User login
Focal Palmoplantar Keratoderma and Gingival Keratosis Caused by a KRT16 Mutation
To the Editor:
Focal palmoplantar keratoderma and gingival keratosis (FPGK)(Online Mendelian Inheritance in Man [OMIM] 148730) is a rare autosomal-dominant syndrome featuring focal, pressure-related, painful palmoplantar keratoderma and gingival hyperkeratosis presenting as leukokeratosis. Focal palmoplantar keratoderma and gingival keratosis was first defined by Gorlin1 in 1976. Since then, only a few cases have been reported, but no causative mutations have been identified.2
Focal pressure-related palmoplantar keratoderma (PPK) and oral hyperkeratosis also are seen in pachyonychia congenita (PC)(OMIM 167200, 615726, 615728, 167210), a rare autosomal-dominant disorder of keratinization characterized by PPK and nail dystrophy. Patients with PC often present with plantar pain; more variable features include oral leukokeratosis, follicular hyperkeratosis, pilosebaceous and epidermal inclusion cysts, hoarseness, hyperhidrosis, and natal teeth. Pachyonychia congenita is caused by mutation in keratin genes KRT6A, KRT6B, KRT16, or KRT17.
Focal palmoplantar keratoderma and gingival keratosis as well as PC are distinct from other forms of PPK with gingival involvement such as
Despite the common features of FPGK and PC, they are considered distinct disorders due to absence of nail changes in FPGK and no prior evidence of a common genetic cause. We present a patient with familial FPGK found by whole exome sequencing to be caused by a mutation in KRT16.
 showing focal palmoplantar keratoderma and gingival keratosis in those heterozygous for KRT16 mutation p.R127H")
The proband was a 57-year-old man born to unrelated parents (Figure 1). He had no skin problems at birth, and his development was normal. He had painful focal keratoderma since childhood that were most prominent at pressure points on the soles and toes (Figure 2A), in addition to gingival hyperkeratosis and oral leukokeratosis (Figure 2B). He had no associated abnormalities of the skin, hair, or teeth and no nail findings (Figure 2C). He reported that his father and 2 of his 3 sisters were affected with similar symptoms. A punch biopsy of the right fifth toe was consistent with verrucous epidermal hyperplasia with perinuclear keratinization in the spinous layer (Figure 3A). A gingival biopsy showed perinuclear eosinophilic globules and basophilic stranding in the cytoplasm (Figure 3B). His older sister had more severe and painful focal keratoderma of the soles, punctate keratoderma of the palms, gingival hyperkeratosis, and leukokeratosis of the tongue.

Whole exome sequencing of the proband revealed a heterozygous missense mutation in KRT16 (c.380G>A, p.R127H, rs57424749). Sanger sequencing confirmed this mutation and showed that it was heterozygous in both of his affected sisters and absent in his unaffected niece (Figure 1). The patient was treated with topical and systemic retinoids, keratolytics, and mechanical removal to moderate effect, with noted improvement in the appearance and associated pain of the plantar keratoderma.

Phenotypic heterogeneity is common in PC, though PC due to KRT6A mutations demonstrates more severe nail disease with oral lesions, cysts, and follicular hyperkeratosis, while PC caused by KRT16 mutations generally presents with more extensive and painful PPK.4KRT16 mutations affecting p.R127 are frequent causes of PC, and genotype-phenotype correlations have been observed. Individuals with p.R127P mutations exhibit more severe disease with earlier age of onset, more extensive nail involvement and oral leukokeratosis, and greater impact on daily quality of life than in individuals with p.R127C mutations.5 Cases of PC with KRT16 p.R127S and p.R127G mutations also have been observed. The KRT16 c.380G>A, p.R127H mutation we documented has been reported in one kindred with PC who presented with PPK, oral leukokeratosis, toenail thickening, and pilosebaceous and follicular hyperkeratosis.6
Although patients with FPGK lack the thickening of fingernails and/or toenails considered a defining feature of PC, the disorders otherwise are phenotypically similar, suggesting the possibility of common pathogenesis. One linkage study of familial FPGK excluded genetic intervals containing type I and type II keratins but was limited to a single small kindred.2 This study and our data together suggest that, similar to PC, there are multiple genes in which mutations cause FPGK.
Murine Krt16 knockouts show distinct phenotypes depending on the mouse strain in which they are propagated, ranging from perinatal lethality to differences in the severity of oral and PPK lesions.7 These observations provide evidence that additional genetic variants contribute to Krt16 phenotypes in mice and suggest the same could be true for humans.
We propose that some cases of FPGK are due to mutations in KRT16 and thus share a genetic pathogenesis with PC, underscoring the utility of whole exome sequencing in providing genetic diagnoses for disorders that are genetically and clinically heterogeneous. Further biologic investigation of phenotypes caused by KRT16 mutation may reveal respective contributions of additional genetic variation and environmental effects to the variable clinical presentations.
- Gorlin RJ. Focal palmoplantar and marginal gingival hyperkeratosis—a syndrome. Birth Defects Orig Artic Ser. 1976;12:239-242.
- Kolde G, Hennies HC, Bethke G, et al. Focal palmoplantar and gingival keratosis: a distinct palmoplantar ectodermal dysplasia with epidermolytic alterations but lack of mutations in known keratins. J Am Acad Dermatol. 2005;52(3 pt 1):403-409.
- Duchatelet S, Hovnanian A. Olmsted syndrome: clinical, molecular and therapeutic aspects. Orphanet J Rare Dis. 2015;10:33.
- Spaunhurst KM, Hogendorf AM, Smith FJ, et al. Pachyonychia congenita patients with mutations in KRT6A have more extensive disease compared with patients who have mutations in KRT16. Br J Dermatol. 2012;166:875-878.
- Fu T, Leachman SA, Wilson NJ, et al. Genotype-phenotype correlations among pachyonychia congenita patients with K16 mutations. J Invest Dermatol. 2011;131:1025-1028.
- Wilson NJ, O’Toole EA, Milstone LM, et al. The molecular genetic analysis of the expanding pachyonychia congenita case collection. Br J Dermatol. 2014;171:343-355.
- Zieman A, Coulombe PA. The keratin 16 null phenotype is modestly impacted by genetic strain background in mice. Exp Dermatol. 2018;27:672-674.
To the Editor:
Focal palmoplantar keratoderma and gingival keratosis (FPGK)(Online Mendelian Inheritance in Man [OMIM] 148730) is a rare autosomal-dominant syndrome featuring focal, pressure-related, painful palmoplantar keratoderma and gingival hyperkeratosis presenting as leukokeratosis. Focal palmoplantar keratoderma and gingival keratosis was first defined by Gorlin1 in 1976. Since then, only a few cases have been reported, but no causative mutations have been identified.2
Focal pressure-related palmoplantar keratoderma (PPK) and oral hyperkeratosis also are seen in pachyonychia congenita (PC)(OMIM 167200, 615726, 615728, 167210), a rare autosomal-dominant disorder of keratinization characterized by PPK and nail dystrophy. Patients with PC often present with plantar pain; more variable features include oral leukokeratosis, follicular hyperkeratosis, pilosebaceous and epidermal inclusion cysts, hoarseness, hyperhidrosis, and natal teeth. Pachyonychia congenita is caused by mutation in keratin genes KRT6A, KRT6B, KRT16, or KRT17.
Focal palmoplantar keratoderma and gingival keratosis as well as PC are distinct from other forms of PPK with gingival involvement such as
Despite the common features of FPGK and PC, they are considered distinct disorders due to absence of nail changes in FPGK and no prior evidence of a common genetic cause. We present a patient with familial FPGK found by whole exome sequencing to be caused by a mutation in KRT16.
The proband was a 57-year-old man born to unrelated parents (Figure 1). He had no skin problems at birth, and his development was normal. He had painful focal keratoderma since childhood that were most prominent at pressure points on the soles and toes (Figure 2A), in addition to gingival hyperkeratosis and oral leukokeratosis (Figure 2B). He had no associated abnormalities of the skin, hair, or teeth and no nail findings (Figure 2C). He reported that his father and 2 of his 3 sisters were affected with similar symptoms. A punch biopsy of the right fifth toe was consistent with verrucous epidermal hyperplasia with perinuclear keratinization in the spinous layer (Figure 3A). A gingival biopsy showed perinuclear eosinophilic globules and basophilic stranding in the cytoplasm (Figure 3B). His older sister had more severe and painful focal keratoderma of the soles, punctate keratoderma of the palms, gingival hyperkeratosis, and leukokeratosis of the tongue.
Whole exome sequencing of the proband revealed a heterozygous missense mutation in KRT16 (c.380G>A, p.R127H, rs57424749). Sanger sequencing confirmed this mutation and showed that it was heterozygous in both of his affected sisters and absent in his unaffected niece (Figure 1). The patient was treated with topical and systemic retinoids, keratolytics, and mechanical removal to moderate effect, with noted improvement in the appearance and associated pain of the plantar keratoderma.
Phenotypic heterogeneity is common in PC, though PC due to KRT6A mutations demonstrates more severe nail disease with oral lesions, cysts, and follicular hyperkeratosis, while PC caused by KRT16 mutations generally presents with more extensive and painful PPK.4KRT16 mutations affecting p.R127 are frequent causes of PC, and genotype-phenotype correlations have been observed. Individuals with p.R127P mutations exhibit more severe disease with earlier age of onset, more extensive nail involvement and oral leukokeratosis, and greater impact on daily quality of life than in individuals with p.R127C mutations.5 Cases of PC with KRT16 p.R127S and p.R127G mutations also have been observed. The KRT16 c.380G>A, p.R127H mutation we documented has been reported in one kindred with PC who presented with PPK, oral leukokeratosis, toenail thickening, and pilosebaceous and follicular hyperkeratosis.6
Although patients with FPGK lack the thickening of fingernails and/or toenails considered a defining feature of PC, the disorders otherwise are phenotypically similar, suggesting the possibility of common pathogenesis. One linkage study of familial FPGK excluded genetic intervals containing type I and type II keratins but was limited to a single small kindred.2 This study and our data together suggest that, similar to PC, there are multiple genes in which mutations cause FPGK.
Murine Krt16 knockouts show distinct phenotypes depending on the mouse strain in which they are propagated, ranging from perinatal lethality to differences in the severity of oral and PPK lesions.7 These observations provide evidence that additional genetic variants contribute to Krt16 phenotypes in mice and suggest the same could be true for humans.
We propose that some cases of FPGK are due to mutations in KRT16 and thus share a genetic pathogenesis with PC, underscoring the utility of whole exome sequencing in providing genetic diagnoses for disorders that are genetically and clinically heterogeneous. Further biologic investigation of phenotypes caused by KRT16 mutation may reveal respective contributions of additional genetic variation and environmental effects to the variable clinical presentations.
To the Editor:
Focal palmoplantar keratoderma and gingival keratosis (FPGK)(Online Mendelian Inheritance in Man [OMIM] 148730) is a rare autosomal-dominant syndrome featuring focal, pressure-related, painful palmoplantar keratoderma and gingival hyperkeratosis presenting as leukokeratosis. Focal palmoplantar keratoderma and gingival keratosis was first defined by Gorlin1 in 1976. Since then, only a few cases have been reported, but no causative mutations have been identified.2
Focal pressure-related palmoplantar keratoderma (PPK) and oral hyperkeratosis also are seen in pachyonychia congenita (PC)(OMIM 167200, 615726, 615728, 167210), a rare autosomal-dominant disorder of keratinization characterized by PPK and nail dystrophy. Patients with PC often present with plantar pain; more variable features include oral leukokeratosis, follicular hyperkeratosis, pilosebaceous and epidermal inclusion cysts, hoarseness, hyperhidrosis, and natal teeth. Pachyonychia congenita is caused by mutation in keratin genes KRT6A, KRT6B, KRT16, or KRT17.
Focal palmoplantar keratoderma and gingival keratosis as well as PC are distinct from other forms of PPK with gingival involvement such as
Despite the common features of FPGK and PC, they are considered distinct disorders due to absence of nail changes in FPGK and no prior evidence of a common genetic cause. We present a patient with familial FPGK found by whole exome sequencing to be caused by a mutation in KRT16.
The proband was a 57-year-old man born to unrelated parents (Figure 1). He had no skin problems at birth, and his development was normal. He had painful focal keratoderma since childhood that were most prominent at pressure points on the soles and toes (Figure 2A), in addition to gingival hyperkeratosis and oral leukokeratosis (Figure 2B). He had no associated abnormalities of the skin, hair, or teeth and no nail findings (Figure 2C). He reported that his father and 2 of his 3 sisters were affected with similar symptoms. A punch biopsy of the right fifth toe was consistent with verrucous epidermal hyperplasia with perinuclear keratinization in the spinous layer (Figure 3A). A gingival biopsy showed perinuclear eosinophilic globules and basophilic stranding in the cytoplasm (Figure 3B). His older sister had more severe and painful focal keratoderma of the soles, punctate keratoderma of the palms, gingival hyperkeratosis, and leukokeratosis of the tongue.
Whole exome sequencing of the proband revealed a heterozygous missense mutation in KRT16 (c.380G>A, p.R127H, rs57424749). Sanger sequencing confirmed this mutation and showed that it was heterozygous in both of his affected sisters and absent in his unaffected niece (Figure 1). The patient was treated with topical and systemic retinoids, keratolytics, and mechanical removal to moderate effect, with noted improvement in the appearance and associated pain of the plantar keratoderma.
Phenotypic heterogeneity is common in PC, though PC due to KRT6A mutations demonstrates more severe nail disease with oral lesions, cysts, and follicular hyperkeratosis, while PC caused by KRT16 mutations generally presents with more extensive and painful PPK.4KRT16 mutations affecting p.R127 are frequent causes of PC, and genotype-phenotype correlations have been observed. Individuals with p.R127P mutations exhibit more severe disease with earlier age of onset, more extensive nail involvement and oral leukokeratosis, and greater impact on daily quality of life than in individuals with p.R127C mutations.5 Cases of PC with KRT16 p.R127S and p.R127G mutations also have been observed. The KRT16 c.380G>A, p.R127H mutation we documented has been reported in one kindred with PC who presented with PPK, oral leukokeratosis, toenail thickening, and pilosebaceous and follicular hyperkeratosis.6
Although patients with FPGK lack the thickening of fingernails and/or toenails considered a defining feature of PC, the disorders otherwise are phenotypically similar, suggesting the possibility of common pathogenesis. One linkage study of familial FPGK excluded genetic intervals containing type I and type II keratins but was limited to a single small kindred.2 This study and our data together suggest that, similar to PC, there are multiple genes in which mutations cause FPGK.
Murine Krt16 knockouts show distinct phenotypes depending on the mouse strain in which they are propagated, ranging from perinatal lethality to differences in the severity of oral and PPK lesions.7 These observations provide evidence that additional genetic variants contribute to Krt16 phenotypes in mice and suggest the same could be true for humans.
We propose that some cases of FPGK are due to mutations in KRT16 and thus share a genetic pathogenesis with PC, underscoring the utility of whole exome sequencing in providing genetic diagnoses for disorders that are genetically and clinically heterogeneous. Further biologic investigation of phenotypes caused by KRT16 mutation may reveal respective contributions of additional genetic variation and environmental effects to the variable clinical presentations.
- Gorlin RJ. Focal palmoplantar and marginal gingival hyperkeratosis—a syndrome. Birth Defects Orig Artic Ser. 1976;12:239-242.
- Kolde G, Hennies HC, Bethke G, et al. Focal palmoplantar and gingival keratosis: a distinct palmoplantar ectodermal dysplasia with epidermolytic alterations but lack of mutations in known keratins. J Am Acad Dermatol. 2005;52(3 pt 1):403-409.
- Duchatelet S, Hovnanian A. Olmsted syndrome: clinical, molecular and therapeutic aspects. Orphanet J Rare Dis. 2015;10:33.
- Spaunhurst KM, Hogendorf AM, Smith FJ, et al. Pachyonychia congenita patients with mutations in KRT6A have more extensive disease compared with patients who have mutations in KRT16. Br J Dermatol. 2012;166:875-878.
- Fu T, Leachman SA, Wilson NJ, et al. Genotype-phenotype correlations among pachyonychia congenita patients with K16 mutations. J Invest Dermatol. 2011;131:1025-1028.
- Wilson NJ, O’Toole EA, Milstone LM, et al. The molecular genetic analysis of the expanding pachyonychia congenita case collection. Br J Dermatol. 2014;171:343-355.
- Zieman A, Coulombe PA. The keratin 16 null phenotype is modestly impacted by genetic strain background in mice. Exp Dermatol. 2018;27:672-674.
- Gorlin RJ. Focal palmoplantar and marginal gingival hyperkeratosis—a syndrome. Birth Defects Orig Artic Ser. 1976;12:239-242.
- Kolde G, Hennies HC, Bethke G, et al. Focal palmoplantar and gingival keratosis: a distinct palmoplantar ectodermal dysplasia with epidermolytic alterations but lack of mutations in known keratins. J Am Acad Dermatol. 2005;52(3 pt 1):403-409.
- Duchatelet S, Hovnanian A. Olmsted syndrome: clinical, molecular and therapeutic aspects. Orphanet J Rare Dis. 2015;10:33.
- Spaunhurst KM, Hogendorf AM, Smith FJ, et al. Pachyonychia congenita patients with mutations in KRT6A have more extensive disease compared with patients who have mutations in KRT16. Br J Dermatol. 2012;166:875-878.
- Fu T, Leachman SA, Wilson NJ, et al. Genotype-phenotype correlations among pachyonychia congenita patients with K16 mutations. J Invest Dermatol. 2011;131:1025-1028.
- Wilson NJ, O’Toole EA, Milstone LM, et al. The molecular genetic analysis of the expanding pachyonychia congenita case collection. Br J Dermatol. 2014;171:343-355.
- Zieman A, Coulombe PA. The keratin 16 null phenotype is modestly impacted by genetic strain background in mice. Exp Dermatol. 2018;27:672-674.
Practice Points
- Focal palmoplantar keratoderma and gingival keratosis (FPGK) is a rare autosomal-dominant syndrome featuring focal, pressure-related, painful palmoplantar keratoderma (PPK) and gingival hyperkeratosis presenting as leukokeratosis.
- Focal pressure-related PPK and oral hyperkeratosis also are seen in pachyonychia congenita (PC), which is caused by mutations in keratin genes and is distinguished from FPGK by characteristic nail changes.
- A shared causative gene suggests that FPGK should be considered part of the PC spectrum.

Postherpetic Isotopic Responses With 3 Simultaneously Occurring Reactions Following Herpes Zoster
Postherpetic isotopic response (PHIR) refers to the occurrence of a second disease manifesting at the site of prior herpes infection. Many forms of PHIR have been described (Table), with postzoster granulomatous dermatitis (eg, granuloma annulare, sarcoidosis, granulomatous vasculitis) being the most common.1 Both primary and metastatic malignancies also can occur at the site of a prior herpes infection. Rarely, multiple types of PHIRs occur simultaneously. We report a case of 3 simultaneously occurring postzoster isotopic responses--granulomatous dermatitis, vasculitis, and chronic lymphocytic leukemia (CLL)--and review the various types of PHIRs.
Case Report
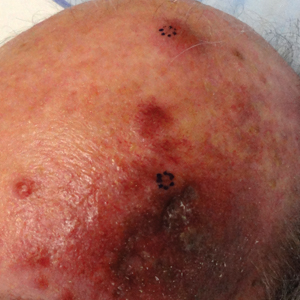
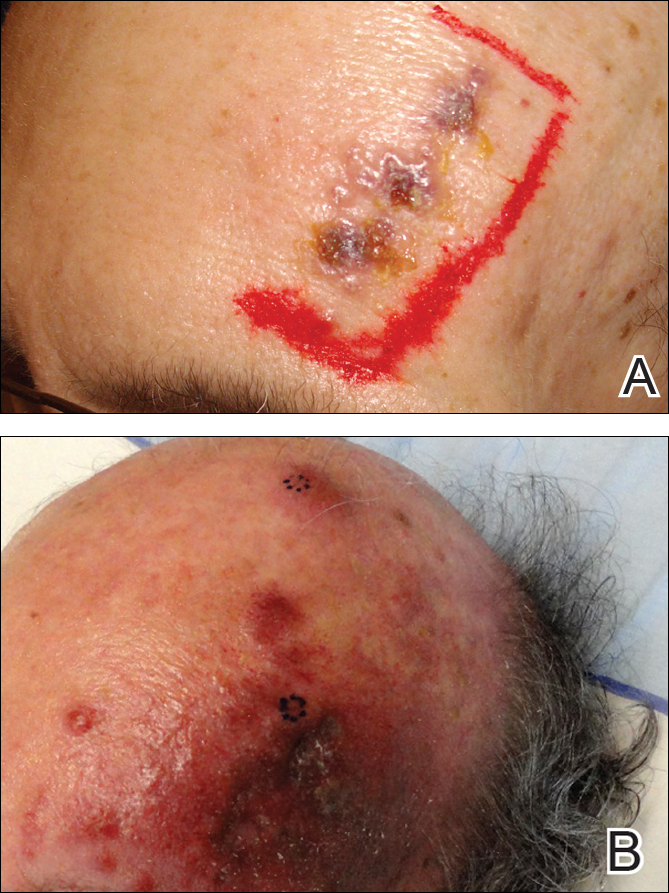
A 55-year-old man with a 4-year history of CLL was admitted to the hospital due to a painful rash on the left side of the face of 2 months' duration. Erythematous to violaceous plaques with surrounding papules and nodules were present on the left side of the forehead and frontal scalp with focal ulceration. Two months prior, the patient had unilateral vesicular lesions in the same distribution (Figure 1A). He initially received a 3-week course of acyclovir for a presumed herpes zoster infection and showed prompt improvement in the vesicular lesions. After resolution of the vesicles, papules and nodules began developing in the prior vesicular areas and he was treated with another course of acyclovir with the addition of clindamycin. When the lesions continued to progress and spread down the left side of the forehead and upper eyelid (Figure 1B), he was admitted to the hospital and assessed by the consultative dermatology team. No fevers, chills, or other systemic symptoms were reported.
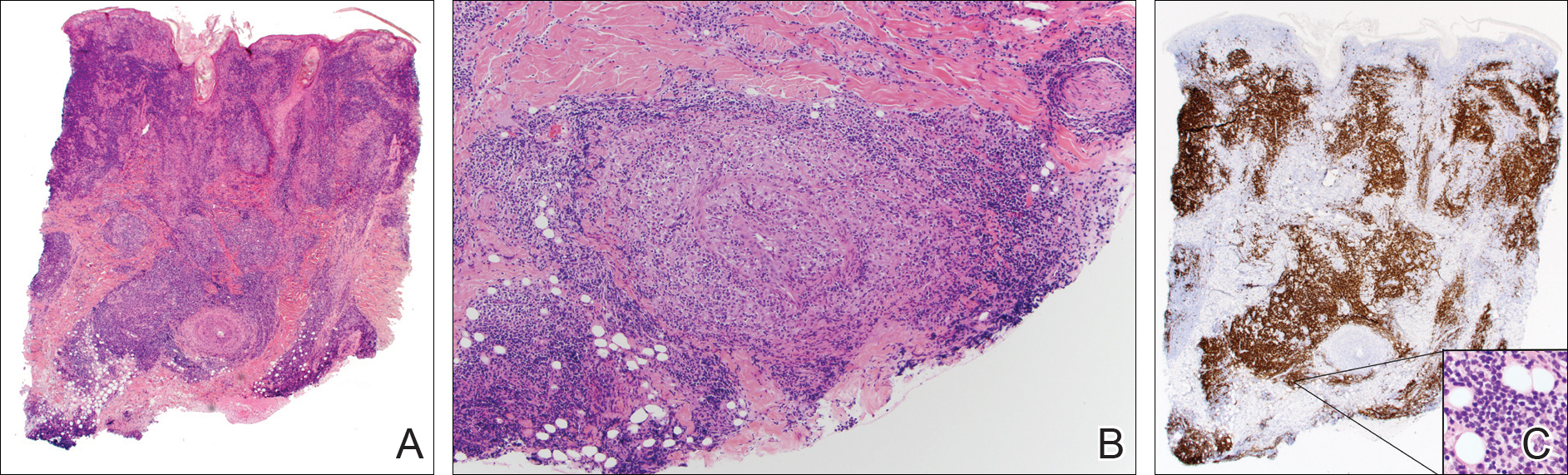
A punch biopsy showed a diffuse lymphocytic infiltrate filling the dermis and extending into the subcutis with nodular collections of histiocytes and some plasma cells scattered throughout (Figure 2A). A medium-vessel vasculitis was present with numerous histiocytes and lymphocytes infiltrating the muscular wall of a blood vessel in the subcutis (Figure 2B). CD3 and CD20 immunostaining showed an overwhelming majority of B cells, some with enlarged atypical nuclei and a smaller number of reactive T lymphocytes (Figure 2C). CD5 and CD43 were diffusely positive in the B cells, confirming the diagnosis of cutaneous CLL. CD23 staining was focally positive. Immunostaining for κ and λ light chains showed a marginal κ predominance. An additional biopsy for tissue culture was negative. A diagnosis of postzoster granulomatous dermatitis with vasculitis and cutaneous CLL was rendered.
Comment
Postherpetic Cutaneous Reactions
Various cutaneous reactions can occur at the site of prior herpes infection. The most frequently reported reactions are granulomatous dermatitides such as granuloma annulare, granulomatous vasculitis, granulomatous folliculitis, sarcoidosis, and nonspecific granulomatous dermatitis.1 Primary cutaneous malignancies and cutaneous metastases, including hematologic malignancies, have also been reported after herpetic infections. In a review of 127 patients with postherpetic cutaneous reactions, 47 had a granulomatous dermatitis, 32 had nonhematologic malignancies, 18 had leukemic or lymphomatous/pseudolymphomatous infiltrates, 10 had acneform lesions, 9 had nongranulomatous dermatitides such as lichen planus and allergic contact dermatitis, and 8 had nonherpetic skin infections; single cases of reactive perforating collagenosis, nodular solar degeneration, and a keloid also were reported.1
Pathogenesis of Cutaneous Reactions
Although postherpetic cutaneous reactions can develop in healthy individuals, they occur more often in immunocompromised patients. Postherpetic isotopic response has been used to describe the development of a nonherpetic disease at the site of prior herpes infection.2 Several different theories have been proposed to explain the pathogenesis of the PHIR, including an unusual delayed-type hypersensitivity reaction to residual viral antigen or host-tissue antigen altered by the virus. This delayed-type hypersensitivity explanation is supported by the presence of helper T cells, activated T lymphocytes, macrophages, varicella major viral envelope glycoproteins, and viral DNA in postherpetic granulomatous lesions3; however, cases that lack detectable virus and viral DNA in these types of lesions also have been reported.4
A second hypothesis proposes that inflammatory or viral-induced alteration of the local microvasculature results in increased site-specific susceptibility to subsequent inflammatory responses and drives these isotopic reactions.2,3 Damage or alteration of local peripheral nerves leading to abnormal release of specific neuromediators involved in regulating cutaneous inflammatory responses also may play a role.5 Varicella-zoster virus utilizes the peripheral nervous system to establish latent infection and can cause destruction of alpha delta and C nerve fibers in the dermis.1 Destruction of nerve fibers may indirectly influence the local immune system by altering the release of neuromediators such as substance P (known to increase blood vessel permeability, increase fibrinolytic activity, and induce mast cell secretion), vasoactive intestinal peptide (enhances monocyte migration, increases histamine release from mast cells, and inhibits natural killer cell activity), calcitonin gene-related peptide (increases vascular permeability, endothelial cell proliferation, and the accumulation of neutrophils), and melanocyte-stimulating hormone (induces anti-inflammatory cytokines). Disruption of the nervous system resulting in an altered local immune response also has been observed in other settings (eg, amputees who develop inflammatory diseases, bacterial and fungal infections, and cutaneous neoplasms confined to stump skin).1
Malignancies in PHIR
The granulomatous inflammation in PHIRs is a nonneoplastic inflammatory reaction with a variable lymphocytic component. Granuloma formation can be seen in both reactive inflammatory infiltrates and in cutaneous involvement of leukemias and lymphomas. Leukemia cutis has been reported in 4% to 20% of patients with CLL/small lymphocytic leukemia.6 In one series of 42 patients with CLL, the malignant cells were confined to the site of postherpetic scars in 14% (6/42) of patients.5 Sixteen percent (7/42) of patients had no prior diagnosis of CLL at the time they developed leukemia cutis, including one patient with leukemia cutis in a postzoster scar. The mechanism involved in the accumulation of neoplastic lymphocytes within postzoster scars has not been fully characterized. The idea that postzoster sites represent a site of least resistance for cutaneous infiltration of CLL due to the changes from prior inflammatory responses has been proposed.7
Combined CLL and granulomatous dermatitis at prior sites of herpes zoster was first reported in 1990.8 In 1995, Cerroni et al9 reported a series of 5 patients with cutaneous CLL following herpes zoster or herpes simplex virus infection. Three of those patients also demonstrated granuloma formation.9 Establishing a new diagnosis of CLL from a biopsy of postzoster granulomatous dermatitis with an associated lymphoid infiltrate also has been reported.10 Cerroni et al9 postulated that cutaneous CLL in post-herpes zoster scars may occur more frequently than reported due to misdiagnoses of CLL as pseudolymphoma. Two additional cases of postherpetic cutaneous CLL and granulomatous dermatitis have been reported since 1995.7,10
Diagnosis of Multiple PHIRs
The presence of 3 concurrent PHIRs is rare. The patient in this report had postzoster cutaneous CLL with an associated granulomatous dermatitis and medium-vessel vasculitis. One other case with these 3 findings was reported by Elgoweini et al.7 Overlooking important diagnoses when multiple findings are present in a biopsy can lead to diagnostic delay and incorrect treatment; we highlighted the importance of careful examination of biopsies in PHIRs to ensure diagnostic accuracy. In cases of postzoster granulomatous dermatitis, assessment of the lymphocytic component should not be overlooked. The presence of a dense lymphocytic infiltrate should raise the possibility of a lymphoproliferative disorder such as CLL, even in patients with no prior history of lymphoma. If initial immunostaining discloses a predominantly B-cell infiltrate, additional immuno-stains (eg, CD5, CD23, CD43) and/or genetic testing for monoclonality should be pursued.
Conclusion
Clinicians and dermatopathologists should be aware of the multiplicity of postherpetic isotopic responses and consider immunohistochemical stains to differentiate between a genuine lymphoma such as CLL and pseudolymphoma in PHIRs with a lymphoid infiltrate.
- Ruocco V, Ruocco E, Ghersetich I, et al. Isotopic response after herpes virus infection: an update. J Am Acad Dermatol. 2002;46:90-94.
- Wolf R, Wolf D, Ruocco E, et al. Wolf's isotopic response. Clin Dermatol. 2011;29:237-240.
- Nikkels AF, Debrus S, Delvenne P, et al. Viral glycoproteins in herpesviridae granulomas. Am J Dermatopathol. 1994;16:588-592.
- Snow J, el-Azhary R, Gibson L, et al. Granulomatous vasculitis associated with herpes virus: a persistent, painful, postherpetic papular eruption. Mayo Clin Proc. 1997;72:851-853.
- Cerroni L, Zenahlik P, Hofler G, et al. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia: a clinicopathologic and prognostic study of 42 patients. Am J Surg Pathol. 1996;20:1000-1010.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Elgoweini M, Blessing K, Jackson R, et al. Coexistent granulomatous vasculitis and leukaemia cutis in a patient with resolving herpes zoster. Clin Exp Dermatol. 2011;36:749-751.
- Pujol RM, Matias-Guiu X, Planaguma M, et al. Chronic lymphocytic leukemia and cutaneous granulomas at sites of herpes zoster scars. Int J Dermatol. 1990;29:652-654.
- Cerroni L, Zenahlik P, Kerl H. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia arising at the site of herpes zoster and herpes simplex scars. Cancer. 1995;76:26-31.
- Trojjet S, Hammami H, Zaraa I, et al. Chronic lymphocytic leukemia revealed by a granulomatous zosteriform eruption. Skinmed. 2012;10:50-52.
Postherpetic isotopic response (PHIR) refers to the occurrence of a second disease manifesting at the site of prior herpes infection. Many forms of PHIR have been described (Table), with postzoster granulomatous dermatitis (eg, granuloma annulare, sarcoidosis, granulomatous vasculitis) being the most common.1 Both primary and metastatic malignancies also can occur at the site of a prior herpes infection. Rarely, multiple types of PHIRs occur simultaneously. We report a case of 3 simultaneously occurring postzoster isotopic responses--granulomatous dermatitis, vasculitis, and chronic lymphocytic leukemia (CLL)--and review the various types of PHIRs.
Case Report
A 55-year-old man with a 4-year history of CLL was admitted to the hospital due to a painful rash on the left side of the face of 2 months' duration. Erythematous to violaceous plaques with surrounding papules and nodules were present on the left side of the forehead and frontal scalp with focal ulceration. Two months prior, the patient had unilateral vesicular lesions in the same distribution (Figure 1A). He initially received a 3-week course of acyclovir for a presumed herpes zoster infection and showed prompt improvement in the vesicular lesions. After resolution of the vesicles, papules and nodules began developing in the prior vesicular areas and he was treated with another course of acyclovir with the addition of clindamycin. When the lesions continued to progress and spread down the left side of the forehead and upper eyelid (Figure 1B), he was admitted to the hospital and assessed by the consultative dermatology team. No fevers, chills, or other systemic symptoms were reported.
A punch biopsy showed a diffuse lymphocytic infiltrate filling the dermis and extending into the subcutis with nodular collections of histiocytes and some plasma cells scattered throughout (Figure 2A). A medium-vessel vasculitis was present with numerous histiocytes and lymphocytes infiltrating the muscular wall of a blood vessel in the subcutis (Figure 2B). CD3 and CD20 immunostaining showed an overwhelming majority of B cells, some with enlarged atypical nuclei and a smaller number of reactive T lymphocytes (Figure 2C). CD5 and CD43 were diffusely positive in the B cells, confirming the diagnosis of cutaneous CLL. CD23 staining was focally positive. Immunostaining for κ and λ light chains showed a marginal κ predominance. An additional biopsy for tissue culture was negative. A diagnosis of postzoster granulomatous dermatitis with vasculitis and cutaneous CLL was rendered.
Comment
Postherpetic Cutaneous Reactions
Various cutaneous reactions can occur at the site of prior herpes infection. The most frequently reported reactions are granulomatous dermatitides such as granuloma annulare, granulomatous vasculitis, granulomatous folliculitis, sarcoidosis, and nonspecific granulomatous dermatitis.1 Primary cutaneous malignancies and cutaneous metastases, including hematologic malignancies, have also been reported after herpetic infections. In a review of 127 patients with postherpetic cutaneous reactions, 47 had a granulomatous dermatitis, 32 had nonhematologic malignancies, 18 had leukemic or lymphomatous/pseudolymphomatous infiltrates, 10 had acneform lesions, 9 had nongranulomatous dermatitides such as lichen planus and allergic contact dermatitis, and 8 had nonherpetic skin infections; single cases of reactive perforating collagenosis, nodular solar degeneration, and a keloid also were reported.1
Pathogenesis of Cutaneous Reactions
Although postherpetic cutaneous reactions can develop in healthy individuals, they occur more often in immunocompromised patients. Postherpetic isotopic response has been used to describe the development of a nonherpetic disease at the site of prior herpes infection.2 Several different theories have been proposed to explain the pathogenesis of the PHIR, including an unusual delayed-type hypersensitivity reaction to residual viral antigen or host-tissue antigen altered by the virus. This delayed-type hypersensitivity explanation is supported by the presence of helper T cells, activated T lymphocytes, macrophages, varicella major viral envelope glycoproteins, and viral DNA in postherpetic granulomatous lesions3; however, cases that lack detectable virus and viral DNA in these types of lesions also have been reported.4
A second hypothesis proposes that inflammatory or viral-induced alteration of the local microvasculature results in increased site-specific susceptibility to subsequent inflammatory responses and drives these isotopic reactions.2,3 Damage or alteration of local peripheral nerves leading to abnormal release of specific neuromediators involved in regulating cutaneous inflammatory responses also may play a role.5 Varicella-zoster virus utilizes the peripheral nervous system to establish latent infection and can cause destruction of alpha delta and C nerve fibers in the dermis.1 Destruction of nerve fibers may indirectly influence the local immune system by altering the release of neuromediators such as substance P (known to increase blood vessel permeability, increase fibrinolytic activity, and induce mast cell secretion), vasoactive intestinal peptide (enhances monocyte migration, increases histamine release from mast cells, and inhibits natural killer cell activity), calcitonin gene-related peptide (increases vascular permeability, endothelial cell proliferation, and the accumulation of neutrophils), and melanocyte-stimulating hormone (induces anti-inflammatory cytokines). Disruption of the nervous system resulting in an altered local immune response also has been observed in other settings (eg, amputees who develop inflammatory diseases, bacterial and fungal infections, and cutaneous neoplasms confined to stump skin).1
Malignancies in PHIR
The granulomatous inflammation in PHIRs is a nonneoplastic inflammatory reaction with a variable lymphocytic component. Granuloma formation can be seen in both reactive inflammatory infiltrates and in cutaneous involvement of leukemias and lymphomas. Leukemia cutis has been reported in 4% to 20% of patients with CLL/small lymphocytic leukemia.6 In one series of 42 patients with CLL, the malignant cells were confined to the site of postherpetic scars in 14% (6/42) of patients.5 Sixteen percent (7/42) of patients had no prior diagnosis of CLL at the time they developed leukemia cutis, including one patient with leukemia cutis in a postzoster scar. The mechanism involved in the accumulation of neoplastic lymphocytes within postzoster scars has not been fully characterized. The idea that postzoster sites represent a site of least resistance for cutaneous infiltration of CLL due to the changes from prior inflammatory responses has been proposed.7
Combined CLL and granulomatous dermatitis at prior sites of herpes zoster was first reported in 1990.8 In 1995, Cerroni et al9 reported a series of 5 patients with cutaneous CLL following herpes zoster or herpes simplex virus infection. Three of those patients also demonstrated granuloma formation.9 Establishing a new diagnosis of CLL from a biopsy of postzoster granulomatous dermatitis with an associated lymphoid infiltrate also has been reported.10 Cerroni et al9 postulated that cutaneous CLL in post-herpes zoster scars may occur more frequently than reported due to misdiagnoses of CLL as pseudolymphoma. Two additional cases of postherpetic cutaneous CLL and granulomatous dermatitis have been reported since 1995.7,10
Diagnosis of Multiple PHIRs
The presence of 3 concurrent PHIRs is rare. The patient in this report had postzoster cutaneous CLL with an associated granulomatous dermatitis and medium-vessel vasculitis. One other case with these 3 findings was reported by Elgoweini et al.7 Overlooking important diagnoses when multiple findings are present in a biopsy can lead to diagnostic delay and incorrect treatment; we highlighted the importance of careful examination of biopsies in PHIRs to ensure diagnostic accuracy. In cases of postzoster granulomatous dermatitis, assessment of the lymphocytic component should not be overlooked. The presence of a dense lymphocytic infiltrate should raise the possibility of a lymphoproliferative disorder such as CLL, even in patients with no prior history of lymphoma. If initial immunostaining discloses a predominantly B-cell infiltrate, additional immuno-stains (eg, CD5, CD23, CD43) and/or genetic testing for monoclonality should be pursued.
Conclusion
Clinicians and dermatopathologists should be aware of the multiplicity of postherpetic isotopic responses and consider immunohistochemical stains to differentiate between a genuine lymphoma such as CLL and pseudolymphoma in PHIRs with a lymphoid infiltrate.
Postherpetic isotopic response (PHIR) refers to the occurrence of a second disease manifesting at the site of prior herpes infection. Many forms of PHIR have been described (Table), with postzoster granulomatous dermatitis (eg, granuloma annulare, sarcoidosis, granulomatous vasculitis) being the most common.1 Both primary and metastatic malignancies also can occur at the site of a prior herpes infection. Rarely, multiple types of PHIRs occur simultaneously. We report a case of 3 simultaneously occurring postzoster isotopic responses--granulomatous dermatitis, vasculitis, and chronic lymphocytic leukemia (CLL)--and review the various types of PHIRs.
Case Report
A 55-year-old man with a 4-year history of CLL was admitted to the hospital due to a painful rash on the left side of the face of 2 months' duration. Erythematous to violaceous plaques with surrounding papules and nodules were present on the left side of the forehead and frontal scalp with focal ulceration. Two months prior, the patient had unilateral vesicular lesions in the same distribution (Figure 1A). He initially received a 3-week course of acyclovir for a presumed herpes zoster infection and showed prompt improvement in the vesicular lesions. After resolution of the vesicles, papules and nodules began developing in the prior vesicular areas and he was treated with another course of acyclovir with the addition of clindamycin. When the lesions continued to progress and spread down the left side of the forehead and upper eyelid (Figure 1B), he was admitted to the hospital and assessed by the consultative dermatology team. No fevers, chills, or other systemic symptoms were reported.
A punch biopsy showed a diffuse lymphocytic infiltrate filling the dermis and extending into the subcutis with nodular collections of histiocytes and some plasma cells scattered throughout (Figure 2A). A medium-vessel vasculitis was present with numerous histiocytes and lymphocytes infiltrating the muscular wall of a blood vessel in the subcutis (Figure 2B). CD3 and CD20 immunostaining showed an overwhelming majority of B cells, some with enlarged atypical nuclei and a smaller number of reactive T lymphocytes (Figure 2C). CD5 and CD43 were diffusely positive in the B cells, confirming the diagnosis of cutaneous CLL. CD23 staining was focally positive. Immunostaining for κ and λ light chains showed a marginal κ predominance. An additional biopsy for tissue culture was negative. A diagnosis of postzoster granulomatous dermatitis with vasculitis and cutaneous CLL was rendered.
Comment
Postherpetic Cutaneous Reactions
Various cutaneous reactions can occur at the site of prior herpes infection. The most frequently reported reactions are granulomatous dermatitides such as granuloma annulare, granulomatous vasculitis, granulomatous folliculitis, sarcoidosis, and nonspecific granulomatous dermatitis.1 Primary cutaneous malignancies and cutaneous metastases, including hematologic malignancies, have also been reported after herpetic infections. In a review of 127 patients with postherpetic cutaneous reactions, 47 had a granulomatous dermatitis, 32 had nonhematologic malignancies, 18 had leukemic or lymphomatous/pseudolymphomatous infiltrates, 10 had acneform lesions, 9 had nongranulomatous dermatitides such as lichen planus and allergic contact dermatitis, and 8 had nonherpetic skin infections; single cases of reactive perforating collagenosis, nodular solar degeneration, and a keloid also were reported.1
Pathogenesis of Cutaneous Reactions
Although postherpetic cutaneous reactions can develop in healthy individuals, they occur more often in immunocompromised patients. Postherpetic isotopic response has been used to describe the development of a nonherpetic disease at the site of prior herpes infection.2 Several different theories have been proposed to explain the pathogenesis of the PHIR, including an unusual delayed-type hypersensitivity reaction to residual viral antigen or host-tissue antigen altered by the virus. This delayed-type hypersensitivity explanation is supported by the presence of helper T cells, activated T lymphocytes, macrophages, varicella major viral envelope glycoproteins, and viral DNA in postherpetic granulomatous lesions3; however, cases that lack detectable virus and viral DNA in these types of lesions also have been reported.4
A second hypothesis proposes that inflammatory or viral-induced alteration of the local microvasculature results in increased site-specific susceptibility to subsequent inflammatory responses and drives these isotopic reactions.2,3 Damage or alteration of local peripheral nerves leading to abnormal release of specific neuromediators involved in regulating cutaneous inflammatory responses also may play a role.5 Varicella-zoster virus utilizes the peripheral nervous system to establish latent infection and can cause destruction of alpha delta and C nerve fibers in the dermis.1 Destruction of nerve fibers may indirectly influence the local immune system by altering the release of neuromediators such as substance P (known to increase blood vessel permeability, increase fibrinolytic activity, and induce mast cell secretion), vasoactive intestinal peptide (enhances monocyte migration, increases histamine release from mast cells, and inhibits natural killer cell activity), calcitonin gene-related peptide (increases vascular permeability, endothelial cell proliferation, and the accumulation of neutrophils), and melanocyte-stimulating hormone (induces anti-inflammatory cytokines). Disruption of the nervous system resulting in an altered local immune response also has been observed in other settings (eg, amputees who develop inflammatory diseases, bacterial and fungal infections, and cutaneous neoplasms confined to stump skin).1
Malignancies in PHIR
The granulomatous inflammation in PHIRs is a nonneoplastic inflammatory reaction with a variable lymphocytic component. Granuloma formation can be seen in both reactive inflammatory infiltrates and in cutaneous involvement of leukemias and lymphomas. Leukemia cutis has been reported in 4% to 20% of patients with CLL/small lymphocytic leukemia.6 In one series of 42 patients with CLL, the malignant cells were confined to the site of postherpetic scars in 14% (6/42) of patients.5 Sixteen percent (7/42) of patients had no prior diagnosis of CLL at the time they developed leukemia cutis, including one patient with leukemia cutis in a postzoster scar. The mechanism involved in the accumulation of neoplastic lymphocytes within postzoster scars has not been fully characterized. The idea that postzoster sites represent a site of least resistance for cutaneous infiltration of CLL due to the changes from prior inflammatory responses has been proposed.7
Combined CLL and granulomatous dermatitis at prior sites of herpes zoster was first reported in 1990.8 In 1995, Cerroni et al9 reported a series of 5 patients with cutaneous CLL following herpes zoster or herpes simplex virus infection. Three of those patients also demonstrated granuloma formation.9 Establishing a new diagnosis of CLL from a biopsy of postzoster granulomatous dermatitis with an associated lymphoid infiltrate also has been reported.10 Cerroni et al9 postulated that cutaneous CLL in post-herpes zoster scars may occur more frequently than reported due to misdiagnoses of CLL as pseudolymphoma. Two additional cases of postherpetic cutaneous CLL and granulomatous dermatitis have been reported since 1995.7,10
Diagnosis of Multiple PHIRs
The presence of 3 concurrent PHIRs is rare. The patient in this report had postzoster cutaneous CLL with an associated granulomatous dermatitis and medium-vessel vasculitis. One other case with these 3 findings was reported by Elgoweini et al.7 Overlooking important diagnoses when multiple findings are present in a biopsy can lead to diagnostic delay and incorrect treatment; we highlighted the importance of careful examination of biopsies in PHIRs to ensure diagnostic accuracy. In cases of postzoster granulomatous dermatitis, assessment of the lymphocytic component should not be overlooked. The presence of a dense lymphocytic infiltrate should raise the possibility of a lymphoproliferative disorder such as CLL, even in patients with no prior history of lymphoma. If initial immunostaining discloses a predominantly B-cell infiltrate, additional immuno-stains (eg, CD5, CD23, CD43) and/or genetic testing for monoclonality should be pursued.
Conclusion
Clinicians and dermatopathologists should be aware of the multiplicity of postherpetic isotopic responses and consider immunohistochemical stains to differentiate between a genuine lymphoma such as CLL and pseudolymphoma in PHIRs with a lymphoid infiltrate.
- Ruocco V, Ruocco E, Ghersetich I, et al. Isotopic response after herpes virus infection: an update. J Am Acad Dermatol. 2002;46:90-94.
- Wolf R, Wolf D, Ruocco E, et al. Wolf's isotopic response. Clin Dermatol. 2011;29:237-240.
- Nikkels AF, Debrus S, Delvenne P, et al. Viral glycoproteins in herpesviridae granulomas. Am J Dermatopathol. 1994;16:588-592.
- Snow J, el-Azhary R, Gibson L, et al. Granulomatous vasculitis associated with herpes virus: a persistent, painful, postherpetic papular eruption. Mayo Clin Proc. 1997;72:851-853.
- Cerroni L, Zenahlik P, Hofler G, et al. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia: a clinicopathologic and prognostic study of 42 patients. Am J Surg Pathol. 1996;20:1000-1010.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Elgoweini M, Blessing K, Jackson R, et al. Coexistent granulomatous vasculitis and leukaemia cutis in a patient with resolving herpes zoster. Clin Exp Dermatol. 2011;36:749-751.
- Pujol RM, Matias-Guiu X, Planaguma M, et al. Chronic lymphocytic leukemia and cutaneous granulomas at sites of herpes zoster scars. Int J Dermatol. 1990;29:652-654.
- Cerroni L, Zenahlik P, Kerl H. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia arising at the site of herpes zoster and herpes simplex scars. Cancer. 1995;76:26-31.
- Trojjet S, Hammami H, Zaraa I, et al. Chronic lymphocytic leukemia revealed by a granulomatous zosteriform eruption. Skinmed. 2012;10:50-52.
- Ruocco V, Ruocco E, Ghersetich I, et al. Isotopic response after herpes virus infection: an update. J Am Acad Dermatol. 2002;46:90-94.
- Wolf R, Wolf D, Ruocco E, et al. Wolf's isotopic response. Clin Dermatol. 2011;29:237-240.
- Nikkels AF, Debrus S, Delvenne P, et al. Viral glycoproteins in herpesviridae granulomas. Am J Dermatopathol. 1994;16:588-592.
- Snow J, el-Azhary R, Gibson L, et al. Granulomatous vasculitis associated with herpes virus: a persistent, painful, postherpetic papular eruption. Mayo Clin Proc. 1997;72:851-853.
- Cerroni L, Zenahlik P, Hofler G, et al. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia: a clinicopathologic and prognostic study of 42 patients. Am J Surg Pathol. 1996;20:1000-1010.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Elgoweini M, Blessing K, Jackson R, et al. Coexistent granulomatous vasculitis and leukaemia cutis in a patient with resolving herpes zoster. Clin Exp Dermatol. 2011;36:749-751.
- Pujol RM, Matias-Guiu X, Planaguma M, et al. Chronic lymphocytic leukemia and cutaneous granulomas at sites of herpes zoster scars. Int J Dermatol. 1990;29:652-654.
- Cerroni L, Zenahlik P, Kerl H. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia arising at the site of herpes zoster and herpes simplex scars. Cancer. 1995;76:26-31.
- Trojjet S, Hammami H, Zaraa I, et al. Chronic lymphocytic leukemia revealed by a granulomatous zosteriform eruption. Skinmed. 2012;10:50-52.
Practice Points
- Multiple diseases may present in prior sites of herpes infection (postherpetic isotopic response).
- Granulomatous dermatitis is the most common postherpetic isotopic response, but other inflammatory, neoplastic, or infectious conditions also occur.
- Multiple conditions may present simultaneously at sites of herpes infection.
- Cutaneous involvement by chronic lymphocytic leukemia (CLL) can be easily overlooked in this setting.