User login
Diagnosing and Classifying Anemia in Adult Primary Care
CE/CME No: CR-1708
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Discuss the importance of diagnosing the type of anemia in order to provide appropriate treatment.
• Describe how the complete blood count and its indices are used to initially determine if an anemia is microcytic, normocytic, or macrocytic.
• List the more common causes of microcytic, normocytic, and macrocytic anemia.
• Discuss addictional laboratory tests that may be used to further assess the cause of anemia.
FACULTY
Jean O’Neil is an Assistant Professor and Coordinator of the Adult Gerontology Acute Care Nurse Practitioner Program in the Patricia A. Chin School of Nursing at California State University, Los Angeles.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through July 31, 2018.
Article begins on next page >>
Anemia affects more than 3 million people in the United States, making it a common problem in primary care practices. Once anemia is detected, clinicians must define the type and identify its underlying cause prior to initiating treatment. In most cases, the cause can be determined using information from the patient history, physical exam, and complete blood count.
Anemia is commonly identified during routine physical exams and laboratory testing.1-3 However, treating anemia can present a challenge for the primary care provider if the immediate cause is not apparent. Iron deficiency is a leading cause of anemia, but simply prescribing an iron supplement without determining the type or the cause of the anemia is not appropriate. Anemia that is misdiagnosed or goes untreated can be associated with a worse prognosis, as well as increased health care costs.4
Primary care providers often manage patients with common types of anemia and refer patients with severe or complex anemia to specialists for further testing and treatment. The most commonly used and cost-effective diagnostic tool for anemia is the complete blood count (CBC).2-6 The CBC provides details that can help the provider determine the type of anemia present, which in turn guides proper diagnostic testing and treatment.
EPIDEMIOLOGY
Anemia involves a reduction in the number of circulating red blood cells, the blood hemoglobin content, or the hematocrit, which leads to impaired delivery of oxygen to the body. Anemia affects more than 2 billion people worldwide, with iron deficiency the most common cause.7 Other leading nutritional causes of anemia include vitamin B12 and folate deficiency.4,7 Approximately 3 to 4 million Americans have anemia in some form, and it affects about 6.6% of men and 12.4% of women.5,8 The prevalence of anemia increases with age. Approximately 11% of men and 10% of women ages 65 or older have anemia, and in men ages 85 or older, prevalence of 20% to 44% has been reported.1,4 Anemia is present in about 3.5% of patients with chronic disease, but only 15% of them receive treatment.4

PATHOPHYSIOLOGY
Blood is composed of water-based plasma (54%), white blood cells and platelets (1%), and red blood cells (45%).5 Hemoglobin, the primary protein of the red blood cell, binds oxygen from the lungs and transports it to the rest of the body. Oxygen is then exchanged for carbon dioxide, which is carried back to the lungs to be exhaled.
Hemoglobin is made up of four globin chains, each containing an iron ion held in a porphyrin ring known as a heme group.5 When the body detects low tissue oxygen, the endothelial cells in the kidneys secrete the hormone erythropoietin (EPO), which stimulates the bone marrow to increase red cell production.5 This feedback loop can be interrupted by renal failure or chronic disease.4 In addition, bone marrow cannot produce enough red blood cells if there are insufficient levels of iron, amino acids, protein, carbohydrates, lipids, folate, and vitamin B12.5 Toxins (eg, lead), some types of cancer (eg, lymphoma), or even common infections (eg, pneumonia) can suppress the bone marrow, causing anemia. The more severe the anemia, the more likely oxygen transport will be compromised and organ failure will ensue.
Mutations affecting the genes that encode the globin chains within hemoglobin can cause one of the more than 600 known hemoglobinopathies (genetic defects of hemoglobin structure), such as sickle cell disease and thalassemias.5,9 While it is important to identify and treat patients with hemoglobinopathies, most anemias have other causes, such as iron deficiency, chronic disease, bone marrow defects, B12 deficiency, renal failure, medications, alcoholism, pregnancy, nutritional intake problems, gastrointestinal malabsorption, and active or recent history of blood loss.5,10
CLINICAL PRESENTATION
There are several signs and symptoms that should lead the primary care provider to suspect anemia (see Table 1).5,6 The severity of these symptoms can vary from mild to very serious. Severe anemia can lead to organ failure and death. However, most patients with anemia are asymptomatic, and anemia is typically detected incidentally during laboratory testing.1,2

Once anemia is confirmed, the evaluation focuses on diagnosing its underlying cause. It should include a thorough patient history and review of systems to ascertain whether the patient has symptoms such as increased fatigue, palpitations, gastrointestinal distress, weakness, or dizziness.
If the provider has access to past CBC results, a comparison of the current and previous results will help determine whether the anemia is acute or chronic. Anemia caused by acute conditions, such as a suspicion of blood loss or bone marrow suppression, must be attended to immediately. A patient with chronic anemia should be carefully monitored and may need follow-up for ongoing treatment. While a provider has more time to work up a patient with chronic anemia, the causes may not be as straightforward.
DIAGNOSIS AND CLASSIFICATION
Anemia in adults is defined as hemoglobin less than 13 g/dL in males and 12 g/dL in females.6 The hemoglobin is part of the complete blood cell report, which also includes the white blood cell count (WBC), red blood cell count (RBC), hematocrit, platelet count, and indices.
When investigating the underlying cause of anemia, the most useful parts of the CBC are the hemoglobin and the mean corpuscular volume (MCV; see Table 2).6,10 The MCV is the average volume of red cells in a specimen. This parameter is used to classify the anemia as microcytic (MCV < 80 fL), normocytic (MCV 80-100 fL), or macrocytic (MCV > 100 fL), which helps to narrow the differential diagnosis and guide any further testing (see Figure).5,6,10

It is important to note that the normal ranges of the CBC parameters differ based on race, with persons of African ancestry having lower normal hemoglobin levels than persons of Caucasian ancestry.10 In addition, laboratories may have slightly different normal values for the CBC based on the equipment they utilize. Therefore, providers must follow their laboratory’s parameters, as well as adjust for the patient’s gender, age, and ethnicity.10
Microcytic Anemia
Iron deficiency
In microcytic anemia, the RBCs are smaller than average (MCV < 80 fL), as well as hypochromic due to lack of hemoglobin.9 Iron deficiency is the most common cause of microcytic anemia worldwide.11,12 Therefore, when a patient has microcytic anemia, a serum ferritin needs to be ordered. Further testing of total iron-binding capacity (TIBC), transferrin saturation, serum iron, and serum receptor levels may be helpful if the ferritin level is between 46-99 ng/mL and anemia due to iron deficiency is not confirmed (see Table 2).12
In iron deficiency anemia, serum ferritin and serum iron levels are low due to lack of iron, but serum TIBC is high.6 The elevated TIBC reflects increased synthesis of transferrin by the liver as it attempts to compensate for the patient’s low serum iron level.9 Since iron levels are controlled by absorption rather than excretion, iron is essentially only depleted from the body through blood loss.12 Therefore, an adult patient who is iron deficient has lost more iron through blood loss than was replaced through nutritional intake and gastrointestinal absorption. In children, increased growth-related iron requirements combined with poor nutritional intake of iron-rich foods is an additional mechanism for iron deficiency.11
Iron def
If the nutritional problem is corrected or the source of bleeding is controlled, treatment with oral or intravenous iron supplements should result in improved serum hemoglobin and reticulocyte counts.13 In the primary care setting, ferrous sulfate 325 mg, which provides 65 mg of elemental iron per tablet, orally three times daily is recommended for adults.13 This gives the patient the recommended dose of approximately 200 mg of elemental iron. Repeat hemoglobin and iron studies should be conducted again in three to six months.12,13
If the patient’s iron deficiency anemia does not improve after oral iron therapy, there may be a source of blood loss the provider missed or a problem with malabsorption of iron, which can be seen in those who have undergone gastric bypass surgery or who have inflammatory bowel disease.13 Such patients should be referred to a specialist, such as a gastroenterologist, for further evaluation.
Thalassemia
Microcytic anemia with normal or elevated serum iron and normal-to-increased serum ferritin can be seen in patients with a type of thalassemia (see Figure).2 Thalassemias are inherited blood disorders that reduce hemoglobin production, leading to microcytosis; they are more common in those of Mediterranean, African, and Southeast Asian descent.2 Red cells in patients with a form of thalassemia are usually very small (microcytic) and have normal or elevated red cell distribution width (RDW).10
Moderate and severe forms of thalassemia can cause anemia. However, thalassemia syndromes that can cause severe (transfusion-dependent) anemia are usually diagnosed in childhood.9 Patients with one of the minor forms of thalassemia typically need minimal to no treatment.5 A patient with significant anemia suspicious for thalassemia should undergo hemoglobin electrophoresis testing to confirm the diagnosis and to determine the type of thalassemia.2 Typically, hemoglobin electrophoresis is normal in α thalassemia and is abnormal in ß thalassemia, as well as other forms of thalassemia. Referral to a hematologist for interpretation of these results and for further evaluation is appropriate.10
Chronic disease
If the patient has microcytic anemia and is not iron deficient or does not have thalassemia, then anemia related to a chronic disease should be considered.5 In such cases, the provider should order a reticulocyte count, which reveals how the bone marrow is responding to the anemia.5 Reticulocytes are immature red cells that have just been released from the bone marrow into the blood stream. The bone marrow increases the release of these cells in response to anemia.6
Any condition that stimulates reticulocyte production or prevents the bone marrow from producing reticulocytes will result in abnormal values (see Table 3). A normal reticulocyte count, expressed as the reticulocyte production index, is between 0.5% and 1.5%.5 The reticulocyte count is low in iron deficiency anemia and diseases that lead to decreased bone marrow production.5,6 Bone marrow suppression can occur in the context of chronic disease, infection, or inflammation. Malignancies are a less common cause for chronic disease microcytic anemia.6

If the cause of the decreased reticulocyte count is iron deficiency anemia, then treatment with iron supplementation should result in an increased reticulocyte count within one week.13 The primary care provider works in conjunction with the specialist to monitor the patient’s anemia when it is due to chronic disease or malignancy.
MACROCYTIC ANEMIA
In macrocytic anemia, the RBCs are larger than normal (MCV > 100 fL). This form of anemia is usually caused by vitamin B12 and folate deficiency, but it can also result from alcoholism, certain medications (eg, chemotherapy, antivirals), bone marrow disorders (eg, leukemia), and liver disease (eg, cirrhosis; see Figure).5,14 Common medications that can cause macrocytosis include the antiseizure drug phenytoin, the antibiotics trimethoprim/sulfamethoxazole and nitrofurantoin, the disease-modifying antirheumatic drug sulfasalazine, and immunosuppressants such as azathioprine.14,15 Antiviral agents, such as reverse transcriptase inhibitors (eg, zidovudine) used to treat HIV infection, can also cause macrocytosis with or without anemia.6,14
Macrocytic anemias caused by low serum levels of B12 and folate usually reflect problems with gastrointestinal malabsorption. For example, gastric bypass or Crohn disease can lead to malabsorption of vitamin B12 and increase a patient’s risk for macrocytic anemia.13
Vitamin B12 deficiency occurs in patients with pernicious anemia because they are missing intrinsic factor, which is necessary to facilitate B12 absorption in the ileum.10 Low vitamin B12 and folate levels also can result from inadequate dietary intake, although this is rare in the United States due to mandatory fortification of certain foods. A diet low in fresh vegetables is the leading cause of folate deficiency. While folate deficiency related to poor nutritional intake can be seen in all age groups, vitamin B12 deficiency more frequently affects the elderly or persons following a strict vegan diet.14
In addition to the fatigue and pallor associated with macrocytic anemia, patients with vitamin B12 deficiency may also have a smooth tongue, peripheral neuropathy, and edema.5,14 Severe vitamin B12 deficiency can lead to subacute combined degeneration of the spinal cord, with demyelination of the dorsal and lateral columns most often occurring in the cervical and thoracic regions.16,17 This spinal cord degeneration can cause paresthesia, muscle spasticity, and ataxia.16
When there is a macrocytic anemia, but the B12 or folate level is only borderline low, additional tests should be performed to help distinguish between B12 and folate deficiency. Both B12 and folate deficiencies can cause elevated homocysteine levels.13 Clinically significant B12 deficiency causes elevation of methylmalonic acid (MMA), whereas folate deficiency does not.13,14 Elevation of MMA can be very sensitive for B12 deficiency but lacks specificity in certain situations, such as pregnancy, renal insufficiency, and advanced age.13,14
Treatment of vitamin B12 and folate deficiencies with supplementation prevents progression of the disease, and has the potential to relieve most of the symptoms. Oral, sublingual, or parenteral vitamin B12 or oral folate supplements can be started in the primary care setting once the provider has identified whether the patient is B12 deficient, folate deficient, or both.
The vitamin B12 dose used for deficiency-induced macrocytic anemia depends on the cause—for example, a temporary condition such as pregnancy versus a lifelong disorder such as pernicious anemia.13 The usual oral dosing regimen is 2 mg/d; if intramuscular injections are used, 50 to 100 mcg are given daily for a week, followed by weekly injections for a month, and then monthly injections of 1 mg for life, if necessary.13 Bone marrow response to supplemental B12 is very rapid, with increased reticulocyte counts seen within four or five days.13
The usual dose for oral folic acid is 1 mg/d as needed.13 Folic acid can be given for folate deficiency only if the vitamin B12 level is normal. Giving folate to a patient with untreated vitamin B12 deficiency can potentially worsen subacute combined degeneration of the spinal cord.13,16
NORMOCYTIC ANEMIA
In normocytic anemia, the hemoglobin is low but the MCV is normal (see Figure).1 The history and physical exam should provide clues about whether the underlying cause of the anemia requires emergent (eg, active bleeding) or nonemergent (eg, anemia of chronic disease) management. Some of the causes of normocytic anemia are active bleeding, pregnancy, malnutrition, renal failure, chronic disease, hemolytic disorders, hypersplenism, congenital disorders, endocrine disorders, infection, and primary bone marrow disorders.1,5 Expanded plasma volume, as seen in pregnancy and overhydration, can also cause normocytic anemia.5 If gastrointestinal bleeding is suspected or the patient reports dark, tarry stools consistent with melena, fecal occult blood testing should be done. A positive result strongly supports gastrointestinal bleeding as the cause of the anemia.18
The reticulocyte count can also be helpful in identifying the cause of this type of anemia. A normocytic anemia with a normal reticulocyte and normal RDW count is usually related to chronic disease.1,10 For example, chronic kidney disease (CKD) is associated with decreased EPO production due to impaired renal function, which leads to reduced erythropoiesis. Decreased EPO prevents the bone marrow from making red blood cells, resulting in anemia. However, a normocytic anemia with an elevated reticulocyte count points to bleeding or hemolysis, as the reticulosis shows that the bone marrow is increasing red cell production to make up for the lost red cells.5
Additional diagnostic laboratory testing for patients with normocytic anemia may involve, for example, creatinine and blood urea nitrogen for patients with CKD, prothrombin time with an INR and liver function tests for patients with liver disease, and urine human chorionic gonadotropin if pregnancy is suspected.
For patients with an infection that is causing severe hemolysis (eg, sepsis due to a ß-hemolytic streptococcal infection), blood cultures should be drawn.5 If red blood cell destruction due to an artificial cardiac valve or an autoimmune disorder is suspected as the cause of the anemia, a hematology consult is needed.1 Anemia caused by disseminated intravascular coagulation or thrombotic thrombocytopenic purpura resulting in hemolysis are usually emergent conditions that require immediate intervention, including hospitalization and management by a hematologist.1
PATIENT EDUCATION
Patients and any accompanying family members should be educated about the signs and symptoms of anemia, the diagnostic testing and treatment regimens specific to their anemia, and medication compliance issues.
For instance, patients who abuse alcohol often have both vitamin B12 and folate deficiencies. If the macrocytosis is caused by alcohol intake, then the provider should educate the patient on the importance of alcohol abstention, as well as refer the patient for rehabilitation and psychologic counseling, as needed. These patients can sometimes recover from macrocytic anemia simply by stopping alcohol intake and improving their nutrition.19 Patients with microcytosis due to iron deficiency anemia should be advised about the importance of good nutrition and compliance with iron supplementation.
Repeat CBCs and a follow-up patient history and physical exam will help the provider assess whether the anemia is resolving. Individualized plans that target the specific type of anemia identified, as well as its underlying cause, are key to successful treatment.
CONCLUSION
When managing a patient with anemia, providers must define the type of anemia present and identify its underlying cause before starting treatment. Clues from the patient’s history, physical exam, and CBC can help isolate the cause of anemia. The MCV is the most helpful of the red blood cell indices because it allows the provider to classify the anemia as microcytic, macrocytic, or normocytic.
In cases in which the anemia is acute or severe—or in which the patient remains anemic even after being treated by the primary care provider—referral to a specialist is appropriate.
1. Brill JR, Baumgardner D. Normocytic anemia. Am Fam Physician. 2000;62(10):2255-2263.
2. Van Vranken M. Evaluation of microcytosis. Am Fam Physician. 2010;82(9):1117-1122.
3. National Institutes of Health/National Heart, Lung, and Blood Institute. How is anemia diagnosed? www.nhlbi.nih.gov/health/health-topics/topics/anemia/diagnosis. Accessed April 28, 2017.
4. Smith RE Jr. The clinical and economic burden of anemia. Am J Manag Care. 2010;16(3):S59-S66.
5. Platt A, Eckman J. Diagnosing anemia. Clinician Reviews. 2006;16(2):44-50.
6. Karnath B. Anemia in the adult patient. Hosp Physician. 2004;40(10):32-36.
7. World Health Organization. Micronutrient deficiencies. www.who.int/nutrition/topics/ida/en/#. Accessed April 29, 2017.
8. US Department of Health and Human Services, Office of Women’s Health. Iron-deficiency anemia. www.womenshealth.gov/publications/our-publications/fact-sheet/anemia.html#a. Accessed April 29, 2017.
9. DeLoughery TG. Microcytic anemia. N Engl J Med. 2014;371(14): 1324-1331.
10. Tefferi A, Hanson CA, Inwards DJ. How to interpret and pursue an abnormal complete blood cell count in adults. Mayo Clin Proc. 2005;80(7):923-936.
11. Camaschella C. Iron-deficiency anemia. N Engl J Med. 2015; 372(19):1832-1843.
12. Killip S, Bennett JM, Chambers MD. Iron deficiency anemia. Am Fam Physician. 2007;75(5):671-678.
13. Rodgers GP, Young N. The Bethesda Handbook of Clinical Hematology. 3rd ed. Baltimore: Lippincott Williams and Wilkins; 2013:1-21.
14. Kaferle J, Strzoda CE. Evaluation of macrocytosis. Am Fam Physician. 2009;79(3):203-208.
15. Hesdorffer CS, Longo DL. Drug-induced megaloblastic anemia. N Engl J Med. 2015;373(17):1649-1658.
16. Vasconcelos OM, Poehm EH, McCarter RJ, et al. Potential outcome factors in subacute combined degeneration. J Gen Intern Med. 2006;21(10):1063-1068.
17. Vide AT, Marques AM, Costa JD. MRI findings in subacute combined degeneration of the spinal cord in a patient with restricted diet. Neurol Sci. 2011;33(3):711-713.
18. Kessenich CR, Cronin K. Fecal occult blood testing in older adult patients with anemia. Nurse Pract. 2013;38(1):6-8.
19. Imashuku S, Kudo N, Kaneda S. Spontaneous resolution of macrocytic anemia: old disease revisited. J Blood Med. 2012;3:45-47.
20. Pagana K, Pagana T, Pagana T. Mosby’s Diagnostic and Laboratory Test Reference. 12th ed. St. Louis, MO: Elsevier Mosby; 2015: 497-501, 785-791, 805-806.
CE/CME No: CR-1708
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Discuss the importance of diagnosing the type of anemia in order to provide appropriate treatment.
• Describe how the complete blood count and its indices are used to initially determine if an anemia is microcytic, normocytic, or macrocytic.
• List the more common causes of microcytic, normocytic, and macrocytic anemia.
• Discuss addictional laboratory tests that may be used to further assess the cause of anemia.
FACULTY
Jean O’Neil is an Assistant Professor and Coordinator of the Adult Gerontology Acute Care Nurse Practitioner Program in the Patricia A. Chin School of Nursing at California State University, Los Angeles.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through July 31, 2018.
Article begins on next page >>
Anemia affects more than 3 million people in the United States, making it a common problem in primary care practices. Once anemia is detected, clinicians must define the type and identify its underlying cause prior to initiating treatment. In most cases, the cause can be determined using information from the patient history, physical exam, and complete blood count.
Anemia is commonly identified during routine physical exams and laboratory testing.1-3 However, treating anemia can present a challenge for the primary care provider if the immediate cause is not apparent. Iron deficiency is a leading cause of anemia, but simply prescribing an iron supplement without determining the type or the cause of the anemia is not appropriate. Anemia that is misdiagnosed or goes untreated can be associated with a worse prognosis, as well as increased health care costs.4
Primary care providers often manage patients with common types of anemia and refer patients with severe or complex anemia to specialists for further testing and treatment. The most commonly used and cost-effective diagnostic tool for anemia is the complete blood count (CBC).2-6 The CBC provides details that can help the provider determine the type of anemia present, which in turn guides proper diagnostic testing and treatment.
EPIDEMIOLOGY
Anemia involves a reduction in the number of circulating red blood cells, the blood hemoglobin content, or the hematocrit, which leads to impaired delivery of oxygen to the body. Anemia affects more than 2 billion people worldwide, with iron deficiency the most common cause.7 Other leading nutritional causes of anemia include vitamin B12 and folate deficiency.4,7 Approximately 3 to 4 million Americans have anemia in some form, and it affects about 6.6% of men and 12.4% of women.5,8 The prevalence of anemia increases with age. Approximately 11% of men and 10% of women ages 65 or older have anemia, and in men ages 85 or older, prevalence of 20% to 44% has been reported.1,4 Anemia is present in about 3.5% of patients with chronic disease, but only 15% of them receive treatment.4
PATHOPHYSIOLOGY
Blood is composed of water-based plasma (54%), white blood cells and platelets (1%), and red blood cells (45%).5 Hemoglobin, the primary protein of the red blood cell, binds oxygen from the lungs and transports it to the rest of the body. Oxygen is then exchanged for carbon dioxide, which is carried back to the lungs to be exhaled.
Hemoglobin is made up of four globin chains, each containing an iron ion held in a porphyrin ring known as a heme group.5 When the body detects low tissue oxygen, the endothelial cells in the kidneys secrete the hormone erythropoietin (EPO), which stimulates the bone marrow to increase red cell production.5 This feedback loop can be interrupted by renal failure or chronic disease.4 In addition, bone marrow cannot produce enough red blood cells if there are insufficient levels of iron, amino acids, protein, carbohydrates, lipids, folate, and vitamin B12.5 Toxins (eg, lead), some types of cancer (eg, lymphoma), or even common infections (eg, pneumonia) can suppress the bone marrow, causing anemia. The more severe the anemia, the more likely oxygen transport will be compromised and organ failure will ensue.
Mutations affecting the genes that encode the globin chains within hemoglobin can cause one of the more than 600 known hemoglobinopathies (genetic defects of hemoglobin structure), such as sickle cell disease and thalassemias.5,9 While it is important to identify and treat patients with hemoglobinopathies, most anemias have other causes, such as iron deficiency, chronic disease, bone marrow defects, B12 deficiency, renal failure, medications, alcoholism, pregnancy, nutritional intake problems, gastrointestinal malabsorption, and active or recent history of blood loss.5,10
CLINICAL PRESENTATION
There are several signs and symptoms that should lead the primary care provider to suspect anemia (see Table 1).5,6 The severity of these symptoms can vary from mild to very serious. Severe anemia can lead to organ failure and death. However, most patients with anemia are asymptomatic, and anemia is typically detected incidentally during laboratory testing.1,2
Once anemia is confirmed, the evaluation focuses on diagnosing its underlying cause. It should include a thorough patient history and review of systems to ascertain whether the patient has symptoms such as increased fatigue, palpitations, gastrointestinal distress, weakness, or dizziness.
If the provider has access to past CBC results, a comparison of the current and previous results will help determine whether the anemia is acute or chronic. Anemia caused by acute conditions, such as a suspicion of blood loss or bone marrow suppression, must be attended to immediately. A patient with chronic anemia should be carefully monitored and may need follow-up for ongoing treatment. While a provider has more time to work up a patient with chronic anemia, the causes may not be as straightforward.
DIAGNOSIS AND CLASSIFICATION
Anemia in adults is defined as hemoglobin less than 13 g/dL in males and 12 g/dL in females.6 The hemoglobin is part of the complete blood cell report, which also includes the white blood cell count (WBC), red blood cell count (RBC), hematocrit, platelet count, and indices.
When investigating the underlying cause of anemia, the most useful parts of the CBC are the hemoglobin and the mean corpuscular volume (MCV; see Table 2).6,10 The MCV is the average volume of red cells in a specimen. This parameter is used to classify the anemia as microcytic (MCV < 80 fL), normocytic (MCV 80-100 fL), or macrocytic (MCV > 100 fL), which helps to narrow the differential diagnosis and guide any further testing (see Figure).5,6,10
It is important to note that the normal ranges of the CBC parameters differ based on race, with persons of African ancestry having lower normal hemoglobin levels than persons of Caucasian ancestry.10 In addition, laboratories may have slightly different normal values for the CBC based on the equipment they utilize. Therefore, providers must follow their laboratory’s parameters, as well as adjust for the patient’s gender, age, and ethnicity.10
Microcytic Anemia
Iron deficiency
In microcytic anemia, the RBCs are smaller than average (MCV < 80 fL), as well as hypochromic due to lack of hemoglobin.9 Iron deficiency is the most common cause of microcytic anemia worldwide.11,12 Therefore, when a patient has microcytic anemia, a serum ferritin needs to be ordered. Further testing of total iron-binding capacity (TIBC), transferrin saturation, serum iron, and serum receptor levels may be helpful if the ferritin level is between 46-99 ng/mL and anemia due to iron deficiency is not confirmed (see Table 2).12
In iron deficiency anemia, serum ferritin and serum iron levels are low due to lack of iron, but serum TIBC is high.6 The elevated TIBC reflects increased synthesis of transferrin by the liver as it attempts to compensate for the patient’s low serum iron level.9 Since iron levels are controlled by absorption rather than excretion, iron is essentially only depleted from the body through blood loss.12 Therefore, an adult patient who is iron deficient has lost more iron through blood loss than was replaced through nutritional intake and gastrointestinal absorption. In children, increased growth-related iron requirements combined with poor nutritional intake of iron-rich foods is an additional mechanism for iron deficiency.11
Iron def
If the nutritional problem is corrected or the source of bleeding is controlled, treatment with oral or intravenous iron supplements should result in improved serum hemoglobin and reticulocyte counts.13 In the primary care setting, ferrous sulfate 325 mg, which provides 65 mg of elemental iron per tablet, orally three times daily is recommended for adults.13 This gives the patient the recommended dose of approximately 200 mg of elemental iron. Repeat hemoglobin and iron studies should be conducted again in three to six months.12,13
If the patient’s iron deficiency anemia does not improve after oral iron therapy, there may be a source of blood loss the provider missed or a problem with malabsorption of iron, which can be seen in those who have undergone gastric bypass surgery or who have inflammatory bowel disease.13 Such patients should be referred to a specialist, such as a gastroenterologist, for further evaluation.
Thalassemia
Microcytic anemia with normal or elevated serum iron and normal-to-increased serum ferritin can be seen in patients with a type of thalassemia (see Figure).2 Thalassemias are inherited blood disorders that reduce hemoglobin production, leading to microcytosis; they are more common in those of Mediterranean, African, and Southeast Asian descent.2 Red cells in patients with a form of thalassemia are usually very small (microcytic) and have normal or elevated red cell distribution width (RDW).10
Moderate and severe forms of thalassemia can cause anemia. However, thalassemia syndromes that can cause severe (transfusion-dependent) anemia are usually diagnosed in childhood.9 Patients with one of the minor forms of thalassemia typically need minimal to no treatment.5 A patient with significant anemia suspicious for thalassemia should undergo hemoglobin electrophoresis testing to confirm the diagnosis and to determine the type of thalassemia.2 Typically, hemoglobin electrophoresis is normal in α thalassemia and is abnormal in ß thalassemia, as well as other forms of thalassemia. Referral to a hematologist for interpretation of these results and for further evaluation is appropriate.10
Chronic disease
If the patient has microcytic anemia and is not iron deficient or does not have thalassemia, then anemia related to a chronic disease should be considered.5 In such cases, the provider should order a reticulocyte count, which reveals how the bone marrow is responding to the anemia.5 Reticulocytes are immature red cells that have just been released from the bone marrow into the blood stream. The bone marrow increases the release of these cells in response to anemia.6
Any condition that stimulates reticulocyte production or prevents the bone marrow from producing reticulocytes will result in abnormal values (see Table 3). A normal reticulocyte count, expressed as the reticulocyte production index, is between 0.5% and 1.5%.5 The reticulocyte count is low in iron deficiency anemia and diseases that lead to decreased bone marrow production.5,6 Bone marrow suppression can occur in the context of chronic disease, infection, or inflammation. Malignancies are a less common cause for chronic disease microcytic anemia.6
If the cause of the decreased reticulocyte count is iron deficiency anemia, then treatment with iron supplementation should result in an increased reticulocyte count within one week.13 The primary care provider works in conjunction with the specialist to monitor the patient’s anemia when it is due to chronic disease or malignancy.
MACROCYTIC ANEMIA
In macrocytic anemia, the RBCs are larger than normal (MCV > 100 fL). This form of anemia is usually caused by vitamin B12 and folate deficiency, but it can also result from alcoholism, certain medications (eg, chemotherapy, antivirals), bone marrow disorders (eg, leukemia), and liver disease (eg, cirrhosis; see Figure).5,14 Common medications that can cause macrocytosis include the antiseizure drug phenytoin, the antibiotics trimethoprim/sulfamethoxazole and nitrofurantoin, the disease-modifying antirheumatic drug sulfasalazine, and immunosuppressants such as azathioprine.14,15 Antiviral agents, such as reverse transcriptase inhibitors (eg, zidovudine) used to treat HIV infection, can also cause macrocytosis with or without anemia.6,14
Macrocytic anemias caused by low serum levels of B12 and folate usually reflect problems with gastrointestinal malabsorption. For example, gastric bypass or Crohn disease can lead to malabsorption of vitamin B12 and increase a patient’s risk for macrocytic anemia.13
Vitamin B12 deficiency occurs in patients with pernicious anemia because they are missing intrinsic factor, which is necessary to facilitate B12 absorption in the ileum.10 Low vitamin B12 and folate levels also can result from inadequate dietary intake, although this is rare in the United States due to mandatory fortification of certain foods. A diet low in fresh vegetables is the leading cause of folate deficiency. While folate deficiency related to poor nutritional intake can be seen in all age groups, vitamin B12 deficiency more frequently affects the elderly or persons following a strict vegan diet.14
In addition to the fatigue and pallor associated with macrocytic anemia, patients with vitamin B12 deficiency may also have a smooth tongue, peripheral neuropathy, and edema.5,14 Severe vitamin B12 deficiency can lead to subacute combined degeneration of the spinal cord, with demyelination of the dorsal and lateral columns most often occurring in the cervical and thoracic regions.16,17 This spinal cord degeneration can cause paresthesia, muscle spasticity, and ataxia.16
When there is a macrocytic anemia, but the B12 or folate level is only borderline low, additional tests should be performed to help distinguish between B12 and folate deficiency. Both B12 and folate deficiencies can cause elevated homocysteine levels.13 Clinically significant B12 deficiency causes elevation of methylmalonic acid (MMA), whereas folate deficiency does not.13,14 Elevation of MMA can be very sensitive for B12 deficiency but lacks specificity in certain situations, such as pregnancy, renal insufficiency, and advanced age.13,14
Treatment of vitamin B12 and folate deficiencies with supplementation prevents progression of the disease, and has the potential to relieve most of the symptoms. Oral, sublingual, or parenteral vitamin B12 or oral folate supplements can be started in the primary care setting once the provider has identified whether the patient is B12 deficient, folate deficient, or both.
The vitamin B12 dose used for deficiency-induced macrocytic anemia depends on the cause—for example, a temporary condition such as pregnancy versus a lifelong disorder such as pernicious anemia.13 The usual oral dosing regimen is 2 mg/d; if intramuscular injections are used, 50 to 100 mcg are given daily for a week, followed by weekly injections for a month, and then monthly injections of 1 mg for life, if necessary.13 Bone marrow response to supplemental B12 is very rapid, with increased reticulocyte counts seen within four or five days.13
The usual dose for oral folic acid is 1 mg/d as needed.13 Folic acid can be given for folate deficiency only if the vitamin B12 level is normal. Giving folate to a patient with untreated vitamin B12 deficiency can potentially worsen subacute combined degeneration of the spinal cord.13,16
NORMOCYTIC ANEMIA
In normocytic anemia, the hemoglobin is low but the MCV is normal (see Figure).1 The history and physical exam should provide clues about whether the underlying cause of the anemia requires emergent (eg, active bleeding) or nonemergent (eg, anemia of chronic disease) management. Some of the causes of normocytic anemia are active bleeding, pregnancy, malnutrition, renal failure, chronic disease, hemolytic disorders, hypersplenism, congenital disorders, endocrine disorders, infection, and primary bone marrow disorders.1,5 Expanded plasma volume, as seen in pregnancy and overhydration, can also cause normocytic anemia.5 If gastrointestinal bleeding is suspected or the patient reports dark, tarry stools consistent with melena, fecal occult blood testing should be done. A positive result strongly supports gastrointestinal bleeding as the cause of the anemia.18
The reticulocyte count can also be helpful in identifying the cause of this type of anemia. A normocytic anemia with a normal reticulocyte and normal RDW count is usually related to chronic disease.1,10 For example, chronic kidney disease (CKD) is associated with decreased EPO production due to impaired renal function, which leads to reduced erythropoiesis. Decreased EPO prevents the bone marrow from making red blood cells, resulting in anemia. However, a normocytic anemia with an elevated reticulocyte count points to bleeding or hemolysis, as the reticulosis shows that the bone marrow is increasing red cell production to make up for the lost red cells.5
Additional diagnostic laboratory testing for patients with normocytic anemia may involve, for example, creatinine and blood urea nitrogen for patients with CKD, prothrombin time with an INR and liver function tests for patients with liver disease, and urine human chorionic gonadotropin if pregnancy is suspected.
For patients with an infection that is causing severe hemolysis (eg, sepsis due to a ß-hemolytic streptococcal infection), blood cultures should be drawn.5 If red blood cell destruction due to an artificial cardiac valve or an autoimmune disorder is suspected as the cause of the anemia, a hematology consult is needed.1 Anemia caused by disseminated intravascular coagulation or thrombotic thrombocytopenic purpura resulting in hemolysis are usually emergent conditions that require immediate intervention, including hospitalization and management by a hematologist.1
PATIENT EDUCATION
Patients and any accompanying family members should be educated about the signs and symptoms of anemia, the diagnostic testing and treatment regimens specific to their anemia, and medication compliance issues.
For instance, patients who abuse alcohol often have both vitamin B12 and folate deficiencies. If the macrocytosis is caused by alcohol intake, then the provider should educate the patient on the importance of alcohol abstention, as well as refer the patient for rehabilitation and psychologic counseling, as needed. These patients can sometimes recover from macrocytic anemia simply by stopping alcohol intake and improving their nutrition.19 Patients with microcytosis due to iron deficiency anemia should be advised about the importance of good nutrition and compliance with iron supplementation.
Repeat CBCs and a follow-up patient history and physical exam will help the provider assess whether the anemia is resolving. Individualized plans that target the specific type of anemia identified, as well as its underlying cause, are key to successful treatment.
CONCLUSION
When managing a patient with anemia, providers must define the type of anemia present and identify its underlying cause before starting treatment. Clues from the patient’s history, physical exam, and CBC can help isolate the cause of anemia. The MCV is the most helpful of the red blood cell indices because it allows the provider to classify the anemia as microcytic, macrocytic, or normocytic.
In cases in which the anemia is acute or severe—or in which the patient remains anemic even after being treated by the primary care provider—referral to a specialist is appropriate.
CE/CME No: CR-1708
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Discuss the importance of diagnosing the type of anemia in order to provide appropriate treatment.
• Describe how the complete blood count and its indices are used to initially determine if an anemia is microcytic, normocytic, or macrocytic.
• List the more common causes of microcytic, normocytic, and macrocytic anemia.
• Discuss addictional laboratory tests that may be used to further assess the cause of anemia.
FACULTY
Jean O’Neil is an Assistant Professor and Coordinator of the Adult Gerontology Acute Care Nurse Practitioner Program in the Patricia A. Chin School of Nursing at California State University, Los Angeles.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid through July 31, 2018.
Article begins on next page >>
Anemia affects more than 3 million people in the United States, making it a common problem in primary care practices. Once anemia is detected, clinicians must define the type and identify its underlying cause prior to initiating treatment. In most cases, the cause can be determined using information from the patient history, physical exam, and complete blood count.
Anemia is commonly identified during routine physical exams and laboratory testing.1-3 However, treating anemia can present a challenge for the primary care provider if the immediate cause is not apparent. Iron deficiency is a leading cause of anemia, but simply prescribing an iron supplement without determining the type or the cause of the anemia is not appropriate. Anemia that is misdiagnosed or goes untreated can be associated with a worse prognosis, as well as increased health care costs.4
Primary care providers often manage patients with common types of anemia and refer patients with severe or complex anemia to specialists for further testing and treatment. The most commonly used and cost-effective diagnostic tool for anemia is the complete blood count (CBC).2-6 The CBC provides details that can help the provider determine the type of anemia present, which in turn guides proper diagnostic testing and treatment.
EPIDEMIOLOGY
Anemia involves a reduction in the number of circulating red blood cells, the blood hemoglobin content, or the hematocrit, which leads to impaired delivery of oxygen to the body. Anemia affects more than 2 billion people worldwide, with iron deficiency the most common cause.7 Other leading nutritional causes of anemia include vitamin B12 and folate deficiency.4,7 Approximately 3 to 4 million Americans have anemia in some form, and it affects about 6.6% of men and 12.4% of women.5,8 The prevalence of anemia increases with age. Approximately 11% of men and 10% of women ages 65 or older have anemia, and in men ages 85 or older, prevalence of 20% to 44% has been reported.1,4 Anemia is present in about 3.5% of patients with chronic disease, but only 15% of them receive treatment.4
PATHOPHYSIOLOGY
Blood is composed of water-based plasma (54%), white blood cells and platelets (1%), and red blood cells (45%).5 Hemoglobin, the primary protein of the red blood cell, binds oxygen from the lungs and transports it to the rest of the body. Oxygen is then exchanged for carbon dioxide, which is carried back to the lungs to be exhaled.
Hemoglobin is made up of four globin chains, each containing an iron ion held in a porphyrin ring known as a heme group.5 When the body detects low tissue oxygen, the endothelial cells in the kidneys secrete the hormone erythropoietin (EPO), which stimulates the bone marrow to increase red cell production.5 This feedback loop can be interrupted by renal failure or chronic disease.4 In addition, bone marrow cannot produce enough red blood cells if there are insufficient levels of iron, amino acids, protein, carbohydrates, lipids, folate, and vitamin B12.5 Toxins (eg, lead), some types of cancer (eg, lymphoma), or even common infections (eg, pneumonia) can suppress the bone marrow, causing anemia. The more severe the anemia, the more likely oxygen transport will be compromised and organ failure will ensue.
Mutations affecting the genes that encode the globin chains within hemoglobin can cause one of the more than 600 known hemoglobinopathies (genetic defects of hemoglobin structure), such as sickle cell disease and thalassemias.5,9 While it is important to identify and treat patients with hemoglobinopathies, most anemias have other causes, such as iron deficiency, chronic disease, bone marrow defects, B12 deficiency, renal failure, medications, alcoholism, pregnancy, nutritional intake problems, gastrointestinal malabsorption, and active or recent history of blood loss.5,10
CLINICAL PRESENTATION
There are several signs and symptoms that should lead the primary care provider to suspect anemia (see Table 1).5,6 The severity of these symptoms can vary from mild to very serious. Severe anemia can lead to organ failure and death. However, most patients with anemia are asymptomatic, and anemia is typically detected incidentally during laboratory testing.1,2
Once anemia is confirmed, the evaluation focuses on diagnosing its underlying cause. It should include a thorough patient history and review of systems to ascertain whether the patient has symptoms such as increased fatigue, palpitations, gastrointestinal distress, weakness, or dizziness.
If the provider has access to past CBC results, a comparison of the current and previous results will help determine whether the anemia is acute or chronic. Anemia caused by acute conditions, such as a suspicion of blood loss or bone marrow suppression, must be attended to immediately. A patient with chronic anemia should be carefully monitored and may need follow-up for ongoing treatment. While a provider has more time to work up a patient with chronic anemia, the causes may not be as straightforward.
DIAGNOSIS AND CLASSIFICATION
Anemia in adults is defined as hemoglobin less than 13 g/dL in males and 12 g/dL in females.6 The hemoglobin is part of the complete blood cell report, which also includes the white blood cell count (WBC), red blood cell count (RBC), hematocrit, platelet count, and indices.
When investigating the underlying cause of anemia, the most useful parts of the CBC are the hemoglobin and the mean corpuscular volume (MCV; see Table 2).6,10 The MCV is the average volume of red cells in a specimen. This parameter is used to classify the anemia as microcytic (MCV < 80 fL), normocytic (MCV 80-100 fL), or macrocytic (MCV > 100 fL), which helps to narrow the differential diagnosis and guide any further testing (see Figure).5,6,10
It is important to note that the normal ranges of the CBC parameters differ based on race, with persons of African ancestry having lower normal hemoglobin levels than persons of Caucasian ancestry.10 In addition, laboratories may have slightly different normal values for the CBC based on the equipment they utilize. Therefore, providers must follow their laboratory’s parameters, as well as adjust for the patient’s gender, age, and ethnicity.10
Microcytic Anemia
Iron deficiency
In microcytic anemia, the RBCs are smaller than average (MCV < 80 fL), as well as hypochromic due to lack of hemoglobin.9 Iron deficiency is the most common cause of microcytic anemia worldwide.11,12 Therefore, when a patient has microcytic anemia, a serum ferritin needs to be ordered. Further testing of total iron-binding capacity (TIBC), transferrin saturation, serum iron, and serum receptor levels may be helpful if the ferritin level is between 46-99 ng/mL and anemia due to iron deficiency is not confirmed (see Table 2).12
In iron deficiency anemia, serum ferritin and serum iron levels are low due to lack of iron, but serum TIBC is high.6 The elevated TIBC reflects increased synthesis of transferrin by the liver as it attempts to compensate for the patient’s low serum iron level.9 Since iron levels are controlled by absorption rather than excretion, iron is essentially only depleted from the body through blood loss.12 Therefore, an adult patient who is iron deficient has lost more iron through blood loss than was replaced through nutritional intake and gastrointestinal absorption. In children, increased growth-related iron requirements combined with poor nutritional intake of iron-rich foods is an additional mechanism for iron deficiency.11
Iron def
If the nutritional problem is corrected or the source of bleeding is controlled, treatment with oral or intravenous iron supplements should result in improved serum hemoglobin and reticulocyte counts.13 In the primary care setting, ferrous sulfate 325 mg, which provides 65 mg of elemental iron per tablet, orally three times daily is recommended for adults.13 This gives the patient the recommended dose of approximately 200 mg of elemental iron. Repeat hemoglobin and iron studies should be conducted again in three to six months.12,13
If the patient’s iron deficiency anemia does not improve after oral iron therapy, there may be a source of blood loss the provider missed or a problem with malabsorption of iron, which can be seen in those who have undergone gastric bypass surgery or who have inflammatory bowel disease.13 Such patients should be referred to a specialist, such as a gastroenterologist, for further evaluation.
Thalassemia
Microcytic anemia with normal or elevated serum iron and normal-to-increased serum ferritin can be seen in patients with a type of thalassemia (see Figure).2 Thalassemias are inherited blood disorders that reduce hemoglobin production, leading to microcytosis; they are more common in those of Mediterranean, African, and Southeast Asian descent.2 Red cells in patients with a form of thalassemia are usually very small (microcytic) and have normal or elevated red cell distribution width (RDW).10
Moderate and severe forms of thalassemia can cause anemia. However, thalassemia syndromes that can cause severe (transfusion-dependent) anemia are usually diagnosed in childhood.9 Patients with one of the minor forms of thalassemia typically need minimal to no treatment.5 A patient with significant anemia suspicious for thalassemia should undergo hemoglobin electrophoresis testing to confirm the diagnosis and to determine the type of thalassemia.2 Typically, hemoglobin electrophoresis is normal in α thalassemia and is abnormal in ß thalassemia, as well as other forms of thalassemia. Referral to a hematologist for interpretation of these results and for further evaluation is appropriate.10
Chronic disease
If the patient has microcytic anemia and is not iron deficient or does not have thalassemia, then anemia related to a chronic disease should be considered.5 In such cases, the provider should order a reticulocyte count, which reveals how the bone marrow is responding to the anemia.5 Reticulocytes are immature red cells that have just been released from the bone marrow into the blood stream. The bone marrow increases the release of these cells in response to anemia.6
Any condition that stimulates reticulocyte production or prevents the bone marrow from producing reticulocytes will result in abnormal values (see Table 3). A normal reticulocyte count, expressed as the reticulocyte production index, is between 0.5% and 1.5%.5 The reticulocyte count is low in iron deficiency anemia and diseases that lead to decreased bone marrow production.5,6 Bone marrow suppression can occur in the context of chronic disease, infection, or inflammation. Malignancies are a less common cause for chronic disease microcytic anemia.6
If the cause of the decreased reticulocyte count is iron deficiency anemia, then treatment with iron supplementation should result in an increased reticulocyte count within one week.13 The primary care provider works in conjunction with the specialist to monitor the patient’s anemia when it is due to chronic disease or malignancy.
MACROCYTIC ANEMIA
In macrocytic anemia, the RBCs are larger than normal (MCV > 100 fL). This form of anemia is usually caused by vitamin B12 and folate deficiency, but it can also result from alcoholism, certain medications (eg, chemotherapy, antivirals), bone marrow disorders (eg, leukemia), and liver disease (eg, cirrhosis; see Figure).5,14 Common medications that can cause macrocytosis include the antiseizure drug phenytoin, the antibiotics trimethoprim/sulfamethoxazole and nitrofurantoin, the disease-modifying antirheumatic drug sulfasalazine, and immunosuppressants such as azathioprine.14,15 Antiviral agents, such as reverse transcriptase inhibitors (eg, zidovudine) used to treat HIV infection, can also cause macrocytosis with or without anemia.6,14
Macrocytic anemias caused by low serum levels of B12 and folate usually reflect problems with gastrointestinal malabsorption. For example, gastric bypass or Crohn disease can lead to malabsorption of vitamin B12 and increase a patient’s risk for macrocytic anemia.13
Vitamin B12 deficiency occurs in patients with pernicious anemia because they are missing intrinsic factor, which is necessary to facilitate B12 absorption in the ileum.10 Low vitamin B12 and folate levels also can result from inadequate dietary intake, although this is rare in the United States due to mandatory fortification of certain foods. A diet low in fresh vegetables is the leading cause of folate deficiency. While folate deficiency related to poor nutritional intake can be seen in all age groups, vitamin B12 deficiency more frequently affects the elderly or persons following a strict vegan diet.14
In addition to the fatigue and pallor associated with macrocytic anemia, patients with vitamin B12 deficiency may also have a smooth tongue, peripheral neuropathy, and edema.5,14 Severe vitamin B12 deficiency can lead to subacute combined degeneration of the spinal cord, with demyelination of the dorsal and lateral columns most often occurring in the cervical and thoracic regions.16,17 This spinal cord degeneration can cause paresthesia, muscle spasticity, and ataxia.16
When there is a macrocytic anemia, but the B12 or folate level is only borderline low, additional tests should be performed to help distinguish between B12 and folate deficiency. Both B12 and folate deficiencies can cause elevated homocysteine levels.13 Clinically significant B12 deficiency causes elevation of methylmalonic acid (MMA), whereas folate deficiency does not.13,14 Elevation of MMA can be very sensitive for B12 deficiency but lacks specificity in certain situations, such as pregnancy, renal insufficiency, and advanced age.13,14
Treatment of vitamin B12 and folate deficiencies with supplementation prevents progression of the disease, and has the potential to relieve most of the symptoms. Oral, sublingual, or parenteral vitamin B12 or oral folate supplements can be started in the primary care setting once the provider has identified whether the patient is B12 deficient, folate deficient, or both.
The vitamin B12 dose used for deficiency-induced macrocytic anemia depends on the cause—for example, a temporary condition such as pregnancy versus a lifelong disorder such as pernicious anemia.13 The usual oral dosing regimen is 2 mg/d; if intramuscular injections are used, 50 to 100 mcg are given daily for a week, followed by weekly injections for a month, and then monthly injections of 1 mg for life, if necessary.13 Bone marrow response to supplemental B12 is very rapid, with increased reticulocyte counts seen within four or five days.13
The usual dose for oral folic acid is 1 mg/d as needed.13 Folic acid can be given for folate deficiency only if the vitamin B12 level is normal. Giving folate to a patient with untreated vitamin B12 deficiency can potentially worsen subacute combined degeneration of the spinal cord.13,16
NORMOCYTIC ANEMIA
In normocytic anemia, the hemoglobin is low but the MCV is normal (see Figure).1 The history and physical exam should provide clues about whether the underlying cause of the anemia requires emergent (eg, active bleeding) or nonemergent (eg, anemia of chronic disease) management. Some of the causes of normocytic anemia are active bleeding, pregnancy, malnutrition, renal failure, chronic disease, hemolytic disorders, hypersplenism, congenital disorders, endocrine disorders, infection, and primary bone marrow disorders.1,5 Expanded plasma volume, as seen in pregnancy and overhydration, can also cause normocytic anemia.5 If gastrointestinal bleeding is suspected or the patient reports dark, tarry stools consistent with melena, fecal occult blood testing should be done. A positive result strongly supports gastrointestinal bleeding as the cause of the anemia.18
The reticulocyte count can also be helpful in identifying the cause of this type of anemia. A normocytic anemia with a normal reticulocyte and normal RDW count is usually related to chronic disease.1,10 For example, chronic kidney disease (CKD) is associated with decreased EPO production due to impaired renal function, which leads to reduced erythropoiesis. Decreased EPO prevents the bone marrow from making red blood cells, resulting in anemia. However, a normocytic anemia with an elevated reticulocyte count points to bleeding or hemolysis, as the reticulosis shows that the bone marrow is increasing red cell production to make up for the lost red cells.5
Additional diagnostic laboratory testing for patients with normocytic anemia may involve, for example, creatinine and blood urea nitrogen for patients with CKD, prothrombin time with an INR and liver function tests for patients with liver disease, and urine human chorionic gonadotropin if pregnancy is suspected.
For patients with an infection that is causing severe hemolysis (eg, sepsis due to a ß-hemolytic streptococcal infection), blood cultures should be drawn.5 If red blood cell destruction due to an artificial cardiac valve or an autoimmune disorder is suspected as the cause of the anemia, a hematology consult is needed.1 Anemia caused by disseminated intravascular coagulation or thrombotic thrombocytopenic purpura resulting in hemolysis are usually emergent conditions that require immediate intervention, including hospitalization and management by a hematologist.1
PATIENT EDUCATION
Patients and any accompanying family members should be educated about the signs and symptoms of anemia, the diagnostic testing and treatment regimens specific to their anemia, and medication compliance issues.
For instance, patients who abuse alcohol often have both vitamin B12 and folate deficiencies. If the macrocytosis is caused by alcohol intake, then the provider should educate the patient on the importance of alcohol abstention, as well as refer the patient for rehabilitation and psychologic counseling, as needed. These patients can sometimes recover from macrocytic anemia simply by stopping alcohol intake and improving their nutrition.19 Patients with microcytosis due to iron deficiency anemia should be advised about the importance of good nutrition and compliance with iron supplementation.
Repeat CBCs and a follow-up patient history and physical exam will help the provider assess whether the anemia is resolving. Individualized plans that target the specific type of anemia identified, as well as its underlying cause, are key to successful treatment.
CONCLUSION
When managing a patient with anemia, providers must define the type of anemia present and identify its underlying cause before starting treatment. Clues from the patient’s history, physical exam, and CBC can help isolate the cause of anemia. The MCV is the most helpful of the red blood cell indices because it allows the provider to classify the anemia as microcytic, macrocytic, or normocytic.
In cases in which the anemia is acute or severe—or in which the patient remains anemic even after being treated by the primary care provider—referral to a specialist is appropriate.
1. Brill JR, Baumgardner D. Normocytic anemia. Am Fam Physician. 2000;62(10):2255-2263.
2. Van Vranken M. Evaluation of microcytosis. Am Fam Physician. 2010;82(9):1117-1122.
3. National Institutes of Health/National Heart, Lung, and Blood Institute. How is anemia diagnosed? www.nhlbi.nih.gov/health/health-topics/topics/anemia/diagnosis. Accessed April 28, 2017.
4. Smith RE Jr. The clinical and economic burden of anemia. Am J Manag Care. 2010;16(3):S59-S66.
5. Platt A, Eckman J. Diagnosing anemia. Clinician Reviews. 2006;16(2):44-50.
6. Karnath B. Anemia in the adult patient. Hosp Physician. 2004;40(10):32-36.
7. World Health Organization. Micronutrient deficiencies. www.who.int/nutrition/topics/ida/en/#. Accessed April 29, 2017.
8. US Department of Health and Human Services, Office of Women’s Health. Iron-deficiency anemia. www.womenshealth.gov/publications/our-publications/fact-sheet/anemia.html#a. Accessed April 29, 2017.
9. DeLoughery TG. Microcytic anemia. N Engl J Med. 2014;371(14): 1324-1331.
10. Tefferi A, Hanson CA, Inwards DJ. How to interpret and pursue an abnormal complete blood cell count in adults. Mayo Clin Proc. 2005;80(7):923-936.
11. Camaschella C. Iron-deficiency anemia. N Engl J Med. 2015; 372(19):1832-1843.
12. Killip S, Bennett JM, Chambers MD. Iron deficiency anemia. Am Fam Physician. 2007;75(5):671-678.
13. Rodgers GP, Young N. The Bethesda Handbook of Clinical Hematology. 3rd ed. Baltimore: Lippincott Williams and Wilkins; 2013:1-21.
14. Kaferle J, Strzoda CE. Evaluation of macrocytosis. Am Fam Physician. 2009;79(3):203-208.
15. Hesdorffer CS, Longo DL. Drug-induced megaloblastic anemia. N Engl J Med. 2015;373(17):1649-1658.
16. Vasconcelos OM, Poehm EH, McCarter RJ, et al. Potential outcome factors in subacute combined degeneration. J Gen Intern Med. 2006;21(10):1063-1068.
17. Vide AT, Marques AM, Costa JD. MRI findings in subacute combined degeneration of the spinal cord in a patient with restricted diet. Neurol Sci. 2011;33(3):711-713.
18. Kessenich CR, Cronin K. Fecal occult blood testing in older adult patients with anemia. Nurse Pract. 2013;38(1):6-8.
19. Imashuku S, Kudo N, Kaneda S. Spontaneous resolution of macrocytic anemia: old disease revisited. J Blood Med. 2012;3:45-47.
20. Pagana K, Pagana T, Pagana T. Mosby’s Diagnostic and Laboratory Test Reference. 12th ed. St. Louis, MO: Elsevier Mosby; 2015: 497-501, 785-791, 805-806.
1. Brill JR, Baumgardner D. Normocytic anemia. Am Fam Physician. 2000;62(10):2255-2263.
2. Van Vranken M. Evaluation of microcytosis. Am Fam Physician. 2010;82(9):1117-1122.
3. National Institutes of Health/National Heart, Lung, and Blood Institute. How is anemia diagnosed? www.nhlbi.nih.gov/health/health-topics/topics/anemia/diagnosis. Accessed April 28, 2017.
4. Smith RE Jr. The clinical and economic burden of anemia. Am J Manag Care. 2010;16(3):S59-S66.
5. Platt A, Eckman J. Diagnosing anemia. Clinician Reviews. 2006;16(2):44-50.
6. Karnath B. Anemia in the adult patient. Hosp Physician. 2004;40(10):32-36.
7. World Health Organization. Micronutrient deficiencies. www.who.int/nutrition/topics/ida/en/#. Accessed April 29, 2017.
8. US Department of Health and Human Services, Office of Women’s Health. Iron-deficiency anemia. www.womenshealth.gov/publications/our-publications/fact-sheet/anemia.html#a. Accessed April 29, 2017.
9. DeLoughery TG. Microcytic anemia. N Engl J Med. 2014;371(14): 1324-1331.
10. Tefferi A, Hanson CA, Inwards DJ. How to interpret and pursue an abnormal complete blood cell count in adults. Mayo Clin Proc. 2005;80(7):923-936.
11. Camaschella C. Iron-deficiency anemia. N Engl J Med. 2015; 372(19):1832-1843.
12. Killip S, Bennett JM, Chambers MD. Iron deficiency anemia. Am Fam Physician. 2007;75(5):671-678.
13. Rodgers GP, Young N. The Bethesda Handbook of Clinical Hematology. 3rd ed. Baltimore: Lippincott Williams and Wilkins; 2013:1-21.
14. Kaferle J, Strzoda CE. Evaluation of macrocytosis. Am Fam Physician. 2009;79(3):203-208.
15. Hesdorffer CS, Longo DL. Drug-induced megaloblastic anemia. N Engl J Med. 2015;373(17):1649-1658.
16. Vasconcelos OM, Poehm EH, McCarter RJ, et al. Potential outcome factors in subacute combined degeneration. J Gen Intern Med. 2006;21(10):1063-1068.
17. Vide AT, Marques AM, Costa JD. MRI findings in subacute combined degeneration of the spinal cord in a patient with restricted diet. Neurol Sci. 2011;33(3):711-713.
18. Kessenich CR, Cronin K. Fecal occult blood testing in older adult patients with anemia. Nurse Pract. 2013;38(1):6-8.
19. Imashuku S, Kudo N, Kaneda S. Spontaneous resolution of macrocytic anemia: old disease revisited. J Blood Med. 2012;3:45-47.
20. Pagana K, Pagana T, Pagana T. Mosby’s Diagnostic and Laboratory Test Reference. 12th ed. St. Louis, MO: Elsevier Mosby; 2015: 497-501, 785-791, 805-806.

July 2017: Click for Credit
Here are 6 articles in the July issue of Clinician Reviews (individual articles are valid for one year from date of publication—expiration dates below):
1. High-dose Oral Vitamin D3 Significantly Reduced Effects of Sunburn
To take the posttest, go to: http://bit.ly/2tmDiKc
Expires May 23, 2018
2. Women Less Likely to Be Diagnosed With Sleep Disorders
To take the posttest, go to: http://bit.ly/2rgLdne
Expires May 30, 2018
3. RA Treatment Delays Raise Risk for Long-term Disability
To take the posttest, go to: http://bit.ly/2tC0IGF
Expires May 30, 2018
4. Target Self-medication of Mood and Anxiety Symptoms
To take the posttest, go to: http://bit.ly/2vy5jel
Expires May 2, 2018
5. Two New Biomarkers for Breast Cancer Show Validity
To take the posttest, go to: http://bit.ly/2ve9H2L
Expires May 2, 2018
6. Time to Therapy for Gram-positive Bacteremia Reduced From 60 Hours to 4 Hours
To take the posttest, go to: http://bit.ly/2ssacIf
Expires May 25, 2018
Here are 6 articles in the July issue of Clinician Reviews (individual articles are valid for one year from date of publication—expiration dates below):
1. High-dose Oral Vitamin D3 Significantly Reduced Effects of Sunburn
To take the posttest, go to: http://bit.ly/2tmDiKc
Expires May 23, 2018
2. Women Less Likely to Be Diagnosed With Sleep Disorders
To take the posttest, go to: http://bit.ly/2rgLdne
Expires May 30, 2018
3. RA Treatment Delays Raise Risk for Long-term Disability
To take the posttest, go to: http://bit.ly/2tC0IGF
Expires May 30, 2018
4. Target Self-medication of Mood and Anxiety Symptoms
To take the posttest, go to: http://bit.ly/2vy5jel
Expires May 2, 2018
5. Two New Biomarkers for Breast Cancer Show Validity
To take the posttest, go to: http://bit.ly/2ve9H2L
Expires May 2, 2018
6. Time to Therapy for Gram-positive Bacteremia Reduced From 60 Hours to 4 Hours
To take the posttest, go to: http://bit.ly/2ssacIf
Expires May 25, 2018
Here are 6 articles in the July issue of Clinician Reviews (individual articles are valid for one year from date of publication—expiration dates below):
1. High-dose Oral Vitamin D3 Significantly Reduced Effects of Sunburn
To take the posttest, go to: http://bit.ly/2tmDiKc
Expires May 23, 2018
2. Women Less Likely to Be Diagnosed With Sleep Disorders
To take the posttest, go to: http://bit.ly/2rgLdne
Expires May 30, 2018
3. RA Treatment Delays Raise Risk for Long-term Disability
To take the posttest, go to: http://bit.ly/2tC0IGF
Expires May 30, 2018
4. Target Self-medication of Mood and Anxiety Symptoms
To take the posttest, go to: http://bit.ly/2vy5jel
Expires May 2, 2018
5. Two New Biomarkers for Breast Cancer Show Validity
To take the posttest, go to: http://bit.ly/2ve9H2L
Expires May 2, 2018
6. Time to Therapy for Gram-positive Bacteremia Reduced From 60 Hours to 4 Hours
To take the posttest, go to: http://bit.ly/2ssacIf
Expires May 25, 2018

Looking for our CE/CME posttests?
You will find a "Login to Take Test" button on each CE/CME activity page. After logging in, you will see a "Take Test" button and will be able to proceed with your posttest and evaluation. We apologize for the temporary suspension in service and thank you for your patience. Please email ahoppel@frontlinemedcom.com if you encounter any additional difficulties.
Transcripts: You can access your certificates for any completed posttest/evaluation by clicking the “My CE/CME Transcript” link in the upper righthand corner of the homepage (www.mdedge.com/clinicianreviews). If prompted, type in the email address you use on our site and click “Look Up” for the fastest access to your transcript. If you experience any difficulties, please email ahoppel@frontlinemedcom.com.
You will find a "Login to Take Test" button on each CE/CME activity page. After logging in, you will see a "Take Test" button and will be able to proceed with your posttest and evaluation. We apologize for the temporary suspension in service and thank you for your patience. Please email ahoppel@frontlinemedcom.com if you encounter any additional difficulties.
Transcripts: You can access your certificates for any completed posttest/evaluation by clicking the “My CE/CME Transcript” link in the upper righthand corner of the homepage (www.mdedge.com/clinicianreviews). If prompted, type in the email address you use on our site and click “Look Up” for the fastest access to your transcript. If you experience any difficulties, please email ahoppel@frontlinemedcom.com.
You will find a "Login to Take Test" button on each CE/CME activity page. After logging in, you will see a "Take Test" button and will be able to proceed with your posttest and evaluation. We apologize for the temporary suspension in service and thank you for your patience. Please email ahoppel@frontlinemedcom.com if you encounter any additional difficulties.
Transcripts: You can access your certificates for any completed posttest/evaluation by clicking the “My CE/CME Transcript” link in the upper righthand corner of the homepage (www.mdedge.com/clinicianreviews). If prompted, type in the email address you use on our site and click “Look Up” for the fastest access to your transcript. If you experience any difficulties, please email ahoppel@frontlinemedcom.com.
Primary Hyperparathyroidism: A Case-based Review
CE/CME No: CR-1705
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Differentiate primary hyperparathyroidism (PHPT) from other causes of hypercalcemia and types of hyerparathyroidism.
• Understand the calcium-parathyroid hormone feedback loop.
• Identify appropriate imaging studies and common laboratory findings in the patient with PHPT.
• Describe the common systemic manifestations of PHPT.
• Discuss medical versus surgical management of the patient with PHPT.
FACULTY
Barbara Austin is a Family Nurse Practitioner at Baptist Primary Care, Jacksonville, Florida, and is pursuing a Doctorate of Nursing Practice (DNP) at Jacksonville University.
The author has no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of May 2017.
Article begins on next page >>
Primary hyperparathyroidism (PHPT) is most often detected as hypercalcemia in an asymptomatic patient during routine blood work. Knowing the appropriate work-up of hypercalcemia is essential, since untreated PHPT can have significant complications affecting multiple organ systems—most notably, renal and musculoskeletal. Parathyroidectomy is curative in up to 95% of cases, but prevention of long-term complications relies on prompt recognition and appropriate follow-up.
Primary hyperparathyroidism (PHPT) is a common endocrine disorder, with a prevalence of approximately 1 to 3 cases per 1,000 persons.1 PHPT results from inappropriate overproduction of parathyroid hormone (PTH), the primary regulator of calcium homeostasis, and is characterized by hypercalcemia in the setting of an elevated or high-normal PTH level. In most cases of PHPT, unregulated PTH production is caused by a single parathyroid adenoma.
PHPT is the most common cause of hypercalcemia in outpatients and is typically diagnosed following incidental discovery during routine blood work in an asymptomatic patient.1,2 It is two to three times more common in women than in men, and incidence increases with age; as such, postmenopausal women are most commonly affected.1,3 PHPT often has an insidious course, and recognition of its clinical manifestations followed by appropriate diagnostic work-up and management are necessary to prevent sequelae.3
PATIENT PRESENTATION
A 68-year-old Caucasian woman presented to her family practice office for a third visit with continued complaints of nontraumatic right lower leg pain. She had previously been diagnosed with tendonitis, which was treated conservatively. The pain failed to improve, and an x-ray was ordered. The x-ray revealed no acute findings but did show osteopenia, prompting an order for a bone mineral density (BMD) test. The BMD test demonstrated osteoporosis, which warranted further investigation. She was prescribed alendronate but refused it, against medical advice, due to concern over potential adverse effects.
Her medical history included hyperlipidemia and hypertension under fair control with lisinopril. She took a low-dose aspirin and flaxseed supplement daily. She also had a history of radiation to the neck, having undergone tonsillar irradiation as a child (a common practice from the 1930s through the 1950s).3 Surgical history included a total hysterectomy with bilateral salpingo-oophorectomy, appendectomy, and tonsillectomy. There was no personal or family history of cancer or endocrine disorders, hypercalcemia, or nephrolithiasis. She was up to date on vaccines and preventive health care measures. Allergies included penicillin and sulfa, both resulting in hives. She was a nonsmoker and did not drink alcohol or engage in illicit drug use.
Review of systems revealed right lower anterior leg pain for four months, characterized as aching, deep, sharp, and throbbing with radiation to the ankle. The pain was worse with activity and prolonged standing; ibuprofen and application of ice provided partial relief. She had experienced some mood changes, including irritability. Physical exam was normal except for dominant right-sided thyromegaly, marked bony point tenderness to the right midshin area, and an antalgic gait.
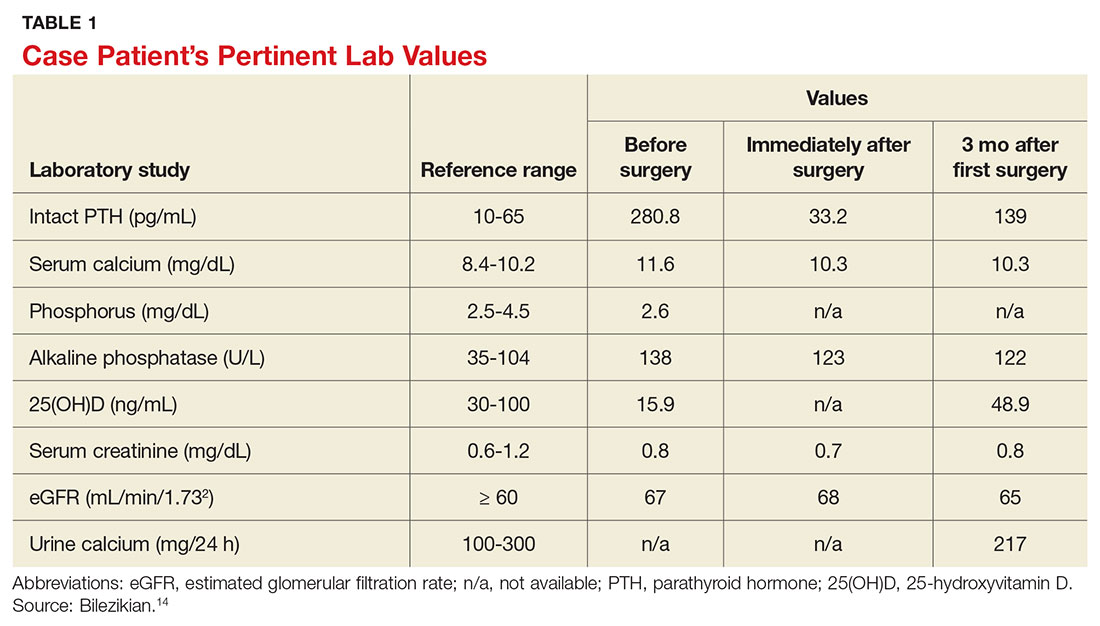
Laboratory work-up demonstrated elevated PTH, alkaline phosphatase, and calcium levels and a low 25-hydroxyvitamin D [25(OH)D] level (see Table 1). MRI of the right lower leg revealed a grade IV stress fracture (see Figure 1). The elevated serum calcium and PTH levels in addition to abnormal bone density findings led to the diagnosis of PHPT. She was referred to endocrinology and orthopedics for management of PHPT and the stress fracture, respectively, and was placed in an orthopedic walking boot for treatment of the midtibial stress fracture.
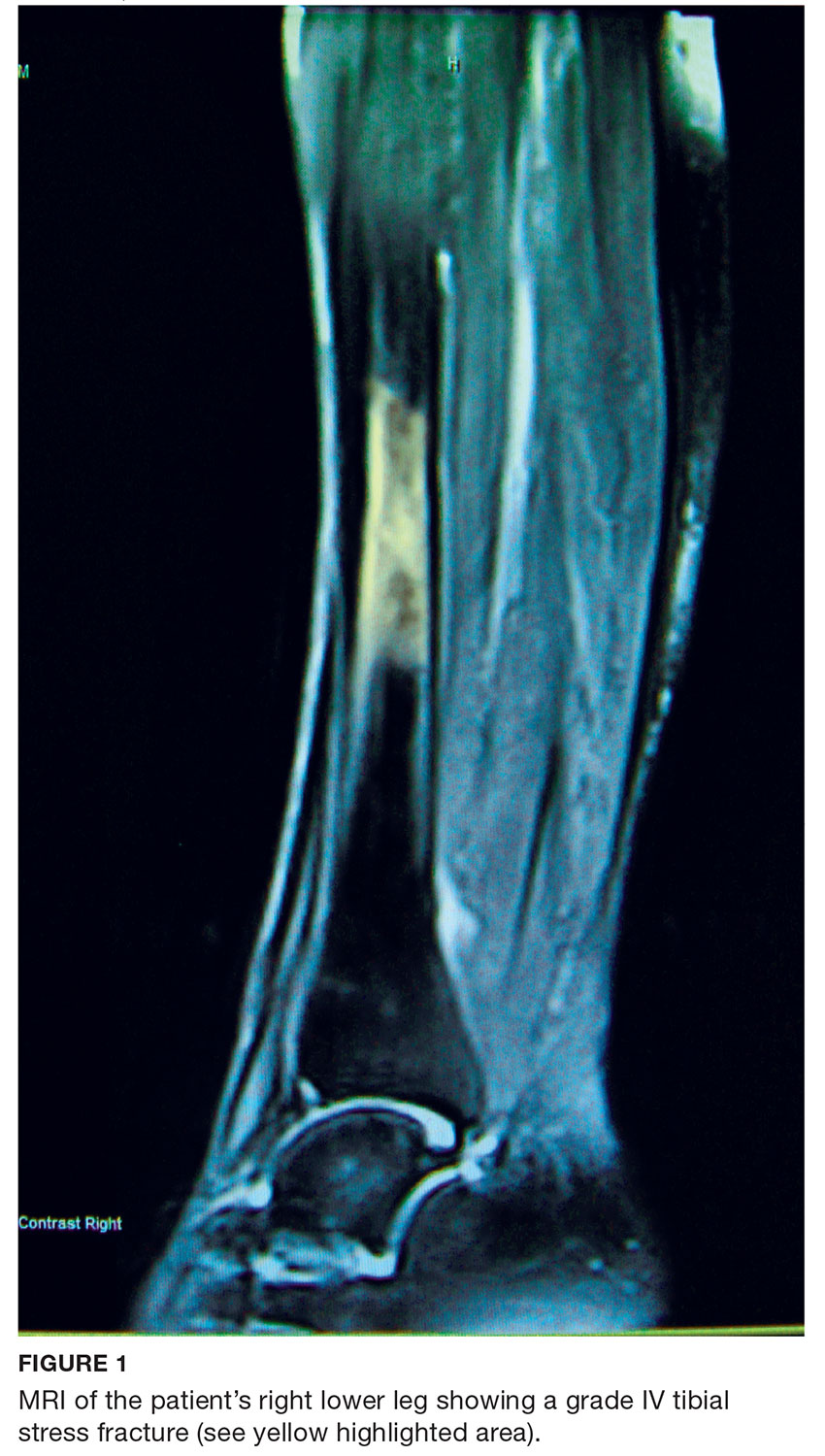
The endocrinologist referred her to an otolaryngologist trained in the surgical management of parathyroid adenomas, who ordered a thyroid ultrasound; this study was inconclusive. Additional imaging, including a Tc-99m sestamibi parathyroid scan and CT with contrast of the soft tissue of the neck, was obtained. The parathyroid scan of the neck and upper chest showed retained activity in the right inferior thyroid pole that was concerning for a parathyroid adenoma (see Figure 2). The CT identified a 1.5-cm parathyroid adenoma off the right inferior pole of the thyroid gland (concordant with the parathyroid scan). A single 300-mg parathyroid adenoma was removed from the right inferior pole of the thyroid. The surgery was deemed successful, with intraoperative normalization of the PTH level.
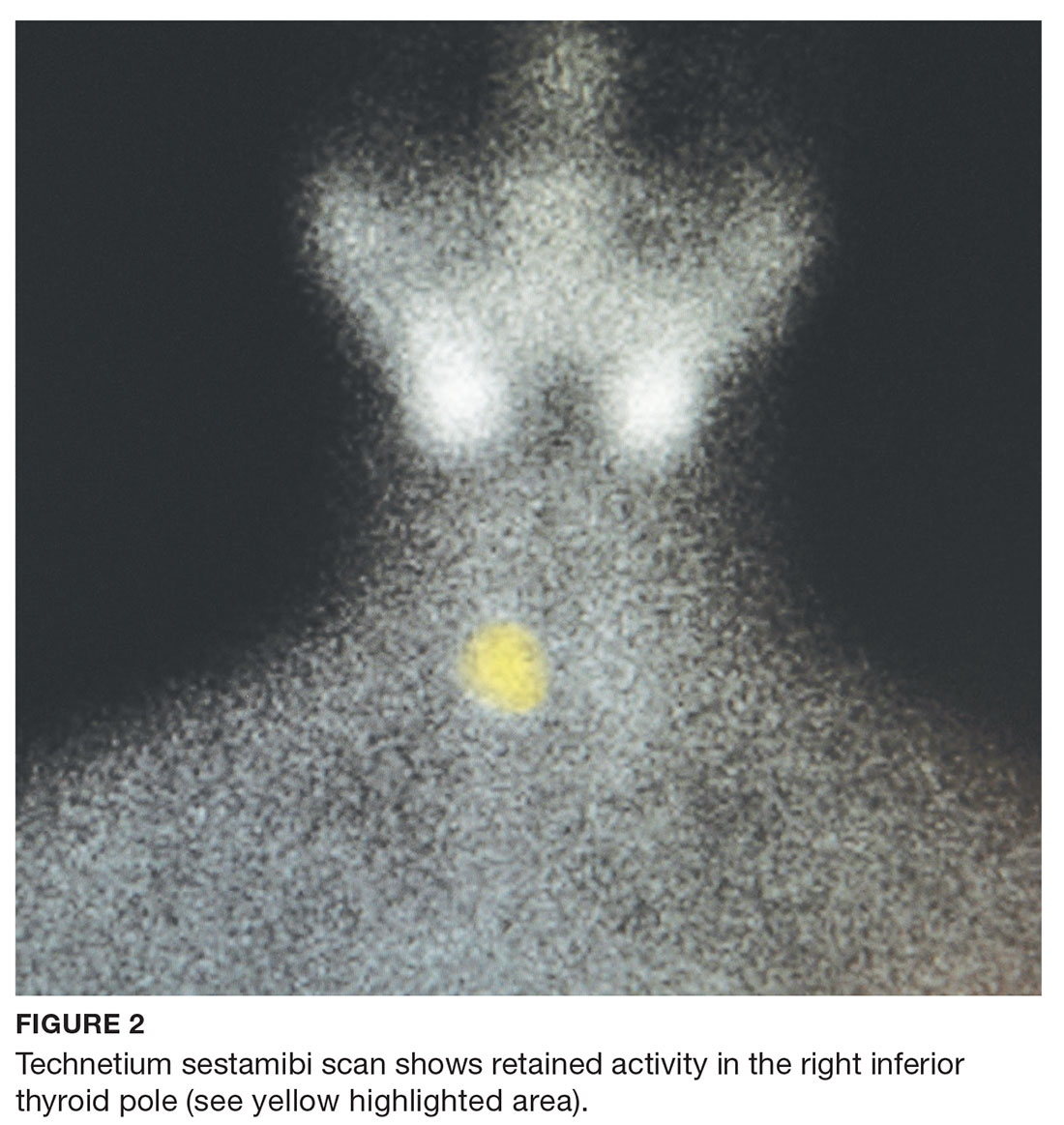
The patient was managed postoperatively by the endocrinologist and was started on calcium and vitamin D supplements. She was prescribed a bisphosphonate, as she had refused to take alendronate following her abnormal BMD test.
DIFFERENTIAL DIAGNOSIS
PHPT and malignancy are the most common causes of hypercalcemia, accounting for 90% of cases.2,4 A less common cause is familial hypocalciuric hypercalcemia (FHH), a rare benign disorder that imitates PHPT.1 FHH is ruled out by measurement of 24-hour urine calcium excretion and is characterized by hypocalciuria, defined as a urine calcium level of less than 100 mg/24 h (reference range, 100-300 mg/24 h).2 Low calcium excretion can also be identified by a calcium-creatinine excretion ratio.2 FHH is a benign autosomal dominant condition caused by a heterozygous mutation of the parathyroid glands’ calcium-sensing receptors.2,5,6 Young adults with FHH are asymptomatic, and mild hypercalcemia and a normal or slightly elevated PTH are the only laboratory findings.4
Measuring PTH levels is key in determining the underlying mechanism of hypercalcemia.2,7 If the hypercalcemia is not PTH-mediated, malignancy and granulomatous diseases such as sarcoidosis must be considered.2,7 PTH is suppressed in malignancy except for rare cases of PTH-producing tumors.4 Bone metastases cause calcium resorption, and sarcoidosis causes an excess of vitamin D, both resulting in hypercalcemia. Lymphomas and sarcoid granulomas express 1α-hydroxylase, an enzyme that increases the conversion of 25(OH)D to 1,25-dihydroxyvitamin D [1,25(OH)2D].2 When malignancy is suspected, it is appropriate to check a 1,25(OH)2D level. Thiazide diuretics, such as hydrochlorothiazide, decrease urinary calcium excretion and may result in mild hypercalcemia.2 Other possibilities in the differential include hypervitaminosis A or D, dehydration, and excess calcium ingestion, but these are less common.6,7
CALCIUM REGULATION
The parathyroid glands stem from four poles on the back of the thyroid gland; there are typically four, but the number can vary from two to 11. Secreted PTH, the primary regulator of calcium homeostasis, maintains calcium levels within a narrow physiologic range.2,8 PTH increases bone resorption, stimulating release of calcium into the blood, and signals the kidneys to increase reabsorption of calcium and excrete phosphorus. It also converts 25(OH)D to 1,25(OH)2 D, the active form of vitamin D that increases gastrointestinal calcium absorption. In a negative feedback loop, PTH secretion is regulated by serum calcium levels, stimulated when levels are low and suppressed when levels are high (see Figure 3).3 Calcium-sensing receptors, located in the chief cells of parathyroid tissue, are essential to calcium homeostasis. These receptors will either increase or decrease PTH release in response to small changes in blood ionized calcium levels. The receptors also play an independent role in the renal tubules by promoting secretion of calcium in the setting of hypercalcemia.5,9 The precise regulation of intracellular and extracellular calcium is necessary for normal functioning of physiologic processes, including bone metabolism, hormone release and regulation, neuromuscular function, and cell signaling.5

PATHOPHYSIOLOGY
Hyperparathyroidism is defined as excess secretion of PTH and is categorized as primary, secondary, or tertiary based on pathophysiologic mechanisms.
Primary hyperparathyroidism
PHPT is defined as PTH levels that are elevated or inappropriately normal in patients with hypercalcemia and no known history of kidney disease.2,6 This occurs when the normal feedback mechanism fails to inhibit excess hormone secretion by one or more of the parathyroid glands.6 With uninhibited PTH secretion, hypercalcemia will result from increased gastrointestinal absorption and bone resorption.
The most common causes of PHPT are an abnormal proliferation of parathyroid cells (parathyroid adenomas) and parathyroid tissue overgrowth (hyperplasia). PHPT may result from a single parathyroid adenoma (80%-90%), multigland hyperplasia (10%-15%), multiple adenomas (2%-3%), or malignancy (< 1%).6,10 Adenomas can occur sporadically or less commonly as part of an inherited syndrome.1 It is estimated that more than 10% of patients with PHPT have a mutation in one of 11 genes associated with PHPT.11 Approximately 5% of PHPT cases are familial, resulting from adenomas or carcinomas associated with mutations in the tumor suppressor genes MEN1 and CDC73 and the RET proto-oncogene.5 Multiple endocrine neoplasia (MEN) syndrome type 1 or 2a is associated with the development of parathyroid adenomas and other endocrine tumors.1,5 Mutations in the CDC73 gene can lead to parathyroid cancer, familial isolated hyperparathyroidism, and familial hyperparathyroidism-jaw tumor syndrome.5 Parathyroid cancer is rare and is linked to a history of radiation to the head and neck.3 Ectopic parathyroid adenomas represent 3% to 4% of all parathyroid adenomas and are often found in the mediastinum.12PHPT is the third most common endocrine disorder, with a prevalence of 1 case per 1,000 men and 2 to 3 cases per 1,000 women.5 Most women with PHPT are postmenopausal and older than 50.1 The condition can occur in younger adults but is rare in childhood and adolescence, with an incidence of 2 to 5 cases per 100,000.13
PHPT affects multiple organ systems, but the most commonly involved are the renal and musculoskeletal systems (see Table 2). The hypersecretory state causes excessive bone resorption and increased osteoclastic activity, resulting in osteoporosis and increased risk for pathologic fractures of the hip, wrist, and spine. The most common osteoporotic fractures are vertebral compression fractures.14 Fractures involving the thoracic spine contribute to the development of kyphosis.15
In the kidney, an increased filtered load of calcium leads to hypercalciuria, precipitation of calcium phosphate in the renal pelvis and collecting ducts, metabolic acidosis, alkaline urine, and hyperphosphaturia. The combination of alkaline urine, hyperphosphaturia, and hypercalciuria leads to the formation of kidney stones.6 Nephrolithiasis and alkaline urine predispose patients to recurrent urinary tract infections and subsequent renal impairment.6 In addition, hypercalcemia impairs the renal collecting system and decreases its response to antidiuretic hormone, resulting in polyuria.6
Secondary hyperparathyroidism
In secondary hyperparathyroidism, calcium levels are either normal or low. Normocalcemic hyperparathyroidism is characterized by normal ionized and total calcium levels and elevated PTH levels; it has no known cause.6 Secondary hyperparathyroidism occurs when excess PTH is excreted as a result of a chronic condition that leads to hypocalcemia. Examples of these disease states include vitamin D deficiency, chronic kidney disease (CKD), and intestinal malabsorption. The most common cause of secondary hyperparathyroidism is CKD; glomerular filtration insufficiency results in hyperphosphatemia, hypocalcemia, and low 1,25(OH)2D, stimulating the release of PTH. Other causes include deficient intake or decreased absorption of calcium or vitamin D; chronic use of medications such as lithium, phenobarbital, or phenytoin; bariatric surgery; celiac disease; and pancreatic disease.4,6,14 Lithium decreases urinary calcium excretion and reduces the sensitivity of the parathyroid gland to calcium.4
Tertiary hyperparathyroidism
Tertiary hyperparathyroidism, marked by hypercalcemia and excessive PTH secretion, can occur after prolonged secondary hyperparathyroidism. In this disorder, persistent parathyroid stimulation leads to gland hyperplasia, resulting in autonomous production of PTH despite correction of calcium levels.6 It most commonly occurs in patients with chronic secondary hyperparathyroidism with renal failure who receive a kidney transplant.2,6 In some cases, parathyroid hyperplasia may not regress after transplantation and parathyroidectomy may be necessary.
EVALUATION AND DIAGNOSTIC WORK-UP
Laboratory tests
Hypercalcemia is the most common initial finding that leads to the diagnosis of PHPT. Elevated serum calcium and PTH is characteristic of the condition. When evaluating a patient with hypercalcemia, the diagnostic work-up includes tests to differentiate between PTH- and non–PTH-mediated causes of elevated calcium (see Table 3).7 Evaluation should begin with measurement of PTH by second- or third-generation immunoassay along with phosphorus, alkaline phosphatase, 25(OH)D, creatinine, estimated glomerular filtration rate (eGFR), and albumin. Additionally, a 24-hour urine collection for calcium, creatinine, and creatinine clearance should be considered in patients with overt nephrolithiasis or nephrocalcinosis. If the urine calcium is > 400 mg/24 h, a renal stone risk profile is indicated because nephrolithiasis is one of the most common complications of PHPT.14 There is a high prevalence of nephrolithiasis in patients with normocalcemic PHPT, even after parathyroidectomy.16 If the 24-hour urine calcium level is low, the diagnosis of FHH is considered. If the urine calcium is high and the intact PTH is elevated or inappropriately normal, the diagnosis of PHPT is considered; urine calcium will be normal in 60% of PHPT cases.4,11

Imaging studies
Imaging is useful for localization of adenomas and abnormal parathyroid tissue to guide surgical planning but is not necessary for diagnosis or medical management. Understanding the strengths and weaknesses of imaging modalities enables the clinician to order the most appropriate option. There are three primary imaging modalities used to locate parathyroid adenoma(s) or aberrant parathyroid tissue: ultrasound, nuclear medicine sestamibi parathyroid scans, and CT. Some clinicians start with an ultrasound, but its operator-dependent results can vary widely; in addition, ultrasound often provides poor anatomic definition and has limited value in locating ectopic parathyroid tissue.17
Nuclear medicine parathyroid scan with technetium-99m sestamibi is a sensitive method for localizing hyperfunctioning, enlarged parathyroid glands or tissue in normal anatomic positions or ectopic locations. Uptake is enhanced and prolonged in parathyroid adenomas as well as in aberrant tissue found in the mediastinum or subclavicular areas. Sestamibi parathyroid scan detects up to 89% of single adenomas, but studies of this imaging modality have demonstrated a wide range of sensitivities (44%-95%).5,17 A drawback of nuclear medicine studies is that they provide little anatomic detail.17 Nonetheless, the ability of the parathyroid scan to locate parathyroid glands has contributed to the success of the minimally invasive parathyroidectomy, and it is considered the most successful imaging modality available.5,10 Identifying the precise location of the parathyroid adenoma is essential for a successful surgical outcome; this is best achieved by combining the sestamibi parathyroid scan with CT.12
Emerging imaging modalities are the multidetector CT (MDCT) and 4D-CT techniques. In an evaluation of the diagnostic accuracy of contrast-enhanced MDCT in the detection of parathyroid adenomas and aberrant parathyroid tissue, MDCT demonstrated the ability to differentiate between adenomas and hyperplasia and display important anatomic structures such as nerves and blood vessels.17 The specificity of MDCT for ruling out abnormal parathyroid tissue was 75%, and the sensitivity for detecting a single adenoma was 80%. Overall, MDCT demonstrated an 88% positive predictive value (PPV) in localizing hyperfunctioning parathyroid glands but showed poor sensitivity in detecting multigland disease.17 The PPV is a key value in determining the ability of an imaging study to precisely locate aberrant parathyroid tissue. MDCT provides detailed definition of anatomy, locating ectopic parathyroid glands in the deeper paraesophageal areas and mediastinum while defining relationships between the tissue and its surrounding vasculature, lymph nodes, and thyroid tissue.17 The 4D-CT technique employs three-dimensional technology and accounts for the movement of the patient’s body over time (the “fourth dimension”). It is an accurate method for identifying parathyroid adenomas but exposes the patient to higher radiation doses.18 The sensitivity of 4D-CT in localizing abnormal parathyroid tissue is comparable to that of MDCT.16,18,19
Additional studies used during the management of the patient with PHPT are BMD testing and renal imaging. Secondary causes of bone loss are responsible for up to 30% of osteoporosis cases in postmenopausal women; one of these causes is PHPT.20 Elevated PTH causes increased bone turnover and results in decreased bone mass with subsequent increased fracture risk.9 Bone density should be measured by dual-energy x-ray absorptiometry (DEXA), and the skeletal survey should include the distal one-third of the radius, hip, and lumbar spine. The distal radius is rich in cortical bone and BMD is often lowest at this site in patients with PHPT, making it the most sensitive DEXA marker for early detection of bone loss.19,21 The hip contains an equal mix of cortical and trabecular bone and is the second most sensitive site for detecting bone loss in PHPT. The spine contains a high proportion of trabecular bone and is the least sensitive site.19,21 Renal imaging studies, including x-ray, ultrasound, and, less frequently, CT of the abdomen and pelvis, are used to assess for nephrolithiasis and nephrocalcinosis.19
TREATMENT/MANAGEMENT
Conservative medical management
PHPT is a complex disease process, and careful evaluation is required when determining whether medical versus surgical management is appropriate. Clinical presentation ranges from no symptoms to multisystem disease. Conservative medical management, which includes regular monitoring, is an acceptable strategy in an asymptomatic patient with a low fracture risk and no nephrolithiasis.1 Conservative care includes maintaining normal dietary calcium intake and adequate hydration, regular exercise, vitamin D supplementation, annual laboratory studies, BMD testing, and the avoidance of thiazide diuretics and lithium.1 Guidelines, from the Fourth International Workshop on the Management of Asymptomatic Primary Hyperparathyroidism, for monitoring asymptomatic PHPT patients recommend
- Annual measurement of serum calcium
- BMD measurement by DEXA every 1 to 2 years
- Annual assessment of eGFR and serum creatinine
- Renal imaging or a 24-h urine stone profile if nephrolithiasis is suspected.19
Long-term medical management of PHPT is difficult because no agents are available to suppress hypercalcemia or completely block PTH release.12
Maintaining serum 25(OH)D at a level > 20 ng/mL significantly reduces PTH secretion, in comparison to levels < 20 ng/mL, and does not aggravate hypercalcemia.22 The Endocrine Society recommends a minimum serum 25(OH)D level of 20 ng/mL and notes that targeting a higher threshold value of 30 ng/mL is reasonable.19 The daily requirement for vitamin D3, 800 IU to 1,000 IU, is a good starting point for supplementation.4 Measurement of 1,25(OH)2D levels lacks value and is not recommended for patients with PHPT. Calcium intake should follow established guidelines and is not limited in PHPT.19
Surgical management
Surgical management is indicated for symptomatic patients.23 Indications include nephrolithiasis, nephrocalcinosis, osteitis fibrosa cystica, or osteoporosis. Surgery is considered appropriate for individuals who do not meet these criteria if there are no medical contraindications.14 The Fourth International Workshop on the Management of Asymptomatic Primary Hyperparathyroidism revised the indications for surgery in 2014 to include asymptomatic patients, since surgery is the only definitive treatment for PHPT. Current guidelines for when to recommend surgery in the asymptomatic patient with PHPT are listed in Table 4.19
Preoperative localization and referral to an experienced surgeon is of utmost importance for a good outcome. An expert surgeon will usually perform a minimally invasive parathyroidectomy (MIP) and obtain intraoperative PTH levels; in some cases, a full neck exploration is necessary. PTH has a half-life of less than five minutes and is an accurate tool for determining whether the culprit gland has been successfully removed.5
Modern imaging studies, less invasive surgical techniques, and intraoperative measurements of PTH have decreased the need to conduct full neck exploration. MIP offers a smaller incision, less tissue dissection, and lower morbidity, and can be offered without the risks associated with general anesthesia.10 The goal of surgery is to restore normocalcemia and, in turn, prevent bone mineral loss and systemic effects of hypercalcemia over the long term.10 Surgical management for an ectopic parathyroid adenoma is controversial because these are often found in the mediastinum, requiring invasive surgery.12
Surgery is curative in up to 95% of cases and has a low rate of complications.2,24 A joint decision regarding treatment options is made among the patient, primary care clinician, and surgeon. Complications include vocal cord paralysis resulting from injury to the recurrent laryngeal nerve, bleeding or hematoma, laryngospasm, symptomatic hypocalcemia, and persistent hyperparathyroidism; seizures are very rare but can occur from transient hypocalcemia and hypomagnesemia.5 PTH levels drop by more than 50% intraoperatively if the procedure is successful; otherwise, exploration for another adenoma is indicated.10 Postoperative calcium and vitamin D supplementation are warranted once lab values are stable.
When surgery is contraindicated/refused
If surgery is indicated but the patient is a poor candidate or refuses surgery, management of hypercalcemia and bone loss with pharmacologic agents is warranted. The calcimimetic cinacalcet is a reasonable medical alternative that has been shown to adequately control hypercalcemia and hypophosphatemia and has proven effective in various patient subgroups.25 This agent is useful in the treatment of patients who are asymptomatic and refuse surgery, patients with refractory PHPT after parathyroidectomy, and patients with contraindications to surgery.24,25 The medication reduces calcium and modestly reduces PTH levels by binding parathyroid calcium-sensing receptors but does not improve bone density.2,12 Cinacalcet is approved by the FDA for use in patients with moderate to severe disease when surgery is contraindicated.24
Treatment options for osteoporosis, vertebral fractures, and progressive bone loss in the patient with PHPT include bisphosphonates. Raloxifene and estrogen replacements may be used in postmenopausal women. Oral bisphosphonates (alendronate or risedronate) are firstline therapies and have been shown to inhibit progression to osteoporosis in PHPT.9,26 They prevent osteoclastic activity, reducing bone resorption and turnover. Contraindications to oral bisphosphonates include esophageal disorders, gastrointestinal intolerance, or inability to follow the dosing requirements. Intravenous zoledronic acid provides an alternative route of administration.
Alendronate has the best evidence for improving bone density and preventing progression to osteoporosis in patients with PHPT, but the medication does not affect calcium or PTH levels.1,19 There is limited data on the effects of combining bisphosphonates with calcimimetics. Raloxifene is a selective estrogen receptor modulator that decreases bone resorption; it is approved for treating osteoporosis and may be used when a patient is not a good candidate for a bisphosphonate.20 Denosumab, currently under study for the treatment of PHPT, is a human monoclonal antibody that improves bone density but does not affect serum calcium.20 Nonpharmacologic therapies include alcohol moderation, decreased caffeine intake, weight-bearing exercise, smoking cessation, adequate hydration, and dietary modifications.20
OUTCOME FOR THE CASE PATIENT
Although PHPT is often discovered incidentally in routine blood work with hypercalcemia, the case patient had developed osteoporosis and a grade IV tibial stress fracture before the diagnosis was made. Following parathyroidectomy, her hypertension worsened, requiring an additional antihypertensive medication. She developed recurrent disease and was referred to a tertiary care center for revision parathyroidectomy due to persistent elevated calcium levels. A 24-hour urine calcium test ruled out concurrent FHH. A full neck exploration was conducted and a 340-mg hypercellular parathyroid gland was removed from the left superior pole. She will be monitored for recurrent disease and will remain on a vitamin D3 supplement and treatment for osteoporosis.
CONCLUSION
Primary care clinicians should have a low threshold for initiating the work-up of mild hypercalcemia in an effort to prevent sequelae. Patient education is essential throughout the process. Understanding the condition and treatment options is necessary for a patient’s active participation in clinical decision making. Conservative management of an asymptomatic patient includes avoiding thiazide diuretics and lithium, staying well hydrated with water, maintaining moderate dietary calcium (1,000-1,200 mg/d) and vitamin D (400-600 IU/d) intake, regular exercise, and appropriate lab and bone density monitoring. Surgical treatment is recommended for symptomatic patients exhibiting decreased bone density, fractures, renal impairment, or nephrolithiasis. Treating bone loss with bisphosphonates and hypercalcemia with calcimimetics is useful. Postmenopausal women may benefit from estrogen therapy or selective estrogen receptor modulators. These agents improve bone density and lower calcium, but are often contraindicated or have adverse effects. Surgery is the only cure.3
1. Turner J. Hypercalcaemia and primary hyperparathyroidism. Medicine. 2009;37(9):461-464.
2. Crowley R, Gittoes N. How to approach hypercalcaemia. Clin Med. 2013;13(3):287-290.
3. Kapustin JF, Schofield DL. Hyperparathyroidism: an incidental finding. Nurse Pract. 2012;37(11):9-14.
4. Cordellat IM. Hyperparathyroidism: primary or secondary disease? Rheumatol Clin. 2012;8(5):287-291.
5. MacKenzie-Feder J, Sirrs S, Anderson D, Sharif J, Khan A. Primary hyperparathyroidism: an overview. Int J Endocrinol. 2011;2011:251410.
6. Brashers VL, Jones RE, Huether SE. Alterations of hormonal regulation. In: McCance KL, Huether SE, eds. Pathophysiology: The Biologic Basis for Disease in Adults and Children. 7th ed. St. Louis, MO: Mosby; 2015:731-733.
7. Osborne JL, Klocko DJ. Woman, 66, with persistent abdominal and back pain. Clinician Reviews. 2014;24(11):34-37, 40.
8. Michels TC, Kelly KM. Parathyroid disorders. Am Fam Physician. 2013;88(4):249-257.
9. Rolighed L, Rejnmark L, Christiansen P. Bone involvement in primary hyperparathyroidism and changes after parathyroidectomy. US Endocrinol. 2013;9(2):181-184.
10. Karahan Ö, Okus A, Sevinç B, et al. Minimally invasive parathyroidectomy under local anesthesia. J Postgrad Med. 2013;59(1):21-24.
11. Fuleihan GE, Silverberg SJ. Primary hyperparathyroidism: diagnosis, differential diagnosis, and evaluation. Up-to-Date. www.uptodate.com/contents/primary-hyperparathyroidism-diagnosis-differential-diagnosis-and-evaluation. Accessed April 20, 2017.
12. Panchani R, Varma T, Goyal A, et al. A challenging case of an ectopic parathyroid adenoma. Indian J Endocrinol Metab. 2012;16:S408-S410.
13. Otasowie J, Hambleton BA. Aggression and homicidal thoughts in a patient with primary hyperparathyroidism: a case report. Br J Medical Pract. 2013;6(4):a630.
14. Bilezikian JP. Primary hyperparathyroidism: new insights, concepts and guidelines. Presented at: American Association of Clinical Endocrinologists 24th Annual Scientific and Clinical Congress; May 13-17, 2015; Nashville, TN. am2015.aace.com/presentations/Friday/F31/PrimaryHyperparathyroidismNew InsightsConceptsandGuidelines.pdf. Accessed April 20, 2017.
15. Crowther-Radulewicz CL, McCance KL. Alterations of musculoskeletal function. In: McCance KL, Huether SE, eds. Pathophysiology: The Biologic Basis for Disease in Adults and Children. 7th ed. St. Louis, MO: Mosby; 2015:1551-1555.
16. Amaral LM, Queiroz DC, Marques TF, et al. Clinical study: normocalcemic versus hypercalcemic primary hyperparathyroidism: more stone than bone? J Osteoporos. 2012;2012:128352.
17. Linda DD, Ng B, Rebello R, et al. The utility of multidetector computed tomography for detection of parathyroid disease in the setting of primary hyperparathyroidism. Can Assoc Radiol J. 2012;63(2):100-108.
18. Bann DV, Zacharia T, Goldenberg D, Goyal, N. Parathyroid localization using 4-D-computed tomography. Ear Nose Throat J. 2015;94(4-5):E55-E57.
19. Bilezikian JP, Brandi ML, Eastell R, et al. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the Fourth International Workshop. J Clin Endocrinol Metab. 2014;99(10):3561-3569.
20. Jeremiah MP, Unwin BK, Greenawald MH, Casiano VE. Diagnosis and management of osteoporosis. Am Fam Physician. 2015;92(4):261-268.
21. Wood K, Dhital S, Chen H, Sippel RS. What is the utility of distal forearm DXA in primary hyperparathyroidism? Oncologist. 2012;17(3):322-325.
22. Jayasena CN, Modi M, Palazzo F, et al. Associations of serum 25-hydroxyvitamin D with circulating PTH, phosphate and calcium in patients with primary hyperparathyroidism. Clin Endocrinol (Oxf). 2013;78(6):838-843.
23. Grey A. Nonsurgical management of mild primary hyperparathyroidism—a reasonable option. Clin Endocrinol. 2012;77(5):639-644.
24. Saponaro F, Faggiano A, Grimaldi F, et al. Cinacalcet in the management of primary hyperparathyroidism: post marketing experience of an Italian multicenter group. Clin Endocrinol (Oxf). 2013;79(1):20-26.
25. Rothe HM, Liangos O, Biggar P, et al. Cinacalcet treatment of primary hyperparathyroidism. Int J Endocrinol. 2011;2011:415719.
26. Farag N, Delbanco T, Strewler GJ. Update: a 64-year-old woman with primary hyperparathyroidism. JAMA. 2008;300(17):2044-2045.
CE/CME No: CR-1705
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Differentiate primary hyperparathyroidism (PHPT) from other causes of hypercalcemia and types of hyerparathyroidism.
• Understand the calcium-parathyroid hormone feedback loop.
• Identify appropriate imaging studies and common laboratory findings in the patient with PHPT.
• Describe the common systemic manifestations of PHPT.
• Discuss medical versus surgical management of the patient with PHPT.
FACULTY
Barbara Austin is a Family Nurse Practitioner at Baptist Primary Care, Jacksonville, Florida, and is pursuing a Doctorate of Nursing Practice (DNP) at Jacksonville University.
The author has no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of May 2017.
Article begins on next page >>
Primary hyperparathyroidism (PHPT) is most often detected as hypercalcemia in an asymptomatic patient during routine blood work. Knowing the appropriate work-up of hypercalcemia is essential, since untreated PHPT can have significant complications affecting multiple organ systems—most notably, renal and musculoskeletal. Parathyroidectomy is curative in up to 95% of cases, but prevention of long-term complications relies on prompt recognition and appropriate follow-up.
Primary hyperparathyroidism (PHPT) is a common endocrine disorder, with a prevalence of approximately 1 to 3 cases per 1,000 persons.1 PHPT results from inappropriate overproduction of parathyroid hormone (PTH), the primary regulator of calcium homeostasis, and is characterized by hypercalcemia in the setting of an elevated or high-normal PTH level. In most cases of PHPT, unregulated PTH production is caused by a single parathyroid adenoma.
PHPT is the most common cause of hypercalcemia in outpatients and is typically diagnosed following incidental discovery during routine blood work in an asymptomatic patient.1,2 It is two to three times more common in women than in men, and incidence increases with age; as such, postmenopausal women are most commonly affected.1,3 PHPT often has an insidious course, and recognition of its clinical manifestations followed by appropriate diagnostic work-up and management are necessary to prevent sequelae.3
PATIENT PRESENTATION
A 68-year-old Caucasian woman presented to her family practice office for a third visit with continued complaints of nontraumatic right lower leg pain. She had previously been diagnosed with tendonitis, which was treated conservatively. The pain failed to improve, and an x-ray was ordered. The x-ray revealed no acute findings but did show osteopenia, prompting an order for a bone mineral density (BMD) test. The BMD test demonstrated osteoporosis, which warranted further investigation. She was prescribed alendronate but refused it, against medical advice, due to concern over potential adverse effects.
Her medical history included hyperlipidemia and hypertension under fair control with lisinopril. She took a low-dose aspirin and flaxseed supplement daily. She also had a history of radiation to the neck, having undergone tonsillar irradiation as a child (a common practice from the 1930s through the 1950s).3 Surgical history included a total hysterectomy with bilateral salpingo-oophorectomy, appendectomy, and tonsillectomy. There was no personal or family history of cancer or endocrine disorders, hypercalcemia, or nephrolithiasis. She was up to date on vaccines and preventive health care measures. Allergies included penicillin and sulfa, both resulting in hives. She was a nonsmoker and did not drink alcohol or engage in illicit drug use.
Review of systems revealed right lower anterior leg pain for four months, characterized as aching, deep, sharp, and throbbing with radiation to the ankle. The pain was worse with activity and prolonged standing; ibuprofen and application of ice provided partial relief. She had experienced some mood changes, including irritability. Physical exam was normal except for dominant right-sided thyromegaly, marked bony point tenderness to the right midshin area, and an antalgic gait.
Laboratory work-up demonstrated elevated PTH, alkaline phosphatase, and calcium levels and a low 25-hydroxyvitamin D [25(OH)D] level (see Table 1). MRI of the right lower leg revealed a grade IV stress fracture (see Figure 1). The elevated serum calcium and PTH levels in addition to abnormal bone density findings led to the diagnosis of PHPT. She was referred to endocrinology and orthopedics for management of PHPT and the stress fracture, respectively, and was placed in an orthopedic walking boot for treatment of the midtibial stress fracture.
The endocrinologist referred her to an otolaryngologist trained in the surgical management of parathyroid adenomas, who ordered a thyroid ultrasound; this study was inconclusive. Additional imaging, including a Tc-99m sestamibi parathyroid scan and CT with contrast of the soft tissue of the neck, was obtained. The parathyroid scan of the neck and upper chest showed retained activity in the right inferior thyroid pole that was concerning for a parathyroid adenoma (see Figure 2). The CT identified a 1.5-cm parathyroid adenoma off the right inferior pole of the thyroid gland (concordant with the parathyroid scan). A single 300-mg parathyroid adenoma was removed from the right inferior pole of the thyroid. The surgery was deemed successful, with intraoperative normalization of the PTH level.
The patient was managed postoperatively by the endocrinologist and was started on calcium and vitamin D supplements. She was prescribed a bisphosphonate, as she had refused to take alendronate following her abnormal BMD test.
DIFFERENTIAL DIAGNOSIS
PHPT and malignancy are the most common causes of hypercalcemia, accounting for 90% of cases.2,4 A less common cause is familial hypocalciuric hypercalcemia (FHH), a rare benign disorder that imitates PHPT.1 FHH is ruled out by measurement of 24-hour urine calcium excretion and is characterized by hypocalciuria, defined as a urine calcium level of less than 100 mg/24 h (reference range, 100-300 mg/24 h).2 Low calcium excretion can also be identified by a calcium-creatinine excretion ratio.2 FHH is a benign autosomal dominant condition caused by a heterozygous mutation of the parathyroid glands’ calcium-sensing receptors.2,5,6 Young adults with FHH are asymptomatic, and mild hypercalcemia and a normal or slightly elevated PTH are the only laboratory findings.4
Measuring PTH levels is key in determining the underlying mechanism of hypercalcemia.2,7 If the hypercalcemia is not PTH-mediated, malignancy and granulomatous diseases such as sarcoidosis must be considered.2,7 PTH is suppressed in malignancy except for rare cases of PTH-producing tumors.4 Bone metastases cause calcium resorption, and sarcoidosis causes an excess of vitamin D, both resulting in hypercalcemia. Lymphomas and sarcoid granulomas express 1α-hydroxylase, an enzyme that increases the conversion of 25(OH)D to 1,25-dihydroxyvitamin D [1,25(OH)2D].2 When malignancy is suspected, it is appropriate to check a 1,25(OH)2D level. Thiazide diuretics, such as hydrochlorothiazide, decrease urinary calcium excretion and may result in mild hypercalcemia.2 Other possibilities in the differential include hypervitaminosis A or D, dehydration, and excess calcium ingestion, but these are less common.6,7
CALCIUM REGULATION
The parathyroid glands stem from four poles on the back of the thyroid gland; there are typically four, but the number can vary from two to 11. Secreted PTH, the primary regulator of calcium homeostasis, maintains calcium levels within a narrow physiologic range.2,8 PTH increases bone resorption, stimulating release of calcium into the blood, and signals the kidneys to increase reabsorption of calcium and excrete phosphorus. It also converts 25(OH)D to 1,25(OH)2 D, the active form of vitamin D that increases gastrointestinal calcium absorption. In a negative feedback loop, PTH secretion is regulated by serum calcium levels, stimulated when levels are low and suppressed when levels are high (see Figure 3).3 Calcium-sensing receptors, located in the chief cells of parathyroid tissue, are essential to calcium homeostasis. These receptors will either increase or decrease PTH release in response to small changes in blood ionized calcium levels. The receptors also play an independent role in the renal tubules by promoting secretion of calcium in the setting of hypercalcemia.5,9 The precise regulation of intracellular and extracellular calcium is necessary for normal functioning of physiologic processes, including bone metabolism, hormone release and regulation, neuromuscular function, and cell signaling.5
PATHOPHYSIOLOGY
Hyperparathyroidism is defined as excess secretion of PTH and is categorized as primary, secondary, or tertiary based on pathophysiologic mechanisms.
Primary hyperparathyroidism
PHPT is defined as PTH levels that are elevated or inappropriately normal in patients with hypercalcemia and no known history of kidney disease.2,6 This occurs when the normal feedback mechanism fails to inhibit excess hormone secretion by one or more of the parathyroid glands.6 With uninhibited PTH secretion, hypercalcemia will result from increased gastrointestinal absorption and bone resorption.
The most common causes of PHPT are an abnormal proliferation of parathyroid cells (parathyroid adenomas) and parathyroid tissue overgrowth (hyperplasia). PHPT may result from a single parathyroid adenoma (80%-90%), multigland hyperplasia (10%-15%), multiple adenomas (2%-3%), or malignancy (< 1%).6,10 Adenomas can occur sporadically or less commonly as part of an inherited syndrome.1 It is estimated that more than 10% of patients with PHPT have a mutation in one of 11 genes associated with PHPT.11 Approximately 5% of PHPT cases are familial, resulting from adenomas or carcinomas associated with mutations in the tumor suppressor genes MEN1 and CDC73 and the RET proto-oncogene.5 Multiple endocrine neoplasia (MEN) syndrome type 1 or 2a is associated with the development of parathyroid adenomas and other endocrine tumors.1,5 Mutations in the CDC73 gene can lead to parathyroid cancer, familial isolated hyperparathyroidism, and familial hyperparathyroidism-jaw tumor syndrome.5 Parathyroid cancer is rare and is linked to a history of radiation to the head and neck.3 Ectopic parathyroid adenomas represent 3% to 4% of all parathyroid adenomas and are often found in the mediastinum.12PHPT is the third most common endocrine disorder, with a prevalence of 1 case per 1,000 men and 2 to 3 cases per 1,000 women.5 Most women with PHPT are postmenopausal and older than 50.1 The condition can occur in younger adults but is rare in childhood and adolescence, with an incidence of 2 to 5 cases per 100,000.13
PHPT affects multiple organ systems, but the most commonly involved are the renal and musculoskeletal systems (see Table 2). The hypersecretory state causes excessive bone resorption and increased osteoclastic activity, resulting in osteoporosis and increased risk for pathologic fractures of the hip, wrist, and spine. The most common osteoporotic fractures are vertebral compression fractures.14 Fractures involving the thoracic spine contribute to the development of kyphosis.15
In the kidney, an increased filtered load of calcium leads to hypercalciuria, precipitation of calcium phosphate in the renal pelvis and collecting ducts, metabolic acidosis, alkaline urine, and hyperphosphaturia. The combination of alkaline urine, hyperphosphaturia, and hypercalciuria leads to the formation of kidney stones.6 Nephrolithiasis and alkaline urine predispose patients to recurrent urinary tract infections and subsequent renal impairment.6 In addition, hypercalcemia impairs the renal collecting system and decreases its response to antidiuretic hormone, resulting in polyuria.6
Secondary hyperparathyroidism
In secondary hyperparathyroidism, calcium levels are either normal or low. Normocalcemic hyperparathyroidism is characterized by normal ionized and total calcium levels and elevated PTH levels; it has no known cause.6 Secondary hyperparathyroidism occurs when excess PTH is excreted as a result of a chronic condition that leads to hypocalcemia. Examples of these disease states include vitamin D deficiency, chronic kidney disease (CKD), and intestinal malabsorption. The most common cause of secondary hyperparathyroidism is CKD; glomerular filtration insufficiency results in hyperphosphatemia, hypocalcemia, and low 1,25(OH)2D, stimulating the release of PTH. Other causes include deficient intake or decreased absorption of calcium or vitamin D; chronic use of medications such as lithium, phenobarbital, or phenytoin; bariatric surgery; celiac disease; and pancreatic disease.4,6,14 Lithium decreases urinary calcium excretion and reduces the sensitivity of the parathyroid gland to calcium.4
Tertiary hyperparathyroidism
Tertiary hyperparathyroidism, marked by hypercalcemia and excessive PTH secretion, can occur after prolonged secondary hyperparathyroidism. In this disorder, persistent parathyroid stimulation leads to gland hyperplasia, resulting in autonomous production of PTH despite correction of calcium levels.6 It most commonly occurs in patients with chronic secondary hyperparathyroidism with renal failure who receive a kidney transplant.2,6 In some cases, parathyroid hyperplasia may not regress after transplantation and parathyroidectomy may be necessary.
EVALUATION AND DIAGNOSTIC WORK-UP
Laboratory tests
Hypercalcemia is the most common initial finding that leads to the diagnosis of PHPT. Elevated serum calcium and PTH is characteristic of the condition. When evaluating a patient with hypercalcemia, the diagnostic work-up includes tests to differentiate between PTH- and non–PTH-mediated causes of elevated calcium (see Table 3).7 Evaluation should begin with measurement of PTH by second- or third-generation immunoassay along with phosphorus, alkaline phosphatase, 25(OH)D, creatinine, estimated glomerular filtration rate (eGFR), and albumin. Additionally, a 24-hour urine collection for calcium, creatinine, and creatinine clearance should be considered in patients with overt nephrolithiasis or nephrocalcinosis. If the urine calcium is > 400 mg/24 h, a renal stone risk profile is indicated because nephrolithiasis is one of the most common complications of PHPT.14 There is a high prevalence of nephrolithiasis in patients with normocalcemic PHPT, even after parathyroidectomy.16 If the 24-hour urine calcium level is low, the diagnosis of FHH is considered. If the urine calcium is high and the intact PTH is elevated or inappropriately normal, the diagnosis of PHPT is considered; urine calcium will be normal in 60% of PHPT cases.4,11
Imaging studies
Imaging is useful for localization of adenomas and abnormal parathyroid tissue to guide surgical planning but is not necessary for diagnosis or medical management. Understanding the strengths and weaknesses of imaging modalities enables the clinician to order the most appropriate option. There are three primary imaging modalities used to locate parathyroid adenoma(s) or aberrant parathyroid tissue: ultrasound, nuclear medicine sestamibi parathyroid scans, and CT. Some clinicians start with an ultrasound, but its operator-dependent results can vary widely; in addition, ultrasound often provides poor anatomic definition and has limited value in locating ectopic parathyroid tissue.17
Nuclear medicine parathyroid scan with technetium-99m sestamibi is a sensitive method for localizing hyperfunctioning, enlarged parathyroid glands or tissue in normal anatomic positions or ectopic locations. Uptake is enhanced and prolonged in parathyroid adenomas as well as in aberrant tissue found in the mediastinum or subclavicular areas. Sestamibi parathyroid scan detects up to 89% of single adenomas, but studies of this imaging modality have demonstrated a wide range of sensitivities (44%-95%).5,17 A drawback of nuclear medicine studies is that they provide little anatomic detail.17 Nonetheless, the ability of the parathyroid scan to locate parathyroid glands has contributed to the success of the minimally invasive parathyroidectomy, and it is considered the most successful imaging modality available.5,10 Identifying the precise location of the parathyroid adenoma is essential for a successful surgical outcome; this is best achieved by combining the sestamibi parathyroid scan with CT.12
Emerging imaging modalities are the multidetector CT (MDCT) and 4D-CT techniques. In an evaluation of the diagnostic accuracy of contrast-enhanced MDCT in the detection of parathyroid adenomas and aberrant parathyroid tissue, MDCT demonstrated the ability to differentiate between adenomas and hyperplasia and display important anatomic structures such as nerves and blood vessels.17 The specificity of MDCT for ruling out abnormal parathyroid tissue was 75%, and the sensitivity for detecting a single adenoma was 80%. Overall, MDCT demonstrated an 88% positive predictive value (PPV) in localizing hyperfunctioning parathyroid glands but showed poor sensitivity in detecting multigland disease.17 The PPV is a key value in determining the ability of an imaging study to precisely locate aberrant parathyroid tissue. MDCT provides detailed definition of anatomy, locating ectopic parathyroid glands in the deeper paraesophageal areas and mediastinum while defining relationships between the tissue and its surrounding vasculature, lymph nodes, and thyroid tissue.17 The 4D-CT technique employs three-dimensional technology and accounts for the movement of the patient’s body over time (the “fourth dimension”). It is an accurate method for identifying parathyroid adenomas but exposes the patient to higher radiation doses.18 The sensitivity of 4D-CT in localizing abnormal parathyroid tissue is comparable to that of MDCT.16,18,19
Additional studies used during the management of the patient with PHPT are BMD testing and renal imaging. Secondary causes of bone loss are responsible for up to 30% of osteoporosis cases in postmenopausal women; one of these causes is PHPT.20 Elevated PTH causes increased bone turnover and results in decreased bone mass with subsequent increased fracture risk.9 Bone density should be measured by dual-energy x-ray absorptiometry (DEXA), and the skeletal survey should include the distal one-third of the radius, hip, and lumbar spine. The distal radius is rich in cortical bone and BMD is often lowest at this site in patients with PHPT, making it the most sensitive DEXA marker for early detection of bone loss.19,21 The hip contains an equal mix of cortical and trabecular bone and is the second most sensitive site for detecting bone loss in PHPT. The spine contains a high proportion of trabecular bone and is the least sensitive site.19,21 Renal imaging studies, including x-ray, ultrasound, and, less frequently, CT of the abdomen and pelvis, are used to assess for nephrolithiasis and nephrocalcinosis.19
TREATMENT/MANAGEMENT
Conservative medical management
PHPT is a complex disease process, and careful evaluation is required when determining whether medical versus surgical management is appropriate. Clinical presentation ranges from no symptoms to multisystem disease. Conservative medical management, which includes regular monitoring, is an acceptable strategy in an asymptomatic patient with a low fracture risk and no nephrolithiasis.1 Conservative care includes maintaining normal dietary calcium intake and adequate hydration, regular exercise, vitamin D supplementation, annual laboratory studies, BMD testing, and the avoidance of thiazide diuretics and lithium.1 Guidelines, from the Fourth International Workshop on the Management of Asymptomatic Primary Hyperparathyroidism, for monitoring asymptomatic PHPT patients recommend
- Annual measurement of serum calcium
- BMD measurement by DEXA every 1 to 2 years
- Annual assessment of eGFR and serum creatinine
- Renal imaging or a 24-h urine stone profile if nephrolithiasis is suspected.19
Long-term medical management of PHPT is difficult because no agents are available to suppress hypercalcemia or completely block PTH release.12
Maintaining serum 25(OH)D at a level > 20 ng/mL significantly reduces PTH secretion, in comparison to levels < 20 ng/mL, and does not aggravate hypercalcemia.22 The Endocrine Society recommends a minimum serum 25(OH)D level of 20 ng/mL and notes that targeting a higher threshold value of 30 ng/mL is reasonable.19 The daily requirement for vitamin D3, 800 IU to 1,000 IU, is a good starting point for supplementation.4 Measurement of 1,25(OH)2D levels lacks value and is not recommended for patients with PHPT. Calcium intake should follow established guidelines and is not limited in PHPT.19
Surgical management
Surgical management is indicated for symptomatic patients.23 Indications include nephrolithiasis, nephrocalcinosis, osteitis fibrosa cystica, or osteoporosis. Surgery is considered appropriate for individuals who do not meet these criteria if there are no medical contraindications.14 The Fourth International Workshop on the Management of Asymptomatic Primary Hyperparathyroidism revised the indications for surgery in 2014 to include asymptomatic patients, since surgery is the only definitive treatment for PHPT. Current guidelines for when to recommend surgery in the asymptomatic patient with PHPT are listed in Table 4.19
Preoperative localization and referral to an experienced surgeon is of utmost importance for a good outcome. An expert surgeon will usually perform a minimally invasive parathyroidectomy (MIP) and obtain intraoperative PTH levels; in some cases, a full neck exploration is necessary. PTH has a half-life of less than five minutes and is an accurate tool for determining whether the culprit gland has been successfully removed.5
Modern imaging studies, less invasive surgical techniques, and intraoperative measurements of PTH have decreased the need to conduct full neck exploration. MIP offers a smaller incision, less tissue dissection, and lower morbidity, and can be offered without the risks associated with general anesthesia.10 The goal of surgery is to restore normocalcemia and, in turn, prevent bone mineral loss and systemic effects of hypercalcemia over the long term.10 Surgical management for an ectopic parathyroid adenoma is controversial because these are often found in the mediastinum, requiring invasive surgery.12
Surgery is curative in up to 95% of cases and has a low rate of complications.2,24 A joint decision regarding treatment options is made among the patient, primary care clinician, and surgeon. Complications include vocal cord paralysis resulting from injury to the recurrent laryngeal nerve, bleeding or hematoma, laryngospasm, symptomatic hypocalcemia, and persistent hyperparathyroidism; seizures are very rare but can occur from transient hypocalcemia and hypomagnesemia.5 PTH levels drop by more than 50% intraoperatively if the procedure is successful; otherwise, exploration for another adenoma is indicated.10 Postoperative calcium and vitamin D supplementation are warranted once lab values are stable.
When surgery is contraindicated/refused
If surgery is indicated but the patient is a poor candidate or refuses surgery, management of hypercalcemia and bone loss with pharmacologic agents is warranted. The calcimimetic cinacalcet is a reasonable medical alternative that has been shown to adequately control hypercalcemia and hypophosphatemia and has proven effective in various patient subgroups.25 This agent is useful in the treatment of patients who are asymptomatic and refuse surgery, patients with refractory PHPT after parathyroidectomy, and patients with contraindications to surgery.24,25 The medication reduces calcium and modestly reduces PTH levels by binding parathyroid calcium-sensing receptors but does not improve bone density.2,12 Cinacalcet is approved by the FDA for use in patients with moderate to severe disease when surgery is contraindicated.24
Treatment options for osteoporosis, vertebral fractures, and progressive bone loss in the patient with PHPT include bisphosphonates. Raloxifene and estrogen replacements may be used in postmenopausal women. Oral bisphosphonates (alendronate or risedronate) are firstline therapies and have been shown to inhibit progression to osteoporosis in PHPT.9,26 They prevent osteoclastic activity, reducing bone resorption and turnover. Contraindications to oral bisphosphonates include esophageal disorders, gastrointestinal intolerance, or inability to follow the dosing requirements. Intravenous zoledronic acid provides an alternative route of administration.
Alendronate has the best evidence for improving bone density and preventing progression to osteoporosis in patients with PHPT, but the medication does not affect calcium or PTH levels.1,19 There is limited data on the effects of combining bisphosphonates with calcimimetics. Raloxifene is a selective estrogen receptor modulator that decreases bone resorption; it is approved for treating osteoporosis and may be used when a patient is not a good candidate for a bisphosphonate.20 Denosumab, currently under study for the treatment of PHPT, is a human monoclonal antibody that improves bone density but does not affect serum calcium.20 Nonpharmacologic therapies include alcohol moderation, decreased caffeine intake, weight-bearing exercise, smoking cessation, adequate hydration, and dietary modifications.20
OUTCOME FOR THE CASE PATIENT
Although PHPT is often discovered incidentally in routine blood work with hypercalcemia, the case patient had developed osteoporosis and a grade IV tibial stress fracture before the diagnosis was made. Following parathyroidectomy, her hypertension worsened, requiring an additional antihypertensive medication. She developed recurrent disease and was referred to a tertiary care center for revision parathyroidectomy due to persistent elevated calcium levels. A 24-hour urine calcium test ruled out concurrent FHH. A full neck exploration was conducted and a 340-mg hypercellular parathyroid gland was removed from the left superior pole. She will be monitored for recurrent disease and will remain on a vitamin D3 supplement and treatment for osteoporosis.
CONCLUSION
Primary care clinicians should have a low threshold for initiating the work-up of mild hypercalcemia in an effort to prevent sequelae. Patient education is essential throughout the process. Understanding the condition and treatment options is necessary for a patient’s active participation in clinical decision making. Conservative management of an asymptomatic patient includes avoiding thiazide diuretics and lithium, staying well hydrated with water, maintaining moderate dietary calcium (1,000-1,200 mg/d) and vitamin D (400-600 IU/d) intake, regular exercise, and appropriate lab and bone density monitoring. Surgical treatment is recommended for symptomatic patients exhibiting decreased bone density, fractures, renal impairment, or nephrolithiasis. Treating bone loss with bisphosphonates and hypercalcemia with calcimimetics is useful. Postmenopausal women may benefit from estrogen therapy or selective estrogen receptor modulators. These agents improve bone density and lower calcium, but are often contraindicated or have adverse effects. Surgery is the only cure.3
CE/CME No: CR-1705
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Differentiate primary hyperparathyroidism (PHPT) from other causes of hypercalcemia and types of hyerparathyroidism.
• Understand the calcium-parathyroid hormone feedback loop.
• Identify appropriate imaging studies and common laboratory findings in the patient with PHPT.
• Describe the common systemic manifestations of PHPT.
• Discuss medical versus surgical management of the patient with PHPT.
FACULTY
Barbara Austin is a Family Nurse Practitioner at Baptist Primary Care, Jacksonville, Florida, and is pursuing a Doctorate of Nursing Practice (DNP) at Jacksonville University.
The author has no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of May 2017.
Article begins on next page >>
Primary hyperparathyroidism (PHPT) is most often detected as hypercalcemia in an asymptomatic patient during routine blood work. Knowing the appropriate work-up of hypercalcemia is essential, since untreated PHPT can have significant complications affecting multiple organ systems—most notably, renal and musculoskeletal. Parathyroidectomy is curative in up to 95% of cases, but prevention of long-term complications relies on prompt recognition and appropriate follow-up.
Primary hyperparathyroidism (PHPT) is a common endocrine disorder, with a prevalence of approximately 1 to 3 cases per 1,000 persons.1 PHPT results from inappropriate overproduction of parathyroid hormone (PTH), the primary regulator of calcium homeostasis, and is characterized by hypercalcemia in the setting of an elevated or high-normal PTH level. In most cases of PHPT, unregulated PTH production is caused by a single parathyroid adenoma.
PHPT is the most common cause of hypercalcemia in outpatients and is typically diagnosed following incidental discovery during routine blood work in an asymptomatic patient.1,2 It is two to three times more common in women than in men, and incidence increases with age; as such, postmenopausal women are most commonly affected.1,3 PHPT often has an insidious course, and recognition of its clinical manifestations followed by appropriate diagnostic work-up and management are necessary to prevent sequelae.3
PATIENT PRESENTATION
A 68-year-old Caucasian woman presented to her family practice office for a third visit with continued complaints of nontraumatic right lower leg pain. She had previously been diagnosed with tendonitis, which was treated conservatively. The pain failed to improve, and an x-ray was ordered. The x-ray revealed no acute findings but did show osteopenia, prompting an order for a bone mineral density (BMD) test. The BMD test demonstrated osteoporosis, which warranted further investigation. She was prescribed alendronate but refused it, against medical advice, due to concern over potential adverse effects.
Her medical history included hyperlipidemia and hypertension under fair control with lisinopril. She took a low-dose aspirin and flaxseed supplement daily. She also had a history of radiation to the neck, having undergone tonsillar irradiation as a child (a common practice from the 1930s through the 1950s).3 Surgical history included a total hysterectomy with bilateral salpingo-oophorectomy, appendectomy, and tonsillectomy. There was no personal or family history of cancer or endocrine disorders, hypercalcemia, or nephrolithiasis. She was up to date on vaccines and preventive health care measures. Allergies included penicillin and sulfa, both resulting in hives. She was a nonsmoker and did not drink alcohol or engage in illicit drug use.
Review of systems revealed right lower anterior leg pain for four months, characterized as aching, deep, sharp, and throbbing with radiation to the ankle. The pain was worse with activity and prolonged standing; ibuprofen and application of ice provided partial relief. She had experienced some mood changes, including irritability. Physical exam was normal except for dominant right-sided thyromegaly, marked bony point tenderness to the right midshin area, and an antalgic gait.
Laboratory work-up demonstrated elevated PTH, alkaline phosphatase, and calcium levels and a low 25-hydroxyvitamin D [25(OH)D] level (see Table 1). MRI of the right lower leg revealed a grade IV stress fracture (see Figure 1). The elevated serum calcium and PTH levels in addition to abnormal bone density findings led to the diagnosis of PHPT. She was referred to endocrinology and orthopedics for management of PHPT and the stress fracture, respectively, and was placed in an orthopedic walking boot for treatment of the midtibial stress fracture.
The endocrinologist referred her to an otolaryngologist trained in the surgical management of parathyroid adenomas, who ordered a thyroid ultrasound; this study was inconclusive. Additional imaging, including a Tc-99m sestamibi parathyroid scan and CT with contrast of the soft tissue of the neck, was obtained. The parathyroid scan of the neck and upper chest showed retained activity in the right inferior thyroid pole that was concerning for a parathyroid adenoma (see Figure 2). The CT identified a 1.5-cm parathyroid adenoma off the right inferior pole of the thyroid gland (concordant with the parathyroid scan). A single 300-mg parathyroid adenoma was removed from the right inferior pole of the thyroid. The surgery was deemed successful, with intraoperative normalization of the PTH level.
The patient was managed postoperatively by the endocrinologist and was started on calcium and vitamin D supplements. She was prescribed a bisphosphonate, as she had refused to take alendronate following her abnormal BMD test.
DIFFERENTIAL DIAGNOSIS
PHPT and malignancy are the most common causes of hypercalcemia, accounting for 90% of cases.2,4 A less common cause is familial hypocalciuric hypercalcemia (FHH), a rare benign disorder that imitates PHPT.1 FHH is ruled out by measurement of 24-hour urine calcium excretion and is characterized by hypocalciuria, defined as a urine calcium level of less than 100 mg/24 h (reference range, 100-300 mg/24 h).2 Low calcium excretion can also be identified by a calcium-creatinine excretion ratio.2 FHH is a benign autosomal dominant condition caused by a heterozygous mutation of the parathyroid glands’ calcium-sensing receptors.2,5,6 Young adults with FHH are asymptomatic, and mild hypercalcemia and a normal or slightly elevated PTH are the only laboratory findings.4
Measuring PTH levels is key in determining the underlying mechanism of hypercalcemia.2,7 If the hypercalcemia is not PTH-mediated, malignancy and granulomatous diseases such as sarcoidosis must be considered.2,7 PTH is suppressed in malignancy except for rare cases of PTH-producing tumors.4 Bone metastases cause calcium resorption, and sarcoidosis causes an excess of vitamin D, both resulting in hypercalcemia. Lymphomas and sarcoid granulomas express 1α-hydroxylase, an enzyme that increases the conversion of 25(OH)D to 1,25-dihydroxyvitamin D [1,25(OH)2D].2 When malignancy is suspected, it is appropriate to check a 1,25(OH)2D level. Thiazide diuretics, such as hydrochlorothiazide, decrease urinary calcium excretion and may result in mild hypercalcemia.2 Other possibilities in the differential include hypervitaminosis A or D, dehydration, and excess calcium ingestion, but these are less common.6,7
CALCIUM REGULATION
The parathyroid glands stem from four poles on the back of the thyroid gland; there are typically four, but the number can vary from two to 11. Secreted PTH, the primary regulator of calcium homeostasis, maintains calcium levels within a narrow physiologic range.2,8 PTH increases bone resorption, stimulating release of calcium into the blood, and signals the kidneys to increase reabsorption of calcium and excrete phosphorus. It also converts 25(OH)D to 1,25(OH)2 D, the active form of vitamin D that increases gastrointestinal calcium absorption. In a negative feedback loop, PTH secretion is regulated by serum calcium levels, stimulated when levels are low and suppressed when levels are high (see Figure 3).3 Calcium-sensing receptors, located in the chief cells of parathyroid tissue, are essential to calcium homeostasis. These receptors will either increase or decrease PTH release in response to small changes in blood ionized calcium levels. The receptors also play an independent role in the renal tubules by promoting secretion of calcium in the setting of hypercalcemia.5,9 The precise regulation of intracellular and extracellular calcium is necessary for normal functioning of physiologic processes, including bone metabolism, hormone release and regulation, neuromuscular function, and cell signaling.5
PATHOPHYSIOLOGY
Hyperparathyroidism is defined as excess secretion of PTH and is categorized as primary, secondary, or tertiary based on pathophysiologic mechanisms.
Primary hyperparathyroidism
PHPT is defined as PTH levels that are elevated or inappropriately normal in patients with hypercalcemia and no known history of kidney disease.2,6 This occurs when the normal feedback mechanism fails to inhibit excess hormone secretion by one or more of the parathyroid glands.6 With uninhibited PTH secretion, hypercalcemia will result from increased gastrointestinal absorption and bone resorption.
The most common causes of PHPT are an abnormal proliferation of parathyroid cells (parathyroid adenomas) and parathyroid tissue overgrowth (hyperplasia). PHPT may result from a single parathyroid adenoma (80%-90%), multigland hyperplasia (10%-15%), multiple adenomas (2%-3%), or malignancy (< 1%).6,10 Adenomas can occur sporadically or less commonly as part of an inherited syndrome.1 It is estimated that more than 10% of patients with PHPT have a mutation in one of 11 genes associated with PHPT.11 Approximately 5% of PHPT cases are familial, resulting from adenomas or carcinomas associated with mutations in the tumor suppressor genes MEN1 and CDC73 and the RET proto-oncogene.5 Multiple endocrine neoplasia (MEN) syndrome type 1 or 2a is associated with the development of parathyroid adenomas and other endocrine tumors.1,5 Mutations in the CDC73 gene can lead to parathyroid cancer, familial isolated hyperparathyroidism, and familial hyperparathyroidism-jaw tumor syndrome.5 Parathyroid cancer is rare and is linked to a history of radiation to the head and neck.3 Ectopic parathyroid adenomas represent 3% to 4% of all parathyroid adenomas and are often found in the mediastinum.12PHPT is the third most common endocrine disorder, with a prevalence of 1 case per 1,000 men and 2 to 3 cases per 1,000 women.5 Most women with PHPT are postmenopausal and older than 50.1 The condition can occur in younger adults but is rare in childhood and adolescence, with an incidence of 2 to 5 cases per 100,000.13
PHPT affects multiple organ systems, but the most commonly involved are the renal and musculoskeletal systems (see Table 2). The hypersecretory state causes excessive bone resorption and increased osteoclastic activity, resulting in osteoporosis and increased risk for pathologic fractures of the hip, wrist, and spine. The most common osteoporotic fractures are vertebral compression fractures.14 Fractures involving the thoracic spine contribute to the development of kyphosis.15
In the kidney, an increased filtered load of calcium leads to hypercalciuria, precipitation of calcium phosphate in the renal pelvis and collecting ducts, metabolic acidosis, alkaline urine, and hyperphosphaturia. The combination of alkaline urine, hyperphosphaturia, and hypercalciuria leads to the formation of kidney stones.6 Nephrolithiasis and alkaline urine predispose patients to recurrent urinary tract infections and subsequent renal impairment.6 In addition, hypercalcemia impairs the renal collecting system and decreases its response to antidiuretic hormone, resulting in polyuria.6
Secondary hyperparathyroidism
In secondary hyperparathyroidism, calcium levels are either normal or low. Normocalcemic hyperparathyroidism is characterized by normal ionized and total calcium levels and elevated PTH levels; it has no known cause.6 Secondary hyperparathyroidism occurs when excess PTH is excreted as a result of a chronic condition that leads to hypocalcemia. Examples of these disease states include vitamin D deficiency, chronic kidney disease (CKD), and intestinal malabsorption. The most common cause of secondary hyperparathyroidism is CKD; glomerular filtration insufficiency results in hyperphosphatemia, hypocalcemia, and low 1,25(OH)2D, stimulating the release of PTH. Other causes include deficient intake or decreased absorption of calcium or vitamin D; chronic use of medications such as lithium, phenobarbital, or phenytoin; bariatric surgery; celiac disease; and pancreatic disease.4,6,14 Lithium decreases urinary calcium excretion and reduces the sensitivity of the parathyroid gland to calcium.4
Tertiary hyperparathyroidism
Tertiary hyperparathyroidism, marked by hypercalcemia and excessive PTH secretion, can occur after prolonged secondary hyperparathyroidism. In this disorder, persistent parathyroid stimulation leads to gland hyperplasia, resulting in autonomous production of PTH despite correction of calcium levels.6 It most commonly occurs in patients with chronic secondary hyperparathyroidism with renal failure who receive a kidney transplant.2,6 In some cases, parathyroid hyperplasia may not regress after transplantation and parathyroidectomy may be necessary.
EVALUATION AND DIAGNOSTIC WORK-UP
Laboratory tests
Hypercalcemia is the most common initial finding that leads to the diagnosis of PHPT. Elevated serum calcium and PTH is characteristic of the condition. When evaluating a patient with hypercalcemia, the diagnostic work-up includes tests to differentiate between PTH- and non–PTH-mediated causes of elevated calcium (see Table 3).7 Evaluation should begin with measurement of PTH by second- or third-generation immunoassay along with phosphorus, alkaline phosphatase, 25(OH)D, creatinine, estimated glomerular filtration rate (eGFR), and albumin. Additionally, a 24-hour urine collection for calcium, creatinine, and creatinine clearance should be considered in patients with overt nephrolithiasis or nephrocalcinosis. If the urine calcium is > 400 mg/24 h, a renal stone risk profile is indicated because nephrolithiasis is one of the most common complications of PHPT.14 There is a high prevalence of nephrolithiasis in patients with normocalcemic PHPT, even after parathyroidectomy.16 If the 24-hour urine calcium level is low, the diagnosis of FHH is considered. If the urine calcium is high and the intact PTH is elevated or inappropriately normal, the diagnosis of PHPT is considered; urine calcium will be normal in 60% of PHPT cases.4,11
Imaging studies
Imaging is useful for localization of adenomas and abnormal parathyroid tissue to guide surgical planning but is not necessary for diagnosis or medical management. Understanding the strengths and weaknesses of imaging modalities enables the clinician to order the most appropriate option. There are three primary imaging modalities used to locate parathyroid adenoma(s) or aberrant parathyroid tissue: ultrasound, nuclear medicine sestamibi parathyroid scans, and CT. Some clinicians start with an ultrasound, but its operator-dependent results can vary widely; in addition, ultrasound often provides poor anatomic definition and has limited value in locating ectopic parathyroid tissue.17
Nuclear medicine parathyroid scan with technetium-99m sestamibi is a sensitive method for localizing hyperfunctioning, enlarged parathyroid glands or tissue in normal anatomic positions or ectopic locations. Uptake is enhanced and prolonged in parathyroid adenomas as well as in aberrant tissue found in the mediastinum or subclavicular areas. Sestamibi parathyroid scan detects up to 89% of single adenomas, but studies of this imaging modality have demonstrated a wide range of sensitivities (44%-95%).5,17 A drawback of nuclear medicine studies is that they provide little anatomic detail.17 Nonetheless, the ability of the parathyroid scan to locate parathyroid glands has contributed to the success of the minimally invasive parathyroidectomy, and it is considered the most successful imaging modality available.5,10 Identifying the precise location of the parathyroid adenoma is essential for a successful surgical outcome; this is best achieved by combining the sestamibi parathyroid scan with CT.12
Emerging imaging modalities are the multidetector CT (MDCT) and 4D-CT techniques. In an evaluation of the diagnostic accuracy of contrast-enhanced MDCT in the detection of parathyroid adenomas and aberrant parathyroid tissue, MDCT demonstrated the ability to differentiate between adenomas and hyperplasia and display important anatomic structures such as nerves and blood vessels.17 The specificity of MDCT for ruling out abnormal parathyroid tissue was 75%, and the sensitivity for detecting a single adenoma was 80%. Overall, MDCT demonstrated an 88% positive predictive value (PPV) in localizing hyperfunctioning parathyroid glands but showed poor sensitivity in detecting multigland disease.17 The PPV is a key value in determining the ability of an imaging study to precisely locate aberrant parathyroid tissue. MDCT provides detailed definition of anatomy, locating ectopic parathyroid glands in the deeper paraesophageal areas and mediastinum while defining relationships between the tissue and its surrounding vasculature, lymph nodes, and thyroid tissue.17 The 4D-CT technique employs three-dimensional technology and accounts for the movement of the patient’s body over time (the “fourth dimension”). It is an accurate method for identifying parathyroid adenomas but exposes the patient to higher radiation doses.18 The sensitivity of 4D-CT in localizing abnormal parathyroid tissue is comparable to that of MDCT.16,18,19
Additional studies used during the management of the patient with PHPT are BMD testing and renal imaging. Secondary causes of bone loss are responsible for up to 30% of osteoporosis cases in postmenopausal women; one of these causes is PHPT.20 Elevated PTH causes increased bone turnover and results in decreased bone mass with subsequent increased fracture risk.9 Bone density should be measured by dual-energy x-ray absorptiometry (DEXA), and the skeletal survey should include the distal one-third of the radius, hip, and lumbar spine. The distal radius is rich in cortical bone and BMD is often lowest at this site in patients with PHPT, making it the most sensitive DEXA marker for early detection of bone loss.19,21 The hip contains an equal mix of cortical and trabecular bone and is the second most sensitive site for detecting bone loss in PHPT. The spine contains a high proportion of trabecular bone and is the least sensitive site.19,21 Renal imaging studies, including x-ray, ultrasound, and, less frequently, CT of the abdomen and pelvis, are used to assess for nephrolithiasis and nephrocalcinosis.19
TREATMENT/MANAGEMENT
Conservative medical management
PHPT is a complex disease process, and careful evaluation is required when determining whether medical versus surgical management is appropriate. Clinical presentation ranges from no symptoms to multisystem disease. Conservative medical management, which includes regular monitoring, is an acceptable strategy in an asymptomatic patient with a low fracture risk and no nephrolithiasis.1 Conservative care includes maintaining normal dietary calcium intake and adequate hydration, regular exercise, vitamin D supplementation, annual laboratory studies, BMD testing, and the avoidance of thiazide diuretics and lithium.1 Guidelines, from the Fourth International Workshop on the Management of Asymptomatic Primary Hyperparathyroidism, for monitoring asymptomatic PHPT patients recommend
- Annual measurement of serum calcium
- BMD measurement by DEXA every 1 to 2 years
- Annual assessment of eGFR and serum creatinine
- Renal imaging or a 24-h urine stone profile if nephrolithiasis is suspected.19
Long-term medical management of PHPT is difficult because no agents are available to suppress hypercalcemia or completely block PTH release.12
Maintaining serum 25(OH)D at a level > 20 ng/mL significantly reduces PTH secretion, in comparison to levels < 20 ng/mL, and does not aggravate hypercalcemia.22 The Endocrine Society recommends a minimum serum 25(OH)D level of 20 ng/mL and notes that targeting a higher threshold value of 30 ng/mL is reasonable.19 The daily requirement for vitamin D3, 800 IU to 1,000 IU, is a good starting point for supplementation.4 Measurement of 1,25(OH)2D levels lacks value and is not recommended for patients with PHPT. Calcium intake should follow established guidelines and is not limited in PHPT.19
Surgical management
Surgical management is indicated for symptomatic patients.23 Indications include nephrolithiasis, nephrocalcinosis, osteitis fibrosa cystica, or osteoporosis. Surgery is considered appropriate for individuals who do not meet these criteria if there are no medical contraindications.14 The Fourth International Workshop on the Management of Asymptomatic Primary Hyperparathyroidism revised the indications for surgery in 2014 to include asymptomatic patients, since surgery is the only definitive treatment for PHPT. Current guidelines for when to recommend surgery in the asymptomatic patient with PHPT are listed in Table 4.19
Preoperative localization and referral to an experienced surgeon is of utmost importance for a good outcome. An expert surgeon will usually perform a minimally invasive parathyroidectomy (MIP) and obtain intraoperative PTH levels; in some cases, a full neck exploration is necessary. PTH has a half-life of less than five minutes and is an accurate tool for determining whether the culprit gland has been successfully removed.5
Modern imaging studies, less invasive surgical techniques, and intraoperative measurements of PTH have decreased the need to conduct full neck exploration. MIP offers a smaller incision, less tissue dissection, and lower morbidity, and can be offered without the risks associated with general anesthesia.10 The goal of surgery is to restore normocalcemia and, in turn, prevent bone mineral loss and systemic effects of hypercalcemia over the long term.10 Surgical management for an ectopic parathyroid adenoma is controversial because these are often found in the mediastinum, requiring invasive surgery.12
Surgery is curative in up to 95% of cases and has a low rate of complications.2,24 A joint decision regarding treatment options is made among the patient, primary care clinician, and surgeon. Complications include vocal cord paralysis resulting from injury to the recurrent laryngeal nerve, bleeding or hematoma, laryngospasm, symptomatic hypocalcemia, and persistent hyperparathyroidism; seizures are very rare but can occur from transient hypocalcemia and hypomagnesemia.5 PTH levels drop by more than 50% intraoperatively if the procedure is successful; otherwise, exploration for another adenoma is indicated.10 Postoperative calcium and vitamin D supplementation are warranted once lab values are stable.
When surgery is contraindicated/refused
If surgery is indicated but the patient is a poor candidate or refuses surgery, management of hypercalcemia and bone loss with pharmacologic agents is warranted. The calcimimetic cinacalcet is a reasonable medical alternative that has been shown to adequately control hypercalcemia and hypophosphatemia and has proven effective in various patient subgroups.25 This agent is useful in the treatment of patients who are asymptomatic and refuse surgery, patients with refractory PHPT after parathyroidectomy, and patients with contraindications to surgery.24,25 The medication reduces calcium and modestly reduces PTH levels by binding parathyroid calcium-sensing receptors but does not improve bone density.2,12 Cinacalcet is approved by the FDA for use in patients with moderate to severe disease when surgery is contraindicated.24
Treatment options for osteoporosis, vertebral fractures, and progressive bone loss in the patient with PHPT include bisphosphonates. Raloxifene and estrogen replacements may be used in postmenopausal women. Oral bisphosphonates (alendronate or risedronate) are firstline therapies and have been shown to inhibit progression to osteoporosis in PHPT.9,26 They prevent osteoclastic activity, reducing bone resorption and turnover. Contraindications to oral bisphosphonates include esophageal disorders, gastrointestinal intolerance, or inability to follow the dosing requirements. Intravenous zoledronic acid provides an alternative route of administration.
Alendronate has the best evidence for improving bone density and preventing progression to osteoporosis in patients with PHPT, but the medication does not affect calcium or PTH levels.1,19 There is limited data on the effects of combining bisphosphonates with calcimimetics. Raloxifene is a selective estrogen receptor modulator that decreases bone resorption; it is approved for treating osteoporosis and may be used when a patient is not a good candidate for a bisphosphonate.20 Denosumab, currently under study for the treatment of PHPT, is a human monoclonal antibody that improves bone density but does not affect serum calcium.20 Nonpharmacologic therapies include alcohol moderation, decreased caffeine intake, weight-bearing exercise, smoking cessation, adequate hydration, and dietary modifications.20
OUTCOME FOR THE CASE PATIENT
Although PHPT is often discovered incidentally in routine blood work with hypercalcemia, the case patient had developed osteoporosis and a grade IV tibial stress fracture before the diagnosis was made. Following parathyroidectomy, her hypertension worsened, requiring an additional antihypertensive medication. She developed recurrent disease and was referred to a tertiary care center for revision parathyroidectomy due to persistent elevated calcium levels. A 24-hour urine calcium test ruled out concurrent FHH. A full neck exploration was conducted and a 340-mg hypercellular parathyroid gland was removed from the left superior pole. She will be monitored for recurrent disease and will remain on a vitamin D3 supplement and treatment for osteoporosis.
CONCLUSION
Primary care clinicians should have a low threshold for initiating the work-up of mild hypercalcemia in an effort to prevent sequelae. Patient education is essential throughout the process. Understanding the condition and treatment options is necessary for a patient’s active participation in clinical decision making. Conservative management of an asymptomatic patient includes avoiding thiazide diuretics and lithium, staying well hydrated with water, maintaining moderate dietary calcium (1,000-1,200 mg/d) and vitamin D (400-600 IU/d) intake, regular exercise, and appropriate lab and bone density monitoring. Surgical treatment is recommended for symptomatic patients exhibiting decreased bone density, fractures, renal impairment, or nephrolithiasis. Treating bone loss with bisphosphonates and hypercalcemia with calcimimetics is useful. Postmenopausal women may benefit from estrogen therapy or selective estrogen receptor modulators. These agents improve bone density and lower calcium, but are often contraindicated or have adverse effects. Surgery is the only cure.3
1. Turner J. Hypercalcaemia and primary hyperparathyroidism. Medicine. 2009;37(9):461-464.
2. Crowley R, Gittoes N. How to approach hypercalcaemia. Clin Med. 2013;13(3):287-290.
3. Kapustin JF, Schofield DL. Hyperparathyroidism: an incidental finding. Nurse Pract. 2012;37(11):9-14.
4. Cordellat IM. Hyperparathyroidism: primary or secondary disease? Rheumatol Clin. 2012;8(5):287-291.
5. MacKenzie-Feder J, Sirrs S, Anderson D, Sharif J, Khan A. Primary hyperparathyroidism: an overview. Int J Endocrinol. 2011;2011:251410.
6. Brashers VL, Jones RE, Huether SE. Alterations of hormonal regulation. In: McCance KL, Huether SE, eds. Pathophysiology: The Biologic Basis for Disease in Adults and Children. 7th ed. St. Louis, MO: Mosby; 2015:731-733.
7. Osborne JL, Klocko DJ. Woman, 66, with persistent abdominal and back pain. Clinician Reviews. 2014;24(11):34-37, 40.
8. Michels TC, Kelly KM. Parathyroid disorders. Am Fam Physician. 2013;88(4):249-257.
9. Rolighed L, Rejnmark L, Christiansen P. Bone involvement in primary hyperparathyroidism and changes after parathyroidectomy. US Endocrinol. 2013;9(2):181-184.
10. Karahan Ö, Okus A, Sevinç B, et al. Minimally invasive parathyroidectomy under local anesthesia. J Postgrad Med. 2013;59(1):21-24.
11. Fuleihan GE, Silverberg SJ. Primary hyperparathyroidism: diagnosis, differential diagnosis, and evaluation. Up-to-Date. www.uptodate.com/contents/primary-hyperparathyroidism-diagnosis-differential-diagnosis-and-evaluation. Accessed April 20, 2017.
12. Panchani R, Varma T, Goyal A, et al. A challenging case of an ectopic parathyroid adenoma. Indian J Endocrinol Metab. 2012;16:S408-S410.
13. Otasowie J, Hambleton BA. Aggression and homicidal thoughts in a patient with primary hyperparathyroidism: a case report. Br J Medical Pract. 2013;6(4):a630.
14. Bilezikian JP. Primary hyperparathyroidism: new insights, concepts and guidelines. Presented at: American Association of Clinical Endocrinologists 24th Annual Scientific and Clinical Congress; May 13-17, 2015; Nashville, TN. am2015.aace.com/presentations/Friday/F31/PrimaryHyperparathyroidismNew InsightsConceptsandGuidelines.pdf. Accessed April 20, 2017.
15. Crowther-Radulewicz CL, McCance KL. Alterations of musculoskeletal function. In: McCance KL, Huether SE, eds. Pathophysiology: The Biologic Basis for Disease in Adults and Children. 7th ed. St. Louis, MO: Mosby; 2015:1551-1555.
16. Amaral LM, Queiroz DC, Marques TF, et al. Clinical study: normocalcemic versus hypercalcemic primary hyperparathyroidism: more stone than bone? J Osteoporos. 2012;2012:128352.
17. Linda DD, Ng B, Rebello R, et al. The utility of multidetector computed tomography for detection of parathyroid disease in the setting of primary hyperparathyroidism. Can Assoc Radiol J. 2012;63(2):100-108.
18. Bann DV, Zacharia T, Goldenberg D, Goyal, N. Parathyroid localization using 4-D-computed tomography. Ear Nose Throat J. 2015;94(4-5):E55-E57.
19. Bilezikian JP, Brandi ML, Eastell R, et al. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the Fourth International Workshop. J Clin Endocrinol Metab. 2014;99(10):3561-3569.
20. Jeremiah MP, Unwin BK, Greenawald MH, Casiano VE. Diagnosis and management of osteoporosis. Am Fam Physician. 2015;92(4):261-268.
21. Wood K, Dhital S, Chen H, Sippel RS. What is the utility of distal forearm DXA in primary hyperparathyroidism? Oncologist. 2012;17(3):322-325.
22. Jayasena CN, Modi M, Palazzo F, et al. Associations of serum 25-hydroxyvitamin D with circulating PTH, phosphate and calcium in patients with primary hyperparathyroidism. Clin Endocrinol (Oxf). 2013;78(6):838-843.
23. Grey A. Nonsurgical management of mild primary hyperparathyroidism—a reasonable option. Clin Endocrinol. 2012;77(5):639-644.
24. Saponaro F, Faggiano A, Grimaldi F, et al. Cinacalcet in the management of primary hyperparathyroidism: post marketing experience of an Italian multicenter group. Clin Endocrinol (Oxf). 2013;79(1):20-26.
25. Rothe HM, Liangos O, Biggar P, et al. Cinacalcet treatment of primary hyperparathyroidism. Int J Endocrinol. 2011;2011:415719.
26. Farag N, Delbanco T, Strewler GJ. Update: a 64-year-old woman with primary hyperparathyroidism. JAMA. 2008;300(17):2044-2045.
1. Turner J. Hypercalcaemia and primary hyperparathyroidism. Medicine. 2009;37(9):461-464.
2. Crowley R, Gittoes N. How to approach hypercalcaemia. Clin Med. 2013;13(3):287-290.
3. Kapustin JF, Schofield DL. Hyperparathyroidism: an incidental finding. Nurse Pract. 2012;37(11):9-14.
4. Cordellat IM. Hyperparathyroidism: primary or secondary disease? Rheumatol Clin. 2012;8(5):287-291.
5. MacKenzie-Feder J, Sirrs S, Anderson D, Sharif J, Khan A. Primary hyperparathyroidism: an overview. Int J Endocrinol. 2011;2011:251410.
6. Brashers VL, Jones RE, Huether SE. Alterations of hormonal regulation. In: McCance KL, Huether SE, eds. Pathophysiology: The Biologic Basis for Disease in Adults and Children. 7th ed. St. Louis, MO: Mosby; 2015:731-733.
7. Osborne JL, Klocko DJ. Woman, 66, with persistent abdominal and back pain. Clinician Reviews. 2014;24(11):34-37, 40.
8. Michels TC, Kelly KM. Parathyroid disorders. Am Fam Physician. 2013;88(4):249-257.
9. Rolighed L, Rejnmark L, Christiansen P. Bone involvement in primary hyperparathyroidism and changes after parathyroidectomy. US Endocrinol. 2013;9(2):181-184.
10. Karahan Ö, Okus A, Sevinç B, et al. Minimally invasive parathyroidectomy under local anesthesia. J Postgrad Med. 2013;59(1):21-24.
11. Fuleihan GE, Silverberg SJ. Primary hyperparathyroidism: diagnosis, differential diagnosis, and evaluation. Up-to-Date. www.uptodate.com/contents/primary-hyperparathyroidism-diagnosis-differential-diagnosis-and-evaluation. Accessed April 20, 2017.
12. Panchani R, Varma T, Goyal A, et al. A challenging case of an ectopic parathyroid adenoma. Indian J Endocrinol Metab. 2012;16:S408-S410.
13. Otasowie J, Hambleton BA. Aggression and homicidal thoughts in a patient with primary hyperparathyroidism: a case report. Br J Medical Pract. 2013;6(4):a630.
14. Bilezikian JP. Primary hyperparathyroidism: new insights, concepts and guidelines. Presented at: American Association of Clinical Endocrinologists 24th Annual Scientific and Clinical Congress; May 13-17, 2015; Nashville, TN. am2015.aace.com/presentations/Friday/F31/PrimaryHyperparathyroidismNew InsightsConceptsandGuidelines.pdf. Accessed April 20, 2017.
15. Crowther-Radulewicz CL, McCance KL. Alterations of musculoskeletal function. In: McCance KL, Huether SE, eds. Pathophysiology: The Biologic Basis for Disease in Adults and Children. 7th ed. St. Louis, MO: Mosby; 2015:1551-1555.
16. Amaral LM, Queiroz DC, Marques TF, et al. Clinical study: normocalcemic versus hypercalcemic primary hyperparathyroidism: more stone than bone? J Osteoporos. 2012;2012:128352.
17. Linda DD, Ng B, Rebello R, et al. The utility of multidetector computed tomography for detection of parathyroid disease in the setting of primary hyperparathyroidism. Can Assoc Radiol J. 2012;63(2):100-108.
18. Bann DV, Zacharia T, Goldenberg D, Goyal, N. Parathyroid localization using 4-D-computed tomography. Ear Nose Throat J. 2015;94(4-5):E55-E57.
19. Bilezikian JP, Brandi ML, Eastell R, et al. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the Fourth International Workshop. J Clin Endocrinol Metab. 2014;99(10):3561-3569.
20. Jeremiah MP, Unwin BK, Greenawald MH, Casiano VE. Diagnosis and management of osteoporosis. Am Fam Physician. 2015;92(4):261-268.
21. Wood K, Dhital S, Chen H, Sippel RS. What is the utility of distal forearm DXA in primary hyperparathyroidism? Oncologist. 2012;17(3):322-325.
22. Jayasena CN, Modi M, Palazzo F, et al. Associations of serum 25-hydroxyvitamin D with circulating PTH, phosphate and calcium in patients with primary hyperparathyroidism. Clin Endocrinol (Oxf). 2013;78(6):838-843.
23. Grey A. Nonsurgical management of mild primary hyperparathyroidism—a reasonable option. Clin Endocrinol. 2012;77(5):639-644.
24. Saponaro F, Faggiano A, Grimaldi F, et al. Cinacalcet in the management of primary hyperparathyroidism: post marketing experience of an Italian multicenter group. Clin Endocrinol (Oxf). 2013;79(1):20-26.
25. Rothe HM, Liangos O, Biggar P, et al. Cinacalcet treatment of primary hyperparathyroidism. Int J Endocrinol. 2011;2011:415719.
26. Farag N, Delbanco T, Strewler GJ. Update: a 64-year-old woman with primary hyperparathyroidism. JAMA. 2008;300(17):2044-2045.
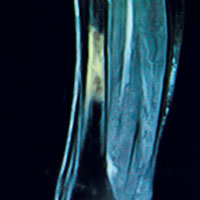
Tetanus: Debilitating Infection
CE/CME No: CR-1704
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Recognize patients who are at risk for tetanus.
• Describe the clinical presentation of tetanus.
• Discuss proper treatment for a patient with tetanus.
• Promote widespread vaccination against tetanus.
FACULTY
Timothy W. Ferrarotti is the Director of Didactic Education and Assistant Professor in the PA Studies Program at the University of Saint Joseph, West Hartford, Connecticut.
The author has no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of April 2017.
Article begins on next page >>
Tetanus is a devastating disease that can be prevented by proper immunization and wound care. Although the incidence is low in the United States due to widespread routine vaccination, immunization coverage remains below target, especially in older adults. Since outcome is influenced by the clinician's ability to make a timely diagnosis and initiate appropriate care, continued appreciation of tetanus is warranted.
Tetanus is a neurologic disorder resulting from infection by the gram-positive, spore-forming anaerobic bacillus Clostridium tetani. The bacterium, in spore form, typically enters the body through a contaminated soft-tissue wound. Ubiquitous in the environment, C tetani spores are found throughout the world—in soil as well as in animal feces and saliva—and are resistant to temperature extremes and antiseptics. Tetanus is infectious but not contagious (not transmitted person-to-person).1 Wounds with devitalized tissue or those supporting anaerobic conditions, such as bites, puncture wounds, burns, and gangrene, are conducive to the development of tetanus. Infection can also occur following dental extractions, abortions, and illicit drug injection.1 Although vaccination programs have decreased the incidence of tetanus in the United States, C tetani infection remains an ongoing clinical concern because the spores are omnipresent, universal vaccination coverage has not been achieved, and vaccine-immunity wanes over time, placing individuals at risk.
PATHOPHYSIOLOGY
C tetani produces two toxins: tetanolysin and tetanospasmin (tetanus toxin). Tetanolysin may have a role in promoting the diffusion of tetanospasmin in soft tissues.2 Tetanospasmin is a highly potent toxin, with a lethal dose in humans of less than 2.5 ng/kg of body weight.3 The toxin enters the peripheral nerve at the site of injury and migrates to the central nervous system (CNS). There, it causes unopposed α-motor neuron firing by preventing the release of inhibitory neurotransmitters such as γ-aminobutyric acid (GABA), resulting in muscle spasms and excess reflexive response to sensory stimuli.4 It also leads to excessive catecholamine release from the adrenal medulla.1
Tetanospasmin binds to neurons in the spinal cord and brainstem. Because toxin binding is irreversible, resolution of tetanus requires the neurons to grow new axon terminals. The effects of tetanus can persist for six to eight weeks until new terminals develop.3,5 Patients often require several weeks of ventilator support during this time.3

EPIDEMIOLOGY
Tetanus continues to be a serious cause of morbidity and mortality worldwide. The majority of cases (80%) occur in Africa and Southeast Asia.6 Incidence is much lower in the United States (0.1 cases per million persons annually) because of widespread vaccination, with only 233 cases of tetanus reported between 2001 and 2008.7 However, in the absence of confirmatory tests, the diagnosis is a clinical one; furthermore, there is no laboratory reporting program for tetanus. As a result, more cases may occur in the US than are detected or reported.
In developed countries, tetanus is primarily a disease of the elderly or the unvaccinated. Older persons, especially non-veterans, are less likely to have received the primary series. Because immunity decreases with age, even those who completed the primary series but have not received booster doses are at increased risk.8 Home-schooled children, who are not subject to school-entry vaccination requirements, are also at risk if unvaccinated.9
Neonatal tetanus is an ongoing problem in undeveloped countries that lack maternal vaccination programs. (Maternal immunization successfully reduces neonatal tetanus via passive immunity, and maternal tetanus via active immunity.) Unvaccinated women who undergo nonmedical abortions or unhygienic childbirth are at increased risk for tetanus.3,10
Other risk factors for tetanus include wound contamination with soil, saliva, or devitalized tissue; injection drug use; and exposure to anthropogenic or natural disasters.1 C tetani spores can contaminate heroin and may grow in abscesses of heroin users.4 Small outbreaks of tetanus among injection drug users have been reported, even among younger adults who had some immunity from childhood vaccination.7,11 In addition, patients with diabetes are at increased risk for tetanus. These patients may have chronic wounds due to slowed healing and poor vascularity, which can lead to lower oxygen tension in their wounds and create an environment conducive to anaerobic infection. These chronic wounds are often ignored as a potential nidus for tetanus; instead, focus is placed on plantar puncture wounds or lacerations.7
Though tetanus risk is greatest for those who were never fully immunized, cases have been reported in persons who were immunized in the remote past but had not received a recent booster. Such cases show that tetanus immunity is not absolute and does wane over time.12-14 Among the 233 tetanus cases reported in the US during 2001-2008, vaccination status was reported for 92. Of these, 24 patients had a complete series and 31 patients had at least one prior dose of tetanus vaccine.7 Furthermore, six cases occurred in patients known to have had the four-dose series and a booster within 10 years of diagnosis. Similarly, a 14-year-old boy who was fully vaccinated developed cephalic tetanus from a stingray wound.14 Given these data, clinicians should not assume that a patient who reports having had “a tetanus shot” is completely protected; a full series and regular boosters are required, and, in rare cases, tetanus can occur despite full vaccination.
PATIENT PRESENTATION AND TETANUS TYPES
The CDC describes tetanus as “the acute onset of hypertonia and/or painful muscle contractions (usually of the muscles of the jaw and neck) and generalized muscle spasms without other apparent medical cause.”15 Clinicians should always consider tetanus in patients with dysphagia and trismus, especially if the patient has a wound, had not received primary vaccination, or has not had a booster in several decades. Tetanus cannot be ruled out based on the lack of a wound, however, since up to 25% of patients who develop tetanus have no obvious site of inoculation.16 The incubation period ranges from 3 to 21 days, with more severe cases having shorter incubation periods (< 8 days).10 The closer the site of inoculation is to the CNS, the more serious the disease usually is—and the shorter the incubation period will be.1
Presentation depends on the time elapsed since inoculation, the severity of illness (determined by the Ablett classification; see Table 1), and the form of tetanus involved. The patient may present early when the infection and toxin are localized to the wound and have not progressed to the CNS (localized tetanus). There may be a wound with signs of infection, including erythema, induration, edema, warmth, tenderness, and drainage. If the injury is on the head or neck, cephalic tetanus may occur, causing the patient to present with painful spasms of the extra-ocular, facial, and/or neck muscles; trismus; dysphagia; or even a Horner-like syndrome. The patient with more advanced, generalized tetanus may have decorticate posturing, abdominal wall rigidity, or opisthotonus.1,2,5,17

Four types of tetanus have been described: generalized, localized, cephalic, and neonatal.
Generalized tetanus is the most common form, accounting for approximately 80% of cases.10 It may involve contractions of the masseter muscles, producing trismus; facial muscles, producing risus sardonicus (sardonic smile); neck and shoulder muscles; abdominal wall muscles, mimicking guarding; and back muscles, producing opisthotonus (arching of the back, neck, and head; see Figure 1) and decorticate posturing (flexion and adduction of the arms, clenched fists, and extension of the lower extremities).1,5,6 Patients with generalized tetanus often exhibit hyperresponsiveness to the environment. As a result, noises and sudden light changes may result in acute spasms. In addition, patients may experience painful spasms when affected muscles are palpated. Affected reflex arcs are usually hyperresponsive to stimuli.1 Intermittent spasms of the thoracic, pharyngeal, and/or laryngeal muscles may cause periods of apnea. Autonomic effects of tetanus mimic those associated with the catecholamine excess of pheochromocytoma. Patients exhibit restlessness, irritability, diaphoresis, fever, excessive salivation, gastric stasis, hypertension, tachycardia, and arrhythmia. There may be interposed hypotension and bradycardia.1,5,17
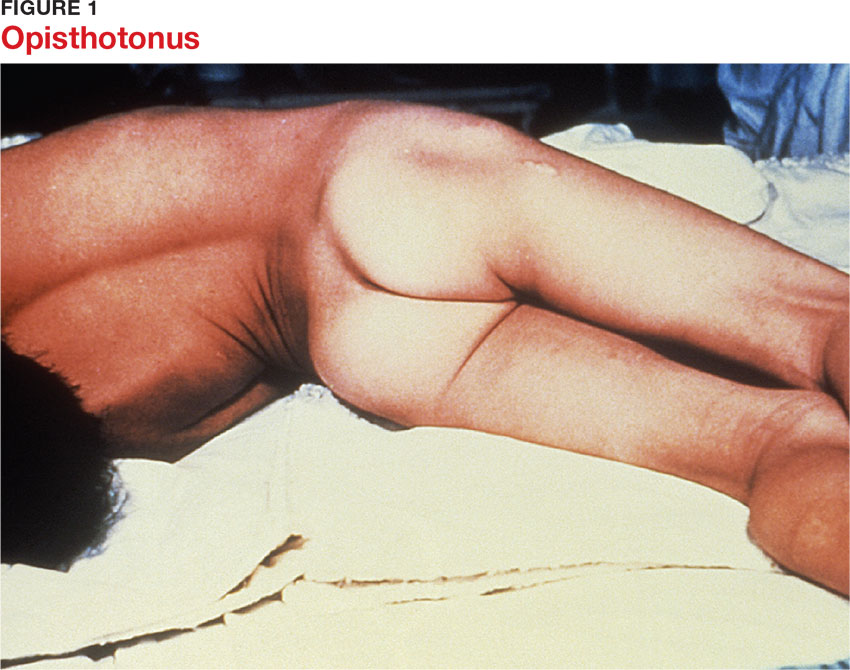
Localized tetanus involves painful spastic contraction of muscles at or near the site of inoculation. It often evolves into generalized tetanus as the toxin spreads further into the CNS.
Cephalic tetanus involves facial and laryngeal muscles. It is rare, accounting for 1% to 3% of tetanus cases.6 Patients may initially have flaccid paralysis, mimicking stroke, rather than spasm, because the toxin has not completely migrated up the peripheral nerve into the CNS. As the toxin enters the CNS and induces the typical spasm (trismus), the diagnosis will be more obvious. The presence of trismus or a subacute wound on the head may be used to discriminate tetanus from stroke. Cephalic tetanus often evolves into generalized tetanus, affecting more of the body in a caudal direction.5
Neonatal tetanus develops within one week after birth. The neonate with tetanus is usually born to a mother lacking immunization. Typically, the infant sucks and feeds for the first couple of days, then develops inability/refusal to suck/feed, has difficulty opening his/her mouth, becomes weak, and develops muscle spasms.3 The affected child may develop risus sardonicus, clenched hands, dorsiflexion of the feet, and opisthotonus.3
DIFFERENTIAL DIAGNOSIS
The clinician should consider other CNS conditions in the differential diagnosis (see Table 2). Although similar to generalized seizures, tetanus causes painful spasms and does not produce a loss of consciousness.1,17 Tetanus, intracranial bleed, and meningitis all can cause meningismus; meningitis, however, is more likely to manifest with other symptoms of infection, such as headache and fever. Although the autonomic dysfunction of tetanus can cause pyrexia, fever would usually coincide with other sympathetic symptoms, such as hypertension, tachycardia, and diaphoresis. Intracranial bleeding tends to have a more rapid onset than tetanus and produces headache and mental status changes. Seventh nerve palsy produces muscle flaccidity, not spasm, and is usually painless unless associated with herpetic inflammation.1,5,6,14,17

Poisoning and medication effects should also be considered. Strychnine poisoning manifests similar to tetanus but occurs without a wound.5 Blood and urine assays for strychnine can be diagnostic. Dystonic reactions resulting from neuroleptic medications—such as phenothiazines—include torticollis, oropharyngeal muscle spasms, and deviation of the eyes. Unlike tetanus, drug-induced dystonia does not cause reflex spasms and often resolves with benztropine or diphenhydramine administration.1 Neuroleptic malignant syndrome can also cause muscular rigidity and autonomic instability, but unlike tetanus, it often causes altered mental status; it should be considered in patients who recently received a causative medication.5,17
Tetanus often manifests with reflexive muscle spasms similar to those seen in electrolyte and acid-base abnormalities. Hypocalcemia may produce a reflexive spasm of the facial muscles when the facial nerve is percussed (Chvostek sign), while alkalemia may produce reflexive spasm of the hand and wrist muscles (Trousseau sign).1 Lab tests can rule out these diagnoses.1,5
A patient with an odontogenic abscess may have pain and muscle spasm/trismus, but the infection is usually easily detected on exam. The clinician should be cautious in attributing the trismus solely to the swelling, however, as C tetani has been found in odontogenic abscesses and the patient may have both.1,17 Peritonsillar abscess will often produce trismus. When abscess is the cause, careful examination of the oropharynx will usually demonstrate tonsillar exudate, hypertrophy, soft tissue erythema, and tenderness, as well as a misplaced uvula.1
DIAGNOSIS
Tetanus is a clinical diagnosis, usually made based on the findings described. Confirmatory lab tests are not readily available. The organism is infrequently recovered in cultures of specimens from suspected wounds (30% of cases).10,11 Serologic testing on specimens drawn before administration of tetanus immunoglobulin (TIG) may indicate very low or undetectable antitetanus antibody levels, but tetanus can still occur when “protective” levels of antibodies are present.11 Detection of tetanus toxin in plasma or a wound with bioassays and polymerase chain reaction might be possible, but these tests are only available in a few settings.3
THE MULTIFACETED CARE PLAN
The primary care provider should refer a patient with suspected tetanus to an emergency department, preferably a tertiary care center with the necessary specialists. Patients are likely to require prolonged hospitalization. In a recent series of tetanus cases in California, the median length of hospitalization was 18 days.12 Treatment is multifaceted; interventions include immunization, wound care, administration of antibiotics and other pharmacologic agents, and supportive therapy (see Table 3).

Immunization
All patients with suspected tetanus should immediately receive both passive (with TIG) and active (tetanus toxoid–containing vaccines) immunization. Because of the extremely high potency of tetanus toxin, the very small amount of toxin that is required to cause tetanus is insufficient to prompt an immune response that would confer immunity. Therefore, treatment is the same regardless of whether the patient had prior disease.10
TIG binds to and neutralizes unbound tetanospasmin, preventing progression of the disease. As noted, TIG will not reverse the binding of the toxin to nerve structures.5 Due to a lack of prospective studies, there is disagreement regarding TIG dosage: Doses as high as 3,000-6,000 U have been recommended, but case studies indicate that the dosage recommended by the CDC (500 U) is likely effective.13 The full CDC recommendation is 500 U of human-derived TIG intramuscularly administered at locations near and away from the wound (but always away from the tetanus toxoid injection site).10,17 Outside the US, equine-based TIG may be the only option. Animal-derived TIG is less desirable because of increased allergy risk; when used, a small amount (0.1 mL) should be first administered as an intradermal test.17
Tetanus toxoid immunization produces active immunity. It is currently available in combination antigen forms (tetanus and diphtheria vaccine [Td], tetanus-diphtheria-acellular pertussis [Tdap] vaccine). The dose of either is 0.5 mL. Patients with tetanus should receive three doses given intramuscularly: immediately, at 4 weeks, and at 6 to 12 months.10
Wound care
Wound care should include incision and drainage, removal of foreign bodies, debridement, and irrigation. These steps are taken in order to ensure an aerobic environment in the wound, ultimately decreasing C tetani survival.1
Antibiotics
The preferred antimicrobial agent for treating tetanus infection is metronidazole 500 mg intravenously (IV) every 6 hours.1,3,17 Penicillin (2 to 4 million U IV every 4 to 6 hours) is effective against C tetani, but it is a GABA-receptor antagonist and may worsen tetanus by further inhibiting the release of GABA.1,14,18 GABA-receptor antagonism may also occur with cephalosporins; however, these broader-spectrum agents may be necessary to treat mixed infections.17 Alternatives include doxycycline, macrolides, and clindamycin.1
Other pharmacologic treatment
Benzodiazepines (eg, diazepam 10 to 30 mg IV) can help control rigidity and muscle spasms and are a mainstay of tetanus treatment.18 Benzodiazepines and propofol both act on GABA receptors, producing sedation in addition to controlling muscle spasms.19 Traditionally, more severe spasms, such as opisthotonus, have required induction of complete paralysis with nondepolarizing paralytics, such as pancuronium or vecuronium. However, paralysis is not optimal therapy since it necessitates sedation, intubation, and mechanical ventilation. Because tetanus does not resolve for 6 to 8 weeks, patients who require mechanical ventilation will also require tracheostomy to prevent laryngotracheal stenosis. Paralysis and mechanical ventilation can also lead to deep venous thrombosis, decubitus ulcers, and pneumonia.5 The ideal treatment would reduce the spasms and autonomic instability without the risks associated with deep sedation and paralysis.5
Other agents used in the treatment of tetanus include magnesium sulfate, which can decrease muscle spasm and ameliorate the effects of autonomic dysfunction, and intrathecal baclofen, which can decrease muscle spasm.19,20 Patients with persistent autonomic dysfunction may require combined α- and ß-adrenergic receptor blockade.1,17-20
Supportive care
It is important to implement supportive care, including limiting auditory and tactile stimulation, as well as providing adequate hydration and nutritional support. IV fluids, parenteral feeding, and enteral feeding are required. Measures should be taken to prevent complications of prolonged immobility, paralysis, and mechanical ventilation, including deep venous thrombosis, pulmonary embolism, and pressure ulcers. The quality of supportive care and the swiftness with which the diagnosis is made and appropriate treatment is initiated are key factors that determine an individual patient’s outcome.21
COMPLICATIONS AND MORTALITY
Tetanus can lead to many complications, including long bone and spine fractures from severe muscle spasms, as well as renal failure and aspiration. Most spinal fractures involve the thoracic spine, but lumbar spine fractures have been reported.22 Burst-type fractures of the vertebrae may cause cauda equina syndrome or directly injure the spinal cord if fragments are retropulsed.22 Persistent muscle spasm can also cause rhabdomyolysis and renal failure. Lab test results, including elevated levels of creatine phosphokinase and myoglobin (rhabdomyolysis) as well as blood urea nitrogen and creatinine (renal failure), can indicate presence of complications. Muscle relaxation and hydration are key to prevention.
Patients with trismus are often unable to swallow and maintain oral hygiene, leading to caries and dental abscess. The trismus itself can also cause dental or jaw fractures.2,13 Aspiration can occur when laryngeal muscles are affected, resulting in pneumonia in 50% to 70% of autopsied cases of tetanus.10 Additionally, the paralyzed patient receiving ventilatory support can develop pneumonia, deep vein thrombosis, and pulmonary embolism.5,13 Neonatal tetanus often results in complications such as cerebral palsy or cognitive delay.1
A number of factors influence the severity and outcome of tetanus. Untreated tetanus is typically fatal, with respiratory failure the most common cause of death in settings where mechanical ventilation is unavailable.1 Where mechanical ventilation is accessible, autonomic dysfunction accounts for most deaths.20 Ventilation aside, the case-fatality rate varies according to the medical system. The rate is often less than 20% where modern ICUs are available but can exceed 50% in undeveloped countries with limited facilities.1,5 A review of outcomes data for 197 of the 233 tetanus cases reported in the US during 2001-2008 (modern medical care was provided in all) showed an overall case-fatality rate of 13.2%.7
Age and vaccination status also affect outcomes, with higher case-fatality rates seen in older (18% in those ≥ 60, 31% in those ≥ 65) and unvaccinated (22%) patients. 7,10 In the study of tetanus cases from 2001-2008, the fatality rate was five times higher in patients ages 65 or older compared with patients younger than 65.7 This study also showed that severity of tetanus may be inversely related to the number of vaccine doses the individual has received, and that having previous vaccination was associated with improved survival, as only four of the 26 deaths occurred in patients with prior vaccination.7
Patients who survive the first two weeks of tetanus have a better chance of recovery. Those with multiple chronic comorbidities, such as chronic obstructive pulmonary disease (COPD), diabetes, or cardiovascular disease, are more likely to die because of the physiologic stress of the illness and its treatment.1,7,12 The provision of ventilator support is more complicated in those with COPD; similarly, the autonomic effects of tetanus can be more problematic for patients with chronic cardiac disease or neurologic complications of chronic diabetes.13
PATIENT EDUCATION
Widespread vaccination against tetanus, which began in the US in the mid-20th century, has greatly reduced disease incidence.7 However, vaccination coverage rates remain below target.
In 2012, only 82.5% of children ages 19 to 35 months received the recommended four doses of diphtheria-tetanus-pertussis (DTaP) vaccine, and 94.3% received at least three doses.23 Only 84.6% of teens ages 13 to 17 years received the primary four doses as well as the recommended booster dose.24 The same year, only 55% of patients ages 65 and older and 64% of adults ages 19 to 64 had received a tetanus booster within the previous 10 years.25
Vaccination rates are lower for black, Hispanic, and Asian adults in the US.25 Clinicians should proactively recommend tetanus booster immunization to all adults.
CONCLUSION
Although few clinicians in developed countries will see a case of tetanus, all should be alert for it. Elderly patients and those not fully vaccinated are at risk. Routine immunization decreases but does not eliminate the risk. Tetanus differs from other illnesses controlled by national immunization efforts in that unvaccinated persons do not benefit from herd immunity, because the disease is not contagious. The diagnosis is clinical and should always be considered in patients with trismus, dysphagia, and/or adrenergic excess. Wounds that place a patient at risk for tetanus involve devitalized tissues and anaerobic conditions. Prompt diagnosis is essential, because it allows for early neutralization of unbound tetanospasmin. Wound care including debridement, antibiotic therapy, control of muscle spasms and the effects of autonomic instability, and airway care are fundamental to the treatment of tetanus.
1. Afshar M, Raju M, Ansell D, Bleck TP. Narrative review: tetanus—a health threat after natural disasters in developing countries. Ann Intern Med. 2011;154(5):329-335.
2. Demir NA, Sumer S, Ural O, et al. An alternative treatment approach in tetanus: botulinum toxin. Trop Doct. 2015;45(1): 46-48.
3. Thwaites CL, Beeching NJ, Newton CR. Maternal and neonatal tetanus. Lancet. 2015;385(9965):362-370.
4. Aronoff DM. Clostridium novyi, sordellii, and tetani: mechanisms of disease. Anaerobe. 2013;24:98-101.
5. Cook TM, Protheroe RT, Handel JM. Tetanus: a review of the literature. Br J Anaesth. 2001;87(3):477-487.
6. Doshi A, Warrell C, Dahdaleh D, Kullmann D. Just a graze? Cephalic tetanus presenting as a stroke mimic. Pract Neurol. 2014;14(1):39-41.
7. CDC. Tetanus surveillance—United States, 2001-2008. MMWR Morb Mortal Wkly Rep. 2011;60(12):365-369.
8. McCabe J, La Varis T, Mason D. Cephalic tetanus complicating geriatric fall. N Z Med J. 2014;127(1400):98-100.
9. Johnson MG, Bradley KK, Mendus S, et al. Vaccine-preventable disease among homeschooled children: two cases of tetanus in Oklahoma. Pediatrics. 2013;132(6):e1686-e1689.
10. CDC. Epidemiology and Prevention of Vaccine-Preventable Diseases. Hamborsky J, Kroger A, Wolfe S, eds. 13th ed. Washington, DC: Public Health Foundation; 2015.
11. Tiwari TS. Chapter 16: Tetanus. In: CDC. Manual for Surveillance of Vaccine-Preventable Diseases. 5th ed. Atlanta, GA: CDC; 2012.
12. Yen C, Murray E, Zipprich J, et al. Missed opportunities for tetanus postexposure prophylaxis—California, January 2008-March 2014. MMWR Morb Mortal Wkly Rep. 2015; 64(9):243-246.
13. Aksoy M, Celik EC, Ahiskalioglu A, Karakaya MA. Tetanus is still a deadly disease: a report of six tetanus cases and reminder of our knowledge. Trop Doct. 2014;44(1):38-42.
14. Felter RA, Zinns LE. Cephalic tetanus in an immunized teenager. Pediatr Emerg Care. 2015;31(7):511-513.
15. CDC. Tetanus (Clostridium tetani) 1996 case definition. www.cdc.gov/nndss/conditions/tetanus/case-definition/1996/. Accessed February 17, 2017.
16. Thwaites CL, Farrar JJ. Preventing and treating tetanus [commentary]. BMJ. 2003;326(7381):117-118.
17. Sexton DJ. Tetanus. UpToDate. www.uptodate.com/contents/tetanus?topicKey=ID%2F5525. Accessed February 17, 2017.
18. Rodrigo C, Fernando D, Rajapakse S. Pharmacological management of tetanus: an evidence-based review. Crit Care. 2014;18(2):217.
19. Santos ML, Mota-Miranda A, Alves-Pereira A, et al. Intrathecal baclofen for the treatment of tetanus. Clin Infect Dis. 2004;38(3):321-328.
20. Thwaites CL, Yen LM, Loan HT, et al. Magnesium sulphate for treatment of severe tetanus: a randomized controlled trial. Lancet. 2006;368:1436-1443.
21. Govindaraj GM, Riyaz A. Current practice in the management of tetanus. Crit Care. 2014;18(3):145.
22. Wilson TJ, Orringer DA, Sullivan SE, Patil PG. An L-2 burst fracture and cauda equina syndrome due to tetanus. J Neurosurg Spine. 2012;16(1):82-85.
23. CDC. National, state and local area vaccination coverage among children aged 19-35 months—United States, 2012. MMWR Morb Mortal Wkly Rep. 2013;62(36):733-740.
24. CDC. National and state vaccination coverage among adolescents aged 13-17 years—United States, 2012. MMWR Morb Mortal Wkly Rep. 2013;62(34):685-693.
25. Williams WW, Lu PJ, O’Halloran A, et al. Noninfluenza vaccination coverage among adults—United States, 2012. MMWR Morb Mortal Wkly Rep. 2014;63(5):95-102.
CE/CME No: CR-1704
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Recognize patients who are at risk for tetanus.
• Describe the clinical presentation of tetanus.
• Discuss proper treatment for a patient with tetanus.
• Promote widespread vaccination against tetanus.
FACULTY
Timothy W. Ferrarotti is the Director of Didactic Education and Assistant Professor in the PA Studies Program at the University of Saint Joseph, West Hartford, Connecticut.
The author has no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of April 2017.
Article begins on next page >>
Tetanus is a devastating disease that can be prevented by proper immunization and wound care. Although the incidence is low in the United States due to widespread routine vaccination, immunization coverage remains below target, especially in older adults. Since outcome is influenced by the clinician's ability to make a timely diagnosis and initiate appropriate care, continued appreciation of tetanus is warranted.
Tetanus is a neurologic disorder resulting from infection by the gram-positive, spore-forming anaerobic bacillus Clostridium tetani. The bacterium, in spore form, typically enters the body through a contaminated soft-tissue wound. Ubiquitous in the environment, C tetani spores are found throughout the world—in soil as well as in animal feces and saliva—and are resistant to temperature extremes and antiseptics. Tetanus is infectious but not contagious (not transmitted person-to-person).1 Wounds with devitalized tissue or those supporting anaerobic conditions, such as bites, puncture wounds, burns, and gangrene, are conducive to the development of tetanus. Infection can also occur following dental extractions, abortions, and illicit drug injection.1 Although vaccination programs have decreased the incidence of tetanus in the United States, C tetani infection remains an ongoing clinical concern because the spores are omnipresent, universal vaccination coverage has not been achieved, and vaccine-immunity wanes over time, placing individuals at risk.
PATHOPHYSIOLOGY
C tetani produces two toxins: tetanolysin and tetanospasmin (tetanus toxin). Tetanolysin may have a role in promoting the diffusion of tetanospasmin in soft tissues.2 Tetanospasmin is a highly potent toxin, with a lethal dose in humans of less than 2.5 ng/kg of body weight.3 The toxin enters the peripheral nerve at the site of injury and migrates to the central nervous system (CNS). There, it causes unopposed α-motor neuron firing by preventing the release of inhibitory neurotransmitters such as γ-aminobutyric acid (GABA), resulting in muscle spasms and excess reflexive response to sensory stimuli.4 It also leads to excessive catecholamine release from the adrenal medulla.1
Tetanospasmin binds to neurons in the spinal cord and brainstem. Because toxin binding is irreversible, resolution of tetanus requires the neurons to grow new axon terminals. The effects of tetanus can persist for six to eight weeks until new terminals develop.3,5 Patients often require several weeks of ventilator support during this time.3
EPIDEMIOLOGY
Tetanus continues to be a serious cause of morbidity and mortality worldwide. The majority of cases (80%) occur in Africa and Southeast Asia.6 Incidence is much lower in the United States (0.1 cases per million persons annually) because of widespread vaccination, with only 233 cases of tetanus reported between 2001 and 2008.7 However, in the absence of confirmatory tests, the diagnosis is a clinical one; furthermore, there is no laboratory reporting program for tetanus. As a result, more cases may occur in the US than are detected or reported.
In developed countries, tetanus is primarily a disease of the elderly or the unvaccinated. Older persons, especially non-veterans, are less likely to have received the primary series. Because immunity decreases with age, even those who completed the primary series but have not received booster doses are at increased risk.8 Home-schooled children, who are not subject to school-entry vaccination requirements, are also at risk if unvaccinated.9
Neonatal tetanus is an ongoing problem in undeveloped countries that lack maternal vaccination programs. (Maternal immunization successfully reduces neonatal tetanus via passive immunity, and maternal tetanus via active immunity.) Unvaccinated women who undergo nonmedical abortions or unhygienic childbirth are at increased risk for tetanus.3,10
Other risk factors for tetanus include wound contamination with soil, saliva, or devitalized tissue; injection drug use; and exposure to anthropogenic or natural disasters.1 C tetani spores can contaminate heroin and may grow in abscesses of heroin users.4 Small outbreaks of tetanus among injection drug users have been reported, even among younger adults who had some immunity from childhood vaccination.7,11 In addition, patients with diabetes are at increased risk for tetanus. These patients may have chronic wounds due to slowed healing and poor vascularity, which can lead to lower oxygen tension in their wounds and create an environment conducive to anaerobic infection. These chronic wounds are often ignored as a potential nidus for tetanus; instead, focus is placed on plantar puncture wounds or lacerations.7
Though tetanus risk is greatest for those who were never fully immunized, cases have been reported in persons who were immunized in the remote past but had not received a recent booster. Such cases show that tetanus immunity is not absolute and does wane over time.12-14 Among the 233 tetanus cases reported in the US during 2001-2008, vaccination status was reported for 92. Of these, 24 patients had a complete series and 31 patients had at least one prior dose of tetanus vaccine.7 Furthermore, six cases occurred in patients known to have had the four-dose series and a booster within 10 years of diagnosis. Similarly, a 14-year-old boy who was fully vaccinated developed cephalic tetanus from a stingray wound.14 Given these data, clinicians should not assume that a patient who reports having had “a tetanus shot” is completely protected; a full series and regular boosters are required, and, in rare cases, tetanus can occur despite full vaccination.
PATIENT PRESENTATION AND TETANUS TYPES
The CDC describes tetanus as “the acute onset of hypertonia and/or painful muscle contractions (usually of the muscles of the jaw and neck) and generalized muscle spasms without other apparent medical cause.”15 Clinicians should always consider tetanus in patients with dysphagia and trismus, especially if the patient has a wound, had not received primary vaccination, or has not had a booster in several decades. Tetanus cannot be ruled out based on the lack of a wound, however, since up to 25% of patients who develop tetanus have no obvious site of inoculation.16 The incubation period ranges from 3 to 21 days, with more severe cases having shorter incubation periods (< 8 days).10 The closer the site of inoculation is to the CNS, the more serious the disease usually is—and the shorter the incubation period will be.1
Presentation depends on the time elapsed since inoculation, the severity of illness (determined by the Ablett classification; see Table 1), and the form of tetanus involved. The patient may present early when the infection and toxin are localized to the wound and have not progressed to the CNS (localized tetanus). There may be a wound with signs of infection, including erythema, induration, edema, warmth, tenderness, and drainage. If the injury is on the head or neck, cephalic tetanus may occur, causing the patient to present with painful spasms of the extra-ocular, facial, and/or neck muscles; trismus; dysphagia; or even a Horner-like syndrome. The patient with more advanced, generalized tetanus may have decorticate posturing, abdominal wall rigidity, or opisthotonus.1,2,5,17
Four types of tetanus have been described: generalized, localized, cephalic, and neonatal.
Generalized tetanus is the most common form, accounting for approximately 80% of cases.10 It may involve contractions of the masseter muscles, producing trismus; facial muscles, producing risus sardonicus (sardonic smile); neck and shoulder muscles; abdominal wall muscles, mimicking guarding; and back muscles, producing opisthotonus (arching of the back, neck, and head; see Figure 1) and decorticate posturing (flexion and adduction of the arms, clenched fists, and extension of the lower extremities).1,5,6 Patients with generalized tetanus often exhibit hyperresponsiveness to the environment. As a result, noises and sudden light changes may result in acute spasms. In addition, patients may experience painful spasms when affected muscles are palpated. Affected reflex arcs are usually hyperresponsive to stimuli.1 Intermittent spasms of the thoracic, pharyngeal, and/or laryngeal muscles may cause periods of apnea. Autonomic effects of tetanus mimic those associated with the catecholamine excess of pheochromocytoma. Patients exhibit restlessness, irritability, diaphoresis, fever, excessive salivation, gastric stasis, hypertension, tachycardia, and arrhythmia. There may be interposed hypotension and bradycardia.1,5,17
Localized tetanus involves painful spastic contraction of muscles at or near the site of inoculation. It often evolves into generalized tetanus as the toxin spreads further into the CNS.
Cephalic tetanus involves facial and laryngeal muscles. It is rare, accounting for 1% to 3% of tetanus cases.6 Patients may initially have flaccid paralysis, mimicking stroke, rather than spasm, because the toxin has not completely migrated up the peripheral nerve into the CNS. As the toxin enters the CNS and induces the typical spasm (trismus), the diagnosis will be more obvious. The presence of trismus or a subacute wound on the head may be used to discriminate tetanus from stroke. Cephalic tetanus often evolves into generalized tetanus, affecting more of the body in a caudal direction.5
Neonatal tetanus develops within one week after birth. The neonate with tetanus is usually born to a mother lacking immunization. Typically, the infant sucks and feeds for the first couple of days, then develops inability/refusal to suck/feed, has difficulty opening his/her mouth, becomes weak, and develops muscle spasms.3 The affected child may develop risus sardonicus, clenched hands, dorsiflexion of the feet, and opisthotonus.3
DIFFERENTIAL DIAGNOSIS
The clinician should consider other CNS conditions in the differential diagnosis (see Table 2). Although similar to generalized seizures, tetanus causes painful spasms and does not produce a loss of consciousness.1,17 Tetanus, intracranial bleed, and meningitis all can cause meningismus; meningitis, however, is more likely to manifest with other symptoms of infection, such as headache and fever. Although the autonomic dysfunction of tetanus can cause pyrexia, fever would usually coincide with other sympathetic symptoms, such as hypertension, tachycardia, and diaphoresis. Intracranial bleeding tends to have a more rapid onset than tetanus and produces headache and mental status changes. Seventh nerve palsy produces muscle flaccidity, not spasm, and is usually painless unless associated with herpetic inflammation.1,5,6,14,17
Poisoning and medication effects should also be considered. Strychnine poisoning manifests similar to tetanus but occurs without a wound.5 Blood and urine assays for strychnine can be diagnostic. Dystonic reactions resulting from neuroleptic medications—such as phenothiazines—include torticollis, oropharyngeal muscle spasms, and deviation of the eyes. Unlike tetanus, drug-induced dystonia does not cause reflex spasms and often resolves with benztropine or diphenhydramine administration.1 Neuroleptic malignant syndrome can also cause muscular rigidity and autonomic instability, but unlike tetanus, it often causes altered mental status; it should be considered in patients who recently received a causative medication.5,17
Tetanus often manifests with reflexive muscle spasms similar to those seen in electrolyte and acid-base abnormalities. Hypocalcemia may produce a reflexive spasm of the facial muscles when the facial nerve is percussed (Chvostek sign), while alkalemia may produce reflexive spasm of the hand and wrist muscles (Trousseau sign).1 Lab tests can rule out these diagnoses.1,5
A patient with an odontogenic abscess may have pain and muscle spasm/trismus, but the infection is usually easily detected on exam. The clinician should be cautious in attributing the trismus solely to the swelling, however, as C tetani has been found in odontogenic abscesses and the patient may have both.1,17 Peritonsillar abscess will often produce trismus. When abscess is the cause, careful examination of the oropharynx will usually demonstrate tonsillar exudate, hypertrophy, soft tissue erythema, and tenderness, as well as a misplaced uvula.1
DIAGNOSIS
Tetanus is a clinical diagnosis, usually made based on the findings described. Confirmatory lab tests are not readily available. The organism is infrequently recovered in cultures of specimens from suspected wounds (30% of cases).10,11 Serologic testing on specimens drawn before administration of tetanus immunoglobulin (TIG) may indicate very low or undetectable antitetanus antibody levels, but tetanus can still occur when “protective” levels of antibodies are present.11 Detection of tetanus toxin in plasma or a wound with bioassays and polymerase chain reaction might be possible, but these tests are only available in a few settings.3
THE MULTIFACETED CARE PLAN
The primary care provider should refer a patient with suspected tetanus to an emergency department, preferably a tertiary care center with the necessary specialists. Patients are likely to require prolonged hospitalization. In a recent series of tetanus cases in California, the median length of hospitalization was 18 days.12 Treatment is multifaceted; interventions include immunization, wound care, administration of antibiotics and other pharmacologic agents, and supportive therapy (see Table 3).
Immunization
All patients with suspected tetanus should immediately receive both passive (with TIG) and active (tetanus toxoid–containing vaccines) immunization. Because of the extremely high potency of tetanus toxin, the very small amount of toxin that is required to cause tetanus is insufficient to prompt an immune response that would confer immunity. Therefore, treatment is the same regardless of whether the patient had prior disease.10
TIG binds to and neutralizes unbound tetanospasmin, preventing progression of the disease. As noted, TIG will not reverse the binding of the toxin to nerve structures.5 Due to a lack of prospective studies, there is disagreement regarding TIG dosage: Doses as high as 3,000-6,000 U have been recommended, but case studies indicate that the dosage recommended by the CDC (500 U) is likely effective.13 The full CDC recommendation is 500 U of human-derived TIG intramuscularly administered at locations near and away from the wound (but always away from the tetanus toxoid injection site).10,17 Outside the US, equine-based TIG may be the only option. Animal-derived TIG is less desirable because of increased allergy risk; when used, a small amount (0.1 mL) should be first administered as an intradermal test.17
Tetanus toxoid immunization produces active immunity. It is currently available in combination antigen forms (tetanus and diphtheria vaccine [Td], tetanus-diphtheria-acellular pertussis [Tdap] vaccine). The dose of either is 0.5 mL. Patients with tetanus should receive three doses given intramuscularly: immediately, at 4 weeks, and at 6 to 12 months.10
Wound care
Wound care should include incision and drainage, removal of foreign bodies, debridement, and irrigation. These steps are taken in order to ensure an aerobic environment in the wound, ultimately decreasing C tetani survival.1
Antibiotics
The preferred antimicrobial agent for treating tetanus infection is metronidazole 500 mg intravenously (IV) every 6 hours.1,3,17 Penicillin (2 to 4 million U IV every 4 to 6 hours) is effective against C tetani, but it is a GABA-receptor antagonist and may worsen tetanus by further inhibiting the release of GABA.1,14,18 GABA-receptor antagonism may also occur with cephalosporins; however, these broader-spectrum agents may be necessary to treat mixed infections.17 Alternatives include doxycycline, macrolides, and clindamycin.1
Other pharmacologic treatment
Benzodiazepines (eg, diazepam 10 to 30 mg IV) can help control rigidity and muscle spasms and are a mainstay of tetanus treatment.18 Benzodiazepines and propofol both act on GABA receptors, producing sedation in addition to controlling muscle spasms.19 Traditionally, more severe spasms, such as opisthotonus, have required induction of complete paralysis with nondepolarizing paralytics, such as pancuronium or vecuronium. However, paralysis is not optimal therapy since it necessitates sedation, intubation, and mechanical ventilation. Because tetanus does not resolve for 6 to 8 weeks, patients who require mechanical ventilation will also require tracheostomy to prevent laryngotracheal stenosis. Paralysis and mechanical ventilation can also lead to deep venous thrombosis, decubitus ulcers, and pneumonia.5 The ideal treatment would reduce the spasms and autonomic instability without the risks associated with deep sedation and paralysis.5
Other agents used in the treatment of tetanus include magnesium sulfate, which can decrease muscle spasm and ameliorate the effects of autonomic dysfunction, and intrathecal baclofen, which can decrease muscle spasm.19,20 Patients with persistent autonomic dysfunction may require combined α- and ß-adrenergic receptor blockade.1,17-20
Supportive care
It is important to implement supportive care, including limiting auditory and tactile stimulation, as well as providing adequate hydration and nutritional support. IV fluids, parenteral feeding, and enteral feeding are required. Measures should be taken to prevent complications of prolonged immobility, paralysis, and mechanical ventilation, including deep venous thrombosis, pulmonary embolism, and pressure ulcers. The quality of supportive care and the swiftness with which the diagnosis is made and appropriate treatment is initiated are key factors that determine an individual patient’s outcome.21
COMPLICATIONS AND MORTALITY
Tetanus can lead to many complications, including long bone and spine fractures from severe muscle spasms, as well as renal failure and aspiration. Most spinal fractures involve the thoracic spine, but lumbar spine fractures have been reported.22 Burst-type fractures of the vertebrae may cause cauda equina syndrome or directly injure the spinal cord if fragments are retropulsed.22 Persistent muscle spasm can also cause rhabdomyolysis and renal failure. Lab test results, including elevated levels of creatine phosphokinase and myoglobin (rhabdomyolysis) as well as blood urea nitrogen and creatinine (renal failure), can indicate presence of complications. Muscle relaxation and hydration are key to prevention.
Patients with trismus are often unable to swallow and maintain oral hygiene, leading to caries and dental abscess. The trismus itself can also cause dental or jaw fractures.2,13 Aspiration can occur when laryngeal muscles are affected, resulting in pneumonia in 50% to 70% of autopsied cases of tetanus.10 Additionally, the paralyzed patient receiving ventilatory support can develop pneumonia, deep vein thrombosis, and pulmonary embolism.5,13 Neonatal tetanus often results in complications such as cerebral palsy or cognitive delay.1
A number of factors influence the severity and outcome of tetanus. Untreated tetanus is typically fatal, with respiratory failure the most common cause of death in settings where mechanical ventilation is unavailable.1 Where mechanical ventilation is accessible, autonomic dysfunction accounts for most deaths.20 Ventilation aside, the case-fatality rate varies according to the medical system. The rate is often less than 20% where modern ICUs are available but can exceed 50% in undeveloped countries with limited facilities.1,5 A review of outcomes data for 197 of the 233 tetanus cases reported in the US during 2001-2008 (modern medical care was provided in all) showed an overall case-fatality rate of 13.2%.7
Age and vaccination status also affect outcomes, with higher case-fatality rates seen in older (18% in those ≥ 60, 31% in those ≥ 65) and unvaccinated (22%) patients. 7,10 In the study of tetanus cases from 2001-2008, the fatality rate was five times higher in patients ages 65 or older compared with patients younger than 65.7 This study also showed that severity of tetanus may be inversely related to the number of vaccine doses the individual has received, and that having previous vaccination was associated with improved survival, as only four of the 26 deaths occurred in patients with prior vaccination.7
Patients who survive the first two weeks of tetanus have a better chance of recovery. Those with multiple chronic comorbidities, such as chronic obstructive pulmonary disease (COPD), diabetes, or cardiovascular disease, are more likely to die because of the physiologic stress of the illness and its treatment.1,7,12 The provision of ventilator support is more complicated in those with COPD; similarly, the autonomic effects of tetanus can be more problematic for patients with chronic cardiac disease or neurologic complications of chronic diabetes.13
PATIENT EDUCATION
Widespread vaccination against tetanus, which began in the US in the mid-20th century, has greatly reduced disease incidence.7 However, vaccination coverage rates remain below target.
In 2012, only 82.5% of children ages 19 to 35 months received the recommended four doses of diphtheria-tetanus-pertussis (DTaP) vaccine, and 94.3% received at least three doses.23 Only 84.6% of teens ages 13 to 17 years received the primary four doses as well as the recommended booster dose.24 The same year, only 55% of patients ages 65 and older and 64% of adults ages 19 to 64 had received a tetanus booster within the previous 10 years.25
Vaccination rates are lower for black, Hispanic, and Asian adults in the US.25 Clinicians should proactively recommend tetanus booster immunization to all adults.
CONCLUSION
Although few clinicians in developed countries will see a case of tetanus, all should be alert for it. Elderly patients and those not fully vaccinated are at risk. Routine immunization decreases but does not eliminate the risk. Tetanus differs from other illnesses controlled by national immunization efforts in that unvaccinated persons do not benefit from herd immunity, because the disease is not contagious. The diagnosis is clinical and should always be considered in patients with trismus, dysphagia, and/or adrenergic excess. Wounds that place a patient at risk for tetanus involve devitalized tissues and anaerobic conditions. Prompt diagnosis is essential, because it allows for early neutralization of unbound tetanospasmin. Wound care including debridement, antibiotic therapy, control of muscle spasms and the effects of autonomic instability, and airway care are fundamental to the treatment of tetanus.
CE/CME No: CR-1704
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Recognize patients who are at risk for tetanus.
• Describe the clinical presentation of tetanus.
• Discuss proper treatment for a patient with tetanus.
• Promote widespread vaccination against tetanus.
FACULTY
Timothy W. Ferrarotti is the Director of Didactic Education and Assistant Professor in the PA Studies Program at the University of Saint Joseph, West Hartford, Connecticut.
The author has no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of April 2017.
Article begins on next page >>
Tetanus is a devastating disease that can be prevented by proper immunization and wound care. Although the incidence is low in the United States due to widespread routine vaccination, immunization coverage remains below target, especially in older adults. Since outcome is influenced by the clinician's ability to make a timely diagnosis and initiate appropriate care, continued appreciation of tetanus is warranted.
Tetanus is a neurologic disorder resulting from infection by the gram-positive, spore-forming anaerobic bacillus Clostridium tetani. The bacterium, in spore form, typically enters the body through a contaminated soft-tissue wound. Ubiquitous in the environment, C tetani spores are found throughout the world—in soil as well as in animal feces and saliva—and are resistant to temperature extremes and antiseptics. Tetanus is infectious but not contagious (not transmitted person-to-person).1 Wounds with devitalized tissue or those supporting anaerobic conditions, such as bites, puncture wounds, burns, and gangrene, are conducive to the development of tetanus. Infection can also occur following dental extractions, abortions, and illicit drug injection.1 Although vaccination programs have decreased the incidence of tetanus in the United States, C tetani infection remains an ongoing clinical concern because the spores are omnipresent, universal vaccination coverage has not been achieved, and vaccine-immunity wanes over time, placing individuals at risk.
PATHOPHYSIOLOGY
C tetani produces two toxins: tetanolysin and tetanospasmin (tetanus toxin). Tetanolysin may have a role in promoting the diffusion of tetanospasmin in soft tissues.2 Tetanospasmin is a highly potent toxin, with a lethal dose in humans of less than 2.5 ng/kg of body weight.3 The toxin enters the peripheral nerve at the site of injury and migrates to the central nervous system (CNS). There, it causes unopposed α-motor neuron firing by preventing the release of inhibitory neurotransmitters such as γ-aminobutyric acid (GABA), resulting in muscle spasms and excess reflexive response to sensory stimuli.4 It also leads to excessive catecholamine release from the adrenal medulla.1
Tetanospasmin binds to neurons in the spinal cord and brainstem. Because toxin binding is irreversible, resolution of tetanus requires the neurons to grow new axon terminals. The effects of tetanus can persist for six to eight weeks until new terminals develop.3,5 Patients often require several weeks of ventilator support during this time.3
EPIDEMIOLOGY
Tetanus continues to be a serious cause of morbidity and mortality worldwide. The majority of cases (80%) occur in Africa and Southeast Asia.6 Incidence is much lower in the United States (0.1 cases per million persons annually) because of widespread vaccination, with only 233 cases of tetanus reported between 2001 and 2008.7 However, in the absence of confirmatory tests, the diagnosis is a clinical one; furthermore, there is no laboratory reporting program for tetanus. As a result, more cases may occur in the US than are detected or reported.
In developed countries, tetanus is primarily a disease of the elderly or the unvaccinated. Older persons, especially non-veterans, are less likely to have received the primary series. Because immunity decreases with age, even those who completed the primary series but have not received booster doses are at increased risk.8 Home-schooled children, who are not subject to school-entry vaccination requirements, are also at risk if unvaccinated.9
Neonatal tetanus is an ongoing problem in undeveloped countries that lack maternal vaccination programs. (Maternal immunization successfully reduces neonatal tetanus via passive immunity, and maternal tetanus via active immunity.) Unvaccinated women who undergo nonmedical abortions or unhygienic childbirth are at increased risk for tetanus.3,10
Other risk factors for tetanus include wound contamination with soil, saliva, or devitalized tissue; injection drug use; and exposure to anthropogenic or natural disasters.1 C tetani spores can contaminate heroin and may grow in abscesses of heroin users.4 Small outbreaks of tetanus among injection drug users have been reported, even among younger adults who had some immunity from childhood vaccination.7,11 In addition, patients with diabetes are at increased risk for tetanus. These patients may have chronic wounds due to slowed healing and poor vascularity, which can lead to lower oxygen tension in their wounds and create an environment conducive to anaerobic infection. These chronic wounds are often ignored as a potential nidus for tetanus; instead, focus is placed on plantar puncture wounds or lacerations.7
Though tetanus risk is greatest for those who were never fully immunized, cases have been reported in persons who were immunized in the remote past but had not received a recent booster. Such cases show that tetanus immunity is not absolute and does wane over time.12-14 Among the 233 tetanus cases reported in the US during 2001-2008, vaccination status was reported for 92. Of these, 24 patients had a complete series and 31 patients had at least one prior dose of tetanus vaccine.7 Furthermore, six cases occurred in patients known to have had the four-dose series and a booster within 10 years of diagnosis. Similarly, a 14-year-old boy who was fully vaccinated developed cephalic tetanus from a stingray wound.14 Given these data, clinicians should not assume that a patient who reports having had “a tetanus shot” is completely protected; a full series and regular boosters are required, and, in rare cases, tetanus can occur despite full vaccination.
PATIENT PRESENTATION AND TETANUS TYPES
The CDC describes tetanus as “the acute onset of hypertonia and/or painful muscle contractions (usually of the muscles of the jaw and neck) and generalized muscle spasms without other apparent medical cause.”15 Clinicians should always consider tetanus in patients with dysphagia and trismus, especially if the patient has a wound, had not received primary vaccination, or has not had a booster in several decades. Tetanus cannot be ruled out based on the lack of a wound, however, since up to 25% of patients who develop tetanus have no obvious site of inoculation.16 The incubation period ranges from 3 to 21 days, with more severe cases having shorter incubation periods (< 8 days).10 The closer the site of inoculation is to the CNS, the more serious the disease usually is—and the shorter the incubation period will be.1
Presentation depends on the time elapsed since inoculation, the severity of illness (determined by the Ablett classification; see Table 1), and the form of tetanus involved. The patient may present early when the infection and toxin are localized to the wound and have not progressed to the CNS (localized tetanus). There may be a wound with signs of infection, including erythema, induration, edema, warmth, tenderness, and drainage. If the injury is on the head or neck, cephalic tetanus may occur, causing the patient to present with painful spasms of the extra-ocular, facial, and/or neck muscles; trismus; dysphagia; or even a Horner-like syndrome. The patient with more advanced, generalized tetanus may have decorticate posturing, abdominal wall rigidity, or opisthotonus.1,2,5,17
Four types of tetanus have been described: generalized, localized, cephalic, and neonatal.
Generalized tetanus is the most common form, accounting for approximately 80% of cases.10 It may involve contractions of the masseter muscles, producing trismus; facial muscles, producing risus sardonicus (sardonic smile); neck and shoulder muscles; abdominal wall muscles, mimicking guarding; and back muscles, producing opisthotonus (arching of the back, neck, and head; see Figure 1) and decorticate posturing (flexion and adduction of the arms, clenched fists, and extension of the lower extremities).1,5,6 Patients with generalized tetanus often exhibit hyperresponsiveness to the environment. As a result, noises and sudden light changes may result in acute spasms. In addition, patients may experience painful spasms when affected muscles are palpated. Affected reflex arcs are usually hyperresponsive to stimuli.1 Intermittent spasms of the thoracic, pharyngeal, and/or laryngeal muscles may cause periods of apnea. Autonomic effects of tetanus mimic those associated with the catecholamine excess of pheochromocytoma. Patients exhibit restlessness, irritability, diaphoresis, fever, excessive salivation, gastric stasis, hypertension, tachycardia, and arrhythmia. There may be interposed hypotension and bradycardia.1,5,17
Localized tetanus involves painful spastic contraction of muscles at or near the site of inoculation. It often evolves into generalized tetanus as the toxin spreads further into the CNS.
Cephalic tetanus involves facial and laryngeal muscles. It is rare, accounting for 1% to 3% of tetanus cases.6 Patients may initially have flaccid paralysis, mimicking stroke, rather than spasm, because the toxin has not completely migrated up the peripheral nerve into the CNS. As the toxin enters the CNS and induces the typical spasm (trismus), the diagnosis will be more obvious. The presence of trismus or a subacute wound on the head may be used to discriminate tetanus from stroke. Cephalic tetanus often evolves into generalized tetanus, affecting more of the body in a caudal direction.5
Neonatal tetanus develops within one week after birth. The neonate with tetanus is usually born to a mother lacking immunization. Typically, the infant sucks and feeds for the first couple of days, then develops inability/refusal to suck/feed, has difficulty opening his/her mouth, becomes weak, and develops muscle spasms.3 The affected child may develop risus sardonicus, clenched hands, dorsiflexion of the feet, and opisthotonus.3
DIFFERENTIAL DIAGNOSIS
The clinician should consider other CNS conditions in the differential diagnosis (see Table 2). Although similar to generalized seizures, tetanus causes painful spasms and does not produce a loss of consciousness.1,17 Tetanus, intracranial bleed, and meningitis all can cause meningismus; meningitis, however, is more likely to manifest with other symptoms of infection, such as headache and fever. Although the autonomic dysfunction of tetanus can cause pyrexia, fever would usually coincide with other sympathetic symptoms, such as hypertension, tachycardia, and diaphoresis. Intracranial bleeding tends to have a more rapid onset than tetanus and produces headache and mental status changes. Seventh nerve palsy produces muscle flaccidity, not spasm, and is usually painless unless associated with herpetic inflammation.1,5,6,14,17
Poisoning and medication effects should also be considered. Strychnine poisoning manifests similar to tetanus but occurs without a wound.5 Blood and urine assays for strychnine can be diagnostic. Dystonic reactions resulting from neuroleptic medications—such as phenothiazines—include torticollis, oropharyngeal muscle spasms, and deviation of the eyes. Unlike tetanus, drug-induced dystonia does not cause reflex spasms and often resolves with benztropine or diphenhydramine administration.1 Neuroleptic malignant syndrome can also cause muscular rigidity and autonomic instability, but unlike tetanus, it often causes altered mental status; it should be considered in patients who recently received a causative medication.5,17
Tetanus often manifests with reflexive muscle spasms similar to those seen in electrolyte and acid-base abnormalities. Hypocalcemia may produce a reflexive spasm of the facial muscles when the facial nerve is percussed (Chvostek sign), while alkalemia may produce reflexive spasm of the hand and wrist muscles (Trousseau sign).1 Lab tests can rule out these diagnoses.1,5
A patient with an odontogenic abscess may have pain and muscle spasm/trismus, but the infection is usually easily detected on exam. The clinician should be cautious in attributing the trismus solely to the swelling, however, as C tetani has been found in odontogenic abscesses and the patient may have both.1,17 Peritonsillar abscess will often produce trismus. When abscess is the cause, careful examination of the oropharynx will usually demonstrate tonsillar exudate, hypertrophy, soft tissue erythema, and tenderness, as well as a misplaced uvula.1
DIAGNOSIS
Tetanus is a clinical diagnosis, usually made based on the findings described. Confirmatory lab tests are not readily available. The organism is infrequently recovered in cultures of specimens from suspected wounds (30% of cases).10,11 Serologic testing on specimens drawn before administration of tetanus immunoglobulin (TIG) may indicate very low or undetectable antitetanus antibody levels, but tetanus can still occur when “protective” levels of antibodies are present.11 Detection of tetanus toxin in plasma or a wound with bioassays and polymerase chain reaction might be possible, but these tests are only available in a few settings.3
THE MULTIFACETED CARE PLAN
The primary care provider should refer a patient with suspected tetanus to an emergency department, preferably a tertiary care center with the necessary specialists. Patients are likely to require prolonged hospitalization. In a recent series of tetanus cases in California, the median length of hospitalization was 18 days.12 Treatment is multifaceted; interventions include immunization, wound care, administration of antibiotics and other pharmacologic agents, and supportive therapy (see Table 3).
Immunization
All patients with suspected tetanus should immediately receive both passive (with TIG) and active (tetanus toxoid–containing vaccines) immunization. Because of the extremely high potency of tetanus toxin, the very small amount of toxin that is required to cause tetanus is insufficient to prompt an immune response that would confer immunity. Therefore, treatment is the same regardless of whether the patient had prior disease.10
TIG binds to and neutralizes unbound tetanospasmin, preventing progression of the disease. As noted, TIG will not reverse the binding of the toxin to nerve structures.5 Due to a lack of prospective studies, there is disagreement regarding TIG dosage: Doses as high as 3,000-6,000 U have been recommended, but case studies indicate that the dosage recommended by the CDC (500 U) is likely effective.13 The full CDC recommendation is 500 U of human-derived TIG intramuscularly administered at locations near and away from the wound (but always away from the tetanus toxoid injection site).10,17 Outside the US, equine-based TIG may be the only option. Animal-derived TIG is less desirable because of increased allergy risk; when used, a small amount (0.1 mL) should be first administered as an intradermal test.17
Tetanus toxoid immunization produces active immunity. It is currently available in combination antigen forms (tetanus and diphtheria vaccine [Td], tetanus-diphtheria-acellular pertussis [Tdap] vaccine). The dose of either is 0.5 mL. Patients with tetanus should receive three doses given intramuscularly: immediately, at 4 weeks, and at 6 to 12 months.10
Wound care
Wound care should include incision and drainage, removal of foreign bodies, debridement, and irrigation. These steps are taken in order to ensure an aerobic environment in the wound, ultimately decreasing C tetani survival.1
Antibiotics
The preferred antimicrobial agent for treating tetanus infection is metronidazole 500 mg intravenously (IV) every 6 hours.1,3,17 Penicillin (2 to 4 million U IV every 4 to 6 hours) is effective against C tetani, but it is a GABA-receptor antagonist and may worsen tetanus by further inhibiting the release of GABA.1,14,18 GABA-receptor antagonism may also occur with cephalosporins; however, these broader-spectrum agents may be necessary to treat mixed infections.17 Alternatives include doxycycline, macrolides, and clindamycin.1
Other pharmacologic treatment
Benzodiazepines (eg, diazepam 10 to 30 mg IV) can help control rigidity and muscle spasms and are a mainstay of tetanus treatment.18 Benzodiazepines and propofol both act on GABA receptors, producing sedation in addition to controlling muscle spasms.19 Traditionally, more severe spasms, such as opisthotonus, have required induction of complete paralysis with nondepolarizing paralytics, such as pancuronium or vecuronium. However, paralysis is not optimal therapy since it necessitates sedation, intubation, and mechanical ventilation. Because tetanus does not resolve for 6 to 8 weeks, patients who require mechanical ventilation will also require tracheostomy to prevent laryngotracheal stenosis. Paralysis and mechanical ventilation can also lead to deep venous thrombosis, decubitus ulcers, and pneumonia.5 The ideal treatment would reduce the spasms and autonomic instability without the risks associated with deep sedation and paralysis.5
Other agents used in the treatment of tetanus include magnesium sulfate, which can decrease muscle spasm and ameliorate the effects of autonomic dysfunction, and intrathecal baclofen, which can decrease muscle spasm.19,20 Patients with persistent autonomic dysfunction may require combined α- and ß-adrenergic receptor blockade.1,17-20
Supportive care
It is important to implement supportive care, including limiting auditory and tactile stimulation, as well as providing adequate hydration and nutritional support. IV fluids, parenteral feeding, and enteral feeding are required. Measures should be taken to prevent complications of prolonged immobility, paralysis, and mechanical ventilation, including deep venous thrombosis, pulmonary embolism, and pressure ulcers. The quality of supportive care and the swiftness with which the diagnosis is made and appropriate treatment is initiated are key factors that determine an individual patient’s outcome.21
COMPLICATIONS AND MORTALITY
Tetanus can lead to many complications, including long bone and spine fractures from severe muscle spasms, as well as renal failure and aspiration. Most spinal fractures involve the thoracic spine, but lumbar spine fractures have been reported.22 Burst-type fractures of the vertebrae may cause cauda equina syndrome or directly injure the spinal cord if fragments are retropulsed.22 Persistent muscle spasm can also cause rhabdomyolysis and renal failure. Lab test results, including elevated levels of creatine phosphokinase and myoglobin (rhabdomyolysis) as well as blood urea nitrogen and creatinine (renal failure), can indicate presence of complications. Muscle relaxation and hydration are key to prevention.
Patients with trismus are often unable to swallow and maintain oral hygiene, leading to caries and dental abscess. The trismus itself can also cause dental or jaw fractures.2,13 Aspiration can occur when laryngeal muscles are affected, resulting in pneumonia in 50% to 70% of autopsied cases of tetanus.10 Additionally, the paralyzed patient receiving ventilatory support can develop pneumonia, deep vein thrombosis, and pulmonary embolism.5,13 Neonatal tetanus often results in complications such as cerebral palsy or cognitive delay.1
A number of factors influence the severity and outcome of tetanus. Untreated tetanus is typically fatal, with respiratory failure the most common cause of death in settings where mechanical ventilation is unavailable.1 Where mechanical ventilation is accessible, autonomic dysfunction accounts for most deaths.20 Ventilation aside, the case-fatality rate varies according to the medical system. The rate is often less than 20% where modern ICUs are available but can exceed 50% in undeveloped countries with limited facilities.1,5 A review of outcomes data for 197 of the 233 tetanus cases reported in the US during 2001-2008 (modern medical care was provided in all) showed an overall case-fatality rate of 13.2%.7
Age and vaccination status also affect outcomes, with higher case-fatality rates seen in older (18% in those ≥ 60, 31% in those ≥ 65) and unvaccinated (22%) patients. 7,10 In the study of tetanus cases from 2001-2008, the fatality rate was five times higher in patients ages 65 or older compared with patients younger than 65.7 This study also showed that severity of tetanus may be inversely related to the number of vaccine doses the individual has received, and that having previous vaccination was associated with improved survival, as only four of the 26 deaths occurred in patients with prior vaccination.7
Patients who survive the first two weeks of tetanus have a better chance of recovery. Those with multiple chronic comorbidities, such as chronic obstructive pulmonary disease (COPD), diabetes, or cardiovascular disease, are more likely to die because of the physiologic stress of the illness and its treatment.1,7,12 The provision of ventilator support is more complicated in those with COPD; similarly, the autonomic effects of tetanus can be more problematic for patients with chronic cardiac disease or neurologic complications of chronic diabetes.13
PATIENT EDUCATION
Widespread vaccination against tetanus, which began in the US in the mid-20th century, has greatly reduced disease incidence.7 However, vaccination coverage rates remain below target.
In 2012, only 82.5% of children ages 19 to 35 months received the recommended four doses of diphtheria-tetanus-pertussis (DTaP) vaccine, and 94.3% received at least three doses.23 Only 84.6% of teens ages 13 to 17 years received the primary four doses as well as the recommended booster dose.24 The same year, only 55% of patients ages 65 and older and 64% of adults ages 19 to 64 had received a tetanus booster within the previous 10 years.25
Vaccination rates are lower for black, Hispanic, and Asian adults in the US.25 Clinicians should proactively recommend tetanus booster immunization to all adults.
CONCLUSION
Although few clinicians in developed countries will see a case of tetanus, all should be alert for it. Elderly patients and those not fully vaccinated are at risk. Routine immunization decreases but does not eliminate the risk. Tetanus differs from other illnesses controlled by national immunization efforts in that unvaccinated persons do not benefit from herd immunity, because the disease is not contagious. The diagnosis is clinical and should always be considered in patients with trismus, dysphagia, and/or adrenergic excess. Wounds that place a patient at risk for tetanus involve devitalized tissues and anaerobic conditions. Prompt diagnosis is essential, because it allows for early neutralization of unbound tetanospasmin. Wound care including debridement, antibiotic therapy, control of muscle spasms and the effects of autonomic instability, and airway care are fundamental to the treatment of tetanus.
1. Afshar M, Raju M, Ansell D, Bleck TP. Narrative review: tetanus—a health threat after natural disasters in developing countries. Ann Intern Med. 2011;154(5):329-335.
2. Demir NA, Sumer S, Ural O, et al. An alternative treatment approach in tetanus: botulinum toxin. Trop Doct. 2015;45(1): 46-48.
3. Thwaites CL, Beeching NJ, Newton CR. Maternal and neonatal tetanus. Lancet. 2015;385(9965):362-370.
4. Aronoff DM. Clostridium novyi, sordellii, and tetani: mechanisms of disease. Anaerobe. 2013;24:98-101.
5. Cook TM, Protheroe RT, Handel JM. Tetanus: a review of the literature. Br J Anaesth. 2001;87(3):477-487.
6. Doshi A, Warrell C, Dahdaleh D, Kullmann D. Just a graze? Cephalic tetanus presenting as a stroke mimic. Pract Neurol. 2014;14(1):39-41.
7. CDC. Tetanus surveillance—United States, 2001-2008. MMWR Morb Mortal Wkly Rep. 2011;60(12):365-369.
8. McCabe J, La Varis T, Mason D. Cephalic tetanus complicating geriatric fall. N Z Med J. 2014;127(1400):98-100.
9. Johnson MG, Bradley KK, Mendus S, et al. Vaccine-preventable disease among homeschooled children: two cases of tetanus in Oklahoma. Pediatrics. 2013;132(6):e1686-e1689.
10. CDC. Epidemiology and Prevention of Vaccine-Preventable Diseases. Hamborsky J, Kroger A, Wolfe S, eds. 13th ed. Washington, DC: Public Health Foundation; 2015.
11. Tiwari TS. Chapter 16: Tetanus. In: CDC. Manual for Surveillance of Vaccine-Preventable Diseases. 5th ed. Atlanta, GA: CDC; 2012.
12. Yen C, Murray E, Zipprich J, et al. Missed opportunities for tetanus postexposure prophylaxis—California, January 2008-March 2014. MMWR Morb Mortal Wkly Rep. 2015; 64(9):243-246.
13. Aksoy M, Celik EC, Ahiskalioglu A, Karakaya MA. Tetanus is still a deadly disease: a report of six tetanus cases and reminder of our knowledge. Trop Doct. 2014;44(1):38-42.
14. Felter RA, Zinns LE. Cephalic tetanus in an immunized teenager. Pediatr Emerg Care. 2015;31(7):511-513.
15. CDC. Tetanus (Clostridium tetani) 1996 case definition. www.cdc.gov/nndss/conditions/tetanus/case-definition/1996/. Accessed February 17, 2017.
16. Thwaites CL, Farrar JJ. Preventing and treating tetanus [commentary]. BMJ. 2003;326(7381):117-118.
17. Sexton DJ. Tetanus. UpToDate. www.uptodate.com/contents/tetanus?topicKey=ID%2F5525. Accessed February 17, 2017.
18. Rodrigo C, Fernando D, Rajapakse S. Pharmacological management of tetanus: an evidence-based review. Crit Care. 2014;18(2):217.
19. Santos ML, Mota-Miranda A, Alves-Pereira A, et al. Intrathecal baclofen for the treatment of tetanus. Clin Infect Dis. 2004;38(3):321-328.
20. Thwaites CL, Yen LM, Loan HT, et al. Magnesium sulphate for treatment of severe tetanus: a randomized controlled trial. Lancet. 2006;368:1436-1443.
21. Govindaraj GM, Riyaz A. Current practice in the management of tetanus. Crit Care. 2014;18(3):145.
22. Wilson TJ, Orringer DA, Sullivan SE, Patil PG. An L-2 burst fracture and cauda equina syndrome due to tetanus. J Neurosurg Spine. 2012;16(1):82-85.
23. CDC. National, state and local area vaccination coverage among children aged 19-35 months—United States, 2012. MMWR Morb Mortal Wkly Rep. 2013;62(36):733-740.
24. CDC. National and state vaccination coverage among adolescents aged 13-17 years—United States, 2012. MMWR Morb Mortal Wkly Rep. 2013;62(34):685-693.
25. Williams WW, Lu PJ, O’Halloran A, et al. Noninfluenza vaccination coverage among adults—United States, 2012. MMWR Morb Mortal Wkly Rep. 2014;63(5):95-102.
1. Afshar M, Raju M, Ansell D, Bleck TP. Narrative review: tetanus—a health threat after natural disasters in developing countries. Ann Intern Med. 2011;154(5):329-335.
2. Demir NA, Sumer S, Ural O, et al. An alternative treatment approach in tetanus: botulinum toxin. Trop Doct. 2015;45(1): 46-48.
3. Thwaites CL, Beeching NJ, Newton CR. Maternal and neonatal tetanus. Lancet. 2015;385(9965):362-370.
4. Aronoff DM. Clostridium novyi, sordellii, and tetani: mechanisms of disease. Anaerobe. 2013;24:98-101.
5. Cook TM, Protheroe RT, Handel JM. Tetanus: a review of the literature. Br J Anaesth. 2001;87(3):477-487.
6. Doshi A, Warrell C, Dahdaleh D, Kullmann D. Just a graze? Cephalic tetanus presenting as a stroke mimic. Pract Neurol. 2014;14(1):39-41.
7. CDC. Tetanus surveillance—United States, 2001-2008. MMWR Morb Mortal Wkly Rep. 2011;60(12):365-369.
8. McCabe J, La Varis T, Mason D. Cephalic tetanus complicating geriatric fall. N Z Med J. 2014;127(1400):98-100.
9. Johnson MG, Bradley KK, Mendus S, et al. Vaccine-preventable disease among homeschooled children: two cases of tetanus in Oklahoma. Pediatrics. 2013;132(6):e1686-e1689.
10. CDC. Epidemiology and Prevention of Vaccine-Preventable Diseases. Hamborsky J, Kroger A, Wolfe S, eds. 13th ed. Washington, DC: Public Health Foundation; 2015.
11. Tiwari TS. Chapter 16: Tetanus. In: CDC. Manual for Surveillance of Vaccine-Preventable Diseases. 5th ed. Atlanta, GA: CDC; 2012.
12. Yen C, Murray E, Zipprich J, et al. Missed opportunities for tetanus postexposure prophylaxis—California, January 2008-March 2014. MMWR Morb Mortal Wkly Rep. 2015; 64(9):243-246.
13. Aksoy M, Celik EC, Ahiskalioglu A, Karakaya MA. Tetanus is still a deadly disease: a report of six tetanus cases and reminder of our knowledge. Trop Doct. 2014;44(1):38-42.
14. Felter RA, Zinns LE. Cephalic tetanus in an immunized teenager. Pediatr Emerg Care. 2015;31(7):511-513.
15. CDC. Tetanus (Clostridium tetani) 1996 case definition. www.cdc.gov/nndss/conditions/tetanus/case-definition/1996/. Accessed February 17, 2017.
16. Thwaites CL, Farrar JJ. Preventing and treating tetanus [commentary]. BMJ. 2003;326(7381):117-118.
17. Sexton DJ. Tetanus. UpToDate. www.uptodate.com/contents/tetanus?topicKey=ID%2F5525. Accessed February 17, 2017.
18. Rodrigo C, Fernando D, Rajapakse S. Pharmacological management of tetanus: an evidence-based review. Crit Care. 2014;18(2):217.
19. Santos ML, Mota-Miranda A, Alves-Pereira A, et al. Intrathecal baclofen for the treatment of tetanus. Clin Infect Dis. 2004;38(3):321-328.
20. Thwaites CL, Yen LM, Loan HT, et al. Magnesium sulphate for treatment of severe tetanus: a randomized controlled trial. Lancet. 2006;368:1436-1443.
21. Govindaraj GM, Riyaz A. Current practice in the management of tetanus. Crit Care. 2014;18(3):145.
22. Wilson TJ, Orringer DA, Sullivan SE, Patil PG. An L-2 burst fracture and cauda equina syndrome due to tetanus. J Neurosurg Spine. 2012;16(1):82-85.
23. CDC. National, state and local area vaccination coverage among children aged 19-35 months—United States, 2012. MMWR Morb Mortal Wkly Rep. 2013;62(36):733-740.
24. CDC. National and state vaccination coverage among adolescents aged 13-17 years—United States, 2012. MMWR Morb Mortal Wkly Rep. 2013;62(34):685-693.
25. Williams WW, Lu PJ, O’Halloran A, et al. Noninfluenza vaccination coverage among adults—United States, 2012. MMWR Morb Mortal Wkly Rep. 2014;63(5):95-102.
Hyperkalemia in Adults: Review of a Common Electrolyte Imbalance
CE/CME No: CR-1703
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Describe the pathophysiology and causes of hyperkalemia.
• Identify patients who are susceptible to hyperkalemia.
• Recognize the clinical sequelae of hyperkalemia.
• Formulate assessment and treatment plans for patients with hyperkalemia.
FACULTY
Melanie Douglas is a Physician Assistant in the Medicine Department at NYU Langone Medical Center in New York, New York. Denise Rizzolo is a Clinical Assistant Professor in the PA Program at Pace University in New York, New York, and Research Director in the Program of PA Studies at Kean University in Union, New Jersey. Danielle Kruger is an Academic Coordinator and Associate Professor in the PA Program at St. John’s University in Queens, New York. The authors have no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of March 2017.
Article begins on next page >>
Hyperkalemia is a common electrolyte disorder associated with life-threatening cardiac arrhythmias. Prompt recognition and appropriate treatment are essential in preventing serious cardiac complications. Although clinical manifestations of hyperkalemia are usually nonspecific or absent, laboratory testing and electrocardiography performed by the astute clinician aware of predisposing risk factors can help direct management.
Potassium is contained mostly in intracellular fluid; only about 2% is found in the extracellular space.1 The average total body potassium is about 50 mEq per kg of body weight (eg, a 70-kg individual has a total body potassium of approximately 3,500 mEq).2 Levels are tightly regulated by alterations in excretion in the distal renal tubule in response to potassium load and balance, and potassium distribution is influenced by insulin, aldosterone, catecholamines, and acid-base status.2 Movement of potassium across cell membranes is driven by the sodium-potassium adenosine triphosphatase (Na-K-ATPase) pump.3 In this article, we use the common serum potassium reference range of 3.5 to 5.0 mEq/L and define hyperkalemia as a serum potassium concentration greater than 5.5 mEq/L.4
Hyperkalemia can lead to life-threatening complications of cardiac arrhythmias, asystole, hypotension, flaccid paralysis, tetany, dyspnea, and altered mental status.5 Among patients with end-stage renal disease (ESRD), hyperkalemia is thought to contribute to 2% to 5% of deaths.6 A retrospective study found that patients with serum potassium levels exceeding 6.0 mEq/L on ICU admission had a significantly higher death rate within 30 days than patients who were normokalemic on presentation.7
RISK FACTORS
It is estimated that more than 35% of patients age 70 and older have chronic kidney disease (CKD) stage 3 or higher.8 Hyperkalemia is closely associated with CKD, increasing linearly in relation to the degree of renal impairment.8 As such, the prevalence of hyperkalemia in older adults is high, and it will increase overall as the US population ages. In a retrospective analysis of veterans older than 65 with CKD stage 3 or higher, the prevalence of hyperkalemia was 2.5%.9 Use of certain medications is also associated with hyperkalemia. Another retrospective study analyzed records obtained from 70,873 patients with CKD (estimated glomerular filtration rate [eGFR] < 60 mL/min/1.73 m2) hospitalized in the Veterans Health Administration system. It found that patients treated with renin-angiotensin-aldosterone system (RAAS) blockers, such as ACE inhibitors (ACEis) or angiotensin-receptor blockers (ARBs), had a higher incidence of hyperkalemia (potassium level ≥ 5.5 mEq/L) than patients not treated with these medications (8.22 vs 1.77 events per 100 patient-months).9,10
POTASSIUM HOMEOSTASIS
Tight control over extracellular potassium is maintained in part by the Na-K-ATPase pump, which uses adenosine triphosphatase to move potassium and sodium ions in opposite directions across cell membranes.3 Specifically, three sodium ions are pumped out of the cell for every two potassium ions pumped in, resulting in a potassium gradient that is partially responsible for maintaining a resting membrane potential. This resting membrane potential, which determines myocardial, skeletal muscle, and nerve cell excitability and signaling, is highly sensitive to changes in the extracellular potassium level.4 Even small extracellular imbalances can induce cell depolarization and evoke an action potential. Increased extracellular potassium concentration decreases the resting membrane potential of the myocardium, shortens repolarization time, and decreases the rate of myocardial cell conduction, and also slows down neuromuscular conduction.11,12
Renal tubular function plays a significant role in potassium homeostasis, with approximately 90% of dietary potassium intake ex
The RAAS is a signal transduction pathway that regulates potassium excretion by the kidneys. Renin is secreted by the kidney in response to low renal perfusion, catecholamines, ß-adrenergic stimulation, potassium and sodium levels, and other factors. Secretion of renin triggers a signaling cascade that eventually results in the release of aldosterone from the adrenal cortex.5 Aldosterone binds to a receptor in the kidney’s collecting ducts where it increases potassium excretion by stimulating sodium reabsorption and fluid retention (see Figure 1).5

CAUSES OF HYPERKALEMIA
The pathophysiology of hyperkalemia generally involves either decreased renal excretion or shifts in extracellular potassium. Causes of hyperkalemia are listed in the Table. Potassium excretion can be disrupted in acute kidney injury (AKI), sepsis, cardiac ischemia, heart failure, diabetic ketoacidosis (DKA), insulin deficiency, tumor lysis syndrome (TLS), sickle cell disease, systemic lupus erythematosus, renal transplant, hepatorenal syndrome, adrenal insufficiency, and obstructive uropathy.15 In addition, certain medications can impair potassium excretion (eg, RAAS blockers, potassium-sparing diuretics in patients with CKD, digoxin toxicity).16 The following sections highlight the pathophysiology and manifestations of more common causes of hyperkalemia.

Renal impairment
Hyperkalemia may be a manifestation of worsening renal function. Potassium excretion is reduced in CKD, and CKD is the most common cause of hyperkalemia due to lower GFR.8,17 Patients with lower GFR tend to be older and male, and frequently have comorbid conditions such as type 2 diabetes, chronic liver disease, and heart failure.17
In CKD, decreased delivery of sodium to the distal tubules and reduced filtration capacity of the kidney diminishes the collecting duct’s ability to excrete potassium in exchange for sodium.2 Metabolic acidosis, which often contributes to AKI or CKD, causes potassium to shift from the intracellular to the extracellular compartment.4 Renal impairment may present clinically with dehydration, oliguria, nausea, vomiting, constipation, altered mental status, or weakness.
Hyperglycemia
Insulin and catecholamines (eg, epinephrine and norepinephrine) drive potassium into cells. Insulin increases potassium uptake into liver and muscle cells.13 A decrease in insulin levels, as may occur in type 2 diabetes or DKA, can cause a buildup of extracellular potassium.4 Also, serum hypertonicity from hyperglycemia results in water movement from the intracellular to the extracellular compartment; this raises the intracellular concentration of potassium, further promoting its movement to the extracellular space.4,14 Patients with hyperglycemia may present with dizziness, polyuria, polydipsia, nausea, vomiting, altered mental status, or fatigue.
Rhabdomyolysis
Rhabdomyolysis is a rapid breakdown of skeletal muscle that results in leakage of cellular contents into the extracellular space.4,18 Causes of rhabdomyolysis include use of medications such as statins, illicit drugs (eg, cocaine), or alcohol; rigorous exercise; and trauma.19
Muscle cell contents that are released into the circulation include potassium and other electrolytes, enzymes (eg, lactate dehydrogenase, aspartate transaminase, aldolase), and myoglobin.19 In rhabdomyolysis, myoglobin accumulation and hypovolemia lead to AKI and hyperkalemia.19 Patients may present with myalgias, extremity paresthesias, generalized weakness, nausea, altered mental status, fever, or darkened urine.18,19
Adrenal insufficiency
During critical illness such as sepsis, adrenal insufficiency can result from destruction of the adrenal glands, leading to hypoaldosteronism.20 Reduced aldosterone in adrenal insufficiency enables sodium and water to be eliminated from the body more easily, but as a result, less potassium gets excreted through the renal system and more is driven into the plasma.15
Acute adrenal insufficiency may manifest with hypotension, nausea, vomiting, or altered mental status, and labwork may reveal hyperkalemia as well as hypoglycemia or hyponatremia. Additionally, long-term glucocorticoid therapy can suppress the hypothalamic-pituitary axis and cause adrenal atrophy; rapid discontinuation of steroids can lead to adrenal insufficiency and hyperkalemia.21
Medications
RAAS blockers reduce CKD progression in patients with an eGFR of 29 mL/min/1.73 m2 or greater.22 Nonetheless, prescribing two or more drugs from the ACEi or ARB classes is not recommended. The Veterans Administration Nephron-Diabetes Trial (VA-NEPHRON-D) was terminated early because patients with stage 3 CKD due to diabetes who received dual ACEi/ARB therapy had higher rates of hyperkalemia but no slowing of CKD.22
Within the RAAS cascade, ACEis block the formation of angiotensin II and ARBs prevent angiotensin II from binding to the adrenal receptor. This impairs renal excretion of potassium and potentially contributes to hyperkalemia.5 Nonetheless, when patients on ACEis or ARBs develop hyperkalemia, aldosterone concentrations usually decrease due to preexisting illnesses (eg, diabetes, heart failure, CKD, AKI) or drug effects (eg, potassium-sparing diuretics, ß-blockers, digoxin).5 Ultimately, a combination of factors resulting from ACEi or ARB therapy causes reductions in renal perfusion and predisposes patients to hyperkalemia.5
NSAIDs may lead to hyperkalemia, as they interfere with prostaglandin release, decrease renal perfusion, and reduce renin and aldosterone levels.22 ß-blockers and tacrolimus inhibit renin release, leading to decreased aldosterone levels.5 Potassium-sparing diuretics block the interaction of aldosterone with the aldosterone receptor in the nephron.5 Digoxin decreases the activity of Na-K-ATPase, diminishing potassium uptake by cells.9 Potassium supplements, often prescribed for patients on diuretics, may contribute to hyperkalemia in patients with CKD. In the hospital setting, potassium tablets or IV formulations are utilized to correct hypokalemia. Especially in patients with CKD, clinicians should prescribe these agents with caution to avoid inducing hyperkalemia. Salt substitutes, which commonly contain potassium chloride, may be appealing to patients concerned about their sodium intake. However, consumption of these substitutes may contribute to hyperkalemia, especially in patients with CKD, heart failure, or type 2 diabetes.23
Tumor lysis syndrome
TLS involves rapid release of electrolytes and other intracellular contents into the extracellular space during the lysis of tumor cells.24 Nucleic acids within DNA strands break down and build up extracellularly, leading to hyperuricemia and often AKI. Potassium and other electrolytes released into the plasma during cell lysis can usually be removed by a healthy renal system. In TLS, however, AKI due to uric acid nephropathy prevents kidneys from removing the excess electrolytes from the bloodstream.24 Patients with rapidly growing hematologic tumors undergoing chemotherapy are especially at risk.
Pseudohyperkalemia
Pseudohyperkalemia is a transiently elevated serum potassium level that erroneously represents the true serum potassium level. It results from hemolysis due to mechanical trauma during the blood draw (eg, a tourniquet tied too tightly or use of a small-bore needle) or during specimen handling afterwards.25 Furthermore, leukocytosis, thrombocytosis, and polycythemia make red blood cells more fragile, increasing the chance of hemolysis and potassium leakage.26 Blood transfusion also can lead to pseudohyperkalemia. When blood is stored, potassium leakage from the cells and cell lysis, along with diminished Na-K-ATPase activity, lead to a buildup of potassium in the medium surrounding the stored red blood cells.27,28 The rise in serum potassium levels post-transfusion is usually transient, as the blood cells redistribute the potassium load once they become metabolically active.27,29
CLINICAL MANIFESTATIONS
Clinical manifestations of mild to moderate hyperkalemia (serum potassium > 5.5 mEq/L but < 6.5 mEq/L) include fatigue, generalized weakness, nausea, vomiting, constipation, and diarrhea.15 In many patients, mild to moderate hyperkalemia may not be associated with any acute symptoms and vital signs may be normal.13 Severe hyperkalemia (serum potassium > 6.5 mEq/L) may present clinically with acute extremity paresthesias, muscle weakness and paralysis, heart palpitations, dyspnea, altered mental status, cardiac arrhythmias, and cardiac arrest.30,31 Irregular heart rhythm, decreased deep tendon reflexes, or decreased strength may be revealed on physical exam.3 Individuals with ESRD on hemodialysis seem to tolerate higher levels of potassium than the general population without displaying clinical symptoms. However, these individuals are still susceptible to the cardiac effects of hyperkalemia.32
INITIAL ASSESSMENT
In assessing hyperkalemia, the clinician must perform a focused history and physical exam and review the patient’s medication list, including supplements and dietary habits that impact potassium intake. Potassium-rich foods include meat, fish, milk, almonds, spinach, cantaloupe, bananas, oranges, mushrooms, and potatoes.33 Hyperkalemia may present in association with various medical emergencies. The clinician should have an index of suspicion, depending on the patient’s overall medical profile and presentation, for emergencies such as cardiac ischemia, sepsis, adrenal crisis, DKA, TLS, and digoxin overdose.
The clinician must identify whether an elevated potassium level requires emergent therapy; assessment of vital signs is paramount in determining this. Orthostatic hypotension and tachycardia may hint that the patient is volume depleted. The patient should be examined for signs of hemodynamic shock with the CAB sequence: circulation, airway, breathing.34 Symptoms such as chest pain, shortness of breath, muscle weakness, paralysis, and altered mental status suggest that an expedited evaluation is warranted.

With a serum potassium level > 5.5 mEq/L, urgent electrocardiography should be performed.26 ECG findings observed with serum potassium levels of 5.5-6.5 mEq/L usually include peaked T-waves and prolonged PR intervals (see Figure 2). With potassium levels > 6.5 mEq/L consistent with further cardiac destabilization, the P-wave flattens then disappears, the QRS complex broadens, and sinus bradycardia or ectopic beats may occur.12,26 ST depression, T-wave inversion, or ST elevation also may be seen.12 With serum potassium levels > 7.5 mEq/L, progressive widening of the QRS complex to a sine-wave with bundle branch blocks or fascicular blocks may occur (see Figure 3).26 Without prompt intervention, ventricular fibrillation may ensue.26

An extensive laboratory workup may be necessary to investigate the etiology; this includes a complete blood count, metabolic panel, liver function tests, cardiac enzymes, blood gas analysis, serum/urine osmolality, urinalysis, urine electrolytes, and toxicology screen.13,26 Arterial blood gas (ABG) analysis may show metabolic acidosis with AKI or DKA, or an elevated lactate may occur with sepsis. In patients with hyperglycemia, besides checking for acidosis, obtaining blood/urine ketone levels and a metabolic panel with anion gap to evaluate for DKA is useful.35
When assessing a patient with an elevated creatinine, the GFR at the time of evaluation should be compared with the patient’s baseline GFR to determine chronicity and duration of his/her kidney disease.36 Obtaining a urinalysis and urine electrolytes in addition to the basic metabolic panel can help narrow the etiology.36 A Foley catheter should be placed in cases of urinary retention because without intervention, urinary obstruction may lead to AKI and hyperkalemia. Myoglobinuria on urinalysis and an elevated creatine kinase are diagnostic markers of rhabdomyolysis.18
TLS should be considered in patients who recently received chemotherapy, especially those with proliferative hematologic malignancies, such as acute lymphoblastic leukemia, acute myeloid leukemia, and Burkitt lymphoma.24 In TLS, bloodwork often reveals hyperkalemia along with AKI, an elevated uric acid level, hyperphosphatemia, and hypocalcemia.24
Patients presenting with hyperkalemia, hypotension, hypoglycemia, and hyponatremia may have adrenal insufficiency.20 If insufficiency is suspected, a cortisol level may be checked during morning hours; a low level is often suggestive of this diagnosis.37 Treatment includes daily doses of steroids, and consultation with an endocrinologist is recommended.37
If an elevated potassium level is not accompanied by renal dysfunction, electrolyte imbalances, ECG changes, or inciting medications, pseudohyperkalemia should be considered.38 A repeat lab sample should be checked. Consider obtaining an ABG analysis, as the shorter time interval between drawing the blood sample and the sample analysis reportedly increases the reliability of the resulting potassium level.38
THERAPY
Emergent
Emergent treatment is needed for severe hyperkalemia (see Figure 4). Any hyperkalemia-inciting medications or potassium supplements should be immediately discontinued.39 IV access and cardiac telemetry monitoring should be promptly applied.26

In cases of severe hyperkalemia that involve cardiac arrhythmias, manifestations on ECG, or risk for arrhythmias, calcium gluconate (10 mL IV over 10 min) should be urgently administered, followed by IV insulin in conjunction with dextrose.26 Calcium chloride should be utilized for hyperkalemia in the context of the advanced cardiac life support (ACLS) protocol for cardiac arrest.26 The patient should remain on cardiac telemetry during this treatment to monitor for ventricular fibrillation or other arrhythmias.15 IV calcium does not lower serum potassium but rather antagonizes the effects of potassium on the cardiac cell membranes, helping to prevent or terminate arrhythmias.15,34 It should be noted, however, that firstline treatment for patients who develop hyperkalemia in the setting of digoxin toxicity involves administration of digoxin-specific antibody, while calcium infusion may be utilized later.34 Alternatively, if the patient is dialysis-dependent with ESRD, dialysis may be considered as a prompt initial treatment, with nephrologist consultation.
Administration of 10 U of regular insulin plus 25 g of 50% dextrose via IV will shift potassium intracellularly (see Figure 4). The dextrose will offset the resultant hypoglycemia.31,34 Of note, this treatment is often firstline for moderate to severe hyperkalemia in patients with a stable cardiac rhythm and ECG. Blood glucose should be monitored with a fingerstick within 30 to 60 minutes of infusion and every hour thereafter for up to six hours following insulin administration.34 Potassium levels should be checked every one to two hours after this treatment step until the serum potassium level stabilizes. Thereafter, recheck the levels every four to six hours to gauge whether further treatment is needed.34
Adjunctive
After performing firstline treatment strategies for severe hyperkalemia, there are alternate therapies to consider that can help lower total body potassium. Nebulized albuterol may be used, which pushes potassium into cells; this works in synergy with insulin and glucose.26,33 Sodium bicarbonate may be effective in cases in which the ABG analysis or labs show metabolic acidosis, as this infusion shifts potassium into cells by increasing the blood pH.33
In patients with dehydration, sepsis, TLS, or rhabdomyolysis, administration of IV fluids to maintain appropriate vascular volume is important. However, excessive fluid resuscitation can result in fluid overload, inducing complications such as respiratory failure and worsened renal function.40 A Foley catheter may be placed for strict intake and output monitoring.
The patient’s volume status must be carefully assessed. Hyperkalemia may present in association with heart failure exacerbation or ascites, which are usually hypervolemic states. Loop diuretics may be used to compensate for volume overload and to help remove potassium from the body, but these medications are contraindicated in anuric patients.13,41
Removing total body potassium
After emergent therapy is carried out, potassium may need to be removed from the body through diuresis, hemodialysis, or potassium binders. Loop diuretics or potassium binders may be used to treat mild to moderate hyperkalemia or to continue to stabilize the potassium level after emergent therapy is carried out. If severe hyperkalemia persists with kidney injury or with absence of urine output, hemodialysis is the therapy of choice.13
The potassium binder sodium polystyrene sulfonate (SPS) exchanges sodium for potassium in the intestine.42 This agent is contraindicated if the patient has intestinal obstruction. SPS’s slow onset of action (two to six hours) makes it ineffective as firstline therapy for severe hyperkalemia.3 In addition, SPS has serious but rare adverse effects, more commonly seen in patients who have uremia after kidney transplant or who have had recent abdominal surgery, bowel injury, or intestinal perforation.41 Adverse effects of SPS include aspiration pneumonitis, upper gastrointestinal injury, colonic necrosis, and rectal stenosis.41 However, there have been documented events of colonic necrosis due to SPS in patients without ESRD who have not had abdominal surgery.43,44 In 2009, the FDA advised against concomitant administration of sorbitol with SPS. However, this drug preparation continues to be the only one stocked by many hospital pharmacies.44 Because SPS has potentially harmful adverse effects and generally is not effective in promptly lowering serum potassium, it is prudent for clinicians to implement other management strategies first.44
MONITORING AT-RISK PATIENTS
Patients with a GFR < 45 mL/min/1.73 m2 and a baseline serum potassium level > 4.5 mEq/L are at risk for hyperkalemia while taking an ACEi or an ARB and should be advised to adhere to a potassium-restrictive diet with frequent laboratory checkups.22 Depending on the serum potassium and GFR levels at checkups, these medication doses may need to be reduced or discontinued altogether.
NEW DRUG DEVELOPMENTS
A potassium binder approved for daily use would benefit patients on aggressive heart failure medication regimens, as hyperkalemia commonly occurs with these regimens. As discussed, the widely available potassium binder SPS has been associated with severe gastrointestinal adverse effects, limiting its potential for routine use.44,45 In clinical trials, new potassium binders patiromer and zirconium cyclosilicate (ZS-9) have demonstrated an ability to maintain normokalemia over weeks of therapy with acceptable adverse effect profiles.45 In 2015, patiromer was approved by the FDA as therapy for hyperkalemia.46 An in-depth discussion, which is outside the scope of this article, will be presented by experts in the April 2017 edition of Renal Consult.
CONCLUSION
The best treatment for hyperkalemia is prevention through close surveillance of at-risk patients. Clinicians should be aware of predisposing risk factors for hyperkalemia, as it can have an insidious onset, with symptoms manifesting only when this electrolyte imbalance becomes life-threatening. It is particularly important to recognize when this condition mandates emergent treatment so that critical cardiac arrhythmias can be prevented.26
1. An JN, Lee JP, Jeon HJ, et al. Severe hyperkalemia requiring hospitalization: predictors of mortality. Crit Care. 2012; 16(6):R225.
2. Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis. Adv Physiol Educ. 2016;40(4):480-490.
3. Medford-Davis L, Rafique Z. Derangements of potassium. Emerg Med Clin North Am. 2014;32(2):329-347.
4. Eleftheriadis T, Leivaditis K, Antoniadi G, Liakopoulos V. Differential diagnosis of hyperkalemia: an update to a complex problem. Hippokratia. 2012;16(4):294-302.
5. Raebel M. Hyperkalemia associated with use of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. Cardiovasc Ther. 2012;30(3):156-166.
6. Korgaonkar S, Tilea A, Gillespie BW, et al. Serum potassium and outcomes in CKD: insights from the RRI-CKD Cohort Study. Clin J Am Soc Nephrol. 2010;5(5):762-769.
7. McMahon GM, Mendu ML, Gibbons FK, Christopher KB. Association between hyperkalemia at critical care initiation and mortality. Intensive Care Med. 2012;38(11):1834-1842.
8. Drawz PE, Babineau DC, Rahman M. Metabolic complications in elderly adults with chronic kidney disease. J Am Geriatr Soc. 2012;60(2):310-315.
9. Sarafidis PA, Georgianos PI, Bakris GL. Advances in treatment of hyperkalemia in chronic kidney disease. Expert Opin Pharmacother. 2015;16(14):2205-2215.
10. Einhorn LM, Zhan M, Hsu VD, et al. The frequency of hyperkalemia and its significance in chronic kidney disease. Arch Intern Med. 2009;169(12):1156-1162.
11. Khanagavi J, Gupta T, Aronow WS, et al. Hyperkalemia among hospitalized patients and association between duration of hyperkalemia and outcomes. Arch Med Sci. 2014;10(2):251-257.
12. Berkova M, Berka Z, Topinkova E. Arrhythmias and ECG changes in life threatening hyperkalemia in older patients treated by potassium sparing drugs. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158(1):84-91.
13. Lehnhardt A, Kemper MJ. Pathogenesis, diagnosis and management of hyperkalemia. Pediatr Nephrol. 2011;26(3):377-384.
14. Palmer BF. A physiologic-based approach to the evaluation of a patient with hyperkalemia. Am J Kidney Dis. 2010;56(2):387-393.
15. Mushiyakh Y, Dangaria H, Qavi S, et al. Treatment and pathogenesis of acute hyperkalemia. J Community Hosp Intern Med Perspect. 2012;1(4):7372.
16. Elliott MJ, Ronksley PE, Clase CM, et al. Management of patients with acute hyperkalemia. CMAJ. 2010;182(15):1631-1635.
17. Wiebe N, Klarenbach SW, Allan GM, et al. Potentially preventable hospitalization as a complication of CKD: a cohort study. Am J Kidney Dis. 2014;64(2):230-238.
18. Zutt R, van der Kooi AJ, Linthorst GE, et al. Rhabdomyolysis: review of the literature. Neuromuscul Disord. 2014;24(8):651-659.
19. Zimmerman JL, Shen MC. Rhabdomyolysis. Chest. 2013; 144(3):1058-1065.
20. Khardori R, Castillo D. Endocrine and metabolic changes during sepsis: an update. Med Clin North Am. 2012;96(6):1095-1105.
21. Raff H, Sharma ST, Nieman LK. Physiological basis for the etiology, diagnosis, and treatment of adrenal disorders: Cushing’s syndrome, adrenal insufficiency, and congenital adrenal hyperplasia. Compr Physiol. 2014;4(20):739-769.
22. Lazich I, Bakris GL. Prediction and management of hyperkalemia across the spectrum of chronic kidney disease. Semin Nephrol. 2014;34(3):333-339.
23. Ayach T, Nappo R, Paugh-Miller J, Ross E. Life-threatening hyperkalemia in a patient with normal renal function. Clin Kidney J. 2014;7(1):49-52.
24. Wilson FP, Berns JS. Tumor lysis syndrome: new challenges and recent advances. Adv Chronic Kidney Dis. 2014;21(1):18-26.
25. Asiryatham JR, Moses V, Bjornson L. Errors in potassium measurement: a laboratory perspective for the clinician. N Am J Med Sci. 2013;5(4):255-259.
26. Pepin J, Shields C. Advances in diagnosis and management of hypokalemic and hyperkalemic emergencies. Emerg Med Pract. 2012;14(2):1-17.
27. Vraets A, Lin Y, Callum JL. Transfusion-associated hyperkalemia. Transfus Med Rev. 2011;25(3):184-196.
28. Aboudara MC, Hurst FP, Abbott KC, Perkins RM. Hyperkalemia after packed red blood cell transfusion in trauma patients. J Trauma. 2008;64(2 suppl):S86-S91.
29. Olson J, Talekar M, Sachdev M, et al. Potassium changes associated with blood transfusion in pediatric patients. Am J Clin Pathol. 2013;139(6):800-805.
30. Chon S, Kwak YH, Hwang SS, et al. Severe hyperkalemia can be detected immediately by quantitative electrocardiography and clinical history in patients with symptomatic or extreme bradycardia: a retrospective cross-sectional study. J Crit Care. 2013;28(6):1112.e7-1112.e13.
31. Viera AJ, Wouk N. Potassium disorders: hypokalemia and hyperkalemia. Am Fam Physician. 2015;92(6):487-495.
32. Sanghavi S, Whitling S, Uribarri J. Potassium balance in dialysis patients. Semin Dial. 2013;26(5):597-603.
33. Crawford AH. Hyperkalemia: Recognition and management of a critical electrolyte disturbance. J Infus Nurs. 2014;37(3):167-175.
34. Maxwell AP, Linden K, O’Donnell S, et al. Management of hyperkalemia. J R Coll Physicians Edinb. 2013;43(3):246-251.
35. Seth P, Kaur H, Kaur M. Clinical profile of diabetic ketoacidosis: a prospective study in a tertiary care hospital. J Clin Diagn Res. 2015;9(6):OC01-OC04.
36. Rahman M, Shad F, Smith M. Acute kidney injury: a guide to diagnosis and management. Am Fam Physician. 2012;86(7): 631-639.
37. Puar TH, Stikkelbroeck NM, Smans LC, et al. Adrenal crisis: still a deadly event in the 21st century. Am J Med. 2016;129(3):339.e1-9.
38. Liamis G, Liberopoulos E, Barkas F, Elisaf M. Spurious electrolyte disorders: a diagnostic challenge for clinicians. Am J Nephrol. 2013;38(1):50-57.
39. Kovesdy CP. Management of hyperkalemia: an update for the internist. Am J Med. 2015;128(12):1281-1287.
40. Labib M, Khalid R, Khan A, Khan S. Volume management in the critically ill patient with acute kidney injury. Crit Care Res Pract. 2013;2013:792830.
41. Watson M, Abbott KC, Yuan CM. Damned if you do, damned if you don’t: potassium binding resins in hyperkalemia. Clin J Am Soc Nephrol. 2010;5(10):1723-1726.
42. Nguyen T, Ondrik D, Zhufyak O, et al. Hyperkalemia and potential pitfalls of sodium polystyrene sulfonate. JAAPA. 2015; 28(3):41-45.
43. McGowan CE, Saha S, Resnick MB, Moss SF. Intestinal necrosis due to sodium polystyrene sulfonate (Kayexalate) in sorbitol. South Med J. 2009;102(5):493-497.
44. Sterns RH, Rojas M, Bernstein P, Chennupati S. Ion-exchange resins for the treatment of hyperkalemia: are they safe and effective? J Am Soc Nephrol. 2010;21:733-735.
45. Pitt B, Bakris GL. New potassium binders for the treatment of hyperkalemia: current data and opportunities for the future. Hypertension. 2015;66(4):731-738.
46. Epstein M, Pitt B. Recent advances in pharmacological treatments of hyperkalemia: focus on patiromer. Expert Opin Pharmacother. 2016;17(10):1435-1448.
CE/CME No: CR-1703
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Describe the pathophysiology and causes of hyperkalemia.
• Identify patients who are susceptible to hyperkalemia.
• Recognize the clinical sequelae of hyperkalemia.
• Formulate assessment and treatment plans for patients with hyperkalemia.
FACULTY
Melanie Douglas is a Physician Assistant in the Medicine Department at NYU Langone Medical Center in New York, New York. Denise Rizzolo is a Clinical Assistant Professor in the PA Program at Pace University in New York, New York, and Research Director in the Program of PA Studies at Kean University in Union, New Jersey. Danielle Kruger is an Academic Coordinator and Associate Professor in the PA Program at St. John’s University in Queens, New York. The authors have no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of March 2017.
Article begins on next page >>
Hyperkalemia is a common electrolyte disorder associated with life-threatening cardiac arrhythmias. Prompt recognition and appropriate treatment are essential in preventing serious cardiac complications. Although clinical manifestations of hyperkalemia are usually nonspecific or absent, laboratory testing and electrocardiography performed by the astute clinician aware of predisposing risk factors can help direct management.
Potassium is contained mostly in intracellular fluid; only about 2% is found in the extracellular space.1 The average total body potassium is about 50 mEq per kg of body weight (eg, a 70-kg individual has a total body potassium of approximately 3,500 mEq).2 Levels are tightly regulated by alterations in excretion in the distal renal tubule in response to potassium load and balance, and potassium distribution is influenced by insulin, aldosterone, catecholamines, and acid-base status.2 Movement of potassium across cell membranes is driven by the sodium-potassium adenosine triphosphatase (Na-K-ATPase) pump.3 In this article, we use the common serum potassium reference range of 3.5 to 5.0 mEq/L and define hyperkalemia as a serum potassium concentration greater than 5.5 mEq/L.4
Hyperkalemia can lead to life-threatening complications of cardiac arrhythmias, asystole, hypotension, flaccid paralysis, tetany, dyspnea, and altered mental status.5 Among patients with end-stage renal disease (ESRD), hyperkalemia is thought to contribute to 2% to 5% of deaths.6 A retrospective study found that patients with serum potassium levels exceeding 6.0 mEq/L on ICU admission had a significantly higher death rate within 30 days than patients who were normokalemic on presentation.7
RISK FACTORS
It is estimated that more than 35% of patients age 70 and older have chronic kidney disease (CKD) stage 3 or higher.8 Hyperkalemia is closely associated with CKD, increasing linearly in relation to the degree of renal impairment.8 As such, the prevalence of hyperkalemia in older adults is high, and it will increase overall as the US population ages. In a retrospective analysis of veterans older than 65 with CKD stage 3 or higher, the prevalence of hyperkalemia was 2.5%.9 Use of certain medications is also associated with hyperkalemia. Another retrospective study analyzed records obtained from 70,873 patients with CKD (estimated glomerular filtration rate [eGFR] < 60 mL/min/1.73 m2) hospitalized in the Veterans Health Administration system. It found that patients treated with renin-angiotensin-aldosterone system (RAAS) blockers, such as ACE inhibitors (ACEis) or angiotensin-receptor blockers (ARBs), had a higher incidence of hyperkalemia (potassium level ≥ 5.5 mEq/L) than patients not treated with these medications (8.22 vs 1.77 events per 100 patient-months).9,10
POTASSIUM HOMEOSTASIS
Tight control over extracellular potassium is maintained in part by the Na-K-ATPase pump, which uses adenosine triphosphatase to move potassium and sodium ions in opposite directions across cell membranes.3 Specifically, three sodium ions are pumped out of the cell for every two potassium ions pumped in, resulting in a potassium gradient that is partially responsible for maintaining a resting membrane potential. This resting membrane potential, which determines myocardial, skeletal muscle, and nerve cell excitability and signaling, is highly sensitive to changes in the extracellular potassium level.4 Even small extracellular imbalances can induce cell depolarization and evoke an action potential. Increased extracellular potassium concentration decreases the resting membrane potential of the myocardium, shortens repolarization time, and decreases the rate of myocardial cell conduction, and also slows down neuromuscular conduction.11,12
Renal tubular function plays a significant role in potassium homeostasis, with approximately 90% of dietary potassium intake ex
The RAAS is a signal transduction pathway that regulates potassium excretion by the kidneys. Renin is secreted by the kidney in response to low renal perfusion, catecholamines, ß-adrenergic stimulation, potassium and sodium levels, and other factors. Secretion of renin triggers a signaling cascade that eventually results in the release of aldosterone from the adrenal cortex.5 Aldosterone binds to a receptor in the kidney’s collecting ducts where it increases potassium excretion by stimulating sodium reabsorption and fluid retention (see Figure 1).5
CAUSES OF HYPERKALEMIA
The pathophysiology of hyperkalemia generally involves either decreased renal excretion or shifts in extracellular potassium. Causes of hyperkalemia are listed in the Table. Potassium excretion can be disrupted in acute kidney injury (AKI), sepsis, cardiac ischemia, heart failure, diabetic ketoacidosis (DKA), insulin deficiency, tumor lysis syndrome (TLS), sickle cell disease, systemic lupus erythematosus, renal transplant, hepatorenal syndrome, adrenal insufficiency, and obstructive uropathy.15 In addition, certain medications can impair potassium excretion (eg, RAAS blockers, potassium-sparing diuretics in patients with CKD, digoxin toxicity).16 The following sections highlight the pathophysiology and manifestations of more common causes of hyperkalemia.
Renal impairment
Hyperkalemia may be a manifestation of worsening renal function. Potassium excretion is reduced in CKD, and CKD is the most common cause of hyperkalemia due to lower GFR.8,17 Patients with lower GFR tend to be older and male, and frequently have comorbid conditions such as type 2 diabetes, chronic liver disease, and heart failure.17
In CKD, decreased delivery of sodium to the distal tubules and reduced filtration capacity of the kidney diminishes the collecting duct’s ability to excrete potassium in exchange for sodium.2 Metabolic acidosis, which often contributes to AKI or CKD, causes potassium to shift from the intracellular to the extracellular compartment.4 Renal impairment may present clinically with dehydration, oliguria, nausea, vomiting, constipation, altered mental status, or weakness.
Hyperglycemia
Insulin and catecholamines (eg, epinephrine and norepinephrine) drive potassium into cells. Insulin increases potassium uptake into liver and muscle cells.13 A decrease in insulin levels, as may occur in type 2 diabetes or DKA, can cause a buildup of extracellular potassium.4 Also, serum hypertonicity from hyperglycemia results in water movement from the intracellular to the extracellular compartment; this raises the intracellular concentration of potassium, further promoting its movement to the extracellular space.4,14 Patients with hyperglycemia may present with dizziness, polyuria, polydipsia, nausea, vomiting, altered mental status, or fatigue.
Rhabdomyolysis
Rhabdomyolysis is a rapid breakdown of skeletal muscle that results in leakage of cellular contents into the extracellular space.4,18 Causes of rhabdomyolysis include use of medications such as statins, illicit drugs (eg, cocaine), or alcohol; rigorous exercise; and trauma.19
Muscle cell contents that are released into the circulation include potassium and other electrolytes, enzymes (eg, lactate dehydrogenase, aspartate transaminase, aldolase), and myoglobin.19 In rhabdomyolysis, myoglobin accumulation and hypovolemia lead to AKI and hyperkalemia.19 Patients may present with myalgias, extremity paresthesias, generalized weakness, nausea, altered mental status, fever, or darkened urine.18,19
Adrenal insufficiency
During critical illness such as sepsis, adrenal insufficiency can result from destruction of the adrenal glands, leading to hypoaldosteronism.20 Reduced aldosterone in adrenal insufficiency enables sodium and water to be eliminated from the body more easily, but as a result, less potassium gets excreted through the renal system and more is driven into the plasma.15
Acute adrenal insufficiency may manifest with hypotension, nausea, vomiting, or altered mental status, and labwork may reveal hyperkalemia as well as hypoglycemia or hyponatremia. Additionally, long-term glucocorticoid therapy can suppress the hypothalamic-pituitary axis and cause adrenal atrophy; rapid discontinuation of steroids can lead to adrenal insufficiency and hyperkalemia.21
Medications
RAAS blockers reduce CKD progression in patients with an eGFR of 29 mL/min/1.73 m2 or greater.22 Nonetheless, prescribing two or more drugs from the ACEi or ARB classes is not recommended. The Veterans Administration Nephron-Diabetes Trial (VA-NEPHRON-D) was terminated early because patients with stage 3 CKD due to diabetes who received dual ACEi/ARB therapy had higher rates of hyperkalemia but no slowing of CKD.22
Within the RAAS cascade, ACEis block the formation of angiotensin II and ARBs prevent angiotensin II from binding to the adrenal receptor. This impairs renal excretion of potassium and potentially contributes to hyperkalemia.5 Nonetheless, when patients on ACEis or ARBs develop hyperkalemia, aldosterone concentrations usually decrease due to preexisting illnesses (eg, diabetes, heart failure, CKD, AKI) or drug effects (eg, potassium-sparing diuretics, ß-blockers, digoxin).5 Ultimately, a combination of factors resulting from ACEi or ARB therapy causes reductions in renal perfusion and predisposes patients to hyperkalemia.5
NSAIDs may lead to hyperkalemia, as they interfere with prostaglandin release, decrease renal perfusion, and reduce renin and aldosterone levels.22 ß-blockers and tacrolimus inhibit renin release, leading to decreased aldosterone levels.5 Potassium-sparing diuretics block the interaction of aldosterone with the aldosterone receptor in the nephron.5 Digoxin decreases the activity of Na-K-ATPase, diminishing potassium uptake by cells.9 Potassium supplements, often prescribed for patients on diuretics, may contribute to hyperkalemia in patients with CKD. In the hospital setting, potassium tablets or IV formulations are utilized to correct hypokalemia. Especially in patients with CKD, clinicians should prescribe these agents with caution to avoid inducing hyperkalemia. Salt substitutes, which commonly contain potassium chloride, may be appealing to patients concerned about their sodium intake. However, consumption of these substitutes may contribute to hyperkalemia, especially in patients with CKD, heart failure, or type 2 diabetes.23
Tumor lysis syndrome
TLS involves rapid release of electrolytes and other intracellular contents into the extracellular space during the lysis of tumor cells.24 Nucleic acids within DNA strands break down and build up extracellularly, leading to hyperuricemia and often AKI. Potassium and other electrolytes released into the plasma during cell lysis can usually be removed by a healthy renal system. In TLS, however, AKI due to uric acid nephropathy prevents kidneys from removing the excess electrolytes from the bloodstream.24 Patients with rapidly growing hematologic tumors undergoing chemotherapy are especially at risk.
Pseudohyperkalemia
Pseudohyperkalemia is a transiently elevated serum potassium level that erroneously represents the true serum potassium level. It results from hemolysis due to mechanical trauma during the blood draw (eg, a tourniquet tied too tightly or use of a small-bore needle) or during specimen handling afterwards.25 Furthermore, leukocytosis, thrombocytosis, and polycythemia make red blood cells more fragile, increasing the chance of hemolysis and potassium leakage.26 Blood transfusion also can lead to pseudohyperkalemia. When blood is stored, potassium leakage from the cells and cell lysis, along with diminished Na-K-ATPase activity, lead to a buildup of potassium in the medium surrounding the stored red blood cells.27,28 The rise in serum potassium levels post-transfusion is usually transient, as the blood cells redistribute the potassium load once they become metabolically active.27,29
CLINICAL MANIFESTATIONS
Clinical manifestations of mild to moderate hyperkalemia (serum potassium > 5.5 mEq/L but < 6.5 mEq/L) include fatigue, generalized weakness, nausea, vomiting, constipation, and diarrhea.15 In many patients, mild to moderate hyperkalemia may not be associated with any acute symptoms and vital signs may be normal.13 Severe hyperkalemia (serum potassium > 6.5 mEq/L) may present clinically with acute extremity paresthesias, muscle weakness and paralysis, heart palpitations, dyspnea, altered mental status, cardiac arrhythmias, and cardiac arrest.30,31 Irregular heart rhythm, decreased deep tendon reflexes, or decreased strength may be revealed on physical exam.3 Individuals with ESRD on hemodialysis seem to tolerate higher levels of potassium than the general population without displaying clinical symptoms. However, these individuals are still susceptible to the cardiac effects of hyperkalemia.32
INITIAL ASSESSMENT
In assessing hyperkalemia, the clinician must perform a focused history and physical exam and review the patient’s medication list, including supplements and dietary habits that impact potassium intake. Potassium-rich foods include meat, fish, milk, almonds, spinach, cantaloupe, bananas, oranges, mushrooms, and potatoes.33 Hyperkalemia may present in association with various medical emergencies. The clinician should have an index of suspicion, depending on the patient’s overall medical profile and presentation, for emergencies such as cardiac ischemia, sepsis, adrenal crisis, DKA, TLS, and digoxin overdose.
The clinician must identify whether an elevated potassium level requires emergent therapy; assessment of vital signs is paramount in determining this. Orthostatic hypotension and tachycardia may hint that the patient is volume depleted. The patient should be examined for signs of hemodynamic shock with the CAB sequence: circulation, airway, breathing.34 Symptoms such as chest pain, shortness of breath, muscle weakness, paralysis, and altered mental status suggest that an expedited evaluation is warranted.
With a serum potassium level > 5.5 mEq/L, urgent electrocardiography should be performed.26 ECG findings observed with serum potassium levels of 5.5-6.5 mEq/L usually include peaked T-waves and prolonged PR intervals (see Figure 2). With potassium levels > 6.5 mEq/L consistent with further cardiac destabilization, the P-wave flattens then disappears, the QRS complex broadens, and sinus bradycardia or ectopic beats may occur.12,26 ST depression, T-wave inversion, or ST elevation also may be seen.12 With serum potassium levels > 7.5 mEq/L, progressive widening of the QRS complex to a sine-wave with bundle branch blocks or fascicular blocks may occur (see Figure 3).26 Without prompt intervention, ventricular fibrillation may ensue.26
An extensive laboratory workup may be necessary to investigate the etiology; this includes a complete blood count, metabolic panel, liver function tests, cardiac enzymes, blood gas analysis, serum/urine osmolality, urinalysis, urine electrolytes, and toxicology screen.13,26 Arterial blood gas (ABG) analysis may show metabolic acidosis with AKI or DKA, or an elevated lactate may occur with sepsis. In patients with hyperglycemia, besides checking for acidosis, obtaining blood/urine ketone levels and a metabolic panel with anion gap to evaluate for DKA is useful.35
When assessing a patient with an elevated creatinine, the GFR at the time of evaluation should be compared with the patient’s baseline GFR to determine chronicity and duration of his/her kidney disease.36 Obtaining a urinalysis and urine electrolytes in addition to the basic metabolic panel can help narrow the etiology.36 A Foley catheter should be placed in cases of urinary retention because without intervention, urinary obstruction may lead to AKI and hyperkalemia. Myoglobinuria on urinalysis and an elevated creatine kinase are diagnostic markers of rhabdomyolysis.18
TLS should be considered in patients who recently received chemotherapy, especially those with proliferative hematologic malignancies, such as acute lymphoblastic leukemia, acute myeloid leukemia, and Burkitt lymphoma.24 In TLS, bloodwork often reveals hyperkalemia along with AKI, an elevated uric acid level, hyperphosphatemia, and hypocalcemia.24
Patients presenting with hyperkalemia, hypotension, hypoglycemia, and hyponatremia may have adrenal insufficiency.20 If insufficiency is suspected, a cortisol level may be checked during morning hours; a low level is often suggestive of this diagnosis.37 Treatment includes daily doses of steroids, and consultation with an endocrinologist is recommended.37
If an elevated potassium level is not accompanied by renal dysfunction, electrolyte imbalances, ECG changes, or inciting medications, pseudohyperkalemia should be considered.38 A repeat lab sample should be checked. Consider obtaining an ABG analysis, as the shorter time interval between drawing the blood sample and the sample analysis reportedly increases the reliability of the resulting potassium level.38
THERAPY
Emergent
Emergent treatment is needed for severe hyperkalemia (see Figure 4). Any hyperkalemia-inciting medications or potassium supplements should be immediately discontinued.39 IV access and cardiac telemetry monitoring should be promptly applied.26
In cases of severe hyperkalemia that involve cardiac arrhythmias, manifestations on ECG, or risk for arrhythmias, calcium gluconate (10 mL IV over 10 min) should be urgently administered, followed by IV insulin in conjunction with dextrose.26 Calcium chloride should be utilized for hyperkalemia in the context of the advanced cardiac life support (ACLS) protocol for cardiac arrest.26 The patient should remain on cardiac telemetry during this treatment to monitor for ventricular fibrillation or other arrhythmias.15 IV calcium does not lower serum potassium but rather antagonizes the effects of potassium on the cardiac cell membranes, helping to prevent or terminate arrhythmias.15,34 It should be noted, however, that firstline treatment for patients who develop hyperkalemia in the setting of digoxin toxicity involves administration of digoxin-specific antibody, while calcium infusion may be utilized later.34 Alternatively, if the patient is dialysis-dependent with ESRD, dialysis may be considered as a prompt initial treatment, with nephrologist consultation.
Administration of 10 U of regular insulin plus 25 g of 50% dextrose via IV will shift potassium intracellularly (see Figure 4). The dextrose will offset the resultant hypoglycemia.31,34 Of note, this treatment is often firstline for moderate to severe hyperkalemia in patients with a stable cardiac rhythm and ECG. Blood glucose should be monitored with a fingerstick within 30 to 60 minutes of infusion and every hour thereafter for up to six hours following insulin administration.34 Potassium levels should be checked every one to two hours after this treatment step until the serum potassium level stabilizes. Thereafter, recheck the levels every four to six hours to gauge whether further treatment is needed.34
Adjunctive
After performing firstline treatment strategies for severe hyperkalemia, there are alternate therapies to consider that can help lower total body potassium. Nebulized albuterol may be used, which pushes potassium into cells; this works in synergy with insulin and glucose.26,33 Sodium bicarbonate may be effective in cases in which the ABG analysis or labs show metabolic acidosis, as this infusion shifts potassium into cells by increasing the blood pH.33
In patients with dehydration, sepsis, TLS, or rhabdomyolysis, administration of IV fluids to maintain appropriate vascular volume is important. However, excessive fluid resuscitation can result in fluid overload, inducing complications such as respiratory failure and worsened renal function.40 A Foley catheter may be placed for strict intake and output monitoring.
The patient’s volume status must be carefully assessed. Hyperkalemia may present in association with heart failure exacerbation or ascites, which are usually hypervolemic states. Loop diuretics may be used to compensate for volume overload and to help remove potassium from the body, but these medications are contraindicated in anuric patients.13,41
Removing total body potassium
After emergent therapy is carried out, potassium may need to be removed from the body through diuresis, hemodialysis, or potassium binders. Loop diuretics or potassium binders may be used to treat mild to moderate hyperkalemia or to continue to stabilize the potassium level after emergent therapy is carried out. If severe hyperkalemia persists with kidney injury or with absence of urine output, hemodialysis is the therapy of choice.13
The potassium binder sodium polystyrene sulfonate (SPS) exchanges sodium for potassium in the intestine.42 This agent is contraindicated if the patient has intestinal obstruction. SPS’s slow onset of action (two to six hours) makes it ineffective as firstline therapy for severe hyperkalemia.3 In addition, SPS has serious but rare adverse effects, more commonly seen in patients who have uremia after kidney transplant or who have had recent abdominal surgery, bowel injury, or intestinal perforation.41 Adverse effects of SPS include aspiration pneumonitis, upper gastrointestinal injury, colonic necrosis, and rectal stenosis.41 However, there have been documented events of colonic necrosis due to SPS in patients without ESRD who have not had abdominal surgery.43,44 In 2009, the FDA advised against concomitant administration of sorbitol with SPS. However, this drug preparation continues to be the only one stocked by many hospital pharmacies.44 Because SPS has potentially harmful adverse effects and generally is not effective in promptly lowering serum potassium, it is prudent for clinicians to implement other management strategies first.44
MONITORING AT-RISK PATIENTS
Patients with a GFR < 45 mL/min/1.73 m2 and a baseline serum potassium level > 4.5 mEq/L are at risk for hyperkalemia while taking an ACEi or an ARB and should be advised to adhere to a potassium-restrictive diet with frequent laboratory checkups.22 Depending on the serum potassium and GFR levels at checkups, these medication doses may need to be reduced or discontinued altogether.
NEW DRUG DEVELOPMENTS
A potassium binder approved for daily use would benefit patients on aggressive heart failure medication regimens, as hyperkalemia commonly occurs with these regimens. As discussed, the widely available potassium binder SPS has been associated with severe gastrointestinal adverse effects, limiting its potential for routine use.44,45 In clinical trials, new potassium binders patiromer and zirconium cyclosilicate (ZS-9) have demonstrated an ability to maintain normokalemia over weeks of therapy with acceptable adverse effect profiles.45 In 2015, patiromer was approved by the FDA as therapy for hyperkalemia.46 An in-depth discussion, which is outside the scope of this article, will be presented by experts in the April 2017 edition of Renal Consult.
CONCLUSION
The best treatment for hyperkalemia is prevention through close surveillance of at-risk patients. Clinicians should be aware of predisposing risk factors for hyperkalemia, as it can have an insidious onset, with symptoms manifesting only when this electrolyte imbalance becomes life-threatening. It is particularly important to recognize when this condition mandates emergent treatment so that critical cardiac arrhythmias can be prevented.26
CE/CME No: CR-1703
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Describe the pathophysiology and causes of hyperkalemia.
• Identify patients who are susceptible to hyperkalemia.
• Recognize the clinical sequelae of hyperkalemia.
• Formulate assessment and treatment plans for patients with hyperkalemia.
FACULTY
Melanie Douglas is a Physician Assistant in the Medicine Department at NYU Langone Medical Center in New York, New York. Denise Rizzolo is a Clinical Assistant Professor in the PA Program at Pace University in New York, New York, and Research Director in the Program of PA Studies at Kean University in Union, New Jersey. Danielle Kruger is an Academic Coordinator and Associate Professor in the PA Program at St. John’s University in Queens, New York. The authors have no financial relationships to disclose.
![]()
ACCREDITATION STATEMENT
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of March 2017.
Article begins on next page >>
Hyperkalemia is a common electrolyte disorder associated with life-threatening cardiac arrhythmias. Prompt recognition and appropriate treatment are essential in preventing serious cardiac complications. Although clinical manifestations of hyperkalemia are usually nonspecific or absent, laboratory testing and electrocardiography performed by the astute clinician aware of predisposing risk factors can help direct management.
Potassium is contained mostly in intracellular fluid; only about 2% is found in the extracellular space.1 The average total body potassium is about 50 mEq per kg of body weight (eg, a 70-kg individual has a total body potassium of approximately 3,500 mEq).2 Levels are tightly regulated by alterations in excretion in the distal renal tubule in response to potassium load and balance, and potassium distribution is influenced by insulin, aldosterone, catecholamines, and acid-base status.2 Movement of potassium across cell membranes is driven by the sodium-potassium adenosine triphosphatase (Na-K-ATPase) pump.3 In this article, we use the common serum potassium reference range of 3.5 to 5.0 mEq/L and define hyperkalemia as a serum potassium concentration greater than 5.5 mEq/L.4
Hyperkalemia can lead to life-threatening complications of cardiac arrhythmias, asystole, hypotension, flaccid paralysis, tetany, dyspnea, and altered mental status.5 Among patients with end-stage renal disease (ESRD), hyperkalemia is thought to contribute to 2% to 5% of deaths.6 A retrospective study found that patients with serum potassium levels exceeding 6.0 mEq/L on ICU admission had a significantly higher death rate within 30 days than patients who were normokalemic on presentation.7
RISK FACTORS
It is estimated that more than 35% of patients age 70 and older have chronic kidney disease (CKD) stage 3 or higher.8 Hyperkalemia is closely associated with CKD, increasing linearly in relation to the degree of renal impairment.8 As such, the prevalence of hyperkalemia in older adults is high, and it will increase overall as the US population ages. In a retrospective analysis of veterans older than 65 with CKD stage 3 or higher, the prevalence of hyperkalemia was 2.5%.9 Use of certain medications is also associated with hyperkalemia. Another retrospective study analyzed records obtained from 70,873 patients with CKD (estimated glomerular filtration rate [eGFR] < 60 mL/min/1.73 m2) hospitalized in the Veterans Health Administration system. It found that patients treated with renin-angiotensin-aldosterone system (RAAS) blockers, such as ACE inhibitors (ACEis) or angiotensin-receptor blockers (ARBs), had a higher incidence of hyperkalemia (potassium level ≥ 5.5 mEq/L) than patients not treated with these medications (8.22 vs 1.77 events per 100 patient-months).9,10
POTASSIUM HOMEOSTASIS
Tight control over extracellular potassium is maintained in part by the Na-K-ATPase pump, which uses adenosine triphosphatase to move potassium and sodium ions in opposite directions across cell membranes.3 Specifically, three sodium ions are pumped out of the cell for every two potassium ions pumped in, resulting in a potassium gradient that is partially responsible for maintaining a resting membrane potential. This resting membrane potential, which determines myocardial, skeletal muscle, and nerve cell excitability and signaling, is highly sensitive to changes in the extracellular potassium level.4 Even small extracellular imbalances can induce cell depolarization and evoke an action potential. Increased extracellular potassium concentration decreases the resting membrane potential of the myocardium, shortens repolarization time, and decreases the rate of myocardial cell conduction, and also slows down neuromuscular conduction.11,12
Renal tubular function plays a significant role in potassium homeostasis, with approximately 90% of dietary potassium intake ex
The RAAS is a signal transduction pathway that regulates potassium excretion by the kidneys. Renin is secreted by the kidney in response to low renal perfusion, catecholamines, ß-adrenergic stimulation, potassium and sodium levels, and other factors. Secretion of renin triggers a signaling cascade that eventually results in the release of aldosterone from the adrenal cortex.5 Aldosterone binds to a receptor in the kidney’s collecting ducts where it increases potassium excretion by stimulating sodium reabsorption and fluid retention (see Figure 1).5
CAUSES OF HYPERKALEMIA
The pathophysiology of hyperkalemia generally involves either decreased renal excretion or shifts in extracellular potassium. Causes of hyperkalemia are listed in the Table. Potassium excretion can be disrupted in acute kidney injury (AKI), sepsis, cardiac ischemia, heart failure, diabetic ketoacidosis (DKA), insulin deficiency, tumor lysis syndrome (TLS), sickle cell disease, systemic lupus erythematosus, renal transplant, hepatorenal syndrome, adrenal insufficiency, and obstructive uropathy.15 In addition, certain medications can impair potassium excretion (eg, RAAS blockers, potassium-sparing diuretics in patients with CKD, digoxin toxicity).16 The following sections highlight the pathophysiology and manifestations of more common causes of hyperkalemia.
Renal impairment
Hyperkalemia may be a manifestation of worsening renal function. Potassium excretion is reduced in CKD, and CKD is the most common cause of hyperkalemia due to lower GFR.8,17 Patients with lower GFR tend to be older and male, and frequently have comorbid conditions such as type 2 diabetes, chronic liver disease, and heart failure.17
In CKD, decreased delivery of sodium to the distal tubules and reduced filtration capacity of the kidney diminishes the collecting duct’s ability to excrete potassium in exchange for sodium.2 Metabolic acidosis, which often contributes to AKI or CKD, causes potassium to shift from the intracellular to the extracellular compartment.4 Renal impairment may present clinically with dehydration, oliguria, nausea, vomiting, constipation, altered mental status, or weakness.
Hyperglycemia
Insulin and catecholamines (eg, epinephrine and norepinephrine) drive potassium into cells. Insulin increases potassium uptake into liver and muscle cells.13 A decrease in insulin levels, as may occur in type 2 diabetes or DKA, can cause a buildup of extracellular potassium.4 Also, serum hypertonicity from hyperglycemia results in water movement from the intracellular to the extracellular compartment; this raises the intracellular concentration of potassium, further promoting its movement to the extracellular space.4,14 Patients with hyperglycemia may present with dizziness, polyuria, polydipsia, nausea, vomiting, altered mental status, or fatigue.
Rhabdomyolysis
Rhabdomyolysis is a rapid breakdown of skeletal muscle that results in leakage of cellular contents into the extracellular space.4,18 Causes of rhabdomyolysis include use of medications such as statins, illicit drugs (eg, cocaine), or alcohol; rigorous exercise; and trauma.19
Muscle cell contents that are released into the circulation include potassium and other electrolytes, enzymes (eg, lactate dehydrogenase, aspartate transaminase, aldolase), and myoglobin.19 In rhabdomyolysis, myoglobin accumulation and hypovolemia lead to AKI and hyperkalemia.19 Patients may present with myalgias, extremity paresthesias, generalized weakness, nausea, altered mental status, fever, or darkened urine.18,19
Adrenal insufficiency
During critical illness such as sepsis, adrenal insufficiency can result from destruction of the adrenal glands, leading to hypoaldosteronism.20 Reduced aldosterone in adrenal insufficiency enables sodium and water to be eliminated from the body more easily, but as a result, less potassium gets excreted through the renal system and more is driven into the plasma.15
Acute adrenal insufficiency may manifest with hypotension, nausea, vomiting, or altered mental status, and labwork may reveal hyperkalemia as well as hypoglycemia or hyponatremia. Additionally, long-term glucocorticoid therapy can suppress the hypothalamic-pituitary axis and cause adrenal atrophy; rapid discontinuation of steroids can lead to adrenal insufficiency and hyperkalemia.21
Medications
RAAS blockers reduce CKD progression in patients with an eGFR of 29 mL/min/1.73 m2 or greater.22 Nonetheless, prescribing two or more drugs from the ACEi or ARB classes is not recommended. The Veterans Administration Nephron-Diabetes Trial (VA-NEPHRON-D) was terminated early because patients with stage 3 CKD due to diabetes who received dual ACEi/ARB therapy had higher rates of hyperkalemia but no slowing of CKD.22
Within the RAAS cascade, ACEis block the formation of angiotensin II and ARBs prevent angiotensin II from binding to the adrenal receptor. This impairs renal excretion of potassium and potentially contributes to hyperkalemia.5 Nonetheless, when patients on ACEis or ARBs develop hyperkalemia, aldosterone concentrations usually decrease due to preexisting illnesses (eg, diabetes, heart failure, CKD, AKI) or drug effects (eg, potassium-sparing diuretics, ß-blockers, digoxin).5 Ultimately, a combination of factors resulting from ACEi or ARB therapy causes reductions in renal perfusion and predisposes patients to hyperkalemia.5
NSAIDs may lead to hyperkalemia, as they interfere with prostaglandin release, decrease renal perfusion, and reduce renin and aldosterone levels.22 ß-blockers and tacrolimus inhibit renin release, leading to decreased aldosterone levels.5 Potassium-sparing diuretics block the interaction of aldosterone with the aldosterone receptor in the nephron.5 Digoxin decreases the activity of Na-K-ATPase, diminishing potassium uptake by cells.9 Potassium supplements, often prescribed for patients on diuretics, may contribute to hyperkalemia in patients with CKD. In the hospital setting, potassium tablets or IV formulations are utilized to correct hypokalemia. Especially in patients with CKD, clinicians should prescribe these agents with caution to avoid inducing hyperkalemia. Salt substitutes, which commonly contain potassium chloride, may be appealing to patients concerned about their sodium intake. However, consumption of these substitutes may contribute to hyperkalemia, especially in patients with CKD, heart failure, or type 2 diabetes.23
Tumor lysis syndrome
TLS involves rapid release of electrolytes and other intracellular contents into the extracellular space during the lysis of tumor cells.24 Nucleic acids within DNA strands break down and build up extracellularly, leading to hyperuricemia and often AKI. Potassium and other electrolytes released into the plasma during cell lysis can usually be removed by a healthy renal system. In TLS, however, AKI due to uric acid nephropathy prevents kidneys from removing the excess electrolytes from the bloodstream.24 Patients with rapidly growing hematologic tumors undergoing chemotherapy are especially at risk.
Pseudohyperkalemia
Pseudohyperkalemia is a transiently elevated serum potassium level that erroneously represents the true serum potassium level. It results from hemolysis due to mechanical trauma during the blood draw (eg, a tourniquet tied too tightly or use of a small-bore needle) or during specimen handling afterwards.25 Furthermore, leukocytosis, thrombocytosis, and polycythemia make red blood cells more fragile, increasing the chance of hemolysis and potassium leakage.26 Blood transfusion also can lead to pseudohyperkalemia. When blood is stored, potassium leakage from the cells and cell lysis, along with diminished Na-K-ATPase activity, lead to a buildup of potassium in the medium surrounding the stored red blood cells.27,28 The rise in serum potassium levels post-transfusion is usually transient, as the blood cells redistribute the potassium load once they become metabolically active.27,29
CLINICAL MANIFESTATIONS
Clinical manifestations of mild to moderate hyperkalemia (serum potassium > 5.5 mEq/L but < 6.5 mEq/L) include fatigue, generalized weakness, nausea, vomiting, constipation, and diarrhea.15 In many patients, mild to moderate hyperkalemia may not be associated with any acute symptoms and vital signs may be normal.13 Severe hyperkalemia (serum potassium > 6.5 mEq/L) may present clinically with acute extremity paresthesias, muscle weakness and paralysis, heart palpitations, dyspnea, altered mental status, cardiac arrhythmias, and cardiac arrest.30,31 Irregular heart rhythm, decreased deep tendon reflexes, or decreased strength may be revealed on physical exam.3 Individuals with ESRD on hemodialysis seem to tolerate higher levels of potassium than the general population without displaying clinical symptoms. However, these individuals are still susceptible to the cardiac effects of hyperkalemia.32
INITIAL ASSESSMENT
In assessing hyperkalemia, the clinician must perform a focused history and physical exam and review the patient’s medication list, including supplements and dietary habits that impact potassium intake. Potassium-rich foods include meat, fish, milk, almonds, spinach, cantaloupe, bananas, oranges, mushrooms, and potatoes.33 Hyperkalemia may present in association with various medical emergencies. The clinician should have an index of suspicion, depending on the patient’s overall medical profile and presentation, for emergencies such as cardiac ischemia, sepsis, adrenal crisis, DKA, TLS, and digoxin overdose.
The clinician must identify whether an elevated potassium level requires emergent therapy; assessment of vital signs is paramount in determining this. Orthostatic hypotension and tachycardia may hint that the patient is volume depleted. The patient should be examined for signs of hemodynamic shock with the CAB sequence: circulation, airway, breathing.34 Symptoms such as chest pain, shortness of breath, muscle weakness, paralysis, and altered mental status suggest that an expedited evaluation is warranted.
With a serum potassium level > 5.5 mEq/L, urgent electrocardiography should be performed.26 ECG findings observed with serum potassium levels of 5.5-6.5 mEq/L usually include peaked T-waves and prolonged PR intervals (see Figure 2). With potassium levels > 6.5 mEq/L consistent with further cardiac destabilization, the P-wave flattens then disappears, the QRS complex broadens, and sinus bradycardia or ectopic beats may occur.12,26 ST depression, T-wave inversion, or ST elevation also may be seen.12 With serum potassium levels > 7.5 mEq/L, progressive widening of the QRS complex to a sine-wave with bundle branch blocks or fascicular blocks may occur (see Figure 3).26 Without prompt intervention, ventricular fibrillation may ensue.26
An extensive laboratory workup may be necessary to investigate the etiology; this includes a complete blood count, metabolic panel, liver function tests, cardiac enzymes, blood gas analysis, serum/urine osmolality, urinalysis, urine electrolytes, and toxicology screen.13,26 Arterial blood gas (ABG) analysis may show metabolic acidosis with AKI or DKA, or an elevated lactate may occur with sepsis. In patients with hyperglycemia, besides checking for acidosis, obtaining blood/urine ketone levels and a metabolic panel with anion gap to evaluate for DKA is useful.35
When assessing a patient with an elevated creatinine, the GFR at the time of evaluation should be compared with the patient’s baseline GFR to determine chronicity and duration of his/her kidney disease.36 Obtaining a urinalysis and urine electrolytes in addition to the basic metabolic panel can help narrow the etiology.36 A Foley catheter should be placed in cases of urinary retention because without intervention, urinary obstruction may lead to AKI and hyperkalemia. Myoglobinuria on urinalysis and an elevated creatine kinase are diagnostic markers of rhabdomyolysis.18
TLS should be considered in patients who recently received chemotherapy, especially those with proliferative hematologic malignancies, such as acute lymphoblastic leukemia, acute myeloid leukemia, and Burkitt lymphoma.24 In TLS, bloodwork often reveals hyperkalemia along with AKI, an elevated uric acid level, hyperphosphatemia, and hypocalcemia.24
Patients presenting with hyperkalemia, hypotension, hypoglycemia, and hyponatremia may have adrenal insufficiency.20 If insufficiency is suspected, a cortisol level may be checked during morning hours; a low level is often suggestive of this diagnosis.37 Treatment includes daily doses of steroids, and consultation with an endocrinologist is recommended.37
If an elevated potassium level is not accompanied by renal dysfunction, electrolyte imbalances, ECG changes, or inciting medications, pseudohyperkalemia should be considered.38 A repeat lab sample should be checked. Consider obtaining an ABG analysis, as the shorter time interval between drawing the blood sample and the sample analysis reportedly increases the reliability of the resulting potassium level.38
THERAPY
Emergent
Emergent treatment is needed for severe hyperkalemia (see Figure 4). Any hyperkalemia-inciting medications or potassium supplements should be immediately discontinued.39 IV access and cardiac telemetry monitoring should be promptly applied.26
In cases of severe hyperkalemia that involve cardiac arrhythmias, manifestations on ECG, or risk for arrhythmias, calcium gluconate (10 mL IV over 10 min) should be urgently administered, followed by IV insulin in conjunction with dextrose.26 Calcium chloride should be utilized for hyperkalemia in the context of the advanced cardiac life support (ACLS) protocol for cardiac arrest.26 The patient should remain on cardiac telemetry during this treatment to monitor for ventricular fibrillation or other arrhythmias.15 IV calcium does not lower serum potassium but rather antagonizes the effects of potassium on the cardiac cell membranes, helping to prevent or terminate arrhythmias.15,34 It should be noted, however, that firstline treatment for patients who develop hyperkalemia in the setting of digoxin toxicity involves administration of digoxin-specific antibody, while calcium infusion may be utilized later.34 Alternatively, if the patient is dialysis-dependent with ESRD, dialysis may be considered as a prompt initial treatment, with nephrologist consultation.
Administration of 10 U of regular insulin plus 25 g of 50% dextrose via IV will shift potassium intracellularly (see Figure 4). The dextrose will offset the resultant hypoglycemia.31,34 Of note, this treatment is often firstline for moderate to severe hyperkalemia in patients with a stable cardiac rhythm and ECG. Blood glucose should be monitored with a fingerstick within 30 to 60 minutes of infusion and every hour thereafter for up to six hours following insulin administration.34 Potassium levels should be checked every one to two hours after this treatment step until the serum potassium level stabilizes. Thereafter, recheck the levels every four to six hours to gauge whether further treatment is needed.34
Adjunctive
After performing firstline treatment strategies for severe hyperkalemia, there are alternate therapies to consider that can help lower total body potassium. Nebulized albuterol may be used, which pushes potassium into cells; this works in synergy with insulin and glucose.26,33 Sodium bicarbonate may be effective in cases in which the ABG analysis or labs show metabolic acidosis, as this infusion shifts potassium into cells by increasing the blood pH.33
In patients with dehydration, sepsis, TLS, or rhabdomyolysis, administration of IV fluids to maintain appropriate vascular volume is important. However, excessive fluid resuscitation can result in fluid overload, inducing complications such as respiratory failure and worsened renal function.40 A Foley catheter may be placed for strict intake and output monitoring.
The patient’s volume status must be carefully assessed. Hyperkalemia may present in association with heart failure exacerbation or ascites, which are usually hypervolemic states. Loop diuretics may be used to compensate for volume overload and to help remove potassium from the body, but these medications are contraindicated in anuric patients.13,41
Removing total body potassium
After emergent therapy is carried out, potassium may need to be removed from the body through diuresis, hemodialysis, or potassium binders. Loop diuretics or potassium binders may be used to treat mild to moderate hyperkalemia or to continue to stabilize the potassium level after emergent therapy is carried out. If severe hyperkalemia persists with kidney injury or with absence of urine output, hemodialysis is the therapy of choice.13
The potassium binder sodium polystyrene sulfonate (SPS) exchanges sodium for potassium in the intestine.42 This agent is contraindicated if the patient has intestinal obstruction. SPS’s slow onset of action (two to six hours) makes it ineffective as firstline therapy for severe hyperkalemia.3 In addition, SPS has serious but rare adverse effects, more commonly seen in patients who have uremia after kidney transplant or who have had recent abdominal surgery, bowel injury, or intestinal perforation.41 Adverse effects of SPS include aspiration pneumonitis, upper gastrointestinal injury, colonic necrosis, and rectal stenosis.41 However, there have been documented events of colonic necrosis due to SPS in patients without ESRD who have not had abdominal surgery.43,44 In 2009, the FDA advised against concomitant administration of sorbitol with SPS. However, this drug preparation continues to be the only one stocked by many hospital pharmacies.44 Because SPS has potentially harmful adverse effects and generally is not effective in promptly lowering serum potassium, it is prudent for clinicians to implement other management strategies first.44
MONITORING AT-RISK PATIENTS
Patients with a GFR < 45 mL/min/1.73 m2 and a baseline serum potassium level > 4.5 mEq/L are at risk for hyperkalemia while taking an ACEi or an ARB and should be advised to adhere to a potassium-restrictive diet with frequent laboratory checkups.22 Depending on the serum potassium and GFR levels at checkups, these medication doses may need to be reduced or discontinued altogether.
NEW DRUG DEVELOPMENTS
A potassium binder approved for daily use would benefit patients on aggressive heart failure medication regimens, as hyperkalemia commonly occurs with these regimens. As discussed, the widely available potassium binder SPS has been associated with severe gastrointestinal adverse effects, limiting its potential for routine use.44,45 In clinical trials, new potassium binders patiromer and zirconium cyclosilicate (ZS-9) have demonstrated an ability to maintain normokalemia over weeks of therapy with acceptable adverse effect profiles.45 In 2015, patiromer was approved by the FDA as therapy for hyperkalemia.46 An in-depth discussion, which is outside the scope of this article, will be presented by experts in the April 2017 edition of Renal Consult.
CONCLUSION
The best treatment for hyperkalemia is prevention through close surveillance of at-risk patients. Clinicians should be aware of predisposing risk factors for hyperkalemia, as it can have an insidious onset, with symptoms manifesting only when this electrolyte imbalance becomes life-threatening. It is particularly important to recognize when this condition mandates emergent treatment so that critical cardiac arrhythmias can be prevented.26
1. An JN, Lee JP, Jeon HJ, et al. Severe hyperkalemia requiring hospitalization: predictors of mortality. Crit Care. 2012; 16(6):R225.
2. Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis. Adv Physiol Educ. 2016;40(4):480-490.
3. Medford-Davis L, Rafique Z. Derangements of potassium. Emerg Med Clin North Am. 2014;32(2):329-347.
4. Eleftheriadis T, Leivaditis K, Antoniadi G, Liakopoulos V. Differential diagnosis of hyperkalemia: an update to a complex problem. Hippokratia. 2012;16(4):294-302.
5. Raebel M. Hyperkalemia associated with use of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. Cardiovasc Ther. 2012;30(3):156-166.
6. Korgaonkar S, Tilea A, Gillespie BW, et al. Serum potassium and outcomes in CKD: insights from the RRI-CKD Cohort Study. Clin J Am Soc Nephrol. 2010;5(5):762-769.
7. McMahon GM, Mendu ML, Gibbons FK, Christopher KB. Association between hyperkalemia at critical care initiation and mortality. Intensive Care Med. 2012;38(11):1834-1842.
8. Drawz PE, Babineau DC, Rahman M. Metabolic complications in elderly adults with chronic kidney disease. J Am Geriatr Soc. 2012;60(2):310-315.
9. Sarafidis PA, Georgianos PI, Bakris GL. Advances in treatment of hyperkalemia in chronic kidney disease. Expert Opin Pharmacother. 2015;16(14):2205-2215.
10. Einhorn LM, Zhan M, Hsu VD, et al. The frequency of hyperkalemia and its significance in chronic kidney disease. Arch Intern Med. 2009;169(12):1156-1162.
11. Khanagavi J, Gupta T, Aronow WS, et al. Hyperkalemia among hospitalized patients and association between duration of hyperkalemia and outcomes. Arch Med Sci. 2014;10(2):251-257.
12. Berkova M, Berka Z, Topinkova E. Arrhythmias and ECG changes in life threatening hyperkalemia in older patients treated by potassium sparing drugs. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158(1):84-91.
13. Lehnhardt A, Kemper MJ. Pathogenesis, diagnosis and management of hyperkalemia. Pediatr Nephrol. 2011;26(3):377-384.
14. Palmer BF. A physiologic-based approach to the evaluation of a patient with hyperkalemia. Am J Kidney Dis. 2010;56(2):387-393.
15. Mushiyakh Y, Dangaria H, Qavi S, et al. Treatment and pathogenesis of acute hyperkalemia. J Community Hosp Intern Med Perspect. 2012;1(4):7372.
16. Elliott MJ, Ronksley PE, Clase CM, et al. Management of patients with acute hyperkalemia. CMAJ. 2010;182(15):1631-1635.
17. Wiebe N, Klarenbach SW, Allan GM, et al. Potentially preventable hospitalization as a complication of CKD: a cohort study. Am J Kidney Dis. 2014;64(2):230-238.
18. Zutt R, van der Kooi AJ, Linthorst GE, et al. Rhabdomyolysis: review of the literature. Neuromuscul Disord. 2014;24(8):651-659.
19. Zimmerman JL, Shen MC. Rhabdomyolysis. Chest. 2013; 144(3):1058-1065.
20. Khardori R, Castillo D. Endocrine and metabolic changes during sepsis: an update. Med Clin North Am. 2012;96(6):1095-1105.
21. Raff H, Sharma ST, Nieman LK. Physiological basis for the etiology, diagnosis, and treatment of adrenal disorders: Cushing’s syndrome, adrenal insufficiency, and congenital adrenal hyperplasia. Compr Physiol. 2014;4(20):739-769.
22. Lazich I, Bakris GL. Prediction and management of hyperkalemia across the spectrum of chronic kidney disease. Semin Nephrol. 2014;34(3):333-339.
23. Ayach T, Nappo R, Paugh-Miller J, Ross E. Life-threatening hyperkalemia in a patient with normal renal function. Clin Kidney J. 2014;7(1):49-52.
24. Wilson FP, Berns JS. Tumor lysis syndrome: new challenges and recent advances. Adv Chronic Kidney Dis. 2014;21(1):18-26.
25. Asiryatham JR, Moses V, Bjornson L. Errors in potassium measurement: a laboratory perspective for the clinician. N Am J Med Sci. 2013;5(4):255-259.
26. Pepin J, Shields C. Advances in diagnosis and management of hypokalemic and hyperkalemic emergencies. Emerg Med Pract. 2012;14(2):1-17.
27. Vraets A, Lin Y, Callum JL. Transfusion-associated hyperkalemia. Transfus Med Rev. 2011;25(3):184-196.
28. Aboudara MC, Hurst FP, Abbott KC, Perkins RM. Hyperkalemia after packed red blood cell transfusion in trauma patients. J Trauma. 2008;64(2 suppl):S86-S91.
29. Olson J, Talekar M, Sachdev M, et al. Potassium changes associated with blood transfusion in pediatric patients. Am J Clin Pathol. 2013;139(6):800-805.
30. Chon S, Kwak YH, Hwang SS, et al. Severe hyperkalemia can be detected immediately by quantitative electrocardiography and clinical history in patients with symptomatic or extreme bradycardia: a retrospective cross-sectional study. J Crit Care. 2013;28(6):1112.e7-1112.e13.
31. Viera AJ, Wouk N. Potassium disorders: hypokalemia and hyperkalemia. Am Fam Physician. 2015;92(6):487-495.
32. Sanghavi S, Whitling S, Uribarri J. Potassium balance in dialysis patients. Semin Dial. 2013;26(5):597-603.
33. Crawford AH. Hyperkalemia: Recognition and management of a critical electrolyte disturbance. J Infus Nurs. 2014;37(3):167-175.
34. Maxwell AP, Linden K, O’Donnell S, et al. Management of hyperkalemia. J R Coll Physicians Edinb. 2013;43(3):246-251.
35. Seth P, Kaur H, Kaur M. Clinical profile of diabetic ketoacidosis: a prospective study in a tertiary care hospital. J Clin Diagn Res. 2015;9(6):OC01-OC04.
36. Rahman M, Shad F, Smith M. Acute kidney injury: a guide to diagnosis and management. Am Fam Physician. 2012;86(7): 631-639.
37. Puar TH, Stikkelbroeck NM, Smans LC, et al. Adrenal crisis: still a deadly event in the 21st century. Am J Med. 2016;129(3):339.e1-9.
38. Liamis G, Liberopoulos E, Barkas F, Elisaf M. Spurious electrolyte disorders: a diagnostic challenge for clinicians. Am J Nephrol. 2013;38(1):50-57.
39. Kovesdy CP. Management of hyperkalemia: an update for the internist. Am J Med. 2015;128(12):1281-1287.
40. Labib M, Khalid R, Khan A, Khan S. Volume management in the critically ill patient with acute kidney injury. Crit Care Res Pract. 2013;2013:792830.
41. Watson M, Abbott KC, Yuan CM. Damned if you do, damned if you don’t: potassium binding resins in hyperkalemia. Clin J Am Soc Nephrol. 2010;5(10):1723-1726.
42. Nguyen T, Ondrik D, Zhufyak O, et al. Hyperkalemia and potential pitfalls of sodium polystyrene sulfonate. JAAPA. 2015; 28(3):41-45.
43. McGowan CE, Saha S, Resnick MB, Moss SF. Intestinal necrosis due to sodium polystyrene sulfonate (Kayexalate) in sorbitol. South Med J. 2009;102(5):493-497.
44. Sterns RH, Rojas M, Bernstein P, Chennupati S. Ion-exchange resins for the treatment of hyperkalemia: are they safe and effective? J Am Soc Nephrol. 2010;21:733-735.
45. Pitt B, Bakris GL. New potassium binders for the treatment of hyperkalemia: current data and opportunities for the future. Hypertension. 2015;66(4):731-738.
46. Epstein M, Pitt B. Recent advances in pharmacological treatments of hyperkalemia: focus on patiromer. Expert Opin Pharmacother. 2016;17(10):1435-1448.
1. An JN, Lee JP, Jeon HJ, et al. Severe hyperkalemia requiring hospitalization: predictors of mortality. Crit Care. 2012; 16(6):R225.
2. Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis. Adv Physiol Educ. 2016;40(4):480-490.
3. Medford-Davis L, Rafique Z. Derangements of potassium. Emerg Med Clin North Am. 2014;32(2):329-347.
4. Eleftheriadis T, Leivaditis K, Antoniadi G, Liakopoulos V. Differential diagnosis of hyperkalemia: an update to a complex problem. Hippokratia. 2012;16(4):294-302.
5. Raebel M. Hyperkalemia associated with use of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. Cardiovasc Ther. 2012;30(3):156-166.
6. Korgaonkar S, Tilea A, Gillespie BW, et al. Serum potassium and outcomes in CKD: insights from the RRI-CKD Cohort Study. Clin J Am Soc Nephrol. 2010;5(5):762-769.
7. McMahon GM, Mendu ML, Gibbons FK, Christopher KB. Association between hyperkalemia at critical care initiation and mortality. Intensive Care Med. 2012;38(11):1834-1842.
8. Drawz PE, Babineau DC, Rahman M. Metabolic complications in elderly adults with chronic kidney disease. J Am Geriatr Soc. 2012;60(2):310-315.
9. Sarafidis PA, Georgianos PI, Bakris GL. Advances in treatment of hyperkalemia in chronic kidney disease. Expert Opin Pharmacother. 2015;16(14):2205-2215.
10. Einhorn LM, Zhan M, Hsu VD, et al. The frequency of hyperkalemia and its significance in chronic kidney disease. Arch Intern Med. 2009;169(12):1156-1162.
11. Khanagavi J, Gupta T, Aronow WS, et al. Hyperkalemia among hospitalized patients and association between duration of hyperkalemia and outcomes. Arch Med Sci. 2014;10(2):251-257.
12. Berkova M, Berka Z, Topinkova E. Arrhythmias and ECG changes in life threatening hyperkalemia in older patients treated by potassium sparing drugs. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158(1):84-91.
13. Lehnhardt A, Kemper MJ. Pathogenesis, diagnosis and management of hyperkalemia. Pediatr Nephrol. 2011;26(3):377-384.
14. Palmer BF. A physiologic-based approach to the evaluation of a patient with hyperkalemia. Am J Kidney Dis. 2010;56(2):387-393.
15. Mushiyakh Y, Dangaria H, Qavi S, et al. Treatment and pathogenesis of acute hyperkalemia. J Community Hosp Intern Med Perspect. 2012;1(4):7372.
16. Elliott MJ, Ronksley PE, Clase CM, et al. Management of patients with acute hyperkalemia. CMAJ. 2010;182(15):1631-1635.
17. Wiebe N, Klarenbach SW, Allan GM, et al. Potentially preventable hospitalization as a complication of CKD: a cohort study. Am J Kidney Dis. 2014;64(2):230-238.
18. Zutt R, van der Kooi AJ, Linthorst GE, et al. Rhabdomyolysis: review of the literature. Neuromuscul Disord. 2014;24(8):651-659.
19. Zimmerman JL, Shen MC. Rhabdomyolysis. Chest. 2013; 144(3):1058-1065.
20. Khardori R, Castillo D. Endocrine and metabolic changes during sepsis: an update. Med Clin North Am. 2012;96(6):1095-1105.
21. Raff H, Sharma ST, Nieman LK. Physiological basis for the etiology, diagnosis, and treatment of adrenal disorders: Cushing’s syndrome, adrenal insufficiency, and congenital adrenal hyperplasia. Compr Physiol. 2014;4(20):739-769.
22. Lazich I, Bakris GL. Prediction and management of hyperkalemia across the spectrum of chronic kidney disease. Semin Nephrol. 2014;34(3):333-339.
23. Ayach T, Nappo R, Paugh-Miller J, Ross E. Life-threatening hyperkalemia in a patient with normal renal function. Clin Kidney J. 2014;7(1):49-52.
24. Wilson FP, Berns JS. Tumor lysis syndrome: new challenges and recent advances. Adv Chronic Kidney Dis. 2014;21(1):18-26.
25. Asiryatham JR, Moses V, Bjornson L. Errors in potassium measurement: a laboratory perspective for the clinician. N Am J Med Sci. 2013;5(4):255-259.
26. Pepin J, Shields C. Advances in diagnosis and management of hypokalemic and hyperkalemic emergencies. Emerg Med Pract. 2012;14(2):1-17.
27. Vraets A, Lin Y, Callum JL. Transfusion-associated hyperkalemia. Transfus Med Rev. 2011;25(3):184-196.
28. Aboudara MC, Hurst FP, Abbott KC, Perkins RM. Hyperkalemia after packed red blood cell transfusion in trauma patients. J Trauma. 2008;64(2 suppl):S86-S91.
29. Olson J, Talekar M, Sachdev M, et al. Potassium changes associated with blood transfusion in pediatric patients. Am J Clin Pathol. 2013;139(6):800-805.
30. Chon S, Kwak YH, Hwang SS, et al. Severe hyperkalemia can be detected immediately by quantitative electrocardiography and clinical history in patients with symptomatic or extreme bradycardia: a retrospective cross-sectional study. J Crit Care. 2013;28(6):1112.e7-1112.e13.
31. Viera AJ, Wouk N. Potassium disorders: hypokalemia and hyperkalemia. Am Fam Physician. 2015;92(6):487-495.
32. Sanghavi S, Whitling S, Uribarri J. Potassium balance in dialysis patients. Semin Dial. 2013;26(5):597-603.
33. Crawford AH. Hyperkalemia: Recognition and management of a critical electrolyte disturbance. J Infus Nurs. 2014;37(3):167-175.
34. Maxwell AP, Linden K, O’Donnell S, et al. Management of hyperkalemia. J R Coll Physicians Edinb. 2013;43(3):246-251.
35. Seth P, Kaur H, Kaur M. Clinical profile of diabetic ketoacidosis: a prospective study in a tertiary care hospital. J Clin Diagn Res. 2015;9(6):OC01-OC04.
36. Rahman M, Shad F, Smith M. Acute kidney injury: a guide to diagnosis and management. Am Fam Physician. 2012;86(7): 631-639.
37. Puar TH, Stikkelbroeck NM, Smans LC, et al. Adrenal crisis: still a deadly event in the 21st century. Am J Med. 2016;129(3):339.e1-9.
38. Liamis G, Liberopoulos E, Barkas F, Elisaf M. Spurious electrolyte disorders: a diagnostic challenge for clinicians. Am J Nephrol. 2013;38(1):50-57.
39. Kovesdy CP. Management of hyperkalemia: an update for the internist. Am J Med. 2015;128(12):1281-1287.
40. Labib M, Khalid R, Khan A, Khan S. Volume management in the critically ill patient with acute kidney injury. Crit Care Res Pract. 2013;2013:792830.
41. Watson M, Abbott KC, Yuan CM. Damned if you do, damned if you don’t: potassium binding resins in hyperkalemia. Clin J Am Soc Nephrol. 2010;5(10):1723-1726.
42. Nguyen T, Ondrik D, Zhufyak O, et al. Hyperkalemia and potential pitfalls of sodium polystyrene sulfonate. JAAPA. 2015; 28(3):41-45.
43. McGowan CE, Saha S, Resnick MB, Moss SF. Intestinal necrosis due to sodium polystyrene sulfonate (Kayexalate) in sorbitol. South Med J. 2009;102(5):493-497.
44. Sterns RH, Rojas M, Bernstein P, Chennupati S. Ion-exchange resins for the treatment of hyperkalemia: are they safe and effective? J Am Soc Nephrol. 2010;21:733-735.
45. Pitt B, Bakris GL. New potassium binders for the treatment of hyperkalemia: current data and opportunities for the future. Hypertension. 2015;66(4):731-738.
46. Epstein M, Pitt B. Recent advances in pharmacological treatments of hyperkalemia: focus on patiromer. Expert Opin Pharmacother. 2016;17(10):1435-1448.

Bipolar Disorder: Recognizing and Treating in Primary Care
CE/CME No: CR-1702
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Define bipolar disorder according to the DSM-5 criteria.
• Recognize how patients with bipolar disorder can present to their primary care provider.
• Discuss how to perform a clinical and psychiatric evaluation on patients with suspected bipolar disorder.
• Describe the therapeutic options for a patient with bipolar disease in a primary care setting.
FACULTY
Jean Covino is a clinical professor at Pace University-Lenox Hill Hospital in New York City, and she practices at the Medemerge Family Practice Center in Green Brook, New Jersey. Jennifer Hofmann is an Associate Clinical Professor at Pace University-Lenox Hill Hospital in New York City.
The authors have no financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of February 2017.
Article begins on next page >>
Primary care clinicians are often the first point of contact for persons with bipolar disorder. Unfortunately, delays in diagnosis are common, as many of these patients are misdiagnosed with unipolar depression on initial presentation. Since an early and accurate diagnosis may reduce the burden of bipolar disorder and improve outcomes, clinicians should be able to recognize its symptoms and initiate treatment of this deceptive disorder.
Bipolar disorder is a chronic mental illness characterized by fluctuations in mood and energy that manifests as recurrent episodes of manic or depressive symptoms. It is estimated that between 10% and 38% of patients with bipolar disorder receive all their mental health care in a primary care setting.1 Although patients with bipolar disorder often initially present to their primary care provider, they frequently go undiagnosed because of the complexity of the disorder’s symptomatology and a low index of suspicion among primary care providers.2 Comorbid medical conditions and psychiatric issues can also lead to misdiagnoses.
Because primary care providers are often the first point of contact for patients with bipolar disorder, they are well positioned to recognize bipolar symptoms early in the course of the illness. The pure subtypes of bipolar disorder include bipolar I and bipolar II. Clinicians who work in a primary care or emergency department setting should be able to recognize and initiate treatment for these two subtypes while the patient is waiting for a psychiatric evaluation. Accurate early diagnosis of this disabling disorder can reduce morbidity and improve outcomes by allowing for appropriate referral, pharmacotherapy, and psychotherapy.
DEFINITION
According to the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders (DSM-5), bipolar disorder is a mood disorder defined by episodes of mania, hypomania, and major depression.3 Patients with bipolar I disorder experience manic episodes and almost always experience major depressive and hypomanic episodes. Bipolar II disorder is marked by at least one hypomanic episode, at least one major depressive episode (MDE), and the absence of manic episodes.3 A manic episode is at least one week of abnormally and continually elevated, expansive, or irritable mood and increased activity or energy accompanied by at least three of the following symptoms (or four if mood is only irritable): inflated self-esteem, decreased need for sleep, increased talkativeness, flight of ideas or racing thoughts, marked distractibility, increased goal-directed activity or agitation, and excessive involvement in dangerous or high-risk activities (eg, reckless spending or increased sexuality). To be considered a manic episode, the mood disturbance must cause marked impairment in social or occupational functioning, result in hospitalization, or involve psychotic features, and the symptoms cannot be attributable to the effects of drugs or medications or another medical condition.3
In a hypomanic episode, the period of elevated or irritable mood lasts for a shorter duration (at least four days); is associated with a clear, uncharacteristic change in functioning; and is observable by others but does not cause marked impairment, need for hospitalization, or psychosis. MDE is defined by the presence of at least five of nine symptoms for a minimum duration of two weeks and a change from previous functioning: depressed mood, markedly decreased interest or pleasure in activities, significant change in weight or appetite, insomnia or hypersomnia, psychomotor agitation or retardation, fatigue or loss of energy, feelings of worthlessness or excessive guilt, decreased ability to think or concentrate or indecisiveness, and recurrent thoughts of death or suicidality (at least one symptom must be depressed mood or loss of interest or pleasure).3
EPIDEMIOLOGY
Bipolar disorder affects men and women equally. It can occur at any age but is seen most commonly in persons younger than 25.4 The mean age at the first manic/hypomanic or major depressive episode was determined to be 18.2 in bipolar I and 20.3 in bipolar II.5 The lifetime prevalence of bipolar disorder in the United States is around 4%, with one study finding prevalence estimates of 1.0% for bipolar I disorder and 1.1% for bipolar II disorder.5
Bipolar disorder is common among primary care patients with depression. Two studies that explored the risk for bipolar disorder among depressed outpatients in primary care settings found that between 20% and 30% of these patients screened positive for bipolar disorder on the Mood Disorders Questionnaire (MDQ), indicating that a more thorough evaluation for bipolar disorder was needed.6,7 A systematic review of the literature found similar rates of positive results on the MDQ screening measure among primary care patients with depression, a trauma exposure, medically unexplained symptoms, or a psychiatric complaint; bipolar disorder was diagnosed with a structured clinical interview in 3% to 9% of these patients.8 Children of parents with bipolar disorder have a 4% to 15% risk for also being affected.4
CLINICAL PRESENTATION IN PRIMARY CARE
Bipolar patients are often depressed or euthymic for a majority of their lives but can also present in a manic or hypomanic state. In primary care settings, these patients often present with depression (including postpartum depression), which can obscure the diagnosis. Misdiagnosis of bipolar disorder as recurrent unipolar depression occurs in 60% of patients seeking treatment for depression.9
Patients with bipolar disorder who present to primary care usually demonstrate a wide range of mood symptomatology other than depression, including mood swings, anxiety, fatigue, sleep disturbances, and the inability to focus or concentrate. Patients can also present in mixed states. These are characterized by elements of irritability, increased energy, and sleeplessness with depressive features.
Several clues that can assist in detecting bipolar disorder relate to age at onset, family history, mood shifts, seasonality, and atypical depressive symptoms (eg, sleep dysregulation and appetite changes). Although the diagnosis of bipolar disorder is commonly delayed by many years, patients often report significant mood symptoms in their early 20s. In a study that used a self-administered questionnaire to assess the experience of persons living with bipolar disorder, 33% of the respondents were younger than 15 when their symptoms first started, 27% were between 15 and 19, and 39% were 20 or older.9 Parental and family history of bipolar disorder increases risk for the disorder in offspring, so a thorough family history is essential when the disorder is suspected.
Aside from the classic presentation defined by the DSM-5 criteria, patients with bipolar disorder can also exhibit other effects of their illness, such as alcohol-related problems and sexually transmitted or drug-related infections. In patients with bipolar disorder, rates of alcohol use range from 21.4% in adults to 54.5% in adolescents and young adults.10 Social history may reveal relationship and marital issues, financial problems, difficulties keeping a job, and legal problems.9,11 Suicide attempts and completed suicides are significantly more common among persons with bipolar disorder than among the general population.12,13
Comorbidity with at least one other disorder is common in bipolar disorder.5 The most common comorbid personality disorder associated with bipolar disorder is borderline personality disorder, which is characterized by ongoing instability in moods and behavior. Persons with this disorder can experience intense episodes of anger, depression, and anxiety that may last from hours to days. The high prevalence of persistent symptoms despite treatment in bipolar disorder and the unstable and partly remitting course of borderline personality make it difficult to distinguish between the two disorders.14 The frequent mood changes that occur with borderline personality disorder may appear to overlap with the mood swings characterizing bipolar disorder, but the mood episodes in borderline personality disorder are of shorter duration than those in bipolar disorder. Other common comorbid disorders seen in patients with bipolar disorder include substance abuse disorders, anxiety disorders (especially panic disorder, generalized anxiety disorder, and obsessive-compulsive disorder), and attention-deficit/hyperactivity disorder.5
Primary care providers should be aware of other common comorbidities that may be present in patients with bipolar disorder. These patients commonly experience medical problems such as diabetes, obesity, and metabolic syndrome, which all lead to increased cardiovascular risk.15-17
CLINICAL EVALUATION
The initial clinical evaluation of the patient should include a thorough medical, social, family, and psychiatric history. Medical conditions that may mimic bipolar disorder include neurologic conditions (eg, partial seizures, neoplasm, strokes, dementia, delirium) and endocrine disorders (eg, Cushing disease, hyperthyroidism/hypothyroidism), as well as vitamin deficiencies (B12, folate, niacin, thiamine) and drug and substance use/misuse (alcohol, drugs including antidepressants and stimulants).4 All patients should have a baseline complete physical examination, including neurologic and mental status examinations. Diagnostic tests to assess for potential differential diagnoses and evaluate baseline levels include the following:
- Basic metabolic panel, including fasting glucose, to evaluate electrolytes and risk for diabetes or Cushing disease, and to assess baseline renal function
- Thyroid function tests
- Complete blood count to assess status prior to anticonvulsant treatment (eg, carbamazepine)
- Pregnancy test if applicable (prior to use of medications)
- Liver function tests to assess baseline measurements prior to use of medications
- Electrocardiography in patients older than 40 to establish baseline and assess QTc interval, especially with use of antipsychotics and carbamazepine
- Urine toxicology screen (to rule out substance abuse).
PSYCHIATRIC EVALUATION
Psychiatric evaluation should focus on age at onset of symptoms, the presence of hypomanic or manic symptoms, prior response to antidepressants, course of the disease including history and duration of depression or manic/hypomanic episodes, and sleep disturbances (increased during depressive episodes and significantly decreased during manic episodes). It is important to assess for a history of self-harm, suicidal ideation, suicide attempts, hospitalizations, legal issues, multiple career shifts, marriage and relationship issues, and smoking and alcohol/substance misuse. Patients with severe manic or depressive episodes may experience psychotic features such as grandiose or paranoid delusions and hallucinations. A history of symptoms from close family members or friends can assist in the diagnosis of bipolar patients.
The use of DSM-5 criteria, as summarized earlier, improves the accuracy of bipolar diagnosis.3 In addition, validated tools are available to help clinicians screen for bipolar disorder, although it is important to remember that a positive screening result is not sufficient to establish a bipolar disorder diagnosis. A widely used instrument that has been validated for screening for bipolar disorder is the MDQ (available at www.dbsalliance.org/pdfs/MDQ.pdf). This self-report questionnaire consists of 15 questions that assess hypomanic or manic symptoms and functional impairment. The first 13 questions of the MDQ screen for a lifetime history of DSM-based hypomanic or manic symptoms. The last two questions ask whether these symptoms occurred at the same time and whether they caused dysfunction in various domains, such as work and family life. The MDQ is considered positive if a patient endorses at least seven of the symptom items, indicates that symptoms have occurred at the same time, and rates their dysfunction in life domains as “moderate” or “serious.” As a screening tool, the MDQ has a reported sensitivity of 73% and a specificity of 90% for bipolar disorder.11 This questionnaire can and should be used by primary care providers to help determine if their patient is at risk and requires a comprehensive evaluation for bipolar disorder.
Notably, even after a clinician has properly diagnosed bipolar disorder, patients and family members are often reluctant to commence treatment due to the stigma associated with mental health disorders.18 To help offset the effects of stigma, patients should be referred for psychologic counseling, including family counseling.
MANAGEMENT
Management of bipolar disorder in the primary care setting includes psychiatric and psychologic counseling referrals. Primary care providers must know the medications used to treat bipolar disorder and their related adverse effects, toxicities, warnings, and drug interactions, as they may treat bipolar patients for other medical conditions. Early diagnosis and treatment/referral can improve prognosis and reduce the risk for relapse and subsequent disability.19 Inpatient management is generally recommended for severe manic episodes, psychotic episodes, patients who present a danger to themselves or others, and patients with suicidal or homicidal ideations/actions.
Medications are the primary treatment for all stages of bipolar disorder, and choice of medications is based on stage, previous response, and adverse effect profiles (see Table 1).2,4,20 Generally, antidepressants (serotonin and norepinephrine reuptake inhibitors [SNRIs] and selective serotonin reuptake inhibitors [SSRIs]) should be avoided or should be used with an effective antimanic/mood stabilizer. Many patients with severe bipolar symptoms require more than two medications, and it is imperative that all primary care providers understand that often one drug alone is not sufficient treatment for patients with bipolar disorder. For less severe manic or hypomanic states, monotherapy with antipsychotics may be effective.

Medications for severe acute manic episodes generally include the mood stabilizers lithium, valproate, or carbamazepine in conjunction with an antipsychotic, such as haloperidol, or an atypical antipsychotic, such as asenapine, aripiprazole, olanzapine, quetiapine, or risperidone.2 The goal of initial therapy in patients with acute mania is rapid resolution of symptoms and restoration of adequate sleep. Lithium has a slower onset of action than valproate and carbamazepine and requires titration and monitoring. Valproate and carbamazepine have a faster onset of action but are less effective than lithium.2 Atypical antipsychotics have a more rapid onset of action than mood stabilizers and are effective in controlling acute manic symptoms, psychosis, and sleep disturbances. Patients with severe acute mania may require hospital admission for stabilization, for their safety and the safety of others.
Acute bipolar depressive episodes can be treated with several different medication options, including combination olanzapine and fluoxetine; the atypical antipsychotic quetiapine; and recently lurasidone, alone or in combination with lithium or valproate. Lamotrigine is more effective for maintenance and prevention of depressive episodes than for treatment of acute episodes, and it is also indicated for treatment of bipolar II. Valproate is more effective than lithium for mixed states and can be titrated more rapidly for faster antimanic effects.4
Generally, due to the high rate of recurrence, maintenance medications should be continued indefinitely. Maintenance medications include the mood stabilizers, lamotrigine, and many of the antipsychotics, including olanzapine.4 Adherence to medications is essential in management of bipolar disorder and can decrease the risk for relapses and destabilization. Poor adherence to medications is common, however, with rates reported at approximately 50%.21 Patient and family education, as well as psychotherapy, can improve adherence rates.2 Primary care providers should educate patients and family members about medication options and adverse effects and must stress the need for adherence to prevent relapse. Providers should also understand the safety profile of mood stabilizers and antipsychotics and the required monitoring of laboratory tests for patients on these medications.
Psychosocial treatments are an elemental component of management. Patients should be referred early for psychologic treatments including, but not limited to, family therapy, group therapy, cognitive-behavioral therapy, and psychotherapy, which have been shown to improve daily functioning, recognition of recurrences, and medication adherence.2 The rate of relapse is significantly lower in patients receiving combination psychotherapy and pharmacotherapy.22
Clinical pearls that every primary care provider should know about bipolar disorder are summarized in Table 2.

CONCLUSION
Given the substantial impact of bipolar disorder on patients and the community, primary care clinicians must maintain a high index of suspicion for this disorder. An early and accurate diagnosis may reduce the burden of bipolar disorder and improve outcomes. However, diagnosing and treating patients with bipolar disorder is challenging for primary care and specialty clinicians alike. In particular, establishing a diagnosis can be difficult, even for the most seasoned clinician, due to the diversity of symptoms. Nonetheless, diagnosing bipolar disorder, initiating treatment, and monitoring and referring patients when necessary are certainly within the purview of the primary care provider.
1. Kilbourne AM, Goodrich DE, O’Donnell AN, Miller CJ. Integrating bipolar disorder management in primary care. Curr Psychiatry Rep. 2012;14:687-695.
2. Culpepper L. The diagnosis and treatment of bipolar disorder: decision making in primary care. Prim Care Companion CNS Disord. 2014;16(3): doi 10.4088/PCC.13r01609.
3. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
4. Price AL, Marzani-Nissen GR. Bipolar disorders: a review. Am Fam Physician. 2012;85:483-493.
5. Merikangas KR, Akiskal HS, Angst J, et al. Lifetime and 12-month prevalence of bipolar spectrum disorder in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2007;64:543-552.
6. Calabrese JR, Muzina DJ, Kemp DE, et al. Predictors of bipolar disorder risk among patients currently treated for major depression. MedGenMed. 2006;8(3):38.
7. Hirschfeld RM, Cass AR, Holt DC, Carlson CA. Screening for bipolar disorder in patients treated for depression in a family medicine clinic. J Am Board Fam Pract. 2005;18(4):233-239.
8. Cerimele JM, Chwastiak LA, Dodson S, Katon WJ. The prevalence of bipolar disorder in primary care patients with depression or other psychiatric complaints: a systematic review. Psychosomatics. 2013;54(6):515-524.
9. Hirschfeld RM, Lewis L, Vornik LA. Perceptions and impact of bipolar disorder: how far have we really come? Results of the National Depressive and Manic-depressive Association 2000 survey of individuals with bipolar disorder. J Clin Psychiatry. 2003; 64:161-174.
10. Pini S, de Queiroz V, Pagnin D, et al. Prevalence and burden of bipolar disorders in European countries. Eur Neuropsychopharmacol. 2005;15(4):425-434.
11. Piver A, Yatham LN, Lam RW. Bipolar spectrum disorders: new perspectives. Can Fam Physician. 2002;48:896-904.
12. Eroglu MZ, Karakus G, Tamam L. Bipolar disorder and suicide. J Psychiatry Neurol Sci. 2013;26:139-147.
13. Simon GE, Hunkeler E, Fireman B, Lee JY, Savarino J. Risk of suicide attempt and suicide death in patients treated for bipolar disorder. Bipolar Disord. 2007;9:526-530.
14. Marcinko D, Vuksan-Cusa B. Borderline personality disorder and bipolar disorder comorbidity in suicidal patients: diagnostic and therapeutic challenges. Psychiatr Danub. 2009;21:386-390.
15. Chwastiak LA, Rosenheck RA, Kazis LE. Association of psychiatric illness and obesity, physical inactivity, and smoking among a national sample of veterans. Psychosomatics. 2011;52:230-236.
16. Vancampfort D, Vansteelandt K, Correll CU, et al. Metabolic syndrome and metabolic abnormalities in bipolar disorder: a meta-analysis of prevalence rates and moderators. Am J Psychiatry. 2013; 170:265-274.
17. Fiedorowicz JG, Solomon DA, Endicott J, et al. Manic/hypomanic symptom burden and cardiovascular mortality in bipolar disorder. Psychosom Med. 2009;71(6):598-606.
18. Hawke LD, Parikh SV, Michalak EE. Stigma and bipolar disorder: A review of the literature. J Affect Disord. 2013; 150:181-191.
19. Berk M, Brnabic A, Dodd S, et al. Does stage of illness impact treatment response in bipolar disorder? Empirical treatment data and their implication for the staging model and early intervention. Bipolar Disord. 2011;13(1):87-98.
20. Tegretol (carbamazepine) [package insert]. East Hanover, NJ: Novartis; 2015. www.pharma.us.novartis.com/product/pi/pdf/tegretol.pdf. Accessed November 18, 2016.
21. Arvilommi P, Suominen K, Mantere O, et al. Predictors of adherence to psychopharmacological and psychosocial treatment in bipolar I or II disorders—an 18-month prospective study. J Affect Disord. 2014;155:110-117.
22. Leclerc E, Mansur RB, Brietzke E. Determinants of adherence to treatment in bipolar disorder: a comprehensive review. J Affect Disord. 2013;149:247-252.
CE/CME No: CR-1702
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Define bipolar disorder according to the DSM-5 criteria.
• Recognize how patients with bipolar disorder can present to their primary care provider.
• Discuss how to perform a clinical and psychiatric evaluation on patients with suspected bipolar disorder.
• Describe the therapeutic options for a patient with bipolar disease in a primary care setting.
FACULTY
Jean Covino is a clinical professor at Pace University-Lenox Hill Hospital in New York City, and she practices at the Medemerge Family Practice Center in Green Brook, New Jersey. Jennifer Hofmann is an Associate Clinical Professor at Pace University-Lenox Hill Hospital in New York City.
The authors have no financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of February 2017.
Article begins on next page >>
Primary care clinicians are often the first point of contact for persons with bipolar disorder. Unfortunately, delays in diagnosis are common, as many of these patients are misdiagnosed with unipolar depression on initial presentation. Since an early and accurate diagnosis may reduce the burden of bipolar disorder and improve outcomes, clinicians should be able to recognize its symptoms and initiate treatment of this deceptive disorder.
Bipolar disorder is a chronic mental illness characterized by fluctuations in mood and energy that manifests as recurrent episodes of manic or depressive symptoms. It is estimated that between 10% and 38% of patients with bipolar disorder receive all their mental health care in a primary care setting.1 Although patients with bipolar disorder often initially present to their primary care provider, they frequently go undiagnosed because of the complexity of the disorder’s symptomatology and a low index of suspicion among primary care providers.2 Comorbid medical conditions and psychiatric issues can also lead to misdiagnoses.
Because primary care providers are often the first point of contact for patients with bipolar disorder, they are well positioned to recognize bipolar symptoms early in the course of the illness. The pure subtypes of bipolar disorder include bipolar I and bipolar II. Clinicians who work in a primary care or emergency department setting should be able to recognize and initiate treatment for these two subtypes while the patient is waiting for a psychiatric evaluation. Accurate early diagnosis of this disabling disorder can reduce morbidity and improve outcomes by allowing for appropriate referral, pharmacotherapy, and psychotherapy.
DEFINITION
According to the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders (DSM-5), bipolar disorder is a mood disorder defined by episodes of mania, hypomania, and major depression.3 Patients with bipolar I disorder experience manic episodes and almost always experience major depressive and hypomanic episodes. Bipolar II disorder is marked by at least one hypomanic episode, at least one major depressive episode (MDE), and the absence of manic episodes.3 A manic episode is at least one week of abnormally and continually elevated, expansive, or irritable mood and increased activity or energy accompanied by at least three of the following symptoms (or four if mood is only irritable): inflated self-esteem, decreased need for sleep, increased talkativeness, flight of ideas or racing thoughts, marked distractibility, increased goal-directed activity or agitation, and excessive involvement in dangerous or high-risk activities (eg, reckless spending or increased sexuality). To be considered a manic episode, the mood disturbance must cause marked impairment in social or occupational functioning, result in hospitalization, or involve psychotic features, and the symptoms cannot be attributable to the effects of drugs or medications or another medical condition.3
In a hypomanic episode, the period of elevated or irritable mood lasts for a shorter duration (at least four days); is associated with a clear, uncharacteristic change in functioning; and is observable by others but does not cause marked impairment, need for hospitalization, or psychosis. MDE is defined by the presence of at least five of nine symptoms for a minimum duration of two weeks and a change from previous functioning: depressed mood, markedly decreased interest or pleasure in activities, significant change in weight or appetite, insomnia or hypersomnia, psychomotor agitation or retardation, fatigue or loss of energy, feelings of worthlessness or excessive guilt, decreased ability to think or concentrate or indecisiveness, and recurrent thoughts of death or suicidality (at least one symptom must be depressed mood or loss of interest or pleasure).3
EPIDEMIOLOGY
Bipolar disorder affects men and women equally. It can occur at any age but is seen most commonly in persons younger than 25.4 The mean age at the first manic/hypomanic or major depressive episode was determined to be 18.2 in bipolar I and 20.3 in bipolar II.5 The lifetime prevalence of bipolar disorder in the United States is around 4%, with one study finding prevalence estimates of 1.0% for bipolar I disorder and 1.1% for bipolar II disorder.5
Bipolar disorder is common among primary care patients with depression. Two studies that explored the risk for bipolar disorder among depressed outpatients in primary care settings found that between 20% and 30% of these patients screened positive for bipolar disorder on the Mood Disorders Questionnaire (MDQ), indicating that a more thorough evaluation for bipolar disorder was needed.6,7 A systematic review of the literature found similar rates of positive results on the MDQ screening measure among primary care patients with depression, a trauma exposure, medically unexplained symptoms, or a psychiatric complaint; bipolar disorder was diagnosed with a structured clinical interview in 3% to 9% of these patients.8 Children of parents with bipolar disorder have a 4% to 15% risk for also being affected.4
CLINICAL PRESENTATION IN PRIMARY CARE
Bipolar patients are often depressed or euthymic for a majority of their lives but can also present in a manic or hypomanic state. In primary care settings, these patients often present with depression (including postpartum depression), which can obscure the diagnosis. Misdiagnosis of bipolar disorder as recurrent unipolar depression occurs in 60% of patients seeking treatment for depression.9
Patients with bipolar disorder who present to primary care usually demonstrate a wide range of mood symptomatology other than depression, including mood swings, anxiety, fatigue, sleep disturbances, and the inability to focus or concentrate. Patients can also present in mixed states. These are characterized by elements of irritability, increased energy, and sleeplessness with depressive features.
Several clues that can assist in detecting bipolar disorder relate to age at onset, family history, mood shifts, seasonality, and atypical depressive symptoms (eg, sleep dysregulation and appetite changes). Although the diagnosis of bipolar disorder is commonly delayed by many years, patients often report significant mood symptoms in their early 20s. In a study that used a self-administered questionnaire to assess the experience of persons living with bipolar disorder, 33% of the respondents were younger than 15 when their symptoms first started, 27% were between 15 and 19, and 39% were 20 or older.9 Parental and family history of bipolar disorder increases risk for the disorder in offspring, so a thorough family history is essential when the disorder is suspected.
Aside from the classic presentation defined by the DSM-5 criteria, patients with bipolar disorder can also exhibit other effects of their illness, such as alcohol-related problems and sexually transmitted or drug-related infections. In patients with bipolar disorder, rates of alcohol use range from 21.4% in adults to 54.5% in adolescents and young adults.10 Social history may reveal relationship and marital issues, financial problems, difficulties keeping a job, and legal problems.9,11 Suicide attempts and completed suicides are significantly more common among persons with bipolar disorder than among the general population.12,13
Comorbidity with at least one other disorder is common in bipolar disorder.5 The most common comorbid personality disorder associated with bipolar disorder is borderline personality disorder, which is characterized by ongoing instability in moods and behavior. Persons with this disorder can experience intense episodes of anger, depression, and anxiety that may last from hours to days. The high prevalence of persistent symptoms despite treatment in bipolar disorder and the unstable and partly remitting course of borderline personality make it difficult to distinguish between the two disorders.14 The frequent mood changes that occur with borderline personality disorder may appear to overlap with the mood swings characterizing bipolar disorder, but the mood episodes in borderline personality disorder are of shorter duration than those in bipolar disorder. Other common comorbid disorders seen in patients with bipolar disorder include substance abuse disorders, anxiety disorders (especially panic disorder, generalized anxiety disorder, and obsessive-compulsive disorder), and attention-deficit/hyperactivity disorder.5
Primary care providers should be aware of other common comorbidities that may be present in patients with bipolar disorder. These patients commonly experience medical problems such as diabetes, obesity, and metabolic syndrome, which all lead to increased cardiovascular risk.15-17
CLINICAL EVALUATION
The initial clinical evaluation of the patient should include a thorough medical, social, family, and psychiatric history. Medical conditions that may mimic bipolar disorder include neurologic conditions (eg, partial seizures, neoplasm, strokes, dementia, delirium) and endocrine disorders (eg, Cushing disease, hyperthyroidism/hypothyroidism), as well as vitamin deficiencies (B12, folate, niacin, thiamine) and drug and substance use/misuse (alcohol, drugs including antidepressants and stimulants).4 All patients should have a baseline complete physical examination, including neurologic and mental status examinations. Diagnostic tests to assess for potential differential diagnoses and evaluate baseline levels include the following:
- Basic metabolic panel, including fasting glucose, to evaluate electrolytes and risk for diabetes or Cushing disease, and to assess baseline renal function
- Thyroid function tests
- Complete blood count to assess status prior to anticonvulsant treatment (eg, carbamazepine)
- Pregnancy test if applicable (prior to use of medications)
- Liver function tests to assess baseline measurements prior to use of medications
- Electrocardiography in patients older than 40 to establish baseline and assess QTc interval, especially with use of antipsychotics and carbamazepine
- Urine toxicology screen (to rule out substance abuse).
PSYCHIATRIC EVALUATION
Psychiatric evaluation should focus on age at onset of symptoms, the presence of hypomanic or manic symptoms, prior response to antidepressants, course of the disease including history and duration of depression or manic/hypomanic episodes, and sleep disturbances (increased during depressive episodes and significantly decreased during manic episodes). It is important to assess for a history of self-harm, suicidal ideation, suicide attempts, hospitalizations, legal issues, multiple career shifts, marriage and relationship issues, and smoking and alcohol/substance misuse. Patients with severe manic or depressive episodes may experience psychotic features such as grandiose or paranoid delusions and hallucinations. A history of symptoms from close family members or friends can assist in the diagnosis of bipolar patients.
The use of DSM-5 criteria, as summarized earlier, improves the accuracy of bipolar diagnosis.3 In addition, validated tools are available to help clinicians screen for bipolar disorder, although it is important to remember that a positive screening result is not sufficient to establish a bipolar disorder diagnosis. A widely used instrument that has been validated for screening for bipolar disorder is the MDQ (available at www.dbsalliance.org/pdfs/MDQ.pdf). This self-report questionnaire consists of 15 questions that assess hypomanic or manic symptoms and functional impairment. The first 13 questions of the MDQ screen for a lifetime history of DSM-based hypomanic or manic symptoms. The last two questions ask whether these symptoms occurred at the same time and whether they caused dysfunction in various domains, such as work and family life. The MDQ is considered positive if a patient endorses at least seven of the symptom items, indicates that symptoms have occurred at the same time, and rates their dysfunction in life domains as “moderate” or “serious.” As a screening tool, the MDQ has a reported sensitivity of 73% and a specificity of 90% for bipolar disorder.11 This questionnaire can and should be used by primary care providers to help determine if their patient is at risk and requires a comprehensive evaluation for bipolar disorder.
Notably, even after a clinician has properly diagnosed bipolar disorder, patients and family members are often reluctant to commence treatment due to the stigma associated with mental health disorders.18 To help offset the effects of stigma, patients should be referred for psychologic counseling, including family counseling.
MANAGEMENT
Management of bipolar disorder in the primary care setting includes psychiatric and psychologic counseling referrals. Primary care providers must know the medications used to treat bipolar disorder and their related adverse effects, toxicities, warnings, and drug interactions, as they may treat bipolar patients for other medical conditions. Early diagnosis and treatment/referral can improve prognosis and reduce the risk for relapse and subsequent disability.19 Inpatient management is generally recommended for severe manic episodes, psychotic episodes, patients who present a danger to themselves or others, and patients with suicidal or homicidal ideations/actions.
Medications are the primary treatment for all stages of bipolar disorder, and choice of medications is based on stage, previous response, and adverse effect profiles (see Table 1).2,4,20 Generally, antidepressants (serotonin and norepinephrine reuptake inhibitors [SNRIs] and selective serotonin reuptake inhibitors [SSRIs]) should be avoided or should be used with an effective antimanic/mood stabilizer. Many patients with severe bipolar symptoms require more than two medications, and it is imperative that all primary care providers understand that often one drug alone is not sufficient treatment for patients with bipolar disorder. For less severe manic or hypomanic states, monotherapy with antipsychotics may be effective.
Medications for severe acute manic episodes generally include the mood stabilizers lithium, valproate, or carbamazepine in conjunction with an antipsychotic, such as haloperidol, or an atypical antipsychotic, such as asenapine, aripiprazole, olanzapine, quetiapine, or risperidone.2 The goal of initial therapy in patients with acute mania is rapid resolution of symptoms and restoration of adequate sleep. Lithium has a slower onset of action than valproate and carbamazepine and requires titration and monitoring. Valproate and carbamazepine have a faster onset of action but are less effective than lithium.2 Atypical antipsychotics have a more rapid onset of action than mood stabilizers and are effective in controlling acute manic symptoms, psychosis, and sleep disturbances. Patients with severe acute mania may require hospital admission for stabilization, for their safety and the safety of others.
Acute bipolar depressive episodes can be treated with several different medication options, including combination olanzapine and fluoxetine; the atypical antipsychotic quetiapine; and recently lurasidone, alone or in combination with lithium or valproate. Lamotrigine is more effective for maintenance and prevention of depressive episodes than for treatment of acute episodes, and it is also indicated for treatment of bipolar II. Valproate is more effective than lithium for mixed states and can be titrated more rapidly for faster antimanic effects.4
Generally, due to the high rate of recurrence, maintenance medications should be continued indefinitely. Maintenance medications include the mood stabilizers, lamotrigine, and many of the antipsychotics, including olanzapine.4 Adherence to medications is essential in management of bipolar disorder and can decrease the risk for relapses and destabilization. Poor adherence to medications is common, however, with rates reported at approximately 50%.21 Patient and family education, as well as psychotherapy, can improve adherence rates.2 Primary care providers should educate patients and family members about medication options and adverse effects and must stress the need for adherence to prevent relapse. Providers should also understand the safety profile of mood stabilizers and antipsychotics and the required monitoring of laboratory tests for patients on these medications.
Psychosocial treatments are an elemental component of management. Patients should be referred early for psychologic treatments including, but not limited to, family therapy, group therapy, cognitive-behavioral therapy, and psychotherapy, which have been shown to improve daily functioning, recognition of recurrences, and medication adherence.2 The rate of relapse is significantly lower in patients receiving combination psychotherapy and pharmacotherapy.22
Clinical pearls that every primary care provider should know about bipolar disorder are summarized in Table 2.
CONCLUSION
Given the substantial impact of bipolar disorder on patients and the community, primary care clinicians must maintain a high index of suspicion for this disorder. An early and accurate diagnosis may reduce the burden of bipolar disorder and improve outcomes. However, diagnosing and treating patients with bipolar disorder is challenging for primary care and specialty clinicians alike. In particular, establishing a diagnosis can be difficult, even for the most seasoned clinician, due to the diversity of symptoms. Nonetheless, diagnosing bipolar disorder, initiating treatment, and monitoring and referring patients when necessary are certainly within the purview of the primary care provider.
CE/CME No: CR-1702
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Define bipolar disorder according to the DSM-5 criteria.
• Recognize how patients with bipolar disorder can present to their primary care provider.
• Discuss how to perform a clinical and psychiatric evaluation on patients with suspected bipolar disorder.
• Describe the therapeutic options for a patient with bipolar disease in a primary care setting.
FACULTY
Jean Covino is a clinical professor at Pace University-Lenox Hill Hospital in New York City, and she practices at the Medemerge Family Practice Center in Green Brook, New Jersey. Jennifer Hofmann is an Associate Clinical Professor at Pace University-Lenox Hill Hospital in New York City.
The authors have no financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of February 2017.
Article begins on next page >>
Primary care clinicians are often the first point of contact for persons with bipolar disorder. Unfortunately, delays in diagnosis are common, as many of these patients are misdiagnosed with unipolar depression on initial presentation. Since an early and accurate diagnosis may reduce the burden of bipolar disorder and improve outcomes, clinicians should be able to recognize its symptoms and initiate treatment of this deceptive disorder.
Bipolar disorder is a chronic mental illness characterized by fluctuations in mood and energy that manifests as recurrent episodes of manic or depressive symptoms. It is estimated that between 10% and 38% of patients with bipolar disorder receive all their mental health care in a primary care setting.1 Although patients with bipolar disorder often initially present to their primary care provider, they frequently go undiagnosed because of the complexity of the disorder’s symptomatology and a low index of suspicion among primary care providers.2 Comorbid medical conditions and psychiatric issues can also lead to misdiagnoses.
Because primary care providers are often the first point of contact for patients with bipolar disorder, they are well positioned to recognize bipolar symptoms early in the course of the illness. The pure subtypes of bipolar disorder include bipolar I and bipolar II. Clinicians who work in a primary care or emergency department setting should be able to recognize and initiate treatment for these two subtypes while the patient is waiting for a psychiatric evaluation. Accurate early diagnosis of this disabling disorder can reduce morbidity and improve outcomes by allowing for appropriate referral, pharmacotherapy, and psychotherapy.
DEFINITION
According to the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders (DSM-5), bipolar disorder is a mood disorder defined by episodes of mania, hypomania, and major depression.3 Patients with bipolar I disorder experience manic episodes and almost always experience major depressive and hypomanic episodes. Bipolar II disorder is marked by at least one hypomanic episode, at least one major depressive episode (MDE), and the absence of manic episodes.3 A manic episode is at least one week of abnormally and continually elevated, expansive, or irritable mood and increased activity or energy accompanied by at least three of the following symptoms (or four if mood is only irritable): inflated self-esteem, decreased need for sleep, increased talkativeness, flight of ideas or racing thoughts, marked distractibility, increased goal-directed activity or agitation, and excessive involvement in dangerous or high-risk activities (eg, reckless spending or increased sexuality). To be considered a manic episode, the mood disturbance must cause marked impairment in social or occupational functioning, result in hospitalization, or involve psychotic features, and the symptoms cannot be attributable to the effects of drugs or medications or another medical condition.3
In a hypomanic episode, the period of elevated or irritable mood lasts for a shorter duration (at least four days); is associated with a clear, uncharacteristic change in functioning; and is observable by others but does not cause marked impairment, need for hospitalization, or psychosis. MDE is defined by the presence of at least five of nine symptoms for a minimum duration of two weeks and a change from previous functioning: depressed mood, markedly decreased interest or pleasure in activities, significant change in weight or appetite, insomnia or hypersomnia, psychomotor agitation or retardation, fatigue or loss of energy, feelings of worthlessness or excessive guilt, decreased ability to think or concentrate or indecisiveness, and recurrent thoughts of death or suicidality (at least one symptom must be depressed mood or loss of interest or pleasure).3
EPIDEMIOLOGY
Bipolar disorder affects men and women equally. It can occur at any age but is seen most commonly in persons younger than 25.4 The mean age at the first manic/hypomanic or major depressive episode was determined to be 18.2 in bipolar I and 20.3 in bipolar II.5 The lifetime prevalence of bipolar disorder in the United States is around 4%, with one study finding prevalence estimates of 1.0% for bipolar I disorder and 1.1% for bipolar II disorder.5
Bipolar disorder is common among primary care patients with depression. Two studies that explored the risk for bipolar disorder among depressed outpatients in primary care settings found that between 20% and 30% of these patients screened positive for bipolar disorder on the Mood Disorders Questionnaire (MDQ), indicating that a more thorough evaluation for bipolar disorder was needed.6,7 A systematic review of the literature found similar rates of positive results on the MDQ screening measure among primary care patients with depression, a trauma exposure, medically unexplained symptoms, or a psychiatric complaint; bipolar disorder was diagnosed with a structured clinical interview in 3% to 9% of these patients.8 Children of parents with bipolar disorder have a 4% to 15% risk for also being affected.4
CLINICAL PRESENTATION IN PRIMARY CARE
Bipolar patients are often depressed or euthymic for a majority of their lives but can also present in a manic or hypomanic state. In primary care settings, these patients often present with depression (including postpartum depression), which can obscure the diagnosis. Misdiagnosis of bipolar disorder as recurrent unipolar depression occurs in 60% of patients seeking treatment for depression.9
Patients with bipolar disorder who present to primary care usually demonstrate a wide range of mood symptomatology other than depression, including mood swings, anxiety, fatigue, sleep disturbances, and the inability to focus or concentrate. Patients can also present in mixed states. These are characterized by elements of irritability, increased energy, and sleeplessness with depressive features.
Several clues that can assist in detecting bipolar disorder relate to age at onset, family history, mood shifts, seasonality, and atypical depressive symptoms (eg, sleep dysregulation and appetite changes). Although the diagnosis of bipolar disorder is commonly delayed by many years, patients often report significant mood symptoms in their early 20s. In a study that used a self-administered questionnaire to assess the experience of persons living with bipolar disorder, 33% of the respondents were younger than 15 when their symptoms first started, 27% were between 15 and 19, and 39% were 20 or older.9 Parental and family history of bipolar disorder increases risk for the disorder in offspring, so a thorough family history is essential when the disorder is suspected.
Aside from the classic presentation defined by the DSM-5 criteria, patients with bipolar disorder can also exhibit other effects of their illness, such as alcohol-related problems and sexually transmitted or drug-related infections. In patients with bipolar disorder, rates of alcohol use range from 21.4% in adults to 54.5% in adolescents and young adults.10 Social history may reveal relationship and marital issues, financial problems, difficulties keeping a job, and legal problems.9,11 Suicide attempts and completed suicides are significantly more common among persons with bipolar disorder than among the general population.12,13
Comorbidity with at least one other disorder is common in bipolar disorder.5 The most common comorbid personality disorder associated with bipolar disorder is borderline personality disorder, which is characterized by ongoing instability in moods and behavior. Persons with this disorder can experience intense episodes of anger, depression, and anxiety that may last from hours to days. The high prevalence of persistent symptoms despite treatment in bipolar disorder and the unstable and partly remitting course of borderline personality make it difficult to distinguish between the two disorders.14 The frequent mood changes that occur with borderline personality disorder may appear to overlap with the mood swings characterizing bipolar disorder, but the mood episodes in borderline personality disorder are of shorter duration than those in bipolar disorder. Other common comorbid disorders seen in patients with bipolar disorder include substance abuse disorders, anxiety disorders (especially panic disorder, generalized anxiety disorder, and obsessive-compulsive disorder), and attention-deficit/hyperactivity disorder.5
Primary care providers should be aware of other common comorbidities that may be present in patients with bipolar disorder. These patients commonly experience medical problems such as diabetes, obesity, and metabolic syndrome, which all lead to increased cardiovascular risk.15-17
CLINICAL EVALUATION
The initial clinical evaluation of the patient should include a thorough medical, social, family, and psychiatric history. Medical conditions that may mimic bipolar disorder include neurologic conditions (eg, partial seizures, neoplasm, strokes, dementia, delirium) and endocrine disorders (eg, Cushing disease, hyperthyroidism/hypothyroidism), as well as vitamin deficiencies (B12, folate, niacin, thiamine) and drug and substance use/misuse (alcohol, drugs including antidepressants and stimulants).4 All patients should have a baseline complete physical examination, including neurologic and mental status examinations. Diagnostic tests to assess for potential differential diagnoses and evaluate baseline levels include the following:
- Basic metabolic panel, including fasting glucose, to evaluate electrolytes and risk for diabetes or Cushing disease, and to assess baseline renal function
- Thyroid function tests
- Complete blood count to assess status prior to anticonvulsant treatment (eg, carbamazepine)
- Pregnancy test if applicable (prior to use of medications)
- Liver function tests to assess baseline measurements prior to use of medications
- Electrocardiography in patients older than 40 to establish baseline and assess QTc interval, especially with use of antipsychotics and carbamazepine
- Urine toxicology screen (to rule out substance abuse).
PSYCHIATRIC EVALUATION
Psychiatric evaluation should focus on age at onset of symptoms, the presence of hypomanic or manic symptoms, prior response to antidepressants, course of the disease including history and duration of depression or manic/hypomanic episodes, and sleep disturbances (increased during depressive episodes and significantly decreased during manic episodes). It is important to assess for a history of self-harm, suicidal ideation, suicide attempts, hospitalizations, legal issues, multiple career shifts, marriage and relationship issues, and smoking and alcohol/substance misuse. Patients with severe manic or depressive episodes may experience psychotic features such as grandiose or paranoid delusions and hallucinations. A history of symptoms from close family members or friends can assist in the diagnosis of bipolar patients.
The use of DSM-5 criteria, as summarized earlier, improves the accuracy of bipolar diagnosis.3 In addition, validated tools are available to help clinicians screen for bipolar disorder, although it is important to remember that a positive screening result is not sufficient to establish a bipolar disorder diagnosis. A widely used instrument that has been validated for screening for bipolar disorder is the MDQ (available at www.dbsalliance.org/pdfs/MDQ.pdf). This self-report questionnaire consists of 15 questions that assess hypomanic or manic symptoms and functional impairment. The first 13 questions of the MDQ screen for a lifetime history of DSM-based hypomanic or manic symptoms. The last two questions ask whether these symptoms occurred at the same time and whether they caused dysfunction in various domains, such as work and family life. The MDQ is considered positive if a patient endorses at least seven of the symptom items, indicates that symptoms have occurred at the same time, and rates their dysfunction in life domains as “moderate” or “serious.” As a screening tool, the MDQ has a reported sensitivity of 73% and a specificity of 90% for bipolar disorder.11 This questionnaire can and should be used by primary care providers to help determine if their patient is at risk and requires a comprehensive evaluation for bipolar disorder.
Notably, even after a clinician has properly diagnosed bipolar disorder, patients and family members are often reluctant to commence treatment due to the stigma associated with mental health disorders.18 To help offset the effects of stigma, patients should be referred for psychologic counseling, including family counseling.
MANAGEMENT
Management of bipolar disorder in the primary care setting includes psychiatric and psychologic counseling referrals. Primary care providers must know the medications used to treat bipolar disorder and their related adverse effects, toxicities, warnings, and drug interactions, as they may treat bipolar patients for other medical conditions. Early diagnosis and treatment/referral can improve prognosis and reduce the risk for relapse and subsequent disability.19 Inpatient management is generally recommended for severe manic episodes, psychotic episodes, patients who present a danger to themselves or others, and patients with suicidal or homicidal ideations/actions.
Medications are the primary treatment for all stages of bipolar disorder, and choice of medications is based on stage, previous response, and adverse effect profiles (see Table 1).2,4,20 Generally, antidepressants (serotonin and norepinephrine reuptake inhibitors [SNRIs] and selective serotonin reuptake inhibitors [SSRIs]) should be avoided or should be used with an effective antimanic/mood stabilizer. Many patients with severe bipolar symptoms require more than two medications, and it is imperative that all primary care providers understand that often one drug alone is not sufficient treatment for patients with bipolar disorder. For less severe manic or hypomanic states, monotherapy with antipsychotics may be effective.
Medications for severe acute manic episodes generally include the mood stabilizers lithium, valproate, or carbamazepine in conjunction with an antipsychotic, such as haloperidol, or an atypical antipsychotic, such as asenapine, aripiprazole, olanzapine, quetiapine, or risperidone.2 The goal of initial therapy in patients with acute mania is rapid resolution of symptoms and restoration of adequate sleep. Lithium has a slower onset of action than valproate and carbamazepine and requires titration and monitoring. Valproate and carbamazepine have a faster onset of action but are less effective than lithium.2 Atypical antipsychotics have a more rapid onset of action than mood stabilizers and are effective in controlling acute manic symptoms, psychosis, and sleep disturbances. Patients with severe acute mania may require hospital admission for stabilization, for their safety and the safety of others.
Acute bipolar depressive episodes can be treated with several different medication options, including combination olanzapine and fluoxetine; the atypical antipsychotic quetiapine; and recently lurasidone, alone or in combination with lithium or valproate. Lamotrigine is more effective for maintenance and prevention of depressive episodes than for treatment of acute episodes, and it is also indicated for treatment of bipolar II. Valproate is more effective than lithium for mixed states and can be titrated more rapidly for faster antimanic effects.4
Generally, due to the high rate of recurrence, maintenance medications should be continued indefinitely. Maintenance medications include the mood stabilizers, lamotrigine, and many of the antipsychotics, including olanzapine.4 Adherence to medications is essential in management of bipolar disorder and can decrease the risk for relapses and destabilization. Poor adherence to medications is common, however, with rates reported at approximately 50%.21 Patient and family education, as well as psychotherapy, can improve adherence rates.2 Primary care providers should educate patients and family members about medication options and adverse effects and must stress the need for adherence to prevent relapse. Providers should also understand the safety profile of mood stabilizers and antipsychotics and the required monitoring of laboratory tests for patients on these medications.
Psychosocial treatments are an elemental component of management. Patients should be referred early for psychologic treatments including, but not limited to, family therapy, group therapy, cognitive-behavioral therapy, and psychotherapy, which have been shown to improve daily functioning, recognition of recurrences, and medication adherence.2 The rate of relapse is significantly lower in patients receiving combination psychotherapy and pharmacotherapy.22
Clinical pearls that every primary care provider should know about bipolar disorder are summarized in Table 2.
CONCLUSION
Given the substantial impact of bipolar disorder on patients and the community, primary care clinicians must maintain a high index of suspicion for this disorder. An early and accurate diagnosis may reduce the burden of bipolar disorder and improve outcomes. However, diagnosing and treating patients with bipolar disorder is challenging for primary care and specialty clinicians alike. In particular, establishing a diagnosis can be difficult, even for the most seasoned clinician, due to the diversity of symptoms. Nonetheless, diagnosing bipolar disorder, initiating treatment, and monitoring and referring patients when necessary are certainly within the purview of the primary care provider.
1. Kilbourne AM, Goodrich DE, O’Donnell AN, Miller CJ. Integrating bipolar disorder management in primary care. Curr Psychiatry Rep. 2012;14:687-695.
2. Culpepper L. The diagnosis and treatment of bipolar disorder: decision making in primary care. Prim Care Companion CNS Disord. 2014;16(3): doi 10.4088/PCC.13r01609.
3. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
4. Price AL, Marzani-Nissen GR. Bipolar disorders: a review. Am Fam Physician. 2012;85:483-493.
5. Merikangas KR, Akiskal HS, Angst J, et al. Lifetime and 12-month prevalence of bipolar spectrum disorder in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2007;64:543-552.
6. Calabrese JR, Muzina DJ, Kemp DE, et al. Predictors of bipolar disorder risk among patients currently treated for major depression. MedGenMed. 2006;8(3):38.
7. Hirschfeld RM, Cass AR, Holt DC, Carlson CA. Screening for bipolar disorder in patients treated for depression in a family medicine clinic. J Am Board Fam Pract. 2005;18(4):233-239.
8. Cerimele JM, Chwastiak LA, Dodson S, Katon WJ. The prevalence of bipolar disorder in primary care patients with depression or other psychiatric complaints: a systematic review. Psychosomatics. 2013;54(6):515-524.
9. Hirschfeld RM, Lewis L, Vornik LA. Perceptions and impact of bipolar disorder: how far have we really come? Results of the National Depressive and Manic-depressive Association 2000 survey of individuals with bipolar disorder. J Clin Psychiatry. 2003; 64:161-174.
10. Pini S, de Queiroz V, Pagnin D, et al. Prevalence and burden of bipolar disorders in European countries. Eur Neuropsychopharmacol. 2005;15(4):425-434.
11. Piver A, Yatham LN, Lam RW. Bipolar spectrum disorders: new perspectives. Can Fam Physician. 2002;48:896-904.
12. Eroglu MZ, Karakus G, Tamam L. Bipolar disorder and suicide. J Psychiatry Neurol Sci. 2013;26:139-147.
13. Simon GE, Hunkeler E, Fireman B, Lee JY, Savarino J. Risk of suicide attempt and suicide death in patients treated for bipolar disorder. Bipolar Disord. 2007;9:526-530.
14. Marcinko D, Vuksan-Cusa B. Borderline personality disorder and bipolar disorder comorbidity in suicidal patients: diagnostic and therapeutic challenges. Psychiatr Danub. 2009;21:386-390.
15. Chwastiak LA, Rosenheck RA, Kazis LE. Association of psychiatric illness and obesity, physical inactivity, and smoking among a national sample of veterans. Psychosomatics. 2011;52:230-236.
16. Vancampfort D, Vansteelandt K, Correll CU, et al. Metabolic syndrome and metabolic abnormalities in bipolar disorder: a meta-analysis of prevalence rates and moderators. Am J Psychiatry. 2013; 170:265-274.
17. Fiedorowicz JG, Solomon DA, Endicott J, et al. Manic/hypomanic symptom burden and cardiovascular mortality in bipolar disorder. Psychosom Med. 2009;71(6):598-606.
18. Hawke LD, Parikh SV, Michalak EE. Stigma and bipolar disorder: A review of the literature. J Affect Disord. 2013; 150:181-191.
19. Berk M, Brnabic A, Dodd S, et al. Does stage of illness impact treatment response in bipolar disorder? Empirical treatment data and their implication for the staging model and early intervention. Bipolar Disord. 2011;13(1):87-98.
20. Tegretol (carbamazepine) [package insert]. East Hanover, NJ: Novartis; 2015. www.pharma.us.novartis.com/product/pi/pdf/tegretol.pdf. Accessed November 18, 2016.
21. Arvilommi P, Suominen K, Mantere O, et al. Predictors of adherence to psychopharmacological and psychosocial treatment in bipolar I or II disorders—an 18-month prospective study. J Affect Disord. 2014;155:110-117.
22. Leclerc E, Mansur RB, Brietzke E. Determinants of adherence to treatment in bipolar disorder: a comprehensive review. J Affect Disord. 2013;149:247-252.
1. Kilbourne AM, Goodrich DE, O’Donnell AN, Miller CJ. Integrating bipolar disorder management in primary care. Curr Psychiatry Rep. 2012;14:687-695.
2. Culpepper L. The diagnosis and treatment of bipolar disorder: decision making in primary care. Prim Care Companion CNS Disord. 2014;16(3): doi 10.4088/PCC.13r01609.
3. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
4. Price AL, Marzani-Nissen GR. Bipolar disorders: a review. Am Fam Physician. 2012;85:483-493.
5. Merikangas KR, Akiskal HS, Angst J, et al. Lifetime and 12-month prevalence of bipolar spectrum disorder in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2007;64:543-552.
6. Calabrese JR, Muzina DJ, Kemp DE, et al. Predictors of bipolar disorder risk among patients currently treated for major depression. MedGenMed. 2006;8(3):38.
7. Hirschfeld RM, Cass AR, Holt DC, Carlson CA. Screening for bipolar disorder in patients treated for depression in a family medicine clinic. J Am Board Fam Pract. 2005;18(4):233-239.
8. Cerimele JM, Chwastiak LA, Dodson S, Katon WJ. The prevalence of bipolar disorder in primary care patients with depression or other psychiatric complaints: a systematic review. Psychosomatics. 2013;54(6):515-524.
9. Hirschfeld RM, Lewis L, Vornik LA. Perceptions and impact of bipolar disorder: how far have we really come? Results of the National Depressive and Manic-depressive Association 2000 survey of individuals with bipolar disorder. J Clin Psychiatry. 2003; 64:161-174.
10. Pini S, de Queiroz V, Pagnin D, et al. Prevalence and burden of bipolar disorders in European countries. Eur Neuropsychopharmacol. 2005;15(4):425-434.
11. Piver A, Yatham LN, Lam RW. Bipolar spectrum disorders: new perspectives. Can Fam Physician. 2002;48:896-904.
12. Eroglu MZ, Karakus G, Tamam L. Bipolar disorder and suicide. J Psychiatry Neurol Sci. 2013;26:139-147.
13. Simon GE, Hunkeler E, Fireman B, Lee JY, Savarino J. Risk of suicide attempt and suicide death in patients treated for bipolar disorder. Bipolar Disord. 2007;9:526-530.
14. Marcinko D, Vuksan-Cusa B. Borderline personality disorder and bipolar disorder comorbidity in suicidal patients: diagnostic and therapeutic challenges. Psychiatr Danub. 2009;21:386-390.
15. Chwastiak LA, Rosenheck RA, Kazis LE. Association of psychiatric illness and obesity, physical inactivity, and smoking among a national sample of veterans. Psychosomatics. 2011;52:230-236.
16. Vancampfort D, Vansteelandt K, Correll CU, et al. Metabolic syndrome and metabolic abnormalities in bipolar disorder: a meta-analysis of prevalence rates and moderators. Am J Psychiatry. 2013; 170:265-274.
17. Fiedorowicz JG, Solomon DA, Endicott J, et al. Manic/hypomanic symptom burden and cardiovascular mortality in bipolar disorder. Psychosom Med. 2009;71(6):598-606.
18. Hawke LD, Parikh SV, Michalak EE. Stigma and bipolar disorder: A review of the literature. J Affect Disord. 2013; 150:181-191.
19. Berk M, Brnabic A, Dodd S, et al. Does stage of illness impact treatment response in bipolar disorder? Empirical treatment data and their implication for the staging model and early intervention. Bipolar Disord. 2011;13(1):87-98.
20. Tegretol (carbamazepine) [package insert]. East Hanover, NJ: Novartis; 2015. www.pharma.us.novartis.com/product/pi/pdf/tegretol.pdf. Accessed November 18, 2016.
21. Arvilommi P, Suominen K, Mantere O, et al. Predictors of adherence to psychopharmacological and psychosocial treatment in bipolar I or II disorders—an 18-month prospective study. J Affect Disord. 2014;155:110-117.
22. Leclerc E, Mansur RB, Brietzke E. Determinants of adherence to treatment in bipolar disorder: a comprehensive review. J Affect Disord. 2013;149:247-252.

January 2017: Click for Credit
Here are 5 articles in the January issue of Clinician Reviews (individual articles are valid for one year from date of publication—expiration dates below):
1. Gluten-free Adherence Triples While Celiac Disease Prevalence Remains Stable
To take the posttest, go to: http://bit.ly/2h2LFDu
Expires September 6, 2017
2. Fluoxetine Appears Safer for Bone Health in At-risk Older Patients
To take the posttest, go to: http://bit.ly/2he1FTD
Expires September 15, 2017
3. High Free T4 Levels Linked to Sudden Cardiac Death
To take the posttest, go to: http://bit.ly/2gMJqUz
Expires September 16, 2017
4. Morning Sickness Linked to Lower Risk for Pregnancy Loss
To take the posttest, go to: http://bit.ly/2uaWMkH
Expires September 26, 2017
5. Anxiety, Depression May Precede Parkinson's by 25 Years
To take the posttest, go to: http://bit.ly/2gMFQtr
Expires September 27, 2017
Here are 5 articles in the January issue of Clinician Reviews (individual articles are valid for one year from date of publication—expiration dates below):
1. Gluten-free Adherence Triples While Celiac Disease Prevalence Remains Stable
To take the posttest, go to: http://bit.ly/2h2LFDu
Expires September 6, 2017
2. Fluoxetine Appears Safer for Bone Health in At-risk Older Patients
To take the posttest, go to: http://bit.ly/2he1FTD
Expires September 15, 2017
3. High Free T4 Levels Linked to Sudden Cardiac Death
To take the posttest, go to: http://bit.ly/2gMJqUz
Expires September 16, 2017
4. Morning Sickness Linked to Lower Risk for Pregnancy Loss
To take the posttest, go to: http://bit.ly/2uaWMkH
Expires September 26, 2017
5. Anxiety, Depression May Precede Parkinson's by 25 Years
To take the posttest, go to: http://bit.ly/2gMFQtr
Expires September 27, 2017
Here are 5 articles in the January issue of Clinician Reviews (individual articles are valid for one year from date of publication—expiration dates below):
1. Gluten-free Adherence Triples While Celiac Disease Prevalence Remains Stable
To take the posttest, go to: http://bit.ly/2h2LFDu
Expires September 6, 2017
2. Fluoxetine Appears Safer for Bone Health in At-risk Older Patients
To take the posttest, go to: http://bit.ly/2he1FTD
Expires September 15, 2017
3. High Free T4 Levels Linked to Sudden Cardiac Death
To take the posttest, go to: http://bit.ly/2gMJqUz
Expires September 16, 2017
4. Morning Sickness Linked to Lower Risk for Pregnancy Loss
To take the posttest, go to: http://bit.ly/2uaWMkH
Expires September 26, 2017
5. Anxiety, Depression May Precede Parkinson's by 25 Years
To take the posttest, go to: http://bit.ly/2gMFQtr
Expires September 27, 2017

Chagas Disease: Creeping into Family Practice in the United States
CE/CME No: CR-1611
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Understand the prevalence and risks of Chagas disease in the United States.
• Explain the pathophysiology of Chagas disease, including the vector and transmission routes of the disease.
• Describe the clinical presentation of both the acute and chronic forms of the disease and learn when to suspect an infection.
• Outline a plan for diagnosis and treatment of Chagas disease.
• Educate women with Chagas disease about the risk of transmission for future offspring.
FACULTY
Jessica McDonald works in the Emergency Medicine Department at Dekalb Medical Center, Atlanta. Jill Mattingly is Academic Coordinator and Clinical Assistant Professor in the Physician Assistant Program at Mercer University, Atlanta.
The authors have no financial relationships to disclose.
![]()
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of November 2016.
Article begins on next page >>
Chagas disease, a parasitic infection, is increasingly being detected in the United States, most likely due to immigration from endemic countries in South and Central America. Approximately 300,000 persons in the US have chronic Chagas disease, and up to 30% of them will develop clinically evident cardiovascular and/or gastrointestinal disease. Here’s practical guidance to help you recognize the features of symptomatic Chagas disease and follow up with appropriate evaluation and management.
Chagas disease, also known as American trypanosomiasis, is caused by the protozoan parasite Trypanosoma cruzi.1 It is most commonly spread by triatomine bugs infected with T cruzi and is endemic in many parts of Mexico and Central and South America.2 Chagas disease was first described in 1909 by Brazilian physician Carlos Chagas.3 Since its discovery, it has often been considered a disease affecting only the poor living in endemic areas of Latin America. However, 6 million to 7 million people are infected with T cruzi worldwide, and estimates suggest that Mexico and the US rank third and seventh, respectively, in the number of persons with T cruzi infection in the Western Hemisphere.1,4
An estimated 300,000 persons in the US have Chagas disease; most of them are not aware that they are infected.5,6 The increasing presence of the disease in the US, which traditionally has been considered a nonendemic area, is due to immigration from endemic areas, with subsequent infections occurring through mechanisms that do not require contact with the triatomine vector (eg, congenital transmission).1 Between 1981 and 2005, more than 7 million people from T cruzi-endemic countries in Latin America moved to the US and became legal residents.3
Early detection and treatment of Chagas disease is important because up to 30% of patients with chronic infection will develop a heart disorder, which can range in severity from conduction system abnormalities to dilated cardiomyopathy.4 In some areas of southern Mexico, Chagas disease is the most common cause of dilated cardiomyopathy.1 Equally concerning is the fact that untreated mothers with Chagas disease can transmit T cruzi to their infants.1,3 An estimated 315 babies are born with congenital Chagas disease each year in the US, an incidence equivalent to that of phenylketonuria.7 It is estimated that congenital transmission is responsible for up to one-quarter of new infections worldwide.1 Unfortunately, obstetricians are not well informed about the risk factors for congenital Chagas disease, and very limited screening of at-risk women is performed. In a 2008 survey exploring health care providers’ knowledge of and understanding about Chagas disease, obstetricians and gynecologists had the greatest knowledge deficits about the disease, although considerable deficits were also seen among other specialties.1
KISSING BUG DISEASE: ETIOLOGY/PATHOPHYSIOLOGY
Exposure to the protozoan parasite T cruzi, the cause of Chagas disease, typically occurs following the bite of a triatomine bug. Also known as “kissing bugs” because they usually bite exposed areas of the skin such as the face, triatomine bugs feed on human blood, typically at night, and act as a vector for the parasite.8 The parasite lives in the feces and urine of the triatomine bugs and is excreted near the bite during or shortly after a blood meal. The bitten person will then unknowingly smear the infected feces into the bite wound, eyes, mouth, or any opening in the skin, which gives the parasites systemic access.4 Once in the host’s bloodstream, the parasite replicates in host cells, a process that ends in cell lysis and hematogenous spread. At this point, the parasites can be seen on peripheral blood smear. Noninfected triatomine insects become infected and continue the cycle when they feed on an infected human host (see Figure 1).3 Persons of lower socioeconomic status living in endemic areas in Latin America are at a higher risk for contracting Chagas disease because “kissing bugs” commonly live in wall or roof cracks of poorly built homes. Populations living in poverty are also at risk due to minimal access to health care and prenatal care.4 Transmission of T cruzi not involving triatomine vectors occurs congenitally or through blood transfusions, consumption of contaminated food, and organ donations.4
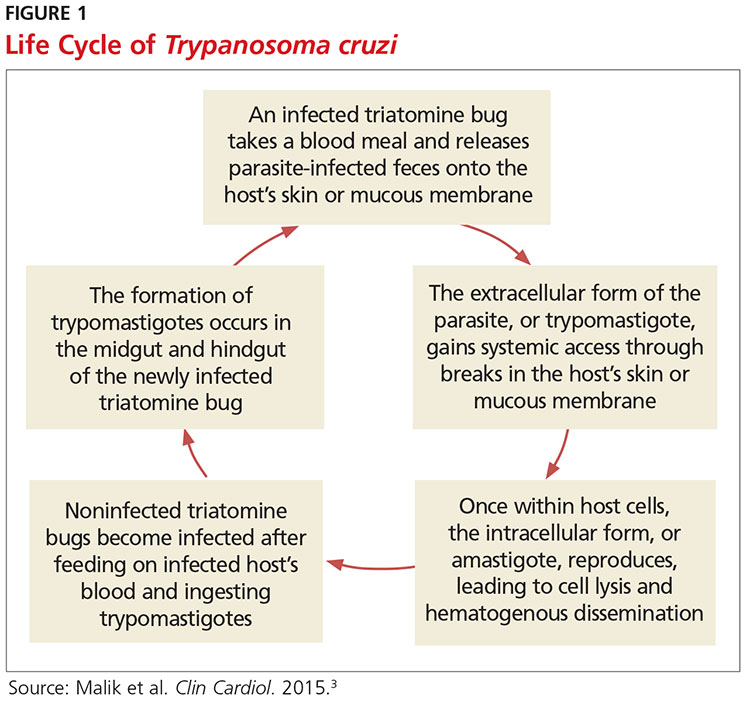
NATURAL HISTORY OF INFECTION AND PATIENT PRESENTATION
Acute phase
Infection with the T cruzi parasite is followed by an asymptomatic incubation period of one to two weeks, which is then followed by an acute phase that can last eight to 12 weeks.5 The acute phase is characterized by a large amount of parasites in the bloodstream (see Table 1). The patient is often asymptomatic but can have nonspecific symptoms such as fever, headache, lymphadenopathy, shortness of breath, myalgia, swelling, and abdominal or chest pain.4 Because symptoms during the acute phase are typically mild, many patients do not seek medical attention until they transition into the chronic phase.4 Infants are more likely to experience severe symptoms, including myocarditis or meningoencephalitis, and thus are more likely to present during the acute phase.9
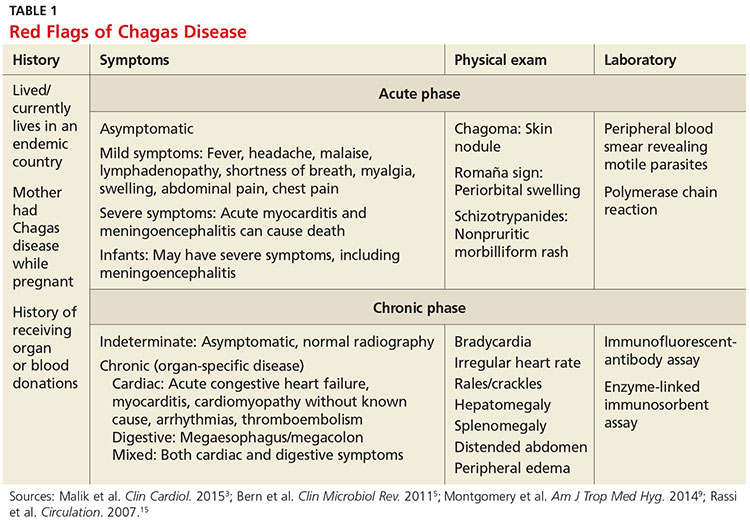
If the patient acquired the infection through an organ transplant, the acute phase symptoms can be delayed, on average, up to 112 days.5 These patients will have more noticeable symptoms, including hepatosplenomegaly, myocarditis, and congestive heart failure. Due to the known risk for transmission through organ transplants, donors are often screened for Chagas disease. Unfortunately, this screening is selective and often inconsistent.5 Therefore, the presence of the previously mentioned symptoms in a person who recently received an organ transplant should raise suspicion of Chagas disease.5
Chronic phase
Patients not treated during the acute phase will pass into the chronic phase of Chagas disease.5 This may occur due to reactivation of T cruzi infection via immunosuppression.9 At this time, the previously asymptomatic patient will have typical signs and symptoms of chronic disease, along with nodules, panniculitis, and myocarditis.4,9,10 During the chronic phase, parasites are undetectable by microscopy, but the patient can still spread the disease to the vector as well as to others congenitally and through organ donation and blood transfusions.5,9
Patients with chronic T cruzi infection who remain without signs or symptoms of infection are considered to have the indeterminate form of chronic disease. Many patients will remain in the indeterminate form throughout their lives, but between 20% and 30% will progress to the determinate form of chronic disease over years to decades.3 The determinate form is characterized by clinically evident disease and is classically divided into cardiac Chagas disease and digestive Chagas disease.5 Symptoms of the chronic phase depend on the genotype of T cruzi that caused the infection. The AG genotype has a higher incidence of digestive disease.11
Cardiac Chagas disease is believed to occur due to parasite invasion and persistence in cardiac tissue, leading to immune-mediated myocardial injury.5 Chagas cardiomyopathy is characterized by chronic myocarditis affecting all cardiac chambers and disturbances in the electrical conduction system; patients also often develop apical aneurysms. Longstanding cardiac Chagas disease can lead to more serious complications, such as episodes of ventricular tachycardia, heart block, thromboembolic phenomena, severe bradycardia, dilated cardiomyopathy, and congestive heart failure. Patients may complain of presyncope, syncope, and episodes of palpitations. They are also at high risk for sudden cardiac death.5 Patients with cardiomyopathy or cardiac insufficiency secondary to Chagas disease have a worse prognosis than those with idiopathic cardiomyopathy or decompensated heart failure due to other etiologies.12
Less common than cardiac Chagas disease, digestive Chagas disease occurs mostly in Argentina, Bolivia, Chile, Paraguay, Uruguay, and parts of Peru and Brazil; it is rarely seen in northern South America, Central America, or Mexico.5 The parasite causes gastrointestinal symptoms by damaging intramural neurons, resulting in denervation of hollow viscera. Since it affects the esophagus and colon, patients may present with dysphagia, odynophagia, cough, reflux, weight loss, constipation, and abdominal pain.5
PHYSICAL EXAMINATION: A CRUCIAL STEP
The physical examination of a patient with suspected Chagas disease can be crucial to the diagnosis. As noted, there are often few specific symptoms or physical exam findings during the acute phase. However, in some patients, swelling and inflammation may be evident at the site of inoculation, often on the face or extremities. This finding is called a chagoma. The Romaña sign, characterized by painless unilateral swelling of the upper and lower eyelid, can also be seen if the infection occurred through the conjunctiva.5 A nonpruritic morbilliform rash, called schizotrypanides, may be a presenting symptom in patients with acute disease.13 Children younger than 2 years of age are at increased risk for a severe acute infection, with signs and symptoms of pericardial effusion, myocarditis, and meningoencephalitis. Children can also develop generalized edema and lymphadenopathy. Those children who develop severe manifestations during acute infection have an increased risk for mortality.5
Chronic chagasic cardiomyopathy may present with signs of left-sided heart failure (pulmonary edema, dyspnea at rest or exertion), biventricular heart failure (hepatomegaly, peripheral edema, jugular venous distention), or thromboembolic events to the brain, lower extremities, and lungs.13 Chronic chagasic megaesophagus may lead to weight loss, esophageal dysmotility, pneumonitis due to aspiration of food trapped in the esophagus and stomach, salivary gland enlargement, and erosive esophagitis, which increases the risk for esophageal cancer. Chronic chagasic megacolon can present as an intestinal obstruction, volvulus, abdominal distention, or fecaloma.13
Clinicians should be alert to the possibility of congenital T cruzi infection in children born to women who emigrated from an endemic area or who visited an area with a high prevalence of Chagas disease. Most newborns with T cruzi infection are asymptomatic, but in some cases a thorough neonatal exam can lead to the diagnosis. Manifestations of symptomatic congenital infection include hepatosplenomegaly, low birth weight, premature birth, and low Apgar scores.5 Lab tests may reveal thrombocytopenia and anemia. Neonates with severe disease may also have respiratory distress, meningoencephalitis, and gastrointestinal problems.5
LABORATORY WORK-UP
Laboratory work-up for Chagas disease depends on the provider’s awareness of the disease and its symptoms. All patients should undergo routine blood work, including complete blood count (CBC) with differential, comprehensive metabolic panel (CMP), and liver function tests to rule out other etiologies that manifest with similar symptoms. If the patient presents during the acute phase, microscopy of blood smears with Giemsa stain should be done to visualize the parasites. In the patient who presents during the chronic phase with cardiac symptoms, measurement of B-type natriuretic peptide, troponin, C-reactive protein, and the erythrocyte sedimentation rate can be used to rule out other differential diagnoses. Electrocardiogram (ECG) may show a right bundle-branch block or left anterior fascicular block.5 Echocardiogram may show left ventricular wall motion abnormalities and/or cardiomyopathy with congestive heart failure.5,10 A work-up for digestive Chagas disease may include a barium swallow, kidney-ureter-bladder x-ray, or MRI/CT of the abdomen.14
DIAGNOSING ACUTE, CHRONIC, AND CONGENITAL CHAGAS
Accurate diagnosis of Chagas disease requires a thorough history and physical exam, as well as a high index of suspicion. Recent travel to an endemic area of Chagas disease in combination with the typical signs and symptoms—such as fever, headache, lymphadenopathy, shortness of breath, myalgia, swelling, and abdominal or chest pain—should prompt the provider to perform more specific tests.4 Inquiry about past medical history, blood transfusions, and surgeries is also imperative to make the correct diagnosis.5
The approach to diagnosis of Chagas disease depends on whether the patient presents during the acute or chronic phase. During the acute phase, the count of the trypomastigote, the mature extracellular form of the parasite T cruzi, is at its highest, making this the best time to obtain an accurate diagnosis if an infection is suspected.3 Microscopy of fresh preparations of anticoagulated blood or buffy coat may show motile parasites.10 Other options include visualization of parasites in a blood smear with Giemsa stain or hemoculture. Hemoculture is a sensitive test but takes several weeks to show growth of the parasites. Therefore, polymerase chain reaction (PCR) assay is the preferred diagnostic test due to its high sensitivity and quick turnaround time.5
Because no diagnostic gold standard exists for chronic disease, confidently diagnosing Chagas in the United States can be difficult.5 Past the acute phase (about three months after infection), microscopy and PCR cannot be used due to low parasitemia. If an infection with T cruzi is suspected but nine to 14 weeks have passed since exposure, serology is the method of choice for diagnosis. The enzyme-linked immunosorbent assay (ELISA) and immunofluorescent-antibody assay (IFA) are most often used to identify immunoglobulin (Ig) G antibodies to the parasite.
The difficulty of diagnosing Chagas disease in the chronic phase lies in the fact that neither ELISA or IFA alone is sensitive or specific enough to confirm the diagnosis.5 In order to make a serologic diagnosis of infection, positive results are needed from two serologic tests based on two different antigens or by using two different techniques (eg, ELISA or IFA). If the two tests are discordant, a third test must be done to determine the patient’s infection status. The radioimmunoprecipitation assay (RIPA) and trypomastigote excreted-secreted antigen immunoblot (TESA-blot) have been traditionally used as confirmatory tests, but even they do not have high sensitivity and specificity. A case of indeterminate Chagas disease is confirmed with positive serologic testing in a patient without symptoms and with normal ECG, chest x-ray, and imaging of the colon and esophagus.15
The preferred protocol for diagnosis of congenital Chagas disease first requires positive serologic testing confirming the infection in the mother (see Figure 2).16 Once that is determined, microscopic and PCR-based examinations of cord blood and peripheral blood specimens are carried out during the first one to two months of the infant’s life.10 PCR is the preferred test for early congenital Chagas disease, recipients of organ transplants, and after accidental exposure since results can determine if the patient is infected earlier than trypomastigotes (developmental stage of trypanosomes) can be seen on a peripheral blood smear.5
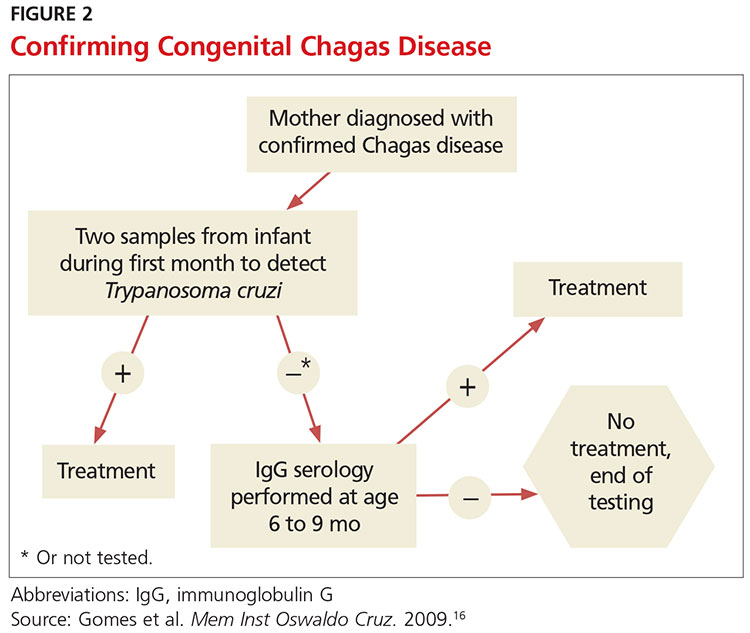
TREATMENT CONSIDERATIONS
If there is a suspicion of Chagas disease, the patient should be referred to an infectious disease specialist for diagnosis and treatment. Nifurtimox and benznidazole are the only drugs that have been shown to improve the course of Chagas disease.5 However, neither drug is approved by the FDA, and both can only be obtained from the CDC, which makes treatment a challenge.9 In addition, up to 30% of patients terminate treatment due to the many adverse effects of these drugs.17
The dosage regimen for nifurtimox is 8-10 mg/kg/d divided into three doses for 90 days.10 Anorexia, weight loss, nausea, vomiting, and abdominal pain occur in up to 70% of patients.5 Irritability, insomnia, disorientation, and tremors can also occur. Neurotoxicity leading to peripheral neuropathy is dose dependent and requires treatment termination.5
Benznidazole is better tolerated and is active against the trypomastigotes as well as the amastigotes or intracellular form of the parasite.10 The dosage regimen for benznidazole is 5-7 mg/kg/d divided into two doses for 60 days.10 Dermatologic reactions such as rash, photosensitivity, and exfoliative dermatitis are the most common adverse effects. Peripheral neuropathy and bone marrow suppression are dose dependent and require therapy cessation.5
The CDC recommends treatment for all cases of acute disease (including congenital disease) regardless of age, and for chronic disease in patients up to age 50 who have not progressed to cardiomyopathy. In patients older than 50, treatment should be determined after weighing the potential risks and benefits (see Table 2).18
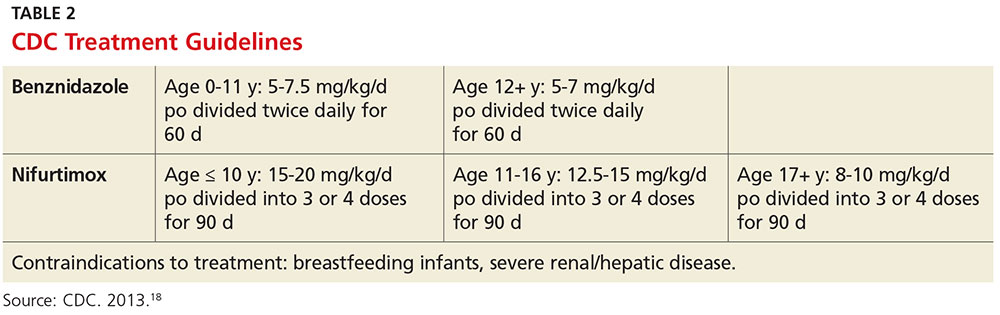
The success of treatment is determined in part by the phase of the disease. Cure rates in patients treated with either nifurtimox or benznidazole during the acute phase range from 65% to 80%.17 Chronic disease shows less of a response to traditional antiparasitic drug regimens, but higher rates of success are seen in younger patients.5 According to current estimates, successful treatment of chronic disease is limited to 15% to 30% patients.17 Treatment of congenital Chagas disease should begin as soon as the diagnosis is confirmed, and cure rates are greater than 90% if patients are treated within the first year of life.10 Treating congenital Chagas disease is important because the infection can be passed to future generations even if the disease never manifests with symptoms.19 However, if an expecting mother has known Chagas disease, antiparasitic medications are not recommended during the pregnancy because of a lack of fetal safety data for the two antiparasitic agents.20 Instead, it is recommended that women of childbearing age be treated before pregnancy, as rates of congenital infection are 25 times lower in women who are treated than in those who are not.21
PRE- AND POSTEXPOSURE PATIENT EDUCATION
Patient education mainly focuses on how to prevent Chagas disease and prognosis once diagnosed. During travel to endemic areas, the use of insecticides and residing in well-built households are the most important prevention measures. No vaccine is available, and primary chemoprophylaxis of persons visiting endemic areas is not recommended due to the low risk for infection and concerns about adverse effects.13
The survival rate of those who remain in the indeterminate phase is the same as that of the general population. However, findings that most strongly predict mortality include ventricular tachycardia, cardiomegaly, congestive heart failure (NYHA class III/IV), left ventricular systolic dysfunction, and male sex.10 Patients diagnosed with Chagas disease should be strongly encouraged not to donate blood or organs.10 Some organ and blood donation organizations selectively or universally screen donated specimens; however, this screening is not required by law.5 Family members of those diagnosed with the disease should also be tested, especially if the patient is a woman who has children or who plans to become pregnant.10
FOLLOW-UP
In patients confirmed to have Chagas disease but without symptoms and a normal ECG, further initial evaluation is not required.10 An annual history, physical exam, and ECG should be done. Those who have symptoms or ECG changes should have a complete cardiac work-up, including a 24-hour ambulatory ECG, exercise stress test, and echocardiogram to determine functional capacity. A barium swallow, barium enema, esophageal manometry, and endoscopy may be indicated in patients with gastrointestinal symptoms of Chagas disease but otherwise are not recommended. Patients taking antiparasitic drugs should have a CBC and CMP at the start of treatment and then bimonthly until the end of treatment to monitor for rare bone marrow suppression. Nifurtimox and benznidazole are also known to be mutagenic and increase the risk for lymphoma in animal studies, but this risk has not been documented in humans.10
CONCLUSION
Chagas disease is considered one of the neglected tropical diseases due to its high prevalence, chronic course, debilitating symptoms, and association with poverty.7 It is evident that incidence and prevalence of Chagas disease in the US are increasing due to recent immigration and mother-to-child transmission. Therefore, family practice clinicians must be able to recognize the red flags that suggest a T cruzi infection.5,9 Enhanced awareness of Chagas disease among health care providers will lead to better symptom control and cure rates for affected patients and may also prevent congenital infections. These efforts could serve to remove Chagas disease from the list of neglected tropical diseases.
1. Hotez PJ, Dumonteil E, Betancourt Cravioto M, et al. An unfolding tragedy of Chagas disease in North America. PLoS Negl Trop Dis. 2013; 7(10):e2300.
2. Verani JR, Seitz A, Gilman RH, et al. Geographic variation in the sensitivity of recombinant antigen-based rapid tests for chronic Trypanosoma cruzi infection. Am J Trop Med Hyg. 2009;80(3):410-415.
3. Malik LH, Singh GD, Amsterdam EA. The epidemiology, clinical manifestations, and management of Chagas heart disease. Clin Cardiol. 2015;38(9):565-569.
4. World Health Organization. Chagas disease (American trypanosomiasis). Fact sheet. Updated March 2016. www.who.int/mediacentre/factsheets/fs340/en/. Accessed October 20, 2016.
5. Bern C, Kjos S, Yabsley MJ, Montgomery SP. Trypanosoma cruzi and Chagas’ disease in the United States. Clin Microbiol Rev. 2011; 24(4):655-681.
6. Stimpert KK, Montgomery SP. Physician awareness of Chagas disease, USA. Emerg Infect Dis. 2010;16(5):871-872.
7. Hotez PJ. Neglected parasitic infections and poverty in the United States. PLoS Negl Trop Dis. 2014;8(9):e3012.
8. Goupil LS, McKerrow JH. Introduction: drug discovery and development for neglected diseases. Chem Rev. 2014;114(22):11131-11137.
9. Montgomery SP, Starr MC, Cantey P, et al. Neglected parasitic infections in the United States: Chagas disease. Am J Trop Med Hyg. 2014; 90(5):814-818.
10. Bern C, Montgomery SP, Herwaldt BL, et al. Evaluation and treatment of Chagas disease in the United States. JAMA. 2007;298(18):2171-2181.
11. de Oliveira AP, Bernardo CR, Camargo AV, et al. Genetic susceptibility to cardiac and digestive clinical forms of chronic Chagas disease: involvement of the CCR5 59029 A/G polymorphism. PLoS One. 2015; 10(11):e0141847.
12. Apt W, Arribada A, Zulantay I, et al. Trypanosoma cruzi burden, genotypes, and clinical evaluation of Chilean patients with chronic Chagas cardiopathy. Parasitol Res. 2015;114(8):3007-3018.
13. Kirchhoff LV. Chagas disease (American trypanosomiasis): Background, pathophysiology, epidemiology. Emedicine.medscape.com. 2015. http://emedicine.medscape.com/article/214581-overview. Accessed October 20, 2016.
14. Knipe H, St-Amant M. Chagas disease. Radiopaedia.org. 2015. http://radiopaedia.org/articles/chagas-disease. Accessed October 20, 2016.
15. Rassi A Jr, Rassi A, Rassi SG. Predictors of mortality in chronic Chagas disease: a systematic review of observational studies. Circulation. 2007;115(9):1101-1108.
16. Gomes YM, Lorena VM, Luquetti AO. Diagnosis of Chagas disease: what has been achieved? What remains to be done with regard to diagnosis and follow up studies? Mem Inst Oswaldo Cruz. 2009; 104(suppl 1):115-121.
17. Molina I, Gómez i Prat J, Salvador F, et al. Randomized trial of posaconazole and benznidazole for chronic Chagas’ disease. N Engl J Med. 2014;370:1899-1908.
18. CDC. Parasites – American trypanosomiasis (also known as Chagas Disease). Antiparasitic Treatment. Resources For Health Professionals. www.cdc.gov/parasites/chagas/health_professionals/tx.html. Accessed October 20, 2016.
19. Carlier Y, Truyens C. Congenital Chagas disease as an ecological model of interactions between Trypanosoma cruzi parasites, pregnant women, placenta and fetuses. Acta Trop. 2015;151:103-115.
20. Moscatelli G, Moroni S, García-Bournissen F, et al. Prevention of congenital Chagas through treatment of girls and women of childbearing age. Mem Inst Oswaldo Cruz. 2015;110(4):507-509.
21. Fabbro D, Danesi E, Olivera V, et al. Trypanocide treatment of women infected with Trypanosoma cruzi and its effect on preventing congenital Chagas. PLoS Negl Trop Dis. 2014;8(11):e3312.
CE/CME No: CR-1611
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Understand the prevalence and risks of Chagas disease in the United States.
• Explain the pathophysiology of Chagas disease, including the vector and transmission routes of the disease.
• Describe the clinical presentation of both the acute and chronic forms of the disease and learn when to suspect an infection.
• Outline a plan for diagnosis and treatment of Chagas disease.
• Educate women with Chagas disease about the risk of transmission for future offspring.
FACULTY
Jessica McDonald works in the Emergency Medicine Department at Dekalb Medical Center, Atlanta. Jill Mattingly is Academic Coordinator and Clinical Assistant Professor in the Physician Assistant Program at Mercer University, Atlanta.
The authors have no financial relationships to disclose.
![]()
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of November 2016.
Article begins on next page >>
Chagas disease, a parasitic infection, is increasingly being detected in the United States, most likely due to immigration from endemic countries in South and Central America. Approximately 300,000 persons in the US have chronic Chagas disease, and up to 30% of them will develop clinically evident cardiovascular and/or gastrointestinal disease. Here’s practical guidance to help you recognize the features of symptomatic Chagas disease and follow up with appropriate evaluation and management.
Chagas disease, also known as American trypanosomiasis, is caused by the protozoan parasite Trypanosoma cruzi.1 It is most commonly spread by triatomine bugs infected with T cruzi and is endemic in many parts of Mexico and Central and South America.2 Chagas disease was first described in 1909 by Brazilian physician Carlos Chagas.3 Since its discovery, it has often been considered a disease affecting only the poor living in endemic areas of Latin America. However, 6 million to 7 million people are infected with T cruzi worldwide, and estimates suggest that Mexico and the US rank third and seventh, respectively, in the number of persons with T cruzi infection in the Western Hemisphere.1,4
An estimated 300,000 persons in the US have Chagas disease; most of them are not aware that they are infected.5,6 The increasing presence of the disease in the US, which traditionally has been considered a nonendemic area, is due to immigration from endemic areas, with subsequent infections occurring through mechanisms that do not require contact with the triatomine vector (eg, congenital transmission).1 Between 1981 and 2005, more than 7 million people from T cruzi-endemic countries in Latin America moved to the US and became legal residents.3
Early detection and treatment of Chagas disease is important because up to 30% of patients with chronic infection will develop a heart disorder, which can range in severity from conduction system abnormalities to dilated cardiomyopathy.4 In some areas of southern Mexico, Chagas disease is the most common cause of dilated cardiomyopathy.1 Equally concerning is the fact that untreated mothers with Chagas disease can transmit T cruzi to their infants.1,3 An estimated 315 babies are born with congenital Chagas disease each year in the US, an incidence equivalent to that of phenylketonuria.7 It is estimated that congenital transmission is responsible for up to one-quarter of new infections worldwide.1 Unfortunately, obstetricians are not well informed about the risk factors for congenital Chagas disease, and very limited screening of at-risk women is performed. In a 2008 survey exploring health care providers’ knowledge of and understanding about Chagas disease, obstetricians and gynecologists had the greatest knowledge deficits about the disease, although considerable deficits were also seen among other specialties.1
KISSING BUG DISEASE: ETIOLOGY/PATHOPHYSIOLOGY
Exposure to the protozoan parasite T cruzi, the cause of Chagas disease, typically occurs following the bite of a triatomine bug. Also known as “kissing bugs” because they usually bite exposed areas of the skin such as the face, triatomine bugs feed on human blood, typically at night, and act as a vector for the parasite.8 The parasite lives in the feces and urine of the triatomine bugs and is excreted near the bite during or shortly after a blood meal. The bitten person will then unknowingly smear the infected feces into the bite wound, eyes, mouth, or any opening in the skin, which gives the parasites systemic access.4 Once in the host’s bloodstream, the parasite replicates in host cells, a process that ends in cell lysis and hematogenous spread. At this point, the parasites can be seen on peripheral blood smear. Noninfected triatomine insects become infected and continue the cycle when they feed on an infected human host (see Figure 1).3 Persons of lower socioeconomic status living in endemic areas in Latin America are at a higher risk for contracting Chagas disease because “kissing bugs” commonly live in wall or roof cracks of poorly built homes. Populations living in poverty are also at risk due to minimal access to health care and prenatal care.4 Transmission of T cruzi not involving triatomine vectors occurs congenitally or through blood transfusions, consumption of contaminated food, and organ donations.4
NATURAL HISTORY OF INFECTION AND PATIENT PRESENTATION
Acute phase
Infection with the T cruzi parasite is followed by an asymptomatic incubation period of one to two weeks, which is then followed by an acute phase that can last eight to 12 weeks.5 The acute phase is characterized by a large amount of parasites in the bloodstream (see Table 1). The patient is often asymptomatic but can have nonspecific symptoms such as fever, headache, lymphadenopathy, shortness of breath, myalgia, swelling, and abdominal or chest pain.4 Because symptoms during the acute phase are typically mild, many patients do not seek medical attention until they transition into the chronic phase.4 Infants are more likely to experience severe symptoms, including myocarditis or meningoencephalitis, and thus are more likely to present during the acute phase.9
If the patient acquired the infection through an organ transplant, the acute phase symptoms can be delayed, on average, up to 112 days.5 These patients will have more noticeable symptoms, including hepatosplenomegaly, myocarditis, and congestive heart failure. Due to the known risk for transmission through organ transplants, donors are often screened for Chagas disease. Unfortunately, this screening is selective and often inconsistent.5 Therefore, the presence of the previously mentioned symptoms in a person who recently received an organ transplant should raise suspicion of Chagas disease.5
Chronic phase
Patients not treated during the acute phase will pass into the chronic phase of Chagas disease.5 This may occur due to reactivation of T cruzi infection via immunosuppression.9 At this time, the previously asymptomatic patient will have typical signs and symptoms of chronic disease, along with nodules, panniculitis, and myocarditis.4,9,10 During the chronic phase, parasites are undetectable by microscopy, but the patient can still spread the disease to the vector as well as to others congenitally and through organ donation and blood transfusions.5,9
Patients with chronic T cruzi infection who remain without signs or symptoms of infection are considered to have the indeterminate form of chronic disease. Many patients will remain in the indeterminate form throughout their lives, but between 20% and 30% will progress to the determinate form of chronic disease over years to decades.3 The determinate form is characterized by clinically evident disease and is classically divided into cardiac Chagas disease and digestive Chagas disease.5 Symptoms of the chronic phase depend on the genotype of T cruzi that caused the infection. The AG genotype has a higher incidence of digestive disease.11
Cardiac Chagas disease is believed to occur due to parasite invasion and persistence in cardiac tissue, leading to immune-mediated myocardial injury.5 Chagas cardiomyopathy is characterized by chronic myocarditis affecting all cardiac chambers and disturbances in the electrical conduction system; patients also often develop apical aneurysms. Longstanding cardiac Chagas disease can lead to more serious complications, such as episodes of ventricular tachycardia, heart block, thromboembolic phenomena, severe bradycardia, dilated cardiomyopathy, and congestive heart failure. Patients may complain of presyncope, syncope, and episodes of palpitations. They are also at high risk for sudden cardiac death.5 Patients with cardiomyopathy or cardiac insufficiency secondary to Chagas disease have a worse prognosis than those with idiopathic cardiomyopathy or decompensated heart failure due to other etiologies.12
Less common than cardiac Chagas disease, digestive Chagas disease occurs mostly in Argentina, Bolivia, Chile, Paraguay, Uruguay, and parts of Peru and Brazil; it is rarely seen in northern South America, Central America, or Mexico.5 The parasite causes gastrointestinal symptoms by damaging intramural neurons, resulting in denervation of hollow viscera. Since it affects the esophagus and colon, patients may present with dysphagia, odynophagia, cough, reflux, weight loss, constipation, and abdominal pain.5
PHYSICAL EXAMINATION: A CRUCIAL STEP
The physical examination of a patient with suspected Chagas disease can be crucial to the diagnosis. As noted, there are often few specific symptoms or physical exam findings during the acute phase. However, in some patients, swelling and inflammation may be evident at the site of inoculation, often on the face or extremities. This finding is called a chagoma. The Romaña sign, characterized by painless unilateral swelling of the upper and lower eyelid, can also be seen if the infection occurred through the conjunctiva.5 A nonpruritic morbilliform rash, called schizotrypanides, may be a presenting symptom in patients with acute disease.13 Children younger than 2 years of age are at increased risk for a severe acute infection, with signs and symptoms of pericardial effusion, myocarditis, and meningoencephalitis. Children can also develop generalized edema and lymphadenopathy. Those children who develop severe manifestations during acute infection have an increased risk for mortality.5
Chronic chagasic cardiomyopathy may present with signs of left-sided heart failure (pulmonary edema, dyspnea at rest or exertion), biventricular heart failure (hepatomegaly, peripheral edema, jugular venous distention), or thromboembolic events to the brain, lower extremities, and lungs.13 Chronic chagasic megaesophagus may lead to weight loss, esophageal dysmotility, pneumonitis due to aspiration of food trapped in the esophagus and stomach, salivary gland enlargement, and erosive esophagitis, which increases the risk for esophageal cancer. Chronic chagasic megacolon can present as an intestinal obstruction, volvulus, abdominal distention, or fecaloma.13
Clinicians should be alert to the possibility of congenital T cruzi infection in children born to women who emigrated from an endemic area or who visited an area with a high prevalence of Chagas disease. Most newborns with T cruzi infection are asymptomatic, but in some cases a thorough neonatal exam can lead to the diagnosis. Manifestations of symptomatic congenital infection include hepatosplenomegaly, low birth weight, premature birth, and low Apgar scores.5 Lab tests may reveal thrombocytopenia and anemia. Neonates with severe disease may also have respiratory distress, meningoencephalitis, and gastrointestinal problems.5
LABORATORY WORK-UP
Laboratory work-up for Chagas disease depends on the provider’s awareness of the disease and its symptoms. All patients should undergo routine blood work, including complete blood count (CBC) with differential, comprehensive metabolic panel (CMP), and liver function tests to rule out other etiologies that manifest with similar symptoms. If the patient presents during the acute phase, microscopy of blood smears with Giemsa stain should be done to visualize the parasites. In the patient who presents during the chronic phase with cardiac symptoms, measurement of B-type natriuretic peptide, troponin, C-reactive protein, and the erythrocyte sedimentation rate can be used to rule out other differential diagnoses. Electrocardiogram (ECG) may show a right bundle-branch block or left anterior fascicular block.5 Echocardiogram may show left ventricular wall motion abnormalities and/or cardiomyopathy with congestive heart failure.5,10 A work-up for digestive Chagas disease may include a barium swallow, kidney-ureter-bladder x-ray, or MRI/CT of the abdomen.14
DIAGNOSING ACUTE, CHRONIC, AND CONGENITAL CHAGAS
Accurate diagnosis of Chagas disease requires a thorough history and physical exam, as well as a high index of suspicion. Recent travel to an endemic area of Chagas disease in combination with the typical signs and symptoms—such as fever, headache, lymphadenopathy, shortness of breath, myalgia, swelling, and abdominal or chest pain—should prompt the provider to perform more specific tests.4 Inquiry about past medical history, blood transfusions, and surgeries is also imperative to make the correct diagnosis.5
The approach to diagnosis of Chagas disease depends on whether the patient presents during the acute or chronic phase. During the acute phase, the count of the trypomastigote, the mature extracellular form of the parasite T cruzi, is at its highest, making this the best time to obtain an accurate diagnosis if an infection is suspected.3 Microscopy of fresh preparations of anticoagulated blood or buffy coat may show motile parasites.10 Other options include visualization of parasites in a blood smear with Giemsa stain or hemoculture. Hemoculture is a sensitive test but takes several weeks to show growth of the parasites. Therefore, polymerase chain reaction (PCR) assay is the preferred diagnostic test due to its high sensitivity and quick turnaround time.5
Because no diagnostic gold standard exists for chronic disease, confidently diagnosing Chagas in the United States can be difficult.5 Past the acute phase (about three months after infection), microscopy and PCR cannot be used due to low parasitemia. If an infection with T cruzi is suspected but nine to 14 weeks have passed since exposure, serology is the method of choice for diagnosis. The enzyme-linked immunosorbent assay (ELISA) and immunofluorescent-antibody assay (IFA) are most often used to identify immunoglobulin (Ig) G antibodies to the parasite.
The difficulty of diagnosing Chagas disease in the chronic phase lies in the fact that neither ELISA or IFA alone is sensitive or specific enough to confirm the diagnosis.5 In order to make a serologic diagnosis of infection, positive results are needed from two serologic tests based on two different antigens or by using two different techniques (eg, ELISA or IFA). If the two tests are discordant, a third test must be done to determine the patient’s infection status. The radioimmunoprecipitation assay (RIPA) and trypomastigote excreted-secreted antigen immunoblot (TESA-blot) have been traditionally used as confirmatory tests, but even they do not have high sensitivity and specificity. A case of indeterminate Chagas disease is confirmed with positive serologic testing in a patient without symptoms and with normal ECG, chest x-ray, and imaging of the colon and esophagus.15
The preferred protocol for diagnosis of congenital Chagas disease first requires positive serologic testing confirming the infection in the mother (see Figure 2).16 Once that is determined, microscopic and PCR-based examinations of cord blood and peripheral blood specimens are carried out during the first one to two months of the infant’s life.10 PCR is the preferred test for early congenital Chagas disease, recipients of organ transplants, and after accidental exposure since results can determine if the patient is infected earlier than trypomastigotes (developmental stage of trypanosomes) can be seen on a peripheral blood smear.5
TREATMENT CONSIDERATIONS
If there is a suspicion of Chagas disease, the patient should be referred to an infectious disease specialist for diagnosis and treatment. Nifurtimox and benznidazole are the only drugs that have been shown to improve the course of Chagas disease.5 However, neither drug is approved by the FDA, and both can only be obtained from the CDC, which makes treatment a challenge.9 In addition, up to 30% of patients terminate treatment due to the many adverse effects of these drugs.17
The dosage regimen for nifurtimox is 8-10 mg/kg/d divided into three doses for 90 days.10 Anorexia, weight loss, nausea, vomiting, and abdominal pain occur in up to 70% of patients.5 Irritability, insomnia, disorientation, and tremors can also occur. Neurotoxicity leading to peripheral neuropathy is dose dependent and requires treatment termination.5
Benznidazole is better tolerated and is active against the trypomastigotes as well as the amastigotes or intracellular form of the parasite.10 The dosage regimen for benznidazole is 5-7 mg/kg/d divided into two doses for 60 days.10 Dermatologic reactions such as rash, photosensitivity, and exfoliative dermatitis are the most common adverse effects. Peripheral neuropathy and bone marrow suppression are dose dependent and require therapy cessation.5
The CDC recommends treatment for all cases of acute disease (including congenital disease) regardless of age, and for chronic disease in patients up to age 50 who have not progressed to cardiomyopathy. In patients older than 50, treatment should be determined after weighing the potential risks and benefits (see Table 2).18
The success of treatment is determined in part by the phase of the disease. Cure rates in patients treated with either nifurtimox or benznidazole during the acute phase range from 65% to 80%.17 Chronic disease shows less of a response to traditional antiparasitic drug regimens, but higher rates of success are seen in younger patients.5 According to current estimates, successful treatment of chronic disease is limited to 15% to 30% patients.17 Treatment of congenital Chagas disease should begin as soon as the diagnosis is confirmed, and cure rates are greater than 90% if patients are treated within the first year of life.10 Treating congenital Chagas disease is important because the infection can be passed to future generations even if the disease never manifests with symptoms.19 However, if an expecting mother has known Chagas disease, antiparasitic medications are not recommended during the pregnancy because of a lack of fetal safety data for the two antiparasitic agents.20 Instead, it is recommended that women of childbearing age be treated before pregnancy, as rates of congenital infection are 25 times lower in women who are treated than in those who are not.21
PRE- AND POSTEXPOSURE PATIENT EDUCATION
Patient education mainly focuses on how to prevent Chagas disease and prognosis once diagnosed. During travel to endemic areas, the use of insecticides and residing in well-built households are the most important prevention measures. No vaccine is available, and primary chemoprophylaxis of persons visiting endemic areas is not recommended due to the low risk for infection and concerns about adverse effects.13
The survival rate of those who remain in the indeterminate phase is the same as that of the general population. However, findings that most strongly predict mortality include ventricular tachycardia, cardiomegaly, congestive heart failure (NYHA class III/IV), left ventricular systolic dysfunction, and male sex.10 Patients diagnosed with Chagas disease should be strongly encouraged not to donate blood or organs.10 Some organ and blood donation organizations selectively or universally screen donated specimens; however, this screening is not required by law.5 Family members of those diagnosed with the disease should also be tested, especially if the patient is a woman who has children or who plans to become pregnant.10
FOLLOW-UP
In patients confirmed to have Chagas disease but without symptoms and a normal ECG, further initial evaluation is not required.10 An annual history, physical exam, and ECG should be done. Those who have symptoms or ECG changes should have a complete cardiac work-up, including a 24-hour ambulatory ECG, exercise stress test, and echocardiogram to determine functional capacity. A barium swallow, barium enema, esophageal manometry, and endoscopy may be indicated in patients with gastrointestinal symptoms of Chagas disease but otherwise are not recommended. Patients taking antiparasitic drugs should have a CBC and CMP at the start of treatment and then bimonthly until the end of treatment to monitor for rare bone marrow suppression. Nifurtimox and benznidazole are also known to be mutagenic and increase the risk for lymphoma in animal studies, but this risk has not been documented in humans.10
CONCLUSION
Chagas disease is considered one of the neglected tropical diseases due to its high prevalence, chronic course, debilitating symptoms, and association with poverty.7 It is evident that incidence and prevalence of Chagas disease in the US are increasing due to recent immigration and mother-to-child transmission. Therefore, family practice clinicians must be able to recognize the red flags that suggest a T cruzi infection.5,9 Enhanced awareness of Chagas disease among health care providers will lead to better symptom control and cure rates for affected patients and may also prevent congenital infections. These efforts could serve to remove Chagas disease from the list of neglected tropical diseases.
CE/CME No: CR-1611
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest and evaluation. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Understand the prevalence and risks of Chagas disease in the United States.
• Explain the pathophysiology of Chagas disease, including the vector and transmission routes of the disease.
• Describe the clinical presentation of both the acute and chronic forms of the disease and learn when to suspect an infection.
• Outline a plan for diagnosis and treatment of Chagas disease.
• Educate women with Chagas disease about the risk of transmission for future offspring.
FACULTY
Jessica McDonald works in the Emergency Medicine Department at Dekalb Medical Center, Atlanta. Jill Mattingly is Academic Coordinator and Clinical Assistant Professor in the Physician Assistant Program at Mercer University, Atlanta.
The authors have no financial relationships to disclose.
![]()
This program has been reviewed and is approved for a maximum of 1.0 hour of American Academy of Physician Assistants (AAPA) Category 1 CME credit by the Physician Assistant Review Panel. [NPs: Both ANCC and the AANP Certification Program recognize AAPA as an approved provider of Category 1 credit.] Approval is valid for one year from the issue date of November 2016.
Article begins on next page >>
Chagas disease, a parasitic infection, is increasingly being detected in the United States, most likely due to immigration from endemic countries in South and Central America. Approximately 300,000 persons in the US have chronic Chagas disease, and up to 30% of them will develop clinically evident cardiovascular and/or gastrointestinal disease. Here’s practical guidance to help you recognize the features of symptomatic Chagas disease and follow up with appropriate evaluation and management.
Chagas disease, also known as American trypanosomiasis, is caused by the protozoan parasite Trypanosoma cruzi.1 It is most commonly spread by triatomine bugs infected with T cruzi and is endemic in many parts of Mexico and Central and South America.2 Chagas disease was first described in 1909 by Brazilian physician Carlos Chagas.3 Since its discovery, it has often been considered a disease affecting only the poor living in endemic areas of Latin America. However, 6 million to 7 million people are infected with T cruzi worldwide, and estimates suggest that Mexico and the US rank third and seventh, respectively, in the number of persons with T cruzi infection in the Western Hemisphere.1,4
An estimated 300,000 persons in the US have Chagas disease; most of them are not aware that they are infected.5,6 The increasing presence of the disease in the US, which traditionally has been considered a nonendemic area, is due to immigration from endemic areas, with subsequent infections occurring through mechanisms that do not require contact with the triatomine vector (eg, congenital transmission).1 Between 1981 and 2005, more than 7 million people from T cruzi-endemic countries in Latin America moved to the US and became legal residents.3
Early detection and treatment of Chagas disease is important because up to 30% of patients with chronic infection will develop a heart disorder, which can range in severity from conduction system abnormalities to dilated cardiomyopathy.4 In some areas of southern Mexico, Chagas disease is the most common cause of dilated cardiomyopathy.1 Equally concerning is the fact that untreated mothers with Chagas disease can transmit T cruzi to their infants.1,3 An estimated 315 babies are born with congenital Chagas disease each year in the US, an incidence equivalent to that of phenylketonuria.7 It is estimated that congenital transmission is responsible for up to one-quarter of new infections worldwide.1 Unfortunately, obstetricians are not well informed about the risk factors for congenital Chagas disease, and very limited screening of at-risk women is performed. In a 2008 survey exploring health care providers’ knowledge of and understanding about Chagas disease, obstetricians and gynecologists had the greatest knowledge deficits about the disease, although considerable deficits were also seen among other specialties.1
KISSING BUG DISEASE: ETIOLOGY/PATHOPHYSIOLOGY
Exposure to the protozoan parasite T cruzi, the cause of Chagas disease, typically occurs following the bite of a triatomine bug. Also known as “kissing bugs” because they usually bite exposed areas of the skin such as the face, triatomine bugs feed on human blood, typically at night, and act as a vector for the parasite.8 The parasite lives in the feces and urine of the triatomine bugs and is excreted near the bite during or shortly after a blood meal. The bitten person will then unknowingly smear the infected feces into the bite wound, eyes, mouth, or any opening in the skin, which gives the parasites systemic access.4 Once in the host’s bloodstream, the parasite replicates in host cells, a process that ends in cell lysis and hematogenous spread. At this point, the parasites can be seen on peripheral blood smear. Noninfected triatomine insects become infected and continue the cycle when they feed on an infected human host (see Figure 1).3 Persons of lower socioeconomic status living in endemic areas in Latin America are at a higher risk for contracting Chagas disease because “kissing bugs” commonly live in wall or roof cracks of poorly built homes. Populations living in poverty are also at risk due to minimal access to health care and prenatal care.4 Transmission of T cruzi not involving triatomine vectors occurs congenitally or through blood transfusions, consumption of contaminated food, and organ donations.4
NATURAL HISTORY OF INFECTION AND PATIENT PRESENTATION
Acute phase
Infection with the T cruzi parasite is followed by an asymptomatic incubation period of one to two weeks, which is then followed by an acute phase that can last eight to 12 weeks.5 The acute phase is characterized by a large amount of parasites in the bloodstream (see Table 1). The patient is often asymptomatic but can have nonspecific symptoms such as fever, headache, lymphadenopathy, shortness of breath, myalgia, swelling, and abdominal or chest pain.4 Because symptoms during the acute phase are typically mild, many patients do not seek medical attention until they transition into the chronic phase.4 Infants are more likely to experience severe symptoms, including myocarditis or meningoencephalitis, and thus are more likely to present during the acute phase.9
If the patient acquired the infection through an organ transplant, the acute phase symptoms can be delayed, on average, up to 112 days.5 These patients will have more noticeable symptoms, including hepatosplenomegaly, myocarditis, and congestive heart failure. Due to the known risk for transmission through organ transplants, donors are often screened for Chagas disease. Unfortunately, this screening is selective and often inconsistent.5 Therefore, the presence of the previously mentioned symptoms in a person who recently received an organ transplant should raise suspicion of Chagas disease.5
Chronic phase
Patients not treated during the acute phase will pass into the chronic phase of Chagas disease.5 This may occur due to reactivation of T cruzi infection via immunosuppression.9 At this time, the previously asymptomatic patient will have typical signs and symptoms of chronic disease, along with nodules, panniculitis, and myocarditis.4,9,10 During the chronic phase, parasites are undetectable by microscopy, but the patient can still spread the disease to the vector as well as to others congenitally and through organ donation and blood transfusions.5,9
Patients with chronic T cruzi infection who remain without signs or symptoms of infection are considered to have the indeterminate form of chronic disease. Many patients will remain in the indeterminate form throughout their lives, but between 20% and 30% will progress to the determinate form of chronic disease over years to decades.3 The determinate form is characterized by clinically evident disease and is classically divided into cardiac Chagas disease and digestive Chagas disease.5 Symptoms of the chronic phase depend on the genotype of T cruzi that caused the infection. The AG genotype has a higher incidence of digestive disease.11
Cardiac Chagas disease is believed to occur due to parasite invasion and persistence in cardiac tissue, leading to immune-mediated myocardial injury.5 Chagas cardiomyopathy is characterized by chronic myocarditis affecting all cardiac chambers and disturbances in the electrical conduction system; patients also often develop apical aneurysms. Longstanding cardiac Chagas disease can lead to more serious complications, such as episodes of ventricular tachycardia, heart block, thromboembolic phenomena, severe bradycardia, dilated cardiomyopathy, and congestive heart failure. Patients may complain of presyncope, syncope, and episodes of palpitations. They are also at high risk for sudden cardiac death.5 Patients with cardiomyopathy or cardiac insufficiency secondary to Chagas disease have a worse prognosis than those with idiopathic cardiomyopathy or decompensated heart failure due to other etiologies.12
Less common than cardiac Chagas disease, digestive Chagas disease occurs mostly in Argentina, Bolivia, Chile, Paraguay, Uruguay, and parts of Peru and Brazil; it is rarely seen in northern South America, Central America, or Mexico.5 The parasite causes gastrointestinal symptoms by damaging intramural neurons, resulting in denervation of hollow viscera. Since it affects the esophagus and colon, patients may present with dysphagia, odynophagia, cough, reflux, weight loss, constipation, and abdominal pain.5
PHYSICAL EXAMINATION: A CRUCIAL STEP
The physical examination of a patient with suspected Chagas disease can be crucial to the diagnosis. As noted, there are often few specific symptoms or physical exam findings during the acute phase. However, in some patients, swelling and inflammation may be evident at the site of inoculation, often on the face or extremities. This finding is called a chagoma. The Romaña sign, characterized by painless unilateral swelling of the upper and lower eyelid, can also be seen if the infection occurred through the conjunctiva.5 A nonpruritic morbilliform rash, called schizotrypanides, may be a presenting symptom in patients with acute disease.13 Children younger than 2 years of age are at increased risk for a severe acute infection, with signs and symptoms of pericardial effusion, myocarditis, and meningoencephalitis. Children can also develop generalized edema and lymphadenopathy. Those children who develop severe manifestations during acute infection have an increased risk for mortality.5
Chronic chagasic cardiomyopathy may present with signs of left-sided heart failure (pulmonary edema, dyspnea at rest or exertion), biventricular heart failure (hepatomegaly, peripheral edema, jugular venous distention), or thromboembolic events to the brain, lower extremities, and lungs.13 Chronic chagasic megaesophagus may lead to weight loss, esophageal dysmotility, pneumonitis due to aspiration of food trapped in the esophagus and stomach, salivary gland enlargement, and erosive esophagitis, which increases the risk for esophageal cancer. Chronic chagasic megacolon can present as an intestinal obstruction, volvulus, abdominal distention, or fecaloma.13
Clinicians should be alert to the possibility of congenital T cruzi infection in children born to women who emigrated from an endemic area or who visited an area with a high prevalence of Chagas disease. Most newborns with T cruzi infection are asymptomatic, but in some cases a thorough neonatal exam can lead to the diagnosis. Manifestations of symptomatic congenital infection include hepatosplenomegaly, low birth weight, premature birth, and low Apgar scores.5 Lab tests may reveal thrombocytopenia and anemia. Neonates with severe disease may also have respiratory distress, meningoencephalitis, and gastrointestinal problems.5
LABORATORY WORK-UP
Laboratory work-up for Chagas disease depends on the provider’s awareness of the disease and its symptoms. All patients should undergo routine blood work, including complete blood count (CBC) with differential, comprehensive metabolic panel (CMP), and liver function tests to rule out other etiologies that manifest with similar symptoms. If the patient presents during the acute phase, microscopy of blood smears with Giemsa stain should be done to visualize the parasites. In the patient who presents during the chronic phase with cardiac symptoms, measurement of B-type natriuretic peptide, troponin, C-reactive protein, and the erythrocyte sedimentation rate can be used to rule out other differential diagnoses. Electrocardiogram (ECG) may show a right bundle-branch block or left anterior fascicular block.5 Echocardiogram may show left ventricular wall motion abnormalities and/or cardiomyopathy with congestive heart failure.5,10 A work-up for digestive Chagas disease may include a barium swallow, kidney-ureter-bladder x-ray, or MRI/CT of the abdomen.14
DIAGNOSING ACUTE, CHRONIC, AND CONGENITAL CHAGAS
Accurate diagnosis of Chagas disease requires a thorough history and physical exam, as well as a high index of suspicion. Recent travel to an endemic area of Chagas disease in combination with the typical signs and symptoms—such as fever, headache, lymphadenopathy, shortness of breath, myalgia, swelling, and abdominal or chest pain—should prompt the provider to perform more specific tests.4 Inquiry about past medical history, blood transfusions, and surgeries is also imperative to make the correct diagnosis.5
The approach to diagnosis of Chagas disease depends on whether the patient presents during the acute or chronic phase. During the acute phase, the count of the trypomastigote, the mature extracellular form of the parasite T cruzi, is at its highest, making this the best time to obtain an accurate diagnosis if an infection is suspected.3 Microscopy of fresh preparations of anticoagulated blood or buffy coat may show motile parasites.10 Other options include visualization of parasites in a blood smear with Giemsa stain or hemoculture. Hemoculture is a sensitive test but takes several weeks to show growth of the parasites. Therefore, polymerase chain reaction (PCR) assay is the preferred diagnostic test due to its high sensitivity and quick turnaround time.5
Because no diagnostic gold standard exists for chronic disease, confidently diagnosing Chagas in the United States can be difficult.5 Past the acute phase (about three months after infection), microscopy and PCR cannot be used due to low parasitemia. If an infection with T cruzi is suspected but nine to 14 weeks have passed since exposure, serology is the method of choice for diagnosis. The enzyme-linked immunosorbent assay (ELISA) and immunofluorescent-antibody assay (IFA) are most often used to identify immunoglobulin (Ig) G antibodies to the parasite.
The difficulty of diagnosing Chagas disease in the chronic phase lies in the fact that neither ELISA or IFA alone is sensitive or specific enough to confirm the diagnosis.5 In order to make a serologic diagnosis of infection, positive results are needed from two serologic tests based on two different antigens or by using two different techniques (eg, ELISA or IFA). If the two tests are discordant, a third test must be done to determine the patient’s infection status. The radioimmunoprecipitation assay (RIPA) and trypomastigote excreted-secreted antigen immunoblot (TESA-blot) have been traditionally used as confirmatory tests, but even they do not have high sensitivity and specificity. A case of indeterminate Chagas disease is confirmed with positive serologic testing in a patient without symptoms and with normal ECG, chest x-ray, and imaging of the colon and esophagus.15
The preferred protocol for diagnosis of congenital Chagas disease first requires positive serologic testing confirming the infection in the mother (see Figure 2).16 Once that is determined, microscopic and PCR-based examinations of cord blood and peripheral blood specimens are carried out during the first one to two months of the infant’s life.10 PCR is the preferred test for early congenital Chagas disease, recipients of organ transplants, and after accidental exposure since results can determine if the patient is infected earlier than trypomastigotes (developmental stage of trypanosomes) can be seen on a peripheral blood smear.5
TREATMENT CONSIDERATIONS
If there is a suspicion of Chagas disease, the patient should be referred to an infectious disease specialist for diagnosis and treatment. Nifurtimox and benznidazole are the only drugs that have been shown to improve the course of Chagas disease.5 However, neither drug is approved by the FDA, and both can only be obtained from the CDC, which makes treatment a challenge.9 In addition, up to 30% of patients terminate treatment due to the many adverse effects of these drugs.17
The dosage regimen for nifurtimox is 8-10 mg/kg/d divided into three doses for 90 days.10 Anorexia, weight loss, nausea, vomiting, and abdominal pain occur in up to 70% of patients.5 Irritability, insomnia, disorientation, and tremors can also occur. Neurotoxicity leading to peripheral neuropathy is dose dependent and requires treatment termination.5
Benznidazole is better tolerated and is active against the trypomastigotes as well as the amastigotes or intracellular form of the parasite.10 The dosage regimen for benznidazole is 5-7 mg/kg/d divided into two doses for 60 days.10 Dermatologic reactions such as rash, photosensitivity, and exfoliative dermatitis are the most common adverse effects. Peripheral neuropathy and bone marrow suppression are dose dependent and require therapy cessation.5
The CDC recommends treatment for all cases of acute disease (including congenital disease) regardless of age, and for chronic disease in patients up to age 50 who have not progressed to cardiomyopathy. In patients older than 50, treatment should be determined after weighing the potential risks and benefits (see Table 2).18
The success of treatment is determined in part by the phase of the disease. Cure rates in patients treated with either nifurtimox or benznidazole during the acute phase range from 65% to 80%.17 Chronic disease shows less of a response to traditional antiparasitic drug regimens, but higher rates of success are seen in younger patients.5 According to current estimates, successful treatment of chronic disease is limited to 15% to 30% patients.17 Treatment of congenital Chagas disease should begin as soon as the diagnosis is confirmed, and cure rates are greater than 90% if patients are treated within the first year of life.10 Treating congenital Chagas disease is important because the infection can be passed to future generations even if the disease never manifests with symptoms.19 However, if an expecting mother has known Chagas disease, antiparasitic medications are not recommended during the pregnancy because of a lack of fetal safety data for the two antiparasitic agents.20 Instead, it is recommended that women of childbearing age be treated before pregnancy, as rates of congenital infection are 25 times lower in women who are treated than in those who are not.21
PRE- AND POSTEXPOSURE PATIENT EDUCATION
Patient education mainly focuses on how to prevent Chagas disease and prognosis once diagnosed. During travel to endemic areas, the use of insecticides and residing in well-built households are the most important prevention measures. No vaccine is available, and primary chemoprophylaxis of persons visiting endemic areas is not recommended due to the low risk for infection and concerns about adverse effects.13
The survival rate of those who remain in the indeterminate phase is the same as that of the general population. However, findings that most strongly predict mortality include ventricular tachycardia, cardiomegaly, congestive heart failure (NYHA class III/IV), left ventricular systolic dysfunction, and male sex.10 Patients diagnosed with Chagas disease should be strongly encouraged not to donate blood or organs.10 Some organ and blood donation organizations selectively or universally screen donated specimens; however, this screening is not required by law.5 Family members of those diagnosed with the disease should also be tested, especially if the patient is a woman who has children or who plans to become pregnant.10
FOLLOW-UP
In patients confirmed to have Chagas disease but without symptoms and a normal ECG, further initial evaluation is not required.10 An annual history, physical exam, and ECG should be done. Those who have symptoms or ECG changes should have a complete cardiac work-up, including a 24-hour ambulatory ECG, exercise stress test, and echocardiogram to determine functional capacity. A barium swallow, barium enema, esophageal manometry, and endoscopy may be indicated in patients with gastrointestinal symptoms of Chagas disease but otherwise are not recommended. Patients taking antiparasitic drugs should have a CBC and CMP at the start of treatment and then bimonthly until the end of treatment to monitor for rare bone marrow suppression. Nifurtimox and benznidazole are also known to be mutagenic and increase the risk for lymphoma in animal studies, but this risk has not been documented in humans.10
CONCLUSION
Chagas disease is considered one of the neglected tropical diseases due to its high prevalence, chronic course, debilitating symptoms, and association with poverty.7 It is evident that incidence and prevalence of Chagas disease in the US are increasing due to recent immigration and mother-to-child transmission. Therefore, family practice clinicians must be able to recognize the red flags that suggest a T cruzi infection.5,9 Enhanced awareness of Chagas disease among health care providers will lead to better symptom control and cure rates for affected patients and may also prevent congenital infections. These efforts could serve to remove Chagas disease from the list of neglected tropical diseases.
1. Hotez PJ, Dumonteil E, Betancourt Cravioto M, et al. An unfolding tragedy of Chagas disease in North America. PLoS Negl Trop Dis. 2013; 7(10):e2300.
2. Verani JR, Seitz A, Gilman RH, et al. Geographic variation in the sensitivity of recombinant antigen-based rapid tests for chronic Trypanosoma cruzi infection. Am J Trop Med Hyg. 2009;80(3):410-415.
3. Malik LH, Singh GD, Amsterdam EA. The epidemiology, clinical manifestations, and management of Chagas heart disease. Clin Cardiol. 2015;38(9):565-569.
4. World Health Organization. Chagas disease (American trypanosomiasis). Fact sheet. Updated March 2016. www.who.int/mediacentre/factsheets/fs340/en/. Accessed October 20, 2016.
5. Bern C, Kjos S, Yabsley MJ, Montgomery SP. Trypanosoma cruzi and Chagas’ disease in the United States. Clin Microbiol Rev. 2011; 24(4):655-681.
6. Stimpert KK, Montgomery SP. Physician awareness of Chagas disease, USA. Emerg Infect Dis. 2010;16(5):871-872.
7. Hotez PJ. Neglected parasitic infections and poverty in the United States. PLoS Negl Trop Dis. 2014;8(9):e3012.
8. Goupil LS, McKerrow JH. Introduction: drug discovery and development for neglected diseases. Chem Rev. 2014;114(22):11131-11137.
9. Montgomery SP, Starr MC, Cantey P, et al. Neglected parasitic infections in the United States: Chagas disease. Am J Trop Med Hyg. 2014; 90(5):814-818.
10. Bern C, Montgomery SP, Herwaldt BL, et al. Evaluation and treatment of Chagas disease in the United States. JAMA. 2007;298(18):2171-2181.
11. de Oliveira AP, Bernardo CR, Camargo AV, et al. Genetic susceptibility to cardiac and digestive clinical forms of chronic Chagas disease: involvement of the CCR5 59029 A/G polymorphism. PLoS One. 2015; 10(11):e0141847.
12. Apt W, Arribada A, Zulantay I, et al. Trypanosoma cruzi burden, genotypes, and clinical evaluation of Chilean patients with chronic Chagas cardiopathy. Parasitol Res. 2015;114(8):3007-3018.
13. Kirchhoff LV. Chagas disease (American trypanosomiasis): Background, pathophysiology, epidemiology. Emedicine.medscape.com. 2015. http://emedicine.medscape.com/article/214581-overview. Accessed October 20, 2016.
14. Knipe H, St-Amant M. Chagas disease. Radiopaedia.org. 2015. http://radiopaedia.org/articles/chagas-disease. Accessed October 20, 2016.
15. Rassi A Jr, Rassi A, Rassi SG. Predictors of mortality in chronic Chagas disease: a systematic review of observational studies. Circulation. 2007;115(9):1101-1108.
16. Gomes YM, Lorena VM, Luquetti AO. Diagnosis of Chagas disease: what has been achieved? What remains to be done with regard to diagnosis and follow up studies? Mem Inst Oswaldo Cruz. 2009; 104(suppl 1):115-121.
17. Molina I, Gómez i Prat J, Salvador F, et al. Randomized trial of posaconazole and benznidazole for chronic Chagas’ disease. N Engl J Med. 2014;370:1899-1908.
18. CDC. Parasites – American trypanosomiasis (also known as Chagas Disease). Antiparasitic Treatment. Resources For Health Professionals. www.cdc.gov/parasites/chagas/health_professionals/tx.html. Accessed October 20, 2016.
19. Carlier Y, Truyens C. Congenital Chagas disease as an ecological model of interactions between Trypanosoma cruzi parasites, pregnant women, placenta and fetuses. Acta Trop. 2015;151:103-115.
20. Moscatelli G, Moroni S, García-Bournissen F, et al. Prevention of congenital Chagas through treatment of girls and women of childbearing age. Mem Inst Oswaldo Cruz. 2015;110(4):507-509.
21. Fabbro D, Danesi E, Olivera V, et al. Trypanocide treatment of women infected with Trypanosoma cruzi and its effect on preventing congenital Chagas. PLoS Negl Trop Dis. 2014;8(11):e3312.
1. Hotez PJ, Dumonteil E, Betancourt Cravioto M, et al. An unfolding tragedy of Chagas disease in North America. PLoS Negl Trop Dis. 2013; 7(10):e2300.
2. Verani JR, Seitz A, Gilman RH, et al. Geographic variation in the sensitivity of recombinant antigen-based rapid tests for chronic Trypanosoma cruzi infection. Am J Trop Med Hyg. 2009;80(3):410-415.
3. Malik LH, Singh GD, Amsterdam EA. The epidemiology, clinical manifestations, and management of Chagas heart disease. Clin Cardiol. 2015;38(9):565-569.
4. World Health Organization. Chagas disease (American trypanosomiasis). Fact sheet. Updated March 2016. www.who.int/mediacentre/factsheets/fs340/en/. Accessed October 20, 2016.
5. Bern C, Kjos S, Yabsley MJ, Montgomery SP. Trypanosoma cruzi and Chagas’ disease in the United States. Clin Microbiol Rev. 2011; 24(4):655-681.
6. Stimpert KK, Montgomery SP. Physician awareness of Chagas disease, USA. Emerg Infect Dis. 2010;16(5):871-872.
7. Hotez PJ. Neglected parasitic infections and poverty in the United States. PLoS Negl Trop Dis. 2014;8(9):e3012.
8. Goupil LS, McKerrow JH. Introduction: drug discovery and development for neglected diseases. Chem Rev. 2014;114(22):11131-11137.
9. Montgomery SP, Starr MC, Cantey P, et al. Neglected parasitic infections in the United States: Chagas disease. Am J Trop Med Hyg. 2014; 90(5):814-818.
10. Bern C, Montgomery SP, Herwaldt BL, et al. Evaluation and treatment of Chagas disease in the United States. JAMA. 2007;298(18):2171-2181.
11. de Oliveira AP, Bernardo CR, Camargo AV, et al. Genetic susceptibility to cardiac and digestive clinical forms of chronic Chagas disease: involvement of the CCR5 59029 A/G polymorphism. PLoS One. 2015; 10(11):e0141847.
12. Apt W, Arribada A, Zulantay I, et al. Trypanosoma cruzi burden, genotypes, and clinical evaluation of Chilean patients with chronic Chagas cardiopathy. Parasitol Res. 2015;114(8):3007-3018.
13. Kirchhoff LV. Chagas disease (American trypanosomiasis): Background, pathophysiology, epidemiology. Emedicine.medscape.com. 2015. http://emedicine.medscape.com/article/214581-overview. Accessed October 20, 2016.
14. Knipe H, St-Amant M. Chagas disease. Radiopaedia.org. 2015. http://radiopaedia.org/articles/chagas-disease. Accessed October 20, 2016.
15. Rassi A Jr, Rassi A, Rassi SG. Predictors of mortality in chronic Chagas disease: a systematic review of observational studies. Circulation. 2007;115(9):1101-1108.
16. Gomes YM, Lorena VM, Luquetti AO. Diagnosis of Chagas disease: what has been achieved? What remains to be done with regard to diagnosis and follow up studies? Mem Inst Oswaldo Cruz. 2009; 104(suppl 1):115-121.
17. Molina I, Gómez i Prat J, Salvador F, et al. Randomized trial of posaconazole and benznidazole for chronic Chagas’ disease. N Engl J Med. 2014;370:1899-1908.
18. CDC. Parasites – American trypanosomiasis (also known as Chagas Disease). Antiparasitic Treatment. Resources For Health Professionals. www.cdc.gov/parasites/chagas/health_professionals/tx.html. Accessed October 20, 2016.
19. Carlier Y, Truyens C. Congenital Chagas disease as an ecological model of interactions between Trypanosoma cruzi parasites, pregnant women, placenta and fetuses. Acta Trop. 2015;151:103-115.
20. Moscatelli G, Moroni S, García-Bournissen F, et al. Prevention of congenital Chagas through treatment of girls and women of childbearing age. Mem Inst Oswaldo Cruz. 2015;110(4):507-509.
21. Fabbro D, Danesi E, Olivera V, et al. Trypanocide treatment of women infected with Trypanosoma cruzi and its effect on preventing congenital Chagas. PLoS Negl Trop Dis. 2014;8(11):e3312.
October 2016: Click for Credit
Here are 5 articles in the October issue of Clinician Reviews (individual articles are valid for one year from date of publication—expiration dates below):
1. Autism Follow-up Screening by PCPs Yields High Accuracy
To take the posttest, go to: http://bit.ly/2bTLhFS
Expires August 19, 2017
VITALS
Key clinical point:
Primary care providers can conduct the M-CHAT/F following a positive M-CHAT screening for autism spectrum disorders.
Major finding:
Primary care providers and trained interviewers agreed 86.6% of the time on the screening results of the M-CHAT/F for ASDs.
Data source:
A cohort study of 5,071 children, mean age 23 months, screened with the M-CHAT, and a subsequent 197 children screened with the M-CHAT/F in 22 Maryland primary care practices.
Disclosures:
The National Institutes of Mental Health funded the research. Dr. Sturner is director of Total Child Health (TCH), a for-profit subsidiary of the Center for Promotion of Child Development through Primary Care, which conducted the study. Barbara Howard, MD, is president of TCH. Tanya Morrel, PhD, is an employee of and stockholder in TCH, and Paul Bergmann has consulted for the company. The remaining authors had no relevant disclosures.
2. Gallstone Disease Boosts Heart Risk
To take the posttest, go to: http://bit.ly/2c7TP7D
Expires August 18, 2017
VITALS
Key clinical point:
Gallstone disease is associated with an increased risk for coronary heart disease; preventing the former can help mitigate chances of developing the latter.
Major finding:
A meta-analysis revealed a 23% increased chance of CHD in gallstone disease patients.
Data source:
A meta-analysis of seven studies involving 842,553 subjects, and a prospective cohort study of 269,142 participants in three separate studies that took place from 1980 to 2011.
Disclosures:
Funding provided by NIH, Boston Obesity Nutrition Research Center, and United States-Israel Binational Science Foundation. The authors had no relevant financial disclosures.
3. New HER2-testing Guidelines Result in More Women Eligible for Directed Treatment
To take the posttest, go to: http://bit.ly/2cd9llO
Expires July 25, 2017
VITALS
Key clinical point:
New IHC and FISH pathology guidelines categorize more breast cancers as "equivocal" regarding HER2 positivity and ultimately lead to identifying more of them as HER2 positive.
Major finding:
By using 2013 guidelines, 358 additional tumors were interpreted as positive, compared with the 2007 guidelines and 298 additional tumors were considered positive, compared with the FDA criteria.
Data source:
A cohort study involving 2,851 breast cancer samples analyzed according to three different pathology guidelines during a 1-year period.
Disclosures:
This study was supported by the Mayo Clinic. Dr. Shah reported having no relevant financial disclosures; his associates reported ties to Merck, Hospira, Ariad Pharmaceuticals, Abbott Molecular, and Genome Diagnostics.
4. Extreme Alcohol Use Worsens HIV Disease
To take the posttest, go to: http://bit.ly/2coIzG3
Expires August 14, 2017
VITALS
Key clinical point:
A pattern of heavy alcohol use over time in HIV-infected patients was associated with accelerated HIV disease progression.
Major finding:
Long-term heavy alcohol use by middle-aged, HIV-infected military veterans was associated with a 1.83-fold increased likelihood of also being in the highest-risk group for accelerated progression of HIV disease.
Data source:
This study included 3,539 U.S. military veterans receiving care for HIV infection at eight VA centers. The impact of their long-term pattern of alcohol use on HIV disease progression was assessed over an 8-year period by annual assessments using validated instruments.
Disclosures:
The presenter reported having no financial conflicts of interest regarding the study, funded by the National Institute on Alcohol Abuse and Alcoholism and the National Institute of Allergy and Infectious Diseases.
5. Weight Loss Boosts TNFis' Psoriatic Arthritis Efficacy
To take the posttest, go to: http://bit.ly/2chD4M1
Expires July 23, 2017
Here are 5 articles in the October issue of Clinician Reviews (individual articles are valid for one year from date of publication—expiration dates below):
1. Autism Follow-up Screening by PCPs Yields High Accuracy
To take the posttest, go to: http://bit.ly/2bTLhFS
Expires August 19, 2017
VITALS
Key clinical point:
Primary care providers can conduct the M-CHAT/F following a positive M-CHAT screening for autism spectrum disorders.
Major finding:
Primary care providers and trained interviewers agreed 86.6% of the time on the screening results of the M-CHAT/F for ASDs.
Data source:
A cohort study of 5,071 children, mean age 23 months, screened with the M-CHAT, and a subsequent 197 children screened with the M-CHAT/F in 22 Maryland primary care practices.
Disclosures:
The National Institutes of Mental Health funded the research. Dr. Sturner is director of Total Child Health (TCH), a for-profit subsidiary of the Center for Promotion of Child Development through Primary Care, which conducted the study. Barbara Howard, MD, is president of TCH. Tanya Morrel, PhD, is an employee of and stockholder in TCH, and Paul Bergmann has consulted for the company. The remaining authors had no relevant disclosures.
2. Gallstone Disease Boosts Heart Risk
To take the posttest, go to: http://bit.ly/2c7TP7D
Expires August 18, 2017
VITALS
Key clinical point:
Gallstone disease is associated with an increased risk for coronary heart disease; preventing the former can help mitigate chances of developing the latter.
Major finding:
A meta-analysis revealed a 23% increased chance of CHD in gallstone disease patients.
Data source:
A meta-analysis of seven studies involving 842,553 subjects, and a prospective cohort study of 269,142 participants in three separate studies that took place from 1980 to 2011.
Disclosures:
Funding provided by NIH, Boston Obesity Nutrition Research Center, and United States-Israel Binational Science Foundation. The authors had no relevant financial disclosures.
3. New HER2-testing Guidelines Result in More Women Eligible for Directed Treatment
To take the posttest, go to: http://bit.ly/2cd9llO
Expires July 25, 2017
VITALS
Key clinical point:
New IHC and FISH pathology guidelines categorize more breast cancers as "equivocal" regarding HER2 positivity and ultimately lead to identifying more of them as HER2 positive.
Major finding:
By using 2013 guidelines, 358 additional tumors were interpreted as positive, compared with the 2007 guidelines and 298 additional tumors were considered positive, compared with the FDA criteria.
Data source:
A cohort study involving 2,851 breast cancer samples analyzed according to three different pathology guidelines during a 1-year period.
Disclosures:
This study was supported by the Mayo Clinic. Dr. Shah reported having no relevant financial disclosures; his associates reported ties to Merck, Hospira, Ariad Pharmaceuticals, Abbott Molecular, and Genome Diagnostics.
4. Extreme Alcohol Use Worsens HIV Disease
To take the posttest, go to: http://bit.ly/2coIzG3
Expires August 14, 2017
VITALS
Key clinical point:
A pattern of heavy alcohol use over time in HIV-infected patients was associated with accelerated HIV disease progression.
Major finding:
Long-term heavy alcohol use by middle-aged, HIV-infected military veterans was associated with a 1.83-fold increased likelihood of also being in the highest-risk group for accelerated progression of HIV disease.
Data source:
This study included 3,539 U.S. military veterans receiving care for HIV infection at eight VA centers. The impact of their long-term pattern of alcohol use on HIV disease progression was assessed over an 8-year period by annual assessments using validated instruments.
Disclosures:
The presenter reported having no financial conflicts of interest regarding the study, funded by the National Institute on Alcohol Abuse and Alcoholism and the National Institute of Allergy and Infectious Diseases.
5. Weight Loss Boosts TNFis' Psoriatic Arthritis Efficacy
To take the posttest, go to: http://bit.ly/2chD4M1
Expires July 23, 2017
Here are 5 articles in the October issue of Clinician Reviews (individual articles are valid for one year from date of publication—expiration dates below):
1. Autism Follow-up Screening by PCPs Yields High Accuracy
To take the posttest, go to: http://bit.ly/2bTLhFS
Expires August 19, 2017
VITALS
Key clinical point:
Primary care providers can conduct the M-CHAT/F following a positive M-CHAT screening for autism spectrum disorders.
Major finding:
Primary care providers and trained interviewers agreed 86.6% of the time on the screening results of the M-CHAT/F for ASDs.
Data source:
A cohort study of 5,071 children, mean age 23 months, screened with the M-CHAT, and a subsequent 197 children screened with the M-CHAT/F in 22 Maryland primary care practices.
Disclosures:
The National Institutes of Mental Health funded the research. Dr. Sturner is director of Total Child Health (TCH), a for-profit subsidiary of the Center for Promotion of Child Development through Primary Care, which conducted the study. Barbara Howard, MD, is president of TCH. Tanya Morrel, PhD, is an employee of and stockholder in TCH, and Paul Bergmann has consulted for the company. The remaining authors had no relevant disclosures.
2. Gallstone Disease Boosts Heart Risk
To take the posttest, go to: http://bit.ly/2c7TP7D
Expires August 18, 2017
VITALS
Key clinical point:
Gallstone disease is associated with an increased risk for coronary heart disease; preventing the former can help mitigate chances of developing the latter.
Major finding:
A meta-analysis revealed a 23% increased chance of CHD in gallstone disease patients.
Data source:
A meta-analysis of seven studies involving 842,553 subjects, and a prospective cohort study of 269,142 participants in three separate studies that took place from 1980 to 2011.
Disclosures:
Funding provided by NIH, Boston Obesity Nutrition Research Center, and United States-Israel Binational Science Foundation. The authors had no relevant financial disclosures.
3. New HER2-testing Guidelines Result in More Women Eligible for Directed Treatment
To take the posttest, go to: http://bit.ly/2cd9llO
Expires July 25, 2017
VITALS
Key clinical point:
New IHC and FISH pathology guidelines categorize more breast cancers as "equivocal" regarding HER2 positivity and ultimately lead to identifying more of them as HER2 positive.
Major finding:
By using 2013 guidelines, 358 additional tumors were interpreted as positive, compared with the 2007 guidelines and 298 additional tumors were considered positive, compared with the FDA criteria.
Data source:
A cohort study involving 2,851 breast cancer samples analyzed according to three different pathology guidelines during a 1-year period.
Disclosures:
This study was supported by the Mayo Clinic. Dr. Shah reported having no relevant financial disclosures; his associates reported ties to Merck, Hospira, Ariad Pharmaceuticals, Abbott Molecular, and Genome Diagnostics.
4. Extreme Alcohol Use Worsens HIV Disease
To take the posttest, go to: http://bit.ly/2coIzG3
Expires August 14, 2017
VITALS
Key clinical point:
A pattern of heavy alcohol use over time in HIV-infected patients was associated with accelerated HIV disease progression.
Major finding:
Long-term heavy alcohol use by middle-aged, HIV-infected military veterans was associated with a 1.83-fold increased likelihood of also being in the highest-risk group for accelerated progression of HIV disease.
Data source:
This study included 3,539 U.S. military veterans receiving care for HIV infection at eight VA centers. The impact of their long-term pattern of alcohol use on HIV disease progression was assessed over an 8-year period by annual assessments using validated instruments.
Disclosures:
The presenter reported having no financial conflicts of interest regarding the study, funded by the National Institute on Alcohol Abuse and Alcoholism and the National Institute of Allergy and Infectious Diseases.
5. Weight Loss Boosts TNFis' Psoriatic Arthritis Efficacy
To take the posttest, go to: http://bit.ly/2chD4M1
Expires July 23, 2017
