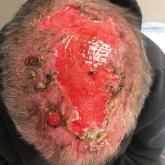
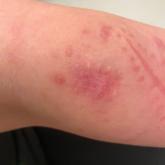
A 7-year-old boy was referred to the dermatology clinic for evaluation of a diffuse pruritic rash of 3 months’ duration. The rash began as scant erythematous papules on the face, and crops of similar lesions later erupted on the trunk, arms, and legs. He was treated previously by a pediatrician for scabies with topical permethrin followed by 2 doses of oral ivermectin 200 μg/kg without improvement. Physical examination revealed innumerable erythematous macules and papules with centrally adherent scaling distributed on the trunk, arms, and legs, as well as scant necrotic papules with a hemorrhagic crust and a peripheral rim of scale.
The Diagnosis: Pityriasis Lichenoides et Varioliformis Acuta
Sectioned punch biopsies were performed on the patient’s right arm. Histopathology showed acanthosis and parakeratosis in the epidermis, with vacuolar degeneration and dyskeratosis in the basal layer. Dermal changes included extravasated red blood cells in the papillary dermis as well as perivascular lymphocytic infiltrates in both the papillary and reticular dermis (Figure). Direct immunofluorescence of a perilesional biopsy using anti–human IgG, IgM, IgA, C3, and fibrin conjugates showed no findings of immune deposition. Biopsy results were consistent with pityriasis lichenoides et varioliformis acuta (PLEVA), and the patient was treated with a 5-day course of oral azithromycin, triamcinolone ointment 0.1% twice daily, and phototherapy with narrowband UVB 3 times weekly. Rapid improvement was noted at 2-month follow-up.
Pityriasis lichenoides et varioliformis acuta is a form of pityriasis lichenoides, a group of inflammatory dermatoses that are characterized clinically by successive crops of morphologically diverse lesions. Epidemiologic studies have shown a slight male predominance. It primarily affects children and young adults, with peak ages of 8 and 32 years in pediatric and adult populations, respectively.1
The pathogenesis of PLEVA remains unclear. An abnormal immune response to Toxoplasma, Epstein-Barr virus, HIV, and other pathogens has been suggested based on serologic evidence of concurrent disease activity with the onset of lesions as well as cutaneous improvement in some patients after treatment of the infection.1 A T-cell lymphoproliferative etiology also has been considered based on histopathologic similarities between PLEVA and lymphomatoid papulosis (LyP) as well as findings of clonality in T-cell receptor gene rearrangement in many patients.1,2 Some clinicians consider LyP and PLEVA as separate entities on one disease spectrum.
Eruptions of PLEVA tend to favor the trunk and proximal extremities. Lesions may begin as macules measuring 2 to 3 mm in diameter that quickly evolve into papules with fine scale that remains attached centrally. Ulcerations with hemorrhagic crusts also may be noted as the lesions progress in stage. The rash may persist for weeks to years, and overlapping crops of macules and papules at varying stages of development may be seen in the same patient.1
Histopathologic findings of PLEVA include spongiosis, dyskeratosis, parakeratosis, and focal keratinocyte necrosis within the epidermis, as well as vacuolar degeneration of the basal layer. Lymphocyte and erythrocyte extravasation may extend into the epidermis. Dermal findings may include edema and wedge-shaped perivascular lymphocytic infiltrates extending into the reticular dermis.1
Histopathology revealed epidermal acanthosis and parakeratosis with vacuolar degeneration as well as dyskeratosis in the basal layer, characteristic of pityriasis lichenoides et varioliformis acuta (H&E, original magnification ×2). Erythrocyte extravasation and perivascular infiltrates in the dermis also were seen.
Important differential diagnoses to consider include LyP, mycosis fungoides (MF), pemphigus foliaceus, and varicella. Lymphomatoid papulosis is a benign CD30+ lymphoproliferative disorder that is characterized by an indolent course of recurrent, often self-resolving papules that occur most frequently on the trunk, arms, and legs of older patients. There are several histologic subtypes of LyP, but the most common (type A) may manifest with wedge-shaped perivascular lymphocytic infiltrates in the dermis, similar to PLEVA. T-cell receptor gene rearrangement studies characteristically reveal clonality in LyP, and clonality has been reported in PLEVA. However, LyP demonstrates a higher cytologic grade and lacks the characteristic parakeratotic scale and superficial dermal microhemorrhage of PLEVA.3
Mycosis fungoides is a malignant lymphoproliferative disorder that is characterized by an indolent clinical course of persistent patches, plaques, or tumors of various sizes that often manifest in non–sun-exposed areas of the skin. Early stages of MF are difficult to detect histologically, but biopsies may show atypical lymphocytes with hyperchromatic, irregularly contoured nuclei arranged along the basal layer of the epidermis. Epidermal aggregates of atypical lymphocytes (also known as Pautrier microabscesses) are considered highly specific for MF. T-cell receptor and immunopathologic studies also are important adjuncts in the diagnosis of MF.4
Pemphigus foliaceus is an autoimmune blistering disease caused by antibodies directed against desmoglein 1, which is found in the granular layer of the epidermis. It manifests with a subtle onset of scattered crusted lesions in the seborrheic areas, such as the scalp, face, chest, and upper back. Histopathologic findings of early blisters may include acantholysis and dyskeratosis in the stratum granulosum as well as vacuolization of the granular layer. The blisters may coalesce into superficial bullae containing fibrin and neutrophils. Immunofluorescence studies that demonstrate intraepidermal C3 and IgG deposition are key to the diagnosis of pemphigus.5
Varicella (also known as chickenpox) manifests with crops of vesicles on an erythematous base in a centripetal distribution favoring the trunk and proximal extremities. It often is preceded by prodromal fever, malaise, and myalgia. Histopathologic evaluation of varicella is uncommon but may reveal acantholysis, multinucleation, and nuclear margination of keratinocytes. Viral culture or nucleic acid amplification testing of lesions can be used to verify the diagnosis.6
Most cases of PLEVA resolve without intervention.7 Treatment is directed at speeding recovery, providing symptomatic relief, and limiting permanent sequelae. Topical steroids often are used to alleviate inflammation and pruritus. Systemic antibiotics such as doxycycline, minocycline, and erythromycin have been used for their anti-inflammatory properties. Phototherapy of various wavelengths, including broadband and narrowband UVB as well as psoralen plus UVA, have led to improvements in affected patients. Refractory disease may warrant consideration of therapy with methotrexate, acitretin, dapsone, or cyclosporine.7
There have been rare reports of PLEVA evolving into its potentially lethal variant, febrile ulceronecrotic Mucha-Habermann disease, which is differentiated by the presence of systemic manifestations, including high fever, sore throat, diarrhea, central nervous system symptoms, abdominal pain, interstitial pneumonitis, splenomegaly, arthritis, sepsis, megaloblastic anemia, or conjunctival ulcers. The orogenital mucosa may be affected. Cutaneous lesions may rapidly progress to large, generalized, coalescent ulcers with necrotic crusts and vasculitic features on biopsy.8 Malignant transformation of PLEVA into LyP or MF rarely may occur and warrants continued follow-up of unresolved lesions.9