User login
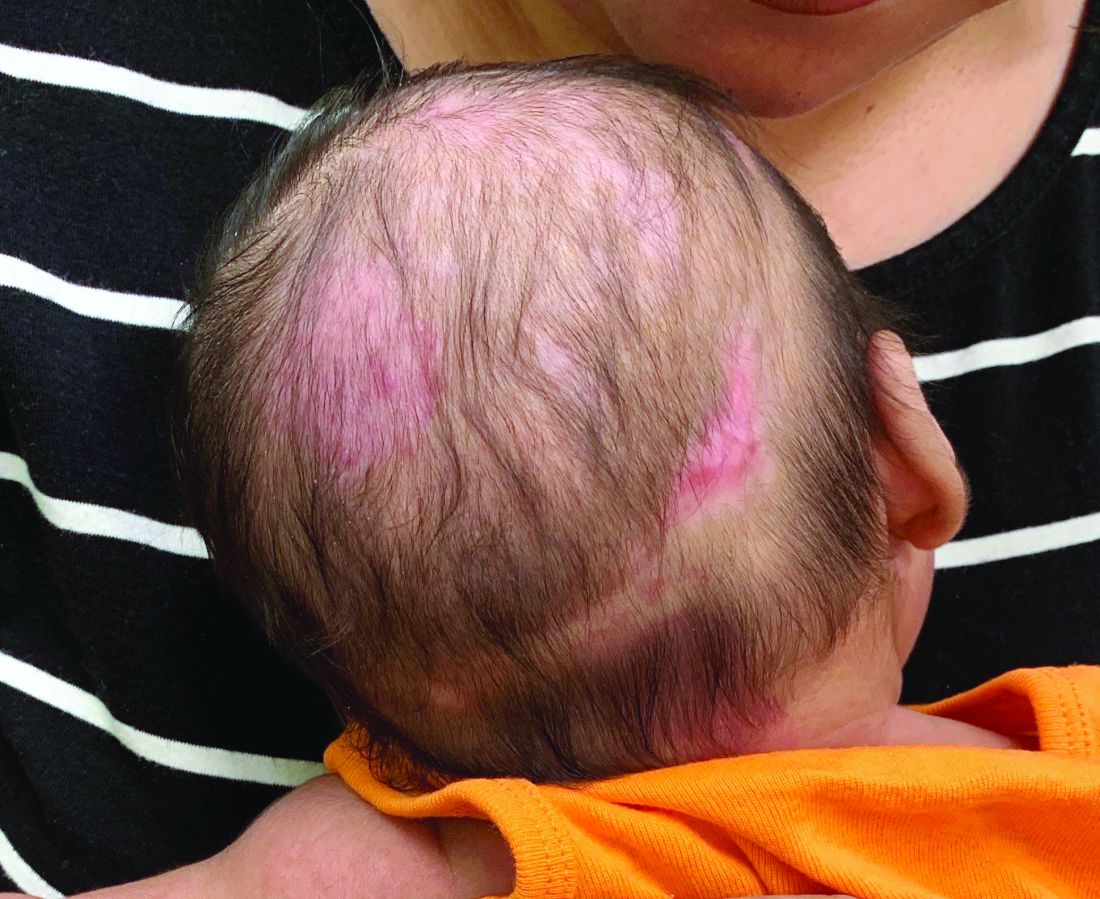
A 2-month-old infant with a scalp rash that appeared after birth
With the perinatal history of prolonged labor and prolonged rupture of membranes, the diagnosis of halo scalp ring was made. This occurs secondary to prolonged pressure of the baby’s scalp with the mother’s pelvic bones, uterus, or cervical area, which causes decreased blood flow to the area, secondary ischemic damage, and in some cases scarring and hair loss.1
The degree of involvement is variable as some babies have mild alopecia and others have severe full-thickness necrosis and scarring. These lesions also can present with associated caput succedaneum and scalp molding, but these were not seen in our patient. Predisposing factors for halo scalp ring include caput succedaneum, prolonged or difficult labor, premature or prolonged rupture of membranes, vaginal delivery, vertex presentation, first delivery, as well as prematurity.2 On physical examination, a semicircular patch of alopecia with associated scarring, crusting, or erythema can be seen in some more severe cases.
The differential diagnosis includes aplasia cutis. In aplasia cutis, there is congenital loss of skin on the affected areas. The scalp usually is affected, but these lesions can occur in any other part of the body. Most patients with aplasia cutis have no other findings, but there are cases that can be associated with other cardiovascular, gastrointestinal, or central nervous system abnormalities. Neonatal lupus also can present with scarring lesions on the scalp, but they usually present a little after birth, mainly affecting the face. The mothers of this children usually have a diagnosis of connective tissue disease and may have positive antibodies to Sjögren’s syndrome antibody A, Sjögren’s syndrome antibody B, or antiribonucleoprotein antibody. Seborrheic dermatitis does not cause scarring alopecia. The lesions present as waxy scaly plaques on the scalp, erythematous waxy plaques behind the ears, face, and folds. Some patients can have hair loss secondary to the inflammation, but the hair grows back once the inflammation is controlled. Dissecting cellulitis is a type of scarring alopecia seen in pubescent and adult individuals. No cases of neonatal dissecting cellulitis have been described.
Halo scalp ring is not associated with any other systemic symptoms or syndromes. Extensive imaging and systemic work-up are not required unless the baby presents with other neurologic symptoms. The areas can be treated with petrolatum and observation as most lesions resolve.
In cases of extensive areas of scarring alopecia, referral to a plastic surgeon can be made to consider tissue expanders or scar revision prior to the child starting school if the lesions are causing psychological stressors.
The true prevalence of this condition is unknown. We believe halo ring alopecia is sometimes not diagnosed, and as lesions tend to resolve, most cases go unreported.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com.
References
1. Arch Pediatr Adolesc Med. 2010;164(7):673.
2. Pediatr Dermatol. 2009 Nov-Dec;26(6):706-8.
3. Dermatol Online J. 2016 Nov 15;22(11).pii:13030/qt7rt592tz.
With the perinatal history of prolonged labor and prolonged rupture of membranes, the diagnosis of halo scalp ring was made. This occurs secondary to prolonged pressure of the baby’s scalp with the mother’s pelvic bones, uterus, or cervical area, which causes decreased blood flow to the area, secondary ischemic damage, and in some cases scarring and hair loss.1
The degree of involvement is variable as some babies have mild alopecia and others have severe full-thickness necrosis and scarring. These lesions also can present with associated caput succedaneum and scalp molding, but these were not seen in our patient. Predisposing factors for halo scalp ring include caput succedaneum, prolonged or difficult labor, premature or prolonged rupture of membranes, vaginal delivery, vertex presentation, first delivery, as well as prematurity.2 On physical examination, a semicircular patch of alopecia with associated scarring, crusting, or erythema can be seen in some more severe cases.
The differential diagnosis includes aplasia cutis. In aplasia cutis, there is congenital loss of skin on the affected areas. The scalp usually is affected, but these lesions can occur in any other part of the body. Most patients with aplasia cutis have no other findings, but there are cases that can be associated with other cardiovascular, gastrointestinal, or central nervous system abnormalities. Neonatal lupus also can present with scarring lesions on the scalp, but they usually present a little after birth, mainly affecting the face. The mothers of this children usually have a diagnosis of connective tissue disease and may have positive antibodies to Sjögren’s syndrome antibody A, Sjögren’s syndrome antibody B, or antiribonucleoprotein antibody. Seborrheic dermatitis does not cause scarring alopecia. The lesions present as waxy scaly plaques on the scalp, erythematous waxy plaques behind the ears, face, and folds. Some patients can have hair loss secondary to the inflammation, but the hair grows back once the inflammation is controlled. Dissecting cellulitis is a type of scarring alopecia seen in pubescent and adult individuals. No cases of neonatal dissecting cellulitis have been described.
Halo scalp ring is not associated with any other systemic symptoms or syndromes. Extensive imaging and systemic work-up are not required unless the baby presents with other neurologic symptoms. The areas can be treated with petrolatum and observation as most lesions resolve.
In cases of extensive areas of scarring alopecia, referral to a plastic surgeon can be made to consider tissue expanders or scar revision prior to the child starting school if the lesions are causing psychological stressors.
The true prevalence of this condition is unknown. We believe halo ring alopecia is sometimes not diagnosed, and as lesions tend to resolve, most cases go unreported.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com.
References
1. Arch Pediatr Adolesc Med. 2010;164(7):673.
2. Pediatr Dermatol. 2009 Nov-Dec;26(6):706-8.
3. Dermatol Online J. 2016 Nov 15;22(11).pii:13030/qt7rt592tz.
With the perinatal history of prolonged labor and prolonged rupture of membranes, the diagnosis of halo scalp ring was made. This occurs secondary to prolonged pressure of the baby’s scalp with the mother’s pelvic bones, uterus, or cervical area, which causes decreased blood flow to the area, secondary ischemic damage, and in some cases scarring and hair loss.1
The degree of involvement is variable as some babies have mild alopecia and others have severe full-thickness necrosis and scarring. These lesions also can present with associated caput succedaneum and scalp molding, but these were not seen in our patient. Predisposing factors for halo scalp ring include caput succedaneum, prolonged or difficult labor, premature or prolonged rupture of membranes, vaginal delivery, vertex presentation, first delivery, as well as prematurity.2 On physical examination, a semicircular patch of alopecia with associated scarring, crusting, or erythema can be seen in some more severe cases.
The differential diagnosis includes aplasia cutis. In aplasia cutis, there is congenital loss of skin on the affected areas. The scalp usually is affected, but these lesions can occur in any other part of the body. Most patients with aplasia cutis have no other findings, but there are cases that can be associated with other cardiovascular, gastrointestinal, or central nervous system abnormalities. Neonatal lupus also can present with scarring lesions on the scalp, but they usually present a little after birth, mainly affecting the face. The mothers of this children usually have a diagnosis of connective tissue disease and may have positive antibodies to Sjögren’s syndrome antibody A, Sjögren’s syndrome antibody B, or antiribonucleoprotein antibody. Seborrheic dermatitis does not cause scarring alopecia. The lesions present as waxy scaly plaques on the scalp, erythematous waxy plaques behind the ears, face, and folds. Some patients can have hair loss secondary to the inflammation, but the hair grows back once the inflammation is controlled. Dissecting cellulitis is a type of scarring alopecia seen in pubescent and adult individuals. No cases of neonatal dissecting cellulitis have been described.
Halo scalp ring is not associated with any other systemic symptoms or syndromes. Extensive imaging and systemic work-up are not required unless the baby presents with other neurologic symptoms. The areas can be treated with petrolatum and observation as most lesions resolve.
In cases of extensive areas of scarring alopecia, referral to a plastic surgeon can be made to consider tissue expanders or scar revision prior to the child starting school if the lesions are causing psychological stressors.
The true prevalence of this condition is unknown. We believe halo ring alopecia is sometimes not diagnosed, and as lesions tend to resolve, most cases go unreported.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com.
References
1. Arch Pediatr Adolesc Med. 2010;164(7):673.
2. Pediatr Dermatol. 2009 Nov-Dec;26(6):706-8.
3. Dermatol Online J. 2016 Nov 15;22(11).pii:13030/qt7rt592tz.
A 2-month-old male is referred to our pediatric dermatology clinic for evaluation of persistent seborrheic dermatitis. The mother reports that he presented with a rash on his scalp a few days after birth. She has been treating the crusted areas with clotrimazole and hydrocortisone and has noted improvement on the crusting, but now is worried that there is some scarring. The affected areas are not bleeding or tender. There are no other rashes elsewhere in the body.
He was born at 36 weeks from a 35-year-old gravida 1 para 0 woman with adequate prenatal care. The mother was diagnosed with preeclampsia and was induced. She had a prolonged labor and had premature rupture of membranes. The baby was delivered via cesarean section because of failure to progress and fetal distress; forceps, vacuum, and a scalp probe were not used during delivery. He was admitted to the neonatal unit for 5 days for sepsis work-up and respiratory distress. No intubation was needed.
Besides the preeclampsia, the mother denied any other medical conditions and was not taking any medications. He has met all developmental milestones for his age. He has no history of seizures.
On physical exam, there are semicircular patches of alopecia on the scalp. Some areas have pink, rubbery plaques with loss of hair follicles. On the frontal scalp, there are waxy plaques.
There is a blanchable violaceous patch on the occiput and there are some erythematous papules on the cheeks.
A healthy 8-year-old boy presents with several skin-colored, round 1-3 mm papules on the nose, forehead, and cheeks
A shave biopsy of one of the lesions was performed that showed a proliferation of nests of basaloid cells on the dermis with palisading and rare vacuolated clear cell change. A rare ductal structure with luminal proteinaceous contents was noted. The findings were consistent with a trichoepithelioma.
Trichoepitheliomas are rare, benign, adnexal skin tumors that can start in early childhood or during puberty. The lesions are most commonly seen in girls as skin color papules on the face, and sometimes on the trunk and the neck. Trichoepitheliomas can appear as a benign single lesion nonfamilial form or as a familial form with multiple lesions.1 Brooke-Spiegler syndrome (BSS) is a rare autosomal dominant condition where affected individuals have multiple trichoepitheliomas, cylindromas, and spiradenomas. Depending on the predominant type of lesion, phenotypic variants include multiple familial trichoepithelioma type 1 and familial cylindromatosis.2 BSS is caused by mutations within CYLD, a tumor-suppressor gene located on chromosome 16q12-q13.3 Our patient presented only with trichoepitheliomas with no other lesions on the scalp, neck, or torso.
Multiple trichoepitheliomas also can be seen in other syndromes including Rombo syndrome, which is characterized by basal cell carcinomas, milia, hypotrichosis, distal vasodilation, and atrophoderma vermiculata; none seen in our patient. Bazex-Dupré-Christol syndrome is an X-linked dominant condition in which affected individuals can present with multiple trichoepitheliomas, as well as milia, hypotrichosis, follicular atrophoderma, and basal cell carcinomas.
The differential diagnosis of skin color papules on the central face on a child should include acne, flat warts, and angiofibromas seen in tuberous sclerosis. Our patient’s lesions were monomorphous, and there were no comedones, pustules, or inflammatory papules characteristic of acne.
He had warts on his hands which could make it suspicious for the face lesions to be verrucous in nature. Flat warts also present as skin color papules, but characteristically are flat, not round and shiny as our patient’s lesions were. Angiofibromas, as seen in individuals with tuberous sclerosis, also can start at an early age in the same location as trichoepitheliomas in BSS, but clinically the lesions are pinker and redder rather than the skin-color, round shape papules characteristic of trichoepitheliomas. Patients may have other findings suggestive of tuberous sclerosis including confetti hypopigmentation, ash leaf spots, shagreen patch, and a history of seizures or developmental delay – none of which were present in our patient. Children with basal cell nevus syndrome can present with skin color to shiny telangiectatic papules (basal cell carcinomas) that can be single or multiple on the face, chest, and back. The lesions usually are not seen in clusters around the nose and central face as seen in patients with BSS. Patients with basal cell nevus syndrome can develop jaw bone cysts, brain tumors (medulloblastoma), and fibromas on the heart or ovaries, palmar pits and be macrocephalic.4
Trichoepitheliomas usually are treated surgically but other nonsurgical removing techniques include laser resurfacing, curettage, and electrocautery.5 Malignant transformation can occur in 5%-10% of the individuals and should be managed by a multidisciplinary team. Topical treatment with sirolimus previously has been reported to be effective in young patients.6
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She said she had no relevant financial disclosures. Email Dr. Matiz at pdnews@mdedge.com.
References
1. Acta Dermatovenerol Croat. 2018 Jun;26(2):162-5.
2. Eur J Med Genet. 2015;58(5):271-8.
3. Am J Dermatopathol. 2014;36(11):868-74.
4. Int J Dermatol. 2016 Apr;55(4):367-75.
5. Int J Dermatol. 2007;46(6):583-6.
6. Dermatol Ther. 2017 Mar. doi: 10.1111/dth.12458.
A shave biopsy of one of the lesions was performed that showed a proliferation of nests of basaloid cells on the dermis with palisading and rare vacuolated clear cell change. A rare ductal structure with luminal proteinaceous contents was noted. The findings were consistent with a trichoepithelioma.
Trichoepitheliomas are rare, benign, adnexal skin tumors that can start in early childhood or during puberty. The lesions are most commonly seen in girls as skin color papules on the face, and sometimes on the trunk and the neck. Trichoepitheliomas can appear as a benign single lesion nonfamilial form or as a familial form with multiple lesions.1 Brooke-Spiegler syndrome (BSS) is a rare autosomal dominant condition where affected individuals have multiple trichoepitheliomas, cylindromas, and spiradenomas. Depending on the predominant type of lesion, phenotypic variants include multiple familial trichoepithelioma type 1 and familial cylindromatosis.2 BSS is caused by mutations within CYLD, a tumor-suppressor gene located on chromosome 16q12-q13.3 Our patient presented only with trichoepitheliomas with no other lesions on the scalp, neck, or torso.
Multiple trichoepitheliomas also can be seen in other syndromes including Rombo syndrome, which is characterized by basal cell carcinomas, milia, hypotrichosis, distal vasodilation, and atrophoderma vermiculata; none seen in our patient. Bazex-Dupré-Christol syndrome is an X-linked dominant condition in which affected individuals can present with multiple trichoepitheliomas, as well as milia, hypotrichosis, follicular atrophoderma, and basal cell carcinomas.
The differential diagnosis of skin color papules on the central face on a child should include acne, flat warts, and angiofibromas seen in tuberous sclerosis. Our patient’s lesions were monomorphous, and there were no comedones, pustules, or inflammatory papules characteristic of acne.
He had warts on his hands which could make it suspicious for the face lesions to be verrucous in nature. Flat warts also present as skin color papules, but characteristically are flat, not round and shiny as our patient’s lesions were. Angiofibromas, as seen in individuals with tuberous sclerosis, also can start at an early age in the same location as trichoepitheliomas in BSS, but clinically the lesions are pinker and redder rather than the skin-color, round shape papules characteristic of trichoepitheliomas. Patients may have other findings suggestive of tuberous sclerosis including confetti hypopigmentation, ash leaf spots, shagreen patch, and a history of seizures or developmental delay – none of which were present in our patient. Children with basal cell nevus syndrome can present with skin color to shiny telangiectatic papules (basal cell carcinomas) that can be single or multiple on the face, chest, and back. The lesions usually are not seen in clusters around the nose and central face as seen in patients with BSS. Patients with basal cell nevus syndrome can develop jaw bone cysts, brain tumors (medulloblastoma), and fibromas on the heart or ovaries, palmar pits and be macrocephalic.4
Trichoepitheliomas usually are treated surgically but other nonsurgical removing techniques include laser resurfacing, curettage, and electrocautery.5 Malignant transformation can occur in 5%-10% of the individuals and should be managed by a multidisciplinary team. Topical treatment with sirolimus previously has been reported to be effective in young patients.6
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She said she had no relevant financial disclosures. Email Dr. Matiz at pdnews@mdedge.com.
References
1. Acta Dermatovenerol Croat. 2018 Jun;26(2):162-5.
2. Eur J Med Genet. 2015;58(5):271-8.
3. Am J Dermatopathol. 2014;36(11):868-74.
4. Int J Dermatol. 2016 Apr;55(4):367-75.
5. Int J Dermatol. 2007;46(6):583-6.
6. Dermatol Ther. 2017 Mar. doi: 10.1111/dth.12458.
A shave biopsy of one of the lesions was performed that showed a proliferation of nests of basaloid cells on the dermis with palisading and rare vacuolated clear cell change. A rare ductal structure with luminal proteinaceous contents was noted. The findings were consistent with a trichoepithelioma.
Trichoepitheliomas are rare, benign, adnexal skin tumors that can start in early childhood or during puberty. The lesions are most commonly seen in girls as skin color papules on the face, and sometimes on the trunk and the neck. Trichoepitheliomas can appear as a benign single lesion nonfamilial form or as a familial form with multiple lesions.1 Brooke-Spiegler syndrome (BSS) is a rare autosomal dominant condition where affected individuals have multiple trichoepitheliomas, cylindromas, and spiradenomas. Depending on the predominant type of lesion, phenotypic variants include multiple familial trichoepithelioma type 1 and familial cylindromatosis.2 BSS is caused by mutations within CYLD, a tumor-suppressor gene located on chromosome 16q12-q13.3 Our patient presented only with trichoepitheliomas with no other lesions on the scalp, neck, or torso.
Multiple trichoepitheliomas also can be seen in other syndromes including Rombo syndrome, which is characterized by basal cell carcinomas, milia, hypotrichosis, distal vasodilation, and atrophoderma vermiculata; none seen in our patient. Bazex-Dupré-Christol syndrome is an X-linked dominant condition in which affected individuals can present with multiple trichoepitheliomas, as well as milia, hypotrichosis, follicular atrophoderma, and basal cell carcinomas.
The differential diagnosis of skin color papules on the central face on a child should include acne, flat warts, and angiofibromas seen in tuberous sclerosis. Our patient’s lesions were monomorphous, and there were no comedones, pustules, or inflammatory papules characteristic of acne.
He had warts on his hands which could make it suspicious for the face lesions to be verrucous in nature. Flat warts also present as skin color papules, but characteristically are flat, not round and shiny as our patient’s lesions were. Angiofibromas, as seen in individuals with tuberous sclerosis, also can start at an early age in the same location as trichoepitheliomas in BSS, but clinically the lesions are pinker and redder rather than the skin-color, round shape papules characteristic of trichoepitheliomas. Patients may have other findings suggestive of tuberous sclerosis including confetti hypopigmentation, ash leaf spots, shagreen patch, and a history of seizures or developmental delay – none of which were present in our patient. Children with basal cell nevus syndrome can present with skin color to shiny telangiectatic papules (basal cell carcinomas) that can be single or multiple on the face, chest, and back. The lesions usually are not seen in clusters around the nose and central face as seen in patients with BSS. Patients with basal cell nevus syndrome can develop jaw bone cysts, brain tumors (medulloblastoma), and fibromas on the heart or ovaries, palmar pits and be macrocephalic.4
Trichoepitheliomas usually are treated surgically but other nonsurgical removing techniques include laser resurfacing, curettage, and electrocautery.5 Malignant transformation can occur in 5%-10% of the individuals and should be managed by a multidisciplinary team. Topical treatment with sirolimus previously has been reported to be effective in young patients.6
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She said she had no relevant financial disclosures. Email Dr. Matiz at pdnews@mdedge.com.
References
1. Acta Dermatovenerol Croat. 2018 Jun;26(2):162-5.
2. Eur J Med Genet. 2015;58(5):271-8.
3. Am J Dermatopathol. 2014;36(11):868-74.
4. Int J Dermatol. 2016 Apr;55(4):367-75.
5. Int J Dermatol. 2007;46(6):583-6.
6. Dermatol Ther. 2017 Mar. doi: 10.1111/dth.12458.
A white 8-year-old boy comes to our pediatric dermatology clinic with his mother for evaluation of acne. The lesions started about a year ago on his nose and now have spread to his cheeks. The bumps are not symptomatic. He has been applying over the counter salicylic acid and benzoyl peroxide gels with no help. The mother reports he has been growing well, denies any growth spurt, no axillary or genital hair or body odor noted.

None of the family members have a history of acne. The mother cannot recall any family members with similar lesions on the face. He has had some warts on his fingers for years and has been treated with over the counter salicylic acid. There is no family history of skin cancer.
On physical exam, he is a healthy young boy with several skin color, round papules 1-3 mm on the nose, forehead, and cheeks. There are no lesions on the scalp. He has abundant brown hair. He has few verrucous papules on the fingers. Axillary and genital hair is not noted. There is no body odor and he is Tanner stage I.
What is your diagnosis?
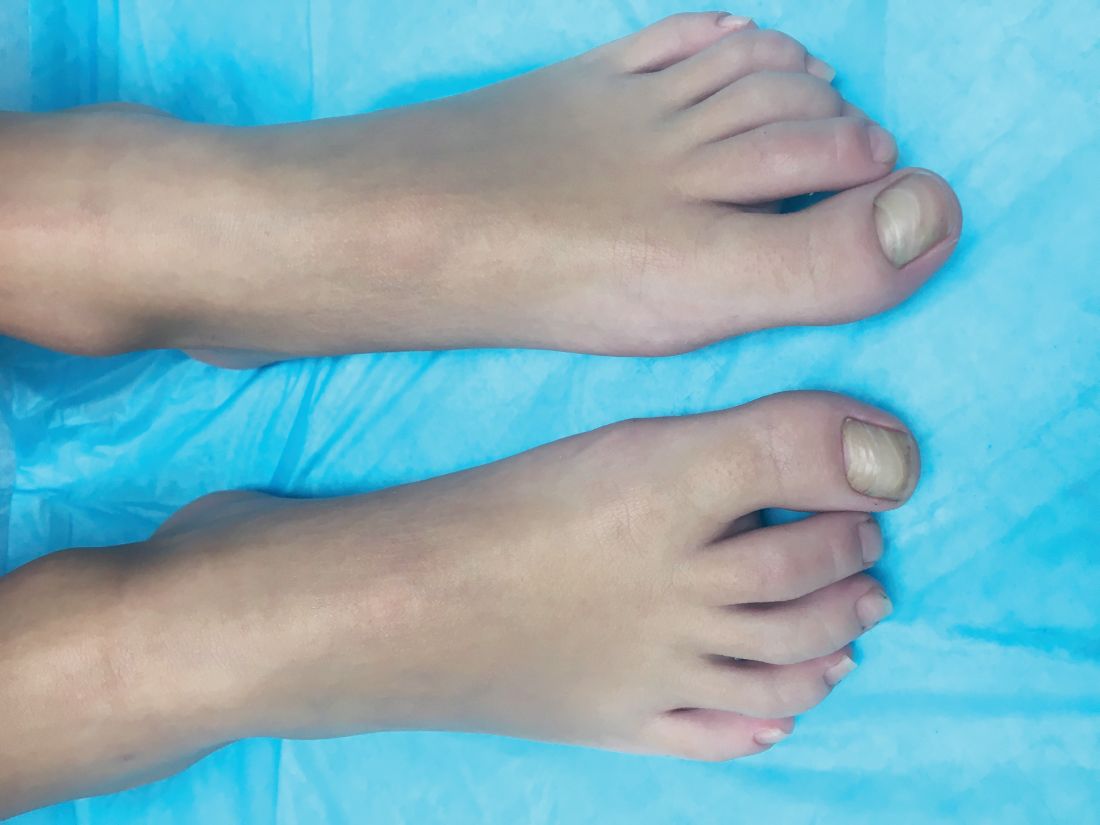
A skin biopsy of one of the lesions on the right toe showed dermal edema with an associated lymphohistiocytic infiltrate. There are scattered areas of perieccrine involvement and areas of vasculitis. Laboratory work up showed a normal complete blood count, a negative antinuclear antibodies (ANA) titer, a negative double-stranded DNA, normal levels of inflammatory markers, and negative cryoglobulins and cold agglutinins. The patient was diagnosed with pernio. The lesions improved within several weeks. She now wears thicker socks when she is ice skating.
Children, women, and the elderly are at a higher risk.1 This condition is frequently described in Northwestern Europe and the United Kingdom, especially in those living in houses without central heating.2
Clinically, the lesions appear a few hours or days after cold exposure on the toes, fingers, and in some unusual cases on the nose and the ears. The lesions present as erythematous to violaceous macules, papules, or nodules that in severe cases may blister and ulcerate. The lesions may be asymptomatic, pruritic, or tender. In children, pernio can be associated with the presence of cryoglobulins, cold agglutinins, anorexia nervosa, and genetic interferonopathy; it may precede the diagnosis of chronic myelomonocytic leukemia and may occur as a presenting sign of a blast crisis in acute lymphoblastic leukemia.3,4 The skin lesions usually resolve within days to a few weeks. Histopathologic analysis shows dermal edema with associated superficial and deep lymphohistiocytic infiltrate and perieccrine involvement.
The differential diagnosis of pernio includes other cold-induced syndromes such as Raynaud’s syndrome, cold panniculitis, cold urticaria, livedo reticularis, acrocyanosis, and chilblain lupus. In chilblain lupus (a form of chronic cutaneous lupus), the lesions may be very similar to pernio but the histopathology is consistent with changes of discoid lupus. Lesions of idiopathic palmoplantar hidradenitis present as erythematous tender nodules on the palms and the soles.5 The lesions can be triggered by vigorous physical activity, exposure to moisture, and excessive sweating. White, blue, and red discoloration of the fingers is seen in Raynaud’s phenomenon rather than the fixed erythematous to violaceous macules, papules, or nodules seen in pernio. Patients with erythromelalgia present with red painful palms and soles triggered by heat and, in contrast to pernio, relieved by cooling. Sweet syndrome, a febrile neutrophilic dermatoses, is characterized by tender erythematous papules and plaques with associated systemic symptoms. These patients may have an associated internal malignancy or infection, or the disorder may be triggered by medications or pregnancy.
Our patient had no systemic symptoms, and the pathology didn’t show any neutrophils. When the diagnosis is in doubt, a skin biopsy may help elucidate the diagnosis.
Once the diagnosis of pernio is made, it is recommended to order a complete blood count to rule out blood malignancies and cryoproteins.
Treatment of this condition consists of rewarming the extremity. If rewarming does not improve the patient’s symptoms, systemic treatment with nifedipine may be warranted.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Dr. Matiz said she had no relevant financial disclosures. Email her at pdnews@mdedge.com.
References
1. Pediatrics. 2005 Sep;116(3):e472-5.
2. Mayo Clin Proc. 2014 Feb;89(2):207-15.
3. Pediatr Dermatol. 2018 Jan;35(1):e74-5.
4. Pediatr Dermatol. 2000 Mar-Apr;17(2):97-9.
5. Eur J Pediatr. 2001 Mar;160(3):189-91.
A skin biopsy of one of the lesions on the right toe showed dermal edema with an associated lymphohistiocytic infiltrate. There are scattered areas of perieccrine involvement and areas of vasculitis. Laboratory work up showed a normal complete blood count, a negative antinuclear antibodies (ANA) titer, a negative double-stranded DNA, normal levels of inflammatory markers, and negative cryoglobulins and cold agglutinins. The patient was diagnosed with pernio. The lesions improved within several weeks. She now wears thicker socks when she is ice skating.
Children, women, and the elderly are at a higher risk.1 This condition is frequently described in Northwestern Europe and the United Kingdom, especially in those living in houses without central heating.2
Clinically, the lesions appear a few hours or days after cold exposure on the toes, fingers, and in some unusual cases on the nose and the ears. The lesions present as erythematous to violaceous macules, papules, or nodules that in severe cases may blister and ulcerate. The lesions may be asymptomatic, pruritic, or tender. In children, pernio can be associated with the presence of cryoglobulins, cold agglutinins, anorexia nervosa, and genetic interferonopathy; it may precede the diagnosis of chronic myelomonocytic leukemia and may occur as a presenting sign of a blast crisis in acute lymphoblastic leukemia.3,4 The skin lesions usually resolve within days to a few weeks. Histopathologic analysis shows dermal edema with associated superficial and deep lymphohistiocytic infiltrate and perieccrine involvement.
The differential diagnosis of pernio includes other cold-induced syndromes such as Raynaud’s syndrome, cold panniculitis, cold urticaria, livedo reticularis, acrocyanosis, and chilblain lupus. In chilblain lupus (a form of chronic cutaneous lupus), the lesions may be very similar to pernio but the histopathology is consistent with changes of discoid lupus. Lesions of idiopathic palmoplantar hidradenitis present as erythematous tender nodules on the palms and the soles.5 The lesions can be triggered by vigorous physical activity, exposure to moisture, and excessive sweating. White, blue, and red discoloration of the fingers is seen in Raynaud’s phenomenon rather than the fixed erythematous to violaceous macules, papules, or nodules seen in pernio. Patients with erythromelalgia present with red painful palms and soles triggered by heat and, in contrast to pernio, relieved by cooling. Sweet syndrome, a febrile neutrophilic dermatoses, is characterized by tender erythematous papules and plaques with associated systemic symptoms. These patients may have an associated internal malignancy or infection, or the disorder may be triggered by medications or pregnancy.
Our patient had no systemic symptoms, and the pathology didn’t show any neutrophils. When the diagnosis is in doubt, a skin biopsy may help elucidate the diagnosis.
Once the diagnosis of pernio is made, it is recommended to order a complete blood count to rule out blood malignancies and cryoproteins.
Treatment of this condition consists of rewarming the extremity. If rewarming does not improve the patient’s symptoms, systemic treatment with nifedipine may be warranted.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Dr. Matiz said she had no relevant financial disclosures. Email her at pdnews@mdedge.com.
References
1. Pediatrics. 2005 Sep;116(3):e472-5.
2. Mayo Clin Proc. 2014 Feb;89(2):207-15.
3. Pediatr Dermatol. 2018 Jan;35(1):e74-5.
4. Pediatr Dermatol. 2000 Mar-Apr;17(2):97-9.
5. Eur J Pediatr. 2001 Mar;160(3):189-91.
A skin biopsy of one of the lesions on the right toe showed dermal edema with an associated lymphohistiocytic infiltrate. There are scattered areas of perieccrine involvement and areas of vasculitis. Laboratory work up showed a normal complete blood count, a negative antinuclear antibodies (ANA) titer, a negative double-stranded DNA, normal levels of inflammatory markers, and negative cryoglobulins and cold agglutinins. The patient was diagnosed with pernio. The lesions improved within several weeks. She now wears thicker socks when she is ice skating.
Children, women, and the elderly are at a higher risk.1 This condition is frequently described in Northwestern Europe and the United Kingdom, especially in those living in houses without central heating.2
Clinically, the lesions appear a few hours or days after cold exposure on the toes, fingers, and in some unusual cases on the nose and the ears. The lesions present as erythematous to violaceous macules, papules, or nodules that in severe cases may blister and ulcerate. The lesions may be asymptomatic, pruritic, or tender. In children, pernio can be associated with the presence of cryoglobulins, cold agglutinins, anorexia nervosa, and genetic interferonopathy; it may precede the diagnosis of chronic myelomonocytic leukemia and may occur as a presenting sign of a blast crisis in acute lymphoblastic leukemia.3,4 The skin lesions usually resolve within days to a few weeks. Histopathologic analysis shows dermal edema with associated superficial and deep lymphohistiocytic infiltrate and perieccrine involvement.
The differential diagnosis of pernio includes other cold-induced syndromes such as Raynaud’s syndrome, cold panniculitis, cold urticaria, livedo reticularis, acrocyanosis, and chilblain lupus. In chilblain lupus (a form of chronic cutaneous lupus), the lesions may be very similar to pernio but the histopathology is consistent with changes of discoid lupus. Lesions of idiopathic palmoplantar hidradenitis present as erythematous tender nodules on the palms and the soles.5 The lesions can be triggered by vigorous physical activity, exposure to moisture, and excessive sweating. White, blue, and red discoloration of the fingers is seen in Raynaud’s phenomenon rather than the fixed erythematous to violaceous macules, papules, or nodules seen in pernio. Patients with erythromelalgia present with red painful palms and soles triggered by heat and, in contrast to pernio, relieved by cooling. Sweet syndrome, a febrile neutrophilic dermatoses, is characterized by tender erythematous papules and plaques with associated systemic symptoms. These patients may have an associated internal malignancy or infection, or the disorder may be triggered by medications or pregnancy.
Our patient had no systemic symptoms, and the pathology didn’t show any neutrophils. When the diagnosis is in doubt, a skin biopsy may help elucidate the diagnosis.
Once the diagnosis of pernio is made, it is recommended to order a complete blood count to rule out blood malignancies and cryoproteins.
Treatment of this condition consists of rewarming the extremity. If rewarming does not improve the patient’s symptoms, systemic treatment with nifedipine may be warranted.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Dr. Matiz said she had no relevant financial disclosures. Email her at pdnews@mdedge.com.
References
1. Pediatrics. 2005 Sep;116(3):e472-5.
2. Mayo Clin Proc. 2014 Feb;89(2):207-15.
3. Pediatr Dermatol. 2018 Jan;35(1):e74-5.
4. Pediatr Dermatol. 2000 Mar-Apr;17(2):97-9.
5. Eur J Pediatr. 2001 Mar;160(3):189-91.
An 8-year-old girl comes to our pediatric dermatology clinic in the company of her mother for evaluation of painless purple spots on her toes. The lesions have been present for about 2 weeks. She has not been treated with any medications or creams. She denies any fevers, weight loss, mouth ulcers, sun sensitivity, joint pain, or any other symptoms. The patient has been a very healthy girl with occasional colds and no recent illnesses. The girl has never been admitted to the hospital. All her vaccinations are up to date. She takes no chronic medications. She lives in San Diego with her parents and two siblings. The girl recently started practicing ice-skating several times a week. There is no family history of any chronic medical conditions. She has no pets.
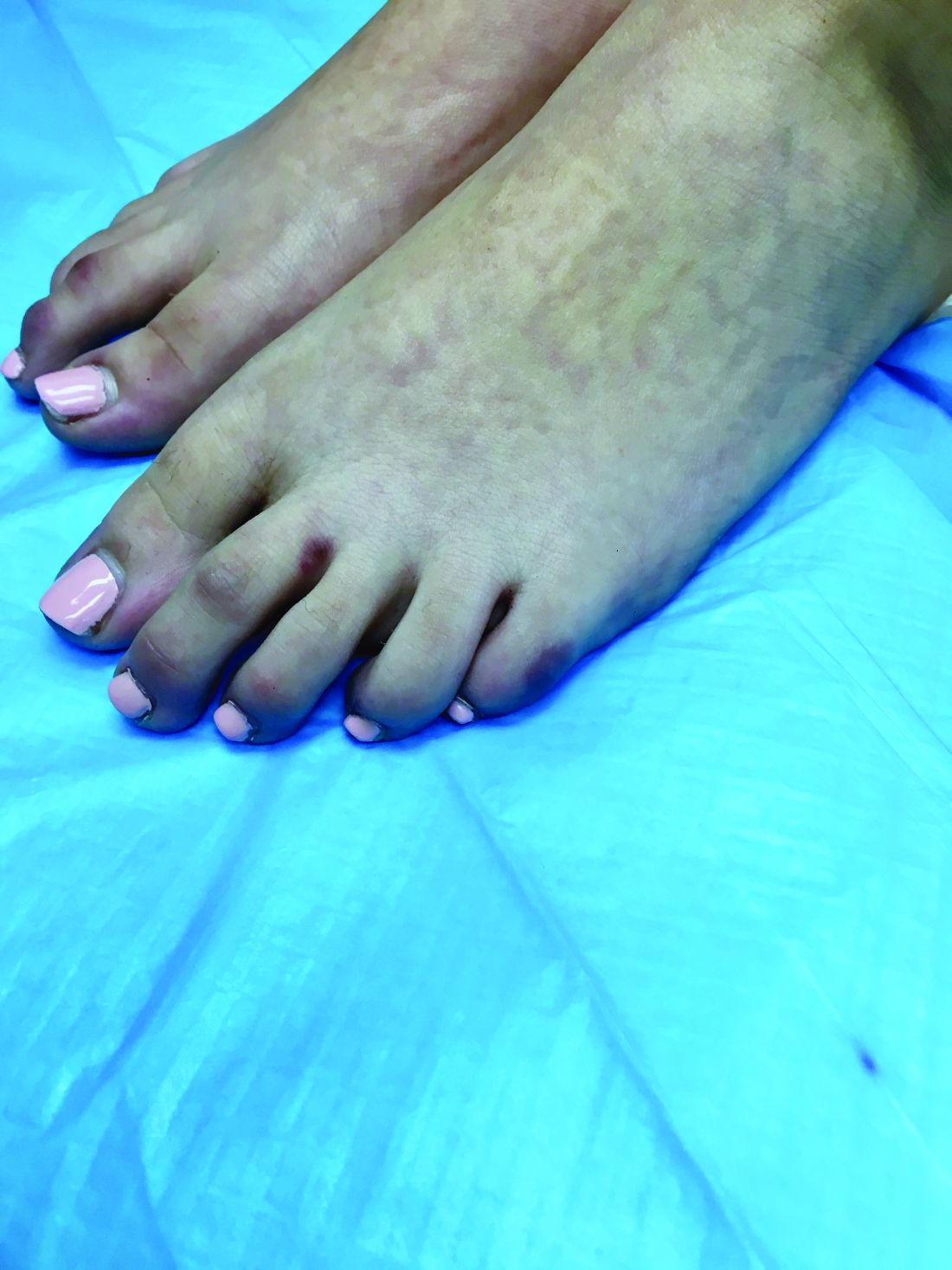
What's your diagnosis?
The patient was diagnosed with lichen spinulosus (LS) based on the physical appearance of the lesions (hyperkeratotic spiny papules forming plaques), the lack of pruritus, and negative personal history of atopic dermatitis.
Lichen spinulosus is an underreported entity, first described in 1908 by Adamson as superficial circumscribed chronic dermatitis in children and adolescents. The median age of presentation is age 16 years. There are several reports of possible associations with systemic infections such as HIV, fungi, and syphilis, as well as chronic diseases such as Crohn’s disease, Hodgkin disease, seborrhea, and secondary to certain medications such as omeprazole. There are no prior reports of infliximab being associated with LS, but it has been reported to cause other lichenoid reactions such as lichen planus and lichen planopilaris.
Clinically the lesions are characterized by asymptomatic, small (1 cm), skin color, hyperkeratotic, follicular papules that coalesce into plaques. The lesions usually occur on the extensor surfaces of the arms, neck, torso, and buttocks. Mild pruritus can occur in some patients.
The lesions in keratosis pilaris can be very similar to lichen spinulosus, but usually they don’t coalesce into plaques and are commonly present on the extensor surfaces of the arms, thighs, and cheeks. Histopathology of both conditions is very similar.
Another condition to consider includes papular eczema. The lesions in papular eczema tend to be pruritic and are not as circumscribed as LS lesions. Papular eczema responds well to the use of topical corticosteroids, while LS lesions usually do not. Lichen nitidus (LN) is characterized by monomorphic, skin color, 1-mm, flat-topped papules. Lesions tend to occur in crops rather than circumscribed papules forming plaques. LN most commonly presents on the extensor surface of the arms, trunk, dorsal hands, and genitalia. Koebner phenomenon is usually seen. Although uncommon in children, a more generalized type of follicular mucinosis can present very similar to lichen spinulosus. A recent study found LS-like lesions with associated hypopigmentation and hair loss should be suspicious for folliculotropic mycosis fungoides.
Keratolytics such as lactic acid, urea, and salicylic acid can help improve LS, although they do not cure it. Other reported treatments include the use of topical adapalene, tacalcitol cream, and tretinoin gel with hydroactive adhesive.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com.
The patient was diagnosed with lichen spinulosus (LS) based on the physical appearance of the lesions (hyperkeratotic spiny papules forming plaques), the lack of pruritus, and negative personal history of atopic dermatitis.
Lichen spinulosus is an underreported entity, first described in 1908 by Adamson as superficial circumscribed chronic dermatitis in children and adolescents. The median age of presentation is age 16 years. There are several reports of possible associations with systemic infections such as HIV, fungi, and syphilis, as well as chronic diseases such as Crohn’s disease, Hodgkin disease, seborrhea, and secondary to certain medications such as omeprazole. There are no prior reports of infliximab being associated with LS, but it has been reported to cause other lichenoid reactions such as lichen planus and lichen planopilaris.
Clinically the lesions are characterized by asymptomatic, small (1 cm), skin color, hyperkeratotic, follicular papules that coalesce into plaques. The lesions usually occur on the extensor surfaces of the arms, neck, torso, and buttocks. Mild pruritus can occur in some patients.
The lesions in keratosis pilaris can be very similar to lichen spinulosus, but usually they don’t coalesce into plaques and are commonly present on the extensor surfaces of the arms, thighs, and cheeks. Histopathology of both conditions is very similar.
Another condition to consider includes papular eczema. The lesions in papular eczema tend to be pruritic and are not as circumscribed as LS lesions. Papular eczema responds well to the use of topical corticosteroids, while LS lesions usually do not. Lichen nitidus (LN) is characterized by monomorphic, skin color, 1-mm, flat-topped papules. Lesions tend to occur in crops rather than circumscribed papules forming plaques. LN most commonly presents on the extensor surface of the arms, trunk, dorsal hands, and genitalia. Koebner phenomenon is usually seen. Although uncommon in children, a more generalized type of follicular mucinosis can present very similar to lichen spinulosus. A recent study found LS-like lesions with associated hypopigmentation and hair loss should be suspicious for folliculotropic mycosis fungoides.
Keratolytics such as lactic acid, urea, and salicylic acid can help improve LS, although they do not cure it. Other reported treatments include the use of topical adapalene, tacalcitol cream, and tretinoin gel with hydroactive adhesive.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com.
The patient was diagnosed with lichen spinulosus (LS) based on the physical appearance of the lesions (hyperkeratotic spiny papules forming plaques), the lack of pruritus, and negative personal history of atopic dermatitis.
Lichen spinulosus is an underreported entity, first described in 1908 by Adamson as superficial circumscribed chronic dermatitis in children and adolescents. The median age of presentation is age 16 years. There are several reports of possible associations with systemic infections such as HIV, fungi, and syphilis, as well as chronic diseases such as Crohn’s disease, Hodgkin disease, seborrhea, and secondary to certain medications such as omeprazole. There are no prior reports of infliximab being associated with LS, but it has been reported to cause other lichenoid reactions such as lichen planus and lichen planopilaris.
Clinically the lesions are characterized by asymptomatic, small (1 cm), skin color, hyperkeratotic, follicular papules that coalesce into plaques. The lesions usually occur on the extensor surfaces of the arms, neck, torso, and buttocks. Mild pruritus can occur in some patients.
The lesions in keratosis pilaris can be very similar to lichen spinulosus, but usually they don’t coalesce into plaques and are commonly present on the extensor surfaces of the arms, thighs, and cheeks. Histopathology of both conditions is very similar.
Another condition to consider includes papular eczema. The lesions in papular eczema tend to be pruritic and are not as circumscribed as LS lesions. Papular eczema responds well to the use of topical corticosteroids, while LS lesions usually do not. Lichen nitidus (LN) is characterized by monomorphic, skin color, 1-mm, flat-topped papules. Lesions tend to occur in crops rather than circumscribed papules forming plaques. LN most commonly presents on the extensor surface of the arms, trunk, dorsal hands, and genitalia. Koebner phenomenon is usually seen. Although uncommon in children, a more generalized type of follicular mucinosis can present very similar to lichen spinulosus. A recent study found LS-like lesions with associated hypopigmentation and hair loss should be suspicious for folliculotropic mycosis fungoides.
Keratolytics such as lactic acid, urea, and salicylic acid can help improve LS, although they do not cure it. Other reported treatments include the use of topical adapalene, tacalcitol cream, and tretinoin gel with hydroactive adhesive.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com.
A 7-year-old male with a history of Crohn's disease presents with 6 months of asymptomatic, bumpy lesions on the torso and extremities. He has been using over-the-counter hydrocortisone and moisturizers without it helping. His Crohn's disease has been controlled with infliximab infusions for 2 years. The mother is concerned the rash could be a side effect of the medication.
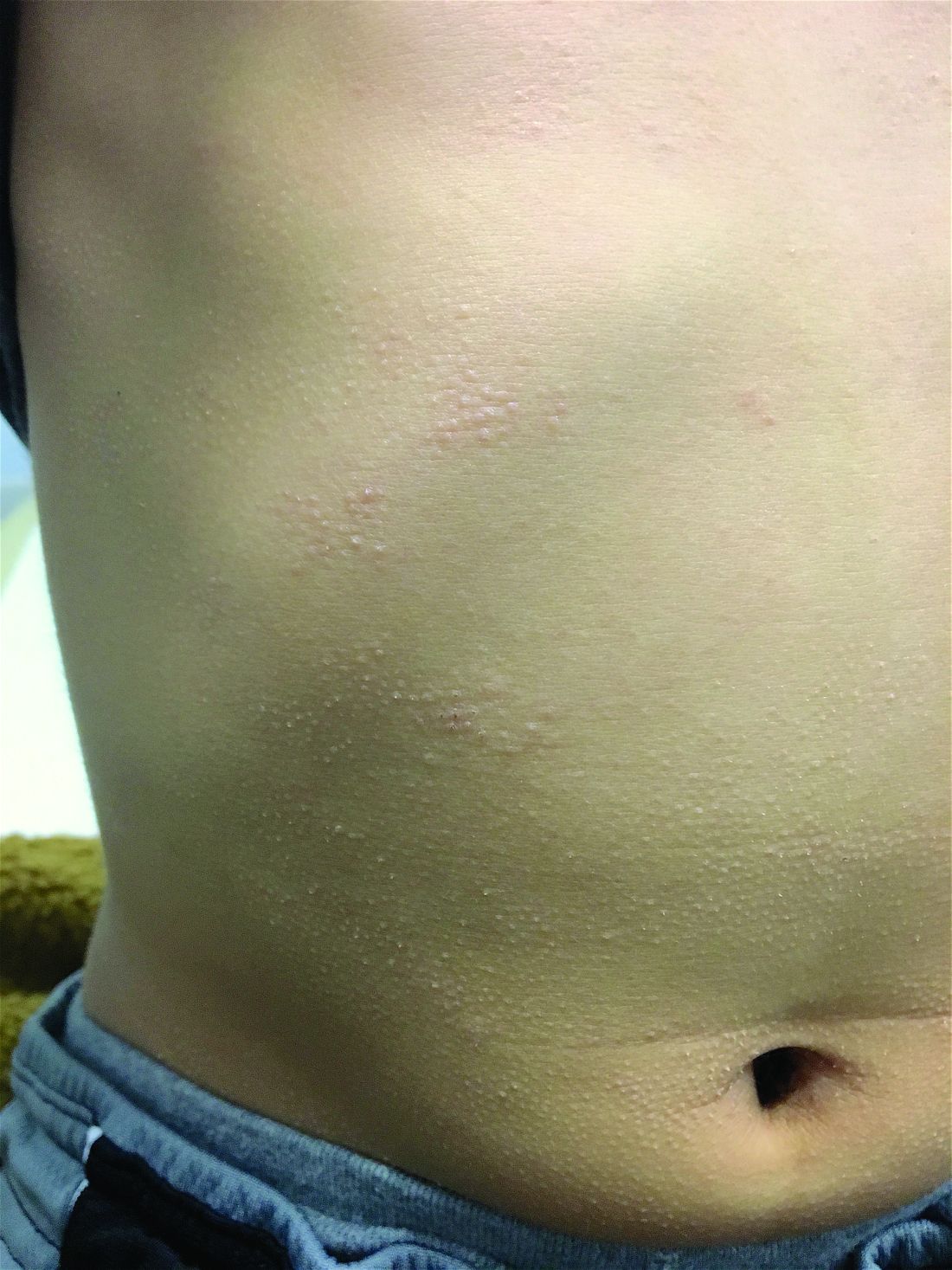
He denies any prior history of atopic dermatitis or psoriasis. The mother had eczema as a child. He has two brothers who have been diagnosed and treated for allergic rhinitis.
On physical examination, he is a thin, pleasant young boy in no distress.
His skin is somewhat dry, and there are several hyperkeratotic follicular papules forming plaques on the torso and extremities. There is no associated hair loss on the affected areas or inflammation noted.
What is your diagnosis? - December 2018
A KOH (potassium hydroxide) test done at the visit was negative as well as a fungal culture of each toenail.
The patient was diagnosed with congenital malalignment of the great toenails (CMGTN) based on history and morphologic appearance.
Congenital malalignment of the great toenails is an underrecognized and underreported nail disorder characterized by lateral deviation of the nail plate, which is not parallel to the longitudinal axis of the distal phalanx.1 The cause is unknown. Some reports suggest a genetic cause being transmitted in an autosomal dominant fashion with variable expression.2 There have been reports of CMGTN in monozygotic and dizygotic twins making this theory likely.3 Other authors consider an external cause such as amniotic bands, neonatal asphyxia, vascular malformations, and uterine pressure. This condition also has been reported in patients with Rubinstein-Taybi syndrome.4
The nail changes can occur at birth but in some cases, such as our patient, the nails become dystrophic months to years after birth. Characteristic nail changes include shorter, discolored, hyperkeratotic nails with transverse groove or ridges. In some cases, the dystrophic nails may cause inflammation and tenderness and is the most common cause of ingrown toenails in children.
The differential diagnosis includes onychomycosis, traumatic nails, nail psoriasis, pachyonychia congenital (PC), and onychomadesis. Onychomycosis can present with white or yellow discoloration of the nail that in some cases can be associated with nail breakage, hyperkeratosis, onycholysis, and subungual debris. Either fungal culture or periodic acid shift stain of nail clippings can help confirm or exclude this diagnosis. Psoriatic nails present with nail pits, oils spots, and onycholysis. Traumatic nail changes may occur from using small shoes and trauma from running or playing soccer, and presents with subungual hemorrhage and nail dystrophy of the first or second toenail. PC is a genetic disorder caused by a mutation in certain keratin proteins of the skin (k6a, k6b, K16 and K17). These patients usually have other skin findings including palmoplantar keratoderma, white plaques on the mouth, and skin cysts (steatocystoma multiplex and vellus hair cysts). Nail changes characteristic of PC includes subungual hyperkeratosis that causes a wedge shape thickening of the nail bed (pincer nails).5 Onychomadesis can be seen after viral infections such as hand-foot-mouth disease or in patients taking chemotherapy drugs that affect nail growth.
CMGTN usually resolves with time, but some patients with severe deviation and paronychia may need surgical correction.6
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com.
References
1. Dermatol Online J. 2014 Jan 15;20(1):21251.
2. J Dtsch Dermatol Ges. 2012 May;10(5):326-30.
3. J Am Acad Dermatol. 2007 Oct;57(4):711-5.
4. Pediatr Dermatol. 2004 Jan-Feb;21(1):44-7.
5. Curr Opin Pediatr. 2014 Aug;26(4):440-5.
6. Skin Appendage Disord. 2018 Oct;4(4):230-5.
A KOH (potassium hydroxide) test done at the visit was negative as well as a fungal culture of each toenail.
The patient was diagnosed with congenital malalignment of the great toenails (CMGTN) based on history and morphologic appearance.
Congenital malalignment of the great toenails is an underrecognized and underreported nail disorder characterized by lateral deviation of the nail plate, which is not parallel to the longitudinal axis of the distal phalanx.1 The cause is unknown. Some reports suggest a genetic cause being transmitted in an autosomal dominant fashion with variable expression.2 There have been reports of CMGTN in monozygotic and dizygotic twins making this theory likely.3 Other authors consider an external cause such as amniotic bands, neonatal asphyxia, vascular malformations, and uterine pressure. This condition also has been reported in patients with Rubinstein-Taybi syndrome.4
The nail changes can occur at birth but in some cases, such as our patient, the nails become dystrophic months to years after birth. Characteristic nail changes include shorter, discolored, hyperkeratotic nails with transverse groove or ridges. In some cases, the dystrophic nails may cause inflammation and tenderness and is the most common cause of ingrown toenails in children.
The differential diagnosis includes onychomycosis, traumatic nails, nail psoriasis, pachyonychia congenital (PC), and onychomadesis. Onychomycosis can present with white or yellow discoloration of the nail that in some cases can be associated with nail breakage, hyperkeratosis, onycholysis, and subungual debris. Either fungal culture or periodic acid shift stain of nail clippings can help confirm or exclude this diagnosis. Psoriatic nails present with nail pits, oils spots, and onycholysis. Traumatic nail changes may occur from using small shoes and trauma from running or playing soccer, and presents with subungual hemorrhage and nail dystrophy of the first or second toenail. PC is a genetic disorder caused by a mutation in certain keratin proteins of the skin (k6a, k6b, K16 and K17). These patients usually have other skin findings including palmoplantar keratoderma, white plaques on the mouth, and skin cysts (steatocystoma multiplex and vellus hair cysts). Nail changes characteristic of PC includes subungual hyperkeratosis that causes a wedge shape thickening of the nail bed (pincer nails).5 Onychomadesis can be seen after viral infections such as hand-foot-mouth disease or in patients taking chemotherapy drugs that affect nail growth.
CMGTN usually resolves with time, but some patients with severe deviation and paronychia may need surgical correction.6
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com.
References
1. Dermatol Online J. 2014 Jan 15;20(1):21251.
2. J Dtsch Dermatol Ges. 2012 May;10(5):326-30.
3. J Am Acad Dermatol. 2007 Oct;57(4):711-5.
4. Pediatr Dermatol. 2004 Jan-Feb;21(1):44-7.
5. Curr Opin Pediatr. 2014 Aug;26(4):440-5.
6. Skin Appendage Disord. 2018 Oct;4(4):230-5.
A KOH (potassium hydroxide) test done at the visit was negative as well as a fungal culture of each toenail.
The patient was diagnosed with congenital malalignment of the great toenails (CMGTN) based on history and morphologic appearance.
Congenital malalignment of the great toenails is an underrecognized and underreported nail disorder characterized by lateral deviation of the nail plate, which is not parallel to the longitudinal axis of the distal phalanx.1 The cause is unknown. Some reports suggest a genetic cause being transmitted in an autosomal dominant fashion with variable expression.2 There have been reports of CMGTN in monozygotic and dizygotic twins making this theory likely.3 Other authors consider an external cause such as amniotic bands, neonatal asphyxia, vascular malformations, and uterine pressure. This condition also has been reported in patients with Rubinstein-Taybi syndrome.4
The nail changes can occur at birth but in some cases, such as our patient, the nails become dystrophic months to years after birth. Characteristic nail changes include shorter, discolored, hyperkeratotic nails with transverse groove or ridges. In some cases, the dystrophic nails may cause inflammation and tenderness and is the most common cause of ingrown toenails in children.
The differential diagnosis includes onychomycosis, traumatic nails, nail psoriasis, pachyonychia congenital (PC), and onychomadesis. Onychomycosis can present with white or yellow discoloration of the nail that in some cases can be associated with nail breakage, hyperkeratosis, onycholysis, and subungual debris. Either fungal culture or periodic acid shift stain of nail clippings can help confirm or exclude this diagnosis. Psoriatic nails present with nail pits, oils spots, and onycholysis. Traumatic nail changes may occur from using small shoes and trauma from running or playing soccer, and presents with subungual hemorrhage and nail dystrophy of the first or second toenail. PC is a genetic disorder caused by a mutation in certain keratin proteins of the skin (k6a, k6b, K16 and K17). These patients usually have other skin findings including palmoplantar keratoderma, white plaques on the mouth, and skin cysts (steatocystoma multiplex and vellus hair cysts). Nail changes characteristic of PC includes subungual hyperkeratosis that causes a wedge shape thickening of the nail bed (pincer nails).5 Onychomadesis can be seen after viral infections such as hand-foot-mouth disease or in patients taking chemotherapy drugs that affect nail growth.
CMGTN usually resolves with time, but some patients with severe deviation and paronychia may need surgical correction.6
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com.
References
1. Dermatol Online J. 2014 Jan 15;20(1):21251.
2. J Dtsch Dermatol Ges. 2012 May;10(5):326-30.
3. J Am Acad Dermatol. 2007 Oct;57(4):711-5.
4. Pediatr Dermatol. 2004 Jan-Feb;21(1):44-7.
5. Curr Opin Pediatr. 2014 Aug;26(4):440-5.
6. Skin Appendage Disord. 2018 Oct;4(4):230-5.
A 4-year-old boy is brought to our pediatric dermatology clinic by his mother with the concern of difficult to treat toenail fungus.
The mother reported that she started noticing the toenail changes at around 8 months of age, and it has been progressively getting worse.
He has been treated with several courses of topical antifungals and 3 months of oral terbinafine without success.
A fungal culture done 1 year prior showed slight growth of Cladosporium Sp., but the nails failed to improve after systemic therapy. He denied any associated pain or inflammation. He likes playing softball and plays soccer sometimes. The mother is very worried because the father also has a history of onychomycosis that he has not been able to clear for years.
On physical exam, he is a very pleasant young boy. His cutaneous exam is normal including hair and teeth except for thickening of the bilateral first toenails associated with transverse ridging and yellow discoloration.
What is the Diagnosis - September 2018
At the visit, the girl’s skin scrapings were analyzed under the microscope with potassium hydroxide (KOH) and no fungal elements were seen. A culture from one of the lesions was positive for methicillin-sensitive Staphylococcus aureus.
She was diagnosed with bullous impetigo (BI).
Impetigo is the most common superficial skin infection and can present as a nonbullous (most common) and bullous (least common) form.1 Nonbullous impetigo is usually caused the Staphylococcus aureus or Streptococcus pyogenes and tends to occur at sites of prior trauma like insect bites, scratches, atopic dermatitis, or varicella. On the other hand, bullous impetigo is caused by the local production of exfoliative toxins (ETA or ETB) by phage group II of Staphylococcus aureus. The exfoliative toxin binds to desmoglin-1, one of the desmosomal proteins of the skin, causing acantholysis at the level of the granular layer and blister formation. Different from nonbullous impetigo, bullous impetigo tends to occur in normal, undamaged skin. Lesions are more common in neonates and young infants but children also can be affected.
The characteristic lesions in bullous impetigo are small blisters that enlarge to 1-cm to 5-cm bullae that easily rupture, leaving an erythematous plaque with a collarette of scale or “double ring scale,” with minimal crust and mild erythema. They commonly occur on the face, trunk, buttocks, and intertriginous areas. The lesions heal within 4-6 weeks, leaving no scarring. Associated systemic symptoms are rare but some patients can present with weakness, fever, and diarrhea. The toxin can disseminate and cause staphylococcal scalded skin syndrome in neonates or older patients with renal failure or immunodeficiency.
The transmission of Staphylococcus aureus can occur from colonized or infected family members, children in contact sports, as well as contact with animals such as dogs, cattle, and poultry.2 Transmission from a pet rabbit also has been reported. In our patient, transmission from her pet hamster could have occurred as the areas on the body where there were lesions were areas where she was holding and cuddling her new pet.
The differential diagnosis of the type of lesions our patient presented with includes tinea corporis, and bullous tinea, which also can be transmitted by animals such as kittens. A KOH analysis ruled out this diagnosis. Tinea skin lesions tend to be more scaly than bullous impetigo lesions, which are more inflamed and crusted. Bullous arthropod reactions should be considered in the differential diagnosis as well. Bullous bite reaction lesions present with tense bullae, as they are subepidermal in nature and are pruritic. Subacute cutaneous lupus lesions present as annular scaly plaques with an erythematous border and central clearing usually in sun exposed areas similar to the distribution of our patient. Severe contact dermatitis reactions also can blister and form similar lesions as seen in our patient but with the difference that our patient didn’t complain of pruritus, which is a characteristic feature of allergic contact dermatitis. In neonates or young infants with bullous lesions other conditions such as herpes simplex infection, epidermolysis bullosa, bullous pemphigoid, linear IgA bullous dermatosis, bullous mastocytosis, and bullous erythema multiforme should be considered in the differential diagnosis.
First line treatment for impetigo consists of the use of topical application of mupirocin (Bactroban) 2% ointment, retapamulin (Altabax) 1% ointment, or fusidic acid 2% cream. A Cochrane review compared systemic versus topical treatment for impetigo concluding that topical treatment with either mupirocin or retapamulin is equally if not more effective than oral antibiotics.3 Ozenoxacin (Xepi), a new nonfluorinated topical quinolone has recently been Food and Drug Administration approved for the treatment of localized impetigo in patients 2 months of age and older.4 When there is treatment failure with topical antibiotics, widespread disease, or systemic symptoms, oral antimicrobials should be consider, such as beta-lactamase–resistant penicillin, first-generation cephalosporins, or clindamycin. The use of bleach baths and general hygiene measures for 4 months can reduce the risks of recurrence in 20% of the patients as noted by a study by Kaplan et al.5
Our patient was treated with oral cephalexin for 7 days as well as topical mupirocin with fast resolution of the lesions. Sadly, the parents gave her hamster pet away.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com
References
1. Am Fam Physician. 2014 Aug 15;90(4):229-35.
2. Zentralbl Bakteriol Mikrobiol Hyg A. 1987 Jun;265(1-2):218-26.
3. Cochrane Database Syst Rev. 2012 Jan 18;1:CD003261.
4. Ann Pharmacother. 2018 Jun 1:1060028018786510.
5. Clin Infect Dis. 2014 Mar;58(5):679-82.
At the visit, the girl’s skin scrapings were analyzed under the microscope with potassium hydroxide (KOH) and no fungal elements were seen. A culture from one of the lesions was positive for methicillin-sensitive Staphylococcus aureus.
She was diagnosed with bullous impetigo (BI).
Impetigo is the most common superficial skin infection and can present as a nonbullous (most common) and bullous (least common) form.1 Nonbullous impetigo is usually caused the Staphylococcus aureus or Streptococcus pyogenes and tends to occur at sites of prior trauma like insect bites, scratches, atopic dermatitis, or varicella. On the other hand, bullous impetigo is caused by the local production of exfoliative toxins (ETA or ETB) by phage group II of Staphylococcus aureus. The exfoliative toxin binds to desmoglin-1, one of the desmosomal proteins of the skin, causing acantholysis at the level of the granular layer and blister formation. Different from nonbullous impetigo, bullous impetigo tends to occur in normal, undamaged skin. Lesions are more common in neonates and young infants but children also can be affected.
The characteristic lesions in bullous impetigo are small blisters that enlarge to 1-cm to 5-cm bullae that easily rupture, leaving an erythematous plaque with a collarette of scale or “double ring scale,” with minimal crust and mild erythema. They commonly occur on the face, trunk, buttocks, and intertriginous areas. The lesions heal within 4-6 weeks, leaving no scarring. Associated systemic symptoms are rare but some patients can present with weakness, fever, and diarrhea. The toxin can disseminate and cause staphylococcal scalded skin syndrome in neonates or older patients with renal failure or immunodeficiency.
The transmission of Staphylococcus aureus can occur from colonized or infected family members, children in contact sports, as well as contact with animals such as dogs, cattle, and poultry.2 Transmission from a pet rabbit also has been reported. In our patient, transmission from her pet hamster could have occurred as the areas on the body where there were lesions were areas where she was holding and cuddling her new pet.
The differential diagnosis of the type of lesions our patient presented with includes tinea corporis, and bullous tinea, which also can be transmitted by animals such as kittens. A KOH analysis ruled out this diagnosis. Tinea skin lesions tend to be more scaly than bullous impetigo lesions, which are more inflamed and crusted. Bullous arthropod reactions should be considered in the differential diagnosis as well. Bullous bite reaction lesions present with tense bullae, as they are subepidermal in nature and are pruritic. Subacute cutaneous lupus lesions present as annular scaly plaques with an erythematous border and central clearing usually in sun exposed areas similar to the distribution of our patient. Severe contact dermatitis reactions also can blister and form similar lesions as seen in our patient but with the difference that our patient didn’t complain of pruritus, which is a characteristic feature of allergic contact dermatitis. In neonates or young infants with bullous lesions other conditions such as herpes simplex infection, epidermolysis bullosa, bullous pemphigoid, linear IgA bullous dermatosis, bullous mastocytosis, and bullous erythema multiforme should be considered in the differential diagnosis.
First line treatment for impetigo consists of the use of topical application of mupirocin (Bactroban) 2% ointment, retapamulin (Altabax) 1% ointment, or fusidic acid 2% cream. A Cochrane review compared systemic versus topical treatment for impetigo concluding that topical treatment with either mupirocin or retapamulin is equally if not more effective than oral antibiotics.3 Ozenoxacin (Xepi), a new nonfluorinated topical quinolone has recently been Food and Drug Administration approved for the treatment of localized impetigo in patients 2 months of age and older.4 When there is treatment failure with topical antibiotics, widespread disease, or systemic symptoms, oral antimicrobials should be consider, such as beta-lactamase–resistant penicillin, first-generation cephalosporins, or clindamycin. The use of bleach baths and general hygiene measures for 4 months can reduce the risks of recurrence in 20% of the patients as noted by a study by Kaplan et al.5
Our patient was treated with oral cephalexin for 7 days as well as topical mupirocin with fast resolution of the lesions. Sadly, the parents gave her hamster pet away.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com
References
1. Am Fam Physician. 2014 Aug 15;90(4):229-35.
2. Zentralbl Bakteriol Mikrobiol Hyg A. 1987 Jun;265(1-2):218-26.
3. Cochrane Database Syst Rev. 2012 Jan 18;1:CD003261.
4. Ann Pharmacother. 2018 Jun 1:1060028018786510.
5. Clin Infect Dis. 2014 Mar;58(5):679-82.
At the visit, the girl’s skin scrapings were analyzed under the microscope with potassium hydroxide (KOH) and no fungal elements were seen. A culture from one of the lesions was positive for methicillin-sensitive Staphylococcus aureus.
She was diagnosed with bullous impetigo (BI).
Impetigo is the most common superficial skin infection and can present as a nonbullous (most common) and bullous (least common) form.1 Nonbullous impetigo is usually caused the Staphylococcus aureus or Streptococcus pyogenes and tends to occur at sites of prior trauma like insect bites, scratches, atopic dermatitis, or varicella. On the other hand, bullous impetigo is caused by the local production of exfoliative toxins (ETA or ETB) by phage group II of Staphylococcus aureus. The exfoliative toxin binds to desmoglin-1, one of the desmosomal proteins of the skin, causing acantholysis at the level of the granular layer and blister formation. Different from nonbullous impetigo, bullous impetigo tends to occur in normal, undamaged skin. Lesions are more common in neonates and young infants but children also can be affected.
The characteristic lesions in bullous impetigo are small blisters that enlarge to 1-cm to 5-cm bullae that easily rupture, leaving an erythematous plaque with a collarette of scale or “double ring scale,” with minimal crust and mild erythema. They commonly occur on the face, trunk, buttocks, and intertriginous areas. The lesions heal within 4-6 weeks, leaving no scarring. Associated systemic symptoms are rare but some patients can present with weakness, fever, and diarrhea. The toxin can disseminate and cause staphylococcal scalded skin syndrome in neonates or older patients with renal failure or immunodeficiency.
The transmission of Staphylococcus aureus can occur from colonized or infected family members, children in contact sports, as well as contact with animals such as dogs, cattle, and poultry.2 Transmission from a pet rabbit also has been reported. In our patient, transmission from her pet hamster could have occurred as the areas on the body where there were lesions were areas where she was holding and cuddling her new pet.
The differential diagnosis of the type of lesions our patient presented with includes tinea corporis, and bullous tinea, which also can be transmitted by animals such as kittens. A KOH analysis ruled out this diagnosis. Tinea skin lesions tend to be more scaly than bullous impetigo lesions, which are more inflamed and crusted. Bullous arthropod reactions should be considered in the differential diagnosis as well. Bullous bite reaction lesions present with tense bullae, as they are subepidermal in nature and are pruritic. Subacute cutaneous lupus lesions present as annular scaly plaques with an erythematous border and central clearing usually in sun exposed areas similar to the distribution of our patient. Severe contact dermatitis reactions also can blister and form similar lesions as seen in our patient but with the difference that our patient didn’t complain of pruritus, which is a characteristic feature of allergic contact dermatitis. In neonates or young infants with bullous lesions other conditions such as herpes simplex infection, epidermolysis bullosa, bullous pemphigoid, linear IgA bullous dermatosis, bullous mastocytosis, and bullous erythema multiforme should be considered in the differential diagnosis.
First line treatment for impetigo consists of the use of topical application of mupirocin (Bactroban) 2% ointment, retapamulin (Altabax) 1% ointment, or fusidic acid 2% cream. A Cochrane review compared systemic versus topical treatment for impetigo concluding that topical treatment with either mupirocin or retapamulin is equally if not more effective than oral antibiotics.3 Ozenoxacin (Xepi), a new nonfluorinated topical quinolone has recently been Food and Drug Administration approved for the treatment of localized impetigo in patients 2 months of age and older.4 When there is treatment failure with topical antibiotics, widespread disease, or systemic symptoms, oral antimicrobials should be consider, such as beta-lactamase–resistant penicillin, first-generation cephalosporins, or clindamycin. The use of bleach baths and general hygiene measures for 4 months can reduce the risks of recurrence in 20% of the patients as noted by a study by Kaplan et al.5
Our patient was treated with oral cephalexin for 7 days as well as topical mupirocin with fast resolution of the lesions. Sadly, the parents gave her hamster pet away.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com
References
1. Am Fam Physician. 2014 Aug 15;90(4):229-35.
2. Zentralbl Bakteriol Mikrobiol Hyg A. 1987 Jun;265(1-2):218-26.
3. Cochrane Database Syst Rev. 2012 Jan 18;1:CD003261.
4. Ann Pharmacother. 2018 Jun 1:1060028018786510.
5. Clin Infect Dis. 2014 Mar;58(5):679-82.
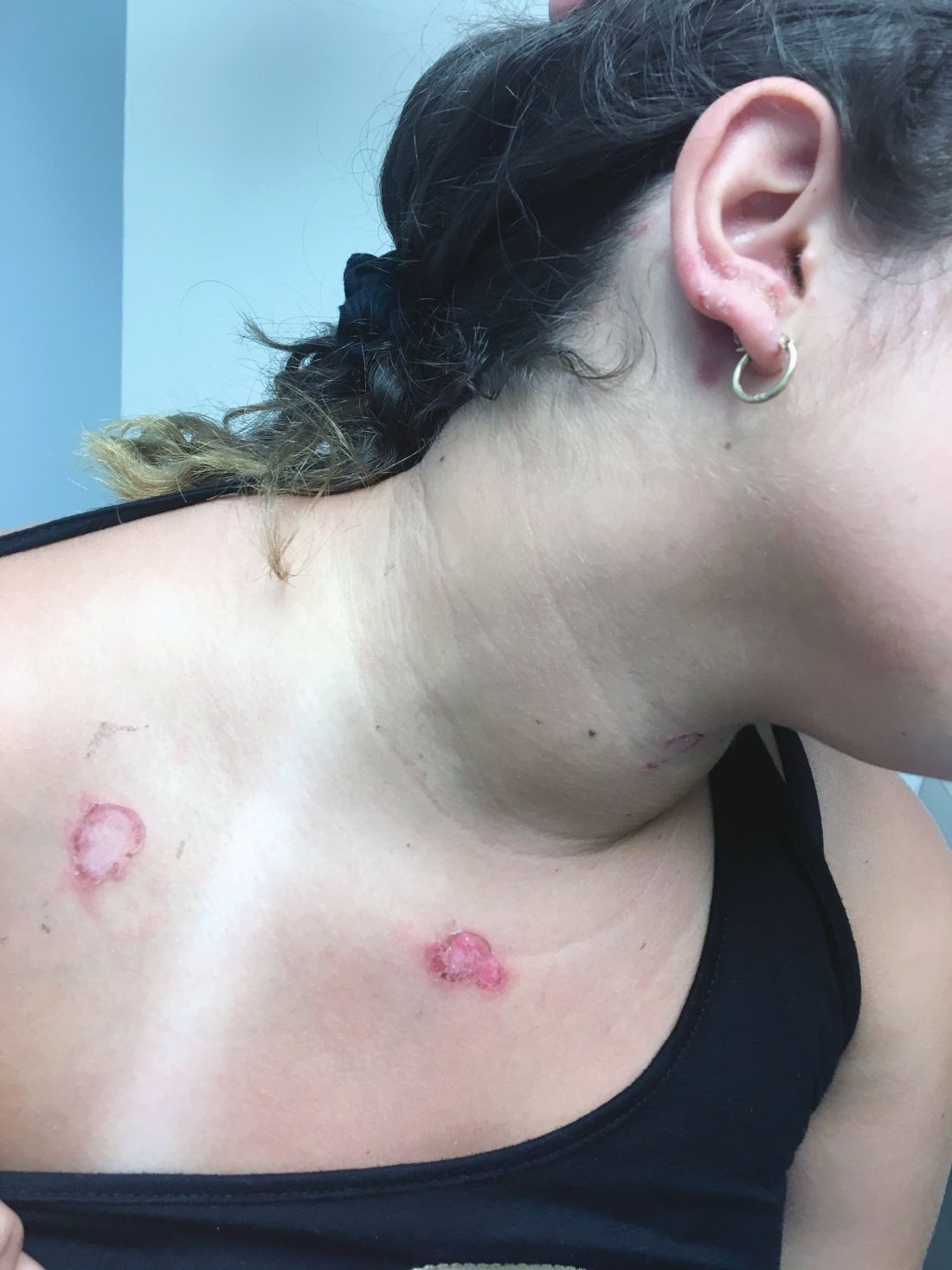
On physical exam, the girl is in no acute distress. Her vital signs are stable, and she has no fever.
On skin examination, she has several erythematous, crusted scaly plaques with double ring of scale on the nose, ears, neck, upper chest, and few on the abdomen. On her left abdomen, there is a small blister. Her seborrheic dermatitis is well controlled with mild erythema behind her ears and minimal scale on her scalp.
What is your diagnosis?
Laboratory work revealed a normal CBC and differential, an elevated C-reactive protein (CRP) and sedimentation rate (ESR), negative antistreptolysin O (ASO) titers, negative pregnancy test, a normal urinalysis, and negative blood, throat, and urine cultures. A chest x-ray also was negative as well as angiotensin-converting enzyme (ACE) levels. Tuberculosis interferon-gamma release essay was negative.
The patient was diagnosed with erythema nodosum (EN), based on physical exam and history of the lesions. In her particular case, infectious causes including streptococcus infection, tuberculosis, and coccidioidomycosis were ruled out. There were no x-ray findings that suggested sarcoidosis and her ACE level was within normal limits. The pregnancy test also was negative. Given her recent start on OCs, this was thought to be the cause of the lesions.
She was treated with elevation, compression stockings, and NSAIDs and discontinuation of OCs. The lesions resolved after 6 weeks leaving bruiselike patches (erythema contusiformis).
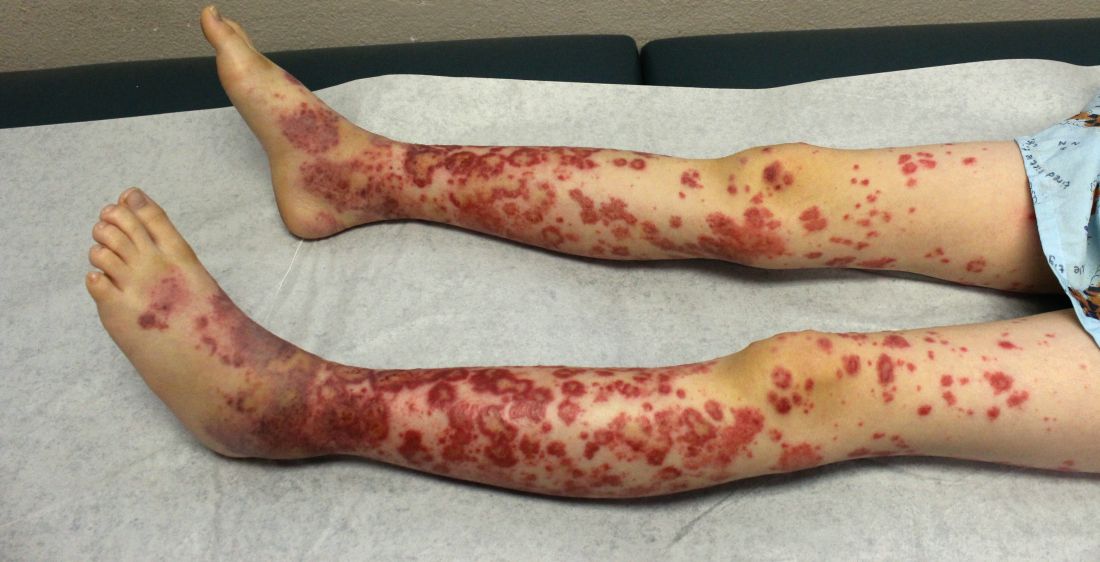
EN is a delayed-type hypersensitivity reaction, causing inflammation on the fat (panniculitis) most commonly on the shins, but it can also occur on the arms, face, neck, and thighs. It is the most common type of panniculitis and is usually seen more often in women from the second to fourth decade of life. Erythematous tender nodules in crops commonly located on the shins are the characteristic physical finding. Systemic symptoms can occur including fever, malaise, and joint pain. The lesions usually last up to 6-8 weeks and may leave bruiselike patches or postinflammatory hyperpigmentation that can take months to improve.1
The diagnosis of EN usually is made by physical examination and natural history. In unusual severe cases or lesions in atypical locations, a skin biopsy is indicated. Histologic examination of one of the lesions reveals a septal panniculitis without vasculitis. Miescher’s radial granulomas (grouped macrophages around neutrophils or septa-like spaces) often are present and are a characteristic feature of EN.
EN can be triggered by different types of infections such as streptococcus, mycoplasma, tuberculosis, or bacterial gastroenteritis; medications such as OCs, sulfonamides, iodides, penicillin, or bromides; medical conditions that include inflammatory bowel disease, pregnancy, or sarcoidosis; or neutrophilic dermatosis and malignancy such as leukemia and Hodgkin disease.2,3 A third of the cases are idiopathic. In children, streptococcal infections are responsible for most cases of EN.4
Recommended work-up to investigate possible triggers includes a CBC with differential, sedimentation rate, CRP, ASO titers or anti-DNase B titers, tuberculin skin test or interferon-gamma TB test and a chest X ray. If there are any other symptoms, physical signs, or risk factors are present for the other not so common causes, further ancillary testing may be warranted.
Erythematous nodules and papules on the shin in children are commonly caused by arthropod bites also known as papular urticaria. These lesions are pruritic rather than tender and usually respond to topical corticosteroids and oral antihistamines. Subcutaneous bacterial, fungal, or atypical mycobacterial infections can present with tender nodules that can ulcerate and drain on the shins, feet, or any other body part. These patients may have a history of immunodeficiency and usually systemic symptoms of infection are present. Cutaneous polyarteritis nodosa (PAN) also can present with tender nodules on the legs but these lesions usually necrose and ulcerate and may be associated with livedo racemosa, a transient or persistent, blotchy, reddish-blue to purple, netlike cyanotic pattern. On pathology, PAN presents with necrotizing medium vessel vasculitis. Malignant nodules also can occur on the shin. Pathology will show atypical cells. Other forms of panniculitis, such as erythema induratum and pancreatic panniculitis, can present with tender nodules but these lesions usually occur on the calves and ulcerate.
Management of EN starts with treating the underlying infection or stopping the causative medication. Initial measures include bed rest, leg elevation, compression bandages, and NSAIDs. Potassium iodide is a very effective therapy as it may control the symptoms within 24 hours. When there is no response to the above, or the patient has severe symptoms, a short course of systemic glucocorticoids can be started. Other medications for recalcitrant or recurrent lesions include colchicine, dapsone, or hydroxychloroquine.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Panniculitis, in “Dermatology,” 3rd ed. (Philadelphia: Elsevier Saunders, 2012, p. 1641).
2. Arthritis Rheum. 2000 Mar;43(3):584-92.
3. J Clin Oncol. 2007 Sep 1;25(25):4011-2.
4. Turk J Pediatr. 2014 Mar-Apr;56(2):144-9.
Laboratory work revealed a normal CBC and differential, an elevated C-reactive protein (CRP) and sedimentation rate (ESR), negative antistreptolysin O (ASO) titers, negative pregnancy test, a normal urinalysis, and negative blood, throat, and urine cultures. A chest x-ray also was negative as well as angiotensin-converting enzyme (ACE) levels. Tuberculosis interferon-gamma release essay was negative.
The patient was diagnosed with erythema nodosum (EN), based on physical exam and history of the lesions. In her particular case, infectious causes including streptococcus infection, tuberculosis, and coccidioidomycosis were ruled out. There were no x-ray findings that suggested sarcoidosis and her ACE level was within normal limits. The pregnancy test also was negative. Given her recent start on OCs, this was thought to be the cause of the lesions.
She was treated with elevation, compression stockings, and NSAIDs and discontinuation of OCs. The lesions resolved after 6 weeks leaving bruiselike patches (erythema contusiformis).
EN is a delayed-type hypersensitivity reaction, causing inflammation on the fat (panniculitis) most commonly on the shins, but it can also occur on the arms, face, neck, and thighs. It is the most common type of panniculitis and is usually seen more often in women from the second to fourth decade of life. Erythematous tender nodules in crops commonly located on the shins are the characteristic physical finding. Systemic symptoms can occur including fever, malaise, and joint pain. The lesions usually last up to 6-8 weeks and may leave bruiselike patches or postinflammatory hyperpigmentation that can take months to improve.1
The diagnosis of EN usually is made by physical examination and natural history. In unusual severe cases or lesions in atypical locations, a skin biopsy is indicated. Histologic examination of one of the lesions reveals a septal panniculitis without vasculitis. Miescher’s radial granulomas (grouped macrophages around neutrophils or septa-like spaces) often are present and are a characteristic feature of EN.
EN can be triggered by different types of infections such as streptococcus, mycoplasma, tuberculosis, or bacterial gastroenteritis; medications such as OCs, sulfonamides, iodides, penicillin, or bromides; medical conditions that include inflammatory bowel disease, pregnancy, or sarcoidosis; or neutrophilic dermatosis and malignancy such as leukemia and Hodgkin disease.2,3 A third of the cases are idiopathic. In children, streptococcal infections are responsible for most cases of EN.4
Recommended work-up to investigate possible triggers includes a CBC with differential, sedimentation rate, CRP, ASO titers or anti-DNase B titers, tuberculin skin test or interferon-gamma TB test and a chest X ray. If there are any other symptoms, physical signs, or risk factors are present for the other not so common causes, further ancillary testing may be warranted.
Erythematous nodules and papules on the shin in children are commonly caused by arthropod bites also known as papular urticaria. These lesions are pruritic rather than tender and usually respond to topical corticosteroids and oral antihistamines. Subcutaneous bacterial, fungal, or atypical mycobacterial infections can present with tender nodules that can ulcerate and drain on the shins, feet, or any other body part. These patients may have a history of immunodeficiency and usually systemic symptoms of infection are present. Cutaneous polyarteritis nodosa (PAN) also can present with tender nodules on the legs but these lesions usually necrose and ulcerate and may be associated with livedo racemosa, a transient or persistent, blotchy, reddish-blue to purple, netlike cyanotic pattern. On pathology, PAN presents with necrotizing medium vessel vasculitis. Malignant nodules also can occur on the shin. Pathology will show atypical cells. Other forms of panniculitis, such as erythema induratum and pancreatic panniculitis, can present with tender nodules but these lesions usually occur on the calves and ulcerate.
Management of EN starts with treating the underlying infection or stopping the causative medication. Initial measures include bed rest, leg elevation, compression bandages, and NSAIDs. Potassium iodide is a very effective therapy as it may control the symptoms within 24 hours. When there is no response to the above, or the patient has severe symptoms, a short course of systemic glucocorticoids can be started. Other medications for recalcitrant or recurrent lesions include colchicine, dapsone, or hydroxychloroquine.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Panniculitis, in “Dermatology,” 3rd ed. (Philadelphia: Elsevier Saunders, 2012, p. 1641).
2. Arthritis Rheum. 2000 Mar;43(3):584-92.
3. J Clin Oncol. 2007 Sep 1;25(25):4011-2.
4. Turk J Pediatr. 2014 Mar-Apr;56(2):144-9.
Laboratory work revealed a normal CBC and differential, an elevated C-reactive protein (CRP) and sedimentation rate (ESR), negative antistreptolysin O (ASO) titers, negative pregnancy test, a normal urinalysis, and negative blood, throat, and urine cultures. A chest x-ray also was negative as well as angiotensin-converting enzyme (ACE) levels. Tuberculosis interferon-gamma release essay was negative.
The patient was diagnosed with erythema nodosum (EN), based on physical exam and history of the lesions. In her particular case, infectious causes including streptococcus infection, tuberculosis, and coccidioidomycosis were ruled out. There were no x-ray findings that suggested sarcoidosis and her ACE level was within normal limits. The pregnancy test also was negative. Given her recent start on OCs, this was thought to be the cause of the lesions.
She was treated with elevation, compression stockings, and NSAIDs and discontinuation of OCs. The lesions resolved after 6 weeks leaving bruiselike patches (erythema contusiformis).
EN is a delayed-type hypersensitivity reaction, causing inflammation on the fat (panniculitis) most commonly on the shins, but it can also occur on the arms, face, neck, and thighs. It is the most common type of panniculitis and is usually seen more often in women from the second to fourth decade of life. Erythematous tender nodules in crops commonly located on the shins are the characteristic physical finding. Systemic symptoms can occur including fever, malaise, and joint pain. The lesions usually last up to 6-8 weeks and may leave bruiselike patches or postinflammatory hyperpigmentation that can take months to improve.1
The diagnosis of EN usually is made by physical examination and natural history. In unusual severe cases or lesions in atypical locations, a skin biopsy is indicated. Histologic examination of one of the lesions reveals a septal panniculitis without vasculitis. Miescher’s radial granulomas (grouped macrophages around neutrophils or septa-like spaces) often are present and are a characteristic feature of EN.
EN can be triggered by different types of infections such as streptococcus, mycoplasma, tuberculosis, or bacterial gastroenteritis; medications such as OCs, sulfonamides, iodides, penicillin, or bromides; medical conditions that include inflammatory bowel disease, pregnancy, or sarcoidosis; or neutrophilic dermatosis and malignancy such as leukemia and Hodgkin disease.2,3 A third of the cases are idiopathic. In children, streptococcal infections are responsible for most cases of EN.4
Recommended work-up to investigate possible triggers includes a CBC with differential, sedimentation rate, CRP, ASO titers or anti-DNase B titers, tuberculin skin test or interferon-gamma TB test and a chest X ray. If there are any other symptoms, physical signs, or risk factors are present for the other not so common causes, further ancillary testing may be warranted.
Erythematous nodules and papules on the shin in children are commonly caused by arthropod bites also known as papular urticaria. These lesions are pruritic rather than tender and usually respond to topical corticosteroids and oral antihistamines. Subcutaneous bacterial, fungal, or atypical mycobacterial infections can present with tender nodules that can ulcerate and drain on the shins, feet, or any other body part. These patients may have a history of immunodeficiency and usually systemic symptoms of infection are present. Cutaneous polyarteritis nodosa (PAN) also can present with tender nodules on the legs but these lesions usually necrose and ulcerate and may be associated with livedo racemosa, a transient or persistent, blotchy, reddish-blue to purple, netlike cyanotic pattern. On pathology, PAN presents with necrotizing medium vessel vasculitis. Malignant nodules also can occur on the shin. Pathology will show atypical cells. Other forms of panniculitis, such as erythema induratum and pancreatic panniculitis, can present with tender nodules but these lesions usually occur on the calves and ulcerate.
Management of EN starts with treating the underlying infection or stopping the causative medication. Initial measures include bed rest, leg elevation, compression bandages, and NSAIDs. Potassium iodide is a very effective therapy as it may control the symptoms within 24 hours. When there is no response to the above, or the patient has severe symptoms, a short course of systemic glucocorticoids can be started. Other medications for recalcitrant or recurrent lesions include colchicine, dapsone, or hydroxychloroquine.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Panniculitis, in “Dermatology,” 3rd ed. (Philadelphia: Elsevier Saunders, 2012, p. 1641).
2. Arthritis Rheum. 2000 Mar;43(3):584-92.
3. J Clin Oncol. 2007 Sep 1;25(25):4011-2.
4. Turk J Pediatr. 2014 Mar-Apr;56(2):144-9.
A 16-year-old female came to the dermatology clinic for acne follow-up. She reported some improvement on her acne since she started taking OCs. She also had been using benzoyl peroxide and tretinoin on her face. In addition to the acne, she also wanted us to check some tender bumps she had been getting on her shins after she came back from a camping trip. Initially she thought they were bug bites, but the lesions were getting larger, more tender, and not improving with diphenhydramine.
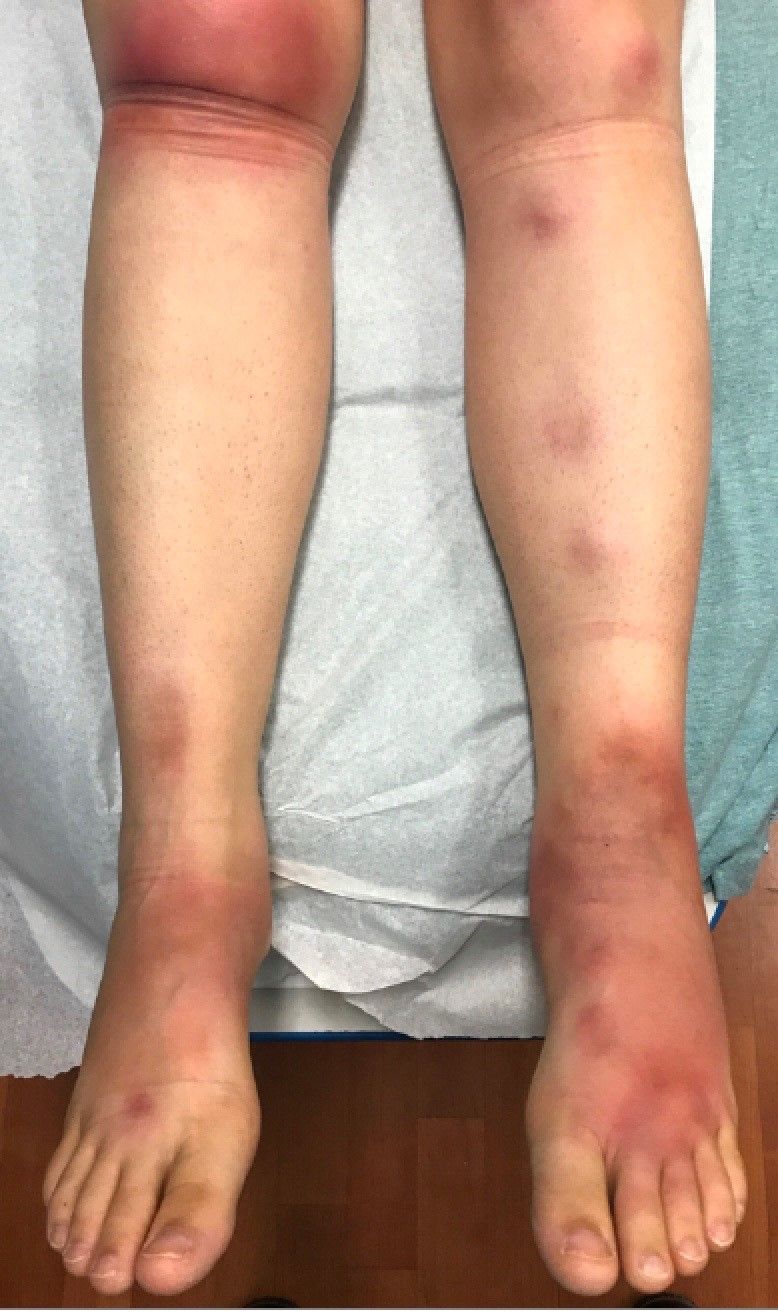
The physical exam did not reveal acute distress. She was afebrile. On skin examination, she had comedones, papules and scars on her face, chest, and back. On her shins there were several erythematous tender nodules and plaques. There was no edema on her legs and pulses were present.
Pediatric Dermatology Consult - June 2018
The patient was diagnosed with granuloma annulare on the basis of history and clinical exam. A potassium hydroxide prep of skin scrapings was performed to rule out tinea corporis, and did not show evidence of fungal elements. The patient was treated with topical betamethasone with partial improvement.
First described as a “ringed eruption of the fingers” by Thomas Colcott Fox in 1895, granuloma annulare (GA) is a relatively common, benign, and self-limited condition whose precise etiology remains unclear. It is characterized commonly by pink to violaceous aciform or annular plaques on clinical examination. In some cases of GA, annular lesions are not present, or may be formed of grouped papules.
GA is characterized histologically by patchy interstitial lymphocytes and histiocytes palisading around mucin. Deep GA, an unusual subtype observed only in children, features a fibrin rather than a mucin core. This granulomatous picture is consistent with a Th1-mediated inflammatory process, and indeed, macrophage tumor necrosis factor production, as well as interleukin-2 and interferon-gamma production have been observed in GA. The reason for this exaggerated Th1 response is unknown, although in susceptible individuals trauma3 (an example of the Koebner phenomenon), arthropod assault,4 and herpes simplex infection5 (an example of Wolf isotopic response) all have been observed to trigger localized and/or generalized GA. Generalized GA has been associated with hyperlipidemia and the human leukocyte antigen–BW35 allele. GA has been described as a paraneoplastic eruption; atypical features such as associated pain or appearance in an uncharacteristic location often are present in such cases.6,7
Diagnosis of typical GA is clinical. If unusual features make you suspect tinea, leprosy, mycosis fungoides, or other annular lesions, then biopsy showing features typical of GA can reveal the correct diagnosis. Biopsy also can help to distinguish papular GA from warts or molluscum contagiosum. If extensive GA are present, then serum lipid testing for hypercholesterolemia or hypertriglyceridemia should be considered.
Other annular and raised lesions are on the differential for GA, but careful attention to the patient’s history and examination can clarify the diagnosis. Urticaria multiforme, a variant of annular urticaria, presents with numerous annular and polycyclic wheals, sometimes with central darkening that may be mistaken for necrosis. This patient did not present with polycyclic wheals. Furthermore, the lesions in urticaria multiforme are typically transient, with individual lesions lasting less than 24 hours, which was not the case with this patient. Wells syndrome, also known as eosinophilic cellulitis, is a condition marked by recurrent episodes of pruritus followed by appearance of edematous, painful, indurated, or edematous papules or plaques, although bullae and vesicles also may be present. The face and extremities are frequently involved and spontaneous resolution typically occurs in 2 months. Annular lesions are possible but papules, plaques, and nodules are more common in Wells syndrome. Annular elastolytic giant cell granuloma (AEGCG), also known as actinic granuloma and Miescher granuloma of the face, is an entity characterized clinically by chronic, persistent, sun-distributed, annular plaques typically seen in older women with significant sun damage. AEGCG is considered by some to be a variant of GA, but if this is the case, it is a distinct subtype with different epidemiologic, clinical, and histopathologic characteristics from GA. Interstitial granulomatous dermatitis is histologically and clinically distinct from GA, presenting as subtly erythematous cords or extensive annular or serpiginous plaques in the axilla, groin, buttocks, or chest, typically in adult patients with rheumatoid arthritis, reactive arthritis, psoriatic arthritis, or ankylosing spondylitis. Tinea corporis, the clinical manifestation of cutaneous dermatophyte infection, may be mistaken for granuloma annulare. However, tinea corporis lesions are scaly, whereas GA does not scale. Histologic examination of tinea corporis reveals hyphae, which are not present in GA.
GA is a relatively common, idiopathic, benign skin disease with numerous annular and papular mimics. Absence of scale, pain, and significant pruritus are important clues to the diagnosis, and biopsy can be helpful when the diagnosis is unclear. Treatment, although not necessary, may be offered using any of a number of modalities. The most consistent and effective healer of GA, however, is time.
References
1. J Am Acad Dermatol. 1980 Sep;3(3):217-30.
2. J Am Acad Dermatol. 2016 Sep;75(3):457-65.
3. Dermatol Online J. 2013 Dec 16;19(12):20719.
4. Acta Derm Venereol. 2008;88(5):519-20.
5. J Cutan Med Surg. 2014 Nov;18(6):413-9.
6. South Med J. 1997 Oct;90(10):1056-9.
7. Am J Dermatopathol. 2003 Apr;25(2):113-6.
8. Am J Clin Dermatol. 2013 Aug;14(4):279-90.
9. Br J Dermatol. 1994 Apr;130(4):494-7.
The patient was diagnosed with granuloma annulare on the basis of history and clinical exam. A potassium hydroxide prep of skin scrapings was performed to rule out tinea corporis, and did not show evidence of fungal elements. The patient was treated with topical betamethasone with partial improvement.
First described as a “ringed eruption of the fingers” by Thomas Colcott Fox in 1895, granuloma annulare (GA) is a relatively common, benign, and self-limited condition whose precise etiology remains unclear. It is characterized commonly by pink to violaceous aciform or annular plaques on clinical examination. In some cases of GA, annular lesions are not present, or may be formed of grouped papules.
GA is characterized histologically by patchy interstitial lymphocytes and histiocytes palisading around mucin. Deep GA, an unusual subtype observed only in children, features a fibrin rather than a mucin core. This granulomatous picture is consistent with a Th1-mediated inflammatory process, and indeed, macrophage tumor necrosis factor production, as well as interleukin-2 and interferon-gamma production have been observed in GA. The reason for this exaggerated Th1 response is unknown, although in susceptible individuals trauma3 (an example of the Koebner phenomenon), arthropod assault,4 and herpes simplex infection5 (an example of Wolf isotopic response) all have been observed to trigger localized and/or generalized GA. Generalized GA has been associated with hyperlipidemia and the human leukocyte antigen–BW35 allele. GA has been described as a paraneoplastic eruption; atypical features such as associated pain or appearance in an uncharacteristic location often are present in such cases.6,7
Diagnosis of typical GA is clinical. If unusual features make you suspect tinea, leprosy, mycosis fungoides, or other annular lesions, then biopsy showing features typical of GA can reveal the correct diagnosis. Biopsy also can help to distinguish papular GA from warts or molluscum contagiosum. If extensive GA are present, then serum lipid testing for hypercholesterolemia or hypertriglyceridemia should be considered.
Other annular and raised lesions are on the differential for GA, but careful attention to the patient’s history and examination can clarify the diagnosis. Urticaria multiforme, a variant of annular urticaria, presents with numerous annular and polycyclic wheals, sometimes with central darkening that may be mistaken for necrosis. This patient did not present with polycyclic wheals. Furthermore, the lesions in urticaria multiforme are typically transient, with individual lesions lasting less than 24 hours, which was not the case with this patient. Wells syndrome, also known as eosinophilic cellulitis, is a condition marked by recurrent episodes of pruritus followed by appearance of edematous, painful, indurated, or edematous papules or plaques, although bullae and vesicles also may be present. The face and extremities are frequently involved and spontaneous resolution typically occurs in 2 months. Annular lesions are possible but papules, plaques, and nodules are more common in Wells syndrome. Annular elastolytic giant cell granuloma (AEGCG), also known as actinic granuloma and Miescher granuloma of the face, is an entity characterized clinically by chronic, persistent, sun-distributed, annular plaques typically seen in older women with significant sun damage. AEGCG is considered by some to be a variant of GA, but if this is the case, it is a distinct subtype with different epidemiologic, clinical, and histopathologic characteristics from GA. Interstitial granulomatous dermatitis is histologically and clinically distinct from GA, presenting as subtly erythematous cords or extensive annular or serpiginous plaques in the axilla, groin, buttocks, or chest, typically in adult patients with rheumatoid arthritis, reactive arthritis, psoriatic arthritis, or ankylosing spondylitis. Tinea corporis, the clinical manifestation of cutaneous dermatophyte infection, may be mistaken for granuloma annulare. However, tinea corporis lesions are scaly, whereas GA does not scale. Histologic examination of tinea corporis reveals hyphae, which are not present in GA.
GA is a relatively common, idiopathic, benign skin disease with numerous annular and papular mimics. Absence of scale, pain, and significant pruritus are important clues to the diagnosis, and biopsy can be helpful when the diagnosis is unclear. Treatment, although not necessary, may be offered using any of a number of modalities. The most consistent and effective healer of GA, however, is time.
References
1. J Am Acad Dermatol. 1980 Sep;3(3):217-30.
2. J Am Acad Dermatol. 2016 Sep;75(3):457-65.
3. Dermatol Online J. 2013 Dec 16;19(12):20719.
4. Acta Derm Venereol. 2008;88(5):519-20.
5. J Cutan Med Surg. 2014 Nov;18(6):413-9.
6. South Med J. 1997 Oct;90(10):1056-9.
7. Am J Dermatopathol. 2003 Apr;25(2):113-6.
8. Am J Clin Dermatol. 2013 Aug;14(4):279-90.
9. Br J Dermatol. 1994 Apr;130(4):494-7.
The patient was diagnosed with granuloma annulare on the basis of history and clinical exam. A potassium hydroxide prep of skin scrapings was performed to rule out tinea corporis, and did not show evidence of fungal elements. The patient was treated with topical betamethasone with partial improvement.
First described as a “ringed eruption of the fingers” by Thomas Colcott Fox in 1895, granuloma annulare (GA) is a relatively common, benign, and self-limited condition whose precise etiology remains unclear. It is characterized commonly by pink to violaceous aciform or annular plaques on clinical examination. In some cases of GA, annular lesions are not present, or may be formed of grouped papules.
GA is characterized histologically by patchy interstitial lymphocytes and histiocytes palisading around mucin. Deep GA, an unusual subtype observed only in children, features a fibrin rather than a mucin core. This granulomatous picture is consistent with a Th1-mediated inflammatory process, and indeed, macrophage tumor necrosis factor production, as well as interleukin-2 and interferon-gamma production have been observed in GA. The reason for this exaggerated Th1 response is unknown, although in susceptible individuals trauma3 (an example of the Koebner phenomenon), arthropod assault,4 and herpes simplex infection5 (an example of Wolf isotopic response) all have been observed to trigger localized and/or generalized GA. Generalized GA has been associated with hyperlipidemia and the human leukocyte antigen–BW35 allele. GA has been described as a paraneoplastic eruption; atypical features such as associated pain or appearance in an uncharacteristic location often are present in such cases.6,7
Diagnosis of typical GA is clinical. If unusual features make you suspect tinea, leprosy, mycosis fungoides, or other annular lesions, then biopsy showing features typical of GA can reveal the correct diagnosis. Biopsy also can help to distinguish papular GA from warts or molluscum contagiosum. If extensive GA are present, then serum lipid testing for hypercholesterolemia or hypertriglyceridemia should be considered.
Other annular and raised lesions are on the differential for GA, but careful attention to the patient’s history and examination can clarify the diagnosis. Urticaria multiforme, a variant of annular urticaria, presents with numerous annular and polycyclic wheals, sometimes with central darkening that may be mistaken for necrosis. This patient did not present with polycyclic wheals. Furthermore, the lesions in urticaria multiforme are typically transient, with individual lesions lasting less than 24 hours, which was not the case with this patient. Wells syndrome, also known as eosinophilic cellulitis, is a condition marked by recurrent episodes of pruritus followed by appearance of edematous, painful, indurated, or edematous papules or plaques, although bullae and vesicles also may be present. The face and extremities are frequently involved and spontaneous resolution typically occurs in 2 months. Annular lesions are possible but papules, plaques, and nodules are more common in Wells syndrome. Annular elastolytic giant cell granuloma (AEGCG), also known as actinic granuloma and Miescher granuloma of the face, is an entity characterized clinically by chronic, persistent, sun-distributed, annular plaques typically seen in older women with significant sun damage. AEGCG is considered by some to be a variant of GA, but if this is the case, it is a distinct subtype with different epidemiologic, clinical, and histopathologic characteristics from GA. Interstitial granulomatous dermatitis is histologically and clinically distinct from GA, presenting as subtly erythematous cords or extensive annular or serpiginous plaques in the axilla, groin, buttocks, or chest, typically in adult patients with rheumatoid arthritis, reactive arthritis, psoriatic arthritis, or ankylosing spondylitis. Tinea corporis, the clinical manifestation of cutaneous dermatophyte infection, may be mistaken for granuloma annulare. However, tinea corporis lesions are scaly, whereas GA does not scale. Histologic examination of tinea corporis reveals hyphae, which are not present in GA.
GA is a relatively common, idiopathic, benign skin disease with numerous annular and papular mimics. Absence of scale, pain, and significant pruritus are important clues to the diagnosis, and biopsy can be helpful when the diagnosis is unclear. Treatment, although not necessary, may be offered using any of a number of modalities. The most consistent and effective healer of GA, however, is time.
References
1. J Am Acad Dermatol. 1980 Sep;3(3):217-30.
2. J Am Acad Dermatol. 2016 Sep;75(3):457-65.
3. Dermatol Online J. 2013 Dec 16;19(12):20719.
4. Acta Derm Venereol. 2008;88(5):519-20.
5. J Cutan Med Surg. 2014 Nov;18(6):413-9.
6. South Med J. 1997 Oct;90(10):1056-9.
7. Am J Dermatopathol. 2003 Apr;25(2):113-6.
8. Am J Clin Dermatol. 2013 Aug;14(4):279-90.
9. Br J Dermatol. 1994 Apr;130(4):494-7.
A 9-year-old girl presented to the dermatology clinic, referred by her pediatrician, for evaluation of asymptomatic lesions on her shins and feet for 2 months. She started developing one lesion over her right shin with other lesions appearing on the opposite leg a few weeks after, and treated the areas initially with an over-the-counter antifungal cream without improvement. She has been healthy and denied any recent fevers or upper respiratory infections, and said she had not taken any medications or vitamin supplements. She reported camping with her father occasionally but denied any bug or tick bites. No other family members were affected. There was no personal or family history of diabetes mellitus or high cholesterol, and there are no pets at home.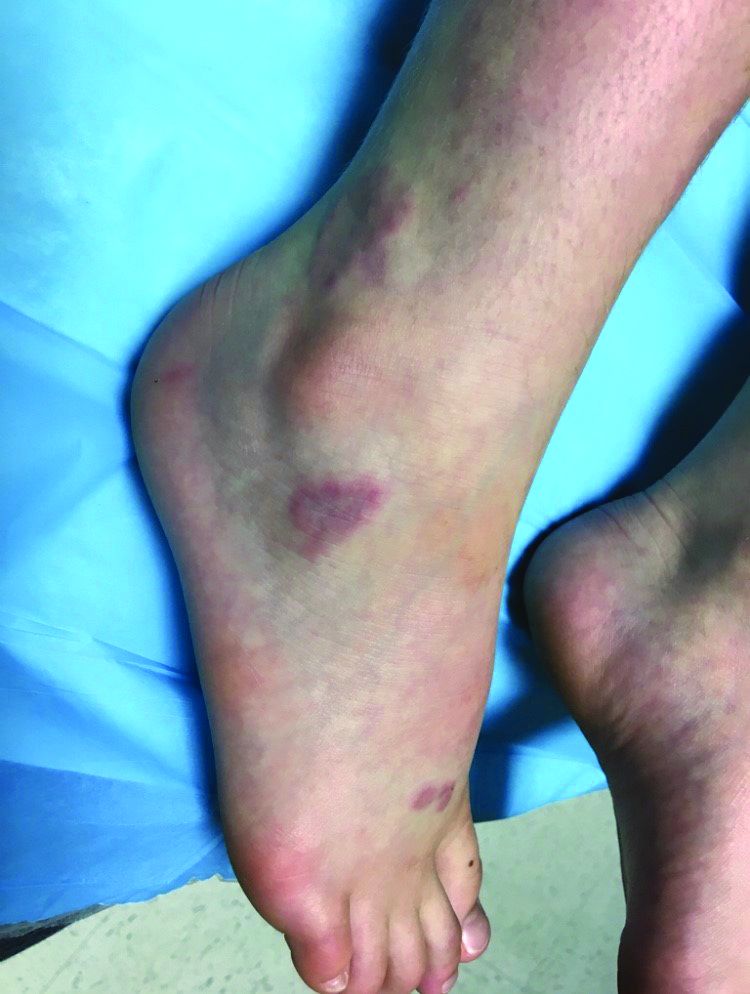
Pediatric Dermatology Consult - February 2018
The patient was diagnosed with pityriasis rosea (PR) on the basis of the clinical findings; a biopsy was not performed. The patient’s pruritus was treated with oral hydroxyzine and topical 1% triamcinolone ointment. She experienced itch relief with these treatments. On follow-up at 3 months, the patient’s lesions had mostly resolved with some postinflammatory hyperpigmentation.
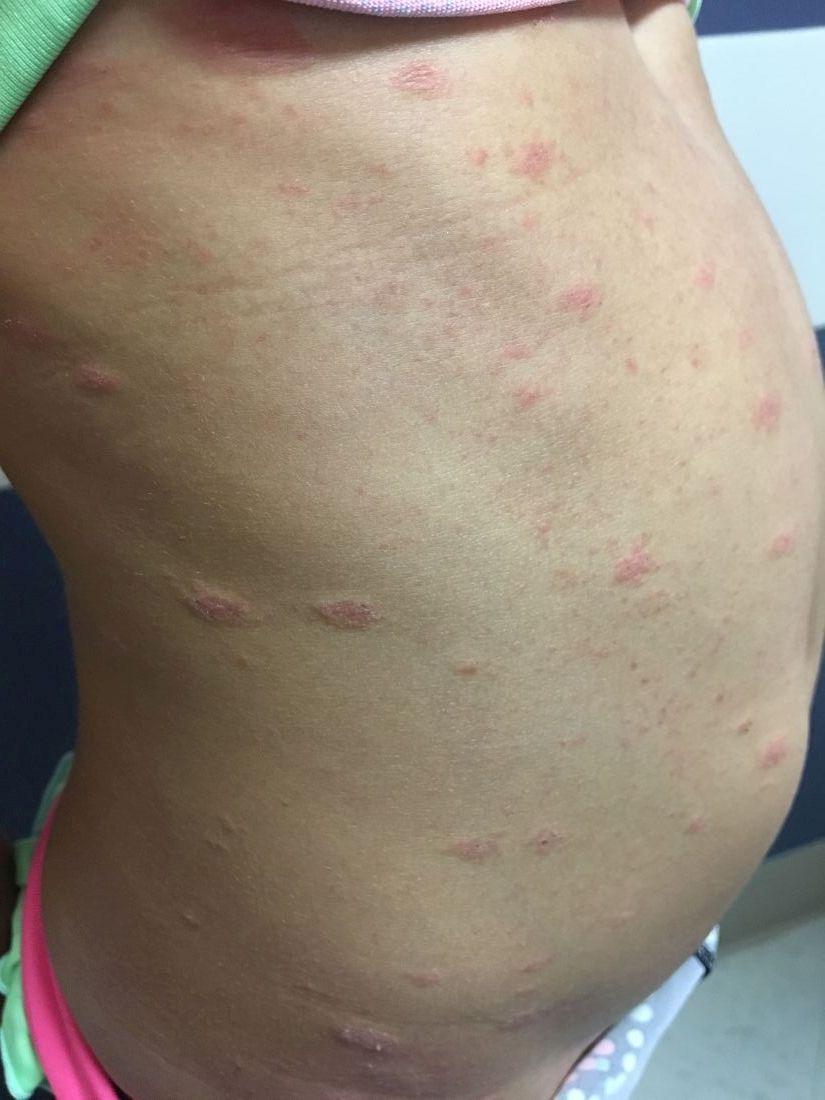
In some patients, flu-like symptoms precede the onset of skin lesions; this has led to speculation regarding a viral etiology for PR. This prodrome, which is present in as many as half of all cases,can include mild headache, low-grade fever, joint aches, or malaise.2 Pityriasis rosea is thought to occur secondary to a systemic activation of human herpesviruses (HHV) 6 and/or HHV-7. Three cases of PR have been reported in the setting of H1N1 influenza virus infection.3 In one small study, HHV-8 was detected by polymerase chain reaction in approximately 20% of biopsy samples of lesional skin in patients with PR.4 However, most research on a viral etiology for pityriasis rosea has focused on HHV-6 and to a lesser extent HHV-7. DNA from both viruses has been isolated from PR lesions, but at varying detection rates.5,6 Furthermore, HHV-7 DNA has been isolated in as many as 14% of normal individuals without pityriasis rosea, suggesting that the presence of this virus on the skin is fairly common.7
Pityriasis rosea occurs in males and females of all ethnicities, with a slight female predominance. It is rare in young children and older adults. Most cases occur in adolescents and in adults in their twenties and early thirties. Cases occur most frequently in fall and spring.8
The herald patch of pityriasis rosea is typically solitary, but cases with multiple herald patches have been described. The herald patch can range in size from 1-10 cm and usually contains the best example of trailing scale – scale seen on the inside edge of the annular lesion. The satellite lesions of pityriasis rosea are typically papules or plaques with a collarette of scale. These lesions usually are oriented along the Langer cleavage lines, giving them a “Christmas tree” configuration when they appear on the posterior trunk.
Mimics
The herald patch of pityriasis rosea can resemble tinea corporis, and if there is any doubt as to the diagnosis, potassium hydroxide examination (also known as a KOH test) and/or fungal culture should be done to rule out a fungal etiology. However, certain features of this case, particularly the subsequent development of satellite lesions, are more consistent with pityriasis rosea.
Secondary syphilis should be considered in patients who are sexually active. The lesions of secondary syphilis are not typically pruritic, and involvement of the palms and soles is common (whereas such involvement is rare in pityriasis rosea).
Like pityriasis rosea, pityriasis lichenoides et varioliformis acuta (PLEVA) is characterized by papular lesions that resolve spontaneously; the lesions of PLEVA usually evolve to vesicular, necrotic, and purpuric papules that take longer to resolve than PR lesions. The lesions of PLEVA are more erythematous, pustular, and crusting than the lesions of pityriasis rosea.
Guttate psoriasis, which occurs following streptococcal pharyngitis in over 50% of patients, does not present with a herald lesion or distribution along Langer’s lines.10 If guttate psoriasis is suspected, rapid streptococcal testing of the throat or perianal area may be considered.
Nummular eczema presents as papules that enlarge to form erythematous, lichenified plaques that measure 1-2 cm in diameter. A relatively sudden eruption, such as this patient’s, would be unusual for nummular eczema. Also, nummular eczema typically occurs on xerotic skin, more often on the extremities than the trunk.
Diagnostic tests, treatment
Most patients do not require specific therapy for pityriasis rosea. Patients should be reassured that PR is typically a self-limited disease without long-term sequelae. Pregnant patients who develop pityriasis rosea in the first trimester may be at higher risk for spontaneous abortion,although data on the subject are sorely lacking.11 Oral antihistamines are useful in reducing pruritus associated with PR, and some patients experience relief by applying a low-potency topical corticosteroid.
In more severe cases, or in cases in which the patient is greatly distressed by the lesions, both broadband and narrowband UVB phototherapy effectively improve severity of lesions and reduces symptoms.12 These observations suggest that moderate sun exposure can help to reduce severity of PR lesions and hasten their resolution, but no studies assessing the effect of sun exposure on pityriasis rosea symptoms have been performed.
Furthermore, the possible role of the HHV-6 in PR has led some investigators to explore the utility of acyclovir in managing pityriasis rosea.13 One group recently found that 400 mg of acyclovir three times per day for 7 days decreased the number of lesions and pruritus associated with pityriasis rosea, compared those seen in controls, at 1-month follow-up.13
Timely recognition of the diagnosis, consideration of mimics, and ample reassurance are appropriate when approaching this disease.
Mr. Kusari is with the division of pediatric and adolescent dermatology at Rady Children’s Hospital, San Diego, and the departments of dermatology and pediatrics, University of California, San Diego. Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. They have no relevant financial disclosures. Email them at pdnews@frontlinemedcom.com.
References
1. Dermatology. 2015;231(1):9-14.
2. World J Clin Cases. 2017 Jun 16;5(6):203-11.
3. Pediatr Dermatol. 2011 May-Jun;28(3):341-2.
4. J Eur Acad Dermatol Venereol. 2006 Jul;20(6):667-71.
5. Dermatology. 1997;195(4):374-8.
6. J Invest Dermatol. 2005 Jun;124(6):1234-40.
7. Arch Dermatol. 1999 Sep;135(9):1070-2.
8. J Am Acad Dermatol. 1982 Jul;7(1):80-9.
9. Iran J Pediatr. 2010 Jun;20(2):237-41.
10. J Pediatr. 1988 Dec;113(6):1037-9.
11. J Am Acad Dermatol. 2008 May;58(5 Suppl 1):S78-83.
12. J Am Acad Dermatol. 1995 Dec;33(6):996-9.
13. Indian Dermatol Online J. 2015 May-Jun;6(3):181-4.
The patient was diagnosed with pityriasis rosea (PR) on the basis of the clinical findings; a biopsy was not performed. The patient’s pruritus was treated with oral hydroxyzine and topical 1% triamcinolone ointment. She experienced itch relief with these treatments. On follow-up at 3 months, the patient’s lesions had mostly resolved with some postinflammatory hyperpigmentation.
In some patients, flu-like symptoms precede the onset of skin lesions; this has led to speculation regarding a viral etiology for PR. This prodrome, which is present in as many as half of all cases,can include mild headache, low-grade fever, joint aches, or malaise.2 Pityriasis rosea is thought to occur secondary to a systemic activation of human herpesviruses (HHV) 6 and/or HHV-7. Three cases of PR have been reported in the setting of H1N1 influenza virus infection.3 In one small study, HHV-8 was detected by polymerase chain reaction in approximately 20% of biopsy samples of lesional skin in patients with PR.4 However, most research on a viral etiology for pityriasis rosea has focused on HHV-6 and to a lesser extent HHV-7. DNA from both viruses has been isolated from PR lesions, but at varying detection rates.5,6 Furthermore, HHV-7 DNA has been isolated in as many as 14% of normal individuals without pityriasis rosea, suggesting that the presence of this virus on the skin is fairly common.7
Pityriasis rosea occurs in males and females of all ethnicities, with a slight female predominance. It is rare in young children and older adults. Most cases occur in adolescents and in adults in their twenties and early thirties. Cases occur most frequently in fall and spring.8
The herald patch of pityriasis rosea is typically solitary, but cases with multiple herald patches have been described. The herald patch can range in size from 1-10 cm and usually contains the best example of trailing scale – scale seen on the inside edge of the annular lesion. The satellite lesions of pityriasis rosea are typically papules or plaques with a collarette of scale. These lesions usually are oriented along the Langer cleavage lines, giving them a “Christmas tree” configuration when they appear on the posterior trunk.
Mimics
The herald patch of pityriasis rosea can resemble tinea corporis, and if there is any doubt as to the diagnosis, potassium hydroxide examination (also known as a KOH test) and/or fungal culture should be done to rule out a fungal etiology. However, certain features of this case, particularly the subsequent development of satellite lesions, are more consistent with pityriasis rosea.
Secondary syphilis should be considered in patients who are sexually active. The lesions of secondary syphilis are not typically pruritic, and involvement of the palms and soles is common (whereas such involvement is rare in pityriasis rosea).
Like pityriasis rosea, pityriasis lichenoides et varioliformis acuta (PLEVA) is characterized by papular lesions that resolve spontaneously; the lesions of PLEVA usually evolve to vesicular, necrotic, and purpuric papules that take longer to resolve than PR lesions. The lesions of PLEVA are more erythematous, pustular, and crusting than the lesions of pityriasis rosea.
Guttate psoriasis, which occurs following streptococcal pharyngitis in over 50% of patients, does not present with a herald lesion or distribution along Langer’s lines.10 If guttate psoriasis is suspected, rapid streptococcal testing of the throat or perianal area may be considered.
Nummular eczema presents as papules that enlarge to form erythematous, lichenified plaques that measure 1-2 cm in diameter. A relatively sudden eruption, such as this patient’s, would be unusual for nummular eczema. Also, nummular eczema typically occurs on xerotic skin, more often on the extremities than the trunk.
Diagnostic tests, treatment
Most patients do not require specific therapy for pityriasis rosea. Patients should be reassured that PR is typically a self-limited disease without long-term sequelae. Pregnant patients who develop pityriasis rosea in the first trimester may be at higher risk for spontaneous abortion,although data on the subject are sorely lacking.11 Oral antihistamines are useful in reducing pruritus associated with PR, and some patients experience relief by applying a low-potency topical corticosteroid.
In more severe cases, or in cases in which the patient is greatly distressed by the lesions, both broadband and narrowband UVB phototherapy effectively improve severity of lesions and reduces symptoms.12 These observations suggest that moderate sun exposure can help to reduce severity of PR lesions and hasten their resolution, but no studies assessing the effect of sun exposure on pityriasis rosea symptoms have been performed.
Furthermore, the possible role of the HHV-6 in PR has led some investigators to explore the utility of acyclovir in managing pityriasis rosea.13 One group recently found that 400 mg of acyclovir three times per day for 7 days decreased the number of lesions and pruritus associated with pityriasis rosea, compared those seen in controls, at 1-month follow-up.13
Timely recognition of the diagnosis, consideration of mimics, and ample reassurance are appropriate when approaching this disease.
Mr. Kusari is with the division of pediatric and adolescent dermatology at Rady Children’s Hospital, San Diego, and the departments of dermatology and pediatrics, University of California, San Diego. Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. They have no relevant financial disclosures. Email them at pdnews@frontlinemedcom.com.
References
1. Dermatology. 2015;231(1):9-14.
2. World J Clin Cases. 2017 Jun 16;5(6):203-11.
3. Pediatr Dermatol. 2011 May-Jun;28(3):341-2.
4. J Eur Acad Dermatol Venereol. 2006 Jul;20(6):667-71.
5. Dermatology. 1997;195(4):374-8.
6. J Invest Dermatol. 2005 Jun;124(6):1234-40.
7. Arch Dermatol. 1999 Sep;135(9):1070-2.
8. J Am Acad Dermatol. 1982 Jul;7(1):80-9.
9. Iran J Pediatr. 2010 Jun;20(2):237-41.
10. J Pediatr. 1988 Dec;113(6):1037-9.
11. J Am Acad Dermatol. 2008 May;58(5 Suppl 1):S78-83.
12. J Am Acad Dermatol. 1995 Dec;33(6):996-9.
13. Indian Dermatol Online J. 2015 May-Jun;6(3):181-4.
The patient was diagnosed with pityriasis rosea (PR) on the basis of the clinical findings; a biopsy was not performed. The patient’s pruritus was treated with oral hydroxyzine and topical 1% triamcinolone ointment. She experienced itch relief with these treatments. On follow-up at 3 months, the patient’s lesions had mostly resolved with some postinflammatory hyperpigmentation.
In some patients, flu-like symptoms precede the onset of skin lesions; this has led to speculation regarding a viral etiology for PR. This prodrome, which is present in as many as half of all cases,can include mild headache, low-grade fever, joint aches, or malaise.2 Pityriasis rosea is thought to occur secondary to a systemic activation of human herpesviruses (HHV) 6 and/or HHV-7. Three cases of PR have been reported in the setting of H1N1 influenza virus infection.3 In one small study, HHV-8 was detected by polymerase chain reaction in approximately 20% of biopsy samples of lesional skin in patients with PR.4 However, most research on a viral etiology for pityriasis rosea has focused on HHV-6 and to a lesser extent HHV-7. DNA from both viruses has been isolated from PR lesions, but at varying detection rates.5,6 Furthermore, HHV-7 DNA has been isolated in as many as 14% of normal individuals without pityriasis rosea, suggesting that the presence of this virus on the skin is fairly common.7
Pityriasis rosea occurs in males and females of all ethnicities, with a slight female predominance. It is rare in young children and older adults. Most cases occur in adolescents and in adults in their twenties and early thirties. Cases occur most frequently in fall and spring.8
The herald patch of pityriasis rosea is typically solitary, but cases with multiple herald patches have been described. The herald patch can range in size from 1-10 cm and usually contains the best example of trailing scale – scale seen on the inside edge of the annular lesion. The satellite lesions of pityriasis rosea are typically papules or plaques with a collarette of scale. These lesions usually are oriented along the Langer cleavage lines, giving them a “Christmas tree” configuration when they appear on the posterior trunk.
Mimics
The herald patch of pityriasis rosea can resemble tinea corporis, and if there is any doubt as to the diagnosis, potassium hydroxide examination (also known as a KOH test) and/or fungal culture should be done to rule out a fungal etiology. However, certain features of this case, particularly the subsequent development of satellite lesions, are more consistent with pityriasis rosea.
Secondary syphilis should be considered in patients who are sexually active. The lesions of secondary syphilis are not typically pruritic, and involvement of the palms and soles is common (whereas such involvement is rare in pityriasis rosea).
Like pityriasis rosea, pityriasis lichenoides et varioliformis acuta (PLEVA) is characterized by papular lesions that resolve spontaneously; the lesions of PLEVA usually evolve to vesicular, necrotic, and purpuric papules that take longer to resolve than PR lesions. The lesions of PLEVA are more erythematous, pustular, and crusting than the lesions of pityriasis rosea.
Guttate psoriasis, which occurs following streptococcal pharyngitis in over 50% of patients, does not present with a herald lesion or distribution along Langer’s lines.10 If guttate psoriasis is suspected, rapid streptococcal testing of the throat or perianal area may be considered.
Nummular eczema presents as papules that enlarge to form erythematous, lichenified plaques that measure 1-2 cm in diameter. A relatively sudden eruption, such as this patient’s, would be unusual for nummular eczema. Also, nummular eczema typically occurs on xerotic skin, more often on the extremities than the trunk.
Diagnostic tests, treatment
Most patients do not require specific therapy for pityriasis rosea. Patients should be reassured that PR is typically a self-limited disease without long-term sequelae. Pregnant patients who develop pityriasis rosea in the first trimester may be at higher risk for spontaneous abortion,although data on the subject are sorely lacking.11 Oral antihistamines are useful in reducing pruritus associated with PR, and some patients experience relief by applying a low-potency topical corticosteroid.
In more severe cases, or in cases in which the patient is greatly distressed by the lesions, both broadband and narrowband UVB phototherapy effectively improve severity of lesions and reduces symptoms.12 These observations suggest that moderate sun exposure can help to reduce severity of PR lesions and hasten their resolution, but no studies assessing the effect of sun exposure on pityriasis rosea symptoms have been performed.
Furthermore, the possible role of the HHV-6 in PR has led some investigators to explore the utility of acyclovir in managing pityriasis rosea.13 One group recently found that 400 mg of acyclovir three times per day for 7 days decreased the number of lesions and pruritus associated with pityriasis rosea, compared those seen in controls, at 1-month follow-up.13
Timely recognition of the diagnosis, consideration of mimics, and ample reassurance are appropriate when approaching this disease.
Mr. Kusari is with the division of pediatric and adolescent dermatology at Rady Children’s Hospital, San Diego, and the departments of dermatology and pediatrics, University of California, San Diego. Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. They have no relevant financial disclosures. Email them at pdnews@frontlinemedcom.com.
References
1. Dermatology. 2015;231(1):9-14.
2. World J Clin Cases. 2017 Jun 16;5(6):203-11.
3. Pediatr Dermatol. 2011 May-Jun;28(3):341-2.
4. J Eur Acad Dermatol Venereol. 2006 Jul;20(6):667-71.
5. Dermatology. 1997;195(4):374-8.
6. J Invest Dermatol. 2005 Jun;124(6):1234-40.
7. Arch Dermatol. 1999 Sep;135(9):1070-2.
8. J Am Acad Dermatol. 1982 Jul;7(1):80-9.
9. Iran J Pediatr. 2010 Jun;20(2):237-41.
10. J Pediatr. 1988 Dec;113(6):1037-9.
11. J Am Acad Dermatol. 2008 May;58(5 Suppl 1):S78-83.
12. J Am Acad Dermatol. 1995 Dec;33(6):996-9.
13. Indian Dermatol Online J. 2015 May-Jun;6(3):181-4.
A 6-year-old female presents to the pediatric dermatology office with a 2-day history of a slightly itchy skin lesion on her back. Her birthday was a week prior, and her mother gave her a new kitten, and since then she has been playing with the kitten daily. She has tried some over-the-counter antifungal cream since the lesion first appeared, but there hasn’t been much improvement. The night prior to presenting to the office, the mother noticed more lesions developing on the child’s torso, and because of this, she became worried.
On physical exam, the patient is well appearing, and vital signs are normal. She has multiple scaly, pink, oval plaques and papules on her torso. There are no oral lesions, and her palms and soles are spared.
Pediatric Dermatology Consult - November 2017
The patient was diagnosed with Henoch-Schönlein purpura (HSP) based on clinical presentation of the lesions and associated symptoms of arthralgia and abdominal pain. Urinalysis was obtained and found to be unremarkable, at presentation and follow-up, and treatment with naproxen 5 mg/kg divided into two doses per day was started for pain relief. A prednisone taper starting at 1 mg/kg per day for 3 weeks also was started due to the presence of severe abdominal pain and bullae on exam. The patient was followed with regular urine studies and blood pressure checks for 2 months, and these also were within normal limits.
HSP, also known as anaphylactoid purpura and immunoglobulin A (IgA) vasculitis, is a small vessel leukocytoclastic vasculitis characterized by the perivascular deposition of IgA1-based immune complexes in the walls of arterioles and postcapillary venules.1 In the vast majority of cases, the condition resolves spontaneously in 4-6 weeks and does not require any specific treatment,2 although NSAIDs and systemic corticosteroids can be used for mild-to-moderate and severe pain, respectively.3
HSP is the most common vasculitis in children, with a peak incidence in boys under the age of 5 years. It occurs worldwide, more commonly among whites and Asians, less commonly among blacks, and recent studies from the Czech Republic,4 Taiwan,5 Spain,6 France,7 South Korea,8 and the United Kingdom9 have shown similar incidence rates of 10-20 per 100,000 children. HSP does occur in adults, but is less common, and is known to carry a worse prognosis – in particular, a higher risk of progression to chronic kidney disease. The disease is more commonly seen in winter months,1 unsurprisingly as upper respiratory tract infections also are more common in these months.10
Pathogenesis
The exact pathogenesis of HSP is the subject of ongoing investigation and continued controversy. Mutations and polymorphisms in mannose-binding lectin, interleukins 1 and 8, vascular endothelial growth factor, and alpha-1-antitrypsin have been associated with HSP.3 Immunoglobulin A (IgA) normally exists in two heavily glycosylated forms – IgA1 and IgA2. Abnormal glycosylation, particularly undergalactosylation, of IgA1, the predominant form of IgA in serum and mucosal secretions, has been linked to HSP.11 HSP has been associated with group A streptococcal infections, Bartonella henselae (cat scratch fever) and numerous drugs,12 although no definitive causal or mechanistic explanation has been identified.
Diagnosis
Two major diagnostic criteria for HSP are widely in use, one developed by the American College of Rheumatology (ACR) in 199013 and the other by the European League Against Rheumatism (EULAR) in 2005.14 Both the ACR and EULAR criteria include acute abdominal pain, purpura, and microscopic evidence of vasculitis. Almost all patients with HSP have cutaneous purpura, and many of these patients have palpable purpura, which is pathognomonic of a leukocytoclastic vasculitis, but palpable purpura is not needed for diagnosis. The ACR criteria additionally include age of 20 years or younger, while the EULAR criteria include arthralgias and the presence of hematuria or proteinuria. Ancillary testing usually is not required to make the diagnosis, but when the diagnosis is not clear histopathologic analysis of a skin sample can identify leukocytoclastic vasculitis. Other laboratory studies that may be needed to rule out other conditions, as well as other organ involvement, include a complete blood count, which can be done to rule out thrombocytopenia as a cause of purpura, a metabolic panel, coagulation studies, occult blood test of stool, abdominal imaging, and urinalysis (UA), which can identify proteinuria or hematuria.
Abdominal pain in HSP is believed to be a result of vasculitis of the gastric, mesenteric, and/or colic vasculature. Bleeding from the inflamed vasculature rarely can lead to gross hematochezia, frank melena, or hematemesis. One serious, potential complication of HSP-related mesenteric vasculitis is intussusception, which is otherwise rare in children older than 2 years. Intussusception should be suspected if features of the classic triad of episodic abdominal pain, sausage-shaped abdominal mass, and currant jelly stool are present. Abdominal ultrasound can help to determine whether intussusception is present.
The purpura in HSP presents in waves or crops, and crops last 5-10 days each. Complete resolution takes 4-6 weeks. If biopsy is desired to confirm the diagnosis, it should be done on a lesion less than 24 hours old. This allows for identification of perivascular IgA on histopathology: beyond 24 hours, IgG and IgM also leak out, contributing to a less specific histopathologic picture.
Accurate diagnosis of HSP is important to guide therapy and anticipate potential complications. Wegener’s granulomatosis (A), also known as granulomatosis with polyangiitis, classically involves the upper and lower respiratory tract and the kidneys, leading to a presentation of epistaxis, cough, and hypertension. It occurs more commonly in adults than children. Finkelstein disease (B), also known as acute hemorrhagic edema of infancy (AHEI), is characterized by the development of petechial, urticarial, or targetoid plaques over 24-48 hours with tender edema and fever in children aged less than 2 years. Unlike HSP, AHEI typically does not involve the gastrointestinal tract, kidneys, or joints. Biopsy of skin lesions of AHEI reveals IgA deposition and leukocytoclastic vasculitis, leading some authors to consider it a closely related entity to HSP. Microscopic polyangiitis (D) is an uncommon pauci-immune vasculitis similar to Wegener’s granulomatosis, but lacking granulomas. It presents typically in the 5th decade of life with fever, fatigue, weight loss, and renal involvement. IgA nephropathy (E), also known as synpharyngitic nephritis and Berger disease, is less likely than HSP to cause a rash, joint pain, or abdominal pain. The nomenclature of HSP (whose alternate name is IgA vasculitis) reflects the multi-organ nature of HSP in comparison to IgA nephropathy, which is more likely to be limited to the kidneys.
Treatment
Aside from intussusception and renal disease, which may result from HSP, treatment is not typically required for HSP as it resolves spontaneously. Patients with significant arthralgias are likely to benefit from NSAIDs such as naproxen 5-20 mg/kg per day, although NSAIDs should be avoided if there is significant renal dysfunction or GI bleeding. Patients with severe abdominal pain or joint pain may be more likely to benefit from oral corticosteroids, particularly prednisone 1-2 mg/kg per day. A meta-analysis showed that corticosteroids significantly reduce the duration of symptoms if given early in the course of disease.15
The prognosis is usually excellent, except for a very small sample of the population (5%) that can develop end-stage renal disease. It is recommended that all children with HSP continue monitoring blood pressure and UA either weekly or biweekly for the first 2 months and then once a month for 6-12 months.16
First described in 1801 by a British physician, HSP is a common and usually self-limited disease for which our understanding has advanced greatly over the past 2 centuries, yet for which many important questions regarding pathophysiology remain unanswered. No diagnostic tests or treatments are needed for the majority of patients. Providers should include HSP in the differential diagnosis for the child with unexplained abdominal pain, renal dysfunction, or nonthrombocytopenic purpura.
Mr. Kusari is a medical student at the University of California, San Diego. Dr. Matiz is a practicing dermatologist at Southern California Permanente Medical Group in La Mesa, California. Dr. Matiz and Mr. Kusari said they had no relevant financial disclosures. Email them at pdnews@frontlinemedcom.com.
References
1. “Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence”, 5th ed. (New York: Elsevier, 2016).
2. Lancet. 2007;369(9566):976-8.
3. “Dermatology”, 3rd ed. (Philadelphia: Elsevier Saunders, 2012).
4. J Rheumatol. 2004 Nov;31(11):2295-9.
5. Rheumatology (Oxford). 2005 May;44(5):618-22.
6. Medicine (Baltimore). 2014 Mar;93(2):106-13.
7. Rheumatology (Oxford). 2017;56(8):1358-66.
8. J Korean Med Sci. 2014 Feb;29(2):198-203.
9. Lancet. 2002 Oct 19;360(9341):1197-202.
10. Rhinology. 2015 Jun;53(2):99-106.
11. PLoS One. 2016 Nov 21;11(11):e0166700.
12. Pediatr Infect Dis J. 2002 Jan;21(1):28-31.
13. Arthritis Rheum. 1990 Aug;33(8):1114-21.
14. Ann Rheum Dis. 2006 Jul;65(7):936-41.
15. Pediatrics. 2007 Nov;120(5):1079-87.
16. Arch Dis Child. 2010 Nov;95(11):877-82.
The patient was diagnosed with Henoch-Schönlein purpura (HSP) based on clinical presentation of the lesions and associated symptoms of arthralgia and abdominal pain. Urinalysis was obtained and found to be unremarkable, at presentation and follow-up, and treatment with naproxen 5 mg/kg divided into two doses per day was started for pain relief. A prednisone taper starting at 1 mg/kg per day for 3 weeks also was started due to the presence of severe abdominal pain and bullae on exam. The patient was followed with regular urine studies and blood pressure checks for 2 months, and these also were within normal limits.
HSP, also known as anaphylactoid purpura and immunoglobulin A (IgA) vasculitis, is a small vessel leukocytoclastic vasculitis characterized by the perivascular deposition of IgA1-based immune complexes in the walls of arterioles and postcapillary venules.1 In the vast majority of cases, the condition resolves spontaneously in 4-6 weeks and does not require any specific treatment,2 although NSAIDs and systemic corticosteroids can be used for mild-to-moderate and severe pain, respectively.3
HSP is the most common vasculitis in children, with a peak incidence in boys under the age of 5 years. It occurs worldwide, more commonly among whites and Asians, less commonly among blacks, and recent studies from the Czech Republic,4 Taiwan,5 Spain,6 France,7 South Korea,8 and the United Kingdom9 have shown similar incidence rates of 10-20 per 100,000 children. HSP does occur in adults, but is less common, and is known to carry a worse prognosis – in particular, a higher risk of progression to chronic kidney disease. The disease is more commonly seen in winter months,1 unsurprisingly as upper respiratory tract infections also are more common in these months.10
Pathogenesis
The exact pathogenesis of HSP is the subject of ongoing investigation and continued controversy. Mutations and polymorphisms in mannose-binding lectin, interleukins 1 and 8, vascular endothelial growth factor, and alpha-1-antitrypsin have been associated with HSP.3 Immunoglobulin A (IgA) normally exists in two heavily glycosylated forms – IgA1 and IgA2. Abnormal glycosylation, particularly undergalactosylation, of IgA1, the predominant form of IgA in serum and mucosal secretions, has been linked to HSP.11 HSP has been associated with group A streptococcal infections, Bartonella henselae (cat scratch fever) and numerous drugs,12 although no definitive causal or mechanistic explanation has been identified.
Diagnosis
Two major diagnostic criteria for HSP are widely in use, one developed by the American College of Rheumatology (ACR) in 199013 and the other by the European League Against Rheumatism (EULAR) in 2005.14 Both the ACR and EULAR criteria include acute abdominal pain, purpura, and microscopic evidence of vasculitis. Almost all patients with HSP have cutaneous purpura, and many of these patients have palpable purpura, which is pathognomonic of a leukocytoclastic vasculitis, but palpable purpura is not needed for diagnosis. The ACR criteria additionally include age of 20 years or younger, while the EULAR criteria include arthralgias and the presence of hematuria or proteinuria. Ancillary testing usually is not required to make the diagnosis, but when the diagnosis is not clear histopathologic analysis of a skin sample can identify leukocytoclastic vasculitis. Other laboratory studies that may be needed to rule out other conditions, as well as other organ involvement, include a complete blood count, which can be done to rule out thrombocytopenia as a cause of purpura, a metabolic panel, coagulation studies, occult blood test of stool, abdominal imaging, and urinalysis (UA), which can identify proteinuria or hematuria.
Abdominal pain in HSP is believed to be a result of vasculitis of the gastric, mesenteric, and/or colic vasculature. Bleeding from the inflamed vasculature rarely can lead to gross hematochezia, frank melena, or hematemesis. One serious, potential complication of HSP-related mesenteric vasculitis is intussusception, which is otherwise rare in children older than 2 years. Intussusception should be suspected if features of the classic triad of episodic abdominal pain, sausage-shaped abdominal mass, and currant jelly stool are present. Abdominal ultrasound can help to determine whether intussusception is present.
The purpura in HSP presents in waves or crops, and crops last 5-10 days each. Complete resolution takes 4-6 weeks. If biopsy is desired to confirm the diagnosis, it should be done on a lesion less than 24 hours old. This allows for identification of perivascular IgA on histopathology: beyond 24 hours, IgG and IgM also leak out, contributing to a less specific histopathologic picture.
Accurate diagnosis of HSP is important to guide therapy and anticipate potential complications. Wegener’s granulomatosis (A), also known as granulomatosis with polyangiitis, classically involves the upper and lower respiratory tract and the kidneys, leading to a presentation of epistaxis, cough, and hypertension. It occurs more commonly in adults than children. Finkelstein disease (B), also known as acute hemorrhagic edema of infancy (AHEI), is characterized by the development of petechial, urticarial, or targetoid plaques over 24-48 hours with tender edema and fever in children aged less than 2 years. Unlike HSP, AHEI typically does not involve the gastrointestinal tract, kidneys, or joints. Biopsy of skin lesions of AHEI reveals IgA deposition and leukocytoclastic vasculitis, leading some authors to consider it a closely related entity to HSP. Microscopic polyangiitis (D) is an uncommon pauci-immune vasculitis similar to Wegener’s granulomatosis, but lacking granulomas. It presents typically in the 5th decade of life with fever, fatigue, weight loss, and renal involvement. IgA nephropathy (E), also known as synpharyngitic nephritis and Berger disease, is less likely than HSP to cause a rash, joint pain, or abdominal pain. The nomenclature of HSP (whose alternate name is IgA vasculitis) reflects the multi-organ nature of HSP in comparison to IgA nephropathy, which is more likely to be limited to the kidneys.
Treatment
Aside from intussusception and renal disease, which may result from HSP, treatment is not typically required for HSP as it resolves spontaneously. Patients with significant arthralgias are likely to benefit from NSAIDs such as naproxen 5-20 mg/kg per day, although NSAIDs should be avoided if there is significant renal dysfunction or GI bleeding. Patients with severe abdominal pain or joint pain may be more likely to benefit from oral corticosteroids, particularly prednisone 1-2 mg/kg per day. A meta-analysis showed that corticosteroids significantly reduce the duration of symptoms if given early in the course of disease.15
The prognosis is usually excellent, except for a very small sample of the population (5%) that can develop end-stage renal disease. It is recommended that all children with HSP continue monitoring blood pressure and UA either weekly or biweekly for the first 2 months and then once a month for 6-12 months.16
First described in 1801 by a British physician, HSP is a common and usually self-limited disease for which our understanding has advanced greatly over the past 2 centuries, yet for which many important questions regarding pathophysiology remain unanswered. No diagnostic tests or treatments are needed for the majority of patients. Providers should include HSP in the differential diagnosis for the child with unexplained abdominal pain, renal dysfunction, or nonthrombocytopenic purpura.
Mr. Kusari is a medical student at the University of California, San Diego. Dr. Matiz is a practicing dermatologist at Southern California Permanente Medical Group in La Mesa, California. Dr. Matiz and Mr. Kusari said they had no relevant financial disclosures. Email them at pdnews@frontlinemedcom.com.
References
1. “Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence”, 5th ed. (New York: Elsevier, 2016).
2. Lancet. 2007;369(9566):976-8.
3. “Dermatology”, 3rd ed. (Philadelphia: Elsevier Saunders, 2012).
4. J Rheumatol. 2004 Nov;31(11):2295-9.
5. Rheumatology (Oxford). 2005 May;44(5):618-22.
6. Medicine (Baltimore). 2014 Mar;93(2):106-13.
7. Rheumatology (Oxford). 2017;56(8):1358-66.
8. J Korean Med Sci. 2014 Feb;29(2):198-203.
9. Lancet. 2002 Oct 19;360(9341):1197-202.
10. Rhinology. 2015 Jun;53(2):99-106.
11. PLoS One. 2016 Nov 21;11(11):e0166700.
12. Pediatr Infect Dis J. 2002 Jan;21(1):28-31.
13. Arthritis Rheum. 1990 Aug;33(8):1114-21.
14. Ann Rheum Dis. 2006 Jul;65(7):936-41.
15. Pediatrics. 2007 Nov;120(5):1079-87.
16. Arch Dis Child. 2010 Nov;95(11):877-82.
The patient was diagnosed with Henoch-Schönlein purpura (HSP) based on clinical presentation of the lesions and associated symptoms of arthralgia and abdominal pain. Urinalysis was obtained and found to be unremarkable, at presentation and follow-up, and treatment with naproxen 5 mg/kg divided into two doses per day was started for pain relief. A prednisone taper starting at 1 mg/kg per day for 3 weeks also was started due to the presence of severe abdominal pain and bullae on exam. The patient was followed with regular urine studies and blood pressure checks for 2 months, and these also were within normal limits.
HSP, also known as anaphylactoid purpura and immunoglobulin A (IgA) vasculitis, is a small vessel leukocytoclastic vasculitis characterized by the perivascular deposition of IgA1-based immune complexes in the walls of arterioles and postcapillary venules.1 In the vast majority of cases, the condition resolves spontaneously in 4-6 weeks and does not require any specific treatment,2 although NSAIDs and systemic corticosteroids can be used for mild-to-moderate and severe pain, respectively.3
HSP is the most common vasculitis in children, with a peak incidence in boys under the age of 5 years. It occurs worldwide, more commonly among whites and Asians, less commonly among blacks, and recent studies from the Czech Republic,4 Taiwan,5 Spain,6 France,7 South Korea,8 and the United Kingdom9 have shown similar incidence rates of 10-20 per 100,000 children. HSP does occur in adults, but is less common, and is known to carry a worse prognosis – in particular, a higher risk of progression to chronic kidney disease. The disease is more commonly seen in winter months,1 unsurprisingly as upper respiratory tract infections also are more common in these months.10
Pathogenesis
The exact pathogenesis of HSP is the subject of ongoing investigation and continued controversy. Mutations and polymorphisms in mannose-binding lectin, interleukins 1 and 8, vascular endothelial growth factor, and alpha-1-antitrypsin have been associated with HSP.3 Immunoglobulin A (IgA) normally exists in two heavily glycosylated forms – IgA1 and IgA2. Abnormal glycosylation, particularly undergalactosylation, of IgA1, the predominant form of IgA in serum and mucosal secretions, has been linked to HSP.11 HSP has been associated with group A streptococcal infections, Bartonella henselae (cat scratch fever) and numerous drugs,12 although no definitive causal or mechanistic explanation has been identified.
Diagnosis
Two major diagnostic criteria for HSP are widely in use, one developed by the American College of Rheumatology (ACR) in 199013 and the other by the European League Against Rheumatism (EULAR) in 2005.14 Both the ACR and EULAR criteria include acute abdominal pain, purpura, and microscopic evidence of vasculitis. Almost all patients with HSP have cutaneous purpura, and many of these patients have palpable purpura, which is pathognomonic of a leukocytoclastic vasculitis, but palpable purpura is not needed for diagnosis. The ACR criteria additionally include age of 20 years or younger, while the EULAR criteria include arthralgias and the presence of hematuria or proteinuria. Ancillary testing usually is not required to make the diagnosis, but when the diagnosis is not clear histopathologic analysis of a skin sample can identify leukocytoclastic vasculitis. Other laboratory studies that may be needed to rule out other conditions, as well as other organ involvement, include a complete blood count, which can be done to rule out thrombocytopenia as a cause of purpura, a metabolic panel, coagulation studies, occult blood test of stool, abdominal imaging, and urinalysis (UA), which can identify proteinuria or hematuria.
Abdominal pain in HSP is believed to be a result of vasculitis of the gastric, mesenteric, and/or colic vasculature. Bleeding from the inflamed vasculature rarely can lead to gross hematochezia, frank melena, or hematemesis. One serious, potential complication of HSP-related mesenteric vasculitis is intussusception, which is otherwise rare in children older than 2 years. Intussusception should be suspected if features of the classic triad of episodic abdominal pain, sausage-shaped abdominal mass, and currant jelly stool are present. Abdominal ultrasound can help to determine whether intussusception is present.
The purpura in HSP presents in waves or crops, and crops last 5-10 days each. Complete resolution takes 4-6 weeks. If biopsy is desired to confirm the diagnosis, it should be done on a lesion less than 24 hours old. This allows for identification of perivascular IgA on histopathology: beyond 24 hours, IgG and IgM also leak out, contributing to a less specific histopathologic picture.
Accurate diagnosis of HSP is important to guide therapy and anticipate potential complications. Wegener’s granulomatosis (A), also known as granulomatosis with polyangiitis, classically involves the upper and lower respiratory tract and the kidneys, leading to a presentation of epistaxis, cough, and hypertension. It occurs more commonly in adults than children. Finkelstein disease (B), also known as acute hemorrhagic edema of infancy (AHEI), is characterized by the development of petechial, urticarial, or targetoid plaques over 24-48 hours with tender edema and fever in children aged less than 2 years. Unlike HSP, AHEI typically does not involve the gastrointestinal tract, kidneys, or joints. Biopsy of skin lesions of AHEI reveals IgA deposition and leukocytoclastic vasculitis, leading some authors to consider it a closely related entity to HSP. Microscopic polyangiitis (D) is an uncommon pauci-immune vasculitis similar to Wegener’s granulomatosis, but lacking granulomas. It presents typically in the 5th decade of life with fever, fatigue, weight loss, and renal involvement. IgA nephropathy (E), also known as synpharyngitic nephritis and Berger disease, is less likely than HSP to cause a rash, joint pain, or abdominal pain. The nomenclature of HSP (whose alternate name is IgA vasculitis) reflects the multi-organ nature of HSP in comparison to IgA nephropathy, which is more likely to be limited to the kidneys.
Treatment
Aside from intussusception and renal disease, which may result from HSP, treatment is not typically required for HSP as it resolves spontaneously. Patients with significant arthralgias are likely to benefit from NSAIDs such as naproxen 5-20 mg/kg per day, although NSAIDs should be avoided if there is significant renal dysfunction or GI bleeding. Patients with severe abdominal pain or joint pain may be more likely to benefit from oral corticosteroids, particularly prednisone 1-2 mg/kg per day. A meta-analysis showed that corticosteroids significantly reduce the duration of symptoms if given early in the course of disease.15
The prognosis is usually excellent, except for a very small sample of the population (5%) that can develop end-stage renal disease. It is recommended that all children with HSP continue monitoring blood pressure and UA either weekly or biweekly for the first 2 months and then once a month for 6-12 months.16
First described in 1801 by a British physician, HSP is a common and usually self-limited disease for which our understanding has advanced greatly over the past 2 centuries, yet for which many important questions regarding pathophysiology remain unanswered. No diagnostic tests or treatments are needed for the majority of patients. Providers should include HSP in the differential diagnosis for the child with unexplained abdominal pain, renal dysfunction, or nonthrombocytopenic purpura.
Mr. Kusari is a medical student at the University of California, San Diego. Dr. Matiz is a practicing dermatologist at Southern California Permanente Medical Group in La Mesa, California. Dr. Matiz and Mr. Kusari said they had no relevant financial disclosures. Email them at pdnews@frontlinemedcom.com.
References
1. “Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence”, 5th ed. (New York: Elsevier, 2016).
2. Lancet. 2007;369(9566):976-8.
3. “Dermatology”, 3rd ed. (Philadelphia: Elsevier Saunders, 2012).
4. J Rheumatol. 2004 Nov;31(11):2295-9.
5. Rheumatology (Oxford). 2005 May;44(5):618-22.
6. Medicine (Baltimore). 2014 Mar;93(2):106-13.
7. Rheumatology (Oxford). 2017;56(8):1358-66.
8. J Korean Med Sci. 2014 Feb;29(2):198-203.
9. Lancet. 2002 Oct 19;360(9341):1197-202.
10. Rhinology. 2015 Jun;53(2):99-106.
11. PLoS One. 2016 Nov 21;11(11):e0166700.
12. Pediatr Infect Dis J. 2002 Jan;21(1):28-31.
13. Arthritis Rheum. 1990 Aug;33(8):1114-21.
14. Ann Rheum Dis. 2006 Jul;65(7):936-41.
15. Pediatrics. 2007 Nov;120(5):1079-87.
16. Arch Dis Child. 2010 Nov;95(11):877-82.
Clinical presentation
A healthy 9-year-old boy presents with 1 week history of a rash that began as “bruises” on both ankles that subsequently ascended over a few days to the proximal lower extremities and upper extremities. The rash has been painful and pruritic at times. The patient’s mother reports regular application of hydrocortisone cream for itch and pain relief, and this has been somewhat successful.
The patient has a history of longstanding constipation and abdominal pain, but over the past week has reported abdominal pain that is different and more severe than his usual abdominal pain. This abdominal pain has limited oral intake over the past 2 days. The patient and family also report bilateral pain of the wrists and elbows, which has limited his daily activities. The patient and mother deny fevers, chills, cough, coryza, and any sick contacts.
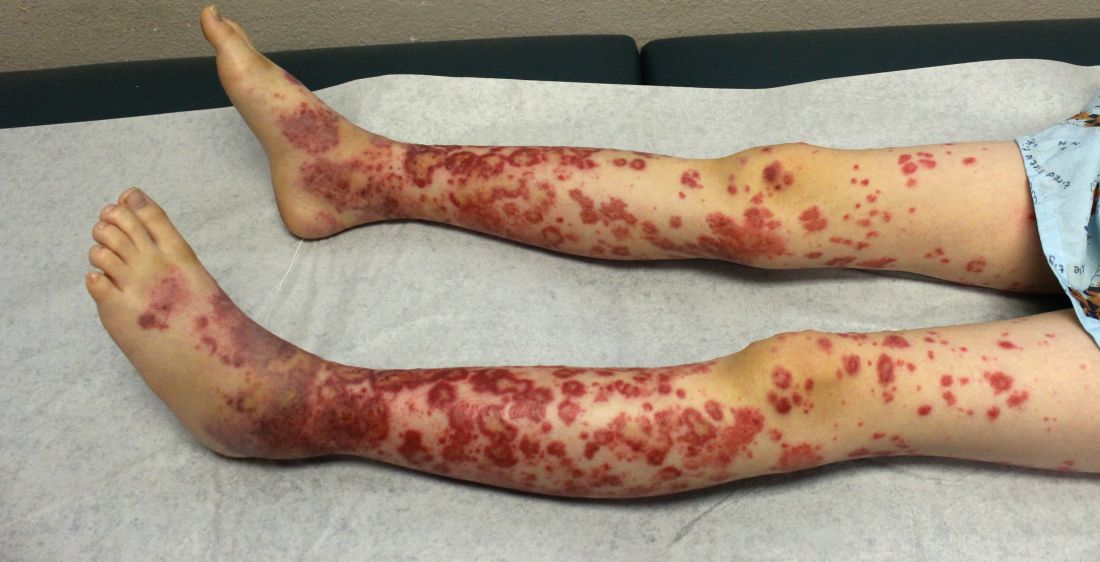
His vital signs are stable. On physical examination there is mild conjunctival injection, no intraoral lesions, and no lymphadenopathy or hepatosplenomegaly. The abdomen is not distended but it is tender to deep palpation. Bowel sounds are present. On skin examination, there are multiple purpuric annular plaques with central clearing, some with bullae and petechiae, on the bilateral buttocks and legs. There is bilateral pedal edema. On the arms, there are a few polymorphic pink and red annular to targetoid plaques.