User login
Protein binding changes and drug interactions: What do we know?
Mr. S, age 47, weighs 209 lb and has a history of seizure disorder, bipolar disorder not otherwise specified, hypertension, and type 2 diabetes mellitus. He presents to the emergency department after not taking his medications for 2 days while on vacation. He has increased energy, decreased sleep, and pressured speech, and insists on walking for up to 10 hours per day “in preparation for a marathon,” even though he has a 4-cm foot ulcer. His family reports that he had been compliant with his medications until the present incident.
Mr. S has no known drug allergies. His medications include oral divalproex sodium delayed release (valproic acid [VPA]), 1,000 mg twice a day, oral lisinopril, 20 mg every morning, and insulin glargine, 22 units subcutaneously every evening.
A complete blood count, basic metabolic panel, creatine kinase level, VPA level, and urine drug screen are ordered. Relevant results include a serum creatinine level of 1.4 mg/dL (normal range: 0.6 to 1.2 mg/dL), a glucose serum level of 188 mg/dL (normal range: 70 to 100 mg/dL), and a VPA level of 23 mcg/mL (therapeutic range: 50 to 125 mcg/mL). A liver function panel is within normal limits: albumin level of 3.9 g/dL, aspartate aminotransferase level of 18 IU/L, and alanine aminotransferase level of 14 IU/L. In light of Mr. S’s seizure history, neurology is consulted and the decision is made to continue treating him with VPA because he has been seizure-free for 4.5 years and this medication has also helped with his bipolar disorder.
Mr. S is admitted to the hospital and his home medications are resumed at the current doses. On hospital Day 3, Mr. S’s VPA level is 62 mcg/mL, his obsession with a marathon has remitted, and his sleep pattern has normalized. Infectious disease and podiatry services are consulted for his diabetic foot infection, which has ulcerated down to the bone. IV ertapenem, 1,000 mg/d, is initiated with plans for debridement the following week. Two days later, Mr. S has a witnessed seizure; his VPA level is 9 mcg/mL.

A common question asked of pharmacists is, “Will protein binding changes affect drug dosages?” In this article, I describe how protein binding changes may occur, and the complexity of the dynamic. Being highly bound to a protein typically does not mean all medications will interact, but some interactions can be important. This article does not cover medications that bind to hormones.
Why is protein binding important? When a medication is bound to plasma protein, it is not free to act. There can be a delay in therapeutic effect (because no drug is available to react), delayed elimination, or possibly displacement of another protein-bound medication. Additionally, medications tend not to cross the blood-brain barrier or be eliminated when bound. For example, if a drug is 99% bound (leaving 1% free) and displacement now leaves 2% of the drug free, this event has doubled the amount of free drug. As the unbound medication is eliminated, the drug that is bound to the protein can act as a reservoir. A dynamic relationship exists between bound drug, unbound drug, and rate of elimination.
Which proteins do drugs commonly bind to? The proteins often associated with binding include albumin, alpha-1-acid glycoprotein (AAG), and lipoproteins. Albumin comprises 60% of total plasma protein in the plasma. Lipoproteins include very high-density lipoprotein (VHDL), high-density lipoprotein (HDL), very low-density lipoprotein (VLDL), and low-density lipoprotein (LDL).1 Medications that bind to lipoproteins include cyclosporine, tacrolimus, and propofol.2
Continued to: What common disease states can cause hypoalbuminemia?
What common disease states can cause hypoalbuminemia? Many disease states can result in low albumin levels. The most common ones are malnutrition, malignancies, stress, injury, burns, pregnancy, and diabetes.3 When there is less albumin to bind to, free drug levels may be increased.
Can AAG levels change with disease states as well? Because AAG accounts for a lower percentage of total plasma protein than albumin, there may be less clinical concern regarding AAG. AAG levels usually do not drop, but instead can become elevated during times of trauma, inflammation, and acute myocardial infarction. This could result in increased binding of the free drug.4Which medications bind to red blood cells (RBCs)? There are several locations for drugs to bind to RBCs, including to hemoglobin and the plasma membrane. Medications that commonly bind to RBCs include barbiturates, chlorpromazine, imipramine, and phenytoin.5
What are common highly-bound medications? The Table1 provides examples of medications that are >90% protein-bound. However, this information may be misleading because many medications are highly bound. Zhang et al1 compiled binding data for 222 drugs, half of which bind 90% to 100%. However, the literature does not indicate that they all have clinically significant interactions. Benet and Hoener6 discuss how factors other than protein binding affect potential drug interactions, and the complexity of the body’s ability to compensate for increased free drug. Medication characteristics that may contribute to producing a significant interaction include, but are not limited to:
- free vs protein-bound drug in the plasma or tissue
- volume of distribution
- organs affected
- hepatic bioavailability
- drug clearance.
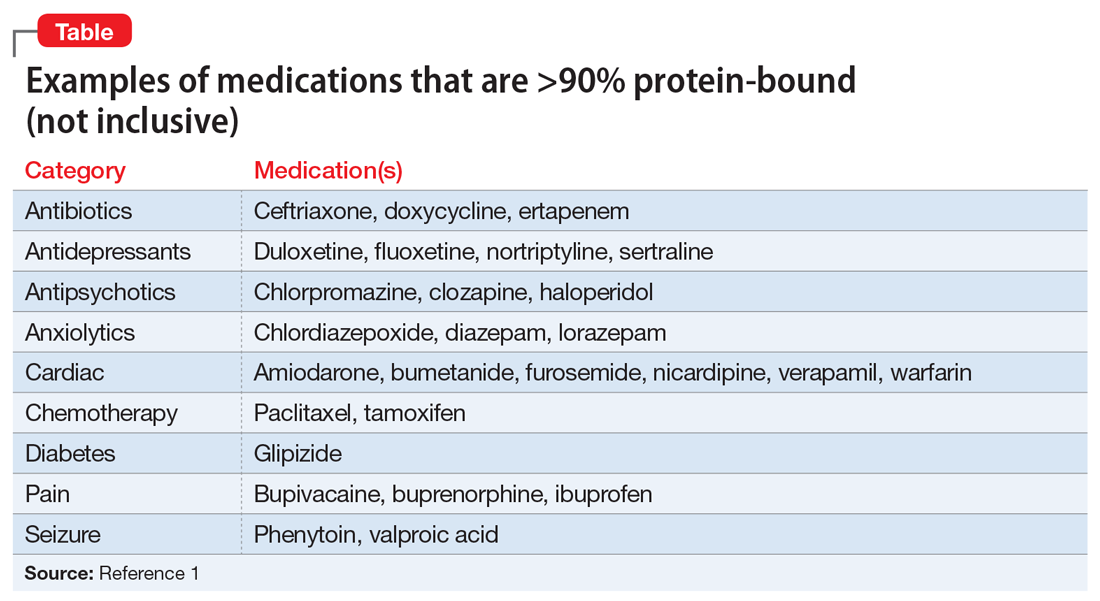
For example, VPA is 93% protein-bound and phenytoin is 91% protein-bound.1 However, this interaction is affected by more than just protein binding. VPA not only displaces the protein-bound phenytoin, but also inhibits its metabolism, which together result in increased free phenytoin levels.
Continued to: Another area of concern is a critically ill patient...
Another area of concern is a critically ill patient who has a change in his or her pH. Medications that are highly bound and have high clearance rates may be affected. This is of particular concern when prescribing antibiotics that are time-dependent, such as beta-lactams.3
What happened to Mr. S? Mr. S likely experienced a drug–drug interaction that resulted in a subtherapeutic VPA level and subsequent seizure. Case reports have shown evidence that the carbapenem class of antibiotics, which includes ertapenem, interacts with VPA.7 Proposed mechanisms include a lowering of VPA serum levels due to a redistribution of the VPA onto the RBCs due to carbapenem. Other theories include the possibility that carbapenems may limit oral VPA absorption, decrease VPA enterohepatic recirculation, and increase VPA metabolism.7 Using VPA and ertapenem together is discouraged because seizures have been reported among patients receiving this combination. If it is medically necessary to administer VPA and ertapenem, closely monitor VPA levels. In Mr. S’s case, another broad-spectrum antibiotic, such as piperacillin-tazobactam, could have been used, for his diabetic foot infection.
While many medications may have high protein binding, there are few clinically important known interactions. However, our understanding of the relationship between protein binding and drug interactions may improve with additional research.
CASE CONTINUED
Under neurology’s care, lacosamide is added for treatment of Mr. S’s seizures. No more seizures are noted during the remainder of his hospitalization. Infectious disease services change his antibiotic to piperacillin-tazobactam. Mr. S continues to progress well and is discharged to a rehabilitation center 2 days later.
Related Resource
- DrugBank. www.drugbank.ca. Canadian Institutes of Health Research.
Drug Brand Names
Amiodarone • Cordarone, Pacerone
Bumetanide • Bumex
Bupivacaine • Marcaine, Sensorcaine
Buprenorphine • Belbuca, Subutex
Ceftriaxone • Rocephin
Chlordiazepoxide • Librium
Chlorpromazine • Thorazine
Clozapine • Clozaril
Cyclosporine • Gengraf, Neoral
Diazepam • Valium
Doxycycline • Acticlate, Doryx
Duloxetine • Cymbalta
Ertapenem • Invanz
Fluoxetine • Prozac, Sarafem
Furosemide • Lasix
Glargine (Insulin) • Lantus, Toujeo
Glipizide • Glucotrol
Haloperidol • Haldol
Ibuprofen • Advil, Motrin
Imipramine • Tofranil
Lacosamide • Vimpat
Lisinopril • Prinivil, Zestril
Lorazepam • Ativan
Nicardipine • Cardene
Nortriptyline • Pamelor
Paclitaxel • Abraxane, Taxol
Phenytoin • Dilantin, Phenytek
Piperacillin-tazobactam • Zosyn
Propofol • Diprivan
Sertraline • Zoloft
Tacrolimus • Prograf
Tamoxifen • Soltamox
Valproic acid • Depakene, Depakote
Verapamil • Calan, Verelan
Warfarin • Coumadin, Jantoven
1. Zhang F, Xue J, Shao J, et al. Compilation of 222 drugs’ plasma protein binding data and guidance for study designs. Drug Discov Today. 2012;17(9-10):475-485.
2. Mehvar R. Role of protein binding in pharmacokinetics. Am J Pharm Edu. 2005;69(5): Article 103;1-8.
3. Roberts JA, Pea F, Lipman J. The clinical relevance of plasma protein binding changes. Clin Pharmacokinet. 2013;52(1):1-8.
4. Schmidt S, Gonzalez D, Derendork H. Significance of protein binding in pharmacokinetics and pharmacodynamics. J Pharm Sci. 2010;99(3):1107-1122.
5. Hinderling P. Red blood cells: a neglected compartment in pharmacokinetics and pharmacodynamics. Pharmacol Rev. 1997;49(3):279-295.
6. Benet LZ, Hoener B. Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther. 2002;71(3):115-121.
7. Park MK, Lim KS, Kim T, et al. Reduced valproic acid serum concentrations due to drug interactions with carbapenem antibiotics: overview of 6 cases. Ther Drug Monit. 2012;34(5):599-603.
Mr. S, age 47, weighs 209 lb and has a history of seizure disorder, bipolar disorder not otherwise specified, hypertension, and type 2 diabetes mellitus. He presents to the emergency department after not taking his medications for 2 days while on vacation. He has increased energy, decreased sleep, and pressured speech, and insists on walking for up to 10 hours per day “in preparation for a marathon,” even though he has a 4-cm foot ulcer. His family reports that he had been compliant with his medications until the present incident.
Mr. S has no known drug allergies. His medications include oral divalproex sodium delayed release (valproic acid [VPA]), 1,000 mg twice a day, oral lisinopril, 20 mg every morning, and insulin glargine, 22 units subcutaneously every evening.
A complete blood count, basic metabolic panel, creatine kinase level, VPA level, and urine drug screen are ordered. Relevant results include a serum creatinine level of 1.4 mg/dL (normal range: 0.6 to 1.2 mg/dL), a glucose serum level of 188 mg/dL (normal range: 70 to 100 mg/dL), and a VPA level of 23 mcg/mL (therapeutic range: 50 to 125 mcg/mL). A liver function panel is within normal limits: albumin level of 3.9 g/dL, aspartate aminotransferase level of 18 IU/L, and alanine aminotransferase level of 14 IU/L. In light of Mr. S’s seizure history, neurology is consulted and the decision is made to continue treating him with VPA because he has been seizure-free for 4.5 years and this medication has also helped with his bipolar disorder.
Mr. S is admitted to the hospital and his home medications are resumed at the current doses. On hospital Day 3, Mr. S’s VPA level is 62 mcg/mL, his obsession with a marathon has remitted, and his sleep pattern has normalized. Infectious disease and podiatry services are consulted for his diabetic foot infection, which has ulcerated down to the bone. IV ertapenem, 1,000 mg/d, is initiated with plans for debridement the following week. Two days later, Mr. S has a witnessed seizure; his VPA level is 9 mcg/mL.
A common question asked of pharmacists is, “Will protein binding changes affect drug dosages?” In this article, I describe how protein binding changes may occur, and the complexity of the dynamic. Being highly bound to a protein typically does not mean all medications will interact, but some interactions can be important. This article does not cover medications that bind to hormones.
Why is protein binding important? When a medication is bound to plasma protein, it is not free to act. There can be a delay in therapeutic effect (because no drug is available to react), delayed elimination, or possibly displacement of another protein-bound medication. Additionally, medications tend not to cross the blood-brain barrier or be eliminated when bound. For example, if a drug is 99% bound (leaving 1% free) and displacement now leaves 2% of the drug free, this event has doubled the amount of free drug. As the unbound medication is eliminated, the drug that is bound to the protein can act as a reservoir. A dynamic relationship exists between bound drug, unbound drug, and rate of elimination.
Which proteins do drugs commonly bind to? The proteins often associated with binding include albumin, alpha-1-acid glycoprotein (AAG), and lipoproteins. Albumin comprises 60% of total plasma protein in the plasma. Lipoproteins include very high-density lipoprotein (VHDL), high-density lipoprotein (HDL), very low-density lipoprotein (VLDL), and low-density lipoprotein (LDL).1 Medications that bind to lipoproteins include cyclosporine, tacrolimus, and propofol.2
Continued to: What common disease states can cause hypoalbuminemia?
What common disease states can cause hypoalbuminemia? Many disease states can result in low albumin levels. The most common ones are malnutrition, malignancies, stress, injury, burns, pregnancy, and diabetes.3 When there is less albumin to bind to, free drug levels may be increased.
Can AAG levels change with disease states as well? Because AAG accounts for a lower percentage of total plasma protein than albumin, there may be less clinical concern regarding AAG. AAG levels usually do not drop, but instead can become elevated during times of trauma, inflammation, and acute myocardial infarction. This could result in increased binding of the free drug.4Which medications bind to red blood cells (RBCs)? There are several locations for drugs to bind to RBCs, including to hemoglobin and the plasma membrane. Medications that commonly bind to RBCs include barbiturates, chlorpromazine, imipramine, and phenytoin.5
What are common highly-bound medications? The Table1 provides examples of medications that are >90% protein-bound. However, this information may be misleading because many medications are highly bound. Zhang et al1 compiled binding data for 222 drugs, half of which bind 90% to 100%. However, the literature does not indicate that they all have clinically significant interactions. Benet and Hoener6 discuss how factors other than protein binding affect potential drug interactions, and the complexity of the body’s ability to compensate for increased free drug. Medication characteristics that may contribute to producing a significant interaction include, but are not limited to:
- free vs protein-bound drug in the plasma or tissue
- volume of distribution
- organs affected
- hepatic bioavailability
- drug clearance.
For example, VPA is 93% protein-bound and phenytoin is 91% protein-bound.1 However, this interaction is affected by more than just protein binding. VPA not only displaces the protein-bound phenytoin, but also inhibits its metabolism, which together result in increased free phenytoin levels.
Continued to: Another area of concern is a critically ill patient...
Another area of concern is a critically ill patient who has a change in his or her pH. Medications that are highly bound and have high clearance rates may be affected. This is of particular concern when prescribing antibiotics that are time-dependent, such as beta-lactams.3
What happened to Mr. S? Mr. S likely experienced a drug–drug interaction that resulted in a subtherapeutic VPA level and subsequent seizure. Case reports have shown evidence that the carbapenem class of antibiotics, which includes ertapenem, interacts with VPA.7 Proposed mechanisms include a lowering of VPA serum levels due to a redistribution of the VPA onto the RBCs due to carbapenem. Other theories include the possibility that carbapenems may limit oral VPA absorption, decrease VPA enterohepatic recirculation, and increase VPA metabolism.7 Using VPA and ertapenem together is discouraged because seizures have been reported among patients receiving this combination. If it is medically necessary to administer VPA and ertapenem, closely monitor VPA levels. In Mr. S’s case, another broad-spectrum antibiotic, such as piperacillin-tazobactam, could have been used, for his diabetic foot infection.
While many medications may have high protein binding, there are few clinically important known interactions. However, our understanding of the relationship between protein binding and drug interactions may improve with additional research.
CASE CONTINUED
Under neurology’s care, lacosamide is added for treatment of Mr. S’s seizures. No more seizures are noted during the remainder of his hospitalization. Infectious disease services change his antibiotic to piperacillin-tazobactam. Mr. S continues to progress well and is discharged to a rehabilitation center 2 days later.
Related Resource
- DrugBank. www.drugbank.ca. Canadian Institutes of Health Research.
Drug Brand Names
Amiodarone • Cordarone, Pacerone
Bumetanide • Bumex
Bupivacaine • Marcaine, Sensorcaine
Buprenorphine • Belbuca, Subutex
Ceftriaxone • Rocephin
Chlordiazepoxide • Librium
Chlorpromazine • Thorazine
Clozapine • Clozaril
Cyclosporine • Gengraf, Neoral
Diazepam • Valium
Doxycycline • Acticlate, Doryx
Duloxetine • Cymbalta
Ertapenem • Invanz
Fluoxetine • Prozac, Sarafem
Furosemide • Lasix
Glargine (Insulin) • Lantus, Toujeo
Glipizide • Glucotrol
Haloperidol • Haldol
Ibuprofen • Advil, Motrin
Imipramine • Tofranil
Lacosamide • Vimpat
Lisinopril • Prinivil, Zestril
Lorazepam • Ativan
Nicardipine • Cardene
Nortriptyline • Pamelor
Paclitaxel • Abraxane, Taxol
Phenytoin • Dilantin, Phenytek
Piperacillin-tazobactam • Zosyn
Propofol • Diprivan
Sertraline • Zoloft
Tacrolimus • Prograf
Tamoxifen • Soltamox
Valproic acid • Depakene, Depakote
Verapamil • Calan, Verelan
Warfarin • Coumadin, Jantoven
Mr. S, age 47, weighs 209 lb and has a history of seizure disorder, bipolar disorder not otherwise specified, hypertension, and type 2 diabetes mellitus. He presents to the emergency department after not taking his medications for 2 days while on vacation. He has increased energy, decreased sleep, and pressured speech, and insists on walking for up to 10 hours per day “in preparation for a marathon,” even though he has a 4-cm foot ulcer. His family reports that he had been compliant with his medications until the present incident.
Mr. S has no known drug allergies. His medications include oral divalproex sodium delayed release (valproic acid [VPA]), 1,000 mg twice a day, oral lisinopril, 20 mg every morning, and insulin glargine, 22 units subcutaneously every evening.
A complete blood count, basic metabolic panel, creatine kinase level, VPA level, and urine drug screen are ordered. Relevant results include a serum creatinine level of 1.4 mg/dL (normal range: 0.6 to 1.2 mg/dL), a glucose serum level of 188 mg/dL (normal range: 70 to 100 mg/dL), and a VPA level of 23 mcg/mL (therapeutic range: 50 to 125 mcg/mL). A liver function panel is within normal limits: albumin level of 3.9 g/dL, aspartate aminotransferase level of 18 IU/L, and alanine aminotransferase level of 14 IU/L. In light of Mr. S’s seizure history, neurology is consulted and the decision is made to continue treating him with VPA because he has been seizure-free for 4.5 years and this medication has also helped with his bipolar disorder.
Mr. S is admitted to the hospital and his home medications are resumed at the current doses. On hospital Day 3, Mr. S’s VPA level is 62 mcg/mL, his obsession with a marathon has remitted, and his sleep pattern has normalized. Infectious disease and podiatry services are consulted for his diabetic foot infection, which has ulcerated down to the bone. IV ertapenem, 1,000 mg/d, is initiated with plans for debridement the following week. Two days later, Mr. S has a witnessed seizure; his VPA level is 9 mcg/mL.
A common question asked of pharmacists is, “Will protein binding changes affect drug dosages?” In this article, I describe how protein binding changes may occur, and the complexity of the dynamic. Being highly bound to a protein typically does not mean all medications will interact, but some interactions can be important. This article does not cover medications that bind to hormones.
Why is protein binding important? When a medication is bound to plasma protein, it is not free to act. There can be a delay in therapeutic effect (because no drug is available to react), delayed elimination, or possibly displacement of another protein-bound medication. Additionally, medications tend not to cross the blood-brain barrier or be eliminated when bound. For example, if a drug is 99% bound (leaving 1% free) and displacement now leaves 2% of the drug free, this event has doubled the amount of free drug. As the unbound medication is eliminated, the drug that is bound to the protein can act as a reservoir. A dynamic relationship exists between bound drug, unbound drug, and rate of elimination.
Which proteins do drugs commonly bind to? The proteins often associated with binding include albumin, alpha-1-acid glycoprotein (AAG), and lipoproteins. Albumin comprises 60% of total plasma protein in the plasma. Lipoproteins include very high-density lipoprotein (VHDL), high-density lipoprotein (HDL), very low-density lipoprotein (VLDL), and low-density lipoprotein (LDL).1 Medications that bind to lipoproteins include cyclosporine, tacrolimus, and propofol.2
Continued to: What common disease states can cause hypoalbuminemia?
What common disease states can cause hypoalbuminemia? Many disease states can result in low albumin levels. The most common ones are malnutrition, malignancies, stress, injury, burns, pregnancy, and diabetes.3 When there is less albumin to bind to, free drug levels may be increased.
Can AAG levels change with disease states as well? Because AAG accounts for a lower percentage of total plasma protein than albumin, there may be less clinical concern regarding AAG. AAG levels usually do not drop, but instead can become elevated during times of trauma, inflammation, and acute myocardial infarction. This could result in increased binding of the free drug.4Which medications bind to red blood cells (RBCs)? There are several locations for drugs to bind to RBCs, including to hemoglobin and the plasma membrane. Medications that commonly bind to RBCs include barbiturates, chlorpromazine, imipramine, and phenytoin.5
What are common highly-bound medications? The Table1 provides examples of medications that are >90% protein-bound. However, this information may be misleading because many medications are highly bound. Zhang et al1 compiled binding data for 222 drugs, half of which bind 90% to 100%. However, the literature does not indicate that they all have clinically significant interactions. Benet and Hoener6 discuss how factors other than protein binding affect potential drug interactions, and the complexity of the body’s ability to compensate for increased free drug. Medication characteristics that may contribute to producing a significant interaction include, but are not limited to:
- free vs protein-bound drug in the plasma or tissue
- volume of distribution
- organs affected
- hepatic bioavailability
- drug clearance.
For example, VPA is 93% protein-bound and phenytoin is 91% protein-bound.1 However, this interaction is affected by more than just protein binding. VPA not only displaces the protein-bound phenytoin, but also inhibits its metabolism, which together result in increased free phenytoin levels.
Continued to: Another area of concern is a critically ill patient...
Another area of concern is a critically ill patient who has a change in his or her pH. Medications that are highly bound and have high clearance rates may be affected. This is of particular concern when prescribing antibiotics that are time-dependent, such as beta-lactams.3
What happened to Mr. S? Mr. S likely experienced a drug–drug interaction that resulted in a subtherapeutic VPA level and subsequent seizure. Case reports have shown evidence that the carbapenem class of antibiotics, which includes ertapenem, interacts with VPA.7 Proposed mechanisms include a lowering of VPA serum levels due to a redistribution of the VPA onto the RBCs due to carbapenem. Other theories include the possibility that carbapenems may limit oral VPA absorption, decrease VPA enterohepatic recirculation, and increase VPA metabolism.7 Using VPA and ertapenem together is discouraged because seizures have been reported among patients receiving this combination. If it is medically necessary to administer VPA and ertapenem, closely monitor VPA levels. In Mr. S’s case, another broad-spectrum antibiotic, such as piperacillin-tazobactam, could have been used, for his diabetic foot infection.
While many medications may have high protein binding, there are few clinically important known interactions. However, our understanding of the relationship between protein binding and drug interactions may improve with additional research.
CASE CONTINUED
Under neurology’s care, lacosamide is added for treatment of Mr. S’s seizures. No more seizures are noted during the remainder of his hospitalization. Infectious disease services change his antibiotic to piperacillin-tazobactam. Mr. S continues to progress well and is discharged to a rehabilitation center 2 days later.
Related Resource
- DrugBank. www.drugbank.ca. Canadian Institutes of Health Research.
Drug Brand Names
Amiodarone • Cordarone, Pacerone
Bumetanide • Bumex
Bupivacaine • Marcaine, Sensorcaine
Buprenorphine • Belbuca, Subutex
Ceftriaxone • Rocephin
Chlordiazepoxide • Librium
Chlorpromazine • Thorazine
Clozapine • Clozaril
Cyclosporine • Gengraf, Neoral
Diazepam • Valium
Doxycycline • Acticlate, Doryx
Duloxetine • Cymbalta
Ertapenem • Invanz
Fluoxetine • Prozac, Sarafem
Furosemide • Lasix
Glargine (Insulin) • Lantus, Toujeo
Glipizide • Glucotrol
Haloperidol • Haldol
Ibuprofen • Advil, Motrin
Imipramine • Tofranil
Lacosamide • Vimpat
Lisinopril • Prinivil, Zestril
Lorazepam • Ativan
Nicardipine • Cardene
Nortriptyline • Pamelor
Paclitaxel • Abraxane, Taxol
Phenytoin • Dilantin, Phenytek
Piperacillin-tazobactam • Zosyn
Propofol • Diprivan
Sertraline • Zoloft
Tacrolimus • Prograf
Tamoxifen • Soltamox
Valproic acid • Depakene, Depakote
Verapamil • Calan, Verelan
Warfarin • Coumadin, Jantoven
1. Zhang F, Xue J, Shao J, et al. Compilation of 222 drugs’ plasma protein binding data and guidance for study designs. Drug Discov Today. 2012;17(9-10):475-485.
2. Mehvar R. Role of protein binding in pharmacokinetics. Am J Pharm Edu. 2005;69(5): Article 103;1-8.
3. Roberts JA, Pea F, Lipman J. The clinical relevance of plasma protein binding changes. Clin Pharmacokinet. 2013;52(1):1-8.
4. Schmidt S, Gonzalez D, Derendork H. Significance of protein binding in pharmacokinetics and pharmacodynamics. J Pharm Sci. 2010;99(3):1107-1122.
5. Hinderling P. Red blood cells: a neglected compartment in pharmacokinetics and pharmacodynamics. Pharmacol Rev. 1997;49(3):279-295.
6. Benet LZ, Hoener B. Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther. 2002;71(3):115-121.
7. Park MK, Lim KS, Kim T, et al. Reduced valproic acid serum concentrations due to drug interactions with carbapenem antibiotics: overview of 6 cases. Ther Drug Monit. 2012;34(5):599-603.
1. Zhang F, Xue J, Shao J, et al. Compilation of 222 drugs’ plasma protein binding data and guidance for study designs. Drug Discov Today. 2012;17(9-10):475-485.
2. Mehvar R. Role of protein binding in pharmacokinetics. Am J Pharm Edu. 2005;69(5): Article 103;1-8.
3. Roberts JA, Pea F, Lipman J. The clinical relevance of plasma protein binding changes. Clin Pharmacokinet. 2013;52(1):1-8.
4. Schmidt S, Gonzalez D, Derendork H. Significance of protein binding in pharmacokinetics and pharmacodynamics. J Pharm Sci. 2010;99(3):1107-1122.
5. Hinderling P. Red blood cells: a neglected compartment in pharmacokinetics and pharmacodynamics. Pharmacol Rev. 1997;49(3):279-295.
6. Benet LZ, Hoener B. Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther. 2002;71(3):115-121.
7. Park MK, Lim KS, Kim T, et al. Reduced valproic acid serum concentrations due to drug interactions with carbapenem antibiotics: overview of 6 cases. Ther Drug Monit. 2012;34(5):599-603.
Managing procedural pain in a patient taking naltrexone

Mr. M, age 55, presents to his primary care physician (PCP) with hematochezia. Mr. M states that for the past week, he has noticed blood upon wiping after a bowel movement and is worried that he might have cancer.
Mr. M has a 10-year history of opioid use disorder as diagnosed by his psychiatrist. He is presently maintained on long-acting injectable naltrexone, 380 mg IM every 4 weeks, and has not used opioids for the past 1.5 years. Mr. M is also taking simvastatin, 40 mg, for dyslipidemia, lisinopril, 5 mg, for hypertension, and cetirizine, 5 mg as needed, for seasonal allergies.
A standard workup including a physical examination and laboratory tests are performed. Mr. M’s PCP would like for him to undergo a colonoscopy to investigate the etiology of the bleeding. In consultation with both the PCP and psychiatrist, the gastroenterologist determines that the colonoscopy can be performed within 48 hours with no changes to Mr. M’s medication regimen. The gastroenterologist utilizes a nonopioid, ketorolac, 30 mg IV, for pain management during the procedure. Diverticula were identified in the lower gastrointestinal tract and are treated endoscopically. Mr. M is successfully withdrawn from sedation with no adverse events or pain and continues to be in opioid remission.
Naltrexone competitively antagonizes opioid receptors with the highest affinity for the µ-opioid receptor. It is approved for treatment of alcohol and opioid dependence following opioid detoxification.1 Its competitive inhibition at the µ-opioid receptor results in the inhibition of exogenous opioid effects. The medication is available as an orally administered tablet as well as a long-acting injection administered intramuscularly (Table 11). The long-acting injection can be useful in patients who have difficulty with adherence, because good adherence to naltrexone is required to maximize efficacy.

Due to its ability to block opioid analgesic effects, naltrexone presents a unique challenge for patients taking it who need to undergo procedures that require pain control. Pharmacologic regimens used during procedures often contain a sedative agent, such as propofol, and an opioid for analgesia. Alternative strategies are needed for patients taking naltrexone who require an opioid analgesic agent for procedures such as colonoscopies.
One strategy could be to withhold naltrexone before the procedure to ensure that the medication will not compete with the opioid agent to relieve pain. This strategy depends on the urgency of the procedure, the formulation of naltrexone being used, and patient-specific factors that may increase the risk for adverse events. For a non-urgent, elective procedure, it may be acceptable to hold oral naltrexone approximately 72 hours before the procedure. However, this is likely not a favorable approach for patients who may be at high risk for relapse or for patients who are receiving the long-acting formulation. Additionally, the use of an opioid agent intra- or post-operatively for pain may increase the risk of relapse. The use of opioids for such procedures may also be more difficult in a patient with a history of opioid abuse or dependence because he or she may have developed tolerance to opioids. Conversely, if a patient has been treated with naltrexone for an extended period, a lack of tolerance may increase the risk of respiratory depression with opioid administration due to upregulation of the opioid receptor.2
Continue to: Nonopioid analgesic agents
Nonopioid analgesic agents
For a patient receiving naltrexone who needs to undergo a procedure, a multidisciplinary consultation between the patient’s psychiatrist and other clinicians is key for providing a regimen that is safe and effective. A nonopioid analgesic agent may be considered to avoid the problematic interactions possible in these patients (Table 23-5). Nonopioid regimens can be utilized alone or in combination, and may include the following3-5:
Ketamine is a non-competitive antagonist at the N-methyl-
Dexmedetomidine is an alpha-2 agonist that can provide sedative and analgesic effects. It can cause procedural hypotension and bradycardia, so caution is advised in patients with cardiac disease and hepatic and/or renal insufficiencies.
Nonsteroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen or ketorolac, inhibit cyclooxygenase enzymes and can be considered in analgesic regimens. However, for most surgical procedures, the increased risk of bleeding due to platelet inhibition is a concern.

Continue to: Acetaminophen
Acetaminophen. Although its full mechanism of action has not been discovered, acetaminophen may also act on the cyclooxygenase pathway to produce analgesia. Compared with the oral formulation, IV acetaminophen is more expensive but may offer certain advantages, including faster plasma peak levels and lower production of acetaminophen’s toxic metabolite, N-acetyl-p-benzoquinone imine. Nonetheless, hepatotoxicity and overdose remain a concern.
The use of nonopioid analgesics during elective procedures that require pain control will allow continued use of an opioid antagonist such as naltrexone, while minimizing the risk for withdrawal or relapse. Their use must be evaluated on a case-by-case basis to ensure maximum safety and efficacy for each patient from both a medical and psychiatric standpoint. Overall, with the proper expertise and consultation, nonopioid pain regimens represent a reasonable alternative to opiates for patients who take naltrexone.
Related Resources
- American Society of Anesthesiologists. Standards guidelines and related resources. https://www.asahq.org/quality-and-practice-management/standards-guidelines-and-related-resources-search.
- American Society of Addiction Medicine. Clinical resources. https://www.asam.org/resources/guidelines-and-consensus-documents.
Drug Brand Names
Acetaminophen • Tylenol
Cetirizine • Zyrtec
Dexmedetomidine • Precedex
Ibuprofen • Caldolor (IV), Motrin (oral)
Ketamine • Ketalar
Ketorolac • Toradol
Lisinopril • Prinivil, Zestril
Naltrexone • ReVia, Vivitrol
Propofol • Diprivan
Simvastatin • Juvisync, Simcor
1. Vivitrol [package insert]. Waltham, MA: Alkermes, Inc.; 2015.
2. Yoburn BC, Duttaroy A, Shah S, et al. Opioid antagonist-induced receptor upregulation: effects of concurrent agonist administration. Brain Res Bull. 1994;33(2):237-240.
3. Vadivelu N, Chang D, Lumermann L, et al. Management of patients on abuse-deterrent opioids in the ambulatory surgery setting. Curr Pain Headache Rep. 2017;21(2):10.
4. Koh W, Nguyen KP, Jahr JS. Intravenous non-opioid analgesia for peri- and postoperative pain management: a scientific review of intravenous acetaminophen and ibuprofen. Korean J Anesthesiol. 2015;68(1):3-12.
5. Kaye AD, Cornett EM, Helander E, et al. An update on nonopioids: intravenous or oral analgesics for perioperative pain management. Anesthesiol Clin. 2017;35(2):e55-e71.
Mr. M, age 55, presents to his primary care physician (PCP) with hematochezia. Mr. M states that for the past week, he has noticed blood upon wiping after a bowel movement and is worried that he might have cancer.
Mr. M has a 10-year history of opioid use disorder as diagnosed by his psychiatrist. He is presently maintained on long-acting injectable naltrexone, 380 mg IM every 4 weeks, and has not used opioids for the past 1.5 years. Mr. M is also taking simvastatin, 40 mg, for dyslipidemia, lisinopril, 5 mg, for hypertension, and cetirizine, 5 mg as needed, for seasonal allergies.
A standard workup including a physical examination and laboratory tests are performed. Mr. M’s PCP would like for him to undergo a colonoscopy to investigate the etiology of the bleeding. In consultation with both the PCP and psychiatrist, the gastroenterologist determines that the colonoscopy can be performed within 48 hours with no changes to Mr. M’s medication regimen. The gastroenterologist utilizes a nonopioid, ketorolac, 30 mg IV, for pain management during the procedure. Diverticula were identified in the lower gastrointestinal tract and are treated endoscopically. Mr. M is successfully withdrawn from sedation with no adverse events or pain and continues to be in opioid remission.
Naltrexone competitively antagonizes opioid receptors with the highest affinity for the µ-opioid receptor. It is approved for treatment of alcohol and opioid dependence following opioid detoxification.1 Its competitive inhibition at the µ-opioid receptor results in the inhibition of exogenous opioid effects. The medication is available as an orally administered tablet as well as a long-acting injection administered intramuscularly (Table 11). The long-acting injection can be useful in patients who have difficulty with adherence, because good adherence to naltrexone is required to maximize efficacy.
Due to its ability to block opioid analgesic effects, naltrexone presents a unique challenge for patients taking it who need to undergo procedures that require pain control. Pharmacologic regimens used during procedures often contain a sedative agent, such as propofol, and an opioid for analgesia. Alternative strategies are needed for patients taking naltrexone who require an opioid analgesic agent for procedures such as colonoscopies.
One strategy could be to withhold naltrexone before the procedure to ensure that the medication will not compete with the opioid agent to relieve pain. This strategy depends on the urgency of the procedure, the formulation of naltrexone being used, and patient-specific factors that may increase the risk for adverse events. For a non-urgent, elective procedure, it may be acceptable to hold oral naltrexone approximately 72 hours before the procedure. However, this is likely not a favorable approach for patients who may be at high risk for relapse or for patients who are receiving the long-acting formulation. Additionally, the use of an opioid agent intra- or post-operatively for pain may increase the risk of relapse. The use of opioids for such procedures may also be more difficult in a patient with a history of opioid abuse or dependence because he or she may have developed tolerance to opioids. Conversely, if a patient has been treated with naltrexone for an extended period, a lack of tolerance may increase the risk of respiratory depression with opioid administration due to upregulation of the opioid receptor.2
Continue to: Nonopioid analgesic agents
Nonopioid analgesic agents
For a patient receiving naltrexone who needs to undergo a procedure, a multidisciplinary consultation between the patient’s psychiatrist and other clinicians is key for providing a regimen that is safe and effective. A nonopioid analgesic agent may be considered to avoid the problematic interactions possible in these patients (Table 23-5). Nonopioid regimens can be utilized alone or in combination, and may include the following3-5:
Ketamine is a non-competitive antagonist at the N-methyl-
Dexmedetomidine is an alpha-2 agonist that can provide sedative and analgesic effects. It can cause procedural hypotension and bradycardia, so caution is advised in patients with cardiac disease and hepatic and/or renal insufficiencies.
Nonsteroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen or ketorolac, inhibit cyclooxygenase enzymes and can be considered in analgesic regimens. However, for most surgical procedures, the increased risk of bleeding due to platelet inhibition is a concern.
Continue to: Acetaminophen
Acetaminophen. Although its full mechanism of action has not been discovered, acetaminophen may also act on the cyclooxygenase pathway to produce analgesia. Compared with the oral formulation, IV acetaminophen is more expensive but may offer certain advantages, including faster plasma peak levels and lower production of acetaminophen’s toxic metabolite, N-acetyl-p-benzoquinone imine. Nonetheless, hepatotoxicity and overdose remain a concern.
The use of nonopioid analgesics during elective procedures that require pain control will allow continued use of an opioid antagonist such as naltrexone, while minimizing the risk for withdrawal or relapse. Their use must be evaluated on a case-by-case basis to ensure maximum safety and efficacy for each patient from both a medical and psychiatric standpoint. Overall, with the proper expertise and consultation, nonopioid pain regimens represent a reasonable alternative to opiates for patients who take naltrexone.
Related Resources
- American Society of Anesthesiologists. Standards guidelines and related resources. https://www.asahq.org/quality-and-practice-management/standards-guidelines-and-related-resources-search.
- American Society of Addiction Medicine. Clinical resources. https://www.asam.org/resources/guidelines-and-consensus-documents.
Drug Brand Names
Acetaminophen • Tylenol
Cetirizine • Zyrtec
Dexmedetomidine • Precedex
Ibuprofen • Caldolor (IV), Motrin (oral)
Ketamine • Ketalar
Ketorolac • Toradol
Lisinopril • Prinivil, Zestril
Naltrexone • ReVia, Vivitrol
Propofol • Diprivan
Simvastatin • Juvisync, Simcor
Mr. M, age 55, presents to his primary care physician (PCP) with hematochezia. Mr. M states that for the past week, he has noticed blood upon wiping after a bowel movement and is worried that he might have cancer.
Mr. M has a 10-year history of opioid use disorder as diagnosed by his psychiatrist. He is presently maintained on long-acting injectable naltrexone, 380 mg IM every 4 weeks, and has not used opioids for the past 1.5 years. Mr. M is also taking simvastatin, 40 mg, for dyslipidemia, lisinopril, 5 mg, for hypertension, and cetirizine, 5 mg as needed, for seasonal allergies.
A standard workup including a physical examination and laboratory tests are performed. Mr. M’s PCP would like for him to undergo a colonoscopy to investigate the etiology of the bleeding. In consultation with both the PCP and psychiatrist, the gastroenterologist determines that the colonoscopy can be performed within 48 hours with no changes to Mr. M’s medication regimen. The gastroenterologist utilizes a nonopioid, ketorolac, 30 mg IV, for pain management during the procedure. Diverticula were identified in the lower gastrointestinal tract and are treated endoscopically. Mr. M is successfully withdrawn from sedation with no adverse events or pain and continues to be in opioid remission.
Naltrexone competitively antagonizes opioid receptors with the highest affinity for the µ-opioid receptor. It is approved for treatment of alcohol and opioid dependence following opioid detoxification.1 Its competitive inhibition at the µ-opioid receptor results in the inhibition of exogenous opioid effects. The medication is available as an orally administered tablet as well as a long-acting injection administered intramuscularly (Table 11). The long-acting injection can be useful in patients who have difficulty with adherence, because good adherence to naltrexone is required to maximize efficacy.
Due to its ability to block opioid analgesic effects, naltrexone presents a unique challenge for patients taking it who need to undergo procedures that require pain control. Pharmacologic regimens used during procedures often contain a sedative agent, such as propofol, and an opioid for analgesia. Alternative strategies are needed for patients taking naltrexone who require an opioid analgesic agent for procedures such as colonoscopies.
One strategy could be to withhold naltrexone before the procedure to ensure that the medication will not compete with the opioid agent to relieve pain. This strategy depends on the urgency of the procedure, the formulation of naltrexone being used, and patient-specific factors that may increase the risk for adverse events. For a non-urgent, elective procedure, it may be acceptable to hold oral naltrexone approximately 72 hours before the procedure. However, this is likely not a favorable approach for patients who may be at high risk for relapse or for patients who are receiving the long-acting formulation. Additionally, the use of an opioid agent intra- or post-operatively for pain may increase the risk of relapse. The use of opioids for such procedures may also be more difficult in a patient with a history of opioid abuse or dependence because he or she may have developed tolerance to opioids. Conversely, if a patient has been treated with naltrexone for an extended period, a lack of tolerance may increase the risk of respiratory depression with opioid administration due to upregulation of the opioid receptor.2
Continue to: Nonopioid analgesic agents
Nonopioid analgesic agents
For a patient receiving naltrexone who needs to undergo a procedure, a multidisciplinary consultation between the patient’s psychiatrist and other clinicians is key for providing a regimen that is safe and effective. A nonopioid analgesic agent may be considered to avoid the problematic interactions possible in these patients (Table 23-5). Nonopioid regimens can be utilized alone or in combination, and may include the following3-5:
Ketamine is a non-competitive antagonist at the N-methyl-
Dexmedetomidine is an alpha-2 agonist that can provide sedative and analgesic effects. It can cause procedural hypotension and bradycardia, so caution is advised in patients with cardiac disease and hepatic and/or renal insufficiencies.
Nonsteroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen or ketorolac, inhibit cyclooxygenase enzymes and can be considered in analgesic regimens. However, for most surgical procedures, the increased risk of bleeding due to platelet inhibition is a concern.
Continue to: Acetaminophen
Acetaminophen. Although its full mechanism of action has not been discovered, acetaminophen may also act on the cyclooxygenase pathway to produce analgesia. Compared with the oral formulation, IV acetaminophen is more expensive but may offer certain advantages, including faster plasma peak levels and lower production of acetaminophen’s toxic metabolite, N-acetyl-p-benzoquinone imine. Nonetheless, hepatotoxicity and overdose remain a concern.
The use of nonopioid analgesics during elective procedures that require pain control will allow continued use of an opioid antagonist such as naltrexone, while minimizing the risk for withdrawal or relapse. Their use must be evaluated on a case-by-case basis to ensure maximum safety and efficacy for each patient from both a medical and psychiatric standpoint. Overall, with the proper expertise and consultation, nonopioid pain regimens represent a reasonable alternative to opiates for patients who take naltrexone.
Related Resources
- American Society of Anesthesiologists. Standards guidelines and related resources. https://www.asahq.org/quality-and-practice-management/standards-guidelines-and-related-resources-search.
- American Society of Addiction Medicine. Clinical resources. https://www.asam.org/resources/guidelines-and-consensus-documents.
Drug Brand Names
Acetaminophen • Tylenol
Cetirizine • Zyrtec
Dexmedetomidine • Precedex
Ibuprofen • Caldolor (IV), Motrin (oral)
Ketamine • Ketalar
Ketorolac • Toradol
Lisinopril • Prinivil, Zestril
Naltrexone • ReVia, Vivitrol
Propofol • Diprivan
Simvastatin • Juvisync, Simcor
1. Vivitrol [package insert]. Waltham, MA: Alkermes, Inc.; 2015.
2. Yoburn BC, Duttaroy A, Shah S, et al. Opioid antagonist-induced receptor upregulation: effects of concurrent agonist administration. Brain Res Bull. 1994;33(2):237-240.
3. Vadivelu N, Chang D, Lumermann L, et al. Management of patients on abuse-deterrent opioids in the ambulatory surgery setting. Curr Pain Headache Rep. 2017;21(2):10.
4. Koh W, Nguyen KP, Jahr JS. Intravenous non-opioid analgesia for peri- and postoperative pain management: a scientific review of intravenous acetaminophen and ibuprofen. Korean J Anesthesiol. 2015;68(1):3-12.
5. Kaye AD, Cornett EM, Helander E, et al. An update on nonopioids: intravenous or oral analgesics for perioperative pain management. Anesthesiol Clin. 2017;35(2):e55-e71.
1. Vivitrol [package insert]. Waltham, MA: Alkermes, Inc.; 2015.
2. Yoburn BC, Duttaroy A, Shah S, et al. Opioid antagonist-induced receptor upregulation: effects of concurrent agonist administration. Brain Res Bull. 1994;33(2):237-240.
3. Vadivelu N, Chang D, Lumermann L, et al. Management of patients on abuse-deterrent opioids in the ambulatory surgery setting. Curr Pain Headache Rep. 2017;21(2):10.
4. Koh W, Nguyen KP, Jahr JS. Intravenous non-opioid analgesia for peri- and postoperative pain management: a scientific review of intravenous acetaminophen and ibuprofen. Korean J Anesthesiol. 2015;68(1):3-12.
5. Kaye AD, Cornett EM, Helander E, et al. An update on nonopioids: intravenous or oral analgesics for perioperative pain management. Anesthesiol Clin. 2017;35(2):e55-e71.
Paliperidone palmitate: Adjusting dosing intervals and measuring serum concentrations

Mr. B, age 27, has a 10-year history of schizophrenia. Last year, he was doing well and working 4 hours/day 3 days/week while taking oral risperidone, 6 mg, at bedtime. However, during the past 2 weeks Mr. B began to have a return of auditory hallucinations and reports that he stopped taking his medication again 6 weeks ago.
As a result, he is started on paliperidone palmitate following the product label’s initiation dosing recommendation. On the first day he is given the first dose of the initiation regimen, 234 mg IM. One week later, the second dose of the initiation regimen, 156 mg IM, is given. One month later, the first maintenance dose of 117 mg IM every 28 days is given. All injections are in his deltoid muscle at his request.
After 3 weeks on the first maintenance dose of 117 mg, the voices begin to bother him again. Subsequently, Mr. B’s maintenance dose is increased first to 156 mg, and for the same problem with breakthrough hallucinations the following month to 234 mg, the maximum dose in the product label. After 6 months of receiving 234 mg IM every 28 days, the auditory hallucinations continue to bother him, but only for a few days prior to his next injection. He misses work 1 or 2 times before each injection.
Can the injection frequency for Mr. B’s paliperidone palmitate, 234 mg IM, in the deltoid muscle be increased to every 21 days to prevent the monthly exacerbations? Yes, the injection frequency can be increased, and doing so will increase the concentrations of paliperidone. The use of long-acting injectable antipsychotics (LAIs) is complicated by the lengthy time needed to reach steady state. In the case of paliperidone palmitate, an initiation regimen was developed that achieves therapeutic concentrations that are close to steady state before the oral antipsychotic’s effects are lost. This initiation strategy avoids the need for oral supplementation to maintain clinical efficacy. However, even using an initiation regimen or a loading dose does not decrease the time to final steady state after a dose adjustment due to the slow absorption of the medication from the injection site. The time to steady state is controlled by “flip-flop” pharmacokinetics. In this kind of pharmacokinetics, which is observed with all LAIs, the absorption rate from the injection site is lower than the elimination rate.1
Cleton et al2 reported the pharmacokinetics of paliperidone palmitate for deltoid and gluteal injection sites. By combining the median data for the deltoid injection route from the article by Cleton et al2 and the dosing from Mr. B’s case, I created a model using superposition of a sixth-degree polynomial fitted to the single dose data. Gluteal injections were not included because their increased complexity is beyond the scope of this article, but the time to maximum concentration (gluteal > deltoid) and peak concentration (deltoid > gluteal) are different for each route. The polynomial was a good fit with the adjusted r2 = 0.976, P < .0001. This model illustrates the paliperidone serum concentrations for Mr. B and is shown in the Figure. As you can see, by Day 9, the serum concentrations had reached the lower limit of the expected range of 20 to 60 ng/mL, shown in the shaded region of the Figure.3

Steady state at the routine maintenance dose of 117 mg every 28 days was never reached as the medication was not sufficient to suppress Mr. B’s hallucinations, and his doses needed to be increased each month. First, Mr. B’s dose was increased to 156 mg and then to the maximum recommended dose of 234 mg every 28 days. Steady state can be considered to have been achieved when 90% of the final steady state is reached after 3.3 half-lives. Because of the flip-flop pharmacokinetics, the important half-life is the absorption half-life of approximately 40 days or 132 days at the same dose. In Mr. B’s case, this was Day 221, where the trough concentration was 35 ng/mL. However, this regimen was still inadequate because he had breakthrough symptoms prior to the next injection.
By decreasing the injection interval from 28 days to 21 days, the concentrations will increase to a new steady state. This will take the same 132 days. With the reduced injection frequency of 21 days, 7 injections will have been given prior to reaching the new steady state. Steady state is not dependent on the number of injections, but only on the absorption half-life. This new steady state trough is substantially higher at 52 ng/mL, but still in the expected range for commonly used doses. Because Mr. B’s hallucinations only appeared at the end of the dosing interval, it is reasonable to expect that his new regimen would be successful in suppressing his hallucinations. However, monitoring for peak-related adverse effects is essential. Based upon controlled clinical trials, the potential dose-related adverse effects of paliperidone include akathisia, other extrapyramidal symptoms, weight gain, and QTc prolongation.
Continue to: Would monitoring a patient's paliperidone serum concentrations be useful?
Would monitoring a patient’s paliperidone serum concentrations be useful? Currently, measuring an individual’s paliperidone serum concentration is generally considered unwarranted.3,4 One of the major reasons is a lack of appropriately designed studies to determine a therapeutic range.5 Flexible dose designs, commonly used in registration studies, cloud the relationships between concentration, time, response, and adverse effects. There are additional problems that are the result of diagnostic heterogeneity and placebo responders. A well-designed study to determine the therapeutic range would have ≥1 fixed dose groups and be diagnostically homogeneous. There are currently only a limited number of clinical laboratories that have implemented suitable assays.
Given the lack of knowledge of a therapeutic range, assured knowledge of nonadherence to LAIs, and the absence of significant drug interactions for paliperidone, there remain a few reasonable justifications for obtaining a patient’s paliperidone serum concentration (Table). If the patient had a good response with mild adverse effects, there is no reason to obtain a paliperidone serum concentration or make any change in the medication or dose. However, if the patient had a good response accompanied by moderate or severe adverse effects, or the patient has a poor response, then obtaining the paliperidone serum concentration could help determine an appropriate course of action.

CASE CONTINUED
After the second dose at the increased frequency on Day 252, the paliperidone serum concentration was maintained above 40 ng/mL. Mr. B continued to tolerate the LAI well and no longer reported any breakthrough hallucinations.
Related Resources
- ARUP Laboratories. Paliperidone, serum or plasma. http://ltd.aruplab.com/tests/pub/2007949.
- LabCorp. Paliperidone Paliperidone (as 9-hydroxyrisperidone), serum or plasma. https://www.labcorp.com/test-menu/38351/paliperidone-as-9-hydroxyrisperidone-serum-or-plasma.
- Janssen Scientific Affairs. Educational dose illustrator. http://www.educationaldoseillustrator.com.
Drug Brand Names
Paliperidone palmitate • Invega Sustenna
Risperidone • Risperdal
1. Jann MW, Ereshefsky L, Saklad SR. Clinical pharmacokinetics of the depot antipsychotics. Clin Pharmacokinet. 1985;10(4):315-333.
2. Cleton A, Rossenu S, Crauwels H, et al. A single-dose, open-label, parallel, randomized, dose-proportionality study of paliperidone after intramuscular injections of paliperidone palmitate in the deltoid or gluteal muscle in patients with schizophrenia. J Clin Pharmacol. 2014;54(9):1048-1057.
3. Taylor D, Paton C, Kapur S. The Maudsley prescribing guidelines in psychiatry. 12th ed. Oxford, UK: John Wiley & Sons, Ltd.; 2015:1-10.
4. Hiemke C, Baumann P, Bergemann N, et al. AGNP consensus guidelines for therapeutic drug monitoring in psychiatry: update 2011. Pharmacopsychiatry. 2011;44(6):195-235.
5. Lopez LV, Kane JM. Plasma levels of second-generation antipsychotics and clinical response in acute psychosis: a review of the literature. Schizophr Res. 2013;147(2-3):368-374.
Mr. B, age 27, has a 10-year history of schizophrenia. Last year, he was doing well and working 4 hours/day 3 days/week while taking oral risperidone, 6 mg, at bedtime. However, during the past 2 weeks Mr. B began to have a return of auditory hallucinations and reports that he stopped taking his medication again 6 weeks ago.
As a result, he is started on paliperidone palmitate following the product label’s initiation dosing recommendation. On the first day he is given the first dose of the initiation regimen, 234 mg IM. One week later, the second dose of the initiation regimen, 156 mg IM, is given. One month later, the first maintenance dose of 117 mg IM every 28 days is given. All injections are in his deltoid muscle at his request.
After 3 weeks on the first maintenance dose of 117 mg, the voices begin to bother him again. Subsequently, Mr. B’s maintenance dose is increased first to 156 mg, and for the same problem with breakthrough hallucinations the following month to 234 mg, the maximum dose in the product label. After 6 months of receiving 234 mg IM every 28 days, the auditory hallucinations continue to bother him, but only for a few days prior to his next injection. He misses work 1 or 2 times before each injection.
Can the injection frequency for Mr. B’s paliperidone palmitate, 234 mg IM, in the deltoid muscle be increased to every 21 days to prevent the monthly exacerbations? Yes, the injection frequency can be increased, and doing so will increase the concentrations of paliperidone. The use of long-acting injectable antipsychotics (LAIs) is complicated by the lengthy time needed to reach steady state. In the case of paliperidone palmitate, an initiation regimen was developed that achieves therapeutic concentrations that are close to steady state before the oral antipsychotic’s effects are lost. This initiation strategy avoids the need for oral supplementation to maintain clinical efficacy. However, even using an initiation regimen or a loading dose does not decrease the time to final steady state after a dose adjustment due to the slow absorption of the medication from the injection site. The time to steady state is controlled by “flip-flop” pharmacokinetics. In this kind of pharmacokinetics, which is observed with all LAIs, the absorption rate from the injection site is lower than the elimination rate.1
Cleton et al2 reported the pharmacokinetics of paliperidone palmitate for deltoid and gluteal injection sites. By combining the median data for the deltoid injection route from the article by Cleton et al2 and the dosing from Mr. B’s case, I created a model using superposition of a sixth-degree polynomial fitted to the single dose data. Gluteal injections were not included because their increased complexity is beyond the scope of this article, but the time to maximum concentration (gluteal > deltoid) and peak concentration (deltoid > gluteal) are different for each route. The polynomial was a good fit with the adjusted r2 = 0.976, P < .0001. This model illustrates the paliperidone serum concentrations for Mr. B and is shown in the Figure. As you can see, by Day 9, the serum concentrations had reached the lower limit of the expected range of 20 to 60 ng/mL, shown in the shaded region of the Figure.3
Steady state at the routine maintenance dose of 117 mg every 28 days was never reached as the medication was not sufficient to suppress Mr. B’s hallucinations, and his doses needed to be increased each month. First, Mr. B’s dose was increased to 156 mg and then to the maximum recommended dose of 234 mg every 28 days. Steady state can be considered to have been achieved when 90% of the final steady state is reached after 3.3 half-lives. Because of the flip-flop pharmacokinetics, the important half-life is the absorption half-life of approximately 40 days or 132 days at the same dose. In Mr. B’s case, this was Day 221, where the trough concentration was 35 ng/mL. However, this regimen was still inadequate because he had breakthrough symptoms prior to the next injection.
By decreasing the injection interval from 28 days to 21 days, the concentrations will increase to a new steady state. This will take the same 132 days. With the reduced injection frequency of 21 days, 7 injections will have been given prior to reaching the new steady state. Steady state is not dependent on the number of injections, but only on the absorption half-life. This new steady state trough is substantially higher at 52 ng/mL, but still in the expected range for commonly used doses. Because Mr. B’s hallucinations only appeared at the end of the dosing interval, it is reasonable to expect that his new regimen would be successful in suppressing his hallucinations. However, monitoring for peak-related adverse effects is essential. Based upon controlled clinical trials, the potential dose-related adverse effects of paliperidone include akathisia, other extrapyramidal symptoms, weight gain, and QTc prolongation.
Continue to: Would monitoring a patient's paliperidone serum concentrations be useful?
Would monitoring a patient’s paliperidone serum concentrations be useful? Currently, measuring an individual’s paliperidone serum concentration is generally considered unwarranted.3,4 One of the major reasons is a lack of appropriately designed studies to determine a therapeutic range.5 Flexible dose designs, commonly used in registration studies, cloud the relationships between concentration, time, response, and adverse effects. There are additional problems that are the result of diagnostic heterogeneity and placebo responders. A well-designed study to determine the therapeutic range would have ≥1 fixed dose groups and be diagnostically homogeneous. There are currently only a limited number of clinical laboratories that have implemented suitable assays.
Given the lack of knowledge of a therapeutic range, assured knowledge of nonadherence to LAIs, and the absence of significant drug interactions for paliperidone, there remain a few reasonable justifications for obtaining a patient’s paliperidone serum concentration (Table). If the patient had a good response with mild adverse effects, there is no reason to obtain a paliperidone serum concentration or make any change in the medication or dose. However, if the patient had a good response accompanied by moderate or severe adverse effects, or the patient has a poor response, then obtaining the paliperidone serum concentration could help determine an appropriate course of action.
CASE CONTINUED
After the second dose at the increased frequency on Day 252, the paliperidone serum concentration was maintained above 40 ng/mL. Mr. B continued to tolerate the LAI well and no longer reported any breakthrough hallucinations.
Related Resources
- ARUP Laboratories. Paliperidone, serum or plasma. http://ltd.aruplab.com/tests/pub/2007949.
- LabCorp. Paliperidone Paliperidone (as 9-hydroxyrisperidone), serum or plasma. https://www.labcorp.com/test-menu/38351/paliperidone-as-9-hydroxyrisperidone-serum-or-plasma.
- Janssen Scientific Affairs. Educational dose illustrator. http://www.educationaldoseillustrator.com.
Drug Brand Names
Paliperidone palmitate • Invega Sustenna
Risperidone • Risperdal
Mr. B, age 27, has a 10-year history of schizophrenia. Last year, he was doing well and working 4 hours/day 3 days/week while taking oral risperidone, 6 mg, at bedtime. However, during the past 2 weeks Mr. B began to have a return of auditory hallucinations and reports that he stopped taking his medication again 6 weeks ago.
As a result, he is started on paliperidone palmitate following the product label’s initiation dosing recommendation. On the first day he is given the first dose of the initiation regimen, 234 mg IM. One week later, the second dose of the initiation regimen, 156 mg IM, is given. One month later, the first maintenance dose of 117 mg IM every 28 days is given. All injections are in his deltoid muscle at his request.
After 3 weeks on the first maintenance dose of 117 mg, the voices begin to bother him again. Subsequently, Mr. B’s maintenance dose is increased first to 156 mg, and for the same problem with breakthrough hallucinations the following month to 234 mg, the maximum dose in the product label. After 6 months of receiving 234 mg IM every 28 days, the auditory hallucinations continue to bother him, but only for a few days prior to his next injection. He misses work 1 or 2 times before each injection.
Can the injection frequency for Mr. B’s paliperidone palmitate, 234 mg IM, in the deltoid muscle be increased to every 21 days to prevent the monthly exacerbations? Yes, the injection frequency can be increased, and doing so will increase the concentrations of paliperidone. The use of long-acting injectable antipsychotics (LAIs) is complicated by the lengthy time needed to reach steady state. In the case of paliperidone palmitate, an initiation regimen was developed that achieves therapeutic concentrations that are close to steady state before the oral antipsychotic’s effects are lost. This initiation strategy avoids the need for oral supplementation to maintain clinical efficacy. However, even using an initiation regimen or a loading dose does not decrease the time to final steady state after a dose adjustment due to the slow absorption of the medication from the injection site. The time to steady state is controlled by “flip-flop” pharmacokinetics. In this kind of pharmacokinetics, which is observed with all LAIs, the absorption rate from the injection site is lower than the elimination rate.1
Cleton et al2 reported the pharmacokinetics of paliperidone palmitate for deltoid and gluteal injection sites. By combining the median data for the deltoid injection route from the article by Cleton et al2 and the dosing from Mr. B’s case, I created a model using superposition of a sixth-degree polynomial fitted to the single dose data. Gluteal injections were not included because their increased complexity is beyond the scope of this article, but the time to maximum concentration (gluteal > deltoid) and peak concentration (deltoid > gluteal) are different for each route. The polynomial was a good fit with the adjusted r2 = 0.976, P < .0001. This model illustrates the paliperidone serum concentrations for Mr. B and is shown in the Figure. As you can see, by Day 9, the serum concentrations had reached the lower limit of the expected range of 20 to 60 ng/mL, shown in the shaded region of the Figure.3
Steady state at the routine maintenance dose of 117 mg every 28 days was never reached as the medication was not sufficient to suppress Mr. B’s hallucinations, and his doses needed to be increased each month. First, Mr. B’s dose was increased to 156 mg and then to the maximum recommended dose of 234 mg every 28 days. Steady state can be considered to have been achieved when 90% of the final steady state is reached after 3.3 half-lives. Because of the flip-flop pharmacokinetics, the important half-life is the absorption half-life of approximately 40 days or 132 days at the same dose. In Mr. B’s case, this was Day 221, where the trough concentration was 35 ng/mL. However, this regimen was still inadequate because he had breakthrough symptoms prior to the next injection.
By decreasing the injection interval from 28 days to 21 days, the concentrations will increase to a new steady state. This will take the same 132 days. With the reduced injection frequency of 21 days, 7 injections will have been given prior to reaching the new steady state. Steady state is not dependent on the number of injections, but only on the absorption half-life. This new steady state trough is substantially higher at 52 ng/mL, but still in the expected range for commonly used doses. Because Mr. B’s hallucinations only appeared at the end of the dosing interval, it is reasonable to expect that his new regimen would be successful in suppressing his hallucinations. However, monitoring for peak-related adverse effects is essential. Based upon controlled clinical trials, the potential dose-related adverse effects of paliperidone include akathisia, other extrapyramidal symptoms, weight gain, and QTc prolongation.
Continue to: Would monitoring a patient's paliperidone serum concentrations be useful?
Would monitoring a patient’s paliperidone serum concentrations be useful? Currently, measuring an individual’s paliperidone serum concentration is generally considered unwarranted.3,4 One of the major reasons is a lack of appropriately designed studies to determine a therapeutic range.5 Flexible dose designs, commonly used in registration studies, cloud the relationships between concentration, time, response, and adverse effects. There are additional problems that are the result of diagnostic heterogeneity and placebo responders. A well-designed study to determine the therapeutic range would have ≥1 fixed dose groups and be diagnostically homogeneous. There are currently only a limited number of clinical laboratories that have implemented suitable assays.
Given the lack of knowledge of a therapeutic range, assured knowledge of nonadherence to LAIs, and the absence of significant drug interactions for paliperidone, there remain a few reasonable justifications for obtaining a patient’s paliperidone serum concentration (Table). If the patient had a good response with mild adverse effects, there is no reason to obtain a paliperidone serum concentration or make any change in the medication or dose. However, if the patient had a good response accompanied by moderate or severe adverse effects, or the patient has a poor response, then obtaining the paliperidone serum concentration could help determine an appropriate course of action.
CASE CONTINUED
After the second dose at the increased frequency on Day 252, the paliperidone serum concentration was maintained above 40 ng/mL. Mr. B continued to tolerate the LAI well and no longer reported any breakthrough hallucinations.
Related Resources
- ARUP Laboratories. Paliperidone, serum or plasma. http://ltd.aruplab.com/tests/pub/2007949.
- LabCorp. Paliperidone Paliperidone (as 9-hydroxyrisperidone), serum or plasma. https://www.labcorp.com/test-menu/38351/paliperidone-as-9-hydroxyrisperidone-serum-or-plasma.
- Janssen Scientific Affairs. Educational dose illustrator. http://www.educationaldoseillustrator.com.
Drug Brand Names
Paliperidone palmitate • Invega Sustenna
Risperidone • Risperdal
1. Jann MW, Ereshefsky L, Saklad SR. Clinical pharmacokinetics of the depot antipsychotics. Clin Pharmacokinet. 1985;10(4):315-333.
2. Cleton A, Rossenu S, Crauwels H, et al. A single-dose, open-label, parallel, randomized, dose-proportionality study of paliperidone after intramuscular injections of paliperidone palmitate in the deltoid or gluteal muscle in patients with schizophrenia. J Clin Pharmacol. 2014;54(9):1048-1057.
3. Taylor D, Paton C, Kapur S. The Maudsley prescribing guidelines in psychiatry. 12th ed. Oxford, UK: John Wiley & Sons, Ltd.; 2015:1-10.
4. Hiemke C, Baumann P, Bergemann N, et al. AGNP consensus guidelines for therapeutic drug monitoring in psychiatry: update 2011. Pharmacopsychiatry. 2011;44(6):195-235.
5. Lopez LV, Kane JM. Plasma levels of second-generation antipsychotics and clinical response in acute psychosis: a review of the literature. Schizophr Res. 2013;147(2-3):368-374.
1. Jann MW, Ereshefsky L, Saklad SR. Clinical pharmacokinetics of the depot antipsychotics. Clin Pharmacokinet. 1985;10(4):315-333.
2. Cleton A, Rossenu S, Crauwels H, et al. A single-dose, open-label, parallel, randomized, dose-proportionality study of paliperidone after intramuscular injections of paliperidone palmitate in the deltoid or gluteal muscle in patients with schizophrenia. J Clin Pharmacol. 2014;54(9):1048-1057.
3. Taylor D, Paton C, Kapur S. The Maudsley prescribing guidelines in psychiatry. 12th ed. Oxford, UK: John Wiley & Sons, Ltd.; 2015:1-10.
4. Hiemke C, Baumann P, Bergemann N, et al. AGNP consensus guidelines for therapeutic drug monitoring in psychiatry: update 2011. Pharmacopsychiatry. 2011;44(6):195-235.
5. Lopez LV, Kane JM. Plasma levels of second-generation antipsychotics and clinical response in acute psychosis: a review of the literature. Schizophr Res. 2013;147(2-3):368-374.
Making sense of CYP2D6 and CYP1A2 genotype vs phenotype

The clinical response to the same dose of a drug may vary among individuals. Cytochrome P450 (CYP) 2D6 and 1A2 are enzymes that metabolize many psychotropic medications. Genetic variations in these enzymes may cause changes in their activity and result in differences in effectiveness and adverse effects. Although pharmacogenetic testing is available for CYP2D6 and CYP1A2, interpretation and clinical application of the results may not be straightforward.
Genetic variations in CYP450 enzymes determine enzymatic activity, which can have a large effect on drug levels, efficacy, and toxicity. However, there are many other important factors that clinicians should consider when trying to predict the effects of medications. While clinicians often focus on a patient’s genotype, this only provides information on a chromosomal level, and this information never changes. In contrast, a patient’s phenotype, or status of metabolism, is subject to change throughout the patient’s life.
Many circumstances influence phenotype, including the use of medications that induce or inhibit CYP450 enzymes, environmental factors, and comorbidities. Phenoconversion occurs when these factors result in a phenotype that is different from that predicted by genotype. Because of the possibility of phenoconversion, knowing a patient’s genotype may be of limited value in making clinical decisions. This article provides guidance on interpreting both the genotype and phenotype of CYP2D6 and CYP1A2. Case 1 and Case 2 illustrate these concepts.

CYP2D6
The enzyme activity of CYP2D6 varies among individuals and may include no activity, decreased activity, normal activity, or increased activity. After obtaining the genotype, the activity level of the CYP2D6 alleles may be determined. The frequency with which certain alleles occur varies with ancestry. More than 100 allelic variants and subvariants have been discovered, and new alleles are continuing to be discovered.1Table 12 lists some of the most common CYP2D6 alleles.

Based on the CYP2D6 enzyme activity determined from the alleles, 4 “traditional” phenotypes can be predicted from the genotype (Table 22). The 7-category phenotypes reported by some laboratory companies provide a more explicit method for reporting phenotypes.

Evidence suggests that, unlike most other CYP450 enzymes, CYP2D6 is not very susceptible to enzyme induction.2 Thus, genetics, rather than drug therapy, accounts for most ultra-rapid CYP2D6 metabolizers. CYP2D6 can be inhibited by the use of medications (Table 32-5) and/or substrates (Table 42,6). Similar to inhibitors, substrates may be saturating high affinity-low capacity enzymes such as CYP2D6, resulting in phenoconversion to poor metabolizers. However, this is unlikely to be the case for substrates of low affinity-high capacity enzymes such as CYP3A4.7 Ultimately, substrates and/or inhibitors of CYP2D6 may result in a phenotype that does not correspond to genotype.

Phenoconversion
Genotyping may not reflect the true prevalence of the CYP2D6 poor metabolizer phenotype when using multiple medications that are substrates and/or inhibitors of CYP2D6.8 In the presence of strong CYP2D6 inhibitors, up to 80% of individuals with a non-poor metabolizer genotype are converted to a poor metabolizer phenotype.8 While the phenotype provides a clearer representation of metabolism status than genotype, this information may not always be available.
Continue to: Determining CYP2D6 phenotype
Determining CYP2D6 phenotype
Risperidone and venlafaxine levels are useful tools for predicting CYP2D6 phenotype.3,8 When a risperidone level is ordered, the results include a risperidone level and a 9-hydroxyrisperidone level. The active metabolite of risperidone is 9-hydroxyrisperidone (
- Ultra-rapid metabolizer: 0.03 (0.02 to 0.06)
- Extensive metabolizer: 0.08 (0.04 to 0.17)
- Intermediate metabolizer: 0.56 (0.30 to 1.0)
- Poor metabolizer: 2.5 (1.8 to 4.1).
Although a R-to-9-OHR concentration ratio >1 generally indicates a poor metabolizer, it could also indicate the presence of a powerful CYP2D6 inhibitor.9
When a venlafaxine level is ordered, the results include a venlafaxine level and an O-desmethylvenlafaxine level. O-desmethylvenlafaxine (

CYP1A2
While the activity of CYP2D6 alleles is determined primarily by genetic factors and medications, the activity of CYP1A2 alleles is largely determined by environmental factors (diet, medications, disease) and genetic variability.2 Consequently, CYP1A2 genotyping may be less clinically useful than CYP2D6 genotyping. The CYP1A2 genotype–phenotype relationship incorporates the degree of allele activity (Table 52), and inducibility in the presence of environmental factors.
Continue to: CYP1A2 inhibiton
CYP1A2 inhibition
A variety of medications and environmental factors may inhibit CYP1A2.
Medications. Medications that may inhibit CYP1A2 include a
Caffeine. A significant increase in caffeine consumption can result in inhibition.3 Among non-tobacco smokers, an increase of 1 cup/d of coffee or 2 cans/d of caffeinated soda would be considered significant.3 However, tobacco smokers would require an increase of 3 cups/d of coffee or 6 cans/d of soda.
Diet. An increase in the daily dietary intake of certain vegetables for 6 days has been shown to result in inhibition.10 Apiaceous (Apiaceae or Umbelliferae) vegetables such as carrots (3/4 cup), celery (1/2 cup), dill (1 teaspoon), parsley (3 tablespoons), and parsnips (1¼ cup) can decrease CYP1A2 activity by approximately 13% to 25%. Allium (Liliaceae) vegetables, such as garlic, leeks, and onions, have no effect on CYP1A2 activity.
Infection. Pneumonia, upper respiratory infections with fever, pyelonephritis or appendicitis, or inflammation are suspected to decrease CYP1A2 activity.8
Continue to: CYP1A2 induction
CYP1A2 induction
A variety of medications and environmental factors may induce CYP1A2.
Medications. Certain medications may induce CYP1A2, including c
Cigarette smoking. A significant increase in smoking after 1 to 3 weeks may decrease drug levels, whereas a significant decrease in smoking after 1 to 3 weeks may result in elevated drug levels.3 Nicotine is not the causative agent of induction, but rather hydrocarbons found in cigarette smoke.11
Diet. An increase in daily dietary intake of certain vegetables for 6 days has been shown to result in induction.3 Brassica (Cruciferae) vegetables such as broccoli (2 cups), cauliflower (1 cup), cabbage (1 cup), and radish sprouts (1/2 cup) have been found to increase CYP1A2 activity by 18% to 37%.10 Grilled meat also plays a role in induction.10
Related Resource
- Ellingrod VL, Ward KM. Using pharmacogenetics guidelines when prescribing: What's available. Current Psychiatry. 2018;17(1):43-46.
Drug Brand Names
Amiodarone • Cordarone, Pacerone
Amitriptyline • Elavil, Endep
Aripiprazole • Abilify
Asenapine • Saphris
Atazanavir • Reyataz
Brexpiprazole • Rexulti
Bupropion • Wellbutrin, Zyban
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Chloroquine • Aralen
Cinacalcet • Sensipar
Ciprofloxacin • Cipro
Citalopram • Celexa
Clozapine • Clozaril
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Diphenhydramine • Benadryl
Doxepin • Silenor
Duloxetine • Cymbalta
Escitalopram • Lexapro
Ethinyl estradiol • Estinyl
Fluoxetine • Prozac
Fluvoxamine • Luvox
Haloperidol • Haldol
Iloperidone • Fanapt
Imatinib • Gleevec
Imipramine • Tofranil
Mirtazapine • Remeron
Nortriptyline • Pamelor
Olanzapine • Zyprexa
Paliperidone • Invega
Paroxetine • Paxil
Perphenazine • Trilafon
Phenytoin • Dilantin
Pimavanserin • Nuplazid
Primidone • Mysoline
Quetiapine • Seroquel
Quinidine • Cardioquin
Rifampin • Rifadin
Risperidone • Risperdal
Sertraline • Zoloft
Terbinafine • Lamisil
Thioridazine • Mellaril
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Vilazodone • Viibryd
Vortioxetine • Trintellix
Ziprasidone • Geodon
1. Pharmacogene Variation Consoritum. CYP2D6 allele nomenclature. https://www.pharmvar.org/gene/CYP2D6. Updated May 22, 2018. Accessed June 11, 2018.
2. Mrazek D. Psychiatric pharmacogenomics. New York, NY: Oxford University Press; 2010:33,42,44,45,85.
3. Spina E, de Leon J. Clinical applications of CYP genotyping in psychiatry. J Neural Transm (Vienna). 2015;122(1):5-28.
4. Adedoyin A, Frye RF, Mauro K, et al. Chloroquine modulation of specific metabolizing enzymes activities: investigation with selective five drug cocktail. Br J Clin Pharmacol. 1998;46(3):215-219.
5. Filppula AM, Laitila J, Neuvonen PJ, et al. Potent mechanism-based inhibition of CYP3A4 by imatinib explains its liability to interact with CYP3A4 substrates. Br J Pharmacol. 2012;165(8):2787-2798.
6. U.S. National Library of Medicine. DailyMed. http://dailymed.nlm.nih.gov/dailymed/about.cfm. Accessed April 26, 2018.
7. Monte AA, Heard KJ, Campbell J, et al. The effect of CYP2D6 drug-drug interactions on hydrocodone effectiveness. Acad Emerg Med. 2014;21(8):879-885.
8. Preskorn SH, Kane CP, Lobello K, et al. Cytochrome P450 2D6 phenoconversion is common in patients being treated for depression: implications for personalized medicine. J Clin Psychiatry. 2013;74(6):614-621.
9. de Leon, J, Susce, MT, Johnson, M, et al. DNA microarray technology in the clinical environment: the AmpliChip CYP450 test for CYP2D6 and CYP2C19 genotyping. CNS Spectr. 2009;14(1):19-34.
10. Lampe JW, King IB, Li S, et al. Brassica vegetables increase and apiaceous vegetables decrease cytochrome P450 1A2 activity in humans: changes in caffeine metabolite ratios in response to controlled vegetable diets. Carcinogenesis. 2000;21(6):1157-1162.
11. Zevin S, Benowitz NL. Drug interaction with tobacco smoking. An update. Clin Pharmacokinet. 1999;36(3):425-438.
The clinical response to the same dose of a drug may vary among individuals. Cytochrome P450 (CYP) 2D6 and 1A2 are enzymes that metabolize many psychotropic medications. Genetic variations in these enzymes may cause changes in their activity and result in differences in effectiveness and adverse effects. Although pharmacogenetic testing is available for CYP2D6 and CYP1A2, interpretation and clinical application of the results may not be straightforward.
Genetic variations in CYP450 enzymes determine enzymatic activity, which can have a large effect on drug levels, efficacy, and toxicity. However, there are many other important factors that clinicians should consider when trying to predict the effects of medications. While clinicians often focus on a patient’s genotype, this only provides information on a chromosomal level, and this information never changes. In contrast, a patient’s phenotype, or status of metabolism, is subject to change throughout the patient’s life.
Many circumstances influence phenotype, including the use of medications that induce or inhibit CYP450 enzymes, environmental factors, and comorbidities. Phenoconversion occurs when these factors result in a phenotype that is different from that predicted by genotype. Because of the possibility of phenoconversion, knowing a patient’s genotype may be of limited value in making clinical decisions. This article provides guidance on interpreting both the genotype and phenotype of CYP2D6 and CYP1A2. Case 1 and Case 2 illustrate these concepts.
CYP2D6
The enzyme activity of CYP2D6 varies among individuals and may include no activity, decreased activity, normal activity, or increased activity. After obtaining the genotype, the activity level of the CYP2D6 alleles may be determined. The frequency with which certain alleles occur varies with ancestry. More than 100 allelic variants and subvariants have been discovered, and new alleles are continuing to be discovered.1Table 12 lists some of the most common CYP2D6 alleles.
Based on the CYP2D6 enzyme activity determined from the alleles, 4 “traditional” phenotypes can be predicted from the genotype (Table 22). The 7-category phenotypes reported by some laboratory companies provide a more explicit method for reporting phenotypes.
Evidence suggests that, unlike most other CYP450 enzymes, CYP2D6 is not very susceptible to enzyme induction.2 Thus, genetics, rather than drug therapy, accounts for most ultra-rapid CYP2D6 metabolizers. CYP2D6 can be inhibited by the use of medications (Table 32-5) and/or substrates (Table 42,6). Similar to inhibitors, substrates may be saturating high affinity-low capacity enzymes such as CYP2D6, resulting in phenoconversion to poor metabolizers. However, this is unlikely to be the case for substrates of low affinity-high capacity enzymes such as CYP3A4.7 Ultimately, substrates and/or inhibitors of CYP2D6 may result in a phenotype that does not correspond to genotype.
Phenoconversion
Genotyping may not reflect the true prevalence of the CYP2D6 poor metabolizer phenotype when using multiple medications that are substrates and/or inhibitors of CYP2D6.8 In the presence of strong CYP2D6 inhibitors, up to 80% of individuals with a non-poor metabolizer genotype are converted to a poor metabolizer phenotype.8 While the phenotype provides a clearer representation of metabolism status than genotype, this information may not always be available.
Continue to: Determining CYP2D6 phenotype
Determining CYP2D6 phenotype
Risperidone and venlafaxine levels are useful tools for predicting CYP2D6 phenotype.3,8 When a risperidone level is ordered, the results include a risperidone level and a 9-hydroxyrisperidone level. The active metabolite of risperidone is 9-hydroxyrisperidone (
- Ultra-rapid metabolizer: 0.03 (0.02 to 0.06)
- Extensive metabolizer: 0.08 (0.04 to 0.17)
- Intermediate metabolizer: 0.56 (0.30 to 1.0)
- Poor metabolizer: 2.5 (1.8 to 4.1).
Although a R-to-9-OHR concentration ratio >1 generally indicates a poor metabolizer, it could also indicate the presence of a powerful CYP2D6 inhibitor.9
When a venlafaxine level is ordered, the results include a venlafaxine level and an O-desmethylvenlafaxine level. O-desmethylvenlafaxine (
CYP1A2
While the activity of CYP2D6 alleles is determined primarily by genetic factors and medications, the activity of CYP1A2 alleles is largely determined by environmental factors (diet, medications, disease) and genetic variability.2 Consequently, CYP1A2 genotyping may be less clinically useful than CYP2D6 genotyping. The CYP1A2 genotype–phenotype relationship incorporates the degree of allele activity (Table 52), and inducibility in the presence of environmental factors.
Continue to: CYP1A2 inhibiton
CYP1A2 inhibition
A variety of medications and environmental factors may inhibit CYP1A2.
Medications. Medications that may inhibit CYP1A2 include a
Caffeine. A significant increase in caffeine consumption can result in inhibition.3 Among non-tobacco smokers, an increase of 1 cup/d of coffee or 2 cans/d of caffeinated soda would be considered significant.3 However, tobacco smokers would require an increase of 3 cups/d of coffee or 6 cans/d of soda.
Diet. An increase in the daily dietary intake of certain vegetables for 6 days has been shown to result in inhibition.10 Apiaceous (Apiaceae or Umbelliferae) vegetables such as carrots (3/4 cup), celery (1/2 cup), dill (1 teaspoon), parsley (3 tablespoons), and parsnips (1¼ cup) can decrease CYP1A2 activity by approximately 13% to 25%. Allium (Liliaceae) vegetables, such as garlic, leeks, and onions, have no effect on CYP1A2 activity.
Infection. Pneumonia, upper respiratory infections with fever, pyelonephritis or appendicitis, or inflammation are suspected to decrease CYP1A2 activity.8
Continue to: CYP1A2 induction
CYP1A2 induction
A variety of medications and environmental factors may induce CYP1A2.
Medications. Certain medications may induce CYP1A2, including c
Cigarette smoking. A significant increase in smoking after 1 to 3 weeks may decrease drug levels, whereas a significant decrease in smoking after 1 to 3 weeks may result in elevated drug levels.3 Nicotine is not the causative agent of induction, but rather hydrocarbons found in cigarette smoke.11
Diet. An increase in daily dietary intake of certain vegetables for 6 days has been shown to result in induction.3 Brassica (Cruciferae) vegetables such as broccoli (2 cups), cauliflower (1 cup), cabbage (1 cup), and radish sprouts (1/2 cup) have been found to increase CYP1A2 activity by 18% to 37%.10 Grilled meat also plays a role in induction.10
Related Resource
- Ellingrod VL, Ward KM. Using pharmacogenetics guidelines when prescribing: What's available. Current Psychiatry. 2018;17(1):43-46.
Drug Brand Names
Amiodarone • Cordarone, Pacerone
Amitriptyline • Elavil, Endep
Aripiprazole • Abilify
Asenapine • Saphris
Atazanavir • Reyataz
Brexpiprazole • Rexulti
Bupropion • Wellbutrin, Zyban
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Chloroquine • Aralen
Cinacalcet • Sensipar
Ciprofloxacin • Cipro
Citalopram • Celexa
Clozapine • Clozaril
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Diphenhydramine • Benadryl
Doxepin • Silenor
Duloxetine • Cymbalta
Escitalopram • Lexapro
Ethinyl estradiol • Estinyl
Fluoxetine • Prozac
Fluvoxamine • Luvox
Haloperidol • Haldol
Iloperidone • Fanapt
Imatinib • Gleevec
Imipramine • Tofranil
Mirtazapine • Remeron
Nortriptyline • Pamelor
Olanzapine • Zyprexa
Paliperidone • Invega
Paroxetine • Paxil
Perphenazine • Trilafon
Phenytoin • Dilantin
Pimavanserin • Nuplazid
Primidone • Mysoline
Quetiapine • Seroquel
Quinidine • Cardioquin
Rifampin • Rifadin
Risperidone • Risperdal
Sertraline • Zoloft
Terbinafine • Lamisil
Thioridazine • Mellaril
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Vilazodone • Viibryd
Vortioxetine • Trintellix
Ziprasidone • Geodon
The clinical response to the same dose of a drug may vary among individuals. Cytochrome P450 (CYP) 2D6 and 1A2 are enzymes that metabolize many psychotropic medications. Genetic variations in these enzymes may cause changes in their activity and result in differences in effectiveness and adverse effects. Although pharmacogenetic testing is available for CYP2D6 and CYP1A2, interpretation and clinical application of the results may not be straightforward.
Genetic variations in CYP450 enzymes determine enzymatic activity, which can have a large effect on drug levels, efficacy, and toxicity. However, there are many other important factors that clinicians should consider when trying to predict the effects of medications. While clinicians often focus on a patient’s genotype, this only provides information on a chromosomal level, and this information never changes. In contrast, a patient’s phenotype, or status of metabolism, is subject to change throughout the patient’s life.
Many circumstances influence phenotype, including the use of medications that induce or inhibit CYP450 enzymes, environmental factors, and comorbidities. Phenoconversion occurs when these factors result in a phenotype that is different from that predicted by genotype. Because of the possibility of phenoconversion, knowing a patient’s genotype may be of limited value in making clinical decisions. This article provides guidance on interpreting both the genotype and phenotype of CYP2D6 and CYP1A2. Case 1 and Case 2 illustrate these concepts.
CYP2D6
The enzyme activity of CYP2D6 varies among individuals and may include no activity, decreased activity, normal activity, or increased activity. After obtaining the genotype, the activity level of the CYP2D6 alleles may be determined. The frequency with which certain alleles occur varies with ancestry. More than 100 allelic variants and subvariants have been discovered, and new alleles are continuing to be discovered.1Table 12 lists some of the most common CYP2D6 alleles.
Based on the CYP2D6 enzyme activity determined from the alleles, 4 “traditional” phenotypes can be predicted from the genotype (Table 22). The 7-category phenotypes reported by some laboratory companies provide a more explicit method for reporting phenotypes.
Evidence suggests that, unlike most other CYP450 enzymes, CYP2D6 is not very susceptible to enzyme induction.2 Thus, genetics, rather than drug therapy, accounts for most ultra-rapid CYP2D6 metabolizers. CYP2D6 can be inhibited by the use of medications (Table 32-5) and/or substrates (Table 42,6). Similar to inhibitors, substrates may be saturating high affinity-low capacity enzymes such as CYP2D6, resulting in phenoconversion to poor metabolizers. However, this is unlikely to be the case for substrates of low affinity-high capacity enzymes such as CYP3A4.7 Ultimately, substrates and/or inhibitors of CYP2D6 may result in a phenotype that does not correspond to genotype.
Phenoconversion
Genotyping may not reflect the true prevalence of the CYP2D6 poor metabolizer phenotype when using multiple medications that are substrates and/or inhibitors of CYP2D6.8 In the presence of strong CYP2D6 inhibitors, up to 80% of individuals with a non-poor metabolizer genotype are converted to a poor metabolizer phenotype.8 While the phenotype provides a clearer representation of metabolism status than genotype, this information may not always be available.
Continue to: Determining CYP2D6 phenotype
Determining CYP2D6 phenotype
Risperidone and venlafaxine levels are useful tools for predicting CYP2D6 phenotype.3,8 When a risperidone level is ordered, the results include a risperidone level and a 9-hydroxyrisperidone level. The active metabolite of risperidone is 9-hydroxyrisperidone (
- Ultra-rapid metabolizer: 0.03 (0.02 to 0.06)
- Extensive metabolizer: 0.08 (0.04 to 0.17)
- Intermediate metabolizer: 0.56 (0.30 to 1.0)
- Poor metabolizer: 2.5 (1.8 to 4.1).
Although a R-to-9-OHR concentration ratio >1 generally indicates a poor metabolizer, it could also indicate the presence of a powerful CYP2D6 inhibitor.9
When a venlafaxine level is ordered, the results include a venlafaxine level and an O-desmethylvenlafaxine level. O-desmethylvenlafaxine (
CYP1A2
While the activity of CYP2D6 alleles is determined primarily by genetic factors and medications, the activity of CYP1A2 alleles is largely determined by environmental factors (diet, medications, disease) and genetic variability.2 Consequently, CYP1A2 genotyping may be less clinically useful than CYP2D6 genotyping. The CYP1A2 genotype–phenotype relationship incorporates the degree of allele activity (Table 52), and inducibility in the presence of environmental factors.
Continue to: CYP1A2 inhibiton
CYP1A2 inhibition
A variety of medications and environmental factors may inhibit CYP1A2.
Medications. Medications that may inhibit CYP1A2 include a
Caffeine. A significant increase in caffeine consumption can result in inhibition.3 Among non-tobacco smokers, an increase of 1 cup/d of coffee or 2 cans/d of caffeinated soda would be considered significant.3 However, tobacco smokers would require an increase of 3 cups/d of coffee or 6 cans/d of soda.
Diet. An increase in the daily dietary intake of certain vegetables for 6 days has been shown to result in inhibition.10 Apiaceous (Apiaceae or Umbelliferae) vegetables such as carrots (3/4 cup), celery (1/2 cup), dill (1 teaspoon), parsley (3 tablespoons), and parsnips (1¼ cup) can decrease CYP1A2 activity by approximately 13% to 25%. Allium (Liliaceae) vegetables, such as garlic, leeks, and onions, have no effect on CYP1A2 activity.
Infection. Pneumonia, upper respiratory infections with fever, pyelonephritis or appendicitis, or inflammation are suspected to decrease CYP1A2 activity.8
Continue to: CYP1A2 induction
CYP1A2 induction
A variety of medications and environmental factors may induce CYP1A2.
Medications. Certain medications may induce CYP1A2, including c
Cigarette smoking. A significant increase in smoking after 1 to 3 weeks may decrease drug levels, whereas a significant decrease in smoking after 1 to 3 weeks may result in elevated drug levels.3 Nicotine is not the causative agent of induction, but rather hydrocarbons found in cigarette smoke.11
Diet. An increase in daily dietary intake of certain vegetables for 6 days has been shown to result in induction.3 Brassica (Cruciferae) vegetables such as broccoli (2 cups), cauliflower (1 cup), cabbage (1 cup), and radish sprouts (1/2 cup) have been found to increase CYP1A2 activity by 18% to 37%.10 Grilled meat also plays a role in induction.10
Related Resource
- Ellingrod VL, Ward KM. Using pharmacogenetics guidelines when prescribing: What's available. Current Psychiatry. 2018;17(1):43-46.
Drug Brand Names
Amiodarone • Cordarone, Pacerone
Amitriptyline • Elavil, Endep
Aripiprazole • Abilify
Asenapine • Saphris
Atazanavir • Reyataz
Brexpiprazole • Rexulti
Bupropion • Wellbutrin, Zyban
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Chloroquine • Aralen
Cinacalcet • Sensipar
Ciprofloxacin • Cipro
Citalopram • Celexa
Clozapine • Clozaril
Desipramine • Norpramin
Desvenlafaxine • Pristiq
Diphenhydramine • Benadryl
Doxepin • Silenor
Duloxetine • Cymbalta
Escitalopram • Lexapro
Ethinyl estradiol • Estinyl
Fluoxetine • Prozac
Fluvoxamine • Luvox
Haloperidol • Haldol
Iloperidone • Fanapt
Imatinib • Gleevec
Imipramine • Tofranil
Mirtazapine • Remeron
Nortriptyline • Pamelor
Olanzapine • Zyprexa
Paliperidone • Invega
Paroxetine • Paxil
Perphenazine • Trilafon
Phenytoin • Dilantin
Pimavanserin • Nuplazid
Primidone • Mysoline
Quetiapine • Seroquel
Quinidine • Cardioquin
Rifampin • Rifadin
Risperidone • Risperdal
Sertraline • Zoloft
Terbinafine • Lamisil
Thioridazine • Mellaril
Trazodone • Desyrel, Oleptro
Venlafaxine • Effexor
Vilazodone • Viibryd
Vortioxetine • Trintellix
Ziprasidone • Geodon
1. Pharmacogene Variation Consoritum. CYP2D6 allele nomenclature. https://www.pharmvar.org/gene/CYP2D6. Updated May 22, 2018. Accessed June 11, 2018.
2. Mrazek D. Psychiatric pharmacogenomics. New York, NY: Oxford University Press; 2010:33,42,44,45,85.
3. Spina E, de Leon J. Clinical applications of CYP genotyping in psychiatry. J Neural Transm (Vienna). 2015;122(1):5-28.
4. Adedoyin A, Frye RF, Mauro K, et al. Chloroquine modulation of specific metabolizing enzymes activities: investigation with selective five drug cocktail. Br J Clin Pharmacol. 1998;46(3):215-219.
5. Filppula AM, Laitila J, Neuvonen PJ, et al. Potent mechanism-based inhibition of CYP3A4 by imatinib explains its liability to interact with CYP3A4 substrates. Br J Pharmacol. 2012;165(8):2787-2798.
6. U.S. National Library of Medicine. DailyMed. http://dailymed.nlm.nih.gov/dailymed/about.cfm. Accessed April 26, 2018.
7. Monte AA, Heard KJ, Campbell J, et al. The effect of CYP2D6 drug-drug interactions on hydrocodone effectiveness. Acad Emerg Med. 2014;21(8):879-885.
8. Preskorn SH, Kane CP, Lobello K, et al. Cytochrome P450 2D6 phenoconversion is common in patients being treated for depression: implications for personalized medicine. J Clin Psychiatry. 2013;74(6):614-621.
9. de Leon, J, Susce, MT, Johnson, M, et al. DNA microarray technology in the clinical environment: the AmpliChip CYP450 test for CYP2D6 and CYP2C19 genotyping. CNS Spectr. 2009;14(1):19-34.
10. Lampe JW, King IB, Li S, et al. Brassica vegetables increase and apiaceous vegetables decrease cytochrome P450 1A2 activity in humans: changes in caffeine metabolite ratios in response to controlled vegetable diets. Carcinogenesis. 2000;21(6):1157-1162.
11. Zevin S, Benowitz NL. Drug interaction with tobacco smoking. An update. Clin Pharmacokinet. 1999;36(3):425-438.
1. Pharmacogene Variation Consoritum. CYP2D6 allele nomenclature. https://www.pharmvar.org/gene/CYP2D6. Updated May 22, 2018. Accessed June 11, 2018.
2. Mrazek D. Psychiatric pharmacogenomics. New York, NY: Oxford University Press; 2010:33,42,44,45,85.
3. Spina E, de Leon J. Clinical applications of CYP genotyping in psychiatry. J Neural Transm (Vienna). 2015;122(1):5-28.
4. Adedoyin A, Frye RF, Mauro K, et al. Chloroquine modulation of specific metabolizing enzymes activities: investigation with selective five drug cocktail. Br J Clin Pharmacol. 1998;46(3):215-219.
5. Filppula AM, Laitila J, Neuvonen PJ, et al. Potent mechanism-based inhibition of CYP3A4 by imatinib explains its liability to interact with CYP3A4 substrates. Br J Pharmacol. 2012;165(8):2787-2798.
6. U.S. National Library of Medicine. DailyMed. http://dailymed.nlm.nih.gov/dailymed/about.cfm. Accessed April 26, 2018.
7. Monte AA, Heard KJ, Campbell J, et al. The effect of CYP2D6 drug-drug interactions on hydrocodone effectiveness. Acad Emerg Med. 2014;21(8):879-885.
8. Preskorn SH, Kane CP, Lobello K, et al. Cytochrome P450 2D6 phenoconversion is common in patients being treated for depression: implications for personalized medicine. J Clin Psychiatry. 2013;74(6):614-621.
9. de Leon, J, Susce, MT, Johnson, M, et al. DNA microarray technology in the clinical environment: the AmpliChip CYP450 test for CYP2D6 and CYP2C19 genotyping. CNS Spectr. 2009;14(1):19-34.
10. Lampe JW, King IB, Li S, et al. Brassica vegetables increase and apiaceous vegetables decrease cytochrome P450 1A2 activity in humans: changes in caffeine metabolite ratios in response to controlled vegetable diets. Carcinogenesis. 2000;21(6):1157-1162.
11. Zevin S, Benowitz NL. Drug interaction with tobacco smoking. An update. Clin Pharmacokinet. 1999;36(3):425-438.
Treating psychosis in patients with HIV/AIDS

Mr. S, age 56, has human immunodeficiency virus (HIV) and schizoaffective disorder. He presents to your clinic with increased auditory hallucinations, disorganized behavior, and worsened tremors that have begun to seriously disrupt his daily life. Mr. S is prescribed risperidone; however, he reports that he has not been taking it lately due to the tremor despite being controlled on his medication regimen for nearly 1 year. His Abnormal Involuntary Movement Scale (AIMS) score reveals an increased wrist rigidity compared with previous clinic visits. Mr. S has a 40 pack-year history of smoking and history of IV drug use. Furthermore, he has a medical history of type 2 diabetes mellitus, hypertension, and hyperlipidemia.
His medication regimen includes atazanavir sulfate, 200 mg/d, ritonavir, 100 mg/d, efavirenz/emtricitabine/tenofovir disoproxil fumarate, 600/200/300 mg/d, risperidone, 6 mg/d, bupropion extended-release, 300 mg/d, gabapentin, 600 mg/d, amlodipine, 5 mg/d, pravastatin, 40 mg/d, metformin, 1000 mg twice daily, and glipizide, 10 mg twice daily. Today, his laboratory findings show that his CD4 count is 405 cell/mm3, and his viral load is <40 copies/mL, indicating his HIV is well managed. A hepatitis C virus antibody test result is negative and serum creatinine level is 1.0 mg/dL. Total cholesterol is 212 mg/dL, high-density lipoprotein cholesterol is 43 mg/dL, low-density lipoprotein cholesterol is 121 mg/dL, and triglycerides level is 238 mg/dL. Electrocardiography reveals a QTc interval of 426 ms. Mr. S’s blood pressure is 105/65 mm Hg. Based on this clinic visit, the treatment team decides to change Mr. S’s antipsychotic.
Psychiatric illness and HIV/AIDS
There is a strong link between mental illness and HIV/AIDS; 50% or more of patients with HIV/AIDS have a comorbid psychiatric disorder.1 The prevalence of mental illness in patients with HIV/AIDS is reported to be 8 times higher than in those without HIV/AIDS.2 Depression, bipolar disorder, anxiety disorders, delirium, substance abuse, and schizophrenia have all been identified in persons receiving highly active antiretroviral therapy (HAART). Patients with HIV/AIDS and psychiatric illness have a decreased quality of life, poor adherence to medications, faster disease progression, and increased mortality. Care of these individuals is complicated by the stigma of HIV/AIDS and the prevalence of the illness in underserved populations, as well as the need for complex medication regimens and the possibility of drug–drug interactions (DDIs).1,2 If left untreated, psychiatric illness in patients with HIV/AIDS may lead to further transmission of HIV as a result of patients engaging in high-risk behaviors, along with poor adherence to HAART.3
Individuals diagnosed with schizophrenia, schizoaffective disorder, and bipolar disorder are at greater risk for HIV infection.3 Patients with HIV/AIDS with primary psychosis may have poor medication adherence rates due to illness-related confusion or paranoia about medications. Furthermore, they may lack the resources to manage the complications and stress related to living with HIV/AIDS.
New-onset, or secondary psychosis, has been reported in individuals with late-stage HIV/AIDS with CD4 counts <200 who have not been diagnosed with a psychotic disorder previously.3 These patients may experience more persecutory and grandiose delusions rather than hallucinations. Neuropsychiatric symptoms in patients with HIV/AIDS may be due to the presence of HIV or other infections in the CNS, tumors, or other inflammatory illnesses. Medications that have been implicated in neuropsychiatric symptoms include efavirenz, rilpivirine, and other HAART regimens; interferon; metoclopramide; corticosteroids; muscle relaxants; and clonidine. It is possible that symptoms may continue even after the medications are discontinued.3
Antipsychotics remain the treatment of choice for psychosis in HIV/AIDS, regardless of the cause of the symptoms. Many factors must be taken into consideration when choosing an antipsychotic, such as DDIs, adverse effect profiles, patient history of antipsychotic use, cost, and patient preference. Here we focus primarily on DDIs and adverse effect profiles.
When treating psychosis in patients with HIV/AIDS, it is crucial to consider potential DDIs. Many antipsychotics and antiretroviral medications utilize cytochrome P450 (CYP) enzymes for their metabolism. The CYP enzyme system is responsible for the oxidative reactions that constitute the phase I reactions necessary for the metabolism of most drugs. Inhibition and induction of CYP enzymes are among the most common causes of pharmacokinetic DDIs. Antipsychotics are predominately metabolized by CYP3A4, CYP1A2, and CYP2D6.4
Continue to: The DDIs arise because...
The DDIs arise because many antiretroviral medications inhibit, or in some cases, induce, these CYP enzymes, thereby altering substrate-drug metabolism. Inhibiting a CYP enzyme pathway can decrease substrate-drug clearance and lead to increased levels of that drug. This, in turn, can cause an increased risk of adverse effects, such as extrapyramidal symptoms (EPS) or QTc prolongation, which are both types of pharmacodynamic DDIs.4-28 However, because antipsychotics often have more than one pathway of metabolism, it can be challenging to understand the full effect of CYP-related DDIs. Furthermore, CYP enzyme inducers can decrease drug levels, and in the case of antipsychotics, lead to subtherapeutic responses.
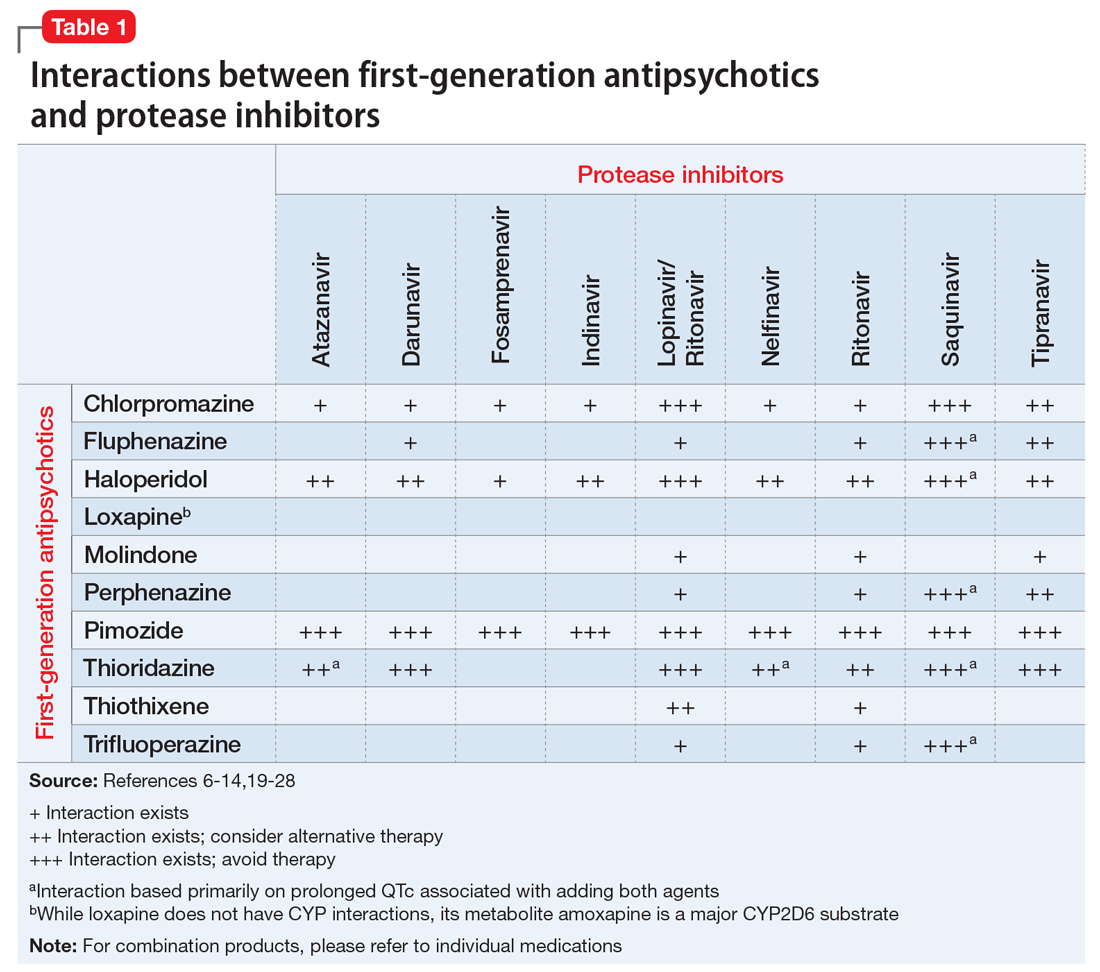
Table 1,6-14,19-28 Table 2,15-28 Table 3,6-14,19-28 and Table 415-28 list many of the known CYP enzyme-related DDIs that may occur with combination antipsychotic and antiretroviral medication therapy and aim to predict CYP induction or inhibition based on a particular combination. The following antiretroviral medications do not have any CYP-related interactions and therefore are not included in the Tables: abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir disoproxil, zidovudine, enfuvirtide, maraviroc, and raltegravir.
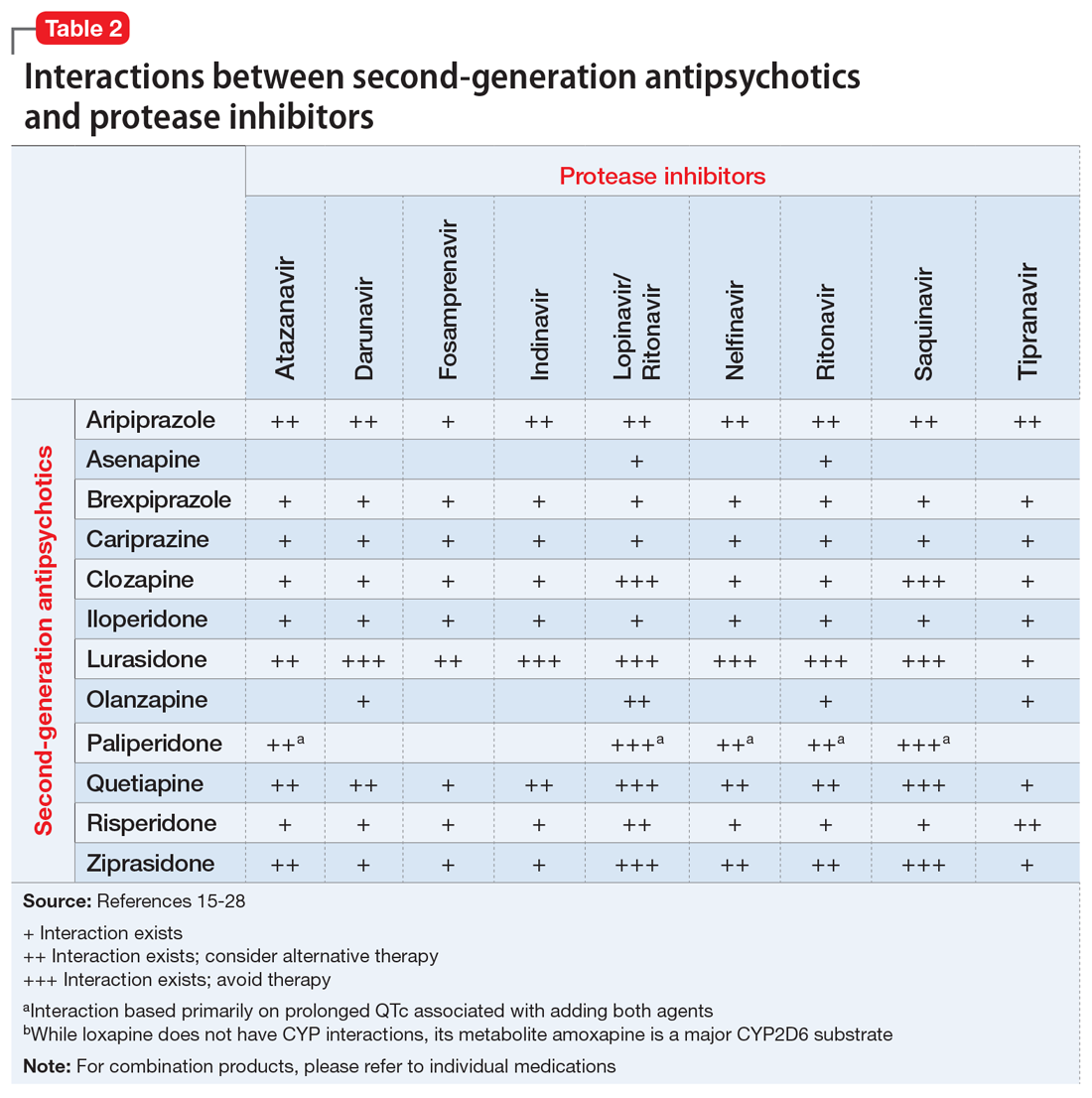
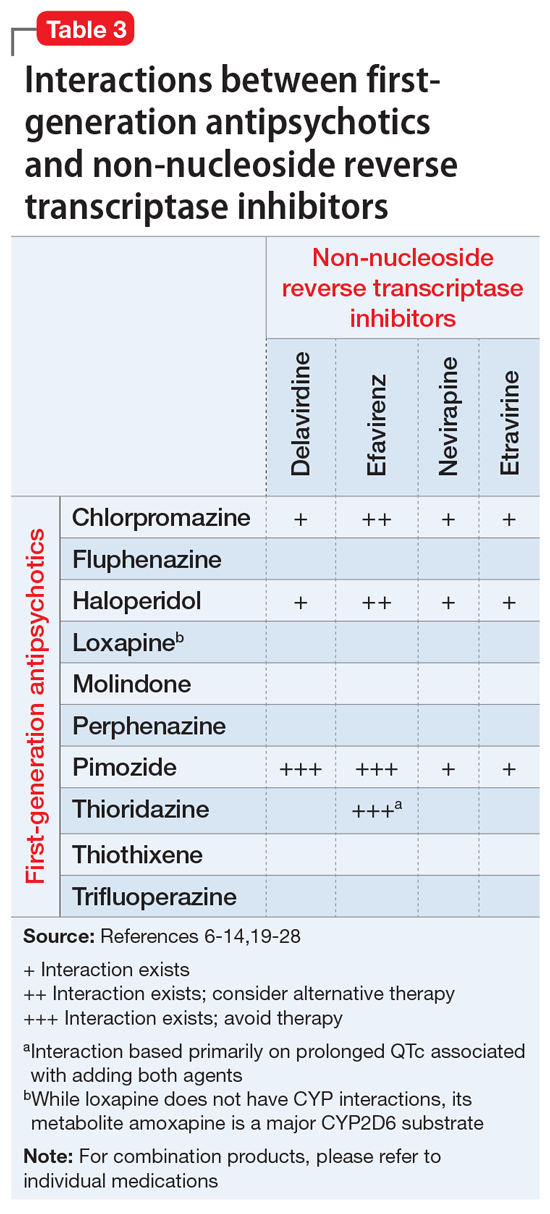
These Tables include the risk ratings for all D-rated (consider alternative therapy) and X-rated (avoid therapy) combinations. The majority of D-rated interactions are caused by CYP inhibition or induction that could potentially lead to altered antipsychotic levels. The majority of X-rated interactions are caused by increased QTc prolongation that may or may not be due to CYP-related DDIs. For example, paliperidone is not believed to be affected by the CYP enzyme system, but it does present a high risk of QTc prolongation on its own. When combined with an antiretroviral that also has a high risk of QTc prolongation, such as lopinavir, then the risk further increases.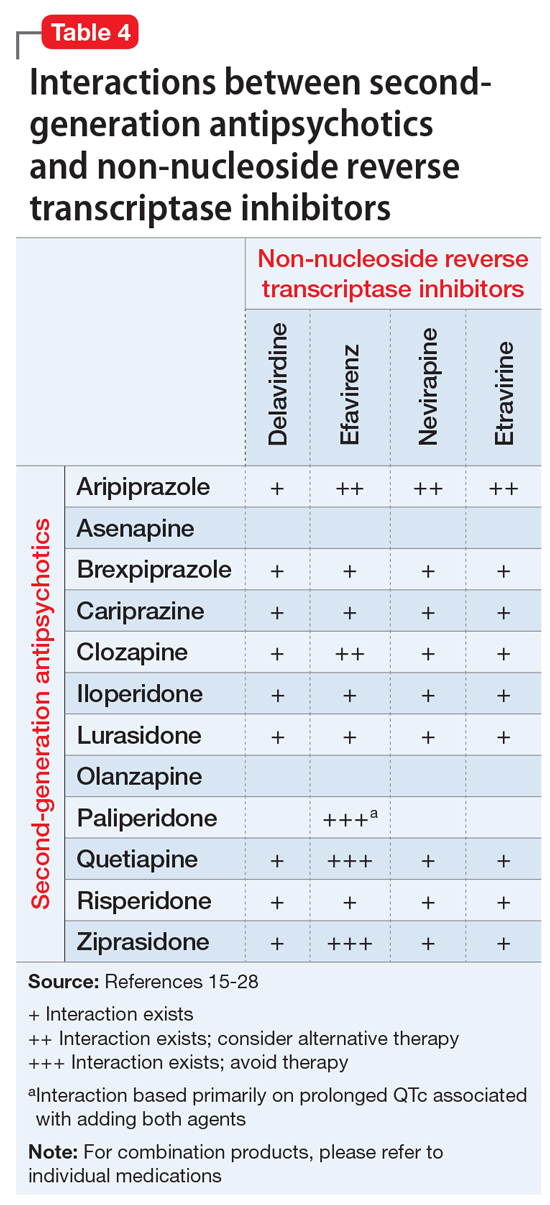
Non-nucleoside reverse transcriptase inhibitors and protease inhibitors (PIs) are the antiretroviral medications most likely to cause DDIs with antipsychotics. Other antiretroviral classes, such as nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs), fusion inhibitors, chemokine receptor 5 inhibitors, and integrase inhibitors, are not associated with CYP-related DDIs.19-28 For the most part, the severity of the CYP-related DDIs have not been well studied; therefore, most recommendations call for closer patient monitoring when combining antiretroviral medications and antipsychotics.6-18 The goal is to monitor for any changes in medication efficacy or adverse effects.
Continue to: Consider adverse effect profiles
Consider adverse effect profiles
When selecting an antipsychotic agent for a patient receiving HIV therapy, also consider adverse effect profiles. The emergence of adverse effects can greatly impact patients’ quality of life, leading to consequences of medication nonadherence and exacerbation of mental illness.
Extrapyramidal symptoms. Patients with HIV have a higher sensitivity to treatment-emergent EPS from antipsychotics.2 This sensitivity is generally thought to arise from the involvement of HIV on the basal ganglia. Historically, psychotic symptoms in HIV have been managed with second-generation antipsychotics (SGAs) at the lowest effective dose because these medications are less likely to cause EPS.1,29 The antipsychotic with the lowest rate of EPS is clozapine, followed by quetiapine, olanzapine, ziprasidone, and aripiprazole. Conversely, high-potency first-generation antipsychotics (FGAs) have the highest rates of EPS, followed by intermediate-potency FGAs and risperidone.30
Metabolic disturbances are another concern with concomitant antipsychotic/antiretroviral therapy. Patients with HIV who are receiving NRTIs or PIs can present with drug-induced lipodystrophy syndrome, which is associated with hyperglycemia, hyperinsulinemia, hyperlipidemia, and hypertension, and ultimately may cause metabolic syndrome.29 The prevalence of metabolic syndrome in patients receiving PI therapy has a vast range—2% to 84%—which can be attributed to inconsistent definitions, criteria, and assessment methodology.29 Use of a PI is considered to be the most prominent risk factor for developing lipodystrophy.29 Among the PIs, metabolic disturbances in regards to lipids are most often seen with lopinavir/ritonavir (LPV/r), saquinavir/ritonavir, tipranavir/ritonavir, and fosamprenavir/ritonavir.31 In comparison with LPV/r, darunavir showed improvement in lipids.32 Atazanavir (ATV) boosted with ritonavir has not shown clinically significant adverse effects on lipids.31 Additionally, amprenavir, LPV/r, and ritonavir demonstrated more glucose uptake inhibition via blockade of the glucose transporter type 4 than ATV.31 Of the NRTIs, lipodystrophy syndrome is most commonly seen with stavudine, which is used minimally in practice.2
The rates of metabolic disturbance with antipsychotic use range from 2% to 36%.2 The American Psychiatric Association recommends selecting one of the SGAs least likely to affect metabolic parameters.29 Aripiprazole and ziprasidone are associated with the lowest risk of weight gain, hyperglycemia, and hyperlipidemia. They are followed by risperidone and quetiapine, which are associated with moderate risk, and then clozapine and olanzapine, which are associated with high risk.2,30,33
Continue to: Management of metabolic adverse effects involves...
Management of metabolic adverse effects involves switching the antiretroviral agent and/or antipsychotic agent to an alternative associated with lower metabolic risk. Antipsychotics with low metabolic risk include aripiprazole, lurasidone, and ziprasidone. Lifestyle modifications are encouraged. Additionally, medication interventions, such as metformin, are also recommended in patients meeting criteria for pre-diabetes or type 2 diabetes mellitus.2 Lipid panels and metabolic parameters should be monitored periodically, according to guidelines.25,34
Bone marrow toxicity and blood dyscrasias. Lastly, consider the risk of bone marrow suppression. Patients receiving clozapine for treatment-resistant schizophrenia should be closely monitored for neutropenia and agranulocytosis. Although zidovudine is rarely used, its use is associated with adverse myelosuppressive effects, and the combination of clozapine and zidovudine could pose danger to the patient.2,35,36
CASE CONTINUED
Because Mr. S’s diagnosis of HIV puts him at a higher risk of developing EPS, and because he is already experiencing increased wrist rigidity, the treatment team decides to switch his antipsychotic therapy to an agent with a lower risk of EPS. His comorbidities, including type 2 diabetes mellitus, hypertension, and hyperlipidemia, are taken into account, and an SGA with a benign metabolic profile is considered. Aripiprazole and ziprasidone are favorable options. However, because efavirenz, ATZ, and ritonavir may cause QTc prolongation, ziprasidone, the SGA with the highest rate of QTc prolongation, is not the preferred option.
Mr. S’s SGA therapy is switched from risperidone to aripiprazole. Because potential CYP-related interactions between aripiprazole and Mr. S’s current antiretroviral therapy could lead to increased aripiprazole levels. Mr. S is started on a low dose (5 mg/d) with the goal to titrate based on response and tolerability. Increased levels of aripiprazole may increase the risk of akathisia, drowsiness, headaches, and fatigue. Mr. S is monitored closely for improvement of EPS, adverse effects of medication, and metabolic parameters. Furthermore, if the treatment team believes there is a more preferred antipsychotic for the patient that it did not prescribe because of the risk of DDIs, it may be worthwhile to consider discussing the HAART regimen with the patient’s infectious disease treatment team.
Continue to: Acknowledgements
Acknowledgements
This material is the result of work supported with resources and the use of facilities at the Chillicothe Veterans Affairs Medical Center in Chillicothe, Ohio. The contents of this paper do not represent the views of the U.S. Department of Veterans Affairs or the U.S. government.
Related Resources
- Cohen MA. HIV: How to provide compassionate care. Current Psychiatry. 2013;12(6):19-23,A,B.
- Khan AY, Zaidi SN. Reducing morbidity and mortality from common medical conditions in schizophrenia. Current Psychiatry. 2016;15(3):30-32,34-38,40.
Drug Brand Names
Abacavir • Ziagen
Amlodipine • Norvasc
Amprenavir • Agenerase
Aripiprazole • Abilify
Asenapine • Saphris
Atazanavir • Reyataz
Brexpiprazole • Rexulti
Bupropion ER • Wellbutrin SR
Cariprazine • Vraylar
Chlorpromazine • Thorazine
Clonidine • Catapres
Clozapine • Clozaril
Darunavir • Prezista
Delavirdine • Rescriptor
Didanosine • Videx EC
Efavirenz • Sustiva
Efavirenz/emtricitabine/tenofovir disoproxil fumarate • Atripla
Enfuvirtide • Fuzeon
Emtricitabine • Emtriva
Etravirine • Intelence
Fluphenazine • Prolixin
Fosamprenavir • Lexiva
Gabapentin • Neurontin
Glipizide • Glucotrol
Haloperidol • Haldol
Iloperidone • Fanapt
Indinavir • Crixivan
Lamivudine • Epivir
Lopinavir/ritonavir • Kaletra
Loxapine • Loxitane
Lurasidone • Latuda
Maraviroc • Selzentry
Metformin • Glucophage
Metoclopramide • Reglan
Molindone • Moban
Nelfinavir • Viracept
Nevirapine • Viramune
Olanzapine • Zyprexa
Paliperidone • Invega
Perphenazine • Trilafon
Pimozide • Orap
Pravastatin • Pravachol
Quetiapine • Seroquel
Raltegravir • Isentress
Rilpivirine • Edurant
Risperidone • Risperdal
Ritonavir • Norvir
Saquinavir • Invirase
Stavudine • Zerit
Tenofovir disoproxil • Viread
Thioridazine • Mellaril
Thiothixene • Navane
Tipranavir • Aptivus
Trifluoperazine • Stelazine
Zidovudine • Retrovir
Ziprasidone • Geodon
1. Freudenreich O, Goforth HW, Cozza KL, et al. Psychiatric treatment of persons with HIV/AIDS: An HIV-psychiatry consensus survey of current practices. Psychosomatics. 2010;51(6):480-488.
2. Hill L, Lee KC. Pharmacotherapy considerations in patients with HIV and psychiatric disorders: Focus on antidepressants and antipsychotics. Ann Pharmacother. 2013;47(1):75-89.
3. Watkins CC, Treisman GJ. Neuropsychiatric complications of aging with HIV. J Neurovirol. 2012;18(4):277-290.
4. Prior TI, Baker GB. Interactions between the cytochrome P450 system and the second-generation antipsychotics. J Psychiatry Neurosci. 2003;28(2):99-112.
5. Ponte ML, Keller GA, Di Girolamo G. Mechanisms of drug induced QT interval prolongation. Curr Drug Saf. 2010;5(1):44-53
6. Reyataz [package insert]. Princeton, NJ: Bristol-Myers Squibb Company; 2017.
7. Prezista [package insert]. Toronto, ON: Janssen Inc.; 2017.
8. Lexiva [package insert]. Research Triangle Park, NC: Viiv Healthcare; 2017
9. Crixivan [package insert]. Whitehouse Station, NJ; Merck; 2016.
10. Kaletra [package insert]. North Chicago, IL: AbbVie Inc; 2017
11. Viracept [package insert]. Kirkland, QC: Pfizer Canada Inc.; 201
12. Norvir tablets and oral solution [package insert]. North Chicago, IL: AbbVie Inc; 2017
13. Invirase [package insert]. South San Francisco, CA: Genentech USA, Inc.; 2016.
14. Aptivus [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals Inc.; 2016.
15. Sustiva [package insert]. Princeton, NJ: Bristol-Myers Squibb Company; 2017
16. Intelence [package insert]. Titusville, NJ: Tibotec Pharmaceuticals; 2014.
17. Viramune [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals Inc.; 2017.
18. Rescriptor [package insert]. Laval, QC: ViiV Healthcare ULC; 2013.
19. Ziagen [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2017.
20. Videx EC [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2015.
21. Emtriva [package insert]. Foster City, CA: Gilead Sciences, Inc.; 2017.
22. Epivir [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2017.
23. Zerit [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2017.
24. Viread [package insert]. Foster City, CA: Gilead Sciences, Inc.; 2017.
25. Retrovir [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2015.
26. Fuzeon [package insert]. South San Francisco, CA: Genentech USA, Inc; 2017.
27. Selzentry [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2016.
28. Isentress [package insert]. Whitehouse Station, NJ: Merck Sharp & Dohme Corp.; 2017.
29. American Psychiatry Association. Practice guidelines for treatment of patients with HIV/AIDS. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/hivaids.pdf. Published 2010. Accessed March 1, 2018.
30. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 Schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
31. Hughes PJ, Cretton-Scott E, Teague A, et al. Protease inhibitors for patients with HIV-1 infection. P T. 2011;36(6):332-336,341-345.
32. Ortiz R, Dejesus E, Khanlou H, et al. Efficacy and safety of once-daily darunavir/ritonavir versus lopinavir/ritonavir in treatment-naive HIV-1-infected patients at week 48. AIDS. 2008;22(12):1389-1397.
33. Leucht S, Cipriani A, Spineli L, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet. 2013;382(9896):951-962.
34. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013;12(9):51-54.
35. Singh D, Goodkin K. Choice of antipsychotic in HIV-infected patients. J Clin Psychiatry. 2007;68(3):479-480.
36. Max B, Sherer R. Management of the adverse effects of antiretroviral therapy and medication adherence. Clin Infect Dis. 2000;30(suppl 2):S96-S116.
Mr. S, age 56, has human immunodeficiency virus (HIV) and schizoaffective disorder. He presents to your clinic with increased auditory hallucinations, disorganized behavior, and worsened tremors that have begun to seriously disrupt his daily life. Mr. S is prescribed risperidone; however, he reports that he has not been taking it lately due to the tremor despite being controlled on his medication regimen for nearly 1 year. His Abnormal Involuntary Movement Scale (AIMS) score reveals an increased wrist rigidity compared with previous clinic visits. Mr. S has a 40 pack-year history of smoking and history of IV drug use. Furthermore, he has a medical history of type 2 diabetes mellitus, hypertension, and hyperlipidemia.
His medication regimen includes atazanavir sulfate, 200 mg/d, ritonavir, 100 mg/d, efavirenz/emtricitabine/tenofovir disoproxil fumarate, 600/200/300 mg/d, risperidone, 6 mg/d, bupropion extended-release, 300 mg/d, gabapentin, 600 mg/d, amlodipine, 5 mg/d, pravastatin, 40 mg/d, metformin, 1000 mg twice daily, and glipizide, 10 mg twice daily. Today, his laboratory findings show that his CD4 count is 405 cell/mm3, and his viral load is <40 copies/mL, indicating his HIV is well managed. A hepatitis C virus antibody test result is negative and serum creatinine level is 1.0 mg/dL. Total cholesterol is 212 mg/dL, high-density lipoprotein cholesterol is 43 mg/dL, low-density lipoprotein cholesterol is 121 mg/dL, and triglycerides level is 238 mg/dL. Electrocardiography reveals a QTc interval of 426 ms. Mr. S’s blood pressure is 105/65 mm Hg. Based on this clinic visit, the treatment team decides to change Mr. S’s antipsychotic.
Psychiatric illness and HIV/AIDS
There is a strong link between mental illness and HIV/AIDS; 50% or more of patients with HIV/AIDS have a comorbid psychiatric disorder.1 The prevalence of mental illness in patients with HIV/AIDS is reported to be 8 times higher than in those without HIV/AIDS.2 Depression, bipolar disorder, anxiety disorders, delirium, substance abuse, and schizophrenia have all been identified in persons receiving highly active antiretroviral therapy (HAART). Patients with HIV/AIDS and psychiatric illness have a decreased quality of life, poor adherence to medications, faster disease progression, and increased mortality. Care of these individuals is complicated by the stigma of HIV/AIDS and the prevalence of the illness in underserved populations, as well as the need for complex medication regimens and the possibility of drug–drug interactions (DDIs).1,2 If left untreated, psychiatric illness in patients with HIV/AIDS may lead to further transmission of HIV as a result of patients engaging in high-risk behaviors, along with poor adherence to HAART.3
Individuals diagnosed with schizophrenia, schizoaffective disorder, and bipolar disorder are at greater risk for HIV infection.3 Patients with HIV/AIDS with primary psychosis may have poor medication adherence rates due to illness-related confusion or paranoia about medications. Furthermore, they may lack the resources to manage the complications and stress related to living with HIV/AIDS.
New-onset, or secondary psychosis, has been reported in individuals with late-stage HIV/AIDS with CD4 counts <200 who have not been diagnosed with a psychotic disorder previously.3 These patients may experience more persecutory and grandiose delusions rather than hallucinations. Neuropsychiatric symptoms in patients with HIV/AIDS may be due to the presence of HIV or other infections in the CNS, tumors, or other inflammatory illnesses. Medications that have been implicated in neuropsychiatric symptoms include efavirenz, rilpivirine, and other HAART regimens; interferon; metoclopramide; corticosteroids; muscle relaxants; and clonidine. It is possible that symptoms may continue even after the medications are discontinued.3
Antipsychotics remain the treatment of choice for psychosis in HIV/AIDS, regardless of the cause of the symptoms. Many factors must be taken into consideration when choosing an antipsychotic, such as DDIs, adverse effect profiles, patient history of antipsychotic use, cost, and patient preference. Here we focus primarily on DDIs and adverse effect profiles.
When treating psychosis in patients with HIV/AIDS, it is crucial to consider potential DDIs. Many antipsychotics and antiretroviral medications utilize cytochrome P450 (CYP) enzymes for their metabolism. The CYP enzyme system is responsible for the oxidative reactions that constitute the phase I reactions necessary for the metabolism of most drugs. Inhibition and induction of CYP enzymes are among the most common causes of pharmacokinetic DDIs. Antipsychotics are predominately metabolized by CYP3A4, CYP1A2, and CYP2D6.4
Continue to: The DDIs arise because...
The DDIs arise because many antiretroviral medications inhibit, or in some cases, induce, these CYP enzymes, thereby altering substrate-drug metabolism. Inhibiting a CYP enzyme pathway can decrease substrate-drug clearance and lead to increased levels of that drug. This, in turn, can cause an increased risk of adverse effects, such as extrapyramidal symptoms (EPS) or QTc prolongation, which are both types of pharmacodynamic DDIs.4-28 However, because antipsychotics often have more than one pathway of metabolism, it can be challenging to understand the full effect of CYP-related DDIs. Furthermore, CYP enzyme inducers can decrease drug levels, and in the case of antipsychotics, lead to subtherapeutic responses.
Table 1,6-14,19-28 Table 2,15-28 Table 3,6-14,19-28 and Table 415-28 list many of the known CYP enzyme-related DDIs that may occur with combination antipsychotic and antiretroviral medication therapy and aim to predict CYP induction or inhibition based on a particular combination. The following antiretroviral medications do not have any CYP-related interactions and therefore are not included in the Tables: abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir disoproxil, zidovudine, enfuvirtide, maraviroc, and raltegravir.
These Tables include the risk ratings for all D-rated (consider alternative therapy) and X-rated (avoid therapy) combinations. The majority of D-rated interactions are caused by CYP inhibition or induction that could potentially lead to altered antipsychotic levels. The majority of X-rated interactions are caused by increased QTc prolongation that may or may not be due to CYP-related DDIs. For example, paliperidone is not believed to be affected by the CYP enzyme system, but it does present a high risk of QTc prolongation on its own. When combined with an antiretroviral that also has a high risk of QTc prolongation, such as lopinavir, then the risk further increases.
Non-nucleoside reverse transcriptase inhibitors and protease inhibitors (PIs) are the antiretroviral medications most likely to cause DDIs with antipsychotics. Other antiretroviral classes, such as nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs), fusion inhibitors, chemokine receptor 5 inhibitors, and integrase inhibitors, are not associated with CYP-related DDIs.19-28 For the most part, the severity of the CYP-related DDIs have not been well studied; therefore, most recommendations call for closer patient monitoring when combining antiretroviral medications and antipsychotics.6-18 The goal is to monitor for any changes in medication efficacy or adverse effects.
Continue to: Consider adverse effect profiles
Consider adverse effect profiles
When selecting an antipsychotic agent for a patient receiving HIV therapy, also consider adverse effect profiles. The emergence of adverse effects can greatly impact patients’ quality of life, leading to consequences of medication nonadherence and exacerbation of mental illness.
Extrapyramidal symptoms. Patients with HIV have a higher sensitivity to treatment-emergent EPS from antipsychotics.2 This sensitivity is generally thought to arise from the involvement of HIV on the basal ganglia. Historically, psychotic symptoms in HIV have been managed with second-generation antipsychotics (SGAs) at the lowest effective dose because these medications are less likely to cause EPS.1,29 The antipsychotic with the lowest rate of EPS is clozapine, followed by quetiapine, olanzapine, ziprasidone, and aripiprazole. Conversely, high-potency first-generation antipsychotics (FGAs) have the highest rates of EPS, followed by intermediate-potency FGAs and risperidone.30
Metabolic disturbances are another concern with concomitant antipsychotic/antiretroviral therapy. Patients with HIV who are receiving NRTIs or PIs can present with drug-induced lipodystrophy syndrome, which is associated with hyperglycemia, hyperinsulinemia, hyperlipidemia, and hypertension, and ultimately may cause metabolic syndrome.29 The prevalence of metabolic syndrome in patients receiving PI therapy has a vast range—2% to 84%—which can be attributed to inconsistent definitions, criteria, and assessment methodology.29 Use of a PI is considered to be the most prominent risk factor for developing lipodystrophy.29 Among the PIs, metabolic disturbances in regards to lipids are most often seen with lopinavir/ritonavir (LPV/r), saquinavir/ritonavir, tipranavir/ritonavir, and fosamprenavir/ritonavir.31 In comparison with LPV/r, darunavir showed improvement in lipids.32 Atazanavir (ATV) boosted with ritonavir has not shown clinically significant adverse effects on lipids.31 Additionally, amprenavir, LPV/r, and ritonavir demonstrated more glucose uptake inhibition via blockade of the glucose transporter type 4 than ATV.31 Of the NRTIs, lipodystrophy syndrome is most commonly seen with stavudine, which is used minimally in practice.2
The rates of metabolic disturbance with antipsychotic use range from 2% to 36%.2 The American Psychiatric Association recommends selecting one of the SGAs least likely to affect metabolic parameters.29 Aripiprazole and ziprasidone are associated with the lowest risk of weight gain, hyperglycemia, and hyperlipidemia. They are followed by risperidone and quetiapine, which are associated with moderate risk, and then clozapine and olanzapine, which are associated with high risk.2,30,33
Continue to: Management of metabolic adverse effects involves...
Management of metabolic adverse effects involves switching the antiretroviral agent and/or antipsychotic agent to an alternative associated with lower metabolic risk. Antipsychotics with low metabolic risk include aripiprazole, lurasidone, and ziprasidone. Lifestyle modifications are encouraged. Additionally, medication interventions, such as metformin, are also recommended in patients meeting criteria for pre-diabetes or type 2 diabetes mellitus.2 Lipid panels and metabolic parameters should be monitored periodically, according to guidelines.25,34
Bone marrow toxicity and blood dyscrasias. Lastly, consider the risk of bone marrow suppression. Patients receiving clozapine for treatment-resistant schizophrenia should be closely monitored for neutropenia and agranulocytosis. Although zidovudine is rarely used, its use is associated with adverse myelosuppressive effects, and the combination of clozapine and zidovudine could pose danger to the patient.2,35,36
CASE CONTINUED
Because Mr. S’s diagnosis of HIV puts him at a higher risk of developing EPS, and because he is already experiencing increased wrist rigidity, the treatment team decides to switch his antipsychotic therapy to an agent with a lower risk of EPS. His comorbidities, including type 2 diabetes mellitus, hypertension, and hyperlipidemia, are taken into account, and an SGA with a benign metabolic profile is considered. Aripiprazole and ziprasidone are favorable options. However, because efavirenz, ATZ, and ritonavir may cause QTc prolongation, ziprasidone, the SGA with the highest rate of QTc prolongation, is not the preferred option.
Mr. S’s SGA therapy is switched from risperidone to aripiprazole. Because potential CYP-related interactions between aripiprazole and Mr. S’s current antiretroviral therapy could lead to increased aripiprazole levels. Mr. S is started on a low dose (5 mg/d) with the goal to titrate based on response and tolerability. Increased levels of aripiprazole may increase the risk of akathisia, drowsiness, headaches, and fatigue. Mr. S is monitored closely for improvement of EPS, adverse effects of medication, and metabolic parameters. Furthermore, if the treatment team believes there is a more preferred antipsychotic for the patient that it did not prescribe because of the risk of DDIs, it may be worthwhile to consider discussing the HAART regimen with the patient’s infectious disease treatment team.
Continue to: Acknowledgements
Acknowledgements
This material is the result of work supported with resources and the use of facilities at the Chillicothe Veterans Affairs Medical Center in Chillicothe, Ohio. The contents of this paper do not represent the views of the U.S. Department of Veterans Affairs or the U.S. government.
Related Resources
- Cohen MA. HIV: How to provide compassionate care. Current Psychiatry. 2013;12(6):19-23,A,B.
- Khan AY, Zaidi SN. Reducing morbidity and mortality from common medical conditions in schizophrenia. Current Psychiatry. 2016;15(3):30-32,34-38,40.
Drug Brand Names
Abacavir • Ziagen
Amlodipine • Norvasc
Amprenavir • Agenerase
Aripiprazole • Abilify
Asenapine • Saphris
Atazanavir • Reyataz
Brexpiprazole • Rexulti
Bupropion ER • Wellbutrin SR
Cariprazine • Vraylar
Chlorpromazine • Thorazine
Clonidine • Catapres
Clozapine • Clozaril
Darunavir • Prezista
Delavirdine • Rescriptor
Didanosine • Videx EC
Efavirenz • Sustiva
Efavirenz/emtricitabine/tenofovir disoproxil fumarate • Atripla
Enfuvirtide • Fuzeon
Emtricitabine • Emtriva
Etravirine • Intelence
Fluphenazine • Prolixin
Fosamprenavir • Lexiva
Gabapentin • Neurontin
Glipizide • Glucotrol
Haloperidol • Haldol
Iloperidone • Fanapt
Indinavir • Crixivan
Lamivudine • Epivir
Lopinavir/ritonavir • Kaletra
Loxapine • Loxitane
Lurasidone • Latuda
Maraviroc • Selzentry
Metformin • Glucophage
Metoclopramide • Reglan
Molindone • Moban
Nelfinavir • Viracept
Nevirapine • Viramune
Olanzapine • Zyprexa
Paliperidone • Invega
Perphenazine • Trilafon
Pimozide • Orap
Pravastatin • Pravachol
Quetiapine • Seroquel
Raltegravir • Isentress
Rilpivirine • Edurant
Risperidone • Risperdal
Ritonavir • Norvir
Saquinavir • Invirase
Stavudine • Zerit
Tenofovir disoproxil • Viread
Thioridazine • Mellaril
Thiothixene • Navane
Tipranavir • Aptivus
Trifluoperazine • Stelazine
Zidovudine • Retrovir
Ziprasidone • Geodon
Mr. S, age 56, has human immunodeficiency virus (HIV) and schizoaffective disorder. He presents to your clinic with increased auditory hallucinations, disorganized behavior, and worsened tremors that have begun to seriously disrupt his daily life. Mr. S is prescribed risperidone; however, he reports that he has not been taking it lately due to the tremor despite being controlled on his medication regimen for nearly 1 year. His Abnormal Involuntary Movement Scale (AIMS) score reveals an increased wrist rigidity compared with previous clinic visits. Mr. S has a 40 pack-year history of smoking and history of IV drug use. Furthermore, he has a medical history of type 2 diabetes mellitus, hypertension, and hyperlipidemia.
His medication regimen includes atazanavir sulfate, 200 mg/d, ritonavir, 100 mg/d, efavirenz/emtricitabine/tenofovir disoproxil fumarate, 600/200/300 mg/d, risperidone, 6 mg/d, bupropion extended-release, 300 mg/d, gabapentin, 600 mg/d, amlodipine, 5 mg/d, pravastatin, 40 mg/d, metformin, 1000 mg twice daily, and glipizide, 10 mg twice daily. Today, his laboratory findings show that his CD4 count is 405 cell/mm3, and his viral load is <40 copies/mL, indicating his HIV is well managed. A hepatitis C virus antibody test result is negative and serum creatinine level is 1.0 mg/dL. Total cholesterol is 212 mg/dL, high-density lipoprotein cholesterol is 43 mg/dL, low-density lipoprotein cholesterol is 121 mg/dL, and triglycerides level is 238 mg/dL. Electrocardiography reveals a QTc interval of 426 ms. Mr. S’s blood pressure is 105/65 mm Hg. Based on this clinic visit, the treatment team decides to change Mr. S’s antipsychotic.
Psychiatric illness and HIV/AIDS
There is a strong link between mental illness and HIV/AIDS; 50% or more of patients with HIV/AIDS have a comorbid psychiatric disorder.1 The prevalence of mental illness in patients with HIV/AIDS is reported to be 8 times higher than in those without HIV/AIDS.2 Depression, bipolar disorder, anxiety disorders, delirium, substance abuse, and schizophrenia have all been identified in persons receiving highly active antiretroviral therapy (HAART). Patients with HIV/AIDS and psychiatric illness have a decreased quality of life, poor adherence to medications, faster disease progression, and increased mortality. Care of these individuals is complicated by the stigma of HIV/AIDS and the prevalence of the illness in underserved populations, as well as the need for complex medication regimens and the possibility of drug–drug interactions (DDIs).1,2 If left untreated, psychiatric illness in patients with HIV/AIDS may lead to further transmission of HIV as a result of patients engaging in high-risk behaviors, along with poor adherence to HAART.3
Individuals diagnosed with schizophrenia, schizoaffective disorder, and bipolar disorder are at greater risk for HIV infection.3 Patients with HIV/AIDS with primary psychosis may have poor medication adherence rates due to illness-related confusion or paranoia about medications. Furthermore, they may lack the resources to manage the complications and stress related to living with HIV/AIDS.
New-onset, or secondary psychosis, has been reported in individuals with late-stage HIV/AIDS with CD4 counts <200 who have not been diagnosed with a psychotic disorder previously.3 These patients may experience more persecutory and grandiose delusions rather than hallucinations. Neuropsychiatric symptoms in patients with HIV/AIDS may be due to the presence of HIV or other infections in the CNS, tumors, or other inflammatory illnesses. Medications that have been implicated in neuropsychiatric symptoms include efavirenz, rilpivirine, and other HAART regimens; interferon; metoclopramide; corticosteroids; muscle relaxants; and clonidine. It is possible that symptoms may continue even after the medications are discontinued.3
Antipsychotics remain the treatment of choice for psychosis in HIV/AIDS, regardless of the cause of the symptoms. Many factors must be taken into consideration when choosing an antipsychotic, such as DDIs, adverse effect profiles, patient history of antipsychotic use, cost, and patient preference. Here we focus primarily on DDIs and adverse effect profiles.
When treating psychosis in patients with HIV/AIDS, it is crucial to consider potential DDIs. Many antipsychotics and antiretroviral medications utilize cytochrome P450 (CYP) enzymes for their metabolism. The CYP enzyme system is responsible for the oxidative reactions that constitute the phase I reactions necessary for the metabolism of most drugs. Inhibition and induction of CYP enzymes are among the most common causes of pharmacokinetic DDIs. Antipsychotics are predominately metabolized by CYP3A4, CYP1A2, and CYP2D6.4
Continue to: The DDIs arise because...
The DDIs arise because many antiretroviral medications inhibit, or in some cases, induce, these CYP enzymes, thereby altering substrate-drug metabolism. Inhibiting a CYP enzyme pathway can decrease substrate-drug clearance and lead to increased levels of that drug. This, in turn, can cause an increased risk of adverse effects, such as extrapyramidal symptoms (EPS) or QTc prolongation, which are both types of pharmacodynamic DDIs.4-28 However, because antipsychotics often have more than one pathway of metabolism, it can be challenging to understand the full effect of CYP-related DDIs. Furthermore, CYP enzyme inducers can decrease drug levels, and in the case of antipsychotics, lead to subtherapeutic responses.
Table 1,6-14,19-28 Table 2,15-28 Table 3,6-14,19-28 and Table 415-28 list many of the known CYP enzyme-related DDIs that may occur with combination antipsychotic and antiretroviral medication therapy and aim to predict CYP induction or inhibition based on a particular combination. The following antiretroviral medications do not have any CYP-related interactions and therefore are not included in the Tables: abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir disoproxil, zidovudine, enfuvirtide, maraviroc, and raltegravir.
These Tables include the risk ratings for all D-rated (consider alternative therapy) and X-rated (avoid therapy) combinations. The majority of D-rated interactions are caused by CYP inhibition or induction that could potentially lead to altered antipsychotic levels. The majority of X-rated interactions are caused by increased QTc prolongation that may or may not be due to CYP-related DDIs. For example, paliperidone is not believed to be affected by the CYP enzyme system, but it does present a high risk of QTc prolongation on its own. When combined with an antiretroviral that also has a high risk of QTc prolongation, such as lopinavir, then the risk further increases.
Non-nucleoside reverse transcriptase inhibitors and protease inhibitors (PIs) are the antiretroviral medications most likely to cause DDIs with antipsychotics. Other antiretroviral classes, such as nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs), fusion inhibitors, chemokine receptor 5 inhibitors, and integrase inhibitors, are not associated with CYP-related DDIs.19-28 For the most part, the severity of the CYP-related DDIs have not been well studied; therefore, most recommendations call for closer patient monitoring when combining antiretroviral medications and antipsychotics.6-18 The goal is to monitor for any changes in medication efficacy or adverse effects.
Continue to: Consider adverse effect profiles
Consider adverse effect profiles
When selecting an antipsychotic agent for a patient receiving HIV therapy, also consider adverse effect profiles. The emergence of adverse effects can greatly impact patients’ quality of life, leading to consequences of medication nonadherence and exacerbation of mental illness.
Extrapyramidal symptoms. Patients with HIV have a higher sensitivity to treatment-emergent EPS from antipsychotics.2 This sensitivity is generally thought to arise from the involvement of HIV on the basal ganglia. Historically, psychotic symptoms in HIV have been managed with second-generation antipsychotics (SGAs) at the lowest effective dose because these medications are less likely to cause EPS.1,29 The antipsychotic with the lowest rate of EPS is clozapine, followed by quetiapine, olanzapine, ziprasidone, and aripiprazole. Conversely, high-potency first-generation antipsychotics (FGAs) have the highest rates of EPS, followed by intermediate-potency FGAs and risperidone.30
Metabolic disturbances are another concern with concomitant antipsychotic/antiretroviral therapy. Patients with HIV who are receiving NRTIs or PIs can present with drug-induced lipodystrophy syndrome, which is associated with hyperglycemia, hyperinsulinemia, hyperlipidemia, and hypertension, and ultimately may cause metabolic syndrome.29 The prevalence of metabolic syndrome in patients receiving PI therapy has a vast range—2% to 84%—which can be attributed to inconsistent definitions, criteria, and assessment methodology.29 Use of a PI is considered to be the most prominent risk factor for developing lipodystrophy.29 Among the PIs, metabolic disturbances in regards to lipids are most often seen with lopinavir/ritonavir (LPV/r), saquinavir/ritonavir, tipranavir/ritonavir, and fosamprenavir/ritonavir.31 In comparison with LPV/r, darunavir showed improvement in lipids.32 Atazanavir (ATV) boosted with ritonavir has not shown clinically significant adverse effects on lipids.31 Additionally, amprenavir, LPV/r, and ritonavir demonstrated more glucose uptake inhibition via blockade of the glucose transporter type 4 than ATV.31 Of the NRTIs, lipodystrophy syndrome is most commonly seen with stavudine, which is used minimally in practice.2
The rates of metabolic disturbance with antipsychotic use range from 2% to 36%.2 The American Psychiatric Association recommends selecting one of the SGAs least likely to affect metabolic parameters.29 Aripiprazole and ziprasidone are associated with the lowest risk of weight gain, hyperglycemia, and hyperlipidemia. They are followed by risperidone and quetiapine, which are associated with moderate risk, and then clozapine and olanzapine, which are associated with high risk.2,30,33
Continue to: Management of metabolic adverse effects involves...
Management of metabolic adverse effects involves switching the antiretroviral agent and/or antipsychotic agent to an alternative associated with lower metabolic risk. Antipsychotics with low metabolic risk include aripiprazole, lurasidone, and ziprasidone. Lifestyle modifications are encouraged. Additionally, medication interventions, such as metformin, are also recommended in patients meeting criteria for pre-diabetes or type 2 diabetes mellitus.2 Lipid panels and metabolic parameters should be monitored periodically, according to guidelines.25,34
Bone marrow toxicity and blood dyscrasias. Lastly, consider the risk of bone marrow suppression. Patients receiving clozapine for treatment-resistant schizophrenia should be closely monitored for neutropenia and agranulocytosis. Although zidovudine is rarely used, its use is associated with adverse myelosuppressive effects, and the combination of clozapine and zidovudine could pose danger to the patient.2,35,36
CASE CONTINUED
Because Mr. S’s diagnosis of HIV puts him at a higher risk of developing EPS, and because he is already experiencing increased wrist rigidity, the treatment team decides to switch his antipsychotic therapy to an agent with a lower risk of EPS. His comorbidities, including type 2 diabetes mellitus, hypertension, and hyperlipidemia, are taken into account, and an SGA with a benign metabolic profile is considered. Aripiprazole and ziprasidone are favorable options. However, because efavirenz, ATZ, and ritonavir may cause QTc prolongation, ziprasidone, the SGA with the highest rate of QTc prolongation, is not the preferred option.
Mr. S’s SGA therapy is switched from risperidone to aripiprazole. Because potential CYP-related interactions between aripiprazole and Mr. S’s current antiretroviral therapy could lead to increased aripiprazole levels. Mr. S is started on a low dose (5 mg/d) with the goal to titrate based on response and tolerability. Increased levels of aripiprazole may increase the risk of akathisia, drowsiness, headaches, and fatigue. Mr. S is monitored closely for improvement of EPS, adverse effects of medication, and metabolic parameters. Furthermore, if the treatment team believes there is a more preferred antipsychotic for the patient that it did not prescribe because of the risk of DDIs, it may be worthwhile to consider discussing the HAART regimen with the patient’s infectious disease treatment team.
Continue to: Acknowledgements
Acknowledgements
This material is the result of work supported with resources and the use of facilities at the Chillicothe Veterans Affairs Medical Center in Chillicothe, Ohio. The contents of this paper do not represent the views of the U.S. Department of Veterans Affairs or the U.S. government.
Related Resources
- Cohen MA. HIV: How to provide compassionate care. Current Psychiatry. 2013;12(6):19-23,A,B.
- Khan AY, Zaidi SN. Reducing morbidity and mortality from common medical conditions in schizophrenia. Current Psychiatry. 2016;15(3):30-32,34-38,40.
Drug Brand Names
Abacavir • Ziagen
Amlodipine • Norvasc
Amprenavir • Agenerase
Aripiprazole • Abilify
Asenapine • Saphris
Atazanavir • Reyataz
Brexpiprazole • Rexulti
Bupropion ER • Wellbutrin SR
Cariprazine • Vraylar
Chlorpromazine • Thorazine
Clonidine • Catapres
Clozapine • Clozaril
Darunavir • Prezista
Delavirdine • Rescriptor
Didanosine • Videx EC
Efavirenz • Sustiva
Efavirenz/emtricitabine/tenofovir disoproxil fumarate • Atripla
Enfuvirtide • Fuzeon
Emtricitabine • Emtriva
Etravirine • Intelence
Fluphenazine • Prolixin
Fosamprenavir • Lexiva
Gabapentin • Neurontin
Glipizide • Glucotrol
Haloperidol • Haldol
Iloperidone • Fanapt
Indinavir • Crixivan
Lamivudine • Epivir
Lopinavir/ritonavir • Kaletra
Loxapine • Loxitane
Lurasidone • Latuda
Maraviroc • Selzentry
Metformin • Glucophage
Metoclopramide • Reglan
Molindone • Moban
Nelfinavir • Viracept
Nevirapine • Viramune
Olanzapine • Zyprexa
Paliperidone • Invega
Perphenazine • Trilafon
Pimozide • Orap
Pravastatin • Pravachol
Quetiapine • Seroquel
Raltegravir • Isentress
Rilpivirine • Edurant
Risperidone • Risperdal
Ritonavir • Norvir
Saquinavir • Invirase
Stavudine • Zerit
Tenofovir disoproxil • Viread
Thioridazine • Mellaril
Thiothixene • Navane
Tipranavir • Aptivus
Trifluoperazine • Stelazine
Zidovudine • Retrovir
Ziprasidone • Geodon
1. Freudenreich O, Goforth HW, Cozza KL, et al. Psychiatric treatment of persons with HIV/AIDS: An HIV-psychiatry consensus survey of current practices. Psychosomatics. 2010;51(6):480-488.
2. Hill L, Lee KC. Pharmacotherapy considerations in patients with HIV and psychiatric disorders: Focus on antidepressants and antipsychotics. Ann Pharmacother. 2013;47(1):75-89.
3. Watkins CC, Treisman GJ. Neuropsychiatric complications of aging with HIV. J Neurovirol. 2012;18(4):277-290.
4. Prior TI, Baker GB. Interactions between the cytochrome P450 system and the second-generation antipsychotics. J Psychiatry Neurosci. 2003;28(2):99-112.
5. Ponte ML, Keller GA, Di Girolamo G. Mechanisms of drug induced QT interval prolongation. Curr Drug Saf. 2010;5(1):44-53
6. Reyataz [package insert]. Princeton, NJ: Bristol-Myers Squibb Company; 2017.
7. Prezista [package insert]. Toronto, ON: Janssen Inc.; 2017.
8. Lexiva [package insert]. Research Triangle Park, NC: Viiv Healthcare; 2017
9. Crixivan [package insert]. Whitehouse Station, NJ; Merck; 2016.
10. Kaletra [package insert]. North Chicago, IL: AbbVie Inc; 2017
11. Viracept [package insert]. Kirkland, QC: Pfizer Canada Inc.; 201
12. Norvir tablets and oral solution [package insert]. North Chicago, IL: AbbVie Inc; 2017
13. Invirase [package insert]. South San Francisco, CA: Genentech USA, Inc.; 2016.
14. Aptivus [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals Inc.; 2016.
15. Sustiva [package insert]. Princeton, NJ: Bristol-Myers Squibb Company; 2017
16. Intelence [package insert]. Titusville, NJ: Tibotec Pharmaceuticals; 2014.
17. Viramune [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals Inc.; 2017.
18. Rescriptor [package insert]. Laval, QC: ViiV Healthcare ULC; 2013.
19. Ziagen [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2017.
20. Videx EC [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2015.
21. Emtriva [package insert]. Foster City, CA: Gilead Sciences, Inc.; 2017.
22. Epivir [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2017.
23. Zerit [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2017.
24. Viread [package insert]. Foster City, CA: Gilead Sciences, Inc.; 2017.
25. Retrovir [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2015.
26. Fuzeon [package insert]. South San Francisco, CA: Genentech USA, Inc; 2017.
27. Selzentry [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2016.
28. Isentress [package insert]. Whitehouse Station, NJ: Merck Sharp & Dohme Corp.; 2017.
29. American Psychiatry Association. Practice guidelines for treatment of patients with HIV/AIDS. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/hivaids.pdf. Published 2010. Accessed March 1, 2018.
30. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 Schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
31. Hughes PJ, Cretton-Scott E, Teague A, et al. Protease inhibitors for patients with HIV-1 infection. P T. 2011;36(6):332-336,341-345.
32. Ortiz R, Dejesus E, Khanlou H, et al. Efficacy and safety of once-daily darunavir/ritonavir versus lopinavir/ritonavir in treatment-naive HIV-1-infected patients at week 48. AIDS. 2008;22(12):1389-1397.
33. Leucht S, Cipriani A, Spineli L, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet. 2013;382(9896):951-962.
34. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013;12(9):51-54.
35. Singh D, Goodkin K. Choice of antipsychotic in HIV-infected patients. J Clin Psychiatry. 2007;68(3):479-480.
36. Max B, Sherer R. Management of the adverse effects of antiretroviral therapy and medication adherence. Clin Infect Dis. 2000;30(suppl 2):S96-S116.
1. Freudenreich O, Goforth HW, Cozza KL, et al. Psychiatric treatment of persons with HIV/AIDS: An HIV-psychiatry consensus survey of current practices. Psychosomatics. 2010;51(6):480-488.
2. Hill L, Lee KC. Pharmacotherapy considerations in patients with HIV and psychiatric disorders: Focus on antidepressants and antipsychotics. Ann Pharmacother. 2013;47(1):75-89.
3. Watkins CC, Treisman GJ. Neuropsychiatric complications of aging with HIV. J Neurovirol. 2012;18(4):277-290.
4. Prior TI, Baker GB. Interactions between the cytochrome P450 system and the second-generation antipsychotics. J Psychiatry Neurosci. 2003;28(2):99-112.
5. Ponte ML, Keller GA, Di Girolamo G. Mechanisms of drug induced QT interval prolongation. Curr Drug Saf. 2010;5(1):44-53
6. Reyataz [package insert]. Princeton, NJ: Bristol-Myers Squibb Company; 2017.
7. Prezista [package insert]. Toronto, ON: Janssen Inc.; 2017.
8. Lexiva [package insert]. Research Triangle Park, NC: Viiv Healthcare; 2017
9. Crixivan [package insert]. Whitehouse Station, NJ; Merck; 2016.
10. Kaletra [package insert]. North Chicago, IL: AbbVie Inc; 2017
11. Viracept [package insert]. Kirkland, QC: Pfizer Canada Inc.; 201
12. Norvir tablets and oral solution [package insert]. North Chicago, IL: AbbVie Inc; 2017
13. Invirase [package insert]. South San Francisco, CA: Genentech USA, Inc.; 2016.
14. Aptivus [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals Inc.; 2016.
15. Sustiva [package insert]. Princeton, NJ: Bristol-Myers Squibb Company; 2017
16. Intelence [package insert]. Titusville, NJ: Tibotec Pharmaceuticals; 2014.
17. Viramune [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals Inc.; 2017.
18. Rescriptor [package insert]. Laval, QC: ViiV Healthcare ULC; 2013.
19. Ziagen [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2017.
20. Videx EC [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2015.
21. Emtriva [package insert]. Foster City, CA: Gilead Sciences, Inc.; 2017.
22. Epivir [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2017.
23. Zerit [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2017.
24. Viread [package insert]. Foster City, CA: Gilead Sciences, Inc.; 2017.
25. Retrovir [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2015.
26. Fuzeon [package insert]. South San Francisco, CA: Genentech USA, Inc; 2017.
27. Selzentry [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2016.
28. Isentress [package insert]. Whitehouse Station, NJ: Merck Sharp & Dohme Corp.; 2017.
29. American Psychiatry Association. Practice guidelines for treatment of patients with HIV/AIDS. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/hivaids.pdf. Published 2010. Accessed March 1, 2018.
30. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 Schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
31. Hughes PJ, Cretton-Scott E, Teague A, et al. Protease inhibitors for patients with HIV-1 infection. P T. 2011;36(6):332-336,341-345.
32. Ortiz R, Dejesus E, Khanlou H, et al. Efficacy and safety of once-daily darunavir/ritonavir versus lopinavir/ritonavir in treatment-naive HIV-1-infected patients at week 48. AIDS. 2008;22(12):1389-1397.
33. Leucht S, Cipriani A, Spineli L, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet. 2013;382(9896):951-962.
34. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013;12(9):51-54.
35. Singh D, Goodkin K. Choice of antipsychotic in HIV-infected patients. J Clin Psychiatry. 2007;68(3):479-480.
36. Max B, Sherer R. Management of the adverse effects of antiretroviral therapy and medication adherence. Clin Infect Dis. 2000;30(suppl 2):S96-S116.