User login
Treatment of Elephantiasic Pretibial Myxedema With Rituximab Therapy
To the Editor:
Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet. Pretibial myxedema occurs in approximately 0.5% to 4.3% of patients with hyperthyroidism.1 Thyroid dermopathy often is thought of as the classic nonpitting PTM with skin induration and color change. However, rarer forms of PTM, including plaque, nodular, and elephantiasic, also are important to note.2
Elephantiasic PTM is extremely rare, occurring in less than 1% of patients with PTM.2 Elephantiasic PTM is characterized by the persistent swelling of 1 or both legs; thickening of the skin overlying the dorsum of the feet, ankles, and toes; and verrucous irregular plaques that often are fleshy and flattened. The clinical differential diagnosis of elephantiasic PTM includes elephantiasis nostra verrucosa, a late-stage complication of chronic lymphedema that can be related to a variety of infectious or noninfectious obstructive processes. Few effective therapeutic modalities exist in the treatment of elephantiasic PTM. We present a case of elephantiasic PTM.
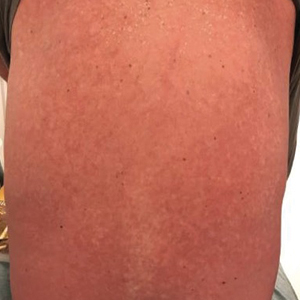
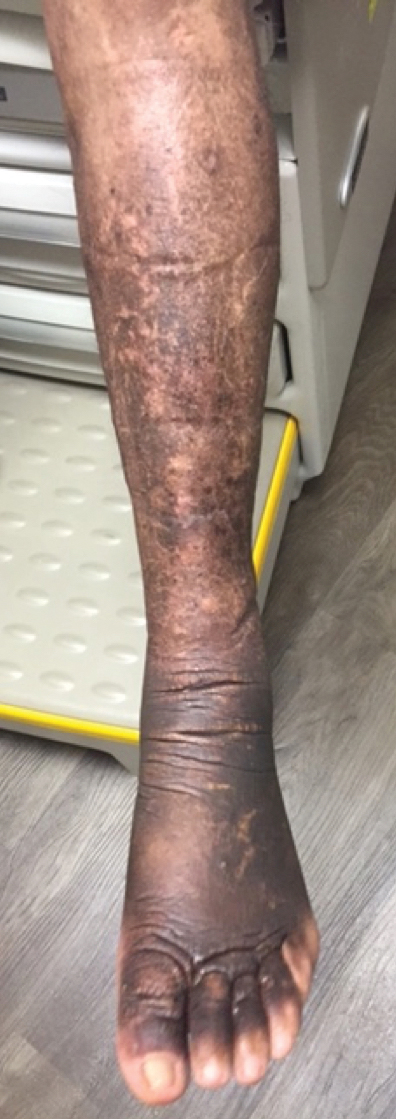
A 59-year-old man presented to dermatology with leonine facies with pronounced glabellar creases and indentations of the earlobes. He had diffuse woody induration, hyperpigmentation, and nonpitting edema of the lower extremities as well as several flesh-colored exophytic nodules scattered throughout the anterior shins and dorsal feet (Figure 1). On the left posterior calf, there was a large, 3-cm, exophytic, firm, flesh-colored nodule. Examination of the hands revealed mild hyperpigmentation of the distal digits, clubbing of the distal phalanges, and cheiroarthropathy.

The patient was diagnosed with Graves disease after experiencing the classic symptoms of hyperthyroidism, including heat intolerance, tremor, palpitations, and anxiety. He received thyroid ablation and subsequently was supplemented with levothyroxine 75 mg daily. Twelve years later, he was diagnosed with Graves ophthalmopathy with ocular proptosis requiring multiple courses of retro-orbital irradiation and surgical procedures for decompression. Approximately 1 year later, he noted increased swelling, firmness, and darkening of the pretibial surfaces. Initially, he was referred to vascular surgery and underwent bilateral saphenous vein ablation. He also was referred to a lymphedema specialist, and workup revealed an unremarkable lymphatic system. Minimal improvement was noted following the saphenous vein ablation, and he subsequently was referred to dermatology for further workup.
At the current presentation, laboratory analysis revealed a low thyrotropin level (0.03 mIU/L [reference range, 0.4–4.2 mIU/L]), and free thyroxine was within reference range. Radiography of the chest was unremarkable; however, radiography of the hand demonstrated arthrosis of the left fifth proximal interphalangeal joint. Nuclear medicine lymphoscintigraphy and lower extremity ultrasonography were unremarkable. Punch biopsies were performed of the left lateral leg and posterior calf. Hematoxylin and eosin staining demonstrated marked mucin deposition extending to the deep dermis along with deep fibroplasia and was read as consistent with PTM. Colloidal iron highlighted prominent mucin within the dermis (Figure 2).
. B, Colloidal iron staining highlighted the prominent mucin within the dermis")
The patient’s medical history, physical examination, laboratory analysis, imaging, and biopsies were considered, and a diagnosis of elephantiasic PTM was made. Minimal improvement was noted with initial therapeutic interventions including compression therapy and application of super high–potency topical corticosteroids. After further evaluation in our multidisciplinary rheumatology-dermatology clinic, the decision was made to initiate rituximab infusions.
Two months after 1 course of rituximab consisting of two 1000-mg infusions separated by 2 weeks, the patient showed substantial clinical improvement. There was striking improvement of the pretibial surfaces with resolution of the exophytic nodules and improvement of the induration (Figure 3). In addition, there was decreased induration of the glabella and earlobes and decreased fullness of the digital pulp on the hands. The patient also reported subjective improvements in mobility.
Our patient demonstrated all 3 aspects of the Diamond triad: PTM, exophthalmos, and acropachy. Patients present with all 3 features in less than 1% of reported cases of Graves disease.3 Although all 3 features are seen together infrequently, thyroid dermopathy and acropachy often are markers of severe Graves ophthalmopathy. In a study of 114 patients with Graves ophthalmopathy, patients who also had dermopathy and acropachy were more likely to have optic neuropathy or require orbital decompression.4
After overcoming the diagnostic dilemma that the elephantiasic presentation of PTM can present, therapeutic management remains a challenge. Heyes et al5 documented the successful treatment of highly recalcitrant elephantiasic PTM with rituximab and plasmapheresis therapy. In this case, a 44-year-old woman with an 11-year history of Graves disease and elephantiasic PTM received 29 rituximab infusions and 241 plasmapheresis treatments over the course of 3.5 years. Her elephantiasic PTM clinically resolved, and she was able to resume daily activities and wear normal shoes after being nonambulatory for years.5
Rituximab is a monoclonal antibody against CD20, a protein found primarily on the surface of B-cell lymphocytes. Although rituximab initially was approved by the US Food and Drug administration for the treatment of malignant lymphoma, it has had an increasing role in the treatment of autoimmune disorders such as rheumatoid arthritis. Rituximab is postulated to target B lymphocytes and halt their progression to plasma cells. By limiting the population of long-lasting, antibody-producing plasma cells and decreasing the autoantibodies that cause many of the symptoms in Graves disease, rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.6
Although the exact mechanism is poorly understood, PTM likely is a sequela of hyperthyroidism because of the expression of thyroid-stimulating hormone receptor proteins found on normal dermal fibroblasts. Thyroid-stimulating hormone receptor autoantibodies are thought to stimulate these fibroblasts to produce glycosaminoglycans. Histopathologically, accumulation of glycosaminoglycans deposited in the reticular dermis with high concentrations of hyaluronic acid is observed in PTM.7
Treatment of elephantiasic PTM remains a therapeutic challenge. Given the rarity of the disease process and limited information on effective therapeutic modalities, rituximab should be viewed as a viable treatment option in the management of recalcitrant elephantiasic PTM.
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446.
- Kakati S, Doley B, Pal S, et al. Elephantiasis nostras verrucosa: a rare thyroid dermopathy in Graves’ disease. J Assoc Physicians India. 2005;53:571-572.
- Anderson CK, Miller OF 3rd. Triad of exophthalmos, pretibial myxedema, and acropachy in a patient with Graves’ disease. J Am Acad Dermatol. 2003;48:970-972.
- Fatourechi V, Bartley GB, Eghbali-Fatourechi GZ, et al. Graves’ dermopathy and acropachy are markers of severe Graves’ ophthalmopathy. Thyroid. 2003;13:1141-1144.
- Heyes C, Nolan R, Leahy M, et al. Treatment‐resistant elephantiasic thyroid dermopathy responding to rituximab and plasmapheresis. Australas J Dermatol. 2012;53:E1-E4.
- Salvi M, Vannucchi G, Campi I, et al. Treatment of Graves’ disease and associated ophthalmopathy with the anti-CD20 monoclonal antibody rituximab: an open study. Eur J Endocrinol. 2007;156:33-40.
- Heufelder AE, Dutton CM, Sarkar G, et al. Detection of TSH receptor RNA in cultured fibroblasts from patients with Graves’ ophthalmopathy and pretibial dermopathy. Thyroid. 1993;3:297-300.
To the Editor:
Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet. Pretibial myxedema occurs in approximately 0.5% to 4.3% of patients with hyperthyroidism.1 Thyroid dermopathy often is thought of as the classic nonpitting PTM with skin induration and color change. However, rarer forms of PTM, including plaque, nodular, and elephantiasic, also are important to note.2
Elephantiasic PTM is extremely rare, occurring in less than 1% of patients with PTM.2 Elephantiasic PTM is characterized by the persistent swelling of 1 or both legs; thickening of the skin overlying the dorsum of the feet, ankles, and toes; and verrucous irregular plaques that often are fleshy and flattened. The clinical differential diagnosis of elephantiasic PTM includes elephantiasis nostra verrucosa, a late-stage complication of chronic lymphedema that can be related to a variety of infectious or noninfectious obstructive processes. Few effective therapeutic modalities exist in the treatment of elephantiasic PTM. We present a case of elephantiasic PTM.
A 59-year-old man presented to dermatology with leonine facies with pronounced glabellar creases and indentations of the earlobes. He had diffuse woody induration, hyperpigmentation, and nonpitting edema of the lower extremities as well as several flesh-colored exophytic nodules scattered throughout the anterior shins and dorsal feet (Figure 1). On the left posterior calf, there was a large, 3-cm, exophytic, firm, flesh-colored nodule. Examination of the hands revealed mild hyperpigmentation of the distal digits, clubbing of the distal phalanges, and cheiroarthropathy.
The patient was diagnosed with Graves disease after experiencing the classic symptoms of hyperthyroidism, including heat intolerance, tremor, palpitations, and anxiety. He received thyroid ablation and subsequently was supplemented with levothyroxine 75 mg daily. Twelve years later, he was diagnosed with Graves ophthalmopathy with ocular proptosis requiring multiple courses of retro-orbital irradiation and surgical procedures for decompression. Approximately 1 year later, he noted increased swelling, firmness, and darkening of the pretibial surfaces. Initially, he was referred to vascular surgery and underwent bilateral saphenous vein ablation. He also was referred to a lymphedema specialist, and workup revealed an unremarkable lymphatic system. Minimal improvement was noted following the saphenous vein ablation, and he subsequently was referred to dermatology for further workup.
At the current presentation, laboratory analysis revealed a low thyrotropin level (0.03 mIU/L [reference range, 0.4–4.2 mIU/L]), and free thyroxine was within reference range. Radiography of the chest was unremarkable; however, radiography of the hand demonstrated arthrosis of the left fifth proximal interphalangeal joint. Nuclear medicine lymphoscintigraphy and lower extremity ultrasonography were unremarkable. Punch biopsies were performed of the left lateral leg and posterior calf. Hematoxylin and eosin staining demonstrated marked mucin deposition extending to the deep dermis along with deep fibroplasia and was read as consistent with PTM. Colloidal iron highlighted prominent mucin within the dermis (Figure 2).
The patient’s medical history, physical examination, laboratory analysis, imaging, and biopsies were considered, and a diagnosis of elephantiasic PTM was made. Minimal improvement was noted with initial therapeutic interventions including compression therapy and application of super high–potency topical corticosteroids. After further evaluation in our multidisciplinary rheumatology-dermatology clinic, the decision was made to initiate rituximab infusions.
Two months after 1 course of rituximab consisting of two 1000-mg infusions separated by 2 weeks, the patient showed substantial clinical improvement. There was striking improvement of the pretibial surfaces with resolution of the exophytic nodules and improvement of the induration (Figure 3). In addition, there was decreased induration of the glabella and earlobes and decreased fullness of the digital pulp on the hands. The patient also reported subjective improvements in mobility.
Our patient demonstrated all 3 aspects of the Diamond triad: PTM, exophthalmos, and acropachy. Patients present with all 3 features in less than 1% of reported cases of Graves disease.3 Although all 3 features are seen together infrequently, thyroid dermopathy and acropachy often are markers of severe Graves ophthalmopathy. In a study of 114 patients with Graves ophthalmopathy, patients who also had dermopathy and acropachy were more likely to have optic neuropathy or require orbital decompression.4
After overcoming the diagnostic dilemma that the elephantiasic presentation of PTM can present, therapeutic management remains a challenge. Heyes et al5 documented the successful treatment of highly recalcitrant elephantiasic PTM with rituximab and plasmapheresis therapy. In this case, a 44-year-old woman with an 11-year history of Graves disease and elephantiasic PTM received 29 rituximab infusions and 241 plasmapheresis treatments over the course of 3.5 years. Her elephantiasic PTM clinically resolved, and she was able to resume daily activities and wear normal shoes after being nonambulatory for years.5
Rituximab is a monoclonal antibody against CD20, a protein found primarily on the surface of B-cell lymphocytes. Although rituximab initially was approved by the US Food and Drug administration for the treatment of malignant lymphoma, it has had an increasing role in the treatment of autoimmune disorders such as rheumatoid arthritis. Rituximab is postulated to target B lymphocytes and halt their progression to plasma cells. By limiting the population of long-lasting, antibody-producing plasma cells and decreasing the autoantibodies that cause many of the symptoms in Graves disease, rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.6
Although the exact mechanism is poorly understood, PTM likely is a sequela of hyperthyroidism because of the expression of thyroid-stimulating hormone receptor proteins found on normal dermal fibroblasts. Thyroid-stimulating hormone receptor autoantibodies are thought to stimulate these fibroblasts to produce glycosaminoglycans. Histopathologically, accumulation of glycosaminoglycans deposited in the reticular dermis with high concentrations of hyaluronic acid is observed in PTM.7
Treatment of elephantiasic PTM remains a therapeutic challenge. Given the rarity of the disease process and limited information on effective therapeutic modalities, rituximab should be viewed as a viable treatment option in the management of recalcitrant elephantiasic PTM.
To the Editor:
Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet. Pretibial myxedema occurs in approximately 0.5% to 4.3% of patients with hyperthyroidism.1 Thyroid dermopathy often is thought of as the classic nonpitting PTM with skin induration and color change. However, rarer forms of PTM, including plaque, nodular, and elephantiasic, also are important to note.2
Elephantiasic PTM is extremely rare, occurring in less than 1% of patients with PTM.2 Elephantiasic PTM is characterized by the persistent swelling of 1 or both legs; thickening of the skin overlying the dorsum of the feet, ankles, and toes; and verrucous irregular plaques that often are fleshy and flattened. The clinical differential diagnosis of elephantiasic PTM includes elephantiasis nostra verrucosa, a late-stage complication of chronic lymphedema that can be related to a variety of infectious or noninfectious obstructive processes. Few effective therapeutic modalities exist in the treatment of elephantiasic PTM. We present a case of elephantiasic PTM.
A 59-year-old man presented to dermatology with leonine facies with pronounced glabellar creases and indentations of the earlobes. He had diffuse woody induration, hyperpigmentation, and nonpitting edema of the lower extremities as well as several flesh-colored exophytic nodules scattered throughout the anterior shins and dorsal feet (Figure 1). On the left posterior calf, there was a large, 3-cm, exophytic, firm, flesh-colored nodule. Examination of the hands revealed mild hyperpigmentation of the distal digits, clubbing of the distal phalanges, and cheiroarthropathy.
The patient was diagnosed with Graves disease after experiencing the classic symptoms of hyperthyroidism, including heat intolerance, tremor, palpitations, and anxiety. He received thyroid ablation and subsequently was supplemented with levothyroxine 75 mg daily. Twelve years later, he was diagnosed with Graves ophthalmopathy with ocular proptosis requiring multiple courses of retro-orbital irradiation and surgical procedures for decompression. Approximately 1 year later, he noted increased swelling, firmness, and darkening of the pretibial surfaces. Initially, he was referred to vascular surgery and underwent bilateral saphenous vein ablation. He also was referred to a lymphedema specialist, and workup revealed an unremarkable lymphatic system. Minimal improvement was noted following the saphenous vein ablation, and he subsequently was referred to dermatology for further workup.
At the current presentation, laboratory analysis revealed a low thyrotropin level (0.03 mIU/L [reference range, 0.4–4.2 mIU/L]), and free thyroxine was within reference range. Radiography of the chest was unremarkable; however, radiography of the hand demonstrated arthrosis of the left fifth proximal interphalangeal joint. Nuclear medicine lymphoscintigraphy and lower extremity ultrasonography were unremarkable. Punch biopsies were performed of the left lateral leg and posterior calf. Hematoxylin and eosin staining demonstrated marked mucin deposition extending to the deep dermis along with deep fibroplasia and was read as consistent with PTM. Colloidal iron highlighted prominent mucin within the dermis (Figure 2).
The patient’s medical history, physical examination, laboratory analysis, imaging, and biopsies were considered, and a diagnosis of elephantiasic PTM was made. Minimal improvement was noted with initial therapeutic interventions including compression therapy and application of super high–potency topical corticosteroids. After further evaluation in our multidisciplinary rheumatology-dermatology clinic, the decision was made to initiate rituximab infusions.
Two months after 1 course of rituximab consisting of two 1000-mg infusions separated by 2 weeks, the patient showed substantial clinical improvement. There was striking improvement of the pretibial surfaces with resolution of the exophytic nodules and improvement of the induration (Figure 3). In addition, there was decreased induration of the glabella and earlobes and decreased fullness of the digital pulp on the hands. The patient also reported subjective improvements in mobility.
Our patient demonstrated all 3 aspects of the Diamond triad: PTM, exophthalmos, and acropachy. Patients present with all 3 features in less than 1% of reported cases of Graves disease.3 Although all 3 features are seen together infrequently, thyroid dermopathy and acropachy often are markers of severe Graves ophthalmopathy. In a study of 114 patients with Graves ophthalmopathy, patients who also had dermopathy and acropachy were more likely to have optic neuropathy or require orbital decompression.4
After overcoming the diagnostic dilemma that the elephantiasic presentation of PTM can present, therapeutic management remains a challenge. Heyes et al5 documented the successful treatment of highly recalcitrant elephantiasic PTM with rituximab and plasmapheresis therapy. In this case, a 44-year-old woman with an 11-year history of Graves disease and elephantiasic PTM received 29 rituximab infusions and 241 plasmapheresis treatments over the course of 3.5 years. Her elephantiasic PTM clinically resolved, and she was able to resume daily activities and wear normal shoes after being nonambulatory for years.5
Rituximab is a monoclonal antibody against CD20, a protein found primarily on the surface of B-cell lymphocytes. Although rituximab initially was approved by the US Food and Drug administration for the treatment of malignant lymphoma, it has had an increasing role in the treatment of autoimmune disorders such as rheumatoid arthritis. Rituximab is postulated to target B lymphocytes and halt their progression to plasma cells. By limiting the population of long-lasting, antibody-producing plasma cells and decreasing the autoantibodies that cause many of the symptoms in Graves disease, rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.6
Although the exact mechanism is poorly understood, PTM likely is a sequela of hyperthyroidism because of the expression of thyroid-stimulating hormone receptor proteins found on normal dermal fibroblasts. Thyroid-stimulating hormone receptor autoantibodies are thought to stimulate these fibroblasts to produce glycosaminoglycans. Histopathologically, accumulation of glycosaminoglycans deposited in the reticular dermis with high concentrations of hyaluronic acid is observed in PTM.7
Treatment of elephantiasic PTM remains a therapeutic challenge. Given the rarity of the disease process and limited information on effective therapeutic modalities, rituximab should be viewed as a viable treatment option in the management of recalcitrant elephantiasic PTM.
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446.
- Kakati S, Doley B, Pal S, et al. Elephantiasis nostras verrucosa: a rare thyroid dermopathy in Graves’ disease. J Assoc Physicians India. 2005;53:571-572.
- Anderson CK, Miller OF 3rd. Triad of exophthalmos, pretibial myxedema, and acropachy in a patient with Graves’ disease. J Am Acad Dermatol. 2003;48:970-972.
- Fatourechi V, Bartley GB, Eghbali-Fatourechi GZ, et al. Graves’ dermopathy and acropachy are markers of severe Graves’ ophthalmopathy. Thyroid. 2003;13:1141-1144.
- Heyes C, Nolan R, Leahy M, et al. Treatment‐resistant elephantiasic thyroid dermopathy responding to rituximab and plasmapheresis. Australas J Dermatol. 2012;53:E1-E4.
- Salvi M, Vannucchi G, Campi I, et al. Treatment of Graves’ disease and associated ophthalmopathy with the anti-CD20 monoclonal antibody rituximab: an open study. Eur J Endocrinol. 2007;156:33-40.
- Heufelder AE, Dutton CM, Sarkar G, et al. Detection of TSH receptor RNA in cultured fibroblasts from patients with Graves’ ophthalmopathy and pretibial dermopathy. Thyroid. 1993;3:297-300.
- Schwartz KM, Fatourechi V, Ahmed DDF, et al. Dermopathy of Graves’ disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab. 2002;87:438-446.
- Kakati S, Doley B, Pal S, et al. Elephantiasis nostras verrucosa: a rare thyroid dermopathy in Graves’ disease. J Assoc Physicians India. 2005;53:571-572.
- Anderson CK, Miller OF 3rd. Triad of exophthalmos, pretibial myxedema, and acropachy in a patient with Graves’ disease. J Am Acad Dermatol. 2003;48:970-972.
- Fatourechi V, Bartley GB, Eghbali-Fatourechi GZ, et al. Graves’ dermopathy and acropachy are markers of severe Graves’ ophthalmopathy. Thyroid. 2003;13:1141-1144.
- Heyes C, Nolan R, Leahy M, et al. Treatment‐resistant elephantiasic thyroid dermopathy responding to rituximab and plasmapheresis. Australas J Dermatol. 2012;53:E1-E4.
- Salvi M, Vannucchi G, Campi I, et al. Treatment of Graves’ disease and associated ophthalmopathy with the anti-CD20 monoclonal antibody rituximab: an open study. Eur J Endocrinol. 2007;156:33-40.
- Heufelder AE, Dutton CM, Sarkar G, et al. Detection of TSH receptor RNA in cultured fibroblasts from patients with Graves’ ophthalmopathy and pretibial dermopathy. Thyroid. 1993;3:297-300.
Practice Points
- Pretibial myxedema (PTM) is bilateral, nonpitting, scaly thickening and induration of the skin that most commonly occurs on the anterior aspects of the legs and feet.
- Although many therapeutic modalities have been described for the management of the elephantiasis variant of PTM, few treatments have shown notable efficacy.
- Rituximab may be an effective therapy to consider in the treatment of elephantiasic PTM.

A Fatal Case of Hemophagocytic Lymphohistiocytosis Secondary to Anti-MDA5–Positive Dermatomyositis
To the Editor:
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by bilateral, symmetrical, proximal muscle weakness and classic cutaneous manifestations.1 Patients with antibodies directed against melanoma differentiation–associated gene 5, MDA5, have a distinct presentation due to vasculopathy with more severe cutaneous ulcerations, palmar papules, alopecia, and an elevated risk of rapidly progressive interstitial lung disease.2 A ferritin level greater than 1600 ng/mL portends an increased risk for pulmonary disease and therefore can be of prognostic value.3 Further, patients with anti-MDA5 DM are at a lower risk of malignancy and are more likely to test negative for antinuclear antibodies in comparison to other patients with DM.2,4
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a potentially lethal condition whereby uncontrolled activation of histiocytes in the reticuloendothelial system causes hemophagocytosis and a hyperinflammatory state. Patients present with fever, splenomegaly, cytopenia, and hyperferritinemia.5 Autoimmune‐associated hemophagocytic syndrome (AAHS) describes HLH that develops in association with autoimmune conditions, most commonly systemic lupus erythematosus and adult-onset Still disease. Cases reported in association with DM exist but are few in number, and there is no standard-of-care treatment.6 We report a case of a woman with anti-MDA5 DM complicated by HLH and DM-associated liver injury.
A 50-year-old woman presented as a direct admit from the rheumatology clinic for diffuse muscle weakness of 8 months’ duration, 40-pound unintentional weight loss, pruritic rash, bilateral joint pains, dry eyes, dry mouth, and altered mental status. Four months prior, she presented to an outside hospital and was given a diagnosis of probable Sjögren syndrome and autoimmune hepatitis vs drug-induced liver injury. At that time, a workup was notable for antibodies against Sjögren syndrome–related antigen A, anti–smooth muscle antibodies, and transaminitis. Ultrasonography of the right upper quadrant revealed hepatic steatosis. The patient was started on oral prednisone and pilocarpine but had been off all medications for 1 month when she presented to our hospital.
On hospital admission, physical examination revealed a violaceous heliotrope rash; a v-sign on the chest; shawl sign; palmar papules with pits at the fingertips; and periungual erythema and ulcerations along the metacarpophalangeal joints, elbows, lateral feet, and upper eyelids (Figure 1). Laboratory workup showed the following results: white blood cell count, 4100/μL (reference range, 4000–11,000/μL); hemoglobin, 11.6 g/dL (reference range, 12–16 g/dL); platelet count, 100,000/μL (reference range, 150,000–450,000/μL); lactate dehydrogenase, 510 U/L (reference range, 80–225 U/L); alkaline phosphatase (ALP), 766 U/L (reference range, 30–120 U/L); alanine aminotransferase (ALT), 88 U/L (reference range, 10–40 U/L); aspartate aminotransferase (AST), 544 U/L (reference range, 10–40 U/L); total bilirubin, 4.2 mg/dL (reference range, 0.3–1.0 mg/dL); direct bilirubin, 3.7 mg/dL (reference range, 0.1–0.3 mg/dL); aldolase, 20.2 U/L (reference range, 1–7.5 U/L), creatine kinase, 180 U/L (reference range, 30–135 U/L); γ-glutamyltransferase (GGT), 2743 U/L (reference range, 8–40 U/L); high sensitivity C-reactive protein, 122.9 mg/L (low-risk reference range, <1.0 mg/L); triglycerides, 534 mg/dL (reference range, <150 mg/dL); ferritin, 3784 ng/mL (reference range, 24–307 ng/mL); antinuclear antibody, negative titer; antimitochondrial antibody, negative titer; soluble IL-2 receptor (CD25), 7000 U/mL (reference range, 189–846 U/mL); anti-Sjögren syndrome–related antigen A antibody, positive.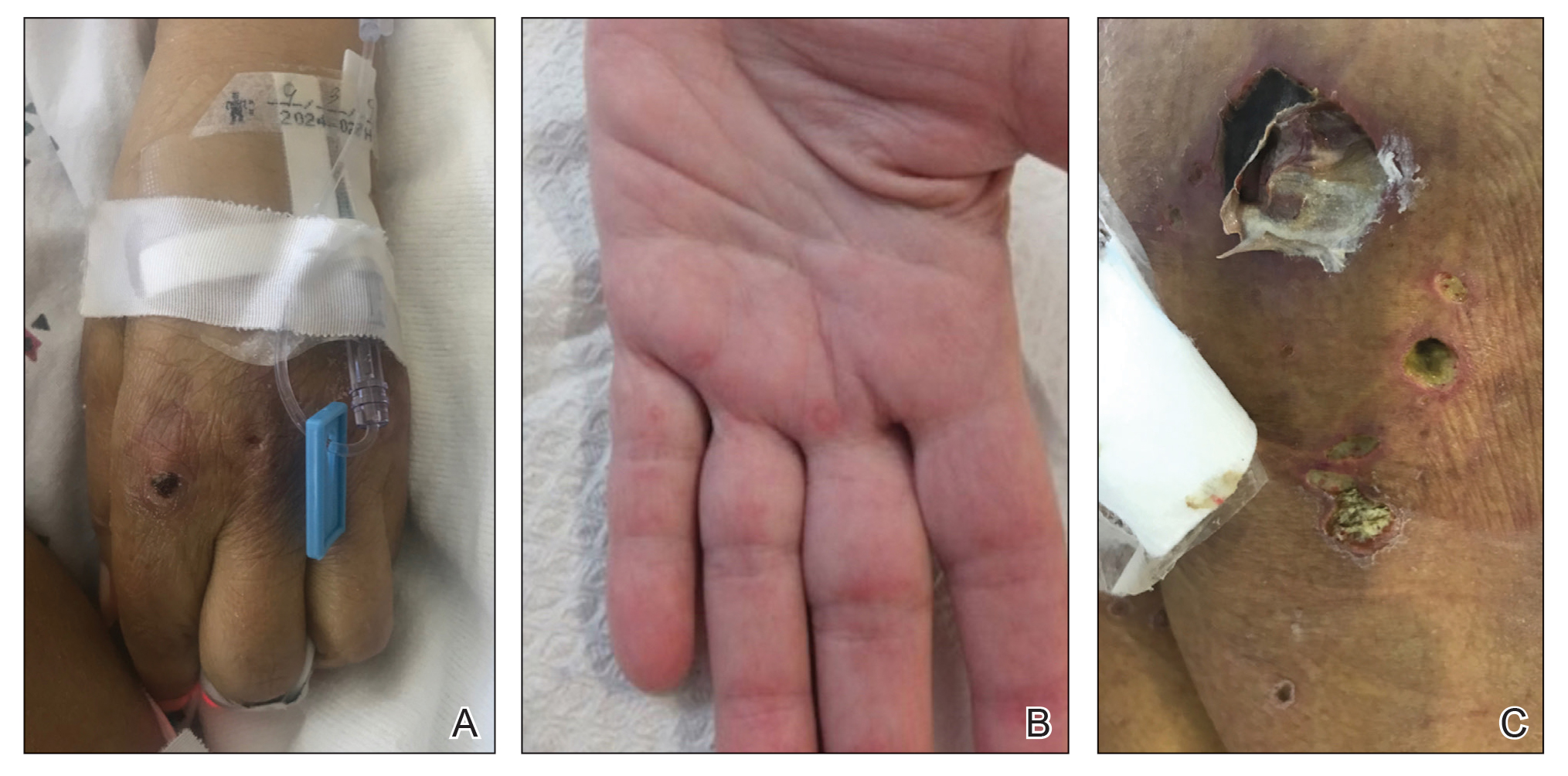
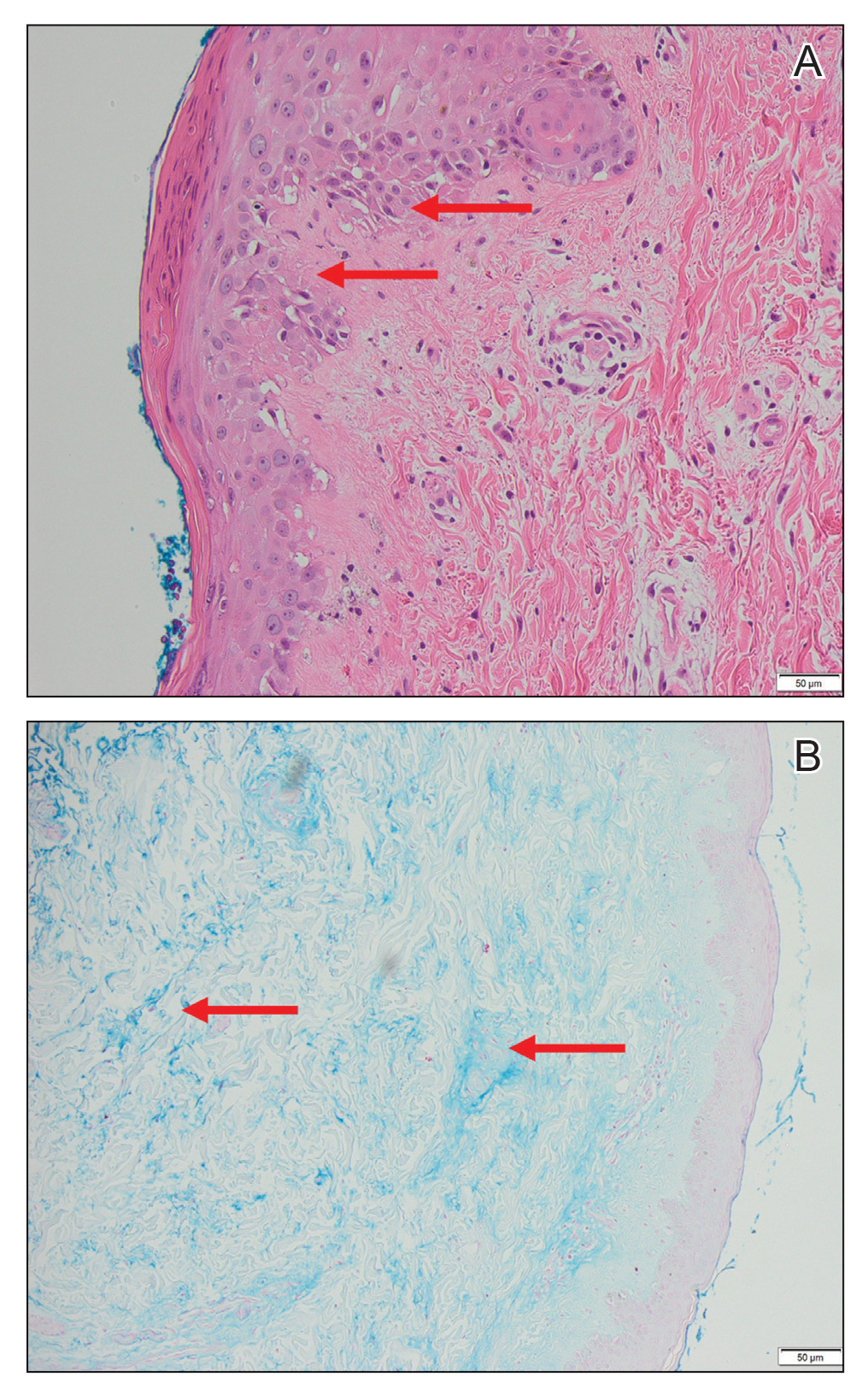
Magnetic resonance imaging of the shoulders showed diffuse soft-tissue edema. Computed tomography (CT) of the chest demonstrated parabronchial thickening and parenchymal bands suggestive of DM. An age-appropriate malignancy workup was negative, and results from a liver biopsy showed diffuse steatosis with no histologic evidence of autoimmune hepatitis. Punch biopsy results from a plaque on the left knee revealed vacuolar interface dermatitis with increased dermal mucin on colloidal iron staining, indicative of connective tissue disease (Figure 2). The patient was treated with intravenous (IV) methylprednisolone 250 mg twice daily for 2 days followed by oral prednisone 50 mg daily with IV immunoglobulin (IVIG) 0.4 mg/kg daily for 5 days. The patient’s symptoms improved, and she was discharged on oral prednisone 50 mg and mycophenolate mofetil 1000 mg twice daily with a plan for outpatient IVIG.
Two days after discharge, the patient was re-admitted for worsening muscle weakness; recalcitrant rash; new-onset hypophonia, dysphagia, and odynophagia; and intermittent fevers. Myositis panel results were positive for MDA5. Additionally, workup for HLH, which was initiated during the first hospital admission, revealed that she met 6 of 8 diagnostic criteria: intermittent fevers (maximum temperature, 38.2 °C), splenomegaly (12.6 cm on CT scan of abdomen), cytopenia in 2 cell lines (anemia, thrombocytopenia), hypertriglyceridemia, hyperferritinemia, and elevated IL-2 receptor (CD25). Based on these findings, the patient was diagnosed with anti-MDA5 DM associated with HLH.
The patient was started on IV methylprednisolone 1000 mg daily and received 1 rituximab infusion. Two days later, she experienced worsening fever with tachycardia, and a chest radiograph showed bibasilar infiltrates concerning for aspiration pneumonia, with sputum cultures growing Staphylococcus aureus. Due to the infection, the dosage of methylprednisolone was decreased to 16 mg 3 times daily and rituximab was stopped. The hematology department was consulted for the patient’s HLH, and due to her profound weakness and sepsis, the decision was made to hold initiation of etoposide, which, in addition to glucocorticoids, is considered first-line therapy for HLH. She subsequently experienced worsening hypoxia requiring intubation and received a second course of IVIG. Two days later, CT of the chest revealed progressive ground-glass opacities in the lower lobes of the lungs. The patient was then started on plasmapheresis every other day, hydroxychloroquine 200 mg daily, and IV methylprednisolone 1000 mg daily. Over the subsequent 6 days, she developed worsening renal failure, liver dysfunction, profound thrombocytopenia (13/μL), and acidemia. After extensive discussion with her family, the patient was transitioned to comfort care, and she died 33 days after the initial admission to our hospital.
Our case is a collection of several rare presentations: anti-MDA5 DM, with HLH and AAHS as complications of anti-MDA5 DM, and DM-associated liver injury. Anti-MDA5 DM is frequently refractory to conventional therapy, including high-dose glucocorticoids, cyclophosphamide, oral tacrolimus, and cyclosporine, and there currently is no single treatment algorithm.2 Lake and colleagues7 highlighted the importance of personalizing treatment of anti-MDA5 DM, as it can be one of the most aggressive rheumatologic diseases. We initially chose to treat our patient with high-dose methylprednisolone, IVIG, and rituximab. Kampylafka et al8 performed a retrospective analysis of the use of IVIG for DM as compared to standard therapy and demonstrated improved muscle and cutaneous involvement from a collection of 50 patients. Case reports have specifically revealed efficacy for the use of IVIG in patients with anti-MDA5 DM.9,10 Additionally, rituximab—an anti–B lymphocyte therapy—has been shown to be an effective supplemental therapy for cases of aggressive anti-MDA5 DM with associated interstitial lung disease, especially when conventional therapy has failed.11,12 Our patient’s sepsis secondary to S aureus pneumonia limited her to only receiving 1 dose of rituximab.
One promising treatment approach for anti-MDA5 DM recently published by Tsuji et al13 involves the use of combination therapy. In this prospective multicenter trial, patients were initially treated with a combination of high-dose glucocorticoids, oral tacrolimus, and IV cyclophosphamide. Plasmapheresis was then started for patients without symptomatic improvement. This method was compared to the more traditional step-up approach of high-dose steroids followed by another immunosuppressant. At 1-year follow-up, the combination therapy group demonstrated an 85% survival rate compared to 33% of historical controls.13
We suspect that our patient developed HLH and AAHS secondary to her underlying anti-MDA5 DM. Kumakura and Murakawa6 reported that among 116 cases of AAHS, 6.9% of cases were associated with DM, most commonly anti-Jo-1 DM. Hemophagocytic lymphohistiocytosis associated with anti-MDA5 DM has been described in only a few cases.14-16 The diagnosis of HLH is critical, as the treatments for HLH and DM differ. Both diseases manifest with hyperferritinemia—greater than 500 ng/mL in the case of HLH and 3784 ng/mL in our patient. Therefore, HLH can be easily overlooked. It is possible the rates of HLH associated with anti-MDA5 DM are higher than reported given their similar presentations.
Analogous to our case, Fujita et al15 reported a case of HLH associated with anti-MDA5 DM successfully treated with IV cyclophosphamide pulse therapy and plasmapheresis. The rationale for using plasmapheresis in anti-MDA5 DM is based on its success in patients with other antibody-mediated conditions such as Goodpasture syndrome and granulomatosis with polyangiitis.7 It is thought to expedite response to traditional treatment, and in the case described by Fujita et al,15 the patient received plasmapheresis 6 times total over the course of 9 days. The patient’s clinical symptoms, as well as platelet levels, liver enzymes, and ferritin value, improved.15 Our patient received 3 days of plasmapheresis with no improvement when the decision was made to discontinue plasmapheresis given her worsening clinical state.
Additionally, our patient had elevated hepatic enzymes (ALT, AST, ALP, GGT), and results of a liver biopsy demonstrated diffuse steatosis. We speculate her transaminitis was a complication of anti-MDA5 DM. Hepatocellular damage accompanying DM has been investigated in multiple studies and is most often defined as an elevated ALT.17-20 Improvement in ALT levels has been seen with DM treatment. However, investigators note that creatine kinase (CK) values often do not correlate with the resolution of the transaminitis, suggesting that CK denotes muscle damage whereas ALT represents separate liver damage.18-21
Nagashima et al22 highlighted that among 50 patients with DM without malignancy, only 20% presented with a transaminitis or elevated bilirubin. However, among those with liver injury, all were positive for antibodies against MDA5.22 The patients with anti-MDA5 DM liver dysfunction had higher ALT, ALP, and GGT levels compared to those without liver dysfunction. Similarly, in a retrospective review of 14 patients with anti-MDA5 DM, Gono and colleagues3 found elevated GGT levels and lower CK levels in comparison to patients with anti-aminoacyl-transfer RNA synthetase DM. Although liver enzymes can be elevated in patients with DM secondary to muscle damage, the authors argue that the specificity of GGT to the liver suggests intrinsic liver damage.3
The mechanism behind liver disease in anti-MDA5 DM is unclear, but it is hypothesized to be similar to nonalcoholic steatohepatitis.22 Other studies have revealed drug-induced hepatitis, hepatic congestion, nonspecific reactive hepatitis, metastatic liver tumor, primary biliary cholangitis, and autoimmune hepatitis as the etiology behind liver disease in their patients with DM.17-19 Liver biopsy results from patients with anti-MDA5 DM most commonly reveal hepatic steatosis, as seen in our patient, as well as hepatocyte ballooning and increased pigmented macrophages.22
We presented a case of anti-MDA5 DM complicated by HLH. Our patient had a fatal outcome despite aggressive treatment with high-dose methylprednisolone, IVIG, rituximab, and plasmapheresis. It is accepted that anti-MDA5 DM affects the lungs and skin, and our patient’s presentation also suggests liver involvement. In our case, onset of symptoms to fatality was approximately 1 year. It is essential to consider the diagnosis of HLH in all cases of anti-MDA5 DM given clinical disease overlap. Our patient could have benefited from earlier disease recognition and thus earlier aggressive therapy.
1. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-347.
2. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785.
3. Gono T, Kawaguchi Y, Satoh T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49:1713-1719.
4. Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34.
5. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. 2017;49:20-26.
6. Kumakura S, Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheum. 2014;66:2297-2307.
7. Lake M, George G, Summer R. Time to personalize the treatment of anti-MDA-5 associated lung disease. Ann Rheum Dis. 2019;78:E52.
8. Kampylafka EI, Kosmidis ML, Panagiotakos DB, et al. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol. 2012;30:397-401.
9. Koguchi-Yoshioka H, Okiyama N, Iwamoto K, et al. Intravenous immunoglobulin contributes to the control of antimelanoma differentiation-associated protein 5 antibody-associated dermatomyositis with palmar violaceous macules/papules. Br J Dermatol. 2017;177:1442-1446.
10. Hamada-Ode K, Taniguchi Y, Kimata T, et al. High-dose intravenous immunoglobulin therapy for rapidly progressive interstitial pneumonitis accompanied by anti-melanoma differentiation-associated gene 5 antibody-positive amyopathic dermatomyositis. Eur J Rheumatol. 2015;2:83-85.
11. So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989.
12. Koichi Y, Aya Y, Megumi U, et al. A case of anti-MDA5-positive rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis ameliorated by rituximab, in addition to standard immunosuppressive treatment. Mod Rheumatol. 2017;27:536-540.
13. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. 2020;72:488-498.
14. Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune-associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246.
15. Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478.
16. Gono T, Miyake K, Kawaguchi Y, et al. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology (Oxford). 2012;51:1336-1338.
17. Wada T, Abe G, Kudou, T, et al. Liver damage in patients with polymyositis and dermatomyositis. Kitasato Med Journal. 2016;46:40-46.
18. Takahashi A, Abe K, Yokokawa J, et al. Clinical features of liver dysfunction in collagen diseases. Hepatol Res. 2010;40:1092-1097.
19. Matsumoto T, Kobayashi S, Shimizu H, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000;20:366-373.
20. Shi Q, Niu J, Huang X, et al. Do muscle enzyme changes forecast liver injury in polymyositis/dermatomyositis patients treated with methylprednisolone and methotrexate? Ann Clin Lab Sci. 2016;46:266-269.
21. Noda S, Asano Y, Tamaki Z, et al. A case of dermatomyositis with “liver disease associated with rheumatoid diseases” positive for anti-liver-kidney microsome-1 antibody. Clin Rheumatol. 2010;29:941-943.
22. Nagashima T, Kamata Y, Iwamoto M, et al. Liver dysfunction in anti-melanoma differentiation-associated gene 5 antibody-positive patients with dermatomyositis. Rheumatol Int. 2019;39:901-909.
To the Editor:
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by bilateral, symmetrical, proximal muscle weakness and classic cutaneous manifestations.1 Patients with antibodies directed against melanoma differentiation–associated gene 5, MDA5, have a distinct presentation due to vasculopathy with more severe cutaneous ulcerations, palmar papules, alopecia, and an elevated risk of rapidly progressive interstitial lung disease.2 A ferritin level greater than 1600 ng/mL portends an increased risk for pulmonary disease and therefore can be of prognostic value.3 Further, patients with anti-MDA5 DM are at a lower risk of malignancy and are more likely to test negative for antinuclear antibodies in comparison to other patients with DM.2,4
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a potentially lethal condition whereby uncontrolled activation of histiocytes in the reticuloendothelial system causes hemophagocytosis and a hyperinflammatory state. Patients present with fever, splenomegaly, cytopenia, and hyperferritinemia.5 Autoimmune‐associated hemophagocytic syndrome (AAHS) describes HLH that develops in association with autoimmune conditions, most commonly systemic lupus erythematosus and adult-onset Still disease. Cases reported in association with DM exist but are few in number, and there is no standard-of-care treatment.6 We report a case of a woman with anti-MDA5 DM complicated by HLH and DM-associated liver injury.
A 50-year-old woman presented as a direct admit from the rheumatology clinic for diffuse muscle weakness of 8 months’ duration, 40-pound unintentional weight loss, pruritic rash, bilateral joint pains, dry eyes, dry mouth, and altered mental status. Four months prior, she presented to an outside hospital and was given a diagnosis of probable Sjögren syndrome and autoimmune hepatitis vs drug-induced liver injury. At that time, a workup was notable for antibodies against Sjögren syndrome–related antigen A, anti–smooth muscle antibodies, and transaminitis. Ultrasonography of the right upper quadrant revealed hepatic steatosis. The patient was started on oral prednisone and pilocarpine but had been off all medications for 1 month when she presented to our hospital.
On hospital admission, physical examination revealed a violaceous heliotrope rash; a v-sign on the chest; shawl sign; palmar papules with pits at the fingertips; and periungual erythema and ulcerations along the metacarpophalangeal joints, elbows, lateral feet, and upper eyelids (Figure 1). Laboratory workup showed the following results: white blood cell count, 4100/μL (reference range, 4000–11,000/μL); hemoglobin, 11.6 g/dL (reference range, 12–16 g/dL); platelet count, 100,000/μL (reference range, 150,000–450,000/μL); lactate dehydrogenase, 510 U/L (reference range, 80–225 U/L); alkaline phosphatase (ALP), 766 U/L (reference range, 30–120 U/L); alanine aminotransferase (ALT), 88 U/L (reference range, 10–40 U/L); aspartate aminotransferase (AST), 544 U/L (reference range, 10–40 U/L); total bilirubin, 4.2 mg/dL (reference range, 0.3–1.0 mg/dL); direct bilirubin, 3.7 mg/dL (reference range, 0.1–0.3 mg/dL); aldolase, 20.2 U/L (reference range, 1–7.5 U/L), creatine kinase, 180 U/L (reference range, 30–135 U/L); γ-glutamyltransferase (GGT), 2743 U/L (reference range, 8–40 U/L); high sensitivity C-reactive protein, 122.9 mg/L (low-risk reference range, <1.0 mg/L); triglycerides, 534 mg/dL (reference range, <150 mg/dL); ferritin, 3784 ng/mL (reference range, 24–307 ng/mL); antinuclear antibody, negative titer; antimitochondrial antibody, negative titer; soluble IL-2 receptor (CD25), 7000 U/mL (reference range, 189–846 U/mL); anti-Sjögren syndrome–related antigen A antibody, positive.
Magnetic resonance imaging of the shoulders showed diffuse soft-tissue edema. Computed tomography (CT) of the chest demonstrated parabronchial thickening and parenchymal bands suggestive of DM. An age-appropriate malignancy workup was negative, and results from a liver biopsy showed diffuse steatosis with no histologic evidence of autoimmune hepatitis. Punch biopsy results from a plaque on the left knee revealed vacuolar interface dermatitis with increased dermal mucin on colloidal iron staining, indicative of connective tissue disease (Figure 2). The patient was treated with intravenous (IV) methylprednisolone 250 mg twice daily for 2 days followed by oral prednisone 50 mg daily with IV immunoglobulin (IVIG) 0.4 mg/kg daily for 5 days. The patient’s symptoms improved, and she was discharged on oral prednisone 50 mg and mycophenolate mofetil 1000 mg twice daily with a plan for outpatient IVIG.
Two days after discharge, the patient was re-admitted for worsening muscle weakness; recalcitrant rash; new-onset hypophonia, dysphagia, and odynophagia; and intermittent fevers. Myositis panel results were positive for MDA5. Additionally, workup for HLH, which was initiated during the first hospital admission, revealed that she met 6 of 8 diagnostic criteria: intermittent fevers (maximum temperature, 38.2 °C), splenomegaly (12.6 cm on CT scan of abdomen), cytopenia in 2 cell lines (anemia, thrombocytopenia), hypertriglyceridemia, hyperferritinemia, and elevated IL-2 receptor (CD25). Based on these findings, the patient was diagnosed with anti-MDA5 DM associated with HLH.
The patient was started on IV methylprednisolone 1000 mg daily and received 1 rituximab infusion. Two days later, she experienced worsening fever with tachycardia, and a chest radiograph showed bibasilar infiltrates concerning for aspiration pneumonia, with sputum cultures growing Staphylococcus aureus. Due to the infection, the dosage of methylprednisolone was decreased to 16 mg 3 times daily and rituximab was stopped. The hematology department was consulted for the patient’s HLH, and due to her profound weakness and sepsis, the decision was made to hold initiation of etoposide, which, in addition to glucocorticoids, is considered first-line therapy for HLH. She subsequently experienced worsening hypoxia requiring intubation and received a second course of IVIG. Two days later, CT of the chest revealed progressive ground-glass opacities in the lower lobes of the lungs. The patient was then started on plasmapheresis every other day, hydroxychloroquine 200 mg daily, and IV methylprednisolone 1000 mg daily. Over the subsequent 6 days, she developed worsening renal failure, liver dysfunction, profound thrombocytopenia (13/μL), and acidemia. After extensive discussion with her family, the patient was transitioned to comfort care, and she died 33 days after the initial admission to our hospital.
Our case is a collection of several rare presentations: anti-MDA5 DM, with HLH and AAHS as complications of anti-MDA5 DM, and DM-associated liver injury. Anti-MDA5 DM is frequently refractory to conventional therapy, including high-dose glucocorticoids, cyclophosphamide, oral tacrolimus, and cyclosporine, and there currently is no single treatment algorithm.2 Lake and colleagues7 highlighted the importance of personalizing treatment of anti-MDA5 DM, as it can be one of the most aggressive rheumatologic diseases. We initially chose to treat our patient with high-dose methylprednisolone, IVIG, and rituximab. Kampylafka et al8 performed a retrospective analysis of the use of IVIG for DM as compared to standard therapy and demonstrated improved muscle and cutaneous involvement from a collection of 50 patients. Case reports have specifically revealed efficacy for the use of IVIG in patients with anti-MDA5 DM.9,10 Additionally, rituximab—an anti–B lymphocyte therapy—has been shown to be an effective supplemental therapy for cases of aggressive anti-MDA5 DM with associated interstitial lung disease, especially when conventional therapy has failed.11,12 Our patient’s sepsis secondary to S aureus pneumonia limited her to only receiving 1 dose of rituximab.
One promising treatment approach for anti-MDA5 DM recently published by Tsuji et al13 involves the use of combination therapy. In this prospective multicenter trial, patients were initially treated with a combination of high-dose glucocorticoids, oral tacrolimus, and IV cyclophosphamide. Plasmapheresis was then started for patients without symptomatic improvement. This method was compared to the more traditional step-up approach of high-dose steroids followed by another immunosuppressant. At 1-year follow-up, the combination therapy group demonstrated an 85% survival rate compared to 33% of historical controls.13
We suspect that our patient developed HLH and AAHS secondary to her underlying anti-MDA5 DM. Kumakura and Murakawa6 reported that among 116 cases of AAHS, 6.9% of cases were associated with DM, most commonly anti-Jo-1 DM. Hemophagocytic lymphohistiocytosis associated with anti-MDA5 DM has been described in only a few cases.14-16 The diagnosis of HLH is critical, as the treatments for HLH and DM differ. Both diseases manifest with hyperferritinemia—greater than 500 ng/mL in the case of HLH and 3784 ng/mL in our patient. Therefore, HLH can be easily overlooked. It is possible the rates of HLH associated with anti-MDA5 DM are higher than reported given their similar presentations.
Analogous to our case, Fujita et al15 reported a case of HLH associated with anti-MDA5 DM successfully treated with IV cyclophosphamide pulse therapy and plasmapheresis. The rationale for using plasmapheresis in anti-MDA5 DM is based on its success in patients with other antibody-mediated conditions such as Goodpasture syndrome and granulomatosis with polyangiitis.7 It is thought to expedite response to traditional treatment, and in the case described by Fujita et al,15 the patient received plasmapheresis 6 times total over the course of 9 days. The patient’s clinical symptoms, as well as platelet levels, liver enzymes, and ferritin value, improved.15 Our patient received 3 days of plasmapheresis with no improvement when the decision was made to discontinue plasmapheresis given her worsening clinical state.
Additionally, our patient had elevated hepatic enzymes (ALT, AST, ALP, GGT), and results of a liver biopsy demonstrated diffuse steatosis. We speculate her transaminitis was a complication of anti-MDA5 DM. Hepatocellular damage accompanying DM has been investigated in multiple studies and is most often defined as an elevated ALT.17-20 Improvement in ALT levels has been seen with DM treatment. However, investigators note that creatine kinase (CK) values often do not correlate with the resolution of the transaminitis, suggesting that CK denotes muscle damage whereas ALT represents separate liver damage.18-21
Nagashima et al22 highlighted that among 50 patients with DM without malignancy, only 20% presented with a transaminitis or elevated bilirubin. However, among those with liver injury, all were positive for antibodies against MDA5.22 The patients with anti-MDA5 DM liver dysfunction had higher ALT, ALP, and GGT levels compared to those without liver dysfunction. Similarly, in a retrospective review of 14 patients with anti-MDA5 DM, Gono and colleagues3 found elevated GGT levels and lower CK levels in comparison to patients with anti-aminoacyl-transfer RNA synthetase DM. Although liver enzymes can be elevated in patients with DM secondary to muscle damage, the authors argue that the specificity of GGT to the liver suggests intrinsic liver damage.3
The mechanism behind liver disease in anti-MDA5 DM is unclear, but it is hypothesized to be similar to nonalcoholic steatohepatitis.22 Other studies have revealed drug-induced hepatitis, hepatic congestion, nonspecific reactive hepatitis, metastatic liver tumor, primary biliary cholangitis, and autoimmune hepatitis as the etiology behind liver disease in their patients with DM.17-19 Liver biopsy results from patients with anti-MDA5 DM most commonly reveal hepatic steatosis, as seen in our patient, as well as hepatocyte ballooning and increased pigmented macrophages.22
We presented a case of anti-MDA5 DM complicated by HLH. Our patient had a fatal outcome despite aggressive treatment with high-dose methylprednisolone, IVIG, rituximab, and plasmapheresis. It is accepted that anti-MDA5 DM affects the lungs and skin, and our patient’s presentation also suggests liver involvement. In our case, onset of symptoms to fatality was approximately 1 year. It is essential to consider the diagnosis of HLH in all cases of anti-MDA5 DM given clinical disease overlap. Our patient could have benefited from earlier disease recognition and thus earlier aggressive therapy.
To the Editor:
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by bilateral, symmetrical, proximal muscle weakness and classic cutaneous manifestations.1 Patients with antibodies directed against melanoma differentiation–associated gene 5, MDA5, have a distinct presentation due to vasculopathy with more severe cutaneous ulcerations, palmar papules, alopecia, and an elevated risk of rapidly progressive interstitial lung disease.2 A ferritin level greater than 1600 ng/mL portends an increased risk for pulmonary disease and therefore can be of prognostic value.3 Further, patients with anti-MDA5 DM are at a lower risk of malignancy and are more likely to test negative for antinuclear antibodies in comparison to other patients with DM.2,4
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a potentially lethal condition whereby uncontrolled activation of histiocytes in the reticuloendothelial system causes hemophagocytosis and a hyperinflammatory state. Patients present with fever, splenomegaly, cytopenia, and hyperferritinemia.5 Autoimmune‐associated hemophagocytic syndrome (AAHS) describes HLH that develops in association with autoimmune conditions, most commonly systemic lupus erythematosus and adult-onset Still disease. Cases reported in association with DM exist but are few in number, and there is no standard-of-care treatment.6 We report a case of a woman with anti-MDA5 DM complicated by HLH and DM-associated liver injury.
A 50-year-old woman presented as a direct admit from the rheumatology clinic for diffuse muscle weakness of 8 months’ duration, 40-pound unintentional weight loss, pruritic rash, bilateral joint pains, dry eyes, dry mouth, and altered mental status. Four months prior, she presented to an outside hospital and was given a diagnosis of probable Sjögren syndrome and autoimmune hepatitis vs drug-induced liver injury. At that time, a workup was notable for antibodies against Sjögren syndrome–related antigen A, anti–smooth muscle antibodies, and transaminitis. Ultrasonography of the right upper quadrant revealed hepatic steatosis. The patient was started on oral prednisone and pilocarpine but had been off all medications for 1 month when she presented to our hospital.
On hospital admission, physical examination revealed a violaceous heliotrope rash; a v-sign on the chest; shawl sign; palmar papules with pits at the fingertips; and periungual erythema and ulcerations along the metacarpophalangeal joints, elbows, lateral feet, and upper eyelids (Figure 1). Laboratory workup showed the following results: white blood cell count, 4100/μL (reference range, 4000–11,000/μL); hemoglobin, 11.6 g/dL (reference range, 12–16 g/dL); platelet count, 100,000/μL (reference range, 150,000–450,000/μL); lactate dehydrogenase, 510 U/L (reference range, 80–225 U/L); alkaline phosphatase (ALP), 766 U/L (reference range, 30–120 U/L); alanine aminotransferase (ALT), 88 U/L (reference range, 10–40 U/L); aspartate aminotransferase (AST), 544 U/L (reference range, 10–40 U/L); total bilirubin, 4.2 mg/dL (reference range, 0.3–1.0 mg/dL); direct bilirubin, 3.7 mg/dL (reference range, 0.1–0.3 mg/dL); aldolase, 20.2 U/L (reference range, 1–7.5 U/L), creatine kinase, 180 U/L (reference range, 30–135 U/L); γ-glutamyltransferase (GGT), 2743 U/L (reference range, 8–40 U/L); high sensitivity C-reactive protein, 122.9 mg/L (low-risk reference range, <1.0 mg/L); triglycerides, 534 mg/dL (reference range, <150 mg/dL); ferritin, 3784 ng/mL (reference range, 24–307 ng/mL); antinuclear antibody, negative titer; antimitochondrial antibody, negative titer; soluble IL-2 receptor (CD25), 7000 U/mL (reference range, 189–846 U/mL); anti-Sjögren syndrome–related antigen A antibody, positive.
Magnetic resonance imaging of the shoulders showed diffuse soft-tissue edema. Computed tomography (CT) of the chest demonstrated parabronchial thickening and parenchymal bands suggestive of DM. An age-appropriate malignancy workup was negative, and results from a liver biopsy showed diffuse steatosis with no histologic evidence of autoimmune hepatitis. Punch biopsy results from a plaque on the left knee revealed vacuolar interface dermatitis with increased dermal mucin on colloidal iron staining, indicative of connective tissue disease (Figure 2). The patient was treated with intravenous (IV) methylprednisolone 250 mg twice daily for 2 days followed by oral prednisone 50 mg daily with IV immunoglobulin (IVIG) 0.4 mg/kg daily for 5 days. The patient’s symptoms improved, and she was discharged on oral prednisone 50 mg and mycophenolate mofetil 1000 mg twice daily with a plan for outpatient IVIG.
Two days after discharge, the patient was re-admitted for worsening muscle weakness; recalcitrant rash; new-onset hypophonia, dysphagia, and odynophagia; and intermittent fevers. Myositis panel results were positive for MDA5. Additionally, workup for HLH, which was initiated during the first hospital admission, revealed that she met 6 of 8 diagnostic criteria: intermittent fevers (maximum temperature, 38.2 °C), splenomegaly (12.6 cm on CT scan of abdomen), cytopenia in 2 cell lines (anemia, thrombocytopenia), hypertriglyceridemia, hyperferritinemia, and elevated IL-2 receptor (CD25). Based on these findings, the patient was diagnosed with anti-MDA5 DM associated with HLH.
The patient was started on IV methylprednisolone 1000 mg daily and received 1 rituximab infusion. Two days later, she experienced worsening fever with tachycardia, and a chest radiograph showed bibasilar infiltrates concerning for aspiration pneumonia, with sputum cultures growing Staphylococcus aureus. Due to the infection, the dosage of methylprednisolone was decreased to 16 mg 3 times daily and rituximab was stopped. The hematology department was consulted for the patient’s HLH, and due to her profound weakness and sepsis, the decision was made to hold initiation of etoposide, which, in addition to glucocorticoids, is considered first-line therapy for HLH. She subsequently experienced worsening hypoxia requiring intubation and received a second course of IVIG. Two days later, CT of the chest revealed progressive ground-glass opacities in the lower lobes of the lungs. The patient was then started on plasmapheresis every other day, hydroxychloroquine 200 mg daily, and IV methylprednisolone 1000 mg daily. Over the subsequent 6 days, she developed worsening renal failure, liver dysfunction, profound thrombocytopenia (13/μL), and acidemia. After extensive discussion with her family, the patient was transitioned to comfort care, and she died 33 days after the initial admission to our hospital.
Our case is a collection of several rare presentations: anti-MDA5 DM, with HLH and AAHS as complications of anti-MDA5 DM, and DM-associated liver injury. Anti-MDA5 DM is frequently refractory to conventional therapy, including high-dose glucocorticoids, cyclophosphamide, oral tacrolimus, and cyclosporine, and there currently is no single treatment algorithm.2 Lake and colleagues7 highlighted the importance of personalizing treatment of anti-MDA5 DM, as it can be one of the most aggressive rheumatologic diseases. We initially chose to treat our patient with high-dose methylprednisolone, IVIG, and rituximab. Kampylafka et al8 performed a retrospective analysis of the use of IVIG for DM as compared to standard therapy and demonstrated improved muscle and cutaneous involvement from a collection of 50 patients. Case reports have specifically revealed efficacy for the use of IVIG in patients with anti-MDA5 DM.9,10 Additionally, rituximab—an anti–B lymphocyte therapy—has been shown to be an effective supplemental therapy for cases of aggressive anti-MDA5 DM with associated interstitial lung disease, especially when conventional therapy has failed.11,12 Our patient’s sepsis secondary to S aureus pneumonia limited her to only receiving 1 dose of rituximab.
One promising treatment approach for anti-MDA5 DM recently published by Tsuji et al13 involves the use of combination therapy. In this prospective multicenter trial, patients were initially treated with a combination of high-dose glucocorticoids, oral tacrolimus, and IV cyclophosphamide. Plasmapheresis was then started for patients without symptomatic improvement. This method was compared to the more traditional step-up approach of high-dose steroids followed by another immunosuppressant. At 1-year follow-up, the combination therapy group demonstrated an 85% survival rate compared to 33% of historical controls.13
We suspect that our patient developed HLH and AAHS secondary to her underlying anti-MDA5 DM. Kumakura and Murakawa6 reported that among 116 cases of AAHS, 6.9% of cases were associated with DM, most commonly anti-Jo-1 DM. Hemophagocytic lymphohistiocytosis associated with anti-MDA5 DM has been described in only a few cases.14-16 The diagnosis of HLH is critical, as the treatments for HLH and DM differ. Both diseases manifest with hyperferritinemia—greater than 500 ng/mL in the case of HLH and 3784 ng/mL in our patient. Therefore, HLH can be easily overlooked. It is possible the rates of HLH associated with anti-MDA5 DM are higher than reported given their similar presentations.
Analogous to our case, Fujita et al15 reported a case of HLH associated with anti-MDA5 DM successfully treated with IV cyclophosphamide pulse therapy and plasmapheresis. The rationale for using plasmapheresis in anti-MDA5 DM is based on its success in patients with other antibody-mediated conditions such as Goodpasture syndrome and granulomatosis with polyangiitis.7 It is thought to expedite response to traditional treatment, and in the case described by Fujita et al,15 the patient received plasmapheresis 6 times total over the course of 9 days. The patient’s clinical symptoms, as well as platelet levels, liver enzymes, and ferritin value, improved.15 Our patient received 3 days of plasmapheresis with no improvement when the decision was made to discontinue plasmapheresis given her worsening clinical state.
Additionally, our patient had elevated hepatic enzymes (ALT, AST, ALP, GGT), and results of a liver biopsy demonstrated diffuse steatosis. We speculate her transaminitis was a complication of anti-MDA5 DM. Hepatocellular damage accompanying DM has been investigated in multiple studies and is most often defined as an elevated ALT.17-20 Improvement in ALT levels has been seen with DM treatment. However, investigators note that creatine kinase (CK) values often do not correlate with the resolution of the transaminitis, suggesting that CK denotes muscle damage whereas ALT represents separate liver damage.18-21
Nagashima et al22 highlighted that among 50 patients with DM without malignancy, only 20% presented with a transaminitis or elevated bilirubin. However, among those with liver injury, all were positive for antibodies against MDA5.22 The patients with anti-MDA5 DM liver dysfunction had higher ALT, ALP, and GGT levels compared to those without liver dysfunction. Similarly, in a retrospective review of 14 patients with anti-MDA5 DM, Gono and colleagues3 found elevated GGT levels and lower CK levels in comparison to patients with anti-aminoacyl-transfer RNA synthetase DM. Although liver enzymes can be elevated in patients with DM secondary to muscle damage, the authors argue that the specificity of GGT to the liver suggests intrinsic liver damage.3
The mechanism behind liver disease in anti-MDA5 DM is unclear, but it is hypothesized to be similar to nonalcoholic steatohepatitis.22 Other studies have revealed drug-induced hepatitis, hepatic congestion, nonspecific reactive hepatitis, metastatic liver tumor, primary biliary cholangitis, and autoimmune hepatitis as the etiology behind liver disease in their patients with DM.17-19 Liver biopsy results from patients with anti-MDA5 DM most commonly reveal hepatic steatosis, as seen in our patient, as well as hepatocyte ballooning and increased pigmented macrophages.22
We presented a case of anti-MDA5 DM complicated by HLH. Our patient had a fatal outcome despite aggressive treatment with high-dose methylprednisolone, IVIG, rituximab, and plasmapheresis. It is accepted that anti-MDA5 DM affects the lungs and skin, and our patient’s presentation also suggests liver involvement. In our case, onset of symptoms to fatality was approximately 1 year. It is essential to consider the diagnosis of HLH in all cases of anti-MDA5 DM given clinical disease overlap. Our patient could have benefited from earlier disease recognition and thus earlier aggressive therapy.
1. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-347.
2. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785.
3. Gono T, Kawaguchi Y, Satoh T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49:1713-1719.
4. Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34.
5. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. 2017;49:20-26.
6. Kumakura S, Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheum. 2014;66:2297-2307.
7. Lake M, George G, Summer R. Time to personalize the treatment of anti-MDA-5 associated lung disease. Ann Rheum Dis. 2019;78:E52.
8. Kampylafka EI, Kosmidis ML, Panagiotakos DB, et al. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol. 2012;30:397-401.
9. Koguchi-Yoshioka H, Okiyama N, Iwamoto K, et al. Intravenous immunoglobulin contributes to the control of antimelanoma differentiation-associated protein 5 antibody-associated dermatomyositis with palmar violaceous macules/papules. Br J Dermatol. 2017;177:1442-1446.
10. Hamada-Ode K, Taniguchi Y, Kimata T, et al. High-dose intravenous immunoglobulin therapy for rapidly progressive interstitial pneumonitis accompanied by anti-melanoma differentiation-associated gene 5 antibody-positive amyopathic dermatomyositis. Eur J Rheumatol. 2015;2:83-85.
11. So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989.
12. Koichi Y, Aya Y, Megumi U, et al. A case of anti-MDA5-positive rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis ameliorated by rituximab, in addition to standard immunosuppressive treatment. Mod Rheumatol. 2017;27:536-540.
13. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. 2020;72:488-498.
14. Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune-associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246.
15. Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478.
16. Gono T, Miyake K, Kawaguchi Y, et al. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology (Oxford). 2012;51:1336-1338.
17. Wada T, Abe G, Kudou, T, et al. Liver damage in patients with polymyositis and dermatomyositis. Kitasato Med Journal. 2016;46:40-46.
18. Takahashi A, Abe K, Yokokawa J, et al. Clinical features of liver dysfunction in collagen diseases. Hepatol Res. 2010;40:1092-1097.
19. Matsumoto T, Kobayashi S, Shimizu H, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000;20:366-373.
20. Shi Q, Niu J, Huang X, et al. Do muscle enzyme changes forecast liver injury in polymyositis/dermatomyositis patients treated with methylprednisolone and methotrexate? Ann Clin Lab Sci. 2016;46:266-269.
21. Noda S, Asano Y, Tamaki Z, et al. A case of dermatomyositis with “liver disease associated with rheumatoid diseases” positive for anti-liver-kidney microsome-1 antibody. Clin Rheumatol. 2010;29:941-943.
22. Nagashima T, Kamata Y, Iwamoto M, et al. Liver dysfunction in anti-melanoma differentiation-associated gene 5 antibody-positive patients with dermatomyositis. Rheumatol Int. 2019;39:901-909.
1. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-347.
2. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785.
3. Gono T, Kawaguchi Y, Satoh T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49:1713-1719.
4. Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34.
5. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. 2017;49:20-26.
6. Kumakura S, Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheum. 2014;66:2297-2307.
7. Lake M, George G, Summer R. Time to personalize the treatment of anti-MDA-5 associated lung disease. Ann Rheum Dis. 2019;78:E52.
8. Kampylafka EI, Kosmidis ML, Panagiotakos DB, et al. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol. 2012;30:397-401.
9. Koguchi-Yoshioka H, Okiyama N, Iwamoto K, et al. Intravenous immunoglobulin contributes to the control of antimelanoma differentiation-associated protein 5 antibody-associated dermatomyositis with palmar violaceous macules/papules. Br J Dermatol. 2017;177:1442-1446.
10. Hamada-Ode K, Taniguchi Y, Kimata T, et al. High-dose intravenous immunoglobulin therapy for rapidly progressive interstitial pneumonitis accompanied by anti-melanoma differentiation-associated gene 5 antibody-positive amyopathic dermatomyositis. Eur J Rheumatol. 2015;2:83-85.
11. So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989.
12. Koichi Y, Aya Y, Megumi U, et al. A case of anti-MDA5-positive rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis ameliorated by rituximab, in addition to standard immunosuppressive treatment. Mod Rheumatol. 2017;27:536-540.
13. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. 2020;72:488-498.
14. Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune-associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246.
15. Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478.
16. Gono T, Miyake K, Kawaguchi Y, et al. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology (Oxford). 2012;51:1336-1338.
17. Wada T, Abe G, Kudou, T, et al. Liver damage in patients with polymyositis and dermatomyositis. Kitasato Med Journal. 2016;46:40-46.
18. Takahashi A, Abe K, Yokokawa J, et al. Clinical features of liver dysfunction in collagen diseases. Hepatol Res. 2010;40:1092-1097.
19. Matsumoto T, Kobayashi S, Shimizu H, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000;20:366-373.
20. Shi Q, Niu J, Huang X, et al. Do muscle enzyme changes forecast liver injury in polymyositis/dermatomyositis patients treated with methylprednisolone and methotrexate? Ann Clin Lab Sci. 2016;46:266-269.
21. Noda S, Asano Y, Tamaki Z, et al. A case of dermatomyositis with “liver disease associated with rheumatoid diseases” positive for anti-liver-kidney microsome-1 antibody. Clin Rheumatol. 2010;29:941-943.
22. Nagashima T, Kamata Y, Iwamoto M, et al. Liver dysfunction in anti-melanoma differentiation-associated gene 5 antibody-positive patients with dermatomyositis. Rheumatol Int. 2019;39:901-909.
PRACTICE POINTS
- Anti-MDA5 (melanoma differentiation–associated gene 5 antibody)–positive dermatomyositis associated with hemophagocytic lymphohistiocytosis is a rare and aggressive condition associated with a poor prognosis, and there is no standard treatment.
- Dermatomyositis-associated liver injury is not well defined.
Efficacy of Etanercept in the Treatment of Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis
Regarded as dermatologic emergencies, Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) represent a spectrum of blistering skin diseases that have a high mortality rate. Because of a misguided immune response to medications or infections, CD8+ T lymphocytes release proinflammatory cytokines, giving rise to the extensive epidermal destruction seen in SJS and TEN. The exact pathogenesis of SJS and TEN is still poorly defined, but studies have proposed that T cells mediate keratinocyte (KC) apoptosis through perforin and granzyme release and activation of the Fas/Fas ligand (FasL). Functioning as a transmembrane death receptor in the tumor necrosis factor (TNF) superfamily, Fas (CD95) activates Fas-associated death domain protein, caspases, and nucleases, resulting in organized cell destruction. Likewise, perforin and granzymes also have been shown to play a similar role in apoptosis via activation of caspases.1
Evidence for the role of TNF-α in SJS and TEN has been supported by findings of elevated levels of TNF-α within the blister fluid, serum, and KC cell surface. Additionally, TNF-α has been shown to upregulate inducible nitric oxide synthase in KCs, causing an accumulation of nitric oxide and subsequent FasL-mediated cell death.1-3 Notably, studies have demonstrated a relative lack of lymphocytes in the tissue of TEN patients despite the extensive destruction that is observed, thus emphasizing the importance of amplification and cell signaling via inflammatory mediators such as TNF-α.1 In this proposed model, T cells release IFN-γ, causing KCs to release TNF-α that subsequently promotes the upregulation of the aforementioned FasL.1 Tumor necrosis factor α also may promote increased MHC class I complex deposition on KC surfaces that may play a role in perforin and granzyme-mediated apoptosis of KCs.1
There is still debate on the standard of care for the treatment of SJS and TEN, attributed to the absence of randomized controlled trials and the rarity of the disease as well as the numerous conflicting studies evaluating potential treatments.1,4 Despite conflicting data to support their use, supportive care and intravenous immunoglobulin (IVIG) continue to be common treatments for SJS and TEN in hospitals worldwide. Elucidation of the role of TNF-α has prompted the use of infliximab and etanercept. In a case series of Italian patients with TEN (average SCORTEN, 3.6) treated with the TNF-α antagonist etanercept, no mortality was observed, which was well below the calculated expected mortality of 46.9%.2 Our retrospective study compared the use of a TNF antagonist to other therapies in the treatment of SJS/TEN. Our data suggest that etanercept is a lifesaving and disease-modifying therapy.
Methods
Twenty-two patients with SJS/TEN were included in this analysis. This included all patients who carried a clinical diagnosis of SJS/TEN with a confirmatory biopsy at our 2 university centers—University of California, Los Angeles, and Keck-LA County-Norris Hospital at the University of Southern California, Los Angeles—from 2013 to 2016. The diagnosis was rendered when a clinical diagnosis of SJS/TEN was given by a dermatologist and a confirmatory biopsy was performed. Every patient given a diagnosis of SJS/TEN at either university system from 2015 onward received an injection of etanercept given the positive results reported by Paradisi et al.2
The 9 patients who presented from 2013 to 2014 to our 2 hospital systems and were given a diagnosis of SJS/TEN received either IVIG or supportive care alone and had an average body surface area (BSA) affected of 23%. The 13 patients who presented from 2015 to 2016 were treated with etanercept in the form of a 50-mg subcutaneous injection given once to the right upper arm. Of this group, 4 patients received dual therapy with both IVIG and etanercept. In the etanercept-treated group (etanercept alone and etanercept plus IVIG), the average BSA affected was 30%. At the time of preliminary diagnosis, all patient medications were evaluated for a possible temporal relationship to the onset of rash and were discontinued if felt to be causative. The causative agent and treatment course for each patient is summarized in Table 1.
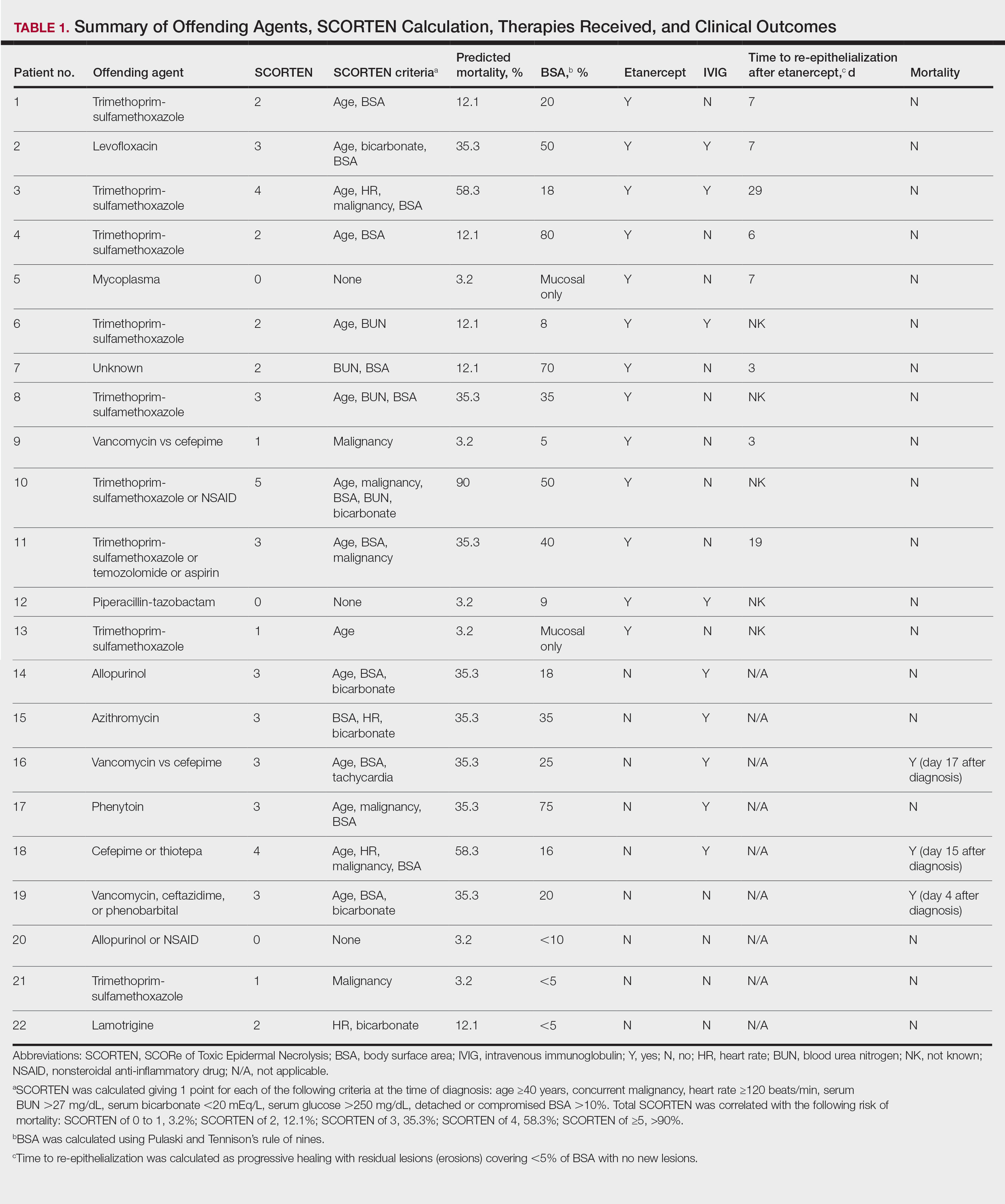
Patients were monitored daily in the hospital for improvement, and time to re-epithelialization was measured. Re-epithelialization was defined as progressive healing with residual lesions (erosions, ulcers, or bullae) covering no more than 5% BSA and was contingent on the patient having no new lesions within 24 hours.5 SCORe of Toxic Epidermal Necrosis (SCORTEN), a validated severity-of-illness score,6 was calculated by giving 1 point for each of the following criteria at the time of diagnosis: age ≥40 years, concurrent malignancy, heart rate ≥120 beats/min, serum blood urea nitrogen >27 mg/dL, serum bicarbonate <20 mEq/L, serum glucose >250 mg/dL, and detached or compromised BSA >10%. The total SCORTEN was correlated with the following risk of mortality as supported by prior validation studies: SCORTEN of 0 to 1, 3.2%; SCORTEN of 2, 12.1%; SCORTEN of 3, 35.3%; SCORTEN of 4, 58.3%; SCORTEN of ≥5, >90%.
Results
A total of 13 patients received etanercept. The mean SCORTEN was 2.2. The observed mortality was 0%, which was markedly lower than the predicted mortality of 24.3% (as determined by linear interpolation). Of this cohort, 9 patients received etanercept alone (mean SCORTEN of 2.1, predicted mortality of 22.9%), whereas 4 patients received a combination of etanercept and IVIG (mean SCORTEN of 2.3, predicted mortality of 27.2%).
The 4 patients who received both etanercept and IVIG received dual therapy for varying reasons. In patient 2 (Table 1), the perceived severity of this case ultimately led to the decision to start IVIG in addition to etanercept, resulting in rapid recovery and discharge after only 1 week of hospitalization. Intravenous immunoglobulin also was given in patient 3 (SCORTEN of 4) and patient 6 (SCORTEN of 2) for progression of disease despite administration of etanercept, with subsequent cessation of progression after the addition of the second agent (IVIG). Patient 12 might have done well on etanercept monotherapy but was administered IVIG as a precautionary measure because of hospital treatment algorithms.
Nine patients did not receive etanercept. Of this group, 5 received IVIG and 4 were managed with supportive care alone. The average SCORTEN for this group was 2.4, only slightly higher than the group that received etanercept (Table 2). The mortality rate in this group was 33%, which was higher than the predicted mortality of 28.1%.
Re-epithelialization data were available for 8 patients who received etanercept. The average time to re-epithelialization for these patients was 8.9 days and ranged from 3 to 19 days. Of these patients, 2 received both IVIG and etanercept, with an average time to re-epithelialization of 13 days. For the 6 patients who received etanercept alone, the average time to re-epithelialization was 7.5 days. Re-epithelialization data were not available for any of the patients who received only IVIG or supportive care but to our recollection ranged from 14 to 21 days.
The clinical course of the 13 patients after the administration of a single dose of etanercept was remarkable, as there was complete absence of mortality and an increase in speed of recovery in most patients receiving this intervention (time to re-epithelialization, 3–19 days). We also observed another interesting trend from our patients treated with etanercept, which was the suggestion that treatment with etanercept may be less effective if IVIG and/or steroids are given prior to etanercept; likewise, treatment is more effective when etanercept is given quickly. For patients 1, 4, 5, 7, 9, and 11 (as shown in Table 1), no prior IVIG therapy or other immunosuppressive therapy had been given before etanercept was administered. In these 6 patients, the average time to re-epithelialization after etanercept administration was 7.5 days; average time to re-epithelialization, unfortunately, is not available for the patients who were not treated with etanercept. In addition, as shown in the Figure, it was noted in some patients that the depth of denudation was markedly more superficial than what would typically be clinically observed with TEN after administration of other immunomodulatory therapies such as IVIG or prednisone or with supportive care alone. In these 2 patients with superficial desquamation—patients 7 and 9—etanercept notably was given within 6 hours of onset of skin pain.
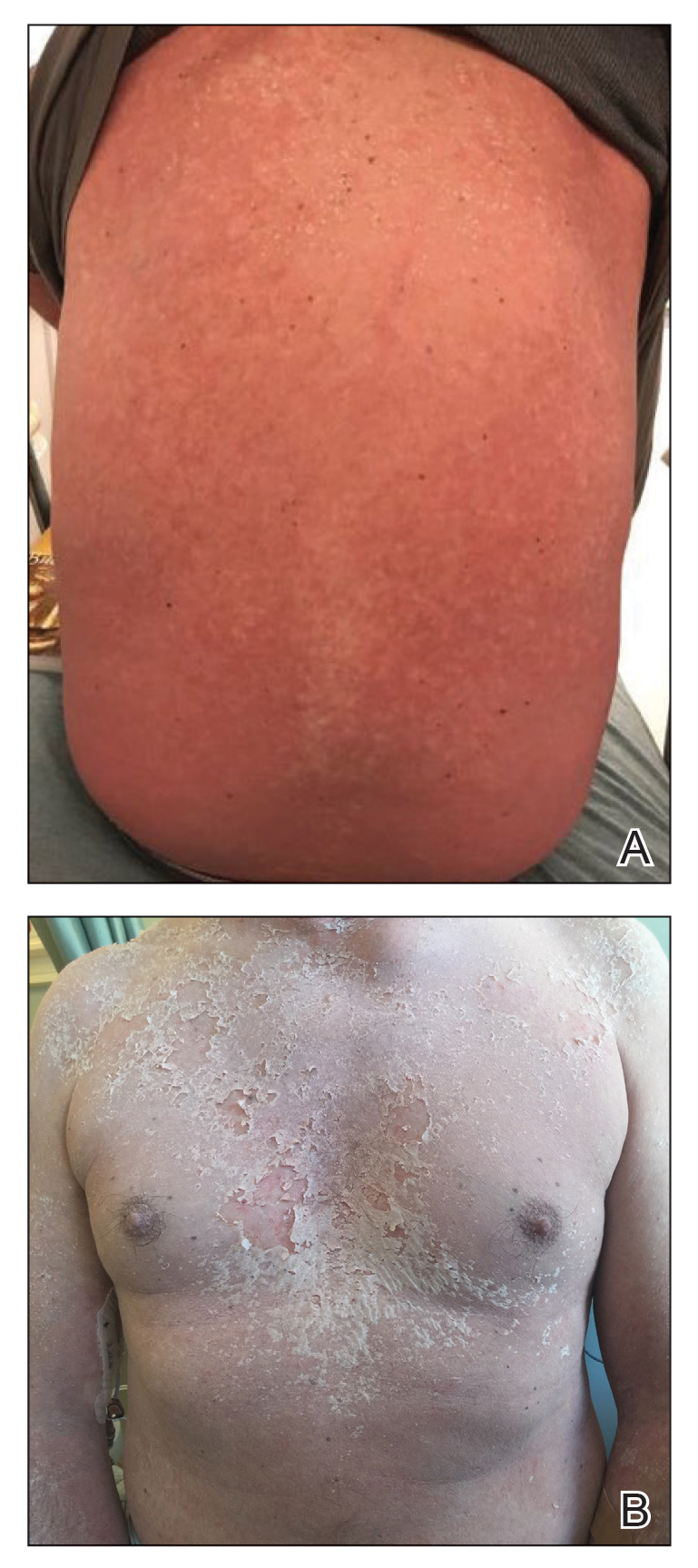
Comment
There is no definitive gold standard treatment of SJS, SJS/TEN overlap, or TEN. However, generally agreed upon management includes immediate discontinuation of the offending medication and supportive therapy with aggressive electrolyte replacement and wound care. Management in a burn unit or intensive care unit is recommended in severe cases. Contention over the efficacy of various medications in the treatment of SJS and TEN continues and largely is due to the rarity of SJS and TEN; studies are small and almost all lack randomization. Therapies that have been used include high-dose steroids, IVIG, plasmapheresis, cyclophosphamide, cyclosporine A, and TNF inhibitors (eg, etanercept, infliximab).1
Evidence for the use of anti–TNF-α antibodies has been limited thus far, with most of the literature focusing on infliximab and etanercept. Adalimumab, a fully humanized clonal antibody, has no reported cases in the dermatologic literature for use in patients with SJS/TEN. Two case reports of adalimumab paradoxically causing SJS have been documented. In both cases, adalimumab was stopped and patients responded to intravenous corticosteroids and infliximab.7,8 Similarly, thalidomide has not proven to be a promising anti–TNF-α agent for the treatment of SJS/TEN. In the only attempted randomized controlled trial for SJS and TEN, thalidomide appeared to increase mortality, eventuating in this trial being terminated prior to the planned end date.9Infliximab and etanercept have several case reports and a few case series highlighting potentially efficacious application of TNF-α inhibitors for the treatment of SJS/TEN.10-13 In 2002, Fischer et al10 reported the first case of TEN treated successfully with a single dose of infliximab 5 mg/kg. Kreft et al14 reported on etoricoxib-induced TEN that was treated with infliximab 5 mg/kg, which led to re-epithelialization within 5 weeks (notably a 5-week re-epithelialization time is not necessarily an improvement).
In 2005, Hunger et al3 demonstrated TNF-α’s release by KCs in the epidermis and by inflammatory cells in the dermis of a TEN patient. Twenty-four hours after the administration of infliximab 5 mg/kg in these patients, TNF-α was found to be below normal and epidermal detachment ceased.3 Wojtkietwicz et al13 demonstrated benefit following an infusion of infliximab 5 mg/kg in a patient whose disease continued to progress despite treatment with dexamethasone and 1.8 g/kg of IVIG.
Then 2 subsequent case series added further support for the efficacy of infliximab in the treatment of TEN. Patmanidis et al15 and Gaitanis et al16 reported similar results in 4 patients, each treated with infliximab 5 mg/kg immediately followed by initiation of high-dose IVIG (2 g/kg over 5 days). Zárate-Correa et al17 reported a 0% mortality rate and near-complete re-epithelialization after 5 to 14 days in 4 patients treated with a single 300-mg dose of infliximab.
However, the success of infliximab in the treatment of TEN has been countered by the pilot study by Paquet et al,18 which compared the efficacy of 150 mg/kg of N-acetylcysteine alone vs adding infliximab 5 mg/kg to treat 10 TEN patients. The study demonstrated no benefit at 48 hours in the group given infliximab, the time frame in which prior case reports touting infliximab’s benefit claimed the benefit was observed. Similarly, there was no effect on mortality for either treatment modality as assessed by illness auxiliary score.18
Evidence in support of the use of etanercept in the treatment of SJS/TEN is mounting, and some centers have begun to use it as the first-choice therapy for SJS/TEN. The first case was reported by Famularo et al,19 in which a patient with TEN was given 2 doses of etanercept 25 mg after failure to improve with prednisolone 1 mg/kg. The patient showed near-complete and rapid re-epithelization in 6 days before death due to disseminated intravascular coagulation 10 days after admission.19 Gubinelli et al20 and Sadighha21 independently reported cases of TEN and TEN/acute generalized exanthematous pustulosis overlap treated with a total of 50 mg of etanercept, demonstrating rapid cessation of lesion progression. Didona et al22 found similar benefit using etanercept 50 mg to treat TEN secondary to rituximab after failure to improve with prednisone and cyclophosphamide. Treatment of TEN with etanercept in an HIV-positive patient also has been reported. Lee et al23 described a patient who was administered 50-mg and 25-mg injections on days 3 and 5 of hospitalization, respectively, with re-epithelialization occurring by day 8. Finally, Owczarczyk-Saczonek et al24 reported a case of SJS in a patient with a 4-year history of etanercept and sulfasalazine treatment of rheumatoid arthritis; sulfasalazine was stopped, but this patient was continued on etanercept until resolution of skin and mucosal symptoms. However, it is important to consider the possibility of publication bias among these cases selected for their positive outcomes.
Perhaps the most compelling literature regarding the use of etanercept for TEN was described in a case series by Paradisi et al.2 This study included 10 patients with TEN, all of whom demonstrated complete re-epithelialization shortly after receiving etanercept 50 mg. Average SCORTEN was 3.6 with a range of 2 to 6. Eight patients in this study had severe comorbidities and all 10 patients survived, with a time to re-epithelialization ranging from 7 to 20 days.2 Additionally, a randomized controlled trial showed that 38 etanercept-treated patients had improved mortality (P=.266) and re-epithelialization time (P=.01) compared to patients treated with intravenous methylprednisolone.25Limitations to our study are similar to other reports of SJS/TEN and included the small number of cases and lack of randomization. Additionally, we do not have data available for all patients for time between onset of disease and treatment initiation. Because of these challenges, data presented in this case series is observational only. Additionally, the patients treated with etanercept alone had a slightly lower SCORTEN compared to the group that received IVIG or supportive care alone (2.1 and 2.4 respectively). However, the etanercept-only group actually had higher involvement of epidermal detachment (33%) compared to the non-etanercept group (23%).
Conclusion
Although treatment with etanercept lacks the support of a randomized controlled trial, similar to all other treatments currently used for SJS and TEN, preliminary reports highlight a benefit in disease progression and improvement in time to re-epithelialization. In particular, if etanercept 50 mg subcutaneously is given as monotherapy or is given early in the disease course (prior to other therapies being attempted and ideally within 6 hours of presentation), our data suggest an even greater trend toward improved mortality and decreased time to re-epithelialization. Additionally, our findings may suggest that in some patients, etanercept monotherapy is not an adequate intervention but the addition of IVIG may be helpful; however, the senior author (S.W.) notes anecdotally that in his experience with the patients treated at the University of California Los Angeles, the order of administration of combination therapies—etanercept followed by IVIG—was important in addition to the choice of therapy. These findings are promising enough to warrant a multicenter randomized controlled trial comparing the efficacy of etanercept to other more commonly used treatments for this spectrum of disease, including IVIG and/or cyclosporine. Based on the data presented in this case series, including the 13 patients who received etanercept and had a 0% mortality rate, etanercept may be viewed as a targeted therapeutic intervention for patients with SJS and TEN.
- Pereira FA, Mudgil AV, Rosmarin DM. Toxic epidermal necrolysis. J Am Acad Dermatol. 2007;56:181-200.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Hunger RE, Hunziker T, Buettiker U, et al. Rapid resolution of toxic epidermal necrolysis with anti-TNF-α treatment. J Allergy Clin Immunol. 2005;116:923-924.
- Worswick S, Cotliar J. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of treatment options. Dermatol Ther. 2011;24:207-218.
- Wallace AB. The exposure treatment of burns. Lancet Lond Engl. 1951;1:501-504.
- Bastuji-Garin S, Fouchard N, Bertocchi M, et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149-153.
- Mounach A, Rezqi A, Nouijai A, et al. Stevens-Johnson syndrome complicating adalimumab therapy in rheumatoid arthritis disease. Rheumatol Int. 2013;33:1351-1353.
- Salama M, Lawrance I-C. Stevens-Johnson syndrome complicating adalimumab therapy in Crohn’s disease. World J Gastroenterol. 2009;15:4449-4452.
- Wolkenstein P, Latarjet J, Roujeau JC, et al. Randomised comparison of thalidomide versus placebo in toxic epidermal necrolysis. Lancet Lond Engl. 1998;352:1586-1589.
- Fischer M, Fiedler E, Marsch WC, et al Antitumour necrosis factor-α antibodies (infliximab) in the treatment of a patient with toxic epidermal necrolysis. Br J Dermatol. 2002;146:707-709.
- Meiss F, Helmbold P, Meykadeh N, et al. Overlap of acute generalized exanthematous pustulosis and toxic epidermal necrolysis: response to antitumour necrosis factor-alpha antibody infliximab: report of three cases. J Eur Acad Dermatol Venereol. 2007;21:717-719.
- Al-Shouli S, Abouchala N, Bogusz MJ, et al. Toxic epidermal necrolysis associated with high intake of sildenafil and its response to infliximab. Acta Derm Venereol. 2005;85:534-535.
- Wojtkiewicz A, Wysocki M, Fortuna J, et al. Beneficial and rapid effect of infliximab on the course of toxic epidermal necrolysis. Acta Derm Venereol. 2008;88:420-421.
- Kreft B, Wohlrab J, Bramsiepe I, et al. Etoricoxib-induced toxic epidermal necrolysis: successful treatment with infliximab. J Dermatol. 2010;37:904-906.
- Patmanidis K, Sidiras A, Dolianitis K, et al. Combination of infliximab and high-dose intravenous immunoglobulin for toxic epidermal necrolysis: successful treatment of an elderly patient. Case Rep Dermatol Med. 2012;2012:915314.
- Gaitanis G, Spyridonos P, Patmanidis K, et al. Treatment of toxic epidermal necrolysis with the combination of infliximab and high-dose intravenous immunoglobulin. Dermatol Basel Switz. 2012;224:134-139.
- Zárate-Correa LC, Carrillo-Gómez DC, Ramírez-Escobar AF, et al. Toxic epidermal necrolysis successfully treated with infliximab. J Investig Allergol Clin Immunol. 2013;23:61-63.
- Paquet P, Jennes S, Rousseau AF, et al. Effect of N-acetylcysteine combined with infliximab on toxic epidermal necrolysis. a proof-of-concept study. Burns J Int Soc Burn Inj. 2014;40:1707-1712.
- Famularo G, Dona BD, Canzona F, et al. Etanercept for toxic epidermal necrolysis. Ann Pharmacother. 2007;41:1083-1084.
- Gubinelli E, Canzona F, Tonanzi T, et al. Toxic epidermal necrolysis successfully treated with etanercept. J Dermatol. 2009;36:150-153.
- Sadighha A. Etanercept in the treatment of a patient with acute generalized exanthematous pustulosis/toxic epidermal necrolysis: definition of a new model based on translational research. Int J Dermatol. 2009;48:913-914.
- Didona D, Paolino G, Garcovich S, et al. Successful use of etanercept in a case of toxic epidermal necrolysis induced by rituximab. J Eur Acad Dermatol Venereol. 2016;30:E83-E84.
- Lee Y-Y, Ko J-H, Wei C-H, et al. Use of etanercept to treat toxic epidermal necrolysis in a human immunodeficiency virus-positive patient. Dermatol Sin. 2013;31:78-81.
- Owczarczyk-Saczonek A, Zdanowska N, Znajewska-Pander A, et al. Stevens-Johnson syndrome in a patient with rheumatoid arthritis during long-term etanercept therapy. J Dermatol Case Rep. 2016;10:14-16.
- Wang CW, Yang LY, Chen CB, et al. Randomized, controlled trial of TNF-α antagonist in CTL mediated severe cutaneous adverse reactions. J Clin Invest. 2018;128:985-996.
Regarded as dermatologic emergencies, Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) represent a spectrum of blistering skin diseases that have a high mortality rate. Because of a misguided immune response to medications or infections, CD8+ T lymphocytes release proinflammatory cytokines, giving rise to the extensive epidermal destruction seen in SJS and TEN. The exact pathogenesis of SJS and TEN is still poorly defined, but studies have proposed that T cells mediate keratinocyte (KC) apoptosis through perforin and granzyme release and activation of the Fas/Fas ligand (FasL). Functioning as a transmembrane death receptor in the tumor necrosis factor (TNF) superfamily, Fas (CD95) activates Fas-associated death domain protein, caspases, and nucleases, resulting in organized cell destruction. Likewise, perforin and granzymes also have been shown to play a similar role in apoptosis via activation of caspases.1
Evidence for the role of TNF-α in SJS and TEN has been supported by findings of elevated levels of TNF-α within the blister fluid, serum, and KC cell surface. Additionally, TNF-α has been shown to upregulate inducible nitric oxide synthase in KCs, causing an accumulation of nitric oxide and subsequent FasL-mediated cell death.1-3 Notably, studies have demonstrated a relative lack of lymphocytes in the tissue of TEN patients despite the extensive destruction that is observed, thus emphasizing the importance of amplification and cell signaling via inflammatory mediators such as TNF-α.1 In this proposed model, T cells release IFN-γ, causing KCs to release TNF-α that subsequently promotes the upregulation of the aforementioned FasL.1 Tumor necrosis factor α also may promote increased MHC class I complex deposition on KC surfaces that may play a role in perforin and granzyme-mediated apoptosis of KCs.1
There is still debate on the standard of care for the treatment of SJS and TEN, attributed to the absence of randomized controlled trials and the rarity of the disease as well as the numerous conflicting studies evaluating potential treatments.1,4 Despite conflicting data to support their use, supportive care and intravenous immunoglobulin (IVIG) continue to be common treatments for SJS and TEN in hospitals worldwide. Elucidation of the role of TNF-α has prompted the use of infliximab and etanercept. In a case series of Italian patients with TEN (average SCORTEN, 3.6) treated with the TNF-α antagonist etanercept, no mortality was observed, which was well below the calculated expected mortality of 46.9%.2 Our retrospective study compared the use of a TNF antagonist to other therapies in the treatment of SJS/TEN. Our data suggest that etanercept is a lifesaving and disease-modifying therapy.
Methods
Twenty-two patients with SJS/TEN were included in this analysis. This included all patients who carried a clinical diagnosis of SJS/TEN with a confirmatory biopsy at our 2 university centers—University of California, Los Angeles, and Keck-LA County-Norris Hospital at the University of Southern California, Los Angeles—from 2013 to 2016. The diagnosis was rendered when a clinical diagnosis of SJS/TEN was given by a dermatologist and a confirmatory biopsy was performed. Every patient given a diagnosis of SJS/TEN at either university system from 2015 onward received an injection of etanercept given the positive results reported by Paradisi et al.2
The 9 patients who presented from 2013 to 2014 to our 2 hospital systems and were given a diagnosis of SJS/TEN received either IVIG or supportive care alone and had an average body surface area (BSA) affected of 23%. The 13 patients who presented from 2015 to 2016 were treated with etanercept in the form of a 50-mg subcutaneous injection given once to the right upper arm. Of this group, 4 patients received dual therapy with both IVIG and etanercept. In the etanercept-treated group (etanercept alone and etanercept plus IVIG), the average BSA affected was 30%. At the time of preliminary diagnosis, all patient medications were evaluated for a possible temporal relationship to the onset of rash and were discontinued if felt to be causative. The causative agent and treatment course for each patient is summarized in Table 1.
Patients were monitored daily in the hospital for improvement, and time to re-epithelialization was measured. Re-epithelialization was defined as progressive healing with residual lesions (erosions, ulcers, or bullae) covering no more than 5% BSA and was contingent on the patient having no new lesions within 24 hours.5 SCORe of Toxic Epidermal Necrosis (SCORTEN), a validated severity-of-illness score,6 was calculated by giving 1 point for each of the following criteria at the time of diagnosis: age ≥40 years, concurrent malignancy, heart rate ≥120 beats/min, serum blood urea nitrogen >27 mg/dL, serum bicarbonate <20 mEq/L, serum glucose >250 mg/dL, and detached or compromised BSA >10%. The total SCORTEN was correlated with the following risk of mortality as supported by prior validation studies: SCORTEN of 0 to 1, 3.2%; SCORTEN of 2, 12.1%; SCORTEN of 3, 35.3%; SCORTEN of 4, 58.3%; SCORTEN of ≥5, >90%.
Results
A total of 13 patients received etanercept. The mean SCORTEN was 2.2. The observed mortality was 0%, which was markedly lower than the predicted mortality of 24.3% (as determined by linear interpolation). Of this cohort, 9 patients received etanercept alone (mean SCORTEN of 2.1, predicted mortality of 22.9%), whereas 4 patients received a combination of etanercept and IVIG (mean SCORTEN of 2.3, predicted mortality of 27.2%).
The 4 patients who received both etanercept and IVIG received dual therapy for varying reasons. In patient 2 (Table 1), the perceived severity of this case ultimately led to the decision to start IVIG in addition to etanercept, resulting in rapid recovery and discharge after only 1 week of hospitalization. Intravenous immunoglobulin also was given in patient 3 (SCORTEN of 4) and patient 6 (SCORTEN of 2) for progression of disease despite administration of etanercept, with subsequent cessation of progression after the addition of the second agent (IVIG). Patient 12 might have done well on etanercept monotherapy but was administered IVIG as a precautionary measure because of hospital treatment algorithms.
Nine patients did not receive etanercept. Of this group, 5 received IVIG and 4 were managed with supportive care alone. The average SCORTEN for this group was 2.4, only slightly higher than the group that received etanercept (Table 2). The mortality rate in this group was 33%, which was higher than the predicted mortality of 28.1%.
Re-epithelialization data were available for 8 patients who received etanercept. The average time to re-epithelialization for these patients was 8.9 days and ranged from 3 to 19 days. Of these patients, 2 received both IVIG and etanercept, with an average time to re-epithelialization of 13 days. For the 6 patients who received etanercept alone, the average time to re-epithelialization was 7.5 days. Re-epithelialization data were not available for any of the patients who received only IVIG or supportive care but to our recollection ranged from 14 to 21 days.
The clinical course of the 13 patients after the administration of a single dose of etanercept was remarkable, as there was complete absence of mortality and an increase in speed of recovery in most patients receiving this intervention (time to re-epithelialization, 3–19 days). We also observed another interesting trend from our patients treated with etanercept, which was the suggestion that treatment with etanercept may be less effective if IVIG and/or steroids are given prior to etanercept; likewise, treatment is more effective when etanercept is given quickly. For patients 1, 4, 5, 7, 9, and 11 (as shown in Table 1), no prior IVIG therapy or other immunosuppressive therapy had been given before etanercept was administered. In these 6 patients, the average time to re-epithelialization after etanercept administration was 7.5 days; average time to re-epithelialization, unfortunately, is not available for the patients who were not treated with etanercept. In addition, as shown in the Figure, it was noted in some patients that the depth of denudation was markedly more superficial than what would typically be clinically observed with TEN after administration of other immunomodulatory therapies such as IVIG or prednisone or with supportive care alone. In these 2 patients with superficial desquamation—patients 7 and 9—etanercept notably was given within 6 hours of onset of skin pain.
Comment
There is no definitive gold standard treatment of SJS, SJS/TEN overlap, or TEN. However, generally agreed upon management includes immediate discontinuation of the offending medication and supportive therapy with aggressive electrolyte replacement and wound care. Management in a burn unit or intensive care unit is recommended in severe cases. Contention over the efficacy of various medications in the treatment of SJS and TEN continues and largely is due to the rarity of SJS and TEN; studies are small and almost all lack randomization. Therapies that have been used include high-dose steroids, IVIG, plasmapheresis, cyclophosphamide, cyclosporine A, and TNF inhibitors (eg, etanercept, infliximab).1
Evidence for the use of anti–TNF-α antibodies has been limited thus far, with most of the literature focusing on infliximab and etanercept. Adalimumab, a fully humanized clonal antibody, has no reported cases in the dermatologic literature for use in patients with SJS/TEN. Two case reports of adalimumab paradoxically causing SJS have been documented. In both cases, adalimumab was stopped and patients responded to intravenous corticosteroids and infliximab.7,8 Similarly, thalidomide has not proven to be a promising anti–TNF-α agent for the treatment of SJS/TEN. In the only attempted randomized controlled trial for SJS and TEN, thalidomide appeared to increase mortality, eventuating in this trial being terminated prior to the planned end date.9Infliximab and etanercept have several case reports and a few case series highlighting potentially efficacious application of TNF-α inhibitors for the treatment of SJS/TEN.10-13 In 2002, Fischer et al10 reported the first case of TEN treated successfully with a single dose of infliximab 5 mg/kg. Kreft et al14 reported on etoricoxib-induced TEN that was treated with infliximab 5 mg/kg, which led to re-epithelialization within 5 weeks (notably a 5-week re-epithelialization time is not necessarily an improvement).
In 2005, Hunger et al3 demonstrated TNF-α’s release by KCs in the epidermis and by inflammatory cells in the dermis of a TEN patient. Twenty-four hours after the administration of infliximab 5 mg/kg in these patients, TNF-α was found to be below normal and epidermal detachment ceased.3 Wojtkietwicz et al13 demonstrated benefit following an infusion of infliximab 5 mg/kg in a patient whose disease continued to progress despite treatment with dexamethasone and 1.8 g/kg of IVIG.
Then 2 subsequent case series added further support for the efficacy of infliximab in the treatment of TEN. Patmanidis et al15 and Gaitanis et al16 reported similar results in 4 patients, each treated with infliximab 5 mg/kg immediately followed by initiation of high-dose IVIG (2 g/kg over 5 days). Zárate-Correa et al17 reported a 0% mortality rate and near-complete re-epithelialization after 5 to 14 days in 4 patients treated with a single 300-mg dose of infliximab.
However, the success of infliximab in the treatment of TEN has been countered by the pilot study by Paquet et al,18 which compared the efficacy of 150 mg/kg of N-acetylcysteine alone vs adding infliximab 5 mg/kg to treat 10 TEN patients. The study demonstrated no benefit at 48 hours in the group given infliximab, the time frame in which prior case reports touting infliximab’s benefit claimed the benefit was observed. Similarly, there was no effect on mortality for either treatment modality as assessed by illness auxiliary score.18
Evidence in support of the use of etanercept in the treatment of SJS/TEN is mounting, and some centers have begun to use it as the first-choice therapy for SJS/TEN. The first case was reported by Famularo et al,19 in which a patient with TEN was given 2 doses of etanercept 25 mg after failure to improve with prednisolone 1 mg/kg. The patient showed near-complete and rapid re-epithelization in 6 days before death due to disseminated intravascular coagulation 10 days after admission.19 Gubinelli et al20 and Sadighha21 independently reported cases of TEN and TEN/acute generalized exanthematous pustulosis overlap treated with a total of 50 mg of etanercept, demonstrating rapid cessation of lesion progression. Didona et al22 found similar benefit using etanercept 50 mg to treat TEN secondary to rituximab after failure to improve with prednisone and cyclophosphamide. Treatment of TEN with etanercept in an HIV-positive patient also has been reported. Lee et al23 described a patient who was administered 50-mg and 25-mg injections on days 3 and 5 of hospitalization, respectively, with re-epithelialization occurring by day 8. Finally, Owczarczyk-Saczonek et al24 reported a case of SJS in a patient with a 4-year history of etanercept and sulfasalazine treatment of rheumatoid arthritis; sulfasalazine was stopped, but this patient was continued on etanercept until resolution of skin and mucosal symptoms. However, it is important to consider the possibility of publication bias among these cases selected for their positive outcomes.
Perhaps the most compelling literature regarding the use of etanercept for TEN was described in a case series by Paradisi et al.2 This study included 10 patients with TEN, all of whom demonstrated complete re-epithelialization shortly after receiving etanercept 50 mg. Average SCORTEN was 3.6 with a range of 2 to 6. Eight patients in this study had severe comorbidities and all 10 patients survived, with a time to re-epithelialization ranging from 7 to 20 days.2 Additionally, a randomized controlled trial showed that 38 etanercept-treated patients had improved mortality (P=.266) and re-epithelialization time (P=.01) compared to patients treated with intravenous methylprednisolone.25Limitations to our study are similar to other reports of SJS/TEN and included the small number of cases and lack of randomization. Additionally, we do not have data available for all patients for time between onset of disease and treatment initiation. Because of these challenges, data presented in this case series is observational only. Additionally, the patients treated with etanercept alone had a slightly lower SCORTEN compared to the group that received IVIG or supportive care alone (2.1 and 2.4 respectively). However, the etanercept-only group actually had higher involvement of epidermal detachment (33%) compared to the non-etanercept group (23%).
Conclusion
Although treatment with etanercept lacks the support of a randomized controlled trial, similar to all other treatments currently used for SJS and TEN, preliminary reports highlight a benefit in disease progression and improvement in time to re-epithelialization. In particular, if etanercept 50 mg subcutaneously is given as monotherapy or is given early in the disease course (prior to other therapies being attempted and ideally within 6 hours of presentation), our data suggest an even greater trend toward improved mortality and decreased time to re-epithelialization. Additionally, our findings may suggest that in some patients, etanercept monotherapy is not an adequate intervention but the addition of IVIG may be helpful; however, the senior author (S.W.) notes anecdotally that in his experience with the patients treated at the University of California Los Angeles, the order of administration of combination therapies—etanercept followed by IVIG—was important in addition to the choice of therapy. These findings are promising enough to warrant a multicenter randomized controlled trial comparing the efficacy of etanercept to other more commonly used treatments for this spectrum of disease, including IVIG and/or cyclosporine. Based on the data presented in this case series, including the 13 patients who received etanercept and had a 0% mortality rate, etanercept may be viewed as a targeted therapeutic intervention for patients with SJS and TEN.
Regarded as dermatologic emergencies, Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) represent a spectrum of blistering skin diseases that have a high mortality rate. Because of a misguided immune response to medications or infections, CD8+ T lymphocytes release proinflammatory cytokines, giving rise to the extensive epidermal destruction seen in SJS and TEN. The exact pathogenesis of SJS and TEN is still poorly defined, but studies have proposed that T cells mediate keratinocyte (KC) apoptosis through perforin and granzyme release and activation of the Fas/Fas ligand (FasL). Functioning as a transmembrane death receptor in the tumor necrosis factor (TNF) superfamily, Fas (CD95) activates Fas-associated death domain protein, caspases, and nucleases, resulting in organized cell destruction. Likewise, perforin and granzymes also have been shown to play a similar role in apoptosis via activation of caspases.1
Evidence for the role of TNF-α in SJS and TEN has been supported by findings of elevated levels of TNF-α within the blister fluid, serum, and KC cell surface. Additionally, TNF-α has been shown to upregulate inducible nitric oxide synthase in KCs, causing an accumulation of nitric oxide and subsequent FasL-mediated cell death.1-3 Notably, studies have demonstrated a relative lack of lymphocytes in the tissue of TEN patients despite the extensive destruction that is observed, thus emphasizing the importance of amplification and cell signaling via inflammatory mediators such as TNF-α.1 In this proposed model, T cells release IFN-γ, causing KCs to release TNF-α that subsequently promotes the upregulation of the aforementioned FasL.1 Tumor necrosis factor α also may promote increased MHC class I complex deposition on KC surfaces that may play a role in perforin and granzyme-mediated apoptosis of KCs.1
There is still debate on the standard of care for the treatment of SJS and TEN, attributed to the absence of randomized controlled trials and the rarity of the disease as well as the numerous conflicting studies evaluating potential treatments.1,4 Despite conflicting data to support their use, supportive care and intravenous immunoglobulin (IVIG) continue to be common treatments for SJS and TEN in hospitals worldwide. Elucidation of the role of TNF-α has prompted the use of infliximab and etanercept. In a case series of Italian patients with TEN (average SCORTEN, 3.6) treated with the TNF-α antagonist etanercept, no mortality was observed, which was well below the calculated expected mortality of 46.9%.2 Our retrospective study compared the use of a TNF antagonist to other therapies in the treatment of SJS/TEN. Our data suggest that etanercept is a lifesaving and disease-modifying therapy.
Methods
Twenty-two patients with SJS/TEN were included in this analysis. This included all patients who carried a clinical diagnosis of SJS/TEN with a confirmatory biopsy at our 2 university centers—University of California, Los Angeles, and Keck-LA County-Norris Hospital at the University of Southern California, Los Angeles—from 2013 to 2016. The diagnosis was rendered when a clinical diagnosis of SJS/TEN was given by a dermatologist and a confirmatory biopsy was performed. Every patient given a diagnosis of SJS/TEN at either university system from 2015 onward received an injection of etanercept given the positive results reported by Paradisi et al.2
The 9 patients who presented from 2013 to 2014 to our 2 hospital systems and were given a diagnosis of SJS/TEN received either IVIG or supportive care alone and had an average body surface area (BSA) affected of 23%. The 13 patients who presented from 2015 to 2016 were treated with etanercept in the form of a 50-mg subcutaneous injection given once to the right upper arm. Of this group, 4 patients received dual therapy with both IVIG and etanercept. In the etanercept-treated group (etanercept alone and etanercept plus IVIG), the average BSA affected was 30%. At the time of preliminary diagnosis, all patient medications were evaluated for a possible temporal relationship to the onset of rash and were discontinued if felt to be causative. The causative agent and treatment course for each patient is summarized in Table 1.
Patients were monitored daily in the hospital for improvement, and time to re-epithelialization was measured. Re-epithelialization was defined as progressive healing with residual lesions (erosions, ulcers, or bullae) covering no more than 5% BSA and was contingent on the patient having no new lesions within 24 hours.5 SCORe of Toxic Epidermal Necrosis (SCORTEN), a validated severity-of-illness score,6 was calculated by giving 1 point for each of the following criteria at the time of diagnosis: age ≥40 years, concurrent malignancy, heart rate ≥120 beats/min, serum blood urea nitrogen >27 mg/dL, serum bicarbonate <20 mEq/L, serum glucose >250 mg/dL, and detached or compromised BSA >10%. The total SCORTEN was correlated with the following risk of mortality as supported by prior validation studies: SCORTEN of 0 to 1, 3.2%; SCORTEN of 2, 12.1%; SCORTEN of 3, 35.3%; SCORTEN of 4, 58.3%; SCORTEN of ≥5, >90%.
Results
A total of 13 patients received etanercept. The mean SCORTEN was 2.2. The observed mortality was 0%, which was markedly lower than the predicted mortality of 24.3% (as determined by linear interpolation). Of this cohort, 9 patients received etanercept alone (mean SCORTEN of 2.1, predicted mortality of 22.9%), whereas 4 patients received a combination of etanercept and IVIG (mean SCORTEN of 2.3, predicted mortality of 27.2%).
The 4 patients who received both etanercept and IVIG received dual therapy for varying reasons. In patient 2 (Table 1), the perceived severity of this case ultimately led to the decision to start IVIG in addition to etanercept, resulting in rapid recovery and discharge after only 1 week of hospitalization. Intravenous immunoglobulin also was given in patient 3 (SCORTEN of 4) and patient 6 (SCORTEN of 2) for progression of disease despite administration of etanercept, with subsequent cessation of progression after the addition of the second agent (IVIG). Patient 12 might have done well on etanercept monotherapy but was administered IVIG as a precautionary measure because of hospital treatment algorithms.
Nine patients did not receive etanercept. Of this group, 5 received IVIG and 4 were managed with supportive care alone. The average SCORTEN for this group was 2.4, only slightly higher than the group that received etanercept (Table 2). The mortality rate in this group was 33%, which was higher than the predicted mortality of 28.1%.
Re-epithelialization data were available for 8 patients who received etanercept. The average time to re-epithelialization for these patients was 8.9 days and ranged from 3 to 19 days. Of these patients, 2 received both IVIG and etanercept, with an average time to re-epithelialization of 13 days. For the 6 patients who received etanercept alone, the average time to re-epithelialization was 7.5 days. Re-epithelialization data were not available for any of the patients who received only IVIG or supportive care but to our recollection ranged from 14 to 21 days.
The clinical course of the 13 patients after the administration of a single dose of etanercept was remarkable, as there was complete absence of mortality and an increase in speed of recovery in most patients receiving this intervention (time to re-epithelialization, 3–19 days). We also observed another interesting trend from our patients treated with etanercept, which was the suggestion that treatment with etanercept may be less effective if IVIG and/or steroids are given prior to etanercept; likewise, treatment is more effective when etanercept is given quickly. For patients 1, 4, 5, 7, 9, and 11 (as shown in Table 1), no prior IVIG therapy or other immunosuppressive therapy had been given before etanercept was administered. In these 6 patients, the average time to re-epithelialization after etanercept administration was 7.5 days; average time to re-epithelialization, unfortunately, is not available for the patients who were not treated with etanercept. In addition, as shown in the Figure, it was noted in some patients that the depth of denudation was markedly more superficial than what would typically be clinically observed with TEN after administration of other immunomodulatory therapies such as IVIG or prednisone or with supportive care alone. In these 2 patients with superficial desquamation—patients 7 and 9—etanercept notably was given within 6 hours of onset of skin pain.
Comment
There is no definitive gold standard treatment of SJS, SJS/TEN overlap, or TEN. However, generally agreed upon management includes immediate discontinuation of the offending medication and supportive therapy with aggressive electrolyte replacement and wound care. Management in a burn unit or intensive care unit is recommended in severe cases. Contention over the efficacy of various medications in the treatment of SJS and TEN continues and largely is due to the rarity of SJS and TEN; studies are small and almost all lack randomization. Therapies that have been used include high-dose steroids, IVIG, plasmapheresis, cyclophosphamide, cyclosporine A, and TNF inhibitors (eg, etanercept, infliximab).1
Evidence for the use of anti–TNF-α antibodies has been limited thus far, with most of the literature focusing on infliximab and etanercept. Adalimumab, a fully humanized clonal antibody, has no reported cases in the dermatologic literature for use in patients with SJS/TEN. Two case reports of adalimumab paradoxically causing SJS have been documented. In both cases, adalimumab was stopped and patients responded to intravenous corticosteroids and infliximab.7,8 Similarly, thalidomide has not proven to be a promising anti–TNF-α agent for the treatment of SJS/TEN. In the only attempted randomized controlled trial for SJS and TEN, thalidomide appeared to increase mortality, eventuating in this trial being terminated prior to the planned end date.9Infliximab and etanercept have several case reports and a few case series highlighting potentially efficacious application of TNF-α inhibitors for the treatment of SJS/TEN.10-13 In 2002, Fischer et al10 reported the first case of TEN treated successfully with a single dose of infliximab 5 mg/kg. Kreft et al14 reported on etoricoxib-induced TEN that was treated with infliximab 5 mg/kg, which led to re-epithelialization within 5 weeks (notably a 5-week re-epithelialization time is not necessarily an improvement).
In 2005, Hunger et al3 demonstrated TNF-α’s release by KCs in the epidermis and by inflammatory cells in the dermis of a TEN patient. Twenty-four hours after the administration of infliximab 5 mg/kg in these patients, TNF-α was found to be below normal and epidermal detachment ceased.3 Wojtkietwicz et al13 demonstrated benefit following an infusion of infliximab 5 mg/kg in a patient whose disease continued to progress despite treatment with dexamethasone and 1.8 g/kg of IVIG.
Then 2 subsequent case series added further support for the efficacy of infliximab in the treatment of TEN. Patmanidis et al15 and Gaitanis et al16 reported similar results in 4 patients, each treated with infliximab 5 mg/kg immediately followed by initiation of high-dose IVIG (2 g/kg over 5 days). Zárate-Correa et al17 reported a 0% mortality rate and near-complete re-epithelialization after 5 to 14 days in 4 patients treated with a single 300-mg dose of infliximab.
However, the success of infliximab in the treatment of TEN has been countered by the pilot study by Paquet et al,18 which compared the efficacy of 150 mg/kg of N-acetylcysteine alone vs adding infliximab 5 mg/kg to treat 10 TEN patients. The study demonstrated no benefit at 48 hours in the group given infliximab, the time frame in which prior case reports touting infliximab’s benefit claimed the benefit was observed. Similarly, there was no effect on mortality for either treatment modality as assessed by illness auxiliary score.18
Evidence in support of the use of etanercept in the treatment of SJS/TEN is mounting, and some centers have begun to use it as the first-choice therapy for SJS/TEN. The first case was reported by Famularo et al,19 in which a patient with TEN was given 2 doses of etanercept 25 mg after failure to improve with prednisolone 1 mg/kg. The patient showed near-complete and rapid re-epithelization in 6 days before death due to disseminated intravascular coagulation 10 days after admission.19 Gubinelli et al20 and Sadighha21 independently reported cases of TEN and TEN/acute generalized exanthematous pustulosis overlap treated with a total of 50 mg of etanercept, demonstrating rapid cessation of lesion progression. Didona et al22 found similar benefit using etanercept 50 mg to treat TEN secondary to rituximab after failure to improve with prednisone and cyclophosphamide. Treatment of TEN with etanercept in an HIV-positive patient also has been reported. Lee et al23 described a patient who was administered 50-mg and 25-mg injections on days 3 and 5 of hospitalization, respectively, with re-epithelialization occurring by day 8. Finally, Owczarczyk-Saczonek et al24 reported a case of SJS in a patient with a 4-year history of etanercept and sulfasalazine treatment of rheumatoid arthritis; sulfasalazine was stopped, but this patient was continued on etanercept until resolution of skin and mucosal symptoms. However, it is important to consider the possibility of publication bias among these cases selected for their positive outcomes.
Perhaps the most compelling literature regarding the use of etanercept for TEN was described in a case series by Paradisi et al.2 This study included 10 patients with TEN, all of whom demonstrated complete re-epithelialization shortly after receiving etanercept 50 mg. Average SCORTEN was 3.6 with a range of 2 to 6. Eight patients in this study had severe comorbidities and all 10 patients survived, with a time to re-epithelialization ranging from 7 to 20 days.2 Additionally, a randomized controlled trial showed that 38 etanercept-treated patients had improved mortality (P=.266) and re-epithelialization time (P=.01) compared to patients treated with intravenous methylprednisolone.25Limitations to our study are similar to other reports of SJS/TEN and included the small number of cases and lack of randomization. Additionally, we do not have data available for all patients for time between onset of disease and treatment initiation. Because of these challenges, data presented in this case series is observational only. Additionally, the patients treated with etanercept alone had a slightly lower SCORTEN compared to the group that received IVIG or supportive care alone (2.1 and 2.4 respectively). However, the etanercept-only group actually had higher involvement of epidermal detachment (33%) compared to the non-etanercept group (23%).
Conclusion
Although treatment with etanercept lacks the support of a randomized controlled trial, similar to all other treatments currently used for SJS and TEN, preliminary reports highlight a benefit in disease progression and improvement in time to re-epithelialization. In particular, if etanercept 50 mg subcutaneously is given as monotherapy or is given early in the disease course (prior to other therapies being attempted and ideally within 6 hours of presentation), our data suggest an even greater trend toward improved mortality and decreased time to re-epithelialization. Additionally, our findings may suggest that in some patients, etanercept monotherapy is not an adequate intervention but the addition of IVIG may be helpful; however, the senior author (S.W.) notes anecdotally that in his experience with the patients treated at the University of California Los Angeles, the order of administration of combination therapies—etanercept followed by IVIG—was important in addition to the choice of therapy. These findings are promising enough to warrant a multicenter randomized controlled trial comparing the efficacy of etanercept to other more commonly used treatments for this spectrum of disease, including IVIG and/or cyclosporine. Based on the data presented in this case series, including the 13 patients who received etanercept and had a 0% mortality rate, etanercept may be viewed as a targeted therapeutic intervention for patients with SJS and TEN.
- Pereira FA, Mudgil AV, Rosmarin DM. Toxic epidermal necrolysis. J Am Acad Dermatol. 2007;56:181-200.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Hunger RE, Hunziker T, Buettiker U, et al. Rapid resolution of toxic epidermal necrolysis with anti-TNF-α treatment. J Allergy Clin Immunol. 2005;116:923-924.
- Worswick S, Cotliar J. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of treatment options. Dermatol Ther. 2011;24:207-218.
- Wallace AB. The exposure treatment of burns. Lancet Lond Engl. 1951;1:501-504.
- Bastuji-Garin S, Fouchard N, Bertocchi M, et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149-153.
- Mounach A, Rezqi A, Nouijai A, et al. Stevens-Johnson syndrome complicating adalimumab therapy in rheumatoid arthritis disease. Rheumatol Int. 2013;33:1351-1353.
- Salama M, Lawrance I-C. Stevens-Johnson syndrome complicating adalimumab therapy in Crohn’s disease. World J Gastroenterol. 2009;15:4449-4452.
- Wolkenstein P, Latarjet J, Roujeau JC, et al. Randomised comparison of thalidomide versus placebo in toxic epidermal necrolysis. Lancet Lond Engl. 1998;352:1586-1589.
- Fischer M, Fiedler E, Marsch WC, et al Antitumour necrosis factor-α antibodies (infliximab) in the treatment of a patient with toxic epidermal necrolysis. Br J Dermatol. 2002;146:707-709.
- Meiss F, Helmbold P, Meykadeh N, et al. Overlap of acute generalized exanthematous pustulosis and toxic epidermal necrolysis: response to antitumour necrosis factor-alpha antibody infliximab: report of three cases. J Eur Acad Dermatol Venereol. 2007;21:717-719.
- Al-Shouli S, Abouchala N, Bogusz MJ, et al. Toxic epidermal necrolysis associated with high intake of sildenafil and its response to infliximab. Acta Derm Venereol. 2005;85:534-535.
- Wojtkiewicz A, Wysocki M, Fortuna J, et al. Beneficial and rapid effect of infliximab on the course of toxic epidermal necrolysis. Acta Derm Venereol. 2008;88:420-421.
- Kreft B, Wohlrab J, Bramsiepe I, et al. Etoricoxib-induced toxic epidermal necrolysis: successful treatment with infliximab. J Dermatol. 2010;37:904-906.
- Patmanidis K, Sidiras A, Dolianitis K, et al. Combination of infliximab and high-dose intravenous immunoglobulin for toxic epidermal necrolysis: successful treatment of an elderly patient. Case Rep Dermatol Med. 2012;2012:915314.
- Gaitanis G, Spyridonos P, Patmanidis K, et al. Treatment of toxic epidermal necrolysis with the combination of infliximab and high-dose intravenous immunoglobulin. Dermatol Basel Switz. 2012;224:134-139.
- Zárate-Correa LC, Carrillo-Gómez DC, Ramírez-Escobar AF, et al. Toxic epidermal necrolysis successfully treated with infliximab. J Investig Allergol Clin Immunol. 2013;23:61-63.
- Paquet P, Jennes S, Rousseau AF, et al. Effect of N-acetylcysteine combined with infliximab on toxic epidermal necrolysis. a proof-of-concept study. Burns J Int Soc Burn Inj. 2014;40:1707-1712.
- Famularo G, Dona BD, Canzona F, et al. Etanercept for toxic epidermal necrolysis. Ann Pharmacother. 2007;41:1083-1084.
- Gubinelli E, Canzona F, Tonanzi T, et al. Toxic epidermal necrolysis successfully treated with etanercept. J Dermatol. 2009;36:150-153.
- Sadighha A. Etanercept in the treatment of a patient with acute generalized exanthematous pustulosis/toxic epidermal necrolysis: definition of a new model based on translational research. Int J Dermatol. 2009;48:913-914.
- Didona D, Paolino G, Garcovich S, et al. Successful use of etanercept in a case of toxic epidermal necrolysis induced by rituximab. J Eur Acad Dermatol Venereol. 2016;30:E83-E84.
- Lee Y-Y, Ko J-H, Wei C-H, et al. Use of etanercept to treat toxic epidermal necrolysis in a human immunodeficiency virus-positive patient. Dermatol Sin. 2013;31:78-81.
- Owczarczyk-Saczonek A, Zdanowska N, Znajewska-Pander A, et al. Stevens-Johnson syndrome in a patient with rheumatoid arthritis during long-term etanercept therapy. J Dermatol Case Rep. 2016;10:14-16.
- Wang CW, Yang LY, Chen CB, et al. Randomized, controlled trial of TNF-α antagonist in CTL mediated severe cutaneous adverse reactions. J Clin Invest. 2018;128:985-996.
- Pereira FA, Mudgil AV, Rosmarin DM. Toxic epidermal necrolysis. J Am Acad Dermatol. 2007;56:181-200.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Hunger RE, Hunziker T, Buettiker U, et al. Rapid resolution of toxic epidermal necrolysis with anti-TNF-α treatment. J Allergy Clin Immunol. 2005;116:923-924.
- Worswick S, Cotliar J. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of treatment options. Dermatol Ther. 2011;24:207-218.
- Wallace AB. The exposure treatment of burns. Lancet Lond Engl. 1951;1:501-504.
- Bastuji-Garin S, Fouchard N, Bertocchi M, et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149-153.
- Mounach A, Rezqi A, Nouijai A, et al. Stevens-Johnson syndrome complicating adalimumab therapy in rheumatoid arthritis disease. Rheumatol Int. 2013;33:1351-1353.
- Salama M, Lawrance I-C. Stevens-Johnson syndrome complicating adalimumab therapy in Crohn’s disease. World J Gastroenterol. 2009;15:4449-4452.
- Wolkenstein P, Latarjet J, Roujeau JC, et al. Randomised comparison of thalidomide versus placebo in toxic epidermal necrolysis. Lancet Lond Engl. 1998;352:1586-1589.
- Fischer M, Fiedler E, Marsch WC, et al Antitumour necrosis factor-α antibodies (infliximab) in the treatment of a patient with toxic epidermal necrolysis. Br J Dermatol. 2002;146:707-709.
- Meiss F, Helmbold P, Meykadeh N, et al. Overlap of acute generalized exanthematous pustulosis and toxic epidermal necrolysis: response to antitumour necrosis factor-alpha antibody infliximab: report of three cases. J Eur Acad Dermatol Venereol. 2007;21:717-719.
- Al-Shouli S, Abouchala N, Bogusz MJ, et al. Toxic epidermal necrolysis associated with high intake of sildenafil and its response to infliximab. Acta Derm Venereol. 2005;85:534-535.
- Wojtkiewicz A, Wysocki M, Fortuna J, et al. Beneficial and rapid effect of infliximab on the course of toxic epidermal necrolysis. Acta Derm Venereol. 2008;88:420-421.
- Kreft B, Wohlrab J, Bramsiepe I, et al. Etoricoxib-induced toxic epidermal necrolysis: successful treatment with infliximab. J Dermatol. 2010;37:904-906.
- Patmanidis K, Sidiras A, Dolianitis K, et al. Combination of infliximab and high-dose intravenous immunoglobulin for toxic epidermal necrolysis: successful treatment of an elderly patient. Case Rep Dermatol Med. 2012;2012:915314.
- Gaitanis G, Spyridonos P, Patmanidis K, et al. Treatment of toxic epidermal necrolysis with the combination of infliximab and high-dose intravenous immunoglobulin. Dermatol Basel Switz. 2012;224:134-139.
- Zárate-Correa LC, Carrillo-Gómez DC, Ramírez-Escobar AF, et al. Toxic epidermal necrolysis successfully treated with infliximab. J Investig Allergol Clin Immunol. 2013;23:61-63.
- Paquet P, Jennes S, Rousseau AF, et al. Effect of N-acetylcysteine combined with infliximab on toxic epidermal necrolysis. a proof-of-concept study. Burns J Int Soc Burn Inj. 2014;40:1707-1712.
- Famularo G, Dona BD, Canzona F, et al. Etanercept for toxic epidermal necrolysis. Ann Pharmacother. 2007;41:1083-1084.
- Gubinelli E, Canzona F, Tonanzi T, et al. Toxic epidermal necrolysis successfully treated with etanercept. J Dermatol. 2009;36:150-153.
- Sadighha A. Etanercept in the treatment of a patient with acute generalized exanthematous pustulosis/toxic epidermal necrolysis: definition of a new model based on translational research. Int J Dermatol. 2009;48:913-914.
- Didona D, Paolino G, Garcovich S, et al. Successful use of etanercept in a case of toxic epidermal necrolysis induced by rituximab. J Eur Acad Dermatol Venereol. 2016;30:E83-E84.
- Lee Y-Y, Ko J-H, Wei C-H, et al. Use of etanercept to treat toxic epidermal necrolysis in a human immunodeficiency virus-positive patient. Dermatol Sin. 2013;31:78-81.
- Owczarczyk-Saczonek A, Zdanowska N, Znajewska-Pander A, et al. Stevens-Johnson syndrome in a patient with rheumatoid arthritis during long-term etanercept therapy. J Dermatol Case Rep. 2016;10:14-16.
- Wang CW, Yang LY, Chen CB, et al. Randomized, controlled trial of TNF-α antagonist in CTL mediated severe cutaneous adverse reactions. J Clin Invest. 2018;128:985-996.
Practice Points
- Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are life-threatening dermatologic emergencies without a universally accepted treatment.
- Results of this study support the use of single-dose subcutaneous etanercept 50 mg as a potentially lifesaving therapy for patients with SJS/TEN.