User login
Pharmacogenetic testing: Navigating through the confusion
Mr. J, age 30, a Black man with major depressive disorder (MDD), has been your patient for the past year. At the time of his diagnosis, Mr. J received sertraline, 100 mg/d, but had little to no improvement. During the past year, he received trials of citalopram and paroxetine, but they were not effective for his recurrent depressive symptoms and/or resulted in significant adverse effects.
During a recent visit, Mr. J asks you about “the genetic tests that help determine which medications will work.” He mentions that his brother had this testing done and that it had “worked for him,” but offers no other details. You research the different testing panels to see which test you might use. After a brief online review, you identify at least 4 different products, and are not sure which test—if any—you should consider.
During the last few years, there has been a rise in commercial pharmacogenetic testing options, including tests available to clinicians at academic medical centers as well as direct-to-consumer testing (Table). Clinician and patient interest regarding pharmacogenetic testing in practice is often followed by the question, “Which test is best?” Although this is a logical question, providing an answer is multifactorial.1-3 Because none of the currently available tests have been compared in head-to-head clinical trials, it is nearly impossible to identify the “best” test.

In this article, we focus on the evidence-based principles that clinicians should consider when adopting pharmacogenetic testing in their practice. We discuss which genes are of most interest when prescribing psychotropic medications, the value of decision support tools, cost considerations, and patient education regarding this type of testing.
Which genes and variants should be tested?
The genes relevant to medication treatment outcomes can be broadly classified into those with pharmacokinetic vs pharmacodynamic effects. Pharmacogenes, such as those coding for the drug-metabolizing enzymes cytochrome P450 (CYP) 1A2, CYP2B6, CYP2C19, CYP2C9, CYP2D6, CYP3A4, and UDP-glucuronosyltransferase (UGT)2B1, may alter the rate at which medications are metabolized, thus varying the serum drug concentration across patients. Variants that impact the function of these enzymes are considered pharmacokinetic. Up to 40% of the variance in patients’ response to antidepressants may be due to variations in the pharmacokinetic genes.4 Alternatively, pharmacodynamic pharmacogenes impact drug action and therefore may affect the degree of receptor activation at a given drug concentration, overall drug efficacy, and/or the occurrence of medication sensitivity. These pharmacogenes may include:
- brain-derived neurotrophic factor (BDNF)
- catechol-O-methyltransferase (COMT)
- human leukocyte antigens A (HLA-A)
- serotonin receptor subtype 2 (HTR2)
- serotonin receptor subtype 2C (HTR2C)
- opioid receptor mu 1 (OPRM1)
- solute carrier family 6 member 4 (SLC6A4).
In articles previously published in
Currently, there is no standardization among commercial pharmacogenetic tests on:
- which genes to test
- which variants specific to a gene need to be included
- how the genetic data is translated to phenotype
- how the phenotype is translated to a treatment recommendation.
Continue to: Due to these factors...
Due to these factors, the FDA has advised clinicians to consult the dosing recommendations provided in a medication’s package insert for information regarding how genetic information should be used in making treatment decisions.2
The value of decision support tools
Researchers have assessed how various manufacturers’ decision support tools (DSTs) (ie, the reports the commercial testing companies send to the clinician who orders the test) agree on genotypes, predicted phenotypes, and medication recommendations.4 Overall, this research found varying levels of disagreement in the medication recommendations of the testing panels they studied, which indicates that not all tests are equivalent or interchangeable.4 Of the actionable recommendations for antidepressants, 16% were conflicting; the recommendations for fluoxetine and imipramine were most frequently in disagreement.4 Similarly, 20% of the actionable antipsychotic advice was conflicting, with the recommendations for aripiprazole and clozapine most frequently in disagreement.4 Researchers also reported a situation in which 4 testing panels agreed on the patient’s phenotyping status for CYP2C19, but the dosing recommendations provided for the CYP2C19 substrate, amitriptyline, differed.4 Thus, it is understandable why DSTs can result in confusion, and why clinicians should use testing panels with recommendations that best align with their individual practices, their patient’s needs, and FDA information.
Additionally, while the genes included on these panels vary, these testing panels also may not evaluate the same variants within a specific gene. These differences may impact the patient’s reported phenotypes and medication recommendations across DSTs. For example, the FDA has recommended HLA gene testing prior to prescribing carbamazepine. However, few of the available tests may include the HLA-B*15:02 variant, which has been associated with carbamazepine-induced severe cutaneous reactions in patients of Asian descent, and fewer may include the HLA-A*31:01 variant, for which testing is recommended prior to prescribing carbamazepine in patients of Caucasian descent.4 Additionally, some of the CYP enzymes—such as CYP2D6*17 and CYP2C19*3 variants, which may be more common in certain populations of patients who are members of ethnic or racial minority groups—may not be consistently included in the various panels. Thus, before deciding on a specific test, clinicians should understand which gene variants are relevant to their patients with regard to race and ethnicity, and key variants for specific medications. Clinicians should refer to FDA guidance and the Clinical Pharmacogenomics Implementation Consortium (CPIC) guidelines to determine the appropriate interpretations of genetic test results.1,2
Despite the disagreement in recommendations from the various testing companies, DSTs are useful and have been shown to facilitate implementation of relevant psychopharmacology dosing guidelines, assist in identifying optimal medication therapy, and improve patient outcomes. A recently published meta-analysis of randomized controlled trials (RCTs) of pharmacogenetic testing found that DSTs improved symptom remission among individuals with MDD by 70%.5 This suggests that pharmacogenetic-guided DSTs may provide superior treatment compared with treatment for DSTs were not used. However, the RCTs in this meta-analysis only included patients who had previously failed an antidepressant trial.5 Therefore, it is currently unknown at what point in care DSTs should be used, and whether they would be more beneficial if they are used when starting a new therapy, or after several trials have failed.
Consider the cost
The cost and availability of pharmacogenetic testing can be an issue when making treatment decisions, and such testing may not be covered by a patient’s insurance plan. Recently, the Centers for Medicare & Medicaid Services announced that Medicare would cover FDA-approved genomic tests that encompass broad gene panels if the evidence supports their use. Similarly, commercial insurers such as UnitedHealthcare have begun to cover some pharmacogenetic tests.6 Medicare or Medicaid plans cover some testing panels’ costs and patients do not incur any out-of-pocket costs; however, some private insurance companies require patients to pay at least a portion of the cost, and many companies offer financial assistance for patients based on income and other factors. Although financial coverage for testing has improved, patients may still face out-of-pocket costs; therefore, clinicians may need to weigh the benefits of pharmacogenetic testing vs its cost.7 Clinicians should also determine what timeline best suits their patient’s financial and clinical needs, and test accordingly.
Continue to: Patient education is critical
Patient education is critical
Although the benefits of using pharmacogenetic testing information when making certain treatment decisions is promising, it is important for both patients and clinicians to understand that test results do not always change therapy. A study on the impact of pharmacogenetic testing on clinical outcomes of patients with MDD found that 79% of patients were already prescribed medications that aligned with recommendations.8 Therefore, switching medications based on the test results of a patient who is doing well clinically is not recommended. However, DSTs may help with clinical decisions for ambiguous cases. For example, if a patient has a genotype and/or phenotype that aligns with medication recommendations, the DST might not be able to identify a better medication to use, but may be able to recommend dosing guidance to improve the tolerability of the patient’s current therapy.6 It is also important to understand that the results of such testing may have a broader use beyond the initial reason for obtaining testing, such as when prescribing a common blood thinner such as warfarin or clopidogrel. However, for many of the pharmacodynamic genes that are included in these panels, their use beyond the treatment of depression may be limited because outcome studies for pharmacodynamic pharmacogenes may vary based on psychiatric diagnosis. Regardless, it may be beneficial to securely save and store patient test results in a standardized place within the medical record for future use.
CASE CONTINUED
You work with Mr. J to help him understand the benefits and limitations associated with pharmacogenetic testing. Assuming Mr. J is comfortable with the costs of obtaining testing, you contact the testing companies you identified to determine the specific pharmacogene variants included on each of these panels, and which would be the most appropriate given his race. If the decision is made to order the testing, provide Mr. J with a copy of his testing report so that he can use this information should he need any additional pharmacotherapy in the future, and also maintain a copy in his patient records using a standardized location for easy future access. If Mr. J is not comfortable with the costs associated with the testing, find out which medication his brother is currently receiving for treatment; this information may help identify a treatment plan for Mr. J.
Impact on practice
As psychiatry continues to gain experience in using pharmacogenetic testing and DSTs to help guide treatments for depression and other disorders, clinicians need to learn about these tools and how to use an evidence-based approach to best implement them in their practice. Many academic medical centers have developed continuing education programs or consult services to help with this.9,10 Just as the choice of which medication to use may be based partly on clinician experience, so too may be which pharmacogenetic test to use.
Bottom Line
Pharmacogenetic tests have not been examined in head-to-head clinical trials, which makes it nearly impossible to identify which test is best to use. Although the testing companies’ decision support tools (DSTs) often disagree in their recommendations, research has shown that using DSTs can facilitate implementation of relevant psychopharmacology dosing guidelines, assist in identifying optimal medication therapy, and improve patient outcomes. Clinicians should use testing panels with recommendations that best align with their individual practices, their patient’s needs, and FDA information.
Related Resources
- PGx Gene-specific information tables. www.pharmgkb.org/page/pgxGeneRef
- Clinical Pharmacogenetics Implementation Consortium. https://cpicpgx.org/guidelines/
Drug Brand Names
Aripiprazole • Abilify
Carbamazepine • Tegretol
Citalopram • Celexa
Clopidogrel • Plavix
Clozapine • Clozaril
Fluoxetine • Prozac
Imipramine • Tofranil
Paroxetine • Paxil
Sertraline • Zoloft
Warfarin • Coumadin, Jantoven
1. Ellingrod, VL. Using pharmacogenetics guidelines when prescribing: what’s available. Current Psychiatry. 2018;17(1):43-46.
2. Ellingrod VL. Pharmacogenomics testing: what the FDA says. Current Psychiatry. 2019;18(4):29-33.
3. Ramsey LB. Pharmacogenetic testing in children: what to test and how to use it. Current Psychiatry. 2018;17(9):30-36.
4. Bousman CA, Dunlop BW. Genotype, phenotype, and medication recommendation agreement among commercial pharmacogenetic-based decision support tools. The Pharmacogenomics Journal. 2018;18(5):613-622. doi:10.1038/s41397-018-0027-3
5. Bousman CA, Arandjelovic K, Mancuso SG, et al. Pharmacogenetic tests and depressive symptom remission: a meta-analysis of randomized controlled trials. Pharmacogenomics. 2019;20(1). doi:10.2217/pgs-2018-0142
6. Nicholson WT, Formea CM, Matey ET, et al. Considerations when applying pharmacogenomics to your practice. Mayo Clin Proc. 2021;96(1);218-230. doi:10.1016/j.mayocp.2020.03.011
7. Krebs K, Milani L. Translating pharmacogenomics into clinical decisions: do not let the perfect be the enemy of the good. Human Genomics. 2019;13(1). doi:10.1186/s40246-019-0229-z
8. Greden JF, Parikh S, Rothschild AJ, et al. Impact of pharmacogenomics on clinical outcomes in major depressive disorder in the GUIDED trial: a large, patient- and rater-blinded, randomized, controlled study. J Psychiatr Res. 2019;111;59-67. doi:10.1016/j.jpsychires.2019.01.003
9. Haga SB. Integrating pharmacogenetic testing into primary care. Expert Review of Precision Medicine and Drug Development. 2017;2(6):327-336. doi:10.1080/23808993.2017.1398046
10. Ward KM, Taubman DS, Pasternak AL, et al. Teaching psychiatric pharmacogenomics effectively: evaluation of a novel interprofessional online course. J Am Coll Clin Pharm. 2021; 4:176-183.
Mr. J, age 30, a Black man with major depressive disorder (MDD), has been your patient for the past year. At the time of his diagnosis, Mr. J received sertraline, 100 mg/d, but had little to no improvement. During the past year, he received trials of citalopram and paroxetine, but they were not effective for his recurrent depressive symptoms and/or resulted in significant adverse effects.
During a recent visit, Mr. J asks you about “the genetic tests that help determine which medications will work.” He mentions that his brother had this testing done and that it had “worked for him,” but offers no other details. You research the different testing panels to see which test you might use. After a brief online review, you identify at least 4 different products, and are not sure which test—if any—you should consider.
During the last few years, there has been a rise in commercial pharmacogenetic testing options, including tests available to clinicians at academic medical centers as well as direct-to-consumer testing (Table). Clinician and patient interest regarding pharmacogenetic testing in practice is often followed by the question, “Which test is best?” Although this is a logical question, providing an answer is multifactorial.1-3 Because none of the currently available tests have been compared in head-to-head clinical trials, it is nearly impossible to identify the “best” test.
In this article, we focus on the evidence-based principles that clinicians should consider when adopting pharmacogenetic testing in their practice. We discuss which genes are of most interest when prescribing psychotropic medications, the value of decision support tools, cost considerations, and patient education regarding this type of testing.
Which genes and variants should be tested?
The genes relevant to medication treatment outcomes can be broadly classified into those with pharmacokinetic vs pharmacodynamic effects. Pharmacogenes, such as those coding for the drug-metabolizing enzymes cytochrome P450 (CYP) 1A2, CYP2B6, CYP2C19, CYP2C9, CYP2D6, CYP3A4, and UDP-glucuronosyltransferase (UGT)2B1, may alter the rate at which medications are metabolized, thus varying the serum drug concentration across patients. Variants that impact the function of these enzymes are considered pharmacokinetic. Up to 40% of the variance in patients’ response to antidepressants may be due to variations in the pharmacokinetic genes.4 Alternatively, pharmacodynamic pharmacogenes impact drug action and therefore may affect the degree of receptor activation at a given drug concentration, overall drug efficacy, and/or the occurrence of medication sensitivity. These pharmacogenes may include:
- brain-derived neurotrophic factor (BDNF)
- catechol-O-methyltransferase (COMT)
- human leukocyte antigens A (HLA-A)
- serotonin receptor subtype 2 (HTR2)
- serotonin receptor subtype 2C (HTR2C)
- opioid receptor mu 1 (OPRM1)
- solute carrier family 6 member 4 (SLC6A4).
In articles previously published in
Currently, there is no standardization among commercial pharmacogenetic tests on:
- which genes to test
- which variants specific to a gene need to be included
- how the genetic data is translated to phenotype
- how the phenotype is translated to a treatment recommendation.
Continue to: Due to these factors...
Due to these factors, the FDA has advised clinicians to consult the dosing recommendations provided in a medication’s package insert for information regarding how genetic information should be used in making treatment decisions.2
The value of decision support tools
Researchers have assessed how various manufacturers’ decision support tools (DSTs) (ie, the reports the commercial testing companies send to the clinician who orders the test) agree on genotypes, predicted phenotypes, and medication recommendations.4 Overall, this research found varying levels of disagreement in the medication recommendations of the testing panels they studied, which indicates that not all tests are equivalent or interchangeable.4 Of the actionable recommendations for antidepressants, 16% were conflicting; the recommendations for fluoxetine and imipramine were most frequently in disagreement.4 Similarly, 20% of the actionable antipsychotic advice was conflicting, with the recommendations for aripiprazole and clozapine most frequently in disagreement.4 Researchers also reported a situation in which 4 testing panels agreed on the patient’s phenotyping status for CYP2C19, but the dosing recommendations provided for the CYP2C19 substrate, amitriptyline, differed.4 Thus, it is understandable why DSTs can result in confusion, and why clinicians should use testing panels with recommendations that best align with their individual practices, their patient’s needs, and FDA information.
Additionally, while the genes included on these panels vary, these testing panels also may not evaluate the same variants within a specific gene. These differences may impact the patient’s reported phenotypes and medication recommendations across DSTs. For example, the FDA has recommended HLA gene testing prior to prescribing carbamazepine. However, few of the available tests may include the HLA-B*15:02 variant, which has been associated with carbamazepine-induced severe cutaneous reactions in patients of Asian descent, and fewer may include the HLA-A*31:01 variant, for which testing is recommended prior to prescribing carbamazepine in patients of Caucasian descent.4 Additionally, some of the CYP enzymes—such as CYP2D6*17 and CYP2C19*3 variants, which may be more common in certain populations of patients who are members of ethnic or racial minority groups—may not be consistently included in the various panels. Thus, before deciding on a specific test, clinicians should understand which gene variants are relevant to their patients with regard to race and ethnicity, and key variants for specific medications. Clinicians should refer to FDA guidance and the Clinical Pharmacogenomics Implementation Consortium (CPIC) guidelines to determine the appropriate interpretations of genetic test results.1,2
Despite the disagreement in recommendations from the various testing companies, DSTs are useful and have been shown to facilitate implementation of relevant psychopharmacology dosing guidelines, assist in identifying optimal medication therapy, and improve patient outcomes. A recently published meta-analysis of randomized controlled trials (RCTs) of pharmacogenetic testing found that DSTs improved symptom remission among individuals with MDD by 70%.5 This suggests that pharmacogenetic-guided DSTs may provide superior treatment compared with treatment for DSTs were not used. However, the RCTs in this meta-analysis only included patients who had previously failed an antidepressant trial.5 Therefore, it is currently unknown at what point in care DSTs should be used, and whether they would be more beneficial if they are used when starting a new therapy, or after several trials have failed.
Consider the cost
The cost and availability of pharmacogenetic testing can be an issue when making treatment decisions, and such testing may not be covered by a patient’s insurance plan. Recently, the Centers for Medicare & Medicaid Services announced that Medicare would cover FDA-approved genomic tests that encompass broad gene panels if the evidence supports their use. Similarly, commercial insurers such as UnitedHealthcare have begun to cover some pharmacogenetic tests.6 Medicare or Medicaid plans cover some testing panels’ costs and patients do not incur any out-of-pocket costs; however, some private insurance companies require patients to pay at least a portion of the cost, and many companies offer financial assistance for patients based on income and other factors. Although financial coverage for testing has improved, patients may still face out-of-pocket costs; therefore, clinicians may need to weigh the benefits of pharmacogenetic testing vs its cost.7 Clinicians should also determine what timeline best suits their patient’s financial and clinical needs, and test accordingly.
Continue to: Patient education is critical
Patient education is critical
Although the benefits of using pharmacogenetic testing information when making certain treatment decisions is promising, it is important for both patients and clinicians to understand that test results do not always change therapy. A study on the impact of pharmacogenetic testing on clinical outcomes of patients with MDD found that 79% of patients were already prescribed medications that aligned with recommendations.8 Therefore, switching medications based on the test results of a patient who is doing well clinically is not recommended. However, DSTs may help with clinical decisions for ambiguous cases. For example, if a patient has a genotype and/or phenotype that aligns with medication recommendations, the DST might not be able to identify a better medication to use, but may be able to recommend dosing guidance to improve the tolerability of the patient’s current therapy.6 It is also important to understand that the results of such testing may have a broader use beyond the initial reason for obtaining testing, such as when prescribing a common blood thinner such as warfarin or clopidogrel. However, for many of the pharmacodynamic genes that are included in these panels, their use beyond the treatment of depression may be limited because outcome studies for pharmacodynamic pharmacogenes may vary based on psychiatric diagnosis. Regardless, it may be beneficial to securely save and store patient test results in a standardized place within the medical record for future use.
CASE CONTINUED
You work with Mr. J to help him understand the benefits and limitations associated with pharmacogenetic testing. Assuming Mr. J is comfortable with the costs of obtaining testing, you contact the testing companies you identified to determine the specific pharmacogene variants included on each of these panels, and which would be the most appropriate given his race. If the decision is made to order the testing, provide Mr. J with a copy of his testing report so that he can use this information should he need any additional pharmacotherapy in the future, and also maintain a copy in his patient records using a standardized location for easy future access. If Mr. J is not comfortable with the costs associated with the testing, find out which medication his brother is currently receiving for treatment; this information may help identify a treatment plan for Mr. J.
Impact on practice
As psychiatry continues to gain experience in using pharmacogenetic testing and DSTs to help guide treatments for depression and other disorders, clinicians need to learn about these tools and how to use an evidence-based approach to best implement them in their practice. Many academic medical centers have developed continuing education programs or consult services to help with this.9,10 Just as the choice of which medication to use may be based partly on clinician experience, so too may be which pharmacogenetic test to use.
Bottom Line
Pharmacogenetic tests have not been examined in head-to-head clinical trials, which makes it nearly impossible to identify which test is best to use. Although the testing companies’ decision support tools (DSTs) often disagree in their recommendations, research has shown that using DSTs can facilitate implementation of relevant psychopharmacology dosing guidelines, assist in identifying optimal medication therapy, and improve patient outcomes. Clinicians should use testing panels with recommendations that best align with their individual practices, their patient’s needs, and FDA information.
Related Resources
- PGx Gene-specific information tables. www.pharmgkb.org/page/pgxGeneRef
- Clinical Pharmacogenetics Implementation Consortium. https://cpicpgx.org/guidelines/
Drug Brand Names
Aripiprazole • Abilify
Carbamazepine • Tegretol
Citalopram • Celexa
Clopidogrel • Plavix
Clozapine • Clozaril
Fluoxetine • Prozac
Imipramine • Tofranil
Paroxetine • Paxil
Sertraline • Zoloft
Warfarin • Coumadin, Jantoven
Mr. J, age 30, a Black man with major depressive disorder (MDD), has been your patient for the past year. At the time of his diagnosis, Mr. J received sertraline, 100 mg/d, but had little to no improvement. During the past year, he received trials of citalopram and paroxetine, but they were not effective for his recurrent depressive symptoms and/or resulted in significant adverse effects.
During a recent visit, Mr. J asks you about “the genetic tests that help determine which medications will work.” He mentions that his brother had this testing done and that it had “worked for him,” but offers no other details. You research the different testing panels to see which test you might use. After a brief online review, you identify at least 4 different products, and are not sure which test—if any—you should consider.
During the last few years, there has been a rise in commercial pharmacogenetic testing options, including tests available to clinicians at academic medical centers as well as direct-to-consumer testing (Table). Clinician and patient interest regarding pharmacogenetic testing in practice is often followed by the question, “Which test is best?” Although this is a logical question, providing an answer is multifactorial.1-3 Because none of the currently available tests have been compared in head-to-head clinical trials, it is nearly impossible to identify the “best” test.
In this article, we focus on the evidence-based principles that clinicians should consider when adopting pharmacogenetic testing in their practice. We discuss which genes are of most interest when prescribing psychotropic medications, the value of decision support tools, cost considerations, and patient education regarding this type of testing.
Which genes and variants should be tested?
The genes relevant to medication treatment outcomes can be broadly classified into those with pharmacokinetic vs pharmacodynamic effects. Pharmacogenes, such as those coding for the drug-metabolizing enzymes cytochrome P450 (CYP) 1A2, CYP2B6, CYP2C19, CYP2C9, CYP2D6, CYP3A4, and UDP-glucuronosyltransferase (UGT)2B1, may alter the rate at which medications are metabolized, thus varying the serum drug concentration across patients. Variants that impact the function of these enzymes are considered pharmacokinetic. Up to 40% of the variance in patients’ response to antidepressants may be due to variations in the pharmacokinetic genes.4 Alternatively, pharmacodynamic pharmacogenes impact drug action and therefore may affect the degree of receptor activation at a given drug concentration, overall drug efficacy, and/or the occurrence of medication sensitivity. These pharmacogenes may include:
- brain-derived neurotrophic factor (BDNF)
- catechol-O-methyltransferase (COMT)
- human leukocyte antigens A (HLA-A)
- serotonin receptor subtype 2 (HTR2)
- serotonin receptor subtype 2C (HTR2C)
- opioid receptor mu 1 (OPRM1)
- solute carrier family 6 member 4 (SLC6A4).
In articles previously published in
Currently, there is no standardization among commercial pharmacogenetic tests on:
- which genes to test
- which variants specific to a gene need to be included
- how the genetic data is translated to phenotype
- how the phenotype is translated to a treatment recommendation.
Continue to: Due to these factors...
Due to these factors, the FDA has advised clinicians to consult the dosing recommendations provided in a medication’s package insert for information regarding how genetic information should be used in making treatment decisions.2
The value of decision support tools
Researchers have assessed how various manufacturers’ decision support tools (DSTs) (ie, the reports the commercial testing companies send to the clinician who orders the test) agree on genotypes, predicted phenotypes, and medication recommendations.4 Overall, this research found varying levels of disagreement in the medication recommendations of the testing panels they studied, which indicates that not all tests are equivalent or interchangeable.4 Of the actionable recommendations for antidepressants, 16% were conflicting; the recommendations for fluoxetine and imipramine were most frequently in disagreement.4 Similarly, 20% of the actionable antipsychotic advice was conflicting, with the recommendations for aripiprazole and clozapine most frequently in disagreement.4 Researchers also reported a situation in which 4 testing panels agreed on the patient’s phenotyping status for CYP2C19, but the dosing recommendations provided for the CYP2C19 substrate, amitriptyline, differed.4 Thus, it is understandable why DSTs can result in confusion, and why clinicians should use testing panels with recommendations that best align with their individual practices, their patient’s needs, and FDA information.
Additionally, while the genes included on these panels vary, these testing panels also may not evaluate the same variants within a specific gene. These differences may impact the patient’s reported phenotypes and medication recommendations across DSTs. For example, the FDA has recommended HLA gene testing prior to prescribing carbamazepine. However, few of the available tests may include the HLA-B*15:02 variant, which has been associated with carbamazepine-induced severe cutaneous reactions in patients of Asian descent, and fewer may include the HLA-A*31:01 variant, for which testing is recommended prior to prescribing carbamazepine in patients of Caucasian descent.4 Additionally, some of the CYP enzymes—such as CYP2D6*17 and CYP2C19*3 variants, which may be more common in certain populations of patients who are members of ethnic or racial minority groups—may not be consistently included in the various panels. Thus, before deciding on a specific test, clinicians should understand which gene variants are relevant to their patients with regard to race and ethnicity, and key variants for specific medications. Clinicians should refer to FDA guidance and the Clinical Pharmacogenomics Implementation Consortium (CPIC) guidelines to determine the appropriate interpretations of genetic test results.1,2
Despite the disagreement in recommendations from the various testing companies, DSTs are useful and have been shown to facilitate implementation of relevant psychopharmacology dosing guidelines, assist in identifying optimal medication therapy, and improve patient outcomes. A recently published meta-analysis of randomized controlled trials (RCTs) of pharmacogenetic testing found that DSTs improved symptom remission among individuals with MDD by 70%.5 This suggests that pharmacogenetic-guided DSTs may provide superior treatment compared with treatment for DSTs were not used. However, the RCTs in this meta-analysis only included patients who had previously failed an antidepressant trial.5 Therefore, it is currently unknown at what point in care DSTs should be used, and whether they would be more beneficial if they are used when starting a new therapy, or after several trials have failed.
Consider the cost
The cost and availability of pharmacogenetic testing can be an issue when making treatment decisions, and such testing may not be covered by a patient’s insurance plan. Recently, the Centers for Medicare & Medicaid Services announced that Medicare would cover FDA-approved genomic tests that encompass broad gene panels if the evidence supports their use. Similarly, commercial insurers such as UnitedHealthcare have begun to cover some pharmacogenetic tests.6 Medicare or Medicaid plans cover some testing panels’ costs and patients do not incur any out-of-pocket costs; however, some private insurance companies require patients to pay at least a portion of the cost, and many companies offer financial assistance for patients based on income and other factors. Although financial coverage for testing has improved, patients may still face out-of-pocket costs; therefore, clinicians may need to weigh the benefits of pharmacogenetic testing vs its cost.7 Clinicians should also determine what timeline best suits their patient’s financial and clinical needs, and test accordingly.
Continue to: Patient education is critical
Patient education is critical
Although the benefits of using pharmacogenetic testing information when making certain treatment decisions is promising, it is important for both patients and clinicians to understand that test results do not always change therapy. A study on the impact of pharmacogenetic testing on clinical outcomes of patients with MDD found that 79% of patients were already prescribed medications that aligned with recommendations.8 Therefore, switching medications based on the test results of a patient who is doing well clinically is not recommended. However, DSTs may help with clinical decisions for ambiguous cases. For example, if a patient has a genotype and/or phenotype that aligns with medication recommendations, the DST might not be able to identify a better medication to use, but may be able to recommend dosing guidance to improve the tolerability of the patient’s current therapy.6 It is also important to understand that the results of such testing may have a broader use beyond the initial reason for obtaining testing, such as when prescribing a common blood thinner such as warfarin or clopidogrel. However, for many of the pharmacodynamic genes that are included in these panels, their use beyond the treatment of depression may be limited because outcome studies for pharmacodynamic pharmacogenes may vary based on psychiatric diagnosis. Regardless, it may be beneficial to securely save and store patient test results in a standardized place within the medical record for future use.
CASE CONTINUED
You work with Mr. J to help him understand the benefits and limitations associated with pharmacogenetic testing. Assuming Mr. J is comfortable with the costs of obtaining testing, you contact the testing companies you identified to determine the specific pharmacogene variants included on each of these panels, and which would be the most appropriate given his race. If the decision is made to order the testing, provide Mr. J with a copy of his testing report so that he can use this information should he need any additional pharmacotherapy in the future, and also maintain a copy in his patient records using a standardized location for easy future access. If Mr. J is not comfortable with the costs associated with the testing, find out which medication his brother is currently receiving for treatment; this information may help identify a treatment plan for Mr. J.
Impact on practice
As psychiatry continues to gain experience in using pharmacogenetic testing and DSTs to help guide treatments for depression and other disorders, clinicians need to learn about these tools and how to use an evidence-based approach to best implement them in their practice. Many academic medical centers have developed continuing education programs or consult services to help with this.9,10 Just as the choice of which medication to use may be based partly on clinician experience, so too may be which pharmacogenetic test to use.
Bottom Line
Pharmacogenetic tests have not been examined in head-to-head clinical trials, which makes it nearly impossible to identify which test is best to use. Although the testing companies’ decision support tools (DSTs) often disagree in their recommendations, research has shown that using DSTs can facilitate implementation of relevant psychopharmacology dosing guidelines, assist in identifying optimal medication therapy, and improve patient outcomes. Clinicians should use testing panels with recommendations that best align with their individual practices, their patient’s needs, and FDA information.
Related Resources
- PGx Gene-specific information tables. www.pharmgkb.org/page/pgxGeneRef
- Clinical Pharmacogenetics Implementation Consortium. https://cpicpgx.org/guidelines/
Drug Brand Names
Aripiprazole • Abilify
Carbamazepine • Tegretol
Citalopram • Celexa
Clopidogrel • Plavix
Clozapine • Clozaril
Fluoxetine • Prozac
Imipramine • Tofranil
Paroxetine • Paxil
Sertraline • Zoloft
Warfarin • Coumadin, Jantoven
1. Ellingrod, VL. Using pharmacogenetics guidelines when prescribing: what’s available. Current Psychiatry. 2018;17(1):43-46.
2. Ellingrod VL. Pharmacogenomics testing: what the FDA says. Current Psychiatry. 2019;18(4):29-33.
3. Ramsey LB. Pharmacogenetic testing in children: what to test and how to use it. Current Psychiatry. 2018;17(9):30-36.
4. Bousman CA, Dunlop BW. Genotype, phenotype, and medication recommendation agreement among commercial pharmacogenetic-based decision support tools. The Pharmacogenomics Journal. 2018;18(5):613-622. doi:10.1038/s41397-018-0027-3
5. Bousman CA, Arandjelovic K, Mancuso SG, et al. Pharmacogenetic tests and depressive symptom remission: a meta-analysis of randomized controlled trials. Pharmacogenomics. 2019;20(1). doi:10.2217/pgs-2018-0142
6. Nicholson WT, Formea CM, Matey ET, et al. Considerations when applying pharmacogenomics to your practice. Mayo Clin Proc. 2021;96(1);218-230. doi:10.1016/j.mayocp.2020.03.011
7. Krebs K, Milani L. Translating pharmacogenomics into clinical decisions: do not let the perfect be the enemy of the good. Human Genomics. 2019;13(1). doi:10.1186/s40246-019-0229-z
8. Greden JF, Parikh S, Rothschild AJ, et al. Impact of pharmacogenomics on clinical outcomes in major depressive disorder in the GUIDED trial: a large, patient- and rater-blinded, randomized, controlled study. J Psychiatr Res. 2019;111;59-67. doi:10.1016/j.jpsychires.2019.01.003
9. Haga SB. Integrating pharmacogenetic testing into primary care. Expert Review of Precision Medicine and Drug Development. 2017;2(6):327-336. doi:10.1080/23808993.2017.1398046
10. Ward KM, Taubman DS, Pasternak AL, et al. Teaching psychiatric pharmacogenomics effectively: evaluation of a novel interprofessional online course. J Am Coll Clin Pharm. 2021; 4:176-183.
1. Ellingrod, VL. Using pharmacogenetics guidelines when prescribing: what’s available. Current Psychiatry. 2018;17(1):43-46.
2. Ellingrod VL. Pharmacogenomics testing: what the FDA says. Current Psychiatry. 2019;18(4):29-33.
3. Ramsey LB. Pharmacogenetic testing in children: what to test and how to use it. Current Psychiatry. 2018;17(9):30-36.
4. Bousman CA, Dunlop BW. Genotype, phenotype, and medication recommendation agreement among commercial pharmacogenetic-based decision support tools. The Pharmacogenomics Journal. 2018;18(5):613-622. doi:10.1038/s41397-018-0027-3
5. Bousman CA, Arandjelovic K, Mancuso SG, et al. Pharmacogenetic tests and depressive symptom remission: a meta-analysis of randomized controlled trials. Pharmacogenomics. 2019;20(1). doi:10.2217/pgs-2018-0142
6. Nicholson WT, Formea CM, Matey ET, et al. Considerations when applying pharmacogenomics to your practice. Mayo Clin Proc. 2021;96(1);218-230. doi:10.1016/j.mayocp.2020.03.011
7. Krebs K, Milani L. Translating pharmacogenomics into clinical decisions: do not let the perfect be the enemy of the good. Human Genomics. 2019;13(1). doi:10.1186/s40246-019-0229-z
8. Greden JF, Parikh S, Rothschild AJ, et al. Impact of pharmacogenomics on clinical outcomes in major depressive disorder in the GUIDED trial: a large, patient- and rater-blinded, randomized, controlled study. J Psychiatr Res. 2019;111;59-67. doi:10.1016/j.jpsychires.2019.01.003
9. Haga SB. Integrating pharmacogenetic testing into primary care. Expert Review of Precision Medicine and Drug Development. 2017;2(6):327-336. doi:10.1080/23808993.2017.1398046
10. Ward KM, Taubman DS, Pasternak AL, et al. Teaching psychiatric pharmacogenomics effectively: evaluation of a novel interprofessional online course. J Am Coll Clin Pharm. 2021; 4:176-183.

Pharmacogenomics testing: What the FDA says

Mr. R, age 30, is referred to you by his primary care physician, who diagnosed him with depression approximately 2 years ago. When he was first diagnosed, Mr. R was prescribed
Mr. R says that based on his primary care physician’s recommendation, he had undergone pharmacogenomics testing to help guide therapy. He presents the results to you, and you notice that he has the cytochrome P450 (CYP) 2C19 *2/*3 genotype and a CYP2D6*4/*5 genotype. Both are associated with a poor metabolism phenotype. Should you use these findings to determine which medication Mr. R should be treated with next?
While the field of pharmacogenomics is not new, within the last few years this science has begun to transition into clinical practice. A recent meta-analysis found support for using pharmacogenomics testing results in clinical practice.1 This study included more than 1,700 patients who took part in 5 controlled trials that randomized participants to either pharmacogenetics-guided or unguided (ie, standard) treatment. Each participant was assessed using the Hamilton Depression Rating Scale-17 (HDRS-17) a minimum of 3 times over a minimum of 8 weeks.1 While the exact inclusion and exclusion criteria for each trial differed, they all defined remission of depression as achieving an HDRS-17 score ≤7. Overall, the authors concluded that based on the random-effects pooled risk ratio, there was a significant association between pharmacogenetics-guided prescribing and remission (relative risk = 1.71, 95% confidence interval [CI], 1.17 to 2.48; P = .005). The results of this meta-analysis are controversial, however, because all 5 studies were industry-funded, and interpretation of the testing results was based on proprietary algorithms.
Experts in the field and professional societies, such as the International Society of Psychiatric Genetics (ISPG), have issued policy statements on genetic testing within psychiatry.2,3 While the ISPG did not necessarily endorse use of pharmacogenomics in practice, they recommended that clinicians follow good medical practice and stay current on changes to drug labeling and adverse event reports.3 The ISPG also noted that useful but not exhaustive lists of pharmacogenetic tests are maintained by the Clinical Pharmacogenetics Implementation Consortium (CPIC) and the US FDA.3
Laboratory developed vs direct-to-consumer tests
In a previous Savvy Psychopharmacology article,4 we had discussed the role of CPIC, but not the role of the FDA. This issue is key because there is a lack of clarity regarding pharmacogenomics tests and whether they are considered Class II devices by the FDA, which would require their review and approval. Until recently, the FDA was fairly quiet regarding pharmacogenomics tests because most of these tests were considered laboratory developed tests, which were regulated under the Clinic Laboratory Improvements Amendments program. The critical distinction of a laboratory developed test is that it is developed and performed in a single laboratory and is offered to patients only when prescribed by a clinician. Due to this distinction, laboratory developed pharmacogenomics tests did not need FDA 510(k) clearance, which is a premarket submission common for medical devices.
Direct-to-consumer pharmacogenomics tests are different in that the FDA has classified these platforms as medical devices; however, they are reviewed by the FDA only if they are being used for moderate- to high-risk medical purposes, or if the results of the testing may have a higher impact on medical care. As part of its review, the FDA examines test accuracy and reliably measures to determine if the measurement is predictive of a certain state of health and supported by what the company claims about the test and how well it works. Additionally, the FDA examines the company-provided descriptive information to ensure that consumers can easily understand it without the help of a clinician.5
Conflicting FDA statements
Recently the FDA issued 2 statements—one a policy statement and the other a safety communication—about laboratory developed tests and direct-to-consumer tests. The statements appear to contradict themselves, despite focusing on using pharmacogenomics testing in practice.
Continue to: The FDA's first statement
The FDA’s first statement. On October 31, 2018, the FDA released a policy statement that they had “permitted marketing, with special controls,” of the Personal Genome Service Pharmacogenetic Reports test through 23andMe (a direct-to-consumer genetic testing company) for 33 different variants within specific pharmacogenomic genes (CYP2C19, CYP2C9, CYP3A5, UGT1A1, DPYD, TPMT, SLC01B1, and CYP2D6) that may impact drug metabolism or response.6 As part of its review of this Personal Genome Service Pharmacogenetic Reports test, the FDA found that the company-provided data showed that the test is accurate and can correctly identify the 33 specific genetic variants. The FDA review also showed that the testing results were reproducible, and the test instructions and reports could be understood by consumers.
While the specific reports related to this testing are not yet available within 23andMe, this approval allows for greater oversight by the FDA with regard to the pharmacogenomics information provided through this company’s Personal Genome Service Pharmacogenetic Reports test. The FDA noted that this approval was only for adults age >185 and that consumers “should not use the test results to stop or change any medication.”6 Further, the FDA stated that the results of the direct-to-consumer test should be confirmed with independent pharmacogenomics testing before making any medical decision. Unfortunately, the FDA did not offer guidance on what would be an appropriate independent pharmacogenomics test, but it did provide a link to a list of FDA-approved nucleic acid–based tests, on which 23andMe’s Personal Genome Service Pharmacogenetic Reports test is included.7
The FDA’s second statement. On November 1, 2018, the FDA issued a separate safety communication that cautioned clinicians and patients that most of the current commercially available testing platforms for pharmacogenomics have not been FDA-reviewed, meaning that they may lack clinical evidence supporting their use.8 Further, the FDA safety communication stated, “Changing drug treatment based on the results from such a genetic test could lead to inappropriate treatment decisions and potentially serious health consequences for the patient.”8
Taken together, these FDA statements appear to support pharmacogenomics testing with approval of the 23andMe’s Personal Genome Service Pharmacogenetic Reports test but warn that the testing results should not be used to make treatment decisions, and that they should be verified. However, the FDA does not offer any guidance on what an appropriate testing platform would be
What the FDA advises
The FDA has provided some guidance to clinicians and patients regarding next steps for patients who are interested in having pharmacogenomics testing or who have already undergone testing. The FDA’s first point is that both clinicians and patients need to be aware that pharmacogenomics testing is not FDA-reviewed, that patients should discuss the results of their testing with their clinicians, and that they should not stop their medication based on the results of the testing. Additionally, the FDA recommends that clinicians and patients should be aware that any claims made by the testing companies regarding the specific effect of a medication may not be supported by evidence. Furthermore, the FDA strongly recommends that clinicians consult the FDA-approved drug label, or the label of the FDA-cleared or FDA-approved genetic test, for information regarding how genetic information should be used in making treatment decisions. The FDA recommends reviewing the Warning section, as well as the Indications and Usage, Dosage and Administration, or Use in Specific Populations sections of the FDA-approved drug labeling.
Continue to: Unfortunately, this information...
Unfortunately, this information might be difficult to locate due to the lack of consistency regarding where it is placed in the FDA-approved drug labeling. The Pharmacogenomics Knowledgebase (https://www.pharmgkb.org/) can help clinicians quickly identify information regarding medications, their metabolic pathways, CPIC dosing guidelines, and the FDA-approved drug labeling information.9 By searching for specific medications within the Pharmacogenomic Knowledge Base, information regarding the FDA-approved drug labeling can be easily found, which is important because currently >120 medications contain pharmacogenomics information in their FDA-approved drug labeling.10
CASE CONTINUED
Overall Mr. R’s pharmacogenomics testing results indicate that he has 2 genotypes that are associated with poor metabolism phenotypes and could result in reduced metabolism of medications that are metabolized by these CYP enzymes, leading to higher blood levels and an increased risk of adverse effects. The Table11 lists pharmacogenomics information from the FDA-approved drug labeling and from the Pharmacogenomics Knowledgebase for both the medications Mr. R has previously been prescribed and for several potential medications to consider.

It would be prudent to first discuss with Mr. R the FDA’s recent policy statement and safety communication. While you could recommend that he pursue additional pharmacogenomics testing, it is unclear which specific laboratory is available to conduct this confirmatory analysis.
Because Mr. R has had unsuccessful trials of several medications that primarily fall in the selective serotonin reuptake inhibitors class, it might be time to consider a medication from a different class. A quick review of the FDA-approved drug labeling for
Related Resources
- Gammal RS, Gardner KN, Burghardt KJ. Where to find guidance on using pharmacogenomics in psychiatric practice. Current Psychiatry. 2016;15(9):93-94.
- Clinical Pharmacogenomics Implementation Consortium. What is CPIC? https://www.pharmgkb.org/page/cpic.
Drug Brand Names
Bupropion • Wellbutrin, Zyban
Citalopram • Celexa
Paroxetine • Paxil
Sertraline • Zoloft
Venlafaxine • Effexor
1. Bousman CA, Arandjelovic K, Mancuso SG, et al. Pharmacogenetic tests and depressive symptom remission: a meta-analysis of randomized controlled trials. Pharmacogenomics. 2019;20(1):37-47.
2. Zubenko GS, Sommer BR, Cohen BM. Pharmacogenetics in psychiatry: a companion, rather than competitor, to protocol-based care-reply. JAMA Psychiatry. 2018;75(10):1090-1091.
3. International Society for Psychiatric Genetics. Genetic testing statement: genetic testing and psychiatric disorders: a statement from the International Society of Psychiatric Genetics. https://ispg.net/genetic-testing-statement/. Revised January 26, 2017. Accessed January 1, 2019.
4. Ellingrod VL, Ward KM. Using pharmacogenetics guidelines when prescribing: what’s available. Current Psychiatry. 2018;17(1):43-46.
5. U.S. Food and Drug Administration. Medical devices: direct-to-consumer tests. https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm624726.htm. Published November 1, 2018. Accessed January 1, 2019.
6. U.S. Food and Drug Administration. FDA news releases: FDA authorizes first direct-to consumer test for detecting variants that may be associated with medication metabolism. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm624753.htm. Published October 31, 2018. Accessed January 1, 2019.
7. U.S. Food and Drug Administration. Medical devices: nucleic acid based tests. https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm330711.htm. Published February 5, 2019. Accessed March 1, 2019.
8. U.S. Food and Drug Administration. Medical devices. The FDA warns against the use of many genetic tests with unapproved claims to predict patient response to specific medications: FDA Safety Communications. https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm624725.htm. Published November 1, 2018. Accessed January 1, 2019.
9. Whirl-Carrillo EM, McDonagh JM, Hebert L, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414-417.
10. U.S. Food and Drug Administration. Drugs. Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm. Published August 3, 2018. Accessed January 1, 2019.
11. U.S. Food and Drug Administration. Drugs@FDA: FDA approved drug products. https://www.accessdata.fda.gov/scripts/cder/daf. Accessed March 4, 2019.
Mr. R, age 30, is referred to you by his primary care physician, who diagnosed him with depression approximately 2 years ago. When he was first diagnosed, Mr. R was prescribed
Mr. R says that based on his primary care physician’s recommendation, he had undergone pharmacogenomics testing to help guide therapy. He presents the results to you, and you notice that he has the cytochrome P450 (CYP) 2C19 *2/*3 genotype and a CYP2D6*4/*5 genotype. Both are associated with a poor metabolism phenotype. Should you use these findings to determine which medication Mr. R should be treated with next?
While the field of pharmacogenomics is not new, within the last few years this science has begun to transition into clinical practice. A recent meta-analysis found support for using pharmacogenomics testing results in clinical practice.1 This study included more than 1,700 patients who took part in 5 controlled trials that randomized participants to either pharmacogenetics-guided or unguided (ie, standard) treatment. Each participant was assessed using the Hamilton Depression Rating Scale-17 (HDRS-17) a minimum of 3 times over a minimum of 8 weeks.1 While the exact inclusion and exclusion criteria for each trial differed, they all defined remission of depression as achieving an HDRS-17 score ≤7. Overall, the authors concluded that based on the random-effects pooled risk ratio, there was a significant association between pharmacogenetics-guided prescribing and remission (relative risk = 1.71, 95% confidence interval [CI], 1.17 to 2.48; P = .005). The results of this meta-analysis are controversial, however, because all 5 studies were industry-funded, and interpretation of the testing results was based on proprietary algorithms.
Experts in the field and professional societies, such as the International Society of Psychiatric Genetics (ISPG), have issued policy statements on genetic testing within psychiatry.2,3 While the ISPG did not necessarily endorse use of pharmacogenomics in practice, they recommended that clinicians follow good medical practice and stay current on changes to drug labeling and adverse event reports.3 The ISPG also noted that useful but not exhaustive lists of pharmacogenetic tests are maintained by the Clinical Pharmacogenetics Implementation Consortium (CPIC) and the US FDA.3
Laboratory developed vs direct-to-consumer tests
In a previous Savvy Psychopharmacology article,4 we had discussed the role of CPIC, but not the role of the FDA. This issue is key because there is a lack of clarity regarding pharmacogenomics tests and whether they are considered Class II devices by the FDA, which would require their review and approval. Until recently, the FDA was fairly quiet regarding pharmacogenomics tests because most of these tests were considered laboratory developed tests, which were regulated under the Clinic Laboratory Improvements Amendments program. The critical distinction of a laboratory developed test is that it is developed and performed in a single laboratory and is offered to patients only when prescribed by a clinician. Due to this distinction, laboratory developed pharmacogenomics tests did not need FDA 510(k) clearance, which is a premarket submission common for medical devices.
Direct-to-consumer pharmacogenomics tests are different in that the FDA has classified these platforms as medical devices; however, they are reviewed by the FDA only if they are being used for moderate- to high-risk medical purposes, or if the results of the testing may have a higher impact on medical care. As part of its review, the FDA examines test accuracy and reliably measures to determine if the measurement is predictive of a certain state of health and supported by what the company claims about the test and how well it works. Additionally, the FDA examines the company-provided descriptive information to ensure that consumers can easily understand it without the help of a clinician.5
Conflicting FDA statements
Recently the FDA issued 2 statements—one a policy statement and the other a safety communication—about laboratory developed tests and direct-to-consumer tests. The statements appear to contradict themselves, despite focusing on using pharmacogenomics testing in practice.
Continue to: The FDA's first statement
The FDA’s first statement. On October 31, 2018, the FDA released a policy statement that they had “permitted marketing, with special controls,” of the Personal Genome Service Pharmacogenetic Reports test through 23andMe (a direct-to-consumer genetic testing company) for 33 different variants within specific pharmacogenomic genes (CYP2C19, CYP2C9, CYP3A5, UGT1A1, DPYD, TPMT, SLC01B1, and CYP2D6) that may impact drug metabolism or response.6 As part of its review of this Personal Genome Service Pharmacogenetic Reports test, the FDA found that the company-provided data showed that the test is accurate and can correctly identify the 33 specific genetic variants. The FDA review also showed that the testing results were reproducible, and the test instructions and reports could be understood by consumers.
While the specific reports related to this testing are not yet available within 23andMe, this approval allows for greater oversight by the FDA with regard to the pharmacogenomics information provided through this company’s Personal Genome Service Pharmacogenetic Reports test. The FDA noted that this approval was only for adults age >185 and that consumers “should not use the test results to stop or change any medication.”6 Further, the FDA stated that the results of the direct-to-consumer test should be confirmed with independent pharmacogenomics testing before making any medical decision. Unfortunately, the FDA did not offer guidance on what would be an appropriate independent pharmacogenomics test, but it did provide a link to a list of FDA-approved nucleic acid–based tests, on which 23andMe’s Personal Genome Service Pharmacogenetic Reports test is included.7
The FDA’s second statement. On November 1, 2018, the FDA issued a separate safety communication that cautioned clinicians and patients that most of the current commercially available testing platforms for pharmacogenomics have not been FDA-reviewed, meaning that they may lack clinical evidence supporting their use.8 Further, the FDA safety communication stated, “Changing drug treatment based on the results from such a genetic test could lead to inappropriate treatment decisions and potentially serious health consequences for the patient.”8
Taken together, these FDA statements appear to support pharmacogenomics testing with approval of the 23andMe’s Personal Genome Service Pharmacogenetic Reports test but warn that the testing results should not be used to make treatment decisions, and that they should be verified. However, the FDA does not offer any guidance on what an appropriate testing platform would be
What the FDA advises
The FDA has provided some guidance to clinicians and patients regarding next steps for patients who are interested in having pharmacogenomics testing or who have already undergone testing. The FDA’s first point is that both clinicians and patients need to be aware that pharmacogenomics testing is not FDA-reviewed, that patients should discuss the results of their testing with their clinicians, and that they should not stop their medication based on the results of the testing. Additionally, the FDA recommends that clinicians and patients should be aware that any claims made by the testing companies regarding the specific effect of a medication may not be supported by evidence. Furthermore, the FDA strongly recommends that clinicians consult the FDA-approved drug label, or the label of the FDA-cleared or FDA-approved genetic test, for information regarding how genetic information should be used in making treatment decisions. The FDA recommends reviewing the Warning section, as well as the Indications and Usage, Dosage and Administration, or Use in Specific Populations sections of the FDA-approved drug labeling.
Continue to: Unfortunately, this information...
Unfortunately, this information might be difficult to locate due to the lack of consistency regarding where it is placed in the FDA-approved drug labeling. The Pharmacogenomics Knowledgebase (https://www.pharmgkb.org/) can help clinicians quickly identify information regarding medications, their metabolic pathways, CPIC dosing guidelines, and the FDA-approved drug labeling information.9 By searching for specific medications within the Pharmacogenomic Knowledge Base, information regarding the FDA-approved drug labeling can be easily found, which is important because currently >120 medications contain pharmacogenomics information in their FDA-approved drug labeling.10
CASE CONTINUED
Overall Mr. R’s pharmacogenomics testing results indicate that he has 2 genotypes that are associated with poor metabolism phenotypes and could result in reduced metabolism of medications that are metabolized by these CYP enzymes, leading to higher blood levels and an increased risk of adverse effects. The Table11 lists pharmacogenomics information from the FDA-approved drug labeling and from the Pharmacogenomics Knowledgebase for both the medications Mr. R has previously been prescribed and for several potential medications to consider.
It would be prudent to first discuss with Mr. R the FDA’s recent policy statement and safety communication. While you could recommend that he pursue additional pharmacogenomics testing, it is unclear which specific laboratory is available to conduct this confirmatory analysis.
Because Mr. R has had unsuccessful trials of several medications that primarily fall in the selective serotonin reuptake inhibitors class, it might be time to consider a medication from a different class. A quick review of the FDA-approved drug labeling for
Related Resources
- Gammal RS, Gardner KN, Burghardt KJ. Where to find guidance on using pharmacogenomics in psychiatric practice. Current Psychiatry. 2016;15(9):93-94.
- Clinical Pharmacogenomics Implementation Consortium. What is CPIC? https://www.pharmgkb.org/page/cpic.
Drug Brand Names
Bupropion • Wellbutrin, Zyban
Citalopram • Celexa
Paroxetine • Paxil
Sertraline • Zoloft
Venlafaxine • Effexor
Mr. R, age 30, is referred to you by his primary care physician, who diagnosed him with depression approximately 2 years ago. When he was first diagnosed, Mr. R was prescribed
Mr. R says that based on his primary care physician’s recommendation, he had undergone pharmacogenomics testing to help guide therapy. He presents the results to you, and you notice that he has the cytochrome P450 (CYP) 2C19 *2/*3 genotype and a CYP2D6*4/*5 genotype. Both are associated with a poor metabolism phenotype. Should you use these findings to determine which medication Mr. R should be treated with next?
While the field of pharmacogenomics is not new, within the last few years this science has begun to transition into clinical practice. A recent meta-analysis found support for using pharmacogenomics testing results in clinical practice.1 This study included more than 1,700 patients who took part in 5 controlled trials that randomized participants to either pharmacogenetics-guided or unguided (ie, standard) treatment. Each participant was assessed using the Hamilton Depression Rating Scale-17 (HDRS-17) a minimum of 3 times over a minimum of 8 weeks.1 While the exact inclusion and exclusion criteria for each trial differed, they all defined remission of depression as achieving an HDRS-17 score ≤7. Overall, the authors concluded that based on the random-effects pooled risk ratio, there was a significant association between pharmacogenetics-guided prescribing and remission (relative risk = 1.71, 95% confidence interval [CI], 1.17 to 2.48; P = .005). The results of this meta-analysis are controversial, however, because all 5 studies were industry-funded, and interpretation of the testing results was based on proprietary algorithms.
Experts in the field and professional societies, such as the International Society of Psychiatric Genetics (ISPG), have issued policy statements on genetic testing within psychiatry.2,3 While the ISPG did not necessarily endorse use of pharmacogenomics in practice, they recommended that clinicians follow good medical practice and stay current on changes to drug labeling and adverse event reports.3 The ISPG also noted that useful but not exhaustive lists of pharmacogenetic tests are maintained by the Clinical Pharmacogenetics Implementation Consortium (CPIC) and the US FDA.3
Laboratory developed vs direct-to-consumer tests
In a previous Savvy Psychopharmacology article,4 we had discussed the role of CPIC, but not the role of the FDA. This issue is key because there is a lack of clarity regarding pharmacogenomics tests and whether they are considered Class II devices by the FDA, which would require their review and approval. Until recently, the FDA was fairly quiet regarding pharmacogenomics tests because most of these tests were considered laboratory developed tests, which were regulated under the Clinic Laboratory Improvements Amendments program. The critical distinction of a laboratory developed test is that it is developed and performed in a single laboratory and is offered to patients only when prescribed by a clinician. Due to this distinction, laboratory developed pharmacogenomics tests did not need FDA 510(k) clearance, which is a premarket submission common for medical devices.
Direct-to-consumer pharmacogenomics tests are different in that the FDA has classified these platforms as medical devices; however, they are reviewed by the FDA only if they are being used for moderate- to high-risk medical purposes, or if the results of the testing may have a higher impact on medical care. As part of its review, the FDA examines test accuracy and reliably measures to determine if the measurement is predictive of a certain state of health and supported by what the company claims about the test and how well it works. Additionally, the FDA examines the company-provided descriptive information to ensure that consumers can easily understand it without the help of a clinician.5
Conflicting FDA statements
Recently the FDA issued 2 statements—one a policy statement and the other a safety communication—about laboratory developed tests and direct-to-consumer tests. The statements appear to contradict themselves, despite focusing on using pharmacogenomics testing in practice.
Continue to: The FDA's first statement
The FDA’s first statement. On October 31, 2018, the FDA released a policy statement that they had “permitted marketing, with special controls,” of the Personal Genome Service Pharmacogenetic Reports test through 23andMe (a direct-to-consumer genetic testing company) for 33 different variants within specific pharmacogenomic genes (CYP2C19, CYP2C9, CYP3A5, UGT1A1, DPYD, TPMT, SLC01B1, and CYP2D6) that may impact drug metabolism or response.6 As part of its review of this Personal Genome Service Pharmacogenetic Reports test, the FDA found that the company-provided data showed that the test is accurate and can correctly identify the 33 specific genetic variants. The FDA review also showed that the testing results were reproducible, and the test instructions and reports could be understood by consumers.
While the specific reports related to this testing are not yet available within 23andMe, this approval allows for greater oversight by the FDA with regard to the pharmacogenomics information provided through this company’s Personal Genome Service Pharmacogenetic Reports test. The FDA noted that this approval was only for adults age >185 and that consumers “should not use the test results to stop or change any medication.”6 Further, the FDA stated that the results of the direct-to-consumer test should be confirmed with independent pharmacogenomics testing before making any medical decision. Unfortunately, the FDA did not offer guidance on what would be an appropriate independent pharmacogenomics test, but it did provide a link to a list of FDA-approved nucleic acid–based tests, on which 23andMe’s Personal Genome Service Pharmacogenetic Reports test is included.7
The FDA’s second statement. On November 1, 2018, the FDA issued a separate safety communication that cautioned clinicians and patients that most of the current commercially available testing platforms for pharmacogenomics have not been FDA-reviewed, meaning that they may lack clinical evidence supporting their use.8 Further, the FDA safety communication stated, “Changing drug treatment based on the results from such a genetic test could lead to inappropriate treatment decisions and potentially serious health consequences for the patient.”8
Taken together, these FDA statements appear to support pharmacogenomics testing with approval of the 23andMe’s Personal Genome Service Pharmacogenetic Reports test but warn that the testing results should not be used to make treatment decisions, and that they should be verified. However, the FDA does not offer any guidance on what an appropriate testing platform would be
What the FDA advises
The FDA has provided some guidance to clinicians and patients regarding next steps for patients who are interested in having pharmacogenomics testing or who have already undergone testing. The FDA’s first point is that both clinicians and patients need to be aware that pharmacogenomics testing is not FDA-reviewed, that patients should discuss the results of their testing with their clinicians, and that they should not stop their medication based on the results of the testing. Additionally, the FDA recommends that clinicians and patients should be aware that any claims made by the testing companies regarding the specific effect of a medication may not be supported by evidence. Furthermore, the FDA strongly recommends that clinicians consult the FDA-approved drug label, or the label of the FDA-cleared or FDA-approved genetic test, for information regarding how genetic information should be used in making treatment decisions. The FDA recommends reviewing the Warning section, as well as the Indications and Usage, Dosage and Administration, or Use in Specific Populations sections of the FDA-approved drug labeling.
Continue to: Unfortunately, this information...
Unfortunately, this information might be difficult to locate due to the lack of consistency regarding where it is placed in the FDA-approved drug labeling. The Pharmacogenomics Knowledgebase (https://www.pharmgkb.org/) can help clinicians quickly identify information regarding medications, their metabolic pathways, CPIC dosing guidelines, and the FDA-approved drug labeling information.9 By searching for specific medications within the Pharmacogenomic Knowledge Base, information regarding the FDA-approved drug labeling can be easily found, which is important because currently >120 medications contain pharmacogenomics information in their FDA-approved drug labeling.10
CASE CONTINUED
Overall Mr. R’s pharmacogenomics testing results indicate that he has 2 genotypes that are associated with poor metabolism phenotypes and could result in reduced metabolism of medications that are metabolized by these CYP enzymes, leading to higher blood levels and an increased risk of adverse effects. The Table11 lists pharmacogenomics information from the FDA-approved drug labeling and from the Pharmacogenomics Knowledgebase for both the medications Mr. R has previously been prescribed and for several potential medications to consider.
It would be prudent to first discuss with Mr. R the FDA’s recent policy statement and safety communication. While you could recommend that he pursue additional pharmacogenomics testing, it is unclear which specific laboratory is available to conduct this confirmatory analysis.
Because Mr. R has had unsuccessful trials of several medications that primarily fall in the selective serotonin reuptake inhibitors class, it might be time to consider a medication from a different class. A quick review of the FDA-approved drug labeling for
Related Resources
- Gammal RS, Gardner KN, Burghardt KJ. Where to find guidance on using pharmacogenomics in psychiatric practice. Current Psychiatry. 2016;15(9):93-94.
- Clinical Pharmacogenomics Implementation Consortium. What is CPIC? https://www.pharmgkb.org/page/cpic.
Drug Brand Names
Bupropion • Wellbutrin, Zyban
Citalopram • Celexa
Paroxetine • Paxil
Sertraline • Zoloft
Venlafaxine • Effexor
1. Bousman CA, Arandjelovic K, Mancuso SG, et al. Pharmacogenetic tests and depressive symptom remission: a meta-analysis of randomized controlled trials. Pharmacogenomics. 2019;20(1):37-47.
2. Zubenko GS, Sommer BR, Cohen BM. Pharmacogenetics in psychiatry: a companion, rather than competitor, to protocol-based care-reply. JAMA Psychiatry. 2018;75(10):1090-1091.
3. International Society for Psychiatric Genetics. Genetic testing statement: genetic testing and psychiatric disorders: a statement from the International Society of Psychiatric Genetics. https://ispg.net/genetic-testing-statement/. Revised January 26, 2017. Accessed January 1, 2019.
4. Ellingrod VL, Ward KM. Using pharmacogenetics guidelines when prescribing: what’s available. Current Psychiatry. 2018;17(1):43-46.
5. U.S. Food and Drug Administration. Medical devices: direct-to-consumer tests. https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm624726.htm. Published November 1, 2018. Accessed January 1, 2019.
6. U.S. Food and Drug Administration. FDA news releases: FDA authorizes first direct-to consumer test for detecting variants that may be associated with medication metabolism. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm624753.htm. Published October 31, 2018. Accessed January 1, 2019.
7. U.S. Food and Drug Administration. Medical devices: nucleic acid based tests. https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm330711.htm. Published February 5, 2019. Accessed March 1, 2019.
8. U.S. Food and Drug Administration. Medical devices. The FDA warns against the use of many genetic tests with unapproved claims to predict patient response to specific medications: FDA Safety Communications. https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm624725.htm. Published November 1, 2018. Accessed January 1, 2019.
9. Whirl-Carrillo EM, McDonagh JM, Hebert L, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414-417.
10. U.S. Food and Drug Administration. Drugs. Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm. Published August 3, 2018. Accessed January 1, 2019.
11. U.S. Food and Drug Administration. Drugs@FDA: FDA approved drug products. https://www.accessdata.fda.gov/scripts/cder/daf. Accessed March 4, 2019.
1. Bousman CA, Arandjelovic K, Mancuso SG, et al. Pharmacogenetic tests and depressive symptom remission: a meta-analysis of randomized controlled trials. Pharmacogenomics. 2019;20(1):37-47.
2. Zubenko GS, Sommer BR, Cohen BM. Pharmacogenetics in psychiatry: a companion, rather than competitor, to protocol-based care-reply. JAMA Psychiatry. 2018;75(10):1090-1091.
3. International Society for Psychiatric Genetics. Genetic testing statement: genetic testing and psychiatric disorders: a statement from the International Society of Psychiatric Genetics. https://ispg.net/genetic-testing-statement/. Revised January 26, 2017. Accessed January 1, 2019.
4. Ellingrod VL, Ward KM. Using pharmacogenetics guidelines when prescribing: what’s available. Current Psychiatry. 2018;17(1):43-46.
5. U.S. Food and Drug Administration. Medical devices: direct-to-consumer tests. https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm624726.htm. Published November 1, 2018. Accessed January 1, 2019.
6. U.S. Food and Drug Administration. FDA news releases: FDA authorizes first direct-to consumer test for detecting variants that may be associated with medication metabolism. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm624753.htm. Published October 31, 2018. Accessed January 1, 2019.
7. U.S. Food and Drug Administration. Medical devices: nucleic acid based tests. https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm330711.htm. Published February 5, 2019. Accessed March 1, 2019.
8. U.S. Food and Drug Administration. Medical devices. The FDA warns against the use of many genetic tests with unapproved claims to predict patient response to specific medications: FDA Safety Communications. https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm624725.htm. Published November 1, 2018. Accessed January 1, 2019.
9. Whirl-Carrillo EM, McDonagh JM, Hebert L, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414-417.
10. U.S. Food and Drug Administration. Drugs. Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm. Published August 3, 2018. Accessed January 1, 2019.
11. U.S. Food and Drug Administration. Drugs@FDA: FDA approved drug products. https://www.accessdata.fda.gov/scripts/cder/daf. Accessed March 4, 2019.

Using pharmacogenetics guidelines when prescribing: What’s available
Ms. C, age 45, has a history of generalized anxiety disorder, which has been controlled for the past 6 weeks with extended-release (ER) venlafaxine, 37.5 mg/d. Previous medication trials included fluvoxamine, 300 mg/d, for 2 weeks; paroxetine, 20 mg/d, for 1 week; sertraline, 100 mg/d, for 1 week; and citalopram, 20 mg/d, for 2 weeks. For each trial, Ms. C was unable to tolerate standard doses because of substantial adverse effects; she complained that her anxiety would significantly worsen with each course of treatment. Although the adverse effects would eventually subside with continued treatment, they appeared to be the dose-limiting factor for treatment, even when much lower doses were started.
Ms. C’s son recently suggested that she undergo pharmacogenomics testing, and she brings in the results of this testing. The report states that Ms. C has cytochrome P450 (CYP) pharmacogenotypes CYP2D6 *5/*9, CYP2C19 *2/*3, CYP2C9 *2/*2, and CYP1A2 *1A/*1F. Ms. C wants to know if these results explain some of the issues she has had with previous medication trials, and if these results mean that she should be taking a different medication.
The human genome project was a vast, international effort to sequence the entire human genome1 and identify individual differences in drug response, which serves as the basis for pharmacogenomics. Since completion of the human genome project in the early 2000s, the field of pharmacogenomics has advanced, and using pharmacogenomic testing to make therapeutic decisions for medication management is becoming commonplace.2 Although this critical change to how medicine is practiced is exciting, implementation of pharmacogenomics into practice has been varied.2 Therefore, having an understanding of the resources available to guide pharmacogenomics into practice is critical, because the FDA now lists >160 medications that include specific pharmacogenomics information within their package insert.3

CPIC provides guidance for implementing pharmacogenomics

In 2000, the National Institutes of Health established the Pharmacogenomics Knowledge Base (PharmGKB) and the Pharmacogenomics Research Network (PGRN). These 2 resources provide information from cutting-edge research on genomic variation and therapeutic and adverse events, as well as practical implementation of this research.4 As part of their partnership, PharmGKB and PGRN established the Clinical Pharmacogenomics Implementation Consortium (CPIC), which has begun to provide clinical practice guidelines for implementing pharmacogenomic results. Although CPIC does not advocate for pharmacogenomics testing as a standard, it recognizes that this testing is becoming more commonplace, and therefore its guidelines can help clinicians make rational prescribing decisions.4
In a recent partnership among several PGRN members, investigators found that 1 out of 4 pharmacogenomic test results had a potential clinically actionable outcome.2 There are currently >43 gene/drug pairs for which CPIC has provided guidelines; however, >200 other gene/drug pairs are being evaluated.5
Table 15 lists the current CPIC gene/drug combinations with accompanying published guidelines that are pertinent to psychiatry. For each of these guidelines, experts reviewed the available literature to provide graded therapeutic recommendations: A (“preponderance of evidence is high or moderate in favor of changing prescribing”), B (“preponderance of evidence is weak with little conflicting data”), and C and D (“evidence levels can vary”).4 Looking at the specific genotypes for Ms. C, we can use the information within the CPIC to assign a drug metabolism phenotype for her genotype combinations (Table 2).6

Consider additional resources
In addition to those from the CPIC, guidelines have been developed by other scientific groups, such as the Dutch Pharmacogenetics Working Group and the European Pharmacogenomics Implementation Consortium. Although most of these guidelines are concordant with CPIC, differences exist, which makes it important to be aware of all available resources.
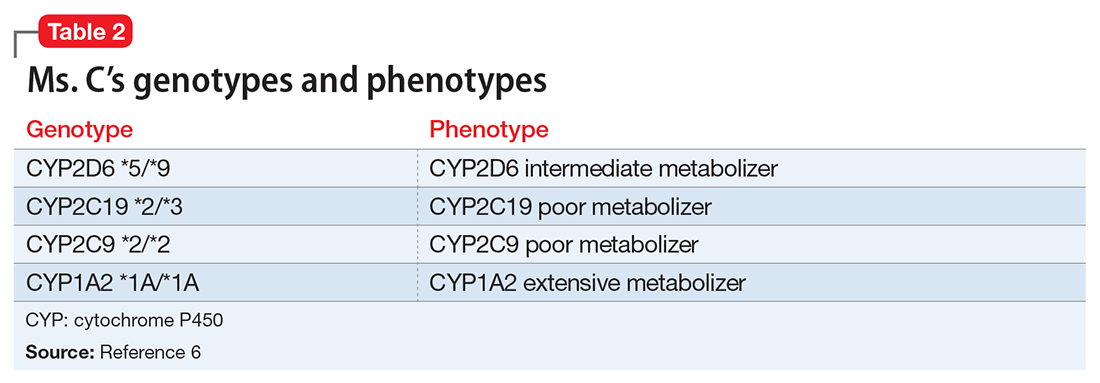
As well as working on the CPIC guidelines, PGRN investigators also provide numerous free online educational resources related to the principles behind pharmacogenomics, including additional resources necessary for systematic implementation. Examples include tables that outline all possible diplotypes (genotypes) for genes in the guidelines and how these are related to the metabolic phenotypes.2,4 Drug metabolizing phenotypes, for example, often are described as poor, intermediate, extensive, and ultra-rapid; in this system, metabolizing ability labeled as poor is less-than-average, and ultra-rapid describes greater-than-average ability. The extensive phenotype is considered average. The data files on the CPIC Web site also can be used as resources to “double check” interpretation results for the diplotype phenotype combinations currently available from various pharmacogenomics companies.7
Based on Ms. C’s presentation, as well as information from the CPIC guidelines, we expect that she might experience substantial adverse effects from most selective serotonin reuptake inhibitors and tricycle antidepressants because of her intermediate metabolizer status for CYP2D6 and poor metabolizer status for CYP2C19. The CPIC’s recommendation for using paroxetine and fluvoxamine in patients with a CYP2D6 intermediate metabolism phenotype is to initiate the recommend starting dose, but acknowledge that reduced metabolic capacity through CYP2D6 may result in higher blood levels and greater probability of adverse drug reactions. For a patient with the CYP2C19 poor metabolizer phenotype, the recommendation is to reduce the starting dose of citalopram or sertraline by 50%, or to prescribe a drug that is not metabolized by CYP2C19.8 Therefore, this pharmacogenomic information may help us understand why Ms. C is unable to tolerate these medications.
Although the CPIC guidelines do not address venlafaxine, the PharmGKB Web site contains literature supporting CYP2D6 as important in venlafaxine metabolism. Current recommendations from the Dutch Pharmacogenetics Working Group Guidelines9 are to either use a non–CYP2D6 metabolized medication or to adjust the dose to clinical response. Because Ms. C has been taking venlafaxine ER for the last 6 weeks and is taking a relatively low but effective dose, our recommendation is to continue current therapy.
It is also important to consider drug interactions when interpreting pharmacogenomic test results. In Ms. C’s case, the impact of a CYP2D6 intermediate metabolism phenotype would be increased if she also was taking a strong CYP2D6 inhibitor such as bupropion. Pharmacogenomics is another clinical tool and discontinuation of an effective treatment that is adequately tolerated should not be done based on pharmacogenomics recommendations alone.
1. Collins FS, Patrinos A, Jordan E, et al. New goals for the U.S. Human Genome Project: 1998-2003. Science. 1998;282(5389):682-689.
2. Luzum JA, Pakyz RE, Elsey AR, et al; Pharmacogenomics Research Network Translational Pharmacogenetics Program. The Pharmacogenomics Research Network Translational Pharmacogenetics Program: outcomes and metrics of pharmacogenetic implementations across diverse healthcare systems. Clin Pharmacol Ther. 2017;102(3):502-510.
3. U.S. Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm. Updated October 3, 2017. Accessed October 23, 2017.
4. Caudle KE, Gammal RS, Whirl-Carrillo M, et al. Evidence and resources to implement pharmacogenetic knowledge for precision medicine. Am J Health Syst Pharm. 2016;73(23):1977-1985.
5. Clinical Pharmacogenomics Implementation Consortium. Genes-drugs. https://cpicpgx.org/genes-drugs. Updated October 2, 2017. Accessed October 23, 2017.
6. PharmGKB. PGx gene-specific information tables. https://www.pharmgkb.org/page/pgxGeneRef. Accessed October 27, 2017.
7. Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414-417.
8. Hicks JK, Bishop JR, Sangkuhl K, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin Pharmacol Ther. 2015;98(2):127-134.
9
Ms. C, age 45, has a history of generalized anxiety disorder, which has been controlled for the past 6 weeks with extended-release (ER) venlafaxine, 37.5 mg/d. Previous medication trials included fluvoxamine, 300 mg/d, for 2 weeks; paroxetine, 20 mg/d, for 1 week; sertraline, 100 mg/d, for 1 week; and citalopram, 20 mg/d, for 2 weeks. For each trial, Ms. C was unable to tolerate standard doses because of substantial adverse effects; she complained that her anxiety would significantly worsen with each course of treatment. Although the adverse effects would eventually subside with continued treatment, they appeared to be the dose-limiting factor for treatment, even when much lower doses were started.
Ms. C’s son recently suggested that she undergo pharmacogenomics testing, and she brings in the results of this testing. The report states that Ms. C has cytochrome P450 (CYP) pharmacogenotypes CYP2D6 *5/*9, CYP2C19 *2/*3, CYP2C9 *2/*2, and CYP1A2 *1A/*1F. Ms. C wants to know if these results explain some of the issues she has had with previous medication trials, and if these results mean that she should be taking a different medication.
The human genome project was a vast, international effort to sequence the entire human genome1 and identify individual differences in drug response, which serves as the basis for pharmacogenomics. Since completion of the human genome project in the early 2000s, the field of pharmacogenomics has advanced, and using pharmacogenomic testing to make therapeutic decisions for medication management is becoming commonplace.2 Although this critical change to how medicine is practiced is exciting, implementation of pharmacogenomics into practice has been varied.2 Therefore, having an understanding of the resources available to guide pharmacogenomics into practice is critical, because the FDA now lists >160 medications that include specific pharmacogenomics information within their package insert.3
CPIC provides guidance for implementing pharmacogenomics
In 2000, the National Institutes of Health established the Pharmacogenomics Knowledge Base (PharmGKB) and the Pharmacogenomics Research Network (PGRN). These 2 resources provide information from cutting-edge research on genomic variation and therapeutic and adverse events, as well as practical implementation of this research.4 As part of their partnership, PharmGKB and PGRN established the Clinical Pharmacogenomics Implementation Consortium (CPIC), which has begun to provide clinical practice guidelines for implementing pharmacogenomic results. Although CPIC does not advocate for pharmacogenomics testing as a standard, it recognizes that this testing is becoming more commonplace, and therefore its guidelines can help clinicians make rational prescribing decisions.4
In a recent partnership among several PGRN members, investigators found that 1 out of 4 pharmacogenomic test results had a potential clinically actionable outcome.2 There are currently >43 gene/drug pairs for which CPIC has provided guidelines; however, >200 other gene/drug pairs are being evaluated.5
Table 15 lists the current CPIC gene/drug combinations with accompanying published guidelines that are pertinent to psychiatry. For each of these guidelines, experts reviewed the available literature to provide graded therapeutic recommendations: A (“preponderance of evidence is high or moderate in favor of changing prescribing”), B (“preponderance of evidence is weak with little conflicting data”), and C and D (“evidence levels can vary”).4 Looking at the specific genotypes for Ms. C, we can use the information within the CPIC to assign a drug metabolism phenotype for her genotype combinations (Table 2).6
Consider additional resources
In addition to those from the CPIC, guidelines have been developed by other scientific groups, such as the Dutch Pharmacogenetics Working Group and the European Pharmacogenomics Implementation Consortium. Although most of these guidelines are concordant with CPIC, differences exist, which makes it important to be aware of all available resources.
As well as working on the CPIC guidelines, PGRN investigators also provide numerous free online educational resources related to the principles behind pharmacogenomics, including additional resources necessary for systematic implementation. Examples include tables that outline all possible diplotypes (genotypes) for genes in the guidelines and how these are related to the metabolic phenotypes.2,4 Drug metabolizing phenotypes, for example, often are described as poor, intermediate, extensive, and ultra-rapid; in this system, metabolizing ability labeled as poor is less-than-average, and ultra-rapid describes greater-than-average ability. The extensive phenotype is considered average. The data files on the CPIC Web site also can be used as resources to “double check” interpretation results for the diplotype phenotype combinations currently available from various pharmacogenomics companies.7
Based on Ms. C’s presentation, as well as information from the CPIC guidelines, we expect that she might experience substantial adverse effects from most selective serotonin reuptake inhibitors and tricycle antidepressants because of her intermediate metabolizer status for CYP2D6 and poor metabolizer status for CYP2C19. The CPIC’s recommendation for using paroxetine and fluvoxamine in patients with a CYP2D6 intermediate metabolism phenotype is to initiate the recommend starting dose, but acknowledge that reduced metabolic capacity through CYP2D6 may result in higher blood levels and greater probability of adverse drug reactions. For a patient with the CYP2C19 poor metabolizer phenotype, the recommendation is to reduce the starting dose of citalopram or sertraline by 50%, or to prescribe a drug that is not metabolized by CYP2C19.8 Therefore, this pharmacogenomic information may help us understand why Ms. C is unable to tolerate these medications.
Although the CPIC guidelines do not address venlafaxine, the PharmGKB Web site contains literature supporting CYP2D6 as important in venlafaxine metabolism. Current recommendations from the Dutch Pharmacogenetics Working Group Guidelines9 are to either use a non–CYP2D6 metabolized medication or to adjust the dose to clinical response. Because Ms. C has been taking venlafaxine ER for the last 6 weeks and is taking a relatively low but effective dose, our recommendation is to continue current therapy.
It is also important to consider drug interactions when interpreting pharmacogenomic test results. In Ms. C’s case, the impact of a CYP2D6 intermediate metabolism phenotype would be increased if she also was taking a strong CYP2D6 inhibitor such as bupropion. Pharmacogenomics is another clinical tool and discontinuation of an effective treatment that is adequately tolerated should not be done based on pharmacogenomics recommendations alone.
Ms. C, age 45, has a history of generalized anxiety disorder, which has been controlled for the past 6 weeks with extended-release (ER) venlafaxine, 37.5 mg/d. Previous medication trials included fluvoxamine, 300 mg/d, for 2 weeks; paroxetine, 20 mg/d, for 1 week; sertraline, 100 mg/d, for 1 week; and citalopram, 20 mg/d, for 2 weeks. For each trial, Ms. C was unable to tolerate standard doses because of substantial adverse effects; she complained that her anxiety would significantly worsen with each course of treatment. Although the adverse effects would eventually subside with continued treatment, they appeared to be the dose-limiting factor for treatment, even when much lower doses were started.
Ms. C’s son recently suggested that she undergo pharmacogenomics testing, and she brings in the results of this testing. The report states that Ms. C has cytochrome P450 (CYP) pharmacogenotypes CYP2D6 *5/*9, CYP2C19 *2/*3, CYP2C9 *2/*2, and CYP1A2 *1A/*1F. Ms. C wants to know if these results explain some of the issues she has had with previous medication trials, and if these results mean that she should be taking a different medication.
The human genome project was a vast, international effort to sequence the entire human genome1 and identify individual differences in drug response, which serves as the basis for pharmacogenomics. Since completion of the human genome project in the early 2000s, the field of pharmacogenomics has advanced, and using pharmacogenomic testing to make therapeutic decisions for medication management is becoming commonplace.2 Although this critical change to how medicine is practiced is exciting, implementation of pharmacogenomics into practice has been varied.2 Therefore, having an understanding of the resources available to guide pharmacogenomics into practice is critical, because the FDA now lists >160 medications that include specific pharmacogenomics information within their package insert.3
CPIC provides guidance for implementing pharmacogenomics
In 2000, the National Institutes of Health established the Pharmacogenomics Knowledge Base (PharmGKB) and the Pharmacogenomics Research Network (PGRN). These 2 resources provide information from cutting-edge research on genomic variation and therapeutic and adverse events, as well as practical implementation of this research.4 As part of their partnership, PharmGKB and PGRN established the Clinical Pharmacogenomics Implementation Consortium (CPIC), which has begun to provide clinical practice guidelines for implementing pharmacogenomic results. Although CPIC does not advocate for pharmacogenomics testing as a standard, it recognizes that this testing is becoming more commonplace, and therefore its guidelines can help clinicians make rational prescribing decisions.4
In a recent partnership among several PGRN members, investigators found that 1 out of 4 pharmacogenomic test results had a potential clinically actionable outcome.2 There are currently >43 gene/drug pairs for which CPIC has provided guidelines; however, >200 other gene/drug pairs are being evaluated.5
Table 15 lists the current CPIC gene/drug combinations with accompanying published guidelines that are pertinent to psychiatry. For each of these guidelines, experts reviewed the available literature to provide graded therapeutic recommendations: A (“preponderance of evidence is high or moderate in favor of changing prescribing”), B (“preponderance of evidence is weak with little conflicting data”), and C and D (“evidence levels can vary”).4 Looking at the specific genotypes for Ms. C, we can use the information within the CPIC to assign a drug metabolism phenotype for her genotype combinations (Table 2).6
Consider additional resources
In addition to those from the CPIC, guidelines have been developed by other scientific groups, such as the Dutch Pharmacogenetics Working Group and the European Pharmacogenomics Implementation Consortium. Although most of these guidelines are concordant with CPIC, differences exist, which makes it important to be aware of all available resources.
As well as working on the CPIC guidelines, PGRN investigators also provide numerous free online educational resources related to the principles behind pharmacogenomics, including additional resources necessary for systematic implementation. Examples include tables that outline all possible diplotypes (genotypes) for genes in the guidelines and how these are related to the metabolic phenotypes.2,4 Drug metabolizing phenotypes, for example, often are described as poor, intermediate, extensive, and ultra-rapid; in this system, metabolizing ability labeled as poor is less-than-average, and ultra-rapid describes greater-than-average ability. The extensive phenotype is considered average. The data files on the CPIC Web site also can be used as resources to “double check” interpretation results for the diplotype phenotype combinations currently available from various pharmacogenomics companies.7
Based on Ms. C’s presentation, as well as information from the CPIC guidelines, we expect that she might experience substantial adverse effects from most selective serotonin reuptake inhibitors and tricycle antidepressants because of her intermediate metabolizer status for CYP2D6 and poor metabolizer status for CYP2C19. The CPIC’s recommendation for using paroxetine and fluvoxamine in patients with a CYP2D6 intermediate metabolism phenotype is to initiate the recommend starting dose, but acknowledge that reduced metabolic capacity through CYP2D6 may result in higher blood levels and greater probability of adverse drug reactions. For a patient with the CYP2C19 poor metabolizer phenotype, the recommendation is to reduce the starting dose of citalopram or sertraline by 50%, or to prescribe a drug that is not metabolized by CYP2C19.8 Therefore, this pharmacogenomic information may help us understand why Ms. C is unable to tolerate these medications.
Although the CPIC guidelines do not address venlafaxine, the PharmGKB Web site contains literature supporting CYP2D6 as important in venlafaxine metabolism. Current recommendations from the Dutch Pharmacogenetics Working Group Guidelines9 are to either use a non–CYP2D6 metabolized medication or to adjust the dose to clinical response. Because Ms. C has been taking venlafaxine ER for the last 6 weeks and is taking a relatively low but effective dose, our recommendation is to continue current therapy.
It is also important to consider drug interactions when interpreting pharmacogenomic test results. In Ms. C’s case, the impact of a CYP2D6 intermediate metabolism phenotype would be increased if she also was taking a strong CYP2D6 inhibitor such as bupropion. Pharmacogenomics is another clinical tool and discontinuation of an effective treatment that is adequately tolerated should not be done based on pharmacogenomics recommendations alone.
1. Collins FS, Patrinos A, Jordan E, et al. New goals for the U.S. Human Genome Project: 1998-2003. Science. 1998;282(5389):682-689.
2. Luzum JA, Pakyz RE, Elsey AR, et al; Pharmacogenomics Research Network Translational Pharmacogenetics Program. The Pharmacogenomics Research Network Translational Pharmacogenetics Program: outcomes and metrics of pharmacogenetic implementations across diverse healthcare systems. Clin Pharmacol Ther. 2017;102(3):502-510.
3. U.S. Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm. Updated October 3, 2017. Accessed October 23, 2017.
4. Caudle KE, Gammal RS, Whirl-Carrillo M, et al. Evidence and resources to implement pharmacogenetic knowledge for precision medicine. Am J Health Syst Pharm. 2016;73(23):1977-1985.
5. Clinical Pharmacogenomics Implementation Consortium. Genes-drugs. https://cpicpgx.org/genes-drugs. Updated October 2, 2017. Accessed October 23, 2017.
6. PharmGKB. PGx gene-specific information tables. https://www.pharmgkb.org/page/pgxGeneRef. Accessed October 27, 2017.
7. Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414-417.
8. Hicks JK, Bishop JR, Sangkuhl K, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin Pharmacol Ther. 2015;98(2):127-134.
9
1. Collins FS, Patrinos A, Jordan E, et al. New goals for the U.S. Human Genome Project: 1998-2003. Science. 1998;282(5389):682-689.
2. Luzum JA, Pakyz RE, Elsey AR, et al; Pharmacogenomics Research Network Translational Pharmacogenetics Program. The Pharmacogenomics Research Network Translational Pharmacogenetics Program: outcomes and metrics of pharmacogenetic implementations across diverse healthcare systems. Clin Pharmacol Ther. 2017;102(3):502-510.
3. U.S. Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm. Updated October 3, 2017. Accessed October 23, 2017.
4. Caudle KE, Gammal RS, Whirl-Carrillo M, et al. Evidence and resources to implement pharmacogenetic knowledge for precision medicine. Am J Health Syst Pharm. 2016;73(23):1977-1985.
5. Clinical Pharmacogenomics Implementation Consortium. Genes-drugs. https://cpicpgx.org/genes-drugs. Updated October 2, 2017. Accessed October 23, 2017.
6. PharmGKB. PGx gene-specific information tables. https://www.pharmgkb.org/page/pgxGeneRef. Accessed October 27, 2017.
7. Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414-417.
8. Hicks JK, Bishop JR, Sangkuhl K, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin Pharmacol Ther. 2015;98(2):127-134.
9

Urine drug screens: When might a test result be false-positive?
Mr. L, age 35, has an appointment at a mental health clinic for ongoing treatment of depression. His medication list includes atorvastatin, bupropion, lisinopril, and cranberry capsules for non-descriptive urinary issues. He has been treated for some time at a different outpatient facility; however he recently moved and changed clinics.
At this visit, his first, Mr. L receives a full physical exam, including a urine drug screen point-of-care (POC) test. He informs the nurse that he has an extensive history of drug abuse: “You name it, I’ve done it.” Although he experimented with many illicit substances, he acknowledges that “downers” were his favorite. He believes that his drug abuse could have caused his depression, but is proud to declare that he has been “clean” for 12 months and his depression is approaching remission.

However, the urine drug screen is positive for amphetamines. Mr. L vehemently swears that the test must be wrong, restating that he has been clean for 12 months. “Besides, I don’t even like ‘uppers’!” Because of Mr. L’s insistence, the clinician does a brief literature search about false-positive results in urine drug screening, which shows that, rarely, bupropion can trigger a false positive in the amphetamine immunoassay.
Could this be a false-positive result? Or is Mr. L not telling the truth?
Because no clinical lab test is perfect, any clinician who runs urine drug screens will encounter a false-positive result. (See the Box,1-3 for discussion of false negatives.) Understanding how each test works—and potential sources of error— can help you evaluate test results and determine the best course of action.

There are 2 main methods involved in urine drug testing: in-office (POC) urine testing and laboratory-based testing. This article describes the differences between these tests and summarizes the potential for false-positive results.
In-office urine testing
POC tests in urine drug screens use a technique called “immunoassay,” which is quantitative and generally will detect the agent in urine for only 3 to 7 days after ingestion.4 This test relies on the principle of competitive binding: If a parent drug or metabolite is present in urine, it will bind to a specific antibody site on the test strip and produce a positive result.5 Other compounds that are similarly “shaped” on a molecular level also can bind to these antibody sites when present in sufficient quantity, producing a “cross reaction,” also called a “false-positive” result. The Table6 lists agents that can cross-react with immunoassay tests. In addition to the cross-reaction, false positives also can occur because of technician or clerical error— making it important to review the process by which the specimen was obtained and tested if a false-positive result is suspected, as in the case described here.7
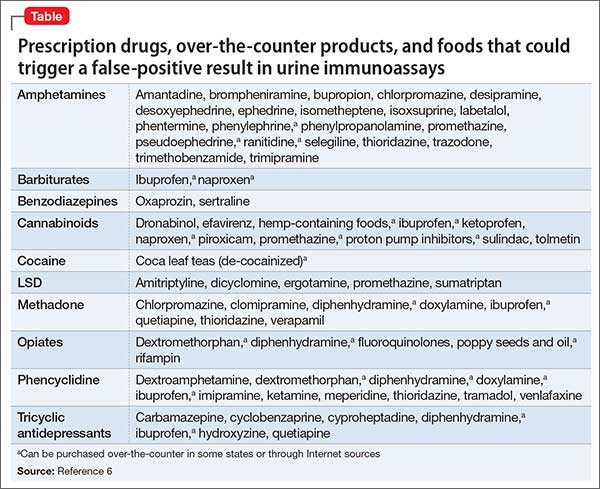
Different POC tests can have varying cross-reactivity patterns, based on the antibody used.8 In general, false positives in immunoassays are rare, but amphetamine and opiate false positives are more common than cocaine metabolite and cannabinoid false positives.9 The odds of a false positive vary, depending on the specificity of the immunoassay used and the substance under detection.6
A study that analyzed 10,000 POC urine drug screens found that 362 specimens tested positive for amphetamines, but that 128 of those did not test positive for amphetamines using more sensitive tests.10 Of these 128 false positives reported, 53 patients were taking bupropion at the time of the test.10 Therefore, clinicians should do a thorough patient medication review at the time of POC urine drug testing. In addition, consider identifying which type of test you are using at your practice site, and ask the manufacturer or lab to provide a list of known possible false positives.
Laboratory-based GC–MS testing
If a false positive is suspected on a POC immunoassay-based urine drug screen, results can be confirmed using gas chromatography–mass spectrometry (GC–MS). Although GC–MS is more accurate than an immunoassay, it also is more expensive and time-consuming.9
GC–MS breaks down a specimen into ionized fragments and separates them based on their mass–charge ratio. Because of this, GC–MS is able to identify the presence of a specific drug (eg, oxycodone) instead of a broad class (eg, opioid). The GC–MS method is a good tool to confirm initial positive screens when their integrity is in question because, unlike POC tests used during an office visit, GC–MS is not influenced by cross-reacting compounds.11-13
GC–MS is not error-free, however. For example, heroin and hydrocodone are metabolized into morphine and hydromorphone, respectively. Depending on when the specimen was collected, the metabolites, not the parents, might be the compounds identified, which might produce confusing results.
Clinical recommendations
When a POC drug screen is positive, confirming the result with GC–MS is good clinical practice. False positives can strain the relationship between patient and provider, thus compromising care. Examining the procedures that were used to obtain the specimen, as well as double-checking POC test results, is, when appropriate, good medicine.
CASE CONTINUED
Because Mr. L is adamant about his sobriety and the fact that his drugs of choice were sedatives, not stimulants, the clinician orders a second drug screen by GC–MS. The second screen is negative for substances of abuse; Mr. L’s clinician concludes that bupropion produced a false-positive result on the POC urine drug screen, confirming Mr. L’s assertions.
Related Resources
• Saitman A, Park HD, Fitzgerald RL. False-positive interferences of common urine drug screen immunoassays: a review. J Anal Toxicol. 2014;38(7):387-396.
• Tenore PL. Advanced urine toxicology testing. J Addict Dis. 2010;29(4):436-448.
Drug Brand Names
Amantadine • Symadine, Symmetrel
Amitriptyline • Elavil
Atorvastatin • Lipitor
Brompheniramine • Dimetane
Bupropion • Wellbutrin, Zyban
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Clomipramine • Anafranil
Cyclobenzaprine • Amrix, Flexeril
Cyproheptadine • Periactin
Desipramine • Nopramin
Desoxyephedrine • Desoxyn
Dextromethorphan • Delsym, Robitussin
Dicyclomine • Bentyl, Dicyclocot
Diphenhydramine • Benadryl, Unisom
Doxylamine • Robitussin, NyQuil
Dronabinol • Marinol
Efavirenz • Sustiva
Ephedrine • Mistol, Va-Tro-Nol
Ergotamine • Ergomar, Cafergot
Hydrocodone • Vicodin
Hydromophone • Dilaudid, Palladone
Hydroxyzine • Atarax, Vistaril
Isometheptene • Amidrine, Migrend
Isoxsuprine • Vasodilan, Vasoprine
Ketoprofen • Orudis, Oruvail
Labetalol • Normodyne, Trandate
Lisinopril • Prinivil, Zestril
Meperidine • Demerol
Naproxen • Aleve, Naprosyn
Oxaprozin • Daypro
Oxycodone • Oxycontin, Percocet, Percodan, Roxicodone
Phentermine • Adipex, Phentrol
Phenylephrine • Sudafed PE, Neo-Synephrine
Piroxicam • Feldene
Promethazine • Phenergan
Pseudoephedrine • Sudafed, Dimetapp
Quetiapine • Seroquel
Ranitidine • Zantac
Rifampin • Rifadin, Rimactane
Selegiline • EMSAM
Sertraline • Zoloft
Sulindac • Clinoril
Sumatriptan • Imitrex
Thioridazine • Mellaril
Tolmetin • Tolectin
Trazodone • Desyrel, Oleptro
Trimethobenzamide • Benzacot, Tigan
Trimipramine • Surmontil
Verapamil • Calan, Isoptin
1. Cobaugh DJ, Gainor C, Gaston CL, et al. The opioid abuse and misuse epidemic: implications for pharmacists in hospitals and health systems. Am J Health Syst Pharm. 2014;71(18):1539-1554.
2. Gilbert JW, Wheeler GR, Mick GE, et al. Importance of urine drug testing in the treatment of chronic noncancer pain: implications of recent medicare policy changes in Kentucky. Pain Physician. 2010;13(2):167-186.
3. Michna E, Jamison RN, Pham LD, et al. Urine toxicology screening among chronic pain patients on opioid therapy: frequency and predictability of abnormal findings. Clin J Pain. 2007;23(2):173-179.
4. U.S. Department of Justice. Fact sheet: drug testing in the criminal justice system. https://www.ncjrs.gov/pdffiles/dtest. pdf. Published March 1992. Accessed July 29, 2015.
5. Australian Diagnostic Services. Technical information: testing principle’s. http://www.australiandrugtesting. com/#!technical-info/c14h4. Accessed November 5, 2014.
6. University of Illinois at Chicago College of Pharmacy. What drugs are likely to interfere with urine drug screens? http://dig.pharm.uic.edu/faq/2011/Feb/faq1.aspx. Accessed November 5, 2014.
7. Wolff K, Farrell M, Marsden J, et al. A review of biological indicators of illicit drug use, practical considerations and clinical usefulness. Addiction. 1999;94(9):1279-1298.
8. Gourlay D, Heit H, Caplan YH. Urine drug testing in primary care – dispelling the myths & designing strategies. PharmaCom Group. http://www.mc.uky.edu/equip-4-pcps/documents/ section8/urine%20drug%20testing%20in%20clinical%20 practice.pdf. Accessed August 6, 2015.
9. Standridge JB, Adams SM, Zotos AP. Urine drug screen: a valuable office procedure. Am Fam Physician. 2010;81(5): 635-640.
10. Casey ER, Scott MG, Tang S, et al. Frequency of false positive amphetamine screens due to bupropion using the Syva EMIT II immunoassay. J Med Toxicol. 2011;7(2):105-108.
11. Casavant MJ. Urine drug screening in adolescents. Pediatr Clin N Am. 2002;49(2):317-327.
12. Shults TF. The medical review officer handbook. 7th ed. Chapel Hill, NC: Quadrangle Research; 1999.
13. Baden LR, Horowitz G, Jacoby H, et al. Quinolones and false-positive urine screening for opiates by immunoassay technology. JAMA. 2001;286(24):3115-3119.
Mr. L, age 35, has an appointment at a mental health clinic for ongoing treatment of depression. His medication list includes atorvastatin, bupropion, lisinopril, and cranberry capsules for non-descriptive urinary issues. He has been treated for some time at a different outpatient facility; however he recently moved and changed clinics.
At this visit, his first, Mr. L receives a full physical exam, including a urine drug screen point-of-care (POC) test. He informs the nurse that he has an extensive history of drug abuse: “You name it, I’ve done it.” Although he experimented with many illicit substances, he acknowledges that “downers” were his favorite. He believes that his drug abuse could have caused his depression, but is proud to declare that he has been “clean” for 12 months and his depression is approaching remission.
However, the urine drug screen is positive for amphetamines. Mr. L vehemently swears that the test must be wrong, restating that he has been clean for 12 months. “Besides, I don’t even like ‘uppers’!” Because of Mr. L’s insistence, the clinician does a brief literature search about false-positive results in urine drug screening, which shows that, rarely, bupropion can trigger a false positive in the amphetamine immunoassay.
Could this be a false-positive result? Or is Mr. L not telling the truth?
Because no clinical lab test is perfect, any clinician who runs urine drug screens will encounter a false-positive result. (See the Box,1-3 for discussion of false negatives.) Understanding how each test works—and potential sources of error— can help you evaluate test results and determine the best course of action.
There are 2 main methods involved in urine drug testing: in-office (POC) urine testing and laboratory-based testing. This article describes the differences between these tests and summarizes the potential for false-positive results.
In-office urine testing
POC tests in urine drug screens use a technique called “immunoassay,” which is quantitative and generally will detect the agent in urine for only 3 to 7 days after ingestion.4 This test relies on the principle of competitive binding: If a parent drug or metabolite is present in urine, it will bind to a specific antibody site on the test strip and produce a positive result.5 Other compounds that are similarly “shaped” on a molecular level also can bind to these antibody sites when present in sufficient quantity, producing a “cross reaction,” also called a “false-positive” result. The Table6 lists agents that can cross-react with immunoassay tests. In addition to the cross-reaction, false positives also can occur because of technician or clerical error— making it important to review the process by which the specimen was obtained and tested if a false-positive result is suspected, as in the case described here.7
Different POC tests can have varying cross-reactivity patterns, based on the antibody used.8 In general, false positives in immunoassays are rare, but amphetamine and opiate false positives are more common than cocaine metabolite and cannabinoid false positives.9 The odds of a false positive vary, depending on the specificity of the immunoassay used and the substance under detection.6
A study that analyzed 10,000 POC urine drug screens found that 362 specimens tested positive for amphetamines, but that 128 of those did not test positive for amphetamines using more sensitive tests.10 Of these 128 false positives reported, 53 patients were taking bupropion at the time of the test.10 Therefore, clinicians should do a thorough patient medication review at the time of POC urine drug testing. In addition, consider identifying which type of test you are using at your practice site, and ask the manufacturer or lab to provide a list of known possible false positives.
Laboratory-based GC–MS testing
If a false positive is suspected on a POC immunoassay-based urine drug screen, results can be confirmed using gas chromatography–mass spectrometry (GC–MS). Although GC–MS is more accurate than an immunoassay, it also is more expensive and time-consuming.9
GC–MS breaks down a specimen into ionized fragments and separates them based on their mass–charge ratio. Because of this, GC–MS is able to identify the presence of a specific drug (eg, oxycodone) instead of a broad class (eg, opioid). The GC–MS method is a good tool to confirm initial positive screens when their integrity is in question because, unlike POC tests used during an office visit, GC–MS is not influenced by cross-reacting compounds.11-13
GC–MS is not error-free, however. For example, heroin and hydrocodone are metabolized into morphine and hydromorphone, respectively. Depending on when the specimen was collected, the metabolites, not the parents, might be the compounds identified, which might produce confusing results.
Clinical recommendations
When a POC drug screen is positive, confirming the result with GC–MS is good clinical practice. False positives can strain the relationship between patient and provider, thus compromising care. Examining the procedures that were used to obtain the specimen, as well as double-checking POC test results, is, when appropriate, good medicine.
CASE CONTINUED
Because Mr. L is adamant about his sobriety and the fact that his drugs of choice were sedatives, not stimulants, the clinician orders a second drug screen by GC–MS. The second screen is negative for substances of abuse; Mr. L’s clinician concludes that bupropion produced a false-positive result on the POC urine drug screen, confirming Mr. L’s assertions.
Related Resources
• Saitman A, Park HD, Fitzgerald RL. False-positive interferences of common urine drug screen immunoassays: a review. J Anal Toxicol. 2014;38(7):387-396.
• Tenore PL. Advanced urine toxicology testing. J Addict Dis. 2010;29(4):436-448.
Drug Brand Names
Amantadine • Symadine, Symmetrel
Amitriptyline • Elavil
Atorvastatin • Lipitor
Brompheniramine • Dimetane
Bupropion • Wellbutrin, Zyban
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Clomipramine • Anafranil
Cyclobenzaprine • Amrix, Flexeril
Cyproheptadine • Periactin
Desipramine • Nopramin
Desoxyephedrine • Desoxyn
Dextromethorphan • Delsym, Robitussin
Dicyclomine • Bentyl, Dicyclocot
Diphenhydramine • Benadryl, Unisom
Doxylamine • Robitussin, NyQuil
Dronabinol • Marinol
Efavirenz • Sustiva
Ephedrine • Mistol, Va-Tro-Nol
Ergotamine • Ergomar, Cafergot
Hydrocodone • Vicodin
Hydromophone • Dilaudid, Palladone
Hydroxyzine • Atarax, Vistaril
Isometheptene • Amidrine, Migrend
Isoxsuprine • Vasodilan, Vasoprine
Ketoprofen • Orudis, Oruvail
Labetalol • Normodyne, Trandate
Lisinopril • Prinivil, Zestril
Meperidine • Demerol
Naproxen • Aleve, Naprosyn
Oxaprozin • Daypro
Oxycodone • Oxycontin, Percocet, Percodan, Roxicodone
Phentermine • Adipex, Phentrol
Phenylephrine • Sudafed PE, Neo-Synephrine
Piroxicam • Feldene
Promethazine • Phenergan
Pseudoephedrine • Sudafed, Dimetapp
Quetiapine • Seroquel
Ranitidine • Zantac
Rifampin • Rifadin, Rimactane
Selegiline • EMSAM
Sertraline • Zoloft
Sulindac • Clinoril
Sumatriptan • Imitrex
Thioridazine • Mellaril
Tolmetin • Tolectin
Trazodone • Desyrel, Oleptro
Trimethobenzamide • Benzacot, Tigan
Trimipramine • Surmontil
Verapamil • Calan, Isoptin
Mr. L, age 35, has an appointment at a mental health clinic for ongoing treatment of depression. His medication list includes atorvastatin, bupropion, lisinopril, and cranberry capsules for non-descriptive urinary issues. He has been treated for some time at a different outpatient facility; however he recently moved and changed clinics.
At this visit, his first, Mr. L receives a full physical exam, including a urine drug screen point-of-care (POC) test. He informs the nurse that he has an extensive history of drug abuse: “You name it, I’ve done it.” Although he experimented with many illicit substances, he acknowledges that “downers” were his favorite. He believes that his drug abuse could have caused his depression, but is proud to declare that he has been “clean” for 12 months and his depression is approaching remission.
However, the urine drug screen is positive for amphetamines. Mr. L vehemently swears that the test must be wrong, restating that he has been clean for 12 months. “Besides, I don’t even like ‘uppers’!” Because of Mr. L’s insistence, the clinician does a brief literature search about false-positive results in urine drug screening, which shows that, rarely, bupropion can trigger a false positive in the amphetamine immunoassay.
Could this be a false-positive result? Or is Mr. L not telling the truth?
Because no clinical lab test is perfect, any clinician who runs urine drug screens will encounter a false-positive result. (See the Box,1-3 for discussion of false negatives.) Understanding how each test works—and potential sources of error— can help you evaluate test results and determine the best course of action.
There are 2 main methods involved in urine drug testing: in-office (POC) urine testing and laboratory-based testing. This article describes the differences between these tests and summarizes the potential for false-positive results.
In-office urine testing
POC tests in urine drug screens use a technique called “immunoassay,” which is quantitative and generally will detect the agent in urine for only 3 to 7 days after ingestion.4 This test relies on the principle of competitive binding: If a parent drug or metabolite is present in urine, it will bind to a specific antibody site on the test strip and produce a positive result.5 Other compounds that are similarly “shaped” on a molecular level also can bind to these antibody sites when present in sufficient quantity, producing a “cross reaction,” also called a “false-positive” result. The Table6 lists agents that can cross-react with immunoassay tests. In addition to the cross-reaction, false positives also can occur because of technician or clerical error— making it important to review the process by which the specimen was obtained and tested if a false-positive result is suspected, as in the case described here.7
Different POC tests can have varying cross-reactivity patterns, based on the antibody used.8 In general, false positives in immunoassays are rare, but amphetamine and opiate false positives are more common than cocaine metabolite and cannabinoid false positives.9 The odds of a false positive vary, depending on the specificity of the immunoassay used and the substance under detection.6
A study that analyzed 10,000 POC urine drug screens found that 362 specimens tested positive for amphetamines, but that 128 of those did not test positive for amphetamines using more sensitive tests.10 Of these 128 false positives reported, 53 patients were taking bupropion at the time of the test.10 Therefore, clinicians should do a thorough patient medication review at the time of POC urine drug testing. In addition, consider identifying which type of test you are using at your practice site, and ask the manufacturer or lab to provide a list of known possible false positives.
Laboratory-based GC–MS testing
If a false positive is suspected on a POC immunoassay-based urine drug screen, results can be confirmed using gas chromatography–mass spectrometry (GC–MS). Although GC–MS is more accurate than an immunoassay, it also is more expensive and time-consuming.9
GC–MS breaks down a specimen into ionized fragments and separates them based on their mass–charge ratio. Because of this, GC–MS is able to identify the presence of a specific drug (eg, oxycodone) instead of a broad class (eg, opioid). The GC–MS method is a good tool to confirm initial positive screens when their integrity is in question because, unlike POC tests used during an office visit, GC–MS is not influenced by cross-reacting compounds.11-13
GC–MS is not error-free, however. For example, heroin and hydrocodone are metabolized into morphine and hydromorphone, respectively. Depending on when the specimen was collected, the metabolites, not the parents, might be the compounds identified, which might produce confusing results.
Clinical recommendations
When a POC drug screen is positive, confirming the result with GC–MS is good clinical practice. False positives can strain the relationship between patient and provider, thus compromising care. Examining the procedures that were used to obtain the specimen, as well as double-checking POC test results, is, when appropriate, good medicine.
CASE CONTINUED
Because Mr. L is adamant about his sobriety and the fact that his drugs of choice were sedatives, not stimulants, the clinician orders a second drug screen by GC–MS. The second screen is negative for substances of abuse; Mr. L’s clinician concludes that bupropion produced a false-positive result on the POC urine drug screen, confirming Mr. L’s assertions.
Related Resources
• Saitman A, Park HD, Fitzgerald RL. False-positive interferences of common urine drug screen immunoassays: a review. J Anal Toxicol. 2014;38(7):387-396.
• Tenore PL. Advanced urine toxicology testing. J Addict Dis. 2010;29(4):436-448.
Drug Brand Names
Amantadine • Symadine, Symmetrel
Amitriptyline • Elavil
Atorvastatin • Lipitor
Brompheniramine • Dimetane
Bupropion • Wellbutrin, Zyban
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Clomipramine • Anafranil
Cyclobenzaprine • Amrix, Flexeril
Cyproheptadine • Periactin
Desipramine • Nopramin
Desoxyephedrine • Desoxyn
Dextromethorphan • Delsym, Robitussin
Dicyclomine • Bentyl, Dicyclocot
Diphenhydramine • Benadryl, Unisom
Doxylamine • Robitussin, NyQuil
Dronabinol • Marinol
Efavirenz • Sustiva
Ephedrine • Mistol, Va-Tro-Nol
Ergotamine • Ergomar, Cafergot
Hydrocodone • Vicodin
Hydromophone • Dilaudid, Palladone
Hydroxyzine • Atarax, Vistaril
Isometheptene • Amidrine, Migrend
Isoxsuprine • Vasodilan, Vasoprine
Ketoprofen • Orudis, Oruvail
Labetalol • Normodyne, Trandate
Lisinopril • Prinivil, Zestril
Meperidine • Demerol
Naproxen • Aleve, Naprosyn
Oxaprozin • Daypro
Oxycodone • Oxycontin, Percocet, Percodan, Roxicodone
Phentermine • Adipex, Phentrol
Phenylephrine • Sudafed PE, Neo-Synephrine
Piroxicam • Feldene
Promethazine • Phenergan
Pseudoephedrine • Sudafed, Dimetapp
Quetiapine • Seroquel
Ranitidine • Zantac
Rifampin • Rifadin, Rimactane
Selegiline • EMSAM
Sertraline • Zoloft
Sulindac • Clinoril
Sumatriptan • Imitrex
Thioridazine • Mellaril
Tolmetin • Tolectin
Trazodone • Desyrel, Oleptro
Trimethobenzamide • Benzacot, Tigan
Trimipramine • Surmontil
Verapamil • Calan, Isoptin
1. Cobaugh DJ, Gainor C, Gaston CL, et al. The opioid abuse and misuse epidemic: implications for pharmacists in hospitals and health systems. Am J Health Syst Pharm. 2014;71(18):1539-1554.
2. Gilbert JW, Wheeler GR, Mick GE, et al. Importance of urine drug testing in the treatment of chronic noncancer pain: implications of recent medicare policy changes in Kentucky. Pain Physician. 2010;13(2):167-186.
3. Michna E, Jamison RN, Pham LD, et al. Urine toxicology screening among chronic pain patients on opioid therapy: frequency and predictability of abnormal findings. Clin J Pain. 2007;23(2):173-179.
4. U.S. Department of Justice. Fact sheet: drug testing in the criminal justice system. https://www.ncjrs.gov/pdffiles/dtest. pdf. Published March 1992. Accessed July 29, 2015.
5. Australian Diagnostic Services. Technical information: testing principle’s. http://www.australiandrugtesting. com/#!technical-info/c14h4. Accessed November 5, 2014.
6. University of Illinois at Chicago College of Pharmacy. What drugs are likely to interfere with urine drug screens? http://dig.pharm.uic.edu/faq/2011/Feb/faq1.aspx. Accessed November 5, 2014.
7. Wolff K, Farrell M, Marsden J, et al. A review of biological indicators of illicit drug use, practical considerations and clinical usefulness. Addiction. 1999;94(9):1279-1298.
8. Gourlay D, Heit H, Caplan YH. Urine drug testing in primary care – dispelling the myths & designing strategies. PharmaCom Group. http://www.mc.uky.edu/equip-4-pcps/documents/ section8/urine%20drug%20testing%20in%20clinical%20 practice.pdf. Accessed August 6, 2015.
9. Standridge JB, Adams SM, Zotos AP. Urine drug screen: a valuable office procedure. Am Fam Physician. 2010;81(5): 635-640.
10. Casey ER, Scott MG, Tang S, et al. Frequency of false positive amphetamine screens due to bupropion using the Syva EMIT II immunoassay. J Med Toxicol. 2011;7(2):105-108.
11. Casavant MJ. Urine drug screening in adolescents. Pediatr Clin N Am. 2002;49(2):317-327.
12. Shults TF. The medical review officer handbook. 7th ed. Chapel Hill, NC: Quadrangle Research; 1999.
13. Baden LR, Horowitz G, Jacoby H, et al. Quinolones and false-positive urine screening for opiates by immunoassay technology. JAMA. 2001;286(24):3115-3119.
1. Cobaugh DJ, Gainor C, Gaston CL, et al. The opioid abuse and misuse epidemic: implications for pharmacists in hospitals and health systems. Am J Health Syst Pharm. 2014;71(18):1539-1554.
2. Gilbert JW, Wheeler GR, Mick GE, et al. Importance of urine drug testing in the treatment of chronic noncancer pain: implications of recent medicare policy changes in Kentucky. Pain Physician. 2010;13(2):167-186.
3. Michna E, Jamison RN, Pham LD, et al. Urine toxicology screening among chronic pain patients on opioid therapy: frequency and predictability of abnormal findings. Clin J Pain. 2007;23(2):173-179.
4. U.S. Department of Justice. Fact sheet: drug testing in the criminal justice system. https://www.ncjrs.gov/pdffiles/dtest. pdf. Published March 1992. Accessed July 29, 2015.
5. Australian Diagnostic Services. Technical information: testing principle’s. http://www.australiandrugtesting. com/#!technical-info/c14h4. Accessed November 5, 2014.
6. University of Illinois at Chicago College of Pharmacy. What drugs are likely to interfere with urine drug screens? http://dig.pharm.uic.edu/faq/2011/Feb/faq1.aspx. Accessed November 5, 2014.
7. Wolff K, Farrell M, Marsden J, et al. A review of biological indicators of illicit drug use, practical considerations and clinical usefulness. Addiction. 1999;94(9):1279-1298.
8. Gourlay D, Heit H, Caplan YH. Urine drug testing in primary care – dispelling the myths & designing strategies. PharmaCom Group. http://www.mc.uky.edu/equip-4-pcps/documents/ section8/urine%20drug%20testing%20in%20clinical%20 practice.pdf. Accessed August 6, 2015.
9. Standridge JB, Adams SM, Zotos AP. Urine drug screen: a valuable office procedure. Am Fam Physician. 2010;81(5): 635-640.
10. Casey ER, Scott MG, Tang S, et al. Frequency of false positive amphetamine screens due to bupropion using the Syva EMIT II immunoassay. J Med Toxicol. 2011;7(2):105-108.
11. Casavant MJ. Urine drug screening in adolescents. Pediatr Clin N Am. 2002;49(2):317-327.
12. Shults TF. The medical review officer handbook. 7th ed. Chapel Hill, NC: Quadrangle Research; 1999.
13. Baden LR, Horowitz G, Jacoby H, et al. Quinolones and false-positive urine screening for opiates by immunoassay technology. JAMA. 2001;286(24):3115-3119.
Avoiding common drug−drug interactions
Mr. T, age 23, was given a diagnosis of bipolar disorder 1 year ago. After he experienced inadequate symptom relief with valproate, you switched him to extended-release lithium, 1,200 mg/d. Mr. T reported improved mood and stability with this medication adjustment. These positive changes led him to resume activities he enjoyed before onset of bipolar disorder, such as running, reading, and going out to dinner with friends.
Now, Mr. T’s mother calls your office to express concern about her son’s slight

hand tremor, which appeared after 2 days of gastrointestinal distress. She tells you that Mr. T sprained his ankle while running 1 week ago and has been taking over-the-counter ibuprofen for pain relief, which he did often in the past.
You suspect that Mr. T is experiencing lithium toxicity as a result of ibuprofen use.
Although mental health providers can easily recognize the drug−drug interaction between lithium and nonsteroidal anti-inflammatory drugs (NSAIDs) that Mr. T experienced, interpreting the safety of a medication regimen with respect to drug− drug interactions before prescribing often is more daunting. This article reviews the basics of drug−drug interactions, while briefly highlighting common examples in psychiatric medicine (Table 11-5). We also provide an outline of additional points to consider when reviewing your patients’ medication regimens and encountering unfamiliar drug−drug interactions.
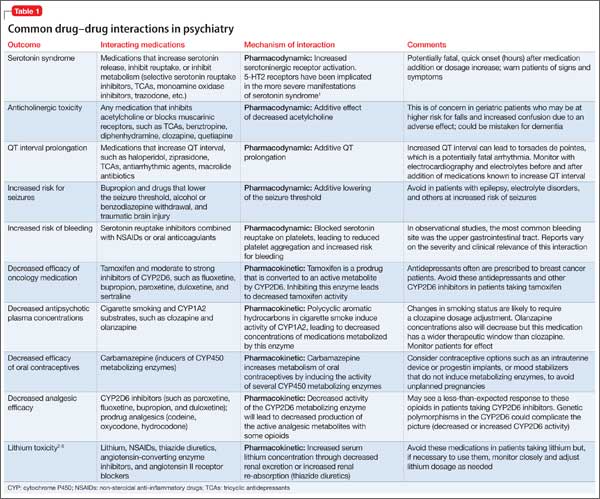
Types of drug−drug interactions
Drug−drug interactions fall into 2 categories: pharmacodynamic (PD) and pharmacokinetic (PK):
• PD interactions are a result of the combined impact of medications on the body when there is no direct effect on absorption, distribution, metabolism, or excretion characteristics, such as 2 medications that act at the same receptor or lead to similar or opposing pharmacologic effects.
• PK interactions occur when a drug affects the absorption, distribution, metabolism, or excretion characteristics of another drug.
Although it is possible that drug−drug interactions will have no clinical effect, when the impact of a PD or PK drug−drug interaction is evident, it likely is the result of additive, synergistic, or antagonistic consequences on the medications’ intended impact or side-effect profile.
Pharmacodynamic interactions
Serotonin syndrome. The potential for serotonin syndrome occurs when medications that increase synaptic serotonin concentration are used concomitantly.1 This can occur through several mechanisms, including increased serotonin release, decreased reuptake, or decreased serotonin metabolism. A high serotonin concentration in the CNS and in the periphery overstimulates serotonin receptors, leading to signs and symptoms that can include diarrhea, fever, delirium, coma, and potentially death.
QT prolongation and anticholinergic toxicity are further examples of additive PD drug−drug interactions. Anticholinergic toxicity is possible when multiple medications contribute to inhibition of the neuro-transmitter acetylcholine at muscarinic receptors. This leads to adverse effects such as dry mouth, constipation, confusion, and urinary retention.
QT prolongation, which can lead to arrhythmia, occurs when a patient is taking several medications that can increase the QT interval. Consider close monitoring and using alternative agents with less potential to increase the QT interval in patients at risk of arrhythmias (geriatric patients, those with an increased QT interval at baseline, etc.).
Decreased seizure threshold. The increased risk of seizures with bupropion and other medications that lower the seizure threshold is another example of an additive PD drug interaction. Bupropion can increase the risk of seizures in a dose-dependent manner, which increases when bupropion is taken with other drugs that lower the seizure threshold.6 Seizure risk associated with alcohol or benzodiazepine withdrawal also may increase the risk for this interaction.
Of note, the increased risk of seizures with the combination of bupropion and alcohol in the absence of withdrawal is not well studied in humans, but positive correlation has been seen in an animal study.6
Decreased platelet function. Another example of a PD drug−drug interaction is increased risk of bleeding when a selective serotonin reuptake inhibitor is used with a NSAID or oral anticoagulant. The proposed mechanism for this interaction is that blocking serotonin reuptake on platelets leads to decreased platelet function and an increased risk for prolonged bleeding.7 This is somewhat controversial because, first, it has been noted that drugs with the highest degree of serotonin reuptake inhibition do not always cause the highest risk of bleeding and, second, most of the evidence for this interaction is from observational studies.7
This potential interaction could be most important for patients who need an antidepressant, are on chronic NSAID or anticoagulant therapy, and are at high risk of bleeding.
Pharmacokinetic interactions
PK interactions in psychiatry often are caused by interference of drug metabolizing enzymes. The cytochrome P450 (CYP450) family of metabolizing enzymes in particular is important to the breakdown of medications in the body. Many drug−drug interactions involve medications that can inhibit or induce metabolism of other drugs through their effect on the CYP450 system.
Inhibition interactions. When a drug’s metabolism is inhibited, the result is usually increased serum concentration of that medication (because of less breakdown) and a more potent impact on the primary mechanism of action or adverse effects. Sometimes, inhibiting metabolism can lead to decreased clinical effect. Tamoxifen (an oral agent used to treat breast cancer) and certain analgesics when used in combination with moderate or strong inhibitors of the CYP2D6 subfamily of CYP450 metabolizing enzymes are 2 examples of metabolism inhibition leading to decreased efficacy.8 Both tamoxifen and the analgesics listed in Table 11-5 are prodrugs; that is, they must be metabolized to be active. When the enzymes that metabolize these drugs into their active form are inhibited, the concentration of active drug decreases.
Induction interactions. Alternatively, there is an increased rate of drug breakdown and resulting decrease in effect when drugs that induce the activity of metabolizing enzymes are used with medications that are substrates of the same enzyme. Carbamazepine is commonly involved in this type of drug interaction because it is a strong inducer of CYP 1A2, 2B6, 2C19, 2C9, and 3A4, and the p-glycoprotein drug efflux pump.9 As a result of this rampant induction, carbamazepine can decrease the serum concentration of oral contraceptives below a reliably effective level. Therefore, it is recommended that women of childbearing potential use other contraceptive methods, such as a progestin implant or an intrauterine device.10
In addition, the polycyclic aromatic hydrocarbons found in cigarettes induce activity of CYP1A2. Patients who smoke and use medications metabolized by this enzyme, such as clozapine and olanzapine, may need a higher dosage.
Drug elimination interactions
The last drug−drug interaction discussed here returns the discussion to Mr. T and involves drug elimination.2 The NSAIDs Mr. T was using for pain likely caused decreased renal excretion of lithium. Because lithium is primarily excreted through the kidneys, Mr. T’s NSAID use, possibly in combination with dehydration caused by gastrointestinal distress, resulted in lithium toxicity. This class of analgesics should be avoided or used cautiously in patients taking lithium.
Clinical applications
The relatively common drug−drug interactions discussed here are just a fraction of the potential interactions mental health practitioners see on a daily basis. Understanding the basics of PD and PK interactions in the setting of patient-specific factors can help to clarify the information found in drug−drug interaction databases, such as Micromedex, Lexicomp, Facts and Comparisons, and Epocrates. Table 2 lists additional insights into drug interactions.
Related Resources
• CredibleMeds. Online resource on QT prolonging drugs. http://crediblemeds.org.
• Madhusoodanan S, Velama U, Parmar J, et al. A current review of cytochrome P450 interactions of psychotropic drugs. Ann Clin Psychiatry. 2014;26(2):120-138.
Drug Brand Names
Benztropine • Cogentin Olanzapine • Zyprexa
Bupropion • Wellbutrin Oxycodone • Oxycontin
Carbamazepine • Tegretol Paroxetine • Paxil
Clozapine • Clozaril Quetiapine • Seroquel
Diphenhydramine • Benadryl Sertraline • Zoloft
Duloxetine • Cymbalta Tamoxifen • Soltamox
Fluoxetine • Prozac Trazodone • Desyrel
Lithium • Eskalith, Lithobid Valproate • Divalproex
Haloperidol • Haldol Ziprasidone • Geodon
Hydrocodone • Vicodin
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ. 2014;348:g1626. doi: 10.1136/bmj.g1626.
2. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
3. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
4. Blanche P, Raynaud E, Kerob D, et al. Lithium intoxication in an elderly patient after combined treatment with losartan. Eur J Clin Pharmacol. 1997;52(6):501.
5. Atacand [package insert]. Wilmington, DE: AstraZeneca LP; 2013.
6. Silverstone PH, Williams R, McMahon L, et al. Alcohol significantly lowers the seizure threshold in mice when co-administered with bupropion hydrochloride. Ann Gen Psychiatry. 2008;7:11.
7. Spina E, Trifirò G, Caraci F. Clinically significant drug interactions with newer antidepressants. CNS Drugs. 2012;26(1):39-67.
8. Ereshefsky L, Sloan DM. Drug-drug interactions with the use of psychotropic medications. CNS Spectr. 2009;14(suppl Q and A forum 8):1-8.
9. Carbamazepine. Drug facts and comparisons database. St. Louis, MO: Wolters Kluwer Health Inc; November 2014.
10. Pennell PB. Pregnancy, epilepsy, and women’s issues. Continuum (Minneap Minn). 2013;19(3 Epilepsy):697-714.
Mr. T, age 23, was given a diagnosis of bipolar disorder 1 year ago. After he experienced inadequate symptom relief with valproate, you switched him to extended-release lithium, 1,200 mg/d. Mr. T reported improved mood and stability with this medication adjustment. These positive changes led him to resume activities he enjoyed before onset of bipolar disorder, such as running, reading, and going out to dinner with friends.
Now, Mr. T’s mother calls your office to express concern about her son’s slight
hand tremor, which appeared after 2 days of gastrointestinal distress. She tells you that Mr. T sprained his ankle while running 1 week ago and has been taking over-the-counter ibuprofen for pain relief, which he did often in the past.
You suspect that Mr. T is experiencing lithium toxicity as a result of ibuprofen use.
Although mental health providers can easily recognize the drug−drug interaction between lithium and nonsteroidal anti-inflammatory drugs (NSAIDs) that Mr. T experienced, interpreting the safety of a medication regimen with respect to drug− drug interactions before prescribing often is more daunting. This article reviews the basics of drug−drug interactions, while briefly highlighting common examples in psychiatric medicine (Table 11-5). We also provide an outline of additional points to consider when reviewing your patients’ medication regimens and encountering unfamiliar drug−drug interactions.
Types of drug−drug interactions
Drug−drug interactions fall into 2 categories: pharmacodynamic (PD) and pharmacokinetic (PK):
• PD interactions are a result of the combined impact of medications on the body when there is no direct effect on absorption, distribution, metabolism, or excretion characteristics, such as 2 medications that act at the same receptor or lead to similar or opposing pharmacologic effects.
• PK interactions occur when a drug affects the absorption, distribution, metabolism, or excretion characteristics of another drug.
Although it is possible that drug−drug interactions will have no clinical effect, when the impact of a PD or PK drug−drug interaction is evident, it likely is the result of additive, synergistic, or antagonistic consequences on the medications’ intended impact or side-effect profile.
Pharmacodynamic interactions
Serotonin syndrome. The potential for serotonin syndrome occurs when medications that increase synaptic serotonin concentration are used concomitantly.1 This can occur through several mechanisms, including increased serotonin release, decreased reuptake, or decreased serotonin metabolism. A high serotonin concentration in the CNS and in the periphery overstimulates serotonin receptors, leading to signs and symptoms that can include diarrhea, fever, delirium, coma, and potentially death.
QT prolongation and anticholinergic toxicity are further examples of additive PD drug−drug interactions. Anticholinergic toxicity is possible when multiple medications contribute to inhibition of the neuro-transmitter acetylcholine at muscarinic receptors. This leads to adverse effects such as dry mouth, constipation, confusion, and urinary retention.
QT prolongation, which can lead to arrhythmia, occurs when a patient is taking several medications that can increase the QT interval. Consider close monitoring and using alternative agents with less potential to increase the QT interval in patients at risk of arrhythmias (geriatric patients, those with an increased QT interval at baseline, etc.).
Decreased seizure threshold. The increased risk of seizures with bupropion and other medications that lower the seizure threshold is another example of an additive PD drug interaction. Bupropion can increase the risk of seizures in a dose-dependent manner, which increases when bupropion is taken with other drugs that lower the seizure threshold.6 Seizure risk associated with alcohol or benzodiazepine withdrawal also may increase the risk for this interaction.
Of note, the increased risk of seizures with the combination of bupropion and alcohol in the absence of withdrawal is not well studied in humans, but positive correlation has been seen in an animal study.6
Decreased platelet function. Another example of a PD drug−drug interaction is increased risk of bleeding when a selective serotonin reuptake inhibitor is used with a NSAID or oral anticoagulant. The proposed mechanism for this interaction is that blocking serotonin reuptake on platelets leads to decreased platelet function and an increased risk for prolonged bleeding.7 This is somewhat controversial because, first, it has been noted that drugs with the highest degree of serotonin reuptake inhibition do not always cause the highest risk of bleeding and, second, most of the evidence for this interaction is from observational studies.7
This potential interaction could be most important for patients who need an antidepressant, are on chronic NSAID or anticoagulant therapy, and are at high risk of bleeding.
Pharmacokinetic interactions
PK interactions in psychiatry often are caused by interference of drug metabolizing enzymes. The cytochrome P450 (CYP450) family of metabolizing enzymes in particular is important to the breakdown of medications in the body. Many drug−drug interactions involve medications that can inhibit or induce metabolism of other drugs through their effect on the CYP450 system.
Inhibition interactions. When a drug’s metabolism is inhibited, the result is usually increased serum concentration of that medication (because of less breakdown) and a more potent impact on the primary mechanism of action or adverse effects. Sometimes, inhibiting metabolism can lead to decreased clinical effect. Tamoxifen (an oral agent used to treat breast cancer) and certain analgesics when used in combination with moderate or strong inhibitors of the CYP2D6 subfamily of CYP450 metabolizing enzymes are 2 examples of metabolism inhibition leading to decreased efficacy.8 Both tamoxifen and the analgesics listed in Table 11-5 are prodrugs; that is, they must be metabolized to be active. When the enzymes that metabolize these drugs into their active form are inhibited, the concentration of active drug decreases.
Induction interactions. Alternatively, there is an increased rate of drug breakdown and resulting decrease in effect when drugs that induce the activity of metabolizing enzymes are used with medications that are substrates of the same enzyme. Carbamazepine is commonly involved in this type of drug interaction because it is a strong inducer of CYP 1A2, 2B6, 2C19, 2C9, and 3A4, and the p-glycoprotein drug efflux pump.9 As a result of this rampant induction, carbamazepine can decrease the serum concentration of oral contraceptives below a reliably effective level. Therefore, it is recommended that women of childbearing potential use other contraceptive methods, such as a progestin implant or an intrauterine device.10
In addition, the polycyclic aromatic hydrocarbons found in cigarettes induce activity of CYP1A2. Patients who smoke and use medications metabolized by this enzyme, such as clozapine and olanzapine, may need a higher dosage.
Drug elimination interactions
The last drug−drug interaction discussed here returns the discussion to Mr. T and involves drug elimination.2 The NSAIDs Mr. T was using for pain likely caused decreased renal excretion of lithium. Because lithium is primarily excreted through the kidneys, Mr. T’s NSAID use, possibly in combination with dehydration caused by gastrointestinal distress, resulted in lithium toxicity. This class of analgesics should be avoided or used cautiously in patients taking lithium.
Clinical applications
The relatively common drug−drug interactions discussed here are just a fraction of the potential interactions mental health practitioners see on a daily basis. Understanding the basics of PD and PK interactions in the setting of patient-specific factors can help to clarify the information found in drug−drug interaction databases, such as Micromedex, Lexicomp, Facts and Comparisons, and Epocrates. Table 2 lists additional insights into drug interactions.
Related Resources
• CredibleMeds. Online resource on QT prolonging drugs. http://crediblemeds.org.
• Madhusoodanan S, Velama U, Parmar J, et al. A current review of cytochrome P450 interactions of psychotropic drugs. Ann Clin Psychiatry. 2014;26(2):120-138.
Drug Brand Names
Benztropine • Cogentin Olanzapine • Zyprexa
Bupropion • Wellbutrin Oxycodone • Oxycontin
Carbamazepine • Tegretol Paroxetine • Paxil
Clozapine • Clozaril Quetiapine • Seroquel
Diphenhydramine • Benadryl Sertraline • Zoloft
Duloxetine • Cymbalta Tamoxifen • Soltamox
Fluoxetine • Prozac Trazodone • Desyrel
Lithium • Eskalith, Lithobid Valproate • Divalproex
Haloperidol • Haldol Ziprasidone • Geodon
Hydrocodone • Vicodin
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Mr. T, age 23, was given a diagnosis of bipolar disorder 1 year ago. After he experienced inadequate symptom relief with valproate, you switched him to extended-release lithium, 1,200 mg/d. Mr. T reported improved mood and stability with this medication adjustment. These positive changes led him to resume activities he enjoyed before onset of bipolar disorder, such as running, reading, and going out to dinner with friends.
Now, Mr. T’s mother calls your office to express concern about her son’s slight
hand tremor, which appeared after 2 days of gastrointestinal distress. She tells you that Mr. T sprained his ankle while running 1 week ago and has been taking over-the-counter ibuprofen for pain relief, which he did often in the past.
You suspect that Mr. T is experiencing lithium toxicity as a result of ibuprofen use.
Although mental health providers can easily recognize the drug−drug interaction between lithium and nonsteroidal anti-inflammatory drugs (NSAIDs) that Mr. T experienced, interpreting the safety of a medication regimen with respect to drug− drug interactions before prescribing often is more daunting. This article reviews the basics of drug−drug interactions, while briefly highlighting common examples in psychiatric medicine (Table 11-5). We also provide an outline of additional points to consider when reviewing your patients’ medication regimens and encountering unfamiliar drug−drug interactions.
Types of drug−drug interactions
Drug−drug interactions fall into 2 categories: pharmacodynamic (PD) and pharmacokinetic (PK):
• PD interactions are a result of the combined impact of medications on the body when there is no direct effect on absorption, distribution, metabolism, or excretion characteristics, such as 2 medications that act at the same receptor or lead to similar or opposing pharmacologic effects.
• PK interactions occur when a drug affects the absorption, distribution, metabolism, or excretion characteristics of another drug.
Although it is possible that drug−drug interactions will have no clinical effect, when the impact of a PD or PK drug−drug interaction is evident, it likely is the result of additive, synergistic, or antagonistic consequences on the medications’ intended impact or side-effect profile.
Pharmacodynamic interactions
Serotonin syndrome. The potential for serotonin syndrome occurs when medications that increase synaptic serotonin concentration are used concomitantly.1 This can occur through several mechanisms, including increased serotonin release, decreased reuptake, or decreased serotonin metabolism. A high serotonin concentration in the CNS and in the periphery overstimulates serotonin receptors, leading to signs and symptoms that can include diarrhea, fever, delirium, coma, and potentially death.
QT prolongation and anticholinergic toxicity are further examples of additive PD drug−drug interactions. Anticholinergic toxicity is possible when multiple medications contribute to inhibition of the neuro-transmitter acetylcholine at muscarinic receptors. This leads to adverse effects such as dry mouth, constipation, confusion, and urinary retention.
QT prolongation, which can lead to arrhythmia, occurs when a patient is taking several medications that can increase the QT interval. Consider close monitoring and using alternative agents with less potential to increase the QT interval in patients at risk of arrhythmias (geriatric patients, those with an increased QT interval at baseline, etc.).
Decreased seizure threshold. The increased risk of seizures with bupropion and other medications that lower the seizure threshold is another example of an additive PD drug interaction. Bupropion can increase the risk of seizures in a dose-dependent manner, which increases when bupropion is taken with other drugs that lower the seizure threshold.6 Seizure risk associated with alcohol or benzodiazepine withdrawal also may increase the risk for this interaction.
Of note, the increased risk of seizures with the combination of bupropion and alcohol in the absence of withdrawal is not well studied in humans, but positive correlation has been seen in an animal study.6
Decreased platelet function. Another example of a PD drug−drug interaction is increased risk of bleeding when a selective serotonin reuptake inhibitor is used with a NSAID or oral anticoagulant. The proposed mechanism for this interaction is that blocking serotonin reuptake on platelets leads to decreased platelet function and an increased risk for prolonged bleeding.7 This is somewhat controversial because, first, it has been noted that drugs with the highest degree of serotonin reuptake inhibition do not always cause the highest risk of bleeding and, second, most of the evidence for this interaction is from observational studies.7
This potential interaction could be most important for patients who need an antidepressant, are on chronic NSAID or anticoagulant therapy, and are at high risk of bleeding.
Pharmacokinetic interactions
PK interactions in psychiatry often are caused by interference of drug metabolizing enzymes. The cytochrome P450 (CYP450) family of metabolizing enzymes in particular is important to the breakdown of medications in the body. Many drug−drug interactions involve medications that can inhibit or induce metabolism of other drugs through their effect on the CYP450 system.
Inhibition interactions. When a drug’s metabolism is inhibited, the result is usually increased serum concentration of that medication (because of less breakdown) and a more potent impact on the primary mechanism of action or adverse effects. Sometimes, inhibiting metabolism can lead to decreased clinical effect. Tamoxifen (an oral agent used to treat breast cancer) and certain analgesics when used in combination with moderate or strong inhibitors of the CYP2D6 subfamily of CYP450 metabolizing enzymes are 2 examples of metabolism inhibition leading to decreased efficacy.8 Both tamoxifen and the analgesics listed in Table 11-5 are prodrugs; that is, they must be metabolized to be active. When the enzymes that metabolize these drugs into their active form are inhibited, the concentration of active drug decreases.
Induction interactions. Alternatively, there is an increased rate of drug breakdown and resulting decrease in effect when drugs that induce the activity of metabolizing enzymes are used with medications that are substrates of the same enzyme. Carbamazepine is commonly involved in this type of drug interaction because it is a strong inducer of CYP 1A2, 2B6, 2C19, 2C9, and 3A4, and the p-glycoprotein drug efflux pump.9 As a result of this rampant induction, carbamazepine can decrease the serum concentration of oral contraceptives below a reliably effective level. Therefore, it is recommended that women of childbearing potential use other contraceptive methods, such as a progestin implant or an intrauterine device.10
In addition, the polycyclic aromatic hydrocarbons found in cigarettes induce activity of CYP1A2. Patients who smoke and use medications metabolized by this enzyme, such as clozapine and olanzapine, may need a higher dosage.
Drug elimination interactions
The last drug−drug interaction discussed here returns the discussion to Mr. T and involves drug elimination.2 The NSAIDs Mr. T was using for pain likely caused decreased renal excretion of lithium. Because lithium is primarily excreted through the kidneys, Mr. T’s NSAID use, possibly in combination with dehydration caused by gastrointestinal distress, resulted in lithium toxicity. This class of analgesics should be avoided or used cautiously in patients taking lithium.
Clinical applications
The relatively common drug−drug interactions discussed here are just a fraction of the potential interactions mental health practitioners see on a daily basis. Understanding the basics of PD and PK interactions in the setting of patient-specific factors can help to clarify the information found in drug−drug interaction databases, such as Micromedex, Lexicomp, Facts and Comparisons, and Epocrates. Table 2 lists additional insights into drug interactions.
Related Resources
• CredibleMeds. Online resource on QT prolonging drugs. http://crediblemeds.org.
• Madhusoodanan S, Velama U, Parmar J, et al. A current review of cytochrome P450 interactions of psychotropic drugs. Ann Clin Psychiatry. 2014;26(2):120-138.
Drug Brand Names
Benztropine • Cogentin Olanzapine • Zyprexa
Bupropion • Wellbutrin Oxycodone • Oxycontin
Carbamazepine • Tegretol Paroxetine • Paxil
Clozapine • Clozaril Quetiapine • Seroquel
Diphenhydramine • Benadryl Sertraline • Zoloft
Duloxetine • Cymbalta Tamoxifen • Soltamox
Fluoxetine • Prozac Trazodone • Desyrel
Lithium • Eskalith, Lithobid Valproate • Divalproex
Haloperidol • Haldol Ziprasidone • Geodon
Hydrocodone • Vicodin
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ. 2014;348:g1626. doi: 10.1136/bmj.g1626.
2. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
3. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
4. Blanche P, Raynaud E, Kerob D, et al. Lithium intoxication in an elderly patient after combined treatment with losartan. Eur J Clin Pharmacol. 1997;52(6):501.
5. Atacand [package insert]. Wilmington, DE: AstraZeneca LP; 2013.
6. Silverstone PH, Williams R, McMahon L, et al. Alcohol significantly lowers the seizure threshold in mice when co-administered with bupropion hydrochloride. Ann Gen Psychiatry. 2008;7:11.
7. Spina E, Trifirò G, Caraci F. Clinically significant drug interactions with newer antidepressants. CNS Drugs. 2012;26(1):39-67.
8. Ereshefsky L, Sloan DM. Drug-drug interactions with the use of psychotropic medications. CNS Spectr. 2009;14(suppl Q and A forum 8):1-8.
9. Carbamazepine. Drug facts and comparisons database. St. Louis, MO: Wolters Kluwer Health Inc; November 2014.
10. Pennell PB. Pregnancy, epilepsy, and women’s issues. Continuum (Minneap Minn). 2013;19(3 Epilepsy):697-714.
1. Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ. 2014;348:g1626. doi: 10.1136/bmj.g1626.
2. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
3. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
4. Blanche P, Raynaud E, Kerob D, et al. Lithium intoxication in an elderly patient after combined treatment with losartan. Eur J Clin Pharmacol. 1997;52(6):501.
5. Atacand [package insert]. Wilmington, DE: AstraZeneca LP; 2013.
6. Silverstone PH, Williams R, McMahon L, et al. Alcohol significantly lowers the seizure threshold in mice when co-administered with bupropion hydrochloride. Ann Gen Psychiatry. 2008;7:11.
7. Spina E, Trifirò G, Caraci F. Clinically significant drug interactions with newer antidepressants. CNS Drugs. 2012;26(1):39-67.
8. Ereshefsky L, Sloan DM. Drug-drug interactions with the use of psychotropic medications. CNS Spectr. 2009;14(suppl Q and A forum 8):1-8.
9. Carbamazepine. Drug facts and comparisons database. St. Louis, MO: Wolters Kluwer Health Inc; November 2014.
10. Pennell PB. Pregnancy, epilepsy, and women’s issues. Continuum (Minneap Minn). 2013;19(3 Epilepsy):697-714.
Do glutamatergic drugs have a role in treating depression?
Mrs. S, age 46, has been struggling to manage depression for 7 years. She completed adequate trials of several selective serotonin reuptake inhibitors and bupropion. Currently, she is taking duloxetine, 60 mg/d, and aripiprazole, 5 mg/d.
At her most recent clinic visit, Mrs. S reports that she is doing “OK,” but that she still feels sad and disengaged most days of the week. She wants to know more about ketamine for treating depression after reading about it on the Internet and hearing it mentioned in a support group she attends. She asks if you think it would work for her, and gives you with a copy of an article about its use in patients with treatment-resistant depression. Mrs. S has no other health conditions and takes a daily vitamin D and calcium supplement.
The monoamine hypothesis of depression postulates that symptoms originate from underactivity of monoamines, such as serotonin, norepinephrine, and dopamine, in the brain. This hypothesis was formulated in the 1960s after researchers observed that monoamine oxidase inhibitors and tricyclic antidepressants relieved depressive symptoms; both were known to increase monoamine concentrations in the synaptic cleft.1

Regrettably, these medications do not adequately relieve depressive symptoms for many people. In fact, symptom remission occurs in only one-third of treated patients.2 This low remission rate reflects a lack of understanding of the pathophysiology of depression, and the need for drugs with unique mechanisms of action.
One of the newest drug targets shown to be relevant in psychiatric illness is the
glutamatergic system. Glutamate is the predominant excitatory neurotransmitter in the CNS, and it is responsible for many key functions, including synaptic plasticity, learning, memory, and locomotion.3 Normally, the glutamatergic system tightly regulates the amount of glutamate in the neuronal synapse via receptors on presynaptic and postsynaptic neurons, as well as on glial cells (Figure). When this equilibrium is disrupted in stressful situations, such as ischemia, trauma, or seizures, excess glutamate is released into the synapse. The resulting glutamatergic hyperactivity can lead to neurotoxicity and cell death when neuronal receptors are activated for an extended period. 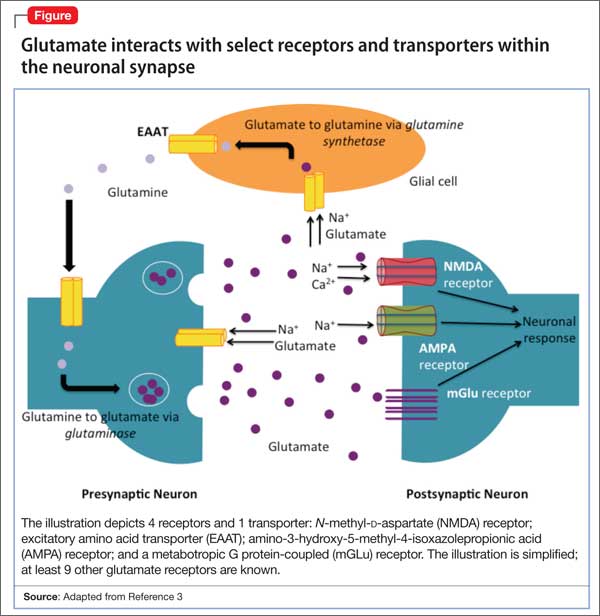
A key component of the glutamatergic system that is responsible for removing excess glutamate from the synapse is membrane-bound transporters, which are similar to serotonin and norepinephrine transporters. These excitatory amino acid transporters (EAATs) are important because glutamate metabolism does not occur within the synapse and EAATS are responsible for removing most of the glutamate from the synapse into glial cells.3
The network of receptors within the synapse that are activated by glutamate is extensive and complex. There are at least 11 glutamate-responsive receptors: 3 are ionotropic action channels, and the remaining 8 are metabotropic G protein-coupled receptors. Previous studies have shown regional changes in glutamate receptors, as well as elevated levels of glutamate, in the brains of patients with major depressive disorder (MDD).4
Ketamine. The ionotropic receptor N-methyl-d-aspartate (NMDA) is one of the most studied glutamate receptors. Pharmacologically, ketamine is a noncompetitive NMDA receptor antagonist that also activates the amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, which is another subtype of ionotropic glutamate receptors. In open-label clinical trials, ketamine has demonstrated rapid antidepressant action in patients with treatment-resistant MDD.4,5
Recently, Murrough et al6 performed the first randomized, psychoactive controlled trial using a single IV infusion of ketamine dosed below anesthesia ranges (0.5 mg/kg), or midazolam (0.045 mg/kg), in patients with treatment-resistant depression who had been antidepressant-free for at least 4 weeks. They found that 24 hours after medication administration, the likelihood of response to ketamine was significantly higher than the response to midazolam (OR: 2.18; 95% CI: 1.21 to 4.14), with a response rate of 64% in the ketamine group and 28% in the midazolam group.6
Psychotropic side effects, such as hallucinations, are a major concern with ketamine tolerability and abuse potential. This is largely because of ketamine’s antagonism of the NMDA receptor, which is a property shared with other abused drugs such as phencyclidine (PCP) and dextromethorphan. In the Murrough et al6 study, there were no reported cases of paranoia or hallucinations, but dissociative symptoms were relatively common (17%).
Although the results in this trial appear encouraging, there are several limitations to using ketamine to treat MDD, especially in an ambulatory setting. Concerns include ketamine’s IV administration, potential for abuse, long-term efficacy, and side-effect profile—particularly psychotic symptoms and hemodynamic changes. An ideal compound would have the rapid efficacy of ketamine, but with a safer side-effect profile, easier administration, and less potential for abuse.
Riluzole also acts on the glutamatergic system, but has not shown antidepressant efficacy as consistently as ketamine. Riluzole is FDA-approved for treating amyotrophic lateral sclerosis.5 Pharmacologically, riluzole is a glutamatergic modulator that increases glutamate reuptake into glial cells, decreases glutamate release, and increases AMPA trafficking. In open-label studies riluzole has shown efficacy in reducing depressive symptoms.4,5 However, when compared with placebo as a means of sustaining treatment response after a 1-time dose of ketamine, riluzole showed was no significant improvement in time to depressive relapse.7
Acamprosate, often used for treating alcohol abuse, is another a drug with glutamatergic activity that has been studied for possible use as an antidepressant.5
A review by Lapidus et al5 has a more extensive listing of current medications and investigational compounds that modulate glutamate transmission, and are of interest for their possible antidepressant activity. Given the relatively new “glutamatergic hypothesis” of depression, it is exciting that so many current and novel glutamatergic drug therapies are being evaluated.
Future of ketamine treatment
Glutamate has been shown to play an important part in the pathophysiology of depression. The rapid antidepressant efficacy of ketamine provides evidence that future medications with glutamate-modulating activity could be useful for patients who struggle to achieve symptom relief using available antidepressants. Several limitations exist regarding ketamine use, and more work in this important therapeutic area needs to be done. This last point is important to remember when speaking with patients such as Mrs. S. Although it is understandable for her to be excited about novel treatment options such as ketamine, stress to her that treating depression with ketamine at this time is strictly investigational, and that the drug needs to be thoroughly evaluated for safety and efficacy before it can be prescribed for this indication.
CASE CONTINUED
Mrs. S realizes that ketamine may not be the best next step for her, and she agrees to explore other approaches to treat her residual depressive symptoms.
Related Resources
• Machado-Vieira R, Ibrahim L, Henter ID, et al. Novel glutamatergic agents for major depressive disorder and bipolar disorder. Pharmacol Biochem Behav. 2012;100(4):678-687.
• Mathews DC, Henter ID, Zarate CA. Targeting the glutamatergic system to treat major depressive disorder: rationale and progress to date. Drugs. 2012;72(10):1313-1333.
Drug Brand Names
Acamprosate • Campral Duloxetine • Cymbalta
Aripiprazole • Abilify Ketamine • Ketalar
Bupropion • Wellbutrin, Zyban Riluzole • Rilutek
Disclosures
The authors report no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products.
1. Niciu MJ, Ionescu DF, Richards EM, et al. Glutamate and its receptors in the pathophysiology and treatment of major depressive disorder. J Neural Transm. 2014;121(8):907-924.
2. Gaynes BN, Dusetzina SB, Ellis AR, et al. Treating depression after initial treatment failure: directly comparing switch and augmenting strategies in STAR*D. J Clin Psychopharmacol. 2012;32(1):114-119.
3. Curry SC, Mills KC, Ruha A, et al. Neurotransmitters and neuromodulators. In: Nelson LS, Lewin NA, Howland MA, et al, eds. Goldfrank’s toxicologic emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:189-220.
4. Zarate C Jr, Machado-Vieira R, Henter I, et al. Glutamatergic modulators: the future of treating mood disorders? Harv Rev Psychiatry. 2010;18(5):293-303.
5. Lapidus KA, Soleimani L, Murrough JW. Novel glutamatergic drugs for the treatment of mood disorders. Neuropsychiatr Dis Treat. 2013;9:1101-1112.
6. Murrough JW, Iosifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134-1142.
7. Ibrahim L, Diazgranados N, Franco-Chaves J, et al. Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add-on riluzole: results from a 4-week, double-blind, placebo-controlled study. Neuropsychopharmacology. 2012;37(6):1526-1533.
Mrs. S, age 46, has been struggling to manage depression for 7 years. She completed adequate trials of several selective serotonin reuptake inhibitors and bupropion. Currently, she is taking duloxetine, 60 mg/d, and aripiprazole, 5 mg/d.
At her most recent clinic visit, Mrs. S reports that she is doing “OK,” but that she still feels sad and disengaged most days of the week. She wants to know more about ketamine for treating depression after reading about it on the Internet and hearing it mentioned in a support group she attends. She asks if you think it would work for her, and gives you with a copy of an article about its use in patients with treatment-resistant depression. Mrs. S has no other health conditions and takes a daily vitamin D and calcium supplement.
The monoamine hypothesis of depression postulates that symptoms originate from underactivity of monoamines, such as serotonin, norepinephrine, and dopamine, in the brain. This hypothesis was formulated in the 1960s after researchers observed that monoamine oxidase inhibitors and tricyclic antidepressants relieved depressive symptoms; both were known to increase monoamine concentrations in the synaptic cleft.1
Regrettably, these medications do not adequately relieve depressive symptoms for many people. In fact, symptom remission occurs in only one-third of treated patients.2 This low remission rate reflects a lack of understanding of the pathophysiology of depression, and the need for drugs with unique mechanisms of action.
One of the newest drug targets shown to be relevant in psychiatric illness is the
glutamatergic system. Glutamate is the predominant excitatory neurotransmitter in the CNS, and it is responsible for many key functions, including synaptic plasticity, learning, memory, and locomotion.3 Normally, the glutamatergic system tightly regulates the amount of glutamate in the neuronal synapse via receptors on presynaptic and postsynaptic neurons, as well as on glial cells (Figure). When this equilibrium is disrupted in stressful situations, such as ischemia, trauma, or seizures, excess glutamate is released into the synapse. The resulting glutamatergic hyperactivity can lead to neurotoxicity and cell death when neuronal receptors are activated for an extended period.
A key component of the glutamatergic system that is responsible for removing excess glutamate from the synapse is membrane-bound transporters, which are similar to serotonin and norepinephrine transporters. These excitatory amino acid transporters (EAATs) are important because glutamate metabolism does not occur within the synapse and EAATS are responsible for removing most of the glutamate from the synapse into glial cells.3
The network of receptors within the synapse that are activated by glutamate is extensive and complex. There are at least 11 glutamate-responsive receptors: 3 are ionotropic action channels, and the remaining 8 are metabotropic G protein-coupled receptors. Previous studies have shown regional changes in glutamate receptors, as well as elevated levels of glutamate, in the brains of patients with major depressive disorder (MDD).4
Ketamine. The ionotropic receptor N-methyl-d-aspartate (NMDA) is one of the most studied glutamate receptors. Pharmacologically, ketamine is a noncompetitive NMDA receptor antagonist that also activates the amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, which is another subtype of ionotropic glutamate receptors. In open-label clinical trials, ketamine has demonstrated rapid antidepressant action in patients with treatment-resistant MDD.4,5
Recently, Murrough et al6 performed the first randomized, psychoactive controlled trial using a single IV infusion of ketamine dosed below anesthesia ranges (0.5 mg/kg), or midazolam (0.045 mg/kg), in patients with treatment-resistant depression who had been antidepressant-free for at least 4 weeks. They found that 24 hours after medication administration, the likelihood of response to ketamine was significantly higher than the response to midazolam (OR: 2.18; 95% CI: 1.21 to 4.14), with a response rate of 64% in the ketamine group and 28% in the midazolam group.6
Psychotropic side effects, such as hallucinations, are a major concern with ketamine tolerability and abuse potential. This is largely because of ketamine’s antagonism of the NMDA receptor, which is a property shared with other abused drugs such as phencyclidine (PCP) and dextromethorphan. In the Murrough et al6 study, there were no reported cases of paranoia or hallucinations, but dissociative symptoms were relatively common (17%).
Although the results in this trial appear encouraging, there are several limitations to using ketamine to treat MDD, especially in an ambulatory setting. Concerns include ketamine’s IV administration, potential for abuse, long-term efficacy, and side-effect profile—particularly psychotic symptoms and hemodynamic changes. An ideal compound would have the rapid efficacy of ketamine, but with a safer side-effect profile, easier administration, and less potential for abuse.
Riluzole also acts on the glutamatergic system, but has not shown antidepressant efficacy as consistently as ketamine. Riluzole is FDA-approved for treating amyotrophic lateral sclerosis.5 Pharmacologically, riluzole is a glutamatergic modulator that increases glutamate reuptake into glial cells, decreases glutamate release, and increases AMPA trafficking. In open-label studies riluzole has shown efficacy in reducing depressive symptoms.4,5 However, when compared with placebo as a means of sustaining treatment response after a 1-time dose of ketamine, riluzole showed was no significant improvement in time to depressive relapse.7
Acamprosate, often used for treating alcohol abuse, is another a drug with glutamatergic activity that has been studied for possible use as an antidepressant.5
A review by Lapidus et al5 has a more extensive listing of current medications and investigational compounds that modulate glutamate transmission, and are of interest for their possible antidepressant activity. Given the relatively new “glutamatergic hypothesis” of depression, it is exciting that so many current and novel glutamatergic drug therapies are being evaluated.
Future of ketamine treatment
Glutamate has been shown to play an important part in the pathophysiology of depression. The rapid antidepressant efficacy of ketamine provides evidence that future medications with glutamate-modulating activity could be useful for patients who struggle to achieve symptom relief using available antidepressants. Several limitations exist regarding ketamine use, and more work in this important therapeutic area needs to be done. This last point is important to remember when speaking with patients such as Mrs. S. Although it is understandable for her to be excited about novel treatment options such as ketamine, stress to her that treating depression with ketamine at this time is strictly investigational, and that the drug needs to be thoroughly evaluated for safety and efficacy before it can be prescribed for this indication.
CASE CONTINUED
Mrs. S realizes that ketamine may not be the best next step for her, and she agrees to explore other approaches to treat her residual depressive symptoms.
Related Resources
• Machado-Vieira R, Ibrahim L, Henter ID, et al. Novel glutamatergic agents for major depressive disorder and bipolar disorder. Pharmacol Biochem Behav. 2012;100(4):678-687.
• Mathews DC, Henter ID, Zarate CA. Targeting the glutamatergic system to treat major depressive disorder: rationale and progress to date. Drugs. 2012;72(10):1313-1333.
Drug Brand Names
Acamprosate • Campral Duloxetine • Cymbalta
Aripiprazole • Abilify Ketamine • Ketalar
Bupropion • Wellbutrin, Zyban Riluzole • Rilutek
Disclosures
The authors report no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products.
Mrs. S, age 46, has been struggling to manage depression for 7 years. She completed adequate trials of several selective serotonin reuptake inhibitors and bupropion. Currently, she is taking duloxetine, 60 mg/d, and aripiprazole, 5 mg/d.
At her most recent clinic visit, Mrs. S reports that she is doing “OK,” but that she still feels sad and disengaged most days of the week. She wants to know more about ketamine for treating depression after reading about it on the Internet and hearing it mentioned in a support group she attends. She asks if you think it would work for her, and gives you with a copy of an article about its use in patients with treatment-resistant depression. Mrs. S has no other health conditions and takes a daily vitamin D and calcium supplement.
The monoamine hypothesis of depression postulates that symptoms originate from underactivity of monoamines, such as serotonin, norepinephrine, and dopamine, in the brain. This hypothesis was formulated in the 1960s after researchers observed that monoamine oxidase inhibitors and tricyclic antidepressants relieved depressive symptoms; both were known to increase monoamine concentrations in the synaptic cleft.1
Regrettably, these medications do not adequately relieve depressive symptoms for many people. In fact, symptom remission occurs in only one-third of treated patients.2 This low remission rate reflects a lack of understanding of the pathophysiology of depression, and the need for drugs with unique mechanisms of action.
One of the newest drug targets shown to be relevant in psychiatric illness is the
glutamatergic system. Glutamate is the predominant excitatory neurotransmitter in the CNS, and it is responsible for many key functions, including synaptic plasticity, learning, memory, and locomotion.3 Normally, the glutamatergic system tightly regulates the amount of glutamate in the neuronal synapse via receptors on presynaptic and postsynaptic neurons, as well as on glial cells (Figure). When this equilibrium is disrupted in stressful situations, such as ischemia, trauma, or seizures, excess glutamate is released into the synapse. The resulting glutamatergic hyperactivity can lead to neurotoxicity and cell death when neuronal receptors are activated for an extended period.
A key component of the glutamatergic system that is responsible for removing excess glutamate from the synapse is membrane-bound transporters, which are similar to serotonin and norepinephrine transporters. These excitatory amino acid transporters (EAATs) are important because glutamate metabolism does not occur within the synapse and EAATS are responsible for removing most of the glutamate from the synapse into glial cells.3
The network of receptors within the synapse that are activated by glutamate is extensive and complex. There are at least 11 glutamate-responsive receptors: 3 are ionotropic action channels, and the remaining 8 are metabotropic G protein-coupled receptors. Previous studies have shown regional changes in glutamate receptors, as well as elevated levels of glutamate, in the brains of patients with major depressive disorder (MDD).4
Ketamine. The ionotropic receptor N-methyl-d-aspartate (NMDA) is one of the most studied glutamate receptors. Pharmacologically, ketamine is a noncompetitive NMDA receptor antagonist that also activates the amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, which is another subtype of ionotropic glutamate receptors. In open-label clinical trials, ketamine has demonstrated rapid antidepressant action in patients with treatment-resistant MDD.4,5
Recently, Murrough et al6 performed the first randomized, psychoactive controlled trial using a single IV infusion of ketamine dosed below anesthesia ranges (0.5 mg/kg), or midazolam (0.045 mg/kg), in patients with treatment-resistant depression who had been antidepressant-free for at least 4 weeks. They found that 24 hours after medication administration, the likelihood of response to ketamine was significantly higher than the response to midazolam (OR: 2.18; 95% CI: 1.21 to 4.14), with a response rate of 64% in the ketamine group and 28% in the midazolam group.6
Psychotropic side effects, such as hallucinations, are a major concern with ketamine tolerability and abuse potential. This is largely because of ketamine’s antagonism of the NMDA receptor, which is a property shared with other abused drugs such as phencyclidine (PCP) and dextromethorphan. In the Murrough et al6 study, there were no reported cases of paranoia or hallucinations, but dissociative symptoms were relatively common (17%).
Although the results in this trial appear encouraging, there are several limitations to using ketamine to treat MDD, especially in an ambulatory setting. Concerns include ketamine’s IV administration, potential for abuse, long-term efficacy, and side-effect profile—particularly psychotic symptoms and hemodynamic changes. An ideal compound would have the rapid efficacy of ketamine, but with a safer side-effect profile, easier administration, and less potential for abuse.
Riluzole also acts on the glutamatergic system, but has not shown antidepressant efficacy as consistently as ketamine. Riluzole is FDA-approved for treating amyotrophic lateral sclerosis.5 Pharmacologically, riluzole is a glutamatergic modulator that increases glutamate reuptake into glial cells, decreases glutamate release, and increases AMPA trafficking. In open-label studies riluzole has shown efficacy in reducing depressive symptoms.4,5 However, when compared with placebo as a means of sustaining treatment response after a 1-time dose of ketamine, riluzole showed was no significant improvement in time to depressive relapse.7
Acamprosate, often used for treating alcohol abuse, is another a drug with glutamatergic activity that has been studied for possible use as an antidepressant.5
A review by Lapidus et al5 has a more extensive listing of current medications and investigational compounds that modulate glutamate transmission, and are of interest for their possible antidepressant activity. Given the relatively new “glutamatergic hypothesis” of depression, it is exciting that so many current and novel glutamatergic drug therapies are being evaluated.
Future of ketamine treatment
Glutamate has been shown to play an important part in the pathophysiology of depression. The rapid antidepressant efficacy of ketamine provides evidence that future medications with glutamate-modulating activity could be useful for patients who struggle to achieve symptom relief using available antidepressants. Several limitations exist regarding ketamine use, and more work in this important therapeutic area needs to be done. This last point is important to remember when speaking with patients such as Mrs. S. Although it is understandable for her to be excited about novel treatment options such as ketamine, stress to her that treating depression with ketamine at this time is strictly investigational, and that the drug needs to be thoroughly evaluated for safety and efficacy before it can be prescribed for this indication.
CASE CONTINUED
Mrs. S realizes that ketamine may not be the best next step for her, and she agrees to explore other approaches to treat her residual depressive symptoms.
Related Resources
• Machado-Vieira R, Ibrahim L, Henter ID, et al. Novel glutamatergic agents for major depressive disorder and bipolar disorder. Pharmacol Biochem Behav. 2012;100(4):678-687.
• Mathews DC, Henter ID, Zarate CA. Targeting the glutamatergic system to treat major depressive disorder: rationale and progress to date. Drugs. 2012;72(10):1313-1333.
Drug Brand Names
Acamprosate • Campral Duloxetine • Cymbalta
Aripiprazole • Abilify Ketamine • Ketalar
Bupropion • Wellbutrin, Zyban Riluzole • Rilutek
Disclosures
The authors report no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products.
1. Niciu MJ, Ionescu DF, Richards EM, et al. Glutamate and its receptors in the pathophysiology and treatment of major depressive disorder. J Neural Transm. 2014;121(8):907-924.
2. Gaynes BN, Dusetzina SB, Ellis AR, et al. Treating depression after initial treatment failure: directly comparing switch and augmenting strategies in STAR*D. J Clin Psychopharmacol. 2012;32(1):114-119.
3. Curry SC, Mills KC, Ruha A, et al. Neurotransmitters and neuromodulators. In: Nelson LS, Lewin NA, Howland MA, et al, eds. Goldfrank’s toxicologic emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:189-220.
4. Zarate C Jr, Machado-Vieira R, Henter I, et al. Glutamatergic modulators: the future of treating mood disorders? Harv Rev Psychiatry. 2010;18(5):293-303.
5. Lapidus KA, Soleimani L, Murrough JW. Novel glutamatergic drugs for the treatment of mood disorders. Neuropsychiatr Dis Treat. 2013;9:1101-1112.
6. Murrough JW, Iosifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134-1142.
7. Ibrahim L, Diazgranados N, Franco-Chaves J, et al. Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add-on riluzole: results from a 4-week, double-blind, placebo-controlled study. Neuropsychopharmacology. 2012;37(6):1526-1533.
1. Niciu MJ, Ionescu DF, Richards EM, et al. Glutamate and its receptors in the pathophysiology and treatment of major depressive disorder. J Neural Transm. 2014;121(8):907-924.
2. Gaynes BN, Dusetzina SB, Ellis AR, et al. Treating depression after initial treatment failure: directly comparing switch and augmenting strategies in STAR*D. J Clin Psychopharmacol. 2012;32(1):114-119.
3. Curry SC, Mills KC, Ruha A, et al. Neurotransmitters and neuromodulators. In: Nelson LS, Lewin NA, Howland MA, et al, eds. Goldfrank’s toxicologic emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:189-220.
4. Zarate C Jr, Machado-Vieira R, Henter I, et al. Glutamatergic modulators: the future of treating mood disorders? Harv Rev Psychiatry. 2010;18(5):293-303.
5. Lapidus KA, Soleimani L, Murrough JW. Novel glutamatergic drugs for the treatment of mood disorders. Neuropsychiatr Dis Treat. 2013;9:1101-1112.
6. Murrough JW, Iosifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134-1142.
7. Ibrahim L, Diazgranados N, Franco-Chaves J, et al. Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add-on riluzole: results from a 4-week, double-blind, placebo-controlled study. Neuropsychopharmacology. 2012;37(6):1526-1533.
Strategies for managing drug-induced tardive dyskinesia
Fifteen years ago, Mr. L, age 40, was given a diagnosis of schizophrenia, which has been treated with haloperidol, 10 mg/d. Approximately 1 year ago, he began experiencing consistent lip smacking, a sign of tardive dyskinesia. Vitamin E was added to the treatment regimen, after which the tardive dyskinesia symptoms resolved.
A few months later, however, those symptoms returned and became worse. In addition to lip smacking, Mr. L now also describes involuntary bilateral twitching and muscle spasms in both legs.
Haloperidol and vitamin E are discontinued and Mr. L is switched to olanzapine, 20 mg/d. Although olanzapine is effective for Mr. L's symptoms of schizophrenia, tardive dyskinesia persists, and he gains 60 pounds and develops diabetes. Olanzapine is discontinued and he begins a trial of risperidone, 4 mg/d.
While on risperidone, blood sugar control, measured by hemoglobin A1c, and insulin resistance improve, but Mr. L continues to have symptoms of tardive dyskinesia. Vitamin E is added again, but is ineffective. The treatment team switches Mr. L to clozapine but symptoms of tardive dyskinesia do not improve.

Extrapyramidal side effects are common with first-generation antipsychotics (FGA) such as haloperidol. Types of antipsychotic-induced movement disorders include dystonias, akathisias, pseudoparkinsonism, and tardive dyskinesia. Of these, tardive dyskinesia is the most concerning because it often is difficult to treat and may be irreversible.
Tardive dyskinesia involves abnormal, involuntary movements, usually involving the face and, sometimes, the limbs. Common symptoms include lip smacking, tongue protrusions, and puffing the cheeks1; severe tardive dyskinesia may affect the larynx and diaphragm, which can be life-threatening. The incidence of tardive dyskinesia is approximately 5% after the first year of FGA treatment and 1% with second-generation antipsychotics (SGAs).2 The risk increases with higher doses and longer duration of treatment, with a prevalence of 20% to 25% with long-term FGA use.3
Treatment strategies
There are no FDA-approved drugs for tardive dyskinesia (Table).4-6 The best strategy is to prevent tardive dyskinesia with judicious use of an antipsychotic. If a patient taking a FGA develops tardive dyskinesia, the first-line treatment is to switch to a SGA. Risperidone, olanzapine, quetiapine, and clozapine have a low risk of tardive dyskinesia. Newer agents, such as lurasidone, asenapine, iloperidone, and aripiprazole, might have a lower risk of tardive dyskinesia, possibly because of differences in dopamine blockage between these agents and FGAs. Clozapine is least likely to cause tardive dyskinesia, but it often is used as a last resort because of the risk of agranulocytosis and the need for frequent tests to measurewhite blood cells.1,4
Other treatments include melatonin, donepezil, vitamin B6, and vitamin E.4 These agents can reduce symptoms, but no large clinical trials have proved that the are efficacious. Last-resort treatments include suppressive treatment using FGAs several times a day, because the constant dopamine blockade may stop symptoms for a short time; this approach is not recommended because it can exacerbate symptoms of tardive dyskinesia.4
Other suppressive treatments used in severe or refractory cases include reserpineand tetrabenazine, which are used off-label and work by blocking monoamine transporters. This blockage results in a reduction in neurotransmitters such as dopamine, which have been implicated in the development of tardive dyskinesia. Compared with tetrabenazine, reserpine has a higher affinity for cells in the periphery and therefore causes side effects such as hypotension and diarrhea.7
Tetrabenazine is indicated for chorea associated with Huntington's disease and is used off-label for treating tardive dyskinesia. Tetrabenazine is thought to work by inhibiting human vesicular monoamine transporters. Blocking these transporters prevents monoamines such as dopamine from entering synaptic vesicles.8 Because of its side-effect profile, lack of large clinical trials, and high cost, tetrabenazine is used as a last-line treatment in severe cases of tardive dyskinesia. Adverse effects include somnolence (31%), insomnia (22%), depression (19%), and akathisia (19%). Tetrabenazine carries a black-box warning for depression and suicidalityand is contraindicated in patients with untreated or inadequately treated depression or who are suicidal.8 Assessing the patient's mental state is important when using this medication.
A review by Chen et al7 found that 9 of 11 studies had positive results for tetrabenazine treatment for tardive dyskinesia. Most of the studies were small and retrospective. The biggest prospective blinded study was a videotaped study by Ondo et al of 20 patients with tardive dyskinesia.9 At least 30 days before beginning the study patients discontinued the medication that caused their tardive dyskinesia and any treatments for tardive dyskinesia. Each patient was videotaped before starting tetrabenazine and an average of 20.3 weeks after starting the drug. Investigators' scores showed an average of 54.2% improvement in movement scores and participants' subjective scores reported an average of 60.4% improvement. One patient withdrew because of somnolence. The remaining 19 patients did not experience more than mild side effects and continued treatment with tetrabenazine after study completion.9
Treatment recommendations
Tardive dyskinesia is a difficult condition to treat; it is best, therefore, to prevent its onset by using the minimally effective antipsychotic dose, by preferential use of an SGA rather than a FGA, and by regular screening for tardive dyskinesia using a standardized rating scale such as the Abnormal Involuntary Movement Scale. Symptoms associated with tardive dyskinesia are more likely to resolve if caught early. If a patient develops tardive dyskinesia while taking a FGA, switching to a SGA may alleviate the symptoms.
Several medications can be used off-label to relieve symptoms, including vitamin E and tetrabenazine, which both have the most-although not considerable-literature-based support. Although some studies show benefit with tetrabenazine for tardive dyskinesia, larger clinical trials are needed to more strongly support its use. Tetrabenazine might be a good option for patients with refractory tardive dyskinesia but, given the associated risk of suicidality and depressive symptoms, careful monitoring of suicide risk is essential and may preclude its use for tardive dyskinesia in patients who are experiencing depressive symptoms.
Related Resources
• National Organization for Rare Disorders. Tardive dyskinesia. www.rarediseases.org/rare-disease-information/rare-diseases/byID/493/viewFullReport.
• Caroff SN, Miller DD, Campbell CE. Is there a rational management strategy for tardive dyskinesia? Current Psychiatry. 2011;10(10):22-32.
Drug Brand Names
Aripiprazole • Abilify Lurasidone • Latuda
Asenapine • Saphris Olanzapine • Zyprexa
Clozapine • Clozaril Quetiapine • Seroquel
Donepezil • Aricept Reserpine • Serpasil
Haloperidol • Haldol Risperidone • Risperdal
Iloperidone • Fanapt Tetrabenazine • Xenazine
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Aia PG, Revuelta GJ, Cloud LJ, et al. Tardive dyskinesia. Curr Treat Options Neurol. 2011;13(3):231-241.
2. Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414-425.
3. Crimson ML, Argo TR, Buckley PF. Schizophrenia. In: DiPiro JT, Talbert RL, Yee GC, et al, eds. Pharmacotherapy: a pathophysiologic approach. 8th ed. New York, NY: McGraw- Hill; 2011:1147-1172.
4. Margolese HC, Chouinard G, Kolivakis TT, et al. Tardive dyskinesia in the era of typical and atypical antipsychotics. Part 2: incidence and management strategies in patients with schizophrenia. Can J Psychiatry. 2005;50(11):703-714.
5. Facts & Comparisons. http://online.factsandcomparisons.com/index.aspx. Accessed November 4, 2012.
6. Natural Standard. http://www.naturalstandard.com/index.asp. Accessed November 4, 2012.
7. Chen JJ, Ondo WG, Dashtipour K, et al. Tetrabenazine for the treatment of hyperkinetic movement disorders: a review of the literature. Clin Ther. 2012;34(7):1487-1504.
8. Xanazine [package insert]. Deerfield, IL: Lundbeck Inc; 2012.
9. Ondo WG, Hanna PA, Jankovic J. Tetrabenazine treatment for tardive dyskinesia: assessment by randomized videotape protocol. Am J Psychiatry. 1999;156(8):1279-1281.
Fifteen years ago, Mr. L, age 40, was given a diagnosis of schizophrenia, which has been treated with haloperidol, 10 mg/d. Approximately 1 year ago, he began experiencing consistent lip smacking, a sign of tardive dyskinesia. Vitamin E was added to the treatment regimen, after which the tardive dyskinesia symptoms resolved.
A few months later, however, those symptoms returned and became worse. In addition to lip smacking, Mr. L now also describes involuntary bilateral twitching and muscle spasms in both legs.
Haloperidol and vitamin E are discontinued and Mr. L is switched to olanzapine, 20 mg/d. Although olanzapine is effective for Mr. L's symptoms of schizophrenia, tardive dyskinesia persists, and he gains 60 pounds and develops diabetes. Olanzapine is discontinued and he begins a trial of risperidone, 4 mg/d.
While on risperidone, blood sugar control, measured by hemoglobin A1c, and insulin resistance improve, but Mr. L continues to have symptoms of tardive dyskinesia. Vitamin E is added again, but is ineffective. The treatment team switches Mr. L to clozapine but symptoms of tardive dyskinesia do not improve.
Extrapyramidal side effects are common with first-generation antipsychotics (FGA) such as haloperidol. Types of antipsychotic-induced movement disorders include dystonias, akathisias, pseudoparkinsonism, and tardive dyskinesia. Of these, tardive dyskinesia is the most concerning because it often is difficult to treat and may be irreversible.
Tardive dyskinesia involves abnormal, involuntary movements, usually involving the face and, sometimes, the limbs. Common symptoms include lip smacking, tongue protrusions, and puffing the cheeks1; severe tardive dyskinesia may affect the larynx and diaphragm, which can be life-threatening. The incidence of tardive dyskinesia is approximately 5% after the first year of FGA treatment and 1% with second-generation antipsychotics (SGAs).2 The risk increases with higher doses and longer duration of treatment, with a prevalence of 20% to 25% with long-term FGA use.3
Treatment strategies
There are no FDA-approved drugs for tardive dyskinesia (Table).4-6 The best strategy is to prevent tardive dyskinesia with judicious use of an antipsychotic. If a patient taking a FGA develops tardive dyskinesia, the first-line treatment is to switch to a SGA. Risperidone, olanzapine, quetiapine, and clozapine have a low risk of tardive dyskinesia. Newer agents, such as lurasidone, asenapine, iloperidone, and aripiprazole, might have a lower risk of tardive dyskinesia, possibly because of differences in dopamine blockage between these agents and FGAs. Clozapine is least likely to cause tardive dyskinesia, but it often is used as a last resort because of the risk of agranulocytosis and the need for frequent tests to measurewhite blood cells.1,4
Other treatments include melatonin, donepezil, vitamin B6, and vitamin E.4 These agents can reduce symptoms, but no large clinical trials have proved that the are efficacious. Last-resort treatments include suppressive treatment using FGAs several times a day, because the constant dopamine blockade may stop symptoms for a short time; this approach is not recommended because it can exacerbate symptoms of tardive dyskinesia.4
Other suppressive treatments used in severe or refractory cases include reserpineand tetrabenazine, which are used off-label and work by blocking monoamine transporters. This blockage results in a reduction in neurotransmitters such as dopamine, which have been implicated in the development of tardive dyskinesia. Compared with tetrabenazine, reserpine has a higher affinity for cells in the periphery and therefore causes side effects such as hypotension and diarrhea.7
Tetrabenazine is indicated for chorea associated with Huntington's disease and is used off-label for treating tardive dyskinesia. Tetrabenazine is thought to work by inhibiting human vesicular monoamine transporters. Blocking these transporters prevents monoamines such as dopamine from entering synaptic vesicles.8 Because of its side-effect profile, lack of large clinical trials, and high cost, tetrabenazine is used as a last-line treatment in severe cases of tardive dyskinesia. Adverse effects include somnolence (31%), insomnia (22%), depression (19%), and akathisia (19%). Tetrabenazine carries a black-box warning for depression and suicidalityand is contraindicated in patients with untreated or inadequately treated depression or who are suicidal.8 Assessing the patient's mental state is important when using this medication.
A review by Chen et al7 found that 9 of 11 studies had positive results for tetrabenazine treatment for tardive dyskinesia. Most of the studies were small and retrospective. The biggest prospective blinded study was a videotaped study by Ondo et al of 20 patients with tardive dyskinesia.9 At least 30 days before beginning the study patients discontinued the medication that caused their tardive dyskinesia and any treatments for tardive dyskinesia. Each patient was videotaped before starting tetrabenazine and an average of 20.3 weeks after starting the drug. Investigators' scores showed an average of 54.2% improvement in movement scores and participants' subjective scores reported an average of 60.4% improvement. One patient withdrew because of somnolence. The remaining 19 patients did not experience more than mild side effects and continued treatment with tetrabenazine after study completion.9
Treatment recommendations
Tardive dyskinesia is a difficult condition to treat; it is best, therefore, to prevent its onset by using the minimally effective antipsychotic dose, by preferential use of an SGA rather than a FGA, and by regular screening for tardive dyskinesia using a standardized rating scale such as the Abnormal Involuntary Movement Scale. Symptoms associated with tardive dyskinesia are more likely to resolve if caught early. If a patient develops tardive dyskinesia while taking a FGA, switching to a SGA may alleviate the symptoms.
Several medications can be used off-label to relieve symptoms, including vitamin E and tetrabenazine, which both have the most-although not considerable-literature-based support. Although some studies show benefit with tetrabenazine for tardive dyskinesia, larger clinical trials are needed to more strongly support its use. Tetrabenazine might be a good option for patients with refractory tardive dyskinesia but, given the associated risk of suicidality and depressive symptoms, careful monitoring of suicide risk is essential and may preclude its use for tardive dyskinesia in patients who are experiencing depressive symptoms.
Related Resources
• National Organization for Rare Disorders. Tardive dyskinesia. www.rarediseases.org/rare-disease-information/rare-diseases/byID/493/viewFullReport.
• Caroff SN, Miller DD, Campbell CE. Is there a rational management strategy for tardive dyskinesia? Current Psychiatry. 2011;10(10):22-32.
Drug Brand Names
Aripiprazole • Abilify Lurasidone • Latuda
Asenapine • Saphris Olanzapine • Zyprexa
Clozapine • Clozaril Quetiapine • Seroquel
Donepezil • Aricept Reserpine • Serpasil
Haloperidol • Haldol Risperidone • Risperdal
Iloperidone • Fanapt Tetrabenazine • Xenazine
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Fifteen years ago, Mr. L, age 40, was given a diagnosis of schizophrenia, which has been treated with haloperidol, 10 mg/d. Approximately 1 year ago, he began experiencing consistent lip smacking, a sign of tardive dyskinesia. Vitamin E was added to the treatment regimen, after which the tardive dyskinesia symptoms resolved.
A few months later, however, those symptoms returned and became worse. In addition to lip smacking, Mr. L now also describes involuntary bilateral twitching and muscle spasms in both legs.
Haloperidol and vitamin E are discontinued and Mr. L is switched to olanzapine, 20 mg/d. Although olanzapine is effective for Mr. L's symptoms of schizophrenia, tardive dyskinesia persists, and he gains 60 pounds and develops diabetes. Olanzapine is discontinued and he begins a trial of risperidone, 4 mg/d.
While on risperidone, blood sugar control, measured by hemoglobin A1c, and insulin resistance improve, but Mr. L continues to have symptoms of tardive dyskinesia. Vitamin E is added again, but is ineffective. The treatment team switches Mr. L to clozapine but symptoms of tardive dyskinesia do not improve.
Extrapyramidal side effects are common with first-generation antipsychotics (FGA) such as haloperidol. Types of antipsychotic-induced movement disorders include dystonias, akathisias, pseudoparkinsonism, and tardive dyskinesia. Of these, tardive dyskinesia is the most concerning because it often is difficult to treat and may be irreversible.
Tardive dyskinesia involves abnormal, involuntary movements, usually involving the face and, sometimes, the limbs. Common symptoms include lip smacking, tongue protrusions, and puffing the cheeks1; severe tardive dyskinesia may affect the larynx and diaphragm, which can be life-threatening. The incidence of tardive dyskinesia is approximately 5% after the first year of FGA treatment and 1% with second-generation antipsychotics (SGAs).2 The risk increases with higher doses and longer duration of treatment, with a prevalence of 20% to 25% with long-term FGA use.3
Treatment strategies
There are no FDA-approved drugs for tardive dyskinesia (Table).4-6 The best strategy is to prevent tardive dyskinesia with judicious use of an antipsychotic. If a patient taking a FGA develops tardive dyskinesia, the first-line treatment is to switch to a SGA. Risperidone, olanzapine, quetiapine, and clozapine have a low risk of tardive dyskinesia. Newer agents, such as lurasidone, asenapine, iloperidone, and aripiprazole, might have a lower risk of tardive dyskinesia, possibly because of differences in dopamine blockage between these agents and FGAs. Clozapine is least likely to cause tardive dyskinesia, but it often is used as a last resort because of the risk of agranulocytosis and the need for frequent tests to measurewhite blood cells.1,4
Other treatments include melatonin, donepezil, vitamin B6, and vitamin E.4 These agents can reduce symptoms, but no large clinical trials have proved that the are efficacious. Last-resort treatments include suppressive treatment using FGAs several times a day, because the constant dopamine blockade may stop symptoms for a short time; this approach is not recommended because it can exacerbate symptoms of tardive dyskinesia.4
Other suppressive treatments used in severe or refractory cases include reserpineand tetrabenazine, which are used off-label and work by blocking monoamine transporters. This blockage results in a reduction in neurotransmitters such as dopamine, which have been implicated in the development of tardive dyskinesia. Compared with tetrabenazine, reserpine has a higher affinity for cells in the periphery and therefore causes side effects such as hypotension and diarrhea.7
Tetrabenazine is indicated for chorea associated with Huntington's disease and is used off-label for treating tardive dyskinesia. Tetrabenazine is thought to work by inhibiting human vesicular monoamine transporters. Blocking these transporters prevents monoamines such as dopamine from entering synaptic vesicles.8 Because of its side-effect profile, lack of large clinical trials, and high cost, tetrabenazine is used as a last-line treatment in severe cases of tardive dyskinesia. Adverse effects include somnolence (31%), insomnia (22%), depression (19%), and akathisia (19%). Tetrabenazine carries a black-box warning for depression and suicidalityand is contraindicated in patients with untreated or inadequately treated depression or who are suicidal.8 Assessing the patient's mental state is important when using this medication.
A review by Chen et al7 found that 9 of 11 studies had positive results for tetrabenazine treatment for tardive dyskinesia. Most of the studies were small and retrospective. The biggest prospective blinded study was a videotaped study by Ondo et al of 20 patients with tardive dyskinesia.9 At least 30 days before beginning the study patients discontinued the medication that caused their tardive dyskinesia and any treatments for tardive dyskinesia. Each patient was videotaped before starting tetrabenazine and an average of 20.3 weeks after starting the drug. Investigators' scores showed an average of 54.2% improvement in movement scores and participants' subjective scores reported an average of 60.4% improvement. One patient withdrew because of somnolence. The remaining 19 patients did not experience more than mild side effects and continued treatment with tetrabenazine after study completion.9
Treatment recommendations
Tardive dyskinesia is a difficult condition to treat; it is best, therefore, to prevent its onset by using the minimally effective antipsychotic dose, by preferential use of an SGA rather than a FGA, and by regular screening for tardive dyskinesia using a standardized rating scale such as the Abnormal Involuntary Movement Scale. Symptoms associated with tardive dyskinesia are more likely to resolve if caught early. If a patient develops tardive dyskinesia while taking a FGA, switching to a SGA may alleviate the symptoms.
Several medications can be used off-label to relieve symptoms, including vitamin E and tetrabenazine, which both have the most-although not considerable-literature-based support. Although some studies show benefit with tetrabenazine for tardive dyskinesia, larger clinical trials are needed to more strongly support its use. Tetrabenazine might be a good option for patients with refractory tardive dyskinesia but, given the associated risk of suicidality and depressive symptoms, careful monitoring of suicide risk is essential and may preclude its use for tardive dyskinesia in patients who are experiencing depressive symptoms.
Related Resources
• National Organization for Rare Disorders. Tardive dyskinesia. www.rarediseases.org/rare-disease-information/rare-diseases/byID/493/viewFullReport.
• Caroff SN, Miller DD, Campbell CE. Is there a rational management strategy for tardive dyskinesia? Current Psychiatry. 2011;10(10):22-32.
Drug Brand Names
Aripiprazole • Abilify Lurasidone • Latuda
Asenapine • Saphris Olanzapine • Zyprexa
Clozapine • Clozaril Quetiapine • Seroquel
Donepezil • Aricept Reserpine • Serpasil
Haloperidol • Haldol Risperidone • Risperdal
Iloperidone • Fanapt Tetrabenazine • Xenazine
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Aia PG, Revuelta GJ, Cloud LJ, et al. Tardive dyskinesia. Curr Treat Options Neurol. 2011;13(3):231-241.
2. Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414-425.
3. Crimson ML, Argo TR, Buckley PF. Schizophrenia. In: DiPiro JT, Talbert RL, Yee GC, et al, eds. Pharmacotherapy: a pathophysiologic approach. 8th ed. New York, NY: McGraw- Hill; 2011:1147-1172.
4. Margolese HC, Chouinard G, Kolivakis TT, et al. Tardive dyskinesia in the era of typical and atypical antipsychotics. Part 2: incidence and management strategies in patients with schizophrenia. Can J Psychiatry. 2005;50(11):703-714.
5. Facts & Comparisons. http://online.factsandcomparisons.com/index.aspx. Accessed November 4, 2012.
6. Natural Standard. http://www.naturalstandard.com/index.asp. Accessed November 4, 2012.
7. Chen JJ, Ondo WG, Dashtipour K, et al. Tetrabenazine for the treatment of hyperkinetic movement disorders: a review of the literature. Clin Ther. 2012;34(7):1487-1504.
8. Xanazine [package insert]. Deerfield, IL: Lundbeck Inc; 2012.
9. Ondo WG, Hanna PA, Jankovic J. Tetrabenazine treatment for tardive dyskinesia: assessment by randomized videotape protocol. Am J Psychiatry. 1999;156(8):1279-1281.
1. Aia PG, Revuelta GJ, Cloud LJ, et al. Tardive dyskinesia. Curr Treat Options Neurol. 2011;13(3):231-241.
2. Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414-425.
3. Crimson ML, Argo TR, Buckley PF. Schizophrenia. In: DiPiro JT, Talbert RL, Yee GC, et al, eds. Pharmacotherapy: a pathophysiologic approach. 8th ed. New York, NY: McGraw- Hill; 2011:1147-1172.
4. Margolese HC, Chouinard G, Kolivakis TT, et al. Tardive dyskinesia in the era of typical and atypical antipsychotics. Part 2: incidence and management strategies in patients with schizophrenia. Can J Psychiatry. 2005;50(11):703-714.
5. Facts & Comparisons. http://online.factsandcomparisons.com/index.aspx. Accessed November 4, 2012.
6. Natural Standard. http://www.naturalstandard.com/index.asp. Accessed November 4, 2012.
7. Chen JJ, Ondo WG, Dashtipour K, et al. Tetrabenazine for the treatment of hyperkinetic movement disorders: a review of the literature. Clin Ther. 2012;34(7):1487-1504.
8. Xanazine [package insert]. Deerfield, IL: Lundbeck Inc; 2012.
9. Ondo WG, Hanna PA, Jankovic J. Tetrabenazine treatment for tardive dyskinesia: assessment by randomized videotape protocol. Am J Psychiatry. 1999;156(8):1279-1281.
SSRIs in pregnancy: What should you tell your depressed patient?
Mrs. D is a 28-year-old married woman who became depressed after her first pregnancy. The depression was treated successfully with paroxetine, 20 mg/d. Before beginning treatment, she reported low mood, spent most of the day in bed, was unable to care for herself, and confessed to thoughts of harming her child.
Mrs. D presents to your clinic asking whether she should continue her selective serotonin reuptake inhibitor (SSRI) because she and her husband are thinking about having a second child. Recently, she tells you, she saw a news article suggesting that antidepressants show little benefit, and she is concerned that her baby might have a heart defect if she continues paroxetine.
Mrs. D wants to discontinue her medication, but her husband thought she should discuss doing so with you first. During this visit she takes a pregnancy test, which is positive. She wants to know what to do.
women experience depression; 3.8% of pregnant women receive an SSRI.1 SSRIs are the most commonly prescribed antidepressants during pregnancy, but their use remains controversial. There is disagreement about the maternal and neonatal risks of untreated depression and SSRI exposure.2-10 Media reports of studies demonstrating adverse effects associated with SSRIs may generate fear among women, possibly prompting them to self-discontinue medication.
Evidence of risks and benefits
Clinicians should be aware of possible adverse effects of SSRI use and untreated depression (Table).2-10 The available data precludes definitive associations between untreated depression and poor outcomes (Box). Studies of SSRI use during pregnancy have shown conflicting results for all potential outcomes. Absolute risk, with the exception of neonatal adaptation syndrome, is estimated to be small. Neonatal adaptation syndrome—which is characterized by jitteriness, poor muscle tone, weak cries, respiratory distress, hypoglycemia, low Apgar scores, and seizures—occurs in 15% to 30% of infants born to mothers taking SSRIs, but it is transient and resolves during the first weeks of life.


Treatment recommendations
Given the conflicting nature of the evidence, treatment plans should be individualized, weighing the risks and benefits of treatment and the patient’s beliefs and psychiatric history. Consider severity of symptoms and history, including effective therapy and history of relapse. For women with mild or moderate depression, cognitive-behavioral therapy might be an appropriate first-line therapy. However, non-pharmacotherapeutic interventions might not relieve severe depression or be available to all women. When discontinuing an SSRI before pregnancy, counsel the patient to not discontinue the medication abruptly and provide an appropriate taper schedule. See Related Resources for detailed recommendations from the American Psychiatric Association and the American College of Obstetricians and Gynecologists.
Reviewing the SSRI literature regarding pregnancy
Sertraline, paroxetine, citalopram, and fluoxetine are the most studied SSRIs during pregnancy; little information is available on escitalopram and fluvoxamine.11 Prescribing preference generally is given to the medications with the most evidence; paroxetine may be an exception. In 2005, the FDA requested a change in paroxetine’s pregnancy category from C to D, indicating that adequate studies demonstrated a risk of congenital cardiac malformations.11 Additional studies have been conducted, and the teratogenicity of paroxetine is debatable. A recent review reports 8 studies that suggest a malformation risk, compared with 15 studies that show no risk.12
The American Academy of Pediatrics considers SSRIs to be compatible with breast-feeding.13 The best-studied drugs include sertraline and paroxetine. Fluoxetine should be avoided when possible because a long elimination half-life can cause the drug to accumulate in the newborn, increasing the risk of irritability, hypertonia, sedation, and poor suckle.7
There is no best SSRI for all pregnant women. Risks and benefits, including previous treatment success and failure, should be taken into account before starting or switching therapy. Whenever possible, consider monotherapy to avoid compounding the risk of harm.
Related Resources
- Yonkers KA, Wisner KL, Stewart DE, et al. The management of depression during pregnancy: a report from the American Psychiatric Association and the American College of Obstetricians and Gynecologists. Gen Hosp Psychiatry. 2009;31:403-413.
- MGH Center for Women’s Mental Health. www.womensmentalhealth.org.
Drug Brand Names
Citalopram • Celexa Escitalopram • Lexapro Fluoxetine • Prozac
Fluvoxamine • Luvox Paroxetine • Paxil Sertraline • Zoloft
Disclosures
Dr. Leino reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products. Dr. Ellingrod receives grant support from the National Institute of Mental Health.
1. Alwan S, Reefhuis J, Rasmussen SA, et al. Patterns of antidepressant medication use among pregnant women in a United States population. J Clin Pharmacol. 2011;51(2):264-270.
2. Domar AD, Moragianni VA, Ryley DA, et al. The risks of selective serotonin reuptake inhibitor use in infertile women: a review of the impact on fertility, pregnancy, neonatal health and beyond. Hum Reprod. 20113;28(1):160-171.
3. Davalos DB, Yadon CA, Tregellas HC. Untreated prenatal maternal depression and the potential risks to offspring: a review. Arch Womens Ment Health. 2012;15(1):1-14.
4. Spinelli M. Antidepressant treatment during pregnancy. Am J Psychiatry. 2012;169(2):121-124.
5. Oyebode F, Rastogi A, Berrisford G, et al. Psychotropics in pregnancy: safety and other considerations. Pharmacol Ther. 2012;135(1):71-77.
6. Byatt N, Deligiannidis KM, Freeman MP. Antidepressant use in pregnancy: a critical review focused on risks and controversies. Acta Psychiatr Scand. 2013;127(2):94-114.
7. Sie SD, Wennink JM, van Driel JJ, et al. Maternal use of SSRIs, SNRIs and NaSSAs: practical recommendations during pregnancy and lactation. Arch Dis Child Fetal Neonatal Ed. 2012;97(6):F472-476.
8. Jimenez-Solem E, Andersen JT, Petersen M, et al. SSRI use during pregnancy and risk of stillbirth and neonatal mortality. Am J Psychiatry. 2013;170(3):299-304.
9. Nikfar S, Rahimi R, Hendoiee N, et al. Increasing the risk of spontaneous abortion and major malformations in newborns following use of serotonin reuptake inhibitors during pregnancy: a systematic review and updated meta-analysis. Daru. 2012;20(1):75.
10. Stephansson O, Kieler H, Haglund B, et al. Selective serotonin reuptake inhibitors during pregnancy and risk of stillbirth and infant mortality. JAMA. 2013;309(1):48-54.
11. U.S. Food and Drug Administration. Public health advisory: paroxetine. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/Public HealthAdvisories/ucm051731.htm. Published December 8, 2005. Accessed September 27, 2013.
12. Koren G, Nordeng H. Antidepressant use during pregnancy: the benefit-risk ratio. Am J Obstet Gynecol. 2012;207(3):157-163.
13. American Academy of Pediatrics Committee on Drugs. Transfer of drugs and other chemicals into human milk. Pediatrics. 2001;108:776-789.
Mrs. D is a 28-year-old married woman who became depressed after her first pregnancy. The depression was treated successfully with paroxetine, 20 mg/d. Before beginning treatment, she reported low mood, spent most of the day in bed, was unable to care for herself, and confessed to thoughts of harming her child.
Mrs. D presents to your clinic asking whether she should continue her selective serotonin reuptake inhibitor (SSRI) because she and her husband are thinking about having a second child. Recently, she tells you, she saw a news article suggesting that antidepressants show little benefit, and she is concerned that her baby might have a heart defect if she continues paroxetine.
Mrs. D wants to discontinue her medication, but her husband thought she should discuss doing so with you first. During this visit she takes a pregnancy test, which is positive. She wants to know what to do.
women experience depression; 3.8% of pregnant women receive an SSRI.1 SSRIs are the most commonly prescribed antidepressants during pregnancy, but their use remains controversial. There is disagreement about the maternal and neonatal risks of untreated depression and SSRI exposure.2-10 Media reports of studies demonstrating adverse effects associated with SSRIs may generate fear among women, possibly prompting them to self-discontinue medication.
Evidence of risks and benefits
Clinicians should be aware of possible adverse effects of SSRI use and untreated depression (Table).2-10 The available data precludes definitive associations between untreated depression and poor outcomes (Box). Studies of SSRI use during pregnancy have shown conflicting results for all potential outcomes. Absolute risk, with the exception of neonatal adaptation syndrome, is estimated to be small. Neonatal adaptation syndrome—which is characterized by jitteriness, poor muscle tone, weak cries, respiratory distress, hypoglycemia, low Apgar scores, and seizures—occurs in 15% to 30% of infants born to mothers taking SSRIs, but it is transient and resolves during the first weeks of life.
Treatment recommendations
Given the conflicting nature of the evidence, treatment plans should be individualized, weighing the risks and benefits of treatment and the patient’s beliefs and psychiatric history. Consider severity of symptoms and history, including effective therapy and history of relapse. For women with mild or moderate depression, cognitive-behavioral therapy might be an appropriate first-line therapy. However, non-pharmacotherapeutic interventions might not relieve severe depression or be available to all women. When discontinuing an SSRI before pregnancy, counsel the patient to not discontinue the medication abruptly and provide an appropriate taper schedule. See Related Resources for detailed recommendations from the American Psychiatric Association and the American College of Obstetricians and Gynecologists.
Reviewing the SSRI literature regarding pregnancy
Sertraline, paroxetine, citalopram, and fluoxetine are the most studied SSRIs during pregnancy; little information is available on escitalopram and fluvoxamine.11 Prescribing preference generally is given to the medications with the most evidence; paroxetine may be an exception. In 2005, the FDA requested a change in paroxetine’s pregnancy category from C to D, indicating that adequate studies demonstrated a risk of congenital cardiac malformations.11 Additional studies have been conducted, and the teratogenicity of paroxetine is debatable. A recent review reports 8 studies that suggest a malformation risk, compared with 15 studies that show no risk.12
The American Academy of Pediatrics considers SSRIs to be compatible with breast-feeding.13 The best-studied drugs include sertraline and paroxetine. Fluoxetine should be avoided when possible because a long elimination half-life can cause the drug to accumulate in the newborn, increasing the risk of irritability, hypertonia, sedation, and poor suckle.7
There is no best SSRI for all pregnant women. Risks and benefits, including previous treatment success and failure, should be taken into account before starting or switching therapy. Whenever possible, consider monotherapy to avoid compounding the risk of harm.
Related Resources
- Yonkers KA, Wisner KL, Stewart DE, et al. The management of depression during pregnancy: a report from the American Psychiatric Association and the American College of Obstetricians and Gynecologists. Gen Hosp Psychiatry. 2009;31:403-413.
- MGH Center for Women’s Mental Health. www.womensmentalhealth.org.
Drug Brand Names
Citalopram • Celexa Escitalopram • Lexapro Fluoxetine • Prozac
Fluvoxamine • Luvox Paroxetine • Paxil Sertraline • Zoloft
Disclosures
Dr. Leino reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products. Dr. Ellingrod receives grant support from the National Institute of Mental Health.
Mrs. D is a 28-year-old married woman who became depressed after her first pregnancy. The depression was treated successfully with paroxetine, 20 mg/d. Before beginning treatment, she reported low mood, spent most of the day in bed, was unable to care for herself, and confessed to thoughts of harming her child.
Mrs. D presents to your clinic asking whether she should continue her selective serotonin reuptake inhibitor (SSRI) because she and her husband are thinking about having a second child. Recently, she tells you, she saw a news article suggesting that antidepressants show little benefit, and she is concerned that her baby might have a heart defect if she continues paroxetine.
Mrs. D wants to discontinue her medication, but her husband thought she should discuss doing so with you first. During this visit she takes a pregnancy test, which is positive. She wants to know what to do.
women experience depression; 3.8% of pregnant women receive an SSRI.1 SSRIs are the most commonly prescribed antidepressants during pregnancy, but their use remains controversial. There is disagreement about the maternal and neonatal risks of untreated depression and SSRI exposure.2-10 Media reports of studies demonstrating adverse effects associated with SSRIs may generate fear among women, possibly prompting them to self-discontinue medication.
Evidence of risks and benefits
Clinicians should be aware of possible adverse effects of SSRI use and untreated depression (Table).2-10 The available data precludes definitive associations between untreated depression and poor outcomes (Box). Studies of SSRI use during pregnancy have shown conflicting results for all potential outcomes. Absolute risk, with the exception of neonatal adaptation syndrome, is estimated to be small. Neonatal adaptation syndrome—which is characterized by jitteriness, poor muscle tone, weak cries, respiratory distress, hypoglycemia, low Apgar scores, and seizures—occurs in 15% to 30% of infants born to mothers taking SSRIs, but it is transient and resolves during the first weeks of life.
Treatment recommendations
Given the conflicting nature of the evidence, treatment plans should be individualized, weighing the risks and benefits of treatment and the patient’s beliefs and psychiatric history. Consider severity of symptoms and history, including effective therapy and history of relapse. For women with mild or moderate depression, cognitive-behavioral therapy might be an appropriate first-line therapy. However, non-pharmacotherapeutic interventions might not relieve severe depression or be available to all women. When discontinuing an SSRI before pregnancy, counsel the patient to not discontinue the medication abruptly and provide an appropriate taper schedule. See Related Resources for detailed recommendations from the American Psychiatric Association and the American College of Obstetricians and Gynecologists.
Reviewing the SSRI literature regarding pregnancy
Sertraline, paroxetine, citalopram, and fluoxetine are the most studied SSRIs during pregnancy; little information is available on escitalopram and fluvoxamine.11 Prescribing preference generally is given to the medications with the most evidence; paroxetine may be an exception. In 2005, the FDA requested a change in paroxetine’s pregnancy category from C to D, indicating that adequate studies demonstrated a risk of congenital cardiac malformations.11 Additional studies have been conducted, and the teratogenicity of paroxetine is debatable. A recent review reports 8 studies that suggest a malformation risk, compared with 15 studies that show no risk.12
The American Academy of Pediatrics considers SSRIs to be compatible with breast-feeding.13 The best-studied drugs include sertraline and paroxetine. Fluoxetine should be avoided when possible because a long elimination half-life can cause the drug to accumulate in the newborn, increasing the risk of irritability, hypertonia, sedation, and poor suckle.7
There is no best SSRI for all pregnant women. Risks and benefits, including previous treatment success and failure, should be taken into account before starting or switching therapy. Whenever possible, consider monotherapy to avoid compounding the risk of harm.
Related Resources
- Yonkers KA, Wisner KL, Stewart DE, et al. The management of depression during pregnancy: a report from the American Psychiatric Association and the American College of Obstetricians and Gynecologists. Gen Hosp Psychiatry. 2009;31:403-413.
- MGH Center for Women’s Mental Health. www.womensmentalhealth.org.
Drug Brand Names
Citalopram • Celexa Escitalopram • Lexapro Fluoxetine • Prozac
Fluvoxamine • Luvox Paroxetine • Paxil Sertraline • Zoloft
Disclosures
Dr. Leino reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products. Dr. Ellingrod receives grant support from the National Institute of Mental Health.
1. Alwan S, Reefhuis J, Rasmussen SA, et al. Patterns of antidepressant medication use among pregnant women in a United States population. J Clin Pharmacol. 2011;51(2):264-270.
2. Domar AD, Moragianni VA, Ryley DA, et al. The risks of selective serotonin reuptake inhibitor use in infertile women: a review of the impact on fertility, pregnancy, neonatal health and beyond. Hum Reprod. 20113;28(1):160-171.
3. Davalos DB, Yadon CA, Tregellas HC. Untreated prenatal maternal depression and the potential risks to offspring: a review. Arch Womens Ment Health. 2012;15(1):1-14.
4. Spinelli M. Antidepressant treatment during pregnancy. Am J Psychiatry. 2012;169(2):121-124.
5. Oyebode F, Rastogi A, Berrisford G, et al. Psychotropics in pregnancy: safety and other considerations. Pharmacol Ther. 2012;135(1):71-77.
6. Byatt N, Deligiannidis KM, Freeman MP. Antidepressant use in pregnancy: a critical review focused on risks and controversies. Acta Psychiatr Scand. 2013;127(2):94-114.
7. Sie SD, Wennink JM, van Driel JJ, et al. Maternal use of SSRIs, SNRIs and NaSSAs: practical recommendations during pregnancy and lactation. Arch Dis Child Fetal Neonatal Ed. 2012;97(6):F472-476.
8. Jimenez-Solem E, Andersen JT, Petersen M, et al. SSRI use during pregnancy and risk of stillbirth and neonatal mortality. Am J Psychiatry. 2013;170(3):299-304.
9. Nikfar S, Rahimi R, Hendoiee N, et al. Increasing the risk of spontaneous abortion and major malformations in newborns following use of serotonin reuptake inhibitors during pregnancy: a systematic review and updated meta-analysis. Daru. 2012;20(1):75.
10. Stephansson O, Kieler H, Haglund B, et al. Selective serotonin reuptake inhibitors during pregnancy and risk of stillbirth and infant mortality. JAMA. 2013;309(1):48-54.
11. U.S. Food and Drug Administration. Public health advisory: paroxetine. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/Public HealthAdvisories/ucm051731.htm. Published December 8, 2005. Accessed September 27, 2013.
12. Koren G, Nordeng H. Antidepressant use during pregnancy: the benefit-risk ratio. Am J Obstet Gynecol. 2012;207(3):157-163.
13. American Academy of Pediatrics Committee on Drugs. Transfer of drugs and other chemicals into human milk. Pediatrics. 2001;108:776-789.
1. Alwan S, Reefhuis J, Rasmussen SA, et al. Patterns of antidepressant medication use among pregnant women in a United States population. J Clin Pharmacol. 2011;51(2):264-270.
2. Domar AD, Moragianni VA, Ryley DA, et al. The risks of selective serotonin reuptake inhibitor use in infertile women: a review of the impact on fertility, pregnancy, neonatal health and beyond. Hum Reprod. 20113;28(1):160-171.
3. Davalos DB, Yadon CA, Tregellas HC. Untreated prenatal maternal depression and the potential risks to offspring: a review. Arch Womens Ment Health. 2012;15(1):1-14.
4. Spinelli M. Antidepressant treatment during pregnancy. Am J Psychiatry. 2012;169(2):121-124.
5. Oyebode F, Rastogi A, Berrisford G, et al. Psychotropics in pregnancy: safety and other considerations. Pharmacol Ther. 2012;135(1):71-77.
6. Byatt N, Deligiannidis KM, Freeman MP. Antidepressant use in pregnancy: a critical review focused on risks and controversies. Acta Psychiatr Scand. 2013;127(2):94-114.
7. Sie SD, Wennink JM, van Driel JJ, et al. Maternal use of SSRIs, SNRIs and NaSSAs: practical recommendations during pregnancy and lactation. Arch Dis Child Fetal Neonatal Ed. 2012;97(6):F472-476.
8. Jimenez-Solem E, Andersen JT, Petersen M, et al. SSRI use during pregnancy and risk of stillbirth and neonatal mortality. Am J Psychiatry. 2013;170(3):299-304.
9. Nikfar S, Rahimi R, Hendoiee N, et al. Increasing the risk of spontaneous abortion and major malformations in newborns following use of serotonin reuptake inhibitors during pregnancy: a systematic review and updated meta-analysis. Daru. 2012;20(1):75.
10. Stephansson O, Kieler H, Haglund B, et al. Selective serotonin reuptake inhibitors during pregnancy and risk of stillbirth and infant mortality. JAMA. 2013;309(1):48-54.
11. U.S. Food and Drug Administration. Public health advisory: paroxetine. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/Public HealthAdvisories/ucm051731.htm. Published December 8, 2005. Accessed September 27, 2013.
12. Koren G, Nordeng H. Antidepressant use during pregnancy: the benefit-risk ratio. Am J Obstet Gynecol. 2012;207(3):157-163.
13. American Academy of Pediatrics Committee on Drugs. Transfer of drugs and other chemicals into human milk. Pediatrics. 2001;108:776-789.
Lithium-induced diabetes insipidus: Prevention and management
Mr. H, age 33, was diagnosed with bipolar I disorder 9 years ago. For the past year, his mood symptoms have been well controlled with lithium 300 mg, 3 times a day, and olanzapine, 20 mg/d. He presents to the outpatient clinic for a routine visit complaining of insomnia, daytime sleepiness, and increased thirst. He also notes that his tremor has become more prominent over the last few weeks. Concerned about his symptoms, Mr. H’s clinician orders a comprehensive laboratory panel (Table).
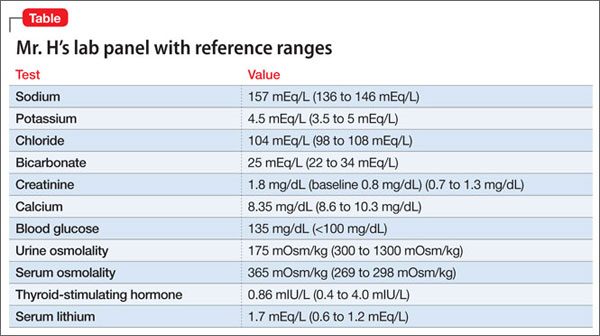
Upon further questioning, Mr. H’s physician determines that his insomnia is caused by nocturnal urination, which is consistent with fluid and electrolyte imbalances seen in Mr. H’s laboratory panel. Mr. H is diagnosed with lithium-induced diabetes insipidus.
Although lithium’s exact mechanism of action is unknown, it is known that lithium can negatively affect the kidneys.1,2 Typically, antidiuretic hormone (ADH) regulates water permeability in the collecting duct of the nephron, allowing water to be reabsorbed through simple diffusion in the kidney’s collecting duct (Figure).3 Chronic lithium use reduces or desensitizes the kidney’s ability to respond to ADH. Resistance to ADH occurs when lithium accumulates in the cells of the collecting duct and inhibits ADH’s ability to increase water permeability. This inhibition can cause some of Mr. H’s symptoms, such as polydipsia and polyuria, and is estimated to occur in approximately 40% of patients receiving long-term lithium therapy.4,5
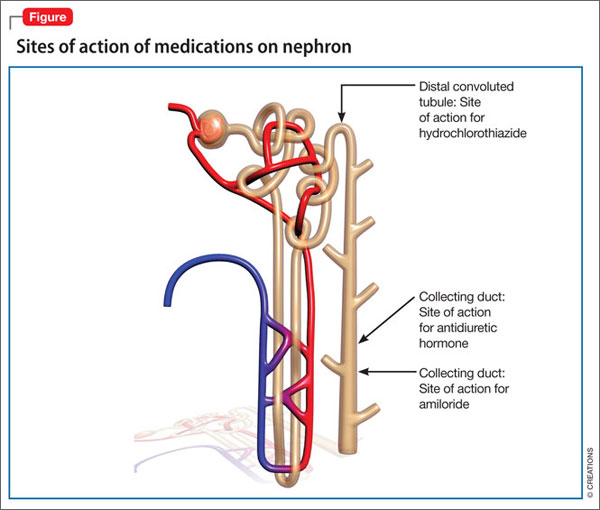
Diagnosis
Diagnosing lithium-induced nephrogenic diabetes insipidus (NDI) begins with a history of the patient’s symptoms and ordering lab tests.5 The next step involves a water restriction test, also known as a thirst test, to measure the patient’s ability to concentrate his or her urine. Baseline serum osmolality and electrolytes are compared with new values obtained after completing the water restriction test. Healthy people will have a 2-to-4-fold increase in urine osmolality compared with patients who have NDI. The last step includes administering desmopressin and differentiates between central diabetes insipidus and NDI.6
After desmopressin use, patients who have central diabetes insipidus will have a >50% increase in urine osmolality, whereas patients who have NDI will have <10% increase in urine osmolality. This distinction is important because patients with central diabetes insipidus might have more severe disease and might not benefit from measures commonly used for lithium-induced NDI.7
Prevention and management
Lithium-induced NDI is thought to be dose-dependent and may be prevented by using the lowest effective dose of lithium for an individual patient. It is important that patients taking lithium receive basic electrolyte, hematologic, liver function, renal function, and thyroid function tests at baseline and every 6 to 12 months after the lithium regimen is stable. Additionally, lithium levels should be monitored frequently. The frequency of these tests may range from twice weekly to every 3 to 4 months or longer, depending on the patient’s condition. This monitoring allows the prescriber to quickly identify emerging adverse effects.
Patients with impaired renal function and those with a urine output >3 liters a day are more susceptible to NDI and require monitoring every 3 months. Also, instruct patients to monitor their urine output and educate them about the dangers of fluid and electrolyte imbalances and the signs and symptoms of NDI, such as excessive thirst and urination.1,2
When a patient experiences lithium-induced NDI, re-evaluate treatment and dosage, including simplifying the dosing regimen or switching to once-daily dosing, usually at bedtime. Once-daily dosing results in a lower overall lithium trough, which might allow the kidneys more “drug-free” time.4,5 Additionally, 12-hour lithium levels are approximately 20% higher with once-daily monitoring; continued monitoring is needed during this switch. Patients who have a moderate or severe form of lithium-induced NDI may need to discontinue lithium altogether. There are several options for treating lithium-induced NDI in patients who need to take lithium. Closely monitor kidney function and lithium routinely with these strategies.
Amiloride. This potassium-sparing diuretic minimizes accumulation of lithium by inhibiting collecting duct sodium channels. Studies have shown that amiloride can decrease mean urine volume, increase urine osmolality, and improve the kidneys’ ability to respond to exogenous arginine vasopressin.8
Thiazide diuretics produce mild sodium depletion, which decreases the distal tubule delivery of sodium, therefore increasing water reabsorption in the collecting duct. Hydrochlorothiazide has been shown to reduce urine output by >50% in patients with NDI on a sodium-restricted diet. Hydrochlorothiazide use requires careful monitoring of potassium and lithium levels. Use of a thiazide diuretic also might warrant decreasing the lithium dose by as much as 50% to prevent toxicity.9,10
Low-sodium diet plus hydrochlorothiazide. This route provides another option to decrease urine output during lithium-induced NDI. A reduction in urine output has been shown to be directly proportional to a decrease in salt intake and excretion. Restricting sodium to <2.3 g/d is an appropriate goal for many patients to prevent reoccurring symptoms, which is more than the 3 g/d average that most Americans consume. Potassium and lithium levels must be monitored closely.9
Nonsteroidal anti-inflammatory drugs (NSAIDs). These drugs’ ability to inhibit prostaglandin synthesis prevents prostaglandins from antagonizing actions of ADH in the kidney. The result is increased urine concentration via the actions of ADH. Indomethacin has a greater effect than ibuprofen in increasing ADH’s actions on the kidney. Use of concomitant NSAIDs with lithium requires close monitoring of renal function tests.11
1. Ecelbarger CA. Lithium treatment and remodeling of the collecting duct. Am J Physiol Renal Physiol. 2006;291(1):F37-38.
2. Christensen BM, Kim YH, Kwon TH, et al. Lithium treatment induces a marked proliferation of primarily principal cells in rat kidney inner medullary collecting duct. Am J Physiol Renal Physiol. 2006;291(1):F39-48.
3. Francis SG, Gardner DG. Basic and clinical endocrinology. 7th ed. New York, NY: McGraw Hill; 2003:154-158.
4. Stone KA. Lithium-induced nephrogenic diabetes insipidus. J Am Board Fam Pract. 1999;12(1):43-47.
5. Grünfeld JP, Rossier BC. Lithium nephrotoxicity revisited. Nat Rev Nephrol. 2009;5(5):270-276.
6. Wesche D, Deen PM, Knoers NV. Congenital nephrogenic diabetes insipidus: the current state of affairs. Pediatr Nephrol. 2012;27(12):2183-2204.
7. Rose BD, Post TW. Clinical physiology of acid-base and electrolyte disorders. 5th ed. New York, NY: McGraw-Hill; 2001:754-759,782-783.
8. Batlle DC, von Riotte AB, Gaviria M, et al. Amelioration of polyuria by amiloride in patients receiving long-term lithium therapy. N Engl J Med. 1985;312(7):408-414.
9. Earley LE, Orloff J. The mechanism of antidiuresis associated with the administration of hydrochlorothiazide to patients with vasopressin-resistant diabetes insipidus. J Clin Invest. 1962;41(11):1988-1997.
10. Kim GH, Lee JW, Oh YK, et al. Antidiuretic effect of hydrochlorothiazide in lithium-induced nephrogenic diabetes insipidus is associated with upregulation of aquaporin-2, Na-Cl co-transporter, and epithelial sodium channel. J Am Soc Nephrol. 2004;15(11):2836-2843.
11. Libber S, Harrison H, Spector D. Treatment of nephrogenic diabetes insipidus with prostaglandin synthesis inhibitors. J Pediatr. 1986;108(2):305-311.
Mr. H, age 33, was diagnosed with bipolar I disorder 9 years ago. For the past year, his mood symptoms have been well controlled with lithium 300 mg, 3 times a day, and olanzapine, 20 mg/d. He presents to the outpatient clinic for a routine visit complaining of insomnia, daytime sleepiness, and increased thirst. He also notes that his tremor has become more prominent over the last few weeks. Concerned about his symptoms, Mr. H’s clinician orders a comprehensive laboratory panel (Table).
Upon further questioning, Mr. H’s physician determines that his insomnia is caused by nocturnal urination, which is consistent with fluid and electrolyte imbalances seen in Mr. H’s laboratory panel. Mr. H is diagnosed with lithium-induced diabetes insipidus.
Although lithium’s exact mechanism of action is unknown, it is known that lithium can negatively affect the kidneys.1,2 Typically, antidiuretic hormone (ADH) regulates water permeability in the collecting duct of the nephron, allowing water to be reabsorbed through simple diffusion in the kidney’s collecting duct (Figure).3 Chronic lithium use reduces or desensitizes the kidney’s ability to respond to ADH. Resistance to ADH occurs when lithium accumulates in the cells of the collecting duct and inhibits ADH’s ability to increase water permeability. This inhibition can cause some of Mr. H’s symptoms, such as polydipsia and polyuria, and is estimated to occur in approximately 40% of patients receiving long-term lithium therapy.4,5
Diagnosis
Diagnosing lithium-induced nephrogenic diabetes insipidus (NDI) begins with a history of the patient’s symptoms and ordering lab tests.5 The next step involves a water restriction test, also known as a thirst test, to measure the patient’s ability to concentrate his or her urine. Baseline serum osmolality and electrolytes are compared with new values obtained after completing the water restriction test. Healthy people will have a 2-to-4-fold increase in urine osmolality compared with patients who have NDI. The last step includes administering desmopressin and differentiates between central diabetes insipidus and NDI.6
After desmopressin use, patients who have central diabetes insipidus will have a >50% increase in urine osmolality, whereas patients who have NDI will have <10% increase in urine osmolality. This distinction is important because patients with central diabetes insipidus might have more severe disease and might not benefit from measures commonly used for lithium-induced NDI.7
Prevention and management
Lithium-induced NDI is thought to be dose-dependent and may be prevented by using the lowest effective dose of lithium for an individual patient. It is important that patients taking lithium receive basic electrolyte, hematologic, liver function, renal function, and thyroid function tests at baseline and every 6 to 12 months after the lithium regimen is stable. Additionally, lithium levels should be monitored frequently. The frequency of these tests may range from twice weekly to every 3 to 4 months or longer, depending on the patient’s condition. This monitoring allows the prescriber to quickly identify emerging adverse effects.
Patients with impaired renal function and those with a urine output >3 liters a day are more susceptible to NDI and require monitoring every 3 months. Also, instruct patients to monitor their urine output and educate them about the dangers of fluid and electrolyte imbalances and the signs and symptoms of NDI, such as excessive thirst and urination.1,2
When a patient experiences lithium-induced NDI, re-evaluate treatment and dosage, including simplifying the dosing regimen or switching to once-daily dosing, usually at bedtime. Once-daily dosing results in a lower overall lithium trough, which might allow the kidneys more “drug-free” time.4,5 Additionally, 12-hour lithium levels are approximately 20% higher with once-daily monitoring; continued monitoring is needed during this switch. Patients who have a moderate or severe form of lithium-induced NDI may need to discontinue lithium altogether. There are several options for treating lithium-induced NDI in patients who need to take lithium. Closely monitor kidney function and lithium routinely with these strategies.
Amiloride. This potassium-sparing diuretic minimizes accumulation of lithium by inhibiting collecting duct sodium channels. Studies have shown that amiloride can decrease mean urine volume, increase urine osmolality, and improve the kidneys’ ability to respond to exogenous arginine vasopressin.8
Thiazide diuretics produce mild sodium depletion, which decreases the distal tubule delivery of sodium, therefore increasing water reabsorption in the collecting duct. Hydrochlorothiazide has been shown to reduce urine output by >50% in patients with NDI on a sodium-restricted diet. Hydrochlorothiazide use requires careful monitoring of potassium and lithium levels. Use of a thiazide diuretic also might warrant decreasing the lithium dose by as much as 50% to prevent toxicity.9,10
Low-sodium diet plus hydrochlorothiazide. This route provides another option to decrease urine output during lithium-induced NDI. A reduction in urine output has been shown to be directly proportional to a decrease in salt intake and excretion. Restricting sodium to <2.3 g/d is an appropriate goal for many patients to prevent reoccurring symptoms, which is more than the 3 g/d average that most Americans consume. Potassium and lithium levels must be monitored closely.9
Nonsteroidal anti-inflammatory drugs (NSAIDs). These drugs’ ability to inhibit prostaglandin synthesis prevents prostaglandins from antagonizing actions of ADH in the kidney. The result is increased urine concentration via the actions of ADH. Indomethacin has a greater effect than ibuprofen in increasing ADH’s actions on the kidney. Use of concomitant NSAIDs with lithium requires close monitoring of renal function tests.11
Mr. H, age 33, was diagnosed with bipolar I disorder 9 years ago. For the past year, his mood symptoms have been well controlled with lithium 300 mg, 3 times a day, and olanzapine, 20 mg/d. He presents to the outpatient clinic for a routine visit complaining of insomnia, daytime sleepiness, and increased thirst. He also notes that his tremor has become more prominent over the last few weeks. Concerned about his symptoms, Mr. H’s clinician orders a comprehensive laboratory panel (Table).
Upon further questioning, Mr. H’s physician determines that his insomnia is caused by nocturnal urination, which is consistent with fluid and electrolyte imbalances seen in Mr. H’s laboratory panel. Mr. H is diagnosed with lithium-induced diabetes insipidus.
Although lithium’s exact mechanism of action is unknown, it is known that lithium can negatively affect the kidneys.1,2 Typically, antidiuretic hormone (ADH) regulates water permeability in the collecting duct of the nephron, allowing water to be reabsorbed through simple diffusion in the kidney’s collecting duct (Figure).3 Chronic lithium use reduces or desensitizes the kidney’s ability to respond to ADH. Resistance to ADH occurs when lithium accumulates in the cells of the collecting duct and inhibits ADH’s ability to increase water permeability. This inhibition can cause some of Mr. H’s symptoms, such as polydipsia and polyuria, and is estimated to occur in approximately 40% of patients receiving long-term lithium therapy.4,5
Diagnosis
Diagnosing lithium-induced nephrogenic diabetes insipidus (NDI) begins with a history of the patient’s symptoms and ordering lab tests.5 The next step involves a water restriction test, also known as a thirst test, to measure the patient’s ability to concentrate his or her urine. Baseline serum osmolality and electrolytes are compared with new values obtained after completing the water restriction test. Healthy people will have a 2-to-4-fold increase in urine osmolality compared with patients who have NDI. The last step includes administering desmopressin and differentiates between central diabetes insipidus and NDI.6
After desmopressin use, patients who have central diabetes insipidus will have a >50% increase in urine osmolality, whereas patients who have NDI will have <10% increase in urine osmolality. This distinction is important because patients with central diabetes insipidus might have more severe disease and might not benefit from measures commonly used for lithium-induced NDI.7
Prevention and management
Lithium-induced NDI is thought to be dose-dependent and may be prevented by using the lowest effective dose of lithium for an individual patient. It is important that patients taking lithium receive basic electrolyte, hematologic, liver function, renal function, and thyroid function tests at baseline and every 6 to 12 months after the lithium regimen is stable. Additionally, lithium levels should be monitored frequently. The frequency of these tests may range from twice weekly to every 3 to 4 months or longer, depending on the patient’s condition. This monitoring allows the prescriber to quickly identify emerging adverse effects.
Patients with impaired renal function and those with a urine output >3 liters a day are more susceptible to NDI and require monitoring every 3 months. Also, instruct patients to monitor their urine output and educate them about the dangers of fluid and electrolyte imbalances and the signs and symptoms of NDI, such as excessive thirst and urination.1,2
When a patient experiences lithium-induced NDI, re-evaluate treatment and dosage, including simplifying the dosing regimen or switching to once-daily dosing, usually at bedtime. Once-daily dosing results in a lower overall lithium trough, which might allow the kidneys more “drug-free” time.4,5 Additionally, 12-hour lithium levels are approximately 20% higher with once-daily monitoring; continued monitoring is needed during this switch. Patients who have a moderate or severe form of lithium-induced NDI may need to discontinue lithium altogether. There are several options for treating lithium-induced NDI in patients who need to take lithium. Closely monitor kidney function and lithium routinely with these strategies.
Amiloride. This potassium-sparing diuretic minimizes accumulation of lithium by inhibiting collecting duct sodium channels. Studies have shown that amiloride can decrease mean urine volume, increase urine osmolality, and improve the kidneys’ ability to respond to exogenous arginine vasopressin.8
Thiazide diuretics produce mild sodium depletion, which decreases the distal tubule delivery of sodium, therefore increasing water reabsorption in the collecting duct. Hydrochlorothiazide has been shown to reduce urine output by >50% in patients with NDI on a sodium-restricted diet. Hydrochlorothiazide use requires careful monitoring of potassium and lithium levels. Use of a thiazide diuretic also might warrant decreasing the lithium dose by as much as 50% to prevent toxicity.9,10
Low-sodium diet plus hydrochlorothiazide. This route provides another option to decrease urine output during lithium-induced NDI. A reduction in urine output has been shown to be directly proportional to a decrease in salt intake and excretion. Restricting sodium to <2.3 g/d is an appropriate goal for many patients to prevent reoccurring symptoms, which is more than the 3 g/d average that most Americans consume. Potassium and lithium levels must be monitored closely.9
Nonsteroidal anti-inflammatory drugs (NSAIDs). These drugs’ ability to inhibit prostaglandin synthesis prevents prostaglandins from antagonizing actions of ADH in the kidney. The result is increased urine concentration via the actions of ADH. Indomethacin has a greater effect than ibuprofen in increasing ADH’s actions on the kidney. Use of concomitant NSAIDs with lithium requires close monitoring of renal function tests.11
1. Ecelbarger CA. Lithium treatment and remodeling of the collecting duct. Am J Physiol Renal Physiol. 2006;291(1):F37-38.
2. Christensen BM, Kim YH, Kwon TH, et al. Lithium treatment induces a marked proliferation of primarily principal cells in rat kidney inner medullary collecting duct. Am J Physiol Renal Physiol. 2006;291(1):F39-48.
3. Francis SG, Gardner DG. Basic and clinical endocrinology. 7th ed. New York, NY: McGraw Hill; 2003:154-158.
4. Stone KA. Lithium-induced nephrogenic diabetes insipidus. J Am Board Fam Pract. 1999;12(1):43-47.
5. Grünfeld JP, Rossier BC. Lithium nephrotoxicity revisited. Nat Rev Nephrol. 2009;5(5):270-276.
6. Wesche D, Deen PM, Knoers NV. Congenital nephrogenic diabetes insipidus: the current state of affairs. Pediatr Nephrol. 2012;27(12):2183-2204.
7. Rose BD, Post TW. Clinical physiology of acid-base and electrolyte disorders. 5th ed. New York, NY: McGraw-Hill; 2001:754-759,782-783.
8. Batlle DC, von Riotte AB, Gaviria M, et al. Amelioration of polyuria by amiloride in patients receiving long-term lithium therapy. N Engl J Med. 1985;312(7):408-414.
9. Earley LE, Orloff J. The mechanism of antidiuresis associated with the administration of hydrochlorothiazide to patients with vasopressin-resistant diabetes insipidus. J Clin Invest. 1962;41(11):1988-1997.
10. Kim GH, Lee JW, Oh YK, et al. Antidiuretic effect of hydrochlorothiazide in lithium-induced nephrogenic diabetes insipidus is associated with upregulation of aquaporin-2, Na-Cl co-transporter, and epithelial sodium channel. J Am Soc Nephrol. 2004;15(11):2836-2843.
11. Libber S, Harrison H, Spector D. Treatment of nephrogenic diabetes insipidus with prostaglandin synthesis inhibitors. J Pediatr. 1986;108(2):305-311.
1. Ecelbarger CA. Lithium treatment and remodeling of the collecting duct. Am J Physiol Renal Physiol. 2006;291(1):F37-38.
2. Christensen BM, Kim YH, Kwon TH, et al. Lithium treatment induces a marked proliferation of primarily principal cells in rat kidney inner medullary collecting duct. Am J Physiol Renal Physiol. 2006;291(1):F39-48.
3. Francis SG, Gardner DG. Basic and clinical endocrinology. 7th ed. New York, NY: McGraw Hill; 2003:154-158.
4. Stone KA. Lithium-induced nephrogenic diabetes insipidus. J Am Board Fam Pract. 1999;12(1):43-47.
5. Grünfeld JP, Rossier BC. Lithium nephrotoxicity revisited. Nat Rev Nephrol. 2009;5(5):270-276.
6. Wesche D, Deen PM, Knoers NV. Congenital nephrogenic diabetes insipidus: the current state of affairs. Pediatr Nephrol. 2012;27(12):2183-2204.
7. Rose BD, Post TW. Clinical physiology of acid-base and electrolyte disorders. 5th ed. New York, NY: McGraw-Hill; 2001:754-759,782-783.
8. Batlle DC, von Riotte AB, Gaviria M, et al. Amelioration of polyuria by amiloride in patients receiving long-term lithium therapy. N Engl J Med. 1985;312(7):408-414.
9. Earley LE, Orloff J. The mechanism of antidiuresis associated with the administration of hydrochlorothiazide to patients with vasopressin-resistant diabetes insipidus. J Clin Invest. 1962;41(11):1988-1997.
10. Kim GH, Lee JW, Oh YK, et al. Antidiuretic effect of hydrochlorothiazide in lithium-induced nephrogenic diabetes insipidus is associated with upregulation of aquaporin-2, Na-Cl co-transporter, and epithelial sodium channel. J Am Soc Nephrol. 2004;15(11):2836-2843.
11. Libber S, Harrison H, Spector D. Treatment of nephrogenic diabetes insipidus with prostaglandin synthesis inhibitors. J Pediatr. 1986;108(2):305-311.