic pain score and sensory deficits, and can detect other potential adverse findings, such as neutropenia.
Total neuropathy score. This commonly used questionnaire measures subjective autonomic, sensory, and motor symptoms on a scale of 0 to 4 for each item, with the individual item scores then summed. A score > 5 indicates CIPN.19 The tested validity of this measure shows that it has an inter-rater reliability of 0.966 and an intra-rater reliability of 0.986.19
Other questionnaires. The Neuropathy Screening Questionnaire, Treatment-Induced Neuropathy Assessment Scale, and Chemotherapy-Induced Peripheral Neuropathy Assessment Tool have been identified as means of understanding what a patient experiences following neurotoxic chemotherapy.18
Pain caused by CIPN can also be assessed with one of several general scales, such as the Neuropathic Pain Scale for Chemotherapy-Induced Neuropathy (NPS-CIN), which identifies a patient’s level of pain on a scale from 0 to 4 on six items: intensity, unpleasantness, sharpness, depth, numbness, and tingling. This scale has been found to be reliable.18
Other scales that can be used are the Neuropathic Pain Symptom Inventory, Patient-Reported Outcomes Measurement Information System: Pain Quality Neuro, and Leeds Assessment of Neuropathic Symptoms and Signs.18
Other diagnostic tests. Tests to determine a chemotherapy patient’s functional ability regarding their extremities include postural stability tests, the Timed Up and Go (TUG) test, the Fullerton Advance Balance (FAB) Scale, the 6-minute walk test, and the grooved pegboard test.
Nerve conduction studies have been identified as useful tools to assess the physiologic function of fibers, but are costly and used most often in research settings.18 Quantitative sensory testing and the Bumps test are used to assess threshold capacities for varying sensations. Nerve-imaging tools, such as high-resolution ultrasonography, magnetic resonance neurography, and positron emission and computed tomography, have been found to be successful in identifying nerve damage.18
Additionally, the accumulation of mitochondrial DNA (mtDNA) in the blood has been identified as a potential biomarker for CIPN following animal trials on rats.69 Researchers conducted a double-blind trial where healthy rats were given doses of paclitaxel, oxaliplatin, and bortezomib and compared to vehicle-treated rats. Researchers found that there was a correlation between the onset of CIPN and levels of mtDNA, with 1-2-fold increases of mtDNA found in paclitaxel and oxaliplatin treated patients (P < 0.01).69 Dysfunctional mitochondria can cause an increase in the activity of reactive oxygen species which results in damage to mtDNA; and abnormal bioenergetics, which may lead to irregular ATP production and result in cellular damage.
Navitoclax. The antineoplastic agent cisplatin is used to treat a variety of cancers, including ovarian, lung, head and neck, testicular, and bladder.20 Using single-cell RNA sequencing of dorsal-root ganglion cells in mouse models that have been given human equivalent doses of cisplatin to induce peripheral neuropathy, a study identified that the drug was upregulating the cyclin-dependent kinase inhibitor 1A gene (CDKN1A) and leading to overproduction of its product, the p21 protein.21 This is due to a cellular response to DNA damage that causes the dorsal-root ganglion sensory neuron to change into a senescence-like state to survive. Subsequently, accumulation of senescent sensory neurons correlates with induction of neuropathic pain and peripheral neuropathy. It has been established, in mouse models, that removing senescent cells has the potential to reduce or reverse peripheral neuropathy associated with cisplatin treatment.21
A study induced irreversible CIPN using cisplatin on mice that were subsequently treated with antineoplastic agent navitoclax (n = 5) or vehicle (n = 10). Using navitoclax, a broad-spectrum senolytic agent, the study examined the dorsal-root ganglia of the mice and found that CIPN was reversed following clearance of senescent cells, with baseline mechanical thresholds able to be reestablished without difference, compared with the control group (P = .7734).22 The investigators found that clearance of senescent cells using navitoclax proved a promising avenue toward mitigating CIPN. More studies should be completed to validate this treatment as an effective preventive.
NGF Monoclonal Antibody (Tanezumab). Tanezumab has been identified as a potential analgesic for CIPN having observed success during animal trials. This monoclonal antibody targets the NGF-TrkA pathway in a dose-dependent manner which results in a reduction of neuronal sensitivity and subsequently neuropathic pain (P < 0.05).70 NGF is a peripheral pain mediator that has functional properties relating to inflammation and neuropathy. Therefore, by targeting this protein and inhibiting its activation, patients could potentially see a dramatic improvement in their quality of life following a CIPN diagnosis. This potential analgesic was observed to be successful for a variety of chemotherapeutic agents including cisplatin, vincristine, and paclitaxel.70
SASP inhibitors. A second possible approach to neutralizing senescent cells would be by inhibiting the senescence-associated secretory phenotype (SASP). This could be accomplished through the use of nuclear factor kappa B inhibitors, mammalian target of rapamycin (mTOR) inhibitors, bromodomain and extra-terminal (BET) inhibitors, and inhibitors of secretory factors, such as interleukin (IL)-6 and tumor necrosis factor (TNF) alpha.23 Rapamycin, an mTOR inhibitor that is already used in clinical settings, has been found to reduce the inflammatory effects of senescent cells, expanding the lifespan of mice.24 JQ1, OTX015, and ARV825 are BET inhibitors that have been found to block bromodomain-containing protein 4, thus inducing senescent cell death.25 IL-6 inhibitors (for example, tocilizumab) and TNF alpha inhibitors (for example, adalimumab) are already used clinically and can mitigate the effects of SASP.23,26 However, further studies are needed to examine potential adverse effects of this type of therapy.
Mitigation of oxaliplatin adverse effects. This platinum-based chemotherapeutic agent associated with peripheral neuropathy is primarily used to treat colorectal cancer and digestive-tract malignancies.27 Oxaliplatin-induced peripheral neuropathy (OIPN) can be acute or chronic, and causes neuropathic pain, autonomic nerve dysfunction, and hypersensitivity to cold, which lead to abnormal nervous system effects, such as peripheral paresthesia.
These symptoms derive from oxaliplatin’s effects on a variety of cellular mechanisms, and differ in chronic and acute OIPN. Acute OIPN includes abnormal changes to sodium, potassium, calcium, and transient receptor potential channels, which lead to dysregulation and dysfunction in peripheral neurons; glia activation associated with dysregulation of pain modulation, by reducing thresholds; and upregulation of the octamer-binding transcription factor (OCT) protein.
Chronic OIPN has been associated with damage to nuclear DNA by platinum adducts, mitochondrial dysfunction (due to oxidative stress), and neuroinflammation caused by glia activation and gut microbiota.28
With increased understanding regarding cellular mechanisms affected in OIPN, treatment options are being established to prevent or reduce its effects. A treatment being tested for the treatment of OIPN is the serotonin and norepinephrine reuptake inhibitor (SSNRI) antidepressant duloxetine.29 In a clinical trial of 40 patients with gastrointestinal cancer, duloxetine was found to reduce cold sensitivity (P = .001), tingling or discomfort of hands (P < .002) and feet (P = .017), and peripheral neuropathic pain (P = .001), and was found to prevent paresthesia (P = .025).29 The SNRI antidepressant venlafaxine has also shown that it can alleviate neuropathic pain and motor neuropathy in clinical trials.30
Antioxidant agents, such as amifostine and calmangafodipir, have also been identified as possible preventive measures against OIPN. Amifostine prevents neuronal hyperactivation and nitrosative stress, while calmangafodipir modulates reactive O2 species, regulates ion channels, and protects axons and the myelin sheath.31,32
Treatments such as riluzole, lidocaine, and pregabalin have all shown promise in reducing the effects of OIPN by their action on potassium, sodium, and calcium channels, respectively.28 A study conducted on mice (n = 565) with OIPN found that riluzole effectively mitigated motor and sensory deficits associated with the use of oxaliplatin.33
TREK-1 and TRAAK, potassium channels that are important for thermal and motor sensitivity, and that act as silencing mechanisms to excitatory stimuli, were shown to degenerate following oxaliplatin treatment, leading to hypersensitivity. Riluzole performs its therapeutic function by activating TREK-1 and TRAAK channels and blocking excessive accumulation of glutamate. Following riluzole treatment, mice were observed to show a significant reduction in sensorimotor deficits. Interestingly, riluzole also aided in reducing depression associated with oxaliplatin (P < .01).33 However, more studies are necessary to ensure the safety and efficacy of riluzole in humans.
Pyridoxine, pyridostigmine for vincristine-induced peripheral neuropathy. Vinca alkaloids have also been identified as chemotherapeutic agents that induce peripheral neuropathy. One such agent, vincristine, which is used primarily to treat leukemia and brain cancer, has been observed to cause peripheral neuropathy, including motor, autonomic, and sensory symptoms, such as abnormal gait, mechanical allodynia, paresthesia, ptosis, and obstipation, and altered perception of stimuli.34,35 These symptoms are caused primarily by the ability of vincristine to activate neuroinflammatory mechanisms in dorsal-root ganglia. This is caused by activation of nucleotide-binding oligomerization domain 3 (NLRP3)-dependent release of IL-1b and subsequent cleavage of gasdermin D and caspase-1 in macrophages (observed in mouse models). Vincristine activates the NLRP3 signaling cascade that results in production of proinflammatory cytokines, thus inducing symptoms of peripheral neuropathy.36
Pyridoxine and pyridostigmine have been introduced as potential treatments for vincristine-induced peripheral neuropathy. Following a clinical trial of pediatric acute lymphoblastic leukemia patients, a study of 23 patients with vincristine-induced peripheral neuropathy found statistical validity for using pyridoxine and pyridostigmine because the drugs improved the neuropathy score (P < .001).37 However, more research is needed before implementing their use in point-of-care settings.
AUTOIMMUNE PERIPHERAL NEUROPATHY
Autoimmune peripheral neuropathies (APNs) occur when the immune system targets peripheral nervous system and its various cells. Although there is a wide range of conditions in this category of peripheral neuropathy, the two most common types – Guillain-Barré syndrome (GBS) and chronic inflammatory demyelinating polyneuropathy (CIDP) – have been targeted for clinical research.
Guillain-Barré syndrome: Diagnostic tools and strategies
Guillain-Barré syndrome encompasses a variety of acute inflammatory polyneuropathies, including axonal motor, sensory, and autonomic neuropathies and Miller Fisher syndrome (MFS).38 In particular, the anti-GQ1b ganglioside antibody is considered archetypical in APNs because it is detected in MFS patients and not found in normal and disease-control samples, which makes it a good clinical marker.39
It is difficult to distinguish GBS from CIDP because the time frame of onset of maximum deficit of neuropathy – 4 weeks – can overlap with subacute CIDP symptoms.40 Current diagnosis is based on elevated levels of cerebrospinal fluid (CSF) proteins, which can increase fourfold 6 weeks into the early phase of disease, and nerve conduction studies.40 However, electrodiagnostic readings and CSF protein levels are normal in 30% to 50% of patients in the first week after onset of disease and must be repeated in weeks that follow.41 A major disadvantage in the workup of suspected GBS is that the syndrome can be confirmed only several weeks after onset of symptoms.
Ultrasonography. A potential new diagnostic tool is serial peripheral nerve ultrasonographic (US) imaging. A pilot study of GBS patients (n = 16) showed that US can detect enlarged nerve cross-sections in median, ulnar, and sural nerves in the first 3 weeks of disease. Imaging performance was consistent with that of nerve conduction studies, and was advantageous because US is easier to perform and for patients to undergo.42
Spinal inflammation. Another study hints at the importance of spinal-root inflammation as an early indicator of disease, especially when nerve conduction study readings are normal.43 Further research is needed to demonstrate the clinical efficacy of this diagnostic method in larger population groups.
Guillain-Barré syndrome: Therapeutic options
The standard of care for GBS in the United States is intravenous immunoglobulin (IVIG) therapy and plasmapheresis, but there is no FDA-approved treatment.44 Although the two treatments have been shown to be equally effective in early stages of disease, early relapses can occur with both. One study found that 20% of patients who underwent plasmapheresis relapsed.40 Because nearly 50% of GBS patients do not respond to IVIG or plasmapheresis, the need is urgent for new therapies to decrease the risk of permanent disability.45
Antibody therapy. Recent developments include the use of monoclonal antibodies against GBS. ANX005 is an immunoglobulin G4 recombinant antibody that inhibits complement component 1q (C1q). Activation of this protein triggers the classical complement cascade, a natural part of the innate immune system that is nonetheless inappropriately activated in some autoimmune diseases, leading to neurodegeneration as a consequence of tissue damage.
ANX005 was found to have high-binding affinity to C1q in human, rat, cynomolgus monkey, and dog sera in nonclinical trials, and demonstrated low cross-reactivity despite being a plasma protein present throughout human tissue. Furthermore, studies show that ANX005 can deplete C1q completely in the CSF of monkeys.46 Phase 1b clinical trials in Bangladesh with GBS patients (n = 23) 18 to 58 years of age against a placebo group (n = 8) indicate that treatment is well tolerated. Drug-related serious adverse events were lacking and subjects’ GBS-Disability Score improved compared with placebo controls at week 1 (r2 = 0.48; P < .0001) and week 8, when an improvement of three or more in the score was observed.40
ANX005 is entering phase 2 trials, which are expected to be completed in 2023.47
Eculizumab. This promising treatment is a monoclonal antibody against C5 convertase, an enzyme that catalyzes formation of C5b-9, a membrane attack complex in nerve membranes. Studies in mouse models showed that treatment could significantly improve symptoms of terminal motor neuropathy and completely block formation of membrane attack complexes.48 Rats in this study were paralyzed by anti-GQ1b antibodies to emulate GBS pathogenesis.
A double-blind, placebo-controlled phase 2 clinical trial in Japan enrolled 34 patients (23 assigned to receive eculizumab; 11, to placebo); all were 18 years old or older and could not walk independently (3-5 on the GBS functional grading scale). Results showed that:
- Sixteen percent more patients receiving eculizumab treatment (n = 14; 42-78 years) than in the placebo group (n = 5; 20-73 years) could walk independently after 4 weeks.
- Fifty-six percent more patients in the functional group (n = 17; 52-90 years) than in the placebo group (n = 2; 20-52 years) could run after 6 months.49 While it is noted that the first portion of the trial failed to meet the predefined significance level, its long-term effects are observed to have therapeutic potential.
Eculizumab is in phase 3 clinical trials with primary data to be released in October 2022.50
Alemtuzumab, which inhibits the CD52 gene, was found to alleviate symptoms and restore strength in a rapidly deteriorating patient with MFS and chronic lymphocytic leukemia. By week 4 of treatment, anti-GQ1B antibodies were eliminated. However, the cause of this patient’s MFS is unclear; recovery might have been the result of multiple factors.51
IgG inhibition. Additional ongoing studies include therapies geared toward the neonatal Fc receptor as a potential clinical target for IgG inhibition.52
Chronic inflammatory demyelinating polyneuropathy (CIDP): Diagnostic tools and strategies
CIDP is the most common chronic APN and shares many similarities with GBS but differs in its responsiveness to corticosteroids, prognosis, and more. Lack of consensus on diagnostic criteria for CIDP has led to reliance on nerve conduction studies and clinical findings for making the diagnosis.53
Guidelines. European Federation of Neurological Societies/Peripheral Nerve Society guidelines have high sensitivity (81%) and specificity (96%) and are utilized as diagnostic criteria for CIDP; however, a survey found that these criteria may be underutilized in clinical practice – which might contribute to a high misdiagnosis rate.54 Furthermore, although current diagnostic methods are dependent on CSF proteins, this disease is lacking a diagnostic biomarker, leading to easy overdiagnosis and unnecessary immunotherapy.55
Electrodiagnostic testing, which is often used, is limited because it cannot evaluate small-fiber nerves, cannot access the CNS adequately, and does not provide a specific diagnosis.56
Sphingomyelin in CSF. Recently, a study in Italy explored the potential of CSF sphingomyelin as a biomarker for CIDP and for GBS. Findings reveal that sphingomyelin levels can be used to diagnose more than 80% of APN cases in the clinical setting. Different levels were identified in GBS, acute inflammatory demyelinating polyneuropathy, and typical and atypical CIDP patients. Additionally, sphingomyelin showed potential to diagnose the correct stage of disease. An increase in sphingomyelin in relapsing CIDP patients was noted, compared with what was seen in controls and stable CIDP patients.57 Larger-scale studies are needed to further test the efficacy of this method.
Chronic inflammatory demyelinating polyneuropathy: Therapeutic options
First-line therapy for CIDP comprises prednisone, 60-100 mg/d, plasmapheresis, and IVIG, all of which have proved effective. Some patients respond better to one treatment than to others40; some have subpar response to all these treatments and are categorized as having refractory CIDP.45
Although there are no newly approved treatments for CIDP, several show promise in ongoing clinical trials.
Rituximab is an anti-CD20 monoclonal antibody being studied in two phase 2 clinical trials of efficacy for refractory CIDP with IgG4 autoantibodies, after showing potential efficacy.58,59
Efgartigimod is an Fc fragment that blocks the neonatal Fc receptor, prevents lysosome degradation of IgGs, and thus allows them to be “recycled.”60 These autoantibodies are crucial in disease pathology because lowering their concentration provides effective therapy.61 Phase 1 trials showed that repeated doses of efgartigimod reduced IgG levels in healthy volunteers by 50%. Repeated dosing lowered IgG levels, on average by 75% in serum, which was an effect that was sustained for an 8-week period.62 Phase 2 trials are recruiting, with a projected primary completion in 2023.
INFECTION-INDUCED PERIPHERAL NEUROPATHY
Infections have been identified as a primary cause of peripheral neuropathy. Infection-induced peripheral neuropathy has been associated with Lyme disease, Epstein-Barr and human immunodeficiency virus (HIV) infection, shingles, hepatitis B and C, diphtheria, leprosy, and rabies.63 Extensive research on peripheral neuropathy has not been completed for most of the diseases, highlighting an unmet need for patients who experience this sequela of infection.
HIV is a well-documented viral cause of peripheral neuropathy. The most common symptom is distal sensory polyneuropathy, which affects more than 50% of patients with HIV.64 The incidence of distal sensory polyneuropathy in HIV has been correlated with the use of antiretroviral therapy – specifically, tenofovir disoproxil fumarate – and with certain proteins secreted by the virus.65 Symptoms include loss of sensory properties, neuropathic pain, and allodynia.66
Diagnostic tools and strategies
Nerve conduction studies have primarily been used to diagnose HIV-induced peripheral neuropathy, as well as electrophysiological testing and noninvasive CCM. These assays can detect changes or abnormalities in large- and small-fiber nerves in HIV infection patients.66
Therapeutic options
Studies in mouse models have illustrated how the Tat protein correlates with induction of motor and sensory distal symmetric polyneuropathy. Expression of Tat can lead to mitochondrial disruption, resulting in degeneration of sensory dorsal root ganglia and subsequent neuropathic pain.67
Pirenzepine. Studies on mice have identified a potential treatment for HIV infection-induced peripheral neuropathy with pirenzepine, targeting the muscarinic subtype-1 receptor. Pirenzepine activates a molecular pathway that promotes neurite growth and mitochondrial function. Researchers found that, following treatment with pirenzepine (n = 6), there was marked reduction in mitochondrial degeneration and HIV-induced distal sensory neuropathy.66 This outcome was due to the ability of pirenzepine to block the effects of Tat protein expression, leading to reversal of its neurodegenerative effects.
Exercise combined with analgesics has also been identified as a potential treatment for alleviating distal sensory polyneuropathy in HIV infection–induced peripheral neuropathy. In a 12-week study, researchers instructed subjects who were receiving a combination of HIV treatments, including tenofovir, lamivudine, and efavirenz, to perform aerobic and resistance exercises. This regimen was intended to improve peripheral nerve-conduction velocity and increase the density of nerve fibers and neurogenic branching.
The study identified baseline pain scores and divided participants into three groups: aerobic exercise (n = 45), resistance exercise (n = 44), and controls (n = 47), for whom the average level of pain was 2 on an ascending scale of 1 to 10. There was significant reduction in pain score in the experimental groups by the end of the study, as well as an increased sensory profile.64 This study has elucidated a pain management therapy for HIV-induced peripheral neuropathy that can prove beneficial for patients.
CRYPTOGENIC SENSORY POLYNEUROPATHY
Also known as idiopathic neuropathy or small-fiber sensory peripheral neuropathy, cryptogenic sensory polyneuropathy (CSPN) affects one-third of patients with peripheral neuropathy, in whom (despite extensive testing) no known cause of their condition is revealed.
Diagnostic tools and strategies
Applicable clinical and laboratory tests of any potential known underlying causes of neuropathy, including diabetes, hereditary disorders, and autoimmune disease, must be performed to rule out those causes and suggest an idiopathic cause.68
Therapeutic options
There are no FDA-approved treatments for CSPN, as most treatments are geared toward neuropathic pain management, rehabilitation, and supportive care.68 Due to a lack of research and data regarding these types of peripheral neuropathies, various studies suggest different first-line therapies. For example, anticonvulsants (pregabalin, gabapentin), antidepressants (duloxetine), and opioid-like compounds (tramadol) are all threapy options to treat DPN.3
Adequate data are lacking to support the efficacy of immunosuppressive therapy in CSPN.
Summing up
The combination of an understanding of a widening range of underlying diseases, advancements in cancer therapies, and the rising prevalence of diabetes have all led to an increasing incidence of peripheral neuropathy. Coupled with the fact that one-third of patients with peripheral neuropathy experience idiopathic neuropathy, this indicates that extensive studies must be undertaken to identify mitigation and prevention strategies for peripheral neuropathy. To summarize the landscape of treatment for peripheral neuropathy:
Diabetic peripheral neuropathy. Treatment for DPN comprises three FDA-approved products: pregabalin, duloxetine, and a higher (8%)-strength capsaicin patch.3 Pain-management therapies also exist to reduce diabetes-induced neuropathic pain, including gabapentin, amitriptyline, and extended-release tapentadol.10
Chemotherapy-induced peripheral neuropathy has yet to be effectively treated in humans; however, many trials are being completed in animals with promising results. Treatment for CIPN has been identified using senolytic agents, such as navitoclax,22 and through inhibition of SASP by a variety of agents, including ARV825, tocilizumab, and adalimumab.23-26
Oxaliplatin-induced peripheral neuropathy. Research has identified a potential preventive agent in duloxetine, with human trials already showing efficacy and safety.29 Animal models have shown progress studying antioxidant agents, such as amifostine31 and calmangafodipir,32 which target ion channels. In a similar mechanism of action, riluzole has been observed to reduce motor and sensory deficits and depression resulting from treatment with oxaliplatin.
Vincristine-induced peripheral neuropathy. Progress has been seen in treating vincristine-induced peripheral neuropathy with pyridoxine and pyridostigmine, which have improved neuropathy scores in trial subjects;37 more studies must be completed before these agents can be established as effective therapy.
Autoimmune PN. There are no FDA-approved drugs to mitigate the peripheral neuropathy induced by GBS and CIDP; however, studies are being conducted to resolve this impediment. Potential treatments, such as ANX005, a recombinant antibody, and eculizumab, a monoclonal antibody, have both shown efficacy in human trials and provide a potential path toward treatment against peripheral neuropathy caused by GBS.47,50 CIDP is currently treated using prednisone, plasmapheresis, and IVIG.40 Clinical trials are studying the efficacy of rituximab and efgartigimod for CIDP.58-60
Infection-induced peripheral neuropathy. Although many infections can induce peripheral neuropathy, HIV is most well documented and therefore was singled out for discussion in this article. Pirenzepine has been shown to promote neurite growth and reduce mitochondrial degeneration – both of which factors are associated with reduction of neuropathic pain.66 Exercise and analgesics have also been found to mitigate the effects of HIV-induced distal sensory neuropathy, with pain scores being reduced.61
Cryptogenic sensory polyneuropathy. Research has yet to identify a causative agent of, or subsequent potential therapy for, CSPN. Increased knowledge about this neuropathy will, it is hoped, bring patients closer to a cure – beyond current pain mitigation strategies with anticonvulsants, antidepressants, and opioid-like compounds.3
Ms. Lee is a first-year master of science candidate in applied life sciences, with an emphasis on infectious diseases, and Mr. Kosacki is a first-year master of science candidate in applied life sciences, with an emphasis on translational research, both at Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences, Claremont, Calif. Dr. Bhandari is professor of clinical sciences and Dr. Tran is professor of clinical sciences, Keck Graduate Institute School of Pharmacy and Health Sciences.
References
1. Barrell K, Smith AG. Peripheral neuropathy. Med Clin North Am. 2019 Mar;103(2):383-97. doi: 10.1016/j.mcna.2018.10.006.
2. Selvarajah D et al. Diabetic peripheral neuropathy: Advances in diagnosis and strategies for screening and early intervention. Lancet Diabetes Endocrinol. 2019 Dec;7(12):938-48. doi: 10.1016/S2213-8587(19)30081-6.
3. Snyder MJ et al. Treating painful diabetic peripheral neuropathy: An update. Am Fam Physician. 2016 Aug;94(3):227-334.
4. Sharma S et al. Assessment of diabetic neuropathy using a point-of-care nerve conduction device shows significant associations with the LDIFLARE method and clinical neuropathy scoring. J Diabetes Sci Technol. 2014 Jan;9(1):123-31. doi: 10.1177/1932296814551044.
5. Zografou I et al. Validation of Neuropad in the assessment of peripheral diabetic neuropathy in patients with diabetes mellitus versus the Michigan Neuropathy Screening Instrument, 10g monofilament application and biothesiometer measurement. Curr Vasc Pharmacol. 2020;18(5):517-22. doi: 10.2174/1570161117666190723155324.
6. Tentolouris N et al. Moisture status of the skin of the feet assessed by the visual test Neuropad correlates with foot ulceration in diabetes. Diabetes Care. 2010;33(5):1112-4. doi: 10.2337/dc09-2027.
7. Mao F et al. Sudoscan is an effective screening method for asymptomatic diabetic neuropathy in Chinese type 2 diabetes mellitus patients. J Diabetes Investig. 2017 May;8(3):363-8. doi: 10.1111/jdi.12575.
8. Kalteniece A et al. Corneal confocal microscopy is a rapid reproducible ophthalmic technique for quantifying corneal nerve abnormalities. PLoS One. 2017 Aug;12(8):e0183040. doi: 10.1371/journal.pone.0183040.
9. Gad H et al. Corneal confocal microscopy for the diagnosis of diabetic peripheral neuropathy: A systematic review and meta-analysis. J Diabetes Investig. 2022 Jan;13(1):134-47. doi: 10.1111/jdi.13643.
10. Pop-Busui R et al. Diabetic neuropathy: A position statement by the American Diabetes Association. Diabetes Care. 2017;40(1):136-54. doi: 10.2337/dc16-2042.
11. Chung YC et al. Calcimimetic restores diabetic peripheral neuropathy by ameliorating apoptosis and improving autophagy. Cell Death Dis. 2018 Nov;9(12):1163. doi: 10.1038/s41419-018-1192-7.
12. Li J et al. Therapeutic effects of moxibustion simultaneously targeting Nrf2 and NF-kB in diabetic peripheral neuropathy. Appl Biochem Biotechnol. 2019 Dec;189(4):1167-82. doi: 10.1007/s12010-019-03052-8.
13. Tan Y et al. Moxibustion for the treatment of diabetic peripheral neuropathy: A systematic review and meta-analysis following PRISMA guidelines. Medicine (Baltimore). 2020 Sep 26;99(39):e22286. doi: 10.1097/MD.0000000000022286.
14. Xie J et al. Protective effect of quercetin on streptozotocin-induced diabetic peripheral neuropathy rats through modulating gut microbiota and reactive oxygen species level. Biomed Pharmacother. 2020 Jul;127:110147. doi: 10.1016/j.biopha.2020.110147.
15. Zhao B et al. Quercetin reduces inflammation in a rat model of diabetic peripheral neuropathy by regulating the TLR4/MyD88/NF-kappa B signalling pathway. Eur J Pharmacol. 2021 Dec;912:174607. doi: 10.1016/j.ejphar.2021.174607.
16. McWhinney SR et al. Platinum neurotoxicity pharmacogenetics. Mol Cancer Ther. 2009;8(1):10-6. doi: 10.1158/1535-7163.MCT-08-0840.
17. Addington J, Freimer M. Chemotherapy-induced peripheral neuropathy: An update on the current understanding. F1000Res. 2016 Jun 22;5:F1000 Faculty Rev-1466. doi: 10.12688/f1000research.8053.1.
18. Lustberg M, Loprinzi C, eds. “Diagnosis, Management and Emerging Strategies for Chemotherapy-Induced Neuropathy: A MASCC Book.” Springer International Publishing; 2021.
19. Cornblath DR et al. Total neuropathy score: Validation and reliability study. Neurology. 1999 Nov;53(8):1660-4. doi: 10.1212/wnl.53.8.1660.
20. Aldossary SA. Review on pharmacology of cisplatin: Clinical use, toxicity and mechanism of resistance of cisplatin. Biomedical and Pharmacology Journal. 2019;12(1):7-15. http://dx.doi.org/10.13005/bpj/1608.
21. Calls A et al. Cisplatin-induced peripheral neuropathy is associated with neuronal senescence-like response. Neuro Oncol. 2021 Jan;23(1):88-99. doi: 10.1093/neuonc/noaa151.
22. Acklin S et al. Depletion of senescent-like neuronal cells alleviates cisplatin-induced peripheral neuropathy in mice. Sci Rep. 2020 Aug;10(1):14170. doi: 10.1038/s41598-020-71042-6.
23. Watanabe S et al. Impact of senescence‐associated secretory phenotype and its potential as a therapeutic target for senescence‐associated diseases. Cancer Sci. 2017 Apr;108(4):563-9. doi: 10.1111/cas.13184.
24. Harrison DE et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009 Jul 16;460(7253):392-5. doi: 10.1038/nature08221.
25. Wakita M et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat Commun. 2020;11(1):1935. doi: 10.1038/s41467-020-15719-6.
26. Prattichizzo F et al. Anti-TNF-alpha treatment modulates SASP and SASP-related microRNAs in endothelial cells and in circulating angiogenic cells. Oncotarget. 2016 Mar 15;7(11):11945-58. doi: 10.18632/oncotarget.7858.
27. Kang L et al. Oxaliplatin-induced peripheral neuropathy: Clinical features, mechanisms, prevention and treatment. J Neurol. 2021 Sep;268(9):3269-82. doi: 10.1007/s00415-020-09942-w.
28. Yang Y et al. Targeting strategies for oxaliplatin-induced peripheral neuropathy: Clinical syndrome, molecular basis, and drug development. J Exp Clin Cancer Res. 2021 Oct 22;40(1):331. doi: 10.1186/s13046-021-02141-z.
29. Rokhsareh S et al. Evaluating the effects of duloxetine on prophylaxis of oxaliplatin-induced peripheral neuropathy in patients with gastrointestinal cancer: A randomized double-blind placebo controlled clinical trial. J Oncol Pharm Pract. 2021 Nov 5;10781552211052646. doi: 10.1177/10781552211052646.
30. Farshchian N et al. Comparative study of the effects of venlafaxine and duloxetine on chemotherapy-induced peripheral neuropathy. Cancer Chemother Pharmacol. 2018 Nov;82(5):787-93. doi: 10.1007/s00280-018-3664-y.
31. Pereira AF et al. Amifostine protects from the peripheral sensory neuropathy induced by oxaliplatin in mice. Braz J Med Biol Res. 2020 Sep 18;53(11):e10263. doi: 10.1590/1414-431X202010263.
32. Glimelius B et al. Persistent prevention of oxaliplatin-induced peripheral neuropathy using calmangafodipir (PledOx®): A placebo-controlled randomised phase II study (PLIANT). Acta Oncol. 2018 Mar;57(3):393-402. doi: 10.1080/0284186X.2017.1398836.
33. Poupon L et al. Targeting the TREK-1 potassium channel via riluzole to eliminate the neuropathic and depressive-like effects of oxaliplatin. Neuropharmacology. 2018 Sep 15;140:43-61. doi: 10.1016/j.neuropharm.2018.07.026.
34. Mora J et al. Next generation ligand binding assays – Review of emerging technologies’ capabilities to enhance throughput and multiplexing. AAPS J. 2014 Nov;16(6):1175-84. doi: 10.1208/s12248-014-9660-1.
35. Starobova H, Vetter I. Pathophysiology of chemotherapy-induced peripheral neuropathy. Front Mol Neurosci. 2017 May 31;10:174. doi: 10.3389/fnmol.2017.00174.
36. Starobova H et al. Vincristine-induced peripheral neuropathy is driven by canonical NLRP3 activation and IL-1-beta release. J Exp Med. 2021;218(5):e20201452. doi: 10.1084/jem.20201452.
37. Aydin Köker S et al. Effect of pyridoxine plus pyridostigmine treatment on vincristine-induced peripheral neuropathy in pediatric patients with acute lymphoblastic leukemia: A single-center experience. Neurol Sci. 2021 Sep;42(9):3681-6. doi: 10.1007/s10072-020-04970-w.
38. Bourque PR et al. Autoimmune peripheral neuropathies. Clin Chim Acta. 2015 Sep 20;449:37-42. doi: 10.1016/j.cca.2015.02.039.
39. Paparounas K. Anti-GQ1b ganglioside antibody in peripheral nervous system disorders: Pathophysiologic role and clinical relevance. Arch Neurol. 2004 Jul;61(7):1013-6. doi: 10.1001/archneur.61.7.1013.
40. Dalakas MC. Autoimmune peripheral neuropathies, in Rich RR et al., eds., “Clinical Immunology.” 5th ed, (Amsterdam: Elsevier, 2019, pp. 903-915.e1). doi: 10.1016/B978-0-7020-6896-6.00067-3
41. Leonhard SE et al. Diagnosis and management of Guillain-Barré syndrome in ten steps. Nat Rev Neurol. 2019;15(11):671-83. doi: 10.1038/s41582-019-0250-9.
42. Razali SNO et al. Serial peripheral nerve ultrasound in Guillain–Barré syndrome. Clin Neurophysiol. 2016 Nov;127(2):1652-6. doi: 10.1016/j.clinph.2015.06.030.
43. Gallardo E et al. Spinal nerve involvement in early Guillain-Barré syndrome: A clinico-electrophysiological, ultrasonographic and pathological study. Clin Neurophysiol. 2015 Apr;126(4):810-9. doi: 10.1016/j.clinph.2014.06.051.
44. Islam Z et al. Inhibition of C1q, initiator of the classical complement cascade, by ANX005 for the treatment of Guillain–Barré syndrome: Results from a phase 1b study (763). Neurology. 2020 Apr;94(15 Suppl):763.
45. Hughes R et al.; . Oral fingolimod for chronic inflammatory demyelinating polyradiculoneuropathy (FORCIDP Trial): A double-blind, multicentre, randomised controlled trial. Lancet Neurol. 2018 Aug;17(8):689-98. doi: 10.1016/S1474-4422(18)30202-3.
46. Lansita JA et al. Nonclinical development of ANX005: A humanized anti-C1q antibody for treatment of autoimmune and neurodegenerative diseases. Int J Toxicol. 2017 Nov/Dec;36(6):449-62. doi: 10.1177/1091581817740873.
47. Annexon Inc. A randomized, double-blind, placebo-controlled phase 2/3 study to evaluate the efficacy, safety, pharmacokinetics, and pharmacodynamics of ANX005 in subjects with Guillain–Barré syndrome. ClinicalTrials.gov Identifier: NCT04701164. Updated Jan 8, 2021. Accessed Feb 23, 2022. https://clinicaltrials.gov/ct2/show/NCT04701164.
48. Halstead SK et al. Eculizumab prevents anti-ganglioside antibody-mediated neuropathy in a murine model. Brain. 2008 May;131(Pt 5):1197-1208. doi: 10.1093/brain/awm316.
49. Misawa S et al. Safety and efficacy of eculizumab in Guillain-Barré syndrome: A multicentre, double-blind, randomised phase 2 trial. Lancet Neurol. 2018 Jun;17(6):519-29. doi: 10.1016/S1474-4422(18)30114-5.
50. Alexion Pharmaceuticals. A phase 3, prospective, multicenter, double blind, randomized, placebo-controlled study to evaluate the efficacy and safety of eculizumab in patients with Guillain–Barré syndrome (GBS). ClinicalTrials.gov Identifier: NCT04752566. Updated Feb 18, 2022. Accessed Feb 23, 2022. https://clinicaltrials.gov/ct2/show/NCT04752566.
51. Tzachanis D et al. Successful treatment of refractory Guillain–Barré syndrome with alemtuzumab in a patient with chronic lymphocytic leukemia. Acta Haematol. 2014 Aug;132(2):240-3. doi: 10.1159/000358292.
52. Satkowiak K, Smith AG. Guillain-Barré syndrome, in Roos KL, ed. “Emergency Neurology.” (Springer, Cham, 2021, pp. 225-50). Accessed Feb 23, 2022. https://doi.org/10.1007/978-3-030-75778-6_12.
53. Gogia B et al. Chronic inflammatory demyelinating polyradiculoneuropathy, in “StatPearls [Internet].” (Treasure Island (Fla.): StatPearls Publishing; 2022 Jan). Updated Nov 22, 2021. Accessed Feb 23, 2022. www.ncbi.nlm.nih.gov/books/NBK563249.
54. Allen JA et al. Challenges in the diagnosis of chronic inflammatory demyelinating polyneuropathy. Brain Behav. 2018 Feb;8(3):e00932. doi: 10.1002/brb3.932.
55. Stino AM et al. Chronic inflammatory demyelinating polyradiculoneuropathy-diagnostic pitfalls and treatment approach. Muscle Nerve. 2021 Feb;63(2):157-69. doi: 10.1002/mus.27046.
56. Ginsberg MR et al. Using and interpreting electrodiagnostic tests. Cleve Clin J Med. 2020 Nov 2;87(11):671-82. doi: 10.3949/ccjm.87a.19154.
57. Capodivento G et al. CSF sphingomyelin: A new biomarker of demyelination in the diagnosis and management of CIDP and GBS. J Neurol Neurosurg Psychiatry. 2021;92(3):303-10. doi: 10.1136/jnnp-2020-324445.
58. Shimizu S et al. Efficacy and safety of rituximab in refractory CIDP with or without IgG4 autoantibodies (RECIPE): Protocol for a double-blind, randomized, placebo-controlled clinical trial. JMIR Res Protoc. 2020 Jan 4;9(4):e17117. doi: 10.2196/17117.
59. Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group. Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barré syndrome. Lancet. 1997;349(9047):225-30.
60. Zuercher AW et al. Next-generation Fc receptor–targeting biologics for autoimmune diseases. Autoimmun Rev. 2019 Oct;18(10):102366. doi: 10.1016/j.autrev.2019.102366.
61. Sesarman A et al. The neonatal Fc receptor as therapeutic target in IgG-mediated autoimmune diseases. Cell Mol Life Sci. 2010 Aug;67(15):2533-50. doi: 10.1007/s00018-010-0318-6.
62. Ulrichts P et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J Clin Invest. 2018 Oct;128(10):4372-86. doi: 10.1172/JCI97911.
63. Peripheral neuropathy [symptoms and causes]. Mayo Clinic [Internet]. Accessed Feb 23, 2022. http://www.mayoclinic.org/diseases-conditions/peripheral-neuropathy/symptoms-causes/syc-20352061.
64. Maharaj SS, Yakasai AM. Does a rehabilitation program of aerobic and progressive resisted exercises influence HIV-induced distal neuropathic pain? Am J Phys Med Rehabil. 2018 May;97(5):364-9. doi: 10.1097/PHM.0000000000000866.
65. Fields JA et al. Tenofovir disoproxil fumarate induces peripheral neuropathy and alters inflammation and mitochondrial biogenesis in the brains of mice. Sci Rep. 2019 Nov 20;9(1):17158. doi: 10.1038/s41598-019-53466-x.
66. Han MM et al. Prevention of HIV-1 TAT protein-induced peripheral neuropathy and mitochondrial disruption by the antimuscarinic pirenzepine. Front Neurol. 2021 Jun 15;12:663373. doi: 10.3389/fneur.2021.663373.
67. Rozzi SJ et al. Human immunodeficiency virus Tat impairs mitochondrial fission in neurons. Cell Death Discov. 2018;4:8. doi: 10.1038/s41420-017-0013-6.
68. Pasnoor M et al. Cryptogenic sensory polyneuropathy. Neurol Clin. 2013 May;31(2):463-76. doi: 10.1016/j.ncl.2013.01.008.
Peripheral neuropathy is becoming an increasing focal point for clinicians when treating patients because of the plethora of causes to which the disorder has been attributed. Characterized by damage to the peripheral nervous system, peripheral neuropathy causes sharp, burning pain; numbness of the extremities that can travel proximally; muscle weakness; and an overall diminished quality of life. Rather than being a self-developing disease, peripheral neuropathy has mostly been identified as a symptom of causative disorders and therapeutic agents – making prevention and treatment extremely important for patients and providers.
The etiology of peripheral neuropathy has been studied thoroughly over the past 2 decades. In this review, we summarize the landscape of peripheral neuropathy, including the more common causative entities; diagnostic tools that can potentially be employed to identify the disorder; and treatments that are in use or being tested to prevent, slow, or reverse the effects of peripheral neuropathy.
DIABETIC PERIPHERAL NEUROPATHY
The most common cause of peripheral neuropathy is diabetes mellitus. Diabetic peripheral neuropathy (DPN) is a symmetrical, length-dependent neuropathy that affects more than 50% of type I and type II diabetes patients.1 Not only is DPN an initiating factor of foot ulcers and nontraumatic lower-limb amputation, but it also leads to a severely lower quality of life, financial burden, and increased risk of death after major surgical procedures.2
Once DPN has progressed significantly, its effects are irreversible; there are no agents capable of reversing or halting DPN past initial stages of disease.3 It is important to detect and treat DPN early on, as it has a favorable prognosis and most DPN-related amputations are preventable.
Diagnosis
Nerve-conduction studies are the preferred diagnostic tool for DPN; however, these studies are costly and difficult to conduct in a clinical setting.2 Currently, such diagnostic tools as the 10-g monofilament and tuning fork are more commonly utilized to detect loss of protective foot sensation to decrease the risk of foot ulceration.2 In addition, other common aspects of diagnosing DPN include assessment of symptoms in the patient’s hands or feet and patient-reported symptoms.
Several diagnostic devices are in experimental stages and have shown potential for utilization in clinical settings.
DPNCheck is a handheld device, with a turnaround time of 3 minutes, that measures sural nerve conduction velocity, which can identify DPN early in asymptomatic cases; and amplitude of sensory-nerve action potentials, which decrease with the degeneration of axons, a clinical characteristic of DPN. In a study of patients with diabetes (n = 162 [type 1, n = 80; type 2, n = 82]) and healthy controls (n = 80), a comparative analysis of DPNCheck and reference techniques showed a strong linear relationship between between clinical neuropathy scores and LDIFLARE (r = 0.64-0.84; P < 0.03), which suggests that the device might be viable in clinical settings.4 LDIFLARE is a method developed to assess axon reflex to detect neuropathy in type 2 diabetes.4
Neuropad, a 10-minute test, measures foot plantar-surface sweat production, indicated by a cobalt compound color change on the device. The test is advantageous because it is highly sensitive – 73% more sensitive than DPNCheck – and does not rely on patient response or require operator training.5 A study of Neuropad showed that a drier foot and, therefore, increased risk of foot ulceration correlated with greater abnormal readings on the device, which might indicate onset of more severe DPN in the future.6
Sudoscan measures sudomotor function in 3 minutes through an electrochemical reaction between stimulated sweat glands and electrodes.2 A study performed in China in patients with type 2 diabetes (n = 394) showed that electrical conductance in the feet is associated with increasing risk and severity of symptoms of DPN in asymptomatic patients (r = 0.98 [95% confidence interval, 0.962-0.993]; P < .01) and might serve as a biomarker of DPN.7
Although these three techniques present favorable data, each is a nerve conduction study that can access only small-fiber nerves. Additional testing is required for larger-fiber nerves that are also affected by DPN.2 Also, some of the studies of these devices have high heterogeneity and a small sample size. Further research utilizing these three methods should include larger sample sizes to appropriately assess any clinically significant patient outcomes.
Corneal confocal microscopy (CCM), another potential technique for DPN screening, is a noninvasive ophthalmic device for assessing corneal small-fiber nerves. A study of patients with diabetes or obesity or both (n = 35) showed high reproducibility of corneal-nerve pathology identification using CCM.8 A larger-scale study showed that CCM can detect a reduction in corneal-nerve parameters in DPN patients, as well as in patients who have yet to develop DPN – thus demonstrating the technique’s ability to detect both early subclinical and established DPN.9 Once CCM is approved as a point-of-care device, it might provide a reliable, sensitive screening method for DPN as an early-intervention tool.
Therapeutic options
The three principal types of treatment for DPN are tricyclic antidepressants, anticonvulsants, and selective serotonin-norepinephrine reuptake inhibitors (SSNRIs). Only three medications are Food and Drug Administration (FDA) approved for the treatment of DPN: pregabalin, duloxetine, and the recently approved capsaicin patch. Some opioid analgesics, including extended-release tapentadol, are FDA approved for DPN-associated neuropathic pain; however, evidence of their efficacy is questionable, and they present a risk of addiction.10 Here, we focus on potential treatments for DPN and DPN-associated neuropathic pain.
Cinacalcet. Several potential treatments have been studied for alleviating DPN symptoms after progression. Cinacalcet is a calcimimetic agent that activates the adenosine monophosphate-activated protein kinase–endothelial nitric oxide synthase pathway, which mediates DPN development. The drug has shown evidence of improving sensorimotor function and restoring nerve function in human Schwann cells expressed in diabetes-induced mice.11 In these animal models, cinacalcet improved tactile response when interventional mice were compared with a control group (P < .01).11 Further research is necessary to determine similar efficacy in human subjects.
Traditional Chinese medicine. Recent studies have focused on traditional Chinese medicine and practice, such as acupuncture and moxibustion, for DPN.
Moxibustion is the technique of burning moxa floss (a plant also known as mugwort) on different points on the body, which is thought to alleviate disease. In a study performed on rats, moxibustion increased nerve velocity (P < .05) and preserved sciatic-nerve ultrastructure.12 Research on the use of moxibustion is preliminary. A meta-analysis of available data found that all clinical studies took place in China, and results were therefore subject to high heterogeneity and small sample size.13 Previously, a lack of high-quality data prevented moxibustion from being considered a potential treatment.3 The technique has demonstrated potential benefit, but larger-scale and more rigorous studies must be utilized to verify its clinical efficacy.
Quercetin. This common dietary flavonoid is in development. In rat models with induced DPN, treatment produced significant neuroprotective effects, such as rescued mechanical withdrawal threshold, lowered nerve densities (P = .0378), and rescued lowered levels of reactive O2 species (P < .0001), which contribute to neurotoxicity in many peripheral neuropathies.14 Another study of the anti-inflammatory effects of quercetin in rat models found significant lowering of inflammatory factors, including proteins encoded by toll-like receptor 4 and MyD88, and protein transcription factor nuclear factor kappa B (P < .001), which can be beneficial in the treatment of DPN.15 Future testing in human subjects might reveal similarly positive effects.
Vitamin B. A systematic review examined the therapeutic effects of vitamin B supplementation on DPN. Through a meta-analysis on 14 studies (N = 997), it was revealed that statistically significant improvements in pain and electrophysiological sensory outcomes were observed after vitamin B supplementation. However, the majority of the studies included in the analysis utilized combination therapies with different vitamins (such as vitamin D) and other vitamin B types. Furthermore, deficiencies in B vitamins – especially folic acid and vitamin B12 – have been observed in diabetic patients, and may be the potential cause of DPN in them. The validity of the studies and their findings are weakened by this observation. Therefore, the clinical efficacy of individual B vitamin supplements must be evaluated in long-term, larger scale future studies that exclude those with B vitamin deficiency and DPN to minimize potential error.71
CHEMOTHERAPY-INDUCED PERIPHERAL NEUROPATHY
Another cause of peripheral neuropathy has been directly linked to particular chemotherapeutic agents. Platinum-based agents have been widely accepted as an ideal solution for slowing tumor progression; however, it has been established that platinum adducts within DNA are the cause of neuronal degeneration – specifically in dorsal-root ganglion neurons of the peripheral nervous system. In a 2010 meta-analysis in the United States, the prevalence of chemotherapy-induced peripheral neuropathy (CIPN) was observed to range from 65% to 75%, depending on the platinum-based agent.16 This problem is often dose-limiting and can lead to cessation of treatment, causing patients physical and financial harm. CIPN can be acute or chronic, and symptoms affect motor, sensory, and autonomic function, which can lead to reduced quality of life.17
Diagnostic tools and strategies
A variety of avenues can be taken to assess whether a patient has CIPN. Because peripheral neuropathy is often subjective, it has been recommended that clinicians use patient-reported outcome measures in this setting, in the form of a questionnaire.
Common toxicity criteria. The most conventional measure of CIPN is the National Cancer Institute’s Common Toxicity Criteria, which grades severity of adverse effects on a scale of 1 to 5 and has been found to be statistically valid.18 This questionnaire assesses a patient’s neuropathic pain score and sensory deficits, and can detect other potential adverse findings, such as neutropenia.
Total neuropathy score. This commonly used questionnaire measures subjective autonomic, sensory, and motor symptoms on a scale of 0 to 4 for each item, with the individual item scores then summed. A score > 5 indicates CIPN.19 The tested validity of this measure shows that it has an inter-rater reliability of 0.966 and an intra-rater reliability of 0.986.19
Other questionnaires. The Neuropathy Screening Questionnaire, Treatment-Induced Neuropathy Assessment Scale, and Chemotherapy-Induced Peripheral Neuropathy Assessment Tool have been identified as means of understanding what a patient experiences following neurotoxic chemotherapy.18
Pain caused by CIPN can also be assessed with one of several general scales, such as the Neuropathic Pain Scale for Chemotherapy-Induced Neuropathy (NPS-CIN), which identifies a patient’s level of pain on a scale from 0 to 4 on six items: intensity, unpleasantness, sharpness, depth, numbness, and tingling. This scale has been found to be reliable.18
Other scales that can be used are the Neuropathic Pain Symptom Inventory, Patient-Reported Outcomes Measurement Information System: Pain Quality Neuro, and Leeds Assessment of Neuropathic Symptoms and Signs.18
Other diagnostic tests. Tests to determine a chemotherapy patient’s functional ability regarding their extremities include postural stability tests, the Timed Up and Go (TUG) test, the Fullerton Advance Balance (FAB) Scale, the 6-minute walk test, and the grooved pegboard test.
Nerve conduction studies have been identified as useful tools to assess the physiologic function of fibers, but are costly and used most often in research settings.18 Quantitative sensory testing and the Bumps test are used to assess threshold capacities for varying sensations. Nerve-imaging tools, such as high-resolution ultrasonography, magnetic resonance neurography, and positron emission and computed tomography, have been found to be successful in identifying nerve damage.18
Additionally, the accumulation of mitochondrial DNA (mtDNA) in the blood has been identified as a potential biomarker for CIPN following animal trials on rats.69 Researchers conducted a double-blind trial where healthy rats were given doses of paclitaxel, oxaliplatin, and bortezomib and compared to vehicle-treated rats. Researchers found that there was a correlation between the onset of CIPN and levels of mtDNA, with 1-2-fold increases of mtDNA found in paclitaxel and oxaliplatin treated patients (P < 0.01).69 Dysfunctional mitochondria can cause an increase in the activity of reactive oxygen species which results in damage to mtDNA; and abnormal bioenergetics, which may lead to irregular ATP production and result in cellular damage.
Navitoclax. The antineoplastic agent cisplatin is used to treat a variety of cancers, including ovarian, lung, head and neck, testicular, and bladder.20 Using single-cell RNA sequencing of dorsal-root ganglion cells in mouse models that have been given human equivalent doses of cisplatin to induce peripheral neuropathy, a study identified that the drug was upregulating the cyclin-dependent kinase inhibitor 1A gene (CDKN1A) and leading to overproduction of its product, the p21 protein.21 This is due to a cellular response to DNA damage that causes the dorsal-root ganglion sensory neuron to change into a senescence-like state to survive. Subsequently, accumulation of senescent sensory neurons correlates with induction of neuropathic pain and peripheral neuropathy. It has been established, in mouse models, that removing senescent cells has the potential to reduce or reverse peripheral neuropathy associated with cisplatin treatment.21
A study induced irreversible CIPN using cisplatin on mice that were subsequently treated with antineoplastic agent navitoclax (n = 5) or vehicle (n = 10). Using navitoclax, a broad-spectrum senolytic agent, the study examined the dorsal-root ganglia of the mice and found that CIPN was reversed following clearance of senescent cells, with baseline mechanical thresholds able to be reestablished without difference, compared with the control group (P = .7734).22 The investigators found that clearance of senescent cells using navitoclax proved a promising avenue toward mitigating CIPN. More studies should be completed to validate this treatment as an effective preventive.
NGF Monoclonal Antibody (Tanezumab). Tanezumab has been identified as a potential analgesic for CIPN having observed success during animal trials. This monoclonal antibody targets the NGF-TrkA pathway in a dose-dependent manner which results in a reduction of neuronal sensitivity and subsequently neuropathic pain (P < 0.05).70 NGF is a peripheral pain mediator that has functional properties relating to inflammation and neuropathy. Therefore, by targeting this protein and inhibiting its activation, patients could potentially see a dramatic improvement in their quality of life following a CIPN diagnosis. This potential analgesic was observed to be successful for a variety of chemotherapeutic agents including cisplatin, vincristine, and paclitaxel.70
SASP inhibitors. A second possible approach to neutralizing senescent cells would be by inhibiting the senescence-associated secretory phenotype (SASP). This could be accomplished through the use of nuclear factor kappa B inhibitors, mammalian target of rapamycin (mTOR) inhibitors, bromodomain and extra-terminal (BET) inhibitors, and inhibitors of secretory factors, such as interleukin (IL)-6 and tumor necrosis factor (TNF) alpha.23 Rapamycin, an mTOR inhibitor that is already used in clinical settings, has been found to reduce the inflammatory effects of senescent cells, expanding the lifespan of mice.24 JQ1, OTX015, and ARV825 are BET inhibitors that have been found to block bromodomain-containing protein 4, thus inducing senescent cell death.25 IL-6 inhibitors (for example, tocilizumab) and TNF alpha inhibitors (for example, adalimumab) are already used clinically and can mitigate the effects of SASP.23,26 However, further studies are needed to examine potential adverse effects of this type of therapy.
Mitigation of oxaliplatin adverse effects. This platinum-based chemotherapeutic agent associated with peripheral neuropathy is primarily used to treat colorectal cancer and digestive-tract malignancies.27 Oxaliplatin-induced peripheral neuropathy (OIPN) can be acute or chronic, and causes neuropathic pain, autonomic nerve dysfunction, and hypersensitivity to cold, which lead to abnormal nervous system effects, such as peripheral paresthesia.
These symptoms derive from oxaliplatin’s effects on a variety of cellular mechanisms, and differ in chronic and acute OIPN. Acute OIPN includes abnormal changes to sodium, potassium, calcium, and transient receptor potential channels, which lead to dysregulation and dysfunction in peripheral neurons; glia activation associated with dysregulation of pain modulation, by reducing thresholds; and upregulation of the octamer-binding transcription factor (OCT) protein.
Chronic OIPN has been associated with damage to nuclear DNA by platinum adducts, mitochondrial dysfunction (due to oxidative stress), and neuroinflammation caused by glia activation and gut microbiota.28
With increased understanding regarding cellular mechanisms affected in OIPN, treatment options are being established to prevent or reduce its effects. A treatment being tested for the treatment of OIPN is the serotonin and norepinephrine reuptake inhibitor (SSNRI) antidepressant duloxetine.29 In a clinical trial of 40 patients with gastrointestinal cancer, duloxetine was found to reduce cold sensitivity (P = .001), tingling or discomfort of hands (P < .002) and feet (P = .017), and peripheral neuropathic pain (P = .001), and was found to prevent paresthesia (P = .025).29 The SNRI antidepressant venlafaxine has also shown that it can alleviate neuropathic pain and motor neuropathy in clinical trials.30
Antioxidant agents, such as amifostine and calmangafodipir, have also been identified as possible preventive measures against OIPN. Amifostine prevents neuronal hyperactivation and nitrosative stress, while calmangafodipir modulates reactive O2 species, regulates ion channels, and protects axons and the myelin sheath.31,32
Treatments such as riluzole, lidocaine, and pregabalin have all shown promise in reducing the effects of OIPN by their action on potassium, sodium, and calcium channels, respectively.28 A study conducted on mice (n = 565) with OIPN found that riluzole effectively mitigated motor and sensory deficits associated with the use of oxaliplatin.33
TREK-1 and TRAAK, potassium channels that are important for thermal and motor sensitivity, and that act as silencing mechanisms to excitatory stimuli, were shown to degenerate following oxaliplatin treatment, leading to hypersensitivity. Riluzole performs its therapeutic function by activating TREK-1 and TRAAK channels and blocking excessive accumulation of glutamate. Following riluzole treatment, mice were observed to show a significant reduction in sensorimotor deficits. Interestingly, riluzole also aided in reducing depression associated with oxaliplatin (P < .01).33 However, more studies are necessary to ensure the safety and efficacy of riluzole in humans.
Pyridoxine, pyridostigmine for vincristine-induced peripheral neuropathy. Vinca alkaloids have also been identified as chemotherapeutic agents that induce peripheral neuropathy. One such agent, vincristine, which is used primarily to treat leukemia and brain cancer, has been observed to cause peripheral neuropathy, including motor, autonomic, and sensory symptoms, such as abnormal gait, mechanical allodynia, paresthesia, ptosis, and obstipation, and altered perception of stimuli.34,35 These symptoms are caused primarily by the ability of vincristine to activate neuroinflammatory mechanisms in dorsal-root ganglia. This is caused by activation of nucleotide-binding oligomerization domain 3 (NLRP3)-dependent release of IL-1b and subsequent cleavage of gasdermin D and caspase-1 in macrophages (observed in mouse models). Vincristine activates the NLRP3 signaling cascade that results in production of proinflammatory cytokines, thus inducing symptoms of peripheral neuropathy.36
Pyridoxine and pyridostigmine have been introduced as potential treatments for vincristine-induced peripheral neuropathy. Following a clinical trial of pediatric acute lymphoblastic leukemia patients, a study of 23 patients with vincristine-induced peripheral neuropathy found statistical validity for using pyridoxine and pyridostigmine because the drugs improved the neuropathy score (P < .001).37 However, more research is needed before implementing their use in point-of-care settings.
AUTOIMMUNE PERIPHERAL NEUROPATHY
Autoimmune peripheral neuropathies (APNs) occur when the immune system targets peripheral nervous system and its various cells. Although there is a wide range of conditions in this category of peripheral neuropathy, the two most common types – Guillain-Barré syndrome (GBS) and chronic inflammatory demyelinating polyneuropathy (CIDP) – have been targeted for clinical research.
Guillain-Barré syndrome: Diagnostic tools and strategies
Guillain-Barré syndrome encompasses a variety of acute inflammatory polyneuropathies, including axonal motor, sensory, and autonomic neuropathies and Miller Fisher syndrome (MFS).38 In particular, the anti-GQ1b ganglioside antibody is considered archetypical in APNs because it is detected in MFS patients and not found in normal and disease-control samples, which makes it a good clinical marker.39
It is difficult to distinguish GBS from CIDP because the time frame of onset of maximum deficit of neuropathy – 4 weeks – can overlap with subacute CIDP symptoms.40 Current diagnosis is based on elevated levels of cerebrospinal fluid (CSF) proteins, which can increase fourfold 6 weeks into the early phase of disease, and nerve conduction studies.40 However, electrodiagnostic readings and CSF protein levels are normal in 30% to 50% of patients in the first week after onset of disease and must be repeated in weeks that follow.41 A major disadvantage in the workup of suspected GBS is that the syndrome can be confirmed only several weeks after onset of symptoms.
Ultrasonography. A potential new diagnostic tool is serial peripheral nerve ultrasonographic (US) imaging. A pilot study of GBS patients (n = 16) showed that US can detect enlarged nerve cross-sections in median, ulnar, and sural nerves in the first 3 weeks of disease. Imaging performance was consistent with that of nerve conduction studies, and was advantageous because US is easier to perform and for patients to undergo.42
Spinal inflammation. Another study hints at the importance of spinal-root inflammation as an early indicator of disease, especially when nerve conduction study readings are normal.43 Further research is needed to demonstrate the clinical efficacy of this diagnostic method in larger population groups.
Guillain-Barré syndrome: Therapeutic options
The standard of care for GBS in the United States is intravenous immunoglobulin (IVIG) therapy and plasmapheresis, but there is no FDA-approved treatment.44 Although the two treatments have been shown to be equally effective in early stages of disease, early relapses can occur with both. One study found that 20% of patients who underwent plasmapheresis relapsed.40 Because nearly 50% of GBS patients do not respond to IVIG or plasmapheresis, the need is urgent for new therapies to decrease the risk of permanent disability.45
Antibody therapy. Recent developments include the use of monoclonal antibodies against GBS. ANX005 is an immunoglobulin G4 recombinant antibody that inhibits complement component 1q (C1q). Activation of this protein triggers the classical complement cascade, a natural part of the innate immune system that is nonetheless inappropriately activated in some autoimmune diseases, leading to neurodegeneration as a consequence of tissue damage.
ANX005 was found to have high-binding affinity to C1q in human, rat, cynomolgus monkey, and dog sera in nonclinical trials, and demonstrated low cross-reactivity despite being a plasma protein present throughout human tissue. Furthermore, studies show that ANX005 can deplete C1q completely in the CSF of monkeys.46 Phase 1b clinical trials in Bangladesh with GBS patients (n = 23) 18 to 58 years of age against a placebo group (n = 8) indicate that treatment is well tolerated. Drug-related serious adverse events were lacking and subjects’ GBS-Disability Score improved compared with placebo controls at week 1 (r2 = 0.48; P < .0001) and week 8, when an improvement of three or more in the score was observed.40
ANX005 is entering phase 2 trials, which are expected to be completed in 2023.47
Eculizumab. This promising treatment is a monoclonal antibody against C5 convertase, an enzyme that catalyzes formation of C5b-9, a membrane attack complex in nerve membranes. Studies in mouse models showed that treatment could significantly improve symptoms of terminal motor neuropathy and completely block formation of membrane attack complexes.48 Rats in this study were paralyzed by anti-GQ1b antibodies to emulate GBS pathogenesis.
A double-blind, placebo-controlled phase 2 clinical trial in Japan enrolled 34 patients (23 assigned to receive eculizumab; 11, to placebo); all were 18 years old or older and could not walk independently (3-5 on the GBS functional grading scale). Results showed that:
- Sixteen percent more patients receiving eculizumab treatment (n = 14; 42-78 years) than in the placebo group (n = 5; 20-73 years) could walk independently after 4 weeks.
- Fifty-six percent more patients in the functional group (n = 17; 52-90 years) than in the placebo group (n = 2; 20-52 years) could run after 6 months.49 While it is noted that the first portion of the trial failed to meet the predefined significance level, its long-term effects are observed to have therapeutic potential.
Eculizumab is in phase 3 clinical trials with primary data to be released in October 2022.50
Alemtuzumab, which inhibits the CD52 gene, was found to alleviate symptoms and restore strength in a rapidly deteriorating patient with MFS and chronic lymphocytic leukemia. By week 4 of treatment, anti-GQ1B antibodies were eliminated. However, the cause of this patient’s MFS is unclear; recovery might have been the result of multiple factors.51
IgG inhibition. Additional ongoing studies include therapies geared toward the neonatal Fc receptor as a potential clinical target for IgG inhibition.52
Chronic inflammatory demyelinating polyneuropathy (CIDP): Diagnostic tools and strategies
CIDP is the most common chronic APN and shares many similarities with GBS but differs in its responsiveness to corticosteroids, prognosis, and more. Lack of consensus on diagnostic criteria for CIDP has led to reliance on nerve conduction studies and clinical findings for making the diagnosis.53
Guidelines. European Federation of Neurological Societies/Peripheral Nerve Society guidelines have high sensitivity (81%) and specificity (96%) and are utilized as diagnostic criteria for CIDP; however, a survey found that these criteria may be underutilized in clinical practice – which might contribute to a high misdiagnosis rate.54 Furthermore, although current diagnostic methods are dependent on CSF proteins, this disease is lacking a diagnostic biomarker, leading to easy overdiagnosis and unnecessary immunotherapy.55
Electrodiagnostic testing, which is often used, is limited because it cannot evaluate small-fiber nerves, cannot access the CNS adequately, and does not provide a specific diagnosis.56
Sphingomyelin in CSF. Recently, a study in Italy explored the potential of CSF sphingomyelin as a biomarker for CIDP and for GBS. Findings reveal that sphingomyelin levels can be used to diagnose more than 80% of APN cases in the clinical setting. Different levels were identified in GBS, acute inflammatory demyelinating polyneuropathy, and typical and atypical CIDP patients. Additionally, sphingomyelin showed potential to diagnose the correct stage of disease. An increase in sphingomyelin in relapsing CIDP patients was noted, compared with what was seen in controls and stable CIDP patients.57 Larger-scale studies are needed to further test the efficacy of this method.
Chronic inflammatory demyelinating polyneuropathy: Therapeutic options
First-line therapy for CIDP comprises prednisone, 60-100 mg/d, plasmapheresis, and IVIG, all of which have proved effective. Some patients respond better to one treatment than to others40; some have subpar response to all these treatments and are categorized as having refractory CIDP.45
Although there are no newly approved treatments for CIDP, several show promise in ongoing clinical trials.
Rituximab is an anti-CD20 monoclonal antibody being studied in two phase 2 clinical trials of efficacy for refractory CIDP with IgG4 autoantibodies, after showing potential efficacy.58,59
Efgartigimod is an Fc fragment that blocks the neonatal Fc receptor, prevents lysosome degradation of IgGs, and thus allows them to be “recycled.”60 These autoantibodies are crucial in disease pathology because lowering their concentration provides effective therapy.61 Phase 1 trials showed that repeated doses of efgartigimod reduced IgG levels in healthy volunteers by 50%. Repeated dosing lowered IgG levels, on average by 75% in serum, which was an effect that was sustained for an 8-week period.62 Phase 2 trials are recruiting, with a projected primary completion in 2023.
INFECTION-INDUCED PERIPHERAL NEUROPATHY
Infections have been identified as a primary cause of peripheral neuropathy. Infection-induced peripheral neuropathy has been associated with Lyme disease, Epstein-Barr and human immunodeficiency virus (HIV) infection, shingles, hepatitis B and C, diphtheria, leprosy, and rabies.63 Extensive research on peripheral neuropathy has not been completed for most of the diseases, highlighting an unmet need for patients who experience this sequela of infection.
HIV is a well-documented viral cause of peripheral neuropathy. The most common symptom is distal sensory polyneuropathy, which affects more than 50% of patients with HIV.64 The incidence of distal sensory polyneuropathy in HIV has been correlated with the use of antiretroviral therapy – specifically, tenofovir disoproxil fumarate – and with certain proteins secreted by the virus.65 Symptoms include loss of sensory properties, neuropathic pain, and allodynia.66
Diagnostic tools and strategies
Nerve conduction studies have primarily been used to diagnose HIV-induced peripheral neuropathy, as well as electrophysiological testing and noninvasive CCM. These assays can detect changes or abnormalities in large- and small-fiber nerves in HIV infection patients.66
Therapeutic options
Studies in mouse models have illustrated how the Tat protein correlates with induction of motor and sensory distal symmetric polyneuropathy. Expression of Tat can lead to mitochondrial disruption, resulting in degeneration of sensory dorsal root ganglia and subsequent neuropathic pain.67
Pirenzepine. Studies on mice have identified a potential treatment for HIV infection-induced peripheral neuropathy with pirenzepine, targeting the muscarinic subtype-1 receptor. Pirenzepine activates a molecular pathway that promotes neurite growth and mitochondrial function. Researchers found that, following treatment with pirenzepine (n = 6), there was marked reduction in mitochondrial degeneration and HIV-induced distal sensory neuropathy.66 This outcome was due to the ability of pirenzepine to block the effects of Tat protein expression, leading to reversal of its neurodegenerative effects.
Exercise combined with analgesics has also been identified as a potential treatment for alleviating distal sensory polyneuropathy in HIV infection–induced peripheral neuropathy. In a 12-week study, researchers instructed subjects who were receiving a combination of HIV treatments, including tenofovir, lamivudine, and efavirenz, to perform aerobic and resistance exercises. This regimen was intended to improve peripheral nerve-conduction velocity and increase the density of nerve fibers and neurogenic branching.
The study identified baseline pain scores and divided participants into three groups: aerobic exercise (n = 45), resistance exercise (n = 44), and controls (n = 47), for whom the average level of pain was 2 on an ascending scale of 1 to 10. There was significant reduction in pain score in the experimental groups by the end of the study, as well as an increased sensory profile.64 This study has elucidated a pain management therapy for HIV-induced peripheral neuropathy that can prove beneficial for patients.
CRYPTOGENIC SENSORY POLYNEUROPATHY
Also known as idiopathic neuropathy or small-fiber sensory peripheral neuropathy, cryptogenic sensory polyneuropathy (CSPN) affects one-third of patients with peripheral neuropathy, in whom (despite extensive testing) no known cause of their condition is revealed.
Diagnostic tools and strategies
Applicable clinical and laboratory tests of any potential known underlying causes of neuropathy, including diabetes, hereditary disorders, and autoimmune disease, must be performed to rule out those causes and suggest an idiopathic cause.68
Therapeutic options
There are no FDA-approved treatments for CSPN, as most treatments are geared toward neuropathic pain management, rehabilitation, and supportive care.68 Due to a lack of research and data regarding these types of peripheral neuropathies, various studies suggest different first-line therapies. For example, anticonvulsants (pregabalin, gabapentin), antidepressants (duloxetine), and opioid-like compounds (tramadol) are all threapy options to treat DPN.3
Adequate data are lacking to support the efficacy of immunosuppressive therapy in CSPN.
Summing up
The combination of an understanding of a widening range of underlying diseases, advancements in cancer therapies, and the rising prevalence of diabetes have all led to an increasing incidence of peripheral neuropathy. Coupled with the fact that one-third of patients with peripheral neuropathy experience idiopathic neuropathy, this indicates that extensive studies must be undertaken to identify mitigation and prevention strategies for peripheral neuropathy. To summarize the landscape of treatment for peripheral neuropathy:
Diabetic peripheral neuropathy. Treatment for DPN comprises three FDA-approved products: pregabalin, duloxetine, and a higher (8%)-strength capsaicin patch.3 Pain-management therapies also exist to reduce diabetes-induced neuropathic pain, including gabapentin, amitriptyline, and extended-release tapentadol.10
Chemotherapy-induced peripheral neuropathy has yet to be effectively treated in humans; however, many trials are being completed in animals with promising results. Treatment for CIPN has been identified using senolytic agents, such as navitoclax,22 and through inhibition of SASP by a variety of agents, including ARV825, tocilizumab, and adalimumab.23-26
Oxaliplatin-induced peripheral neuropathy. Research has identified a potential preventive agent in duloxetine, with human trials already showing efficacy and safety.29 Animal models have shown progress studying antioxidant agents, such as amifostine31 and calmangafodipir,32 which target ion channels. In a similar mechanism of action, riluzole has been observed to reduce motor and sensory deficits and depression resulting from treatment with oxaliplatin.
Vincristine-induced peripheral neuropathy. Progress has been seen in treating vincristine-induced peripheral neuropathy with pyridoxine and pyridostigmine, which have improved neuropathy scores in trial subjects;37 more studies must be completed before these agents can be established as effective therapy.
Autoimmune PN. There are no FDA-approved drugs to mitigate the peripheral neuropathy induced by GBS and CIDP; however, studies are being conducted to resolve this impediment. Potential treatments, such as ANX005, a recombinant antibody, and eculizumab, a monoclonal antibody, have both shown efficacy in human trials and provide a potential path toward treatment against peripheral neuropathy caused by GBS.47,50 CIDP is currently treated using prednisone, plasmapheresis, and IVIG.40 Clinical trials are studying the efficacy of rituximab and efgartigimod for CIDP.58-60
Infection-induced peripheral neuropathy. Although many infections can induce peripheral neuropathy, HIV is most well documented and therefore was singled out for discussion in this article. Pirenzepine has been shown to promote neurite growth and reduce mitochondrial degeneration – both of which factors are associated with reduction of neuropathic pain.66 Exercise and analgesics have also been found to mitigate the effects of HIV-induced distal sensory neuropathy, with pain scores being reduced.61
Cryptogenic sensory polyneuropathy. Research has yet to identify a causative agent of, or subsequent potential therapy for, CSPN. Increased knowledge about this neuropathy will, it is hoped, bring patients closer to a cure – beyond current pain mitigation strategies with anticonvulsants, antidepressants, and opioid-like compounds.3
Ms. Lee is a first-year master of science candidate in applied life sciences, with an emphasis on infectious diseases, and Mr. Kosacki is a first-year master of science candidate in applied life sciences, with an emphasis on translational research, both at Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences, Claremont, Calif. Dr. Bhandari is professor of clinical sciences and Dr. Tran is professor of clinical sciences, Keck Graduate Institute School of Pharmacy and Health Sciences.
References
1. Barrell K, Smith AG. Peripheral neuropathy. Med Clin North Am. 2019 Mar;103(2):383-97. doi: 10.1016/j.mcna.2018.10.006.
2. Selvarajah D et al. Diabetic peripheral neuropathy: Advances in diagnosis and strategies for screening and early intervention. Lancet Diabetes Endocrinol. 2019 Dec;7(12):938-48. doi: 10.1016/S2213-8587(19)30081-6.
3. Snyder MJ et al. Treating painful diabetic peripheral neuropathy: An update. Am Fam Physician. 2016 Aug;94(3):227-334.
4. Sharma S et al. Assessment of diabetic neuropathy using a point-of-care nerve conduction device shows significant associations with the LDIFLARE method and clinical neuropathy scoring. J Diabetes Sci Technol. 2014 Jan;9(1):123-31. doi: 10.1177/1932296814551044.
5. Zografou I et al. Validation of Neuropad in the assessment of peripheral diabetic neuropathy in patients with diabetes mellitus versus the Michigan Neuropathy Screening Instrument, 10g monofilament application and biothesiometer measurement. Curr Vasc Pharmacol. 2020;18(5):517-22. doi: 10.2174/1570161117666190723155324.
6. Tentolouris N et al. Moisture status of the skin of the feet assessed by the visual test Neuropad correlates with foot ulceration in diabetes. Diabetes Care. 2010;33(5):1112-4. doi: 10.2337/dc09-2027.
7. Mao F et al. Sudoscan is an effective screening method for asymptomatic diabetic neuropathy in Chinese type 2 diabetes mellitus patients. J Diabetes Investig. 2017 May;8(3):363-8. doi: 10.1111/jdi.12575.
8. Kalteniece A et al. Corneal confocal microscopy is a rapid reproducible ophthalmic technique for quantifying corneal nerve abnormalities. PLoS One. 2017 Aug;12(8):e0183040. doi: 10.1371/journal.pone.0183040.
9. Gad H et al. Corneal confocal microscopy for the diagnosis of diabetic peripheral neuropathy: A systematic review and meta-analysis. J Diabetes Investig. 2022 Jan;13(1):134-47. doi: 10.1111/jdi.13643.
10. Pop-Busui R et al. Diabetic neuropathy: A position statement by the American Diabetes Association. Diabetes Care. 2017;40(1):136-54. doi: 10.2337/dc16-2042.
11. Chung YC et al. Calcimimetic restores diabetic peripheral neuropathy by ameliorating apoptosis and improving autophagy. Cell Death Dis. 2018 Nov;9(12):1163. doi: 10.1038/s41419-018-1192-7.
12. Li J et al. Therapeutic effects of moxibustion simultaneously targeting Nrf2 and NF-kB in diabetic peripheral neuropathy. Appl Biochem Biotechnol. 2019 Dec;189(4):1167-82. doi: 10.1007/s12010-019-03052-8.
13. Tan Y et al. Moxibustion for the treatment of diabetic peripheral neuropathy: A systematic review and meta-analysis following PRISMA guidelines. Medicine (Baltimore). 2020 Sep 26;99(39):e22286. doi: 10.1097/MD.0000000000022286.
14. Xie J et al. Protective effect of quercetin on streptozotocin-induced diabetic peripheral neuropathy rats through modulating gut microbiota and reactive oxygen species level. Biomed Pharmacother. 2020 Jul;127:110147. doi: 10.1016/j.biopha.2020.110147.
15. Zhao B et al. Quercetin reduces inflammation in a rat model of diabetic peripheral neuropathy by regulating the TLR4/MyD88/NF-kappa B signalling pathway. Eur J Pharmacol. 2021 Dec;912:174607. doi: 10.1016/j.ejphar.2021.174607.
16. McWhinney SR et al. Platinum neurotoxicity pharmacogenetics. Mol Cancer Ther. 2009;8(1):10-6. doi: 10.1158/1535-7163.MCT-08-0840.
17. Addington J, Freimer M. Chemotherapy-induced peripheral neuropathy: An update on the current understanding. F1000Res. 2016 Jun 22;5:F1000 Faculty Rev-1466. doi: 10.12688/f1000research.8053.1.
18. Lustberg M, Loprinzi C, eds. “Diagnosis, Management and Emerging Strategies for Chemotherapy-Induced Neuropathy: A MASCC Book.” Springer International Publishing; 2021.
19. Cornblath DR et al. Total neuropathy score: Validation and reliability study. Neurology. 1999 Nov;53(8):1660-4. doi: 10.1212/wnl.53.8.1660.
20. Aldossary SA. Review on pharmacology of cisplatin: Clinical use, toxicity and mechanism of resistance of cisplatin. Biomedical and Pharmacology Journal. 2019;12(1):7-15. http://dx.doi.org/10.13005/bpj/1608.
21. Calls A et al. Cisplatin-induced peripheral neuropathy is associated with neuronal senescence-like response. Neuro Oncol. 2021 Jan;23(1):88-99. doi: 10.1093/neuonc/noaa151.
22. Acklin S et al. Depletion of senescent-like neuronal cells alleviates cisplatin-induced peripheral neuropathy in mice. Sci Rep. 2020 Aug;10(1):14170. doi: 10.1038/s41598-020-71042-6.
23. Watanabe S et al. Impact of senescence‐associated secretory phenotype and its potential as a therapeutic target for senescence‐associated diseases. Cancer Sci. 2017 Apr;108(4):563-9. doi: 10.1111/cas.13184.
24. Harrison DE et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009 Jul 16;460(7253):392-5. doi: 10.1038/nature08221.
25. Wakita M et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat Commun. 2020;11(1):1935. doi: 10.1038/s41467-020-15719-6.
26. Prattichizzo F et al. Anti-TNF-alpha treatment modulates SASP and SASP-related microRNAs in endothelial cells and in circulating angiogenic cells. Oncotarget. 2016 Mar 15;7(11):11945-58. doi: 10.18632/oncotarget.7858.
27. Kang L et al. Oxaliplatin-induced peripheral neuropathy: Clinical features, mechanisms, prevention and treatment. J Neurol. 2021 Sep;268(9):3269-82. doi: 10.1007/s00415-020-09942-w.
28. Yang Y et al. Targeting strategies for oxaliplatin-induced peripheral neuropathy: Clinical syndrome, molecular basis, and drug development. J Exp Clin Cancer Res. 2021 Oct 22;40(1):331. doi: 10.1186/s13046-021-02141-z.
29. Rokhsareh S et al. Evaluating the effects of duloxetine on prophylaxis of oxaliplatin-induced peripheral neuropathy in patients with gastrointestinal cancer: A randomized double-blind placebo controlled clinical trial. J Oncol Pharm Pract. 2021 Nov 5;10781552211052646. doi: 10.1177/10781552211052646.
30. Farshchian N et al. Comparative study of the effects of venlafaxine and duloxetine on chemotherapy-induced peripheral neuropathy. Cancer Chemother Pharmacol. 2018 Nov;82(5):787-93. doi: 10.1007/s00280-018-3664-y.
31. Pereira AF et al. Amifostine protects from the peripheral sensory neuropathy induced by oxaliplatin in mice. Braz J Med Biol Res. 2020 Sep 18;53(11):e10263. doi: 10.1590/1414-431X202010263.
32. Glimelius B et al. Persistent prevention of oxaliplatin-induced peripheral neuropathy using calmangafodipir (PledOx®): A placebo-controlled randomised phase II study (PLIANT). Acta Oncol. 2018 Mar;57(3):393-402. doi: 10.1080/0284186X.2017.1398836.
33. Poupon L et al. Targeting the TREK-1 potassium channel via riluzole to eliminate the neuropathic and depressive-like effects of oxaliplatin. Neuropharmacology. 2018 Sep 15;140:43-61. doi: 10.1016/j.neuropharm.2018.07.026.
34. Mora J et al. Next generation ligand binding assays – Review of emerging technologies’ capabilities to enhance throughput and multiplexing. AAPS J. 2014 Nov;16(6):1175-84. doi: 10.1208/s12248-014-9660-1.
35. Starobova H, Vetter I. Pathophysiology of chemotherapy-induced peripheral neuropathy. Front Mol Neurosci. 2017 May 31;10:174. doi: 10.3389/fnmol.2017.00174.
36. Starobova H et al. Vincristine-induced peripheral neuropathy is driven by canonical NLRP3 activation and IL-1-beta release. J Exp Med. 2021;218(5):e20201452. doi: 10.1084/jem.20201452.
37. Aydin Köker S et al. Effect of pyridoxine plus pyridostigmine treatment on vincristine-induced peripheral neuropathy in pediatric patients with acute lymphoblastic leukemia: A single-center experience. Neurol Sci. 2021 Sep;42(9):3681-6. doi: 10.1007/s10072-020-04970-w.
38. Bourque PR et al. Autoimmune peripheral neuropathies. Clin Chim Acta. 2015 Sep 20;449:37-42. doi: 10.1016/j.cca.2015.02.039.
39. Paparounas K. Anti-GQ1b ganglioside antibody in peripheral nervous system disorders: Pathophysiologic role and clinical relevance. Arch Neurol. 2004 Jul;61(7):1013-6. doi: 10.1001/archneur.61.7.1013.
40. Dalakas MC. Autoimmune peripheral neuropathies, in Rich RR et al., eds., “Clinical Immunology.” 5th ed, (Amsterdam: Elsevier, 2019, pp. 903-915.e1). doi: 10.1016/B978-0-7020-6896-6.00067-3
41. Leonhard SE et al. Diagnosis and management of Guillain-Barré syndrome in ten steps. Nat Rev Neurol. 2019;15(11):671-83. doi: 10.1038/s41582-019-0250-9.
42. Razali SNO et al. Serial peripheral nerve ultrasound in Guillain–Barré syndrome. Clin Neurophysiol. 2016 Nov;127(2):1652-6. doi: 10.1016/j.clinph.2015.06.030.
43. Gallardo E et al. Spinal nerve involvement in early Guillain-Barré syndrome: A clinico-electrophysiological, ultrasonographic and pathological study. Clin Neurophysiol. 2015 Apr;126(4):810-9. doi: 10.1016/j.clinph.2014.06.051.
44. Islam Z et al. Inhibition of C1q, initiator of the classical complement cascade, by ANX005 for the treatment of Guillain–Barré syndrome: Results from a phase 1b study (763). Neurology. 2020 Apr;94(15 Suppl):763.
45. Hughes R et al.; . Oral fingolimod for chronic inflammatory demyelinating polyradiculoneuropathy (FORCIDP Trial): A double-blind, multicentre, randomised controlled trial. Lancet Neurol. 2018 Aug;17(8):689-98. doi: 10.1016/S1474-4422(18)30202-3.
46. Lansita JA et al. Nonclinical development of ANX005: A humanized anti-C1q antibody for treatment of autoimmune and neurodegenerative diseases. Int J Toxicol. 2017 Nov/Dec;36(6):449-62. doi: 10.1177/1091581817740873.
47. Annexon Inc. A randomized, double-blind, placebo-controlled phase 2/3 study to evaluate the efficacy, safety, pharmacokinetics, and pharmacodynamics of ANX005 in subjects with Guillain–Barré syndrome. ClinicalTrials.gov Identifier: NCT04701164. Updated Jan 8, 2021. Accessed Feb 23, 2022. https://clinicaltrials.gov/ct2/show/NCT04701164.
48. Halstead SK et al. Eculizumab prevents anti-ganglioside antibody-mediated neuropathy in a murine model. Brain. 2008 May;131(Pt 5):1197-1208. doi: 10.1093/brain/awm316.
49. Misawa S et al. Safety and efficacy of eculizumab in Guillain-Barré syndrome: A multicentre, double-blind, randomised phase 2 trial. Lancet Neurol. 2018 Jun;17(6):519-29. doi: 10.1016/S1474-4422(18)30114-5.
50. Alexion Pharmaceuticals. A phase 3, prospective, multicenter, double blind, randomized, placebo-controlled study to evaluate the efficacy and safety of eculizumab in patients with Guillain–Barré syndrome (GBS). ClinicalTrials.gov Identifier: NCT04752566. Updated Feb 18, 2022. Accessed Feb 23, 2022. https://clinicaltrials.gov/ct2/show/NCT04752566.
51. Tzachanis D et al. Successful treatment of refractory Guillain–Barré syndrome with alemtuzumab in a patient with chronic lymphocytic leukemia. Acta Haematol. 2014 Aug;132(2):240-3. doi: 10.1159/000358292.
52. Satkowiak K, Smith AG. Guillain-Barré syndrome, in Roos KL, ed. “Emergency Neurology.” (Springer, Cham, 2021, pp. 225-50). Accessed Feb 23, 2022. https://doi.org/10.1007/978-3-030-75778-6_12.
53. Gogia B et al. Chronic inflammatory demyelinating polyradiculoneuropathy, in “StatPearls [Internet].” (Treasure Island (Fla.): StatPearls Publishing; 2022 Jan). Updated Nov 22, 2021. Accessed Feb 23, 2022. www.ncbi.nlm.nih.gov/books/NBK563249.
54. Allen JA et al. Challenges in the diagnosis of chronic inflammatory demyelinating polyneuropathy. Brain Behav. 2018 Feb;8(3):e00932. doi: 10.1002/brb3.932.
55. Stino AM et al. Chronic inflammatory demyelinating polyradiculoneuropathy-diagnostic pitfalls and treatment approach. Muscle Nerve. 2021 Feb;63(2):157-69. doi: 10.1002/mus.27046.
56. Ginsberg MR et al. Using and interpreting electrodiagnostic tests. Cleve Clin J Med. 2020 Nov 2;87(11):671-82. doi: 10.3949/ccjm.87a.19154.
57. Capodivento G et al. CSF sphingomyelin: A new biomarker of demyelination in the diagnosis and management of CIDP and GBS. J Neurol Neurosurg Psychiatry. 2021;92(3):303-10. doi: 10.1136/jnnp-2020-324445.
58. Shimizu S et al. Efficacy and safety of rituximab in refractory CIDP with or without IgG4 autoantibodies (RECIPE): Protocol for a double-blind, randomized, placebo-controlled clinical trial. JMIR Res Protoc. 2020 Jan 4;9(4):e17117. doi: 10.2196/17117.
59. Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group. Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barré syndrome. Lancet. 1997;349(9047):225-30.
60. Zuercher AW et al. Next-generation Fc receptor–targeting biologics for autoimmune diseases. Autoimmun Rev. 2019 Oct;18(10):102366. doi: 10.1016/j.autrev.2019.102366.
61. Sesarman A et al. The neonatal Fc receptor as therapeutic target in IgG-mediated autoimmune diseases. Cell Mol Life Sci. 2010 Aug;67(15):2533-50. doi: 10.1007/s00018-010-0318-6.
62. Ulrichts P et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J Clin Invest. 2018 Oct;128(10):4372-86. doi: 10.1172/JCI97911.
63. Peripheral neuropathy [symptoms and causes]. Mayo Clinic [Internet]. Accessed Feb 23, 2022. http://www.mayoclinic.org/diseases-conditions/peripheral-neuropathy/symptoms-causes/syc-20352061.
64. Maharaj SS, Yakasai AM. Does a rehabilitation program of aerobic and progressive resisted exercises influence HIV-induced distal neuropathic pain? Am J Phys Med Rehabil. 2018 May;97(5):364-9. doi: 10.1097/PHM.0000000000000866.
65. Fields JA et al. Tenofovir disoproxil fumarate induces peripheral neuropathy and alters inflammation and mitochondrial biogenesis in the brains of mice. Sci Rep. 2019 Nov 20;9(1):17158. doi: 10.1038/s41598-019-53466-x.
66. Han MM et al. Prevention of HIV-1 TAT protein-induced peripheral neuropathy and mitochondrial disruption by the antimuscarinic pirenzepine. Front Neurol. 2021 Jun 15;12:663373. doi: 10.3389/fneur.2021.663373.
67. Rozzi SJ et al. Human immunodeficiency virus Tat impairs mitochondrial fission in neurons. Cell Death Discov. 2018;4:8. doi: 10.1038/s41420-017-0013-6.
68. Pasnoor M et al. Cryptogenic sensory polyneuropathy. Neurol Clin. 2013 May;31(2):463-76. doi: 10.1016/j.ncl.2013.01.008.
Spinal muscular atrophy: Patient care in the age of genetically targeted therapy
In 2016, the U.S. Food and Drug Administration approved nusinersen, the first treatment for spinal muscular atrophy (SMA). Until then, SMA had a mortality rate nearly double that of the general population.1 Two-thirds of patients were symptomatic within 6 months of birth and, in the absence of mechanical ventilation and other support, had a nearly 100% mortality rate by age 2.2
Five years later, there are three approved treatments for SMA, all of which have been shown to slow or even halt disease progression in many patients. Neurologists, whose SMA patient population once consisted almost entirely of children, are now treating more adults with the disease. Indeed, more than half of all people alive with SMA in the United States today are adults, according to Cure SMA.
“Managing SMA used to be clinic follow-ups where we were doing our best supportive care and watching people fall apart before our eyes,” said John Brandsema, MD, a physician and neuromuscular section head at the Children’s Hospital of Philadelphia. “Today, what we see in the vast majority of people is that they are either the same as they were before – which is completely against the natural history of this disease and something to be celebrated – or that people are really better with their function. It totally changes everything in the clinic.”
Among those changes are a more proactive approach to rehabilitation and an even greater emphasis on personalized medicine and multidisciplinary care. But there is also a need for updated treatment guidelines, a new classification system to measure disease severity, specific biomarkers to guide therapy choices, more data on long-term efficacy of existing therapeutics, new medications to complement those therapies, and a deeper understanding of a disease that may have treatment options but still has no cure.
Advances in early diagnosis
Patients with SMA lack a working copy of the survival motor neuron 1 (SMN1) gene, which provides instructions for producing a protein called SMN that is critical for the maintenance and function of motor neurons. Without this protein, motor neurons eventually die, causing debilitating and progressive muscle weakness that affects the ability to walk, eat, and breathe. SMA is rare, affecting about 1 in 10,000 newborns.
In approximately 96% of patients, SMA is caused by homozygous loss of the SMN1 gene. People with SMA have at least one copy of the SMN2 gene, sometimes called a “backup” gene, that also produces SMN protein. However, a single nucleotide difference between SMN2 and SMN1 causes about 90% of the protein produced by SMN2 to be truncated and less stable. Even with multiple copies of SMN2 present, as is the case with many infants with SMA, the amount of functional protein produced isn’t enough to compensate for the loss of SMN1.3
All three approved medications are SMN up-regulators and work to increase the amount of functional SMN protein. Starting these medications early, even before symptoms present, is critical to preserve motor function. Early treatment depends on early diagnosis, which became more widespread after 2018 when SMA was added to the federally Recommended Uniform Screening Panel for newborns. As of July 1, 2022, 47 states have incorporated SMA newborn screening into their state panel, ensuring that 97% of all infants born in the United States undergo SMA screening shortly after birth. Screening in the remaining states – Hawaii, Nevada, and South Carolina – and Washington, D.C. is expected by mid-2023.
SMA newborn screening is a PCR-based assay that detects homozygous SMN1 gene deletion found in about 95% of all people with SMA. The remaining 5% of cases are caused by various genetic mutations that can only be detected with gene sequencing. In these cases, and in children who don’t undergo SMA newborn screening, the disease is usually identified when symptoms are noticed by a parent, pediatrician, or primary care provider. But a study found that in 2018 only 52.7% of pediatricians correctly identified genetic testing as a requirement for a definitive diagnosis of SMA; in 2019, with a larger sample size, that number decreased to 45%.4 The lack of awareness of diagnostic requirements for SMA could contribute to delays in diagnosis, said Mary Schroth, MD, chief medical officer for Cure SMA and a coauthor of the study.
“In our world, suspicion of SMA in an infant is an emergency situation,” Dr. Schroth said. “These babies need to be referred immediately and have genetic testing so that treatment can begin as soon as possible.”
Based on the study findings, Dr. Schroth and others with Cure SMA launched a new tool in 2021 designed to help pediatricians, primary care physicians, and parents identify early signs of SMA, so that a referral to a pediatric neurologist happens quickly. Called SMArt Moves, the educational resource features videos and a checklist to help increase early detection in infants who had a negative SMA newborn screening result or did not receive SMA screening at birth.5
Who to treat, when, and with which treatment
For many patients, having multiple effective treatment options means that SMA is no longer a fatal disease in early childhood, but one that can be managed into adolescence and adulthood. The question for clinicians is, who do they treat, when, and with which treatment?
Studies have long shown that the number of copies of the backup gene that a patient has is inversely associated with disease severity.6 In 2018, a group of SMA experts published a treatment algorithm to help guide decision-making following a positive SMA newborn screening.7 The treatment guidelines were updated in 2020 based on clinical trial data for presymptomatic infants, and current recommendations include immediate treatment for infants with two to four copies of the SMN2 gene.8 For patients with only one copy of SMN2, most of whom will likely be symptomatic at birth, the guidelines recommend that treatment decisions be made jointly between the clinician and the family.7,8
Some suggest that the number of SMN2 copies a patient has should also be a factor in determining phenotype, which has started a conversation on the development of a new classification system.9 The original classification system for disease severity – Types 0-4 – was based on age of onset and degree of motor function achieved, with Type 0 developing prenatally and being the most severe and Type 4 developing in adulthood. Type 1 is the most common, affecting more than half of all people with SMA, followed by Types 2-4. In 2018, updated consensus care guidelines offered a revised classification system that better reflected disease progression in the age of therapy. The functional motor outcomes include nonsitters (historically Type I), sitters (historically Type 2/3), and walkers (historically Type 3/4).10,11 These guidelines are a start, but clinicians say more revision is needed.
“Types 1, 2, 3, 4 were based on function – getting to a certain point and then losing it, but now that we can treat this disease, people will shift categories based on therapeutic response or based on normal development that is possible now that the neurologic piece has been stabilized,” Dr. Brandsema said. “We need to completely change our thinking around all these different aspects of SMA management.”
While discussions of a new classification system for SMA are underway, another effort to update treatment recommendations is closer to completion. Led by Cure SMA, a group of about 50 physician experts in the United States and Europe who specialize in SMA are revising guidelines for diagnosis and treatment, the first time the recommendations have been updated since 2018. The updated recommendations, which should be published later this year, will focus on diagnosis and treatment considerations.
“We have three treatments that are available, and there are specific FDA indications for each of those, but it’s not totally clear just how those medications should be used or applied to different clinical situations,” said Dr. Schroth. “We’re in a rapid phase of learning right now in the SMA community, trying to understand how these treatments alter physiology and disease outcomes and how to best use the tools that we now have available to us. In parallel with clinical treatments, we have to be doing the best care we can to optimize the outcomes for those treatments.”
Research advances in 2021
Although all three drugs approved to treat SMA – nusinersen (Spinraza; Biogen), onasemnogene abeparvovec-xioi gene replacement therapy (Zolgensma; Novartis Gene Therapies), and risdiplam (Evrysdi, Genentech/Roche) – are highly effective, there are still unanswered questions and unmet needs. New research findings from 2021 focused on higher dosing, different drug-delivery methods, combination therapy, and complementary therapeutics to address SMA comorbidities.
Higher-dose nusinersen. The first drug approved to treat SMA, nusinersen is an antisense oligonucleotide approved for all ages and all SMA types. It works by altering splicing of the SMN2 gene pre-mRNA to make more complete SMN protein. Given as an intrathecal (IT) injection, four “loading doses” are administered within the first 2 months of treatment, followed by a maintenance dose every 4 months for the duration of the individual’s life.
Reports from patients of waning effects of nusinersen just prior to follow-up treatment have led some clinicians to ask if a higher dose may be needed. A study underway seeks to address that issue.
DEVOTE is a phase 2/3 trial to study the safety and efficacy of high-dose nusinersen in patients with SMA. Preliminary findings reported in 2021 found no adverse events among patients treated with 28 mg of nusinersen for 161-257 days.12 Another analysis from this trial found that higher doses are associated with greater decrease of plasma phosphorylated neurofilament heavy chain (pNF-H) levels in patients with SMA and may lead to clinically meaningful improvement in motor function beyond that observed with the approved 12 mg dose.13 The trial is ongoing.
Another trial, ASCEND, is a phase 3B study assessing higher dose nusinersen in patients previously treated with risdiplam. Recruitment for that trial began in October 2021.
Long-term efficacy and IT administration of SMA therapy. Several studies are looking at the long-term efficacy and alternate routes of administration of onasemnogene abeparvovec and other SMA therapies.
A one-time gene replacement therapy delivered via an IV infusion replaces the function of the missing or nonworking SMN1 gene with a new, working copy of the SMN1 gene. FDA approved in 2019, it is authorized for use in patients with SMA up to 2 years of age.
The latest data from an ongoing, long-term follow-up safety study of onasemnogene abeparvovec, published in May 2021, suggest that the treatment’s effects persist more than 5 years after treatment. Researchers followed 13 infants with symptomatic SMA type 1 since the beginning of the phase 1 clinical trial of the gene transfer therapy. All patients who received the therapeutic dose maintained their baseline motor function, and two of the patients actually improved without other SMN-targeted treatment. At a median 6.2 years after they received treatment, all were alive and none needed permanent ventilation.14
After a 2-year hold by the FDA, a study of IT administration of onasemnogene abeparvovec is now enrolling patients. Citing concerns from animal studies that IT administration might result in dorsal root ganglia injury, the FDA issued a partial hold on the STRONG trial in 2019. Following positive study results in nonhuman primates, the FDA announced the trial can continue. Novartis is launching a new phase 3 STEER trial to test the drug delivered intrathecally in patients aged 2-18 years with Type 2 SMA. IT administration could allow the gene therapy to be used safely and effectively in more patients with SMA.
Efficacy of risdiplam in more patients. The first oral treatment for SMA was approved by the FDA in 2020. It’s given once per day in patients with SMA of all ages and disease types. The drug increases functional SMN protein production by the SMN2 gene.
A July 2021 publication of the results of the FIREFISH study found that infants with Type I SMA treated with risdiplam for 12 months were significantly more likely to achieve motor milestones, such as sitting without support, compared with untreated infants with Type 1 SMA.15 Risdiplam is also effective in older patients with Type 2 or 3 SMA, according to results published in December from the SUNFISH clinical trial.16 Another study, RAINBOWFISH, is studying safety and efficacy at 24 months in presymptomatic infants started on treatment at up to 6 weeks of age.
The efficacy of risdiplam in previously treated patients is the subject of JEWELFISH, an ongoing study in patients 6 months to 60 years with SMA. Preliminary data presented at the 2020 Virtual SMA Research and Clinical Care Meeting suggest treatment with risdiplam led to a median two-fold increase in the amount of blood SMN protein levels after 4 weeks, which was sustained for at least 24 months.17
Combination therapy. Among the more eagerly awaited results are those from studies of combination therapies, including those that combine approved SMN up-regulators with new non–SMN-targeted therapeutics.
“We’re seeing that while these three approved therapies have dramatic results, especially for infants who are treated presymptomatically, there are still unmet medical needs in those patients, particularly for older teens and adults whose disease may have progressed before they were able to start therapy,” said Jackie Glascock, PhD, vice president of research for Cure SMA.
Of particular interest are studies of myostatin inhibitors, therapeutics that block the production of the protein myostatin. Myostatin acts on muscle cells to reduce muscle growth. Animal studies suggest that inhibiting myostatin increases muscle mass, which could be important in patients with muscle loss due to SMA.
Three experimental myostatin inhibitors are currently in clinical trials. MANATEE is a global phase 2-3 trial that aims to evaluate the safety and efficacy of the antimyostatin antibody GYM329 (RO7204239) in combination with risdiplam. SAPPHIRE is a phase 3 trial of apitegromab (SRK-015) in combination with nusinersen or risdiplam. RESILIANT is a phase 3 trial of tadefgrobep alfa in combination with other treatments.
A trial is underway to study the efficacy and safety of nusinersen in patients with persistent symptoms of SMA after treatment with the gene therapy. The phase 4 study, RESPOND, is enrolling children aged 2-36 months.
What’s needed next
Despite the advances in treatment and patient care, Dr. Brandsema, Dr. Schroth, and Dr. Glascock note that there remain unmet needs in the SMA community in a variety of areas.
Increased focus on adults with SMA. Before nusinersen, treatment of SMA mainly involved treating its symptoms. Many patients stopped seeing their neurologist, relying more heavily on pulmonary care specialists and/or primary care providers to address breathing, nutrition, and mobility problems. “Now with the approval of these treatments, they’re coming back to see their neurologists and are becoming more visible in the SMA community,” Dr. Schroth said.
Despite this re-emergence, a 2020 meta-analysis of studies on adults with SMA found a paucity of data on physical and occupational therapy, respiratory management, mental health care, and palliative care.18
“There is just so much work we need to do in the area of adult clinical care of SMA.”
Treatment algorithms. While the development of the newborn screening algorithm and revised patient care guidelines are helpful resources, clinicians still face uncertainty when choosing which therapy will work best for their patients. Treatment algorithms that help clinicians figure out what therapy or combination of therapies will offer the best outcomes for individual patients are desperately needed, Dr. Brandsema said.
“Each person’s experience of this disease is so unique to the individual based partly on their genetics and partly on the factors about what got them into care and how compliant they are with everything we’re trying to do to help them,” he said. “Biomarkers would help clinicians create personalized treatment plans for each patient.”
More basic science. While scientists have a good understanding of the SMN gene, there are many unanswered questions about the function of the SMN protein and its relationship to motor neuron loss. SMN is a ubiquitously expressed protein, and its function in other cell types is largely unknown. Despite all of the research advances, there is much basic science left to be done.
“We are strongly advocating to regulatory authorities that these aren’t cures and we need to continue to invest in the basic research,” Dr. Glascock said. “These biological questions that pertain to SMN and its function and expression really drive drug development. I really think that understanding those pathways better will lead us to more druggable targets.”
Two deaths from liver failure linked to spinal muscular atrophy drug
Two children taking the gene therapy drug onasemnogene abeparvovec (Zolgensma, Novartis) for spinal muscular atrophy (SMA) have died from acute liver failure, according to a statement issued by the drug’s manufacturer.
The patients were 4 months and 28 months of age and lived in Russia and Kazakhstan. They died 5-6 weeks after infusion with Zolgensma and approximately 1-10 days after the initiation of a corticosteroid taper.
These are the first known fatal cases of acute liver failure associated with the drug, which the company notes was a known side effect included in the product label and in a boxed warning in the United States.
“Following two recent patient fatalities, and in alignment with health authorities, we will be updating the labeling to specify that fatal acute liver failure has been reported,” the statement reads.
“While this is important safety information, it is not a new safety signal,” it adds.
Rare genetic disorder
SMA is a rare genetic disorder that affects about 1 in 10,000 newborns. Patients with SMA lack a working copy of the survival motor neuron 1 (SMN1) gene, which encodes a protein called SMN that is critical for the maintenance and function of motor neurons.
Without this protein, motor neurons eventually die, causing debilitating and progressive muscle weakness that affects the ability to walk, eat, and breathe.
Zolgensma, a one-time gene replacement therapy delivered via intravenous infusion, replaces the function of the missing or nonworking SMN1 gene with a new, working copy of the SMN1 gene.
The first gene therapy treatment for SMA, it was approved by the U.S. Food and Drug Administration in 2019 for patients with SMA up to 2 years of age. It is also the most expensive drug in the world, costing about $2.1 million for a one-time treatment.
“We have notified health authorities in all markets where Zolgensma is used, including the FDA, and are communicating to relevant healthcare professionals as an additional step in markets where this action is supported by health authorities,” the manufacturer’s statement says.
Studies have suggested that the treatment’s effects persist more than 5 years after infusion.
Clinical trials currently underway by Novartis are studying the drug’s long-term efficacy and safety and its potential use in older patients.
The company is also leading the phase 3 clinical trial STEER to test intrathecal (IT) administration of the drug in patients ages 2-18 years who have type 2 SMA.
That trial began late last year after the FDA lifted a 2-year partial hold on an earlier study. The FDA halted the STRONG trial in 2019, citing concerns from animal studies that IT administration may result in dorsal root ganglia injury. The partial hold was released last fall following positive study results in nonhuman primates.
None of the current trials will be affected by the two deaths reported, according to a Novartis spokesperson.
Kelli Whitlock Burton is a staff writer/reporter for Medscape Neurology and MDedge Neurology.
References
1. Viscidi E et al. Comparative all-cause mortality among a large population of patients with spinal muscular atrophy versus matched controls. Neurol Ther. 2022 Mar;11(1):449-457. doi: 10.1007/s40120-021-00307-7.
2. Finkel RS et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. 2014 Aug 26;83(9):810-817. doi: 10.1212/WNL.0000000000000741.
3. Klotz J et al. Advances in the therapy of spinal muscular atrophy. J Pediatr. 2021 Sep;236:13-20.e1. doi: 10.1016/j.jpeds.2021.06.033.
4. Curry M et al. Awareness screening and referral patterns among pediatricians in the United States related to early clinical features of spinal muscular atrophy (SMA). BMC Pediatr. 2021 May;21(1):236. doi: 10.1186/s12887-021-02692-2.
5. SMArt Moves. https://smartmoves.curesma.org/
6. Swoboda KJ et al. Natural history of denervation in SMA: Relation to age, SMN2 copy number, and function. Ann Neurol. 2005 May;57(5):704-12. doi: 10.1002/ana.20473.
7. Glascock J et al. Treatment algorithm for infants diagnosed with spinal muscular atrophy through newborn screening. J Neuromuscul Dis. 2018;5(2):145-158. doi: 10.3233/JND-180304.
8. Glascock J et al. Revised recommendations for the treatment of infants diagnosed with spinal muscular atrophy via newborn screening who have 4 copies of SMN2. J Neuromuscul Dis. 2020;7(2):97-100. doi: 10.3233/JND-190468.
9. Talbot K, Tizzano EF. The clinical landscape for SMA in a new therapeutic era. Gene Ther. 2017 Sep;24(9):529-533. doi: 10.1038/gt.2017.52.
10. Mercuri E et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018 Feb;28(2):103-115. doi: 10.1016/j.nmd.2017.11.005.
11. Finkel RS et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. 2018 Mar;28(3):197-207. doi: 10.1016/j.nmd.2017.11.004.
12. Pascual SI et al. Ongoing phase 2/3 DEVOTE (232SM203) randomized, controlled study to explore high-dose nusinersen in SMA: Part A interim results and Part B enrollment update. Presented at MDA Clinical and Scientific Conference 2021, Mar 15-18.
13. Finkel RS et al. Scientific rationale for a higher dose of nusinersen. Presented at 2021 Cure SMA Annual Meeting, Jun 9-11. Abstract P46.
14. Mendell JR et al. Five-year extension results of the phase 1 START trial of onasemnogene abeparvovec in spinal muscular atrophy. JAMA Neurol. 2021 Jul;78(7):834-841. doi: 10.1001/jamaneurol.2021.1272.
15. Darras BT et al. Risdiplam-treated infants with type 1 spinal muscular atrophy versus historical controls. N Engl J Med. 2021 Jul 29;385(5):427-435. doi: 10.1056/NEJMoa2102047.
16. Mercuri E et al. Safety and efficacy of once-daily risdiplam in type 2 and non-ambulant type 3 spinal muscular atrophy (SUNFISH part 2): A phase 3, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2022 Jan;21(1):42-52. doi: 10.1016/S1474-4422(21)00367-7. Erratum in: Lancet Neurol. 2022 Feb;21(2):e2. doi: 10.1016/S1474-4422(22)00006-0. Correction in: Lancet Neurol. 2022 Mar;21(3):e3. doi: 10.1016/S1474-4422(22)00038-2.
17. Genentech announces 2-year risdiplam data from SUNFISH and new data from JEWELFISH in infants, children and adults with SMA. https://www.curesma.org/genentech-risdiplam-data-conference-2020/
18. Wan HWY et al. Health, wellbeing and lived experiences of adults with SMA: a scoping systematic review. Orphanet J Rare Dis. 2020;15(1):70. doi: 10.1186/s13023-020-1339-3.
In 2016, the U.S. Food and Drug Administration approved nusinersen, the first treatment for spinal muscular atrophy (SMA). Until then, SMA had a mortality rate nearly double that of the general population.1 Two-thirds of patients were symptomatic within 6 months of birth and, in the absence of mechanical ventilation and other support, had a nearly 100% mortality rate by age 2.2
Five years later, there are three approved treatments for SMA, all of which have been shown to slow or even halt disease progression in many patients. Neurologists, whose SMA patient population once consisted almost entirely of children, are now treating more adults with the disease. Indeed, more than half of all people alive with SMA in the United States today are adults, according to Cure SMA.
“Managing SMA used to be clinic follow-ups where we were doing our best supportive care and watching people fall apart before our eyes,” said John Brandsema, MD, a physician and neuromuscular section head at the Children’s Hospital of Philadelphia. “Today, what we see in the vast majority of people is that they are either the same as they were before – which is completely against the natural history of this disease and something to be celebrated – or that people are really better with their function. It totally changes everything in the clinic.”
Among those changes are a more proactive approach to rehabilitation and an even greater emphasis on personalized medicine and multidisciplinary care. But there is also a need for updated treatment guidelines, a new classification system to measure disease severity, specific biomarkers to guide therapy choices, more data on long-term efficacy of existing therapeutics, new medications to complement those therapies, and a deeper understanding of a disease that may have treatment options but still has no cure.
Advances in early diagnosis
Patients with SMA lack a working copy of the survival motor neuron 1 (SMN1) gene, which provides instructions for producing a protein called SMN that is critical for the maintenance and function of motor neurons. Without this protein, motor neurons eventually die, causing debilitating and progressive muscle weakness that affects the ability to walk, eat, and breathe. SMA is rare, affecting about 1 in 10,000 newborns.
In approximately 96% of patients, SMA is caused by homozygous loss of the SMN1 gene. People with SMA have at least one copy of the SMN2 gene, sometimes called a “backup” gene, that also produces SMN protein. However, a single nucleotide difference between SMN2 and SMN1 causes about 90% of the protein produced by SMN2 to be truncated and less stable. Even with multiple copies of SMN2 present, as is the case with many infants with SMA, the amount of functional protein produced isn’t enough to compensate for the loss of SMN1.3
All three approved medications are SMN up-regulators and work to increase the amount of functional SMN protein. Starting these medications early, even before symptoms present, is critical to preserve motor function. Early treatment depends on early diagnosis, which became more widespread after 2018 when SMA was added to the federally Recommended Uniform Screening Panel for newborns. As of July 1, 2022, 47 states have incorporated SMA newborn screening into their state panel, ensuring that 97% of all infants born in the United States undergo SMA screening shortly after birth. Screening in the remaining states – Hawaii, Nevada, and South Carolina – and Washington, D.C. is expected by mid-2023.
SMA newborn screening is a PCR-based assay that detects homozygous SMN1 gene deletion found in about 95% of all people with SMA. The remaining 5% of cases are caused by various genetic mutations that can only be detected with gene sequencing. In these cases, and in children who don’t undergo SMA newborn screening, the disease is usually identified when symptoms are noticed by a parent, pediatrician, or primary care provider. But a study found that in 2018 only 52.7% of pediatricians correctly identified genetic testing as a requirement for a definitive diagnosis of SMA; in 2019, with a larger sample size, that number decreased to 45%.4 The lack of awareness of diagnostic requirements for SMA could contribute to delays in diagnosis, said Mary Schroth, MD, chief medical officer for Cure SMA and a coauthor of the study.
“In our world, suspicion of SMA in an infant is an emergency situation,” Dr. Schroth said. “These babies need to be referred immediately and have genetic testing so that treatment can begin as soon as possible.”
Based on the study findings, Dr. Schroth and others with Cure SMA launched a new tool in 2021 designed to help pediatricians, primary care physicians, and parents identify early signs of SMA, so that a referral to a pediatric neurologist happens quickly. Called SMArt Moves, the educational resource features videos and a checklist to help increase early detection in infants who had a negative SMA newborn screening result or did not receive SMA screening at birth.5
Who to treat, when, and with which treatment
For many patients, having multiple effective treatment options means that SMA is no longer a fatal disease in early childhood, but one that can be managed into adolescence and adulthood. The question for clinicians is, who do they treat, when, and with which treatment?
Studies have long shown that the number of copies of the backup gene that a patient has is inversely associated with disease severity.6 In 2018, a group of SMA experts published a treatment algorithm to help guide decision-making following a positive SMA newborn screening.7 The treatment guidelines were updated in 2020 based on clinical trial data for presymptomatic infants, and current recommendations include immediate treatment for infants with two to four copies of the SMN2 gene.8 For patients with only one copy of SMN2, most of whom will likely be symptomatic at birth, the guidelines recommend that treatment decisions be made jointly between the clinician and the family.7,8
Some suggest that the number of SMN2 copies a patient has should also be a factor in determining phenotype, which has started a conversation on the development of a new classification system.9 The original classification system for disease severity – Types 0-4 – was based on age of onset and degree of motor function achieved, with Type 0 developing prenatally and being the most severe and Type 4 developing in adulthood. Type 1 is the most common, affecting more than half of all people with SMA, followed by Types 2-4. In 2018, updated consensus care guidelines offered a revised classification system that better reflected disease progression in the age of therapy. The functional motor outcomes include nonsitters (historically Type I), sitters (historically Type 2/3), and walkers (historically Type 3/4).10,11 These guidelines are a start, but clinicians say more revision is needed.
“Types 1, 2, 3, 4 were based on function – getting to a certain point and then losing it, but now that we can treat this disease, people will shift categories based on therapeutic response or based on normal development that is possible now that the neurologic piece has been stabilized,” Dr. Brandsema said. “We need to completely change our thinking around all these different aspects of SMA management.”
While discussions of a new classification system for SMA are underway, another effort to update treatment recommendations is closer to completion. Led by Cure SMA, a group of about 50 physician experts in the United States and Europe who specialize in SMA are revising guidelines for diagnosis and treatment, the first time the recommendations have been updated since 2018. The updated recommendations, which should be published later this year, will focus on diagnosis and treatment considerations.
“We have three treatments that are available, and there are specific FDA indications for each of those, but it’s not totally clear just how those medications should be used or applied to different clinical situations,” said Dr. Schroth. “We’re in a rapid phase of learning right now in the SMA community, trying to understand how these treatments alter physiology and disease outcomes and how to best use the tools that we now have available to us. In parallel with clinical treatments, we have to be doing the best care we can to optimize the outcomes for those treatments.”
Research advances in 2021
Although all three drugs approved to treat SMA – nusinersen (Spinraza; Biogen), onasemnogene abeparvovec-xioi gene replacement therapy (Zolgensma; Novartis Gene Therapies), and risdiplam (Evrysdi, Genentech/Roche) – are highly effective, there are still unanswered questions and unmet needs. New research findings from 2021 focused on higher dosing, different drug-delivery methods, combination therapy, and complementary therapeutics to address SMA comorbidities.
Higher-dose nusinersen. The first drug approved to treat SMA, nusinersen is an antisense oligonucleotide approved for all ages and all SMA types. It works by altering splicing of the SMN2 gene pre-mRNA to make more complete SMN protein. Given as an intrathecal (IT) injection, four “loading doses” are administered within the first 2 months of treatment, followed by a maintenance dose every 4 months for the duration of the individual’s life.
Reports from patients of waning effects of nusinersen just prior to follow-up treatment have led some clinicians to ask if a higher dose may be needed. A study underway seeks to address that issue.
DEVOTE is a phase 2/3 trial to study the safety and efficacy of high-dose nusinersen in patients with SMA. Preliminary findings reported in 2021 found no adverse events among patients treated with 28 mg of nusinersen for 161-257 days.12 Another analysis from this trial found that higher doses are associated with greater decrease of plasma phosphorylated neurofilament heavy chain (pNF-H) levels in patients with SMA and may lead to clinically meaningful improvement in motor function beyond that observed with the approved 12 mg dose.13 The trial is ongoing.
Another trial, ASCEND, is a phase 3B study assessing higher dose nusinersen in patients previously treated with risdiplam. Recruitment for that trial began in October 2021.
Long-term efficacy and IT administration of SMA therapy. Several studies are looking at the long-term efficacy and alternate routes of administration of onasemnogene abeparvovec and other SMA therapies.
A one-time gene replacement therapy delivered via an IV infusion replaces the function of the missing or nonworking SMN1 gene with a new, working copy of the SMN1 gene. FDA approved in 2019, it is authorized for use in patients with SMA up to 2 years of age.
The latest data from an ongoing, long-term follow-up safety study of onasemnogene abeparvovec, published in May 2021, suggest that the treatment’s effects persist more than 5 years after treatment. Researchers followed 13 infants with symptomatic SMA type 1 since the beginning of the phase 1 clinical trial of the gene transfer therapy. All patients who received the therapeutic dose maintained their baseline motor function, and two of the patients actually improved without other SMN-targeted treatment. At a median 6.2 years after they received treatment, all were alive and none needed permanent ventilation.14
After a 2-year hold by the FDA, a study of IT administration of onasemnogene abeparvovec is now enrolling patients. Citing concerns from animal studies that IT administration might result in dorsal root ganglia injury, the FDA issued a partial hold on the STRONG trial in 2019. Following positive study results in nonhuman primates, the FDA announced the trial can continue. Novartis is launching a new phase 3 STEER trial to test the drug delivered intrathecally in patients aged 2-18 years with Type 2 SMA. IT administration could allow the gene therapy to be used safely and effectively in more patients with SMA.
Efficacy of risdiplam in more patients. The first oral treatment for SMA was approved by the FDA in 2020. It’s given once per day in patients with SMA of all ages and disease types. The drug increases functional SMN protein production by the SMN2 gene.
A July 2021 publication of the results of the FIREFISH study found that infants with Type I SMA treated with risdiplam for 12 months were significantly more likely to achieve motor milestones, such as sitting without support, compared with untreated infants with Type 1 SMA.15 Risdiplam is also effective in older patients with Type 2 or 3 SMA, according to results published in December from the SUNFISH clinical trial.16 Another study, RAINBOWFISH, is studying safety and efficacy at 24 months in presymptomatic infants started on treatment at up to 6 weeks of age.
The efficacy of risdiplam in previously treated patients is the subject of JEWELFISH, an ongoing study in patients 6 months to 60 years with SMA. Preliminary data presented at the 2020 Virtual SMA Research and Clinical Care Meeting suggest treatment with risdiplam led to a median two-fold increase in the amount of blood SMN protein levels after 4 weeks, which was sustained for at least 24 months.17
Combination therapy. Among the more eagerly awaited results are those from studies of combination therapies, including those that combine approved SMN up-regulators with new non–SMN-targeted therapeutics.
“We’re seeing that while these three approved therapies have dramatic results, especially for infants who are treated presymptomatically, there are still unmet medical needs in those patients, particularly for older teens and adults whose disease may have progressed before they were able to start therapy,” said Jackie Glascock, PhD, vice president of research for Cure SMA.
Of particular interest are studies of myostatin inhibitors, therapeutics that block the production of the protein myostatin. Myostatin acts on muscle cells to reduce muscle growth. Animal studies suggest that inhibiting myostatin increases muscle mass, which could be important in patients with muscle loss due to SMA.
Three experimental myostatin inhibitors are currently in clinical trials. MANATEE is a global phase 2-3 trial that aims to evaluate the safety and efficacy of the antimyostatin antibody GYM329 (RO7204239) in combination with risdiplam. SAPPHIRE is a phase 3 trial of apitegromab (SRK-015) in combination with nusinersen or risdiplam. RESILIANT is a phase 3 trial of tadefgrobep alfa in combination with other treatments.
A trial is underway to study the efficacy and safety of nusinersen in patients with persistent symptoms of SMA after treatment with the gene therapy. The phase 4 study, RESPOND, is enrolling children aged 2-36 months.
What’s needed next
Despite the advances in treatment and patient care, Dr. Brandsema, Dr. Schroth, and Dr. Glascock note that there remain unmet needs in the SMA community in a variety of areas.
Increased focus on adults with SMA. Before nusinersen, treatment of SMA mainly involved treating its symptoms. Many patients stopped seeing their neurologist, relying more heavily on pulmonary care specialists and/or primary care providers to address breathing, nutrition, and mobility problems. “Now with the approval of these treatments, they’re coming back to see their neurologists and are becoming more visible in the SMA community,” Dr. Schroth said.
Despite this re-emergence, a 2020 meta-analysis of studies on adults with SMA found a paucity of data on physical and occupational therapy, respiratory management, mental health care, and palliative care.18
“There is just so much work we need to do in the area of adult clinical care of SMA.”
Treatment algorithms. While the development of the newborn screening algorithm and revised patient care guidelines are helpful resources, clinicians still face uncertainty when choosing which therapy will work best for their patients. Treatment algorithms that help clinicians figure out what therapy or combination of therapies will offer the best outcomes for individual patients are desperately needed, Dr. Brandsema said.
“Each person’s experience of this disease is so unique to the individual based partly on their genetics and partly on the factors about what got them into care and how compliant they are with everything we’re trying to do to help them,” he said. “Biomarkers would help clinicians create personalized treatment plans for each patient.”
More basic science. While scientists have a good understanding of the SMN gene, there are many unanswered questions about the function of the SMN protein and its relationship to motor neuron loss. SMN is a ubiquitously expressed protein, and its function in other cell types is largely unknown. Despite all of the research advances, there is much basic science left to be done.
“We are strongly advocating to regulatory authorities that these aren’t cures and we need to continue to invest in the basic research,” Dr. Glascock said. “These biological questions that pertain to SMN and its function and expression really drive drug development. I really think that understanding those pathways better will lead us to more druggable targets.”
Two deaths from liver failure linked to spinal muscular atrophy drug
Two children taking the gene therapy drug onasemnogene abeparvovec (Zolgensma, Novartis) for spinal muscular atrophy (SMA) have died from acute liver failure, according to a statement issued by the drug’s manufacturer.
The patients were 4 months and 28 months of age and lived in Russia and Kazakhstan. They died 5-6 weeks after infusion with Zolgensma and approximately 1-10 days after the initiation of a corticosteroid taper.
These are the first known fatal cases of acute liver failure associated with the drug, which the company notes was a known side effect included in the product label and in a boxed warning in the United States.
“Following two recent patient fatalities, and in alignment with health authorities, we will be updating the labeling to specify that fatal acute liver failure has been reported,” the statement reads.
“While this is important safety information, it is not a new safety signal,” it adds.
Rare genetic disorder
SMA is a rare genetic disorder that affects about 1 in 10,000 newborns. Patients with SMA lack a working copy of the survival motor neuron 1 (SMN1) gene, which encodes a protein called SMN that is critical for the maintenance and function of motor neurons.
Without this protein, motor neurons eventually die, causing debilitating and progressive muscle weakness that affects the ability to walk, eat, and breathe.
Zolgensma, a one-time gene replacement therapy delivered via intravenous infusion, replaces the function of the missing or nonworking SMN1 gene with a new, working copy of the SMN1 gene.
The first gene therapy treatment for SMA, it was approved by the U.S. Food and Drug Administration in 2019 for patients with SMA up to 2 years of age. It is also the most expensive drug in the world, costing about $2.1 million for a one-time treatment.
“We have notified health authorities in all markets where Zolgensma is used, including the FDA, and are communicating to relevant healthcare professionals as an additional step in markets where this action is supported by health authorities,” the manufacturer’s statement says.
Studies have suggested that the treatment’s effects persist more than 5 years after infusion.
Clinical trials currently underway by Novartis are studying the drug’s long-term efficacy and safety and its potential use in older patients.
The company is also leading the phase 3 clinical trial STEER to test intrathecal (IT) administration of the drug in patients ages 2-18 years who have type 2 SMA.
That trial began late last year after the FDA lifted a 2-year partial hold on an earlier study. The FDA halted the STRONG trial in 2019, citing concerns from animal studies that IT administration may result in dorsal root ganglia injury. The partial hold was released last fall following positive study results in nonhuman primates.
None of the current trials will be affected by the two deaths reported, according to a Novartis spokesperson.
Kelli Whitlock Burton is a staff writer/reporter for Medscape Neurology and MDedge Neurology.
References
1. Viscidi E et al. Comparative all-cause mortality among a large population of patients with spinal muscular atrophy versus matched controls. Neurol Ther. 2022 Mar;11(1):449-457. doi: 10.1007/s40120-021-00307-7.
2. Finkel RS et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. 2014 Aug 26;83(9):810-817. doi: 10.1212/WNL.0000000000000741.
3. Klotz J et al. Advances in the therapy of spinal muscular atrophy. J Pediatr. 2021 Sep;236:13-20.e1. doi: 10.1016/j.jpeds.2021.06.033.
4. Curry M et al. Awareness screening and referral patterns among pediatricians in the United States related to early clinical features of spinal muscular atrophy (SMA). BMC Pediatr. 2021 May;21(1):236. doi: 10.1186/s12887-021-02692-2.
5. SMArt Moves. https://smartmoves.curesma.org/
6. Swoboda KJ et al. Natural history of denervation in SMA: Relation to age, SMN2 copy number, and function. Ann Neurol. 2005 May;57(5):704-12. doi: 10.1002/ana.20473.
7. Glascock J et al. Treatment algorithm for infants diagnosed with spinal muscular atrophy through newborn screening. J Neuromuscul Dis. 2018;5(2):145-158. doi: 10.3233/JND-180304.
8. Glascock J et al. Revised recommendations for the treatment of infants diagnosed with spinal muscular atrophy via newborn screening who have 4 copies of SMN2. J Neuromuscul Dis. 2020;7(2):97-100. doi: 10.3233/JND-190468.
9. Talbot K, Tizzano EF. The clinical landscape for SMA in a new therapeutic era. Gene Ther. 2017 Sep;24(9):529-533. doi: 10.1038/gt.2017.52.
10. Mercuri E et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018 Feb;28(2):103-115. doi: 10.1016/j.nmd.2017.11.005.
11. Finkel RS et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. 2018 Mar;28(3):197-207. doi: 10.1016/j.nmd.2017.11.004.
12. Pascual SI et al. Ongoing phase 2/3 DEVOTE (232SM203) randomized, controlled study to explore high-dose nusinersen in SMA: Part A interim results and Part B enrollment update. Presented at MDA Clinical and Scientific Conference 2021, Mar 15-18.
13. Finkel RS et al. Scientific rationale for a higher dose of nusinersen. Presented at 2021 Cure SMA Annual Meeting, Jun 9-11. Abstract P46.
14. Mendell JR et al. Five-year extension results of the phase 1 START trial of onasemnogene abeparvovec in spinal muscular atrophy. JAMA Neurol. 2021 Jul;78(7):834-841. doi: 10.1001/jamaneurol.2021.1272.
15. Darras BT et al. Risdiplam-treated infants with type 1 spinal muscular atrophy versus historical controls. N Engl J Med. 2021 Jul 29;385(5):427-435. doi: 10.1056/NEJMoa2102047.
16. Mercuri E et al. Safety and efficacy of once-daily risdiplam in type 2 and non-ambulant type 3 spinal muscular atrophy (SUNFISH part 2): A phase 3, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2022 Jan;21(1):42-52. doi: 10.1016/S1474-4422(21)00367-7. Erratum in: Lancet Neurol. 2022 Feb;21(2):e2. doi: 10.1016/S1474-4422(22)00006-0. Correction in: Lancet Neurol. 2022 Mar;21(3):e3. doi: 10.1016/S1474-4422(22)00038-2.
17. Genentech announces 2-year risdiplam data from SUNFISH and new data from JEWELFISH in infants, children and adults with SMA. https://www.curesma.org/genentech-risdiplam-data-conference-2020/
18. Wan HWY et al. Health, wellbeing and lived experiences of adults with SMA: a scoping systematic review. Orphanet J Rare Dis. 2020;15(1):70. doi: 10.1186/s13023-020-1339-3.
In 2016, the U.S. Food and Drug Administration approved nusinersen, the first treatment for spinal muscular atrophy (SMA). Until then, SMA had a mortality rate nearly double that of the general population.1 Two-thirds of patients were symptomatic within 6 months of birth and, in the absence of mechanical ventilation and other support, had a nearly 100% mortality rate by age 2.2
Five years later, there are three approved treatments for SMA, all of which have been shown to slow or even halt disease progression in many patients. Neurologists, whose SMA patient population once consisted almost entirely of children, are now treating more adults with the disease. Indeed, more than half of all people alive with SMA in the United States today are adults, according to Cure SMA.
“Managing SMA used to be clinic follow-ups where we were doing our best supportive care and watching people fall apart before our eyes,” said John Brandsema, MD, a physician and neuromuscular section head at the Children’s Hospital of Philadelphia. “Today, what we see in the vast majority of people is that they are either the same as they were before – which is completely against the natural history of this disease and something to be celebrated – or that people are really better with their function. It totally changes everything in the clinic.”
Among those changes are a more proactive approach to rehabilitation and an even greater emphasis on personalized medicine and multidisciplinary care. But there is also a need for updated treatment guidelines, a new classification system to measure disease severity, specific biomarkers to guide therapy choices, more data on long-term efficacy of existing therapeutics, new medications to complement those therapies, and a deeper understanding of a disease that may have treatment options but still has no cure.
Advances in early diagnosis
Patients with SMA lack a working copy of the survival motor neuron 1 (SMN1) gene, which provides instructions for producing a protein called SMN that is critical for the maintenance and function of motor neurons. Without this protein, motor neurons eventually die, causing debilitating and progressive muscle weakness that affects the ability to walk, eat, and breathe. SMA is rare, affecting about 1 in 10,000 newborns.
In approximately 96% of patients, SMA is caused by homozygous loss of the SMN1 gene. People with SMA have at least one copy of the SMN2 gene, sometimes called a “backup” gene, that also produces SMN protein. However, a single nucleotide difference between SMN2 and SMN1 causes about 90% of the protein produced by SMN2 to be truncated and less stable. Even with multiple copies of SMN2 present, as is the case with many infants with SMA, the amount of functional protein produced isn’t enough to compensate for the loss of SMN1.3
All three approved medications are SMN up-regulators and work to increase the amount of functional SMN protein. Starting these medications early, even before symptoms present, is critical to preserve motor function. Early treatment depends on early diagnosis, which became more widespread after 2018 when SMA was added to the federally Recommended Uniform Screening Panel for newborns. As of July 1, 2022, 47 states have incorporated SMA newborn screening into their state panel, ensuring that 97% of all infants born in the United States undergo SMA screening shortly after birth. Screening in the remaining states – Hawaii, Nevada, and South Carolina – and Washington, D.C. is expected by mid-2023.
SMA newborn screening is a PCR-based assay that detects homozygous SMN1 gene deletion found in about 95% of all people with SMA. The remaining 5% of cases are caused by various genetic mutations that can only be detected with gene sequencing. In these cases, and in children who don’t undergo SMA newborn screening, the disease is usually identified when symptoms are noticed by a parent, pediatrician, or primary care provider. But a study found that in 2018 only 52.7% of pediatricians correctly identified genetic testing as a requirement for a definitive diagnosis of SMA; in 2019, with a larger sample size, that number decreased to 45%.4 The lack of awareness of diagnostic requirements for SMA could contribute to delays in diagnosis, said Mary Schroth, MD, chief medical officer for Cure SMA and a coauthor of the study.
“In our world, suspicion of SMA in an infant is an emergency situation,” Dr. Schroth said. “These babies need to be referred immediately and have genetic testing so that treatment can begin as soon as possible.”
Based on the study findings, Dr. Schroth and others with Cure SMA launched a new tool in 2021 designed to help pediatricians, primary care physicians, and parents identify early signs of SMA, so that a referral to a pediatric neurologist happens quickly. Called SMArt Moves, the educational resource features videos and a checklist to help increase early detection in infants who had a negative SMA newborn screening result or did not receive SMA screening at birth.5
Who to treat, when, and with which treatment
For many patients, having multiple effective treatment options means that SMA is no longer a fatal disease in early childhood, but one that can be managed into adolescence and adulthood. The question for clinicians is, who do they treat, when, and with which treatment?
Studies have long shown that the number of copies of the backup gene that a patient has is inversely associated with disease severity.6 In 2018, a group of SMA experts published a treatment algorithm to help guide decision-making following a positive SMA newborn screening.7 The treatment guidelines were updated in 2020 based on clinical trial data for presymptomatic infants, and current recommendations include immediate treatment for infants with two to four copies of the SMN2 gene.8 For patients with only one copy of SMN2, most of whom will likely be symptomatic at birth, the guidelines recommend that treatment decisions be made jointly between the clinician and the family.7,8
Some suggest that the number of SMN2 copies a patient has should also be a factor in determining phenotype, which has started a conversation on the development of a new classification system.9 The original classification system for disease severity – Types 0-4 – was based on age of onset and degree of motor function achieved, with Type 0 developing prenatally and being the most severe and Type 4 developing in adulthood. Type 1 is the most common, affecting more than half of all people with SMA, followed by Types 2-4. In 2018, updated consensus care guidelines offered a revised classification system that better reflected disease progression in the age of therapy. The functional motor outcomes include nonsitters (historically Type I), sitters (historically Type 2/3), and walkers (historically Type 3/4).10,11 These guidelines are a start, but clinicians say more revision is needed.
“Types 1, 2, 3, 4 were based on function – getting to a certain point and then losing it, but now that we can treat this disease, people will shift categories based on therapeutic response or based on normal development that is possible now that the neurologic piece has been stabilized,” Dr. Brandsema said. “We need to completely change our thinking around all these different aspects of SMA management.”
While discussions of a new classification system for SMA are underway, another effort to update treatment recommendations is closer to completion. Led by Cure SMA, a group of about 50 physician experts in the United States and Europe who specialize in SMA are revising guidelines for diagnosis and treatment, the first time the recommendations have been updated since 2018. The updated recommendations, which should be published later this year, will focus on diagnosis and treatment considerations.
“We have three treatments that are available, and there are specific FDA indications for each of those, but it’s not totally clear just how those medications should be used or applied to different clinical situations,” said Dr. Schroth. “We’re in a rapid phase of learning right now in the SMA community, trying to understand how these treatments alter physiology and disease outcomes and how to best use the tools that we now have available to us. In parallel with clinical treatments, we have to be doing the best care we can to optimize the outcomes for those treatments.”
Research advances in 2021
Although all three drugs approved to treat SMA – nusinersen (Spinraza; Biogen), onasemnogene abeparvovec-xioi gene replacement therapy (Zolgensma; Novartis Gene Therapies), and risdiplam (Evrysdi, Genentech/Roche) – are highly effective, there are still unanswered questions and unmet needs. New research findings from 2021 focused on higher dosing, different drug-delivery methods, combination therapy, and complementary therapeutics to address SMA comorbidities.
Higher-dose nusinersen. The first drug approved to treat SMA, nusinersen is an antisense oligonucleotide approved for all ages and all SMA types. It works by altering splicing of the SMN2 gene pre-mRNA to make more complete SMN protein. Given as an intrathecal (IT) injection, four “loading doses” are administered within the first 2 months of treatment, followed by a maintenance dose every 4 months for the duration of the individual’s life.
Reports from patients of waning effects of nusinersen just prior to follow-up treatment have led some clinicians to ask if a higher dose may be needed. A study underway seeks to address that issue.
DEVOTE is a phase 2/3 trial to study the safety and efficacy of high-dose nusinersen in patients with SMA. Preliminary findings reported in 2021 found no adverse events among patients treated with 28 mg of nusinersen for 161-257 days.12 Another analysis from this trial found that higher doses are associated with greater decrease of plasma phosphorylated neurofilament heavy chain (pNF-H) levels in patients with SMA and may lead to clinically meaningful improvement in motor function beyond that observed with the approved 12 mg dose.13 The trial is ongoing.
Another trial, ASCEND, is a phase 3B study assessing higher dose nusinersen in patients previously treated with risdiplam. Recruitment for that trial began in October 2021.
Long-term efficacy and IT administration of SMA therapy. Several studies are looking at the long-term efficacy and alternate routes of administration of onasemnogene abeparvovec and other SMA therapies.
A one-time gene replacement therapy delivered via an IV infusion replaces the function of the missing or nonworking SMN1 gene with a new, working copy of the SMN1 gene. FDA approved in 2019, it is authorized for use in patients with SMA up to 2 years of age.
The latest data from an ongoing, long-term follow-up safety study of onasemnogene abeparvovec, published in May 2021, suggest that the treatment’s effects persist more than 5 years after treatment. Researchers followed 13 infants with symptomatic SMA type 1 since the beginning of the phase 1 clinical trial of the gene transfer therapy. All patients who received the therapeutic dose maintained their baseline motor function, and two of the patients actually improved without other SMN-targeted treatment. At a median 6.2 years after they received treatment, all were alive and none needed permanent ventilation.14
After a 2-year hold by the FDA, a study of IT administration of onasemnogene abeparvovec is now enrolling patients. Citing concerns from animal studies that IT administration might result in dorsal root ganglia injury, the FDA issued a partial hold on the STRONG trial in 2019. Following positive study results in nonhuman primates, the FDA announced the trial can continue. Novartis is launching a new phase 3 STEER trial to test the drug delivered intrathecally in patients aged 2-18 years with Type 2 SMA. IT administration could allow the gene therapy to be used safely and effectively in more patients with SMA.
Efficacy of risdiplam in more patients. The first oral treatment for SMA was approved by the FDA in 2020. It’s given once per day in patients with SMA of all ages and disease types. The drug increases functional SMN protein production by the SMN2 gene.
A July 2021 publication of the results of the FIREFISH study found that infants with Type I SMA treated with risdiplam for 12 months were significantly more likely to achieve motor milestones, such as sitting without support, compared with untreated infants with Type 1 SMA.15 Risdiplam is also effective in older patients with Type 2 or 3 SMA, according to results published in December from the SUNFISH clinical trial.16 Another study, RAINBOWFISH, is studying safety and efficacy at 24 months in presymptomatic infants started on treatment at up to 6 weeks of age.
The efficacy of risdiplam in previously treated patients is the subject of JEWELFISH, an ongoing study in patients 6 months to 60 years with SMA. Preliminary data presented at the 2020 Virtual SMA Research and Clinical Care Meeting suggest treatment with risdiplam led to a median two-fold increase in the amount of blood SMN protein levels after 4 weeks, which was sustained for at least 24 months.17
Combination therapy. Among the more eagerly awaited results are those from studies of combination therapies, including those that combine approved SMN up-regulators with new non–SMN-targeted therapeutics.
“We’re seeing that while these three approved therapies have dramatic results, especially for infants who are treated presymptomatically, there are still unmet medical needs in those patients, particularly for older teens and adults whose disease may have progressed before they were able to start therapy,” said Jackie Glascock, PhD, vice president of research for Cure SMA.
Of particular interest are studies of myostatin inhibitors, therapeutics that block the production of the protein myostatin. Myostatin acts on muscle cells to reduce muscle growth. Animal studies suggest that inhibiting myostatin increases muscle mass, which could be important in patients with muscle loss due to SMA.
Three experimental myostatin inhibitors are currently in clinical trials. MANATEE is a global phase 2-3 trial that aims to evaluate the safety and efficacy of the antimyostatin antibody GYM329 (RO7204239) in combination with risdiplam. SAPPHIRE is a phase 3 trial of apitegromab (SRK-015) in combination with nusinersen or risdiplam. RESILIANT is a phase 3 trial of tadefgrobep alfa in combination with other treatments.
A trial is underway to study the efficacy and safety of nusinersen in patients with persistent symptoms of SMA after treatment with the gene therapy. The phase 4 study, RESPOND, is enrolling children aged 2-36 months.
What’s needed next
Despite the advances in treatment and patient care, Dr. Brandsema, Dr. Schroth, and Dr. Glascock note that there remain unmet needs in the SMA community in a variety of areas.
Increased focus on adults with SMA. Before nusinersen, treatment of SMA mainly involved treating its symptoms. Many patients stopped seeing their neurologist, relying more heavily on pulmonary care specialists and/or primary care providers to address breathing, nutrition, and mobility problems. “Now with the approval of these treatments, they’re coming back to see their neurologists and are becoming more visible in the SMA community,” Dr. Schroth said.
Despite this re-emergence, a 2020 meta-analysis of studies on adults with SMA found a paucity of data on physical and occupational therapy, respiratory management, mental health care, and palliative care.18
“There is just so much work we need to do in the area of adult clinical care of SMA.”
Treatment algorithms. While the development of the newborn screening algorithm and revised patient care guidelines are helpful resources, clinicians still face uncertainty when choosing which therapy will work best for their patients. Treatment algorithms that help clinicians figure out what therapy or combination of therapies will offer the best outcomes for individual patients are desperately needed, Dr. Brandsema said.
“Each person’s experience of this disease is so unique to the individual based partly on their genetics and partly on the factors about what got them into care and how compliant they are with everything we’re trying to do to help them,” he said. “Biomarkers would help clinicians create personalized treatment plans for each patient.”
More basic science. While scientists have a good understanding of the SMN gene, there are many unanswered questions about the function of the SMN protein and its relationship to motor neuron loss. SMN is a ubiquitously expressed protein, and its function in other cell types is largely unknown. Despite all of the research advances, there is much basic science left to be done.
“We are strongly advocating to regulatory authorities that these aren’t cures and we need to continue to invest in the basic research,” Dr. Glascock said. “These biological questions that pertain to SMN and its function and expression really drive drug development. I really think that understanding those pathways better will lead us to more druggable targets.”
Two deaths from liver failure linked to spinal muscular atrophy drug
Two children taking the gene therapy drug onasemnogene abeparvovec (Zolgensma, Novartis) for spinal muscular atrophy (SMA) have died from acute liver failure, according to a statement issued by the drug’s manufacturer.
The patients were 4 months and 28 months of age and lived in Russia and Kazakhstan. They died 5-6 weeks after infusion with Zolgensma and approximately 1-10 days after the initiation of a corticosteroid taper.
These are the first known fatal cases of acute liver failure associated with the drug, which the company notes was a known side effect included in the product label and in a boxed warning in the United States.
“Following two recent patient fatalities, and in alignment with health authorities, we will be updating the labeling to specify that fatal acute liver failure has been reported,” the statement reads.
“While this is important safety information, it is not a new safety signal,” it adds.
Rare genetic disorder
SMA is a rare genetic disorder that affects about 1 in 10,000 newborns. Patients with SMA lack a working copy of the survival motor neuron 1 (SMN1) gene, which encodes a protein called SMN that is critical for the maintenance and function of motor neurons.
Without this protein, motor neurons eventually die, causing debilitating and progressive muscle weakness that affects the ability to walk, eat, and breathe.
Zolgensma, a one-time gene replacement therapy delivered via intravenous infusion, replaces the function of the missing or nonworking SMN1 gene with a new, working copy of the SMN1 gene.
The first gene therapy treatment for SMA, it was approved by the U.S. Food and Drug Administration in 2019 for patients with SMA up to 2 years of age. It is also the most expensive drug in the world, costing about $2.1 million for a one-time treatment.
“We have notified health authorities in all markets where Zolgensma is used, including the FDA, and are communicating to relevant healthcare professionals as an additional step in markets where this action is supported by health authorities,” the manufacturer’s statement says.
Studies have suggested that the treatment’s effects persist more than 5 years after infusion.
Clinical trials currently underway by Novartis are studying the drug’s long-term efficacy and safety and its potential use in older patients.
The company is also leading the phase 3 clinical trial STEER to test intrathecal (IT) administration of the drug in patients ages 2-18 years who have type 2 SMA.
That trial began late last year after the FDA lifted a 2-year partial hold on an earlier study. The FDA halted the STRONG trial in 2019, citing concerns from animal studies that IT administration may result in dorsal root ganglia injury. The partial hold was released last fall following positive study results in nonhuman primates.
None of the current trials will be affected by the two deaths reported, according to a Novartis spokesperson.
Kelli Whitlock Burton is a staff writer/reporter for Medscape Neurology and MDedge Neurology.
References
1. Viscidi E et al. Comparative all-cause mortality among a large population of patients with spinal muscular atrophy versus matched controls. Neurol Ther. 2022 Mar;11(1):449-457. doi: 10.1007/s40120-021-00307-7.
2. Finkel RS et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. 2014 Aug 26;83(9):810-817. doi: 10.1212/WNL.0000000000000741.
3. Klotz J et al. Advances in the therapy of spinal muscular atrophy. J Pediatr. 2021 Sep;236:13-20.e1. doi: 10.1016/j.jpeds.2021.06.033.
4. Curry M et al. Awareness screening and referral patterns among pediatricians in the United States related to early clinical features of spinal muscular atrophy (SMA). BMC Pediatr. 2021 May;21(1):236. doi: 10.1186/s12887-021-02692-2.
5. SMArt Moves. https://smartmoves.curesma.org/
6. Swoboda KJ et al. Natural history of denervation in SMA: Relation to age, SMN2 copy number, and function. Ann Neurol. 2005 May;57(5):704-12. doi: 10.1002/ana.20473.
7. Glascock J et al. Treatment algorithm for infants diagnosed with spinal muscular atrophy through newborn screening. J Neuromuscul Dis. 2018;5(2):145-158. doi: 10.3233/JND-180304.
8. Glascock J et al. Revised recommendations for the treatment of infants diagnosed with spinal muscular atrophy via newborn screening who have 4 copies of SMN2. J Neuromuscul Dis. 2020;7(2):97-100. doi: 10.3233/JND-190468.
9. Talbot K, Tizzano EF. The clinical landscape for SMA in a new therapeutic era. Gene Ther. 2017 Sep;24(9):529-533. doi: 10.1038/gt.2017.52.
10. Mercuri E et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018 Feb;28(2):103-115. doi: 10.1016/j.nmd.2017.11.005.
11. Finkel RS et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. 2018 Mar;28(3):197-207. doi: 10.1016/j.nmd.2017.11.004.
12. Pascual SI et al. Ongoing phase 2/3 DEVOTE (232SM203) randomized, controlled study to explore high-dose nusinersen in SMA: Part A interim results and Part B enrollment update. Presented at MDA Clinical and Scientific Conference 2021, Mar 15-18.
13. Finkel RS et al. Scientific rationale for a higher dose of nusinersen. Presented at 2021 Cure SMA Annual Meeting, Jun 9-11. Abstract P46.
14. Mendell JR et al. Five-year extension results of the phase 1 START trial of onasemnogene abeparvovec in spinal muscular atrophy. JAMA Neurol. 2021 Jul;78(7):834-841. doi: 10.1001/jamaneurol.2021.1272.
15. Darras BT et al. Risdiplam-treated infants with type 1 spinal muscular atrophy versus historical controls. N Engl J Med. 2021 Jul 29;385(5):427-435. doi: 10.1056/NEJMoa2102047.
16. Mercuri E et al. Safety and efficacy of once-daily risdiplam in type 2 and non-ambulant type 3 spinal muscular atrophy (SUNFISH part 2): A phase 3, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2022 Jan;21(1):42-52. doi: 10.1016/S1474-4422(21)00367-7. Erratum in: Lancet Neurol. 2022 Feb;21(2):e2. doi: 10.1016/S1474-4422(22)00006-0. Correction in: Lancet Neurol. 2022 Mar;21(3):e3. doi: 10.1016/S1474-4422(22)00038-2.
17. Genentech announces 2-year risdiplam data from SUNFISH and new data from JEWELFISH in infants, children and adults with SMA. https://www.curesma.org/genentech-risdiplam-data-conference-2020/
18. Wan HWY et al. Health, wellbeing and lived experiences of adults with SMA: a scoping systematic review. Orphanet J Rare Dis. 2020;15(1):70. doi: 10.1186/s13023-020-1339-3.
2022 Rare Neurological Disease Special Report

INTRODUCTION
Editor’s note
By Glenn S. Williams
Our 7th annual issue highlights several notable FDA approvals in 2021 and early 2022, emerging genetic therapies for monogenetic disorders, and recent advances in rare disease diagnosis and testing.
A note from NORD
By Peter L. Saltonstall
As we approach NORD’s 40th anniversary, it is astonishing to realize how far we all have come since the early 1980s, when rare disease patients and their medical providers were essentially on their own to navigate the challenging waters of rare disease diagnosis and treatment.
CLINICAL REVIEWS
Health care providers should have higher suspicion for rare diseases
By Jeff Craven
Learning to recognize when a cluster of symptoms doesn’t fit a pattern is important, as patients and their providers tend to gravitate toward diagnoses they are used to seeing, rather than suspecting a disease outside a usual pattern.
The paradox of Pompe disease
By Jennie Smith
For nearly 2 decades, patients with even the most severe genetic mutations have been surviving on therapy. But clinicians must now contend with previously unknown manifestations of this rare muscular disease.
Rett syndrome: Looking to the future and the promise of gene therapy
By Courtney S. Ambrose and Barbara J. Bailus, PhD
Like many monogenic disorders, Rett syndrome is entering an exciting stage – at which the words “treatment” and “cure” can be spoken with intent and conviction, not just hopeful optimism.
Rare disease patient advocacy groups empowered by data
By Theodore Bosworth
On the IAMRARE platform, patient advocacy organizations are trained to conduct observational research and host natural history.
Myasthenia gravis: Finding strength in treatment options
By Peter van der Eb; Scarlet Toruno, MS; and Jason Laird, DMSc, MHS, MBA, PA-C
Although the treatment of myasthenia gravis might have once been considered stagnant, newer expert consensus and novel research are generating optimism for innovative therapies.
Spinal muscular atrophy: Patient care in the age of genetically targeted therapy
By Kelli Whitlock Burton
Newly available treatments have changed the natural history of SMA. Newborn screening, updated treatment guidelines, and treatment algorithms have likewise changed what can be clinically done for patients with SMA, but still questions remain.
The broad and challenging – but promising – landscape of peripheral neuropathy
By Yun Seo Lee; Jonathan Kosacki; Kanika Bhandari, PharmD; Amanda Tran, PharmD
This review of peripheral neuropathy summarizes the more common causative entities, diagnostic tools that can potentially be employed to identify the disorder, and treatments that are in use or being tested to prevent, slow, or reverse its effects.
NORD Rare Disease Centers of Excellence: A new network seeks to break down barriers in rare disease care
By Jennie Smith
“The goal was to find places that could help with unanswered questions, whether diagnostic questions or treatment questions. To identify places where a patient could reasonably expect to go and have a deeper dive – maybe an interdisciplinary deep dive.”
Staying alert for patients with narcolepsy
By Erik Greb
The chronic neurologic disorder entails not only excessive sleepiness but also social and professional challenges.
ONLINE-ONLY CONTENT
Novel gene-based therapies for neuromuscular diseases
By Nahomi Yewhalashet, MBS, and Larry J. Davis, PharmD
Novel gene-based therapies show significant potential for transforming the treatment of neuromuscular diseases such as amyotrophic lateral sclerosis, spinal muscular atrophy, and Duchenne muscular dystrophy.
The urgent need to diagnose Sanfilippo syndrome at an early age
By Theodore Bosworth
The quest for effective treatments is dependent on enrolling more children at a very young age, before loss of milestones.
INTRODUCTION
Editor’s note
By Glenn S. Williams
Our 7th annual issue highlights several notable FDA approvals in 2021 and early 2022, emerging genetic therapies for monogenetic disorders, and recent advances in rare disease diagnosis and testing.
A note from NORD
By Peter L. Saltonstall
As we approach NORD’s 40th anniversary, it is astonishing to realize how far we all have come since the early 1980s, when rare disease patients and their medical providers were essentially on their own to navigate the challenging waters of rare disease diagnosis and treatment.
CLINICAL REVIEWS
Health care providers should have higher suspicion for rare diseases
By Jeff Craven
Learning to recognize when a cluster of symptoms doesn’t fit a pattern is important, as patients and their providers tend to gravitate toward diagnoses they are used to seeing, rather than suspecting a disease outside a usual pattern.
The paradox of Pompe disease
By Jennie Smith
For nearly 2 decades, patients with even the most severe genetic mutations have been surviving on therapy. But clinicians must now contend with previously unknown manifestations of this rare muscular disease.
Rett syndrome: Looking to the future and the promise of gene therapy
By Courtney S. Ambrose and Barbara J. Bailus, PhD
Like many monogenic disorders, Rett syndrome is entering an exciting stage – at which the words “treatment” and “cure” can be spoken with intent and conviction, not just hopeful optimism.
Rare disease patient advocacy groups empowered by data
By Theodore Bosworth
On the IAMRARE platform, patient advocacy organizations are trained to conduct observational research and host natural history.
Myasthenia gravis: Finding strength in treatment options
By Peter van der Eb; Scarlet Toruno, MS; and Jason Laird, DMSc, MHS, MBA, PA-C
Although the treatment of myasthenia gravis might have once been considered stagnant, newer expert consensus and novel research are generating optimism for innovative therapies.
Spinal muscular atrophy: Patient care in the age of genetically targeted therapy
By Kelli Whitlock Burton
Newly available treatments have changed the natural history of SMA. Newborn screening, updated treatment guidelines, and treatment algorithms have likewise changed what can be clinically done for patients with SMA, but still questions remain.
The broad and challenging – but promising – landscape of peripheral neuropathy
By Yun Seo Lee; Jonathan Kosacki; Kanika Bhandari, PharmD; Amanda Tran, PharmD
This review of peripheral neuropathy summarizes the more common causative entities, diagnostic tools that can potentially be employed to identify the disorder, and treatments that are in use or being tested to prevent, slow, or reverse its effects.
NORD Rare Disease Centers of Excellence: A new network seeks to break down barriers in rare disease care
By Jennie Smith
“The goal was to find places that could help with unanswered questions, whether diagnostic questions or treatment questions. To identify places where a patient could reasonably expect to go and have a deeper dive – maybe an interdisciplinary deep dive.”
Staying alert for patients with narcolepsy
By Erik Greb
The chronic neurologic disorder entails not only excessive sleepiness but also social and professional challenges.
ONLINE-ONLY CONTENT
Novel gene-based therapies for neuromuscular diseases
By Nahomi Yewhalashet, MBS, and Larry J. Davis, PharmD
Novel gene-based therapies show significant potential for transforming the treatment of neuromuscular diseases such as amyotrophic lateral sclerosis, spinal muscular atrophy, and Duchenne muscular dystrophy.
The urgent need to diagnose Sanfilippo syndrome at an early age
By Theodore Bosworth
The quest for effective treatments is dependent on enrolling more children at a very young age, before loss of milestones.
INTRODUCTION
Editor’s note
By Glenn S. Williams
Our 7th annual issue highlights several notable FDA approvals in 2021 and early 2022, emerging genetic therapies for monogenetic disorders, and recent advances in rare disease diagnosis and testing.
A note from NORD
By Peter L. Saltonstall
As we approach NORD’s 40th anniversary, it is astonishing to realize how far we all have come since the early 1980s, when rare disease patients and their medical providers were essentially on their own to navigate the challenging waters of rare disease diagnosis and treatment.
CLINICAL REVIEWS
Health care providers should have higher suspicion for rare diseases
By Jeff Craven
Learning to recognize when a cluster of symptoms doesn’t fit a pattern is important, as patients and their providers tend to gravitate toward diagnoses they are used to seeing, rather than suspecting a disease outside a usual pattern.
The paradox of Pompe disease
By Jennie Smith
For nearly 2 decades, patients with even the most severe genetic mutations have been surviving on therapy. But clinicians must now contend with previously unknown manifestations of this rare muscular disease.
Rett syndrome: Looking to the future and the promise of gene therapy
By Courtney S. Ambrose and Barbara J. Bailus, PhD
Like many monogenic disorders, Rett syndrome is entering an exciting stage – at which the words “treatment” and “cure” can be spoken with intent and conviction, not just hopeful optimism.
Rare disease patient advocacy groups empowered by data
By Theodore Bosworth
On the IAMRARE platform, patient advocacy organizations are trained to conduct observational research and host natural history.
Myasthenia gravis: Finding strength in treatment options
By Peter van der Eb; Scarlet Toruno, MS; and Jason Laird, DMSc, MHS, MBA, PA-C
Although the treatment of myasthenia gravis might have once been considered stagnant, newer expert consensus and novel research are generating optimism for innovative therapies.
Spinal muscular atrophy: Patient care in the age of genetically targeted therapy
By Kelli Whitlock Burton
Newly available treatments have changed the natural history of SMA. Newborn screening, updated treatment guidelines, and treatment algorithms have likewise changed what can be clinically done for patients with SMA, but still questions remain.
The broad and challenging – but promising – landscape of peripheral neuropathy
By Yun Seo Lee; Jonathan Kosacki; Kanika Bhandari, PharmD; Amanda Tran, PharmD
This review of peripheral neuropathy summarizes the more common causative entities, diagnostic tools that can potentially be employed to identify the disorder, and treatments that are in use or being tested to prevent, slow, or reverse its effects.
NORD Rare Disease Centers of Excellence: A new network seeks to break down barriers in rare disease care
By Jennie Smith
“The goal was to find places that could help with unanswered questions, whether diagnostic questions or treatment questions. To identify places where a patient could reasonably expect to go and have a deeper dive – maybe an interdisciplinary deep dive.”
Staying alert for patients with narcolepsy
By Erik Greb
The chronic neurologic disorder entails not only excessive sleepiness but also social and professional challenges.
ONLINE-ONLY CONTENT
Novel gene-based therapies for neuromuscular diseases
By Nahomi Yewhalashet, MBS, and Larry J. Davis, PharmD
Novel gene-based therapies show significant potential for transforming the treatment of neuromuscular diseases such as amyotrophic lateral sclerosis, spinal muscular atrophy, and Duchenne muscular dystrophy.
The urgent need to diagnose Sanfilippo syndrome at an early age
By Theodore Bosworth
The quest for effective treatments is dependent on enrolling more children at a very young age, before loss of milestones.
Rare disease patient advocacy groups empowered by data
With the goal of advancing treatment of rare neurological diseases – or rare diseases of any type – the National Organization for Rare Disorders (NORD) has launched innovative new research initiatives in recent years to help patient advocacy organizations develop a precious asset: data to support better understanding of diseases and research that might lead to life-altering diagnostics or treatments.

“Most rare diseases still don’t have approved therapies, and the problem is often a lack of the basic information needed to advance research,” explained Aliza Fink, DSc, the director of research programs at NORD. “Our goal is to help patient organizations play a key role in the collection, analysis, and sharing of data to support better understanding of how a disease presents, its natural history, the types and severity of symptoms, and other unanswered questions.”
Over the past 2 decades, the Internet, social media, and other communications resources have provided patient organizations with unprecedented reach. As a result, these organizations are in a unique position to connect patients and caregivers around the world – those dealing with even the rarest of rare diseases – and become a repository of information on the disease and the patient experience.
Since the late 1980s, NORD has had a research grants program, and the grants this program provides to academic researchers have led to numerous significant discoveries and publications, as well as to two products that ultimately were approved by FDA. More recently, however, NORD’s research programs have been expanded to include an initiative known as IAMRARE, in which patient advocacy organizations are trained to conduct observational research and host natural history studies and registries on a platform developed by NORD.
“We work with the patient groups to determine what types of data would be most important to drive research, help develop the methodology for data collection, and advise them on protocols for supporting the quality and integrity of the data,” Dr. Fink said. “By systematically collecting data from the patients and families they serve, these groups are in a position to contribute enormously to understanding the disease and advancing research.”
NORD also helps with the practical aspects of conducting research of sufficient quality to be publishable, such as providing groups with guidelines and best practices for developing medical advisory committees, creating templates and materials to streamline their project’s submission to institutional review boards, ensuring data security and privacy in accordance with Health Insurance Portability and Accountability Act criteria, and developing other expected standards for data collection and analysis.
Unlike even academic medical centers with an interest in a given rare disease, leading patient advocacy groups for these specific disorders have unmatched access to affected patients and families. This includes patients being managed in diverse settings or those not yet receiving care at all. By harnessing this patient population to record the signs, symptoms, disease course, and other information, the patient advocacy groups can contribute greatly to the pool of available data and ultimately what is known about the disease.
Data empowers research
While NORD helps groups through the IAMRARE program to become research-ready and guides them in developing research protocols and goals, the data are ultimately owned by the patient advocacy groups themselves. This helps to ensure that the patient voice is heard. By controlling data collection and dissemination, the advocacy groups can take a leading role in defining the goals of research, including what outcome measures are important to them and what they agree are the most promising avenues for research to achieve those goals.
“By collecting the data to understand the disease, it sets the stage for the next steps in research,” explained Debbie Drell, the director of membership for NORD. She noted that IAMRARE has grown steadily since its inception in 2014 and that there are now close to 40 advocacy groups participating.
The value of this initiative is not difficult to grasp. Even though direct participation in research was not generally part of the agenda for some advocacy groups when IAMRARE was conceived, Ms. Drell said that this initiative is a compelling perk of becoming involved with NORD. Groups that elect to become research-ready in order to participate in IAMRARE fall into a category of membership that requires specific organizational structures – such as a medical advisory board – and NORD provides templates and guidance to help them meet these qualifications to successfully become research-ready.
Collaboration leads to progress
NORD was founded by an ad hoc committee of patient organizations that played a key role in enactment of the Orphan Drug Act nearly 40 years ago. Shortly after the Orphan Drug Act was passed by Congress and signed into law by President Ronald Reagan in 1983, the ad hoc committee formally united to create NORD to continue the momentum of this initial collaboration and support the rare disease community. According to Mary Dunkle, a senior advisor at NORD, passage of the Orphan Drug Act, which is widely considered a major driver of progress in development of treatments for rare diseases, made the advantages of their cooperation clear.
“The groups had so many issues in common across the spectrum of diseases that they decided to continue their collaboration,” she explained. ”They realized that, while each disease is rare, the challenges they present to patients, families, clinicians, and researchers have many similarities.”
The definition of rare disease, according to the National Institutes of Health, is a disorder that affects fewer than 200,000 people in the United States. More than 7,000 such disorders have been identified. Approximately one-third of rare diseases are neurological. Whether neurological or affecting different or multiple organ systems, most – perhaps 75%-80% – involve a genetic component, according to Ms. Dunkle.
Research reaps rewards
Altogether, today there are more than 1,000 patient organizations that provide various types of support and services for patients and caregivers affected by rare diseases. Approximately one-third of these organizations are members of NORD. For organizations that don’t yet meet the membership criteria or for other reasons have not yet formally joined NORD, there are still many opportunities to get involved and to learn best practices to strengthen their governance, infrastructure, and capacity to support their members.
Of these, the IAMRARE program is one of the best examples of ways to get involved. Beyond the many other ways these groups help patients and families cope with challenging diseases, participation in research takes rare disorder advocacy to a different level. Objective data can attract the attention of those with the resources to further study the disease, while also giving advocacy groups a seat at the table when researchers or industry become interested.
“Why create a registry? It removes competition between academic centers or industry working on their own. It creates one central source for data-sharing, and the advantage is that advocacy groups have a trusted relationship with the patient community because they are not-for-profit, community-run, and patient-driven,” Ms. Drell explained.
The registry platforms developed for IAMRARE are customizable. With guidance from NORD, the advocacy groups themselves decide what data to collect and what questions they wish to answer, according to Dr. Fink. Once the registries are created, patients and caregivers participate by responding to survey questions on disease onset, progression over time, types and severity of symptoms, and other topics. The data can be de-identified for research purposes. The advocacy groups decide how and when to share the data, including whether to publish findings.
“Some of the groups have been very successful in getting the data published and leveraging their results to drive research forward, but there is variability in the extent of dissemination across the groups,” said Dr. Fink. She noted that many of the registries that NORD has helped set up involve groups whose officers have had little or no prior research experience.
“We have advocacy groups that have had biomedical researchers on staff and other groups that are coming to research completely new,” Dr. Fink said. In trying to get them up to speed on quality data collection, “We try to meet them where they are,” she added, indicating that leading groups to a research-ready status is not just about logistics but can sometimes involve an organizational reorientation.
The examples of peer-reviewed publications that can be directly traced to IAMRARE registries are growing. One example is a registry on Prader-Willi syndrome, which is a complex neurodevelopmental disorder characterized by failure to thrive and by multiple endocrine abnormalities. The registry was developed in NORD’s IAMRARE program by the Foundation for Prader-Willi Research, a nonprofit created in 2003 by parents of children with this disorder.
By 2019, when the first data from the Global Prader-Willi Syndrome Registry were published, they drew from 23,550 surveys completed for 1,696 separate cases of the disorder in 37 countries. The surveys provided some preliminary findings on demographics and on the genetic subtypes most commonly encountered, as well as simply proof that the registry was viable. From its inception in 2015, a significant proportion of the Prader-Willi population in the United States had been enrolled, according to the study authors. With time, the serial accumulation of more data on more cases will be invaluable for documenting disease characteristics. It will be a constantly maturing resource even after fundamental questions on disease impact and prognosis are addressed.
Data accumulation
Only about 10% of rare diseases currently have approved treatments, but there is widespread belief in the rare community that collecting and analyzing the data that can promote understanding of the biology of the disease and identify therapeutic targets could accelerate the development of treatments for diseases that currently have none.
Therefore, data accumulation has become central to the mission of NORD. In addition to IAMRARE, the organization has embarked on several other important initiatives in data accumulation for rare diseases. One is the Rare Disease Cures Accelerator – Data and Analytics Platform (RDCA-DAP), an initiative in which NORD is partnering with the Critical Path Institute. The goal of this program is to gather disparate pools of existing data in a standardized format to increase their power.
“With funding from the Food and Drug Administration, we have helped to support this platform, which is designed specifically to provide a centralized structure for combining and sharing of data,” according to Dr. Fink. In RDCA-DAP, patient-level data is being assembled from a variety of resources, including academic centers, industry, registries, observational studies, and clinical trials. The program was launched in September 2021. In some cases, gaining access to data includes resolving privacy issues or addressing the proprietary concerns of those who currently have the data, but the value of the combined data is a compelling argument for participation.
“What we are trying to do is pull together the data from their current silos into one platform, and then make it generally available,” said Dr. Fink. As with IAMRARE, RDCA-DAP offers enormous potential.
“The primary challenge for those studying rare diseases is the small numbers of patients. Randomized clinical trials for some of these diseases are simply not feasible because there are not enough subjects to power two study arms,” said Dr. Fink in explaining why NORD has turned to novel strategies for data generation. One strategy for maximizing the potential value of data from these small populations of patients is data-sharing. For RDCA-DAP, data access will be open to all stakeholders after scientific review and approval.
“Anyone can get an account and request data from the platform,” said Dr. Fink, who expects this to spur more and novel types of research in rare disorders.
Another example of recent NORD initiatives to advance research and understanding of rare diseases is a study of metachromatic leukodystrophy (MLD) that is now enrolling patients, which also represents a partnership with the FDA. For this study, which is known as the HOME study, NORD hosts a platform where patients and caregivers enter data to capture the natural history of this disease. All MLD patients, even if they are already participating in a clinical trial or another registry, are invited. As with the IAMRARE registries, surveys capture patient or caregiver responses entered from a computer or smart device.
“We have always believed that the fact that so many rare diseases don’t have treatments or are not even being studied by researchers doesn’t reflect a lack of interest among academic or industry researchers. Rather, it reflects a lack of data to support research and to provide a fundamental understanding of the disease,” Dr. Fink said. “If NORD’s expanded research programs can draw the patient community together to provide that crucially needed data, we will have provided an important and essential service to patients, patient organizations, and researchers alike.”
Theodore Bosworth is a freelance journalist and editor specializing in medicine and health.
With the goal of advancing treatment of rare neurological diseases – or rare diseases of any type – the National Organization for Rare Disorders (NORD) has launched innovative new research initiatives in recent years to help patient advocacy organizations develop a precious asset: data to support better understanding of diseases and research that might lead to life-altering diagnostics or treatments.
“Most rare diseases still don’t have approved therapies, and the problem is often a lack of the basic information needed to advance research,” explained Aliza Fink, DSc, the director of research programs at NORD. “Our goal is to help patient organizations play a key role in the collection, analysis, and sharing of data to support better understanding of how a disease presents, its natural history, the types and severity of symptoms, and other unanswered questions.”
Over the past 2 decades, the Internet, social media, and other communications resources have provided patient organizations with unprecedented reach. As a result, these organizations are in a unique position to connect patients and caregivers around the world – those dealing with even the rarest of rare diseases – and become a repository of information on the disease and the patient experience.
Since the late 1980s, NORD has had a research grants program, and the grants this program provides to academic researchers have led to numerous significant discoveries and publications, as well as to two products that ultimately were approved by FDA. More recently, however, NORD’s research programs have been expanded to include an initiative known as IAMRARE, in which patient advocacy organizations are trained to conduct observational research and host natural history studies and registries on a platform developed by NORD.
“We work with the patient groups to determine what types of data would be most important to drive research, help develop the methodology for data collection, and advise them on protocols for supporting the quality and integrity of the data,” Dr. Fink said. “By systematically collecting data from the patients and families they serve, these groups are in a position to contribute enormously to understanding the disease and advancing research.”
NORD also helps with the practical aspects of conducting research of sufficient quality to be publishable, such as providing groups with guidelines and best practices for developing medical advisory committees, creating templates and materials to streamline their project’s submission to institutional review boards, ensuring data security and privacy in accordance with Health Insurance Portability and Accountability Act criteria, and developing other expected standards for data collection and analysis.
Unlike even academic medical centers with an interest in a given rare disease, leading patient advocacy groups for these specific disorders have unmatched access to affected patients and families. This includes patients being managed in diverse settings or those not yet receiving care at all. By harnessing this patient population to record the signs, symptoms, disease course, and other information, the patient advocacy groups can contribute greatly to the pool of available data and ultimately what is known about the disease.
Data empowers research
While NORD helps groups through the IAMRARE program to become research-ready and guides them in developing research protocols and goals, the data are ultimately owned by the patient advocacy groups themselves. This helps to ensure that the patient voice is heard. By controlling data collection and dissemination, the advocacy groups can take a leading role in defining the goals of research, including what outcome measures are important to them and what they agree are the most promising avenues for research to achieve those goals.
“By collecting the data to understand the disease, it sets the stage for the next steps in research,” explained Debbie Drell, the director of membership for NORD. She noted that IAMRARE has grown steadily since its inception in 2014 and that there are now close to 40 advocacy groups participating.
The value of this initiative is not difficult to grasp. Even though direct participation in research was not generally part of the agenda for some advocacy groups when IAMRARE was conceived, Ms. Drell said that this initiative is a compelling perk of becoming involved with NORD. Groups that elect to become research-ready in order to participate in IAMRARE fall into a category of membership that requires specific organizational structures – such as a medical advisory board – and NORD provides templates and guidance to help them meet these qualifications to successfully become research-ready.
Collaboration leads to progress
NORD was founded by an ad hoc committee of patient organizations that played a key role in enactment of the Orphan Drug Act nearly 40 years ago. Shortly after the Orphan Drug Act was passed by Congress and signed into law by President Ronald Reagan in 1983, the ad hoc committee formally united to create NORD to continue the momentum of this initial collaboration and support the rare disease community. According to Mary Dunkle, a senior advisor at NORD, passage of the Orphan Drug Act, which is widely considered a major driver of progress in development of treatments for rare diseases, made the advantages of their cooperation clear.
“The groups had so many issues in common across the spectrum of diseases that they decided to continue their collaboration,” she explained. ”They realized that, while each disease is rare, the challenges they present to patients, families, clinicians, and researchers have many similarities.”
The definition of rare disease, according to the National Institutes of Health, is a disorder that affects fewer than 200,000 people in the United States. More than 7,000 such disorders have been identified. Approximately one-third of rare diseases are neurological. Whether neurological or affecting different or multiple organ systems, most – perhaps 75%-80% – involve a genetic component, according to Ms. Dunkle.
Research reaps rewards
Altogether, today there are more than 1,000 patient organizations that provide various types of support and services for patients and caregivers affected by rare diseases. Approximately one-third of these organizations are members of NORD. For organizations that don’t yet meet the membership criteria or for other reasons have not yet formally joined NORD, there are still many opportunities to get involved and to learn best practices to strengthen their governance, infrastructure, and capacity to support their members.
Of these, the IAMRARE program is one of the best examples of ways to get involved. Beyond the many other ways these groups help patients and families cope with challenging diseases, participation in research takes rare disorder advocacy to a different level. Objective data can attract the attention of those with the resources to further study the disease, while also giving advocacy groups a seat at the table when researchers or industry become interested.
“Why create a registry? It removes competition between academic centers or industry working on their own. It creates one central source for data-sharing, and the advantage is that advocacy groups have a trusted relationship with the patient community because they are not-for-profit, community-run, and patient-driven,” Ms. Drell explained.
The registry platforms developed for IAMRARE are customizable. With guidance from NORD, the advocacy groups themselves decide what data to collect and what questions they wish to answer, according to Dr. Fink. Once the registries are created, patients and caregivers participate by responding to survey questions on disease onset, progression over time, types and severity of symptoms, and other topics. The data can be de-identified for research purposes. The advocacy groups decide how and when to share the data, including whether to publish findings.
“Some of the groups have been very successful in getting the data published and leveraging their results to drive research forward, but there is variability in the extent of dissemination across the groups,” said Dr. Fink. She noted that many of the registries that NORD has helped set up involve groups whose officers have had little or no prior research experience.
“We have advocacy groups that have had biomedical researchers on staff and other groups that are coming to research completely new,” Dr. Fink said. In trying to get them up to speed on quality data collection, “We try to meet them where they are,” she added, indicating that leading groups to a research-ready status is not just about logistics but can sometimes involve an organizational reorientation.
The examples of peer-reviewed publications that can be directly traced to IAMRARE registries are growing. One example is a registry on Prader-Willi syndrome, which is a complex neurodevelopmental disorder characterized by failure to thrive and by multiple endocrine abnormalities. The registry was developed in NORD’s IAMRARE program by the Foundation for Prader-Willi Research, a nonprofit created in 2003 by parents of children with this disorder.
By 2019, when the first data from the Global Prader-Willi Syndrome Registry were published, they drew from 23,550 surveys completed for 1,696 separate cases of the disorder in 37 countries. The surveys provided some preliminary findings on demographics and on the genetic subtypes most commonly encountered, as well as simply proof that the registry was viable. From its inception in 2015, a significant proportion of the Prader-Willi population in the United States had been enrolled, according to the study authors. With time, the serial accumulation of more data on more cases will be invaluable for documenting disease characteristics. It will be a constantly maturing resource even after fundamental questions on disease impact and prognosis are addressed.
Data accumulation
Only about 10% of rare diseases currently have approved treatments, but there is widespread belief in the rare community that collecting and analyzing the data that can promote understanding of the biology of the disease and identify therapeutic targets could accelerate the development of treatments for diseases that currently have none.
Therefore, data accumulation has become central to the mission of NORD. In addition to IAMRARE, the organization has embarked on several other important initiatives in data accumulation for rare diseases. One is the Rare Disease Cures Accelerator – Data and Analytics Platform (RDCA-DAP), an initiative in which NORD is partnering with the Critical Path Institute. The goal of this program is to gather disparate pools of existing data in a standardized format to increase their power.
“With funding from the Food and Drug Administration, we have helped to support this platform, which is designed specifically to provide a centralized structure for combining and sharing of data,” according to Dr. Fink. In RDCA-DAP, patient-level data is being assembled from a variety of resources, including academic centers, industry, registries, observational studies, and clinical trials. The program was launched in September 2021. In some cases, gaining access to data includes resolving privacy issues or addressing the proprietary concerns of those who currently have the data, but the value of the combined data is a compelling argument for participation.
“What we are trying to do is pull together the data from their current silos into one platform, and then make it generally available,” said Dr. Fink. As with IAMRARE, RDCA-DAP offers enormous potential.
“The primary challenge for those studying rare diseases is the small numbers of patients. Randomized clinical trials for some of these diseases are simply not feasible because there are not enough subjects to power two study arms,” said Dr. Fink in explaining why NORD has turned to novel strategies for data generation. One strategy for maximizing the potential value of data from these small populations of patients is data-sharing. For RDCA-DAP, data access will be open to all stakeholders after scientific review and approval.
“Anyone can get an account and request data from the platform,” said Dr. Fink, who expects this to spur more and novel types of research in rare disorders.
Another example of recent NORD initiatives to advance research and understanding of rare diseases is a study of metachromatic leukodystrophy (MLD) that is now enrolling patients, which also represents a partnership with the FDA. For this study, which is known as the HOME study, NORD hosts a platform where patients and caregivers enter data to capture the natural history of this disease. All MLD patients, even if they are already participating in a clinical trial or another registry, are invited. As with the IAMRARE registries, surveys capture patient or caregiver responses entered from a computer or smart device.
“We have always believed that the fact that so many rare diseases don’t have treatments or are not even being studied by researchers doesn’t reflect a lack of interest among academic or industry researchers. Rather, it reflects a lack of data to support research and to provide a fundamental understanding of the disease,” Dr. Fink said. “If NORD’s expanded research programs can draw the patient community together to provide that crucially needed data, we will have provided an important and essential service to patients, patient organizations, and researchers alike.”
Theodore Bosworth is a freelance journalist and editor specializing in medicine and health.
With the goal of advancing treatment of rare neurological diseases – or rare diseases of any type – the National Organization for Rare Disorders (NORD) has launched innovative new research initiatives in recent years to help patient advocacy organizations develop a precious asset: data to support better understanding of diseases and research that might lead to life-altering diagnostics or treatments.
“Most rare diseases still don’t have approved therapies, and the problem is often a lack of the basic information needed to advance research,” explained Aliza Fink, DSc, the director of research programs at NORD. “Our goal is to help patient organizations play a key role in the collection, analysis, and sharing of data to support better understanding of how a disease presents, its natural history, the types and severity of symptoms, and other unanswered questions.”
Over the past 2 decades, the Internet, social media, and other communications resources have provided patient organizations with unprecedented reach. As a result, these organizations are in a unique position to connect patients and caregivers around the world – those dealing with even the rarest of rare diseases – and become a repository of information on the disease and the patient experience.
Since the late 1980s, NORD has had a research grants program, and the grants this program provides to academic researchers have led to numerous significant discoveries and publications, as well as to two products that ultimately were approved by FDA. More recently, however, NORD’s research programs have been expanded to include an initiative known as IAMRARE, in which patient advocacy organizations are trained to conduct observational research and host natural history studies and registries on a platform developed by NORD.
“We work with the patient groups to determine what types of data would be most important to drive research, help develop the methodology for data collection, and advise them on protocols for supporting the quality and integrity of the data,” Dr. Fink said. “By systematically collecting data from the patients and families they serve, these groups are in a position to contribute enormously to understanding the disease and advancing research.”
NORD also helps with the practical aspects of conducting research of sufficient quality to be publishable, such as providing groups with guidelines and best practices for developing medical advisory committees, creating templates and materials to streamline their project’s submission to institutional review boards, ensuring data security and privacy in accordance with Health Insurance Portability and Accountability Act criteria, and developing other expected standards for data collection and analysis.
Unlike even academic medical centers with an interest in a given rare disease, leading patient advocacy groups for these specific disorders have unmatched access to affected patients and families. This includes patients being managed in diverse settings or those not yet receiving care at all. By harnessing this patient population to record the signs, symptoms, disease course, and other information, the patient advocacy groups can contribute greatly to the pool of available data and ultimately what is known about the disease.
Data empowers research
While NORD helps groups through the IAMRARE program to become research-ready and guides them in developing research protocols and goals, the data are ultimately owned by the patient advocacy groups themselves. This helps to ensure that the patient voice is heard. By controlling data collection and dissemination, the advocacy groups can take a leading role in defining the goals of research, including what outcome measures are important to them and what they agree are the most promising avenues for research to achieve those goals.
“By collecting the data to understand the disease, it sets the stage for the next steps in research,” explained Debbie Drell, the director of membership for NORD. She noted that IAMRARE has grown steadily since its inception in 2014 and that there are now close to 40 advocacy groups participating.
The value of this initiative is not difficult to grasp. Even though direct participation in research was not generally part of the agenda for some advocacy groups when IAMRARE was conceived, Ms. Drell said that this initiative is a compelling perk of becoming involved with NORD. Groups that elect to become research-ready in order to participate in IAMRARE fall into a category of membership that requires specific organizational structures – such as a medical advisory board – and NORD provides templates and guidance to help them meet these qualifications to successfully become research-ready.
Collaboration leads to progress
NORD was founded by an ad hoc committee of patient organizations that played a key role in enactment of the Orphan Drug Act nearly 40 years ago. Shortly after the Orphan Drug Act was passed by Congress and signed into law by President Ronald Reagan in 1983, the ad hoc committee formally united to create NORD to continue the momentum of this initial collaboration and support the rare disease community. According to Mary Dunkle, a senior advisor at NORD, passage of the Orphan Drug Act, which is widely considered a major driver of progress in development of treatments for rare diseases, made the advantages of their cooperation clear.
“The groups had so many issues in common across the spectrum of diseases that they decided to continue their collaboration,” she explained. ”They realized that, while each disease is rare, the challenges they present to patients, families, clinicians, and researchers have many similarities.”
The definition of rare disease, according to the National Institutes of Health, is a disorder that affects fewer than 200,000 people in the United States. More than 7,000 such disorders have been identified. Approximately one-third of rare diseases are neurological. Whether neurological or affecting different or multiple organ systems, most – perhaps 75%-80% – involve a genetic component, according to Ms. Dunkle.
Research reaps rewards
Altogether, today there are more than 1,000 patient organizations that provide various types of support and services for patients and caregivers affected by rare diseases. Approximately one-third of these organizations are members of NORD. For organizations that don’t yet meet the membership criteria or for other reasons have not yet formally joined NORD, there are still many opportunities to get involved and to learn best practices to strengthen their governance, infrastructure, and capacity to support their members.
Of these, the IAMRARE program is one of the best examples of ways to get involved. Beyond the many other ways these groups help patients and families cope with challenging diseases, participation in research takes rare disorder advocacy to a different level. Objective data can attract the attention of those with the resources to further study the disease, while also giving advocacy groups a seat at the table when researchers or industry become interested.
“Why create a registry? It removes competition between academic centers or industry working on their own. It creates one central source for data-sharing, and the advantage is that advocacy groups have a trusted relationship with the patient community because they are not-for-profit, community-run, and patient-driven,” Ms. Drell explained.
The registry platforms developed for IAMRARE are customizable. With guidance from NORD, the advocacy groups themselves decide what data to collect and what questions they wish to answer, according to Dr. Fink. Once the registries are created, patients and caregivers participate by responding to survey questions on disease onset, progression over time, types and severity of symptoms, and other topics. The data can be de-identified for research purposes. The advocacy groups decide how and when to share the data, including whether to publish findings.
“Some of the groups have been very successful in getting the data published and leveraging their results to drive research forward, but there is variability in the extent of dissemination across the groups,” said Dr. Fink. She noted that many of the registries that NORD has helped set up involve groups whose officers have had little or no prior research experience.
“We have advocacy groups that have had biomedical researchers on staff and other groups that are coming to research completely new,” Dr. Fink said. In trying to get them up to speed on quality data collection, “We try to meet them where they are,” she added, indicating that leading groups to a research-ready status is not just about logistics but can sometimes involve an organizational reorientation.
The examples of peer-reviewed publications that can be directly traced to IAMRARE registries are growing. One example is a registry on Prader-Willi syndrome, which is a complex neurodevelopmental disorder characterized by failure to thrive and by multiple endocrine abnormalities. The registry was developed in NORD’s IAMRARE program by the Foundation for Prader-Willi Research, a nonprofit created in 2003 by parents of children with this disorder.
By 2019, when the first data from the Global Prader-Willi Syndrome Registry were published, they drew from 23,550 surveys completed for 1,696 separate cases of the disorder in 37 countries. The surveys provided some preliminary findings on demographics and on the genetic subtypes most commonly encountered, as well as simply proof that the registry was viable. From its inception in 2015, a significant proportion of the Prader-Willi population in the United States had been enrolled, according to the study authors. With time, the serial accumulation of more data on more cases will be invaluable for documenting disease characteristics. It will be a constantly maturing resource even after fundamental questions on disease impact and prognosis are addressed.
Data accumulation
Only about 10% of rare diseases currently have approved treatments, but there is widespread belief in the rare community that collecting and analyzing the data that can promote understanding of the biology of the disease and identify therapeutic targets could accelerate the development of treatments for diseases that currently have none.
Therefore, data accumulation has become central to the mission of NORD. In addition to IAMRARE, the organization has embarked on several other important initiatives in data accumulation for rare diseases. One is the Rare Disease Cures Accelerator – Data and Analytics Platform (RDCA-DAP), an initiative in which NORD is partnering with the Critical Path Institute. The goal of this program is to gather disparate pools of existing data in a standardized format to increase their power.
“With funding from the Food and Drug Administration, we have helped to support this platform, which is designed specifically to provide a centralized structure for combining and sharing of data,” according to Dr. Fink. In RDCA-DAP, patient-level data is being assembled from a variety of resources, including academic centers, industry, registries, observational studies, and clinical trials. The program was launched in September 2021. In some cases, gaining access to data includes resolving privacy issues or addressing the proprietary concerns of those who currently have the data, but the value of the combined data is a compelling argument for participation.
“What we are trying to do is pull together the data from their current silos into one platform, and then make it generally available,” said Dr. Fink. As with IAMRARE, RDCA-DAP offers enormous potential.
“The primary challenge for those studying rare diseases is the small numbers of patients. Randomized clinical trials for some of these diseases are simply not feasible because there are not enough subjects to power two study arms,” said Dr. Fink in explaining why NORD has turned to novel strategies for data generation. One strategy for maximizing the potential value of data from these small populations of patients is data-sharing. For RDCA-DAP, data access will be open to all stakeholders after scientific review and approval.
“Anyone can get an account and request data from the platform,” said Dr. Fink, who expects this to spur more and novel types of research in rare disorders.
Another example of recent NORD initiatives to advance research and understanding of rare diseases is a study of metachromatic leukodystrophy (MLD) that is now enrolling patients, which also represents a partnership with the FDA. For this study, which is known as the HOME study, NORD hosts a platform where patients and caregivers enter data to capture the natural history of this disease. All MLD patients, even if they are already participating in a clinical trial or another registry, are invited. As with the IAMRARE registries, surveys capture patient or caregiver responses entered from a computer or smart device.
“We have always believed that the fact that so many rare diseases don’t have treatments or are not even being studied by researchers doesn’t reflect a lack of interest among academic or industry researchers. Rather, it reflects a lack of data to support research and to provide a fundamental understanding of the disease,” Dr. Fink said. “If NORD’s expanded research programs can draw the patient community together to provide that crucially needed data, we will have provided an important and essential service to patients, patient organizations, and researchers alike.”
Theodore Bosworth is a freelance journalist and editor specializing in medicine and health.
Rett syndrome: Looking to the future and the promise of gene therapy
The dream of curing genetic disorders has been a persistent but elusive goal, even before the human genome was mapped. Once mapping of the human genome was complete in 2001, an entirely new avenue of potential treatments and cures for genetic diseases and disorders was opened.1,2

The disorders best suited for targeted gene therapy are monogenic; however, tools and delivery methods for editing the human genome were limited and difficult to apply, until the advent of the CRISPR system in 2012.3 CRISPR (an acronym of clustered regularly interspaced short palindromic repeats) has changed the way in which gene therapy strategies are pursued, with dozens of companies leveraging a variety of platforms to create potentially life-changing therapies for devastating rare diseases and disorders.
One of the rare monogenic disorders that is embracing multiple gene therapy approaches is Rett syndrome, a rare, debilitating neurodevelopmental disorder. In this review, we explore the molecular cause of Rett syndrome, disease presentation, current treatments, ongoing clinical trials, and therapies that are on the horizon.
Underlying molecular cause
Rett syndrome is caused by mutations in, or the absence of, the MECP2 gene, which produces methyl-CpG binding protein 2 (MECP2). The syndrome was first described clinically in 1954 by the Austrian physician Andreas Rett; it would take until 1982 before the disorder was officially named, eponymously, in a seminal paper by Hagberg.4 After Hagberg’s characterization, Rett syndrome became the predominant global clinical diagnosis identified among cognitively impaired females, with an incidence of 1 in every 10,000 to 15,000.4

In 1999, mutations in, and deletions of, MECP2 were identified as the cause of Rett syndrome.4,5 MECP2 is located on the X chromosome, in the Xq28 region, making Rett syndrome an X-linked dominant disorder.6 Rett syndrome is seen predominantly in females who are mosaic for mutant or deleted MECP2. Random X chromosome inactivation results in some cells expressing the mutant MECP2 allele and other cells expressing the normal functioning MECP2 allele; the percentage of cells expressing the normal allele correlates with the degree of syndrome severity.7-9
The incidence of Rett syndrome is much lower in males, in whom the syndrome was originally thought to be lethal; many observed male cases are either mosaic or occur in XXY males.10,11
Approximately 95% of cases of Rett syndrome are due to de novo mutations in MECP2, with a handful of specific mutations and large deletions accounting for more than 85% of cases.12 The fact that Rett syndrome is monogenic and most cases are caused by, in total, only a handful of mutations or deletions makes the syndrome a promising candidate for gene therapy.
At the molecular level, it has been observed that the MECP2 mutations of Rett syndrome lead to loss of gene function, thus disrupting the ability of the MECP2 nuclear protein to regulate global gene transcription through its binding to methylated DNA sites.12 A large percentage of these missense and nonsense mutations lead to a truncated or nonfunctional protein.12
One of the ways in which MECP2 regulates transcription is as a component of heterochromatin condensates and by separation of heterochromatin and euchromatin.13-15 It has been observed that the cells of Rett syndrome patients have an altered chromatin state, potentially contributing to transcriptional dysregulation.16,17 Several mutations observed in Rett syndrome patients occur in crucial domains for heterochromatin condensate formation, which helps explain this cellular phenotype.13 Introduction of a engineered “mini” MECP2 in a murine model of Rett syndrome has resulted in partial rescue of heterochromatin condensate formation and transcriptional regulation – fostering the hypothesis that correcting those genetic changes could lead to a potential therapy.18
Beyond the role of MECP2 in heterochromatin condensate formation, the gene interacts with more than 40 proteins that have diverse roles in cellular function, epigenetic modulation, and neuronal development. This volume of interactions contributes to MECP2 being a global gene regulatory protein that has far-reaching effects on transcriptional regulation across the genome.19-22
Epigenetic dysregulation has been associated with neurodevelopmental and neuropsychiatric disorders.23 Both insulin-like growth factor 1 (IGF-1) and brain-derived neurotrophic factor are transcriptional targets of MECP2, and are involved in neuronal differentiation, synaptic function, and neurite outgrowth.12 This helps explain the neurodevelopmental phenotypes observed in MECP2-mutated patients.
Notably, although Rett syndrome patients experience neurodevelopmental phenotypes at the cellular level, neuronal death is not readily observed. That observation provides hope that an interventional therapy after onset of symptoms might still be of benefit.
Presentation
Early neurotypical development. A hallmark of Rett syndrome is neurotypical physical and mental development until 6 to 24 months of age.
Stagnation is the first stage of the syndrome, involving a small but rapid decline in habitual milestones, such motor and language skills.12 Subtle signs, such as microcephaly and hypotonia, can also arise at this time but might be missed.24
Rapid regression follows stagnation. Speech and motor delays and impaired gait and breathing occur;12,25 purposeful hand skills are lost, replaced by repetitive hand-wringing movements that are a hallmark of the syndrome.12,24 Seizures are observed; they become more common during the next stage.12
Plateau. Language advances can be observed, but further deficits are seen in motor skills and hand coordination.12
Late motor deterioration stage. Late physical deficits develop, leading to lifelong impairments. The physical deficits observed are the result of severe muscle weakness, usually resulting in wheelchair dependency.12
Plateau. Patients then reach a second plateau. Regression stops; deficient physical and cognitive states stabilize and are maintained.25
At all stages of Rett syndrome, the following are observed:
- Gastrointestinal problems.
- Sleep disturbances.
- Abnormal cardiorespiratory coupling.
- Greater-than-expected mortality.12
Final regression. The patient is fully dependent for the rest of their lifespan, partially due to seizure activity.26,27
A life-changing diagnosis
A diagnosis of Rett syndrome is life-changing for a patient’s family; access to supportive groups of other patients and their families is extremely beneficial. Two helpful organizations – the Rett Syndrome Research Trust28 and International Rett Syndrome Foundation,29 – offer patient support and community and fund research.
Because X chromosome inactivation is random in Rett syndrome, the individual patient can present with a wide variety of phenotypic combinations – making the patient, and their needs, unique.12 During stages of regression, patients often experience emotional dysregulation and anxiety, which is attributable to their increasing physical difficulties.30 They often exhibit combinations of uncontrolled movements, including repetitive rocking, scratching, and self-injurious behavior.30 For most, regression subsides after the first 5 years of alternating development and regression; after that, their ultimate symptoms persist for life.25
As patients mature, they need to be monitored for proper nutrition and scoliosis.25 As adults, they are at risk of pneumonia, respiratory distress, status epilepticus, osteopenia, and lack of adequate food or water because of impaired ability to feed.25
The lifespan of Rett syndrome patients has increased, thanks to improvements in health care, advances in technology, and early genetic testing, which allows for earlier diagnosis, intervention, and management of symptoms.
Current treatments
When a female patient presents with regression and loss of milestones, sequencing of MECP2 is performed to verify whether Rett syndrome is the cause, by detecting any of the known mutations. Multiplex ligation-dependent probe amplification is also performed to detect major deletions.25
All available treatments for Rett syndrome are symptomatic; intensive early intervention is practiced.31 Multidisciplinary management – medical, psychiatric, and physical – is introduced almost immediately after diagnosis. Following diagnosis, patients are prescribed anti-seizure, sleep, and anxiety medications.31 Electroencephalography can be performed to identify seizure type. Neuromuscular blockage drugs can be prescribed to help with gait and stereotypic hand movements.25
Handguards or splints to the elbows can be prescribed by an occupational therapist to improve hand movement.25 Physical therapy can improve mobility; hydrotherapy and hippotherapy have been successful in helping to maintain mobility and muscle support.32,33 Nutritional management is implemented to control caloric intake and maintain the vitamin D level.31 Some patients experience constipation and urinary retention, putting them at risk of nephrolithiasis.
Once the signs and symptoms of Rett syndrome progress, and milestones regress to a certain point, patients need constant, full-time care for the rest of their lives.34 As symptomatic interventions have greatly improved patient outcomes and it has been shown that about 70% can reach adulthood with a potential lifespan of about 50 years.25
Although there is no cure for Rett syndrome and treatments are symptomatic, ongoing studies – both clinical and preclinical – offer promise that treatments will be developed that work at molecular and genetic levels.
Clinical trials
Advances in understanding of Rett syndrome have led to many therapies in clinical trials, several of which show promise.
Trofinetide. One of the most promising targets for downstream therapy, mentioned earlier, is IGF-1, which was the target of a successful phase 3 clinical trial, LAVENDER (sponsored by Acadia Pharmaceuticals).35,36 This trial studied trofinetide, a synthetic IGF-1 analog that inhibits neuroinflammation, restores glial function, corrects synaptic deficiencies, and regulates oxidative stress response.12,37,38 Initial results from phase 2 and phase 3 trials indicate improved scores for treated patients in the Rett syndrome Behaviour Questionnaire (RSBQ) and Clinical Global Impression–Improvement (CGI-I) scores, while also showing improvements in the Communication and Symbolic Behavior Scales Developmental Profile Infant–Toddler Checklist–Social composite score.36,39
The most common adverse events seen with trofinetide were diarrhea and vomiting.
Acadia Pharmaceuticals has filed for approval of a new drug application for trofinetide with the Food and Drug Administration, for which the company has been granted Fast Track Status and orphan drug designations. Most (95%) subjects in the phase 3 LAVENDER trial elected to continue taking trofinetide in the subsequent open-label Lilac and Lilac-2 extension studies.36 A current open-label phase 2/3 trial is recruiting patients 2 to 5 years of age to evaluate trofinetide.40 It is expected that, in the near future, this could be a drug given to Rett patients as an FDA-approved treatment.
Blarcamesine. Another small molecule drug, blarcamesine (also known as ANAVEX2-73), a sigma-1 receptor agonist, produced promising results in phase 2 clinical trials in adult Rett syndrome patients. The drug is in a phase 2/3 clinical trial for pediatric Rett syndrome patients (sponsored by Anavex Life Sciences).41-43
Phase 2 results indicated statistically significant and clinically meaningful improvement in RSBQ and CGI-I scores with blarcamesine. Improvement was initially observed within 4 weeks after the start of treatment and was sustained throughout the study. The drug was shown to be well tolerated, with minimal adverse effects; no serious adverse events were recorded. These results were observed in adult patients, demonstrating that improvements in Rett syndrome are possible even after regression.
Blarcamesine activates the sigma 1 receptor, which is pivotal to restoring cellular homeostasis and restoring neuroplasticity – deficiencies of which have been linked to autophagy and glutamate toxicity. The drug has also been explored as a potential treatment for other neurological disorders.44-47 Improvements in blarcamesine-treated patients further correlated with lower levels of glutamate in cerebrospinal fluid, which is a Rett syndrome biomarker, supporting the proposition that behavioral improvements were due to drug intervention.48,49 The phase 2 trial was modified into a phase 3 trial and additional endpoints were added.41-43
All patients in the phase 2 adult trial elected to continue in the extension study.
Based on these promising data, Anavex is pursuing an approval pathway for adult patients, while continuing dosage optimization phase 2/3 trials and recruitment for a pediatric trial.42,43
Is the future about gene therapy?
TSHA-102 (miniMECP2). Taysha Gene Therapies is developing a promising gene therapy, TSHA-102, for Rett syndrome, and is aiming to begin phase 1/2 clinical trials in 2022.50 The technology for this therapy relies on the delivery of a fragment of MECP2 (known as miniMECP2), which is regulated by a built-in microRNA regulator (miR-responsive auto-regulatory element, or miRARE) to help ameliorate MECP2 dosage toxicity. (Overexpression of MECP2 is toxic to neurons, which has made traditional [so to speak] gene replacement therapy difficult in Rett syndrome: Levels of MECP2 need to be tightly regulated, and the Taysha microRNA technology regulates levels of miniMECP2, thus reducing toxicity.)
The Taysha microRNA technology has yielded promising results in mouse studies for Rett syndrome; results indicate a lengthening of lifespan and delayed onset of gait abnormalities.51 TSHA-102 is in the preclinical stage but offers promise that it will be the first gene therapy for Rett syndrome to enter clinical trials.
As the field of gene therapy advances, several promising technologies are on the horizon that could potentially have disease-altering impacts on Rett syndrome. These therapies are divided into two broad categories: those at the gene level and those at the transcription and protein level. A few of these approaches are highlighted below.
Gene replacement involves adding a full or partial copy of MECP2 to neuronal cells. This type of therapy presents challenges, from delivery of the new gene to dosage concerns, because MECP2 can be toxic if overexpressed.52-54 Groundbreaking work was done in mouse models involving truncated MECP2, exhibiting phenotypic rescue and validating the gene-replacement approach.18 This strategy is being pursued by Neurogene, which has a uinique technology that allows for tuning of the gene’s expression to get the correct protein levels in the patient. Promising data was presented this year at the American Society of Gene and Cell Therapy conference.55
Early gene replacement therapy studies also laid the foundation for the minMECP2 and microRNA approach being used by Taysha Gene Therapies (discussed above).51
“Correcting” DNA mutations. A different approach at the genetic level involves “correcting” mutations in MECP2 at the DNA level. This is possible because, in a large subset of Rett syndrome patients who have the same missense or nonsense mutations, by using CRISPR, a gene editing tool (discussed above) a single base pair can be corrected.56,57 Previous research, in a Rett syndrome-model of induced pluripotent stem cells, showed that this type of editing is possible – and effective.52 An approach with particular promise involves use of a class of CRISPR proteins known as base editors that are able to specifically alter a single base of DNA.57 The technique has the potential to address many of the mutations seen in Rett syndrome; research on this type of technology is being pursued by Beam Therapeutics, and has the potential to impact Rett syndrome.58
Another promising “correction” approach is exonic editing, in which a much larger section of DNA – potentially, exons 3 and 4, which, taken together, comprise 97% of known MECP2 mutations seen in Rett syndrome – are replaced.59
In both CRISPR and exonic editing therapeutic approaches, endogenous levels of MECP2 expression would be maintained. Of note, both approaches are being pursued for use in treating other genetic disorders, which provides a boost in scaling-up work on addressing safety and efficacy concerns that accompany gene-editing approaches.58 One advantage to the DNA correction approach is that is has the potential to be a “one-and-done” treatment.
“Correcting” RNA. Beyond directly editing DNA, several therapeutic approaches are exploring the ability to edit RNA or to provide the protein directly to cells as enzyme replacement therapy. Such an RNA correction strategy leverages a technology that takes advantage of cells’ natural RNA editor, known as adenosine deaminase acting on RNA (ADAR), which corrects errors in cells’ RNA by providing specific guides to the cell. ADAR can be targeted to fix mutations in the MECP2 RNA transcript, resulting in a “corrected” MECP2 protein.60,61 This technology has delivered promising proof-of-concept evidence in cells and in murine models, and is in the therapeutic pipeline at VICO Therapeutics.62
Shape Therapeutics has also leveraged ADAR to “correct” mutated RNA; Rett syndrome is among the top priorities in the company’s pipeline.
Worth noting is that there are several advantages to the “correction” approach:
- Leveraging internal repair mechanisms minimizes the immune response.
- The flexibility of correction means that it can be used to address many of the mutations that cause Rett syndrome.63
Enzyme replacement therapy, in which the MECP2 protein produced by MECP2 would be directly replaced, is being explored in Rett syndrome patients. This technology has been used successfully in Pompe disease; however, Rett syndrome presents its own challenge because MECP2 needs to be delivered to the brain and neuronal cells.64
Where does this work stand? The technologies described in this section are in preclinical stages of study. Nonetheless, it is expected that several will enter human clinical trials during the next 5 years.
Conclusion
A diagnosis of Rett syndrome is a life-altering event for patients and their family. But there is more hope than ever for effective therapies and, eventually, a cure.
Multiple late-stage clinical trials in progress are demonstrating promising results from therapeutic products, with minimal adverse events. Remarkably, these interventions have delivered improvements to adult patients after regression has stabilized. With rapid progress being made in the field of gene therapy, several technologies for which are focused on Rett syndrome, a hopeful picture is emerging: that therapeutic intervention will be possible before regression, thus effectively treating and, potentially, even curing Rett syndrome.
The landscape is broadening. Add to this hope for approved therapies is the fact that Rett syndrome isn’t the only target being pursued with such strategies; in fact, researchers in the larger field of neurodevelopmental disorder study are working together to find common solutions to shared challenges – from how therapies are designed and delivered to how toxicity is minimized. Much of what is being explored in the Rett syndrome field is also under investigation in other neurodevelopmental syndromes, including Angelman, Prader-Willi, chromosome 15q11.2-13.1 duplication (dup15q), and Fragile X syndrome. This kind of parallel investigation benefits all parties and optimizes a treatment platform so that it can be applied across more than a single disorder.
Like many monogenic disorders, Rett syndrome is entering an exciting stage – at which the words “treatment” and “cure” can be spoken with intent and vision, not just wide-eyed optimism. These words portend real promise for patients who carry the weight of a diagnosis of Rett syndrome, and for their families.
Ms. Ambrose is a student in the master’s of science in human genetics and genomic data analytics program, Keck Graduate Institute, Claremont, Calif. Dr. Bailus is an assistant professor of genetics, Keck Graduate Institute. The authors report no conflict of interest related to this article.
References
1. Lander ES et al; . Initial sequencing and analysis of the human genome. Nature. 2001 Feb 15;409(6822):860-921. doi: 10.1038/35057062.
2. Venter JC et al. The sequence of the human genome. Science. 2001 Feb 16;291(5507):1304-51. doi: 10.1126/science.1058040.
3. Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012 Aug 17;337(6096):816-21. doi: 10.1126/science.1225829.
4. Percy A. The American history of Rett syndrome. Pediatr Neurol. 2014 Jan;50(1):1-3. doi: 10.1016/j.pediatrneurol.2013.08.018.
5. Amir RE et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999 Oct;23(2):185-8. doi: 10.1038/13810.
6. Pitzianti MB et al. Rett syndrome in males: The different clinical course in two brothers with the same microduplication MECP2 Xq28. Int J Environ Res Public Health. 2019 Aug;16(17):3075. doi: 10.3390/ijerph16173075.
7. Ribeiro MC, MacDonald JL. Sex differences in Mecp2-mutant Rett syndrome model mice and the impact of cellular mosaicism in phenotype development. Brain Res. 2020 Feb 15;1729:146644. doi: 10.1016/j.brainres.2019.146644.
8. Bao X et al. X chromosome inactivation in Rett syndrome and its correlations with MECP2 mutations and phenotype. J Child Neurol. 2008 Jan;23(1):22-5. doi: 10.1177/0883073807307077.
9. Knudsen GPS et al. Increased skewing of X chromosome inactivation in Rett syndrome patients and their mothers. Eur J Hum Genet. 2006 Jul;14(11):1189-94. doi: 10.1038/sj.ejhg.5201682.
10. Chahil G et al. Rett syndrome in males: A case report and review of literature. Cureus. 2018;10(10):e3414. doi: 10.7759/cureus.3414.
11. Reichow B et al. Brief report: Systematic review of Rett syndrome in males. J Autism Dev Disord. 2015 Oct;45(10):3377-83. doi: 10.1007/s10803-015-2519-1.
12. Vashi N, Justice MJ. Treating Rett syndrome: From mouse models to human therapies. Mamm Genome. 2019 Jun;30(5-6):90-110. doi: 10.1007/s00335-019-09793-5.
13. Li CH et al. MeCP2 links heterochromatin condensates and neurodevelopmental disease. Nature. 2020 Oct;586(7829):440-4. doi: 10.1038/s41586-020-2574-4.
14. Schmidt A et al. MeCP2 and chromatin compartmentalization. Cells. 2020 Apr;9(4):878. doi: 10.3390/cells9040878.
15. Wang L et al. Rett syndrome-causing mutations compromise MeCP2-mediated liquid-liquid phase separation of chromatin. Cell Res. 2020 May;30(5):393-407. doi: 10.1038/s41422-020-0288-7.
16. Lin P et al. Transcriptome analysis of human brain tissue identifies reduced expression of complement complex C1Q genes in Rett syndrome. BMC Genomics. 2016;17:427. doi: 10.1186/s12864-016-2746-7.
17. Tudor M et al. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc Natl Acad Sci U S A. 2002 Nov 26;99(24):15536-41. doi: 10.1073/pnas.242566899.
18. Tillotson R et al. Radically truncated MeCP2 rescues Rett syndrome–like neurological defects. Nature. 2017 Oct 19;550(7676):398-401. doi: 10.1038/nature24058.
19. Connolly DR, Zhou Z. Genomic insights into MeCP2 function: A role for the maintenance of chromatin architecture. Curr Opin Neurobiol. 2019 Dec;59:174-9. doi: 10.1016/j.conb.2019.07.002.
20. Johnson BS et al. Biotin tagging of MeCP2 in mice reveals contextual insights into the Rett syndrome transcriptome. Nat Med. 2017 Oct;23(10):1203-14. doi: 10.1038/nm.4406.
21. Gabel HW et al. Disruption of DNA-methylation–dependent long gene repression in Rett syndrome. Nature. 2015 Jun 4;522(7554):89-93. doi: 10.1038/nature14319.
22. Lyst MJ, Bird A. Rett syndrome: A complex disorder with simple roots. Nat Rev Genet. 2015 May;16(5):261-75. doi: 10.1038/nrg3897.
23. Kuehner JN et al. Epigenetic regulations in neuropsychiatric disorders. Front Genet. 2019 Apr 4;10:268. doi: 10.3389/fgene.2019.00268.
24. Pejhan S, Rastegar M. Role of DNA methyl-CpG-binding protein MeCP2 in Rett syndrome pathobiology and mechanism of disease. Biomolecules. 2021 Jan;11(1):75. doi: 10.3390/biom11010075.
25. Fu C et al. Consensus guidelines on managing Rett syndrome across the lifespan. BMJ Paediatr Open. 2020;4(1):e000717. doi: 10.1136/bmjpo-2020-000717.
26. Operto FF et al. Epilepsy and genetic in Rett syndrome: A review. Brain Behav. 2019 May;9(5):e01250. doi: 10.1002/brb3.1250.
27. Nissenkorn A et al. Epilepsy in Rett syndrome – The experience of a National Rett Center. Epilepsia. 2010 Jul;51(7):1252-8. doi: 10.1111/j.1528-1167.2010.02597.x.
28. Welcome to the Rett cure community. Rett Syndrome Research Trust [Internet]. Updated Feb 8, 2022. Accessed Feb 23, 2022. https://reverserett.org.
29. About Rett syndrome. International Rett Syndrome Foundation [Internet]. Updated Jan 4, 2022. Accessed Feb 23, 2022. http://www.rettsyndrome.org.
30. Singh J, Santosh P. Key issues in Rett syndrome: Emotional, behavioural and autonomic dysregulation (EBAD) – A target for clinical trials. Orphanet J Rare Dis. 2018 Jul 31;13(1):128. doi: 10.1186/s13023-018-0873-8.
31. Banerjee A et al. Towards a better diagnosis and treatment of Rett syndrome: A model synaptic disorder. Brain. 2019 Feb 1;142(2):239-48. doi: 10.1093/brain/awy323.
32. Ager S et al. Parental experiences of scoliosis management in Rett syndrome. Disabil Rehabil. 2009 Sep 19;31(23):1917-24. doi: 10.1080/09638280902846392.
33. Budden SS. Management of Rett syndrome: A ten year experience. Neuropediatrics. 1995;26(2):75-7. doi: 10.1055/s-2007-979727.
34. Ip JPK et al. Rett syndrome: Insights into genetic, molecular and circuit mechanisms. Nat Rev Neurosci. 2018 Jun;19(6):368-82. doi: 10.1038/s41583-018-0006-3.
35. Acadia Pharmaceuticals Inc. Study of trofinetide for the treatment of girls and women with Rett syndrome (LAVENDER™). ClinicalTrials.gov identifier: NCT04181723. Updated Feb 17, 2022. Accessed Feb 23, 2022. https://clinicaltrials.gov/ct2/show/NCT04181723.
36. Acadia Pharmaceuticals announces positive top-line results from the pivotal phase 3 LAVENDER trial of trofinetide in Rett syndrome. Press release. Acadia Pharmaceuticals Inc. Dec 6, 2021. Accessed Feb 23, 2022. https://ir.acadia-pharm.com/news-releases/news-release-details/acadia-pharmaceuticals-announces-positive-top-line-results-1.
37. Copping NA et al. Emerging gene and small molecule therapies for the neurodevelopmental disorder Angelman syndrome. Neurotherapeutics. 2021 Jul;18(3):1535-47. doi: 10.1007/s13311-021-01082-x.
38. Riikonen R. Insulin-like growth factors in the pathogenesis of neurological diseases in children. Int J Mol Sci. 2017 Sep;18(10):2056. doi: 10.3390/ijms18102056.
39. Glaze DG et al; Rett 002 Study Group. Double-blind, randomized, placebo-controlled study of trofinetide in pediatric Rett syndrome. Neurology. 2019 April 16;92(16):e1912-e1925. doi: 10.1212/WNL.0000000000007316.
40. Acadia Pharmaceuticals Inc. An open-label study of trofinetide for the treatment of girls two to five years of age who have Rett syndrome (DAFFODIL™). ClinicalTrials.gov Identifier: NCT04988867. Updated Jan 24, 2022. Accessed Feb 23, 2022. https://clinicaltrials.gov/ct2/show/NCT04988867.
41. Anavex Life Sciences announces ANAVEX®2-73 meets primary and secondary endpoints in clinical trial. Press release. Anavex Life Sciences Corp. Dec 15, 2020. Accessed Feb 23, 2022. http://www.anavex.com/post/anavex-life-sciences-announces-anavex-2-73-meets-primary-and-secondary-endpoints-in-clinical-trial.
42. Anavex Life Sciences Corp. ANAVEX2-73 study in patients with Rett syndrome (AVATAR). ClinicalTrials.gov Identifier: NCT03941444. Updated Jan 27, 2022. Accessed Feb 23, 2022. https://clinicaltrials.gov/ct2/show/NCT03941444.
43. Anavex Life Sciences Corp. ANAVEX2-73 study in pediatric patients with Rett syndrome (EXCELLENCE). ClinicalTrials.gov Identifier: NCT04304482. Updated Sep 28, 2021. Accessed Feb 23, 2022. http://www.clinicaltrials.gov/ct2/show/NCT04304482.
44.Christ MG et al. The Sigma-1 receptor at the crossroad of proteostasis, neurodegeneration, and autophagy. Trends Neurosci. 2020 Feb;43(2):79-81. doi: 10.1016/j.tins.2019.12.002.
45. Kaufmann WE et al. ANAVEX®2-73 (blarcamesine), a sigma-1 receptor agonist, ameliorates neurologic impairments in a mouse model of Rett syndrome. Pharmacol Biochem Behav. 2019 Dec;187:172796. doi: 10.1016/j.pbb.2019.172796.
46. Brimson JM et al. Dipentylammonium binds to the sigma-1 receptor and protects against glutamate toxicity, attenuates dopamine toxicity and potentiates neurite outgrowth in various cultured cell lines. Neurotox Res. 2018 Aug;34(2):263-72. doi: 10.1007/s12640-018-9883-5.
47. Kourrich S et al. The sigma-1 receptor: roles in neuronal plasticity and disease. Trends Neurosci. 2012 Dec;35(12):762-71. doi: 10.1016/j.tins.2012.09.007.
48. Lappalainen R, Riikonen RS. High levels of cerebrospinal fluid glutamate in Rett syndrome. Pediatr Neurol. 1996 Oct;15(3):213-6. doi: 10.1016/s0887-8994(96)00218-4.
49. Hamberger A et al. Elevated CSF glutamate in Rett syndrome. Neuropediatrics. 1992;23(4):212-3. doi: 10.1055/s-2008-1071344.
50. Inacio P. FDA acts to support development of potential gene therapy, TSHA-102. Rett Syndrome News [Internet]. Oct 16, 2020. Accessed Feb 23, 2022. https://rettsyndromenews.com/2020/10/16/fda-grants-orphan-drug-rare-pediatric-disease-status-to-tsha-102-potential-rett-gene-therapy.
51. Sinnett SE et al. Engineered microRNA-based regulatory element permits safe high-dose miniMECP2 gene therapy in Rett mice. Brain. 2021 Nov 29;144(10):3005-19. doi: 10.1093/brain/awab182.
52. Le TTH et al. Efficient and precise CRISPR/Cas9-mediated MECP2 modifications in human-induced pluripotent stem cells. Front Genet. 2019 Jul 2;10:625. doi: 10.3389/fgene.2019.00625.
53. Koerner MV et al. Toxicity of overexpressed MeCP2 is independent of HDAC3 activity. Genes Dev. 2018;32(23-24):1514-24. doi: 10.1101/gad.320325.118.
54. Heckman LD et al. Rett-causing mutations reveal two domains critical for MeCP2 function and for toxicity in MECP2 duplication syndrome mice. Elife. 2014;3:e02676. doi: 10.7554/eLife.02676.
55. Neurogene announces new development program in Rett syndrome utilizing novel EXACT technology platform [Internet]. Accessed Aug 12, 2022. https://www.neurogene.com/press-releases/neurogene-announces-new-development-program-in-rett-syndrome-utilizing-novel-exact-technology-platform/
56. Anzalone AV et al. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. 2020 Jul;38(7):824-44. doi: 10.1038/s41587-020-0561-9.
57. Gaudelli NM, Komor AC, Rees HA, et al. Programmable base editing of A●T to G●C in genomic DNA without DNA cleavage. Nature. 2017 Nov 23;551(7681):464-71. doi: 10.1038/nature24644.
58. Coenraads M. How RSRT is driving the search for a Rett cure. Rett Syndrome Research Trust [Internet]. Dec 7, 2021. Accessed Feb 23, 2022. https://rettnews.org/articles/how-rsrt-is-driving-the-search-for-a-rett-cure.
59. Cutting-edge technologies to repair the underlying mutations that cause Rett. Rett Syndrome Research Trust [Internet]. Updated Nov 3, 2021. Accessed Feb 23, 2022. https://reverserett.org/research/cures/gene-editing.
60. Sinnamon JR et al. In vivo repair of a protein underlying a neurological disorder by programmable RNA editing. Cell Rep. 2020 Jul 14;32(2):107878. doi: 10.1016/j.celrep.2020.107878.
61. Sinnamon JR et al. Site-directed RNA repair of endogenous Mecp2 RNA in neurons. Proc Natl Acad Sci U S A. 2017 Oct 31;114(44):E9395-E9402. doi: 10.1073/pnas.1715320114.
62. Pipeline. VICO Therapeutics [Internet]. Updated Nov 5, 2021. Accessed Feb 23, 2022. https://vicotx.com/pipeline.
63. Therapeutics platform. Shape Therapeutics [Internet]. Updated Feb 20, 2021. Accessed Feb 23, 2022.
https://live-shapetx.pantheonsite.io/therapeutics-platform.
64. Koeberl DD et al. Glycogen storage disease types I and II: Treatment updates. J Inherit Metab Dis. 2007 Apr;30(2):159-64. doi: 10.1007/s10545-007-0519-9.
The dream of curing genetic disorders has been a persistent but elusive goal, even before the human genome was mapped. Once mapping of the human genome was complete in 2001, an entirely new avenue of potential treatments and cures for genetic diseases and disorders was opened.1,2
The disorders best suited for targeted gene therapy are monogenic; however, tools and delivery methods for editing the human genome were limited and difficult to apply, until the advent of the CRISPR system in 2012.3 CRISPR (an acronym of clustered regularly interspaced short palindromic repeats) has changed the way in which gene therapy strategies are pursued, with dozens of companies leveraging a variety of platforms to create potentially life-changing therapies for devastating rare diseases and disorders.
One of the rare monogenic disorders that is embracing multiple gene therapy approaches is Rett syndrome, a rare, debilitating neurodevelopmental disorder. In this review, we explore the molecular cause of Rett syndrome, disease presentation, current treatments, ongoing clinical trials, and therapies that are on the horizon.
Underlying molecular cause
Rett syndrome is caused by mutations in, or the absence of, the MECP2 gene, which produces methyl-CpG binding protein 2 (MECP2). The syndrome was first described clinically in 1954 by the Austrian physician Andreas Rett; it would take until 1982 before the disorder was officially named, eponymously, in a seminal paper by Hagberg.4 After Hagberg’s characterization, Rett syndrome became the predominant global clinical diagnosis identified among cognitively impaired females, with an incidence of 1 in every 10,000 to 15,000.4
In 1999, mutations in, and deletions of, MECP2 were identified as the cause of Rett syndrome.4,5 MECP2 is located on the X chromosome, in the Xq28 region, making Rett syndrome an X-linked dominant disorder.6 Rett syndrome is seen predominantly in females who are mosaic for mutant or deleted MECP2. Random X chromosome inactivation results in some cells expressing the mutant MECP2 allele and other cells expressing the normal functioning MECP2 allele; the percentage of cells expressing the normal allele correlates with the degree of syndrome severity.7-9
The incidence of Rett syndrome is much lower in males, in whom the syndrome was originally thought to be lethal; many observed male cases are either mosaic or occur in XXY males.10,11
Approximately 95% of cases of Rett syndrome are due to de novo mutations in MECP2, with a handful of specific mutations and large deletions accounting for more than 85% of cases.12 The fact that Rett syndrome is monogenic and most cases are caused by, in total, only a handful of mutations or deletions makes the syndrome a promising candidate for gene therapy.
At the molecular level, it has been observed that the MECP2 mutations of Rett syndrome lead to loss of gene function, thus disrupting the ability of the MECP2 nuclear protein to regulate global gene transcription through its binding to methylated DNA sites.12 A large percentage of these missense and nonsense mutations lead to a truncated or nonfunctional protein.12
One of the ways in which MECP2 regulates transcription is as a component of heterochromatin condensates and by separation of heterochromatin and euchromatin.13-15 It has been observed that the cells of Rett syndrome patients have an altered chromatin state, potentially contributing to transcriptional dysregulation.16,17 Several mutations observed in Rett syndrome patients occur in crucial domains for heterochromatin condensate formation, which helps explain this cellular phenotype.13 Introduction of a engineered “mini” MECP2 in a murine model of Rett syndrome has resulted in partial rescue of heterochromatin condensate formation and transcriptional regulation – fostering the hypothesis that correcting those genetic changes could lead to a potential therapy.18
Beyond the role of MECP2 in heterochromatin condensate formation, the gene interacts with more than 40 proteins that have diverse roles in cellular function, epigenetic modulation, and neuronal development. This volume of interactions contributes to MECP2 being a global gene regulatory protein that has far-reaching effects on transcriptional regulation across the genome.19-22
Epigenetic dysregulation has been associated with neurodevelopmental and neuropsychiatric disorders.23 Both insulin-like growth factor 1 (IGF-1) and brain-derived neurotrophic factor are transcriptional targets of MECP2, and are involved in neuronal differentiation, synaptic function, and neurite outgrowth.12 This helps explain the neurodevelopmental phenotypes observed in MECP2-mutated patients.
Notably, although Rett syndrome patients experience neurodevelopmental phenotypes at the cellular level, neuronal death is not readily observed. That observation provides hope that an interventional therapy after onset of symptoms might still be of benefit.
Presentation
Early neurotypical development. A hallmark of Rett syndrome is neurotypical physical and mental development until 6 to 24 months of age.
Stagnation is the first stage of the syndrome, involving a small but rapid decline in habitual milestones, such motor and language skills.12 Subtle signs, such as microcephaly and hypotonia, can also arise at this time but might be missed.24
Rapid regression follows stagnation. Speech and motor delays and impaired gait and breathing occur;12,25 purposeful hand skills are lost, replaced by repetitive hand-wringing movements that are a hallmark of the syndrome.12,24 Seizures are observed; they become more common during the next stage.12
Plateau. Language advances can be observed, but further deficits are seen in motor skills and hand coordination.12
Late motor deterioration stage. Late physical deficits develop, leading to lifelong impairments. The physical deficits observed are the result of severe muscle weakness, usually resulting in wheelchair dependency.12
Plateau. Patients then reach a second plateau. Regression stops; deficient physical and cognitive states stabilize and are maintained.25
At all stages of Rett syndrome, the following are observed:
- Gastrointestinal problems.
- Sleep disturbances.
- Abnormal cardiorespiratory coupling.
- Greater-than-expected mortality.12
Final regression. The patient is fully dependent for the rest of their lifespan, partially due to seizure activity.26,27
A life-changing diagnosis
A diagnosis of Rett syndrome is life-changing for a patient’s family; access to supportive groups of other patients and their families is extremely beneficial. Two helpful organizations – the Rett Syndrome Research Trust28 and International Rett Syndrome Foundation,29 – offer patient support and community and fund research.
Because X chromosome inactivation is random in Rett syndrome, the individual patient can present with a wide variety of phenotypic combinations – making the patient, and their needs, unique.12 During stages of regression, patients often experience emotional dysregulation and anxiety, which is attributable to their increasing physical difficulties.30 They often exhibit combinations of uncontrolled movements, including repetitive rocking, scratching, and self-injurious behavior.30 For most, regression subsides after the first 5 years of alternating development and regression; after that, their ultimate symptoms persist for life.25
As patients mature, they need to be monitored for proper nutrition and scoliosis.25 As adults, they are at risk of pneumonia, respiratory distress, status epilepticus, osteopenia, and lack of adequate food or water because of impaired ability to feed.25
The lifespan of Rett syndrome patients has increased, thanks to improvements in health care, advances in technology, and early genetic testing, which allows for earlier diagnosis, intervention, and management of symptoms.
Current treatments
When a female patient presents with regression and loss of milestones, sequencing of MECP2 is performed to verify whether Rett syndrome is the cause, by detecting any of the known mutations. Multiplex ligation-dependent probe amplification is also performed to detect major deletions.25
All available treatments for Rett syndrome are symptomatic; intensive early intervention is practiced.31 Multidisciplinary management – medical, psychiatric, and physical – is introduced almost immediately after diagnosis. Following diagnosis, patients are prescribed anti-seizure, sleep, and anxiety medications.31 Electroencephalography can be performed to identify seizure type. Neuromuscular blockage drugs can be prescribed to help with gait and stereotypic hand movements.25
Handguards or splints to the elbows can be prescribed by an occupational therapist to improve hand movement.25 Physical therapy can improve mobility; hydrotherapy and hippotherapy have been successful in helping to maintain mobility and muscle support.32,33 Nutritional management is implemented to control caloric intake and maintain the vitamin D level.31 Some patients experience constipation and urinary retention, putting them at risk of nephrolithiasis.
Once the signs and symptoms of Rett syndrome progress, and milestones regress to a certain point, patients need constant, full-time care for the rest of their lives.34 As symptomatic interventions have greatly improved patient outcomes and it has been shown that about 70% can reach adulthood with a potential lifespan of about 50 years.25
Although there is no cure for Rett syndrome and treatments are symptomatic, ongoing studies – both clinical and preclinical – offer promise that treatments will be developed that work at molecular and genetic levels.
Clinical trials
Advances in understanding of Rett syndrome have led to many therapies in clinical trials, several of which show promise.
Trofinetide. One of the most promising targets for downstream therapy, mentioned earlier, is IGF-1, which was the target of a successful phase 3 clinical trial, LAVENDER (sponsored by Acadia Pharmaceuticals).35,36 This trial studied trofinetide, a synthetic IGF-1 analog that inhibits neuroinflammation, restores glial function, corrects synaptic deficiencies, and regulates oxidative stress response.12,37,38 Initial results from phase 2 and phase 3 trials indicate improved scores for treated patients in the Rett syndrome Behaviour Questionnaire (RSBQ) and Clinical Global Impression–Improvement (CGI-I) scores, while also showing improvements in the Communication and Symbolic Behavior Scales Developmental Profile Infant–Toddler Checklist–Social composite score.36,39
The most common adverse events seen with trofinetide were diarrhea and vomiting.
Acadia Pharmaceuticals has filed for approval of a new drug application for trofinetide with the Food and Drug Administration, for which the company has been granted Fast Track Status and orphan drug designations. Most (95%) subjects in the phase 3 LAVENDER trial elected to continue taking trofinetide in the subsequent open-label Lilac and Lilac-2 extension studies.36 A current open-label phase 2/3 trial is recruiting patients 2 to 5 years of age to evaluate trofinetide.40 It is expected that, in the near future, this could be a drug given to Rett patients as an FDA-approved treatment.
Blarcamesine. Another small molecule drug, blarcamesine (also known as ANAVEX2-73), a sigma-1 receptor agonist, produced promising results in phase 2 clinical trials in adult Rett syndrome patients. The drug is in a phase 2/3 clinical trial for pediatric Rett syndrome patients (sponsored by Anavex Life Sciences).41-43
Phase 2 results indicated statistically significant and clinically meaningful improvement in RSBQ and CGI-I scores with blarcamesine. Improvement was initially observed within 4 weeks after the start of treatment and was sustained throughout the study. The drug was shown to be well tolerated, with minimal adverse effects; no serious adverse events were recorded. These results were observed in adult patients, demonstrating that improvements in Rett syndrome are possible even after regression.
Blarcamesine activates the sigma 1 receptor, which is pivotal to restoring cellular homeostasis and restoring neuroplasticity – deficiencies of which have been linked to autophagy and glutamate toxicity. The drug has also been explored as a potential treatment for other neurological disorders.44-47 Improvements in blarcamesine-treated patients further correlated with lower levels of glutamate in cerebrospinal fluid, which is a Rett syndrome biomarker, supporting the proposition that behavioral improvements were due to drug intervention.48,49 The phase 2 trial was modified into a phase 3 trial and additional endpoints were added.41-43
All patients in the phase 2 adult trial elected to continue in the extension study.
Based on these promising data, Anavex is pursuing an approval pathway for adult patients, while continuing dosage optimization phase 2/3 trials and recruitment for a pediatric trial.42,43
Is the future about gene therapy?
TSHA-102 (miniMECP2). Taysha Gene Therapies is developing a promising gene therapy, TSHA-102, for Rett syndrome, and is aiming to begin phase 1/2 clinical trials in 2022.50 The technology for this therapy relies on the delivery of a fragment of MECP2 (known as miniMECP2), which is regulated by a built-in microRNA regulator (miR-responsive auto-regulatory element, or miRARE) to help ameliorate MECP2 dosage toxicity. (Overexpression of MECP2 is toxic to neurons, which has made traditional [so to speak] gene replacement therapy difficult in Rett syndrome: Levels of MECP2 need to be tightly regulated, and the Taysha microRNA technology regulates levels of miniMECP2, thus reducing toxicity.)
The Taysha microRNA technology has yielded promising results in mouse studies for Rett syndrome; results indicate a lengthening of lifespan and delayed onset of gait abnormalities.51 TSHA-102 is in the preclinical stage but offers promise that it will be the first gene therapy for Rett syndrome to enter clinical trials.
As the field of gene therapy advances, several promising technologies are on the horizon that could potentially have disease-altering impacts on Rett syndrome. These therapies are divided into two broad categories: those at the gene level and those at the transcription and protein level. A few of these approaches are highlighted below.
Gene replacement involves adding a full or partial copy of MECP2 to neuronal cells. This type of therapy presents challenges, from delivery of the new gene to dosage concerns, because MECP2 can be toxic if overexpressed.52-54 Groundbreaking work was done in mouse models involving truncated MECP2, exhibiting phenotypic rescue and validating the gene-replacement approach.18 This strategy is being pursued by Neurogene, which has a uinique technology that allows for tuning of the gene’s expression to get the correct protein levels in the patient. Promising data was presented this year at the American Society of Gene and Cell Therapy conference.55
Early gene replacement therapy studies also laid the foundation for the minMECP2 and microRNA approach being used by Taysha Gene Therapies (discussed above).51
“Correcting” DNA mutations. A different approach at the genetic level involves “correcting” mutations in MECP2 at the DNA level. This is possible because, in a large subset of Rett syndrome patients who have the same missense or nonsense mutations, by using CRISPR, a gene editing tool (discussed above) a single base pair can be corrected.56,57 Previous research, in a Rett syndrome-model of induced pluripotent stem cells, showed that this type of editing is possible – and effective.52 An approach with particular promise involves use of a class of CRISPR proteins known as base editors that are able to specifically alter a single base of DNA.57 The technique has the potential to address many of the mutations seen in Rett syndrome; research on this type of technology is being pursued by Beam Therapeutics, and has the potential to impact Rett syndrome.58
Another promising “correction” approach is exonic editing, in which a much larger section of DNA – potentially, exons 3 and 4, which, taken together, comprise 97% of known MECP2 mutations seen in Rett syndrome – are replaced.59
In both CRISPR and exonic editing therapeutic approaches, endogenous levels of MECP2 expression would be maintained. Of note, both approaches are being pursued for use in treating other genetic disorders, which provides a boost in scaling-up work on addressing safety and efficacy concerns that accompany gene-editing approaches.58 One advantage to the DNA correction approach is that is has the potential to be a “one-and-done” treatment.
“Correcting” RNA. Beyond directly editing DNA, several therapeutic approaches are exploring the ability to edit RNA or to provide the protein directly to cells as enzyme replacement therapy. Such an RNA correction strategy leverages a technology that takes advantage of cells’ natural RNA editor, known as adenosine deaminase acting on RNA (ADAR), which corrects errors in cells’ RNA by providing specific guides to the cell. ADAR can be targeted to fix mutations in the MECP2 RNA transcript, resulting in a “corrected” MECP2 protein.60,61 This technology has delivered promising proof-of-concept evidence in cells and in murine models, and is in the therapeutic pipeline at VICO Therapeutics.62
Shape Therapeutics has also leveraged ADAR to “correct” mutated RNA; Rett syndrome is among the top priorities in the company’s pipeline.
Worth noting is that there are several advantages to the “correction” approach:
- Leveraging internal repair mechanisms minimizes the immune response.
- The flexibility of correction means that it can be used to address many of the mutations that cause Rett syndrome.63
Enzyme replacement therapy, in which the MECP2 protein produced by MECP2 would be directly replaced, is being explored in Rett syndrome patients. This technology has been used successfully in Pompe disease; however, Rett syndrome presents its own challenge because MECP2 needs to be delivered to the brain and neuronal cells.64
Where does this work stand? The technologies described in this section are in preclinical stages of study. Nonetheless, it is expected that several will enter human clinical trials during the next 5 years.
Conclusion
A diagnosis of Rett syndrome is a life-altering event for patients and their family. But there is more hope than ever for effective therapies and, eventually, a cure.
Multiple late-stage clinical trials in progress are demonstrating promising results from therapeutic products, with minimal adverse events. Remarkably, these interventions have delivered improvements to adult patients after regression has stabilized. With rapid progress being made in the field of gene therapy, several technologies for which are focused on Rett syndrome, a hopeful picture is emerging: that therapeutic intervention will be possible before regression, thus effectively treating and, potentially, even curing Rett syndrome.
The landscape is broadening. Add to this hope for approved therapies is the fact that Rett syndrome isn’t the only target being pursued with such strategies; in fact, researchers in the larger field of neurodevelopmental disorder study are working together to find common solutions to shared challenges – from how therapies are designed and delivered to how toxicity is minimized. Much of what is being explored in the Rett syndrome field is also under investigation in other neurodevelopmental syndromes, including Angelman, Prader-Willi, chromosome 15q11.2-13.1 duplication (dup15q), and Fragile X syndrome. This kind of parallel investigation benefits all parties and optimizes a treatment platform so that it can be applied across more than a single disorder.
Like many monogenic disorders, Rett syndrome is entering an exciting stage – at which the words “treatment” and “cure” can be spoken with intent and vision, not just wide-eyed optimism. These words portend real promise for patients who carry the weight of a diagnosis of Rett syndrome, and for their families.
Ms. Ambrose is a student in the master’s of science in human genetics and genomic data analytics program, Keck Graduate Institute, Claremont, Calif. Dr. Bailus is an assistant professor of genetics, Keck Graduate Institute. The authors report no conflict of interest related to this article.
References
1. Lander ES et al; . Initial sequencing and analysis of the human genome. Nature. 2001 Feb 15;409(6822):860-921. doi: 10.1038/35057062.
2. Venter JC et al. The sequence of the human genome. Science. 2001 Feb 16;291(5507):1304-51. doi: 10.1126/science.1058040.
3. Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012 Aug 17;337(6096):816-21. doi: 10.1126/science.1225829.
4. Percy A. The American history of Rett syndrome. Pediatr Neurol. 2014 Jan;50(1):1-3. doi: 10.1016/j.pediatrneurol.2013.08.018.
5. Amir RE et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999 Oct;23(2):185-8. doi: 10.1038/13810.
6. Pitzianti MB et al. Rett syndrome in males: The different clinical course in two brothers with the same microduplication MECP2 Xq28. Int J Environ Res Public Health. 2019 Aug;16(17):3075. doi: 10.3390/ijerph16173075.
7. Ribeiro MC, MacDonald JL. Sex differences in Mecp2-mutant Rett syndrome model mice and the impact of cellular mosaicism in phenotype development. Brain Res. 2020 Feb 15;1729:146644. doi: 10.1016/j.brainres.2019.146644.
8. Bao X et al. X chromosome inactivation in Rett syndrome and its correlations with MECP2 mutations and phenotype. J Child Neurol. 2008 Jan;23(1):22-5. doi: 10.1177/0883073807307077.
9. Knudsen GPS et al. Increased skewing of X chromosome inactivation in Rett syndrome patients and their mothers. Eur J Hum Genet. 2006 Jul;14(11):1189-94. doi: 10.1038/sj.ejhg.5201682.
10. Chahil G et al. Rett syndrome in males: A case report and review of literature. Cureus. 2018;10(10):e3414. doi: 10.7759/cureus.3414.
11. Reichow B et al. Brief report: Systematic review of Rett syndrome in males. J Autism Dev Disord. 2015 Oct;45(10):3377-83. doi: 10.1007/s10803-015-2519-1.
12. Vashi N, Justice MJ. Treating Rett syndrome: From mouse models to human therapies. Mamm Genome. 2019 Jun;30(5-6):90-110. doi: 10.1007/s00335-019-09793-5.
13. Li CH et al. MeCP2 links heterochromatin condensates and neurodevelopmental disease. Nature. 2020 Oct;586(7829):440-4. doi: 10.1038/s41586-020-2574-4.
14. Schmidt A et al. MeCP2 and chromatin compartmentalization. Cells. 2020 Apr;9(4):878. doi: 10.3390/cells9040878.
15. Wang L et al. Rett syndrome-causing mutations compromise MeCP2-mediated liquid-liquid phase separation of chromatin. Cell Res. 2020 May;30(5):393-407. doi: 10.1038/s41422-020-0288-7.
16. Lin P et al. Transcriptome analysis of human brain tissue identifies reduced expression of complement complex C1Q genes in Rett syndrome. BMC Genomics. 2016;17:427. doi: 10.1186/s12864-016-2746-7.
17. Tudor M et al. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc Natl Acad Sci U S A. 2002 Nov 26;99(24):15536-41. doi: 10.1073/pnas.242566899.
18. Tillotson R et al. Radically truncated MeCP2 rescues Rett syndrome–like neurological defects. Nature. 2017 Oct 19;550(7676):398-401. doi: 10.1038/nature24058.
19. Connolly DR, Zhou Z. Genomic insights into MeCP2 function: A role for the maintenance of chromatin architecture. Curr Opin Neurobiol. 2019 Dec;59:174-9. doi: 10.1016/j.conb.2019.07.002.
20. Johnson BS et al. Biotin tagging of MeCP2 in mice reveals contextual insights into the Rett syndrome transcriptome. Nat Med. 2017 Oct;23(10):1203-14. doi: 10.1038/nm.4406.
21. Gabel HW et al. Disruption of DNA-methylation–dependent long gene repression in Rett syndrome. Nature. 2015 Jun 4;522(7554):89-93. doi: 10.1038/nature14319.
22. Lyst MJ, Bird A. Rett syndrome: A complex disorder with simple roots. Nat Rev Genet. 2015 May;16(5):261-75. doi: 10.1038/nrg3897.
23. Kuehner JN et al. Epigenetic regulations in neuropsychiatric disorders. Front Genet. 2019 Apr 4;10:268. doi: 10.3389/fgene.2019.00268.
24. Pejhan S, Rastegar M. Role of DNA methyl-CpG-binding protein MeCP2 in Rett syndrome pathobiology and mechanism of disease. Biomolecules. 2021 Jan;11(1):75. doi: 10.3390/biom11010075.
25. Fu C et al. Consensus guidelines on managing Rett syndrome across the lifespan. BMJ Paediatr Open. 2020;4(1):e000717. doi: 10.1136/bmjpo-2020-000717.
26. Operto FF et al. Epilepsy and genetic in Rett syndrome: A review. Brain Behav. 2019 May;9(5):e01250. doi: 10.1002/brb3.1250.
27. Nissenkorn A et al. Epilepsy in Rett syndrome – The experience of a National Rett Center. Epilepsia. 2010 Jul;51(7):1252-8. doi: 10.1111/j.1528-1167.2010.02597.x.
28. Welcome to the Rett cure community. Rett Syndrome Research Trust [Internet]. Updated Feb 8, 2022. Accessed Feb 23, 2022. https://reverserett.org.
29. About Rett syndrome. International Rett Syndrome Foundation [Internet]. Updated Jan 4, 2022. Accessed Feb 23, 2022. http://www.rettsyndrome.org.
30. Singh J, Santosh P. Key issues in Rett syndrome: Emotional, behavioural and autonomic dysregulation (EBAD) – A target for clinical trials. Orphanet J Rare Dis. 2018 Jul 31;13(1):128. doi: 10.1186/s13023-018-0873-8.
31. Banerjee A et al. Towards a better diagnosis and treatment of Rett syndrome: A model synaptic disorder. Brain. 2019 Feb 1;142(2):239-48. doi: 10.1093/brain/awy323.
32. Ager S et al. Parental experiences of scoliosis management in Rett syndrome. Disabil Rehabil. 2009 Sep 19;31(23):1917-24. doi: 10.1080/09638280902846392.
33. Budden SS. Management of Rett syndrome: A ten year experience. Neuropediatrics. 1995;26(2):75-7. doi: 10.1055/s-2007-979727.
34. Ip JPK et al. Rett syndrome: Insights into genetic, molecular and circuit mechanisms. Nat Rev Neurosci. 2018 Jun;19(6):368-82. doi: 10.1038/s41583-018-0006-3.
35. Acadia Pharmaceuticals Inc. Study of trofinetide for the treatment of girls and women with Rett syndrome (LAVENDER™). ClinicalTrials.gov identifier: NCT04181723. Updated Feb 17, 2022. Accessed Feb 23, 2022. https://clinicaltrials.gov/ct2/show/NCT04181723.
36. Acadia Pharmaceuticals announces positive top-line results from the pivotal phase 3 LAVENDER trial of trofinetide in Rett syndrome. Press release. Acadia Pharmaceuticals Inc. Dec 6, 2021. Accessed Feb 23, 2022. https://ir.acadia-pharm.com/news-releases/news-release-details/acadia-pharmaceuticals-announces-positive-top-line-results-1.
37. Copping NA et al. Emerging gene and small molecule therapies for the neurodevelopmental disorder Angelman syndrome. Neurotherapeutics. 2021 Jul;18(3):1535-47. doi: 10.1007/s13311-021-01082-x.
38. Riikonen R. Insulin-like growth factors in the pathogenesis of neurological diseases in children. Int J Mol Sci. 2017 Sep;18(10):2056. doi: 10.3390/ijms18102056.
39. Glaze DG et al; Rett 002 Study Group. Double-blind, randomized, placebo-controlled study of trofinetide in pediatric Rett syndrome. Neurology. 2019 April 16;92(16):e1912-e1925. doi: 10.1212/WNL.0000000000007316.
40. Acadia Pharmaceuticals Inc. An open-label study of trofinetide for the treatment of girls two to five years of age who have Rett syndrome (DAFFODIL™). ClinicalTrials.gov Identifier: NCT04988867. Updated Jan 24, 2022. Accessed Feb 23, 2022. https://clinicaltrials.gov/ct2/show/NCT04988867.
41. Anavex Life Sciences announces ANAVEX®2-73 meets primary and secondary endpoints in clinical trial. Press release. Anavex Life Sciences Corp. Dec 15, 2020. Accessed Feb 23, 2022. http://www.anavex.com/post/anavex-life-sciences-announces-anavex-2-73-meets-primary-and-secondary-endpoints-in-clinical-trial.
42. Anavex Life Sciences Corp. ANAVEX2-73 study in patients with Rett syndrome (AVATAR). ClinicalTrials.gov Identifier: NCT03941444. Updated Jan 27, 2022. Accessed Feb 23, 2022. https://clinicaltrials.gov/ct2/show/NCT03941444.
43. Anavex Life Sciences Corp. ANAVEX2-73 study in pediatric patients with Rett syndrome (EXCELLENCE). ClinicalTrials.gov Identifier: NCT04304482. Updated Sep 28, 2021. Accessed Feb 23, 2022. http://www.clinicaltrials.gov/ct2/show/NCT04304482.
44.Christ MG et al. The Sigma-1 receptor at the crossroad of proteostasis, neurodegeneration, and autophagy. Trends Neurosci. 2020 Feb;43(2):79-81. doi: 10.1016/j.tins.2019.12.002.
45. Kaufmann WE et al. ANAVEX®2-73 (blarcamesine), a sigma-1 receptor agonist, ameliorates neurologic impairments in a mouse model of Rett syndrome. Pharmacol Biochem Behav. 2019 Dec;187:172796. doi: 10.1016/j.pbb.2019.172796.
46. Brimson JM et al. Dipentylammonium binds to the sigma-1 receptor and protects against glutamate toxicity, attenuates dopamine toxicity and potentiates neurite outgrowth in various cultured cell lines. Neurotox Res. 2018 Aug;34(2):263-72. doi: 10.1007/s12640-018-9883-5.
47. Kourrich S et al. The sigma-1 receptor: roles in neuronal plasticity and disease. Trends Neurosci. 2012 Dec;35(12):762-71. doi: 10.1016/j.tins.2012.09.007.
48. Lappalainen R, Riikonen RS. High levels of cerebrospinal fluid glutamate in Rett syndrome. Pediatr Neurol. 1996 Oct;15(3):213-6. doi: 10.1016/s0887-8994(96)00218-4.
49. Hamberger A et al. Elevated CSF glutamate in Rett syndrome. Neuropediatrics. 1992;23(4):212-3. doi: 10.1055/s-2008-1071344.
50. Inacio P. FDA acts to support development of potential gene therapy, TSHA-102. Rett Syndrome News [Internet]. Oct 16, 2020. Accessed Feb 23, 2022. https://rettsyndromenews.com/2020/10/16/fda-grants-orphan-drug-rare-pediatric-disease-status-to-tsha-102-potential-rett-gene-therapy.
51. Sinnett SE et al. Engineered microRNA-based regulatory element permits safe high-dose miniMECP2 gene therapy in Rett mice. Brain. 2021 Nov 29;144(10):3005-19. doi: 10.1093/brain/awab182.
52. Le TTH et al. Efficient and precise CRISPR/Cas9-mediated MECP2 modifications in human-induced pluripotent stem cells. Front Genet. 2019 Jul 2;10:625. doi: 10.3389/fgene.2019.00625.
53. Koerner MV et al. Toxicity of overexpressed MeCP2 is independent of HDAC3 activity. Genes Dev. 2018;32(23-24):1514-24. doi: 10.1101/gad.320325.118.
54. Heckman LD et al. Rett-causing mutations reveal two domains critical for MeCP2 function and for toxicity in MECP2 duplication syndrome mice. Elife. 2014;3:e02676. doi: 10.7554/eLife.02676.
55. Neurogene announces new development program in Rett syndrome utilizing novel EXACT technology platform [Internet]. Accessed Aug 12, 2022. https://www.neurogene.com/press-releases/neurogene-announces-new-development-program-in-rett-syndrome-utilizing-novel-exact-technology-platform/
56. Anzalone AV et al. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. 2020 Jul;38(7):824-44. doi: 10.1038/s41587-020-0561-9.
57. Gaudelli NM, Komor AC, Rees HA, et al. Programmable base editing of A●T to G●C in genomic DNA without DNA cleavage. Nature. 2017 Nov 23;551(7681):464-71. doi: 10.1038/nature24644.
58. Coenraads M. How RSRT is driving the search for a Rett cure. Rett Syndrome Research Trust [Internet]. Dec 7, 2021. Accessed Feb 23, 2022. https://rettnews.org/articles/how-rsrt-is-driving-the-search-for-a-rett-cure.
59. Cutting-edge technologies to repair the underlying mutations that cause Rett. Rett Syndrome Research Trust [Internet]. Updated Nov 3, 2021. Accessed Feb 23, 2022. https://reverserett.org/research/cures/gene-editing.
60. Sinnamon JR et al. In vivo repair of a protein underlying a neurological disorder by programmable RNA editing. Cell Rep. 2020 Jul 14;32(2):107878. doi: 10.1016/j.celrep.2020.107878.
61. Sinnamon JR et al. Site-directed RNA repair of endogenous Mecp2 RNA in neurons. Proc Natl Acad Sci U S A. 2017 Oct 31;114(44):E9395-E9402. doi: 10.1073/pnas.1715320114.
62. Pipeline. VICO Therapeutics [Internet]. Updated Nov 5, 2021. Accessed Feb 23, 2022. https://vicotx.com/pipeline.
63. Therapeutics platform. Shape Therapeutics [Internet]. Updated Feb 20, 2021. Accessed Feb 23, 2022.
https://live-shapetx.pantheonsite.io/therapeutics-platform.
64. Koeberl DD et al. Glycogen storage disease types I and II: Treatment updates. J Inherit Metab Dis. 2007 Apr;30(2):159-64. doi: 10.1007/s10545-007-0519-9.
The dream of curing genetic disorders has been a persistent but elusive goal, even before the human genome was mapped. Once mapping of the human genome was complete in 2001, an entirely new avenue of potential treatments and cures for genetic diseases and disorders was opened.1,2
The disorders best suited for targeted gene therapy are monogenic; however, tools and delivery methods for editing the human genome were limited and difficult to apply, until the advent of the CRISPR system in 2012.3 CRISPR (an acronym of clustered regularly interspaced short palindromic repeats) has changed the way in which gene therapy strategies are pursued, with dozens of companies leveraging a variety of platforms to create potentially life-changing therapies for devastating rare diseases and disorders.
One of the rare monogenic disorders that is embracing multiple gene therapy approaches is Rett syndrome, a rare, debilitating neurodevelopmental disorder. In this review, we explore the molecular cause of Rett syndrome, disease presentation, current treatments, ongoing clinical trials, and therapies that are on the horizon.
Underlying molecular cause
Rett syndrome is caused by mutations in, or the absence of, the MECP2 gene, which produces methyl-CpG binding protein 2 (MECP2). The syndrome was first described clinically in 1954 by the Austrian physician Andreas Rett; it would take until 1982 before the disorder was officially named, eponymously, in a seminal paper by Hagberg.4 After Hagberg’s characterization, Rett syndrome became the predominant global clinical diagnosis identified among cognitively impaired females, with an incidence of 1 in every 10,000 to 15,000.4
In 1999, mutations in, and deletions of, MECP2 were identified as the cause of Rett syndrome.4,5 MECP2 is located on the X chromosome, in the Xq28 region, making Rett syndrome an X-linked dominant disorder.6 Rett syndrome is seen predominantly in females who are mosaic for mutant or deleted MECP2. Random X chromosome inactivation results in some cells expressing the mutant MECP2 allele and other cells expressing the normal functioning MECP2 allele; the percentage of cells expressing the normal allele correlates with the degree of syndrome severity.7-9
The incidence of Rett syndrome is much lower in males, in whom the syndrome was originally thought to be lethal; many observed male cases are either mosaic or occur in XXY males.10,11
Approximately 95% of cases of Rett syndrome are due to de novo mutations in MECP2, with a handful of specific mutations and large deletions accounting for more than 85% of cases.12 The fact that Rett syndrome is monogenic and most cases are caused by, in total, only a handful of mutations or deletions makes the syndrome a promising candidate for gene therapy.
At the molecular level, it has been observed that the MECP2 mutations of Rett syndrome lead to loss of gene function, thus disrupting the ability of the MECP2 nuclear protein to regulate global gene transcription through its binding to methylated DNA sites.12 A large percentage of these missense and nonsense mutations lead to a truncated or nonfunctional protein.12
One of the ways in which MECP2 regulates transcription is as a component of heterochromatin condensates and by separation of heterochromatin and euchromatin.13-15 It has been observed that the cells of Rett syndrome patients have an altered chromatin state, potentially contributing to transcriptional dysregulation.16,17 Several mutations observed in Rett syndrome patients occur in crucial domains for heterochromatin condensate formation, which helps explain this cellular phenotype.13 Introduction of a engineered “mini” MECP2 in a murine model of Rett syndrome has resulted in partial rescue of heterochromatin condensate formation and transcriptional regulation – fostering the hypothesis that correcting those genetic changes could lead to a potential therapy.18
Beyond the role of MECP2 in heterochromatin condensate formation, the gene interacts with more than 40 proteins that have diverse roles in cellular function, epigenetic modulation, and neuronal development. This volume of interactions contributes to MECP2 being a global gene regulatory protein that has far-reaching effects on transcriptional regulation across the genome.19-22
Epigenetic dysregulation has been associated with neurodevelopmental and neuropsychiatric disorders.23 Both insulin-like growth factor 1 (IGF-1) and brain-derived neurotrophic factor are transcriptional targets of MECP2, and are involved in neuronal differentiation, synaptic function, and neurite outgrowth.12 This helps explain the neurodevelopmental phenotypes observed in MECP2-mutated patients.
Notably, although Rett syndrome patients experience neurodevelopmental phenotypes at the cellular level, neuronal death is not readily observed. That observation provides hope that an interventional therapy after onset of symptoms might still be of benefit.
Presentation
Early neurotypical development. A hallmark of Rett syndrome is neurotypical physical and mental development until 6 to 24 months of age.
Stagnation is the first stage of the syndrome, involving a small but rapid decline in habitual milestones, such motor and language skills.12 Subtle signs, such as microcephaly and hypotonia, can also arise at this time but might be missed.24
Rapid regression follows stagnation. Speech and motor delays and impaired gait and breathing occur;12,25 purposeful hand skills are lost, replaced by repetitive hand-wringing movements that are a hallmark of the syndrome.12,24 Seizures are observed; they become more common during the next stage.12
Plateau. Language advances can be observed, but further deficits are seen in motor skills and hand coordination.12
Late motor deterioration stage. Late physical deficits develop, leading to lifelong impairments. The physical deficits observed are the result of severe muscle weakness, usually resulting in wheelchair dependency.12
Plateau. Patients then reach a second plateau. Regression stops; deficient physical and cognitive states stabilize and are maintained.25
At all stages of Rett syndrome, the following are observed:
- Gastrointestinal problems.
- Sleep disturbances.
- Abnormal cardiorespiratory coupling.
- Greater-than-expected mortality.12
Final regression. The patient is fully dependent for the rest of their lifespan, partially due to seizure activity.26,27
A life-changing diagnosis
A diagnosis of Rett syndrome is life-changing for a patient’s family; access to supportive groups of other patients and their families is extremely beneficial. Two helpful organizations – the Rett Syndrome Research Trust28 and International Rett Syndrome Foundation,29 – offer patient support and community and fund research.
Because X chromosome inactivation is random in Rett syndrome, the individual patient can present with a wide variety of phenotypic combinations – making the patient, and their needs, unique.12 During stages of regression, patients often experience emotional dysregulation and anxiety, which is attributable to their increasing physical difficulties.30 They often exhibit combinations of uncontrolled movements, including repetitive rocking, scratching, and self-injurious behavior.30 For most, regression subsides after the first 5 years of alternating development and regression; after that, their ultimate symptoms persist for life.25
As patients mature, they need to be monitored for proper nutrition and scoliosis.25 As adults, they are at risk of pneumonia, respiratory distress, status epilepticus, osteopenia, and lack of adequate food or water because of impaired ability to feed.25
The lifespan of Rett syndrome patients has increased, thanks to improvements in health care, advances in technology, and early genetic testing, which allows for earlier diagnosis, intervention, and management of symptoms.
Current treatments
When a female patient presents with regression and loss of milestones, sequencing of MECP2 is performed to verify whether Rett syndrome is the cause, by detecting any of the known mutations. Multiplex ligation-dependent probe amplification is also performed to detect major deletions.25
All available treatments for Rett syndrome are symptomatic; intensive early intervention is practiced.31 Multidisciplinary management – medical, psychiatric, and physical – is introduced almost immediately after diagnosis. Following diagnosis, patients are prescribed anti-seizure, sleep, and anxiety medications.31 Electroencephalography can be performed to identify seizure type. Neuromuscular blockage drugs can be prescribed to help with gait and stereotypic hand movements.25
Handguards or splints to the elbows can be prescribed by an occupational therapist to improve hand movement.25 Physical therapy can improve mobility; hydrotherapy and hippotherapy have been successful in helping to maintain mobility and muscle support.32,33 Nutritional management is implemented to control caloric intake and maintain the vitamin D level.31 Some patients experience constipation and urinary retention, putting them at risk of nephrolithiasis.
Once the signs and symptoms of Rett syndrome progress, and milestones regress to a certain point, patients need constant, full-time care for the rest of their lives.34 As symptomatic interventions have greatly improved patient outcomes and it has been shown that about 70% can reach adulthood with a potential lifespan of about 50 years.25
Although there is no cure for Rett syndrome and treatments are symptomatic, ongoing studies – both clinical and preclinical – offer promise that treatments will be developed that work at molecular and genetic levels.
Clinical trials
Advances in understanding of Rett syndrome have led to many therapies in clinical trials, several of which show promise.
Trofinetide. One of the most promising targets for downstream therapy, mentioned earlier, is IGF-1, which was the target of a successful phase 3 clinical trial, LAVENDER (sponsored by Acadia Pharmaceuticals).35,36 This trial studied trofinetide, a synthetic IGF-1 analog that inhibits neuroinflammation, restores glial function, corrects synaptic deficiencies, and regulates oxidative stress response.12,37,38 Initial results from phase 2 and phase 3 trials indicate improved scores for treated patients in the Rett syndrome Behaviour Questionnaire (RSBQ) and Clinical Global Impression–Improvement (CGI-I) scores, while also showing improvements in the Communication and Symbolic Behavior Scales Developmental Profile Infant–Toddler Checklist–Social composite score.36,39
The most common adverse events seen with trofinetide were diarrhea and vomiting.
Acadia Pharmaceuticals has filed for approval of a new drug application for trofinetide with the Food and Drug Administration, for which the company has been granted Fast Track Status and orphan drug designations. Most (95%) subjects in the phase 3 LAVENDER trial elected to continue taking trofinetide in the subsequent open-label Lilac and Lilac-2 extension studies.36 A current open-label phase 2/3 trial is recruiting patients 2 to 5 years of age to evaluate trofinetide.40 It is expected that, in the near future, this could be a drug given to Rett patients as an FDA-approved treatment.
Blarcamesine. Another small molecule drug, blarcamesine (also known as ANAVEX2-73), a sigma-1 receptor agonist, produced promising results in phase 2 clinical trials in adult Rett syndrome patients. The drug is in a phase 2/3 clinical trial for pediatric Rett syndrome patients (sponsored by Anavex Life Sciences).41-43
Phase 2 results indicated statistically significant and clinically meaningful improvement in RSBQ and CGI-I scores with blarcamesine. Improvement was initially observed within 4 weeks after the start of treatment and was sustained throughout the study. The drug was shown to be well tolerated, with minimal adverse effects; no serious adverse events were recorded. These results were observed in adult patients, demonstrating that improvements in Rett syndrome are possible even after regression.
Blarcamesine activates the sigma 1 receptor, which is pivotal to restoring cellular homeostasis and restoring neuroplasticity – deficiencies of which have been linked to autophagy and glutamate toxicity. The drug has also been explored as a potential treatment for other neurological disorders.44-47 Improvements in blarcamesine-treated patients further correlated with lower levels of glutamate in cerebrospinal fluid, which is a Rett syndrome biomarker, supporting the proposition that behavioral improvements were due to drug intervention.48,49 The phase 2 trial was modified into a phase 3 trial and additional endpoints were added.41-43
All patients in the phase 2 adult trial elected to continue in the extension study.
Based on these promising data, Anavex is pursuing an approval pathway for adult patients, while continuing dosage optimization phase 2/3 trials and recruitment for a pediatric trial.42,43
Is the future about gene therapy?
TSHA-102 (miniMECP2). Taysha Gene Therapies is developing a promising gene therapy, TSHA-102, for Rett syndrome, and is aiming to begin phase 1/2 clinical trials in 2022.50 The technology for this therapy relies on the delivery of a fragment of MECP2 (known as miniMECP2), which is regulated by a built-in microRNA regulator (miR-responsive auto-regulatory element, or miRARE) to help ameliorate MECP2 dosage toxicity. (Overexpression of MECP2 is toxic to neurons, which has made traditional [so to speak] gene replacement therapy difficult in Rett syndrome: Levels of MECP2 need to be tightly regulated, and the Taysha microRNA technology regulates levels of miniMECP2, thus reducing toxicity.)
The Taysha microRNA technology has yielded promising results in mouse studies for Rett syndrome; results indicate a lengthening of lifespan and delayed onset of gait abnormalities.51 TSHA-102 is in the preclinical stage but offers promise that it will be the first gene therapy for Rett syndrome to enter clinical trials.
As the field of gene therapy advances, several promising technologies are on the horizon that could potentially have disease-altering impacts on Rett syndrome. These therapies are divided into two broad categories: those at the gene level and those at the transcription and protein level. A few of these approaches are highlighted below.
Gene replacement involves adding a full or partial copy of MECP2 to neuronal cells. This type of therapy presents challenges, from delivery of the new gene to dosage concerns, because MECP2 can be toxic if overexpressed.52-54 Groundbreaking work was done in mouse models involving truncated MECP2, exhibiting phenotypic rescue and validating the gene-replacement approach.18 This strategy is being pursued by Neurogene, which has a uinique technology that allows for tuning of the gene’s expression to get the correct protein levels in the patient. Promising data was presented this year at the American Society of Gene and Cell Therapy conference.55
Early gene replacement therapy studies also laid the foundation for the minMECP2 and microRNA approach being used by Taysha Gene Therapies (discussed above).51
“Correcting” DNA mutations. A different approach at the genetic level involves “correcting” mutations in MECP2 at the DNA level. This is possible because, in a large subset of Rett syndrome patients who have the same missense or nonsense mutations, by using CRISPR, a gene editing tool (discussed above) a single base pair can be corrected.56,57 Previous research, in a Rett syndrome-model of induced pluripotent stem cells, showed that this type of editing is possible – and effective.52 An approach with particular promise involves use of a class of CRISPR proteins known as base editors that are able to specifically alter a single base of DNA.57 The technique has the potential to address many of the mutations seen in Rett syndrome; research on this type of technology is being pursued by Beam Therapeutics, and has the potential to impact Rett syndrome.58
Another promising “correction” approach is exonic editing, in which a much larger section of DNA – potentially, exons 3 and 4, which, taken together, comprise 97% of known MECP2 mutations seen in Rett syndrome – are replaced.59
In both CRISPR and exonic editing therapeutic approaches, endogenous levels of MECP2 expression would be maintained. Of note, both approaches are being pursued for use in treating other genetic disorders, which provides a boost in scaling-up work on addressing safety and efficacy concerns that accompany gene-editing approaches.58 One advantage to the DNA correction approach is that is has the potential to be a “one-and-done” treatment.
“Correcting” RNA. Beyond directly editing DNA, several therapeutic approaches are exploring the ability to edit RNA or to provide the protein directly to cells as enzyme replacement therapy. Such an RNA correction strategy leverages a technology that takes advantage of cells’ natural RNA editor, known as adenosine deaminase acting on RNA (ADAR), which corrects errors in cells’ RNA by providing specific guides to the cell. ADAR can be targeted to fix mutations in the MECP2 RNA transcript, resulting in a “corrected” MECP2 protein.60,61 This technology has delivered promising proof-of-concept evidence in cells and in murine models, and is in the therapeutic pipeline at VICO Therapeutics.62
Shape Therapeutics has also leveraged ADAR to “correct” mutated RNA; Rett syndrome is among the top priorities in the company’s pipeline.
Worth noting is that there are several advantages to the “correction” approach:
- Leveraging internal repair mechanisms minimizes the immune response.
- The flexibility of correction means that it can be used to address many of the mutations that cause Rett syndrome.63
Enzyme replacement therapy, in which the MECP2 protein produced by MECP2 would be directly replaced, is being explored in Rett syndrome patients. This technology has been used successfully in Pompe disease; however, Rett syndrome presents its own challenge because MECP2 needs to be delivered to the brain and neuronal cells.64
Where does this work stand? The technologies described in this section are in preclinical stages of study. Nonetheless, it is expected that several will enter human clinical trials during the next 5 years.
Conclusion
A diagnosis of Rett syndrome is a life-altering event for patients and their family. But there is more hope than ever for effective therapies and, eventually, a cure.
Multiple late-stage clinical trials in progress are demonstrating promising results from therapeutic products, with minimal adverse events. Remarkably, these interventions have delivered improvements to adult patients after regression has stabilized. With rapid progress being made in the field of gene therapy, several technologies for which are focused on Rett syndrome, a hopeful picture is emerging: that therapeutic intervention will be possible before regression, thus effectively treating and, potentially, even curing Rett syndrome.
The landscape is broadening. Add to this hope for approved therapies is the fact that Rett syndrome isn’t the only target being pursued with such strategies; in fact, researchers in the larger field of neurodevelopmental disorder study are working together to find common solutions to shared challenges – from how therapies are designed and delivered to how toxicity is minimized. Much of what is being explored in the Rett syndrome field is also under investigation in other neurodevelopmental syndromes, including Angelman, Prader-Willi, chromosome 15q11.2-13.1 duplication (dup15q), and Fragile X syndrome. This kind of parallel investigation benefits all parties and optimizes a treatment platform so that it can be applied across more than a single disorder.
Like many monogenic disorders, Rett syndrome is entering an exciting stage – at which the words “treatment” and “cure” can be spoken with intent and vision, not just wide-eyed optimism. These words portend real promise for patients who carry the weight of a diagnosis of Rett syndrome, and for their families.
Ms. Ambrose is a student in the master’s of science in human genetics and genomic data analytics program, Keck Graduate Institute, Claremont, Calif. Dr. Bailus is an assistant professor of genetics, Keck Graduate Institute. The authors report no conflict of interest related to this article.
References
1. Lander ES et al; . Initial sequencing and analysis of the human genome. Nature. 2001 Feb 15;409(6822):860-921. doi: 10.1038/35057062.
2. Venter JC et al. The sequence of the human genome. Science. 2001 Feb 16;291(5507):1304-51. doi: 10.1126/science.1058040.
3. Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012 Aug 17;337(6096):816-21. doi: 10.1126/science.1225829.
4. Percy A. The American history of Rett syndrome. Pediatr Neurol. 2014 Jan;50(1):1-3. doi: 10.1016/j.pediatrneurol.2013.08.018.
5. Amir RE et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999 Oct;23(2):185-8. doi: 10.1038/13810.
6. Pitzianti MB et al. Rett syndrome in males: The different clinical course in two brothers with the same microduplication MECP2 Xq28. Int J Environ Res Public Health. 2019 Aug;16(17):3075. doi: 10.3390/ijerph16173075.
7. Ribeiro MC, MacDonald JL. Sex differences in Mecp2-mutant Rett syndrome model mice and the impact of cellular mosaicism in phenotype development. Brain Res. 2020 Feb 15;1729:146644. doi: 10.1016/j.brainres.2019.146644.
8. Bao X et al. X chromosome inactivation in Rett syndrome and its correlations with MECP2 mutations and phenotype. J Child Neurol. 2008 Jan;23(1):22-5. doi: 10.1177/0883073807307077.
9. Knudsen GPS et al. Increased skewing of X chromosome inactivation in Rett syndrome patients and their mothers. Eur J Hum Genet. 2006 Jul;14(11):1189-94. doi: 10.1038/sj.ejhg.5201682.
10. Chahil G et al. Rett syndrome in males: A case report and review of literature. Cureus. 2018;10(10):e3414. doi: 10.7759/cureus.3414.
11. Reichow B et al. Brief report: Systematic review of Rett syndrome in males. J Autism Dev Disord. 2015 Oct;45(10):3377-83. doi: 10.1007/s10803-015-2519-1.
12. Vashi N, Justice MJ. Treating Rett syndrome: From mouse models to human therapies. Mamm Genome. 2019 Jun;30(5-6):90-110. doi: 10.1007/s00335-019-09793-5.
13. Li CH et al. MeCP2 links heterochromatin condensates and neurodevelopmental disease. Nature. 2020 Oct;586(7829):440-4. doi: 10.1038/s41586-020-2574-4.
14. Schmidt A et al. MeCP2 and chromatin compartmentalization. Cells. 2020 Apr;9(4):878. doi: 10.3390/cells9040878.
15. Wang L et al. Rett syndrome-causing mutations compromise MeCP2-mediated liquid-liquid phase separation of chromatin. Cell Res. 2020 May;30(5):393-407. doi: 10.1038/s41422-020-0288-7.
16. Lin P et al. Transcriptome analysis of human brain tissue identifies reduced expression of complement complex C1Q genes in Rett syndrome. BMC Genomics. 2016;17:427. doi: 10.1186/s12864-016-2746-7.
17. Tudor M et al. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc Natl Acad Sci U S A. 2002 Nov 26;99(24):15536-41. doi: 10.1073/pnas.242566899.
18. Tillotson R et al. Radically truncated MeCP2 rescues Rett syndrome–like neurological defects. Nature. 2017 Oct 19;550(7676):398-401. doi: 10.1038/nature24058.
19. Connolly DR, Zhou Z. Genomic insights into MeCP2 function: A role for the maintenance of chromatin architecture. Curr Opin Neurobiol. 2019 Dec;59:174-9. doi: 10.1016/j.conb.2019.07.002.
20. Johnson BS et al. Biotin tagging of MeCP2 in mice reveals contextual insights into the Rett syndrome transcriptome. Nat Med. 2017 Oct;23(10):1203-14. doi: 10.1038/nm.4406.
21. Gabel HW et al. Disruption of DNA-methylation–dependent long gene repression in Rett syndrome. Nature. 2015 Jun 4;522(7554):89-93. doi: 10.1038/nature14319.
22. Lyst MJ, Bird A. Rett syndrome: A complex disorder with simple roots. Nat Rev Genet. 2015 May;16(5):261-75. doi: 10.1038/nrg3897.
23. Kuehner JN et al. Epigenetic regulations in neuropsychiatric disorders. Front Genet. 2019 Apr 4;10:268. doi: 10.3389/fgene.2019.00268.
24. Pejhan S, Rastegar M. Role of DNA methyl-CpG-binding protein MeCP2 in Rett syndrome pathobiology and mechanism of disease. Biomolecules. 2021 Jan;11(1):75. doi: 10.3390/biom11010075.
25. Fu C et al. Consensus guidelines on managing Rett syndrome across the lifespan. BMJ Paediatr Open. 2020;4(1):e000717. doi: 10.1136/bmjpo-2020-000717.
26. Operto FF et al. Epilepsy and genetic in Rett syndrome: A review. Brain Behav. 2019 May;9(5):e01250. doi: 10.1002/brb3.1250.
27. Nissenkorn A et al. Epilepsy in Rett syndrome – The experience of a National Rett Center. Epilepsia. 2010 Jul;51(7):1252-8. doi: 10.1111/j.1528-1167.2010.02597.x.
28. Welcome to the Rett cure community. Rett Syndrome Research Trust [Internet]. Updated Feb 8, 2022. Accessed Feb 23, 2022. https://reverserett.org.
29. About Rett syndrome. International Rett Syndrome Foundation [Internet]. Updated Jan 4, 2022. Accessed Feb 23, 2022. http://www.rettsyndrome.org.
30. Singh J, Santosh P. Key issues in Rett syndrome: Emotional, behavioural and autonomic dysregulation (EBAD) – A target for clinical trials. Orphanet J Rare Dis. 2018 Jul 31;13(1):128. doi: 10.1186/s13023-018-0873-8.
31. Banerjee A et al. Towards a better diagnosis and treatment of Rett syndrome: A model synaptic disorder. Brain. 2019 Feb 1;142(2):239-48. doi: 10.1093/brain/awy323.
32. Ager S et al. Parental experiences of scoliosis management in Rett syndrome. Disabil Rehabil. 2009 Sep 19;31(23):1917-24. doi: 10.1080/09638280902846392.
33. Budden SS. Management of Rett syndrome: A ten year experience. Neuropediatrics. 1995;26(2):75-7. doi: 10.1055/s-2007-979727.
34. Ip JPK et al. Rett syndrome: Insights into genetic, molecular and circuit mechanisms. Nat Rev Neurosci. 2018 Jun;19(6):368-82. doi: 10.1038/s41583-018-0006-3.
35. Acadia Pharmaceuticals Inc. Study of trofinetide for the treatment of girls and women with Rett syndrome (LAVENDER™). ClinicalTrials.gov identifier: NCT04181723. Updated Feb 17, 2022. Accessed Feb 23, 2022. https://clinicaltrials.gov/ct2/show/NCT04181723.
36. Acadia Pharmaceuticals announces positive top-line results from the pivotal phase 3 LAVENDER trial of trofinetide in Rett syndrome. Press release. Acadia Pharmaceuticals Inc. Dec 6, 2021. Accessed Feb 23, 2022. https://ir.acadia-pharm.com/news-releases/news-release-details/acadia-pharmaceuticals-announces-positive-top-line-results-1.
37. Copping NA et al. Emerging gene and small molecule therapies for the neurodevelopmental disorder Angelman syndrome. Neurotherapeutics. 2021 Jul;18(3):1535-47. doi: 10.1007/s13311-021-01082-x.
38. Riikonen R. Insulin-like growth factors in the pathogenesis of neurological diseases in children. Int J Mol Sci. 2017 Sep;18(10):2056. doi: 10.3390/ijms18102056.
39. Glaze DG et al; Rett 002 Study Group. Double-blind, randomized, placebo-controlled study of trofinetide in pediatric Rett syndrome. Neurology. 2019 April 16;92(16):e1912-e1925. doi: 10.1212/WNL.0000000000007316.
40. Acadia Pharmaceuticals Inc. An open-label study of trofinetide for the treatment of girls two to five years of age who have Rett syndrome (DAFFODIL™). ClinicalTrials.gov Identifier: NCT04988867. Updated Jan 24, 2022. Accessed Feb 23, 2022. https://clinicaltrials.gov/ct2/show/NCT04988867.
41. Anavex Life Sciences announces ANAVEX®2-73 meets primary and secondary endpoints in clinical trial. Press release. Anavex Life Sciences Corp. Dec 15, 2020. Accessed Feb 23, 2022. http://www.anavex.com/post/anavex-life-sciences-announces-anavex-2-73-meets-primary-and-secondary-endpoints-in-clinical-trial.
42. Anavex Life Sciences Corp. ANAVEX2-73 study in patients with Rett syndrome (AVATAR). ClinicalTrials.gov Identifier: NCT03941444. Updated Jan 27, 2022. Accessed Feb 23, 2022. https://clinicaltrials.gov/ct2/show/NCT03941444.
43. Anavex Life Sciences Corp. ANAVEX2-73 study in pediatric patients with Rett syndrome (EXCELLENCE). ClinicalTrials.gov Identifier: NCT04304482. Updated Sep 28, 2021. Accessed Feb 23, 2022. http://www.clinicaltrials.gov/ct2/show/NCT04304482.
44.Christ MG et al. The Sigma-1 receptor at the crossroad of proteostasis, neurodegeneration, and autophagy. Trends Neurosci. 2020 Feb;43(2):79-81. doi: 10.1016/j.tins.2019.12.002.
45. Kaufmann WE et al. ANAVEX®2-73 (blarcamesine), a sigma-1 receptor agonist, ameliorates neurologic impairments in a mouse model of Rett syndrome. Pharmacol Biochem Behav. 2019 Dec;187:172796. doi: 10.1016/j.pbb.2019.172796.
46. Brimson JM et al. Dipentylammonium binds to the sigma-1 receptor and protects against glutamate toxicity, attenuates dopamine toxicity and potentiates neurite outgrowth in various cultured cell lines. Neurotox Res. 2018 Aug;34(2):263-72. doi: 10.1007/s12640-018-9883-5.
47. Kourrich S et al. The sigma-1 receptor: roles in neuronal plasticity and disease. Trends Neurosci. 2012 Dec;35(12):762-71. doi: 10.1016/j.tins.2012.09.007.
48. Lappalainen R, Riikonen RS. High levels of cerebrospinal fluid glutamate in Rett syndrome. Pediatr Neurol. 1996 Oct;15(3):213-6. doi: 10.1016/s0887-8994(96)00218-4.
49. Hamberger A et al. Elevated CSF glutamate in Rett syndrome. Neuropediatrics. 1992;23(4):212-3. doi: 10.1055/s-2008-1071344.
50. Inacio P. FDA acts to support development of potential gene therapy, TSHA-102. Rett Syndrome News [Internet]. Oct 16, 2020. Accessed Feb 23, 2022. https://rettsyndromenews.com/2020/10/16/fda-grants-orphan-drug-rare-pediatric-disease-status-to-tsha-102-potential-rett-gene-therapy.
51. Sinnett SE et al. Engineered microRNA-based regulatory element permits safe high-dose miniMECP2 gene therapy in Rett mice. Brain. 2021 Nov 29;144(10):3005-19. doi: 10.1093/brain/awab182.
52. Le TTH et al. Efficient and precise CRISPR/Cas9-mediated MECP2 modifications in human-induced pluripotent stem cells. Front Genet. 2019 Jul 2;10:625. doi: 10.3389/fgene.2019.00625.
53. Koerner MV et al. Toxicity of overexpressed MeCP2 is independent of HDAC3 activity. Genes Dev. 2018;32(23-24):1514-24. doi: 10.1101/gad.320325.118.
54. Heckman LD et al. Rett-causing mutations reveal two domains critical for MeCP2 function and for toxicity in MECP2 duplication syndrome mice. Elife. 2014;3:e02676. doi: 10.7554/eLife.02676.
55. Neurogene announces new development program in Rett syndrome utilizing novel EXACT technology platform [Internet]. Accessed Aug 12, 2022. https://www.neurogene.com/press-releases/neurogene-announces-new-development-program-in-rett-syndrome-utilizing-novel-exact-technology-platform/
56. Anzalone AV et al. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. 2020 Jul;38(7):824-44. doi: 10.1038/s41587-020-0561-9.
57. Gaudelli NM, Komor AC, Rees HA, et al. Programmable base editing of A●T to G●C in genomic DNA without DNA cleavage. Nature. 2017 Nov 23;551(7681):464-71. doi: 10.1038/nature24644.
58. Coenraads M. How RSRT is driving the search for a Rett cure. Rett Syndrome Research Trust [Internet]. Dec 7, 2021. Accessed Feb 23, 2022. https://rettnews.org/articles/how-rsrt-is-driving-the-search-for-a-rett-cure.
59. Cutting-edge technologies to repair the underlying mutations that cause Rett. Rett Syndrome Research Trust [Internet]. Updated Nov 3, 2021. Accessed Feb 23, 2022. https://reverserett.org/research/cures/gene-editing.
60. Sinnamon JR et al. In vivo repair of a protein underlying a neurological disorder by programmable RNA editing. Cell Rep. 2020 Jul 14;32(2):107878. doi: 10.1016/j.celrep.2020.107878.
61. Sinnamon JR et al. Site-directed RNA repair of endogenous Mecp2 RNA in neurons. Proc Natl Acad Sci U S A. 2017 Oct 31;114(44):E9395-E9402. doi: 10.1073/pnas.1715320114.
62. Pipeline. VICO Therapeutics [Internet]. Updated Nov 5, 2021. Accessed Feb 23, 2022. https://vicotx.com/pipeline.
63. Therapeutics platform. Shape Therapeutics [Internet]. Updated Feb 20, 2021. Accessed Feb 23, 2022.
https://live-shapetx.pantheonsite.io/therapeutics-platform.
64. Koeberl DD et al. Glycogen storage disease types I and II: Treatment updates. J Inherit Metab Dis. 2007 Apr;30(2):159-64. doi: 10.1007/s10545-007-0519-9.
The paradox of Pompe disease
Until 2006, when a breakthrough therapy first made treatment possible, Pompe disease was a little-known metabolic myopathy fatal to infants. Those with later-onset disease experienced progressive, often severe disability into adulthood.
In this rare autosomal recessive disorder, which occurs in approximately one in 40,000 births worldwide, a deficiency or absence of the enzyme acid alpha-glucosidase causes glycogen to build up in the lysosomes of cells. While many tissues are affected, skeletal and cardiac muscle see the earliest involvement, with muscle hypotonia, cardiomyopathy, and breathing difficulties (mainly due to diaphragm weakness) comprising the hallmark symptoms of the infantile form. Muscle weakness and progressive respiratory failure are prominent in later-onset disease.

The spectrum of severity and age of onset in Pompe disease is linked to combinations of mutations on the GAA gene, some of which destroy the body’s ability to produce acid alpha-glucosidase whereas others merely hamper it. Less enzyme produced in the body generally corresponds with more severe disease activity.
The most severe end of the disease spectrum, or “classic infantile Pompe disease,” presents at birth and is recognized in early infancy. Until treatment with enzyme replacement therapy (ERT) became available, patients usually died of cardiorespiratory failure within their first year of life. With therapy, patients have survived into their 20s and beyond. Late-onset disease is a far broader category in which patients can present at any time from their first year, including into middle age.
The emergence in 2006 of alglucosidase alfa (Lumizyme, Sanofi Genzyme), an ERT used long-term to improve survival and slow progression in children and adults, resulted in a boom of research interest, a push to timelier diagnosis, and – with patients living longer – a more thorough understanding of the natural history of Pompe disease. In addition to the usual clinical picture of progressive muscle weakness, difficulty breathing, and cardiomyopathy, investigators are seeing nervous system involvement in patients with Pompe disease.
To learn more, Neurology Reviews talked to two global experts in Pompe disease: Priya Kishnani, MD, of Duke University in Durham, N.C., and Antonio Toscano, MD, of the University of Messina, in Messina, Italy.
Diagnosis: Still room to improve
“Most neurologists will encounter a patient with Pompe disease,” said Dr. Kishnani, who has been working with Pompe for her entire career as a pediatrician and medical geneticist, treating patients of all ages and disease phenotypes.
“In newborns, diagnosis is more straightforward, because you’ve got an enlarged heart,” she said. And thanks to efforts of researchers like Dr. Kishnani and Pompe advocacy groups, Pompe disease is now a part of the RUSP (Recommended Uniform Screening Panel) for newborns; currently 28 U.S. states are screening for Pompe disease.
“The challenge really is for the later-onset cases, which are 80% of all cases,” Dr. Kishnani said.
Previously, muscle biopsies were the first step toward diagnosis. Dried blot spot assays to detect enzyme deficiency have since become the standard, along with other biochemical tests. Confirmation of the diagnosis is through gene sequencing panels to detect GAA mutations.
“Now that there is a treatment for Pompe disease and the availability of blood-based testing, many previously undiagnosed patients with limb girdle weakness are evaluated and the diagnostic odyssey ends,” Dr. Kishnani said. “But there is still a diagnostic delay, and many cases remain undiagnosed.”
Routine blood tests for creatine kinase and for liver enzymes can help point to Pompe disease. But elevated liver enzymes are often misinterpreted. “It’s about the ratios,” Dr. Kishnani said. “ALT is usually much more elevated if it is coming from a liver cause, and AST is usually higher than ALT if it is coming from muscle. But patients often end up getting a liver biopsy due to so-called elevated liver enzymes. As the workup continues, it is often later recognized that the source of the elevated enzymes is muscle involvement, and a referral to the geneticist or neurologist is made. Only then is appropriate testing to confirm a diagnosis initiated.”
Dr. Toscano, a neurologist who specializes in Pompe disease and other myopathies and who has published on tools for diagnosing late-onset Pompe disease,1 said that clinicians should be vigilant when evaluating any patient with limb girdle weakness and elevated creatine kinase (CK) – “especially if the CK is under 2,000,” he said, “because it is very rare that patients with Pompe disease have a more elevated CK than that.”
“Elevated CK, myalgia, and exercise intolerance” should prompt clinicians to suspect Pompe disease in a patient of any age, Dr. Toscano said. “When you come across this, you should be very persistent and get to the end of the story.”
Dr. Toscano noted that the blood spot assay, while an important early step, is not fully diagnostic, “because you can have false positives.” The molecular GAA assay is used to confirm Pompe disease. But detecting pathogenic variants on the GAA gene – of which there are more than 500 – can be more complicated than it sounds. Whereas two mutations are required for Pompe disease, sometimes only one can be detected. Dr. Toscano said he also treated some patients for Pompe with only one known mutation but with unequivocal clinical and biochemical aspects of Pompe disease.
While delays in diagnosis for late-onset Pompe disease remain significant -- between 5 and 6 years on average for older patients, and up to 20 years for those with pediatric onset – both Dr. Kishnani and Dr. Toscano said they perceive them to be improving. With McArdle disease, another inherited glycogen storage disorder that is more common than Pompe disease but for which there is no treatment, “the delay is nearly 12 years,” Dr. Toscano said.
ERT: The sooner the better
Enzyme replacement therapy is indicated for all patients with Pompe disease. Currently two are commercially available: alglucosidase alfa (Lumizyme, Sanofi Genzyme), indicated for all forms of Pompe disease, and avalglucosidase alfa-ngpt (Nexviazyme, Sanofi Genzyme), approved in 2021 for later-onset Pompe, though its indications have yet to be fully defined.
The semimonthly infusions represent, to date, the only disease-modifying therapies commercially available. Enzyme replacement therapy can reverse cardiac damage seen in infants and allow them to meet developmental milestones previously unthinkable. In adults, it can slow progression, though many treated patients will still develop chronic disability and require a wheelchair, respiratory support, or both. “The phenotype of the patients we are seeing today is not as involved as it was prior to enzyme therapy,” said Dr. Kishnani, who was part of the research team that developed ERT and launched the first clinical trials. “This is across the disease spectrum.”
But optimal management means more than just getting a patient on therapy fast, Dr. Kishnani said.
“Very often the thinking is if the patient is on ERT, we’ve done right by the patient. Aspects we don’t look at enough include: Are we monitoring these patients well? Are patients being followed by a multidisciplinary team that includes cardiology, physical therapy, and pulmonary medicine? Are we doing appropriate musculoskeletal assessments? They might have sleep hypoventilation. The BiPap settings may not be correct. Or they have not been assessed for antibodies,” she said.
Many infants with severe phenotypes, notably those who produce no enzyme naturally, will develop immune reactions to the exogenous enzyme therapy. High antibody titers also have been seen and are associated with poor therapeutic response. While this is very clear in the infantile setting, late-onset patients also develop antibodies in response to ERT. In one study in 64 patients,2 Dr. Toscano and his colleagues saw that antibodies may affect clinical response during the first 3 years of treatment, while a small study3 by Dr. Kishnani’s group saw clinical decline associated with high antibody titers in patients with late-onset disease.
While the relationship of specific titers to therapeutic response remains unclear, it is important to consider antibodies, along with other factors, in the monitoring of patients with Pompe disease. “We need to always ask, if a patient is falling behind, what could be the reason?” Dr. Kishnani said. “These are the things we as clinicians can do to improve or enhance the impact of ERT.”
Dr. Toscano noted that a common misconception about late-onset Pompe disease is that cardiac manifestations are minimal or absent, whereas as many as about 20% of patients will have heart problems and need to be carefully monitored.
Neurological manifestations
With patients surviving longer on ERT, researchers have been able to develop a deeper understanding of the natural history of Pompe disease. Increasingly, they are seeing it as a multisystem disease that includes central nervous system involvement.
“Is Pompe an overt neurodegenerative disease? I would say no,” Dr. Kishnani said. “But there is a neurological component that we’ve got to understand and follow more.”
Glycogen accumulation, she noted, has been found in anterior horn cells, motor neurons, and other parts of the brain. “We have been doing MRIs on children with infantile Pompe, and we have seen some white matter hyperintensities. The clinical significance of this finding is still emerging. Sometimes it is present, but the child is cognitively intact. We have had college graduates who have white matter hyperintensities. So putting it in context will be important. But we know that glycogen is ubiquitous, and autopsy studies have shown that it is present in the brain.”
In recent years, Dr. Toscano’s group has investigated neurovascular complications of Pompe in late-onset patients. “This was something that really surprised us because for several years we have investigated mainly heart, muscle, or respiratory manifestations of the disease, but the central nervous system was really neglected,” he said.
“Occasionally we did some brain MRIs and we found in even young patients some ischemic areas. We thought this was related to slowed circulation – that blood vessels in these patients are weak because they are impaired by glycogen accumulation.” Dr. Toscano and his colleagues followed that observation with a study of late-onset patients,4 in which they found that more than half had cerebrovascular abnormalities. “Even in, say, patients 30 to 35 years old we saw this – it’s unusual to have a vascular disorder at that age.”
Dr. Toscano and his colleagues also reported cerebral aneurysms5 in patients with Pompe disease and have recommended that clinicians conduct MRI or cerebral angiograms on patients as part of routine follow-up. Blood pressure in Pompe patients should be carefully watched and managed with antihypertensive medication as needed, he said.
Part of the problem is that the proteins in ERT are not able to cross the blood-brain barrier, Dr. Toscano noted, adding that researchers are investigating other treatments that can.
Pompe disease as a research model
The successful development of ERT for Pompe disease marked a boom in research interest into not just Pompe – for which several experimental therapies are currently in the pipeline – but for other myopathies and glycogen storage disorders.
“I think that Pompe has served as a template both as a muscle disease and a lysosomal storage disease, and so some of our learnings from Pompe have been applied across different diseases,” Dr. Kishnani said.
Studies in spinal muscular atrophy, for example, “in some ways mirrored what was done for Pompe – treatment trials were initiated in babies at the most severe end of the disease population with infantile disease, and used similar clinical trial endpoints,” Dr. Kishnani said. “Even for the later-onset end of the spectrum, the endpoints we used in Pompe for muscle strength and function have been relevant to many other neuromuscular disorders.”
Pompe disease research also ushered in a new appreciation of immune responses in protein replacement therapies, Dr. Kishnani noted.
“In the field today, you hear the term cross-reactive immunological material, or CRIM, all the time,” she said. “But when we first started talking about it in the space of Pompe disease, there was a lot of scientific debate about what the significance of CRIM-negative status was in relationship to the risk for development of high and sustained antibody titer and a poor clinical response. To understand this involved a lot of going back to the data and digging into the small subset of nonresponders. One of the powers of rare disease research is that every patient matters, and it’s important to understand what’s going on at the patient level rather than just the group data level.”
A robust pipeline
The decade and a half since the advent of ERT has seen what Dr. Toscano described as “an explosion of interest” in Pompe disease.
“We’re seeing an extraordinary number of papers on everything from clinical, biomarkers, genetics, and rehabilitation – this disease is now considered from every point of view, and this is very important for patients,” Dr. Toscano said. Alongside this has come industry interest in this rare disease, with several companies investigating a range of treatment approaches.
The existence of a treatment, “while not perfect,” he said, “has interested the patient associations and doctors to try and improve service to patients. Patients with Pompe disease are well attended, probably more so than patients with degenerative disorders in which there is no therapy.”
Last year the second ERT, avalglucosidase alfa (Nexviazyme, Sanofi Genzyme) was approved by the U.S. Food and Drug Administration to treat late-onset Pompe disease. The drug, currently being investigated in infants as well, was designed to improve the delivery of the therapeutic enzyme to muscles and enhance glycogen clearance, and results from ongoing trials suggest some functional and clinical benefit over standard ERT.
Other drugs in development for Pompe disease include substrate reduction therapies, which aim to reduce the storage of glycogen in cells, and therapies that improve residual function of mutant GAA enzyme in the body. These and other therapies in development have the potential to modify nervous system manifestations of Pompe disease.6
Because a single gene is implicated in Pompe disease, it has long been considered a good candidate for gene therapies that prompt the body to make stable enzyme. Seven companies are now investigating gene therapies in Pompe disease.7 Some of these deliver to skeletal muscles and others aim for the liver, where proteins are synthesized and secreted and adverse immune responses might be more easily mitigated. Other gene therapies use an ex vivo approach, removing and replacing cells in bone marrow.
Dr. Kishnani’s research group at Duke University is leading a small clinical trial in late-onset patients of a GAA gene transfer to the liver using adeno-associated virus (AAV) vectors.8
“We have started AAV gene therapy trials in in adults with Pompe disease and will later evaluate children because ERT is available as a standard of care, and so from a safety perspective this makes the most sense,” Dr. Kishnani said. “We do have challenges in the field of gene therapy, but I think if we are able to overcome the immune responses, and … to treat at a lower dose, there’s a very good pathway forward.”
Dr. Toscano and Dr. Kishnani have received reimbursement from Sanofi and other manufacturers for participation on advisory boards and as speakers.
Jennie Smith is a freelance journalist and editor specializing in medicine and health.
References
1. Musumeci O, Toscano A. Diagnostic tools in late onset Pompe disease (LOPD). Ann Transl Med. 2019 Jul;7(13):286. doi: 10.21037/atm.2019.06.60.
2. Filosto M et al. Assessing the role of anti rh-GAA in modulating response to ERT in a late-onset Pompe disease cohort from the Italian GSDII Study Group. Adv Ther. 2019 May;36(5):1177-1189. doi: 10.1007/s12325-019-00926-5.
3. Patel TT et al. The impact of antibodies in late-onset Pompe disease: A case series and literature review. Mol Genet Metab. 2012 Jul;106(3):301-9. doi: 10.1016/j.ymgme.2012.04.027.
4. Montagnese F et al. Intracranial arterial abnormalities in patients with late onset Pompe disease (LOPD). J Inherit Metab Dis. 2016 May;39(3):391-398. doi: 10.1007/s10545-015-9913-x.
5. Musumeci O et al. Central nervous system involvement in late-onset Pompe disease: Clues from neuroimaging and neuropsychological analysis. Eur J Neurol. 2019 Mar;26(3):442-e35. doi: 10.1111/ene.13835.
6. Edelmann MJ, Maegawa GHB. CNS-targeting therapies for lysosomal storage diseases: Current advances and challenges. Front Mol Biosci. 2020 Nov 12;7:559804. doi: 10.3389/fmolb.2020.559804
7. Ronzitti G et al. Progress and challenges of gene therapy for Pompe disease. Ann Transl Med. 2019 Jul;7(13):287. doi: 10.21037/atm.2019.04.67.
8. Kishnani PS, Koeberl DD. Liver depot gene therapy for Pompe disease. Ann Transl Med. 2019 Jul;7(13):288. doi: 10.21037/atm.2019.05.02.
Until 2006, when a breakthrough therapy first made treatment possible, Pompe disease was a little-known metabolic myopathy fatal to infants. Those with later-onset disease experienced progressive, often severe disability into adulthood.
In this rare autosomal recessive disorder, which occurs in approximately one in 40,000 births worldwide, a deficiency or absence of the enzyme acid alpha-glucosidase causes glycogen to build up in the lysosomes of cells. While many tissues are affected, skeletal and cardiac muscle see the earliest involvement, with muscle hypotonia, cardiomyopathy, and breathing difficulties (mainly due to diaphragm weakness) comprising the hallmark symptoms of the infantile form. Muscle weakness and progressive respiratory failure are prominent in later-onset disease.
The spectrum of severity and age of onset in Pompe disease is linked to combinations of mutations on the GAA gene, some of which destroy the body’s ability to produce acid alpha-glucosidase whereas others merely hamper it. Less enzyme produced in the body generally corresponds with more severe disease activity.
The most severe end of the disease spectrum, or “classic infantile Pompe disease,” presents at birth and is recognized in early infancy. Until treatment with enzyme replacement therapy (ERT) became available, patients usually died of cardiorespiratory failure within their first year of life. With therapy, patients have survived into their 20s and beyond. Late-onset disease is a far broader category in which patients can present at any time from their first year, including into middle age.
The emergence in 2006 of alglucosidase alfa (Lumizyme, Sanofi Genzyme), an ERT used long-term to improve survival and slow progression in children and adults, resulted in a boom of research interest, a push to timelier diagnosis, and – with patients living longer – a more thorough understanding of the natural history of Pompe disease. In addition to the usual clinical picture of progressive muscle weakness, difficulty breathing, and cardiomyopathy, investigators are seeing nervous system involvement in patients with Pompe disease.
To learn more, Neurology Reviews talked to two global experts in Pompe disease: Priya Kishnani, MD, of Duke University in Durham, N.C., and Antonio Toscano, MD, of the University of Messina, in Messina, Italy.
Diagnosis: Still room to improve
“Most neurologists will encounter a patient with Pompe disease,” said Dr. Kishnani, who has been working with Pompe for her entire career as a pediatrician and medical geneticist, treating patients of all ages and disease phenotypes.
“In newborns, diagnosis is more straightforward, because you’ve got an enlarged heart,” she said. And thanks to efforts of researchers like Dr. Kishnani and Pompe advocacy groups, Pompe disease is now a part of the RUSP (Recommended Uniform Screening Panel) for newborns; currently 28 U.S. states are screening for Pompe disease.
“The challenge really is for the later-onset cases, which are 80% of all cases,” Dr. Kishnani said.
Previously, muscle biopsies were the first step toward diagnosis. Dried blot spot assays to detect enzyme deficiency have since become the standard, along with other biochemical tests. Confirmation of the diagnosis is through gene sequencing panels to detect GAA mutations.
“Now that there is a treatment for Pompe disease and the availability of blood-based testing, many previously undiagnosed patients with limb girdle weakness are evaluated and the diagnostic odyssey ends,” Dr. Kishnani said. “But there is still a diagnostic delay, and many cases remain undiagnosed.”
Routine blood tests for creatine kinase and for liver enzymes can help point to Pompe disease. But elevated liver enzymes are often misinterpreted. “It’s about the ratios,” Dr. Kishnani said. “ALT is usually much more elevated if it is coming from a liver cause, and AST is usually higher than ALT if it is coming from muscle. But patients often end up getting a liver biopsy due to so-called elevated liver enzymes. As the workup continues, it is often later recognized that the source of the elevated enzymes is muscle involvement, and a referral to the geneticist or neurologist is made. Only then is appropriate testing to confirm a diagnosis initiated.”
Dr. Toscano, a neurologist who specializes in Pompe disease and other myopathies and who has published on tools for diagnosing late-onset Pompe disease,1 said that clinicians should be vigilant when evaluating any patient with limb girdle weakness and elevated creatine kinase (CK) – “especially if the CK is under 2,000,” he said, “because it is very rare that patients with Pompe disease have a more elevated CK than that.”
“Elevated CK, myalgia, and exercise intolerance” should prompt clinicians to suspect Pompe disease in a patient of any age, Dr. Toscano said. “When you come across this, you should be very persistent and get to the end of the story.”
Dr. Toscano noted that the blood spot assay, while an important early step, is not fully diagnostic, “because you can have false positives.” The molecular GAA assay is used to confirm Pompe disease. But detecting pathogenic variants on the GAA gene – of which there are more than 500 – can be more complicated than it sounds. Whereas two mutations are required for Pompe disease, sometimes only one can be detected. Dr. Toscano said he also treated some patients for Pompe with only one known mutation but with unequivocal clinical and biochemical aspects of Pompe disease.
While delays in diagnosis for late-onset Pompe disease remain significant -- between 5 and 6 years on average for older patients, and up to 20 years for those with pediatric onset – both Dr. Kishnani and Dr. Toscano said they perceive them to be improving. With McArdle disease, another inherited glycogen storage disorder that is more common than Pompe disease but for which there is no treatment, “the delay is nearly 12 years,” Dr. Toscano said.
ERT: The sooner the better
Enzyme replacement therapy is indicated for all patients with Pompe disease. Currently two are commercially available: alglucosidase alfa (Lumizyme, Sanofi Genzyme), indicated for all forms of Pompe disease, and avalglucosidase alfa-ngpt (Nexviazyme, Sanofi Genzyme), approved in 2021 for later-onset Pompe, though its indications have yet to be fully defined.
The semimonthly infusions represent, to date, the only disease-modifying therapies commercially available. Enzyme replacement therapy can reverse cardiac damage seen in infants and allow them to meet developmental milestones previously unthinkable. In adults, it can slow progression, though many treated patients will still develop chronic disability and require a wheelchair, respiratory support, or both. “The phenotype of the patients we are seeing today is not as involved as it was prior to enzyme therapy,” said Dr. Kishnani, who was part of the research team that developed ERT and launched the first clinical trials. “This is across the disease spectrum.”
But optimal management means more than just getting a patient on therapy fast, Dr. Kishnani said.
“Very often the thinking is if the patient is on ERT, we’ve done right by the patient. Aspects we don’t look at enough include: Are we monitoring these patients well? Are patients being followed by a multidisciplinary team that includes cardiology, physical therapy, and pulmonary medicine? Are we doing appropriate musculoskeletal assessments? They might have sleep hypoventilation. The BiPap settings may not be correct. Or they have not been assessed for antibodies,” she said.
Many infants with severe phenotypes, notably those who produce no enzyme naturally, will develop immune reactions to the exogenous enzyme therapy. High antibody titers also have been seen and are associated with poor therapeutic response. While this is very clear in the infantile setting, late-onset patients also develop antibodies in response to ERT. In one study in 64 patients,2 Dr. Toscano and his colleagues saw that antibodies may affect clinical response during the first 3 years of treatment, while a small study3 by Dr. Kishnani’s group saw clinical decline associated with high antibody titers in patients with late-onset disease.
While the relationship of specific titers to therapeutic response remains unclear, it is important to consider antibodies, along with other factors, in the monitoring of patients with Pompe disease. “We need to always ask, if a patient is falling behind, what could be the reason?” Dr. Kishnani said. “These are the things we as clinicians can do to improve or enhance the impact of ERT.”
Dr. Toscano noted that a common misconception about late-onset Pompe disease is that cardiac manifestations are minimal or absent, whereas as many as about 20% of patients will have heart problems and need to be carefully monitored.
Neurological manifestations
With patients surviving longer on ERT, researchers have been able to develop a deeper understanding of the natural history of Pompe disease. Increasingly, they are seeing it as a multisystem disease that includes central nervous system involvement.
“Is Pompe an overt neurodegenerative disease? I would say no,” Dr. Kishnani said. “But there is a neurological component that we’ve got to understand and follow more.”
Glycogen accumulation, she noted, has been found in anterior horn cells, motor neurons, and other parts of the brain. “We have been doing MRIs on children with infantile Pompe, and we have seen some white matter hyperintensities. The clinical significance of this finding is still emerging. Sometimes it is present, but the child is cognitively intact. We have had college graduates who have white matter hyperintensities. So putting it in context will be important. But we know that glycogen is ubiquitous, and autopsy studies have shown that it is present in the brain.”
In recent years, Dr. Toscano’s group has investigated neurovascular complications of Pompe in late-onset patients. “This was something that really surprised us because for several years we have investigated mainly heart, muscle, or respiratory manifestations of the disease, but the central nervous system was really neglected,” he said.
“Occasionally we did some brain MRIs and we found in even young patients some ischemic areas. We thought this was related to slowed circulation – that blood vessels in these patients are weak because they are impaired by glycogen accumulation.” Dr. Toscano and his colleagues followed that observation with a study of late-onset patients,4 in which they found that more than half had cerebrovascular abnormalities. “Even in, say, patients 30 to 35 years old we saw this – it’s unusual to have a vascular disorder at that age.”
Dr. Toscano and his colleagues also reported cerebral aneurysms5 in patients with Pompe disease and have recommended that clinicians conduct MRI or cerebral angiograms on patients as part of routine follow-up. Blood pressure in Pompe patients should be carefully watched and managed with antihypertensive medication as needed, he said.
Part of the problem is that the proteins in ERT are not able to cross the blood-brain barrier, Dr. Toscano noted, adding that researchers are investigating other treatments that can.
Pompe disease as a research model
The successful development of ERT for Pompe disease marked a boom in research interest into not just Pompe – for which several experimental therapies are currently in the pipeline – but for other myopathies and glycogen storage disorders.
“I think that Pompe has served as a template both as a muscle disease and a lysosomal storage disease, and so some of our learnings from Pompe have been applied across different diseases,” Dr. Kishnani said.
Studies in spinal muscular atrophy, for example, “in some ways mirrored what was done for Pompe – treatment trials were initiated in babies at the most severe end of the disease population with infantile disease, and used similar clinical trial endpoints,” Dr. Kishnani said. “Even for the later-onset end of the spectrum, the endpoints we used in Pompe for muscle strength and function have been relevant to many other neuromuscular disorders.”
Pompe disease research also ushered in a new appreciation of immune responses in protein replacement therapies, Dr. Kishnani noted.
“In the field today, you hear the term cross-reactive immunological material, or CRIM, all the time,” she said. “But when we first started talking about it in the space of Pompe disease, there was a lot of scientific debate about what the significance of CRIM-negative status was in relationship to the risk for development of high and sustained antibody titer and a poor clinical response. To understand this involved a lot of going back to the data and digging into the small subset of nonresponders. One of the powers of rare disease research is that every patient matters, and it’s important to understand what’s going on at the patient level rather than just the group data level.”
A robust pipeline
The decade and a half since the advent of ERT has seen what Dr. Toscano described as “an explosion of interest” in Pompe disease.
“We’re seeing an extraordinary number of papers on everything from clinical, biomarkers, genetics, and rehabilitation – this disease is now considered from every point of view, and this is very important for patients,” Dr. Toscano said. Alongside this has come industry interest in this rare disease, with several companies investigating a range of treatment approaches.
The existence of a treatment, “while not perfect,” he said, “has interested the patient associations and doctors to try and improve service to patients. Patients with Pompe disease are well attended, probably more so than patients with degenerative disorders in which there is no therapy.”
Last year the second ERT, avalglucosidase alfa (Nexviazyme, Sanofi Genzyme) was approved by the U.S. Food and Drug Administration to treat late-onset Pompe disease. The drug, currently being investigated in infants as well, was designed to improve the delivery of the therapeutic enzyme to muscles and enhance glycogen clearance, and results from ongoing trials suggest some functional and clinical benefit over standard ERT.
Other drugs in development for Pompe disease include substrate reduction therapies, which aim to reduce the storage of glycogen in cells, and therapies that improve residual function of mutant GAA enzyme in the body. These and other therapies in development have the potential to modify nervous system manifestations of Pompe disease.6
Because a single gene is implicated in Pompe disease, it has long been considered a good candidate for gene therapies that prompt the body to make stable enzyme. Seven companies are now investigating gene therapies in Pompe disease.7 Some of these deliver to skeletal muscles and others aim for the liver, where proteins are synthesized and secreted and adverse immune responses might be more easily mitigated. Other gene therapies use an ex vivo approach, removing and replacing cells in bone marrow.
Dr. Kishnani’s research group at Duke University is leading a small clinical trial in late-onset patients of a GAA gene transfer to the liver using adeno-associated virus (AAV) vectors.8
“We have started AAV gene therapy trials in in adults with Pompe disease and will later evaluate children because ERT is available as a standard of care, and so from a safety perspective this makes the most sense,” Dr. Kishnani said. “We do have challenges in the field of gene therapy, but I think if we are able to overcome the immune responses, and … to treat at a lower dose, there’s a very good pathway forward.”
Dr. Toscano and Dr. Kishnani have received reimbursement from Sanofi and other manufacturers for participation on advisory boards and as speakers.
Jennie Smith is a freelance journalist and editor specializing in medicine and health.
References
1. Musumeci O, Toscano A. Diagnostic tools in late onset Pompe disease (LOPD). Ann Transl Med. 2019 Jul;7(13):286. doi: 10.21037/atm.2019.06.60.
2. Filosto M et al. Assessing the role of anti rh-GAA in modulating response to ERT in a late-onset Pompe disease cohort from the Italian GSDII Study Group. Adv Ther. 2019 May;36(5):1177-1189. doi: 10.1007/s12325-019-00926-5.
3. Patel TT et al. The impact of antibodies in late-onset Pompe disease: A case series and literature review. Mol Genet Metab. 2012 Jul;106(3):301-9. doi: 10.1016/j.ymgme.2012.04.027.
4. Montagnese F et al. Intracranial arterial abnormalities in patients with late onset Pompe disease (LOPD). J Inherit Metab Dis. 2016 May;39(3):391-398. doi: 10.1007/s10545-015-9913-x.
5. Musumeci O et al. Central nervous system involvement in late-onset Pompe disease: Clues from neuroimaging and neuropsychological analysis. Eur J Neurol. 2019 Mar;26(3):442-e35. doi: 10.1111/ene.13835.
6. Edelmann MJ, Maegawa GHB. CNS-targeting therapies for lysosomal storage diseases: Current advances and challenges. Front Mol Biosci. 2020 Nov 12;7:559804. doi: 10.3389/fmolb.2020.559804
7. Ronzitti G et al. Progress and challenges of gene therapy for Pompe disease. Ann Transl Med. 2019 Jul;7(13):287. doi: 10.21037/atm.2019.04.67.
8. Kishnani PS, Koeberl DD. Liver depot gene therapy for Pompe disease. Ann Transl Med. 2019 Jul;7(13):288. doi: 10.21037/atm.2019.05.02.
Until 2006, when a breakthrough therapy first made treatment possible, Pompe disease was a little-known metabolic myopathy fatal to infants. Those with later-onset disease experienced progressive, often severe disability into adulthood.
In this rare autosomal recessive disorder, which occurs in approximately one in 40,000 births worldwide, a deficiency or absence of the enzyme acid alpha-glucosidase causes glycogen to build up in the lysosomes of cells. While many tissues are affected, skeletal and cardiac muscle see the earliest involvement, with muscle hypotonia, cardiomyopathy, and breathing difficulties (mainly due to diaphragm weakness) comprising the hallmark symptoms of the infantile form. Muscle weakness and progressive respiratory failure are prominent in later-onset disease.
The spectrum of severity and age of onset in Pompe disease is linked to combinations of mutations on the GAA gene, some of which destroy the body’s ability to produce acid alpha-glucosidase whereas others merely hamper it. Less enzyme produced in the body generally corresponds with more severe disease activity.
The most severe end of the disease spectrum, or “classic infantile Pompe disease,” presents at birth and is recognized in early infancy. Until treatment with enzyme replacement therapy (ERT) became available, patients usually died of cardiorespiratory failure within their first year of life. With therapy, patients have survived into their 20s and beyond. Late-onset disease is a far broader category in which patients can present at any time from their first year, including into middle age.
The emergence in 2006 of alglucosidase alfa (Lumizyme, Sanofi Genzyme), an ERT used long-term to improve survival and slow progression in children and adults, resulted in a boom of research interest, a push to timelier diagnosis, and – with patients living longer – a more thorough understanding of the natural history of Pompe disease. In addition to the usual clinical picture of progressive muscle weakness, difficulty breathing, and cardiomyopathy, investigators are seeing nervous system involvement in patients with Pompe disease.
To learn more, Neurology Reviews talked to two global experts in Pompe disease: Priya Kishnani, MD, of Duke University in Durham, N.C., and Antonio Toscano, MD, of the University of Messina, in Messina, Italy.
Diagnosis: Still room to improve
“Most neurologists will encounter a patient with Pompe disease,” said Dr. Kishnani, who has been working with Pompe for her entire career as a pediatrician and medical geneticist, treating patients of all ages and disease phenotypes.
“In newborns, diagnosis is more straightforward, because you’ve got an enlarged heart,” she said. And thanks to efforts of researchers like Dr. Kishnani and Pompe advocacy groups, Pompe disease is now a part of the RUSP (Recommended Uniform Screening Panel) for newborns; currently 28 U.S. states are screening for Pompe disease.
“The challenge really is for the later-onset cases, which are 80% of all cases,” Dr. Kishnani said.
Previously, muscle biopsies were the first step toward diagnosis. Dried blot spot assays to detect enzyme deficiency have since become the standard, along with other biochemical tests. Confirmation of the diagnosis is through gene sequencing panels to detect GAA mutations.
“Now that there is a treatment for Pompe disease and the availability of blood-based testing, many previously undiagnosed patients with limb girdle weakness are evaluated and the diagnostic odyssey ends,” Dr. Kishnani said. “But there is still a diagnostic delay, and many cases remain undiagnosed.”
Routine blood tests for creatine kinase and for liver enzymes can help point to Pompe disease. But elevated liver enzymes are often misinterpreted. “It’s about the ratios,” Dr. Kishnani said. “ALT is usually much more elevated if it is coming from a liver cause, and AST is usually higher than ALT if it is coming from muscle. But patients often end up getting a liver biopsy due to so-called elevated liver enzymes. As the workup continues, it is often later recognized that the source of the elevated enzymes is muscle involvement, and a referral to the geneticist or neurologist is made. Only then is appropriate testing to confirm a diagnosis initiated.”
Dr. Toscano, a neurologist who specializes in Pompe disease and other myopathies and who has published on tools for diagnosing late-onset Pompe disease,1 said that clinicians should be vigilant when evaluating any patient with limb girdle weakness and elevated creatine kinase (CK) – “especially if the CK is under 2,000,” he said, “because it is very rare that patients with Pompe disease have a more elevated CK than that.”
“Elevated CK, myalgia, and exercise intolerance” should prompt clinicians to suspect Pompe disease in a patient of any age, Dr. Toscano said. “When you come across this, you should be very persistent and get to the end of the story.”
Dr. Toscano noted that the blood spot assay, while an important early step, is not fully diagnostic, “because you can have false positives.” The molecular GAA assay is used to confirm Pompe disease. But detecting pathogenic variants on the GAA gene – of which there are more than 500 – can be more complicated than it sounds. Whereas two mutations are required for Pompe disease, sometimes only one can be detected. Dr. Toscano said he also treated some patients for Pompe with only one known mutation but with unequivocal clinical and biochemical aspects of Pompe disease.
While delays in diagnosis for late-onset Pompe disease remain significant -- between 5 and 6 years on average for older patients, and up to 20 years for those with pediatric onset – both Dr. Kishnani and Dr. Toscano said they perceive them to be improving. With McArdle disease, another inherited glycogen storage disorder that is more common than Pompe disease but for which there is no treatment, “the delay is nearly 12 years,” Dr. Toscano said.
ERT: The sooner the better
Enzyme replacement therapy is indicated for all patients with Pompe disease. Currently two are commercially available: alglucosidase alfa (Lumizyme, Sanofi Genzyme), indicated for all forms of Pompe disease, and avalglucosidase alfa-ngpt (Nexviazyme, Sanofi Genzyme), approved in 2021 for later-onset Pompe, though its indications have yet to be fully defined.
The semimonthly infusions represent, to date, the only disease-modifying therapies commercially available. Enzyme replacement therapy can reverse cardiac damage seen in infants and allow them to meet developmental milestones previously unthinkable. In adults, it can slow progression, though many treated patients will still develop chronic disability and require a wheelchair, respiratory support, or both. “The phenotype of the patients we are seeing today is not as involved as it was prior to enzyme therapy,” said Dr. Kishnani, who was part of the research team that developed ERT and launched the first clinical trials. “This is across the disease spectrum.”
But optimal management means more than just getting a patient on therapy fast, Dr. Kishnani said.
“Very often the thinking is if the patient is on ERT, we’ve done right by the patient. Aspects we don’t look at enough include: Are we monitoring these patients well? Are patients being followed by a multidisciplinary team that includes cardiology, physical therapy, and pulmonary medicine? Are we doing appropriate musculoskeletal assessments? They might have sleep hypoventilation. The BiPap settings may not be correct. Or they have not been assessed for antibodies,” she said.
Many infants with severe phenotypes, notably those who produce no enzyme naturally, will develop immune reactions to the exogenous enzyme therapy. High antibody titers also have been seen and are associated with poor therapeutic response. While this is very clear in the infantile setting, late-onset patients also develop antibodies in response to ERT. In one study in 64 patients,2 Dr. Toscano and his colleagues saw that antibodies may affect clinical response during the first 3 years of treatment, while a small study3 by Dr. Kishnani’s group saw clinical decline associated with high antibody titers in patients with late-onset disease.
While the relationship of specific titers to therapeutic response remains unclear, it is important to consider antibodies, along with other factors, in the monitoring of patients with Pompe disease. “We need to always ask, if a patient is falling behind, what could be the reason?” Dr. Kishnani said. “These are the things we as clinicians can do to improve or enhance the impact of ERT.”
Dr. Toscano noted that a common misconception about late-onset Pompe disease is that cardiac manifestations are minimal or absent, whereas as many as about 20% of patients will have heart problems and need to be carefully monitored.
Neurological manifestations
With patients surviving longer on ERT, researchers have been able to develop a deeper understanding of the natural history of Pompe disease. Increasingly, they are seeing it as a multisystem disease that includes central nervous system involvement.
“Is Pompe an overt neurodegenerative disease? I would say no,” Dr. Kishnani said. “But there is a neurological component that we’ve got to understand and follow more.”
Glycogen accumulation, she noted, has been found in anterior horn cells, motor neurons, and other parts of the brain. “We have been doing MRIs on children with infantile Pompe, and we have seen some white matter hyperintensities. The clinical significance of this finding is still emerging. Sometimes it is present, but the child is cognitively intact. We have had college graduates who have white matter hyperintensities. So putting it in context will be important. But we know that glycogen is ubiquitous, and autopsy studies have shown that it is present in the brain.”
In recent years, Dr. Toscano’s group has investigated neurovascular complications of Pompe in late-onset patients. “This was something that really surprised us because for several years we have investigated mainly heart, muscle, or respiratory manifestations of the disease, but the central nervous system was really neglected,” he said.
“Occasionally we did some brain MRIs and we found in even young patients some ischemic areas. We thought this was related to slowed circulation – that blood vessels in these patients are weak because they are impaired by glycogen accumulation.” Dr. Toscano and his colleagues followed that observation with a study of late-onset patients,4 in which they found that more than half had cerebrovascular abnormalities. “Even in, say, patients 30 to 35 years old we saw this – it’s unusual to have a vascular disorder at that age.”
Dr. Toscano and his colleagues also reported cerebral aneurysms5 in patients with Pompe disease and have recommended that clinicians conduct MRI or cerebral angiograms on patients as part of routine follow-up. Blood pressure in Pompe patients should be carefully watched and managed with antihypertensive medication as needed, he said.
Part of the problem is that the proteins in ERT are not able to cross the blood-brain barrier, Dr. Toscano noted, adding that researchers are investigating other treatments that can.
Pompe disease as a research model
The successful development of ERT for Pompe disease marked a boom in research interest into not just Pompe – for which several experimental therapies are currently in the pipeline – but for other myopathies and glycogen storage disorders.
“I think that Pompe has served as a template both as a muscle disease and a lysosomal storage disease, and so some of our learnings from Pompe have been applied across different diseases,” Dr. Kishnani said.
Studies in spinal muscular atrophy, for example, “in some ways mirrored what was done for Pompe – treatment trials were initiated in babies at the most severe end of the disease population with infantile disease, and used similar clinical trial endpoints,” Dr. Kishnani said. “Even for the later-onset end of the spectrum, the endpoints we used in Pompe for muscle strength and function have been relevant to many other neuromuscular disorders.”
Pompe disease research also ushered in a new appreciation of immune responses in protein replacement therapies, Dr. Kishnani noted.
“In the field today, you hear the term cross-reactive immunological material, or CRIM, all the time,” she said. “But when we first started talking about it in the space of Pompe disease, there was a lot of scientific debate about what the significance of CRIM-negative status was in relationship to the risk for development of high and sustained antibody titer and a poor clinical response. To understand this involved a lot of going back to the data and digging into the small subset of nonresponders. One of the powers of rare disease research is that every patient matters, and it’s important to understand what’s going on at the patient level rather than just the group data level.”
A robust pipeline
The decade and a half since the advent of ERT has seen what Dr. Toscano described as “an explosion of interest” in Pompe disease.
“We’re seeing an extraordinary number of papers on everything from clinical, biomarkers, genetics, and rehabilitation – this disease is now considered from every point of view, and this is very important for patients,” Dr. Toscano said. Alongside this has come industry interest in this rare disease, with several companies investigating a range of treatment approaches.
The existence of a treatment, “while not perfect,” he said, “has interested the patient associations and doctors to try and improve service to patients. Patients with Pompe disease are well attended, probably more so than patients with degenerative disorders in which there is no therapy.”
Last year the second ERT, avalglucosidase alfa (Nexviazyme, Sanofi Genzyme) was approved by the U.S. Food and Drug Administration to treat late-onset Pompe disease. The drug, currently being investigated in infants as well, was designed to improve the delivery of the therapeutic enzyme to muscles and enhance glycogen clearance, and results from ongoing trials suggest some functional and clinical benefit over standard ERT.
Other drugs in development for Pompe disease include substrate reduction therapies, which aim to reduce the storage of glycogen in cells, and therapies that improve residual function of mutant GAA enzyme in the body. These and other therapies in development have the potential to modify nervous system manifestations of Pompe disease.6
Because a single gene is implicated in Pompe disease, it has long been considered a good candidate for gene therapies that prompt the body to make stable enzyme. Seven companies are now investigating gene therapies in Pompe disease.7 Some of these deliver to skeletal muscles and others aim for the liver, where proteins are synthesized and secreted and adverse immune responses might be more easily mitigated. Other gene therapies use an ex vivo approach, removing and replacing cells in bone marrow.
Dr. Kishnani’s research group at Duke University is leading a small clinical trial in late-onset patients of a GAA gene transfer to the liver using adeno-associated virus (AAV) vectors.8
“We have started AAV gene therapy trials in in adults with Pompe disease and will later evaluate children because ERT is available as a standard of care, and so from a safety perspective this makes the most sense,” Dr. Kishnani said. “We do have challenges in the field of gene therapy, but I think if we are able to overcome the immune responses, and … to treat at a lower dose, there’s a very good pathway forward.”
Dr. Toscano and Dr. Kishnani have received reimbursement from Sanofi and other manufacturers for participation on advisory boards and as speakers.
Jennie Smith is a freelance journalist and editor specializing in medicine and health.
References
1. Musumeci O, Toscano A. Diagnostic tools in late onset Pompe disease (LOPD). Ann Transl Med. 2019 Jul;7(13):286. doi: 10.21037/atm.2019.06.60.
2. Filosto M et al. Assessing the role of anti rh-GAA in modulating response to ERT in a late-onset Pompe disease cohort from the Italian GSDII Study Group. Adv Ther. 2019 May;36(5):1177-1189. doi: 10.1007/s12325-019-00926-5.
3. Patel TT et al. The impact of antibodies in late-onset Pompe disease: A case series and literature review. Mol Genet Metab. 2012 Jul;106(3):301-9. doi: 10.1016/j.ymgme.2012.04.027.
4. Montagnese F et al. Intracranial arterial abnormalities in patients with late onset Pompe disease (LOPD). J Inherit Metab Dis. 2016 May;39(3):391-398. doi: 10.1007/s10545-015-9913-x.
5. Musumeci O et al. Central nervous system involvement in late-onset Pompe disease: Clues from neuroimaging and neuropsychological analysis. Eur J Neurol. 2019 Mar;26(3):442-e35. doi: 10.1111/ene.13835.
6. Edelmann MJ, Maegawa GHB. CNS-targeting therapies for lysosomal storage diseases: Current advances and challenges. Front Mol Biosci. 2020 Nov 12;7:559804. doi: 10.3389/fmolb.2020.559804
7. Ronzitti G et al. Progress and challenges of gene therapy for Pompe disease. Ann Transl Med. 2019 Jul;7(13):287. doi: 10.21037/atm.2019.04.67.
8. Kishnani PS, Koeberl DD. Liver depot gene therapy for Pompe disease. Ann Transl Med. 2019 Jul;7(13):288. doi: 10.21037/atm.2019.05.02.