User login
Strategies for management of intermittent fasting in patients with diabetes
Islam is the second most common religion in the world, and there are 1.6 billion Muslims, many in areas where diabetes is prevalent. Each year observant Muslims fast during the daylight hours for the holy month of Ramadan. It is estimated that 50 million diabetic people fast between dawn and sundown during Ramadan, and Muslims are not the only group of patients who fast for religious or other reasons. It is important for healthcare providers to guide patients with diabetes in avoiding problems related to prolonged fasting.
In this issue of the Cleveland Clinic Journal of Medicine, Drs. A.V. and Zagar address management of diabetes specifically relating to Ramadan fasting, with considerations that also apply to other diabetic patients who fast for religious or for medical reasons.
Fortunately, we now have antihyperglycemic agents that are unlikely to cause hypoglycemia if used alone or in combination, as long as the regimen does not include insulin or a sulfonylurea. These include:
- Metformin and thiazolidinediones (pioglitazone and rosiglitazone), which improve insulin sensitivity
- Glucagon-like peptide 1 (GLP-1) agonists (exenatide, liraglutide, dulaglutide, and albaglutide), which facilitate insulin release in a glucose-dependent fashion
- Dipeptidyl peptidase 4 inhibitors (sitagliptin, saxagliptin, alogliptin, and linagliptin), which augment endogenous incretin hormones, primarily GLP-1, and also facilitate insulin production in a glucose-dependent fashion
- Alpha glucosidase inhibitors (acarbose and miglitol), which slow carbohydrate absorption.
Introduced in recent years, the sodium-glucose cotransporter 2 (SGLT-2) inhibitors canagliflozin, dapagliflozin, and empagliflozin lower blood glucose by reducing the renal threshold for reabsorption of glucose, coupled with reabsorption of sodium leading to daily urinary excretion of about 200 calories. These agents alone or taken with any of the agents above should not cause hypoglycemia. However, they can lead to dehydration if fasting precludes the intake of water as well as food.
The primary concern during fasting is hypoglycemia when diabetes regimens involve insulin or insulin secretagogues, most commonly sulfonylureas. Long-acting basal insulin should not require adjustment during fasting if the dose is not excessive. The amount and timing of short-acting analogues administered before meals should be adjusted to the timing of meals, and doses should be adjusted proportionally to the anticipated carbohydrate intake. Premixed insulins such as intermediate-acting (protamine suspension) insulin and a short-acting insulin in 70/30, 75/25, or 50/50 ratios should be avoided. They do not lend themselves to changes in timing, and the short-acting component is fixed and cannot be changed for varied intake without changing the intermediate-acting portion, which functions as the basal insulin.
Sulfonylurea doses can be reduced or the larger dose moved to before the evening meal, but these agents still pose a risk of hypoglycemia during fasting hours. And as Drs. A.V. and Zagar state, glimepiride, glipizide, and gliclazide are the only agents in the class that should be considered; glyburide (ie, glibenclamide) poses too great a risk of hypoglycemia. On the other hand, the short-acting secretagogue nateglinide can be used safely before meals without much risk of hypoglycemia.
We have focused primarily on hypoglycemia risk. But if antihyperglycemic agents are halted completely or if the reduction is too severe, patients are at risk for hyperglycemia and even diabetic ketoacidosis. Careful monitoring of blood glucose levels during the fasting period is most important for patients taking agents that can cause hypoglycemia, and patients should be advised to break the fast if dangerously low glycemic levels occur. Similarly, if severe hyperglycemia or ketoacidosis develops, patients should be advised to seek medical advice promptly.
Islam is the second most common religion in the world, and there are 1.6 billion Muslims, many in areas where diabetes is prevalent. Each year observant Muslims fast during the daylight hours for the holy month of Ramadan. It is estimated that 50 million diabetic people fast between dawn and sundown during Ramadan, and Muslims are not the only group of patients who fast for religious or other reasons. It is important for healthcare providers to guide patients with diabetes in avoiding problems related to prolonged fasting.
In this issue of the Cleveland Clinic Journal of Medicine, Drs. A.V. and Zagar address management of diabetes specifically relating to Ramadan fasting, with considerations that also apply to other diabetic patients who fast for religious or for medical reasons.
Fortunately, we now have antihyperglycemic agents that are unlikely to cause hypoglycemia if used alone or in combination, as long as the regimen does not include insulin or a sulfonylurea. These include:
- Metformin and thiazolidinediones (pioglitazone and rosiglitazone), which improve insulin sensitivity
- Glucagon-like peptide 1 (GLP-1) agonists (exenatide, liraglutide, dulaglutide, and albaglutide), which facilitate insulin release in a glucose-dependent fashion
- Dipeptidyl peptidase 4 inhibitors (sitagliptin, saxagliptin, alogliptin, and linagliptin), which augment endogenous incretin hormones, primarily GLP-1, and also facilitate insulin production in a glucose-dependent fashion
- Alpha glucosidase inhibitors (acarbose and miglitol), which slow carbohydrate absorption.
Introduced in recent years, the sodium-glucose cotransporter 2 (SGLT-2) inhibitors canagliflozin, dapagliflozin, and empagliflozin lower blood glucose by reducing the renal threshold for reabsorption of glucose, coupled with reabsorption of sodium leading to daily urinary excretion of about 200 calories. These agents alone or taken with any of the agents above should not cause hypoglycemia. However, they can lead to dehydration if fasting precludes the intake of water as well as food.
The primary concern during fasting is hypoglycemia when diabetes regimens involve insulin or insulin secretagogues, most commonly sulfonylureas. Long-acting basal insulin should not require adjustment during fasting if the dose is not excessive. The amount and timing of short-acting analogues administered before meals should be adjusted to the timing of meals, and doses should be adjusted proportionally to the anticipated carbohydrate intake. Premixed insulins such as intermediate-acting (protamine suspension) insulin and a short-acting insulin in 70/30, 75/25, or 50/50 ratios should be avoided. They do not lend themselves to changes in timing, and the short-acting component is fixed and cannot be changed for varied intake without changing the intermediate-acting portion, which functions as the basal insulin.
Sulfonylurea doses can be reduced or the larger dose moved to before the evening meal, but these agents still pose a risk of hypoglycemia during fasting hours. And as Drs. A.V. and Zagar state, glimepiride, glipizide, and gliclazide are the only agents in the class that should be considered; glyburide (ie, glibenclamide) poses too great a risk of hypoglycemia. On the other hand, the short-acting secretagogue nateglinide can be used safely before meals without much risk of hypoglycemia.
We have focused primarily on hypoglycemia risk. But if antihyperglycemic agents are halted completely or if the reduction is too severe, patients are at risk for hyperglycemia and even diabetic ketoacidosis. Careful monitoring of blood glucose levels during the fasting period is most important for patients taking agents that can cause hypoglycemia, and patients should be advised to break the fast if dangerously low glycemic levels occur. Similarly, if severe hyperglycemia or ketoacidosis develops, patients should be advised to seek medical advice promptly.
Islam is the second most common religion in the world, and there are 1.6 billion Muslims, many in areas where diabetes is prevalent. Each year observant Muslims fast during the daylight hours for the holy month of Ramadan. It is estimated that 50 million diabetic people fast between dawn and sundown during Ramadan, and Muslims are not the only group of patients who fast for religious or other reasons. It is important for healthcare providers to guide patients with diabetes in avoiding problems related to prolonged fasting.
In this issue of the Cleveland Clinic Journal of Medicine, Drs. A.V. and Zagar address management of diabetes specifically relating to Ramadan fasting, with considerations that also apply to other diabetic patients who fast for religious or for medical reasons.
Fortunately, we now have antihyperglycemic agents that are unlikely to cause hypoglycemia if used alone or in combination, as long as the regimen does not include insulin or a sulfonylurea. These include:
- Metformin and thiazolidinediones (pioglitazone and rosiglitazone), which improve insulin sensitivity
- Glucagon-like peptide 1 (GLP-1) agonists (exenatide, liraglutide, dulaglutide, and albaglutide), which facilitate insulin release in a glucose-dependent fashion
- Dipeptidyl peptidase 4 inhibitors (sitagliptin, saxagliptin, alogliptin, and linagliptin), which augment endogenous incretin hormones, primarily GLP-1, and also facilitate insulin production in a glucose-dependent fashion
- Alpha glucosidase inhibitors (acarbose and miglitol), which slow carbohydrate absorption.
Introduced in recent years, the sodium-glucose cotransporter 2 (SGLT-2) inhibitors canagliflozin, dapagliflozin, and empagliflozin lower blood glucose by reducing the renal threshold for reabsorption of glucose, coupled with reabsorption of sodium leading to daily urinary excretion of about 200 calories. These agents alone or taken with any of the agents above should not cause hypoglycemia. However, they can lead to dehydration if fasting precludes the intake of water as well as food.
The primary concern during fasting is hypoglycemia when diabetes regimens involve insulin or insulin secretagogues, most commonly sulfonylureas. Long-acting basal insulin should not require adjustment during fasting if the dose is not excessive. The amount and timing of short-acting analogues administered before meals should be adjusted to the timing of meals, and doses should be adjusted proportionally to the anticipated carbohydrate intake. Premixed insulins such as intermediate-acting (protamine suspension) insulin and a short-acting insulin in 70/30, 75/25, or 50/50 ratios should be avoided. They do not lend themselves to changes in timing, and the short-acting component is fixed and cannot be changed for varied intake without changing the intermediate-acting portion, which functions as the basal insulin.
Sulfonylurea doses can be reduced or the larger dose moved to before the evening meal, but these agents still pose a risk of hypoglycemia during fasting hours. And as Drs. A.V. and Zagar state, glimepiride, glipizide, and gliclazide are the only agents in the class that should be considered; glyburide (ie, glibenclamide) poses too great a risk of hypoglycemia. On the other hand, the short-acting secretagogue nateglinide can be used safely before meals without much risk of hypoglycemia.
We have focused primarily on hypoglycemia risk. But if antihyperglycemic agents are halted completely or if the reduction is too severe, patients are at risk for hyperglycemia and even diabetic ketoacidosis. Careful monitoring of blood glucose levels during the fasting period is most important for patients taking agents that can cause hypoglycemia, and patients should be advised to break the fast if dangerously low glycemic levels occur. Similarly, if severe hyperglycemia or ketoacidosis develops, patients should be advised to seek medical advice promptly.

Progressive muscle weakness: More there than meets the eye
Our patient, a 56-year-old woman, presents with proximal muscle weakness in all four limbs. It started a few months ago and has gradually become severe, so that she now has difficulty rising from a seated position and has trouble opening jars. She has fallen several times. She says she has no muscle pain, difficulty swallowing, or difficulty breathing.
She sought medical attention at another hospital and was found to be hypothyroid, with a thyrotropin (thyroid-stimulating hormone [TSH]) level of 38 μU/mL (reference range 0.4–5.5), for which she was started on levothyroxine (Synthroid) 100 μg daily. She also had a low serum potassium level, for which potassium supplements and spironolactone (Aldactone) were started. She was taking furosemide (Lasix) 20 mg/day at the time.
Despite the thyroid replacement therapy, she continued to become weaker and had more falls. She also noticed a new, nonpainful rash on her lower abdomen.
Review of systems
- Night sweats
- Leg swelling
- Puffiness and discoloration around the eyes, with easy bruisability.
Medical history
- Diabetes mellitus
- Seizures in the 1970s
- Resection of a thymic tumor in 2003 (the exact pathology is unknown)
- Cirrhosis of unknown etiology
- No known history of hypertension
- No history of alcohol or intravenous drug use
- Quit smoking many years ago
- Coronary artery bypass surgery in 2003
- One sibling with myasthenia gravis.
Medications
- Levothyroxine
- Rosuvastatin (Crestor)
- Omeprazole (Prilosec)
- Spironolactone
- Furosemide
- Potassium chloride
- Metoprolol tartrate (Lopressor)
- Metformin (Glucophage)
- Ramipril (Altace).
Physical examination
She is hemodynamically stable and is not hypertensive. Her thyroid is not enlarged. Her lungs are clear to auscultation. Her heart sounds are normal, except for a nonradiating pansystolic murmur most audible at the apex.
Her abdomen is soft and is not distended. Her abdominal rash has a dermatomal distribution consistent with an L1 distribution, with vesicles over an erythematous base. Purpuric lesions are noted over her lower extremities.
Her leg strength is 3 on a scale of 5 on both sides; her arm strength is normal. Ankle and knee reflexes are absent bilaterally.
Initial laboratory analysis
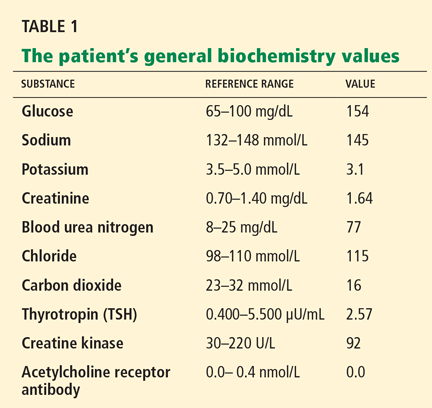
PROGRESSIVE MUSCLE WEAKNESS
1. What are possible causes of her muscle weakness?
- Myasthenia gravis
- Hypothyroidism
- Dermatomyositis-polymyositis
- Drug-induced myopathy
- Cushing syndrome
- All of the above
All of these are potential causes of muscle weakness.
Myasthenia gravis
Myasthenia gravis, an autoimmune disease, can affect people of all ages and either sex. It presents with muscle weakness and fatigability, which characteristically fluctuate during the day. Some patients present in crisis with respiratory failure, which may require ventilatory support.1,2
Myasthenia gravis is characterized by auto-antibodies against the postsynaptic membrane of the neuromuscular junction. Most patients have antibodies to the extracellular portion of the acetylcholine receptor; a small number of patients have antibodies against a muscle-specific tyrosine kinase that interacts with this receptor.
About 15% of patients with myasthenia gravis have a thymoma thought to be involved in the pathogenesis of the disease. Treatments include immune suppressive therapy and thymectomy.
Our patient has a history of thymic lesion resection, but her antibody workup for myasthenia gravis was negative.
Hypothyroidism
Hypothyroidism, the most common disorder of the thyroid gland, is especially prevalent in women.3 Its common symptoms include fatigue, exercise intolerance, muscle weakness, cramps, and stiffness.
Both the TSH and the free thyroxine (T4) level must be measured to diagnose hypothyroidism. This information can also help differentiate primary hypothyroidism (ie, due to a defect in the thyroid gland) from secondary hypothyroidism (ie, due to a defect in the pituitary gland). Elevated TSH with low free T4 levels indicates primary thyroid failure, whereas the combination of a normal or low TSH and a low free T4 usually indicates pituitary failure. Subclinical hypothyroidism is characterized by mildly to moderately elevated TSH, but total T4 and free T4 values are still within the reference range. Replacement therapy is with levothyroxine.3–6
Our patient has a history of hypothyroidism, which could explain her muscle weakness, but she is currently on replacement therapy, and her TSH level on admission was normal.
Dermatomyositis-polymyositis
Dermatomyositis-polymyositis is characterized by proximal muscle weakness, creatine kinase elevation, erythema on sun-exposed skin, heliotrope rash, and Gottron papules. It occurs mostly in women after the second decade of life. Some medications have been implicated in its pathogenesis, such as statins, fibrates, hydroxyurea, penicillamine, and omeprazole (Prilosec).7
In a middle-aged patient, this diagnosis should prompt a search for cancer, especially of the gastrointestinal system, breast, and lung.8 Cancer can arise up to 3 years after the diagnosis of dermatomyositis or polymyositis.
Antisynthetase antibody syndrome is suspected if the patient is positive for antisynthetase antibody and has the following manifestations: acute onset of disease, constitutional symptoms, interstitial lung disease, inflammatory arthritis, mechanic’s hands (thickened, cracked skin on the palmar aspect of the thumb and index finger), and Raynaud phenomenon.4,8,9
The diagnosis is made by a thorough clinical evaluation. Electromyography can show an inflammatory pattern of myopathy. The gold standard test for this diagnosis is muscle biopsy.
Our patient has a normal creatine kinase level, which excludes the diagnosis of dermatomyositis-polymyositis.
Statin-induced myopathy
Up to 10% of patients taking statins develop myalgia. Rhabdomyolysis, the extreme form of myopathy, is rare.
The exact mechanism of statin-induced myopathy remains unclear; mitochondrial dysfunction, cholesterol composition of cell membranes, and coenzyme Q10 deficiency have been proposed.
Risk factors for statin-induced myopathy include female sex, older age, higher doses of statins, a family history of statin-induced myopathy, and hypothyroidism. Drugs that increase the risk include fibric acid derivatives, macrolides, and amiodarone (Cordarone). If a statin and any of the above drugs are both required, certain statins—ie, pravastatin (Pravachol) and rosuvastatin—are recommended, since they are the statins least likely to cause rhabdomyolysis.5,7,10–12
The combination of fluvastatin (Lescol) and gemfibrozil (Lopid) has also been found to be safe.13 In a crossover study in 17 patients, no significant difference was seen in the area under the curve for plasma concentration over time, in the maximum plasma concentration, or in the time to maximum concentration with the combination vs with each drug alone.13
Our patient is taking a statin and has hypothyroidism, which increases the risk of statin-induced myopathy. However, her creatine kinase level is normal.
Cushing syndrome
Cushing syndrome (hypercortisolism) is one of the most challenging endocrine diseases to diagnose. Most of its clinical features overlap with those of common diseases, and some patients have an atypical clinical presentation with only isolated symptoms. Further, its presentation can be subtle, with weight gain, amenorrhea, muscle weakness, and easy bruisability. Acne, moon facies, plethora, abdominal striae, and purpura are other common signs. It is three to 10 times more common in women than in men.
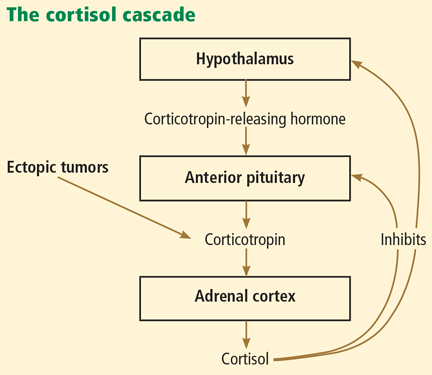
Cushing syndrome can be classified according to whether or not the excess cortisol secretion depends on corticotropin (formerly called adrenocorticotropic hormone or ACTH) (Figure 1). In corticotropin-dependent cases, the most common cause is pituitary adenoma. (When Cushing syndrome is due to excessive pituitary secretion of corticotropin, which in turn stimulates the adrenal glands to secrete excessive amounts of cortisol, it is called Cushing disease). Other causes of corticotropin-dependent Cushing syndrome are ectopic corticotropin-producing tumors such as carcinoid tumors or medullary thyroid cancers. Corticotropin-independent Cushing syndrome can be caused by adrenal adenomas, adrenal carcinoma, and bilateral primary micronodular or macronodular adrenocortical hyperplasia.14–17
However, the most common cause of Cushing syndrome is glucocorticoid therapy.
BACK TO OUR PATIENT: HER CONDITION DETERIORATES
Our patient’s physical condition deteriorates, she develops respiratory distress, and she is admitted to the medical intensive care unit. Her mental status also deteriorates, and she becomes lethargic and unresponsive.
She is intubated to protect her airway. After this, she develops hypotension that does not respond to fluid resuscitation and that requires vasopressors. Her condition continues to worsen as she develops acute kidney injury and disseminated intravascular coagulation. Her vesicular rash becomes more widespread, involving the entire trunk.
A workup for sepsis is initiated, but her initial blood and urine cultures are negative. Chest radiography does not reveal any infiltrates. No other source of an infection is found.
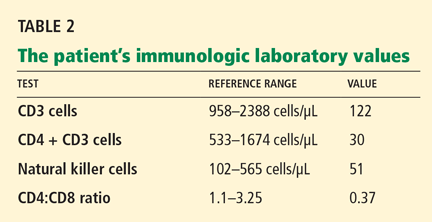
Varicella zoster is isolated on viral culture of a specimen obtained from the rash, and a polymerase chain reaction test of her blood shows cytomegalovirus DNA (64,092 copies per mL). Immune suppression is suspected, so a CD4 count is ordered (Table 2). Serologic tests for human immunodeficiency virus are negative.
What could have caused our patient to have muscle weakness in addition to disseminated zoster with cytomegalovirus viremia?
The diagnosis here is Cushing syndrome.
HOW TO TEST FOR CUSHING SYNDROME
2. In any practice, you may meet many perimenopausal women who have complaints of weight gain, amenorrhea, and acne. How can you determine if this is Cushing syndrome? What are the screening tests?
- 24-Hour urinary cortisol excretion
- A late-night salivary cortisol level
- A low-dose dexamethasone suppression test
- All of the above
- None of the above
Any of the tests listed here can be used to determine whether this is truly Cushing syndrome.
24-Hour urinary cortisol excretion has a reference range of 20 to 100 μg/24 hours. However, results may be falsely high in patients who are depressed or who abuse alcohol.
The late-night salivary cortisol level is another useful test.14,16,18 Patients with Cushing syndrome are found to have high late-night salivary cortisol levels as compared with normal people, indicating the loss of natural circadian rhythm.14,16,18
The low-dose dexamethasone suppression test, as first described by Liddle in 1960,19 involved giving dexamethasone 0.5 mg by mouth every 6 hours for 48 hours and measuring the serum cortisol level 6 hours after the last dose. In healthy people, this low dose of dexamethasone suppresses the production of corticotropin by the pituitary gland and in turn the production of cortisol, but in patients with Cushing syndrome the cortisol level remains high. An alternative is the overnight 1-mg dexamethasone suppression test—ie, giving 1 mg of dexamethasone at 11:00 pm and measuring the serum cortisol level early the next morning. Failure of the cortisol level to drop to less than 1.8 μg/dL suggests Cushing syndrome and warrants a complete evaluation for it.
Confirmatory testing is sometimes needed if patients have mild abnormalities in their screening tests. A combination low-dose dexamethasone suppression test and corticotropin-releasing hormone test can be used to differentiate Cushing syndrome from pseudo-Cushing syndrome. This is performed by giving dexamethasone orally 0.5 mg every 6 hours for 48 hours and then giving ovine-sequence corticotropin-releasing hormone 1 μg/kg intravenously 2 hours after the last dose of dexamethasone. The plasma cortisol value 15 minutes after the dose of corticotropin-releasing hormone is greater than 1.4 μg/dL (38 nmol/L) in patients with Cushing syndrome but remains low in patients with pseudo-Cushing syndrome.

Is this corticotropin-dependent or corticotropin-independent?
Once Cushing syndrome is diagnosed by one of the screening methods described above, the source of the excess glucocorticoids needs to be determined. Measuring the serum corticotropin level early in the morning would be the next step.
A low corticotropin level (< 10 pg/mL) indicates a corticotropin-independent source, most likely in the adrenal glands. Hence, computed tomography or magnetic resonance imaging (MRI) of the adrenal glands is warranted. Of note: adrenal incidentalomas are quite common, present in 5% of the general population, and a lesion on the adrenal gland does not prove that the patient has primary adrenal disease.16,20
IS THE EXCESS CORTICOTROPIN FROM A PITUITARY OR AN ECTOPIC SOURCE?
3. If the corticotropin level is elevated, how can you determine if it is from the pituitary or from an ectopic source?
- MRI of the pituitary gland
- High-dose dexamethasone suppression test
- Corticotropin-releasing hormone stimulation test
- Bilateral inferior petrosal sinus sampling
If the corticotropin level is high (> 10 pg/mL), it is of paramount importance to determine whether the corticotropin comes from the pituitary gland or from an ectopic source.
MRI of the pituitary gland should be done in patients with suspected corticotropin-dependent Cushing syndrome. However, MRI may be negative in 50% of patients with Cushing disease, and it should therefore not be used for screening. In addition, 10% of the population may have pituitary incidentalomas on MRI.
Most cases of corticotropin-dependent Cushing syndrome are caused by microadenomas (smaller than 1 cm), while a few cases are caused by macroadenomas (larger than 1 cm). If a microadenoma is found on MRI, further testing with bilateral inferior petrosal sinus sampling is recommended (described below); if a macroadenoma is found, then no further testing is required.21,22 In fact, patients who have biochemical findings compatible with Cushing disease (ie, due to an overactive pituitary) and who have an adenoma larger than 6 mm do not require further evaluation.23
A high-dose dexamethasone suppression test involves giving 8 mg of dexamethasone in the evening and measuring the cortisol level the next morning. If the cortisol level declines to 50% of the baseline level after this dose, this suggests a pituitary cause.
Corticotropin-releasing hormone stimulation testing. In most cases of pituitary tumors and a few cases of ectopic corticotropin-secreting tumors, giving corticotropin-releasing hormone leads to an increase in serum corticotropin and cortisol levels. In contrast, these levels do not respond to corticotropin-releasing hormone stimulation if the problem is in the adrenal gland. The test is performed by giving 1 μg/kg or 100 μg synthetic or human corticotropin-releasing hormone. A 35% to 50% increase above baseline in corticotropin suggests a pituitary cause.23
Bilateral inferior petrosal sinus sampling can be used to confirm a pituitary source, as it is the gold standard for differentiating ectopic from pituitary corticotropin production. Once this is confirmed, a neurosurgical consult is warranted.16,18
This procedure is usually done by advancing a sheath from the femoral vein to reach the inferior petrosal sinuses. Blood samples are obtained from both the inferior petrosal sinuses and from a peripheral vein to measure corticotropin levels before and after giving corticotropin-releasing hormone (1 μg/kg). Before corticotropin-releasing hormone is given, a gradient of central-peripheral corticotropin levels of 2.0 or greater indicates a pituitary source. With ectopic corticotropin production, the corticotropin gradient is usually less than 1.5. Corticotropin-releasing hormone is given to increase the sensitivity: after it is given, a gradient of 3.0 or greater is considered indicative of Cushing disease.24
If the corticotropin level is elevated and the above tests indicate ectopic production, the source should be sought. The most common site of ectopic corticotropin production is the chest. Common causes are bronchial, thymic, and pancreatic carcinoid tumors. Other causes are small-cell lung cancer, medullary cell cancer, and pheochromocytoma.15,18,25
BACK TO OUR PATIENT
Our patient’s further laboratory results are listed in Table 3.
She has elevated 24-hour urinary cortisol excretion, consistent with Cushing syndrome. Her corticotropin level is elevated, which rules out an adrenal cause. Her 5-HIAA (a serotonin breakdown product) and calcitonin levels are also elevated, suggesting either medullary thyroid cancer or a carcinoid tumor. She also has a mild elevation of dehydroepiandrosterone sulfate, which is consistent with corticotropin-dependent Cushing syndrome.
Our patient’s elevated levels of cortisol were the cause of her muscle weakness and severe immune deficiency, which in turn led to cytomegalovirus viremia and sepsis. Cushing syndrome usually causes hypertension, especially in cases of ectopic corticotropin production. However, our patient was normotensive on admission and then developed cytomegalovirus sepsis, which led to hypotension and shock.
Immune suppression is a well-known effect of glucocorticoids.26–28 Kronfol et al28 found that CD4 and CD8 counts and the CD4-to-CD8 ratio were low in patients with Cushing syndrome, and natural killer cell activity was suppressed. Opportunistic infections have been described in patients with Cushing syndrome.26,27,29
MANAGEMENT OF CUSHING SYNDROME
Management of Cushing syndrome should be tailored after determining its source.
A neurosurgical consultation is warranted in cases of pituitary adenoma, with surgical resection of the adrenal source or ectopic tumor if feasible.25
Medical management is recommended if surgical resection is not possible.30,31 Several drugs can be used to inhibit cortisol synthesis in this situation.30,32
Adrenal-acting agents
Aminoglutethimide (Cytadren) acts by blocking the conversion of cholesterol to pregnenolone, a precursor of cortisol. The dosage is 250 mg twice or three times a day. This drug is no longer available in the United States.
Ketoconazole (Nizoral) inhibits side-chain cleavage, 11-beta hydroxylase, and 17-alpha hydroxylase, thus inhibiting cortisol synthesis; it also inhibits corticotropin secretion. The dosage is 200 to 400 mg three times a day.
Metyrapone (Metopirone) blocks 11-beta-hydroxylation of deoxycortisol, the reaction that produces cortisol. The dosage is 500 to 750 mg three times a day. This drug can be obtained only from the manufacturer and only on a named-patient basis.
Etomidate (Amidate), an anesthetic drug, also blocks 11-beta-hydroxylation of deoxycortisol. It is given intravenously at a rate of 0.3 mg/kg per hour.
Centrally acting agents
Cabergoline (Dostinex). It is believed that corticotropin-producing pituitary tumors express D2 receptors. Cabergoline is a dopamine agonist that has been used in patients with Cushing disease. The dosage is 0.5 to 7 mg/week.
Pasireotide is still investigational. It is a somatostatin receptor agonist given subcutaneously for 15 consecutive days to patients with Cushing disease.
Glucocorticoid receptor antagonist
Mifepristone (Mifeprex) is a progesterone receptor and glucocorticoid II receptor antagonist that is being investigated in the treatment of persistent or recurrent Cushing disease. It is not yet approved by the US Food and Drug Administration for this indication.
BACK TO OUR PATIENT
The patient was too ill to undergo additional imaging, including octreotide scanning to identify an ectopic corticotropin-secreting tumor. She was medically treated with intravenous etomidate to reduce her cortisol level.30,31
Unfortunately, our patient died of multiorgan failure. The exact site of her ectopic corticotropin-producing tumor was never identified, and no autopsy was done.
- Meriggioli MN. Myasthenia gravis with anti-acetylcholine receptor antibodies. Front Neurol Neurosci 2009; 26:94–108.
- Gilhus NE. Autoimmune myasthenia gravis. Expert Rev Neurother 2009; 9:351–358.
- Heitman B, Irizarry A. Hypothyroidism: common complaints, perplexing diagnosis. Nurse Pract 1995; 20:54–60.
- Brick JE, Brick JF, Elnicki DM. Musculoskeletal disorders. When are they caused by hormone imbalance? Postgrad Med 1991; 90:129–132,135–136.
- Bar SL, Holmes DT, Frohlich J. Asymptomatic hypothyroidism and statin-induced myopathy. Can Fam Physician 2007; 53:428–431.
- McDermott MT. In the clinic. Hypothyroidism. Ann Intern Med 2009; 151:ITC61.
- Klopstock T. Drug-induced myopathies. Curr Opin Neurol 2008; 21:590–595.
- Dimachkie MM, Barohn RJ. Idiopathic inflammatory myopathies. Front Neurol Neurosci 2009; 26:126–146.
- Joseph A, Brasington R, Kahl L, Ranganathan P, Cheng TP, Atkinson J. Immunologic rheumatic disorders. J Allergy Clin Immunol 2010; 125(suppl 2):S204–S215.
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med 2009; 150:858–868.
- Kiernan TJ, Rochford M, McDermott JH. Simvastatin induced rhabdomyolysis and an important clinical link with hypothyroidism. Int J Cardiol 2007; 119:374–376.
- Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA 2003; 289:1681–1690.
- Spence JD, Munoz CE, Hendricks L, Latchinian L, Khouri HE. Pharmacokinetics of the combination of fluvastatin and gemfibrozil. Am J Cardiol 1995; 76:80A–83A.
- Boscaro M, Arnaldi G. Approach to the patient with possible Cushing’s syndrome. J Clin Endocrinol Metab 2009; 94:3121–3131.
- Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, Nieman LK. Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab 2005; 90:4955–4962.
- Pecori Giraldi F. Recent challenges in the diagnosis of Cushing’s syndrome. Horm Res 2009; 71(suppl 1):123–127.
- von Mach MA, Kann P, Piepkorn B, Bruder S, Beyer J. [Cushing’s syndrome caused by paraneoplastic ACTH secretion 11 years after occurrence of a medullary thyroid carcinoma]. Dtsch Med Wochenschr 2002; 127:850–852.
- Beauregard C, Dickstein G, Lacroix A. Classic and recent etiologies of Cushing’s syndrome: diagnosis and therapy. Treat Endocrinol 2002; 1:79–94.
- Liddle GW. Tests of pituitary-adrenal suppressibility in the diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab 1960; 20:1539–1560.
- Louiset E, Gobet F, Libé R, et al. ACTH-independent Cushing’s syndrome with bilateral micronodular adrenal hyperplasia and ectopic adrenocortical adenoma. J Clin Endocrinol Metab 2010; 95:18–24.
- Andrioli M, Pecori Giraldi F, De Martin M, Cattaneo A, Carzaniga C, Cavagnini F. Differential diagnosis of ACTH-dependent hypercortisolism: imaging versus laboratory. Pituitary 2009; 12:294–296.
- Sahdev A, Reznek RH, Evanson J, Grossman AB. Imaging in Cushing’s syndrome. Arq Bras Endocrinol Metabol 2007; 51:1319–1328.
- Arnaldi G, Angeli A, Atkinson AB, et al. Diagnosis and complications of Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab 2003; 88:5593–5602.
- Lad SP, Patil CG, Laws ER, Katznelson L. The role of inferior petrosal sinus sampling in the diagnostic localization of Cushing’s disease. Neurosurg Focus 2007; 23:E2.
- Bhansali A, Walia R, Rana SS, et al. Ectopic Cushing’s syndrome: experience from a tertiary care centre. Indian J Med Res 2009; 129:33–41.
- Arlt A, Harbeck B, Anlauf M, et al. Fatal Pneumocystis jirovecii pneumonia in a case of ectopic Cushing’s syndrome due to neuroendocrine carcinoma of the kidney. Exp Clin Endocrinol Diabetes 2008; 116:515–519.
- Graham BS, Tucker WS. Opportunistic infections in endogenous Cushing’s syndrome. Ann Intern Med 1984; 101:334–338.
- Kronfol Z, Starkman M, Schteingart DE, Singh V, Zhang Q, Hill E. Immune regulation in Cushing’s syndrome: relationship to hypothalamic-pituitary-adrenal axis hormones. Psychoneuroendocrinology 1996; 21:599–608.
- Sepkowitz KA. Opportunistic infections in patients with and patients without acquired immunodeficiency syndrome. Clin Infect Dis 2002; 34:1098–1107.
- Schteingart DE. Drugs in the medical treatment of Cushing’s syndrome. Expert Opin Emerg Drugs 2009; 14:661–671.
- Shalet S, Mukherjee A. Pharmacological treatment of hypercortisolism. Curr Opin Endocrinol Diabetes Obes 2008; 15:234–238.
- Arnaldi G, Boscaro M. Pasireotide for the treatment of Cushing’s disease. Expert Opin Investig Drugs 2010; 19:889–898.
Our patient, a 56-year-old woman, presents with proximal muscle weakness in all four limbs. It started a few months ago and has gradually become severe, so that she now has difficulty rising from a seated position and has trouble opening jars. She has fallen several times. She says she has no muscle pain, difficulty swallowing, or difficulty breathing.
She sought medical attention at another hospital and was found to be hypothyroid, with a thyrotropin (thyroid-stimulating hormone [TSH]) level of 38 μU/mL (reference range 0.4–5.5), for which she was started on levothyroxine (Synthroid) 100 μg daily. She also had a low serum potassium level, for which potassium supplements and spironolactone (Aldactone) were started. She was taking furosemide (Lasix) 20 mg/day at the time.
Despite the thyroid replacement therapy, she continued to become weaker and had more falls. She also noticed a new, nonpainful rash on her lower abdomen.
Review of systems
- Night sweats
- Leg swelling
- Puffiness and discoloration around the eyes, with easy bruisability.
Medical history
- Diabetes mellitus
- Seizures in the 1970s
- Resection of a thymic tumor in 2003 (the exact pathology is unknown)
- Cirrhosis of unknown etiology
- No known history of hypertension
- No history of alcohol or intravenous drug use
- Quit smoking many years ago
- Coronary artery bypass surgery in 2003
- One sibling with myasthenia gravis.
Medications
- Levothyroxine
- Rosuvastatin (Crestor)
- Omeprazole (Prilosec)
- Spironolactone
- Furosemide
- Potassium chloride
- Metoprolol tartrate (Lopressor)
- Metformin (Glucophage)
- Ramipril (Altace).
Physical examination
She is hemodynamically stable and is not hypertensive. Her thyroid is not enlarged. Her lungs are clear to auscultation. Her heart sounds are normal, except for a nonradiating pansystolic murmur most audible at the apex.
Her abdomen is soft and is not distended. Her abdominal rash has a dermatomal distribution consistent with an L1 distribution, with vesicles over an erythematous base. Purpuric lesions are noted over her lower extremities.
Her leg strength is 3 on a scale of 5 on both sides; her arm strength is normal. Ankle and knee reflexes are absent bilaterally.
Initial laboratory analysis
PROGRESSIVE MUSCLE WEAKNESS
1. What are possible causes of her muscle weakness?
- Myasthenia gravis
- Hypothyroidism
- Dermatomyositis-polymyositis
- Drug-induced myopathy
- Cushing syndrome
- All of the above
All of these are potential causes of muscle weakness.
Myasthenia gravis
Myasthenia gravis, an autoimmune disease, can affect people of all ages and either sex. It presents with muscle weakness and fatigability, which characteristically fluctuate during the day. Some patients present in crisis with respiratory failure, which may require ventilatory support.1,2
Myasthenia gravis is characterized by auto-antibodies against the postsynaptic membrane of the neuromuscular junction. Most patients have antibodies to the extracellular portion of the acetylcholine receptor; a small number of patients have antibodies against a muscle-specific tyrosine kinase that interacts with this receptor.
About 15% of patients with myasthenia gravis have a thymoma thought to be involved in the pathogenesis of the disease. Treatments include immune suppressive therapy and thymectomy.
Our patient has a history of thymic lesion resection, but her antibody workup for myasthenia gravis was negative.
Hypothyroidism
Hypothyroidism, the most common disorder of the thyroid gland, is especially prevalent in women.3 Its common symptoms include fatigue, exercise intolerance, muscle weakness, cramps, and stiffness.
Both the TSH and the free thyroxine (T4) level must be measured to diagnose hypothyroidism. This information can also help differentiate primary hypothyroidism (ie, due to a defect in the thyroid gland) from secondary hypothyroidism (ie, due to a defect in the pituitary gland). Elevated TSH with low free T4 levels indicates primary thyroid failure, whereas the combination of a normal or low TSH and a low free T4 usually indicates pituitary failure. Subclinical hypothyroidism is characterized by mildly to moderately elevated TSH, but total T4 and free T4 values are still within the reference range. Replacement therapy is with levothyroxine.3–6
Our patient has a history of hypothyroidism, which could explain her muscle weakness, but she is currently on replacement therapy, and her TSH level on admission was normal.
Dermatomyositis-polymyositis
Dermatomyositis-polymyositis is characterized by proximal muscle weakness, creatine kinase elevation, erythema on sun-exposed skin, heliotrope rash, and Gottron papules. It occurs mostly in women after the second decade of life. Some medications have been implicated in its pathogenesis, such as statins, fibrates, hydroxyurea, penicillamine, and omeprazole (Prilosec).7
In a middle-aged patient, this diagnosis should prompt a search for cancer, especially of the gastrointestinal system, breast, and lung.8 Cancer can arise up to 3 years after the diagnosis of dermatomyositis or polymyositis.
Antisynthetase antibody syndrome is suspected if the patient is positive for antisynthetase antibody and has the following manifestations: acute onset of disease, constitutional symptoms, interstitial lung disease, inflammatory arthritis, mechanic’s hands (thickened, cracked skin on the palmar aspect of the thumb and index finger), and Raynaud phenomenon.4,8,9
The diagnosis is made by a thorough clinical evaluation. Electromyography can show an inflammatory pattern of myopathy. The gold standard test for this diagnosis is muscle biopsy.
Our patient has a normal creatine kinase level, which excludes the diagnosis of dermatomyositis-polymyositis.
Statin-induced myopathy
Up to 10% of patients taking statins develop myalgia. Rhabdomyolysis, the extreme form of myopathy, is rare.
The exact mechanism of statin-induced myopathy remains unclear; mitochondrial dysfunction, cholesterol composition of cell membranes, and coenzyme Q10 deficiency have been proposed.
Risk factors for statin-induced myopathy include female sex, older age, higher doses of statins, a family history of statin-induced myopathy, and hypothyroidism. Drugs that increase the risk include fibric acid derivatives, macrolides, and amiodarone (Cordarone). If a statin and any of the above drugs are both required, certain statins—ie, pravastatin (Pravachol) and rosuvastatin—are recommended, since they are the statins least likely to cause rhabdomyolysis.5,7,10–12
The combination of fluvastatin (Lescol) and gemfibrozil (Lopid) has also been found to be safe.13 In a crossover study in 17 patients, no significant difference was seen in the area under the curve for plasma concentration over time, in the maximum plasma concentration, or in the time to maximum concentration with the combination vs with each drug alone.13
Our patient is taking a statin and has hypothyroidism, which increases the risk of statin-induced myopathy. However, her creatine kinase level is normal.
Cushing syndrome
Cushing syndrome (hypercortisolism) is one of the most challenging endocrine diseases to diagnose. Most of its clinical features overlap with those of common diseases, and some patients have an atypical clinical presentation with only isolated symptoms. Further, its presentation can be subtle, with weight gain, amenorrhea, muscle weakness, and easy bruisability. Acne, moon facies, plethora, abdominal striae, and purpura are other common signs. It is three to 10 times more common in women than in men.
Cushing syndrome can be classified according to whether or not the excess cortisol secretion depends on corticotropin (formerly called adrenocorticotropic hormone or ACTH) (Figure 1). In corticotropin-dependent cases, the most common cause is pituitary adenoma. (When Cushing syndrome is due to excessive pituitary secretion of corticotropin, which in turn stimulates the adrenal glands to secrete excessive amounts of cortisol, it is called Cushing disease). Other causes of corticotropin-dependent Cushing syndrome are ectopic corticotropin-producing tumors such as carcinoid tumors or medullary thyroid cancers. Corticotropin-independent Cushing syndrome can be caused by adrenal adenomas, adrenal carcinoma, and bilateral primary micronodular or macronodular adrenocortical hyperplasia.14–17
However, the most common cause of Cushing syndrome is glucocorticoid therapy.
BACK TO OUR PATIENT: HER CONDITION DETERIORATES
Our patient’s physical condition deteriorates, she develops respiratory distress, and she is admitted to the medical intensive care unit. Her mental status also deteriorates, and she becomes lethargic and unresponsive.
She is intubated to protect her airway. After this, she develops hypotension that does not respond to fluid resuscitation and that requires vasopressors. Her condition continues to worsen as she develops acute kidney injury and disseminated intravascular coagulation. Her vesicular rash becomes more widespread, involving the entire trunk.
A workup for sepsis is initiated, but her initial blood and urine cultures are negative. Chest radiography does not reveal any infiltrates. No other source of an infection is found.
Varicella zoster is isolated on viral culture of a specimen obtained from the rash, and a polymerase chain reaction test of her blood shows cytomegalovirus DNA (64,092 copies per mL). Immune suppression is suspected, so a CD4 count is ordered (Table 2). Serologic tests for human immunodeficiency virus are negative.
What could have caused our patient to have muscle weakness in addition to disseminated zoster with cytomegalovirus viremia?
The diagnosis here is Cushing syndrome.
HOW TO TEST FOR CUSHING SYNDROME
2. In any practice, you may meet many perimenopausal women who have complaints of weight gain, amenorrhea, and acne. How can you determine if this is Cushing syndrome? What are the screening tests?
- 24-Hour urinary cortisol excretion
- A late-night salivary cortisol level
- A low-dose dexamethasone suppression test
- All of the above
- None of the above
Any of the tests listed here can be used to determine whether this is truly Cushing syndrome.
24-Hour urinary cortisol excretion has a reference range of 20 to 100 μg/24 hours. However, results may be falsely high in patients who are depressed or who abuse alcohol.
The late-night salivary cortisol level is another useful test.14,16,18 Patients with Cushing syndrome are found to have high late-night salivary cortisol levels as compared with normal people, indicating the loss of natural circadian rhythm.14,16,18
The low-dose dexamethasone suppression test, as first described by Liddle in 1960,19 involved giving dexamethasone 0.5 mg by mouth every 6 hours for 48 hours and measuring the serum cortisol level 6 hours after the last dose. In healthy people, this low dose of dexamethasone suppresses the production of corticotropin by the pituitary gland and in turn the production of cortisol, but in patients with Cushing syndrome the cortisol level remains high. An alternative is the overnight 1-mg dexamethasone suppression test—ie, giving 1 mg of dexamethasone at 11:00 pm and measuring the serum cortisol level early the next morning. Failure of the cortisol level to drop to less than 1.8 μg/dL suggests Cushing syndrome and warrants a complete evaluation for it.
Confirmatory testing is sometimes needed if patients have mild abnormalities in their screening tests. A combination low-dose dexamethasone suppression test and corticotropin-releasing hormone test can be used to differentiate Cushing syndrome from pseudo-Cushing syndrome. This is performed by giving dexamethasone orally 0.5 mg every 6 hours for 48 hours and then giving ovine-sequence corticotropin-releasing hormone 1 μg/kg intravenously 2 hours after the last dose of dexamethasone. The plasma cortisol value 15 minutes after the dose of corticotropin-releasing hormone is greater than 1.4 μg/dL (38 nmol/L) in patients with Cushing syndrome but remains low in patients with pseudo-Cushing syndrome.
Is this corticotropin-dependent or corticotropin-independent?
Once Cushing syndrome is diagnosed by one of the screening methods described above, the source of the excess glucocorticoids needs to be determined. Measuring the serum corticotropin level early in the morning would be the next step.
A low corticotropin level (< 10 pg/mL) indicates a corticotropin-independent source, most likely in the adrenal glands. Hence, computed tomography or magnetic resonance imaging (MRI) of the adrenal glands is warranted. Of note: adrenal incidentalomas are quite common, present in 5% of the general population, and a lesion on the adrenal gland does not prove that the patient has primary adrenal disease.16,20
IS THE EXCESS CORTICOTROPIN FROM A PITUITARY OR AN ECTOPIC SOURCE?
3. If the corticotropin level is elevated, how can you determine if it is from the pituitary or from an ectopic source?
- MRI of the pituitary gland
- High-dose dexamethasone suppression test
- Corticotropin-releasing hormone stimulation test
- Bilateral inferior petrosal sinus sampling
If the corticotropin level is high (> 10 pg/mL), it is of paramount importance to determine whether the corticotropin comes from the pituitary gland or from an ectopic source.
MRI of the pituitary gland should be done in patients with suspected corticotropin-dependent Cushing syndrome. However, MRI may be negative in 50% of patients with Cushing disease, and it should therefore not be used for screening. In addition, 10% of the population may have pituitary incidentalomas on MRI.
Most cases of corticotropin-dependent Cushing syndrome are caused by microadenomas (smaller than 1 cm), while a few cases are caused by macroadenomas (larger than 1 cm). If a microadenoma is found on MRI, further testing with bilateral inferior petrosal sinus sampling is recommended (described below); if a macroadenoma is found, then no further testing is required.21,22 In fact, patients who have biochemical findings compatible with Cushing disease (ie, due to an overactive pituitary) and who have an adenoma larger than 6 mm do not require further evaluation.23
A high-dose dexamethasone suppression test involves giving 8 mg of dexamethasone in the evening and measuring the cortisol level the next morning. If the cortisol level declines to 50% of the baseline level after this dose, this suggests a pituitary cause.
Corticotropin-releasing hormone stimulation testing. In most cases of pituitary tumors and a few cases of ectopic corticotropin-secreting tumors, giving corticotropin-releasing hormone leads to an increase in serum corticotropin and cortisol levels. In contrast, these levels do not respond to corticotropin-releasing hormone stimulation if the problem is in the adrenal gland. The test is performed by giving 1 μg/kg or 100 μg synthetic or human corticotropin-releasing hormone. A 35% to 50% increase above baseline in corticotropin suggests a pituitary cause.23
Bilateral inferior petrosal sinus sampling can be used to confirm a pituitary source, as it is the gold standard for differentiating ectopic from pituitary corticotropin production. Once this is confirmed, a neurosurgical consult is warranted.16,18
This procedure is usually done by advancing a sheath from the femoral vein to reach the inferior petrosal sinuses. Blood samples are obtained from both the inferior petrosal sinuses and from a peripheral vein to measure corticotropin levels before and after giving corticotropin-releasing hormone (1 μg/kg). Before corticotropin-releasing hormone is given, a gradient of central-peripheral corticotropin levels of 2.0 or greater indicates a pituitary source. With ectopic corticotropin production, the corticotropin gradient is usually less than 1.5. Corticotropin-releasing hormone is given to increase the sensitivity: after it is given, a gradient of 3.0 or greater is considered indicative of Cushing disease.24
If the corticotropin level is elevated and the above tests indicate ectopic production, the source should be sought. The most common site of ectopic corticotropin production is the chest. Common causes are bronchial, thymic, and pancreatic carcinoid tumors. Other causes are small-cell lung cancer, medullary cell cancer, and pheochromocytoma.15,18,25
BACK TO OUR PATIENT
Our patient’s further laboratory results are listed in Table 3.
She has elevated 24-hour urinary cortisol excretion, consistent with Cushing syndrome. Her corticotropin level is elevated, which rules out an adrenal cause. Her 5-HIAA (a serotonin breakdown product) and calcitonin levels are also elevated, suggesting either medullary thyroid cancer or a carcinoid tumor. She also has a mild elevation of dehydroepiandrosterone sulfate, which is consistent with corticotropin-dependent Cushing syndrome.
Our patient’s elevated levels of cortisol were the cause of her muscle weakness and severe immune deficiency, which in turn led to cytomegalovirus viremia and sepsis. Cushing syndrome usually causes hypertension, especially in cases of ectopic corticotropin production. However, our patient was normotensive on admission and then developed cytomegalovirus sepsis, which led to hypotension and shock.
Immune suppression is a well-known effect of glucocorticoids.26–28 Kronfol et al28 found that CD4 and CD8 counts and the CD4-to-CD8 ratio were low in patients with Cushing syndrome, and natural killer cell activity was suppressed. Opportunistic infections have been described in patients with Cushing syndrome.26,27,29
MANAGEMENT OF CUSHING SYNDROME
Management of Cushing syndrome should be tailored after determining its source.
A neurosurgical consultation is warranted in cases of pituitary adenoma, with surgical resection of the adrenal source or ectopic tumor if feasible.25
Medical management is recommended if surgical resection is not possible.30,31 Several drugs can be used to inhibit cortisol synthesis in this situation.30,32
Adrenal-acting agents
Aminoglutethimide (Cytadren) acts by blocking the conversion of cholesterol to pregnenolone, a precursor of cortisol. The dosage is 250 mg twice or three times a day. This drug is no longer available in the United States.
Ketoconazole (Nizoral) inhibits side-chain cleavage, 11-beta hydroxylase, and 17-alpha hydroxylase, thus inhibiting cortisol synthesis; it also inhibits corticotropin secretion. The dosage is 200 to 400 mg three times a day.
Metyrapone (Metopirone) blocks 11-beta-hydroxylation of deoxycortisol, the reaction that produces cortisol. The dosage is 500 to 750 mg three times a day. This drug can be obtained only from the manufacturer and only on a named-patient basis.
Etomidate (Amidate), an anesthetic drug, also blocks 11-beta-hydroxylation of deoxycortisol. It is given intravenously at a rate of 0.3 mg/kg per hour.
Centrally acting agents
Cabergoline (Dostinex). It is believed that corticotropin-producing pituitary tumors express D2 receptors. Cabergoline is a dopamine agonist that has been used in patients with Cushing disease. The dosage is 0.5 to 7 mg/week.
Pasireotide is still investigational. It is a somatostatin receptor agonist given subcutaneously for 15 consecutive days to patients with Cushing disease.
Glucocorticoid receptor antagonist
Mifepristone (Mifeprex) is a progesterone receptor and glucocorticoid II receptor antagonist that is being investigated in the treatment of persistent or recurrent Cushing disease. It is not yet approved by the US Food and Drug Administration for this indication.
BACK TO OUR PATIENT
The patient was too ill to undergo additional imaging, including octreotide scanning to identify an ectopic corticotropin-secreting tumor. She was medically treated with intravenous etomidate to reduce her cortisol level.30,31
Unfortunately, our patient died of multiorgan failure. The exact site of her ectopic corticotropin-producing tumor was never identified, and no autopsy was done.
Our patient, a 56-year-old woman, presents with proximal muscle weakness in all four limbs. It started a few months ago and has gradually become severe, so that she now has difficulty rising from a seated position and has trouble opening jars. She has fallen several times. She says she has no muscle pain, difficulty swallowing, or difficulty breathing.
She sought medical attention at another hospital and was found to be hypothyroid, with a thyrotropin (thyroid-stimulating hormone [TSH]) level of 38 μU/mL (reference range 0.4–5.5), for which she was started on levothyroxine (Synthroid) 100 μg daily. She also had a low serum potassium level, for which potassium supplements and spironolactone (Aldactone) were started. She was taking furosemide (Lasix) 20 mg/day at the time.
Despite the thyroid replacement therapy, she continued to become weaker and had more falls. She also noticed a new, nonpainful rash on her lower abdomen.
Review of systems
- Night sweats
- Leg swelling
- Puffiness and discoloration around the eyes, with easy bruisability.
Medical history
- Diabetes mellitus
- Seizures in the 1970s
- Resection of a thymic tumor in 2003 (the exact pathology is unknown)
- Cirrhosis of unknown etiology
- No known history of hypertension
- No history of alcohol or intravenous drug use
- Quit smoking many years ago
- Coronary artery bypass surgery in 2003
- One sibling with myasthenia gravis.
Medications
- Levothyroxine
- Rosuvastatin (Crestor)
- Omeprazole (Prilosec)
- Spironolactone
- Furosemide
- Potassium chloride
- Metoprolol tartrate (Lopressor)
- Metformin (Glucophage)
- Ramipril (Altace).
Physical examination
She is hemodynamically stable and is not hypertensive. Her thyroid is not enlarged. Her lungs are clear to auscultation. Her heart sounds are normal, except for a nonradiating pansystolic murmur most audible at the apex.
Her abdomen is soft and is not distended. Her abdominal rash has a dermatomal distribution consistent with an L1 distribution, with vesicles over an erythematous base. Purpuric lesions are noted over her lower extremities.
Her leg strength is 3 on a scale of 5 on both sides; her arm strength is normal. Ankle and knee reflexes are absent bilaterally.
Initial laboratory analysis
PROGRESSIVE MUSCLE WEAKNESS
1. What are possible causes of her muscle weakness?
- Myasthenia gravis
- Hypothyroidism
- Dermatomyositis-polymyositis
- Drug-induced myopathy
- Cushing syndrome
- All of the above
All of these are potential causes of muscle weakness.
Myasthenia gravis
Myasthenia gravis, an autoimmune disease, can affect people of all ages and either sex. It presents with muscle weakness and fatigability, which characteristically fluctuate during the day. Some patients present in crisis with respiratory failure, which may require ventilatory support.1,2
Myasthenia gravis is characterized by auto-antibodies against the postsynaptic membrane of the neuromuscular junction. Most patients have antibodies to the extracellular portion of the acetylcholine receptor; a small number of patients have antibodies against a muscle-specific tyrosine kinase that interacts with this receptor.
About 15% of patients with myasthenia gravis have a thymoma thought to be involved in the pathogenesis of the disease. Treatments include immune suppressive therapy and thymectomy.
Our patient has a history of thymic lesion resection, but her antibody workup for myasthenia gravis was negative.
Hypothyroidism
Hypothyroidism, the most common disorder of the thyroid gland, is especially prevalent in women.3 Its common symptoms include fatigue, exercise intolerance, muscle weakness, cramps, and stiffness.
Both the TSH and the free thyroxine (T4) level must be measured to diagnose hypothyroidism. This information can also help differentiate primary hypothyroidism (ie, due to a defect in the thyroid gland) from secondary hypothyroidism (ie, due to a defect in the pituitary gland). Elevated TSH with low free T4 levels indicates primary thyroid failure, whereas the combination of a normal or low TSH and a low free T4 usually indicates pituitary failure. Subclinical hypothyroidism is characterized by mildly to moderately elevated TSH, but total T4 and free T4 values are still within the reference range. Replacement therapy is with levothyroxine.3–6
Our patient has a history of hypothyroidism, which could explain her muscle weakness, but she is currently on replacement therapy, and her TSH level on admission was normal.
Dermatomyositis-polymyositis
Dermatomyositis-polymyositis is characterized by proximal muscle weakness, creatine kinase elevation, erythema on sun-exposed skin, heliotrope rash, and Gottron papules. It occurs mostly in women after the second decade of life. Some medications have been implicated in its pathogenesis, such as statins, fibrates, hydroxyurea, penicillamine, and omeprazole (Prilosec).7
In a middle-aged patient, this diagnosis should prompt a search for cancer, especially of the gastrointestinal system, breast, and lung.8 Cancer can arise up to 3 years after the diagnosis of dermatomyositis or polymyositis.
Antisynthetase antibody syndrome is suspected if the patient is positive for antisynthetase antibody and has the following manifestations: acute onset of disease, constitutional symptoms, interstitial lung disease, inflammatory arthritis, mechanic’s hands (thickened, cracked skin on the palmar aspect of the thumb and index finger), and Raynaud phenomenon.4,8,9
The diagnosis is made by a thorough clinical evaluation. Electromyography can show an inflammatory pattern of myopathy. The gold standard test for this diagnosis is muscle biopsy.
Our patient has a normal creatine kinase level, which excludes the diagnosis of dermatomyositis-polymyositis.
Statin-induced myopathy
Up to 10% of patients taking statins develop myalgia. Rhabdomyolysis, the extreme form of myopathy, is rare.
The exact mechanism of statin-induced myopathy remains unclear; mitochondrial dysfunction, cholesterol composition of cell membranes, and coenzyme Q10 deficiency have been proposed.
Risk factors for statin-induced myopathy include female sex, older age, higher doses of statins, a family history of statin-induced myopathy, and hypothyroidism. Drugs that increase the risk include fibric acid derivatives, macrolides, and amiodarone (Cordarone). If a statin and any of the above drugs are both required, certain statins—ie, pravastatin (Pravachol) and rosuvastatin—are recommended, since they are the statins least likely to cause rhabdomyolysis.5,7,10–12
The combination of fluvastatin (Lescol) and gemfibrozil (Lopid) has also been found to be safe.13 In a crossover study in 17 patients, no significant difference was seen in the area under the curve for plasma concentration over time, in the maximum plasma concentration, or in the time to maximum concentration with the combination vs with each drug alone.13
Our patient is taking a statin and has hypothyroidism, which increases the risk of statin-induced myopathy. However, her creatine kinase level is normal.
Cushing syndrome
Cushing syndrome (hypercortisolism) is one of the most challenging endocrine diseases to diagnose. Most of its clinical features overlap with those of common diseases, and some patients have an atypical clinical presentation with only isolated symptoms. Further, its presentation can be subtle, with weight gain, amenorrhea, muscle weakness, and easy bruisability. Acne, moon facies, plethora, abdominal striae, and purpura are other common signs. It is three to 10 times more common in women than in men.
Cushing syndrome can be classified according to whether or not the excess cortisol secretion depends on corticotropin (formerly called adrenocorticotropic hormone or ACTH) (Figure 1). In corticotropin-dependent cases, the most common cause is pituitary adenoma. (When Cushing syndrome is due to excessive pituitary secretion of corticotropin, which in turn stimulates the adrenal glands to secrete excessive amounts of cortisol, it is called Cushing disease). Other causes of corticotropin-dependent Cushing syndrome are ectopic corticotropin-producing tumors such as carcinoid tumors or medullary thyroid cancers. Corticotropin-independent Cushing syndrome can be caused by adrenal adenomas, adrenal carcinoma, and bilateral primary micronodular or macronodular adrenocortical hyperplasia.14–17
However, the most common cause of Cushing syndrome is glucocorticoid therapy.
BACK TO OUR PATIENT: HER CONDITION DETERIORATES
Our patient’s physical condition deteriorates, she develops respiratory distress, and she is admitted to the medical intensive care unit. Her mental status also deteriorates, and she becomes lethargic and unresponsive.
She is intubated to protect her airway. After this, she develops hypotension that does not respond to fluid resuscitation and that requires vasopressors. Her condition continues to worsen as she develops acute kidney injury and disseminated intravascular coagulation. Her vesicular rash becomes more widespread, involving the entire trunk.
A workup for sepsis is initiated, but her initial blood and urine cultures are negative. Chest radiography does not reveal any infiltrates. No other source of an infection is found.
Varicella zoster is isolated on viral culture of a specimen obtained from the rash, and a polymerase chain reaction test of her blood shows cytomegalovirus DNA (64,092 copies per mL). Immune suppression is suspected, so a CD4 count is ordered (Table 2). Serologic tests for human immunodeficiency virus are negative.
What could have caused our patient to have muscle weakness in addition to disseminated zoster with cytomegalovirus viremia?
The diagnosis here is Cushing syndrome.
HOW TO TEST FOR CUSHING SYNDROME
2. In any practice, you may meet many perimenopausal women who have complaints of weight gain, amenorrhea, and acne. How can you determine if this is Cushing syndrome? What are the screening tests?
- 24-Hour urinary cortisol excretion
- A late-night salivary cortisol level
- A low-dose dexamethasone suppression test
- All of the above
- None of the above
Any of the tests listed here can be used to determine whether this is truly Cushing syndrome.
24-Hour urinary cortisol excretion has a reference range of 20 to 100 μg/24 hours. However, results may be falsely high in patients who are depressed or who abuse alcohol.
The late-night salivary cortisol level is another useful test.14,16,18 Patients with Cushing syndrome are found to have high late-night salivary cortisol levels as compared with normal people, indicating the loss of natural circadian rhythm.14,16,18
The low-dose dexamethasone suppression test, as first described by Liddle in 1960,19 involved giving dexamethasone 0.5 mg by mouth every 6 hours for 48 hours and measuring the serum cortisol level 6 hours after the last dose. In healthy people, this low dose of dexamethasone suppresses the production of corticotropin by the pituitary gland and in turn the production of cortisol, but in patients with Cushing syndrome the cortisol level remains high. An alternative is the overnight 1-mg dexamethasone suppression test—ie, giving 1 mg of dexamethasone at 11:00 pm and measuring the serum cortisol level early the next morning. Failure of the cortisol level to drop to less than 1.8 μg/dL suggests Cushing syndrome and warrants a complete evaluation for it.
Confirmatory testing is sometimes needed if patients have mild abnormalities in their screening tests. A combination low-dose dexamethasone suppression test and corticotropin-releasing hormone test can be used to differentiate Cushing syndrome from pseudo-Cushing syndrome. This is performed by giving dexamethasone orally 0.5 mg every 6 hours for 48 hours and then giving ovine-sequence corticotropin-releasing hormone 1 μg/kg intravenously 2 hours after the last dose of dexamethasone. The plasma cortisol value 15 minutes after the dose of corticotropin-releasing hormone is greater than 1.4 μg/dL (38 nmol/L) in patients with Cushing syndrome but remains low in patients with pseudo-Cushing syndrome.
Is this corticotropin-dependent or corticotropin-independent?
Once Cushing syndrome is diagnosed by one of the screening methods described above, the source of the excess glucocorticoids needs to be determined. Measuring the serum corticotropin level early in the morning would be the next step.
A low corticotropin level (< 10 pg/mL) indicates a corticotropin-independent source, most likely in the adrenal glands. Hence, computed tomography or magnetic resonance imaging (MRI) of the adrenal glands is warranted. Of note: adrenal incidentalomas are quite common, present in 5% of the general population, and a lesion on the adrenal gland does not prove that the patient has primary adrenal disease.16,20
IS THE EXCESS CORTICOTROPIN FROM A PITUITARY OR AN ECTOPIC SOURCE?
3. If the corticotropin level is elevated, how can you determine if it is from the pituitary or from an ectopic source?
- MRI of the pituitary gland
- High-dose dexamethasone suppression test
- Corticotropin-releasing hormone stimulation test
- Bilateral inferior petrosal sinus sampling
If the corticotropin level is high (> 10 pg/mL), it is of paramount importance to determine whether the corticotropin comes from the pituitary gland or from an ectopic source.
MRI of the pituitary gland should be done in patients with suspected corticotropin-dependent Cushing syndrome. However, MRI may be negative in 50% of patients with Cushing disease, and it should therefore not be used for screening. In addition, 10% of the population may have pituitary incidentalomas on MRI.
Most cases of corticotropin-dependent Cushing syndrome are caused by microadenomas (smaller than 1 cm), while a few cases are caused by macroadenomas (larger than 1 cm). If a microadenoma is found on MRI, further testing with bilateral inferior petrosal sinus sampling is recommended (described below); if a macroadenoma is found, then no further testing is required.21,22 In fact, patients who have biochemical findings compatible with Cushing disease (ie, due to an overactive pituitary) and who have an adenoma larger than 6 mm do not require further evaluation.23
A high-dose dexamethasone suppression test involves giving 8 mg of dexamethasone in the evening and measuring the cortisol level the next morning. If the cortisol level declines to 50% of the baseline level after this dose, this suggests a pituitary cause.
Corticotropin-releasing hormone stimulation testing. In most cases of pituitary tumors and a few cases of ectopic corticotropin-secreting tumors, giving corticotropin-releasing hormone leads to an increase in serum corticotropin and cortisol levels. In contrast, these levels do not respond to corticotropin-releasing hormone stimulation if the problem is in the adrenal gland. The test is performed by giving 1 μg/kg or 100 μg synthetic or human corticotropin-releasing hormone. A 35% to 50% increase above baseline in corticotropin suggests a pituitary cause.23
Bilateral inferior petrosal sinus sampling can be used to confirm a pituitary source, as it is the gold standard for differentiating ectopic from pituitary corticotropin production. Once this is confirmed, a neurosurgical consult is warranted.16,18
This procedure is usually done by advancing a sheath from the femoral vein to reach the inferior petrosal sinuses. Blood samples are obtained from both the inferior petrosal sinuses and from a peripheral vein to measure corticotropin levels before and after giving corticotropin-releasing hormone (1 μg/kg). Before corticotropin-releasing hormone is given, a gradient of central-peripheral corticotropin levels of 2.0 or greater indicates a pituitary source. With ectopic corticotropin production, the corticotropin gradient is usually less than 1.5. Corticotropin-releasing hormone is given to increase the sensitivity: after it is given, a gradient of 3.0 or greater is considered indicative of Cushing disease.24
If the corticotropin level is elevated and the above tests indicate ectopic production, the source should be sought. The most common site of ectopic corticotropin production is the chest. Common causes are bronchial, thymic, and pancreatic carcinoid tumors. Other causes are small-cell lung cancer, medullary cell cancer, and pheochromocytoma.15,18,25
BACK TO OUR PATIENT
Our patient’s further laboratory results are listed in Table 3.
She has elevated 24-hour urinary cortisol excretion, consistent with Cushing syndrome. Her corticotropin level is elevated, which rules out an adrenal cause. Her 5-HIAA (a serotonin breakdown product) and calcitonin levels are also elevated, suggesting either medullary thyroid cancer or a carcinoid tumor. She also has a mild elevation of dehydroepiandrosterone sulfate, which is consistent with corticotropin-dependent Cushing syndrome.
Our patient’s elevated levels of cortisol were the cause of her muscle weakness and severe immune deficiency, which in turn led to cytomegalovirus viremia and sepsis. Cushing syndrome usually causes hypertension, especially in cases of ectopic corticotropin production. However, our patient was normotensive on admission and then developed cytomegalovirus sepsis, which led to hypotension and shock.
Immune suppression is a well-known effect of glucocorticoids.26–28 Kronfol et al28 found that CD4 and CD8 counts and the CD4-to-CD8 ratio were low in patients with Cushing syndrome, and natural killer cell activity was suppressed. Opportunistic infections have been described in patients with Cushing syndrome.26,27,29
MANAGEMENT OF CUSHING SYNDROME
Management of Cushing syndrome should be tailored after determining its source.
A neurosurgical consultation is warranted in cases of pituitary adenoma, with surgical resection of the adrenal source or ectopic tumor if feasible.25
Medical management is recommended if surgical resection is not possible.30,31 Several drugs can be used to inhibit cortisol synthesis in this situation.30,32
Adrenal-acting agents
Aminoglutethimide (Cytadren) acts by blocking the conversion of cholesterol to pregnenolone, a precursor of cortisol. The dosage is 250 mg twice or three times a day. This drug is no longer available in the United States.
Ketoconazole (Nizoral) inhibits side-chain cleavage, 11-beta hydroxylase, and 17-alpha hydroxylase, thus inhibiting cortisol synthesis; it also inhibits corticotropin secretion. The dosage is 200 to 400 mg three times a day.
Metyrapone (Metopirone) blocks 11-beta-hydroxylation of deoxycortisol, the reaction that produces cortisol. The dosage is 500 to 750 mg three times a day. This drug can be obtained only from the manufacturer and only on a named-patient basis.
Etomidate (Amidate), an anesthetic drug, also blocks 11-beta-hydroxylation of deoxycortisol. It is given intravenously at a rate of 0.3 mg/kg per hour.
Centrally acting agents
Cabergoline (Dostinex). It is believed that corticotropin-producing pituitary tumors express D2 receptors. Cabergoline is a dopamine agonist that has been used in patients with Cushing disease. The dosage is 0.5 to 7 mg/week.
Pasireotide is still investigational. It is a somatostatin receptor agonist given subcutaneously for 15 consecutive days to patients with Cushing disease.
Glucocorticoid receptor antagonist
Mifepristone (Mifeprex) is a progesterone receptor and glucocorticoid II receptor antagonist that is being investigated in the treatment of persistent or recurrent Cushing disease. It is not yet approved by the US Food and Drug Administration for this indication.
BACK TO OUR PATIENT
The patient was too ill to undergo additional imaging, including octreotide scanning to identify an ectopic corticotropin-secreting tumor. She was medically treated with intravenous etomidate to reduce her cortisol level.30,31
Unfortunately, our patient died of multiorgan failure. The exact site of her ectopic corticotropin-producing tumor was never identified, and no autopsy was done.
- Meriggioli MN. Myasthenia gravis with anti-acetylcholine receptor antibodies. Front Neurol Neurosci 2009; 26:94–108.
- Gilhus NE. Autoimmune myasthenia gravis. Expert Rev Neurother 2009; 9:351–358.
- Heitman B, Irizarry A. Hypothyroidism: common complaints, perplexing diagnosis. Nurse Pract 1995; 20:54–60.
- Brick JE, Brick JF, Elnicki DM. Musculoskeletal disorders. When are they caused by hormone imbalance? Postgrad Med 1991; 90:129–132,135–136.
- Bar SL, Holmes DT, Frohlich J. Asymptomatic hypothyroidism and statin-induced myopathy. Can Fam Physician 2007; 53:428–431.
- McDermott MT. In the clinic. Hypothyroidism. Ann Intern Med 2009; 151:ITC61.
- Klopstock T. Drug-induced myopathies. Curr Opin Neurol 2008; 21:590–595.
- Dimachkie MM, Barohn RJ. Idiopathic inflammatory myopathies. Front Neurol Neurosci 2009; 26:126–146.
- Joseph A, Brasington R, Kahl L, Ranganathan P, Cheng TP, Atkinson J. Immunologic rheumatic disorders. J Allergy Clin Immunol 2010; 125(suppl 2):S204–S215.
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med 2009; 150:858–868.
- Kiernan TJ, Rochford M, McDermott JH. Simvastatin induced rhabdomyolysis and an important clinical link with hypothyroidism. Int J Cardiol 2007; 119:374–376.
- Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA 2003; 289:1681–1690.
- Spence JD, Munoz CE, Hendricks L, Latchinian L, Khouri HE. Pharmacokinetics of the combination of fluvastatin and gemfibrozil. Am J Cardiol 1995; 76:80A–83A.
- Boscaro M, Arnaldi G. Approach to the patient with possible Cushing’s syndrome. J Clin Endocrinol Metab 2009; 94:3121–3131.
- Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, Nieman LK. Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab 2005; 90:4955–4962.
- Pecori Giraldi F. Recent challenges in the diagnosis of Cushing’s syndrome. Horm Res 2009; 71(suppl 1):123–127.
- von Mach MA, Kann P, Piepkorn B, Bruder S, Beyer J. [Cushing’s syndrome caused by paraneoplastic ACTH secretion 11 years after occurrence of a medullary thyroid carcinoma]. Dtsch Med Wochenschr 2002; 127:850–852.
- Beauregard C, Dickstein G, Lacroix A. Classic and recent etiologies of Cushing’s syndrome: diagnosis and therapy. Treat Endocrinol 2002; 1:79–94.
- Liddle GW. Tests of pituitary-adrenal suppressibility in the diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab 1960; 20:1539–1560.
- Louiset E, Gobet F, Libé R, et al. ACTH-independent Cushing’s syndrome with bilateral micronodular adrenal hyperplasia and ectopic adrenocortical adenoma. J Clin Endocrinol Metab 2010; 95:18–24.
- Andrioli M, Pecori Giraldi F, De Martin M, Cattaneo A, Carzaniga C, Cavagnini F. Differential diagnosis of ACTH-dependent hypercortisolism: imaging versus laboratory. Pituitary 2009; 12:294–296.
- Sahdev A, Reznek RH, Evanson J, Grossman AB. Imaging in Cushing’s syndrome. Arq Bras Endocrinol Metabol 2007; 51:1319–1328.
- Arnaldi G, Angeli A, Atkinson AB, et al. Diagnosis and complications of Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab 2003; 88:5593–5602.
- Lad SP, Patil CG, Laws ER, Katznelson L. The role of inferior petrosal sinus sampling in the diagnostic localization of Cushing’s disease. Neurosurg Focus 2007; 23:E2.
- Bhansali A, Walia R, Rana SS, et al. Ectopic Cushing’s syndrome: experience from a tertiary care centre. Indian J Med Res 2009; 129:33–41.
- Arlt A, Harbeck B, Anlauf M, et al. Fatal Pneumocystis jirovecii pneumonia in a case of ectopic Cushing’s syndrome due to neuroendocrine carcinoma of the kidney. Exp Clin Endocrinol Diabetes 2008; 116:515–519.
- Graham BS, Tucker WS. Opportunistic infections in endogenous Cushing’s syndrome. Ann Intern Med 1984; 101:334–338.
- Kronfol Z, Starkman M, Schteingart DE, Singh V, Zhang Q, Hill E. Immune regulation in Cushing’s syndrome: relationship to hypothalamic-pituitary-adrenal axis hormones. Psychoneuroendocrinology 1996; 21:599–608.
- Sepkowitz KA. Opportunistic infections in patients with and patients without acquired immunodeficiency syndrome. Clin Infect Dis 2002; 34:1098–1107.
- Schteingart DE. Drugs in the medical treatment of Cushing’s syndrome. Expert Opin Emerg Drugs 2009; 14:661–671.
- Shalet S, Mukherjee A. Pharmacological treatment of hypercortisolism. Curr Opin Endocrinol Diabetes Obes 2008; 15:234–238.
- Arnaldi G, Boscaro M. Pasireotide for the treatment of Cushing’s disease. Expert Opin Investig Drugs 2010; 19:889–898.
- Meriggioli MN. Myasthenia gravis with anti-acetylcholine receptor antibodies. Front Neurol Neurosci 2009; 26:94–108.
- Gilhus NE. Autoimmune myasthenia gravis. Expert Rev Neurother 2009; 9:351–358.
- Heitman B, Irizarry A. Hypothyroidism: common complaints, perplexing diagnosis. Nurse Pract 1995; 20:54–60.
- Brick JE, Brick JF, Elnicki DM. Musculoskeletal disorders. When are they caused by hormone imbalance? Postgrad Med 1991; 90:129–132,135–136.
- Bar SL, Holmes DT, Frohlich J. Asymptomatic hypothyroidism and statin-induced myopathy. Can Fam Physician 2007; 53:428–431.
- McDermott MT. In the clinic. Hypothyroidism. Ann Intern Med 2009; 151:ITC61.
- Klopstock T. Drug-induced myopathies. Curr Opin Neurol 2008; 21:590–595.
- Dimachkie MM, Barohn RJ. Idiopathic inflammatory myopathies. Front Neurol Neurosci 2009; 26:126–146.
- Joseph A, Brasington R, Kahl L, Ranganathan P, Cheng TP, Atkinson J. Immunologic rheumatic disorders. J Allergy Clin Immunol 2010; 125(suppl 2):S204–S215.
- Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med 2009; 150:858–868.
- Kiernan TJ, Rochford M, McDermott JH. Simvastatin induced rhabdomyolysis and an important clinical link with hypothyroidism. Int J Cardiol 2007; 119:374–376.
- Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA 2003; 289:1681–1690.
- Spence JD, Munoz CE, Hendricks L, Latchinian L, Khouri HE. Pharmacokinetics of the combination of fluvastatin and gemfibrozil. Am J Cardiol 1995; 76:80A–83A.
- Boscaro M, Arnaldi G. Approach to the patient with possible Cushing’s syndrome. J Clin Endocrinol Metab 2009; 94:3121–3131.
- Ilias I, Torpy DJ, Pacak K, Mullen N, Wesley RA, Nieman LK. Cushing’s syndrome due to ectopic corticotropin secretion: twenty years’ experience at the National Institutes of Health. J Clin Endocrinol Metab 2005; 90:4955–4962.
- Pecori Giraldi F. Recent challenges in the diagnosis of Cushing’s syndrome. Horm Res 2009; 71(suppl 1):123–127.
- von Mach MA, Kann P, Piepkorn B, Bruder S, Beyer J. [Cushing’s syndrome caused by paraneoplastic ACTH secretion 11 years after occurrence of a medullary thyroid carcinoma]. Dtsch Med Wochenschr 2002; 127:850–852.
- Beauregard C, Dickstein G, Lacroix A. Classic and recent etiologies of Cushing’s syndrome: diagnosis and therapy. Treat Endocrinol 2002; 1:79–94.
- Liddle GW. Tests of pituitary-adrenal suppressibility in the diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab 1960; 20:1539–1560.
- Louiset E, Gobet F, Libé R, et al. ACTH-independent Cushing’s syndrome with bilateral micronodular adrenal hyperplasia and ectopic adrenocortical adenoma. J Clin Endocrinol Metab 2010; 95:18–24.
- Andrioli M, Pecori Giraldi F, De Martin M, Cattaneo A, Carzaniga C, Cavagnini F. Differential diagnosis of ACTH-dependent hypercortisolism: imaging versus laboratory. Pituitary 2009; 12:294–296.
- Sahdev A, Reznek RH, Evanson J, Grossman AB. Imaging in Cushing’s syndrome. Arq Bras Endocrinol Metabol 2007; 51:1319–1328.
- Arnaldi G, Angeli A, Atkinson AB, et al. Diagnosis and complications of Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab 2003; 88:5593–5602.
- Lad SP, Patil CG, Laws ER, Katznelson L. The role of inferior petrosal sinus sampling in the diagnostic localization of Cushing’s disease. Neurosurg Focus 2007; 23:E2.
- Bhansali A, Walia R, Rana SS, et al. Ectopic Cushing’s syndrome: experience from a tertiary care centre. Indian J Med Res 2009; 129:33–41.
- Arlt A, Harbeck B, Anlauf M, et al. Fatal Pneumocystis jirovecii pneumonia in a case of ectopic Cushing’s syndrome due to neuroendocrine carcinoma of the kidney. Exp Clin Endocrinol Diabetes 2008; 116:515–519.
- Graham BS, Tucker WS. Opportunistic infections in endogenous Cushing’s syndrome. Ann Intern Med 1984; 101:334–338.
- Kronfol Z, Starkman M, Schteingart DE, Singh V, Zhang Q, Hill E. Immune regulation in Cushing’s syndrome: relationship to hypothalamic-pituitary-adrenal axis hormones. Psychoneuroendocrinology 1996; 21:599–608.
- Sepkowitz KA. Opportunistic infections in patients with and patients without acquired immunodeficiency syndrome. Clin Infect Dis 2002; 34:1098–1107.
- Schteingart DE. Drugs in the medical treatment of Cushing’s syndrome. Expert Opin Emerg Drugs 2009; 14:661–671.
- Shalet S, Mukherjee A. Pharmacological treatment of hypercortisolism. Curr Opin Endocrinol Diabetes Obes 2008; 15:234–238.
- Arnaldi G, Boscaro M. Pasireotide for the treatment of Cushing’s disease. Expert Opin Investig Drugs 2010; 19:889–898.
Do incretin drugs for type 2 diabetes increase the risk of acute pancreatitis?
Probably not. Although cases of acute pancreatitis have occurred in patients taking these drugs, cases have been reported in patients taking other drugs as well. Furthermore, the incidence of acute pancreatitis is higher in patients with type 2 diabetes (for which incretin-type drugs are indicated) than in the general population, regardless of treatment.
INCRETINS, A NEW CLASS OF DRUGS FOR TYPE 2 DIABETES
Incretins are hormones secreted by the small intestine in response to glucose in food. Glucagon-like peptide 1 (GLP-1) is an endogenous incretin that stimulates insulin secretion, suppresses glucagon secretion, and delays gastric emptying.
Current incretin-based therapies for type 2 diabetes include two types of agents. First are drugs that mimic the action of native GLP-1, such as the injectable GLP-1 analogues exenatide (Byetta) and liraglutide (Victoza). Second are agents that interfere with the metabolism of native GLP-1, mainly by inhibiting the endogenous enzyme dipeptidyl peptidase 4 (DPP-4), thus extending the life of native GLP-1. Two DPP-4 inhibitors pertinent to this discussion are saxagliptin (Onglyza) and sitagliptin (Januvia), both of which are taken orally.
The question has been raised whether incretin-based therapy causes pancreatitis. The package inserts for exenatide and sitagliptin have been updated to reflect this possibility, thus causing concern to practitioners. Is this concern warranted?
MANY DRUGS ARE ASSOCIATED WITH ACUTE PANCREATIS
In a review published in 2005, Trivedi and Pitchumoni1 reported that, of the top 100 prescribed drugs in the United States, 44 had been associated with acute pancreatitis. These agents included over-the-counter drugs such as acetaminophen (Tylenol), common antibiotics such as trimethoprim-sulfamethoxazole (Bactrim) and erythromycin, and drugs used to treat acquired immunodeficiency syndrome and cancer. No clear pathophysiologic basis connects these agents.
In 2002, Blomgren et al2 suggested that glyburide (Micronase) use might be a risk factor for acute pancreatitis, and that the risk of pancreatitis is higher if the body mass index is 30 kg/m2 or more. In 2008, more concern was raised with a report of hemorrhagic or necrotizing pancreatitis in six patients taking exenatide, two of whom died.3 And more recently, reports of 88 cases of acute pancreatitis (including 2 cases of hemorrhagic or necrotizing pancreatitis) from October 2006 to February 2009 in patients taking sitagliptin or the sitagliptin-metformin combination Janumet4 prompted a revision of the package inserts.
Do these cases represent unexpected toxicities not appreciated in premarket clinical trials, or are they to be expected in the population treated with these agents as greater numbers are exposed?
TYPE 2 DIABETES ALSO POSES A RISK OF PANCREATITIS
A number of comorbidities associated with type 2 diabetes predispose to pancreatitis, particularly hypertriglyceridemia and gallbladder disease.5–7 People with diabetes can also be exposed to alcohol or other drugs reported to be associated with pancreatitis.
What is the risk of pancreatitis in patients with type 2 diabetes? Is there evidence of a greater risk when incretin-based drugs are used to control hyperglycemia rather than other agents?
Pancreatitis appears to be increasingly prevalent in the general population in western countries. Some 60% to 80% of cases are attributed to alcohol or gallstones, but 20% do not have a clear cause.
In 2009, a new cause of acute pancreatitis was introduced when Frulloni et al8 reported that a novel antibody that recognizes epitopes shared with Helicobacter pylori was associated with autoimmune pancreatitis. H pylori is a common gastrointestinal organism, found in diabetic and nondiabetic patients, and it may well account for what has up to now been considered idiopathic pancreatitis.
Type 2 diabetes is associated with obesity and hyperlipidemia, each of which has been considered a putative risk factor for pancreatits.5–7
Noel et al9 examined the risk of pancreatitis in patients with type 2 diabetes in a large insurance database (29,332,477 covered lives). They identified people with type 2 diabetes and those without diabetes eligible for coverage by the plan, using medical and pharmacy claims from January 1, 1999, to December 31, 2005. The authors also used medical claims to identify episodes of acute pancreatitis and gallbladder disease. They found that the risk of acute pancreatitis was 2.8 times higher in the overall diabetic cohort than in the nondiabetic cohort, and five times higher in the youngest diabetic cohort (ages 18 to 44) than in nondiabetic people of the same age. The risk was three times higher in diabetic men than in nondiabetic men, and 2.6 times higher in diabetic women than in nondiabetic women.
The time period examined in this study is fortuitous, since exenatide was approved in June 2005 and had very little market penetration during its first 6 months, corresponding to the last 6 months of the study period. Sitagliptin, the first DPP-4 inhibitor, had not yet reached the market.
Noel et al9 also found that the risk of biliary disease in patients with diabetes was 1.9 times higher than in those without diabetes. The relative risk of gallbladder disease was proportionally greater in a younger population with diabetes than in the population without diabetes, in whom the risk of gallbladder disease increases with age. Cholelithiasis was believed to be the underlying cause in at least 50% of the cases of pancreatitis.
PANCREATITIS AND INCRETIN-BASED THERAPIES
The estimated risk of acute pancreatitis in the population at large is reported as 0.33 to 0.44 events per 1,000 adults per year10; 15% to 20% of cases are considered severe, and 2% to 4% result in death.5,10 A relatively small number (1%–2%) are believed to be drug-induced.10
Exenatide. In the exenatide development program, six cases of acute pancreatitis were observed in about 3,489 subject-years of exposure (1.7 per 1,000 subject-years), compared with one case in about 336 subject-years with placebo (3.0 per 1,000 subject-years) and one case in about 497 subject-years (2.0 per 1,000 subject-years) with insulin.11
Sitagliptin. Dore et al12 examined claims from another database for the period of June 2005 through June 2008 to look specifically at the risk with incretin-based therapies. This database included 27,996 people starting exenatide and 16,276 people starting sitagliptin, matched with people with type 2 diabetes taking metformin (Glucophage) or glyburide. Over a period of 1 year, 0.13% of exenatide users and 0.12% of sitagliptin users suffered acute pancreatitis. The risk of pancreatitis was comparable in each group:
- For exenatide, relative risk (RR) 1.0, 95% confidence interval (CI) 0.6 to 1.7, compared with metformin or glyburide
- For sitagliptin, RR 1.0, 95% CI 0.5 to 2.0.
Saxagliptin. In clinical trials of saxagliptin, the incidence of pancreatitis was 0.2% in 3,422 patients receiving saxagliptin and 0.2% in 1,066 controls,13 similar to the rates for sitagliptin and exenatide.
Liraglutide appeared to be associated with a risk of acute pancreatitis, with seven cases in 3,900 patients receiving liraglutide vs one case in a patient taking another diabetes drug.14 This rate is similar to that reported in exenatide clinical trials, suggesting that pancreatitis has been underreported in the comparator subjects. We need more experience to see if this agent really poses more risk than other antidiabetic therapies.
As new antidiabetic agents enter the market and their use becomes common, it would not be surprising to see rates of pancreatitis similar to those reported by Blomgren et al2 in 2002, when glyburide was becoming a mainstay of therapy for type 2 diabetes.
- Trivedi CD, Pitchumoni CS. Drug-induced pancreatitis: an update. J Clin Gastroenterol 2005; 39:709–716.
- Blomgren KB, Sundström A, Steineck G, Wiholm BE. Obesity and treatment of diabetes with glyburide may both be risk factors for acute pancreatitis. Diabetes Care 2002; 25:298–302.
- US Food and Drug Administration. Information for healthcare professionals: exenatide (marketed as Byetta)—8/2008 update. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm124713.htm. Accessed July 1, 2010.
- US Food and Drug Administration. Information for healthcare professionals—acute pancreatitis and sitagliptin (marketed as Januvia and Janumet). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm183764.htm. Accessed July 1, 2010.
- Forsmark CE, Baillie J; AGA Institute Clinical Practice and Economics Committee. AGA Institute technical review on acute pancreatitis. Gastroenterology 2007; 132:2022–2044.
- Pagliarulo M, Fornari F, Fraquelli M, et al. Gallstone disease and related risk factors in a large cohort of diabetic patients. Dig Liver Dis 2004; 36:130–134.
- Field AE, Coakley EH, Must A, et al. Impact of overweight on the risk of developing common chronic diseases during a 10-year period. Arch Intern Med 2001; 161:1581–1586.
- Frulloni L, Lunardi C, Simone R, et al. Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med 2009; 361:2135–2142.
- Noel RA, Braun DK, Patterson RE, Bloomgren GL. Increased risk of acute pancreatitis and biliary disease observed in patients with type 2 diabetes: a retrospective cohort study. Diabetes Care 2009; 32:834–838.
- Whitcomb DC. Clinical practice. Acute pancreatitis. N Engl J Med 2006; 354:2142–2150.
- Data on file, Amylin Pharmaceuticals, Inc. and Eli Lilly.
- Dore DD, Seeger JD, Arnold Chan K. Use of a claims-based active drug safety surveillance system to assess the risk of acute pancreatitis with exenatide or sitagliptin compared to metformin or glyburide. Curr Med Res Opin 2009; 25:1019–1027.
- US Food and Drug Administration. Controlled Phase 2b/3 Pooled Population—Day 120 Update. http://www.fda.gov/downloads/AdvisoryCommittees/Committees-MeetingMaterials/Drugs/EndocrinologicandMetabolic-DrugsAdvisoryCommittee/UCM149589. Accessed July 4, 2010.
- US Food and Drug Administration. Questions and answers—safety requirements for Victoza (liraglutide). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrug-SafetyInformationforPatientsandProviders/ucm198543.htm. Accessed July 4, 2010.
Probably not. Although cases of acute pancreatitis have occurred in patients taking these drugs, cases have been reported in patients taking other drugs as well. Furthermore, the incidence of acute pancreatitis is higher in patients with type 2 diabetes (for which incretin-type drugs are indicated) than in the general population, regardless of treatment.
INCRETINS, A NEW CLASS OF DRUGS FOR TYPE 2 DIABETES
Incretins are hormones secreted by the small intestine in response to glucose in food. Glucagon-like peptide 1 (GLP-1) is an endogenous incretin that stimulates insulin secretion, suppresses glucagon secretion, and delays gastric emptying.
Current incretin-based therapies for type 2 diabetes include two types of agents. First are drugs that mimic the action of native GLP-1, such as the injectable GLP-1 analogues exenatide (Byetta) and liraglutide (Victoza). Second are agents that interfere with the metabolism of native GLP-1, mainly by inhibiting the endogenous enzyme dipeptidyl peptidase 4 (DPP-4), thus extending the life of native GLP-1. Two DPP-4 inhibitors pertinent to this discussion are saxagliptin (Onglyza) and sitagliptin (Januvia), both of which are taken orally.
The question has been raised whether incretin-based therapy causes pancreatitis. The package inserts for exenatide and sitagliptin have been updated to reflect this possibility, thus causing concern to practitioners. Is this concern warranted?
MANY DRUGS ARE ASSOCIATED WITH ACUTE PANCREATIS
In a review published in 2005, Trivedi and Pitchumoni1 reported that, of the top 100 prescribed drugs in the United States, 44 had been associated with acute pancreatitis. These agents included over-the-counter drugs such as acetaminophen (Tylenol), common antibiotics such as trimethoprim-sulfamethoxazole (Bactrim) and erythromycin, and drugs used to treat acquired immunodeficiency syndrome and cancer. No clear pathophysiologic basis connects these agents.
In 2002, Blomgren et al2 suggested that glyburide (Micronase) use might be a risk factor for acute pancreatitis, and that the risk of pancreatitis is higher if the body mass index is 30 kg/m2 or more. In 2008, more concern was raised with a report of hemorrhagic or necrotizing pancreatitis in six patients taking exenatide, two of whom died.3 And more recently, reports of 88 cases of acute pancreatitis (including 2 cases of hemorrhagic or necrotizing pancreatitis) from October 2006 to February 2009 in patients taking sitagliptin or the sitagliptin-metformin combination Janumet4 prompted a revision of the package inserts.
Do these cases represent unexpected toxicities not appreciated in premarket clinical trials, or are they to be expected in the population treated with these agents as greater numbers are exposed?
TYPE 2 DIABETES ALSO POSES A RISK OF PANCREATITIS
A number of comorbidities associated with type 2 diabetes predispose to pancreatitis, particularly hypertriglyceridemia and gallbladder disease.5–7 People with diabetes can also be exposed to alcohol or other drugs reported to be associated with pancreatitis.
What is the risk of pancreatitis in patients with type 2 diabetes? Is there evidence of a greater risk when incretin-based drugs are used to control hyperglycemia rather than other agents?
Pancreatitis appears to be increasingly prevalent in the general population in western countries. Some 60% to 80% of cases are attributed to alcohol or gallstones, but 20% do not have a clear cause.
In 2009, a new cause of acute pancreatitis was introduced when Frulloni et al8 reported that a novel antibody that recognizes epitopes shared with Helicobacter pylori was associated with autoimmune pancreatitis. H pylori is a common gastrointestinal organism, found in diabetic and nondiabetic patients, and it may well account for what has up to now been considered idiopathic pancreatitis.
Type 2 diabetes is associated with obesity and hyperlipidemia, each of which has been considered a putative risk factor for pancreatits.5–7
Noel et al9 examined the risk of pancreatitis in patients with type 2 diabetes in a large insurance database (29,332,477 covered lives). They identified people with type 2 diabetes and those without diabetes eligible for coverage by the plan, using medical and pharmacy claims from January 1, 1999, to December 31, 2005. The authors also used medical claims to identify episodes of acute pancreatitis and gallbladder disease. They found that the risk of acute pancreatitis was 2.8 times higher in the overall diabetic cohort than in the nondiabetic cohort, and five times higher in the youngest diabetic cohort (ages 18 to 44) than in nondiabetic people of the same age. The risk was three times higher in diabetic men than in nondiabetic men, and 2.6 times higher in diabetic women than in nondiabetic women.
The time period examined in this study is fortuitous, since exenatide was approved in June 2005 and had very little market penetration during its first 6 months, corresponding to the last 6 months of the study period. Sitagliptin, the first DPP-4 inhibitor, had not yet reached the market.
Noel et al9 also found that the risk of biliary disease in patients with diabetes was 1.9 times higher than in those without diabetes. The relative risk of gallbladder disease was proportionally greater in a younger population with diabetes than in the population without diabetes, in whom the risk of gallbladder disease increases with age. Cholelithiasis was believed to be the underlying cause in at least 50% of the cases of pancreatitis.
PANCREATITIS AND INCRETIN-BASED THERAPIES
The estimated risk of acute pancreatitis in the population at large is reported as 0.33 to 0.44 events per 1,000 adults per year10; 15% to 20% of cases are considered severe, and 2% to 4% result in death.5,10 A relatively small number (1%–2%) are believed to be drug-induced.10
Exenatide. In the exenatide development program, six cases of acute pancreatitis were observed in about 3,489 subject-years of exposure (1.7 per 1,000 subject-years), compared with one case in about 336 subject-years with placebo (3.0 per 1,000 subject-years) and one case in about 497 subject-years (2.0 per 1,000 subject-years) with insulin.11
Sitagliptin. Dore et al12 examined claims from another database for the period of June 2005 through June 2008 to look specifically at the risk with incretin-based therapies. This database included 27,996 people starting exenatide and 16,276 people starting sitagliptin, matched with people with type 2 diabetes taking metformin (Glucophage) or glyburide. Over a period of 1 year, 0.13% of exenatide users and 0.12% of sitagliptin users suffered acute pancreatitis. The risk of pancreatitis was comparable in each group:
- For exenatide, relative risk (RR) 1.0, 95% confidence interval (CI) 0.6 to 1.7, compared with metformin or glyburide
- For sitagliptin, RR 1.0, 95% CI 0.5 to 2.0.
Saxagliptin. In clinical trials of saxagliptin, the incidence of pancreatitis was 0.2% in 3,422 patients receiving saxagliptin and 0.2% in 1,066 controls,13 similar to the rates for sitagliptin and exenatide.
Liraglutide appeared to be associated with a risk of acute pancreatitis, with seven cases in 3,900 patients receiving liraglutide vs one case in a patient taking another diabetes drug.14 This rate is similar to that reported in exenatide clinical trials, suggesting that pancreatitis has been underreported in the comparator subjects. We need more experience to see if this agent really poses more risk than other antidiabetic therapies.
As new antidiabetic agents enter the market and their use becomes common, it would not be surprising to see rates of pancreatitis similar to those reported by Blomgren et al2 in 2002, when glyburide was becoming a mainstay of therapy for type 2 diabetes.
Probably not. Although cases of acute pancreatitis have occurred in patients taking these drugs, cases have been reported in patients taking other drugs as well. Furthermore, the incidence of acute pancreatitis is higher in patients with type 2 diabetes (for which incretin-type drugs are indicated) than in the general population, regardless of treatment.
INCRETINS, A NEW CLASS OF DRUGS FOR TYPE 2 DIABETES
Incretins are hormones secreted by the small intestine in response to glucose in food. Glucagon-like peptide 1 (GLP-1) is an endogenous incretin that stimulates insulin secretion, suppresses glucagon secretion, and delays gastric emptying.
Current incretin-based therapies for type 2 diabetes include two types of agents. First are drugs that mimic the action of native GLP-1, such as the injectable GLP-1 analogues exenatide (Byetta) and liraglutide (Victoza). Second are agents that interfere with the metabolism of native GLP-1, mainly by inhibiting the endogenous enzyme dipeptidyl peptidase 4 (DPP-4), thus extending the life of native GLP-1. Two DPP-4 inhibitors pertinent to this discussion are saxagliptin (Onglyza) and sitagliptin (Januvia), both of which are taken orally.
The question has been raised whether incretin-based therapy causes pancreatitis. The package inserts for exenatide and sitagliptin have been updated to reflect this possibility, thus causing concern to practitioners. Is this concern warranted?
MANY DRUGS ARE ASSOCIATED WITH ACUTE PANCREATIS
In a review published in 2005, Trivedi and Pitchumoni1 reported that, of the top 100 prescribed drugs in the United States, 44 had been associated with acute pancreatitis. These agents included over-the-counter drugs such as acetaminophen (Tylenol), common antibiotics such as trimethoprim-sulfamethoxazole (Bactrim) and erythromycin, and drugs used to treat acquired immunodeficiency syndrome and cancer. No clear pathophysiologic basis connects these agents.
In 2002, Blomgren et al2 suggested that glyburide (Micronase) use might be a risk factor for acute pancreatitis, and that the risk of pancreatitis is higher if the body mass index is 30 kg/m2 or more. In 2008, more concern was raised with a report of hemorrhagic or necrotizing pancreatitis in six patients taking exenatide, two of whom died.3 And more recently, reports of 88 cases of acute pancreatitis (including 2 cases of hemorrhagic or necrotizing pancreatitis) from October 2006 to February 2009 in patients taking sitagliptin or the sitagliptin-metformin combination Janumet4 prompted a revision of the package inserts.
Do these cases represent unexpected toxicities not appreciated in premarket clinical trials, or are they to be expected in the population treated with these agents as greater numbers are exposed?
TYPE 2 DIABETES ALSO POSES A RISK OF PANCREATITIS
A number of comorbidities associated with type 2 diabetes predispose to pancreatitis, particularly hypertriglyceridemia and gallbladder disease.5–7 People with diabetes can also be exposed to alcohol or other drugs reported to be associated with pancreatitis.
What is the risk of pancreatitis in patients with type 2 diabetes? Is there evidence of a greater risk when incretin-based drugs are used to control hyperglycemia rather than other agents?
Pancreatitis appears to be increasingly prevalent in the general population in western countries. Some 60% to 80% of cases are attributed to alcohol or gallstones, but 20% do not have a clear cause.
In 2009, a new cause of acute pancreatitis was introduced when Frulloni et al8 reported that a novel antibody that recognizes epitopes shared with Helicobacter pylori was associated with autoimmune pancreatitis. H pylori is a common gastrointestinal organism, found in diabetic and nondiabetic patients, and it may well account for what has up to now been considered idiopathic pancreatitis.
Type 2 diabetes is associated with obesity and hyperlipidemia, each of which has been considered a putative risk factor for pancreatits.5–7
Noel et al9 examined the risk of pancreatitis in patients with type 2 diabetes in a large insurance database (29,332,477 covered lives). They identified people with type 2 diabetes and those without diabetes eligible for coverage by the plan, using medical and pharmacy claims from January 1, 1999, to December 31, 2005. The authors also used medical claims to identify episodes of acute pancreatitis and gallbladder disease. They found that the risk of acute pancreatitis was 2.8 times higher in the overall diabetic cohort than in the nondiabetic cohort, and five times higher in the youngest diabetic cohort (ages 18 to 44) than in nondiabetic people of the same age. The risk was three times higher in diabetic men than in nondiabetic men, and 2.6 times higher in diabetic women than in nondiabetic women.
The time period examined in this study is fortuitous, since exenatide was approved in June 2005 and had very little market penetration during its first 6 months, corresponding to the last 6 months of the study period. Sitagliptin, the first DPP-4 inhibitor, had not yet reached the market.
Noel et al9 also found that the risk of biliary disease in patients with diabetes was 1.9 times higher than in those without diabetes. The relative risk of gallbladder disease was proportionally greater in a younger population with diabetes than in the population without diabetes, in whom the risk of gallbladder disease increases with age. Cholelithiasis was believed to be the underlying cause in at least 50% of the cases of pancreatitis.
PANCREATITIS AND INCRETIN-BASED THERAPIES
The estimated risk of acute pancreatitis in the population at large is reported as 0.33 to 0.44 events per 1,000 adults per year10; 15% to 20% of cases are considered severe, and 2% to 4% result in death.5,10 A relatively small number (1%–2%) are believed to be drug-induced.10
Exenatide. In the exenatide development program, six cases of acute pancreatitis were observed in about 3,489 subject-years of exposure (1.7 per 1,000 subject-years), compared with one case in about 336 subject-years with placebo (3.0 per 1,000 subject-years) and one case in about 497 subject-years (2.0 per 1,000 subject-years) with insulin.11
Sitagliptin. Dore et al12 examined claims from another database for the period of June 2005 through June 2008 to look specifically at the risk with incretin-based therapies. This database included 27,996 people starting exenatide and 16,276 people starting sitagliptin, matched with people with type 2 diabetes taking metformin (Glucophage) or glyburide. Over a period of 1 year, 0.13% of exenatide users and 0.12% of sitagliptin users suffered acute pancreatitis. The risk of pancreatitis was comparable in each group:
- For exenatide, relative risk (RR) 1.0, 95% confidence interval (CI) 0.6 to 1.7, compared with metformin or glyburide
- For sitagliptin, RR 1.0, 95% CI 0.5 to 2.0.
Saxagliptin. In clinical trials of saxagliptin, the incidence of pancreatitis was 0.2% in 3,422 patients receiving saxagliptin and 0.2% in 1,066 controls,13 similar to the rates for sitagliptin and exenatide.
Liraglutide appeared to be associated with a risk of acute pancreatitis, with seven cases in 3,900 patients receiving liraglutide vs one case in a patient taking another diabetes drug.14 This rate is similar to that reported in exenatide clinical trials, suggesting that pancreatitis has been underreported in the comparator subjects. We need more experience to see if this agent really poses more risk than other antidiabetic therapies.
As new antidiabetic agents enter the market and their use becomes common, it would not be surprising to see rates of pancreatitis similar to those reported by Blomgren et al2 in 2002, when glyburide was becoming a mainstay of therapy for type 2 diabetes.
- Trivedi CD, Pitchumoni CS. Drug-induced pancreatitis: an update. J Clin Gastroenterol 2005; 39:709–716.
- Blomgren KB, Sundström A, Steineck G, Wiholm BE. Obesity and treatment of diabetes with glyburide may both be risk factors for acute pancreatitis. Diabetes Care 2002; 25:298–302.
- US Food and Drug Administration. Information for healthcare professionals: exenatide (marketed as Byetta)—8/2008 update. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm124713.htm. Accessed July 1, 2010.
- US Food and Drug Administration. Information for healthcare professionals—acute pancreatitis and sitagliptin (marketed as Januvia and Janumet). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm183764.htm. Accessed July 1, 2010.
- Forsmark CE, Baillie J; AGA Institute Clinical Practice and Economics Committee. AGA Institute technical review on acute pancreatitis. Gastroenterology 2007; 132:2022–2044.
- Pagliarulo M, Fornari F, Fraquelli M, et al. Gallstone disease and related risk factors in a large cohort of diabetic patients. Dig Liver Dis 2004; 36:130–134.
- Field AE, Coakley EH, Must A, et al. Impact of overweight on the risk of developing common chronic diseases during a 10-year period. Arch Intern Med 2001; 161:1581–1586.
- Frulloni L, Lunardi C, Simone R, et al. Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med 2009; 361:2135–2142.
- Noel RA, Braun DK, Patterson RE, Bloomgren GL. Increased risk of acute pancreatitis and biliary disease observed in patients with type 2 diabetes: a retrospective cohort study. Diabetes Care 2009; 32:834–838.
- Whitcomb DC. Clinical practice. Acute pancreatitis. N Engl J Med 2006; 354:2142–2150.
- Data on file, Amylin Pharmaceuticals, Inc. and Eli Lilly.
- Dore DD, Seeger JD, Arnold Chan K. Use of a claims-based active drug safety surveillance system to assess the risk of acute pancreatitis with exenatide or sitagliptin compared to metformin or glyburide. Curr Med Res Opin 2009; 25:1019–1027.
- US Food and Drug Administration. Controlled Phase 2b/3 Pooled Population—Day 120 Update. http://www.fda.gov/downloads/AdvisoryCommittees/Committees-MeetingMaterials/Drugs/EndocrinologicandMetabolic-DrugsAdvisoryCommittee/UCM149589. Accessed July 4, 2010.
- US Food and Drug Administration. Questions and answers—safety requirements for Victoza (liraglutide). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrug-SafetyInformationforPatientsandProviders/ucm198543.htm. Accessed July 4, 2010.
- Trivedi CD, Pitchumoni CS. Drug-induced pancreatitis: an update. J Clin Gastroenterol 2005; 39:709–716.
- Blomgren KB, Sundström A, Steineck G, Wiholm BE. Obesity and treatment of diabetes with glyburide may both be risk factors for acute pancreatitis. Diabetes Care 2002; 25:298–302.
- US Food and Drug Administration. Information for healthcare professionals: exenatide (marketed as Byetta)—8/2008 update. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm124713.htm. Accessed July 1, 2010.
- US Food and Drug Administration. Information for healthcare professionals—acute pancreatitis and sitagliptin (marketed as Januvia and Janumet). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm183764.htm. Accessed July 1, 2010.
- Forsmark CE, Baillie J; AGA Institute Clinical Practice and Economics Committee. AGA Institute technical review on acute pancreatitis. Gastroenterology 2007; 132:2022–2044.
- Pagliarulo M, Fornari F, Fraquelli M, et al. Gallstone disease and related risk factors in a large cohort of diabetic patients. Dig Liver Dis 2004; 36:130–134.
- Field AE, Coakley EH, Must A, et al. Impact of overweight on the risk of developing common chronic diseases during a 10-year period. Arch Intern Med 2001; 161:1581–1586.
- Frulloni L, Lunardi C, Simone R, et al. Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med 2009; 361:2135–2142.
- Noel RA, Braun DK, Patterson RE, Bloomgren GL. Increased risk of acute pancreatitis and biliary disease observed in patients with type 2 diabetes: a retrospective cohort study. Diabetes Care 2009; 32:834–838.
- Whitcomb DC. Clinical practice. Acute pancreatitis. N Engl J Med 2006; 354:2142–2150.
- Data on file, Amylin Pharmaceuticals, Inc. and Eli Lilly.
- Dore DD, Seeger JD, Arnold Chan K. Use of a claims-based active drug safety surveillance system to assess the risk of acute pancreatitis with exenatide or sitagliptin compared to metformin or glyburide. Curr Med Res Opin 2009; 25:1019–1027.
- US Food and Drug Administration. Controlled Phase 2b/3 Pooled Population—Day 120 Update. http://www.fda.gov/downloads/AdvisoryCommittees/Committees-MeetingMaterials/Drugs/EndocrinologicandMetabolic-DrugsAdvisoryCommittee/UCM149589. Accessed July 4, 2010.
- US Food and Drug Administration. Questions and answers—safety requirements for Victoza (liraglutide). http://www.fda.gov/Drugs/DrugSafety/PostmarketDrug-SafetyInformationforPatientsandProviders/ucm198543.htm. Accessed July 4, 2010.