User login
Valproic acid-induced hyperammonemic encephalopathy

Mrs. C, age 75, is transferred to our inpatient medical/surgical hospital from a psychiatric hospital after presenting with shortness of breath and altered mental status.
Eight days earlier, Mrs. C had been admitted to the psychiatric hospital for bipolar mania with psychotic features. While there, Mrs. C received quetiapine, 400 mg nightly, and an initial valproic acid (VPA) dosage of 500 mg 2 times daily. While receiving VPA 500 mg 2 times daily, her VPA total level was 62 µg/mL, which is on the lower end of the therapeutic range (50 to 125 µg/mL). This prompted the team at the psychiatric hospital to increase her VPA dosage to 500 mg 3 times daily the day before she was transferred to our hospital.
At our hospital, she is found to be in hypoxic respiratory failure secondary to pneumonia. Upon admission, her laboratory data show evidence of infection and anemia and she also has an
From hospital Day 3 to Day 6, Mrs. C experiences gradual improvement in her respiratory and mental status. However, on hospital Day 7, she has extreme somnolence and altered mental status without respiratory involvement. Our team suspects VPA toxicity and/or VPA-induced hyperammonemic encephalopathy (VHE).
VPA-induced hyperammonemia
Hyperammonemia can occur in individuals receiving VPA and is most often asymptomatic. However, elevations in ammonia may lead to VHE, which is a rare but serious adverse effect. VHE has been reported early in treatment, in acute VPA overdose, and in chronic VPA use despite normal doses and levels.1 It also can occur in the absence of clinical and laboratory evidence of hepatotoxicity. VHE is associated with significant morbidity and CNS damage. Symptoms of VHE include vomiting, lethargy, and confusion. If left untreated, VHE can lead to coma and death.
Mechanism of VHE. The exact mechanism of VHE is unknown.1-3 Ammonia is a toxic base produced by deamination of amino acids. The liver eliminates ammonia via the urea cycle.2 Valproic acid metabolites, propionate and 4-en-VPA, can directly inhibit N-acetyl glutamate, which can disrupt the urea cycle, leading to elevated ammonia levels.3 Long-term or high-dose VPA can lead to carnitine deficiency, primarily by inhibiting its biosynthesis and depleting stores.4 Carnitine deficiency leads to disturbances in mitochondrial function, causing inhibition of the urea cycle and increasing ammonia. CNS toxicity due to hyperammonemia is thought to be due to activation of glutamate receptors.3
Risk factors. Co-administration of other antiepileptic drugs (AEDs) with VPA is a risk factor for VHE.1,5 This happens because enzyme-inducing AEDs such as phenytoin, phenobarbital, and carbamazepine can increase toxic metabolites of VPA, which can lead to hyperammonemia. Topiramate can also inhibit the urea cycle, leading to increased ammonia levels. Additionally, co-administration of VPA with quetiapine, paliperidone, risperidone, or aripiprazole has been reported to increase the risk of VHE.1,5 Intellectual disability, carnitine deficiency, low albumin, and abnormal liver function have also been reported to increase the risk of VHE.1,5
Continue to: Diagnosis and management
Diagnosis and management. If a patient receiving VPA is experiencing nausea, fatigue, or somnolence, it is important to check the patient’s ammonia level (normal range: 11 to 32 µmol/L) and VPA total levels (therapeutic range: 50 to 125 µg/mL). Consider checking a VPA free level, especially in geriatric patients or patients who have low albumin; the therapeutic range of VPA free is 6 to 22 µg/mL.3 If the ammonia level is elevated, discontinue VPA immediately (Table).1-3 Clinicians may also elect to prescribe lactulose until ammonia levels return to normal range. Adding levocarnitine may also help, although evidence is limited to small case series or retrospective studies.3 Currently, there is no known advantage in combining lactulose and levocarnitine to address VHE. Severe cases of VHE (ammonia levels >400 µmol/L) may require hemodialysis.1

Prevention. Strategies to prevent VHE include avoiding polypharmacy, especially concurrent use of enzyme-inducing AEDs and possibly second-generation antipsychotics. Additionally, VPA should not be used in individuals with urea cycle disorders. It is unknown if levocarnitine supplementation is preventive, but this approach has been suggested.3
CASE CONTINUED
Mrs. C has several possible risk factors for VHE, including co-administration of quetiapine and VPA, and a low albumin level. A further laboratory workup for Mrs. C reveals a VPA free level of 19 µg/mL (21.1% free), a VPA total level of 90 µg/mL, and an ammonia level of 79 µmol/L, confirming our suspicions regarding VHE. We determine that Mrs. C’s altered mental status is likely due her elevated ammonia levels, because the infection had been improving in the days leading up to the sudden, extreme somnolence.
VPA is immediately stopped and Mrs. C receives 1 dose of lactulose. The following day, Mrs. C’s mental status improves, and her ammonia levels return to normal. On hospital Day 9, she is transferred back to the psychiatric facility for management of manic and psychotic symptoms.
Related Resources
- Brown LM, Cupples N, Moore TA. Levocarnitine for valproate-induced hyperammonemia in the psychiatric setting: a case series and literature review. Ment Health Clin. 2018;8(3):148-154.
- Aires CCP, van Cruchten A, Ijlat L, et al. New insights on the mechanisms of valproate-induced hyperammonemia: inhibition of hepatic N-acetylglutamate synthase activity by valproyl-CoA. J Hepatol. 2011;55(2):426-434.
Drug Brand Names
Aripiprazole • Abilify
Carbamazepine • Tegretol
Lactulose • Enulose
Levocarnitine • Carnitine, Carnitor
Levofloxacin • Levaquin IV
Paliperidone • Invega
Phenobarbital • Luminal
Phenytoin • Dilantin
Quetiapine • Seroquel
Risperidone • Risperdal
Topiramate • Topamax
Valproic acid • Depakene
1. Chopra A, Kolla BP, Mansukhani MP, et al. Valproate-induced hyperammonemic encephalopathy: an update on risk factors, clinical correlates, and management. Gen Hosp Psychiatry. 2012;34(3):290-298.
2. Kowalski PC, Dowben JS, Keltner NL. Ammonium: the deadly toxin you don’t want to miss when using mood stabilizers. Perspect Psychiatr Care. 2013;49(4):221-225.
3. Baddour E, Tewksbury A, Stauner N. Valproic acid-induced hyper ammonemia: incidence, clinical significance, and treatment management. Ment Health Clin. 2018;8(2):73-77.
4. Raskind JY, El-Chaar GM. The role of carnitine supplementation during valproic acid therapy. Ann Pharmacother. 2000;34(5):630-638. 5. Tseng YL, Huang CR, Lin CH, et al. Risk factors of hyperammonemia in patients with epilepsy. Medicine (Baltimore). 2014;93(11):e66. doi: 10.1097/MD.0000000000000066.
Mrs. C, age 75, is transferred to our inpatient medical/surgical hospital from a psychiatric hospital after presenting with shortness of breath and altered mental status.
Eight days earlier, Mrs. C had been admitted to the psychiatric hospital for bipolar mania with psychotic features. While there, Mrs. C received quetiapine, 400 mg nightly, and an initial valproic acid (VPA) dosage of 500 mg 2 times daily. While receiving VPA 500 mg 2 times daily, her VPA total level was 62 µg/mL, which is on the lower end of the therapeutic range (50 to 125 µg/mL). This prompted the team at the psychiatric hospital to increase her VPA dosage to 500 mg 3 times daily the day before she was transferred to our hospital.
At our hospital, she is found to be in hypoxic respiratory failure secondary to pneumonia. Upon admission, her laboratory data show evidence of infection and anemia and she also has an
From hospital Day 3 to Day 6, Mrs. C experiences gradual improvement in her respiratory and mental status. However, on hospital Day 7, she has extreme somnolence and altered mental status without respiratory involvement. Our team suspects VPA toxicity and/or VPA-induced hyperammonemic encephalopathy (VHE).
VPA-induced hyperammonemia
Hyperammonemia can occur in individuals receiving VPA and is most often asymptomatic. However, elevations in ammonia may lead to VHE, which is a rare but serious adverse effect. VHE has been reported early in treatment, in acute VPA overdose, and in chronic VPA use despite normal doses and levels.1 It also can occur in the absence of clinical and laboratory evidence of hepatotoxicity. VHE is associated with significant morbidity and CNS damage. Symptoms of VHE include vomiting, lethargy, and confusion. If left untreated, VHE can lead to coma and death.
Mechanism of VHE. The exact mechanism of VHE is unknown.1-3 Ammonia is a toxic base produced by deamination of amino acids. The liver eliminates ammonia via the urea cycle.2 Valproic acid metabolites, propionate and 4-en-VPA, can directly inhibit N-acetyl glutamate, which can disrupt the urea cycle, leading to elevated ammonia levels.3 Long-term or high-dose VPA can lead to carnitine deficiency, primarily by inhibiting its biosynthesis and depleting stores.4 Carnitine deficiency leads to disturbances in mitochondrial function, causing inhibition of the urea cycle and increasing ammonia. CNS toxicity due to hyperammonemia is thought to be due to activation of glutamate receptors.3
Risk factors. Co-administration of other antiepileptic drugs (AEDs) with VPA is a risk factor for VHE.1,5 This happens because enzyme-inducing AEDs such as phenytoin, phenobarbital, and carbamazepine can increase toxic metabolites of VPA, which can lead to hyperammonemia. Topiramate can also inhibit the urea cycle, leading to increased ammonia levels. Additionally, co-administration of VPA with quetiapine, paliperidone, risperidone, or aripiprazole has been reported to increase the risk of VHE.1,5 Intellectual disability, carnitine deficiency, low albumin, and abnormal liver function have also been reported to increase the risk of VHE.1,5
Continue to: Diagnosis and management
Diagnosis and management. If a patient receiving VPA is experiencing nausea, fatigue, or somnolence, it is important to check the patient’s ammonia level (normal range: 11 to 32 µmol/L) and VPA total levels (therapeutic range: 50 to 125 µg/mL). Consider checking a VPA free level, especially in geriatric patients or patients who have low albumin; the therapeutic range of VPA free is 6 to 22 µg/mL.3 If the ammonia level is elevated, discontinue VPA immediately (Table).1-3 Clinicians may also elect to prescribe lactulose until ammonia levels return to normal range. Adding levocarnitine may also help, although evidence is limited to small case series or retrospective studies.3 Currently, there is no known advantage in combining lactulose and levocarnitine to address VHE. Severe cases of VHE (ammonia levels >400 µmol/L) may require hemodialysis.1
Prevention. Strategies to prevent VHE include avoiding polypharmacy, especially concurrent use of enzyme-inducing AEDs and possibly second-generation antipsychotics. Additionally, VPA should not be used in individuals with urea cycle disorders. It is unknown if levocarnitine supplementation is preventive, but this approach has been suggested.3
CASE CONTINUED
Mrs. C has several possible risk factors for VHE, including co-administration of quetiapine and VPA, and a low albumin level. A further laboratory workup for Mrs. C reveals a VPA free level of 19 µg/mL (21.1% free), a VPA total level of 90 µg/mL, and an ammonia level of 79 µmol/L, confirming our suspicions regarding VHE. We determine that Mrs. C’s altered mental status is likely due her elevated ammonia levels, because the infection had been improving in the days leading up to the sudden, extreme somnolence.
VPA is immediately stopped and Mrs. C receives 1 dose of lactulose. The following day, Mrs. C’s mental status improves, and her ammonia levels return to normal. On hospital Day 9, she is transferred back to the psychiatric facility for management of manic and psychotic symptoms.
Related Resources
- Brown LM, Cupples N, Moore TA. Levocarnitine for valproate-induced hyperammonemia in the psychiatric setting: a case series and literature review. Ment Health Clin. 2018;8(3):148-154.
- Aires CCP, van Cruchten A, Ijlat L, et al. New insights on the mechanisms of valproate-induced hyperammonemia: inhibition of hepatic N-acetylglutamate synthase activity by valproyl-CoA. J Hepatol. 2011;55(2):426-434.
Drug Brand Names
Aripiprazole • Abilify
Carbamazepine • Tegretol
Lactulose • Enulose
Levocarnitine • Carnitine, Carnitor
Levofloxacin • Levaquin IV
Paliperidone • Invega
Phenobarbital • Luminal
Phenytoin • Dilantin
Quetiapine • Seroquel
Risperidone • Risperdal
Topiramate • Topamax
Valproic acid • Depakene
Mrs. C, age 75, is transferred to our inpatient medical/surgical hospital from a psychiatric hospital after presenting with shortness of breath and altered mental status.
Eight days earlier, Mrs. C had been admitted to the psychiatric hospital for bipolar mania with psychotic features. While there, Mrs. C received quetiapine, 400 mg nightly, and an initial valproic acid (VPA) dosage of 500 mg 2 times daily. While receiving VPA 500 mg 2 times daily, her VPA total level was 62 µg/mL, which is on the lower end of the therapeutic range (50 to 125 µg/mL). This prompted the team at the psychiatric hospital to increase her VPA dosage to 500 mg 3 times daily the day before she was transferred to our hospital.
At our hospital, she is found to be in hypoxic respiratory failure secondary to pneumonia. Upon admission, her laboratory data show evidence of infection and anemia and she also has an
From hospital Day 3 to Day 6, Mrs. C experiences gradual improvement in her respiratory and mental status. However, on hospital Day 7, she has extreme somnolence and altered mental status without respiratory involvement. Our team suspects VPA toxicity and/or VPA-induced hyperammonemic encephalopathy (VHE).
VPA-induced hyperammonemia
Hyperammonemia can occur in individuals receiving VPA and is most often asymptomatic. However, elevations in ammonia may lead to VHE, which is a rare but serious adverse effect. VHE has been reported early in treatment, in acute VPA overdose, and in chronic VPA use despite normal doses and levels.1 It also can occur in the absence of clinical and laboratory evidence of hepatotoxicity. VHE is associated with significant morbidity and CNS damage. Symptoms of VHE include vomiting, lethargy, and confusion. If left untreated, VHE can lead to coma and death.
Mechanism of VHE. The exact mechanism of VHE is unknown.1-3 Ammonia is a toxic base produced by deamination of amino acids. The liver eliminates ammonia via the urea cycle.2 Valproic acid metabolites, propionate and 4-en-VPA, can directly inhibit N-acetyl glutamate, which can disrupt the urea cycle, leading to elevated ammonia levels.3 Long-term or high-dose VPA can lead to carnitine deficiency, primarily by inhibiting its biosynthesis and depleting stores.4 Carnitine deficiency leads to disturbances in mitochondrial function, causing inhibition of the urea cycle and increasing ammonia. CNS toxicity due to hyperammonemia is thought to be due to activation of glutamate receptors.3
Risk factors. Co-administration of other antiepileptic drugs (AEDs) with VPA is a risk factor for VHE.1,5 This happens because enzyme-inducing AEDs such as phenytoin, phenobarbital, and carbamazepine can increase toxic metabolites of VPA, which can lead to hyperammonemia. Topiramate can also inhibit the urea cycle, leading to increased ammonia levels. Additionally, co-administration of VPA with quetiapine, paliperidone, risperidone, or aripiprazole has been reported to increase the risk of VHE.1,5 Intellectual disability, carnitine deficiency, low albumin, and abnormal liver function have also been reported to increase the risk of VHE.1,5
Continue to: Diagnosis and management
Diagnosis and management. If a patient receiving VPA is experiencing nausea, fatigue, or somnolence, it is important to check the patient’s ammonia level (normal range: 11 to 32 µmol/L) and VPA total levels (therapeutic range: 50 to 125 µg/mL). Consider checking a VPA free level, especially in geriatric patients or patients who have low albumin; the therapeutic range of VPA free is 6 to 22 µg/mL.3 If the ammonia level is elevated, discontinue VPA immediately (Table).1-3 Clinicians may also elect to prescribe lactulose until ammonia levels return to normal range. Adding levocarnitine may also help, although evidence is limited to small case series or retrospective studies.3 Currently, there is no known advantage in combining lactulose and levocarnitine to address VHE. Severe cases of VHE (ammonia levels >400 µmol/L) may require hemodialysis.1
Prevention. Strategies to prevent VHE include avoiding polypharmacy, especially concurrent use of enzyme-inducing AEDs and possibly second-generation antipsychotics. Additionally, VPA should not be used in individuals with urea cycle disorders. It is unknown if levocarnitine supplementation is preventive, but this approach has been suggested.3
CASE CONTINUED
Mrs. C has several possible risk factors for VHE, including co-administration of quetiapine and VPA, and a low albumin level. A further laboratory workup for Mrs. C reveals a VPA free level of 19 µg/mL (21.1% free), a VPA total level of 90 µg/mL, and an ammonia level of 79 µmol/L, confirming our suspicions regarding VHE. We determine that Mrs. C’s altered mental status is likely due her elevated ammonia levels, because the infection had been improving in the days leading up to the sudden, extreme somnolence.
VPA is immediately stopped and Mrs. C receives 1 dose of lactulose. The following day, Mrs. C’s mental status improves, and her ammonia levels return to normal. On hospital Day 9, she is transferred back to the psychiatric facility for management of manic and psychotic symptoms.
Related Resources
- Brown LM, Cupples N, Moore TA. Levocarnitine for valproate-induced hyperammonemia in the psychiatric setting: a case series and literature review. Ment Health Clin. 2018;8(3):148-154.
- Aires CCP, van Cruchten A, Ijlat L, et al. New insights on the mechanisms of valproate-induced hyperammonemia: inhibition of hepatic N-acetylglutamate synthase activity by valproyl-CoA. J Hepatol. 2011;55(2):426-434.
Drug Brand Names
Aripiprazole • Abilify
Carbamazepine • Tegretol
Lactulose • Enulose
Levocarnitine • Carnitine, Carnitor
Levofloxacin • Levaquin IV
Paliperidone • Invega
Phenobarbital • Luminal
Phenytoin • Dilantin
Quetiapine • Seroquel
Risperidone • Risperdal
Topiramate • Topamax
Valproic acid • Depakene
1. Chopra A, Kolla BP, Mansukhani MP, et al. Valproate-induced hyperammonemic encephalopathy: an update on risk factors, clinical correlates, and management. Gen Hosp Psychiatry. 2012;34(3):290-298.
2. Kowalski PC, Dowben JS, Keltner NL. Ammonium: the deadly toxin you don’t want to miss when using mood stabilizers. Perspect Psychiatr Care. 2013;49(4):221-225.
3. Baddour E, Tewksbury A, Stauner N. Valproic acid-induced hyper ammonemia: incidence, clinical significance, and treatment management. Ment Health Clin. 2018;8(2):73-77.
4. Raskind JY, El-Chaar GM. The role of carnitine supplementation during valproic acid therapy. Ann Pharmacother. 2000;34(5):630-638. 5. Tseng YL, Huang CR, Lin CH, et al. Risk factors of hyperammonemia in patients with epilepsy. Medicine (Baltimore). 2014;93(11):e66. doi: 10.1097/MD.0000000000000066.
1. Chopra A, Kolla BP, Mansukhani MP, et al. Valproate-induced hyperammonemic encephalopathy: an update on risk factors, clinical correlates, and management. Gen Hosp Psychiatry. 2012;34(3):290-298.
2. Kowalski PC, Dowben JS, Keltner NL. Ammonium: the deadly toxin you don’t want to miss when using mood stabilizers. Perspect Psychiatr Care. 2013;49(4):221-225.
3. Baddour E, Tewksbury A, Stauner N. Valproic acid-induced hyper ammonemia: incidence, clinical significance, and treatment management. Ment Health Clin. 2018;8(2):73-77.
4. Raskind JY, El-Chaar GM. The role of carnitine supplementation during valproic acid therapy. Ann Pharmacother. 2000;34(5):630-638. 5. Tseng YL, Huang CR, Lin CH, et al. Risk factors of hyperammonemia in patients with epilepsy. Medicine (Baltimore). 2014;93(11):e66. doi: 10.1097/MD.0000000000000066.

Effects of psychotropic medications on thyroid function

Ms. L, age 53, presents to an inpatient psychiatric unit with depression, difficulty concentrating, fatigue, cognitive blunting, loss of appetite, increased alcohol intake, and recent suicidal ideation. Her symptoms began 3 months ago and gradually worsened. Her medical and psychiatric history is significant for hypertension, fibromyalgia, and chronic pain (back and neck), major depressive disorder (MDD; recurrent, severe), and generalized anxiety disorder (GAD). Ms. L’s current medication regimen includes lisinopril, 40 mg daily; fluoxetine, 60 mg daily; mirtazapine, 30 mg at bedtime; gabapentin, 300 mg twice daily; alprazolam, 0.5 mg twice daily as needed for anxiety; and oral docusate, 100 mg twice daily as needed. Her blood pressure is 124/85 mm Hg, heart rate is 66 beats per minute, and an electrocardiogram is normal. Laboratory workup reveals a potassium level of 4.4 mEq/L, blood urea nitrogen level of 20 mg/dL, serum creatinine level of 0.8 mg/dL, estimated creatinine clearance of 89.6 mL/min, free triiodothyronine (T3) levels of 2.7 pg/mL, thyroid-stimulating hormone (TSH) level of 7.68 mIU/L, free thyroxine (T4) level of 1.3 ng/dL, and blood ethanol level <10 mg/dL. In addition to the symptoms Ms. L initially described, a review of systems reveals word-finding difficulty, cold intolerance, constipation, hair loss, brittle nails, and dry skin.
To target Ms. L’s MDD, GAD, fibromyalgia, and chronic pain, fluoxetine, 60 mg daily is cross titrated beginning on Day 1 to duloxetine, 60 mg twice daily, over 4 days. Mirtazapine is decreased on Day 3 to 7.5 mg at bedtime to target Ms. L’s sleep and appetite. Due to the presence of several symptoms associated with hypothyroidism and a slightly elevated TSH level, on Day 6 we initiate adjunctive levothyroxine, 50 mcg daily each morning to target symptomatic subclinical hypothyroidism, and to potentially augment the other medications prescribed to address Ms. L’s MDD.
Thyroid hormone function is a complex physiological process controlled through the hypothalamic-pituitary-thyroid (HPT) axis. Psychotropic medications can impact thyroid hormone function and contribute to aberrations in thyroid physiology.1 Because patients with mental illness may require multiple psychotropic medications, it is imperative to understand the potential effects of these agents.
Antidepressants can induce hypothyroidism along multiple points of hormonal synthesis and iodine utilization. Tricyclic antidepressants have been implicated in the development of drug-iodide complexes, thus reducing biologically active iodine.2 Tricyclic antidepressants also can bind thyroid peroxidase, an enzyme necessary in the production of T4 and T3, altering hormonal production, resulting in a hypothyroid state.1 Non-tricyclic antidepressants (ie, selective serotonin reuptake inhibitors [SSRIs] and non-SSRIs [including serotonin-norepinephrine reuptake inhibitors and mirtazapine]) have also been implicated in thyroid dysfunction. Selective serotonin reuptake inhibitors have the propensity to induce hypothyroidism through inhibition of thyroid hormones T4 and T3.1,3 This inhibition is not always seen with concurrent reductions in TSH levels. Conversely, non-SSRIs can influence thyroid hormone levels with great variation, leading to thyroid hormone levels that are increased, decreased, or unchanged.1 Patients with a history of thyroid dysfunction should receive close thyroid function monitoring, especially while taking antidepressants.
Antipsychotics have a proclivity to induce hypothyroidism by means similar to antidepressants via hormonal manipulation and immunogenicity. Phenothiazines impact thyroid function through hormonal activation and degradation, and induction of autoimmunity.1 Autoimmunity may develop by means of antibody production or antigen immunization through the major histocompatibility complex.2 Other first-generation antipsychotics (FGAs) (eg, haloperidol and loxapine) are known to antagonize dopamine receptors in the tuberoinfundibular pathway, resulting in increased prolactin levels. Hyperprolactinemia may result in increased TSH levels through HPT axis activation.1 Additionally, FGAs can induce an immunogenic effect through production of antithyroid antibodies.1 Similar to FGAs, second-generation antipsychotics (SGAs) can increase TSH levels through hyperprolactinemia. Further research focused on SGAs is needed to determine how profound this effect may be.
The Table1 outlines considerations for modifying psychotropic therapy based on the presence of concurrent thyroid dysfunction. Thyroid function should be routinely assessed in patients treated with antipsychotics.

Mood stabilizers are capable of altering thyroid function and inducing a hypothyroid state. Lithium has been implicated in both hypothyroidism and hyperthyroidism due to its inhibition of hormonal secretion, and toxicity to thyroid cells with chronic use, respectively.1,4 Hypothyroidism can develop shortly after initiating lithium; women tend to have a greater predilection for thyroid dysfunction than men.1 Carbamazepine (CBZ) can reduce thyroid hormone levels without having a direct effect on TSH or thyroid dysfunction.1 As with lithium, women tend to be more susceptible to this effect. Valproic acid (VPA) has been shown to either increase, decrease, or have no impact on thyroid hormone levels, with little effect on TSH.1 When VPA is given in combination with CBZ, significant reductions in thyroid levels with a concurrent increase in TSH can occur.1 In patients with preexisting thyroid dysfunction, the combination of VPA and CBZ should be used with caution.
Continue to: CASE
CASE CONTINUED
By Day 8, Ms. L reports less fatigue, clearer thinking, improved concentration, and less pain. She also no longer reports suicidal ideation, and demonstrates improved appetite and mood. She is discharged on Day 9 of her hospitalization.
The treatment team refers Ms. L for outpatient follow-up in 4 weeks, with a goal TSH level <3.0. Unfortunately, the effects of levothyroxine on Ms. L’s TSH level could not be determined during her hospital stay, and she has not returned to the facility since the initial presentation.
Thyroid function and mood
Ms. L’s case illustrates how thyroid function, pain, cognition, and mood may be interconnected. It is important to address all potential underlying comorbidities and establish appropriate outpatient care and follow-up so that patients may experience a more robust recovery. Further, this case highlights the importance of ruling out other potential medical causes of MDD during the initial diagnosis, and during times of recurrence or relapse, especially when a recent stressor, medication changes, or medication nonadherence cannot be identified as potential contributors.
Related Resources
- Cojić M, Cvejanov-Kezunović L. Subclinical hypothyroidism – whether and when to start treatment? Open Access Maced J Med Sci. 2017;5(7):1042-1046.
- Garber JR, Cobin RH, Gharib H, et al. Clinical practice guidelines for hypothyroidism in adults: cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Thyroid. 2012;22(12):1200-1235.
- Iosifescu DV. ‘Supercharge’ antidepressants by adding thyroid hormones. Current Psychiatry. 2006;5(7):15-20,25.
Drug Brand Names
Alprazolam • Xanax
Aripiprazole • Abilify
Bupropion • Wellbutrin
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Clozapine • Clozaril
Duloxetine • Cymbalta
Fluoxetine • Prozac
Fluphenazine • Prolixin
Gabapentin • Neurontin
Haloperidol • Haldol
Levothyroxine • Synthroid
Lisinopril • Prinivil, Zestril
Lithium • Eskalith, Lithobid
Loxapine • Loxitane
Mirtazapine • Remeron
Quetiapine • Seroquel
Risperidone • Risperdal
Thioridazine • Mellaril
Valproic acid • Depakote
1. Bou Khalil R, Richa S. Thyroid adverse effect of psychotropic drugs: a review. Clin Neuropharm. 2001;34(6):248-255.
2. Sauvage MF, Marquet P, Rousseau A, et al. Relationship between psychotropic drugs and thyroid function: a review. Toxicol Appl Pharmacol. 1998;149(2):127-135.
3. Shelton RC, Winn S, Ekhatore N, et al. The effects of antidepressants on the thyroid axis in depression. Biol Psychiatry. 1993;33(2):120-126.
4. Kundra P, Burman KD. The effect of medications on thyroid function tests. Med Clin North Am. 2012;96(2):283-295.
Ms. L, age 53, presents to an inpatient psychiatric unit with depression, difficulty concentrating, fatigue, cognitive blunting, loss of appetite, increased alcohol intake, and recent suicidal ideation. Her symptoms began 3 months ago and gradually worsened. Her medical and psychiatric history is significant for hypertension, fibromyalgia, and chronic pain (back and neck), major depressive disorder (MDD; recurrent, severe), and generalized anxiety disorder (GAD). Ms. L’s current medication regimen includes lisinopril, 40 mg daily; fluoxetine, 60 mg daily; mirtazapine, 30 mg at bedtime; gabapentin, 300 mg twice daily; alprazolam, 0.5 mg twice daily as needed for anxiety; and oral docusate, 100 mg twice daily as needed. Her blood pressure is 124/85 mm Hg, heart rate is 66 beats per minute, and an electrocardiogram is normal. Laboratory workup reveals a potassium level of 4.4 mEq/L, blood urea nitrogen level of 20 mg/dL, serum creatinine level of 0.8 mg/dL, estimated creatinine clearance of 89.6 mL/min, free triiodothyronine (T3) levels of 2.7 pg/mL, thyroid-stimulating hormone (TSH) level of 7.68 mIU/L, free thyroxine (T4) level of 1.3 ng/dL, and blood ethanol level <10 mg/dL. In addition to the symptoms Ms. L initially described, a review of systems reveals word-finding difficulty, cold intolerance, constipation, hair loss, brittle nails, and dry skin.
To target Ms. L’s MDD, GAD, fibromyalgia, and chronic pain, fluoxetine, 60 mg daily is cross titrated beginning on Day 1 to duloxetine, 60 mg twice daily, over 4 days. Mirtazapine is decreased on Day 3 to 7.5 mg at bedtime to target Ms. L’s sleep and appetite. Due to the presence of several symptoms associated with hypothyroidism and a slightly elevated TSH level, on Day 6 we initiate adjunctive levothyroxine, 50 mcg daily each morning to target symptomatic subclinical hypothyroidism, and to potentially augment the other medications prescribed to address Ms. L’s MDD.
Thyroid hormone function is a complex physiological process controlled through the hypothalamic-pituitary-thyroid (HPT) axis. Psychotropic medications can impact thyroid hormone function and contribute to aberrations in thyroid physiology.1 Because patients with mental illness may require multiple psychotropic medications, it is imperative to understand the potential effects of these agents.
Antidepressants can induce hypothyroidism along multiple points of hormonal synthesis and iodine utilization. Tricyclic antidepressants have been implicated in the development of drug-iodide complexes, thus reducing biologically active iodine.2 Tricyclic antidepressants also can bind thyroid peroxidase, an enzyme necessary in the production of T4 and T3, altering hormonal production, resulting in a hypothyroid state.1 Non-tricyclic antidepressants (ie, selective serotonin reuptake inhibitors [SSRIs] and non-SSRIs [including serotonin-norepinephrine reuptake inhibitors and mirtazapine]) have also been implicated in thyroid dysfunction. Selective serotonin reuptake inhibitors have the propensity to induce hypothyroidism through inhibition of thyroid hormones T4 and T3.1,3 This inhibition is not always seen with concurrent reductions in TSH levels. Conversely, non-SSRIs can influence thyroid hormone levels with great variation, leading to thyroid hormone levels that are increased, decreased, or unchanged.1 Patients with a history of thyroid dysfunction should receive close thyroid function monitoring, especially while taking antidepressants.
Antipsychotics have a proclivity to induce hypothyroidism by means similar to antidepressants via hormonal manipulation and immunogenicity. Phenothiazines impact thyroid function through hormonal activation and degradation, and induction of autoimmunity.1 Autoimmunity may develop by means of antibody production or antigen immunization through the major histocompatibility complex.2 Other first-generation antipsychotics (FGAs) (eg, haloperidol and loxapine) are known to antagonize dopamine receptors in the tuberoinfundibular pathway, resulting in increased prolactin levels. Hyperprolactinemia may result in increased TSH levels through HPT axis activation.1 Additionally, FGAs can induce an immunogenic effect through production of antithyroid antibodies.1 Similar to FGAs, second-generation antipsychotics (SGAs) can increase TSH levels through hyperprolactinemia. Further research focused on SGAs is needed to determine how profound this effect may be.
The Table1 outlines considerations for modifying psychotropic therapy based on the presence of concurrent thyroid dysfunction. Thyroid function should be routinely assessed in patients treated with antipsychotics.
Mood stabilizers are capable of altering thyroid function and inducing a hypothyroid state. Lithium has been implicated in both hypothyroidism and hyperthyroidism due to its inhibition of hormonal secretion, and toxicity to thyroid cells with chronic use, respectively.1,4 Hypothyroidism can develop shortly after initiating lithium; women tend to have a greater predilection for thyroid dysfunction than men.1 Carbamazepine (CBZ) can reduce thyroid hormone levels without having a direct effect on TSH or thyroid dysfunction.1 As with lithium, women tend to be more susceptible to this effect. Valproic acid (VPA) has been shown to either increase, decrease, or have no impact on thyroid hormone levels, with little effect on TSH.1 When VPA is given in combination with CBZ, significant reductions in thyroid levels with a concurrent increase in TSH can occur.1 In patients with preexisting thyroid dysfunction, the combination of VPA and CBZ should be used with caution.
Continue to: CASE
CASE CONTINUED
By Day 8, Ms. L reports less fatigue, clearer thinking, improved concentration, and less pain. She also no longer reports suicidal ideation, and demonstrates improved appetite and mood. She is discharged on Day 9 of her hospitalization.
The treatment team refers Ms. L for outpatient follow-up in 4 weeks, with a goal TSH level <3.0. Unfortunately, the effects of levothyroxine on Ms. L’s TSH level could not be determined during her hospital stay, and she has not returned to the facility since the initial presentation.
Thyroid function and mood
Ms. L’s case illustrates how thyroid function, pain, cognition, and mood may be interconnected. It is important to address all potential underlying comorbidities and establish appropriate outpatient care and follow-up so that patients may experience a more robust recovery. Further, this case highlights the importance of ruling out other potential medical causes of MDD during the initial diagnosis, and during times of recurrence or relapse, especially when a recent stressor, medication changes, or medication nonadherence cannot be identified as potential contributors.
Related Resources
- Cojić M, Cvejanov-Kezunović L. Subclinical hypothyroidism – whether and when to start treatment? Open Access Maced J Med Sci. 2017;5(7):1042-1046.
- Garber JR, Cobin RH, Gharib H, et al. Clinical practice guidelines for hypothyroidism in adults: cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Thyroid. 2012;22(12):1200-1235.
- Iosifescu DV. ‘Supercharge’ antidepressants by adding thyroid hormones. Current Psychiatry. 2006;5(7):15-20,25.
Drug Brand Names
Alprazolam • Xanax
Aripiprazole • Abilify
Bupropion • Wellbutrin
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Clozapine • Clozaril
Duloxetine • Cymbalta
Fluoxetine • Prozac
Fluphenazine • Prolixin
Gabapentin • Neurontin
Haloperidol • Haldol
Levothyroxine • Synthroid
Lisinopril • Prinivil, Zestril
Lithium • Eskalith, Lithobid
Loxapine • Loxitane
Mirtazapine • Remeron
Quetiapine • Seroquel
Risperidone • Risperdal
Thioridazine • Mellaril
Valproic acid • Depakote
Ms. L, age 53, presents to an inpatient psychiatric unit with depression, difficulty concentrating, fatigue, cognitive blunting, loss of appetite, increased alcohol intake, and recent suicidal ideation. Her symptoms began 3 months ago and gradually worsened. Her medical and psychiatric history is significant for hypertension, fibromyalgia, and chronic pain (back and neck), major depressive disorder (MDD; recurrent, severe), and generalized anxiety disorder (GAD). Ms. L’s current medication regimen includes lisinopril, 40 mg daily; fluoxetine, 60 mg daily; mirtazapine, 30 mg at bedtime; gabapentin, 300 mg twice daily; alprazolam, 0.5 mg twice daily as needed for anxiety; and oral docusate, 100 mg twice daily as needed. Her blood pressure is 124/85 mm Hg, heart rate is 66 beats per minute, and an electrocardiogram is normal. Laboratory workup reveals a potassium level of 4.4 mEq/L, blood urea nitrogen level of 20 mg/dL, serum creatinine level of 0.8 mg/dL, estimated creatinine clearance of 89.6 mL/min, free triiodothyronine (T3) levels of 2.7 pg/mL, thyroid-stimulating hormone (TSH) level of 7.68 mIU/L, free thyroxine (T4) level of 1.3 ng/dL, and blood ethanol level <10 mg/dL. In addition to the symptoms Ms. L initially described, a review of systems reveals word-finding difficulty, cold intolerance, constipation, hair loss, brittle nails, and dry skin.
To target Ms. L’s MDD, GAD, fibromyalgia, and chronic pain, fluoxetine, 60 mg daily is cross titrated beginning on Day 1 to duloxetine, 60 mg twice daily, over 4 days. Mirtazapine is decreased on Day 3 to 7.5 mg at bedtime to target Ms. L’s sleep and appetite. Due to the presence of several symptoms associated with hypothyroidism and a slightly elevated TSH level, on Day 6 we initiate adjunctive levothyroxine, 50 mcg daily each morning to target symptomatic subclinical hypothyroidism, and to potentially augment the other medications prescribed to address Ms. L’s MDD.
Thyroid hormone function is a complex physiological process controlled through the hypothalamic-pituitary-thyroid (HPT) axis. Psychotropic medications can impact thyroid hormone function and contribute to aberrations in thyroid physiology.1 Because patients with mental illness may require multiple psychotropic medications, it is imperative to understand the potential effects of these agents.
Antidepressants can induce hypothyroidism along multiple points of hormonal synthesis and iodine utilization. Tricyclic antidepressants have been implicated in the development of drug-iodide complexes, thus reducing biologically active iodine.2 Tricyclic antidepressants also can bind thyroid peroxidase, an enzyme necessary in the production of T4 and T3, altering hormonal production, resulting in a hypothyroid state.1 Non-tricyclic antidepressants (ie, selective serotonin reuptake inhibitors [SSRIs] and non-SSRIs [including serotonin-norepinephrine reuptake inhibitors and mirtazapine]) have also been implicated in thyroid dysfunction. Selective serotonin reuptake inhibitors have the propensity to induce hypothyroidism through inhibition of thyroid hormones T4 and T3.1,3 This inhibition is not always seen with concurrent reductions in TSH levels. Conversely, non-SSRIs can influence thyroid hormone levels with great variation, leading to thyroid hormone levels that are increased, decreased, or unchanged.1 Patients with a history of thyroid dysfunction should receive close thyroid function monitoring, especially while taking antidepressants.
Antipsychotics have a proclivity to induce hypothyroidism by means similar to antidepressants via hormonal manipulation and immunogenicity. Phenothiazines impact thyroid function through hormonal activation and degradation, and induction of autoimmunity.1 Autoimmunity may develop by means of antibody production or antigen immunization through the major histocompatibility complex.2 Other first-generation antipsychotics (FGAs) (eg, haloperidol and loxapine) are known to antagonize dopamine receptors in the tuberoinfundibular pathway, resulting in increased prolactin levels. Hyperprolactinemia may result in increased TSH levels through HPT axis activation.1 Additionally, FGAs can induce an immunogenic effect through production of antithyroid antibodies.1 Similar to FGAs, second-generation antipsychotics (SGAs) can increase TSH levels through hyperprolactinemia. Further research focused on SGAs is needed to determine how profound this effect may be.
The Table1 outlines considerations for modifying psychotropic therapy based on the presence of concurrent thyroid dysfunction. Thyroid function should be routinely assessed in patients treated with antipsychotics.
Mood stabilizers are capable of altering thyroid function and inducing a hypothyroid state. Lithium has been implicated in both hypothyroidism and hyperthyroidism due to its inhibition of hormonal secretion, and toxicity to thyroid cells with chronic use, respectively.1,4 Hypothyroidism can develop shortly after initiating lithium; women tend to have a greater predilection for thyroid dysfunction than men.1 Carbamazepine (CBZ) can reduce thyroid hormone levels without having a direct effect on TSH or thyroid dysfunction.1 As with lithium, women tend to be more susceptible to this effect. Valproic acid (VPA) has been shown to either increase, decrease, or have no impact on thyroid hormone levels, with little effect on TSH.1 When VPA is given in combination with CBZ, significant reductions in thyroid levels with a concurrent increase in TSH can occur.1 In patients with preexisting thyroid dysfunction, the combination of VPA and CBZ should be used with caution.
Continue to: CASE
CASE CONTINUED
By Day 8, Ms. L reports less fatigue, clearer thinking, improved concentration, and less pain. She also no longer reports suicidal ideation, and demonstrates improved appetite and mood. She is discharged on Day 9 of her hospitalization.
The treatment team refers Ms. L for outpatient follow-up in 4 weeks, with a goal TSH level <3.0. Unfortunately, the effects of levothyroxine on Ms. L’s TSH level could not be determined during her hospital stay, and she has not returned to the facility since the initial presentation.
Thyroid function and mood
Ms. L’s case illustrates how thyroid function, pain, cognition, and mood may be interconnected. It is important to address all potential underlying comorbidities and establish appropriate outpatient care and follow-up so that patients may experience a more robust recovery. Further, this case highlights the importance of ruling out other potential medical causes of MDD during the initial diagnosis, and during times of recurrence or relapse, especially when a recent stressor, medication changes, or medication nonadherence cannot be identified as potential contributors.
Related Resources
- Cojić M, Cvejanov-Kezunović L. Subclinical hypothyroidism – whether and when to start treatment? Open Access Maced J Med Sci. 2017;5(7):1042-1046.
- Garber JR, Cobin RH, Gharib H, et al. Clinical practice guidelines for hypothyroidism in adults: cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Thyroid. 2012;22(12):1200-1235.
- Iosifescu DV. ‘Supercharge’ antidepressants by adding thyroid hormones. Current Psychiatry. 2006;5(7):15-20,25.
Drug Brand Names
Alprazolam • Xanax
Aripiprazole • Abilify
Bupropion • Wellbutrin
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Clozapine • Clozaril
Duloxetine • Cymbalta
Fluoxetine • Prozac
Fluphenazine • Prolixin
Gabapentin • Neurontin
Haloperidol • Haldol
Levothyroxine • Synthroid
Lisinopril • Prinivil, Zestril
Lithium • Eskalith, Lithobid
Loxapine • Loxitane
Mirtazapine • Remeron
Quetiapine • Seroquel
Risperidone • Risperdal
Thioridazine • Mellaril
Valproic acid • Depakote
1. Bou Khalil R, Richa S. Thyroid adverse effect of psychotropic drugs: a review. Clin Neuropharm. 2001;34(6):248-255.
2. Sauvage MF, Marquet P, Rousseau A, et al. Relationship between psychotropic drugs and thyroid function: a review. Toxicol Appl Pharmacol. 1998;149(2):127-135.
3. Shelton RC, Winn S, Ekhatore N, et al. The effects of antidepressants on the thyroid axis in depression. Biol Psychiatry. 1993;33(2):120-126.
4. Kundra P, Burman KD. The effect of medications on thyroid function tests. Med Clin North Am. 2012;96(2):283-295.
1. Bou Khalil R, Richa S. Thyroid adverse effect of psychotropic drugs: a review. Clin Neuropharm. 2001;34(6):248-255.
2. Sauvage MF, Marquet P, Rousseau A, et al. Relationship between psychotropic drugs and thyroid function: a review. Toxicol Appl Pharmacol. 1998;149(2):127-135.
3. Shelton RC, Winn S, Ekhatore N, et al. The effects of antidepressants on the thyroid axis in depression. Biol Psychiatry. 1993;33(2):120-126.
4. Kundra P, Burman KD. The effect of medications on thyroid function tests. Med Clin North Am. 2012;96(2):283-295.

Serotonin syndrome: How to keep your patients safe

Mr. S, age 55, comes to your clinic as a walk-in for management of major depressive disorder, insomnia, and migraines. He also has tobacco use disorder and hypertension. Several days ago, Mr. S had visited the clinic because he was continuing to experience depressive symptoms, so his sertraline was increased from 100 to 200 mg/d. His current medication regimen includes sertraline 200 mg/d, trazodone 100 mg/d, lisinopril 10 mg/d, and sumatriptan, 100 mg as needed for migraine. He says last week he used 4 or 5 doses of sumatriptan because he experienced several migraines. Mr. S also reports occasionally taking 2 tablets of trazodone instead of 1 on nights that he has trouble falling asleep.
Today, Mr. S presents with a low-grade fever, diarrhea, internal restlessness, and a racing heartbeat that started shortly after his last visit. During physical examination, he exhibits slow, continuous lateral eye movements. His vital signs are markedly elevated: blood pressure, 175/85 mm Hg; heart rate, 110 beats per minute; and temperature, 39°C (102.2°F). Based on his presentation, the treatment team decides to send Mr. S to urgent care for closer monitoring.
Serotonin syndrome is a drug-induced syndrome caused by overstimulation of serotonin receptors. The syndrome is characterized by a classic clinical triad consisting of mental status changes, autonomic hyperactivity, and neuromuscular abnormalities. The clinical presentation is highly variable, and the severity ranges from mild to life-threatening.1-3 The incidence and prevalence of serotonin syndrome has not been well defined.3 Serotonin syndrome may be underreported because mild cases are often overlooked due to nonspecific symptoms. In addition, lack of physician awareness of drug–drug interactions, signs and symptoms, and differential diagnoses may result in underdiagnosis or misdiagnosis.1-3
What causes it?
Serotonin syndrome is usually a consequence of a drug–drug interaction between 2 or more serotonergic agents.4 Serotonin syndrome may result following medication misuse, overdose, initiation of a serotonergic agent, or increase in the dose of a currently prescribed serotonergic agent.3,4 In addition to medication classes and specific agents, Table 12-5 lists the drug mechanisms associated with serotonin syndrome:
- inhibition of serotonin reuptake
- inhibition of serotonin metabolism
- increased serotonin synthesis
- agonism of the serotonin receptor.

The amount of serotonergic activity most likely to cause serotonin syndrome is unclear.4
Pathophysiology. Serotonin, also known as 5-hydroxytryptamine (5-HT), is a metabolite of the amino acid tryptophan. This neurotransmitter is located in both the CNS and the periphery. Regulation of the serotonergic system begins in the presynaptic neurons with decarboxylation and hydroxylation of tryptophan resulting in serotonin synthesis. Once serotonin is produced, it is released into the synaptic cleft, where it binds to serotonin receptors.1,4,5 After receptor binding, serotonin reuptake occurs in the presynaptic neurons, where it can be metabolized by the monoamine oxidase enzyme. Finally, the metabolites are excreted in the urine. Serotonin syndrome results when this regulatory system is disrupted due to hyperstimulation of the postsynaptic serotonin receptors, mainly via agonism of the 5-HT2A and 5-HT1A receptors.1,4,5
Continue to: A nonspecific presentation
A nonspecific presentation
Unfortunately, many of the symptoms of serotonin syndrome are nonspecific, and the severity varies among patients.2,3 The onset of symptoms usually occurs within 6 to 8 hours after ingestion of a serotonergic agent.5 It is important to immediately recognize the symptoms (Table 22-5) and formulate a differential diagnosis because sudden progression of symptoms is common and may lead to life-threatening circumstances.1,3

In mild cases of serotonin syndrome, patients may have a low-grade fever or be afebrile. Hyperthermia tends to be present in moderate and severe cases, with temperatures >41°C (105.8°F) during life-threatening cases. Diaphoresis and tachycardia may be present regardless of severity. Additional autonomic irregularities include hypertension, tachypnea, nausea, vomiting, diarrhea, and hyperactive bowel sounds. In terms of neuromuscular abnormalities, hyperreflexia is a primary concern, as well as myoclonus. As the severity progresses to life-threatening, the clonus may convert from inducible to spontaneous and slow, continuous lateral eye movements may be present. Additional neuromuscular symptoms include tremor, akathisia, and muscle rigidity.1,3-5
Common mental status changes during mild cases include restlessness and anxiety. Abnormal mentation during moderate cases may present as increased hypervigilance and agitation, and this may advance to delirium or coma in severe cases. As the severity intensifies, the risk of developing additional physiological complications also increases. Rhabdomyolysis may occur due to muscle damage and myoglobinuria secondary to hyperreflexia, myoclonus, hypertonicity, and muscle rigidity. Muscle breakdown may then progress to further complications, such as renal failure. In rare instances, serotonin syndrome can result in seizures or death.1,3-5
Medication history tips off the diagnosis
The first step in diagnosing serotonin syndrome is to conduct a thorough review of the patient’s medication history, specifically taking into account any recent exposure to serotonergic agents.3,5 It is important to ask about prescription medications as well as over-the-counter products, herbal supplements, and illicit substances.1,4 When reviewing the medication history, investigate whether there may have been a recent change in therapy with serotonergic agents. Also, determine when the patient’s symptoms began in relation to exposure to serotonergic agents.4

After the medication review, conduct a thorough physical and neurologic examination to identify current symptoms and severity.1,3 No specific laboratory test is available to definitively confirm the diagnosis of serotonin syndrome.1,4 Monitoring of serum serotonin is not recommended because the levels do not correlate with symptom severity.3 The recommended diagnostic tool is the Hunter Serotonin Toxicity Criteria (Figure1,3).3,4 Historically, the Sternbach’s Diagnostic Criteria for serotonin syndrome were used for diagnosis; however, the Hunter Serotonin Toxicity Criteria are more sensitive (96% vs 75%) and more specific (97% vs 84%) than the Sternbach’s Diagnostic Criteria for serotonin syndrome.1,3-5
Continue to: In addition to using the proper diagnostic tool...
In addition to using the proper diagnostic tool, conduct a differential diagnosis to rule out other drug-induced syndromes, such as anticholinergic toxidrome, neuroleptic malignant syndrome, or malignant hyperthermia.1,3,5 Autonomic instability, including hypertension, tachycardia, tachypnea, and hyperthermia, may be present in all of the aforementioned drug-induced syndromes.1 As a result, the clinician must monitor for other symptoms that may differentiate the disease states to establish a clear diagnosis.
Discontinue agents, offer supportive care
There are no official published guidelines for managing serotonin syndrome.5 Regardless of the severity of a patient’s presentation, all serotonergic agents should be discontinued immediately. In addition, supportive care should be initiated for symptom management. Intravenous fluid replacement is recommended for hydration and to treat hyperthermia. External cooling may also be warranted to reduce body temperatures. Vital signs should be stabilized with appropriate pharmacotherapy.1,3-5
Benzodiazepines are considered a mainstay for relief of agitation during serotonin syndrome of any severity. In life-threatening cases—which are characterized by hyperthermia >41°C (105.8°F)—sedation, paralysis, and intubation may be necessary to maintain the airway, breathing, and circulation.1,3-5 Because treatment of hyperthermia requires elimination of hyperreflexia, paralysis is recommended.1 Nondepolarizing neuromuscular blocking agents, such as vecuronium, are preferred over depolarizing agents due to their decreased potential for rhabdomyolysis.1,3
Cyproheptadine, a histamine-1 receptor antagonist and a 5-HT2A receptor antagonist, is recommended for off-label treatment of serotonin syndrome to help decrease the intensity of symptoms. This should be initiated as a single dose of 12 mg followed by 2 mg every 2 hours until symptoms improve.1,3,5 After stabilization, a maintenance dose of 8 mg every 6 hours is recommended. Doses should not exceed the maximum recommended dose of 0.5 mg/kg/d.1,3,6 The most common adverse reactions associated with cyproheptadine are sedation and anticholinergic adverse effects.1,4,6
Antipsychotics, such as olanzapine and chlorpromazine, have been considered treatment alternatives due to their associated 5-HT2A receptor antagonism. However, there is limited data supporting such use.1,4 Antipsychotics should be used with caution because neuroleptic malignant syndrome may be mistaken for serotonin syndrome. Use of antipyretics is not recommended for treating fever and hyperthermia because the increase in body temperature is secondary to excessive muscle activity rather than dysfunction of the hypothalamic temperature set point.1,3,5 Physical restraints are also not recommended because their use may provoke further hyperthermia and increase the risk of rhabdomyolysis.3,5
Continue to: Ultimately, the duration of treatment...
Ultimately, the duration of treatment will be influenced by the pharmacokinetics of the serotonergic agents that induced the serotonin syndrome. Following resolution, retrial of the offending serotonergic agents should be carefully assessed. A retrial should only be considered after an adequate washout period has been observed, and clinicians should consider utilizing lower doses.2,5
Take steps for prevention
Patients at highest risk of developing serotonin syndrome are those who have multiple comorbidities that result in treatment with multiple serotonergic agents.3 Clinicians and patients alike need to be educated about the signs and symptoms of serotonin syndrome to promote early recognition. Also consider modifying your prescribing practices to minimize the use of multiple serotonergic agents. When switching between serotonergic agents, institute safe washout periods. Encourage patients to adhere to their prescribed medication regimens. Using electronic ordering systems can help detect drug–drug interactions.1,3 Prophylaxis with cyproheptadine may be considered in high-risk patients; however, no clinical trials have been conducted to evaluate using cyproheptadine to prevent serotonin syndrome.7
CASE CONTINUED
Upon further assessment in urgent care, Mr. S is found to have muscle rigidity in addition to ocular clonus and a temperature >38°C (100.4°F). Because Mr. S’s symptoms coincide with a recent increase of sertraline and increased use of both trazodone and sumatriptan, he meets Hunter Serotonin Toxicity Criteria. Therefore, his symptoms are likely related to excessive increase in serotonergic activity. Mr. S is admitted to the hospital for closer monitoring, and his sertraline, trazodone, and sumatriptan are held. He receives IV fluids for several days as well as cyproheptadine, 8 mg every 6 hours after stabilization, until his symptoms resolve. On Day 4, Mr. S no longer experiences diarrhea and internal restlessness. His vital signs return to normal, and as a result of symptom resolution, he is discharged from the hospital. The treatment team discusses changing his medication regimen to avoid multiple serotonergic agents. Mr. S is switched from sertraline to bupropion XL, 150 mg/d. Sumatriptan, 100 mg/d as needed, is continued for acute migraine treatment. Trazodone is discontinued and replaced with melatonin, 3 mg/d. The team also counsels Mr. S on the importance of proper adherence to his medication regimen. He is advised to return to the clinic in 2 weeks for reassessment of safety and efficacy.
Related Resource
- Turner AH, Kim JJ, McCarron RM. Differentiating serotonin syndrome and neuroleptic malignant syndrome. Current Psychiatry. 2019;18(2):30-36.
Drug Brand Names
Almotriptan • Axert
Buprenorphine • Subutex
Bupropion • Wellbutrin, Zyban
Buspirone • BuSpar
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Cyproheptadine • Periactin
Eletriptan • Relpax
Frovatriptan • Frova
Granisetron • Kytril
Lisinopril • Prinivil, Zestril
Meperidine • Demerol
Methadone • Dolophine, Methadose
Metoclopramide • Reglan
Mirtazapine • Remeron
Naratriptan • Amerge
Olanzapine • Zyprexa
Ondansetron • Zofran
Rizatriptan • Maxalt
Sertraline • Zoloft
Sumatriptan • Imitrex tablets
Tapentadol • Nucynta
Tramadol • Conzip
Trazodone • Desyrel, Oleptro
Valproic acid • Depakene, Depakote
Vecuronium • Norcuron
Zolmitriptan • Zomig
1. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112-1120.
2. Beakley BD, Kaye AM, Kaye AD. Tramadol, pharmacology, side effects, and serotonin syndrome: a review. Pain Physician. 2015;18(4):395-400.
3. Wang RZ, Vashistha V, Kaur S, et al. Serotonin syndrome: preventing, recognizing, and treating it. Cleve Clin J Med. 2016;83(11):810-817.
4. Bartlett D. Drug-induced serotonin syndrome. Crit Care Nurse. 2017;37(1):49-54.
5. Frank C. Recognition and treatment of serotonin syndrome. Can Fam Physician. 2008;54(7):988-992.
6. Cyproheptadine hydrochloride tablets [package insert]. Hayward, CA: Impax Generics; 2017.
7. Deardorff OG, Khan T, Kulkarni G, et al. Serotonin syndrome: prophylactic treatment with cyproheptadine. Prim Care Companion CNS Disord. 2016;18(4). doi: 10.4088/PCC.16br01966.
Mr. S, age 55, comes to your clinic as a walk-in for management of major depressive disorder, insomnia, and migraines. He also has tobacco use disorder and hypertension. Several days ago, Mr. S had visited the clinic because he was continuing to experience depressive symptoms, so his sertraline was increased from 100 to 200 mg/d. His current medication regimen includes sertraline 200 mg/d, trazodone 100 mg/d, lisinopril 10 mg/d, and sumatriptan, 100 mg as needed for migraine. He says last week he used 4 or 5 doses of sumatriptan because he experienced several migraines. Mr. S also reports occasionally taking 2 tablets of trazodone instead of 1 on nights that he has trouble falling asleep.
Today, Mr. S presents with a low-grade fever, diarrhea, internal restlessness, and a racing heartbeat that started shortly after his last visit. During physical examination, he exhibits slow, continuous lateral eye movements. His vital signs are markedly elevated: blood pressure, 175/85 mm Hg; heart rate, 110 beats per minute; and temperature, 39°C (102.2°F). Based on his presentation, the treatment team decides to send Mr. S to urgent care for closer monitoring.
Serotonin syndrome is a drug-induced syndrome caused by overstimulation of serotonin receptors. The syndrome is characterized by a classic clinical triad consisting of mental status changes, autonomic hyperactivity, and neuromuscular abnormalities. The clinical presentation is highly variable, and the severity ranges from mild to life-threatening.1-3 The incidence and prevalence of serotonin syndrome has not been well defined.3 Serotonin syndrome may be underreported because mild cases are often overlooked due to nonspecific symptoms. In addition, lack of physician awareness of drug–drug interactions, signs and symptoms, and differential diagnoses may result in underdiagnosis or misdiagnosis.1-3
What causes it?
Serotonin syndrome is usually a consequence of a drug–drug interaction between 2 or more serotonergic agents.4 Serotonin syndrome may result following medication misuse, overdose, initiation of a serotonergic agent, or increase in the dose of a currently prescribed serotonergic agent.3,4 In addition to medication classes and specific agents, Table 12-5 lists the drug mechanisms associated with serotonin syndrome:
- inhibition of serotonin reuptake
- inhibition of serotonin metabolism
- increased serotonin synthesis
- agonism of the serotonin receptor.
The amount of serotonergic activity most likely to cause serotonin syndrome is unclear.4
Pathophysiology. Serotonin, also known as 5-hydroxytryptamine (5-HT), is a metabolite of the amino acid tryptophan. This neurotransmitter is located in both the CNS and the periphery. Regulation of the serotonergic system begins in the presynaptic neurons with decarboxylation and hydroxylation of tryptophan resulting in serotonin synthesis. Once serotonin is produced, it is released into the synaptic cleft, where it binds to serotonin receptors.1,4,5 After receptor binding, serotonin reuptake occurs in the presynaptic neurons, where it can be metabolized by the monoamine oxidase enzyme. Finally, the metabolites are excreted in the urine. Serotonin syndrome results when this regulatory system is disrupted due to hyperstimulation of the postsynaptic serotonin receptors, mainly via agonism of the 5-HT2A and 5-HT1A receptors.1,4,5
Continue to: A nonspecific presentation
A nonspecific presentation
Unfortunately, many of the symptoms of serotonin syndrome are nonspecific, and the severity varies among patients.2,3 The onset of symptoms usually occurs within 6 to 8 hours after ingestion of a serotonergic agent.5 It is important to immediately recognize the symptoms (Table 22-5) and formulate a differential diagnosis because sudden progression of symptoms is common and may lead to life-threatening circumstances.1,3
In mild cases of serotonin syndrome, patients may have a low-grade fever or be afebrile. Hyperthermia tends to be present in moderate and severe cases, with temperatures >41°C (105.8°F) during life-threatening cases. Diaphoresis and tachycardia may be present regardless of severity. Additional autonomic irregularities include hypertension, tachypnea, nausea, vomiting, diarrhea, and hyperactive bowel sounds. In terms of neuromuscular abnormalities, hyperreflexia is a primary concern, as well as myoclonus. As the severity progresses to life-threatening, the clonus may convert from inducible to spontaneous and slow, continuous lateral eye movements may be present. Additional neuromuscular symptoms include tremor, akathisia, and muscle rigidity.1,3-5
Common mental status changes during mild cases include restlessness and anxiety. Abnormal mentation during moderate cases may present as increased hypervigilance and agitation, and this may advance to delirium or coma in severe cases. As the severity intensifies, the risk of developing additional physiological complications also increases. Rhabdomyolysis may occur due to muscle damage and myoglobinuria secondary to hyperreflexia, myoclonus, hypertonicity, and muscle rigidity. Muscle breakdown may then progress to further complications, such as renal failure. In rare instances, serotonin syndrome can result in seizures or death.1,3-5
Medication history tips off the diagnosis
The first step in diagnosing serotonin syndrome is to conduct a thorough review of the patient’s medication history, specifically taking into account any recent exposure to serotonergic agents.3,5 It is important to ask about prescription medications as well as over-the-counter products, herbal supplements, and illicit substances.1,4 When reviewing the medication history, investigate whether there may have been a recent change in therapy with serotonergic agents. Also, determine when the patient’s symptoms began in relation to exposure to serotonergic agents.4
After the medication review, conduct a thorough physical and neurologic examination to identify current symptoms and severity.1,3 No specific laboratory test is available to definitively confirm the diagnosis of serotonin syndrome.1,4 Monitoring of serum serotonin is not recommended because the levels do not correlate with symptom severity.3 The recommended diagnostic tool is the Hunter Serotonin Toxicity Criteria (Figure1,3).3,4 Historically, the Sternbach’s Diagnostic Criteria for serotonin syndrome were used for diagnosis; however, the Hunter Serotonin Toxicity Criteria are more sensitive (96% vs 75%) and more specific (97% vs 84%) than the Sternbach’s Diagnostic Criteria for serotonin syndrome.1,3-5
Continue to: In addition to using the proper diagnostic tool...
In addition to using the proper diagnostic tool, conduct a differential diagnosis to rule out other drug-induced syndromes, such as anticholinergic toxidrome, neuroleptic malignant syndrome, or malignant hyperthermia.1,3,5 Autonomic instability, including hypertension, tachycardia, tachypnea, and hyperthermia, may be present in all of the aforementioned drug-induced syndromes.1 As a result, the clinician must monitor for other symptoms that may differentiate the disease states to establish a clear diagnosis.
Discontinue agents, offer supportive care
There are no official published guidelines for managing serotonin syndrome.5 Regardless of the severity of a patient’s presentation, all serotonergic agents should be discontinued immediately. In addition, supportive care should be initiated for symptom management. Intravenous fluid replacement is recommended for hydration and to treat hyperthermia. External cooling may also be warranted to reduce body temperatures. Vital signs should be stabilized with appropriate pharmacotherapy.1,3-5
Benzodiazepines are considered a mainstay for relief of agitation during serotonin syndrome of any severity. In life-threatening cases—which are characterized by hyperthermia >41°C (105.8°F)—sedation, paralysis, and intubation may be necessary to maintain the airway, breathing, and circulation.1,3-5 Because treatment of hyperthermia requires elimination of hyperreflexia, paralysis is recommended.1 Nondepolarizing neuromuscular blocking agents, such as vecuronium, are preferred over depolarizing agents due to their decreased potential for rhabdomyolysis.1,3
Cyproheptadine, a histamine-1 receptor antagonist and a 5-HT2A receptor antagonist, is recommended for off-label treatment of serotonin syndrome to help decrease the intensity of symptoms. This should be initiated as a single dose of 12 mg followed by 2 mg every 2 hours until symptoms improve.1,3,5 After stabilization, a maintenance dose of 8 mg every 6 hours is recommended. Doses should not exceed the maximum recommended dose of 0.5 mg/kg/d.1,3,6 The most common adverse reactions associated with cyproheptadine are sedation and anticholinergic adverse effects.1,4,6
Antipsychotics, such as olanzapine and chlorpromazine, have been considered treatment alternatives due to their associated 5-HT2A receptor antagonism. However, there is limited data supporting such use.1,4 Antipsychotics should be used with caution because neuroleptic malignant syndrome may be mistaken for serotonin syndrome. Use of antipyretics is not recommended for treating fever and hyperthermia because the increase in body temperature is secondary to excessive muscle activity rather than dysfunction of the hypothalamic temperature set point.1,3,5 Physical restraints are also not recommended because their use may provoke further hyperthermia and increase the risk of rhabdomyolysis.3,5
Continue to: Ultimately, the duration of treatment...
Ultimately, the duration of treatment will be influenced by the pharmacokinetics of the serotonergic agents that induced the serotonin syndrome. Following resolution, retrial of the offending serotonergic agents should be carefully assessed. A retrial should only be considered after an adequate washout period has been observed, and clinicians should consider utilizing lower doses.2,5
Take steps for prevention
Patients at highest risk of developing serotonin syndrome are those who have multiple comorbidities that result in treatment with multiple serotonergic agents.3 Clinicians and patients alike need to be educated about the signs and symptoms of serotonin syndrome to promote early recognition. Also consider modifying your prescribing practices to minimize the use of multiple serotonergic agents. When switching between serotonergic agents, institute safe washout periods. Encourage patients to adhere to their prescribed medication regimens. Using electronic ordering systems can help detect drug–drug interactions.1,3 Prophylaxis with cyproheptadine may be considered in high-risk patients; however, no clinical trials have been conducted to evaluate using cyproheptadine to prevent serotonin syndrome.7
CASE CONTINUED
Upon further assessment in urgent care, Mr. S is found to have muscle rigidity in addition to ocular clonus and a temperature >38°C (100.4°F). Because Mr. S’s symptoms coincide with a recent increase of sertraline and increased use of both trazodone and sumatriptan, he meets Hunter Serotonin Toxicity Criteria. Therefore, his symptoms are likely related to excessive increase in serotonergic activity. Mr. S is admitted to the hospital for closer monitoring, and his sertraline, trazodone, and sumatriptan are held. He receives IV fluids for several days as well as cyproheptadine, 8 mg every 6 hours after stabilization, until his symptoms resolve. On Day 4, Mr. S no longer experiences diarrhea and internal restlessness. His vital signs return to normal, and as a result of symptom resolution, he is discharged from the hospital. The treatment team discusses changing his medication regimen to avoid multiple serotonergic agents. Mr. S is switched from sertraline to bupropion XL, 150 mg/d. Sumatriptan, 100 mg/d as needed, is continued for acute migraine treatment. Trazodone is discontinued and replaced with melatonin, 3 mg/d. The team also counsels Mr. S on the importance of proper adherence to his medication regimen. He is advised to return to the clinic in 2 weeks for reassessment of safety and efficacy.
Related Resource
- Turner AH, Kim JJ, McCarron RM. Differentiating serotonin syndrome and neuroleptic malignant syndrome. Current Psychiatry. 2019;18(2):30-36.
Drug Brand Names
Almotriptan • Axert
Buprenorphine • Subutex
Bupropion • Wellbutrin, Zyban
Buspirone • BuSpar
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Cyproheptadine • Periactin
Eletriptan • Relpax
Frovatriptan • Frova
Granisetron • Kytril
Lisinopril • Prinivil, Zestril
Meperidine • Demerol
Methadone • Dolophine, Methadose
Metoclopramide • Reglan
Mirtazapine • Remeron
Naratriptan • Amerge
Olanzapine • Zyprexa
Ondansetron • Zofran
Rizatriptan • Maxalt
Sertraline • Zoloft
Sumatriptan • Imitrex tablets
Tapentadol • Nucynta
Tramadol • Conzip
Trazodone • Desyrel, Oleptro
Valproic acid • Depakene, Depakote
Vecuronium • Norcuron
Zolmitriptan • Zomig
Mr. S, age 55, comes to your clinic as a walk-in for management of major depressive disorder, insomnia, and migraines. He also has tobacco use disorder and hypertension. Several days ago, Mr. S had visited the clinic because he was continuing to experience depressive symptoms, so his sertraline was increased from 100 to 200 mg/d. His current medication regimen includes sertraline 200 mg/d, trazodone 100 mg/d, lisinopril 10 mg/d, and sumatriptan, 100 mg as needed for migraine. He says last week he used 4 or 5 doses of sumatriptan because he experienced several migraines. Mr. S also reports occasionally taking 2 tablets of trazodone instead of 1 on nights that he has trouble falling asleep.
Today, Mr. S presents with a low-grade fever, diarrhea, internal restlessness, and a racing heartbeat that started shortly after his last visit. During physical examination, he exhibits slow, continuous lateral eye movements. His vital signs are markedly elevated: blood pressure, 175/85 mm Hg; heart rate, 110 beats per minute; and temperature, 39°C (102.2°F). Based on his presentation, the treatment team decides to send Mr. S to urgent care for closer monitoring.
Serotonin syndrome is a drug-induced syndrome caused by overstimulation of serotonin receptors. The syndrome is characterized by a classic clinical triad consisting of mental status changes, autonomic hyperactivity, and neuromuscular abnormalities. The clinical presentation is highly variable, and the severity ranges from mild to life-threatening.1-3 The incidence and prevalence of serotonin syndrome has not been well defined.3 Serotonin syndrome may be underreported because mild cases are often overlooked due to nonspecific symptoms. In addition, lack of physician awareness of drug–drug interactions, signs and symptoms, and differential diagnoses may result in underdiagnosis or misdiagnosis.1-3
What causes it?
Serotonin syndrome is usually a consequence of a drug–drug interaction between 2 or more serotonergic agents.4 Serotonin syndrome may result following medication misuse, overdose, initiation of a serotonergic agent, or increase in the dose of a currently prescribed serotonergic agent.3,4 In addition to medication classes and specific agents, Table 12-5 lists the drug mechanisms associated with serotonin syndrome:
- inhibition of serotonin reuptake
- inhibition of serotonin metabolism
- increased serotonin synthesis
- agonism of the serotonin receptor.
The amount of serotonergic activity most likely to cause serotonin syndrome is unclear.4
Pathophysiology. Serotonin, also known as 5-hydroxytryptamine (5-HT), is a metabolite of the amino acid tryptophan. This neurotransmitter is located in both the CNS and the periphery. Regulation of the serotonergic system begins in the presynaptic neurons with decarboxylation and hydroxylation of tryptophan resulting in serotonin synthesis. Once serotonin is produced, it is released into the synaptic cleft, where it binds to serotonin receptors.1,4,5 After receptor binding, serotonin reuptake occurs in the presynaptic neurons, where it can be metabolized by the monoamine oxidase enzyme. Finally, the metabolites are excreted in the urine. Serotonin syndrome results when this regulatory system is disrupted due to hyperstimulation of the postsynaptic serotonin receptors, mainly via agonism of the 5-HT2A and 5-HT1A receptors.1,4,5
Continue to: A nonspecific presentation
A nonspecific presentation
Unfortunately, many of the symptoms of serotonin syndrome are nonspecific, and the severity varies among patients.2,3 The onset of symptoms usually occurs within 6 to 8 hours after ingestion of a serotonergic agent.5 It is important to immediately recognize the symptoms (Table 22-5) and formulate a differential diagnosis because sudden progression of symptoms is common and may lead to life-threatening circumstances.1,3
In mild cases of serotonin syndrome, patients may have a low-grade fever or be afebrile. Hyperthermia tends to be present in moderate and severe cases, with temperatures >41°C (105.8°F) during life-threatening cases. Diaphoresis and tachycardia may be present regardless of severity. Additional autonomic irregularities include hypertension, tachypnea, nausea, vomiting, diarrhea, and hyperactive bowel sounds. In terms of neuromuscular abnormalities, hyperreflexia is a primary concern, as well as myoclonus. As the severity progresses to life-threatening, the clonus may convert from inducible to spontaneous and slow, continuous lateral eye movements may be present. Additional neuromuscular symptoms include tremor, akathisia, and muscle rigidity.1,3-5
Common mental status changes during mild cases include restlessness and anxiety. Abnormal mentation during moderate cases may present as increased hypervigilance and agitation, and this may advance to delirium or coma in severe cases. As the severity intensifies, the risk of developing additional physiological complications also increases. Rhabdomyolysis may occur due to muscle damage and myoglobinuria secondary to hyperreflexia, myoclonus, hypertonicity, and muscle rigidity. Muscle breakdown may then progress to further complications, such as renal failure. In rare instances, serotonin syndrome can result in seizures or death.1,3-5
Medication history tips off the diagnosis
The first step in diagnosing serotonin syndrome is to conduct a thorough review of the patient’s medication history, specifically taking into account any recent exposure to serotonergic agents.3,5 It is important to ask about prescription medications as well as over-the-counter products, herbal supplements, and illicit substances.1,4 When reviewing the medication history, investigate whether there may have been a recent change in therapy with serotonergic agents. Also, determine when the patient’s symptoms began in relation to exposure to serotonergic agents.4
After the medication review, conduct a thorough physical and neurologic examination to identify current symptoms and severity.1,3 No specific laboratory test is available to definitively confirm the diagnosis of serotonin syndrome.1,4 Monitoring of serum serotonin is not recommended because the levels do not correlate with symptom severity.3 The recommended diagnostic tool is the Hunter Serotonin Toxicity Criteria (Figure1,3).3,4 Historically, the Sternbach’s Diagnostic Criteria for serotonin syndrome were used for diagnosis; however, the Hunter Serotonin Toxicity Criteria are more sensitive (96% vs 75%) and more specific (97% vs 84%) than the Sternbach’s Diagnostic Criteria for serotonin syndrome.1,3-5
Continue to: In addition to using the proper diagnostic tool...
In addition to using the proper diagnostic tool, conduct a differential diagnosis to rule out other drug-induced syndromes, such as anticholinergic toxidrome, neuroleptic malignant syndrome, or malignant hyperthermia.1,3,5 Autonomic instability, including hypertension, tachycardia, tachypnea, and hyperthermia, may be present in all of the aforementioned drug-induced syndromes.1 As a result, the clinician must monitor for other symptoms that may differentiate the disease states to establish a clear diagnosis.
Discontinue agents, offer supportive care
There are no official published guidelines for managing serotonin syndrome.5 Regardless of the severity of a patient’s presentation, all serotonergic agents should be discontinued immediately. In addition, supportive care should be initiated for symptom management. Intravenous fluid replacement is recommended for hydration and to treat hyperthermia. External cooling may also be warranted to reduce body temperatures. Vital signs should be stabilized with appropriate pharmacotherapy.1,3-5
Benzodiazepines are considered a mainstay for relief of agitation during serotonin syndrome of any severity. In life-threatening cases—which are characterized by hyperthermia >41°C (105.8°F)—sedation, paralysis, and intubation may be necessary to maintain the airway, breathing, and circulation.1,3-5 Because treatment of hyperthermia requires elimination of hyperreflexia, paralysis is recommended.1 Nondepolarizing neuromuscular blocking agents, such as vecuronium, are preferred over depolarizing agents due to their decreased potential for rhabdomyolysis.1,3
Cyproheptadine, a histamine-1 receptor antagonist and a 5-HT2A receptor antagonist, is recommended for off-label treatment of serotonin syndrome to help decrease the intensity of symptoms. This should be initiated as a single dose of 12 mg followed by 2 mg every 2 hours until symptoms improve.1,3,5 After stabilization, a maintenance dose of 8 mg every 6 hours is recommended. Doses should not exceed the maximum recommended dose of 0.5 mg/kg/d.1,3,6 The most common adverse reactions associated with cyproheptadine are sedation and anticholinergic adverse effects.1,4,6
Antipsychotics, such as olanzapine and chlorpromazine, have been considered treatment alternatives due to their associated 5-HT2A receptor antagonism. However, there is limited data supporting such use.1,4 Antipsychotics should be used with caution because neuroleptic malignant syndrome may be mistaken for serotonin syndrome. Use of antipyretics is not recommended for treating fever and hyperthermia because the increase in body temperature is secondary to excessive muscle activity rather than dysfunction of the hypothalamic temperature set point.1,3,5 Physical restraints are also not recommended because their use may provoke further hyperthermia and increase the risk of rhabdomyolysis.3,5
Continue to: Ultimately, the duration of treatment...
Ultimately, the duration of treatment will be influenced by the pharmacokinetics of the serotonergic agents that induced the serotonin syndrome. Following resolution, retrial of the offending serotonergic agents should be carefully assessed. A retrial should only be considered after an adequate washout period has been observed, and clinicians should consider utilizing lower doses.2,5
Take steps for prevention
Patients at highest risk of developing serotonin syndrome are those who have multiple comorbidities that result in treatment with multiple serotonergic agents.3 Clinicians and patients alike need to be educated about the signs and symptoms of serotonin syndrome to promote early recognition. Also consider modifying your prescribing practices to minimize the use of multiple serotonergic agents. When switching between serotonergic agents, institute safe washout periods. Encourage patients to adhere to their prescribed medication regimens. Using electronic ordering systems can help detect drug–drug interactions.1,3 Prophylaxis with cyproheptadine may be considered in high-risk patients; however, no clinical trials have been conducted to evaluate using cyproheptadine to prevent serotonin syndrome.7
CASE CONTINUED
Upon further assessment in urgent care, Mr. S is found to have muscle rigidity in addition to ocular clonus and a temperature >38°C (100.4°F). Because Mr. S’s symptoms coincide with a recent increase of sertraline and increased use of both trazodone and sumatriptan, he meets Hunter Serotonin Toxicity Criteria. Therefore, his symptoms are likely related to excessive increase in serotonergic activity. Mr. S is admitted to the hospital for closer monitoring, and his sertraline, trazodone, and sumatriptan are held. He receives IV fluids for several days as well as cyproheptadine, 8 mg every 6 hours after stabilization, until his symptoms resolve. On Day 4, Mr. S no longer experiences diarrhea and internal restlessness. His vital signs return to normal, and as a result of symptom resolution, he is discharged from the hospital. The treatment team discusses changing his medication regimen to avoid multiple serotonergic agents. Mr. S is switched from sertraline to bupropion XL, 150 mg/d. Sumatriptan, 100 mg/d as needed, is continued for acute migraine treatment. Trazodone is discontinued and replaced with melatonin, 3 mg/d. The team also counsels Mr. S on the importance of proper adherence to his medication regimen. He is advised to return to the clinic in 2 weeks for reassessment of safety and efficacy.
Related Resource
- Turner AH, Kim JJ, McCarron RM. Differentiating serotonin syndrome and neuroleptic malignant syndrome. Current Psychiatry. 2019;18(2):30-36.
Drug Brand Names
Almotriptan • Axert
Buprenorphine • Subutex
Bupropion • Wellbutrin, Zyban
Buspirone • BuSpar
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Cyproheptadine • Periactin
Eletriptan • Relpax
Frovatriptan • Frova
Granisetron • Kytril
Lisinopril • Prinivil, Zestril
Meperidine • Demerol
Methadone • Dolophine, Methadose
Metoclopramide • Reglan
Mirtazapine • Remeron
Naratriptan • Amerge
Olanzapine • Zyprexa
Ondansetron • Zofran
Rizatriptan • Maxalt
Sertraline • Zoloft
Sumatriptan • Imitrex tablets
Tapentadol • Nucynta
Tramadol • Conzip
Trazodone • Desyrel, Oleptro
Valproic acid • Depakene, Depakote
Vecuronium • Norcuron
Zolmitriptan • Zomig
1. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112-1120.
2. Beakley BD, Kaye AM, Kaye AD. Tramadol, pharmacology, side effects, and serotonin syndrome: a review. Pain Physician. 2015;18(4):395-400.
3. Wang RZ, Vashistha V, Kaur S, et al. Serotonin syndrome: preventing, recognizing, and treating it. Cleve Clin J Med. 2016;83(11):810-817.
4. Bartlett D. Drug-induced serotonin syndrome. Crit Care Nurse. 2017;37(1):49-54.
5. Frank C. Recognition and treatment of serotonin syndrome. Can Fam Physician. 2008;54(7):988-992.
6. Cyproheptadine hydrochloride tablets [package insert]. Hayward, CA: Impax Generics; 2017.
7. Deardorff OG, Khan T, Kulkarni G, et al. Serotonin syndrome: prophylactic treatment with cyproheptadine. Prim Care Companion CNS Disord. 2016;18(4). doi: 10.4088/PCC.16br01966.
1. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112-1120.
2. Beakley BD, Kaye AM, Kaye AD. Tramadol, pharmacology, side effects, and serotonin syndrome: a review. Pain Physician. 2015;18(4):395-400.
3. Wang RZ, Vashistha V, Kaur S, et al. Serotonin syndrome: preventing, recognizing, and treating it. Cleve Clin J Med. 2016;83(11):810-817.
4. Bartlett D. Drug-induced serotonin syndrome. Crit Care Nurse. 2017;37(1):49-54.
5. Frank C. Recognition and treatment of serotonin syndrome. Can Fam Physician. 2008;54(7):988-992.
6. Cyproheptadine hydrochloride tablets [package insert]. Hayward, CA: Impax Generics; 2017.
7. Deardorff OG, Khan T, Kulkarni G, et al. Serotonin syndrome: prophylactic treatment with cyproheptadine. Prim Care Companion CNS Disord. 2016;18(4). doi: 10.4088/PCC.16br01966.

Pharmacogenomics testing: What the FDA says

Mr. R, age 30, is referred to you by his primary care physician, who diagnosed him with depression approximately 2 years ago. When he was first diagnosed, Mr. R was prescribed
Mr. R says that based on his primary care physician’s recommendation, he had undergone pharmacogenomics testing to help guide therapy. He presents the results to you, and you notice that he has the cytochrome P450 (CYP) 2C19 *2/*3 genotype and a CYP2D6*4/*5 genotype. Both are associated with a poor metabolism phenotype. Should you use these findings to determine which medication Mr. R should be treated with next?
While the field of pharmacogenomics is not new, within the last few years this science has begun to transition into clinical practice. A recent meta-analysis found support for using pharmacogenomics testing results in clinical practice.1 This study included more than 1,700 patients who took part in 5 controlled trials that randomized participants to either pharmacogenetics-guided or unguided (ie, standard) treatment. Each participant was assessed using the Hamilton Depression Rating Scale-17 (HDRS-17) a minimum of 3 times over a minimum of 8 weeks.1 While the exact inclusion and exclusion criteria for each trial differed, they all defined remission of depression as achieving an HDRS-17 score ≤7. Overall, the authors concluded that based on the random-effects pooled risk ratio, there was a significant association between pharmacogenetics-guided prescribing and remission (relative risk = 1.71, 95% confidence interval [CI], 1.17 to 2.48; P = .005). The results of this meta-analysis are controversial, however, because all 5 studies were industry-funded, and interpretation of the testing results was based on proprietary algorithms.
Experts in the field and professional societies, such as the International Society of Psychiatric Genetics (ISPG), have issued policy statements on genetic testing within psychiatry.2,3 While the ISPG did not necessarily endorse use of pharmacogenomics in practice, they recommended that clinicians follow good medical practice and stay current on changes to drug labeling and adverse event reports.3 The ISPG also noted that useful but not exhaustive lists of pharmacogenetic tests are maintained by the Clinical Pharmacogenetics Implementation Consortium (CPIC) and the US FDA.3
Laboratory developed vs direct-to-consumer tests
In a previous Savvy Psychopharmacology article,4 we had discussed the role of CPIC, but not the role of the FDA. This issue is key because there is a lack of clarity regarding pharmacogenomics tests and whether they are considered Class II devices by the FDA, which would require their review and approval. Until recently, the FDA was fairly quiet regarding pharmacogenomics tests because most of these tests were considered laboratory developed tests, which were regulated under the Clinic Laboratory Improvements Amendments program. The critical distinction of a laboratory developed test is that it is developed and performed in a single laboratory and is offered to patients only when prescribed by a clinician. Due to this distinction, laboratory developed pharmacogenomics tests did not need FDA 510(k) clearance, which is a premarket submission common for medical devices.
Direct-to-consumer pharmacogenomics tests are different in that the FDA has classified these platforms as medical devices; however, they are reviewed by the FDA only if they are being used for moderate- to high-risk medical purposes, or if the results of the testing may have a higher impact on medical care. As part of its review, the FDA examines test accuracy and reliably measures to determine if the measurement is predictive of a certain state of health and supported by what the company claims about the test and how well it works. Additionally, the FDA examines the company-provided descriptive information to ensure that consumers can easily understand it without the help of a clinician.5
Conflicting FDA statements
Recently the FDA issued 2 statements—one a policy statement and the other a safety communication—about laboratory developed tests and direct-to-consumer tests. The statements appear to contradict themselves, despite focusing on using pharmacogenomics testing in practice.
Continue to: The FDA's first statement
The FDA’s first statement. On October 31, 2018, the FDA released a policy statement that they had “permitted marketing, with special controls,” of the Personal Genome Service Pharmacogenetic Reports test through 23andMe (a direct-to-consumer genetic testing company) for 33 different variants within specific pharmacogenomic genes (CYP2C19, CYP2C9, CYP3A5, UGT1A1, DPYD, TPMT, SLC01B1, and CYP2D6) that may impact drug metabolism or response.6 As part of its review of this Personal Genome Service Pharmacogenetic Reports test, the FDA found that the company-provided data showed that the test is accurate and can correctly identify the 33 specific genetic variants. The FDA review also showed that the testing results were reproducible, and the test instructions and reports could be understood by consumers.
While the specific reports related to this testing are not yet available within 23andMe, this approval allows for greater oversight by the FDA with regard to the pharmacogenomics information provided through this company’s Personal Genome Service Pharmacogenetic Reports test. The FDA noted that this approval was only for adults age >185 and that consumers “should not use the test results to stop or change any medication.”6 Further, the FDA stated that the results of the direct-to-consumer test should be confirmed with independent pharmacogenomics testing before making any medical decision. Unfortunately, the FDA did not offer guidance on what would be an appropriate independent pharmacogenomics test, but it did provide a link to a list of FDA-approved nucleic acid–based tests, on which 23andMe’s Personal Genome Service Pharmacogenetic Reports test is included.7
The FDA’s second statement. On November 1, 2018, the FDA issued a separate safety communication that cautioned clinicians and patients that most of the current commercially available testing platforms for pharmacogenomics have not been FDA-reviewed, meaning that they may lack clinical evidence supporting their use.8 Further, the FDA safety communication stated, “Changing drug treatment based on the results from such a genetic test could lead to inappropriate treatment decisions and potentially serious health consequences for the patient.”8
Taken together, these FDA statements appear to support pharmacogenomics testing with approval of the 23andMe’s Personal Genome Service Pharmacogenetic Reports test but warn that the testing results should not be used to make treatment decisions, and that they should be verified. However, the FDA does not offer any guidance on what an appropriate testing platform would be
What the FDA advises
The FDA has provided some guidance to clinicians and patients regarding next steps for patients who are interested in having pharmacogenomics testing or who have already undergone testing. The FDA’s first point is that both clinicians and patients need to be aware that pharmacogenomics testing is not FDA-reviewed, that patients should discuss the results of their testing with their clinicians, and that they should not stop their medication based on the results of the testing. Additionally, the FDA recommends that clinicians and patients should be aware that any claims made by the testing companies regarding the specific effect of a medication may not be supported by evidence. Furthermore, the FDA strongly recommends that clinicians consult the FDA-approved drug label, or the label of the FDA-cleared or FDA-approved genetic test, for information regarding how genetic information should be used in making treatment decisions. The FDA recommends reviewing the Warning section, as well as the Indications and Usage, Dosage and Administration, or Use in Specific Populations sections of the FDA-approved drug labeling.
Continue to: Unfortunately, this information...
Unfortunately, this information might be difficult to locate due to the lack of consistency regarding where it is placed in the FDA-approved drug labeling. The Pharmacogenomics Knowledgebase (https://www.pharmgkb.org/) can help clinicians quickly identify information regarding medications, their metabolic pathways, CPIC dosing guidelines, and the FDA-approved drug labeling information.9 By searching for specific medications within the Pharmacogenomic Knowledge Base, information regarding the FDA-approved drug labeling can be easily found, which is important because currently >120 medications contain pharmacogenomics information in their FDA-approved drug labeling.10
CASE CONTINUED
Overall Mr. R’s pharmacogenomics testing results indicate that he has 2 genotypes that are associated with poor metabolism phenotypes and could result in reduced metabolism of medications that are metabolized by these CYP enzymes, leading to higher blood levels and an increased risk of adverse effects. The Table11 lists pharmacogenomics information from the FDA-approved drug labeling and from the Pharmacogenomics Knowledgebase for both the medications Mr. R has previously been prescribed and for several potential medications to consider.

It would be prudent to first discuss with Mr. R the FDA’s recent policy statement and safety communication. While you could recommend that he pursue additional pharmacogenomics testing, it is unclear which specific laboratory is available to conduct this confirmatory analysis.
Because Mr. R has had unsuccessful trials of several medications that primarily fall in the selective serotonin reuptake inhibitors class, it might be time to consider a medication from a different class. A quick review of the FDA-approved drug labeling for
Related Resources
- Gammal RS, Gardner KN, Burghardt KJ. Where to find guidance on using pharmacogenomics in psychiatric practice. Current Psychiatry. 2016;15(9):93-94.
- Clinical Pharmacogenomics Implementation Consortium. What is CPIC? https://www.pharmgkb.org/page/cpic.
Drug Brand Names
Bupropion • Wellbutrin, Zyban
Citalopram • Celexa
Paroxetine • Paxil
Sertraline • Zoloft
Venlafaxine • Effexor
1. Bousman CA, Arandjelovic K, Mancuso SG, et al. Pharmacogenetic tests and depressive symptom remission: a meta-analysis of randomized controlled trials. Pharmacogenomics. 2019;20(1):37-47.
2. Zubenko GS, Sommer BR, Cohen BM. Pharmacogenetics in psychiatry: a companion, rather than competitor, to protocol-based care-reply. JAMA Psychiatry. 2018;75(10):1090-1091.
3. International Society for Psychiatric Genetics. Genetic testing statement: genetic testing and psychiatric disorders: a statement from the International Society of Psychiatric Genetics. https://ispg.net/genetic-testing-statement/. Revised January 26, 2017. Accessed January 1, 2019.
4. Ellingrod VL, Ward KM. Using pharmacogenetics guidelines when prescribing: what’s available. Current Psychiatry. 2018;17(1):43-46.
5. U.S. Food and Drug Administration. Medical devices: direct-to-consumer tests. https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm624726.htm. Published November 1, 2018. Accessed January 1, 2019.
6. U.S. Food and Drug Administration. FDA news releases: FDA authorizes first direct-to consumer test for detecting variants that may be associated with medication metabolism. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm624753.htm. Published October 31, 2018. Accessed January 1, 2019.
7. U.S. Food and Drug Administration. Medical devices: nucleic acid based tests. https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm330711.htm. Published February 5, 2019. Accessed March 1, 2019.
8. U.S. Food and Drug Administration. Medical devices. The FDA warns against the use of many genetic tests with unapproved claims to predict patient response to specific medications: FDA Safety Communications. https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm624725.htm. Published November 1, 2018. Accessed January 1, 2019.
9. Whirl-Carrillo EM, McDonagh JM, Hebert L, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414-417.
10. U.S. Food and Drug Administration. Drugs. Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm. Published August 3, 2018. Accessed January 1, 2019.
11. U.S. Food and Drug Administration. Drugs@FDA: FDA approved drug products. https://www.accessdata.fda.gov/scripts/cder/daf. Accessed March 4, 2019.
Mr. R, age 30, is referred to you by his primary care physician, who diagnosed him with depression approximately 2 years ago. When he was first diagnosed, Mr. R was prescribed
Mr. R says that based on his primary care physician’s recommendation, he had undergone pharmacogenomics testing to help guide therapy. He presents the results to you, and you notice that he has the cytochrome P450 (CYP) 2C19 *2/*3 genotype and a CYP2D6*4/*5 genotype. Both are associated with a poor metabolism phenotype. Should you use these findings to determine which medication Mr. R should be treated with next?
While the field of pharmacogenomics is not new, within the last few years this science has begun to transition into clinical practice. A recent meta-analysis found support for using pharmacogenomics testing results in clinical practice.1 This study included more than 1,700 patients who took part in 5 controlled trials that randomized participants to either pharmacogenetics-guided or unguided (ie, standard) treatment. Each participant was assessed using the Hamilton Depression Rating Scale-17 (HDRS-17) a minimum of 3 times over a minimum of 8 weeks.1 While the exact inclusion and exclusion criteria for each trial differed, they all defined remission of depression as achieving an HDRS-17 score ≤7. Overall, the authors concluded that based on the random-effects pooled risk ratio, there was a significant association between pharmacogenetics-guided prescribing and remission (relative risk = 1.71, 95% confidence interval [CI], 1.17 to 2.48; P = .005). The results of this meta-analysis are controversial, however, because all 5 studies were industry-funded, and interpretation of the testing results was based on proprietary algorithms.
Experts in the field and professional societies, such as the International Society of Psychiatric Genetics (ISPG), have issued policy statements on genetic testing within psychiatry.2,3 While the ISPG did not necessarily endorse use of pharmacogenomics in practice, they recommended that clinicians follow good medical practice and stay current on changes to drug labeling and adverse event reports.3 The ISPG also noted that useful but not exhaustive lists of pharmacogenetic tests are maintained by the Clinical Pharmacogenetics Implementation Consortium (CPIC) and the US FDA.3
Laboratory developed vs direct-to-consumer tests
In a previous Savvy Psychopharmacology article,4 we had discussed the role of CPIC, but not the role of the FDA. This issue is key because there is a lack of clarity regarding pharmacogenomics tests and whether they are considered Class II devices by the FDA, which would require their review and approval. Until recently, the FDA was fairly quiet regarding pharmacogenomics tests because most of these tests were considered laboratory developed tests, which were regulated under the Clinic Laboratory Improvements Amendments program. The critical distinction of a laboratory developed test is that it is developed and performed in a single laboratory and is offered to patients only when prescribed by a clinician. Due to this distinction, laboratory developed pharmacogenomics tests did not need FDA 510(k) clearance, which is a premarket submission common for medical devices.
Direct-to-consumer pharmacogenomics tests are different in that the FDA has classified these platforms as medical devices; however, they are reviewed by the FDA only if they are being used for moderate- to high-risk medical purposes, or if the results of the testing may have a higher impact on medical care. As part of its review, the FDA examines test accuracy and reliably measures to determine if the measurement is predictive of a certain state of health and supported by what the company claims about the test and how well it works. Additionally, the FDA examines the company-provided descriptive information to ensure that consumers can easily understand it without the help of a clinician.5
Conflicting FDA statements
Recently the FDA issued 2 statements—one a policy statement and the other a safety communication—about laboratory developed tests and direct-to-consumer tests. The statements appear to contradict themselves, despite focusing on using pharmacogenomics testing in practice.
Continue to: The FDA's first statement
The FDA’s first statement. On October 31, 2018, the FDA released a policy statement that they had “permitted marketing, with special controls,” of the Personal Genome Service Pharmacogenetic Reports test through 23andMe (a direct-to-consumer genetic testing company) for 33 different variants within specific pharmacogenomic genes (CYP2C19, CYP2C9, CYP3A5, UGT1A1, DPYD, TPMT, SLC01B1, and CYP2D6) that may impact drug metabolism or response.6 As part of its review of this Personal Genome Service Pharmacogenetic Reports test, the FDA found that the company-provided data showed that the test is accurate and can correctly identify the 33 specific genetic variants. The FDA review also showed that the testing results were reproducible, and the test instructions and reports could be understood by consumers.
While the specific reports related to this testing are not yet available within 23andMe, this approval allows for greater oversight by the FDA with regard to the pharmacogenomics information provided through this company’s Personal Genome Service Pharmacogenetic Reports test. The FDA noted that this approval was only for adults age >185 and that consumers “should not use the test results to stop or change any medication.”6 Further, the FDA stated that the results of the direct-to-consumer test should be confirmed with independent pharmacogenomics testing before making any medical decision. Unfortunately, the FDA did not offer guidance on what would be an appropriate independent pharmacogenomics test, but it did provide a link to a list of FDA-approved nucleic acid–based tests, on which 23andMe’s Personal Genome Service Pharmacogenetic Reports test is included.7
The FDA’s second statement. On November 1, 2018, the FDA issued a separate safety communication that cautioned clinicians and patients that most of the current commercially available testing platforms for pharmacogenomics have not been FDA-reviewed, meaning that they may lack clinical evidence supporting their use.8 Further, the FDA safety communication stated, “Changing drug treatment based on the results from such a genetic test could lead to inappropriate treatment decisions and potentially serious health consequences for the patient.”8
Taken together, these FDA statements appear to support pharmacogenomics testing with approval of the 23andMe’s Personal Genome Service Pharmacogenetic Reports test but warn that the testing results should not be used to make treatment decisions, and that they should be verified. However, the FDA does not offer any guidance on what an appropriate testing platform would be
What the FDA advises
The FDA has provided some guidance to clinicians and patients regarding next steps for patients who are interested in having pharmacogenomics testing or who have already undergone testing. The FDA’s first point is that both clinicians and patients need to be aware that pharmacogenomics testing is not FDA-reviewed, that patients should discuss the results of their testing with their clinicians, and that they should not stop their medication based on the results of the testing. Additionally, the FDA recommends that clinicians and patients should be aware that any claims made by the testing companies regarding the specific effect of a medication may not be supported by evidence. Furthermore, the FDA strongly recommends that clinicians consult the FDA-approved drug label, or the label of the FDA-cleared or FDA-approved genetic test, for information regarding how genetic information should be used in making treatment decisions. The FDA recommends reviewing the Warning section, as well as the Indications and Usage, Dosage and Administration, or Use in Specific Populations sections of the FDA-approved drug labeling.
Continue to: Unfortunately, this information...
Unfortunately, this information might be difficult to locate due to the lack of consistency regarding where it is placed in the FDA-approved drug labeling. The Pharmacogenomics Knowledgebase (https://www.pharmgkb.org/) can help clinicians quickly identify information regarding medications, their metabolic pathways, CPIC dosing guidelines, and the FDA-approved drug labeling information.9 By searching for specific medications within the Pharmacogenomic Knowledge Base, information regarding the FDA-approved drug labeling can be easily found, which is important because currently >120 medications contain pharmacogenomics information in their FDA-approved drug labeling.10
CASE CONTINUED
Overall Mr. R’s pharmacogenomics testing results indicate that he has 2 genotypes that are associated with poor metabolism phenotypes and could result in reduced metabolism of medications that are metabolized by these CYP enzymes, leading to higher blood levels and an increased risk of adverse effects. The Table11 lists pharmacogenomics information from the FDA-approved drug labeling and from the Pharmacogenomics Knowledgebase for both the medications Mr. R has previously been prescribed and for several potential medications to consider.
It would be prudent to first discuss with Mr. R the FDA’s recent policy statement and safety communication. While you could recommend that he pursue additional pharmacogenomics testing, it is unclear which specific laboratory is available to conduct this confirmatory analysis.
Because Mr. R has had unsuccessful trials of several medications that primarily fall in the selective serotonin reuptake inhibitors class, it might be time to consider a medication from a different class. A quick review of the FDA-approved drug labeling for
Related Resources
- Gammal RS, Gardner KN, Burghardt KJ. Where to find guidance on using pharmacogenomics in psychiatric practice. Current Psychiatry. 2016;15(9):93-94.
- Clinical Pharmacogenomics Implementation Consortium. What is CPIC? https://www.pharmgkb.org/page/cpic.
Drug Brand Names
Bupropion • Wellbutrin, Zyban
Citalopram • Celexa
Paroxetine • Paxil
Sertraline • Zoloft
Venlafaxine • Effexor
Mr. R, age 30, is referred to you by his primary care physician, who diagnosed him with depression approximately 2 years ago. When he was first diagnosed, Mr. R was prescribed
Mr. R says that based on his primary care physician’s recommendation, he had undergone pharmacogenomics testing to help guide therapy. He presents the results to you, and you notice that he has the cytochrome P450 (CYP) 2C19 *2/*3 genotype and a CYP2D6*4/*5 genotype. Both are associated with a poor metabolism phenotype. Should you use these findings to determine which medication Mr. R should be treated with next?
While the field of pharmacogenomics is not new, within the last few years this science has begun to transition into clinical practice. A recent meta-analysis found support for using pharmacogenomics testing results in clinical practice.1 This study included more than 1,700 patients who took part in 5 controlled trials that randomized participants to either pharmacogenetics-guided or unguided (ie, standard) treatment. Each participant was assessed using the Hamilton Depression Rating Scale-17 (HDRS-17) a minimum of 3 times over a minimum of 8 weeks.1 While the exact inclusion and exclusion criteria for each trial differed, they all defined remission of depression as achieving an HDRS-17 score ≤7. Overall, the authors concluded that based on the random-effects pooled risk ratio, there was a significant association between pharmacogenetics-guided prescribing and remission (relative risk = 1.71, 95% confidence interval [CI], 1.17 to 2.48; P = .005). The results of this meta-analysis are controversial, however, because all 5 studies were industry-funded, and interpretation of the testing results was based on proprietary algorithms.
Experts in the field and professional societies, such as the International Society of Psychiatric Genetics (ISPG), have issued policy statements on genetic testing within psychiatry.2,3 While the ISPG did not necessarily endorse use of pharmacogenomics in practice, they recommended that clinicians follow good medical practice and stay current on changes to drug labeling and adverse event reports.3 The ISPG also noted that useful but not exhaustive lists of pharmacogenetic tests are maintained by the Clinical Pharmacogenetics Implementation Consortium (CPIC) and the US FDA.3
Laboratory developed vs direct-to-consumer tests
In a previous Savvy Psychopharmacology article,4 we had discussed the role of CPIC, but not the role of the FDA. This issue is key because there is a lack of clarity regarding pharmacogenomics tests and whether they are considered Class II devices by the FDA, which would require their review and approval. Until recently, the FDA was fairly quiet regarding pharmacogenomics tests because most of these tests were considered laboratory developed tests, which were regulated under the Clinic Laboratory Improvements Amendments program. The critical distinction of a laboratory developed test is that it is developed and performed in a single laboratory and is offered to patients only when prescribed by a clinician. Due to this distinction, laboratory developed pharmacogenomics tests did not need FDA 510(k) clearance, which is a premarket submission common for medical devices.
Direct-to-consumer pharmacogenomics tests are different in that the FDA has classified these platforms as medical devices; however, they are reviewed by the FDA only if they are being used for moderate- to high-risk medical purposes, or if the results of the testing may have a higher impact on medical care. As part of its review, the FDA examines test accuracy and reliably measures to determine if the measurement is predictive of a certain state of health and supported by what the company claims about the test and how well it works. Additionally, the FDA examines the company-provided descriptive information to ensure that consumers can easily understand it without the help of a clinician.5
Conflicting FDA statements
Recently the FDA issued 2 statements—one a policy statement and the other a safety communication—about laboratory developed tests and direct-to-consumer tests. The statements appear to contradict themselves, despite focusing on using pharmacogenomics testing in practice.
Continue to: The FDA's first statement
The FDA’s first statement. On October 31, 2018, the FDA released a policy statement that they had “permitted marketing, with special controls,” of the Personal Genome Service Pharmacogenetic Reports test through 23andMe (a direct-to-consumer genetic testing company) for 33 different variants within specific pharmacogenomic genes (CYP2C19, CYP2C9, CYP3A5, UGT1A1, DPYD, TPMT, SLC01B1, and CYP2D6) that may impact drug metabolism or response.6 As part of its review of this Personal Genome Service Pharmacogenetic Reports test, the FDA found that the company-provided data showed that the test is accurate and can correctly identify the 33 specific genetic variants. The FDA review also showed that the testing results were reproducible, and the test instructions and reports could be understood by consumers.
While the specific reports related to this testing are not yet available within 23andMe, this approval allows for greater oversight by the FDA with regard to the pharmacogenomics information provided through this company’s Personal Genome Service Pharmacogenetic Reports test. The FDA noted that this approval was only for adults age >185 and that consumers “should not use the test results to stop or change any medication.”6 Further, the FDA stated that the results of the direct-to-consumer test should be confirmed with independent pharmacogenomics testing before making any medical decision. Unfortunately, the FDA did not offer guidance on what would be an appropriate independent pharmacogenomics test, but it did provide a link to a list of FDA-approved nucleic acid–based tests, on which 23andMe’s Personal Genome Service Pharmacogenetic Reports test is included.7
The FDA’s second statement. On November 1, 2018, the FDA issued a separate safety communication that cautioned clinicians and patients that most of the current commercially available testing platforms for pharmacogenomics have not been FDA-reviewed, meaning that they may lack clinical evidence supporting their use.8 Further, the FDA safety communication stated, “Changing drug treatment based on the results from such a genetic test could lead to inappropriate treatment decisions and potentially serious health consequences for the patient.”8
Taken together, these FDA statements appear to support pharmacogenomics testing with approval of the 23andMe’s Personal Genome Service Pharmacogenetic Reports test but warn that the testing results should not be used to make treatment decisions, and that they should be verified. However, the FDA does not offer any guidance on what an appropriate testing platform would be
What the FDA advises
The FDA has provided some guidance to clinicians and patients regarding next steps for patients who are interested in having pharmacogenomics testing or who have already undergone testing. The FDA’s first point is that both clinicians and patients need to be aware that pharmacogenomics testing is not FDA-reviewed, that patients should discuss the results of their testing with their clinicians, and that they should not stop their medication based on the results of the testing. Additionally, the FDA recommends that clinicians and patients should be aware that any claims made by the testing companies regarding the specific effect of a medication may not be supported by evidence. Furthermore, the FDA strongly recommends that clinicians consult the FDA-approved drug label, or the label of the FDA-cleared or FDA-approved genetic test, for information regarding how genetic information should be used in making treatment decisions. The FDA recommends reviewing the Warning section, as well as the Indications and Usage, Dosage and Administration, or Use in Specific Populations sections of the FDA-approved drug labeling.
Continue to: Unfortunately, this information...
Unfortunately, this information might be difficult to locate due to the lack of consistency regarding where it is placed in the FDA-approved drug labeling. The Pharmacogenomics Knowledgebase (https://www.pharmgkb.org/) can help clinicians quickly identify information regarding medications, their metabolic pathways, CPIC dosing guidelines, and the FDA-approved drug labeling information.9 By searching for specific medications within the Pharmacogenomic Knowledge Base, information regarding the FDA-approved drug labeling can be easily found, which is important because currently >120 medications contain pharmacogenomics information in their FDA-approved drug labeling.10
CASE CONTINUED
Overall Mr. R’s pharmacogenomics testing results indicate that he has 2 genotypes that are associated with poor metabolism phenotypes and could result in reduced metabolism of medications that are metabolized by these CYP enzymes, leading to higher blood levels and an increased risk of adverse effects. The Table11 lists pharmacogenomics information from the FDA-approved drug labeling and from the Pharmacogenomics Knowledgebase for both the medications Mr. R has previously been prescribed and for several potential medications to consider.
It would be prudent to first discuss with Mr. R the FDA’s recent policy statement and safety communication. While you could recommend that he pursue additional pharmacogenomics testing, it is unclear which specific laboratory is available to conduct this confirmatory analysis.
Because Mr. R has had unsuccessful trials of several medications that primarily fall in the selective serotonin reuptake inhibitors class, it might be time to consider a medication from a different class. A quick review of the FDA-approved drug labeling for
Related Resources
- Gammal RS, Gardner KN, Burghardt KJ. Where to find guidance on using pharmacogenomics in psychiatric practice. Current Psychiatry. 2016;15(9):93-94.
- Clinical Pharmacogenomics Implementation Consortium. What is CPIC? https://www.pharmgkb.org/page/cpic.
Drug Brand Names
Bupropion • Wellbutrin, Zyban
Citalopram • Celexa
Paroxetine • Paxil
Sertraline • Zoloft
Venlafaxine • Effexor
1. Bousman CA, Arandjelovic K, Mancuso SG, et al. Pharmacogenetic tests and depressive symptom remission: a meta-analysis of randomized controlled trials. Pharmacogenomics. 2019;20(1):37-47.
2. Zubenko GS, Sommer BR, Cohen BM. Pharmacogenetics in psychiatry: a companion, rather than competitor, to protocol-based care-reply. JAMA Psychiatry. 2018;75(10):1090-1091.
3. International Society for Psychiatric Genetics. Genetic testing statement: genetic testing and psychiatric disorders: a statement from the International Society of Psychiatric Genetics. https://ispg.net/genetic-testing-statement/. Revised January 26, 2017. Accessed January 1, 2019.
4. Ellingrod VL, Ward KM. Using pharmacogenetics guidelines when prescribing: what’s available. Current Psychiatry. 2018;17(1):43-46.
5. U.S. Food and Drug Administration. Medical devices: direct-to-consumer tests. https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm624726.htm. Published November 1, 2018. Accessed January 1, 2019.
6. U.S. Food and Drug Administration. FDA news releases: FDA authorizes first direct-to consumer test for detecting variants that may be associated with medication metabolism. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm624753.htm. Published October 31, 2018. Accessed January 1, 2019.
7. U.S. Food and Drug Administration. Medical devices: nucleic acid based tests. https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm330711.htm. Published February 5, 2019. Accessed March 1, 2019.
8. U.S. Food and Drug Administration. Medical devices. The FDA warns against the use of many genetic tests with unapproved claims to predict patient response to specific medications: FDA Safety Communications. https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm624725.htm. Published November 1, 2018. Accessed January 1, 2019.
9. Whirl-Carrillo EM, McDonagh JM, Hebert L, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414-417.
10. U.S. Food and Drug Administration. Drugs. Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm. Published August 3, 2018. Accessed January 1, 2019.
11. U.S. Food and Drug Administration. Drugs@FDA: FDA approved drug products. https://www.accessdata.fda.gov/scripts/cder/daf. Accessed March 4, 2019.
1. Bousman CA, Arandjelovic K, Mancuso SG, et al. Pharmacogenetic tests and depressive symptom remission: a meta-analysis of randomized controlled trials. Pharmacogenomics. 2019;20(1):37-47.
2. Zubenko GS, Sommer BR, Cohen BM. Pharmacogenetics in psychiatry: a companion, rather than competitor, to protocol-based care-reply. JAMA Psychiatry. 2018;75(10):1090-1091.
3. International Society for Psychiatric Genetics. Genetic testing statement: genetic testing and psychiatric disorders: a statement from the International Society of Psychiatric Genetics. https://ispg.net/genetic-testing-statement/. Revised January 26, 2017. Accessed January 1, 2019.
4. Ellingrod VL, Ward KM. Using pharmacogenetics guidelines when prescribing: what’s available. Current Psychiatry. 2018;17(1):43-46.
5. U.S. Food and Drug Administration. Medical devices: direct-to-consumer tests. https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm624726.htm. Published November 1, 2018. Accessed January 1, 2019.
6. U.S. Food and Drug Administration. FDA news releases: FDA authorizes first direct-to consumer test for detecting variants that may be associated with medication metabolism. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm624753.htm. Published October 31, 2018. Accessed January 1, 2019.
7. U.S. Food and Drug Administration. Medical devices: nucleic acid based tests. https://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm330711.htm. Published February 5, 2019. Accessed March 1, 2019.
8. U.S. Food and Drug Administration. Medical devices. The FDA warns against the use of many genetic tests with unapproved claims to predict patient response to specific medications: FDA Safety Communications. https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm624725.htm. Published November 1, 2018. Accessed January 1, 2019.
9. Whirl-Carrillo EM, McDonagh JM, Hebert L, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414-417.
10. U.S. Food and Drug Administration. Drugs. Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm. Published August 3, 2018. Accessed January 1, 2019.
11. U.S. Food and Drug Administration. Drugs@FDA: FDA approved drug products. https://www.accessdata.fda.gov/scripts/cder/daf. Accessed March 4, 2019.

Psychotropic-induced hyponatremia

Hyponatremia is a common, multifactorial clinical condition. Hyponatremia is usually defined as a plasma sodium level <135 mmol/L; however, some studies define it as a level <130 mmol/L. Hyponatremia results from the inability of the kidney to excrete a sufficient amount of fluid, or is due to excessive fluid intake. Increases in osmolality stimulate thirst and result in increased fluid intake. This increase in osmolality is recognized by the osmoreceptors located in the hypothalamus, which release antidiuretic hormone (ADH). Antidiuretic hormone works on the collecting ducts within the kidneys, triggering increased fluid reabsorption resulting in decreased fluid loss and a reduction in thirst.
The syndrome of inappropriate antidiuretic hormone (SIADH) occurs when there is persistent ADH stimulation resulting in hyponatremia. SIADH commonly presents as euvolemic hyponatremia. Common diagnostic criteria for SIADH are listed in Table 1.1

Medications are a major cause of SIADH, and psychotropics are a primary offender. Most of the data for drug-induced SIADH come from case reports and small case series, such as those described in Table 2.2-4 The extent to which each psychotropic class causes SIADH remains unknown. In this article, we focus on 3 classes of psychotropics, and their role in causing SIADH.

Antidepressants
There is a fair amount of data associating antidepressants with SIADH. The incidence of SIADH with selective serotonin reuptake inhibitors (SSRIs) varies greatly among studies, from .06% to 40%.5-12 This wide variation is due to the way each study defined hyponatremia. A higher incidence was found when hyponatremia was defined as <135 mmol/L as opposed to <130 mmol/L. A large cohort study of SSRIs found that there was an increased risk with fluoxetine, escitalopram, and citalopram (.078% to .085%) vs paroxetine and sertraline (.033% to .053%).13 Studies comparing the incidence of SIADH with SSRIs and serotonin-norepinephrine reuptake inhibitors (SNRIs) found that the rates were equal or slightly higher with the SNRI venlafaxine.13 SNRIs as a group have an estimated incidence of .08% to 4%, based on studies that defined hyponatremia as <130 mmol/L.13,14 Tricyclic antidepressants have an estimated incidence of .005% to 16.7%, based on a retrospective study that reviewed 15 studies and 100 case reports.15 Mirtazapine and bupropion do not have enough evidence to obtain a true definition of incidence; case reports for these drugs suggest a causal link for hyponatremia. Table 37,9,12-15 provides an overview of the incidence rate of hyponatremia for select antidepressants. It is clear that a more stringent cutoff for hyponatremia (<130 mmol/L) reduces the incidence rates. More evidence is needed to identify the true incidence and prevalence of SIADH with these agents.

Antipsychotics
Compared with antidepressants, there’s less evidence linking SIADH with antipsychotics; this data come mainly from case reports and observational studies. Serrano et al16 reported on a cross-sectional study that included 88 patients receiving clozapine, 61 patients receiving other atypical antipsychotics, 23 patients receiving typical antipsychotics, and 11 patients receiving both typical and atypical antipsychotics. They reported incidence rates of 3.4% for clozapine, 4.9% for atypical antipsychotics, 26.1% for typical antipsychotics, and 9.1% for the group receiving both typical and atypical antipsychotics.16 The primary theory for the decreased incidence of SIADH with use of atypical antipsychotics is related to decreased rates of psychogenic polydipsia leading to lower incidence of hyponatremia.
Mood stabilizers
Several studies have associated carbamazepine/oxcarbazepine, valproic acid, and lamotrigine with SIADH.17-23 Studies show incidence rates ranging from 4.8% to 41.5% for these medications. Carbamazepine appears to have the highest incidence of SIADH. A limitation of these studies is the small sample sizes, which ranged from 12 to 60 participants.
Pathophysiology
The kidneys are responsible for maintaining homeostasis between bodily fluids and serum sodium levels. ADH, which is produced by the hypothalamus, plays a significant role in fluid balance, thirst, and fluid retention. Inappropriate and continuous secretion of ADH, despite normal or high fluid status, results in hyposmolality and hyponatremia. The specific mechanisms by which psychotropic medications cause SIADH are listed in Table 4.24

Diagnosis
Diagnosis of SIADH can be complex because there are many clinical reasons a patient may have hyponatremia. For example, SIADH and psychogenic polydipsia both result in hyponatremia, and sometimes the 2 conditions can be difficult to distinguish. Hyponatremia is typically discovered by routine blood testing if the patient is asymptomatic. Table 525 highlights the major laboratory markers that distinguish SIADH and psychogenic polydipsia.

Continue to: Treatment
Treatment
The primary treatment for SIADH is cessation of the offending agent. Based on the patient’s clinical presentation, free water restriction (.5 to 1 L/d) can be implemented to increase serum sodium levels. If the patient is having neurologic complications due to the severity of hyponatremia, correction with hypertonic saline is indicated. Upon resolution, the recommended course of action is to switch to a medication in a different class. Re-challenging the patient with the same medication is not recommended unless there is no other alternative class of medication.24 Table 626 highlights other causes of hyponatremia, what laboratory markers to assess, and how to treat high-risk individuals.

Hyponatremia is a complex medical complication that can be life-threatening. Psychotropics are a relatively common cause of hyponatremia, specifically SIADH. Older adults appear to be at highest risk, as most case reports are in patients age ≥65. Patients who are prescribed psychotropics should be treated with the lowest effective dose and monitored for signs and symptoms of hyponatremia throughout therapy.
Related Resources
- Spasovski G, Vanholder R, Allolio B, et al. Clinical practice guidelines on diagnosis and treatment of hyponatremia. Eur J Endocrinol. 2014;170(3):G1-G47.
- Verbalis JG, Goldsmith SR, Greenberg A, et al. Diagnosis, evaluation, and treatment of hyponatremia: expert panel recommendations. Am J Med. 2013;126(10 Suppl 1):S1-S42.
Drug Brand Names
Bupropion • Wellbutrin
Carbamazepine • Tegretol
Citalopram • Celexa
Clozapine • Clozaril
Escitalopram • Lexapro
Fluoxetine • Prozac
Haloperidol • Haldol
Lamotrigine • Lamictal
Levathyroxine • Levothroid
Mirtazapine • Remeron
Oxcarbazepine • Trileptal
Paroxetine • Paxil
Quetiapine • Seroquel
Risperidone • Risperdal
Sertraline • Zoloft
Valproic acid • Depakote
Venlafaxine • Effexor
1. Sahay M, Sahay R. Hyponatremia: a practical approach. Indian J Endocrinol Metab. 2014;18(6):760-771.
2. Kenes MT, Hamblin S, Tumuluri SS, et al. Syndrome of inappropriate antidiuretic hormone in a patient receiving high-dose haloperidol and quetiapine therapy. J Neuropsychiatry Clin Neurosci. 2016;28(2):e29-e30. doi: 10.1176/appi.neuropsych.15110392.
3. Twardowschy CA, Bertolucci CB, Gracia Cde M, et al. Severe hyponatremia and the syndrome of inappropriate secretion of antidiuretic hormone (SIADH) associated with fluoxetine: case report. Arq Neuropsiquiatr. 2006;64(1):142-145.
4. Patel KR, Meesala A, Stanilla JK. Sodium valproate–induced hyponatremia: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(5):PCC.09100941. doi: 10.4088/PCC.09100941.
5. Pillans PI, Coulter DM. Fluoxetine and hyponatraemia—a potential hazard in the elderly. N Z Med J. 1994;107(973):85‑86.
6. Strachan J, Shepherd J. Hyponatraemia associated with the use of selective serotonin reuptake inhibitors. Aust N Z J Psychiatry. 1998;32(2):295‑298.
7. Bouman WP, Pinner G, Johnson H. Incidence of selective serotonin reuptake inhibitor (SSRI) induced hyponatraemia due to the syndrome of inappropriate antidiuretic hormone (SIADH) secretion in the elderly. Int J Geriatr Psychiatry. 1998;13(1):12‑15.
8. Wilkinson TJ, Begg EJ, Winter AC, et al. Incidence and risk factors for hyponatraemia following treatment with fluoxetine or paroxetine in elderly people. Br J Clin Pharmacol. 1999;47(2):211‑217.
9. Kirby D, Harrigan S, Ames D. Hyponatraemia in elderly psychiatric patients treated with selective serotonin reuptake inhibitors and venlafaxine: a retrospective controlled study in an inpatient unit. Int J Geriatr Psychiatry. 2002;17(3):231‑237.
10. Wee R, Lim WK. Selective serotonin re‑uptake inhibitors (SSRIs) and hyponatraemia in the elderly. Int J Geriatr Psychiatry. 2004;19(6):590‑591.
11. Jung YE, Jun TY, Kim KS, et al. Hyponatremia associated with selective serotonin reuptake inhibitors, mirtazapine, and venlafaxine in Korean patients with major depressive disorder. Int J Clin Pharmacol Ther. 2011;49(7):437‑443.
12. Letmaier M, Painold A, Holl AK, et al. Hyponatremia during psychopharmacological treatment: Results of a drug surveillance program. Int J Neuropsychopharmacol. 2012;15(6):739‑748.
13. Coupland CA, Dhiman P, Barton G, et al. A study of the safety and harms of antidepressant drugs for older people: a cohort study using a large primary care database. Health Technol Assess. 2011;15(28):1‑202, iii‑iv.
14. Leah-Møller KB, Hansen AH, Torstensson M, et al. Antidepressants and the risk of hyponatremia: a Danish register-based population study. BMJ Open. 2016;6(5):e011200. doi: 10.1136/bmjopen-2016-011200.
15. De Picker LD, Van Den Eede F, Dumont G, et al. Antidepressants and the risk of hyponatremia: a class by class review of literature. Psychosomatics. 2014;55(6):536-547.
16. Serrano A, Rangel N, Carrizo E, et al. Safety of long-term clozapine administration. Frequency of cardiomyopathy and hyponatraemia: two cross-sectional, naturalistic studies. Aust N Z J Psychiatry. 2014;48(2):183‑192.
17. Uhde TW, Post RM. Effects of carbamazepine on serum electrolytes: clinical and theoretical implications. J Clin Psychopharmacol. 1983;3(2):103‑106.
18. Lahr MB. Hyponatremia during carbamazepine therapy. Clin Pharmacol Ther. 1985;37(6):693‑696.
19. Joffe RT, Post RM, Uhde TW. Effects of carbamazepine on serum electrolytes in affectively ill patients. Psychol Med. 1986;16(2):331‑335.
20. Vieweg V, Glick JL, Herring S, et al. Absence of carbamazepine‑induced hyponatremia among patients also given lithium. Am J Psychiatry. 1987;144(7):943‑947.
21. Yassa R, Iskandar H, Nastase C, et al. Carbamazepine and hyponatremia in patients with affective disorder. Am J Psychiatry. 1988;145(3):339‑342.
22. Kastner T, Friedman DL, Pond WS. Carbamazepine‑induced hyponatremia in patients with mental retardation. Am J Ment Retard. 1992;96(5):536‑540.
23. Kelly BD, Hillery J. Hyponatremia during carbamazepine therapy in patients with intellectual disability. J Intellect Disabil Res. 2001;45(Pt 2):152‑156.
24. Sahoo S, Grover S. Hyponatremia and psychotropics. J Geriatr Ment Health. 2016;3(2):108-122.
25. Siragy HM. Hyponatremia, fluid-electrolyte disorders and the syndrome of inappropriate antidiuretic hormone secretion: diagnosis and treatment options. Endocr Pract. 2006;12(4):446-457.
26. Braun M, Barstow CH, Pyzocha NJ. Diagnosis and management of sodium disorders: hyponatremia and hypernatremia. Am Fam Physician. 2015;91(5):299-307.
Hyponatremia is a common, multifactorial clinical condition. Hyponatremia is usually defined as a plasma sodium level <135 mmol/L; however, some studies define it as a level <130 mmol/L. Hyponatremia results from the inability of the kidney to excrete a sufficient amount of fluid, or is due to excessive fluid intake. Increases in osmolality stimulate thirst and result in increased fluid intake. This increase in osmolality is recognized by the osmoreceptors located in the hypothalamus, which release antidiuretic hormone (ADH). Antidiuretic hormone works on the collecting ducts within the kidneys, triggering increased fluid reabsorption resulting in decreased fluid loss and a reduction in thirst.
The syndrome of inappropriate antidiuretic hormone (SIADH) occurs when there is persistent ADH stimulation resulting in hyponatremia. SIADH commonly presents as euvolemic hyponatremia. Common diagnostic criteria for SIADH are listed in Table 1.1
Medications are a major cause of SIADH, and psychotropics are a primary offender. Most of the data for drug-induced SIADH come from case reports and small case series, such as those described in Table 2.2-4 The extent to which each psychotropic class causes SIADH remains unknown. In this article, we focus on 3 classes of psychotropics, and their role in causing SIADH.
Antidepressants
There is a fair amount of data associating antidepressants with SIADH. The incidence of SIADH with selective serotonin reuptake inhibitors (SSRIs) varies greatly among studies, from .06% to 40%.5-12 This wide variation is due to the way each study defined hyponatremia. A higher incidence was found when hyponatremia was defined as <135 mmol/L as opposed to <130 mmol/L. A large cohort study of SSRIs found that there was an increased risk with fluoxetine, escitalopram, and citalopram (.078% to .085%) vs paroxetine and sertraline (.033% to .053%).13 Studies comparing the incidence of SIADH with SSRIs and serotonin-norepinephrine reuptake inhibitors (SNRIs) found that the rates were equal or slightly higher with the SNRI venlafaxine.13 SNRIs as a group have an estimated incidence of .08% to 4%, based on studies that defined hyponatremia as <130 mmol/L.13,14 Tricyclic antidepressants have an estimated incidence of .005% to 16.7%, based on a retrospective study that reviewed 15 studies and 100 case reports.15 Mirtazapine and bupropion do not have enough evidence to obtain a true definition of incidence; case reports for these drugs suggest a causal link for hyponatremia. Table 37,9,12-15 provides an overview of the incidence rate of hyponatremia for select antidepressants. It is clear that a more stringent cutoff for hyponatremia (<130 mmol/L) reduces the incidence rates. More evidence is needed to identify the true incidence and prevalence of SIADH with these agents.
Antipsychotics
Compared with antidepressants, there’s less evidence linking SIADH with antipsychotics; this data come mainly from case reports and observational studies. Serrano et al16 reported on a cross-sectional study that included 88 patients receiving clozapine, 61 patients receiving other atypical antipsychotics, 23 patients receiving typical antipsychotics, and 11 patients receiving both typical and atypical antipsychotics. They reported incidence rates of 3.4% for clozapine, 4.9% for atypical antipsychotics, 26.1% for typical antipsychotics, and 9.1% for the group receiving both typical and atypical antipsychotics.16 The primary theory for the decreased incidence of SIADH with use of atypical antipsychotics is related to decreased rates of psychogenic polydipsia leading to lower incidence of hyponatremia.
Mood stabilizers
Several studies have associated carbamazepine/oxcarbazepine, valproic acid, and lamotrigine with SIADH.17-23 Studies show incidence rates ranging from 4.8% to 41.5% for these medications. Carbamazepine appears to have the highest incidence of SIADH. A limitation of these studies is the small sample sizes, which ranged from 12 to 60 participants.
Pathophysiology
The kidneys are responsible for maintaining homeostasis between bodily fluids and serum sodium levels. ADH, which is produced by the hypothalamus, plays a significant role in fluid balance, thirst, and fluid retention. Inappropriate and continuous secretion of ADH, despite normal or high fluid status, results in hyposmolality and hyponatremia. The specific mechanisms by which psychotropic medications cause SIADH are listed in Table 4.24
Diagnosis
Diagnosis of SIADH can be complex because there are many clinical reasons a patient may have hyponatremia. For example, SIADH and psychogenic polydipsia both result in hyponatremia, and sometimes the 2 conditions can be difficult to distinguish. Hyponatremia is typically discovered by routine blood testing if the patient is asymptomatic. Table 525 highlights the major laboratory markers that distinguish SIADH and psychogenic polydipsia.
Continue to: Treatment
Treatment
The primary treatment for SIADH is cessation of the offending agent. Based on the patient’s clinical presentation, free water restriction (.5 to 1 L/d) can be implemented to increase serum sodium levels. If the patient is having neurologic complications due to the severity of hyponatremia, correction with hypertonic saline is indicated. Upon resolution, the recommended course of action is to switch to a medication in a different class. Re-challenging the patient with the same medication is not recommended unless there is no other alternative class of medication.24 Table 626 highlights other causes of hyponatremia, what laboratory markers to assess, and how to treat high-risk individuals.
Hyponatremia is a complex medical complication that can be life-threatening. Psychotropics are a relatively common cause of hyponatremia, specifically SIADH. Older adults appear to be at highest risk, as most case reports are in patients age ≥65. Patients who are prescribed psychotropics should be treated with the lowest effective dose and monitored for signs and symptoms of hyponatremia throughout therapy.
Related Resources
- Spasovski G, Vanholder R, Allolio B, et al. Clinical practice guidelines on diagnosis and treatment of hyponatremia. Eur J Endocrinol. 2014;170(3):G1-G47.
- Verbalis JG, Goldsmith SR, Greenberg A, et al. Diagnosis, evaluation, and treatment of hyponatremia: expert panel recommendations. Am J Med. 2013;126(10 Suppl 1):S1-S42.
Drug Brand Names
Bupropion • Wellbutrin
Carbamazepine • Tegretol
Citalopram • Celexa
Clozapine • Clozaril
Escitalopram • Lexapro
Fluoxetine • Prozac
Haloperidol • Haldol
Lamotrigine • Lamictal
Levathyroxine • Levothroid
Mirtazapine • Remeron
Oxcarbazepine • Trileptal
Paroxetine • Paxil
Quetiapine • Seroquel
Risperidone • Risperdal
Sertraline • Zoloft
Valproic acid • Depakote
Venlafaxine • Effexor
Hyponatremia is a common, multifactorial clinical condition. Hyponatremia is usually defined as a plasma sodium level <135 mmol/L; however, some studies define it as a level <130 mmol/L. Hyponatremia results from the inability of the kidney to excrete a sufficient amount of fluid, or is due to excessive fluid intake. Increases in osmolality stimulate thirst and result in increased fluid intake. This increase in osmolality is recognized by the osmoreceptors located in the hypothalamus, which release antidiuretic hormone (ADH). Antidiuretic hormone works on the collecting ducts within the kidneys, triggering increased fluid reabsorption resulting in decreased fluid loss and a reduction in thirst.
The syndrome of inappropriate antidiuretic hormone (SIADH) occurs when there is persistent ADH stimulation resulting in hyponatremia. SIADH commonly presents as euvolemic hyponatremia. Common diagnostic criteria for SIADH are listed in Table 1.1
Medications are a major cause of SIADH, and psychotropics are a primary offender. Most of the data for drug-induced SIADH come from case reports and small case series, such as those described in Table 2.2-4 The extent to which each psychotropic class causes SIADH remains unknown. In this article, we focus on 3 classes of psychotropics, and their role in causing SIADH.
Antidepressants
There is a fair amount of data associating antidepressants with SIADH. The incidence of SIADH with selective serotonin reuptake inhibitors (SSRIs) varies greatly among studies, from .06% to 40%.5-12 This wide variation is due to the way each study defined hyponatremia. A higher incidence was found when hyponatremia was defined as <135 mmol/L as opposed to <130 mmol/L. A large cohort study of SSRIs found that there was an increased risk with fluoxetine, escitalopram, and citalopram (.078% to .085%) vs paroxetine and sertraline (.033% to .053%).13 Studies comparing the incidence of SIADH with SSRIs and serotonin-norepinephrine reuptake inhibitors (SNRIs) found that the rates were equal or slightly higher with the SNRI venlafaxine.13 SNRIs as a group have an estimated incidence of .08% to 4%, based on studies that defined hyponatremia as <130 mmol/L.13,14 Tricyclic antidepressants have an estimated incidence of .005% to 16.7%, based on a retrospective study that reviewed 15 studies and 100 case reports.15 Mirtazapine and bupropion do not have enough evidence to obtain a true definition of incidence; case reports for these drugs suggest a causal link for hyponatremia. Table 37,9,12-15 provides an overview of the incidence rate of hyponatremia for select antidepressants. It is clear that a more stringent cutoff for hyponatremia (<130 mmol/L) reduces the incidence rates. More evidence is needed to identify the true incidence and prevalence of SIADH with these agents.
Antipsychotics
Compared with antidepressants, there’s less evidence linking SIADH with antipsychotics; this data come mainly from case reports and observational studies. Serrano et al16 reported on a cross-sectional study that included 88 patients receiving clozapine, 61 patients receiving other atypical antipsychotics, 23 patients receiving typical antipsychotics, and 11 patients receiving both typical and atypical antipsychotics. They reported incidence rates of 3.4% for clozapine, 4.9% for atypical antipsychotics, 26.1% for typical antipsychotics, and 9.1% for the group receiving both typical and atypical antipsychotics.16 The primary theory for the decreased incidence of SIADH with use of atypical antipsychotics is related to decreased rates of psychogenic polydipsia leading to lower incidence of hyponatremia.
Mood stabilizers
Several studies have associated carbamazepine/oxcarbazepine, valproic acid, and lamotrigine with SIADH.17-23 Studies show incidence rates ranging from 4.8% to 41.5% for these medications. Carbamazepine appears to have the highest incidence of SIADH. A limitation of these studies is the small sample sizes, which ranged from 12 to 60 participants.
Pathophysiology
The kidneys are responsible for maintaining homeostasis between bodily fluids and serum sodium levels. ADH, which is produced by the hypothalamus, plays a significant role in fluid balance, thirst, and fluid retention. Inappropriate and continuous secretion of ADH, despite normal or high fluid status, results in hyposmolality and hyponatremia. The specific mechanisms by which psychotropic medications cause SIADH are listed in Table 4.24
Diagnosis
Diagnosis of SIADH can be complex because there are many clinical reasons a patient may have hyponatremia. For example, SIADH and psychogenic polydipsia both result in hyponatremia, and sometimes the 2 conditions can be difficult to distinguish. Hyponatremia is typically discovered by routine blood testing if the patient is asymptomatic. Table 525 highlights the major laboratory markers that distinguish SIADH and psychogenic polydipsia.
Continue to: Treatment
Treatment
The primary treatment for SIADH is cessation of the offending agent. Based on the patient’s clinical presentation, free water restriction (.5 to 1 L/d) can be implemented to increase serum sodium levels. If the patient is having neurologic complications due to the severity of hyponatremia, correction with hypertonic saline is indicated. Upon resolution, the recommended course of action is to switch to a medication in a different class. Re-challenging the patient with the same medication is not recommended unless there is no other alternative class of medication.24 Table 626 highlights other causes of hyponatremia, what laboratory markers to assess, and how to treat high-risk individuals.
Hyponatremia is a complex medical complication that can be life-threatening. Psychotropics are a relatively common cause of hyponatremia, specifically SIADH. Older adults appear to be at highest risk, as most case reports are in patients age ≥65. Patients who are prescribed psychotropics should be treated with the lowest effective dose and monitored for signs and symptoms of hyponatremia throughout therapy.
Related Resources
- Spasovski G, Vanholder R, Allolio B, et al. Clinical practice guidelines on diagnosis and treatment of hyponatremia. Eur J Endocrinol. 2014;170(3):G1-G47.
- Verbalis JG, Goldsmith SR, Greenberg A, et al. Diagnosis, evaluation, and treatment of hyponatremia: expert panel recommendations. Am J Med. 2013;126(10 Suppl 1):S1-S42.
Drug Brand Names
Bupropion • Wellbutrin
Carbamazepine • Tegretol
Citalopram • Celexa
Clozapine • Clozaril
Escitalopram • Lexapro
Fluoxetine • Prozac
Haloperidol • Haldol
Lamotrigine • Lamictal
Levathyroxine • Levothroid
Mirtazapine • Remeron
Oxcarbazepine • Trileptal
Paroxetine • Paxil
Quetiapine • Seroquel
Risperidone • Risperdal
Sertraline • Zoloft
Valproic acid • Depakote
Venlafaxine • Effexor
1. Sahay M, Sahay R. Hyponatremia: a practical approach. Indian J Endocrinol Metab. 2014;18(6):760-771.
2. Kenes MT, Hamblin S, Tumuluri SS, et al. Syndrome of inappropriate antidiuretic hormone in a patient receiving high-dose haloperidol and quetiapine therapy. J Neuropsychiatry Clin Neurosci. 2016;28(2):e29-e30. doi: 10.1176/appi.neuropsych.15110392.
3. Twardowschy CA, Bertolucci CB, Gracia Cde M, et al. Severe hyponatremia and the syndrome of inappropriate secretion of antidiuretic hormone (SIADH) associated with fluoxetine: case report. Arq Neuropsiquiatr. 2006;64(1):142-145.
4. Patel KR, Meesala A, Stanilla JK. Sodium valproate–induced hyponatremia: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(5):PCC.09100941. doi: 10.4088/PCC.09100941.
5. Pillans PI, Coulter DM. Fluoxetine and hyponatraemia—a potential hazard in the elderly. N Z Med J. 1994;107(973):85‑86.
6. Strachan J, Shepherd J. Hyponatraemia associated with the use of selective serotonin reuptake inhibitors. Aust N Z J Psychiatry. 1998;32(2):295‑298.
7. Bouman WP, Pinner G, Johnson H. Incidence of selective serotonin reuptake inhibitor (SSRI) induced hyponatraemia due to the syndrome of inappropriate antidiuretic hormone (SIADH) secretion in the elderly. Int J Geriatr Psychiatry. 1998;13(1):12‑15.
8. Wilkinson TJ, Begg EJ, Winter AC, et al. Incidence and risk factors for hyponatraemia following treatment with fluoxetine or paroxetine in elderly people. Br J Clin Pharmacol. 1999;47(2):211‑217.
9. Kirby D, Harrigan S, Ames D. Hyponatraemia in elderly psychiatric patients treated with selective serotonin reuptake inhibitors and venlafaxine: a retrospective controlled study in an inpatient unit. Int J Geriatr Psychiatry. 2002;17(3):231‑237.
10. Wee R, Lim WK. Selective serotonin re‑uptake inhibitors (SSRIs) and hyponatraemia in the elderly. Int J Geriatr Psychiatry. 2004;19(6):590‑591.
11. Jung YE, Jun TY, Kim KS, et al. Hyponatremia associated with selective serotonin reuptake inhibitors, mirtazapine, and venlafaxine in Korean patients with major depressive disorder. Int J Clin Pharmacol Ther. 2011;49(7):437‑443.
12. Letmaier M, Painold A, Holl AK, et al. Hyponatremia during psychopharmacological treatment: Results of a drug surveillance program. Int J Neuropsychopharmacol. 2012;15(6):739‑748.
13. Coupland CA, Dhiman P, Barton G, et al. A study of the safety and harms of antidepressant drugs for older people: a cohort study using a large primary care database. Health Technol Assess. 2011;15(28):1‑202, iii‑iv.
14. Leah-Møller KB, Hansen AH, Torstensson M, et al. Antidepressants and the risk of hyponatremia: a Danish register-based population study. BMJ Open. 2016;6(5):e011200. doi: 10.1136/bmjopen-2016-011200.
15. De Picker LD, Van Den Eede F, Dumont G, et al. Antidepressants and the risk of hyponatremia: a class by class review of literature. Psychosomatics. 2014;55(6):536-547.
16. Serrano A, Rangel N, Carrizo E, et al. Safety of long-term clozapine administration. Frequency of cardiomyopathy and hyponatraemia: two cross-sectional, naturalistic studies. Aust N Z J Psychiatry. 2014;48(2):183‑192.
17. Uhde TW, Post RM. Effects of carbamazepine on serum electrolytes: clinical and theoretical implications. J Clin Psychopharmacol. 1983;3(2):103‑106.
18. Lahr MB. Hyponatremia during carbamazepine therapy. Clin Pharmacol Ther. 1985;37(6):693‑696.
19. Joffe RT, Post RM, Uhde TW. Effects of carbamazepine on serum electrolytes in affectively ill patients. Psychol Med. 1986;16(2):331‑335.
20. Vieweg V, Glick JL, Herring S, et al. Absence of carbamazepine‑induced hyponatremia among patients also given lithium. Am J Psychiatry. 1987;144(7):943‑947.
21. Yassa R, Iskandar H, Nastase C, et al. Carbamazepine and hyponatremia in patients with affective disorder. Am J Psychiatry. 1988;145(3):339‑342.
22. Kastner T, Friedman DL, Pond WS. Carbamazepine‑induced hyponatremia in patients with mental retardation. Am J Ment Retard. 1992;96(5):536‑540.
23. Kelly BD, Hillery J. Hyponatremia during carbamazepine therapy in patients with intellectual disability. J Intellect Disabil Res. 2001;45(Pt 2):152‑156.
24. Sahoo S, Grover S. Hyponatremia and psychotropics. J Geriatr Ment Health. 2016;3(2):108-122.
25. Siragy HM. Hyponatremia, fluid-electrolyte disorders and the syndrome of inappropriate antidiuretic hormone secretion: diagnosis and treatment options. Endocr Pract. 2006;12(4):446-457.
26. Braun M, Barstow CH, Pyzocha NJ. Diagnosis and management of sodium disorders: hyponatremia and hypernatremia. Am Fam Physician. 2015;91(5):299-307.
1. Sahay M, Sahay R. Hyponatremia: a practical approach. Indian J Endocrinol Metab. 2014;18(6):760-771.
2. Kenes MT, Hamblin S, Tumuluri SS, et al. Syndrome of inappropriate antidiuretic hormone in a patient receiving high-dose haloperidol and quetiapine therapy. J Neuropsychiatry Clin Neurosci. 2016;28(2):e29-e30. doi: 10.1176/appi.neuropsych.15110392.
3. Twardowschy CA, Bertolucci CB, Gracia Cde M, et al. Severe hyponatremia and the syndrome of inappropriate secretion of antidiuretic hormone (SIADH) associated with fluoxetine: case report. Arq Neuropsiquiatr. 2006;64(1):142-145.
4. Patel KR, Meesala A, Stanilla JK. Sodium valproate–induced hyponatremia: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(5):PCC.09100941. doi: 10.4088/PCC.09100941.
5. Pillans PI, Coulter DM. Fluoxetine and hyponatraemia—a potential hazard in the elderly. N Z Med J. 1994;107(973):85‑86.
6. Strachan J, Shepherd J. Hyponatraemia associated with the use of selective serotonin reuptake inhibitors. Aust N Z J Psychiatry. 1998;32(2):295‑298.
7. Bouman WP, Pinner G, Johnson H. Incidence of selective serotonin reuptake inhibitor (SSRI) induced hyponatraemia due to the syndrome of inappropriate antidiuretic hormone (SIADH) secretion in the elderly. Int J Geriatr Psychiatry. 1998;13(1):12‑15.
8. Wilkinson TJ, Begg EJ, Winter AC, et al. Incidence and risk factors for hyponatraemia following treatment with fluoxetine or paroxetine in elderly people. Br J Clin Pharmacol. 1999;47(2):211‑217.
9. Kirby D, Harrigan S, Ames D. Hyponatraemia in elderly psychiatric patients treated with selective serotonin reuptake inhibitors and venlafaxine: a retrospective controlled study in an inpatient unit. Int J Geriatr Psychiatry. 2002;17(3):231‑237.
10. Wee R, Lim WK. Selective serotonin re‑uptake inhibitors (SSRIs) and hyponatraemia in the elderly. Int J Geriatr Psychiatry. 2004;19(6):590‑591.
11. Jung YE, Jun TY, Kim KS, et al. Hyponatremia associated with selective serotonin reuptake inhibitors, mirtazapine, and venlafaxine in Korean patients with major depressive disorder. Int J Clin Pharmacol Ther. 2011;49(7):437‑443.
12. Letmaier M, Painold A, Holl AK, et al. Hyponatremia during psychopharmacological treatment: Results of a drug surveillance program. Int J Neuropsychopharmacol. 2012;15(6):739‑748.
13. Coupland CA, Dhiman P, Barton G, et al. A study of the safety and harms of antidepressant drugs for older people: a cohort study using a large primary care database. Health Technol Assess. 2011;15(28):1‑202, iii‑iv.
14. Leah-Møller KB, Hansen AH, Torstensson M, et al. Antidepressants and the risk of hyponatremia: a Danish register-based population study. BMJ Open. 2016;6(5):e011200. doi: 10.1136/bmjopen-2016-011200.
15. De Picker LD, Van Den Eede F, Dumont G, et al. Antidepressants and the risk of hyponatremia: a class by class review of literature. Psychosomatics. 2014;55(6):536-547.
16. Serrano A, Rangel N, Carrizo E, et al. Safety of long-term clozapine administration. Frequency of cardiomyopathy and hyponatraemia: two cross-sectional, naturalistic studies. Aust N Z J Psychiatry. 2014;48(2):183‑192.
17. Uhde TW, Post RM. Effects of carbamazepine on serum electrolytes: clinical and theoretical implications. J Clin Psychopharmacol. 1983;3(2):103‑106.
18. Lahr MB. Hyponatremia during carbamazepine therapy. Clin Pharmacol Ther. 1985;37(6):693‑696.
19. Joffe RT, Post RM, Uhde TW. Effects of carbamazepine on serum electrolytes in affectively ill patients. Psychol Med. 1986;16(2):331‑335.
20. Vieweg V, Glick JL, Herring S, et al. Absence of carbamazepine‑induced hyponatremia among patients also given lithium. Am J Psychiatry. 1987;144(7):943‑947.
21. Yassa R, Iskandar H, Nastase C, et al. Carbamazepine and hyponatremia in patients with affective disorder. Am J Psychiatry. 1988;145(3):339‑342.
22. Kastner T, Friedman DL, Pond WS. Carbamazepine‑induced hyponatremia in patients with mental retardation. Am J Ment Retard. 1992;96(5):536‑540.
23. Kelly BD, Hillery J. Hyponatremia during carbamazepine therapy in patients with intellectual disability. J Intellect Disabil Res. 2001;45(Pt 2):152‑156.
24. Sahoo S, Grover S. Hyponatremia and psychotropics. J Geriatr Ment Health. 2016;3(2):108-122.
25. Siragy HM. Hyponatremia, fluid-electrolyte disorders and the syndrome of inappropriate antidiuretic hormone secretion: diagnosis and treatment options. Endocr Pract. 2006;12(4):446-457.
26. Braun M, Barstow CH, Pyzocha NJ. Diagnosis and management of sodium disorders: hyponatremia and hypernatremia. Am Fam Physician. 2015;91(5):299-307.
Catatonia: Recognition, management, and prevention of complications
Mr. W, age 50, who has been diagnosed with hypertension and catatonia associated with schizophrenia, is brought to the emergency department by his case manager for evaluation of increasing disorganization, inability to function, and nonadherence to medications. He has not been bathing, eating, or drinking. During the admission interview, he is mute, and is noted to have purposeless activity, alternating between rocking from leg to leg to pacing in circles. At times Mr. W holds a rigid, prayer-type posture with his arms. Negativism is present, primarily opposition to interviewer requests.
Previously stable on
On the inpatient psychiatry unit, Mr. W continues to be mute, staying in bed except to use the bathroom. He refuses all food and fluids. The team initiates subcutaneous

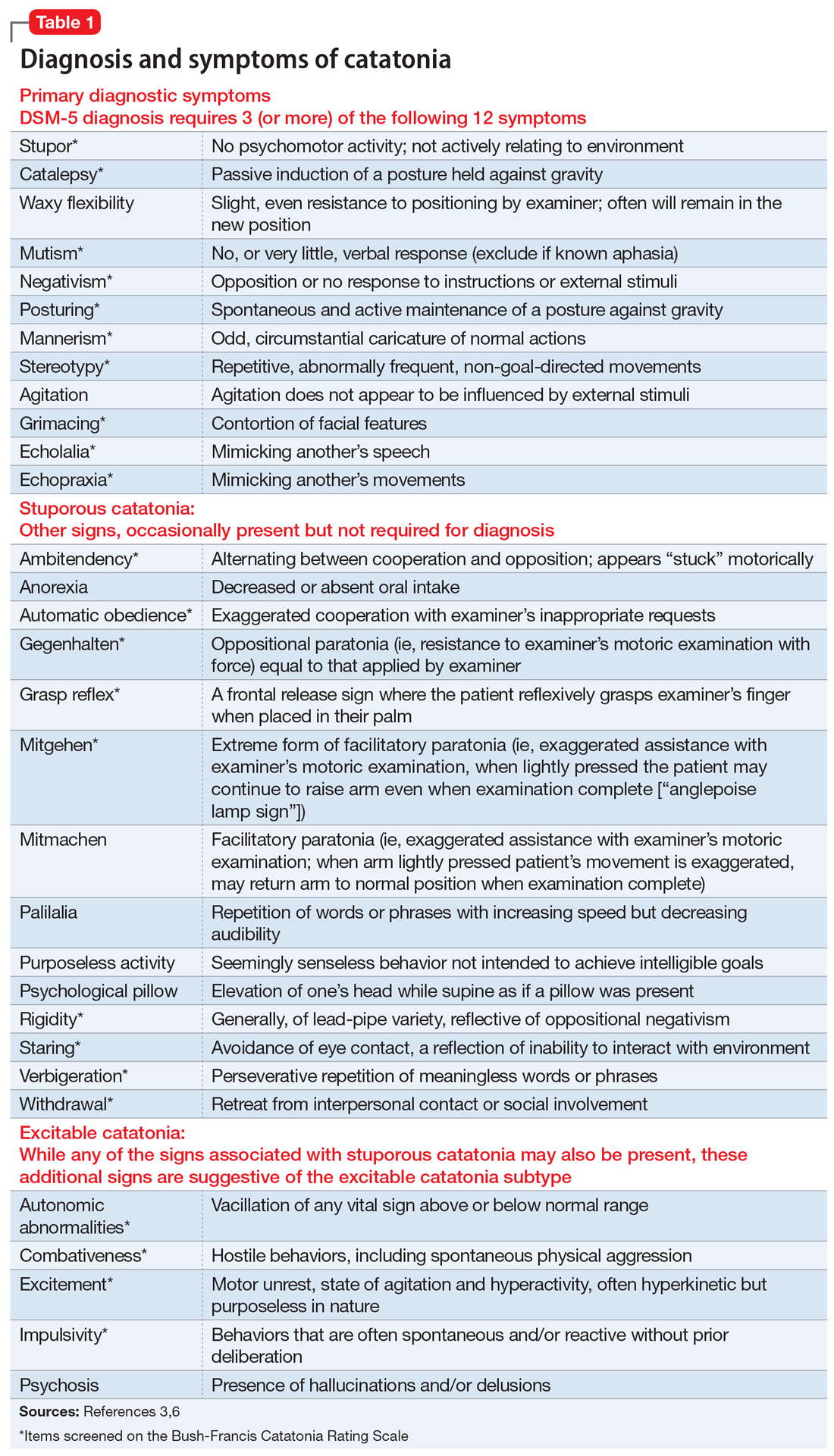
Continue to: Medical complications can be fatal
Medical complications can be fatal

Treatment usually starts with lorazepam
Benzodiazepines are a first-line option for the management of catatonia.2,5 Controversy exists as to effectiveness of different routes of administration. Generally, IV lorazepam is preferred due to its ease of administration, fast onset, and longer duration of action.1 Some inpatient psychiatric units are unable to administer IV benzodiazepines; in these scenarios, IM administration is preferred to oral benzodiazepines.
The initial lorazepam challenge dose should be 2 mg. A positive response to the lorazepam challenge often confirms the catatonia diagnosis.2,7 This challenge should be followed by maintenance doses ranging from 6 to 8 mg/d in divided doses (3 or 4 times a day). Higher doses (up to 24 mg/d) are sometimes used.2,5,8 A recent case report described catatonia remission using lorazepam, 28 mg/d, after unsuccessful ECT.9 The lorazepam dose prior to ECT was 8 mg/d.9 Response is usually seen within 3 to 7 days of an adequate dose.2,8 Parenteral lorazepam typically is continued for several days before converting to oral lorazepam.1 Approximately 70% to 80% of patients with catatonia will show improvement in symptoms with lorazepam.2,7,8
The optimal duration of benzodiazepine treatment is unclear.2 In some cases, once remission of the underlying illness is achieved, benzodiazepines are discontinued.2 However, in other cases, symptoms of catatonia may emerge when lorazepam is tapered, therefore suggesting the need for a longer duration of treatment.2 Despite this high rate of improvement, many patients ultimately receive ECT due to unsustained response or to prevent future episodes of catatonia.
A recent review of 60 Turkish patients with catatonia found 91.7% (n = 55) received oral lorazepam (up to 15 mg/d) as the first-line therapy.7 Improvement was seen in 23.7% (n = 13) of patients treated with lorazepam, yet 70% (n = 42) showed either no response or partial response, and ultimately received ECT in combination with lorazepam.7 The lower improvement rate seen in this review may be secondary to the use of oral lorazepam instead of parenteral, or may highlight the frequency in which patients ultimately go on to receive ECT.
Continue to: ECT
ECT. If high doses of benzodiazepines are not effective within 48 to 72 hours, ECT should be considered.1,7 ECT should be considered sooner for patients with life-threatening catatonia or those who present with excited features or malignant catatonia.1,2,7 In patients with catatonia, ECT response rates range from 80% to 100%.2,7 Unal et al7 reported a 100% response rate if ECT was used as the first-line treatment (n = 5), and a 92.9% (n = 39) response rate after adding ECT to lorazepam. Lorazepam may interfere with the seizure threshold, but if indicated, this medication can be continued.2 A minimum of 6 ECT treatments are suggested; however, as many as 20 treatments have been needed.1 Mr. W required a total of 18 ECT treatments. In some cases, maintenance ECT may be required.2
Antipsychotics. Discontinuation of antipsychotics is generally encouraged in patients presenting with catatonia.2,7,8 Antipsychotics carry a risk of potentially worsening catatonia, conversion to malignant catatonia, or precipitation of NMS; therefore, carefully weigh the risks vs benefits.1,2 If catatonia is secondary to psychosis, as in Mr. W’s case, antipsychotics may be considered once catatonia improves.2 If an antipsychotic is warranted, consider aripiprazole (because of its D2 partial agonist activity) or low-dose olanzapine.1,2 If catatonia is secondary to clozapine withdrawal, the initial therapy should be clozapine re-initiation.1 Although high-potency agents, such as haloperidol and risperidone, typically are not preferred, risperidone was restarted for Mr. W because of his history of response to and tolerability of this medication during a previous catatonic episode.
Other treatments. In a recent review, Beach et al1 described the use of additional agents, mostly in a small number of positive case reports, for managing catatonia. These included:
- zolpidem (zolpidem 10 mg as a challenge test, and doses of ≤40 mg/d)
- the N-methyl-
D -aspartic acid antagonists amantadine (100 to 600 mg/d) or memantine (5 to 20 mg/d) - carbidopa/levodopa
- methylphenidate
- antiepileptics (eg, carbamazepine, topiramate, and divalproex sodium)
- anticholinergics.1,2
Lithium has been used in attempts to prevent recurrent catatonia with limited success.2 There are also a few reports of using transcranial magnetic stimulation (TMS) to manage catatonia.1
Beach et al1 proposed a treatment algorithm in which IV lorazepam (Step 1) and ECT (Step 2) remain the preferred treatments. Next, for Step 3 consider a glutamate antagonist (amantadine or memantine), followed by an antiepileptic (Step 4), and lastly an atypical antipsychotic (aripiprazole, olanzapine, or clozapine) in combination with lorazepam (Step 5).
When indicated, don’t delay ECT
Initial management of catatonia is with a benzodiazepine challenge. Ultimately, the gold-standard treatment of catatonia that does not improve with benzodiazepines is ECT, and ECT should be implemented as soon as it is clear that pharmacotherapy is less than fully effective. Consider ECT initially in life-threatening cases and for patients with malignant catatonia. Although additional agents and TMS have been explored, these should be reserved for patients who fail to respond to, or who are not candidates for, benzodiazepines or ECT.
CASE CONTINUED
After 5 ECT treatments, Mr. W says a few words, but he communicates primarily with gestures (primarily waving people away). After 10 to 12 ECT treatments, Mr. W becomes more interactive and conversant, and his nutrition improves; however, he still exhibits symptoms of catatonia and is not at baseline. He undergoes a total of 18 ECT treatments. Antipsychotics were initially discontinued; however, given Mr. W’s improvement with ECT and the presence of auditory hallucinations, oral risperidone is restarted and titrated to 2 mg, 2 times a day, and he is transitioned back to paliperidone palmitate before he is discharged. Lorazepam is tapered and discontinued. Mr. W is discharged back to his nursing home and is interactive (laughing and joking with family) and attending to his activities of daily living. Unfortunately, Mr. W did not followup with the recommendation for maintenance ECT, and adherence to paliperidone palmitate injections is unknown. Mr. W presented to our facility again 6 months later with symptoms of catatonia and ultimately transferred to a state hospital.
Related Resources
- Fink M, Taylor MA. Catatonia: A clinician’s guide to diagnosis and treatment. New York, NY: Cambridge University Press; 2006. • Carroll BT, Spiegel DR. Catatonia on the consultation liaison service and other clinical settings. Hauppauge, NY: Nova Science Pub Inc.; 2016.
- Benarous X, Raffin M, Ferrafiat V, et al. Catatonia in children and adolescents: new perspectives. Schizophr Res. 2018;200:56-67.
- Malignant Hyperthermia Association of the United States. What is NMSIS? http://www.mhaus.org/nmsis/about-us/ what-is-nmsis/.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Asenapine • Saphris
Carbamazepine • Carbatrol, Tegretol
Carbidopa/Levodopa • Sinemet
Citalopram • Celexa
Clozapine • Clozaril
Divalproex Sodium • Depakote
Enoxaparin • Lovenox
Fluoxetine • Prozac
Haloperidol • Haldol
Lithium • Eskalith, Lithobid
Lorazepam • Ativan
Lurasidone • Latuda
Memantine • Namenda
Methylphenidate • Concerta, Ritalin
Mirtazapine • Remeron
Olanzapine • Zyprexa
Paliperidone palmitate • Invega Sustenna
Quetiapine • Seroquel
Risperidone • Risperdal
Risperidone long-acting injection • Risperdal Consta
Topiramate • Topamax
Zolpidem • Ambien
1. Beach SR, Gomez-Bernal F, Huffman JC, et al. Alternative treatment strategies for catatonia: a systematic review. Gen Hosp Psychiatry. 2017;48:1-19.
2. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:1-6.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome: focus on treatment and rechallenge. Ann Pharmacother. 2016;50(11):973-981.
5. Ohi K, Kuwata A, Shimada T, et al. Response to benzodiazepines and clinical course in malignant catatonia associated with schizophrenia: a case report. Medicine (Baltimore). 2017;96(16):e6566. doi: 10.1097/MD.0000000000006566.
6. Bush G, Fink M, Petrides G, et al. Catatonia I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
7. Unal A, Altindag A, Demir B, et al. The use of lorazepam and electroconvulsive therapy in the treatment of catatonia: treatment characteristics and outcomes in 60 patients. J ECT. 2017;33(4):290-293.
8. Fink M, Taylor MA. Neuroleptic malignant syndrome is malignant catatonia, warranting treatments efficacious for catatonia. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):1182-1183.
9. van der Markt A, Heller HM, van Exel E. A woman with catatonia, what to do after ECT fails: a case report. J ECT. 2016;32(3):e6-7. doi: 10.1097/YCT.0000000000000290.
Mr. W, age 50, who has been diagnosed with hypertension and catatonia associated with schizophrenia, is brought to the emergency department by his case manager for evaluation of increasing disorganization, inability to function, and nonadherence to medications. He has not been bathing, eating, or drinking. During the admission interview, he is mute, and is noted to have purposeless activity, alternating between rocking from leg to leg to pacing in circles. At times Mr. W holds a rigid, prayer-type posture with his arms. Negativism is present, primarily opposition to interviewer requests.
Previously stable on
On the inpatient psychiatry unit, Mr. W continues to be mute, staying in bed except to use the bathroom. He refuses all food and fluids. The team initiates subcutaneous
Continue to: Medical complications can be fatal
Medical complications can be fatal
Treatment usually starts with lorazepam
Benzodiazepines are a first-line option for the management of catatonia.2,5 Controversy exists as to effectiveness of different routes of administration. Generally, IV lorazepam is preferred due to its ease of administration, fast onset, and longer duration of action.1 Some inpatient psychiatric units are unable to administer IV benzodiazepines; in these scenarios, IM administration is preferred to oral benzodiazepines.
The initial lorazepam challenge dose should be 2 mg. A positive response to the lorazepam challenge often confirms the catatonia diagnosis.2,7 This challenge should be followed by maintenance doses ranging from 6 to 8 mg/d in divided doses (3 or 4 times a day). Higher doses (up to 24 mg/d) are sometimes used.2,5,8 A recent case report described catatonia remission using lorazepam, 28 mg/d, after unsuccessful ECT.9 The lorazepam dose prior to ECT was 8 mg/d.9 Response is usually seen within 3 to 7 days of an adequate dose.2,8 Parenteral lorazepam typically is continued for several days before converting to oral lorazepam.1 Approximately 70% to 80% of patients with catatonia will show improvement in symptoms with lorazepam.2,7,8
The optimal duration of benzodiazepine treatment is unclear.2 In some cases, once remission of the underlying illness is achieved, benzodiazepines are discontinued.2 However, in other cases, symptoms of catatonia may emerge when lorazepam is tapered, therefore suggesting the need for a longer duration of treatment.2 Despite this high rate of improvement, many patients ultimately receive ECT due to unsustained response or to prevent future episodes of catatonia.
A recent review of 60 Turkish patients with catatonia found 91.7% (n = 55) received oral lorazepam (up to 15 mg/d) as the first-line therapy.7 Improvement was seen in 23.7% (n = 13) of patients treated with lorazepam, yet 70% (n = 42) showed either no response or partial response, and ultimately received ECT in combination with lorazepam.7 The lower improvement rate seen in this review may be secondary to the use of oral lorazepam instead of parenteral, or may highlight the frequency in which patients ultimately go on to receive ECT.
Continue to: ECT
ECT. If high doses of benzodiazepines are not effective within 48 to 72 hours, ECT should be considered.1,7 ECT should be considered sooner for patients with life-threatening catatonia or those who present with excited features or malignant catatonia.1,2,7 In patients with catatonia, ECT response rates range from 80% to 100%.2,7 Unal et al7 reported a 100% response rate if ECT was used as the first-line treatment (n = 5), and a 92.9% (n = 39) response rate after adding ECT to lorazepam. Lorazepam may interfere with the seizure threshold, but if indicated, this medication can be continued.2 A minimum of 6 ECT treatments are suggested; however, as many as 20 treatments have been needed.1 Mr. W required a total of 18 ECT treatments. In some cases, maintenance ECT may be required.2
Antipsychotics. Discontinuation of antipsychotics is generally encouraged in patients presenting with catatonia.2,7,8 Antipsychotics carry a risk of potentially worsening catatonia, conversion to malignant catatonia, or precipitation of NMS; therefore, carefully weigh the risks vs benefits.1,2 If catatonia is secondary to psychosis, as in Mr. W’s case, antipsychotics may be considered once catatonia improves.2 If an antipsychotic is warranted, consider aripiprazole (because of its D2 partial agonist activity) or low-dose olanzapine.1,2 If catatonia is secondary to clozapine withdrawal, the initial therapy should be clozapine re-initiation.1 Although high-potency agents, such as haloperidol and risperidone, typically are not preferred, risperidone was restarted for Mr. W because of his history of response to and tolerability of this medication during a previous catatonic episode.
Other treatments. In a recent review, Beach et al1 described the use of additional agents, mostly in a small number of positive case reports, for managing catatonia. These included:
- zolpidem (zolpidem 10 mg as a challenge test, and doses of ≤40 mg/d)
- the N-methyl-
D -aspartic acid antagonists amantadine (100 to 600 mg/d) or memantine (5 to 20 mg/d) - carbidopa/levodopa
- methylphenidate
- antiepileptics (eg, carbamazepine, topiramate, and divalproex sodium)
- anticholinergics.1,2
Lithium has been used in attempts to prevent recurrent catatonia with limited success.2 There are also a few reports of using transcranial magnetic stimulation (TMS) to manage catatonia.1
Beach et al1 proposed a treatment algorithm in which IV lorazepam (Step 1) and ECT (Step 2) remain the preferred treatments. Next, for Step 3 consider a glutamate antagonist (amantadine or memantine), followed by an antiepileptic (Step 4), and lastly an atypical antipsychotic (aripiprazole, olanzapine, or clozapine) in combination with lorazepam (Step 5).
When indicated, don’t delay ECT
Initial management of catatonia is with a benzodiazepine challenge. Ultimately, the gold-standard treatment of catatonia that does not improve with benzodiazepines is ECT, and ECT should be implemented as soon as it is clear that pharmacotherapy is less than fully effective. Consider ECT initially in life-threatening cases and for patients with malignant catatonia. Although additional agents and TMS have been explored, these should be reserved for patients who fail to respond to, or who are not candidates for, benzodiazepines or ECT.
CASE CONTINUED
After 5 ECT treatments, Mr. W says a few words, but he communicates primarily with gestures (primarily waving people away). After 10 to 12 ECT treatments, Mr. W becomes more interactive and conversant, and his nutrition improves; however, he still exhibits symptoms of catatonia and is not at baseline. He undergoes a total of 18 ECT treatments. Antipsychotics were initially discontinued; however, given Mr. W’s improvement with ECT and the presence of auditory hallucinations, oral risperidone is restarted and titrated to 2 mg, 2 times a day, and he is transitioned back to paliperidone palmitate before he is discharged. Lorazepam is tapered and discontinued. Mr. W is discharged back to his nursing home and is interactive (laughing and joking with family) and attending to his activities of daily living. Unfortunately, Mr. W did not followup with the recommendation for maintenance ECT, and adherence to paliperidone palmitate injections is unknown. Mr. W presented to our facility again 6 months later with symptoms of catatonia and ultimately transferred to a state hospital.
Related Resources
- Fink M, Taylor MA. Catatonia: A clinician’s guide to diagnosis and treatment. New York, NY: Cambridge University Press; 2006. • Carroll BT, Spiegel DR. Catatonia on the consultation liaison service and other clinical settings. Hauppauge, NY: Nova Science Pub Inc.; 2016.
- Benarous X, Raffin M, Ferrafiat V, et al. Catatonia in children and adolescents: new perspectives. Schizophr Res. 2018;200:56-67.
- Malignant Hyperthermia Association of the United States. What is NMSIS? http://www.mhaus.org/nmsis/about-us/ what-is-nmsis/.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Asenapine • Saphris
Carbamazepine • Carbatrol, Tegretol
Carbidopa/Levodopa • Sinemet
Citalopram • Celexa
Clozapine • Clozaril
Divalproex Sodium • Depakote
Enoxaparin • Lovenox
Fluoxetine • Prozac
Haloperidol • Haldol
Lithium • Eskalith, Lithobid
Lorazepam • Ativan
Lurasidone • Latuda
Memantine • Namenda
Methylphenidate • Concerta, Ritalin
Mirtazapine • Remeron
Olanzapine • Zyprexa
Paliperidone palmitate • Invega Sustenna
Quetiapine • Seroquel
Risperidone • Risperdal
Risperidone long-acting injection • Risperdal Consta
Topiramate • Topamax
Zolpidem • Ambien
Mr. W, age 50, who has been diagnosed with hypertension and catatonia associated with schizophrenia, is brought to the emergency department by his case manager for evaluation of increasing disorganization, inability to function, and nonadherence to medications. He has not been bathing, eating, or drinking. During the admission interview, he is mute, and is noted to have purposeless activity, alternating between rocking from leg to leg to pacing in circles. At times Mr. W holds a rigid, prayer-type posture with his arms. Negativism is present, primarily opposition to interviewer requests.
Previously stable on
On the inpatient psychiatry unit, Mr. W continues to be mute, staying in bed except to use the bathroom. He refuses all food and fluids. The team initiates subcutaneous
Continue to: Medical complications can be fatal
Medical complications can be fatal
Treatment usually starts with lorazepam
Benzodiazepines are a first-line option for the management of catatonia.2,5 Controversy exists as to effectiveness of different routes of administration. Generally, IV lorazepam is preferred due to its ease of administration, fast onset, and longer duration of action.1 Some inpatient psychiatric units are unable to administer IV benzodiazepines; in these scenarios, IM administration is preferred to oral benzodiazepines.
The initial lorazepam challenge dose should be 2 mg. A positive response to the lorazepam challenge often confirms the catatonia diagnosis.2,7 This challenge should be followed by maintenance doses ranging from 6 to 8 mg/d in divided doses (3 or 4 times a day). Higher doses (up to 24 mg/d) are sometimes used.2,5,8 A recent case report described catatonia remission using lorazepam, 28 mg/d, after unsuccessful ECT.9 The lorazepam dose prior to ECT was 8 mg/d.9 Response is usually seen within 3 to 7 days of an adequate dose.2,8 Parenteral lorazepam typically is continued for several days before converting to oral lorazepam.1 Approximately 70% to 80% of patients with catatonia will show improvement in symptoms with lorazepam.2,7,8
The optimal duration of benzodiazepine treatment is unclear.2 In some cases, once remission of the underlying illness is achieved, benzodiazepines are discontinued.2 However, in other cases, symptoms of catatonia may emerge when lorazepam is tapered, therefore suggesting the need for a longer duration of treatment.2 Despite this high rate of improvement, many patients ultimately receive ECT due to unsustained response or to prevent future episodes of catatonia.
A recent review of 60 Turkish patients with catatonia found 91.7% (n = 55) received oral lorazepam (up to 15 mg/d) as the first-line therapy.7 Improvement was seen in 23.7% (n = 13) of patients treated with lorazepam, yet 70% (n = 42) showed either no response or partial response, and ultimately received ECT in combination with lorazepam.7 The lower improvement rate seen in this review may be secondary to the use of oral lorazepam instead of parenteral, or may highlight the frequency in which patients ultimately go on to receive ECT.
Continue to: ECT
ECT. If high doses of benzodiazepines are not effective within 48 to 72 hours, ECT should be considered.1,7 ECT should be considered sooner for patients with life-threatening catatonia or those who present with excited features or malignant catatonia.1,2,7 In patients with catatonia, ECT response rates range from 80% to 100%.2,7 Unal et al7 reported a 100% response rate if ECT was used as the first-line treatment (n = 5), and a 92.9% (n = 39) response rate after adding ECT to lorazepam. Lorazepam may interfere with the seizure threshold, but if indicated, this medication can be continued.2 A minimum of 6 ECT treatments are suggested; however, as many as 20 treatments have been needed.1 Mr. W required a total of 18 ECT treatments. In some cases, maintenance ECT may be required.2
Antipsychotics. Discontinuation of antipsychotics is generally encouraged in patients presenting with catatonia.2,7,8 Antipsychotics carry a risk of potentially worsening catatonia, conversion to malignant catatonia, or precipitation of NMS; therefore, carefully weigh the risks vs benefits.1,2 If catatonia is secondary to psychosis, as in Mr. W’s case, antipsychotics may be considered once catatonia improves.2 If an antipsychotic is warranted, consider aripiprazole (because of its D2 partial agonist activity) or low-dose olanzapine.1,2 If catatonia is secondary to clozapine withdrawal, the initial therapy should be clozapine re-initiation.1 Although high-potency agents, such as haloperidol and risperidone, typically are not preferred, risperidone was restarted for Mr. W because of his history of response to and tolerability of this medication during a previous catatonic episode.
Other treatments. In a recent review, Beach et al1 described the use of additional agents, mostly in a small number of positive case reports, for managing catatonia. These included:
- zolpidem (zolpidem 10 mg as a challenge test, and doses of ≤40 mg/d)
- the N-methyl-
D -aspartic acid antagonists amantadine (100 to 600 mg/d) or memantine (5 to 20 mg/d) - carbidopa/levodopa
- methylphenidate
- antiepileptics (eg, carbamazepine, topiramate, and divalproex sodium)
- anticholinergics.1,2
Lithium has been used in attempts to prevent recurrent catatonia with limited success.2 There are also a few reports of using transcranial magnetic stimulation (TMS) to manage catatonia.1
Beach et al1 proposed a treatment algorithm in which IV lorazepam (Step 1) and ECT (Step 2) remain the preferred treatments. Next, for Step 3 consider a glutamate antagonist (amantadine or memantine), followed by an antiepileptic (Step 4), and lastly an atypical antipsychotic (aripiprazole, olanzapine, or clozapine) in combination with lorazepam (Step 5).
When indicated, don’t delay ECT
Initial management of catatonia is with a benzodiazepine challenge. Ultimately, the gold-standard treatment of catatonia that does not improve with benzodiazepines is ECT, and ECT should be implemented as soon as it is clear that pharmacotherapy is less than fully effective. Consider ECT initially in life-threatening cases and for patients with malignant catatonia. Although additional agents and TMS have been explored, these should be reserved for patients who fail to respond to, or who are not candidates for, benzodiazepines or ECT.
CASE CONTINUED
After 5 ECT treatments, Mr. W says a few words, but he communicates primarily with gestures (primarily waving people away). After 10 to 12 ECT treatments, Mr. W becomes more interactive and conversant, and his nutrition improves; however, he still exhibits symptoms of catatonia and is not at baseline. He undergoes a total of 18 ECT treatments. Antipsychotics were initially discontinued; however, given Mr. W’s improvement with ECT and the presence of auditory hallucinations, oral risperidone is restarted and titrated to 2 mg, 2 times a day, and he is transitioned back to paliperidone palmitate before he is discharged. Lorazepam is tapered and discontinued. Mr. W is discharged back to his nursing home and is interactive (laughing and joking with family) and attending to his activities of daily living. Unfortunately, Mr. W did not followup with the recommendation for maintenance ECT, and adherence to paliperidone palmitate injections is unknown. Mr. W presented to our facility again 6 months later with symptoms of catatonia and ultimately transferred to a state hospital.
Related Resources
- Fink M, Taylor MA. Catatonia: A clinician’s guide to diagnosis and treatment. New York, NY: Cambridge University Press; 2006. • Carroll BT, Spiegel DR. Catatonia on the consultation liaison service and other clinical settings. Hauppauge, NY: Nova Science Pub Inc.; 2016.
- Benarous X, Raffin M, Ferrafiat V, et al. Catatonia in children and adolescents: new perspectives. Schizophr Res. 2018;200:56-67.
- Malignant Hyperthermia Association of the United States. What is NMSIS? http://www.mhaus.org/nmsis/about-us/ what-is-nmsis/.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Asenapine • Saphris
Carbamazepine • Carbatrol, Tegretol
Carbidopa/Levodopa • Sinemet
Citalopram • Celexa
Clozapine • Clozaril
Divalproex Sodium • Depakote
Enoxaparin • Lovenox
Fluoxetine • Prozac
Haloperidol • Haldol
Lithium • Eskalith, Lithobid
Lorazepam • Ativan
Lurasidone • Latuda
Memantine • Namenda
Methylphenidate • Concerta, Ritalin
Mirtazapine • Remeron
Olanzapine • Zyprexa
Paliperidone palmitate • Invega Sustenna
Quetiapine • Seroquel
Risperidone • Risperdal
Risperidone long-acting injection • Risperdal Consta
Topiramate • Topamax
Zolpidem • Ambien
1. Beach SR, Gomez-Bernal F, Huffman JC, et al. Alternative treatment strategies for catatonia: a systematic review. Gen Hosp Psychiatry. 2017;48:1-19.
2. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:1-6.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome: focus on treatment and rechallenge. Ann Pharmacother. 2016;50(11):973-981.
5. Ohi K, Kuwata A, Shimada T, et al. Response to benzodiazepines and clinical course in malignant catatonia associated with schizophrenia: a case report. Medicine (Baltimore). 2017;96(16):e6566. doi: 10.1097/MD.0000000000006566.
6. Bush G, Fink M, Petrides G, et al. Catatonia I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
7. Unal A, Altindag A, Demir B, et al. The use of lorazepam and electroconvulsive therapy in the treatment of catatonia: treatment characteristics and outcomes in 60 patients. J ECT. 2017;33(4):290-293.
8. Fink M, Taylor MA. Neuroleptic malignant syndrome is malignant catatonia, warranting treatments efficacious for catatonia. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):1182-1183.
9. van der Markt A, Heller HM, van Exel E. A woman with catatonia, what to do after ECT fails: a case report. J ECT. 2016;32(3):e6-7. doi: 10.1097/YCT.0000000000000290.
1. Beach SR, Gomez-Bernal F, Huffman JC, et al. Alternative treatment strategies for catatonia: a systematic review. Gen Hosp Psychiatry. 2017;48:1-19.
2. Sienaert P, Dhossche DM, Vancampfort D, et al. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:1-6.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Pileggi DJ, Cook AM. Neuroleptic malignant syndrome: focus on treatment and rechallenge. Ann Pharmacother. 2016;50(11):973-981.
5. Ohi K, Kuwata A, Shimada T, et al. Response to benzodiazepines and clinical course in malignant catatonia associated with schizophrenia: a case report. Medicine (Baltimore). 2017;96(16):e6566. doi: 10.1097/MD.0000000000006566.
6. Bush G, Fink M, Petrides G, et al. Catatonia I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
7. Unal A, Altindag A, Demir B, et al. The use of lorazepam and electroconvulsive therapy in the treatment of catatonia: treatment characteristics and outcomes in 60 patients. J ECT. 2017;33(4):290-293.
8. Fink M, Taylor MA. Neuroleptic malignant syndrome is malignant catatonia, warranting treatments efficacious for catatonia. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):1182-1183.
9. van der Markt A, Heller HM, van Exel E. A woman with catatonia, what to do after ECT fails: a case report. J ECT. 2016;32(3):e6-7. doi: 10.1097/YCT.0000000000000290.