User login
Myocardial fibrosis assessment fine-tunes ICD selection
SNOWMASS, COLO. – Detection of myocardial midwall fibrosis via cardiovascular magnetic resonance in patients with nonischemic dilated cardiomyopathy provides prognostic information independent of left ventricular ejection fraction.
"Low LVEF and fibrosis uniquely identify the need for an ICD [implantable cardioverter-defibrillator]. This is new information that helps refine our understanding of whom it is that’s uniquely at risk. We’ve always had these troublesome questions in patients with nonischemic heart failure," Dr. Clyde W. Yancy observed at the Annual Cardiovascular Conference at Snowmass.
He highlighted what he called "very provocative data" in a recent study led by Dr. Sanjay K. Prasad of Royal Brompton Hospital in London. The investigators evaluated 472 consecutive patients with nonischemic dilated cardiomyopathy using late gadolinium cardiovascular magnetic resonance (CMR). Thirty percent of them were found to have midwall fibrosis. Their all-cause mortality rate during a median 5.3 years of prospective follow-up was 26.8%, compared with 10.6% in the 330 patients without fibrosis. An arrhythmic composite event comprising sudden cardiac death (SCD), aborted SCD, or sustained ventricular tachycardia occurred in 29.6% of the group with fibrosis vs. 7% of patients without fibrosis.

In a multivariate analysis adjusted for LVEF and other prognostic factors, the presence of midwall fibrosis was independently associated with a 2.43-fold increased risk of all-cause mortality, a 3.2-fold greater risk of cardiovascular mortality or heart transplantation, a 4.6-fold increase in SCD or aborted SCD, and a 1.6-fold increased likelihood of a composite of heart failure hospitalization, mortality from heart failure, or cardiac transplantation (JAMA 2013;309:896-908).
Dr. Yancy, who chaired the writing committee for the 2013 ACC/AHA Guideline for the Management of Heart Failure, noted that the guidelines grant a strong Class I/Level of Evidence B recommendation for implantation of an ICD for primary prevention in nonischemic dilated cardiomyopathy patients who are in New York Heart Association functional class II or III and have an LVEF of 35% or less. But there is a pressing need for refined implantation criteria. Basing the ICD decision on only these criteria results in a low rate of appropriate shocks, a high frequency of inappropriate shocks, and exclusion from device therapy of a group of patients with a high relative risk of SCD.
The British study provides reason for optimism in this regard. Using a greater than 15% estimated SCD risk based upon LVEF plus midwall fibrosis as a proposed indication for ICD implantation, the investigators found that an additional 12 patients in their cohort would receive an ICD and 43 others would now avoid ICD implantation, noted Dr. Yancy, professor of medicine and of medical social sciences and chief of cardiology at Northwestern University, Chicago.
He reported having no financial conflicts.
SNOWMASS, COLO. – Detection of myocardial midwall fibrosis via cardiovascular magnetic resonance in patients with nonischemic dilated cardiomyopathy provides prognostic information independent of left ventricular ejection fraction.
"Low LVEF and fibrosis uniquely identify the need for an ICD [implantable cardioverter-defibrillator]. This is new information that helps refine our understanding of whom it is that’s uniquely at risk. We’ve always had these troublesome questions in patients with nonischemic heart failure," Dr. Clyde W. Yancy observed at the Annual Cardiovascular Conference at Snowmass.
He highlighted what he called "very provocative data" in a recent study led by Dr. Sanjay K. Prasad of Royal Brompton Hospital in London. The investigators evaluated 472 consecutive patients with nonischemic dilated cardiomyopathy using late gadolinium cardiovascular magnetic resonance (CMR). Thirty percent of them were found to have midwall fibrosis. Their all-cause mortality rate during a median 5.3 years of prospective follow-up was 26.8%, compared with 10.6% in the 330 patients without fibrosis. An arrhythmic composite event comprising sudden cardiac death (SCD), aborted SCD, or sustained ventricular tachycardia occurred in 29.6% of the group with fibrosis vs. 7% of patients without fibrosis.
In a multivariate analysis adjusted for LVEF and other prognostic factors, the presence of midwall fibrosis was independently associated with a 2.43-fold increased risk of all-cause mortality, a 3.2-fold greater risk of cardiovascular mortality or heart transplantation, a 4.6-fold increase in SCD or aborted SCD, and a 1.6-fold increased likelihood of a composite of heart failure hospitalization, mortality from heart failure, or cardiac transplantation (JAMA 2013;309:896-908).
Dr. Yancy, who chaired the writing committee for the 2013 ACC/AHA Guideline for the Management of Heart Failure, noted that the guidelines grant a strong Class I/Level of Evidence B recommendation for implantation of an ICD for primary prevention in nonischemic dilated cardiomyopathy patients who are in New York Heart Association functional class II or III and have an LVEF of 35% or less. But there is a pressing need for refined implantation criteria. Basing the ICD decision on only these criteria results in a low rate of appropriate shocks, a high frequency of inappropriate shocks, and exclusion from device therapy of a group of patients with a high relative risk of SCD.
The British study provides reason for optimism in this regard. Using a greater than 15% estimated SCD risk based upon LVEF plus midwall fibrosis as a proposed indication for ICD implantation, the investigators found that an additional 12 patients in their cohort would receive an ICD and 43 others would now avoid ICD implantation, noted Dr. Yancy, professor of medicine and of medical social sciences and chief of cardiology at Northwestern University, Chicago.
He reported having no financial conflicts.
SNOWMASS, COLO. – Detection of myocardial midwall fibrosis via cardiovascular magnetic resonance in patients with nonischemic dilated cardiomyopathy provides prognostic information independent of left ventricular ejection fraction.
"Low LVEF and fibrosis uniquely identify the need for an ICD [implantable cardioverter-defibrillator]. This is new information that helps refine our understanding of whom it is that’s uniquely at risk. We’ve always had these troublesome questions in patients with nonischemic heart failure," Dr. Clyde W. Yancy observed at the Annual Cardiovascular Conference at Snowmass.
He highlighted what he called "very provocative data" in a recent study led by Dr. Sanjay K. Prasad of Royal Brompton Hospital in London. The investigators evaluated 472 consecutive patients with nonischemic dilated cardiomyopathy using late gadolinium cardiovascular magnetic resonance (CMR). Thirty percent of them were found to have midwall fibrosis. Their all-cause mortality rate during a median 5.3 years of prospective follow-up was 26.8%, compared with 10.6% in the 330 patients without fibrosis. An arrhythmic composite event comprising sudden cardiac death (SCD), aborted SCD, or sustained ventricular tachycardia occurred in 29.6% of the group with fibrosis vs. 7% of patients without fibrosis.
In a multivariate analysis adjusted for LVEF and other prognostic factors, the presence of midwall fibrosis was independently associated with a 2.43-fold increased risk of all-cause mortality, a 3.2-fold greater risk of cardiovascular mortality or heart transplantation, a 4.6-fold increase in SCD or aborted SCD, and a 1.6-fold increased likelihood of a composite of heart failure hospitalization, mortality from heart failure, or cardiac transplantation (JAMA 2013;309:896-908).
Dr. Yancy, who chaired the writing committee for the 2013 ACC/AHA Guideline for the Management of Heart Failure, noted that the guidelines grant a strong Class I/Level of Evidence B recommendation for implantation of an ICD for primary prevention in nonischemic dilated cardiomyopathy patients who are in New York Heart Association functional class II or III and have an LVEF of 35% or less. But there is a pressing need for refined implantation criteria. Basing the ICD decision on only these criteria results in a low rate of appropriate shocks, a high frequency of inappropriate shocks, and exclusion from device therapy of a group of patients with a high relative risk of SCD.
The British study provides reason for optimism in this regard. Using a greater than 15% estimated SCD risk based upon LVEF plus midwall fibrosis as a proposed indication for ICD implantation, the investigators found that an additional 12 patients in their cohort would receive an ICD and 43 others would now avoid ICD implantation, noted Dr. Yancy, professor of medicine and of medical social sciences and chief of cardiology at Northwestern University, Chicago.
He reported having no financial conflicts.
EXPERT ANALYSIS FROM THE CARDIOVASCULAR CONFERENCE AT SNOWMASS

Palliative care shortens ICU, hospital stays, review data show
SAN FRANCISCO – Palliative care in the intensive care unit reduces the length of stay in the ICU and the hospital without changing mortality rates or family satisfaction, according to a review of the literature.
Although measurements of family satisfaction overall didn’t change much from palliative care of a loved one in the ICU, some measures of components of satisfaction increased with palliative care, such as improved communication with the physician, better consensus around the goals of care, and decreased anxiety and depression in family members, reported Dr. Rebecca A. Aslakson of Johns Hopkins University, Baltimore.
The findings have been submitted for publication, she said at the Critical Care Congress, sponsored by the Society for Critical Care Medicine.
Dr. Aslakson and her associates were unable to perform a formal meta-analysis of the 37 published trials of palliative care in the ICU because of the heterogeneity of the studies, which looked at more than 40 different outcomes. Instead, their systematic review grouped results under four outcomes that commonly were measured, and assessed those either by the number of studies or by the number of patients studied.
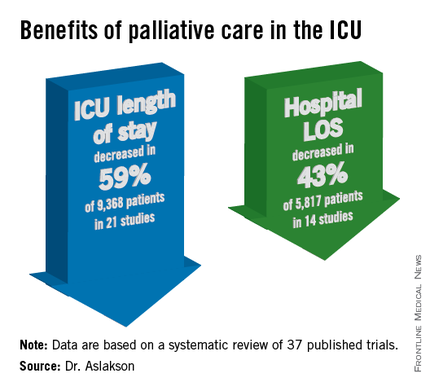
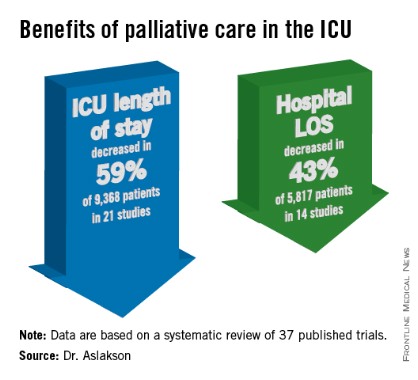
ICU length of stay decreased with palliative care in 13 of 21 studies (62%) that used this outcome and in 59% of 9,368 patients in those studies. Hospital length of stay decreased with palliative care in 8 of 14 studies (57%) and in 43% of 5,817 patients. Family satisfaction did not decrease in any studies or families and increased in only 1 of 14 studies (7%) and in 2% of families of 4,927 patients, Dr. Aslakson reported.
Mortality rates did not change with palliative care in 14 of 16 studies (88%) that assessed mortality and in 57% of 5,969 patients in those studies. Mortality increased in one small study (6%) and decreased in one larger study (6%).
"Talking about big-picture issues and goals of care doesn’t lead to people dying," Dr. Aslakson said. "No harm came in any of these studies." Some separate studies of palliative care outside of ICUs reported that this increases hope, "because people feel that they have more control over their choices and what’s happening to their loved ones," she added.
Integrative vs. consultative model
Dr. Aslakson and her associates also reviewed studies based on whether the interventions used integrative or consultative models of palliative care.
Generally, consultative models bring outsiders into the ICU to help provide palliative care, and integrative models train the ICU team to be the palliative care providers. In reality, the two models may overlap. For this review, the investigators applied mutually exclusive definitions to 36 of the studies. In 18 studies of integrative interventions, members of the ICU team were the only caregivers in face-to-face interactions with the patient and families. In 18 studies of consultative interventions, palliative care providers included others besides the ICU team.
In the studies of integrative palliative care, ICU length of stay decreased with palliative care in four of nine studies (44%) that measured this outcome and in 52% of 6,963 patients in those studies, she reported. Hospital length of stay decreased in two of five studies (40%) and in 24% of 3,812 patients. Family satisfaction changed in none of 15 studies, and mortality decreased in 1 of 5 studies (20%) and in 34% of 3,807 patients.
In the studies of consultative care, ICU length of stay decreased with palliative care in 9 of 12 studies (75%) that measured this outcome and in 79% of 2,405 patients in those studies. Hospital length of stay decreased in six of nine studies (67%) and in 79% of 2,005 patients. Family satisfaction increased in one of four studies (25%) and in 21% of 429 patients. Mortality increased in 1 of 11 studies (9%) and in 5% of 2,162 patients.
One model isn’t necessarily better than the other, Dr. Aslakson said. Integrative palliative care may work best in a closed ICU with perhaps four or five intensivists in a relatively small unit. An integrative approach can be much more difficult in open or semiopen ICUs that have "40 different doctors floating around," she said. "We tried that in my unit, and it didn’t work that well."

Different ICUs need palliative care models that fit them. "Look at your unit, the way it works, and who the providers are, then look at the literature and see what matches that and what might work for your unit," she said.
Outcomes of improved communication
A previous, separate review of the medical literature identified 21 controlled trials of 16 interventions to improve communication in ICUs between families and care providers. Overall, the interventions improved emotional outcomes for families and reduced ICU length of stay and treatment intensity (Chest 2011;139:543-54), she noted.
Yet another prior review of the literature reported that interventions to promote family meetings, use empathetic communication skills, and employ palliative care consultations improved family satisfaction and reduced ICU length of stay and the adverse effects of family bereavement (Curr. Opin. Crit. Care 2009;15:569-77).
Dr. Aslakson reported having no financial disclosures.
sboschert@frontlinemedcom.com
On Twitter @sherryboschert
SAN FRANCISCO – Palliative care in the intensive care unit reduces the length of stay in the ICU and the hospital without changing mortality rates or family satisfaction, according to a review of the literature.
Although measurements of family satisfaction overall didn’t change much from palliative care of a loved one in the ICU, some measures of components of satisfaction increased with palliative care, such as improved communication with the physician, better consensus around the goals of care, and decreased anxiety and depression in family members, reported Dr. Rebecca A. Aslakson of Johns Hopkins University, Baltimore.
The findings have been submitted for publication, she said at the Critical Care Congress, sponsored by the Society for Critical Care Medicine.
Dr. Aslakson and her associates were unable to perform a formal meta-analysis of the 37 published trials of palliative care in the ICU because of the heterogeneity of the studies, which looked at more than 40 different outcomes. Instead, their systematic review grouped results under four outcomes that commonly were measured, and assessed those either by the number of studies or by the number of patients studied.
ICU length of stay decreased with palliative care in 13 of 21 studies (62%) that used this outcome and in 59% of 9,368 patients in those studies. Hospital length of stay decreased with palliative care in 8 of 14 studies (57%) and in 43% of 5,817 patients. Family satisfaction did not decrease in any studies or families and increased in only 1 of 14 studies (7%) and in 2% of families of 4,927 patients, Dr. Aslakson reported.
Mortality rates did not change with palliative care in 14 of 16 studies (88%) that assessed mortality and in 57% of 5,969 patients in those studies. Mortality increased in one small study (6%) and decreased in one larger study (6%).
"Talking about big-picture issues and goals of care doesn’t lead to people dying," Dr. Aslakson said. "No harm came in any of these studies." Some separate studies of palliative care outside of ICUs reported that this increases hope, "because people feel that they have more control over their choices and what’s happening to their loved ones," she added.
Integrative vs. consultative model
Dr. Aslakson and her associates also reviewed studies based on whether the interventions used integrative or consultative models of palliative care.
Generally, consultative models bring outsiders into the ICU to help provide palliative care, and integrative models train the ICU team to be the palliative care providers. In reality, the two models may overlap. For this review, the investigators applied mutually exclusive definitions to 36 of the studies. In 18 studies of integrative interventions, members of the ICU team were the only caregivers in face-to-face interactions with the patient and families. In 18 studies of consultative interventions, palliative care providers included others besides the ICU team.
In the studies of integrative palliative care, ICU length of stay decreased with palliative care in four of nine studies (44%) that measured this outcome and in 52% of 6,963 patients in those studies, she reported. Hospital length of stay decreased in two of five studies (40%) and in 24% of 3,812 patients. Family satisfaction changed in none of 15 studies, and mortality decreased in 1 of 5 studies (20%) and in 34% of 3,807 patients.
In the studies of consultative care, ICU length of stay decreased with palliative care in 9 of 12 studies (75%) that measured this outcome and in 79% of 2,405 patients in those studies. Hospital length of stay decreased in six of nine studies (67%) and in 79% of 2,005 patients. Family satisfaction increased in one of four studies (25%) and in 21% of 429 patients. Mortality increased in 1 of 11 studies (9%) and in 5% of 2,162 patients.
One model isn’t necessarily better than the other, Dr. Aslakson said. Integrative palliative care may work best in a closed ICU with perhaps four or five intensivists in a relatively small unit. An integrative approach can be much more difficult in open or semiopen ICUs that have "40 different doctors floating around," she said. "We tried that in my unit, and it didn’t work that well."
Different ICUs need palliative care models that fit them. "Look at your unit, the way it works, and who the providers are, then look at the literature and see what matches that and what might work for your unit," she said.
Outcomes of improved communication
A previous, separate review of the medical literature identified 21 controlled trials of 16 interventions to improve communication in ICUs between families and care providers. Overall, the interventions improved emotional outcomes for families and reduced ICU length of stay and treatment intensity (Chest 2011;139:543-54), she noted.
Yet another prior review of the literature reported that interventions to promote family meetings, use empathetic communication skills, and employ palliative care consultations improved family satisfaction and reduced ICU length of stay and the adverse effects of family bereavement (Curr. Opin. Crit. Care 2009;15:569-77).
Dr. Aslakson reported having no financial disclosures.
sboschert@frontlinemedcom.com
On Twitter @sherryboschert
SAN FRANCISCO – Palliative care in the intensive care unit reduces the length of stay in the ICU and the hospital without changing mortality rates or family satisfaction, according to a review of the literature.
Although measurements of family satisfaction overall didn’t change much from palliative care of a loved one in the ICU, some measures of components of satisfaction increased with palliative care, such as improved communication with the physician, better consensus around the goals of care, and decreased anxiety and depression in family members, reported Dr. Rebecca A. Aslakson of Johns Hopkins University, Baltimore.
The findings have been submitted for publication, she said at the Critical Care Congress, sponsored by the Society for Critical Care Medicine.
Dr. Aslakson and her associates were unable to perform a formal meta-analysis of the 37 published trials of palliative care in the ICU because of the heterogeneity of the studies, which looked at more than 40 different outcomes. Instead, their systematic review grouped results under four outcomes that commonly were measured, and assessed those either by the number of studies or by the number of patients studied.
ICU length of stay decreased with palliative care in 13 of 21 studies (62%) that used this outcome and in 59% of 9,368 patients in those studies. Hospital length of stay decreased with palliative care in 8 of 14 studies (57%) and in 43% of 5,817 patients. Family satisfaction did not decrease in any studies or families and increased in only 1 of 14 studies (7%) and in 2% of families of 4,927 patients, Dr. Aslakson reported.
Mortality rates did not change with palliative care in 14 of 16 studies (88%) that assessed mortality and in 57% of 5,969 patients in those studies. Mortality increased in one small study (6%) and decreased in one larger study (6%).
"Talking about big-picture issues and goals of care doesn’t lead to people dying," Dr. Aslakson said. "No harm came in any of these studies." Some separate studies of palliative care outside of ICUs reported that this increases hope, "because people feel that they have more control over their choices and what’s happening to their loved ones," she added.
Integrative vs. consultative model
Dr. Aslakson and her associates also reviewed studies based on whether the interventions used integrative or consultative models of palliative care.
Generally, consultative models bring outsiders into the ICU to help provide palliative care, and integrative models train the ICU team to be the palliative care providers. In reality, the two models may overlap. For this review, the investigators applied mutually exclusive definitions to 36 of the studies. In 18 studies of integrative interventions, members of the ICU team were the only caregivers in face-to-face interactions with the patient and families. In 18 studies of consultative interventions, palliative care providers included others besides the ICU team.
In the studies of integrative palliative care, ICU length of stay decreased with palliative care in four of nine studies (44%) that measured this outcome and in 52% of 6,963 patients in those studies, she reported. Hospital length of stay decreased in two of five studies (40%) and in 24% of 3,812 patients. Family satisfaction changed in none of 15 studies, and mortality decreased in 1 of 5 studies (20%) and in 34% of 3,807 patients.
In the studies of consultative care, ICU length of stay decreased with palliative care in 9 of 12 studies (75%) that measured this outcome and in 79% of 2,405 patients in those studies. Hospital length of stay decreased in six of nine studies (67%) and in 79% of 2,005 patients. Family satisfaction increased in one of four studies (25%) and in 21% of 429 patients. Mortality increased in 1 of 11 studies (9%) and in 5% of 2,162 patients.
One model isn’t necessarily better than the other, Dr. Aslakson said. Integrative palliative care may work best in a closed ICU with perhaps four or five intensivists in a relatively small unit. An integrative approach can be much more difficult in open or semiopen ICUs that have "40 different doctors floating around," she said. "We tried that in my unit, and it didn’t work that well."
Different ICUs need palliative care models that fit them. "Look at your unit, the way it works, and who the providers are, then look at the literature and see what matches that and what might work for your unit," she said.
Outcomes of improved communication
A previous, separate review of the medical literature identified 21 controlled trials of 16 interventions to improve communication in ICUs between families and care providers. Overall, the interventions improved emotional outcomes for families and reduced ICU length of stay and treatment intensity (Chest 2011;139:543-54), she noted.
Yet another prior review of the literature reported that interventions to promote family meetings, use empathetic communication skills, and employ palliative care consultations improved family satisfaction and reduced ICU length of stay and the adverse effects of family bereavement (Curr. Opin. Crit. Care 2009;15:569-77).
Dr. Aslakson reported having no financial disclosures.
sboschert@frontlinemedcom.com
On Twitter @sherryboschert
AT THE CRITICAL CARE CONGRESS
Mini-VADs could transform heart failure therapy
SNOWMASS, COLO. – Survival in patients with advanced heart failure who receive a left ventricular assist device as destination therapy or as a bridge to transplant has increased dramatically in recent years – and the best may be yet to come.
"There is a robust pipeline of mini-VADs [mini–ventricular assist devices] providing partial circulatory support that should allow wider applicability," Dr. Michael J. Mack said at the Annual Cardiovascular Conference at Snowmass.
This new generation of smaller, ever-more-high-tech VADs now entering clinical trials is made up of devices designed for implantation via a shorter, less invasive, off-pump hybrid surgical procedure, which should reduce operative morbidity and mortality. The rechargeable battery will also be implanted. Some of the devices are even earmarked for implantation nonsurgically in the cath lab, explained Dr. Mack, a cardiothoracic surgeon who is medical director of the Baylor Health Care System in Plano, Tex.

The ease of implantation of these next-gen mini-VADs makes them suitable for use at an earlier stage in the development of heart failure than the two left ventricular assist devices (LVADs) now approved for long-term use in the United States. The expectation is that this earlier mechanical intervention might further improve patient outcomes, he added.
Two-year survival following implantation of the HeartMate II, the current workhorse LVAD, is 65%, the same as with heart transplantation. To put that in perspective, in the REMATCH trial, which more than a decade ago led to Food and Drug Administration (FDA) approval of the first LVAD as long-term therapy, 2-year survival with device therapy was just 25%.
Today at many top institutions, Dr. Mack said, the number of LVADs implanted as destination therapy – that is, as an alternative to heart transplantation – now dwarfs the number of heart transplants done. This reflects a widespread recognition that the organ donor pool is unlikely to grow larger despite many efforts. Indeed, the number of heart transplants performed in North America has remained constant at roughly 2,300 per year for the past 20 years.
The other LVAD approved as destination therapy or bridge to transplant is the HeartWare VAD. The FDA granted approval based upon the results of the 30-center, prospective ADVANCE trial, which demonstrated that the HeartWare VAD’s performance was noninferior to that of the HeartMate II (Circulation 2012;125:3191-200).
Both LVADs are small continuous-flow devices with only a single moving part. They are quiet and energy efficient. Their simplicity of design makes them less susceptible to mechanical wear and much more reliable than earlier-generation LVADs. Three-year device replacement rates are now less than 10%, according to Dr. Mack.
But the current LVADs have significant shortcomings. In a worldwide series of close to 6,000 patients who have received a device as destination therapy or bridge to transplant, the 3-year combined rate of bleeding, stroke, infection, device malfunction, or death was 86%. The 3-year stroke incidence was 19% (J. Heart Lung Transpl. 2013;32:141-56).
Bleeding is a major problem with these devices for three reasons. Virtually all LVAD recipients develop acquired von Willebrand’s disease. Moreover, patients are on warfarin because of their high stroke risk. And gastrointestinal bleeding is a significant problem due to the angiodysplasia and arteriovenous malformations that result from the nonpulsatile continuous blood flow.
Among the mini-VADs well along in the developmental pipeline are the MicroMed HeartAssist 5, which is 71 x 30 mm in size and weighs only 92 g. Also in development are the HeartWare Miniaturized VAD (MVAD), which is about the size of a golf ball, and the HeartMate III.
The device drawing the most interest from cardiac surgeons, however, is the CircuLite Synergy mini-VAD, which is the size of an AA battery. It’s implanted off-pump with a small right thoracotomy for left atrial access. The device sits in a pocket in the right deltoid-pectoral groove for axillary artery access. The inflow is from the left atrium, with outflow into the right subclavian artery.
This device will undergo further modification to a totally endovascular concept that avoids the right thoracotomy. Inflow will be through the subclavian vein via a transseptal puncture approach, with outflow into the subclavian artery.
"The idea is that this will become a cath lab procedure. The device will sit in a pocket similar to a pacemaker," Dr. Mack explained.
Although the future looks bright for mini-VADs, plenty of key questions about them still await answers, the surgeon noted. Among them: Will partial circulatory support be sufficient for all patients with advanced heart failure? Will it be possible to predict which patients with New York Heart Association class III heart failure are destined to progress to advanced disease, and if so, will early implantation of a partial support device in these less-sick patients have a favorable effect on their quality of life? Might it slow progression of heart failure, and perhaps even induce remission?
The answers are expected to come from ongoing and planned clinical trials of these investigational devices.

Dr. Clyde W. Yancy, who chaired the writing committee for the 2013 ACC/AHA (American College of Cardiology/American Heart Association) Guideline for the Management of Heart Failure (J. Am. Coll. Cardiol. 2013;62:e147-239), noted that the guidelines endorse LVADs as destination therapy or bridge to transplant or recovery in carefully selected patients with advanced heart failure with reduced ejection fraction, with a class IIa/level of evidence B recommendation.
"Mechanical circulatory support is no longer a Hail Mary pass. This is no longer an experimental intervention. This really should be incorporated in our usual thought processes in advanced heart failure," said Dr. Yancy, professor of medicine and of medical social sciences and chief of cardiology at Northwestern University, Chicago.
He noted that the guidelines list a number of clinical events and other findings that are useful in identifying patients who have progressed to advanced heart failure, at which point he said it’s time to communicate with a center that can evaluate the patient for a possible LVAD or heart transplantation. These indicators include two or more hospitalizations or emergency department visits for heart failure in the past year, progressive deterioration in renal function, intolerance to beta-blocker therapy, unexplained weight loss, or inability to walk one block on level ground because of fatigue or shortness of breath.
Dr. Mack and Dr. Yancy reported having no financial conflicts.
SNOWMASS, COLO. – Survival in patients with advanced heart failure who receive a left ventricular assist device as destination therapy or as a bridge to transplant has increased dramatically in recent years – and the best may be yet to come.
"There is a robust pipeline of mini-VADs [mini–ventricular assist devices] providing partial circulatory support that should allow wider applicability," Dr. Michael J. Mack said at the Annual Cardiovascular Conference at Snowmass.
This new generation of smaller, ever-more-high-tech VADs now entering clinical trials is made up of devices designed for implantation via a shorter, less invasive, off-pump hybrid surgical procedure, which should reduce operative morbidity and mortality. The rechargeable battery will also be implanted. Some of the devices are even earmarked for implantation nonsurgically in the cath lab, explained Dr. Mack, a cardiothoracic surgeon who is medical director of the Baylor Health Care System in Plano, Tex.
The ease of implantation of these next-gen mini-VADs makes them suitable for use at an earlier stage in the development of heart failure than the two left ventricular assist devices (LVADs) now approved for long-term use in the United States. The expectation is that this earlier mechanical intervention might further improve patient outcomes, he added.
Two-year survival following implantation of the HeartMate II, the current workhorse LVAD, is 65%, the same as with heart transplantation. To put that in perspective, in the REMATCH trial, which more than a decade ago led to Food and Drug Administration (FDA) approval of the first LVAD as long-term therapy, 2-year survival with device therapy was just 25%.
Today at many top institutions, Dr. Mack said, the number of LVADs implanted as destination therapy – that is, as an alternative to heart transplantation – now dwarfs the number of heart transplants done. This reflects a widespread recognition that the organ donor pool is unlikely to grow larger despite many efforts. Indeed, the number of heart transplants performed in North America has remained constant at roughly 2,300 per year for the past 20 years.
The other LVAD approved as destination therapy or bridge to transplant is the HeartWare VAD. The FDA granted approval based upon the results of the 30-center, prospective ADVANCE trial, which demonstrated that the HeartWare VAD’s performance was noninferior to that of the HeartMate II (Circulation 2012;125:3191-200).
Both LVADs are small continuous-flow devices with only a single moving part. They are quiet and energy efficient. Their simplicity of design makes them less susceptible to mechanical wear and much more reliable than earlier-generation LVADs. Three-year device replacement rates are now less than 10%, according to Dr. Mack.
But the current LVADs have significant shortcomings. In a worldwide series of close to 6,000 patients who have received a device as destination therapy or bridge to transplant, the 3-year combined rate of bleeding, stroke, infection, device malfunction, or death was 86%. The 3-year stroke incidence was 19% (J. Heart Lung Transpl. 2013;32:141-56).
Bleeding is a major problem with these devices for three reasons. Virtually all LVAD recipients develop acquired von Willebrand’s disease. Moreover, patients are on warfarin because of their high stroke risk. And gastrointestinal bleeding is a significant problem due to the angiodysplasia and arteriovenous malformations that result from the nonpulsatile continuous blood flow.
Among the mini-VADs well along in the developmental pipeline are the MicroMed HeartAssist 5, which is 71 x 30 mm in size and weighs only 92 g. Also in development are the HeartWare Miniaturized VAD (MVAD), which is about the size of a golf ball, and the HeartMate III.
The device drawing the most interest from cardiac surgeons, however, is the CircuLite Synergy mini-VAD, which is the size of an AA battery. It’s implanted off-pump with a small right thoracotomy for left atrial access. The device sits in a pocket in the right deltoid-pectoral groove for axillary artery access. The inflow is from the left atrium, with outflow into the right subclavian artery.
This device will undergo further modification to a totally endovascular concept that avoids the right thoracotomy. Inflow will be through the subclavian vein via a transseptal puncture approach, with outflow into the subclavian artery.
"The idea is that this will become a cath lab procedure. The device will sit in a pocket similar to a pacemaker," Dr. Mack explained.
Although the future looks bright for mini-VADs, plenty of key questions about them still await answers, the surgeon noted. Among them: Will partial circulatory support be sufficient for all patients with advanced heart failure? Will it be possible to predict which patients with New York Heart Association class III heart failure are destined to progress to advanced disease, and if so, will early implantation of a partial support device in these less-sick patients have a favorable effect on their quality of life? Might it slow progression of heart failure, and perhaps even induce remission?
The answers are expected to come from ongoing and planned clinical trials of these investigational devices.
Dr. Clyde W. Yancy, who chaired the writing committee for the 2013 ACC/AHA (American College of Cardiology/American Heart Association) Guideline for the Management of Heart Failure (J. Am. Coll. Cardiol. 2013;62:e147-239), noted that the guidelines endorse LVADs as destination therapy or bridge to transplant or recovery in carefully selected patients with advanced heart failure with reduced ejection fraction, with a class IIa/level of evidence B recommendation.
"Mechanical circulatory support is no longer a Hail Mary pass. This is no longer an experimental intervention. This really should be incorporated in our usual thought processes in advanced heart failure," said Dr. Yancy, professor of medicine and of medical social sciences and chief of cardiology at Northwestern University, Chicago.
He noted that the guidelines list a number of clinical events and other findings that are useful in identifying patients who have progressed to advanced heart failure, at which point he said it’s time to communicate with a center that can evaluate the patient for a possible LVAD or heart transplantation. These indicators include two or more hospitalizations or emergency department visits for heart failure in the past year, progressive deterioration in renal function, intolerance to beta-blocker therapy, unexplained weight loss, or inability to walk one block on level ground because of fatigue or shortness of breath.
Dr. Mack and Dr. Yancy reported having no financial conflicts.
SNOWMASS, COLO. – Survival in patients with advanced heart failure who receive a left ventricular assist device as destination therapy or as a bridge to transplant has increased dramatically in recent years – and the best may be yet to come.
"There is a robust pipeline of mini-VADs [mini–ventricular assist devices] providing partial circulatory support that should allow wider applicability," Dr. Michael J. Mack said at the Annual Cardiovascular Conference at Snowmass.
This new generation of smaller, ever-more-high-tech VADs now entering clinical trials is made up of devices designed for implantation via a shorter, less invasive, off-pump hybrid surgical procedure, which should reduce operative morbidity and mortality. The rechargeable battery will also be implanted. Some of the devices are even earmarked for implantation nonsurgically in the cath lab, explained Dr. Mack, a cardiothoracic surgeon who is medical director of the Baylor Health Care System in Plano, Tex.
The ease of implantation of these next-gen mini-VADs makes them suitable for use at an earlier stage in the development of heart failure than the two left ventricular assist devices (LVADs) now approved for long-term use in the United States. The expectation is that this earlier mechanical intervention might further improve patient outcomes, he added.
Two-year survival following implantation of the HeartMate II, the current workhorse LVAD, is 65%, the same as with heart transplantation. To put that in perspective, in the REMATCH trial, which more than a decade ago led to Food and Drug Administration (FDA) approval of the first LVAD as long-term therapy, 2-year survival with device therapy was just 25%.
Today at many top institutions, Dr. Mack said, the number of LVADs implanted as destination therapy – that is, as an alternative to heart transplantation – now dwarfs the number of heart transplants done. This reflects a widespread recognition that the organ donor pool is unlikely to grow larger despite many efforts. Indeed, the number of heart transplants performed in North America has remained constant at roughly 2,300 per year for the past 20 years.
The other LVAD approved as destination therapy or bridge to transplant is the HeartWare VAD. The FDA granted approval based upon the results of the 30-center, prospective ADVANCE trial, which demonstrated that the HeartWare VAD’s performance was noninferior to that of the HeartMate II (Circulation 2012;125:3191-200).
Both LVADs are small continuous-flow devices with only a single moving part. They are quiet and energy efficient. Their simplicity of design makes them less susceptible to mechanical wear and much more reliable than earlier-generation LVADs. Three-year device replacement rates are now less than 10%, according to Dr. Mack.
But the current LVADs have significant shortcomings. In a worldwide series of close to 6,000 patients who have received a device as destination therapy or bridge to transplant, the 3-year combined rate of bleeding, stroke, infection, device malfunction, or death was 86%. The 3-year stroke incidence was 19% (J. Heart Lung Transpl. 2013;32:141-56).
Bleeding is a major problem with these devices for three reasons. Virtually all LVAD recipients develop acquired von Willebrand’s disease. Moreover, patients are on warfarin because of their high stroke risk. And gastrointestinal bleeding is a significant problem due to the angiodysplasia and arteriovenous malformations that result from the nonpulsatile continuous blood flow.
Among the mini-VADs well along in the developmental pipeline are the MicroMed HeartAssist 5, which is 71 x 30 mm in size and weighs only 92 g. Also in development are the HeartWare Miniaturized VAD (MVAD), which is about the size of a golf ball, and the HeartMate III.
The device drawing the most interest from cardiac surgeons, however, is the CircuLite Synergy mini-VAD, which is the size of an AA battery. It’s implanted off-pump with a small right thoracotomy for left atrial access. The device sits in a pocket in the right deltoid-pectoral groove for axillary artery access. The inflow is from the left atrium, with outflow into the right subclavian artery.
This device will undergo further modification to a totally endovascular concept that avoids the right thoracotomy. Inflow will be through the subclavian vein via a transseptal puncture approach, with outflow into the subclavian artery.
"The idea is that this will become a cath lab procedure. The device will sit in a pocket similar to a pacemaker," Dr. Mack explained.
Although the future looks bright for mini-VADs, plenty of key questions about them still await answers, the surgeon noted. Among them: Will partial circulatory support be sufficient for all patients with advanced heart failure? Will it be possible to predict which patients with New York Heart Association class III heart failure are destined to progress to advanced disease, and if so, will early implantation of a partial support device in these less-sick patients have a favorable effect on their quality of life? Might it slow progression of heart failure, and perhaps even induce remission?
The answers are expected to come from ongoing and planned clinical trials of these investigational devices.
Dr. Clyde W. Yancy, who chaired the writing committee for the 2013 ACC/AHA (American College of Cardiology/American Heart Association) Guideline for the Management of Heart Failure (J. Am. Coll. Cardiol. 2013;62:e147-239), noted that the guidelines endorse LVADs as destination therapy or bridge to transplant or recovery in carefully selected patients with advanced heart failure with reduced ejection fraction, with a class IIa/level of evidence B recommendation.
"Mechanical circulatory support is no longer a Hail Mary pass. This is no longer an experimental intervention. This really should be incorporated in our usual thought processes in advanced heart failure," said Dr. Yancy, professor of medicine and of medical social sciences and chief of cardiology at Northwestern University, Chicago.
He noted that the guidelines list a number of clinical events and other findings that are useful in identifying patients who have progressed to advanced heart failure, at which point he said it’s time to communicate with a center that can evaluate the patient for a possible LVAD or heart transplantation. These indicators include two or more hospitalizations or emergency department visits for heart failure in the past year, progressive deterioration in renal function, intolerance to beta-blocker therapy, unexplained weight loss, or inability to walk one block on level ground because of fatigue or shortness of breath.
Dr. Mack and Dr. Yancy reported having no financial conflicts.
EXPERT ANALYSIS FROM THE CARDIOVASCULAR CONFERENCE AT SNOWMASS

FDA to probe saxagliptin’s heart failure risk
The U.S. Food and Drug Administration will investigate the possible association between treatment with the type 2 diabetes drug saxagliptin and an increased rate of hospitalizations for heart failure using data collected by the drug’s manufacturer during clinical trials. The agency released a "safety announcement" on Feb.11 that it had requested these data from Bristol-Myers Squibb and AstraZeneca, the two companies that together market saxagliptin as Onglyza and Kombiglyze XR.
The agency based its request on results reported last year in the SAVOR (Saxagliptin Assessment of Vascular Outcomes Recorded in Patients With Diabetes Mellitus)–TIMI 53 (Thrombolysis in Myocardial Infarction) trial, a trial with more than 16,000 patients with type 2 diabetes designed to test the cardiovascular safety of saxagliptin (N. Engl. J. Med. 2013;369:1317-26).
Although treatment with the drug produced no excess in the rate of the primary endpoint of all cardiovascular events, compared with a control group, the results showed a statistically significant, 0.7% absolute increase in the rate of hospitalizations for heart failure in the saxagliptin-treated patients, compared with those not on the drug, a 27% relative increase.
While presenting more detailed data on the heart failure hospitalization effect in a poster during the American Heart Association’s Scientific Sessions last November, Dr. Benjamin M. Scirica, lead author for the study, called the effect by saxagliptin on raising heart failure hospitalizations "real."
"Every drug has adverse effects. With these data, the calculation about saxagliptin can be made to a much more certain level. Clinicians need to weigh a patient’s risk factors, which drugs they can or cannot take, and a drug’s overall safety for ischemic events," said Dr. Scirica while presenting his poster last November. "At this point, we have no definitive data to a priori say don’t give saxagliptin to patients with heart failure."
The manufacturers are expected to submit the saxagliptin-trial data to the FDA by March 2014, after which the agency said it will "conduct a thorough analysis and report our findings publically."
The FDA’s statement added this caution to patients and physicians about its action:
"At this time, we consider information from the New England Journal of Medicine study to be preliminary. Our analysis of the saxagliptin clinical trial data is part of a broader evaluation of all type 2 diabetes drug therapies and cardiovascular risk. Patients should not stop taking saxagliptin and should speak with their health care professionals about any questions or concerns. Health care professionals should continue to follow the prescribing recommendations in the drug labels." The FDA also urged health care professionals and patients to report side effects involving saxagliptin products to the FDA MedWatch program.
Dr. Scirica said that he had been a consultant to Bristol-Myers Squibb and to several other drug companies and that he has received research grants from Bristol-Myers Squibb and AstraZeneca and from several other companies.
On Twitter @mitchelzoler
The U.S. Food and Drug Administration will investigate the possible association between treatment with the type 2 diabetes drug saxagliptin and an increased rate of hospitalizations for heart failure using data collected by the drug’s manufacturer during clinical trials. The agency released a "safety announcement" on Feb.11 that it had requested these data from Bristol-Myers Squibb and AstraZeneca, the two companies that together market saxagliptin as Onglyza and Kombiglyze XR.
The agency based its request on results reported last year in the SAVOR (Saxagliptin Assessment of Vascular Outcomes Recorded in Patients With Diabetes Mellitus)–TIMI 53 (Thrombolysis in Myocardial Infarction) trial, a trial with more than 16,000 patients with type 2 diabetes designed to test the cardiovascular safety of saxagliptin (N. Engl. J. Med. 2013;369:1317-26).
Although treatment with the drug produced no excess in the rate of the primary endpoint of all cardiovascular events, compared with a control group, the results showed a statistically significant, 0.7% absolute increase in the rate of hospitalizations for heart failure in the saxagliptin-treated patients, compared with those not on the drug, a 27% relative increase.
While presenting more detailed data on the heart failure hospitalization effect in a poster during the American Heart Association’s Scientific Sessions last November, Dr. Benjamin M. Scirica, lead author for the study, called the effect by saxagliptin on raising heart failure hospitalizations "real."
"Every drug has adverse effects. With these data, the calculation about saxagliptin can be made to a much more certain level. Clinicians need to weigh a patient’s risk factors, which drugs they can or cannot take, and a drug’s overall safety for ischemic events," said Dr. Scirica while presenting his poster last November. "At this point, we have no definitive data to a priori say don’t give saxagliptin to patients with heart failure."
The manufacturers are expected to submit the saxagliptin-trial data to the FDA by March 2014, after which the agency said it will "conduct a thorough analysis and report our findings publically."
The FDA’s statement added this caution to patients and physicians about its action:
"At this time, we consider information from the New England Journal of Medicine study to be preliminary. Our analysis of the saxagliptin clinical trial data is part of a broader evaluation of all type 2 diabetes drug therapies and cardiovascular risk. Patients should not stop taking saxagliptin and should speak with their health care professionals about any questions or concerns. Health care professionals should continue to follow the prescribing recommendations in the drug labels." The FDA also urged health care professionals and patients to report side effects involving saxagliptin products to the FDA MedWatch program.
Dr. Scirica said that he had been a consultant to Bristol-Myers Squibb and to several other drug companies and that he has received research grants from Bristol-Myers Squibb and AstraZeneca and from several other companies.
On Twitter @mitchelzoler
The U.S. Food and Drug Administration will investigate the possible association between treatment with the type 2 diabetes drug saxagliptin and an increased rate of hospitalizations for heart failure using data collected by the drug’s manufacturer during clinical trials. The agency released a "safety announcement" on Feb.11 that it had requested these data from Bristol-Myers Squibb and AstraZeneca, the two companies that together market saxagliptin as Onglyza and Kombiglyze XR.
The agency based its request on results reported last year in the SAVOR (Saxagliptin Assessment of Vascular Outcomes Recorded in Patients With Diabetes Mellitus)–TIMI 53 (Thrombolysis in Myocardial Infarction) trial, a trial with more than 16,000 patients with type 2 diabetes designed to test the cardiovascular safety of saxagliptin (N. Engl. J. Med. 2013;369:1317-26).
Although treatment with the drug produced no excess in the rate of the primary endpoint of all cardiovascular events, compared with a control group, the results showed a statistically significant, 0.7% absolute increase in the rate of hospitalizations for heart failure in the saxagliptin-treated patients, compared with those not on the drug, a 27% relative increase.
While presenting more detailed data on the heart failure hospitalization effect in a poster during the American Heart Association’s Scientific Sessions last November, Dr. Benjamin M. Scirica, lead author for the study, called the effect by saxagliptin on raising heart failure hospitalizations "real."
"Every drug has adverse effects. With these data, the calculation about saxagliptin can be made to a much more certain level. Clinicians need to weigh a patient’s risk factors, which drugs they can or cannot take, and a drug’s overall safety for ischemic events," said Dr. Scirica while presenting his poster last November. "At this point, we have no definitive data to a priori say don’t give saxagliptin to patients with heart failure."
The manufacturers are expected to submit the saxagliptin-trial data to the FDA by March 2014, after which the agency said it will "conduct a thorough analysis and report our findings publically."
The FDA’s statement added this caution to patients and physicians about its action:
"At this time, we consider information from the New England Journal of Medicine study to be preliminary. Our analysis of the saxagliptin clinical trial data is part of a broader evaluation of all type 2 diabetes drug therapies and cardiovascular risk. Patients should not stop taking saxagliptin and should speak with their health care professionals about any questions or concerns. Health care professionals should continue to follow the prescribing recommendations in the drug labels." The FDA also urged health care professionals and patients to report side effects involving saxagliptin products to the FDA MedWatch program.
Dr. Scirica said that he had been a consultant to Bristol-Myers Squibb and to several other drug companies and that he has received research grants from Bristol-Myers Squibb and AstraZeneca and from several other companies.
On Twitter @mitchelzoler
CoreValve holds size advantage for U.S. TAVR
When the Food and Drug Administration in January granted marketing approval to a second transcatheter aortic valve replacement system for inoperable patients with aortic stenosis, the CoreValve marketed by Medtronic, the new valve conceded a greater than 2-year head start to the first system on the U.S. market, Sapien marketed by Edwards.
But cardiologists see that 2-year edge in familiarity eclipsed for at least some patients by two major advantages that CoreValve currently holds over Sapien: delivery via a significantly thinner sheath, and the option of larger-diameter valves that allow replacement in patients with a wider aortic annulus.

The CoreValve delivery sheath is 18 French, compared with a 22F or 24F size for the Sapien transcatheter aortic valve replacement (TAVR) with U.S. approval, and the Sapien valves come in diameters of 23 and 26 mm, compared with options of 23, 26, 29, and 31 mm for the CoreValve.
"CoreValve is the device of choice for patients with smaller vessel sizes. Sapien has been a wonderful device to use, and we have so much experience with it, but the smaller CoreValve size will allow many more patients to be done with a transfemoral approach," said Dr. Peter C. Block, a professor of medicine at Emory University in Atlanta and an interventional cardiologist who performs TAVR.
"More patients will qualify for TAVR and will be treated transfemorally with the larger valve diameters and smaller sheath size," agreed Dr. Mauricio G. Cohen, director of cardiac catheterization at the University of Miami and TAVR interventionalist. Another potential positive of having CoreValve on the U.S. market is that competition between the two options will likely drive down the cost of a TAVR system, which until now has run more than $30,000, Dr. Cohen said in an interview.
CoreValve received FDA approval less than 3 months after researchers first reported data from CoreValve’s U.S. Pivotal Trial Extreme Risk Iliofemoral Study last October at the TCT (Transcatheter Cardiovascular Therapeutics) annual meeting. In that study, 471 inoperable aortic stenosis patients had a 26% 1-year rate of death or major stroke, substantially surpassing the 43% rate that the study set up as the target for superiority, reported Dr. Jeffrey J. Popma, lead investigator on the study.
Dr. Popma warned against comparing CoreValve’s efficacy and safety performance in the trial and the Sapien system’s performance in its pivotal trial in inoperable patients, the PARTNER cohort B trial (N. Engl. J. Med. 2010;363:1597-607). "It’s very difficult to make cross-trial comparison," he said in an interview, a limitation also noted by Dr. Cohen and Dr. Block. But Dr. Popma highlighted the 2.4% 30-day stroke rate in the pivotal trial, and a 1.8% 30-day stroke rate seen with CoreValve in inoperable patients in a continued access program at the trial’s study sites. He also highlighted the 11% rate of moderate paravalvular aortic regurgitation after 30 days that dropped to a 4% rate after 1 year.
Perhaps the biggest downside of CoreValve’s performance in the pivotal trial was that 22% of patients required a permanent pacemaker implant within the first 30 days, increasing to 27% of patients with 1-year follow-up. Increased risk for a pacemaker is an inherent downside of CoreValve because of its longer size compared with the Sapien valve and how the CoreValve sits in the aortic annulus. The CoreValve is designed for supravalvular placement and anchoring in the left ventricular outflow tract near the left bundle branch that can result in mechanical irritation and arrhythmia with the need for pacing, explained Dr. Popma, professor of medicine at Harvard University and an interventional cardiologist at Beth Israel Deaconess Medical Center, Boston.
"I think our pacemaker rate was very acceptable. I don’t think it will ever be as low as with Sapien, but it’s a worthwhile trade-off because the CoreValve functions well and results in a low rate of paravalvular regurgitation," he said.
Dr. Popma also stressed that 1-year mortality was no greater among the patients who required a pacemaker implant in the pivotal trial. A subgroup analysis of results from the trial to try to identify which patients had the greatest risk for needing a pacemaker after a CoreValve implant has not yet finished, he said. It’s possible that certain patients with preexisting conduction abnormalities, such as a right bundle branch block coupled with a left anterior fascicular block, have the greatest vulnerability.
Patients for whom the Sapien system remains ideal are those with a narrow sinus of Valsalva, because the longer CoreValve frame crosses the sinus and may compromise coronary blood flow in patients with a narrow sinus, Dr. Popma said.
The choice between CoreValve and Sapien systems will grow even more complicated for U.S. cardiologists and surgeons when the Sapien XT valve system receives FDA marketing approval, likely later this year. The Sapien XT delivery sheath matches the 18F size of CoreValve and will also come in a 29-mm size, blunting two of CoreValves main advantages.
Medtronic also faces charges of patent infringement by its CoreValve in a court case initiated by Edwards. In mid-January, a jury in a U.S. District Court assessed a penalty of $394 million against Medtronic. Edwards is also seeking a court-ordered halt to U.S. marketing of CoreValve. But Medtronic* is appealing the jury verdict and continues to fight the injunction, and a company spokesperson said in an interview that the legal maneuverings will likely take at least another year to fully resolve. In the meantime, Medtronic began U.S. distribution of the CoreValve on Jan. 17.
Dr. Block said that his institution received a research grant to participate in Sapien trials. Dr. Cohen said that he has been a consultant to Medtronic and Edwards. Dr. Popma* said that his institution received research support from Medtronic and that he has been a consultant to and received research support from Boston Scientific.
On Twitter @mitchelzoler
*CORRECTION, 1/30/2014: In an earlier version of this article, the appelant was misidentified.
When the Food and Drug Administration in January granted marketing approval to a second transcatheter aortic valve replacement system for inoperable patients with aortic stenosis, the CoreValve marketed by Medtronic, the new valve conceded a greater than 2-year head start to the first system on the U.S. market, Sapien marketed by Edwards.
But cardiologists see that 2-year edge in familiarity eclipsed for at least some patients by two major advantages that CoreValve currently holds over Sapien: delivery via a significantly thinner sheath, and the option of larger-diameter valves that allow replacement in patients with a wider aortic annulus.
The CoreValve delivery sheath is 18 French, compared with a 22F or 24F size for the Sapien transcatheter aortic valve replacement (TAVR) with U.S. approval, and the Sapien valves come in diameters of 23 and 26 mm, compared with options of 23, 26, 29, and 31 mm for the CoreValve.
"CoreValve is the device of choice for patients with smaller vessel sizes. Sapien has been a wonderful device to use, and we have so much experience with it, but the smaller CoreValve size will allow many more patients to be done with a transfemoral approach," said Dr. Peter C. Block, a professor of medicine at Emory University in Atlanta and an interventional cardiologist who performs TAVR.
"More patients will qualify for TAVR and will be treated transfemorally with the larger valve diameters and smaller sheath size," agreed Dr. Mauricio G. Cohen, director of cardiac catheterization at the University of Miami and TAVR interventionalist. Another potential positive of having CoreValve on the U.S. market is that competition between the two options will likely drive down the cost of a TAVR system, which until now has run more than $30,000, Dr. Cohen said in an interview.
CoreValve received FDA approval less than 3 months after researchers first reported data from CoreValve’s U.S. Pivotal Trial Extreme Risk Iliofemoral Study last October at the TCT (Transcatheter Cardiovascular Therapeutics) annual meeting. In that study, 471 inoperable aortic stenosis patients had a 26% 1-year rate of death or major stroke, substantially surpassing the 43% rate that the study set up as the target for superiority, reported Dr. Jeffrey J. Popma, lead investigator on the study.
Dr. Popma warned against comparing CoreValve’s efficacy and safety performance in the trial and the Sapien system’s performance in its pivotal trial in inoperable patients, the PARTNER cohort B trial (N. Engl. J. Med. 2010;363:1597-607). "It’s very difficult to make cross-trial comparison," he said in an interview, a limitation also noted by Dr. Cohen and Dr. Block. But Dr. Popma highlighted the 2.4% 30-day stroke rate in the pivotal trial, and a 1.8% 30-day stroke rate seen with CoreValve in inoperable patients in a continued access program at the trial’s study sites. He also highlighted the 11% rate of moderate paravalvular aortic regurgitation after 30 days that dropped to a 4% rate after 1 year.
Perhaps the biggest downside of CoreValve’s performance in the pivotal trial was that 22% of patients required a permanent pacemaker implant within the first 30 days, increasing to 27% of patients with 1-year follow-up. Increased risk for a pacemaker is an inherent downside of CoreValve because of its longer size compared with the Sapien valve and how the CoreValve sits in the aortic annulus. The CoreValve is designed for supravalvular placement and anchoring in the left ventricular outflow tract near the left bundle branch that can result in mechanical irritation and arrhythmia with the need for pacing, explained Dr. Popma, professor of medicine at Harvard University and an interventional cardiologist at Beth Israel Deaconess Medical Center, Boston.
"I think our pacemaker rate was very acceptable. I don’t think it will ever be as low as with Sapien, but it’s a worthwhile trade-off because the CoreValve functions well and results in a low rate of paravalvular regurgitation," he said.
Dr. Popma also stressed that 1-year mortality was no greater among the patients who required a pacemaker implant in the pivotal trial. A subgroup analysis of results from the trial to try to identify which patients had the greatest risk for needing a pacemaker after a CoreValve implant has not yet finished, he said. It’s possible that certain patients with preexisting conduction abnormalities, such as a right bundle branch block coupled with a left anterior fascicular block, have the greatest vulnerability.
Patients for whom the Sapien system remains ideal are those with a narrow sinus of Valsalva, because the longer CoreValve frame crosses the sinus and may compromise coronary blood flow in patients with a narrow sinus, Dr. Popma said.
The choice between CoreValve and Sapien systems will grow even more complicated for U.S. cardiologists and surgeons when the Sapien XT valve system receives FDA marketing approval, likely later this year. The Sapien XT delivery sheath matches the 18F size of CoreValve and will also come in a 29-mm size, blunting two of CoreValves main advantages.
Medtronic also faces charges of patent infringement by its CoreValve in a court case initiated by Edwards. In mid-January, a jury in a U.S. District Court assessed a penalty of $394 million against Medtronic. Edwards is also seeking a court-ordered halt to U.S. marketing of CoreValve. But Medtronic* is appealing the jury verdict and continues to fight the injunction, and a company spokesperson said in an interview that the legal maneuverings will likely take at least another year to fully resolve. In the meantime, Medtronic began U.S. distribution of the CoreValve on Jan. 17.
Dr. Block said that his institution received a research grant to participate in Sapien trials. Dr. Cohen said that he has been a consultant to Medtronic and Edwards. Dr. Popma* said that his institution received research support from Medtronic and that he has been a consultant to and received research support from Boston Scientific.
On Twitter @mitchelzoler
*CORRECTION, 1/30/2014: In an earlier version of this article, the appelant was misidentified.
When the Food and Drug Administration in January granted marketing approval to a second transcatheter aortic valve replacement system for inoperable patients with aortic stenosis, the CoreValve marketed by Medtronic, the new valve conceded a greater than 2-year head start to the first system on the U.S. market, Sapien marketed by Edwards.
But cardiologists see that 2-year edge in familiarity eclipsed for at least some patients by two major advantages that CoreValve currently holds over Sapien: delivery via a significantly thinner sheath, and the option of larger-diameter valves that allow replacement in patients with a wider aortic annulus.
The CoreValve delivery sheath is 18 French, compared with a 22F or 24F size for the Sapien transcatheter aortic valve replacement (TAVR) with U.S. approval, and the Sapien valves come in diameters of 23 and 26 mm, compared with options of 23, 26, 29, and 31 mm for the CoreValve.
"CoreValve is the device of choice for patients with smaller vessel sizes. Sapien has been a wonderful device to use, and we have so much experience with it, but the smaller CoreValve size will allow many more patients to be done with a transfemoral approach," said Dr. Peter C. Block, a professor of medicine at Emory University in Atlanta and an interventional cardiologist who performs TAVR.
"More patients will qualify for TAVR and will be treated transfemorally with the larger valve diameters and smaller sheath size," agreed Dr. Mauricio G. Cohen, director of cardiac catheterization at the University of Miami and TAVR interventionalist. Another potential positive of having CoreValve on the U.S. market is that competition between the two options will likely drive down the cost of a TAVR system, which until now has run more than $30,000, Dr. Cohen said in an interview.
CoreValve received FDA approval less than 3 months after researchers first reported data from CoreValve’s U.S. Pivotal Trial Extreme Risk Iliofemoral Study last October at the TCT (Transcatheter Cardiovascular Therapeutics) annual meeting. In that study, 471 inoperable aortic stenosis patients had a 26% 1-year rate of death or major stroke, substantially surpassing the 43% rate that the study set up as the target for superiority, reported Dr. Jeffrey J. Popma, lead investigator on the study.
Dr. Popma warned against comparing CoreValve’s efficacy and safety performance in the trial and the Sapien system’s performance in its pivotal trial in inoperable patients, the PARTNER cohort B trial (N. Engl. J. Med. 2010;363:1597-607). "It’s very difficult to make cross-trial comparison," he said in an interview, a limitation also noted by Dr. Cohen and Dr. Block. But Dr. Popma highlighted the 2.4% 30-day stroke rate in the pivotal trial, and a 1.8% 30-day stroke rate seen with CoreValve in inoperable patients in a continued access program at the trial’s study sites. He also highlighted the 11% rate of moderate paravalvular aortic regurgitation after 30 days that dropped to a 4% rate after 1 year.
Perhaps the biggest downside of CoreValve’s performance in the pivotal trial was that 22% of patients required a permanent pacemaker implant within the first 30 days, increasing to 27% of patients with 1-year follow-up. Increased risk for a pacemaker is an inherent downside of CoreValve because of its longer size compared with the Sapien valve and how the CoreValve sits in the aortic annulus. The CoreValve is designed for supravalvular placement and anchoring in the left ventricular outflow tract near the left bundle branch that can result in mechanical irritation and arrhythmia with the need for pacing, explained Dr. Popma, professor of medicine at Harvard University and an interventional cardiologist at Beth Israel Deaconess Medical Center, Boston.
"I think our pacemaker rate was very acceptable. I don’t think it will ever be as low as with Sapien, but it’s a worthwhile trade-off because the CoreValve functions well and results in a low rate of paravalvular regurgitation," he said.
Dr. Popma also stressed that 1-year mortality was no greater among the patients who required a pacemaker implant in the pivotal trial. A subgroup analysis of results from the trial to try to identify which patients had the greatest risk for needing a pacemaker after a CoreValve implant has not yet finished, he said. It’s possible that certain patients with preexisting conduction abnormalities, such as a right bundle branch block coupled with a left anterior fascicular block, have the greatest vulnerability.
Patients for whom the Sapien system remains ideal are those with a narrow sinus of Valsalva, because the longer CoreValve frame crosses the sinus and may compromise coronary blood flow in patients with a narrow sinus, Dr. Popma said.
The choice between CoreValve and Sapien systems will grow even more complicated for U.S. cardiologists and surgeons when the Sapien XT valve system receives FDA marketing approval, likely later this year. The Sapien XT delivery sheath matches the 18F size of CoreValve and will also come in a 29-mm size, blunting two of CoreValves main advantages.
Medtronic also faces charges of patent infringement by its CoreValve in a court case initiated by Edwards. In mid-January, a jury in a U.S. District Court assessed a penalty of $394 million against Medtronic. Edwards is also seeking a court-ordered halt to U.S. marketing of CoreValve. But Medtronic* is appealing the jury verdict and continues to fight the injunction, and a company spokesperson said in an interview that the legal maneuverings will likely take at least another year to fully resolve. In the meantime, Medtronic began U.S. distribution of the CoreValve on Jan. 17.
Dr. Block said that his institution received a research grant to participate in Sapien trials. Dr. Cohen said that he has been a consultant to Medtronic and Edwards. Dr. Popma* said that his institution received research support from Medtronic and that he has been a consultant to and received research support from Boston Scientific.
On Twitter @mitchelzoler
*CORRECTION, 1/30/2014: In an earlier version of this article, the appelant was misidentified.

Inpatient safety efforts yield mixed results
Adverse events decreased over the last decade for Medicare inpatients with acute myocardial infarction or heart failure but barely changed for those with pneumonia or conditions requiring surgery, according to an analysis of a Medicare database.
The improvements likely translated to 81,000 fewer adverse events for patients with acute MI (AMI) and heart failure (HF) from 2010 to 2011 alone, according to the study published Jan. 22 in the New England Journal of Medicine.
"Although this suggests that national efforts focused on patient safety have made some inroads, the lack of reductions across the board is disappointing," Yun Wang, Ph.D., of the Harvard School of Public Health, Boston, and his colleagues wrote.
The researchers examined whether hospitalized patients are any better off in light of the current focus on patient safety, including the launch of initiatives such as the American College of Surgeons’ National Surgical Quality Improvement Program and the federal government’s Surgical Infection Prevention Project.
They used three composite outcomes measures: the rate of occurrence for adverse events for which patients were at risk (for instance, only patients receiving warfarin were at risk for warfarin-related events); the proportion of patients with one or more adverse events; and the number of adverse events per 1,000 hospitalizations. They analyzed data on 61,523 patients who were discharged from 4,372 hospitals; the data were extracted from the Medicare Patient Safety Monitoring System database (N. Engl. J. Med. 2014:370;341-51).
The 61,523 patients included 11,399 with AMI, 15,374 with HF, 18,269 with pneumonia, and 16,481 with conditions requiring surgery. Postsurgical patients largely were being treated for joint replacement procedures and other osteoarthritis-related conditions, femur fracture, colon cancer, post-AMI procedures, or other forms of chronic ischemic heart disease.
From 2005-2006 to 2010-2011, AMI and HF patients saw a 1.3 percentage point decline in the rate of adverse events, from 5% to 3.7%. The proportion who had one or more such events decreased from 26% to 19%. The number of adverse events per 1,000 hospitalizations declined from 402 to 262 for AMI patients and from 235 to 167 for HF patients.
Infection-related and drug-related adverse events declined significantly in heart attack and HF patients. There was also a substantial improvement in postprocedure events in HF patients.
Postsurgical patients experienced slight increases in all three outcomes measures, in particular, increases in infection-related and postprocedural events such as venous thromboembolism, and cardiac and catheter-related events. The number of events per 1,000 hospitalizations for pneumonia patients increased insignificantly from 216 to 223. For postsurgical patients, the number of events increased insignificantly from 352/1,000 to 368/1,000.
Patients who had adverse events had significantly longer hospital stays and were at higher risk for death. As the number of adverse events increased, so did the risk of death.
The authors noted that declines in events for AMI and HF patients might be a reflection of the numerous efforts and initiatives to improve care in those two conditions.
But they also found that concerted efforts to improve safety did not necessarily work. There was an increase in pressure ulcers in postsurgical patients, and no decline in ventilator-associated pneumonia in most patients, even though there have been initiatives focused on those conditions.
"Our finding of an increased adverse-event rate among surgical patients indicates a continuing challenge and identifies an important target for patient-safety initiatives," the researchers said.
The study was supported by the Agency for Healthcare Research and Quality as well as academic and federal grants. Dr. Wang was a consultant to and other researchers were employees of the research firm Qualidigm and participated in the analysis. One of the researchers, Dr. Harlan M. Krumholz, disclosed being on a scientific advisory board for UnitedHealthcare and receiving grant money from Medtronic.
On Twitter @aliciaault
Adverse events decreased over the last decade for Medicare inpatients with acute myocardial infarction or heart failure but barely changed for those with pneumonia or conditions requiring surgery, according to an analysis of a Medicare database.
The improvements likely translated to 81,000 fewer adverse events for patients with acute MI (AMI) and heart failure (HF) from 2010 to 2011 alone, according to the study published Jan. 22 in the New England Journal of Medicine.
"Although this suggests that national efforts focused on patient safety have made some inroads, the lack of reductions across the board is disappointing," Yun Wang, Ph.D., of the Harvard School of Public Health, Boston, and his colleagues wrote.
The researchers examined whether hospitalized patients are any better off in light of the current focus on patient safety, including the launch of initiatives such as the American College of Surgeons’ National Surgical Quality Improvement Program and the federal government’s Surgical Infection Prevention Project.
They used three composite outcomes measures: the rate of occurrence for adverse events for which patients were at risk (for instance, only patients receiving warfarin were at risk for warfarin-related events); the proportion of patients with one or more adverse events; and the number of adverse events per 1,000 hospitalizations. They analyzed data on 61,523 patients who were discharged from 4,372 hospitals; the data were extracted from the Medicare Patient Safety Monitoring System database (N. Engl. J. Med. 2014:370;341-51).
The 61,523 patients included 11,399 with AMI, 15,374 with HF, 18,269 with pneumonia, and 16,481 with conditions requiring surgery. Postsurgical patients largely were being treated for joint replacement procedures and other osteoarthritis-related conditions, femur fracture, colon cancer, post-AMI procedures, or other forms of chronic ischemic heart disease.
From 2005-2006 to 2010-2011, AMI and HF patients saw a 1.3 percentage point decline in the rate of adverse events, from 5% to 3.7%. The proportion who had one or more such events decreased from 26% to 19%. The number of adverse events per 1,000 hospitalizations declined from 402 to 262 for AMI patients and from 235 to 167 for HF patients.
Infection-related and drug-related adverse events declined significantly in heart attack and HF patients. There was also a substantial improvement in postprocedure events in HF patients.
Postsurgical patients experienced slight increases in all three outcomes measures, in particular, increases in infection-related and postprocedural events such as venous thromboembolism, and cardiac and catheter-related events. The number of events per 1,000 hospitalizations for pneumonia patients increased insignificantly from 216 to 223. For postsurgical patients, the number of events increased insignificantly from 352/1,000 to 368/1,000.
Patients who had adverse events had significantly longer hospital stays and were at higher risk for death. As the number of adverse events increased, so did the risk of death.
The authors noted that declines in events for AMI and HF patients might be a reflection of the numerous efforts and initiatives to improve care in those two conditions.
But they also found that concerted efforts to improve safety did not necessarily work. There was an increase in pressure ulcers in postsurgical patients, and no decline in ventilator-associated pneumonia in most patients, even though there have been initiatives focused on those conditions.
"Our finding of an increased adverse-event rate among surgical patients indicates a continuing challenge and identifies an important target for patient-safety initiatives," the researchers said.
The study was supported by the Agency for Healthcare Research and Quality as well as academic and federal grants. Dr. Wang was a consultant to and other researchers were employees of the research firm Qualidigm and participated in the analysis. One of the researchers, Dr. Harlan M. Krumholz, disclosed being on a scientific advisory board for UnitedHealthcare and receiving grant money from Medtronic.
On Twitter @aliciaault
Adverse events decreased over the last decade for Medicare inpatients with acute myocardial infarction or heart failure but barely changed for those with pneumonia or conditions requiring surgery, according to an analysis of a Medicare database.
The improvements likely translated to 81,000 fewer adverse events for patients with acute MI (AMI) and heart failure (HF) from 2010 to 2011 alone, according to the study published Jan. 22 in the New England Journal of Medicine.
"Although this suggests that national efforts focused on patient safety have made some inroads, the lack of reductions across the board is disappointing," Yun Wang, Ph.D., of the Harvard School of Public Health, Boston, and his colleagues wrote.
The researchers examined whether hospitalized patients are any better off in light of the current focus on patient safety, including the launch of initiatives such as the American College of Surgeons’ National Surgical Quality Improvement Program and the federal government’s Surgical Infection Prevention Project.
They used three composite outcomes measures: the rate of occurrence for adverse events for which patients were at risk (for instance, only patients receiving warfarin were at risk for warfarin-related events); the proportion of patients with one or more adverse events; and the number of adverse events per 1,000 hospitalizations. They analyzed data on 61,523 patients who were discharged from 4,372 hospitals; the data were extracted from the Medicare Patient Safety Monitoring System database (N. Engl. J. Med. 2014:370;341-51).
The 61,523 patients included 11,399 with AMI, 15,374 with HF, 18,269 with pneumonia, and 16,481 with conditions requiring surgery. Postsurgical patients largely were being treated for joint replacement procedures and other osteoarthritis-related conditions, femur fracture, colon cancer, post-AMI procedures, or other forms of chronic ischemic heart disease.
From 2005-2006 to 2010-2011, AMI and HF patients saw a 1.3 percentage point decline in the rate of adverse events, from 5% to 3.7%. The proportion who had one or more such events decreased from 26% to 19%. The number of adverse events per 1,000 hospitalizations declined from 402 to 262 for AMI patients and from 235 to 167 for HF patients.
Infection-related and drug-related adverse events declined significantly in heart attack and HF patients. There was also a substantial improvement in postprocedure events in HF patients.
Postsurgical patients experienced slight increases in all three outcomes measures, in particular, increases in infection-related and postprocedural events such as venous thromboembolism, and cardiac and catheter-related events. The number of events per 1,000 hospitalizations for pneumonia patients increased insignificantly from 216 to 223. For postsurgical patients, the number of events increased insignificantly from 352/1,000 to 368/1,000.
Patients who had adverse events had significantly longer hospital stays and were at higher risk for death. As the number of adverse events increased, so did the risk of death.
The authors noted that declines in events for AMI and HF patients might be a reflection of the numerous efforts and initiatives to improve care in those two conditions.
But they also found that concerted efforts to improve safety did not necessarily work. There was an increase in pressure ulcers in postsurgical patients, and no decline in ventilator-associated pneumonia in most patients, even though there have been initiatives focused on those conditions.
"Our finding of an increased adverse-event rate among surgical patients indicates a continuing challenge and identifies an important target for patient-safety initiatives," the researchers said.
The study was supported by the Agency for Healthcare Research and Quality as well as academic and federal grants. Dr. Wang was a consultant to and other researchers were employees of the research firm Qualidigm and participated in the analysis. One of the researchers, Dr. Harlan M. Krumholz, disclosed being on a scientific advisory board for UnitedHealthcare and receiving grant money from Medtronic.
On Twitter @aliciaault
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Major finding: At least 81,000 adverse events were avoided in a single year in patients hospitalized for acute MI and heart failure.
Data source: A retrospective analysis of data from 61,523 patients in the Medicare Patient Safety Monitoring System.
Disclosures: The study was supported by the Agency for Healthcare Research and Quality as well as academic and federal grants. Dr. Wang was a consultant to and other researchers were employees of the research firm Qualidigm and participated in the analysis. One of the researchers, Dr. Harlan M. Krumholz, disclosed being on a scientific advisory board for UnitedHealthcare and receiving grant money from Medtronic.
CRT shown highly cost effective in mild heart failure
DALLAS – Cardiac resynchronization therapy demonstrated a robust cost-effectiveness of $18,275 per quality-adjusted life-year gained in an analysis from the landmark REVERSE trial.
REVERSE (Resynchronization Reverses Remodeling in Systolic Left Ventricular Dysfunction) was the first large, international, randomized, double-blind clinical trial to establish that cardiac resynchronization therapy (CRT) is clinically beneficial in patients with mild heart failure (J. Am. Coll. Cardiol. 2008;52:1834-43). The incremental cost-effectiveness ratio (ICER) of $18,275 per quality-adjusted life-year (QALY) gained in the new secondary analysis is well under the figure of $50,000 per QALY gained generally accepted as the benchmark defining cost-effectiveness, Dr. Michael R. Gold noted at the American Heart Association scientific sessions.

He presented an analysis based on an economic model that utilized 5-year follow-up data from REVERSE to predict the impact of CRT on life-years and costs out to 10 years. REVERSE included 610 patients with New York Heart Association (NYHA) class I or II heart failure, a QRS interval of 120 milliseconds or more, and a left ventricular ejection fraction of 40% or less, all of whom were on optimal medical therapy. Participants were implanted with a CRT device, which was placed in either the on or off position for the first year in North American patients or the first 2 years in Europeans, after which everyone’s device was switched on for the remainder of follow-up.
The model projected that patients with CRT turned on would gain an extra 0.83 years of life compared with those without active CRT. The ICER ratio of $18,275 per QALY gained through CRT was determined by applying costs based on national Medicare rates to the reduced heart failure hospitalization rate resulting from the device therapy, explained Dr. Gold, professor of medicine and director of cardiology at the Medical University of South Carolina, Charleston.
CRT was found to be highly cost effective in all patient subgroups examined in the analysis. For example, the projected ICER with cardiac resynchronization therapy switched on in patients with heart failure of ischemic etiology was $15,648 per QALY. In those with left bundle branch block, it was $22,086 per QALY.
Significantly fewer patients were projected to progress to NYHA class III heart failure over the course of 10 years with CRT on than off, by a margin of 18.7% to 28.1%. From the 5-year follow-up data, it was estimated that at 10 years 31% of the CRT-on group would be in NYHA class I, compared with 22.3% of controls with CRT off.
A combined CRT/implantable cardioverter-defibrillator (CRT-D) with both functions switched on had an ICER of $13,050 per QALY gained compared with CRT only.
On the basis of the results of REVERSE and the Resynchronization/Defibrillation in Ambulatory Heart Failure Trial (RAFT, N. Engl. J. Med. 2010;363:2385-95), in 2012 the Food and Drug Administration expanded the indication for Medtronic’s CRT-D device to include the treatment of patients with NYHA class II heart failure with a left ventricular ejection fraction of 30% or less, left bundle branch block, and a QRS duration of at least 130 milliseconds. The new secondary analysis of REVERSE demonstrates that CRT is not only clinically effective in patients with mild heart failure, as recognized by the FDA, it is highly cost effective as well, according to Dr. Gold.
The REVERSE trial was sponsored by Medtronic. Dr. Gold is a consultant to Medtronic and Boston Scientific.
DALLAS – Cardiac resynchronization therapy demonstrated a robust cost-effectiveness of $18,275 per quality-adjusted life-year gained in an analysis from the landmark REVERSE trial.
REVERSE (Resynchronization Reverses Remodeling in Systolic Left Ventricular Dysfunction) was the first large, international, randomized, double-blind clinical trial to establish that cardiac resynchronization therapy (CRT) is clinically beneficial in patients with mild heart failure (J. Am. Coll. Cardiol. 2008;52:1834-43). The incremental cost-effectiveness ratio (ICER) of $18,275 per quality-adjusted life-year (QALY) gained in the new secondary analysis is well under the figure of $50,000 per QALY gained generally accepted as the benchmark defining cost-effectiveness, Dr. Michael R. Gold noted at the American Heart Association scientific sessions.
He presented an analysis based on an economic model that utilized 5-year follow-up data from REVERSE to predict the impact of CRT on life-years and costs out to 10 years. REVERSE included 610 patients with New York Heart Association (NYHA) class I or II heart failure, a QRS interval of 120 milliseconds or more, and a left ventricular ejection fraction of 40% or less, all of whom were on optimal medical therapy. Participants were implanted with a CRT device, which was placed in either the on or off position for the first year in North American patients or the first 2 years in Europeans, after which everyone’s device was switched on for the remainder of follow-up.
The model projected that patients with CRT turned on would gain an extra 0.83 years of life compared with those without active CRT. The ICER ratio of $18,275 per QALY gained through CRT was determined by applying costs based on national Medicare rates to the reduced heart failure hospitalization rate resulting from the device therapy, explained Dr. Gold, professor of medicine and director of cardiology at the Medical University of South Carolina, Charleston.
CRT was found to be highly cost effective in all patient subgroups examined in the analysis. For example, the projected ICER with cardiac resynchronization therapy switched on in patients with heart failure of ischemic etiology was $15,648 per QALY. In those with left bundle branch block, it was $22,086 per QALY.
Significantly fewer patients were projected to progress to NYHA class III heart failure over the course of 10 years with CRT on than off, by a margin of 18.7% to 28.1%. From the 5-year follow-up data, it was estimated that at 10 years 31% of the CRT-on group would be in NYHA class I, compared with 22.3% of controls with CRT off.
A combined CRT/implantable cardioverter-defibrillator (CRT-D) with both functions switched on had an ICER of $13,050 per QALY gained compared with CRT only.
On the basis of the results of REVERSE and the Resynchronization/Defibrillation in Ambulatory Heart Failure Trial (RAFT, N. Engl. J. Med. 2010;363:2385-95), in 2012 the Food and Drug Administration expanded the indication for Medtronic’s CRT-D device to include the treatment of patients with NYHA class II heart failure with a left ventricular ejection fraction of 30% or less, left bundle branch block, and a QRS duration of at least 130 milliseconds. The new secondary analysis of REVERSE demonstrates that CRT is not only clinically effective in patients with mild heart failure, as recognized by the FDA, it is highly cost effective as well, according to Dr. Gold.
The REVERSE trial was sponsored by Medtronic. Dr. Gold is a consultant to Medtronic and Boston Scientific.
DALLAS – Cardiac resynchronization therapy demonstrated a robust cost-effectiveness of $18,275 per quality-adjusted life-year gained in an analysis from the landmark REVERSE trial.
REVERSE (Resynchronization Reverses Remodeling in Systolic Left Ventricular Dysfunction) was the first large, international, randomized, double-blind clinical trial to establish that cardiac resynchronization therapy (CRT) is clinically beneficial in patients with mild heart failure (J. Am. Coll. Cardiol. 2008;52:1834-43). The incremental cost-effectiveness ratio (ICER) of $18,275 per quality-adjusted life-year (QALY) gained in the new secondary analysis is well under the figure of $50,000 per QALY gained generally accepted as the benchmark defining cost-effectiveness, Dr. Michael R. Gold noted at the American Heart Association scientific sessions.
He presented an analysis based on an economic model that utilized 5-year follow-up data from REVERSE to predict the impact of CRT on life-years and costs out to 10 years. REVERSE included 610 patients with New York Heart Association (NYHA) class I or II heart failure, a QRS interval of 120 milliseconds or more, and a left ventricular ejection fraction of 40% or less, all of whom were on optimal medical therapy. Participants were implanted with a CRT device, which was placed in either the on or off position for the first year in North American patients or the first 2 years in Europeans, after which everyone’s device was switched on for the remainder of follow-up.
The model projected that patients with CRT turned on would gain an extra 0.83 years of life compared with those without active CRT. The ICER ratio of $18,275 per QALY gained through CRT was determined by applying costs based on national Medicare rates to the reduced heart failure hospitalization rate resulting from the device therapy, explained Dr. Gold, professor of medicine and director of cardiology at the Medical University of South Carolina, Charleston.
CRT was found to be highly cost effective in all patient subgroups examined in the analysis. For example, the projected ICER with cardiac resynchronization therapy switched on in patients with heart failure of ischemic etiology was $15,648 per QALY. In those with left bundle branch block, it was $22,086 per QALY.
Significantly fewer patients were projected to progress to NYHA class III heart failure over the course of 10 years with CRT on than off, by a margin of 18.7% to 28.1%. From the 5-year follow-up data, it was estimated that at 10 years 31% of the CRT-on group would be in NYHA class I, compared with 22.3% of controls with CRT off.
A combined CRT/implantable cardioverter-defibrillator (CRT-D) with both functions switched on had an ICER of $13,050 per QALY gained compared with CRT only.
On the basis of the results of REVERSE and the Resynchronization/Defibrillation in Ambulatory Heart Failure Trial (RAFT, N. Engl. J. Med. 2010;363:2385-95), in 2012 the Food and Drug Administration expanded the indication for Medtronic’s CRT-D device to include the treatment of patients with NYHA class II heart failure with a left ventricular ejection fraction of 30% or less, left bundle branch block, and a QRS duration of at least 130 milliseconds. The new secondary analysis of REVERSE demonstrates that CRT is not only clinically effective in patients with mild heart failure, as recognized by the FDA, it is highly cost effective as well, according to Dr. Gold.
The REVERSE trial was sponsored by Medtronic. Dr. Gold is a consultant to Medtronic and Boston Scientific.
AT THE AHA SCIENTIFIC SESSIONS
Major finding: Implantation of a cardiac resynchronization therapy device in patients with mild heart failure on optimal medical therapy had a projected cost-effectiveness of $18,275 per quality-adjusted life-year gained, compared with controls on optimal medical therapy alone.
Data source: This cost-benefit analysis utilized 5-year follow-up data from the randomized, double-blind prospective REVERSE trial, in which 610 patients with mild heart failure were implanted with a cardiac resynchronization therapy device placed in the on or off position.
Disclosures: The study was sponsored by Medtronic. The presenter is a consultant to Medtronic and Boston Scientific.

Repositionable aortic valve meets performance goals
SAN FRANCISCO – A mean aortic valve pressure gradient of 11.5 mm Hg 30 days after transcatheter aortic valve replacement with the investigational repositionable Lotus valve in 120 patients was significantly better than the performance goal of 18 mm Hg, a study showed.
Approximately 4% of patients in the prospective registry study died within the 30 days and 2% had a disabling stroke, both of which could be considered low rates, Dr. Ian T. Meredith said at the Transcatheter Cardiovascular Therapeutics annual meeting. Another 4% of patients developed nondisabling strokes by the 30-day follow-up in an intent-to-treat analysis.
Aortic regurgitation rates were "negligible" after the procedure, said Dr. Meredith of Monash Medical Centre, Clayton, Australia, and professor of cardiology at Monash University, Melbourne.
Before valve replacement, 15% of patients had moderate or severe aortic regurgitation. Thirty days later, none had severe and 1% had moderate aortic regurgitation in the REPRISE II study (Repositionable Percutaneous Replacement of Stenotic Aortic Valve Through Implantation of Lotus Valve System – Evaluation of Safety and Performance).

Mild aortic regurgitation rates went from 44% at baseline to 17% at 30 days. Trace regurgitation was seen in 21% at baseline and at 30 days of follow-up.
The primary endpoints in the study were mean aortic valve pressure gradient at 30 days and 30-day all-cause mortality. The mean aortic gradient decreased from 46 mm Hg at baseline to 12.1 mm Hg at discharge and 11.5 mm Hg at 30 days. The mean effective orifice area increased from 0.7 cm2 at baseline to 1.6 cm2 at discharge and 1.7 cm2 at 30 days. Clinical event rates were similar to those reported in other valve trials, he said.
The 14-center study enrolled patients between October 2012 and April 2013 who were aged at least 70 years and rated New York Heart Association class II or greater. They had an aortic valve area less than 1 cm2, an aortic annulus measuring 19-27 mm, and a mean pressure gradient greater than 40 mm Hg or a jet velocity greater than 4 m/sec. Patients also had a Society of Thoracic Surgeons score of at least 8% and/or high surgical risk due to frailty or comorbidities.
The Lotus valve can be assessed in position before release, shortened or expanded mechanically, and, if desired, repositioned or fully resheathed and retrieved. It includes patented adaptive seal technology on the outside surfaces of the valve to fill up irregular surfaces of the fractured native aortic valve and reduce paravalvular leakage, Dr. Meredith said. The study used two valve sizes, 23 mm and 27 mm.
All 120 Lotus valves were successfully placed. Repositionings of 31 valves were all successful, and attempted retrievals of 6 valves were all successful. "As a consequence of this, there was no valve migration, no embolization, no ectopic valve deployment, and absolutely no need to put a second valve inside a valve for incorrect positioning or aortic regurgitation," he said at the meeting, cosponsored by the American College of Cardiology.
New pacemakers were needed in 29% (34 patients), mainly for third-degree atrioventricular block in 30 patients. "Perhaps the most important determinant of new pacemaker insertion was oversizing of the left ventricular outflow tract or the annulus due to the fact that only two valve sizes were available for this study," Dr. Meredith said. He predicted that this high rate of pacemaker implantation could be halved when more Lotus valve sizes and a more flexible catheter are available for future trials.
"We use the Lotus valve. The beauty of the valve is it can be repositioned and retrieved," Dr. A. Pieter Kappetein said at a press briefing. "You can plant the valve with a lot of confidence for the operator. At every stage of the procedure, you can control. I think that is the beauty of this valve," said Dr. Kappetein, professor of thoracic surgery at Erasmus University, Rotterdam, the Netherlands.
The Lotus valve system is available at selected centers in Europe and is not yet approved in the United States or Japan. The investigators plan to conduct a 130-patient extension of REPRISE II and a 1,000-patient pivotal trial (REPRISE III) to seek Food and Drug Administration approval.
Boston Scientific, which makes the Lotus valve system, funded the study. Dr. Meredith reported financial associations with Boston Scientific and Medtronic.
On Twitter @sherryboschert
SAN FRANCISCO – A mean aortic valve pressure gradient of 11.5 mm Hg 30 days after transcatheter aortic valve replacement with the investigational repositionable Lotus valve in 120 patients was significantly better than the performance goal of 18 mm Hg, a study showed.
Approximately 4% of patients in the prospective registry study died within the 30 days and 2% had a disabling stroke, both of which could be considered low rates, Dr. Ian T. Meredith said at the Transcatheter Cardiovascular Therapeutics annual meeting. Another 4% of patients developed nondisabling strokes by the 30-day follow-up in an intent-to-treat analysis.
Aortic regurgitation rates were "negligible" after the procedure, said Dr. Meredith of Monash Medical Centre, Clayton, Australia, and professor of cardiology at Monash University, Melbourne.
Before valve replacement, 15% of patients had moderate or severe aortic regurgitation. Thirty days later, none had severe and 1% had moderate aortic regurgitation in the REPRISE II study (Repositionable Percutaneous Replacement of Stenotic Aortic Valve Through Implantation of Lotus Valve System – Evaluation of Safety and Performance).
Mild aortic regurgitation rates went from 44% at baseline to 17% at 30 days. Trace regurgitation was seen in 21% at baseline and at 30 days of follow-up.
The primary endpoints in the study were mean aortic valve pressure gradient at 30 days and 30-day all-cause mortality. The mean aortic gradient decreased from 46 mm Hg at baseline to 12.1 mm Hg at discharge and 11.5 mm Hg at 30 days. The mean effective orifice area increased from 0.7 cm2 at baseline to 1.6 cm2 at discharge and 1.7 cm2 at 30 days. Clinical event rates were similar to those reported in other valve trials, he said.
The 14-center study enrolled patients between October 2012 and April 2013 who were aged at least 70 years and rated New York Heart Association class II or greater. They had an aortic valve area less than 1 cm2, an aortic annulus measuring 19-27 mm, and a mean pressure gradient greater than 40 mm Hg or a jet velocity greater than 4 m/sec. Patients also had a Society of Thoracic Surgeons score of at least 8% and/or high surgical risk due to frailty or comorbidities.
The Lotus valve can be assessed in position before release, shortened or expanded mechanically, and, if desired, repositioned or fully resheathed and retrieved. It includes patented adaptive seal technology on the outside surfaces of the valve to fill up irregular surfaces of the fractured native aortic valve and reduce paravalvular leakage, Dr. Meredith said. The study used two valve sizes, 23 mm and 27 mm.
All 120 Lotus valves were successfully placed. Repositionings of 31 valves were all successful, and attempted retrievals of 6 valves were all successful. "As a consequence of this, there was no valve migration, no embolization, no ectopic valve deployment, and absolutely no need to put a second valve inside a valve for incorrect positioning or aortic regurgitation," he said at the meeting, cosponsored by the American College of Cardiology.
New pacemakers were needed in 29% (34 patients), mainly for third-degree atrioventricular block in 30 patients. "Perhaps the most important determinant of new pacemaker insertion was oversizing of the left ventricular outflow tract or the annulus due to the fact that only two valve sizes were available for this study," Dr. Meredith said. He predicted that this high rate of pacemaker implantation could be halved when more Lotus valve sizes and a more flexible catheter are available for future trials.
"We use the Lotus valve. The beauty of the valve is it can be repositioned and retrieved," Dr. A. Pieter Kappetein said at a press briefing. "You can plant the valve with a lot of confidence for the operator. At every stage of the procedure, you can control. I think that is the beauty of this valve," said Dr. Kappetein, professor of thoracic surgery at Erasmus University, Rotterdam, the Netherlands.
The Lotus valve system is available at selected centers in Europe and is not yet approved in the United States or Japan. The investigators plan to conduct a 130-patient extension of REPRISE II and a 1,000-patient pivotal trial (REPRISE III) to seek Food and Drug Administration approval.
Boston Scientific, which makes the Lotus valve system, funded the study. Dr. Meredith reported financial associations with Boston Scientific and Medtronic.
On Twitter @sherryboschert
SAN FRANCISCO – A mean aortic valve pressure gradient of 11.5 mm Hg 30 days after transcatheter aortic valve replacement with the investigational repositionable Lotus valve in 120 patients was significantly better than the performance goal of 18 mm Hg, a study showed.
Approximately 4% of patients in the prospective registry study died within the 30 days and 2% had a disabling stroke, both of which could be considered low rates, Dr. Ian T. Meredith said at the Transcatheter Cardiovascular Therapeutics annual meeting. Another 4% of patients developed nondisabling strokes by the 30-day follow-up in an intent-to-treat analysis.
Aortic regurgitation rates were "negligible" after the procedure, said Dr. Meredith of Monash Medical Centre, Clayton, Australia, and professor of cardiology at Monash University, Melbourne.
Before valve replacement, 15% of patients had moderate or severe aortic regurgitation. Thirty days later, none had severe and 1% had moderate aortic regurgitation in the REPRISE II study (Repositionable Percutaneous Replacement of Stenotic Aortic Valve Through Implantation of Lotus Valve System – Evaluation of Safety and Performance).
Mild aortic regurgitation rates went from 44% at baseline to 17% at 30 days. Trace regurgitation was seen in 21% at baseline and at 30 days of follow-up.
The primary endpoints in the study were mean aortic valve pressure gradient at 30 days and 30-day all-cause mortality. The mean aortic gradient decreased from 46 mm Hg at baseline to 12.1 mm Hg at discharge and 11.5 mm Hg at 30 days. The mean effective orifice area increased from 0.7 cm2 at baseline to 1.6 cm2 at discharge and 1.7 cm2 at 30 days. Clinical event rates were similar to those reported in other valve trials, he said.
The 14-center study enrolled patients between October 2012 and April 2013 who were aged at least 70 years and rated New York Heart Association class II or greater. They had an aortic valve area less than 1 cm2, an aortic annulus measuring 19-27 mm, and a mean pressure gradient greater than 40 mm Hg or a jet velocity greater than 4 m/sec. Patients also had a Society of Thoracic Surgeons score of at least 8% and/or high surgical risk due to frailty or comorbidities.
The Lotus valve can be assessed in position before release, shortened or expanded mechanically, and, if desired, repositioned or fully resheathed and retrieved. It includes patented adaptive seal technology on the outside surfaces of the valve to fill up irregular surfaces of the fractured native aortic valve and reduce paravalvular leakage, Dr. Meredith said. The study used two valve sizes, 23 mm and 27 mm.
All 120 Lotus valves were successfully placed. Repositionings of 31 valves were all successful, and attempted retrievals of 6 valves were all successful. "As a consequence of this, there was no valve migration, no embolization, no ectopic valve deployment, and absolutely no need to put a second valve inside a valve for incorrect positioning or aortic regurgitation," he said at the meeting, cosponsored by the American College of Cardiology.
New pacemakers were needed in 29% (34 patients), mainly for third-degree atrioventricular block in 30 patients. "Perhaps the most important determinant of new pacemaker insertion was oversizing of the left ventricular outflow tract or the annulus due to the fact that only two valve sizes were available for this study," Dr. Meredith said. He predicted that this high rate of pacemaker implantation could be halved when more Lotus valve sizes and a more flexible catheter are available for future trials.
"We use the Lotus valve. The beauty of the valve is it can be repositioned and retrieved," Dr. A. Pieter Kappetein said at a press briefing. "You can plant the valve with a lot of confidence for the operator. At every stage of the procedure, you can control. I think that is the beauty of this valve," said Dr. Kappetein, professor of thoracic surgery at Erasmus University, Rotterdam, the Netherlands.
The Lotus valve system is available at selected centers in Europe and is not yet approved in the United States or Japan. The investigators plan to conduct a 130-patient extension of REPRISE II and a 1,000-patient pivotal trial (REPRISE III) to seek Food and Drug Administration approval.
Boston Scientific, which makes the Lotus valve system, funded the study. Dr. Meredith reported financial associations with Boston Scientific and Medtronic.
On Twitter @sherryboschert
AT TCT 2013
Major finding: The 30-day mean aortic valve pressure gradient was 11.5 mm Hg, and 4% of patients died within 30 days.
Data source: A prospective registry study of 120 patients with aortic stenosis and high surgery risk who underwent transcatheter aortic valve replacement with the Lotus valve system.
Disclosures: Boston Scientific, which makes the Lotus valve system, funded the study. Dr. Meredith reported financial associations with Boston Scientific and Medtronic.

But will renal denervation work in heart failure?
The release of top-line data from the sponsor of SYMPLICITY HTN-3, a large study of renal denervation in hypertension, provided a shock wave to the cardiovascular community.
To some degree, the findings of the study are not surprising. As always, patients who are in clinical trials do well, in this case the sham-operated patients who perhaps at long last were subjected to a focused effort at blood pressure lowering. We will have to wait a while to evaluate the data including subgroup analyses and substudies, but the much ballyhooed effects seen in earlier studies (Lancet 2009;373:1275-81; Lancet 2010;376:1903-9), which did not include a sham-operated arm, now seem to be a series of false leads.
In reading the obituaries, it sounds as if both the company and researchers have quickly opted to move on. But they shouldn’t. There is ample reason to believe that renal denervation may mitigate cardiorenal syndrome, a clinically challenging state that is associated with higher mortality in patients with heart failure. There are a number of small studies underway, the most prominent of which, PRESERVE (Promotion of Renal Sodium Excretion by Renal Sympathetic Denervation in Congestive Heart Failure), is supported by the National Heart, Lung, and Blood Institute. These studies should move forward. It would be a disservice to patients and medical science if one negative study in hypertension, albeit a big one, fails.
Will renal denervation work in heart failure? The studies must go on.
Dr. Hauptman is professor of internal medicine and assistant dean of clinical-translational research at Saint Louis University and director of heart failure at Saint Louis University Hospital. He is an associate editor for Circulation: Heart Failure and blogs while staring out his office window at the Arch.
The release of top-line data from the sponsor of SYMPLICITY HTN-3, a large study of renal denervation in hypertension, provided a shock wave to the cardiovascular community.
To some degree, the findings of the study are not surprising. As always, patients who are in clinical trials do well, in this case the sham-operated patients who perhaps at long last were subjected to a focused effort at blood pressure lowering. We will have to wait a while to evaluate the data including subgroup analyses and substudies, but the much ballyhooed effects seen in earlier studies (Lancet 2009;373:1275-81; Lancet 2010;376:1903-9), which did not include a sham-operated arm, now seem to be a series of false leads.
In reading the obituaries, it sounds as if both the company and researchers have quickly opted to move on. But they shouldn’t. There is ample reason to believe that renal denervation may mitigate cardiorenal syndrome, a clinically challenging state that is associated with higher mortality in patients with heart failure. There are a number of small studies underway, the most prominent of which, PRESERVE (Promotion of Renal Sodium Excretion by Renal Sympathetic Denervation in Congestive Heart Failure), is supported by the National Heart, Lung, and Blood Institute. These studies should move forward. It would be a disservice to patients and medical science if one negative study in hypertension, albeit a big one, fails.
Will renal denervation work in heart failure? The studies must go on.
Dr. Hauptman is professor of internal medicine and assistant dean of clinical-translational research at Saint Louis University and director of heart failure at Saint Louis University Hospital. He is an associate editor for Circulation: Heart Failure and blogs while staring out his office window at the Arch.
The release of top-line data from the sponsor of SYMPLICITY HTN-3, a large study of renal denervation in hypertension, provided a shock wave to the cardiovascular community.
To some degree, the findings of the study are not surprising. As always, patients who are in clinical trials do well, in this case the sham-operated patients who perhaps at long last were subjected to a focused effort at blood pressure lowering. We will have to wait a while to evaluate the data including subgroup analyses and substudies, but the much ballyhooed effects seen in earlier studies (Lancet 2009;373:1275-81; Lancet 2010;376:1903-9), which did not include a sham-operated arm, now seem to be a series of false leads.
In reading the obituaries, it sounds as if both the company and researchers have quickly opted to move on. But they shouldn’t. There is ample reason to believe that renal denervation may mitigate cardiorenal syndrome, a clinically challenging state that is associated with higher mortality in patients with heart failure. There are a number of small studies underway, the most prominent of which, PRESERVE (Promotion of Renal Sodium Excretion by Renal Sympathetic Denervation in Congestive Heart Failure), is supported by the National Heart, Lung, and Blood Institute. These studies should move forward. It would be a disservice to patients and medical science if one negative study in hypertension, albeit a big one, fails.
Will renal denervation work in heart failure? The studies must go on.
Dr. Hauptman is professor of internal medicine and assistant dean of clinical-translational research at Saint Louis University and director of heart failure at Saint Louis University Hospital. He is an associate editor for Circulation: Heart Failure and blogs while staring out his office window at the Arch.
TOPCAT is a dog
Another trial in HFpEF, another surprise? To the contrary, the TOPCAT data on heart failure with preserved ejection fraction were in line with prior trials in the diastolic heart failure space. At long last, heart failure hospitalizations, if not mortality, were decreased. Of course, that finding may not be reproducible in the real world. If spironolactone becomes widely used in patients with preserved LV function, does anyone think that hyperkalemia won’t be a problem? It was after the Randomized Aldactone Evaluation Study (RALES) was published (N. Engl. J. Med. 1999;341:709-17; N. Engl. J. Med. 2004;351:543-51).
TOPCAT (Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist) also had the usual array of strange subgroups based on site: Eastern European sites were outliers this time with an unexpectedly low event rate in the placebo arm. (I’ll have more on this issue – the negative aspects of international randomized clinical trials – in some future column.) But the bottom line remains the same. There is little reason to think that our approach to HFpEF patients will significantly change. Indeed, if a pharmaceutical company had a new drug for HFpEF, I would advise them to think very carefully about whether it was worth the financial risk of conducting a large outcomes trial. Even if the trial name had no embedded CAT, odds are the trial would still be a dog.
Dr. Hauptman is professor of internal medicine and assistant dean of clinical-translational research at Saint Louis University and director of heart failure at Saint Louis University Hospital. He is an associate editor for Circulation: Heart Failure and blogs while staring out his office window at the Arch. Write to Dr. Hauptman at cardnews@frontlinemedcom.com.
Another trial in HFpEF, another surprise? To the contrary, the TOPCAT data on heart failure with preserved ejection fraction were in line with prior trials in the diastolic heart failure space. At long last, heart failure hospitalizations, if not mortality, were decreased. Of course, that finding may not be reproducible in the real world. If spironolactone becomes widely used in patients with preserved LV function, does anyone think that hyperkalemia won’t be a problem? It was after the Randomized Aldactone Evaluation Study (RALES) was published (N. Engl. J. Med. 1999;341:709-17; N. Engl. J. Med. 2004;351:543-51).
TOPCAT (Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist) also had the usual array of strange subgroups based on site: Eastern European sites were outliers this time with an unexpectedly low event rate in the placebo arm. (I’ll have more on this issue – the negative aspects of international randomized clinical trials – in some future column.) But the bottom line remains the same. There is little reason to think that our approach to HFpEF patients will significantly change. Indeed, if a pharmaceutical company had a new drug for HFpEF, I would advise them to think very carefully about whether it was worth the financial risk of conducting a large outcomes trial. Even if the trial name had no embedded CAT, odds are the trial would still be a dog.
Dr. Hauptman is professor of internal medicine and assistant dean of clinical-translational research at Saint Louis University and director of heart failure at Saint Louis University Hospital. He is an associate editor for Circulation: Heart Failure and blogs while staring out his office window at the Arch. Write to Dr. Hauptman at cardnews@frontlinemedcom.com.
Another trial in HFpEF, another surprise? To the contrary, the TOPCAT data on heart failure with preserved ejection fraction were in line with prior trials in the diastolic heart failure space. At long last, heart failure hospitalizations, if not mortality, were decreased. Of course, that finding may not be reproducible in the real world. If spironolactone becomes widely used in patients with preserved LV function, does anyone think that hyperkalemia won’t be a problem? It was after the Randomized Aldactone Evaluation Study (RALES) was published (N. Engl. J. Med. 1999;341:709-17; N. Engl. J. Med. 2004;351:543-51).
TOPCAT (Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist) also had the usual array of strange subgroups based on site: Eastern European sites were outliers this time with an unexpectedly low event rate in the placebo arm. (I’ll have more on this issue – the negative aspects of international randomized clinical trials – in some future column.) But the bottom line remains the same. There is little reason to think that our approach to HFpEF patients will significantly change. Indeed, if a pharmaceutical company had a new drug for HFpEF, I would advise them to think very carefully about whether it was worth the financial risk of conducting a large outcomes trial. Even if the trial name had no embedded CAT, odds are the trial would still be a dog.
Dr. Hauptman is professor of internal medicine and assistant dean of clinical-translational research at Saint Louis University and director of heart failure at Saint Louis University Hospital. He is an associate editor for Circulation: Heart Failure and blogs while staring out his office window at the Arch. Write to Dr. Hauptman at cardnews@frontlinemedcom.com.