User login
CCSs have increased risk of hypertension
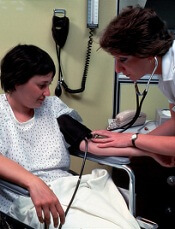
A study of childhood cancer survivors (CCSs) suggests these individuals have an increased risk of developing hypertension as adults.
The CCSs studied had more than double the rate of hypertension observed in the matched general population.
Sex, age, race, and weight were all significantly associated with hypertension among CCSs, but most treatment types were not.
The exception was nephrectomy, which was associated with an increased risk of hypertension.
Todd M. Gibson, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues conducted this research and reported the results in Cancer Epidemiology, Biomarkers & Prevention.
“High blood pressure is an important modifiable risk factor that increases the risk of heart problems in everyone,” Dr Gibson said. “Research has shown that high blood pressure can have an even greater negative impact on survivors of childhood cancer who were treated with cardiotoxic therapies such as anthracyclines or chest radiation.”
To assess the prevalence of hypertension among CCSs, Dr Gibson and his colleagues examined 3016 adults who were 10-year survivors of childhood cancers. The subjects were enrolled in the St. Jude Lifetime Cohort Study, which provides ongoing medical assessments of CCSs to advance knowledge of their long-term health outcomes.
The subjects’ mean age at the initial study assessment was 32, and 52% were male. Most (83%) were non-Hispanic white, 14% were non-Hispanic black, 2% were Hispanic, and 1% were “other.”
Thirty-seven percent of subjects had leukemia, 12% had Hodgkin lymphoma, and 7% had non-Hodgkin lymphoma.
Eighty-six percent of subjects had received chemotherapy, and 59% received radiation.
Results
Subjects were considered to have hypertension if their systolic blood pressure was 140 or greater, their diastolic blood pressure was 90 or greater, or if they had been previously diagnosed with hypertension and were taking antihypertensive medication.
The prevalence of hypertension was 2.6 times higher among CCSs than expected, based on age-, sex-, race- and body mass index-specific rates in the general population.
In addition, the incidence of hypertension increased for CCSs over time. Thirteen percent of CCSs had hypertension at age 30, 37% had it at age 40, and more than 70% had it at age 50.
Dr Gibson said rates of hypertension in CCSs matched rates in the general population of people about a decade older.
The researchers identified several factors that were significantly associated with hypertension among CCSs, including:
- Male sex (odd ratio [OR], 1.38; 95% CI, 1.14–1.67)
- Non-Hispanic black race (OR, 1.66; 95% CI, 1.28–2.16)
- Older age at assessment (OR per 1 year of age, 1.10; 95% CI, 1.08–1.11)
- Being overweight (OR, 1.58; 95% CI, 1.21–2.07)
- Obesity (OR, 3.02; 95% CI, 2.34–3.88).
Exposure to any type of radiation or chemotherapy was not significantly associated with hypertension, but nephrectomy was (OR, 1.68; 95% CI, 1.11–2.53).
Dr Gibson said the lack of an association between hypertension and radiation/chemotherapy was surprising. It suggests the connection between childhood cancer survival and adult hypertension is multifactorial and worthy of future research.
In the meantime, he said, clinicians should be mindful that CCSs are more likely than the general public to develop hypertension.
“The good news is that, unlike prior cancer therapy, high blood pressure is a modifiable risk factor,” Dr Gibson noted. “Research is needed to identify effective interventions to prevent hypertension in survivors, but our results emphasize the importance of blood pressure surveillance and management.”
Dr Gibson said a limitation of this study is that it was based on blood pressure measurements taken at a single study visit. A clinical diagnosis of hypertension typically requires measurements taken at multiple intervals.
In addition, the St. Jude Lifetime Cohort is a group of CCSs who undergo frequent clinical follow-up, so its participants may have benefited from being monitored and may therefore be in better health than CCSs who have less comprehensive follow-up. ![]()
A study of childhood cancer survivors (CCSs) suggests these individuals have an increased risk of developing hypertension as adults.
The CCSs studied had more than double the rate of hypertension observed in the matched general population.
Sex, age, race, and weight were all significantly associated with hypertension among CCSs, but most treatment types were not.
The exception was nephrectomy, which was associated with an increased risk of hypertension.
Todd M. Gibson, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues conducted this research and reported the results in Cancer Epidemiology, Biomarkers & Prevention.
“High blood pressure is an important modifiable risk factor that increases the risk of heart problems in everyone,” Dr Gibson said. “Research has shown that high blood pressure can have an even greater negative impact on survivors of childhood cancer who were treated with cardiotoxic therapies such as anthracyclines or chest radiation.”
To assess the prevalence of hypertension among CCSs, Dr Gibson and his colleagues examined 3016 adults who were 10-year survivors of childhood cancers. The subjects were enrolled in the St. Jude Lifetime Cohort Study, which provides ongoing medical assessments of CCSs to advance knowledge of their long-term health outcomes.
The subjects’ mean age at the initial study assessment was 32, and 52% were male. Most (83%) were non-Hispanic white, 14% were non-Hispanic black, 2% were Hispanic, and 1% were “other.”
Thirty-seven percent of subjects had leukemia, 12% had Hodgkin lymphoma, and 7% had non-Hodgkin lymphoma.
Eighty-six percent of subjects had received chemotherapy, and 59% received radiation.
Results
Subjects were considered to have hypertension if their systolic blood pressure was 140 or greater, their diastolic blood pressure was 90 or greater, or if they had been previously diagnosed with hypertension and were taking antihypertensive medication.
The prevalence of hypertension was 2.6 times higher among CCSs than expected, based on age-, sex-, race- and body mass index-specific rates in the general population.
In addition, the incidence of hypertension increased for CCSs over time. Thirteen percent of CCSs had hypertension at age 30, 37% had it at age 40, and more than 70% had it at age 50.
Dr Gibson said rates of hypertension in CCSs matched rates in the general population of people about a decade older.
The researchers identified several factors that were significantly associated with hypertension among CCSs, including:
- Male sex (odd ratio [OR], 1.38; 95% CI, 1.14–1.67)
- Non-Hispanic black race (OR, 1.66; 95% CI, 1.28–2.16)
- Older age at assessment (OR per 1 year of age, 1.10; 95% CI, 1.08–1.11)
- Being overweight (OR, 1.58; 95% CI, 1.21–2.07)
- Obesity (OR, 3.02; 95% CI, 2.34–3.88).
Exposure to any type of radiation or chemotherapy was not significantly associated with hypertension, but nephrectomy was (OR, 1.68; 95% CI, 1.11–2.53).
Dr Gibson said the lack of an association between hypertension and radiation/chemotherapy was surprising. It suggests the connection between childhood cancer survival and adult hypertension is multifactorial and worthy of future research.
In the meantime, he said, clinicians should be mindful that CCSs are more likely than the general public to develop hypertension.
“The good news is that, unlike prior cancer therapy, high blood pressure is a modifiable risk factor,” Dr Gibson noted. “Research is needed to identify effective interventions to prevent hypertension in survivors, but our results emphasize the importance of blood pressure surveillance and management.”
Dr Gibson said a limitation of this study is that it was based on blood pressure measurements taken at a single study visit. A clinical diagnosis of hypertension typically requires measurements taken at multiple intervals.
In addition, the St. Jude Lifetime Cohort is a group of CCSs who undergo frequent clinical follow-up, so its participants may have benefited from being monitored and may therefore be in better health than CCSs who have less comprehensive follow-up. ![]()
A study of childhood cancer survivors (CCSs) suggests these individuals have an increased risk of developing hypertension as adults.
The CCSs studied had more than double the rate of hypertension observed in the matched general population.
Sex, age, race, and weight were all significantly associated with hypertension among CCSs, but most treatment types were not.
The exception was nephrectomy, which was associated with an increased risk of hypertension.
Todd M. Gibson, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues conducted this research and reported the results in Cancer Epidemiology, Biomarkers & Prevention.
“High blood pressure is an important modifiable risk factor that increases the risk of heart problems in everyone,” Dr Gibson said. “Research has shown that high blood pressure can have an even greater negative impact on survivors of childhood cancer who were treated with cardiotoxic therapies such as anthracyclines or chest radiation.”
To assess the prevalence of hypertension among CCSs, Dr Gibson and his colleagues examined 3016 adults who were 10-year survivors of childhood cancers. The subjects were enrolled in the St. Jude Lifetime Cohort Study, which provides ongoing medical assessments of CCSs to advance knowledge of their long-term health outcomes.
The subjects’ mean age at the initial study assessment was 32, and 52% were male. Most (83%) were non-Hispanic white, 14% were non-Hispanic black, 2% were Hispanic, and 1% were “other.”
Thirty-seven percent of subjects had leukemia, 12% had Hodgkin lymphoma, and 7% had non-Hodgkin lymphoma.
Eighty-six percent of subjects had received chemotherapy, and 59% received radiation.
Results
Subjects were considered to have hypertension if their systolic blood pressure was 140 or greater, their diastolic blood pressure was 90 or greater, or if they had been previously diagnosed with hypertension and were taking antihypertensive medication.
The prevalence of hypertension was 2.6 times higher among CCSs than expected, based on age-, sex-, race- and body mass index-specific rates in the general population.
In addition, the incidence of hypertension increased for CCSs over time. Thirteen percent of CCSs had hypertension at age 30, 37% had it at age 40, and more than 70% had it at age 50.
Dr Gibson said rates of hypertension in CCSs matched rates in the general population of people about a decade older.
The researchers identified several factors that were significantly associated with hypertension among CCSs, including:
- Male sex (odd ratio [OR], 1.38; 95% CI, 1.14–1.67)
- Non-Hispanic black race (OR, 1.66; 95% CI, 1.28–2.16)
- Older age at assessment (OR per 1 year of age, 1.10; 95% CI, 1.08–1.11)
- Being overweight (OR, 1.58; 95% CI, 1.21–2.07)
- Obesity (OR, 3.02; 95% CI, 2.34–3.88).
Exposure to any type of radiation or chemotherapy was not significantly associated with hypertension, but nephrectomy was (OR, 1.68; 95% CI, 1.11–2.53).
Dr Gibson said the lack of an association between hypertension and radiation/chemotherapy was surprising. It suggests the connection between childhood cancer survival and adult hypertension is multifactorial and worthy of future research.
In the meantime, he said, clinicians should be mindful that CCSs are more likely than the general public to develop hypertension.
“The good news is that, unlike prior cancer therapy, high blood pressure is a modifiable risk factor,” Dr Gibson noted. “Research is needed to identify effective interventions to prevent hypertension in survivors, but our results emphasize the importance of blood pressure surveillance and management.”
Dr Gibson said a limitation of this study is that it was based on blood pressure measurements taken at a single study visit. A clinical diagnosis of hypertension typically requires measurements taken at multiple intervals.
In addition, the St. Jude Lifetime Cohort is a group of CCSs who undergo frequent clinical follow-up, so its participants may have benefited from being monitored and may therefore be in better health than CCSs who have less comprehensive follow-up. ![]()
Method identifies effective treatments for leukemias, lymphomas

An ex vivo drug screening method can reveal optimal therapies for patients with hematologic malignancies, according to research published in The Lancet Haematology.
Researchers used a method called pharmacoscopy to measure single-cell responses to possible treatments in samples from patients with leukemias and lymphomas.
The team then used these results to guide treatment decisions and found that pharmacoscopy-guided treatment greatly improved response rates and progression-free survival (PFS).
“Having a robust, fast, and reliable predictive test at our disposal during the patient treatment process, especially at the time of relapse where a new intervention must be selected quickly, will change how medical doctors prioritize drugs to use for late-stage patients,” said study author Philipp Staber, MD, of Medical University of Vienna in Austria.
With pharmacoscopy, hundreds of drug options can be pre-tested ex vivo in small liquid biopsy samples collected from individual patients. The effects of each drug on the individual cells are quantified using high-throughput and high-content automated confocal microscopy.
In combination with specially developed analysis methods, machine learning, and other algorithms, pharmacoscopy allows quantification of never-before visualized phenotypes. The method was first described last April in Nature Chemical Biology.
Now, Dr Staber and his colleagues have reported, in The Lancet Haematology, an interim analysis of the first clinical trial testing pharmacoscopy-guided treatment.
There were 17 evaluable patients, all of whom had aggressive hematologic malignancies. This included diffuse large B-cell lymphoma (n=6), acute myeloid leukemia (n=3), B-cell acute lymphoblastic leukemia (n=2), precursor B-cell lymphoblastic lymphoma (n=1), peripheral T-cell lymphoma (n=1), primary mediastinal B-cell lymphoma (n=1), T-cell lymphoblastic lymphoma (n=1), follicular lymphoma (n=1), and T-cell prolymphocytic leukemia (n=1).
The researchers compared outcomes with pharmacoscopy-guided treatment to outcomes with the most recent regimen on which the patient had progressed.
The overall response rate was 88% with pharmacoscopy-guided treatment and 24% with the patients’ most recent previous treatment regimen (odds ratio=24.38; 95%, CI 3.99–125.4; P=0.0013).
None of the patients had progressive disease as their best overall response when they received pharmacoscopy-guided treatment. However, 7 patients had progressive disease in response to their most recent prior regimen.
At the time of analysis, 8 patients (47%) still had ongoing responses after pharmacoscopy-guided treatment.
In addition, pharmacoscopy-guided treatment significantly improved PFS. The median PFS was 22.6 weeks with pharmacoscopy-guided treatment and 5.7 weeks with the most recent prior regimen (hazard ratio=3.14; 95%, CI 1.37–7.22; P=0.0075).
“Evidence that the pharmacoscopy approach is helpful for clinical evaluation of therapy is wonderful,” said study author Giulio Superti-Furga, PhD, of CeMM Research Center for Molecular Medicine in Vienna, Austria.
“Single-cell functional analysis of primary material gives unprecedented resolution and precision that we are sure to further develop in the future to address yet more diseases.” ![]()
An ex vivo drug screening method can reveal optimal therapies for patients with hematologic malignancies, according to research published in The Lancet Haematology.
Researchers used a method called pharmacoscopy to measure single-cell responses to possible treatments in samples from patients with leukemias and lymphomas.
The team then used these results to guide treatment decisions and found that pharmacoscopy-guided treatment greatly improved response rates and progression-free survival (PFS).
“Having a robust, fast, and reliable predictive test at our disposal during the patient treatment process, especially at the time of relapse where a new intervention must be selected quickly, will change how medical doctors prioritize drugs to use for late-stage patients,” said study author Philipp Staber, MD, of Medical University of Vienna in Austria.
With pharmacoscopy, hundreds of drug options can be pre-tested ex vivo in small liquid biopsy samples collected from individual patients. The effects of each drug on the individual cells are quantified using high-throughput and high-content automated confocal microscopy.
In combination with specially developed analysis methods, machine learning, and other algorithms, pharmacoscopy allows quantification of never-before visualized phenotypes. The method was first described last April in Nature Chemical Biology.
Now, Dr Staber and his colleagues have reported, in The Lancet Haematology, an interim analysis of the first clinical trial testing pharmacoscopy-guided treatment.
There were 17 evaluable patients, all of whom had aggressive hematologic malignancies. This included diffuse large B-cell lymphoma (n=6), acute myeloid leukemia (n=3), B-cell acute lymphoblastic leukemia (n=2), precursor B-cell lymphoblastic lymphoma (n=1), peripheral T-cell lymphoma (n=1), primary mediastinal B-cell lymphoma (n=1), T-cell lymphoblastic lymphoma (n=1), follicular lymphoma (n=1), and T-cell prolymphocytic leukemia (n=1).
The researchers compared outcomes with pharmacoscopy-guided treatment to outcomes with the most recent regimen on which the patient had progressed.
The overall response rate was 88% with pharmacoscopy-guided treatment and 24% with the patients’ most recent previous treatment regimen (odds ratio=24.38; 95%, CI 3.99–125.4; P=0.0013).
None of the patients had progressive disease as their best overall response when they received pharmacoscopy-guided treatment. However, 7 patients had progressive disease in response to their most recent prior regimen.
At the time of analysis, 8 patients (47%) still had ongoing responses after pharmacoscopy-guided treatment.
In addition, pharmacoscopy-guided treatment significantly improved PFS. The median PFS was 22.6 weeks with pharmacoscopy-guided treatment and 5.7 weeks with the most recent prior regimen (hazard ratio=3.14; 95%, CI 1.37–7.22; P=0.0075).
“Evidence that the pharmacoscopy approach is helpful for clinical evaluation of therapy is wonderful,” said study author Giulio Superti-Furga, PhD, of CeMM Research Center for Molecular Medicine in Vienna, Austria.
“Single-cell functional analysis of primary material gives unprecedented resolution and precision that we are sure to further develop in the future to address yet more diseases.” ![]()
An ex vivo drug screening method can reveal optimal therapies for patients with hematologic malignancies, according to research published in The Lancet Haematology.
Researchers used a method called pharmacoscopy to measure single-cell responses to possible treatments in samples from patients with leukemias and lymphomas.
The team then used these results to guide treatment decisions and found that pharmacoscopy-guided treatment greatly improved response rates and progression-free survival (PFS).
“Having a robust, fast, and reliable predictive test at our disposal during the patient treatment process, especially at the time of relapse where a new intervention must be selected quickly, will change how medical doctors prioritize drugs to use for late-stage patients,” said study author Philipp Staber, MD, of Medical University of Vienna in Austria.
With pharmacoscopy, hundreds of drug options can be pre-tested ex vivo in small liquid biopsy samples collected from individual patients. The effects of each drug on the individual cells are quantified using high-throughput and high-content automated confocal microscopy.
In combination with specially developed analysis methods, machine learning, and other algorithms, pharmacoscopy allows quantification of never-before visualized phenotypes. The method was first described last April in Nature Chemical Biology.
Now, Dr Staber and his colleagues have reported, in The Lancet Haematology, an interim analysis of the first clinical trial testing pharmacoscopy-guided treatment.
There were 17 evaluable patients, all of whom had aggressive hematologic malignancies. This included diffuse large B-cell lymphoma (n=6), acute myeloid leukemia (n=3), B-cell acute lymphoblastic leukemia (n=2), precursor B-cell lymphoblastic lymphoma (n=1), peripheral T-cell lymphoma (n=1), primary mediastinal B-cell lymphoma (n=1), T-cell lymphoblastic lymphoma (n=1), follicular lymphoma (n=1), and T-cell prolymphocytic leukemia (n=1).
The researchers compared outcomes with pharmacoscopy-guided treatment to outcomes with the most recent regimen on which the patient had progressed.
The overall response rate was 88% with pharmacoscopy-guided treatment and 24% with the patients’ most recent previous treatment regimen (odds ratio=24.38; 95%, CI 3.99–125.4; P=0.0013).
None of the patients had progressive disease as their best overall response when they received pharmacoscopy-guided treatment. However, 7 patients had progressive disease in response to their most recent prior regimen.
At the time of analysis, 8 patients (47%) still had ongoing responses after pharmacoscopy-guided treatment.
In addition, pharmacoscopy-guided treatment significantly improved PFS. The median PFS was 22.6 weeks with pharmacoscopy-guided treatment and 5.7 weeks with the most recent prior regimen (hazard ratio=3.14; 95%, CI 1.37–7.22; P=0.0075).
“Evidence that the pharmacoscopy approach is helpful for clinical evaluation of therapy is wonderful,” said study author Giulio Superti-Furga, PhD, of CeMM Research Center for Molecular Medicine in Vienna, Austria.
“Single-cell functional analysis of primary material gives unprecedented resolution and precision that we are sure to further develop in the future to address yet more diseases.” ![]()
FDA expands approval of obinutuzumab

The US Food and Drug Administration (FDA) has expanded the approved use of obinutuzumab (Gazyva®).
The drug is now approved for use in combination with chemotherapy to treat patients with previously untreated follicular lymphoma (FL) that is advanced (stage II bulky, stage III, or stage IV) .
In patients who respond to this treatment, obinutuzumab monotherapy can be given as maintenance.
The FDA granted this new approval of obinutuzumab to Genentech, Inc. The application for obinutuzumab in this indication received priority review.
The latest FDA approval means obinutuzumab is available in the US for the following indications:
- In combination with chlorambucil to treat chronic lymphocytic leukemia (CLL) in adults who have not had previous CLL treatment
- In combination with bendamustine, followed by obinutuzumab alone, to treat FL in adults who did not respond to a rituximab-containing regimen or whose FL returned after such treatment
- In combination with chemotherapy, followed by obinutuzumab alone in responders, to treat stage II bulky, stage III, or stage IV FL in adults who have not had previous FL treatment.
Phase 3 results
The latest approval of obinutuzumab is based on results from the phase 3 GALLIUM study, which were presented at the 2016 ASH Annual Meeting and published in NEJM in October.
The following are updated data from the obinutuzumab prescribing information.
GALLIUM included 1385 patients with previously untreated non-Hodgkin lymphoma, and 1202 of these patients had advanced FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone), CVP (cyclophosphamide, vincristine, and prednisolone), and bendamustine.
At a median observation time of 38 months, the overall response rate was 91% in the obinutuzumab arm and 88% in the rituximab arm. The complete response rates were 28% and 27%, respectively.
The median progression-free survival was not reached in either arm. The hazard ratio, for obinutuzumab compared to rituximab, was 0.72 (95% CI, 0.56-0.93, P=0.0118).
Safety was evaluated based on all 1385 patients in the study, 86% of whom had previously untreated FL and 14% of whom had marginal zone lymphoma.
Serious adverse events (AEs) occurred in 50% of patients in the obinutuzumab arm and 43% in the rituximab arm. Fatal AEs occurred in 5% and 4%, respectively. Infections and second malignancies were the leading causes of these deaths.
The most common AEs (incidence ≥ 20%) observed at least 2% more patients in the obinutuzumab arm were infusion-related reactions, neutropenia, upper respiratory tract infection, cough, constipation, and diarrhea.
The most common grade 3 to 5 AEs (incidence ≥ 5%) observed more frequently in the obinutuzumab arm were neutropenia, infusion-related reactions, febrile neutropenia, and thrombocytopenia. ![]()
The US Food and Drug Administration (FDA) has expanded the approved use of obinutuzumab (Gazyva®).
The drug is now approved for use in combination with chemotherapy to treat patients with previously untreated follicular lymphoma (FL) that is advanced (stage II bulky, stage III, or stage IV) .
In patients who respond to this treatment, obinutuzumab monotherapy can be given as maintenance.
The FDA granted this new approval of obinutuzumab to Genentech, Inc. The application for obinutuzumab in this indication received priority review.
The latest FDA approval means obinutuzumab is available in the US for the following indications:
- In combination with chlorambucil to treat chronic lymphocytic leukemia (CLL) in adults who have not had previous CLL treatment
- In combination with bendamustine, followed by obinutuzumab alone, to treat FL in adults who did not respond to a rituximab-containing regimen or whose FL returned after such treatment
- In combination with chemotherapy, followed by obinutuzumab alone in responders, to treat stage II bulky, stage III, or stage IV FL in adults who have not had previous FL treatment.
Phase 3 results
The latest approval of obinutuzumab is based on results from the phase 3 GALLIUM study, which were presented at the 2016 ASH Annual Meeting and published in NEJM in October.
The following are updated data from the obinutuzumab prescribing information.
GALLIUM included 1385 patients with previously untreated non-Hodgkin lymphoma, and 1202 of these patients had advanced FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone), CVP (cyclophosphamide, vincristine, and prednisolone), and bendamustine.
At a median observation time of 38 months, the overall response rate was 91% in the obinutuzumab arm and 88% in the rituximab arm. The complete response rates were 28% and 27%, respectively.
The median progression-free survival was not reached in either arm. The hazard ratio, for obinutuzumab compared to rituximab, was 0.72 (95% CI, 0.56-0.93, P=0.0118).
Safety was evaluated based on all 1385 patients in the study, 86% of whom had previously untreated FL and 14% of whom had marginal zone lymphoma.
Serious adverse events (AEs) occurred in 50% of patients in the obinutuzumab arm and 43% in the rituximab arm. Fatal AEs occurred in 5% and 4%, respectively. Infections and second malignancies were the leading causes of these deaths.
The most common AEs (incidence ≥ 20%) observed at least 2% more patients in the obinutuzumab arm were infusion-related reactions, neutropenia, upper respiratory tract infection, cough, constipation, and diarrhea.
The most common grade 3 to 5 AEs (incidence ≥ 5%) observed more frequently in the obinutuzumab arm were neutropenia, infusion-related reactions, febrile neutropenia, and thrombocytopenia. ![]()
The US Food and Drug Administration (FDA) has expanded the approved use of obinutuzumab (Gazyva®).
The drug is now approved for use in combination with chemotherapy to treat patients with previously untreated follicular lymphoma (FL) that is advanced (stage II bulky, stage III, or stage IV) .
In patients who respond to this treatment, obinutuzumab monotherapy can be given as maintenance.
The FDA granted this new approval of obinutuzumab to Genentech, Inc. The application for obinutuzumab in this indication received priority review.
The latest FDA approval means obinutuzumab is available in the US for the following indications:
- In combination with chlorambucil to treat chronic lymphocytic leukemia (CLL) in adults who have not had previous CLL treatment
- In combination with bendamustine, followed by obinutuzumab alone, to treat FL in adults who did not respond to a rituximab-containing regimen or whose FL returned after such treatment
- In combination with chemotherapy, followed by obinutuzumab alone in responders, to treat stage II bulky, stage III, or stage IV FL in adults who have not had previous FL treatment.
Phase 3 results
The latest approval of obinutuzumab is based on results from the phase 3 GALLIUM study, which were presented at the 2016 ASH Annual Meeting and published in NEJM in October.
The following are updated data from the obinutuzumab prescribing information.
GALLIUM included 1385 patients with previously untreated non-Hodgkin lymphoma, and 1202 of these patients had advanced FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone), CVP (cyclophosphamide, vincristine, and prednisolone), and bendamustine.
At a median observation time of 38 months, the overall response rate was 91% in the obinutuzumab arm and 88% in the rituximab arm. The complete response rates were 28% and 27%, respectively.
The median progression-free survival was not reached in either arm. The hazard ratio, for obinutuzumab compared to rituximab, was 0.72 (95% CI, 0.56-0.93, P=0.0118).
Safety was evaluated based on all 1385 patients in the study, 86% of whom had previously untreated FL and 14% of whom had marginal zone lymphoma.
Serious adverse events (AEs) occurred in 50% of patients in the obinutuzumab arm and 43% in the rituximab arm. Fatal AEs occurred in 5% and 4%, respectively. Infections and second malignancies were the leading causes of these deaths.
The most common AEs (incidence ≥ 20%) observed at least 2% more patients in the obinutuzumab arm were infusion-related reactions, neutropenia, upper respiratory tract infection, cough, constipation, and diarrhea.
The most common grade 3 to 5 AEs (incidence ≥ 5%) observed more frequently in the obinutuzumab arm were neutropenia, infusion-related reactions, febrile neutropenia, and thrombocytopenia. ![]()
CHMP wants to expand use of BV to include CTCL

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for brentuximab vedotin (BV, Adcetris).
The CHMP is recommending authorization of BV to treat adults with CD30+ cutaneous T-cell lymphoma (CTCL) who have received at least 1 prior systemic therapy.
The CHMP’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The EC previously approved BV to treat:
- Adults with relapsed or refractory CD30+ Hodgkin lymphoma (HL) following autologous stem cell transplant (ASCT) or following at least 2 prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- Adults with CD30+ HL at increased risk of relapse or progression following ASCT
- Adults with relapsed or refractory systemic anaplastic large-cell lymphoma.
The CHMP’s recommendation to approve BV for CTCL is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in July 2015 and August 2015.
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for brentuximab vedotin (BV, Adcetris).
The CHMP is recommending authorization of BV to treat adults with CD30+ cutaneous T-cell lymphoma (CTCL) who have received at least 1 prior systemic therapy.
The CHMP’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The EC previously approved BV to treat:
- Adults with relapsed or refractory CD30+ Hodgkin lymphoma (HL) following autologous stem cell transplant (ASCT) or following at least 2 prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- Adults with CD30+ HL at increased risk of relapse or progression following ASCT
- Adults with relapsed or refractory systemic anaplastic large-cell lymphoma.
The CHMP’s recommendation to approve BV for CTCL is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in July 2015 and August 2015.
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for brentuximab vedotin (BV, Adcetris).
The CHMP is recommending authorization of BV to treat adults with CD30+ cutaneous T-cell lymphoma (CTCL) who have received at least 1 prior systemic therapy.
The CHMP’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The EC previously approved BV to treat:
- Adults with relapsed or refractory CD30+ Hodgkin lymphoma (HL) following autologous stem cell transplant (ASCT) or following at least 2 prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- Adults with CD30+ HL at increased risk of relapse or progression following ASCT
- Adults with relapsed or refractory systemic anaplastic large-cell lymphoma.
The CHMP’s recommendation to approve BV for CTCL is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in July 2015 and August 2015.
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June. ![]()
FDA approves brentuximab vedotin for pcALCL, MF

The US Food and Drug Administration (FDA) has expanded the approved use of brentuximab vedotin (BV, ADCETRIS).
BV is now approved for adults with primary cutaneous anaplastic large-cell lymphoma (pcALCL) and CD30-expressing mycosis fungoides (MF) who have received prior systemic therapy.
This is the fourth FDA-approved indication for BV. The drug has regular approval for 2 indications in classical Hodgkin lymphoma and accelerated approval for the treatment of systemic ALCL.
In November 2016, the FDA granted BV breakthrough therapy designation for the treatment of patients with pcALCL and CD30-expressing MF who require systemic therapy and have received one prior systemic therapy. The agency also granted the supplemental biologics license application priority review.
The approval for BV in pcALCL and CD30-expressing MF is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Phase 3 trial
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June.
There were 128 patients in the intent-to-treat and safety populations. Sixty-four patients (48 with MF and 16 with pcALCL) were randomized to receive BV at 1.8 mg/kg every 3 weeks for up to 48 weeks.
The other 64 patients (49 with MF and 15 with pcALCL) were randomized to receive standard of care (SOC)—methotrexate at 5 mg to 50 mg weekly or bexarotene at a target dose of 300 mg/m² daily for up to 48 weeks.
The study’s primary endpoint was the rate of objective response lasting at least 4 months (ORR4). The ORR4 rate was significantly higher with BV than with SOC—56.3% and 12.5%, respectively (P<0.0001).
For patients with MF, the ORR4 was 50% with BV and 10% with SOC. For patients with pcALCL, the ORR4 was 75% with BV and 20% with SOC.
Overall, the complete response (CR) rates were 15.6% in the BV arm and 1.6% in the SOC arm (P=0.0046).
For patients with MF, the CR rate was 10% with BV and 0% with SOC. For patients with pcALCL, the CR rate was 31% with BV and 7% with SOC.
Progression-free survival (PFS) was significantly longer in the BV arm than the SOC arm. The median PFS was 16.7 months and 3.5 months, respectively. The hazard ratio was 0.270 (P<0.0001).
For patients with MF, the median PFS was 15.9 months with BV and 3.5 months with SOC. For patients with pcALCL, the median PFS was 27.5 months with BV and 5.3 months with SOC.
The most common adverse events (AEs) of any grade (occurring in 15% or more of patients in the BV and SOC arms, respectively) were peripheral neuropathy (67% and 6%), nausea (36% and 13%), diarrhea (29% and 6%), fatigue (29% and 27%), vomiting (17% and 5%), alopecia (15% and 3%), pruritus (17% and 13%), pyrexia (17% and 18%), decreased appetite (15% and 5%), and hypertriglyceridemia (2% and 18%).
Phase 2 trials
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in 2015.
The first study was published in July of that year. The trial enrolled 32 patients with MF or Sézary syndrome. Thirty patients were evaluable for efficacy, and more than half had received 3 or more prior systemic therapies.
Patients received BV (1.8 mg/kg) every 3 weeks for a maximum of 16 doses. The primary endpoint was objective clinical response rate.
Seventy percent of patients (21/30) achieved an objective response across all stages of disease. One patient had a CR, 20 had a partial response, 4 had stable disease, 5 had progressive disease, and 2 were not evaluable for response.
The most common related AEs of any grade were peripheral neuropathy (66%), fatigue (47%), nausea (28%), hair loss (22%), and neutropenia (19%). Grade 3/4 related AEs included neutropenia (n=4), rash (n=3), and peripheral neuropathy (n=1).
The second phase 2 trial was published in August 2015. This trial enrolled CD30-positive patients with lymphomatoid papulosis (LyP), pcALCL, and MF.
Fifty-four patients were enrolled, and 48 were evaluable at the time of analysis. Patients had received an infusion of BV (1.8 mg/kg) every 21 days.
Seventy-three percent of patients (35/48) achieved an objective response, including 100% (20/20) with LyP and/or pcALCL and 54% (15/28) with MF. The CR rate was 35% (n=17).
The most common AEs were peripheral neuropathy (67%), fatigue (35%), skin rash (24%), diarrhea (15%), muscle pain (17%), localized skin infection (15%), neutropenia (15%), and hair loss (11%).
Grade 3/4 AEs included neutropenia (n=3), nausea (n=2), unstable angina or myocardial infarction (n=2), infection (n=2), joint pain (n=2), fatigue (n=1), deep vein thrombosis (n=1), pulmonary embolism (n=1), aminotransferase elevation (n=1), and dehydration (n=1). ![]()
The US Food and Drug Administration (FDA) has expanded the approved use of brentuximab vedotin (BV, ADCETRIS).
BV is now approved for adults with primary cutaneous anaplastic large-cell lymphoma (pcALCL) and CD30-expressing mycosis fungoides (MF) who have received prior systemic therapy.
This is the fourth FDA-approved indication for BV. The drug has regular approval for 2 indications in classical Hodgkin lymphoma and accelerated approval for the treatment of systemic ALCL.
In November 2016, the FDA granted BV breakthrough therapy designation for the treatment of patients with pcALCL and CD30-expressing MF who require systemic therapy and have received one prior systemic therapy. The agency also granted the supplemental biologics license application priority review.
The approval for BV in pcALCL and CD30-expressing MF is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Phase 3 trial
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June.
There were 128 patients in the intent-to-treat and safety populations. Sixty-four patients (48 with MF and 16 with pcALCL) were randomized to receive BV at 1.8 mg/kg every 3 weeks for up to 48 weeks.
The other 64 patients (49 with MF and 15 with pcALCL) were randomized to receive standard of care (SOC)—methotrexate at 5 mg to 50 mg weekly or bexarotene at a target dose of 300 mg/m² daily for up to 48 weeks.
The study’s primary endpoint was the rate of objective response lasting at least 4 months (ORR4). The ORR4 rate was significantly higher with BV than with SOC—56.3% and 12.5%, respectively (P<0.0001).
For patients with MF, the ORR4 was 50% with BV and 10% with SOC. For patients with pcALCL, the ORR4 was 75% with BV and 20% with SOC.
Overall, the complete response (CR) rates were 15.6% in the BV arm and 1.6% in the SOC arm (P=0.0046).
For patients with MF, the CR rate was 10% with BV and 0% with SOC. For patients with pcALCL, the CR rate was 31% with BV and 7% with SOC.
Progression-free survival (PFS) was significantly longer in the BV arm than the SOC arm. The median PFS was 16.7 months and 3.5 months, respectively. The hazard ratio was 0.270 (P<0.0001).
For patients with MF, the median PFS was 15.9 months with BV and 3.5 months with SOC. For patients with pcALCL, the median PFS was 27.5 months with BV and 5.3 months with SOC.
The most common adverse events (AEs) of any grade (occurring in 15% or more of patients in the BV and SOC arms, respectively) were peripheral neuropathy (67% and 6%), nausea (36% and 13%), diarrhea (29% and 6%), fatigue (29% and 27%), vomiting (17% and 5%), alopecia (15% and 3%), pruritus (17% and 13%), pyrexia (17% and 18%), decreased appetite (15% and 5%), and hypertriglyceridemia (2% and 18%).
Phase 2 trials
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in 2015.
The first study was published in July of that year. The trial enrolled 32 patients with MF or Sézary syndrome. Thirty patients were evaluable for efficacy, and more than half had received 3 or more prior systemic therapies.
Patients received BV (1.8 mg/kg) every 3 weeks for a maximum of 16 doses. The primary endpoint was objective clinical response rate.
Seventy percent of patients (21/30) achieved an objective response across all stages of disease. One patient had a CR, 20 had a partial response, 4 had stable disease, 5 had progressive disease, and 2 were not evaluable for response.
The most common related AEs of any grade were peripheral neuropathy (66%), fatigue (47%), nausea (28%), hair loss (22%), and neutropenia (19%). Grade 3/4 related AEs included neutropenia (n=4), rash (n=3), and peripheral neuropathy (n=1).
The second phase 2 trial was published in August 2015. This trial enrolled CD30-positive patients with lymphomatoid papulosis (LyP), pcALCL, and MF.
Fifty-four patients were enrolled, and 48 were evaluable at the time of analysis. Patients had received an infusion of BV (1.8 mg/kg) every 21 days.
Seventy-three percent of patients (35/48) achieved an objective response, including 100% (20/20) with LyP and/or pcALCL and 54% (15/28) with MF. The CR rate was 35% (n=17).
The most common AEs were peripheral neuropathy (67%), fatigue (35%), skin rash (24%), diarrhea (15%), muscle pain (17%), localized skin infection (15%), neutropenia (15%), and hair loss (11%).
Grade 3/4 AEs included neutropenia (n=3), nausea (n=2), unstable angina or myocardial infarction (n=2), infection (n=2), joint pain (n=2), fatigue (n=1), deep vein thrombosis (n=1), pulmonary embolism (n=1), aminotransferase elevation (n=1), and dehydration (n=1). ![]()
The US Food and Drug Administration (FDA) has expanded the approved use of brentuximab vedotin (BV, ADCETRIS).
BV is now approved for adults with primary cutaneous anaplastic large-cell lymphoma (pcALCL) and CD30-expressing mycosis fungoides (MF) who have received prior systemic therapy.
This is the fourth FDA-approved indication for BV. The drug has regular approval for 2 indications in classical Hodgkin lymphoma and accelerated approval for the treatment of systemic ALCL.
In November 2016, the FDA granted BV breakthrough therapy designation for the treatment of patients with pcALCL and CD30-expressing MF who require systemic therapy and have received one prior systemic therapy. The agency also granted the supplemental biologics license application priority review.
The approval for BV in pcALCL and CD30-expressing MF is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Phase 3 trial
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June.
There were 128 patients in the intent-to-treat and safety populations. Sixty-four patients (48 with MF and 16 with pcALCL) were randomized to receive BV at 1.8 mg/kg every 3 weeks for up to 48 weeks.
The other 64 patients (49 with MF and 15 with pcALCL) were randomized to receive standard of care (SOC)—methotrexate at 5 mg to 50 mg weekly or bexarotene at a target dose of 300 mg/m² daily for up to 48 weeks.
The study’s primary endpoint was the rate of objective response lasting at least 4 months (ORR4). The ORR4 rate was significantly higher with BV than with SOC—56.3% and 12.5%, respectively (P<0.0001).
For patients with MF, the ORR4 was 50% with BV and 10% with SOC. For patients with pcALCL, the ORR4 was 75% with BV and 20% with SOC.
Overall, the complete response (CR) rates were 15.6% in the BV arm and 1.6% in the SOC arm (P=0.0046).
For patients with MF, the CR rate was 10% with BV and 0% with SOC. For patients with pcALCL, the CR rate was 31% with BV and 7% with SOC.
Progression-free survival (PFS) was significantly longer in the BV arm than the SOC arm. The median PFS was 16.7 months and 3.5 months, respectively. The hazard ratio was 0.270 (P<0.0001).
For patients with MF, the median PFS was 15.9 months with BV and 3.5 months with SOC. For patients with pcALCL, the median PFS was 27.5 months with BV and 5.3 months with SOC.
The most common adverse events (AEs) of any grade (occurring in 15% or more of patients in the BV and SOC arms, respectively) were peripheral neuropathy (67% and 6%), nausea (36% and 13%), diarrhea (29% and 6%), fatigue (29% and 27%), vomiting (17% and 5%), alopecia (15% and 3%), pruritus (17% and 13%), pyrexia (17% and 18%), decreased appetite (15% and 5%), and hypertriglyceridemia (2% and 18%).
Phase 2 trials
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in 2015.
The first study was published in July of that year. The trial enrolled 32 patients with MF or Sézary syndrome. Thirty patients were evaluable for efficacy, and more than half had received 3 or more prior systemic therapies.
Patients received BV (1.8 mg/kg) every 3 weeks for a maximum of 16 doses. The primary endpoint was objective clinical response rate.
Seventy percent of patients (21/30) achieved an objective response across all stages of disease. One patient had a CR, 20 had a partial response, 4 had stable disease, 5 had progressive disease, and 2 were not evaluable for response.
The most common related AEs of any grade were peripheral neuropathy (66%), fatigue (47%), nausea (28%), hair loss (22%), and neutropenia (19%). Grade 3/4 related AEs included neutropenia (n=4), rash (n=3), and peripheral neuropathy (n=1).
The second phase 2 trial was published in August 2015. This trial enrolled CD30-positive patients with lymphomatoid papulosis (LyP), pcALCL, and MF.
Fifty-four patients were enrolled, and 48 were evaluable at the time of analysis. Patients had received an infusion of BV (1.8 mg/kg) every 21 days.
Seventy-three percent of patients (35/48) achieved an objective response, including 100% (20/20) with LyP and/or pcALCL and 54% (15/28) with MF. The CR rate was 35% (n=17).
The most common AEs were peripheral neuropathy (67%), fatigue (35%), skin rash (24%), diarrhea (15%), muscle pain (17%), localized skin infection (15%), neutropenia (15%), and hair loss (11%).
Grade 3/4 AEs included neutropenia (n=3), nausea (n=2), unstable angina or myocardial infarction (n=2), infection (n=2), joint pain (n=2), fatigue (n=1), deep vein thrombosis (n=1), pulmonary embolism (n=1), aminotransferase elevation (n=1), and dehydration (n=1). ![]()
Ibrutinib sustains efficacy in CLL at 4-year follow-up

NEW YORK, NY—The 4-year follow-up of the RESONATE trial suggests ibrutinib may provide long-term efficacy in previously treated patients with chronic lymphocytic leukemia (CLL).
The median progression-free survival (PFS) has not yet been reached in this trial, regardless of high-risk cytogenetics, according to Jennifer Brown, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
She presented the update at Lymphoma & Myeloma 2017. The follow-up study was awarded the best clinical CLL abstract of the meeting.
In the phase 3 RESONATE study, investigators compared ibrutinib—the first-in-class, once-daily, oral inhibitor of Bruton tyrosine kinase—to ofatumumab in previously treated CLL/small lymphocytic lymphoma (SLL).
The primary analysis showed ibrutinib significantly improved survival, with a 78% reduction in the risk of progression and a 57% reduction in the risk of death.
The phase 3 trial randomized 195 CLL/SLL patients to oral ibrutinib at 420 mg once daily and 196 patients to intravenous ofatumumab at an initial dose of 300 mg followed by 2000 mg for 11 doses over 24 weeks.
One hundred thirty-three patients progressed on ofatumumab and crossed over to receive once-daily ibrutinib.
Patient characteristics
In each arm, the median patient age was 67, more than half of patients had an ECOG status of 1, and more than half had advanced-stage disease.
High-risk genetic abnormalities were common, Dr Brown said, with deletion 11q in a third of patients in the ibrutinib arm and 31% in the ofatumumab arm. Another third in each arm had deletion 17p, while 51% in the ibrutinib arm and 46% in the ofatumumab arm had TP53 mutation.
About a quarter of the patients in each arm had complex karyotype, and 73% and 63% in the ibrutinib and ofatumumab arms, respectively, were IGHV-unmutated.
Survival
Ibrutinib significantly extended PFS compared with ofatumumab. At a median follow-up for ibrutinib of 44 months (range, 0.33 – 53), ibrutinib led to an 87% reduction in the risk of progression or death. The 3-year PFS rate was 59% with ibrutinib and 3% with ofatumumab.
Ibrutinib conferred a benefit in PFS across all baseline patient characteristics.
Among ibrutinib-treated patients, the 3-year PFS was 53% for patients with deletion 17p, 66% for those with deletion 11q but not deletion 17p, and 58% for those with neither abnormality.
Dr Brown noted how closely complex karyotype associates with high-risk cytogenetics. Forty-two percent of patients with 17p deletion had a complex karyotype, as did 23% of patients with 11q deletion and 15% of patients with neither 17p nor 11q deletion.
For IGHV-mutation status, Dr Brown said there is no difference in PFS with this degree of follow-up.
In terms of TP53 mutation status, Dr Brown pointed out a trend toward a worse PFS in those patients with the mutation.
“We actually looked by individual p53 mutation versus 17p deletion, versus both, versus neither, in the 2-year follow-up paper and found that p53 with 17p, both abnormalities, did have worse PFS than neither,” she said.
“This may require further follow-up because we do know that most 17p patients also have a p53 mutation, particularly in the relapsed setting.”
As expected, Dr Brown said, those patients with more than 2 prior therapies had a worse PFS compared to patients with 2 or fewer prior therapies.
Multivariate analysis demonstrated that more than 2 prior lines of therapy or an elevated ß2 microglobulin were associated with decreased PFS with ibrutinib.
When the investigators adjusted the overall survival data for cross-over, ibrutinib was projected to continue the overall survival benefit compared with ofatumumab, with a hazard ratio of 0.37.
Response rates
Dr Brown noted that, early on, there’s quite a significant rate of partial response with lymphocytosis observed in patients on ibrutinib.
This “diminishes dramatically,” she said, but about 5% of patients at 3 and 4 years still have ongoing lymphocytosis.
“Similarly, initially, there’s a very low rate of complete remission, which has risen steadily to 9% at this follow-up,” she said.
And the overall response rate is 91%.
Treatment exposure and toxicity
The median duration of ibrutinib treatment is 41 months, and 46% of patients continue on treatment. Twenty-seven percent of patients discontinued due to progression, and 12% because of adverse events (AEs).
Of the 53 patients who discontinued therapy, 14 had transformation as their primary reason, 9 with diffuse large B-cell lymphoma, 3 with Hodgkin disease, and 2 with prolymphocytic lymphoma.
The most frequent AEs leading to discontinuation included pneumonia (n=3), anemia (n=2), thrombocytopenia (n=2), diarrhea (n=2), and anal incontinence (n=2).
AEs leading to discontinuation decreased over time—6% in year 0 to 1 and 4% in years 2 to 3.
“The most frequent cumulative AEs are similar to what we’ve seen in most prior studies,” Dr Brown said, including diarrhea, fatigue, and cough.
In terms of grade 3 or higher AEs, about a quarter of patients had neutropenia, 17% had pneumonia, and 8% had hypertension.
Six percent of patients had major hemorrhage, and all-grade atrial fibrillation occurred in 11% of patients.
“Now, many of the grade 3 and higher AEs did decline over time during the study,” Dr Brown noted. “You can see this is quite evident for neutropenia as well as pneumonia, and all infections declined from year 1 to subsequent years.”
Hypertension, in contrast, has been fairly steady over the later years, she said, and atrial fibrillation is highest in the first 6 months but then continues at a low rate thereafter.
The investigators believe these long-term results demonstrate that ibrutinib is tolerable and continues to show sustained efficacy in previously treated and high-genomic-risk patients with CLL. In addition, no long-term safety signals have emerged.
This study was sponsored by Pharmacyclics, LLC, an AbbVie company. ![]()
NEW YORK, NY—The 4-year follow-up of the RESONATE trial suggests ibrutinib may provide long-term efficacy in previously treated patients with chronic lymphocytic leukemia (CLL).
The median progression-free survival (PFS) has not yet been reached in this trial, regardless of high-risk cytogenetics, according to Jennifer Brown, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
She presented the update at Lymphoma & Myeloma 2017. The follow-up study was awarded the best clinical CLL abstract of the meeting.
In the phase 3 RESONATE study, investigators compared ibrutinib—the first-in-class, once-daily, oral inhibitor of Bruton tyrosine kinase—to ofatumumab in previously treated CLL/small lymphocytic lymphoma (SLL).
The primary analysis showed ibrutinib significantly improved survival, with a 78% reduction in the risk of progression and a 57% reduction in the risk of death.
The phase 3 trial randomized 195 CLL/SLL patients to oral ibrutinib at 420 mg once daily and 196 patients to intravenous ofatumumab at an initial dose of 300 mg followed by 2000 mg for 11 doses over 24 weeks.
One hundred thirty-three patients progressed on ofatumumab and crossed over to receive once-daily ibrutinib.
Patient characteristics
In each arm, the median patient age was 67, more than half of patients had an ECOG status of 1, and more than half had advanced-stage disease.
High-risk genetic abnormalities were common, Dr Brown said, with deletion 11q in a third of patients in the ibrutinib arm and 31% in the ofatumumab arm. Another third in each arm had deletion 17p, while 51% in the ibrutinib arm and 46% in the ofatumumab arm had TP53 mutation.
About a quarter of the patients in each arm had complex karyotype, and 73% and 63% in the ibrutinib and ofatumumab arms, respectively, were IGHV-unmutated.
Survival
Ibrutinib significantly extended PFS compared with ofatumumab. At a median follow-up for ibrutinib of 44 months (range, 0.33 – 53), ibrutinib led to an 87% reduction in the risk of progression or death. The 3-year PFS rate was 59% with ibrutinib and 3% with ofatumumab.
Ibrutinib conferred a benefit in PFS across all baseline patient characteristics.
Among ibrutinib-treated patients, the 3-year PFS was 53% for patients with deletion 17p, 66% for those with deletion 11q but not deletion 17p, and 58% for those with neither abnormality.
Dr Brown noted how closely complex karyotype associates with high-risk cytogenetics. Forty-two percent of patients with 17p deletion had a complex karyotype, as did 23% of patients with 11q deletion and 15% of patients with neither 17p nor 11q deletion.
For IGHV-mutation status, Dr Brown said there is no difference in PFS with this degree of follow-up.
In terms of TP53 mutation status, Dr Brown pointed out a trend toward a worse PFS in those patients with the mutation.
“We actually looked by individual p53 mutation versus 17p deletion, versus both, versus neither, in the 2-year follow-up paper and found that p53 with 17p, both abnormalities, did have worse PFS than neither,” she said.
“This may require further follow-up because we do know that most 17p patients also have a p53 mutation, particularly in the relapsed setting.”
As expected, Dr Brown said, those patients with more than 2 prior therapies had a worse PFS compared to patients with 2 or fewer prior therapies.
Multivariate analysis demonstrated that more than 2 prior lines of therapy or an elevated ß2 microglobulin were associated with decreased PFS with ibrutinib.
When the investigators adjusted the overall survival data for cross-over, ibrutinib was projected to continue the overall survival benefit compared with ofatumumab, with a hazard ratio of 0.37.
Response rates
Dr Brown noted that, early on, there’s quite a significant rate of partial response with lymphocytosis observed in patients on ibrutinib.
This “diminishes dramatically,” she said, but about 5% of patients at 3 and 4 years still have ongoing lymphocytosis.
“Similarly, initially, there’s a very low rate of complete remission, which has risen steadily to 9% at this follow-up,” she said.
And the overall response rate is 91%.
Treatment exposure and toxicity
The median duration of ibrutinib treatment is 41 months, and 46% of patients continue on treatment. Twenty-seven percent of patients discontinued due to progression, and 12% because of adverse events (AEs).
Of the 53 patients who discontinued therapy, 14 had transformation as their primary reason, 9 with diffuse large B-cell lymphoma, 3 with Hodgkin disease, and 2 with prolymphocytic lymphoma.
The most frequent AEs leading to discontinuation included pneumonia (n=3), anemia (n=2), thrombocytopenia (n=2), diarrhea (n=2), and anal incontinence (n=2).
AEs leading to discontinuation decreased over time—6% in year 0 to 1 and 4% in years 2 to 3.
“The most frequent cumulative AEs are similar to what we’ve seen in most prior studies,” Dr Brown said, including diarrhea, fatigue, and cough.
In terms of grade 3 or higher AEs, about a quarter of patients had neutropenia, 17% had pneumonia, and 8% had hypertension.
Six percent of patients had major hemorrhage, and all-grade atrial fibrillation occurred in 11% of patients.
“Now, many of the grade 3 and higher AEs did decline over time during the study,” Dr Brown noted. “You can see this is quite evident for neutropenia as well as pneumonia, and all infections declined from year 1 to subsequent years.”
Hypertension, in contrast, has been fairly steady over the later years, she said, and atrial fibrillation is highest in the first 6 months but then continues at a low rate thereafter.
The investigators believe these long-term results demonstrate that ibrutinib is tolerable and continues to show sustained efficacy in previously treated and high-genomic-risk patients with CLL. In addition, no long-term safety signals have emerged.
This study was sponsored by Pharmacyclics, LLC, an AbbVie company. ![]()
NEW YORK, NY—The 4-year follow-up of the RESONATE trial suggests ibrutinib may provide long-term efficacy in previously treated patients with chronic lymphocytic leukemia (CLL).
The median progression-free survival (PFS) has not yet been reached in this trial, regardless of high-risk cytogenetics, according to Jennifer Brown, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
She presented the update at Lymphoma & Myeloma 2017. The follow-up study was awarded the best clinical CLL abstract of the meeting.
In the phase 3 RESONATE study, investigators compared ibrutinib—the first-in-class, once-daily, oral inhibitor of Bruton tyrosine kinase—to ofatumumab in previously treated CLL/small lymphocytic lymphoma (SLL).
The primary analysis showed ibrutinib significantly improved survival, with a 78% reduction in the risk of progression and a 57% reduction in the risk of death.
The phase 3 trial randomized 195 CLL/SLL patients to oral ibrutinib at 420 mg once daily and 196 patients to intravenous ofatumumab at an initial dose of 300 mg followed by 2000 mg for 11 doses over 24 weeks.
One hundred thirty-three patients progressed on ofatumumab and crossed over to receive once-daily ibrutinib.
Patient characteristics
In each arm, the median patient age was 67, more than half of patients had an ECOG status of 1, and more than half had advanced-stage disease.
High-risk genetic abnormalities were common, Dr Brown said, with deletion 11q in a third of patients in the ibrutinib arm and 31% in the ofatumumab arm. Another third in each arm had deletion 17p, while 51% in the ibrutinib arm and 46% in the ofatumumab arm had TP53 mutation.
About a quarter of the patients in each arm had complex karyotype, and 73% and 63% in the ibrutinib and ofatumumab arms, respectively, were IGHV-unmutated.
Survival
Ibrutinib significantly extended PFS compared with ofatumumab. At a median follow-up for ibrutinib of 44 months (range, 0.33 – 53), ibrutinib led to an 87% reduction in the risk of progression or death. The 3-year PFS rate was 59% with ibrutinib and 3% with ofatumumab.
Ibrutinib conferred a benefit in PFS across all baseline patient characteristics.
Among ibrutinib-treated patients, the 3-year PFS was 53% for patients with deletion 17p, 66% for those with deletion 11q but not deletion 17p, and 58% for those with neither abnormality.
Dr Brown noted how closely complex karyotype associates with high-risk cytogenetics. Forty-two percent of patients with 17p deletion had a complex karyotype, as did 23% of patients with 11q deletion and 15% of patients with neither 17p nor 11q deletion.
For IGHV-mutation status, Dr Brown said there is no difference in PFS with this degree of follow-up.
In terms of TP53 mutation status, Dr Brown pointed out a trend toward a worse PFS in those patients with the mutation.
“We actually looked by individual p53 mutation versus 17p deletion, versus both, versus neither, in the 2-year follow-up paper and found that p53 with 17p, both abnormalities, did have worse PFS than neither,” she said.
“This may require further follow-up because we do know that most 17p patients also have a p53 mutation, particularly in the relapsed setting.”
As expected, Dr Brown said, those patients with more than 2 prior therapies had a worse PFS compared to patients with 2 or fewer prior therapies.
Multivariate analysis demonstrated that more than 2 prior lines of therapy or an elevated ß2 microglobulin were associated with decreased PFS with ibrutinib.
When the investigators adjusted the overall survival data for cross-over, ibrutinib was projected to continue the overall survival benefit compared with ofatumumab, with a hazard ratio of 0.37.
Response rates
Dr Brown noted that, early on, there’s quite a significant rate of partial response with lymphocytosis observed in patients on ibrutinib.
This “diminishes dramatically,” she said, but about 5% of patients at 3 and 4 years still have ongoing lymphocytosis.
“Similarly, initially, there’s a very low rate of complete remission, which has risen steadily to 9% at this follow-up,” she said.
And the overall response rate is 91%.
Treatment exposure and toxicity
The median duration of ibrutinib treatment is 41 months, and 46% of patients continue on treatment. Twenty-seven percent of patients discontinued due to progression, and 12% because of adverse events (AEs).
Of the 53 patients who discontinued therapy, 14 had transformation as their primary reason, 9 with diffuse large B-cell lymphoma, 3 with Hodgkin disease, and 2 with prolymphocytic lymphoma.
The most frequent AEs leading to discontinuation included pneumonia (n=3), anemia (n=2), thrombocytopenia (n=2), diarrhea (n=2), and anal incontinence (n=2).
AEs leading to discontinuation decreased over time—6% in year 0 to 1 and 4% in years 2 to 3.
“The most frequent cumulative AEs are similar to what we’ve seen in most prior studies,” Dr Brown said, including diarrhea, fatigue, and cough.
In terms of grade 3 or higher AEs, about a quarter of patients had neutropenia, 17% had pneumonia, and 8% had hypertension.
Six percent of patients had major hemorrhage, and all-grade atrial fibrillation occurred in 11% of patients.
“Now, many of the grade 3 and higher AEs did decline over time during the study,” Dr Brown noted. “You can see this is quite evident for neutropenia as well as pneumonia, and all infections declined from year 1 to subsequent years.”
Hypertension, in contrast, has been fairly steady over the later years, she said, and atrial fibrillation is highest in the first 6 months but then continues at a low rate thereafter.
The investigators believe these long-term results demonstrate that ibrutinib is tolerable and continues to show sustained efficacy in previously treated and high-genomic-risk patients with CLL. In addition, no long-term safety signals have emerged.
This study was sponsored by Pharmacyclics, LLC, an AbbVie company. ![]()
Cancer drug costs increasing despite competition

Cancer drug costs in the US increase substantially after launch, regardless of competition, according to a study published in the Journal of Clinical Oncology.*
Researchers studied 24 cancer drugs approved over the last 20 years and found a mean cumulative cost increase of about 37%, or 19% when adjusted for inflation.
Among drugs approved to treat hematologic malignancies, the greatest inflation-adjusted price increases were for arsenic trioxide (57%), nelarabine (55%), and rituximab (49%).
The lowest inflation-adjusted price increases were for ofatumumab (8%), clofarabine (8%), and liposomal vincristine (18%).
For this study, Daniel A. Goldstein, MD, of Emory University in Atlanta, Georgia, and his colleagues measured the monthly price trajectories of 24 cancer drugs approved by the US Food and Drug Administration. This included 10 drugs approved to treat hematologic malignancies between 1997 and 2011.
To account for discounts and rebates, the researchers used the average sales prices published by the Centers for Medicare and Medicaid Services and adjusted to general and health-related inflation rates. For each drug, the researchers calculated the cumulative and annual drug cost changes.
Results
The mean follow-up was 8 years. The mean cumulative cost increase for all 24 drugs was +36.5% (95% CI, 24.7% to 48.3%).
The general inflation-adjusted increase was +19.1% (95% CI, 11.0% to 27.2%), and the health-related inflation-adjusted increase was +8.4% (95% CI, 1.4% to 15.4%).
Only 1 of the 24 drugs studied had a price decrease over time. That drug is ziv-aflibercept, which was approved to treat metastatic colorectal cancer in 2012.
Ziv-aflibercept was launched with an annual price exceeding $110,000. After public outcry, the drug’s manufacturer, Sanofi, cut the price in half. By the end of the study’s follow-up period in 2017, the cost of ziv-aflibercept had decreased 13% (inflation-adjusted decrease of 15%, health-related inflation-adjusted decrease of 20%).
Cost changes for the drugs approved to treat hematologic malignancies are listed in the following table.
| Drug (indication, approval date, years of follow-up) | Mean monthly cost at launch | Mean annual cost change (SD) | Cumulative cost change | General and health-related inflation-adjusted change, respectively |
| Arsenic trioxide (APL, 2000, 12) | $11,455 | +6% (4) | +95% | +57%, +39% |
| Bendamustine (CLL, NHL, 2008, 8) | $6924 | +5% (5) | +50% | +32%, +21% |
| Bortezomib (MM, MCL, 2003, 12) | $5490 | +4% (3) | +63% | +31%, +16% |
| Brentuximab (lymphoma, 2011, 4) | $19,482 | +8% (0.1) | +35% | +29%, +22% |
| Clofarabine (ALL, 2004, 11) | $56,486 | +3% (3) | +31% | +8%, -4% |
| Liposomal vincristine (ALL, 2012, 3) | $34,602 | +8% (0.5) | +21% | +18%, +14% |
| Nelarabine (ALL, lymphoma, 2005, 10) | $18,513 | +6% (2) | +83% | +55%, +39% |
| Ofatumumab (CLL, 2009, 6) | $4538 | +3% (2) | +17% | +8%, -0.5% |
| Pralatrexate (lymphoma, 2009, 6) | $31,684 | +6% (4) | +43% | +31%, +21% |
| Rituximab (NHL, CLL, 1997, 12) | $4111 | +5% (0.5) | +85% | +49%, +32% |
Abbreviations: ALL, acute lymphoblastic leukemia; APL, acute promyelocytic leukemia; CLL, chronic lymphocytic leukemia; MCL, mantle cell lymphoma; MM, multiple myeloma; NHL, non-Hodgkin lymphoma; SD, standard deviation.
The researchers noted that there was a steady increase in drug costs over the study period, regardless of whether a drug was granted a new supplemental indication, the drug had a new off-label indication, or a competitor drug was approved.
The only variable that was significantly associated with price change was the amount of time that had elapsed from a drug’s launch.
This association was significant in models in which the researchers used prices adjusted to inflation (P=0.002) and health-related inflation (P=0.023). However, it was not significant when the researchers used the actual drug price (P=0.085). ![]()
*Data in the abstract differ from data in the body of the JCO paper. This article includes data from the body of the JCO paper.
Cancer drug costs in the US increase substantially after launch, regardless of competition, according to a study published in the Journal of Clinical Oncology.*
Researchers studied 24 cancer drugs approved over the last 20 years and found a mean cumulative cost increase of about 37%, or 19% when adjusted for inflation.
Among drugs approved to treat hematologic malignancies, the greatest inflation-adjusted price increases were for arsenic trioxide (57%), nelarabine (55%), and rituximab (49%).
The lowest inflation-adjusted price increases were for ofatumumab (8%), clofarabine (8%), and liposomal vincristine (18%).
For this study, Daniel A. Goldstein, MD, of Emory University in Atlanta, Georgia, and his colleagues measured the monthly price trajectories of 24 cancer drugs approved by the US Food and Drug Administration. This included 10 drugs approved to treat hematologic malignancies between 1997 and 2011.
To account for discounts and rebates, the researchers used the average sales prices published by the Centers for Medicare and Medicaid Services and adjusted to general and health-related inflation rates. For each drug, the researchers calculated the cumulative and annual drug cost changes.
Results
The mean follow-up was 8 years. The mean cumulative cost increase for all 24 drugs was +36.5% (95% CI, 24.7% to 48.3%).
The general inflation-adjusted increase was +19.1% (95% CI, 11.0% to 27.2%), and the health-related inflation-adjusted increase was +8.4% (95% CI, 1.4% to 15.4%).
Only 1 of the 24 drugs studied had a price decrease over time. That drug is ziv-aflibercept, which was approved to treat metastatic colorectal cancer in 2012.
Ziv-aflibercept was launched with an annual price exceeding $110,000. After public outcry, the drug’s manufacturer, Sanofi, cut the price in half. By the end of the study’s follow-up period in 2017, the cost of ziv-aflibercept had decreased 13% (inflation-adjusted decrease of 15%, health-related inflation-adjusted decrease of 20%).
Cost changes for the drugs approved to treat hematologic malignancies are listed in the following table.
| Drug (indication, approval date, years of follow-up) | Mean monthly cost at launch | Mean annual cost change (SD) | Cumulative cost change | General and health-related inflation-adjusted change, respectively |
| Arsenic trioxide (APL, 2000, 12) | $11,455 | +6% (4) | +95% | +57%, +39% |
| Bendamustine (CLL, NHL, 2008, 8) | $6924 | +5% (5) | +50% | +32%, +21% |
| Bortezomib (MM, MCL, 2003, 12) | $5490 | +4% (3) | +63% | +31%, +16% |
| Brentuximab (lymphoma, 2011, 4) | $19,482 | +8% (0.1) | +35% | +29%, +22% |
| Clofarabine (ALL, 2004, 11) | $56,486 | +3% (3) | +31% | +8%, -4% |
| Liposomal vincristine (ALL, 2012, 3) | $34,602 | +8% (0.5) | +21% | +18%, +14% |
| Nelarabine (ALL, lymphoma, 2005, 10) | $18,513 | +6% (2) | +83% | +55%, +39% |
| Ofatumumab (CLL, 2009, 6) | $4538 | +3% (2) | +17% | +8%, -0.5% |
| Pralatrexate (lymphoma, 2009, 6) | $31,684 | +6% (4) | +43% | +31%, +21% |
| Rituximab (NHL, CLL, 1997, 12) | $4111 | +5% (0.5) | +85% | +49%, +32% |
Abbreviations: ALL, acute lymphoblastic leukemia; APL, acute promyelocytic leukemia; CLL, chronic lymphocytic leukemia; MCL, mantle cell lymphoma; MM, multiple myeloma; NHL, non-Hodgkin lymphoma; SD, standard deviation.
The researchers noted that there was a steady increase in drug costs over the study period, regardless of whether a drug was granted a new supplemental indication, the drug had a new off-label indication, or a competitor drug was approved.
The only variable that was significantly associated with price change was the amount of time that had elapsed from a drug’s launch.
This association was significant in models in which the researchers used prices adjusted to inflation (P=0.002) and health-related inflation (P=0.023). However, it was not significant when the researchers used the actual drug price (P=0.085). ![]()
*Data in the abstract differ from data in the body of the JCO paper. This article includes data from the body of the JCO paper.
Cancer drug costs in the US increase substantially after launch, regardless of competition, according to a study published in the Journal of Clinical Oncology.*
Researchers studied 24 cancer drugs approved over the last 20 years and found a mean cumulative cost increase of about 37%, or 19% when adjusted for inflation.
Among drugs approved to treat hematologic malignancies, the greatest inflation-adjusted price increases were for arsenic trioxide (57%), nelarabine (55%), and rituximab (49%).
The lowest inflation-adjusted price increases were for ofatumumab (8%), clofarabine (8%), and liposomal vincristine (18%).
For this study, Daniel A. Goldstein, MD, of Emory University in Atlanta, Georgia, and his colleagues measured the monthly price trajectories of 24 cancer drugs approved by the US Food and Drug Administration. This included 10 drugs approved to treat hematologic malignancies between 1997 and 2011.
To account for discounts and rebates, the researchers used the average sales prices published by the Centers for Medicare and Medicaid Services and adjusted to general and health-related inflation rates. For each drug, the researchers calculated the cumulative and annual drug cost changes.
Results
The mean follow-up was 8 years. The mean cumulative cost increase for all 24 drugs was +36.5% (95% CI, 24.7% to 48.3%).
The general inflation-adjusted increase was +19.1% (95% CI, 11.0% to 27.2%), and the health-related inflation-adjusted increase was +8.4% (95% CI, 1.4% to 15.4%).
Only 1 of the 24 drugs studied had a price decrease over time. That drug is ziv-aflibercept, which was approved to treat metastatic colorectal cancer in 2012.
Ziv-aflibercept was launched with an annual price exceeding $110,000. After public outcry, the drug’s manufacturer, Sanofi, cut the price in half. By the end of the study’s follow-up period in 2017, the cost of ziv-aflibercept had decreased 13% (inflation-adjusted decrease of 15%, health-related inflation-adjusted decrease of 20%).
Cost changes for the drugs approved to treat hematologic malignancies are listed in the following table.
| Drug (indication, approval date, years of follow-up) | Mean monthly cost at launch | Mean annual cost change (SD) | Cumulative cost change | General and health-related inflation-adjusted change, respectively |
| Arsenic trioxide (APL, 2000, 12) | $11,455 | +6% (4) | +95% | +57%, +39% |
| Bendamustine (CLL, NHL, 2008, 8) | $6924 | +5% (5) | +50% | +32%, +21% |
| Bortezomib (MM, MCL, 2003, 12) | $5490 | +4% (3) | +63% | +31%, +16% |
| Brentuximab (lymphoma, 2011, 4) | $19,482 | +8% (0.1) | +35% | +29%, +22% |
| Clofarabine (ALL, 2004, 11) | $56,486 | +3% (3) | +31% | +8%, -4% |
| Liposomal vincristine (ALL, 2012, 3) | $34,602 | +8% (0.5) | +21% | +18%, +14% |
| Nelarabine (ALL, lymphoma, 2005, 10) | $18,513 | +6% (2) | +83% | +55%, +39% |
| Ofatumumab (CLL, 2009, 6) | $4538 | +3% (2) | +17% | +8%, -0.5% |
| Pralatrexate (lymphoma, 2009, 6) | $31,684 | +6% (4) | +43% | +31%, +21% |
| Rituximab (NHL, CLL, 1997, 12) | $4111 | +5% (0.5) | +85% | +49%, +32% |
Abbreviations: ALL, acute lymphoblastic leukemia; APL, acute promyelocytic leukemia; CLL, chronic lymphocytic leukemia; MCL, mantle cell lymphoma; MM, multiple myeloma; NHL, non-Hodgkin lymphoma; SD, standard deviation.
The researchers noted that there was a steady increase in drug costs over the study period, regardless of whether a drug was granted a new supplemental indication, the drug had a new off-label indication, or a competitor drug was approved.
The only variable that was significantly associated with price change was the amount of time that had elapsed from a drug’s launch.
This association was significant in models in which the researchers used prices adjusted to inflation (P=0.002) and health-related inflation (P=0.023). However, it was not significant when the researchers used the actual drug price (P=0.085).
*Data in the abstract differ from data in the body of the JCO paper. This article includes data from the body of the JCO paper.
FDA approves drug to treat rel/ref MCL
The US Food and Drug Administration (FDA) has granted accelerated approval to the BTK inhibitor acalabrutinib (Calquence, formerly ACP-196).
The drug is now approved to treat adults with mantle cell lymphoma (MCL) who have received at least 1 prior therapy.
The FDA’s accelerated approval pathway is used for drugs intended to treat serious conditions where there is unmet medical need and when said drugs have demonstrated effects that suggest they will provide a clinical benefit to patients.
This means further study is required to verify and describe the anticipated clinical benefits of acalabrutinib, which was approved based on the overall response rate observed in a phase 2 trial.
The company developing acalabrutinib, AstraZeneca Pharmaceuticals LP, is currently conducting the necessary additional research.
The FDA previously granted AstraZeneca priority review, breakthrough therapy, and orphan drug designations for acalabrutinib as a treatment for MCL.
Phase 2 trial
The FDA approved acalabrutinib based on results of the phase 2 ACE-LY-004 trial. This single-arm trial enrolled 124 adults with relapsed or refractory MCL.
According to AstraZeneca, acalabrutinib produced an overall response rate of 80%, with 40% of patients achieving a complete response and 40% experiencing a partial response.
The most common adverse events (AEs) of any grade (occurring in at least 20% of patients) were anemia (46%), thrombocytopenia (44%), headache (39%), neutropenia (36%), diarrhea (31%), fatigue (28%), myalgia (21%), and bruising (21%).
Dosage reductions due to AEs occurred in 1.6% of patients. Discontinuations due to AEs occurred in 6.5% of patients. Increases in creatinine 1.5 to 3 times the upper limit of normal occurred in 4.8% of patients.
According to AstraZeneca, full results from ACE-LY-004 have been submitted for presentation at an upcoming medical meeting.
This will be the first MCL trial data to be presented from the acalabrutinib development program, which includes both monotherapy and combination therapies in hematologic and solid tumor malignancies.
The US Food and Drug Administration (FDA) has granted accelerated approval to the BTK inhibitor acalabrutinib (Calquence, formerly ACP-196).
The drug is now approved to treat adults with mantle cell lymphoma (MCL) who have received at least 1 prior therapy.
The FDA’s accelerated approval pathway is used for drugs intended to treat serious conditions where there is unmet medical need and when said drugs have demonstrated effects that suggest they will provide a clinical benefit to patients.
This means further study is required to verify and describe the anticipated clinical benefits of acalabrutinib, which was approved based on the overall response rate observed in a phase 2 trial.
The company developing acalabrutinib, AstraZeneca Pharmaceuticals LP, is currently conducting the necessary additional research.
The FDA previously granted AstraZeneca priority review, breakthrough therapy, and orphan drug designations for acalabrutinib as a treatment for MCL.
Phase 2 trial
The FDA approved acalabrutinib based on results of the phase 2 ACE-LY-004 trial. This single-arm trial enrolled 124 adults with relapsed or refractory MCL.
According to AstraZeneca, acalabrutinib produced an overall response rate of 80%, with 40% of patients achieving a complete response and 40% experiencing a partial response.
The most common adverse events (AEs) of any grade (occurring in at least 20% of patients) were anemia (46%), thrombocytopenia (44%), headache (39%), neutropenia (36%), diarrhea (31%), fatigue (28%), myalgia (21%), and bruising (21%).
Dosage reductions due to AEs occurred in 1.6% of patients. Discontinuations due to AEs occurred in 6.5% of patients. Increases in creatinine 1.5 to 3 times the upper limit of normal occurred in 4.8% of patients.
According to AstraZeneca, full results from ACE-LY-004 have been submitted for presentation at an upcoming medical meeting.
This will be the first MCL trial data to be presented from the acalabrutinib development program, which includes both monotherapy and combination therapies in hematologic and solid tumor malignancies.
The US Food and Drug Administration (FDA) has granted accelerated approval to the BTK inhibitor acalabrutinib (Calquence, formerly ACP-196).
The drug is now approved to treat adults with mantle cell lymphoma (MCL) who have received at least 1 prior therapy.
The FDA’s accelerated approval pathway is used for drugs intended to treat serious conditions where there is unmet medical need and when said drugs have demonstrated effects that suggest they will provide a clinical benefit to patients.
This means further study is required to verify and describe the anticipated clinical benefits of acalabrutinib, which was approved based on the overall response rate observed in a phase 2 trial.
The company developing acalabrutinib, AstraZeneca Pharmaceuticals LP, is currently conducting the necessary additional research.
The FDA previously granted AstraZeneca priority review, breakthrough therapy, and orphan drug designations for acalabrutinib as a treatment for MCL.
Phase 2 trial
The FDA approved acalabrutinib based on results of the phase 2 ACE-LY-004 trial. This single-arm trial enrolled 124 adults with relapsed or refractory MCL.
According to AstraZeneca, acalabrutinib produced an overall response rate of 80%, with 40% of patients achieving a complete response and 40% experiencing a partial response.
The most common adverse events (AEs) of any grade (occurring in at least 20% of patients) were anemia (46%), thrombocytopenia (44%), headache (39%), neutropenia (36%), diarrhea (31%), fatigue (28%), myalgia (21%), and bruising (21%).
Dosage reductions due to AEs occurred in 1.6% of patients. Discontinuations due to AEs occurred in 6.5% of patients. Increases in creatinine 1.5 to 3 times the upper limit of normal occurred in 4.8% of patients.
According to AstraZeneca, full results from ACE-LY-004 have been submitted for presentation at an upcoming medical meeting.
This will be the first MCL trial data to be presented from the acalabrutinib development program, which includes both monotherapy and combination therapies in hematologic and solid tumor malignancies.
CCSs more likely to stay at jobs to keep health insurance

Survey results suggest childhood cancer survivors (CCSs) in the US are more likely than individuals without a history of cancer to experience “job lock,” or staying at a job to keep work-related health insurance.
CCSs are also more likely than individuals without a history of cancer to report problems paying medical bills and being denied health insurance.
Anne Kirchhoff, PhD, of Huntsman Cancer Institute at the University of Utah in Salt Lake City, and her colleagues reported these findings in JAMA Oncology.
The researchers analyzed 394 CCSs from pediatric oncology institutions across the US, along with 128 of their siblings who had no history of cancer. All study participants worked 35 hours or more per week.
The most common cancer diagnosis among CCSs was leukemia (35.4%), followed by Hodgkin lymphoma (14.9%). Most patients had undergone chemotherapy (77.2%), radiotherapy (63.9%), and surgery (81.1%).
Overall, sociodemographic and clinical characteristics were similar between CCSs and siblings. However, CCSs were more likely than siblings to have severe, disabling, or life-threatening chronic conditions—33.9% and 17.7%, respectively (P<0.001).
Most CCSs (88.0%) and siblings (88.5%) had employer-sponsored health insurance. Three percent of siblings and 5.3% of CCSs had individual insurance; 1.9% and 2.3%, respectively, had public insurance; and 6.7% and 4.4%, respectively, were uninsured.
Results
CCSs were more likely than siblings to report:
- Job lock—23.2% and 16.9%, respectively (P=0.16)
- Problems paying medical bills—20.1% and 12.9%, respectively (P=0.09)
- Denial of health insurance—13.4% and 1.8%, respectively (P<0.001).
In a multivariable analysis, insurance denial remained significantly more common among CCSs than siblings (relative risk [RR]=7.38).
In another multivariable analysis, 38% of CCSs with a previous insurance denial reported job lock, compared with 20% of those who never experienced insurance denial (RR=1.60). And 44% of CCSs who reported problems paying their medical bills also reported job lock, compared to 16% of those who had no problems paying medical bills (RR=2.43).
The researchers also found that female CCSs (RR=1.70) and CCSs with severe, disabling, or life-threatening chronic conditions (RR=1.72) were more likely to report job lock.
“This information gives us a feel for high-risk groups of survivors who may need more information about insurance,” Dr Kirchhoff said. “Many people experience a gap in education and literacy around insurance, and it’s important for people to understand their options—even those who are employed and consistently had access to insurance through work. We want to know what their concerns are so we can help patients and survivors. Getting healthcare should not be a worry for cancer survivors.”
“Survivors have been through a lot when they were younger and understand the importance of making sure they can get healthcare when they need it. I think a lot of them also saw what their parents and families went through in terms of the financial stress and burden of dealing with a health crisis. So they’re just primed to understand the importance of health insurance.”
Dr Kirchhoff noted that this study was conducted as the Affordable Care Act was rolling out. Therefore, she would like to do a follow-up study to see if the insurance exchanges and Medicaid expansion lessened job-related insurance worries.
Survey results suggest childhood cancer survivors (CCSs) in the US are more likely than individuals without a history of cancer to experience “job lock,” or staying at a job to keep work-related health insurance.
CCSs are also more likely than individuals without a history of cancer to report problems paying medical bills and being denied health insurance.
Anne Kirchhoff, PhD, of Huntsman Cancer Institute at the University of Utah in Salt Lake City, and her colleagues reported these findings in JAMA Oncology.
The researchers analyzed 394 CCSs from pediatric oncology institutions across the US, along with 128 of their siblings who had no history of cancer. All study participants worked 35 hours or more per week.
The most common cancer diagnosis among CCSs was leukemia (35.4%), followed by Hodgkin lymphoma (14.9%). Most patients had undergone chemotherapy (77.2%), radiotherapy (63.9%), and surgery (81.1%).
Overall, sociodemographic and clinical characteristics were similar between CCSs and siblings. However, CCSs were more likely than siblings to have severe, disabling, or life-threatening chronic conditions—33.9% and 17.7%, respectively (P<0.001).
Most CCSs (88.0%) and siblings (88.5%) had employer-sponsored health insurance. Three percent of siblings and 5.3% of CCSs had individual insurance; 1.9% and 2.3%, respectively, had public insurance; and 6.7% and 4.4%, respectively, were uninsured.
Results
CCSs were more likely than siblings to report:
- Job lock—23.2% and 16.9%, respectively (P=0.16)
- Problems paying medical bills—20.1% and 12.9%, respectively (P=0.09)
- Denial of health insurance—13.4% and 1.8%, respectively (P<0.001).
In a multivariable analysis, insurance denial remained significantly more common among CCSs than siblings (relative risk [RR]=7.38).
In another multivariable analysis, 38% of CCSs with a previous insurance denial reported job lock, compared with 20% of those who never experienced insurance denial (RR=1.60). And 44% of CCSs who reported problems paying their medical bills also reported job lock, compared to 16% of those who had no problems paying medical bills (RR=2.43).
The researchers also found that female CCSs (RR=1.70) and CCSs with severe, disabling, or life-threatening chronic conditions (RR=1.72) were more likely to report job lock.
“This information gives us a feel for high-risk groups of survivors who may need more information about insurance,” Dr Kirchhoff said. “Many people experience a gap in education and literacy around insurance, and it’s important for people to understand their options—even those who are employed and consistently had access to insurance through work. We want to know what their concerns are so we can help patients and survivors. Getting healthcare should not be a worry for cancer survivors.”
“Survivors have been through a lot when they were younger and understand the importance of making sure they can get healthcare when they need it. I think a lot of them also saw what their parents and families went through in terms of the financial stress and burden of dealing with a health crisis. So they’re just primed to understand the importance of health insurance.”
Dr Kirchhoff noted that this study was conducted as the Affordable Care Act was rolling out. Therefore, she would like to do a follow-up study to see if the insurance exchanges and Medicaid expansion lessened job-related insurance worries.
Survey results suggest childhood cancer survivors (CCSs) in the US are more likely than individuals without a history of cancer to experience “job lock,” or staying at a job to keep work-related health insurance.
CCSs are also more likely than individuals without a history of cancer to report problems paying medical bills and being denied health insurance.
Anne Kirchhoff, PhD, of Huntsman Cancer Institute at the University of Utah in Salt Lake City, and her colleagues reported these findings in JAMA Oncology.
The researchers analyzed 394 CCSs from pediatric oncology institutions across the US, along with 128 of their siblings who had no history of cancer. All study participants worked 35 hours or more per week.
The most common cancer diagnosis among CCSs was leukemia (35.4%), followed by Hodgkin lymphoma (14.9%). Most patients had undergone chemotherapy (77.2%), radiotherapy (63.9%), and surgery (81.1%).
Overall, sociodemographic and clinical characteristics were similar between CCSs and siblings. However, CCSs were more likely than siblings to have severe, disabling, or life-threatening chronic conditions—33.9% and 17.7%, respectively (P<0.001).
Most CCSs (88.0%) and siblings (88.5%) had employer-sponsored health insurance. Three percent of siblings and 5.3% of CCSs had individual insurance; 1.9% and 2.3%, respectively, had public insurance; and 6.7% and 4.4%, respectively, were uninsured.
Results
CCSs were more likely than siblings to report:
- Job lock—23.2% and 16.9%, respectively (P=0.16)
- Problems paying medical bills—20.1% and 12.9%, respectively (P=0.09)
- Denial of health insurance—13.4% and 1.8%, respectively (P<0.001).
In a multivariable analysis, insurance denial remained significantly more common among CCSs than siblings (relative risk [RR]=7.38).
In another multivariable analysis, 38% of CCSs with a previous insurance denial reported job lock, compared with 20% of those who never experienced insurance denial (RR=1.60). And 44% of CCSs who reported problems paying their medical bills also reported job lock, compared to 16% of those who had no problems paying medical bills (RR=2.43).
The researchers also found that female CCSs (RR=1.70) and CCSs with severe, disabling, or life-threatening chronic conditions (RR=1.72) were more likely to report job lock.
“This information gives us a feel for high-risk groups of survivors who may need more information about insurance,” Dr Kirchhoff said. “Many people experience a gap in education and literacy around insurance, and it’s important for people to understand their options—even those who are employed and consistently had access to insurance through work. We want to know what their concerns are so we can help patients and survivors. Getting healthcare should not be a worry for cancer survivors.”
“Survivors have been through a lot when they were younger and understand the importance of making sure they can get healthcare when they need it. I think a lot of them also saw what their parents and families went through in terms of the financial stress and burden of dealing with a health crisis. So they’re just primed to understand the importance of health insurance.”
Dr Kirchhoff noted that this study was conducted as the Affordable Care Act was rolling out. Therefore, she would like to do a follow-up study to see if the insurance exchanges and Medicaid expansion lessened job-related insurance worries.
Drug receives breakthrough designation for DLBCL

The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to MOR208, an Fc-enhanced monoclonal antibody directed against CD19.
The designation is for MOR208 to be used in combination with lenalidomide to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who are not eligible for high-dose chemotherapy and autologous stem cell transplant.
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
The breakthrough designation for MOR208 is based on preliminary data from the ongoing phase 2 L-MIND study (NCT02399085).
In this trial, researchers are evaluating MOR208 in combination with lenalidomide in patients with relapsed/refractory DLBCL who are ineligible for high-dose chemotherapy and autologous stem cell transplant.
Preliminary data from this trial were presented at ASCO 2017.* Of the 44 patients enrolled at the data cut-off, 34 were evaluable for efficacy.
The objective response rate was 56% (19/34), and the complete response rate was 32% (11/34). Sixteen of the 19 responders were still on study at the data cut-off point.
The most frequent adverse events of grade 3 or higher were neutropenia (32%), thrombocytopenia (9%), and leukopenia (9%). As of the data cut-off, 27% of patients required a reduction of the lenalidomide dose due to side effects.
“We expect to report further data from our ongoing phase 2 L-MIND trial with MOR208 plus lenalidomide at this year’s American Society of Hematology conference in December,” said Malte Peters, chief development officer of MorphoSys AG, the company developing MOR208.
“In addition, we are currently evaluating MOR208 in combination with bendamustine in our phase 3 B-MIND trial.”
The B-MIND study is designed to compare MOR208 plus bendamustine to rituximab plus bendamustine in patients with relapsed/refractory DLBCL who are not eligible for high-dose chemotherapy and autologous stem cell transplant.
*Data in the abstract differ from the data presented at the meeting.
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to MOR208, an Fc-enhanced monoclonal antibody directed against CD19.
The designation is for MOR208 to be used in combination with lenalidomide to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who are not eligible for high-dose chemotherapy and autologous stem cell transplant.
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
The breakthrough designation for MOR208 is based on preliminary data from the ongoing phase 2 L-MIND study (NCT02399085).
In this trial, researchers are evaluating MOR208 in combination with lenalidomide in patients with relapsed/refractory DLBCL who are ineligible for high-dose chemotherapy and autologous stem cell transplant.
Preliminary data from this trial were presented at ASCO 2017.* Of the 44 patients enrolled at the data cut-off, 34 were evaluable for efficacy.
The objective response rate was 56% (19/34), and the complete response rate was 32% (11/34). Sixteen of the 19 responders were still on study at the data cut-off point.
The most frequent adverse events of grade 3 or higher were neutropenia (32%), thrombocytopenia (9%), and leukopenia (9%). As of the data cut-off, 27% of patients required a reduction of the lenalidomide dose due to side effects.
“We expect to report further data from our ongoing phase 2 L-MIND trial with MOR208 plus lenalidomide at this year’s American Society of Hematology conference in December,” said Malte Peters, chief development officer of MorphoSys AG, the company developing MOR208.
“In addition, we are currently evaluating MOR208 in combination with bendamustine in our phase 3 B-MIND trial.”
The B-MIND study is designed to compare MOR208 plus bendamustine to rituximab plus bendamustine in patients with relapsed/refractory DLBCL who are not eligible for high-dose chemotherapy and autologous stem cell transplant.
*Data in the abstract differ from the data presented at the meeting.
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to MOR208, an Fc-enhanced monoclonal antibody directed against CD19.
The designation is for MOR208 to be used in combination with lenalidomide to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who are not eligible for high-dose chemotherapy and autologous stem cell transplant.
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
The breakthrough designation for MOR208 is based on preliminary data from the ongoing phase 2 L-MIND study (NCT02399085).
In this trial, researchers are evaluating MOR208 in combination with lenalidomide in patients with relapsed/refractory DLBCL who are ineligible for high-dose chemotherapy and autologous stem cell transplant.
Preliminary data from this trial were presented at ASCO 2017.* Of the 44 patients enrolled at the data cut-off, 34 were evaluable for efficacy.
The objective response rate was 56% (19/34), and the complete response rate was 32% (11/34). Sixteen of the 19 responders were still on study at the data cut-off point.
The most frequent adverse events of grade 3 or higher were neutropenia (32%), thrombocytopenia (9%), and leukopenia (9%). As of the data cut-off, 27% of patients required a reduction of the lenalidomide dose due to side effects.
“We expect to report further data from our ongoing phase 2 L-MIND trial with MOR208 plus lenalidomide at this year’s American Society of Hematology conference in December,” said Malte Peters, chief development officer of MorphoSys AG, the company developing MOR208.
“In addition, we are currently evaluating MOR208 in combination with bendamustine in our phase 3 B-MIND trial.”
The B-MIND study is designed to compare MOR208 plus bendamustine to rituximab plus bendamustine in patients with relapsed/refractory DLBCL who are not eligible for high-dose chemotherapy and autologous stem cell transplant.
*Data in the abstract differ from the data presented at the meeting.