User login
Pacritinib bests BAT despite study truncation

SAN DIEGO—The JAK2/FLT3 inhibitor pacritinib significantly reduces spleen volume and symptoms in patients with myelofibrosis and low platelet counts,
compared to best available therapy (BAT), according to results of the PERSIST-2 trial.
In this phase 3 trial, BAT included the JAK1/2 inhibitor ruxolitinib. And pacritinib demonstrated benefits over BAT despite a truncated trial.
The US Food and Drug Administration (FDA) placed PERSIST-2 on clinical hold in February 2016 due to concerns over interim survival results, bleeding, and cardiovascular events.
Patients randomized at least 22 weeks prior to the clinical hold contributed data to the week 24 endpoint, the results of which were presented at the 2016 ASH Annual Meeting.
John Mascarenhas, MD, of Icahn School of Medicine at Mount Sinai in New York, New York, delivered the results as a late-breaking abstract (LBA-5).
Ruxolitinib is FDA-approved to treat myelofibrosis, but it is associated with dose-limiting cytopenias and is not indicated for patients with platelet counts less than 50,000/μL.
The earlier PERSIST-1 trial demonstrated sustained spleen volume reduction (SVR) and symptom control with pacritinib therapy regardless of baseline platelet count, but it did not include ruxolitinib in the BAT arm.
PERSIST-2 study design
Patients were randomized on a 1:1:1 basis to pacritinib at 400 mg once daily (PAC QD), pacritinib at 200 mg twice daily (PAC BID), or BAT, including ruxolitinib.
Patients had to have primary or secondary myelofibrosis and platelet counts of 100,000/μL or less. Patients were allowed to have had prior JAK2 inhibitors.
Crossover from BAT was allowed after progression at any time or at week 24.
The study had 2 primary endpoints: percent of patients achieving a 35% or greater SVR and the percent of patients achieving a 50% or more reduction in total symptom score (TSS) by MPN-SAF TSS 2.0.
The primary study objective was the efficacy of pooled QD and BID pacritinib compared to BAT. The secondary objective was the efficacy of QD or BD separately compared to BAT.
Patient demographics
The study randomized 311 patients, and 221 were included in the intent-to-treat efficacy population.
The efficacy population consisted of all patients randomized prior to September 7, 2015, which allowed their data to be included in the week 24 endpoint analysis prior to the clinical hold.
The PAC QD arm consisted of 75 patients with a median age of 69 (range, 39-85). Seventy-one percent were 65 or older, and about half were male.
About three-quarters had an ECOG performance status of 0-1, 61% had primary myelofibrosis, 21% had primary polycythemia vera (PPV), and 17% had primary essential thrombocythemia (PET). About half were DIPSS Intermediate-2 risk, 80% were JAK2V617F positive, and 51% had a platelet count less than 50,000/μL.
The PAC BID arm consisted of 74 patients with a median age of 67 (range, 39-85). Sixty-two percent were 65 or older, and 65% were male.
Eighty-eight percent had an ECOG performance status of 0-1, 74% had primary myelofibrosis, 19% had PPV, and 7% had PET. About half were DIPSS Intermediate-2 risk, 80% were JAK2V617F positive, and 42% had a platelet count less than 50,000/μL.
The BAT arm consisted of 72 patients with a median age of 69 (range, 32-83). Seventy-one percent were 65 or older, and about half were male.
Three-quarters had an ECOG performance status of 0-1, 60% had primary myelofibrosis, 22% had PPV, and 18% had PET. About half were DIPSS Intermediate-2 risk, 71% were JAK2V617F positive, and 44% had a platelet count less than 50,000/µL.
Prior ruxolitinib therapy was consistent across the arms—41.3% (PAC QD), 42% (PAC BID), and 46% (BAT).
“Now, it’s important to note,” Dr Mascarenhas said, “that the most common BAT was ruxolitinib, 45%, and hydroxyurea, 19%. And 19% of BAT patients actually had no treatment, watch and wait. This highlights the fact that this is an area where there is really no other viable therapeutic option for these patients.”
Efficacy
Pacritinib-treated patients had significantly greater spleen reduction from baseline to week 24 than BAT-treated patients, with 18% (QD+BID), 15% (QD), and 22% (BID) achieving 35% or more SVR compared to 3% in the BAT arm.
Pacritinib-treated patients also experienced greater TSS reduction, with 25% (QD+BID), 17% (QD), and 32% (BID) reporting 50% or more reduction in TSS compared to 14% in the BAT arm. However, only the PAC BID arm was significantly different from BAT.
SVR in all subgroups—age, gender, JAK2 mutation status, prior treatment, platelet count, hemoglobin, peripheral blasts, and white blood cell count—demonstrated superiority for pacritinib.
Median changes in individual symptom scores were also better in the pacritinib arms than in the BAT arm in almost every category—tiredness, early satiety, abdominal discomfort, inactivity, night sweats, bone pain, and pain under ribs on the left side.
Pruritus was the only category in which BAT was superior, and that was compared to the QD arm and not the BID arm.
The majority of patients who stopped pacritinib therapy were taken off due to the clinical hold.
There were no significant differences between the groups in overall survival. Hazard ratios for overall survival (95% confidence intervals) were 0.68 (0.30, 1.53) for PAC BID vs BAT, 1.18 (0.57, 2.44) for PAC QD vs BAT, and 0.61 (0.27, 1.35) for PAC BID vs QD.
PAC BID maintained this survival advantage compared with BAT across nearly all demographic and myelofibrosis-associated risk factors.
Patients who were red blood cell transfusion-dependent at baseline experienced a statistically significant decrease in transfusion frequency from baseline to week 24 in both the QD and BID pacritinib arms compared with BAT.
And thrombocytopenia was not a significant factor for patients who were receiving pacritinib and had platelet counts less than 50,000/μL.
Toxicity
The most common treatment-emergent adverse events (AEs) associated with pacritinib were diarrhea, nausea, vomiting, anemia, and thrombocytopenia. They were generally less frequent for BID compared with QD administration.
The most common serious adverse events (SAEs)—occurring in 5% of patients or more in any arms—were anemia (5%, 8%, and 3%), thrombocytopenia (2%, 6%, and 2%), pneumonia (5%, 6%, and 4%), and acute renal failure (5%, 2%, and 2%) in the QD, BID, and BAT arms, respectively.
SAEs of interest included congestive heart failure, atrial fibrillation, cardiac arrest, epistaxis, and subdural hematoma, which occurred in 3% or fewer patients in any arm.
“They [the SAEs] were relatively infrequent, and there was not a clear signal of toxicity,” Dr Mascarenhas said.
Deaths
Deaths in the intent-to-treat evaluable population were censored at the time of full clinical hold.
For the entire enrolled population, 15/104 deaths occurred in the QD arm, 10/107 in the BID arm, and 14/100 in the BAT arm.
After the full clinical hold, 7, 10, and 6 deaths occurred in the QD, BID, and BAT arms, respectively.
Seven of 20 patients died after crossover to pacritinib. Five were due to AEs—3 cardiac, 1 bleeding, and 1 other.
“It’s important to note that progression of disease was the leading cause of death in the PAC BID arm,” Dr Mascarenhas noted. “This is after the patients stopped the drug [when the trial was on clinical hold].”
Conclusions
Despite study truncation, pacritinib (QD+BID) was significantly more effective than BAT for SVR (P=0.001) and trended toward improved TSS (P=0.079).
Pacritinib BID appeared more effective than QD versus BAT for SVR and TSS, and pacritinib BID appeared to have a better benefit/risk profile than BAT.
“This is a well-tolerated drug in many respects,” Dr Mascarenhas said. “This is a patient population that is quite ill, low platelets, poor outcome, and they did pretty well.”
When asked about the future of pacritinib, Dr Mascarenhas said he believes the benefit-to-risk ratio is in favor of the drug.
“Pacritinib offers patients in this vulnerable situation an opportunity for symptom relief, basically spleen and symptoms,” he said. “So I think this is a drug that will hopefully move forward.”
Dr Mascarenhas disclosed research funding from CTI Biopharma, the sponsor of the trial. ![]()
SAN DIEGO—The JAK2/FLT3 inhibitor pacritinib significantly reduces spleen volume and symptoms in patients with myelofibrosis and low platelet counts,
compared to best available therapy (BAT), according to results of the PERSIST-2 trial.
In this phase 3 trial, BAT included the JAK1/2 inhibitor ruxolitinib. And pacritinib demonstrated benefits over BAT despite a truncated trial.
The US Food and Drug Administration (FDA) placed PERSIST-2 on clinical hold in February 2016 due to concerns over interim survival results, bleeding, and cardiovascular events.
Patients randomized at least 22 weeks prior to the clinical hold contributed data to the week 24 endpoint, the results of which were presented at the 2016 ASH Annual Meeting.
John Mascarenhas, MD, of Icahn School of Medicine at Mount Sinai in New York, New York, delivered the results as a late-breaking abstract (LBA-5).
Ruxolitinib is FDA-approved to treat myelofibrosis, but it is associated with dose-limiting cytopenias and is not indicated for patients with platelet counts less than 50,000/μL.
The earlier PERSIST-1 trial demonstrated sustained spleen volume reduction (SVR) and symptom control with pacritinib therapy regardless of baseline platelet count, but it did not include ruxolitinib in the BAT arm.
PERSIST-2 study design
Patients were randomized on a 1:1:1 basis to pacritinib at 400 mg once daily (PAC QD), pacritinib at 200 mg twice daily (PAC BID), or BAT, including ruxolitinib.
Patients had to have primary or secondary myelofibrosis and platelet counts of 100,000/μL or less. Patients were allowed to have had prior JAK2 inhibitors.
Crossover from BAT was allowed after progression at any time or at week 24.
The study had 2 primary endpoints: percent of patients achieving a 35% or greater SVR and the percent of patients achieving a 50% or more reduction in total symptom score (TSS) by MPN-SAF TSS 2.0.
The primary study objective was the efficacy of pooled QD and BID pacritinib compared to BAT. The secondary objective was the efficacy of QD or BD separately compared to BAT.
Patient demographics
The study randomized 311 patients, and 221 were included in the intent-to-treat efficacy population.
The efficacy population consisted of all patients randomized prior to September 7, 2015, which allowed their data to be included in the week 24 endpoint analysis prior to the clinical hold.
The PAC QD arm consisted of 75 patients with a median age of 69 (range, 39-85). Seventy-one percent were 65 or older, and about half were male.
About three-quarters had an ECOG performance status of 0-1, 61% had primary myelofibrosis, 21% had primary polycythemia vera (PPV), and 17% had primary essential thrombocythemia (PET). About half were DIPSS Intermediate-2 risk, 80% were JAK2V617F positive, and 51% had a platelet count less than 50,000/μL.
The PAC BID arm consisted of 74 patients with a median age of 67 (range, 39-85). Sixty-two percent were 65 or older, and 65% were male.
Eighty-eight percent had an ECOG performance status of 0-1, 74% had primary myelofibrosis, 19% had PPV, and 7% had PET. About half were DIPSS Intermediate-2 risk, 80% were JAK2V617F positive, and 42% had a platelet count less than 50,000/μL.
The BAT arm consisted of 72 patients with a median age of 69 (range, 32-83). Seventy-one percent were 65 or older, and about half were male.
Three-quarters had an ECOG performance status of 0-1, 60% had primary myelofibrosis, 22% had PPV, and 18% had PET. About half were DIPSS Intermediate-2 risk, 71% were JAK2V617F positive, and 44% had a platelet count less than 50,000/µL.
Prior ruxolitinib therapy was consistent across the arms—41.3% (PAC QD), 42% (PAC BID), and 46% (BAT).
“Now, it’s important to note,” Dr Mascarenhas said, “that the most common BAT was ruxolitinib, 45%, and hydroxyurea, 19%. And 19% of BAT patients actually had no treatment, watch and wait. This highlights the fact that this is an area where there is really no other viable therapeutic option for these patients.”
Efficacy
Pacritinib-treated patients had significantly greater spleen reduction from baseline to week 24 than BAT-treated patients, with 18% (QD+BID), 15% (QD), and 22% (BID) achieving 35% or more SVR compared to 3% in the BAT arm.
Pacritinib-treated patients also experienced greater TSS reduction, with 25% (QD+BID), 17% (QD), and 32% (BID) reporting 50% or more reduction in TSS compared to 14% in the BAT arm. However, only the PAC BID arm was significantly different from BAT.
SVR in all subgroups—age, gender, JAK2 mutation status, prior treatment, platelet count, hemoglobin, peripheral blasts, and white blood cell count—demonstrated superiority for pacritinib.
Median changes in individual symptom scores were also better in the pacritinib arms than in the BAT arm in almost every category—tiredness, early satiety, abdominal discomfort, inactivity, night sweats, bone pain, and pain under ribs on the left side.
Pruritus was the only category in which BAT was superior, and that was compared to the QD arm and not the BID arm.
The majority of patients who stopped pacritinib therapy were taken off due to the clinical hold.
There were no significant differences between the groups in overall survival. Hazard ratios for overall survival (95% confidence intervals) were 0.68 (0.30, 1.53) for PAC BID vs BAT, 1.18 (0.57, 2.44) for PAC QD vs BAT, and 0.61 (0.27, 1.35) for PAC BID vs QD.
PAC BID maintained this survival advantage compared with BAT across nearly all demographic and myelofibrosis-associated risk factors.
Patients who were red blood cell transfusion-dependent at baseline experienced a statistically significant decrease in transfusion frequency from baseline to week 24 in both the QD and BID pacritinib arms compared with BAT.
And thrombocytopenia was not a significant factor for patients who were receiving pacritinib and had platelet counts less than 50,000/μL.
Toxicity
The most common treatment-emergent adverse events (AEs) associated with pacritinib were diarrhea, nausea, vomiting, anemia, and thrombocytopenia. They were generally less frequent for BID compared with QD administration.
The most common serious adverse events (SAEs)—occurring in 5% of patients or more in any arms—were anemia (5%, 8%, and 3%), thrombocytopenia (2%, 6%, and 2%), pneumonia (5%, 6%, and 4%), and acute renal failure (5%, 2%, and 2%) in the QD, BID, and BAT arms, respectively.
SAEs of interest included congestive heart failure, atrial fibrillation, cardiac arrest, epistaxis, and subdural hematoma, which occurred in 3% or fewer patients in any arm.
“They [the SAEs] were relatively infrequent, and there was not a clear signal of toxicity,” Dr Mascarenhas said.
Deaths
Deaths in the intent-to-treat evaluable population were censored at the time of full clinical hold.
For the entire enrolled population, 15/104 deaths occurred in the QD arm, 10/107 in the BID arm, and 14/100 in the BAT arm.
After the full clinical hold, 7, 10, and 6 deaths occurred in the QD, BID, and BAT arms, respectively.
Seven of 20 patients died after crossover to pacritinib. Five were due to AEs—3 cardiac, 1 bleeding, and 1 other.
“It’s important to note that progression of disease was the leading cause of death in the PAC BID arm,” Dr Mascarenhas noted. “This is after the patients stopped the drug [when the trial was on clinical hold].”
Conclusions
Despite study truncation, pacritinib (QD+BID) was significantly more effective than BAT for SVR (P=0.001) and trended toward improved TSS (P=0.079).
Pacritinib BID appeared more effective than QD versus BAT for SVR and TSS, and pacritinib BID appeared to have a better benefit/risk profile than BAT.
“This is a well-tolerated drug in many respects,” Dr Mascarenhas said. “This is a patient population that is quite ill, low platelets, poor outcome, and they did pretty well.”
When asked about the future of pacritinib, Dr Mascarenhas said he believes the benefit-to-risk ratio is in favor of the drug.
“Pacritinib offers patients in this vulnerable situation an opportunity for symptom relief, basically spleen and symptoms,” he said. “So I think this is a drug that will hopefully move forward.”
Dr Mascarenhas disclosed research funding from CTI Biopharma, the sponsor of the trial. ![]()
SAN DIEGO—The JAK2/FLT3 inhibitor pacritinib significantly reduces spleen volume and symptoms in patients with myelofibrosis and low platelet counts,
compared to best available therapy (BAT), according to results of the PERSIST-2 trial.
In this phase 3 trial, BAT included the JAK1/2 inhibitor ruxolitinib. And pacritinib demonstrated benefits over BAT despite a truncated trial.
The US Food and Drug Administration (FDA) placed PERSIST-2 on clinical hold in February 2016 due to concerns over interim survival results, bleeding, and cardiovascular events.
Patients randomized at least 22 weeks prior to the clinical hold contributed data to the week 24 endpoint, the results of which were presented at the 2016 ASH Annual Meeting.
John Mascarenhas, MD, of Icahn School of Medicine at Mount Sinai in New York, New York, delivered the results as a late-breaking abstract (LBA-5).
Ruxolitinib is FDA-approved to treat myelofibrosis, but it is associated with dose-limiting cytopenias and is not indicated for patients with platelet counts less than 50,000/μL.
The earlier PERSIST-1 trial demonstrated sustained spleen volume reduction (SVR) and symptom control with pacritinib therapy regardless of baseline platelet count, but it did not include ruxolitinib in the BAT arm.
PERSIST-2 study design
Patients were randomized on a 1:1:1 basis to pacritinib at 400 mg once daily (PAC QD), pacritinib at 200 mg twice daily (PAC BID), or BAT, including ruxolitinib.
Patients had to have primary or secondary myelofibrosis and platelet counts of 100,000/μL or less. Patients were allowed to have had prior JAK2 inhibitors.
Crossover from BAT was allowed after progression at any time or at week 24.
The study had 2 primary endpoints: percent of patients achieving a 35% or greater SVR and the percent of patients achieving a 50% or more reduction in total symptom score (TSS) by MPN-SAF TSS 2.0.
The primary study objective was the efficacy of pooled QD and BID pacritinib compared to BAT. The secondary objective was the efficacy of QD or BD separately compared to BAT.
Patient demographics
The study randomized 311 patients, and 221 were included in the intent-to-treat efficacy population.
The efficacy population consisted of all patients randomized prior to September 7, 2015, which allowed their data to be included in the week 24 endpoint analysis prior to the clinical hold.
The PAC QD arm consisted of 75 patients with a median age of 69 (range, 39-85). Seventy-one percent were 65 or older, and about half were male.
About three-quarters had an ECOG performance status of 0-1, 61% had primary myelofibrosis, 21% had primary polycythemia vera (PPV), and 17% had primary essential thrombocythemia (PET). About half were DIPSS Intermediate-2 risk, 80% were JAK2V617F positive, and 51% had a platelet count less than 50,000/μL.
The PAC BID arm consisted of 74 patients with a median age of 67 (range, 39-85). Sixty-two percent were 65 or older, and 65% were male.
Eighty-eight percent had an ECOG performance status of 0-1, 74% had primary myelofibrosis, 19% had PPV, and 7% had PET. About half were DIPSS Intermediate-2 risk, 80% were JAK2V617F positive, and 42% had a platelet count less than 50,000/μL.
The BAT arm consisted of 72 patients with a median age of 69 (range, 32-83). Seventy-one percent were 65 or older, and about half were male.
Three-quarters had an ECOG performance status of 0-1, 60% had primary myelofibrosis, 22% had PPV, and 18% had PET. About half were DIPSS Intermediate-2 risk, 71% were JAK2V617F positive, and 44% had a platelet count less than 50,000/µL.
Prior ruxolitinib therapy was consistent across the arms—41.3% (PAC QD), 42% (PAC BID), and 46% (BAT).
“Now, it’s important to note,” Dr Mascarenhas said, “that the most common BAT was ruxolitinib, 45%, and hydroxyurea, 19%. And 19% of BAT patients actually had no treatment, watch and wait. This highlights the fact that this is an area where there is really no other viable therapeutic option for these patients.”
Efficacy
Pacritinib-treated patients had significantly greater spleen reduction from baseline to week 24 than BAT-treated patients, with 18% (QD+BID), 15% (QD), and 22% (BID) achieving 35% or more SVR compared to 3% in the BAT arm.
Pacritinib-treated patients also experienced greater TSS reduction, with 25% (QD+BID), 17% (QD), and 32% (BID) reporting 50% or more reduction in TSS compared to 14% in the BAT arm. However, only the PAC BID arm was significantly different from BAT.
SVR in all subgroups—age, gender, JAK2 mutation status, prior treatment, platelet count, hemoglobin, peripheral blasts, and white blood cell count—demonstrated superiority for pacritinib.
Median changes in individual symptom scores were also better in the pacritinib arms than in the BAT arm in almost every category—tiredness, early satiety, abdominal discomfort, inactivity, night sweats, bone pain, and pain under ribs on the left side.
Pruritus was the only category in which BAT was superior, and that was compared to the QD arm and not the BID arm.
The majority of patients who stopped pacritinib therapy were taken off due to the clinical hold.
There were no significant differences between the groups in overall survival. Hazard ratios for overall survival (95% confidence intervals) were 0.68 (0.30, 1.53) for PAC BID vs BAT, 1.18 (0.57, 2.44) for PAC QD vs BAT, and 0.61 (0.27, 1.35) for PAC BID vs QD.
PAC BID maintained this survival advantage compared with BAT across nearly all demographic and myelofibrosis-associated risk factors.
Patients who were red blood cell transfusion-dependent at baseline experienced a statistically significant decrease in transfusion frequency from baseline to week 24 in both the QD and BID pacritinib arms compared with BAT.
And thrombocytopenia was not a significant factor for patients who were receiving pacritinib and had platelet counts less than 50,000/μL.
Toxicity
The most common treatment-emergent adverse events (AEs) associated with pacritinib were diarrhea, nausea, vomiting, anemia, and thrombocytopenia. They were generally less frequent for BID compared with QD administration.
The most common serious adverse events (SAEs)—occurring in 5% of patients or more in any arms—were anemia (5%, 8%, and 3%), thrombocytopenia (2%, 6%, and 2%), pneumonia (5%, 6%, and 4%), and acute renal failure (5%, 2%, and 2%) in the QD, BID, and BAT arms, respectively.
SAEs of interest included congestive heart failure, atrial fibrillation, cardiac arrest, epistaxis, and subdural hematoma, which occurred in 3% or fewer patients in any arm.
“They [the SAEs] were relatively infrequent, and there was not a clear signal of toxicity,” Dr Mascarenhas said.
Deaths
Deaths in the intent-to-treat evaluable population were censored at the time of full clinical hold.
For the entire enrolled population, 15/104 deaths occurred in the QD arm, 10/107 in the BID arm, and 14/100 in the BAT arm.
After the full clinical hold, 7, 10, and 6 deaths occurred in the QD, BID, and BAT arms, respectively.
Seven of 20 patients died after crossover to pacritinib. Five were due to AEs—3 cardiac, 1 bleeding, and 1 other.
“It’s important to note that progression of disease was the leading cause of death in the PAC BID arm,” Dr Mascarenhas noted. “This is after the patients stopped the drug [when the trial was on clinical hold].”
Conclusions
Despite study truncation, pacritinib (QD+BID) was significantly more effective than BAT for SVR (P=0.001) and trended toward improved TSS (P=0.079).
Pacritinib BID appeared more effective than QD versus BAT for SVR and TSS, and pacritinib BID appeared to have a better benefit/risk profile than BAT.
“This is a well-tolerated drug in many respects,” Dr Mascarenhas said. “This is a patient population that is quite ill, low platelets, poor outcome, and they did pretty well.”
When asked about the future of pacritinib, Dr Mascarenhas said he believes the benefit-to-risk ratio is in favor of the drug.
“Pacritinib offers patients in this vulnerable situation an opportunity for symptom relief, basically spleen and symptoms,” he said. “So I think this is a drug that will hopefully move forward.”
Dr Mascarenhas disclosed research funding from CTI Biopharma, the sponsor of the trial. ![]()
MPNs limit daily activities and ability to work

2016 ASH Annual Meeting
SAN DIEGO—Many patients living with myeloproliferative neoplasms (MPNs) have a high burden of disease that affects their quality of life and limits their ability to work, according to research presented at the 2016 ASH Annual Meeting.
Researchers conducted the first-ever international survey of MPN patients, including those with myelofibrosis (MF), polycythemia vera (PV), and essential thrombocythemia (ET).
Patients reported a high prevalence of symptoms, an increase in the number of symptoms from diagnosis, and reductions in emotional well-being, quality of life, and ability to work.
“The survey results help paint the full picture of the impact of these diseases,” said Claire Harrison, DM, of Guy’s and St. Thomas’ NHS Foundation Trust in London, UK.
“The dominating symptom is fatigue, which validates what we are seeing in the clinic. The novel findings regarding the impact on patient’s work life show the consequences of MPNs.”
Dr Harrison and her colleagues reported these findings at the meeting as (abstract 4267*).
The international MPN LANDMARK survey included 699 patients with MPNs (174 with MF, 223 with PV, and 302 with ET), and physicians who treated these conditions across Germany, Italy, UK, Japan, Canada, and Australia.
Patients completed an online questionnaire to measure MPN-related symptoms experienced over the past year and the impact of their condition on quality of life and ability to work.
Patients reported that their disease negatively impacted their ability to complete daily activities by 40%. Patients also noted 35% impairment in their capacity to work. Patients who missed work over the last 7 days due to illness reported missing an average of 3.1 hours due to their disease and/or symptom burden.
“For one quarter of PV patients, the disease stopped them from working, and another one quarter voluntarily reduced their work time,” Dr Harrison said. “This was surprising. Even though their disease was managed, it impacted their work life.”
She noted that many of her PV patients are young, working professionals.
Results also showed that about three-quarters of patients who experienced symptoms suffered a significant reduction in quality of life due to symptoms (83% of MF patients, 72% of PV patients, and 74% of ET patients).
These results are consistent with those from a previous US LANDMARK survey of MPN patients.
The most commonly reported symptom in the last 12 months was fatigue (54% of MF patients, 45% of PV patients, and 64% of ET patients). This was also the symptom patients stated they most wanted to resolve.
“Patients stated doctors often don’t ask enough about the details of symptoms,” Dr Harrison said.
In addition to physical symptoms, about one-third of patients felt anxious or worried about their disease (34% of MF patients, 29% of PV patients, and 26% of ET patients).
“We found a mismatch in aspirations of MPN patients and their physicians,” Dr Harrison said. “For example, physicians said it was important to teach PV patients to prevent blood clots. PV patients were more concerned with having a better quality of life and slowing disease progression.”
A comprehensive symptom assessment is important to help physicians understand other hardships of these diseases, she said, noting “in our service, we enlist nutritionists and psychologists and also use physiotherapists and occupational therapists.” ![]()
*Information presented at the meeting differs from the abstract.
2016 ASH Annual Meeting
SAN DIEGO—Many patients living with myeloproliferative neoplasms (MPNs) have a high burden of disease that affects their quality of life and limits their ability to work, according to research presented at the 2016 ASH Annual Meeting.
Researchers conducted the first-ever international survey of MPN patients, including those with myelofibrosis (MF), polycythemia vera (PV), and essential thrombocythemia (ET).
Patients reported a high prevalence of symptoms, an increase in the number of symptoms from diagnosis, and reductions in emotional well-being, quality of life, and ability to work.
“The survey results help paint the full picture of the impact of these diseases,” said Claire Harrison, DM, of Guy’s and St. Thomas’ NHS Foundation Trust in London, UK.
“The dominating symptom is fatigue, which validates what we are seeing in the clinic. The novel findings regarding the impact on patient’s work life show the consequences of MPNs.”
Dr Harrison and her colleagues reported these findings at the meeting as (abstract 4267*).
The international MPN LANDMARK survey included 699 patients with MPNs (174 with MF, 223 with PV, and 302 with ET), and physicians who treated these conditions across Germany, Italy, UK, Japan, Canada, and Australia.
Patients completed an online questionnaire to measure MPN-related symptoms experienced over the past year and the impact of their condition on quality of life and ability to work.
Patients reported that their disease negatively impacted their ability to complete daily activities by 40%. Patients also noted 35% impairment in their capacity to work. Patients who missed work over the last 7 days due to illness reported missing an average of 3.1 hours due to their disease and/or symptom burden.
“For one quarter of PV patients, the disease stopped them from working, and another one quarter voluntarily reduced their work time,” Dr Harrison said. “This was surprising. Even though their disease was managed, it impacted their work life.”
She noted that many of her PV patients are young, working professionals.
Results also showed that about three-quarters of patients who experienced symptoms suffered a significant reduction in quality of life due to symptoms (83% of MF patients, 72% of PV patients, and 74% of ET patients).
These results are consistent with those from a previous US LANDMARK survey of MPN patients.
The most commonly reported symptom in the last 12 months was fatigue (54% of MF patients, 45% of PV patients, and 64% of ET patients). This was also the symptom patients stated they most wanted to resolve.
“Patients stated doctors often don’t ask enough about the details of symptoms,” Dr Harrison said.
In addition to physical symptoms, about one-third of patients felt anxious or worried about their disease (34% of MF patients, 29% of PV patients, and 26% of ET patients).
“We found a mismatch in aspirations of MPN patients and their physicians,” Dr Harrison said. “For example, physicians said it was important to teach PV patients to prevent blood clots. PV patients were more concerned with having a better quality of life and slowing disease progression.”
A comprehensive symptom assessment is important to help physicians understand other hardships of these diseases, she said, noting “in our service, we enlist nutritionists and psychologists and also use physiotherapists and occupational therapists.” ![]()
*Information presented at the meeting differs from the abstract.
2016 ASH Annual Meeting
SAN DIEGO—Many patients living with myeloproliferative neoplasms (MPNs) have a high burden of disease that affects their quality of life and limits their ability to work, according to research presented at the 2016 ASH Annual Meeting.
Researchers conducted the first-ever international survey of MPN patients, including those with myelofibrosis (MF), polycythemia vera (PV), and essential thrombocythemia (ET).
Patients reported a high prevalence of symptoms, an increase in the number of symptoms from diagnosis, and reductions in emotional well-being, quality of life, and ability to work.
“The survey results help paint the full picture of the impact of these diseases,” said Claire Harrison, DM, of Guy’s and St. Thomas’ NHS Foundation Trust in London, UK.
“The dominating symptom is fatigue, which validates what we are seeing in the clinic. The novel findings regarding the impact on patient’s work life show the consequences of MPNs.”
Dr Harrison and her colleagues reported these findings at the meeting as (abstract 4267*).
The international MPN LANDMARK survey included 699 patients with MPNs (174 with MF, 223 with PV, and 302 with ET), and physicians who treated these conditions across Germany, Italy, UK, Japan, Canada, and Australia.
Patients completed an online questionnaire to measure MPN-related symptoms experienced over the past year and the impact of their condition on quality of life and ability to work.
Patients reported that their disease negatively impacted their ability to complete daily activities by 40%. Patients also noted 35% impairment in their capacity to work. Patients who missed work over the last 7 days due to illness reported missing an average of 3.1 hours due to their disease and/or symptom burden.
“For one quarter of PV patients, the disease stopped them from working, and another one quarter voluntarily reduced their work time,” Dr Harrison said. “This was surprising. Even though their disease was managed, it impacted their work life.”
She noted that many of her PV patients are young, working professionals.
Results also showed that about three-quarters of patients who experienced symptoms suffered a significant reduction in quality of life due to symptoms (83% of MF patients, 72% of PV patients, and 74% of ET patients).
These results are consistent with those from a previous US LANDMARK survey of MPN patients.
The most commonly reported symptom in the last 12 months was fatigue (54% of MF patients, 45% of PV patients, and 64% of ET patients). This was also the symptom patients stated they most wanted to resolve.
“Patients stated doctors often don’t ask enough about the details of symptoms,” Dr Harrison said.
In addition to physical symptoms, about one-third of patients felt anxious or worried about their disease (34% of MF patients, 29% of PV patients, and 26% of ET patients).
“We found a mismatch in aspirations of MPN patients and their physicians,” Dr Harrison said. “For example, physicians said it was important to teach PV patients to prevent blood clots. PV patients were more concerned with having a better quality of life and slowing disease progression.”
A comprehensive symptom assessment is important to help physicians understand other hardships of these diseases, she said, noting “in our service, we enlist nutritionists and psychologists and also use physiotherapists and occupational therapists.” ![]()
*Information presented at the meeting differs from the abstract.
Drug produces mixed results in myelofibrosis
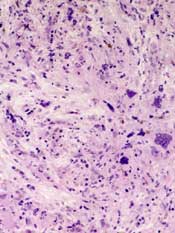
Two phase 3 trials have shown mixed results in myelofibrosis (MF) patients receiving the JAK inhibitor momelotinib, according to Gilead Sciences, Inc., the company developing the drug.
In the SIMPLIFY-1 study, momelotinib proved non-inferior to ruxolitinib when it came to the study’s primary endpoint but not its key secondary
endpoint.
In the SIMPLIFY-2 trial, momelotinib was not superior to best available therapy (BAT) with regard to the primary endpoint.
However, there were differences in favor of momelotinib when it came to some secondary endpoints.
“The results from both the SIMPLIFY-1 and SIMPLIFY-2 studies indicate that momelotinib provides some treatment benefit, including benefit on anemia-related endpoints,” said Norbert Bischofberger, PhD, executive vice president of research and development and chief scientific officer at Gilead Sciences, Inc.
“We plan to discuss these results with regulatory authorities to determine the next steps.”
About the studies
The SIMPLIFY studies are randomized, phase 3 trials designed to evaluate momelotinib in patients with primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF. The trials have the same primary and secondary endpoints.
The primary efficacy endpoint is splenic response rate at week 24 (SRR24), defined as the proportion of patients achieving a ≥ 35% reduction in spleen volume at week 24, as measured by MRI or CT scan.
Secondary endpoints include:
- Response rate in total symptom score (TSS) at week 24, defined as the proportion of patients achieving ≥ 50% reduction in symptoms, as measured by the modified Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score diary
- The proportion of patients who are transfusion-independent at week 24, defined as no red blood cell transfusion and no hemoglobin level below 8 g/dL in the prior 12 weeks
- The proportion who are transfusion-dependent at week 24, defined as at least 4 units of red blood cell transfusion or hemoglobin level below 8 g/dL in the prior 8 weeks
- The rate of red blood cell transfusion through week 24.
SIMPLIFY-1 trial
In SIMPLIFY-1, a double-blind, active-controlled study, 432 MF patients who had not previously been treated with a JAK inhibitor were randomized (1:1) to receive momelotinib or ruxolitinib for 24 weeks.
The study achieved its pre-specified primary endpoint of non-inferiority to ruxolitinib for SRR24. The incidence of SRR24 was 26.5% in the momelotinib arm and 29.0% in the ruxolitinib arm (95% CI: -11.2% to +5.6%; P=0.011).
However, non-inferiority was not achieved for the key secondary endpoint of response rate in TSS.
Greater improvements in all 3 anemia-related secondary endpoints—transfusion independence, transfusion dependence, and transfusion rate—were observed in patients receiving momelotinib compared to ruxolitinib.
However, because the TSS response rate did not meet the non-inferiority test, formal sequential statistical testing was not undertaken for these 3 secondary endpoints.
During 24 weeks of treatment in SIMPLIFY-1, the most frequent adverse events in patients receiving momelotinib were thrombocytopenia, diarrhea, headache, dizziness, and nausea.
The most frequent adverse events in patients receiving ruxolitinib were anemia, thrombocytopenia, diarrhea, headache, and dizziness.
Ten percent of patients receiving momelotinib reported peripheral neuropathy (any grade), compared to 5% of ruxolitinib-treated patients. There was no grade 3 or higher peripheral neuropathy in momelotinib-treated patients, but there was 1 case in the ruxolitinib arm.
SIMPLIFY-2 trial
In SIMPLIFY-2, 156 patients previously treated with, but not refractory to, ruxolitinib were randomized (2:1) to receive momelotinib or BAT for 24 weeks.
Eighty-eight percent of patients randomized to the BAT arm continued to receive ruxolitinib. The remainder of patients received chemotherapy, interferon, corticosteroids, other therapies, or some combination thereof.
The study’s primary endpoint was not met. Momelotinib did not prove superior to BAT with regard to SRR24. The incidence of SRR24 was 6.7% in the momelotinib arm and 5.8% in the BAT arm (95% CI: -8.9% to +10.2%; P=0.90).
Differences in favor of momelotinib were observed for the secondary endpoints of TSS and transfusion independence. However, formal sequential statistical testing was not undertaken because the primary superiority endpoint was not achieved.
Gilead did not release safety data from this trial. The company said detailed results from both SIMPLIFY studies will be submitted for presentation at upcoming scientific conferences. ![]()
Two phase 3 trials have shown mixed results in myelofibrosis (MF) patients receiving the JAK inhibitor momelotinib, according to Gilead Sciences, Inc., the company developing the drug.
In the SIMPLIFY-1 study, momelotinib proved non-inferior to ruxolitinib when it came to the study’s primary endpoint but not its key secondary
endpoint.
In the SIMPLIFY-2 trial, momelotinib was not superior to best available therapy (BAT) with regard to the primary endpoint.
However, there were differences in favor of momelotinib when it came to some secondary endpoints.
“The results from both the SIMPLIFY-1 and SIMPLIFY-2 studies indicate that momelotinib provides some treatment benefit, including benefit on anemia-related endpoints,” said Norbert Bischofberger, PhD, executive vice president of research and development and chief scientific officer at Gilead Sciences, Inc.
“We plan to discuss these results with regulatory authorities to determine the next steps.”
About the studies
The SIMPLIFY studies are randomized, phase 3 trials designed to evaluate momelotinib in patients with primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF. The trials have the same primary and secondary endpoints.
The primary efficacy endpoint is splenic response rate at week 24 (SRR24), defined as the proportion of patients achieving a ≥ 35% reduction in spleen volume at week 24, as measured by MRI or CT scan.
Secondary endpoints include:
- Response rate in total symptom score (TSS) at week 24, defined as the proportion of patients achieving ≥ 50% reduction in symptoms, as measured by the modified Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score diary
- The proportion of patients who are transfusion-independent at week 24, defined as no red blood cell transfusion and no hemoglobin level below 8 g/dL in the prior 12 weeks
- The proportion who are transfusion-dependent at week 24, defined as at least 4 units of red blood cell transfusion or hemoglobin level below 8 g/dL in the prior 8 weeks
- The rate of red blood cell transfusion through week 24.
SIMPLIFY-1 trial
In SIMPLIFY-1, a double-blind, active-controlled study, 432 MF patients who had not previously been treated with a JAK inhibitor were randomized (1:1) to receive momelotinib or ruxolitinib for 24 weeks.
The study achieved its pre-specified primary endpoint of non-inferiority to ruxolitinib for SRR24. The incidence of SRR24 was 26.5% in the momelotinib arm and 29.0% in the ruxolitinib arm (95% CI: -11.2% to +5.6%; P=0.011).
However, non-inferiority was not achieved for the key secondary endpoint of response rate in TSS.
Greater improvements in all 3 anemia-related secondary endpoints—transfusion independence, transfusion dependence, and transfusion rate—were observed in patients receiving momelotinib compared to ruxolitinib.
However, because the TSS response rate did not meet the non-inferiority test, formal sequential statistical testing was not undertaken for these 3 secondary endpoints.
During 24 weeks of treatment in SIMPLIFY-1, the most frequent adverse events in patients receiving momelotinib were thrombocytopenia, diarrhea, headache, dizziness, and nausea.
The most frequent adverse events in patients receiving ruxolitinib were anemia, thrombocytopenia, diarrhea, headache, and dizziness.
Ten percent of patients receiving momelotinib reported peripheral neuropathy (any grade), compared to 5% of ruxolitinib-treated patients. There was no grade 3 or higher peripheral neuropathy in momelotinib-treated patients, but there was 1 case in the ruxolitinib arm.
SIMPLIFY-2 trial
In SIMPLIFY-2, 156 patients previously treated with, but not refractory to, ruxolitinib were randomized (2:1) to receive momelotinib or BAT for 24 weeks.
Eighty-eight percent of patients randomized to the BAT arm continued to receive ruxolitinib. The remainder of patients received chemotherapy, interferon, corticosteroids, other therapies, or some combination thereof.
The study’s primary endpoint was not met. Momelotinib did not prove superior to BAT with regard to SRR24. The incidence of SRR24 was 6.7% in the momelotinib arm and 5.8% in the BAT arm (95% CI: -8.9% to +10.2%; P=0.90).
Differences in favor of momelotinib were observed for the secondary endpoints of TSS and transfusion independence. However, formal sequential statistical testing was not undertaken because the primary superiority endpoint was not achieved.
Gilead did not release safety data from this trial. The company said detailed results from both SIMPLIFY studies will be submitted for presentation at upcoming scientific conferences. ![]()
Two phase 3 trials have shown mixed results in myelofibrosis (MF) patients receiving the JAK inhibitor momelotinib, according to Gilead Sciences, Inc., the company developing the drug.
In the SIMPLIFY-1 study, momelotinib proved non-inferior to ruxolitinib when it came to the study’s primary endpoint but not its key secondary
endpoint.
In the SIMPLIFY-2 trial, momelotinib was not superior to best available therapy (BAT) with regard to the primary endpoint.
However, there were differences in favor of momelotinib when it came to some secondary endpoints.
“The results from both the SIMPLIFY-1 and SIMPLIFY-2 studies indicate that momelotinib provides some treatment benefit, including benefit on anemia-related endpoints,” said Norbert Bischofberger, PhD, executive vice president of research and development and chief scientific officer at Gilead Sciences, Inc.
“We plan to discuss these results with regulatory authorities to determine the next steps.”
About the studies
The SIMPLIFY studies are randomized, phase 3 trials designed to evaluate momelotinib in patients with primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF. The trials have the same primary and secondary endpoints.
The primary efficacy endpoint is splenic response rate at week 24 (SRR24), defined as the proportion of patients achieving a ≥ 35% reduction in spleen volume at week 24, as measured by MRI or CT scan.
Secondary endpoints include:
- Response rate in total symptom score (TSS) at week 24, defined as the proportion of patients achieving ≥ 50% reduction in symptoms, as measured by the modified Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score diary
- The proportion of patients who are transfusion-independent at week 24, defined as no red blood cell transfusion and no hemoglobin level below 8 g/dL in the prior 12 weeks
- The proportion who are transfusion-dependent at week 24, defined as at least 4 units of red blood cell transfusion or hemoglobin level below 8 g/dL in the prior 8 weeks
- The rate of red blood cell transfusion through week 24.
SIMPLIFY-1 trial
In SIMPLIFY-1, a double-blind, active-controlled study, 432 MF patients who had not previously been treated with a JAK inhibitor were randomized (1:1) to receive momelotinib or ruxolitinib for 24 weeks.
The study achieved its pre-specified primary endpoint of non-inferiority to ruxolitinib for SRR24. The incidence of SRR24 was 26.5% in the momelotinib arm and 29.0% in the ruxolitinib arm (95% CI: -11.2% to +5.6%; P=0.011).
However, non-inferiority was not achieved for the key secondary endpoint of response rate in TSS.
Greater improvements in all 3 anemia-related secondary endpoints—transfusion independence, transfusion dependence, and transfusion rate—were observed in patients receiving momelotinib compared to ruxolitinib.
However, because the TSS response rate did not meet the non-inferiority test, formal sequential statistical testing was not undertaken for these 3 secondary endpoints.
During 24 weeks of treatment in SIMPLIFY-1, the most frequent adverse events in patients receiving momelotinib were thrombocytopenia, diarrhea, headache, dizziness, and nausea.
The most frequent adverse events in patients receiving ruxolitinib were anemia, thrombocytopenia, diarrhea, headache, and dizziness.
Ten percent of patients receiving momelotinib reported peripheral neuropathy (any grade), compared to 5% of ruxolitinib-treated patients. There was no grade 3 or higher peripheral neuropathy in momelotinib-treated patients, but there was 1 case in the ruxolitinib arm.
SIMPLIFY-2 trial
In SIMPLIFY-2, 156 patients previously treated with, but not refractory to, ruxolitinib were randomized (2:1) to receive momelotinib or BAT for 24 weeks.
Eighty-eight percent of patients randomized to the BAT arm continued to receive ruxolitinib. The remainder of patients received chemotherapy, interferon, corticosteroids, other therapies, or some combination thereof.
The study’s primary endpoint was not met. Momelotinib did not prove superior to BAT with regard to SRR24. The incidence of SRR24 was 6.7% in the momelotinib arm and 5.8% in the BAT arm (95% CI: -8.9% to +10.2%; P=0.90).
Differences in favor of momelotinib were observed for the secondary endpoints of TSS and transfusion independence. However, formal sequential statistical testing was not undertaken because the primary superiority endpoint was not achieved.
Gilead did not release safety data from this trial. The company said detailed results from both SIMPLIFY studies will be submitted for presentation at upcoming scientific conferences. ![]()
MRI detects early stages of MF in mice

Magnetic resonance imaging (MRI) can effectively detect myelofibrosis (MF) in a mouse model, according to research published in the journal Blood Cancer.
In fact, researchers found that MRI could detect early and late stages of primary MF.
The researchers believe this discovery could potentially change the way MF is diagnosed, as MRI might be used to help physicians decide if or where to biopsy.
Katya Ravid, PhD, of Boston University School of Medicine in Massachusetts, and her colleagues conducted this research, aiming to determine whether T2-weighted MRI could detect bone marrow fibrosis in a mouse model of primary MF.
The team looked specifically at how effectively MRI could detect MF during the pre-fibrotic stage (when mice were less than 16 weeks old), when the mice had early MF (16 to 36 weeks old), and once the mice had overt MF (older than 36 weeks).
The researchers found that MRI could detect MF at the pre-fibrotic stage as well as detecting progressive MF.
The team said they observed a clear, bright signal that allowed them to differentiate mice with MF from healthy control mice.
The researchers proposed that the abundance of large megakaryocytes contributed to the bright signal they observed, since, in T2-weighted MR images, increased water/proton content, as in increased cellularity, yields high MR-signal intensity.
The team said this study provides proof of concept that T2-weighted MRI can detect primary MF in the early and late stages. ![]()
Magnetic resonance imaging (MRI) can effectively detect myelofibrosis (MF) in a mouse model, according to research published in the journal Blood Cancer.
In fact, researchers found that MRI could detect early and late stages of primary MF.
The researchers believe this discovery could potentially change the way MF is diagnosed, as MRI might be used to help physicians decide if or where to biopsy.
Katya Ravid, PhD, of Boston University School of Medicine in Massachusetts, and her colleagues conducted this research, aiming to determine whether T2-weighted MRI could detect bone marrow fibrosis in a mouse model of primary MF.
The team looked specifically at how effectively MRI could detect MF during the pre-fibrotic stage (when mice were less than 16 weeks old), when the mice had early MF (16 to 36 weeks old), and once the mice had overt MF (older than 36 weeks).
The researchers found that MRI could detect MF at the pre-fibrotic stage as well as detecting progressive MF.
The team said they observed a clear, bright signal that allowed them to differentiate mice with MF from healthy control mice.
The researchers proposed that the abundance of large megakaryocytes contributed to the bright signal they observed, since, in T2-weighted MR images, increased water/proton content, as in increased cellularity, yields high MR-signal intensity.
The team said this study provides proof of concept that T2-weighted MRI can detect primary MF in the early and late stages. ![]()
Magnetic resonance imaging (MRI) can effectively detect myelofibrosis (MF) in a mouse model, according to research published in the journal Blood Cancer.
In fact, researchers found that MRI could detect early and late stages of primary MF.
The researchers believe this discovery could potentially change the way MF is diagnosed, as MRI might be used to help physicians decide if or where to biopsy.
Katya Ravid, PhD, of Boston University School of Medicine in Massachusetts, and her colleagues conducted this research, aiming to determine whether T2-weighted MRI could detect bone marrow fibrosis in a mouse model of primary MF.
The team looked specifically at how effectively MRI could detect MF during the pre-fibrotic stage (when mice were less than 16 weeks old), when the mice had early MF (16 to 36 weeks old), and once the mice had overt MF (older than 36 weeks).
The researchers found that MRI could detect MF at the pre-fibrotic stage as well as detecting progressive MF.
The team said they observed a clear, bright signal that allowed them to differentiate mice with MF from healthy control mice.
The researchers proposed that the abundance of large megakaryocytes contributed to the bright signal they observed, since, in T2-weighted MR images, increased water/proton content, as in increased cellularity, yields high MR-signal intensity.
The team said this study provides proof of concept that T2-weighted MRI can detect primary MF in the early and late stages. ![]()
FDA grants priority review for midostaurin
The US Food and Drug Administration (FDA) has granted priority review for the new drug application for midostaurin (PKC412) as a treatment for advanced systemic mastocytosis (SM) and newly diagnosed, FLT3-mutated acute myeloid leukemia (AML).
The FDA has also accepted for review the premarket approval application for the midostaurin FLT3 companion diagnostic, which is designed to help identify patients who may have a FLT3 mutation and could potentially benefit from treatment with midostaurin.
Midostaurin is being developed by Novartis. The companion diagnostic is being developed by Novartis and Invivoscribe Technologies, Inc.
About priority review
The FDA grants priority review to applications for therapies that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it. The goal in the standard review process is to take action within 10 months.
About midostaurin
Midostaurin is an oral, multi-targeted kinase inhibitor. The drug was granted breakthrough therapy designation by the FDA earlier this year for newly diagnosed, FLT3-mutated AML.
According to Novartis, the new drug application submission for midostaurin includes data from the largest clinical trials conducted to date in advanced SM and newly diagnosed, FLT3-mutated AML.
Midostaurin in AML
In the phase 3 RATIFY trial, researchers compared midostaurin plus standard chemotherapy to placebo plus standard chemotherapy in adults younger than 60 with FLT3-mutated AML. Results from this trial were presented at the 2015 ASH Annual Meeting.
Patients in the midostaurin arm experienced a statistically significant improvement in overall survival, with a 23% reduction in risk of death compared to the placebo arm (hazard ratio=0.77, P=0.0074).
There was no significant difference in the overall rate of grade 3 or higher hematologic and non-hematologic adverse events in midostaurin arm and the placebo arm. Similarly, there was no significant difference in treatment-related deaths between the arms.
Midostaurin in SM
Data from the phase 2 study of midostaurin in patients with advanced SM were published in NEJM in June.
The drug produced a 60% overall response rate, and the median duration of response was 24.1 months.
Fifty-six percent of patients required dose reductions due to toxic effects, but 32% of these patients were able to return to the starting dose of midostaurin.
Access to midostaurin
Since midostaurin remains investigational, both within the US and globally, Novartis opened a Global Individual Patient Program (compassionate use program) and, in the US, an Expanded Treatment Protocol, to provide access to midostaurin for eligible patients with newly diagnosed AML and advanced SM.
Physicians who want to request midostaurin for eligible patients can contact a Novartis medical representative in their respective countries. In the US, physicians can call 1-888-NOW-NOVA (1-888-669-6682) for more information. ![]()
The US Food and Drug Administration (FDA) has granted priority review for the new drug application for midostaurin (PKC412) as a treatment for advanced systemic mastocytosis (SM) and newly diagnosed, FLT3-mutated acute myeloid leukemia (AML).
The FDA has also accepted for review the premarket approval application for the midostaurin FLT3 companion diagnostic, which is designed to help identify patients who may have a FLT3 mutation and could potentially benefit from treatment with midostaurin.
Midostaurin is being developed by Novartis. The companion diagnostic is being developed by Novartis and Invivoscribe Technologies, Inc.
About priority review
The FDA grants priority review to applications for therapies that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it. The goal in the standard review process is to take action within 10 months.
About midostaurin
Midostaurin is an oral, multi-targeted kinase inhibitor. The drug was granted breakthrough therapy designation by the FDA earlier this year for newly diagnosed, FLT3-mutated AML.
According to Novartis, the new drug application submission for midostaurin includes data from the largest clinical trials conducted to date in advanced SM and newly diagnosed, FLT3-mutated AML.
Midostaurin in AML
In the phase 3 RATIFY trial, researchers compared midostaurin plus standard chemotherapy to placebo plus standard chemotherapy in adults younger than 60 with FLT3-mutated AML. Results from this trial were presented at the 2015 ASH Annual Meeting.
Patients in the midostaurin arm experienced a statistically significant improvement in overall survival, with a 23% reduction in risk of death compared to the placebo arm (hazard ratio=0.77, P=0.0074).
There was no significant difference in the overall rate of grade 3 or higher hematologic and non-hematologic adverse events in midostaurin arm and the placebo arm. Similarly, there was no significant difference in treatment-related deaths between the arms.
Midostaurin in SM
Data from the phase 2 study of midostaurin in patients with advanced SM were published in NEJM in June.
The drug produced a 60% overall response rate, and the median duration of response was 24.1 months.
Fifty-six percent of patients required dose reductions due to toxic effects, but 32% of these patients were able to return to the starting dose of midostaurin.
Access to midostaurin
Since midostaurin remains investigational, both within the US and globally, Novartis opened a Global Individual Patient Program (compassionate use program) and, in the US, an Expanded Treatment Protocol, to provide access to midostaurin for eligible patients with newly diagnosed AML and advanced SM.
Physicians who want to request midostaurin for eligible patients can contact a Novartis medical representative in their respective countries. In the US, physicians can call 1-888-NOW-NOVA (1-888-669-6682) for more information. ![]()
The US Food and Drug Administration (FDA) has granted priority review for the new drug application for midostaurin (PKC412) as a treatment for advanced systemic mastocytosis (SM) and newly diagnosed, FLT3-mutated acute myeloid leukemia (AML).
The FDA has also accepted for review the premarket approval application for the midostaurin FLT3 companion diagnostic, which is designed to help identify patients who may have a FLT3 mutation and could potentially benefit from treatment with midostaurin.
Midostaurin is being developed by Novartis. The companion diagnostic is being developed by Novartis and Invivoscribe Technologies, Inc.
About priority review
The FDA grants priority review to applications for therapies that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
The agency’s goal is to take action on a priority review application within 6 months of receiving it. The goal in the standard review process is to take action within 10 months.
About midostaurin
Midostaurin is an oral, multi-targeted kinase inhibitor. The drug was granted breakthrough therapy designation by the FDA earlier this year for newly diagnosed, FLT3-mutated AML.
According to Novartis, the new drug application submission for midostaurin includes data from the largest clinical trials conducted to date in advanced SM and newly diagnosed, FLT3-mutated AML.
Midostaurin in AML
In the phase 3 RATIFY trial, researchers compared midostaurin plus standard chemotherapy to placebo plus standard chemotherapy in adults younger than 60 with FLT3-mutated AML. Results from this trial were presented at the 2015 ASH Annual Meeting.
Patients in the midostaurin arm experienced a statistically significant improvement in overall survival, with a 23% reduction in risk of death compared to the placebo arm (hazard ratio=0.77, P=0.0074).
There was no significant difference in the overall rate of grade 3 or higher hematologic and non-hematologic adverse events in midostaurin arm and the placebo arm. Similarly, there was no significant difference in treatment-related deaths between the arms.
Midostaurin in SM
Data from the phase 2 study of midostaurin in patients with advanced SM were published in NEJM in June.
The drug produced a 60% overall response rate, and the median duration of response was 24.1 months.
Fifty-six percent of patients required dose reductions due to toxic effects, but 32% of these patients were able to return to the starting dose of midostaurin.
Access to midostaurin
Since midostaurin remains investigational, both within the US and globally, Novartis opened a Global Individual Patient Program (compassionate use program) and, in the US, an Expanded Treatment Protocol, to provide access to midostaurin for eligible patients with newly diagnosed AML and advanced SM.
Physicians who want to request midostaurin for eligible patients can contact a Novartis medical representative in their respective countries. In the US, physicians can call 1-888-NOW-NOVA (1-888-669-6682) for more information. ![]()
Study provides new insight into RBC production

Andes who suffers from
chronic mountain sickness
Photo courtesy of
UC San Diego Health
Findings from a study of people living at high altitude have implications for the treatment of red blood cell (RBC) disorders such as anemia and polycythemia, according to researchers.
To better understand why some people adapt well to life at high altitude while others don’t, the researchers studied RBCs derived from representatives of both groups who were living in the Andes Mountains.
The study revealed that high-altitude dwellers prone to chronic mountain sickness produce massive amounts of RBCs thanks to overproduction of the enzyme SENP1.
The researchers reported these findings in the Journal of Experimental Medicine.
“In addition to improving the health of millions of people around the world who live above 8000 feet, information on how Andeans have adapted—or not adapted—to high-altitude life might teach us how to speed up red blood cell production at lower altitudes, such as in anemia or when blood transfusions are needed rapidly,” said study author Gabriel Haddad, MD, of the University of California San Diego School of Medicine.
Dr Haddad and his colleagues noted that chronic mountain sickness affects approximately 20% of people who live at high altitudes, and a critical aspect of the condition is polycythemia.
Some extra RBCs can be beneficial in high-altitude, low-oxygen environments by helping to keep blood oxygenated. However, too many RBCs can increase the risk of heart attack and stroke, even in young adults.
For this study, the researchers collected skin cells from people living in the Andes Mountains—4 who were healthy and 5 who suffer from chronic mountain sickness—plus an additional 3 healthy people who live at sea level and served as controls.
To produce enough RBCs from each participant to study them in the lab, the researchers converted the skin cells into induced pluripotent stem cells (iPSCs).
Then, adding a cocktail of growth factors and other molecules, the team coaxed the iPSCs to differentiate into RBCs. Multiple samples were tested for each person, for a total of at least 24 iPSC lines.
The researchers exposed the RBCs to low-oxygen conditions that mimic high altitude—5% oxygen—for 3 weeks.
RBCs from healthy sea-level or high-altitude-dwelling donors increased a little or not at all. In contrast, RBC counts from high-altitude dwellers with chronic mountain sickness increased 60-fold.
This result led the researchers to question why people with chronic mountain sickness produce so many extra RBCs in response to low oxygen.
In a previous study, the team had compared the genomes of high-altitude dwellers with and without chronic mountain sickness. This revealed a gene that varied between the 2 groups—SENP1, which is increased in low-oxygen situations in people with chronic mountain sickness but not in healthy individuals.
In the current study, the researchers set out to determine if SENP1 plays a role in high-altitude adaptation.
The team inhibited the SENP1 gene in iPSCs from patients with chronic mountain sickness. As a result, excessive RBC production was reduced by more than 90%.
When the researchers added extra SENP1 to healthy, adapted highlander iPSCs, RBC production increased 30-fold, nearly recapitulating that seen in patients with chronic mountains sickness.
Further experiments revealed how SENP1 affects RBC production. Elevated levels of the enzyme in chronic mountain sickness boost levels of several other proteins that promote cell division and survival, including VEGF, GATA1, and Bcl-xL.
“We’re interested in determining the early steps in this process—how low oxygen triggers SENP1 in the first place,” said study author Priti Azad, PhD, of the University of California San Diego School of Medicine.
“We are also investigating how existing altitude sickness medications, such as Diamox, work and whether or not it’s through this same mechanism.” ![]()
Andes who suffers from
chronic mountain sickness
Photo courtesy of
UC San Diego Health
Findings from a study of people living at high altitude have implications for the treatment of red blood cell (RBC) disorders such as anemia and polycythemia, according to researchers.
To better understand why some people adapt well to life at high altitude while others don’t, the researchers studied RBCs derived from representatives of both groups who were living in the Andes Mountains.
The study revealed that high-altitude dwellers prone to chronic mountain sickness produce massive amounts of RBCs thanks to overproduction of the enzyme SENP1.
The researchers reported these findings in the Journal of Experimental Medicine.
“In addition to improving the health of millions of people around the world who live above 8000 feet, information on how Andeans have adapted—or not adapted—to high-altitude life might teach us how to speed up red blood cell production at lower altitudes, such as in anemia or when blood transfusions are needed rapidly,” said study author Gabriel Haddad, MD, of the University of California San Diego School of Medicine.
Dr Haddad and his colleagues noted that chronic mountain sickness affects approximately 20% of people who live at high altitudes, and a critical aspect of the condition is polycythemia.
Some extra RBCs can be beneficial in high-altitude, low-oxygen environments by helping to keep blood oxygenated. However, too many RBCs can increase the risk of heart attack and stroke, even in young adults.
For this study, the researchers collected skin cells from people living in the Andes Mountains—4 who were healthy and 5 who suffer from chronic mountain sickness—plus an additional 3 healthy people who live at sea level and served as controls.
To produce enough RBCs from each participant to study them in the lab, the researchers converted the skin cells into induced pluripotent stem cells (iPSCs).
Then, adding a cocktail of growth factors and other molecules, the team coaxed the iPSCs to differentiate into RBCs. Multiple samples were tested for each person, for a total of at least 24 iPSC lines.
The researchers exposed the RBCs to low-oxygen conditions that mimic high altitude—5% oxygen—for 3 weeks.
RBCs from healthy sea-level or high-altitude-dwelling donors increased a little or not at all. In contrast, RBC counts from high-altitude dwellers with chronic mountain sickness increased 60-fold.
This result led the researchers to question why people with chronic mountain sickness produce so many extra RBCs in response to low oxygen.
In a previous study, the team had compared the genomes of high-altitude dwellers with and without chronic mountain sickness. This revealed a gene that varied between the 2 groups—SENP1, which is increased in low-oxygen situations in people with chronic mountain sickness but not in healthy individuals.
In the current study, the researchers set out to determine if SENP1 plays a role in high-altitude adaptation.
The team inhibited the SENP1 gene in iPSCs from patients with chronic mountain sickness. As a result, excessive RBC production was reduced by more than 90%.
When the researchers added extra SENP1 to healthy, adapted highlander iPSCs, RBC production increased 30-fold, nearly recapitulating that seen in patients with chronic mountains sickness.
Further experiments revealed how SENP1 affects RBC production. Elevated levels of the enzyme in chronic mountain sickness boost levels of several other proteins that promote cell division and survival, including VEGF, GATA1, and Bcl-xL.
“We’re interested in determining the early steps in this process—how low oxygen triggers SENP1 in the first place,” said study author Priti Azad, PhD, of the University of California San Diego School of Medicine.
“We are also investigating how existing altitude sickness medications, such as Diamox, work and whether or not it’s through this same mechanism.” ![]()
Andes who suffers from
chronic mountain sickness
Photo courtesy of
UC San Diego Health
Findings from a study of people living at high altitude have implications for the treatment of red blood cell (RBC) disorders such as anemia and polycythemia, according to researchers.
To better understand why some people adapt well to life at high altitude while others don’t, the researchers studied RBCs derived from representatives of both groups who were living in the Andes Mountains.
The study revealed that high-altitude dwellers prone to chronic mountain sickness produce massive amounts of RBCs thanks to overproduction of the enzyme SENP1.
The researchers reported these findings in the Journal of Experimental Medicine.
“In addition to improving the health of millions of people around the world who live above 8000 feet, information on how Andeans have adapted—or not adapted—to high-altitude life might teach us how to speed up red blood cell production at lower altitudes, such as in anemia or when blood transfusions are needed rapidly,” said study author Gabriel Haddad, MD, of the University of California San Diego School of Medicine.
Dr Haddad and his colleagues noted that chronic mountain sickness affects approximately 20% of people who live at high altitudes, and a critical aspect of the condition is polycythemia.
Some extra RBCs can be beneficial in high-altitude, low-oxygen environments by helping to keep blood oxygenated. However, too many RBCs can increase the risk of heart attack and stroke, even in young adults.
For this study, the researchers collected skin cells from people living in the Andes Mountains—4 who were healthy and 5 who suffer from chronic mountain sickness—plus an additional 3 healthy people who live at sea level and served as controls.
To produce enough RBCs from each participant to study them in the lab, the researchers converted the skin cells into induced pluripotent stem cells (iPSCs).
Then, adding a cocktail of growth factors and other molecules, the team coaxed the iPSCs to differentiate into RBCs. Multiple samples were tested for each person, for a total of at least 24 iPSC lines.
The researchers exposed the RBCs to low-oxygen conditions that mimic high altitude—5% oxygen—for 3 weeks.
RBCs from healthy sea-level or high-altitude-dwelling donors increased a little or not at all. In contrast, RBC counts from high-altitude dwellers with chronic mountain sickness increased 60-fold.
This result led the researchers to question why people with chronic mountain sickness produce so many extra RBCs in response to low oxygen.
In a previous study, the team had compared the genomes of high-altitude dwellers with and without chronic mountain sickness. This revealed a gene that varied between the 2 groups—SENP1, which is increased in low-oxygen situations in people with chronic mountain sickness but not in healthy individuals.
In the current study, the researchers set out to determine if SENP1 plays a role in high-altitude adaptation.
The team inhibited the SENP1 gene in iPSCs from patients with chronic mountain sickness. As a result, excessive RBC production was reduced by more than 90%.
When the researchers added extra SENP1 to healthy, adapted highlander iPSCs, RBC production increased 30-fold, nearly recapitulating that seen in patients with chronic mountains sickness.
Further experiments revealed how SENP1 affects RBC production. Elevated levels of the enzyme in chronic mountain sickness boost levels of several other proteins that promote cell division and survival, including VEGF, GATA1, and Bcl-xL.
“We’re interested in determining the early steps in this process—how low oxygen triggers SENP1 in the first place,” said study author Priti Azad, PhD, of the University of California San Diego School of Medicine.
“We are also investigating how existing altitude sickness medications, such as Diamox, work and whether or not it’s through this same mechanism.” ![]()
Explaining the development of MPNs, leukemia
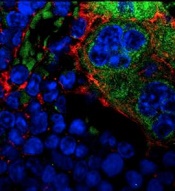
MSPCs with mutant PTPN11
(red) and monocytes (green).
Image courtesy of
Dong et al, Nature 2016
New research published in Nature has shown how certain mutations drive the development of myeloproliferative neoplasms (MPNs) and leukemia.
Investigators discovered that PTPN11 activating mutations promote the development and progression of MPNs through “profound detrimental effects” on hematopoietic stem cells (HSCs).
The team also identified a potential method of treating MPNs in patients with Noonan syndrome.
Noonan syndrome can be caused by mutations in several genes, but the most common is PTPN11. Children with Noonan syndrome are known to have an increased risk of developing MPNs/leukemia.
Previous research had established that mutations in PTPN11 have a conventional cell-autonomous effect on HSC growth.
In the current study, investigators showed that PTPN11 mutations also affect mesenchymal stem/progenitor cells (MSPCs) and osteoprogenitors.
The mutations cause over-production of the CC chemokine CCL3, which attracts monocytes into the HSCs’ niches. The monocytes make inflammatory molecules that stimulate the HSCs to differentiate and proliferate, leading to MPNs and leukemia.
“We have identified CCL3 as a potential therapeutic target for controlling leukemic progression in Noonan syndrome and for improving stem cell transplantation therapy in Noonan syndrome-associated leukemias,” said study author Cheng-Kui Qu, MD, PhD, of Emory University School of Medicine in Atlanta, Georgia.
Dr Qu and his colleagues began this research intending to investigate the effects of PTPN11 mutations in the nervous system. The team developed genetically engineered mice that had altered PTPN11 in neural cells.
The mice all developed a condition resembling an MPN at an early age. It turned out that the mice had changes in the PTPN11 gene in their MSPCs and osteoprogenitors (in addition to their neural cells) but not in their HSCs.
The investigators found the MPN in these PTPN11-mutant mice can be treated in the short-term by HSC transplant, but the condition comes back within months.
However, drugs counteracting CCL3 successfully reversed MPN phenotypes. One of the drugs is the CCR5 antagonist maraviroc, which is approved in the US to combat HIV infection, and another is the CCR1 antagonist BX471.
The investigators noted that other Noonan syndrome mutations, in genes besides PTPN11, need to be assessed for their effects on MPN/leukemia formation. ![]()
MSPCs with mutant PTPN11
(red) and monocytes (green).
Image courtesy of
Dong et al, Nature 2016
New research published in Nature has shown how certain mutations drive the development of myeloproliferative neoplasms (MPNs) and leukemia.
Investigators discovered that PTPN11 activating mutations promote the development and progression of MPNs through “profound detrimental effects” on hematopoietic stem cells (HSCs).
The team also identified a potential method of treating MPNs in patients with Noonan syndrome.
Noonan syndrome can be caused by mutations in several genes, but the most common is PTPN11. Children with Noonan syndrome are known to have an increased risk of developing MPNs/leukemia.
Previous research had established that mutations in PTPN11 have a conventional cell-autonomous effect on HSC growth.
In the current study, investigators showed that PTPN11 mutations also affect mesenchymal stem/progenitor cells (MSPCs) and osteoprogenitors.
The mutations cause over-production of the CC chemokine CCL3, which attracts monocytes into the HSCs’ niches. The monocytes make inflammatory molecules that stimulate the HSCs to differentiate and proliferate, leading to MPNs and leukemia.
“We have identified CCL3 as a potential therapeutic target for controlling leukemic progression in Noonan syndrome and for improving stem cell transplantation therapy in Noonan syndrome-associated leukemias,” said study author Cheng-Kui Qu, MD, PhD, of Emory University School of Medicine in Atlanta, Georgia.
Dr Qu and his colleagues began this research intending to investigate the effects of PTPN11 mutations in the nervous system. The team developed genetically engineered mice that had altered PTPN11 in neural cells.
The mice all developed a condition resembling an MPN at an early age. It turned out that the mice had changes in the PTPN11 gene in their MSPCs and osteoprogenitors (in addition to their neural cells) but not in their HSCs.
The investigators found the MPN in these PTPN11-mutant mice can be treated in the short-term by HSC transplant, but the condition comes back within months.
However, drugs counteracting CCL3 successfully reversed MPN phenotypes. One of the drugs is the CCR5 antagonist maraviroc, which is approved in the US to combat HIV infection, and another is the CCR1 antagonist BX471.
The investigators noted that other Noonan syndrome mutations, in genes besides PTPN11, need to be assessed for their effects on MPN/leukemia formation. ![]()
MSPCs with mutant PTPN11
(red) and monocytes (green).
Image courtesy of
Dong et al, Nature 2016
New research published in Nature has shown how certain mutations drive the development of myeloproliferative neoplasms (MPNs) and leukemia.
Investigators discovered that PTPN11 activating mutations promote the development and progression of MPNs through “profound detrimental effects” on hematopoietic stem cells (HSCs).
The team also identified a potential method of treating MPNs in patients with Noonan syndrome.
Noonan syndrome can be caused by mutations in several genes, but the most common is PTPN11. Children with Noonan syndrome are known to have an increased risk of developing MPNs/leukemia.
Previous research had established that mutations in PTPN11 have a conventional cell-autonomous effect on HSC growth.
In the current study, investigators showed that PTPN11 mutations also affect mesenchymal stem/progenitor cells (MSPCs) and osteoprogenitors.
The mutations cause over-production of the CC chemokine CCL3, which attracts monocytes into the HSCs’ niches. The monocytes make inflammatory molecules that stimulate the HSCs to differentiate and proliferate, leading to MPNs and leukemia.
“We have identified CCL3 as a potential therapeutic target for controlling leukemic progression in Noonan syndrome and for improving stem cell transplantation therapy in Noonan syndrome-associated leukemias,” said study author Cheng-Kui Qu, MD, PhD, of Emory University School of Medicine in Atlanta, Georgia.
Dr Qu and his colleagues began this research intending to investigate the effects of PTPN11 mutations in the nervous system. The team developed genetically engineered mice that had altered PTPN11 in neural cells.
The mice all developed a condition resembling an MPN at an early age. It turned out that the mice had changes in the PTPN11 gene in their MSPCs and osteoprogenitors (in addition to their neural cells) but not in their HSCs.
The investigators found the MPN in these PTPN11-mutant mice can be treated in the short-term by HSC transplant, but the condition comes back within months.
However, drugs counteracting CCL3 successfully reversed MPN phenotypes. One of the drugs is the CCR5 antagonist maraviroc, which is approved in the US to combat HIV infection, and another is the CCR1 antagonist BX471.
The investigators noted that other Noonan syndrome mutations, in genes besides PTPN11, need to be assessed for their effects on MPN/leukemia formation.
MF assessment and treatment an ‘evolution’ from the past

© ASCO/Zach Boyden-Holmes
NEW YORK—The 11th NCCN Congress: Hematologic Malignancies coincided with the release of the inaugural edition of the NCCN treatment guideline on myeloproliferative neoplasms (MPNs), and Ruben A. Mesa, MD, took the opportunity to discuss the framework of the document and the evolving management of MPNs.
Dr Mesa, of the Mayo Clinic Cancer Center in Arizona, is the chair of the MPN guideline committee.
This initial version of the guideline focuses on the workup and diagnosis of primary myelofibrosis (MF), post-polycythemia vera MF, and post-essential thrombocythemia MF, and treatment guidelines for MF.
The committee decided to first tackle the treatment guidelines in MF because, as Dr Mesa explained, MF is “the most severe of the MPNs with the most unmet needs in terms of guidance.”
Treatment guidelines for polycythemia vera (PV) and essential thrombocythemia (ET) will be forthcoming in 2017, he said, as well as diagnosis and treatment of atypical MPNs, such as hypereosinophilic syndrome and systemic mast cell disease.
Workup of an MPN
The guideline committee focused on information regarding diagnosis—the time and place to consider bone marrow biopsies, when to use cytogenetics, and when to perform next-generation sequencing.
They stressed the importance of quantifying disease burden and utilizing the now-validated symptom assessment tools, particularly in MF.
“Finally, we leveraged the many prognostic scoring systems that have been developed for these diseases, particularly IPSS for myelofibrosis at diagnosis or the DIPSS and DIPSS-plus at subsequent time points,” Dr Mesa said.
In addition to clinical-based prognostic scoring systems, information regarding molecular features and their impact is evolving, he said.
The guideline provides a table of mutations with prognostic significance. JAK2, MPL, or CALR mutations are “brighter mutations,” he said, while adverse prognostic markers include ASXL1, EZH2, IDH1/2, SRSF2, and TP53.
“All of these have some significant prognostic implications in primary myelofibrosis, specifically,” he said.
Assessing MPN burden
“When treating these patients, it’s important to be mindful of the overall burden of the disease,” Dr Mesa said.
He called this emphasis on disease burden “an evolution from the past, where therapy was either supportive or primarily focused on prevention of thrombosis with ET or PV.”
Practitioners need to be additionally mindful of the risk of vascular events, progression, the impact of cytopenias, splenomegaly, the burden of symptoms, and the baseline degree of comorbidities.
“[W]e encourage the use of validated symptom tools that have been used now in the majority of clinical trials in this setting,” Dr Mesa added.
The MPN-10 assessment tool, included in the guideline, evaluates 10 symptoms—early satiety, abdominal discomfort, inactivity, problems concentrating, numbness/tingling, night sweats, itching, bone pain, fever, and unintentional weight loss—on a scale of 0 to 10.
Patients with MF are the most symptomatic, Dr Mesa commented, although “it is notable how frequent symptoms are present in patients with PV and ET.”
Response criteria
For a complete response, individuals must have marked improvement to near normalization in both bone marrow and peripheral blood in addition to resolution of their disease symptoms.
Partial response is basically “just shy” of the complete response level, Dr Mesa said, with bone marrow resolution not being required.
The guideline also outlines response criteria for progressive, stable, and relapsed disease, clinical improvement, and anemia, spleen, and symptom response.
“But I’ll highlight that the majority of the responses that are received currently with medical therapy are in the area of clinical improvement,” Dr Mesa said.
Treatment guidance
Specific treatment guidelines for low-risk, intermediate-1 risk, intermediate-2- or high-risk MF from the new guideline have been described in an earlier article and will not be discussed here.
Of note, however, Dr Mesa explained that ruxolitinib is a very important part of the treatment guideline because it is the only therapy approved by the US Food and Drug Administration (FDA) to treat MF.
“Over time, it’s been shown that there is an improvement in survival [with ruxolitinib],” Dr Mesa said, perhaps because of a decrease with treatment in the morbidity and the debilitation of patients.
Investigators presented the 5-year update of this information at ASCO 2016 and reported that patients maintained reduction in splenomegaly up through 5 years, both in those randomized to ruxolitinib and those crossing over from placebo.
“Long-term, we clearly looked to see whether there was a signal of new onset or late onset toxicities,” Dr Mesa said, which largely was not the case. "Toxicities have been well described in earlier studies, primarily around anemia, thrombocytopenia, and mild constitutional symptoms.”
However, long-term therapy increases the rate of development of shingles and non-melanoma skin cancer. Rates of transformation to acute myeloid leukemia were consistent with those published for similar patient populations with MF.
“If I see a patient stable on ruxolitinib who has a marked drop in their counts later on in the course of their disease,” Dr Mesa added, “I’m certainly suspicious of progression.”
He also works them up for other causes of anemia if it evolves out of the blue.
Support in MF-related anemia
“With anemia, we’re mindful of iron stores, EPO level, and being certain there’s not the presence of hemolysis or other contributors,” Dr Mesa said.
The guideline recommends stratifying patients based on serum EPO levels—those with less than 500 mU/mL and those with 500 mU/mL and higher.
“If we lower the EPO level, the greater the likelihood of response,” Dr Mesa said. “In my experience, the lower the transfusion burden, the greater the likelihood of response.”
Other than erythropoiesis-stimulating agents for individuals with lower serum levels, androgens and immunomodulatory drugs for those with higher levels have some benefit.
But “they all have their limitations,” Dr Mesa said, and “they tend to range in benefit from 20% to 30%.”
The future
Dr Mesa discussed a few agents in the pipeline that “might impact these guidelines,” such as the JAK2/FLT3 inhibitor pacritinib and the JAK1/JAK2 inhibitor momelotinib.
Pacritinib had a positive phase 3 study, but the mortality rate was higher than expected, and it was put on an FDA hold.
Data from a second phase 3 study (PERSIST-1) will be reviewed in the aggregate to evaluate the benefit of pacritinib and whether the mortality rate was associated with drug-related side effects or adverse patient selection, “which we suspect might be the case,” Dr Mesa said.
“This agent may come off hold, depending upon the data of that second phase 3 study,” he added.
Momelotinib is also in an advanced phase 3 program with 2 trials underway. Both trials have completed accrual.
One is an upfront study of momelotinib versus ruxolitinib (NCT01969838). The goal is to reduce anemia without inferiority of splenomegaly and MPN symptoms.
The other phase 3 momelotinib trial is a second-line study versus best alternative therapy, including ruxoltinib (NCT02101268).
Combination studies are ongoing with a ruxolitinib base and a variety of secondary agents, including danazol, pomalidomide, PEG IFN α2a, 5-AZA, panobinostat, BKM-120, and LDE-225. All agents appear to achieve improvement in splenomegaly and symptoms.
But “incremental benefit over ruxolitinib alone is not yet clear,” Dr Mesa said. “I would say there’s not yet a recommended off-label combination which is widely being used.”
Dr Mesa also highlighted PRM-151, an antifibrosing drug that had a favorable early stage study with several doses, and the telomerase inhibitor imetelstat, which had a deep set of molecular responses in about a third of patients with MF. Imetelstat is currently being evaluated in the second-line setting (NCT02426086).
Regarding the possible positioning of new therapies, Dr Mesa believes “momelotinib and pacritinib may play a role in front-line, depending on the final data from those studies.”
“In second-line for MF—this is where most of the activity is in the trials,” he said. “Momelotinib, pacritinib, PRM-151, and imetelstat have possibilities.”
© ASCO/Zach Boyden-Holmes
NEW YORK—The 11th NCCN Congress: Hematologic Malignancies coincided with the release of the inaugural edition of the NCCN treatment guideline on myeloproliferative neoplasms (MPNs), and Ruben A. Mesa, MD, took the opportunity to discuss the framework of the document and the evolving management of MPNs.
Dr Mesa, of the Mayo Clinic Cancer Center in Arizona, is the chair of the MPN guideline committee.
This initial version of the guideline focuses on the workup and diagnosis of primary myelofibrosis (MF), post-polycythemia vera MF, and post-essential thrombocythemia MF, and treatment guidelines for MF.
The committee decided to first tackle the treatment guidelines in MF because, as Dr Mesa explained, MF is “the most severe of the MPNs with the most unmet needs in terms of guidance.”
Treatment guidelines for polycythemia vera (PV) and essential thrombocythemia (ET) will be forthcoming in 2017, he said, as well as diagnosis and treatment of atypical MPNs, such as hypereosinophilic syndrome and systemic mast cell disease.
Workup of an MPN
The guideline committee focused on information regarding diagnosis—the time and place to consider bone marrow biopsies, when to use cytogenetics, and when to perform next-generation sequencing.
They stressed the importance of quantifying disease burden and utilizing the now-validated symptom assessment tools, particularly in MF.
“Finally, we leveraged the many prognostic scoring systems that have been developed for these diseases, particularly IPSS for myelofibrosis at diagnosis or the DIPSS and DIPSS-plus at subsequent time points,” Dr Mesa said.
In addition to clinical-based prognostic scoring systems, information regarding molecular features and their impact is evolving, he said.
The guideline provides a table of mutations with prognostic significance. JAK2, MPL, or CALR mutations are “brighter mutations,” he said, while adverse prognostic markers include ASXL1, EZH2, IDH1/2, SRSF2, and TP53.
“All of these have some significant prognostic implications in primary myelofibrosis, specifically,” he said.
Assessing MPN burden
“When treating these patients, it’s important to be mindful of the overall burden of the disease,” Dr Mesa said.
He called this emphasis on disease burden “an evolution from the past, where therapy was either supportive or primarily focused on prevention of thrombosis with ET or PV.”
Practitioners need to be additionally mindful of the risk of vascular events, progression, the impact of cytopenias, splenomegaly, the burden of symptoms, and the baseline degree of comorbidities.
“[W]e encourage the use of validated symptom tools that have been used now in the majority of clinical trials in this setting,” Dr Mesa added.
The MPN-10 assessment tool, included in the guideline, evaluates 10 symptoms—early satiety, abdominal discomfort, inactivity, problems concentrating, numbness/tingling, night sweats, itching, bone pain, fever, and unintentional weight loss—on a scale of 0 to 10.
Patients with MF are the most symptomatic, Dr Mesa commented, although “it is notable how frequent symptoms are present in patients with PV and ET.”
Response criteria
For a complete response, individuals must have marked improvement to near normalization in both bone marrow and peripheral blood in addition to resolution of their disease symptoms.
Partial response is basically “just shy” of the complete response level, Dr Mesa said, with bone marrow resolution not being required.
The guideline also outlines response criteria for progressive, stable, and relapsed disease, clinical improvement, and anemia, spleen, and symptom response.
“But I’ll highlight that the majority of the responses that are received currently with medical therapy are in the area of clinical improvement,” Dr Mesa said.
Treatment guidance
Specific treatment guidelines for low-risk, intermediate-1 risk, intermediate-2- or high-risk MF from the new guideline have been described in an earlier article and will not be discussed here.
Of note, however, Dr Mesa explained that ruxolitinib is a very important part of the treatment guideline because it is the only therapy approved by the US Food and Drug Administration (FDA) to treat MF.
“Over time, it’s been shown that there is an improvement in survival [with ruxolitinib],” Dr Mesa said, perhaps because of a decrease with treatment in the morbidity and the debilitation of patients.
Investigators presented the 5-year update of this information at ASCO 2016 and reported that patients maintained reduction in splenomegaly up through 5 years, both in those randomized to ruxolitinib and those crossing over from placebo.
“Long-term, we clearly looked to see whether there was a signal of new onset or late onset toxicities,” Dr Mesa said, which largely was not the case. "Toxicities have been well described in earlier studies, primarily around anemia, thrombocytopenia, and mild constitutional symptoms.”
However, long-term therapy increases the rate of development of shingles and non-melanoma skin cancer. Rates of transformation to acute myeloid leukemia were consistent with those published for similar patient populations with MF.
“If I see a patient stable on ruxolitinib who has a marked drop in their counts later on in the course of their disease,” Dr Mesa added, “I’m certainly suspicious of progression.”
He also works them up for other causes of anemia if it evolves out of the blue.
Support in MF-related anemia
“With anemia, we’re mindful of iron stores, EPO level, and being certain there’s not the presence of hemolysis or other contributors,” Dr Mesa said.
The guideline recommends stratifying patients based on serum EPO levels—those with less than 500 mU/mL and those with 500 mU/mL and higher.
“If we lower the EPO level, the greater the likelihood of response,” Dr Mesa said. “In my experience, the lower the transfusion burden, the greater the likelihood of response.”
Other than erythropoiesis-stimulating agents for individuals with lower serum levels, androgens and immunomodulatory drugs for those with higher levels have some benefit.
But “they all have their limitations,” Dr Mesa said, and “they tend to range in benefit from 20% to 30%.”
The future
Dr Mesa discussed a few agents in the pipeline that “might impact these guidelines,” such as the JAK2/FLT3 inhibitor pacritinib and the JAK1/JAK2 inhibitor momelotinib.
Pacritinib had a positive phase 3 study, but the mortality rate was higher than expected, and it was put on an FDA hold.
Data from a second phase 3 study (PERSIST-1) will be reviewed in the aggregate to evaluate the benefit of pacritinib and whether the mortality rate was associated with drug-related side effects or adverse patient selection, “which we suspect might be the case,” Dr Mesa said.
“This agent may come off hold, depending upon the data of that second phase 3 study,” he added.
Momelotinib is also in an advanced phase 3 program with 2 trials underway. Both trials have completed accrual.
One is an upfront study of momelotinib versus ruxolitinib (NCT01969838). The goal is to reduce anemia without inferiority of splenomegaly and MPN symptoms.
The other phase 3 momelotinib trial is a second-line study versus best alternative therapy, including ruxoltinib (NCT02101268).
Combination studies are ongoing with a ruxolitinib base and a variety of secondary agents, including danazol, pomalidomide, PEG IFN α2a, 5-AZA, panobinostat, BKM-120, and LDE-225. All agents appear to achieve improvement in splenomegaly and symptoms.
But “incremental benefit over ruxolitinib alone is not yet clear,” Dr Mesa said. “I would say there’s not yet a recommended off-label combination which is widely being used.”
Dr Mesa also highlighted PRM-151, an antifibrosing drug that had a favorable early stage study with several doses, and the telomerase inhibitor imetelstat, which had a deep set of molecular responses in about a third of patients with MF. Imetelstat is currently being evaluated in the second-line setting (NCT02426086).
Regarding the possible positioning of new therapies, Dr Mesa believes “momelotinib and pacritinib may play a role in front-line, depending on the final data from those studies.”
“In second-line for MF—this is where most of the activity is in the trials,” he said. “Momelotinib, pacritinib, PRM-151, and imetelstat have possibilities.”
© ASCO/Zach Boyden-Holmes
NEW YORK—The 11th NCCN Congress: Hematologic Malignancies coincided with the release of the inaugural edition of the NCCN treatment guideline on myeloproliferative neoplasms (MPNs), and Ruben A. Mesa, MD, took the opportunity to discuss the framework of the document and the evolving management of MPNs.
Dr Mesa, of the Mayo Clinic Cancer Center in Arizona, is the chair of the MPN guideline committee.
This initial version of the guideline focuses on the workup and diagnosis of primary myelofibrosis (MF), post-polycythemia vera MF, and post-essential thrombocythemia MF, and treatment guidelines for MF.
The committee decided to first tackle the treatment guidelines in MF because, as Dr Mesa explained, MF is “the most severe of the MPNs with the most unmet needs in terms of guidance.”
Treatment guidelines for polycythemia vera (PV) and essential thrombocythemia (ET) will be forthcoming in 2017, he said, as well as diagnosis and treatment of atypical MPNs, such as hypereosinophilic syndrome and systemic mast cell disease.
Workup of an MPN
The guideline committee focused on information regarding diagnosis—the time and place to consider bone marrow biopsies, when to use cytogenetics, and when to perform next-generation sequencing.
They stressed the importance of quantifying disease burden and utilizing the now-validated symptom assessment tools, particularly in MF.
“Finally, we leveraged the many prognostic scoring systems that have been developed for these diseases, particularly IPSS for myelofibrosis at diagnosis or the DIPSS and DIPSS-plus at subsequent time points,” Dr Mesa said.
In addition to clinical-based prognostic scoring systems, information regarding molecular features and their impact is evolving, he said.
The guideline provides a table of mutations with prognostic significance. JAK2, MPL, or CALR mutations are “brighter mutations,” he said, while adverse prognostic markers include ASXL1, EZH2, IDH1/2, SRSF2, and TP53.
“All of these have some significant prognostic implications in primary myelofibrosis, specifically,” he said.
Assessing MPN burden
“When treating these patients, it’s important to be mindful of the overall burden of the disease,” Dr Mesa said.
He called this emphasis on disease burden “an evolution from the past, where therapy was either supportive or primarily focused on prevention of thrombosis with ET or PV.”
Practitioners need to be additionally mindful of the risk of vascular events, progression, the impact of cytopenias, splenomegaly, the burden of symptoms, and the baseline degree of comorbidities.
“[W]e encourage the use of validated symptom tools that have been used now in the majority of clinical trials in this setting,” Dr Mesa added.
The MPN-10 assessment tool, included in the guideline, evaluates 10 symptoms—early satiety, abdominal discomfort, inactivity, problems concentrating, numbness/tingling, night sweats, itching, bone pain, fever, and unintentional weight loss—on a scale of 0 to 10.
Patients with MF are the most symptomatic, Dr Mesa commented, although “it is notable how frequent symptoms are present in patients with PV and ET.”
Response criteria
For a complete response, individuals must have marked improvement to near normalization in both bone marrow and peripheral blood in addition to resolution of their disease symptoms.
Partial response is basically “just shy” of the complete response level, Dr Mesa said, with bone marrow resolution not being required.
The guideline also outlines response criteria for progressive, stable, and relapsed disease, clinical improvement, and anemia, spleen, and symptom response.
“But I’ll highlight that the majority of the responses that are received currently with medical therapy are in the area of clinical improvement,” Dr Mesa said.
Treatment guidance
Specific treatment guidelines for low-risk, intermediate-1 risk, intermediate-2- or high-risk MF from the new guideline have been described in an earlier article and will not be discussed here.
Of note, however, Dr Mesa explained that ruxolitinib is a very important part of the treatment guideline because it is the only therapy approved by the US Food and Drug Administration (FDA) to treat MF.
“Over time, it’s been shown that there is an improvement in survival [with ruxolitinib],” Dr Mesa said, perhaps because of a decrease with treatment in the morbidity and the debilitation of patients.
Investigators presented the 5-year update of this information at ASCO 2016 and reported that patients maintained reduction in splenomegaly up through 5 years, both in those randomized to ruxolitinib and those crossing over from placebo.
“Long-term, we clearly looked to see whether there was a signal of new onset or late onset toxicities,” Dr Mesa said, which largely was not the case. "Toxicities have been well described in earlier studies, primarily around anemia, thrombocytopenia, and mild constitutional symptoms.”
However, long-term therapy increases the rate of development of shingles and non-melanoma skin cancer. Rates of transformation to acute myeloid leukemia were consistent with those published for similar patient populations with MF.
“If I see a patient stable on ruxolitinib who has a marked drop in their counts later on in the course of their disease,” Dr Mesa added, “I’m certainly suspicious of progression.”
He also works them up for other causes of anemia if it evolves out of the blue.
Support in MF-related anemia
“With anemia, we’re mindful of iron stores, EPO level, and being certain there’s not the presence of hemolysis or other contributors,” Dr Mesa said.
The guideline recommends stratifying patients based on serum EPO levels—those with less than 500 mU/mL and those with 500 mU/mL and higher.
“If we lower the EPO level, the greater the likelihood of response,” Dr Mesa said. “In my experience, the lower the transfusion burden, the greater the likelihood of response.”
Other than erythropoiesis-stimulating agents for individuals with lower serum levels, androgens and immunomodulatory drugs for those with higher levels have some benefit.
But “they all have their limitations,” Dr Mesa said, and “they tend to range in benefit from 20% to 30%.”
The future
Dr Mesa discussed a few agents in the pipeline that “might impact these guidelines,” such as the JAK2/FLT3 inhibitor pacritinib and the JAK1/JAK2 inhibitor momelotinib.
Pacritinib had a positive phase 3 study, but the mortality rate was higher than expected, and it was put on an FDA hold.
Data from a second phase 3 study (PERSIST-1) will be reviewed in the aggregate to evaluate the benefit of pacritinib and whether the mortality rate was associated with drug-related side effects or adverse patient selection, “which we suspect might be the case,” Dr Mesa said.
“This agent may come off hold, depending upon the data of that second phase 3 study,” he added.
Momelotinib is also in an advanced phase 3 program with 2 trials underway. Both trials have completed accrual.
One is an upfront study of momelotinib versus ruxolitinib (NCT01969838). The goal is to reduce anemia without inferiority of splenomegaly and MPN symptoms.
The other phase 3 momelotinib trial is a second-line study versus best alternative therapy, including ruxoltinib (NCT02101268).
Combination studies are ongoing with a ruxolitinib base and a variety of secondary agents, including danazol, pomalidomide, PEG IFN α2a, 5-AZA, panobinostat, BKM-120, and LDE-225. All agents appear to achieve improvement in splenomegaly and symptoms.
But “incremental benefit over ruxolitinib alone is not yet clear,” Dr Mesa said. “I would say there’s not yet a recommended off-label combination which is widely being used.”
Dr Mesa also highlighted PRM-151, an antifibrosing drug that had a favorable early stage study with several doses, and the telomerase inhibitor imetelstat, which had a deep set of molecular responses in about a third of patients with MF. Imetelstat is currently being evaluated in the second-line setting (NCT02426086).
Regarding the possible positioning of new therapies, Dr Mesa believes “momelotinib and pacritinib may play a role in front-line, depending on the final data from those studies.”
“In second-line for MF—this is where most of the activity is in the trials,” he said. “Momelotinib, pacritinib, PRM-151, and imetelstat have possibilities.”
NCCN releases guidelines for managing MF

The National Comprehensive Cancer Network (NCCN) has published new clinical practice guidelines for myeloproliferative neoplasms (MPNs).
The current guidelines focus on the management of patients with myelofibrosis (MF), but NCCN said recommendations for managing essential thrombocythemia (ET) and polycythemia vera (PV) will be included in subsequent versions of the NCCN guidelines for MPNs.
The new guidelines provide recommendations on the workup and diagnosis of primary MF, post-ET MF, and post-PV MF.
The document also includes recommendations for managing MF-associated anemia, supportive care options, and treatment recommendations according to a patient’s risk group.
Treatment of low-risk MF
The guidelines recommend that patients with low-risk, asymptomatic MF be observed or enrolled in a clinical trial. They should be monitored for progression every 3 to 6 months.
Patients with low-risk, symptomatic MF should receive ruxolitinib or interferons or be enrolled on a clinical trial. They should be monitored for progression every 3 to 6 months.
If patients respond to treatment, they should continue to receive it. If they do not respond or lose their response, they should receive ruxolitinib or interferons or be enrolled on a clinical trial. If these patients progress, they should be treated according to their risk category.
Intermediate-1-risk MF
Patients with intermediate-1-risk MF should be assessed for symptom burden and observed if asymptomatic. If symptomatic, they should receive ruxolitinib, be enrolled on a clinical trial, or undergo allogeneic hematopoietic stem cell transplant (HSCT).
Patients should be monitored for response and progression every 3 to 6 months. If they respond to treatment, they should continue to receive it.
If patients do not respond or lose their response, they should be observed, receive ruxolitinib, be enrolled on a clinical trial, or undergo allogeneic HSCT. If these patients progress, they should be treated according to their risk category.
Intermediate-2- or high-risk MF
Patients who are transplant candidates should receive allogeneic HSCT.
Patients who are ineligible for transplant should be assessed for symptom burden. Those with a platelet count of 50,000 or lower should be considered for a clinical trial.
Those with higher platelet counts should receive ruxolitinib or be enrolled on a clinical trial. They should be monitored for response or progression every 3 to 6 months.
If these patients respond to treatment, they should continue on that treatment. If they do not respond or lose their response, the patients should be monitored for progression.
If they progress and are candidates for transplant, these patients should receive hypomethylating agents or chemotherapy to induce remission, followed by HSCT.
If the patients progress and are ineligible for transplant, they should be enrolled on a clinical trial or receive hypomethylating agents or chemotherapy.
For patients who are ineligible for transplant and only have symptomatic anemia, they should receive treatment to manage that anemia. The guidelines list a range of treatment options.
“The management of MPNs has been variable in the past and largely driven by review articles and individual opinions,” said Ruben A. Mesa, MD, chair of the NCCN Guidelines Panel for MPN.
“The NCCN Guidelines Panel for MPN hopes these inaugural guidelines will help leverage the evidence base in MPN care for clear, well-informed treatment guidelines to hopefully improve quality of care and provide better outcomes for patients with MPN.”
Dr Mesa is scheduled to present the new NCCN guidelines during the NCCN 11th Annual Congress: Hematologic Malignancies™ on September 30, in a session titled, “Myeloprofilerative Neoplasms and Myelofibrosis: Evolving Management.”
The National Comprehensive Cancer Network (NCCN) has published new clinical practice guidelines for myeloproliferative neoplasms (MPNs).
The current guidelines focus on the management of patients with myelofibrosis (MF), but NCCN said recommendations for managing essential thrombocythemia (ET) and polycythemia vera (PV) will be included in subsequent versions of the NCCN guidelines for MPNs.
The new guidelines provide recommendations on the workup and diagnosis of primary MF, post-ET MF, and post-PV MF.
The document also includes recommendations for managing MF-associated anemia, supportive care options, and treatment recommendations according to a patient’s risk group.
Treatment of low-risk MF
The guidelines recommend that patients with low-risk, asymptomatic MF be observed or enrolled in a clinical trial. They should be monitored for progression every 3 to 6 months.
Patients with low-risk, symptomatic MF should receive ruxolitinib or interferons or be enrolled on a clinical trial. They should be monitored for progression every 3 to 6 months.
If patients respond to treatment, they should continue to receive it. If they do not respond or lose their response, they should receive ruxolitinib or interferons or be enrolled on a clinical trial. If these patients progress, they should be treated according to their risk category.
Intermediate-1-risk MF
Patients with intermediate-1-risk MF should be assessed for symptom burden and observed if asymptomatic. If symptomatic, they should receive ruxolitinib, be enrolled on a clinical trial, or undergo allogeneic hematopoietic stem cell transplant (HSCT).
Patients should be monitored for response and progression every 3 to 6 months. If they respond to treatment, they should continue to receive it.
If patients do not respond or lose their response, they should be observed, receive ruxolitinib, be enrolled on a clinical trial, or undergo allogeneic HSCT. If these patients progress, they should be treated according to their risk category.
Intermediate-2- or high-risk MF
Patients who are transplant candidates should receive allogeneic HSCT.
Patients who are ineligible for transplant should be assessed for symptom burden. Those with a platelet count of 50,000 or lower should be considered for a clinical trial.
Those with higher platelet counts should receive ruxolitinib or be enrolled on a clinical trial. They should be monitored for response or progression every 3 to 6 months.
If these patients respond to treatment, they should continue on that treatment. If they do not respond or lose their response, the patients should be monitored for progression.
If they progress and are candidates for transplant, these patients should receive hypomethylating agents or chemotherapy to induce remission, followed by HSCT.
If the patients progress and are ineligible for transplant, they should be enrolled on a clinical trial or receive hypomethylating agents or chemotherapy.
For patients who are ineligible for transplant and only have symptomatic anemia, they should receive treatment to manage that anemia. The guidelines list a range of treatment options.
“The management of MPNs has been variable in the past and largely driven by review articles and individual opinions,” said Ruben A. Mesa, MD, chair of the NCCN Guidelines Panel for MPN.
“The NCCN Guidelines Panel for MPN hopes these inaugural guidelines will help leverage the evidence base in MPN care for clear, well-informed treatment guidelines to hopefully improve quality of care and provide better outcomes for patients with MPN.”
Dr Mesa is scheduled to present the new NCCN guidelines during the NCCN 11th Annual Congress: Hematologic Malignancies™ on September 30, in a session titled, “Myeloprofilerative Neoplasms and Myelofibrosis: Evolving Management.”
The National Comprehensive Cancer Network (NCCN) has published new clinical practice guidelines for myeloproliferative neoplasms (MPNs).
The current guidelines focus on the management of patients with myelofibrosis (MF), but NCCN said recommendations for managing essential thrombocythemia (ET) and polycythemia vera (PV) will be included in subsequent versions of the NCCN guidelines for MPNs.
The new guidelines provide recommendations on the workup and diagnosis of primary MF, post-ET MF, and post-PV MF.
The document also includes recommendations for managing MF-associated anemia, supportive care options, and treatment recommendations according to a patient’s risk group.
Treatment of low-risk MF
The guidelines recommend that patients with low-risk, asymptomatic MF be observed or enrolled in a clinical trial. They should be monitored for progression every 3 to 6 months.
Patients with low-risk, symptomatic MF should receive ruxolitinib or interferons or be enrolled on a clinical trial. They should be monitored for progression every 3 to 6 months.
If patients respond to treatment, they should continue to receive it. If they do not respond or lose their response, they should receive ruxolitinib or interferons or be enrolled on a clinical trial. If these patients progress, they should be treated according to their risk category.
Intermediate-1-risk MF
Patients with intermediate-1-risk MF should be assessed for symptom burden and observed if asymptomatic. If symptomatic, they should receive ruxolitinib, be enrolled on a clinical trial, or undergo allogeneic hematopoietic stem cell transplant (HSCT).
Patients should be monitored for response and progression every 3 to 6 months. If they respond to treatment, they should continue to receive it.
If patients do not respond or lose their response, they should be observed, receive ruxolitinib, be enrolled on a clinical trial, or undergo allogeneic HSCT. If these patients progress, they should be treated according to their risk category.
Intermediate-2- or high-risk MF
Patients who are transplant candidates should receive allogeneic HSCT.
Patients who are ineligible for transplant should be assessed for symptom burden. Those with a platelet count of 50,000 or lower should be considered for a clinical trial.
Those with higher platelet counts should receive ruxolitinib or be enrolled on a clinical trial. They should be monitored for response or progression every 3 to 6 months.
If these patients respond to treatment, they should continue on that treatment. If they do not respond or lose their response, the patients should be monitored for progression.
If they progress and are candidates for transplant, these patients should receive hypomethylating agents or chemotherapy to induce remission, followed by HSCT.
If the patients progress and are ineligible for transplant, they should be enrolled on a clinical trial or receive hypomethylating agents or chemotherapy.
For patients who are ineligible for transplant and only have symptomatic anemia, they should receive treatment to manage that anemia. The guidelines list a range of treatment options.
“The management of MPNs has been variable in the past and largely driven by review articles and individual opinions,” said Ruben A. Mesa, MD, chair of the NCCN Guidelines Panel for MPN.
“The NCCN Guidelines Panel for MPN hopes these inaugural guidelines will help leverage the evidence base in MPN care for clear, well-informed treatment guidelines to hopefully improve quality of care and provide better outcomes for patients with MPN.”
Dr Mesa is scheduled to present the new NCCN guidelines during the NCCN 11th Annual Congress: Hematologic Malignancies™ on September 30, in a session titled, “Myeloprofilerative Neoplasms and Myelofibrosis: Evolving Management.”
Teva launches generic imatinib tablets in US

Photo by Steven Harbour
Teva Pharmaceutical Industries Ltd. has announced the US launch of imatinib mesylate, the generic equivalent of Novartis’s Gleevec®, in 100 mg and 400 mg tablets.
In the US, imatinib is approved to treat newly diagnosed Philadelphia-chromosome-positive (Ph+) chronic myeloid leukemia in chronic phase, blast crisis, and accelerated phase, as well as Ph+
chronic myeloid leukemia in chronic phase after failure of interferon-alpha therapy.
Imatinib is also approved to treat adults with relapsed or refractory Ph+ acute lymphoblastic leukemia, adults with myelodysplastic syndromes or myeloproliferative neoplasms associated with platelet-derived growth factor receptor gene re-arrangements, and adults with aggressive systemic mastocytosis without the D816V c-Kit mutation or with unknown c-Kit mutational status.
In addition, imatinib is approved to treat adults with hypereosinophilic syndrome and/or chronic eosinophilic leukemia (regardless of whether they have the FIP1L1-PDGFRα fusion kinase) and adults with unresectable, recurrent, and/or metastatic dermatofibrosarcoma protuberans.
Finally, the drug is approved as an adjuvant treatment following complete gross resection of Kit (CD117)-positive gastrointestinal stromal tumors in adults.
For more details on imatinib, see the full prescribing information.
Photo by Steven Harbour
Teva Pharmaceutical Industries Ltd. has announced the US launch of imatinib mesylate, the generic equivalent of Novartis’s Gleevec®, in 100 mg and 400 mg tablets.
In the US, imatinib is approved to treat newly diagnosed Philadelphia-chromosome-positive (Ph+) chronic myeloid leukemia in chronic phase, blast crisis, and accelerated phase, as well as Ph+
chronic myeloid leukemia in chronic phase after failure of interferon-alpha therapy.
Imatinib is also approved to treat adults with relapsed or refractory Ph+ acute lymphoblastic leukemia, adults with myelodysplastic syndromes or myeloproliferative neoplasms associated with platelet-derived growth factor receptor gene re-arrangements, and adults with aggressive systemic mastocytosis without the D816V c-Kit mutation or with unknown c-Kit mutational status.
In addition, imatinib is approved to treat adults with hypereosinophilic syndrome and/or chronic eosinophilic leukemia (regardless of whether they have the FIP1L1-PDGFRα fusion kinase) and adults with unresectable, recurrent, and/or metastatic dermatofibrosarcoma protuberans.
Finally, the drug is approved as an adjuvant treatment following complete gross resection of Kit (CD117)-positive gastrointestinal stromal tumors in adults.
For more details on imatinib, see the full prescribing information.
Photo by Steven Harbour
Teva Pharmaceutical Industries Ltd. has announced the US launch of imatinib mesylate, the generic equivalent of Novartis’s Gleevec®, in 100 mg and 400 mg tablets.
In the US, imatinib is approved to treat newly diagnosed Philadelphia-chromosome-positive (Ph+) chronic myeloid leukemia in chronic phase, blast crisis, and accelerated phase, as well as Ph+
chronic myeloid leukemia in chronic phase after failure of interferon-alpha therapy.
Imatinib is also approved to treat adults with relapsed or refractory Ph+ acute lymphoblastic leukemia, adults with myelodysplastic syndromes or myeloproliferative neoplasms associated with platelet-derived growth factor receptor gene re-arrangements, and adults with aggressive systemic mastocytosis without the D816V c-Kit mutation or with unknown c-Kit mutational status.
In addition, imatinib is approved to treat adults with hypereosinophilic syndrome and/or chronic eosinophilic leukemia (regardless of whether they have the FIP1L1-PDGFRα fusion kinase) and adults with unresectable, recurrent, and/or metastatic dermatofibrosarcoma protuberans.
Finally, the drug is approved as an adjuvant treatment following complete gross resection of Kit (CD117)-positive gastrointestinal stromal tumors in adults.
For more details on imatinib, see the full prescribing information.