User login
Nonuremic Calciphylaxis Triggered by Rapid Weight Loss and Hypotension
Calciphylaxis, otherwise known as calcific uremic arteriolopathy, is characterized by calcification of the tunica media of the small- to medium-sized blood vessels of the dermis and subcutis, leading to ischemia and necrosis.1 It is a deadly disease with a 1-year mortality rate of more than 50%.2 End-stage renal disease (ESRD) is the most common risk factor for calciphylaxis, with a prevalence of 1% to 4% of hemodialysis patients with calciphylaxis in the United States.2-5 However, nonuremic calciphylaxis (NUC) has been increasingly reported in the literature and has risk factors other than ESRD, including but not limited to obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, and underlying malignancy.3,6-9 Triggers for calciphylaxis in at-risk patients include use of corticosteroids or warfarin, iron or albumin infusions, and rapid weight loss.3,6,9-11 We report an unusual case of NUC that most likely was triggered by rapid weight loss and hypotension in a patient with multiple risk factors for calciphylaxis.
Case Report
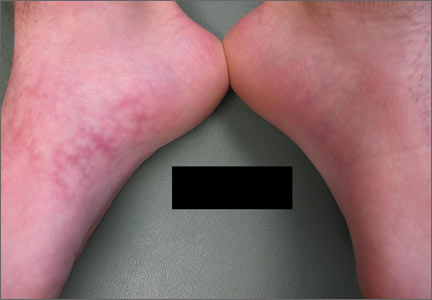
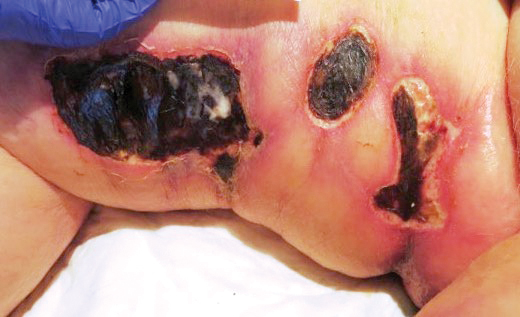
A 75-year-old white woman with history of morbid obesity (body mass index, 40 kg/m2), unexplained weight loss of 70 lb over the last year, and polymyalgia rheumatica requiring chronic prednisone therapy presented with painful lesions on the thighs, buttocks, and right shoulder of 4 months’ duration. She had multiple hospital admissions preceding the onset of lesions for severe infections resulting in sepsis with hypotension, including Enterococcus faecalis endocarditis, extended-spectrum beta-lactamase bacteremia, and Pseudomonas aeruginosa pneumonia. Physical examination revealed large well-demarcated ulcers and necrotic eschars with surrounding violaceous induration and stellate erythema on the anterior, medial, and posterior thighs and buttocks that were exquisitely tender (Figures 1 and 2).

Notable laboratory results included hypoalbuminemia (1.3 g/dL [reference range, 3.5–5.0 g/dL]) with normal renal function, a corrected calcium level of 9.7 mg/dL (reference range, 8.2–10.2 mg/dL), a serum phosphorus level of 3.5 mg/dL (reference range, 2.3–4.7 mg/dL), a calcium-phosphate product of 27.3 mg2/dL2 (reference range, <55 mg2/dL2), and a parathyroid hormone level of 49.3 pg/mL (reference range, 10–65 pg/mL). Antinuclear antibodies were negative. A hypercoagulability evaluation showed normal protein C and S levels, negative lupus anticoagulant, and negative anticardiolipin antibodies.
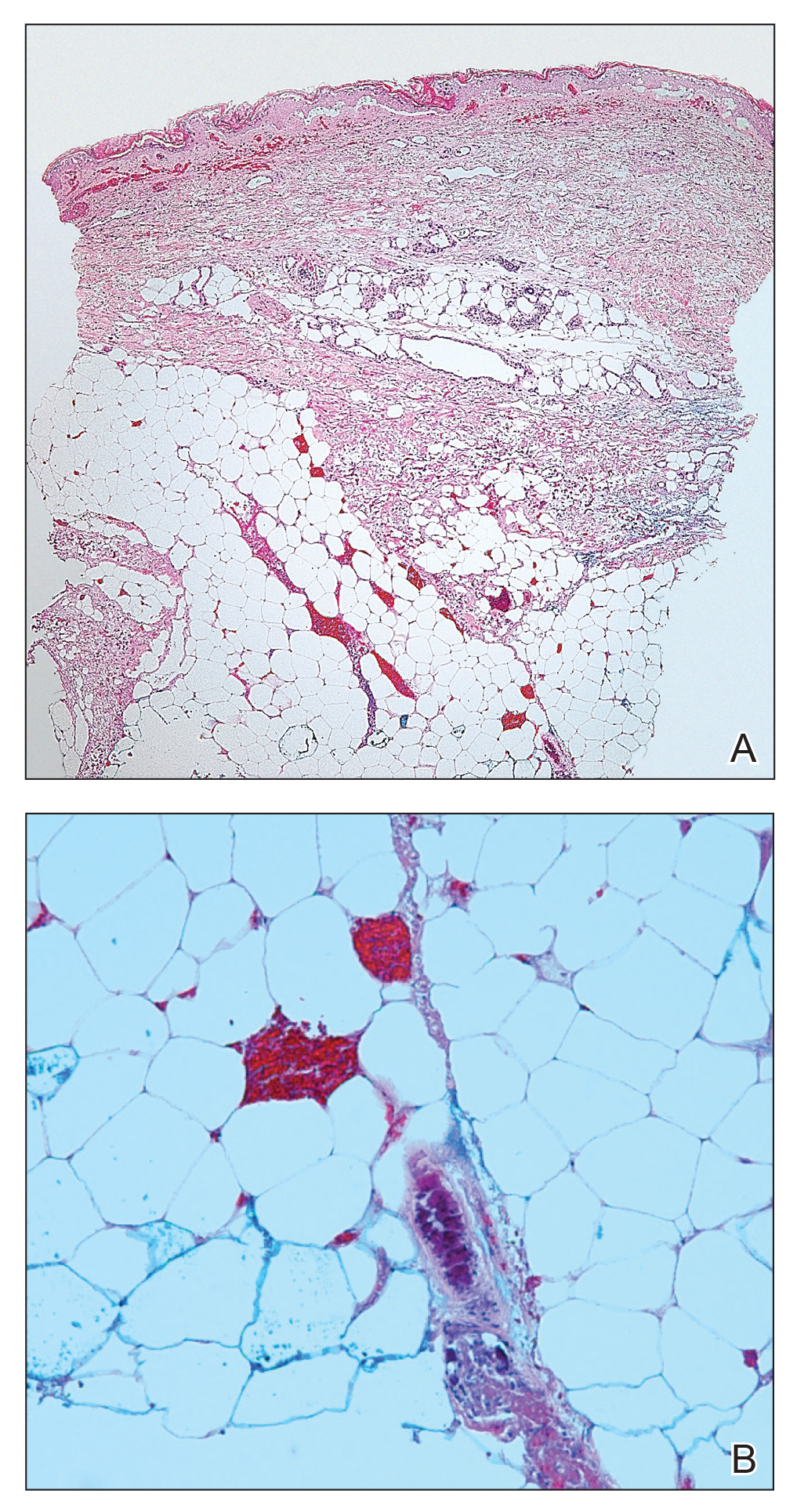
Telescoping punch biopsies of the indurated borders of the eschars showed prominent calcification of the small- and medium-sized vessels in the mid and deep dermis, intravascular thrombi, and necrosis of the epidermis and subcutaneous fat consistent with calciphylaxis (Figure 3).
After the diagnosis of calciphylaxis was made, the patient was treated with intravenous sodium thiosulfate 25 mg 3 times weekly and alendronate 70 mg weekly. Daily arterial blood gas studies did not detect metabolic acidosis during the patient’s sodium thiosulfate therapy. The wounds were debrided, and we attempted to slowly taper the patient off the oral prednisone. Unfortunately, her condition slowly deteriorated secondary to sepsis, resulting in septic shock. The patient died 3 weeks after the diagnosis of calciphylaxis was made. At the time of diagnosis, the patient had a poor prognosis and notable risk for sepsis due to the large eschars on the thighs and abdomen as well as her relative immunosuppression due to chronic prednisone use.
Comment
Background on Calciphylaxis
Calciphylaxis is a rare but deadly disease that affects both ESRD patients receiving dialysis and patients without ESRD who have known risk factors for calciphylaxis, including female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.3,6-9,11 Although the molecular pathogenesis of calciphylaxis is not completely understood, it is believed to be caused by local deposition of calcium in the tunica media of small- to medium-sized arterioles and venules in the skin.12 This deposition leads to intimal proliferation and progressive narrowing of the vessels with resultant thrombosis, ischemia, and necrosis. The cutaneous manifestations and histopathology of calciphylaxis classically follow its pathogenesis. Calciphylaxis typically presents with livedo reticularis as vessels narrow and then progresses to purpura, bullae, necrosis, and eschar formation with the onset of acute thrombosis and ischemia. Histopathology is characterized by small- and medium-sized vessel calcification and thrombus, dermal necrosis, and septal panniculitis, though the histology can be highly variable.12 Unfortunately, the already poor prognosis for calciphylaxis worsens when lesions become either ulcerative or present on the proximal extremities and trunk.4,13 Sepsis is the leading cause of death in calciphylaxis patients, affecting more than 50% of patients.2,3,14 The differential diagnoses for calciphylactic-appearing lesions include warfarin-induced skin necrosis, disseminated intravascular coagulation, pyoderma gangrenosum, cholesterol emboli, and various vasculitides and coagulopathies.
Risk Factors
Our case demonstrates the importance of risk factor minimization, trigger avoidance, and early intervention due to the high mortality rate of calciphylaxis. Selye et al15 coined the term calciphylaxis in 1961 based on experiments that induced calciphylaxis in rat models. Their research concluded that there were certain sensitizers (ie, risk factors) that predisposed patients to medial calcium deposition in blood vessels and other challengers (ie, triggers) that acted as inciting events to calcium deposition. Our patient presented with multiple known risk factors for calciphylaxis, including obesity (body mass index, 40 kg/m2), female gender, white race, hypoalbuminemia, and chronic corticosteroid use.16 In the presence of a milieu of risk factors, the patient’s rapid weight loss and episodes of hypotension likely were triggers for calciphylaxis.
Other case reports in the literature have suggested weight loss as a trigger for NUC. One morbidly obese patient with inactive rheumatoid arthritis had onset of calciphylaxis lesions after unintentional weight loss of approximately 50% body weight in 1 year17; however, the weight loss does not have to be drastic to trigger calciphylaxis. Another study of 16 patients with uremic calciphylaxis found that 7 of 16 (44%) patients lost 10 to 50 kg in the 6 months prior to calciphylaxis onset.14 One proposed mechanism by Munavalli et al10 is that elevated levels of matrix metalloproteinases during catabolic weight loss states enhance the deposition of calcium into elastic fibers of small vessels. The authors found elevated serum levels of matrix metalloproteinases in their patients with NUC induced by rapid weight loss.10
A meta-analysis by Nigwekar et al3 found a history of prior corticosteroid use in 61% (22/36) of NUC cases reviewed. However, it is unclear whether it is the use of corticosteroids or chronic inflammation that is implicated in NUC pathogenesis. Chronic inflammation causes downregulation of anticalcification signaling pathways.18-20 The role of 2 vascular calcification inhibitors has been evaluated in the pathogenesis of calciphylaxis: fetuin-A and matrix gla protein (MGP).21 The activity of these proteins is decreased not only in calciphylaxis but also in other inflammatory states and chronic renal failure.18-20 One study found lower fetuin-A levels in 312 hemodialysis patients compared to healthy controls and an association between low fetuin-A levels and increased C-reactive protein levels.22 Reduced fetuin-A and MGP levels may be the result of several calciphylaxis risk factors. Warfarin is believed to trigger calciphylaxis via inhibition of gamma-carboxylation of MGP, which is necessary for its anticalcification activity.23 Hypoalbuminemia and alcoholic liver disease also are risk factors that may be explained by the fact that fetuin-A is synthesized in the liver.24 Therefore, liver disease results in decreased production of fetuin-A that is permissive to vascular calcification in calciphylaxis patients.
There have been other reports of calciphylaxis patients who were originally hospitalized due to hypotension, which may serve as a trigger for calciphylaxis onset.25 Because calciphylaxis lesions are more likely to occur in the fatty areas of the abdomen and proximal thighs where blood flow is slower, hypotension likely accentuates the slowing of blood flow and subsequent blood vessel calcification. This theory is supported by studies showing that established calciphylactic lesions worsen more quickly in the presence of systemic hypotension.26 One patient with ESRD and calciphylaxis of the breasts had consistent systolic blood pressure readings in the high 60s to low 70s between dialysis sessions.27 Due to this association, we recommend that patients with calciphylaxis have close blood pressure monitoring to aid in preventing disease progression.28
Management
Calciphylaxis treatment has not yet been standardized, as it is an uncommon disease whose pathogenesis is not fully understood. Current management strategies aim to normalize metabolic abnormalities such as hypercalcemia if they are present and remove inciting agents such as warfarin and corticosteroids.29 Other medical treatments that have been successfully used include sodium thiosulfate, oral steroids, and adjunctive bisphosphonates.29-31 Sodium thiosulfate is known to cause metabolic acidosis by generating thiosulfuric acid in vivo in patients with or without renal disease; therefore, patients on sodium thiosulfate therapy should be monitored for development of metabolic acidosis and treated with oral sodium bicarbonate or dialysis as needed.30,32 Wound care also is an important element of calciphylaxis treatment; however, the debridement of wounds is controversial. Some argue that dry intact eschars serve to protect against sepsis, which is the leading cause of death in calciphylaxis.2,14,33 In contrast, a retrospective study of 63 calciphylaxis patients found a 1-year survival rate of 61.6% in 17 patients receiving wound debridement vs 27.4% in 46 patients who did not.2 The current consensus is that debridement should be considered on a case-by-case basis, factoring in the presence of wound infection, size of wounds, stability of eschars, and treatment goals of the patient.34 Future studies should be aimed at this issue, with special focus on how these factors and the decision to debride or not impact patient outcomes.
Conclusion
Calciphylaxis is a potentially fatal disease that impacts both patients with ESRD and those with nonuremic risk factors. The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature. In such cases, patients often have multiple risk factors, including obesity, primary hyperparathyroidism, alcoholic liver disease, and underlying malignancy, among others. Certain triggers for onset of calciphylaxis should be avoided in at-risk patients, including the use of corticosteroids or warfarin; iron and albumin infusions; hypotension; and rapid weight loss. Our fatal case of NUC is a reminder to dermatologists treating at-risk patients to avoid these triggers and to keep calciphylaxis in the differential diagnosis when encountering early lesions such as livedo reticularis, as progression of these lesions has a 1-year mortality rate of more than 50% with the therapies being utilized at this time.
- Au S, Crawford RI. Three-dimensional analysis of a calciphylaxis plaque: clues to pathogenesis. J Am Acad Dermatol. 2007;47:53-57.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Angelis M, Wong LL, Myers SA, et al. Calciphylaxis in patients on hemodialysis: a prevalence study. Surgery. 1997;122:1083-1090.
- Chavel SM, Taraszka KS, Schaffer JV, et al. Calciphylaxis associated with acute, reversible renal failure in the setting of alcoholic cirrhosis. J Am Acad Dermatol. 2004;50:125-128.
- Bosler DS, Amin MB, Gulli F, et al. Unusual case of calciphylaxis associated with metastatic breast carcinoma. Am J Dermatopathol. 2007;29:400-403.
- Buxtorf K, Cerottini JP, Panizzon RG. Lower limb skin ulcerations, intravascular calcifications and sensorimotor polyneuropathy: calciphylaxis as part of a hyperparathyroidism? Dermatology. 1999;198:423-425.
- Brouns K, Verbeken E, Degreef H, et al. Fatal calciphylaxis in two patients with giant cell arteritis. Clin Rheumatol. 2007;26:836-840.
- Munavalli G, Reisenauer A, Moses M, et al. Weight loss-induced calciphylaxis: potential role of matrix metalloproteinases. J Dermatol. 2003;30:915-919.
- Bae GH, Nambudiri VE, Bach DQ, et al. Rapidly progressive nonuremic calciphylaxis in setting of warfarin. Am J Med. 2015;128:E19-E21.
- Essary LR, Wick MR. Cutaneous calciphylaxis. an underrecognized clinicopathologic entity. Am J Clin Pathol. 2000;113:280-287.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Coates T, Kirkland GS, Dymock RB, et al. Cutaneous necrosis from calcific uremic arteriolopathy. Am J Kidney Dis. 1998;32:384-391.
- Selye H, Gentile G, Prioreschi P. Cutaneous molt induced by calciphylaxis in the rat. Science. 1961;134:1876-1877.
- Kalajian AH, Malhotra PS, Callen JP, et al. Calciphylaxis with normal renal and parathyroid function: not as rare as previously believed. Arch Dermatol. 2009;145:451-458.
- Malabu U, Roberts L, Sangla K. Calciphylaxis in a morbidly obese woman with rheumatoid arthritis presenting with severe weight loss and vitamin D deficiency. Endocr Pract. 2011;17:104-108.
- Schäfer C, Heiss A, Schwarz A, et al. The serum protein alpha 2–Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Cozzolino M, Galassi A, Biondi ML, et al. Serum fetuin-A levels link inflammation and cardiovascular calcification in hemodialysis patients. Am J Nephrol. 2006;26:423-429.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Weenig RH. Pathogenesis of calciphylaxis: Hans Selye to nuclear factor kappa-B. J Am Acad Dermatol. 2008;58:458-471.
- Ketteler M, Bongartz P, Westenfeld R, et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet. 2003;361:827-833.
- Wallin R, Cain D, Sane DC. Matrix Gla protein synthesis and gamma-carboxylation in the aortic vessel wall and proliferating vascular smooth muscle cells a cell system which resembles the system in bone cells. Thromb Haemost. 1999;82:1764-1767.
- Sowers KM, Hayden MR. Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches. Oxid Med Cell Longev. 2010;3:109-121.
- Allegretti AS, Nazarian RM, Goverman J, et al. Calciphylaxis: a rare but fatal delayed complication of Roux-en-Y gastric bypass surgery. Am J Kidney Dis. 2014;64:274-277.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial. 2002;15:172-186.
- Gupta D, Tadros R, Mazumdar A, et al. Breast lesions with intractable pain in end-stage renal disease: calciphylaxis with chronic hypotensive dermatopathy related watershed breast lesions. J Palliat Med. 2013;16:551-554.
- Janigan DT, Hirsch DJ, Klassen GA, et al. Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis. 2000;35:588-597.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Bourgeois P, De Haes P. Sodium thiosulfate as a treatment for calciphylaxis: a case series. J Dermatolog Treat. 2016;27:520-524.
- Biswas A, Walsh NM, Tremaine R. A case of nonuremic calciphylaxis treated effectively with systemic corticosteroids. J Cutan Med Surg. 2016;20:275-278.
- Selk N, Rodby, RA. Unexpectedly severe metabolic acidosis associated with sodium thiosulfate therapy in a patient with calcific uremic arteriolopathy. Semin Dial. 2011;24:85-88.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage. 2004:50:64-66, 68-70.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
Calciphylaxis, otherwise known as calcific uremic arteriolopathy, is characterized by calcification of the tunica media of the small- to medium-sized blood vessels of the dermis and subcutis, leading to ischemia and necrosis.1 It is a deadly disease with a 1-year mortality rate of more than 50%.2 End-stage renal disease (ESRD) is the most common risk factor for calciphylaxis, with a prevalence of 1% to 4% of hemodialysis patients with calciphylaxis in the United States.2-5 However, nonuremic calciphylaxis (NUC) has been increasingly reported in the literature and has risk factors other than ESRD, including but not limited to obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, and underlying malignancy.3,6-9 Triggers for calciphylaxis in at-risk patients include use of corticosteroids or warfarin, iron or albumin infusions, and rapid weight loss.3,6,9-11 We report an unusual case of NUC that most likely was triggered by rapid weight loss and hypotension in a patient with multiple risk factors for calciphylaxis.
Case Report
A 75-year-old white woman with history of morbid obesity (body mass index, 40 kg/m2), unexplained weight loss of 70 lb over the last year, and polymyalgia rheumatica requiring chronic prednisone therapy presented with painful lesions on the thighs, buttocks, and right shoulder of 4 months’ duration. She had multiple hospital admissions preceding the onset of lesions for severe infections resulting in sepsis with hypotension, including Enterococcus faecalis endocarditis, extended-spectrum beta-lactamase bacteremia, and Pseudomonas aeruginosa pneumonia. Physical examination revealed large well-demarcated ulcers and necrotic eschars with surrounding violaceous induration and stellate erythema on the anterior, medial, and posterior thighs and buttocks that were exquisitely tender (Figures 1 and 2).
Notable laboratory results included hypoalbuminemia (1.3 g/dL [reference range, 3.5–5.0 g/dL]) with normal renal function, a corrected calcium level of 9.7 mg/dL (reference range, 8.2–10.2 mg/dL), a serum phosphorus level of 3.5 mg/dL (reference range, 2.3–4.7 mg/dL), a calcium-phosphate product of 27.3 mg2/dL2 (reference range, <55 mg2/dL2), and a parathyroid hormone level of 49.3 pg/mL (reference range, 10–65 pg/mL). Antinuclear antibodies were negative. A hypercoagulability evaluation showed normal protein C and S levels, negative lupus anticoagulant, and negative anticardiolipin antibodies.
Telescoping punch biopsies of the indurated borders of the eschars showed prominent calcification of the small- and medium-sized vessels in the mid and deep dermis, intravascular thrombi, and necrosis of the epidermis and subcutaneous fat consistent with calciphylaxis (Figure 3).
After the diagnosis of calciphylaxis was made, the patient was treated with intravenous sodium thiosulfate 25 mg 3 times weekly and alendronate 70 mg weekly. Daily arterial blood gas studies did not detect metabolic acidosis during the patient’s sodium thiosulfate therapy. The wounds were debrided, and we attempted to slowly taper the patient off the oral prednisone. Unfortunately, her condition slowly deteriorated secondary to sepsis, resulting in septic shock. The patient died 3 weeks after the diagnosis of calciphylaxis was made. At the time of diagnosis, the patient had a poor prognosis and notable risk for sepsis due to the large eschars on the thighs and abdomen as well as her relative immunosuppression due to chronic prednisone use.
Comment
Background on Calciphylaxis
Calciphylaxis is a rare but deadly disease that affects both ESRD patients receiving dialysis and patients without ESRD who have known risk factors for calciphylaxis, including female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.3,6-9,11 Although the molecular pathogenesis of calciphylaxis is not completely understood, it is believed to be caused by local deposition of calcium in the tunica media of small- to medium-sized arterioles and venules in the skin.12 This deposition leads to intimal proliferation and progressive narrowing of the vessels with resultant thrombosis, ischemia, and necrosis. The cutaneous manifestations and histopathology of calciphylaxis classically follow its pathogenesis. Calciphylaxis typically presents with livedo reticularis as vessels narrow and then progresses to purpura, bullae, necrosis, and eschar formation with the onset of acute thrombosis and ischemia. Histopathology is characterized by small- and medium-sized vessel calcification and thrombus, dermal necrosis, and septal panniculitis, though the histology can be highly variable.12 Unfortunately, the already poor prognosis for calciphylaxis worsens when lesions become either ulcerative or present on the proximal extremities and trunk.4,13 Sepsis is the leading cause of death in calciphylaxis patients, affecting more than 50% of patients.2,3,14 The differential diagnoses for calciphylactic-appearing lesions include warfarin-induced skin necrosis, disseminated intravascular coagulation, pyoderma gangrenosum, cholesterol emboli, and various vasculitides and coagulopathies.
Risk Factors
Our case demonstrates the importance of risk factor minimization, trigger avoidance, and early intervention due to the high mortality rate of calciphylaxis. Selye et al15 coined the term calciphylaxis in 1961 based on experiments that induced calciphylaxis in rat models. Their research concluded that there were certain sensitizers (ie, risk factors) that predisposed patients to medial calcium deposition in blood vessels and other challengers (ie, triggers) that acted as inciting events to calcium deposition. Our patient presented with multiple known risk factors for calciphylaxis, including obesity (body mass index, 40 kg/m2), female gender, white race, hypoalbuminemia, and chronic corticosteroid use.16 In the presence of a milieu of risk factors, the patient’s rapid weight loss and episodes of hypotension likely were triggers for calciphylaxis.
Other case reports in the literature have suggested weight loss as a trigger for NUC. One morbidly obese patient with inactive rheumatoid arthritis had onset of calciphylaxis lesions after unintentional weight loss of approximately 50% body weight in 1 year17; however, the weight loss does not have to be drastic to trigger calciphylaxis. Another study of 16 patients with uremic calciphylaxis found that 7 of 16 (44%) patients lost 10 to 50 kg in the 6 months prior to calciphylaxis onset.14 One proposed mechanism by Munavalli et al10 is that elevated levels of matrix metalloproteinases during catabolic weight loss states enhance the deposition of calcium into elastic fibers of small vessels. The authors found elevated serum levels of matrix metalloproteinases in their patients with NUC induced by rapid weight loss.10
A meta-analysis by Nigwekar et al3 found a history of prior corticosteroid use in 61% (22/36) of NUC cases reviewed. However, it is unclear whether it is the use of corticosteroids or chronic inflammation that is implicated in NUC pathogenesis. Chronic inflammation causes downregulation of anticalcification signaling pathways.18-20 The role of 2 vascular calcification inhibitors has been evaluated in the pathogenesis of calciphylaxis: fetuin-A and matrix gla protein (MGP).21 The activity of these proteins is decreased not only in calciphylaxis but also in other inflammatory states and chronic renal failure.18-20 One study found lower fetuin-A levels in 312 hemodialysis patients compared to healthy controls and an association between low fetuin-A levels and increased C-reactive protein levels.22 Reduced fetuin-A and MGP levels may be the result of several calciphylaxis risk factors. Warfarin is believed to trigger calciphylaxis via inhibition of gamma-carboxylation of MGP, which is necessary for its anticalcification activity.23 Hypoalbuminemia and alcoholic liver disease also are risk factors that may be explained by the fact that fetuin-A is synthesized in the liver.24 Therefore, liver disease results in decreased production of fetuin-A that is permissive to vascular calcification in calciphylaxis patients.
There have been other reports of calciphylaxis patients who were originally hospitalized due to hypotension, which may serve as a trigger for calciphylaxis onset.25 Because calciphylaxis lesions are more likely to occur in the fatty areas of the abdomen and proximal thighs where blood flow is slower, hypotension likely accentuates the slowing of blood flow and subsequent blood vessel calcification. This theory is supported by studies showing that established calciphylactic lesions worsen more quickly in the presence of systemic hypotension.26 One patient with ESRD and calciphylaxis of the breasts had consistent systolic blood pressure readings in the high 60s to low 70s between dialysis sessions.27 Due to this association, we recommend that patients with calciphylaxis have close blood pressure monitoring to aid in preventing disease progression.28
Management
Calciphylaxis treatment has not yet been standardized, as it is an uncommon disease whose pathogenesis is not fully understood. Current management strategies aim to normalize metabolic abnormalities such as hypercalcemia if they are present and remove inciting agents such as warfarin and corticosteroids.29 Other medical treatments that have been successfully used include sodium thiosulfate, oral steroids, and adjunctive bisphosphonates.29-31 Sodium thiosulfate is known to cause metabolic acidosis by generating thiosulfuric acid in vivo in patients with or without renal disease; therefore, patients on sodium thiosulfate therapy should be monitored for development of metabolic acidosis and treated with oral sodium bicarbonate or dialysis as needed.30,32 Wound care also is an important element of calciphylaxis treatment; however, the debridement of wounds is controversial. Some argue that dry intact eschars serve to protect against sepsis, which is the leading cause of death in calciphylaxis.2,14,33 In contrast, a retrospective study of 63 calciphylaxis patients found a 1-year survival rate of 61.6% in 17 patients receiving wound debridement vs 27.4% in 46 patients who did not.2 The current consensus is that debridement should be considered on a case-by-case basis, factoring in the presence of wound infection, size of wounds, stability of eschars, and treatment goals of the patient.34 Future studies should be aimed at this issue, with special focus on how these factors and the decision to debride or not impact patient outcomes.
Conclusion
Calciphylaxis is a potentially fatal disease that impacts both patients with ESRD and those with nonuremic risk factors. The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature. In such cases, patients often have multiple risk factors, including obesity, primary hyperparathyroidism, alcoholic liver disease, and underlying malignancy, among others. Certain triggers for onset of calciphylaxis should be avoided in at-risk patients, including the use of corticosteroids or warfarin; iron and albumin infusions; hypotension; and rapid weight loss. Our fatal case of NUC is a reminder to dermatologists treating at-risk patients to avoid these triggers and to keep calciphylaxis in the differential diagnosis when encountering early lesions such as livedo reticularis, as progression of these lesions has a 1-year mortality rate of more than 50% with the therapies being utilized at this time.
Calciphylaxis, otherwise known as calcific uremic arteriolopathy, is characterized by calcification of the tunica media of the small- to medium-sized blood vessels of the dermis and subcutis, leading to ischemia and necrosis.1 It is a deadly disease with a 1-year mortality rate of more than 50%.2 End-stage renal disease (ESRD) is the most common risk factor for calciphylaxis, with a prevalence of 1% to 4% of hemodialysis patients with calciphylaxis in the United States.2-5 However, nonuremic calciphylaxis (NUC) has been increasingly reported in the literature and has risk factors other than ESRD, including but not limited to obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, and underlying malignancy.3,6-9 Triggers for calciphylaxis in at-risk patients include use of corticosteroids or warfarin, iron or albumin infusions, and rapid weight loss.3,6,9-11 We report an unusual case of NUC that most likely was triggered by rapid weight loss and hypotension in a patient with multiple risk factors for calciphylaxis.
Case Report
A 75-year-old white woman with history of morbid obesity (body mass index, 40 kg/m2), unexplained weight loss of 70 lb over the last year, and polymyalgia rheumatica requiring chronic prednisone therapy presented with painful lesions on the thighs, buttocks, and right shoulder of 4 months’ duration. She had multiple hospital admissions preceding the onset of lesions for severe infections resulting in sepsis with hypotension, including Enterococcus faecalis endocarditis, extended-spectrum beta-lactamase bacteremia, and Pseudomonas aeruginosa pneumonia. Physical examination revealed large well-demarcated ulcers and necrotic eschars with surrounding violaceous induration and stellate erythema on the anterior, medial, and posterior thighs and buttocks that were exquisitely tender (Figures 1 and 2).
Notable laboratory results included hypoalbuminemia (1.3 g/dL [reference range, 3.5–5.0 g/dL]) with normal renal function, a corrected calcium level of 9.7 mg/dL (reference range, 8.2–10.2 mg/dL), a serum phosphorus level of 3.5 mg/dL (reference range, 2.3–4.7 mg/dL), a calcium-phosphate product of 27.3 mg2/dL2 (reference range, <55 mg2/dL2), and a parathyroid hormone level of 49.3 pg/mL (reference range, 10–65 pg/mL). Antinuclear antibodies were negative. A hypercoagulability evaluation showed normal protein C and S levels, negative lupus anticoagulant, and negative anticardiolipin antibodies.
Telescoping punch biopsies of the indurated borders of the eschars showed prominent calcification of the small- and medium-sized vessels in the mid and deep dermis, intravascular thrombi, and necrosis of the epidermis and subcutaneous fat consistent with calciphylaxis (Figure 3).
After the diagnosis of calciphylaxis was made, the patient was treated with intravenous sodium thiosulfate 25 mg 3 times weekly and alendronate 70 mg weekly. Daily arterial blood gas studies did not detect metabolic acidosis during the patient’s sodium thiosulfate therapy. The wounds were debrided, and we attempted to slowly taper the patient off the oral prednisone. Unfortunately, her condition slowly deteriorated secondary to sepsis, resulting in septic shock. The patient died 3 weeks after the diagnosis of calciphylaxis was made. At the time of diagnosis, the patient had a poor prognosis and notable risk for sepsis due to the large eschars on the thighs and abdomen as well as her relative immunosuppression due to chronic prednisone use.
Comment
Background on Calciphylaxis
Calciphylaxis is a rare but deadly disease that affects both ESRD patients receiving dialysis and patients without ESRD who have known risk factors for calciphylaxis, including female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.3,6-9,11 Although the molecular pathogenesis of calciphylaxis is not completely understood, it is believed to be caused by local deposition of calcium in the tunica media of small- to medium-sized arterioles and venules in the skin.12 This deposition leads to intimal proliferation and progressive narrowing of the vessels with resultant thrombosis, ischemia, and necrosis. The cutaneous manifestations and histopathology of calciphylaxis classically follow its pathogenesis. Calciphylaxis typically presents with livedo reticularis as vessels narrow and then progresses to purpura, bullae, necrosis, and eschar formation with the onset of acute thrombosis and ischemia. Histopathology is characterized by small- and medium-sized vessel calcification and thrombus, dermal necrosis, and septal panniculitis, though the histology can be highly variable.12 Unfortunately, the already poor prognosis for calciphylaxis worsens when lesions become either ulcerative or present on the proximal extremities and trunk.4,13 Sepsis is the leading cause of death in calciphylaxis patients, affecting more than 50% of patients.2,3,14 The differential diagnoses for calciphylactic-appearing lesions include warfarin-induced skin necrosis, disseminated intravascular coagulation, pyoderma gangrenosum, cholesterol emboli, and various vasculitides and coagulopathies.
Risk Factors
Our case demonstrates the importance of risk factor minimization, trigger avoidance, and early intervention due to the high mortality rate of calciphylaxis. Selye et al15 coined the term calciphylaxis in 1961 based on experiments that induced calciphylaxis in rat models. Their research concluded that there were certain sensitizers (ie, risk factors) that predisposed patients to medial calcium deposition in blood vessels and other challengers (ie, triggers) that acted as inciting events to calcium deposition. Our patient presented with multiple known risk factors for calciphylaxis, including obesity (body mass index, 40 kg/m2), female gender, white race, hypoalbuminemia, and chronic corticosteroid use.16 In the presence of a milieu of risk factors, the patient’s rapid weight loss and episodes of hypotension likely were triggers for calciphylaxis.
Other case reports in the literature have suggested weight loss as a trigger for NUC. One morbidly obese patient with inactive rheumatoid arthritis had onset of calciphylaxis lesions after unintentional weight loss of approximately 50% body weight in 1 year17; however, the weight loss does not have to be drastic to trigger calciphylaxis. Another study of 16 patients with uremic calciphylaxis found that 7 of 16 (44%) patients lost 10 to 50 kg in the 6 months prior to calciphylaxis onset.14 One proposed mechanism by Munavalli et al10 is that elevated levels of matrix metalloproteinases during catabolic weight loss states enhance the deposition of calcium into elastic fibers of small vessels. The authors found elevated serum levels of matrix metalloproteinases in their patients with NUC induced by rapid weight loss.10
A meta-analysis by Nigwekar et al3 found a history of prior corticosteroid use in 61% (22/36) of NUC cases reviewed. However, it is unclear whether it is the use of corticosteroids or chronic inflammation that is implicated in NUC pathogenesis. Chronic inflammation causes downregulation of anticalcification signaling pathways.18-20 The role of 2 vascular calcification inhibitors has been evaluated in the pathogenesis of calciphylaxis: fetuin-A and matrix gla protein (MGP).21 The activity of these proteins is decreased not only in calciphylaxis but also in other inflammatory states and chronic renal failure.18-20 One study found lower fetuin-A levels in 312 hemodialysis patients compared to healthy controls and an association between low fetuin-A levels and increased C-reactive protein levels.22 Reduced fetuin-A and MGP levels may be the result of several calciphylaxis risk factors. Warfarin is believed to trigger calciphylaxis via inhibition of gamma-carboxylation of MGP, which is necessary for its anticalcification activity.23 Hypoalbuminemia and alcoholic liver disease also are risk factors that may be explained by the fact that fetuin-A is synthesized in the liver.24 Therefore, liver disease results in decreased production of fetuin-A that is permissive to vascular calcification in calciphylaxis patients.
There have been other reports of calciphylaxis patients who were originally hospitalized due to hypotension, which may serve as a trigger for calciphylaxis onset.25 Because calciphylaxis lesions are more likely to occur in the fatty areas of the abdomen and proximal thighs where blood flow is slower, hypotension likely accentuates the slowing of blood flow and subsequent blood vessel calcification. This theory is supported by studies showing that established calciphylactic lesions worsen more quickly in the presence of systemic hypotension.26 One patient with ESRD and calciphylaxis of the breasts had consistent systolic blood pressure readings in the high 60s to low 70s between dialysis sessions.27 Due to this association, we recommend that patients with calciphylaxis have close blood pressure monitoring to aid in preventing disease progression.28
Management
Calciphylaxis treatment has not yet been standardized, as it is an uncommon disease whose pathogenesis is not fully understood. Current management strategies aim to normalize metabolic abnormalities such as hypercalcemia if they are present and remove inciting agents such as warfarin and corticosteroids.29 Other medical treatments that have been successfully used include sodium thiosulfate, oral steroids, and adjunctive bisphosphonates.29-31 Sodium thiosulfate is known to cause metabolic acidosis by generating thiosulfuric acid in vivo in patients with or without renal disease; therefore, patients on sodium thiosulfate therapy should be monitored for development of metabolic acidosis and treated with oral sodium bicarbonate or dialysis as needed.30,32 Wound care also is an important element of calciphylaxis treatment; however, the debridement of wounds is controversial. Some argue that dry intact eschars serve to protect against sepsis, which is the leading cause of death in calciphylaxis.2,14,33 In contrast, a retrospective study of 63 calciphylaxis patients found a 1-year survival rate of 61.6% in 17 patients receiving wound debridement vs 27.4% in 46 patients who did not.2 The current consensus is that debridement should be considered on a case-by-case basis, factoring in the presence of wound infection, size of wounds, stability of eschars, and treatment goals of the patient.34 Future studies should be aimed at this issue, with special focus on how these factors and the decision to debride or not impact patient outcomes.
Conclusion
Calciphylaxis is a potentially fatal disease that impacts both patients with ESRD and those with nonuremic risk factors. The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature. In such cases, patients often have multiple risk factors, including obesity, primary hyperparathyroidism, alcoholic liver disease, and underlying malignancy, among others. Certain triggers for onset of calciphylaxis should be avoided in at-risk patients, including the use of corticosteroids or warfarin; iron and albumin infusions; hypotension; and rapid weight loss. Our fatal case of NUC is a reminder to dermatologists treating at-risk patients to avoid these triggers and to keep calciphylaxis in the differential diagnosis when encountering early lesions such as livedo reticularis, as progression of these lesions has a 1-year mortality rate of more than 50% with the therapies being utilized at this time.
- Au S, Crawford RI. Three-dimensional analysis of a calciphylaxis plaque: clues to pathogenesis. J Am Acad Dermatol. 2007;47:53-57.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Angelis M, Wong LL, Myers SA, et al. Calciphylaxis in patients on hemodialysis: a prevalence study. Surgery. 1997;122:1083-1090.
- Chavel SM, Taraszka KS, Schaffer JV, et al. Calciphylaxis associated with acute, reversible renal failure in the setting of alcoholic cirrhosis. J Am Acad Dermatol. 2004;50:125-128.
- Bosler DS, Amin MB, Gulli F, et al. Unusual case of calciphylaxis associated with metastatic breast carcinoma. Am J Dermatopathol. 2007;29:400-403.
- Buxtorf K, Cerottini JP, Panizzon RG. Lower limb skin ulcerations, intravascular calcifications and sensorimotor polyneuropathy: calciphylaxis as part of a hyperparathyroidism? Dermatology. 1999;198:423-425.
- Brouns K, Verbeken E, Degreef H, et al. Fatal calciphylaxis in two patients with giant cell arteritis. Clin Rheumatol. 2007;26:836-840.
- Munavalli G, Reisenauer A, Moses M, et al. Weight loss-induced calciphylaxis: potential role of matrix metalloproteinases. J Dermatol. 2003;30:915-919.
- Bae GH, Nambudiri VE, Bach DQ, et al. Rapidly progressive nonuremic calciphylaxis in setting of warfarin. Am J Med. 2015;128:E19-E21.
- Essary LR, Wick MR. Cutaneous calciphylaxis. an underrecognized clinicopathologic entity. Am J Clin Pathol. 2000;113:280-287.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Coates T, Kirkland GS, Dymock RB, et al. Cutaneous necrosis from calcific uremic arteriolopathy. Am J Kidney Dis. 1998;32:384-391.
- Selye H, Gentile G, Prioreschi P. Cutaneous molt induced by calciphylaxis in the rat. Science. 1961;134:1876-1877.
- Kalajian AH, Malhotra PS, Callen JP, et al. Calciphylaxis with normal renal and parathyroid function: not as rare as previously believed. Arch Dermatol. 2009;145:451-458.
- Malabu U, Roberts L, Sangla K. Calciphylaxis in a morbidly obese woman with rheumatoid arthritis presenting with severe weight loss and vitamin D deficiency. Endocr Pract. 2011;17:104-108.
- Schäfer C, Heiss A, Schwarz A, et al. The serum protein alpha 2–Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Cozzolino M, Galassi A, Biondi ML, et al. Serum fetuin-A levels link inflammation and cardiovascular calcification in hemodialysis patients. Am J Nephrol. 2006;26:423-429.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Weenig RH. Pathogenesis of calciphylaxis: Hans Selye to nuclear factor kappa-B. J Am Acad Dermatol. 2008;58:458-471.
- Ketteler M, Bongartz P, Westenfeld R, et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet. 2003;361:827-833.
- Wallin R, Cain D, Sane DC. Matrix Gla protein synthesis and gamma-carboxylation in the aortic vessel wall and proliferating vascular smooth muscle cells a cell system which resembles the system in bone cells. Thromb Haemost. 1999;82:1764-1767.
- Sowers KM, Hayden MR. Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches. Oxid Med Cell Longev. 2010;3:109-121.
- Allegretti AS, Nazarian RM, Goverman J, et al. Calciphylaxis: a rare but fatal delayed complication of Roux-en-Y gastric bypass surgery. Am J Kidney Dis. 2014;64:274-277.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial. 2002;15:172-186.
- Gupta D, Tadros R, Mazumdar A, et al. Breast lesions with intractable pain in end-stage renal disease: calciphylaxis with chronic hypotensive dermatopathy related watershed breast lesions. J Palliat Med. 2013;16:551-554.
- Janigan DT, Hirsch DJ, Klassen GA, et al. Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis. 2000;35:588-597.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Bourgeois P, De Haes P. Sodium thiosulfate as a treatment for calciphylaxis: a case series. J Dermatolog Treat. 2016;27:520-524.
- Biswas A, Walsh NM, Tremaine R. A case of nonuremic calciphylaxis treated effectively with systemic corticosteroids. J Cutan Med Surg. 2016;20:275-278.
- Selk N, Rodby, RA. Unexpectedly severe metabolic acidosis associated with sodium thiosulfate therapy in a patient with calcific uremic arteriolopathy. Semin Dial. 2011;24:85-88.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage. 2004:50:64-66, 68-70.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
- Au S, Crawford RI. Three-dimensional analysis of a calciphylaxis plaque: clues to pathogenesis. J Am Acad Dermatol. 2007;47:53-57.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Angelis M, Wong LL, Myers SA, et al. Calciphylaxis in patients on hemodialysis: a prevalence study. Surgery. 1997;122:1083-1090.
- Chavel SM, Taraszka KS, Schaffer JV, et al. Calciphylaxis associated with acute, reversible renal failure in the setting of alcoholic cirrhosis. J Am Acad Dermatol. 2004;50:125-128.
- Bosler DS, Amin MB, Gulli F, et al. Unusual case of calciphylaxis associated with metastatic breast carcinoma. Am J Dermatopathol. 2007;29:400-403.
- Buxtorf K, Cerottini JP, Panizzon RG. Lower limb skin ulcerations, intravascular calcifications and sensorimotor polyneuropathy: calciphylaxis as part of a hyperparathyroidism? Dermatology. 1999;198:423-425.
- Brouns K, Verbeken E, Degreef H, et al. Fatal calciphylaxis in two patients with giant cell arteritis. Clin Rheumatol. 2007;26:836-840.
- Munavalli G, Reisenauer A, Moses M, et al. Weight loss-induced calciphylaxis: potential role of matrix metalloproteinases. J Dermatol. 2003;30:915-919.
- Bae GH, Nambudiri VE, Bach DQ, et al. Rapidly progressive nonuremic calciphylaxis in setting of warfarin. Am J Med. 2015;128:E19-E21.
- Essary LR, Wick MR. Cutaneous calciphylaxis. an underrecognized clinicopathologic entity. Am J Clin Pathol. 2000;113:280-287.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Coates T, Kirkland GS, Dymock RB, et al. Cutaneous necrosis from calcific uremic arteriolopathy. Am J Kidney Dis. 1998;32:384-391.
- Selye H, Gentile G, Prioreschi P. Cutaneous molt induced by calciphylaxis in the rat. Science. 1961;134:1876-1877.
- Kalajian AH, Malhotra PS, Callen JP, et al. Calciphylaxis with normal renal and parathyroid function: not as rare as previously believed. Arch Dermatol. 2009;145:451-458.
- Malabu U, Roberts L, Sangla K. Calciphylaxis in a morbidly obese woman with rheumatoid arthritis presenting with severe weight loss and vitamin D deficiency. Endocr Pract. 2011;17:104-108.
- Schäfer C, Heiss A, Schwarz A, et al. The serum protein alpha 2–Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Cozzolino M, Galassi A, Biondi ML, et al. Serum fetuin-A levels link inflammation and cardiovascular calcification in hemodialysis patients. Am J Nephrol. 2006;26:423-429.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Weenig RH. Pathogenesis of calciphylaxis: Hans Selye to nuclear factor kappa-B. J Am Acad Dermatol. 2008;58:458-471.
- Ketteler M, Bongartz P, Westenfeld R, et al. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: a cross-sectional study. Lancet. 2003;361:827-833.
- Wallin R, Cain D, Sane DC. Matrix Gla protein synthesis and gamma-carboxylation in the aortic vessel wall and proliferating vascular smooth muscle cells a cell system which resembles the system in bone cells. Thromb Haemost. 1999;82:1764-1767.
- Sowers KM, Hayden MR. Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches. Oxid Med Cell Longev. 2010;3:109-121.
- Allegretti AS, Nazarian RM, Goverman J, et al. Calciphylaxis: a rare but fatal delayed complication of Roux-en-Y gastric bypass surgery. Am J Kidney Dis. 2014;64:274-277.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial. 2002;15:172-186.
- Gupta D, Tadros R, Mazumdar A, et al. Breast lesions with intractable pain in end-stage renal disease: calciphylaxis with chronic hypotensive dermatopathy related watershed breast lesions. J Palliat Med. 2013;16:551-554.
- Janigan DT, Hirsch DJ, Klassen GA, et al. Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis. 2000;35:588-597.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Bourgeois P, De Haes P. Sodium thiosulfate as a treatment for calciphylaxis: a case series. J Dermatolog Treat. 2016;27:520-524.
- Biswas A, Walsh NM, Tremaine R. A case of nonuremic calciphylaxis treated effectively with systemic corticosteroids. J Cutan Med Surg. 2016;20:275-278.
- Selk N, Rodby, RA. Unexpectedly severe metabolic acidosis associated with sodium thiosulfate therapy in a patient with calcific uremic arteriolopathy. Semin Dial. 2011;24:85-88.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage. 2004:50:64-66, 68-70.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
Practice Points
- Calciphylaxis is a potentially fatal disease caused by metastatic calcification of cutaneous small- and medium-sized blood vessels leading to ischemia and necrosis.
- Calciphylaxis most commonly is seen in patients with renal disease requiring dialysis, but it also may be triggered by nonuremic causes in patients with known risk factors for calciphylaxis.
- Risk factors for calciphylaxis include female gender, white race, obesity, alcoholic liver disease, primary hyperparathyroidism, connective tissue disease, underlying malignancy, protein C or S deficiency, corticosteroid use, warfarin use, diabetes, iron or albumin infusions, and rapid weight loss.
- The term calcific uremic arteriolopathy should be disregarded, as nonuremic causes are being reported with increased frequency in the literature.
Cutaneous Metastasis of Endometrial Carcinoma: An Unusual and Dramatic Presentation
Case Report
A 62-year-old woman presented with multiple large friable tumors of the abdominal panniculus. The patient also reported an unintentional 75-lb weight loss over the last 9 months as well as vaginal bleeding and fecal discharge from the vagina of 2 weeks’ duration. The patient had a surgical and medical history of a robotic-assisted hysterectomy and bilateral salpingo-oophorectomy performed 4 years prior to presentation. Final surgical pathology showed complex atypical endometrial hyperplasia with no adenocarcinoma identified.
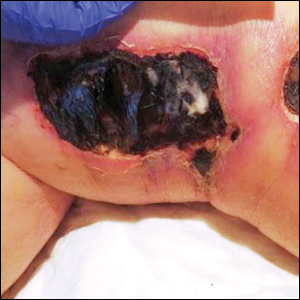
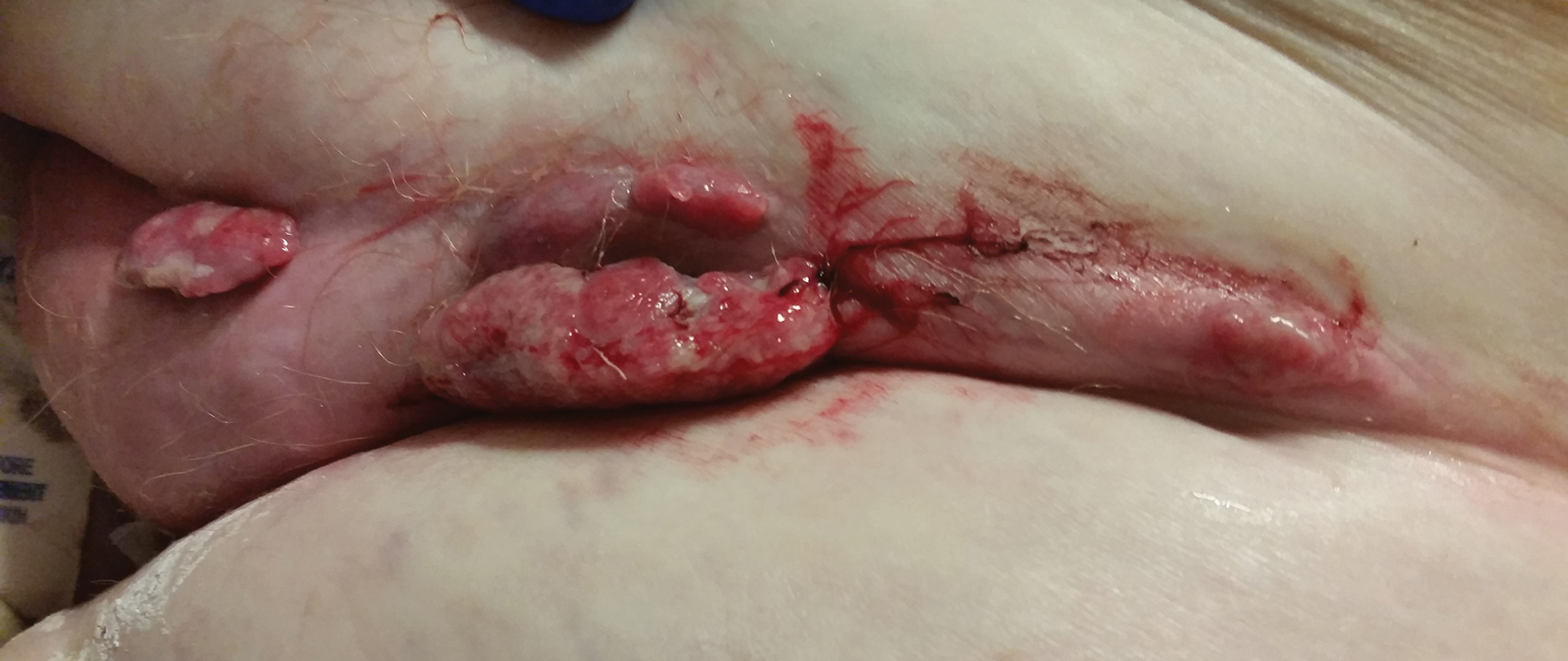
Physical examination revealed multiple large, friable, exophytic tumors of the left side of the lower abdominal panniculus within close vicinity of the patient’s abdominal hysterectomy scars (Figure 1). The largest lesion measured approximately 6 cm in length. Laboratory values were elevated for carcinoembryonic antigen (5.9 ng/mL [reference range, <3.0 ng/mL]) and cancer antigen 125 (202 U/mL [reference range, <35 U/mL]). Computed tomography of the abdomen and pelvis revealed diffuse metastatic disease.
Comment
Incidence and Pathogenesis
Endometrial carcinoma is the most common gynecologic malignancy in the United States, but it rarely progresses to disseminated disease because of routine gynecologic examinations and the low threshold for surgical intervention. Cutaneous metastases represent one of the rarest presentations of disseminated disease, occurring in only 0.8% of those diagnosed with endometrial carcinoma.1 Cutaneous metastases occur almost exclusively in women older than 50 years and typically appear several months to years after hysterectomy. Although the exact pathogenesis is unknown, it is theorized that small foci of malignant cells may be seeded during surgery, leading to visceral and cutaneous involvement.
Clinical Presentation
Lesions vary morphologically, most commonly presenting as nonspecific, painless, hemorrhagic nodules. Lesions typically present in areas of direct local extension; prior radiotherapy; or areas of initial surgery, as was the case with our patient.2 Approximately 20 cases of umbilical involvement (Sister Mary Joseph nodule) have been reported in the literature. These cases are thought to occur from direct local spread of disease from the peritoneum.3 Hematogenous and lymphatic spread to distant sites such as the scalp and mandible also have been reported. More than 50% of patients will have underlying visceral metastatic disease at the time of diagnosis.3
Histopathologic Findings
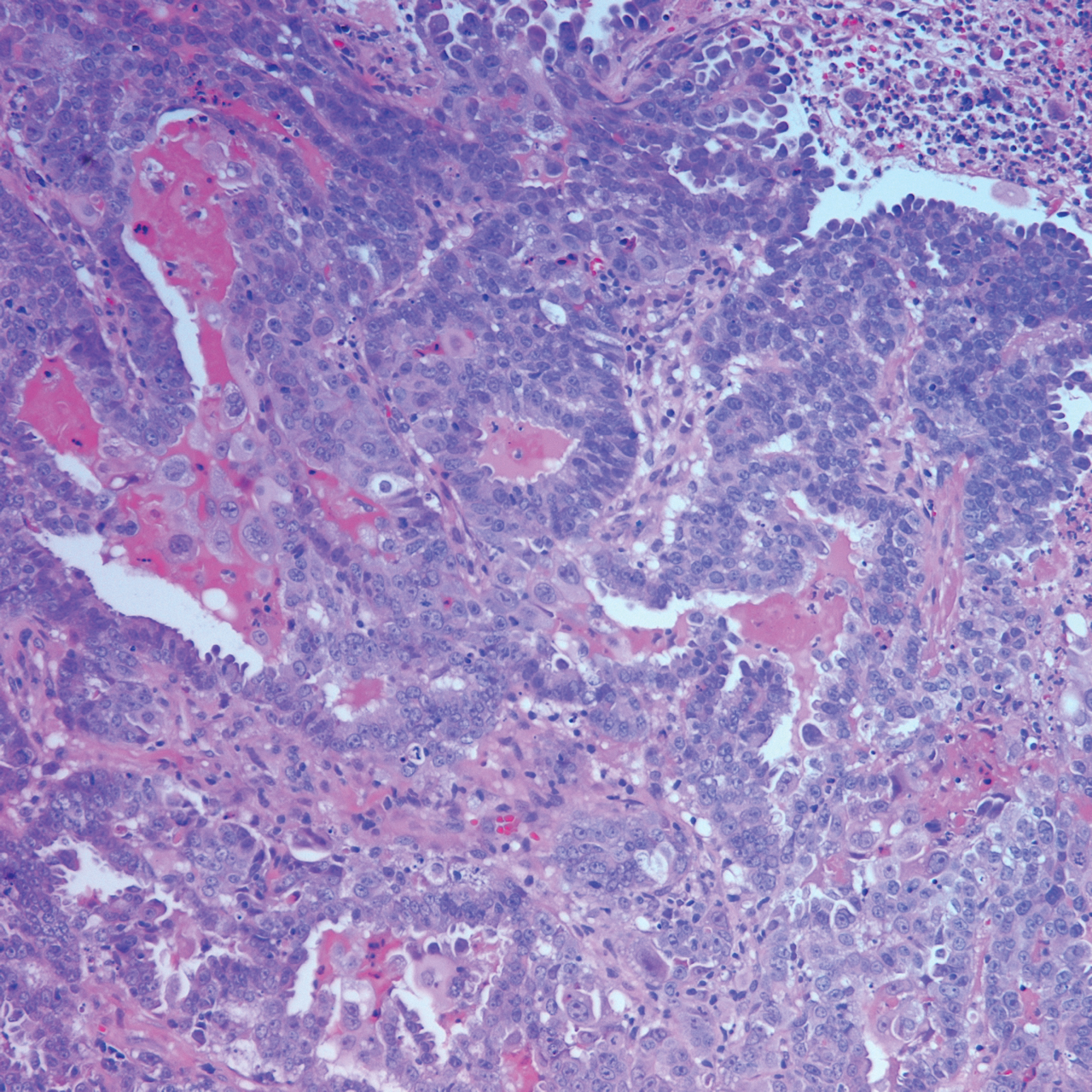
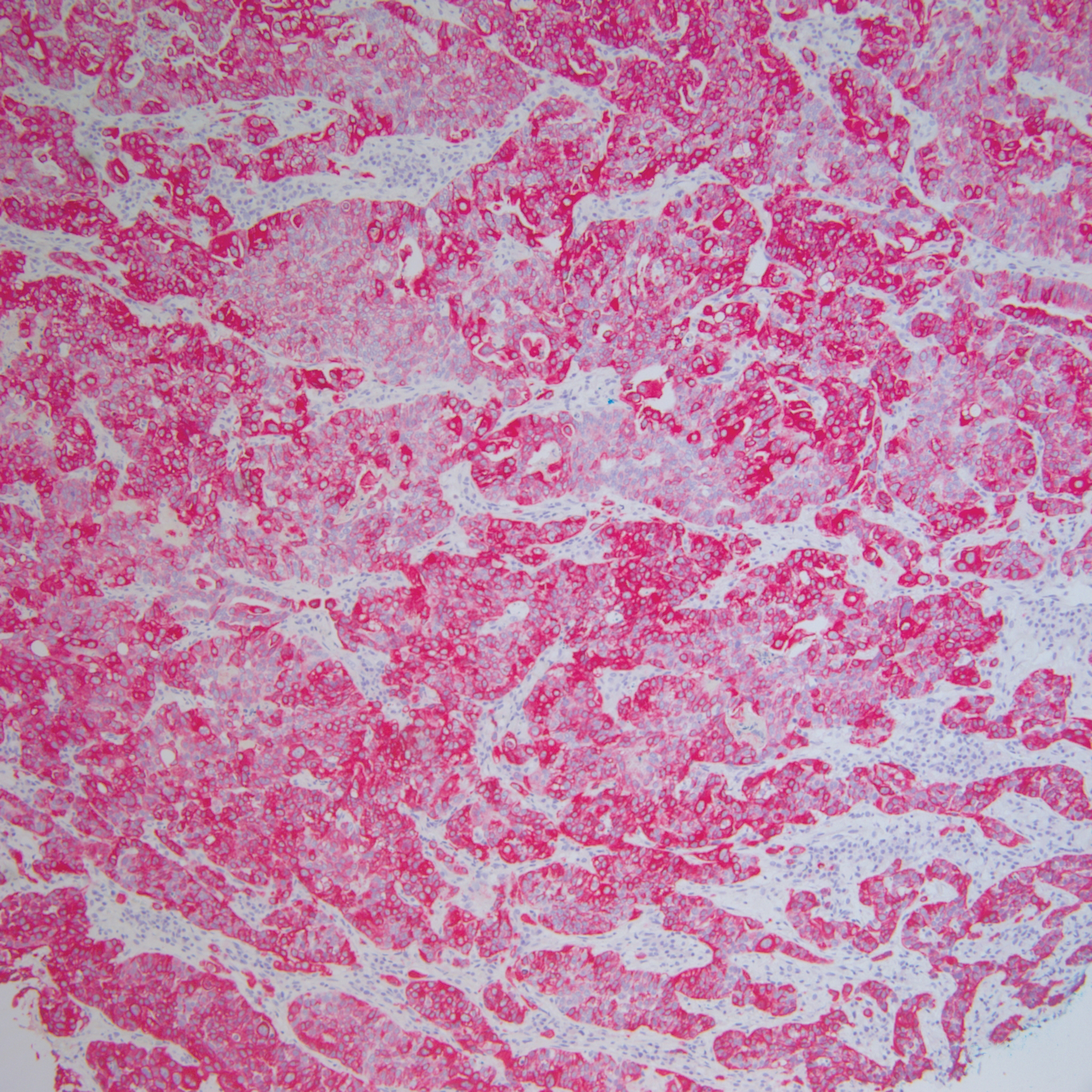
Histopathology varies with the morphology of the underlying primary tumor, with endometrioid adenocarcinoma being the most common form associated with cutaneous metastasis, as was the case with our patient.4 Histology is characterized by dermal proliferation of atypical glandular epithelium with diffuse hemorrhage. Staining typically is positive for CK7 and negative for CK20 and CDX2.5 Histopathology and immunohistochemical staining are not specific for diagnosis and must be correlated with clinical history.
Management and Prognosis
Similar to cutaneous metastasis in other internal malignancies, prognosis is poor, as widespread dissemination of the underlying malignancy typically is present. Mean life expectancy is 4 to 12 months.6 Treatment is primarily palliative, as chemotherapy and radiotherapy are largely ineffective.
Conclusion
Our patient represents a dramatic form of cutaneous extension of a common disease. Dermatologists often are consulted because of the nonspecific nature of the lesions and must be conscious of this entity. As with other cutaneous metastases, a thorough medical and surgical history in conjunction with histopathology are necessary for an accurate diagnosis.
- Atallah D, el Kassis N, Lutfallah F, et al. Cutaneous metastasis in endometrial cancer: once in a blue moon—case report. World J Surg Oncol. 2014;12:86.
- Temkin SM, Hellman M, Lee YC, et al. Surgical resection of vulvar metastases of endometrial cancer: a presentation of two cases. J Low Genit Tract Dis. 2007;11:118-121.
- Kushner DM, Lurain JR, Fu TS, et al. Endometrial adenocarcinoma metastatic to the scalp: case report and literature review. Gynecol Oncol. 1997;65:530-533.
- El M’rabet FZ, Hottinger A, George AC. Cutaneous metastasis of endometrial carcinoma: a case report and literature review. J Clin Gynecol Obstet. 2012;1:19-23.
- Stonard CM, Manek S. Cutaneous metastasis from an endometrial carcinoma: a case history and review of the literature. Histopathology. 2003;43:201-203
- Damewood MD, Rosenshein NB, Grumbine FC, et al. Cutaneous metastasis of endometrial carcinoma. Cancer. 1980;46:1471-1477.
Case Report
A 62-year-old woman presented with multiple large friable tumors of the abdominal panniculus. The patient also reported an unintentional 75-lb weight loss over the last 9 months as well as vaginal bleeding and fecal discharge from the vagina of 2 weeks’ duration. The patient had a surgical and medical history of a robotic-assisted hysterectomy and bilateral salpingo-oophorectomy performed 4 years prior to presentation. Final surgical pathology showed complex atypical endometrial hyperplasia with no adenocarcinoma identified.
Physical examination revealed multiple large, friable, exophytic tumors of the left side of the lower abdominal panniculus within close vicinity of the patient’s abdominal hysterectomy scars (Figure 1). The largest lesion measured approximately 6 cm in length. Laboratory values were elevated for carcinoembryonic antigen (5.9 ng/mL [reference range, <3.0 ng/mL]) and cancer antigen 125 (202 U/mL [reference range, <35 U/mL]). Computed tomography of the abdomen and pelvis revealed diffuse metastatic disease.
Comment
Incidence and Pathogenesis
Endometrial carcinoma is the most common gynecologic malignancy in the United States, but it rarely progresses to disseminated disease because of routine gynecologic examinations and the low threshold for surgical intervention. Cutaneous metastases represent one of the rarest presentations of disseminated disease, occurring in only 0.8% of those diagnosed with endometrial carcinoma.1 Cutaneous metastases occur almost exclusively in women older than 50 years and typically appear several months to years after hysterectomy. Although the exact pathogenesis is unknown, it is theorized that small foci of malignant cells may be seeded during surgery, leading to visceral and cutaneous involvement.
Clinical Presentation
Lesions vary morphologically, most commonly presenting as nonspecific, painless, hemorrhagic nodules. Lesions typically present in areas of direct local extension; prior radiotherapy; or areas of initial surgery, as was the case with our patient.2 Approximately 20 cases of umbilical involvement (Sister Mary Joseph nodule) have been reported in the literature. These cases are thought to occur from direct local spread of disease from the peritoneum.3 Hematogenous and lymphatic spread to distant sites such as the scalp and mandible also have been reported. More than 50% of patients will have underlying visceral metastatic disease at the time of diagnosis.3
Histopathologic Findings
Histopathology varies with the morphology of the underlying primary tumor, with endometrioid adenocarcinoma being the most common form associated with cutaneous metastasis, as was the case with our patient.4 Histology is characterized by dermal proliferation of atypical glandular epithelium with diffuse hemorrhage. Staining typically is positive for CK7 and negative for CK20 and CDX2.5 Histopathology and immunohistochemical staining are not specific for diagnosis and must be correlated with clinical history.
Management and Prognosis
Similar to cutaneous metastasis in other internal malignancies, prognosis is poor, as widespread dissemination of the underlying malignancy typically is present. Mean life expectancy is 4 to 12 months.6 Treatment is primarily palliative, as chemotherapy and radiotherapy are largely ineffective.
Conclusion
Our patient represents a dramatic form of cutaneous extension of a common disease. Dermatologists often are consulted because of the nonspecific nature of the lesions and must be conscious of this entity. As with other cutaneous metastases, a thorough medical and surgical history in conjunction with histopathology are necessary for an accurate diagnosis.
Case Report
A 62-year-old woman presented with multiple large friable tumors of the abdominal panniculus. The patient also reported an unintentional 75-lb weight loss over the last 9 months as well as vaginal bleeding and fecal discharge from the vagina of 2 weeks’ duration. The patient had a surgical and medical history of a robotic-assisted hysterectomy and bilateral salpingo-oophorectomy performed 4 years prior to presentation. Final surgical pathology showed complex atypical endometrial hyperplasia with no adenocarcinoma identified.
Physical examination revealed multiple large, friable, exophytic tumors of the left side of the lower abdominal panniculus within close vicinity of the patient’s abdominal hysterectomy scars (Figure 1). The largest lesion measured approximately 6 cm in length. Laboratory values were elevated for carcinoembryonic antigen (5.9 ng/mL [reference range, <3.0 ng/mL]) and cancer antigen 125 (202 U/mL [reference range, <35 U/mL]). Computed tomography of the abdomen and pelvis revealed diffuse metastatic disease.
Comment
Incidence and Pathogenesis
Endometrial carcinoma is the most common gynecologic malignancy in the United States, but it rarely progresses to disseminated disease because of routine gynecologic examinations and the low threshold for surgical intervention. Cutaneous metastases represent one of the rarest presentations of disseminated disease, occurring in only 0.8% of those diagnosed with endometrial carcinoma.1 Cutaneous metastases occur almost exclusively in women older than 50 years and typically appear several months to years after hysterectomy. Although the exact pathogenesis is unknown, it is theorized that small foci of malignant cells may be seeded during surgery, leading to visceral and cutaneous involvement.
Clinical Presentation
Lesions vary morphologically, most commonly presenting as nonspecific, painless, hemorrhagic nodules. Lesions typically present in areas of direct local extension; prior radiotherapy; or areas of initial surgery, as was the case with our patient.2 Approximately 20 cases of umbilical involvement (Sister Mary Joseph nodule) have been reported in the literature. These cases are thought to occur from direct local spread of disease from the peritoneum.3 Hematogenous and lymphatic spread to distant sites such as the scalp and mandible also have been reported. More than 50% of patients will have underlying visceral metastatic disease at the time of diagnosis.3
Histopathologic Findings
Histopathology varies with the morphology of the underlying primary tumor, with endometrioid adenocarcinoma being the most common form associated with cutaneous metastasis, as was the case with our patient.4 Histology is characterized by dermal proliferation of atypical glandular epithelium with diffuse hemorrhage. Staining typically is positive for CK7 and negative for CK20 and CDX2.5 Histopathology and immunohistochemical staining are not specific for diagnosis and must be correlated with clinical history.
Management and Prognosis
Similar to cutaneous metastasis in other internal malignancies, prognosis is poor, as widespread dissemination of the underlying malignancy typically is present. Mean life expectancy is 4 to 12 months.6 Treatment is primarily palliative, as chemotherapy and radiotherapy are largely ineffective.
Conclusion
Our patient represents a dramatic form of cutaneous extension of a common disease. Dermatologists often are consulted because of the nonspecific nature of the lesions and must be conscious of this entity. As with other cutaneous metastases, a thorough medical and surgical history in conjunction with histopathology are necessary for an accurate diagnosis.
- Atallah D, el Kassis N, Lutfallah F, et al. Cutaneous metastasis in endometrial cancer: once in a blue moon—case report. World J Surg Oncol. 2014;12:86.
- Temkin SM, Hellman M, Lee YC, et al. Surgical resection of vulvar metastases of endometrial cancer: a presentation of two cases. J Low Genit Tract Dis. 2007;11:118-121.
- Kushner DM, Lurain JR, Fu TS, et al. Endometrial adenocarcinoma metastatic to the scalp: case report and literature review. Gynecol Oncol. 1997;65:530-533.
- El M’rabet FZ, Hottinger A, George AC. Cutaneous metastasis of endometrial carcinoma: a case report and literature review. J Clin Gynecol Obstet. 2012;1:19-23.
- Stonard CM, Manek S. Cutaneous metastasis from an endometrial carcinoma: a case history and review of the literature. Histopathology. 2003;43:201-203
- Damewood MD, Rosenshein NB, Grumbine FC, et al. Cutaneous metastasis of endometrial carcinoma. Cancer. 1980;46:1471-1477.
- Atallah D, el Kassis N, Lutfallah F, et al. Cutaneous metastasis in endometrial cancer: once in a blue moon—case report. World J Surg Oncol. 2014;12:86.
- Temkin SM, Hellman M, Lee YC, et al. Surgical resection of vulvar metastases of endometrial cancer: a presentation of two cases. J Low Genit Tract Dis. 2007;11:118-121.
- Kushner DM, Lurain JR, Fu TS, et al. Endometrial adenocarcinoma metastatic to the scalp: case report and literature review. Gynecol Oncol. 1997;65:530-533.
- El M’rabet FZ, Hottinger A, George AC. Cutaneous metastasis of endometrial carcinoma: a case report and literature review. J Clin Gynecol Obstet. 2012;1:19-23.
- Stonard CM, Manek S. Cutaneous metastasis from an endometrial carcinoma: a case history and review of the literature. Histopathology. 2003;43:201-203
- Damewood MD, Rosenshein NB, Grumbine FC, et al. Cutaneous metastasis of endometrial carcinoma. Cancer. 1980;46:1471-1477.
Practice Points
- Cutaneous metastases of endometrial carcinoma are extremely rare and typically present in areas of direct local spread.
- As with other cutaneous metastases, lesions often are nonspecific, making history and histopathology essential for diagnosis.
Primary Cutaneous Epstein-Barr Virus–Positive Diffuse Large B-Cell Lymphoma: A Rare and Aggressive Cutaneous Lymphoma
Cutaneous B-cell lymphomas represent a group of lymphomas derived from B lymphocytes in various stages of differentiation. The skin can be the site of primary or secondary involvement of any of the B-cell lymphomas. Primary cutaneous B-cell lymphomas present in the skin without evidence of extracutaneous disease at the time of diagnosis.1 The World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues recognizes 5 distinct primary cutaneous B-cell lymphoma subtypes: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone lymphoma; primary cutaneous diffuse large B-cell lymphoma (DLBCL), leg type; DLBCL, not otherwise specified; and intravascular DLBCL.1-3 The DLBCL, not otherwise specified, category includes less common provisional entities with insufficient evidence to be recognized as distinct diseases. Epstein-Barr virus (EBV)–positive DLBCL is a rare subtype in this group.4
This article reviews the different clinicopathologic subtypes of primary cutaneous B-cell lymphoma. It also serves to help dermatologists recognize primary cutaneous EBV-positive DLBCL as a rare and aggressive form of this disease.
Case Report
An 84-year-old white man presented with a pruritic eruption on the arms, legs, back, neck, and face of 5 months’ duration. His medical history was notable for prostate cancer that was successfully treated with radiation therapy 6 years prior. The patient denied any constitutional symptoms such as fever, chills, night sweats, or weight loss, and review of systems was negative. The patient was taking prednisone, which alleviated the pruritus, but the lesions persisted.
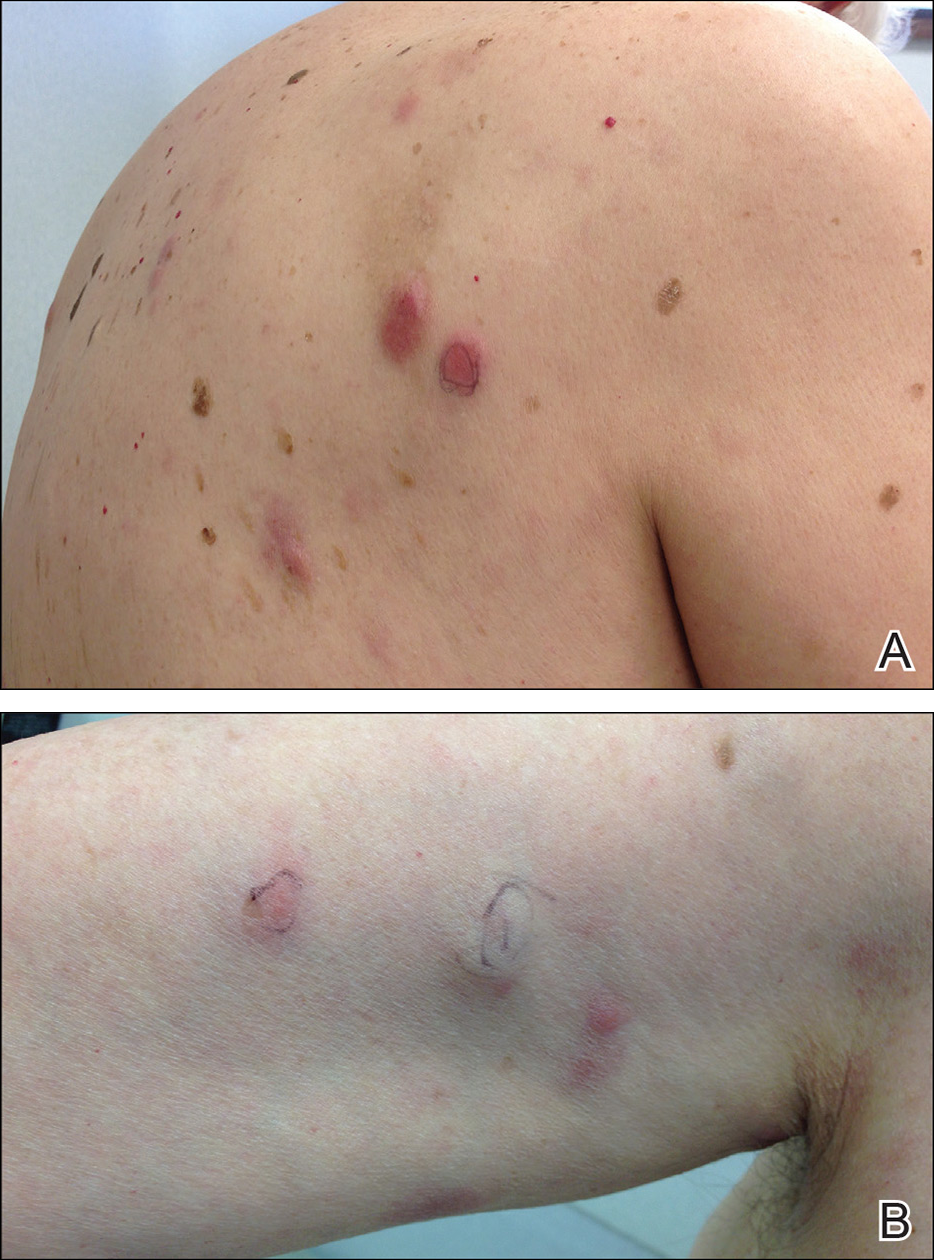
Physical examination revealed multiple pink to erythematous papules and subcutaneous nodules involving the face, neck, back, arms, and legs (Figure 1). No scale, crust, or ulceration was present. Palpation of the cervical, supraclavicular, axillary, and inguinal lymph nodes was negative for lymphadenopathy.
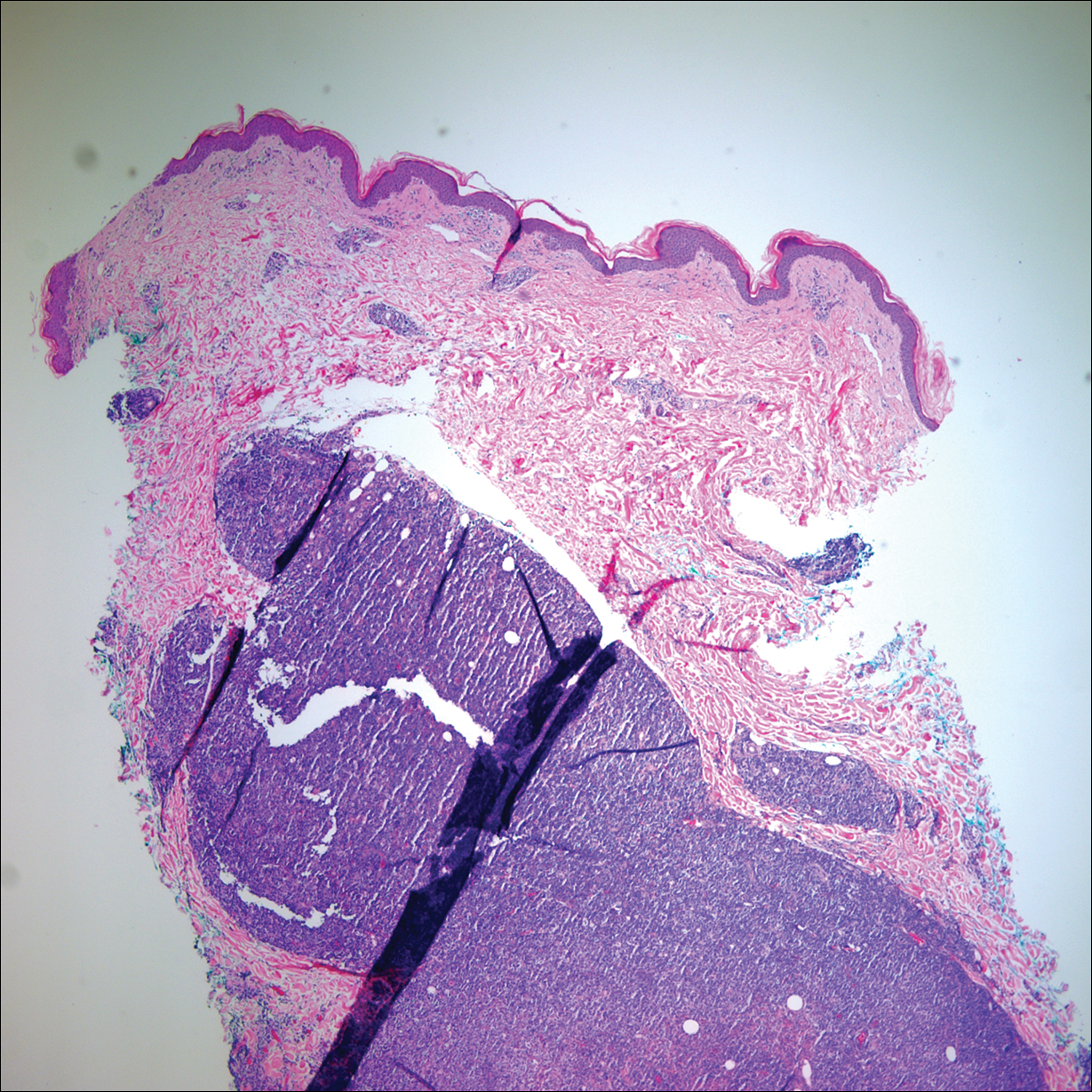
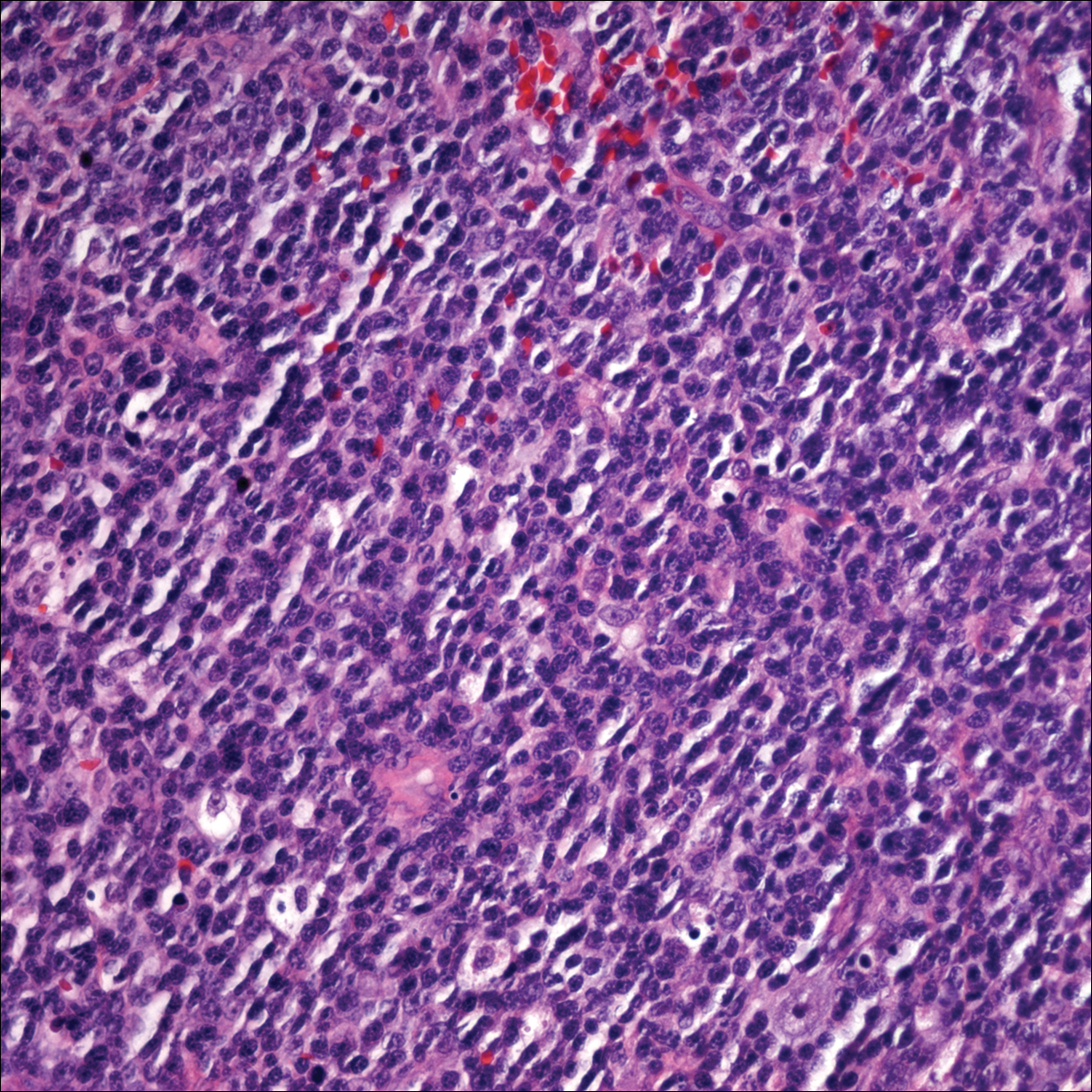
Punch biopsies of representative lesions on the upper back and right arm revealed diffuse and nodular infiltrates of large atypical lymphoid cells with scattered centroblasts and immunoblasts (Figures 2 and 3). Immunohistochemical staining demonstrated CD79, MUM-1, and EBV-encoded RNA positivity among the neoplastic cells. The Ki-67 proliferative index was greater than 90%. The neoplastic cells were negative for CD5, CD10, CD20, CD21, CD30, CD56, CD123, CD138, PAX5, C-MYC, BCL-2, BCL-6, cyclin D1, TCL-1A, and terminal deoxynucleotidyl transferase
A peripheral blood smear did not show evidence of a B-cell lymphoproliferative process. A bone marrow biopsy was performed and did not show evidence of B-cell lymphoid neoplasia but did show reactive lymphoid aggregates composed of CD4+ and CD10+ T cells. Peripheral blood T-cell rearrangement and JAK2 were negative.
Based on clinical and histologic findings, the patient was diagnosed with primary cutaneous EBV-positive DLBCL. The patient was started on CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy for treatment of this aggressive cutaneous lymphoma, which initially resulted in clinical improvement of the lesions and complete involution of the subcutaneous nodules. After the sixth cycle of CHOP, he developed faintly erythematous indurated papules on the upper arms, chest, and back. Biopsy confirmed recurrence of the EBV-positive cutaneous lymphoma, and he started salvage chemotherapy with gemcitabine, oxaliplatin, and rituximab every 2 weeks; however, 4 months later (9 months after the initial presentation) he died from complications of the disease.
Comment
Etiology
Epstein-Barr virus–positive DLBCL, also called EBV-positive DLBCL of the elderly, was initially described in 2003 by Oyama et al5 and was included as a provisional entity in the 2008 World Health Organization classification system as a rare subtype of the DLBCL, not otherwise specified, category.2 It is defined as an EBV-positive monoclonal large B-cell proliferation that occurs in immunocompetent patients older than 50 years.6 Epstein-Barr virus is a human herpesvirus that demonstrates tropism for lymphocytes and survives in human hosts by establishing latency in B cells. Under normal immune conditions, the proliferation of EBV-infected B cells is prevented by cytotoxic T cells.7 It is important to recognize that patients with EBV-positive DLBCL do not have a known immunodeficiency state; therefore, it has been postulated that EBV-positive DLBCL might be caused by age-related senescence of the immune system.4,8
Epidemiology and Clinical Features
Epstein-Barr virus–positive DLBCL is more common in Asian countries than in Western countries, and there is a slight male predominance.6 A majority of patients present with extranodal disease at the time of diagnosis, and the skin is the most common extranodal site of involvement.6,9 Rare cases of primary cutaneous involvement also have been described.7,9,10 Cutaneous manifestations include erythematous papules and subcutaneous nodules. Other sites of extranodal involvement include the lungs, oral cavity, pharynx, gastrointestinal tract, and bone marrow.8,9 However, EBV-positive DLBCL is an aggressive lymphoma and prognosis is poor irrespective of the primary site of involvement.
Histopathology
Two morphologic subtypes can be seen on histology. The polymorphic pattern is characterized by a broad range of B-cell maturation with admixed reactive cells (eg, lymphocytes, histiocytes, plasma cells). The monomorphic or large-cell pattern is characterized by monotonous sheets of large transformed B cells.4,11 Many cases show both histologic patterns, and these morphologic variants do not impart any clinical or prognostic significance. Regardless of the histologic subtype, the neoplastic cells express pan B-cell antigens (eg, CD19, CD20, CD79a, PAX5), as well as MUM-1, BCL-2, and EBV-encoded RNA.4 Cases with plasmablastic features, as in our patient, may show weak or absent CD20 staining.12 Detection of EBV by in situ hybridization is required for the diagnosis.
Diagnosis
Workup for a suspected cutaneous lymphoma should include a complete history and physical examination; laboratory studies; and relevant imaging evaluation such as computed tomography of the chest, abdomen, and pelvis with or without whole-body positron emission tomography. A bone marrow biopsy and aspirate also should be performed in all cutaneous lymphomas with intermediate to aggressive clinical behavior. Accurate staging evaluation is integral to confirm the absence of extracutaneous involvement and to provide prognostic and anatomic information for the appropriate selection of treatment.13
Prognosis and Management
Primary cutaneous lymphomas tend to have different clinical behaviors and prognoses compared to histologically similar systemic lymphomas; therefore, different therapeutic strategies are warranted.14 Epstein-Barr virus–positive DLBCL has an aggressive clinical course with a median survival of 2 years.8 Patients with EBV-positive DLBCL have a poorer overall survival and treatment response when compared to patients with EBV-negative DLBCLs.4 Primary cutaneous B-cell lymphomas with indolent behavior, such as primary cutaneous marginal zone lymphoma and primary cutaneous follicle center lymphoma, can be treated with surgical excision, radiation therapy, or observation.15 No standard treatment exists for EBV-positive DLBCL, but R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone), which is the standard treatment of primary cutaneous DLBCL, leg type, may provide a survival benefit.13,15 Further studies are required to determine optimal treatment strategies.
Conclusion
Although rare, EBV-positive DLBCL is an important entity to consider when evaluating a patient with a suspected primary cutaneous lymphoma. Workup to rule out an underlying systemic lymphoma with relevant laboratory evaluation, imaging studies, and bone marrow biopsy is critical. Prognosis is poor and treatment is difficult, as standard treatment protocols have yet to be determined.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nakmura S, Jaffe ES, Swerdlow SH. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2008:243-244.
- Kempf W, Sander CA. Classification of cutaneous lymphomas—an update. Histopathology. 2010;56:57-70.
- Castillo JJ, Beltran BE, Miranda RN, et al. Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: what we know so far. Oncologist. 2011;16:87-96.
- Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16-26.
- Ok CY, Papathomas TG, Medeiros LJ, et al. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328-340.
- Tokuda Y, Fukushima M, Nakazawa K, et al. A case of primary Epstein-Barr virus-associated cutaneous diffuse large B-cell lymphoma unassociated with iatrogenic or endogenous immune dysregulation. J Cutan Pathol. 2008;35:666-671.
- Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124-5132.
- Eminger LA, Hall LD, Hesterman KS, et al. Epstein-Barr virus: dermatologic associations and implications. J Am Acad Dermatol. 2015;72:21-34.
- Martin B, Whittaker S, Morris S, et al. A case of primary cutaneous senile EBV-related diffuse large B-cell lymphoma. Am J Dermatopathol. 2010;32:190-193.
- Gibson SE, Hsi ED. Epstein-Barr virus-positive B-cell lymphoma of the elderly at a United States tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol. 2009;40:653-661.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-329.e13; quiz 341-342.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:343.e1-343.e11; quiz 355-356.
Cutaneous B-cell lymphomas represent a group of lymphomas derived from B lymphocytes in various stages of differentiation. The skin can be the site of primary or secondary involvement of any of the B-cell lymphomas. Primary cutaneous B-cell lymphomas present in the skin without evidence of extracutaneous disease at the time of diagnosis.1 The World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues recognizes 5 distinct primary cutaneous B-cell lymphoma subtypes: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone lymphoma; primary cutaneous diffuse large B-cell lymphoma (DLBCL), leg type; DLBCL, not otherwise specified; and intravascular DLBCL.1-3 The DLBCL, not otherwise specified, category includes less common provisional entities with insufficient evidence to be recognized as distinct diseases. Epstein-Barr virus (EBV)–positive DLBCL is a rare subtype in this group.4
This article reviews the different clinicopathologic subtypes of primary cutaneous B-cell lymphoma. It also serves to help dermatologists recognize primary cutaneous EBV-positive DLBCL as a rare and aggressive form of this disease.
Case Report
An 84-year-old white man presented with a pruritic eruption on the arms, legs, back, neck, and face of 5 months’ duration. His medical history was notable for prostate cancer that was successfully treated with radiation therapy 6 years prior. The patient denied any constitutional symptoms such as fever, chills, night sweats, or weight loss, and review of systems was negative. The patient was taking prednisone, which alleviated the pruritus, but the lesions persisted.
Physical examination revealed multiple pink to erythematous papules and subcutaneous nodules involving the face, neck, back, arms, and legs (Figure 1). No scale, crust, or ulceration was present. Palpation of the cervical, supraclavicular, axillary, and inguinal lymph nodes was negative for lymphadenopathy.
Punch biopsies of representative lesions on the upper back and right arm revealed diffuse and nodular infiltrates of large atypical lymphoid cells with scattered centroblasts and immunoblasts (Figures 2 and 3). Immunohistochemical staining demonstrated CD79, MUM-1, and EBV-encoded RNA positivity among the neoplastic cells. The Ki-67 proliferative index was greater than 90%. The neoplastic cells were negative for CD5, CD10, CD20, CD21, CD30, CD56, CD123, CD138, PAX5, C-MYC, BCL-2, BCL-6, cyclin D1, TCL-1A, and terminal deoxynucleotidyl transferase
A peripheral blood smear did not show evidence of a B-cell lymphoproliferative process. A bone marrow biopsy was performed and did not show evidence of B-cell lymphoid neoplasia but did show reactive lymphoid aggregates composed of CD4+ and CD10+ T cells. Peripheral blood T-cell rearrangement and JAK2 were negative.
Based on clinical and histologic findings, the patient was diagnosed with primary cutaneous EBV-positive DLBCL. The patient was started on CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy for treatment of this aggressive cutaneous lymphoma, which initially resulted in clinical improvement of the lesions and complete involution of the subcutaneous nodules. After the sixth cycle of CHOP, he developed faintly erythematous indurated papules on the upper arms, chest, and back. Biopsy confirmed recurrence of the EBV-positive cutaneous lymphoma, and he started salvage chemotherapy with gemcitabine, oxaliplatin, and rituximab every 2 weeks; however, 4 months later (9 months after the initial presentation) he died from complications of the disease.
Comment
Etiology
Epstein-Barr virus–positive DLBCL, also called EBV-positive DLBCL of the elderly, was initially described in 2003 by Oyama et al5 and was included as a provisional entity in the 2008 World Health Organization classification system as a rare subtype of the DLBCL, not otherwise specified, category.2 It is defined as an EBV-positive monoclonal large B-cell proliferation that occurs in immunocompetent patients older than 50 years.6 Epstein-Barr virus is a human herpesvirus that demonstrates tropism for lymphocytes and survives in human hosts by establishing latency in B cells. Under normal immune conditions, the proliferation of EBV-infected B cells is prevented by cytotoxic T cells.7 It is important to recognize that patients with EBV-positive DLBCL do not have a known immunodeficiency state; therefore, it has been postulated that EBV-positive DLBCL might be caused by age-related senescence of the immune system.4,8
Epidemiology and Clinical Features
Epstein-Barr virus–positive DLBCL is more common in Asian countries than in Western countries, and there is a slight male predominance.6 A majority of patients present with extranodal disease at the time of diagnosis, and the skin is the most common extranodal site of involvement.6,9 Rare cases of primary cutaneous involvement also have been described.7,9,10 Cutaneous manifestations include erythematous papules and subcutaneous nodules. Other sites of extranodal involvement include the lungs, oral cavity, pharynx, gastrointestinal tract, and bone marrow.8,9 However, EBV-positive DLBCL is an aggressive lymphoma and prognosis is poor irrespective of the primary site of involvement.
Histopathology
Two morphologic subtypes can be seen on histology. The polymorphic pattern is characterized by a broad range of B-cell maturation with admixed reactive cells (eg, lymphocytes, histiocytes, plasma cells). The monomorphic or large-cell pattern is characterized by monotonous sheets of large transformed B cells.4,11 Many cases show both histologic patterns, and these morphologic variants do not impart any clinical or prognostic significance. Regardless of the histologic subtype, the neoplastic cells express pan B-cell antigens (eg, CD19, CD20, CD79a, PAX5), as well as MUM-1, BCL-2, and EBV-encoded RNA.4 Cases with plasmablastic features, as in our patient, may show weak or absent CD20 staining.12 Detection of EBV by in situ hybridization is required for the diagnosis.
Diagnosis
Workup for a suspected cutaneous lymphoma should include a complete history and physical examination; laboratory studies; and relevant imaging evaluation such as computed tomography of the chest, abdomen, and pelvis with or without whole-body positron emission tomography. A bone marrow biopsy and aspirate also should be performed in all cutaneous lymphomas with intermediate to aggressive clinical behavior. Accurate staging evaluation is integral to confirm the absence of extracutaneous involvement and to provide prognostic and anatomic information for the appropriate selection of treatment.13
Prognosis and Management
Primary cutaneous lymphomas tend to have different clinical behaviors and prognoses compared to histologically similar systemic lymphomas; therefore, different therapeutic strategies are warranted.14 Epstein-Barr virus–positive DLBCL has an aggressive clinical course with a median survival of 2 years.8 Patients with EBV-positive DLBCL have a poorer overall survival and treatment response when compared to patients with EBV-negative DLBCLs.4 Primary cutaneous B-cell lymphomas with indolent behavior, such as primary cutaneous marginal zone lymphoma and primary cutaneous follicle center lymphoma, can be treated with surgical excision, radiation therapy, or observation.15 No standard treatment exists for EBV-positive DLBCL, but R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone), which is the standard treatment of primary cutaneous DLBCL, leg type, may provide a survival benefit.13,15 Further studies are required to determine optimal treatment strategies.
Conclusion
Although rare, EBV-positive DLBCL is an important entity to consider when evaluating a patient with a suspected primary cutaneous lymphoma. Workup to rule out an underlying systemic lymphoma with relevant laboratory evaluation, imaging studies, and bone marrow biopsy is critical. Prognosis is poor and treatment is difficult, as standard treatment protocols have yet to be determined.
Cutaneous B-cell lymphomas represent a group of lymphomas derived from B lymphocytes in various stages of differentiation. The skin can be the site of primary or secondary involvement of any of the B-cell lymphomas. Primary cutaneous B-cell lymphomas present in the skin without evidence of extracutaneous disease at the time of diagnosis.1 The World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues recognizes 5 distinct primary cutaneous B-cell lymphoma subtypes: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone lymphoma; primary cutaneous diffuse large B-cell lymphoma (DLBCL), leg type; DLBCL, not otherwise specified; and intravascular DLBCL.1-3 The DLBCL, not otherwise specified, category includes less common provisional entities with insufficient evidence to be recognized as distinct diseases. Epstein-Barr virus (EBV)–positive DLBCL is a rare subtype in this group.4
This article reviews the different clinicopathologic subtypes of primary cutaneous B-cell lymphoma. It also serves to help dermatologists recognize primary cutaneous EBV-positive DLBCL as a rare and aggressive form of this disease.
Case Report
An 84-year-old white man presented with a pruritic eruption on the arms, legs, back, neck, and face of 5 months’ duration. His medical history was notable for prostate cancer that was successfully treated with radiation therapy 6 years prior. The patient denied any constitutional symptoms such as fever, chills, night sweats, or weight loss, and review of systems was negative. The patient was taking prednisone, which alleviated the pruritus, but the lesions persisted.
Physical examination revealed multiple pink to erythematous papules and subcutaneous nodules involving the face, neck, back, arms, and legs (Figure 1). No scale, crust, or ulceration was present. Palpation of the cervical, supraclavicular, axillary, and inguinal lymph nodes was negative for lymphadenopathy.
Punch biopsies of representative lesions on the upper back and right arm revealed diffuse and nodular infiltrates of large atypical lymphoid cells with scattered centroblasts and immunoblasts (Figures 2 and 3). Immunohistochemical staining demonstrated CD79, MUM-1, and EBV-encoded RNA positivity among the neoplastic cells. The Ki-67 proliferative index was greater than 90%. The neoplastic cells were negative for CD5, CD10, CD20, CD21, CD30, CD56, CD123, CD138, PAX5, C-MYC, BCL-2, BCL-6, cyclin D1, TCL-1A, and terminal deoxynucleotidyl transferase
A peripheral blood smear did not show evidence of a B-cell lymphoproliferative process. A bone marrow biopsy was performed and did not show evidence of B-cell lymphoid neoplasia but did show reactive lymphoid aggregates composed of CD4+ and CD10+ T cells. Peripheral blood T-cell rearrangement and JAK2 were negative.
Based on clinical and histologic findings, the patient was diagnosed with primary cutaneous EBV-positive DLBCL. The patient was started on CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy for treatment of this aggressive cutaneous lymphoma, which initially resulted in clinical improvement of the lesions and complete involution of the subcutaneous nodules. After the sixth cycle of CHOP, he developed faintly erythematous indurated papules on the upper arms, chest, and back. Biopsy confirmed recurrence of the EBV-positive cutaneous lymphoma, and he started salvage chemotherapy with gemcitabine, oxaliplatin, and rituximab every 2 weeks; however, 4 months later (9 months after the initial presentation) he died from complications of the disease.
Comment
Etiology
Epstein-Barr virus–positive DLBCL, also called EBV-positive DLBCL of the elderly, was initially described in 2003 by Oyama et al5 and was included as a provisional entity in the 2008 World Health Organization classification system as a rare subtype of the DLBCL, not otherwise specified, category.2 It is defined as an EBV-positive monoclonal large B-cell proliferation that occurs in immunocompetent patients older than 50 years.6 Epstein-Barr virus is a human herpesvirus that demonstrates tropism for lymphocytes and survives in human hosts by establishing latency in B cells. Under normal immune conditions, the proliferation of EBV-infected B cells is prevented by cytotoxic T cells.7 It is important to recognize that patients with EBV-positive DLBCL do not have a known immunodeficiency state; therefore, it has been postulated that EBV-positive DLBCL might be caused by age-related senescence of the immune system.4,8
Epidemiology and Clinical Features
Epstein-Barr virus–positive DLBCL is more common in Asian countries than in Western countries, and there is a slight male predominance.6 A majority of patients present with extranodal disease at the time of diagnosis, and the skin is the most common extranodal site of involvement.6,9 Rare cases of primary cutaneous involvement also have been described.7,9,10 Cutaneous manifestations include erythematous papules and subcutaneous nodules. Other sites of extranodal involvement include the lungs, oral cavity, pharynx, gastrointestinal tract, and bone marrow.8,9 However, EBV-positive DLBCL is an aggressive lymphoma and prognosis is poor irrespective of the primary site of involvement.
Histopathology
Two morphologic subtypes can be seen on histology. The polymorphic pattern is characterized by a broad range of B-cell maturation with admixed reactive cells (eg, lymphocytes, histiocytes, plasma cells). The monomorphic or large-cell pattern is characterized by monotonous sheets of large transformed B cells.4,11 Many cases show both histologic patterns, and these morphologic variants do not impart any clinical or prognostic significance. Regardless of the histologic subtype, the neoplastic cells express pan B-cell antigens (eg, CD19, CD20, CD79a, PAX5), as well as MUM-1, BCL-2, and EBV-encoded RNA.4 Cases with plasmablastic features, as in our patient, may show weak or absent CD20 staining.12 Detection of EBV by in situ hybridization is required for the diagnosis.
Diagnosis
Workup for a suspected cutaneous lymphoma should include a complete history and physical examination; laboratory studies; and relevant imaging evaluation such as computed tomography of the chest, abdomen, and pelvis with or without whole-body positron emission tomography. A bone marrow biopsy and aspirate also should be performed in all cutaneous lymphomas with intermediate to aggressive clinical behavior. Accurate staging evaluation is integral to confirm the absence of extracutaneous involvement and to provide prognostic and anatomic information for the appropriate selection of treatment.13
Prognosis and Management
Primary cutaneous lymphomas tend to have different clinical behaviors and prognoses compared to histologically similar systemic lymphomas; therefore, different therapeutic strategies are warranted.14 Epstein-Barr virus–positive DLBCL has an aggressive clinical course with a median survival of 2 years.8 Patients with EBV-positive DLBCL have a poorer overall survival and treatment response when compared to patients with EBV-negative DLBCLs.4 Primary cutaneous B-cell lymphomas with indolent behavior, such as primary cutaneous marginal zone lymphoma and primary cutaneous follicle center lymphoma, can be treated with surgical excision, radiation therapy, or observation.15 No standard treatment exists for EBV-positive DLBCL, but R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone), which is the standard treatment of primary cutaneous DLBCL, leg type, may provide a survival benefit.13,15 Further studies are required to determine optimal treatment strategies.
Conclusion
Although rare, EBV-positive DLBCL is an important entity to consider when evaluating a patient with a suspected primary cutaneous lymphoma. Workup to rule out an underlying systemic lymphoma with relevant laboratory evaluation, imaging studies, and bone marrow biopsy is critical. Prognosis is poor and treatment is difficult, as standard treatment protocols have yet to be determined.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nakmura S, Jaffe ES, Swerdlow SH. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2008:243-244.
- Kempf W, Sander CA. Classification of cutaneous lymphomas—an update. Histopathology. 2010;56:57-70.
- Castillo JJ, Beltran BE, Miranda RN, et al. Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: what we know so far. Oncologist. 2011;16:87-96.
- Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16-26.
- Ok CY, Papathomas TG, Medeiros LJ, et al. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328-340.
- Tokuda Y, Fukushima M, Nakazawa K, et al. A case of primary Epstein-Barr virus-associated cutaneous diffuse large B-cell lymphoma unassociated with iatrogenic or endogenous immune dysregulation. J Cutan Pathol. 2008;35:666-671.
- Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124-5132.
- Eminger LA, Hall LD, Hesterman KS, et al. Epstein-Barr virus: dermatologic associations and implications. J Am Acad Dermatol. 2015;72:21-34.
- Martin B, Whittaker S, Morris S, et al. A case of primary cutaneous senile EBV-related diffuse large B-cell lymphoma. Am J Dermatopathol. 2010;32:190-193.
- Gibson SE, Hsi ED. Epstein-Barr virus-positive B-cell lymphoma of the elderly at a United States tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol. 2009;40:653-661.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-329.e13; quiz 341-342.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:343.e1-343.e11; quiz 355-356.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nakmura S, Jaffe ES, Swerdlow SH. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2008:243-244.
- Kempf W, Sander CA. Classification of cutaneous lymphomas—an update. Histopathology. 2010;56:57-70.
- Castillo JJ, Beltran BE, Miranda RN, et al. Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: what we know so far. Oncologist. 2011;16:87-96.
- Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16-26.
- Ok CY, Papathomas TG, Medeiros LJ, et al. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328-340.
- Tokuda Y, Fukushima M, Nakazawa K, et al. A case of primary Epstein-Barr virus-associated cutaneous diffuse large B-cell lymphoma unassociated with iatrogenic or endogenous immune dysregulation. J Cutan Pathol. 2008;35:666-671.
- Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124-5132.
- Eminger LA, Hall LD, Hesterman KS, et al. Epstein-Barr virus: dermatologic associations and implications. J Am Acad Dermatol. 2015;72:21-34.
- Martin B, Whittaker S, Morris S, et al. A case of primary cutaneous senile EBV-related diffuse large B-cell lymphoma. Am J Dermatopathol. 2010;32:190-193.
- Gibson SE, Hsi ED. Epstein-Barr virus-positive B-cell lymphoma of the elderly at a United States tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol. 2009;40:653-661.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-329.e13; quiz 341-342.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:343.e1-343.e11; quiz 355-356.
Practice Points
- Primary cutaneous lymphomas are malignant lymphomas confined to the skin.
- Complete staging workup is necessary to rule out secondary involvement of the skin from a nodal lymphoma.
- Epstein-Barr virus-positive diffuse large B-cell lymphoma is a rare and aggressive primary cutaneous lymphoma.
Reticulated erythematous patch on teenager’s foot
An 18-year-old Caucasian male sought care for an ill-defined reticulated patch on his right plantar arch (FIGURE 1). The patient said that the lesion had gradually appeared 2 years earlier, had grown slowly, and was occasionally itchy. Physical exam revealed a lacy violaceous, hyperpigmented, reticulated patch that was blanchable and nontender to palpation.
Our patient denied having a history of trauma to the area or a coagulation or connective tissue disorder. The lesion didn’t vary with temperature or season, and there were no known triggers. The patient’s left plantar arch was unchanged.

WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Erythema ab igne
Upon further questioning, the patient acknowledged that he occasionally rested his bare feet around a portable heater under his desk while using his computer for a few hours each day (FIGURE 2). He often kept his right foot on the heater while he let his left foot rest on the ground. A punch biopsy was performed; the findings, when combined with the patient’s report of having exposed his foot to heat, supported the diagnosis of erythema ab igne (EAI).

EAI commonly presents as an asymptomatic reticulated erythematous to violaceous patch in an area of the body that has been in contact with heat.1 It originally was described on the bilateral anterior lower extremities after prolonged exposure to burning stoves or open fires.1 With the advent of central heating, these presentations have decreased, but there has been a resurgence of EAI with atypical distributions as a result of evolving technology and new heating sources. Reported causes of EAI include heating pads,1,2 laptop computers3 (FIGURE 3), car seat heaters,4 hot water bottles, popcorn bags, cell phones,5 and space heaters that have resulted in patches on the breast, thighs, arms, and, in our patient, foot.1-5
Blood work, biopsy can help narrow the differential
The differential for EAI includes livedo reticularis, livedo racemosa, cutis marmorata, and cutis marmorata telangiectasia. Livedo reticularis can be associated with autoimmune conditions and coagulopathies. Livedo racemosa is a typical sign of Sneddon’s syndrome and can be seen in up to 70% of patients with antiphospholipid-antibody syndrome and systemic lupus erythematosus. Diagnosis of these conditions is confirmed by elevated coagulation factors, presence of autoimmune antibodies, or history of cerebrovascular accident.6 These tests would be normal in EAI.
Histopathologic changes observed in EAI include an atrophic epidermis with an interface dermatitis, vasodilation, and dermal pigmentation. Necrotic keratinocytes and focal hyperkeratosis can be noted, along with squamous atypia. Although these changes are nonspecific, they can be used to confirm an EAI diagnosis in patients for whom the affected area has been exposed to a heat source.
Histologically, EAI is similar to actinic keratosis, with epidermal changes showing squamous atypia.2 Due to the similarities, these lesions are sometimes referred to as “thermal keratosis.” Some researchers have suggested that the thermal heat may induce epithelial changes in the same way that ultraviolet light produces epithelial changes.7
Rarely, EAI can turn into cancer. There have been a few reported cases of EAI transforming into squamous cell carcinoma or Merkel cell carcinoma; squamous cell carcinoma is more common, and tends to occur after a long latent period (up to 30 years).7-9 EAI lesions often begin as a chronic ulcer and tend not to heal. If the lesion continues to evolve (ie, ulcerate), a biopsy may be warranted to rule out a malignant transformation.
Eliminate heat exposure, consider a topical treatment
Treatment of acute EAI involves eliminating the offending heat source. The hyperpigmentation will slowly resolve over months to years.4 Persistent exposure to heat sources can lead to chronic EAI, which is more difficult to eliminate.
Because hyperpigmentation can be visually unappealing and emotionally distressing, some patients prefer active treatment. EAI has been effectively treated with 4% hydroquinone topical cream twice a day and tretinoin topical cream at night.2,10,11 Lesions that have epithelial atypia have improved with 5-fluorouracil topical cream.7
EAI also has been successfully treated with laser therapy with the 1064-nm Q-switched Nd:YAG laser with low fluence at 2-week intervals.9
Our patient declined topical therapy. He improved after a few months of avoiding the heater under his desk.
CORRESPONDENCE
Megan Morrison, DO, 5333 McAuley Drive Suite R-5003, Ypsilanti, MI 48197; memorrison10@gmail.com
1. Huynh N, Sarma D, Huerter C. Erythema ab igne: a case report and review of the literature. Cutis. 2011;88:290-292.
2. Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162:77-78.
3. Fu LW, Vender R. Erythema ab igne caused by laptop computer gaming—a case report. Int J Dermatol. 2012;51:716-717.
4. Brodell D, Mostow EN. Automobile seat heater-induced erythema ab igne. Arch Dermtol. 2012;148:264-265.
5. Dela Rosa K, Satter EK. Erythematous patches on the chest. Arch Dermatol. 2012;148:113-118.
6. Uthman IW, Khamashta MA. Livedo racemosa: a striking dermatological sign for antiphospholipid syndrome. J Rheumatol. 2006;33:2379-2382.
7. Bilic M, Adams B. Erythema ab igne induced by a laptop computer. J Am Acad Dermatol. 2004;50:973-974.
8. Jones CS, Tyring SK, Lee PC, et al. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1998;124:110-113.
9. Cho S, Jung JY, Lee JH. Erythema ab igne successfully treated using 1,064-nm Q-switched neodymium-doped yttrium aluminum garnet laser with low fluence. Dermatol Surg. 2011;37:551-553.
10. Cardona LFC, Parsons AC, Sangueza OP. Erythematous lesions on the back of a man: challenge. Erythema ab igne. Am J Dermatopathol. 2011;33:185,199.
11. Sahl WJ, Taira JW. Erythema ab igne: treatment with 5-fluorouracil cream. J Am Acad Dermatol. 1992;27:109-110.
An 18-year-old Caucasian male sought care for an ill-defined reticulated patch on his right plantar arch (FIGURE 1). The patient said that the lesion had gradually appeared 2 years earlier, had grown slowly, and was occasionally itchy. Physical exam revealed a lacy violaceous, hyperpigmented, reticulated patch that was blanchable and nontender to palpation.
Our patient denied having a history of trauma to the area or a coagulation or connective tissue disorder. The lesion didn’t vary with temperature or season, and there were no known triggers. The patient’s left plantar arch was unchanged.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Erythema ab igne
Upon further questioning, the patient acknowledged that he occasionally rested his bare feet around a portable heater under his desk while using his computer for a few hours each day (FIGURE 2). He often kept his right foot on the heater while he let his left foot rest on the ground. A punch biopsy was performed; the findings, when combined with the patient’s report of having exposed his foot to heat, supported the diagnosis of erythema ab igne (EAI).
EAI commonly presents as an asymptomatic reticulated erythematous to violaceous patch in an area of the body that has been in contact with heat.1 It originally was described on the bilateral anterior lower extremities after prolonged exposure to burning stoves or open fires.1 With the advent of central heating, these presentations have decreased, but there has been a resurgence of EAI with atypical distributions as a result of evolving technology and new heating sources. Reported causes of EAI include heating pads,1,2 laptop computers3 (FIGURE 3), car seat heaters,4 hot water bottles, popcorn bags, cell phones,5 and space heaters that have resulted in patches on the breast, thighs, arms, and, in our patient, foot.1-5
Blood work, biopsy can help narrow the differential
The differential for EAI includes livedo reticularis, livedo racemosa, cutis marmorata, and cutis marmorata telangiectasia. Livedo reticularis can be associated with autoimmune conditions and coagulopathies. Livedo racemosa is a typical sign of Sneddon’s syndrome and can be seen in up to 70% of patients with antiphospholipid-antibody syndrome and systemic lupus erythematosus. Diagnosis of these conditions is confirmed by elevated coagulation factors, presence of autoimmune antibodies, or history of cerebrovascular accident.6 These tests would be normal in EAI.
Histopathologic changes observed in EAI include an atrophic epidermis with an interface dermatitis, vasodilation, and dermal pigmentation. Necrotic keratinocytes and focal hyperkeratosis can be noted, along with squamous atypia. Although these changes are nonspecific, they can be used to confirm an EAI diagnosis in patients for whom the affected area has been exposed to a heat source.
Histologically, EAI is similar to actinic keratosis, with epidermal changes showing squamous atypia.2 Due to the similarities, these lesions are sometimes referred to as “thermal keratosis.” Some researchers have suggested that the thermal heat may induce epithelial changes in the same way that ultraviolet light produces epithelial changes.7
Rarely, EAI can turn into cancer. There have been a few reported cases of EAI transforming into squamous cell carcinoma or Merkel cell carcinoma; squamous cell carcinoma is more common, and tends to occur after a long latent period (up to 30 years).7-9 EAI lesions often begin as a chronic ulcer and tend not to heal. If the lesion continues to evolve (ie, ulcerate), a biopsy may be warranted to rule out a malignant transformation.
Eliminate heat exposure, consider a topical treatment
Treatment of acute EAI involves eliminating the offending heat source. The hyperpigmentation will slowly resolve over months to years.4 Persistent exposure to heat sources can lead to chronic EAI, which is more difficult to eliminate.
Because hyperpigmentation can be visually unappealing and emotionally distressing, some patients prefer active treatment. EAI has been effectively treated with 4% hydroquinone topical cream twice a day and tretinoin topical cream at night.2,10,11 Lesions that have epithelial atypia have improved with 5-fluorouracil topical cream.7
EAI also has been successfully treated with laser therapy with the 1064-nm Q-switched Nd:YAG laser with low fluence at 2-week intervals.9
Our patient declined topical therapy. He improved after a few months of avoiding the heater under his desk.
CORRESPONDENCE
Megan Morrison, DO, 5333 McAuley Drive Suite R-5003, Ypsilanti, MI 48197; memorrison10@gmail.com
An 18-year-old Caucasian male sought care for an ill-defined reticulated patch on his right plantar arch (FIGURE 1). The patient said that the lesion had gradually appeared 2 years earlier, had grown slowly, and was occasionally itchy. Physical exam revealed a lacy violaceous, hyperpigmented, reticulated patch that was blanchable and nontender to palpation.
Our patient denied having a history of trauma to the area or a coagulation or connective tissue disorder. The lesion didn’t vary with temperature or season, and there were no known triggers. The patient’s left plantar arch was unchanged.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Erythema ab igne
Upon further questioning, the patient acknowledged that he occasionally rested his bare feet around a portable heater under his desk while using his computer for a few hours each day (FIGURE 2). He often kept his right foot on the heater while he let his left foot rest on the ground. A punch biopsy was performed; the findings, when combined with the patient’s report of having exposed his foot to heat, supported the diagnosis of erythema ab igne (EAI).
EAI commonly presents as an asymptomatic reticulated erythematous to violaceous patch in an area of the body that has been in contact with heat.1 It originally was described on the bilateral anterior lower extremities after prolonged exposure to burning stoves or open fires.1 With the advent of central heating, these presentations have decreased, but there has been a resurgence of EAI with atypical distributions as a result of evolving technology and new heating sources. Reported causes of EAI include heating pads,1,2 laptop computers3 (FIGURE 3), car seat heaters,4 hot water bottles, popcorn bags, cell phones,5 and space heaters that have resulted in patches on the breast, thighs, arms, and, in our patient, foot.1-5
Blood work, biopsy can help narrow the differential
The differential for EAI includes livedo reticularis, livedo racemosa, cutis marmorata, and cutis marmorata telangiectasia. Livedo reticularis can be associated with autoimmune conditions and coagulopathies. Livedo racemosa is a typical sign of Sneddon’s syndrome and can be seen in up to 70% of patients with antiphospholipid-antibody syndrome and systemic lupus erythematosus. Diagnosis of these conditions is confirmed by elevated coagulation factors, presence of autoimmune antibodies, or history of cerebrovascular accident.6 These tests would be normal in EAI.
Histopathologic changes observed in EAI include an atrophic epidermis with an interface dermatitis, vasodilation, and dermal pigmentation. Necrotic keratinocytes and focal hyperkeratosis can be noted, along with squamous atypia. Although these changes are nonspecific, they can be used to confirm an EAI diagnosis in patients for whom the affected area has been exposed to a heat source.
Histologically, EAI is similar to actinic keratosis, with epidermal changes showing squamous atypia.2 Due to the similarities, these lesions are sometimes referred to as “thermal keratosis.” Some researchers have suggested that the thermal heat may induce epithelial changes in the same way that ultraviolet light produces epithelial changes.7
Rarely, EAI can turn into cancer. There have been a few reported cases of EAI transforming into squamous cell carcinoma or Merkel cell carcinoma; squamous cell carcinoma is more common, and tends to occur after a long latent period (up to 30 years).7-9 EAI lesions often begin as a chronic ulcer and tend not to heal. If the lesion continues to evolve (ie, ulcerate), a biopsy may be warranted to rule out a malignant transformation.
Eliminate heat exposure, consider a topical treatment
Treatment of acute EAI involves eliminating the offending heat source. The hyperpigmentation will slowly resolve over months to years.4 Persistent exposure to heat sources can lead to chronic EAI, which is more difficult to eliminate.
Because hyperpigmentation can be visually unappealing and emotionally distressing, some patients prefer active treatment. EAI has been effectively treated with 4% hydroquinone topical cream twice a day and tretinoin topical cream at night.2,10,11 Lesions that have epithelial atypia have improved with 5-fluorouracil topical cream.7
EAI also has been successfully treated with laser therapy with the 1064-nm Q-switched Nd:YAG laser with low fluence at 2-week intervals.9
Our patient declined topical therapy. He improved after a few months of avoiding the heater under his desk.
CORRESPONDENCE
Megan Morrison, DO, 5333 McAuley Drive Suite R-5003, Ypsilanti, MI 48197; memorrison10@gmail.com
1. Huynh N, Sarma D, Huerter C. Erythema ab igne: a case report and review of the literature. Cutis. 2011;88:290-292.
2. Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162:77-78.
3. Fu LW, Vender R. Erythema ab igne caused by laptop computer gaming—a case report. Int J Dermatol. 2012;51:716-717.
4. Brodell D, Mostow EN. Automobile seat heater-induced erythema ab igne. Arch Dermtol. 2012;148:264-265.
5. Dela Rosa K, Satter EK. Erythematous patches on the chest. Arch Dermatol. 2012;148:113-118.
6. Uthman IW, Khamashta MA. Livedo racemosa: a striking dermatological sign for antiphospholipid syndrome. J Rheumatol. 2006;33:2379-2382.
7. Bilic M, Adams B. Erythema ab igne induced by a laptop computer. J Am Acad Dermatol. 2004;50:973-974.
8. Jones CS, Tyring SK, Lee PC, et al. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1998;124:110-113.
9. Cho S, Jung JY, Lee JH. Erythema ab igne successfully treated using 1,064-nm Q-switched neodymium-doped yttrium aluminum garnet laser with low fluence. Dermatol Surg. 2011;37:551-553.
10. Cardona LFC, Parsons AC, Sangueza OP. Erythematous lesions on the back of a man: challenge. Erythema ab igne. Am J Dermatopathol. 2011;33:185,199.
11. Sahl WJ, Taira JW. Erythema ab igne: treatment with 5-fluorouracil cream. J Am Acad Dermatol. 1992;27:109-110.
1. Huynh N, Sarma D, Huerter C. Erythema ab igne: a case report and review of the literature. Cutis. 2011;88:290-292.
2. Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162:77-78.
3. Fu LW, Vender R. Erythema ab igne caused by laptop computer gaming—a case report. Int J Dermatol. 2012;51:716-717.
4. Brodell D, Mostow EN. Automobile seat heater-induced erythema ab igne. Arch Dermtol. 2012;148:264-265.
5. Dela Rosa K, Satter EK. Erythematous patches on the chest. Arch Dermatol. 2012;148:113-118.
6. Uthman IW, Khamashta MA. Livedo racemosa: a striking dermatological sign for antiphospholipid syndrome. J Rheumatol. 2006;33:2379-2382.
7. Bilic M, Adams B. Erythema ab igne induced by a laptop computer. J Am Acad Dermatol. 2004;50:973-974.
8. Jones CS, Tyring SK, Lee PC, et al. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1998;124:110-113.
9. Cho S, Jung JY, Lee JH. Erythema ab igne successfully treated using 1,064-nm Q-switched neodymium-doped yttrium aluminum garnet laser with low fluence. Dermatol Surg. 2011;37:551-553.
10. Cardona LFC, Parsons AC, Sangueza OP. Erythematous lesions on the back of a man: challenge. Erythema ab igne. Am J Dermatopathol. 2011;33:185,199.
11. Sahl WJ, Taira JW. Erythema ab igne: treatment with 5-fluorouracil cream. J Am Acad Dermatol. 1992;27:109-110.