User login
Evidence Needing a Lift
In this issue of the Journal of Hospital Medicine, Hansen and colleagues provide a first, early look at the effectiveness of the BOOST intervention to reduce 30‐day readmissions among hospitalized patients.[1] BOOST[2] is 1 of a number of care transition improvement methodologies that have been applied to the problem of readmissions, each of which has evidence to support its effectiveness in its initial settings[3, 4] but has proven to be difficult to translate to other sites.[5, 6, 7]
BOOST stands in contrast with other, largely research protocol‐derived, programs in that it allows sites to tailor adoption of recommendations to local contexts and is therefore potentially more feasible to implement. Feasibility and practicality has led BOOST to be adopted in large national settings, even if it has had little evidence to support its effectiveness to date.
Given the nonstandardized and ad hoc nature of most multicenter collaboratives generally, and the flexibility of the BOOST model specifically, the BOOST authors are to be commended for undertaking any evaluation at all. Perhaps, not surprisingly, they encountered many of the problems associated with a multicenter studydropout of sites, problematic data, and limited evidence for adoption of the intervention at participating hospitals. Although these represent real‐world experiences of a quality‐improvement program, as a group they pose a number of problems that limit the study's robustness, and generate important caveats that readers should use to temper their interpretation of the authors' findings.
The first caveat relates to the substantial number of sites that either dropped out of BOOST or failed to submit data after enlisting in the collaborative. Although this may be common in quality improvement collaboratives, similar problems would not be permissible in a trial of a new drug or device. Dropout and selected ability to contribute data suggest that the ability to fully adopt BOOST may not be universal, and raises the possibility of bias, because the least successful sites may have had less interest in remaining engaged and submitting data.
The second caveat relates to how readmission rates were assessed. Because sites provided rates of readmissions at the unit level rather than the actual counts of admissions or readmissions, the authors were unable to conduct statistical analyses typically performed for these interventions, such as time series or difference‐in‐difference analyses. More importantly, one cannot discern whether their results are driven by a small absolute but large relative change in the number of readmissions at small sites. That is, large percentage changes of low statistical significance could have misleadingly affected the overall results. Conversely, we cannot identify large sites where a similar relative reduction could be statistically significant and more broadly interpreted as representing the real effectiveness of BOOST efforts.
The third caveat is in regard to the data describing the sites' performance. The effectiveness of BOOST in this analysis varied greatly among sites, with only 1 site showing a strong reduction in readmission rate, and nearly all others showing no statistical improvements. In fact, it appears that their overall results were almost entirely driven by the improvements at that 1 site.
Variable effectiveness of an intervention can be related to variable adoption or contextual factors (such as availability of personnel to implement the program). Although these authors have data on BOOST programmatic adoption, they do not have qualitative data on local barriers and facilitators to BOOST implementation, which at this stage of evaluation would be particularly valuable in understanding the results. Analyzing site‐level effectiveness is of growing relevance to multicenter quality improvement collaboratives,[8, 9] but this evaluation provides little insight into reasons for variable success across institutions.
Finally, their study design does not allow us to understand a number of key questions. How many patients were involved in the intervention? How many patients received all BOOST‐recommended interventions? Which of these interventions seemed most effective in which patients? To what degree did patient severity of illness, cognitive status, social supports, or access to primary care influence readmission risk? Such information would help frame cost‐effective deployment of BOOST or related tools.
In the end, it seems unlikely that this iteration of the BOOST program produced broad reductions in readmission rates. Having said this, the authors provide the necessary start down the road toward a fuller understanding of real‐world efforts to reduce readmissions. Stated alternately, the nuances and flaws of this study provide ample fodder for others working in the field. BOOST is in good stead with other care transition models that have not translated well from their initial research environment to real‐world practices. The question now is: Do any of these interventions actually work in clinical practice settings, and will we ever know? Even more fundamentally, how important and meaningful are these hospital‐based care transition interventions? Where is the engagement with primary care? Where are the primary care outcomes? Does BOOST truly impact outcomes other than readmission?[10]
Doing high‐quality research in the context of a rapidly evolving quality improvement program is hard. Doing it at more than 1 site is harder. BOOST's flexibility is both a great source of strength and a clear challenge to rigorous evaluation. However, when the costs of care transition programs are so high, and the potential consequences of high readmission rates are so great for patients and for hospitals, the need to address these issues with real data and better evidence is paramount. We look forward to the next phase of BOOST and to the growth and refinement of the evidence base for how to improve care coordination and transitions effectively.
- , , , et al. Project BOOST: effectiveness of a multihospital effort to reduce rehospitalization. J Hosp Med. 2013;8:421–427.
- , . BOOSTing the hospital discharge. J Hosp Med. 2009;4:209–210.
- , , , et al. A reengineered hospital discharge program to decrease rehospitalization: a randomized trial. Ann Intern Med. 2009;150:178–187.
- , , , et al. Comprehensive discharge planning and home follow‐up of hospitalized elders: a randomized clinical trial. JAMA. 1999;281:613–620.
- , , , et al. Effectiveness and cost of a transitional care program for heart failure: a prospective study with concurrent controls. Arch Intern Med. 2011;171:1238–1243.
- . Hospitals question Medicare rules on readmissions. New York Times. March 29, 2013. Available at: http://www.nytimes.com/2013/03/30/business/hospitals‐question‐fairness‐of‐new‐medicare‐rules.html?pagewanted=all
In this issue of the Journal of Hospital Medicine, Hansen and colleagues provide a first, early look at the effectiveness of the BOOST intervention to reduce 30‐day readmissions among hospitalized patients.[1] BOOST[2] is 1 of a number of care transition improvement methodologies that have been applied to the problem of readmissions, each of which has evidence to support its effectiveness in its initial settings[3, 4] but has proven to be difficult to translate to other sites.[5, 6, 7]
BOOST stands in contrast with other, largely research protocol‐derived, programs in that it allows sites to tailor adoption of recommendations to local contexts and is therefore potentially more feasible to implement. Feasibility and practicality has led BOOST to be adopted in large national settings, even if it has had little evidence to support its effectiveness to date.
Given the nonstandardized and ad hoc nature of most multicenter collaboratives generally, and the flexibility of the BOOST model specifically, the BOOST authors are to be commended for undertaking any evaluation at all. Perhaps, not surprisingly, they encountered many of the problems associated with a multicenter studydropout of sites, problematic data, and limited evidence for adoption of the intervention at participating hospitals. Although these represent real‐world experiences of a quality‐improvement program, as a group they pose a number of problems that limit the study's robustness, and generate important caveats that readers should use to temper their interpretation of the authors' findings.
The first caveat relates to the substantial number of sites that either dropped out of BOOST or failed to submit data after enlisting in the collaborative. Although this may be common in quality improvement collaboratives, similar problems would not be permissible in a trial of a new drug or device. Dropout and selected ability to contribute data suggest that the ability to fully adopt BOOST may not be universal, and raises the possibility of bias, because the least successful sites may have had less interest in remaining engaged and submitting data.
The second caveat relates to how readmission rates were assessed. Because sites provided rates of readmissions at the unit level rather than the actual counts of admissions or readmissions, the authors were unable to conduct statistical analyses typically performed for these interventions, such as time series or difference‐in‐difference analyses. More importantly, one cannot discern whether their results are driven by a small absolute but large relative change in the number of readmissions at small sites. That is, large percentage changes of low statistical significance could have misleadingly affected the overall results. Conversely, we cannot identify large sites where a similar relative reduction could be statistically significant and more broadly interpreted as representing the real effectiveness of BOOST efforts.
The third caveat is in regard to the data describing the sites' performance. The effectiveness of BOOST in this analysis varied greatly among sites, with only 1 site showing a strong reduction in readmission rate, and nearly all others showing no statistical improvements. In fact, it appears that their overall results were almost entirely driven by the improvements at that 1 site.
Variable effectiveness of an intervention can be related to variable adoption or contextual factors (such as availability of personnel to implement the program). Although these authors have data on BOOST programmatic adoption, they do not have qualitative data on local barriers and facilitators to BOOST implementation, which at this stage of evaluation would be particularly valuable in understanding the results. Analyzing site‐level effectiveness is of growing relevance to multicenter quality improvement collaboratives,[8, 9] but this evaluation provides little insight into reasons for variable success across institutions.
Finally, their study design does not allow us to understand a number of key questions. How many patients were involved in the intervention? How many patients received all BOOST‐recommended interventions? Which of these interventions seemed most effective in which patients? To what degree did patient severity of illness, cognitive status, social supports, or access to primary care influence readmission risk? Such information would help frame cost‐effective deployment of BOOST or related tools.
In the end, it seems unlikely that this iteration of the BOOST program produced broad reductions in readmission rates. Having said this, the authors provide the necessary start down the road toward a fuller understanding of real‐world efforts to reduce readmissions. Stated alternately, the nuances and flaws of this study provide ample fodder for others working in the field. BOOST is in good stead with other care transition models that have not translated well from their initial research environment to real‐world practices. The question now is: Do any of these interventions actually work in clinical practice settings, and will we ever know? Even more fundamentally, how important and meaningful are these hospital‐based care transition interventions? Where is the engagement with primary care? Where are the primary care outcomes? Does BOOST truly impact outcomes other than readmission?[10]
Doing high‐quality research in the context of a rapidly evolving quality improvement program is hard. Doing it at more than 1 site is harder. BOOST's flexibility is both a great source of strength and a clear challenge to rigorous evaluation. However, when the costs of care transition programs are so high, and the potential consequences of high readmission rates are so great for patients and for hospitals, the need to address these issues with real data and better evidence is paramount. We look forward to the next phase of BOOST and to the growth and refinement of the evidence base for how to improve care coordination and transitions effectively.
In this issue of the Journal of Hospital Medicine, Hansen and colleagues provide a first, early look at the effectiveness of the BOOST intervention to reduce 30‐day readmissions among hospitalized patients.[1] BOOST[2] is 1 of a number of care transition improvement methodologies that have been applied to the problem of readmissions, each of which has evidence to support its effectiveness in its initial settings[3, 4] but has proven to be difficult to translate to other sites.[5, 6, 7]
BOOST stands in contrast with other, largely research protocol‐derived, programs in that it allows sites to tailor adoption of recommendations to local contexts and is therefore potentially more feasible to implement. Feasibility and practicality has led BOOST to be adopted in large national settings, even if it has had little evidence to support its effectiveness to date.
Given the nonstandardized and ad hoc nature of most multicenter collaboratives generally, and the flexibility of the BOOST model specifically, the BOOST authors are to be commended for undertaking any evaluation at all. Perhaps, not surprisingly, they encountered many of the problems associated with a multicenter studydropout of sites, problematic data, and limited evidence for adoption of the intervention at participating hospitals. Although these represent real‐world experiences of a quality‐improvement program, as a group they pose a number of problems that limit the study's robustness, and generate important caveats that readers should use to temper their interpretation of the authors' findings.
The first caveat relates to the substantial number of sites that either dropped out of BOOST or failed to submit data after enlisting in the collaborative. Although this may be common in quality improvement collaboratives, similar problems would not be permissible in a trial of a new drug or device. Dropout and selected ability to contribute data suggest that the ability to fully adopt BOOST may not be universal, and raises the possibility of bias, because the least successful sites may have had less interest in remaining engaged and submitting data.
The second caveat relates to how readmission rates were assessed. Because sites provided rates of readmissions at the unit level rather than the actual counts of admissions or readmissions, the authors were unable to conduct statistical analyses typically performed for these interventions, such as time series or difference‐in‐difference analyses. More importantly, one cannot discern whether their results are driven by a small absolute but large relative change in the number of readmissions at small sites. That is, large percentage changes of low statistical significance could have misleadingly affected the overall results. Conversely, we cannot identify large sites where a similar relative reduction could be statistically significant and more broadly interpreted as representing the real effectiveness of BOOST efforts.
The third caveat is in regard to the data describing the sites' performance. The effectiveness of BOOST in this analysis varied greatly among sites, with only 1 site showing a strong reduction in readmission rate, and nearly all others showing no statistical improvements. In fact, it appears that their overall results were almost entirely driven by the improvements at that 1 site.
Variable effectiveness of an intervention can be related to variable adoption or contextual factors (such as availability of personnel to implement the program). Although these authors have data on BOOST programmatic adoption, they do not have qualitative data on local barriers and facilitators to BOOST implementation, which at this stage of evaluation would be particularly valuable in understanding the results. Analyzing site‐level effectiveness is of growing relevance to multicenter quality improvement collaboratives,[8, 9] but this evaluation provides little insight into reasons for variable success across institutions.
Finally, their study design does not allow us to understand a number of key questions. How many patients were involved in the intervention? How many patients received all BOOST‐recommended interventions? Which of these interventions seemed most effective in which patients? To what degree did patient severity of illness, cognitive status, social supports, or access to primary care influence readmission risk? Such information would help frame cost‐effective deployment of BOOST or related tools.
In the end, it seems unlikely that this iteration of the BOOST program produced broad reductions in readmission rates. Having said this, the authors provide the necessary start down the road toward a fuller understanding of real‐world efforts to reduce readmissions. Stated alternately, the nuances and flaws of this study provide ample fodder for others working in the field. BOOST is in good stead with other care transition models that have not translated well from their initial research environment to real‐world practices. The question now is: Do any of these interventions actually work in clinical practice settings, and will we ever know? Even more fundamentally, how important and meaningful are these hospital‐based care transition interventions? Where is the engagement with primary care? Where are the primary care outcomes? Does BOOST truly impact outcomes other than readmission?[10]
Doing high‐quality research in the context of a rapidly evolving quality improvement program is hard. Doing it at more than 1 site is harder. BOOST's flexibility is both a great source of strength and a clear challenge to rigorous evaluation. However, when the costs of care transition programs are so high, and the potential consequences of high readmission rates are so great for patients and for hospitals, the need to address these issues with real data and better evidence is paramount. We look forward to the next phase of BOOST and to the growth and refinement of the evidence base for how to improve care coordination and transitions effectively.
- , , , et al. Project BOOST: effectiveness of a multihospital effort to reduce rehospitalization. J Hosp Med. 2013;8:421–427.
- , . BOOSTing the hospital discharge. J Hosp Med. 2009;4:209–210.
- , , , et al. A reengineered hospital discharge program to decrease rehospitalization: a randomized trial. Ann Intern Med. 2009;150:178–187.
- , , , et al. Comprehensive discharge planning and home follow‐up of hospitalized elders: a randomized clinical trial. JAMA. 1999;281:613–620.
- , , , et al. Effectiveness and cost of a transitional care program for heart failure: a prospective study with concurrent controls. Arch Intern Med. 2011;171:1238–1243.
- . Hospitals question Medicare rules on readmissions. New York Times. March 29, 2013. Available at: http://www.nytimes.com/2013/03/30/business/hospitals‐question‐fairness‐of‐new‐medicare‐rules.html?pagewanted=all
- , , , et al. Project BOOST: effectiveness of a multihospital effort to reduce rehospitalization. J Hosp Med. 2013;8:421–427.
- , . BOOSTing the hospital discharge. J Hosp Med. 2009;4:209–210.
- , , , et al. A reengineered hospital discharge program to decrease rehospitalization: a randomized trial. Ann Intern Med. 2009;150:178–187.
- , , , et al. Comprehensive discharge planning and home follow‐up of hospitalized elders: a randomized clinical trial. JAMA. 1999;281:613–620.
- , , , et al. Effectiveness and cost of a transitional care program for heart failure: a prospective study with concurrent controls. Arch Intern Med. 2011;171:1238–1243.
- . Hospitals question Medicare rules on readmissions. New York Times. March 29, 2013. Available at: http://www.nytimes.com/2013/03/30/business/hospitals‐question‐fairness‐of‐new‐medicare‐rules.html?pagewanted=all
Management of Blood Pressure after Stroke
Hospitalists are on the front lines of care for patients with cerebrovascular accidents. After the first steps in acute stroke management occur in the emergency room, care is frequently transferred to the hospitalist. This review focuses on the evidence‐based management of blood pressure following acute ischemic stroke. Management of blood pressure after stroke is still controversial, and current consensus statement guidelines acknowledge that optimal treatment has not yet been established.1, 2 As such, it is essential to understand the changes in normal homeostatic physiologic processes that occur after stroke and their subsequent effects on neurologic function. Only then can the appropriate blood pressure target and antihypertensive regimen be chosen.
Physiology of Cerebral Perfusion
Once a stroke has occurred and perfusion to a section of brain tissue has been acutely compromised, systemic pressure tends to rise. This rise is presumably due in part to increased adrenergic tone and activation of the renin‐aldosterone system and potentially is part of Cushing's reflex in cases in which intracranial pressure is elevated.3 The increase in mean arterial pressure may represent a protective response. In the first major study on the topic, in 1981, on admission for stroke mean blood pressure was 163/90 mm Hg for patients without a history of hypertension and 214/118 mm Hg for patients who had been treated for hypertension previously.4 This rise in blood pressure in response to endogenous mechanisms is attenuated over the first 24 hours, and even without intervention, blood pressure tends to fall spontaneously over the next 10 days.47
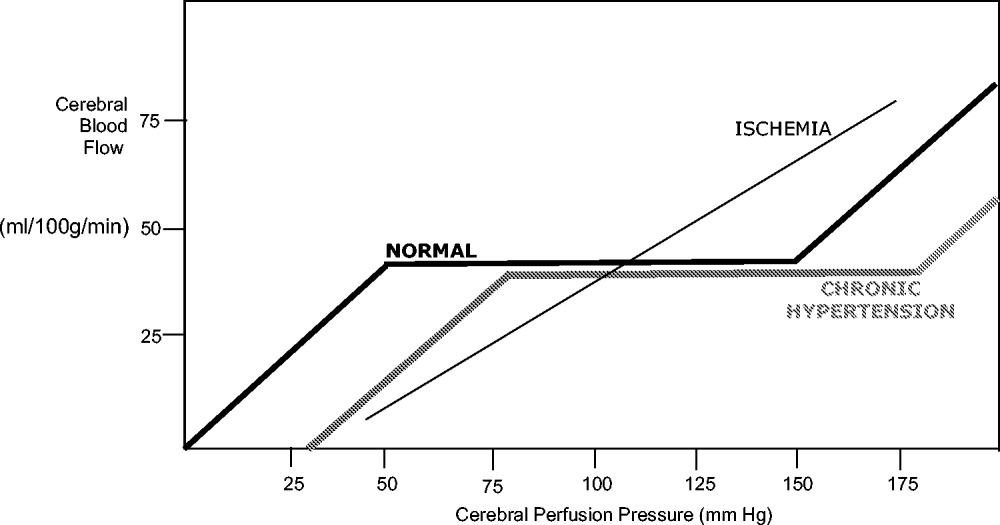
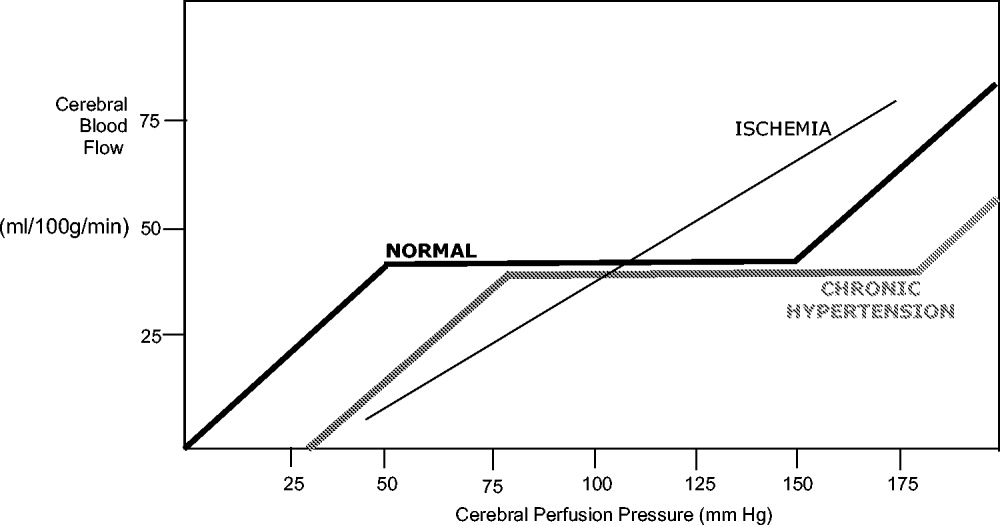
Under normal circumstances, cerebral blood flow (CBF) is tightly autoregulated across a wide range of cerebral perfusion pressures by alteration in cerebrovascular resistance via arteriolar constriction.89 This allows CBF to remain constant even if cerebral perfusion pressure (CPP) fluctuates from 60 to 150 mm Hg.9 In patients with chronic hypertension, autoregulation works best with blood pressure in a higher range because of vascular smooth muscle hypertrophy and structural changes in the cerebral vessels (see Table 1).
| Cerebral blood flow (CBF) of 50‐70 mL/100 g/minnormal |
| CBF of 20‐50 mL/100 g/minreduced flow compensated for by increased oxygen extraction |
| CBF of 15‐20 mL/100 g/minneuronal quiescence |
| CBF < 15 mL/100 g/minneuronal death |
After an acute ischemic stroke, autoregulation is lost, and local CBF becomes linearly associated with cerebral perfusion pressure. Loss of autoregulation occurs because of several local factors as a result of the infarct. Acidosis and hypoxia in the region of the stroke lead to vasodilatation in the perfusing vessels.9 This may improve local circulation in collateral vessels as a response to the obstruction in the primary supplying artery, but the consequence of maximal vasodilatation is loss of the ability to autoregulate. Normal CPP is driven by the mean arterial pressure minus the intracranial pressure. However, if intracranial vessels have lost the ability to accommodate changes in perfusion pressure, then blood flow to the area of injury becomes linearly correlated with mean arterial pressure.
Surrounding a central core of irreversible necrosis may be a zone of at‐risk tissue that is susceptible to reduction below the threshold of viability in response to any decrement in systemic mean blood pressure.11 This critical concept in stroke management is referred to as the peri‐infarct penumbra (see Fig. 1 and Table 2).
| Mean arterial pressure (MAP) = ⅔ diastolic blood pressure (DBP) + ⅓ systolic blood pressure (SBP) |
| Cerebral perfusion pressure (CPP) = mean arterial pressure (MAP) intracranial pressure (ICP) |
| Cerebral blood flow (CBF) = cerebral perfusion pressure (CPP)/cerebrovascular resistance (CVR) |
Blood Pressure Management after Stroke
The presence of a systemic blood pressure‐dependent peri‐infarct penumbra that might be compromised by blood pressure reduction and thus extend the infarct is the principal argument for allowing permissive hypertension. This is bolstered by the observation that decreases in blood pressure in the first 24 hours are associated with a significant risk of poor neurologic outcome.6 In their an observational study Vlcek et al. found that a greater than 25% drop in diastolic blood pressure (DBP) was associated with a 4‐fold risk of severe disability.5 In a 2003 observational study Oliveria et al. found that reduction in systolic blood pressure (SBP) in the first 24 hours was independently associated with poor outcomes, with a doubling of the risk for poor outcomes for every 10% drop in systolic blood pressure.6 In both these studies, the worsened outcome did not depend on whether the drop was spontaneous or induced by medications. Case reports suggest that large drops in blood pressure can be catastrophic and that even moderate lowering of blood pressure after acute stroke can be associated with clinical deterioration.8, 12
The primary argument for lowering blood pressure after an acute stroke hinges on secondary prevention of new ischemic events, minimization of cerebral edema, and prevention of hemorrhagic conversion. It has long been recognized that hypertension itself is a risk factor for stroke13 and that reduction in chronic hypertension is part of secondary prevention for cerebrovascular accidents.14 Further, hypertension is the most common risk factor for intracerebral hemorrhage, and it would stand to reason that damage to the brain parenchyma from ischemic stroke would increase the risk of pressure‐induced bleeding in an acute setting.15
Because there are compelling theoretical arguments both for and against lowering blood pressure after an acute stroke, it is necessary to look at the results of randomized clinical trials for guidance in weighing the risks and benefits. The overall goal of blood pressure management is to maximize perfusion to the ischemic penumbra while minimizing the hypertensive risk of hemorrhagic transformation.
Lisk et al. conducted a small randomized trial in 1993 of antihypertensive therapy versus placebo after ischemic stroke looking at SPECT perfusion and found lower CBF if mean arterial pressure (MAP) dropped more than 16%.16 This is in contrast with the results of a 1997 randomized trial of perindopril after cerebrovascular accident that found no decrease in Doppler CBF despite a 10% decrease in blood pressure in the active treatment group.17 Neither study was powered to detect significant differences in clinical outcome.
Early randomized controlled trials of antihypertensive agents after acute stroke investigated neuroprotection via mechanisms other than the antihypertensive effect. Nimodipine, a dihydropyridine derivative thought to prevent neuronal death via blockade of calcium channels, has been studied extensively.1820 A meta‐analysis of the 9 early trials of nimodipine, from 1988 to 1992, suggested that nimodipine was potentially beneficial in neurologic score and functional outcome only if used within the first 12 hours and that it could be harmful if started after 24 hours.21 Two additional studies were done in 1994 using both intravenous and oral nimodipine formulations. They demonstrated worsened neurologic function and higher mortality, respectively, which was hypothesized to be a result of the detrimental hemodynamic effects of nimodipine.18, 22
The BEST trial used beta‐blockers in the early period after acute stroke and failed to find benefit, whereas the FIST trial had similar results with the calcium channel blocker flunarizine.23, 24 The ACCESS trial in 2003 was a prospective, randomized, controlled trial using oral candesartan in the first 24 hours after stroke.25 It did find improved mortality in the candesartan arm after 1 year, yet there were no significant differences in blood pressure between candesartan and placebo. Thus, the improved outcome was presumed to be a result of mechanisms other than antihypertensive effect.
Some significant caveats should be kept in mind when interpreting the results of these studies. None of the trials was designed to titrate blood pressure to a prespecified goal in a prospective randomized fashion. It is also difficult to tease out the effect of the active medical intervention from the effect of spontaneous drops in blood pressure, and many trials did not find a significant difference in blood pressure between the medication and placebo groups. A large randomized trial to evaluate interventions to a predefined blood pressure target is needed.
The Cochrane Stroke Group reviewed 32 trials involving 5368 patients and concluded there was not enough evidence to reliably evaluate the effect of altering blood pressure on outcomes.3
Recommendations for Treatment of Hypertension in Acute Stroke
The decision of when to initiate antihypertensive therapy has not been clearly delineated. However, most experts agree that blood pressure targets need to take into account whether thrombolytics are used. The available data provide little evidence that lowering blood pressure decreases adverse events; however, there is some evidence that lowering blood pressure can worsen outcomes by expanding the area of ischemia. Thus, in treating most acute ischemic strokes, antihypertensive therapy can and should be withheld. The exception to this is stroke in a patient with comorbid hypertensive organ damage such as myocardial infarction, aortic dissection, acute hypertensive renal failure, pulmonary edema, or encephalopathy. The consensus statement of the American Stroke Association is to withhold treatment until blood pressure exceeds 220/120 mm Hg,1 roughly corresponding to a MAP of 150 mm Hg, the normal upper limit of cerebral autoregulation. Treatment optimally should be titratable in order to avoid overcorrection, preferably with minimal cerebral venodilatation effect. The parenteral agents labetolol, fenoldopam, nicardipine, and nitroprusside are most commonly used. A disadvantage of nitrates or nitroprusside is that their venodilating effect may raise intracranial pressure. Clonidine may induce central nervous system depression, which can complicate interpretation of the mental status of a patient with an acute neurologic event. Sublingual nifedipine should be avoided because of its well‐documented tendency to cause overcorrection of blood pressure.26 No large direct comparison trial has evaluated which of these antihypertensive agents is superior in clinical outcomes. For the hospitalist, choice of antihypertensive agent should be driven by a need for rapid control of blood pressure to target without overcorrection (see Table 3). Issues such as intracranial pressure, heart rate, and comorbidities may also play a role in choice of medication.
| Not eligible for thrombolytic therapy | |
|---|---|
| |
| Systolic < 220 or diastolic < 120 | Observe unless other end‐organ involvement (aortic dissection, acute myocardial infarction, pulmonary edema, hypertensive encephalopathy) |
| Systolic > 220 or diastolic 121‐140 | Aim for 10%‐15% reduction in blood pressure |
| Labetalol 10‐20 mg IV over 1‐2 minutes; repeat or double every 10 minutes; maximum 300 mg | |
| or | |
| Nicardipine 5 mg/hour IV; titrate by 2.5 mg every 5 minutes; maximum 15 mg/hour | |
| or | |
| Nitroprusside 0.5 g/kg/minute IV; titrate to goal | |
| Diastolic > 140 | Aim for 10%‐15% reduction in blood pressure |
| Nitroprusside 0.5 g/kg/minute IV; titrate to desired blood pressure | |
| Eligible for thrombolytic therapy | |
| Pretreatment | |
| Systolic > 185 or diastolic > 110 | Labetalol 10‐20mg IV |
| May repeat 1 or Nitropaste 1‐2 inches | |
| During and after tPA | |
| Systolic 180‐230 or Diastolic 105‐120 | Labetalol 10 mg |
| May repeat or double labetalol every 10‐20 minutes to a maximum dose of 300 mg or give bolus and start drip at 2‐8 mg/minute | |
| Systolic > 230 or diastolic 121‐140 | Labetalol 10mg IV |
| May repeat or double labetalol every 10‐20 minutes to a maximum dose of 300 mg or give bolus and start drip at 2‐8 mg/minute or | |
| Nicardipine 5 mg/hour; titrate by 2.5 mg every 5 minutes; maximum 15 mg/hour | |
| If not at goal with labetalol or nicardipine, consider nitroprusside | |
| Diastolic>140 | Nitroprusside 0.5 g/kg/minute IV infusion; titrate to desired blood pressure |
The risk of hemorrhagic conversion is greater when thrombolysis is used for the treatment of cerebrovascular accident, in the NINDS trial rising from 0.6% in patients receiving a placebo to 6.4% in patients receiving tPA.27 Given that the goal of thrombolysis is to restore perfusion through the previously blocked vessel and that the risk of hemorrhage rises after thrombolysis, the balance between maintaining CPP and decreasing the risk of bleeding shifts toward maintaining a lower blood pressure postthrombolysis. The NINDS thrombolytic trial required patients to have a blood pressure less than 185/110 mm Hg to be included.27 Once a thrombolytic had been administered, the permitted maximum blood pressure dropped to 180/105 mm Hg. Blood pressure needs to be monitored closely, initially every 15 minutes for 2 hours, then every 30 minutes for the next 6 hours, and then hourly until the end of the first 24 hours. After this, blood pressure monitoring intervals can be more spaced out depending on the need for active antihypertensive therapy.28 However, arterial punctures for invasive monitoring are not recommended if thrombolytics are administered. Review of the experience of the NINDS trial suggests that approximately one third of patients receiving thrombolytics will require pharmacologic therapy to reach the recommended blood pressure goals in the first 24 hours.28
Although guidelines for the management of blood pressure after cerebrovascular accident from major organizations such as the American Stroke Association and the European Stroke Initiative are published and widely quoted in reviews of stroke management, physician adherence to these guidelines is poor.1, 2933 A 2002 review of prescribing practices in a Canadian hospital found excessive reduction of blood pressure in 60% of patients where nitroglycerin was used, and sublingual nifedipine was still the second most commonly prescribed medication.34 A similar 2004 study by Lindenauer et al. found that only 26% of patients who had antihypertensive medication initiated in the hospital met consensus guidelines for therapy.35
Induced Hypertension
Hypotension should be avoided after acute stroke. Observational studies have demonstrated an increased mortality rate of patients who present with hypotension.42 Patients with acute ischemic stroke and loss of autoregulation may have impaired tolerance to even mild levels of hypotension.8 First‐line therapy for hypotension after stroke is volume resuscitation and optimization of cardiac function by correcting arrhythmias.1 If this should fail, vasopressor support is advisable to avoid ongoing hypotension and cerebral hypoperfusion.
The same physiologic arguments that favor permissively managing high blood pressure and avoiding hypotension raise the question of whether inducing hypertension through the use of vasopressors might improve outcomes of stroke. This hypothesis has been bolstered by animal studies and imaging data that suggest improved perfusion to the area of injury with vasopressor‐induced hypertension.3638 However, there are concerns that this strategy may increase edema and hemorrhage and create the potential for vasopressor‐induced myocardial ischemia or arrhythmias. Although the results of small trials have appeared promising, particularly for patients with large vessel stenosis, at this point induced hypertension is still considered experimental until the results of larger randomized, controlled trials provide greater support for this procedure.1, 3941
Chronic Blood Pressure Control for Secondary Prevention of Stroke
Multiple trials have demonstrated that interventions to treat chronic hypertension can reduce the rate of future strokes.14, 43, 44 The PROGRESS trial demonstrated that blood pressure reduction with combination therapy decreased stroke recurrence by 43%.43 Clearly long‐term blood pressure control is a vital component of secondary prevention of stroke. Based on the results of the PROGRESS and ACCESS trials, it is suggested that angiotensin‐converting enzyme (ACE) inhibitors or angiotensin receptor blockers, potentially with additional diuretic therapy, are beneficial in the treatment of chronic hypertension following cerebrovascular accident.25, 42 The issue of optimal medication class is not settled. Recommendations are to treat chronically elevated blood pressure to lower and maintain it at less than 140/80 mm Hg. Because there is no single threshold level of blood pressure beyond which the risk of cerebrovascular disease increases, reduction below this cut point may still offer incremental benefit.13, 45, 46 The timing of initiation of therapy to reach secondary‐prevention goals is not addressed in current guidelines, although some authors recommend waiting days to weeks after an acute stroke before starting therapy.1, 37 For a patient who remains hypertensive at discharge, it is incumbent on the hospitalist to communicate the plan for initiation of antihypertensive agents to the patient and the primary care physician in order to ensure that this critical secondary prevention measure is addressed.
Areas of Ambiguity and Need for Future Research
Within an evidence‐guided practice, there are still many areas for which the evidence to date does not answer important clinical questions about patient management. Whether a patient's prestroke baseline blood pressure should be considered in individualizing blood pressure goals is unclear, as is the question of whether to continue or hold previous chronic antihypertensive therapy. Given the many stroke subtypes, from tiny lacunar infarcts with little collateralization to massive malignant middle‐cerebral infarctions, should blood pressure management be individualized according to the likely size and duration of the peri‐infarct penumbra? Research is needed to determine whether reducing blood pressure to a specific target will improve outcome and the optimal timing of therapy to achieve secondary prevention goals. Finally, using the concept of the penumbra as the theoretical physiologic basis for current blood pressure recommendations, there exists a potential for rapidly evolving neuroimaging techniques to help define the presence, size, and duration of the penumbra to guide these decisions.
CONCLUSIONS
Management of blood pressure after acute ischemic stroke differs from that of many other hypertensive conditions because of the need to preserve perfusion of the peri‐infarct penumbra. Evidence to date suggests little benefit and potential harm from acutely lowering blood pressure after stroke, although there have not been large randomized trials examining outcomes when blood pressure is lowered to a prespecified goal. Current consensus guidelines suggest that blood pressure may be managed permissively up to 220/120 mm Hg when thrombolysis is not administered and to 180/105 mm Hg when thrombolysis is performed. Data suggest that low‐dose antihypertensive therapy such as ACE inhibitors or angiotensin receptor blockers may be safe in the acute setting if pressure is reduced by less than 10%‐15%.17, 25 However, the evidence is not sufficiently strong for this practice to be routinely recommended. At this point, it is recommended that hypotension be aggressively treated; however, induced hypertension remains a promising but unproven therapy. Evidence is strong that long‐term blood pressure control is key to secondary prevention of stroke, but the timing of its initiation remains poorly determined.
- ,,, et al.Guidelines for the early management of patients with ischemic stroke.Stroke.2003;34:1056–1083.
- ,,,.Guidelines for the early management of patients with ischemic stroke: 2005 guidelines update.Stroke.2005;36:916–921.
- Blood Pressure in Acute Stroke Collaboration (BASC).Vasoactive drugs for acute strokeCochrane Database Syst Rev.2004;1:CD002839.
- ,.Blood pressure after stroke.JAMA.1981;246:2177–2180.
- ,,,,,.Association between course of blood pressure within the first 24 hours and functional recovery after acute ischemic stroke.Ann Emerg Med.2003;42:619–626.
- ,,,,,.Detrimental effect of blood pressure reduction in the first 24 hours of acute stroke onset.Neurology.2003;61:1047–1050.
- ,,,, et al.Blood pressure during the first minutes of focal cerebral ischemia.Ann Emerg Med.1993;22:1438–1443.
- ,,,,.Stroke precipitated by moderate blood pressure reduction.J Emerg Med.2000;19:339–346.
- .Hemodynamics and metabolism in ischemic cerebrovascular disease.Neurologic Clinics.1992;10(1):31–48.
- ,.The 5 Ps of acute ischemic stroke treatment: parenchyma, pipes, perfusion, penumbra, and prevention of complications.South Med J.2003;96:336–341.
- .Viability Thresholds and the Penumbra of Focal Ischemia.Annals of Neurology.1994;36:557–565.
- ,,.Hazards of therapy for excessive hypertension in acute stroke.Acta Med Scand.1980;207:253–257.
- Prospective Studies Collaboration.Age‐specific relevance of usual blood pressure to vascular mortality: a meta‐analysis of individual data for one million adults in 61 prospective studies.Lancet.2002;360:1903–1913.
- ,,,.Secondary prevention of ischemic cerebrovascular disease. What is the evidence?Angiology.2005;56:539–552.
- ,,.Intracerebral hemorrhage.Emerg Med Clin North Am.2002;20:631–655.
- ,,, et al.Should hypertension be treated after acute stroke?Arch Neurol.1993;50:855–862.
- ,,.Perindopril reduces blood pressure but not cerebral blood flow in patients with recent cerebral ischemic stroke.Stroke.1997;28:580–583.
- ,,,,.Intravenous Nimodipine West European Stroke Trial (INWEST) of nimodipine in the treatment of acute ischaemic stroke.Cerebrovasc Dis.1994;4:204–210.
- The American Nimodipine Study Group.Clinical trial of nimodipine in acute ischemic stroke.Stroke.1992;23(1):3–8.
- ,,, et al.Placebo‐controlled trial of nimodipine in the treatment of acute ischemic cerebral infarction.Stroke.1990;21:1023–1028.
- ,,, et al.Meta‐analysis of oral nimodipine trials in acute ischemic stroke.Cerebrovasc Dis.1994;4:197–203.
- ,,, et al.A randomized, double‐blind, placebo‐controlled trial of nimodipine in acute ischemic hemispheric stroke.Stroke.1994;25:1348–1353.
- ,,,.Low dose B blockade in acute stroke (“BEST” trial): an evaluationBMJ.1988;296:737–741.
- ,,, et al.Flunarizine in stroke treatment (FIST): a double‐blind, placebo‐controlled trial in Scandinavia and the Netherlands.Acta Neurol Scand.1996;93:56–60.
- ,,, et al.The ACCESS study: Evaluation of acute candesartan cilexetil therapy in stroke survivors.Stroke.2003;34:1699–1703.
- .FDA gives Calcium channel blockers clean bill of health but warns of short‐acting nifedipine hazards.JAMA.1996;275:1638.
- The National Institute of Neurological Disorders and Stroke rt‐PA Stroke Study Group.Tissue plasminogen activator for acute ischemic stroke.N Engl J Med.1995;333:1581–1587.
- ,,, et al.Hypertension and its treatment in the NINDS rt‐PA Stroke Trial.Stroke.1998;29:1504–1509.
- ,.Optimizing blood pressure in neurologic emergencies.Neurocrit Care.2004;1:287–299.
- .Management of acute stroke.South M J.2003;96:380–385.
- .Management of acute stroke.Lancet Neurol.2002;1(1):41–50.
- ,.Acute ischemic stroke: emergent evaluation and management.Emerg Med Clin North Am.2002;20:609–630.
- ,.Post‐emergency department management of stroke.Emerg Med Clin North Am.2002;20:687–702.
- ,,.Blood pressure management in acute stroke: comparison of current guidelines with prescribing patterns.Can J Neurol Sci.2002;29(2):125–131.
- ,,,,,.Use of antihypertensive agents in the management of patients with acute ischemic stroke.Neurology.2004;63:318–323.
- ,,,.Induced hypertension during ischemia reduces infarct area after temporary middle cerebral artery occlusion in rats.Surg Neurol.1996;46:229–234.
- ,,,.Effects of induced hypertension on intracranial pressure and flow velocities of the middle cerebral arteries in patients with large hemispheric stroke.Stroke.2002;33:998–1004.
- ,,,.Induced hypertension improves cerebral blood flow in acute ischemic stroke.Neurology.2005;64:1979.
- ,,,,.A pilot study of drug induced hypertension for treatment of acute stroke.Neurology.2001;56:1210–1213.
- ,,,,,.Pharmacologic elevation of blood pressure in acute stroke: clinical effects and safety.Stroke.1997;28:2133–2138.
- ,,,,.Feasibility and safety of norephinephrine‐induced arterial hypertension in acute ischemic stroke.Neurology.2004;62:1193–1195.
- ,,,,.Initial emergency department blood pressure as predictor of survival after acute ischemic stroke.Neurology.2005;65:1179–1183.
- The PROGRESS Collaborative Group.Randomized trial of a perindopril‐based blood‐pressure‐lowering regimen among 6105 individuals with previous stroke of transient ischaemic attack.Lancet.2001;358:1033–1041.
- ,,, et al.Effect of treating isolated systolic hypertension on the risk of developing various types and subtypes of stroke.JAMA.2000;284:465–471.
- ,.Treatment of chronic hypertension for the prevention of stroke.South Med J.2003;96:359–362.
- ,,,.Epidemiologic assessment of the role of blood pressure in stroke: the Framingham Study.JAMA.1996;276:1269–1278.
Hospitalists are on the front lines of care for patients with cerebrovascular accidents. After the first steps in acute stroke management occur in the emergency room, care is frequently transferred to the hospitalist. This review focuses on the evidence‐based management of blood pressure following acute ischemic stroke. Management of blood pressure after stroke is still controversial, and current consensus statement guidelines acknowledge that optimal treatment has not yet been established.1, 2 As such, it is essential to understand the changes in normal homeostatic physiologic processes that occur after stroke and their subsequent effects on neurologic function. Only then can the appropriate blood pressure target and antihypertensive regimen be chosen.
Physiology of Cerebral Perfusion
Once a stroke has occurred and perfusion to a section of brain tissue has been acutely compromised, systemic pressure tends to rise. This rise is presumably due in part to increased adrenergic tone and activation of the renin‐aldosterone system and potentially is part of Cushing's reflex in cases in which intracranial pressure is elevated.3 The increase in mean arterial pressure may represent a protective response. In the first major study on the topic, in 1981, on admission for stroke mean blood pressure was 163/90 mm Hg for patients without a history of hypertension and 214/118 mm Hg for patients who had been treated for hypertension previously.4 This rise in blood pressure in response to endogenous mechanisms is attenuated over the first 24 hours, and even without intervention, blood pressure tends to fall spontaneously over the next 10 days.47
Under normal circumstances, cerebral blood flow (CBF) is tightly autoregulated across a wide range of cerebral perfusion pressures by alteration in cerebrovascular resistance via arteriolar constriction.89 This allows CBF to remain constant even if cerebral perfusion pressure (CPP) fluctuates from 60 to 150 mm Hg.9 In patients with chronic hypertension, autoregulation works best with blood pressure in a higher range because of vascular smooth muscle hypertrophy and structural changes in the cerebral vessels (see Table 1).
| Cerebral blood flow (CBF) of 50‐70 mL/100 g/minnormal |
| CBF of 20‐50 mL/100 g/minreduced flow compensated for by increased oxygen extraction |
| CBF of 15‐20 mL/100 g/minneuronal quiescence |
| CBF < 15 mL/100 g/minneuronal death |
After an acute ischemic stroke, autoregulation is lost, and local CBF becomes linearly associated with cerebral perfusion pressure. Loss of autoregulation occurs because of several local factors as a result of the infarct. Acidosis and hypoxia in the region of the stroke lead to vasodilatation in the perfusing vessels.9 This may improve local circulation in collateral vessels as a response to the obstruction in the primary supplying artery, but the consequence of maximal vasodilatation is loss of the ability to autoregulate. Normal CPP is driven by the mean arterial pressure minus the intracranial pressure. However, if intracranial vessels have lost the ability to accommodate changes in perfusion pressure, then blood flow to the area of injury becomes linearly correlated with mean arterial pressure.
Surrounding a central core of irreversible necrosis may be a zone of at‐risk tissue that is susceptible to reduction below the threshold of viability in response to any decrement in systemic mean blood pressure.11 This critical concept in stroke management is referred to as the peri‐infarct penumbra (see Fig. 1 and Table 2).
| Mean arterial pressure (MAP) = ⅔ diastolic blood pressure (DBP) + ⅓ systolic blood pressure (SBP) |
| Cerebral perfusion pressure (CPP) = mean arterial pressure (MAP) intracranial pressure (ICP) |
| Cerebral blood flow (CBF) = cerebral perfusion pressure (CPP)/cerebrovascular resistance (CVR) |
Blood Pressure Management after Stroke
The presence of a systemic blood pressure‐dependent peri‐infarct penumbra that might be compromised by blood pressure reduction and thus extend the infarct is the principal argument for allowing permissive hypertension. This is bolstered by the observation that decreases in blood pressure in the first 24 hours are associated with a significant risk of poor neurologic outcome.6 In their an observational study Vlcek et al. found that a greater than 25% drop in diastolic blood pressure (DBP) was associated with a 4‐fold risk of severe disability.5 In a 2003 observational study Oliveria et al. found that reduction in systolic blood pressure (SBP) in the first 24 hours was independently associated with poor outcomes, with a doubling of the risk for poor outcomes for every 10% drop in systolic blood pressure.6 In both these studies, the worsened outcome did not depend on whether the drop was spontaneous or induced by medications. Case reports suggest that large drops in blood pressure can be catastrophic and that even moderate lowering of blood pressure after acute stroke can be associated with clinical deterioration.8, 12
The primary argument for lowering blood pressure after an acute stroke hinges on secondary prevention of new ischemic events, minimization of cerebral edema, and prevention of hemorrhagic conversion. It has long been recognized that hypertension itself is a risk factor for stroke13 and that reduction in chronic hypertension is part of secondary prevention for cerebrovascular accidents.14 Further, hypertension is the most common risk factor for intracerebral hemorrhage, and it would stand to reason that damage to the brain parenchyma from ischemic stroke would increase the risk of pressure‐induced bleeding in an acute setting.15
Because there are compelling theoretical arguments both for and against lowering blood pressure after an acute stroke, it is necessary to look at the results of randomized clinical trials for guidance in weighing the risks and benefits. The overall goal of blood pressure management is to maximize perfusion to the ischemic penumbra while minimizing the hypertensive risk of hemorrhagic transformation.
Lisk et al. conducted a small randomized trial in 1993 of antihypertensive therapy versus placebo after ischemic stroke looking at SPECT perfusion and found lower CBF if mean arterial pressure (MAP) dropped more than 16%.16 This is in contrast with the results of a 1997 randomized trial of perindopril after cerebrovascular accident that found no decrease in Doppler CBF despite a 10% decrease in blood pressure in the active treatment group.17 Neither study was powered to detect significant differences in clinical outcome.
Early randomized controlled trials of antihypertensive agents after acute stroke investigated neuroprotection via mechanisms other than the antihypertensive effect. Nimodipine, a dihydropyridine derivative thought to prevent neuronal death via blockade of calcium channels, has been studied extensively.1820 A meta‐analysis of the 9 early trials of nimodipine, from 1988 to 1992, suggested that nimodipine was potentially beneficial in neurologic score and functional outcome only if used within the first 12 hours and that it could be harmful if started after 24 hours.21 Two additional studies were done in 1994 using both intravenous and oral nimodipine formulations. They demonstrated worsened neurologic function and higher mortality, respectively, which was hypothesized to be a result of the detrimental hemodynamic effects of nimodipine.18, 22
The BEST trial used beta‐blockers in the early period after acute stroke and failed to find benefit, whereas the FIST trial had similar results with the calcium channel blocker flunarizine.23, 24 The ACCESS trial in 2003 was a prospective, randomized, controlled trial using oral candesartan in the first 24 hours after stroke.25 It did find improved mortality in the candesartan arm after 1 year, yet there were no significant differences in blood pressure between candesartan and placebo. Thus, the improved outcome was presumed to be a result of mechanisms other than antihypertensive effect.
Some significant caveats should be kept in mind when interpreting the results of these studies. None of the trials was designed to titrate blood pressure to a prespecified goal in a prospective randomized fashion. It is also difficult to tease out the effect of the active medical intervention from the effect of spontaneous drops in blood pressure, and many trials did not find a significant difference in blood pressure between the medication and placebo groups. A large randomized trial to evaluate interventions to a predefined blood pressure target is needed.
The Cochrane Stroke Group reviewed 32 trials involving 5368 patients and concluded there was not enough evidence to reliably evaluate the effect of altering blood pressure on outcomes.3
Recommendations for Treatment of Hypertension in Acute Stroke
The decision of when to initiate antihypertensive therapy has not been clearly delineated. However, most experts agree that blood pressure targets need to take into account whether thrombolytics are used. The available data provide little evidence that lowering blood pressure decreases adverse events; however, there is some evidence that lowering blood pressure can worsen outcomes by expanding the area of ischemia. Thus, in treating most acute ischemic strokes, antihypertensive therapy can and should be withheld. The exception to this is stroke in a patient with comorbid hypertensive organ damage such as myocardial infarction, aortic dissection, acute hypertensive renal failure, pulmonary edema, or encephalopathy. The consensus statement of the American Stroke Association is to withhold treatment until blood pressure exceeds 220/120 mm Hg,1 roughly corresponding to a MAP of 150 mm Hg, the normal upper limit of cerebral autoregulation. Treatment optimally should be titratable in order to avoid overcorrection, preferably with minimal cerebral venodilatation effect. The parenteral agents labetolol, fenoldopam, nicardipine, and nitroprusside are most commonly used. A disadvantage of nitrates or nitroprusside is that their venodilating effect may raise intracranial pressure. Clonidine may induce central nervous system depression, which can complicate interpretation of the mental status of a patient with an acute neurologic event. Sublingual nifedipine should be avoided because of its well‐documented tendency to cause overcorrection of blood pressure.26 No large direct comparison trial has evaluated which of these antihypertensive agents is superior in clinical outcomes. For the hospitalist, choice of antihypertensive agent should be driven by a need for rapid control of blood pressure to target without overcorrection (see Table 3). Issues such as intracranial pressure, heart rate, and comorbidities may also play a role in choice of medication.
| Not eligible for thrombolytic therapy | |
|---|---|
| |
| Systolic < 220 or diastolic < 120 | Observe unless other end‐organ involvement (aortic dissection, acute myocardial infarction, pulmonary edema, hypertensive encephalopathy) |
| Systolic > 220 or diastolic 121‐140 | Aim for 10%‐15% reduction in blood pressure |
| Labetalol 10‐20 mg IV over 1‐2 minutes; repeat or double every 10 minutes; maximum 300 mg | |
| or | |
| Nicardipine 5 mg/hour IV; titrate by 2.5 mg every 5 minutes; maximum 15 mg/hour | |
| or | |
| Nitroprusside 0.5 g/kg/minute IV; titrate to goal | |
| Diastolic > 140 | Aim for 10%‐15% reduction in blood pressure |
| Nitroprusside 0.5 g/kg/minute IV; titrate to desired blood pressure | |
| Eligible for thrombolytic therapy | |
| Pretreatment | |
| Systolic > 185 or diastolic > 110 | Labetalol 10‐20mg IV |
| May repeat 1 or Nitropaste 1‐2 inches | |
| During and after tPA | |
| Systolic 180‐230 or Diastolic 105‐120 | Labetalol 10 mg |
| May repeat or double labetalol every 10‐20 minutes to a maximum dose of 300 mg or give bolus and start drip at 2‐8 mg/minute | |
| Systolic > 230 or diastolic 121‐140 | Labetalol 10mg IV |
| May repeat or double labetalol every 10‐20 minutes to a maximum dose of 300 mg or give bolus and start drip at 2‐8 mg/minute or | |
| Nicardipine 5 mg/hour; titrate by 2.5 mg every 5 minutes; maximum 15 mg/hour | |
| If not at goal with labetalol or nicardipine, consider nitroprusside | |
| Diastolic>140 | Nitroprusside 0.5 g/kg/minute IV infusion; titrate to desired blood pressure |
The risk of hemorrhagic conversion is greater when thrombolysis is used for the treatment of cerebrovascular accident, in the NINDS trial rising from 0.6% in patients receiving a placebo to 6.4% in patients receiving tPA.27 Given that the goal of thrombolysis is to restore perfusion through the previously blocked vessel and that the risk of hemorrhage rises after thrombolysis, the balance between maintaining CPP and decreasing the risk of bleeding shifts toward maintaining a lower blood pressure postthrombolysis. The NINDS thrombolytic trial required patients to have a blood pressure less than 185/110 mm Hg to be included.27 Once a thrombolytic had been administered, the permitted maximum blood pressure dropped to 180/105 mm Hg. Blood pressure needs to be monitored closely, initially every 15 minutes for 2 hours, then every 30 minutes for the next 6 hours, and then hourly until the end of the first 24 hours. After this, blood pressure monitoring intervals can be more spaced out depending on the need for active antihypertensive therapy.28 However, arterial punctures for invasive monitoring are not recommended if thrombolytics are administered. Review of the experience of the NINDS trial suggests that approximately one third of patients receiving thrombolytics will require pharmacologic therapy to reach the recommended blood pressure goals in the first 24 hours.28
Although guidelines for the management of blood pressure after cerebrovascular accident from major organizations such as the American Stroke Association and the European Stroke Initiative are published and widely quoted in reviews of stroke management, physician adherence to these guidelines is poor.1, 2933 A 2002 review of prescribing practices in a Canadian hospital found excessive reduction of blood pressure in 60% of patients where nitroglycerin was used, and sublingual nifedipine was still the second most commonly prescribed medication.34 A similar 2004 study by Lindenauer et al. found that only 26% of patients who had antihypertensive medication initiated in the hospital met consensus guidelines for therapy.35
Induced Hypertension
Hypotension should be avoided after acute stroke. Observational studies have demonstrated an increased mortality rate of patients who present with hypotension.42 Patients with acute ischemic stroke and loss of autoregulation may have impaired tolerance to even mild levels of hypotension.8 First‐line therapy for hypotension after stroke is volume resuscitation and optimization of cardiac function by correcting arrhythmias.1 If this should fail, vasopressor support is advisable to avoid ongoing hypotension and cerebral hypoperfusion.
The same physiologic arguments that favor permissively managing high blood pressure and avoiding hypotension raise the question of whether inducing hypertension through the use of vasopressors might improve outcomes of stroke. This hypothesis has been bolstered by animal studies and imaging data that suggest improved perfusion to the area of injury with vasopressor‐induced hypertension.3638 However, there are concerns that this strategy may increase edema and hemorrhage and create the potential for vasopressor‐induced myocardial ischemia or arrhythmias. Although the results of small trials have appeared promising, particularly for patients with large vessel stenosis, at this point induced hypertension is still considered experimental until the results of larger randomized, controlled trials provide greater support for this procedure.1, 3941
Chronic Blood Pressure Control for Secondary Prevention of Stroke
Multiple trials have demonstrated that interventions to treat chronic hypertension can reduce the rate of future strokes.14, 43, 44 The PROGRESS trial demonstrated that blood pressure reduction with combination therapy decreased stroke recurrence by 43%.43 Clearly long‐term blood pressure control is a vital component of secondary prevention of stroke. Based on the results of the PROGRESS and ACCESS trials, it is suggested that angiotensin‐converting enzyme (ACE) inhibitors or angiotensin receptor blockers, potentially with additional diuretic therapy, are beneficial in the treatment of chronic hypertension following cerebrovascular accident.25, 42 The issue of optimal medication class is not settled. Recommendations are to treat chronically elevated blood pressure to lower and maintain it at less than 140/80 mm Hg. Because there is no single threshold level of blood pressure beyond which the risk of cerebrovascular disease increases, reduction below this cut point may still offer incremental benefit.13, 45, 46 The timing of initiation of therapy to reach secondary‐prevention goals is not addressed in current guidelines, although some authors recommend waiting days to weeks after an acute stroke before starting therapy.1, 37 For a patient who remains hypertensive at discharge, it is incumbent on the hospitalist to communicate the plan for initiation of antihypertensive agents to the patient and the primary care physician in order to ensure that this critical secondary prevention measure is addressed.
Areas of Ambiguity and Need for Future Research
Within an evidence‐guided practice, there are still many areas for which the evidence to date does not answer important clinical questions about patient management. Whether a patient's prestroke baseline blood pressure should be considered in individualizing blood pressure goals is unclear, as is the question of whether to continue or hold previous chronic antihypertensive therapy. Given the many stroke subtypes, from tiny lacunar infarcts with little collateralization to massive malignant middle‐cerebral infarctions, should blood pressure management be individualized according to the likely size and duration of the peri‐infarct penumbra? Research is needed to determine whether reducing blood pressure to a specific target will improve outcome and the optimal timing of therapy to achieve secondary prevention goals. Finally, using the concept of the penumbra as the theoretical physiologic basis for current blood pressure recommendations, there exists a potential for rapidly evolving neuroimaging techniques to help define the presence, size, and duration of the penumbra to guide these decisions.
CONCLUSIONS
Management of blood pressure after acute ischemic stroke differs from that of many other hypertensive conditions because of the need to preserve perfusion of the peri‐infarct penumbra. Evidence to date suggests little benefit and potential harm from acutely lowering blood pressure after stroke, although there have not been large randomized trials examining outcomes when blood pressure is lowered to a prespecified goal. Current consensus guidelines suggest that blood pressure may be managed permissively up to 220/120 mm Hg when thrombolysis is not administered and to 180/105 mm Hg when thrombolysis is performed. Data suggest that low‐dose antihypertensive therapy such as ACE inhibitors or angiotensin receptor blockers may be safe in the acute setting if pressure is reduced by less than 10%‐15%.17, 25 However, the evidence is not sufficiently strong for this practice to be routinely recommended. At this point, it is recommended that hypotension be aggressively treated; however, induced hypertension remains a promising but unproven therapy. Evidence is strong that long‐term blood pressure control is key to secondary prevention of stroke, but the timing of its initiation remains poorly determined.
Hospitalists are on the front lines of care for patients with cerebrovascular accidents. After the first steps in acute stroke management occur in the emergency room, care is frequently transferred to the hospitalist. This review focuses on the evidence‐based management of blood pressure following acute ischemic stroke. Management of blood pressure after stroke is still controversial, and current consensus statement guidelines acknowledge that optimal treatment has not yet been established.1, 2 As such, it is essential to understand the changes in normal homeostatic physiologic processes that occur after stroke and their subsequent effects on neurologic function. Only then can the appropriate blood pressure target and antihypertensive regimen be chosen.
Physiology of Cerebral Perfusion
Once a stroke has occurred and perfusion to a section of brain tissue has been acutely compromised, systemic pressure tends to rise. This rise is presumably due in part to increased adrenergic tone and activation of the renin‐aldosterone system and potentially is part of Cushing's reflex in cases in which intracranial pressure is elevated.3 The increase in mean arterial pressure may represent a protective response. In the first major study on the topic, in 1981, on admission for stroke mean blood pressure was 163/90 mm Hg for patients without a history of hypertension and 214/118 mm Hg for patients who had been treated for hypertension previously.4 This rise in blood pressure in response to endogenous mechanisms is attenuated over the first 24 hours, and even without intervention, blood pressure tends to fall spontaneously over the next 10 days.47
Under normal circumstances, cerebral blood flow (CBF) is tightly autoregulated across a wide range of cerebral perfusion pressures by alteration in cerebrovascular resistance via arteriolar constriction.89 This allows CBF to remain constant even if cerebral perfusion pressure (CPP) fluctuates from 60 to 150 mm Hg.9 In patients with chronic hypertension, autoregulation works best with blood pressure in a higher range because of vascular smooth muscle hypertrophy and structural changes in the cerebral vessels (see Table 1).
| Cerebral blood flow (CBF) of 50‐70 mL/100 g/minnormal |
| CBF of 20‐50 mL/100 g/minreduced flow compensated for by increased oxygen extraction |
| CBF of 15‐20 mL/100 g/minneuronal quiescence |
| CBF < 15 mL/100 g/minneuronal death |
After an acute ischemic stroke, autoregulation is lost, and local CBF becomes linearly associated with cerebral perfusion pressure. Loss of autoregulation occurs because of several local factors as a result of the infarct. Acidosis and hypoxia in the region of the stroke lead to vasodilatation in the perfusing vessels.9 This may improve local circulation in collateral vessels as a response to the obstruction in the primary supplying artery, but the consequence of maximal vasodilatation is loss of the ability to autoregulate. Normal CPP is driven by the mean arterial pressure minus the intracranial pressure. However, if intracranial vessels have lost the ability to accommodate changes in perfusion pressure, then blood flow to the area of injury becomes linearly correlated with mean arterial pressure.
Surrounding a central core of irreversible necrosis may be a zone of at‐risk tissue that is susceptible to reduction below the threshold of viability in response to any decrement in systemic mean blood pressure.11 This critical concept in stroke management is referred to as the peri‐infarct penumbra (see Fig. 1 and Table 2).
| Mean arterial pressure (MAP) = ⅔ diastolic blood pressure (DBP) + ⅓ systolic blood pressure (SBP) |
| Cerebral perfusion pressure (CPP) = mean arterial pressure (MAP) intracranial pressure (ICP) |
| Cerebral blood flow (CBF) = cerebral perfusion pressure (CPP)/cerebrovascular resistance (CVR) |
Blood Pressure Management after Stroke
The presence of a systemic blood pressure‐dependent peri‐infarct penumbra that might be compromised by blood pressure reduction and thus extend the infarct is the principal argument for allowing permissive hypertension. This is bolstered by the observation that decreases in blood pressure in the first 24 hours are associated with a significant risk of poor neurologic outcome.6 In their an observational study Vlcek et al. found that a greater than 25% drop in diastolic blood pressure (DBP) was associated with a 4‐fold risk of severe disability.5 In a 2003 observational study Oliveria et al. found that reduction in systolic blood pressure (SBP) in the first 24 hours was independently associated with poor outcomes, with a doubling of the risk for poor outcomes for every 10% drop in systolic blood pressure.6 In both these studies, the worsened outcome did not depend on whether the drop was spontaneous or induced by medications. Case reports suggest that large drops in blood pressure can be catastrophic and that even moderate lowering of blood pressure after acute stroke can be associated with clinical deterioration.8, 12
The primary argument for lowering blood pressure after an acute stroke hinges on secondary prevention of new ischemic events, minimization of cerebral edema, and prevention of hemorrhagic conversion. It has long been recognized that hypertension itself is a risk factor for stroke13 and that reduction in chronic hypertension is part of secondary prevention for cerebrovascular accidents.14 Further, hypertension is the most common risk factor for intracerebral hemorrhage, and it would stand to reason that damage to the brain parenchyma from ischemic stroke would increase the risk of pressure‐induced bleeding in an acute setting.15
Because there are compelling theoretical arguments both for and against lowering blood pressure after an acute stroke, it is necessary to look at the results of randomized clinical trials for guidance in weighing the risks and benefits. The overall goal of blood pressure management is to maximize perfusion to the ischemic penumbra while minimizing the hypertensive risk of hemorrhagic transformation.
Lisk et al. conducted a small randomized trial in 1993 of antihypertensive therapy versus placebo after ischemic stroke looking at SPECT perfusion and found lower CBF if mean arterial pressure (MAP) dropped more than 16%.16 This is in contrast with the results of a 1997 randomized trial of perindopril after cerebrovascular accident that found no decrease in Doppler CBF despite a 10% decrease in blood pressure in the active treatment group.17 Neither study was powered to detect significant differences in clinical outcome.
Early randomized controlled trials of antihypertensive agents after acute stroke investigated neuroprotection via mechanisms other than the antihypertensive effect. Nimodipine, a dihydropyridine derivative thought to prevent neuronal death via blockade of calcium channels, has been studied extensively.1820 A meta‐analysis of the 9 early trials of nimodipine, from 1988 to 1992, suggested that nimodipine was potentially beneficial in neurologic score and functional outcome only if used within the first 12 hours and that it could be harmful if started after 24 hours.21 Two additional studies were done in 1994 using both intravenous and oral nimodipine formulations. They demonstrated worsened neurologic function and higher mortality, respectively, which was hypothesized to be a result of the detrimental hemodynamic effects of nimodipine.18, 22
The BEST trial used beta‐blockers in the early period after acute stroke and failed to find benefit, whereas the FIST trial had similar results with the calcium channel blocker flunarizine.23, 24 The ACCESS trial in 2003 was a prospective, randomized, controlled trial using oral candesartan in the first 24 hours after stroke.25 It did find improved mortality in the candesartan arm after 1 year, yet there were no significant differences in blood pressure between candesartan and placebo. Thus, the improved outcome was presumed to be a result of mechanisms other than antihypertensive effect.
Some significant caveats should be kept in mind when interpreting the results of these studies. None of the trials was designed to titrate blood pressure to a prespecified goal in a prospective randomized fashion. It is also difficult to tease out the effect of the active medical intervention from the effect of spontaneous drops in blood pressure, and many trials did not find a significant difference in blood pressure between the medication and placebo groups. A large randomized trial to evaluate interventions to a predefined blood pressure target is needed.
The Cochrane Stroke Group reviewed 32 trials involving 5368 patients and concluded there was not enough evidence to reliably evaluate the effect of altering blood pressure on outcomes.3
Recommendations for Treatment of Hypertension in Acute Stroke
The decision of when to initiate antihypertensive therapy has not been clearly delineated. However, most experts agree that blood pressure targets need to take into account whether thrombolytics are used. The available data provide little evidence that lowering blood pressure decreases adverse events; however, there is some evidence that lowering blood pressure can worsen outcomes by expanding the area of ischemia. Thus, in treating most acute ischemic strokes, antihypertensive therapy can and should be withheld. The exception to this is stroke in a patient with comorbid hypertensive organ damage such as myocardial infarction, aortic dissection, acute hypertensive renal failure, pulmonary edema, or encephalopathy. The consensus statement of the American Stroke Association is to withhold treatment until blood pressure exceeds 220/120 mm Hg,1 roughly corresponding to a MAP of 150 mm Hg, the normal upper limit of cerebral autoregulation. Treatment optimally should be titratable in order to avoid overcorrection, preferably with minimal cerebral venodilatation effect. The parenteral agents labetolol, fenoldopam, nicardipine, and nitroprusside are most commonly used. A disadvantage of nitrates or nitroprusside is that their venodilating effect may raise intracranial pressure. Clonidine may induce central nervous system depression, which can complicate interpretation of the mental status of a patient with an acute neurologic event. Sublingual nifedipine should be avoided because of its well‐documented tendency to cause overcorrection of blood pressure.26 No large direct comparison trial has evaluated which of these antihypertensive agents is superior in clinical outcomes. For the hospitalist, choice of antihypertensive agent should be driven by a need for rapid control of blood pressure to target without overcorrection (see Table 3). Issues such as intracranial pressure, heart rate, and comorbidities may also play a role in choice of medication.
| Not eligible for thrombolytic therapy | |
|---|---|
| |
| Systolic < 220 or diastolic < 120 | Observe unless other end‐organ involvement (aortic dissection, acute myocardial infarction, pulmonary edema, hypertensive encephalopathy) |
| Systolic > 220 or diastolic 121‐140 | Aim for 10%‐15% reduction in blood pressure |
| Labetalol 10‐20 mg IV over 1‐2 minutes; repeat or double every 10 minutes; maximum 300 mg | |
| or | |
| Nicardipine 5 mg/hour IV; titrate by 2.5 mg every 5 minutes; maximum 15 mg/hour | |
| or | |
| Nitroprusside 0.5 g/kg/minute IV; titrate to goal | |
| Diastolic > 140 | Aim for 10%‐15% reduction in blood pressure |
| Nitroprusside 0.5 g/kg/minute IV; titrate to desired blood pressure | |
| Eligible for thrombolytic therapy | |
| Pretreatment | |
| Systolic > 185 or diastolic > 110 | Labetalol 10‐20mg IV |
| May repeat 1 or Nitropaste 1‐2 inches | |
| During and after tPA | |
| Systolic 180‐230 or Diastolic 105‐120 | Labetalol 10 mg |
| May repeat or double labetalol every 10‐20 minutes to a maximum dose of 300 mg or give bolus and start drip at 2‐8 mg/minute | |
| Systolic > 230 or diastolic 121‐140 | Labetalol 10mg IV |
| May repeat or double labetalol every 10‐20 minutes to a maximum dose of 300 mg or give bolus and start drip at 2‐8 mg/minute or | |
| Nicardipine 5 mg/hour; titrate by 2.5 mg every 5 minutes; maximum 15 mg/hour | |
| If not at goal with labetalol or nicardipine, consider nitroprusside | |
| Diastolic>140 | Nitroprusside 0.5 g/kg/minute IV infusion; titrate to desired blood pressure |
The risk of hemorrhagic conversion is greater when thrombolysis is used for the treatment of cerebrovascular accident, in the NINDS trial rising from 0.6% in patients receiving a placebo to 6.4% in patients receiving tPA.27 Given that the goal of thrombolysis is to restore perfusion through the previously blocked vessel and that the risk of hemorrhage rises after thrombolysis, the balance between maintaining CPP and decreasing the risk of bleeding shifts toward maintaining a lower blood pressure postthrombolysis. The NINDS thrombolytic trial required patients to have a blood pressure less than 185/110 mm Hg to be included.27 Once a thrombolytic had been administered, the permitted maximum blood pressure dropped to 180/105 mm Hg. Blood pressure needs to be monitored closely, initially every 15 minutes for 2 hours, then every 30 minutes for the next 6 hours, and then hourly until the end of the first 24 hours. After this, blood pressure monitoring intervals can be more spaced out depending on the need for active antihypertensive therapy.28 However, arterial punctures for invasive monitoring are not recommended if thrombolytics are administered. Review of the experience of the NINDS trial suggests that approximately one third of patients receiving thrombolytics will require pharmacologic therapy to reach the recommended blood pressure goals in the first 24 hours.28
Although guidelines for the management of blood pressure after cerebrovascular accident from major organizations such as the American Stroke Association and the European Stroke Initiative are published and widely quoted in reviews of stroke management, physician adherence to these guidelines is poor.1, 2933 A 2002 review of prescribing practices in a Canadian hospital found excessive reduction of blood pressure in 60% of patients where nitroglycerin was used, and sublingual nifedipine was still the second most commonly prescribed medication.34 A similar 2004 study by Lindenauer et al. found that only 26% of patients who had antihypertensive medication initiated in the hospital met consensus guidelines for therapy.35
Induced Hypertension
Hypotension should be avoided after acute stroke. Observational studies have demonstrated an increased mortality rate of patients who present with hypotension.42 Patients with acute ischemic stroke and loss of autoregulation may have impaired tolerance to even mild levels of hypotension.8 First‐line therapy for hypotension after stroke is volume resuscitation and optimization of cardiac function by correcting arrhythmias.1 If this should fail, vasopressor support is advisable to avoid ongoing hypotension and cerebral hypoperfusion.
The same physiologic arguments that favor permissively managing high blood pressure and avoiding hypotension raise the question of whether inducing hypertension through the use of vasopressors might improve outcomes of stroke. This hypothesis has been bolstered by animal studies and imaging data that suggest improved perfusion to the area of injury with vasopressor‐induced hypertension.3638 However, there are concerns that this strategy may increase edema and hemorrhage and create the potential for vasopressor‐induced myocardial ischemia or arrhythmias. Although the results of small trials have appeared promising, particularly for patients with large vessel stenosis, at this point induced hypertension is still considered experimental until the results of larger randomized, controlled trials provide greater support for this procedure.1, 3941
Chronic Blood Pressure Control for Secondary Prevention of Stroke
Multiple trials have demonstrated that interventions to treat chronic hypertension can reduce the rate of future strokes.14, 43, 44 The PROGRESS trial demonstrated that blood pressure reduction with combination therapy decreased stroke recurrence by 43%.43 Clearly long‐term blood pressure control is a vital component of secondary prevention of stroke. Based on the results of the PROGRESS and ACCESS trials, it is suggested that angiotensin‐converting enzyme (ACE) inhibitors or angiotensin receptor blockers, potentially with additional diuretic therapy, are beneficial in the treatment of chronic hypertension following cerebrovascular accident.25, 42 The issue of optimal medication class is not settled. Recommendations are to treat chronically elevated blood pressure to lower and maintain it at less than 140/80 mm Hg. Because there is no single threshold level of blood pressure beyond which the risk of cerebrovascular disease increases, reduction below this cut point may still offer incremental benefit.13, 45, 46 The timing of initiation of therapy to reach secondary‐prevention goals is not addressed in current guidelines, although some authors recommend waiting days to weeks after an acute stroke before starting therapy.1, 37 For a patient who remains hypertensive at discharge, it is incumbent on the hospitalist to communicate the plan for initiation of antihypertensive agents to the patient and the primary care physician in order to ensure that this critical secondary prevention measure is addressed.
Areas of Ambiguity and Need for Future Research
Within an evidence‐guided practice, there are still many areas for which the evidence to date does not answer important clinical questions about patient management. Whether a patient's prestroke baseline blood pressure should be considered in individualizing blood pressure goals is unclear, as is the question of whether to continue or hold previous chronic antihypertensive therapy. Given the many stroke subtypes, from tiny lacunar infarcts with little collateralization to massive malignant middle‐cerebral infarctions, should blood pressure management be individualized according to the likely size and duration of the peri‐infarct penumbra? Research is needed to determine whether reducing blood pressure to a specific target will improve outcome and the optimal timing of therapy to achieve secondary prevention goals. Finally, using the concept of the penumbra as the theoretical physiologic basis for current blood pressure recommendations, there exists a potential for rapidly evolving neuroimaging techniques to help define the presence, size, and duration of the penumbra to guide these decisions.
CONCLUSIONS
Management of blood pressure after acute ischemic stroke differs from that of many other hypertensive conditions because of the need to preserve perfusion of the peri‐infarct penumbra. Evidence to date suggests little benefit and potential harm from acutely lowering blood pressure after stroke, although there have not been large randomized trials examining outcomes when blood pressure is lowered to a prespecified goal. Current consensus guidelines suggest that blood pressure may be managed permissively up to 220/120 mm Hg when thrombolysis is not administered and to 180/105 mm Hg when thrombolysis is performed. Data suggest that low‐dose antihypertensive therapy such as ACE inhibitors or angiotensin receptor blockers may be safe in the acute setting if pressure is reduced by less than 10%‐15%.17, 25 However, the evidence is not sufficiently strong for this practice to be routinely recommended. At this point, it is recommended that hypotension be aggressively treated; however, induced hypertension remains a promising but unproven therapy. Evidence is strong that long‐term blood pressure control is key to secondary prevention of stroke, but the timing of its initiation remains poorly determined.
- ,,, et al.Guidelines for the early management of patients with ischemic stroke.Stroke.2003;34:1056–1083.
- ,,,.Guidelines for the early management of patients with ischemic stroke: 2005 guidelines update.Stroke.2005;36:916–921.
- Blood Pressure in Acute Stroke Collaboration (BASC).Vasoactive drugs for acute strokeCochrane Database Syst Rev.2004;1:CD002839.
- ,.Blood pressure after stroke.JAMA.1981;246:2177–2180.
- ,,,,,.Association between course of blood pressure within the first 24 hours and functional recovery after acute ischemic stroke.Ann Emerg Med.2003;42:619–626.
- ,,,,,.Detrimental effect of blood pressure reduction in the first 24 hours of acute stroke onset.Neurology.2003;61:1047–1050.
- ,,,, et al.Blood pressure during the first minutes of focal cerebral ischemia.Ann Emerg Med.1993;22:1438–1443.
- ,,,,.Stroke precipitated by moderate blood pressure reduction.J Emerg Med.2000;19:339–346.
- .Hemodynamics and metabolism in ischemic cerebrovascular disease.Neurologic Clinics.1992;10(1):31–48.
- ,.The 5 Ps of acute ischemic stroke treatment: parenchyma, pipes, perfusion, penumbra, and prevention of complications.South Med J.2003;96:336–341.
- .Viability Thresholds and the Penumbra of Focal Ischemia.Annals of Neurology.1994;36:557–565.
- ,,.Hazards of therapy for excessive hypertension in acute stroke.Acta Med Scand.1980;207:253–257.
- Prospective Studies Collaboration.Age‐specific relevance of usual blood pressure to vascular mortality: a meta‐analysis of individual data for one million adults in 61 prospective studies.Lancet.2002;360:1903–1913.
- ,,,.Secondary prevention of ischemic cerebrovascular disease. What is the evidence?Angiology.2005;56:539–552.
- ,,.Intracerebral hemorrhage.Emerg Med Clin North Am.2002;20:631–655.
- ,,, et al.Should hypertension be treated after acute stroke?Arch Neurol.1993;50:855–862.
- ,,.Perindopril reduces blood pressure but not cerebral blood flow in patients with recent cerebral ischemic stroke.Stroke.1997;28:580–583.
- ,,,,.Intravenous Nimodipine West European Stroke Trial (INWEST) of nimodipine in the treatment of acute ischaemic stroke.Cerebrovasc Dis.1994;4:204–210.
- The American Nimodipine Study Group.Clinical trial of nimodipine in acute ischemic stroke.Stroke.1992;23(1):3–8.
- ,,, et al.Placebo‐controlled trial of nimodipine in the treatment of acute ischemic cerebral infarction.Stroke.1990;21:1023–1028.
- ,,, et al.Meta‐analysis of oral nimodipine trials in acute ischemic stroke.Cerebrovasc Dis.1994;4:197–203.
- ,,, et al.A randomized, double‐blind, placebo‐controlled trial of nimodipine in acute ischemic hemispheric stroke.Stroke.1994;25:1348–1353.
- ,,,.Low dose B blockade in acute stroke (“BEST” trial): an evaluationBMJ.1988;296:737–741.
- ,,, et al.Flunarizine in stroke treatment (FIST): a double‐blind, placebo‐controlled trial in Scandinavia and the Netherlands.Acta Neurol Scand.1996;93:56–60.
- ,,, et al.The ACCESS study: Evaluation of acute candesartan cilexetil therapy in stroke survivors.Stroke.2003;34:1699–1703.
- .FDA gives Calcium channel blockers clean bill of health but warns of short‐acting nifedipine hazards.JAMA.1996;275:1638.
- The National Institute of Neurological Disorders and Stroke rt‐PA Stroke Study Group.Tissue plasminogen activator for acute ischemic stroke.N Engl J Med.1995;333:1581–1587.
- ,,, et al.Hypertension and its treatment in the NINDS rt‐PA Stroke Trial.Stroke.1998;29:1504–1509.
- ,.Optimizing blood pressure in neurologic emergencies.Neurocrit Care.2004;1:287–299.
- .Management of acute stroke.South M J.2003;96:380–385.
- .Management of acute stroke.Lancet Neurol.2002;1(1):41–50.
- ,.Acute ischemic stroke: emergent evaluation and management.Emerg Med Clin North Am.2002;20:609–630.
- ,.Post‐emergency department management of stroke.Emerg Med Clin North Am.2002;20:687–702.
- ,,.Blood pressure management in acute stroke: comparison of current guidelines with prescribing patterns.Can J Neurol Sci.2002;29(2):125–131.
- ,,,,,.Use of antihypertensive agents in the management of patients with acute ischemic stroke.Neurology.2004;63:318–323.
- ,,,.Induced hypertension during ischemia reduces infarct area after temporary middle cerebral artery occlusion in rats.Surg Neurol.1996;46:229–234.
- ,,,.Effects of induced hypertension on intracranial pressure and flow velocities of the middle cerebral arteries in patients with large hemispheric stroke.Stroke.2002;33:998–1004.
- ,,,.Induced hypertension improves cerebral blood flow in acute ischemic stroke.Neurology.2005;64:1979.
- ,,,,.A pilot study of drug induced hypertension for treatment of acute stroke.Neurology.2001;56:1210–1213.
- ,,,,,.Pharmacologic elevation of blood pressure in acute stroke: clinical effects and safety.Stroke.1997;28:2133–2138.
- ,,,,.Feasibility and safety of norephinephrine‐induced arterial hypertension in acute ischemic stroke.Neurology.2004;62:1193–1195.
- ,,,,.Initial emergency department blood pressure as predictor of survival after acute ischemic stroke.Neurology.2005;65:1179–1183.
- The PROGRESS Collaborative Group.Randomized trial of a perindopril‐based blood‐pressure‐lowering regimen among 6105 individuals with previous stroke of transient ischaemic attack.Lancet.2001;358:1033–1041.
- ,,, et al.Effect of treating isolated systolic hypertension on the risk of developing various types and subtypes of stroke.JAMA.2000;284:465–471.
- ,.Treatment of chronic hypertension for the prevention of stroke.South Med J.2003;96:359–362.
- ,,,.Epidemiologic assessment of the role of blood pressure in stroke: the Framingham Study.JAMA.1996;276:1269–1278.
- ,,, et al.Guidelines for the early management of patients with ischemic stroke.Stroke.2003;34:1056–1083.
- ,,,.Guidelines for the early management of patients with ischemic stroke: 2005 guidelines update.Stroke.2005;36:916–921.
- Blood Pressure in Acute Stroke Collaboration (BASC).Vasoactive drugs for acute strokeCochrane Database Syst Rev.2004;1:CD002839.
- ,.Blood pressure after stroke.JAMA.1981;246:2177–2180.
- ,,,,,.Association between course of blood pressure within the first 24 hours and functional recovery after acute ischemic stroke.Ann Emerg Med.2003;42:619–626.
- ,,,,,.Detrimental effect of blood pressure reduction in the first 24 hours of acute stroke onset.Neurology.2003;61:1047–1050.
- ,,,, et al.Blood pressure during the first minutes of focal cerebral ischemia.Ann Emerg Med.1993;22:1438–1443.
- ,,,,.Stroke precipitated by moderate blood pressure reduction.J Emerg Med.2000;19:339–346.
- .Hemodynamics and metabolism in ischemic cerebrovascular disease.Neurologic Clinics.1992;10(1):31–48.
- ,.The 5 Ps of acute ischemic stroke treatment: parenchyma, pipes, perfusion, penumbra, and prevention of complications.South Med J.2003;96:336–341.
- .Viability Thresholds and the Penumbra of Focal Ischemia.Annals of Neurology.1994;36:557–565.
- ,,.Hazards of therapy for excessive hypertension in acute stroke.Acta Med Scand.1980;207:253–257.
- Prospective Studies Collaboration.Age‐specific relevance of usual blood pressure to vascular mortality: a meta‐analysis of individual data for one million adults in 61 prospective studies.Lancet.2002;360:1903–1913.
- ,,,.Secondary prevention of ischemic cerebrovascular disease. What is the evidence?Angiology.2005;56:539–552.
- ,,.Intracerebral hemorrhage.Emerg Med Clin North Am.2002;20:631–655.
- ,,, et al.Should hypertension be treated after acute stroke?Arch Neurol.1993;50:855–862.
- ,,.Perindopril reduces blood pressure but not cerebral blood flow in patients with recent cerebral ischemic stroke.Stroke.1997;28:580–583.
- ,,,,.Intravenous Nimodipine West European Stroke Trial (INWEST) of nimodipine in the treatment of acute ischaemic stroke.Cerebrovasc Dis.1994;4:204–210.
- The American Nimodipine Study Group.Clinical trial of nimodipine in acute ischemic stroke.Stroke.1992;23(1):3–8.
- ,,, et al.Placebo‐controlled trial of nimodipine in the treatment of acute ischemic cerebral infarction.Stroke.1990;21:1023–1028.
- ,,, et al.Meta‐analysis of oral nimodipine trials in acute ischemic stroke.Cerebrovasc Dis.1994;4:197–203.
- ,,, et al.A randomized, double‐blind, placebo‐controlled trial of nimodipine in acute ischemic hemispheric stroke.Stroke.1994;25:1348–1353.
- ,,,.Low dose B blockade in acute stroke (“BEST” trial): an evaluationBMJ.1988;296:737–741.
- ,,, et al.Flunarizine in stroke treatment (FIST): a double‐blind, placebo‐controlled trial in Scandinavia and the Netherlands.Acta Neurol Scand.1996;93:56–60.
- ,,, et al.The ACCESS study: Evaluation of acute candesartan cilexetil therapy in stroke survivors.Stroke.2003;34:1699–1703.
- .FDA gives Calcium channel blockers clean bill of health but warns of short‐acting nifedipine hazards.JAMA.1996;275:1638.
- The National Institute of Neurological Disorders and Stroke rt‐PA Stroke Study Group.Tissue plasminogen activator for acute ischemic stroke.N Engl J Med.1995;333:1581–1587.
- ,,, et al.Hypertension and its treatment in the NINDS rt‐PA Stroke Trial.Stroke.1998;29:1504–1509.
- ,.Optimizing blood pressure in neurologic emergencies.Neurocrit Care.2004;1:287–299.
- .Management of acute stroke.South M J.2003;96:380–385.
- .Management of acute stroke.Lancet Neurol.2002;1(1):41–50.
- ,.Acute ischemic stroke: emergent evaluation and management.Emerg Med Clin North Am.2002;20:609–630.
- ,.Post‐emergency department management of stroke.Emerg Med Clin North Am.2002;20:687–702.
- ,,.Blood pressure management in acute stroke: comparison of current guidelines with prescribing patterns.Can J Neurol Sci.2002;29(2):125–131.
- ,,,,,.Use of antihypertensive agents in the management of patients with acute ischemic stroke.Neurology.2004;63:318–323.
- ,,,.Induced hypertension during ischemia reduces infarct area after temporary middle cerebral artery occlusion in rats.Surg Neurol.1996;46:229–234.
- ,,,.Effects of induced hypertension on intracranial pressure and flow velocities of the middle cerebral arteries in patients with large hemispheric stroke.Stroke.2002;33:998–1004.
- ,,,.Induced hypertension improves cerebral blood flow in acute ischemic stroke.Neurology.2005;64:1979.
- ,,,,.A pilot study of drug induced hypertension for treatment of acute stroke.Neurology.2001;56:1210–1213.
- ,,,,,.Pharmacologic elevation of blood pressure in acute stroke: clinical effects and safety.Stroke.1997;28:2133–2138.
- ,,,,.Feasibility and safety of norephinephrine‐induced arterial hypertension in acute ischemic stroke.Neurology.2004;62:1193–1195.
- ,,,,.Initial emergency department blood pressure as predictor of survival after acute ischemic stroke.Neurology.2005;65:1179–1183.
- The PROGRESS Collaborative Group.Randomized trial of a perindopril‐based blood‐pressure‐lowering regimen among 6105 individuals with previous stroke of transient ischaemic attack.Lancet.2001;358:1033–1041.
- ,,, et al.Effect of treating isolated systolic hypertension on the risk of developing various types and subtypes of stroke.JAMA.2000;284:465–471.
- ,.Treatment of chronic hypertension for the prevention of stroke.South Med J.2003;96:359–362.
- ,,,.Epidemiologic assessment of the role of blood pressure in stroke: the Framingham Study.JAMA.1996;276:1269–1278.