User login
FDA Reviewing Possible Heart Failure Risk with Pramipexole
Pramipexole, the dopamine agonist approved for treating Parkinson’s disease and restless legs syndrome, may be associated with an increased risk for heart failure, according to a statement issued by the Food and Drug Administration.
"Results of recent studies suggest a potential risk of heart failure that needs further review of available data," but the FDA has not concluded that the drug increases the risk of heart failure, the FDA said in the Sept. 19 statement.
The FDA is currently working with Boehringer Ingelheim, which markets pramipexole as Mirapex, to investigate this association further and will provide an update when more information is available.
The studies include a pooled analysis of randomized clinical trials, which found more cases of heart failure (12 of 4,157 patients) among patients treated with pramipexole than among those on placebo (4 of 2,820). The difference between groups, however, was not statistically significant.
In two epidemiologic studies using European data, though, the increased risk of heart failure associated with pramipexole was statistically significant. In one of the studies, a case control study using a database of patients aged 40-89 years treated with anti-parkinsonian drugs, the risk of heart failure associated with any use of a dopamine agonist compared with no use was increased by almost 60% (relative risk, 1.58). The heart failure risk associated with use of pramipexole (RR, 1.86) and with another dopamine agonist, cabergoline (RR, 2.07), were each higher compared with no use of these drugs.
In the other epidemiologic study, current use of pramipexole was associated with an increased risk of heart failure, compared with levodopa. The risk was increased in the first 3 months of treatment and among patients aged 80 years and older, but was no higher among people who had been treated with pramipexole for more than 3 months.
This last finding is "difficult to explain," since heart failure is a chronic condition, and the studies had limitations, which "make it difficult to determine whether excess heart failure was related to Mirapex use or other influencing factors," the FDA statement said.
The agency advises that health care professionals continue following recommendations in the pramipexole label and that patients continue to take the medication as directed.
Pramipexole was approved in July 1997.
The full FDA notice is available here. Serious adverse events associated with pramipexole should be reported to the FDA’s MedWatch program by clicking here or by calling 800-332-1088.
Pramipexole, the dopamine agonist approved for treating Parkinson’s disease and restless legs syndrome, may be associated with an increased risk for heart failure, according to a statement issued by the Food and Drug Administration.
"Results of recent studies suggest a potential risk of heart failure that needs further review of available data," but the FDA has not concluded that the drug increases the risk of heart failure, the FDA said in the Sept. 19 statement.
The FDA is currently working with Boehringer Ingelheim, which markets pramipexole as Mirapex, to investigate this association further and will provide an update when more information is available.
The studies include a pooled analysis of randomized clinical trials, which found more cases of heart failure (12 of 4,157 patients) among patients treated with pramipexole than among those on placebo (4 of 2,820). The difference between groups, however, was not statistically significant.
In two epidemiologic studies using European data, though, the increased risk of heart failure associated with pramipexole was statistically significant. In one of the studies, a case control study using a database of patients aged 40-89 years treated with anti-parkinsonian drugs, the risk of heart failure associated with any use of a dopamine agonist compared with no use was increased by almost 60% (relative risk, 1.58). The heart failure risk associated with use of pramipexole (RR, 1.86) and with another dopamine agonist, cabergoline (RR, 2.07), were each higher compared with no use of these drugs.
In the other epidemiologic study, current use of pramipexole was associated with an increased risk of heart failure, compared with levodopa. The risk was increased in the first 3 months of treatment and among patients aged 80 years and older, but was no higher among people who had been treated with pramipexole for more than 3 months.
This last finding is "difficult to explain," since heart failure is a chronic condition, and the studies had limitations, which "make it difficult to determine whether excess heart failure was related to Mirapex use or other influencing factors," the FDA statement said.
The agency advises that health care professionals continue following recommendations in the pramipexole label and that patients continue to take the medication as directed.
Pramipexole was approved in July 1997.
The full FDA notice is available here. Serious adverse events associated with pramipexole should be reported to the FDA’s MedWatch program by clicking here or by calling 800-332-1088.
Pramipexole, the dopamine agonist approved for treating Parkinson’s disease and restless legs syndrome, may be associated with an increased risk for heart failure, according to a statement issued by the Food and Drug Administration.
"Results of recent studies suggest a potential risk of heart failure that needs further review of available data," but the FDA has not concluded that the drug increases the risk of heart failure, the FDA said in the Sept. 19 statement.
The FDA is currently working with Boehringer Ingelheim, which markets pramipexole as Mirapex, to investigate this association further and will provide an update when more information is available.
The studies include a pooled analysis of randomized clinical trials, which found more cases of heart failure (12 of 4,157 patients) among patients treated with pramipexole than among those on placebo (4 of 2,820). The difference between groups, however, was not statistically significant.
In two epidemiologic studies using European data, though, the increased risk of heart failure associated with pramipexole was statistically significant. In one of the studies, a case control study using a database of patients aged 40-89 years treated with anti-parkinsonian drugs, the risk of heart failure associated with any use of a dopamine agonist compared with no use was increased by almost 60% (relative risk, 1.58). The heart failure risk associated with use of pramipexole (RR, 1.86) and with another dopamine agonist, cabergoline (RR, 2.07), were each higher compared with no use of these drugs.
In the other epidemiologic study, current use of pramipexole was associated with an increased risk of heart failure, compared with levodopa. The risk was increased in the first 3 months of treatment and among patients aged 80 years and older, but was no higher among people who had been treated with pramipexole for more than 3 months.
This last finding is "difficult to explain," since heart failure is a chronic condition, and the studies had limitations, which "make it difficult to determine whether excess heart failure was related to Mirapex use or other influencing factors," the FDA statement said.
The agency advises that health care professionals continue following recommendations in the pramipexole label and that patients continue to take the medication as directed.
Pramipexole was approved in July 1997.
The full FDA notice is available here. Serious adverse events associated with pramipexole should be reported to the FDA’s MedWatch program by clicking here or by calling 800-332-1088.
FDA Panel Votes Against Approval of Hyponatremia Drug
SILVER SPRING, MD. – A Food and Drug Administration advisory panel has recommended against the approval of lixivaptan, an orally administered vasopressin-2 receptor antagonist, for treating hyponatremia in two groups of adult patients at a meeting on Sept. 13.
The manufacturer, Cardiokine Biopharma, has proposed that lixivaptan be approved for the treatment of hypervolemic hyponatremia associated with heart failure, and for the treatment of euvolemic hyponatremia associated with syndrome of inappropriate antidiuretic hormone (SIADH). The company submitted the results of three phase III placebo-controlled trials that evaluated the changes from baseline in serum sodium at day 7 of treatment, the primary efficacy end point. The company proposed that for the SIADH indication, treatment could be initiated in an outpatient setting, which would be a potential benefit over existing treatments, including the only approved orally administered vasopressin-2 receptor antagonist that is approved for hyponatremia, tolvaptan (Samsca). Tolvaptan is approved for inpatients with diagnoses that include SIADH, heart failure, and cirrhosis, and is initiated in the hospital. The other approved "vaptan" – zomivaptan (Vaprisol), approved in 2005 – is administered intravenously.
Lixivaptan acts specifically on the vasopressin-2 receptor in the kidneys, "causing water to be excreted while sparing sodium without affecting other electrolytes," according to Cardiokine. While the changes in serum sodium from baseline to day 7 among treated patients over placebo were statistically significant, the improvements were modest and were not associated with improved cognitive function, the FDA reviewers pointed out.
In the study of patients with hypervolemic hyponatremia hospitalized with New York Heart Association (NYHA) class III or IV heart failure (652 patients), the increase in serum sodium at day 7 was 1.2 mEq/L greater among treated patients, compared with those on placebo. The mortality rate was 17.7% among those who had at least one dose of lixivaptan, compared with 14.3% on placebo; most deaths were related to heart failure, according to Cardiokine, which was recently acquired by Cornerstone Therapeutics.
In the two other studies, comprising a total of 312 patients with SIADH, the increases in serum sodium at day 7 over placebo were 2.2 mEq/L and 2.4 mEq/L.
But at the meeting of the FDA’s Cardiovascular and Renal Drugs Advisory Committee, the panel unanimously voted against approval of lixivaptan for the heart failure indication, because its members were not convinced that the modest effect of treatment on hyponatremia observed in the study of heart failure patients provided evidence that it benefited patients. The panel also agreed that more data on how improvements in serum sodium translated to clinical benefits, such as quality of life improvements, were needed. Another concern was that mortality was higher among the patients treated with lixivaptan than in those on placebo, particularly early in treatment.
The panel voted 5 to 3 against approval for treatment of hyponatremia in patients with SIADH, with clinicians voting on both sides of this question.
Dr. Linda Fried, professor of medicine at the VA Health Care System in Pittsburgh, voted in favor of approval because she said she believed that the vaptans directly affect pathophysiology in SIADH, and because she did not see a safety signal.
But Dr. Mori Krantz, associate professor of cardiology at the University of Colorado, Denver, voted against approval, because there were not enough data to show whether the improvements with treatment were clinically meaningful. He said that a larger study evaluating the effects of improvements in serum sodium with treatment on quality of life and other measures was needed, "to at least give clinicians some sense of security that they are helping patients."
Most panelists questioned the feasibility of initiating treatment as an outpatient, in most clinical settings.
The FDA is expected to make a decision on approval by Oct. 29.
Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel has recommended against the approval of lixivaptan, an orally administered vasopressin-2 receptor antagonist, for treating hyponatremia in two groups of adult patients at a meeting on Sept. 13.
The manufacturer, Cardiokine Biopharma, has proposed that lixivaptan be approved for the treatment of hypervolemic hyponatremia associated with heart failure, and for the treatment of euvolemic hyponatremia associated with syndrome of inappropriate antidiuretic hormone (SIADH). The company submitted the results of three phase III placebo-controlled trials that evaluated the changes from baseline in serum sodium at day 7 of treatment, the primary efficacy end point. The company proposed that for the SIADH indication, treatment could be initiated in an outpatient setting, which would be a potential benefit over existing treatments, including the only approved orally administered vasopressin-2 receptor antagonist that is approved for hyponatremia, tolvaptan (Samsca). Tolvaptan is approved for inpatients with diagnoses that include SIADH, heart failure, and cirrhosis, and is initiated in the hospital. The other approved "vaptan" – zomivaptan (Vaprisol), approved in 2005 – is administered intravenously.
Lixivaptan acts specifically on the vasopressin-2 receptor in the kidneys, "causing water to be excreted while sparing sodium without affecting other electrolytes," according to Cardiokine. While the changes in serum sodium from baseline to day 7 among treated patients over placebo were statistically significant, the improvements were modest and were not associated with improved cognitive function, the FDA reviewers pointed out.
In the study of patients with hypervolemic hyponatremia hospitalized with New York Heart Association (NYHA) class III or IV heart failure (652 patients), the increase in serum sodium at day 7 was 1.2 mEq/L greater among treated patients, compared with those on placebo. The mortality rate was 17.7% among those who had at least one dose of lixivaptan, compared with 14.3% on placebo; most deaths were related to heart failure, according to Cardiokine, which was recently acquired by Cornerstone Therapeutics.
In the two other studies, comprising a total of 312 patients with SIADH, the increases in serum sodium at day 7 over placebo were 2.2 mEq/L and 2.4 mEq/L.
But at the meeting of the FDA’s Cardiovascular and Renal Drugs Advisory Committee, the panel unanimously voted against approval of lixivaptan for the heart failure indication, because its members were not convinced that the modest effect of treatment on hyponatremia observed in the study of heart failure patients provided evidence that it benefited patients. The panel also agreed that more data on how improvements in serum sodium translated to clinical benefits, such as quality of life improvements, were needed. Another concern was that mortality was higher among the patients treated with lixivaptan than in those on placebo, particularly early in treatment.
The panel voted 5 to 3 against approval for treatment of hyponatremia in patients with SIADH, with clinicians voting on both sides of this question.
Dr. Linda Fried, professor of medicine at the VA Health Care System in Pittsburgh, voted in favor of approval because she said she believed that the vaptans directly affect pathophysiology in SIADH, and because she did not see a safety signal.
But Dr. Mori Krantz, associate professor of cardiology at the University of Colorado, Denver, voted against approval, because there were not enough data to show whether the improvements with treatment were clinically meaningful. He said that a larger study evaluating the effects of improvements in serum sodium with treatment on quality of life and other measures was needed, "to at least give clinicians some sense of security that they are helping patients."
Most panelists questioned the feasibility of initiating treatment as an outpatient, in most clinical settings.
The FDA is expected to make a decision on approval by Oct. 29.
Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel has recommended against the approval of lixivaptan, an orally administered vasopressin-2 receptor antagonist, for treating hyponatremia in two groups of adult patients at a meeting on Sept. 13.
The manufacturer, Cardiokine Biopharma, has proposed that lixivaptan be approved for the treatment of hypervolemic hyponatremia associated with heart failure, and for the treatment of euvolemic hyponatremia associated with syndrome of inappropriate antidiuretic hormone (SIADH). The company submitted the results of three phase III placebo-controlled trials that evaluated the changes from baseline in serum sodium at day 7 of treatment, the primary efficacy end point. The company proposed that for the SIADH indication, treatment could be initiated in an outpatient setting, which would be a potential benefit over existing treatments, including the only approved orally administered vasopressin-2 receptor antagonist that is approved for hyponatremia, tolvaptan (Samsca). Tolvaptan is approved for inpatients with diagnoses that include SIADH, heart failure, and cirrhosis, and is initiated in the hospital. The other approved "vaptan" – zomivaptan (Vaprisol), approved in 2005 – is administered intravenously.
Lixivaptan acts specifically on the vasopressin-2 receptor in the kidneys, "causing water to be excreted while sparing sodium without affecting other electrolytes," according to Cardiokine. While the changes in serum sodium from baseline to day 7 among treated patients over placebo were statistically significant, the improvements were modest and were not associated with improved cognitive function, the FDA reviewers pointed out.
In the study of patients with hypervolemic hyponatremia hospitalized with New York Heart Association (NYHA) class III or IV heart failure (652 patients), the increase in serum sodium at day 7 was 1.2 mEq/L greater among treated patients, compared with those on placebo. The mortality rate was 17.7% among those who had at least one dose of lixivaptan, compared with 14.3% on placebo; most deaths were related to heart failure, according to Cardiokine, which was recently acquired by Cornerstone Therapeutics.
In the two other studies, comprising a total of 312 patients with SIADH, the increases in serum sodium at day 7 over placebo were 2.2 mEq/L and 2.4 mEq/L.
But at the meeting of the FDA’s Cardiovascular and Renal Drugs Advisory Committee, the panel unanimously voted against approval of lixivaptan for the heart failure indication, because its members were not convinced that the modest effect of treatment on hyponatremia observed in the study of heart failure patients provided evidence that it benefited patients. The panel also agreed that more data on how improvements in serum sodium translated to clinical benefits, such as quality of life improvements, were needed. Another concern was that mortality was higher among the patients treated with lixivaptan than in those on placebo, particularly early in treatment.
The panel voted 5 to 3 against approval for treatment of hyponatremia in patients with SIADH, with clinicians voting on both sides of this question.
Dr. Linda Fried, professor of medicine at the VA Health Care System in Pittsburgh, voted in favor of approval because she said she believed that the vaptans directly affect pathophysiology in SIADH, and because she did not see a safety signal.
But Dr. Mori Krantz, associate professor of cardiology at the University of Colorado, Denver, voted against approval, because there were not enough data to show whether the improvements with treatment were clinically meaningful. He said that a larger study evaluating the effects of improvements in serum sodium with treatment on quality of life and other measures was needed, "to at least give clinicians some sense of security that they are helping patients."
Most panelists questioned the feasibility of initiating treatment as an outpatient, in most clinical settings.
The FDA is expected to make a decision on approval by Oct. 29.
Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but not at this meeting.
Nurse-Led Case Management Cuts HF Readmissions, Mortality
For patients discharged after hospitalization for heart failure, case-management interventions led by a specialist nurse reduce HF-related readmissions, HF-related mortality, and all-cause mortality, according to an update published online Sept. 11 in the Cochrane Database of Systematic Reviews.

The initial Cochrane Review that addressed the effectiveness of three different types of follow-up care (case-management, clinic-based, and multidisciplinary) after hospitalization for HF was published in 2005, and found mixed and inconclusive results for all three (Cochrane Database Syst. Rev. 2005 8;(2):CD002752).
Since then, several new clinical trials have been performed, and "there is now strong evidence that case-management interventions are associated with a substantial, statistically significant reduction in all-cause mortality" as well as HF mortality and rehospitalization, said Dr. Andrea Takeda of Queen Mary University, London, and her associates in the Cochrane Collaboration.
In contrast, the evidence doesn’t support interventions in which the major component is follow-up in a hospital HF clinic. And the evidence for multidisciplinary interventions is too sparse to be conclusive because only two high-quality studies have examined these approaches, the investigators wrote (Cochrane Database of Systematic Reviews 2012 [doi:101002/14651858.CD002752.pub3]).
Case-management programs led by personnel other than nurses specializing in HF, such as hospital-based or community pharmacists, nonspecialist nurses, or interdisciplinary teams, were not as successful as those led by specialist nurses, they added.
Dr. Takeda and her colleagues reviewed 25 randomized clinical trials of at least 6 months’ duration that compared the three types of interventions against usual care in 5,942 adults who had been hospitalized for HF. They excluded interventions that focused on patient education only, exercise only, telemedicine (which was examined in a separate Cochrane Review), or cardiac rehabilitation.
Most of the clinical trials had 100-350 study subjects, but some had fewer than 100 and one had over 1,000 subjects. They were conducted in Europe, the United States, Canada, Australia, and China.
Seventeen studies assessed case-management interventions. Pooling the results demonstrated that these interventions produced a substantial and highly significant reduction in all-cause mortality, HF-related mortality, and HF-related rehospitalization at 12 months. Although the overall impact of the interventions on inpatient days was "not clear," the researchers wrote, there was "a strong suggestion" that these interventions decreased hospital length of stay.
However, case-management interventions did not appear to improve event-free survival. They also did not appear to improve health-related quality of life, but few studies examined this outcome and those that did suffered from high dropout rates, so the data were inconclusive.
Only six clinical trials involving 1,486 patients assessed clinic-based interventions. They showed no reduction in readmissions, mortality, inpatient days, event-free survival, or health-related quality of life.
Only two clinical trials involving 403 patients assessed multidisciplinary programs. The data were inadequate to establish conclusive results, Dr. Takeda and her associates wrote.
In a post hoc analysis, the intensity of various interventions did not appear to influence their effectiveness. Studies of the most intensive, moderately intensive, and low-intensity interventions all showed some reductions in mortality and rehospitalization, the researchers added.
No financial conflicts of interest were reported.
For patients discharged after hospitalization for heart failure, case-management interventions led by a specialist nurse reduce HF-related readmissions, HF-related mortality, and all-cause mortality, according to an update published online Sept. 11 in the Cochrane Database of Systematic Reviews.
The initial Cochrane Review that addressed the effectiveness of three different types of follow-up care (case-management, clinic-based, and multidisciplinary) after hospitalization for HF was published in 2005, and found mixed and inconclusive results for all three (Cochrane Database Syst. Rev. 2005 8;(2):CD002752).
Since then, several new clinical trials have been performed, and "there is now strong evidence that case-management interventions are associated with a substantial, statistically significant reduction in all-cause mortality" as well as HF mortality and rehospitalization, said Dr. Andrea Takeda of Queen Mary University, London, and her associates in the Cochrane Collaboration.
In contrast, the evidence doesn’t support interventions in which the major component is follow-up in a hospital HF clinic. And the evidence for multidisciplinary interventions is too sparse to be conclusive because only two high-quality studies have examined these approaches, the investigators wrote (Cochrane Database of Systematic Reviews 2012 [doi:101002/14651858.CD002752.pub3]).
Case-management programs led by personnel other than nurses specializing in HF, such as hospital-based or community pharmacists, nonspecialist nurses, or interdisciplinary teams, were not as successful as those led by specialist nurses, they added.
Dr. Takeda and her colleagues reviewed 25 randomized clinical trials of at least 6 months’ duration that compared the three types of interventions against usual care in 5,942 adults who had been hospitalized for HF. They excluded interventions that focused on patient education only, exercise only, telemedicine (which was examined in a separate Cochrane Review), or cardiac rehabilitation.
Most of the clinical trials had 100-350 study subjects, but some had fewer than 100 and one had over 1,000 subjects. They were conducted in Europe, the United States, Canada, Australia, and China.
Seventeen studies assessed case-management interventions. Pooling the results demonstrated that these interventions produced a substantial and highly significant reduction in all-cause mortality, HF-related mortality, and HF-related rehospitalization at 12 months. Although the overall impact of the interventions on inpatient days was "not clear," the researchers wrote, there was "a strong suggestion" that these interventions decreased hospital length of stay.
However, case-management interventions did not appear to improve event-free survival. They also did not appear to improve health-related quality of life, but few studies examined this outcome and those that did suffered from high dropout rates, so the data were inconclusive.
Only six clinical trials involving 1,486 patients assessed clinic-based interventions. They showed no reduction in readmissions, mortality, inpatient days, event-free survival, or health-related quality of life.
Only two clinical trials involving 403 patients assessed multidisciplinary programs. The data were inadequate to establish conclusive results, Dr. Takeda and her associates wrote.
In a post hoc analysis, the intensity of various interventions did not appear to influence their effectiveness. Studies of the most intensive, moderately intensive, and low-intensity interventions all showed some reductions in mortality and rehospitalization, the researchers added.
No financial conflicts of interest were reported.
For patients discharged after hospitalization for heart failure, case-management interventions led by a specialist nurse reduce HF-related readmissions, HF-related mortality, and all-cause mortality, according to an update published online Sept. 11 in the Cochrane Database of Systematic Reviews.
The initial Cochrane Review that addressed the effectiveness of three different types of follow-up care (case-management, clinic-based, and multidisciplinary) after hospitalization for HF was published in 2005, and found mixed and inconclusive results for all three (Cochrane Database Syst. Rev. 2005 8;(2):CD002752).
Since then, several new clinical trials have been performed, and "there is now strong evidence that case-management interventions are associated with a substantial, statistically significant reduction in all-cause mortality" as well as HF mortality and rehospitalization, said Dr. Andrea Takeda of Queen Mary University, London, and her associates in the Cochrane Collaboration.
In contrast, the evidence doesn’t support interventions in which the major component is follow-up in a hospital HF clinic. And the evidence for multidisciplinary interventions is too sparse to be conclusive because only two high-quality studies have examined these approaches, the investigators wrote (Cochrane Database of Systematic Reviews 2012 [doi:101002/14651858.CD002752.pub3]).
Case-management programs led by personnel other than nurses specializing in HF, such as hospital-based or community pharmacists, nonspecialist nurses, or interdisciplinary teams, were not as successful as those led by specialist nurses, they added.
Dr. Takeda and her colleagues reviewed 25 randomized clinical trials of at least 6 months’ duration that compared the three types of interventions against usual care in 5,942 adults who had been hospitalized for HF. They excluded interventions that focused on patient education only, exercise only, telemedicine (which was examined in a separate Cochrane Review), or cardiac rehabilitation.
Most of the clinical trials had 100-350 study subjects, but some had fewer than 100 and one had over 1,000 subjects. They were conducted in Europe, the United States, Canada, Australia, and China.
Seventeen studies assessed case-management interventions. Pooling the results demonstrated that these interventions produced a substantial and highly significant reduction in all-cause mortality, HF-related mortality, and HF-related rehospitalization at 12 months. Although the overall impact of the interventions on inpatient days was "not clear," the researchers wrote, there was "a strong suggestion" that these interventions decreased hospital length of stay.
However, case-management interventions did not appear to improve event-free survival. They also did not appear to improve health-related quality of life, but few studies examined this outcome and those that did suffered from high dropout rates, so the data were inconclusive.
Only six clinical trials involving 1,486 patients assessed clinic-based interventions. They showed no reduction in readmissions, mortality, inpatient days, event-free survival, or health-related quality of life.
Only two clinical trials involving 403 patients assessed multidisciplinary programs. The data were inadequate to establish conclusive results, Dr. Takeda and her associates wrote.
In a post hoc analysis, the intensity of various interventions did not appear to influence their effectiveness. Studies of the most intensive, moderately intensive, and low-intensity interventions all showed some reductions in mortality and rehospitalization, the researchers added.
No financial conflicts of interest were reported.
FROM THE COCHRANE COLLABORATION
Major Finding: Case-management interventions led by a specialist nurse produced a substantial and highly significant reduction in all-cause mortality, HF-mortality, an HF readmissions at 12 months.
Data Source: Results were taken from a systematic review of the literature and meta-analysis of 25 randomized clinical trials involving 5,942 adults previously hospitalized for HF, which compared the effectiveness of various follow-up interventions in reducing HF mortality, HF readmissions, and all-cause mortality.
Disclosures: No financial conflicts of interest were reported.

Evidence Mounts on Heart Failure After Trastuzumab in Breast Cancer Survivors
Trastuzumab, whether given with or without anthracyline-based chemotherapy, was associated with significant increases in heart failure and cardiomyopathy in a large population-based, retrospective cohort study of women treated for breast cancer.
In the "real world" study, which used data from the health maintenance organization Cancer Research Network, the adjusted risk of heart failure (HF) and/or cardiomyopathy increased fourfold among women who received trastuzumab (Herceptin) alone and sevenfold in those who received anthracycline plus trastuzumab, compared with women who received no chemotherapy.
In all, the risk of anthracycline-associated HF/cardiomyopathy among women younger than 65 years was similar to results from randomized clinical trials, while the trastuzumab-associated HF/cardiomyopathy risk – whether administered alone or following an anthracycline – was greater than that previously reported.
"Our results highlight the importance of generalizability in applying clinical trial findings to community settings; although similar to clinical trial results, these population-based results cannot be attributed to any single patient in clinical practice," wrote Erin J. Aiello Bowles of Group Health Research Institute, Seattle, and her associates.
Randomized trials have typically excluded older women and those with major comorbidities, and therefore the association between the two agents and HF/cardiomyopathy in this population is not well understood, Ms. Bowles and her associates noted (J. Natl. Cancer Inst. 2012 Aug. 30 [doi:10.1093/jnci/djs317]).
The current study population comprised 12,500 women diagnosed with incident, invasive breast cancer from Jan. 1, 1999, through Dec. 31, 2007, at eight integrated Cancer Research Network health systems. Women diagnosed with HF/cardiomyopathy prior to breast cancer diagnosis or initiation of chemotherapy were excluded. They had a mean age of 60 years (range, 22-99), and 85.8% were white.
In a median follow-up of 4.4 years, nearly half (46.5%) had received no chemotherapy. Just under a third, 29.6%, had received anthracycline-based chemotherapy alone, 0.9% received trastuzumab-based therapy without anthracycline, 3.5% received anthracycline plus trastuzumab, and 19.5% received other chemotherapy.
Compared with the women who received other chemotherapy or no chemotherapy, the women who received anthracycline alone or anthracycline plus trastuzumab were younger, diagnosed at later stages, had fewer comorbidities, and were slightly more likely to receive radiation therapy. These findings "suggest substantial individualization of adjuvant chemotherapy administration by age and comorbidity in community practice," Ms. Bowles and her associates wrote.
The incidence of HF/cardiomyopathy increased with increasing follow-up time for all of the chemotherapy types, but to a greater degree with trastuzumab. The cumulative HF/cardiomyopathy incidence increased from 1.2% at year 1 to 4.3% at year 5 for the anthracycline recipients, which was similar to the increase from 1.3% to 4.5% for those on other chemotherapies. For those with no chemotherapy, the cumulative incidence rose from 0.9% to 3.1%.
In contrast, the cumulative HF/cardiomyopathy incidence among recipients of anthracycline plus trastuzumab was 6.2% after 1 year of follow-up and continued to increase to 20.1% by 5 years.
Compared with no chemotherapy, the risk of incident HF/cardiomyopathy among all women was statistically significantly increased for anthracycline alone (adjusted hazard ratio, 1.40), trastuzumab without anthracycline (HR, 4.12), anthracycline plus trastuzumab (HR, 7.19), and other chemotherapy (HR, 1.49), the investigators said.
The 5-year cumulative incidence for HF/cardiomyopathy associated with each of the chemotherapies was greater in the older age groups, but the hazard ratios for HF/cardiomyopathy associated with chemotherapy use decreased with increasing age. For example, the hazard ratio for HF/cardiomyopathy associated with anthracycline use alone was statistically significant among women younger than 55 years (HR, 2.52) but not among women 55-64 years (HR, 1.61) or older.
According to Ms. Bowles and her associates, this study demonstrates the importance of observational comparative safety and effectiveness studies in providing complementary data to those obtained in clinical trials. "Observational studies allow for estimation of risks and benefits in community practice, which includes patients who may not be eligible for clinical trials. Clinical trials may provide more relevant estimates for patients who are eligible candidates, but many people are not and still receive these treatments in community practice," they wrote.
This work was supported by the National Cancer Institute through an administrative supplement to the Cancer Research Network. Ms. Bowles did not report any financial disclosures, but one of her coauthors, Dr. Larry A. Allen, has received consulting fees from Amgen, Janssen Scientific Affairs, and the Robert Wood Johnson Foundation.
The development of trastuzumab for the treatment of nonmetastatic invasive breast cancer tumors expressing human epidermal growth factor receptor 2 has been an important step in the field of personalized medicine.
Multiple trials have documented clinically and statistically significant improvements in overall and disease-free survival for women receiving adjuvant treatment including trastuzumab, compared with those receiving other chemotherapy agents. Unlike most new cancer chemotherapies, trastuzumab’s use in early-stage HER2-positive breast cancer also appears to be cost effective (Ann. Pharmacother. 2009;43:296-303).
This benefit, however, has been offset by safety concerns, with a pooled analysis of the early breast cancer trials suggesting a fivefold increase in risk of congestive heart failure for women who receive trastuzumab versus those who do not, with consistent results across varying treatment regimens (Cochrane Database Syst. Rev. 2012;4:CD006243).
The current study adds an additional year of follow-up to previous trials, during which the incidence of congestive heart failure continues to increase with no indication of a plateau. This justifies long-term surveillance for congestive heart failure in women who have received trastuzumab, as well as extended follow-up of women enrolled in trials.
Although most previous randomized clinical trials have included anthracycline in both treatment and comparison arms, the current observational study found that, in real life, about 40% of women undergoing chemotherapy received regimens excluding anthracycline, probably due in part to the older age and higher comorbidity burden relative to participants in clinical trials.
Finally, this study raises concern in that roughly a quarter of the women who received adjuvant trastuzumab had been treated well before the publication of peer-reviewed results of relevant randomized controlled trials. Clinicians may have been basing their decision on findings from trials in women with metastatic breast cancer and preliminary data from the early breast cancer trials. The interim findings, however, were based on follow-up periods too short to account for the longer life expectancy of women with early breast cancer relative to women with metastatic disease.
There have been many instances in which new treatments disseminate based on preliminary reports of benefit, only to be withdrawn after additional safety data become available. Patients, clinicians, and researchers must temper their enthusiasm about the benefits of new cancer therapies with the recognition that estimates of the long-term risk of adverse events are based on short-term observations among carefully selected clinical trial participants.
Ann M. Geiger, Ph.D., is with the division of public health sciences at Wake Forest University, Winston Salem, N.C. She said that she has no conflicts of interest.
The development of trastuzumab for the treatment of nonmetastatic invasive breast cancer tumors expressing human epidermal growth factor receptor 2 has been an important step in the field of personalized medicine.
Multiple trials have documented clinically and statistically significant improvements in overall and disease-free survival for women receiving adjuvant treatment including trastuzumab, compared with those receiving other chemotherapy agents. Unlike most new cancer chemotherapies, trastuzumab’s use in early-stage HER2-positive breast cancer also appears to be cost effective (Ann. Pharmacother. 2009;43:296-303).
This benefit, however, has been offset by safety concerns, with a pooled analysis of the early breast cancer trials suggesting a fivefold increase in risk of congestive heart failure for women who receive trastuzumab versus those who do not, with consistent results across varying treatment regimens (Cochrane Database Syst. Rev. 2012;4:CD006243).
The current study adds an additional year of follow-up to previous trials, during which the incidence of congestive heart failure continues to increase with no indication of a plateau. This justifies long-term surveillance for congestive heart failure in women who have received trastuzumab, as well as extended follow-up of women enrolled in trials.
Although most previous randomized clinical trials have included anthracycline in both treatment and comparison arms, the current observational study found that, in real life, about 40% of women undergoing chemotherapy received regimens excluding anthracycline, probably due in part to the older age and higher comorbidity burden relative to participants in clinical trials.
Finally, this study raises concern in that roughly a quarter of the women who received adjuvant trastuzumab had been treated well before the publication of peer-reviewed results of relevant randomized controlled trials. Clinicians may have been basing their decision on findings from trials in women with metastatic breast cancer and preliminary data from the early breast cancer trials. The interim findings, however, were based on follow-up periods too short to account for the longer life expectancy of women with early breast cancer relative to women with metastatic disease.
There have been many instances in which new treatments disseminate based on preliminary reports of benefit, only to be withdrawn after additional safety data become available. Patients, clinicians, and researchers must temper their enthusiasm about the benefits of new cancer therapies with the recognition that estimates of the long-term risk of adverse events are based on short-term observations among carefully selected clinical trial participants.
Ann M. Geiger, Ph.D., is with the division of public health sciences at Wake Forest University, Winston Salem, N.C. She said that she has no conflicts of interest.
The development of trastuzumab for the treatment of nonmetastatic invasive breast cancer tumors expressing human epidermal growth factor receptor 2 has been an important step in the field of personalized medicine.
Multiple trials have documented clinically and statistically significant improvements in overall and disease-free survival for women receiving adjuvant treatment including trastuzumab, compared with those receiving other chemotherapy agents. Unlike most new cancer chemotherapies, trastuzumab’s use in early-stage HER2-positive breast cancer also appears to be cost effective (Ann. Pharmacother. 2009;43:296-303).
This benefit, however, has been offset by safety concerns, with a pooled analysis of the early breast cancer trials suggesting a fivefold increase in risk of congestive heart failure for women who receive trastuzumab versus those who do not, with consistent results across varying treatment regimens (Cochrane Database Syst. Rev. 2012;4:CD006243).
The current study adds an additional year of follow-up to previous trials, during which the incidence of congestive heart failure continues to increase with no indication of a plateau. This justifies long-term surveillance for congestive heart failure in women who have received trastuzumab, as well as extended follow-up of women enrolled in trials.
Although most previous randomized clinical trials have included anthracycline in both treatment and comparison arms, the current observational study found that, in real life, about 40% of women undergoing chemotherapy received regimens excluding anthracycline, probably due in part to the older age and higher comorbidity burden relative to participants in clinical trials.
Finally, this study raises concern in that roughly a quarter of the women who received adjuvant trastuzumab had been treated well before the publication of peer-reviewed results of relevant randomized controlled trials. Clinicians may have been basing their decision on findings from trials in women with metastatic breast cancer and preliminary data from the early breast cancer trials. The interim findings, however, were based on follow-up periods too short to account for the longer life expectancy of women with early breast cancer relative to women with metastatic disease.
There have been many instances in which new treatments disseminate based on preliminary reports of benefit, only to be withdrawn after additional safety data become available. Patients, clinicians, and researchers must temper their enthusiasm about the benefits of new cancer therapies with the recognition that estimates of the long-term risk of adverse events are based on short-term observations among carefully selected clinical trial participants.
Ann M. Geiger, Ph.D., is with the division of public health sciences at Wake Forest University, Winston Salem, N.C. She said that she has no conflicts of interest.
Trastuzumab, whether given with or without anthracyline-based chemotherapy, was associated with significant increases in heart failure and cardiomyopathy in a large population-based, retrospective cohort study of women treated for breast cancer.
In the "real world" study, which used data from the health maintenance organization Cancer Research Network, the adjusted risk of heart failure (HF) and/or cardiomyopathy increased fourfold among women who received trastuzumab (Herceptin) alone and sevenfold in those who received anthracycline plus trastuzumab, compared with women who received no chemotherapy.
In all, the risk of anthracycline-associated HF/cardiomyopathy among women younger than 65 years was similar to results from randomized clinical trials, while the trastuzumab-associated HF/cardiomyopathy risk – whether administered alone or following an anthracycline – was greater than that previously reported.
"Our results highlight the importance of generalizability in applying clinical trial findings to community settings; although similar to clinical trial results, these population-based results cannot be attributed to any single patient in clinical practice," wrote Erin J. Aiello Bowles of Group Health Research Institute, Seattle, and her associates.
Randomized trials have typically excluded older women and those with major comorbidities, and therefore the association between the two agents and HF/cardiomyopathy in this population is not well understood, Ms. Bowles and her associates noted (J. Natl. Cancer Inst. 2012 Aug. 30 [doi:10.1093/jnci/djs317]).
The current study population comprised 12,500 women diagnosed with incident, invasive breast cancer from Jan. 1, 1999, through Dec. 31, 2007, at eight integrated Cancer Research Network health systems. Women diagnosed with HF/cardiomyopathy prior to breast cancer diagnosis or initiation of chemotherapy were excluded. They had a mean age of 60 years (range, 22-99), and 85.8% were white.
In a median follow-up of 4.4 years, nearly half (46.5%) had received no chemotherapy. Just under a third, 29.6%, had received anthracycline-based chemotherapy alone, 0.9% received trastuzumab-based therapy without anthracycline, 3.5% received anthracycline plus trastuzumab, and 19.5% received other chemotherapy.
Compared with the women who received other chemotherapy or no chemotherapy, the women who received anthracycline alone or anthracycline plus trastuzumab were younger, diagnosed at later stages, had fewer comorbidities, and were slightly more likely to receive radiation therapy. These findings "suggest substantial individualization of adjuvant chemotherapy administration by age and comorbidity in community practice," Ms. Bowles and her associates wrote.
The incidence of HF/cardiomyopathy increased with increasing follow-up time for all of the chemotherapy types, but to a greater degree with trastuzumab. The cumulative HF/cardiomyopathy incidence increased from 1.2% at year 1 to 4.3% at year 5 for the anthracycline recipients, which was similar to the increase from 1.3% to 4.5% for those on other chemotherapies. For those with no chemotherapy, the cumulative incidence rose from 0.9% to 3.1%.
In contrast, the cumulative HF/cardiomyopathy incidence among recipients of anthracycline plus trastuzumab was 6.2% after 1 year of follow-up and continued to increase to 20.1% by 5 years.
Compared with no chemotherapy, the risk of incident HF/cardiomyopathy among all women was statistically significantly increased for anthracycline alone (adjusted hazard ratio, 1.40), trastuzumab without anthracycline (HR, 4.12), anthracycline plus trastuzumab (HR, 7.19), and other chemotherapy (HR, 1.49), the investigators said.
The 5-year cumulative incidence for HF/cardiomyopathy associated with each of the chemotherapies was greater in the older age groups, but the hazard ratios for HF/cardiomyopathy associated with chemotherapy use decreased with increasing age. For example, the hazard ratio for HF/cardiomyopathy associated with anthracycline use alone was statistically significant among women younger than 55 years (HR, 2.52) but not among women 55-64 years (HR, 1.61) or older.
According to Ms. Bowles and her associates, this study demonstrates the importance of observational comparative safety and effectiveness studies in providing complementary data to those obtained in clinical trials. "Observational studies allow for estimation of risks and benefits in community practice, which includes patients who may not be eligible for clinical trials. Clinical trials may provide more relevant estimates for patients who are eligible candidates, but many people are not and still receive these treatments in community practice," they wrote.
This work was supported by the National Cancer Institute through an administrative supplement to the Cancer Research Network. Ms. Bowles did not report any financial disclosures, but one of her coauthors, Dr. Larry A. Allen, has received consulting fees from Amgen, Janssen Scientific Affairs, and the Robert Wood Johnson Foundation.
Trastuzumab, whether given with or without anthracyline-based chemotherapy, was associated with significant increases in heart failure and cardiomyopathy in a large population-based, retrospective cohort study of women treated for breast cancer.
In the "real world" study, which used data from the health maintenance organization Cancer Research Network, the adjusted risk of heart failure (HF) and/or cardiomyopathy increased fourfold among women who received trastuzumab (Herceptin) alone and sevenfold in those who received anthracycline plus trastuzumab, compared with women who received no chemotherapy.
In all, the risk of anthracycline-associated HF/cardiomyopathy among women younger than 65 years was similar to results from randomized clinical trials, while the trastuzumab-associated HF/cardiomyopathy risk – whether administered alone or following an anthracycline – was greater than that previously reported.
"Our results highlight the importance of generalizability in applying clinical trial findings to community settings; although similar to clinical trial results, these population-based results cannot be attributed to any single patient in clinical practice," wrote Erin J. Aiello Bowles of Group Health Research Institute, Seattle, and her associates.
Randomized trials have typically excluded older women and those with major comorbidities, and therefore the association between the two agents and HF/cardiomyopathy in this population is not well understood, Ms. Bowles and her associates noted (J. Natl. Cancer Inst. 2012 Aug. 30 [doi:10.1093/jnci/djs317]).
The current study population comprised 12,500 women diagnosed with incident, invasive breast cancer from Jan. 1, 1999, through Dec. 31, 2007, at eight integrated Cancer Research Network health systems. Women diagnosed with HF/cardiomyopathy prior to breast cancer diagnosis or initiation of chemotherapy were excluded. They had a mean age of 60 years (range, 22-99), and 85.8% were white.
In a median follow-up of 4.4 years, nearly half (46.5%) had received no chemotherapy. Just under a third, 29.6%, had received anthracycline-based chemotherapy alone, 0.9% received trastuzumab-based therapy without anthracycline, 3.5% received anthracycline plus trastuzumab, and 19.5% received other chemotherapy.
Compared with the women who received other chemotherapy or no chemotherapy, the women who received anthracycline alone or anthracycline plus trastuzumab were younger, diagnosed at later stages, had fewer comorbidities, and were slightly more likely to receive radiation therapy. These findings "suggest substantial individualization of adjuvant chemotherapy administration by age and comorbidity in community practice," Ms. Bowles and her associates wrote.
The incidence of HF/cardiomyopathy increased with increasing follow-up time for all of the chemotherapy types, but to a greater degree with trastuzumab. The cumulative HF/cardiomyopathy incidence increased from 1.2% at year 1 to 4.3% at year 5 for the anthracycline recipients, which was similar to the increase from 1.3% to 4.5% for those on other chemotherapies. For those with no chemotherapy, the cumulative incidence rose from 0.9% to 3.1%.
In contrast, the cumulative HF/cardiomyopathy incidence among recipients of anthracycline plus trastuzumab was 6.2% after 1 year of follow-up and continued to increase to 20.1% by 5 years.
Compared with no chemotherapy, the risk of incident HF/cardiomyopathy among all women was statistically significantly increased for anthracycline alone (adjusted hazard ratio, 1.40), trastuzumab without anthracycline (HR, 4.12), anthracycline plus trastuzumab (HR, 7.19), and other chemotherapy (HR, 1.49), the investigators said.
The 5-year cumulative incidence for HF/cardiomyopathy associated with each of the chemotherapies was greater in the older age groups, but the hazard ratios for HF/cardiomyopathy associated with chemotherapy use decreased with increasing age. For example, the hazard ratio for HF/cardiomyopathy associated with anthracycline use alone was statistically significant among women younger than 55 years (HR, 2.52) but not among women 55-64 years (HR, 1.61) or older.
According to Ms. Bowles and her associates, this study demonstrates the importance of observational comparative safety and effectiveness studies in providing complementary data to those obtained in clinical trials. "Observational studies allow for estimation of risks and benefits in community practice, which includes patients who may not be eligible for clinical trials. Clinical trials may provide more relevant estimates for patients who are eligible candidates, but many people are not and still receive these treatments in community practice," they wrote.
This work was supported by the National Cancer Institute through an administrative supplement to the Cancer Research Network. Ms. Bowles did not report any financial disclosures, but one of her coauthors, Dr. Larry A. Allen, has received consulting fees from Amgen, Janssen Scientific Affairs, and the Robert Wood Johnson Foundation.
FROM THE JOURNAL OF THE NATIONAL CANCER INSTITUTE
Major Finding: The adjusted risk of heart failure (HF) and/or cardiomyopathy increased fourfold among women who received trastuzumab alone and sevenfold in those who received anthracycline plus trastuzumab, compared with women who received no chemotherapy.
Data Source: The "real world" study used data on 12,500 women from the health maintenance organization Cancer Research Network.
Disclosures: This work was supported by the National Cancer Institute through an administrative supplement to the Cancer Research Network. Ms. Bowles did not report any financial disclosures, but coauthor Dr. Larry A. Allen has received consulting fees from Amgen, Janssen Scientific Affairs, and the Robert Wood Johnson Foundation.
Third Universal MI Definition Unveiled
MUNICH – A new universal definition of myocardial infarction has been unveiled, sparked by the development of ever more sensitive cardiac biomarker assays and imaging techniques.
These assays, including the new high-sensitivity cardiac troponin (cTn) assays available throughout Europe and awaiting approval in the United States, have created confusion in the diagnosis of myocardial infarction because they detect small cTn elevations associated with many other clinical conditions such as heart failure, arrhythmias, and pulmonary embolism that are not MIs, but rather myocardial injury with necrosis.

"I think there has been a little bit of a problem in the past where we’ve had too many infarctions [diagnosed] ... where there is some damage or injury to the myocardial cells," said document task force cochair Dr. Kristian Thygeysen, who presented the third universal MI definition at the annual meeting of the European Society of Cardiology (ESC).
The expert consensus document, developed by the ESC, American College of Cardiology (ACC), American Heart Association (AHA), and World Heart Federation (WHF), maintains the pathological definition of acute MI as myocardial cell death due to prolonged myocardial ischemia, but goes on to refine the definition of MI in five settings, including the controversial area of MIs associated with revascularization procedures.

MI in the PCI Setting
An MI related to percutaneous coronary intervention (PCI) is defined as an elevation of cTn values more than five times the 99th percentile upper reference limit (URL) in the first 48 hours after a procedure in patients with normal baseline troponin values, or a rise of cTn values of more than 20% in patients with elevated baseline levels that are stable or falling.
It also requires one of the following events: symptoms suggestive of myocardial ischemia, new ischemic ECG changes, angiographic findings consistent with a procedural complication, or imaging evidence of new loss of viable myocardium or new regional wall motion abnormality.
In the previous 2007 document, the troponin threshold had been more than three times the 99th percentile, and was raised based on new prognostic information from long-term follow-up of patients undergoing PCI showing that there is unavoidable injury associated with the procedure, document task force codirector Dr. Joseph Alpert said during the presentation.
CABG-Related MI
Similarly, the 2012 version raises the troponin threshold for MI related to coronary artery bypass graft surgery from five times the 99th percentile URL in the 2007 document to 10 times the 99th percentile in patients with normal cTn baseline values.
It also requires one of the following: new pathological Q waves or new left bundle branch block (LBBB), angiographically documented new graft or new native coronary artery occlusion, or imaging evidence of new loss of viable myocardium or new regional wall motion abnormality.
Once again, the decision to raise the troponin threshold was made because there is unavoidable injury to the heart during CABG from needle sticks, handling of the heart, and the myocardial preservation procedure, said Dr. Alpert, a professor of medicine at the University of Arizona, Tucson.
Many Sources of Heart Injury
"The problem that every clinician – not just cardiologists, but internists and surgeons – is having with these troponin tests, and particularly with the high-sensitivity test, is that it turns out we’re finding that there are lots and lots of people having heart injuries," he said in an interview. "We’ve known for decades that it’s not uncommon for a very sick patient to have liver injuries, but now we’re saying, ‘My goodness, they’re having heart injuries, and these injuries are not MIs, or at least we have no evidence there is ischemia.’ "
The updated guideline points out that novel procedures such as transcatheter aortic valve implantation or mitral clip may also cause myocardial injury with necrosis, and that "it is likely that, similarly to CABG, the more marked the elevation of the biomarker values, the worse the prognosis – but data on that are not available.&qu
Although high-sensitivity troponin assays are not yet approved in the United States, it is only a matter of time before they are and the financial battle heats up over the distinction between myocardial injury and MI, according to Dr. Alpert. The reason is that there is currently no reimbursement code for patients with myocardial injury, who require substantial time and resources that currently are not being reimbursed.
"We’re pushing to get that code, because when you have an elevated troponin it means something, and it always means something not good," he said in the interview.
Cardiac troponin (I or T) is the preferred biomarker for the definition of acute MI, although less sensitive biomarkers such as the creatine kinase-MB (CKMB) mass can still be used when cardiac troponin is not available, said Dr. Thygesen, with the department of cardiological medicine, Aarhus (Denmark) University.
The criteria for an acute MI include detection of a rise and/or fall of cardiac biomarker values exceeding the 99th percentile URL, plus at least one of the following:
• Symptoms of ischemia.
• New or presumably new significant ST-segment/T wave changes or new LBBB.
• Development of pathological Q waves in the ECG.
• Imaging evidence of new loss of viable myocardium or new regional wall motion abnormality.
• Identification of an intracoronary thrombus by angiography or autopsy.
The new MI definition is expected to become the gold standard for diagnosis and to be adopted by the U.S. Food and Drug Administration for use in clinical trial protocols accepted by the agency. This is significant because it will help standardize the way MI is defined in clinical trials, making comparisons between studies more meaningful, Dr. Thygesen observed.
The expert consensus document, as well as pocket versions, are available on the websites of the ESC, ACC, AHA, and World Heart Federation.
The document is also being copublished in five journals: the Journal of the American College of Cardiology, European Heart Journal, Circulation, Global Heart, and Nature Reviews of Cardiology.
Dr. Thygesen reported no conflicts of interest. Dr. Alpert reported consulting for several pharmaceutical firms as well as the North American Center for Continuing Medical Education.
MUNICH – A new universal definition of myocardial infarction has been unveiled, sparked by the development of ever more sensitive cardiac biomarker assays and imaging techniques.
These assays, including the new high-sensitivity cardiac troponin (cTn) assays available throughout Europe and awaiting approval in the United States, have created confusion in the diagnosis of myocardial infarction because they detect small cTn elevations associated with many other clinical conditions such as heart failure, arrhythmias, and pulmonary embolism that are not MIs, but rather myocardial injury with necrosis.
"I think there has been a little bit of a problem in the past where we’ve had too many infarctions [diagnosed] ... where there is some damage or injury to the myocardial cells," said document task force cochair Dr. Kristian Thygeysen, who presented the third universal MI definition at the annual meeting of the European Society of Cardiology (ESC).
The expert consensus document, developed by the ESC, American College of Cardiology (ACC), American Heart Association (AHA), and World Heart Federation (WHF), maintains the pathological definition of acute MI as myocardial cell death due to prolonged myocardial ischemia, but goes on to refine the definition of MI in five settings, including the controversial area of MIs associated with revascularization procedures.
MI in the PCI Setting
An MI related to percutaneous coronary intervention (PCI) is defined as an elevation of cTn values more than five times the 99th percentile upper reference limit (URL) in the first 48 hours after a procedure in patients with normal baseline troponin values, or a rise of cTn values of more than 20% in patients with elevated baseline levels that are stable or falling.
It also requires one of the following events: symptoms suggestive of myocardial ischemia, new ischemic ECG changes, angiographic findings consistent with a procedural complication, or imaging evidence of new loss of viable myocardium or new regional wall motion abnormality.
In the previous 2007 document, the troponin threshold had been more than three times the 99th percentile, and was raised based on new prognostic information from long-term follow-up of patients undergoing PCI showing that there is unavoidable injury associated with the procedure, document task force codirector Dr. Joseph Alpert said during the presentation.
CABG-Related MI
Similarly, the 2012 version raises the troponin threshold for MI related to coronary artery bypass graft surgery from five times the 99th percentile URL in the 2007 document to 10 times the 99th percentile in patients with normal cTn baseline values.
It also requires one of the following: new pathological Q waves or new left bundle branch block (LBBB), angiographically documented new graft or new native coronary artery occlusion, or imaging evidence of new loss of viable myocardium or new regional wall motion abnormality.
Once again, the decision to raise the troponin threshold was made because there is unavoidable injury to the heart during CABG from needle sticks, handling of the heart, and the myocardial preservation procedure, said Dr. Alpert, a professor of medicine at the University of Arizona, Tucson.
Many Sources of Heart Injury
"The problem that every clinician – not just cardiologists, but internists and surgeons – is having with these troponin tests, and particularly with the high-sensitivity test, is that it turns out we’re finding that there are lots and lots of people having heart injuries," he said in an interview. "We’ve known for decades that it’s not uncommon for a very sick patient to have liver injuries, but now we’re saying, ‘My goodness, they’re having heart injuries, and these injuries are not MIs, or at least we have no evidence there is ischemia.’ "
The updated guideline points out that novel procedures such as transcatheter aortic valve implantation or mitral clip may also cause myocardial injury with necrosis, and that "it is likely that, similarly to CABG, the more marked the elevation of the biomarker values, the worse the prognosis – but data on that are not available.&qu
Although high-sensitivity troponin assays are not yet approved in the United States, it is only a matter of time before they are and the financial battle heats up over the distinction between myocardial injury and MI, according to Dr. Alpert. The reason is that there is currently no reimbursement code for patients with myocardial injury, who require substantial time and resources that currently are not being reimbursed.
"We’re pushing to get that code, because when you have an elevated troponin it means something, and it always means something not good," he said in the interview.
Cardiac troponin (I or T) is the preferred biomarker for the definition of acute MI, although less sensitive biomarkers such as the creatine kinase-MB (CKMB) mass can still be used when cardiac troponin is not available, said Dr. Thygesen, with the department of cardiological medicine, Aarhus (Denmark) University.
The criteria for an acute MI include detection of a rise and/or fall of cardiac biomarker values exceeding the 99th percentile URL, plus at least one of the following:
• Symptoms of ischemia.
• New or presumably new significant ST-segment/T wave changes or new LBBB.
• Development of pathological Q waves in the ECG.
• Imaging evidence of new loss of viable myocardium or new regional wall motion abnormality.
• Identification of an intracoronary thrombus by angiography or autopsy.
The new MI definition is expected to become the gold standard for diagnosis and to be adopted by the U.S. Food and Drug Administration for use in clinical trial protocols accepted by the agency. This is significant because it will help standardize the way MI is defined in clinical trials, making comparisons between studies more meaningful, Dr. Thygesen observed.
The expert consensus document, as well as pocket versions, are available on the websites of the ESC, ACC, AHA, and World Heart Federation.
The document is also being copublished in five journals: the Journal of the American College of Cardiology, European Heart Journal, Circulation, Global Heart, and Nature Reviews of Cardiology.
Dr. Thygesen reported no conflicts of interest. Dr. Alpert reported consulting for several pharmaceutical firms as well as the North American Center for Continuing Medical Education.
MUNICH – A new universal definition of myocardial infarction has been unveiled, sparked by the development of ever more sensitive cardiac biomarker assays and imaging techniques.
These assays, including the new high-sensitivity cardiac troponin (cTn) assays available throughout Europe and awaiting approval in the United States, have created confusion in the diagnosis of myocardial infarction because they detect small cTn elevations associated with many other clinical conditions such as heart failure, arrhythmias, and pulmonary embolism that are not MIs, but rather myocardial injury with necrosis.
"I think there has been a little bit of a problem in the past where we’ve had too many infarctions [diagnosed] ... where there is some damage or injury to the myocardial cells," said document task force cochair Dr. Kristian Thygeysen, who presented the third universal MI definition at the annual meeting of the European Society of Cardiology (ESC).
The expert consensus document, developed by the ESC, American College of Cardiology (ACC), American Heart Association (AHA), and World Heart Federation (WHF), maintains the pathological definition of acute MI as myocardial cell death due to prolonged myocardial ischemia, but goes on to refine the definition of MI in five settings, including the controversial area of MIs associated with revascularization procedures.
MI in the PCI Setting
An MI related to percutaneous coronary intervention (PCI) is defined as an elevation of cTn values more than five times the 99th percentile upper reference limit (URL) in the first 48 hours after a procedure in patients with normal baseline troponin values, or a rise of cTn values of more than 20% in patients with elevated baseline levels that are stable or falling.
It also requires one of the following events: symptoms suggestive of myocardial ischemia, new ischemic ECG changes, angiographic findings consistent with a procedural complication, or imaging evidence of new loss of viable myocardium or new regional wall motion abnormality.
In the previous 2007 document, the troponin threshold had been more than three times the 99th percentile, and was raised based on new prognostic information from long-term follow-up of patients undergoing PCI showing that there is unavoidable injury associated with the procedure, document task force codirector Dr. Joseph Alpert said during the presentation.
CABG-Related MI
Similarly, the 2012 version raises the troponin threshold for MI related to coronary artery bypass graft surgery from five times the 99th percentile URL in the 2007 document to 10 times the 99th percentile in patients with normal cTn baseline values.
It also requires one of the following: new pathological Q waves or new left bundle branch block (LBBB), angiographically documented new graft or new native coronary artery occlusion, or imaging evidence of new loss of viable myocardium or new regional wall motion abnormality.
Once again, the decision to raise the troponin threshold was made because there is unavoidable injury to the heart during CABG from needle sticks, handling of the heart, and the myocardial preservation procedure, said Dr. Alpert, a professor of medicine at the University of Arizona, Tucson.
Many Sources of Heart Injury
"The problem that every clinician – not just cardiologists, but internists and surgeons – is having with these troponin tests, and particularly with the high-sensitivity test, is that it turns out we’re finding that there are lots and lots of people having heart injuries," he said in an interview. "We’ve known for decades that it’s not uncommon for a very sick patient to have liver injuries, but now we’re saying, ‘My goodness, they’re having heart injuries, and these injuries are not MIs, or at least we have no evidence there is ischemia.’ "
The updated guideline points out that novel procedures such as transcatheter aortic valve implantation or mitral clip may also cause myocardial injury with necrosis, and that "it is likely that, similarly to CABG, the more marked the elevation of the biomarker values, the worse the prognosis – but data on that are not available.&qu
Although high-sensitivity troponin assays are not yet approved in the United States, it is only a matter of time before they are and the financial battle heats up over the distinction between myocardial injury and MI, according to Dr. Alpert. The reason is that there is currently no reimbursement code for patients with myocardial injury, who require substantial time and resources that currently are not being reimbursed.
"We’re pushing to get that code, because when you have an elevated troponin it means something, and it always means something not good," he said in the interview.
Cardiac troponin (I or T) is the preferred biomarker for the definition of acute MI, although less sensitive biomarkers such as the creatine kinase-MB (CKMB) mass can still be used when cardiac troponin is not available, said Dr. Thygesen, with the department of cardiological medicine, Aarhus (Denmark) University.
The criteria for an acute MI include detection of a rise and/or fall of cardiac biomarker values exceeding the 99th percentile URL, plus at least one of the following:
• Symptoms of ischemia.
• New or presumably new significant ST-segment/T wave changes or new LBBB.
• Development of pathological Q waves in the ECG.
• Imaging evidence of new loss of viable myocardium or new regional wall motion abnormality.
• Identification of an intracoronary thrombus by angiography or autopsy.
The new MI definition is expected to become the gold standard for diagnosis and to be adopted by the U.S. Food and Drug Administration for use in clinical trial protocols accepted by the agency. This is significant because it will help standardize the way MI is defined in clinical trials, making comparisons between studies more meaningful, Dr. Thygesen observed.
The expert consensus document, as well as pocket versions, are available on the websites of the ESC, ACC, AHA, and World Heart Federation.
The document is also being copublished in five journals: the Journal of the American College of Cardiology, European Heart Journal, Circulation, Global Heart, and Nature Reviews of Cardiology.
Dr. Thygesen reported no conflicts of interest. Dr. Alpert reported consulting for several pharmaceutical firms as well as the North American Center for Continuing Medical Education.
AT THE ANNUAL MEETING OF THE EUROPEAN SOCIETY OF CARDIOLOGY

LCZ696 Promising in Subset of Heart Failure Patients
Heart failure patients with preserved ejection who received an investigational agent LCA696 experienced a significant reduction of NT-proBNP at 12 weeks, compared with those who received valsartan, a randomized, multicenter, phase II trial demonstrated.
In addition, a reduction in left atrial size at 36 weeks was observed in patients in the LCA696 group, compared with those who received the angiotensin receptor blocker, researchers led by Dr. Scott Solomon of the cardiovascular division at Brigham and Women’s Hospital, Boston, reported in a study published online Aug. 26, 2012, in the Lancet. The study was simultaneously presented at the annual congress of the European Society of Cardiology.
"Present treatment of heart failure with preserved ejection fraction remains both symptom based and empiric, with no specific treatment approved for this indication," the researchers wrote. "Although ACE inhibitors and ARBs have been associated with symptom improvement, increased functional capacity, and reduction in admission to hospital in these patients, existing guidelines state that no treatment has convincingly been shown to reduce morbidity or mortality.
In a phase II trial known as PARAMOUNT, Dr. Solomon and his associates at 65 centers in 13 countries set out to assess the efficacy and safety of LCZ696 in 301 heart failure patients with preserved ejection fraction. LCZ696 is a first-in-class angiotensin receptor neprilysin inhibitor that includes valsartan. The researchers hypothesized that LCA696 works by augmenting the active natriuretic peptides to improve myocardial relaxation and reduce hypertrophy. They recruited patients from November 2009 through March 2011.
Patients were eligible for PARAMOUNT if they were at least 40 years of age, had New York Heart Association class II-III heart failure, a left ventricular ejection fraction (LVEF) of 45% or higher, and an NT-proBNP greater than 400 pg/mL (Lancet 2012 Aug. 26 [http://dx.doi.org/10.1016/S0140-6736(12)61227-6]).
After randomization, 149 patients were started on 50 mg LCZ696 twice daily and 152 were started on 40 mg valsartan twice daily and titrated to their final doses of 200 mg LCZ696 twice daily or 160 mg valsartan twice daily over a period of 2-4 weeks. All patients were treated for 36 weeks; the primary end point was change in NT-proBNP from baseline to 12 weeks. This end point was selected "because raised natriuretic peptide concentrations are associated with adverse outcomes in patients with heart failure, including those with preserved ejection fraction, and reductions in NT-proBNP have been associated with improved outcomes in heart failure," the researchers explained.
Dr. Solomon and his associates reported complete data from 134 patients in the LCZ696 group and 132 patients in the valsartan group. The mean age of patients in both groups was 71 years and more than half (57%) were women. The change in NT-proBNP from baseline to week 12 was significantly reduced in patients in the LCZ696 group (from 783 to 605 pg/mL), compared with patients in the valsartan group (from 862 to 835 pg/mL). This translated into a significant ratio of change between the two treatment groups of 0.77. By week 36, the differences in NT-proBNP between the two groups were no longer significant.
After 36 weeks of treatment, patients in the LCZ696 group experienced significant reductions in left atrial volume and in left atrial dimension, compared with their counterparts in the valsartan group. "Left atrial size has been one of the most powerful predictors of outcome in heart failure, including heart failure with preserved ejection fraction," the researchers wrote. "The reported reduction in left atrial size offers support to the notion that LCZ696 had a sustained physiological benefit to 36 weeks."
LCZ696 was well tolerated and the proportion of patients who experienced one or more serious adverse events was similar between the two groups (15% among those in the LCZ696 group vs. 20% among those in the valsartan group).
Novartis funded the study. Dr. Solomon and six of his coauthors disclosed having received research support from, and have consulted for, Novartis, which is developing LCZ696. Three other coauthors are employed by the company.
The positive signals from PARAMOUNT will surely trigger a definitive trial. However, what will the comparator be? Valsartan, a drug not known to be effective for heart failure with preserved ejection fraction? This comparison would show whether there was an advantage to adding a neprilysin inhibitor, but would not provide evidence that valsartan was useful in patients with heart failure with preserved ejection fraction. ACE inhibitors, which seem to have some effect in disease with preserved ejection fraction? An increase in diuretic dose – perhaps the best method of improving symptoms in a congested patient? Or simply placebo? A placebo-controlled design would be the easiest to interpret, but could be confounded by the widespread use of rennin-angiotensin-aldosterone system antagonists in patients with heart failure with preserved ejection fraction, often for problems such as hypertension and peripheral edema. Such background treatment might not easily be withdrawn, rendering enrollment difficult.
Another trial, in patients with heart failure with reduced ejection fraction and raised plasma natriuretic peptides, will show whether LCZ696 is superior to enalapril. If trials in disease with both preserved and reduced ejection fraction are positive (and use the same comparator), cardiac phenotype could become less important than plasma concentration of natriuretic peptides for management of heart failure. However, if LCZ696 proves ineffective in heart failure with preserved ejection fraction, then more attention should be paid to targeting of comorbid disease, to the individual phenotypes, to the causes underlying disease with preserved ejection fraction, or to the aging process itself, which could be the ultimate determinant of prognosis in these patients.
Dr. John G.F. Cleland and Dr. Andrew L. Clark are with the department of cardiology at Castle Hill Hospital in Kingston-Upon-Hull, United Kingdom. These remarks were adapted from a editorial accompanying the PARAMOUNT report (Lancet 2012 Aug. 26 [ http://dx.doi.org/10.1016/ S0140-6736(12)61349-X ]). Dr. Cleland disclosed that he has received honoraria from Novartis. Dr. Clark stated that he has no relevant financial conflicts of interest.
The positive signals from PARAMOUNT will surely trigger a definitive trial. However, what will the comparator be? Valsartan, a drug not known to be effective for heart failure with preserved ejection fraction? This comparison would show whether there was an advantage to adding a neprilysin inhibitor, but would not provide evidence that valsartan was useful in patients with heart failure with preserved ejection fraction. ACE inhibitors, which seem to have some effect in disease with preserved ejection fraction? An increase in diuretic dose – perhaps the best method of improving symptoms in a congested patient? Or simply placebo? A placebo-controlled design would be the easiest to interpret, but could be confounded by the widespread use of rennin-angiotensin-aldosterone system antagonists in patients with heart failure with preserved ejection fraction, often for problems such as hypertension and peripheral edema. Such background treatment might not easily be withdrawn, rendering enrollment difficult.
Another trial, in patients with heart failure with reduced ejection fraction and raised plasma natriuretic peptides, will show whether LCZ696 is superior to enalapril. If trials in disease with both preserved and reduced ejection fraction are positive (and use the same comparator), cardiac phenotype could become less important than plasma concentration of natriuretic peptides for management of heart failure. However, if LCZ696 proves ineffective in heart failure with preserved ejection fraction, then more attention should be paid to targeting of comorbid disease, to the individual phenotypes, to the causes underlying disease with preserved ejection fraction, or to the aging process itself, which could be the ultimate determinant of prognosis in these patients.
Dr. John G.F. Cleland and Dr. Andrew L. Clark are with the department of cardiology at Castle Hill Hospital in Kingston-Upon-Hull, United Kingdom. These remarks were adapted from a editorial accompanying the PARAMOUNT report (Lancet 2012 Aug. 26 [ http://dx.doi.org/10.1016/ S0140-6736(12)61349-X ]). Dr. Cleland disclosed that he has received honoraria from Novartis. Dr. Clark stated that he has no relevant financial conflicts of interest.
The positive signals from PARAMOUNT will surely trigger a definitive trial. However, what will the comparator be? Valsartan, a drug not known to be effective for heart failure with preserved ejection fraction? This comparison would show whether there was an advantage to adding a neprilysin inhibitor, but would not provide evidence that valsartan was useful in patients with heart failure with preserved ejection fraction. ACE inhibitors, which seem to have some effect in disease with preserved ejection fraction? An increase in diuretic dose – perhaps the best method of improving symptoms in a congested patient? Or simply placebo? A placebo-controlled design would be the easiest to interpret, but could be confounded by the widespread use of rennin-angiotensin-aldosterone system antagonists in patients with heart failure with preserved ejection fraction, often for problems such as hypertension and peripheral edema. Such background treatment might not easily be withdrawn, rendering enrollment difficult.
Another trial, in patients with heart failure with reduced ejection fraction and raised plasma natriuretic peptides, will show whether LCZ696 is superior to enalapril. If trials in disease with both preserved and reduced ejection fraction are positive (and use the same comparator), cardiac phenotype could become less important than plasma concentration of natriuretic peptides for management of heart failure. However, if LCZ696 proves ineffective in heart failure with preserved ejection fraction, then more attention should be paid to targeting of comorbid disease, to the individual phenotypes, to the causes underlying disease with preserved ejection fraction, or to the aging process itself, which could be the ultimate determinant of prognosis in these patients.
Dr. John G.F. Cleland and Dr. Andrew L. Clark are with the department of cardiology at Castle Hill Hospital in Kingston-Upon-Hull, United Kingdom. These remarks were adapted from a editorial accompanying the PARAMOUNT report (Lancet 2012 Aug. 26 [ http://dx.doi.org/10.1016/ S0140-6736(12)61349-X ]). Dr. Cleland disclosed that he has received honoraria from Novartis. Dr. Clark stated that he has no relevant financial conflicts of interest.
Heart failure patients with preserved ejection who received an investigational agent LCA696 experienced a significant reduction of NT-proBNP at 12 weeks, compared with those who received valsartan, a randomized, multicenter, phase II trial demonstrated.
In addition, a reduction in left atrial size at 36 weeks was observed in patients in the LCA696 group, compared with those who received the angiotensin receptor blocker, researchers led by Dr. Scott Solomon of the cardiovascular division at Brigham and Women’s Hospital, Boston, reported in a study published online Aug. 26, 2012, in the Lancet. The study was simultaneously presented at the annual congress of the European Society of Cardiology.
"Present treatment of heart failure with preserved ejection fraction remains both symptom based and empiric, with no specific treatment approved for this indication," the researchers wrote. "Although ACE inhibitors and ARBs have been associated with symptom improvement, increased functional capacity, and reduction in admission to hospital in these patients, existing guidelines state that no treatment has convincingly been shown to reduce morbidity or mortality.
In a phase II trial known as PARAMOUNT, Dr. Solomon and his associates at 65 centers in 13 countries set out to assess the efficacy and safety of LCZ696 in 301 heart failure patients with preserved ejection fraction. LCZ696 is a first-in-class angiotensin receptor neprilysin inhibitor that includes valsartan. The researchers hypothesized that LCA696 works by augmenting the active natriuretic peptides to improve myocardial relaxation and reduce hypertrophy. They recruited patients from November 2009 through March 2011.
Patients were eligible for PARAMOUNT if they were at least 40 years of age, had New York Heart Association class II-III heart failure, a left ventricular ejection fraction (LVEF) of 45% or higher, and an NT-proBNP greater than 400 pg/mL (Lancet 2012 Aug. 26 [http://dx.doi.org/10.1016/S0140-6736(12)61227-6]).
After randomization, 149 patients were started on 50 mg LCZ696 twice daily and 152 were started on 40 mg valsartan twice daily and titrated to their final doses of 200 mg LCZ696 twice daily or 160 mg valsartan twice daily over a period of 2-4 weeks. All patients were treated for 36 weeks; the primary end point was change in NT-proBNP from baseline to 12 weeks. This end point was selected "because raised natriuretic peptide concentrations are associated with adverse outcomes in patients with heart failure, including those with preserved ejection fraction, and reductions in NT-proBNP have been associated with improved outcomes in heart failure," the researchers explained.
Dr. Solomon and his associates reported complete data from 134 patients in the LCZ696 group and 132 patients in the valsartan group. The mean age of patients in both groups was 71 years and more than half (57%) were women. The change in NT-proBNP from baseline to week 12 was significantly reduced in patients in the LCZ696 group (from 783 to 605 pg/mL), compared with patients in the valsartan group (from 862 to 835 pg/mL). This translated into a significant ratio of change between the two treatment groups of 0.77. By week 36, the differences in NT-proBNP between the two groups were no longer significant.
After 36 weeks of treatment, patients in the LCZ696 group experienced significant reductions in left atrial volume and in left atrial dimension, compared with their counterparts in the valsartan group. "Left atrial size has been one of the most powerful predictors of outcome in heart failure, including heart failure with preserved ejection fraction," the researchers wrote. "The reported reduction in left atrial size offers support to the notion that LCZ696 had a sustained physiological benefit to 36 weeks."
LCZ696 was well tolerated and the proportion of patients who experienced one or more serious adverse events was similar between the two groups (15% among those in the LCZ696 group vs. 20% among those in the valsartan group).
Novartis funded the study. Dr. Solomon and six of his coauthors disclosed having received research support from, and have consulted for, Novartis, which is developing LCZ696. Three other coauthors are employed by the company.
Heart failure patients with preserved ejection who received an investigational agent LCA696 experienced a significant reduction of NT-proBNP at 12 weeks, compared with those who received valsartan, a randomized, multicenter, phase II trial demonstrated.
In addition, a reduction in left atrial size at 36 weeks was observed in patients in the LCA696 group, compared with those who received the angiotensin receptor blocker, researchers led by Dr. Scott Solomon of the cardiovascular division at Brigham and Women’s Hospital, Boston, reported in a study published online Aug. 26, 2012, in the Lancet. The study was simultaneously presented at the annual congress of the European Society of Cardiology.
"Present treatment of heart failure with preserved ejection fraction remains both symptom based and empiric, with no specific treatment approved for this indication," the researchers wrote. "Although ACE inhibitors and ARBs have been associated with symptom improvement, increased functional capacity, and reduction in admission to hospital in these patients, existing guidelines state that no treatment has convincingly been shown to reduce morbidity or mortality.
In a phase II trial known as PARAMOUNT, Dr. Solomon and his associates at 65 centers in 13 countries set out to assess the efficacy and safety of LCZ696 in 301 heart failure patients with preserved ejection fraction. LCZ696 is a first-in-class angiotensin receptor neprilysin inhibitor that includes valsartan. The researchers hypothesized that LCA696 works by augmenting the active natriuretic peptides to improve myocardial relaxation and reduce hypertrophy. They recruited patients from November 2009 through March 2011.
Patients were eligible for PARAMOUNT if they were at least 40 years of age, had New York Heart Association class II-III heart failure, a left ventricular ejection fraction (LVEF) of 45% or higher, and an NT-proBNP greater than 400 pg/mL (Lancet 2012 Aug. 26 [http://dx.doi.org/10.1016/S0140-6736(12)61227-6]).
After randomization, 149 patients were started on 50 mg LCZ696 twice daily and 152 were started on 40 mg valsartan twice daily and titrated to their final doses of 200 mg LCZ696 twice daily or 160 mg valsartan twice daily over a period of 2-4 weeks. All patients were treated for 36 weeks; the primary end point was change in NT-proBNP from baseline to 12 weeks. This end point was selected "because raised natriuretic peptide concentrations are associated with adverse outcomes in patients with heart failure, including those with preserved ejection fraction, and reductions in NT-proBNP have been associated with improved outcomes in heart failure," the researchers explained.
Dr. Solomon and his associates reported complete data from 134 patients in the LCZ696 group and 132 patients in the valsartan group. The mean age of patients in both groups was 71 years and more than half (57%) were women. The change in NT-proBNP from baseline to week 12 was significantly reduced in patients in the LCZ696 group (from 783 to 605 pg/mL), compared with patients in the valsartan group (from 862 to 835 pg/mL). This translated into a significant ratio of change between the two treatment groups of 0.77. By week 36, the differences in NT-proBNP between the two groups were no longer significant.
After 36 weeks of treatment, patients in the LCZ696 group experienced significant reductions in left atrial volume and in left atrial dimension, compared with their counterparts in the valsartan group. "Left atrial size has been one of the most powerful predictors of outcome in heart failure, including heart failure with preserved ejection fraction," the researchers wrote. "The reported reduction in left atrial size offers support to the notion that LCZ696 had a sustained physiological benefit to 36 weeks."
LCZ696 was well tolerated and the proportion of patients who experienced one or more serious adverse events was similar between the two groups (15% among those in the LCZ696 group vs. 20% among those in the valsartan group).
Novartis funded the study. Dr. Solomon and six of his coauthors disclosed having received research support from, and have consulted for, Novartis, which is developing LCZ696. Three other coauthors are employed by the company.
FROM THE LANCET
Major Finding: The change in NT-proBNP from baseline to week 12 was significantly reduced in patients in the LCZ696 group (from 783 to 605 pg/mL), compared with patients in the valsartan group (from 862 to 835 pg/mL). This translated into a ratio of change of 0.77.
Data Source: Data are from a phase II randomized trial of 301 heart failure patients with preserved ejection fraction that compared the angiotensin receptor neprilysin inhibitor LCZ696 with valsartan.
Disclosures: Novartis funded the study. Dr. Solomon and six of his coauthors disclosed having received research support from, and have consulted for, Novartis. Three other coauthors are employed by the company.
Rethinking Resynchronization: Why Women Fare Better Than Men
MIAMI BEACH – When it comes to heart failure, women tend to respond better to cardiac resynchronization therapy than do men, but for a long time no one knew why.
Turns out women have some distinct physiologic advantages, specifically more left bundle branch block and more nonischemic cardiomyopathy. Each drives a better response to cardiac resynchronization therapy (CRT), said Dr. David G. Strauss, a medical officer at the Food and Drug Administration’s Center for Devices and Radiologic Health in Silver Spring, Md.

In addition, timing of a specific electrical component within the heart triggers a third important distinction. Women benefit significantly from CRT even when their QRS duration is shorter than 150 msec. This supports the need for new gender-specific criteria for left bundle branch block and, ultimately, better identification of patients most likely to benefit from CRT device placement, Dr. Strauss said at the annual meeting of the International Society on Hypertension in Blacks.
"This is important because different professional societies recently have recommended that CRT be given to patients with QRS duration of 150 msec or more, and primarily in class II heart failure," he said. However, this cutoff "would exclude women with left bundle branch block and QRS duration 130-149 msec that derived significant benefit in MADIT-CRT" (Circulation 2011;123:1061-72).
In MADIT-CRT (Multicenter Automatic Defibrillator Implantation Trial–Cardiac Resynchronization Therapy), 1,820 patients were randomly assigned to receive CRT with an implantable cardioverter-defibrillator (ICD) or an ICD alone. In men with QRS duration of 130-140 msec in the study, there was no benefit from CRT and a trend toward harm associated with the device (hazard ratio, 1.69), Dr. Strauss said.
In contrast, women with QRS durations of 130-140 msec "had very significant benefit from CRT with a hazard ratio of 0.20, indicating 80% decrease in heart failure hospitalization or death," Dr. Strauss said.
More nonischemic cardiomyopathy among women translates to less myocardial scar and, ultimately, to a better CRT response. Scar tissue, no matter how it is electrically activated, does not contract, Dr. Strauss said.
Presence of left bundle branch block conferred a significant benefit in terms of reduced heart failure hospitalizations and deaths with use of CRT in the MADIT-CRT research. Conversely, participants without left bundle branch block realized no clinical advantage with CRT.
Be careful, however, to correctly identify left bundle branch block, Dr. Strauss said. Research suggests that one third of patients diagnosed with left bundle branch block by conventional electrocardiographic criteria are misdiagnosed (Am. J. Cardiol. 2011;107:927-34). Patients with intraventricular conduction delays, such as those caused by left ventricular hypertrophy and left ventricular dilation who do not have a blocked left bundle branch, can still display a prolonged QRS duration. This duration can be in the range generally considered diagnostic of left bundle branch block.
New criteria could parlay into better patient selection in the future, Dr. Strauss said. "CRT has been shown to improve heart failure symptoms, reduce heart failure hospitalization, and reduce mortality. However, not all patients benefit and significant risks exist." For example, addition of a CRT device with a left ventricular lead is associated with more complications, compared with ICD alone. "Thus there is a need for better patient risk stratification and patient identification criteria."
Based on results of electrical activation mapping and simulation studies, "My colleagues and I proposed stricter left bundle branch block criteria," he said. For example, they propose QRS duration of 130 msec or greater in women and 140 msec greater in men to replace the conventional 120 msec definition of left bundle branch block in either gender.
Another proposed new requirement is mid-QRS notching in at least two of the electrocardiographic leads of I, aVL, V1, V2, V5, or V6. This is important for diagnosing left bundle branch block because it reflects two events, Dr. Strauss said. There is an initial notch when electrical activation reaches the endocardium of the left ventricle and a second notch when activation reaches the lateral wall opposite the septum.
Other researchers support changing the current criteria for left bundle branch block. In a study of 111 patients, researchers found meeting what they termed "false" criteria was associated with a fourfold higher rate of heart failure hospitalization or death, compared with those meeting strict criteria (Pacing Clin. Electrophysiol. 2012;35:927-34).
CRT is FDA approved for class 3 or 4 heart failure, both of ischemic and nonischemic etiology, with a left ventricular ejection fraction less than 35% and a QRS duration over 120 msec. The FDA also approved CRT for class I ischemic or class II (ischemic or nonischemic) heart failure with an ejection fraction less than 30%, a QRS duration over 130 msec, and left bundle branch block.
Dr. Strauss reported no relevant financial disclosures.
MIAMI BEACH – When it comes to heart failure, women tend to respond better to cardiac resynchronization therapy than do men, but for a long time no one knew why.
Turns out women have some distinct physiologic advantages, specifically more left bundle branch block and more nonischemic cardiomyopathy. Each drives a better response to cardiac resynchronization therapy (CRT), said Dr. David G. Strauss, a medical officer at the Food and Drug Administration’s Center for Devices and Radiologic Health in Silver Spring, Md.
In addition, timing of a specific electrical component within the heart triggers a third important distinction. Women benefit significantly from CRT even when their QRS duration is shorter than 150 msec. This supports the need for new gender-specific criteria for left bundle branch block and, ultimately, better identification of patients most likely to benefit from CRT device placement, Dr. Strauss said at the annual meeting of the International Society on Hypertension in Blacks.
"This is important because different professional societies recently have recommended that CRT be given to patients with QRS duration of 150 msec or more, and primarily in class II heart failure," he said. However, this cutoff "would exclude women with left bundle branch block and QRS duration 130-149 msec that derived significant benefit in MADIT-CRT" (Circulation 2011;123:1061-72).
In MADIT-CRT (Multicenter Automatic Defibrillator Implantation Trial–Cardiac Resynchronization Therapy), 1,820 patients were randomly assigned to receive CRT with an implantable cardioverter-defibrillator (ICD) or an ICD alone. In men with QRS duration of 130-140 msec in the study, there was no benefit from CRT and a trend toward harm associated with the device (hazard ratio, 1.69), Dr. Strauss said.
In contrast, women with QRS durations of 130-140 msec "had very significant benefit from CRT with a hazard ratio of 0.20, indicating 80% decrease in heart failure hospitalization or death," Dr. Strauss said.
More nonischemic cardiomyopathy among women translates to less myocardial scar and, ultimately, to a better CRT response. Scar tissue, no matter how it is electrically activated, does not contract, Dr. Strauss said.
Presence of left bundle branch block conferred a significant benefit in terms of reduced heart failure hospitalizations and deaths with use of CRT in the MADIT-CRT research. Conversely, participants without left bundle branch block realized no clinical advantage with CRT.
Be careful, however, to correctly identify left bundle branch block, Dr. Strauss said. Research suggests that one third of patients diagnosed with left bundle branch block by conventional electrocardiographic criteria are misdiagnosed (Am. J. Cardiol. 2011;107:927-34). Patients with intraventricular conduction delays, such as those caused by left ventricular hypertrophy and left ventricular dilation who do not have a blocked left bundle branch, can still display a prolonged QRS duration. This duration can be in the range generally considered diagnostic of left bundle branch block.
New criteria could parlay into better patient selection in the future, Dr. Strauss said. "CRT has been shown to improve heart failure symptoms, reduce heart failure hospitalization, and reduce mortality. However, not all patients benefit and significant risks exist." For example, addition of a CRT device with a left ventricular lead is associated with more complications, compared with ICD alone. "Thus there is a need for better patient risk stratification and patient identification criteria."
Based on results of electrical activation mapping and simulation studies, "My colleagues and I proposed stricter left bundle branch block criteria," he said. For example, they propose QRS duration of 130 msec or greater in women and 140 msec greater in men to replace the conventional 120 msec definition of left bundle branch block in either gender.
Another proposed new requirement is mid-QRS notching in at least two of the electrocardiographic leads of I, aVL, V1, V2, V5, or V6. This is important for diagnosing left bundle branch block because it reflects two events, Dr. Strauss said. There is an initial notch when electrical activation reaches the endocardium of the left ventricle and a second notch when activation reaches the lateral wall opposite the septum.
Other researchers support changing the current criteria for left bundle branch block. In a study of 111 patients, researchers found meeting what they termed "false" criteria was associated with a fourfold higher rate of heart failure hospitalization or death, compared with those meeting strict criteria (Pacing Clin. Electrophysiol. 2012;35:927-34).
CRT is FDA approved for class 3 or 4 heart failure, both of ischemic and nonischemic etiology, with a left ventricular ejection fraction less than 35% and a QRS duration over 120 msec. The FDA also approved CRT for class I ischemic or class II (ischemic or nonischemic) heart failure with an ejection fraction less than 30%, a QRS duration over 130 msec, and left bundle branch block.
Dr. Strauss reported no relevant financial disclosures.
MIAMI BEACH – When it comes to heart failure, women tend to respond better to cardiac resynchronization therapy than do men, but for a long time no one knew why.
Turns out women have some distinct physiologic advantages, specifically more left bundle branch block and more nonischemic cardiomyopathy. Each drives a better response to cardiac resynchronization therapy (CRT), said Dr. David G. Strauss, a medical officer at the Food and Drug Administration’s Center for Devices and Radiologic Health in Silver Spring, Md.
In addition, timing of a specific electrical component within the heart triggers a third important distinction. Women benefit significantly from CRT even when their QRS duration is shorter than 150 msec. This supports the need for new gender-specific criteria for left bundle branch block and, ultimately, better identification of patients most likely to benefit from CRT device placement, Dr. Strauss said at the annual meeting of the International Society on Hypertension in Blacks.
"This is important because different professional societies recently have recommended that CRT be given to patients with QRS duration of 150 msec or more, and primarily in class II heart failure," he said. However, this cutoff "would exclude women with left bundle branch block and QRS duration 130-149 msec that derived significant benefit in MADIT-CRT" (Circulation 2011;123:1061-72).
In MADIT-CRT (Multicenter Automatic Defibrillator Implantation Trial–Cardiac Resynchronization Therapy), 1,820 patients were randomly assigned to receive CRT with an implantable cardioverter-defibrillator (ICD) or an ICD alone. In men with QRS duration of 130-140 msec in the study, there was no benefit from CRT and a trend toward harm associated with the device (hazard ratio, 1.69), Dr. Strauss said.
In contrast, women with QRS durations of 130-140 msec "had very significant benefit from CRT with a hazard ratio of 0.20, indicating 80% decrease in heart failure hospitalization or death," Dr. Strauss said.
More nonischemic cardiomyopathy among women translates to less myocardial scar and, ultimately, to a better CRT response. Scar tissue, no matter how it is electrically activated, does not contract, Dr. Strauss said.
Presence of left bundle branch block conferred a significant benefit in terms of reduced heart failure hospitalizations and deaths with use of CRT in the MADIT-CRT research. Conversely, participants without left bundle branch block realized no clinical advantage with CRT.
Be careful, however, to correctly identify left bundle branch block, Dr. Strauss said. Research suggests that one third of patients diagnosed with left bundle branch block by conventional electrocardiographic criteria are misdiagnosed (Am. J. Cardiol. 2011;107:927-34). Patients with intraventricular conduction delays, such as those caused by left ventricular hypertrophy and left ventricular dilation who do not have a blocked left bundle branch, can still display a prolonged QRS duration. This duration can be in the range generally considered diagnostic of left bundle branch block.
New criteria could parlay into better patient selection in the future, Dr. Strauss said. "CRT has been shown to improve heart failure symptoms, reduce heart failure hospitalization, and reduce mortality. However, not all patients benefit and significant risks exist." For example, addition of a CRT device with a left ventricular lead is associated with more complications, compared with ICD alone. "Thus there is a need for better patient risk stratification and patient identification criteria."
Based on results of electrical activation mapping and simulation studies, "My colleagues and I proposed stricter left bundle branch block criteria," he said. For example, they propose QRS duration of 130 msec or greater in women and 140 msec greater in men to replace the conventional 120 msec definition of left bundle branch block in either gender.
Another proposed new requirement is mid-QRS notching in at least two of the electrocardiographic leads of I, aVL, V1, V2, V5, or V6. This is important for diagnosing left bundle branch block because it reflects two events, Dr. Strauss said. There is an initial notch when electrical activation reaches the endocardium of the left ventricle and a second notch when activation reaches the lateral wall opposite the septum.
Other researchers support changing the current criteria for left bundle branch block. In a study of 111 patients, researchers found meeting what they termed "false" criteria was associated with a fourfold higher rate of heart failure hospitalization or death, compared with those meeting strict criteria (Pacing Clin. Electrophysiol. 2012;35:927-34).
CRT is FDA approved for class 3 or 4 heart failure, both of ischemic and nonischemic etiology, with a left ventricular ejection fraction less than 35% and a QRS duration over 120 msec. The FDA also approved CRT for class I ischemic or class II (ischemic or nonischemic) heart failure with an ejection fraction less than 30%, a QRS duration over 130 msec, and left bundle branch block.
Dr. Strauss reported no relevant financial disclosures.
AT THE ANNUAL MEETING OF THE INTERNATIONAL SOCIETY ON HYPERTENSION IN BLACKS

LVADs for Severe Heart Failure Gradually Take Hold
A sea change in management of severe heart failure began two and a half years ago, in January 2010, when the Food and Drug Administration approved U.S. marketing of the HeartMate II continuous-flow, left ventricular assist device for destination therapy. Patients, cardiologists, and cardiothoracic surgeons at last had a reasonably durable, effective, and relatively safe alternative to heart transplant to offer patients at the end stage of deteriorating heart function.
The Heart Mate II quickly supplanted the prior-generation, pulsatile-flow devices, and 2010 also saw a 10-fold spike in the number of left ventricular assist devices (LVADs) placed in U.S. patients as destination therapy.
During the first half of 2011, 6-month survival among U.S. patients with a continuous-flow LVAD (which means the HeartMate II, the only continuous-flow device on the U.S. market) was 89%, and 12-month survival during 2010 was 81% (J. Heart Lung Transplant 2012;31:117-26, putting the continuous-flow LVAD in at least the same ballpark as heart transplant, which has a 5-year survival of about 80% and a median survival of about 10 years in U.S. patients.
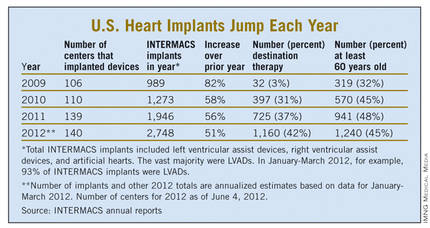
But despite estimates that many thousands, if not tens of thousands, of Americans meet the clinical criteria to qualify for placement of an LVAD, the reality is that in the 30 months since the HeartMate II became available for routine destination therapy through mid-2012, only about 4,600 were placed in U.S. patients. In addition, just slightly more than a third of those patients, roughly 1,700, received their LVAD explicitly for destination therapy.
Even in 2012, a majority of patients who received an LVAD got their device with the understanding that it was as a bridge to transplant, an LVAD placed with temporary intent to help patients survive and improve clinically until their LVAD could be swapped out and replaced by a transplanted heart.
LVAD Numbers Up, but Still Low
If the availability of the HeartMate II for destination therapy in routine practice was a revolution for the management of many patients with New York Heart Association class IV heart failure – which is how many experts in this field view the device – it has so far been a revolution played out in slow motion. In large part that’s because the field started small, and stayed small until recently. At the end of 2008, fewer than 1,000 patients had ever received a ventricular assist device or a total artificial heart. Even though the number nearly doubled in 2009, by decade’s end the cumulative total still remained under 2,000.

Despite the low numbers compared with the estimated need, many physicians and surgeons who specialize in advanced heart failure say they are satisfied with the pace at which continuous-flow LVADs have entered patients over the past 2 years. Though they acknowledge that the number of potential U.S. candidates for an LVAD undoubtedly is far larger than the nearly 4,600 who received one since early 2010, they note that the implantation rate grew in a steady and robust way during the past 30 months, as has the number of centers performing the surgery. With a better-than-50% year-over-year growth spurt in both 2010 and 2011, and with that pace continuing into the first months of 2012, the 140 U.S. centers that now place LVADs are on track to do nearly 3,000 this year (although only 42% were for destination therapy during January-March 2012).
"You probably don’t want it to increase too rapidly, because it is still high risk and expensive," said Dr. David E. Lanfear, a cardiologist specializing in advanced heart failure and transplantation at Henry Ford Hospital in Detroit. LVAD placements "have not reached the maximum number of patients who they could help, but you need to be cautious because it requires specialized expertise to do it properly. I wouldn’t expect that [during 2 years] it would immediately reach maximum use. It’s still in the growth phase. There are a lot of patients out there, so it will continue to grow; I’m not concerned that it’s not growing fast enough," he said in an interview.
An LVAD or a Heart Transplant?
Experts also note an important shift in attitude about the role for LVADs, a change that has brought them to the brink of replacing transplanted hearts as the default treatment for patients with end-stage heart failure.

"The current outcomes with HeartMate II have already changed the field. We published a paper last year that showed there was no difference between LVADs and transplant in costs and outcomes at 1 year (Ann. Thorac. Surg. 2011;91:1330-4)," said Dr. Mark S. Slaughter, professor of surgery and chief of thoracic and cardiovascular surgery at the University of Louisville (Ky.). Between that and the competitive mortality benefit from LVADs "we need to rethink the role of transplant. In appropriately selected patients, we can achieve outstanding long-term results with limited adverse events."
In their 2011 paper, Dr. Slaughter and his associates put the pending change more bluntly: "As outcomes for continuous-flow LVADs and heart transplantation converge, the therapy of heart transplantation could emerge as salvage therapy for major device-related complications or dysfunction or progressive right heart failure as opposed to the default option for all patients who are eligible for transplant."
Despite their huge promise, some experts have lingering concerns about the current generation of continuous-flow LVADs that make them reluctant to say that heart transplant has unquestionably stepped down from its pedestal as the gold standard treatment for patients with severe, end-stage heart failure.

"There is no question that [current continuous-flow LVADs] are very good technology. But whether they are great technology remains to be proven," said Dr. James K. Kirklin, professor of surgery and director of the division of cardiothoracic surgery at the University of Alabama at Birmingham. "We still have important concerns about things like drive-line infections and thromboembolic complications. Neither rate is terribly high, but they are high enough to be a problem if you apply LVADs to patients who are doing okay. We still have important doubts about whether we can really mimic the quality of life" achieved by transplant, he said in an interview.
LVAD placement also poses an operative risk, which may be as high as 10%, said Dr. David Taylor, a cardiologist and advanced heart failure specialist at the Cleveland Clinic. "If the risk was 2% or 3%, I’d say do it, but a risk closer to 10% makes you hesitate.
"There are a lot of really sick, elderly patients with heart failure who could unquestionably benefit from an LVAD. But as soon as you push the envelope [by treating sicker patients] you increase mortality. A large group of heart failure patients are very ill with chronic disease and feel terrible, and these are the patients where you’d see the greatest benefit, but we can’t afford to do that. We can’t afford to allocate this resource to patients with a 30% mortality risk, because that would limit our ability to extend it to other patients," Dr. Kirklin said in an interview.
"Most patients appreciate an LVAD and like it, but they still look forward to getting a transplant," said Dr. Stephen H. Bailey, director of cardiothoracic surgery at Allegheny General Hospital in Pittsburgh. "In July 2012, while the margin between LVAD and transplant is narrowing, transplant is still the gold standard for definitive treatment. Most patients still favor transplant, but that might change. It’s not quite there yet that an LVAD is equal to a transplant for the long term. It also requires getting rid of the drive line. That would be a game changer. It would also help if we could reduce gastrointestinal bleeds, but that is more of a nuisance."
Part of the bleeding problem comes from anticoagulation treatment to prevent clots from forming in the LVAD, but another facet is an acquired Von Willebrand factor deficiency caused by absent pulsatile blood flow, Dr. Bailey said. Early clinical results suggest that allowing the heart to beat every few seconds, by adjusting the LVAD’s continuous flow rate, can minimize the acquired deficiency.
Which INTERMACS Level?
The most widely used gauge physicians and surgeons have for assessing LVAD candidates is their level in a seven-step sequence created by INTERMACS (the Interagency Registry for Mechanically Assisted Circulatory Support), which subdivides a patient’s descent from advanced New York Heart Association class III heart failure through the strata of class IV disease. (See table.) The most severe INTERMACS stage, level 1 patients (also known as "crash and burn") are those in cardiogenic shock. During the past year or two, about 16% of U.S. LVAD recipients have been level 1 patients, a percentage that should ideally drop much closer to zero, experts say.

But beyond the maxim that an advanced heart failure patient should get an LVAD before reaching level 1, opinions vary on the best target.
"Everyone agrees that inotrope-dependent patients [profile 2 and 3] should get an LVAD," said Dr. Lanfear. "But you definitely should not wait until INTERMACS 1 or 2. Everyone tries hard to treat level 3 and 4 patients. Level 5 is controversial. We need more data about these clearly less- sick patients."
"INTERMACS 2 and 3 is the sweet spot. Patients in INTERMACS 4, 5, or 6 are not as motivated to be connected to a device and undergo big open heart surgery," said Dr. Bailey.
"When patients are on the cusp of inotrope dependence [before they reach level 3], I start to think about implanting," said Dr. Jeffrey J. Teuteberg, a cardiologist and associate director of the cardiac transplant program at the University of Pittsburgh. "It’s nice to get patients who are pre–inotrope dependent – profile 4, 5, or 6 – but are symptom limited and quality of life limited. For destination therapy, you want patients with function and good quality of life. Mechanical support provides more than just improved survival; it also reduces adverse events and raises quality of life."
"For level 2 patients, there is no question that an LVAD as bridge to transplant is better than continued medical treatment," said Dr. Taylor. "For level 3 patients, it’s a little trickier, but I think the majority would say that if a patient is stable and inotropic dependent, use an LVAD unless you believe a transplant will occur quickly. An LVAD would reduce the risk for developing cardiogenic shock. The patient who does the best with an LVAD is the one who is [relatively] healthy when implanted, but the patient who needs it most is the one who is literally dying."
"Everyone knows that if you take patients who are sicker [for LVADs], you’ll have trouble reaching 80% survival after 2 years. But there is not yet enough confidence in the treatment to extend it to INTERMACS level 4, 5, or 6 patients," said Dr. Kirklin. "That’s where the big potential is. The action currently is in INTERMACS 2 and 3. For patients who are INTERMACS 3, there is no question that if they can’t get a transplant they need a VAD. The sweet spot will be patients at INTERMACS 4."
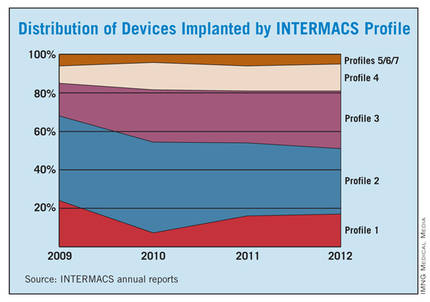
The INTERMACS registry numbers show that the field is currently stuck when it comes to INTERMACS levels, with the greatest number of LVADs going into level 2 patients, who received 34% of U.S LVADS during January-March 2012. The 34% level in early 2012 was down from 38% of LVAD recipients in 2011 and 47% in 2010, but growth in less-severe levels has been slow. At the start of 2012, 30% of LVAD recipients were at level 3, a small increase from the 27% rate in 2011 and 2010. Level 4 patients constituted 14% of LVAD recipients in early 2012, essentially unchanged from the prior 2 years, and level 5, 6, or 7 patients have consistently been a small slice of the U.S. LVAD pie, roughly 5% of recipients each year.
Beyond the INTERMACS Level
Heart failure specialists now recognize that INTERMACS level tells just part of the story.
"INTERMACS profiles depend on heart failure symptoms but not comorbidities. Severe diabetes, obstructive pulmonary disease, cardiorenal syndrome, chronic malnourishment, morbid obesity, and other factors all fall outside the INTERMACS profile criteria," said Dr. Kirklin.
He and his associates are formulating a risk assessment equation that will take comorbidities into account for a more global patient assessment. The most recently published INTERMACS registry analysis, which he first authored using data through the end of June 2011 with a total of 4,366 patients who received left ventricular support since 2006 (J. Heart Lung Transplant 2012;31:117-26), identified several comorbidity markers that each significantly linked with increased mortality. For example, a 1-unit increase in bilirubin linked with a 10% boost in mortality, a 1-unit increase in creatinine raised mortality by 16%, and a 0.5-unit increase in body surface area linked with a 48% rise in deaths. One of the strongest risk factors was being at INTERMACS level 1, which linked with a more-than-threefold higher mortality rate.
But more work must be done before the risk assessment formula is ready for clinical use. Right now, the formula "is not very reliable yet, because the maximum patient follow-up is 2 years. We need a little more follow-up," Dr. Kirklin said.
Growing the LVAD Numbers
Further growth in LVAD placements will happen on two fronts: broader use in patients at INTERMACS levels 2 and 3, and possibly level 4, where a strong consensus exists for LVAD support; and new evidence to document efficacy and safety in patients with less-severe disease at INTERMACS levels 4, 5, 6, and 7.
For the existing population, it’s a matter of physician and patient awareness. "We need to educate physicians that this is an option," said Dr. Lanfear. "Awareness is lacking. There is a lot of heart failure out there that is underrecognized and underreferred. Neither patients nor their physicians realize how sick they are, and that they are at a high enough risk to justify this."
Furthermore, there are parts of the country where the technology is underused. "In areas without a big center nearby, some physicians may not recognize patients who are sick enough to be LVAD candidates. We are trying to spread the word on who these patients are and when they should be referred."
According to Dr. Teuteberg, major warning signs that should flag patients who are potential LVAD candidates include advanced symptoms, increasing numbers of hospitalizations for heart failure, dwindling responses to ACE inhibitors and beta-blockers, increasing dosages of diuretics, a persistently high serum level of brain natriuretic peptide (BNP) despite good medical treatment, and lack of response to cardiac resynchronization therapy.
Expansion of the evidence base to show LVAD benefits in patients with INTERMACS level 5, 6, or 7 disease depends on a trial just starting, the REVIVE-IT (Randomized Evaluation of VAD Intervention Before Inotropic Therapy) study that’s set to enroll about 100 patients. But at press time, REVIVE-IT had not yet begun, posing doubts about LVAD use in the study’s targeted patients. "We haven’t really demonstrated reproducibly good survival [with LVADs] to compete with medical therapy in level 5 and 6 patients," said Dr. Kirklin. "The FDA put the study on hold while they reflected on that."
Making LVADs Better
With fast-paced technological advancement, continuous-flow LVADs will continue to evolve and improve. In June, results appeared on a new continuous-flow LVAD, the HeartWare device (Circulation 2012;125:3191-200), and last April an FDA advisory committee recommended that the agency approve the HeartWare LVAD for use as a bridge to (a trial testing the HeartWare LVAD for destination therapy is ongoing).
But no one interviewed for this article anticipates that the HeartWare LVAD will be a major advance. "Fundamentally, the major components and the implant technique are the same for the two devices," the HeartWare and the HeartMate II, said Dr. Slaughter. The HeartWare LVAD is smaller and designed to be placed completely in the pericardial space, but any clinical advantages based on these differences remain to be proved, he said in an interview.
A more meaningful improvement in LVAD design is in the works, and may reach initial clinical testing within a couple of years: a fully implantable LVAD with no transcutaneous drive line, a part that is subject to infection, prevents patients from submerging, and physically and psychologically limits patients by tethering them to equipment. "If there were one thing that could make a dramatic difference, it would be getting rid of the drive line. That is the Holy Grail for the field," said Dr. Bailey.
When a fully implantable LVAD becomes available for routine use, it will complete the LVAD revolution and help device therapy for advanced heart failure reach its full potential.
Dr. Lanfear has received research support and has received honoraria as a speaker for Thoratec, the company that markets the HeartMate II, and has received research support from HeartWare, the company developing the HeartWare LVAD. Dr. Slaughter has had contracts for services to Thoratec and HeartWare. Dr. Kirklin, Dr. Bailey, Dr. Teuteberg, and Dr. Taylor said that they had no disclosures.
*CORRECTION 8/10/12: The credit for the photo with the caption "The next LVAD in line, HeartWare, is smaller and dwells in the pericardial space" was misstated and should have been ©2012 HeartWare International, Inc.
A sea change in management of severe heart failure began two and a half years ago, in January 2010, when the Food and Drug Administration approved U.S. marketing of the HeartMate II continuous-flow, left ventricular assist device for destination therapy. Patients, cardiologists, and cardiothoracic surgeons at last had a reasonably durable, effective, and relatively safe alternative to heart transplant to offer patients at the end stage of deteriorating heart function.
The Heart Mate II quickly supplanted the prior-generation, pulsatile-flow devices, and 2010 also saw a 10-fold spike in the number of left ventricular assist devices (LVADs) placed in U.S. patients as destination therapy.
During the first half of 2011, 6-month survival among U.S. patients with a continuous-flow LVAD (which means the HeartMate II, the only continuous-flow device on the U.S. market) was 89%, and 12-month survival during 2010 was 81% (J. Heart Lung Transplant 2012;31:117-26, putting the continuous-flow LVAD in at least the same ballpark as heart transplant, which has a 5-year survival of about 80% and a median survival of about 10 years in U.S. patients.
But despite estimates that many thousands, if not tens of thousands, of Americans meet the clinical criteria to qualify for placement of an LVAD, the reality is that in the 30 months since the HeartMate II became available for routine destination therapy through mid-2012, only about 4,600 were placed in U.S. patients. In addition, just slightly more than a third of those patients, roughly 1,700, received their LVAD explicitly for destination therapy.
Even in 2012, a majority of patients who received an LVAD got their device with the understanding that it was as a bridge to transplant, an LVAD placed with temporary intent to help patients survive and improve clinically until their LVAD could be swapped out and replaced by a transplanted heart.
LVAD Numbers Up, but Still Low
If the availability of the HeartMate II for destination therapy in routine practice was a revolution for the management of many patients with New York Heart Association class IV heart failure – which is how many experts in this field view the device – it has so far been a revolution played out in slow motion. In large part that’s because the field started small, and stayed small until recently. At the end of 2008, fewer than 1,000 patients had ever received a ventricular assist device or a total artificial heart. Even though the number nearly doubled in 2009, by decade’s end the cumulative total still remained under 2,000.
Despite the low numbers compared with the estimated need, many physicians and surgeons who specialize in advanced heart failure say they are satisfied with the pace at which continuous-flow LVADs have entered patients over the past 2 years. Though they acknowledge that the number of potential U.S. candidates for an LVAD undoubtedly is far larger than the nearly 4,600 who received one since early 2010, they note that the implantation rate grew in a steady and robust way during the past 30 months, as has the number of centers performing the surgery. With a better-than-50% year-over-year growth spurt in both 2010 and 2011, and with that pace continuing into the first months of 2012, the 140 U.S. centers that now place LVADs are on track to do nearly 3,000 this year (although only 42% were for destination therapy during January-March 2012).
"You probably don’t want it to increase too rapidly, because it is still high risk and expensive," said Dr. David E. Lanfear, a cardiologist specializing in advanced heart failure and transplantation at Henry Ford Hospital in Detroit. LVAD placements "have not reached the maximum number of patients who they could help, but you need to be cautious because it requires specialized expertise to do it properly. I wouldn’t expect that [during 2 years] it would immediately reach maximum use. It’s still in the growth phase. There are a lot of patients out there, so it will continue to grow; I’m not concerned that it’s not growing fast enough," he said in an interview.
An LVAD or a Heart Transplant?
Experts also note an important shift in attitude about the role for LVADs, a change that has brought them to the brink of replacing transplanted hearts as the default treatment for patients with end-stage heart failure.
"The current outcomes with HeartMate II have already changed the field. We published a paper last year that showed there was no difference between LVADs and transplant in costs and outcomes at 1 year (Ann. Thorac. Surg. 2011;91:1330-4)," said Dr. Mark S. Slaughter, professor of surgery and chief of thoracic and cardiovascular surgery at the University of Louisville (Ky.). Between that and the competitive mortality benefit from LVADs "we need to rethink the role of transplant. In appropriately selected patients, we can achieve outstanding long-term results with limited adverse events."
In their 2011 paper, Dr. Slaughter and his associates put the pending change more bluntly: "As outcomes for continuous-flow LVADs and heart transplantation converge, the therapy of heart transplantation could emerge as salvage therapy for major device-related complications or dysfunction or progressive right heart failure as opposed to the default option for all patients who are eligible for transplant."
Despite their huge promise, some experts have lingering concerns about the current generation of continuous-flow LVADs that make them reluctant to say that heart transplant has unquestionably stepped down from its pedestal as the gold standard treatment for patients with severe, end-stage heart failure.
"There is no question that [current continuous-flow LVADs] are very good technology. But whether they are great technology remains to be proven," said Dr. James K. Kirklin, professor of surgery and director of the division of cardiothoracic surgery at the University of Alabama at Birmingham. "We still have important concerns about things like drive-line infections and thromboembolic complications. Neither rate is terribly high, but they are high enough to be a problem if you apply LVADs to patients who are doing okay. We still have important doubts about whether we can really mimic the quality of life" achieved by transplant, he said in an interview.
LVAD placement also poses an operative risk, which may be as high as 10%, said Dr. David Taylor, a cardiologist and advanced heart failure specialist at the Cleveland Clinic. "If the risk was 2% or 3%, I’d say do it, but a risk closer to 10% makes you hesitate.
"There are a lot of really sick, elderly patients with heart failure who could unquestionably benefit from an LVAD. But as soon as you push the envelope [by treating sicker patients] you increase mortality. A large group of heart failure patients are very ill with chronic disease and feel terrible, and these are the patients where you’d see the greatest benefit, but we can’t afford to do that. We can’t afford to allocate this resource to patients with a 30% mortality risk, because that would limit our ability to extend it to other patients," Dr. Kirklin said in an interview.
"Most patients appreciate an LVAD and like it, but they still look forward to getting a transplant," said Dr. Stephen H. Bailey, director of cardiothoracic surgery at Allegheny General Hospital in Pittsburgh. "In July 2012, while the margin between LVAD and transplant is narrowing, transplant is still the gold standard for definitive treatment. Most patients still favor transplant, but that might change. It’s not quite there yet that an LVAD is equal to a transplant for the long term. It also requires getting rid of the drive line. That would be a game changer. It would also help if we could reduce gastrointestinal bleeds, but that is more of a nuisance."
Part of the bleeding problem comes from anticoagulation treatment to prevent clots from forming in the LVAD, but another facet is an acquired Von Willebrand factor deficiency caused by absent pulsatile blood flow, Dr. Bailey said. Early clinical results suggest that allowing the heart to beat every few seconds, by adjusting the LVAD’s continuous flow rate, can minimize the acquired deficiency.
Which INTERMACS Level?
The most widely used gauge physicians and surgeons have for assessing LVAD candidates is their level in a seven-step sequence created by INTERMACS (the Interagency Registry for Mechanically Assisted Circulatory Support), which subdivides a patient’s descent from advanced New York Heart Association class III heart failure through the strata of class IV disease. (See table.) The most severe INTERMACS stage, level 1 patients (also known as "crash and burn") are those in cardiogenic shock. During the past year or two, about 16% of U.S. LVAD recipients have been level 1 patients, a percentage that should ideally drop much closer to zero, experts say.
But beyond the maxim that an advanced heart failure patient should get an LVAD before reaching level 1, opinions vary on the best target.
"Everyone agrees that inotrope-dependent patients [profile 2 and 3] should get an LVAD," said Dr. Lanfear. "But you definitely should not wait until INTERMACS 1 or 2. Everyone tries hard to treat level 3 and 4 patients. Level 5 is controversial. We need more data about these clearly less- sick patients."
"INTERMACS 2 and 3 is the sweet spot. Patients in INTERMACS 4, 5, or 6 are not as motivated to be connected to a device and undergo big open heart surgery," said Dr. Bailey.
"When patients are on the cusp of inotrope dependence [before they reach level 3], I start to think about implanting," said Dr. Jeffrey J. Teuteberg, a cardiologist and associate director of the cardiac transplant program at the University of Pittsburgh. "It’s nice to get patients who are pre–inotrope dependent – profile 4, 5, or 6 – but are symptom limited and quality of life limited. For destination therapy, you want patients with function and good quality of life. Mechanical support provides more than just improved survival; it also reduces adverse events and raises quality of life."
"For level 2 patients, there is no question that an LVAD as bridge to transplant is better than continued medical treatment," said Dr. Taylor. "For level 3 patients, it’s a little trickier, but I think the majority would say that if a patient is stable and inotropic dependent, use an LVAD unless you believe a transplant will occur quickly. An LVAD would reduce the risk for developing cardiogenic shock. The patient who does the best with an LVAD is the one who is [relatively] healthy when implanted, but the patient who needs it most is the one who is literally dying."
"Everyone knows that if you take patients who are sicker [for LVADs], you’ll have trouble reaching 80% survival after 2 years. But there is not yet enough confidence in the treatment to extend it to INTERMACS level 4, 5, or 6 patients," said Dr. Kirklin. "That’s where the big potential is. The action currently is in INTERMACS 2 and 3. For patients who are INTERMACS 3, there is no question that if they can’t get a transplant they need a VAD. The sweet spot will be patients at INTERMACS 4."
The INTERMACS registry numbers show that the field is currently stuck when it comes to INTERMACS levels, with the greatest number of LVADs going into level 2 patients, who received 34% of U.S LVADS during January-March 2012. The 34% level in early 2012 was down from 38% of LVAD recipients in 2011 and 47% in 2010, but growth in less-severe levels has been slow. At the start of 2012, 30% of LVAD recipients were at level 3, a small increase from the 27% rate in 2011 and 2010. Level 4 patients constituted 14% of LVAD recipients in early 2012, essentially unchanged from the prior 2 years, and level 5, 6, or 7 patients have consistently been a small slice of the U.S. LVAD pie, roughly 5% of recipients each year.
Beyond the INTERMACS Level
Heart failure specialists now recognize that INTERMACS level tells just part of the story.
"INTERMACS profiles depend on heart failure symptoms but not comorbidities. Severe diabetes, obstructive pulmonary disease, cardiorenal syndrome, chronic malnourishment, morbid obesity, and other factors all fall outside the INTERMACS profile criteria," said Dr. Kirklin.
He and his associates are formulating a risk assessment equation that will take comorbidities into account for a more global patient assessment. The most recently published INTERMACS registry analysis, which he first authored using data through the end of June 2011 with a total of 4,366 patients who received left ventricular support since 2006 (J. Heart Lung Transplant 2012;31:117-26), identified several comorbidity markers that each significantly linked with increased mortality. For example, a 1-unit increase in bilirubin linked with a 10% boost in mortality, a 1-unit increase in creatinine raised mortality by 16%, and a 0.5-unit increase in body surface area linked with a 48% rise in deaths. One of the strongest risk factors was being at INTERMACS level 1, which linked with a more-than-threefold higher mortality rate.
But more work must be done before the risk assessment formula is ready for clinical use. Right now, the formula "is not very reliable yet, because the maximum patient follow-up is 2 years. We need a little more follow-up," Dr. Kirklin said.
Growing the LVAD Numbers
Further growth in LVAD placements will happen on two fronts: broader use in patients at INTERMACS levels 2 and 3, and possibly level 4, where a strong consensus exists for LVAD support; and new evidence to document efficacy and safety in patients with less-severe disease at INTERMACS levels 4, 5, 6, and 7.
For the existing population, it’s a matter of physician and patient awareness. "We need to educate physicians that this is an option," said Dr. Lanfear. "Awareness is lacking. There is a lot of heart failure out there that is underrecognized and underreferred. Neither patients nor their physicians realize how sick they are, and that they are at a high enough risk to justify this."
Furthermore, there are parts of the country where the technology is underused. "In areas without a big center nearby, some physicians may not recognize patients who are sick enough to be LVAD candidates. We are trying to spread the word on who these patients are and when they should be referred."
According to Dr. Teuteberg, major warning signs that should flag patients who are potential LVAD candidates include advanced symptoms, increasing numbers of hospitalizations for heart failure, dwindling responses to ACE inhibitors and beta-blockers, increasing dosages of diuretics, a persistently high serum level of brain natriuretic peptide (BNP) despite good medical treatment, and lack of response to cardiac resynchronization therapy.
Expansion of the evidence base to show LVAD benefits in patients with INTERMACS level 5, 6, or 7 disease depends on a trial just starting, the REVIVE-IT (Randomized Evaluation of VAD Intervention Before Inotropic Therapy) study that’s set to enroll about 100 patients. But at press time, REVIVE-IT had not yet begun, posing doubts about LVAD use in the study’s targeted patients. "We haven’t really demonstrated reproducibly good survival [with LVADs] to compete with medical therapy in level 5 and 6 patients," said Dr. Kirklin. "The FDA put the study on hold while they reflected on that."
Making LVADs Better
With fast-paced technological advancement, continuous-flow LVADs will continue to evolve and improve. In June, results appeared on a new continuous-flow LVAD, the HeartWare device (Circulation 2012;125:3191-200), and last April an FDA advisory committee recommended that the agency approve the HeartWare LVAD for use as a bridge to (a trial testing the HeartWare LVAD for destination therapy is ongoing).
But no one interviewed for this article anticipates that the HeartWare LVAD will be a major advance. "Fundamentally, the major components and the implant technique are the same for the two devices," the HeartWare and the HeartMate II, said Dr. Slaughter. The HeartWare LVAD is smaller and designed to be placed completely in the pericardial space, but any clinical advantages based on these differences remain to be proved, he said in an interview.
A more meaningful improvement in LVAD design is in the works, and may reach initial clinical testing within a couple of years: a fully implantable LVAD with no transcutaneous drive line, a part that is subject to infection, prevents patients from submerging, and physically and psychologically limits patients by tethering them to equipment. "If there were one thing that could make a dramatic difference, it would be getting rid of the drive line. That is the Holy Grail for the field," said Dr. Bailey.
When a fully implantable LVAD becomes available for routine use, it will complete the LVAD revolution and help device therapy for advanced heart failure reach its full potential.
Dr. Lanfear has received research support and has received honoraria as a speaker for Thoratec, the company that markets the HeartMate II, and has received research support from HeartWare, the company developing the HeartWare LVAD. Dr. Slaughter has had contracts for services to Thoratec and HeartWare. Dr. Kirklin, Dr. Bailey, Dr. Teuteberg, and Dr. Taylor said that they had no disclosures.
*CORRECTION 8/10/12: The credit for the photo with the caption "The next LVAD in line, HeartWare, is smaller and dwells in the pericardial space" was misstated and should have been ©2012 HeartWare International, Inc.
A sea change in management of severe heart failure began two and a half years ago, in January 2010, when the Food and Drug Administration approved U.S. marketing of the HeartMate II continuous-flow, left ventricular assist device for destination therapy. Patients, cardiologists, and cardiothoracic surgeons at last had a reasonably durable, effective, and relatively safe alternative to heart transplant to offer patients at the end stage of deteriorating heart function.
The Heart Mate II quickly supplanted the prior-generation, pulsatile-flow devices, and 2010 also saw a 10-fold spike in the number of left ventricular assist devices (LVADs) placed in U.S. patients as destination therapy.
During the first half of 2011, 6-month survival among U.S. patients with a continuous-flow LVAD (which means the HeartMate II, the only continuous-flow device on the U.S. market) was 89%, and 12-month survival during 2010 was 81% (J. Heart Lung Transplant 2012;31:117-26, putting the continuous-flow LVAD in at least the same ballpark as heart transplant, which has a 5-year survival of about 80% and a median survival of about 10 years in U.S. patients.
But despite estimates that many thousands, if not tens of thousands, of Americans meet the clinical criteria to qualify for placement of an LVAD, the reality is that in the 30 months since the HeartMate II became available for routine destination therapy through mid-2012, only about 4,600 were placed in U.S. patients. In addition, just slightly more than a third of those patients, roughly 1,700, received their LVAD explicitly for destination therapy.
Even in 2012, a majority of patients who received an LVAD got their device with the understanding that it was as a bridge to transplant, an LVAD placed with temporary intent to help patients survive and improve clinically until their LVAD could be swapped out and replaced by a transplanted heart.
LVAD Numbers Up, but Still Low
If the availability of the HeartMate II for destination therapy in routine practice was a revolution for the management of many patients with New York Heart Association class IV heart failure – which is how many experts in this field view the device – it has so far been a revolution played out in slow motion. In large part that’s because the field started small, and stayed small until recently. At the end of 2008, fewer than 1,000 patients had ever received a ventricular assist device or a total artificial heart. Even though the number nearly doubled in 2009, by decade’s end the cumulative total still remained under 2,000.
Despite the low numbers compared with the estimated need, many physicians and surgeons who specialize in advanced heart failure say they are satisfied with the pace at which continuous-flow LVADs have entered patients over the past 2 years. Though they acknowledge that the number of potential U.S. candidates for an LVAD undoubtedly is far larger than the nearly 4,600 who received one since early 2010, they note that the implantation rate grew in a steady and robust way during the past 30 months, as has the number of centers performing the surgery. With a better-than-50% year-over-year growth spurt in both 2010 and 2011, and with that pace continuing into the first months of 2012, the 140 U.S. centers that now place LVADs are on track to do nearly 3,000 this year (although only 42% were for destination therapy during January-March 2012).
"You probably don’t want it to increase too rapidly, because it is still high risk and expensive," said Dr. David E. Lanfear, a cardiologist specializing in advanced heart failure and transplantation at Henry Ford Hospital in Detroit. LVAD placements "have not reached the maximum number of patients who they could help, but you need to be cautious because it requires specialized expertise to do it properly. I wouldn’t expect that [during 2 years] it would immediately reach maximum use. It’s still in the growth phase. There are a lot of patients out there, so it will continue to grow; I’m not concerned that it’s not growing fast enough," he said in an interview.
An LVAD or a Heart Transplant?
Experts also note an important shift in attitude about the role for LVADs, a change that has brought them to the brink of replacing transplanted hearts as the default treatment for patients with end-stage heart failure.
"The current outcomes with HeartMate II have already changed the field. We published a paper last year that showed there was no difference between LVADs and transplant in costs and outcomes at 1 year (Ann. Thorac. Surg. 2011;91:1330-4)," said Dr. Mark S. Slaughter, professor of surgery and chief of thoracic and cardiovascular surgery at the University of Louisville (Ky.). Between that and the competitive mortality benefit from LVADs "we need to rethink the role of transplant. In appropriately selected patients, we can achieve outstanding long-term results with limited adverse events."
In their 2011 paper, Dr. Slaughter and his associates put the pending change more bluntly: "As outcomes for continuous-flow LVADs and heart transplantation converge, the therapy of heart transplantation could emerge as salvage therapy for major device-related complications or dysfunction or progressive right heart failure as opposed to the default option for all patients who are eligible for transplant."
Despite their huge promise, some experts have lingering concerns about the current generation of continuous-flow LVADs that make them reluctant to say that heart transplant has unquestionably stepped down from its pedestal as the gold standard treatment for patients with severe, end-stage heart failure.
"There is no question that [current continuous-flow LVADs] are very good technology. But whether they are great technology remains to be proven," said Dr. James K. Kirklin, professor of surgery and director of the division of cardiothoracic surgery at the University of Alabama at Birmingham. "We still have important concerns about things like drive-line infections and thromboembolic complications. Neither rate is terribly high, but they are high enough to be a problem if you apply LVADs to patients who are doing okay. We still have important doubts about whether we can really mimic the quality of life" achieved by transplant, he said in an interview.
LVAD placement also poses an operative risk, which may be as high as 10%, said Dr. David Taylor, a cardiologist and advanced heart failure specialist at the Cleveland Clinic. "If the risk was 2% or 3%, I’d say do it, but a risk closer to 10% makes you hesitate.
"There are a lot of really sick, elderly patients with heart failure who could unquestionably benefit from an LVAD. But as soon as you push the envelope [by treating sicker patients] you increase mortality. A large group of heart failure patients are very ill with chronic disease and feel terrible, and these are the patients where you’d see the greatest benefit, but we can’t afford to do that. We can’t afford to allocate this resource to patients with a 30% mortality risk, because that would limit our ability to extend it to other patients," Dr. Kirklin said in an interview.
"Most patients appreciate an LVAD and like it, but they still look forward to getting a transplant," said Dr. Stephen H. Bailey, director of cardiothoracic surgery at Allegheny General Hospital in Pittsburgh. "In July 2012, while the margin between LVAD and transplant is narrowing, transplant is still the gold standard for definitive treatment. Most patients still favor transplant, but that might change. It’s not quite there yet that an LVAD is equal to a transplant for the long term. It also requires getting rid of the drive line. That would be a game changer. It would also help if we could reduce gastrointestinal bleeds, but that is more of a nuisance."
Part of the bleeding problem comes from anticoagulation treatment to prevent clots from forming in the LVAD, but another facet is an acquired Von Willebrand factor deficiency caused by absent pulsatile blood flow, Dr. Bailey said. Early clinical results suggest that allowing the heart to beat every few seconds, by adjusting the LVAD’s continuous flow rate, can minimize the acquired deficiency.
Which INTERMACS Level?
The most widely used gauge physicians and surgeons have for assessing LVAD candidates is their level in a seven-step sequence created by INTERMACS (the Interagency Registry for Mechanically Assisted Circulatory Support), which subdivides a patient’s descent from advanced New York Heart Association class III heart failure through the strata of class IV disease. (See table.) The most severe INTERMACS stage, level 1 patients (also known as "crash and burn") are those in cardiogenic shock. During the past year or two, about 16% of U.S. LVAD recipients have been level 1 patients, a percentage that should ideally drop much closer to zero, experts say.
But beyond the maxim that an advanced heart failure patient should get an LVAD before reaching level 1, opinions vary on the best target.
"Everyone agrees that inotrope-dependent patients [profile 2 and 3] should get an LVAD," said Dr. Lanfear. "But you definitely should not wait until INTERMACS 1 or 2. Everyone tries hard to treat level 3 and 4 patients. Level 5 is controversial. We need more data about these clearly less- sick patients."
"INTERMACS 2 and 3 is the sweet spot. Patients in INTERMACS 4, 5, or 6 are not as motivated to be connected to a device and undergo big open heart surgery," said Dr. Bailey.
"When patients are on the cusp of inotrope dependence [before they reach level 3], I start to think about implanting," said Dr. Jeffrey J. Teuteberg, a cardiologist and associate director of the cardiac transplant program at the University of Pittsburgh. "It’s nice to get patients who are pre–inotrope dependent – profile 4, 5, or 6 – but are symptom limited and quality of life limited. For destination therapy, you want patients with function and good quality of life. Mechanical support provides more than just improved survival; it also reduces adverse events and raises quality of life."
"For level 2 patients, there is no question that an LVAD as bridge to transplant is better than continued medical treatment," said Dr. Taylor. "For level 3 patients, it’s a little trickier, but I think the majority would say that if a patient is stable and inotropic dependent, use an LVAD unless you believe a transplant will occur quickly. An LVAD would reduce the risk for developing cardiogenic shock. The patient who does the best with an LVAD is the one who is [relatively] healthy when implanted, but the patient who needs it most is the one who is literally dying."
"Everyone knows that if you take patients who are sicker [for LVADs], you’ll have trouble reaching 80% survival after 2 years. But there is not yet enough confidence in the treatment to extend it to INTERMACS level 4, 5, or 6 patients," said Dr. Kirklin. "That’s where the big potential is. The action currently is in INTERMACS 2 and 3. For patients who are INTERMACS 3, there is no question that if they can’t get a transplant they need a VAD. The sweet spot will be patients at INTERMACS 4."
The INTERMACS registry numbers show that the field is currently stuck when it comes to INTERMACS levels, with the greatest number of LVADs going into level 2 patients, who received 34% of U.S LVADS during January-March 2012. The 34% level in early 2012 was down from 38% of LVAD recipients in 2011 and 47% in 2010, but growth in less-severe levels has been slow. At the start of 2012, 30% of LVAD recipients were at level 3, a small increase from the 27% rate in 2011 and 2010. Level 4 patients constituted 14% of LVAD recipients in early 2012, essentially unchanged from the prior 2 years, and level 5, 6, or 7 patients have consistently been a small slice of the U.S. LVAD pie, roughly 5% of recipients each year.
Beyond the INTERMACS Level
Heart failure specialists now recognize that INTERMACS level tells just part of the story.
"INTERMACS profiles depend on heart failure symptoms but not comorbidities. Severe diabetes, obstructive pulmonary disease, cardiorenal syndrome, chronic malnourishment, morbid obesity, and other factors all fall outside the INTERMACS profile criteria," said Dr. Kirklin.
He and his associates are formulating a risk assessment equation that will take comorbidities into account for a more global patient assessment. The most recently published INTERMACS registry analysis, which he first authored using data through the end of June 2011 with a total of 4,366 patients who received left ventricular support since 2006 (J. Heart Lung Transplant 2012;31:117-26), identified several comorbidity markers that each significantly linked with increased mortality. For example, a 1-unit increase in bilirubin linked with a 10% boost in mortality, a 1-unit increase in creatinine raised mortality by 16%, and a 0.5-unit increase in body surface area linked with a 48% rise in deaths. One of the strongest risk factors was being at INTERMACS level 1, which linked with a more-than-threefold higher mortality rate.
But more work must be done before the risk assessment formula is ready for clinical use. Right now, the formula "is not very reliable yet, because the maximum patient follow-up is 2 years. We need a little more follow-up," Dr. Kirklin said.
Growing the LVAD Numbers
Further growth in LVAD placements will happen on two fronts: broader use in patients at INTERMACS levels 2 and 3, and possibly level 4, where a strong consensus exists for LVAD support; and new evidence to document efficacy and safety in patients with less-severe disease at INTERMACS levels 4, 5, 6, and 7.
For the existing population, it’s a matter of physician and patient awareness. "We need to educate physicians that this is an option," said Dr. Lanfear. "Awareness is lacking. There is a lot of heart failure out there that is underrecognized and underreferred. Neither patients nor their physicians realize how sick they are, and that they are at a high enough risk to justify this."
Furthermore, there are parts of the country where the technology is underused. "In areas without a big center nearby, some physicians may not recognize patients who are sick enough to be LVAD candidates. We are trying to spread the word on who these patients are and when they should be referred."
According to Dr. Teuteberg, major warning signs that should flag patients who are potential LVAD candidates include advanced symptoms, increasing numbers of hospitalizations for heart failure, dwindling responses to ACE inhibitors and beta-blockers, increasing dosages of diuretics, a persistently high serum level of brain natriuretic peptide (BNP) despite good medical treatment, and lack of response to cardiac resynchronization therapy.
Expansion of the evidence base to show LVAD benefits in patients with INTERMACS level 5, 6, or 7 disease depends on a trial just starting, the REVIVE-IT (Randomized Evaluation of VAD Intervention Before Inotropic Therapy) study that’s set to enroll about 100 patients. But at press time, REVIVE-IT had not yet begun, posing doubts about LVAD use in the study’s targeted patients. "We haven’t really demonstrated reproducibly good survival [with LVADs] to compete with medical therapy in level 5 and 6 patients," said Dr. Kirklin. "The FDA put the study on hold while they reflected on that."
Making LVADs Better
With fast-paced technological advancement, continuous-flow LVADs will continue to evolve and improve. In June, results appeared on a new continuous-flow LVAD, the HeartWare device (Circulation 2012;125:3191-200), and last April an FDA advisory committee recommended that the agency approve the HeartWare LVAD for use as a bridge to (a trial testing the HeartWare LVAD for destination therapy is ongoing).
But no one interviewed for this article anticipates that the HeartWare LVAD will be a major advance. "Fundamentally, the major components and the implant technique are the same for the two devices," the HeartWare and the HeartMate II, said Dr. Slaughter. The HeartWare LVAD is smaller and designed to be placed completely in the pericardial space, but any clinical advantages based on these differences remain to be proved, he said in an interview.
A more meaningful improvement in LVAD design is in the works, and may reach initial clinical testing within a couple of years: a fully implantable LVAD with no transcutaneous drive line, a part that is subject to infection, prevents patients from submerging, and physically and psychologically limits patients by tethering them to equipment. "If there were one thing that could make a dramatic difference, it would be getting rid of the drive line. That is the Holy Grail for the field," said Dr. Bailey.
When a fully implantable LVAD becomes available for routine use, it will complete the LVAD revolution and help device therapy for advanced heart failure reach its full potential.
Dr. Lanfear has received research support and has received honoraria as a speaker for Thoratec, the company that markets the HeartMate II, and has received research support from HeartWare, the company developing the HeartWare LVAD. Dr. Slaughter has had contracts for services to Thoratec and HeartWare. Dr. Kirklin, Dr. Bailey, Dr. Teuteberg, and Dr. Taylor said that they had no disclosures.
*CORRECTION 8/10/12: The credit for the photo with the caption "The next LVAD in line, HeartWare, is smaller and dwells in the pericardial space" was misstated and should have been ©2012 HeartWare International, Inc.
Forgoing Defibrillation Testing at ICD Insertion Found to Be Safe
Patients who did not undergo defibrillation testing during the insertion of their first implantable cardioverter defibrillator had outcomes similar to those who did undergo defibrillation testing of the device in the largest study to date comparing the two approaches, which was reported online August 1 in the Journal of the American College of Cardiology.

This finding supports a strategy of omitting defibrillation testing in most such patients – a strategy that clinicians are already adopting in increasing numbers, said Dr. Michele Brignole, chief of cardiology at Ospedale del Tigullio, Lavagna, Italy, and his associates.
Defibrillation testing has been considered a standard procedure at ICD insertion "to ensure adequate sensing of ventricular fibrillation, appropriate connection of high-voltage electrodes, and the ability of the device to terminate VF with a shock. Nevertheless, implant techniques and technology have evolved in recent years, and deviations from this clinical practice are frequent," the investigators noted.
To assess the safety of omitting this step in the implantation process, Dr. Brignole and his colleagues performed SAFE-ICD (Safety of Two Strategies of ICD Management at Implantation), a prospective study of 2,120 consecutive procedures in adults at 41 Italian medical centers. The treating physicians were allowed either to perform or not perform defibrillation testing according to their standard practice; patients were followed for 2 years.
The frequency of performing defibrillation testing varied widely among the different medical centers, with some of them conducting the test in all patients and others doing so in no patients. Overall, 836 study subjects (39%) underwent defibrillation testing during insertion of their ICD, and the remaining 1,284 (61%) did not.
The primary end point was a composite of severe implant-related complications periprocedurally plus serious events during follow-up, such as sudden cardiac death or resuscitation after delivery of ineffective but appropriate ICD shocks.
This end point was reached in 18 patients who underwent defibrillation testing and 16 who did not. The estimated yearly incidence of this composite end point was 1.15% with defibrillation testing and 0.68% without it, a "negligible" difference.
In addition, 2-year all-cause mortality was not significantly lower for patients who underwent defibrillation testing (12.9%) than for those who did not (14.6%).
During follow-up, the devices delivered appropriate and effective shocks in a similar proportion of patients in the two study groups.
These findings indicate that "the clinical relevance of defibrillation testing is limited, thus supporting the practice of omitting [it] at implant," Dr. Brignole and his associates said (J. Am. Coll. Cardiol. 2012 Aug. 1 [doi:10.1016/j.jacc.2012.05.014]).
ICD recipients "are very well protected from sudden cardiac death irrespective of performing defibrillation testing or not," the authors said. Moreover, performing defibrillation testing is not likely to decrease the rate of sudden cardiac death to a clinically relevant degree, below the already low 1% rate observed in this study population, they added.
The strengths of this study included its large population that represented the general ICD population in Western countries, its very low (3%) dropout rate, and the use of any commercially available ICD devices.
However, the study was limited in that the unexpectedly low incidence of sudden cardiac death may have been insufficient to show a true difference between the two study groups.
The observational design of SAFE-ICD, unlike that of a randomized clinical trial, "does not allow us to draw a definitive conclusion" as to the safety of omitting defibrillation testing. But there is such a large, prospective, multicenter, randomized clinical trial – SIMPLE (Shockless Implant Evaluation), taking place now – that should provide a definitive answer, the researchers said.
This study was funded by Boston Scientific. One of Dr. Brignole’s associates is an employee of Boston Scientific, and others reported ties to Boston Scientific and Medtronic. Dr. Estes reported ties to Boston Scientific, Medtronic, and St. Jude Medical.
The study by Dr. Brignole and colleagues makes a "meaningful contribution to the collective evidence that routine determination of defibrillation thresholds has more risks than benefits for many patients at the time of initial ICD insertion," said Dr. N.A. Mark Estes III.
However, the absence of randomization resulted in confounding differences in the clinical profiles of the patients who were enrolled in the two strategies, he added. For example, patients who underwent defibrillation testing had lower rates of atrial fibrillation, New York Heart Association class III and IV heart failure, and use of diuretics and digoxin. In addition, as the authors pointed out, the lower-than-anticipated number of patients reaching the primary end point rendered the study underpowered to detect a difference between the two groups.
Thus, this observational study cannot provide definitive proof that omitting this step is clinically justified, so it is not yet time to abandon the practice. "Implanting physicians will have to decide on the basis of the best available data, their experience, and judgment whether to omit defibrillation testing selectively in low-risk patients," Dr. Estes noted.
Dr. Estes is professor of medicine at Tufts University and director of the cardiac arrhythmia center at Tufts Medical Center, Boston. He reported ties to Boston Scientific, Medtronic, and St. Jude Medical. These remarks were taken from his editorial comment accompanying Dr. Brignole’s report (J. Am. Coll. Cardiol. 2012 Aug. 1 [doi:10.1016/j.jacc.2012.05.016]).
The study by Dr. Brignole and colleagues makes a "meaningful contribution to the collective evidence that routine determination of defibrillation thresholds has more risks than benefits for many patients at the time of initial ICD insertion," said Dr. N.A. Mark Estes III.
However, the absence of randomization resulted in confounding differences in the clinical profiles of the patients who were enrolled in the two strategies, he added. For example, patients who underwent defibrillation testing had lower rates of atrial fibrillation, New York Heart Association class III and IV heart failure, and use of diuretics and digoxin. In addition, as the authors pointed out, the lower-than-anticipated number of patients reaching the primary end point rendered the study underpowered to detect a difference between the two groups.
Thus, this observational study cannot provide definitive proof that omitting this step is clinically justified, so it is not yet time to abandon the practice. "Implanting physicians will have to decide on the basis of the best available data, their experience, and judgment whether to omit defibrillation testing selectively in low-risk patients," Dr. Estes noted.
Dr. Estes is professor of medicine at Tufts University and director of the cardiac arrhythmia center at Tufts Medical Center, Boston. He reported ties to Boston Scientific, Medtronic, and St. Jude Medical. These remarks were taken from his editorial comment accompanying Dr. Brignole’s report (J. Am. Coll. Cardiol. 2012 Aug. 1 [doi:10.1016/j.jacc.2012.05.016]).
The study by Dr. Brignole and colleagues makes a "meaningful contribution to the collective evidence that routine determination of defibrillation thresholds has more risks than benefits for many patients at the time of initial ICD insertion," said Dr. N.A. Mark Estes III.
However, the absence of randomization resulted in confounding differences in the clinical profiles of the patients who were enrolled in the two strategies, he added. For example, patients who underwent defibrillation testing had lower rates of atrial fibrillation, New York Heart Association class III and IV heart failure, and use of diuretics and digoxin. In addition, as the authors pointed out, the lower-than-anticipated number of patients reaching the primary end point rendered the study underpowered to detect a difference between the two groups.
Thus, this observational study cannot provide definitive proof that omitting this step is clinically justified, so it is not yet time to abandon the practice. "Implanting physicians will have to decide on the basis of the best available data, their experience, and judgment whether to omit defibrillation testing selectively in low-risk patients," Dr. Estes noted.
Dr. Estes is professor of medicine at Tufts University and director of the cardiac arrhythmia center at Tufts Medical Center, Boston. He reported ties to Boston Scientific, Medtronic, and St. Jude Medical. These remarks were taken from his editorial comment accompanying Dr. Brignole’s report (J. Am. Coll. Cardiol. 2012 Aug. 1 [doi:10.1016/j.jacc.2012.05.016]).
Patients who did not undergo defibrillation testing during the insertion of their first implantable cardioverter defibrillator had outcomes similar to those who did undergo defibrillation testing of the device in the largest study to date comparing the two approaches, which was reported online August 1 in the Journal of the American College of Cardiology.
This finding supports a strategy of omitting defibrillation testing in most such patients – a strategy that clinicians are already adopting in increasing numbers, said Dr. Michele Brignole, chief of cardiology at Ospedale del Tigullio, Lavagna, Italy, and his associates.
Defibrillation testing has been considered a standard procedure at ICD insertion "to ensure adequate sensing of ventricular fibrillation, appropriate connection of high-voltage electrodes, and the ability of the device to terminate VF with a shock. Nevertheless, implant techniques and technology have evolved in recent years, and deviations from this clinical practice are frequent," the investigators noted.
To assess the safety of omitting this step in the implantation process, Dr. Brignole and his colleagues performed SAFE-ICD (Safety of Two Strategies of ICD Management at Implantation), a prospective study of 2,120 consecutive procedures in adults at 41 Italian medical centers. The treating physicians were allowed either to perform or not perform defibrillation testing according to their standard practice; patients were followed for 2 years.
The frequency of performing defibrillation testing varied widely among the different medical centers, with some of them conducting the test in all patients and others doing so in no patients. Overall, 836 study subjects (39%) underwent defibrillation testing during insertion of their ICD, and the remaining 1,284 (61%) did not.
The primary end point was a composite of severe implant-related complications periprocedurally plus serious events during follow-up, such as sudden cardiac death or resuscitation after delivery of ineffective but appropriate ICD shocks.
This end point was reached in 18 patients who underwent defibrillation testing and 16 who did not. The estimated yearly incidence of this composite end point was 1.15% with defibrillation testing and 0.68% without it, a "negligible" difference.
In addition, 2-year all-cause mortality was not significantly lower for patients who underwent defibrillation testing (12.9%) than for those who did not (14.6%).
During follow-up, the devices delivered appropriate and effective shocks in a similar proportion of patients in the two study groups.
These findings indicate that "the clinical relevance of defibrillation testing is limited, thus supporting the practice of omitting [it] at implant," Dr. Brignole and his associates said (J. Am. Coll. Cardiol. 2012 Aug. 1 [doi:10.1016/j.jacc.2012.05.014]).
ICD recipients "are very well protected from sudden cardiac death irrespective of performing defibrillation testing or not," the authors said. Moreover, performing defibrillation testing is not likely to decrease the rate of sudden cardiac death to a clinically relevant degree, below the already low 1% rate observed in this study population, they added.
The strengths of this study included its large population that represented the general ICD population in Western countries, its very low (3%) dropout rate, and the use of any commercially available ICD devices.
However, the study was limited in that the unexpectedly low incidence of sudden cardiac death may have been insufficient to show a true difference between the two study groups.
The observational design of SAFE-ICD, unlike that of a randomized clinical trial, "does not allow us to draw a definitive conclusion" as to the safety of omitting defibrillation testing. But there is such a large, prospective, multicenter, randomized clinical trial – SIMPLE (Shockless Implant Evaluation), taking place now – that should provide a definitive answer, the researchers said.
This study was funded by Boston Scientific. One of Dr. Brignole’s associates is an employee of Boston Scientific, and others reported ties to Boston Scientific and Medtronic. Dr. Estes reported ties to Boston Scientific, Medtronic, and St. Jude Medical.
Patients who did not undergo defibrillation testing during the insertion of their first implantable cardioverter defibrillator had outcomes similar to those who did undergo defibrillation testing of the device in the largest study to date comparing the two approaches, which was reported online August 1 in the Journal of the American College of Cardiology.
This finding supports a strategy of omitting defibrillation testing in most such patients – a strategy that clinicians are already adopting in increasing numbers, said Dr. Michele Brignole, chief of cardiology at Ospedale del Tigullio, Lavagna, Italy, and his associates.
Defibrillation testing has been considered a standard procedure at ICD insertion "to ensure adequate sensing of ventricular fibrillation, appropriate connection of high-voltage electrodes, and the ability of the device to terminate VF with a shock. Nevertheless, implant techniques and technology have evolved in recent years, and deviations from this clinical practice are frequent," the investigators noted.
To assess the safety of omitting this step in the implantation process, Dr. Brignole and his colleagues performed SAFE-ICD (Safety of Two Strategies of ICD Management at Implantation), a prospective study of 2,120 consecutive procedures in adults at 41 Italian medical centers. The treating physicians were allowed either to perform or not perform defibrillation testing according to their standard practice; patients were followed for 2 years.
The frequency of performing defibrillation testing varied widely among the different medical centers, with some of them conducting the test in all patients and others doing so in no patients. Overall, 836 study subjects (39%) underwent defibrillation testing during insertion of their ICD, and the remaining 1,284 (61%) did not.
The primary end point was a composite of severe implant-related complications periprocedurally plus serious events during follow-up, such as sudden cardiac death or resuscitation after delivery of ineffective but appropriate ICD shocks.
This end point was reached in 18 patients who underwent defibrillation testing and 16 who did not. The estimated yearly incidence of this composite end point was 1.15% with defibrillation testing and 0.68% without it, a "negligible" difference.
In addition, 2-year all-cause mortality was not significantly lower for patients who underwent defibrillation testing (12.9%) than for those who did not (14.6%).
During follow-up, the devices delivered appropriate and effective shocks in a similar proportion of patients in the two study groups.
These findings indicate that "the clinical relevance of defibrillation testing is limited, thus supporting the practice of omitting [it] at implant," Dr. Brignole and his associates said (J. Am. Coll. Cardiol. 2012 Aug. 1 [doi:10.1016/j.jacc.2012.05.014]).
ICD recipients "are very well protected from sudden cardiac death irrespective of performing defibrillation testing or not," the authors said. Moreover, performing defibrillation testing is not likely to decrease the rate of sudden cardiac death to a clinically relevant degree, below the already low 1% rate observed in this study population, they added.
The strengths of this study included its large population that represented the general ICD population in Western countries, its very low (3%) dropout rate, and the use of any commercially available ICD devices.
However, the study was limited in that the unexpectedly low incidence of sudden cardiac death may have been insufficient to show a true difference between the two study groups.
The observational design of SAFE-ICD, unlike that of a randomized clinical trial, "does not allow us to draw a definitive conclusion" as to the safety of omitting defibrillation testing. But there is such a large, prospective, multicenter, randomized clinical trial – SIMPLE (Shockless Implant Evaluation), taking place now – that should provide a definitive answer, the researchers said.
This study was funded by Boston Scientific. One of Dr. Brignole’s associates is an employee of Boston Scientific, and others reported ties to Boston Scientific and Medtronic. Dr. Estes reported ties to Boston Scientific, Medtronic, and St. Jude Medical.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY
Major Finding: The annual incidence of the primary end point – sudden cardiac death or severe complications either periprocedurally or during follow-up – was 1.15% with defibrillation testing and 0.68% without it.
Data Source: SAFE-ICD was a prospective observational study of 836 adults who underwent defibrillation testing at ICD implantation and 1,284 who did not, and who were followed for sudden cardiac death and other adverse outcomes for 2 years.
Disclosures: This study was funded by Boston Scientific. One of Dr. Brignole’s associates is an employee of Boston Scientific, and others reported ties to Boston Scientific and Medtronic.

Exercise Cuts Depressive Symptoms in Heart Failure Patients
Aerobic exercise for 90-120 minutes per week modestly reduces depressive symptoms in patients with heart failure, according to a report in the Aug. 1 issue of JAMA.
After 3 months of a supervised exercise program, patients who exercised showed a small but significant reduction in depression scores, compared with patients who received usual care. After an additional 9 months of home exercise, that difference persisted.

The difference was characterized as modest, "and the clinical significance of this small improvement is not known," said James A. Blumenthal, Ph.D., of Duke University, Durham, N.C., and his associates.
However, because the difference was consistent over a full year, it appears to be robust and "is likely to be associated with better social functioning and higher quality of life," they noted.
As many as 40% of patients with heart failure have been reported to have comorbid depression, and as many as 75% of heart failure patients have been reported to have elevated depressive symptoms. But few randomized trials have examined treatment of depression in this patient population.
HF-ACTION (Heart Failure–A Controlled Trial Investigating Outcomes of Exercise Training) found that adding exercise to usual care modestly reduced rates of death from any cause and hospitalization for any cause. Dr. Blumenthal and his colleagues reported on an ancillary study to HF-ACTION in which they further assessed the effects of exercise on depressive symptoms.
The study subjects were 2,322 patients whose HF failed to improve after at least 6 weeks of optimal therapy at 82 medical centers in the United States, Canada, and France. All the subjects had undergone exercise stress testing and had completed the Beck Depression Inventory II at baseline in 2003-2007. The median age was 59 years (range, 19-91 years).
The BDI-II is a self-reported measure of depressive symptoms with a possible score ranging from 0 (no depressive symptoms) to 63 (maximal depressive symptoms). A score of 14 or higher reflects clinically significant depressive symptoms. A total of 28% of the study subjects scored in this range at baseline.
A total of 1,164 subjects were randomly assigned to usual care (the control group) and 1,158 to usual care plus three supervised sessions per week on a treadmill or stationary cycle for 3 months. The exercise group was then given the exercise equipment of their choice to take home and was encouraged to increase to 90 min/week of exercise until they reached 1 year from baseline.
At 3 months, the mean BDI-II score was 8.95 for the exercise group and 9.70 for the control group. At 1 year, the mean scores were 8.86 and 9.54, respectively. Both of these differences are statistically significant, although small, the investigators said (JAMA 2012;308:465-74).
The results were more robust for the subgroup of 653 patients who scored 14 or higher on the BDI-II at baseline. At 3 months, the mean score in the exercise group was 16.66, compared with a mean score of 17.98 in the control group. At 1 year, the mean scores were 15.85 and 17.34, respectively.
In addition, the amount of exercise was inversely associated with the reduction in depressive symptoms. "Compared with a participant reporting no exercise, a participant reporting 90 minutes of exercise per week could be expected to have a 1.55-point lower BDI-II score at 3 months and a 1.67-point lower BDI-II score at 12 months," Dr. Blumenthal and his associates said.
Such a difference "is comparable with placebo-control trials involving patients with major depressive disorder," they added.
"We also observed that elevated depressive symptoms were associated with more than a 20% increase in risk for all-cause mortality and hospitalizations and that the increased risk was independent of antidepressant use and established risk factors in patients with heart failure, including age and disease severity.
"These data add to the evidence suggesting that elevated depressive symptoms, without necessarily meeting diagnostic criteria for major depressive disorder, are associated with increased risk for adverse clinical events," the researchers said.
The study findings also support the recent recommendation by the American Heart Association that all patients with cardiac disease be routinely assessed for depression, they added.
The study results also demonstrated that HF patients whose depression worsened over time were at particularly increased risk for hospitalization and death. This highlights the need to not only reduce depressive symptoms but also to prevent the worsening of existing depressive symptoms, Dr. Blumenthal and his colleagues said.
The study had two important limitations.
First, only about 40% of the patients assigned to exercise were fully adherent to the program and actually exercised 90-120 minutes per week. And 40%-50% of the patients in the control reported that they had actually exercised to some degree during follow-up. Such unintended crossover might have adversely affected the study findings.
Second, the study could not rule out the possibility that patients with more severe depressive symptoms were less likely to exercise, "so it is not clear whether exercise resulted in less depression or if depression resulted in less exercise," the researchers said.
Aerobic exercise for 90-120 minutes per week modestly reduces depressive symptoms in patients with heart failure, according to a report in the Aug. 1 issue of JAMA.
After 3 months of a supervised exercise program, patients who exercised showed a small but significant reduction in depression scores, compared with patients who received usual care. After an additional 9 months of home exercise, that difference persisted.
The difference was characterized as modest, "and the clinical significance of this small improvement is not known," said James A. Blumenthal, Ph.D., of Duke University, Durham, N.C., and his associates.
However, because the difference was consistent over a full year, it appears to be robust and "is likely to be associated with better social functioning and higher quality of life," they noted.
As many as 40% of patients with heart failure have been reported to have comorbid depression, and as many as 75% of heart failure patients have been reported to have elevated depressive symptoms. But few randomized trials have examined treatment of depression in this patient population.
HF-ACTION (Heart Failure–A Controlled Trial Investigating Outcomes of Exercise Training) found that adding exercise to usual care modestly reduced rates of death from any cause and hospitalization for any cause. Dr. Blumenthal and his colleagues reported on an ancillary study to HF-ACTION in which they further assessed the effects of exercise on depressive symptoms.
The study subjects were 2,322 patients whose HF failed to improve after at least 6 weeks of optimal therapy at 82 medical centers in the United States, Canada, and France. All the subjects had undergone exercise stress testing and had completed the Beck Depression Inventory II at baseline in 2003-2007. The median age was 59 years (range, 19-91 years).
The BDI-II is a self-reported measure of depressive symptoms with a possible score ranging from 0 (no depressive symptoms) to 63 (maximal depressive symptoms). A score of 14 or higher reflects clinically significant depressive symptoms. A total of 28% of the study subjects scored in this range at baseline.
A total of 1,164 subjects were randomly assigned to usual care (the control group) and 1,158 to usual care plus three supervised sessions per week on a treadmill or stationary cycle for 3 months. The exercise group was then given the exercise equipment of their choice to take home and was encouraged to increase to 90 min/week of exercise until they reached 1 year from baseline.
At 3 months, the mean BDI-II score was 8.95 for the exercise group and 9.70 for the control group. At 1 year, the mean scores were 8.86 and 9.54, respectively. Both of these differences are statistically significant, although small, the investigators said (JAMA 2012;308:465-74).
The results were more robust for the subgroup of 653 patients who scored 14 or higher on the BDI-II at baseline. At 3 months, the mean score in the exercise group was 16.66, compared with a mean score of 17.98 in the control group. At 1 year, the mean scores were 15.85 and 17.34, respectively.
In addition, the amount of exercise was inversely associated with the reduction in depressive symptoms. "Compared with a participant reporting no exercise, a participant reporting 90 minutes of exercise per week could be expected to have a 1.55-point lower BDI-II score at 3 months and a 1.67-point lower BDI-II score at 12 months," Dr. Blumenthal and his associates said.
Such a difference "is comparable with placebo-control trials involving patients with major depressive disorder," they added.
"We also observed that elevated depressive symptoms were associated with more than a 20% increase in risk for all-cause mortality and hospitalizations and that the increased risk was independent of antidepressant use and established risk factors in patients with heart failure, including age and disease severity.
"These data add to the evidence suggesting that elevated depressive symptoms, without necessarily meeting diagnostic criteria for major depressive disorder, are associated with increased risk for adverse clinical events," the researchers said.
The study findings also support the recent recommendation by the American Heart Association that all patients with cardiac disease be routinely assessed for depression, they added.
The study results also demonstrated that HF patients whose depression worsened over time were at particularly increased risk for hospitalization and death. This highlights the need to not only reduce depressive symptoms but also to prevent the worsening of existing depressive symptoms, Dr. Blumenthal and his colleagues said.
The study had two important limitations.
First, only about 40% of the patients assigned to exercise were fully adherent to the program and actually exercised 90-120 minutes per week. And 40%-50% of the patients in the control reported that they had actually exercised to some degree during follow-up. Such unintended crossover might have adversely affected the study findings.
Second, the study could not rule out the possibility that patients with more severe depressive symptoms were less likely to exercise, "so it is not clear whether exercise resulted in less depression or if depression resulted in less exercise," the researchers said.
Aerobic exercise for 90-120 minutes per week modestly reduces depressive symptoms in patients with heart failure, according to a report in the Aug. 1 issue of JAMA.
After 3 months of a supervised exercise program, patients who exercised showed a small but significant reduction in depression scores, compared with patients who received usual care. After an additional 9 months of home exercise, that difference persisted.
The difference was characterized as modest, "and the clinical significance of this small improvement is not known," said James A. Blumenthal, Ph.D., of Duke University, Durham, N.C., and his associates.
However, because the difference was consistent over a full year, it appears to be robust and "is likely to be associated with better social functioning and higher quality of life," they noted.
As many as 40% of patients with heart failure have been reported to have comorbid depression, and as many as 75% of heart failure patients have been reported to have elevated depressive symptoms. But few randomized trials have examined treatment of depression in this patient population.
HF-ACTION (Heart Failure–A Controlled Trial Investigating Outcomes of Exercise Training) found that adding exercise to usual care modestly reduced rates of death from any cause and hospitalization for any cause. Dr. Blumenthal and his colleagues reported on an ancillary study to HF-ACTION in which they further assessed the effects of exercise on depressive symptoms.
The study subjects were 2,322 patients whose HF failed to improve after at least 6 weeks of optimal therapy at 82 medical centers in the United States, Canada, and France. All the subjects had undergone exercise stress testing and had completed the Beck Depression Inventory II at baseline in 2003-2007. The median age was 59 years (range, 19-91 years).
The BDI-II is a self-reported measure of depressive symptoms with a possible score ranging from 0 (no depressive symptoms) to 63 (maximal depressive symptoms). A score of 14 or higher reflects clinically significant depressive symptoms. A total of 28% of the study subjects scored in this range at baseline.
A total of 1,164 subjects were randomly assigned to usual care (the control group) and 1,158 to usual care plus three supervised sessions per week on a treadmill or stationary cycle for 3 months. The exercise group was then given the exercise equipment of their choice to take home and was encouraged to increase to 90 min/week of exercise until they reached 1 year from baseline.
At 3 months, the mean BDI-II score was 8.95 for the exercise group and 9.70 for the control group. At 1 year, the mean scores were 8.86 and 9.54, respectively. Both of these differences are statistically significant, although small, the investigators said (JAMA 2012;308:465-74).
The results were more robust for the subgroup of 653 patients who scored 14 or higher on the BDI-II at baseline. At 3 months, the mean score in the exercise group was 16.66, compared with a mean score of 17.98 in the control group. At 1 year, the mean scores were 15.85 and 17.34, respectively.
In addition, the amount of exercise was inversely associated with the reduction in depressive symptoms. "Compared with a participant reporting no exercise, a participant reporting 90 minutes of exercise per week could be expected to have a 1.55-point lower BDI-II score at 3 months and a 1.67-point lower BDI-II score at 12 months," Dr. Blumenthal and his associates said.
Such a difference "is comparable with placebo-control trials involving patients with major depressive disorder," they added.
"We also observed that elevated depressive symptoms were associated with more than a 20% increase in risk for all-cause mortality and hospitalizations and that the increased risk was independent of antidepressant use and established risk factors in patients with heart failure, including age and disease severity.
"These data add to the evidence suggesting that elevated depressive symptoms, without necessarily meeting diagnostic criteria for major depressive disorder, are associated with increased risk for adverse clinical events," the researchers said.
The study findings also support the recent recommendation by the American Heart Association that all patients with cardiac disease be routinely assessed for depression, they added.
The study results also demonstrated that HF patients whose depression worsened over time were at particularly increased risk for hospitalization and death. This highlights the need to not only reduce depressive symptoms but also to prevent the worsening of existing depressive symptoms, Dr. Blumenthal and his colleagues said.
The study had two important limitations.
First, only about 40% of the patients assigned to exercise were fully adherent to the program and actually exercised 90-120 minutes per week. And 40%-50% of the patients in the control reported that they had actually exercised to some degree during follow-up. Such unintended crossover might have adversely affected the study findings.
Second, the study could not rule out the possibility that patients with more severe depressive symptoms were less likely to exercise, "so it is not clear whether exercise resulted in less depression or if depression resulted in less exercise," the researchers said.
FROM JAMA
Major Finding: Mean scores on the BDI-II were modestly but significantly lower in 1,158 heart failure patients who exercised on a treadmill or stationary bike for 90 min/week than for 1,164 controls.
Data Source: An ancillary study of the HF-ACTION randomized clinical trial involving HF patients at 82 medical centers who were followed at 3 and 12 months.
Disclosures: This study was funded by the National Heart, Lung, and Blood Institute. Dr. Blumenthal reported no conflicts of interest, and an associate reported ties to numerous industry sources.
