User login
Obstructive Sleep Apnea: Evaluation & Management
CE/CME No: CR-1508
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Explain the diagnostic criteria and clinical presentation of obstructive sleep apnea (OSA).
• Discuss the common screening questionnaires used in clinical practice to identify patients at risk for OSA.
• Describe the common comorbidities associated with OSA.
• Identify the features of pharyngeal structures used in the modified Mallampati classification.
• Know how to provide support and education for the patient in regard to treatment options, weight loss, residual daytime sleepiness, and smoking and alcohol cessation.
FACULTY
Bonnie Dadig is the Chair and Program Director of the Georgia Regents University (GRU) Physician Assistant Department and a PA in the GRU Department of Family Medicine outpatient clinic, Augusta. Morgan Edwards is a recently graduated PA student from GRU. The authors have no financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of August 2015.
Article begins on next page >>
Nearly 22 million Americans are affected by obstructive sleep apnea (OSA), making it the most common sleep disorder. Patients with undiagnosed and untreated OSA are at increased risk for cardiovascular and cerebrovascular health consequences and excessive daytime sleepiness. Here’s how you can address the symptoms and complications that make OSA a major public health concern, and as a result, decrease economic burdens and increase quality of life.
Obstructive sleep apnea (OSA) is an increasingly common diagnosis, with nearly 22 million Americans currently affected.1 In 2010, the diagnosis of OSA was recorded in 5.8 million office visits made by patients ages 20 or older—a 427% increase since 1999.2 While nearly 50% of patients in primary care settings complain of sleep disturbances, up to 80% of OSA cases remain undiagnosed.1,3 The high prevalence of sleep disorders in both the general population and in primary care settings, along with the underdiagnosis of OSA, illustrates the essential role of primary care providers in recognizing the signs and symptoms of OSA and evaluating patients for this condition.1-3
BACKGROUND AND PATHOPHYSIOLOGY
OSA is the most common sleep disorder. Characterized by recurrent episodes of upper airway obstruction during sleep, it results in significantly reduced airflow (hypopnea) or complete cessation of breathing (apnea) for intervals lasting ≥ 10 seconds.4,5 The total number of apneas or hypopneas per hour of sleep—the apnea-hypopnea index (AHI)—is one domain that can be used to classify the severity of OSA (see Table 1).5
Because the human upper airway lacks rigid or bony support, under normal circumstances, the pharyngeal dilator muscles act to oppose the negative pressure within the upper airway during inspiration when awake. And although the striated nature of the pharyngeal dilator muscles leads to a reduction in airway muscle tone during sleep, enough opposition is maintained to prevent airway collapse. In OSA patients, however, the reduced pharyngeal muscle tone fails to offset the negative airway pressure generated during inspiration while asleep, causing recurrent airway obstructions.6 In addition, anatomically narrowed upper airways can further impede airway patency during sleep.4 Causes include macroglossia and/or tonsillar hypertrophy, obesity with fat tissue deposits in the neck that compress the airway, and bony craniofacial abnormalities such as retrognathia.7
Disruptions in breathing from apneas and hypopneas result in hypercapnia and hypoxia, which trigger arousal centers in the central nervous system to increase sympathetic activity. As a result, the patient briefly awakens from sleep, which increases respiratory and pharyngeal dilator muscle tone and ultimately restores airway patency, and ventilation resumes.4
The sympathetic response associated with apneic and hypopneic episodes causes concomitant cardiac stimulation. This leads to transient vasoconstriction and elevations in heart rate and blood pressure. Vascular changes occur over time and result in persistent hypertension and other cardiovascular impairments.4
Undiagnosed and untreated OSA predisposes individuals to a multitude of health consequences and ultimately results in substantial public health and economic burdens. A recent study estimated that the economic cost of moderate-to-severe OSA in the United States is between $65 billion and $165 billion annually.8 Patients with OSA are at increased risk for complications such as systemic and pulmonary hypertension, congestive heart failure, cardiac arrhythmias, coronary artery disease, myocardial infarction, cerebrovascular accident, diabetes, and metabolic syndrome.9 Excessive daytime sleepiness from OSA has also been shown to contribute to increased rates of motor vehicle accidents, poor job performance, impaired cognitive function, and decreased quality-of-life measures.9
Continue for clinical evaluation >>
CLINICAL EVALUATION
Patient History
Clinicians become aware of patients with OSA when they present with associated symptoms or complications. These symptoms are encountered in several settings—as a part of the routine health maintenance examination, as the patient’s presenting complaint, or as a part of the evaluation for another complaint or medical condition.
Regardless of how the symptoms are discovered, all patients with symptoms of and/or positive risk factors for OSA should be assessed via a comprehensive sleep oriented history and physical examination. Symptoms suggestive of OSA include witnessed or reported apneas, snoring, gasping/choking at night, excessive daytime sleepiness not explained by other factors, severity of sleepiness (as determined by the Epworth Sleepiness Scale), nonrefreshing sleep, sleep fragmentation/maintenance insomnia, nocturia, morning headaches, decreased concentration, memory loss, decreased libido, and irritability.10 A recently published study found that nocturnal gasping or choking is the most reliable indicator for identifying patients with OSA.11

The clinical presentation of OSA in certain populations may differ from that of typical adult OSA, requiring a higher degree of clinical suspicion. For instance, pediatric OSA should be suspected in any child who presents with enuresis, poor school performance, failure to thrive, attention-deficit/hyperactivity disorder, or learning disabilities.9 Alternatively, females with OSA have a propensity to emphasize complaints of tiredness, fatigue, or lack of energy rather than report excessive sleepiness per se. These complaints frequently misdirect clinicians toward psychiatric and endocrine diagnoses, without considering the possibility of a sleep disorder.12
Risk Factors
In addition to symptomatology, there are several risk factors associated with OSA that should be considered. The most important are male gender, increasing age (40 to 70), obesity, large neck circumference, and craniofacial and upper airway abnormalities.13 Among the intermediate risk factors are family history of OSA, pregnancy, and Hispanic, Asian, and African-American ethnicity.
Other risk factors associated with OSA include cigarette smoking, alcohol use before sleep, and use of sedative medications.13 Comorbid medical conditions have also been shown to exacerbate and to contribute to OSA; these include GERD, diabetes, congestive heart failure, treatment-refractory hypertension, hypothyroidism, acromegaly, atrial fibrillation, stroke, Down syndrome (due to macroglossia), and pulmonary hypertension.4,10
Screening Questionnaires
Several questionnaires have been validated to screen for OSA. They can be used clinically as part of the sleep-oriented history to quantify a patient’s subjective complaint of sleepiness; however, they are typically more helpful for ruling out sleep apnea than for diagnosing it.11 Two of the most commonly utilized are the Epworth Sleepiness Scale (ESS; http://epworthsleepinessscale.com/epworth-sleepiness-scale.pdf) and the Berlin Questionnaire (www.sleepapnea.org/assets/files/pdf/Berlin%20Questionnaire.pdf).
The ESS was developed to identify patients with excessive daytime sleepiness. It asks patients to rate—on a scale of 0 (would never doze) to 4 (high chance of dozing)—how likely they are to doze off or fall asleep during eight everyday scenarios (ie, sitting and reading; watching TV; sitting inactive in a public place; being a passenger in a vehicle for one or more hours; lying down in the afternoon; sitting and talking to someone; sitting quietly after lunch; or while stopped for a few minutes while driving in traffic).14 The total score can range from 0 to 24. Patients scoring higher than 10 should be referred to a sleep specialist. The ESS can be used to detect symptoms suggestive of OSA; however, it is not specific to OSA and was designed to screen for all sleep disorders.14
In contrast, the Berlin Questionnaire consists of 10 questions that quantify and qualify a patient’s snoring and sleepiness, and it was developed specifically to evaluate for OSA. The questionnaire measures three categories of symptoms: snoring, fatigue, and blood pressure and BMI. A patient is deemed at high risk for OSA when scoring positive in two or more symptom categories, and at low risk when scoring positive in one category.15 However, when OSA is defined as an AHI of ≥ 5 events/hr, the Berlin Questionnaire is 80% sensitive and 46% specific for OSA.11 Therefore, screening questionnaires are useful as supplementary tools but are of limited utility as the sole method for evaluating and diagnosing OSA.
Physical Examination
Physical examination should include assessment for risk factors and comorbidities associated with OSA; this includes measurement of blood pressure, blood glucose, and neck circumference; calculation of BMI and waist-to-hip ratio; and assessment for craniofacial abnormalities. Additionally, cardiovascular, respiratory, and neurologic systems should be evaluated for potential alterations due to OSA.10
Physical exam findings suggestive of OSA are BMI ≥ 30, neck circumference > 17 in for males and > 16 in for females, and craniofacial and upper airway abnormalities. Such anatomic variations include retrognathia, macroglossia (eg, Down syndrome patients), tonsillar hypertrophy (eg, pediatric patients), lateral peritonsillar narrowing, high-arched or narrow hard palate, enlarged uvula, nasal polyps, nasal deviation, turbinate hypertrophy, and oropharynx anatomy consistent with modified Mallampati class III or IV.10 Although OSA is more prevalent in obese patients (those with a BMI ≥ 30), it should be noted that 14.6% of OSA patients are normal weight (BMI < 25) and 34.5%, overweight (BMI 25 to 30).16
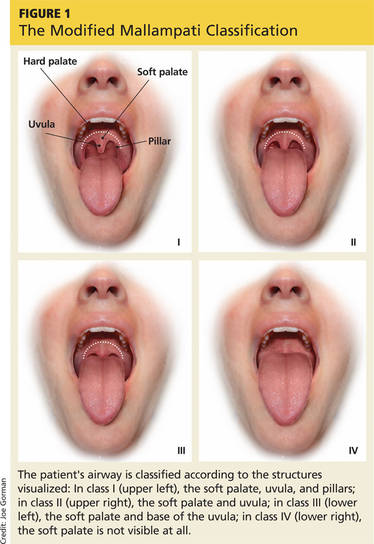
The modified Mallampati classification is a straightforward scoring method that was designed to estimate the difficulty of oral intubation based on the extent of mouth opening relative to the size of the tongue.17 According to the protocol set forth by Mallampati and colleagues, assessment is performed with the patient sitting upright with the head in a neutral position, the mouth open, and the tongue maximally protruded without the patient speaking or saying “ahh.”18 The examiner then inspects the pharyngeal structures and classifies the patient’s airway according to the structures visualized. Class I is present when the soft palate, uvula, and pillars are visible; class II when the soft palate and uvula are visible; class III when the soft palate and base of the uvula are visible; class IV when the soft palate is not visible at all (see Figure 1).17,18
An alternative method for observing the pharyngeal structures is via nasopharyngoscopy.19 This procedure can be done in the office setting and utilizes a flexible scope inserted into the nasal cavity and advanced into the pharynx. By providing direct visualization of the upper airway structures, nasopharyngoscopy allows the operator to determine the extent and the location of airway collapse. Imaging by nasopharyngoscopy does not involve any radiation exposure and can be done while the patient is sedated or awake.19
LABORATORY WORKUP AND DIAGNOSIS
Patients deemed high-risk by the sleep-oriented history and physical examination should be subsequently referred to a sleep specialist for objective testing to establish the diagnosis and determine the severity of OSA. Despite the utilization of screening questionnaires, currently there is no consensus on risk stratification to determine when to refer patients with suspected OSA. In other words, no clear thresholds exist on whether a patient is at “low risk” or at “high risk” for OSA based on history and physical examination findings. Therefore, referral is made at the clinician’s discretion, based on the number and severity of OSA symptoms and risk factors.
There are two standard objective sleep study tests: in-laboratory polysomnography (PSG) and home-based sleep studies. The in-laboratory PSG is the gold standard for the diagnosis of OSA.4 The following physiologic signals are measured and interpreted during in-laboratory PSG: electroencephalogram, electro-oculogram, chin electromyogram, airflow, oxygen saturation, respiratory effort, and electrocardiogram.4
Because PSG is resource intensive, home-based sleep studies are an acceptable alternative; however, they should only be considered in patients with a high probability of moderate-to-severe OSA without comorbid sleep disorders or medical conditions. During home-based sleep studies, airflow, respiratory effort, and blood oxygenation are documented.10
The diagnosis of OSA is validated if the AHI on PSG or on home-based sleep studies is > 15 events/h or > 5 events/h with any of the following coexisting signs or symptoms: excessive daytime sleepiness; unintentional sleep episodes during wakefulness; unrefreshing sleep; fatigue; insomnia; waking up holding breath, gasping, or choking; or loud snoring, breathing interruptions, or both, described by the bed partner.10 In addition to diagnosing OSA, PSG and home-based sleep studies are useful in ruling out a variety of conditions that similarly cause excessive daytime sleepiness due to disrupted sleep.
The differential diagnosis of OSA includes, but is not limited to, periodic limb movement disorder, restless legs syndrome, narcolepsy, central sleep apnea, circadian rhythm sleep-wake disorders, respiratory diseases (ie, COPD, asthma), primary snoring, depression, and substance abuse.20
Continue for treatment options >>
TREATMENT OPTIONS
Because OSA is a chronic condition with no definitive cure, lifelong management and follow-up are needed to evaluate treatment adherence/response and the development or resolution of comorbidities. Treatment options for OSA consist primarily of medical (positive airway pressure, use of oral appliances, and pharmacotherapy), behavioral (weight loss, positional therapy, and substance avoidance), and surgical approaches. The patient should actively participate in the treatment and management of this chronic condition.10
Positive Airway Pressure
Regardless of OSA severity, the treatment of choice is positive airway pressure (PAP) during sleep, with continuous PAP (CPAP) being the recommended mode of delivery. PAP is delivered via a face or nasal mask attached to a machine that maintains a constant upper airway pressure, which in turn prevents pharyngeal collapse during sleep. As a result, the upper airway remains patent, and apneas and hypopneas are inhibited.4
PAP can be delivered in three different modes: CPAP, bilevel (BPAP), or autotitrating (APAP). CPAP provides PAP at a fixed rate throughout the respiratory cycle. In contrast, BPAP delivers a preset PAP that is greater during inspiration than during expiration. BPAP is used to alleviate the discomfort of exhaling against the fixed airway pressure of traditional CPAP by delivering lower pressure during expiration than during inspiration. APAP delivers fluctuating positive airway pressures according to changes in the patient’s breathing patterns and is used in patients with complex breathing patterns. CPAP is the recommended mode of airway pressure delivery; however, BPAP or APAP can be considered in patients who cannot tolerate CPAP.10,21
The amount of pressure needed to maintain airway patency is determined during PSG, and the CPAP system is titrated up to this optimal level. Patient adherence is a commonly encountered problem with CPAP therapy due to adverse effects, such as skin irritation, misfit of mask, dry mouth, nasal congestion, and rhinorrhea. Humidification, nasal saline sprays, and mask resizing can help to alleviate such symptoms.4 Follow-up in the early stages should be frequent enough to troubleshoot and remediate problems with the PAP mask, the machine, and adherence. Long-term follow-up for maintenance, adherence, and signs and symptoms of worsening OSA should be scheduled yearly.10
Oral Appliances
Oral appliances (OAs) are an alternative treatment option for OSA patients who are nonadherent with CPAP, fail CPAP or behavioral therapies, experience adverse effects from CPAP, or prefer OAs. Two types of OAs exist: mandibular repositioning appliances and tongue retaining devices. The former anteriorly displaces the mandible relative to the maxilla; the latter anteriorly displaces the tongue only. Regardless of the type selected, OAs are designed to increase the diameter of the pharynx and decrease pharyngeal collapsibility.10
Although less efficacious than traditional CPAP therapy, OAs may be considered in patients with mild-to-moderate OSA.10 Patients with severe OSA should not be offered OAs as a treatment option. Current evidence suggests that OAs can substantially reduce the severity of OSA as well as daytime sleepiness. Additionally, OAs are superior to CPAP in patient preference, which could improve long-term patient adherence to treatment.22
Prior to initiating therapy with an OA, patients should have a dental examination to assess candidacy for, and proper fit of, an OA. Regular follow-ups are required initially to ensure proper fit and comfort of the device. Patients should be instructed to make an office visit if they experience adverse effects such as temporomandibular joint discomfort, tooth tenderness, excessive salivation, and gum irritation.22 Once final adjustments have been made to the fit of the OA, a PSG should be performed to ensure maximal therapeutic efficacy. Follow-up with a dental specialist is advised every six months for the first year and yearly thereafter.10
Pharmacology
According to the practice parameters of the American Academy of Sleep Medicine (AASM), there are currently no pharmacologic agents that prevent or overcome upper airway obstruction well enough to be considered a primary treatment option for OSA.23 The most widely studied pharmacologic agents—selective serotonergic reuptake inhibitors, protriptyline, methylxanthine derivatives (aminophylline and theophylline), estrogen therapy (with or without progesterone), supplemental oxygen, and short-acting nasal decongestants—are not recommended for the primary treatment of OSA.23 Pharmacologic therapy is therefore most beneficial when used as adjuvant therapy (which targets a residual symptom) rather than primary therapy (which targets the underlying cause of the disorder).
There are currently two FDA-approved medications for the treatment of residual excessive daytime sleepiness in patients who are receiving adequate primary treatment (eg, PAP, OAs) and who do not have any other identifiable cause for the sleepiness: modafinil and armodafinil.23,24 The exact mechanism of action of these agents has not been fully characterized, but they are known to act on the central nervous system to enhance alertness (likely by enhancing dopamine signaling).24
Prior to prescribing these medications, other causes of sleepiness must be ruled out; these include suboptimal adherence with PAP, poorly fitting PAP mask, poor sleep hygiene, insufficient sleep, other sleep disorders, and depression.10 Patients should be informed that these drugs should be used in conjunction with pre-existing CPAP therapy. Common adverse effects of modafinil include headache, dizziness, gastrointestinal upset, and insomnia. This medication should be used cautiously in patients with a history of cardiovascular and/or psychologic problems because it can cause serious adverse effects, including chest pain, palpitations, hypertension, hallucinations, aggression, anxiety, depression, and severe skin reactions, such as Stevens-Johnson syndrome and toxic epidermal necrolysis.4,24
Weight Loss
Because obesity is one of the most prevalent risk factors for OSA, weight loss should be recommended for all overweight and obese OSA patients.13,23 Weight loss can be achieved by several different methods, including exercise programs, dietary programs, a combination of the two, and bariatric surgery. A recent meta-analysis found that exercise alone is less successful in reducing AHI compared to dietary modifications; however, a combination of exercise and dietary changes is superior to both individual interventions in reducing AHI.25 This meta-analysis also found that weight-loss interventions improve, but fail to normalize, AHI.25 Thus, weight-loss interventions should be an adjuvant therapy in the treatment of OSA.23
Another recent meta-analysis found that lifestyle interventions, specifically exercise training, might improve OSA severity and symptoms even in the absence of significant weight loss. According to this study, exercise training produced a 32% reduction in AHI in OSA patients and a 42% reduction in AHI when compared to OSA patients who did not partake in exercise training.26 Additionally, OSA patients experienced significant improvements in cardiorespiratory fitness, daytime sleepiness, and sleep efficiency. Because there was no significant reduction in BMI with exercise training, it is likely that there are additional benefits of exercise training that are independent of the effects on weight loss.26
Positional Therapy
Approximately 50% to 60% of OSA patients have positional OSA, which is classified as an increase of 50% or more in the AHI when sleeping in the supine position compared with nonsupine sleep positions.27 It is thought that positional therapy, consisting of any technique that maintains the patient in a nonsupine sleep position, decreases the tendency for the tongue to prolapse and for the airway to collapse.27 Several positional devices exist, including pillows, tennis balls, backpacks, and vibratory chest alarms. Additionally, sleeping in the lateral recumbent position can function as a form of positional therapy.4,21
According to the AASM, less obese, younger patients and those with less severe OSA are more likely to achieve a normalized AHI by sleeping in a nonsupine position; thus, these patients are more likely to benefit from positional therapy than their OSA counterparts.23,27 A recent study found that CPAP therapy is superior to positional therapy in reducing apneic episodes (ie, AHI) in patients with positional OSA.27 Thus, positional therapy is an effective secondary treatment that can be used as an adjuvant therapy to CPAP in patients with mild positional OSA.23,27
Substance Avoidance
All patients with OSA should be advised to avoid alcohol, as it has been shown to increase the duration and frequency of apneic episodes and lower arterial oxyhemoglobin saturation during sleep.28 Not only does alcohol exacerbate preexisting OSA, but it has also been shown to induce frank OSA in patients who snore but do not have OSA at baseline.28 Sedative medications (eg, benzodiazepines) have similar effects on the central nervous system and should also be avoided if possible. Both of these substances have inhibitory actions on the central nervous system, thereby relaxing the pharyngeal muscles and promoting upper airway collapse during sleep.
Because cigarette smoking is an independent risk factor for snoring and thus a presumed risk factor for OSA, smoking cessation is a necessary component of the treatment of OSA.13,29 Cigarette smoking is thought to increase airway inflammation, exacerbate preexisting lung conditions such as COPD and asthma, and induce sleep fragmentation due to the effects of nicotine withdrawal during sleep.29 Alcohol, sedative medications, and cigarettes have all been shown to exacerbate OSA; thus, patient education about the harmful effects of these substances and the benefits of avoiding them is a crucial aspect of OSA therapy.
Surgical Therapies
When both CPAP and OA are inadequate in the management of OSA and/or when obstructive or functional anatomic abnormalities exist, upper airway surgery can be considered for patients. There is currently no consensus about the role of surgery in OSA patients, nor are there clear guidelines or screening questionnaires that accurately predict which patients will benefit the most from upper airway surgery. The appropriate surgical procedure depends on the site of anatomic abnormality and could be a nasal, oral, oropharyngeal, nasopharyngeal, hypopharyngeal, laryngeal, or global airway procedure.10 Some of the most common surgical procedures for the management of OSA are described below.
Uvulopalatopharyngoplasty (UPPP). This surgical procedure involves resecting the entire uvula and the obstructive portion of the soft palate while resizing and reorienting the tonsillar pillars. As a sole procedure, with or without tonsillectomy, UPPP does not reliably normalize the AHI when treating moderate-to-severe OSA.30
Radiofrequency ablation (RFA). RFA is a less invasive variation of UPPP that involves using a temperature-controlled probe to deliver energy to the upper airway tissue (typically the tongue base and/or the soft palate) in an effort to induce palatal stiffening. This procedure can be considered in patients with mild-to-moderate OSA who cannot tolerate or are unwilling to adhere to PAP or OA therapies.30
Maxillo-mandibular advancement (MMA). This operation involves indirect advancement of the anterior pharyngeal tissues (ie, the soft palate, tongue base, and suprahyoid musculature) via their attachment to the maxilla, mandible, and hyoid bone. The simultaneous advancement of the maxilla and mandible are accomplished by sagittal split osteotomies that are stabilized with plates, screws, or bone grafts.31 This surgery is designed to enlarge the retrolingual airway and provide some advancement of the retropalate without directly manipulating the pharyngeal tissues. MMA is indicated for patients with severe OSA who cannot tolerate or are unwilling to adhere to PAP therapy or in whom OAs have been found ineffective.30
Tracheostomy. This procedure consists of creating an airway through the anterior neck into the upper trachea. This opening bypasses the entire upper airway obstruction and thus is 100% effective in curing OSA. However, tracheostomy is typically last-line due to the resulting undesirable alterations in the patient’s physical appearance and to the risks associated with the procedure.5 According to the AASM, this operation should only be considered when other options do not exist, have failed, or are refused, or when this operation is deemed necessary by clinical urgency.30
Hypoglossal nerve stimulation. This therapy uses a surgically implanted device, approved by the FDA in 2014, to detect the patient’s breathing pattern and stimulate the hypoglossal nerve, causing tongue protrusion during inspiration. Tongue movement is controlled, and thus, the airway remains patent during inspiration.32 It is indicated for use in patients with moderate-to-severe OSA who have refused CPAP or for whom CPAP treatment has been unsuccessful.
According to a recently published study, a 68% decrease in median AHI at 12 months postimplantation was reported among those receiving hypoglossal nerve stimulation.33 Additionally, OSA patients subjectively reported decreased sleepiness and an increased quality of life compared to baseline.
The most common adverse effects were transient tongue weakness postoperatively, discomfort related to stimulation, and tongue soreness.33 Its use is contraindicated in patients who are pregnant or plan to become pregnant and in those who will require MRI, who have other implantable devices that may interact with the stimulation system, who have any condition that may affect neurologic control of the upper airway, and who have any anatomic abnormalities that may prevent effective performance of the upper airway stimulation (eg, the presence of complete concentric collapse at the retropalatal airway during endoscopy).32,33 Although its use is promising for the treatment of OSA, hypoglossal nerve stimulation is still a novel option that is not yet readily employed.33
OUTCOMES ASSESSMENT
Clinical judgment should be used to determine the appropriate treatment(s) based on OSA severity and patient preference. Regardless of the treatment option chosen, all OSA patients should have an outcomes assessment performed after the initiation of therapy. Indicators to monitor include resolution of sleepiness, OSA-specific quality-of-life measures, adherence to therapy, avoidance of factors exacerbating OSA (eg, alcohol, tobacco, sedative medications), amount of sleep being obtained, sleep hygiene practices, weight loss, and patient and spousal satisfaction.10 Patients with all levels of OSA severity should receive ongoing management to ensure long-term resolution of symptoms and adherence to treatment. Improvements in primary care treatment, follow-up, and outcomes evaluation are becoming increasingly important to address the symptoms and complications that make OSA a major public health concern.
1. American Sleep Apnea Association. A very short course on sleep apnea. www.sleepapnea.org/i-am-a-health-care-professional.html. Accessed July 21, 2015.
2. Ford ES, Wheaton AG, Cunningham TJ, et al. Trends in outpatient visits for insomnia, sleep apnea, and prescriptions for sleep medications among US adults: findings from the National Ambulatory Medical Care Survey 1999-2010. Sleep. 2014;37(8):1283-1293.
3. NIH State-of-the-Science Conference Statement on Manifestations and Management of Chronic Insomnia in Adults. NIH Consens Sci Statements. 2005;22(2):1-30.
4. Carlucci M, Smith M, Corbridge SJ. Poor sleep, hazardous breathing: an overview of obstructive sleep apnea. Nurse Pract. 2013;38(3):20-28.
5. Institute for Clinical Systems Improvement. Health Care Guideline: Diagnosis and Treatment of Obstructive Sleep Apnea. 6th ed. Bloomington, MN: Institute for Clinical Systems Improvement; 2008.
6. Douglas NJ. Sleep apnea. Harrison’s Principles of Internal Medicine. 18th ed. New York, NY: McGraw-Hill; 2012:2186-2188.
7. US Department of Health and Human Services, National Heart, Lung, and Blood Institute. What causes sleep apnea? www.nhlbi.nih.gov/health/health-topics/topics/sleepapnea/causes.html. Accessed uly 21, 2015.
8. Harvard Medical School, Division of Sleep Medicine. The price of fatigue: the surprising economic costs of unmanaged sleep apnea. December 2010. https://sleep.med.harvard.edu/file_download/100. Accessed July 21, 2015.
9. Pagel JF. Obstructive sleep apnea (OSA) in primary care: evidence-based practice. J Am Board Fam Med. 2007;20(4):392-398.
10. Epstein LJ, Kristo D, Strollo PJ, et al; Adult Obstructive Sleep Apnea Task Force of the American Academy of Sleep Medicine. Clinical guideline for the evaluation, management, and long-term care of obstructive sleep apnea in adults. J Clin Sleep Med. 2009;5(3):263-276.
11. Myers KA, Mrkobrada M, Simel DL. Does this patient have obstructive sleep apnea: the rational clinical examination systematic review. JAMA. 2013;310(7):731-741.
12. Ye L, Pien GW, Weaver TE. Gender differences in the clinical manifestation of obstructive sleep apnea. Sleep Med. 2009;10(10):1075-1084.
13. Young T, Skatrud J, Peppard PE. Risk factors for obstructive sleep apnea in adults. JAMA. 2004;291(16):2013-2016.
14. Johns MW. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep. 1991;14(6):540-545.
15. Netzer NC, Stoohs RA, Netzer CM, et al. Using the Berlin Questionnaire to identify patients at risk for the sleep apnea syndrome. Ann Intern Med. 1999;131(7):485-491.
16. Tufik S, Santos-Silva R, Taddei JA, Bittencourt LR. Obstructive sleep apnea syndrome in the Sao Paulo Epidemiologic Sleep Study. Sleep Med. 2010;11(5):441-446.
17. Samsoon GL, Young JR. Difficult tracheal intubation: a retrospective study. Anaesthesia. 1987;42:487-490.
18. Mallampati SR, Gatt SP, Gugino LD, et al. A clinical sign to predict difficult tracheal intubation: a prospective study. Can Anaesth Soc J. 1985;32(4):429-434.
19. Gregorio MG, Jacomelli M, Figueiredo AC, et al. Evaluation of airway obstruction by nasopharyngoscopy: comparison of the Müller maneuver versus induced sleep. Braz J Otorhinolaryngol. 2007;73(5):618-622.
20. Kline LR. Clinical presentation and diagnosis of obstructive sleep apnea in adults. Up-to-Date. www.uptodate.com/contents/clinical-presentation-and-diagnosis-of-obstructive-sleep-apnea-in-adults. Accessed July 21, 2015.
21. Kryger MH, Malhotra A. Management of obstructive sleep apnea in adults. Up-to-Date. www.uptodate.com/contents/management-of-obstructive-sleep-apnea-in-adults. Accessed July 21, 2015.
22. Lim J, Lasserson TJ, Fleetham J, Wright JJ. Oral appliances for obstructive sleep apneoa. Cochrane Database Syst Rev. 2006;1:CD004435.
23. Morgenthaler TI, Kapen S, Lee-Chiong T, et al; Standards of Practice Committee; American Academy of Sleep Medicine. Practice parameters for the medical therapy of obstructive sleep apnea. Sleep. 2006;29(8): 1031-1035.
24. Pepin JL. Evaluation and management of residual sleepiness in obstructive sleep apnea. Up-to-Date. www.uptodate.com/contents/evaluation-and-management-of-residual-sleepiness-in-obstructive-sleep-apnea. Accessed July 21, 2015.
25. Araghi MH, Chen YF, Jagielski A, et al. Effectiveness of lifestyle interventions on obstructive sleep apnea (OSA): systematic review and meta-analysis. Sleep. 2013;36(10):1553-1562.
26. Iftikhar IH, Kline CE, Youngstedt SD. Effects of exercise training on sleep apnea: a meta-analysis. Lung. 2014;192(1):175-184.
27. Ha SC, Hirai HW, Tsoi KK. Comparison of positional therapy versus continuous positive airway pressure in patients with positional obstructive sleep apnea: a meta-analysis of randomized trials. Sleep Med Rev. 2014; 18(1):19-24.
28. Issa FG, Sullivan CE. Alcohol, snoring, and sleep apnea. J Neurol Neurosurg Psychiatry. 1982;45(4):353-359.
29. Lin Y, Li QY, Zhang XJ. Interaction between smoking and obstructive sleep apnea: not just participants. Chin Med J. 2012;125(17):3150-3156.
30. Aurora RN, Casey KR, Kristo D, et al; American Academy of Sleep Medicine. Practice parameters for the surgical modifications of the upper airway for obstructive sleep apnea in adults. Sleep. 2010;33(10):1408-1413.
31. Caples SM, Rowley JA, Prinsell JR, et al. Surgical modifications of the upper airway for obstructive sleep apnea in adults: a systematic review and meta-analysis. Sleep. 2010;33(10):1396-1407.
32. US Food and Drug Administration. Recently-approved devices. Inspire upper airway stimulation - P130008. www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/Recently-ApprovedDevices/ucm398321.htm. Accessed July 21, 2015.
33. Strollo PJ, Soose RJ, Maurer JT, et al. Upper-airway stimulation for obstructive sleep apnea. N Engl J Med. 2014;370(2):139-149.
CE/CME No: CR-1508
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Explain the diagnostic criteria and clinical presentation of obstructive sleep apnea (OSA).
• Discuss the common screening questionnaires used in clinical practice to identify patients at risk for OSA.
• Describe the common comorbidities associated with OSA.
• Identify the features of pharyngeal structures used in the modified Mallampati classification.
• Know how to provide support and education for the patient in regard to treatment options, weight loss, residual daytime sleepiness, and smoking and alcohol cessation.
FACULTY
Bonnie Dadig is the Chair and Program Director of the Georgia Regents University (GRU) Physician Assistant Department and a PA in the GRU Department of Family Medicine outpatient clinic, Augusta. Morgan Edwards is a recently graduated PA student from GRU. The authors have no financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of August 2015.
Article begins on next page >>
Nearly 22 million Americans are affected by obstructive sleep apnea (OSA), making it the most common sleep disorder. Patients with undiagnosed and untreated OSA are at increased risk for cardiovascular and cerebrovascular health consequences and excessive daytime sleepiness. Here’s how you can address the symptoms and complications that make OSA a major public health concern, and as a result, decrease economic burdens and increase quality of life.
Obstructive sleep apnea (OSA) is an increasingly common diagnosis, with nearly 22 million Americans currently affected.1 In 2010, the diagnosis of OSA was recorded in 5.8 million office visits made by patients ages 20 or older—a 427% increase since 1999.2 While nearly 50% of patients in primary care settings complain of sleep disturbances, up to 80% of OSA cases remain undiagnosed.1,3 The high prevalence of sleep disorders in both the general population and in primary care settings, along with the underdiagnosis of OSA, illustrates the essential role of primary care providers in recognizing the signs and symptoms of OSA and evaluating patients for this condition.1-3
BACKGROUND AND PATHOPHYSIOLOGY
OSA is the most common sleep disorder. Characterized by recurrent episodes of upper airway obstruction during sleep, it results in significantly reduced airflow (hypopnea) or complete cessation of breathing (apnea) for intervals lasting ≥ 10 seconds.4,5 The total number of apneas or hypopneas per hour of sleep—the apnea-hypopnea index (AHI)—is one domain that can be used to classify the severity of OSA (see Table 1).5
Because the human upper airway lacks rigid or bony support, under normal circumstances, the pharyngeal dilator muscles act to oppose the negative pressure within the upper airway during inspiration when awake. And although the striated nature of the pharyngeal dilator muscles leads to a reduction in airway muscle tone during sleep, enough opposition is maintained to prevent airway collapse. In OSA patients, however, the reduced pharyngeal muscle tone fails to offset the negative airway pressure generated during inspiration while asleep, causing recurrent airway obstructions.6 In addition, anatomically narrowed upper airways can further impede airway patency during sleep.4 Causes include macroglossia and/or tonsillar hypertrophy, obesity with fat tissue deposits in the neck that compress the airway, and bony craniofacial abnormalities such as retrognathia.7
Disruptions in breathing from apneas and hypopneas result in hypercapnia and hypoxia, which trigger arousal centers in the central nervous system to increase sympathetic activity. As a result, the patient briefly awakens from sleep, which increases respiratory and pharyngeal dilator muscle tone and ultimately restores airway patency, and ventilation resumes.4
The sympathetic response associated with apneic and hypopneic episodes causes concomitant cardiac stimulation. This leads to transient vasoconstriction and elevations in heart rate and blood pressure. Vascular changes occur over time and result in persistent hypertension and other cardiovascular impairments.4
Undiagnosed and untreated OSA predisposes individuals to a multitude of health consequences and ultimately results in substantial public health and economic burdens. A recent study estimated that the economic cost of moderate-to-severe OSA in the United States is between $65 billion and $165 billion annually.8 Patients with OSA are at increased risk for complications such as systemic and pulmonary hypertension, congestive heart failure, cardiac arrhythmias, coronary artery disease, myocardial infarction, cerebrovascular accident, diabetes, and metabolic syndrome.9 Excessive daytime sleepiness from OSA has also been shown to contribute to increased rates of motor vehicle accidents, poor job performance, impaired cognitive function, and decreased quality-of-life measures.9
Continue for clinical evaluation >>
CLINICAL EVALUATION
Patient History
Clinicians become aware of patients with OSA when they present with associated symptoms or complications. These symptoms are encountered in several settings—as a part of the routine health maintenance examination, as the patient’s presenting complaint, or as a part of the evaluation for another complaint or medical condition.
Regardless of how the symptoms are discovered, all patients with symptoms of and/or positive risk factors for OSA should be assessed via a comprehensive sleep oriented history and physical examination. Symptoms suggestive of OSA include witnessed or reported apneas, snoring, gasping/choking at night, excessive daytime sleepiness not explained by other factors, severity of sleepiness (as determined by the Epworth Sleepiness Scale), nonrefreshing sleep, sleep fragmentation/maintenance insomnia, nocturia, morning headaches, decreased concentration, memory loss, decreased libido, and irritability.10 A recently published study found that nocturnal gasping or choking is the most reliable indicator for identifying patients with OSA.11
The clinical presentation of OSA in certain populations may differ from that of typical adult OSA, requiring a higher degree of clinical suspicion. For instance, pediatric OSA should be suspected in any child who presents with enuresis, poor school performance, failure to thrive, attention-deficit/hyperactivity disorder, or learning disabilities.9 Alternatively, females with OSA have a propensity to emphasize complaints of tiredness, fatigue, or lack of energy rather than report excessive sleepiness per se. These complaints frequently misdirect clinicians toward psychiatric and endocrine diagnoses, without considering the possibility of a sleep disorder.12
Risk Factors
In addition to symptomatology, there are several risk factors associated with OSA that should be considered. The most important are male gender, increasing age (40 to 70), obesity, large neck circumference, and craniofacial and upper airway abnormalities.13 Among the intermediate risk factors are family history of OSA, pregnancy, and Hispanic, Asian, and African-American ethnicity.
Other risk factors associated with OSA include cigarette smoking, alcohol use before sleep, and use of sedative medications.13 Comorbid medical conditions have also been shown to exacerbate and to contribute to OSA; these include GERD, diabetes, congestive heart failure, treatment-refractory hypertension, hypothyroidism, acromegaly, atrial fibrillation, stroke, Down syndrome (due to macroglossia), and pulmonary hypertension.4,10
Screening Questionnaires
Several questionnaires have been validated to screen for OSA. They can be used clinically as part of the sleep-oriented history to quantify a patient’s subjective complaint of sleepiness; however, they are typically more helpful for ruling out sleep apnea than for diagnosing it.11 Two of the most commonly utilized are the Epworth Sleepiness Scale (ESS; http://epworthsleepinessscale.com/epworth-sleepiness-scale.pdf) and the Berlin Questionnaire (www.sleepapnea.org/assets/files/pdf/Berlin%20Questionnaire.pdf).
The ESS was developed to identify patients with excessive daytime sleepiness. It asks patients to rate—on a scale of 0 (would never doze) to 4 (high chance of dozing)—how likely they are to doze off or fall asleep during eight everyday scenarios (ie, sitting and reading; watching TV; sitting inactive in a public place; being a passenger in a vehicle for one or more hours; lying down in the afternoon; sitting and talking to someone; sitting quietly after lunch; or while stopped for a few minutes while driving in traffic).14 The total score can range from 0 to 24. Patients scoring higher than 10 should be referred to a sleep specialist. The ESS can be used to detect symptoms suggestive of OSA; however, it is not specific to OSA and was designed to screen for all sleep disorders.14
In contrast, the Berlin Questionnaire consists of 10 questions that quantify and qualify a patient’s snoring and sleepiness, and it was developed specifically to evaluate for OSA. The questionnaire measures three categories of symptoms: snoring, fatigue, and blood pressure and BMI. A patient is deemed at high risk for OSA when scoring positive in two or more symptom categories, and at low risk when scoring positive in one category.15 However, when OSA is defined as an AHI of ≥ 5 events/hr, the Berlin Questionnaire is 80% sensitive and 46% specific for OSA.11 Therefore, screening questionnaires are useful as supplementary tools but are of limited utility as the sole method for evaluating and diagnosing OSA.
Physical Examination
Physical examination should include assessment for risk factors and comorbidities associated with OSA; this includes measurement of blood pressure, blood glucose, and neck circumference; calculation of BMI and waist-to-hip ratio; and assessment for craniofacial abnormalities. Additionally, cardiovascular, respiratory, and neurologic systems should be evaluated for potential alterations due to OSA.10
Physical exam findings suggestive of OSA are BMI ≥ 30, neck circumference > 17 in for males and > 16 in for females, and craniofacial and upper airway abnormalities. Such anatomic variations include retrognathia, macroglossia (eg, Down syndrome patients), tonsillar hypertrophy (eg, pediatric patients), lateral peritonsillar narrowing, high-arched or narrow hard palate, enlarged uvula, nasal polyps, nasal deviation, turbinate hypertrophy, and oropharynx anatomy consistent with modified Mallampati class III or IV.10 Although OSA is more prevalent in obese patients (those with a BMI ≥ 30), it should be noted that 14.6% of OSA patients are normal weight (BMI < 25) and 34.5%, overweight (BMI 25 to 30).16
The modified Mallampati classification is a straightforward scoring method that was designed to estimate the difficulty of oral intubation based on the extent of mouth opening relative to the size of the tongue.17 According to the protocol set forth by Mallampati and colleagues, assessment is performed with the patient sitting upright with the head in a neutral position, the mouth open, and the tongue maximally protruded without the patient speaking or saying “ahh.”18 The examiner then inspects the pharyngeal structures and classifies the patient’s airway according to the structures visualized. Class I is present when the soft palate, uvula, and pillars are visible; class II when the soft palate and uvula are visible; class III when the soft palate and base of the uvula are visible; class IV when the soft palate is not visible at all (see Figure 1).17,18
An alternative method for observing the pharyngeal structures is via nasopharyngoscopy.19 This procedure can be done in the office setting and utilizes a flexible scope inserted into the nasal cavity and advanced into the pharynx. By providing direct visualization of the upper airway structures, nasopharyngoscopy allows the operator to determine the extent and the location of airway collapse. Imaging by nasopharyngoscopy does not involve any radiation exposure and can be done while the patient is sedated or awake.19
LABORATORY WORKUP AND DIAGNOSIS
Patients deemed high-risk by the sleep-oriented history and physical examination should be subsequently referred to a sleep specialist for objective testing to establish the diagnosis and determine the severity of OSA. Despite the utilization of screening questionnaires, currently there is no consensus on risk stratification to determine when to refer patients with suspected OSA. In other words, no clear thresholds exist on whether a patient is at “low risk” or at “high risk” for OSA based on history and physical examination findings. Therefore, referral is made at the clinician’s discretion, based on the number and severity of OSA symptoms and risk factors.
There are two standard objective sleep study tests: in-laboratory polysomnography (PSG) and home-based sleep studies. The in-laboratory PSG is the gold standard for the diagnosis of OSA.4 The following physiologic signals are measured and interpreted during in-laboratory PSG: electroencephalogram, electro-oculogram, chin electromyogram, airflow, oxygen saturation, respiratory effort, and electrocardiogram.4
Because PSG is resource intensive, home-based sleep studies are an acceptable alternative; however, they should only be considered in patients with a high probability of moderate-to-severe OSA without comorbid sleep disorders or medical conditions. During home-based sleep studies, airflow, respiratory effort, and blood oxygenation are documented.10
The diagnosis of OSA is validated if the AHI on PSG or on home-based sleep studies is > 15 events/h or > 5 events/h with any of the following coexisting signs or symptoms: excessive daytime sleepiness; unintentional sleep episodes during wakefulness; unrefreshing sleep; fatigue; insomnia; waking up holding breath, gasping, or choking; or loud snoring, breathing interruptions, or both, described by the bed partner.10 In addition to diagnosing OSA, PSG and home-based sleep studies are useful in ruling out a variety of conditions that similarly cause excessive daytime sleepiness due to disrupted sleep.
The differential diagnosis of OSA includes, but is not limited to, periodic limb movement disorder, restless legs syndrome, narcolepsy, central sleep apnea, circadian rhythm sleep-wake disorders, respiratory diseases (ie, COPD, asthma), primary snoring, depression, and substance abuse.20
Continue for treatment options >>
TREATMENT OPTIONS
Because OSA is a chronic condition with no definitive cure, lifelong management and follow-up are needed to evaluate treatment adherence/response and the development or resolution of comorbidities. Treatment options for OSA consist primarily of medical (positive airway pressure, use of oral appliances, and pharmacotherapy), behavioral (weight loss, positional therapy, and substance avoidance), and surgical approaches. The patient should actively participate in the treatment and management of this chronic condition.10
Positive Airway Pressure
Regardless of OSA severity, the treatment of choice is positive airway pressure (PAP) during sleep, with continuous PAP (CPAP) being the recommended mode of delivery. PAP is delivered via a face or nasal mask attached to a machine that maintains a constant upper airway pressure, which in turn prevents pharyngeal collapse during sleep. As a result, the upper airway remains patent, and apneas and hypopneas are inhibited.4
PAP can be delivered in three different modes: CPAP, bilevel (BPAP), or autotitrating (APAP). CPAP provides PAP at a fixed rate throughout the respiratory cycle. In contrast, BPAP delivers a preset PAP that is greater during inspiration than during expiration. BPAP is used to alleviate the discomfort of exhaling against the fixed airway pressure of traditional CPAP by delivering lower pressure during expiration than during inspiration. APAP delivers fluctuating positive airway pressures according to changes in the patient’s breathing patterns and is used in patients with complex breathing patterns. CPAP is the recommended mode of airway pressure delivery; however, BPAP or APAP can be considered in patients who cannot tolerate CPAP.10,21
The amount of pressure needed to maintain airway patency is determined during PSG, and the CPAP system is titrated up to this optimal level. Patient adherence is a commonly encountered problem with CPAP therapy due to adverse effects, such as skin irritation, misfit of mask, dry mouth, nasal congestion, and rhinorrhea. Humidification, nasal saline sprays, and mask resizing can help to alleviate such symptoms.4 Follow-up in the early stages should be frequent enough to troubleshoot and remediate problems with the PAP mask, the machine, and adherence. Long-term follow-up for maintenance, adherence, and signs and symptoms of worsening OSA should be scheduled yearly.10
Oral Appliances
Oral appliances (OAs) are an alternative treatment option for OSA patients who are nonadherent with CPAP, fail CPAP or behavioral therapies, experience adverse effects from CPAP, or prefer OAs. Two types of OAs exist: mandibular repositioning appliances and tongue retaining devices. The former anteriorly displaces the mandible relative to the maxilla; the latter anteriorly displaces the tongue only. Regardless of the type selected, OAs are designed to increase the diameter of the pharynx and decrease pharyngeal collapsibility.10
Although less efficacious than traditional CPAP therapy, OAs may be considered in patients with mild-to-moderate OSA.10 Patients with severe OSA should not be offered OAs as a treatment option. Current evidence suggests that OAs can substantially reduce the severity of OSA as well as daytime sleepiness. Additionally, OAs are superior to CPAP in patient preference, which could improve long-term patient adherence to treatment.22
Prior to initiating therapy with an OA, patients should have a dental examination to assess candidacy for, and proper fit of, an OA. Regular follow-ups are required initially to ensure proper fit and comfort of the device. Patients should be instructed to make an office visit if they experience adverse effects such as temporomandibular joint discomfort, tooth tenderness, excessive salivation, and gum irritation.22 Once final adjustments have been made to the fit of the OA, a PSG should be performed to ensure maximal therapeutic efficacy. Follow-up with a dental specialist is advised every six months for the first year and yearly thereafter.10
Pharmacology
According to the practice parameters of the American Academy of Sleep Medicine (AASM), there are currently no pharmacologic agents that prevent or overcome upper airway obstruction well enough to be considered a primary treatment option for OSA.23 The most widely studied pharmacologic agents—selective serotonergic reuptake inhibitors, protriptyline, methylxanthine derivatives (aminophylline and theophylline), estrogen therapy (with or without progesterone), supplemental oxygen, and short-acting nasal decongestants—are not recommended for the primary treatment of OSA.23 Pharmacologic therapy is therefore most beneficial when used as adjuvant therapy (which targets a residual symptom) rather than primary therapy (which targets the underlying cause of the disorder).
There are currently two FDA-approved medications for the treatment of residual excessive daytime sleepiness in patients who are receiving adequate primary treatment (eg, PAP, OAs) and who do not have any other identifiable cause for the sleepiness: modafinil and armodafinil.23,24 The exact mechanism of action of these agents has not been fully characterized, but they are known to act on the central nervous system to enhance alertness (likely by enhancing dopamine signaling).24
Prior to prescribing these medications, other causes of sleepiness must be ruled out; these include suboptimal adherence with PAP, poorly fitting PAP mask, poor sleep hygiene, insufficient sleep, other sleep disorders, and depression.10 Patients should be informed that these drugs should be used in conjunction with pre-existing CPAP therapy. Common adverse effects of modafinil include headache, dizziness, gastrointestinal upset, and insomnia. This medication should be used cautiously in patients with a history of cardiovascular and/or psychologic problems because it can cause serious adverse effects, including chest pain, palpitations, hypertension, hallucinations, aggression, anxiety, depression, and severe skin reactions, such as Stevens-Johnson syndrome and toxic epidermal necrolysis.4,24
Weight Loss
Because obesity is one of the most prevalent risk factors for OSA, weight loss should be recommended for all overweight and obese OSA patients.13,23 Weight loss can be achieved by several different methods, including exercise programs, dietary programs, a combination of the two, and bariatric surgery. A recent meta-analysis found that exercise alone is less successful in reducing AHI compared to dietary modifications; however, a combination of exercise and dietary changes is superior to both individual interventions in reducing AHI.25 This meta-analysis also found that weight-loss interventions improve, but fail to normalize, AHI.25 Thus, weight-loss interventions should be an adjuvant therapy in the treatment of OSA.23
Another recent meta-analysis found that lifestyle interventions, specifically exercise training, might improve OSA severity and symptoms even in the absence of significant weight loss. According to this study, exercise training produced a 32% reduction in AHI in OSA patients and a 42% reduction in AHI when compared to OSA patients who did not partake in exercise training.26 Additionally, OSA patients experienced significant improvements in cardiorespiratory fitness, daytime sleepiness, and sleep efficiency. Because there was no significant reduction in BMI with exercise training, it is likely that there are additional benefits of exercise training that are independent of the effects on weight loss.26
Positional Therapy
Approximately 50% to 60% of OSA patients have positional OSA, which is classified as an increase of 50% or more in the AHI when sleeping in the supine position compared with nonsupine sleep positions.27 It is thought that positional therapy, consisting of any technique that maintains the patient in a nonsupine sleep position, decreases the tendency for the tongue to prolapse and for the airway to collapse.27 Several positional devices exist, including pillows, tennis balls, backpacks, and vibratory chest alarms. Additionally, sleeping in the lateral recumbent position can function as a form of positional therapy.4,21
According to the AASM, less obese, younger patients and those with less severe OSA are more likely to achieve a normalized AHI by sleeping in a nonsupine position; thus, these patients are more likely to benefit from positional therapy than their OSA counterparts.23,27 A recent study found that CPAP therapy is superior to positional therapy in reducing apneic episodes (ie, AHI) in patients with positional OSA.27 Thus, positional therapy is an effective secondary treatment that can be used as an adjuvant therapy to CPAP in patients with mild positional OSA.23,27
Substance Avoidance
All patients with OSA should be advised to avoid alcohol, as it has been shown to increase the duration and frequency of apneic episodes and lower arterial oxyhemoglobin saturation during sleep.28 Not only does alcohol exacerbate preexisting OSA, but it has also been shown to induce frank OSA in patients who snore but do not have OSA at baseline.28 Sedative medications (eg, benzodiazepines) have similar effects on the central nervous system and should also be avoided if possible. Both of these substances have inhibitory actions on the central nervous system, thereby relaxing the pharyngeal muscles and promoting upper airway collapse during sleep.
Because cigarette smoking is an independent risk factor for snoring and thus a presumed risk factor for OSA, smoking cessation is a necessary component of the treatment of OSA.13,29 Cigarette smoking is thought to increase airway inflammation, exacerbate preexisting lung conditions such as COPD and asthma, and induce sleep fragmentation due to the effects of nicotine withdrawal during sleep.29 Alcohol, sedative medications, and cigarettes have all been shown to exacerbate OSA; thus, patient education about the harmful effects of these substances and the benefits of avoiding them is a crucial aspect of OSA therapy.
Surgical Therapies
When both CPAP and OA are inadequate in the management of OSA and/or when obstructive or functional anatomic abnormalities exist, upper airway surgery can be considered for patients. There is currently no consensus about the role of surgery in OSA patients, nor are there clear guidelines or screening questionnaires that accurately predict which patients will benefit the most from upper airway surgery. The appropriate surgical procedure depends on the site of anatomic abnormality and could be a nasal, oral, oropharyngeal, nasopharyngeal, hypopharyngeal, laryngeal, or global airway procedure.10 Some of the most common surgical procedures for the management of OSA are described below.
Uvulopalatopharyngoplasty (UPPP). This surgical procedure involves resecting the entire uvula and the obstructive portion of the soft palate while resizing and reorienting the tonsillar pillars. As a sole procedure, with or without tonsillectomy, UPPP does not reliably normalize the AHI when treating moderate-to-severe OSA.30
Radiofrequency ablation (RFA). RFA is a less invasive variation of UPPP that involves using a temperature-controlled probe to deliver energy to the upper airway tissue (typically the tongue base and/or the soft palate) in an effort to induce palatal stiffening. This procedure can be considered in patients with mild-to-moderate OSA who cannot tolerate or are unwilling to adhere to PAP or OA therapies.30
Maxillo-mandibular advancement (MMA). This operation involves indirect advancement of the anterior pharyngeal tissues (ie, the soft palate, tongue base, and suprahyoid musculature) via their attachment to the maxilla, mandible, and hyoid bone. The simultaneous advancement of the maxilla and mandible are accomplished by sagittal split osteotomies that are stabilized with plates, screws, or bone grafts.31 This surgery is designed to enlarge the retrolingual airway and provide some advancement of the retropalate without directly manipulating the pharyngeal tissues. MMA is indicated for patients with severe OSA who cannot tolerate or are unwilling to adhere to PAP therapy or in whom OAs have been found ineffective.30
Tracheostomy. This procedure consists of creating an airway through the anterior neck into the upper trachea. This opening bypasses the entire upper airway obstruction and thus is 100% effective in curing OSA. However, tracheostomy is typically last-line due to the resulting undesirable alterations in the patient’s physical appearance and to the risks associated with the procedure.5 According to the AASM, this operation should only be considered when other options do not exist, have failed, or are refused, or when this operation is deemed necessary by clinical urgency.30
Hypoglossal nerve stimulation. This therapy uses a surgically implanted device, approved by the FDA in 2014, to detect the patient’s breathing pattern and stimulate the hypoglossal nerve, causing tongue protrusion during inspiration. Tongue movement is controlled, and thus, the airway remains patent during inspiration.32 It is indicated for use in patients with moderate-to-severe OSA who have refused CPAP or for whom CPAP treatment has been unsuccessful.
According to a recently published study, a 68% decrease in median AHI at 12 months postimplantation was reported among those receiving hypoglossal nerve stimulation.33 Additionally, OSA patients subjectively reported decreased sleepiness and an increased quality of life compared to baseline.
The most common adverse effects were transient tongue weakness postoperatively, discomfort related to stimulation, and tongue soreness.33 Its use is contraindicated in patients who are pregnant or plan to become pregnant and in those who will require MRI, who have other implantable devices that may interact with the stimulation system, who have any condition that may affect neurologic control of the upper airway, and who have any anatomic abnormalities that may prevent effective performance of the upper airway stimulation (eg, the presence of complete concentric collapse at the retropalatal airway during endoscopy).32,33 Although its use is promising for the treatment of OSA, hypoglossal nerve stimulation is still a novel option that is not yet readily employed.33
OUTCOMES ASSESSMENT
Clinical judgment should be used to determine the appropriate treatment(s) based on OSA severity and patient preference. Regardless of the treatment option chosen, all OSA patients should have an outcomes assessment performed after the initiation of therapy. Indicators to monitor include resolution of sleepiness, OSA-specific quality-of-life measures, adherence to therapy, avoidance of factors exacerbating OSA (eg, alcohol, tobacco, sedative medications), amount of sleep being obtained, sleep hygiene practices, weight loss, and patient and spousal satisfaction.10 Patients with all levels of OSA severity should receive ongoing management to ensure long-term resolution of symptoms and adherence to treatment. Improvements in primary care treatment, follow-up, and outcomes evaluation are becoming increasingly important to address the symptoms and complications that make OSA a major public health concern.
CE/CME No: CR-1508
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Explain the diagnostic criteria and clinical presentation of obstructive sleep apnea (OSA).
• Discuss the common screening questionnaires used in clinical practice to identify patients at risk for OSA.
• Describe the common comorbidities associated with OSA.
• Identify the features of pharyngeal structures used in the modified Mallampati classification.
• Know how to provide support and education for the patient in regard to treatment options, weight loss, residual daytime sleepiness, and smoking and alcohol cessation.
FACULTY
Bonnie Dadig is the Chair and Program Director of the Georgia Regents University (GRU) Physician Assistant Department and a PA in the GRU Department of Family Medicine outpatient clinic, Augusta. Morgan Edwards is a recently graduated PA student from GRU. The authors have no financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of August 2015.
Article begins on next page >>
Nearly 22 million Americans are affected by obstructive sleep apnea (OSA), making it the most common sleep disorder. Patients with undiagnosed and untreated OSA are at increased risk for cardiovascular and cerebrovascular health consequences and excessive daytime sleepiness. Here’s how you can address the symptoms and complications that make OSA a major public health concern, and as a result, decrease economic burdens and increase quality of life.
Obstructive sleep apnea (OSA) is an increasingly common diagnosis, with nearly 22 million Americans currently affected.1 In 2010, the diagnosis of OSA was recorded in 5.8 million office visits made by patients ages 20 or older—a 427% increase since 1999.2 While nearly 50% of patients in primary care settings complain of sleep disturbances, up to 80% of OSA cases remain undiagnosed.1,3 The high prevalence of sleep disorders in both the general population and in primary care settings, along with the underdiagnosis of OSA, illustrates the essential role of primary care providers in recognizing the signs and symptoms of OSA and evaluating patients for this condition.1-3
BACKGROUND AND PATHOPHYSIOLOGY
OSA is the most common sleep disorder. Characterized by recurrent episodes of upper airway obstruction during sleep, it results in significantly reduced airflow (hypopnea) or complete cessation of breathing (apnea) for intervals lasting ≥ 10 seconds.4,5 The total number of apneas or hypopneas per hour of sleep—the apnea-hypopnea index (AHI)—is one domain that can be used to classify the severity of OSA (see Table 1).5
Because the human upper airway lacks rigid or bony support, under normal circumstances, the pharyngeal dilator muscles act to oppose the negative pressure within the upper airway during inspiration when awake. And although the striated nature of the pharyngeal dilator muscles leads to a reduction in airway muscle tone during sleep, enough opposition is maintained to prevent airway collapse. In OSA patients, however, the reduced pharyngeal muscle tone fails to offset the negative airway pressure generated during inspiration while asleep, causing recurrent airway obstructions.6 In addition, anatomically narrowed upper airways can further impede airway patency during sleep.4 Causes include macroglossia and/or tonsillar hypertrophy, obesity with fat tissue deposits in the neck that compress the airway, and bony craniofacial abnormalities such as retrognathia.7
Disruptions in breathing from apneas and hypopneas result in hypercapnia and hypoxia, which trigger arousal centers in the central nervous system to increase sympathetic activity. As a result, the patient briefly awakens from sleep, which increases respiratory and pharyngeal dilator muscle tone and ultimately restores airway patency, and ventilation resumes.4
The sympathetic response associated with apneic and hypopneic episodes causes concomitant cardiac stimulation. This leads to transient vasoconstriction and elevations in heart rate and blood pressure. Vascular changes occur over time and result in persistent hypertension and other cardiovascular impairments.4
Undiagnosed and untreated OSA predisposes individuals to a multitude of health consequences and ultimately results in substantial public health and economic burdens. A recent study estimated that the economic cost of moderate-to-severe OSA in the United States is between $65 billion and $165 billion annually.8 Patients with OSA are at increased risk for complications such as systemic and pulmonary hypertension, congestive heart failure, cardiac arrhythmias, coronary artery disease, myocardial infarction, cerebrovascular accident, diabetes, and metabolic syndrome.9 Excessive daytime sleepiness from OSA has also been shown to contribute to increased rates of motor vehicle accidents, poor job performance, impaired cognitive function, and decreased quality-of-life measures.9
Continue for clinical evaluation >>
CLINICAL EVALUATION
Patient History
Clinicians become aware of patients with OSA when they present with associated symptoms or complications. These symptoms are encountered in several settings—as a part of the routine health maintenance examination, as the patient’s presenting complaint, or as a part of the evaluation for another complaint or medical condition.
Regardless of how the symptoms are discovered, all patients with symptoms of and/or positive risk factors for OSA should be assessed via a comprehensive sleep oriented history and physical examination. Symptoms suggestive of OSA include witnessed or reported apneas, snoring, gasping/choking at night, excessive daytime sleepiness not explained by other factors, severity of sleepiness (as determined by the Epworth Sleepiness Scale), nonrefreshing sleep, sleep fragmentation/maintenance insomnia, nocturia, morning headaches, decreased concentration, memory loss, decreased libido, and irritability.10 A recently published study found that nocturnal gasping or choking is the most reliable indicator for identifying patients with OSA.11
The clinical presentation of OSA in certain populations may differ from that of typical adult OSA, requiring a higher degree of clinical suspicion. For instance, pediatric OSA should be suspected in any child who presents with enuresis, poor school performance, failure to thrive, attention-deficit/hyperactivity disorder, or learning disabilities.9 Alternatively, females with OSA have a propensity to emphasize complaints of tiredness, fatigue, or lack of energy rather than report excessive sleepiness per se. These complaints frequently misdirect clinicians toward psychiatric and endocrine diagnoses, without considering the possibility of a sleep disorder.12
Risk Factors
In addition to symptomatology, there are several risk factors associated with OSA that should be considered. The most important are male gender, increasing age (40 to 70), obesity, large neck circumference, and craniofacial and upper airway abnormalities.13 Among the intermediate risk factors are family history of OSA, pregnancy, and Hispanic, Asian, and African-American ethnicity.
Other risk factors associated with OSA include cigarette smoking, alcohol use before sleep, and use of sedative medications.13 Comorbid medical conditions have also been shown to exacerbate and to contribute to OSA; these include GERD, diabetes, congestive heart failure, treatment-refractory hypertension, hypothyroidism, acromegaly, atrial fibrillation, stroke, Down syndrome (due to macroglossia), and pulmonary hypertension.4,10
Screening Questionnaires
Several questionnaires have been validated to screen for OSA. They can be used clinically as part of the sleep-oriented history to quantify a patient’s subjective complaint of sleepiness; however, they are typically more helpful for ruling out sleep apnea than for diagnosing it.11 Two of the most commonly utilized are the Epworth Sleepiness Scale (ESS; http://epworthsleepinessscale.com/epworth-sleepiness-scale.pdf) and the Berlin Questionnaire (www.sleepapnea.org/assets/files/pdf/Berlin%20Questionnaire.pdf).
The ESS was developed to identify patients with excessive daytime sleepiness. It asks patients to rate—on a scale of 0 (would never doze) to 4 (high chance of dozing)—how likely they are to doze off or fall asleep during eight everyday scenarios (ie, sitting and reading; watching TV; sitting inactive in a public place; being a passenger in a vehicle for one or more hours; lying down in the afternoon; sitting and talking to someone; sitting quietly after lunch; or while stopped for a few minutes while driving in traffic).14 The total score can range from 0 to 24. Patients scoring higher than 10 should be referred to a sleep specialist. The ESS can be used to detect symptoms suggestive of OSA; however, it is not specific to OSA and was designed to screen for all sleep disorders.14
In contrast, the Berlin Questionnaire consists of 10 questions that quantify and qualify a patient’s snoring and sleepiness, and it was developed specifically to evaluate for OSA. The questionnaire measures three categories of symptoms: snoring, fatigue, and blood pressure and BMI. A patient is deemed at high risk for OSA when scoring positive in two or more symptom categories, and at low risk when scoring positive in one category.15 However, when OSA is defined as an AHI of ≥ 5 events/hr, the Berlin Questionnaire is 80% sensitive and 46% specific for OSA.11 Therefore, screening questionnaires are useful as supplementary tools but are of limited utility as the sole method for evaluating and diagnosing OSA.
Physical Examination
Physical examination should include assessment for risk factors and comorbidities associated with OSA; this includes measurement of blood pressure, blood glucose, and neck circumference; calculation of BMI and waist-to-hip ratio; and assessment for craniofacial abnormalities. Additionally, cardiovascular, respiratory, and neurologic systems should be evaluated for potential alterations due to OSA.10
Physical exam findings suggestive of OSA are BMI ≥ 30, neck circumference > 17 in for males and > 16 in for females, and craniofacial and upper airway abnormalities. Such anatomic variations include retrognathia, macroglossia (eg, Down syndrome patients), tonsillar hypertrophy (eg, pediatric patients), lateral peritonsillar narrowing, high-arched or narrow hard palate, enlarged uvula, nasal polyps, nasal deviation, turbinate hypertrophy, and oropharynx anatomy consistent with modified Mallampati class III or IV.10 Although OSA is more prevalent in obese patients (those with a BMI ≥ 30), it should be noted that 14.6% of OSA patients are normal weight (BMI < 25) and 34.5%, overweight (BMI 25 to 30).16
The modified Mallampati classification is a straightforward scoring method that was designed to estimate the difficulty of oral intubation based on the extent of mouth opening relative to the size of the tongue.17 According to the protocol set forth by Mallampati and colleagues, assessment is performed with the patient sitting upright with the head in a neutral position, the mouth open, and the tongue maximally protruded without the patient speaking or saying “ahh.”18 The examiner then inspects the pharyngeal structures and classifies the patient’s airway according to the structures visualized. Class I is present when the soft palate, uvula, and pillars are visible; class II when the soft palate and uvula are visible; class III when the soft palate and base of the uvula are visible; class IV when the soft palate is not visible at all (see Figure 1).17,18
An alternative method for observing the pharyngeal structures is via nasopharyngoscopy.19 This procedure can be done in the office setting and utilizes a flexible scope inserted into the nasal cavity and advanced into the pharynx. By providing direct visualization of the upper airway structures, nasopharyngoscopy allows the operator to determine the extent and the location of airway collapse. Imaging by nasopharyngoscopy does not involve any radiation exposure and can be done while the patient is sedated or awake.19
LABORATORY WORKUP AND DIAGNOSIS
Patients deemed high-risk by the sleep-oriented history and physical examination should be subsequently referred to a sleep specialist for objective testing to establish the diagnosis and determine the severity of OSA. Despite the utilization of screening questionnaires, currently there is no consensus on risk stratification to determine when to refer patients with suspected OSA. In other words, no clear thresholds exist on whether a patient is at “low risk” or at “high risk” for OSA based on history and physical examination findings. Therefore, referral is made at the clinician’s discretion, based on the number and severity of OSA symptoms and risk factors.
There are two standard objective sleep study tests: in-laboratory polysomnography (PSG) and home-based sleep studies. The in-laboratory PSG is the gold standard for the diagnosis of OSA.4 The following physiologic signals are measured and interpreted during in-laboratory PSG: electroencephalogram, electro-oculogram, chin electromyogram, airflow, oxygen saturation, respiratory effort, and electrocardiogram.4
Because PSG is resource intensive, home-based sleep studies are an acceptable alternative; however, they should only be considered in patients with a high probability of moderate-to-severe OSA without comorbid sleep disorders or medical conditions. During home-based sleep studies, airflow, respiratory effort, and blood oxygenation are documented.10
The diagnosis of OSA is validated if the AHI on PSG or on home-based sleep studies is > 15 events/h or > 5 events/h with any of the following coexisting signs or symptoms: excessive daytime sleepiness; unintentional sleep episodes during wakefulness; unrefreshing sleep; fatigue; insomnia; waking up holding breath, gasping, or choking; or loud snoring, breathing interruptions, or both, described by the bed partner.10 In addition to diagnosing OSA, PSG and home-based sleep studies are useful in ruling out a variety of conditions that similarly cause excessive daytime sleepiness due to disrupted sleep.
The differential diagnosis of OSA includes, but is not limited to, periodic limb movement disorder, restless legs syndrome, narcolepsy, central sleep apnea, circadian rhythm sleep-wake disorders, respiratory diseases (ie, COPD, asthma), primary snoring, depression, and substance abuse.20
Continue for treatment options >>
TREATMENT OPTIONS
Because OSA is a chronic condition with no definitive cure, lifelong management and follow-up are needed to evaluate treatment adherence/response and the development or resolution of comorbidities. Treatment options for OSA consist primarily of medical (positive airway pressure, use of oral appliances, and pharmacotherapy), behavioral (weight loss, positional therapy, and substance avoidance), and surgical approaches. The patient should actively participate in the treatment and management of this chronic condition.10
Positive Airway Pressure
Regardless of OSA severity, the treatment of choice is positive airway pressure (PAP) during sleep, with continuous PAP (CPAP) being the recommended mode of delivery. PAP is delivered via a face or nasal mask attached to a machine that maintains a constant upper airway pressure, which in turn prevents pharyngeal collapse during sleep. As a result, the upper airway remains patent, and apneas and hypopneas are inhibited.4
PAP can be delivered in three different modes: CPAP, bilevel (BPAP), or autotitrating (APAP). CPAP provides PAP at a fixed rate throughout the respiratory cycle. In contrast, BPAP delivers a preset PAP that is greater during inspiration than during expiration. BPAP is used to alleviate the discomfort of exhaling against the fixed airway pressure of traditional CPAP by delivering lower pressure during expiration than during inspiration. APAP delivers fluctuating positive airway pressures according to changes in the patient’s breathing patterns and is used in patients with complex breathing patterns. CPAP is the recommended mode of airway pressure delivery; however, BPAP or APAP can be considered in patients who cannot tolerate CPAP.10,21
The amount of pressure needed to maintain airway patency is determined during PSG, and the CPAP system is titrated up to this optimal level. Patient adherence is a commonly encountered problem with CPAP therapy due to adverse effects, such as skin irritation, misfit of mask, dry mouth, nasal congestion, and rhinorrhea. Humidification, nasal saline sprays, and mask resizing can help to alleviate such symptoms.4 Follow-up in the early stages should be frequent enough to troubleshoot and remediate problems with the PAP mask, the machine, and adherence. Long-term follow-up for maintenance, adherence, and signs and symptoms of worsening OSA should be scheduled yearly.10
Oral Appliances
Oral appliances (OAs) are an alternative treatment option for OSA patients who are nonadherent with CPAP, fail CPAP or behavioral therapies, experience adverse effects from CPAP, or prefer OAs. Two types of OAs exist: mandibular repositioning appliances and tongue retaining devices. The former anteriorly displaces the mandible relative to the maxilla; the latter anteriorly displaces the tongue only. Regardless of the type selected, OAs are designed to increase the diameter of the pharynx and decrease pharyngeal collapsibility.10
Although less efficacious than traditional CPAP therapy, OAs may be considered in patients with mild-to-moderate OSA.10 Patients with severe OSA should not be offered OAs as a treatment option. Current evidence suggests that OAs can substantially reduce the severity of OSA as well as daytime sleepiness. Additionally, OAs are superior to CPAP in patient preference, which could improve long-term patient adherence to treatment.22
Prior to initiating therapy with an OA, patients should have a dental examination to assess candidacy for, and proper fit of, an OA. Regular follow-ups are required initially to ensure proper fit and comfort of the device. Patients should be instructed to make an office visit if they experience adverse effects such as temporomandibular joint discomfort, tooth tenderness, excessive salivation, and gum irritation.22 Once final adjustments have been made to the fit of the OA, a PSG should be performed to ensure maximal therapeutic efficacy. Follow-up with a dental specialist is advised every six months for the first year and yearly thereafter.10
Pharmacology
According to the practice parameters of the American Academy of Sleep Medicine (AASM), there are currently no pharmacologic agents that prevent or overcome upper airway obstruction well enough to be considered a primary treatment option for OSA.23 The most widely studied pharmacologic agents—selective serotonergic reuptake inhibitors, protriptyline, methylxanthine derivatives (aminophylline and theophylline), estrogen therapy (with or without progesterone), supplemental oxygen, and short-acting nasal decongestants—are not recommended for the primary treatment of OSA.23 Pharmacologic therapy is therefore most beneficial when used as adjuvant therapy (which targets a residual symptom) rather than primary therapy (which targets the underlying cause of the disorder).
There are currently two FDA-approved medications for the treatment of residual excessive daytime sleepiness in patients who are receiving adequate primary treatment (eg, PAP, OAs) and who do not have any other identifiable cause for the sleepiness: modafinil and armodafinil.23,24 The exact mechanism of action of these agents has not been fully characterized, but they are known to act on the central nervous system to enhance alertness (likely by enhancing dopamine signaling).24
Prior to prescribing these medications, other causes of sleepiness must be ruled out; these include suboptimal adherence with PAP, poorly fitting PAP mask, poor sleep hygiene, insufficient sleep, other sleep disorders, and depression.10 Patients should be informed that these drugs should be used in conjunction with pre-existing CPAP therapy. Common adverse effects of modafinil include headache, dizziness, gastrointestinal upset, and insomnia. This medication should be used cautiously in patients with a history of cardiovascular and/or psychologic problems because it can cause serious adverse effects, including chest pain, palpitations, hypertension, hallucinations, aggression, anxiety, depression, and severe skin reactions, such as Stevens-Johnson syndrome and toxic epidermal necrolysis.4,24
Weight Loss
Because obesity is one of the most prevalent risk factors for OSA, weight loss should be recommended for all overweight and obese OSA patients.13,23 Weight loss can be achieved by several different methods, including exercise programs, dietary programs, a combination of the two, and bariatric surgery. A recent meta-analysis found that exercise alone is less successful in reducing AHI compared to dietary modifications; however, a combination of exercise and dietary changes is superior to both individual interventions in reducing AHI.25 This meta-analysis also found that weight-loss interventions improve, but fail to normalize, AHI.25 Thus, weight-loss interventions should be an adjuvant therapy in the treatment of OSA.23
Another recent meta-analysis found that lifestyle interventions, specifically exercise training, might improve OSA severity and symptoms even in the absence of significant weight loss. According to this study, exercise training produced a 32% reduction in AHI in OSA patients and a 42% reduction in AHI when compared to OSA patients who did not partake in exercise training.26 Additionally, OSA patients experienced significant improvements in cardiorespiratory fitness, daytime sleepiness, and sleep efficiency. Because there was no significant reduction in BMI with exercise training, it is likely that there are additional benefits of exercise training that are independent of the effects on weight loss.26
Positional Therapy
Approximately 50% to 60% of OSA patients have positional OSA, which is classified as an increase of 50% or more in the AHI when sleeping in the supine position compared with nonsupine sleep positions.27 It is thought that positional therapy, consisting of any technique that maintains the patient in a nonsupine sleep position, decreases the tendency for the tongue to prolapse and for the airway to collapse.27 Several positional devices exist, including pillows, tennis balls, backpacks, and vibratory chest alarms. Additionally, sleeping in the lateral recumbent position can function as a form of positional therapy.4,21
According to the AASM, less obese, younger patients and those with less severe OSA are more likely to achieve a normalized AHI by sleeping in a nonsupine position; thus, these patients are more likely to benefit from positional therapy than their OSA counterparts.23,27 A recent study found that CPAP therapy is superior to positional therapy in reducing apneic episodes (ie, AHI) in patients with positional OSA.27 Thus, positional therapy is an effective secondary treatment that can be used as an adjuvant therapy to CPAP in patients with mild positional OSA.23,27
Substance Avoidance
All patients with OSA should be advised to avoid alcohol, as it has been shown to increase the duration and frequency of apneic episodes and lower arterial oxyhemoglobin saturation during sleep.28 Not only does alcohol exacerbate preexisting OSA, but it has also been shown to induce frank OSA in patients who snore but do not have OSA at baseline.28 Sedative medications (eg, benzodiazepines) have similar effects on the central nervous system and should also be avoided if possible. Both of these substances have inhibitory actions on the central nervous system, thereby relaxing the pharyngeal muscles and promoting upper airway collapse during sleep.
Because cigarette smoking is an independent risk factor for snoring and thus a presumed risk factor for OSA, smoking cessation is a necessary component of the treatment of OSA.13,29 Cigarette smoking is thought to increase airway inflammation, exacerbate preexisting lung conditions such as COPD and asthma, and induce sleep fragmentation due to the effects of nicotine withdrawal during sleep.29 Alcohol, sedative medications, and cigarettes have all been shown to exacerbate OSA; thus, patient education about the harmful effects of these substances and the benefits of avoiding them is a crucial aspect of OSA therapy.
Surgical Therapies
When both CPAP and OA are inadequate in the management of OSA and/or when obstructive or functional anatomic abnormalities exist, upper airway surgery can be considered for patients. There is currently no consensus about the role of surgery in OSA patients, nor are there clear guidelines or screening questionnaires that accurately predict which patients will benefit the most from upper airway surgery. The appropriate surgical procedure depends on the site of anatomic abnormality and could be a nasal, oral, oropharyngeal, nasopharyngeal, hypopharyngeal, laryngeal, or global airway procedure.10 Some of the most common surgical procedures for the management of OSA are described below.
Uvulopalatopharyngoplasty (UPPP). This surgical procedure involves resecting the entire uvula and the obstructive portion of the soft palate while resizing and reorienting the tonsillar pillars. As a sole procedure, with or without tonsillectomy, UPPP does not reliably normalize the AHI when treating moderate-to-severe OSA.30
Radiofrequency ablation (RFA). RFA is a less invasive variation of UPPP that involves using a temperature-controlled probe to deliver energy to the upper airway tissue (typically the tongue base and/or the soft palate) in an effort to induce palatal stiffening. This procedure can be considered in patients with mild-to-moderate OSA who cannot tolerate or are unwilling to adhere to PAP or OA therapies.30
Maxillo-mandibular advancement (MMA). This operation involves indirect advancement of the anterior pharyngeal tissues (ie, the soft palate, tongue base, and suprahyoid musculature) via their attachment to the maxilla, mandible, and hyoid bone. The simultaneous advancement of the maxilla and mandible are accomplished by sagittal split osteotomies that are stabilized with plates, screws, or bone grafts.31 This surgery is designed to enlarge the retrolingual airway and provide some advancement of the retropalate without directly manipulating the pharyngeal tissues. MMA is indicated for patients with severe OSA who cannot tolerate or are unwilling to adhere to PAP therapy or in whom OAs have been found ineffective.30
Tracheostomy. This procedure consists of creating an airway through the anterior neck into the upper trachea. This opening bypasses the entire upper airway obstruction and thus is 100% effective in curing OSA. However, tracheostomy is typically last-line due to the resulting undesirable alterations in the patient’s physical appearance and to the risks associated with the procedure.5 According to the AASM, this operation should only be considered when other options do not exist, have failed, or are refused, or when this operation is deemed necessary by clinical urgency.30
Hypoglossal nerve stimulation. This therapy uses a surgically implanted device, approved by the FDA in 2014, to detect the patient’s breathing pattern and stimulate the hypoglossal nerve, causing tongue protrusion during inspiration. Tongue movement is controlled, and thus, the airway remains patent during inspiration.32 It is indicated for use in patients with moderate-to-severe OSA who have refused CPAP or for whom CPAP treatment has been unsuccessful.
According to a recently published study, a 68% decrease in median AHI at 12 months postimplantation was reported among those receiving hypoglossal nerve stimulation.33 Additionally, OSA patients subjectively reported decreased sleepiness and an increased quality of life compared to baseline.
The most common adverse effects were transient tongue weakness postoperatively, discomfort related to stimulation, and tongue soreness.33 Its use is contraindicated in patients who are pregnant or plan to become pregnant and in those who will require MRI, who have other implantable devices that may interact with the stimulation system, who have any condition that may affect neurologic control of the upper airway, and who have any anatomic abnormalities that may prevent effective performance of the upper airway stimulation (eg, the presence of complete concentric collapse at the retropalatal airway during endoscopy).32,33 Although its use is promising for the treatment of OSA, hypoglossal nerve stimulation is still a novel option that is not yet readily employed.33
OUTCOMES ASSESSMENT
Clinical judgment should be used to determine the appropriate treatment(s) based on OSA severity and patient preference. Regardless of the treatment option chosen, all OSA patients should have an outcomes assessment performed after the initiation of therapy. Indicators to monitor include resolution of sleepiness, OSA-specific quality-of-life measures, adherence to therapy, avoidance of factors exacerbating OSA (eg, alcohol, tobacco, sedative medications), amount of sleep being obtained, sleep hygiene practices, weight loss, and patient and spousal satisfaction.10 Patients with all levels of OSA severity should receive ongoing management to ensure long-term resolution of symptoms and adherence to treatment. Improvements in primary care treatment, follow-up, and outcomes evaluation are becoming increasingly important to address the symptoms and complications that make OSA a major public health concern.
1. American Sleep Apnea Association. A very short course on sleep apnea. www.sleepapnea.org/i-am-a-health-care-professional.html. Accessed July 21, 2015.
2. Ford ES, Wheaton AG, Cunningham TJ, et al. Trends in outpatient visits for insomnia, sleep apnea, and prescriptions for sleep medications among US adults: findings from the National Ambulatory Medical Care Survey 1999-2010. Sleep. 2014;37(8):1283-1293.
3. NIH State-of-the-Science Conference Statement on Manifestations and Management of Chronic Insomnia in Adults. NIH Consens Sci Statements. 2005;22(2):1-30.
4. Carlucci M, Smith M, Corbridge SJ. Poor sleep, hazardous breathing: an overview of obstructive sleep apnea. Nurse Pract. 2013;38(3):20-28.
5. Institute for Clinical Systems Improvement. Health Care Guideline: Diagnosis and Treatment of Obstructive Sleep Apnea. 6th ed. Bloomington, MN: Institute for Clinical Systems Improvement; 2008.
6. Douglas NJ. Sleep apnea. Harrison’s Principles of Internal Medicine. 18th ed. New York, NY: McGraw-Hill; 2012:2186-2188.
7. US Department of Health and Human Services, National Heart, Lung, and Blood Institute. What causes sleep apnea? www.nhlbi.nih.gov/health/health-topics/topics/sleepapnea/causes.html. Accessed uly 21, 2015.
8. Harvard Medical School, Division of Sleep Medicine. The price of fatigue: the surprising economic costs of unmanaged sleep apnea. December 2010. https://sleep.med.harvard.edu/file_download/100. Accessed July 21, 2015.
9. Pagel JF. Obstructive sleep apnea (OSA) in primary care: evidence-based practice. J Am Board Fam Med. 2007;20(4):392-398.
10. Epstein LJ, Kristo D, Strollo PJ, et al; Adult Obstructive Sleep Apnea Task Force of the American Academy of Sleep Medicine. Clinical guideline for the evaluation, management, and long-term care of obstructive sleep apnea in adults. J Clin Sleep Med. 2009;5(3):263-276.
11. Myers KA, Mrkobrada M, Simel DL. Does this patient have obstructive sleep apnea: the rational clinical examination systematic review. JAMA. 2013;310(7):731-741.
12. Ye L, Pien GW, Weaver TE. Gender differences in the clinical manifestation of obstructive sleep apnea. Sleep Med. 2009;10(10):1075-1084.
13. Young T, Skatrud J, Peppard PE. Risk factors for obstructive sleep apnea in adults. JAMA. 2004;291(16):2013-2016.
14. Johns MW. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep. 1991;14(6):540-545.
15. Netzer NC, Stoohs RA, Netzer CM, et al. Using the Berlin Questionnaire to identify patients at risk for the sleep apnea syndrome. Ann Intern Med. 1999;131(7):485-491.
16. Tufik S, Santos-Silva R, Taddei JA, Bittencourt LR. Obstructive sleep apnea syndrome in the Sao Paulo Epidemiologic Sleep Study. Sleep Med. 2010;11(5):441-446.
17. Samsoon GL, Young JR. Difficult tracheal intubation: a retrospective study. Anaesthesia. 1987;42:487-490.
18. Mallampati SR, Gatt SP, Gugino LD, et al. A clinical sign to predict difficult tracheal intubation: a prospective study. Can Anaesth Soc J. 1985;32(4):429-434.
19. Gregorio MG, Jacomelli M, Figueiredo AC, et al. Evaluation of airway obstruction by nasopharyngoscopy: comparison of the Müller maneuver versus induced sleep. Braz J Otorhinolaryngol. 2007;73(5):618-622.
20. Kline LR. Clinical presentation and diagnosis of obstructive sleep apnea in adults. Up-to-Date. www.uptodate.com/contents/clinical-presentation-and-diagnosis-of-obstructive-sleep-apnea-in-adults. Accessed July 21, 2015.
21. Kryger MH, Malhotra A. Management of obstructive sleep apnea in adults. Up-to-Date. www.uptodate.com/contents/management-of-obstructive-sleep-apnea-in-adults. Accessed July 21, 2015.
22. Lim J, Lasserson TJ, Fleetham J, Wright JJ. Oral appliances for obstructive sleep apneoa. Cochrane Database Syst Rev. 2006;1:CD004435.
23. Morgenthaler TI, Kapen S, Lee-Chiong T, et al; Standards of Practice Committee; American Academy of Sleep Medicine. Practice parameters for the medical therapy of obstructive sleep apnea. Sleep. 2006;29(8): 1031-1035.
24. Pepin JL. Evaluation and management of residual sleepiness in obstructive sleep apnea. Up-to-Date. www.uptodate.com/contents/evaluation-and-management-of-residual-sleepiness-in-obstructive-sleep-apnea. Accessed July 21, 2015.
25. Araghi MH, Chen YF, Jagielski A, et al. Effectiveness of lifestyle interventions on obstructive sleep apnea (OSA): systematic review and meta-analysis. Sleep. 2013;36(10):1553-1562.
26. Iftikhar IH, Kline CE, Youngstedt SD. Effects of exercise training on sleep apnea: a meta-analysis. Lung. 2014;192(1):175-184.
27. Ha SC, Hirai HW, Tsoi KK. Comparison of positional therapy versus continuous positive airway pressure in patients with positional obstructive sleep apnea: a meta-analysis of randomized trials. Sleep Med Rev. 2014; 18(1):19-24.
28. Issa FG, Sullivan CE. Alcohol, snoring, and sleep apnea. J Neurol Neurosurg Psychiatry. 1982;45(4):353-359.
29. Lin Y, Li QY, Zhang XJ. Interaction between smoking and obstructive sleep apnea: not just participants. Chin Med J. 2012;125(17):3150-3156.
30. Aurora RN, Casey KR, Kristo D, et al; American Academy of Sleep Medicine. Practice parameters for the surgical modifications of the upper airway for obstructive sleep apnea in adults. Sleep. 2010;33(10):1408-1413.
31. Caples SM, Rowley JA, Prinsell JR, et al. Surgical modifications of the upper airway for obstructive sleep apnea in adults: a systematic review and meta-analysis. Sleep. 2010;33(10):1396-1407.
32. US Food and Drug Administration. Recently-approved devices. Inspire upper airway stimulation - P130008. www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/Recently-ApprovedDevices/ucm398321.htm. Accessed July 21, 2015.
33. Strollo PJ, Soose RJ, Maurer JT, et al. Upper-airway stimulation for obstructive sleep apnea. N Engl J Med. 2014;370(2):139-149.
1. American Sleep Apnea Association. A very short course on sleep apnea. www.sleepapnea.org/i-am-a-health-care-professional.html. Accessed July 21, 2015.
2. Ford ES, Wheaton AG, Cunningham TJ, et al. Trends in outpatient visits for insomnia, sleep apnea, and prescriptions for sleep medications among US adults: findings from the National Ambulatory Medical Care Survey 1999-2010. Sleep. 2014;37(8):1283-1293.
3. NIH State-of-the-Science Conference Statement on Manifestations and Management of Chronic Insomnia in Adults. NIH Consens Sci Statements. 2005;22(2):1-30.
4. Carlucci M, Smith M, Corbridge SJ. Poor sleep, hazardous breathing: an overview of obstructive sleep apnea. Nurse Pract. 2013;38(3):20-28.
5. Institute for Clinical Systems Improvement. Health Care Guideline: Diagnosis and Treatment of Obstructive Sleep Apnea. 6th ed. Bloomington, MN: Institute for Clinical Systems Improvement; 2008.
6. Douglas NJ. Sleep apnea. Harrison’s Principles of Internal Medicine. 18th ed. New York, NY: McGraw-Hill; 2012:2186-2188.
7. US Department of Health and Human Services, National Heart, Lung, and Blood Institute. What causes sleep apnea? www.nhlbi.nih.gov/health/health-topics/topics/sleepapnea/causes.html. Accessed uly 21, 2015.
8. Harvard Medical School, Division of Sleep Medicine. The price of fatigue: the surprising economic costs of unmanaged sleep apnea. December 2010. https://sleep.med.harvard.edu/file_download/100. Accessed July 21, 2015.
9. Pagel JF. Obstructive sleep apnea (OSA) in primary care: evidence-based practice. J Am Board Fam Med. 2007;20(4):392-398.
10. Epstein LJ, Kristo D, Strollo PJ, et al; Adult Obstructive Sleep Apnea Task Force of the American Academy of Sleep Medicine. Clinical guideline for the evaluation, management, and long-term care of obstructive sleep apnea in adults. J Clin Sleep Med. 2009;5(3):263-276.
11. Myers KA, Mrkobrada M, Simel DL. Does this patient have obstructive sleep apnea: the rational clinical examination systematic review. JAMA. 2013;310(7):731-741.
12. Ye L, Pien GW, Weaver TE. Gender differences in the clinical manifestation of obstructive sleep apnea. Sleep Med. 2009;10(10):1075-1084.
13. Young T, Skatrud J, Peppard PE. Risk factors for obstructive sleep apnea in adults. JAMA. 2004;291(16):2013-2016.
14. Johns MW. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep. 1991;14(6):540-545.
15. Netzer NC, Stoohs RA, Netzer CM, et al. Using the Berlin Questionnaire to identify patients at risk for the sleep apnea syndrome. Ann Intern Med. 1999;131(7):485-491.
16. Tufik S, Santos-Silva R, Taddei JA, Bittencourt LR. Obstructive sleep apnea syndrome in the Sao Paulo Epidemiologic Sleep Study. Sleep Med. 2010;11(5):441-446.
17. Samsoon GL, Young JR. Difficult tracheal intubation: a retrospective study. Anaesthesia. 1987;42:487-490.
18. Mallampati SR, Gatt SP, Gugino LD, et al. A clinical sign to predict difficult tracheal intubation: a prospective study. Can Anaesth Soc J. 1985;32(4):429-434.
19. Gregorio MG, Jacomelli M, Figueiredo AC, et al. Evaluation of airway obstruction by nasopharyngoscopy: comparison of the Müller maneuver versus induced sleep. Braz J Otorhinolaryngol. 2007;73(5):618-622.
20. Kline LR. Clinical presentation and diagnosis of obstructive sleep apnea in adults. Up-to-Date. www.uptodate.com/contents/clinical-presentation-and-diagnosis-of-obstructive-sleep-apnea-in-adults. Accessed July 21, 2015.
21. Kryger MH, Malhotra A. Management of obstructive sleep apnea in adults. Up-to-Date. www.uptodate.com/contents/management-of-obstructive-sleep-apnea-in-adults. Accessed July 21, 2015.
22. Lim J, Lasserson TJ, Fleetham J, Wright JJ. Oral appliances for obstructive sleep apneoa. Cochrane Database Syst Rev. 2006;1:CD004435.
23. Morgenthaler TI, Kapen S, Lee-Chiong T, et al; Standards of Practice Committee; American Academy of Sleep Medicine. Practice parameters for the medical therapy of obstructive sleep apnea. Sleep. 2006;29(8): 1031-1035.
24. Pepin JL. Evaluation and management of residual sleepiness in obstructive sleep apnea. Up-to-Date. www.uptodate.com/contents/evaluation-and-management-of-residual-sleepiness-in-obstructive-sleep-apnea. Accessed July 21, 2015.
25. Araghi MH, Chen YF, Jagielski A, et al. Effectiveness of lifestyle interventions on obstructive sleep apnea (OSA): systematic review and meta-analysis. Sleep. 2013;36(10):1553-1562.
26. Iftikhar IH, Kline CE, Youngstedt SD. Effects of exercise training on sleep apnea: a meta-analysis. Lung. 2014;192(1):175-184.
27. Ha SC, Hirai HW, Tsoi KK. Comparison of positional therapy versus continuous positive airway pressure in patients with positional obstructive sleep apnea: a meta-analysis of randomized trials. Sleep Med Rev. 2014; 18(1):19-24.
28. Issa FG, Sullivan CE. Alcohol, snoring, and sleep apnea. J Neurol Neurosurg Psychiatry. 1982;45(4):353-359.
29. Lin Y, Li QY, Zhang XJ. Interaction between smoking and obstructive sleep apnea: not just participants. Chin Med J. 2012;125(17):3150-3156.
30. Aurora RN, Casey KR, Kristo D, et al; American Academy of Sleep Medicine. Practice parameters for the surgical modifications of the upper airway for obstructive sleep apnea in adults. Sleep. 2010;33(10):1408-1413.
31. Caples SM, Rowley JA, Prinsell JR, et al. Surgical modifications of the upper airway for obstructive sleep apnea in adults: a systematic review and meta-analysis. Sleep. 2010;33(10):1396-1407.
32. US Food and Drug Administration. Recently-approved devices. Inspire upper airway stimulation - P130008. www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/Recently-ApprovedDevices/ucm398321.htm. Accessed July 21, 2015.
33. Strollo PJ, Soose RJ, Maurer JT, et al. Upper-airway stimulation for obstructive sleep apnea. N Engl J Med. 2014;370(2):139-149.
Gout: A Clinical Overview
Gouty arthritis is a common form of inflammatory arthritis, occurring more frequently in men than in women. The condition has a male–female ratio of 3 or 4 to 1, although that ratio narrows as adults age; because the uricosuric effects of estrogen decline with menopause, the risk for gout increases in postmenopausal women.1,2 Mean age at disease onset is 40 to 60 in men,3,4 with onset in women averaging seven years later.5
Apart from the pain and loss of function associated with this disorder of purine metabolism6 and the risk for a chronic form of the disease, gout is almost universally linked with serious comorbidities that require timely intervention. These include hypertension, dyslipidemia, hyperglycemia and diabetes, obesity, metabolic syndrome, cardiovascular disease (CVD), renal insufficiency, and coronary heart disease (CHD).2,3,7 The presence of gout is independently associated with a risk for acute myocardial infarction (AMI) and increased rates of all-cause mortality.3,8-10
EPIDEMIOLOGY
In 2007, the National Arthritis Data Workgroup11,12 estimated that about three million US adults had had “self-reported gout” in the previous year. An estimated six million US adults have been diagnosed with gout,2,11 and its incidence and prevalence are increasing. The incidence of primary gout more than doubled between 1977-1978 and 1995-1996,13 especially affecting the aging population. The prevalence of gout among 1,000 managed care patients ages 65 to 74 increased by at least 30% between 1990 and 1999, while prevalence among those older than 75 almost doubled during this same period.14
Numerous factors appear to contribute to these trends, including aging of the population, dietary trends (ie, increased consumption of red meat, organ food, game, and shellfish and reduced consumption of low-fat dairy products), presence of certain comorbid conditions (ie, hypertension, dyslipidemia, diabetes, metabolic syndrome, end-stage renal disease), the increasing prevalence of obesity in younger adults, use of specific prescription medications, and increased incidence of organ transplantation.1,7,8,15-19
The body’s underexcretion or overproduction of uric acid (a byproduct of purine metabolism12) can lead to hyperuricemia. This condition, defined as a serum urate level exceeding 7.0 mg/dL in men or 6.0 mg/dL in women20,21 (levels above 9.0 mg/dL are considered very high22), is the primary risk factor for gout.8,23,24 As with gout, the incidence of hyperuricemia has increased in recent years,20 with researchers attributing the trend to worldwide popularization of the Westernized diet (particularly use of high-fructose corn syrup20,25) and increased use of certain medications, including thiazide diuretics, cyclosporine, and low-dose aspirin.2,20,25,26
As serum urate levels rise, the patient with hyperuricemia may experience urate supersaturation, often followed by crystallization of the excess urate into monosodium urate (MSU) crystals. Subsequently, circulating MSU crystals may deposit in body tissues, especially in the joint spaces. The body’s ensuing inflammatory response to the MSU deposits is gout.20
In addition to hyperuricemia, risk factors for gout include a high-purine diet, habitual alcohol consumption (especially beer and fortified wines27), diuretic therapy (particularly in patients with heart failure or renal insufficiency), obesity, hypertension, and high levels of fructose consumption.7,28 Additionally, cyclosporine use in an organ transplant recipient, poorly controlled uric acid levels, and a long history of gout increase the patient’s risk for chronic tophaceous gout.24,29 Tophi may be more common in a patient with a history of organ transplantation.16
Genetic variants are currently being investigated to possibly identify a predisposition to gout. The most significant genetic factors appear to involve mechanisms that regulate serum uric acid levels—particularly urate underexcretion.23 Other factors that contribute to underexcretion or overproduction of uric acid are shown in Table 1.1,15,26,30-33
A dynamic relationship exists between gout and a number of pathologic processes. According to researchers investigating nearly 178,000 patients with gout in a managed care database, 36% had hypertension, 27% had dyslipidemia, and 15% had diabetes.8 In a smaller cohort study conducted in Spain and Mexico, it was demonstrated that 93% of patients with gout had one or more associated diseases, in order of decreasing frequency: hypertriglyceridemia, obesity, hypertension, metabolic syndrome, hyperglycemia, chronic renal failure, diabetes, and ischemic heart disease.3
Of particular clinical importance in this study was a finding that the first gout attack generally preceded the diagnosis of the associated diseases.3 Thus, a diagnosis of gout should lead the primary care provider to discuss modifiable risk factors with the patient—but also to investigate for comorbid illnesses that may require timely management.2
In a 12-year-long prospective study of more than 50,000 men participating in the Health Professionals Follow-Up Study,9 it was found that men with gout had a 28% increased risk for all-cause mortality, a 38% increased risk for CVD-related death, and a 55% increased risk for CHD-related death, compared with men who did not have gout (excluding other risk factors).9 Similarly, researchers for the Multiple Risk Factor Intervention Trial10 demonstrated a clinically significant association between gout and an increased risk for AMI: 10.5% of men with gout, compared with 8.43% of men without gout, had an AMI during mean follow-up of 6.5 years.10
THE STAGES
The four stages of gout are asymptomatic hyperuricemia, acute gout, intercritical gout, and chronic tophaceous gout.20
Only a small percentage (0.5% to 4.5%) of patients with asymptomatic hyperuricemia will develop acute gout.28 Nevertheless, any patient with serum urate greater than 6.8 mg/dL is at risk for the deposition of MSU crystals into body tissues and the potential associated organ damage—even patients without symptoms. There is currently no evidence-based method to determine which patients with asymptomatic hyperuricemia will experience disease progression.16
Acute gout develops when deposition of MSU crystals in the joints initiates an inflammatory response. In the typical history, the patient experiences sudden-onset severe pain, swelling, and erythema. The pain often starts in the middle of the night or early morning,34 waking the patient from sleep and peaking within 24 hours of onset. At this time, the patient is often unable to bear weight comfortably on the affected joint. The patient may also report fever and flu-like malaise resulting from the release of interleukin 1- (IL-1), IL-1 receptor, cytokines, and prostaglandins.16,24,35 Usually in these early attacks, symptoms resolve spontaneously within three to 14 days.16,24
After resolution of an acute attack, the patient enters the intercritical stage, another asymptomatic stage that may last for months or years—or indefinitely. During the intercritical stage, MSU crystal deposition continues, adding crystals in and around the affected joint or joints, possibly continuing to inflict damage (in some patients, substantial), and in many cases resulting in additional attacks and pain.16 Any subsequent acute gout attacks the patient may experience are likely to last longer than the initial attack and to involve additional joints or tendons.24
Some patients, especially those who do not receive adequate treatment for hyperuricemia,2 progress to develop chronic tophaceous gout. This is a deforming disease process in which the joints may become stiff and swollen, and subcutaneous nodules or whitish-yellow intradermal deposits may be present under taut skin, anywhere in the body.16
PATIENT PRESENTATION AND HISTORY
Typically, a patient with gout will present with a chief complaint of a painful, tender, inflamed joint (classically described in Latin as calor, rubor, dolor, et tumor6). However, clinicians must also be aware of unusual presentations and consider gout in the differential whenever a patient with a history of gout or pertinent risk factors presents with unexplained clinical findings.32 The history of present illness will vary according to the stage of the disease.
About 90% of recognized initial attacks of gout are monoarticular, usually occurring in one of the lower extremities.16 While the first metatarsophalangeal (MTP) joint is affected in about 50% of gout cases (podagra, the Greek term for gout),2,35 eventually patients with gout have a 90% chance of involvement with the MTP joint (see Figure 1). According to Zhang et al,26 patients with hyperuricemia and an affected MTP joint have an 82% chance of having gout.2,26
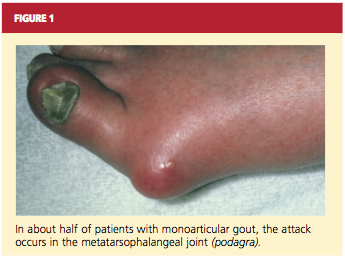
Because such a large proportion of patients have the classic presentation of rapid-onset warmth, redness, and tenderness at the MTP, knee, or ankle and surrounding soft tissue, cases with a differing presentation are likely to be misdiagnosed or overlooked, or a correct diagnosis is delayed.26 In many documented cases, gout was the ultimate diagnosis—but one that was reached only incidentally because of unusual clinical presentation, ranging from entrapment neuropathy to a pancreatic mass.32
The presence of hyperuricemia and other risk factors must be investigated. Also relevant in the history of an acute gout attack may be a preceding event that has caused damage or stress to the joint, such as infection, trauma, or surgery. Other possible triggers for an attack include alcohol ingestion, acidosis, use of IV contrast media, diuretic therapy, chemotherapy, recent hospitalization or surgery, and initiation or termination of urate-lowering therapy with the xanthine oxidase inhibitor allopurinol.2,30 According to Primatesta et al,8 the risk for flares is increased in patients with cardiometabolic comorbidities.
The medication history of a patient with gout may include low-dose aspirin (but not standard-dose aspirin, which is uricosuric2), diuretics, cyclosporine, cytotoxic agents, and vitamin B12, which may contribute to hyperuricemia.24 Additionally, ethambutol, pyrazinamide, levodopa, nicotinic acid, didanosine, niacin, and warfarin may raise uric acid levels.15,30,33
Medical history should include a thorough assessment of the comorbidities associated with gout. In addition to the conditions mentioned previously, patients with a history of polycystic kidney disease, dehydration, lactic acidosis, hyperparathyroidism, toxemia of pregnancy, hypothyroidism, or sarcoidosis may have elevated urate levels due to underexcretion—and thus may be vulnerable to gout.30 History of gout in a first-degree relative is associated with an increased risk for gout.36
The social history should address alcohol use or abuse. The clinician should also inquire about how gout is impacting the daily life of the patient. Diet and exercise habits should be assessed37 (see “Patient Education,” below).
PHYSICAL EXAMINATION
The physical exam begins with evaluation of the skin and extremities for the classic features of gout. Affected joints will be exquisitely tender, and patients may be febrile. Most cases are monoarticular, but polyarticular involvement is likely in patients with advanced disease (and can, particularly in women, be mistaken for rheumatoid arthritis).2,16 In patients with chronic tophaceous gout, there may be whitish-yellow skin deposits, subcutaneous nodules, and areas of taut skin. The lower-extremity joints and tendons, as well as the wrists, fingers, and elbows, are commonly affected16 (see Figure 2).
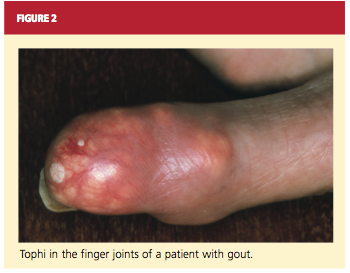
DIAGNOSIS
Diagnostic criteria that are currently available (and have long been in use) include the American College of Rheumatology/American Rheumatism Association (ACR/ARA) preliminary criteria,34 the New York criteria,21 and the Rome criteria.38 The specifics of each are listed in Table 2.21,34,38,39
The gold standard for gout diagnosis is detection of MSU crystals in a sample of synovial fluid aspirated from the affected joint or from a tophus and examined by polarized light microscopy.24 This is of significant importance to the clinician who is faced with a questionable diagnosis.16 However, crystal visualization is not ordinarily available to the primary care clinician,2,17,40 and it is not always necessary if a careful history and physical exam are conducted in a patient with hyperuricemia or other risk factors for gout. A presumptive diagnosis may be acceptable in a patient with the classic presentation of acute gout: rapid onset of severe pain in a swollen, erythematous joint and symptoms peaking within 24 hours. The presence of tophi is pathognomonic for chronic tophaceous gout.41
In cases of questionable or unusual manifestation of gout, however, various imaging techniques and crystal visualization may be indicated.32
In order to compare the effectiveness of the latter technique with conventional diagnostic criteria for gout, Malik et al39 conducted a pilot study involving 82 patients who had undergone synovial fluid analysis with polarized light microscopy. Patients were surveyed about the clinical features of their disease, as listed in the three standard sets of criteria for diagnosis of gout. Compared with the “gold standard” of urate crystal detection (which is one of the Rome criteria38), the study authors found the ACR/ARA preliminary criteria,34 the New York criteria,21 and the Rome criteria38 generally unsatisfactory.
In the study, among patients with confirmed presence of MSU crystals:
• 87% reported more than one attack of acute arthritis (ACR/ARA34)
• 86% reported monoarthritis attack (ACR/ARA34)
• 89% had hyperuricemia (ACR/ARA34 and Rome,38 with the latter giving effective, specific parameters)
• 100% had negative results on joint fluid culture (ACR/ARA34)
• 90% reported an attack starting at night (ACR/ARA34).
The positive predictive values for these signs and symptoms are 38%, 39%, 74%, 50%, and 45%, respectively, according to Malik et al.39 The presence of tophi (cited by all three sets of criteria but “proven or suspected” in the ACR/ARA34) had the highest positive predictive value for gout (91%) and a likelihood ratio of 15.56, which was at least three times higher than any of the other listed criteria. A verified response to colchicine, one of the New York criteria,21 had the second highest positive predictive value at 86%.39
In summary, the ACR/ARA,34 the New York,21 and the Rome criteria38 had specificity of 79%, 83%, and 89%, respectively; sensitivity of 70%, 70%, and 67%, respectively; and positive predictive values for gout of 66%, 70%, and 77%, respectively. The Rome criteria38 had the highest specificity and highest positive predictive value, perhaps making them most helpful for clinicians who lack access to synovial fluid analysis.
DIFFERENTIAL DIAGNOSIS
Conditions to be considered and ruled out before a diagnosis of gout can be made are:
• Pseudogout
• Septic arthritis
• Psoriatic arthritis
• Rheumatoid arthritis
• Erosive osteoarthritis
• Bacterial cellulitis
• Sarcoid arthropathy.16,28,42,43
Unlike gout (in which compensated polarized light microscopy reveals needle-shaped urate crystals with strong negative birefringence), pseudogout is characterized by calcium pyrophosphate dihydrate crystals; these are rhomboid-shaped, with weak positive birefringence.42 Additionally, radiographic imaging will reveal soft tissue swelling and chondrocalcinosis of the joint in pseudogout.44
The patient with septic arthritis, most likely affecting the knee, will have a white blood cell (WBC) count exceeding 50,000/mm3 and a positive culture of the synovial fluid, with absence of crystals.28
Chronic tophaceous gout can mimic rheumatoid arthritis in appearance and joint distribution, and patients affected by either condition may develop a positive rheumatoid factor. Examination of synovial fluid for MSU crystals and radiographic imaging will be of value in making a distinction.
Osteoarthritis is usually evidenced by joint space narrowing on x-ray.42
Bacterial cellulitis will present similarly to gout, but the erythema of bacterial cellulitis will more likely extend beyond the involved joint.16
Sarcoid arthropathy often presents as a polyarthritis, as in advanced gouty arthritis. However, in sarcoid arthropathy, serum calcium and angiotensin-converting enzyme will likely be elevated.43 Synovial or tendon sheath biopsy will show non-caseating granulomas, which are the hallmark for sarcoid disease. Additionally, joint fluid analysis will demonstrate a predominance of mononuclear or polymorphonuclear cells.43
Diagnostic Tests
Diagnostic tests to consider are analysis and culture of the synovial fluid, complete blood count (CBC), blood urea nitrogen (BUN), creatinine, radiography, ultrasonography, serum uric acid, and blood culture if septic arthritis is suspected.28 While serum urate levels may be normal during an acute gout attack, measurement may still be helpful for comparison, since elevation is a likely finding two weeks after an attack—if the patient was, in fact, experiencing an acute gout attack.42
Since renal dialysis increases the risk for gout, pseudogout, and septic arthritis, synovial fluid analysis is essential in patients undergoing renal dialysis.42
Various imaging techniques may aid in confirming a diagnosis of gout and monitoring its progression, but further studies are needed to more clearly define the role of these techniques in management of gout.45 Plain radiographic evidence of asymmetric swelling in a joint (one of the ACR/ARA preliminary criteria34) was shown to have a 60% positive predictive value for a diagnosis of gout.39 Late in the disease process, an affected joint may be affected by characteristic “punched out” intra-articular lesions, with a normal amount of joint space.45
Ultrasound is a safe and inexpensive test that can reveal soft tissue edema and increased vascularity during an acute gout attack. Chronic changes include the double contour sign and tophus-like lesions surrounded by a thin, anechoic rim.45
CT will also show tophi and bony erosion. While CT is more specific than other techniques, it is also more expensive and exposes the patient to increased radiation. MRI can help monitor the complications of gout, especially entrapment neuropathies.45
TREATMENT/MANAGEMENT
According to current evidence, treatment is not indicated for asymptomatic hyperuricemia.16
Acute Gout Management
Pharmacologic treatments available for an acute gout attack include NSAIDs, colchicine, and local or systemic corticosteroids.24,46 At the onset of an attack, patients should start high-dose NSAID therapy, and continue for two to three days after symptoms are resolved.6 Oral indomethacin (50 mg tid) or oral ibuprofen (800 mg tid) are both reasonable options.6 It may be prudent to consider a proton pump inhibitor (eg, omeprazole) to protect the gastric mucosa in patients who are susceptible to gastrointestinal problems.27
In addition to high-dose NSAID therapy, adding colchicine (1.2 mg by mouth at onset of symptoms, followed by 0.6 mg one hour later) has proven to be effective in relieving the symptoms of gout, but its serious gastrointestinal adverse effects, particularly diarrhea, must be considered.6,47
In patients with monoarticular gout who cannot tolerate NSAIDs, intra-articular aspiration and corticosteroid injections may provide relief.
Long-acting triamcinolone, administered by intra-articular injection, has been found to relieve pain and inflammation in patients with gout. Septic arthritis must be ruled out by way of joint aspiration and culture before injection of corticosteroids.47
Oral or IM-administered corticosteroids may be considered for patients with polyarticular involvement. Prednisone (60 mg/d, tapered over 10 days) is an appropriate option for outpatients or inpatients; methylprednisone (80 to 120 mg IM) may be suitable for inpatients.6 Again, septic arthritis must be ruled out before corticosteroids are administered.47
For the patient who is currently taking a thiazide diuretic for hypertension, substituting a different medication may be warranted; the angiotensin receptor blocker losartan, for example, has uricosuric action.27,29,47 Nonpharmacologic strategies, such as rest, ice, elevation, and avoiding trauma to the affected joint, are also recommended.27
Of note, allopurinol therapy should be neither initiated nor discontinued during an acute gout attack.27
Management of Chronic and Intercritical Gout
Urate-lowering therapy, such as allopurinol (50 to 300 mg/d29), should be considered for patients who experience frequent attacks (ie, three or more per year), patients with chronic tophi, patients with radiographically demonstrated joint damage,47 or patients with a documented state of uric acid overproduction.29
Allopurinol dosage should be adjusted based on creatinine clearance; dosing as high as 800 mg/d has been recommended in patients with normal renal function.2 Again, allopurinol should never be started or discontinued during an acute attack,27 because abrupt fluctuations in uric acid levels may heighten the inflammation. The target serum urate level is 6.0 mg/dL.29
Febuxostat, which received FDA approval in 2009, was the first oral urate-lowering treatment to be approved since the 1960s. Like allopurinol, this nonpurine xanthine oxidase inhibitor blocks uric acid synthesis.48,49 In a trial reported by Becker et al,50 67% of patients who took febuxostat 80 mg/d reached the target serum urate level (ie, < 6.0 mg/dL), compared with 45% of those who took 40 mg/d of febuxostat and 42% of those taking 300 mg/d of allopurinol. While incidence of adverse events was low in all treatment groups, Hu and Tomlinson51 report that febuxostat is tolerable in patients who are hypersensitive to allopurinol. As with other urate-lowering medications, gout flares are common during the early period of febuxostat use.51
For patients with gout that does not respond to conventional urate-lowering therapy, new options are being introduced. Two agents, each a recombinant form of the enzyme urate oxidase, are designed to convert uric acid into allantoin, which can then be excreted in the urine. Late in 2010, one of these agents, pegloticase, was approved for use in patients with refractory gout.48 In one clinical trial, tophi were reported dissolved in 40% of patients who took pegloticase, but 58% of patients did not achieve the targeted response (ie, serum urate < 6.0 mg/dL), and 77% of patients experienced gout flares.52 Infusion reactions occurred in 26% to 31% of patients, and Reinders and Jansen52 recommended the clinical evaluation of glucocorticoids and other anti-inflammatory agents to prevent the formation of antibodies involved in these reactions.
The second agent, rasburicase, has been approved for treatment and prevention of acute hyperuricemia in adult cancer patients. Rasburicase is now being investigated for use in patients with nonresponsive tophaceous gout.53-55 It can be administered in the form of monthly infusions.54
Patient Education
Educating the patient about modifiable risk factors, such as diet, alcohol consumption, and adherence to the medication regimen, should be a priority.
Patients should be encouraged to target and maintain an ideal body weight, through diet and moderate physical exercise, as a strategy to normalize serum urate levels.27,47 However, they should be advised to avoid “crash dieting,” as this may precipitate a gout attack.27 In the recommended low-purine diet, consumption of red meat and shellfish is restricted,17 whereas consumption of soy, nonfat milk and other low-fat dairy products, cherries and other fruits, and increased vegetable protein is encouraged.31,37 Consumption of alcohol, especially beer and fortified wines, should be limited.27,47
Avoiding trauma to joints affected by gout (including the stress of bearing excess weight) can help patients limit future attacks.7,27
CONCLUSION
Patients with gout often have the characteristic presentation of an acutely tender, inflamed joint, but since gout is a systemic disorder, the clinician must also consider the possibility of gout in almost any organ system. Gout is a common disease, and its diagnosis can alert the astute clinician to investigate for certain metabolic disorders requiring intervention. Hyperlipidemia, metabolic syndrome, hypertension, chronic kidney disease, obesity, cardiovascular disease, and diabetes are all conditions associated with gout.
Recognizing the opportunity to offer preventive care measures and recommend lifestyle modifications to the patient with gout allows the clinician to play an important role in the patient’s care.
REFERENCES
1. Bhole V, de Vera M, Rahman MM, et al. Epidemiology of gout in women: fifty-two–year followup of a prospective cohort. Arthritis Rheum. 2010;62(4):1069-1076.
2. Neogi T. Clinical practice: gout. N Engl J Med. 2011;364(5):443-452.
3. Hernández-Cuevas CB, Roque LH, Huerta-Sil G, et al. First acute gout attacks commonly precede features of the metabolic syndrome. J Clin Rheumatol. 2009;15(2):65-67.
4. Louthrenoo W, Kasitanon N, Sukitawut W, Wichainun R. A clinical study of crystal-proven gouty arthritis in a university hospital. J Med Assoc Thai. 2003;86(9):868-875.
5. De Souza AW, Fernandes V, Ferrari AJ. Female gout: clinical and laboratory features. J Rheumatol. 2005;32(11):2186-2188.
6. Kurakula PC, Keenan RT. Diagnosis and management of gout: an update. J Musculoskel Med. 2010;27(10). www.musculoskeletalnet work.com/display/article/1145622/1692895. Accessed June 14, 2011.
7. Choi HK, Atkinson K, Karlson EW, Curhan G. Obesity, weight change, hypertension, diuretic use, and risk of gout in men: the Health Professionals Follow-up Study. Arch Intern Med. 2005;165(7):742-748.
8. Primatesta P, Plana E, Rothenbacher D. Gout treatment and comorbidities: a retrospective cohort study in a large US managed care population. BMC Musculoskelet Disord. 2011 May 20;12(1):103. [Epub ahead of print]
9. Choi HK, Curhan G. Independent impact of gout on mortality and risk for coronary heart disease. Circulation. 2007;116(8):894-900.
10. Krishnan E, Baker JF, Furst DE, Schumacher HR. Gout and the risk of acute myocardial infarction. Arthritis Rheum. 2006;54(8):2688-2696.
11. Lawrence RC, Felson DT, Helmick CG, et al; National Arthritis Data Workgroup. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008;58(1):26-35.
12. CDC. Gout. www.cdc.gov/arthritis/basics/gout.htm. Accessed June 14, 2011.
13. Arromdee E, Michet CJ, Crowson CS, et al. Epidemiology of gout: is the incidence rising? J Rheumatol. 2002;29(11):2403-2406.
14. Wallace KL, Riedel AA, Joseph-Ridge N, Wortmann R. Increasing prevalence of gout and hyperuricemia over 10 years among older adults in a managed care population. J Rheumatol. 2004; 31(8):1582-1587.
15. Weaver AL. Epidemiology of gout. Cleve Clin J Med. 2008;75 suppl 5:S9-S12.
16. Mandell BF. Clinical manifestations of hyperuricemia and gout. Cleve Clin J Med. 2008;75 suppl 5:S5-S8.
17. Choi HK, Atkinson K, Karlson EW, et al. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med. 2004;350(11):1093-1103.
18. Demarco MA, Maynard JW, Huizinga MM, et al. Younger age at gout onset is related to obesity in a community-based cohort. Arthritis Care Res (Hoboken). 2011 Apr 11; [Epub ahead of print].
19. Brook RA, Forsythe A, Smeeding JE, Lawrence Edwards N. Chronic gout: epidemiology, disease progression treatment and disease burden. Curr Med Res Opin. 2010;26(12):2813-2821.
20. Sachs L, Batra KL, Zimmermann B. Medical implications of hyperuricemia. Med Health R I. 2009;92(11):353-355.
21. Kellgren JH, Jeffrey MR, Ball J, eds. The Epidemiology of Chronic Rheumatism: Atlas of Standard Radiographs of Arthritis. Oxford: Blackwell; 1963:327.
22. Wu EQ, Patel PA, Mody RR, et al. Frequency, risk, and cost of gout-related episodes among the elderly: does serum uric acid level matter? J Rheumatol. 2009;36(5):1032-1040.
23. Riches PL, Wright AF, Ralston SH. Recent insights into the pathogenesis of hyperuricaemia and gout. Hum Mol Genet. 2009;18(R2):R177-R184.
24. Schumacher HR Jr. The pathogenesis of gout. Cleve Clin J Med. 2008;75 suppl 5:S2-S4.
25. Nakagawa T, Hu H, Zharikov S, et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Renal Physiol. 2006;290(3):F625-F631.
26. Zhang W, Doherty M, Pascual E, et al; EULAR (European League Against Rheumatism) Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part I: Diagnosis. Ann Rheum Dis. 2006;65(10):1301-1311.
27. Jordan KM, Cameron JS, Snaith M, et al. British Society for Rheumatology and British Health Professionals in Rheumatology guideline for the management of gout. Rheumatology. 2007;46(8):1372-1374.
28. Eggebeen AT. Gout: an update. Am Fam Physician. 2007;76(6):801-808.
29. Terkeltaub RA. Gout. N Engl J Med. 2003; 349(17):1647-1655.
30. Harris MD, Siegel LB, Alloway JA. Gout and hyperuricemia. Am Fam Physician. 1999;59(4): 925-934.
31. Schlesinger N. Dietary factors and hyperuricemia. Curr Pharm Des. 2005;11(32):4133-4138.
32. Ning TC, Keenan RT. Unusual presentations of gout. Curr Opin Rheumatol. 2010;22(2): 181-187.
33. Menon RK, Mikhailidis DP, Bell JL, et al. Warfarin administration increases uric acid concentrations in plasma. Clin Chem. 1986;32(8):
1557-1559.
34. Wallace SL, Robinson H, Masi AT, et al. Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum. 1977;20(3):895-900.
35. Martinon F, Glincher LH. Gout: new insights into an old disease. J Clin Invest. 2006;116 (8):2073-2075.
36. Zampogna G, Andracco R, Parodi M, Cimmino MA. Clinical features of gout in a cohort of Italian patients [in Italian]. Reumatismo. 2009; 61(1):41-47.
37. Choi HK. A prescription for lifestyle change in patients with hyperuricemia and gout. Curr Opin Rheumatol. 2010;22(2):165-172.
38. Bennett PH, Wood PH, eds. Population studies of the rheumatic diseases: proceedings of the Third International Symposium; June 5-10, 1966; New York, NY. Amsterdam: Excerpta Medica Foundation; 1968:457-458.
39. Malik A, Schumacher HR, Dinnella JE, Clayburne GM. Clinical diagnostic criteria for gout: comparison with the gold standard of synovial fluid crystal analysis. J Clin Rheumatol. 2009;15 (1):22-24.
40. Wijnands JMA, Boonen A, Arts ICW, et al. Large epidemiologic studies of gout: challenges in diagnosis and diagnostic criteria. Curr Rheumatol Rep. 2011;13(2):167-174.
41. Dodd LG, Major NM. Fine-needle aspiration cytology of articular and periarticular lesions. Cancer. 2002;96(3):157-165.
42. Dore RK. The gout diagnosis. Cleve Clin J Med. 2008;75 suppl 5:S17-S21.
43. Pettersson T. Sarcoid and erythema nodosum arthropathies. Baillieres Best Pract Res Clin Rheumatol. 2000;14(3):461-476.
44. Córdoba-Fernández A, Rayo-Rosado R. Pseudogout of the first metatarsophalangeal joint associated with hallux valgus: an atypical bilateral case. J Am Podiatr Med Assoc. 2010;100(2):138-142.
45. Dalbeth N, McQueen FM. Use of imaging to evaluate gout and other crystal deposition disorders. Curr Opin Rheumatol. 2009;21(2):124-131.
46. Wu EQ, Forsythe A, Guérin A, et al. Comorbidity burden healthcare resource utilization, and costs in chronic gout patients refractory to conventional urate-lowering therapy. Am J Ther. 2011 Feb 10; [Epub ahead of print].
47. Zhang W, Doherty M, Pascual E, et al; EULAR (European League Against Rheumatism) Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part II: Management. Ann Rheum Dis. 2006;65(10):1312-1324.
48. Schlesinger N, Yasothan U, Kirkpatrick P. Pegloticase [published correction appears in Nat Rev Drug Discov. 2011;10(2):156]. Nat Rev Drug Discov. 2011;10(1):17-18.
49. Pascual E, Sivera F, Yasothan U, Kurkpatrick P. Febuxostat. Nat Rev Drug Discov. 2009;8(3): 191-192.
50. Becker MA, Schumacher HR, Espinoza LR, et al. The urate-lowering efficacy and safety of febuxostat in the treatment of the hyperuricemia of gout; the CONFIRMS trial. Arthritis Res Ther. 2010;12(2):R63.
51. Hu M, Tomlinson B. Febuxostat in the management of hyperuricemia and chronic gout: a review. Ther Clin Risk Manag. 2008;4(6):1209-1220.
52. Reinders MK, Jansen TL. New advances in the treatment of gout: review of pegloticase. Ther Clin Risk Manag. 2010;6:543-550.
53. Cammalleri L, Malaguarnera M. Rasburicase represents a new tool for hyperuricemia in tumor lysis syndrome and in gout. Int J Med Sci. 2007; 4(2):83-93.
54. Richette P, Brière C, Hoenen-Clavert V, et al. Rasburicase for topaceous gout not treatable with allopurinol: an exploratory study. J Rheumatol. 2007;34(10):2093-2098.
55. Moolenburgh JD, Reinders MK, Jansen TL. Rasburicase treatment in severe tophaceous gout: a novel therapeutic option. Clin Rheumatol. 2006;25(5):749-752.
Gouty arthritis is a common form of inflammatory arthritis, occurring more frequently in men than in women. The condition has a male–female ratio of 3 or 4 to 1, although that ratio narrows as adults age; because the uricosuric effects of estrogen decline with menopause, the risk for gout increases in postmenopausal women.1,2 Mean age at disease onset is 40 to 60 in men,3,4 with onset in women averaging seven years later.5
Apart from the pain and loss of function associated with this disorder of purine metabolism6 and the risk for a chronic form of the disease, gout is almost universally linked with serious comorbidities that require timely intervention. These include hypertension, dyslipidemia, hyperglycemia and diabetes, obesity, metabolic syndrome, cardiovascular disease (CVD), renal insufficiency, and coronary heart disease (CHD).2,3,7 The presence of gout is independently associated with a risk for acute myocardial infarction (AMI) and increased rates of all-cause mortality.3,8-10
EPIDEMIOLOGY
In 2007, the National Arthritis Data Workgroup11,12 estimated that about three million US adults had had “self-reported gout” in the previous year. An estimated six million US adults have been diagnosed with gout,2,11 and its incidence and prevalence are increasing. The incidence of primary gout more than doubled between 1977-1978 and 1995-1996,13 especially affecting the aging population. The prevalence of gout among 1,000 managed care patients ages 65 to 74 increased by at least 30% between 1990 and 1999, while prevalence among those older than 75 almost doubled during this same period.14
Numerous factors appear to contribute to these trends, including aging of the population, dietary trends (ie, increased consumption of red meat, organ food, game, and shellfish and reduced consumption of low-fat dairy products), presence of certain comorbid conditions (ie, hypertension, dyslipidemia, diabetes, metabolic syndrome, end-stage renal disease), the increasing prevalence of obesity in younger adults, use of specific prescription medications, and increased incidence of organ transplantation.1,7,8,15-19
The body’s underexcretion or overproduction of uric acid (a byproduct of purine metabolism12) can lead to hyperuricemia. This condition, defined as a serum urate level exceeding 7.0 mg/dL in men or 6.0 mg/dL in women20,21 (levels above 9.0 mg/dL are considered very high22), is the primary risk factor for gout.8,23,24 As with gout, the incidence of hyperuricemia has increased in recent years,20 with researchers attributing the trend to worldwide popularization of the Westernized diet (particularly use of high-fructose corn syrup20,25) and increased use of certain medications, including thiazide diuretics, cyclosporine, and low-dose aspirin.2,20,25,26
As serum urate levels rise, the patient with hyperuricemia may experience urate supersaturation, often followed by crystallization of the excess urate into monosodium urate (MSU) crystals. Subsequently, circulating MSU crystals may deposit in body tissues, especially in the joint spaces. The body’s ensuing inflammatory response to the MSU deposits is gout.20
In addition to hyperuricemia, risk factors for gout include a high-purine diet, habitual alcohol consumption (especially beer and fortified wines27), diuretic therapy (particularly in patients with heart failure or renal insufficiency), obesity, hypertension, and high levels of fructose consumption.7,28 Additionally, cyclosporine use in an organ transplant recipient, poorly controlled uric acid levels, and a long history of gout increase the patient’s risk for chronic tophaceous gout.24,29 Tophi may be more common in a patient with a history of organ transplantation.16
Genetic variants are currently being investigated to possibly identify a predisposition to gout. The most significant genetic factors appear to involve mechanisms that regulate serum uric acid levels—particularly urate underexcretion.23 Other factors that contribute to underexcretion or overproduction of uric acid are shown in Table 1.1,15,26,30-33
A dynamic relationship exists between gout and a number of pathologic processes. According to researchers investigating nearly 178,000 patients with gout in a managed care database, 36% had hypertension, 27% had dyslipidemia, and 15% had diabetes.8 In a smaller cohort study conducted in Spain and Mexico, it was demonstrated that 93% of patients with gout had one or more associated diseases, in order of decreasing frequency: hypertriglyceridemia, obesity, hypertension, metabolic syndrome, hyperglycemia, chronic renal failure, diabetes, and ischemic heart disease.3
Of particular clinical importance in this study was a finding that the first gout attack generally preceded the diagnosis of the associated diseases.3 Thus, a diagnosis of gout should lead the primary care provider to discuss modifiable risk factors with the patient—but also to investigate for comorbid illnesses that may require timely management.2
In a 12-year-long prospective study of more than 50,000 men participating in the Health Professionals Follow-Up Study,9 it was found that men with gout had a 28% increased risk for all-cause mortality, a 38% increased risk for CVD-related death, and a 55% increased risk for CHD-related death, compared with men who did not have gout (excluding other risk factors).9 Similarly, researchers for the Multiple Risk Factor Intervention Trial10 demonstrated a clinically significant association between gout and an increased risk for AMI: 10.5% of men with gout, compared with 8.43% of men without gout, had an AMI during mean follow-up of 6.5 years.10
THE STAGES
The four stages of gout are asymptomatic hyperuricemia, acute gout, intercritical gout, and chronic tophaceous gout.20
Only a small percentage (0.5% to 4.5%) of patients with asymptomatic hyperuricemia will develop acute gout.28 Nevertheless, any patient with serum urate greater than 6.8 mg/dL is at risk for the deposition of MSU crystals into body tissues and the potential associated organ damage—even patients without symptoms. There is currently no evidence-based method to determine which patients with asymptomatic hyperuricemia will experience disease progression.16
Acute gout develops when deposition of MSU crystals in the joints initiates an inflammatory response. In the typical history, the patient experiences sudden-onset severe pain, swelling, and erythema. The pain often starts in the middle of the night or early morning,34 waking the patient from sleep and peaking within 24 hours of onset. At this time, the patient is often unable to bear weight comfortably on the affected joint. The patient may also report fever and flu-like malaise resulting from the release of interleukin 1- (IL-1), IL-1 receptor, cytokines, and prostaglandins.16,24,35 Usually in these early attacks, symptoms resolve spontaneously within three to 14 days.16,24
After resolution of an acute attack, the patient enters the intercritical stage, another asymptomatic stage that may last for months or years—or indefinitely. During the intercritical stage, MSU crystal deposition continues, adding crystals in and around the affected joint or joints, possibly continuing to inflict damage (in some patients, substantial), and in many cases resulting in additional attacks and pain.16 Any subsequent acute gout attacks the patient may experience are likely to last longer than the initial attack and to involve additional joints or tendons.24
Some patients, especially those who do not receive adequate treatment for hyperuricemia,2 progress to develop chronic tophaceous gout. This is a deforming disease process in which the joints may become stiff and swollen, and subcutaneous nodules or whitish-yellow intradermal deposits may be present under taut skin, anywhere in the body.16
PATIENT PRESENTATION AND HISTORY
Typically, a patient with gout will present with a chief complaint of a painful, tender, inflamed joint (classically described in Latin as calor, rubor, dolor, et tumor6). However, clinicians must also be aware of unusual presentations and consider gout in the differential whenever a patient with a history of gout or pertinent risk factors presents with unexplained clinical findings.32 The history of present illness will vary according to the stage of the disease.
About 90% of recognized initial attacks of gout are monoarticular, usually occurring in one of the lower extremities.16 While the first metatarsophalangeal (MTP) joint is affected in about 50% of gout cases (podagra, the Greek term for gout),2,35 eventually patients with gout have a 90% chance of involvement with the MTP joint (see Figure 1). According to Zhang et al,26 patients with hyperuricemia and an affected MTP joint have an 82% chance of having gout.2,26
Because such a large proportion of patients have the classic presentation of rapid-onset warmth, redness, and tenderness at the MTP, knee, or ankle and surrounding soft tissue, cases with a differing presentation are likely to be misdiagnosed or overlooked, or a correct diagnosis is delayed.26 In many documented cases, gout was the ultimate diagnosis—but one that was reached only incidentally because of unusual clinical presentation, ranging from entrapment neuropathy to a pancreatic mass.32
The presence of hyperuricemia and other risk factors must be investigated. Also relevant in the history of an acute gout attack may be a preceding event that has caused damage or stress to the joint, such as infection, trauma, or surgery. Other possible triggers for an attack include alcohol ingestion, acidosis, use of IV contrast media, diuretic therapy, chemotherapy, recent hospitalization or surgery, and initiation or termination of urate-lowering therapy with the xanthine oxidase inhibitor allopurinol.2,30 According to Primatesta et al,8 the risk for flares is increased in patients with cardiometabolic comorbidities.
The medication history of a patient with gout may include low-dose aspirin (but not standard-dose aspirin, which is uricosuric2), diuretics, cyclosporine, cytotoxic agents, and vitamin B12, which may contribute to hyperuricemia.24 Additionally, ethambutol, pyrazinamide, levodopa, nicotinic acid, didanosine, niacin, and warfarin may raise uric acid levels.15,30,33
Medical history should include a thorough assessment of the comorbidities associated with gout. In addition to the conditions mentioned previously, patients with a history of polycystic kidney disease, dehydration, lactic acidosis, hyperparathyroidism, toxemia of pregnancy, hypothyroidism, or sarcoidosis may have elevated urate levels due to underexcretion—and thus may be vulnerable to gout.30 History of gout in a first-degree relative is associated with an increased risk for gout.36
The social history should address alcohol use or abuse. The clinician should also inquire about how gout is impacting the daily life of the patient. Diet and exercise habits should be assessed37 (see “Patient Education,” below).
PHYSICAL EXAMINATION
The physical exam begins with evaluation of the skin and extremities for the classic features of gout. Affected joints will be exquisitely tender, and patients may be febrile. Most cases are monoarticular, but polyarticular involvement is likely in patients with advanced disease (and can, particularly in women, be mistaken for rheumatoid arthritis).2,16 In patients with chronic tophaceous gout, there may be whitish-yellow skin deposits, subcutaneous nodules, and areas of taut skin. The lower-extremity joints and tendons, as well as the wrists, fingers, and elbows, are commonly affected16 (see Figure 2).
DIAGNOSIS
Diagnostic criteria that are currently available (and have long been in use) include the American College of Rheumatology/American Rheumatism Association (ACR/ARA) preliminary criteria,34 the New York criteria,21 and the Rome criteria.38 The specifics of each are listed in Table 2.21,34,38,39
The gold standard for gout diagnosis is detection of MSU crystals in a sample of synovial fluid aspirated from the affected joint or from a tophus and examined by polarized light microscopy.24 This is of significant importance to the clinician who is faced with a questionable diagnosis.16 However, crystal visualization is not ordinarily available to the primary care clinician,2,17,40 and it is not always necessary if a careful history and physical exam are conducted in a patient with hyperuricemia or other risk factors for gout. A presumptive diagnosis may be acceptable in a patient with the classic presentation of acute gout: rapid onset of severe pain in a swollen, erythematous joint and symptoms peaking within 24 hours. The presence of tophi is pathognomonic for chronic tophaceous gout.41
In cases of questionable or unusual manifestation of gout, however, various imaging techniques and crystal visualization may be indicated.32
In order to compare the effectiveness of the latter technique with conventional diagnostic criteria for gout, Malik et al39 conducted a pilot study involving 82 patients who had undergone synovial fluid analysis with polarized light microscopy. Patients were surveyed about the clinical features of their disease, as listed in the three standard sets of criteria for diagnosis of gout. Compared with the “gold standard” of urate crystal detection (which is one of the Rome criteria38), the study authors found the ACR/ARA preliminary criteria,34 the New York criteria,21 and the Rome criteria38 generally unsatisfactory.
In the study, among patients with confirmed presence of MSU crystals:
• 87% reported more than one attack of acute arthritis (ACR/ARA34)
• 86% reported monoarthritis attack (ACR/ARA34)
• 89% had hyperuricemia (ACR/ARA34 and Rome,38 with the latter giving effective, specific parameters)
• 100% had negative results on joint fluid culture (ACR/ARA34)
• 90% reported an attack starting at night (ACR/ARA34).
The positive predictive values for these signs and symptoms are 38%, 39%, 74%, 50%, and 45%, respectively, according to Malik et al.39 The presence of tophi (cited by all three sets of criteria but “proven or suspected” in the ACR/ARA34) had the highest positive predictive value for gout (91%) and a likelihood ratio of 15.56, which was at least three times higher than any of the other listed criteria. A verified response to colchicine, one of the New York criteria,21 had the second highest positive predictive value at 86%.39
In summary, the ACR/ARA,34 the New York,21 and the Rome criteria38 had specificity of 79%, 83%, and 89%, respectively; sensitivity of 70%, 70%, and 67%, respectively; and positive predictive values for gout of 66%, 70%, and 77%, respectively. The Rome criteria38 had the highest specificity and highest positive predictive value, perhaps making them most helpful for clinicians who lack access to synovial fluid analysis.
DIFFERENTIAL DIAGNOSIS
Conditions to be considered and ruled out before a diagnosis of gout can be made are:
• Pseudogout
• Septic arthritis
• Psoriatic arthritis
• Rheumatoid arthritis
• Erosive osteoarthritis
• Bacterial cellulitis
• Sarcoid arthropathy.16,28,42,43
Unlike gout (in which compensated polarized light microscopy reveals needle-shaped urate crystals with strong negative birefringence), pseudogout is characterized by calcium pyrophosphate dihydrate crystals; these are rhomboid-shaped, with weak positive birefringence.42 Additionally, radiographic imaging will reveal soft tissue swelling and chondrocalcinosis of the joint in pseudogout.44
The patient with septic arthritis, most likely affecting the knee, will have a white blood cell (WBC) count exceeding 50,000/mm3 and a positive culture of the synovial fluid, with absence of crystals.28
Chronic tophaceous gout can mimic rheumatoid arthritis in appearance and joint distribution, and patients affected by either condition may develop a positive rheumatoid factor. Examination of synovial fluid for MSU crystals and radiographic imaging will be of value in making a distinction.
Osteoarthritis is usually evidenced by joint space narrowing on x-ray.42
Bacterial cellulitis will present similarly to gout, but the erythema of bacterial cellulitis will more likely extend beyond the involved joint.16
Sarcoid arthropathy often presents as a polyarthritis, as in advanced gouty arthritis. However, in sarcoid arthropathy, serum calcium and angiotensin-converting enzyme will likely be elevated.43 Synovial or tendon sheath biopsy will show non-caseating granulomas, which are the hallmark for sarcoid disease. Additionally, joint fluid analysis will demonstrate a predominance of mononuclear or polymorphonuclear cells.43
Diagnostic Tests
Diagnostic tests to consider are analysis and culture of the synovial fluid, complete blood count (CBC), blood urea nitrogen (BUN), creatinine, radiography, ultrasonography, serum uric acid, and blood culture if septic arthritis is suspected.28 While serum urate levels may be normal during an acute gout attack, measurement may still be helpful for comparison, since elevation is a likely finding two weeks after an attack—if the patient was, in fact, experiencing an acute gout attack.42
Since renal dialysis increases the risk for gout, pseudogout, and septic arthritis, synovial fluid analysis is essential in patients undergoing renal dialysis.42
Various imaging techniques may aid in confirming a diagnosis of gout and monitoring its progression, but further studies are needed to more clearly define the role of these techniques in management of gout.45 Plain radiographic evidence of asymmetric swelling in a joint (one of the ACR/ARA preliminary criteria34) was shown to have a 60% positive predictive value for a diagnosis of gout.39 Late in the disease process, an affected joint may be affected by characteristic “punched out” intra-articular lesions, with a normal amount of joint space.45
Ultrasound is a safe and inexpensive test that can reveal soft tissue edema and increased vascularity during an acute gout attack. Chronic changes include the double contour sign and tophus-like lesions surrounded by a thin, anechoic rim.45
CT will also show tophi and bony erosion. While CT is more specific than other techniques, it is also more expensive and exposes the patient to increased radiation. MRI can help monitor the complications of gout, especially entrapment neuropathies.45
TREATMENT/MANAGEMENT
According to current evidence, treatment is not indicated for asymptomatic hyperuricemia.16
Acute Gout Management
Pharmacologic treatments available for an acute gout attack include NSAIDs, colchicine, and local or systemic corticosteroids.24,46 At the onset of an attack, patients should start high-dose NSAID therapy, and continue for two to three days after symptoms are resolved.6 Oral indomethacin (50 mg tid) or oral ibuprofen (800 mg tid) are both reasonable options.6 It may be prudent to consider a proton pump inhibitor (eg, omeprazole) to protect the gastric mucosa in patients who are susceptible to gastrointestinal problems.27
In addition to high-dose NSAID therapy, adding colchicine (1.2 mg by mouth at onset of symptoms, followed by 0.6 mg one hour later) has proven to be effective in relieving the symptoms of gout, but its serious gastrointestinal adverse effects, particularly diarrhea, must be considered.6,47
In patients with monoarticular gout who cannot tolerate NSAIDs, intra-articular aspiration and corticosteroid injections may provide relief.
Long-acting triamcinolone, administered by intra-articular injection, has been found to relieve pain and inflammation in patients with gout. Septic arthritis must be ruled out by way of joint aspiration and culture before injection of corticosteroids.47
Oral or IM-administered corticosteroids may be considered for patients with polyarticular involvement. Prednisone (60 mg/d, tapered over 10 days) is an appropriate option for outpatients or inpatients; methylprednisone (80 to 120 mg IM) may be suitable for inpatients.6 Again, septic arthritis must be ruled out before corticosteroids are administered.47
For the patient who is currently taking a thiazide diuretic for hypertension, substituting a different medication may be warranted; the angiotensin receptor blocker losartan, for example, has uricosuric action.27,29,47 Nonpharmacologic strategies, such as rest, ice, elevation, and avoiding trauma to the affected joint, are also recommended.27
Of note, allopurinol therapy should be neither initiated nor discontinued during an acute gout attack.27
Management of Chronic and Intercritical Gout
Urate-lowering therapy, such as allopurinol (50 to 300 mg/d29), should be considered for patients who experience frequent attacks (ie, three or more per year), patients with chronic tophi, patients with radiographically demonstrated joint damage,47 or patients with a documented state of uric acid overproduction.29
Allopurinol dosage should be adjusted based on creatinine clearance; dosing as high as 800 mg/d has been recommended in patients with normal renal function.2 Again, allopurinol should never be started or discontinued during an acute attack,27 because abrupt fluctuations in uric acid levels may heighten the inflammation. The target serum urate level is 6.0 mg/dL.29
Febuxostat, which received FDA approval in 2009, was the first oral urate-lowering treatment to be approved since the 1960s. Like allopurinol, this nonpurine xanthine oxidase inhibitor blocks uric acid synthesis.48,49 In a trial reported by Becker et al,50 67% of patients who took febuxostat 80 mg/d reached the target serum urate level (ie, < 6.0 mg/dL), compared with 45% of those who took 40 mg/d of febuxostat and 42% of those taking 300 mg/d of allopurinol. While incidence of adverse events was low in all treatment groups, Hu and Tomlinson51 report that febuxostat is tolerable in patients who are hypersensitive to allopurinol. As with other urate-lowering medications, gout flares are common during the early period of febuxostat use.51
For patients with gout that does not respond to conventional urate-lowering therapy, new options are being introduced. Two agents, each a recombinant form of the enzyme urate oxidase, are designed to convert uric acid into allantoin, which can then be excreted in the urine. Late in 2010, one of these agents, pegloticase, was approved for use in patients with refractory gout.48 In one clinical trial, tophi were reported dissolved in 40% of patients who took pegloticase, but 58% of patients did not achieve the targeted response (ie, serum urate < 6.0 mg/dL), and 77% of patients experienced gout flares.52 Infusion reactions occurred in 26% to 31% of patients, and Reinders and Jansen52 recommended the clinical evaluation of glucocorticoids and other anti-inflammatory agents to prevent the formation of antibodies involved in these reactions.
The second agent, rasburicase, has been approved for treatment and prevention of acute hyperuricemia in adult cancer patients. Rasburicase is now being investigated for use in patients with nonresponsive tophaceous gout.53-55 It can be administered in the form of monthly infusions.54
Patient Education
Educating the patient about modifiable risk factors, such as diet, alcohol consumption, and adherence to the medication regimen, should be a priority.
Patients should be encouraged to target and maintain an ideal body weight, through diet and moderate physical exercise, as a strategy to normalize serum urate levels.27,47 However, they should be advised to avoid “crash dieting,” as this may precipitate a gout attack.27 In the recommended low-purine diet, consumption of red meat and shellfish is restricted,17 whereas consumption of soy, nonfat milk and other low-fat dairy products, cherries and other fruits, and increased vegetable protein is encouraged.31,37 Consumption of alcohol, especially beer and fortified wines, should be limited.27,47
Avoiding trauma to joints affected by gout (including the stress of bearing excess weight) can help patients limit future attacks.7,27
CONCLUSION
Patients with gout often have the characteristic presentation of an acutely tender, inflamed joint, but since gout is a systemic disorder, the clinician must also consider the possibility of gout in almost any organ system. Gout is a common disease, and its diagnosis can alert the astute clinician to investigate for certain metabolic disorders requiring intervention. Hyperlipidemia, metabolic syndrome, hypertension, chronic kidney disease, obesity, cardiovascular disease, and diabetes are all conditions associated with gout.
Recognizing the opportunity to offer preventive care measures and recommend lifestyle modifications to the patient with gout allows the clinician to play an important role in the patient’s care.
REFERENCES
1. Bhole V, de Vera M, Rahman MM, et al. Epidemiology of gout in women: fifty-two–year followup of a prospective cohort. Arthritis Rheum. 2010;62(4):1069-1076.
2. Neogi T. Clinical practice: gout. N Engl J Med. 2011;364(5):443-452.
3. Hernández-Cuevas CB, Roque LH, Huerta-Sil G, et al. First acute gout attacks commonly precede features of the metabolic syndrome. J Clin Rheumatol. 2009;15(2):65-67.
4. Louthrenoo W, Kasitanon N, Sukitawut W, Wichainun R. A clinical study of crystal-proven gouty arthritis in a university hospital. J Med Assoc Thai. 2003;86(9):868-875.
5. De Souza AW, Fernandes V, Ferrari AJ. Female gout: clinical and laboratory features. J Rheumatol. 2005;32(11):2186-2188.
6. Kurakula PC, Keenan RT. Diagnosis and management of gout: an update. J Musculoskel Med. 2010;27(10). www.musculoskeletalnet work.com/display/article/1145622/1692895. Accessed June 14, 2011.
7. Choi HK, Atkinson K, Karlson EW, Curhan G. Obesity, weight change, hypertension, diuretic use, and risk of gout in men: the Health Professionals Follow-up Study. Arch Intern Med. 2005;165(7):742-748.
8. Primatesta P, Plana E, Rothenbacher D. Gout treatment and comorbidities: a retrospective cohort study in a large US managed care population. BMC Musculoskelet Disord. 2011 May 20;12(1):103. [Epub ahead of print]
9. Choi HK, Curhan G. Independent impact of gout on mortality and risk for coronary heart disease. Circulation. 2007;116(8):894-900.
10. Krishnan E, Baker JF, Furst DE, Schumacher HR. Gout and the risk of acute myocardial infarction. Arthritis Rheum. 2006;54(8):2688-2696.
11. Lawrence RC, Felson DT, Helmick CG, et al; National Arthritis Data Workgroup. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008;58(1):26-35.
12. CDC. Gout. www.cdc.gov/arthritis/basics/gout.htm. Accessed June 14, 2011.
13. Arromdee E, Michet CJ, Crowson CS, et al. Epidemiology of gout: is the incidence rising? J Rheumatol. 2002;29(11):2403-2406.
14. Wallace KL, Riedel AA, Joseph-Ridge N, Wortmann R. Increasing prevalence of gout and hyperuricemia over 10 years among older adults in a managed care population. J Rheumatol. 2004; 31(8):1582-1587.
15. Weaver AL. Epidemiology of gout. Cleve Clin J Med. 2008;75 suppl 5:S9-S12.
16. Mandell BF. Clinical manifestations of hyperuricemia and gout. Cleve Clin J Med. 2008;75 suppl 5:S5-S8.
17. Choi HK, Atkinson K, Karlson EW, et al. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med. 2004;350(11):1093-1103.
18. Demarco MA, Maynard JW, Huizinga MM, et al. Younger age at gout onset is related to obesity in a community-based cohort. Arthritis Care Res (Hoboken). 2011 Apr 11; [Epub ahead of print].
19. Brook RA, Forsythe A, Smeeding JE, Lawrence Edwards N. Chronic gout: epidemiology, disease progression treatment and disease burden. Curr Med Res Opin. 2010;26(12):2813-2821.
20. Sachs L, Batra KL, Zimmermann B. Medical implications of hyperuricemia. Med Health R I. 2009;92(11):353-355.
21. Kellgren JH, Jeffrey MR, Ball J, eds. The Epidemiology of Chronic Rheumatism: Atlas of Standard Radiographs of Arthritis. Oxford: Blackwell; 1963:327.
22. Wu EQ, Patel PA, Mody RR, et al. Frequency, risk, and cost of gout-related episodes among the elderly: does serum uric acid level matter? J Rheumatol. 2009;36(5):1032-1040.
23. Riches PL, Wright AF, Ralston SH. Recent insights into the pathogenesis of hyperuricaemia and gout. Hum Mol Genet. 2009;18(R2):R177-R184.
24. Schumacher HR Jr. The pathogenesis of gout. Cleve Clin J Med. 2008;75 suppl 5:S2-S4.
25. Nakagawa T, Hu H, Zharikov S, et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Renal Physiol. 2006;290(3):F625-F631.
26. Zhang W, Doherty M, Pascual E, et al; EULAR (European League Against Rheumatism) Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part I: Diagnosis. Ann Rheum Dis. 2006;65(10):1301-1311.
27. Jordan KM, Cameron JS, Snaith M, et al. British Society for Rheumatology and British Health Professionals in Rheumatology guideline for the management of gout. Rheumatology. 2007;46(8):1372-1374.
28. Eggebeen AT. Gout: an update. Am Fam Physician. 2007;76(6):801-808.
29. Terkeltaub RA. Gout. N Engl J Med. 2003; 349(17):1647-1655.
30. Harris MD, Siegel LB, Alloway JA. Gout and hyperuricemia. Am Fam Physician. 1999;59(4): 925-934.
31. Schlesinger N. Dietary factors and hyperuricemia. Curr Pharm Des. 2005;11(32):4133-4138.
32. Ning TC, Keenan RT. Unusual presentations of gout. Curr Opin Rheumatol. 2010;22(2): 181-187.
33. Menon RK, Mikhailidis DP, Bell JL, et al. Warfarin administration increases uric acid concentrations in plasma. Clin Chem. 1986;32(8):
1557-1559.
34. Wallace SL, Robinson H, Masi AT, et al. Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum. 1977;20(3):895-900.
35. Martinon F, Glincher LH. Gout: new insights into an old disease. J Clin Invest. 2006;116 (8):2073-2075.
36. Zampogna G, Andracco R, Parodi M, Cimmino MA. Clinical features of gout in a cohort of Italian patients [in Italian]. Reumatismo. 2009; 61(1):41-47.
37. Choi HK. A prescription for lifestyle change in patients with hyperuricemia and gout. Curr Opin Rheumatol. 2010;22(2):165-172.
38. Bennett PH, Wood PH, eds. Population studies of the rheumatic diseases: proceedings of the Third International Symposium; June 5-10, 1966; New York, NY. Amsterdam: Excerpta Medica Foundation; 1968:457-458.
39. Malik A, Schumacher HR, Dinnella JE, Clayburne GM. Clinical diagnostic criteria for gout: comparison with the gold standard of synovial fluid crystal analysis. J Clin Rheumatol. 2009;15 (1):22-24.
40. Wijnands JMA, Boonen A, Arts ICW, et al. Large epidemiologic studies of gout: challenges in diagnosis and diagnostic criteria. Curr Rheumatol Rep. 2011;13(2):167-174.
41. Dodd LG, Major NM. Fine-needle aspiration cytology of articular and periarticular lesions. Cancer. 2002;96(3):157-165.
42. Dore RK. The gout diagnosis. Cleve Clin J Med. 2008;75 suppl 5:S17-S21.
43. Pettersson T. Sarcoid and erythema nodosum arthropathies. Baillieres Best Pract Res Clin Rheumatol. 2000;14(3):461-476.
44. Córdoba-Fernández A, Rayo-Rosado R. Pseudogout of the first metatarsophalangeal joint associated with hallux valgus: an atypical bilateral case. J Am Podiatr Med Assoc. 2010;100(2):138-142.
45. Dalbeth N, McQueen FM. Use of imaging to evaluate gout and other crystal deposition disorders. Curr Opin Rheumatol. 2009;21(2):124-131.
46. Wu EQ, Forsythe A, Guérin A, et al. Comorbidity burden healthcare resource utilization, and costs in chronic gout patients refractory to conventional urate-lowering therapy. Am J Ther. 2011 Feb 10; [Epub ahead of print].
47. Zhang W, Doherty M, Pascual E, et al; EULAR (European League Against Rheumatism) Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part II: Management. Ann Rheum Dis. 2006;65(10):1312-1324.
48. Schlesinger N, Yasothan U, Kirkpatrick P. Pegloticase [published correction appears in Nat Rev Drug Discov. 2011;10(2):156]. Nat Rev Drug Discov. 2011;10(1):17-18.
49. Pascual E, Sivera F, Yasothan U, Kurkpatrick P. Febuxostat. Nat Rev Drug Discov. 2009;8(3): 191-192.
50. Becker MA, Schumacher HR, Espinoza LR, et al. The urate-lowering efficacy and safety of febuxostat in the treatment of the hyperuricemia of gout; the CONFIRMS trial. Arthritis Res Ther. 2010;12(2):R63.
51. Hu M, Tomlinson B. Febuxostat in the management of hyperuricemia and chronic gout: a review. Ther Clin Risk Manag. 2008;4(6):1209-1220.
52. Reinders MK, Jansen TL. New advances in the treatment of gout: review of pegloticase. Ther Clin Risk Manag. 2010;6:543-550.
53. Cammalleri L, Malaguarnera M. Rasburicase represents a new tool for hyperuricemia in tumor lysis syndrome and in gout. Int J Med Sci. 2007; 4(2):83-93.
54. Richette P, Brière C, Hoenen-Clavert V, et al. Rasburicase for topaceous gout not treatable with allopurinol: an exploratory study. J Rheumatol. 2007;34(10):2093-2098.
55. Moolenburgh JD, Reinders MK, Jansen TL. Rasburicase treatment in severe tophaceous gout: a novel therapeutic option. Clin Rheumatol. 2006;25(5):749-752.
Gouty arthritis is a common form of inflammatory arthritis, occurring more frequently in men than in women. The condition has a male–female ratio of 3 or 4 to 1, although that ratio narrows as adults age; because the uricosuric effects of estrogen decline with menopause, the risk for gout increases in postmenopausal women.1,2 Mean age at disease onset is 40 to 60 in men,3,4 with onset in women averaging seven years later.5
Apart from the pain and loss of function associated with this disorder of purine metabolism6 and the risk for a chronic form of the disease, gout is almost universally linked with serious comorbidities that require timely intervention. These include hypertension, dyslipidemia, hyperglycemia and diabetes, obesity, metabolic syndrome, cardiovascular disease (CVD), renal insufficiency, and coronary heart disease (CHD).2,3,7 The presence of gout is independently associated with a risk for acute myocardial infarction (AMI) and increased rates of all-cause mortality.3,8-10
EPIDEMIOLOGY
In 2007, the National Arthritis Data Workgroup11,12 estimated that about three million US adults had had “self-reported gout” in the previous year. An estimated six million US adults have been diagnosed with gout,2,11 and its incidence and prevalence are increasing. The incidence of primary gout more than doubled between 1977-1978 and 1995-1996,13 especially affecting the aging population. The prevalence of gout among 1,000 managed care patients ages 65 to 74 increased by at least 30% between 1990 and 1999, while prevalence among those older than 75 almost doubled during this same period.14
Numerous factors appear to contribute to these trends, including aging of the population, dietary trends (ie, increased consumption of red meat, organ food, game, and shellfish and reduced consumption of low-fat dairy products), presence of certain comorbid conditions (ie, hypertension, dyslipidemia, diabetes, metabolic syndrome, end-stage renal disease), the increasing prevalence of obesity in younger adults, use of specific prescription medications, and increased incidence of organ transplantation.1,7,8,15-19
The body’s underexcretion or overproduction of uric acid (a byproduct of purine metabolism12) can lead to hyperuricemia. This condition, defined as a serum urate level exceeding 7.0 mg/dL in men or 6.0 mg/dL in women20,21 (levels above 9.0 mg/dL are considered very high22), is the primary risk factor for gout.8,23,24 As with gout, the incidence of hyperuricemia has increased in recent years,20 with researchers attributing the trend to worldwide popularization of the Westernized diet (particularly use of high-fructose corn syrup20,25) and increased use of certain medications, including thiazide diuretics, cyclosporine, and low-dose aspirin.2,20,25,26
As serum urate levels rise, the patient with hyperuricemia may experience urate supersaturation, often followed by crystallization of the excess urate into monosodium urate (MSU) crystals. Subsequently, circulating MSU crystals may deposit in body tissues, especially in the joint spaces. The body’s ensuing inflammatory response to the MSU deposits is gout.20
In addition to hyperuricemia, risk factors for gout include a high-purine diet, habitual alcohol consumption (especially beer and fortified wines27), diuretic therapy (particularly in patients with heart failure or renal insufficiency), obesity, hypertension, and high levels of fructose consumption.7,28 Additionally, cyclosporine use in an organ transplant recipient, poorly controlled uric acid levels, and a long history of gout increase the patient’s risk for chronic tophaceous gout.24,29 Tophi may be more common in a patient with a history of organ transplantation.16
Genetic variants are currently being investigated to possibly identify a predisposition to gout. The most significant genetic factors appear to involve mechanisms that regulate serum uric acid levels—particularly urate underexcretion.23 Other factors that contribute to underexcretion or overproduction of uric acid are shown in Table 1.1,15,26,30-33
A dynamic relationship exists between gout and a number of pathologic processes. According to researchers investigating nearly 178,000 patients with gout in a managed care database, 36% had hypertension, 27% had dyslipidemia, and 15% had diabetes.8 In a smaller cohort study conducted in Spain and Mexico, it was demonstrated that 93% of patients with gout had one or more associated diseases, in order of decreasing frequency: hypertriglyceridemia, obesity, hypertension, metabolic syndrome, hyperglycemia, chronic renal failure, diabetes, and ischemic heart disease.3
Of particular clinical importance in this study was a finding that the first gout attack generally preceded the diagnosis of the associated diseases.3 Thus, a diagnosis of gout should lead the primary care provider to discuss modifiable risk factors with the patient—but also to investigate for comorbid illnesses that may require timely management.2
In a 12-year-long prospective study of more than 50,000 men participating in the Health Professionals Follow-Up Study,9 it was found that men with gout had a 28% increased risk for all-cause mortality, a 38% increased risk for CVD-related death, and a 55% increased risk for CHD-related death, compared with men who did not have gout (excluding other risk factors).9 Similarly, researchers for the Multiple Risk Factor Intervention Trial10 demonstrated a clinically significant association between gout and an increased risk for AMI: 10.5% of men with gout, compared with 8.43% of men without gout, had an AMI during mean follow-up of 6.5 years.10
THE STAGES
The four stages of gout are asymptomatic hyperuricemia, acute gout, intercritical gout, and chronic tophaceous gout.20
Only a small percentage (0.5% to 4.5%) of patients with asymptomatic hyperuricemia will develop acute gout.28 Nevertheless, any patient with serum urate greater than 6.8 mg/dL is at risk for the deposition of MSU crystals into body tissues and the potential associated organ damage—even patients without symptoms. There is currently no evidence-based method to determine which patients with asymptomatic hyperuricemia will experience disease progression.16
Acute gout develops when deposition of MSU crystals in the joints initiates an inflammatory response. In the typical history, the patient experiences sudden-onset severe pain, swelling, and erythema. The pain often starts in the middle of the night or early morning,34 waking the patient from sleep and peaking within 24 hours of onset. At this time, the patient is often unable to bear weight comfortably on the affected joint. The patient may also report fever and flu-like malaise resulting from the release of interleukin 1- (IL-1), IL-1 receptor, cytokines, and prostaglandins.16,24,35 Usually in these early attacks, symptoms resolve spontaneously within three to 14 days.16,24
After resolution of an acute attack, the patient enters the intercritical stage, another asymptomatic stage that may last for months or years—or indefinitely. During the intercritical stage, MSU crystal deposition continues, adding crystals in and around the affected joint or joints, possibly continuing to inflict damage (in some patients, substantial), and in many cases resulting in additional attacks and pain.16 Any subsequent acute gout attacks the patient may experience are likely to last longer than the initial attack and to involve additional joints or tendons.24
Some patients, especially those who do not receive adequate treatment for hyperuricemia,2 progress to develop chronic tophaceous gout. This is a deforming disease process in which the joints may become stiff and swollen, and subcutaneous nodules or whitish-yellow intradermal deposits may be present under taut skin, anywhere in the body.16
PATIENT PRESENTATION AND HISTORY
Typically, a patient with gout will present with a chief complaint of a painful, tender, inflamed joint (classically described in Latin as calor, rubor, dolor, et tumor6). However, clinicians must also be aware of unusual presentations and consider gout in the differential whenever a patient with a history of gout or pertinent risk factors presents with unexplained clinical findings.32 The history of present illness will vary according to the stage of the disease.
About 90% of recognized initial attacks of gout are monoarticular, usually occurring in one of the lower extremities.16 While the first metatarsophalangeal (MTP) joint is affected in about 50% of gout cases (podagra, the Greek term for gout),2,35 eventually patients with gout have a 90% chance of involvement with the MTP joint (see Figure 1). According to Zhang et al,26 patients with hyperuricemia and an affected MTP joint have an 82% chance of having gout.2,26
Because such a large proportion of patients have the classic presentation of rapid-onset warmth, redness, and tenderness at the MTP, knee, or ankle and surrounding soft tissue, cases with a differing presentation are likely to be misdiagnosed or overlooked, or a correct diagnosis is delayed.26 In many documented cases, gout was the ultimate diagnosis—but one that was reached only incidentally because of unusual clinical presentation, ranging from entrapment neuropathy to a pancreatic mass.32
The presence of hyperuricemia and other risk factors must be investigated. Also relevant in the history of an acute gout attack may be a preceding event that has caused damage or stress to the joint, such as infection, trauma, or surgery. Other possible triggers for an attack include alcohol ingestion, acidosis, use of IV contrast media, diuretic therapy, chemotherapy, recent hospitalization or surgery, and initiation or termination of urate-lowering therapy with the xanthine oxidase inhibitor allopurinol.2,30 According to Primatesta et al,8 the risk for flares is increased in patients with cardiometabolic comorbidities.
The medication history of a patient with gout may include low-dose aspirin (but not standard-dose aspirin, which is uricosuric2), diuretics, cyclosporine, cytotoxic agents, and vitamin B12, which may contribute to hyperuricemia.24 Additionally, ethambutol, pyrazinamide, levodopa, nicotinic acid, didanosine, niacin, and warfarin may raise uric acid levels.15,30,33
Medical history should include a thorough assessment of the comorbidities associated with gout. In addition to the conditions mentioned previously, patients with a history of polycystic kidney disease, dehydration, lactic acidosis, hyperparathyroidism, toxemia of pregnancy, hypothyroidism, or sarcoidosis may have elevated urate levels due to underexcretion—and thus may be vulnerable to gout.30 History of gout in a first-degree relative is associated with an increased risk for gout.36
The social history should address alcohol use or abuse. The clinician should also inquire about how gout is impacting the daily life of the patient. Diet and exercise habits should be assessed37 (see “Patient Education,” below).
PHYSICAL EXAMINATION
The physical exam begins with evaluation of the skin and extremities for the classic features of gout. Affected joints will be exquisitely tender, and patients may be febrile. Most cases are monoarticular, but polyarticular involvement is likely in patients with advanced disease (and can, particularly in women, be mistaken for rheumatoid arthritis).2,16 In patients with chronic tophaceous gout, there may be whitish-yellow skin deposits, subcutaneous nodules, and areas of taut skin. The lower-extremity joints and tendons, as well as the wrists, fingers, and elbows, are commonly affected16 (see Figure 2).
DIAGNOSIS
Diagnostic criteria that are currently available (and have long been in use) include the American College of Rheumatology/American Rheumatism Association (ACR/ARA) preliminary criteria,34 the New York criteria,21 and the Rome criteria.38 The specifics of each are listed in Table 2.21,34,38,39
The gold standard for gout diagnosis is detection of MSU crystals in a sample of synovial fluid aspirated from the affected joint or from a tophus and examined by polarized light microscopy.24 This is of significant importance to the clinician who is faced with a questionable diagnosis.16 However, crystal visualization is not ordinarily available to the primary care clinician,2,17,40 and it is not always necessary if a careful history and physical exam are conducted in a patient with hyperuricemia or other risk factors for gout. A presumptive diagnosis may be acceptable in a patient with the classic presentation of acute gout: rapid onset of severe pain in a swollen, erythematous joint and symptoms peaking within 24 hours. The presence of tophi is pathognomonic for chronic tophaceous gout.41
In cases of questionable or unusual manifestation of gout, however, various imaging techniques and crystal visualization may be indicated.32
In order to compare the effectiveness of the latter technique with conventional diagnostic criteria for gout, Malik et al39 conducted a pilot study involving 82 patients who had undergone synovial fluid analysis with polarized light microscopy. Patients were surveyed about the clinical features of their disease, as listed in the three standard sets of criteria for diagnosis of gout. Compared with the “gold standard” of urate crystal detection (which is one of the Rome criteria38), the study authors found the ACR/ARA preliminary criteria,34 the New York criteria,21 and the Rome criteria38 generally unsatisfactory.
In the study, among patients with confirmed presence of MSU crystals:
• 87% reported more than one attack of acute arthritis (ACR/ARA34)
• 86% reported monoarthritis attack (ACR/ARA34)
• 89% had hyperuricemia (ACR/ARA34 and Rome,38 with the latter giving effective, specific parameters)
• 100% had negative results on joint fluid culture (ACR/ARA34)
• 90% reported an attack starting at night (ACR/ARA34).
The positive predictive values for these signs and symptoms are 38%, 39%, 74%, 50%, and 45%, respectively, according to Malik et al.39 The presence of tophi (cited by all three sets of criteria but “proven or suspected” in the ACR/ARA34) had the highest positive predictive value for gout (91%) and a likelihood ratio of 15.56, which was at least three times higher than any of the other listed criteria. A verified response to colchicine, one of the New York criteria,21 had the second highest positive predictive value at 86%.39
In summary, the ACR/ARA,34 the New York,21 and the Rome criteria38 had specificity of 79%, 83%, and 89%, respectively; sensitivity of 70%, 70%, and 67%, respectively; and positive predictive values for gout of 66%, 70%, and 77%, respectively. The Rome criteria38 had the highest specificity and highest positive predictive value, perhaps making them most helpful for clinicians who lack access to synovial fluid analysis.
DIFFERENTIAL DIAGNOSIS
Conditions to be considered and ruled out before a diagnosis of gout can be made are:
• Pseudogout
• Septic arthritis
• Psoriatic arthritis
• Rheumatoid arthritis
• Erosive osteoarthritis
• Bacterial cellulitis
• Sarcoid arthropathy.16,28,42,43
Unlike gout (in which compensated polarized light microscopy reveals needle-shaped urate crystals with strong negative birefringence), pseudogout is characterized by calcium pyrophosphate dihydrate crystals; these are rhomboid-shaped, with weak positive birefringence.42 Additionally, radiographic imaging will reveal soft tissue swelling and chondrocalcinosis of the joint in pseudogout.44
The patient with septic arthritis, most likely affecting the knee, will have a white blood cell (WBC) count exceeding 50,000/mm3 and a positive culture of the synovial fluid, with absence of crystals.28
Chronic tophaceous gout can mimic rheumatoid arthritis in appearance and joint distribution, and patients affected by either condition may develop a positive rheumatoid factor. Examination of synovial fluid for MSU crystals and radiographic imaging will be of value in making a distinction.
Osteoarthritis is usually evidenced by joint space narrowing on x-ray.42
Bacterial cellulitis will present similarly to gout, but the erythema of bacterial cellulitis will more likely extend beyond the involved joint.16
Sarcoid arthropathy often presents as a polyarthritis, as in advanced gouty arthritis. However, in sarcoid arthropathy, serum calcium and angiotensin-converting enzyme will likely be elevated.43 Synovial or tendon sheath biopsy will show non-caseating granulomas, which are the hallmark for sarcoid disease. Additionally, joint fluid analysis will demonstrate a predominance of mononuclear or polymorphonuclear cells.43
Diagnostic Tests
Diagnostic tests to consider are analysis and culture of the synovial fluid, complete blood count (CBC), blood urea nitrogen (BUN), creatinine, radiography, ultrasonography, serum uric acid, and blood culture if septic arthritis is suspected.28 While serum urate levels may be normal during an acute gout attack, measurement may still be helpful for comparison, since elevation is a likely finding two weeks after an attack—if the patient was, in fact, experiencing an acute gout attack.42
Since renal dialysis increases the risk for gout, pseudogout, and septic arthritis, synovial fluid analysis is essential in patients undergoing renal dialysis.42
Various imaging techniques may aid in confirming a diagnosis of gout and monitoring its progression, but further studies are needed to more clearly define the role of these techniques in management of gout.45 Plain radiographic evidence of asymmetric swelling in a joint (one of the ACR/ARA preliminary criteria34) was shown to have a 60% positive predictive value for a diagnosis of gout.39 Late in the disease process, an affected joint may be affected by characteristic “punched out” intra-articular lesions, with a normal amount of joint space.45
Ultrasound is a safe and inexpensive test that can reveal soft tissue edema and increased vascularity during an acute gout attack. Chronic changes include the double contour sign and tophus-like lesions surrounded by a thin, anechoic rim.45
CT will also show tophi and bony erosion. While CT is more specific than other techniques, it is also more expensive and exposes the patient to increased radiation. MRI can help monitor the complications of gout, especially entrapment neuropathies.45
TREATMENT/MANAGEMENT
According to current evidence, treatment is not indicated for asymptomatic hyperuricemia.16
Acute Gout Management
Pharmacologic treatments available for an acute gout attack include NSAIDs, colchicine, and local or systemic corticosteroids.24,46 At the onset of an attack, patients should start high-dose NSAID therapy, and continue for two to three days after symptoms are resolved.6 Oral indomethacin (50 mg tid) or oral ibuprofen (800 mg tid) are both reasonable options.6 It may be prudent to consider a proton pump inhibitor (eg, omeprazole) to protect the gastric mucosa in patients who are susceptible to gastrointestinal problems.27
In addition to high-dose NSAID therapy, adding colchicine (1.2 mg by mouth at onset of symptoms, followed by 0.6 mg one hour later) has proven to be effective in relieving the symptoms of gout, but its serious gastrointestinal adverse effects, particularly diarrhea, must be considered.6,47
In patients with monoarticular gout who cannot tolerate NSAIDs, intra-articular aspiration and corticosteroid injections may provide relief.
Long-acting triamcinolone, administered by intra-articular injection, has been found to relieve pain and inflammation in patients with gout. Septic arthritis must be ruled out by way of joint aspiration and culture before injection of corticosteroids.47
Oral or IM-administered corticosteroids may be considered for patients with polyarticular involvement. Prednisone (60 mg/d, tapered over 10 days) is an appropriate option for outpatients or inpatients; methylprednisone (80 to 120 mg IM) may be suitable for inpatients.6 Again, septic arthritis must be ruled out before corticosteroids are administered.47
For the patient who is currently taking a thiazide diuretic for hypertension, substituting a different medication may be warranted; the angiotensin receptor blocker losartan, for example, has uricosuric action.27,29,47 Nonpharmacologic strategies, such as rest, ice, elevation, and avoiding trauma to the affected joint, are also recommended.27
Of note, allopurinol therapy should be neither initiated nor discontinued during an acute gout attack.27
Management of Chronic and Intercritical Gout
Urate-lowering therapy, such as allopurinol (50 to 300 mg/d29), should be considered for patients who experience frequent attacks (ie, three or more per year), patients with chronic tophi, patients with radiographically demonstrated joint damage,47 or patients with a documented state of uric acid overproduction.29
Allopurinol dosage should be adjusted based on creatinine clearance; dosing as high as 800 mg/d has been recommended in patients with normal renal function.2 Again, allopurinol should never be started or discontinued during an acute attack,27 because abrupt fluctuations in uric acid levels may heighten the inflammation. The target serum urate level is 6.0 mg/dL.29
Febuxostat, which received FDA approval in 2009, was the first oral urate-lowering treatment to be approved since the 1960s. Like allopurinol, this nonpurine xanthine oxidase inhibitor blocks uric acid synthesis.48,49 In a trial reported by Becker et al,50 67% of patients who took febuxostat 80 mg/d reached the target serum urate level (ie, < 6.0 mg/dL), compared with 45% of those who took 40 mg/d of febuxostat and 42% of those taking 300 mg/d of allopurinol. While incidence of adverse events was low in all treatment groups, Hu and Tomlinson51 report that febuxostat is tolerable in patients who are hypersensitive to allopurinol. As with other urate-lowering medications, gout flares are common during the early period of febuxostat use.51
For patients with gout that does not respond to conventional urate-lowering therapy, new options are being introduced. Two agents, each a recombinant form of the enzyme urate oxidase, are designed to convert uric acid into allantoin, which can then be excreted in the urine. Late in 2010, one of these agents, pegloticase, was approved for use in patients with refractory gout.48 In one clinical trial, tophi were reported dissolved in 40% of patients who took pegloticase, but 58% of patients did not achieve the targeted response (ie, serum urate < 6.0 mg/dL), and 77% of patients experienced gout flares.52 Infusion reactions occurred in 26% to 31% of patients, and Reinders and Jansen52 recommended the clinical evaluation of glucocorticoids and other anti-inflammatory agents to prevent the formation of antibodies involved in these reactions.
The second agent, rasburicase, has been approved for treatment and prevention of acute hyperuricemia in adult cancer patients. Rasburicase is now being investigated for use in patients with nonresponsive tophaceous gout.53-55 It can be administered in the form of monthly infusions.54
Patient Education
Educating the patient about modifiable risk factors, such as diet, alcohol consumption, and adherence to the medication regimen, should be a priority.
Patients should be encouraged to target and maintain an ideal body weight, through diet and moderate physical exercise, as a strategy to normalize serum urate levels.27,47 However, they should be advised to avoid “crash dieting,” as this may precipitate a gout attack.27 In the recommended low-purine diet, consumption of red meat and shellfish is restricted,17 whereas consumption of soy, nonfat milk and other low-fat dairy products, cherries and other fruits, and increased vegetable protein is encouraged.31,37 Consumption of alcohol, especially beer and fortified wines, should be limited.27,47
Avoiding trauma to joints affected by gout (including the stress of bearing excess weight) can help patients limit future attacks.7,27
CONCLUSION
Patients with gout often have the characteristic presentation of an acutely tender, inflamed joint, but since gout is a systemic disorder, the clinician must also consider the possibility of gout in almost any organ system. Gout is a common disease, and its diagnosis can alert the astute clinician to investigate for certain metabolic disorders requiring intervention. Hyperlipidemia, metabolic syndrome, hypertension, chronic kidney disease, obesity, cardiovascular disease, and diabetes are all conditions associated with gout.
Recognizing the opportunity to offer preventive care measures and recommend lifestyle modifications to the patient with gout allows the clinician to play an important role in the patient’s care.
REFERENCES
1. Bhole V, de Vera M, Rahman MM, et al. Epidemiology of gout in women: fifty-two–year followup of a prospective cohort. Arthritis Rheum. 2010;62(4):1069-1076.
2. Neogi T. Clinical practice: gout. N Engl J Med. 2011;364(5):443-452.
3. Hernández-Cuevas CB, Roque LH, Huerta-Sil G, et al. First acute gout attacks commonly precede features of the metabolic syndrome. J Clin Rheumatol. 2009;15(2):65-67.
4. Louthrenoo W, Kasitanon N, Sukitawut W, Wichainun R. A clinical study of crystal-proven gouty arthritis in a university hospital. J Med Assoc Thai. 2003;86(9):868-875.
5. De Souza AW, Fernandes V, Ferrari AJ. Female gout: clinical and laboratory features. J Rheumatol. 2005;32(11):2186-2188.
6. Kurakula PC, Keenan RT. Diagnosis and management of gout: an update. J Musculoskel Med. 2010;27(10). www.musculoskeletalnet work.com/display/article/1145622/1692895. Accessed June 14, 2011.
7. Choi HK, Atkinson K, Karlson EW, Curhan G. Obesity, weight change, hypertension, diuretic use, and risk of gout in men: the Health Professionals Follow-up Study. Arch Intern Med. 2005;165(7):742-748.
8. Primatesta P, Plana E, Rothenbacher D. Gout treatment and comorbidities: a retrospective cohort study in a large US managed care population. BMC Musculoskelet Disord. 2011 May 20;12(1):103. [Epub ahead of print]
9. Choi HK, Curhan G. Independent impact of gout on mortality and risk for coronary heart disease. Circulation. 2007;116(8):894-900.
10. Krishnan E, Baker JF, Furst DE, Schumacher HR. Gout and the risk of acute myocardial infarction. Arthritis Rheum. 2006;54(8):2688-2696.
11. Lawrence RC, Felson DT, Helmick CG, et al; National Arthritis Data Workgroup. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008;58(1):26-35.
12. CDC. Gout. www.cdc.gov/arthritis/basics/gout.htm. Accessed June 14, 2011.
13. Arromdee E, Michet CJ, Crowson CS, et al. Epidemiology of gout: is the incidence rising? J Rheumatol. 2002;29(11):2403-2406.
14. Wallace KL, Riedel AA, Joseph-Ridge N, Wortmann R. Increasing prevalence of gout and hyperuricemia over 10 years among older adults in a managed care population. J Rheumatol. 2004; 31(8):1582-1587.
15. Weaver AL. Epidemiology of gout. Cleve Clin J Med. 2008;75 suppl 5:S9-S12.
16. Mandell BF. Clinical manifestations of hyperuricemia and gout. Cleve Clin J Med. 2008;75 suppl 5:S5-S8.
17. Choi HK, Atkinson K, Karlson EW, et al. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med. 2004;350(11):1093-1103.
18. Demarco MA, Maynard JW, Huizinga MM, et al. Younger age at gout onset is related to obesity in a community-based cohort. Arthritis Care Res (Hoboken). 2011 Apr 11; [Epub ahead of print].
19. Brook RA, Forsythe A, Smeeding JE, Lawrence Edwards N. Chronic gout: epidemiology, disease progression treatment and disease burden. Curr Med Res Opin. 2010;26(12):2813-2821.
20. Sachs L, Batra KL, Zimmermann B. Medical implications of hyperuricemia. Med Health R I. 2009;92(11):353-355.
21. Kellgren JH, Jeffrey MR, Ball J, eds. The Epidemiology of Chronic Rheumatism: Atlas of Standard Radiographs of Arthritis. Oxford: Blackwell; 1963:327.
22. Wu EQ, Patel PA, Mody RR, et al. Frequency, risk, and cost of gout-related episodes among the elderly: does serum uric acid level matter? J Rheumatol. 2009;36(5):1032-1040.
23. Riches PL, Wright AF, Ralston SH. Recent insights into the pathogenesis of hyperuricaemia and gout. Hum Mol Genet. 2009;18(R2):R177-R184.
24. Schumacher HR Jr. The pathogenesis of gout. Cleve Clin J Med. 2008;75 suppl 5:S2-S4.
25. Nakagawa T, Hu H, Zharikov S, et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Renal Physiol. 2006;290(3):F625-F631.
26. Zhang W, Doherty M, Pascual E, et al; EULAR (European League Against Rheumatism) Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part I: Diagnosis. Ann Rheum Dis. 2006;65(10):1301-1311.
27. Jordan KM, Cameron JS, Snaith M, et al. British Society for Rheumatology and British Health Professionals in Rheumatology guideline for the management of gout. Rheumatology. 2007;46(8):1372-1374.
28. Eggebeen AT. Gout: an update. Am Fam Physician. 2007;76(6):801-808.
29. Terkeltaub RA. Gout. N Engl J Med. 2003; 349(17):1647-1655.
30. Harris MD, Siegel LB, Alloway JA. Gout and hyperuricemia. Am Fam Physician. 1999;59(4): 925-934.
31. Schlesinger N. Dietary factors and hyperuricemia. Curr Pharm Des. 2005;11(32):4133-4138.
32. Ning TC, Keenan RT. Unusual presentations of gout. Curr Opin Rheumatol. 2010;22(2): 181-187.
33. Menon RK, Mikhailidis DP, Bell JL, et al. Warfarin administration increases uric acid concentrations in plasma. Clin Chem. 1986;32(8):
1557-1559.
34. Wallace SL, Robinson H, Masi AT, et al. Preliminary criteria for the classification of the acute arthritis of primary gout. Arthritis Rheum. 1977;20(3):895-900.
35. Martinon F, Glincher LH. Gout: new insights into an old disease. J Clin Invest. 2006;116 (8):2073-2075.
36. Zampogna G, Andracco R, Parodi M, Cimmino MA. Clinical features of gout in a cohort of Italian patients [in Italian]. Reumatismo. 2009; 61(1):41-47.
37. Choi HK. A prescription for lifestyle change in patients with hyperuricemia and gout. Curr Opin Rheumatol. 2010;22(2):165-172.
38. Bennett PH, Wood PH, eds. Population studies of the rheumatic diseases: proceedings of the Third International Symposium; June 5-10, 1966; New York, NY. Amsterdam: Excerpta Medica Foundation; 1968:457-458.
39. Malik A, Schumacher HR, Dinnella JE, Clayburne GM. Clinical diagnostic criteria for gout: comparison with the gold standard of synovial fluid crystal analysis. J Clin Rheumatol. 2009;15 (1):22-24.
40. Wijnands JMA, Boonen A, Arts ICW, et al. Large epidemiologic studies of gout: challenges in diagnosis and diagnostic criteria. Curr Rheumatol Rep. 2011;13(2):167-174.
41. Dodd LG, Major NM. Fine-needle aspiration cytology of articular and periarticular lesions. Cancer. 2002;96(3):157-165.
42. Dore RK. The gout diagnosis. Cleve Clin J Med. 2008;75 suppl 5:S17-S21.
43. Pettersson T. Sarcoid and erythema nodosum arthropathies. Baillieres Best Pract Res Clin Rheumatol. 2000;14(3):461-476.
44. Córdoba-Fernández A, Rayo-Rosado R. Pseudogout of the first metatarsophalangeal joint associated with hallux valgus: an atypical bilateral case. J Am Podiatr Med Assoc. 2010;100(2):138-142.
45. Dalbeth N, McQueen FM. Use of imaging to evaluate gout and other crystal deposition disorders. Curr Opin Rheumatol. 2009;21(2):124-131.
46. Wu EQ, Forsythe A, Guérin A, et al. Comorbidity burden healthcare resource utilization, and costs in chronic gout patients refractory to conventional urate-lowering therapy. Am J Ther. 2011 Feb 10; [Epub ahead of print].
47. Zhang W, Doherty M, Pascual E, et al; EULAR (European League Against Rheumatism) Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part II: Management. Ann Rheum Dis. 2006;65(10):1312-1324.
48. Schlesinger N, Yasothan U, Kirkpatrick P. Pegloticase [published correction appears in Nat Rev Drug Discov. 2011;10(2):156]. Nat Rev Drug Discov. 2011;10(1):17-18.
49. Pascual E, Sivera F, Yasothan U, Kurkpatrick P. Febuxostat. Nat Rev Drug Discov. 2009;8(3): 191-192.
50. Becker MA, Schumacher HR, Espinoza LR, et al. The urate-lowering efficacy and safety of febuxostat in the treatment of the hyperuricemia of gout; the CONFIRMS trial. Arthritis Res Ther. 2010;12(2):R63.
51. Hu M, Tomlinson B. Febuxostat in the management of hyperuricemia and chronic gout: a review. Ther Clin Risk Manag. 2008;4(6):1209-1220.
52. Reinders MK, Jansen TL. New advances in the treatment of gout: review of pegloticase. Ther Clin Risk Manag. 2010;6:543-550.
53. Cammalleri L, Malaguarnera M. Rasburicase represents a new tool for hyperuricemia in tumor lysis syndrome and in gout. Int J Med Sci. 2007; 4(2):83-93.
54. Richette P, Brière C, Hoenen-Clavert V, et al. Rasburicase for topaceous gout not treatable with allopurinol: an exploratory study. J Rheumatol. 2007;34(10):2093-2098.
55. Moolenburgh JD, Reinders MK, Jansen TL. Rasburicase treatment in severe tophaceous gout: a novel therapeutic option. Clin Rheumatol. 2006;25(5):749-752.