To the Editor:
Henoch-Schönlein purpura (HSP)(also known as IgA vasculitis) is a small vessel vasculitis characterized by deposition of IgA in small vessels, resulting in the development of purpura on the legs. Based on the European Alliance of Associations for Rheumatology criteria,1 the patient also must have at least 1 of the following: arthritis, arthralgia, abdominal pain, leukocytoclastic vasculitis with IgA deposition, or kidney involvement. The disease can be triggered by infection—with more than 75% of patients reporting an antecedent upper respiratory tract infection2—as well as medications, circulating immune complexes, certain foods, vaccines, and rarely cancer.3,4 The disease more commonly occurs in children but also can affect adults.
Several cases of HSP have been reported following COVID-19 vaccination.5 We report a case of HSP developing days after the messenger RNA Pfizer-BioNTech COVID-19 vaccine booster that was associated with anti-Smith and anti–double-stranded DNA (dsDNA) antibodies as well as antineutrophil cytoplasmic antibodies (ANCAs).
A 24-year-old man presented to dermatology with a rash of 3 weeks’ duration that first appeared 1 week after receiving his second booster of the messenger RNA Pfizer-BioNTech COVID-19 vaccine. Physical examination revealed petechiae with nonblanching erythematous macules and papules covering the legs below the knees (Figure 1) as well as the back of the right arm. A few days later, he developed arthralgia in the knees, hands, and feet. The patient denied any recent infections as well as respiratory and urinary tract symptoms. Approximately 10 days after the rash appeared, he developed epigastric abdominal pain that gradually worsened and sought care from his primary care physician, who ordered computed tomography and referred him for endoscopy. Computed tomography with and without contrast was suspicious for colitis. Colonoscopy and endoscopy were unremarkable. Laboratory tests were notable for elevated white blood cell count (17.08×103/µL [reference range, 3.66–10.60×103/µL]), serum IgA (437 mg/dL [reference range, 70–400 mg/dL]), C-reactive protein (1.5 mg/dL [reference range, <0.5 mg/dL]), anti-Smith antibody (28.1 CU [reference range, <20 CU), positive antinuclear antibody with titer (1:160 [reference range, <1:80]), anti-dsDNA (40.4 IU/mL [reference range, <27 IU/mL]), and cytoplasmic ANCA (c-ANCA) titer (1:320 [reference range, <1:20]). Blood urea nitrogen, creatinine, and estimated glomerular filtration rate were all within reference range. Urinalysis with microscopic examination was notable for 2 to 5 red blood cells per high-power field (reference range, 0) and proteinuria of 1+ (reference range, negative for protein).
The patient’s rash progressively worsened over the next few weeks, spreading proximally on the legs to the buttocks and the back of both elbows. A repeat complete blood cell count showed resolution of the leukocytosis. Two biopsies were taken from a lesion on the left proximal thigh: 1 for hematoxylin and eosin stain for histopathologic examination and 1 for direct immunofluorescence examination.
The patient was preliminarily diagnosed with HSP, and dermatology prescribed oral tofacitinib 5 mg twice daily for 5 days, which was supposed to be increased to 10 mg twice daily on the sixth day of treatment; however, the patient discontinued the medication after 4 days based on his primary care physician’s recommendation due to clotting concerns. The rash and arthralgia temporarily improved for 1 week, then relapsed.
Histopathology revealed neutrophils surrounding and infiltrating small dermal blood vessel walls as well as associated neutrophilic debris and erythrocytes, consistent with leukocytoclastic vasculitis (Figure 2). Direct immunofluorescence was negative for IgA antibodies. His primary care physician, in consultation with his dermatologist, then started the patient on oral prednisone 70 mg once daily for 7 days with a plan to taper. Three days after prednisone was started, the arthralgia and abdominal pain resolved, and the rash became lighter in color. After 1 week, the rash resolved completely.
Due to the unusual antibodies, the patient was referred to a rheumatologist, who repeated the blood tests approximately 1 week after the patient started prednisone. The tests were negative for anti-Smith, anti-dsDNA, and c-ANCA but showed an elevated atypical perinuclear ANCA (p-ANCA) titer of 1:80 (reference range [negative], <1:20). A repeat urinalysis was unremarkable. The patient slowly tapered the prednisone over the course of 3 months and was subsequently lost to follow-up. The rash and other symptoms had not recurred as of the patient’s last physician contact. The most recent laboratory results showed a white blood cell count of 14.0×103/µL (reference range, 3.4–10.8×103/µL), likely due to the prednisone; blood urea nitrogen, creatinine, and estimated glomerular filtration rate were within reference range. The urinalysis was notable for occult blood and was negative for protein. C-reactive protein was 1 mg/dL (reference range, 0–10 mg/dL); p-ANCA, c-ANCA, and atypical p-ANCA, as well as antinuclear antibody, were negative. As of his last follow-up, the patient felt well.
The major differential diagnoses for our patient included HSP, ANCA vasculitis, and systemic lupus erythematosus. Although ANCA vasculitis has been reported after SARS-CoV-2 infection,6 the lack of pulmonary symptoms made this diagnosis unlikely.7 Although our patient initially had elevated anti-Smith and anti-dsDNA antibodies as well as mild renal involvement, he fulfilled at most only 2 of the 11 criteria necessary for diagnosing lupus: malar rash, discoid rash (includes alopecia), photosensitivity, ocular ulcers, nonerosive arthritis, serositis, renal disorder (protein >500 mg/24 h, red blood cells, casts), neurologic disorder (seizures, psychosis), hematologic disorders (hemolytic anemia, leukopenia), ANA, and immunologic disorder (anti-Smith). Four of the 11 criteria are necessary for the diagnosis of lupus.8
Torraca et al7 reported a case of HSP with positive c-ANCA (1:640) in a patient lacking pulmonary symptoms who was diagnosed with HSP. Cytoplasmic ANCA is not a typical finding in HSP. However, the additional findings of anti-Smith, anti-dsDNA, and mildly elevated atypical p-ANCA antibodies in our patient were unexpected and could be explained by the proposed pathogenesis of HSP—an overzealous immune response resulting in aberrant antibody complex deposition with ensuing complement activation.5,9 Production of these additional antibodies could be part of the overzealous response to COVID-19 vaccination.
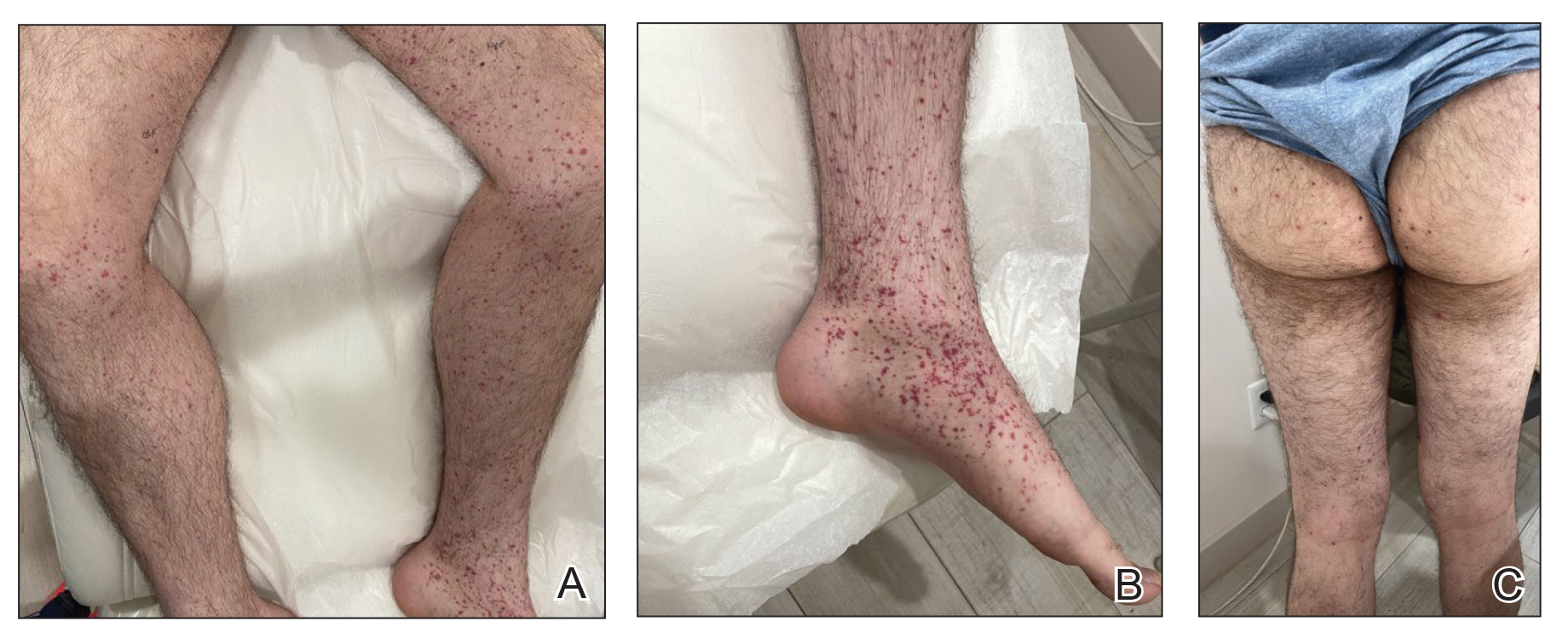
FIGURE 1. A–C, Macules and papules on the legs, foot, and buttocks, respectively, consistent with Henoch-Schönlein purpura.
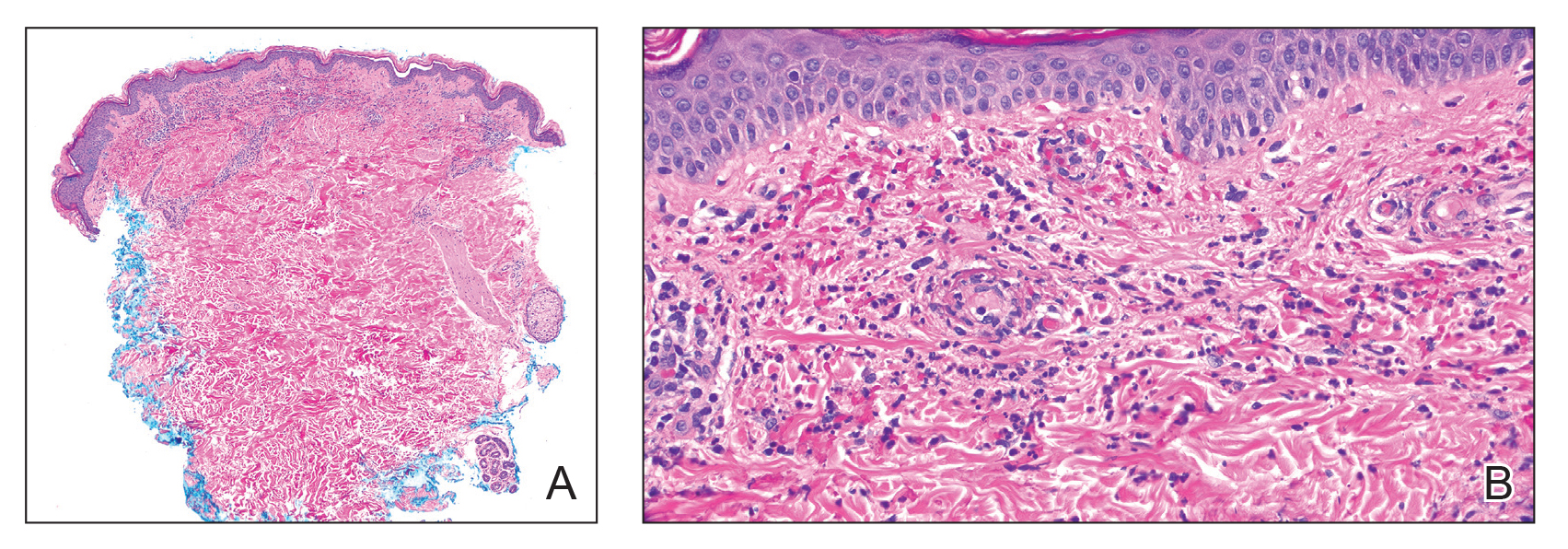
FIGURE 2. A and B, Biopsy of a purpuric papule revealed leukocytoclastic vasculitis depicted by small blood vessel damage with neutrophilic debris and erythrocytes as well as neutrophils surrounding and infiltrating its walls (H&E, original magnifications ×40 and ×400), consistent with leukocytoclastic vasculitis.
Of all the COVID-19 vaccines, messenger RNA–based vaccines have been associated with the majority of cutaneous reactions, including local injection-site reactions (most common), delayed local reactions, urticaria, angioedema, morbilliform eruption, herpes zoster eruption, bullous eruptions, dermal filler reactions, chilblains, and pityriasis rosea. Less common reactions have included acute generalized exanthematous pustulosis, Stevens-Johnson syndrome, erythema multiforme, Sweet Syndrome, lichen planus, papulovesicular eruptions, pityriasis rosea–like eruptions, generalized annular lesions, facial pustular neutrophilic eruptions, and flares of underlying autoimmune skin conditions.10 Multiple cases of HSP have been reported following COVID-19 vaccination from all the major vaccine companies.5
In our patient, laboratory tests were repeated by a rheumatologist and were negative for anti-Smith and anti-dsDNA antibodies as well as c-ANCA, most likely because he started taking prednisone approximately 1 week prior, which may have resulted in decreased antibodies. Also, the patient’s symptoms resolved after 1 week of steroid therapy. Therefore, the diagnosis is most consistent with HSP associated with COVID-19 vaccination. The clinical presentation, microscopic hematuria and proteinuria, and histopathology were consistent with the European Alliance of Associations for Rheumatology criteria for HSP.1
Although direct immunofluorescence typically is positive for IgA deposition on biopsies, it can be negative for IgA, especially in lesions that are biopsied more than 7 days after their appearance, as shown in our case; a negative IgA on immunofluorescence does not rule out HSP.4 Elevated serum IgA is seen in more than 50% of cases of HSP.11 Although the disease typically is self-limited, glucocorticoids are used if the disease course is prolonged or if there is evidence of kidney involvement.9 The unique combination of anti-Smith and anti-dsDNA antibodies as well as ANCAs associated with HSP with negative IgA on direct immunofluorescence has been reported with lupus.12 Clinicians should be aware of COVID-19 vaccine–associated HSP that is negative for IgA deposition and positive for anti-Smith and anti-dsDNA antibodies as well as ANCAs.
Acknowledgment—We thank our patient for granting permission to publish this information.