User login
Probiotics improve nonmotor symptoms of Parkinson’s
COPENHAGEN – results of a new randomized trial show.
Participants taking the probiotic also saw a reduced delay in “time to on” of treatment with levodopa, thus reducing the delay until effectiveness of the treatment, said study presenter Valentina Leta, MD, PhD, department of neurosciences, King’s College London Institute of Psychiatry, Psychology and Neuroscience.
Dr. Leta presented the findings at the International Congress of Parkinson’s Disease and Movement Disorders.
“Virtually every person with Parkinson’s might have some degree of gastrointestinal dysfunction, and virtually the entire tract might be affected, from the mouth to the rectum,” Dr. Leta told attendees of the congress.
A number of different mechanisms have been associated with this gastrointestinal dysfunction, she noted, including proinflammatory changes in the gut microbiota, so a modulatory intervention “could be a therapeutic strategy for Parkinson’s disease.”
However, “despite numerous preclinical studies showing potential beneficial effects on a variety of pathological mechanisms involved in Parkinson’s disease, the clinical evidence is limited ... to the treatment of constipation,” she explained.
The team therefore conducted a multicenter, randomized, double-blind, placebo-controlled trial, in which patients with both Parkinson’s disease and constipation, based on the Rome IV criteria, were randomly assigned to receive a probiotic or placebo for 3 months.
The probiotic used was a liquid formulation (Symprove) and contained four strains: Lacticaseibacillus rhamnosus, Enterococcus faecium, Lactobacillus acidophilus, and Lactiplantibacillus plantarum.
A total of 74 patients were randomly assigned to the two study arms. The two groups were well matched for sociodemographics, Parkinson’s disease, and constipation-related characteristics, Dr. Leta reported, and only 3 patients in each arm discontinued the study. The probiotic intervention had a “good tolerability and safety profile, with a similar number of adverse events between the two groups, and no serious adverse events.”
Increase in healthy bacteria
The study met its primary outcome of changes in gut microbiome at the end of the 12-week intervention, as measured on shallow shotgun sequencing.
The probiotic was associated with a “statistically significant increase of the abundance of bacteria which are known to have beneficial health related properties, such as Odoribacteraceae,” Dr. Leta said.
This bacterium is “known to be reduced in people with Parkinson’s disease,” she explained, “and is involved in the production of short-chain fatty acids, which are known to have beneficial health-related properties.”
The secondary endpoint of the study included changes in motor and nonmotor symptoms, and the probiotic was associated with a significant improvement in the “time to on” with levodopa treatment, shortening this period from an average of 31.43 minutes at baseline to 23.95 minutes at the postintervention assessment (P < .027).
There was also a significant improvement in the Non-Motor Symptoms Scale (NMSS) score between baseline and the postintervention assessment in patients given the probiotic, from 70.71 to 61.34 (P = .005).
This, Dr. Leta observed, was “driven by improvements in the sleep, fatigue, and gastrointestinal domains.”
No such significant improvements were observed in the placebo arm.
Probiotics ‘hot topic’ among patients
Claudia Trenkwalder, MD, full professor of neurology at University Medical Center Goettingen (Germany), said in an interview that the use of probiotics is a “hot topic in Parkinson’s disease research, especially among patients.”
Dr. Trenkwalder, who was not involved in the study, noted that Lactobacillus strains “are established in Parkinson’s disease constipation treatment, with randomized controlled trials showing a significant improvement in constipation.
“Therefore, this is a useful treatment. The question here is: Do we have additional effects that can be measured in the microbiome and in clinical symptomatology?”
The trial showed that the probiotic studied “did alter the microbiome and did improve the constipation,” said Dr. Trenkwalder; however, the current data cannot prove whether the probiotic influenced the symptoms of Parkinson’s disease because the improvement in NMSS scores “is driven by the improvement in constipation.”
This, she argued, could have resulted in better absorption of levodopa.
A dietitian in the audience agreed. She asked whether the probiotic was doing anything “besides improving constipation,” adding that the resulting increased ability to absorb levodopa is also “going to help your sleep.”
Beyond constipation?
Dr. Leta replied that “we can assume that there is a link between the reduction in the ‘time to on’ and the improvement in constipation. We are doing some analyses in terms of levodopa pharmacokinetics to really understand the mechanisms behind this result.”
Although the improvement in constipation is “one of the possible hypotheses for the improvement in ‘time to on,’” she continued, “there is a more speculative one” in which the probiotics are modulating inflammatory parameters that could contribute to the improvement in sleep.
Veronica Bruno, MD, MPH, assistant professor in the department of clinical neurosciences at the University of Calgary (Alta.), commented in a press release that there has been “increasing interest” in examining the relationship between gut dysbiosis and the “gut-brain axis” in Parkinson’s disease.
The current study “stands out as a significant contribution to this area of study,” she said.
“While the implications of the observed changes in gut microbiota remain a captivating realm for further investigation, a particularly noteworthy finding revolves around the reduction in the ‘time to on’ observed within the active treatment group.”
Dr. Bruno said that shortening of the time to on “holds promise for substantial enhancements in patients’ lives” by reducing “difficult ‘off’ intervals and enhancing overall well-being.”
The study was funded by the UK National Institute for Health Research Mental Health Biomedical Research Centre and Dementia Unit at South London and Maudsley NHS Foundation Trust, and King’s College London. No relevant financial relationships were declared.
A version of this article first appeared on Medscape.com.
COPENHAGEN – results of a new randomized trial show.
Participants taking the probiotic also saw a reduced delay in “time to on” of treatment with levodopa, thus reducing the delay until effectiveness of the treatment, said study presenter Valentina Leta, MD, PhD, department of neurosciences, King’s College London Institute of Psychiatry, Psychology and Neuroscience.
Dr. Leta presented the findings at the International Congress of Parkinson’s Disease and Movement Disorders.
“Virtually every person with Parkinson’s might have some degree of gastrointestinal dysfunction, and virtually the entire tract might be affected, from the mouth to the rectum,” Dr. Leta told attendees of the congress.
A number of different mechanisms have been associated with this gastrointestinal dysfunction, she noted, including proinflammatory changes in the gut microbiota, so a modulatory intervention “could be a therapeutic strategy for Parkinson’s disease.”
However, “despite numerous preclinical studies showing potential beneficial effects on a variety of pathological mechanisms involved in Parkinson’s disease, the clinical evidence is limited ... to the treatment of constipation,” she explained.
The team therefore conducted a multicenter, randomized, double-blind, placebo-controlled trial, in which patients with both Parkinson’s disease and constipation, based on the Rome IV criteria, were randomly assigned to receive a probiotic or placebo for 3 months.
The probiotic used was a liquid formulation (Symprove) and contained four strains: Lacticaseibacillus rhamnosus, Enterococcus faecium, Lactobacillus acidophilus, and Lactiplantibacillus plantarum.
A total of 74 patients were randomly assigned to the two study arms. The two groups were well matched for sociodemographics, Parkinson’s disease, and constipation-related characteristics, Dr. Leta reported, and only 3 patients in each arm discontinued the study. The probiotic intervention had a “good tolerability and safety profile, with a similar number of adverse events between the two groups, and no serious adverse events.”
Increase in healthy bacteria
The study met its primary outcome of changes in gut microbiome at the end of the 12-week intervention, as measured on shallow shotgun sequencing.
The probiotic was associated with a “statistically significant increase of the abundance of bacteria which are known to have beneficial health related properties, such as Odoribacteraceae,” Dr. Leta said.
This bacterium is “known to be reduced in people with Parkinson’s disease,” she explained, “and is involved in the production of short-chain fatty acids, which are known to have beneficial health-related properties.”
The secondary endpoint of the study included changes in motor and nonmotor symptoms, and the probiotic was associated with a significant improvement in the “time to on” with levodopa treatment, shortening this period from an average of 31.43 minutes at baseline to 23.95 minutes at the postintervention assessment (P < .027).
There was also a significant improvement in the Non-Motor Symptoms Scale (NMSS) score between baseline and the postintervention assessment in patients given the probiotic, from 70.71 to 61.34 (P = .005).
This, Dr. Leta observed, was “driven by improvements in the sleep, fatigue, and gastrointestinal domains.”
No such significant improvements were observed in the placebo arm.
Probiotics ‘hot topic’ among patients
Claudia Trenkwalder, MD, full professor of neurology at University Medical Center Goettingen (Germany), said in an interview that the use of probiotics is a “hot topic in Parkinson’s disease research, especially among patients.”
Dr. Trenkwalder, who was not involved in the study, noted that Lactobacillus strains “are established in Parkinson’s disease constipation treatment, with randomized controlled trials showing a significant improvement in constipation.
“Therefore, this is a useful treatment. The question here is: Do we have additional effects that can be measured in the microbiome and in clinical symptomatology?”
The trial showed that the probiotic studied “did alter the microbiome and did improve the constipation,” said Dr. Trenkwalder; however, the current data cannot prove whether the probiotic influenced the symptoms of Parkinson’s disease because the improvement in NMSS scores “is driven by the improvement in constipation.”
This, she argued, could have resulted in better absorption of levodopa.
A dietitian in the audience agreed. She asked whether the probiotic was doing anything “besides improving constipation,” adding that the resulting increased ability to absorb levodopa is also “going to help your sleep.”
Beyond constipation?
Dr. Leta replied that “we can assume that there is a link between the reduction in the ‘time to on’ and the improvement in constipation. We are doing some analyses in terms of levodopa pharmacokinetics to really understand the mechanisms behind this result.”
Although the improvement in constipation is “one of the possible hypotheses for the improvement in ‘time to on,’” she continued, “there is a more speculative one” in which the probiotics are modulating inflammatory parameters that could contribute to the improvement in sleep.
Veronica Bruno, MD, MPH, assistant professor in the department of clinical neurosciences at the University of Calgary (Alta.), commented in a press release that there has been “increasing interest” in examining the relationship between gut dysbiosis and the “gut-brain axis” in Parkinson’s disease.
The current study “stands out as a significant contribution to this area of study,” she said.
“While the implications of the observed changes in gut microbiota remain a captivating realm for further investigation, a particularly noteworthy finding revolves around the reduction in the ‘time to on’ observed within the active treatment group.”
Dr. Bruno said that shortening of the time to on “holds promise for substantial enhancements in patients’ lives” by reducing “difficult ‘off’ intervals and enhancing overall well-being.”
The study was funded by the UK National Institute for Health Research Mental Health Biomedical Research Centre and Dementia Unit at South London and Maudsley NHS Foundation Trust, and King’s College London. No relevant financial relationships were declared.
A version of this article first appeared on Medscape.com.
COPENHAGEN – results of a new randomized trial show.
Participants taking the probiotic also saw a reduced delay in “time to on” of treatment with levodopa, thus reducing the delay until effectiveness of the treatment, said study presenter Valentina Leta, MD, PhD, department of neurosciences, King’s College London Institute of Psychiatry, Psychology and Neuroscience.
Dr. Leta presented the findings at the International Congress of Parkinson’s Disease and Movement Disorders.
“Virtually every person with Parkinson’s might have some degree of gastrointestinal dysfunction, and virtually the entire tract might be affected, from the mouth to the rectum,” Dr. Leta told attendees of the congress.
A number of different mechanisms have been associated with this gastrointestinal dysfunction, she noted, including proinflammatory changes in the gut microbiota, so a modulatory intervention “could be a therapeutic strategy for Parkinson’s disease.”
However, “despite numerous preclinical studies showing potential beneficial effects on a variety of pathological mechanisms involved in Parkinson’s disease, the clinical evidence is limited ... to the treatment of constipation,” she explained.
The team therefore conducted a multicenter, randomized, double-blind, placebo-controlled trial, in which patients with both Parkinson’s disease and constipation, based on the Rome IV criteria, were randomly assigned to receive a probiotic or placebo for 3 months.
The probiotic used was a liquid formulation (Symprove) and contained four strains: Lacticaseibacillus rhamnosus, Enterococcus faecium, Lactobacillus acidophilus, and Lactiplantibacillus plantarum.
A total of 74 patients were randomly assigned to the two study arms. The two groups were well matched for sociodemographics, Parkinson’s disease, and constipation-related characteristics, Dr. Leta reported, and only 3 patients in each arm discontinued the study. The probiotic intervention had a “good tolerability and safety profile, with a similar number of adverse events between the two groups, and no serious adverse events.”
Increase in healthy bacteria
The study met its primary outcome of changes in gut microbiome at the end of the 12-week intervention, as measured on shallow shotgun sequencing.
The probiotic was associated with a “statistically significant increase of the abundance of bacteria which are known to have beneficial health related properties, such as Odoribacteraceae,” Dr. Leta said.
This bacterium is “known to be reduced in people with Parkinson’s disease,” she explained, “and is involved in the production of short-chain fatty acids, which are known to have beneficial health-related properties.”
The secondary endpoint of the study included changes in motor and nonmotor symptoms, and the probiotic was associated with a significant improvement in the “time to on” with levodopa treatment, shortening this period from an average of 31.43 minutes at baseline to 23.95 minutes at the postintervention assessment (P < .027).
There was also a significant improvement in the Non-Motor Symptoms Scale (NMSS) score between baseline and the postintervention assessment in patients given the probiotic, from 70.71 to 61.34 (P = .005).
This, Dr. Leta observed, was “driven by improvements in the sleep, fatigue, and gastrointestinal domains.”
No such significant improvements were observed in the placebo arm.
Probiotics ‘hot topic’ among patients
Claudia Trenkwalder, MD, full professor of neurology at University Medical Center Goettingen (Germany), said in an interview that the use of probiotics is a “hot topic in Parkinson’s disease research, especially among patients.”
Dr. Trenkwalder, who was not involved in the study, noted that Lactobacillus strains “are established in Parkinson’s disease constipation treatment, with randomized controlled trials showing a significant improvement in constipation.
“Therefore, this is a useful treatment. The question here is: Do we have additional effects that can be measured in the microbiome and in clinical symptomatology?”
The trial showed that the probiotic studied “did alter the microbiome and did improve the constipation,” said Dr. Trenkwalder; however, the current data cannot prove whether the probiotic influenced the symptoms of Parkinson’s disease because the improvement in NMSS scores “is driven by the improvement in constipation.”
This, she argued, could have resulted in better absorption of levodopa.
A dietitian in the audience agreed. She asked whether the probiotic was doing anything “besides improving constipation,” adding that the resulting increased ability to absorb levodopa is also “going to help your sleep.”
Beyond constipation?
Dr. Leta replied that “we can assume that there is a link between the reduction in the ‘time to on’ and the improvement in constipation. We are doing some analyses in terms of levodopa pharmacokinetics to really understand the mechanisms behind this result.”
Although the improvement in constipation is “one of the possible hypotheses for the improvement in ‘time to on,’” she continued, “there is a more speculative one” in which the probiotics are modulating inflammatory parameters that could contribute to the improvement in sleep.
Veronica Bruno, MD, MPH, assistant professor in the department of clinical neurosciences at the University of Calgary (Alta.), commented in a press release that there has been “increasing interest” in examining the relationship between gut dysbiosis and the “gut-brain axis” in Parkinson’s disease.
The current study “stands out as a significant contribution to this area of study,” she said.
“While the implications of the observed changes in gut microbiota remain a captivating realm for further investigation, a particularly noteworthy finding revolves around the reduction in the ‘time to on’ observed within the active treatment group.”
Dr. Bruno said that shortening of the time to on “holds promise for substantial enhancements in patients’ lives” by reducing “difficult ‘off’ intervals and enhancing overall well-being.”
The study was funded by the UK National Institute for Health Research Mental Health Biomedical Research Centre and Dementia Unit at South London and Maudsley NHS Foundation Trust, and King’s College London. No relevant financial relationships were declared.
A version of this article first appeared on Medscape.com.
AT MDS 2023
Why genetic testing may be our best shot at progress in Parkinson’s disease
In 2017, Sanofi Genzyme launched a phase 2 clinical trial of a drug designed to target a specific genetic mutation in some patients with Parkinson’s disease. Researchers hoped the drug would slow or even stop disease progression.
Like many before it, the trial yielded disappointing results and the company shut it down in 2021. It was the latest in a string of unsuccessful clinical trials testing disease-modifying Parkinson’s disease drugs.
Although it failed, the Sanofi Genzyme study was different: It was the first to enroll patients with Parkinson’s disease who had a specific genotype and marked the earliest days of precision medicine and gene-specific drug development for the disease.
Once thought to play only a small role in a small number of patients with Parkinson’s disease,
“We’re about to enter this era of precision medicine for Parkinson’s disease, which makes genetic testing important,” said James Beck, PhD, senior vice president and chief scientific officer for the Parkinson’s Foundation.
“A number of companies have clinical trials or are in preparation for clinical trials to test some specific therapies that would depend upon people having a specific genetic mutation,” he said.
Today, at least four clinical trials of drugs that target specific Parkinson’s disease-related gene variants on LRRK2 and GBA are under way, and more are in the pipeline. Whether these drugs will be effective at modifying the course of the disease remains to be seen. First, the trials must enroll enough patients. And therein lies the challenge: Genetic testing isn’t part of routine Parkinson’s disease care and isn’t covered by most insurance policies. Most patients don’t know their genotype.
It’s a significant roadblock to the future of a precision medicine approach that is based on a patient’s individual genotype, which some experts argue offers the best shot at slowing disease progression.
“To enroll in clinical trials for precision drugs people with Parkinson’s disease have to be aware of their genetic status,” said Roy N. Alcalay, MD, chief of the movement disorders division at Tel Aviv Medical Center in Israel and part-time associate professor at Columbia University in New York. “How can a person with Parkinson’s and a LRRK2 mutation join a precision medicine trial for LRRK2 if she does not know she is a LRRK2 carrier?”
Free genetic testing
Previous studies have shown that some genetic variants increase the risk for Parkinson’s disease after exposure to environmental factors such as pesticides. Research has also shown that a patient’s genotype can predict survival time and that certain medications may prove more effective at slowing disease progression in patients with specific genotypes. All of this points to a significant role for genetics in a disorder that is rapidly increasing.
This makes expanding patient access to genetic testing even more important, Dr. Alcalay said, noting that it’s equally important that patients are informed of their genotype, something that doesn’t usually happen in blinded clinical trials.
To that end, Dr. Alcalay hopes a national genetics study he is leading will address access and need-to-know issues. PD GENEration, a project launched in 2019 by the Parkinson’s Foundation, offers patients free genetic testing for seven clinically relevant Parkinson’s disease-related genes.
Testing is done at home or in a nearby clinic and the results are shared with patients during a free genetic counseling session and with site investigators. Patient samples are stored in a genetic data bank that is open to researchers around the world.
“We surveyed clinical trialists in the Parkinson’s disease field prior to initiation of PD GENEration and estimated that over 90% of people with Parkinson’s disease prior to the effort were not aware of their genetic status,” Dr. Alcalay said.
“I think precision medicine in Parkinson’s disease will not happen without PD GENEration or similar efforts.”
‘Overwhelming’ patient interest
Participants in the study are screened for variants in seven genes known to be involved in Parkinson’s disease risk: GBA, LRRK2, PRKN, PINK1, SNCA, PARK7, and VPS35.
In less than 3 years, the study has already produced what is thought to be the largest genetic data bank of sequenced sets of Parkinson’s disease-risk genes made accessible to patients. Since the end of 2020, the first year of patient enrollment, the number of participants has increased from 676 to 10,515 and the number of participating clinical sites rose from 12 to 101.
The foundation has spent nearly $20 million on the project so far and plans to spend another $10 million to reach a goal of 15,000 patients. The study, which is funded by private donors, is so successful that the foundation has had to scale back enrollment.
“When we were at a peak, we had over 700 participants enrolling each month,” Dr. Beck said. Beginning in April, the program capped new sign-ups to 200 patients per month and created a waiting list for future enrollment. The waiting list is hundreds of patients long.
“The participants’ response to enroll in PD GENEration demonstrates there is an overwhelming interest by people with Parkinson’s disease to learn more about their genetic risk factors,” Dr. Alcalay said.
A research driver
Nearly 60% of participants enrolled so far are male and close to 80% are White. The average age is 69 years and 44% were diagnosed in the past 5 years. Close to 75% had never participated in a clinical trial.
Nearly 13% have tested positive for mutations on at least one of the seven target genes. Previous studies had suggested genetics were involved in only about 10% of cases.
The majority of those with positive results had early-onset Parkinson’s disease, high-risk ancestry, or a first-degree relative with the disease. However, 9% of people who tested positive weren’t in any of those categories.
Genetic information collected by the project is shared with the Global Parkinson’s Genetics Program (GP2), a resource program of the Aligning Science Across Parkinson’s initiative that is focused on the disease’s genetic architecture. Researchers around the world have access to GP2 data to study known gene variants and identify new ones.
PD GENEration participants can choose to be notified if they are carriers of gene variants discovered in the future.
“All DNA samples shared by participants are undergoing research-grade testing,” Dr. Beck said. “Not only do we want to be able to inform people with Parkinson’s disease about their genetic status, but we also want to be able to use this precious resource to further drive research into the genetics of Parkinson’s disease.”
Early success
Patient recruitment has long been one of the biggest challenges to any clinical trial’s success. Research suggests that 90% of all clinical trials fail to reach recruitment milestones in their allotted time frame and two-thirds of multicenter trials fold because too few patients sign up. Data from the Parkinson’s Foundation show that only about 1% of all patients with Parkinson’s disease participate in clinical trials.
Increasing those numbers is the primary goal of PD GENEration, Dr. Beck said. And there’s evidence it’s already paying off.
Earlier this year, one of the program’s participating clinical sites, Intermountain Health, in Salt Lake City, Utah, joined a phase 2 clinical trial of an experimental drug that targets a mutation on the GBA1 gene.
“One of the reasons we were able to participate was when we got the call about joining, we were able to say that we had patients with that specific gene mutation, and we could only say that because the patients had been genotyped through PD GENEration,” said Kathleen E. McKee, MD, director of movement disorders, associate medical director of neurosciences research, and PD GENEration principal investigator at Intermountain Health.
Since 2021, Dr. McKee has enrolled hundreds of patients in the foundation’s gene study and hopes to enroll even more. Few patients turn down the opportunity to participate, she added. Knowing their genotype has proven empowering for her patients, most of whom could not afford genetic testing on their own.
“Previously I would tell patients this is not going to change your immediate management,” Dr. McKee said. “Now I tell my patients that these trials are out there, it may actually change how I treat you and what I recommend.”
A version of this article appeared on Medscape.com.
In 2017, Sanofi Genzyme launched a phase 2 clinical trial of a drug designed to target a specific genetic mutation in some patients with Parkinson’s disease. Researchers hoped the drug would slow or even stop disease progression.
Like many before it, the trial yielded disappointing results and the company shut it down in 2021. It was the latest in a string of unsuccessful clinical trials testing disease-modifying Parkinson’s disease drugs.
Although it failed, the Sanofi Genzyme study was different: It was the first to enroll patients with Parkinson’s disease who had a specific genotype and marked the earliest days of precision medicine and gene-specific drug development for the disease.
Once thought to play only a small role in a small number of patients with Parkinson’s disease,
“We’re about to enter this era of precision medicine for Parkinson’s disease, which makes genetic testing important,” said James Beck, PhD, senior vice president and chief scientific officer for the Parkinson’s Foundation.
“A number of companies have clinical trials or are in preparation for clinical trials to test some specific therapies that would depend upon people having a specific genetic mutation,” he said.
Today, at least four clinical trials of drugs that target specific Parkinson’s disease-related gene variants on LRRK2 and GBA are under way, and more are in the pipeline. Whether these drugs will be effective at modifying the course of the disease remains to be seen. First, the trials must enroll enough patients. And therein lies the challenge: Genetic testing isn’t part of routine Parkinson’s disease care and isn’t covered by most insurance policies. Most patients don’t know their genotype.
It’s a significant roadblock to the future of a precision medicine approach that is based on a patient’s individual genotype, which some experts argue offers the best shot at slowing disease progression.
“To enroll in clinical trials for precision drugs people with Parkinson’s disease have to be aware of their genetic status,” said Roy N. Alcalay, MD, chief of the movement disorders division at Tel Aviv Medical Center in Israel and part-time associate professor at Columbia University in New York. “How can a person with Parkinson’s and a LRRK2 mutation join a precision medicine trial for LRRK2 if she does not know she is a LRRK2 carrier?”
Free genetic testing
Previous studies have shown that some genetic variants increase the risk for Parkinson’s disease after exposure to environmental factors such as pesticides. Research has also shown that a patient’s genotype can predict survival time and that certain medications may prove more effective at slowing disease progression in patients with specific genotypes. All of this points to a significant role for genetics in a disorder that is rapidly increasing.
This makes expanding patient access to genetic testing even more important, Dr. Alcalay said, noting that it’s equally important that patients are informed of their genotype, something that doesn’t usually happen in blinded clinical trials.
To that end, Dr. Alcalay hopes a national genetics study he is leading will address access and need-to-know issues. PD GENEration, a project launched in 2019 by the Parkinson’s Foundation, offers patients free genetic testing for seven clinically relevant Parkinson’s disease-related genes.
Testing is done at home or in a nearby clinic and the results are shared with patients during a free genetic counseling session and with site investigators. Patient samples are stored in a genetic data bank that is open to researchers around the world.
“We surveyed clinical trialists in the Parkinson’s disease field prior to initiation of PD GENEration and estimated that over 90% of people with Parkinson’s disease prior to the effort were not aware of their genetic status,” Dr. Alcalay said.
“I think precision medicine in Parkinson’s disease will not happen without PD GENEration or similar efforts.”
‘Overwhelming’ patient interest
Participants in the study are screened for variants in seven genes known to be involved in Parkinson’s disease risk: GBA, LRRK2, PRKN, PINK1, SNCA, PARK7, and VPS35.
In less than 3 years, the study has already produced what is thought to be the largest genetic data bank of sequenced sets of Parkinson’s disease-risk genes made accessible to patients. Since the end of 2020, the first year of patient enrollment, the number of participants has increased from 676 to 10,515 and the number of participating clinical sites rose from 12 to 101.
The foundation has spent nearly $20 million on the project so far and plans to spend another $10 million to reach a goal of 15,000 patients. The study, which is funded by private donors, is so successful that the foundation has had to scale back enrollment.
“When we were at a peak, we had over 700 participants enrolling each month,” Dr. Beck said. Beginning in April, the program capped new sign-ups to 200 patients per month and created a waiting list for future enrollment. The waiting list is hundreds of patients long.
“The participants’ response to enroll in PD GENEration demonstrates there is an overwhelming interest by people with Parkinson’s disease to learn more about their genetic risk factors,” Dr. Alcalay said.
A research driver
Nearly 60% of participants enrolled so far are male and close to 80% are White. The average age is 69 years and 44% were diagnosed in the past 5 years. Close to 75% had never participated in a clinical trial.
Nearly 13% have tested positive for mutations on at least one of the seven target genes. Previous studies had suggested genetics were involved in only about 10% of cases.
The majority of those with positive results had early-onset Parkinson’s disease, high-risk ancestry, or a first-degree relative with the disease. However, 9% of people who tested positive weren’t in any of those categories.
Genetic information collected by the project is shared with the Global Parkinson’s Genetics Program (GP2), a resource program of the Aligning Science Across Parkinson’s initiative that is focused on the disease’s genetic architecture. Researchers around the world have access to GP2 data to study known gene variants and identify new ones.
PD GENEration participants can choose to be notified if they are carriers of gene variants discovered in the future.
“All DNA samples shared by participants are undergoing research-grade testing,” Dr. Beck said. “Not only do we want to be able to inform people with Parkinson’s disease about their genetic status, but we also want to be able to use this precious resource to further drive research into the genetics of Parkinson’s disease.”
Early success
Patient recruitment has long been one of the biggest challenges to any clinical trial’s success. Research suggests that 90% of all clinical trials fail to reach recruitment milestones in their allotted time frame and two-thirds of multicenter trials fold because too few patients sign up. Data from the Parkinson’s Foundation show that only about 1% of all patients with Parkinson’s disease participate in clinical trials.
Increasing those numbers is the primary goal of PD GENEration, Dr. Beck said. And there’s evidence it’s already paying off.
Earlier this year, one of the program’s participating clinical sites, Intermountain Health, in Salt Lake City, Utah, joined a phase 2 clinical trial of an experimental drug that targets a mutation on the GBA1 gene.
“One of the reasons we were able to participate was when we got the call about joining, we were able to say that we had patients with that specific gene mutation, and we could only say that because the patients had been genotyped through PD GENEration,” said Kathleen E. McKee, MD, director of movement disorders, associate medical director of neurosciences research, and PD GENEration principal investigator at Intermountain Health.
Since 2021, Dr. McKee has enrolled hundreds of patients in the foundation’s gene study and hopes to enroll even more. Few patients turn down the opportunity to participate, she added. Knowing their genotype has proven empowering for her patients, most of whom could not afford genetic testing on their own.
“Previously I would tell patients this is not going to change your immediate management,” Dr. McKee said. “Now I tell my patients that these trials are out there, it may actually change how I treat you and what I recommend.”
A version of this article appeared on Medscape.com.
In 2017, Sanofi Genzyme launched a phase 2 clinical trial of a drug designed to target a specific genetic mutation in some patients with Parkinson’s disease. Researchers hoped the drug would slow or even stop disease progression.
Like many before it, the trial yielded disappointing results and the company shut it down in 2021. It was the latest in a string of unsuccessful clinical trials testing disease-modifying Parkinson’s disease drugs.
Although it failed, the Sanofi Genzyme study was different: It was the first to enroll patients with Parkinson’s disease who had a specific genotype and marked the earliest days of precision medicine and gene-specific drug development for the disease.
Once thought to play only a small role in a small number of patients with Parkinson’s disease,
“We’re about to enter this era of precision medicine for Parkinson’s disease, which makes genetic testing important,” said James Beck, PhD, senior vice president and chief scientific officer for the Parkinson’s Foundation.
“A number of companies have clinical trials or are in preparation for clinical trials to test some specific therapies that would depend upon people having a specific genetic mutation,” he said.
Today, at least four clinical trials of drugs that target specific Parkinson’s disease-related gene variants on LRRK2 and GBA are under way, and more are in the pipeline. Whether these drugs will be effective at modifying the course of the disease remains to be seen. First, the trials must enroll enough patients. And therein lies the challenge: Genetic testing isn’t part of routine Parkinson’s disease care and isn’t covered by most insurance policies. Most patients don’t know their genotype.
It’s a significant roadblock to the future of a precision medicine approach that is based on a patient’s individual genotype, which some experts argue offers the best shot at slowing disease progression.
“To enroll in clinical trials for precision drugs people with Parkinson’s disease have to be aware of their genetic status,” said Roy N. Alcalay, MD, chief of the movement disorders division at Tel Aviv Medical Center in Israel and part-time associate professor at Columbia University in New York. “How can a person with Parkinson’s and a LRRK2 mutation join a precision medicine trial for LRRK2 if she does not know she is a LRRK2 carrier?”
Free genetic testing
Previous studies have shown that some genetic variants increase the risk for Parkinson’s disease after exposure to environmental factors such as pesticides. Research has also shown that a patient’s genotype can predict survival time and that certain medications may prove more effective at slowing disease progression in patients with specific genotypes. All of this points to a significant role for genetics in a disorder that is rapidly increasing.
This makes expanding patient access to genetic testing even more important, Dr. Alcalay said, noting that it’s equally important that patients are informed of their genotype, something that doesn’t usually happen in blinded clinical trials.
To that end, Dr. Alcalay hopes a national genetics study he is leading will address access and need-to-know issues. PD GENEration, a project launched in 2019 by the Parkinson’s Foundation, offers patients free genetic testing for seven clinically relevant Parkinson’s disease-related genes.
Testing is done at home or in a nearby clinic and the results are shared with patients during a free genetic counseling session and with site investigators. Patient samples are stored in a genetic data bank that is open to researchers around the world.
“We surveyed clinical trialists in the Parkinson’s disease field prior to initiation of PD GENEration and estimated that over 90% of people with Parkinson’s disease prior to the effort were not aware of their genetic status,” Dr. Alcalay said.
“I think precision medicine in Parkinson’s disease will not happen without PD GENEration or similar efforts.”
‘Overwhelming’ patient interest
Participants in the study are screened for variants in seven genes known to be involved in Parkinson’s disease risk: GBA, LRRK2, PRKN, PINK1, SNCA, PARK7, and VPS35.
In less than 3 years, the study has already produced what is thought to be the largest genetic data bank of sequenced sets of Parkinson’s disease-risk genes made accessible to patients. Since the end of 2020, the first year of patient enrollment, the number of participants has increased from 676 to 10,515 and the number of participating clinical sites rose from 12 to 101.
The foundation has spent nearly $20 million on the project so far and plans to spend another $10 million to reach a goal of 15,000 patients. The study, which is funded by private donors, is so successful that the foundation has had to scale back enrollment.
“When we were at a peak, we had over 700 participants enrolling each month,” Dr. Beck said. Beginning in April, the program capped new sign-ups to 200 patients per month and created a waiting list for future enrollment. The waiting list is hundreds of patients long.
“The participants’ response to enroll in PD GENEration demonstrates there is an overwhelming interest by people with Parkinson’s disease to learn more about their genetic risk factors,” Dr. Alcalay said.
A research driver
Nearly 60% of participants enrolled so far are male and close to 80% are White. The average age is 69 years and 44% were diagnosed in the past 5 years. Close to 75% had never participated in a clinical trial.
Nearly 13% have tested positive for mutations on at least one of the seven target genes. Previous studies had suggested genetics were involved in only about 10% of cases.
The majority of those with positive results had early-onset Parkinson’s disease, high-risk ancestry, or a first-degree relative with the disease. However, 9% of people who tested positive weren’t in any of those categories.
Genetic information collected by the project is shared with the Global Parkinson’s Genetics Program (GP2), a resource program of the Aligning Science Across Parkinson’s initiative that is focused on the disease’s genetic architecture. Researchers around the world have access to GP2 data to study known gene variants and identify new ones.
PD GENEration participants can choose to be notified if they are carriers of gene variants discovered in the future.
“All DNA samples shared by participants are undergoing research-grade testing,” Dr. Beck said. “Not only do we want to be able to inform people with Parkinson’s disease about their genetic status, but we also want to be able to use this precious resource to further drive research into the genetics of Parkinson’s disease.”
Early success
Patient recruitment has long been one of the biggest challenges to any clinical trial’s success. Research suggests that 90% of all clinical trials fail to reach recruitment milestones in their allotted time frame and two-thirds of multicenter trials fold because too few patients sign up. Data from the Parkinson’s Foundation show that only about 1% of all patients with Parkinson’s disease participate in clinical trials.
Increasing those numbers is the primary goal of PD GENEration, Dr. Beck said. And there’s evidence it’s already paying off.
Earlier this year, one of the program’s participating clinical sites, Intermountain Health, in Salt Lake City, Utah, joined a phase 2 clinical trial of an experimental drug that targets a mutation on the GBA1 gene.
“One of the reasons we were able to participate was when we got the call about joining, we were able to say that we had patients with that specific gene mutation, and we could only say that because the patients had been genotyped through PD GENEration,” said Kathleen E. McKee, MD, director of movement disorders, associate medical director of neurosciences research, and PD GENEration principal investigator at Intermountain Health.
Since 2021, Dr. McKee has enrolled hundreds of patients in the foundation’s gene study and hopes to enroll even more. Few patients turn down the opportunity to participate, she added. Knowing their genotype has proven empowering for her patients, most of whom could not afford genetic testing on their own.
“Previously I would tell patients this is not going to change your immediate management,” Dr. McKee said. “Now I tell my patients that these trials are out there, it may actually change how I treat you and what I recommend.”
A version of this article appeared on Medscape.com.
Can a repurposed Parkinson’s drug slow ALS progression?
However, at least one expert believes the study has “significant flaws.”
Investigators randomly assigned 20 individuals with sporadic ALS to receive either ropinirole or placebo for 24 weeks. During the double-blind period, there was no difference between the groups in terms of decline in functional status.
However, during a further open-label extension period, the ropinirole group showed significant suppression of functional decline and an average of an additional 7 months of progression-free survival.
The researchers were able to predict clinical responsiveness to ropinirole in vitro by analyzing motor neurons derived from participants’ stem cells.
“We found that ropinirole is safe and tolerable for ALS patients and shows therapeutic promise at helping them sustain daily activity and muscle strength,” first author Satoru Morimoto, MD, of the department of physiology, Keio University School of Medicine, Tokyo, said in a news release.
The study was published online in Cell Stem Cell.
Feasibility study
“ALS is totally incurable and it’s a very difficult disease to treat,” senior author Hideyuki Okano, MD, PhD, professor, department of physiology, Keio University, said in the news release.
Preclinical animal models have “limited translational potential” for identifying drug candidates, but induced pluripotent stem cell (iPSC)–derived motor neurons (MNs) from ALS patients can “overcome these limitations for drug screening,” the authors write.
“We previously identified ropinirole [a dopamine D2 receptor agonist] as a potential anti-ALS drug in vitro by iPSC drug discovery,” Dr. Okano said.
The current trial was a randomized, placebo-controlled phase 1/2a feasibility trial that evaluated the safety, tolerability, and efficacy of ropinirole in patients with ALS, using several parameters:
- The revised ALS functional rating scale (ALSFRS-R) score.
- Composite functional endpoints.
- Event-free survival.
- Time to ≤ 50% forced vital capacity (FVC).
The trial consisted of a 12-week run-in period, a 24-week double-blind period, an open-label extension period that lasted from 4 to 24 weeks, and a 4-week follow-up period after administration.
Thirteen patients were assigned to receive ropinirole (23.1% women; mean age, 65.2 ± 12.6 years; 7.7% with clinically definite and 76.9% with clinically probable ALS); seven were assigned to receive placebo (57.1% women; mean age, 66.3 ± 7.5 years; 14.3% with clinically definite and 85.7% with clinically probable ALS).
Of the treatment group, 30.8% had a bulbar onset lesion vs. 57.1% in the placebo group. At baseline, the mean FVC was 94.4% ± 14.9 and 81.5% ± 23.2 in the ropinirole and placebo groups, respectively. The mean body mass index (BMI) was 22.91 ± 3.82 and 19.69 ± 2.63, respectively.
Of the participants,12 in the ropinirole and six in the control group completed the full 24-week treatment protocol; 12 in the ropinirole and five in the placebo group completed the open-label extension (participants who had received placebo were switched to the active drug).
However only seven participants in the ropinirole group and one participant in the placebo group completed the full 1-year trial.
‘Striking correlation’
“During the double-blind period, muscle strength and daily activity were maintained, but a decline in the ALSFRS-R … was not different from that in the placebo group,” the researchers write.
In the open-label extension period, the ropinirole group showed “significant suppression of ALSFRS-R decline,” with an ALSFRS-R score change of only 7.75 (95% confidence interval, 10.66-4.63) for the treatment group vs. 17.51 (95% CI, 22.46-12.56) for the placebo group.
The researchers used the assessment of function and survival (CAFS) score, which adjusts the ALSFRS-R score against mortality, to see whether functional benefits translated into improved survival.
The score “favored ropinirole” in the open-extension period and the entire treatment period but not in the double-blind period.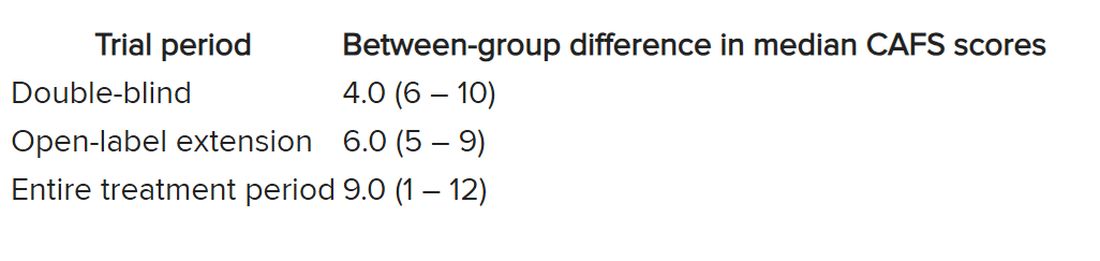
Disease progression events occurred in 7 of 7 (100%) participants in the placebo group and 7 of 13 (54%) in the ropinirole group, “suggesting a twofold decrease in disease progression” in the treatment group.
The ropinirole group experienced an additional 27.9 weeks of disease progression–free survival, compared with the placebo group.
“No participant discontinued treatment because of adverse experiences in either treatment group,” the authors report.
The analysis of iPSC-derived motor neurons from participants showed dopamine D2 receptor expression, as well as the potential involvement of the cholesterol pathway SREBP2 in the therapeutic effects of ropinirole. Lipid peroxide was also identified as a good “surrogate clinical marker to assess disease progression and drug efficacy.”
“We found a very striking correlation between a patient’s clinical response and the response of their motor neurons in vitro,” said Dr. Morimoto. “Patients whose motor neurons responded robustly to ropinirole in vitro had a much slower clinical disease progression with ropinirole treatment, while suboptimal responders showed much more rapid disease progression, despite taking ropinirole.”
Limitations include “small sample sizes and high attrition rates in the open-label extension period,” so “further validation” is required, the authors state.
Significant flaws
Commenting for this article, Carmel Armon, MD, MHS, professor of neurology, Loma Linda (Calif.) University, said the study “falls short of being a credible 1/2a clinical trial.”
Although the “intentions were good and the design not unusual,” the two groups were not “balanced on risk factors for faster progressing disease.” Rather, the placebo group was “tilted towards faster progressing disease” because there were more clinically definite and probable ALS patients in the placebo group than the treatment group, and there were more patients with bulbar onset.
Participants in the placebo group also had shorter median disease duration, lower BMI, and lower FVC, noted Dr. Armon, who was not involved with the study.
And only 1 in 7 control patients completed the open-label extension, compared with 7 of 13 patients in the intervention group.
“With these limitations, I would be disinclined to rely on the findings to justify a larger clinical trial,” Dr. Armon concluded.
The trial was sponsored by K Pharma. The study drug, active drugs, and placebo were supplied free of charge by GlaxoSmithKline K.K. Dr. Okano received grants from JSPS and AMED and grants and personal fees from K Pharma during the conduct of the study and personal fees from Sanbio, outside the submitted work. Dr. Okano has a patent on a therapeutic agent for ALS and composition for treatment licensed to K Pharma. The other authors’ disclosures and additional information are available in the original article. Dr. Armon reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
However, at least one expert believes the study has “significant flaws.”
Investigators randomly assigned 20 individuals with sporadic ALS to receive either ropinirole or placebo for 24 weeks. During the double-blind period, there was no difference between the groups in terms of decline in functional status.
However, during a further open-label extension period, the ropinirole group showed significant suppression of functional decline and an average of an additional 7 months of progression-free survival.
The researchers were able to predict clinical responsiveness to ropinirole in vitro by analyzing motor neurons derived from participants’ stem cells.
“We found that ropinirole is safe and tolerable for ALS patients and shows therapeutic promise at helping them sustain daily activity and muscle strength,” first author Satoru Morimoto, MD, of the department of physiology, Keio University School of Medicine, Tokyo, said in a news release.
The study was published online in Cell Stem Cell.
Feasibility study
“ALS is totally incurable and it’s a very difficult disease to treat,” senior author Hideyuki Okano, MD, PhD, professor, department of physiology, Keio University, said in the news release.
Preclinical animal models have “limited translational potential” for identifying drug candidates, but induced pluripotent stem cell (iPSC)–derived motor neurons (MNs) from ALS patients can “overcome these limitations for drug screening,” the authors write.
“We previously identified ropinirole [a dopamine D2 receptor agonist] as a potential anti-ALS drug in vitro by iPSC drug discovery,” Dr. Okano said.
The current trial was a randomized, placebo-controlled phase 1/2a feasibility trial that evaluated the safety, tolerability, and efficacy of ropinirole in patients with ALS, using several parameters:
- The revised ALS functional rating scale (ALSFRS-R) score.
- Composite functional endpoints.
- Event-free survival.
- Time to ≤ 50% forced vital capacity (FVC).
The trial consisted of a 12-week run-in period, a 24-week double-blind period, an open-label extension period that lasted from 4 to 24 weeks, and a 4-week follow-up period after administration.
Thirteen patients were assigned to receive ropinirole (23.1% women; mean age, 65.2 ± 12.6 years; 7.7% with clinically definite and 76.9% with clinically probable ALS); seven were assigned to receive placebo (57.1% women; mean age, 66.3 ± 7.5 years; 14.3% with clinically definite and 85.7% with clinically probable ALS).
Of the treatment group, 30.8% had a bulbar onset lesion vs. 57.1% in the placebo group. At baseline, the mean FVC was 94.4% ± 14.9 and 81.5% ± 23.2 in the ropinirole and placebo groups, respectively. The mean body mass index (BMI) was 22.91 ± 3.82 and 19.69 ± 2.63, respectively.
Of the participants,12 in the ropinirole and six in the control group completed the full 24-week treatment protocol; 12 in the ropinirole and five in the placebo group completed the open-label extension (participants who had received placebo were switched to the active drug).
However only seven participants in the ropinirole group and one participant in the placebo group completed the full 1-year trial.
‘Striking correlation’
“During the double-blind period, muscle strength and daily activity were maintained, but a decline in the ALSFRS-R … was not different from that in the placebo group,” the researchers write.
In the open-label extension period, the ropinirole group showed “significant suppression of ALSFRS-R decline,” with an ALSFRS-R score change of only 7.75 (95% confidence interval, 10.66-4.63) for the treatment group vs. 17.51 (95% CI, 22.46-12.56) for the placebo group.
The researchers used the assessment of function and survival (CAFS) score, which adjusts the ALSFRS-R score against mortality, to see whether functional benefits translated into improved survival.
The score “favored ropinirole” in the open-extension period and the entire treatment period but not in the double-blind period.
Disease progression events occurred in 7 of 7 (100%) participants in the placebo group and 7 of 13 (54%) in the ropinirole group, “suggesting a twofold decrease in disease progression” in the treatment group.
The ropinirole group experienced an additional 27.9 weeks of disease progression–free survival, compared with the placebo group.
“No participant discontinued treatment because of adverse experiences in either treatment group,” the authors report.
The analysis of iPSC-derived motor neurons from participants showed dopamine D2 receptor expression, as well as the potential involvement of the cholesterol pathway SREBP2 in the therapeutic effects of ropinirole. Lipid peroxide was also identified as a good “surrogate clinical marker to assess disease progression and drug efficacy.”
“We found a very striking correlation between a patient’s clinical response and the response of their motor neurons in vitro,” said Dr. Morimoto. “Patients whose motor neurons responded robustly to ropinirole in vitro had a much slower clinical disease progression with ropinirole treatment, while suboptimal responders showed much more rapid disease progression, despite taking ropinirole.”
Limitations include “small sample sizes and high attrition rates in the open-label extension period,” so “further validation” is required, the authors state.
Significant flaws
Commenting for this article, Carmel Armon, MD, MHS, professor of neurology, Loma Linda (Calif.) University, said the study “falls short of being a credible 1/2a clinical trial.”
Although the “intentions were good and the design not unusual,” the two groups were not “balanced on risk factors for faster progressing disease.” Rather, the placebo group was “tilted towards faster progressing disease” because there were more clinically definite and probable ALS patients in the placebo group than the treatment group, and there were more patients with bulbar onset.
Participants in the placebo group also had shorter median disease duration, lower BMI, and lower FVC, noted Dr. Armon, who was not involved with the study.
And only 1 in 7 control patients completed the open-label extension, compared with 7 of 13 patients in the intervention group.
“With these limitations, I would be disinclined to rely on the findings to justify a larger clinical trial,” Dr. Armon concluded.
The trial was sponsored by K Pharma. The study drug, active drugs, and placebo were supplied free of charge by GlaxoSmithKline K.K. Dr. Okano received grants from JSPS and AMED and grants and personal fees from K Pharma during the conduct of the study and personal fees from Sanbio, outside the submitted work. Dr. Okano has a patent on a therapeutic agent for ALS and composition for treatment licensed to K Pharma. The other authors’ disclosures and additional information are available in the original article. Dr. Armon reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
However, at least one expert believes the study has “significant flaws.”
Investigators randomly assigned 20 individuals with sporadic ALS to receive either ropinirole or placebo for 24 weeks. During the double-blind period, there was no difference between the groups in terms of decline in functional status.
However, during a further open-label extension period, the ropinirole group showed significant suppression of functional decline and an average of an additional 7 months of progression-free survival.
The researchers were able to predict clinical responsiveness to ropinirole in vitro by analyzing motor neurons derived from participants’ stem cells.
“We found that ropinirole is safe and tolerable for ALS patients and shows therapeutic promise at helping them sustain daily activity and muscle strength,” first author Satoru Morimoto, MD, of the department of physiology, Keio University School of Medicine, Tokyo, said in a news release.
The study was published online in Cell Stem Cell.
Feasibility study
“ALS is totally incurable and it’s a very difficult disease to treat,” senior author Hideyuki Okano, MD, PhD, professor, department of physiology, Keio University, said in the news release.
Preclinical animal models have “limited translational potential” for identifying drug candidates, but induced pluripotent stem cell (iPSC)–derived motor neurons (MNs) from ALS patients can “overcome these limitations for drug screening,” the authors write.
“We previously identified ropinirole [a dopamine D2 receptor agonist] as a potential anti-ALS drug in vitro by iPSC drug discovery,” Dr. Okano said.
The current trial was a randomized, placebo-controlled phase 1/2a feasibility trial that evaluated the safety, tolerability, and efficacy of ropinirole in patients with ALS, using several parameters:
- The revised ALS functional rating scale (ALSFRS-R) score.
- Composite functional endpoints.
- Event-free survival.
- Time to ≤ 50% forced vital capacity (FVC).
The trial consisted of a 12-week run-in period, a 24-week double-blind period, an open-label extension period that lasted from 4 to 24 weeks, and a 4-week follow-up period after administration.
Thirteen patients were assigned to receive ropinirole (23.1% women; mean age, 65.2 ± 12.6 years; 7.7% with clinically definite and 76.9% with clinically probable ALS); seven were assigned to receive placebo (57.1% women; mean age, 66.3 ± 7.5 years; 14.3% with clinically definite and 85.7% with clinically probable ALS).
Of the treatment group, 30.8% had a bulbar onset lesion vs. 57.1% in the placebo group. At baseline, the mean FVC was 94.4% ± 14.9 and 81.5% ± 23.2 in the ropinirole and placebo groups, respectively. The mean body mass index (BMI) was 22.91 ± 3.82 and 19.69 ± 2.63, respectively.
Of the participants,12 in the ropinirole and six in the control group completed the full 24-week treatment protocol; 12 in the ropinirole and five in the placebo group completed the open-label extension (participants who had received placebo were switched to the active drug).
However only seven participants in the ropinirole group and one participant in the placebo group completed the full 1-year trial.
‘Striking correlation’
“During the double-blind period, muscle strength and daily activity were maintained, but a decline in the ALSFRS-R … was not different from that in the placebo group,” the researchers write.
In the open-label extension period, the ropinirole group showed “significant suppression of ALSFRS-R decline,” with an ALSFRS-R score change of only 7.75 (95% confidence interval, 10.66-4.63) for the treatment group vs. 17.51 (95% CI, 22.46-12.56) for the placebo group.
The researchers used the assessment of function and survival (CAFS) score, which adjusts the ALSFRS-R score against mortality, to see whether functional benefits translated into improved survival.
The score “favored ropinirole” in the open-extension period and the entire treatment period but not in the double-blind period.
Disease progression events occurred in 7 of 7 (100%) participants in the placebo group and 7 of 13 (54%) in the ropinirole group, “suggesting a twofold decrease in disease progression” in the treatment group.
The ropinirole group experienced an additional 27.9 weeks of disease progression–free survival, compared with the placebo group.
“No participant discontinued treatment because of adverse experiences in either treatment group,” the authors report.
The analysis of iPSC-derived motor neurons from participants showed dopamine D2 receptor expression, as well as the potential involvement of the cholesterol pathway SREBP2 in the therapeutic effects of ropinirole. Lipid peroxide was also identified as a good “surrogate clinical marker to assess disease progression and drug efficacy.”
“We found a very striking correlation between a patient’s clinical response and the response of their motor neurons in vitro,” said Dr. Morimoto. “Patients whose motor neurons responded robustly to ropinirole in vitro had a much slower clinical disease progression with ropinirole treatment, while suboptimal responders showed much more rapid disease progression, despite taking ropinirole.”
Limitations include “small sample sizes and high attrition rates in the open-label extension period,” so “further validation” is required, the authors state.
Significant flaws
Commenting for this article, Carmel Armon, MD, MHS, professor of neurology, Loma Linda (Calif.) University, said the study “falls short of being a credible 1/2a clinical trial.”
Although the “intentions were good and the design not unusual,” the two groups were not “balanced on risk factors for faster progressing disease.” Rather, the placebo group was “tilted towards faster progressing disease” because there were more clinically definite and probable ALS patients in the placebo group than the treatment group, and there were more patients with bulbar onset.
Participants in the placebo group also had shorter median disease duration, lower BMI, and lower FVC, noted Dr. Armon, who was not involved with the study.
And only 1 in 7 control patients completed the open-label extension, compared with 7 of 13 patients in the intervention group.
“With these limitations, I would be disinclined to rely on the findings to justify a larger clinical trial,” Dr. Armon concluded.
The trial was sponsored by K Pharma. The study drug, active drugs, and placebo were supplied free of charge by GlaxoSmithKline K.K. Dr. Okano received grants from JSPS and AMED and grants and personal fees from K Pharma during the conduct of the study and personal fees from Sanbio, outside the submitted work. Dr. Okano has a patent on a therapeutic agent for ALS and composition for treatment licensed to K Pharma. The other authors’ disclosures and additional information are available in the original article. Dr. Armon reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM CELL STEM CELL
Novel cannabis oil curbs tics in severe Tourette’s
(TS), results of a double-blind, placebo-controlled, crossover study show.
“In a methodologically robust manner (and independent of any drug company sponsorship), we provide evidence for the effectiveness of repeated dosing with THC:CBD vs. placebo in tic suppression, as well as reduction of comorbid anxiety and obsessive-compulsive disorder in severe TS,” neuropsychiatrist and lead investigator Philip Mosley, PhD, said in an interview.
The results offer support to people with TS who “want to approach their doctor to try medicinal cannabis when other drugs have not worked or are intolerable,” said Dr. Mosley, of the Wesley Research Institute and QIMR Berghofer Medical Research Institute, Herston, Australia.
The study was published online in NEJM Evidence.
A viable treatment option
Twenty-two adults (mean age, 31 years) with severe TS received THC:CBD oil titrated upward over 6 weeks to a daily dose of 20 mg of THC and 20 mg of CBD, followed by a 6-week course of placebo (or vice versa). Six participants had not previously used cannabis.
The primary outcome was the total tic score on the Yale Global Tic Severity Scale (YGTSS; range 0 to 50 with higher scores = greater tic severity).
The mean baseline YGTSS total tic score was 35.7. At 6 weeks, the reduction in total tic score was 8.9 with THC:CBD vs. 2.5 with placebo.
A linear mixed-effects model (intention-to-treat) showed a significant interaction of treatment and visit number (P = .008), indicating a greater decrease (improvement) in tic score over time with THC:CBD, the study team reported.
On average, the magnitude of the tic reduction was “moderate” and comparable to the effect observed with existing treatments such as antipsychotic agents, the investigators noted.
THC:CBD also led to a reduction in other symptoms associated with TS, particularly symptoms of OCD and anxiety.
The symptomatic response to THC:CBD correlated with serum metabolites of the cannabinoids, further supporting a biological relationship, the researchers noted.
There were no serious adverse events. Adverse effects with THC:CBD were generally mild. The most common adverse effect was cognitive difficulties, including slowed mentation, memory lapses, and poor concentration.
“Like many studies of psychoactive compounds, blinding among participants was a problem,” the researchers noted. Despite best efforts to conceal treatment allocation and match placebo to the active agent in terms of color and smell, most participants were able to correctly guess their treatment order.
Based on the findings in this small trial, larger and longer trials of THC:CBD in TS are warranted, they concluded.
“We need a plurality of treatment options in Tourette syndrome. For some, antipsychotics are effective tic-suppressing agents but for many these benefits are complicated by side effects such as weight gain & sedation,” Dr. Mosley tweeted. “Cannabinoids are a biologically plausible therapeutic agent. The body’s own ‘endocannabinoid’ receptors are concentrated in the basal ganglia – the neuroanatomical nexus of TS.”
The study was funded by the Wesley Medical Research Institute, Brisbane, and the Lambert Initiative for Cannabinoid Therapeutics, a philanthropically funded research organization at the University of Sydney, Australia. Dr. Mosley reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
(TS), results of a double-blind, placebo-controlled, crossover study show.
“In a methodologically robust manner (and independent of any drug company sponsorship), we provide evidence for the effectiveness of repeated dosing with THC:CBD vs. placebo in tic suppression, as well as reduction of comorbid anxiety and obsessive-compulsive disorder in severe TS,” neuropsychiatrist and lead investigator Philip Mosley, PhD, said in an interview.
The results offer support to people with TS who “want to approach their doctor to try medicinal cannabis when other drugs have not worked or are intolerable,” said Dr. Mosley, of the Wesley Research Institute and QIMR Berghofer Medical Research Institute, Herston, Australia.
The study was published online in NEJM Evidence.
A viable treatment option
Twenty-two adults (mean age, 31 years) with severe TS received THC:CBD oil titrated upward over 6 weeks to a daily dose of 20 mg of THC and 20 mg of CBD, followed by a 6-week course of placebo (or vice versa). Six participants had not previously used cannabis.
The primary outcome was the total tic score on the Yale Global Tic Severity Scale (YGTSS; range 0 to 50 with higher scores = greater tic severity).
The mean baseline YGTSS total tic score was 35.7. At 6 weeks, the reduction in total tic score was 8.9 with THC:CBD vs. 2.5 with placebo.
A linear mixed-effects model (intention-to-treat) showed a significant interaction of treatment and visit number (P = .008), indicating a greater decrease (improvement) in tic score over time with THC:CBD, the study team reported.
On average, the magnitude of the tic reduction was “moderate” and comparable to the effect observed with existing treatments such as antipsychotic agents, the investigators noted.
THC:CBD also led to a reduction in other symptoms associated with TS, particularly symptoms of OCD and anxiety.
The symptomatic response to THC:CBD correlated with serum metabolites of the cannabinoids, further supporting a biological relationship, the researchers noted.
There were no serious adverse events. Adverse effects with THC:CBD were generally mild. The most common adverse effect was cognitive difficulties, including slowed mentation, memory lapses, and poor concentration.
“Like many studies of psychoactive compounds, blinding among participants was a problem,” the researchers noted. Despite best efforts to conceal treatment allocation and match placebo to the active agent in terms of color and smell, most participants were able to correctly guess their treatment order.
Based on the findings in this small trial, larger and longer trials of THC:CBD in TS are warranted, they concluded.
“We need a plurality of treatment options in Tourette syndrome. For some, antipsychotics are effective tic-suppressing agents but for many these benefits are complicated by side effects such as weight gain & sedation,” Dr. Mosley tweeted. “Cannabinoids are a biologically plausible therapeutic agent. The body’s own ‘endocannabinoid’ receptors are concentrated in the basal ganglia – the neuroanatomical nexus of TS.”
The study was funded by the Wesley Medical Research Institute, Brisbane, and the Lambert Initiative for Cannabinoid Therapeutics, a philanthropically funded research organization at the University of Sydney, Australia. Dr. Mosley reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
(TS), results of a double-blind, placebo-controlled, crossover study show.
“In a methodologically robust manner (and independent of any drug company sponsorship), we provide evidence for the effectiveness of repeated dosing with THC:CBD vs. placebo in tic suppression, as well as reduction of comorbid anxiety and obsessive-compulsive disorder in severe TS,” neuropsychiatrist and lead investigator Philip Mosley, PhD, said in an interview.
The results offer support to people with TS who “want to approach their doctor to try medicinal cannabis when other drugs have not worked or are intolerable,” said Dr. Mosley, of the Wesley Research Institute and QIMR Berghofer Medical Research Institute, Herston, Australia.
The study was published online in NEJM Evidence.
A viable treatment option
Twenty-two adults (mean age, 31 years) with severe TS received THC:CBD oil titrated upward over 6 weeks to a daily dose of 20 mg of THC and 20 mg of CBD, followed by a 6-week course of placebo (or vice versa). Six participants had not previously used cannabis.
The primary outcome was the total tic score on the Yale Global Tic Severity Scale (YGTSS; range 0 to 50 with higher scores = greater tic severity).
The mean baseline YGTSS total tic score was 35.7. At 6 weeks, the reduction in total tic score was 8.9 with THC:CBD vs. 2.5 with placebo.
A linear mixed-effects model (intention-to-treat) showed a significant interaction of treatment and visit number (P = .008), indicating a greater decrease (improvement) in tic score over time with THC:CBD, the study team reported.
On average, the magnitude of the tic reduction was “moderate” and comparable to the effect observed with existing treatments such as antipsychotic agents, the investigators noted.
THC:CBD also led to a reduction in other symptoms associated with TS, particularly symptoms of OCD and anxiety.
The symptomatic response to THC:CBD correlated with serum metabolites of the cannabinoids, further supporting a biological relationship, the researchers noted.
There were no serious adverse events. Adverse effects with THC:CBD were generally mild. The most common adverse effect was cognitive difficulties, including slowed mentation, memory lapses, and poor concentration.
“Like many studies of psychoactive compounds, blinding among participants was a problem,” the researchers noted. Despite best efforts to conceal treatment allocation and match placebo to the active agent in terms of color and smell, most participants were able to correctly guess their treatment order.
Based on the findings in this small trial, larger and longer trials of THC:CBD in TS are warranted, they concluded.
“We need a plurality of treatment options in Tourette syndrome. For some, antipsychotics are effective tic-suppressing agents but for many these benefits are complicated by side effects such as weight gain & sedation,” Dr. Mosley tweeted. “Cannabinoids are a biologically plausible therapeutic agent. The body’s own ‘endocannabinoid’ receptors are concentrated in the basal ganglia – the neuroanatomical nexus of TS.”
The study was funded by the Wesley Medical Research Institute, Brisbane, and the Lambert Initiative for Cannabinoid Therapeutics, a philanthropically funded research organization at the University of Sydney, Australia. Dr. Mosley reports no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM NEJM EVIDENCE
Common gut bacteria linked to Parkinson’s disease
, a small study suggests.
Environmental factors as well as genetics are also suspected to play a role in PD etiology, although the exact cause remains unknown.
“Our findings indicate that specific strains of Desulfovibrio bacteria are likely to cause Parkinson’s disease,” study investigator Per Erik Saris, PhD, from the University of Helsinki, Finland, says in a news release.
The study was published online in Frontiers in Cellular and Infection Microbiology.
Screen and treat?
It builds on earlier work by the researchers that showed that Desulfovibrio bacteria were more prevalent and more abundant in quantity in patients with PD, especially patients with more severe disease, than in healthy individuals.
Desulfovibrio is a genus of gram-negative bacteria commonly found in aquatic environments in which levels of organic material are elevated, as well as in waterlogged soils.
In their latest study, Dr. Saris and colleagues looked for Desulfovibrio species in fecal samples from 10 patients with PD and their healthy spouses. Isolated Desulfovibrio strains were fed to a strain of Caenorhabditis elegans roundworms that expressed human alpha-syn fused with yellow fluorescent protein.
They found that worms fed Desulfovibrio bacteria from patients with PD harbored significantly more (P < .001) and larger alpha-syn aggregates (P < .001) than worms fed Desulfovibrio bacteria from healthy individuals or worms fed Escherichia coli strains.
In addition, worms fed Desulfovibrio strains from patients with PD died in significantly higher quantities than worms fed E. coli bacteria (P < .01).
Desulfovibrio strains isolated from patients with PD and strains isolated from healthy individuals appear to have different traits. Comparative genomics studies are needed to identify genetic differences and pathogenic genes from Desulfovibrio strains from patients with PD, the researchers note.
“Taking into account that aggregation of alpha-syn is a hallmark of PD, the ability of Desulfovibrio bacteria to induce alpha-syn aggregation in large numbers and sizes, as demonstrated in the present study, provides further evidence for the pathogenic role of Desulfovibrio bacteria in PD, as previously suggested,” they add.
The findings highlight the potential for screening and targeted removal of harmful Desulfovibrio bacteria, Dr. Saris suggests in the news release.
No clinical implications
In a comment, James Beck, PhD, chief scientific officer at the Parkinson’s Foundation, cautioned that “this research is in a very early stage, uses a nonvertebrate animal model, and the number of participants is small.
“Understanding the role of the gut microbiome in influencing PD is in its infancy. These are important steps to determining what – if any – link may be between gut bacteria and PD,” Dr. Beck said.
“Right now, there are no implications for the screening/treatment of carriers,” Dr. Beck said.
“It seems that a lot of people, whether with PD or not, harbor Desulfovibrio bacteria in their gut. More research is needed to understand what is different between the Desulfovibrio bacteria of people with PD vs. those who do not have PD,” Dr. Beck added.
The study was supported by the Magnus Ehrnrooth Foundation and the Jane and Aatos Erkko Foundation. Dr. Saris and Dr. Beck have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, a small study suggests.
Environmental factors as well as genetics are also suspected to play a role in PD etiology, although the exact cause remains unknown.
“Our findings indicate that specific strains of Desulfovibrio bacteria are likely to cause Parkinson’s disease,” study investigator Per Erik Saris, PhD, from the University of Helsinki, Finland, says in a news release.
The study was published online in Frontiers in Cellular and Infection Microbiology.
Screen and treat?
It builds on earlier work by the researchers that showed that Desulfovibrio bacteria were more prevalent and more abundant in quantity in patients with PD, especially patients with more severe disease, than in healthy individuals.
Desulfovibrio is a genus of gram-negative bacteria commonly found in aquatic environments in which levels of organic material are elevated, as well as in waterlogged soils.
In their latest study, Dr. Saris and colleagues looked for Desulfovibrio species in fecal samples from 10 patients with PD and their healthy spouses. Isolated Desulfovibrio strains were fed to a strain of Caenorhabditis elegans roundworms that expressed human alpha-syn fused with yellow fluorescent protein.
They found that worms fed Desulfovibrio bacteria from patients with PD harbored significantly more (P < .001) and larger alpha-syn aggregates (P < .001) than worms fed Desulfovibrio bacteria from healthy individuals or worms fed Escherichia coli strains.
In addition, worms fed Desulfovibrio strains from patients with PD died in significantly higher quantities than worms fed E. coli bacteria (P < .01).
Desulfovibrio strains isolated from patients with PD and strains isolated from healthy individuals appear to have different traits. Comparative genomics studies are needed to identify genetic differences and pathogenic genes from Desulfovibrio strains from patients with PD, the researchers note.
“Taking into account that aggregation of alpha-syn is a hallmark of PD, the ability of Desulfovibrio bacteria to induce alpha-syn aggregation in large numbers and sizes, as demonstrated in the present study, provides further evidence for the pathogenic role of Desulfovibrio bacteria in PD, as previously suggested,” they add.
The findings highlight the potential for screening and targeted removal of harmful Desulfovibrio bacteria, Dr. Saris suggests in the news release.
No clinical implications
In a comment, James Beck, PhD, chief scientific officer at the Parkinson’s Foundation, cautioned that “this research is in a very early stage, uses a nonvertebrate animal model, and the number of participants is small.
“Understanding the role of the gut microbiome in influencing PD is in its infancy. These are important steps to determining what – if any – link may be between gut bacteria and PD,” Dr. Beck said.
“Right now, there are no implications for the screening/treatment of carriers,” Dr. Beck said.
“It seems that a lot of people, whether with PD or not, harbor Desulfovibrio bacteria in their gut. More research is needed to understand what is different between the Desulfovibrio bacteria of people with PD vs. those who do not have PD,” Dr. Beck added.
The study was supported by the Magnus Ehrnrooth Foundation and the Jane and Aatos Erkko Foundation. Dr. Saris and Dr. Beck have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, a small study suggests.
Environmental factors as well as genetics are also suspected to play a role in PD etiology, although the exact cause remains unknown.
“Our findings indicate that specific strains of Desulfovibrio bacteria are likely to cause Parkinson’s disease,” study investigator Per Erik Saris, PhD, from the University of Helsinki, Finland, says in a news release.
The study was published online in Frontiers in Cellular and Infection Microbiology.
Screen and treat?
It builds on earlier work by the researchers that showed that Desulfovibrio bacteria were more prevalent and more abundant in quantity in patients with PD, especially patients with more severe disease, than in healthy individuals.
Desulfovibrio is a genus of gram-negative bacteria commonly found in aquatic environments in which levels of organic material are elevated, as well as in waterlogged soils.
In their latest study, Dr. Saris and colleagues looked for Desulfovibrio species in fecal samples from 10 patients with PD and their healthy spouses. Isolated Desulfovibrio strains were fed to a strain of Caenorhabditis elegans roundworms that expressed human alpha-syn fused with yellow fluorescent protein.
They found that worms fed Desulfovibrio bacteria from patients with PD harbored significantly more (P < .001) and larger alpha-syn aggregates (P < .001) than worms fed Desulfovibrio bacteria from healthy individuals or worms fed Escherichia coli strains.
In addition, worms fed Desulfovibrio strains from patients with PD died in significantly higher quantities than worms fed E. coli bacteria (P < .01).
Desulfovibrio strains isolated from patients with PD and strains isolated from healthy individuals appear to have different traits. Comparative genomics studies are needed to identify genetic differences and pathogenic genes from Desulfovibrio strains from patients with PD, the researchers note.
“Taking into account that aggregation of alpha-syn is a hallmark of PD, the ability of Desulfovibrio bacteria to induce alpha-syn aggregation in large numbers and sizes, as demonstrated in the present study, provides further evidence for the pathogenic role of Desulfovibrio bacteria in PD, as previously suggested,” they add.
The findings highlight the potential for screening and targeted removal of harmful Desulfovibrio bacteria, Dr. Saris suggests in the news release.
No clinical implications
In a comment, James Beck, PhD, chief scientific officer at the Parkinson’s Foundation, cautioned that “this research is in a very early stage, uses a nonvertebrate animal model, and the number of participants is small.
“Understanding the role of the gut microbiome in influencing PD is in its infancy. These are important steps to determining what – if any – link may be between gut bacteria and PD,” Dr. Beck said.
“Right now, there are no implications for the screening/treatment of carriers,” Dr. Beck said.
“It seems that a lot of people, whether with PD or not, harbor Desulfovibrio bacteria in their gut. More research is needed to understand what is different between the Desulfovibrio bacteria of people with PD vs. those who do not have PD,” Dr. Beck added.
The study was supported by the Magnus Ehrnrooth Foundation and the Jane and Aatos Erkko Foundation. Dr. Saris and Dr. Beck have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM FRONTIERS IN CELLULAR AND INFECTION MICROBIOLOGY
Rheumatoid arthritis linked to increased Parkinson’s risk
Claims data in 55,000 patients with RA and 273,000 age- and sex-matched controls show that those with RA were 1.74 times more likely than controls to be diagnosed with PD.
“If patients with rheumatoid arthritis begin exhibiting motor symptoms such as muscle rigidity, tremors, or slowed movement, it is imperative that they be evaluated by a qualified neurologist to rule out the possibility of developing Parkinson’s disease,” study investigator Hyungjin Kim, MD, PhD, told this news organization.
Dr. Kim is an associate professor in the department of medical humanities at Sungkyunkwan University School of Medicine in Seoul, South Korea.
The findings were published online in JAMA Neurology.
Conflicting findings
The investigators note that a number of studies have examined the link between RA and PD, with conflicting results – one even showing a 35% reduced risk for PD for individuals with RA. A more recent population-based study in Taiwan showed a 37% higher rate of PD in patients with rheumatic disease.
However, previous studies did not control for important variables such as body mass index or diabetes.
For the current study, the investigators analyzed claims on about 55,000 patients diagnosed with RA between 2010 and 2017, with follow-up until 2019, and compared the outcomes of this group vs. those of 273,000 controls.
The mean age of claimants was 58 years, and 75% were female.
Results showed that those diagnosed with seropositive RA were about twice as likely as controls to be diagnosed with PD. Those with seronegative RA were 1.2 times as likely as controls to be diagnosed with PD.
Dr. Kim noted that although the pathogenic link between RA and PD remains elusive, inflammation probably plays an important role. “Inflammatory cytokines such as tumor necrosis factor alpha and interleukin-6, which are increased in RA patients, can induce microglial activation, leading to neuroinflammation,” he stated.
“These inflammatory cytokines are known to be associated with the dysfunction and degeneration of nigral dopaminergic neurons, which are important in the pathogenesis of PD,” he added.
The investigators noted that patients with RA may have been subject to more frequent health care services than controls and so were more likely to obtain a PD diagnosis.
Another possibility was that because patients with health check-ups were included in the analysis, the findings may have been biased toward those who were older and who had a higher income.
Dr. Kim noted that additional research is required to clarify the pathogenic connection between RA and PD.
“Moreover, additional studies are necessary to explore the potential influence of novel therapeutic treatments for RA on Parkinson’s disease susceptibility in patients with RA,” he said.
Commenting on the findings for this news organization, David Sulzer, PhD, professor of psychiatry, neurology, and pharmacology at Columbia University in New York, said that the study adds to the growing body of evidence showing there is an autoimmune component to PD.
Dr. Sulzer pointed to data in several papers he published with others to this effect, including one showing higher rates of PD in people with inflammatory bowel disease.
The study had no specific funding. The study investigators and Dr. Sulzer report no relevant disclosures.
A version of this article first appeared on Medscape.com.
Claims data in 55,000 patients with RA and 273,000 age- and sex-matched controls show that those with RA were 1.74 times more likely than controls to be diagnosed with PD.
“If patients with rheumatoid arthritis begin exhibiting motor symptoms such as muscle rigidity, tremors, or slowed movement, it is imperative that they be evaluated by a qualified neurologist to rule out the possibility of developing Parkinson’s disease,” study investigator Hyungjin Kim, MD, PhD, told this news organization.
Dr. Kim is an associate professor in the department of medical humanities at Sungkyunkwan University School of Medicine in Seoul, South Korea.
The findings were published online in JAMA Neurology.
Conflicting findings
The investigators note that a number of studies have examined the link between RA and PD, with conflicting results – one even showing a 35% reduced risk for PD for individuals with RA. A more recent population-based study in Taiwan showed a 37% higher rate of PD in patients with rheumatic disease.
However, previous studies did not control for important variables such as body mass index or diabetes.
For the current study, the investigators analyzed claims on about 55,000 patients diagnosed with RA between 2010 and 2017, with follow-up until 2019, and compared the outcomes of this group vs. those of 273,000 controls.
The mean age of claimants was 58 years, and 75% were female.
Results showed that those diagnosed with seropositive RA were about twice as likely as controls to be diagnosed with PD. Those with seronegative RA were 1.2 times as likely as controls to be diagnosed with PD.
Dr. Kim noted that although the pathogenic link between RA and PD remains elusive, inflammation probably plays an important role. “Inflammatory cytokines such as tumor necrosis factor alpha and interleukin-6, which are increased in RA patients, can induce microglial activation, leading to neuroinflammation,” he stated.
“These inflammatory cytokines are known to be associated with the dysfunction and degeneration of nigral dopaminergic neurons, which are important in the pathogenesis of PD,” he added.
The investigators noted that patients with RA may have been subject to more frequent health care services than controls and so were more likely to obtain a PD diagnosis.
Another possibility was that because patients with health check-ups were included in the analysis, the findings may have been biased toward those who were older and who had a higher income.
Dr. Kim noted that additional research is required to clarify the pathogenic connection between RA and PD.
“Moreover, additional studies are necessary to explore the potential influence of novel therapeutic treatments for RA on Parkinson’s disease susceptibility in patients with RA,” he said.
Commenting on the findings for this news organization, David Sulzer, PhD, professor of psychiatry, neurology, and pharmacology at Columbia University in New York, said that the study adds to the growing body of evidence showing there is an autoimmune component to PD.
Dr. Sulzer pointed to data in several papers he published with others to this effect, including one showing higher rates of PD in people with inflammatory bowel disease.
The study had no specific funding. The study investigators and Dr. Sulzer report no relevant disclosures.
A version of this article first appeared on Medscape.com.
Claims data in 55,000 patients with RA and 273,000 age- and sex-matched controls show that those with RA were 1.74 times more likely than controls to be diagnosed with PD.
“If patients with rheumatoid arthritis begin exhibiting motor symptoms such as muscle rigidity, tremors, or slowed movement, it is imperative that they be evaluated by a qualified neurologist to rule out the possibility of developing Parkinson’s disease,” study investigator Hyungjin Kim, MD, PhD, told this news organization.
Dr. Kim is an associate professor in the department of medical humanities at Sungkyunkwan University School of Medicine in Seoul, South Korea.
The findings were published online in JAMA Neurology.
Conflicting findings
The investigators note that a number of studies have examined the link between RA and PD, with conflicting results – one even showing a 35% reduced risk for PD for individuals with RA. A more recent population-based study in Taiwan showed a 37% higher rate of PD in patients with rheumatic disease.
However, previous studies did not control for important variables such as body mass index or diabetes.
For the current study, the investigators analyzed claims on about 55,000 patients diagnosed with RA between 2010 and 2017, with follow-up until 2019, and compared the outcomes of this group vs. those of 273,000 controls.
The mean age of claimants was 58 years, and 75% were female.
Results showed that those diagnosed with seropositive RA were about twice as likely as controls to be diagnosed with PD. Those with seronegative RA were 1.2 times as likely as controls to be diagnosed with PD.
Dr. Kim noted that although the pathogenic link between RA and PD remains elusive, inflammation probably plays an important role. “Inflammatory cytokines such as tumor necrosis factor alpha and interleukin-6, which are increased in RA patients, can induce microglial activation, leading to neuroinflammation,” he stated.
“These inflammatory cytokines are known to be associated with the dysfunction and degeneration of nigral dopaminergic neurons, which are important in the pathogenesis of PD,” he added.
The investigators noted that patients with RA may have been subject to more frequent health care services than controls and so were more likely to obtain a PD diagnosis.
Another possibility was that because patients with health check-ups were included in the analysis, the findings may have been biased toward those who were older and who had a higher income.
Dr. Kim noted that additional research is required to clarify the pathogenic connection between RA and PD.
“Moreover, additional studies are necessary to explore the potential influence of novel therapeutic treatments for RA on Parkinson’s disease susceptibility in patients with RA,” he said.
Commenting on the findings for this news organization, David Sulzer, PhD, professor of psychiatry, neurology, and pharmacology at Columbia University in New York, said that the study adds to the growing body of evidence showing there is an autoimmune component to PD.
Dr. Sulzer pointed to data in several papers he published with others to this effect, including one showing higher rates of PD in people with inflammatory bowel disease.
The study had no specific funding. The study investigators and Dr. Sulzer report no relevant disclosures.
A version of this article first appeared on Medscape.com.
FROM JAMA NEUROLOGY
Novel levodopa delivery system promises continuous dosing without surgery or pump
BOSTON – , according to an early clinical experience described in the Emerging Science session at the 2023 annual meeting of the American Academy of Neurology.

On this device, the attenuation of levodopa fluctuations “translated into dramatic improvements in clinical behavior, including highly significant reductions in OFF time and an increase in ON time with no dyskinesias,” reported C. Warren Olanow, MD, who is a chairman emeritus of the department of neurology at the Icahn School of Medicine at Mount Sinai, New York, and now an employee of the company developing this new device.
A novel strategy
Numerous studies have demonstrated that reductions in the troughs of plasma levodopa associated with oral dosing result in longer ON time with fewer dyskinesias, according to Dr. Olanow, who explained this has led to strategies for numerous strategies to achieve continuous delivery. A device that delivers levodopa into the stomach through a surgically implanted catheter has already received regulatory approval. Other devices delivering levodopa subcutaneously are in development, but Dr. Olanow said each of these has had limitations.
“The problem with these approaches is they are associated with potentially serious side effects and they require the patient to wear a cumbersome device,” he explained. Relative to the subcutaneous delivery systems, which have been associated with injection site reactions that include painful nodules, and the surgically implanted devices, which also require an external pump, the latest strategy avoids both disadvantages.
Called DopaFuse, the experimental device is designed to deliver the levodopa and carbidopa into the mouth through a micropump within a wearable retainer. Dr. Olanow said that previous experimental studies demonstrated that small doses of levodopa delivered by mouth to the gastrointestinal system reduce levodopa plasma variability. This early clinical study supports that premise. Levodopa delivered into the mouth by way of a propellant in the retainer-mounted pump improved clinical endpoints.
Encouraging trial results
In the study, 16 patients between the ages of 30 and 75 with Parkinson’s disease were enrolled. On day 1, they received an oral dose of levodopa/carbidopa consistent with their current treatment. On day 2, levodopa/carbidopa was delivered through the retainer-mounted device at equivalent doses. On day 3, they received a single morning oral dose and the received the remainder of their levodopa/carbidopa regimen through the device. On days 4 to 14, they received treatment in the same schedule as day 3.
When pharmacokinetics of levodopa on day 3 were compared with those on day 1, the fluctuation index and coefficient of levodopa concentration variability was reduced to a degree that was highly statistically significant (P < .0001). This, in turn, correlated with “striking” reductions in OFF time with equally statistically significant improvement in ON time and ON time without dyskinesias, according to Dr. Olanow.
Relative to an OFF time of 3.2 hours on day 1, the OFF time of 1.6 hours on day 3 represented a 50% reduction (P < .0001). ON time improved from 12.8 hours to 14.5 hours (P < .001). ON time without dyskinesias improved numerically from 8.8 hours to 9.6 hours.
“There were also improvements in activities of daily living when patients were on DopaFuse, which is a hard endpoint to reach in a study with such a small sample size,” Dr. Olanow reported.
There were no serious adverse events. Three patients reported vomiting and two patients each reported headache, but these events were mild and all resolved within a day. Three patients reported buccal lesions, but these also resolved within a day.
“Some patients reported trouble with speaking in the beginning but at the end of the study, patients were reporting that it was easier to speak because of the motor improvements,” Dr. Olanow said.
Overall, the device was well tolerated by the subjects, providing the evidence for the next stages of clinical studies, reported Dr. Olanow.
“If this turns out to be what we hope it is, it will allow us to deliver levodopa without motor complications, without need for a surgical procedure, and without the risk of subcutaneous lesions,” Dr. Olanow said.
More delivery strategies are needed
This device is in an early phase of development, but several specialists in Parkinson’s disease agreed that there is a need for more strategies to provide continuous levodopa in patients with advancing symptoms. Stuart Isaacson, MD, director, Parkinson’s Disease and Movement Disorders Center of Boca Raton, Fla., is among them.
“Novel delivery devices that can provide more continuous levodopa delivery would be an important therapeutic advance,” Dr. Isaacson said. He called levodopa “the cornerstone of treatment through the course of Parkinson’s disease,” but more physiologic dosing in advancing disease has been a challenge.
“While there are many therapies currently available to manage OFF time, many people living with Parkinson’s disease continue to spend only half of their waking day with good ON time,” he added.
The currently approved method of delivering continuous levodopa through a surgically placed catheter into the gastrointestinal system is effective, but has limitations, according to Aaron L. Ellenbogen, MD, a neurologist at Beaumont Hospital, Farmington Hills, Mich.
“One of the challenges with the current treatment landscape of Parkinson’s disease is that medication can be absorbed variably through the gastrointestinal system,” he said. “As the disease progresses, this often becomes more troublesome.” Although this new device is likely to share this issue, Dr. Ellenbogen said that several devices might be useful to match patients with the one that works best for them.
Dr. Olanow is the founder and CEO of Clintrex Research Corporation, through which he also serves as chief medical officer of SynAgile, the company developing DopaFuse. Dr. Isaacson has financial relationships with more than 30 companies, including those that produce levodopa and levodopa delivery systems. Dr. Ellenbogen has financial relationships with Allergan, Acorda, Supernus, and Teva.
BOSTON – , according to an early clinical experience described in the Emerging Science session at the 2023 annual meeting of the American Academy of Neurology.
On this device, the attenuation of levodopa fluctuations “translated into dramatic improvements in clinical behavior, including highly significant reductions in OFF time and an increase in ON time with no dyskinesias,” reported C. Warren Olanow, MD, who is a chairman emeritus of the department of neurology at the Icahn School of Medicine at Mount Sinai, New York, and now an employee of the company developing this new device.
A novel strategy
Numerous studies have demonstrated that reductions in the troughs of plasma levodopa associated with oral dosing result in longer ON time with fewer dyskinesias, according to Dr. Olanow, who explained this has led to strategies for numerous strategies to achieve continuous delivery. A device that delivers levodopa into the stomach through a surgically implanted catheter has already received regulatory approval. Other devices delivering levodopa subcutaneously are in development, but Dr. Olanow said each of these has had limitations.
“The problem with these approaches is they are associated with potentially serious side effects and they require the patient to wear a cumbersome device,” he explained. Relative to the subcutaneous delivery systems, which have been associated with injection site reactions that include painful nodules, and the surgically implanted devices, which also require an external pump, the latest strategy avoids both disadvantages.
Called DopaFuse, the experimental device is designed to deliver the levodopa and carbidopa into the mouth through a micropump within a wearable retainer. Dr. Olanow said that previous experimental studies demonstrated that small doses of levodopa delivered by mouth to the gastrointestinal system reduce levodopa plasma variability. This early clinical study supports that premise. Levodopa delivered into the mouth by way of a propellant in the retainer-mounted pump improved clinical endpoints.
Encouraging trial results
In the study, 16 patients between the ages of 30 and 75 with Parkinson’s disease were enrolled. On day 1, they received an oral dose of levodopa/carbidopa consistent with their current treatment. On day 2, levodopa/carbidopa was delivered through the retainer-mounted device at equivalent doses. On day 3, they received a single morning oral dose and the received the remainder of their levodopa/carbidopa regimen through the device. On days 4 to 14, they received treatment in the same schedule as day 3.
When pharmacokinetics of levodopa on day 3 were compared with those on day 1, the fluctuation index and coefficient of levodopa concentration variability was reduced to a degree that was highly statistically significant (P < .0001). This, in turn, correlated with “striking” reductions in OFF time with equally statistically significant improvement in ON time and ON time without dyskinesias, according to Dr. Olanow.
Relative to an OFF time of 3.2 hours on day 1, the OFF time of 1.6 hours on day 3 represented a 50% reduction (P < .0001). ON time improved from 12.8 hours to 14.5 hours (P < .001). ON time without dyskinesias improved numerically from 8.8 hours to 9.6 hours.
“There were also improvements in activities of daily living when patients were on DopaFuse, which is a hard endpoint to reach in a study with such a small sample size,” Dr. Olanow reported.
There were no serious adverse events. Three patients reported vomiting and two patients each reported headache, but these events were mild and all resolved within a day. Three patients reported buccal lesions, but these also resolved within a day.
“Some patients reported trouble with speaking in the beginning but at the end of the study, patients were reporting that it was easier to speak because of the motor improvements,” Dr. Olanow said.
Overall, the device was well tolerated by the subjects, providing the evidence for the next stages of clinical studies, reported Dr. Olanow.
“If this turns out to be what we hope it is, it will allow us to deliver levodopa without motor complications, without need for a surgical procedure, and without the risk of subcutaneous lesions,” Dr. Olanow said.
More delivery strategies are needed
This device is in an early phase of development, but several specialists in Parkinson’s disease agreed that there is a need for more strategies to provide continuous levodopa in patients with advancing symptoms. Stuart Isaacson, MD, director, Parkinson’s Disease and Movement Disorders Center of Boca Raton, Fla., is among them.
“Novel delivery devices that can provide more continuous levodopa delivery would be an important therapeutic advance,” Dr. Isaacson said. He called levodopa “the cornerstone of treatment through the course of Parkinson’s disease,” but more physiologic dosing in advancing disease has been a challenge.
“While there are many therapies currently available to manage OFF time, many people living with Parkinson’s disease continue to spend only half of their waking day with good ON time,” he added.
The currently approved method of delivering continuous levodopa through a surgically placed catheter into the gastrointestinal system is effective, but has limitations, according to Aaron L. Ellenbogen, MD, a neurologist at Beaumont Hospital, Farmington Hills, Mich.
“One of the challenges with the current treatment landscape of Parkinson’s disease is that medication can be absorbed variably through the gastrointestinal system,” he said. “As the disease progresses, this often becomes more troublesome.” Although this new device is likely to share this issue, Dr. Ellenbogen said that several devices might be useful to match patients with the one that works best for them.
Dr. Olanow is the founder and CEO of Clintrex Research Corporation, through which he also serves as chief medical officer of SynAgile, the company developing DopaFuse. Dr. Isaacson has financial relationships with more than 30 companies, including those that produce levodopa and levodopa delivery systems. Dr. Ellenbogen has financial relationships with Allergan, Acorda, Supernus, and Teva.
BOSTON – , according to an early clinical experience described in the Emerging Science session at the 2023 annual meeting of the American Academy of Neurology.
On this device, the attenuation of levodopa fluctuations “translated into dramatic improvements in clinical behavior, including highly significant reductions in OFF time and an increase in ON time with no dyskinesias,” reported C. Warren Olanow, MD, who is a chairman emeritus of the department of neurology at the Icahn School of Medicine at Mount Sinai, New York, and now an employee of the company developing this new device.
A novel strategy
Numerous studies have demonstrated that reductions in the troughs of plasma levodopa associated with oral dosing result in longer ON time with fewer dyskinesias, according to Dr. Olanow, who explained this has led to strategies for numerous strategies to achieve continuous delivery. A device that delivers levodopa into the stomach through a surgically implanted catheter has already received regulatory approval. Other devices delivering levodopa subcutaneously are in development, but Dr. Olanow said each of these has had limitations.
“The problem with these approaches is they are associated with potentially serious side effects and they require the patient to wear a cumbersome device,” he explained. Relative to the subcutaneous delivery systems, which have been associated with injection site reactions that include painful nodules, and the surgically implanted devices, which also require an external pump, the latest strategy avoids both disadvantages.
Called DopaFuse, the experimental device is designed to deliver the levodopa and carbidopa into the mouth through a micropump within a wearable retainer. Dr. Olanow said that previous experimental studies demonstrated that small doses of levodopa delivered by mouth to the gastrointestinal system reduce levodopa plasma variability. This early clinical study supports that premise. Levodopa delivered into the mouth by way of a propellant in the retainer-mounted pump improved clinical endpoints.
Encouraging trial results
In the study, 16 patients between the ages of 30 and 75 with Parkinson’s disease were enrolled. On day 1, they received an oral dose of levodopa/carbidopa consistent with their current treatment. On day 2, levodopa/carbidopa was delivered through the retainer-mounted device at equivalent doses. On day 3, they received a single morning oral dose and the received the remainder of their levodopa/carbidopa regimen through the device. On days 4 to 14, they received treatment in the same schedule as day 3.
When pharmacokinetics of levodopa on day 3 were compared with those on day 1, the fluctuation index and coefficient of levodopa concentration variability was reduced to a degree that was highly statistically significant (P < .0001). This, in turn, correlated with “striking” reductions in OFF time with equally statistically significant improvement in ON time and ON time without dyskinesias, according to Dr. Olanow.
Relative to an OFF time of 3.2 hours on day 1, the OFF time of 1.6 hours on day 3 represented a 50% reduction (P < .0001). ON time improved from 12.8 hours to 14.5 hours (P < .001). ON time without dyskinesias improved numerically from 8.8 hours to 9.6 hours.
“There were also improvements in activities of daily living when patients were on DopaFuse, which is a hard endpoint to reach in a study with such a small sample size,” Dr. Olanow reported.
There were no serious adverse events. Three patients reported vomiting and two patients each reported headache, but these events were mild and all resolved within a day. Three patients reported buccal lesions, but these also resolved within a day.
“Some patients reported trouble with speaking in the beginning but at the end of the study, patients were reporting that it was easier to speak because of the motor improvements,” Dr. Olanow said.
Overall, the device was well tolerated by the subjects, providing the evidence for the next stages of clinical studies, reported Dr. Olanow.
“If this turns out to be what we hope it is, it will allow us to deliver levodopa without motor complications, without need for a surgical procedure, and without the risk of subcutaneous lesions,” Dr. Olanow said.
More delivery strategies are needed
This device is in an early phase of development, but several specialists in Parkinson’s disease agreed that there is a need for more strategies to provide continuous levodopa in patients with advancing symptoms. Stuart Isaacson, MD, director, Parkinson’s Disease and Movement Disorders Center of Boca Raton, Fla., is among them.
“Novel delivery devices that can provide more continuous levodopa delivery would be an important therapeutic advance,” Dr. Isaacson said. He called levodopa “the cornerstone of treatment through the course of Parkinson’s disease,” but more physiologic dosing in advancing disease has been a challenge.
“While there are many therapies currently available to manage OFF time, many people living with Parkinson’s disease continue to spend only half of their waking day with good ON time,” he added.
The currently approved method of delivering continuous levodopa through a surgically placed catheter into the gastrointestinal system is effective, but has limitations, according to Aaron L. Ellenbogen, MD, a neurologist at Beaumont Hospital, Farmington Hills, Mich.
“One of the challenges with the current treatment landscape of Parkinson’s disease is that medication can be absorbed variably through the gastrointestinal system,” he said. “As the disease progresses, this often becomes more troublesome.” Although this new device is likely to share this issue, Dr. Ellenbogen said that several devices might be useful to match patients with the one that works best for them.
Dr. Olanow is the founder and CEO of Clintrex Research Corporation, through which he also serves as chief medical officer of SynAgile, the company developing DopaFuse. Dr. Isaacson has financial relationships with more than 30 companies, including those that produce levodopa and levodopa delivery systems. Dr. Ellenbogen has financial relationships with Allergan, Acorda, Supernus, and Teva.
FROM AAN 2023
Therapy to reverse muscle dystrophies shows promise
BOSTON – Becker (BMD) and Duchenne muscle dystrophy (DMD) progress largely from irreversible contraction-induced injury of skeletal muscles, making the very positive interim results of an early-phase trial with a drug that prevents these injuries worth attention.
The phase 1b data in BMD, presented at the 2023 annual meeting of the American Academy of Neurology, were sufficiently promising that controlled phase 2 trials in both BMD and DMD are already enrolling, reported Joanne Donovan, MD, PhD, an adjunct professor at Boston University and chief medical officer of Edgewise Therapeutics, the company developing the drug.
Phase 1 study
Early phase studies are largely focused on safety, but the
Moreover, the evidence of a clinical effect was achieved in adult patients with a North Star Ambulatory Assessment (NSAA) score of 15, signifying advanced disease. Only 12 patients were enrolled and there were no controls, but objective evidence of a favorable effect was generated by highly significant reductions in creatine kinase (CK) and fast skeletal muscle (TNNI2) troponin, which are both biomarkers commonly used to track muscular dystrophy progression.
In patients with BMD or DMD, a lack of dystrophin is a key pathogenic feature, according to Dr. Donovan. She explained that dystrophin in muscles connects contractile proteins to membranes and surrounding matrix. In the presence of dystrophin, muscle fibers support each other, but when this protein is absent, contraction causes injury.
The drug in development, currently identified as EDG-5506, is a selective fast myosin inhibitor. This agent was shown to prevent the muscle injury caused by lack of dystrophin in animal models of muscular dystrophy and is now showing the same effect in humans. Preservation of muscle is critical to preventing BMD and DMD progression according to several sets of data, according to Dr. Donovan.
For one, it has been shown that BMD or DMD patients with relatively preserved function as defined by a NSAA score above 32 have minimal muscle damage. As NSAA scores fall below 32 points, muscle mass diminishes and fat accumulates. In natural history studies of BMD, there is a 1.2-point decline in NSAA score over 5 years, and this tracks with muscle loss and not with other variables, such as patient age.
“Progression depends on the degree of muscle loss,” Dr. Donovan stated, providing the rationale for moving forward with EDG-5506.
Proof of concept
In experimental studies, modulation of fast myelin provided complete protection against muscle injury while preserving its contractile function, and this translated into protection against loss of function. Phase 1 studies in BMD patients and healthy controls have already provided evidence that EDG-5506 is well tolerated and safe, but the new phase 1b provides a proof of concept for its ability to inhibit muscle injury in BMD patients.
In this study, called ARCH, 12 adults 18 years of age or older with a dystrophin mutation and a BMD phenotype who could complete a 100-meter timed test were enrolled. The median age at entry was 32 years. Several patients had participated in a previous phase 1 safety study. The daily starting dose of 10 mg was increased from 10 mg to 15 mg at 2 months. The dose was increased again to 20 mg at 6 months, but the data presented by Dr. Donovan were restricted to the first 6 months.
At the interim 6-month analysis, creatine kinase was reduced by 40% and TINN2 was reduced by 84% (both P < 0.001). The significant reductions in these biomarkers and others, such as myoglobin, were mostly achieved within the first month, although further reductions were observed for some biomarkers subsequently.
The NSAA score at 6 months improved on average by about 1 point on treatment. Natural history studies of BMD predict a 1-point reduction in NSAA score over this period of time. The modest improvements from baseline in pain scores at 1 month were sustained at 6 months.
On the basis of a proteomic analysis, 125 proteins mostly associated with metabolic pathways consistent with muscle injury were found to be altered in BMD patients relative to healthy controls. The majority of these proteins, whether assessed collectively or individually, normalized after 1 to 2 months of treatment with EDG-5506 and have remained stable during follow-up to date, according to Dr. Donovan.
As in previous studies, the drug was well tolerated. The three most common treatment-emergent events were dizziness, somnolence, and headache. Each was reported by about 25% of patients, but no patient discontinued therapy as a result of adverse events.
Findings deemed ‘a big deal’
These data, despite the small number of patients in the study and the limited follow-up, “are a big deal,” according to Nicholas E. Johnson, MD, division chief, neuromuscular disorders, Virginia Commonwealth University, Richmond. He pointed out that there are no effective treatments currently for BMD, and the mechanism of action is plausible.
“I am excited about the potential of this treatment, although we clearly need longer follow-up and more patients evaluated on this treatment,” Dr. Johnson said. He said that clinicians with BMD patients should be aware of the phase 2 trial that is now recruiting adult subjects.
“Becker muscular dystrophy is highly disabling. As disease advances, most patients have very limited function,” said Dr. Johnson, emphasizing the urgent unmet need for an effective therapy.
Dr. Donovan is a full time employee of Edgewise Therapeutics, which funded this study. Dr. Johnson has financial relationships with Acceleron, Arthex, AveXis, Avidity, Biogen, Dyne Therapeutics, Entrada, Juvena, ML Bio, Sarepta Therapeutics, Triplet Therapeutics, and Vertex Pharma.
BOSTON – Becker (BMD) and Duchenne muscle dystrophy (DMD) progress largely from irreversible contraction-induced injury of skeletal muscles, making the very positive interim results of an early-phase trial with a drug that prevents these injuries worth attention.
The phase 1b data in BMD, presented at the 2023 annual meeting of the American Academy of Neurology, were sufficiently promising that controlled phase 2 trials in both BMD and DMD are already enrolling, reported Joanne Donovan, MD, PhD, an adjunct professor at Boston University and chief medical officer of Edgewise Therapeutics, the company developing the drug.
Phase 1 study
Early phase studies are largely focused on safety, but the
Moreover, the evidence of a clinical effect was achieved in adult patients with a North Star Ambulatory Assessment (NSAA) score of 15, signifying advanced disease. Only 12 patients were enrolled and there were no controls, but objective evidence of a favorable effect was generated by highly significant reductions in creatine kinase (CK) and fast skeletal muscle (TNNI2) troponin, which are both biomarkers commonly used to track muscular dystrophy progression.
In patients with BMD or DMD, a lack of dystrophin is a key pathogenic feature, according to Dr. Donovan. She explained that dystrophin in muscles connects contractile proteins to membranes and surrounding matrix. In the presence of dystrophin, muscle fibers support each other, but when this protein is absent, contraction causes injury.
The drug in development, currently identified as EDG-5506, is a selective fast myosin inhibitor. This agent was shown to prevent the muscle injury caused by lack of dystrophin in animal models of muscular dystrophy and is now showing the same effect in humans. Preservation of muscle is critical to preventing BMD and DMD progression according to several sets of data, according to Dr. Donovan.
For one, it has been shown that BMD or DMD patients with relatively preserved function as defined by a NSAA score above 32 have minimal muscle damage. As NSAA scores fall below 32 points, muscle mass diminishes and fat accumulates. In natural history studies of BMD, there is a 1.2-point decline in NSAA score over 5 years, and this tracks with muscle loss and not with other variables, such as patient age.
“Progression depends on the degree of muscle loss,” Dr. Donovan stated, providing the rationale for moving forward with EDG-5506.
Proof of concept
In experimental studies, modulation of fast myelin provided complete protection against muscle injury while preserving its contractile function, and this translated into protection against loss of function. Phase 1 studies in BMD patients and healthy controls have already provided evidence that EDG-5506 is well tolerated and safe, but the new phase 1b provides a proof of concept for its ability to inhibit muscle injury in BMD patients.
In this study, called ARCH, 12 adults 18 years of age or older with a dystrophin mutation and a BMD phenotype who could complete a 100-meter timed test were enrolled. The median age at entry was 32 years. Several patients had participated in a previous phase 1 safety study. The daily starting dose of 10 mg was increased from 10 mg to 15 mg at 2 months. The dose was increased again to 20 mg at 6 months, but the data presented by Dr. Donovan were restricted to the first 6 months.
At the interim 6-month analysis, creatine kinase was reduced by 40% and TINN2 was reduced by 84% (both P < 0.001). The significant reductions in these biomarkers and others, such as myoglobin, were mostly achieved within the first month, although further reductions were observed for some biomarkers subsequently.
The NSAA score at 6 months improved on average by about 1 point on treatment. Natural history studies of BMD predict a 1-point reduction in NSAA score over this period of time. The modest improvements from baseline in pain scores at 1 month were sustained at 6 months.
On the basis of a proteomic analysis, 125 proteins mostly associated with metabolic pathways consistent with muscle injury were found to be altered in BMD patients relative to healthy controls. The majority of these proteins, whether assessed collectively or individually, normalized after 1 to 2 months of treatment with EDG-5506 and have remained stable during follow-up to date, according to Dr. Donovan.
As in previous studies, the drug was well tolerated. The three most common treatment-emergent events were dizziness, somnolence, and headache. Each was reported by about 25% of patients, but no patient discontinued therapy as a result of adverse events.
Findings deemed ‘a big deal’
These data, despite the small number of patients in the study and the limited follow-up, “are a big deal,” according to Nicholas E. Johnson, MD, division chief, neuromuscular disorders, Virginia Commonwealth University, Richmond. He pointed out that there are no effective treatments currently for BMD, and the mechanism of action is plausible.
“I am excited about the potential of this treatment, although we clearly need longer follow-up and more patients evaluated on this treatment,” Dr. Johnson said. He said that clinicians with BMD patients should be aware of the phase 2 trial that is now recruiting adult subjects.
“Becker muscular dystrophy is highly disabling. As disease advances, most patients have very limited function,” said Dr. Johnson, emphasizing the urgent unmet need for an effective therapy.
Dr. Donovan is a full time employee of Edgewise Therapeutics, which funded this study. Dr. Johnson has financial relationships with Acceleron, Arthex, AveXis, Avidity, Biogen, Dyne Therapeutics, Entrada, Juvena, ML Bio, Sarepta Therapeutics, Triplet Therapeutics, and Vertex Pharma.
BOSTON – Becker (BMD) and Duchenne muscle dystrophy (DMD) progress largely from irreversible contraction-induced injury of skeletal muscles, making the very positive interim results of an early-phase trial with a drug that prevents these injuries worth attention.
The phase 1b data in BMD, presented at the 2023 annual meeting of the American Academy of Neurology, were sufficiently promising that controlled phase 2 trials in both BMD and DMD are already enrolling, reported Joanne Donovan, MD, PhD, an adjunct professor at Boston University and chief medical officer of Edgewise Therapeutics, the company developing the drug.
Phase 1 study
Early phase studies are largely focused on safety, but the
Moreover, the evidence of a clinical effect was achieved in adult patients with a North Star Ambulatory Assessment (NSAA) score of 15, signifying advanced disease. Only 12 patients were enrolled and there were no controls, but objective evidence of a favorable effect was generated by highly significant reductions in creatine kinase (CK) and fast skeletal muscle (TNNI2) troponin, which are both biomarkers commonly used to track muscular dystrophy progression.
In patients with BMD or DMD, a lack of dystrophin is a key pathogenic feature, according to Dr. Donovan. She explained that dystrophin in muscles connects contractile proteins to membranes and surrounding matrix. In the presence of dystrophin, muscle fibers support each other, but when this protein is absent, contraction causes injury.
The drug in development, currently identified as EDG-5506, is a selective fast myosin inhibitor. This agent was shown to prevent the muscle injury caused by lack of dystrophin in animal models of muscular dystrophy and is now showing the same effect in humans. Preservation of muscle is critical to preventing BMD and DMD progression according to several sets of data, according to Dr. Donovan.
For one, it has been shown that BMD or DMD patients with relatively preserved function as defined by a NSAA score above 32 have minimal muscle damage. As NSAA scores fall below 32 points, muscle mass diminishes and fat accumulates. In natural history studies of BMD, there is a 1.2-point decline in NSAA score over 5 years, and this tracks with muscle loss and not with other variables, such as patient age.
“Progression depends on the degree of muscle loss,” Dr. Donovan stated, providing the rationale for moving forward with EDG-5506.
Proof of concept
In experimental studies, modulation of fast myelin provided complete protection against muscle injury while preserving its contractile function, and this translated into protection against loss of function. Phase 1 studies in BMD patients and healthy controls have already provided evidence that EDG-5506 is well tolerated and safe, but the new phase 1b provides a proof of concept for its ability to inhibit muscle injury in BMD patients.
In this study, called ARCH, 12 adults 18 years of age or older with a dystrophin mutation and a BMD phenotype who could complete a 100-meter timed test were enrolled. The median age at entry was 32 years. Several patients had participated in a previous phase 1 safety study. The daily starting dose of 10 mg was increased from 10 mg to 15 mg at 2 months. The dose was increased again to 20 mg at 6 months, but the data presented by Dr. Donovan were restricted to the first 6 months.
At the interim 6-month analysis, creatine kinase was reduced by 40% and TINN2 was reduced by 84% (both P < 0.001). The significant reductions in these biomarkers and others, such as myoglobin, were mostly achieved within the first month, although further reductions were observed for some biomarkers subsequently.
The NSAA score at 6 months improved on average by about 1 point on treatment. Natural history studies of BMD predict a 1-point reduction in NSAA score over this period of time. The modest improvements from baseline in pain scores at 1 month were sustained at 6 months.
On the basis of a proteomic analysis, 125 proteins mostly associated with metabolic pathways consistent with muscle injury were found to be altered in BMD patients relative to healthy controls. The majority of these proteins, whether assessed collectively or individually, normalized after 1 to 2 months of treatment with EDG-5506 and have remained stable during follow-up to date, according to Dr. Donovan.
As in previous studies, the drug was well tolerated. The three most common treatment-emergent events were dizziness, somnolence, and headache. Each was reported by about 25% of patients, but no patient discontinued therapy as a result of adverse events.
Findings deemed ‘a big deal’
These data, despite the small number of patients in the study and the limited follow-up, “are a big deal,” according to Nicholas E. Johnson, MD, division chief, neuromuscular disorders, Virginia Commonwealth University, Richmond. He pointed out that there are no effective treatments currently for BMD, and the mechanism of action is plausible.
“I am excited about the potential of this treatment, although we clearly need longer follow-up and more patients evaluated on this treatment,” Dr. Johnson said. He said that clinicians with BMD patients should be aware of the phase 2 trial that is now recruiting adult subjects.
“Becker muscular dystrophy is highly disabling. As disease advances, most patients have very limited function,” said Dr. Johnson, emphasizing the urgent unmet need for an effective therapy.
Dr. Donovan is a full time employee of Edgewise Therapeutics, which funded this study. Dr. Johnson has financial relationships with Acceleron, Arthex, AveXis, Avidity, Biogen, Dyne Therapeutics, Entrada, Juvena, ML Bio, Sarepta Therapeutics, Triplet Therapeutics, and Vertex Pharma.
FROM AAN 2023
Phase 3 study of new levodopa/carbidopa delivery system meets all efficacy endpoints
BOSTON – presented at the 2023 annual meeting of the American Academy of Neurology.
When compared with optimized oral immediate-release medication, the delivery system, called ND0612 (NeuroDerm, Rehovot, Israel), improved ON time without troublesome dyskinesias while improving symptoms according to ratings from both patients and clinicians, according to Alberto J. Espay, MD, professor of neurology and director of the Gardner Family Center for Parkinson’s Disease and Movement Disorders, University of Cincinnati.
The new delivery system addresses the challenge of reducing the variability in levodopa plasma concentrations, a major factor in motor fluctuations and diminishing benefit from orally administered drug, according to Dr. Espay. He said that continuous infusion strategies have long been sought as a method to preserve levodopa efficacy.
BouNDless findings
There were two phases to this multinational trial, called BouNDless. In the first, an open-label run-in phase, 381 patients with Parkinson’s disease were dose titrated for optimization of oral immediate-release levodopa and carbidopa. They were then optimized for the same drugs delivered with ND0612. The study was conducted over 12 weeks; 122 patients left the study after this phase due to adverse events, lack of efficacy, or withdrawal of consent.
In the second phase, the 259 remaining patients were randomized to the continuous infusion arm or to immediate release oral therapy. In this double-blind, double-dummy phase, those randomized to the ND0612 infusion also received oral placebos. Those randomized to oral therapy received a placebo infusion. Efficacy and safety were assessed at the end of 12 weeks.
At the end of phase 1, the ON time increased by about 3 hours when levodopa-carbidopa dosing was optimized on either delivery method. Dr. Espay attributed the improvement to the value of optimized dosing even in patients with relatively advanced disease.
However, for the purposes of the double-blind comparison, this improvement in ON time provided a new baseline for comparison of the two delivery methods. This is important for interpreting the primary result, which was a 1.72-hour difference in ON time at the end of the study. The difference was created when ON time was maintained with ND0612 continuous drug delivery but eroded in the group randomized to oral immediate-release treatment.
Several secondary endpoints supported the greater efficacy of continuous subcutaneous delivery. These included lower OFF time (0.50 vs. 1.90 hours), less accumulation of disability on the United Parkinson’s Disease Rating Scale part II-M-EDL (-0.30 vs. +2.75 points), and greater improvement on the Patient Global Impression of Change (+0.31 vs. +0.70 points), and the Clinical Global Impression of change (+0.31 vs. +0.77 points). The differences were highly statistically significant (all P < .0001).
The patients participating in the double-blind phase of the study were similar with a mean age of 63.5 years in both groups and time since Parkinson’s disease diagnosis (> 9 years). The median ON time without troublesome dyskinesias was about 12 hours at baseline in both groups and the median OFF time was about 3.5 hours.
The higher rate of treatment-related adverse events in the ND0612 group (67.2% vs. 52.7%) was largely explained by the greater rate of infusion site reactions (57.0% vs. 42.7%). The rates of severe reactions in the two groups were the same (0.8%), but both mild (43.8% vs. 36.6%) and moderate (12.5% vs. 5.3%) reactions occurred more commonly in the group receiving active therapy.
“Infusion reactions are the Achilles heel of all subcutaneous therapies,” acknowledged Dr. Espay, who expects other infusion systems in development to share this risk. He suggested that the clinical impact can be attenuated to some degree by rotating infusion sites.
BeyoND extension study
Data from an open-label extension (OLE) of the phase 2b BeyoND trial were also presented at the AAN meeting and generated generally similar results. Largely a safety study, there was no active control in the initial BeyoND or the BeyoND OLE. In BeyoND, the improvement in ON time from baseline was even greater than that seen in BouNDless, but, again, the optimization of dosing in the BouNDless run-in established a greater baseline of disease control.
In the OLE of BeyoND, presented by Aaron Ellenbogen, DO, a neurologist in Farmington, Mich., one of the notable findings was the retention of patients. After 2 years of follow-up, 82% completed at least 2 years of follow-up and 66.7% have now remained on treatment for at least 3 years. Dr. Ellenbogen maintains that this retention rate provides compelling evidence of a favorable benefit-to-risk ratio.
Fulfilling an unmet need
The favorable efficacy data from this trial represent “a big advance,” according to Ihtsham Ul Haq, MD, chief, movement disorders division, University of Miami, who was reached for comment. He noted that continuous infusion delivery has been anticipated for some time, and he expects these types of systems to fulfill an unmet need.
“This will be a useful option in a carefully selected group of patients,” said Dr. Haq, who considers the types of improvement in ON time to be highly clinically meaningful.
However, he cautioned that the nodules created by injection site reactions might limit the utility of this treatment option in at least some patients. Wearing the external device might also be a limiting factor for some patients.
In complex Parkinson’s disease, a stage that can be reached fairly rapidly in some patients but might take 15 years or more in others, all of the options involve a careful benefit-to-risk calculation, according to Dr. Haq. Deep brain stimulation is among the most effective options, but continuous infusion might appeal to some patients for delaying this procedure or as an alternative.
“We need multiple options for these types of patients, and it appears that continuous infusion will be one of them,” Dr. Haq said.
Dr. Espay has financial relationships with Acadia, Acorda, Amneal, AskBio, Bexion, Kyowa Kirin, Neuroderm, Neurocrine, and Sunovion. Dr. Ellenbogen has financial relationships with Allergan, Acorda, Supernus, and Teva. Dr. Haq reports no potential conflicts of interest.
BOSTON – presented at the 2023 annual meeting of the American Academy of Neurology.
When compared with optimized oral immediate-release medication, the delivery system, called ND0612 (NeuroDerm, Rehovot, Israel), improved ON time without troublesome dyskinesias while improving symptoms according to ratings from both patients and clinicians, according to Alberto J. Espay, MD, professor of neurology and director of the Gardner Family Center for Parkinson’s Disease and Movement Disorders, University of Cincinnati.
The new delivery system addresses the challenge of reducing the variability in levodopa plasma concentrations, a major factor in motor fluctuations and diminishing benefit from orally administered drug, according to Dr. Espay. He said that continuous infusion strategies have long been sought as a method to preserve levodopa efficacy.
BouNDless findings
There were two phases to this multinational trial, called BouNDless. In the first, an open-label run-in phase, 381 patients with Parkinson’s disease were dose titrated for optimization of oral immediate-release levodopa and carbidopa. They were then optimized for the same drugs delivered with ND0612. The study was conducted over 12 weeks; 122 patients left the study after this phase due to adverse events, lack of efficacy, or withdrawal of consent.
In the second phase, the 259 remaining patients were randomized to the continuous infusion arm or to immediate release oral therapy. In this double-blind, double-dummy phase, those randomized to the ND0612 infusion also received oral placebos. Those randomized to oral therapy received a placebo infusion. Efficacy and safety were assessed at the end of 12 weeks.
At the end of phase 1, the ON time increased by about 3 hours when levodopa-carbidopa dosing was optimized on either delivery method. Dr. Espay attributed the improvement to the value of optimized dosing even in patients with relatively advanced disease.
However, for the purposes of the double-blind comparison, this improvement in ON time provided a new baseline for comparison of the two delivery methods. This is important for interpreting the primary result, which was a 1.72-hour difference in ON time at the end of the study. The difference was created when ON time was maintained with ND0612 continuous drug delivery but eroded in the group randomized to oral immediate-release treatment.
Several secondary endpoints supported the greater efficacy of continuous subcutaneous delivery. These included lower OFF time (0.50 vs. 1.90 hours), less accumulation of disability on the United Parkinson’s Disease Rating Scale part II-M-EDL (-0.30 vs. +2.75 points), and greater improvement on the Patient Global Impression of Change (+0.31 vs. +0.70 points), and the Clinical Global Impression of change (+0.31 vs. +0.77 points). The differences were highly statistically significant (all P < .0001).
The patients participating in the double-blind phase of the study were similar with a mean age of 63.5 years in both groups and time since Parkinson’s disease diagnosis (> 9 years). The median ON time without troublesome dyskinesias was about 12 hours at baseline in both groups and the median OFF time was about 3.5 hours.
The higher rate of treatment-related adverse events in the ND0612 group (67.2% vs. 52.7%) was largely explained by the greater rate of infusion site reactions (57.0% vs. 42.7%). The rates of severe reactions in the two groups were the same (0.8%), but both mild (43.8% vs. 36.6%) and moderate (12.5% vs. 5.3%) reactions occurred more commonly in the group receiving active therapy.
“Infusion reactions are the Achilles heel of all subcutaneous therapies,” acknowledged Dr. Espay, who expects other infusion systems in development to share this risk. He suggested that the clinical impact can be attenuated to some degree by rotating infusion sites.
BeyoND extension study
Data from an open-label extension (OLE) of the phase 2b BeyoND trial were also presented at the AAN meeting and generated generally similar results. Largely a safety study, there was no active control in the initial BeyoND or the BeyoND OLE. In BeyoND, the improvement in ON time from baseline was even greater than that seen in BouNDless, but, again, the optimization of dosing in the BouNDless run-in established a greater baseline of disease control.
In the OLE of BeyoND, presented by Aaron Ellenbogen, DO, a neurologist in Farmington, Mich., one of the notable findings was the retention of patients. After 2 years of follow-up, 82% completed at least 2 years of follow-up and 66.7% have now remained on treatment for at least 3 years. Dr. Ellenbogen maintains that this retention rate provides compelling evidence of a favorable benefit-to-risk ratio.
Fulfilling an unmet need
The favorable efficacy data from this trial represent “a big advance,” according to Ihtsham Ul Haq, MD, chief, movement disorders division, University of Miami, who was reached for comment. He noted that continuous infusion delivery has been anticipated for some time, and he expects these types of systems to fulfill an unmet need.
“This will be a useful option in a carefully selected group of patients,” said Dr. Haq, who considers the types of improvement in ON time to be highly clinically meaningful.
However, he cautioned that the nodules created by injection site reactions might limit the utility of this treatment option in at least some patients. Wearing the external device might also be a limiting factor for some patients.
In complex Parkinson’s disease, a stage that can be reached fairly rapidly in some patients but might take 15 years or more in others, all of the options involve a careful benefit-to-risk calculation, according to Dr. Haq. Deep brain stimulation is among the most effective options, but continuous infusion might appeal to some patients for delaying this procedure or as an alternative.
“We need multiple options for these types of patients, and it appears that continuous infusion will be one of them,” Dr. Haq said.
Dr. Espay has financial relationships with Acadia, Acorda, Amneal, AskBio, Bexion, Kyowa Kirin, Neuroderm, Neurocrine, and Sunovion. Dr. Ellenbogen has financial relationships with Allergan, Acorda, Supernus, and Teva. Dr. Haq reports no potential conflicts of interest.
BOSTON – presented at the 2023 annual meeting of the American Academy of Neurology.
When compared with optimized oral immediate-release medication, the delivery system, called ND0612 (NeuroDerm, Rehovot, Israel), improved ON time without troublesome dyskinesias while improving symptoms according to ratings from both patients and clinicians, according to Alberto J. Espay, MD, professor of neurology and director of the Gardner Family Center for Parkinson’s Disease and Movement Disorders, University of Cincinnati.
The new delivery system addresses the challenge of reducing the variability in levodopa plasma concentrations, a major factor in motor fluctuations and diminishing benefit from orally administered drug, according to Dr. Espay. He said that continuous infusion strategies have long been sought as a method to preserve levodopa efficacy.
BouNDless findings
There were two phases to this multinational trial, called BouNDless. In the first, an open-label run-in phase, 381 patients with Parkinson’s disease were dose titrated for optimization of oral immediate-release levodopa and carbidopa. They were then optimized for the same drugs delivered with ND0612. The study was conducted over 12 weeks; 122 patients left the study after this phase due to adverse events, lack of efficacy, or withdrawal of consent.
In the second phase, the 259 remaining patients were randomized to the continuous infusion arm or to immediate release oral therapy. In this double-blind, double-dummy phase, those randomized to the ND0612 infusion also received oral placebos. Those randomized to oral therapy received a placebo infusion. Efficacy and safety were assessed at the end of 12 weeks.
At the end of phase 1, the ON time increased by about 3 hours when levodopa-carbidopa dosing was optimized on either delivery method. Dr. Espay attributed the improvement to the value of optimized dosing even in patients with relatively advanced disease.
However, for the purposes of the double-blind comparison, this improvement in ON time provided a new baseline for comparison of the two delivery methods. This is important for interpreting the primary result, which was a 1.72-hour difference in ON time at the end of the study. The difference was created when ON time was maintained with ND0612 continuous drug delivery but eroded in the group randomized to oral immediate-release treatment.
Several secondary endpoints supported the greater efficacy of continuous subcutaneous delivery. These included lower OFF time (0.50 vs. 1.90 hours), less accumulation of disability on the United Parkinson’s Disease Rating Scale part II-M-EDL (-0.30 vs. +2.75 points), and greater improvement on the Patient Global Impression of Change (+0.31 vs. +0.70 points), and the Clinical Global Impression of change (+0.31 vs. +0.77 points). The differences were highly statistically significant (all P < .0001).
The patients participating in the double-blind phase of the study were similar with a mean age of 63.5 years in both groups and time since Parkinson’s disease diagnosis (> 9 years). The median ON time without troublesome dyskinesias was about 12 hours at baseline in both groups and the median OFF time was about 3.5 hours.
The higher rate of treatment-related adverse events in the ND0612 group (67.2% vs. 52.7%) was largely explained by the greater rate of infusion site reactions (57.0% vs. 42.7%). The rates of severe reactions in the two groups were the same (0.8%), but both mild (43.8% vs. 36.6%) and moderate (12.5% vs. 5.3%) reactions occurred more commonly in the group receiving active therapy.
“Infusion reactions are the Achilles heel of all subcutaneous therapies,” acknowledged Dr. Espay, who expects other infusion systems in development to share this risk. He suggested that the clinical impact can be attenuated to some degree by rotating infusion sites.
BeyoND extension study
Data from an open-label extension (OLE) of the phase 2b BeyoND trial were also presented at the AAN meeting and generated generally similar results. Largely a safety study, there was no active control in the initial BeyoND or the BeyoND OLE. In BeyoND, the improvement in ON time from baseline was even greater than that seen in BouNDless, but, again, the optimization of dosing in the BouNDless run-in established a greater baseline of disease control.
In the OLE of BeyoND, presented by Aaron Ellenbogen, DO, a neurologist in Farmington, Mich., one of the notable findings was the retention of patients. After 2 years of follow-up, 82% completed at least 2 years of follow-up and 66.7% have now remained on treatment for at least 3 years. Dr. Ellenbogen maintains that this retention rate provides compelling evidence of a favorable benefit-to-risk ratio.
Fulfilling an unmet need
The favorable efficacy data from this trial represent “a big advance,” according to Ihtsham Ul Haq, MD, chief, movement disorders division, University of Miami, who was reached for comment. He noted that continuous infusion delivery has been anticipated for some time, and he expects these types of systems to fulfill an unmet need.
“This will be a useful option in a carefully selected group of patients,” said Dr. Haq, who considers the types of improvement in ON time to be highly clinically meaningful.
However, he cautioned that the nodules created by injection site reactions might limit the utility of this treatment option in at least some patients. Wearing the external device might also be a limiting factor for some patients.
In complex Parkinson’s disease, a stage that can be reached fairly rapidly in some patients but might take 15 years or more in others, all of the options involve a careful benefit-to-risk calculation, according to Dr. Haq. Deep brain stimulation is among the most effective options, but continuous infusion might appeal to some patients for delaying this procedure or as an alternative.
“We need multiple options for these types of patients, and it appears that continuous infusion will be one of them,” Dr. Haq said.
Dr. Espay has financial relationships with Acadia, Acorda, Amneal, AskBio, Bexion, Kyowa Kirin, Neuroderm, Neurocrine, and Sunovion. Dr. Ellenbogen has financial relationships with Allergan, Acorda, Supernus, and Teva. Dr. Haq reports no potential conflicts of interest.
FROM AAN 2023
New assay hailed as a game changer for early Parkinson’s diagnosis
, and provides information on molecular subtypes, new research indicates.
“Identifying an effective biomarker for Parkinson’s disease pathology could have profound implications for the way we treat the condition, potentially making it possible to diagnose people earlier, identify the best treatments for different subsets of patients, and speed up clinical trials,” the study’s co-lead author Andrew Siderowf, MD, of the University of Pennsylvania, Philadelphia, said in a news release.
“Our findings suggest that the αSyn-SAA technique is highly accurate at detecting the biomarker for Parkinson’s disease regardless of the clinical features, making it possible to accurately diagnose the disease in patients at early stages,” added co-lead author Luis Concha-Marambio, PhD, director of research and development at Amprion, San Diego, Calif.
The study was published online in The Lancet Neurology.
‘New era’ in Parkinson’s disease
The researchers assessed the usefulness of αSyn-SAA in a cross-sectional analysis of 1,123 participants in the Parkinson’s Progression Markers Initiative (PPMI) cohort from 33 participating academic neurology outpatient practices in 12 countries.
The cohort included individuals with sporadic Parkinson’s disease from LRRK2 or GBA variants, healthy controls, individuals with clinical syndromes prodromal to Parkinson’s disease (rapid eye movement sleep behavior disorder [RBD] or hyposmia), and nonmanifesting carriers of LRRK2 and GBA variants. Cerebrospinal fluid (CSF) samples from each participant were analyzed using αSyn-SAA.
Overall, αSyn-SAA differentiated Parkinson’s disease from healthy controls with 87.7% sensitivity and 96.3% specificity.
Sensitivity of the assay varied across subgroups based on genetic and clinical features. Among genetic Parkinson’s disease subgroups, sensitivity was highest for GBA Parkinson’s disease (95.9%), followed by sporadic Parkinson’s disease (93.3%), and lowest for LRRK2 Parkinson’s disease (67.5%). Among clinical features, hyposmia was the most robust predictor of a positive assay result.
Among all Parkinson’s disease cases with hyposmia, the sensitivity of the assay was 97.2%, compared with 63.0% for Parkinson’s disease without olfactory dysfunction. Combining genetic and clinical features, the sensitivity of positive αSyn-SAA in sporadic Parkinson’s disease with olfactory deficit was 98.6%, compared with 78.3% in sporadic Parkinson’s disease without hyposmia. Most prodromal participants (86%) with RBD and hyposmia had positive αSyn-SAA results, indicating they had α-synuclein aggregates despite not yet being diagnosed with Parkinson’s disease.
Among those recruited based on their loss of smell, 89% (16 of 18 participants) had positive αSyn-SAA results. Similarly, in those with RBD, positive αSyn-SAA results were present in 85% of cases (28 of 33). No other clinical features were associated with a positive αSyn-SAA result.
In participants who carried LRRK2 or GBA variants but had no Parkinson’s disease diagnosis or prodromal symptoms (nonmanifesting carriers), 9% (14 of 159) and 7% (11 of 151), respectively, had positive αSyn-SAA results.
To date, this is the largest analysis of α-Syn-SAA for the biochemical diagnosis of Parkinson’s disease, the researchers said.
The results show that the assay classifies people with Parkinson’s disease with “high sensitivity and specificity, provides information about molecular heterogeneity, and detects prodromal individuals before diagnosis,” they wrote.
“These findings suggest a crucial role for the α-synuclein SAA in therapeutic development, both to identify pathologically defined subgroups of people with Parkinson’s disease and to establish biomarker-defined at-risk cohorts,” they added.
Amprion has commercialized the assay (SYNTap test), which can be ordered online.
‘Seminal development’
The authors of an accompanying editorial noted the study “lays the foundation for a biological diagnosis” of Parkinson’s disease. “We have entered a new era of biomarker and treatment development for Parkinson’s disease. The possibility of detecting a misfolded α-synuclein, the pathological hallmark of Parkinson’s disease, by employing an SSA, is a seminal development,” wrote Daniela Berg, MD, PhD, and Christine Klein, MD, with University Hospital Schleswig-Holstein, Germany.
“However, to fully leverage the enormous potential of the α-synuclein seed amplification, the test would have to be performed in blood rather than the CSF, a less invasive approach that has proven to be viable,” they added.
“Although the blood-based method needs to be further elaborated for scalability, α-synuclein SAA is a game changer in Parkinson’s disease diagnostics, research, and treatment trials,” they concluded.
The study was funded by The Michael J. Fox Foundation for Parkinson’s Research and a consortium of more than 40 private and philanthropic partners. Dr. Siderowf has declared consulting for Merck and Parkinson Study Group, and receiving honoraria from Bial. A full list of author disclosures is available with the original article. Dr. Berg and Dr. Klein have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, and provides information on molecular subtypes, new research indicates.
“Identifying an effective biomarker for Parkinson’s disease pathology could have profound implications for the way we treat the condition, potentially making it possible to diagnose people earlier, identify the best treatments for different subsets of patients, and speed up clinical trials,” the study’s co-lead author Andrew Siderowf, MD, of the University of Pennsylvania, Philadelphia, said in a news release.
“Our findings suggest that the αSyn-SAA technique is highly accurate at detecting the biomarker for Parkinson’s disease regardless of the clinical features, making it possible to accurately diagnose the disease in patients at early stages,” added co-lead author Luis Concha-Marambio, PhD, director of research and development at Amprion, San Diego, Calif.
The study was published online in The Lancet Neurology.
‘New era’ in Parkinson’s disease
The researchers assessed the usefulness of αSyn-SAA in a cross-sectional analysis of 1,123 participants in the Parkinson’s Progression Markers Initiative (PPMI) cohort from 33 participating academic neurology outpatient practices in 12 countries.
The cohort included individuals with sporadic Parkinson’s disease from LRRK2 or GBA variants, healthy controls, individuals with clinical syndromes prodromal to Parkinson’s disease (rapid eye movement sleep behavior disorder [RBD] or hyposmia), and nonmanifesting carriers of LRRK2 and GBA variants. Cerebrospinal fluid (CSF) samples from each participant were analyzed using αSyn-SAA.
Overall, αSyn-SAA differentiated Parkinson’s disease from healthy controls with 87.7% sensitivity and 96.3% specificity.
Sensitivity of the assay varied across subgroups based on genetic and clinical features. Among genetic Parkinson’s disease subgroups, sensitivity was highest for GBA Parkinson’s disease (95.9%), followed by sporadic Parkinson’s disease (93.3%), and lowest for LRRK2 Parkinson’s disease (67.5%). Among clinical features, hyposmia was the most robust predictor of a positive assay result.
Among all Parkinson’s disease cases with hyposmia, the sensitivity of the assay was 97.2%, compared with 63.0% for Parkinson’s disease without olfactory dysfunction. Combining genetic and clinical features, the sensitivity of positive αSyn-SAA in sporadic Parkinson’s disease with olfactory deficit was 98.6%, compared with 78.3% in sporadic Parkinson’s disease without hyposmia. Most prodromal participants (86%) with RBD and hyposmia had positive αSyn-SAA results, indicating they had α-synuclein aggregates despite not yet being diagnosed with Parkinson’s disease.
Among those recruited based on their loss of smell, 89% (16 of 18 participants) had positive αSyn-SAA results. Similarly, in those with RBD, positive αSyn-SAA results were present in 85% of cases (28 of 33). No other clinical features were associated with a positive αSyn-SAA result.
In participants who carried LRRK2 or GBA variants but had no Parkinson’s disease diagnosis or prodromal symptoms (nonmanifesting carriers), 9% (14 of 159) and 7% (11 of 151), respectively, had positive αSyn-SAA results.
To date, this is the largest analysis of α-Syn-SAA for the biochemical diagnosis of Parkinson’s disease, the researchers said.
The results show that the assay classifies people with Parkinson’s disease with “high sensitivity and specificity, provides information about molecular heterogeneity, and detects prodromal individuals before diagnosis,” they wrote.
“These findings suggest a crucial role for the α-synuclein SAA in therapeutic development, both to identify pathologically defined subgroups of people with Parkinson’s disease and to establish biomarker-defined at-risk cohorts,” they added.
Amprion has commercialized the assay (SYNTap test), which can be ordered online.
‘Seminal development’
The authors of an accompanying editorial noted the study “lays the foundation for a biological diagnosis” of Parkinson’s disease. “We have entered a new era of biomarker and treatment development for Parkinson’s disease. The possibility of detecting a misfolded α-synuclein, the pathological hallmark of Parkinson’s disease, by employing an SSA, is a seminal development,” wrote Daniela Berg, MD, PhD, and Christine Klein, MD, with University Hospital Schleswig-Holstein, Germany.
“However, to fully leverage the enormous potential of the α-synuclein seed amplification, the test would have to be performed in blood rather than the CSF, a less invasive approach that has proven to be viable,” they added.
“Although the blood-based method needs to be further elaborated for scalability, α-synuclein SAA is a game changer in Parkinson’s disease diagnostics, research, and treatment trials,” they concluded.
The study was funded by The Michael J. Fox Foundation for Parkinson’s Research and a consortium of more than 40 private and philanthropic partners. Dr. Siderowf has declared consulting for Merck and Parkinson Study Group, and receiving honoraria from Bial. A full list of author disclosures is available with the original article. Dr. Berg and Dr. Klein have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, and provides information on molecular subtypes, new research indicates.
“Identifying an effective biomarker for Parkinson’s disease pathology could have profound implications for the way we treat the condition, potentially making it possible to diagnose people earlier, identify the best treatments for different subsets of patients, and speed up clinical trials,” the study’s co-lead author Andrew Siderowf, MD, of the University of Pennsylvania, Philadelphia, said in a news release.
“Our findings suggest that the αSyn-SAA technique is highly accurate at detecting the biomarker for Parkinson’s disease regardless of the clinical features, making it possible to accurately diagnose the disease in patients at early stages,” added co-lead author Luis Concha-Marambio, PhD, director of research and development at Amprion, San Diego, Calif.
The study was published online in The Lancet Neurology.
‘New era’ in Parkinson’s disease
The researchers assessed the usefulness of αSyn-SAA in a cross-sectional analysis of 1,123 participants in the Parkinson’s Progression Markers Initiative (PPMI) cohort from 33 participating academic neurology outpatient practices in 12 countries.
The cohort included individuals with sporadic Parkinson’s disease from LRRK2 or GBA variants, healthy controls, individuals with clinical syndromes prodromal to Parkinson’s disease (rapid eye movement sleep behavior disorder [RBD] or hyposmia), and nonmanifesting carriers of LRRK2 and GBA variants. Cerebrospinal fluid (CSF) samples from each participant were analyzed using αSyn-SAA.
Overall, αSyn-SAA differentiated Parkinson’s disease from healthy controls with 87.7% sensitivity and 96.3% specificity.
Sensitivity of the assay varied across subgroups based on genetic and clinical features. Among genetic Parkinson’s disease subgroups, sensitivity was highest for GBA Parkinson’s disease (95.9%), followed by sporadic Parkinson’s disease (93.3%), and lowest for LRRK2 Parkinson’s disease (67.5%). Among clinical features, hyposmia was the most robust predictor of a positive assay result.
Among all Parkinson’s disease cases with hyposmia, the sensitivity of the assay was 97.2%, compared with 63.0% for Parkinson’s disease without olfactory dysfunction. Combining genetic and clinical features, the sensitivity of positive αSyn-SAA in sporadic Parkinson’s disease with olfactory deficit was 98.6%, compared with 78.3% in sporadic Parkinson’s disease without hyposmia. Most prodromal participants (86%) with RBD and hyposmia had positive αSyn-SAA results, indicating they had α-synuclein aggregates despite not yet being diagnosed with Parkinson’s disease.
Among those recruited based on their loss of smell, 89% (16 of 18 participants) had positive αSyn-SAA results. Similarly, in those with RBD, positive αSyn-SAA results were present in 85% of cases (28 of 33). No other clinical features were associated with a positive αSyn-SAA result.
In participants who carried LRRK2 or GBA variants but had no Parkinson’s disease diagnosis or prodromal symptoms (nonmanifesting carriers), 9% (14 of 159) and 7% (11 of 151), respectively, had positive αSyn-SAA results.
To date, this is the largest analysis of α-Syn-SAA for the biochemical diagnosis of Parkinson’s disease, the researchers said.
The results show that the assay classifies people with Parkinson’s disease with “high sensitivity and specificity, provides information about molecular heterogeneity, and detects prodromal individuals before diagnosis,” they wrote.
“These findings suggest a crucial role for the α-synuclein SAA in therapeutic development, both to identify pathologically defined subgroups of people with Parkinson’s disease and to establish biomarker-defined at-risk cohorts,” they added.
Amprion has commercialized the assay (SYNTap test), which can be ordered online.
‘Seminal development’
The authors of an accompanying editorial noted the study “lays the foundation for a biological diagnosis” of Parkinson’s disease. “We have entered a new era of biomarker and treatment development for Parkinson’s disease. The possibility of detecting a misfolded α-synuclein, the pathological hallmark of Parkinson’s disease, by employing an SSA, is a seminal development,” wrote Daniela Berg, MD, PhD, and Christine Klein, MD, with University Hospital Schleswig-Holstein, Germany.
“However, to fully leverage the enormous potential of the α-synuclein seed amplification, the test would have to be performed in blood rather than the CSF, a less invasive approach that has proven to be viable,” they added.
“Although the blood-based method needs to be further elaborated for scalability, α-synuclein SAA is a game changer in Parkinson’s disease diagnostics, research, and treatment trials,” they concluded.
The study was funded by The Michael J. Fox Foundation for Parkinson’s Research and a consortium of more than 40 private and philanthropic partners. Dr. Siderowf has declared consulting for Merck and Parkinson Study Group, and receiving honoraria from Bial. A full list of author disclosures is available with the original article. Dr. Berg and Dr. Klein have reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM THE LANCET NEUROLOGY