User login
Promising New Approaches for Testicular and Prostate Cancer
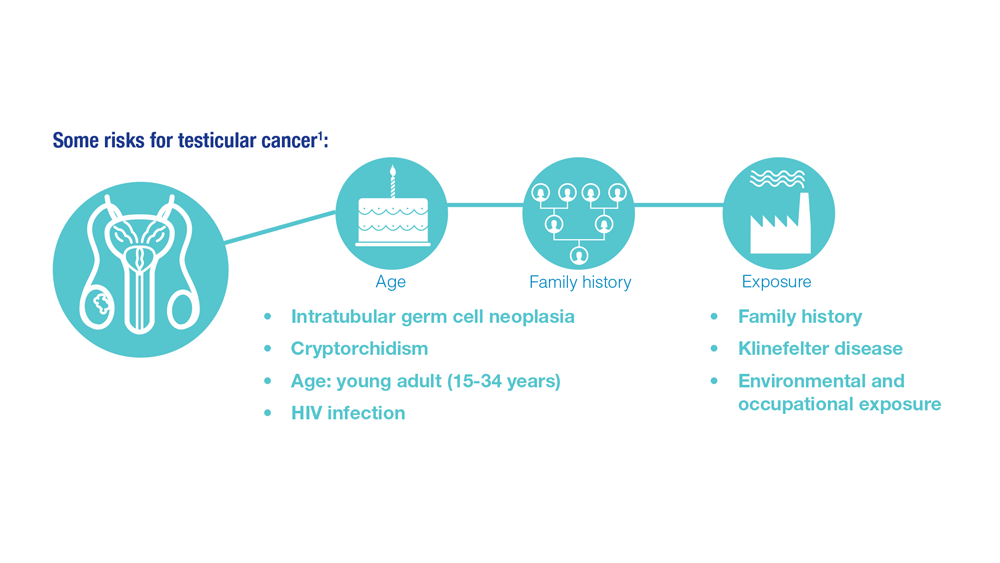
- Risk factors for testicular cancer. American Cancer Society. Updated May 17, 2018. Accessed December 15, 2022. https://www.cancer.org/cancer/testicular-cancer/causes-risks-prevention/risk-factors.html
- Chovanec M, Cheng L. BMJ. 2022;379:e070499. doi:10.1136/bmj-2022-070499
- Tavares NT et al. J Pathol. 2022. doi:10.1002/path.6037
- Bryant AK et al. JAMA Oncol. 2022;e224319. doi:10.1001/jamaoncol.2022.4319
- Kabasakal L et al. Nucl Med Commun. 2017;38(2):149-155. doi:10.1097/MNM.0000000000000617
- Sartor O et al; VISION Investigators. N Engl J Med. 2021;385(12):1091-1103. doi:10.1056/NEJMoa2107322
- Rowe SP et al. Annu Rev Med. 2019;70:461-477. doi:10.1146/annurev-med-062117-073027
- Pomykala KL et al. Eur Urol Oncol. 2022;S2588-9311(22)00177-8. doi:10.1016/j.euo.2022.10.007
- Keam SJ. Mol Diagn Ther. 2022;26(4):467-475. doi:10.1007/s40291-022-00594-2
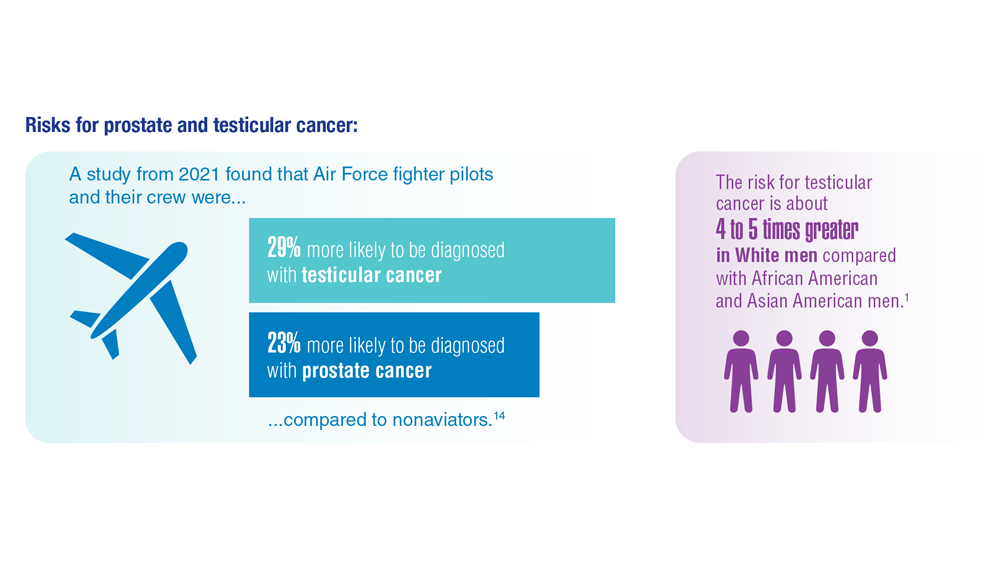
- Lovejoy LA et al. Mil Med. 2022:usac297. doi:10.1093/milmed/usac297
- Smith ZL et al. Med Clin North Am. 2018;102(2):251-264. doi:10.1016/j.mcna.2017.10.003
- Hohnloser JH et al. Eur J Med Res.1996;1(11):509-514.
- Johns Hopkins Medicine website. Testicular Cancer tumor Markers. Accessed December 2022. https://www.hopkinsmedicine.org/health/conditions-and-diseases/testicular-cancer/testicular-cancer-tumor-markers
- Webber BJ et al. J Occup Environ Med. 2022;64(1):71-78. doi:10.1097/JOM.0000000000002353
- Risk factors for testicular cancer. American Cancer Society. Updated May 17, 2018. Accessed December 15, 2022. https://www.cancer.org/cancer/testicular-cancer/causes-risks-prevention/risk-factors.html
- Chovanec M, Cheng L. BMJ. 2022;379:e070499. doi:10.1136/bmj-2022-070499
- Tavares NT et al. J Pathol. 2022. doi:10.1002/path.6037
- Bryant AK et al. JAMA Oncol. 2022;e224319. doi:10.1001/jamaoncol.2022.4319
- Kabasakal L et al. Nucl Med Commun. 2017;38(2):149-155. doi:10.1097/MNM.0000000000000617
- Sartor O et al; VISION Investigators. N Engl J Med. 2021;385(12):1091-1103. doi:10.1056/NEJMoa2107322
- Rowe SP et al. Annu Rev Med. 2019;70:461-477. doi:10.1146/annurev-med-062117-073027
- Pomykala KL et al. Eur Urol Oncol. 2022;S2588-9311(22)00177-8. doi:10.1016/j.euo.2022.10.007
- Keam SJ. Mol Diagn Ther. 2022;26(4):467-475. doi:10.1007/s40291-022-00594-2
- Lovejoy LA et al. Mil Med. 2022:usac297. doi:10.1093/milmed/usac297
- Smith ZL et al. Med Clin North Am. 2018;102(2):251-264. doi:10.1016/j.mcna.2017.10.003
- Hohnloser JH et al. Eur J Med Res.1996;1(11):509-514.
- Johns Hopkins Medicine website. Testicular Cancer tumor Markers. Accessed December 2022. https://www.hopkinsmedicine.org/health/conditions-and-diseases/testicular-cancer/testicular-cancer-tumor-markers
- Webber BJ et al. J Occup Environ Med. 2022;64(1):71-78. doi:10.1097/JOM.0000000000002353
- Risk factors for testicular cancer. American Cancer Society. Updated May 17, 2018. Accessed December 15, 2022. https://www.cancer.org/cancer/testicular-cancer/causes-risks-prevention/risk-factors.html
- Chovanec M, Cheng L. BMJ. 2022;379:e070499. doi:10.1136/bmj-2022-070499
- Tavares NT et al. J Pathol. 2022. doi:10.1002/path.6037
- Bryant AK et al. JAMA Oncol. 2022;e224319. doi:10.1001/jamaoncol.2022.4319
- Kabasakal L et al. Nucl Med Commun. 2017;38(2):149-155. doi:10.1097/MNM.0000000000000617
- Sartor O et al; VISION Investigators. N Engl J Med. 2021;385(12):1091-1103. doi:10.1056/NEJMoa2107322
- Rowe SP et al. Annu Rev Med. 2019;70:461-477. doi:10.1146/annurev-med-062117-073027
- Pomykala KL et al. Eur Urol Oncol. 2022;S2588-9311(22)00177-8. doi:10.1016/j.euo.2022.10.007
- Keam SJ. Mol Diagn Ther. 2022;26(4):467-475. doi:10.1007/s40291-022-00594-2
- Lovejoy LA et al. Mil Med. 2022:usac297. doi:10.1093/milmed/usac297
- Smith ZL et al. Med Clin North Am. 2018;102(2):251-264. doi:10.1016/j.mcna.2017.10.003
- Hohnloser JH et al. Eur J Med Res.1996;1(11):509-514.
- Johns Hopkins Medicine website. Testicular Cancer tumor Markers. Accessed December 2022. https://www.hopkinsmedicine.org/health/conditions-and-diseases/testicular-cancer/testicular-cancer-tumor-markers
- Webber BJ et al. J Occup Environ Med. 2022;64(1):71-78. doi:10.1097/JOM.0000000000002353


Ordering and Interpreting Precision Oncology Studies for Adults With Advanced Solid Tumors: A Primer
The ability to find and target specific biomarkers in the DNA of advanced cancers is rapidly changing options and outcomes for patients with locally advanced and metastatic solid tumors. This strategy is the basis for precision oncology, defined here as using predictive biomarkers from tumor and/or germline sequencing to guide therapies. This article focuses specifically on the use of DNA sequencing to find those biomarkers and provides guidance about which test is optimal in a specific situation, as well as interpretation of the results. We emphasize the identification of biomarkers that provide adult patients with advanced solid tumors access to therapies that would not be an option had sequencing not been performed and that have the potential for significant clinical benefit. The best approach is to have an expert team with experience in precision oncology to assist in the interpretation of results.
Which test?
Deciding what test of the array of assays available to use and which tissue to test can be overwhelming, and uncertainty may prevent oncology practitioners from ordering germline or somatic sequencing. For the purposes of this article, we will focus on DNA sequencing for inherited/germline alterations (including mutations, copy number changes, or fusions), which may inform treatment, or alterations that arise in the process of carcinogenesis and tumor evolution (somatic alterations in tumor DNA). This focus is not meant to exclude any specific test but to focus on DNA-based tests in patients with locally advanced or metastatic malignancy.
Germline Testing
Germline testing is the sequencing of inherited DNA in noncancerous cells to find alterations that may play a role in the development of cancers and are actionable in some cases. Germline alterations can inform therapeutic decisions, predict future cancer risk, and provide information that can help family members to better manage their risks of malignancy. Detailed discussions of the importance of germline testing to inform cancer surveillance, risk-reducing interventions, and the testing of relatives to determine who carries inherited alterations (cascade testing) is extremely important with several advantages and is covered in a number of excellent reviews elsewhere.1-3 Testing of germline DNA in patients with a metastatic malignancy can provide treatment options otherwise not available for patients, particularly for BRCA1/2 and Lynch syndrome–related cancers. Recent studies have shown that 10 to 15% of patients with advanced malignancies of many types have a pathogenic germline alteration.4,5
Germline DNA is usually acquired from peripheral blood, a buccal swab, or saliva collection and is therefore readily available. This is advantageous because it does not require a biopsy to identify relevant alterations. Germline testing is also less susceptible to the rare situations in which artifacts occur in formalin-fixed tissues and obscure relevant alterations.
The cost of germline testing varies, but most commercial vendor assays for germline testing are significantly less expensive than the cost of somatic testing. The disadvantages include the inability of germline testing to find any alterations that arise solely in tumor tissue and the smaller gene panels included in germline testing as compared to somatic testing panels. Other considerations relate to the inherited nature of pathogenic germline variants and its implications for family members that may affect the patient’s psychosocial health and potentially change the family dynamics.
Deciding who is appropriate for germline testing and when to perform the testing should be individualized to the patient’s wishes and disease status. Treatment planning may be less complicated if testing has been performed and germline status is known. In some cases urgent germline testing is indicated to inform pending procedures and/or surgical decisions for risk reduction, including more extensive tissue resection, such as the removal of additional organs or contralateral tissue. A minor point regarding germline testing is that the DNA of patients with hematologic malignancies may be difficult to sequence because of sample contamination by the circulating malignancy. For this reason, most laboratories will not accept peripheral blood or saliva samples for germline testing in patients with active hematologic malignancies; they often require DNA from another source such as fibroblasts from a skin biopsy or cells from a muscle biopsy. Germline testing is recommended for all patients with metastatic prostate cancer, as well as any patient with any stage of pancreatic cancer or ovarian cancer and patients with breast cancer diagnosed at age ≤ 45 years. More detailed criteria for who is appropriate for germline testing outside of these groups can be found in the appropriate National Comprehensive Cancer Network (NCCN) guidelines.6-8 In patients with some malignancies such as prostate and pancreatic cancer, approximately half of patients who have a BRCA-related cancer developed that malignancy because of a germline BRCA alteration.9-11 Testing germline DNA is therefore an easy way to quickly find almost half of all targetable alterations with a treatment approved by the US Food and Drug Administration (FDA) and at low cost, with the added benefit of providing critical information for families who may be unaware that members carry a relevant pathogenic germline alteration. In those families, cascade testing can provide surveillance and intervention strategies that can be lifesaving.
A related and particularly relevant question is when should a result found on a somatic testing panel prompt follow-up germline testing? Some institutions have algorithms in place to automate referral for germline testing based on specific genetic criteria.12 Excellent reviews are available that outline the following considerations in more detail.13 Typically, somatic testing results that would trigger follow-up germline testing would be truncating or deleterious or likely deleterious mutations per germline datasets in high-risk genes associated with highly penetrant autosomal dominant conditions (BRCA1, BRCA2, PALB2, MLH1, MSH2, and MSH6), selected moderate-risk genes (BRIP1, RAD51C, and RAD51D), and specific variants with a high probability of being germline because they are common germline founder mutations. Although the actionability and significance of specific genes remains a matter of some discussion, generally finding a somatic pathogenic sequencing result included in the 59-gene list of the American College of Medical Genetics and Genomics (ACMG) guidelines would be an indication for germline testing. Another indication for germline testing would be finding genes with germline mutations for which the NCCN has specific management guidelines, or the presence of alterations consistent with known founder mutations.14 When a patient’s tumor has microsatellite instability or is hypermutated (defined as > 10 mutations per megabase), a search for germline alterations is warranted given that about 15% of these patients with these tumors carry a Lynch syndrome gene.15 Genes that are commonly found as somatic alterations alone (eg, TP53 or APC) are generally not an indication for germline testing unless family history is compelling.
Although some clinicians use the variant allele fraction in the somatic sequencing report to decide whether to conduct germline testing, this approach is suboptimal, as allele fraction may be confounded by assay conditions and a high allele fraction may be found in pure tumors with loss of heterozygosity (LOH) of the other allele. There is also evidence that for a variety of reasons, somatic sequencing panels do not always detect germline alterations in somatic tissues.16 Reasons for this may include discordance between the genes being tested in the germline vs the somatic panel, technical differences such as interference of formalin-fixed paraffin-embedded (FFPE) artifact with detecting the germline variant, lack of expertise in germline variant interpretation among laboratories doing tumor-only sequencing, and, in rare cases, large deletions in tumor tissue masking a germline point mutation.
Variant Interpretation of Germline Testing
A general understanding of the terminology used for germline variant interpretation allows for the ordering health care practitioner (HCP) to provide the best quality care and an appreciation for the limitations of current molecular testing. Not all variants are associated with disease; the clinical significance of a genetic variant falls on a spectrum. The criteria for determining pathogenicity differ between molecular laboratories, but most are influenced by the standards and guidelines set forth by the ACMG.14 The clinical molecular laboratory determines variant classification, and a detailed discussion is beyond the scope of this primer. In brief, variant classification is based on evidence of varying strength in different categories including population data, computational and predictive data, functional data, segregation data, de novo data, allelic data, and information from various databases. The ACMG has proposed a 5-tiered classification system, by which most molecular laboratories adhere to in their genetic test reports (Table 1).14
Pathogenic and likely pathogenic variants are clinically actionable, whereas variants of uncertain significance (VUS) require additional data and/or functional studies before making clinical decisions. Depending on the clinical context and existing supporting evidence, it may be prudent to continue monitoring for worsening or new signs of disease in patients with one or more VUS while additional efforts are underway to understand the variant’s significance.

In some cases, variants are reclassified, which may alter the management and treatment of patients. Reclassification can occur with VUS, and in rare instances, can also occur with variants previously classified as pathogenic/likely pathogenic or benign/likely benign. In such a case, the reporting laboratory will typically make concerted efforts to alert the ordering HCP. However, variant reclassifications are not always communicated to the care team. Thus, it is important to periodically contact the molecular laboratory of interest to obtain updated test interpretations.
Somatic Testing
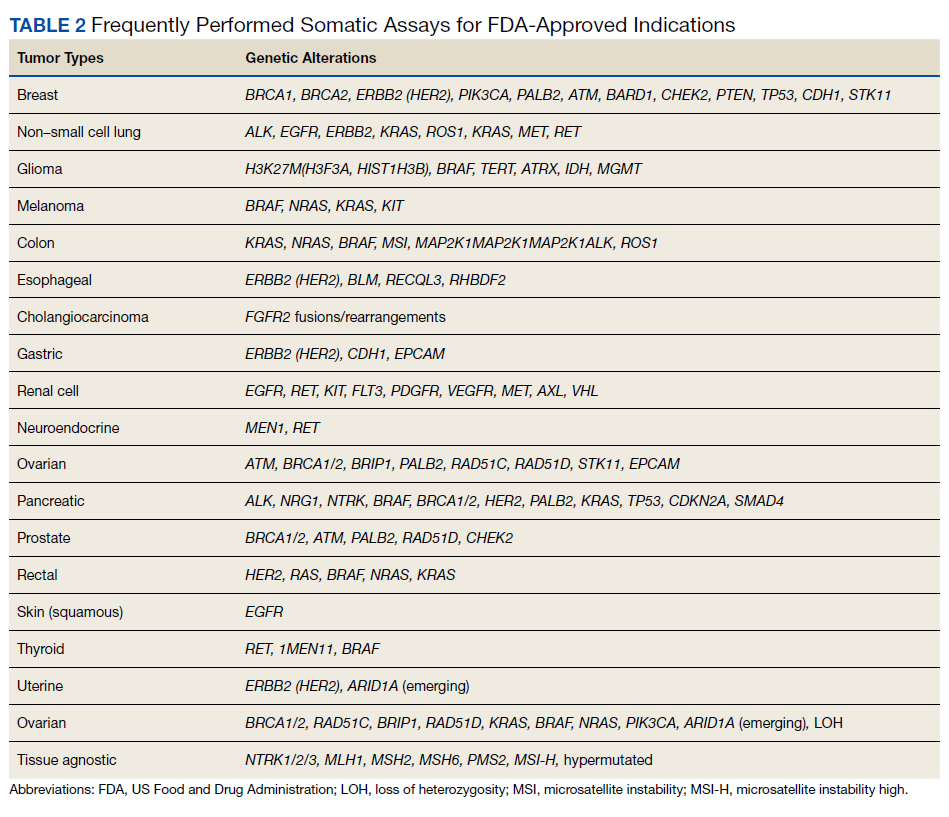
Testing of somatic (tumor) tissue is critical and is the approach most commonly taken in medical oncology (Table 2).
The advantages of primary tumor are that it is usually in hand as a diagnostic biopsy, acquisition is standard of care, and several targetable alterations are truncal, defined as driver mutations present at the time of tumor development. Also, the potential that the tumor arose in the background of a predisposing germline alteration can be suggested by sequencing primary tumor as discussed above. Moreover, sequencing the primary tumor can be done at any time unless the biopsy sample is considered too old or degraded (per specific platform requirements). The information gained can be used to anticipate additional treatment options that are relevant when patients experience disease progression. Disadvantages include the problem that primary specimens may be old or have limited tumor content, both of which increase the likelihood that sequencing will not be technically successful.
Alterations that are targetable and arise as a result of either treatment pressure or clonal evolution are considered evolutionary. If evolutionary alterations are the main focus for sequencing, then metastasis biopsy or ctDNA are better choices. The advantages of a metastasis biopsy are that tissue is contemporary, tumor content may be higher than in primary tumor, and both truncal and evolutionary alterations can be detected.
For specific tumors, continued analysis of evolving genomic alterations can play a critical role in management. In non–small cell lung cancer (NSCLC), somatic testing is conducted again at progression on repeat biopsies to evaluate for emerging resistance mutations. In epidermal growth factor receptor (EGFR)–mutated lung cancer, the resistance mutation, exon 20 p.T790M (point mutation), can present in patients after treatment with first- or second-generation EGFR tyrosine kinase inhibitors (TKI). Even in patients who are treated with the third-generation EGFR TKI osimertinib that can treat T790M-mutated lung cancer, multiple possible evolutionary mutations can occur at progression, including other EGFR mutations, MET/HER2 amplification, and BRAF V600E, to name a few.20 Resistance mechanisms develop due to treatment selection pressure and the molecular heterogeneity seen in lung cancer.
Disadvantages for metastatic biopsy include the inability to safely access a metastatic site, the time considerations for preauthorization and arrangement of biopsy, and a lower-than-average likelihood of successful sequencing from sites such as bone.21,22 In addition, there is some concern that a single metastatic site may not capture all relevant alterations for multiple reasons, including tumor heterogeneity.
Significant advances in the past decade have dramatically improved the ability to use ctDNA to guide therapy. Advantages include ease of acquisition as acquiring a sample requires only a blood draw, and the potential that the pool of ctDNA is a better reflection of the relevant biology as it potentially reflects all metastatic tissues. Disadvantages are that sequencing attempts may not be productive if the sample is acquired at a time when the tumor is either quiescent or tumor burden is so low that only limited amounts of DNA are being shed. Performing ctDNA analysis when a tumor is not progressing is less likely to be productive for a number of tumor types.23,24 Sequencing ctDNA is also more susceptible than sequencing tumor biopsies to detection of alterations that are not from the tumor of interest but from clonal hematopoiesis of indeterminate potential (CHIP) or other clonal hematopoietic disorders (see Confounders section below).
Selecting the Tissue
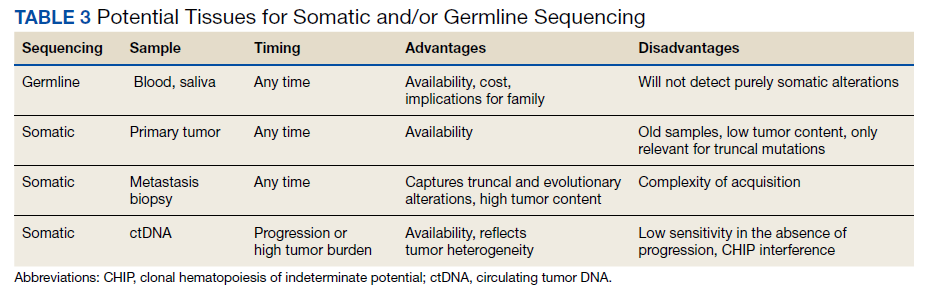
Deciding on the tissue to analyze is a critical part of the decision process (Table 3). If the primary tumor tissue is old the likelihood of productive sequencing is lower, although age alone is not the only consideration and the methods of fixation may be just as relevant.
For prostate cancer in particular, the ability to successfully sequence primary tumor tissue decreases as the amount of tumor decreases in low-volume biopsies such as prostate needle biopsies. Generally, if tumor content is < 10% of the biopsy specimen, then sequencing is less likely to be productive.25 Also, if the alteration of interest is not known to be truncal, then a relevant target might be missed by sequencing tissue that does not reflect current biology. Metastasis biopsy may be the most appropriate tissue, particularly if this specimen has already been acquired. As above, a metastasis biopsy may have a higher tumor content, and it should reflect relevant biology if it is recent. However, bone biopsies have a relatively low yield for successful sequencing, so a soft tissue lesion (eg, liver or lymph node metastasis) is generally preferred.
The inability to safely access tissue is often a consideration. Proximity to vital structures such as large blood vessels or the potential for significant morbidity in the event of a complication (liver or lung biopsies, particularly in patients on anticoagulation medications) may make the risk/benefit ratio too high. The inability to conduct somatic testing has been reported to often be due to inadequate tissue sampling.26 ctDNA is an attractive alternative but should typically be drawn when a tumor is progressing with a reasonable tumor burden that is more likely to be shedding DNA. Performing ctDNA analysis in patients without obvious radiographic metastasis or in patients whose tumor is under good control is unlikely to produce interpretable results.
Interpreting the Results
The intent of sequencing tumor tissue is to identify alterations that are biologically important and may provide a point of therapeutic leverage. However, deciding which alterations are relevant is not always straightforward. For example, any normal individual genome contains around 10,000 missense variants, hundreds of insertion/deletion variants, and dozens of protein-truncating variants. Distinguishing these alterations, which are part of the individual, from those that are tumor-specific and have functional significance can be difficult in the absence of paired sequencing of both normal and tissue samples.
Specific Alterations
Although most commercial vendors provide important information in sequencing reports to assist oncology HCPs in deciding which alterations are relevant, the reports are not always clear. In many cases the report will specifically indicate whether the alteration has been reported previously as pathogenic or benign. However, some platforms will report alterations that are not known to be drivers of tumor biology. It is critical to be aware that if variants are not reported as pathogenic, they should not be assumed to be pathogenic simply because they are included in the report. Alterations more likely to be drivers of relevant biology are those that change gene and protein structure and include frameshift (fs*), nonsense (denoted by sequence ending in “X” or “*”), or specific fusions or insertions/deletions (indel) that occur in important domains of the gene.
For some genes, only specific alterations are targetable and not all alterations have the same effect on protein function. Although overexpression of certain genes and proteins are actionable (eg, HER2), amplification of a gene does not necessarily indicate that it is targetable. In NSCLC, specific alterations convey sensitivity to targeted therapies. For example, in EGFR-mutated NSCLC, the sensitizing mutations to EGFR TKIs are exon19 deletions and exon 21 L858R point mutations (the most common mutations), as well as less common mutations found in exon 18-21. Exon 20 mutations, however, are not responsive to EGFR TKIs with a few exceptions.27 Patients who have tumors that do not harbor a sensitizing EGFR mutation should not be treated with an EGFR TKI. In a variety of solid tumors, gene fusions of the NTRK 1/2/3, act as oncogenic drivers. The chromosomal fusion events involving the carboxy-terminal kinase domain of TRK and upstream amino-terminal partners lead to overexpression of the chimeric proteins tropomyosin receptor kinase (TRK) A/B/C, resulting in constitutively active, ligand-independent downstream signaling. In patients with NTRK 1/2/3 gene fusions, larotrectinib and entrectinib, small molecule inhibitors to TRK, have shown antitumor activity.28,29 No alterations beyond these fusions are known to be targetable.
Allele Fraction
Knowing the fraction (or proportion) of the alteration of interest in the sequenced tissue relative to the estimated tumor content can assist in decision making. Not all platforms will provide this information, which is referred to as mutation allele fraction (MAF) or variant allele fraction (VAF), but sometimes will provide it on request. Platforms will usually provide an estimate of the percent tumor in the tissue being sampled if it is from a biopsy. If the MAF is around 50% in the sequenced tissue (including ctDNA), then there is a reasonable chance that it is a germline variant. However, there are nuances as germline alterations in some genes, such as BRCA1/2, can be accompanied by loss of the other allele of the gene (LOH). In that case, if most of the circulating DNA is from tumor, then the MAF can be > 50%.
If there are 2 alterations of the same gene with MAF percentages that are each half of the total percent tumor, there is a high likelihood of biallelic alteration. These sorts of paired alterations or one mutation with apparent LOH or copy loss would again indicate a high likelihood that the alteration is in fact pathogenic and a relevant driver. Not all pathogenic alterations have to be biallelic to be driver mutations but in BRCA1/2, or mismatch repair deficiency genes, the presence of biallelic alterations increases the likelihood of their being pathogenic.
Tumors that are hypermutated—containing sometimes hundreds of mutations per megabyte of DNA—can be particularly complicated to interpret, because the likelihood increases that many of the alterations are a function of the hypermutation and not a driver mutation. This is particularly important when there are concurrent mutations in mismatch repair genes and genes, such as BRCA1/2. If the tumor is
Confounders
In some situations, interpretation can be particularly challenging. For example, several alterations for which there are FDA on-label indications (such as ATM or BRCA2) can be detected in ctDNA that may not be due to the tumor but to CHIP. CHIP represents hematopoietic clones that are dysplastic as a result of exposure to DNA-damaging agents (eg, platinum chemotherapy) or as a result of aging and arise when mutations in hematopoietic stem cells provide a competitive advantage.31 The most common CHIP clones that can be detected are DNMT3A, ASXL1, or TET2; because these alterations are not targetable, their importance lies primarily in whether patients have evidence of hematologic abnormality, which might represent an evolving hematopoietic disorder. Because CHIP alterations can overlap with somatic alterations for which FDA-approved drugs exist, such as ATM or CHEK2 (olaparib for prostate cancer) and BRCA2 (poly-ADP-ribose polymerase inhibitors in a range of indications) there is concern that CHIP might result in patient harm from inappropriate treatment of CHIP rather than the tumor, with no likelihood that the treatment would affect the tumor, causing treatment delays.32 General considerations for deciding whether an alteration represents CHIP include excluding alteration in which the VAF is < 1% and when the VAF in the alteration of interest is < 20% of the estimated tumor fraction in the sample. Exceptions to this are found in patients with true myelodysplasia or chronic lymphocytic leukemia, in whom the VAF can be well over 50% because of circulating tumor burden. The only way to be certain that an alteration detected on ctDNA reflects tumor rather than CHIP is to utilize an assay with matched tumor-normal sequencing.
Resources for Assistance
For oncology HCPs, perhaps the best resource to help in selecting and interpreting the appropriate testing is through a dedicated molecular oncology tumor board and subject matter experts who contribute to those tumor boards. In the US Department of Veterans Affairs, the national precision oncology program and its affiliated clinical services, such as the option to order a national consultation and molecular tumor board education, are easily accessible to all HCPs (www.cancer.va.gov). Many commercial vendors provide support to assist with questions of interpretation and to inform clinical decision-making. Other resources that can assist with deciding whether an alteration is pathogenic include extensive curated databases such as ClinVar (www.ncbi.nlm.nih.gov/clinvar) and the Human Genetic Mutation Database (www.hgmd.cf.ac.uk/ac/index.php) for germline alterations or COSMIC (cancer.sanger.ac.uk/cosmic) for somatic alterations. OncoKB (www.oncokb.org) is a resource for assistance in defining levels of evidence for the use of agents to target specific alterations and to assist in assigning pathogenicity to specific alterations. Additional educational resources for training in genomics and genetics are also included in the Appendix.
The rapid growth in technology and ability to enhance understanding of relevant tumor biology continues to improve the therapeutic landscape for men and women dealing with malignancy and our ability to find targetable genetic alterations with the potential for meaningful clinical benefit.
Acknowledgments
Dedicated to Neil Spector.
1. Domchek SM, Mardis E, Carlisle JW, Owonikoko TK. Integrating genetic and genomic testing into oncology practice. Am Soc Clin Oncol Educ Book. 2020;40:e259-e263. doi:10.1200/EDBK_280607
2. Stoffel EM, Carethers JM. Current approaches to germline cancer genetic testing. Annu Rev Med. 2020;71:85-102. doi:10.1146/annurev-med-052318-101009
3. Lappalainen T, Scott AJ, Brandt M, Hall IM. Genomic analysis in the age of human genome sequencing. Cell. 2019;177(1):70-84. doi:10.1016/j.cell.2019.02.032
4. Samadder NJ, Riegert-Johnson D, Boardman L, et al. Comparison of universal genetic testing vs guideline-directed targeted testing for patients with hereditary cancer syndrome. JAMA Oncol. 2021;7(2):230-237. doi:10.1001/jamaoncol.2020.6252
5. Schneider BP, Stout L, Philips S, et al. Implications of incidental germline findings identified in the context of clinical whole exome sequencing for guiding cancer therapy. JCO Precis Oncol. 2020;4:1109-1121. doi:10.1200/PO.19.00354
6. National Comprehensive Cancer Network. Pancreatic cancer (Version 1.2022). Updated February 24, 2022. Accessed April 13, 2022. https://www.nccn.org/professionals/physician_gls/pdf/pancreatic.pdf
7. National Comprehensive Cancer Network. Prostate cancer (Version 3.2022). Updated January 10, 2022. Accessed April 13, 2022. https://www.nccn.org/professionals/physician_gls/pdf/prostate.pdf
8. National Comprehensive Cancer Network. Genetic/familial high-risk assessment: breast, ovarian, and pancreatic (Version 2.2022). Updated March 9, 2022. Accessed April 13, 2022. https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf
9. Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215-1228. doi:10.1016/j.cell.2015.05.001
10. Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375(5):443-453. doi:10.1056/NEJMoa1603144
11. Cancer Genome Atlas Research Network. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017;32(2):185-203.e13. doi:10.1016/j.ccell.2017.07.007
12. Clark DF, Maxwell KN, Powers J, et al. Identification and confirmation of potentially actionable germline mutations in tumor-only genomic sequencing. JCO Precis Oncol. 2019;3:PO.19.00076. doi:10.1200/PO.19.00076
13. DeLeonardis K, Hogan L, Cannistra SA, Rangachari D, Tung N. When should tumor genomic profiling prompt consideration of germline testing? J Oncol Pract. 2019;15(9):465-473. doi:10.1200/JOP.19.00201
14. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi:10.1038/gim.2015.30
15. Latham A, Srinivasan P, Kemel Y, et al. Microsatellite instability is associated with the presence of Lynch syndrome pan-cancer. J Clin Oncol. 2019;37(4):286-295. doi:10.1200/JCO.18.00283
16. Lincoln SE, Nussbaum RL, Kurian AW, et al. Yield and utility of germline testing following tumor sequencing in patients with cancer. JAMA Netw Open. 2020;3(10):e2019452. doi:10.1001/jamanetworkopen.2020.19452
17. National Comprehensive Cancer Network. Non-small cell lung cancer (Version: 3.2022). Updated March 16, 2022. Accessed April 13, 2022. https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf
18. National Comprehensive Cancer Network. Colon cancer (Version 1.2022). February 25, 2022. Accessed April 13, 2022. https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf
19. National Comprehensive Cancer Network. Melanoma: cutaneous (Version 3.2022). April 11, 2022. Accessed April 13, 2022. https://www.nccn.org/professionals/physician_gls/pdf/cutaneous_melanoma.pdf
20. Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer. 2019;121(9):725-737. doi:10.1038/s41416-019-0573-8
21. Zheng G, Lin MT, Lokhandwala PM, et al. Clinical mutational profiling of bone metastases of lung and colon carcinoma and malignant melanoma using next-generation sequencing. Cancer Cytopathol. 2016;124(10):744-753. doi:10.1002/cncy.21743
22. Spritzer CE, Afonso PD, Vinson EN, et al. Bone marrow biopsy: RNA isolation with expression profiling in men with metastatic castration-resistant prostate cancer—factors affecting diagnostic success. Radiology. 2013;269(3):816-823. doi:10.1148/radiol.13121782
23. Schweizer MT, Gulati R, Beightol M, et al. Clinical determinants for successful circulating tumor DNA analysis in prostate cancer. Prostate. 2019;79(7):701-708. doi:10.1002/pros.23778
24. Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6(224):224ra224. doi:10.1126/scitranslmed.3007094
25. Pritchard CC, Salipante SJ, Koehler K, et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn. 2014;16(1):56-67. doi:10.1016/j.jmoldx.2013.08.004
26. Gutierrez ME, Choi K, Lanman RB, et al. Genomic profiling of advanced non-small cell lung cancer in community settings: gaps and opportunities. Clin Lung Cancer. 2017;18(6):651-659. doi:10.1016/j.cllc.2017.04.004
27. Malapelle U, Pilotto S, Passiglia F, et al. Dealing with NSCLC EGFR mutation testing and treatment: a comprehensive review with an Italian real-world perspective. Crit Rev Oncol Hematol. 2021;160:103300. doi:10.1016/j.critrevonc.2021.103300
28. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. 2018;378(8):731-739. doi:10.1056/NEJMoa1714448
29. Doebele RC, Drilon A, Paz-Ares L, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020;21(2):271-282. doi:10.1016/S1470-2045(19)30691-6
30. Jonsson P, Bandlamudi C, Cheng ML, et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature. 2019;571(7766):576-579. doi:10.1038/s41586-019-1382-1
31. Steensma DP. Clinical consequences of clonal hematopoiesis of indeterminate potential. Hematology Am Soc Hematol Educ Program. 2018;2018(1):264-269. doi:10.1182/asheducation-2018.1.264
32. Jensen K, Konnick EQ, Schweizer MT, et al. Association of clonal hematopoiesis in DNA repair genes with prostate cancer plasma cell-free DNA testing interference. JAMA Oncol. 2021;7(1):107-110. doi:10.1001/jamaoncol.2020.5161
The ability to find and target specific biomarkers in the DNA of advanced cancers is rapidly changing options and outcomes for patients with locally advanced and metastatic solid tumors. This strategy is the basis for precision oncology, defined here as using predictive biomarkers from tumor and/or germline sequencing to guide therapies. This article focuses specifically on the use of DNA sequencing to find those biomarkers and provides guidance about which test is optimal in a specific situation, as well as interpretation of the results. We emphasize the identification of biomarkers that provide adult patients with advanced solid tumors access to therapies that would not be an option had sequencing not been performed and that have the potential for significant clinical benefit. The best approach is to have an expert team with experience in precision oncology to assist in the interpretation of results.
Which test?
Deciding what test of the array of assays available to use and which tissue to test can be overwhelming, and uncertainty may prevent oncology practitioners from ordering germline or somatic sequencing. For the purposes of this article, we will focus on DNA sequencing for inherited/germline alterations (including mutations, copy number changes, or fusions), which may inform treatment, or alterations that arise in the process of carcinogenesis and tumor evolution (somatic alterations in tumor DNA). This focus is not meant to exclude any specific test but to focus on DNA-based tests in patients with locally advanced or metastatic malignancy.
Germline Testing
Germline testing is the sequencing of inherited DNA in noncancerous cells to find alterations that may play a role in the development of cancers and are actionable in some cases. Germline alterations can inform therapeutic decisions, predict future cancer risk, and provide information that can help family members to better manage their risks of malignancy. Detailed discussions of the importance of germline testing to inform cancer surveillance, risk-reducing interventions, and the testing of relatives to determine who carries inherited alterations (cascade testing) is extremely important with several advantages and is covered in a number of excellent reviews elsewhere.1-3 Testing of germline DNA in patients with a metastatic malignancy can provide treatment options otherwise not available for patients, particularly for BRCA1/2 and Lynch syndrome–related cancers. Recent studies have shown that 10 to 15% of patients with advanced malignancies of many types have a pathogenic germline alteration.4,5
Germline DNA is usually acquired from peripheral blood, a buccal swab, or saliva collection and is therefore readily available. This is advantageous because it does not require a biopsy to identify relevant alterations. Germline testing is also less susceptible to the rare situations in which artifacts occur in formalin-fixed tissues and obscure relevant alterations.
The cost of germline testing varies, but most commercial vendor assays for germline testing are significantly less expensive than the cost of somatic testing. The disadvantages include the inability of germline testing to find any alterations that arise solely in tumor tissue and the smaller gene panels included in germline testing as compared to somatic testing panels. Other considerations relate to the inherited nature of pathogenic germline variants and its implications for family members that may affect the patient’s psychosocial health and potentially change the family dynamics.
Deciding who is appropriate for germline testing and when to perform the testing should be individualized to the patient’s wishes and disease status. Treatment planning may be less complicated if testing has been performed and germline status is known. In some cases urgent germline testing is indicated to inform pending procedures and/or surgical decisions for risk reduction, including more extensive tissue resection, such as the removal of additional organs or contralateral tissue. A minor point regarding germline testing is that the DNA of patients with hematologic malignancies may be difficult to sequence because of sample contamination by the circulating malignancy. For this reason, most laboratories will not accept peripheral blood or saliva samples for germline testing in patients with active hematologic malignancies; they often require DNA from another source such as fibroblasts from a skin biopsy or cells from a muscle biopsy. Germline testing is recommended for all patients with metastatic prostate cancer, as well as any patient with any stage of pancreatic cancer or ovarian cancer and patients with breast cancer diagnosed at age ≤ 45 years. More detailed criteria for who is appropriate for germline testing outside of these groups can be found in the appropriate National Comprehensive Cancer Network (NCCN) guidelines.6-8 In patients with some malignancies such as prostate and pancreatic cancer, approximately half of patients who have a BRCA-related cancer developed that malignancy because of a germline BRCA alteration.9-11 Testing germline DNA is therefore an easy way to quickly find almost half of all targetable alterations with a treatment approved by the US Food and Drug Administration (FDA) and at low cost, with the added benefit of providing critical information for families who may be unaware that members carry a relevant pathogenic germline alteration. In those families, cascade testing can provide surveillance and intervention strategies that can be lifesaving.
A related and particularly relevant question is when should a result found on a somatic testing panel prompt follow-up germline testing? Some institutions have algorithms in place to automate referral for germline testing based on specific genetic criteria.12 Excellent reviews are available that outline the following considerations in more detail.13 Typically, somatic testing results that would trigger follow-up germline testing would be truncating or deleterious or likely deleterious mutations per germline datasets in high-risk genes associated with highly penetrant autosomal dominant conditions (BRCA1, BRCA2, PALB2, MLH1, MSH2, and MSH6), selected moderate-risk genes (BRIP1, RAD51C, and RAD51D), and specific variants with a high probability of being germline because they are common germline founder mutations. Although the actionability and significance of specific genes remains a matter of some discussion, generally finding a somatic pathogenic sequencing result included in the 59-gene list of the American College of Medical Genetics and Genomics (ACMG) guidelines would be an indication for germline testing. Another indication for germline testing would be finding genes with germline mutations for which the NCCN has specific management guidelines, or the presence of alterations consistent with known founder mutations.14 When a patient’s tumor has microsatellite instability or is hypermutated (defined as > 10 mutations per megabase), a search for germline alterations is warranted given that about 15% of these patients with these tumors carry a Lynch syndrome gene.15 Genes that are commonly found as somatic alterations alone (eg, TP53 or APC) are generally not an indication for germline testing unless family history is compelling.
Although some clinicians use the variant allele fraction in the somatic sequencing report to decide whether to conduct germline testing, this approach is suboptimal, as allele fraction may be confounded by assay conditions and a high allele fraction may be found in pure tumors with loss of heterozygosity (LOH) of the other allele. There is also evidence that for a variety of reasons, somatic sequencing panels do not always detect germline alterations in somatic tissues.16 Reasons for this may include discordance between the genes being tested in the germline vs the somatic panel, technical differences such as interference of formalin-fixed paraffin-embedded (FFPE) artifact with detecting the germline variant, lack of expertise in germline variant interpretation among laboratories doing tumor-only sequencing, and, in rare cases, large deletions in tumor tissue masking a germline point mutation.
Variant Interpretation of Germline Testing
A general understanding of the terminology used for germline variant interpretation allows for the ordering health care practitioner (HCP) to provide the best quality care and an appreciation for the limitations of current molecular testing. Not all variants are associated with disease; the clinical significance of a genetic variant falls on a spectrum. The criteria for determining pathogenicity differ between molecular laboratories, but most are influenced by the standards and guidelines set forth by the ACMG.14 The clinical molecular laboratory determines variant classification, and a detailed discussion is beyond the scope of this primer. In brief, variant classification is based on evidence of varying strength in different categories including population data, computational and predictive data, functional data, segregation data, de novo data, allelic data, and information from various databases. The ACMG has proposed a 5-tiered classification system, by which most molecular laboratories adhere to in their genetic test reports (Table 1).14
Pathogenic and likely pathogenic variants are clinically actionable, whereas variants of uncertain significance (VUS) require additional data and/or functional studies before making clinical decisions. Depending on the clinical context and existing supporting evidence, it may be prudent to continue monitoring for worsening or new signs of disease in patients with one or more VUS while additional efforts are underway to understand the variant’s significance.
In some cases, variants are reclassified, which may alter the management and treatment of patients. Reclassification can occur with VUS, and in rare instances, can also occur with variants previously classified as pathogenic/likely pathogenic or benign/likely benign. In such a case, the reporting laboratory will typically make concerted efforts to alert the ordering HCP. However, variant reclassifications are not always communicated to the care team. Thus, it is important to periodically contact the molecular laboratory of interest to obtain updated test interpretations.
Somatic Testing
Testing of somatic (tumor) tissue is critical and is the approach most commonly taken in medical oncology (Table 2).
The advantages of primary tumor are that it is usually in hand as a diagnostic biopsy, acquisition is standard of care, and several targetable alterations are truncal, defined as driver mutations present at the time of tumor development. Also, the potential that the tumor arose in the background of a predisposing germline alteration can be suggested by sequencing primary tumor as discussed above. Moreover, sequencing the primary tumor can be done at any time unless the biopsy sample is considered too old or degraded (per specific platform requirements). The information gained can be used to anticipate additional treatment options that are relevant when patients experience disease progression. Disadvantages include the problem that primary specimens may be old or have limited tumor content, both of which increase the likelihood that sequencing will not be technically successful.
Alterations that are targetable and arise as a result of either treatment pressure or clonal evolution are considered evolutionary. If evolutionary alterations are the main focus for sequencing, then metastasis biopsy or ctDNA are better choices. The advantages of a metastasis biopsy are that tissue is contemporary, tumor content may be higher than in primary tumor, and both truncal and evolutionary alterations can be detected.
For specific tumors, continued analysis of evolving genomic alterations can play a critical role in management. In non–small cell lung cancer (NSCLC), somatic testing is conducted again at progression on repeat biopsies to evaluate for emerging resistance mutations. In epidermal growth factor receptor (EGFR)–mutated lung cancer, the resistance mutation, exon 20 p.T790M (point mutation), can present in patients after treatment with first- or second-generation EGFR tyrosine kinase inhibitors (TKI). Even in patients who are treated with the third-generation EGFR TKI osimertinib that can treat T790M-mutated lung cancer, multiple possible evolutionary mutations can occur at progression, including other EGFR mutations, MET/HER2 amplification, and BRAF V600E, to name a few.20 Resistance mechanisms develop due to treatment selection pressure and the molecular heterogeneity seen in lung cancer.
Disadvantages for metastatic biopsy include the inability to safely access a metastatic site, the time considerations for preauthorization and arrangement of biopsy, and a lower-than-average likelihood of successful sequencing from sites such as bone.21,22 In addition, there is some concern that a single metastatic site may not capture all relevant alterations for multiple reasons, including tumor heterogeneity.
Significant advances in the past decade have dramatically improved the ability to use ctDNA to guide therapy. Advantages include ease of acquisition as acquiring a sample requires only a blood draw, and the potential that the pool of ctDNA is a better reflection of the relevant biology as it potentially reflects all metastatic tissues. Disadvantages are that sequencing attempts may not be productive if the sample is acquired at a time when the tumor is either quiescent or tumor burden is so low that only limited amounts of DNA are being shed. Performing ctDNA analysis when a tumor is not progressing is less likely to be productive for a number of tumor types.23,24 Sequencing ctDNA is also more susceptible than sequencing tumor biopsies to detection of alterations that are not from the tumor of interest but from clonal hematopoiesis of indeterminate potential (CHIP) or other clonal hematopoietic disorders (see Confounders section below).
Selecting the Tissue
Deciding on the tissue to analyze is a critical part of the decision process (Table 3). If the primary tumor tissue is old the likelihood of productive sequencing is lower, although age alone is not the only consideration and the methods of fixation may be just as relevant.
For prostate cancer in particular, the ability to successfully sequence primary tumor tissue decreases as the amount of tumor decreases in low-volume biopsies such as prostate needle biopsies. Generally, if tumor content is < 10% of the biopsy specimen, then sequencing is less likely to be productive.25 Also, if the alteration of interest is not known to be truncal, then a relevant target might be missed by sequencing tissue that does not reflect current biology. Metastasis biopsy may be the most appropriate tissue, particularly if this specimen has already been acquired. As above, a metastasis biopsy may have a higher tumor content, and it should reflect relevant biology if it is recent. However, bone biopsies have a relatively low yield for successful sequencing, so a soft tissue lesion (eg, liver or lymph node metastasis) is generally preferred.
The inability to safely access tissue is often a consideration. Proximity to vital structures such as large blood vessels or the potential for significant morbidity in the event of a complication (liver or lung biopsies, particularly in patients on anticoagulation medications) may make the risk/benefit ratio too high. The inability to conduct somatic testing has been reported to often be due to inadequate tissue sampling.26 ctDNA is an attractive alternative but should typically be drawn when a tumor is progressing with a reasonable tumor burden that is more likely to be shedding DNA. Performing ctDNA analysis in patients without obvious radiographic metastasis or in patients whose tumor is under good control is unlikely to produce interpretable results.
Interpreting the Results
The intent of sequencing tumor tissue is to identify alterations that are biologically important and may provide a point of therapeutic leverage. However, deciding which alterations are relevant is not always straightforward. For example, any normal individual genome contains around 10,000 missense variants, hundreds of insertion/deletion variants, and dozens of protein-truncating variants. Distinguishing these alterations, which are part of the individual, from those that are tumor-specific and have functional significance can be difficult in the absence of paired sequencing of both normal and tissue samples.
Specific Alterations
Although most commercial vendors provide important information in sequencing reports to assist oncology HCPs in deciding which alterations are relevant, the reports are not always clear. In many cases the report will specifically indicate whether the alteration has been reported previously as pathogenic or benign. However, some platforms will report alterations that are not known to be drivers of tumor biology. It is critical to be aware that if variants are not reported as pathogenic, they should not be assumed to be pathogenic simply because they are included in the report. Alterations more likely to be drivers of relevant biology are those that change gene and protein structure and include frameshift (fs*), nonsense (denoted by sequence ending in “X” or “*”), or specific fusions or insertions/deletions (indel) that occur in important domains of the gene.
For some genes, only specific alterations are targetable and not all alterations have the same effect on protein function. Although overexpression of certain genes and proteins are actionable (eg, HER2), amplification of a gene does not necessarily indicate that it is targetable. In NSCLC, specific alterations convey sensitivity to targeted therapies. For example, in EGFR-mutated NSCLC, the sensitizing mutations to EGFR TKIs are exon19 deletions and exon 21 L858R point mutations (the most common mutations), as well as less common mutations found in exon 18-21. Exon 20 mutations, however, are not responsive to EGFR TKIs with a few exceptions.27 Patients who have tumors that do not harbor a sensitizing EGFR mutation should not be treated with an EGFR TKI. In a variety of solid tumors, gene fusions of the NTRK 1/2/3, act as oncogenic drivers. The chromosomal fusion events involving the carboxy-terminal kinase domain of TRK and upstream amino-terminal partners lead to overexpression of the chimeric proteins tropomyosin receptor kinase (TRK) A/B/C, resulting in constitutively active, ligand-independent downstream signaling. In patients with NTRK 1/2/3 gene fusions, larotrectinib and entrectinib, small molecule inhibitors to TRK, have shown antitumor activity.28,29 No alterations beyond these fusions are known to be targetable.
Allele Fraction
Knowing the fraction (or proportion) of the alteration of interest in the sequenced tissue relative to the estimated tumor content can assist in decision making. Not all platforms will provide this information, which is referred to as mutation allele fraction (MAF) or variant allele fraction (VAF), but sometimes will provide it on request. Platforms will usually provide an estimate of the percent tumor in the tissue being sampled if it is from a biopsy. If the MAF is around 50% in the sequenced tissue (including ctDNA), then there is a reasonable chance that it is a germline variant. However, there are nuances as germline alterations in some genes, such as BRCA1/2, can be accompanied by loss of the other allele of the gene (LOH). In that case, if most of the circulating DNA is from tumor, then the MAF can be > 50%.
If there are 2 alterations of the same gene with MAF percentages that are each half of the total percent tumor, there is a high likelihood of biallelic alteration. These sorts of paired alterations or one mutation with apparent LOH or copy loss would again indicate a high likelihood that the alteration is in fact pathogenic and a relevant driver. Not all pathogenic alterations have to be biallelic to be driver mutations but in BRCA1/2, or mismatch repair deficiency genes, the presence of biallelic alterations increases the likelihood of their being pathogenic.
Tumors that are hypermutated—containing sometimes hundreds of mutations per megabyte of DNA—can be particularly complicated to interpret, because the likelihood increases that many of the alterations are a function of the hypermutation and not a driver mutation. This is particularly important when there are concurrent mutations in mismatch repair genes and genes, such as BRCA1/2. If the tumor is
Confounders
In some situations, interpretation can be particularly challenging. For example, several alterations for which there are FDA on-label indications (such as ATM or BRCA2) can be detected in ctDNA that may not be due to the tumor but to CHIP. CHIP represents hematopoietic clones that are dysplastic as a result of exposure to DNA-damaging agents (eg, platinum chemotherapy) or as a result of aging and arise when mutations in hematopoietic stem cells provide a competitive advantage.31 The most common CHIP clones that can be detected are DNMT3A, ASXL1, or TET2; because these alterations are not targetable, their importance lies primarily in whether patients have evidence of hematologic abnormality, which might represent an evolving hematopoietic disorder. Because CHIP alterations can overlap with somatic alterations for which FDA-approved drugs exist, such as ATM or CHEK2 (olaparib for prostate cancer) and BRCA2 (poly-ADP-ribose polymerase inhibitors in a range of indications) there is concern that CHIP might result in patient harm from inappropriate treatment of CHIP rather than the tumor, with no likelihood that the treatment would affect the tumor, causing treatment delays.32 General considerations for deciding whether an alteration represents CHIP include excluding alteration in which the VAF is < 1% and when the VAF in the alteration of interest is < 20% of the estimated tumor fraction in the sample. Exceptions to this are found in patients with true myelodysplasia or chronic lymphocytic leukemia, in whom the VAF can be well over 50% because of circulating tumor burden. The only way to be certain that an alteration detected on ctDNA reflects tumor rather than CHIP is to utilize an assay with matched tumor-normal sequencing.
Resources for Assistance
For oncology HCPs, perhaps the best resource to help in selecting and interpreting the appropriate testing is through a dedicated molecular oncology tumor board and subject matter experts who contribute to those tumor boards. In the US Department of Veterans Affairs, the national precision oncology program and its affiliated clinical services, such as the option to order a national consultation and molecular tumor board education, are easily accessible to all HCPs (www.cancer.va.gov). Many commercial vendors provide support to assist with questions of interpretation and to inform clinical decision-making. Other resources that can assist with deciding whether an alteration is pathogenic include extensive curated databases such as ClinVar (www.ncbi.nlm.nih.gov/clinvar) and the Human Genetic Mutation Database (www.hgmd.cf.ac.uk/ac/index.php) for germline alterations or COSMIC (cancer.sanger.ac.uk/cosmic) for somatic alterations. OncoKB (www.oncokb.org) is a resource for assistance in defining levels of evidence for the use of agents to target specific alterations and to assist in assigning pathogenicity to specific alterations. Additional educational resources for training in genomics and genetics are also included in the Appendix.
The rapid growth in technology and ability to enhance understanding of relevant tumor biology continues to improve the therapeutic landscape for men and women dealing with malignancy and our ability to find targetable genetic alterations with the potential for meaningful clinical benefit.
Acknowledgments
Dedicated to Neil Spector.
The ability to find and target specific biomarkers in the DNA of advanced cancers is rapidly changing options and outcomes for patients with locally advanced and metastatic solid tumors. This strategy is the basis for precision oncology, defined here as using predictive biomarkers from tumor and/or germline sequencing to guide therapies. This article focuses specifically on the use of DNA sequencing to find those biomarkers and provides guidance about which test is optimal in a specific situation, as well as interpretation of the results. We emphasize the identification of biomarkers that provide adult patients with advanced solid tumors access to therapies that would not be an option had sequencing not been performed and that have the potential for significant clinical benefit. The best approach is to have an expert team with experience in precision oncology to assist in the interpretation of results.
Which test?
Deciding what test of the array of assays available to use and which tissue to test can be overwhelming, and uncertainty may prevent oncology practitioners from ordering germline or somatic sequencing. For the purposes of this article, we will focus on DNA sequencing for inherited/germline alterations (including mutations, copy number changes, or fusions), which may inform treatment, or alterations that arise in the process of carcinogenesis and tumor evolution (somatic alterations in tumor DNA). This focus is not meant to exclude any specific test but to focus on DNA-based tests in patients with locally advanced or metastatic malignancy.
Germline Testing
Germline testing is the sequencing of inherited DNA in noncancerous cells to find alterations that may play a role in the development of cancers and are actionable in some cases. Germline alterations can inform therapeutic decisions, predict future cancer risk, and provide information that can help family members to better manage their risks of malignancy. Detailed discussions of the importance of germline testing to inform cancer surveillance, risk-reducing interventions, and the testing of relatives to determine who carries inherited alterations (cascade testing) is extremely important with several advantages and is covered in a number of excellent reviews elsewhere.1-3 Testing of germline DNA in patients with a metastatic malignancy can provide treatment options otherwise not available for patients, particularly for BRCA1/2 and Lynch syndrome–related cancers. Recent studies have shown that 10 to 15% of patients with advanced malignancies of many types have a pathogenic germline alteration.4,5
Germline DNA is usually acquired from peripheral blood, a buccal swab, or saliva collection and is therefore readily available. This is advantageous because it does not require a biopsy to identify relevant alterations. Germline testing is also less susceptible to the rare situations in which artifacts occur in formalin-fixed tissues and obscure relevant alterations.
The cost of germline testing varies, but most commercial vendor assays for germline testing are significantly less expensive than the cost of somatic testing. The disadvantages include the inability of germline testing to find any alterations that arise solely in tumor tissue and the smaller gene panels included in germline testing as compared to somatic testing panels. Other considerations relate to the inherited nature of pathogenic germline variants and its implications for family members that may affect the patient’s psychosocial health and potentially change the family dynamics.
Deciding who is appropriate for germline testing and when to perform the testing should be individualized to the patient’s wishes and disease status. Treatment planning may be less complicated if testing has been performed and germline status is known. In some cases urgent germline testing is indicated to inform pending procedures and/or surgical decisions for risk reduction, including more extensive tissue resection, such as the removal of additional organs or contralateral tissue. A minor point regarding germline testing is that the DNA of patients with hematologic malignancies may be difficult to sequence because of sample contamination by the circulating malignancy. For this reason, most laboratories will not accept peripheral blood or saliva samples for germline testing in patients with active hematologic malignancies; they often require DNA from another source such as fibroblasts from a skin biopsy or cells from a muscle biopsy. Germline testing is recommended for all patients with metastatic prostate cancer, as well as any patient with any stage of pancreatic cancer or ovarian cancer and patients with breast cancer diagnosed at age ≤ 45 years. More detailed criteria for who is appropriate for germline testing outside of these groups can be found in the appropriate National Comprehensive Cancer Network (NCCN) guidelines.6-8 In patients with some malignancies such as prostate and pancreatic cancer, approximately half of patients who have a BRCA-related cancer developed that malignancy because of a germline BRCA alteration.9-11 Testing germline DNA is therefore an easy way to quickly find almost half of all targetable alterations with a treatment approved by the US Food and Drug Administration (FDA) and at low cost, with the added benefit of providing critical information for families who may be unaware that members carry a relevant pathogenic germline alteration. In those families, cascade testing can provide surveillance and intervention strategies that can be lifesaving.
A related and particularly relevant question is when should a result found on a somatic testing panel prompt follow-up germline testing? Some institutions have algorithms in place to automate referral for germline testing based on specific genetic criteria.12 Excellent reviews are available that outline the following considerations in more detail.13 Typically, somatic testing results that would trigger follow-up germline testing would be truncating or deleterious or likely deleterious mutations per germline datasets in high-risk genes associated with highly penetrant autosomal dominant conditions (BRCA1, BRCA2, PALB2, MLH1, MSH2, and MSH6), selected moderate-risk genes (BRIP1, RAD51C, and RAD51D), and specific variants with a high probability of being germline because they are common germline founder mutations. Although the actionability and significance of specific genes remains a matter of some discussion, generally finding a somatic pathogenic sequencing result included in the 59-gene list of the American College of Medical Genetics and Genomics (ACMG) guidelines would be an indication for germline testing. Another indication for germline testing would be finding genes with germline mutations for which the NCCN has specific management guidelines, or the presence of alterations consistent with known founder mutations.14 When a patient’s tumor has microsatellite instability or is hypermutated (defined as > 10 mutations per megabase), a search for germline alterations is warranted given that about 15% of these patients with these tumors carry a Lynch syndrome gene.15 Genes that are commonly found as somatic alterations alone (eg, TP53 or APC) are generally not an indication for germline testing unless family history is compelling.
Although some clinicians use the variant allele fraction in the somatic sequencing report to decide whether to conduct germline testing, this approach is suboptimal, as allele fraction may be confounded by assay conditions and a high allele fraction may be found in pure tumors with loss of heterozygosity (LOH) of the other allele. There is also evidence that for a variety of reasons, somatic sequencing panels do not always detect germline alterations in somatic tissues.16 Reasons for this may include discordance between the genes being tested in the germline vs the somatic panel, technical differences such as interference of formalin-fixed paraffin-embedded (FFPE) artifact with detecting the germline variant, lack of expertise in germline variant interpretation among laboratories doing tumor-only sequencing, and, in rare cases, large deletions in tumor tissue masking a germline point mutation.
Variant Interpretation of Germline Testing
A general understanding of the terminology used for germline variant interpretation allows for the ordering health care practitioner (HCP) to provide the best quality care and an appreciation for the limitations of current molecular testing. Not all variants are associated with disease; the clinical significance of a genetic variant falls on a spectrum. The criteria for determining pathogenicity differ between molecular laboratories, but most are influenced by the standards and guidelines set forth by the ACMG.14 The clinical molecular laboratory determines variant classification, and a detailed discussion is beyond the scope of this primer. In brief, variant classification is based on evidence of varying strength in different categories including population data, computational and predictive data, functional data, segregation data, de novo data, allelic data, and information from various databases. The ACMG has proposed a 5-tiered classification system, by which most molecular laboratories adhere to in their genetic test reports (Table 1).14
Pathogenic and likely pathogenic variants are clinically actionable, whereas variants of uncertain significance (VUS) require additional data and/or functional studies before making clinical decisions. Depending on the clinical context and existing supporting evidence, it may be prudent to continue monitoring for worsening or new signs of disease in patients with one or more VUS while additional efforts are underway to understand the variant’s significance.
In some cases, variants are reclassified, which may alter the management and treatment of patients. Reclassification can occur with VUS, and in rare instances, can also occur with variants previously classified as pathogenic/likely pathogenic or benign/likely benign. In such a case, the reporting laboratory will typically make concerted efforts to alert the ordering HCP. However, variant reclassifications are not always communicated to the care team. Thus, it is important to periodically contact the molecular laboratory of interest to obtain updated test interpretations.
Somatic Testing
Testing of somatic (tumor) tissue is critical and is the approach most commonly taken in medical oncology (Table 2).
The advantages of primary tumor are that it is usually in hand as a diagnostic biopsy, acquisition is standard of care, and several targetable alterations are truncal, defined as driver mutations present at the time of tumor development. Also, the potential that the tumor arose in the background of a predisposing germline alteration can be suggested by sequencing primary tumor as discussed above. Moreover, sequencing the primary tumor can be done at any time unless the biopsy sample is considered too old or degraded (per specific platform requirements). The information gained can be used to anticipate additional treatment options that are relevant when patients experience disease progression. Disadvantages include the problem that primary specimens may be old or have limited tumor content, both of which increase the likelihood that sequencing will not be technically successful.
Alterations that are targetable and arise as a result of either treatment pressure or clonal evolution are considered evolutionary. If evolutionary alterations are the main focus for sequencing, then metastasis biopsy or ctDNA are better choices. The advantages of a metastasis biopsy are that tissue is contemporary, tumor content may be higher than in primary tumor, and both truncal and evolutionary alterations can be detected.
For specific tumors, continued analysis of evolving genomic alterations can play a critical role in management. In non–small cell lung cancer (NSCLC), somatic testing is conducted again at progression on repeat biopsies to evaluate for emerging resistance mutations. In epidermal growth factor receptor (EGFR)–mutated lung cancer, the resistance mutation, exon 20 p.T790M (point mutation), can present in patients after treatment with first- or second-generation EGFR tyrosine kinase inhibitors (TKI). Even in patients who are treated with the third-generation EGFR TKI osimertinib that can treat T790M-mutated lung cancer, multiple possible evolutionary mutations can occur at progression, including other EGFR mutations, MET/HER2 amplification, and BRAF V600E, to name a few.20 Resistance mechanisms develop due to treatment selection pressure and the molecular heterogeneity seen in lung cancer.
Disadvantages for metastatic biopsy include the inability to safely access a metastatic site, the time considerations for preauthorization and arrangement of biopsy, and a lower-than-average likelihood of successful sequencing from sites such as bone.21,22 In addition, there is some concern that a single metastatic site may not capture all relevant alterations for multiple reasons, including tumor heterogeneity.
Significant advances in the past decade have dramatically improved the ability to use ctDNA to guide therapy. Advantages include ease of acquisition as acquiring a sample requires only a blood draw, and the potential that the pool of ctDNA is a better reflection of the relevant biology as it potentially reflects all metastatic tissues. Disadvantages are that sequencing attempts may not be productive if the sample is acquired at a time when the tumor is either quiescent or tumor burden is so low that only limited amounts of DNA are being shed. Performing ctDNA analysis when a tumor is not progressing is less likely to be productive for a number of tumor types.23,24 Sequencing ctDNA is also more susceptible than sequencing tumor biopsies to detection of alterations that are not from the tumor of interest but from clonal hematopoiesis of indeterminate potential (CHIP) or other clonal hematopoietic disorders (see Confounders section below).
Selecting the Tissue
Deciding on the tissue to analyze is a critical part of the decision process (Table 3). If the primary tumor tissue is old the likelihood of productive sequencing is lower, although age alone is not the only consideration and the methods of fixation may be just as relevant.
For prostate cancer in particular, the ability to successfully sequence primary tumor tissue decreases as the amount of tumor decreases in low-volume biopsies such as prostate needle biopsies. Generally, if tumor content is < 10% of the biopsy specimen, then sequencing is less likely to be productive.25 Also, if the alteration of interest is not known to be truncal, then a relevant target might be missed by sequencing tissue that does not reflect current biology. Metastasis biopsy may be the most appropriate tissue, particularly if this specimen has already been acquired. As above, a metastasis biopsy may have a higher tumor content, and it should reflect relevant biology if it is recent. However, bone biopsies have a relatively low yield for successful sequencing, so a soft tissue lesion (eg, liver or lymph node metastasis) is generally preferred.
The inability to safely access tissue is often a consideration. Proximity to vital structures such as large blood vessels or the potential for significant morbidity in the event of a complication (liver or lung biopsies, particularly in patients on anticoagulation medications) may make the risk/benefit ratio too high. The inability to conduct somatic testing has been reported to often be due to inadequate tissue sampling.26 ctDNA is an attractive alternative but should typically be drawn when a tumor is progressing with a reasonable tumor burden that is more likely to be shedding DNA. Performing ctDNA analysis in patients without obvious radiographic metastasis or in patients whose tumor is under good control is unlikely to produce interpretable results.
Interpreting the Results
The intent of sequencing tumor tissue is to identify alterations that are biologically important and may provide a point of therapeutic leverage. However, deciding which alterations are relevant is not always straightforward. For example, any normal individual genome contains around 10,000 missense variants, hundreds of insertion/deletion variants, and dozens of protein-truncating variants. Distinguishing these alterations, which are part of the individual, from those that are tumor-specific and have functional significance can be difficult in the absence of paired sequencing of both normal and tissue samples.
Specific Alterations
Although most commercial vendors provide important information in sequencing reports to assist oncology HCPs in deciding which alterations are relevant, the reports are not always clear. In many cases the report will specifically indicate whether the alteration has been reported previously as pathogenic or benign. However, some platforms will report alterations that are not known to be drivers of tumor biology. It is critical to be aware that if variants are not reported as pathogenic, they should not be assumed to be pathogenic simply because they are included in the report. Alterations more likely to be drivers of relevant biology are those that change gene and protein structure and include frameshift (fs*), nonsense (denoted by sequence ending in “X” or “*”), or specific fusions or insertions/deletions (indel) that occur in important domains of the gene.
For some genes, only specific alterations are targetable and not all alterations have the same effect on protein function. Although overexpression of certain genes and proteins are actionable (eg, HER2), amplification of a gene does not necessarily indicate that it is targetable. In NSCLC, specific alterations convey sensitivity to targeted therapies. For example, in EGFR-mutated NSCLC, the sensitizing mutations to EGFR TKIs are exon19 deletions and exon 21 L858R point mutations (the most common mutations), as well as less common mutations found in exon 18-21. Exon 20 mutations, however, are not responsive to EGFR TKIs with a few exceptions.27 Patients who have tumors that do not harbor a sensitizing EGFR mutation should not be treated with an EGFR TKI. In a variety of solid tumors, gene fusions of the NTRK 1/2/3, act as oncogenic drivers. The chromosomal fusion events involving the carboxy-terminal kinase domain of TRK and upstream amino-terminal partners lead to overexpression of the chimeric proteins tropomyosin receptor kinase (TRK) A/B/C, resulting in constitutively active, ligand-independent downstream signaling. In patients with NTRK 1/2/3 gene fusions, larotrectinib and entrectinib, small molecule inhibitors to TRK, have shown antitumor activity.28,29 No alterations beyond these fusions are known to be targetable.
Allele Fraction
Knowing the fraction (or proportion) of the alteration of interest in the sequenced tissue relative to the estimated tumor content can assist in decision making. Not all platforms will provide this information, which is referred to as mutation allele fraction (MAF) or variant allele fraction (VAF), but sometimes will provide it on request. Platforms will usually provide an estimate of the percent tumor in the tissue being sampled if it is from a biopsy. If the MAF is around 50% in the sequenced tissue (including ctDNA), then there is a reasonable chance that it is a germline variant. However, there are nuances as germline alterations in some genes, such as BRCA1/2, can be accompanied by loss of the other allele of the gene (LOH). In that case, if most of the circulating DNA is from tumor, then the MAF can be > 50%.
If there are 2 alterations of the same gene with MAF percentages that are each half of the total percent tumor, there is a high likelihood of biallelic alteration. These sorts of paired alterations or one mutation with apparent LOH or copy loss would again indicate a high likelihood that the alteration is in fact pathogenic and a relevant driver. Not all pathogenic alterations have to be biallelic to be driver mutations but in BRCA1/2, or mismatch repair deficiency genes, the presence of biallelic alterations increases the likelihood of their being pathogenic.
Tumors that are hypermutated—containing sometimes hundreds of mutations per megabyte of DNA—can be particularly complicated to interpret, because the likelihood increases that many of the alterations are a function of the hypermutation and not a driver mutation. This is particularly important when there are concurrent mutations in mismatch repair genes and genes, such as BRCA1/2. If the tumor is
Confounders
In some situations, interpretation can be particularly challenging. For example, several alterations for which there are FDA on-label indications (such as ATM or BRCA2) can be detected in ctDNA that may not be due to the tumor but to CHIP. CHIP represents hematopoietic clones that are dysplastic as a result of exposure to DNA-damaging agents (eg, platinum chemotherapy) or as a result of aging and arise when mutations in hematopoietic stem cells provide a competitive advantage.31 The most common CHIP clones that can be detected are DNMT3A, ASXL1, or TET2; because these alterations are not targetable, their importance lies primarily in whether patients have evidence of hematologic abnormality, which might represent an evolving hematopoietic disorder. Because CHIP alterations can overlap with somatic alterations for which FDA-approved drugs exist, such as ATM or CHEK2 (olaparib for prostate cancer) and BRCA2 (poly-ADP-ribose polymerase inhibitors in a range of indications) there is concern that CHIP might result in patient harm from inappropriate treatment of CHIP rather than the tumor, with no likelihood that the treatment would affect the tumor, causing treatment delays.32 General considerations for deciding whether an alteration represents CHIP include excluding alteration in which the VAF is < 1% and when the VAF in the alteration of interest is < 20% of the estimated tumor fraction in the sample. Exceptions to this are found in patients with true myelodysplasia or chronic lymphocytic leukemia, in whom the VAF can be well over 50% because of circulating tumor burden. The only way to be certain that an alteration detected on ctDNA reflects tumor rather than CHIP is to utilize an assay with matched tumor-normal sequencing.
Resources for Assistance
For oncology HCPs, perhaps the best resource to help in selecting and interpreting the appropriate testing is through a dedicated molecular oncology tumor board and subject matter experts who contribute to those tumor boards. In the US Department of Veterans Affairs, the national precision oncology program and its affiliated clinical services, such as the option to order a national consultation and molecular tumor board education, are easily accessible to all HCPs (www.cancer.va.gov). Many commercial vendors provide support to assist with questions of interpretation and to inform clinical decision-making. Other resources that can assist with deciding whether an alteration is pathogenic include extensive curated databases such as ClinVar (www.ncbi.nlm.nih.gov/clinvar) and the Human Genetic Mutation Database (www.hgmd.cf.ac.uk/ac/index.php) for germline alterations or COSMIC (cancer.sanger.ac.uk/cosmic) for somatic alterations. OncoKB (www.oncokb.org) is a resource for assistance in defining levels of evidence for the use of agents to target specific alterations and to assist in assigning pathogenicity to specific alterations. Additional educational resources for training in genomics and genetics are also included in the Appendix.
The rapid growth in technology and ability to enhance understanding of relevant tumor biology continues to improve the therapeutic landscape for men and women dealing with malignancy and our ability to find targetable genetic alterations with the potential for meaningful clinical benefit.
Acknowledgments
Dedicated to Neil Spector.
1. Domchek SM, Mardis E, Carlisle JW, Owonikoko TK. Integrating genetic and genomic testing into oncology practice. Am Soc Clin Oncol Educ Book. 2020;40:e259-e263. doi:10.1200/EDBK_280607
2. Stoffel EM, Carethers JM. Current approaches to germline cancer genetic testing. Annu Rev Med. 2020;71:85-102. doi:10.1146/annurev-med-052318-101009
3. Lappalainen T, Scott AJ, Brandt M, Hall IM. Genomic analysis in the age of human genome sequencing. Cell. 2019;177(1):70-84. doi:10.1016/j.cell.2019.02.032
4. Samadder NJ, Riegert-Johnson D, Boardman L, et al. Comparison of universal genetic testing vs guideline-directed targeted testing for patients with hereditary cancer syndrome. JAMA Oncol. 2021;7(2):230-237. doi:10.1001/jamaoncol.2020.6252
5. Schneider BP, Stout L, Philips S, et al. Implications of incidental germline findings identified in the context of clinical whole exome sequencing for guiding cancer therapy. JCO Precis Oncol. 2020;4:1109-1121. doi:10.1200/PO.19.00354
6. National Comprehensive Cancer Network. Pancreatic cancer (Version 1.2022). Updated February 24, 2022. Accessed April 13, 2022. https://www.nccn.org/professionals/physician_gls/pdf/pancreatic.pdf
7. National Comprehensive Cancer Network. Prostate cancer (Version 3.2022). Updated January 10, 2022. Accessed April 13, 2022. https://www.nccn.org/professionals/physician_gls/pdf/prostate.pdf
8. National Comprehensive Cancer Network. Genetic/familial high-risk assessment: breast, ovarian, and pancreatic (Version 2.2022). Updated March 9, 2022. Accessed April 13, 2022. https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf
9. Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215-1228. doi:10.1016/j.cell.2015.05.001
10. Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375(5):443-453. doi:10.1056/NEJMoa1603144
11. Cancer Genome Atlas Research Network. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017;32(2):185-203.e13. doi:10.1016/j.ccell.2017.07.007
12. Clark DF, Maxwell KN, Powers J, et al. Identification and confirmation of potentially actionable germline mutations in tumor-only genomic sequencing. JCO Precis Oncol. 2019;3:PO.19.00076. doi:10.1200/PO.19.00076
13. DeLeonardis K, Hogan L, Cannistra SA, Rangachari D, Tung N. When should tumor genomic profiling prompt consideration of germline testing? J Oncol Pract. 2019;15(9):465-473. doi:10.1200/JOP.19.00201
14. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi:10.1038/gim.2015.30
15. Latham A, Srinivasan P, Kemel Y, et al. Microsatellite instability is associated with the presence of Lynch syndrome pan-cancer. J Clin Oncol. 2019;37(4):286-295. doi:10.1200/JCO.18.00283
16. Lincoln SE, Nussbaum RL, Kurian AW, et al. Yield and utility of germline testing following tumor sequencing in patients with cancer. JAMA Netw Open. 2020;3(10):e2019452. doi:10.1001/jamanetworkopen.2020.19452
17. National Comprehensive Cancer Network. Non-small cell lung cancer (Version: 3.2022). Updated March 16, 2022. Accessed April 13, 2022. https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf
18. National Comprehensive Cancer Network. Colon cancer (Version 1.2022). February 25, 2022. Accessed April 13, 2022. https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf
19. National Comprehensive Cancer Network. Melanoma: cutaneous (Version 3.2022). April 11, 2022. Accessed April 13, 2022. https://www.nccn.org/professionals/physician_gls/pdf/cutaneous_melanoma.pdf
20. Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer. 2019;121(9):725-737. doi:10.1038/s41416-019-0573-8
21. Zheng G, Lin MT, Lokhandwala PM, et al. Clinical mutational profiling of bone metastases of lung and colon carcinoma and malignant melanoma using next-generation sequencing. Cancer Cytopathol. 2016;124(10):744-753. doi:10.1002/cncy.21743
22. Spritzer CE, Afonso PD, Vinson EN, et al. Bone marrow biopsy: RNA isolation with expression profiling in men with metastatic castration-resistant prostate cancer—factors affecting diagnostic success. Radiology. 2013;269(3):816-823. doi:10.1148/radiol.13121782
23. Schweizer MT, Gulati R, Beightol M, et al. Clinical determinants for successful circulating tumor DNA analysis in prostate cancer. Prostate. 2019;79(7):701-708. doi:10.1002/pros.23778
24. Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6(224):224ra224. doi:10.1126/scitranslmed.3007094
25. Pritchard CC, Salipante SJ, Koehler K, et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn. 2014;16(1):56-67. doi:10.1016/j.jmoldx.2013.08.004
26. Gutierrez ME, Choi K, Lanman RB, et al. Genomic profiling of advanced non-small cell lung cancer in community settings: gaps and opportunities. Clin Lung Cancer. 2017;18(6):651-659. doi:10.1016/j.cllc.2017.04.004
27. Malapelle U, Pilotto S, Passiglia F, et al. Dealing with NSCLC EGFR mutation testing and treatment: a comprehensive review with an Italian real-world perspective. Crit Rev Oncol Hematol. 2021;160:103300. doi:10.1016/j.critrevonc.2021.103300
28. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. 2018;378(8):731-739. doi:10.1056/NEJMoa1714448
29. Doebele RC, Drilon A, Paz-Ares L, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020;21(2):271-282. doi:10.1016/S1470-2045(19)30691-6
30. Jonsson P, Bandlamudi C, Cheng ML, et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature. 2019;571(7766):576-579. doi:10.1038/s41586-019-1382-1
31. Steensma DP. Clinical consequences of clonal hematopoiesis of indeterminate potential. Hematology Am Soc Hematol Educ Program. 2018;2018(1):264-269. doi:10.1182/asheducation-2018.1.264
32. Jensen K, Konnick EQ, Schweizer MT, et al. Association of clonal hematopoiesis in DNA repair genes with prostate cancer plasma cell-free DNA testing interference. JAMA Oncol. 2021;7(1):107-110. doi:10.1001/jamaoncol.2020.5161
1. Domchek SM, Mardis E, Carlisle JW, Owonikoko TK. Integrating genetic and genomic testing into oncology practice. Am Soc Clin Oncol Educ Book. 2020;40:e259-e263. doi:10.1200/EDBK_280607
2. Stoffel EM, Carethers JM. Current approaches to germline cancer genetic testing. Annu Rev Med. 2020;71:85-102. doi:10.1146/annurev-med-052318-101009
3. Lappalainen T, Scott AJ, Brandt M, Hall IM. Genomic analysis in the age of human genome sequencing. Cell. 2019;177(1):70-84. doi:10.1016/j.cell.2019.02.032
4. Samadder NJ, Riegert-Johnson D, Boardman L, et al. Comparison of universal genetic testing vs guideline-directed targeted testing for patients with hereditary cancer syndrome. JAMA Oncol. 2021;7(2):230-237. doi:10.1001/jamaoncol.2020.6252
5. Schneider BP, Stout L, Philips S, et al. Implications of incidental germline findings identified in the context of clinical whole exome sequencing for guiding cancer therapy. JCO Precis Oncol. 2020;4:1109-1121. doi:10.1200/PO.19.00354
6. National Comprehensive Cancer Network. Pancreatic cancer (Version 1.2022). Updated February 24, 2022. Accessed April 13, 2022. https://www.nccn.org/professionals/physician_gls/pdf/pancreatic.pdf
7. National Comprehensive Cancer Network. Prostate cancer (Version 3.2022). Updated January 10, 2022. Accessed April 13, 2022. https://www.nccn.org/professionals/physician_gls/pdf/prostate.pdf
8. National Comprehensive Cancer Network. Genetic/familial high-risk assessment: breast, ovarian, and pancreatic (Version 2.2022). Updated March 9, 2022. Accessed April 13, 2022. https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf
9. Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215-1228. doi:10.1016/j.cell.2015.05.001
10. Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375(5):443-453. doi:10.1056/NEJMoa1603144
11. Cancer Genome Atlas Research Network. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017;32(2):185-203.e13. doi:10.1016/j.ccell.2017.07.007
12. Clark DF, Maxwell KN, Powers J, et al. Identification and confirmation of potentially actionable germline mutations in tumor-only genomic sequencing. JCO Precis Oncol. 2019;3:PO.19.00076. doi:10.1200/PO.19.00076
13. DeLeonardis K, Hogan L, Cannistra SA, Rangachari D, Tung N. When should tumor genomic profiling prompt consideration of germline testing? J Oncol Pract. 2019;15(9):465-473. doi:10.1200/JOP.19.00201
14. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi:10.1038/gim.2015.30
15. Latham A, Srinivasan P, Kemel Y, et al. Microsatellite instability is associated with the presence of Lynch syndrome pan-cancer. J Clin Oncol. 2019;37(4):286-295. doi:10.1200/JCO.18.00283
16. Lincoln SE, Nussbaum RL, Kurian AW, et al. Yield and utility of germline testing following tumor sequencing in patients with cancer. JAMA Netw Open. 2020;3(10):e2019452. doi:10.1001/jamanetworkopen.2020.19452
17. National Comprehensive Cancer Network. Non-small cell lung cancer (Version: 3.2022). Updated March 16, 2022. Accessed April 13, 2022. https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf
18. National Comprehensive Cancer Network. Colon cancer (Version 1.2022). February 25, 2022. Accessed April 13, 2022. https://www.nccn.org/professionals/physician_gls/pdf/colon.pdf
19. National Comprehensive Cancer Network. Melanoma: cutaneous (Version 3.2022). April 11, 2022. Accessed April 13, 2022. https://www.nccn.org/professionals/physician_gls/pdf/cutaneous_melanoma.pdf
20. Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer. 2019;121(9):725-737. doi:10.1038/s41416-019-0573-8
21. Zheng G, Lin MT, Lokhandwala PM, et al. Clinical mutational profiling of bone metastases of lung and colon carcinoma and malignant melanoma using next-generation sequencing. Cancer Cytopathol. 2016;124(10):744-753. doi:10.1002/cncy.21743
22. Spritzer CE, Afonso PD, Vinson EN, et al. Bone marrow biopsy: RNA isolation with expression profiling in men with metastatic castration-resistant prostate cancer—factors affecting diagnostic success. Radiology. 2013;269(3):816-823. doi:10.1148/radiol.13121782
23. Schweizer MT, Gulati R, Beightol M, et al. Clinical determinants for successful circulating tumor DNA analysis in prostate cancer. Prostate. 2019;79(7):701-708. doi:10.1002/pros.23778
24. Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6(224):224ra224. doi:10.1126/scitranslmed.3007094
25. Pritchard CC, Salipante SJ, Koehler K, et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn. 2014;16(1):56-67. doi:10.1016/j.jmoldx.2013.08.004
26. Gutierrez ME, Choi K, Lanman RB, et al. Genomic profiling of advanced non-small cell lung cancer in community settings: gaps and opportunities. Clin Lung Cancer. 2017;18(6):651-659. doi:10.1016/j.cllc.2017.04.004
27. Malapelle U, Pilotto S, Passiglia F, et al. Dealing with NSCLC EGFR mutation testing and treatment: a comprehensive review with an Italian real-world perspective. Crit Rev Oncol Hematol. 2021;160:103300. doi:10.1016/j.critrevonc.2021.103300
28. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. 2018;378(8):731-739. doi:10.1056/NEJMoa1714448
29. Doebele RC, Drilon A, Paz-Ares L, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020;21(2):271-282. doi:10.1016/S1470-2045(19)30691-6
30. Jonsson P, Bandlamudi C, Cheng ML, et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature. 2019;571(7766):576-579. doi:10.1038/s41586-019-1382-1
31. Steensma DP. Clinical consequences of clonal hematopoiesis of indeterminate potential. Hematology Am Soc Hematol Educ Program. 2018;2018(1):264-269. doi:10.1182/asheducation-2018.1.264
32. Jensen K, Konnick EQ, Schweizer MT, et al. Association of clonal hematopoiesis in DNA repair genes with prostate cancer plasma cell-free DNA testing interference. JAMA Oncol. 2021;7(1):107-110. doi:10.1001/jamaoncol.2020.5161
The Precision Oncology Program for Cancer of the Prostate (POPCaP) Network: A Veterans Affairs/Prostate Cancer Foundation Collaboration(FULL)
The US Department of Veterans Affairs (VA) is home to the Veterans Health Administration (VHA), which delivers care at 1,255 health care facilities, including 170 medical centers. The VA serves 6 million veterans each year and is the largest integrated provider of cancer care in the US. The system uses a single, enterprise-wide electronic health record. The detailed curation of clinical outcomes, laboratory results, and radiology is used in VA efforts to improve oncology outcomes for veterans. The VA also has a National Precision Oncology Program (NPOP), which offers system-wide DNA sequencing for veterans with cancer. Given its size, integration, and capabilities, the VA is an ideal setting for rapid learning cycles of testing and implementing best practices at scale.
Prostate cancer is the most common malignancy affecting men in the US. It is the most commonly-diagnosed solid tumor in the VA, and in 2014, there were 11,376 prostate cancer diagnoses in the VA.1 The clinical characteristics and treatment of veterans with prostate cancer largely parallel the broader population of men in the US.1 Although the majority of men diagnosed with prostate cancer have disease localized to the prostate, an important minority develop metastatic disease, which represents a risk for substantial morbidity and is the lethal form of the disease. Research has yielded transformative advances in the care of men with metastatic prostate cancer, including drugs targeting the testosterone/androgen signaling axis, taxane chemotherapy, the radionuclide radium-223, and a dendritic cell vaccine. Unfortunately, the magnitude and duration of response to these therapies varies widely, and determining the biology relevant to an individual patient that would better inform their treatment decisions is a critical next step. As the ability to interrogate the cancer genome has improved, relevant drivers of tumorigenesis and predictive biomarkers are being identified rapidly, and oncology care has evolved from a one-size-fits-all approach to a precision approach, which uses these biomarkers to assist in therapeutic decision making.
Precision Oncology for Prostate Cancer
A series of studies interrogating the genomics of metastatic prostate cancer have been critical to defining the relevance of precision oncology for prostate cancer. Most of what is known about the genomics of prostate cancer has been derived from analysis of samples from the prostate itself. These samples may not reflect the biology of metastasis and genetic evolution in response to treatment pressure, so the genomic alterations in metastatic disease remained incompletely characterized. Two large research teams supported by grants from the American Association for Cancer Research, Stand Up 2 Cancer, and Prostate Cancer Foundation (PCF) focused their efforts on sampling and analyzing metastatic tissue to define the most relevant genomic alterations in advanced prostate cancer.
These efforts defined a broad range of relatively common alterations in the androgen receptor, as well as the tumor suppressors TP53 and PTEN.2,3 Important subsets of less common alterations in pathways that were potentially targetable were also found, including new alterations in PIK3CA/B, BRAF/RAF1, and β-catenin. Most surprisingly, alterations of DNA repair pathways, including mismatch repair and homologous recombination were found in 20% of tumors, and half of these tumors contained germline alterations. The same groups performed a follow up analysis of germline DNA from men with metastatic prostate cancer, which confirmed that 12% of these patients carry a pathogenic germline alteration in a DNA repair pathway gene.4 These efforts immediately invigorated precision oncology clinical trials for prostate cancer and spurred an effort to find the molecular alterations that could be leveraged to improve care for men with advanced prostate cancer.
Targetable Alterations
Currently a number of genomic alterations are immediately actionable. There are several agents approved by the US Food and Drug Administration (FDA) that exploit these Achilles heels of prostate cancer. Mismatch repair deficiency occurs when any of a group of genes responsible for proofreading the fidelity of DNA replication is compromised by mutation or deletion. Imperfect reading and correction subsequently lead to many DNA mutations in a tissue (hypermutation), which then increases the risk of developing malignancy. If a defective gene in the mismatch repair pathway is inherited, a patient has a genetic predisposition to specific malignancies that are part of the Lynch syndrome.5 Prostate cancer is a relatively rare manifestation of Lynch syndrome, although it is considered one of the malignancies in the Lynch syndrome spectrum.6
Alteration of one of the mismatch repair genes also can occur spontaneously in a tumor, resulting in the same high frequency of spontaneous DNA mutations. Overall, between 3% and 5% of metastatic prostate cancers contain mismatch repair deficiency. The majority of these cases are a result of spontaneous loss or mutation of the relevant gene, but 1 in 5 of these tumors occurs as a component of Lynch syndrome.7 Identification of mismatch repair deficiency is critical because the resulting hypermutation makes these tumors particularly susceptible to intervention with immunotherapy. Up to half of patients with metastatic prostate cancer can have durable responses. This finding is consistent with the experience treating other malignancies with mismatch repair deficiency.8 Although screening for mismatch repair deficiency is standard of care for patients with malignancies such as colorectal cancer, few patients with prostate cancer may receive the mismatch repair deficiency screening (based on unpublished data). In contrast, screening is routine for patients with adenocarcinoma of the lung because their proportion of ROS1 and ALK alterations is similar to the frequency of mismatch repair deficiency when compared with patients with prostate cancer.9
Homologous recombination is another mechanism by which cells repair DNA damage and is responsible for repairing double strand breaks, the type of DNA damage most likely to lead to carcinogenesis. In advanced prostate cancer, BRCA2, ATM, BRCA1 and other members of the Fanconi Anemia/BRCA gene family are altered 20% of the time. These genes also are the most common germline alterations implicated in the development of prostate cancer.2,10 Prostate cancer is considered a BRCA-related cancer much like breast, ovarian, and pancreatic cancers. Defects in homologous recombination repair make BRCA-altered prostate cancers susceptible to DNA damaging chemotherapy, such as platinum and to the use of poly–(adenosine diphosphate–ribose) polymerase (PARP) inhibitors because cancer cells then accumulate cytotoxic and apoptotic levels of DNA.11
In May 2020, the FDA approved the use of PARP inhibitors for the treatment of prostate cancers that contain BRCA and other DNA repair alterations. Rucaparib received accelerated approval for the treatment of prostate cancers containing BRCA alterations and olaparib received full approval for treatment of prostate cancers containing an array of alterations in DNA repair genes.12,13 Both approvals were the direct result of the cited landmark studies that demonstrated the frequency of these alterations in advanced prostate cancer.2,3
Beyond mismatch and homologous recombination repair, there are a large number of potentially targetable alterations found in advanced prostate cancer. It is thus critical that we put systems into place both to find germline and somatic alterations that will inform a veteran’s clinical care and to provide veterans access to precision oncology clinical trials.
The POPCaP Network
Because prostate cancer is such a significant issue in the VA and best practices for precision oncology can be implemented broadly once defined as successful, the PCF and the VA formed a collaboration to support a network of centers that would focus on implementing a comprehensive strategy for precision oncology in prostate cancer. There are currently 11 centers in the Precision Oncology Program for Cancer of the Prostate (POPCaP) network (Figure). These centers are tasked with comprehensively sequencing germline and somatic tissue from veterans with metastatic prostate cancer to find alterations, which could provide access to treatments that would otherwise not be available or appropriate.
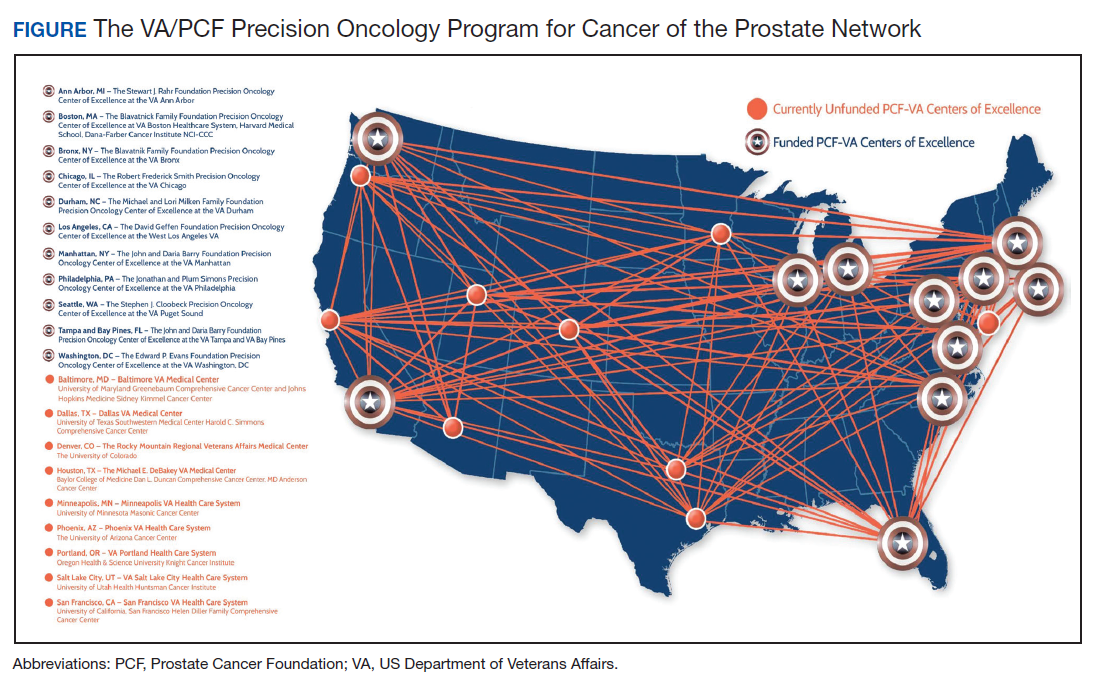
The network is collaborating with NPOP, which provides clinical grade tumor gene panel sequencing for veterans with prostate cancer from > 90% of VA medical centers. POPCaP also partners with the University of Washington to use its OncoPlex gene panel and University of Michigan to use the Oncomine panel to define the best platform for defining targetable alterations for veterans with prostate cancer. Investigators participate in a monthly molecular oncology tumor board continuing medical education-accredited program, which provides guidance and education across the VA about the evidence available to assist in decision making for veterans sequenced through NPOP and the academic platforms. These efforts leverage VA’s partnership with IBM Watson for Genomics to annotate DNA sequencing results to provide clinicians with potential therapeutic options for veterans.
A clinical trials mechanism is embedded in POPCaP to broaden treatment options, improve care for men with prostate cancer, and leverage the sequencing efforts in the network. The Prostate Cancer Analysis for Therapy Choice (PATCH) clinical trials network employs an umbrella study approach whereby alterations are identified through sequencing and veterans are given access to studies embedded at sites across the network. Graff and Huang provide a detailed description of the PATCH network and its potential as a multisite clinical trials mechanism.14 For studies within the network, funds can be provided to support travel to participate in clinical trials for veterans who would be eligible for study but do not live in a catchment for a network site. POPCaP also leverages both the resources of the National Cancer Institute (NCI)-designated cancer centers that are VA academic affiliates, as well as a VA/NCI partnership (NAVIGATE) to increase veteran access to NCI cutting-edge clinical trials.
The network has regular teleconference meetings of the investigators, coordinators, and stakeholders and face-to-face meetings, which are coordinated around other national meetings. These meetings enable investigators to work collaboratively to advance current knowledge in prostate cancer through the application of complementary and synergistic research approaches. Since research plays a critical role within the learning health care system, POPCaP investigators are working to optimize the transfer of knowledge from the clinic to the bench and back to the clinic. In this regard, investigators from network sites have organized themselves into working groups to focus on multiple critical aspects of research and care within the network, including sequencing, phenotyping, health services, health disparities, and a network biorepository.
VA Office of Research and Development
With support from the VA Office of Research and Development, there are research efforts focused on the development of data analytics to identify veterans with metastatic prostate cancer within the electronic health record to ensure access to appropriate testing, treatment, and clinical trials. This will optimize tracking and continuous quality improvement in precision oncology. The Office of Research and Development also supports the use of artificial intelligence to identify predictive markers for diagnosis, prognosis, therapeutic response and patient stratification. POPCaP investigators, along with other investigators from across the VA, conduct research that continually improves the care of veterans with prostate cancer. POPCaP has a special focus on prostate cancer among African Americans, who are disproportionately affected by the disease and well represented in VA. The efforts of the working groups, the research studies and the network as a whole also serve to recruit both junior and senior investigators to the VA in order to support the VA research enterprise.
Active collaborations between the network and other elements of VA include efforts to optimize germline testing and genetic counseling in prostate cancer through the Genomic Medicine Service, which provides telehealth genetic counseling throughout the VA. POPCaP pilots innovative approaches to increase access to clinical genetics and genetic counseling services to support the volume of genetic testing of veterans with cancer. Current National Comprehensive Cancer Network (NCCN) guidelines recommend germline testing for all men with metastatic prostate cancer, which can efficiently identify the roughly 10% of veterans with metastatic disease who carry a germline alteration and provide them with access to studies, FDA-approved treatments, while also offering critical health care information to family members who may also carry a pathogenic germline alteration.
Million Veteran Program
The Million Veteran Program (MVP) has collected > 825,000 germline DNA samples from an anticipated enrollment of > 1 million veterans in one of the most ambitious genetic research efforts to correlate how germline DNA interacts with lifestyle, medications and military exposure to affect health and illness (www.research.va.gov/mvp). MVP is a racially and ethnically diverse veteran cohort that is roughly 20% African American and 7% Hispanic. More than 40,000 of the participants have had prostate cancer, one third of whom are African Americans, giving researchers unprecedented ability to discover factors that impact the development and treatment of the disease in this population. In particular, MVP will provide unique insights into the genetic mutations that drive the development of aggressive prostate cancer in all male veterans, including African Americans. These discoveries will undoubtedly lead to improved screening of and treatment for prostate cancer.
In order to demonstrate clinical utility as well as the infrastructure needs to scale up within the VHA, MVP has launched a pilot project that offers to return clinically actionable genetic results to MVP participants with metastatic prostate cancer, opening the door to new therapies to improve the length and quality of these veterans’ lives. Importantly, the pilot includes cascade testing in family members of enrolled veterans. Given that the original MVP consent did not allow for return of results, and MVP genetic testing is research grade, veterans who volunteer will provide a second consent and undergo clinical genetic testing to confirm the variants. Results from this pilot study also will inform expansion of VA precision oncology efforts for patients with other cancers such as breast cancer or ovarian cancer, where the specific genetic mutations are known to play a role, (eg, BRCA2). In addition, through an interagency agreement with the US Department of Energy (DOE), MVP is leveraging DOE expertise and high-performance computing capabilities to identify clinical and genetic risk factors for prostate cancer that will progress to metastatic disease.
This active research collaboration between POPCaP, MVP, and the Genomic Medicine Service will identify germline BRCA alterations from MVP participants with metastatic prostate cancer and give them access to therapies that may provide better outcomes and access to genetic testing for their family members.
Future Directions
The POPCaP network and its partnership with VA clinical and research efforts is anticipated to provide important insights into barriers and solutions to the implementation of precision oncology for prostate cancer across the VA. These lessons learned may also be relevant for precision oncology care in other settings. As an example, the role of germline testing and genetic counseling is growing more relevant in precision oncology, yet it is clear that the number of men and women dealing with malignancy who actually receive counseling and testing is suboptimal in most health care systems.14 Optimizing the quality and efficiency of oncogenetics within the VA system in a manner that gives access to these services for every veteran in urban or rural environments is an important goal.
The VA has done extensive work in teleoncology and the Genomic Medicine Service provides telehealth genetic counseling service to 90 VA medical facilities nationwide. Expanding on this model to create a distributed network system across the country is an opportunity that will continue to raise VA profile as a leader in this area while providing increased access to genetic services.
Finally, the clinical trials network within POPCaP already has provided valuable insights into how research efforts that originate within the VA can leverage the VA’s strengths. The use of the NPOP centralized sequencing platform to identify potentially targetable alterations across medical centers provides the potential to bring critical access to research to veterans where they live through virtual clinical trials. The VA has a centralized institutional review board that can service large multisite study participation efficiently across the VA. The promise of virtual clinical trials to interrogate relatively rare biomarkers would benefit from institution of a virtual clinical trials workflow. In theory patients with a potentially targetable biomarker could be identified through the centralized DNA sequencing platform and a clinical trial team of virtual investigators and research coordinators would work with health care providers at sites for study startup and performance. Efforts to design and implement this approach are actively being pursued.
The goal of the VA/PCF POPCaP network is to make certain that every veteran has access to appropriate genetic and genomic testing and that the results are utilized so that veterans with targetable alterations receive the best clinical care and have access to clinical trials that could benefit them individually while advancing knowledge that benefits all.
1. Montgomery B, Williams C. Prostate cancer federal health care data trends. https://www.mdedge.com/fedprac/article/208077/oncology/prostate-cancer-federal-health-care-data-trends. Published September 1, 2019. Accessed July 16, 2020.
2. Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer [published correction appears in Cell. 2015 Jul 16;162(2):454]. Cell. 2015;161(5):1215-1228. doi:10.1016/j.cell.2015.05.001
3. Quigley DA, Dang HX, Zhao SG, et al. Genomic hallmarks and structural variation in metastatic prostate cancer [published correction appears in Cell. 2018 Oct 18;175(3):889]. Cell. 2018;174(3):758-769.e9. doi:10.1016/j.cell.2018.06.039
4. Pritchard CC, Offit K, Nelson PS. DNA-repair gene mutations in metastatic prostate cancer. N Engl J Med. 2016;375(18):1804-1805. doi:10.1056/NEJMc1611137
5. Guillem JG. Molecular diagnosis of hereditary nonpolyposis colon cancer. N Engl J Med. 1998;339(13):924-925. doi:10.1056/nejm199809243391316
6. Ryan S, Jenkins MA, Win AK. Risk of prostate cancer in Lynch syndrome: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2014;23(3):437-449. doi:10.1158/1055-9965.EPI-13-1165
7. Abida W, Cheng ML, Armenia J, et al. Analysis of the prevalence of microsatellite instability in prostate cancer and response to immune checkpoint blockade. JAMA Oncol. 2019;5(4):471-478. doi:10.1001/jamaoncol.2018.5801
8. Graham LS, Montgomery B, Cheng HH, et al. Mismatch repair deficiency in metastatic prostate cancer: Response to PD-1 blockade and standard therapies. PLoS One. 2020;15(5):e0233260. Published 2020 May 26. doi:10.1371/journal.pone.0233260
9. Yu HA, Planchard D, Lovly CM. Sequencing therapy for genetically defined subgroups of non-small cell lung cancer. Am Soc Clin Oncol Educ Book. 2018;38:726-739. doi:10.1200/EDBK_201331
10. Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375(5):443-453. doi:10.1056/NEJMoa1603144
11. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917-921. doi:10.1038/nature03445
12. Abida W, Campbell D, Patnaik A, et al. Preliminary results from the TRITON2 study of rucaparib in patients with DNA damage repair deficiency metastatic, castration resistant prostate cancer: updated analyses. Ann Oncol. 2019;30(suppl 5): v325-v355. doi:10.1093/annonc/mdz248
13. de Bono J, Mateo J, Fizazi K, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382(22):2091-2102. doi:10.1056/NEJMoa1911440
14. Graff JN, Huang GD. Leveraging Veterans Health Administration clinical and research resources to accelerate discovery and testing in precision oncology. Fed Pract. 2020;37(suppl 4):S62-S67. doi: 10.12788/fp.0028
The US Department of Veterans Affairs (VA) is home to the Veterans Health Administration (VHA), which delivers care at 1,255 health care facilities, including 170 medical centers. The VA serves 6 million veterans each year and is the largest integrated provider of cancer care in the US. The system uses a single, enterprise-wide electronic health record. The detailed curation of clinical outcomes, laboratory results, and radiology is used in VA efforts to improve oncology outcomes for veterans. The VA also has a National Precision Oncology Program (NPOP), which offers system-wide DNA sequencing for veterans with cancer. Given its size, integration, and capabilities, the VA is an ideal setting for rapid learning cycles of testing and implementing best practices at scale.
Prostate cancer is the most common malignancy affecting men in the US. It is the most commonly-diagnosed solid tumor in the VA, and in 2014, there were 11,376 prostate cancer diagnoses in the VA.1 The clinical characteristics and treatment of veterans with prostate cancer largely parallel the broader population of men in the US.1 Although the majority of men diagnosed with prostate cancer have disease localized to the prostate, an important minority develop metastatic disease, which represents a risk for substantial morbidity and is the lethal form of the disease. Research has yielded transformative advances in the care of men with metastatic prostate cancer, including drugs targeting the testosterone/androgen signaling axis, taxane chemotherapy, the radionuclide radium-223, and a dendritic cell vaccine. Unfortunately, the magnitude and duration of response to these therapies varies widely, and determining the biology relevant to an individual patient that would better inform their treatment decisions is a critical next step. As the ability to interrogate the cancer genome has improved, relevant drivers of tumorigenesis and predictive biomarkers are being identified rapidly, and oncology care has evolved from a one-size-fits-all approach to a precision approach, which uses these biomarkers to assist in therapeutic decision making.
Precision Oncology for Prostate Cancer
A series of studies interrogating the genomics of metastatic prostate cancer have been critical to defining the relevance of precision oncology for prostate cancer. Most of what is known about the genomics of prostate cancer has been derived from analysis of samples from the prostate itself. These samples may not reflect the biology of metastasis and genetic evolution in response to treatment pressure, so the genomic alterations in metastatic disease remained incompletely characterized. Two large research teams supported by grants from the American Association for Cancer Research, Stand Up 2 Cancer, and Prostate Cancer Foundation (PCF) focused their efforts on sampling and analyzing metastatic tissue to define the most relevant genomic alterations in advanced prostate cancer.
These efforts defined a broad range of relatively common alterations in the androgen receptor, as well as the tumor suppressors TP53 and PTEN.2,3 Important subsets of less common alterations in pathways that were potentially targetable were also found, including new alterations in PIK3CA/B, BRAF/RAF1, and β-catenin. Most surprisingly, alterations of DNA repair pathways, including mismatch repair and homologous recombination were found in 20% of tumors, and half of these tumors contained germline alterations. The same groups performed a follow up analysis of germline DNA from men with metastatic prostate cancer, which confirmed that 12% of these patients carry a pathogenic germline alteration in a DNA repair pathway gene.4 These efforts immediately invigorated precision oncology clinical trials for prostate cancer and spurred an effort to find the molecular alterations that could be leveraged to improve care for men with advanced prostate cancer.
Targetable Alterations
Currently a number of genomic alterations are immediately actionable. There are several agents approved by the US Food and Drug Administration (FDA) that exploit these Achilles heels of prostate cancer. Mismatch repair deficiency occurs when any of a group of genes responsible for proofreading the fidelity of DNA replication is compromised by mutation or deletion. Imperfect reading and correction subsequently lead to many DNA mutations in a tissue (hypermutation), which then increases the risk of developing malignancy. If a defective gene in the mismatch repair pathway is inherited, a patient has a genetic predisposition to specific malignancies that are part of the Lynch syndrome.5 Prostate cancer is a relatively rare manifestation of Lynch syndrome, although it is considered one of the malignancies in the Lynch syndrome spectrum.6
Alteration of one of the mismatch repair genes also can occur spontaneously in a tumor, resulting in the same high frequency of spontaneous DNA mutations. Overall, between 3% and 5% of metastatic prostate cancers contain mismatch repair deficiency. The majority of these cases are a result of spontaneous loss or mutation of the relevant gene, but 1 in 5 of these tumors occurs as a component of Lynch syndrome.7 Identification of mismatch repair deficiency is critical because the resulting hypermutation makes these tumors particularly susceptible to intervention with immunotherapy. Up to half of patients with metastatic prostate cancer can have durable responses. This finding is consistent with the experience treating other malignancies with mismatch repair deficiency.8 Although screening for mismatch repair deficiency is standard of care for patients with malignancies such as colorectal cancer, few patients with prostate cancer may receive the mismatch repair deficiency screening (based on unpublished data). In contrast, screening is routine for patients with adenocarcinoma of the lung because their proportion of ROS1 and ALK alterations is similar to the frequency of mismatch repair deficiency when compared with patients with prostate cancer.9
Homologous recombination is another mechanism by which cells repair DNA damage and is responsible for repairing double strand breaks, the type of DNA damage most likely to lead to carcinogenesis. In advanced prostate cancer, BRCA2, ATM, BRCA1 and other members of the Fanconi Anemia/BRCA gene family are altered 20% of the time. These genes also are the most common germline alterations implicated in the development of prostate cancer.2,10 Prostate cancer is considered a BRCA-related cancer much like breast, ovarian, and pancreatic cancers. Defects in homologous recombination repair make BRCA-altered prostate cancers susceptible to DNA damaging chemotherapy, such as platinum and to the use of poly–(adenosine diphosphate–ribose) polymerase (PARP) inhibitors because cancer cells then accumulate cytotoxic and apoptotic levels of DNA.11
In May 2020, the FDA approved the use of PARP inhibitors for the treatment of prostate cancers that contain BRCA and other DNA repair alterations. Rucaparib received accelerated approval for the treatment of prostate cancers containing BRCA alterations and olaparib received full approval for treatment of prostate cancers containing an array of alterations in DNA repair genes.12,13 Both approvals were the direct result of the cited landmark studies that demonstrated the frequency of these alterations in advanced prostate cancer.2,3
Beyond mismatch and homologous recombination repair, there are a large number of potentially targetable alterations found in advanced prostate cancer. It is thus critical that we put systems into place both to find germline and somatic alterations that will inform a veteran’s clinical care and to provide veterans access to precision oncology clinical trials.
The POPCaP Network
Because prostate cancer is such a significant issue in the VA and best practices for precision oncology can be implemented broadly once defined as successful, the PCF and the VA formed a collaboration to support a network of centers that would focus on implementing a comprehensive strategy for precision oncology in prostate cancer. There are currently 11 centers in the Precision Oncology Program for Cancer of the Prostate (POPCaP) network (Figure). These centers are tasked with comprehensively sequencing germline and somatic tissue from veterans with metastatic prostate cancer to find alterations, which could provide access to treatments that would otherwise not be available or appropriate.
The network is collaborating with NPOP, which provides clinical grade tumor gene panel sequencing for veterans with prostate cancer from > 90% of VA medical centers. POPCaP also partners with the University of Washington to use its OncoPlex gene panel and University of Michigan to use the Oncomine panel to define the best platform for defining targetable alterations for veterans with prostate cancer. Investigators participate in a monthly molecular oncology tumor board continuing medical education-accredited program, which provides guidance and education across the VA about the evidence available to assist in decision making for veterans sequenced through NPOP and the academic platforms. These efforts leverage VA’s partnership with IBM Watson for Genomics to annotate DNA sequencing results to provide clinicians with potential therapeutic options for veterans.
A clinical trials mechanism is embedded in POPCaP to broaden treatment options, improve care for men with prostate cancer, and leverage the sequencing efforts in the network. The Prostate Cancer Analysis for Therapy Choice (PATCH) clinical trials network employs an umbrella study approach whereby alterations are identified through sequencing and veterans are given access to studies embedded at sites across the network. Graff and Huang provide a detailed description of the PATCH network and its potential as a multisite clinical trials mechanism.14 For studies within the network, funds can be provided to support travel to participate in clinical trials for veterans who would be eligible for study but do not live in a catchment for a network site. POPCaP also leverages both the resources of the National Cancer Institute (NCI)-designated cancer centers that are VA academic affiliates, as well as a VA/NCI partnership (NAVIGATE) to increase veteran access to NCI cutting-edge clinical trials.
The network has regular teleconference meetings of the investigators, coordinators, and stakeholders and face-to-face meetings, which are coordinated around other national meetings. These meetings enable investigators to work collaboratively to advance current knowledge in prostate cancer through the application of complementary and synergistic research approaches. Since research plays a critical role within the learning health care system, POPCaP investigators are working to optimize the transfer of knowledge from the clinic to the bench and back to the clinic. In this regard, investigators from network sites have organized themselves into working groups to focus on multiple critical aspects of research and care within the network, including sequencing, phenotyping, health services, health disparities, and a network biorepository.
VA Office of Research and Development
With support from the VA Office of Research and Development, there are research efforts focused on the development of data analytics to identify veterans with metastatic prostate cancer within the electronic health record to ensure access to appropriate testing, treatment, and clinical trials. This will optimize tracking and continuous quality improvement in precision oncology. The Office of Research and Development also supports the use of artificial intelligence to identify predictive markers for diagnosis, prognosis, therapeutic response and patient stratification. POPCaP investigators, along with other investigators from across the VA, conduct research that continually improves the care of veterans with prostate cancer. POPCaP has a special focus on prostate cancer among African Americans, who are disproportionately affected by the disease and well represented in VA. The efforts of the working groups, the research studies and the network as a whole also serve to recruit both junior and senior investigators to the VA in order to support the VA research enterprise.
Active collaborations between the network and other elements of VA include efforts to optimize germline testing and genetic counseling in prostate cancer through the Genomic Medicine Service, which provides telehealth genetic counseling throughout the VA. POPCaP pilots innovative approaches to increase access to clinical genetics and genetic counseling services to support the volume of genetic testing of veterans with cancer. Current National Comprehensive Cancer Network (NCCN) guidelines recommend germline testing for all men with metastatic prostate cancer, which can efficiently identify the roughly 10% of veterans with metastatic disease who carry a germline alteration and provide them with access to studies, FDA-approved treatments, while also offering critical health care information to family members who may also carry a pathogenic germline alteration.
Million Veteran Program
The Million Veteran Program (MVP) has collected > 825,000 germline DNA samples from an anticipated enrollment of > 1 million veterans in one of the most ambitious genetic research efforts to correlate how germline DNA interacts with lifestyle, medications and military exposure to affect health and illness (www.research.va.gov/mvp). MVP is a racially and ethnically diverse veteran cohort that is roughly 20% African American and 7% Hispanic. More than 40,000 of the participants have had prostate cancer, one third of whom are African Americans, giving researchers unprecedented ability to discover factors that impact the development and treatment of the disease in this population. In particular, MVP will provide unique insights into the genetic mutations that drive the development of aggressive prostate cancer in all male veterans, including African Americans. These discoveries will undoubtedly lead to improved screening of and treatment for prostate cancer.
In order to demonstrate clinical utility as well as the infrastructure needs to scale up within the VHA, MVP has launched a pilot project that offers to return clinically actionable genetic results to MVP participants with metastatic prostate cancer, opening the door to new therapies to improve the length and quality of these veterans’ lives. Importantly, the pilot includes cascade testing in family members of enrolled veterans. Given that the original MVP consent did not allow for return of results, and MVP genetic testing is research grade, veterans who volunteer will provide a second consent and undergo clinical genetic testing to confirm the variants. Results from this pilot study also will inform expansion of VA precision oncology efforts for patients with other cancers such as breast cancer or ovarian cancer, where the specific genetic mutations are known to play a role, (eg, BRCA2). In addition, through an interagency agreement with the US Department of Energy (DOE), MVP is leveraging DOE expertise and high-performance computing capabilities to identify clinical and genetic risk factors for prostate cancer that will progress to metastatic disease.
This active research collaboration between POPCaP, MVP, and the Genomic Medicine Service will identify germline BRCA alterations from MVP participants with metastatic prostate cancer and give them access to therapies that may provide better outcomes and access to genetic testing for their family members.
Future Directions
The POPCaP network and its partnership with VA clinical and research efforts is anticipated to provide important insights into barriers and solutions to the implementation of precision oncology for prostate cancer across the VA. These lessons learned may also be relevant for precision oncology care in other settings. As an example, the role of germline testing and genetic counseling is growing more relevant in precision oncology, yet it is clear that the number of men and women dealing with malignancy who actually receive counseling and testing is suboptimal in most health care systems.14 Optimizing the quality and efficiency of oncogenetics within the VA system in a manner that gives access to these services for every veteran in urban or rural environments is an important goal.
The VA has done extensive work in teleoncology and the Genomic Medicine Service provides telehealth genetic counseling service to 90 VA medical facilities nationwide. Expanding on this model to create a distributed network system across the country is an opportunity that will continue to raise VA profile as a leader in this area while providing increased access to genetic services.
Finally, the clinical trials network within POPCaP already has provided valuable insights into how research efforts that originate within the VA can leverage the VA’s strengths. The use of the NPOP centralized sequencing platform to identify potentially targetable alterations across medical centers provides the potential to bring critical access to research to veterans where they live through virtual clinical trials. The VA has a centralized institutional review board that can service large multisite study participation efficiently across the VA. The promise of virtual clinical trials to interrogate relatively rare biomarkers would benefit from institution of a virtual clinical trials workflow. In theory patients with a potentially targetable biomarker could be identified through the centralized DNA sequencing platform and a clinical trial team of virtual investigators and research coordinators would work with health care providers at sites for study startup and performance. Efforts to design and implement this approach are actively being pursued.
The goal of the VA/PCF POPCaP network is to make certain that every veteran has access to appropriate genetic and genomic testing and that the results are utilized so that veterans with targetable alterations receive the best clinical care and have access to clinical trials that could benefit them individually while advancing knowledge that benefits all.
The US Department of Veterans Affairs (VA) is home to the Veterans Health Administration (VHA), which delivers care at 1,255 health care facilities, including 170 medical centers. The VA serves 6 million veterans each year and is the largest integrated provider of cancer care in the US. The system uses a single, enterprise-wide electronic health record. The detailed curation of clinical outcomes, laboratory results, and radiology is used in VA efforts to improve oncology outcomes for veterans. The VA also has a National Precision Oncology Program (NPOP), which offers system-wide DNA sequencing for veterans with cancer. Given its size, integration, and capabilities, the VA is an ideal setting for rapid learning cycles of testing and implementing best practices at scale.
Prostate cancer is the most common malignancy affecting men in the US. It is the most commonly-diagnosed solid tumor in the VA, and in 2014, there were 11,376 prostate cancer diagnoses in the VA.1 The clinical characteristics and treatment of veterans with prostate cancer largely parallel the broader population of men in the US.1 Although the majority of men diagnosed with prostate cancer have disease localized to the prostate, an important minority develop metastatic disease, which represents a risk for substantial morbidity and is the lethal form of the disease. Research has yielded transformative advances in the care of men with metastatic prostate cancer, including drugs targeting the testosterone/androgen signaling axis, taxane chemotherapy, the radionuclide radium-223, and a dendritic cell vaccine. Unfortunately, the magnitude and duration of response to these therapies varies widely, and determining the biology relevant to an individual patient that would better inform their treatment decisions is a critical next step. As the ability to interrogate the cancer genome has improved, relevant drivers of tumorigenesis and predictive biomarkers are being identified rapidly, and oncology care has evolved from a one-size-fits-all approach to a precision approach, which uses these biomarkers to assist in therapeutic decision making.
Precision Oncology for Prostate Cancer
A series of studies interrogating the genomics of metastatic prostate cancer have been critical to defining the relevance of precision oncology for prostate cancer. Most of what is known about the genomics of prostate cancer has been derived from analysis of samples from the prostate itself. These samples may not reflect the biology of metastasis and genetic evolution in response to treatment pressure, so the genomic alterations in metastatic disease remained incompletely characterized. Two large research teams supported by grants from the American Association for Cancer Research, Stand Up 2 Cancer, and Prostate Cancer Foundation (PCF) focused their efforts on sampling and analyzing metastatic tissue to define the most relevant genomic alterations in advanced prostate cancer.
These efforts defined a broad range of relatively common alterations in the androgen receptor, as well as the tumor suppressors TP53 and PTEN.2,3 Important subsets of less common alterations in pathways that were potentially targetable were also found, including new alterations in PIK3CA/B, BRAF/RAF1, and β-catenin. Most surprisingly, alterations of DNA repair pathways, including mismatch repair and homologous recombination were found in 20% of tumors, and half of these tumors contained germline alterations. The same groups performed a follow up analysis of germline DNA from men with metastatic prostate cancer, which confirmed that 12% of these patients carry a pathogenic germline alteration in a DNA repair pathway gene.4 These efforts immediately invigorated precision oncology clinical trials for prostate cancer and spurred an effort to find the molecular alterations that could be leveraged to improve care for men with advanced prostate cancer.
Targetable Alterations
Currently a number of genomic alterations are immediately actionable. There are several agents approved by the US Food and Drug Administration (FDA) that exploit these Achilles heels of prostate cancer. Mismatch repair deficiency occurs when any of a group of genes responsible for proofreading the fidelity of DNA replication is compromised by mutation or deletion. Imperfect reading and correction subsequently lead to many DNA mutations in a tissue (hypermutation), which then increases the risk of developing malignancy. If a defective gene in the mismatch repair pathway is inherited, a patient has a genetic predisposition to specific malignancies that are part of the Lynch syndrome.5 Prostate cancer is a relatively rare manifestation of Lynch syndrome, although it is considered one of the malignancies in the Lynch syndrome spectrum.6
Alteration of one of the mismatch repair genes also can occur spontaneously in a tumor, resulting in the same high frequency of spontaneous DNA mutations. Overall, between 3% and 5% of metastatic prostate cancers contain mismatch repair deficiency. The majority of these cases are a result of spontaneous loss or mutation of the relevant gene, but 1 in 5 of these tumors occurs as a component of Lynch syndrome.7 Identification of mismatch repair deficiency is critical because the resulting hypermutation makes these tumors particularly susceptible to intervention with immunotherapy. Up to half of patients with metastatic prostate cancer can have durable responses. This finding is consistent with the experience treating other malignancies with mismatch repair deficiency.8 Although screening for mismatch repair deficiency is standard of care for patients with malignancies such as colorectal cancer, few patients with prostate cancer may receive the mismatch repair deficiency screening (based on unpublished data). In contrast, screening is routine for patients with adenocarcinoma of the lung because their proportion of ROS1 and ALK alterations is similar to the frequency of mismatch repair deficiency when compared with patients with prostate cancer.9
Homologous recombination is another mechanism by which cells repair DNA damage and is responsible for repairing double strand breaks, the type of DNA damage most likely to lead to carcinogenesis. In advanced prostate cancer, BRCA2, ATM, BRCA1 and other members of the Fanconi Anemia/BRCA gene family are altered 20% of the time. These genes also are the most common germline alterations implicated in the development of prostate cancer.2,10 Prostate cancer is considered a BRCA-related cancer much like breast, ovarian, and pancreatic cancers. Defects in homologous recombination repair make BRCA-altered prostate cancers susceptible to DNA damaging chemotherapy, such as platinum and to the use of poly–(adenosine diphosphate–ribose) polymerase (PARP) inhibitors because cancer cells then accumulate cytotoxic and apoptotic levels of DNA.11
In May 2020, the FDA approved the use of PARP inhibitors for the treatment of prostate cancers that contain BRCA and other DNA repair alterations. Rucaparib received accelerated approval for the treatment of prostate cancers containing BRCA alterations and olaparib received full approval for treatment of prostate cancers containing an array of alterations in DNA repair genes.12,13 Both approvals were the direct result of the cited landmark studies that demonstrated the frequency of these alterations in advanced prostate cancer.2,3
Beyond mismatch and homologous recombination repair, there are a large number of potentially targetable alterations found in advanced prostate cancer. It is thus critical that we put systems into place both to find germline and somatic alterations that will inform a veteran’s clinical care and to provide veterans access to precision oncology clinical trials.
The POPCaP Network
Because prostate cancer is such a significant issue in the VA and best practices for precision oncology can be implemented broadly once defined as successful, the PCF and the VA formed a collaboration to support a network of centers that would focus on implementing a comprehensive strategy for precision oncology in prostate cancer. There are currently 11 centers in the Precision Oncology Program for Cancer of the Prostate (POPCaP) network (Figure). These centers are tasked with comprehensively sequencing germline and somatic tissue from veterans with metastatic prostate cancer to find alterations, which could provide access to treatments that would otherwise not be available or appropriate.
The network is collaborating with NPOP, which provides clinical grade tumor gene panel sequencing for veterans with prostate cancer from > 90% of VA medical centers. POPCaP also partners with the University of Washington to use its OncoPlex gene panel and University of Michigan to use the Oncomine panel to define the best platform for defining targetable alterations for veterans with prostate cancer. Investigators participate in a monthly molecular oncology tumor board continuing medical education-accredited program, which provides guidance and education across the VA about the evidence available to assist in decision making for veterans sequenced through NPOP and the academic platforms. These efforts leverage VA’s partnership with IBM Watson for Genomics to annotate DNA sequencing results to provide clinicians with potential therapeutic options for veterans.
A clinical trials mechanism is embedded in POPCaP to broaden treatment options, improve care for men with prostate cancer, and leverage the sequencing efforts in the network. The Prostate Cancer Analysis for Therapy Choice (PATCH) clinical trials network employs an umbrella study approach whereby alterations are identified through sequencing and veterans are given access to studies embedded at sites across the network. Graff and Huang provide a detailed description of the PATCH network and its potential as a multisite clinical trials mechanism.14 For studies within the network, funds can be provided to support travel to participate in clinical trials for veterans who would be eligible for study but do not live in a catchment for a network site. POPCaP also leverages both the resources of the National Cancer Institute (NCI)-designated cancer centers that are VA academic affiliates, as well as a VA/NCI partnership (NAVIGATE) to increase veteran access to NCI cutting-edge clinical trials.
The network has regular teleconference meetings of the investigators, coordinators, and stakeholders and face-to-face meetings, which are coordinated around other national meetings. These meetings enable investigators to work collaboratively to advance current knowledge in prostate cancer through the application of complementary and synergistic research approaches. Since research plays a critical role within the learning health care system, POPCaP investigators are working to optimize the transfer of knowledge from the clinic to the bench and back to the clinic. In this regard, investigators from network sites have organized themselves into working groups to focus on multiple critical aspects of research and care within the network, including sequencing, phenotyping, health services, health disparities, and a network biorepository.
VA Office of Research and Development
With support from the VA Office of Research and Development, there are research efforts focused on the development of data analytics to identify veterans with metastatic prostate cancer within the electronic health record to ensure access to appropriate testing, treatment, and clinical trials. This will optimize tracking and continuous quality improvement in precision oncology. The Office of Research and Development also supports the use of artificial intelligence to identify predictive markers for diagnosis, prognosis, therapeutic response and patient stratification. POPCaP investigators, along with other investigators from across the VA, conduct research that continually improves the care of veterans with prostate cancer. POPCaP has a special focus on prostate cancer among African Americans, who are disproportionately affected by the disease and well represented in VA. The efforts of the working groups, the research studies and the network as a whole also serve to recruit both junior and senior investigators to the VA in order to support the VA research enterprise.
Active collaborations between the network and other elements of VA include efforts to optimize germline testing and genetic counseling in prostate cancer through the Genomic Medicine Service, which provides telehealth genetic counseling throughout the VA. POPCaP pilots innovative approaches to increase access to clinical genetics and genetic counseling services to support the volume of genetic testing of veterans with cancer. Current National Comprehensive Cancer Network (NCCN) guidelines recommend germline testing for all men with metastatic prostate cancer, which can efficiently identify the roughly 10% of veterans with metastatic disease who carry a germline alteration and provide them with access to studies, FDA-approved treatments, while also offering critical health care information to family members who may also carry a pathogenic germline alteration.
Million Veteran Program
The Million Veteran Program (MVP) has collected > 825,000 germline DNA samples from an anticipated enrollment of > 1 million veterans in one of the most ambitious genetic research efforts to correlate how germline DNA interacts with lifestyle, medications and military exposure to affect health and illness (www.research.va.gov/mvp). MVP is a racially and ethnically diverse veteran cohort that is roughly 20% African American and 7% Hispanic. More than 40,000 of the participants have had prostate cancer, one third of whom are African Americans, giving researchers unprecedented ability to discover factors that impact the development and treatment of the disease in this population. In particular, MVP will provide unique insights into the genetic mutations that drive the development of aggressive prostate cancer in all male veterans, including African Americans. These discoveries will undoubtedly lead to improved screening of and treatment for prostate cancer.
In order to demonstrate clinical utility as well as the infrastructure needs to scale up within the VHA, MVP has launched a pilot project that offers to return clinically actionable genetic results to MVP participants with metastatic prostate cancer, opening the door to new therapies to improve the length and quality of these veterans’ lives. Importantly, the pilot includes cascade testing in family members of enrolled veterans. Given that the original MVP consent did not allow for return of results, and MVP genetic testing is research grade, veterans who volunteer will provide a second consent and undergo clinical genetic testing to confirm the variants. Results from this pilot study also will inform expansion of VA precision oncology efforts for patients with other cancers such as breast cancer or ovarian cancer, where the specific genetic mutations are known to play a role, (eg, BRCA2). In addition, through an interagency agreement with the US Department of Energy (DOE), MVP is leveraging DOE expertise and high-performance computing capabilities to identify clinical and genetic risk factors for prostate cancer that will progress to metastatic disease.
This active research collaboration between POPCaP, MVP, and the Genomic Medicine Service will identify germline BRCA alterations from MVP participants with metastatic prostate cancer and give them access to therapies that may provide better outcomes and access to genetic testing for their family members.
Future Directions
The POPCaP network and its partnership with VA clinical and research efforts is anticipated to provide important insights into barriers and solutions to the implementation of precision oncology for prostate cancer across the VA. These lessons learned may also be relevant for precision oncology care in other settings. As an example, the role of germline testing and genetic counseling is growing more relevant in precision oncology, yet it is clear that the number of men and women dealing with malignancy who actually receive counseling and testing is suboptimal in most health care systems.14 Optimizing the quality and efficiency of oncogenetics within the VA system in a manner that gives access to these services for every veteran in urban or rural environments is an important goal.
The VA has done extensive work in teleoncology and the Genomic Medicine Service provides telehealth genetic counseling service to 90 VA medical facilities nationwide. Expanding on this model to create a distributed network system across the country is an opportunity that will continue to raise VA profile as a leader in this area while providing increased access to genetic services.
Finally, the clinical trials network within POPCaP already has provided valuable insights into how research efforts that originate within the VA can leverage the VA’s strengths. The use of the NPOP centralized sequencing platform to identify potentially targetable alterations across medical centers provides the potential to bring critical access to research to veterans where they live through virtual clinical trials. The VA has a centralized institutional review board that can service large multisite study participation efficiently across the VA. The promise of virtual clinical trials to interrogate relatively rare biomarkers would benefit from institution of a virtual clinical trials workflow. In theory patients with a potentially targetable biomarker could be identified through the centralized DNA sequencing platform and a clinical trial team of virtual investigators and research coordinators would work with health care providers at sites for study startup and performance. Efforts to design and implement this approach are actively being pursued.
The goal of the VA/PCF POPCaP network is to make certain that every veteran has access to appropriate genetic and genomic testing and that the results are utilized so that veterans with targetable alterations receive the best clinical care and have access to clinical trials that could benefit them individually while advancing knowledge that benefits all.
1. Montgomery B, Williams C. Prostate cancer federal health care data trends. https://www.mdedge.com/fedprac/article/208077/oncology/prostate-cancer-federal-health-care-data-trends. Published September 1, 2019. Accessed July 16, 2020.
2. Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer [published correction appears in Cell. 2015 Jul 16;162(2):454]. Cell. 2015;161(5):1215-1228. doi:10.1016/j.cell.2015.05.001
3. Quigley DA, Dang HX, Zhao SG, et al. Genomic hallmarks and structural variation in metastatic prostate cancer [published correction appears in Cell. 2018 Oct 18;175(3):889]. Cell. 2018;174(3):758-769.e9. doi:10.1016/j.cell.2018.06.039
4. Pritchard CC, Offit K, Nelson PS. DNA-repair gene mutations in metastatic prostate cancer. N Engl J Med. 2016;375(18):1804-1805. doi:10.1056/NEJMc1611137
5. Guillem JG. Molecular diagnosis of hereditary nonpolyposis colon cancer. N Engl J Med. 1998;339(13):924-925. doi:10.1056/nejm199809243391316
6. Ryan S, Jenkins MA, Win AK. Risk of prostate cancer in Lynch syndrome: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2014;23(3):437-449. doi:10.1158/1055-9965.EPI-13-1165
7. Abida W, Cheng ML, Armenia J, et al. Analysis of the prevalence of microsatellite instability in prostate cancer and response to immune checkpoint blockade. JAMA Oncol. 2019;5(4):471-478. doi:10.1001/jamaoncol.2018.5801
8. Graham LS, Montgomery B, Cheng HH, et al. Mismatch repair deficiency in metastatic prostate cancer: Response to PD-1 blockade and standard therapies. PLoS One. 2020;15(5):e0233260. Published 2020 May 26. doi:10.1371/journal.pone.0233260
9. Yu HA, Planchard D, Lovly CM. Sequencing therapy for genetically defined subgroups of non-small cell lung cancer. Am Soc Clin Oncol Educ Book. 2018;38:726-739. doi:10.1200/EDBK_201331
10. Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375(5):443-453. doi:10.1056/NEJMoa1603144
11. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917-921. doi:10.1038/nature03445
12. Abida W, Campbell D, Patnaik A, et al. Preliminary results from the TRITON2 study of rucaparib in patients with DNA damage repair deficiency metastatic, castration resistant prostate cancer: updated analyses. Ann Oncol. 2019;30(suppl 5): v325-v355. doi:10.1093/annonc/mdz248
13. de Bono J, Mateo J, Fizazi K, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382(22):2091-2102. doi:10.1056/NEJMoa1911440
14. Graff JN, Huang GD. Leveraging Veterans Health Administration clinical and research resources to accelerate discovery and testing in precision oncology. Fed Pract. 2020;37(suppl 4):S62-S67. doi: 10.12788/fp.0028
1. Montgomery B, Williams C. Prostate cancer federal health care data trends. https://www.mdedge.com/fedprac/article/208077/oncology/prostate-cancer-federal-health-care-data-trends. Published September 1, 2019. Accessed July 16, 2020.
2. Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer [published correction appears in Cell. 2015 Jul 16;162(2):454]. Cell. 2015;161(5):1215-1228. doi:10.1016/j.cell.2015.05.001
3. Quigley DA, Dang HX, Zhao SG, et al. Genomic hallmarks and structural variation in metastatic prostate cancer [published correction appears in Cell. 2018 Oct 18;175(3):889]. Cell. 2018;174(3):758-769.e9. doi:10.1016/j.cell.2018.06.039
4. Pritchard CC, Offit K, Nelson PS. DNA-repair gene mutations in metastatic prostate cancer. N Engl J Med. 2016;375(18):1804-1805. doi:10.1056/NEJMc1611137
5. Guillem JG. Molecular diagnosis of hereditary nonpolyposis colon cancer. N Engl J Med. 1998;339(13):924-925. doi:10.1056/nejm199809243391316
6. Ryan S, Jenkins MA, Win AK. Risk of prostate cancer in Lynch syndrome: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2014;23(3):437-449. doi:10.1158/1055-9965.EPI-13-1165
7. Abida W, Cheng ML, Armenia J, et al. Analysis of the prevalence of microsatellite instability in prostate cancer and response to immune checkpoint blockade. JAMA Oncol. 2019;5(4):471-478. doi:10.1001/jamaoncol.2018.5801
8. Graham LS, Montgomery B, Cheng HH, et al. Mismatch repair deficiency in metastatic prostate cancer: Response to PD-1 blockade and standard therapies. PLoS One. 2020;15(5):e0233260. Published 2020 May 26. doi:10.1371/journal.pone.0233260
9. Yu HA, Planchard D, Lovly CM. Sequencing therapy for genetically defined subgroups of non-small cell lung cancer. Am Soc Clin Oncol Educ Book. 2018;38:726-739. doi:10.1200/EDBK_201331
10. Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375(5):443-453. doi:10.1056/NEJMoa1603144
11. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917-921. doi:10.1038/nature03445
12. Abida W, Campbell D, Patnaik A, et al. Preliminary results from the TRITON2 study of rucaparib in patients with DNA damage repair deficiency metastatic, castration resistant prostate cancer: updated analyses. Ann Oncol. 2019;30(suppl 5): v325-v355. doi:10.1093/annonc/mdz248
13. de Bono J, Mateo J, Fizazi K, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382(22):2091-2102. doi:10.1056/NEJMoa1911440
14. Graff JN, Huang GD. Leveraging Veterans Health Administration clinical and research resources to accelerate discovery and testing in precision oncology. Fed Pract. 2020;37(suppl 4):S62-S67. doi: 10.12788/fp.0028
DNA Repair Gene Variants in Patients With Prostate Cancer Achieving Durable Clinical Benefit With PARP Inhibitors
BACKGROUND: PARP inhibitors (PARPi’s) were recently approved for the treatment of metastatic prostate cancer among patients harboring mutations in an array of genes responsible for DNA repair. We sought to identify whether a subset of these genes correlates with response to treatment more frequently than others. Consequently, an evaluation of the specific DNA repair genotypes associated with durable clinical benefit (DCB) using real-world patient data was undertaken.
METHODS: The U.S. Department of Veterans Affairs (VA) National Precision Oncology Program’s (NPOP) database and Corporate Data Warehouse (CDW) were reviewed to select patients who (1) carried a diagnosis of prostate cancer, (2) successfully underwent tumor DNA sequencing through NPOP, (3) were prescribed olaparib, rucaparib, nirapib, and/or talazaporib for their prostate cancer between July 2016 and February 2020, and (4) and achieved DCB, defined as no progression in prostate-specific antigen (PSA) for at least 6 months following PARPi initiation without concurrent systemic or non-systemic therapies other than androgen-deprivation. The DNA repair gene variants and orders placed for NPOP consultative support were reviewed.
RESULTS: Of the 44 prostate cancer patients treated with a PARPi, 6 (13.6%) had tumor DNA sequencing through NPOP and had achieved DCB. Five patients were treated with olaparib and 1 with rucaparib. The median PSA progression-free survival was 8.9 (interquartile range = 8.5 – 11.2) months among these selected patients. Regarding gene variants, 5 patients had 7 BRCA2 mutations, including 4 frameshift, 1 nonsense, 1 single nucleotide variant, and 1 splice site. One patient had frameshift and missense ATM mutations. Referrals to the NPOP consult service were ordered for 2 out of the 5 patients with BRCA2 mutations achieving DCB.
CONCLUSIONS: Within the VA’s NPOP, the presence of BRCA2 gene variants was the most common finding from tumor DNA sequencing among patients with prostate cancer achieving DCB with a PARPi. Further analysis of the genotypes of all patients treated with PARPi in NPOP to assess the differential impact of BRCA2 mutations is needed to confirm the clinical implication of this finding.
BACKGROUND: PARP inhibitors (PARPi’s) were recently approved for the treatment of metastatic prostate cancer among patients harboring mutations in an array of genes responsible for DNA repair. We sought to identify whether a subset of these genes correlates with response to treatment more frequently than others. Consequently, an evaluation of the specific DNA repair genotypes associated with durable clinical benefit (DCB) using real-world patient data was undertaken.
METHODS: The U.S. Department of Veterans Affairs (VA) National Precision Oncology Program’s (NPOP) database and Corporate Data Warehouse (CDW) were reviewed to select patients who (1) carried a diagnosis of prostate cancer, (2) successfully underwent tumor DNA sequencing through NPOP, (3) were prescribed olaparib, rucaparib, nirapib, and/or talazaporib for their prostate cancer between July 2016 and February 2020, and (4) and achieved DCB, defined as no progression in prostate-specific antigen (PSA) for at least 6 months following PARPi initiation without concurrent systemic or non-systemic therapies other than androgen-deprivation. The DNA repair gene variants and orders placed for NPOP consultative support were reviewed.
RESULTS: Of the 44 prostate cancer patients treated with a PARPi, 6 (13.6%) had tumor DNA sequencing through NPOP and had achieved DCB. Five patients were treated with olaparib and 1 with rucaparib. The median PSA progression-free survival was 8.9 (interquartile range = 8.5 – 11.2) months among these selected patients. Regarding gene variants, 5 patients had 7 BRCA2 mutations, including 4 frameshift, 1 nonsense, 1 single nucleotide variant, and 1 splice site. One patient had frameshift and missense ATM mutations. Referrals to the NPOP consult service were ordered for 2 out of the 5 patients with BRCA2 mutations achieving DCB.
CONCLUSIONS: Within the VA’s NPOP, the presence of BRCA2 gene variants was the most common finding from tumor DNA sequencing among patients with prostate cancer achieving DCB with a PARPi. Further analysis of the genotypes of all patients treated with PARPi in NPOP to assess the differential impact of BRCA2 mutations is needed to confirm the clinical implication of this finding.
BACKGROUND: PARP inhibitors (PARPi’s) were recently approved for the treatment of metastatic prostate cancer among patients harboring mutations in an array of genes responsible for DNA repair. We sought to identify whether a subset of these genes correlates with response to treatment more frequently than others. Consequently, an evaluation of the specific DNA repair genotypes associated with durable clinical benefit (DCB) using real-world patient data was undertaken.
METHODS: The U.S. Department of Veterans Affairs (VA) National Precision Oncology Program’s (NPOP) database and Corporate Data Warehouse (CDW) were reviewed to select patients who (1) carried a diagnosis of prostate cancer, (2) successfully underwent tumor DNA sequencing through NPOP, (3) were prescribed olaparib, rucaparib, nirapib, and/or talazaporib for their prostate cancer between July 2016 and February 2020, and (4) and achieved DCB, defined as no progression in prostate-specific antigen (PSA) for at least 6 months following PARPi initiation without concurrent systemic or non-systemic therapies other than androgen-deprivation. The DNA repair gene variants and orders placed for NPOP consultative support were reviewed.
RESULTS: Of the 44 prostate cancer patients treated with a PARPi, 6 (13.6%) had tumor DNA sequencing through NPOP and had achieved DCB. Five patients were treated with olaparib and 1 with rucaparib. The median PSA progression-free survival was 8.9 (interquartile range = 8.5 – 11.2) months among these selected patients. Regarding gene variants, 5 patients had 7 BRCA2 mutations, including 4 frameshift, 1 nonsense, 1 single nucleotide variant, and 1 splice site. One patient had frameshift and missense ATM mutations. Referrals to the NPOP consult service were ordered for 2 out of the 5 patients with BRCA2 mutations achieving DCB.
CONCLUSIONS: Within the VA’s NPOP, the presence of BRCA2 gene variants was the most common finding from tumor DNA sequencing among patients with prostate cancer achieving DCB with a PARPi. Further analysis of the genotypes of all patients treated with PARPi in NPOP to assess the differential impact of BRCA2 mutations is needed to confirm the clinical implication of this finding.
VHA Practice Guideline Recommendations for Diffuse Gliomas (FULL)
Over the past few decades, our understanding of the molecular underpinning of primary neoplasms of the central nervous system (CNS) has progressed substantially. Thanks in large part to this expansion in our knowledge base, the World Health Organization (WHO) has recently updated its classification of tumors of the CNS.1 One of the key elements of this update was the inclusion of molecular diagnostic criteria for the classification of infiltrating gliomas. While the previous classification system was based upon histologic subtypes of the tumor (astrocytoma, oligodendroglioma, and oligoastrocytoma), the revised classification system incorporates molecular testing to establish the genetic characteristics of the tumor to reach a final integrated diagnosis.
In this article, we present 3 cases to highlight some of these recent changes in the WHO diagnostic categories of primary CNS tumors and to illustrate the role of specific molecular tests in reaching a final integrated diagnosis. We then propose a clinical practice guideline for the Veterans Health Administration (VHA) that recommends use of molecular testing for veterans as part of the diagnostic workup of primary CNS neoplasms.
Purpose
In 2013 the VHA National Director of Pathology & Laboratory Medicine Services (P&LMS) chartered a national molecular genetics pathology workgroup (MGPW) that was charged with 4 specific tasks: (1) Provide recommendations about the effective use of molecular genetic testing for veterans; (2) Promote increased quality and availability of molecular testing within the VHA; (3) Encourage internal referral testing; and (4) Create an organizational structure and policies for molecular genetic testing and laboratory developed tests. The workgroup is currently composed of 4 subcommittees: genetic medicine, hematopathology, pharmacogenomics, and molecular oncology. The molecular oncology subcommittee is focused upon molecular genetic testing for solid tumors.
This article is intended to be the first of several publications from the molecular oncology subcommittee of the MGPW that address some of the aforementioned tasks. Similar to the recent publication from the hematopathology subcommittee of the MGPW, this article focuses on CNS neoplasms.2
Scope of Problem
The incidence of tumors of the CNS in the US population varies among age groups. It is the most common solid tumor in children aged < 14 years and represents a significant cause of mortality across all age groups.3 Of CNS tumors, diffuse gliomas comprise about 20% of the tumors and more than 70% of the primary malignant CNS tumors.3 Analysis of the VA Central Cancer Registry data from 2010 to 2014 identified 1,186 veterans (about 237 veterans per year) who were diagnosed with diffuse gliomas. (Lynch, Kulich, Colman, unpublished data, February 2018). While the majority (nearly 80%) of these cases were glioblastomas (GBMs), unfortunately a majority of these cases did not undergo molecular testing (Lynch, Kulich, Colman, unpublished data, February 2018).
Although this low rate of testing may be in part reflective of the period from which these data were gleaned (ie, prior to the WHO release of their updated the classification of tumors of the CNS), it is important to raise VA practitioners’ awareness of these recent changes to ensure that veterans receive the proper diagnosis and treatment for their disease. Thus, while the number of veterans diagnosed with diffuse gliomas within the VHA is relatively small in comparison to other malignancies, such as prostatic adenocarcinomas and lung carcinomas, the majority of diffuse gliomas do not seem to be receiving the molecular testing that would be necessary for (1) appropriate classification under the recently revised WHO recommendations; and (2) making important treatment decisions.
Case Presentations
Case 1. A veteran of the Gulf War presented with a 3-month history of possible narcoleptic events associated with a motor vehicle accident. Magnetic resonance imaging (MRI) revealed a large left frontal mass lesion with minimal surrounding edema without appreciable contrast enhancement (Figures 1A, 1B, and 1C). 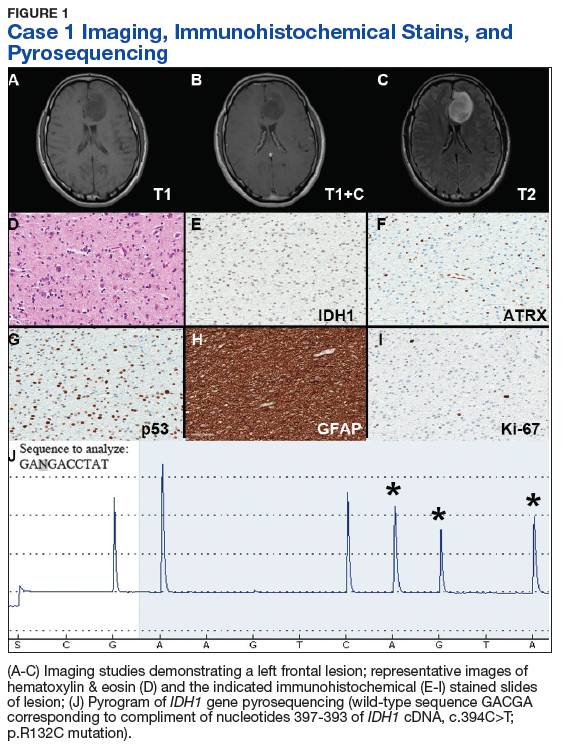
Neither mitotic figures nor endothelial proliferation were identified. Immunohistochemical stains revealed a lack of R132H mutant IDH1 protein expression, a loss of nuclear staining for ATRX protein within a substantial number of cells, and a clonal pattern of p53 protein overexpression (Figures 1E, 1F, and 1G). The lesion demonstrated diffuse glial fibrillary acidic protein (GFAP) immunoreactivity and a low proliferation index (as determined by Ki-67 staining; estimated at less than 5%) (Figures 1H and 1I).
Based upon these results, an initial morphologic diagnosis of diffuse glioma was issued, and tissue was subjected to a variety of nucleic acid-based tests. While fluorescence in situ hybridization (FISH) studies were negative for 1p/19q codeletion, pyrosequencing analysis revealed the presence of a c.394C>T (R132C) mutation of the IDH1 gene (Figure 1J). The University of Pittsburgh Medical Center’s GlioSeq targeted next-generation sequence (NGS) analysis confirmed the presence of the c.394C > T mutation in IDH1 gene.4 Based upon this additional information, a final integrated morphologic and molecular diagnosis of diffuse astrocytoma, IDH-mutant was rendered.
Case 2. A Vietnam War veteran presented with a 6-week history of new onset falls with associated left lower extremity weakness. A MRI revealed a right frontoparietal mass lesion with surrounding edema without appreciable contrast enhancement (Figures 2A, 2B, and 2C). 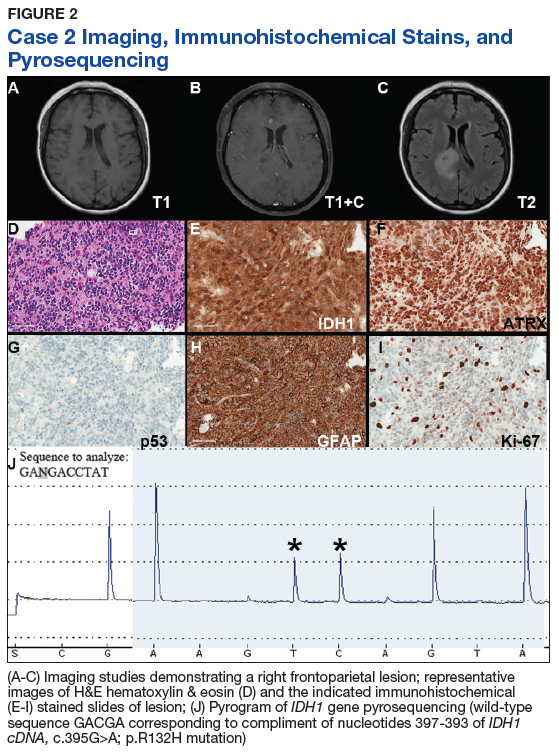
Immunohistochemical stains revealed R132H mutant IDH1 protein expression, retention of nuclear staining for ATRX protein, the lack of a clonal pattern of p53 protein overexpression, diffuse GFAP immunoreactivity, and a proliferation index (as determined by Ki-67 staining) focally approaching 20% (Figures 2E, 2F, 2G, 2H and 2I).
Based upon these results, an initial morphologic diagnosis of diffuse (high grade) glioma was issued, and tissue was subjected to a variety of nucleic acid-based tests. The FISH studies were positive for 1p/19q codeletion, and pyrosequencing analysis confirmed the immunohistochemical findings of a c.395G>A (R132H) mutation of the IDH1 gene (Figure 2J). GlioSeq targeted NGS analysis confirmed the presence of the c.395G>A mutation in the IDH1 gene, a mutation in the telomerase reverse transcriptase (TERT) promoter, and possible decreased copy number of the CIC (chromosome 1p) and FUBP1 (chromosome 19q) genes.
A final integrated morphologic and molecular diagnosis of anaplastic oligodendroglioma, IDH-mutant and 1p/19q-codeleted was rendered based on the additional information. With this final diagnosis, methylation analysis of the MGMT gene promoter, which was performed for prognostic and predictive purposes, was identified in this case.5,6
Case 3. A veteran of the Vietnam War presented with a new onset seizure. A MRI revealed a focally contrast-enhancing mass with surrounding edema within the left frontal lobe (Figures 3A, 3B, and 3C). 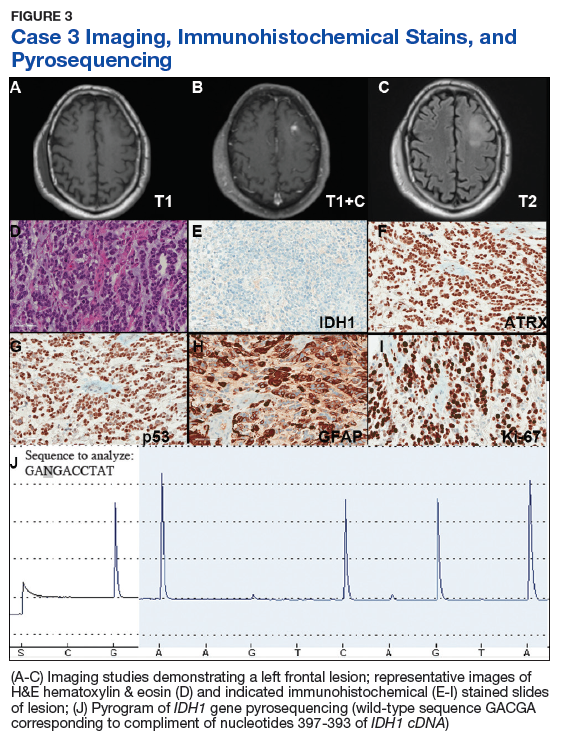
Hematoxylin and eosin (H&E) stained sections following formalin fixation and paraffin embedding demonstrated similar findings (Figure 3D), and while mitotic figures were readily identified, areas of necrosis were not identified and endothelial proliferation was not a prominent feature. Immunohistochemical stains revealed no evidence of R132H mutant IDH1 protein expression, retention of nuclear staining for ATRX protein, a clonal pattern of p53 protein overexpression, patchy GFAP immunoreactivity, and a proliferation index (as determined by Ki-67 staining) focally approaching 50% (Figures 3E, 3F, 3G, 3H, and 3I).
Based upon these results, an initial morphologic diagnosis of diffuse (high grade) glioma was issued, and the tissue was subjected to a variety of nucleic acid-based tests. The FISH studies were negative for EGFR gene amplification and 1p/19q codeletion, although a gain of the long arm of chromosome 1 was detected. Pyrosequencing analysis for mutations in codon 132 of the IDH1 gene revealed no mutations (Figure 3J). GlioSeq targeted NGS analysis identified mutations within the NF1, TP53, and PIK3CA genes without evidence of mutations in the IDH1, IDH2, ATRX, H3F3A, or EGFR genes or the TERT promoter. Based upon this additional information, a final integrated morphologic and molecular diagnosis of GBM, IDH wild-type was issued. The MGMT gene promoter was negative for methylation, a finding that has prognostic and predictive impact with regard to treatment with temazolamide.7-9
New Diffuse Glioma Classification
Since the issuance of the previous edition of the WHO classification of CNS tumors in 2007, several sentinel discoveries have been made that have advanced our understanding of the underlying biology of primary CNS neoplasms. Since a detailed review of these findings is beyond the scope and purpose of this manuscript and salient reviews on the topic can be found elsewhere, we will focus on the molecular findings that have been incorporated into the recently revised WHO classification.10 The importance of providing such information for proper patient management is illustrated by the recent acknowledgement by the American Academy of Neurology that molecular testing of brain tumors is a specific area in which there is a need for quality improvement.11 Therefore, it is critical that these underlying molecular abnormalities are identified to allow for proper classification and treatment of diffuse gliomas in the veteran population.
As noted previously, based on VA cancer registry data, diffuse gliomas are the most commonly encountered primary CNS cancers in the veteran population. Several of the aforementioned seminal discoveries have been incorporated into the updated classification of diffuse gliomas. While the recently updated WHO classification allows for the assignment of “not otherwise specified (NOS)” diagnostic designation, this category must be limited to cases where there is insufficient data to allow for a more precise classification due to sample limitations and not simply due to a failure of VA pathology laboratories to pursue the appropriate diagnostic testing.
Figure 4 presents the recommended diagnostic workflow for the workup of diffuse gliomas. As illustrated in the above cases, a variety of different methodologies, including immunohistochemical, FISH, loss of heterozygosity analysis, traditional and NGS may be applied when elucidating the status of molecular events at critical diagnostic branch points. 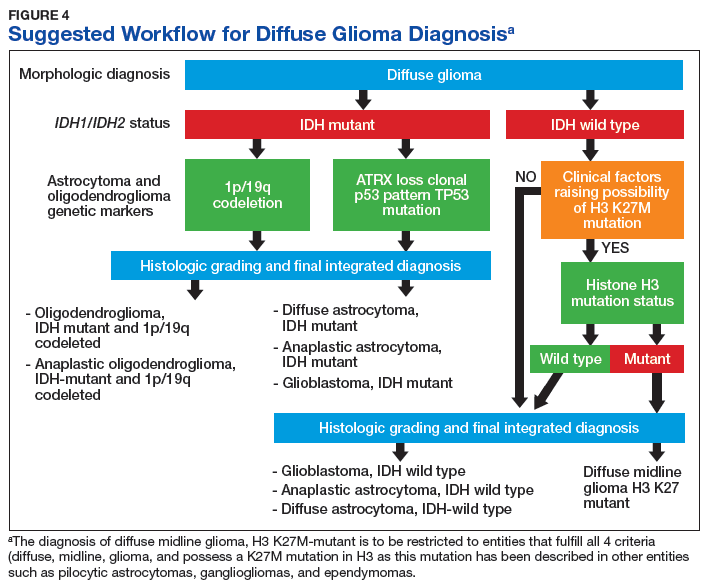
Diagnostic Uses of Molecular Testing
While the case studies in this article demonstrate the use of ancillary testing and provide a suggested strategy for properly subclassifying diffuse gliomas, inherent in this strategy is the assumption that, based upon the initial clinical and pathologic information available, one can accurately categorize the lesion as a diffuse glioma. In reality, such a distinction is not always a straightforward endeavor. It is well recognized that a proportion of low-grade, typically radiologically circumscribed, CNS neoplasms, such as pilocytic astrocytomas and glioneuronal tumors, may infiltrate the surrounding brain parenchyma. In addition, many of these low-grade CNS neoplasms also may have growth patterns that are shared with diffuse gliomas, a diagnostic challenge that often can be further hampered by the inherent limitations involved in obtaining adequate samples for diagnosis from the CNS.
Although there are limitations and caveats, molecular diagnostic testing may be invaluable in properly classifying CNS tumors in such situations. The finding of mutations in the IDH1 or IDH2 genes has been shown to be very valuable in distinguishing low-grade diffuse glioma from both nonneoplastic and low-grade circumscribed neuroepithelial neoplasms that may exhibit growth patterns that can mimic those of diffuse gliomas.15-17 Conversely, finding abnormalities in the BRAF gene in a brain neoplasm that has a low-grade morphology suggests that the lesion may represent one of these low-grade lesions such as a pleomorphic xanthoastrocytoma, pilocytic astrocytoma, or mixed neuronal-glial tumor as opposed to a diffuse glioma.18,19
Depending upon the environment in which one practices, small biopsy specimens may be prevalent, and unfortunately, it is not uncommon to obtain a biopsy that exhibits a histologic growth pattern that is discordant from what one would predict based on the clinical context and imaging findings. Molecular testing may be useful in resolving discordances in such situations. If a biopsy of a ring-enhancing lesion demonstrates a diffuse glioma that doesn’t meet WHO grade IV criteria, applying methodologies that look for genetic features commonly encountered in high-grade astrocytomas may identify genetic abnormalities that suggest a more aggressive lesion than is indicated by the histologic findings. The presence of genetic abnormalities such as homozygous deletion of the CDKN2A gene, TERT promoter mutation, loss of heterozygosity of chromosome 10q and/or phosphatase and tensin homolog (PTEN) mutations, EGFR gene amplification or the presence of the EGFR variant III are a few findings that would suggest the aforementioned sample may represent an undersampling of a higher grade diffuse astrocytoma, which would be important information to convey to the treating clinicians.20-26
Testing In the VA
The goals of the MPWG include promoting increased quality and availability of genetic testing within the VHA as well as encouraging internal referral testing. An informal survey of the chiefs of VA Pathology and Laboratory Medicine Services was conducted in November of 2017 in an attempt to identify internal VA pathology laboratories currently conducting testing that may be of use in the workup of diffuse gliomas (Table 1). 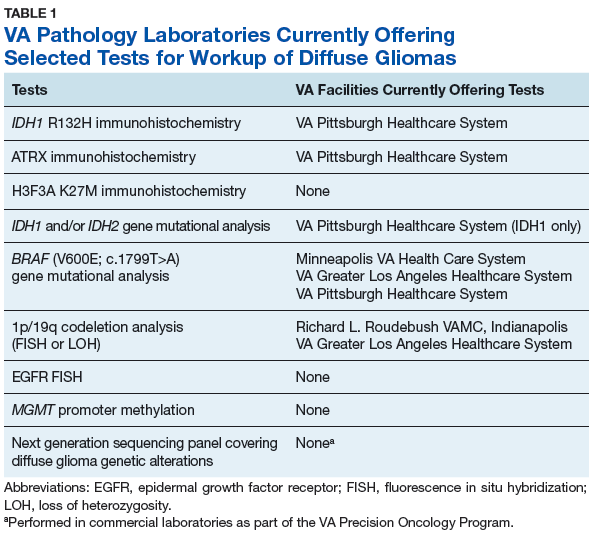
The VA currently offers NGS panels for patients with advanced-stage malignancies under the auspices of the Precision Oncology Program, whose reports provide both (1) mutational analyses for genes such as TP53, ATRX, NF1, BRAF, PTEN, TERT IDH1, and IDH2 that may be useful in the proper classifying of high-grade diffuse gliomas; and (2) information regarding clinical trials for which the veteran may be eligible for based on their glioma’s mutational profile. Interested VA providers should visit tinyurl.com/precisiononcology/ for more information about this program. Finally, although internal testing within VA laboratories is recommended to allow for the development of more cost-effective testing, testing may be performed through many nationally contracted reference laboratories.
Conclusion
In light of the recent progress made in our understanding of the molecular events of gliomagenesis, the way we diagnose diffuse gliomas within the CNS has undergone a major paradigm shift. While histology still plays a critical role in the process, we believe that additional ancillary testing is a requirement for all diffuse gliomas diagnosed within VA pathology laboratories. In the context of recently encountered cases, we have provided a recommended workflow highlighting the testing that can be performed to allow for the proper diagnosis of our veterans with diffuse gliomas (Figure 4).
Unless limited by the amount of tissue available for such tests, ancillary testing must be performed on all diffuse gliomas diagnosed within the VA system to ensure proper diagnosis and treatment of our veterans with diffuse gliomas. 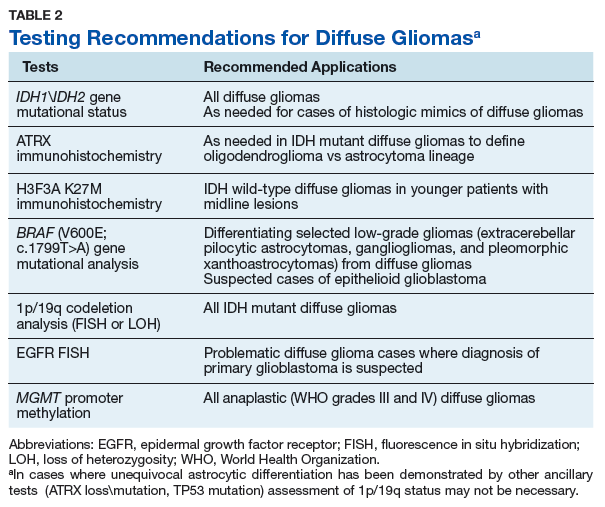

Acknowledgments
The authors thank Dr. Craig M. Horbinski (Feinberg School of Medicine, Northwestern University) and Dr. Geoffrey H. Murdoch (University of Pittsburgh) for their constructive criticism of the manuscript. We also thank the following individuals for past service as members of the molecular oncology subcommittee of the MGPW: Dr. George Ansstas (Washington University School of Medicine), Dr. Osssama Hemadeh (Bay Pines VA Health Care System), Dr. James Herman (VA Pittsburgh Healthcare System), and Dr. Ryan Phan (formerly of the VA Greater Los Angeles Healthcare System) as well as the members of the Veterans Administration pathology and laboratory medicine service molecular genetics pathology workgroup.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies.
Dr. Kulich is the Acting Chief of Pathology and Laboratory Medicine Service at VA Pittsburgh Healthcare System and member of the Division of Neuropathology at University of Pittsburgh Department of Pathology, Dr. Duvvuri is an Otolaryngologist at VA Pittsburgh Healthcare System, and Dr. Passero is the Section Chief of Hematology\Oncology at VA Pittsburgh Healthcare System in Pennsylvania. Dr. Becker is an Oncologist at VA-New York Harbor Healthcare System. Dr. Dacic is a Pathologist at University of Pittsburgh Department of Pathology in Pennsylvania. Dr. Ehsan is Chief of Pathology and Laboratory Medicine Services at the South Texas Veterans Healthcare System in San Antonio. Dr. Gutkin is the former Chief of Pathology and Laboratory Medicine Service at VA Pittsburgh Healthcare System. Dr. Hou is a Pathologist at St. Louis VA Medical Center in Missouri. Dr. Icardi is the VA National Director of Pathology and Laboratory Medicine Services. Dr. Lyle is a Pathologist at Bay Pine Health Care System in Florida. Dr. Lynch is an Investigator at VA Salt Lake Health Care System Informatics and Computing Infrastructure. Dr. Montgomery is an Oncologist at VA Puget Sound Health Care System, in Seattle, Washington. Dr. Przygodzki is the Director of Genomic Medicine Implementation and Associate Director of Genomic Medicine for the VA. Dr. Colman is a Neuro-Oncologist at George E. Wahlen VA Medical Center and the Director of Medical Neuro-Oncology at the Huntsman Cancer Institute, Salt Lake City, Utah.
Correspondence: Dr. Kulich (scott.kulich@va.gov)
1. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803-820.
2. Wang-Rodriguez J, Yunes A, Phan R, et al. The challenges of precision medicine and new advances in molecular diagnostic testing in hematolymphoid malignancies: impact on the VHA. Fed Pract. 2017;34(suppl 5):S38-S49.
3. Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol. 2017;19(suppl 5):v1-v88.
4. Nikiforova MN, Wald AI, Melan MA, et al. Targeted next-generation sequencing panel (GlioSeq) provides comprehensive genetic profiling of central nervous system tumors. Neuro Oncol. 2016;18(3)379-387.
5. Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90(19):1473-1479.
6. van den Bent MJ, Erdem-Eraslan L, Idbaih A, et al. MGMT-STP27 methylation status as predictive marker for response to PCV in anaplastic oligodendrogliomas and oligoastrocytomas. A report from EORTC study 26951. Clin Cancer Res. 2013;19(19):5513-5522.
7. Stupp R, Hegi ME, Mason WP, et al; European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459-466.
8. Malmstrom A, Gronberg BH, Marosi C, et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol. 2012;13(9):916-926.
9. van den Bent MJ, Kros JM. Predictive and prognostic markers in neuro-oncology. J Neuropathol Exp Neurol. 2007;66(12):1074-1081.
10. Chen R, Smith-Cohn M, Cohen AL, Colman H. Glioma subclassifications and their clinical significance. Neurotherapeutics. 2017;14(2):284-297.
11. Jordan JT, Sanders AE, Armstrong T, et al. Quality improvement in neurology: neuro-oncology quality measurement set. Neurology. 2018;90(14):652-658.
12. Chen L, Voronovich Z, Clark K, et al. Predicting the likelihood of an isocitrate dehydrogenase 1 or 2 mutation in diagnoses of infiltrative glioma. Neuro Oncol. 2014;16(11):1478-1483.
13. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997-1003.
14. Wick W, Platten M, Meisner C, et al; NOA-08 Study Group of Neuro-oncology Working Group (NOA) of German Cancer Society. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012;13(7):707-715.
15. Horbinski C, Kofler J, Kelly LM, Murdoch GH, Nikiforova MN. Diagnostic use of IDH1/2 mutation analysis in routine clinical testing of formalin-fixed, paraffin-embedded glioma tissues. J Neuropathol Exp Neurol. 2009;68(12):1319-1325.
16. Camelo-Piragua S, Jansen M, Ganguly A, Kim JC, Louis DN, Nutt CL. Mutant IDH1-specific immunohistochemistry distinguishes diffuse astrocytoma from astrocytosis. Acta Neuropathol. 2010;119(4):509-511.
17. Horbinski C, Kofler J, Yeaney G, et al. Isocitrate dehydrogenase 1 analysis differentiates gangliogliomas from infiltrative gliomas. Brain Pathol. 2011;21(5):564-574.
18. Berghoff AS, Preusser M. BRAF alterations in brain tumours: molecular pathology and therapeutic opportunities. Curr Opin Neurol. 2014;27(6):689-696.
19. Korshunov A, Meyer J, Capper D, et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol. 2009;118(3):401-405.
20. Fuller CE, Schmidt RE, Roth KA, et al. Clinical utility of fluorescence in situ hybridization (FISH) in morphologically ambiguous gliomas with hybrid oligodendroglial/astrocytic features. J Neuropathol Exp Neurol. 2003;62(11):1118-1128.
21. Horbinski C. Practical molecular diagnostics in neuropathology: making a tough job a little easier. Semin Diagn Pathol. 2010;27(2):105-113.
22. Fuller GN, Bigner SH. Amplified cellular oncogenes in neoplasms of the human central nervous system. Mutat Res. 1992;276(3):299-306.
23. Brennan CW, Verhaak RG, McKenna A, et al; TCGA Research Network. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462-477.
24. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015;129(6):829-848.
25. Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110(15):6021-6026.
26. Nikiforova MN, Hamilton RL. Molecular diagnostics of gliomas. Arch Pathol Lab Med. 2011;135(5):558-568.
Over the past few decades, our understanding of the molecular underpinning of primary neoplasms of the central nervous system (CNS) has progressed substantially. Thanks in large part to this expansion in our knowledge base, the World Health Organization (WHO) has recently updated its classification of tumors of the CNS.1 One of the key elements of this update was the inclusion of molecular diagnostic criteria for the classification of infiltrating gliomas. While the previous classification system was based upon histologic subtypes of the tumor (astrocytoma, oligodendroglioma, and oligoastrocytoma), the revised classification system incorporates molecular testing to establish the genetic characteristics of the tumor to reach a final integrated diagnosis.
In this article, we present 3 cases to highlight some of these recent changes in the WHO diagnostic categories of primary CNS tumors and to illustrate the role of specific molecular tests in reaching a final integrated diagnosis. We then propose a clinical practice guideline for the Veterans Health Administration (VHA) that recommends use of molecular testing for veterans as part of the diagnostic workup of primary CNS neoplasms.
Purpose
In 2013 the VHA National Director of Pathology & Laboratory Medicine Services (P&LMS) chartered a national molecular genetics pathology workgroup (MGPW) that was charged with 4 specific tasks: (1) Provide recommendations about the effective use of molecular genetic testing for veterans; (2) Promote increased quality and availability of molecular testing within the VHA; (3) Encourage internal referral testing; and (4) Create an organizational structure and policies for molecular genetic testing and laboratory developed tests. The workgroup is currently composed of 4 subcommittees: genetic medicine, hematopathology, pharmacogenomics, and molecular oncology. The molecular oncology subcommittee is focused upon molecular genetic testing for solid tumors.
This article is intended to be the first of several publications from the molecular oncology subcommittee of the MGPW that address some of the aforementioned tasks. Similar to the recent publication from the hematopathology subcommittee of the MGPW, this article focuses on CNS neoplasms.2
Scope of Problem
The incidence of tumors of the CNS in the US population varies among age groups. It is the most common solid tumor in children aged < 14 years and represents a significant cause of mortality across all age groups.3 Of CNS tumors, diffuse gliomas comprise about 20% of the tumors and more than 70% of the primary malignant CNS tumors.3 Analysis of the VA Central Cancer Registry data from 2010 to 2014 identified 1,186 veterans (about 237 veterans per year) who were diagnosed with diffuse gliomas. (Lynch, Kulich, Colman, unpublished data, February 2018). While the majority (nearly 80%) of these cases were glioblastomas (GBMs), unfortunately a majority of these cases did not undergo molecular testing (Lynch, Kulich, Colman, unpublished data, February 2018).
Although this low rate of testing may be in part reflective of the period from which these data were gleaned (ie, prior to the WHO release of their updated the classification of tumors of the CNS), it is important to raise VA practitioners’ awareness of these recent changes to ensure that veterans receive the proper diagnosis and treatment for their disease. Thus, while the number of veterans diagnosed with diffuse gliomas within the VHA is relatively small in comparison to other malignancies, such as prostatic adenocarcinomas and lung carcinomas, the majority of diffuse gliomas do not seem to be receiving the molecular testing that would be necessary for (1) appropriate classification under the recently revised WHO recommendations; and (2) making important treatment decisions.
Case Presentations
Case 1. A veteran of the Gulf War presented with a 3-month history of possible narcoleptic events associated with a motor vehicle accident. Magnetic resonance imaging (MRI) revealed a large left frontal mass lesion with minimal surrounding edema without appreciable contrast enhancement (Figures 1A, 1B, and 1C).
Neither mitotic figures nor endothelial proliferation were identified. Immunohistochemical stains revealed a lack of R132H mutant IDH1 protein expression, a loss of nuclear staining for ATRX protein within a substantial number of cells, and a clonal pattern of p53 protein overexpression (Figures 1E, 1F, and 1G). The lesion demonstrated diffuse glial fibrillary acidic protein (GFAP) immunoreactivity and a low proliferation index (as determined by Ki-67 staining; estimated at less than 5%) (Figures 1H and 1I).
Based upon these results, an initial morphologic diagnosis of diffuse glioma was issued, and tissue was subjected to a variety of nucleic acid-based tests. While fluorescence in situ hybridization (FISH) studies were negative for 1p/19q codeletion, pyrosequencing analysis revealed the presence of a c.394C>T (R132C) mutation of the IDH1 gene (Figure 1J). The University of Pittsburgh Medical Center’s GlioSeq targeted next-generation sequence (NGS) analysis confirmed the presence of the c.394C > T mutation in IDH1 gene.4 Based upon this additional information, a final integrated morphologic and molecular diagnosis of diffuse astrocytoma, IDH-mutant was rendered.
Case 2. A Vietnam War veteran presented with a 6-week history of new onset falls with associated left lower extremity weakness. A MRI revealed a right frontoparietal mass lesion with surrounding edema without appreciable contrast enhancement (Figures 2A, 2B, and 2C).
Immunohistochemical stains revealed R132H mutant IDH1 protein expression, retention of nuclear staining for ATRX protein, the lack of a clonal pattern of p53 protein overexpression, diffuse GFAP immunoreactivity, and a proliferation index (as determined by Ki-67 staining) focally approaching 20% (Figures 2E, 2F, 2G, 2H and 2I).
Based upon these results, an initial morphologic diagnosis of diffuse (high grade) glioma was issued, and tissue was subjected to a variety of nucleic acid-based tests. The FISH studies were positive for 1p/19q codeletion, and pyrosequencing analysis confirmed the immunohistochemical findings of a c.395G>A (R132H) mutation of the IDH1 gene (Figure 2J). GlioSeq targeted NGS analysis confirmed the presence of the c.395G>A mutation in the IDH1 gene, a mutation in the telomerase reverse transcriptase (TERT) promoter, and possible decreased copy number of the CIC (chromosome 1p) and FUBP1 (chromosome 19q) genes.
A final integrated morphologic and molecular diagnosis of anaplastic oligodendroglioma, IDH-mutant and 1p/19q-codeleted was rendered based on the additional information. With this final diagnosis, methylation analysis of the MGMT gene promoter, which was performed for prognostic and predictive purposes, was identified in this case.5,6
Case 3. A veteran of the Vietnam War presented with a new onset seizure. A MRI revealed a focally contrast-enhancing mass with surrounding edema within the left frontal lobe (Figures 3A, 3B, and 3C).
Hematoxylin and eosin (H&E) stained sections following formalin fixation and paraffin embedding demonstrated similar findings (Figure 3D), and while mitotic figures were readily identified, areas of necrosis were not identified and endothelial proliferation was not a prominent feature. Immunohistochemical stains revealed no evidence of R132H mutant IDH1 protein expression, retention of nuclear staining for ATRX protein, a clonal pattern of p53 protein overexpression, patchy GFAP immunoreactivity, and a proliferation index (as determined by Ki-67 staining) focally approaching 50% (Figures 3E, 3F, 3G, 3H, and 3I).
Based upon these results, an initial morphologic diagnosis of diffuse (high grade) glioma was issued, and the tissue was subjected to a variety of nucleic acid-based tests. The FISH studies were negative for EGFR gene amplification and 1p/19q codeletion, although a gain of the long arm of chromosome 1 was detected. Pyrosequencing analysis for mutations in codon 132 of the IDH1 gene revealed no mutations (Figure 3J). GlioSeq targeted NGS analysis identified mutations within the NF1, TP53, and PIK3CA genes without evidence of mutations in the IDH1, IDH2, ATRX, H3F3A, or EGFR genes or the TERT promoter. Based upon this additional information, a final integrated morphologic and molecular diagnosis of GBM, IDH wild-type was issued. The MGMT gene promoter was negative for methylation, a finding that has prognostic and predictive impact with regard to treatment with temazolamide.7-9
New Diffuse Glioma Classification
Since the issuance of the previous edition of the WHO classification of CNS tumors in 2007, several sentinel discoveries have been made that have advanced our understanding of the underlying biology of primary CNS neoplasms. Since a detailed review of these findings is beyond the scope and purpose of this manuscript and salient reviews on the topic can be found elsewhere, we will focus on the molecular findings that have been incorporated into the recently revised WHO classification.10 The importance of providing such information for proper patient management is illustrated by the recent acknowledgement by the American Academy of Neurology that molecular testing of brain tumors is a specific area in which there is a need for quality improvement.11 Therefore, it is critical that these underlying molecular abnormalities are identified to allow for proper classification and treatment of diffuse gliomas in the veteran population.
As noted previously, based on VA cancer registry data, diffuse gliomas are the most commonly encountered primary CNS cancers in the veteran population. Several of the aforementioned seminal discoveries have been incorporated into the updated classification of diffuse gliomas. While the recently updated WHO classification allows for the assignment of “not otherwise specified (NOS)” diagnostic designation, this category must be limited to cases where there is insufficient data to allow for a more precise classification due to sample limitations and not simply due to a failure of VA pathology laboratories to pursue the appropriate diagnostic testing.
Figure 4 presents the recommended diagnostic workflow for the workup of diffuse gliomas. As illustrated in the above cases, a variety of different methodologies, including immunohistochemical, FISH, loss of heterozygosity analysis, traditional and NGS may be applied when elucidating the status of molecular events at critical diagnostic branch points.
Diagnostic Uses of Molecular Testing
While the case studies in this article demonstrate the use of ancillary testing and provide a suggested strategy for properly subclassifying diffuse gliomas, inherent in this strategy is the assumption that, based upon the initial clinical and pathologic information available, one can accurately categorize the lesion as a diffuse glioma. In reality, such a distinction is not always a straightforward endeavor. It is well recognized that a proportion of low-grade, typically radiologically circumscribed, CNS neoplasms, such as pilocytic astrocytomas and glioneuronal tumors, may infiltrate the surrounding brain parenchyma. In addition, many of these low-grade CNS neoplasms also may have growth patterns that are shared with diffuse gliomas, a diagnostic challenge that often can be further hampered by the inherent limitations involved in obtaining adequate samples for diagnosis from the CNS.
Although there are limitations and caveats, molecular diagnostic testing may be invaluable in properly classifying CNS tumors in such situations. The finding of mutations in the IDH1 or IDH2 genes has been shown to be very valuable in distinguishing low-grade diffuse glioma from both nonneoplastic and low-grade circumscribed neuroepithelial neoplasms that may exhibit growth patterns that can mimic those of diffuse gliomas.15-17 Conversely, finding abnormalities in the BRAF gene in a brain neoplasm that has a low-grade morphology suggests that the lesion may represent one of these low-grade lesions such as a pleomorphic xanthoastrocytoma, pilocytic astrocytoma, or mixed neuronal-glial tumor as opposed to a diffuse glioma.18,19
Depending upon the environment in which one practices, small biopsy specimens may be prevalent, and unfortunately, it is not uncommon to obtain a biopsy that exhibits a histologic growth pattern that is discordant from what one would predict based on the clinical context and imaging findings. Molecular testing may be useful in resolving discordances in such situations. If a biopsy of a ring-enhancing lesion demonstrates a diffuse glioma that doesn’t meet WHO grade IV criteria, applying methodologies that look for genetic features commonly encountered in high-grade astrocytomas may identify genetic abnormalities that suggest a more aggressive lesion than is indicated by the histologic findings. The presence of genetic abnormalities such as homozygous deletion of the CDKN2A gene, TERT promoter mutation, loss of heterozygosity of chromosome 10q and/or phosphatase and tensin homolog (PTEN) mutations, EGFR gene amplification or the presence of the EGFR variant III are a few findings that would suggest the aforementioned sample may represent an undersampling of a higher grade diffuse astrocytoma, which would be important information to convey to the treating clinicians.20-26
Testing In the VA
The goals of the MPWG include promoting increased quality and availability of genetic testing within the VHA as well as encouraging internal referral testing. An informal survey of the chiefs of VA Pathology and Laboratory Medicine Services was conducted in November of 2017 in an attempt to identify internal VA pathology laboratories currently conducting testing that may be of use in the workup of diffuse gliomas (Table 1).
The VA currently offers NGS panels for patients with advanced-stage malignancies under the auspices of the Precision Oncology Program, whose reports provide both (1) mutational analyses for genes such as TP53, ATRX, NF1, BRAF, PTEN, TERT IDH1, and IDH2 that may be useful in the proper classifying of high-grade diffuse gliomas; and (2) information regarding clinical trials for which the veteran may be eligible for based on their glioma’s mutational profile. Interested VA providers should visit tinyurl.com/precisiononcology/ for more information about this program. Finally, although internal testing within VA laboratories is recommended to allow for the development of more cost-effective testing, testing may be performed through many nationally contracted reference laboratories.
Conclusion
In light of the recent progress made in our understanding of the molecular events of gliomagenesis, the way we diagnose diffuse gliomas within the CNS has undergone a major paradigm shift. While histology still plays a critical role in the process, we believe that additional ancillary testing is a requirement for all diffuse gliomas diagnosed within VA pathology laboratories. In the context of recently encountered cases, we have provided a recommended workflow highlighting the testing that can be performed to allow for the proper diagnosis of our veterans with diffuse gliomas (Figure 4).
Unless limited by the amount of tissue available for such tests, ancillary testing must be performed on all diffuse gliomas diagnosed within the VA system to ensure proper diagnosis and treatment of our veterans with diffuse gliomas.
Acknowledgments
The authors thank Dr. Craig M. Horbinski (Feinberg School of Medicine, Northwestern University) and Dr. Geoffrey H. Murdoch (University of Pittsburgh) for their constructive criticism of the manuscript. We also thank the following individuals for past service as members of the molecular oncology subcommittee of the MGPW: Dr. George Ansstas (Washington University School of Medicine), Dr. Osssama Hemadeh (Bay Pines VA Health Care System), Dr. James Herman (VA Pittsburgh Healthcare System), and Dr. Ryan Phan (formerly of the VA Greater Los Angeles Healthcare System) as well as the members of the Veterans Administration pathology and laboratory medicine service molecular genetics pathology workgroup.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies.
Dr. Kulich is the Acting Chief of Pathology and Laboratory Medicine Service at VA Pittsburgh Healthcare System and member of the Division of Neuropathology at University of Pittsburgh Department of Pathology, Dr. Duvvuri is an Otolaryngologist at VA Pittsburgh Healthcare System, and Dr. Passero is the Section Chief of Hematology\Oncology at VA Pittsburgh Healthcare System in Pennsylvania. Dr. Becker is an Oncologist at VA-New York Harbor Healthcare System. Dr. Dacic is a Pathologist at University of Pittsburgh Department of Pathology in Pennsylvania. Dr. Ehsan is Chief of Pathology and Laboratory Medicine Services at the South Texas Veterans Healthcare System in San Antonio. Dr. Gutkin is the former Chief of Pathology and Laboratory Medicine Service at VA Pittsburgh Healthcare System. Dr. Hou is a Pathologist at St. Louis VA Medical Center in Missouri. Dr. Icardi is the VA National Director of Pathology and Laboratory Medicine Services. Dr. Lyle is a Pathologist at Bay Pine Health Care System in Florida. Dr. Lynch is an Investigator at VA Salt Lake Health Care System Informatics and Computing Infrastructure. Dr. Montgomery is an Oncologist at VA Puget Sound Health Care System, in Seattle, Washington. Dr. Przygodzki is the Director of Genomic Medicine Implementation and Associate Director of Genomic Medicine for the VA. Dr. Colman is a Neuro-Oncologist at George E. Wahlen VA Medical Center and the Director of Medical Neuro-Oncology at the Huntsman Cancer Institute, Salt Lake City, Utah.
Correspondence: Dr. Kulich (scott.kulich@va.gov)
Over the past few decades, our understanding of the molecular underpinning of primary neoplasms of the central nervous system (CNS) has progressed substantially. Thanks in large part to this expansion in our knowledge base, the World Health Organization (WHO) has recently updated its classification of tumors of the CNS.1 One of the key elements of this update was the inclusion of molecular diagnostic criteria for the classification of infiltrating gliomas. While the previous classification system was based upon histologic subtypes of the tumor (astrocytoma, oligodendroglioma, and oligoastrocytoma), the revised classification system incorporates molecular testing to establish the genetic characteristics of the tumor to reach a final integrated diagnosis.
In this article, we present 3 cases to highlight some of these recent changes in the WHO diagnostic categories of primary CNS tumors and to illustrate the role of specific molecular tests in reaching a final integrated diagnosis. We then propose a clinical practice guideline for the Veterans Health Administration (VHA) that recommends use of molecular testing for veterans as part of the diagnostic workup of primary CNS neoplasms.
Purpose
In 2013 the VHA National Director of Pathology & Laboratory Medicine Services (P&LMS) chartered a national molecular genetics pathology workgroup (MGPW) that was charged with 4 specific tasks: (1) Provide recommendations about the effective use of molecular genetic testing for veterans; (2) Promote increased quality and availability of molecular testing within the VHA; (3) Encourage internal referral testing; and (4) Create an organizational structure and policies for molecular genetic testing and laboratory developed tests. The workgroup is currently composed of 4 subcommittees: genetic medicine, hematopathology, pharmacogenomics, and molecular oncology. The molecular oncology subcommittee is focused upon molecular genetic testing for solid tumors.
This article is intended to be the first of several publications from the molecular oncology subcommittee of the MGPW that address some of the aforementioned tasks. Similar to the recent publication from the hematopathology subcommittee of the MGPW, this article focuses on CNS neoplasms.2
Scope of Problem
The incidence of tumors of the CNS in the US population varies among age groups. It is the most common solid tumor in children aged < 14 years and represents a significant cause of mortality across all age groups.3 Of CNS tumors, diffuse gliomas comprise about 20% of the tumors and more than 70% of the primary malignant CNS tumors.3 Analysis of the VA Central Cancer Registry data from 2010 to 2014 identified 1,186 veterans (about 237 veterans per year) who were diagnosed with diffuse gliomas. (Lynch, Kulich, Colman, unpublished data, February 2018). While the majority (nearly 80%) of these cases were glioblastomas (GBMs), unfortunately a majority of these cases did not undergo molecular testing (Lynch, Kulich, Colman, unpublished data, February 2018).
Although this low rate of testing may be in part reflective of the period from which these data were gleaned (ie, prior to the WHO release of their updated the classification of tumors of the CNS), it is important to raise VA practitioners’ awareness of these recent changes to ensure that veterans receive the proper diagnosis and treatment for their disease. Thus, while the number of veterans diagnosed with diffuse gliomas within the VHA is relatively small in comparison to other malignancies, such as prostatic adenocarcinomas and lung carcinomas, the majority of diffuse gliomas do not seem to be receiving the molecular testing that would be necessary for (1) appropriate classification under the recently revised WHO recommendations; and (2) making important treatment decisions.
Case Presentations
Case 1. A veteran of the Gulf War presented with a 3-month history of possible narcoleptic events associated with a motor vehicle accident. Magnetic resonance imaging (MRI) revealed a large left frontal mass lesion with minimal surrounding edema without appreciable contrast enhancement (Figures 1A, 1B, and 1C).
Neither mitotic figures nor endothelial proliferation were identified. Immunohistochemical stains revealed a lack of R132H mutant IDH1 protein expression, a loss of nuclear staining for ATRX protein within a substantial number of cells, and a clonal pattern of p53 protein overexpression (Figures 1E, 1F, and 1G). The lesion demonstrated diffuse glial fibrillary acidic protein (GFAP) immunoreactivity and a low proliferation index (as determined by Ki-67 staining; estimated at less than 5%) (Figures 1H and 1I).
Based upon these results, an initial morphologic diagnosis of diffuse glioma was issued, and tissue was subjected to a variety of nucleic acid-based tests. While fluorescence in situ hybridization (FISH) studies were negative for 1p/19q codeletion, pyrosequencing analysis revealed the presence of a c.394C>T (R132C) mutation of the IDH1 gene (Figure 1J). The University of Pittsburgh Medical Center’s GlioSeq targeted next-generation sequence (NGS) analysis confirmed the presence of the c.394C > T mutation in IDH1 gene.4 Based upon this additional information, a final integrated morphologic and molecular diagnosis of diffuse astrocytoma, IDH-mutant was rendered.
Case 2. A Vietnam War veteran presented with a 6-week history of new onset falls with associated left lower extremity weakness. A MRI revealed a right frontoparietal mass lesion with surrounding edema without appreciable contrast enhancement (Figures 2A, 2B, and 2C).
Immunohistochemical stains revealed R132H mutant IDH1 protein expression, retention of nuclear staining for ATRX protein, the lack of a clonal pattern of p53 protein overexpression, diffuse GFAP immunoreactivity, and a proliferation index (as determined by Ki-67 staining) focally approaching 20% (Figures 2E, 2F, 2G, 2H and 2I).
Based upon these results, an initial morphologic diagnosis of diffuse (high grade) glioma was issued, and tissue was subjected to a variety of nucleic acid-based tests. The FISH studies were positive for 1p/19q codeletion, and pyrosequencing analysis confirmed the immunohistochemical findings of a c.395G>A (R132H) mutation of the IDH1 gene (Figure 2J). GlioSeq targeted NGS analysis confirmed the presence of the c.395G>A mutation in the IDH1 gene, a mutation in the telomerase reverse transcriptase (TERT) promoter, and possible decreased copy number of the CIC (chromosome 1p) and FUBP1 (chromosome 19q) genes.
A final integrated morphologic and molecular diagnosis of anaplastic oligodendroglioma, IDH-mutant and 1p/19q-codeleted was rendered based on the additional information. With this final diagnosis, methylation analysis of the MGMT gene promoter, which was performed for prognostic and predictive purposes, was identified in this case.5,6
Case 3. A veteran of the Vietnam War presented with a new onset seizure. A MRI revealed a focally contrast-enhancing mass with surrounding edema within the left frontal lobe (Figures 3A, 3B, and 3C).
Hematoxylin and eosin (H&E) stained sections following formalin fixation and paraffin embedding demonstrated similar findings (Figure 3D), and while mitotic figures were readily identified, areas of necrosis were not identified and endothelial proliferation was not a prominent feature. Immunohistochemical stains revealed no evidence of R132H mutant IDH1 protein expression, retention of nuclear staining for ATRX protein, a clonal pattern of p53 protein overexpression, patchy GFAP immunoreactivity, and a proliferation index (as determined by Ki-67 staining) focally approaching 50% (Figures 3E, 3F, 3G, 3H, and 3I).
Based upon these results, an initial morphologic diagnosis of diffuse (high grade) glioma was issued, and the tissue was subjected to a variety of nucleic acid-based tests. The FISH studies were negative for EGFR gene amplification and 1p/19q codeletion, although a gain of the long arm of chromosome 1 was detected. Pyrosequencing analysis for mutations in codon 132 of the IDH1 gene revealed no mutations (Figure 3J). GlioSeq targeted NGS analysis identified mutations within the NF1, TP53, and PIK3CA genes without evidence of mutations in the IDH1, IDH2, ATRX, H3F3A, or EGFR genes or the TERT promoter. Based upon this additional information, a final integrated morphologic and molecular diagnosis of GBM, IDH wild-type was issued. The MGMT gene promoter was negative for methylation, a finding that has prognostic and predictive impact with regard to treatment with temazolamide.7-9
New Diffuse Glioma Classification
Since the issuance of the previous edition of the WHO classification of CNS tumors in 2007, several sentinel discoveries have been made that have advanced our understanding of the underlying biology of primary CNS neoplasms. Since a detailed review of these findings is beyond the scope and purpose of this manuscript and salient reviews on the topic can be found elsewhere, we will focus on the molecular findings that have been incorporated into the recently revised WHO classification.10 The importance of providing such information for proper patient management is illustrated by the recent acknowledgement by the American Academy of Neurology that molecular testing of brain tumors is a specific area in which there is a need for quality improvement.11 Therefore, it is critical that these underlying molecular abnormalities are identified to allow for proper classification and treatment of diffuse gliomas in the veteran population.
As noted previously, based on VA cancer registry data, diffuse gliomas are the most commonly encountered primary CNS cancers in the veteran population. Several of the aforementioned seminal discoveries have been incorporated into the updated classification of diffuse gliomas. While the recently updated WHO classification allows for the assignment of “not otherwise specified (NOS)” diagnostic designation, this category must be limited to cases where there is insufficient data to allow for a more precise classification due to sample limitations and not simply due to a failure of VA pathology laboratories to pursue the appropriate diagnostic testing.
Figure 4 presents the recommended diagnostic workflow for the workup of diffuse gliomas. As illustrated in the above cases, a variety of different methodologies, including immunohistochemical, FISH, loss of heterozygosity analysis, traditional and NGS may be applied when elucidating the status of molecular events at critical diagnostic branch points.
Diagnostic Uses of Molecular Testing
While the case studies in this article demonstrate the use of ancillary testing and provide a suggested strategy for properly subclassifying diffuse gliomas, inherent in this strategy is the assumption that, based upon the initial clinical and pathologic information available, one can accurately categorize the lesion as a diffuse glioma. In reality, such a distinction is not always a straightforward endeavor. It is well recognized that a proportion of low-grade, typically radiologically circumscribed, CNS neoplasms, such as pilocytic astrocytomas and glioneuronal tumors, may infiltrate the surrounding brain parenchyma. In addition, many of these low-grade CNS neoplasms also may have growth patterns that are shared with diffuse gliomas, a diagnostic challenge that often can be further hampered by the inherent limitations involved in obtaining adequate samples for diagnosis from the CNS.
Although there are limitations and caveats, molecular diagnostic testing may be invaluable in properly classifying CNS tumors in such situations. The finding of mutations in the IDH1 or IDH2 genes has been shown to be very valuable in distinguishing low-grade diffuse glioma from both nonneoplastic and low-grade circumscribed neuroepithelial neoplasms that may exhibit growth patterns that can mimic those of diffuse gliomas.15-17 Conversely, finding abnormalities in the BRAF gene in a brain neoplasm that has a low-grade morphology suggests that the lesion may represent one of these low-grade lesions such as a pleomorphic xanthoastrocytoma, pilocytic astrocytoma, or mixed neuronal-glial tumor as opposed to a diffuse glioma.18,19
Depending upon the environment in which one practices, small biopsy specimens may be prevalent, and unfortunately, it is not uncommon to obtain a biopsy that exhibits a histologic growth pattern that is discordant from what one would predict based on the clinical context and imaging findings. Molecular testing may be useful in resolving discordances in such situations. If a biopsy of a ring-enhancing lesion demonstrates a diffuse glioma that doesn’t meet WHO grade IV criteria, applying methodologies that look for genetic features commonly encountered in high-grade astrocytomas may identify genetic abnormalities that suggest a more aggressive lesion than is indicated by the histologic findings. The presence of genetic abnormalities such as homozygous deletion of the CDKN2A gene, TERT promoter mutation, loss of heterozygosity of chromosome 10q and/or phosphatase and tensin homolog (PTEN) mutations, EGFR gene amplification or the presence of the EGFR variant III are a few findings that would suggest the aforementioned sample may represent an undersampling of a higher grade diffuse astrocytoma, which would be important information to convey to the treating clinicians.20-26
Testing In the VA
The goals of the MPWG include promoting increased quality and availability of genetic testing within the VHA as well as encouraging internal referral testing. An informal survey of the chiefs of VA Pathology and Laboratory Medicine Services was conducted in November of 2017 in an attempt to identify internal VA pathology laboratories currently conducting testing that may be of use in the workup of diffuse gliomas (Table 1).
The VA currently offers NGS panels for patients with advanced-stage malignancies under the auspices of the Precision Oncology Program, whose reports provide both (1) mutational analyses for genes such as TP53, ATRX, NF1, BRAF, PTEN, TERT IDH1, and IDH2 that may be useful in the proper classifying of high-grade diffuse gliomas; and (2) information regarding clinical trials for which the veteran may be eligible for based on their glioma’s mutational profile. Interested VA providers should visit tinyurl.com/precisiononcology/ for more information about this program. Finally, although internal testing within VA laboratories is recommended to allow for the development of more cost-effective testing, testing may be performed through many nationally contracted reference laboratories.
Conclusion
In light of the recent progress made in our understanding of the molecular events of gliomagenesis, the way we diagnose diffuse gliomas within the CNS has undergone a major paradigm shift. While histology still plays a critical role in the process, we believe that additional ancillary testing is a requirement for all diffuse gliomas diagnosed within VA pathology laboratories. In the context of recently encountered cases, we have provided a recommended workflow highlighting the testing that can be performed to allow for the proper diagnosis of our veterans with diffuse gliomas (Figure 4).
Unless limited by the amount of tissue available for such tests, ancillary testing must be performed on all diffuse gliomas diagnosed within the VA system to ensure proper diagnosis and treatment of our veterans with diffuse gliomas.
Acknowledgments
The authors thank Dr. Craig M. Horbinski (Feinberg School of Medicine, Northwestern University) and Dr. Geoffrey H. Murdoch (University of Pittsburgh) for their constructive criticism of the manuscript. We also thank the following individuals for past service as members of the molecular oncology subcommittee of the MGPW: Dr. George Ansstas (Washington University School of Medicine), Dr. Osssama Hemadeh (Bay Pines VA Health Care System), Dr. James Herman (VA Pittsburgh Healthcare System), and Dr. Ryan Phan (formerly of the VA Greater Los Angeles Healthcare System) as well as the members of the Veterans Administration pathology and laboratory medicine service molecular genetics pathology workgroup.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies.
Dr. Kulich is the Acting Chief of Pathology and Laboratory Medicine Service at VA Pittsburgh Healthcare System and member of the Division of Neuropathology at University of Pittsburgh Department of Pathology, Dr. Duvvuri is an Otolaryngologist at VA Pittsburgh Healthcare System, and Dr. Passero is the Section Chief of Hematology\Oncology at VA Pittsburgh Healthcare System in Pennsylvania. Dr. Becker is an Oncologist at VA-New York Harbor Healthcare System. Dr. Dacic is a Pathologist at University of Pittsburgh Department of Pathology in Pennsylvania. Dr. Ehsan is Chief of Pathology and Laboratory Medicine Services at the South Texas Veterans Healthcare System in San Antonio. Dr. Gutkin is the former Chief of Pathology and Laboratory Medicine Service at VA Pittsburgh Healthcare System. Dr. Hou is a Pathologist at St. Louis VA Medical Center in Missouri. Dr. Icardi is the VA National Director of Pathology and Laboratory Medicine Services. Dr. Lyle is a Pathologist at Bay Pine Health Care System in Florida. Dr. Lynch is an Investigator at VA Salt Lake Health Care System Informatics and Computing Infrastructure. Dr. Montgomery is an Oncologist at VA Puget Sound Health Care System, in Seattle, Washington. Dr. Przygodzki is the Director of Genomic Medicine Implementation and Associate Director of Genomic Medicine for the VA. Dr. Colman is a Neuro-Oncologist at George E. Wahlen VA Medical Center and the Director of Medical Neuro-Oncology at the Huntsman Cancer Institute, Salt Lake City, Utah.
Correspondence: Dr. Kulich (scott.kulich@va.gov)
1. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803-820.
2. Wang-Rodriguez J, Yunes A, Phan R, et al. The challenges of precision medicine and new advances in molecular diagnostic testing in hematolymphoid malignancies: impact on the VHA. Fed Pract. 2017;34(suppl 5):S38-S49.
3. Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol. 2017;19(suppl 5):v1-v88.
4. Nikiforova MN, Wald AI, Melan MA, et al. Targeted next-generation sequencing panel (GlioSeq) provides comprehensive genetic profiling of central nervous system tumors. Neuro Oncol. 2016;18(3)379-387.
5. Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90(19):1473-1479.
6. van den Bent MJ, Erdem-Eraslan L, Idbaih A, et al. MGMT-STP27 methylation status as predictive marker for response to PCV in anaplastic oligodendrogliomas and oligoastrocytomas. A report from EORTC study 26951. Clin Cancer Res. 2013;19(19):5513-5522.
7. Stupp R, Hegi ME, Mason WP, et al; European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459-466.
8. Malmstrom A, Gronberg BH, Marosi C, et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol. 2012;13(9):916-926.
9. van den Bent MJ, Kros JM. Predictive and prognostic markers in neuro-oncology. J Neuropathol Exp Neurol. 2007;66(12):1074-1081.
10. Chen R, Smith-Cohn M, Cohen AL, Colman H. Glioma subclassifications and their clinical significance. Neurotherapeutics. 2017;14(2):284-297.
11. Jordan JT, Sanders AE, Armstrong T, et al. Quality improvement in neurology: neuro-oncology quality measurement set. Neurology. 2018;90(14):652-658.
12. Chen L, Voronovich Z, Clark K, et al. Predicting the likelihood of an isocitrate dehydrogenase 1 or 2 mutation in diagnoses of infiltrative glioma. Neuro Oncol. 2014;16(11):1478-1483.
13. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997-1003.
14. Wick W, Platten M, Meisner C, et al; NOA-08 Study Group of Neuro-oncology Working Group (NOA) of German Cancer Society. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012;13(7):707-715.
15. Horbinski C, Kofler J, Kelly LM, Murdoch GH, Nikiforova MN. Diagnostic use of IDH1/2 mutation analysis in routine clinical testing of formalin-fixed, paraffin-embedded glioma tissues. J Neuropathol Exp Neurol. 2009;68(12):1319-1325.
16. Camelo-Piragua S, Jansen M, Ganguly A, Kim JC, Louis DN, Nutt CL. Mutant IDH1-specific immunohistochemistry distinguishes diffuse astrocytoma from astrocytosis. Acta Neuropathol. 2010;119(4):509-511.
17. Horbinski C, Kofler J, Yeaney G, et al. Isocitrate dehydrogenase 1 analysis differentiates gangliogliomas from infiltrative gliomas. Brain Pathol. 2011;21(5):564-574.
18. Berghoff AS, Preusser M. BRAF alterations in brain tumours: molecular pathology and therapeutic opportunities. Curr Opin Neurol. 2014;27(6):689-696.
19. Korshunov A, Meyer J, Capper D, et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol. 2009;118(3):401-405.
20. Fuller CE, Schmidt RE, Roth KA, et al. Clinical utility of fluorescence in situ hybridization (FISH) in morphologically ambiguous gliomas with hybrid oligodendroglial/astrocytic features. J Neuropathol Exp Neurol. 2003;62(11):1118-1128.
21. Horbinski C. Practical molecular diagnostics in neuropathology: making a tough job a little easier. Semin Diagn Pathol. 2010;27(2):105-113.
22. Fuller GN, Bigner SH. Amplified cellular oncogenes in neoplasms of the human central nervous system. Mutat Res. 1992;276(3):299-306.
23. Brennan CW, Verhaak RG, McKenna A, et al; TCGA Research Network. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462-477.
24. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015;129(6):829-848.
25. Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110(15):6021-6026.
26. Nikiforova MN, Hamilton RL. Molecular diagnostics of gliomas. Arch Pathol Lab Med. 2011;135(5):558-568.
1. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803-820.
2. Wang-Rodriguez J, Yunes A, Phan R, et al. The challenges of precision medicine and new advances in molecular diagnostic testing in hematolymphoid malignancies: impact on the VHA. Fed Pract. 2017;34(suppl 5):S38-S49.
3. Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol. 2017;19(suppl 5):v1-v88.
4. Nikiforova MN, Wald AI, Melan MA, et al. Targeted next-generation sequencing panel (GlioSeq) provides comprehensive genetic profiling of central nervous system tumors. Neuro Oncol. 2016;18(3)379-387.
5. Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90(19):1473-1479.
6. van den Bent MJ, Erdem-Eraslan L, Idbaih A, et al. MGMT-STP27 methylation status as predictive marker for response to PCV in anaplastic oligodendrogliomas and oligoastrocytomas. A report from EORTC study 26951. Clin Cancer Res. 2013;19(19):5513-5522.
7. Stupp R, Hegi ME, Mason WP, et al; European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459-466.
8. Malmstrom A, Gronberg BH, Marosi C, et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol. 2012;13(9):916-926.
9. van den Bent MJ, Kros JM. Predictive and prognostic markers in neuro-oncology. J Neuropathol Exp Neurol. 2007;66(12):1074-1081.
10. Chen R, Smith-Cohn M, Cohen AL, Colman H. Glioma subclassifications and their clinical significance. Neurotherapeutics. 2017;14(2):284-297.
11. Jordan JT, Sanders AE, Armstrong T, et al. Quality improvement in neurology: neuro-oncology quality measurement set. Neurology. 2018;90(14):652-658.
12. Chen L, Voronovich Z, Clark K, et al. Predicting the likelihood of an isocitrate dehydrogenase 1 or 2 mutation in diagnoses of infiltrative glioma. Neuro Oncol. 2014;16(11):1478-1483.
13. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997-1003.
14. Wick W, Platten M, Meisner C, et al; NOA-08 Study Group of Neuro-oncology Working Group (NOA) of German Cancer Society. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012;13(7):707-715.
15. Horbinski C, Kofler J, Kelly LM, Murdoch GH, Nikiforova MN. Diagnostic use of IDH1/2 mutation analysis in routine clinical testing of formalin-fixed, paraffin-embedded glioma tissues. J Neuropathol Exp Neurol. 2009;68(12):1319-1325.
16. Camelo-Piragua S, Jansen M, Ganguly A, Kim JC, Louis DN, Nutt CL. Mutant IDH1-specific immunohistochemistry distinguishes diffuse astrocytoma from astrocytosis. Acta Neuropathol. 2010;119(4):509-511.
17. Horbinski C, Kofler J, Yeaney G, et al. Isocitrate dehydrogenase 1 analysis differentiates gangliogliomas from infiltrative gliomas. Brain Pathol. 2011;21(5):564-574.
18. Berghoff AS, Preusser M. BRAF alterations in brain tumours: molecular pathology and therapeutic opportunities. Curr Opin Neurol. 2014;27(6):689-696.
19. Korshunov A, Meyer J, Capper D, et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol. 2009;118(3):401-405.
20. Fuller CE, Schmidt RE, Roth KA, et al. Clinical utility of fluorescence in situ hybridization (FISH) in morphologically ambiguous gliomas with hybrid oligodendroglial/astrocytic features. J Neuropathol Exp Neurol. 2003;62(11):1118-1128.
21. Horbinski C. Practical molecular diagnostics in neuropathology: making a tough job a little easier. Semin Diagn Pathol. 2010;27(2):105-113.
22. Fuller GN, Bigner SH. Amplified cellular oncogenes in neoplasms of the human central nervous system. Mutat Res. 1992;276(3):299-306.
23. Brennan CW, Verhaak RG, McKenna A, et al; TCGA Research Network. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462-477.
24. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015;129(6):829-848.
25. Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110(15):6021-6026.
26. Nikiforova MN, Hamilton RL. Molecular diagnostics of gliomas. Arch Pathol Lab Med. 2011;135(5):558-568.