User login
First-line ambrisentan plus tadalafil halved PAH events
First-line combination therapy with ambrisentan and tadalafil cut the rate of clinical events in pulmonary arterial hypertension (PAH) by half, compared with monotherapy using either drug, in an international phase 3-4 clinical trial reported online Aug. 27 in the New England Journal of Medicine.
Ambrisentan, a selective endothelin-A-receptor antagonist, and tadalafil, a phosphodiesterase type 5 inhibitor, target different intracellular pathways known to have dysfunctional signaling in PAH, so researchers expected them to have an additive effect when combined. The study findings support the rationale of targeting multiple affected pathways early in the course of PAH, rather than following the traditional approach of sequentially adding newer agents to established background therapy, said Dr. Nazzareno Galie of the department of experimental, diagnostic, and specialty medicine, University of Bologna (Italy), and his associates.

The 4-year, industry-sponsored trial involved 500 adults treated at 120 medical centers in 14 countries for PAH with World Health Organization functional class II or III symptoms. It included patients whose disorder was idiopathic; hereditary; or associated with connective tissue disease, drugs or toxins, stable HIV infection, or repaired congenital heart defects.
The mean age of participants was 54.4 years, and 78% were women. The mean pulmonary artery pressure was 48.7 mm Hg, and mean 6-minute walk distance was 353 m at baseline. A total of 253 patients were randomly assigned to receive oral, once-daily combination therapy, 126 to receive ambrisentan plus placebo, and 121 to receive tadalafil plus placebo. They were assessed at monthly intervals during the 24-week treatment period and were allowed to continue therapy indefinitely. The mean duration of use of the study medication was 517 days. Patients were followed up a final time 1 month after taking their last dose of study medication.
The primary efficacy endpoint was the first event of clinical failure, which was a composite of death, hospitalization for worsening PAH, disease progression, or unsatisfactory long-term treatment response. Only 18% of the combination-therapy group reached this endpoint, compared with 34% of the ambrisentan group, 28% of the tadalafil group, and 31% of the pooled-monotherapy group. The hazard ratios for the primary endpoint were 0.50 for the combination therapy versus pooled monotherapy, 0.48 for combination therapy versus ambrisentan alone, and 0.53 for combination therapy versus tadalafil alone.
This treatment benefit was mainly driven by one component of the combined endpoint: The rate of hospitalization for worsening PAH was three times higher with the two monotherapies (12%) than with combination therapy (4%). Improvement in the secondary endpoints of change in N-terminal pro–brain natriuretic peptide level, the percentage of participants with a satisfactory treatment response, and change in 6-minute walk distance all significantly favored the combination therapy, Dr. Galie and his associates said (N Engl J Med. 2015 Aug 27. doi: 10.1056/NEJMoa1413687).It is important to note, however, that “despite improvements in a variety of factors with combination therapy, we found no significant difference in WHO functional class among the study groups at week 24,” they wrote.
The combination of ambrisentan and tadalafil produced more adverse effects than either monotherapy, but the rate of discontinuation of a study drug and the rate of serious adverse events were similar across the three study groups. The most frequent adverse effects were peripheral edema, headache, nasal congestion, and anemia.
The AMBITION study was funded by Gilead Sciences and GlaxoSmithKline, which designed the trial, collected and analyzed the data, and wrote the report in conjunction with the authors. Gilead Sciences, GlaxoSmithKline, and Eli Lilly provided the study drugs. Dr. Galie reported receiving grants and personal fees from GlaxoSmithKline, Actelion, Bayer, and Pfizer; his associates reported ties to numerous industry sources.
First-line combination therapy with ambrisentan and tadalafil cut the rate of clinical events in pulmonary arterial hypertension (PAH) by half, compared with monotherapy using either drug, in an international phase 3-4 clinical trial reported online Aug. 27 in the New England Journal of Medicine.
Ambrisentan, a selective endothelin-A-receptor antagonist, and tadalafil, a phosphodiesterase type 5 inhibitor, target different intracellular pathways known to have dysfunctional signaling in PAH, so researchers expected them to have an additive effect when combined. The study findings support the rationale of targeting multiple affected pathways early in the course of PAH, rather than following the traditional approach of sequentially adding newer agents to established background therapy, said Dr. Nazzareno Galie of the department of experimental, diagnostic, and specialty medicine, University of Bologna (Italy), and his associates.
The 4-year, industry-sponsored trial involved 500 adults treated at 120 medical centers in 14 countries for PAH with World Health Organization functional class II or III symptoms. It included patients whose disorder was idiopathic; hereditary; or associated with connective tissue disease, drugs or toxins, stable HIV infection, or repaired congenital heart defects.
The mean age of participants was 54.4 years, and 78% were women. The mean pulmonary artery pressure was 48.7 mm Hg, and mean 6-minute walk distance was 353 m at baseline. A total of 253 patients were randomly assigned to receive oral, once-daily combination therapy, 126 to receive ambrisentan plus placebo, and 121 to receive tadalafil plus placebo. They were assessed at monthly intervals during the 24-week treatment period and were allowed to continue therapy indefinitely. The mean duration of use of the study medication was 517 days. Patients were followed up a final time 1 month after taking their last dose of study medication.
The primary efficacy endpoint was the first event of clinical failure, which was a composite of death, hospitalization for worsening PAH, disease progression, or unsatisfactory long-term treatment response. Only 18% of the combination-therapy group reached this endpoint, compared with 34% of the ambrisentan group, 28% of the tadalafil group, and 31% of the pooled-monotherapy group. The hazard ratios for the primary endpoint were 0.50 for the combination therapy versus pooled monotherapy, 0.48 for combination therapy versus ambrisentan alone, and 0.53 for combination therapy versus tadalafil alone.
This treatment benefit was mainly driven by one component of the combined endpoint: The rate of hospitalization for worsening PAH was three times higher with the two monotherapies (12%) than with combination therapy (4%). Improvement in the secondary endpoints of change in N-terminal pro–brain natriuretic peptide level, the percentage of participants with a satisfactory treatment response, and change in 6-minute walk distance all significantly favored the combination therapy, Dr. Galie and his associates said (N Engl J Med. 2015 Aug 27. doi: 10.1056/NEJMoa1413687).It is important to note, however, that “despite improvements in a variety of factors with combination therapy, we found no significant difference in WHO functional class among the study groups at week 24,” they wrote.
The combination of ambrisentan and tadalafil produced more adverse effects than either monotherapy, but the rate of discontinuation of a study drug and the rate of serious adverse events were similar across the three study groups. The most frequent adverse effects were peripheral edema, headache, nasal congestion, and anemia.
The AMBITION study was funded by Gilead Sciences and GlaxoSmithKline, which designed the trial, collected and analyzed the data, and wrote the report in conjunction with the authors. Gilead Sciences, GlaxoSmithKline, and Eli Lilly provided the study drugs. Dr. Galie reported receiving grants and personal fees from GlaxoSmithKline, Actelion, Bayer, and Pfizer; his associates reported ties to numerous industry sources.
First-line combination therapy with ambrisentan and tadalafil cut the rate of clinical events in pulmonary arterial hypertension (PAH) by half, compared with monotherapy using either drug, in an international phase 3-4 clinical trial reported online Aug. 27 in the New England Journal of Medicine.
Ambrisentan, a selective endothelin-A-receptor antagonist, and tadalafil, a phosphodiesterase type 5 inhibitor, target different intracellular pathways known to have dysfunctional signaling in PAH, so researchers expected them to have an additive effect when combined. The study findings support the rationale of targeting multiple affected pathways early in the course of PAH, rather than following the traditional approach of sequentially adding newer agents to established background therapy, said Dr. Nazzareno Galie of the department of experimental, diagnostic, and specialty medicine, University of Bologna (Italy), and his associates.
The 4-year, industry-sponsored trial involved 500 adults treated at 120 medical centers in 14 countries for PAH with World Health Organization functional class II or III symptoms. It included patients whose disorder was idiopathic; hereditary; or associated with connective tissue disease, drugs or toxins, stable HIV infection, or repaired congenital heart defects.
The mean age of participants was 54.4 years, and 78% were women. The mean pulmonary artery pressure was 48.7 mm Hg, and mean 6-minute walk distance was 353 m at baseline. A total of 253 patients were randomly assigned to receive oral, once-daily combination therapy, 126 to receive ambrisentan plus placebo, and 121 to receive tadalafil plus placebo. They were assessed at monthly intervals during the 24-week treatment period and were allowed to continue therapy indefinitely. The mean duration of use of the study medication was 517 days. Patients were followed up a final time 1 month after taking their last dose of study medication.
The primary efficacy endpoint was the first event of clinical failure, which was a composite of death, hospitalization for worsening PAH, disease progression, or unsatisfactory long-term treatment response. Only 18% of the combination-therapy group reached this endpoint, compared with 34% of the ambrisentan group, 28% of the tadalafil group, and 31% of the pooled-monotherapy group. The hazard ratios for the primary endpoint were 0.50 for the combination therapy versus pooled monotherapy, 0.48 for combination therapy versus ambrisentan alone, and 0.53 for combination therapy versus tadalafil alone.
This treatment benefit was mainly driven by one component of the combined endpoint: The rate of hospitalization for worsening PAH was three times higher with the two monotherapies (12%) than with combination therapy (4%). Improvement in the secondary endpoints of change in N-terminal pro–brain natriuretic peptide level, the percentage of participants with a satisfactory treatment response, and change in 6-minute walk distance all significantly favored the combination therapy, Dr. Galie and his associates said (N Engl J Med. 2015 Aug 27. doi: 10.1056/NEJMoa1413687).It is important to note, however, that “despite improvements in a variety of factors with combination therapy, we found no significant difference in WHO functional class among the study groups at week 24,” they wrote.
The combination of ambrisentan and tadalafil produced more adverse effects than either monotherapy, but the rate of discontinuation of a study drug and the rate of serious adverse events were similar across the three study groups. The most frequent adverse effects were peripheral edema, headache, nasal congestion, and anemia.
The AMBITION study was funded by Gilead Sciences and GlaxoSmithKline, which designed the trial, collected and analyzed the data, and wrote the report in conjunction with the authors. Gilead Sciences, GlaxoSmithKline, and Eli Lilly provided the study drugs. Dr. Galie reported receiving grants and personal fees from GlaxoSmithKline, Actelion, Bayer, and Pfizer; his associates reported ties to numerous industry sources.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: First-line combination therapy with ambrisentan plus tadalafil cut the rate of clinical events in pulmonary arterial hypertension by half, compared with either monotherapy.
Major finding: Only 18% of the combination-therapy group reached the primary efficacy endpoint of clinical failure, compared with 34% of the ambrisentan group, 28% of the tadalafil group, and 31% of the pooled-monotherapy group.
Data source: An international, randomized, double-blind phase 3-4 clinical trial involving 500 men and women with previously untreated PAH.
Disclosures: The AMBITION study was funded by Gilead Sciences and GlaxoSmithKline, which designed the trial, collected and analyzed the data, and wrote the report in conjunction with the authors. Gilead Sciences, GlaxoSmithKline, and Eli Lilly provided the study drugs. Dr. Galie reported receiving grants and personal fees from GlaxoSmithKline, Actelion, Bayer, and Pfizer; his associates reported ties to numerous industry sources.

ICDs in the elderly
There has been a spate of recent publications dealing with the lack of implantable cardioverter-defibrillator implantations in the elderly. The most recent indicates that only 8% of patients over age 75 years who have experienced a non–ST-segment elevation myocardial infarction (NSTEMI) or STEMI receive an ICD largely due to the restriction of implantation within 40 days of the incident infarction because of the delayed repair of ventricular function after an MI and the uncertainty of ejection fraction measurements during this period. That article indicates that despite this delay, patients who receive an ICD had an improved survival over the subsequent 2 years, compared with those who did not (JAMA. 2015;313[24]:2433-40). Some of my octogenarian friends might question whether improved survival and the prevention of sudden death is a benefit at that age.
An accompanying editorial suggests that a closer follow-up of these patients is warranted in order to improve the rate of implantation in those patients who are discharged so that they do not “fall through the cracks” and are lost to follow-up (JAMA. 2015:313[24]:2429-30).
Although there has been a tapering off of the explosive use of ICDs in the United States, almost a quarter million devices were implanted between 2010 and 2011 (Heart Rhythm. 2013 Apr;10[4]:e59-65). The U.S. number is roughly four times that of Western Europe. Primary prevention was the reason for implantation in 73.8%, and half of the implantations were in patients over 65; 29% were in the 70-79 age group, and 14% were octogenarians. A history of New York Heart Association class II-IV heart failure was present in 82% and a myocardial infarction in 49%.
There are of course other reasons why the elderly may not receive an ICD. The most frequent are the presence of concomitant diseases like stroke, cancer, and chronic renal disease. However, many elderly do not wish to have their survival tied to a device that they have no control over. Some may view sudden death as an acceptable mortality outcome considering other alternatives. Certainly, adverse lifestyle changes at advanced age may be a reason for the reluctance to choose an ICD.
The application of our new technologies like ICDs, catheter-implanted aortic valves, and mini–left ventricular assist devices have been remarkably successful and have brought lifesaving interventions to thousands of patients. Their relative ease of application has led to a casualness in regard to appropriateness in patients with concomitant diseases, and particularly in the elderly.
The definition of who is elderly has changed dramatically in clinical trials from the mid–20th century when those studies excluded patients over 65. In today’s world, the definition of the elderly has become a slippery slope, as there is no age limit and the inclusion of octogenarians is not unusual. The observation that elderly postinfarction patients with decreased ejection fraction can experience improved survival needs to be evaluated in the light of important considerations of current and expected quality of life in individual patients, as well as their own mortality expectations.
Most of the studies examining implantation rates, extensively analyze the effect of comorbidity and cost benefit of implantation. They rarely deal with how quality of life of the patient and their own mortality expectation impacts on the decision for ICD implantation. Once implanted, removal of the device is often impossible and when possible, as with an ICD, raise important and difficult ethical questions for patient, family, and doctor.
For those of us who are octogenarians and treat octogenarians, these issues are first and foremost in the decision process. The decision to use these devices for the relatively short-term benefit may become an onerous burden for the very elderly whose future quality of life can become abruptly abbreviated by the aging process. Easy exodus from life by sudden death may unfortunately be prolonged by an ICD.
Dr. Goldstein, medical editor of Cardiology News, is professor of medicine at Wayne State University and division head emeritus of cardiovascular medicine at Henry Ford Hospital, both in Detroit. He is on data safety monitoring committees for the National Institutes of Health and several pharmaceutical companies.
There has been a spate of recent publications dealing with the lack of implantable cardioverter-defibrillator implantations in the elderly. The most recent indicates that only 8% of patients over age 75 years who have experienced a non–ST-segment elevation myocardial infarction (NSTEMI) or STEMI receive an ICD largely due to the restriction of implantation within 40 days of the incident infarction because of the delayed repair of ventricular function after an MI and the uncertainty of ejection fraction measurements during this period. That article indicates that despite this delay, patients who receive an ICD had an improved survival over the subsequent 2 years, compared with those who did not (JAMA. 2015;313[24]:2433-40). Some of my octogenarian friends might question whether improved survival and the prevention of sudden death is a benefit at that age.
An accompanying editorial suggests that a closer follow-up of these patients is warranted in order to improve the rate of implantation in those patients who are discharged so that they do not “fall through the cracks” and are lost to follow-up (JAMA. 2015:313[24]:2429-30).
Although there has been a tapering off of the explosive use of ICDs in the United States, almost a quarter million devices were implanted between 2010 and 2011 (Heart Rhythm. 2013 Apr;10[4]:e59-65). The U.S. number is roughly four times that of Western Europe. Primary prevention was the reason for implantation in 73.8%, and half of the implantations were in patients over 65; 29% were in the 70-79 age group, and 14% were octogenarians. A history of New York Heart Association class II-IV heart failure was present in 82% and a myocardial infarction in 49%.
There are of course other reasons why the elderly may not receive an ICD. The most frequent are the presence of concomitant diseases like stroke, cancer, and chronic renal disease. However, many elderly do not wish to have their survival tied to a device that they have no control over. Some may view sudden death as an acceptable mortality outcome considering other alternatives. Certainly, adverse lifestyle changes at advanced age may be a reason for the reluctance to choose an ICD.
The application of our new technologies like ICDs, catheter-implanted aortic valves, and mini–left ventricular assist devices have been remarkably successful and have brought lifesaving interventions to thousands of patients. Their relative ease of application has led to a casualness in regard to appropriateness in patients with concomitant diseases, and particularly in the elderly.
The definition of who is elderly has changed dramatically in clinical trials from the mid–20th century when those studies excluded patients over 65. In today’s world, the definition of the elderly has become a slippery slope, as there is no age limit and the inclusion of octogenarians is not unusual. The observation that elderly postinfarction patients with decreased ejection fraction can experience improved survival needs to be evaluated in the light of important considerations of current and expected quality of life in individual patients, as well as their own mortality expectations.
Most of the studies examining implantation rates, extensively analyze the effect of comorbidity and cost benefit of implantation. They rarely deal with how quality of life of the patient and their own mortality expectation impacts on the decision for ICD implantation. Once implanted, removal of the device is often impossible and when possible, as with an ICD, raise important and difficult ethical questions for patient, family, and doctor.
For those of us who are octogenarians and treat octogenarians, these issues are first and foremost in the decision process. The decision to use these devices for the relatively short-term benefit may become an onerous burden for the very elderly whose future quality of life can become abruptly abbreviated by the aging process. Easy exodus from life by sudden death may unfortunately be prolonged by an ICD.
Dr. Goldstein, medical editor of Cardiology News, is professor of medicine at Wayne State University and division head emeritus of cardiovascular medicine at Henry Ford Hospital, both in Detroit. He is on data safety monitoring committees for the National Institutes of Health and several pharmaceutical companies.
There has been a spate of recent publications dealing with the lack of implantable cardioverter-defibrillator implantations in the elderly. The most recent indicates that only 8% of patients over age 75 years who have experienced a non–ST-segment elevation myocardial infarction (NSTEMI) or STEMI receive an ICD largely due to the restriction of implantation within 40 days of the incident infarction because of the delayed repair of ventricular function after an MI and the uncertainty of ejection fraction measurements during this period. That article indicates that despite this delay, patients who receive an ICD had an improved survival over the subsequent 2 years, compared with those who did not (JAMA. 2015;313[24]:2433-40). Some of my octogenarian friends might question whether improved survival and the prevention of sudden death is a benefit at that age.
An accompanying editorial suggests that a closer follow-up of these patients is warranted in order to improve the rate of implantation in those patients who are discharged so that they do not “fall through the cracks” and are lost to follow-up (JAMA. 2015:313[24]:2429-30).
Although there has been a tapering off of the explosive use of ICDs in the United States, almost a quarter million devices were implanted between 2010 and 2011 (Heart Rhythm. 2013 Apr;10[4]:e59-65). The U.S. number is roughly four times that of Western Europe. Primary prevention was the reason for implantation in 73.8%, and half of the implantations were in patients over 65; 29% were in the 70-79 age group, and 14% were octogenarians. A history of New York Heart Association class II-IV heart failure was present in 82% and a myocardial infarction in 49%.
There are of course other reasons why the elderly may not receive an ICD. The most frequent are the presence of concomitant diseases like stroke, cancer, and chronic renal disease. However, many elderly do not wish to have their survival tied to a device that they have no control over. Some may view sudden death as an acceptable mortality outcome considering other alternatives. Certainly, adverse lifestyle changes at advanced age may be a reason for the reluctance to choose an ICD.
The application of our new technologies like ICDs, catheter-implanted aortic valves, and mini–left ventricular assist devices have been remarkably successful and have brought lifesaving interventions to thousands of patients. Their relative ease of application has led to a casualness in regard to appropriateness in patients with concomitant diseases, and particularly in the elderly.
The definition of who is elderly has changed dramatically in clinical trials from the mid–20th century when those studies excluded patients over 65. In today’s world, the definition of the elderly has become a slippery slope, as there is no age limit and the inclusion of octogenarians is not unusual. The observation that elderly postinfarction patients with decreased ejection fraction can experience improved survival needs to be evaluated in the light of important considerations of current and expected quality of life in individual patients, as well as their own mortality expectations.
Most of the studies examining implantation rates, extensively analyze the effect of comorbidity and cost benefit of implantation. They rarely deal with how quality of life of the patient and their own mortality expectation impacts on the decision for ICD implantation. Once implanted, removal of the device is often impossible and when possible, as with an ICD, raise important and difficult ethical questions for patient, family, and doctor.
For those of us who are octogenarians and treat octogenarians, these issues are first and foremost in the decision process. The decision to use these devices for the relatively short-term benefit may become an onerous burden for the very elderly whose future quality of life can become abruptly abbreviated by the aging process. Easy exodus from life by sudden death may unfortunately be prolonged by an ICD.
Dr. Goldstein, medical editor of Cardiology News, is professor of medicine at Wayne State University and division head emeritus of cardiovascular medicine at Henry Ford Hospital, both in Detroit. He is on data safety monitoring committees for the National Institutes of Health and several pharmaceutical companies.

U-shaped link between physical activity and heart failure
A longitudinal cohort study shows a U-shaped relationship between total physical activity and heart failure risk in men.
The 15-year study of 33,012 men, average age 60 years at baseline, showed that those who engaged in the highest levels and intensity of total physical activity had a 31% greater risk of heart failure than did those in the median activity category.

However, men who undertook the lowest levels of physical activity had up to a 69% greater risk of heart failure compared to the median activity group, according to the results, published online Aug. 12 in JACC: Heart Failure.
“The U-shaped relationship between exercise levels and the likelihood of subsequent heart failure is a unique finding and will stimulate further research in the important field of prevention,” said Dr. Christopher O’Connor, editor-in-chief of JACC: Heart Failure, who was not involved in the study.
The questionnaire-based study involved of 3,609 heart failure events, which included 3,190 first events of heart failure hospitalizations and 419 deaths from heart failure.
The study authors assigned intensity scores – defined as metabolic equivalents (MET) hours/day – to each type of physical activity, then calculated a total daily physical activity score for each individual by multiplying the intensity scores by reported duration of each activity.
Watching TV, reading, and sleep were assigned the lowest intensity MET scores, walking or bicycling were in the mid-range, and exercise was assigned the highest MET scores.
Walking or bicycling at least 20 minutes a day were associated with the greatest reductions in the risk of heart failure – 21% – compared to not walking or biking, and this type and level of activity was linked to an 8-month delay in the onset of heart failure among those who engaged in it.
The investigators also found no differences between the two groups in terms of age or education level, and the exclusion of study participants who developed heart failure in the 3 first years of follow-up did not impact results.
“When examining long-term behavior regarding walking or biking and HF [heart failure] risk, the results suggested that more recent active behavior in this PA [physical activity] domain may be more important for HF protection than past PA levels,” wrote Dr. Iffat Rahman and colleagues at the Karolinska Institute, Stockholm.
Exercising for more than 1 hour per week was linked with a 14% reduction in risk, but work occupation, household work, and physical inactivity did not affect heart failure risk (JACC Heart Fail. 2015 Aug 12. doi:10.1016/j.jchf.2015.05.006).
While previous studies have shown that lower levels of physical activity increase the risk of heart failure, the authors said this was the first to show an increase in risk among individuals who undertake very high levels of physical activity.
“It is possible that substantial increase of pumping of blood by the heart could damage the cardiac muscle fibers causing damage in the myocardium,” the authors wrote.
“Moreover, adverse cardiovascular outcomes could potentially be attributed to increased oxidative stress, arterial stiffness, and coronary artery calcification.”
A similar study looking at the relationship between physical activity and heart failure in women did not find an increased risk with very high levels of physical activity, suggesting a differential effect between men and women.
There were no conflicts of interest declared.
This study reminds us that we still know relatively little about how variations in physical activity and exercise “dose” might impact disease onset, and further information is needed about whether or not exercise and physical activity confer different levels of immunity based on the type and volume of exercise, as well as race, gender, and the presence of comorbidities.
However, for the vast majority of the patients we counsel about exercise and disease prevention, recommending the current exercise guidelines of 150 minutes or more of moderate intensity exercise per week is prudent.
Dr. Steven J. Keteyian and Dr. Clinton A. Brawner are from the division of cardiovascular medicine at Henry Ford Hospital, Detroit. These comments are taken from an editorial (JACC Heart Fail. 2015 Aug 12. doi:10.1016/j.jchf.2015.06.002). No conflicts of interest were declared.
This study reminds us that we still know relatively little about how variations in physical activity and exercise “dose” might impact disease onset, and further information is needed about whether or not exercise and physical activity confer different levels of immunity based on the type and volume of exercise, as well as race, gender, and the presence of comorbidities.
However, for the vast majority of the patients we counsel about exercise and disease prevention, recommending the current exercise guidelines of 150 minutes or more of moderate intensity exercise per week is prudent.
Dr. Steven J. Keteyian and Dr. Clinton A. Brawner are from the division of cardiovascular medicine at Henry Ford Hospital, Detroit. These comments are taken from an editorial (JACC Heart Fail. 2015 Aug 12. doi:10.1016/j.jchf.2015.06.002). No conflicts of interest were declared.
This study reminds us that we still know relatively little about how variations in physical activity and exercise “dose” might impact disease onset, and further information is needed about whether or not exercise and physical activity confer different levels of immunity based on the type and volume of exercise, as well as race, gender, and the presence of comorbidities.
However, for the vast majority of the patients we counsel about exercise and disease prevention, recommending the current exercise guidelines of 150 minutes or more of moderate intensity exercise per week is prudent.
Dr. Steven J. Keteyian and Dr. Clinton A. Brawner are from the division of cardiovascular medicine at Henry Ford Hospital, Detroit. These comments are taken from an editorial (JACC Heart Fail. 2015 Aug 12. doi:10.1016/j.jchf.2015.06.002). No conflicts of interest were declared.
A longitudinal cohort study shows a U-shaped relationship between total physical activity and heart failure risk in men.
The 15-year study of 33,012 men, average age 60 years at baseline, showed that those who engaged in the highest levels and intensity of total physical activity had a 31% greater risk of heart failure than did those in the median activity category.
However, men who undertook the lowest levels of physical activity had up to a 69% greater risk of heart failure compared to the median activity group, according to the results, published online Aug. 12 in JACC: Heart Failure.
“The U-shaped relationship between exercise levels and the likelihood of subsequent heart failure is a unique finding and will stimulate further research in the important field of prevention,” said Dr. Christopher O’Connor, editor-in-chief of JACC: Heart Failure, who was not involved in the study.
The questionnaire-based study involved of 3,609 heart failure events, which included 3,190 first events of heart failure hospitalizations and 419 deaths from heart failure.
The study authors assigned intensity scores – defined as metabolic equivalents (MET) hours/day – to each type of physical activity, then calculated a total daily physical activity score for each individual by multiplying the intensity scores by reported duration of each activity.
Watching TV, reading, and sleep were assigned the lowest intensity MET scores, walking or bicycling were in the mid-range, and exercise was assigned the highest MET scores.
Walking or bicycling at least 20 minutes a day were associated with the greatest reductions in the risk of heart failure – 21% – compared to not walking or biking, and this type and level of activity was linked to an 8-month delay in the onset of heart failure among those who engaged in it.
The investigators also found no differences between the two groups in terms of age or education level, and the exclusion of study participants who developed heart failure in the 3 first years of follow-up did not impact results.
“When examining long-term behavior regarding walking or biking and HF [heart failure] risk, the results suggested that more recent active behavior in this PA [physical activity] domain may be more important for HF protection than past PA levels,” wrote Dr. Iffat Rahman and colleagues at the Karolinska Institute, Stockholm.
Exercising for more than 1 hour per week was linked with a 14% reduction in risk, but work occupation, household work, and physical inactivity did not affect heart failure risk (JACC Heart Fail. 2015 Aug 12. doi:10.1016/j.jchf.2015.05.006).
While previous studies have shown that lower levels of physical activity increase the risk of heart failure, the authors said this was the first to show an increase in risk among individuals who undertake very high levels of physical activity.
“It is possible that substantial increase of pumping of blood by the heart could damage the cardiac muscle fibers causing damage in the myocardium,” the authors wrote.
“Moreover, adverse cardiovascular outcomes could potentially be attributed to increased oxidative stress, arterial stiffness, and coronary artery calcification.”
A similar study looking at the relationship between physical activity and heart failure in women did not find an increased risk with very high levels of physical activity, suggesting a differential effect between men and women.
There were no conflicts of interest declared.
A longitudinal cohort study shows a U-shaped relationship between total physical activity and heart failure risk in men.
The 15-year study of 33,012 men, average age 60 years at baseline, showed that those who engaged in the highest levels and intensity of total physical activity had a 31% greater risk of heart failure than did those in the median activity category.
However, men who undertook the lowest levels of physical activity had up to a 69% greater risk of heart failure compared to the median activity group, according to the results, published online Aug. 12 in JACC: Heart Failure.
“The U-shaped relationship between exercise levels and the likelihood of subsequent heart failure is a unique finding and will stimulate further research in the important field of prevention,” said Dr. Christopher O’Connor, editor-in-chief of JACC: Heart Failure, who was not involved in the study.
The questionnaire-based study involved of 3,609 heart failure events, which included 3,190 first events of heart failure hospitalizations and 419 deaths from heart failure.
The study authors assigned intensity scores – defined as metabolic equivalents (MET) hours/day – to each type of physical activity, then calculated a total daily physical activity score for each individual by multiplying the intensity scores by reported duration of each activity.
Watching TV, reading, and sleep were assigned the lowest intensity MET scores, walking or bicycling were in the mid-range, and exercise was assigned the highest MET scores.
Walking or bicycling at least 20 minutes a day were associated with the greatest reductions in the risk of heart failure – 21% – compared to not walking or biking, and this type and level of activity was linked to an 8-month delay in the onset of heart failure among those who engaged in it.
The investigators also found no differences between the two groups in terms of age or education level, and the exclusion of study participants who developed heart failure in the 3 first years of follow-up did not impact results.
“When examining long-term behavior regarding walking or biking and HF [heart failure] risk, the results suggested that more recent active behavior in this PA [physical activity] domain may be more important for HF protection than past PA levels,” wrote Dr. Iffat Rahman and colleagues at the Karolinska Institute, Stockholm.
Exercising for more than 1 hour per week was linked with a 14% reduction in risk, but work occupation, household work, and physical inactivity did not affect heart failure risk (JACC Heart Fail. 2015 Aug 12. doi:10.1016/j.jchf.2015.05.006).
While previous studies have shown that lower levels of physical activity increase the risk of heart failure, the authors said this was the first to show an increase in risk among individuals who undertake very high levels of physical activity.
“It is possible that substantial increase of pumping of blood by the heart could damage the cardiac muscle fibers causing damage in the myocardium,” the authors wrote.
“Moreover, adverse cardiovascular outcomes could potentially be attributed to increased oxidative stress, arterial stiffness, and coronary artery calcification.”
A similar study looking at the relationship between physical activity and heart failure in women did not find an increased risk with very high levels of physical activity, suggesting a differential effect between men and women.
There were no conflicts of interest declared.
FROM JACC: HEART FAILURE
Key clinical point: A study has found a U-shaped association between total physical activity and heart failure risk in men.
Major finding: Men who engaged in the highest levels and intensity of total physical activity had a 31% greater risk of heart failure than did those in the median activity category.
Data source: A longitudinal cohort study of 33,012 men.
Disclosures: There were no conflicts of interest declared.

High troponin T level doubles CVD risk
An abnormal troponin T level of 14 ng/L or higher, as measured using a high-sensitivity assay, doubles the risk of cardiovascular events and death among patients who have stable ischemic heart disease and type 2 diabetes, according to a report published online Aug. 13 in the New England Journal of Medicine.
Moreover, an increase of more than 25% in troponin T level during the course of 1 year predicts a worse outcome than do stable or decreasing troponin T levels in this patient population, significantly increasing the rates of death from cardiovascular causes, MI, or stroke. These findings “raise the possibility that serial measurements of troponin concentration may improve its prognostic value, and that persistently elevated and increasing troponin concentrations may be the best predictor of adverse outcomes,” said Dr. Brendan M. Everett of the divisions of cardiovascular medicine and preventive medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston.
These findings have particular significance because the prevalence of elevated troponin T was fully 40% in this study, noted Dr. Everett and his associates.
To examine the possible relationship between elevated troponin T and adverse cardiovascular outcomes in patients who had both stable ischemic heart disease and diabetes, the investigators performed a post hoc analysis of data gathered in the Bypass Angioplasty Revascularization Investigation in Type 2 Diabetes (BARI 2D) clinical trial, which compared outcomes between patients who underwent preventive percutaneous coronary intervention or CABG plus intensive medical therapy against those who received intensive medical therapy alone.
For their ancillary study, Dr. Everett and his colleagues focused on 2,285 of these participants who had high-sensitivity assays to measure very low cardiac troponin levels in plasma samples and who were followed for a mean of 5 years. At baseline, 897 (40%) of these patients had troponin T levels of 14 ng/L or higher, the current cutoff point for both men and women.
The 5-year incidence of the composite outcome of death from cardiovascular causes, MI, or stroke was 27.1% in patients with elevated troponin T at baseline, compared with 12.9% in those with normal troponin T. The between-groups differences in each of the individual components of this composite outcome were of similar magnitude, as were the between-group differences in the secondary outcomes of death from any cause and heart failure (N Engl J Med. 2015 Aug 13 [doi:10.1056/NEJMoa1415921]).
This association remained robust after the data were adjusted to account for numerous potentially confounding factors such as traditional CV risk factors, a history of MI, a history of heart failure, the severity of diabetes, glomerular filtration rate, ECG abnormalities, the number of coronary lesions, and the presence of an abnormal ejection fraction.
In a further analysis that divided patients into five groups according to their troponin T levels, adverse event rates were substantially higher than average only in the highest two categories: 14.0-23.0 ng/L and 23.0 ng/L and higher.
In another analysis, the investigators assessed outcomes in a subgroup of 1,984 participants who underwent troponin T testing both at baseline and 1 year later. Patients whose levels increased by more than 25% during that year – about 7% of this subgroup – showed significantly increased risk of all adverse outcomes, compared with patients whose troponin T levels either remained stable or decreased.
Prompt revascularization via percutaneous coronary intervention or coronary artery bypass surgery did not lower the risk of any adverse outcomes in patients who had elevated troponin T. It is therefore crucial that the troponin T assay, already in widespread use, not be used to justify revascularization procedures, the researchers wrote. At least on the basis of this study’s findings, such procedures appear to offer little benefit, they noted.
The National Heart, Lung, and Blood Institute, the National Institute of Diabetes and Digestive and Kidney Diseases, and Roche Diagnostics supported the study. Dr. Everett reported ties with Roche, Novartis, and Genzyme; his associates reported ties to numerous industry sources.
The findings of this study add to the accumulating data that suggest cardiac troponin testing may become routine for risk stratification across the entire spectrum of ischemic heart disease.
However, it is important to note that interpreting elevations in troponin T is a challenge in patients who have kidney impairment, because the problem may impair renal clearance of troponin. In addition, kidney disease may be a concomitant risk factor for ongoing subclinical thrombosis, which could be one of the pathological mechanisms underlying troponin elevation in patients with stable ischemic heart disease.
Chiara Melloni, M.D., and Matthew T. Roe, M.D., are at Duke Clinical Research Institute, Durham, N.C. Their financial disclosures are available at NEJM.org. Dr. Melloni and Dr. Roe made these remarks in an editorial accompanying Dr. Everett’s report (N Engl J Med. 2015 Aug 13 [doi:10.1056/NEJMe1506298]).
The findings of this study add to the accumulating data that suggest cardiac troponin testing may become routine for risk stratification across the entire spectrum of ischemic heart disease.
However, it is important to note that interpreting elevations in troponin T is a challenge in patients who have kidney impairment, because the problem may impair renal clearance of troponin. In addition, kidney disease may be a concomitant risk factor for ongoing subclinical thrombosis, which could be one of the pathological mechanisms underlying troponin elevation in patients with stable ischemic heart disease.
Chiara Melloni, M.D., and Matthew T. Roe, M.D., are at Duke Clinical Research Institute, Durham, N.C. Their financial disclosures are available at NEJM.org. Dr. Melloni and Dr. Roe made these remarks in an editorial accompanying Dr. Everett’s report (N Engl J Med. 2015 Aug 13 [doi:10.1056/NEJMe1506298]).
The findings of this study add to the accumulating data that suggest cardiac troponin testing may become routine for risk stratification across the entire spectrum of ischemic heart disease.
However, it is important to note that interpreting elevations in troponin T is a challenge in patients who have kidney impairment, because the problem may impair renal clearance of troponin. In addition, kidney disease may be a concomitant risk factor for ongoing subclinical thrombosis, which could be one of the pathological mechanisms underlying troponin elevation in patients with stable ischemic heart disease.
Chiara Melloni, M.D., and Matthew T. Roe, M.D., are at Duke Clinical Research Institute, Durham, N.C. Their financial disclosures are available at NEJM.org. Dr. Melloni and Dr. Roe made these remarks in an editorial accompanying Dr. Everett’s report (N Engl J Med. 2015 Aug 13 [doi:10.1056/NEJMe1506298]).
An abnormal troponin T level of 14 ng/L or higher, as measured using a high-sensitivity assay, doubles the risk of cardiovascular events and death among patients who have stable ischemic heart disease and type 2 diabetes, according to a report published online Aug. 13 in the New England Journal of Medicine.
Moreover, an increase of more than 25% in troponin T level during the course of 1 year predicts a worse outcome than do stable or decreasing troponin T levels in this patient population, significantly increasing the rates of death from cardiovascular causes, MI, or stroke. These findings “raise the possibility that serial measurements of troponin concentration may improve its prognostic value, and that persistently elevated and increasing troponin concentrations may be the best predictor of adverse outcomes,” said Dr. Brendan M. Everett of the divisions of cardiovascular medicine and preventive medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston.
These findings have particular significance because the prevalence of elevated troponin T was fully 40% in this study, noted Dr. Everett and his associates.
To examine the possible relationship between elevated troponin T and adverse cardiovascular outcomes in patients who had both stable ischemic heart disease and diabetes, the investigators performed a post hoc analysis of data gathered in the Bypass Angioplasty Revascularization Investigation in Type 2 Diabetes (BARI 2D) clinical trial, which compared outcomes between patients who underwent preventive percutaneous coronary intervention or CABG plus intensive medical therapy against those who received intensive medical therapy alone.
For their ancillary study, Dr. Everett and his colleagues focused on 2,285 of these participants who had high-sensitivity assays to measure very low cardiac troponin levels in plasma samples and who were followed for a mean of 5 years. At baseline, 897 (40%) of these patients had troponin T levels of 14 ng/L or higher, the current cutoff point for both men and women.
The 5-year incidence of the composite outcome of death from cardiovascular causes, MI, or stroke was 27.1% in patients with elevated troponin T at baseline, compared with 12.9% in those with normal troponin T. The between-groups differences in each of the individual components of this composite outcome were of similar magnitude, as were the between-group differences in the secondary outcomes of death from any cause and heart failure (N Engl J Med. 2015 Aug 13 [doi:10.1056/NEJMoa1415921]).
This association remained robust after the data were adjusted to account for numerous potentially confounding factors such as traditional CV risk factors, a history of MI, a history of heart failure, the severity of diabetes, glomerular filtration rate, ECG abnormalities, the number of coronary lesions, and the presence of an abnormal ejection fraction.
In a further analysis that divided patients into five groups according to their troponin T levels, adverse event rates were substantially higher than average only in the highest two categories: 14.0-23.0 ng/L and 23.0 ng/L and higher.
In another analysis, the investigators assessed outcomes in a subgroup of 1,984 participants who underwent troponin T testing both at baseline and 1 year later. Patients whose levels increased by more than 25% during that year – about 7% of this subgroup – showed significantly increased risk of all adverse outcomes, compared with patients whose troponin T levels either remained stable or decreased.
Prompt revascularization via percutaneous coronary intervention or coronary artery bypass surgery did not lower the risk of any adverse outcomes in patients who had elevated troponin T. It is therefore crucial that the troponin T assay, already in widespread use, not be used to justify revascularization procedures, the researchers wrote. At least on the basis of this study’s findings, such procedures appear to offer little benefit, they noted.
The National Heart, Lung, and Blood Institute, the National Institute of Diabetes and Digestive and Kidney Diseases, and Roche Diagnostics supported the study. Dr. Everett reported ties with Roche, Novartis, and Genzyme; his associates reported ties to numerous industry sources.
An abnormal troponin T level of 14 ng/L or higher, as measured using a high-sensitivity assay, doubles the risk of cardiovascular events and death among patients who have stable ischemic heart disease and type 2 diabetes, according to a report published online Aug. 13 in the New England Journal of Medicine.
Moreover, an increase of more than 25% in troponin T level during the course of 1 year predicts a worse outcome than do stable or decreasing troponin T levels in this patient population, significantly increasing the rates of death from cardiovascular causes, MI, or stroke. These findings “raise the possibility that serial measurements of troponin concentration may improve its prognostic value, and that persistently elevated and increasing troponin concentrations may be the best predictor of adverse outcomes,” said Dr. Brendan M. Everett of the divisions of cardiovascular medicine and preventive medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston.
These findings have particular significance because the prevalence of elevated troponin T was fully 40% in this study, noted Dr. Everett and his associates.
To examine the possible relationship between elevated troponin T and adverse cardiovascular outcomes in patients who had both stable ischemic heart disease and diabetes, the investigators performed a post hoc analysis of data gathered in the Bypass Angioplasty Revascularization Investigation in Type 2 Diabetes (BARI 2D) clinical trial, which compared outcomes between patients who underwent preventive percutaneous coronary intervention or CABG plus intensive medical therapy against those who received intensive medical therapy alone.
For their ancillary study, Dr. Everett and his colleagues focused on 2,285 of these participants who had high-sensitivity assays to measure very low cardiac troponin levels in plasma samples and who were followed for a mean of 5 years. At baseline, 897 (40%) of these patients had troponin T levels of 14 ng/L or higher, the current cutoff point for both men and women.
The 5-year incidence of the composite outcome of death from cardiovascular causes, MI, or stroke was 27.1% in patients with elevated troponin T at baseline, compared with 12.9% in those with normal troponin T. The between-groups differences in each of the individual components of this composite outcome were of similar magnitude, as were the between-group differences in the secondary outcomes of death from any cause and heart failure (N Engl J Med. 2015 Aug 13 [doi:10.1056/NEJMoa1415921]).
This association remained robust after the data were adjusted to account for numerous potentially confounding factors such as traditional CV risk factors, a history of MI, a history of heart failure, the severity of diabetes, glomerular filtration rate, ECG abnormalities, the number of coronary lesions, and the presence of an abnormal ejection fraction.
In a further analysis that divided patients into five groups according to their troponin T levels, adverse event rates were substantially higher than average only in the highest two categories: 14.0-23.0 ng/L and 23.0 ng/L and higher.
In another analysis, the investigators assessed outcomes in a subgroup of 1,984 participants who underwent troponin T testing both at baseline and 1 year later. Patients whose levels increased by more than 25% during that year – about 7% of this subgroup – showed significantly increased risk of all adverse outcomes, compared with patients whose troponin T levels either remained stable or decreased.
Prompt revascularization via percutaneous coronary intervention or coronary artery bypass surgery did not lower the risk of any adverse outcomes in patients who had elevated troponin T. It is therefore crucial that the troponin T assay, already in widespread use, not be used to justify revascularization procedures, the researchers wrote. At least on the basis of this study’s findings, such procedures appear to offer little benefit, they noted.
The National Heart, Lung, and Blood Institute, the National Institute of Diabetes and Digestive and Kidney Diseases, and Roche Diagnostics supported the study. Dr. Everett reported ties with Roche, Novartis, and Genzyme; his associates reported ties to numerous industry sources.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Troponin T levels of 14 ng/L or higher double the risk of CVD events and death in patients with stable ischemic heart disease and concomitant type 2 diabetes.
Major finding: The 5-year incidence of the composite outcome of death from cardiovascular causes, MI, or stroke was 27.1% in patients with elevated troponin T at baseline, compared with 12.9% in those with normal troponin T.
Data source: A post hoc analysis of data in the Bypass Angioplasty Revascularization Type 2 Diabetes (BARI 2D) trial of 2,285 participants who were followed for 5 years.
Disclosures: The National Heart, Lung, and Blood Institute, the National Institute of Diabetes and Digestive and Kidney Diseases, and Roche Diagnostics supported the study. Dr. Everett reported ties with Roche, Novartis, and Genzyme; his associates reported ties to numerous industry sources.
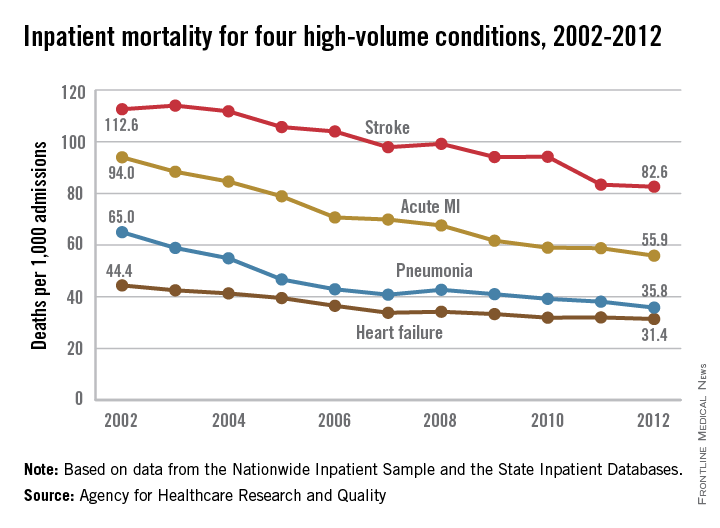
Inpatient mortality down for high-volume conditions
Inpatient mortality for pneumonia, acute MI, heart failure, and stroke each fell significantly from 2002 to 2012, the Agency for Healthcare Research and Quality reported.
Over that period, mortality among adults hospitalized with pneumonia went from 65 per 1,000 admissions to 35.8 per 1,000 for a drop of 45% – largest of the four high-volume conditions. Corresponding declines for the others were 41% for acute MI, 29% for heart failure, and 27% for stroke, the AHRQ noted.

Since “death following discharge from a hospital is not reflected in these data,” the report said, measures of inpatient mortality “can reflect both improvements in health care and shifts in where end-of-life care takes place over time.”
The estimates in the report are based on data from the Nationwide Inpatient Sample (2002-2011) and State Inpatient Databases (2012).
Inpatient mortality for pneumonia, acute MI, heart failure, and stroke each fell significantly from 2002 to 2012, the Agency for Healthcare Research and Quality reported.
Over that period, mortality among adults hospitalized with pneumonia went from 65 per 1,000 admissions to 35.8 per 1,000 for a drop of 45% – largest of the four high-volume conditions. Corresponding declines for the others were 41% for acute MI, 29% for heart failure, and 27% for stroke, the AHRQ noted.
Since “death following discharge from a hospital is not reflected in these data,” the report said, measures of inpatient mortality “can reflect both improvements in health care and shifts in where end-of-life care takes place over time.”
The estimates in the report are based on data from the Nationwide Inpatient Sample (2002-2011) and State Inpatient Databases (2012).
Inpatient mortality for pneumonia, acute MI, heart failure, and stroke each fell significantly from 2002 to 2012, the Agency for Healthcare Research and Quality reported.
Over that period, mortality among adults hospitalized with pneumonia went from 65 per 1,000 admissions to 35.8 per 1,000 for a drop of 45% – largest of the four high-volume conditions. Corresponding declines for the others were 41% for acute MI, 29% for heart failure, and 27% for stroke, the AHRQ noted.
Since “death following discharge from a hospital is not reflected in these data,” the report said, measures of inpatient mortality “can reflect both improvements in health care and shifts in where end-of-life care takes place over time.”
The estimates in the report are based on data from the Nationwide Inpatient Sample (2002-2011) and State Inpatient Databases (2012).
LVEF improvements over time in ICD recipients tied to lower mortality
In the one-quarter of heart failure patients who receive an implantable cardioverter defibrillator for primary prevention and whose left ventricular ejection fraction improves more than 35%, both mortality and appropriate ICD shocks are decreased, according to a report published online July 27 in Journal of the American College of Cardiology.
This raises the question of whether such patients’ risk for sudden cardiac death still warrants replacement of the ICD generator years later, especially among those whose devices have never needed to deliver a shock, said Yiyi Zhang, Ph.D., of the Welch Center for Prevention, Epidemiology, and Clinical Research, Johns Hopkins University, Baltimore, and her associates.
To examine this issue, the investigators analyzed data from PROSE-ICD (Prospective Observational Study of Implantable Cardioverter-Defibrillators), in which patients with systolic heart failure received primary-prevention ICDs at four U.S. cardiology centers after an initial LVEF assessment. For their study, Dr. Zhang and her associates focused on 538 of these study participants whose LVEF was reassessed at least once during roughly 5 years of follow-up.
About 57% of the study subjects were white and 70% were men. The average age at baseline was 59 years.
LVEF improved after ICD implantation in 215 (40%) of the participants, including 134 patients (25%) in whom it improved to greater than 35%. These patients were at significantly reduced risk of all-cause mortality and of requiring ICD shocks, compared with patients whose LVEF was either unchanged (47%) or decreased (13%) after ICD implantation, the investigators said. In a Cox regression model adjusted for age, sex, race, baseline LVEF, and stratified by enrollment center, the hazard ratio for all-cause mortality was 0.31, and that for an appropriate shock was 0.33 (J. Am. Coll. Cardiol. 2015 July 27 [doi:10.1016/j.jacc.2015.05.057]).
The mode of death could not be determined in many cases because records were unreliable for patients who died out of hospital, so the researchers couldn’t examine any association between LVEF changes and cardiac-specific mortality.
These study results are consistent with those of several previous studies, Dr. Zhang and her associates noted.
“Findings from our study indicate that repeated LVEF assessment after ICD implantation can provide additional prognostic information and may also allow for more informed decision making regarding ICD generator replacement, especially in patients whose LVEF improved significantly,” they said.
Further studies in larger populations that have more frequent LVEF reassessments are needed to establish whether ICD generator replacement has a positive or negative impact on this patient population, and to better guide clinicians in deciding whether ICD generator replacement should be deferred in individual patients, the investigators added.
Dr. Zhang and colleagues have conducted a meticulous analysis and made an important contribution to a critical area of patient care.
However, even though the findings were consistent with those of previous studies and even though this is the largest series of ICD recipients with improved LVEF done to date, it included only 134 such patients. These are small numbers, and the results should be interpreted with caution.
The essential question for physicians – helping patients decide if the benefit of continued ICD therapy is worth the risk – requires longer-term follow-up in a considerably larger study population.
Dr. Kristen K. Patton is in the division of cardiology at the University of Washington, Seattle. She reported having no relevant financial disclosures. Dr. Patton made these remarks in an editorial comment accompanying Dr. Zhang’s report (J. Am. Coll. Cardiol. 2015 July 27 [doi:10.1016/j.jacc.2015.06.015]).
Dr. Zhang and colleagues have conducted a meticulous analysis and made an important contribution to a critical area of patient care.
However, even though the findings were consistent with those of previous studies and even though this is the largest series of ICD recipients with improved LVEF done to date, it included only 134 such patients. These are small numbers, and the results should be interpreted with caution.
The essential question for physicians – helping patients decide if the benefit of continued ICD therapy is worth the risk – requires longer-term follow-up in a considerably larger study population.
Dr. Kristen K. Patton is in the division of cardiology at the University of Washington, Seattle. She reported having no relevant financial disclosures. Dr. Patton made these remarks in an editorial comment accompanying Dr. Zhang’s report (J. Am. Coll. Cardiol. 2015 July 27 [doi:10.1016/j.jacc.2015.06.015]).
Dr. Zhang and colleagues have conducted a meticulous analysis and made an important contribution to a critical area of patient care.
However, even though the findings were consistent with those of previous studies and even though this is the largest series of ICD recipients with improved LVEF done to date, it included only 134 such patients. These are small numbers, and the results should be interpreted with caution.
The essential question for physicians – helping patients decide if the benefit of continued ICD therapy is worth the risk – requires longer-term follow-up in a considerably larger study population.
Dr. Kristen K. Patton is in the division of cardiology at the University of Washington, Seattle. She reported having no relevant financial disclosures. Dr. Patton made these remarks in an editorial comment accompanying Dr. Zhang’s report (J. Am. Coll. Cardiol. 2015 July 27 [doi:10.1016/j.jacc.2015.06.015]).
In the one-quarter of heart failure patients who receive an implantable cardioverter defibrillator for primary prevention and whose left ventricular ejection fraction improves more than 35%, both mortality and appropriate ICD shocks are decreased, according to a report published online July 27 in Journal of the American College of Cardiology.
This raises the question of whether such patients’ risk for sudden cardiac death still warrants replacement of the ICD generator years later, especially among those whose devices have never needed to deliver a shock, said Yiyi Zhang, Ph.D., of the Welch Center for Prevention, Epidemiology, and Clinical Research, Johns Hopkins University, Baltimore, and her associates.
To examine this issue, the investigators analyzed data from PROSE-ICD (Prospective Observational Study of Implantable Cardioverter-Defibrillators), in which patients with systolic heart failure received primary-prevention ICDs at four U.S. cardiology centers after an initial LVEF assessment. For their study, Dr. Zhang and her associates focused on 538 of these study participants whose LVEF was reassessed at least once during roughly 5 years of follow-up.
About 57% of the study subjects were white and 70% were men. The average age at baseline was 59 years.
LVEF improved after ICD implantation in 215 (40%) of the participants, including 134 patients (25%) in whom it improved to greater than 35%. These patients were at significantly reduced risk of all-cause mortality and of requiring ICD shocks, compared with patients whose LVEF was either unchanged (47%) or decreased (13%) after ICD implantation, the investigators said. In a Cox regression model adjusted for age, sex, race, baseline LVEF, and stratified by enrollment center, the hazard ratio for all-cause mortality was 0.31, and that for an appropriate shock was 0.33 (J. Am. Coll. Cardiol. 2015 July 27 [doi:10.1016/j.jacc.2015.05.057]).
The mode of death could not be determined in many cases because records were unreliable for patients who died out of hospital, so the researchers couldn’t examine any association between LVEF changes and cardiac-specific mortality.
These study results are consistent with those of several previous studies, Dr. Zhang and her associates noted.
“Findings from our study indicate that repeated LVEF assessment after ICD implantation can provide additional prognostic information and may also allow for more informed decision making regarding ICD generator replacement, especially in patients whose LVEF improved significantly,” they said.
Further studies in larger populations that have more frequent LVEF reassessments are needed to establish whether ICD generator replacement has a positive or negative impact on this patient population, and to better guide clinicians in deciding whether ICD generator replacement should be deferred in individual patients, the investigators added.
In the one-quarter of heart failure patients who receive an implantable cardioverter defibrillator for primary prevention and whose left ventricular ejection fraction improves more than 35%, both mortality and appropriate ICD shocks are decreased, according to a report published online July 27 in Journal of the American College of Cardiology.
This raises the question of whether such patients’ risk for sudden cardiac death still warrants replacement of the ICD generator years later, especially among those whose devices have never needed to deliver a shock, said Yiyi Zhang, Ph.D., of the Welch Center for Prevention, Epidemiology, and Clinical Research, Johns Hopkins University, Baltimore, and her associates.
To examine this issue, the investigators analyzed data from PROSE-ICD (Prospective Observational Study of Implantable Cardioverter-Defibrillators), in which patients with systolic heart failure received primary-prevention ICDs at four U.S. cardiology centers after an initial LVEF assessment. For their study, Dr. Zhang and her associates focused on 538 of these study participants whose LVEF was reassessed at least once during roughly 5 years of follow-up.
About 57% of the study subjects were white and 70% were men. The average age at baseline was 59 years.
LVEF improved after ICD implantation in 215 (40%) of the participants, including 134 patients (25%) in whom it improved to greater than 35%. These patients were at significantly reduced risk of all-cause mortality and of requiring ICD shocks, compared with patients whose LVEF was either unchanged (47%) or decreased (13%) after ICD implantation, the investigators said. In a Cox regression model adjusted for age, sex, race, baseline LVEF, and stratified by enrollment center, the hazard ratio for all-cause mortality was 0.31, and that for an appropriate shock was 0.33 (J. Am. Coll. Cardiol. 2015 July 27 [doi:10.1016/j.jacc.2015.05.057]).
The mode of death could not be determined in many cases because records were unreliable for patients who died out of hospital, so the researchers couldn’t examine any association between LVEF changes and cardiac-specific mortality.
These study results are consistent with those of several previous studies, Dr. Zhang and her associates noted.
“Findings from our study indicate that repeated LVEF assessment after ICD implantation can provide additional prognostic information and may also allow for more informed decision making regarding ICD generator replacement, especially in patients whose LVEF improved significantly,” they said.
Further studies in larger populations that have more frequent LVEF reassessments are needed to establish whether ICD generator replacement has a positive or negative impact on this patient population, and to better guide clinicians in deciding whether ICD generator replacement should be deferred in individual patients, the investigators added.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY
Key clinical point: LVEF improves to greater than 35% in about a quarter of patients with heart failure who receive ICDs for primary prevention, and that change is associated with lower mortality and fewer inappropriate shocks.
Major finding: In HF patients whose LVEF improved after ICD implantation (40%), all-cause mortality and appropriate shocks were reduced by nearly 70%.
Data source: A secondary analysis of data from PROSE-ICD, a multicenter prospective observational study in 538 HF patients with ICDs whose LVEF was assessed at least once during 10 years of follow-up.
Disclosures: This study was supported by the Donald W. Reynolds Foundation and the National Institutes of Health. Dr. Zhang reported having no relevant financial disclosures; Dr. Zhang’s associates reported ties to Medtronic, Boston Scientific, Biotronik, and St. Jude Medical.
Patiromer cuts potassium in diabetic CKD with hyperkalemia
Patiromer, an orally administered potassium-binding polymer, significantly decreased serum potassium in adults who had diabetic kidney disease with hyperkalemia in a phase II study funded by and conducted with the manufacturer.
Patiromer consists of tiny, smooth beads in a liquid suspension and works by binding potassium throughout the GI tract, allowing it to be excreted in the feces. After preliminary studies demonstrated the agent’s usefulness in reducing hyperkalemia for up to 12 weeks in high-risk patients, researchers performed this open-label, uncontrolled trial to determine optimal dosing and to assess its longer-term safety. The findings were reported online July 14 in JAMA.
All four doses of patiromer studied significantly reduced serum potassium, beginning within 48 hours of the initial dose and continuing through all 52 weeks of treatment. The proportion of patients whose potassium levels reached target range at all scheduled visits ranged from 83% to 93% in those who had mild hyperkalemia at baseline and from 77% to 95% in those who had moderate hyperkalemia at baseline. Hyperkalemia quickly reappeared when patiromer treatment ended, reverting to baseline levels, wrote Dr. George L. Bakris of the ASH Comprehensive Hypertension Center, division of endocrinology, diabetes, and metabolism, University of Chicago, and his associates.
Study participants were 222 white patients with mild and 84 with moderate hyperkalemia (mean age, 66 years). All had type 2 diabetes and hypertension, and approximately one-third had heart failure; 65% had stage 3 and 22% had stage 4 chronic kidney disease (CKD), All were taking an ACE inhibitor, an angiotensin II receptor blocker, or both, with or without spironolactone. At 48 medical centers in five European countries, they were randomly assigned to receive one of four doses of patiromer that could be titrated up or down as needed for 4 weeks. Patients then entered an 8-week treatment phase, after which they continued maintenance therapy for a further 40 weeks (JAMA 2015 July 14 [doi:10.1001/jama.2015.7446]).
The optimal doses of patiromer were found to be the two lowest doses assessed in this study, 8.4 g daily and 16.8 g daily, which significantly decreased serum potassium without provoking hypokalemia. These doses will now be tested in a phase III study.
The most frequent treatment-related adverse events in the phase II study were nonsevere hypomagnesemia (7.2%), constipation (4.6%), and diarrhea (2.7%). A total of 28 patients (9.2%) developed at least one adverse event prompting them to discontinue patiromer, including worsening of CKD (which was considered unrelated to treatment), hypokalemia (5 patients), and one hypertensive crisis (also unrelated to treatment). No dose-related edema was noted, and there were no clinically relevant changes in serum calcium or phosphate levels.
The findings of Bakris et al. have the potential to fundamentally change the current treatment approach for hyperkalemia.
Relypsa, the manufacturer of patiromer, filed a New Drug Application with the Food and Drug Administration, and the agent will likely be approved for use in the United States by the end of October. The FDA should consider mandating a sizable postmarketing trial and safety surveillance program to clearly establish the agent’s safety and effectiveness for the hard endpoints that patients care about: halting the progression of chronic kidney disease and thus deferring dialysis, and improving heart failure. Otherwise the manufacturer may not be motivated to conduct such crucial trials, especially if it could instead spend those dollars on marketing and company-directed contract research.
Wolfgang C. Winkelmayer, M.D., Sc.D., is in the nephrology section at Baylor University, Houston, and is an associate editor at JAMA. He reported serving as an adviser or consultant to Amgen, AstraZeneca, Bayer, Keryx, Medgenics, Medtronic, Mitsubishi Tanabe, and Rockwell Pharma. Dr. Winkelmayer made these remarks in an editorial accompanying Dr. Bakris’ report (JAMA 2015 July 14;314:129-30).
The findings of Bakris et al. have the potential to fundamentally change the current treatment approach for hyperkalemia.
Relypsa, the manufacturer of patiromer, filed a New Drug Application with the Food and Drug Administration, and the agent will likely be approved for use in the United States by the end of October. The FDA should consider mandating a sizable postmarketing trial and safety surveillance program to clearly establish the agent’s safety and effectiveness for the hard endpoints that patients care about: halting the progression of chronic kidney disease and thus deferring dialysis, and improving heart failure. Otherwise the manufacturer may not be motivated to conduct such crucial trials, especially if it could instead spend those dollars on marketing and company-directed contract research.
Wolfgang C. Winkelmayer, M.D., Sc.D., is in the nephrology section at Baylor University, Houston, and is an associate editor at JAMA. He reported serving as an adviser or consultant to Amgen, AstraZeneca, Bayer, Keryx, Medgenics, Medtronic, Mitsubishi Tanabe, and Rockwell Pharma. Dr. Winkelmayer made these remarks in an editorial accompanying Dr. Bakris’ report (JAMA 2015 July 14;314:129-30).
The findings of Bakris et al. have the potential to fundamentally change the current treatment approach for hyperkalemia.
Relypsa, the manufacturer of patiromer, filed a New Drug Application with the Food and Drug Administration, and the agent will likely be approved for use in the United States by the end of October. The FDA should consider mandating a sizable postmarketing trial and safety surveillance program to clearly establish the agent’s safety and effectiveness for the hard endpoints that patients care about: halting the progression of chronic kidney disease and thus deferring dialysis, and improving heart failure. Otherwise the manufacturer may not be motivated to conduct such crucial trials, especially if it could instead spend those dollars on marketing and company-directed contract research.
Wolfgang C. Winkelmayer, M.D., Sc.D., is in the nephrology section at Baylor University, Houston, and is an associate editor at JAMA. He reported serving as an adviser or consultant to Amgen, AstraZeneca, Bayer, Keryx, Medgenics, Medtronic, Mitsubishi Tanabe, and Rockwell Pharma. Dr. Winkelmayer made these remarks in an editorial accompanying Dr. Bakris’ report (JAMA 2015 July 14;314:129-30).
Patiromer, an orally administered potassium-binding polymer, significantly decreased serum potassium in adults who had diabetic kidney disease with hyperkalemia in a phase II study funded by and conducted with the manufacturer.
Patiromer consists of tiny, smooth beads in a liquid suspension and works by binding potassium throughout the GI tract, allowing it to be excreted in the feces. After preliminary studies demonstrated the agent’s usefulness in reducing hyperkalemia for up to 12 weeks in high-risk patients, researchers performed this open-label, uncontrolled trial to determine optimal dosing and to assess its longer-term safety. The findings were reported online July 14 in JAMA.
All four doses of patiromer studied significantly reduced serum potassium, beginning within 48 hours of the initial dose and continuing through all 52 weeks of treatment. The proportion of patients whose potassium levels reached target range at all scheduled visits ranged from 83% to 93% in those who had mild hyperkalemia at baseline and from 77% to 95% in those who had moderate hyperkalemia at baseline. Hyperkalemia quickly reappeared when patiromer treatment ended, reverting to baseline levels, wrote Dr. George L. Bakris of the ASH Comprehensive Hypertension Center, division of endocrinology, diabetes, and metabolism, University of Chicago, and his associates.
Study participants were 222 white patients with mild and 84 with moderate hyperkalemia (mean age, 66 years). All had type 2 diabetes and hypertension, and approximately one-third had heart failure; 65% had stage 3 and 22% had stage 4 chronic kidney disease (CKD), All were taking an ACE inhibitor, an angiotensin II receptor blocker, or both, with or without spironolactone. At 48 medical centers in five European countries, they were randomly assigned to receive one of four doses of patiromer that could be titrated up or down as needed for 4 weeks. Patients then entered an 8-week treatment phase, after which they continued maintenance therapy for a further 40 weeks (JAMA 2015 July 14 [doi:10.1001/jama.2015.7446]).
The optimal doses of patiromer were found to be the two lowest doses assessed in this study, 8.4 g daily and 16.8 g daily, which significantly decreased serum potassium without provoking hypokalemia. These doses will now be tested in a phase III study.
The most frequent treatment-related adverse events in the phase II study were nonsevere hypomagnesemia (7.2%), constipation (4.6%), and diarrhea (2.7%). A total of 28 patients (9.2%) developed at least one adverse event prompting them to discontinue patiromer, including worsening of CKD (which was considered unrelated to treatment), hypokalemia (5 patients), and one hypertensive crisis (also unrelated to treatment). No dose-related edema was noted, and there were no clinically relevant changes in serum calcium or phosphate levels.
Patiromer, an orally administered potassium-binding polymer, significantly decreased serum potassium in adults who had diabetic kidney disease with hyperkalemia in a phase II study funded by and conducted with the manufacturer.
Patiromer consists of tiny, smooth beads in a liquid suspension and works by binding potassium throughout the GI tract, allowing it to be excreted in the feces. After preliminary studies demonstrated the agent’s usefulness in reducing hyperkalemia for up to 12 weeks in high-risk patients, researchers performed this open-label, uncontrolled trial to determine optimal dosing and to assess its longer-term safety. The findings were reported online July 14 in JAMA.
All four doses of patiromer studied significantly reduced serum potassium, beginning within 48 hours of the initial dose and continuing through all 52 weeks of treatment. The proportion of patients whose potassium levels reached target range at all scheduled visits ranged from 83% to 93% in those who had mild hyperkalemia at baseline and from 77% to 95% in those who had moderate hyperkalemia at baseline. Hyperkalemia quickly reappeared when patiromer treatment ended, reverting to baseline levels, wrote Dr. George L. Bakris of the ASH Comprehensive Hypertension Center, division of endocrinology, diabetes, and metabolism, University of Chicago, and his associates.
Study participants were 222 white patients with mild and 84 with moderate hyperkalemia (mean age, 66 years). All had type 2 diabetes and hypertension, and approximately one-third had heart failure; 65% had stage 3 and 22% had stage 4 chronic kidney disease (CKD), All were taking an ACE inhibitor, an angiotensin II receptor blocker, or both, with or without spironolactone. At 48 medical centers in five European countries, they were randomly assigned to receive one of four doses of patiromer that could be titrated up or down as needed for 4 weeks. Patients then entered an 8-week treatment phase, after which they continued maintenance therapy for a further 40 weeks (JAMA 2015 July 14 [doi:10.1001/jama.2015.7446]).
The optimal doses of patiromer were found to be the two lowest doses assessed in this study, 8.4 g daily and 16.8 g daily, which significantly decreased serum potassium without provoking hypokalemia. These doses will now be tested in a phase III study.
The most frequent treatment-related adverse events in the phase II study were nonsevere hypomagnesemia (7.2%), constipation (4.6%), and diarrhea (2.7%). A total of 28 patients (9.2%) developed at least one adverse event prompting them to discontinue patiromer, including worsening of CKD (which was considered unrelated to treatment), hypokalemia (5 patients), and one hypertensive crisis (also unrelated to treatment). No dose-related edema was noted, and there were no clinically relevant changes in serum calcium or phosphate levels.
FROM JAMA
Key clinical point: The oral potassium-binding polymer patiromer decreased serum potassium in patients who had diabetic kidney disease with hyperkalemia.
Major finding: The proportion of patients whose potassium levels remained within target range throughout 1 year of treatment was 83%-93% in those who had mild hyperkalemia at baseline and 77%-95% in those who had moderate hyperkalemia at baseline.
Data source: A multicenter open-label, noncontrolled phase II, randomized trial involving 306 adults with diabetic kidney disease and mild to moderate hyperkalemia treated for 1 year.
Disclosures: This study was funded by Relypsa, maker of patiromer. Relypsa also was involved in designing and conducting the study; collecting, analyzing, and interpreting the data; and preparing the manuscript. Dr. Bakris reported receiving personal fees from AbbVie, Takeda. Medtronic, Relypsa, Janssen, Daiichi-Sankyo, Novartis, and Bayer, as well as grants from Takeda. His associates reported ties to numerous industry sources.
Doxorubicin, radiation doses predict heart risk in lymphoma survivors
Adult lymphoma survivors who were treated with autologous hematopoietic stem-cell transplantation had a greater than sixfold increased risk of left ventricular systolic dysfunction compared with controls, according to a study published online in the Journal of Clinical Oncology.
Among 274 adult survivors of Hodgkin or non-Hodgkin lymphoma, 16% had left ventricular systolic dysfunction (LVSD): 11% had overt heart failure (HF) and 5% had asymptomatic LVSD, defined as a left ventricular ejection fraction of less than 50%.Heart symptoms were significantly associated with exposure to doxorubicin at a cumulative dose of 300 mg/m2 or more and with cardiac radiation therapy of more than 30 Gy. Recognizing these patient risk factors allows for more intensive follow-up with the goal of “identification and early treatment of asymptomatic LVSD [which] may prevent the development of HF,” wrote Dr. Klaus Murbraech of Oslo University Hospital and his colleagues (J. Clin. Oncol. 2015 July 13 [doi:10.1200/JCO.2015.60.8125]).
The investigators observed no association between lower-dose cardiac radiation therapy and LVSD. There was only a marginally significant association between the presence of two or more traditional cardiovascular disease risk factors and LVSD.
The cross-sectional multicenter cohort study is the first to assess the prevalence of LVSD, according to Dr. Murbraech and his colleagues. The study included adult survivors of Hodgkin or non-Hodgkin lymphoma, median age 56 years, who underwent autologous stem-cell transplants in Norway from 1987 to 2008. The median observation time was 13 years (range, 4-34 years). The control group consisted of initially healthy patients in an echocardiographic follow-up study. Controls were matched to patients based on age, sex, systolic blood pressure, and body mass index.
The study was supported by the South-Eastern Norway Regional Health Authority and Extrastiftelsen. Dr. Murbraech reported having no disclosures.
Adult lymphoma survivors who were treated with autologous hematopoietic stem-cell transplantation had a greater than sixfold increased risk of left ventricular systolic dysfunction compared with controls, according to a study published online in the Journal of Clinical Oncology.
Among 274 adult survivors of Hodgkin or non-Hodgkin lymphoma, 16% had left ventricular systolic dysfunction (LVSD): 11% had overt heart failure (HF) and 5% had asymptomatic LVSD, defined as a left ventricular ejection fraction of less than 50%.Heart symptoms were significantly associated with exposure to doxorubicin at a cumulative dose of 300 mg/m2 or more and with cardiac radiation therapy of more than 30 Gy. Recognizing these patient risk factors allows for more intensive follow-up with the goal of “identification and early treatment of asymptomatic LVSD [which] may prevent the development of HF,” wrote Dr. Klaus Murbraech of Oslo University Hospital and his colleagues (J. Clin. Oncol. 2015 July 13 [doi:10.1200/JCO.2015.60.8125]).
The investigators observed no association between lower-dose cardiac radiation therapy and LVSD. There was only a marginally significant association between the presence of two or more traditional cardiovascular disease risk factors and LVSD.
The cross-sectional multicenter cohort study is the first to assess the prevalence of LVSD, according to Dr. Murbraech and his colleagues. The study included adult survivors of Hodgkin or non-Hodgkin lymphoma, median age 56 years, who underwent autologous stem-cell transplants in Norway from 1987 to 2008. The median observation time was 13 years (range, 4-34 years). The control group consisted of initially healthy patients in an echocardiographic follow-up study. Controls were matched to patients based on age, sex, systolic blood pressure, and body mass index.
The study was supported by the South-Eastern Norway Regional Health Authority and Extrastiftelsen. Dr. Murbraech reported having no disclosures.
Adult lymphoma survivors who were treated with autologous hematopoietic stem-cell transplantation had a greater than sixfold increased risk of left ventricular systolic dysfunction compared with controls, according to a study published online in the Journal of Clinical Oncology.
Among 274 adult survivors of Hodgkin or non-Hodgkin lymphoma, 16% had left ventricular systolic dysfunction (LVSD): 11% had overt heart failure (HF) and 5% had asymptomatic LVSD, defined as a left ventricular ejection fraction of less than 50%.Heart symptoms were significantly associated with exposure to doxorubicin at a cumulative dose of 300 mg/m2 or more and with cardiac radiation therapy of more than 30 Gy. Recognizing these patient risk factors allows for more intensive follow-up with the goal of “identification and early treatment of asymptomatic LVSD [which] may prevent the development of HF,” wrote Dr. Klaus Murbraech of Oslo University Hospital and his colleagues (J. Clin. Oncol. 2015 July 13 [doi:10.1200/JCO.2015.60.8125]).
The investigators observed no association between lower-dose cardiac radiation therapy and LVSD. There was only a marginally significant association between the presence of two or more traditional cardiovascular disease risk factors and LVSD.
The cross-sectional multicenter cohort study is the first to assess the prevalence of LVSD, according to Dr. Murbraech and his colleagues. The study included adult survivors of Hodgkin or non-Hodgkin lymphoma, median age 56 years, who underwent autologous stem-cell transplants in Norway from 1987 to 2008. The median observation time was 13 years (range, 4-34 years). The control group consisted of initially healthy patients in an echocardiographic follow-up study. Controls were matched to patients based on age, sex, systolic blood pressure, and body mass index.
The study was supported by the South-Eastern Norway Regional Health Authority and Extrastiftelsen. Dr. Murbraech reported having no disclosures.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Lymphoma survivors treated with autologous hematopoietic stem-cell transplantation (auto-HSC) had a significantly higher risk of left ventricular systolic dysfunction than did controls.
Major finding: Treatment with at least 300 mg/m2 cumulative of doxorubicin and with over 30 Gy of cardiac radiation therapy were independent risk factors for LVSD.
Data source: A cross-sectional multicenter cohort study of 274 Hodgkin or non-Hodgkin lymphoma survivors.
Disclosures: Supported by the South-Eastern Norway Regional Health Authority and Extrastiftelsen. Dr. Murbraech reported having no disclosures.
FDA approval of sacubitril-valsartan combo opens ‘new chapter’ for heart failure
A combination of a new drug, sacubitril, a neprilysin inhibitor, and the angiotensin receptor blocker valsartan has been approved for treating heart failure, providing what experts are describing as a major advance in the treatment of heart failure.
The approval, announced by the Food and Drug Administration on July 7, was based on the results of the PARADIGM-HF study of about 8,400 patients with class II-IV heart failure and an ejection fraction of 40% or less. The study showed that the combination reduced the risk of cardiovascular death and hospitalization for heart failure by 20% and reduced the risk for all-cause mortality by 16%, compared with enalapril, with a favorable adverse event profile.
The approved indication for sacubitril-valsartan is to “reduce the risk of cardiovascular death and hospitalization for heart failure in patients with chronic heart failure [New York Heart Association class II-IV] and reduced ejection fraction,” according to the prescribing information. The indications section includes the statement that it “is usually administered in conjunction with other heart failure therapies, in place of an ACE inhibitor or other ARB [angiotensin II receptor blocker].” It is contraindicated for use with ACE inhibitors, and when patients on an ACE inhibitor are switched to this drug a washout period of 36 hours before starting treatment is recommended.
Previously called LCZ696, the combination tablet formulation will be marketed by Novartis as Entresto, and will be available in three dosage strengths of sacubitril-valsartan: 24/26 mg, 49/51 mg, and 97/103 mg; in published studies of the combination, these doses are referred to as 50 mg, 100 mg, and 200 mg, respectively. The target dose is 97/103 mg twice a day.

“Many of us see this as a real sea change in heart failure management; we’re all eager to start using this new medicine in our patients, and I think clinicians will very rapidly get a feel for this drug,” Dr. Scott D. Solomon, professor of medicine, Harvard Medical School, Boston, and one of the authors of the study, said in an interview. Referring to the trial results, he noted that for at least 10 years, there has not been a new drug for heart failure that has had this impact on mortality, “so this is really quite an extraordinary finding.”
The availability of a new drug class, a neprilysin inhibitor, “opens up a new chapter in our treatment of heart failure,” said Dr. Mariell Jessup, professor of medicine at the University of Pennsylvania, Philadelphia, who was not involved in the study. “I’m very excited that we now have this new drug for use in our patients,” she said, pointing out that with the exception of ivabradine (Corlanor), approved in April, there has not been a new type of drug approved for heart failure in a long time.

Neprilysin is an endopeptidase that degrades several endogenous vasoactive peptides. Sacubitril “inhibits the breakdown of natriuretic peptides, among other vasoactive compounds, so the drug simultaneously blocks one of the neurohormonal systems that is abnormally activated in the setting of heart failure, and it also augments one of the neurohormonal systems that can be beneficial in patients with heart failure,” said Dr. Solomon, who is also director of noninvasive cardiology at Brigham and Women’s Hospital, Boston.
In PARADIGM-HF, conducted to determine if sacubitril-valsartan was superior to treatment with an ACE inhibitor, the recognized standard treatment for heart failure, a composite of death from cardiovascular causes or a first hospitalization for heart failure (the primary outcome), was 21.8% among those randomized to the combination vs. 26.5% of those on enalapril after a median 27-month follow-up, a highly statistically significant difference that represented a 20% reduced risk over the comparator (N. Engl. J. Med. 371:993-1004). A 20% reduced risk over enalapril was seen for the two components individually in the study, which was stopped early because of the magnitude of the effect over enalapril at a dose of the ACE inhibitor that the investigators pointed out had been shown to reduce mortality, compared with placebo.
Hypotension and nonserious cases of angioedema were higher among those on sacubitril-valsartan, while renal impairment, hyperkalemia, and cough were higher in the enalapril-treated patients.
As with ACE inhibitors, there is a small risk for angioedema, and while the number of cases was low in the study, physicians need to be aware of this issue, Dr. Solomon said. “To use this effectively, patients who are on ACE inhibitors or ARBs will need to be stopped, given 36 hours to wash out, and then started on this new agent,” he said.
In an interview, Dr. Jessup said that she hopes that physicians approach this new drug with the same caution they have with other drugs for patients with systolic heart failure and encouraged clinicians to carefully read the study and familiarize themselves with the prescribing information. Patients should not be taken off an ACE inhibitor and immediately switched to Entresto, she said.
She referred to the “famous” Canadian study that identified a significant increase in spironolactone prescriptions and in the rates of hyperkalemia and hyperkalemia-associated deaths after the positive Randomized Aldactone Evaluation Study (RALES) results were published in 1999 (N. Engl. J. Med. 2004;351:543-51). After the study, which found significant improvements in morbidity and mortality in patients with severe heart failure, was published, “physicians immediately started to put their patients on spironolactone and there was a spike in hyperkalemia and in deaths,” she noted.
The Entresto label includes a boxed warning about the risk of fetal toxicity, and the FDA statement recommends that health care professionals counsel patients about the risks to an unborn baby. One of the company’s postmarketing requirements is to conduct an epidemiologic study evaluating the incidence of angioedema in black patients treated with the combination, compared with another drug, according to the FDA’s approval letter.
The PARADIGM-HF results were reported in August 2014 at the European Society of Cardiology annual meeting in Barcelona. Dr. Solomon said that a large international study evaluating Entresto in patients with heart failure and preserved ejection fraction is currently underway.
The cost per day of Entresto is $12.50 (the wholesale acquisition cost), and the company anticipates that the product will be available in most pharmacies within weeks of the approval date, according to a Novartis spokesperson.
Valsartan, approved in 2001, is marketed as Diovan by Novartis and is available in generic form from Ranbaxy.
The PARADIGM-HF trial was funded by Novartis. Dr. Solomon, a member of the executive committee for the study, has received research support from Novartis for the conduct of this and other studies, and has served as a consultant to the company. Dr. Jessup had no related disclosures.
A combination of a new drug, sacubitril, a neprilysin inhibitor, and the angiotensin receptor blocker valsartan has been approved for treating heart failure, providing what experts are describing as a major advance in the treatment of heart failure.
The approval, announced by the Food and Drug Administration on July 7, was based on the results of the PARADIGM-HF study of about 8,400 patients with class II-IV heart failure and an ejection fraction of 40% or less. The study showed that the combination reduced the risk of cardiovascular death and hospitalization for heart failure by 20% and reduced the risk for all-cause mortality by 16%, compared with enalapril, with a favorable adverse event profile.
The approved indication for sacubitril-valsartan is to “reduce the risk of cardiovascular death and hospitalization for heart failure in patients with chronic heart failure [New York Heart Association class II-IV] and reduced ejection fraction,” according to the prescribing information. The indications section includes the statement that it “is usually administered in conjunction with other heart failure therapies, in place of an ACE inhibitor or other ARB [angiotensin II receptor blocker].” It is contraindicated for use with ACE inhibitors, and when patients on an ACE inhibitor are switched to this drug a washout period of 36 hours before starting treatment is recommended.
Previously called LCZ696, the combination tablet formulation will be marketed by Novartis as Entresto, and will be available in three dosage strengths of sacubitril-valsartan: 24/26 mg, 49/51 mg, and 97/103 mg; in published studies of the combination, these doses are referred to as 50 mg, 100 mg, and 200 mg, respectively. The target dose is 97/103 mg twice a day.
“Many of us see this as a real sea change in heart failure management; we’re all eager to start using this new medicine in our patients, and I think clinicians will very rapidly get a feel for this drug,” Dr. Scott D. Solomon, professor of medicine, Harvard Medical School, Boston, and one of the authors of the study, said in an interview. Referring to the trial results, he noted that for at least 10 years, there has not been a new drug for heart failure that has had this impact on mortality, “so this is really quite an extraordinary finding.”
The availability of a new drug class, a neprilysin inhibitor, “opens up a new chapter in our treatment of heart failure,” said Dr. Mariell Jessup, professor of medicine at the University of Pennsylvania, Philadelphia, who was not involved in the study. “I’m very excited that we now have this new drug for use in our patients,” she said, pointing out that with the exception of ivabradine (Corlanor), approved in April, there has not been a new type of drug approved for heart failure in a long time.
Neprilysin is an endopeptidase that degrades several endogenous vasoactive peptides. Sacubitril “inhibits the breakdown of natriuretic peptides, among other vasoactive compounds, so the drug simultaneously blocks one of the neurohormonal systems that is abnormally activated in the setting of heart failure, and it also augments one of the neurohormonal systems that can be beneficial in patients with heart failure,” said Dr. Solomon, who is also director of noninvasive cardiology at Brigham and Women’s Hospital, Boston.
In PARADIGM-HF, conducted to determine if sacubitril-valsartan was superior to treatment with an ACE inhibitor, the recognized standard treatment for heart failure, a composite of death from cardiovascular causes or a first hospitalization for heart failure (the primary outcome), was 21.8% among those randomized to the combination vs. 26.5% of those on enalapril after a median 27-month follow-up, a highly statistically significant difference that represented a 20% reduced risk over the comparator (N. Engl. J. Med. 371:993-1004). A 20% reduced risk over enalapril was seen for the two components individually in the study, which was stopped early because of the magnitude of the effect over enalapril at a dose of the ACE inhibitor that the investigators pointed out had been shown to reduce mortality, compared with placebo.
Hypotension and nonserious cases of angioedema were higher among those on sacubitril-valsartan, while renal impairment, hyperkalemia, and cough were higher in the enalapril-treated patients.
As with ACE inhibitors, there is a small risk for angioedema, and while the number of cases was low in the study, physicians need to be aware of this issue, Dr. Solomon said. “To use this effectively, patients who are on ACE inhibitors or ARBs will need to be stopped, given 36 hours to wash out, and then started on this new agent,” he said.
In an interview, Dr. Jessup said that she hopes that physicians approach this new drug with the same caution they have with other drugs for patients with systolic heart failure and encouraged clinicians to carefully read the study and familiarize themselves with the prescribing information. Patients should not be taken off an ACE inhibitor and immediately switched to Entresto, she said.
She referred to the “famous” Canadian study that identified a significant increase in spironolactone prescriptions and in the rates of hyperkalemia and hyperkalemia-associated deaths after the positive Randomized Aldactone Evaluation Study (RALES) results were published in 1999 (N. Engl. J. Med. 2004;351:543-51). After the study, which found significant improvements in morbidity and mortality in patients with severe heart failure, was published, “physicians immediately started to put their patients on spironolactone and there was a spike in hyperkalemia and in deaths,” she noted.
The Entresto label includes a boxed warning about the risk of fetal toxicity, and the FDA statement recommends that health care professionals counsel patients about the risks to an unborn baby. One of the company’s postmarketing requirements is to conduct an epidemiologic study evaluating the incidence of angioedema in black patients treated with the combination, compared with another drug, according to the FDA’s approval letter.
The PARADIGM-HF results were reported in August 2014 at the European Society of Cardiology annual meeting in Barcelona. Dr. Solomon said that a large international study evaluating Entresto in patients with heart failure and preserved ejection fraction is currently underway.
The cost per day of Entresto is $12.50 (the wholesale acquisition cost), and the company anticipates that the product will be available in most pharmacies within weeks of the approval date, according to a Novartis spokesperson.
Valsartan, approved in 2001, is marketed as Diovan by Novartis and is available in generic form from Ranbaxy.
The PARADIGM-HF trial was funded by Novartis. Dr. Solomon, a member of the executive committee for the study, has received research support from Novartis for the conduct of this and other studies, and has served as a consultant to the company. Dr. Jessup had no related disclosures.
A combination of a new drug, sacubitril, a neprilysin inhibitor, and the angiotensin receptor blocker valsartan has been approved for treating heart failure, providing what experts are describing as a major advance in the treatment of heart failure.
The approval, announced by the Food and Drug Administration on July 7, was based on the results of the PARADIGM-HF study of about 8,400 patients with class II-IV heart failure and an ejection fraction of 40% or less. The study showed that the combination reduced the risk of cardiovascular death and hospitalization for heart failure by 20% and reduced the risk for all-cause mortality by 16%, compared with enalapril, with a favorable adverse event profile.
The approved indication for sacubitril-valsartan is to “reduce the risk of cardiovascular death and hospitalization for heart failure in patients with chronic heart failure [New York Heart Association class II-IV] and reduced ejection fraction,” according to the prescribing information. The indications section includes the statement that it “is usually administered in conjunction with other heart failure therapies, in place of an ACE inhibitor or other ARB [angiotensin II receptor blocker].” It is contraindicated for use with ACE inhibitors, and when patients on an ACE inhibitor are switched to this drug a washout period of 36 hours before starting treatment is recommended.
Previously called LCZ696, the combination tablet formulation will be marketed by Novartis as Entresto, and will be available in three dosage strengths of sacubitril-valsartan: 24/26 mg, 49/51 mg, and 97/103 mg; in published studies of the combination, these doses are referred to as 50 mg, 100 mg, and 200 mg, respectively. The target dose is 97/103 mg twice a day.
“Many of us see this as a real sea change in heart failure management; we’re all eager to start using this new medicine in our patients, and I think clinicians will very rapidly get a feel for this drug,” Dr. Scott D. Solomon, professor of medicine, Harvard Medical School, Boston, and one of the authors of the study, said in an interview. Referring to the trial results, he noted that for at least 10 years, there has not been a new drug for heart failure that has had this impact on mortality, “so this is really quite an extraordinary finding.”
The availability of a new drug class, a neprilysin inhibitor, “opens up a new chapter in our treatment of heart failure,” said Dr. Mariell Jessup, professor of medicine at the University of Pennsylvania, Philadelphia, who was not involved in the study. “I’m very excited that we now have this new drug for use in our patients,” she said, pointing out that with the exception of ivabradine (Corlanor), approved in April, there has not been a new type of drug approved for heart failure in a long time.
Neprilysin is an endopeptidase that degrades several endogenous vasoactive peptides. Sacubitril “inhibits the breakdown of natriuretic peptides, among other vasoactive compounds, so the drug simultaneously blocks one of the neurohormonal systems that is abnormally activated in the setting of heart failure, and it also augments one of the neurohormonal systems that can be beneficial in patients with heart failure,” said Dr. Solomon, who is also director of noninvasive cardiology at Brigham and Women’s Hospital, Boston.
In PARADIGM-HF, conducted to determine if sacubitril-valsartan was superior to treatment with an ACE inhibitor, the recognized standard treatment for heart failure, a composite of death from cardiovascular causes or a first hospitalization for heart failure (the primary outcome), was 21.8% among those randomized to the combination vs. 26.5% of those on enalapril after a median 27-month follow-up, a highly statistically significant difference that represented a 20% reduced risk over the comparator (N. Engl. J. Med. 371:993-1004). A 20% reduced risk over enalapril was seen for the two components individually in the study, which was stopped early because of the magnitude of the effect over enalapril at a dose of the ACE inhibitor that the investigators pointed out had been shown to reduce mortality, compared with placebo.
Hypotension and nonserious cases of angioedema were higher among those on sacubitril-valsartan, while renal impairment, hyperkalemia, and cough were higher in the enalapril-treated patients.
As with ACE inhibitors, there is a small risk for angioedema, and while the number of cases was low in the study, physicians need to be aware of this issue, Dr. Solomon said. “To use this effectively, patients who are on ACE inhibitors or ARBs will need to be stopped, given 36 hours to wash out, and then started on this new agent,” he said.
In an interview, Dr. Jessup said that she hopes that physicians approach this new drug with the same caution they have with other drugs for patients with systolic heart failure and encouraged clinicians to carefully read the study and familiarize themselves with the prescribing information. Patients should not be taken off an ACE inhibitor and immediately switched to Entresto, she said.
She referred to the “famous” Canadian study that identified a significant increase in spironolactone prescriptions and in the rates of hyperkalemia and hyperkalemia-associated deaths after the positive Randomized Aldactone Evaluation Study (RALES) results were published in 1999 (N. Engl. J. Med. 2004;351:543-51). After the study, which found significant improvements in morbidity and mortality in patients with severe heart failure, was published, “physicians immediately started to put their patients on spironolactone and there was a spike in hyperkalemia and in deaths,” she noted.
The Entresto label includes a boxed warning about the risk of fetal toxicity, and the FDA statement recommends that health care professionals counsel patients about the risks to an unborn baby. One of the company’s postmarketing requirements is to conduct an epidemiologic study evaluating the incidence of angioedema in black patients treated with the combination, compared with another drug, according to the FDA’s approval letter.
The PARADIGM-HF results were reported in August 2014 at the European Society of Cardiology annual meeting in Barcelona. Dr. Solomon said that a large international study evaluating Entresto in patients with heart failure and preserved ejection fraction is currently underway.
The cost per day of Entresto is $12.50 (the wholesale acquisition cost), and the company anticipates that the product will be available in most pharmacies within weeks of the approval date, according to a Novartis spokesperson.
Valsartan, approved in 2001, is marketed as Diovan by Novartis and is available in generic form from Ranbaxy.
The PARADIGM-HF trial was funded by Novartis. Dr. Solomon, a member of the executive committee for the study, has received research support from Novartis for the conduct of this and other studies, and has served as a consultant to the company. Dr. Jessup had no related disclosures.

Two U.S. transcatheter valve approvals reshape TAVR
The nature of U.S. transcatheter aortic valve replacement (TAVR) shifted dramatically in mid-June when the Food and Drug Administration, in two separate actions timed just days apart, approved marketing of next-generation models for the only two transcatheter aortic valve replacement systems on the U.S. market.
For inoperable or high-risk patients in came the Edwards Sapien 3 and the CoreValve Evolut R systems, and out went the Sapien XT and the original CoreValve. In the process the transcatheter aortic valve replacement (TAVR) procedures available to American patients on a routine basis became smaller – meaning more likely to use transfemoral approaches, less likely to cause substantial paravalvular leak, less likely to cause stroke, and in the case of the Evolut system also became repositionable and less likely to result in the need for a pacemaker.
With approvals of these two latest-generation devices, in both cases based on follow-up data of only 30 days, “we have really expanded the types of patients who can be treated with TAVR in the U.S., and we can do it with better products and get better outcomes. I am thrilled this technology will be available to patients,” said Dr. Jeffrey J. Popma, a lead investigator for the Evolut U.S. pivotal trial and professor of medicine at Harvard University in Boston.

The pair of approvals were also notable as clear demonstration that the FDA was willing to base its decisions on 30-day follow-up data, a step closer to the approval model it applies to heart valves placed using open surgery. Perhaps most attention-grabbing of all, the FDA based its approval of the Evolut system on data from a total of 151 patients, with 60 coming from the ex-U.S. trial designed to secure a CE mark in Europe, and the other 91 patients the first ones enrolled in what was designed to be the U.S. pivotal trial for Evolut, with a planned enrollment of 250 patients that had almost fully filled by mid-June.
“This was a really good week for the FDA,” said Dr. Popma. “The FDA approved devices with reasonable assurance of safety and efficacy but without the burden of requiring 1 or more years follow-up. We are seeing a changed FDA,” he declared in an interview.
Although the full dataset for all 151 patients that the FDA used to approve the self-expanding Evolut system has not yet been released, investigators from the U.S. trial say data from their first 91 patients looked similar to the 60 patients in the CE trial, the results of which appeared in a poster in March at the annual scientific sessions of the American College of Cardiology.
The CE trial enrolled 60 symptomatic extreme- or high-risk patients at six centers in Australia, New Zealand, and the United Kingdom, who averaged 83 years old and had an average Society of Thoracic Surgeons predicted mortality estimate of 7%. Fifty-nine of the 60 patients (98%) had their TAVR done via the transfemoral route, and during 30-day follow-up no patients died and none had a stroke. The researchers identified a moderate or severe paravalvular leak in 3% of patients after 30 days, and 12% of patients had a new need for a pacemaker. By comparison, in the CoreValve pivotal trial for the first-generation version of this self-expanding valve, death occurred in 3% of patients after 30 days, major strokes in 4%, moderate or severe paravalvular leaks occurred in 9%, and 20% of patients by that point had required placement of a new pacemaker (N. Engl. J. Med. 2014;370:1790-8).

The striking reductions in more severe paravalvular leaks and in the need for a pacemaker likely related at least in part to the ability to reposition the Evolut valve during placement as long as the valve was not more than 80% deployed and as long as retrieval was not attempted more than three times.
Repositioning is “huge” said Dr. Mathew R. Williams, chief of adult cardiac surgery and director of interventional cardiology at New York University, and one of two lead investigators on the U.S. CoreValve Evolut R pivotal trial. “We don’t need to use it most of the time, but having that ability reduces stress and helps make the operator more comfortable,” he said in an interview. The more optimized positioning allowed by the recapture feature likely helped minimize both paravalvular leaks and the valve’s ability to trigger an arrhythmia and need for pacing. In the CE trial the operator used the repositioning function in 15 patients (25%) for a total of 22 repositioning events.
Like Dr. Popma, Dr. Williams welcomed the June 17 approval of the Sapien 3 TAVR system, based on 30-day outcomes from all 583 patients enrolled in the U.S. pivotal trial. Results from that study became public last March in a late-breaker report at the American College of Cardiology meeting.

Sapien 3 treatment of high-risk, symptomatic patients, who averaged 83 years old and had a STS predicted mortality rate of 8.6%, resulted in a 2% mortality rate, a 1.5% stroke rate, a 3% rate of moderate or severe paravalvular leaks, and 13% of patients had a new need for a pacemaker, Dr. Susheel Kodali reported when he presented these data in March. Trial investigators were able to place the Sapien 3 aortic valve via a transfemoral approach in 491 of the 583 patients (84%) enrolled in the study. The adverse outcomes with Sapien 3 contrasted with rates for the prior Sapien XT TAVR system of 3.5% for 30-day mortality, 4.3% for strokes, and a 24% rate of moderate or severe paravalvular leak, said Dr. Kodali, director of the heart valve program at Columbia University in New York.
While the Sapien 3 and Evolut systems appeared to produce roughly similar outcomes for parameters like 30-day mortality, stroke, and paravalvular leak rates, with both systems outperforming what they displaced, the Evolut has two important differences, compared with Sapien 3. First, the Evolut is slightly smaller, with the ability to traverse arteries as narrow as 5 mm, whereas the minimum vessel size for Sapien 3 passage is 5.5 mm; second, Evolute is repositionable, while Sapien 3 is not.
The CoreValve Evolut R trials were sponsored by Medtronic. Dr. Popma has received research grants from and served as a speaker on behalf of Medtronic. He has also been a speaker for Abbott Vascular, Boston Scientific, Covidien, Cook Medical, and Direct Flow Medical and has received research support from five other companies. Dr. Williams has been a consultant to and received research support from Medtronic and has also been a consultant to Edwards and Direct Flow Medical. The Sapien 3 trial was sponsored by Edwards. Dr. Kodali has received research support from Edwards, has an equity interest in Thubrikar Aortic Valve, has received honoraria from St. Jude, and has served on the steering committee for trials sponsored by Claret Medical and Meril.
On Twitter @mitchelzoler
The nature of U.S. transcatheter aortic valve replacement (TAVR) shifted dramatically in mid-June when the Food and Drug Administration, in two separate actions timed just days apart, approved marketing of next-generation models for the only two transcatheter aortic valve replacement systems on the U.S. market.
For inoperable or high-risk patients in came the Edwards Sapien 3 and the CoreValve Evolut R systems, and out went the Sapien XT and the original CoreValve. In the process the transcatheter aortic valve replacement (TAVR) procedures available to American patients on a routine basis became smaller – meaning more likely to use transfemoral approaches, less likely to cause substantial paravalvular leak, less likely to cause stroke, and in the case of the Evolut system also became repositionable and less likely to result in the need for a pacemaker.
With approvals of these two latest-generation devices, in both cases based on follow-up data of only 30 days, “we have really expanded the types of patients who can be treated with TAVR in the U.S., and we can do it with better products and get better outcomes. I am thrilled this technology will be available to patients,” said Dr. Jeffrey J. Popma, a lead investigator for the Evolut U.S. pivotal trial and professor of medicine at Harvard University in Boston.
The pair of approvals were also notable as clear demonstration that the FDA was willing to base its decisions on 30-day follow-up data, a step closer to the approval model it applies to heart valves placed using open surgery. Perhaps most attention-grabbing of all, the FDA based its approval of the Evolut system on data from a total of 151 patients, with 60 coming from the ex-U.S. trial designed to secure a CE mark in Europe, and the other 91 patients the first ones enrolled in what was designed to be the U.S. pivotal trial for Evolut, with a planned enrollment of 250 patients that had almost fully filled by mid-June.
“This was a really good week for the FDA,” said Dr. Popma. “The FDA approved devices with reasonable assurance of safety and efficacy but without the burden of requiring 1 or more years follow-up. We are seeing a changed FDA,” he declared in an interview.
Although the full dataset for all 151 patients that the FDA used to approve the self-expanding Evolut system has not yet been released, investigators from the U.S. trial say data from their first 91 patients looked similar to the 60 patients in the CE trial, the results of which appeared in a poster in March at the annual scientific sessions of the American College of Cardiology.
The CE trial enrolled 60 symptomatic extreme- or high-risk patients at six centers in Australia, New Zealand, and the United Kingdom, who averaged 83 years old and had an average Society of Thoracic Surgeons predicted mortality estimate of 7%. Fifty-nine of the 60 patients (98%) had their TAVR done via the transfemoral route, and during 30-day follow-up no patients died and none had a stroke. The researchers identified a moderate or severe paravalvular leak in 3% of patients after 30 days, and 12% of patients had a new need for a pacemaker. By comparison, in the CoreValve pivotal trial for the first-generation version of this self-expanding valve, death occurred in 3% of patients after 30 days, major strokes in 4%, moderate or severe paravalvular leaks occurred in 9%, and 20% of patients by that point had required placement of a new pacemaker (N. Engl. J. Med. 2014;370:1790-8).
The striking reductions in more severe paravalvular leaks and in the need for a pacemaker likely related at least in part to the ability to reposition the Evolut valve during placement as long as the valve was not more than 80% deployed and as long as retrieval was not attempted more than three times.
Repositioning is “huge” said Dr. Mathew R. Williams, chief of adult cardiac surgery and director of interventional cardiology at New York University, and one of two lead investigators on the U.S. CoreValve Evolut R pivotal trial. “We don’t need to use it most of the time, but having that ability reduces stress and helps make the operator more comfortable,” he said in an interview. The more optimized positioning allowed by the recapture feature likely helped minimize both paravalvular leaks and the valve’s ability to trigger an arrhythmia and need for pacing. In the CE trial the operator used the repositioning function in 15 patients (25%) for a total of 22 repositioning events.
Like Dr. Popma, Dr. Williams welcomed the June 17 approval of the Sapien 3 TAVR system, based on 30-day outcomes from all 583 patients enrolled in the U.S. pivotal trial. Results from that study became public last March in a late-breaker report at the American College of Cardiology meeting.
Sapien 3 treatment of high-risk, symptomatic patients, who averaged 83 years old and had a STS predicted mortality rate of 8.6%, resulted in a 2% mortality rate, a 1.5% stroke rate, a 3% rate of moderate or severe paravalvular leaks, and 13% of patients had a new need for a pacemaker, Dr. Susheel Kodali reported when he presented these data in March. Trial investigators were able to place the Sapien 3 aortic valve via a transfemoral approach in 491 of the 583 patients (84%) enrolled in the study. The adverse outcomes with Sapien 3 contrasted with rates for the prior Sapien XT TAVR system of 3.5% for 30-day mortality, 4.3% for strokes, and a 24% rate of moderate or severe paravalvular leak, said Dr. Kodali, director of the heart valve program at Columbia University in New York.
While the Sapien 3 and Evolut systems appeared to produce roughly similar outcomes for parameters like 30-day mortality, stroke, and paravalvular leak rates, with both systems outperforming what they displaced, the Evolut has two important differences, compared with Sapien 3. First, the Evolut is slightly smaller, with the ability to traverse arteries as narrow as 5 mm, whereas the minimum vessel size for Sapien 3 passage is 5.5 mm; second, Evolute is repositionable, while Sapien 3 is not.
The CoreValve Evolut R trials were sponsored by Medtronic. Dr. Popma has received research grants from and served as a speaker on behalf of Medtronic. He has also been a speaker for Abbott Vascular, Boston Scientific, Covidien, Cook Medical, and Direct Flow Medical and has received research support from five other companies. Dr. Williams has been a consultant to and received research support from Medtronic and has also been a consultant to Edwards and Direct Flow Medical. The Sapien 3 trial was sponsored by Edwards. Dr. Kodali has received research support from Edwards, has an equity interest in Thubrikar Aortic Valve, has received honoraria from St. Jude, and has served on the steering committee for trials sponsored by Claret Medical and Meril.
On Twitter @mitchelzoler
The nature of U.S. transcatheter aortic valve replacement (TAVR) shifted dramatically in mid-June when the Food and Drug Administration, in two separate actions timed just days apart, approved marketing of next-generation models for the only two transcatheter aortic valve replacement systems on the U.S. market.
For inoperable or high-risk patients in came the Edwards Sapien 3 and the CoreValve Evolut R systems, and out went the Sapien XT and the original CoreValve. In the process the transcatheter aortic valve replacement (TAVR) procedures available to American patients on a routine basis became smaller – meaning more likely to use transfemoral approaches, less likely to cause substantial paravalvular leak, less likely to cause stroke, and in the case of the Evolut system also became repositionable and less likely to result in the need for a pacemaker.
With approvals of these two latest-generation devices, in both cases based on follow-up data of only 30 days, “we have really expanded the types of patients who can be treated with TAVR in the U.S., and we can do it with better products and get better outcomes. I am thrilled this technology will be available to patients,” said Dr. Jeffrey J. Popma, a lead investigator for the Evolut U.S. pivotal trial and professor of medicine at Harvard University in Boston.
The pair of approvals were also notable as clear demonstration that the FDA was willing to base its decisions on 30-day follow-up data, a step closer to the approval model it applies to heart valves placed using open surgery. Perhaps most attention-grabbing of all, the FDA based its approval of the Evolut system on data from a total of 151 patients, with 60 coming from the ex-U.S. trial designed to secure a CE mark in Europe, and the other 91 patients the first ones enrolled in what was designed to be the U.S. pivotal trial for Evolut, with a planned enrollment of 250 patients that had almost fully filled by mid-June.
“This was a really good week for the FDA,” said Dr. Popma. “The FDA approved devices with reasonable assurance of safety and efficacy but without the burden of requiring 1 or more years follow-up. We are seeing a changed FDA,” he declared in an interview.
Although the full dataset for all 151 patients that the FDA used to approve the self-expanding Evolut system has not yet been released, investigators from the U.S. trial say data from their first 91 patients looked similar to the 60 patients in the CE trial, the results of which appeared in a poster in March at the annual scientific sessions of the American College of Cardiology.
The CE trial enrolled 60 symptomatic extreme- or high-risk patients at six centers in Australia, New Zealand, and the United Kingdom, who averaged 83 years old and had an average Society of Thoracic Surgeons predicted mortality estimate of 7%. Fifty-nine of the 60 patients (98%) had their TAVR done via the transfemoral route, and during 30-day follow-up no patients died and none had a stroke. The researchers identified a moderate or severe paravalvular leak in 3% of patients after 30 days, and 12% of patients had a new need for a pacemaker. By comparison, in the CoreValve pivotal trial for the first-generation version of this self-expanding valve, death occurred in 3% of patients after 30 days, major strokes in 4%, moderate or severe paravalvular leaks occurred in 9%, and 20% of patients by that point had required placement of a new pacemaker (N. Engl. J. Med. 2014;370:1790-8).
The striking reductions in more severe paravalvular leaks and in the need for a pacemaker likely related at least in part to the ability to reposition the Evolut valve during placement as long as the valve was not more than 80% deployed and as long as retrieval was not attempted more than three times.
Repositioning is “huge” said Dr. Mathew R. Williams, chief of adult cardiac surgery and director of interventional cardiology at New York University, and one of two lead investigators on the U.S. CoreValve Evolut R pivotal trial. “We don’t need to use it most of the time, but having that ability reduces stress and helps make the operator more comfortable,” he said in an interview. The more optimized positioning allowed by the recapture feature likely helped minimize both paravalvular leaks and the valve’s ability to trigger an arrhythmia and need for pacing. In the CE trial the operator used the repositioning function in 15 patients (25%) for a total of 22 repositioning events.
Like Dr. Popma, Dr. Williams welcomed the June 17 approval of the Sapien 3 TAVR system, based on 30-day outcomes from all 583 patients enrolled in the U.S. pivotal trial. Results from that study became public last March in a late-breaker report at the American College of Cardiology meeting.
Sapien 3 treatment of high-risk, symptomatic patients, who averaged 83 years old and had a STS predicted mortality rate of 8.6%, resulted in a 2% mortality rate, a 1.5% stroke rate, a 3% rate of moderate or severe paravalvular leaks, and 13% of patients had a new need for a pacemaker, Dr. Susheel Kodali reported when he presented these data in March. Trial investigators were able to place the Sapien 3 aortic valve via a transfemoral approach in 491 of the 583 patients (84%) enrolled in the study. The adverse outcomes with Sapien 3 contrasted with rates for the prior Sapien XT TAVR system of 3.5% for 30-day mortality, 4.3% for strokes, and a 24% rate of moderate or severe paravalvular leak, said Dr. Kodali, director of the heart valve program at Columbia University in New York.
While the Sapien 3 and Evolut systems appeared to produce roughly similar outcomes for parameters like 30-day mortality, stroke, and paravalvular leak rates, with both systems outperforming what they displaced, the Evolut has two important differences, compared with Sapien 3. First, the Evolut is slightly smaller, with the ability to traverse arteries as narrow as 5 mm, whereas the minimum vessel size for Sapien 3 passage is 5.5 mm; second, Evolute is repositionable, while Sapien 3 is not.
The CoreValve Evolut R trials were sponsored by Medtronic. Dr. Popma has received research grants from and served as a speaker on behalf of Medtronic. He has also been a speaker for Abbott Vascular, Boston Scientific, Covidien, Cook Medical, and Direct Flow Medical and has received research support from five other companies. Dr. Williams has been a consultant to and received research support from Medtronic and has also been a consultant to Edwards and Direct Flow Medical. The Sapien 3 trial was sponsored by Edwards. Dr. Kodali has received research support from Edwards, has an equity interest in Thubrikar Aortic Valve, has received honoraria from St. Jude, and has served on the steering committee for trials sponsored by Claret Medical and Meril.
On Twitter @mitchelzoler
