User login
Mycobacterium haemophilum: A Challenging Treatment Dilemma in an Immunocompromised Patient
To the Editor:
The increase in nontuberculous mycobacteria (NTM) infections over the last 3 decades likely is multifaceted, including increased clinical awareness, improved laboratory diagnostics, growing numbers of immunocompromised patients, and an aging population.1,2 Historically, the majority of mycobacteria-related diseases are due to Mycobacterium tuberculosis, Mycobacterium bovis, and Mycobacterium leprae.3
Mycobacterium haemophilum is a slow-growing acid-fast bacillus (AFB) that differs from other Mycobacterium species in that it requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. As these requirements for growth are not standard for AFB cultures, M haemophilum infection may be underrecognized and underreported.3Mycobacterium haemophilum infections largely are cutaneous and generally are seen in AIDS patients and bone marrow transplant recipients who are iatrogenically immunosuppressed.4,5 No species-specific treatment guidelines exist2; however, triple-drug therapy combining a macrolide, rifamycin, and a quinolone for a minimum of 12 months often is recommended.
A 64-year-old man with a history of coronary artery disease, hypertension, hyperlipidemia, and acute myelogenous leukemia (AML) underwent allogenic stem cell transplantation. His posttransplant course was complicated by multiple deep vein thromboses, hypogammaglobulinemia, and graft-vs-host disease (GVHD) of the skin and gastrointestinal tract that manifested as chronic diarrhea, which was managed with chronic prednisone. Thirteen months after the transplant, the patient presented to his outpatient oncologist (M.K.) for evaluation of painless, nonpruritic, erythematous papules and nodules that had emerged on the right side of the chest, right arm, and left leg of approximately 2 weeks’ duration.
On review of systems by oncology, the patient denied any fevers, chills, or night sweats but noted chronic loose nonbloody stools without abdominal pain, likely related to the GVHD. The patient’s medications included prednisone 20 mg once daily, fluconazole, amitriptyline, atovaquone, budesonide, dabigatran, metoprolol, pantoprazole, rosuvastatin, senna glycoside, spironolactone, tramadol, and valacyclovir.
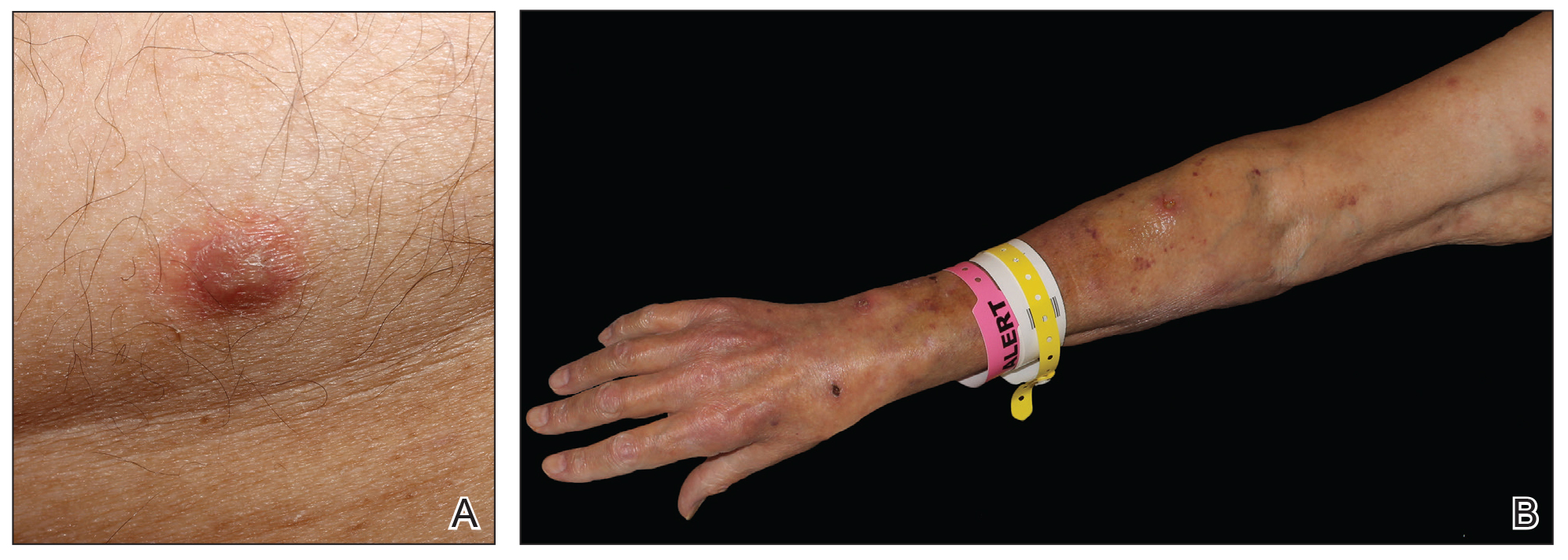
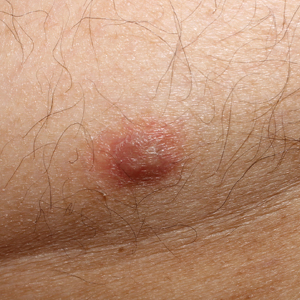
Physical examination revealed multiple singular erythematous nodules on the right side of the chest (Figure 1A), right arm (Figure 1B), and left leg. There was no regional lymphadenopathy. The patient was afebrile and hemodynamically stable. A biopsy of the arm performed to rule out leukemia cutis revealed a granulomatous dermatitis with numerous AFB (Figures 2A and 2B), which were confirmed on Ziehl-Neelsen staining (Figures 2C and 2D). The presence of AFB raised concern for a disseminated mycobacterial infection. The patient was admitted to our institution approximately 1 week after the outpatient biopsy was performed. He was evaluated by infectious diseases (B.H.) and was recommended for repeat biopsy with AFB culture and for initiation of intravenous antibiotics.

The patient was evaluated by the dermatology consultation service on hospital day 1. At the time of consultation, the lesions were still painless but had enlarged. Two new satellite lesions were noted on his other extremities. Due to the widespread distribution of the lesions, there was concern for disseminated disease. The relatively rapid onset of new lesions increased concern for infection with rapid-growing mycobacteria, including Mycobacterium abscessus, Mycobacterium fortuitum, and Mycobacterium chelonae. A detailed history revealed that the patient’s wife had a fish tank, which supported the inclusion of Mycobacterium marinum in the differential; however, further questioning revealed that the patient never came in contact with the aquarium water. The initial outpatient biopsy had not been sent for culture. Following inpatient biopsy, the patient was initiated on empiric antimycobacterials, including imipenem, amikacin, clarithromycin, and levofloxacin. Computed tomography of the head was negative for cerebral involvement.
Acid-fast bacilli blood cultures were drawn per the recommendation from infectious diseases in an attempt to confirm disseminated disease; however, blood cultures remained negative. Tissue biopsy from the right arm was sent for AFB staining and culture. Many AFB were identified on microscopy, and growth was observed in the mycobacterial growth indicator tube after 6 days of incubation. The DNA probe was negative for M tuberculosis complex or Mycobacterium avium complex.
The patient was discharged on hospital day 6 on empiric therapy for rapid-growing mycobacteria while cultures were pending. The empiric regimen included intravenous imipenem 1 g every 6 hours, intravenous amikacin 1 g once daily, clarithromycin 500 mg every 12 hours, and levofloxacin 750 mg once daily. All solid media cultures were negative at the time of discharge.
The biopsy specimen proved difficult to culture on solid media using traditional methods. Three weeks after the inpatient biopsy, the microbiology laboratory reported that growth was observed on solid media that was incubated at 30°C and supplemented with iron. These findings were not characteristic of a rapidly growing mycobacteria (eg, M fortuitum, M chelonae, M abscessus) or M marinum but raised concern for infectionwith M haemophilum. Antimycobacterial treatment was adjusted to amikacin, clarithromycin, levofloxacin, and rifabutin.
Six weeks after the inpatient skin biopsy, final speciation confirmed infection with M haemophilum. The isolate proved susceptible to amikacin (minimal inhibitory concentration [MIC], 16), clarithromycin (MIC, 0.12), linezolid (MIC, <1), moxifloxacin (MIC, 0.5), rifabutin (MIC, <0.25), and trimethoprim-sulfamethoxazole (MIC, 0.5/9.5). The isolate was resistant to ciprofloxacin (MIC, 4), ethambutol (MIC, >16), and rifampin (MIC, 2). Based on these findings, an infectious disease specialist modified the treatment regimen to azithromycin 600 mg once daily, moxifloxacin 400 mg once daily, and rifabutin 300 mg once daily. Azithromycin was substituted for clarithromycin in an attempt to minimize the gastrointestinal side effects of the antibiotics. The infectious disease specialist was concerned that the clarithromycin could exacerbate the patient’s chronic GVHD-associated diarrhea, which posed a challenge to the oncologist, who was attempting to manage the patient’s GVHD and minimize the use of additional prednisone. At the time of this change, the patient was doing well clinically and denied any active skin lesions.
Four months later, he developed new left-sided neck swelling. Computed tomography revealed nonspecific enhancement involving the skin and superficial subcutaneous tissues in the left anterior neck. He was referred to otolaryngology given concern for recurrent infection vs leukemia cutis. He underwent excisional biopsy. Pathology was negative for malignancy but demonstrated subcutaneous necrotizing granulomatous inflammation with a positive AFB stain. Tissue AFB cultures revealed moderate AFB on direct stain, but there was no AFB growth at 12 weeks. Clarithromycin was restarted in place of azithromycin to increase the potency of the antimycobacterial regimen. Cultures from this neck biopsy were negative after 12 weeks of incubation.
In addition to this change in antibiotic coverage, the patient’s medical oncologist tapered the patient’s immunosuppression considerably. The patient subsequently completed 12 months of therapy with clarithromycin, moxifloxacin, and rifabutin starting from the time of the neck biopsy. He remained free of recurrence of mycobacterial infection for nearly 2 years until he died from an unrelated illness.
Nontuberculous mycobacteria are an ubiquitous environmental group.2 Sources include soil and natural water (M avium), fish tanks and swimming pools (M marinum), and tap water and occasionally domestic animals (Mycobacterium kansasii). Additionally, rapidly growing NTM such as M abscessus, M chelonae, and M fortuitum have been isolated from soil and natural water supplies.3
Mycobacterium haemophilum is a fastidious organism with a predilection for skin of the chest and extremities. Iatrogenically or inherently immunocompromised patients are most commonly affected6-11; however, there also have been reports in healthy patients.12,13 Infections typically present as painless erythematous papules or nodules that eventually suppurate, ulcerate, and become painful. Presentations involving Fitz-Hugh–Curtis syndrome,13 new B-cell lymphoma,10 and lymphadenitis12 also have been described. Beyond cutaneous involvement, M haemophilum has been cultured from bone, the synovium, the lungs, and the central nervous system.4,9 The majority of morbidities occur in patients with lung involvement.4 Therefore, even patients presenting with isolated cutaneous disease require close follow-up.
Mycobacterium haemophilum is a slowly proliferating organism that is unable to grow in standard egg-potato (Lowenstein-Jensen) medium or agar base (Middlebrook 7H10 or 7H11 agar) without iron supplementation (ferric ammonium citrate, hemin, or hemoglobin). It also requires temperatures of 30°C to 32°C for growth. Its iron requisite is unique, but species such as M marinum and Mycobacterium ulcerans also share reduced temperature requirements. Without a high index of suspicion, growth often is absent because standard Mycobacterium culture techniques will not foster organism growth. Our case demonstrated that special culture instructions must be relayed to the laboratory, even in the face of positive AFB smears. Failure to request hemin and modified incubation temperatures may have contributed to the negative AFB blood culture in our patient.
Due to the relatively rare incidence of M haemophilum infection, there are no known randomized controlled trials guiding antibiotic regimens. Infectious disease specialists often treat empirically with triple-drug therapy derived from locally reported species susceptibilities. The largest case series to date did not identify resistance to amikacin, ciprofloxacin, or clarithromycin.4 Our case identified a novel finding of ciprofloxacin and rifampin resistance, which may highlight the emergence of a newly resistant strain of M haemophilum. Of note, one case of rifampin resistance has been reported, but the culture was drawn from a postmortem specimen in the setting of previously rifampin-sensitive isolates.4 Empiric therapies should be guided by hospital susceptibility reports and expert consultation.
Coinfection with 2 or more NTM—including M tuberculosis, M leprae, and M fortuitum—has been reported.8,14 Temporally distinct coinfections with M leprae and M haemophilum also have been described.15 Thus, practitioners should have a low threshold for repeat cultures in the context of new cutaneous nodules or granulomas, not only to detect concomitant infections but also to identify resistance patterns that might explain recurrent or recalcitrant disease. Immune reconstitution inflammatory syndrome also must be considered with new or worsening lesions, especially in the first months of therapy, as this is a common occurrence when immunosuppressive regimens are tapered to help manage infections.
In conclusion, M haemophilum is an underrecognized infection that presents as cutaneous nodules or lymphadenitis in immunocompromised or healthy individuals. Diagnosis requires a high index of suspicion because its unique growth requirements necessitate special laboratory techniques. Our case represents a classic presentation of this NTM infection in a patient with AML following allogenic stem cell transplantation. Repeat cultures, workup of potentially disseminated infections, and close follow-up are requisite to minimizing morbidity and mortality. A multidisciplinary approach involving infectious disease, medical oncology, radiology, and dermatology best manages this type of infection.
- Sheu LC, Tran TM, Jarlsberg LG, et al. Non-tuberculous mycobacterial infections at San Francisco General Hospital. Clin Respir J. 2015;9:436-442.
- Knoll BM. Update on nontuberculous mycobacterial infections in solid organ and hematopoietic stem cell transplant recipients. Curr Infect Dis Rep. 2014;16:421.
- Diagnosis and treatment of disease caused by nontuberculous mycobacteria. this official statement of the American Thoracic Society was approved by the Board of Directors, March 1997. Medical Section of the American Lung Association. Am J Respir Crit Care Med. 1997;156(2 pt 2):S1-S25.
- Shah MK, Sebti A, Kiehn TE, et al. Mycobacterium haemophilum in immunocompromised patients. Clin Infect Dis. 2001;33:330-337.
- Griffiths DE, Aksamit T, Brown-Elliott BA. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Copeland NK, Arora NS, Ferguson TM. Mycobacterium haemophilum masquerading as leprosy in a renal transplant patient [published online November 28, 2013]. Case Rep Dermatol Med. 2013;2013:793127.
- Aslam A, Green RL, Motta L, et al. Cutaneous Mycobacterium haemophilum infection in a patient receiving infliximab for psoriasis. Br J Dermatol. 2013;168:446-447.
- Agrawal S, Sharma A. Dual mycobacterial infection in the setting of leflunomide treatment for rheumatoid arthritis. Ann Rheum Dis. 2007;66:277.
- Buppajarntham A, Apisarnthanarak A, Rutjanawech S, et al. Central nervous system infection due to Mycobacterium haemophilum in a patient with acquired immunodeficiency syndrome. Int J STD AIDS. 2015;26:288-290.
- Doherty T, Lynn M, Cavazza A, et al. Mycobacterium haemophilum as the initial presentation of a B-cell lymphoma in a liver transplant patient [published online January 12, 2014]. Case Rep Rheumatol. 2014;2014:742978.
- Ducharlet K, Murphy C, Tan SJ, et al. Recurrent Mycobacterium haemophilum in a renal transplant recipient. Nephrology (Carlton). 2014;(19 suppl 1):14-17.
- Dawson DJ, Blacklock ZM, Kane DW. Mycobacterium haemophilum causing lymphadenitis in an otherwise healthy child. Med J Aust. 1981;2:289-290.
- Jang HY, Burbelo PD, Chae YS, et al. Nontuberculous mycobacterial infection in a clinical presentation of Fitz-Hugh-Curtis syndrome: a case report with multigene diagnostic approach. BMC Womens Health. 2014;14:95.
- Scollard DM, Stryjewska BM, Prestigiacomo JF, et al. Hansen’s disease (leprosy) complicated by secondary mycobacterial infection. J Am Acad Dermatol. 2011;64:593-596.
- SoRelle JA, Beal SG, Scollard DM, et al. Mycobacterium leprae and Mycobacterium haemophilum co-infection in an iatrogenically immunosuppressed patient. Diagn Microbiol Infect Dis. 2014;78:494-496.
To the Editor:
The increase in nontuberculous mycobacteria (NTM) infections over the last 3 decades likely is multifaceted, including increased clinical awareness, improved laboratory diagnostics, growing numbers of immunocompromised patients, and an aging population.1,2 Historically, the majority of mycobacteria-related diseases are due to Mycobacterium tuberculosis, Mycobacterium bovis, and Mycobacterium leprae.3
Mycobacterium haemophilum is a slow-growing acid-fast bacillus (AFB) that differs from other Mycobacterium species in that it requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. As these requirements for growth are not standard for AFB cultures, M haemophilum infection may be underrecognized and underreported.3Mycobacterium haemophilum infections largely are cutaneous and generally are seen in AIDS patients and bone marrow transplant recipients who are iatrogenically immunosuppressed.4,5 No species-specific treatment guidelines exist2; however, triple-drug therapy combining a macrolide, rifamycin, and a quinolone for a minimum of 12 months often is recommended.
A 64-year-old man with a history of coronary artery disease, hypertension, hyperlipidemia, and acute myelogenous leukemia (AML) underwent allogenic stem cell transplantation. His posttransplant course was complicated by multiple deep vein thromboses, hypogammaglobulinemia, and graft-vs-host disease (GVHD) of the skin and gastrointestinal tract that manifested as chronic diarrhea, which was managed with chronic prednisone. Thirteen months after the transplant, the patient presented to his outpatient oncologist (M.K.) for evaluation of painless, nonpruritic, erythematous papules and nodules that had emerged on the right side of the chest, right arm, and left leg of approximately 2 weeks’ duration.
On review of systems by oncology, the patient denied any fevers, chills, or night sweats but noted chronic loose nonbloody stools without abdominal pain, likely related to the GVHD. The patient’s medications included prednisone 20 mg once daily, fluconazole, amitriptyline, atovaquone, budesonide, dabigatran, metoprolol, pantoprazole, rosuvastatin, senna glycoside, spironolactone, tramadol, and valacyclovir.
Physical examination revealed multiple singular erythematous nodules on the right side of the chest (Figure 1A), right arm (Figure 1B), and left leg. There was no regional lymphadenopathy. The patient was afebrile and hemodynamically stable. A biopsy of the arm performed to rule out leukemia cutis revealed a granulomatous dermatitis with numerous AFB (Figures 2A and 2B), which were confirmed on Ziehl-Neelsen staining (Figures 2C and 2D). The presence of AFB raised concern for a disseminated mycobacterial infection. The patient was admitted to our institution approximately 1 week after the outpatient biopsy was performed. He was evaluated by infectious diseases (B.H.) and was recommended for repeat biopsy with AFB culture and for initiation of intravenous antibiotics.
The patient was evaluated by the dermatology consultation service on hospital day 1. At the time of consultation, the lesions were still painless but had enlarged. Two new satellite lesions were noted on his other extremities. Due to the widespread distribution of the lesions, there was concern for disseminated disease. The relatively rapid onset of new lesions increased concern for infection with rapid-growing mycobacteria, including Mycobacterium abscessus, Mycobacterium fortuitum, and Mycobacterium chelonae. A detailed history revealed that the patient’s wife had a fish tank, which supported the inclusion of Mycobacterium marinum in the differential; however, further questioning revealed that the patient never came in contact with the aquarium water. The initial outpatient biopsy had not been sent for culture. Following inpatient biopsy, the patient was initiated on empiric antimycobacterials, including imipenem, amikacin, clarithromycin, and levofloxacin. Computed tomography of the head was negative for cerebral involvement.
Acid-fast bacilli blood cultures were drawn per the recommendation from infectious diseases in an attempt to confirm disseminated disease; however, blood cultures remained negative. Tissue biopsy from the right arm was sent for AFB staining and culture. Many AFB were identified on microscopy, and growth was observed in the mycobacterial growth indicator tube after 6 days of incubation. The DNA probe was negative for M tuberculosis complex or Mycobacterium avium complex.
The patient was discharged on hospital day 6 on empiric therapy for rapid-growing mycobacteria while cultures were pending. The empiric regimen included intravenous imipenem 1 g every 6 hours, intravenous amikacin 1 g once daily, clarithromycin 500 mg every 12 hours, and levofloxacin 750 mg once daily. All solid media cultures were negative at the time of discharge.
The biopsy specimen proved difficult to culture on solid media using traditional methods. Three weeks after the inpatient biopsy, the microbiology laboratory reported that growth was observed on solid media that was incubated at 30°C and supplemented with iron. These findings were not characteristic of a rapidly growing mycobacteria (eg, M fortuitum, M chelonae, M abscessus) or M marinum but raised concern for infectionwith M haemophilum. Antimycobacterial treatment was adjusted to amikacin, clarithromycin, levofloxacin, and rifabutin.
Six weeks after the inpatient skin biopsy, final speciation confirmed infection with M haemophilum. The isolate proved susceptible to amikacin (minimal inhibitory concentration [MIC], 16), clarithromycin (MIC, 0.12), linezolid (MIC, <1), moxifloxacin (MIC, 0.5), rifabutin (MIC, <0.25), and trimethoprim-sulfamethoxazole (MIC, 0.5/9.5). The isolate was resistant to ciprofloxacin (MIC, 4), ethambutol (MIC, >16), and rifampin (MIC, 2). Based on these findings, an infectious disease specialist modified the treatment regimen to azithromycin 600 mg once daily, moxifloxacin 400 mg once daily, and rifabutin 300 mg once daily. Azithromycin was substituted for clarithromycin in an attempt to minimize the gastrointestinal side effects of the antibiotics. The infectious disease specialist was concerned that the clarithromycin could exacerbate the patient’s chronic GVHD-associated diarrhea, which posed a challenge to the oncologist, who was attempting to manage the patient’s GVHD and minimize the use of additional prednisone. At the time of this change, the patient was doing well clinically and denied any active skin lesions.
Four months later, he developed new left-sided neck swelling. Computed tomography revealed nonspecific enhancement involving the skin and superficial subcutaneous tissues in the left anterior neck. He was referred to otolaryngology given concern for recurrent infection vs leukemia cutis. He underwent excisional biopsy. Pathology was negative for malignancy but demonstrated subcutaneous necrotizing granulomatous inflammation with a positive AFB stain. Tissue AFB cultures revealed moderate AFB on direct stain, but there was no AFB growth at 12 weeks. Clarithromycin was restarted in place of azithromycin to increase the potency of the antimycobacterial regimen. Cultures from this neck biopsy were negative after 12 weeks of incubation.
In addition to this change in antibiotic coverage, the patient’s medical oncologist tapered the patient’s immunosuppression considerably. The patient subsequently completed 12 months of therapy with clarithromycin, moxifloxacin, and rifabutin starting from the time of the neck biopsy. He remained free of recurrence of mycobacterial infection for nearly 2 years until he died from an unrelated illness.
Nontuberculous mycobacteria are an ubiquitous environmental group.2 Sources include soil and natural water (M avium), fish tanks and swimming pools (M marinum), and tap water and occasionally domestic animals (Mycobacterium kansasii). Additionally, rapidly growing NTM such as M abscessus, M chelonae, and M fortuitum have been isolated from soil and natural water supplies.3
Mycobacterium haemophilum is a fastidious organism with a predilection for skin of the chest and extremities. Iatrogenically or inherently immunocompromised patients are most commonly affected6-11; however, there also have been reports in healthy patients.12,13 Infections typically present as painless erythematous papules or nodules that eventually suppurate, ulcerate, and become painful. Presentations involving Fitz-Hugh–Curtis syndrome,13 new B-cell lymphoma,10 and lymphadenitis12 also have been described. Beyond cutaneous involvement, M haemophilum has been cultured from bone, the synovium, the lungs, and the central nervous system.4,9 The majority of morbidities occur in patients with lung involvement.4 Therefore, even patients presenting with isolated cutaneous disease require close follow-up.
Mycobacterium haemophilum is a slowly proliferating organism that is unable to grow in standard egg-potato (Lowenstein-Jensen) medium or agar base (Middlebrook 7H10 or 7H11 agar) without iron supplementation (ferric ammonium citrate, hemin, or hemoglobin). It also requires temperatures of 30°C to 32°C for growth. Its iron requisite is unique, but species such as M marinum and Mycobacterium ulcerans also share reduced temperature requirements. Without a high index of suspicion, growth often is absent because standard Mycobacterium culture techniques will not foster organism growth. Our case demonstrated that special culture instructions must be relayed to the laboratory, even in the face of positive AFB smears. Failure to request hemin and modified incubation temperatures may have contributed to the negative AFB blood culture in our patient.
Due to the relatively rare incidence of M haemophilum infection, there are no known randomized controlled trials guiding antibiotic regimens. Infectious disease specialists often treat empirically with triple-drug therapy derived from locally reported species susceptibilities. The largest case series to date did not identify resistance to amikacin, ciprofloxacin, or clarithromycin.4 Our case identified a novel finding of ciprofloxacin and rifampin resistance, which may highlight the emergence of a newly resistant strain of M haemophilum. Of note, one case of rifampin resistance has been reported, but the culture was drawn from a postmortem specimen in the setting of previously rifampin-sensitive isolates.4 Empiric therapies should be guided by hospital susceptibility reports and expert consultation.
Coinfection with 2 or more NTM—including M tuberculosis, M leprae, and M fortuitum—has been reported.8,14 Temporally distinct coinfections with M leprae and M haemophilum also have been described.15 Thus, practitioners should have a low threshold for repeat cultures in the context of new cutaneous nodules or granulomas, not only to detect concomitant infections but also to identify resistance patterns that might explain recurrent or recalcitrant disease. Immune reconstitution inflammatory syndrome also must be considered with new or worsening lesions, especially in the first months of therapy, as this is a common occurrence when immunosuppressive regimens are tapered to help manage infections.
In conclusion, M haemophilum is an underrecognized infection that presents as cutaneous nodules or lymphadenitis in immunocompromised or healthy individuals. Diagnosis requires a high index of suspicion because its unique growth requirements necessitate special laboratory techniques. Our case represents a classic presentation of this NTM infection in a patient with AML following allogenic stem cell transplantation. Repeat cultures, workup of potentially disseminated infections, and close follow-up are requisite to minimizing morbidity and mortality. A multidisciplinary approach involving infectious disease, medical oncology, radiology, and dermatology best manages this type of infection.
To the Editor:
The increase in nontuberculous mycobacteria (NTM) infections over the last 3 decades likely is multifaceted, including increased clinical awareness, improved laboratory diagnostics, growing numbers of immunocompromised patients, and an aging population.1,2 Historically, the majority of mycobacteria-related diseases are due to Mycobacterium tuberculosis, Mycobacterium bovis, and Mycobacterium leprae.3
Mycobacterium haemophilum is a slow-growing acid-fast bacillus (AFB) that differs from other Mycobacterium species in that it requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. As these requirements for growth are not standard for AFB cultures, M haemophilum infection may be underrecognized and underreported.3Mycobacterium haemophilum infections largely are cutaneous and generally are seen in AIDS patients and bone marrow transplant recipients who are iatrogenically immunosuppressed.4,5 No species-specific treatment guidelines exist2; however, triple-drug therapy combining a macrolide, rifamycin, and a quinolone for a minimum of 12 months often is recommended.
A 64-year-old man with a history of coronary artery disease, hypertension, hyperlipidemia, and acute myelogenous leukemia (AML) underwent allogenic stem cell transplantation. His posttransplant course was complicated by multiple deep vein thromboses, hypogammaglobulinemia, and graft-vs-host disease (GVHD) of the skin and gastrointestinal tract that manifested as chronic diarrhea, which was managed with chronic prednisone. Thirteen months after the transplant, the patient presented to his outpatient oncologist (M.K.) for evaluation of painless, nonpruritic, erythematous papules and nodules that had emerged on the right side of the chest, right arm, and left leg of approximately 2 weeks’ duration.
On review of systems by oncology, the patient denied any fevers, chills, or night sweats but noted chronic loose nonbloody stools without abdominal pain, likely related to the GVHD. The patient’s medications included prednisone 20 mg once daily, fluconazole, amitriptyline, atovaquone, budesonide, dabigatran, metoprolol, pantoprazole, rosuvastatin, senna glycoside, spironolactone, tramadol, and valacyclovir.
Physical examination revealed multiple singular erythematous nodules on the right side of the chest (Figure 1A), right arm (Figure 1B), and left leg. There was no regional lymphadenopathy. The patient was afebrile and hemodynamically stable. A biopsy of the arm performed to rule out leukemia cutis revealed a granulomatous dermatitis with numerous AFB (Figures 2A and 2B), which were confirmed on Ziehl-Neelsen staining (Figures 2C and 2D). The presence of AFB raised concern for a disseminated mycobacterial infection. The patient was admitted to our institution approximately 1 week after the outpatient biopsy was performed. He was evaluated by infectious diseases (B.H.) and was recommended for repeat biopsy with AFB culture and for initiation of intravenous antibiotics.
The patient was evaluated by the dermatology consultation service on hospital day 1. At the time of consultation, the lesions were still painless but had enlarged. Two new satellite lesions were noted on his other extremities. Due to the widespread distribution of the lesions, there was concern for disseminated disease. The relatively rapid onset of new lesions increased concern for infection with rapid-growing mycobacteria, including Mycobacterium abscessus, Mycobacterium fortuitum, and Mycobacterium chelonae. A detailed history revealed that the patient’s wife had a fish tank, which supported the inclusion of Mycobacterium marinum in the differential; however, further questioning revealed that the patient never came in contact with the aquarium water. The initial outpatient biopsy had not been sent for culture. Following inpatient biopsy, the patient was initiated on empiric antimycobacterials, including imipenem, amikacin, clarithromycin, and levofloxacin. Computed tomography of the head was negative for cerebral involvement.
Acid-fast bacilli blood cultures were drawn per the recommendation from infectious diseases in an attempt to confirm disseminated disease; however, blood cultures remained negative. Tissue biopsy from the right arm was sent for AFB staining and culture. Many AFB were identified on microscopy, and growth was observed in the mycobacterial growth indicator tube after 6 days of incubation. The DNA probe was negative for M tuberculosis complex or Mycobacterium avium complex.
The patient was discharged on hospital day 6 on empiric therapy for rapid-growing mycobacteria while cultures were pending. The empiric regimen included intravenous imipenem 1 g every 6 hours, intravenous amikacin 1 g once daily, clarithromycin 500 mg every 12 hours, and levofloxacin 750 mg once daily. All solid media cultures were negative at the time of discharge.
The biopsy specimen proved difficult to culture on solid media using traditional methods. Three weeks after the inpatient biopsy, the microbiology laboratory reported that growth was observed on solid media that was incubated at 30°C and supplemented with iron. These findings were not characteristic of a rapidly growing mycobacteria (eg, M fortuitum, M chelonae, M abscessus) or M marinum but raised concern for infectionwith M haemophilum. Antimycobacterial treatment was adjusted to amikacin, clarithromycin, levofloxacin, and rifabutin.
Six weeks after the inpatient skin biopsy, final speciation confirmed infection with M haemophilum. The isolate proved susceptible to amikacin (minimal inhibitory concentration [MIC], 16), clarithromycin (MIC, 0.12), linezolid (MIC, <1), moxifloxacin (MIC, 0.5), rifabutin (MIC, <0.25), and trimethoprim-sulfamethoxazole (MIC, 0.5/9.5). The isolate was resistant to ciprofloxacin (MIC, 4), ethambutol (MIC, >16), and rifampin (MIC, 2). Based on these findings, an infectious disease specialist modified the treatment regimen to azithromycin 600 mg once daily, moxifloxacin 400 mg once daily, and rifabutin 300 mg once daily. Azithromycin was substituted for clarithromycin in an attempt to minimize the gastrointestinal side effects of the antibiotics. The infectious disease specialist was concerned that the clarithromycin could exacerbate the patient’s chronic GVHD-associated diarrhea, which posed a challenge to the oncologist, who was attempting to manage the patient’s GVHD and minimize the use of additional prednisone. At the time of this change, the patient was doing well clinically and denied any active skin lesions.
Four months later, he developed new left-sided neck swelling. Computed tomography revealed nonspecific enhancement involving the skin and superficial subcutaneous tissues in the left anterior neck. He was referred to otolaryngology given concern for recurrent infection vs leukemia cutis. He underwent excisional biopsy. Pathology was negative for malignancy but demonstrated subcutaneous necrotizing granulomatous inflammation with a positive AFB stain. Tissue AFB cultures revealed moderate AFB on direct stain, but there was no AFB growth at 12 weeks. Clarithromycin was restarted in place of azithromycin to increase the potency of the antimycobacterial regimen. Cultures from this neck biopsy were negative after 12 weeks of incubation.
In addition to this change in antibiotic coverage, the patient’s medical oncologist tapered the patient’s immunosuppression considerably. The patient subsequently completed 12 months of therapy with clarithromycin, moxifloxacin, and rifabutin starting from the time of the neck biopsy. He remained free of recurrence of mycobacterial infection for nearly 2 years until he died from an unrelated illness.
Nontuberculous mycobacteria are an ubiquitous environmental group.2 Sources include soil and natural water (M avium), fish tanks and swimming pools (M marinum), and tap water and occasionally domestic animals (Mycobacterium kansasii). Additionally, rapidly growing NTM such as M abscessus, M chelonae, and M fortuitum have been isolated from soil and natural water supplies.3
Mycobacterium haemophilum is a fastidious organism with a predilection for skin of the chest and extremities. Iatrogenically or inherently immunocompromised patients are most commonly affected6-11; however, there also have been reports in healthy patients.12,13 Infections typically present as painless erythematous papules or nodules that eventually suppurate, ulcerate, and become painful. Presentations involving Fitz-Hugh–Curtis syndrome,13 new B-cell lymphoma,10 and lymphadenitis12 also have been described. Beyond cutaneous involvement, M haemophilum has been cultured from bone, the synovium, the lungs, and the central nervous system.4,9 The majority of morbidities occur in patients with lung involvement.4 Therefore, even patients presenting with isolated cutaneous disease require close follow-up.
Mycobacterium haemophilum is a slowly proliferating organism that is unable to grow in standard egg-potato (Lowenstein-Jensen) medium or agar base (Middlebrook 7H10 or 7H11 agar) without iron supplementation (ferric ammonium citrate, hemin, or hemoglobin). It also requires temperatures of 30°C to 32°C for growth. Its iron requisite is unique, but species such as M marinum and Mycobacterium ulcerans also share reduced temperature requirements. Without a high index of suspicion, growth often is absent because standard Mycobacterium culture techniques will not foster organism growth. Our case demonstrated that special culture instructions must be relayed to the laboratory, even in the face of positive AFB smears. Failure to request hemin and modified incubation temperatures may have contributed to the negative AFB blood culture in our patient.
Due to the relatively rare incidence of M haemophilum infection, there are no known randomized controlled trials guiding antibiotic regimens. Infectious disease specialists often treat empirically with triple-drug therapy derived from locally reported species susceptibilities. The largest case series to date did not identify resistance to amikacin, ciprofloxacin, or clarithromycin.4 Our case identified a novel finding of ciprofloxacin and rifampin resistance, which may highlight the emergence of a newly resistant strain of M haemophilum. Of note, one case of rifampin resistance has been reported, but the culture was drawn from a postmortem specimen in the setting of previously rifampin-sensitive isolates.4 Empiric therapies should be guided by hospital susceptibility reports and expert consultation.
Coinfection with 2 or more NTM—including M tuberculosis, M leprae, and M fortuitum—has been reported.8,14 Temporally distinct coinfections with M leprae and M haemophilum also have been described.15 Thus, practitioners should have a low threshold for repeat cultures in the context of new cutaneous nodules or granulomas, not only to detect concomitant infections but also to identify resistance patterns that might explain recurrent or recalcitrant disease. Immune reconstitution inflammatory syndrome also must be considered with new or worsening lesions, especially in the first months of therapy, as this is a common occurrence when immunosuppressive regimens are tapered to help manage infections.
In conclusion, M haemophilum is an underrecognized infection that presents as cutaneous nodules or lymphadenitis in immunocompromised or healthy individuals. Diagnosis requires a high index of suspicion because its unique growth requirements necessitate special laboratory techniques. Our case represents a classic presentation of this NTM infection in a patient with AML following allogenic stem cell transplantation. Repeat cultures, workup of potentially disseminated infections, and close follow-up are requisite to minimizing morbidity and mortality. A multidisciplinary approach involving infectious disease, medical oncology, radiology, and dermatology best manages this type of infection.
- Sheu LC, Tran TM, Jarlsberg LG, et al. Non-tuberculous mycobacterial infections at San Francisco General Hospital. Clin Respir J. 2015;9:436-442.
- Knoll BM. Update on nontuberculous mycobacterial infections in solid organ and hematopoietic stem cell transplant recipients. Curr Infect Dis Rep. 2014;16:421.
- Diagnosis and treatment of disease caused by nontuberculous mycobacteria. this official statement of the American Thoracic Society was approved by the Board of Directors, March 1997. Medical Section of the American Lung Association. Am J Respir Crit Care Med. 1997;156(2 pt 2):S1-S25.
- Shah MK, Sebti A, Kiehn TE, et al. Mycobacterium haemophilum in immunocompromised patients. Clin Infect Dis. 2001;33:330-337.
- Griffiths DE, Aksamit T, Brown-Elliott BA. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Copeland NK, Arora NS, Ferguson TM. Mycobacterium haemophilum masquerading as leprosy in a renal transplant patient [published online November 28, 2013]. Case Rep Dermatol Med. 2013;2013:793127.
- Aslam A, Green RL, Motta L, et al. Cutaneous Mycobacterium haemophilum infection in a patient receiving infliximab for psoriasis. Br J Dermatol. 2013;168:446-447.
- Agrawal S, Sharma A. Dual mycobacterial infection in the setting of leflunomide treatment for rheumatoid arthritis. Ann Rheum Dis. 2007;66:277.
- Buppajarntham A, Apisarnthanarak A, Rutjanawech S, et al. Central nervous system infection due to Mycobacterium haemophilum in a patient with acquired immunodeficiency syndrome. Int J STD AIDS. 2015;26:288-290.
- Doherty T, Lynn M, Cavazza A, et al. Mycobacterium haemophilum as the initial presentation of a B-cell lymphoma in a liver transplant patient [published online January 12, 2014]. Case Rep Rheumatol. 2014;2014:742978.
- Ducharlet K, Murphy C, Tan SJ, et al. Recurrent Mycobacterium haemophilum in a renal transplant recipient. Nephrology (Carlton). 2014;(19 suppl 1):14-17.
- Dawson DJ, Blacklock ZM, Kane DW. Mycobacterium haemophilum causing lymphadenitis in an otherwise healthy child. Med J Aust. 1981;2:289-290.
- Jang HY, Burbelo PD, Chae YS, et al. Nontuberculous mycobacterial infection in a clinical presentation of Fitz-Hugh-Curtis syndrome: a case report with multigene diagnostic approach. BMC Womens Health. 2014;14:95.
- Scollard DM, Stryjewska BM, Prestigiacomo JF, et al. Hansen’s disease (leprosy) complicated by secondary mycobacterial infection. J Am Acad Dermatol. 2011;64:593-596.
- SoRelle JA, Beal SG, Scollard DM, et al. Mycobacterium leprae and Mycobacterium haemophilum co-infection in an iatrogenically immunosuppressed patient. Diagn Microbiol Infect Dis. 2014;78:494-496.
- Sheu LC, Tran TM, Jarlsberg LG, et al. Non-tuberculous mycobacterial infections at San Francisco General Hospital. Clin Respir J. 2015;9:436-442.
- Knoll BM. Update on nontuberculous mycobacterial infections in solid organ and hematopoietic stem cell transplant recipients. Curr Infect Dis Rep. 2014;16:421.
- Diagnosis and treatment of disease caused by nontuberculous mycobacteria. this official statement of the American Thoracic Society was approved by the Board of Directors, March 1997. Medical Section of the American Lung Association. Am J Respir Crit Care Med. 1997;156(2 pt 2):S1-S25.
- Shah MK, Sebti A, Kiehn TE, et al. Mycobacterium haemophilum in immunocompromised patients. Clin Infect Dis. 2001;33:330-337.
- Griffiths DE, Aksamit T, Brown-Elliott BA. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Copeland NK, Arora NS, Ferguson TM. Mycobacterium haemophilum masquerading as leprosy in a renal transplant patient [published online November 28, 2013]. Case Rep Dermatol Med. 2013;2013:793127.
- Aslam A, Green RL, Motta L, et al. Cutaneous Mycobacterium haemophilum infection in a patient receiving infliximab for psoriasis. Br J Dermatol. 2013;168:446-447.
- Agrawal S, Sharma A. Dual mycobacterial infection in the setting of leflunomide treatment for rheumatoid arthritis. Ann Rheum Dis. 2007;66:277.
- Buppajarntham A, Apisarnthanarak A, Rutjanawech S, et al. Central nervous system infection due to Mycobacterium haemophilum in a patient with acquired immunodeficiency syndrome. Int J STD AIDS. 2015;26:288-290.
- Doherty T, Lynn M, Cavazza A, et al. Mycobacterium haemophilum as the initial presentation of a B-cell lymphoma in a liver transplant patient [published online January 12, 2014]. Case Rep Rheumatol. 2014;2014:742978.
- Ducharlet K, Murphy C, Tan SJ, et al. Recurrent Mycobacterium haemophilum in a renal transplant recipient. Nephrology (Carlton). 2014;(19 suppl 1):14-17.
- Dawson DJ, Blacklock ZM, Kane DW. Mycobacterium haemophilum causing lymphadenitis in an otherwise healthy child. Med J Aust. 1981;2:289-290.
- Jang HY, Burbelo PD, Chae YS, et al. Nontuberculous mycobacterial infection in a clinical presentation of Fitz-Hugh-Curtis syndrome: a case report with multigene diagnostic approach. BMC Womens Health. 2014;14:95.
- Scollard DM, Stryjewska BM, Prestigiacomo JF, et al. Hansen’s disease (leprosy) complicated by secondary mycobacterial infection. J Am Acad Dermatol. 2011;64:593-596.
- SoRelle JA, Beal SG, Scollard DM, et al. Mycobacterium leprae and Mycobacterium haemophilum co-infection in an iatrogenically immunosuppressed patient. Diagn Microbiol Infect Dis. 2014;78:494-496.
Practice Points
- Mycobacterium haemophilum is a slow-growing acid-fast bacillus that requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. Because these requirements for growth are not standard for acid-fast bacteria cultures, M haemophilum infection may be underrecognized and underreported.
- There are no species-specific treatment guidelines, but extended course of treatment with multiple active antibacterials typically is recommended.
Facial Involvement in Progressive Macular Hypomelanosis
Progressive macular hypomelanosis (PMH) is a noninflammatory skin disorder characterized by ill-defined, nummular, hypopigmented, and nonscaly macules. Historically, various names have been used to describe this entity. Several of these terms, including cutis trunci variata and nummular and confluent hypomelanosis of the trunk, reflected its predominantly truncal distribution.1,2 Less frequently, involvement on the neck, buttocks, and arms and legs has been noted.1,2 A lack of facial involvement previously has been highlighted as a key clinical feature of PMH.3
Progressive macular hypomelanosis is a diagnosis of exclusion. Hypopigmented diseases commonly considered in the differential include those caused by fungi and yeasts (eg, tinea versicolor, seborrheic dermatitis), inflammatory skin disorders (eg, pityriasis alba, postinflammatory dyschromia), and mycosis fungoides (MF) as well as leprosy.
The hypopigmented macules of PMH have nonspecific histopathologic findings; lesional skin often shows minimal alterations as compared to normal skin. A sparse perivascular lymphocytic infiltrate often is observed,4,5 and at times, a decrease in epidermal melanin content can be detected.1-3,6,7
We report 4 cases with considerable facial involvement of hypopigmented macules that were determined to be consistent with PMH. We propose that characteristic macules that are not clinically or histopathologically consistent with other disease entities are compatible with a diagnosis of PMH, regardless of the distribution. A diagnosis of PMH should be considered in the differential when there are suggestive facial lesions in addition to truncal lesions.
Case Reports
Patient 1
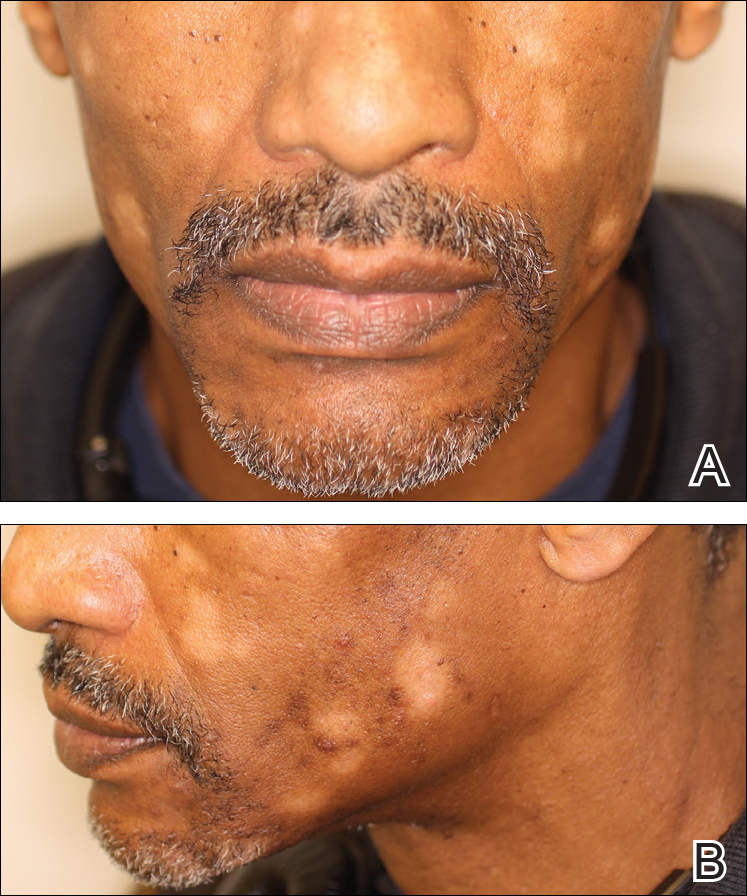
A 40-year-old man presented with hypopigmented macules on the face (Figure 1), trunk, chest, arms, and legs of 2 years’ duration. The lesions were asymptomatic and had started on the forehead as hypopigmented macules, then progressed to the trunk, arms, and legs. The patient denied any prior rash, injury, or hyperpigmentation associated with the distribution of the lesions.
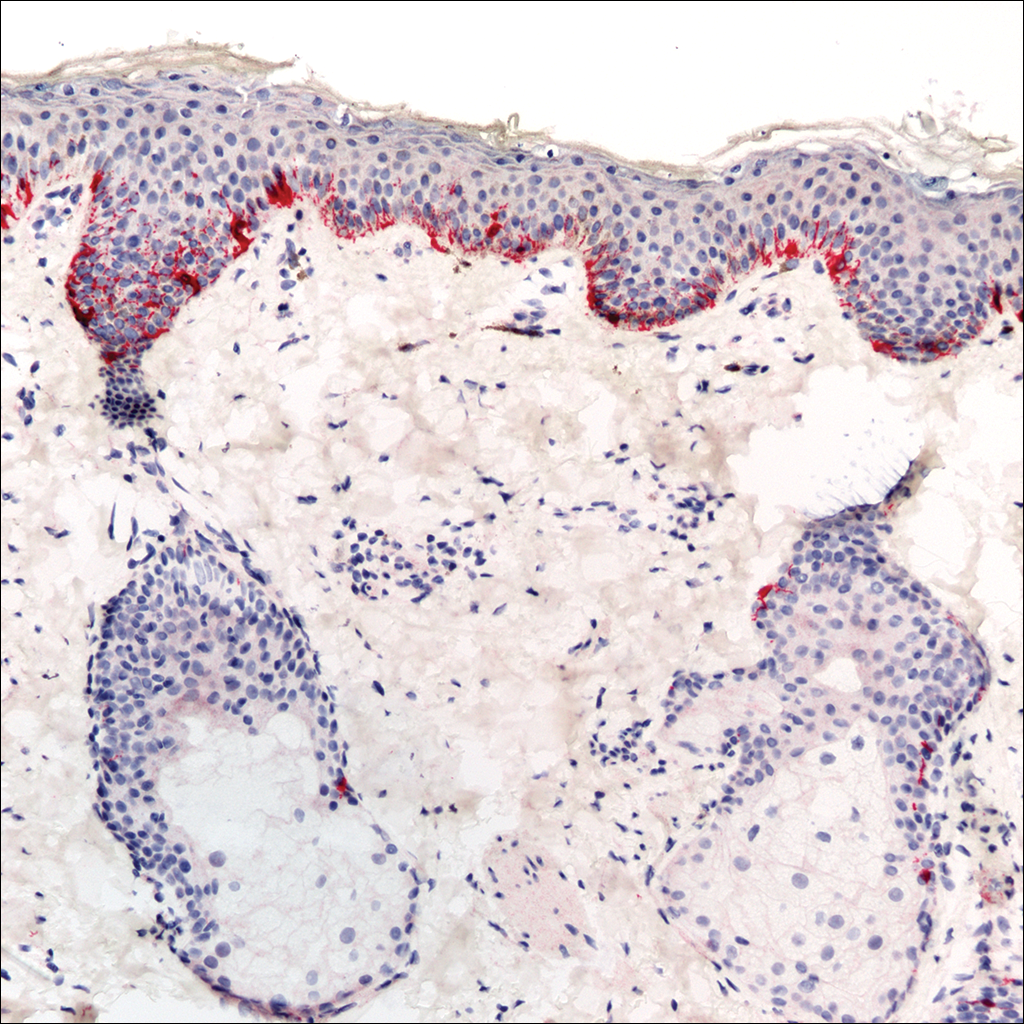
A rapid plasma reagin (RPR) test was conducted to rule out secondary syphilis and was nonreactive. During a series of clinical encounters over several months, a total of 5 biopsies of lesions on the face and back were performed. All specimens contained mild mononuclear perivascular inflammation (Figure 2). In some foci, staining for Melan-A revealed a decrease in epidermal melanocytes (Figure 3). Periodic acid–Schiff staining performed on one section revealed a few pityriasis spores but no hyphal elements, suggesting colonization rather than infection.

The patient initially was started on tacrolimus ointment 0.1% once daily and narrowband UVB phototherapy twice weekly for 3 months without benefit. A diagnosis of tinea versicolor was revisited and the patient was switched to ketoconazole shampoo 1% two to 3 times weekly on the face, trunk, arms, and legs for 10 to 15 minutes prior to rinsing, and ketoconazole cream 2% was applied twice daily to the affected areas for 2 months without notable improvement. Once-weekly 150-mg pulse doses of oral fluconazole for 8 weeks were started but proved equally ineffective. Antibiotic therapy aimed at eradicating Propionibacterium acnes was considered following a provisional diagnosis of PMH after the patient failed 5 months of therapy for tinea versicolor.
Patient 2
A 54-year-old man presented with hypopigmented to depigmented nonscaly macules on the face, trunk, chest, and arms of several months’ duration. The patient initially noted hypopigmentation on the face that gradually spread to the rest of the body. The patient denied any prior rash or hyperpigmentation in the affected areas. At the initial visit to our clinic, a potassium hydroxide (KOH) preparation of the face and back was positive for tinea versicolor. The patient was treated with ketoconazole shampoo 1% two to 3 times weekly for several weeks on the scalp, face, trunk, arms, and legs for 10 to 15 minutes prior to rinsing and 2 total doses of oral fluconazole 150 mg taken 1 week apart.
Three months later the patient returned with no improvement of the existing lesions and with progression of the disease to previously uninvolved areas of the trunk, arms, and legs. Biopsy of a facial lesion was performed, and laboratory studies including RPR, thyroid-stimulating hormone, and antinuclear antibody tests were conducted to screen for possible systemic disease. Microscopic analysis of the biopsied facial lesion revealed a sparse perivascular infiltrate of lymphocytes and plasma cells but no evidence of yeast or hyphal elements. Melan-A staining did not reveal a decreased number of epidermal melanocytes. All laboratory studies were negative or within normal limits. Desonide ointment 0.05% was prescribed to relieve the patient’s occasional pruritus. Although the patient’s symptoms resolved, the hypopigmented macules continued to progress, making a diagnosis of PMH more likely given the lack of improvement on treatment for tinea versicolor. Pimecrolimus cream 1% was started with discontinuation of desonide for steroid-sparing therapy.
Patient 3
A 63-year-old man presented with progressive nonscaly and asymptomatic hypopigmented macules on the face, trunk, abdomen, and back of 5 years’ duration. He first noted lesions on the abdomen and they subsequently spread to the rest of the body. The patient denied any prior rash, hyperpigmentation, or other lesions in the involved areas.
One year prior to the current presentation, KOH scrapings from the lesions performed by an outside physician were negative. During his initial visit to our clinic, an abdominal biopsy was performed, and histopathologic analysis showed postinflammatory pigmentary alteration; however, the patient denied any prior history of rash or injury in the distribution of the lesions that would correlate with the histopathologic findings of postinflammatory pigmentation. Because the histopathologic findings showed postinflammatory pigmentary alteration, additional stains including Melan-A were not performed.
The patient was provisionally treated with ketoconazole shampoo 1% two to 3 times weekly on the face, trunk, arms, and legs for 10 to 15 minutes prior to rinsing and ketoconazole cream 2% twice daily to the affected areas. After several months on this regimen, the patient did not report any improvement. An abdominal skin biopsy was again performed and revealed similar histopathology. Periodic acid–Schiff staining was negative for fungus. A diagnosis of PMH was made, and the patient was started on benzoyl peroxide wash 5% and clindamycin lotion.
Patient 4
A 45-year-old woman presented with hypopigmented, nonscaly macules on the face, neck, chest, trunk, and back. She first noted the lesions on the face and trunk more than 8 years prior, and they subsequently progressed. Potassium hydroxide scrapings performed on the lesions at the current presentation were negative, and a skin biopsy from the neck revealed postinflammatory pigmentary alteration, although the patient had no history of rash or injury in the areas in which the lesions were distributed.
Fontana-Masson and Melan-A staining of the skin biopsy of the neck revealed a normal distribution of melanocytes and pigment at the dermoepidermal junction. An RPR test was nonreactive. A diagnosis of PMH was made, and the patient was started on benzoyl peroxide wash 5% and clindamycin phosphate lotion 1%.
Comment
The 4 cases of PMH reported here showed extensive facial involvement in addition to the characteristic hypopigmented lesions on the trunk, arms, and legs. It is unclear why the lesions in these patients had a predominantly facial distribution. Involvement of the face in PMH has not been commonly reported in the literature. Martínez-Martínez et al3 reported 12 PMH patients with lesions only presenting in lumbar and abdominal distributions. Kim et al8 presented a series of 23 PMH patients treated with narrowband UVB in whom 56% (9/16) saw repigmentation in 90% of the lesions following treatment. The most commonly affected area was the lower back, followed by the abdomen, upper back, chest, sacral region, flank, and shoulders, respectively.8 In a review by Relyveld et al,1 PMH is described as a predominantly truncal disease that can occasionally extend to the neck, face, and proximal arms and legs; however, no specific cases were reported.
Previous case series have reported PMH primarily in adolescents and young adults, with mean ages ranging from 26 to 30 years.1,3 The 4 patients reported here were older, ranging in age from 40 to 65 years. This discrepancy in age may contribute to the facial distribution encountered in this patient population; however, given the small number of patients in our case series, such extrapolation is premature. Most recently, Westerhof et al6 demonstrated a relationship between the presence of P acnes, a common skin commensal of the face, and the hypopigmented macules of PMH. The investigators suggested that some strains of P acnes produce a factor that is yet to be identified that interferes with melanogenesis. The response of PMH lesions to topical treatments such as benzoyl peroxide, clindamycin, and phototherapy has lent credence to the potential etiologic role of P acnes in this condition.9,10 The interplay between age, PMH distribution, and P acnes requires further investigation.
The biopsies in our 4 patients were consistent with the nonspecific histopathologic characteristics of PMH lesions. Biopsies in all 4 patients revealed a sparse perivascular lymphocytic infiltrate, and in 2 of the cases, postinflammatory pigmentary alteration was noted. Such changes often are described in PMH lesions.4,5 In other cases detailed in the literature, lesional and nonlesional skin often are indistinguishable on hematoxylin and eosin staining.11 In the 3 patients for whom we performed additional immunohistochemical studies, results were mixed: Melan-A staining revealed a decreased number of melanocytes in Patient 1 but not in Patients 2 or 4. Many reported cases in the literature have not demonstrated a decrease in melanocyte density but instead show a decrease in melanin content in lesional skin.1-3,6,7 Although additional stains performed in Patient 4 revealed neither a decrease in the number of melanocytes nor a decrease in the melanin content, such histopathologic findings of PMH often are subtle. Additional stains were not performed in Patient 3. More studies are needed to characterize the immunohistochemical staining patterns of lesional skin in patients with PMH.
Tinea versicolor, pityriasis alba, mycosis fungoides, sarcoidosis, leprosy, and syphilis typically are included in the differential diagnosis for PMH. Tinea versicolor traditionally is diagnosed based on the combination of irregular hypopigmented or hyperpigmented scaly macules and a KOH preparation that is positive for hyphae and spores. Similar to PMH, tinea versicolor is most often found on the trunk, but unusual cases have been reported involving the face.12
Patient 2 reflected how it can be difficult diagnostically to distinguish between tinea versicolor and PMH. Although this patient initially had a KOH scraping suggestive for tinea versicolor, adequate treatment with oral fluconazole and ketoconazole shampoo did not result in improvement. The hypopigmented lesions in this patient continued to progress despite therapy. Additionally, his hypopigmented to depigmented nonscaly macules were more clinically consistent with the characteristic description of lesion configuration in PMH than with the irregular, more sharply defined, asymmetric, and scaly spots of tinea versicolor. Furthermore, the inflammatory findings on biopsy favored a diagnosis of PMH.
Pityriasis alba, most frequently presents on the face in the form of hypopigmented, sometimes slightly scaly macules but also can occur on the body. It usually occurs in younger patients who often have an atopic diathesis. Histologic findings generally are nonspecific, but discrete eczematous changes can sometimes be appreciated in the epidermis and dermis. None of our patients had histories suggestive of an atopic diathesis or lesion distributions typical of pityriasis alba. Histologic findings also were more consistent with PMH than pityriasis alba.
A diagnosis of patch-stage hypopigmented MF should also be entertained in patients with hypopigmented macules, as it can appear similar to the lesions of PMH. Hypopigmented MF often is associated with subtle atrophy, scaling, poikiloderma, and erythema. These features were not present in the 4 cases presented here. Histologically, atypical lymphocytes with prominent epidermotropism and tagging of the epidermis by large lymphocytic infiltrates are seen in cases of hypopigmented MF. These findings were not present in biopsies from our patients.
Hypopigmented sarcoidosis, leprosy, and syphilis are other systemic diseases associated with hypopigmented lesions. Histologically, noncaseasting granulomas in the dermis or subcutaneous tissue would favor a diagnosis of sarcoidosis over PMH. In patients who live in endemic areas, a diagnosis of leprosy for an anesthetic hypopigmented lesion would be higher in the differential. Finally, it is important to rule out secondary syphilis when diagnosing PMH. Known as the great imitator, secondary syphilis may present in a patient in the form of hypopigmented macules. Patients 1, 2, and 4 had nonreactive RPR tests; unfortunately, RPR was not checked in Patient 3. He denied all risk factors for syphilis.
Various topical and oral treatments were prescribed for each patient, but so far none have been unequivocally effective. In the literature, there are reports supporting the efficacy of topical antimicrobial agents targeting P acnes.9,10 One case report noted improvement in a patient with PMH after isotretinoin use.13 Phototherapy also has been reported to improve PMH in several case reports4-8; however, consistent response to these therapies has not been documented. Unfortunately for patients with a diagnosis of PMH, a lack of effective treatment options often exists.
This series of 4 cases highlights the importance of considering PMH in the differential of hypopigmented macules, even when they appear predominantly on the face.
- Relyveld G, Menke H, Westerhof W. Progressive macular hypomelanosis: an overview. Am J Clin Dermatol. 2007;8:13-19.
- Hwang SW, Hong SK, Kim SH, et al. Progressive macular hypomelanosis in Korean patients: a clinicopathologic study. Ann Dermatol. 2009;21:261-267.
- Martinéz-Martinéz ML, Azaña-Defez JM, Rodríguez-Vázquez M, et al. Progressive macular hypomelanosis. Pediatr Dermatol. 2012;29:460-462.
- Montero LC, Belinchonón I, Toledo F, et al. Progressive macular hypomelanosis, excellent response with narrow-band ultraviolet B phototherapy. Photodermatol Photoimmunol Photomed. 2011;27:162-163.
- Choi YJ, Hann SK. Two cases of progressive macular hypomelanosis of the trunk. Korean J Dermatol. 2000;38:655-658.
- Westerhof W, Rlyveld G, Kingswijk M, et al. Propionibacterium acnes and the pathogenesis of progressive macular hypomelanosis. Arch Dermatol. 2004;140:210-214.
- Wu SG, Xu AE, Song XZ, et al. Clinical, pathologic, and ultrastructural studies of progressive macular hypomelanosis. Int J Dermatol. 2010;29:1127-1132.
- Kim MB, Kim GW, Cho HH, et al. Narrowband UVB treatment of progressive macular hypomelanosis. J Am Acad Dermatol. 2012;66:598-605.
- Revlyveld GN, Menkie HE, Westerhof W. Benzoyl peroxide/clindamycin/UVA is more effective than fluticasone/UVA in progressive macular hypomelanosis: a randomized study. Am J Clin Dermatol. 2006;55:836-843.
- Santos JB, Almeida OL, Silva LM, et al. Efficacy of topical combination of benzoyl peroxide 5% and clindamcyin 1% for the treatment of progressive macular hypomelanosis: a randomized, doubleblind, placebo-controlled trial [in Portuguese]. An Bras Dermatol. 2011;86:50-54.
- Kumarasinghe SP, Tan SH, Thng S, et al. Progressive macular hypomelanosis in Singapore: a clinico-pathological study. Int J Dermatol. 2006;45:737-742.
- Terragni L, Lasagni A, Oriani A. Pityriasis versicolor of the face. Mycoses. 1991;34:345-347.
- Kim YK, Lee DY, Lee, JY, et al. Progressive macular hypomelanosis showing excellent response to oral isotretinoin [published online June 23, 2012]. J Dermatol. 2012;39:937-938.
Progressive macular hypomelanosis (PMH) is a noninflammatory skin disorder characterized by ill-defined, nummular, hypopigmented, and nonscaly macules. Historically, various names have been used to describe this entity. Several of these terms, including cutis trunci variata and nummular and confluent hypomelanosis of the trunk, reflected its predominantly truncal distribution.1,2 Less frequently, involvement on the neck, buttocks, and arms and legs has been noted.1,2 A lack of facial involvement previously has been highlighted as a key clinical feature of PMH.3
Progressive macular hypomelanosis is a diagnosis of exclusion. Hypopigmented diseases commonly considered in the differential include those caused by fungi and yeasts (eg, tinea versicolor, seborrheic dermatitis), inflammatory skin disorders (eg, pityriasis alba, postinflammatory dyschromia), and mycosis fungoides (MF) as well as leprosy.
The hypopigmented macules of PMH have nonspecific histopathologic findings; lesional skin often shows minimal alterations as compared to normal skin. A sparse perivascular lymphocytic infiltrate often is observed,4,5 and at times, a decrease in epidermal melanin content can be detected.1-3,6,7
We report 4 cases with considerable facial involvement of hypopigmented macules that were determined to be consistent with PMH. We propose that characteristic macules that are not clinically or histopathologically consistent with other disease entities are compatible with a diagnosis of PMH, regardless of the distribution. A diagnosis of PMH should be considered in the differential when there are suggestive facial lesions in addition to truncal lesions.
Case Reports
Patient 1
A 40-year-old man presented with hypopigmented macules on the face (Figure 1), trunk, chest, arms, and legs of 2 years’ duration. The lesions were asymptomatic and had started on the forehead as hypopigmented macules, then progressed to the trunk, arms, and legs. The patient denied any prior rash, injury, or hyperpigmentation associated with the distribution of the lesions.
A rapid plasma reagin (RPR) test was conducted to rule out secondary syphilis and was nonreactive. During a series of clinical encounters over several months, a total of 5 biopsies of lesions on the face and back were performed. All specimens contained mild mononuclear perivascular inflammation (Figure 2). In some foci, staining for Melan-A revealed a decrease in epidermal melanocytes (Figure 3). Periodic acid–Schiff staining performed on one section revealed a few pityriasis spores but no hyphal elements, suggesting colonization rather than infection.
The patient initially was started on tacrolimus ointment 0.1% once daily and narrowband UVB phototherapy twice weekly for 3 months without benefit. A diagnosis of tinea versicolor was revisited and the patient was switched to ketoconazole shampoo 1% two to 3 times weekly on the face, trunk, arms, and legs for 10 to 15 minutes prior to rinsing, and ketoconazole cream 2% was applied twice daily to the affected areas for 2 months without notable improvement. Once-weekly 150-mg pulse doses of oral fluconazole for 8 weeks were started but proved equally ineffective. Antibiotic therapy aimed at eradicating Propionibacterium acnes was considered following a provisional diagnosis of PMH after the patient failed 5 months of therapy for tinea versicolor.
Patient 2
A 54-year-old man presented with hypopigmented to depigmented nonscaly macules on the face, trunk, chest, and arms of several months’ duration. The patient initially noted hypopigmentation on the face that gradually spread to the rest of the body. The patient denied any prior rash or hyperpigmentation in the affected areas. At the initial visit to our clinic, a potassium hydroxide (KOH) preparation of the face and back was positive for tinea versicolor. The patient was treated with ketoconazole shampoo 1% two to 3 times weekly for several weeks on the scalp, face, trunk, arms, and legs for 10 to 15 minutes prior to rinsing and 2 total doses of oral fluconazole 150 mg taken 1 week apart.
Three months later the patient returned with no improvement of the existing lesions and with progression of the disease to previously uninvolved areas of the trunk, arms, and legs. Biopsy of a facial lesion was performed, and laboratory studies including RPR, thyroid-stimulating hormone, and antinuclear antibody tests were conducted to screen for possible systemic disease. Microscopic analysis of the biopsied facial lesion revealed a sparse perivascular infiltrate of lymphocytes and plasma cells but no evidence of yeast or hyphal elements. Melan-A staining did not reveal a decreased number of epidermal melanocytes. All laboratory studies were negative or within normal limits. Desonide ointment 0.05% was prescribed to relieve the patient’s occasional pruritus. Although the patient’s symptoms resolved, the hypopigmented macules continued to progress, making a diagnosis of PMH more likely given the lack of improvement on treatment for tinea versicolor. Pimecrolimus cream 1% was started with discontinuation of desonide for steroid-sparing therapy.
Patient 3
A 63-year-old man presented with progressive nonscaly and asymptomatic hypopigmented macules on the face, trunk, abdomen, and back of 5 years’ duration. He first noted lesions on the abdomen and they subsequently spread to the rest of the body. The patient denied any prior rash, hyperpigmentation, or other lesions in the involved areas.
One year prior to the current presentation, KOH scrapings from the lesions performed by an outside physician were negative. During his initial visit to our clinic, an abdominal biopsy was performed, and histopathologic analysis showed postinflammatory pigmentary alteration; however, the patient denied any prior history of rash or injury in the distribution of the lesions that would correlate with the histopathologic findings of postinflammatory pigmentation. Because the histopathologic findings showed postinflammatory pigmentary alteration, additional stains including Melan-A were not performed.
The patient was provisionally treated with ketoconazole shampoo 1% two to 3 times weekly on the face, trunk, arms, and legs for 10 to 15 minutes prior to rinsing and ketoconazole cream 2% twice daily to the affected areas. After several months on this regimen, the patient did not report any improvement. An abdominal skin biopsy was again performed and revealed similar histopathology. Periodic acid–Schiff staining was negative for fungus. A diagnosis of PMH was made, and the patient was started on benzoyl peroxide wash 5% and clindamycin lotion.
Patient 4
A 45-year-old woman presented with hypopigmented, nonscaly macules on the face, neck, chest, trunk, and back. She first noted the lesions on the face and trunk more than 8 years prior, and they subsequently progressed. Potassium hydroxide scrapings performed on the lesions at the current presentation were negative, and a skin biopsy from the neck revealed postinflammatory pigmentary alteration, although the patient had no history of rash or injury in the areas in which the lesions were distributed.
Fontana-Masson and Melan-A staining of the skin biopsy of the neck revealed a normal distribution of melanocytes and pigment at the dermoepidermal junction. An RPR test was nonreactive. A diagnosis of PMH was made, and the patient was started on benzoyl peroxide wash 5% and clindamycin phosphate lotion 1%.
Comment
The 4 cases of PMH reported here showed extensive facial involvement in addition to the characteristic hypopigmented lesions on the trunk, arms, and legs. It is unclear why the lesions in these patients had a predominantly facial distribution. Involvement of the face in PMH has not been commonly reported in the literature. Martínez-Martínez et al3 reported 12 PMH patients with lesions only presenting in lumbar and abdominal distributions. Kim et al8 presented a series of 23 PMH patients treated with narrowband UVB in whom 56% (9/16) saw repigmentation in 90% of the lesions following treatment. The most commonly affected area was the lower back, followed by the abdomen, upper back, chest, sacral region, flank, and shoulders, respectively.8 In a review by Relyveld et al,1 PMH is described as a predominantly truncal disease that can occasionally extend to the neck, face, and proximal arms and legs; however, no specific cases were reported.
Previous case series have reported PMH primarily in adolescents and young adults, with mean ages ranging from 26 to 30 years.1,3 The 4 patients reported here were older, ranging in age from 40 to 65 years. This discrepancy in age may contribute to the facial distribution encountered in this patient population; however, given the small number of patients in our case series, such extrapolation is premature. Most recently, Westerhof et al6 demonstrated a relationship between the presence of P acnes, a common skin commensal of the face, and the hypopigmented macules of PMH. The investigators suggested that some strains of P acnes produce a factor that is yet to be identified that interferes with melanogenesis. The response of PMH lesions to topical treatments such as benzoyl peroxide, clindamycin, and phototherapy has lent credence to the potential etiologic role of P acnes in this condition.9,10 The interplay between age, PMH distribution, and P acnes requires further investigation.
The biopsies in our 4 patients were consistent with the nonspecific histopathologic characteristics of PMH lesions. Biopsies in all 4 patients revealed a sparse perivascular lymphocytic infiltrate, and in 2 of the cases, postinflammatory pigmentary alteration was noted. Such changes often are described in PMH lesions.4,5 In other cases detailed in the literature, lesional and nonlesional skin often are indistinguishable on hematoxylin and eosin staining.11 In the 3 patients for whom we performed additional immunohistochemical studies, results were mixed: Melan-A staining revealed a decreased number of melanocytes in Patient 1 but not in Patients 2 or 4. Many reported cases in the literature have not demonstrated a decrease in melanocyte density but instead show a decrease in melanin content in lesional skin.1-3,6,7 Although additional stains performed in Patient 4 revealed neither a decrease in the number of melanocytes nor a decrease in the melanin content, such histopathologic findings of PMH often are subtle. Additional stains were not performed in Patient 3. More studies are needed to characterize the immunohistochemical staining patterns of lesional skin in patients with PMH.
Tinea versicolor, pityriasis alba, mycosis fungoides, sarcoidosis, leprosy, and syphilis typically are included in the differential diagnosis for PMH. Tinea versicolor traditionally is diagnosed based on the combination of irregular hypopigmented or hyperpigmented scaly macules and a KOH preparation that is positive for hyphae and spores. Similar to PMH, tinea versicolor is most often found on the trunk, but unusual cases have been reported involving the face.12
Patient 2 reflected how it can be difficult diagnostically to distinguish between tinea versicolor and PMH. Although this patient initially had a KOH scraping suggestive for tinea versicolor, adequate treatment with oral fluconazole and ketoconazole shampoo did not result in improvement. The hypopigmented lesions in this patient continued to progress despite therapy. Additionally, his hypopigmented to depigmented nonscaly macules were more clinically consistent with the characteristic description of lesion configuration in PMH than with the irregular, more sharply defined, asymmetric, and scaly spots of tinea versicolor. Furthermore, the inflammatory findings on biopsy favored a diagnosis of PMH.
Pityriasis alba, most frequently presents on the face in the form of hypopigmented, sometimes slightly scaly macules but also can occur on the body. It usually occurs in younger patients who often have an atopic diathesis. Histologic findings generally are nonspecific, but discrete eczematous changes can sometimes be appreciated in the epidermis and dermis. None of our patients had histories suggestive of an atopic diathesis or lesion distributions typical of pityriasis alba. Histologic findings also were more consistent with PMH than pityriasis alba.
A diagnosis of patch-stage hypopigmented MF should also be entertained in patients with hypopigmented macules, as it can appear similar to the lesions of PMH. Hypopigmented MF often is associated with subtle atrophy, scaling, poikiloderma, and erythema. These features were not present in the 4 cases presented here. Histologically, atypical lymphocytes with prominent epidermotropism and tagging of the epidermis by large lymphocytic infiltrates are seen in cases of hypopigmented MF. These findings were not present in biopsies from our patients.
Hypopigmented sarcoidosis, leprosy, and syphilis are other systemic diseases associated with hypopigmented lesions. Histologically, noncaseasting granulomas in the dermis or subcutaneous tissue would favor a diagnosis of sarcoidosis over PMH. In patients who live in endemic areas, a diagnosis of leprosy for an anesthetic hypopigmented lesion would be higher in the differential. Finally, it is important to rule out secondary syphilis when diagnosing PMH. Known as the great imitator, secondary syphilis may present in a patient in the form of hypopigmented macules. Patients 1, 2, and 4 had nonreactive RPR tests; unfortunately, RPR was not checked in Patient 3. He denied all risk factors for syphilis.
Various topical and oral treatments were prescribed for each patient, but so far none have been unequivocally effective. In the literature, there are reports supporting the efficacy of topical antimicrobial agents targeting P acnes.9,10 One case report noted improvement in a patient with PMH after isotretinoin use.13 Phototherapy also has been reported to improve PMH in several case reports4-8; however, consistent response to these therapies has not been documented. Unfortunately for patients with a diagnosis of PMH, a lack of effective treatment options often exists.
This series of 4 cases highlights the importance of considering PMH in the differential of hypopigmented macules, even when they appear predominantly on the face.
Progressive macular hypomelanosis (PMH) is a noninflammatory skin disorder characterized by ill-defined, nummular, hypopigmented, and nonscaly macules. Historically, various names have been used to describe this entity. Several of these terms, including cutis trunci variata and nummular and confluent hypomelanosis of the trunk, reflected its predominantly truncal distribution.1,2 Less frequently, involvement on the neck, buttocks, and arms and legs has been noted.1,2 A lack of facial involvement previously has been highlighted as a key clinical feature of PMH.3
Progressive macular hypomelanosis is a diagnosis of exclusion. Hypopigmented diseases commonly considered in the differential include those caused by fungi and yeasts (eg, tinea versicolor, seborrheic dermatitis), inflammatory skin disorders (eg, pityriasis alba, postinflammatory dyschromia), and mycosis fungoides (MF) as well as leprosy.
The hypopigmented macules of PMH have nonspecific histopathologic findings; lesional skin often shows minimal alterations as compared to normal skin. A sparse perivascular lymphocytic infiltrate often is observed,4,5 and at times, a decrease in epidermal melanin content can be detected.1-3,6,7
We report 4 cases with considerable facial involvement of hypopigmented macules that were determined to be consistent with PMH. We propose that characteristic macules that are not clinically or histopathologically consistent with other disease entities are compatible with a diagnosis of PMH, regardless of the distribution. A diagnosis of PMH should be considered in the differential when there are suggestive facial lesions in addition to truncal lesions.
Case Reports
Patient 1
A 40-year-old man presented with hypopigmented macules on the face (Figure 1), trunk, chest, arms, and legs of 2 years’ duration. The lesions were asymptomatic and had started on the forehead as hypopigmented macules, then progressed to the trunk, arms, and legs. The patient denied any prior rash, injury, or hyperpigmentation associated with the distribution of the lesions.
A rapid plasma reagin (RPR) test was conducted to rule out secondary syphilis and was nonreactive. During a series of clinical encounters over several months, a total of 5 biopsies of lesions on the face and back were performed. All specimens contained mild mononuclear perivascular inflammation (Figure 2). In some foci, staining for Melan-A revealed a decrease in epidermal melanocytes (Figure 3). Periodic acid–Schiff staining performed on one section revealed a few pityriasis spores but no hyphal elements, suggesting colonization rather than infection.
The patient initially was started on tacrolimus ointment 0.1% once daily and narrowband UVB phototherapy twice weekly for 3 months without benefit. A diagnosis of tinea versicolor was revisited and the patient was switched to ketoconazole shampoo 1% two to 3 times weekly on the face, trunk, arms, and legs for 10 to 15 minutes prior to rinsing, and ketoconazole cream 2% was applied twice daily to the affected areas for 2 months without notable improvement. Once-weekly 150-mg pulse doses of oral fluconazole for 8 weeks were started but proved equally ineffective. Antibiotic therapy aimed at eradicating Propionibacterium acnes was considered following a provisional diagnosis of PMH after the patient failed 5 months of therapy for tinea versicolor.
Patient 2
A 54-year-old man presented with hypopigmented to depigmented nonscaly macules on the face, trunk, chest, and arms of several months’ duration. The patient initially noted hypopigmentation on the face that gradually spread to the rest of the body. The patient denied any prior rash or hyperpigmentation in the affected areas. At the initial visit to our clinic, a potassium hydroxide (KOH) preparation of the face and back was positive for tinea versicolor. The patient was treated with ketoconazole shampoo 1% two to 3 times weekly for several weeks on the scalp, face, trunk, arms, and legs for 10 to 15 minutes prior to rinsing and 2 total doses of oral fluconazole 150 mg taken 1 week apart.
Three months later the patient returned with no improvement of the existing lesions and with progression of the disease to previously uninvolved areas of the trunk, arms, and legs. Biopsy of a facial lesion was performed, and laboratory studies including RPR, thyroid-stimulating hormone, and antinuclear antibody tests were conducted to screen for possible systemic disease. Microscopic analysis of the biopsied facial lesion revealed a sparse perivascular infiltrate of lymphocytes and plasma cells but no evidence of yeast or hyphal elements. Melan-A staining did not reveal a decreased number of epidermal melanocytes. All laboratory studies were negative or within normal limits. Desonide ointment 0.05% was prescribed to relieve the patient’s occasional pruritus. Although the patient’s symptoms resolved, the hypopigmented macules continued to progress, making a diagnosis of PMH more likely given the lack of improvement on treatment for tinea versicolor. Pimecrolimus cream 1% was started with discontinuation of desonide for steroid-sparing therapy.
Patient 3
A 63-year-old man presented with progressive nonscaly and asymptomatic hypopigmented macules on the face, trunk, abdomen, and back of 5 years’ duration. He first noted lesions on the abdomen and they subsequently spread to the rest of the body. The patient denied any prior rash, hyperpigmentation, or other lesions in the involved areas.
One year prior to the current presentation, KOH scrapings from the lesions performed by an outside physician were negative. During his initial visit to our clinic, an abdominal biopsy was performed, and histopathologic analysis showed postinflammatory pigmentary alteration; however, the patient denied any prior history of rash or injury in the distribution of the lesions that would correlate with the histopathologic findings of postinflammatory pigmentation. Because the histopathologic findings showed postinflammatory pigmentary alteration, additional stains including Melan-A were not performed.
The patient was provisionally treated with ketoconazole shampoo 1% two to 3 times weekly on the face, trunk, arms, and legs for 10 to 15 minutes prior to rinsing and ketoconazole cream 2% twice daily to the affected areas. After several months on this regimen, the patient did not report any improvement. An abdominal skin biopsy was again performed and revealed similar histopathology. Periodic acid–Schiff staining was negative for fungus. A diagnosis of PMH was made, and the patient was started on benzoyl peroxide wash 5% and clindamycin lotion.
Patient 4
A 45-year-old woman presented with hypopigmented, nonscaly macules on the face, neck, chest, trunk, and back. She first noted the lesions on the face and trunk more than 8 years prior, and they subsequently progressed. Potassium hydroxide scrapings performed on the lesions at the current presentation were negative, and a skin biopsy from the neck revealed postinflammatory pigmentary alteration, although the patient had no history of rash or injury in the areas in which the lesions were distributed.
Fontana-Masson and Melan-A staining of the skin biopsy of the neck revealed a normal distribution of melanocytes and pigment at the dermoepidermal junction. An RPR test was nonreactive. A diagnosis of PMH was made, and the patient was started on benzoyl peroxide wash 5% and clindamycin phosphate lotion 1%.
Comment
The 4 cases of PMH reported here showed extensive facial involvement in addition to the characteristic hypopigmented lesions on the trunk, arms, and legs. It is unclear why the lesions in these patients had a predominantly facial distribution. Involvement of the face in PMH has not been commonly reported in the literature. Martínez-Martínez et al3 reported 12 PMH patients with lesions only presenting in lumbar and abdominal distributions. Kim et al8 presented a series of 23 PMH patients treated with narrowband UVB in whom 56% (9/16) saw repigmentation in 90% of the lesions following treatment. The most commonly affected area was the lower back, followed by the abdomen, upper back, chest, sacral region, flank, and shoulders, respectively.8 In a review by Relyveld et al,1 PMH is described as a predominantly truncal disease that can occasionally extend to the neck, face, and proximal arms and legs; however, no specific cases were reported.
Previous case series have reported PMH primarily in adolescents and young adults, with mean ages ranging from 26 to 30 years.1,3 The 4 patients reported here were older, ranging in age from 40 to 65 years. This discrepancy in age may contribute to the facial distribution encountered in this patient population; however, given the small number of patients in our case series, such extrapolation is premature. Most recently, Westerhof et al6 demonstrated a relationship between the presence of P acnes, a common skin commensal of the face, and the hypopigmented macules of PMH. The investigators suggested that some strains of P acnes produce a factor that is yet to be identified that interferes with melanogenesis. The response of PMH lesions to topical treatments such as benzoyl peroxide, clindamycin, and phototherapy has lent credence to the potential etiologic role of P acnes in this condition.9,10 The interplay between age, PMH distribution, and P acnes requires further investigation.
The biopsies in our 4 patients were consistent with the nonspecific histopathologic characteristics of PMH lesions. Biopsies in all 4 patients revealed a sparse perivascular lymphocytic infiltrate, and in 2 of the cases, postinflammatory pigmentary alteration was noted. Such changes often are described in PMH lesions.4,5 In other cases detailed in the literature, lesional and nonlesional skin often are indistinguishable on hematoxylin and eosin staining.11 In the 3 patients for whom we performed additional immunohistochemical studies, results were mixed: Melan-A staining revealed a decreased number of melanocytes in Patient 1 but not in Patients 2 or 4. Many reported cases in the literature have not demonstrated a decrease in melanocyte density but instead show a decrease in melanin content in lesional skin.1-3,6,7 Although additional stains performed in Patient 4 revealed neither a decrease in the number of melanocytes nor a decrease in the melanin content, such histopathologic findings of PMH often are subtle. Additional stains were not performed in Patient 3. More studies are needed to characterize the immunohistochemical staining patterns of lesional skin in patients with PMH.
Tinea versicolor, pityriasis alba, mycosis fungoides, sarcoidosis, leprosy, and syphilis typically are included in the differential diagnosis for PMH. Tinea versicolor traditionally is diagnosed based on the combination of irregular hypopigmented or hyperpigmented scaly macules and a KOH preparation that is positive for hyphae and spores. Similar to PMH, tinea versicolor is most often found on the trunk, but unusual cases have been reported involving the face.12
Patient 2 reflected how it can be difficult diagnostically to distinguish between tinea versicolor and PMH. Although this patient initially had a KOH scraping suggestive for tinea versicolor, adequate treatment with oral fluconazole and ketoconazole shampoo did not result in improvement. The hypopigmented lesions in this patient continued to progress despite therapy. Additionally, his hypopigmented to depigmented nonscaly macules were more clinically consistent with the characteristic description of lesion configuration in PMH than with the irregular, more sharply defined, asymmetric, and scaly spots of tinea versicolor. Furthermore, the inflammatory findings on biopsy favored a diagnosis of PMH.
Pityriasis alba, most frequently presents on the face in the form of hypopigmented, sometimes slightly scaly macules but also can occur on the body. It usually occurs in younger patients who often have an atopic diathesis. Histologic findings generally are nonspecific, but discrete eczematous changes can sometimes be appreciated in the epidermis and dermis. None of our patients had histories suggestive of an atopic diathesis or lesion distributions typical of pityriasis alba. Histologic findings also were more consistent with PMH than pityriasis alba.
A diagnosis of patch-stage hypopigmented MF should also be entertained in patients with hypopigmented macules, as it can appear similar to the lesions of PMH. Hypopigmented MF often is associated with subtle atrophy, scaling, poikiloderma, and erythema. These features were not present in the 4 cases presented here. Histologically, atypical lymphocytes with prominent epidermotropism and tagging of the epidermis by large lymphocytic infiltrates are seen in cases of hypopigmented MF. These findings were not present in biopsies from our patients.
Hypopigmented sarcoidosis, leprosy, and syphilis are other systemic diseases associated with hypopigmented lesions. Histologically, noncaseasting granulomas in the dermis or subcutaneous tissue would favor a diagnosis of sarcoidosis over PMH. In patients who live in endemic areas, a diagnosis of leprosy for an anesthetic hypopigmented lesion would be higher in the differential. Finally, it is important to rule out secondary syphilis when diagnosing PMH. Known as the great imitator, secondary syphilis may present in a patient in the form of hypopigmented macules. Patients 1, 2, and 4 had nonreactive RPR tests; unfortunately, RPR was not checked in Patient 3. He denied all risk factors for syphilis.
Various topical and oral treatments were prescribed for each patient, but so far none have been unequivocally effective. In the literature, there are reports supporting the efficacy of topical antimicrobial agents targeting P acnes.9,10 One case report noted improvement in a patient with PMH after isotretinoin use.13 Phototherapy also has been reported to improve PMH in several case reports4-8; however, consistent response to these therapies has not been documented. Unfortunately for patients with a diagnosis of PMH, a lack of effective treatment options often exists.
This series of 4 cases highlights the importance of considering PMH in the differential of hypopigmented macules, even when they appear predominantly on the face.
- Relyveld G, Menke H, Westerhof W. Progressive macular hypomelanosis: an overview. Am J Clin Dermatol. 2007;8:13-19.
- Hwang SW, Hong SK, Kim SH, et al. Progressive macular hypomelanosis in Korean patients: a clinicopathologic study. Ann Dermatol. 2009;21:261-267.
- Martinéz-Martinéz ML, Azaña-Defez JM, Rodríguez-Vázquez M, et al. Progressive macular hypomelanosis. Pediatr Dermatol. 2012;29:460-462.
- Montero LC, Belinchonón I, Toledo F, et al. Progressive macular hypomelanosis, excellent response with narrow-band ultraviolet B phototherapy. Photodermatol Photoimmunol Photomed. 2011;27:162-163.
- Choi YJ, Hann SK. Two cases of progressive macular hypomelanosis of the trunk. Korean J Dermatol. 2000;38:655-658.
- Westerhof W, Rlyveld G, Kingswijk M, et al. Propionibacterium acnes and the pathogenesis of progressive macular hypomelanosis. Arch Dermatol. 2004;140:210-214.
- Wu SG, Xu AE, Song XZ, et al. Clinical, pathologic, and ultrastructural studies of progressive macular hypomelanosis. Int J Dermatol. 2010;29:1127-1132.
- Kim MB, Kim GW, Cho HH, et al. Narrowband UVB treatment of progressive macular hypomelanosis. J Am Acad Dermatol. 2012;66:598-605.
- Revlyveld GN, Menkie HE, Westerhof W. Benzoyl peroxide/clindamycin/UVA is more effective than fluticasone/UVA in progressive macular hypomelanosis: a randomized study. Am J Clin Dermatol. 2006;55:836-843.
- Santos JB, Almeida OL, Silva LM, et al. Efficacy of topical combination of benzoyl peroxide 5% and clindamcyin 1% for the treatment of progressive macular hypomelanosis: a randomized, doubleblind, placebo-controlled trial [in Portuguese]. An Bras Dermatol. 2011;86:50-54.
- Kumarasinghe SP, Tan SH, Thng S, et al. Progressive macular hypomelanosis in Singapore: a clinico-pathological study. Int J Dermatol. 2006;45:737-742.
- Terragni L, Lasagni A, Oriani A. Pityriasis versicolor of the face. Mycoses. 1991;34:345-347.
- Kim YK, Lee DY, Lee, JY, et al. Progressive macular hypomelanosis showing excellent response to oral isotretinoin [published online June 23, 2012]. J Dermatol. 2012;39:937-938.
- Relyveld G, Menke H, Westerhof W. Progressive macular hypomelanosis: an overview. Am J Clin Dermatol. 2007;8:13-19.
- Hwang SW, Hong SK, Kim SH, et al. Progressive macular hypomelanosis in Korean patients: a clinicopathologic study. Ann Dermatol. 2009;21:261-267.
- Martinéz-Martinéz ML, Azaña-Defez JM, Rodríguez-Vázquez M, et al. Progressive macular hypomelanosis. Pediatr Dermatol. 2012;29:460-462.
- Montero LC, Belinchonón I, Toledo F, et al. Progressive macular hypomelanosis, excellent response with narrow-band ultraviolet B phototherapy. Photodermatol Photoimmunol Photomed. 2011;27:162-163.
- Choi YJ, Hann SK. Two cases of progressive macular hypomelanosis of the trunk. Korean J Dermatol. 2000;38:655-658.
- Westerhof W, Rlyveld G, Kingswijk M, et al. Propionibacterium acnes and the pathogenesis of progressive macular hypomelanosis. Arch Dermatol. 2004;140:210-214.
- Wu SG, Xu AE, Song XZ, et al. Clinical, pathologic, and ultrastructural studies of progressive macular hypomelanosis. Int J Dermatol. 2010;29:1127-1132.
- Kim MB, Kim GW, Cho HH, et al. Narrowband UVB treatment of progressive macular hypomelanosis. J Am Acad Dermatol. 2012;66:598-605.
- Revlyveld GN, Menkie HE, Westerhof W. Benzoyl peroxide/clindamycin/UVA is more effective than fluticasone/UVA in progressive macular hypomelanosis: a randomized study. Am J Clin Dermatol. 2006;55:836-843.
- Santos JB, Almeida OL, Silva LM, et al. Efficacy of topical combination of benzoyl peroxide 5% and clindamcyin 1% for the treatment of progressive macular hypomelanosis: a randomized, doubleblind, placebo-controlled trial [in Portuguese]. An Bras Dermatol. 2011;86:50-54.
- Kumarasinghe SP, Tan SH, Thng S, et al. Progressive macular hypomelanosis in Singapore: a clinico-pathological study. Int J Dermatol. 2006;45:737-742.
- Terragni L, Lasagni A, Oriani A. Pityriasis versicolor of the face. Mycoses. 1991;34:345-347.
- Kim YK, Lee DY, Lee, JY, et al. Progressive macular hypomelanosis showing excellent response to oral isotretinoin [published online June 23, 2012]. J Dermatol. 2012;39:937-938.
Practice Points
- Progressive macular hypomelanosis should be considered in the differential diagnosis for hypopigmented facial lesions.
- Progressive macular hypomelanosis proves to be a diagnosis of exclusion.
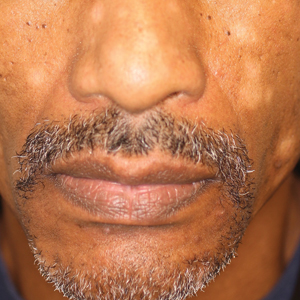
Subungual Onycholemmal Cyst of the Toenail Mimicking Subungual Melanoma
Case Report
A 23-year-old woman presented with a horizontal split along the midline of the right great toenail associated with some tenderness of 2 to 3 months’ duration. Approximately 5 years prior, she noticed a bluish-colored area under the nail that had been steadily increasing in size. She denied a history of trauma, drainage, or bleeding. There was no history of other nail abnormalities. Her medications and personal, family, and social history were noncontributory.
Physical examination of the right great toenail revealed a horizontal split of the nail plate with a bluish hue visible under the nail plate (Figure 1A). The remaining toenails and fingernails were normal. A punch biopsy of the nail bed was performed with a presumptive clinical diagnosis of subungual melanoma versus melanocytic nevus versus cyst (Figure 1B). Nail plate avulsion revealed a blackened nail bed dotted with areas of bluish color and a red friable nodule present focally. Upon further inspection, extension was apparent into the distal matrix.
Histopathologic examination revealed a cystic structure with an epithelial lining mostly reminiscent of an isthmus catagen cyst admixed with the presence of both an intermittent focal granular layer and an eosinophilic cuticle surrounding pink laminated keratin, most consistent with a diagnosis of subungual onycholemmal cyst (SOC)(Figure 2). A reexcision was performed with removal of half of the nail bed, including a portion of the distal matrix extending inferiorly to the bone. Variably sized, epithelium-lined, keratin-filled cystic structures emanated from the nail bed epithelium. There were foci of hemorrhage and granulation tissue secondary to cyst rupture (Figure 3). The defect healed by secondary intention. No clinical evidence of recurrence was seen at 6-month follow-up.

Subungual onycholemmal cysts, also known as subungual epidermoid cysts or subungual epidermoid inclusions, are rare and distinctive nail abnormalities occurring in the dermis of the nail bed. We present a case of an SOC in a toenail mimicking subungual malignant melanoma.
Originally described by Samman1 in 1959, SOCs were attributed to trauma to the nail with resultant implantation of the epidermis into the deeper tissue. Lewin2,3 examined 90 postmortem fingernail and nail bed samples and found 8 subungual epidermoid cysts associated with clubbing of the fingernails. He postulated that the early pathogenesis of clubbing involved dermal fibroblast proliferation in the nail bed, leading to sequestration of nail bed epithelium into the dermis with resultant cyst formation. Microscopic subungual cysts also were identified in normal-appearing nails without evidence of trauma, thought to have arisen from the tips of the nail bed rete ridges by a process of bulbous proliferation rather than sequestration. These findings in normal nails suggest that SOCs may represent a more common entity than previously recognized.
It is imperative to recognize the presence of nail unit tumors early because of the risk for permanent nail plate dystrophy and the possibility of a malignant tumor.4,5 Subungual onycholemmal cysts may present with a wide spectrum of clinical findings including marked subungual hyperkeratosis, onychodystrophy, ridging, nail bed pigmentation, clubbing, thickening, or less often a normal-appearing nail. Based on reported cases, several trends are evident. Although nail dystrophy is most often asymptomatic, pain is not uncommon.5,6 It most commonly involves single digits, predominantly thumbs and great toenails.7,8 This predilection suggests that trauma or other local factors may be involved in its pathogenesis. Of note, trauma to the nail may occur years before the development of the lesions or it may not be recalled at all.
Diagnosis requires a degree of clinical suspicion and a nail bed biopsy with partial or total nail plate avulsion to visualize the pathologic portion of the nail bed. Because surgical intervention may lead to the implantation of epithelium, recurrences after nail biopsy or excision may occur.
In contrast to epidermal inclusion cysts arising in the skin, most SOCs do not have a granular layer.9 Hair and nails represent analogous differentiation products of the ectoderm. The nail matrix is homologous to portions of the hair matrix, while the nail bed epithelium is comparable to the outer root sheath of the hair follicle.7 Subungual onycholemmal cysts originate from the nail bed epithelium, which keratinizes in the absence of a granular layer, similar to the follicular isthmus outer root sheath. Thus, SOCs are comparable to the outer root sheath–derived isthmus-catagen cysts because of their abrupt central keratinization.8
Subungual onycholemmal cysts also must be distinguished from slowly growing malignant tumors of the nail bed epithelium, referred to as onycholemmal carcinomas by Alessi et al.10 This entity characteristically presents in elderly patients as a slowly growing, circumscribed, subungual discoloration that may ulcerate, destroying the nail apparatus and penetrating the phalangeal bone. On histopathology, it is characterized by small cysts filled with eosinophilic keratin devoid of a granular layer and lined by atypical squamous epithelium accompanied by solid nests and strands of atypical keratinocytes within the dermis.11 When a cystic component and clear cells predominate, the designation of malignant proliferating onycholemmal cyst has been applied. Its infiltrative growth pattern with destruction of the underlying bone makes it an important entity to exclude when considering the differential diagnosis of tumors of the nail bed.
Subungual melanomas comprise only 1% to 3% of malignant melanomas and 85% are initially misdiagnosed due to their rarity and nonspecific variable presentation. Aside from clinical evidence of Hutchinson sign in the early stages in almost all cases, accurate diagnosis of subungual melanoma and differentiation from SOCs relies on histopathology. A biopsy is necessary to make the diagnosis, but even microscopic findings may be nonspecific during the early stages.
Conclusion
We report a case of a 23-year-old woman with horizontal ridging and tenderness of the right great toenail associated with pigmentation of 5 years’ duration due to an SOC. The etiology of these subungual cysts, with or without nail abnormalities, still remains unclear. Its predilection for the thumbs and great toenails suggests that trauma or other local factors may be involved in its pathogenesis. Because of the rarity of this entity, there are no guidelines for surgical treatment. Subungual onycholemmal cysts may be an underrecognized and more common entity that must be considered when discussing tumors of the nail unit.
- Samman PD. The human toe nail. its genesis and blood supply. Br J Dermatol. 1959;71:296-302.
- Lewin K. The normal fingernail. Br J Dermatol. 1965;77:421-430.
- Lewin K. Subungual epidermoid inclusions. Br J Dermatol. 1969;81:671-675.
- Dominguez-Cherit J, Chanussot-Deprez C, Maria-Sarti H, et al. Nail unit tumors: a study of 234 patients in the dermatology department of the “Dr. Manuel Gea González” General Hospital in Mexico City. Dermatol Surg. 2008;34:1363-1371.
- Sáez-de-Ocariz MM, Domínguez-Cherit J, García-Corona C. Subungual epidermoid cysts. Int J Dermatol. 2001;40:524-526.
- Molly DO, Herbert K. Subungual epidermoid cyst. J Hand Surg Br. 2006;31:345.
- Telang GH, Jellinek N. Multiple calcified subungual epidermoid inclusions. J Am Acad Dermatol. 2007;56:336-339.
- Fanti PA, Tosti A. Subungual epidermoid inclusions: report of 8 cases. Dermatologica. 1989;178:209-212.
- Takiyoshi N, Nakano H, Matsuzaki T, et al. An eclipse in the subungual space: a diagnostic sign for a subungual epidermal cyst? Br J Dermatol. 2009;161:962-963.
- Alessi E, Coggi A, Gianotti R, et al. Onycholemmal carcinoma. Am J Dermatopathol. 2004;26:397-402.
- Inaoki M, Makino E, Adachi M, et al. Onycholemmal carcinoma. J Cutan Pathol. 2006;33:577-580.
Case Report
A 23-year-old woman presented with a horizontal split along the midline of the right great toenail associated with some tenderness of 2 to 3 months’ duration. Approximately 5 years prior, she noticed a bluish-colored area under the nail that had been steadily increasing in size. She denied a history of trauma, drainage, or bleeding. There was no history of other nail abnormalities. Her medications and personal, family, and social history were noncontributory.
Physical examination of the right great toenail revealed a horizontal split of the nail plate with a bluish hue visible under the nail plate (Figure 1A). The remaining toenails and fingernails were normal. A punch biopsy of the nail bed was performed with a presumptive clinical diagnosis of subungual melanoma versus melanocytic nevus versus cyst (Figure 1B). Nail plate avulsion revealed a blackened nail bed dotted with areas of bluish color and a red friable nodule present focally. Upon further inspection, extension was apparent into the distal matrix.
Histopathologic examination revealed a cystic structure with an epithelial lining mostly reminiscent of an isthmus catagen cyst admixed with the presence of both an intermittent focal granular layer and an eosinophilic cuticle surrounding pink laminated keratin, most consistent with a diagnosis of subungual onycholemmal cyst (SOC)(Figure 2). A reexcision was performed with removal of half of the nail bed, including a portion of the distal matrix extending inferiorly to the bone. Variably sized, epithelium-lined, keratin-filled cystic structures emanated from the nail bed epithelium. There were foci of hemorrhage and granulation tissue secondary to cyst rupture (Figure 3). The defect healed by secondary intention. No clinical evidence of recurrence was seen at 6-month follow-up.
Subungual onycholemmal cysts, also known as subungual epidermoid cysts or subungual epidermoid inclusions, are rare and distinctive nail abnormalities occurring in the dermis of the nail bed. We present a case of an SOC in a toenail mimicking subungual malignant melanoma.
Originally described by Samman1 in 1959, SOCs were attributed to trauma to the nail with resultant implantation of the epidermis into the deeper tissue. Lewin2,3 examined 90 postmortem fingernail and nail bed samples and found 8 subungual epidermoid cysts associated with clubbing of the fingernails. He postulated that the early pathogenesis of clubbing involved dermal fibroblast proliferation in the nail bed, leading to sequestration of nail bed epithelium into the dermis with resultant cyst formation. Microscopic subungual cysts also were identified in normal-appearing nails without evidence of trauma, thought to have arisen from the tips of the nail bed rete ridges by a process of bulbous proliferation rather than sequestration. These findings in normal nails suggest that SOCs may represent a more common entity than previously recognized.
It is imperative to recognize the presence of nail unit tumors early because of the risk for permanent nail plate dystrophy and the possibility of a malignant tumor.4,5 Subungual onycholemmal cysts may present with a wide spectrum of clinical findings including marked subungual hyperkeratosis, onychodystrophy, ridging, nail bed pigmentation, clubbing, thickening, or less often a normal-appearing nail. Based on reported cases, several trends are evident. Although nail dystrophy is most often asymptomatic, pain is not uncommon.5,6 It most commonly involves single digits, predominantly thumbs and great toenails.7,8 This predilection suggests that trauma or other local factors may be involved in its pathogenesis. Of note, trauma to the nail may occur years before the development of the lesions or it may not be recalled at all.
Diagnosis requires a degree of clinical suspicion and a nail bed biopsy with partial or total nail plate avulsion to visualize the pathologic portion of the nail bed. Because surgical intervention may lead to the implantation of epithelium, recurrences after nail biopsy or excision may occur.
In contrast to epidermal inclusion cysts arising in the skin, most SOCs do not have a granular layer.9 Hair and nails represent analogous differentiation products of the ectoderm. The nail matrix is homologous to portions of the hair matrix, while the nail bed epithelium is comparable to the outer root sheath of the hair follicle.7 Subungual onycholemmal cysts originate from the nail bed epithelium, which keratinizes in the absence of a granular layer, similar to the follicular isthmus outer root sheath. Thus, SOCs are comparable to the outer root sheath–derived isthmus-catagen cysts because of their abrupt central keratinization.8
Subungual onycholemmal cysts also must be distinguished from slowly growing malignant tumors of the nail bed epithelium, referred to as onycholemmal carcinomas by Alessi et al.10 This entity characteristically presents in elderly patients as a slowly growing, circumscribed, subungual discoloration that may ulcerate, destroying the nail apparatus and penetrating the phalangeal bone. On histopathology, it is characterized by small cysts filled with eosinophilic keratin devoid of a granular layer and lined by atypical squamous epithelium accompanied by solid nests and strands of atypical keratinocytes within the dermis.11 When a cystic component and clear cells predominate, the designation of malignant proliferating onycholemmal cyst has been applied. Its infiltrative growth pattern with destruction of the underlying bone makes it an important entity to exclude when considering the differential diagnosis of tumors of the nail bed.
Subungual melanomas comprise only 1% to 3% of malignant melanomas and 85% are initially misdiagnosed due to their rarity and nonspecific variable presentation. Aside from clinical evidence of Hutchinson sign in the early stages in almost all cases, accurate diagnosis of subungual melanoma and differentiation from SOCs relies on histopathology. A biopsy is necessary to make the diagnosis, but even microscopic findings may be nonspecific during the early stages.
Conclusion
We report a case of a 23-year-old woman with horizontal ridging and tenderness of the right great toenail associated with pigmentation of 5 years’ duration due to an SOC. The etiology of these subungual cysts, with or without nail abnormalities, still remains unclear. Its predilection for the thumbs and great toenails suggests that trauma or other local factors may be involved in its pathogenesis. Because of the rarity of this entity, there are no guidelines for surgical treatment. Subungual onycholemmal cysts may be an underrecognized and more common entity that must be considered when discussing tumors of the nail unit.
Case Report
A 23-year-old woman presented with a horizontal split along the midline of the right great toenail associated with some tenderness of 2 to 3 months’ duration. Approximately 5 years prior, she noticed a bluish-colored area under the nail that had been steadily increasing in size. She denied a history of trauma, drainage, or bleeding. There was no history of other nail abnormalities. Her medications and personal, family, and social history were noncontributory.
Physical examination of the right great toenail revealed a horizontal split of the nail plate with a bluish hue visible under the nail plate (Figure 1A). The remaining toenails and fingernails were normal. A punch biopsy of the nail bed was performed with a presumptive clinical diagnosis of subungual melanoma versus melanocytic nevus versus cyst (Figure 1B). Nail plate avulsion revealed a blackened nail bed dotted with areas of bluish color and a red friable nodule present focally. Upon further inspection, extension was apparent into the distal matrix.
Histopathologic examination revealed a cystic structure with an epithelial lining mostly reminiscent of an isthmus catagen cyst admixed with the presence of both an intermittent focal granular layer and an eosinophilic cuticle surrounding pink laminated keratin, most consistent with a diagnosis of subungual onycholemmal cyst (SOC)(Figure 2). A reexcision was performed with removal of half of the nail bed, including a portion of the distal matrix extending inferiorly to the bone. Variably sized, epithelium-lined, keratin-filled cystic structures emanated from the nail bed epithelium. There were foci of hemorrhage and granulation tissue secondary to cyst rupture (Figure 3). The defect healed by secondary intention. No clinical evidence of recurrence was seen at 6-month follow-up.
Subungual onycholemmal cysts, also known as subungual epidermoid cysts or subungual epidermoid inclusions, are rare and distinctive nail abnormalities occurring in the dermis of the nail bed. We present a case of an SOC in a toenail mimicking subungual malignant melanoma.
Originally described by Samman1 in 1959, SOCs were attributed to trauma to the nail with resultant implantation of the epidermis into the deeper tissue. Lewin2,3 examined 90 postmortem fingernail and nail bed samples and found 8 subungual epidermoid cysts associated with clubbing of the fingernails. He postulated that the early pathogenesis of clubbing involved dermal fibroblast proliferation in the nail bed, leading to sequestration of nail bed epithelium into the dermis with resultant cyst formation. Microscopic subungual cysts also were identified in normal-appearing nails without evidence of trauma, thought to have arisen from the tips of the nail bed rete ridges by a process of bulbous proliferation rather than sequestration. These findings in normal nails suggest that SOCs may represent a more common entity than previously recognized.
It is imperative to recognize the presence of nail unit tumors early because of the risk for permanent nail plate dystrophy and the possibility of a malignant tumor.4,5 Subungual onycholemmal cysts may present with a wide spectrum of clinical findings including marked subungual hyperkeratosis, onychodystrophy, ridging, nail bed pigmentation, clubbing, thickening, or less often a normal-appearing nail. Based on reported cases, several trends are evident. Although nail dystrophy is most often asymptomatic, pain is not uncommon.5,6 It most commonly involves single digits, predominantly thumbs and great toenails.7,8 This predilection suggests that trauma or other local factors may be involved in its pathogenesis. Of note, trauma to the nail may occur years before the development of the lesions or it may not be recalled at all.
Diagnosis requires a degree of clinical suspicion and a nail bed biopsy with partial or total nail plate avulsion to visualize the pathologic portion of the nail bed. Because surgical intervention may lead to the implantation of epithelium, recurrences after nail biopsy or excision may occur.
In contrast to epidermal inclusion cysts arising in the skin, most SOCs do not have a granular layer.9 Hair and nails represent analogous differentiation products of the ectoderm. The nail matrix is homologous to portions of the hair matrix, while the nail bed epithelium is comparable to the outer root sheath of the hair follicle.7 Subungual onycholemmal cysts originate from the nail bed epithelium, which keratinizes in the absence of a granular layer, similar to the follicular isthmus outer root sheath. Thus, SOCs are comparable to the outer root sheath–derived isthmus-catagen cysts because of their abrupt central keratinization.8
Subungual onycholemmal cysts also must be distinguished from slowly growing malignant tumors of the nail bed epithelium, referred to as onycholemmal carcinomas by Alessi et al.10 This entity characteristically presents in elderly patients as a slowly growing, circumscribed, subungual discoloration that may ulcerate, destroying the nail apparatus and penetrating the phalangeal bone. On histopathology, it is characterized by small cysts filled with eosinophilic keratin devoid of a granular layer and lined by atypical squamous epithelium accompanied by solid nests and strands of atypical keratinocytes within the dermis.11 When a cystic component and clear cells predominate, the designation of malignant proliferating onycholemmal cyst has been applied. Its infiltrative growth pattern with destruction of the underlying bone makes it an important entity to exclude when considering the differential diagnosis of tumors of the nail bed.
Subungual melanomas comprise only 1% to 3% of malignant melanomas and 85% are initially misdiagnosed due to their rarity and nonspecific variable presentation. Aside from clinical evidence of Hutchinson sign in the early stages in almost all cases, accurate diagnosis of subungual melanoma and differentiation from SOCs relies on histopathology. A biopsy is necessary to make the diagnosis, but even microscopic findings may be nonspecific during the early stages.
Conclusion
We report a case of a 23-year-old woman with horizontal ridging and tenderness of the right great toenail associated with pigmentation of 5 years’ duration due to an SOC. The etiology of these subungual cysts, with or without nail abnormalities, still remains unclear. Its predilection for the thumbs and great toenails suggests that trauma or other local factors may be involved in its pathogenesis. Because of the rarity of this entity, there are no guidelines for surgical treatment. Subungual onycholemmal cysts may be an underrecognized and more common entity that must be considered when discussing tumors of the nail unit.
- Samman PD. The human toe nail. its genesis and blood supply. Br J Dermatol. 1959;71:296-302.
- Lewin K. The normal fingernail. Br J Dermatol. 1965;77:421-430.
- Lewin K. Subungual epidermoid inclusions. Br J Dermatol. 1969;81:671-675.
- Dominguez-Cherit J, Chanussot-Deprez C, Maria-Sarti H, et al. Nail unit tumors: a study of 234 patients in the dermatology department of the “Dr. Manuel Gea González” General Hospital in Mexico City. Dermatol Surg. 2008;34:1363-1371.
- Sáez-de-Ocariz MM, Domínguez-Cherit J, García-Corona C. Subungual epidermoid cysts. Int J Dermatol. 2001;40:524-526.
- Molly DO, Herbert K. Subungual epidermoid cyst. J Hand Surg Br. 2006;31:345.
- Telang GH, Jellinek N. Multiple calcified subungual epidermoid inclusions. J Am Acad Dermatol. 2007;56:336-339.
- Fanti PA, Tosti A. Subungual epidermoid inclusions: report of 8 cases. Dermatologica. 1989;178:209-212.
- Takiyoshi N, Nakano H, Matsuzaki T, et al. An eclipse in the subungual space: a diagnostic sign for a subungual epidermal cyst? Br J Dermatol. 2009;161:962-963.
- Alessi E, Coggi A, Gianotti R, et al. Onycholemmal carcinoma. Am J Dermatopathol. 2004;26:397-402.
- Inaoki M, Makino E, Adachi M, et al. Onycholemmal carcinoma. J Cutan Pathol. 2006;33:577-580.
- Samman PD. The human toe nail. its genesis and blood supply. Br J Dermatol. 1959;71:296-302.
- Lewin K. The normal fingernail. Br J Dermatol. 1965;77:421-430.
- Lewin K. Subungual epidermoid inclusions. Br J Dermatol. 1969;81:671-675.
- Dominguez-Cherit J, Chanussot-Deprez C, Maria-Sarti H, et al. Nail unit tumors: a study of 234 patients in the dermatology department of the “Dr. Manuel Gea González” General Hospital in Mexico City. Dermatol Surg. 2008;34:1363-1371.
- Sáez-de-Ocariz MM, Domínguez-Cherit J, García-Corona C. Subungual epidermoid cysts. Int J Dermatol. 2001;40:524-526.
- Molly DO, Herbert K. Subungual epidermoid cyst. J Hand Surg Br. 2006;31:345.
- Telang GH, Jellinek N. Multiple calcified subungual epidermoid inclusions. J Am Acad Dermatol. 2007;56:336-339.
- Fanti PA, Tosti A. Subungual epidermoid inclusions: report of 8 cases. Dermatologica. 1989;178:209-212.
- Takiyoshi N, Nakano H, Matsuzaki T, et al. An eclipse in the subungual space: a diagnostic sign for a subungual epidermal cyst? Br J Dermatol. 2009;161:962-963.
- Alessi E, Coggi A, Gianotti R, et al. Onycholemmal carcinoma. Am J Dermatopathol. 2004;26:397-402.
- Inaoki M, Makino E, Adachi M, et al. Onycholemmal carcinoma. J Cutan Pathol. 2006;33:577-580.
Practice Points
- Trauma to the nail may occur years before the development of subungual onycholemmal cysts or it may not be recalled at all.
- Diagnosis requires a degree of clinical suspicion and a nail bed biopsy.
- Subungual onycholemmal cysts must be distinguished from slowly growing malignant tumors of the nail bed epithelium.