User login
The Molting Man: Anasarca-Induced Full-Body Desquamation
Edema blisters are a common but often underreported entity most commonly seen on the lower extremities in the setting of acute edema. 1 Reported risk factors and associations include chronic venous insufficiency, congestive heart failure, hereditary angioedema, and medications (eg, amlodipine). 1,2 We report a newly described variant that we have termed anasarca-induced desquamation in which a patient sloughed the entire cutaneous surface of the body after gaining almost 40 pounds over 5 days.
Case Report
A 50-year-old man without a home was found minimally responsive in a yard. His core body temperature was 25.5 °C. He was profoundly acidotic (pH, <6.733 [reference range, 7.35–7.45]; lactic acid, 20.5 mmol/L [reference range, 0.5–2.2 mmol/L]) at admission. His medical history was notable for diabetes mellitus, hypertension, alcohol abuse, and pulmonary embolism. The patient was resuscitated with rewarming and intravenous fluids in the setting of acute renal insufficiency. By day 5 of the hospital stay, he had a net positive intake of 21.8 L and an 18-kg (39.7-lb) weight gain.

Dermatology was consulted for skin sloughing. Physical examination revealed nonpainful desquamation of the vermilion lip, periorbital skin, right shoulder, and hips without notable mucosal changes. Two 4-mm punch biopsies of the shoulder revealed an intracorneal split with desquamation of the stratum corneum and a mild dermal lymphocytic infiltrate, consistent with exfoliation secondary to edema or staphylococcal scalded skin syndrome (Figure 1). No staphylococcal growth was noted on blood, urine, nasal, wound, and ocular cultures throughout the hospital stay.

As the patient’s anasarca improved with diuretics and continuous renal replacement therapy, the entire cutaneous surface—head to toe—underwent desquamation, including the palms and soles. He was managed with supportive skin care. The anasarca healed completely with residual hypopigmentation (Figures 2 and 3).

Comment
Anasarca-induced desquamation represents a more diffuse form of a known entity: edema blisters. Occurring most commonly in the setting of acute exacerbation of chronic venous insufficiency, edema blisters can mimic other vesiculobullous conditions, such as bullous pemphigoid and herpes zoster.3
Pathogenesis of Edema Blisters—Edema develops in the skin when the capillary filtration rate, determined by the hydrostatic and oncotic pressures of the capillaries and interstitium, exceeds venous and lymphatic drainage. The appearance of edema blisters in the acute setting likely is related to the speed at which edema develops in skin.1 Although edema blisters often are described as tense, there is a paucity of histologic data at the anatomical level of split in the skin.In our patient, desquamation was within the stratum corneum and likely multifactorial. His weight gain of nearly 40 lb, the result of intravenous instillation of fluids and low urine output, was undeniably a contributing factor. The anasarca was aggravated by hypoalbuminemia (2.1 g/dL) in the setting of known liver disease. Other possible contributing factors were hypotension, which required vasopressor therapy that led to hypoperfusion of the skin, and treatment of hypothermia, with resulting reactive vasodilation and capillary leak.
Management—Treatment of acute edema blisters is focused on the underlying cause of the edema. In a study of 13 patients with edema blisters, all had blisters on the legs that resolved with treatment, such as diuretics or compression therapy.1
Anasarca-induced desquamation is an inherently benign condition that mimics potentially fatal disorders, such as Stevens-Johnson syndrome, staphylococcal scalded skin syndrome, and toxic shock syndrome. Therefore, patients presenting with diffuse superficial desquamation should be assessed for the mucosal changes of Stevens-Johnson syndrome and a history of acute edema in the affected areas to avoid potentially harmful empiric treatments, such as corticosteroids and intravenous antibiotics.
Conclusion
Anasarca-induced desquamation represents a more diffuse form of edema blisters. This desquamation can mimic a potentially fatal rash, such as Stevens-Johnson syndrome and staphylococcal scalded skin syndrome.
- Bhushan M, Chalmers RJ, Cox NH. Acute oedema blisters: a report of 13 cases. Br J Dermatol. 2001;144:580-582. doi:10.1046/j.1365-2133.2001.04087.x
- Fabiani J, Bork K. Acute edema blisters on a skin swelling: an unusual manifestation of hereditary angioedema. Acta Derm Venereol. 2016;96:556-557. doi:10.2340/00015555-2252
- Chen SX, Cohen PR. Edema bullae mimicking disseminated herpes zoster. Cureus. 2017;9:E1780. doi:10.7759/cureus.1780
Edema blisters are a common but often underreported entity most commonly seen on the lower extremities in the setting of acute edema. 1 Reported risk factors and associations include chronic venous insufficiency, congestive heart failure, hereditary angioedema, and medications (eg, amlodipine). 1,2 We report a newly described variant that we have termed anasarca-induced desquamation in which a patient sloughed the entire cutaneous surface of the body after gaining almost 40 pounds over 5 days.
Case Report
A 50-year-old man without a home was found minimally responsive in a yard. His core body temperature was 25.5 °C. He was profoundly acidotic (pH, <6.733 [reference range, 7.35–7.45]; lactic acid, 20.5 mmol/L [reference range, 0.5–2.2 mmol/L]) at admission. His medical history was notable for diabetes mellitus, hypertension, alcohol abuse, and pulmonary embolism. The patient was resuscitated with rewarming and intravenous fluids in the setting of acute renal insufficiency. By day 5 of the hospital stay, he had a net positive intake of 21.8 L and an 18-kg (39.7-lb) weight gain.
Dermatology was consulted for skin sloughing. Physical examination revealed nonpainful desquamation of the vermilion lip, periorbital skin, right shoulder, and hips without notable mucosal changes. Two 4-mm punch biopsies of the shoulder revealed an intracorneal split with desquamation of the stratum corneum and a mild dermal lymphocytic infiltrate, consistent with exfoliation secondary to edema or staphylococcal scalded skin syndrome (Figure 1). No staphylococcal growth was noted on blood, urine, nasal, wound, and ocular cultures throughout the hospital stay.
As the patient’s anasarca improved with diuretics and continuous renal replacement therapy, the entire cutaneous surface—head to toe—underwent desquamation, including the palms and soles. He was managed with supportive skin care. The anasarca healed completely with residual hypopigmentation (Figures 2 and 3).
Comment
Anasarca-induced desquamation represents a more diffuse form of a known entity: edema blisters. Occurring most commonly in the setting of acute exacerbation of chronic venous insufficiency, edema blisters can mimic other vesiculobullous conditions, such as bullous pemphigoid and herpes zoster.3
Pathogenesis of Edema Blisters—Edema develops in the skin when the capillary filtration rate, determined by the hydrostatic and oncotic pressures of the capillaries and interstitium, exceeds venous and lymphatic drainage. The appearance of edema blisters in the acute setting likely is related to the speed at which edema develops in skin.1 Although edema blisters often are described as tense, there is a paucity of histologic data at the anatomical level of split in the skin.In our patient, desquamation was within the stratum corneum and likely multifactorial. His weight gain of nearly 40 lb, the result of intravenous instillation of fluids and low urine output, was undeniably a contributing factor. The anasarca was aggravated by hypoalbuminemia (2.1 g/dL) in the setting of known liver disease. Other possible contributing factors were hypotension, which required vasopressor therapy that led to hypoperfusion of the skin, and treatment of hypothermia, with resulting reactive vasodilation and capillary leak.
Management—Treatment of acute edema blisters is focused on the underlying cause of the edema. In a study of 13 patients with edema blisters, all had blisters on the legs that resolved with treatment, such as diuretics or compression therapy.1
Anasarca-induced desquamation is an inherently benign condition that mimics potentially fatal disorders, such as Stevens-Johnson syndrome, staphylococcal scalded skin syndrome, and toxic shock syndrome. Therefore, patients presenting with diffuse superficial desquamation should be assessed for the mucosal changes of Stevens-Johnson syndrome and a history of acute edema in the affected areas to avoid potentially harmful empiric treatments, such as corticosteroids and intravenous antibiotics.
Conclusion
Anasarca-induced desquamation represents a more diffuse form of edema blisters. This desquamation can mimic a potentially fatal rash, such as Stevens-Johnson syndrome and staphylococcal scalded skin syndrome.
Edema blisters are a common but often underreported entity most commonly seen on the lower extremities in the setting of acute edema. 1 Reported risk factors and associations include chronic venous insufficiency, congestive heart failure, hereditary angioedema, and medications (eg, amlodipine). 1,2 We report a newly described variant that we have termed anasarca-induced desquamation in which a patient sloughed the entire cutaneous surface of the body after gaining almost 40 pounds over 5 days.
Case Report
A 50-year-old man without a home was found minimally responsive in a yard. His core body temperature was 25.5 °C. He was profoundly acidotic (pH, <6.733 [reference range, 7.35–7.45]; lactic acid, 20.5 mmol/L [reference range, 0.5–2.2 mmol/L]) at admission. His medical history was notable for diabetes mellitus, hypertension, alcohol abuse, and pulmonary embolism. The patient was resuscitated with rewarming and intravenous fluids in the setting of acute renal insufficiency. By day 5 of the hospital stay, he had a net positive intake of 21.8 L and an 18-kg (39.7-lb) weight gain.
Dermatology was consulted for skin sloughing. Physical examination revealed nonpainful desquamation of the vermilion lip, periorbital skin, right shoulder, and hips without notable mucosal changes. Two 4-mm punch biopsies of the shoulder revealed an intracorneal split with desquamation of the stratum corneum and a mild dermal lymphocytic infiltrate, consistent with exfoliation secondary to edema or staphylococcal scalded skin syndrome (Figure 1). No staphylococcal growth was noted on blood, urine, nasal, wound, and ocular cultures throughout the hospital stay.
As the patient’s anasarca improved with diuretics and continuous renal replacement therapy, the entire cutaneous surface—head to toe—underwent desquamation, including the palms and soles. He was managed with supportive skin care. The anasarca healed completely with residual hypopigmentation (Figures 2 and 3).
Comment
Anasarca-induced desquamation represents a more diffuse form of a known entity: edema blisters. Occurring most commonly in the setting of acute exacerbation of chronic venous insufficiency, edema blisters can mimic other vesiculobullous conditions, such as bullous pemphigoid and herpes zoster.3
Pathogenesis of Edema Blisters—Edema develops in the skin when the capillary filtration rate, determined by the hydrostatic and oncotic pressures of the capillaries and interstitium, exceeds venous and lymphatic drainage. The appearance of edema blisters in the acute setting likely is related to the speed at which edema develops in skin.1 Although edema blisters often are described as tense, there is a paucity of histologic data at the anatomical level of split in the skin.In our patient, desquamation was within the stratum corneum and likely multifactorial. His weight gain of nearly 40 lb, the result of intravenous instillation of fluids and low urine output, was undeniably a contributing factor. The anasarca was aggravated by hypoalbuminemia (2.1 g/dL) in the setting of known liver disease. Other possible contributing factors were hypotension, which required vasopressor therapy that led to hypoperfusion of the skin, and treatment of hypothermia, with resulting reactive vasodilation and capillary leak.
Management—Treatment of acute edema blisters is focused on the underlying cause of the edema. In a study of 13 patients with edema blisters, all had blisters on the legs that resolved with treatment, such as diuretics or compression therapy.1
Anasarca-induced desquamation is an inherently benign condition that mimics potentially fatal disorders, such as Stevens-Johnson syndrome, staphylococcal scalded skin syndrome, and toxic shock syndrome. Therefore, patients presenting with diffuse superficial desquamation should be assessed for the mucosal changes of Stevens-Johnson syndrome and a history of acute edema in the affected areas to avoid potentially harmful empiric treatments, such as corticosteroids and intravenous antibiotics.
Conclusion
Anasarca-induced desquamation represents a more diffuse form of edema blisters. This desquamation can mimic a potentially fatal rash, such as Stevens-Johnson syndrome and staphylococcal scalded skin syndrome.
- Bhushan M, Chalmers RJ, Cox NH. Acute oedema blisters: a report of 13 cases. Br J Dermatol. 2001;144:580-582. doi:10.1046/j.1365-2133.2001.04087.x
- Fabiani J, Bork K. Acute edema blisters on a skin swelling: an unusual manifestation of hereditary angioedema. Acta Derm Venereol. 2016;96:556-557. doi:10.2340/00015555-2252
- Chen SX, Cohen PR. Edema bullae mimicking disseminated herpes zoster. Cureus. 2017;9:E1780. doi:10.7759/cureus.1780
- Bhushan M, Chalmers RJ, Cox NH. Acute oedema blisters: a report of 13 cases. Br J Dermatol. 2001;144:580-582. doi:10.1046/j.1365-2133.2001.04087.x
- Fabiani J, Bork K. Acute edema blisters on a skin swelling: an unusual manifestation of hereditary angioedema. Acta Derm Venereol. 2016;96:556-557. doi:10.2340/00015555-2252
- Chen SX, Cohen PR. Edema bullae mimicking disseminated herpes zoster. Cureus. 2017;9:E1780. doi:10.7759/cureus.1780
Practice Points
- The appearance of anasarca-induced desquamation can be similar to staphylococcal scalded skin syndrome and Stevens-Johnson syndrome.
- Histopathologic evaluation of this condition shows desquamation localized to the stratum corneum without epidermal necrosis.
- Careful evaluation, including bacterial culture, is required to rule out an infectious cause.
- Early diagnosis of anasarca-induced desquamation reduces the potential for providing harmful empiric treatment, such as systemic steroids and intravenous antibiotics, especially in patients known to have comorbidities.

Nonuremic Calciphylaxis Triggered by Rapid Weight Loss and Hypotension
Calciphylaxis, otherwise known as calcific uremic arteriolopathy, is characterized by calcification of the tunica media of the small- to medium-sized blood vessels of the dermis and subcutis, leading to ischemia and necrosis.1 It is a deadly disease with a 1-year mortality rate of more than 50%.2 End-stage renal disease (ESRD) is the most common risk factor for calciphylaxis, with a prevalence of 1% to 4% of hemodialysis patients with calciphylaxis in the United States.2-5 However, nonuremic calciphylaxis (NUC) has been increasingly reported in the literature and has risk factors other than ESRD, including but not limited to obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, and underlying malignancy.3,6-9 Triggers for calciphylaxis in at-risk patients include use of corticosteroids or warfarin, iron or albumin infusions, and rapid weight loss.3,6,9-11 We report an unusual case of NUC that most likely was triggered by rapid weight loss and hypotension in a patient with multiple risk factors for calciphylaxis.
Case Report
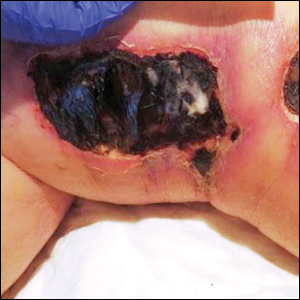
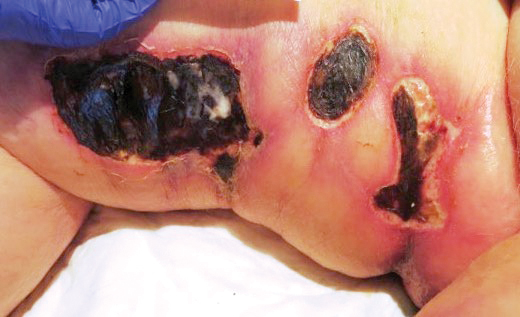
A 75-year-old white woman with history of morbid obesity (body mass index, 40 kg/m2), unexplained weight loss of 70 lb over the last year, and polymyalgia rheumatica requiring chronic prednisone therapy presented with painful lesions on the thighs, buttocks, and right shoulder of 4 months’ duration. She had multiple hospital admissions preceding the onset of lesions for severe infections resulting in sepsis with hypotension, including Enterococcus faecalis endocarditis, extended-spectrum beta-lactamase bacteremia, and Pseudomonas aeruginosa pneumonia. Physical examination revealed large well-demarcated ulcers and necrotic eschars with surrounding violaceous induration and stellate erythema on the anterior, medial, and posterior thighs and buttocks that were exquisitely tender (Figures 1 and 2).

Notable laboratory results included hypoalbuminemia (1.3 g/dL [reference range, 3.5–5.0 g/dL]) with normal renal function, a corrected calcium level of 9.7 mg/dL (reference range, 8.2–10.2 mg/dL), a serum phosphorus level of 3.5 mg/dL (reference range, 2.3–4.7 mg/dL), a calcium-phosphate product of 27.3 mg2/dL2 (reference range, <55 mg2/dL2), and a parathyroid hormone level of 49.3 pg/mL (reference range, 10–65 pg/mL). Antinuclear antibodies were negative. A hypercoagulability evaluation showed normal protein C and S levels, negative lupus anticoagulant, and negative anticardiolipin antibodies.
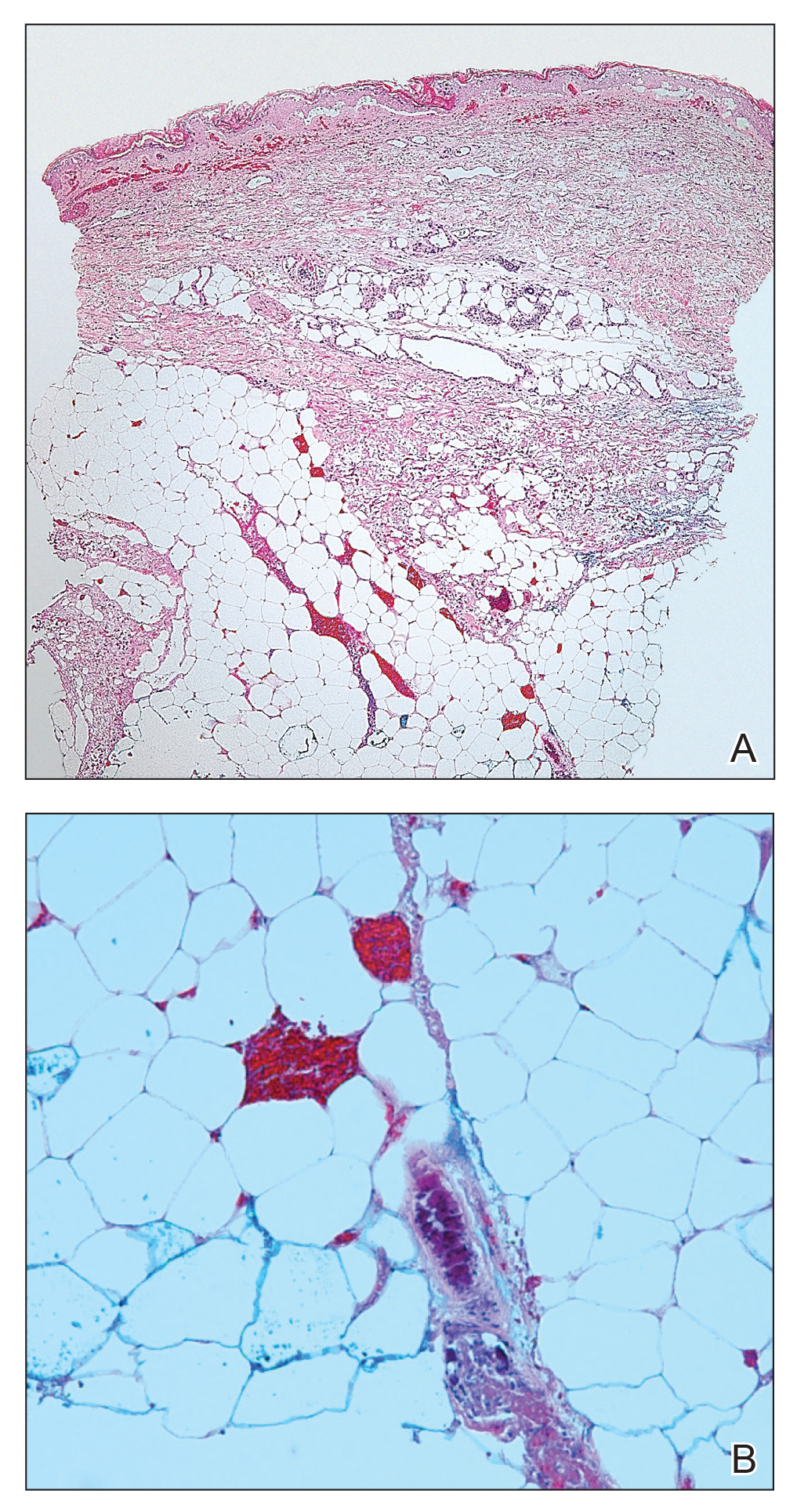
Telescoping punch biopsies of the indurated borders of the eschars showed prominent calcification of the small- and medium-sized vessels in the mid and deep dermis, intravascular thrombi, and necrosis of the epidermis and subcutaneous fat consistent with calciphylaxis (Figure 3).
After the diagnosis of calciphylaxis was made, the patient was treated with intravenous sodium thiosulfate 25 mg 3 times weekly and alendronate 70 mg weekly. Daily arterial blood gas studies did not detect metabolic acidosis during the patient’s sodium thiosulfate therapy. The wounds were debrided, and we attempted to slowly taper the patient off the oral prednisone. Unfortunately, her condition slowly deteriorated secondary to sepsis, resulting in septic shock. The patient died 3 weeks after the diagnosis of calciphylaxis was made. At the time of diagnosis, the patient had a poor prognosis and notable risk for sepsis due to the large eschars on the thighs and abdomen as well as her relative immunosuppression due to chronic prednisone use.
Comment
Background on Calciphylaxis
Calciphylaxis is a rare but deadly disease that affects both ESRD patients receiving dialysis and patients without ESRD who have known risk factors for calciphylaxis, including female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.3,6-9,11 Although the molecular pathogenesis of calciphylaxis is not completely understood, it is believed to be caused by local deposition of calcium in the tunica media of small- to medium-sized arterioles and venules in the skin.12 This deposition leads to intimal proliferation and progressive narrowing of the vessels with resultant thrombosis, ischemia, and necrosis. The cutaneous manifestations and histopathology of calciphylaxis classically follow its pathogenesis. Calciphylaxis typically presents with livedo reticularis as vessels narrow and then progresses to purpura, bullae, necrosis, and eschar formation with the onset of acute thrombosis and ischemia. Histopathology is characterized by small- and medium-sized vessel calcification and thrombus, dermal necrosis, and septal panniculitis, though the histology can be highly variable.12 Unfortunately, the already poor prognosis for calciphylaxis worsens when lesions become either ulcerative or present on the proximal extremities and trunk.4,13 Sepsis is the leading cause of death in calciphylaxis patients, affecting more than 50% of patients.2,3,14 The differential diagnoses for calciphylactic-appearing lesions include warfarin-induced skin necrosis, disseminated intravascular coagulation, pyoderma gangrenosum, cholesterol emboli, and various vasculitides and coagulopathies.
Risk Factors
Our case demonstrates the importance of risk factor minimization, trigger avoidance, and early intervention due to the high mortality rate of calciphylaxis. Selye et al15 coined the term calciphylaxis in 1961 based on experiments that induced calciphylaxis in rat models. Their research concluded that there were certain sensitizers (ie, risk factors) that predisposed patients to medial calcium deposition in blood vessels and other challengers (ie, triggers) that acted as inciting events to calcium deposition. Our patient presented with multiple known risk factors for calciphylaxis, including obesity (body mass index, 40 kg/m2), female gender, white race, hypoalbuminemia, and chronic corticosteroid use.16 In the presence of a milieu of risk factors, the patient’s rapid weight loss and episodes of hypotension likely were triggers for calciphylaxis.
Other case reports in the literature have suggested weight loss as a trigger for NUC. One morbidly obese patient with inactive rheumatoid arthritis had onset of calciphylaxis lesions after unintentional weight loss of approximately 50% body weight in 1 year17; however, the weight loss does not have to be drastic to trigger calciphylaxis. Another study of 16 patients with uremic calciphylaxis found that 7 of 16 (44%) patients lost 10 to 50 kg in the 6 months prior to calciphylaxis onset.14 One proposed mechanism by Munavalli et al10 is that elevated levels of matrix metalloproteinases during catabolic weight loss states enhance the deposition of calcium into elastic fibers of small vessels. The authors found elevated serum levels of matrix metalloproteinases in their patients with NUC induced by rapid weight loss.10
A meta-analysis by Nigwekar et al3 found a history of prior corticosteroid use in 61% (22/36) of NUC cases reviewed. However, it is unclear whether it is the use of corticosteroids or chronic inflammation that is implicated in NUC pathogenesis. Chronic inflammation causes downregulation of anticalcification signaling pathways.18-20 The role of 2 vascular calcification inhibitors has been evaluated in the pathogenesis of calciphylaxis: fetuin-A and matrix gla protein (MGP).21 The activity of these proteins is decreased not only in calciphylaxis but also in other inflammatory states and chronic renal failure.18-20 One study found lower fetuin-A levels in 312 hemodialysis patients compared to healthy controls and an association between low fetuin-A levels and increased C-reactive protein levels.22 Reduced fetuin-A and MGP levels may be the result of several calciphylaxis risk factors. Warfarin is believed to trigger calciphylaxis via inhibition of gamma-carboxylation of MGP, which is necessary for its anticalcification activity.23 Hypoalbuminemia and alcoholic liver disease also are risk factors that may be explained by the fact that fetuin-A is synthesized in the liver.24 Therefore, liver disease results in decreased production of fetuin-A that is permissive to vascular calcification in calciphylaxis patients.
There have been other reports of calciphylaxis patients who were originally hospitalized due to hypotension, which may serve as a trigger for calciphylaxis onset.25 Because calciphylaxis lesions are more likely to occur in the fatty areas of the abdomen and proximal thighs where blood flow is slower, hypotension likely accentuates the slowing of blood flow and subsequent blood vessel calcification. This theory is supported by studies showing that established calciphylactic lesions worsen more quickly in the presence of systemic hypotension.26 One patient with ESRD and calciphylaxis of the breasts had consistent systolic blood pressure readings in the high 60s to low 70s between dialysis sessions.27 Due to this association, we recommend that patients with calciphylaxis have close blood pressure monitoring to aid in preventing disease progression.28
Management
Calciphylaxis treatment has not yet been standardized, as it is an uncommon disease whose pathogenesis is not fully understood. Current management strategies aim to normalize metabolic abnormalities such as hypercalcemia if they are present and remove inciting agents such as warfarin and corticosteroids.29 Other medical treatments that have been successfully used include sodium thiosulfate, oral steroids, and adjunctive bisphosphonates.29-31 Sodium thiosulfate is known to cause metabolic acidosis by generating thiosulfuric acid in vivo in patients with or without renal disease; therefore, patients on sodium thiosulfate therapy should be monitored for development of metabolic acidosis and treated with oral sodium bicarbonate or dialysis as needed.30,32 Wound care also is an important element of calciphylaxis treatment; however, the debridement of wounds is controversial. Some argue that dry intact eschars serve to protect against sepsis, which is the leading cause of death in calciphylaxis.2,14,33 In contrast, a retrospective study of 63 calciphylaxis patients found a 1-year survival rate of 61.6% in 17 patients receiving wound debridement vs 27.4% in 46 patients who did not.2 The current consensus is that debridement should be considered on a case-by-case basis, factoring in the presence of wound infection, size of wounds, stability of eschars, and treatment goals of the patient.34 Future studies should be aimed at this issue, with special focus on how these factors and the decision to debride or not impact patient outcomes.
Conclusion
Calciphylaxis is a potentially fatal disease that impacts both patients with ESRD and those with nonuremic risk factors. The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature. In such cases, patients often have multiple risk factors, including obesity, primary hyperparathyroidism, alcoholic liver disease, and underlying malignancy, among others. Certain triggers for onset of calciphylaxis should be avoided in at-risk patients, including the use of corticosteroids or warfarin; iron and albumin infusions; hypotension; and rapid weight loss. Our fatal case of NUC is a reminder to dermatologists treating at-risk patients to avoid these triggers and to keep calciphylaxis in the differential diagnosis when encountering early lesions such as livedo reticularis, as progression of these lesions has a 1-year mortality rate of more than 50% with the therapies being utilized at this time.
- Au S, Crawford RI. Three-dimensional analysis of a calciphylaxis plaque: clues to pathogenesis. J Am Acad Dermatol. 2007;47:53-57.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Angelis M, Wong LL, Myers SA, et al. Calciphylaxis in patients on hemodialysis: a prevalence study. Surgery. 1997;122:1083-1090.
- Chavel SM, Taraszka KS, Schaffer JV, et al. Calciphylaxis associated with acute, reversible renal failure in the setting of alcoholic cirrhosis. J Am Acad Dermatol. 2004;50:125-128.
- Bosler DS, Amin MB, Gulli F, et al. Unusual case of calciphylaxis associated with metastatic breast carcinoma. Am J Dermatopathol. 2007;29:400-403.
- Buxtorf K, Cerottini JP, Panizzon RG. Lower limb skin ulcerations, intravascular calcifications and sensorimotor polyneuropathy: calciphylaxis as part of a hyperparathyroidism? Dermatology. 1999;198:423-425.
- Brouns K, Verbeken E, Degreef H, et al. Fatal calciphylaxis in two patients with giant cell arteritis. Clin Rheumatol. 2007;26:836-840.
- Munavalli G, Reisenauer A, Moses M, et al. Weight loss-induced calciphylaxis: potential role of matrix metalloproteinases. J Dermatol. 2003;30:915-919.
- Bae GH, Nambudiri VE, Bach DQ, et al. Rapidly progressive nonuremic calciphylaxis in setting of warfarin. Am J Med. 2015;128:E19-E21.
- Essary LR, Wick MR. Cutaneous calciphylaxis. an underrecognized clinicopathologic entity. Am J Clin Pathol. 2000;113:280-287.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Coates T, Kirkland GS, Dymock RB, et al. Cutaneous necrosis from calcific uremic arteriolopathy. Am J Kidney Dis. 1998;32:384-391.
- Selye H, Gentile G, Prioreschi P. Cutaneous molt induced by calciphylaxis in the rat. Science. 1961;134:1876-1877.
- Kalajian AH, Malhotra PS, Callen JP, et al. Calciphylaxis with normal renal and parathyroid function: not as rare as previously believed. Arch Dermatol. 2009;145:451-458.
- Malabu U, Roberts L, Sangla K. Calciphylaxis in a morbidly obese woman with rheumatoid arthritis presenting with severe weight loss and vitamin D deficiency. Endocr Pract. 2011;17:104-108.
- Schäfer C, Heiss A, Schwarz A, et al. The serum protein alpha 2–Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Cozzolino M, Galassi A, Biondi ML, et al. Serum fetuin-A levels link inflammation and cardiovascular calcification in hemodialysis patients. Am J Nephrol. 2006;26:423-429.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Weenig RH. Pathogenesis of calciphylaxis: Hans Selye to nuclear factor kappa-B. J Am Acad Dermatol. 2008;58:458-471.
- Ketteler M, Bongartz P, Westenfeld R, et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet. 2003;361:827-833.
- Wallin R, Cain D, Sane DC. Matrix Gla protein synthesis and gamma-carboxylation in the aortic vessel wall and proliferating vascular smooth muscle cells a cell system which resembles the system in bone cells. Thromb Haemost. 1999;82:1764-1767.
- Sowers KM, Hayden MR. Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches. Oxid Med Cell Longev. 2010;3:109-121.
- Allegretti AS, Nazarian RM, Goverman J, et al. Calciphylaxis: a rare but fatal delayed complication of Roux-en-Y gastric bypass surgery. Am J Kidney Dis. 2014;64:274-277.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial. 2002;15:172-186.
- Gupta D, Tadros R, Mazumdar A, et al. Breast lesions with intractable pain in end-stage renal disease: calciphylaxis with chronic hypotensive dermatopathy related watershed breast lesions. J Palliat Med. 2013;16:551-554.
- Janigan DT, Hirsch DJ, Klassen GA, et al. Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis. 2000;35:588-597.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Bourgeois P, De Haes P. Sodium thiosulfate as a treatment for calciphylaxis: a case series. J Dermatolog Treat. 2016;27:520-524.
- Biswas A, Walsh NM, Tremaine R. A case of nonuremic calciphylaxis treated effectively with systemic corticosteroids. J Cutan Med Surg. 2016;20:275-278.
- Selk N, Rodby, RA. Unexpectedly severe metabolic acidosis associated with sodium thiosulfate therapy in a patient with calcific uremic arteriolopathy. Semin Dial. 2011;24:85-88.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage. 2004:50:64-66, 68-70.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
Calciphylaxis, otherwise known as calcific uremic arteriolopathy, is characterized by calcification of the tunica media of the small- to medium-sized blood vessels of the dermis and subcutis, leading to ischemia and necrosis.1 It is a deadly disease with a 1-year mortality rate of more than 50%.2 End-stage renal disease (ESRD) is the most common risk factor for calciphylaxis, with a prevalence of 1% to 4% of hemodialysis patients with calciphylaxis in the United States.2-5 However, nonuremic calciphylaxis (NUC) has been increasingly reported in the literature and has risk factors other than ESRD, including but not limited to obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, and underlying malignancy.3,6-9 Triggers for calciphylaxis in at-risk patients include use of corticosteroids or warfarin, iron or albumin infusions, and rapid weight loss.3,6,9-11 We report an unusual case of NUC that most likely was triggered by rapid weight loss and hypotension in a patient with multiple risk factors for calciphylaxis.
Case Report
A 75-year-old white woman with history of morbid obesity (body mass index, 40 kg/m2), unexplained weight loss of 70 lb over the last year, and polymyalgia rheumatica requiring chronic prednisone therapy presented with painful lesions on the thighs, buttocks, and right shoulder of 4 months’ duration. She had multiple hospital admissions preceding the onset of lesions for severe infections resulting in sepsis with hypotension, including Enterococcus faecalis endocarditis, extended-spectrum beta-lactamase bacteremia, and Pseudomonas aeruginosa pneumonia. Physical examination revealed large well-demarcated ulcers and necrotic eschars with surrounding violaceous induration and stellate erythema on the anterior, medial, and posterior thighs and buttocks that were exquisitely tender (Figures 1 and 2).
Notable laboratory results included hypoalbuminemia (1.3 g/dL [reference range, 3.5–5.0 g/dL]) with normal renal function, a corrected calcium level of 9.7 mg/dL (reference range, 8.2–10.2 mg/dL), a serum phosphorus level of 3.5 mg/dL (reference range, 2.3–4.7 mg/dL), a calcium-phosphate product of 27.3 mg2/dL2 (reference range, <55 mg2/dL2), and a parathyroid hormone level of 49.3 pg/mL (reference range, 10–65 pg/mL). Antinuclear antibodies were negative. A hypercoagulability evaluation showed normal protein C and S levels, negative lupus anticoagulant, and negative anticardiolipin antibodies.
Telescoping punch biopsies of the indurated borders of the eschars showed prominent calcification of the small- and medium-sized vessels in the mid and deep dermis, intravascular thrombi, and necrosis of the epidermis and subcutaneous fat consistent with calciphylaxis (Figure 3).
After the diagnosis of calciphylaxis was made, the patient was treated with intravenous sodium thiosulfate 25 mg 3 times weekly and alendronate 70 mg weekly. Daily arterial blood gas studies did not detect metabolic acidosis during the patient’s sodium thiosulfate therapy. The wounds were debrided, and we attempted to slowly taper the patient off the oral prednisone. Unfortunately, her condition slowly deteriorated secondary to sepsis, resulting in septic shock. The patient died 3 weeks after the diagnosis of calciphylaxis was made. At the time of diagnosis, the patient had a poor prognosis and notable risk for sepsis due to the large eschars on the thighs and abdomen as well as her relative immunosuppression due to chronic prednisone use.
Comment
Background on Calciphylaxis
Calciphylaxis is a rare but deadly disease that affects both ESRD patients receiving dialysis and patients without ESRD who have known risk factors for calciphylaxis, including female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.3,6-9,11 Although the molecular pathogenesis of calciphylaxis is not completely understood, it is believed to be caused by local deposition of calcium in the tunica media of small- to medium-sized arterioles and venules in the skin.12 This deposition leads to intimal proliferation and progressive narrowing of the vessels with resultant thrombosis, ischemia, and necrosis. The cutaneous manifestations and histopathology of calciphylaxis classically follow its pathogenesis. Calciphylaxis typically presents with livedo reticularis as vessels narrow and then progresses to purpura, bullae, necrosis, and eschar formation with the onset of acute thrombosis and ischemia. Histopathology is characterized by small- and medium-sized vessel calcification and thrombus, dermal necrosis, and septal panniculitis, though the histology can be highly variable.12 Unfortunately, the already poor prognosis for calciphylaxis worsens when lesions become either ulcerative or present on the proximal extremities and trunk.4,13 Sepsis is the leading cause of death in calciphylaxis patients, affecting more than 50% of patients.2,3,14 The differential diagnoses for calciphylactic-appearing lesions include warfarin-induced skin necrosis, disseminated intravascular coagulation, pyoderma gangrenosum, cholesterol emboli, and various vasculitides and coagulopathies.
Risk Factors
Our case demonstrates the importance of risk factor minimization, trigger avoidance, and early intervention due to the high mortality rate of calciphylaxis. Selye et al15 coined the term calciphylaxis in 1961 based on experiments that induced calciphylaxis in rat models. Their research concluded that there were certain sensitizers (ie, risk factors) that predisposed patients to medial calcium deposition in blood vessels and other challengers (ie, triggers) that acted as inciting events to calcium deposition. Our patient presented with multiple known risk factors for calciphylaxis, including obesity (body mass index, 40 kg/m2), female gender, white race, hypoalbuminemia, and chronic corticosteroid use.16 In the presence of a milieu of risk factors, the patient’s rapid weight loss and episodes of hypotension likely were triggers for calciphylaxis.
Other case reports in the literature have suggested weight loss as a trigger for NUC. One morbidly obese patient with inactive rheumatoid arthritis had onset of calciphylaxis lesions after unintentional weight loss of approximately 50% body weight in 1 year17; however, the weight loss does not have to be drastic to trigger calciphylaxis. Another study of 16 patients with uremic calciphylaxis found that 7 of 16 (44%) patients lost 10 to 50 kg in the 6 months prior to calciphylaxis onset.14 One proposed mechanism by Munavalli et al10 is that elevated levels of matrix metalloproteinases during catabolic weight loss states enhance the deposition of calcium into elastic fibers of small vessels. The authors found elevated serum levels of matrix metalloproteinases in their patients with NUC induced by rapid weight loss.10
A meta-analysis by Nigwekar et al3 found a history of prior corticosteroid use in 61% (22/36) of NUC cases reviewed. However, it is unclear whether it is the use of corticosteroids or chronic inflammation that is implicated in NUC pathogenesis. Chronic inflammation causes downregulation of anticalcification signaling pathways.18-20 The role of 2 vascular calcification inhibitors has been evaluated in the pathogenesis of calciphylaxis: fetuin-A and matrix gla protein (MGP).21 The activity of these proteins is decreased not only in calciphylaxis but also in other inflammatory states and chronic renal failure.18-20 One study found lower fetuin-A levels in 312 hemodialysis patients compared to healthy controls and an association between low fetuin-A levels and increased C-reactive protein levels.22 Reduced fetuin-A and MGP levels may be the result of several calciphylaxis risk factors. Warfarin is believed to trigger calciphylaxis via inhibition of gamma-carboxylation of MGP, which is necessary for its anticalcification activity.23 Hypoalbuminemia and alcoholic liver disease also are risk factors that may be explained by the fact that fetuin-A is synthesized in the liver.24 Therefore, liver disease results in decreased production of fetuin-A that is permissive to vascular calcification in calciphylaxis patients.
There have been other reports of calciphylaxis patients who were originally hospitalized due to hypotension, which may serve as a trigger for calciphylaxis onset.25 Because calciphylaxis lesions are more likely to occur in the fatty areas of the abdomen and proximal thighs where blood flow is slower, hypotension likely accentuates the slowing of blood flow and subsequent blood vessel calcification. This theory is supported by studies showing that established calciphylactic lesions worsen more quickly in the presence of systemic hypotension.26 One patient with ESRD and calciphylaxis of the breasts had consistent systolic blood pressure readings in the high 60s to low 70s between dialysis sessions.27 Due to this association, we recommend that patients with calciphylaxis have close blood pressure monitoring to aid in preventing disease progression.28
Management
Calciphylaxis treatment has not yet been standardized, as it is an uncommon disease whose pathogenesis is not fully understood. Current management strategies aim to normalize metabolic abnormalities such as hypercalcemia if they are present and remove inciting agents such as warfarin and corticosteroids.29 Other medical treatments that have been successfully used include sodium thiosulfate, oral steroids, and adjunctive bisphosphonates.29-31 Sodium thiosulfate is known to cause metabolic acidosis by generating thiosulfuric acid in vivo in patients with or without renal disease; therefore, patients on sodium thiosulfate therapy should be monitored for development of metabolic acidosis and treated with oral sodium bicarbonate or dialysis as needed.30,32 Wound care also is an important element of calciphylaxis treatment; however, the debridement of wounds is controversial. Some argue that dry intact eschars serve to protect against sepsis, which is the leading cause of death in calciphylaxis.2,14,33 In contrast, a retrospective study of 63 calciphylaxis patients found a 1-year survival rate of 61.6% in 17 patients receiving wound debridement vs 27.4% in 46 patients who did not.2 The current consensus is that debridement should be considered on a case-by-case basis, factoring in the presence of wound infection, size of wounds, stability of eschars, and treatment goals of the patient.34 Future studies should be aimed at this issue, with special focus on how these factors and the decision to debride or not impact patient outcomes.
Conclusion
Calciphylaxis is a potentially fatal disease that impacts both patients with ESRD and those with nonuremic risk factors. The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature. In such cases, patients often have multiple risk factors, including obesity, primary hyperparathyroidism, alcoholic liver disease, and underlying malignancy, among others. Certain triggers for onset of calciphylaxis should be avoided in at-risk patients, including the use of corticosteroids or warfarin; iron and albumin infusions; hypotension; and rapid weight loss. Our fatal case of NUC is a reminder to dermatologists treating at-risk patients to avoid these triggers and to keep calciphylaxis in the differential diagnosis when encountering early lesions such as livedo reticularis, as progression of these lesions has a 1-year mortality rate of more than 50% with the therapies being utilized at this time.
Calciphylaxis, otherwise known as calcific uremic arteriolopathy, is characterized by calcification of the tunica media of the small- to medium-sized blood vessels of the dermis and subcutis, leading to ischemia and necrosis.1 It is a deadly disease with a 1-year mortality rate of more than 50%.2 End-stage renal disease (ESRD) is the most common risk factor for calciphylaxis, with a prevalence of 1% to 4% of hemodialysis patients with calciphylaxis in the United States.2-5 However, nonuremic calciphylaxis (NUC) has been increasingly reported in the literature and has risk factors other than ESRD, including but not limited to obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, and underlying malignancy.3,6-9 Triggers for calciphylaxis in at-risk patients include use of corticosteroids or warfarin, iron or albumin infusions, and rapid weight loss.3,6,9-11 We report an unusual case of NUC that most likely was triggered by rapid weight loss and hypotension in a patient with multiple risk factors for calciphylaxis.
Case Report
A 75-year-old white woman with history of morbid obesity (body mass index, 40 kg/m2), unexplained weight loss of 70 lb over the last year, and polymyalgia rheumatica requiring chronic prednisone therapy presented with painful lesions on the thighs, buttocks, and right shoulder of 4 months’ duration. She had multiple hospital admissions preceding the onset of lesions for severe infections resulting in sepsis with hypotension, including Enterococcus faecalis endocarditis, extended-spectrum beta-lactamase bacteremia, and Pseudomonas aeruginosa pneumonia. Physical examination revealed large well-demarcated ulcers and necrotic eschars with surrounding violaceous induration and stellate erythema on the anterior, medial, and posterior thighs and buttocks that were exquisitely tender (Figures 1 and 2).
Notable laboratory results included hypoalbuminemia (1.3 g/dL [reference range, 3.5–5.0 g/dL]) with normal renal function, a corrected calcium level of 9.7 mg/dL (reference range, 8.2–10.2 mg/dL), a serum phosphorus level of 3.5 mg/dL (reference range, 2.3–4.7 mg/dL), a calcium-phosphate product of 27.3 mg2/dL2 (reference range, <55 mg2/dL2), and a parathyroid hormone level of 49.3 pg/mL (reference range, 10–65 pg/mL). Antinuclear antibodies were negative. A hypercoagulability evaluation showed normal protein C and S levels, negative lupus anticoagulant, and negative anticardiolipin antibodies.
Telescoping punch biopsies of the indurated borders of the eschars showed prominent calcification of the small- and medium-sized vessels in the mid and deep dermis, intravascular thrombi, and necrosis of the epidermis and subcutaneous fat consistent with calciphylaxis (Figure 3).
After the diagnosis of calciphylaxis was made, the patient was treated with intravenous sodium thiosulfate 25 mg 3 times weekly and alendronate 70 mg weekly. Daily arterial blood gas studies did not detect metabolic acidosis during the patient’s sodium thiosulfate therapy. The wounds were debrided, and we attempted to slowly taper the patient off the oral prednisone. Unfortunately, her condition slowly deteriorated secondary to sepsis, resulting in septic shock. The patient died 3 weeks after the diagnosis of calciphylaxis was made. At the time of diagnosis, the patient had a poor prognosis and notable risk for sepsis due to the large eschars on the thighs and abdomen as well as her relative immunosuppression due to chronic prednisone use.
Comment
Background on Calciphylaxis
Calciphylaxis is a rare but deadly disease that affects both ESRD patients receiving dialysis and patients without ESRD who have known risk factors for calciphylaxis, including female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.3,6-9,11 Although the molecular pathogenesis of calciphylaxis is not completely understood, it is believed to be caused by local deposition of calcium in the tunica media of small- to medium-sized arterioles and venules in the skin.12 This deposition leads to intimal proliferation and progressive narrowing of the vessels with resultant thrombosis, ischemia, and necrosis. The cutaneous manifestations and histopathology of calciphylaxis classically follow its pathogenesis. Calciphylaxis typically presents with livedo reticularis as vessels narrow and then progresses to purpura, bullae, necrosis, and eschar formation with the onset of acute thrombosis and ischemia. Histopathology is characterized by small- and medium-sized vessel calcification and thrombus, dermal necrosis, and septal panniculitis, though the histology can be highly variable.12 Unfortunately, the already poor prognosis for calciphylaxis worsens when lesions become either ulcerative or present on the proximal extremities and trunk.4,13 Sepsis is the leading cause of death in calciphylaxis patients, affecting more than 50% of patients.2,3,14 The differential diagnoses for calciphylactic-appearing lesions include warfarin-induced skin necrosis, disseminated intravascular coagulation, pyoderma gangrenosum, cholesterol emboli, and various vasculitides and coagulopathies.
Risk Factors
Our case demonstrates the importance of risk factor minimization, trigger avoidance, and early intervention due to the high mortality rate of calciphylaxis. Selye et al15 coined the term calciphylaxis in 1961 based on experiments that induced calciphylaxis in rat models. Their research concluded that there were certain sensitizers (ie, risk factors) that predisposed patients to medial calcium deposition in blood vessels and other challengers (ie, triggers) that acted as inciting events to calcium deposition. Our patient presented with multiple known risk factors for calciphylaxis, including obesity (body mass index, 40 kg/m2), female gender, white race, hypoalbuminemia, and chronic corticosteroid use.16 In the presence of a milieu of risk factors, the patient’s rapid weight loss and episodes of hypotension likely were triggers for calciphylaxis.
Other case reports in the literature have suggested weight loss as a trigger for NUC. One morbidly obese patient with inactive rheumatoid arthritis had onset of calciphylaxis lesions after unintentional weight loss of approximately 50% body weight in 1 year17; however, the weight loss does not have to be drastic to trigger calciphylaxis. Another study of 16 patients with uremic calciphylaxis found that 7 of 16 (44%) patients lost 10 to 50 kg in the 6 months prior to calciphylaxis onset.14 One proposed mechanism by Munavalli et al10 is that elevated levels of matrix metalloproteinases during catabolic weight loss states enhance the deposition of calcium into elastic fibers of small vessels. The authors found elevated serum levels of matrix metalloproteinases in their patients with NUC induced by rapid weight loss.10
A meta-analysis by Nigwekar et al3 found a history of prior corticosteroid use in 61% (22/36) of NUC cases reviewed. However, it is unclear whether it is the use of corticosteroids or chronic inflammation that is implicated in NUC pathogenesis. Chronic inflammation causes downregulation of anticalcification signaling pathways.18-20 The role of 2 vascular calcification inhibitors has been evaluated in the pathogenesis of calciphylaxis: fetuin-A and matrix gla protein (MGP).21 The activity of these proteins is decreased not only in calciphylaxis but also in other inflammatory states and chronic renal failure.18-20 One study found lower fetuin-A levels in 312 hemodialysis patients compared to healthy controls and an association between low fetuin-A levels and increased C-reactive protein levels.22 Reduced fetuin-A and MGP levels may be the result of several calciphylaxis risk factors. Warfarin is believed to trigger calciphylaxis via inhibition of gamma-carboxylation of MGP, which is necessary for its anticalcification activity.23 Hypoalbuminemia and alcoholic liver disease also are risk factors that may be explained by the fact that fetuin-A is synthesized in the liver.24 Therefore, liver disease results in decreased production of fetuin-A that is permissive to vascular calcification in calciphylaxis patients.
There have been other reports of calciphylaxis patients who were originally hospitalized due to hypotension, which may serve as a trigger for calciphylaxis onset.25 Because calciphylaxis lesions are more likely to occur in the fatty areas of the abdomen and proximal thighs where blood flow is slower, hypotension likely accentuates the slowing of blood flow and subsequent blood vessel calcification. This theory is supported by studies showing that established calciphylactic lesions worsen more quickly in the presence of systemic hypotension.26 One patient with ESRD and calciphylaxis of the breasts had consistent systolic blood pressure readings in the high 60s to low 70s between dialysis sessions.27 Due to this association, we recommend that patients with calciphylaxis have close blood pressure monitoring to aid in preventing disease progression.28
Management
Calciphylaxis treatment has not yet been standardized, as it is an uncommon disease whose pathogenesis is not fully understood. Current management strategies aim to normalize metabolic abnormalities such as hypercalcemia if they are present and remove inciting agents such as warfarin and corticosteroids.29 Other medical treatments that have been successfully used include sodium thiosulfate, oral steroids, and adjunctive bisphosphonates.29-31 Sodium thiosulfate is known to cause metabolic acidosis by generating thiosulfuric acid in vivo in patients with or without renal disease; therefore, patients on sodium thiosulfate therapy should be monitored for development of metabolic acidosis and treated with oral sodium bicarbonate or dialysis as needed.30,32 Wound care also is an important element of calciphylaxis treatment; however, the debridement of wounds is controversial. Some argue that dry intact eschars serve to protect against sepsis, which is the leading cause of death in calciphylaxis.2,14,33 In contrast, a retrospective study of 63 calciphylaxis patients found a 1-year survival rate of 61.6% in 17 patients receiving wound debridement vs 27.4% in 46 patients who did not.2 The current consensus is that debridement should be considered on a case-by-case basis, factoring in the presence of wound infection, size of wounds, stability of eschars, and treatment goals of the patient.34 Future studies should be aimed at this issue, with special focus on how these factors and the decision to debride or not impact patient outcomes.
Conclusion
Calciphylaxis is a potentially fatal disease that impacts both patients with ESRD and those with nonuremic risk factors. The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature. In such cases, patients often have multiple risk factors, including obesity, primary hyperparathyroidism, alcoholic liver disease, and underlying malignancy, among others. Certain triggers for onset of calciphylaxis should be avoided in at-risk patients, including the use of corticosteroids or warfarin; iron and albumin infusions; hypotension; and rapid weight loss. Our fatal case of NUC is a reminder to dermatologists treating at-risk patients to avoid these triggers and to keep calciphylaxis in the differential diagnosis when encountering early lesions such as livedo reticularis, as progression of these lesions has a 1-year mortality rate of more than 50% with the therapies being utilized at this time.
- Au S, Crawford RI. Three-dimensional analysis of a calciphylaxis plaque: clues to pathogenesis. J Am Acad Dermatol. 2007;47:53-57.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Angelis M, Wong LL, Myers SA, et al. Calciphylaxis in patients on hemodialysis: a prevalence study. Surgery. 1997;122:1083-1090.
- Chavel SM, Taraszka KS, Schaffer JV, et al. Calciphylaxis associated with acute, reversible renal failure in the setting of alcoholic cirrhosis. J Am Acad Dermatol. 2004;50:125-128.
- Bosler DS, Amin MB, Gulli F, et al. Unusual case of calciphylaxis associated with metastatic breast carcinoma. Am J Dermatopathol. 2007;29:400-403.
- Buxtorf K, Cerottini JP, Panizzon RG. Lower limb skin ulcerations, intravascular calcifications and sensorimotor polyneuropathy: calciphylaxis as part of a hyperparathyroidism? Dermatology. 1999;198:423-425.
- Brouns K, Verbeken E, Degreef H, et al. Fatal calciphylaxis in two patients with giant cell arteritis. Clin Rheumatol. 2007;26:836-840.
- Munavalli G, Reisenauer A, Moses M, et al. Weight loss-induced calciphylaxis: potential role of matrix metalloproteinases. J Dermatol. 2003;30:915-919.
- Bae GH, Nambudiri VE, Bach DQ, et al. Rapidly progressive nonuremic calciphylaxis in setting of warfarin. Am J Med. 2015;128:E19-E21.
- Essary LR, Wick MR. Cutaneous calciphylaxis. an underrecognized clinicopathologic entity. Am J Clin Pathol. 2000;113:280-287.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Coates T, Kirkland GS, Dymock RB, et al. Cutaneous necrosis from calcific uremic arteriolopathy. Am J Kidney Dis. 1998;32:384-391.
- Selye H, Gentile G, Prioreschi P. Cutaneous molt induced by calciphylaxis in the rat. Science. 1961;134:1876-1877.
- Kalajian AH, Malhotra PS, Callen JP, et al. Calciphylaxis with normal renal and parathyroid function: not as rare as previously believed. Arch Dermatol. 2009;145:451-458.
- Malabu U, Roberts L, Sangla K. Calciphylaxis in a morbidly obese woman with rheumatoid arthritis presenting with severe weight loss and vitamin D deficiency. Endocr Pract. 2011;17:104-108.
- Schäfer C, Heiss A, Schwarz A, et al. The serum protein alpha 2–Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Cozzolino M, Galassi A, Biondi ML, et al. Serum fetuin-A levels link inflammation and cardiovascular calcification in hemodialysis patients. Am J Nephrol. 2006;26:423-429.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Weenig RH. Pathogenesis of calciphylaxis: Hans Selye to nuclear factor kappa-B. J Am Acad Dermatol. 2008;58:458-471.
- Ketteler M, Bongartz P, Westenfeld R, et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet. 2003;361:827-833.
- Wallin R, Cain D, Sane DC. Matrix Gla protein synthesis and gamma-carboxylation in the aortic vessel wall and proliferating vascular smooth muscle cells a cell system which resembles the system in bone cells. Thromb Haemost. 1999;82:1764-1767.
- Sowers KM, Hayden MR. Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches. Oxid Med Cell Longev. 2010;3:109-121.
- Allegretti AS, Nazarian RM, Goverman J, et al. Calciphylaxis: a rare but fatal delayed complication of Roux-en-Y gastric bypass surgery. Am J Kidney Dis. 2014;64:274-277.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial. 2002;15:172-186.
- Gupta D, Tadros R, Mazumdar A, et al. Breast lesions with intractable pain in end-stage renal disease: calciphylaxis with chronic hypotensive dermatopathy related watershed breast lesions. J Palliat Med. 2013;16:551-554.
- Janigan DT, Hirsch DJ, Klassen GA, et al. Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis. 2000;35:588-597.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Bourgeois P, De Haes P. Sodium thiosulfate as a treatment for calciphylaxis: a case series. J Dermatolog Treat. 2016;27:520-524.
- Biswas A, Walsh NM, Tremaine R. A case of nonuremic calciphylaxis treated effectively with systemic corticosteroids. J Cutan Med Surg. 2016;20:275-278.
- Selk N, Rodby, RA. Unexpectedly severe metabolic acidosis associated with sodium thiosulfate therapy in a patient with calcific uremic arteriolopathy. Semin Dial. 2011;24:85-88.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage. 2004:50:64-66, 68-70.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
- Au S, Crawford RI. Three-dimensional analysis of a calciphylaxis plaque: clues to pathogenesis. J Am Acad Dermatol. 2007;47:53-57.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Angelis M, Wong LL, Myers SA, et al. Calciphylaxis in patients on hemodialysis: a prevalence study. Surgery. 1997;122:1083-1090.
- Chavel SM, Taraszka KS, Schaffer JV, et al. Calciphylaxis associated with acute, reversible renal failure in the setting of alcoholic cirrhosis. J Am Acad Dermatol. 2004;50:125-128.
- Bosler DS, Amin MB, Gulli F, et al. Unusual case of calciphylaxis associated with metastatic breast carcinoma. Am J Dermatopathol. 2007;29:400-403.
- Buxtorf K, Cerottini JP, Panizzon RG. Lower limb skin ulcerations, intravascular calcifications and sensorimotor polyneuropathy: calciphylaxis as part of a hyperparathyroidism? Dermatology. 1999;198:423-425.
- Brouns K, Verbeken E, Degreef H, et al. Fatal calciphylaxis in two patients with giant cell arteritis. Clin Rheumatol. 2007;26:836-840.
- Munavalli G, Reisenauer A, Moses M, et al. Weight loss-induced calciphylaxis: potential role of matrix metalloproteinases. J Dermatol. 2003;30:915-919.
- Bae GH, Nambudiri VE, Bach DQ, et al. Rapidly progressive nonuremic calciphylaxis in setting of warfarin. Am J Med. 2015;128:E19-E21.
- Essary LR, Wick MR. Cutaneous calciphylaxis. an underrecognized clinicopathologic entity. Am J Clin Pathol. 2000;113:280-287.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Coates T, Kirkland GS, Dymock RB, et al. Cutaneous necrosis from calcific uremic arteriolopathy. Am J Kidney Dis. 1998;32:384-391.
- Selye H, Gentile G, Prioreschi P. Cutaneous molt induced by calciphylaxis in the rat. Science. 1961;134:1876-1877.
- Kalajian AH, Malhotra PS, Callen JP, et al. Calciphylaxis with normal renal and parathyroid function: not as rare as previously believed. Arch Dermatol. 2009;145:451-458.
- Malabu U, Roberts L, Sangla K. Calciphylaxis in a morbidly obese woman with rheumatoid arthritis presenting with severe weight loss and vitamin D deficiency. Endocr Pract. 2011;17:104-108.
- Schäfer C, Heiss A, Schwarz A, et al. The serum protein alpha 2–Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Cozzolino M, Galassi A, Biondi ML, et al. Serum fetuin-A levels link inflammation and cardiovascular calcification in hemodialysis patients. Am J Nephrol. 2006;26:423-429.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Weenig RH. Pathogenesis of calciphylaxis: Hans Selye to nuclear factor kappa-B. J Am Acad Dermatol. 2008;58:458-471.
- Ketteler M, Bongartz P, Westenfeld R, et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet. 2003;361:827-833.
- Wallin R, Cain D, Sane DC. Matrix Gla protein synthesis and gamma-carboxylation in the aortic vessel wall and proliferating vascular smooth muscle cells a cell system which resembles the system in bone cells. Thromb Haemost. 1999;82:1764-1767.
- Sowers KM, Hayden MR. Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches. Oxid Med Cell Longev. 2010;3:109-121.
- Allegretti AS, Nazarian RM, Goverman J, et al. Calciphylaxis: a rare but fatal delayed complication of Roux-en-Y gastric bypass surgery. Am J Kidney Dis. 2014;64:274-277.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial. 2002;15:172-186.
- Gupta D, Tadros R, Mazumdar A, et al. Breast lesions with intractable pain in end-stage renal disease: calciphylaxis with chronic hypotensive dermatopathy related watershed breast lesions. J Palliat Med. 2013;16:551-554.
- Janigan DT, Hirsch DJ, Klassen GA, et al. Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis. 2000;35:588-597.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Bourgeois P, De Haes P. Sodium thiosulfate as a treatment for calciphylaxis: a case series. J Dermatolog Treat. 2016;27:520-524.
- Biswas A, Walsh NM, Tremaine R. A case of nonuremic calciphylaxis treated effectively with systemic corticosteroids. J Cutan Med Surg. 2016;20:275-278.
- Selk N, Rodby, RA. Unexpectedly severe metabolic acidosis associated with sodium thiosulfate therapy in a patient with calcific uremic arteriolopathy. Semin Dial. 2011;24:85-88.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage. 2004:50:64-66, 68-70.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
Practice Points
- Calciphylaxis is a potentially fatal disease caused by metastatic calcification of cutaneous small- and medium-sized blood vessels leading to ischemia and necrosis.
- Calciphylaxis most commonly is seen in patients with renal disease requiring dialysis, but it also may be triggered by nonuremic causes in patients with known risk factors for calciphylaxis.
- Risk factors for calciphylaxis include female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.
- The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature.