To the Editor:
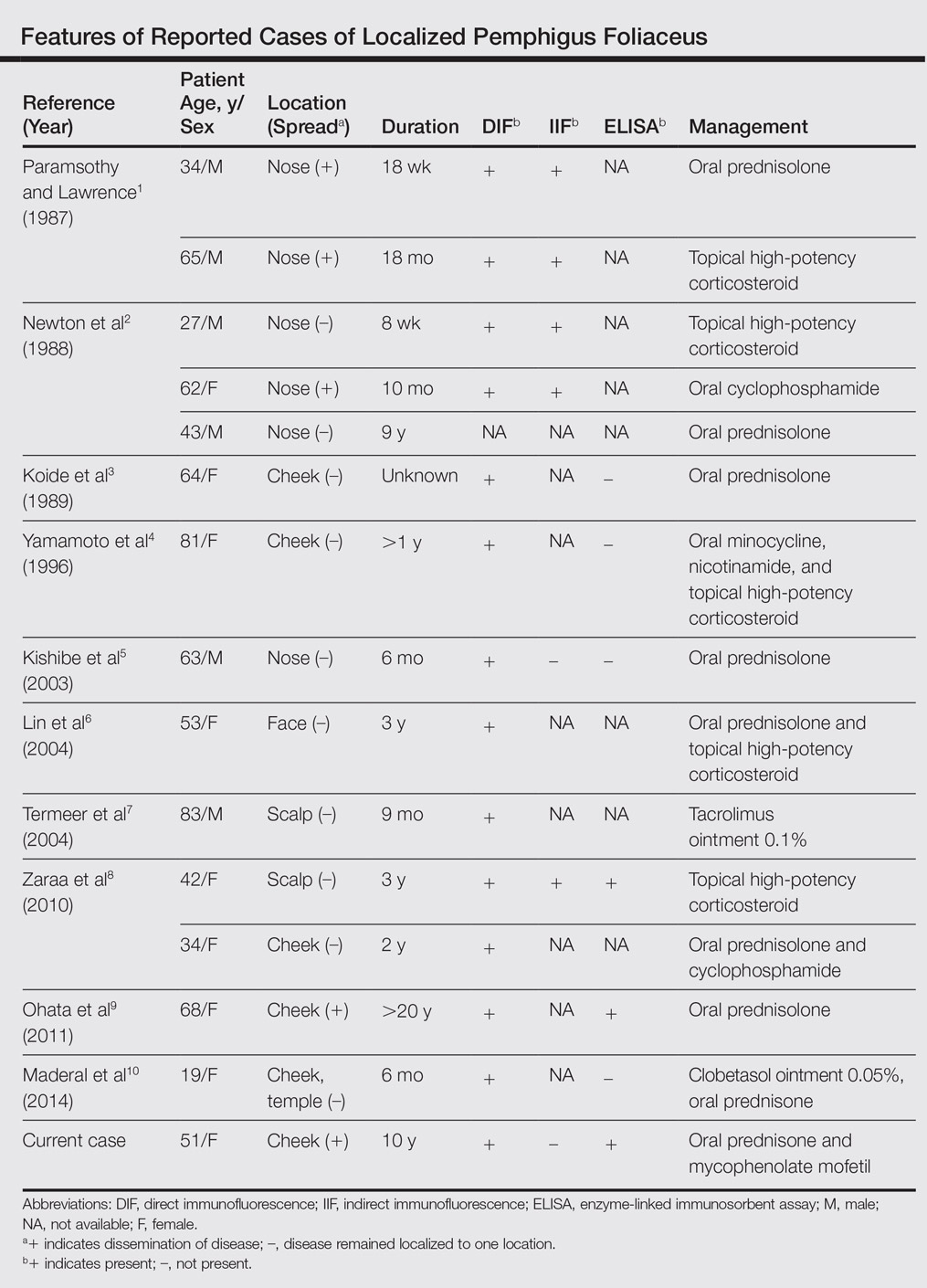
Pemphigus foliaceus is a rare autoimmune blistering disorder that typically presents with crusted scaly erosions in a seborrheic distribution. We describe a case of pemphigus foliaceus localized to the right cheek of 10 years’ duration that spread to other areas. With a PubMed search of articles indexed for MEDLINE yielding only 14 cases of localized pemphigus foliaceus (Table), it represents an extremely rare entity that often is a diagnostic challenge and may be a harbinger for disseminated disease months to years after the inciting lesion appears.
A 51-year-old woman presented with an asymptomatic cutaneous eruption that had remained localized to the right cheek for 10 years before it increased in size and new lesions developed on the left cheek, chest, and upper back. No inciting factors, such as contactants, insect bites, infections, medications, or recent travel were identified. On physical examination a well-demarcated, hypertrophic, verrucouslike plaque with central pink atrophy and exfoliative scale involved the right malar and submalar regions but spared the mucocutaneous junctions of the face (Figure 1). Subtle dark brown papules, some with overlying scale, speckled the left cheek, right jawline, chest, and upper back. The oral cavity was clear.
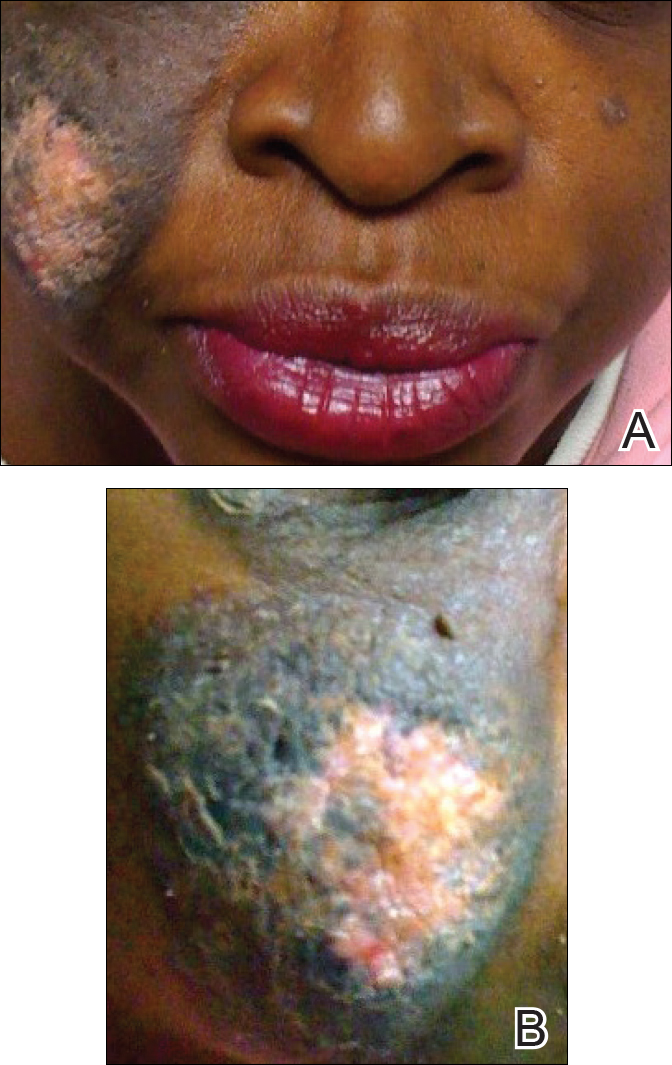
Figure 1. A well-demarcated, hypertrophic, dark brown to grayish plaque with central pink atrophy and exfoliative scale involving the right malar and submalar regions (A and B). On the left cheek, a subtle dark brown scaly papule was noted (A).
Leading differentials included hypertrophic discoid lupus erythematosus and pemphigus vegetans. Other considerations included sarcoidosis, granuloma faciale, lupus vulgaris, disseminated coccidioidomycosis or blastomycosis, and squamous cell carcinoma.
An initial biopsy revealed a lymphocytic lichenoid dermatitis with epidermal hyperplasia and scattered eosinophils for which the following differentials were provided: insect bite, hypertrophic lichen planus, prurigo nodularis superimposed on rosacea, and allergic contact dermatitis. Under these histologic diagnoses, tacrolimus ointment 0.03%, topical mid-potency corticosteroid, and a combination of oral doxycycline and metronidazole gel 1% were prescribed but failed to ameliorate her condition.
Because the clinical differentials were vast and noncorrelative with the original pathology, additional biopsies were performed: one from the edge of the large malar plaque, which was transected for hematoxylin and eosin (H&E) and tissue cultures; one perilesional to the large malar plaque for direct immunofluorescence (DIF); and one from the papule on the right jawline for H&E. Tissue cultures were negative for fungal and mycobacterial organisms. Both specimens submitted for H&E showed the prominent epidermal hyperplasia and lymphocytic dermal infiltrate noted on the original H&E but also demonstrated intragranular acantholysis (Figure 2). The DIF revealed intercellular IgG and C3 deposition throughout the epidermis (Figure 3). Indirect immunofluorescence was negative, but enzyme-linked immunosorbent assay detected circulating antidesmoglein-1 but not antidesmoglein-3 autoantibodies. Other serologies including antinuclear antibody, anti–double-stranded DNA, antihistone, anti–Sjögren syndrome A, and anti–Sjögren syndrome B antibodies were negative.
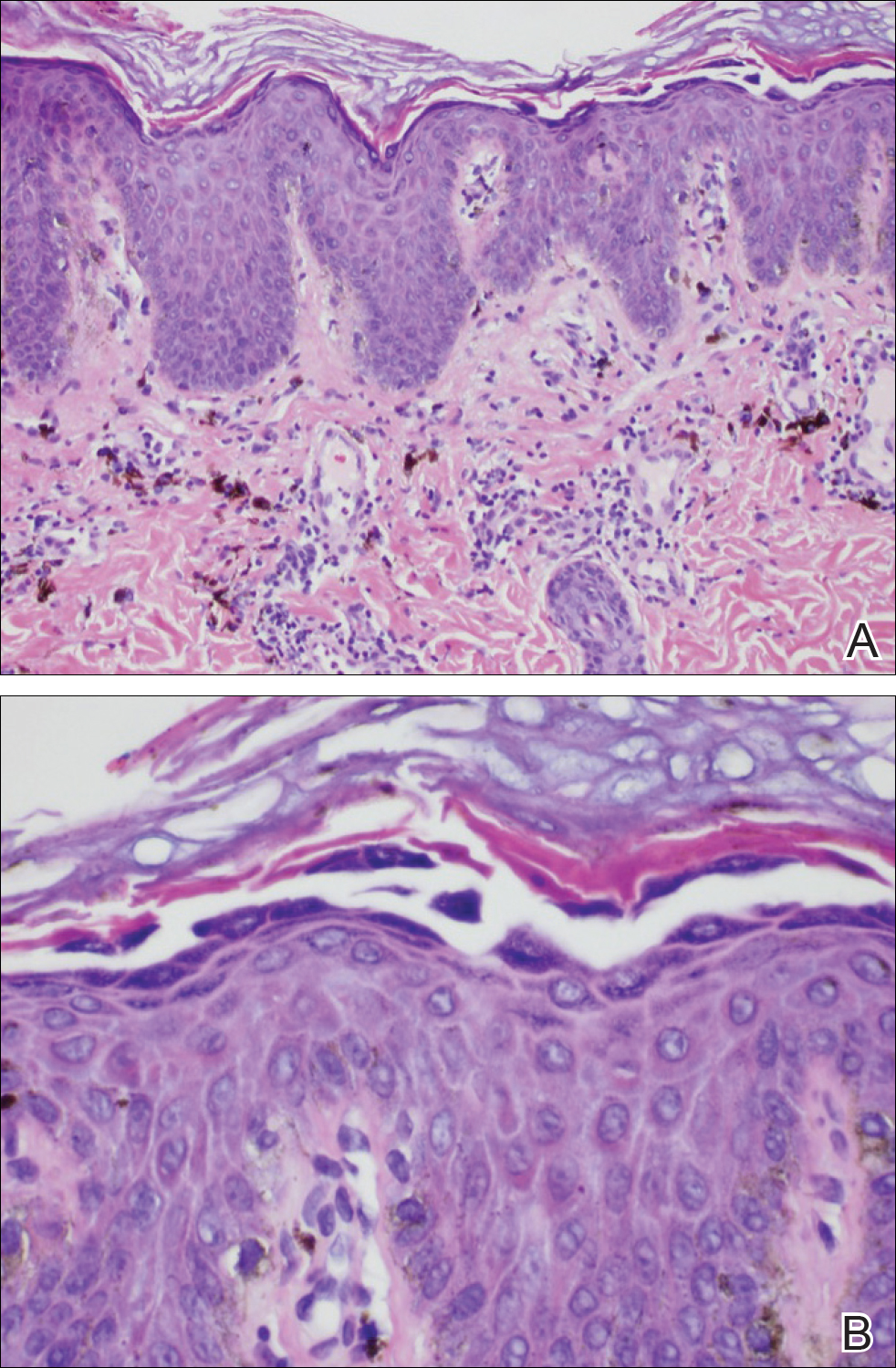
Figure 2. Intragranular acantholysis (A and B)(H&E, original magnifications ×100 and ×200).

Figure 3. Direct immunofluorescence demonstrating intercellular IgG and C3 deposition throughout the epidermis.
The diagnosis of localized pemphigus foliaceus was made and management with oral prednisone and mycophenolate mofetil resulted in improvement within weeks.
Localized pemphigus foliaceus is extremely rare with only 14 cases reported in the literature (Table).1-10 Its diagnosis is challenging, as the clinical presentation simulates various entities and the histological features and serological markers are difficult to capture.
Localized pemphigus foliaceus typically presents as an isolated, erythematous, scaly, crusted plaque involving the nose, cheek, or scalp and may mimic several conditions including contact dermatitis, seborrheic dermatitis, rosacea, cutaneous sarcoidosis, discoid lupus erythematosus, lupus vulgaris, impetigo contagiosa, solar keratosis, and nonmelanoma skin cancer.1-10
The predilection for sun-exposed areas suggests UV radiation may induce binding of antidesmoglein-1 autoantibodies with subsequent cytokine-mediated inflammation and acantholysis at these sites.11-13 Similarly, the immunomodulatory agent imiquimod has been reported to induce pemphigus foliaceus at its application sites.6
When pemphigus foliaceus is clinically discernible, the histology and DIF are in accordance with the clinical diagnosis 53.8% of the time.13 In cases of localized pemphigus foliaceus in which the diagnosis is more elusive, many biopsies often are needed to capture the characteristic intragranular acantholysis; this feature often is so subtle that unless the diagnosis is suspected, it is underappreciated or undetectable. In chronic lesions, it may be masked by secondary changes such as acanthosis, hyperkeratosis, and parakeratosis.14
In pemphigus foliaceus, detection of circulating antidesmoglein-1 autoantibodies by enzyme-linked immunosorbent assay is slightly more sensitive and specific compared to indirect immunofluorescence, but both correlate with disease activity.15,16 The low or absent autoantibody titers in localized pemphigus foliaceus may reflect its limited involvement, but dissemination of the disease with subsequent elevation of autoantibody titers may occur months to years after initial presentation,1,2,9 as was the case with our patient.
The majority of localized pemphigus foliaceus cases require systemic prednisone, sometimes in conjunction with nonsteroidal immunosuppressants or topical high-potency corticosteroids.1-3,5,6,8-10 One case was efficaciously managed with tacrolimus ointment 0.1%.7
Localized pemphigus foliaceus is a rare and challenging entity that must be a diagnostic consideration for any chronic focal plaque on the face or scalp, as it may herald disseminated disease.