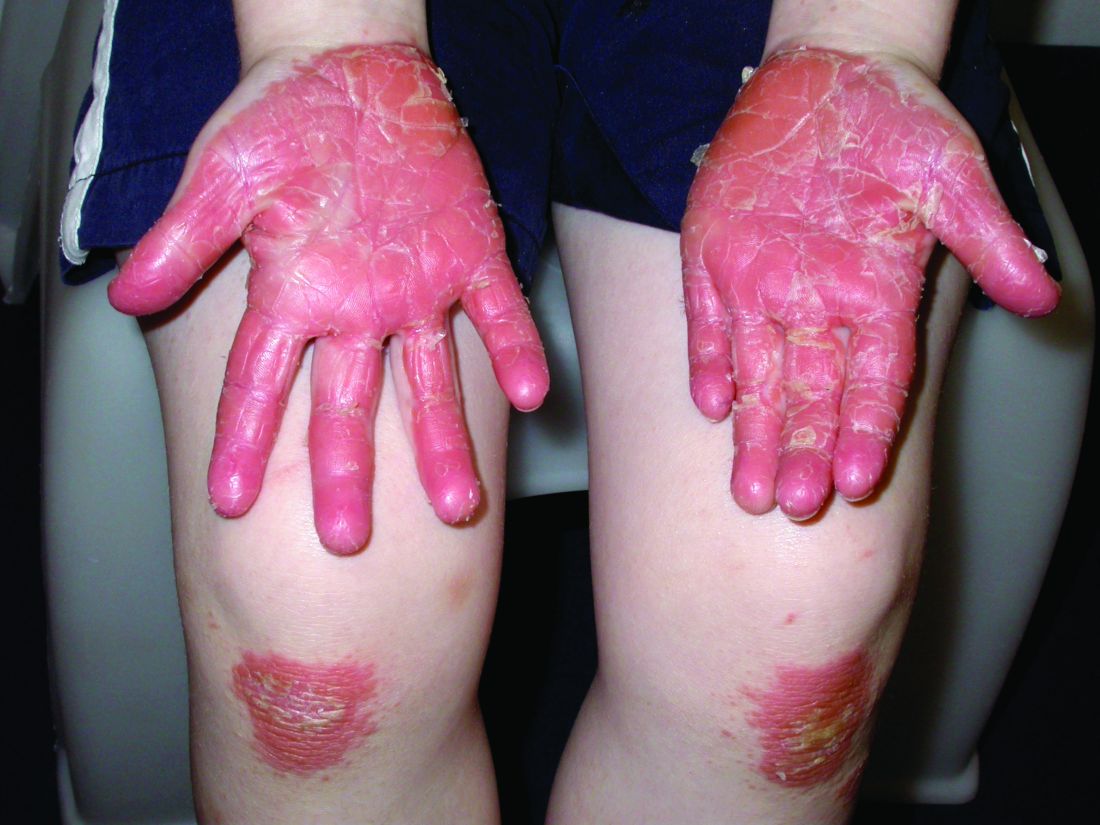
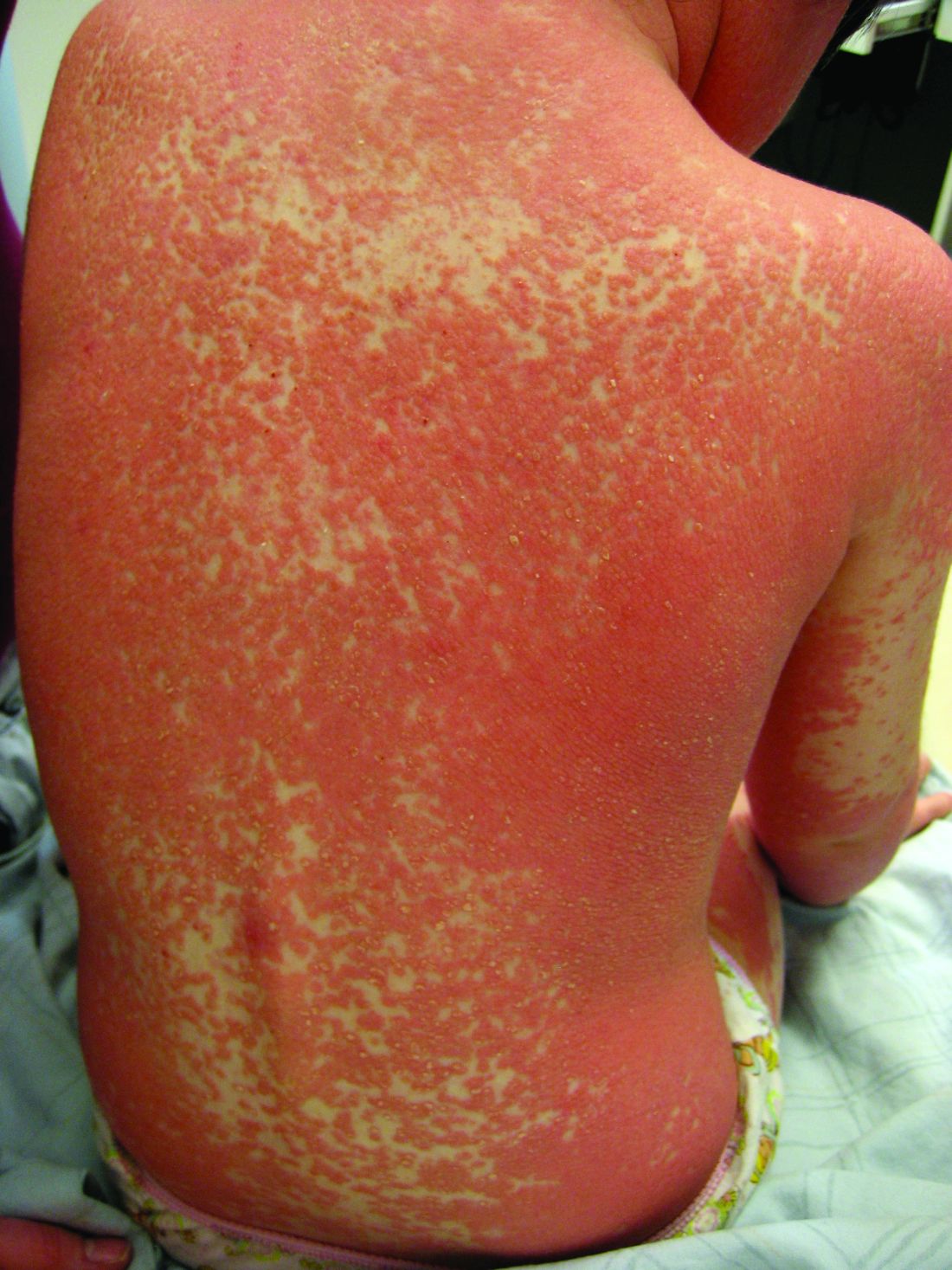
Pityriasis rubra pilaris (PRP) is the name given to a heterogeneous group of rare inflammatory papulosquamous dermatoses. There are six sub-types that can present with various skin findings, however, the cardinal features across sub-types include well-defined, red-orange hued plaques with varying scale, palmoplantar keratoderma, and follicular keratosis. In the more generalized subtypes, there is a characteristic feature of intervening areas of unaffected skin often referred to as “islands of sparing.” The plaques may cover the entire body or just parts of the body such as the elbows and knees, palms and soles. Lesions are generally asymptomatic; occasionally patients complain of mild pruritus.
 Photos courtesy of Robert Silverman, MD
Photos courtesy of Robert Silverman, MD
The etiology and pathophysiology of this group of disorders is not well understood. However, there are several hypotheses including dysfunction in vitamin A metabolism, autoimmune dysregulation, as well as environmental and immunologic triggers such as infection and ultraviolet exposure. Although most cases are sporadic, genetics do seem to play a role in the development of some cases. Caspase recruitment domain-containing protein 14 (CARD14) mutations are seen in familial PRP, and occasionally in patients with sporadic PRP, with gain of function mutations. Interestingly, CARD14 mutations are also associated with psoriasis in some individuals.1 The type-VI PRP variant has been associated with HIV, although this is incredibly rare in pediatrics.2
PRP shows significant clinical diversity, with six subtypes defined by age of onset, distribution, and appearance of lesions, and presence of HIV. This includes type I (classical adult onset), type II (atypical adult onset), type III (classical juvenile onset), type IV (circumscribed juvenile onset), type V (atypical juvenile onset), and type VI (HIV-associated). As mentioned earlier, shared features that appear across subtypes in variable degrees include red-orange papules and plaques, hyperkeratotic follicular papules, and palmoplantar hyperkeratosis.
Of the six subtypes, type III, IV, and V occur in the pediatric population. Type III, classic juvenile PRP, typically occurs within the first 2 years of life or in adolescence. Only 10% of cases fall into this category. It shares similar features to type I PRP including red-orange plaques; islands of sparing, perifollicular hyperkeratotic papules; waxy palmoplantar keratoderma; and the distribution of affected skin is more diffuse overall. While some children clear within a few years, more recent studies stress a more prolonged course similar to the type IV variant.2
Type-IV PRP, also known as circumscribed juvenile PRP, is a focal variant, usually seen in prepubertal children and making up 25% of total cases. Clinically, these patients tend to have sharply demarcated grouped erythematous, follicular papules on the elbows, knees and over bony prominences.2
Type-V PRP is an atypical generalized juvenile variant which affects 5% of patients. It is a non-remitting hereditary condition with classic characteristics similar to type III with additional scleroderma-like changes involving the palms and soles.2

Dr. Alexis Tracy
Diagnosis of PRP is based on clinical recognition and biopsy can be important to secure a diagnosis.
PRP, in many cases is self-limited and asymptomatic, and therefore does not necessarily require treatment. In other patients treatment can be challenging, and referral to a pediatric dermatology specialist is reasonable. Most practitioners recommend combination therapy with topical agents (emollients, topical corticosteroids, tazarotene, topical calcineurin inhibitors, and keratolytic agents such as urea, salicylic acid, or alpha-hydroxy acids) for symptomatic management and systemic therapies (methotrexate, isotretinoin) aimed at reducing inflammation. There is some data that CARD14-associated PRP can respond well to targeted biologic therapies.1

Dr. Lawrence F. Eichenfield
The subtypes of PRP can present in a myriad of ways and often the disease is misdiagnosed. Depending on the particular subtype and findings present, the differential can vary considerably. Commonly, physicians need to consider: psoriasis, seborrheic dermatitis, atopic dermatitis, ichthyoses, and other conditions which can cause erythroderma.3 The characteristic red-orange color and variable associated edema helps to distinguish keratoderma of PRP from psoriasis, atopic dermatitis, ichthyosis, and hereditary palmoplantar keratoderma. Scalp involvement of PRP should be differentiated from the waxy scale of seborrheic dermatitis and the well demarcated silvery scale of psoriasis. History alone may assist in distinguishing PRP from other major causes of generalized erythroderma, although biopsy is warranted in these cases.
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. Dr. Tracy is a research fellow in pediatric dermatology at Rady Children’s Hospital-San Diego and the University of California, San Diego. They have no relevant financial disclosures. Email them at pdnews@mdedge.com.
References
1. J Am Acad Dermatol. 2018 Sep;79(3):487-94.
2. “Pityriasis Rubra Pilaris” (Treasure Island, Fla.: StatPearls Publishing, July 20, 2019). 3. JAMA Dermatol. 2016 Jun 1;152(6):670-5.