User login
VIDEO: About 1 in 20 ALS patients in Washington state chose assisted suicide
BOSTON – A new study estimates that 3.4%-6.7% of amyotrophic lateral sclerosis (ALS) patients in Washington state sought to commit physician-assisted suicide over a 5-year period.
The rate is many times higher than that among cancer patients in the state, researchers found. They also discovered that ALS patients were significantly more likely than were other terminally ill people to use the deadly medication after getting prescriptions for it.
The findings appear to reflect the unique hopelessness facing ALS patients. “They’re not afforded as much denial of decline and death as are patients with other terminal illnesses,” said Linda Ganzini, MD, MPH, a professor of psychiatry and medicine at Oregon Health & Science University, Portland, who has studied end of life in ALS patients.
“Many cancer patients, even in the final days of life, receive treatments that they hope will extend their lives,” she said in an interview after reviewing the study findings. “In contrast, treatments for ALS are minimally effective.”
Physician-assisted suicide is legal in California, Colorado, the District of Columbia, Montana, Oregon, Vermont, and Washington.
A team led by Leo H. Wang, MD, PhD, of the University of Washington, Seattle, examined the medical records of 39 ALS patients who sought medication to end their lives at three hospitals in Seattle from March 2009 to Dec. 31, 2014.
Washington’s Death with Dignity (DWD) law, which went into effect in 2009, allows physicians to prescribe lethal medication if the patient has a terminal illness and a prognosis of less than 6 months to live as judged by two physicians.
The researchers reported their findings, a follow-up to a previous study (Neurology. 2016 Nov 15;87[20]:2117-22), at the annual meeting of the American Academy of Neurology.
The median age of the ALS patients at symptom onset was 64 (range, 42-83), and a median of 712 days passed (range, 207-2,407) from the date of diagnosis to date of prescription for lethal medication.
The median time from prescription to death was 22 days, with at least one patient dying immediately (range, 0-386 days). All 39 patients had limb involvement, and 82%-92% had bulbar involvement, dysarthria, dysphagia, and/or dyspnea.
The researchers estimate that 3.4%-6.7% of 1,146 ALS patients in Washington who died over the time period of the study sought a physician-assisted death. The 3.4% figure assumes that the 39 patients at the three hospitals make up all the ALS patients who received medication prescriptions. The 6.7% figure assumes that all patients with neurodegenerative disease who sought DWD in the state over that period had ALS.
“Similarly, 5% (92 of 1,795) of Oregon ALS patient who died sought medication under DWD between 1998 and 2014,” Dr. Wang said. “This is slightly increased compared to the percentage during the first decade, following enactment of the Oregon law (1998-2007), when 2.7% (26 of 962) of ALS patients died using DWD medication.”
Using Washington state data, researchers also estimated that 0.6% of 73,319 cancer patients and 0.2% of 298,178 people in the state who died of all causes sought DWD over the study period.
A total of 30 (77%) ALS patients who received the deadly prescriptions chose to take them, compared with 67% of all-cause patients who took advantage of the DWD law and 60% of cancer patients.
All 30 patients died. The nine who chose to not take the prescribed medication died after a median of 76 days. The patients who did not take the medication were more likely to be married (88% vs. 69%), to be college educated (100% vs. 74%), and to use a motorized wheelchair (78% vs. 31%).
Those who chose to not take the prescribed medication were also less motivated by loss of dignity (63% vs. 93% among those who took the medication) and by being a burden on others (25% vs. 66%). They were more likely to identify themselves as religious (80% vs. 35%).
Multiple factors may explain why ALS patients made different choices regarding the deadly drugs, lead study author Dr. Wang said in an interview. “We thought that the loss of communication may have played a role based on our finding, as most patients who followed through had more substantial trouble speaking,” he said. “For the patients who ultimately did not choose to take the medication, we found more of them had stronger religious beliefs than those who did not.”
As for pain, he reported that it was not a major issue. “Only about 10% of ALS patients were worried about pain, as opposed to 30% of the general Death with Dignity patients,” he said.
Dr. Ganzini noted that some patients who seek the prescribed drugs “want reassurance that, if their quality of life becomes unbearable, they have the option of physician-assisted death. But, they continue to cope and find reasons to live. As such, they ultimately die of their disease without taking the medications. Others lose the ability to ingest the medications, often because of sudden worsening of their disease.”
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
No specific funding was reported. Dr. Ganzini and Dr. Wang had no disclosures.
BOSTON – A new study estimates that 3.4%-6.7% of amyotrophic lateral sclerosis (ALS) patients in Washington state sought to commit physician-assisted suicide over a 5-year period.
The rate is many times higher than that among cancer patients in the state, researchers found. They also discovered that ALS patients were significantly more likely than were other terminally ill people to use the deadly medication after getting prescriptions for it.
The findings appear to reflect the unique hopelessness facing ALS patients. “They’re not afforded as much denial of decline and death as are patients with other terminal illnesses,” said Linda Ganzini, MD, MPH, a professor of psychiatry and medicine at Oregon Health & Science University, Portland, who has studied end of life in ALS patients.
“Many cancer patients, even in the final days of life, receive treatments that they hope will extend their lives,” she said in an interview after reviewing the study findings. “In contrast, treatments for ALS are minimally effective.”
Physician-assisted suicide is legal in California, Colorado, the District of Columbia, Montana, Oregon, Vermont, and Washington.
A team led by Leo H. Wang, MD, PhD, of the University of Washington, Seattle, examined the medical records of 39 ALS patients who sought medication to end their lives at three hospitals in Seattle from March 2009 to Dec. 31, 2014.
Washington’s Death with Dignity (DWD) law, which went into effect in 2009, allows physicians to prescribe lethal medication if the patient has a terminal illness and a prognosis of less than 6 months to live as judged by two physicians.
The researchers reported their findings, a follow-up to a previous study (Neurology. 2016 Nov 15;87[20]:2117-22), at the annual meeting of the American Academy of Neurology.
The median age of the ALS patients at symptom onset was 64 (range, 42-83), and a median of 712 days passed (range, 207-2,407) from the date of diagnosis to date of prescription for lethal medication.
The median time from prescription to death was 22 days, with at least one patient dying immediately (range, 0-386 days). All 39 patients had limb involvement, and 82%-92% had bulbar involvement, dysarthria, dysphagia, and/or dyspnea.
The researchers estimate that 3.4%-6.7% of 1,146 ALS patients in Washington who died over the time period of the study sought a physician-assisted death. The 3.4% figure assumes that the 39 patients at the three hospitals make up all the ALS patients who received medication prescriptions. The 6.7% figure assumes that all patients with neurodegenerative disease who sought DWD in the state over that period had ALS.
“Similarly, 5% (92 of 1,795) of Oregon ALS patient who died sought medication under DWD between 1998 and 2014,” Dr. Wang said. “This is slightly increased compared to the percentage during the first decade, following enactment of the Oregon law (1998-2007), when 2.7% (26 of 962) of ALS patients died using DWD medication.”
Using Washington state data, researchers also estimated that 0.6% of 73,319 cancer patients and 0.2% of 298,178 people in the state who died of all causes sought DWD over the study period.
A total of 30 (77%) ALS patients who received the deadly prescriptions chose to take them, compared with 67% of all-cause patients who took advantage of the DWD law and 60% of cancer patients.
All 30 patients died. The nine who chose to not take the prescribed medication died after a median of 76 days. The patients who did not take the medication were more likely to be married (88% vs. 69%), to be college educated (100% vs. 74%), and to use a motorized wheelchair (78% vs. 31%).
Those who chose to not take the prescribed medication were also less motivated by loss of dignity (63% vs. 93% among those who took the medication) and by being a burden on others (25% vs. 66%). They were more likely to identify themselves as religious (80% vs. 35%).
Multiple factors may explain why ALS patients made different choices regarding the deadly drugs, lead study author Dr. Wang said in an interview. “We thought that the loss of communication may have played a role based on our finding, as most patients who followed through had more substantial trouble speaking,” he said. “For the patients who ultimately did not choose to take the medication, we found more of them had stronger religious beliefs than those who did not.”
As for pain, he reported that it was not a major issue. “Only about 10% of ALS patients were worried about pain, as opposed to 30% of the general Death with Dignity patients,” he said.
Dr. Ganzini noted that some patients who seek the prescribed drugs “want reassurance that, if their quality of life becomes unbearable, they have the option of physician-assisted death. But, they continue to cope and find reasons to live. As such, they ultimately die of their disease without taking the medications. Others lose the ability to ingest the medications, often because of sudden worsening of their disease.”
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
No specific funding was reported. Dr. Ganzini and Dr. Wang had no disclosures.
BOSTON – A new study estimates that 3.4%-6.7% of amyotrophic lateral sclerosis (ALS) patients in Washington state sought to commit physician-assisted suicide over a 5-year period.
The rate is many times higher than that among cancer patients in the state, researchers found. They also discovered that ALS patients were significantly more likely than were other terminally ill people to use the deadly medication after getting prescriptions for it.
The findings appear to reflect the unique hopelessness facing ALS patients. “They’re not afforded as much denial of decline and death as are patients with other terminal illnesses,” said Linda Ganzini, MD, MPH, a professor of psychiatry and medicine at Oregon Health & Science University, Portland, who has studied end of life in ALS patients.
“Many cancer patients, even in the final days of life, receive treatments that they hope will extend their lives,” she said in an interview after reviewing the study findings. “In contrast, treatments for ALS are minimally effective.”
Physician-assisted suicide is legal in California, Colorado, the District of Columbia, Montana, Oregon, Vermont, and Washington.
A team led by Leo H. Wang, MD, PhD, of the University of Washington, Seattle, examined the medical records of 39 ALS patients who sought medication to end their lives at three hospitals in Seattle from March 2009 to Dec. 31, 2014.
Washington’s Death with Dignity (DWD) law, which went into effect in 2009, allows physicians to prescribe lethal medication if the patient has a terminal illness and a prognosis of less than 6 months to live as judged by two physicians.
The researchers reported their findings, a follow-up to a previous study (Neurology. 2016 Nov 15;87[20]:2117-22), at the annual meeting of the American Academy of Neurology.
The median age of the ALS patients at symptom onset was 64 (range, 42-83), and a median of 712 days passed (range, 207-2,407) from the date of diagnosis to date of prescription for lethal medication.
The median time from prescription to death was 22 days, with at least one patient dying immediately (range, 0-386 days). All 39 patients had limb involvement, and 82%-92% had bulbar involvement, dysarthria, dysphagia, and/or dyspnea.
The researchers estimate that 3.4%-6.7% of 1,146 ALS patients in Washington who died over the time period of the study sought a physician-assisted death. The 3.4% figure assumes that the 39 patients at the three hospitals make up all the ALS patients who received medication prescriptions. The 6.7% figure assumes that all patients with neurodegenerative disease who sought DWD in the state over that period had ALS.
“Similarly, 5% (92 of 1,795) of Oregon ALS patient who died sought medication under DWD between 1998 and 2014,” Dr. Wang said. “This is slightly increased compared to the percentage during the first decade, following enactment of the Oregon law (1998-2007), when 2.7% (26 of 962) of ALS patients died using DWD medication.”
Using Washington state data, researchers also estimated that 0.6% of 73,319 cancer patients and 0.2% of 298,178 people in the state who died of all causes sought DWD over the study period.
A total of 30 (77%) ALS patients who received the deadly prescriptions chose to take them, compared with 67% of all-cause patients who took advantage of the DWD law and 60% of cancer patients.
All 30 patients died. The nine who chose to not take the prescribed medication died after a median of 76 days. The patients who did not take the medication were more likely to be married (88% vs. 69%), to be college educated (100% vs. 74%), and to use a motorized wheelchair (78% vs. 31%).
Those who chose to not take the prescribed medication were also less motivated by loss of dignity (63% vs. 93% among those who took the medication) and by being a burden on others (25% vs. 66%). They were more likely to identify themselves as religious (80% vs. 35%).
Multiple factors may explain why ALS patients made different choices regarding the deadly drugs, lead study author Dr. Wang said in an interview. “We thought that the loss of communication may have played a role based on our finding, as most patients who followed through had more substantial trouble speaking,” he said. “For the patients who ultimately did not choose to take the medication, we found more of them had stronger religious beliefs than those who did not.”
As for pain, he reported that it was not a major issue. “Only about 10% of ALS patients were worried about pain, as opposed to 30% of the general Death with Dignity patients,” he said.
Dr. Ganzini noted that some patients who seek the prescribed drugs “want reassurance that, if their quality of life becomes unbearable, they have the option of physician-assisted death. But, they continue to cope and find reasons to live. As such, they ultimately die of their disease without taking the medications. Others lose the ability to ingest the medications, often because of sudden worsening of their disease.”
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
No specific funding was reported. Dr. Ganzini and Dr. Wang had no disclosures.
At AAN 2017
Key clinical point:
Major finding: An estimated 3.4%-6.7% of ALS patients in Washington state sought physician-assisted death, and 77% took the prescribed deadly medication, a higher rate than all-cause (67%) and cancer patients (60%).
Data source: Analysis of 39 ALS patients who sought deadly medication from three Seattle hospitals from March 2009 to Dec. 31, 2014.
Disclosures: No specific funding was reported, and Dr. Wang had no disclosures.

VIDEO: Big research trials at AAN bring up important cost decisions
BOSTON – Some of the most influential clinical research reports coming out of the annual meeting of the American Academy of Neurology raise questions on how neurologists will strike a balance between the improved efficacy and safety of drugs in new therapeutic classes and their affordability for patients.
Natalia Rost, MD, vice chair of the AAN Science Committee, discussed phase III clinical trials (ARISE and STRIVE) in episodic migraine with erenumab, an investigational humanized monoclonal antibody against calcitonin gene-related peptide receptor; phase III clinical trials (ENDEAR and CHERISH) of the antisense oligonucleotide drug nusinersen (Spinraza) that was approved by the Food and Drug Administration for spinal muscular atrophy in late 2016; as well as phase III trials of a pharmaceutical-grade extract of the cannabis-derived compound cannabidiol in patients with Dravet syndrome and Lennox-Gastaut syndrome.
Erenumab and nusinersen are “disease-specific targeted biologics” that have been developed over decades to target a specific disease pathway, and hence translate into high prices, Dr. Rost said in a video interview at the meeting.
“How you value the cost of a drug against improvement in a physiological outcome is very difficult to measure,” she noted, for relatively small gains in reducing migraine days per month and improvements in functional outcome and disability against placebo.
But this calculation is different with the potentially lifesaving effects of nusinersen for spinal muscular atrophy patients, in which “we’re not talking about days of improvement, we’re talking about days of life,” said Dr. Rost, director of acute stroke services at Massachusetts General Hospital, Boston. “And so that becomes an ethical dilemma in terms of the cost of administration, who is paying for the drug, and how this is covered. Whom do you offer treatment to?”
The development of cannabidiol as a potential adjunctive treatment for Dravet and Lennox-Gastaut syndromes is a welcome addition to the armamentarium against these conditions, Dr. Rost added, because it offers an alternative to the unregulated use of herbal medications and supplements – particularly cannabis in its various forms – that patients ask about but are difficult to dose consistently and to ensure a pharmaceutical-grade level of purity.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
BOSTON – Some of the most influential clinical research reports coming out of the annual meeting of the American Academy of Neurology raise questions on how neurologists will strike a balance between the improved efficacy and safety of drugs in new therapeutic classes and their affordability for patients.
Natalia Rost, MD, vice chair of the AAN Science Committee, discussed phase III clinical trials (ARISE and STRIVE) in episodic migraine with erenumab, an investigational humanized monoclonal antibody against calcitonin gene-related peptide receptor; phase III clinical trials (ENDEAR and CHERISH) of the antisense oligonucleotide drug nusinersen (Spinraza) that was approved by the Food and Drug Administration for spinal muscular atrophy in late 2016; as well as phase III trials of a pharmaceutical-grade extract of the cannabis-derived compound cannabidiol in patients with Dravet syndrome and Lennox-Gastaut syndrome.
Erenumab and nusinersen are “disease-specific targeted biologics” that have been developed over decades to target a specific disease pathway, and hence translate into high prices, Dr. Rost said in a video interview at the meeting.
“How you value the cost of a drug against improvement in a physiological outcome is very difficult to measure,” she noted, for relatively small gains in reducing migraine days per month and improvements in functional outcome and disability against placebo.
But this calculation is different with the potentially lifesaving effects of nusinersen for spinal muscular atrophy patients, in which “we’re not talking about days of improvement, we’re talking about days of life,” said Dr. Rost, director of acute stroke services at Massachusetts General Hospital, Boston. “And so that becomes an ethical dilemma in terms of the cost of administration, who is paying for the drug, and how this is covered. Whom do you offer treatment to?”
The development of cannabidiol as a potential adjunctive treatment for Dravet and Lennox-Gastaut syndromes is a welcome addition to the armamentarium against these conditions, Dr. Rost added, because it offers an alternative to the unregulated use of herbal medications and supplements – particularly cannabis in its various forms – that patients ask about but are difficult to dose consistently and to ensure a pharmaceutical-grade level of purity.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
BOSTON – Some of the most influential clinical research reports coming out of the annual meeting of the American Academy of Neurology raise questions on how neurologists will strike a balance between the improved efficacy and safety of drugs in new therapeutic classes and their affordability for patients.
Natalia Rost, MD, vice chair of the AAN Science Committee, discussed phase III clinical trials (ARISE and STRIVE) in episodic migraine with erenumab, an investigational humanized monoclonal antibody against calcitonin gene-related peptide receptor; phase III clinical trials (ENDEAR and CHERISH) of the antisense oligonucleotide drug nusinersen (Spinraza) that was approved by the Food and Drug Administration for spinal muscular atrophy in late 2016; as well as phase III trials of a pharmaceutical-grade extract of the cannabis-derived compound cannabidiol in patients with Dravet syndrome and Lennox-Gastaut syndrome.
Erenumab and nusinersen are “disease-specific targeted biologics” that have been developed over decades to target a specific disease pathway, and hence translate into high prices, Dr. Rost said in a video interview at the meeting.
“How you value the cost of a drug against improvement in a physiological outcome is very difficult to measure,” she noted, for relatively small gains in reducing migraine days per month and improvements in functional outcome and disability against placebo.
But this calculation is different with the potentially lifesaving effects of nusinersen for spinal muscular atrophy patients, in which “we’re not talking about days of improvement, we’re talking about days of life,” said Dr. Rost, director of acute stroke services at Massachusetts General Hospital, Boston. “And so that becomes an ethical dilemma in terms of the cost of administration, who is paying for the drug, and how this is covered. Whom do you offer treatment to?”
The development of cannabidiol as a potential adjunctive treatment for Dravet and Lennox-Gastaut syndromes is a welcome addition to the armamentarium against these conditions, Dr. Rost added, because it offers an alternative to the unregulated use of herbal medications and supplements – particularly cannabis in its various forms – that patients ask about but are difficult to dose consistently and to ensure a pharmaceutical-grade level of purity.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT AAN 2017

AAN spotlights spinal muscular atrophy clinical research
A variety of plenary and emerging science sessions at this year’s annual meeting of the American Academy of Neurology in Boston will highlight clinical research efforts to treat children with spinal muscular atrophy.
At the Hot Topics Plenary Session on April 22, Claudia A. Chiriboga, MD, of Columbia University, New York, will discuss the results of clinical trials involving antisense oligonucleotide treatments for spinal muscular atrophy (SMA), including the recently approved nusinersen (Spinraza), which promotes transcription of the full-length survival motor neuron (SMN) protein from the SMN2 gene.
In the first of two reports on new clinical research about nusinersen, Nancy L. Kuntz, MD, of Ann & Robert H. Lurie Children’s Hospital of Chicago will present the initial interim efficacy and safety findings from the phase III international ENDEAR study on April 24 at the Contemporary Clinical Issues Plenary Session. The study of 122 infants with SMA is comparing intrathecal administration of nusinersen against a sham procedure of a small needle prick on the lower back to look for differences at day 402 in the primary outcome of the percentage of patients who attain motor milestones as assessed by section 2 of the Hammersmith Infant Neurological Examination or the time to death or need for respiratory intervention. Charlotte J. Sumner, MD, of Johns Hopkins University, Baltimore, will discuss the study following Dr. Kuntz’s presentation.
The second nusinersen trial to be reported at the meeting will describe interim results of the drug’s efficacy and safety in children with later-onset SMA in the phase III CHERISH study. At the Emerging Science Platform Session on April 25, Richard S. Finkel, MD, of Nemours Children’s Hospital in Orlando, Fla., will discuss how the primary outcome of the Hammersmith Functional Motor Scale–Expanded score changed from baseline to 15 months following intrathecal injection or a sham procedure in children aged 2-12 years.
An investigational SMA type 1 treatment just beginning testing in clinical trials will also receive attention in a plenary session and a platform session. In the Clinical Trials Plenary Session on April 25, Jerry R. Mendell, MD, of Nationwide Children’s Hospital, Columbus, Ohio, will report on the first gene therapy trial for SMA type 1, a phase I trial of AVXS-101, which delivers the SMN gene in a AAV9 viral vector that is able to cross the blood-brain barrier. The primary objective of the trial is to assess safety of a single intravenous dose. The secondary objectives include survival (avoidance of death/permanent-ventilation) and the ability to sit unassisted. Other analyses of data from the phase I trial will be reported during the “Motor Neuron Diseases: Biomarkers, Outcome Measures, and Therapeutics,” platform session on April 24, including the evaluation of preexisting anti-AAV9 antibodies and the proportion of patients who achieve CHOP-INTEND scores of 50 and above and sit unassisted.
A variety of plenary and emerging science sessions at this year’s annual meeting of the American Academy of Neurology in Boston will highlight clinical research efforts to treat children with spinal muscular atrophy.
At the Hot Topics Plenary Session on April 22, Claudia A. Chiriboga, MD, of Columbia University, New York, will discuss the results of clinical trials involving antisense oligonucleotide treatments for spinal muscular atrophy (SMA), including the recently approved nusinersen (Spinraza), which promotes transcription of the full-length survival motor neuron (SMN) protein from the SMN2 gene.
In the first of two reports on new clinical research about nusinersen, Nancy L. Kuntz, MD, of Ann & Robert H. Lurie Children’s Hospital of Chicago will present the initial interim efficacy and safety findings from the phase III international ENDEAR study on April 24 at the Contemporary Clinical Issues Plenary Session. The study of 122 infants with SMA is comparing intrathecal administration of nusinersen against a sham procedure of a small needle prick on the lower back to look for differences at day 402 in the primary outcome of the percentage of patients who attain motor milestones as assessed by section 2 of the Hammersmith Infant Neurological Examination or the time to death or need for respiratory intervention. Charlotte J. Sumner, MD, of Johns Hopkins University, Baltimore, will discuss the study following Dr. Kuntz’s presentation.
The second nusinersen trial to be reported at the meeting will describe interim results of the drug’s efficacy and safety in children with later-onset SMA in the phase III CHERISH study. At the Emerging Science Platform Session on April 25, Richard S. Finkel, MD, of Nemours Children’s Hospital in Orlando, Fla., will discuss how the primary outcome of the Hammersmith Functional Motor Scale–Expanded score changed from baseline to 15 months following intrathecal injection or a sham procedure in children aged 2-12 years.
An investigational SMA type 1 treatment just beginning testing in clinical trials will also receive attention in a plenary session and a platform session. In the Clinical Trials Plenary Session on April 25, Jerry R. Mendell, MD, of Nationwide Children’s Hospital, Columbus, Ohio, will report on the first gene therapy trial for SMA type 1, a phase I trial of AVXS-101, which delivers the SMN gene in a AAV9 viral vector that is able to cross the blood-brain barrier. The primary objective of the trial is to assess safety of a single intravenous dose. The secondary objectives include survival (avoidance of death/permanent-ventilation) and the ability to sit unassisted. Other analyses of data from the phase I trial will be reported during the “Motor Neuron Diseases: Biomarkers, Outcome Measures, and Therapeutics,” platform session on April 24, including the evaluation of preexisting anti-AAV9 antibodies and the proportion of patients who achieve CHOP-INTEND scores of 50 and above and sit unassisted.
A variety of plenary and emerging science sessions at this year’s annual meeting of the American Academy of Neurology in Boston will highlight clinical research efforts to treat children with spinal muscular atrophy.
At the Hot Topics Plenary Session on April 22, Claudia A. Chiriboga, MD, of Columbia University, New York, will discuss the results of clinical trials involving antisense oligonucleotide treatments for spinal muscular atrophy (SMA), including the recently approved nusinersen (Spinraza), which promotes transcription of the full-length survival motor neuron (SMN) protein from the SMN2 gene.
In the first of two reports on new clinical research about nusinersen, Nancy L. Kuntz, MD, of Ann & Robert H. Lurie Children’s Hospital of Chicago will present the initial interim efficacy and safety findings from the phase III international ENDEAR study on April 24 at the Contemporary Clinical Issues Plenary Session. The study of 122 infants with SMA is comparing intrathecal administration of nusinersen against a sham procedure of a small needle prick on the lower back to look for differences at day 402 in the primary outcome of the percentage of patients who attain motor milestones as assessed by section 2 of the Hammersmith Infant Neurological Examination or the time to death or need for respiratory intervention. Charlotte J. Sumner, MD, of Johns Hopkins University, Baltimore, will discuss the study following Dr. Kuntz’s presentation.
The second nusinersen trial to be reported at the meeting will describe interim results of the drug’s efficacy and safety in children with later-onset SMA in the phase III CHERISH study. At the Emerging Science Platform Session on April 25, Richard S. Finkel, MD, of Nemours Children’s Hospital in Orlando, Fla., will discuss how the primary outcome of the Hammersmith Functional Motor Scale–Expanded score changed from baseline to 15 months following intrathecal injection or a sham procedure in children aged 2-12 years.
An investigational SMA type 1 treatment just beginning testing in clinical trials will also receive attention in a plenary session and a platform session. In the Clinical Trials Plenary Session on April 25, Jerry R. Mendell, MD, of Nationwide Children’s Hospital, Columbus, Ohio, will report on the first gene therapy trial for SMA type 1, a phase I trial of AVXS-101, which delivers the SMN gene in a AAV9 viral vector that is able to cross the blood-brain barrier. The primary objective of the trial is to assess safety of a single intravenous dose. The secondary objectives include survival (avoidance of death/permanent-ventilation) and the ability to sit unassisted. Other analyses of data from the phase I trial will be reported during the “Motor Neuron Diseases: Biomarkers, Outcome Measures, and Therapeutics,” platform session on April 24, including the evaluation of preexisting anti-AAV9 antibodies and the proportion of patients who achieve CHOP-INTEND scores of 50 and above and sit unassisted.
Novel protein biomarker could accelerate ALS research
A protein found in the cerebrospinal fluid and peripheral blood cells of patients with a common type of amyotrophic lateral sclerosis may serve as a “pharmacodynamic marker,” providing a mechanism to assess RNA-based therapies that are now in clinical trials.
The protein, poly(GP), is expressed in patients who have a mutation in the gene chromosome 9 open reading frame 72 (C9ORF72), which causes one type of amyotrophic lateral sclerosis (ALS). This means that detecting poly(GP) also, eventually, may help to identify “presymptomatic individuals who are expected to benefit from early therapeutic interventions,” wrote the authors of a newly published paper (Sci Transl Med. 2017;9[383]:eaai7866).
Tania Gendron, PhD, of the department of neuroscience at the Mayo Clinic, Jacksonville, Fla., and her colleagues noted that poly(GP) was found in the cerebrospinal fluid (CSF) of both symptomatic and asymptomatic patients who carried the C9ORF72 mutation. The mutation can cause ALS, and is also associated with frontotemporal dementia (FTD) in a pattern with incomplete overlap with ALS.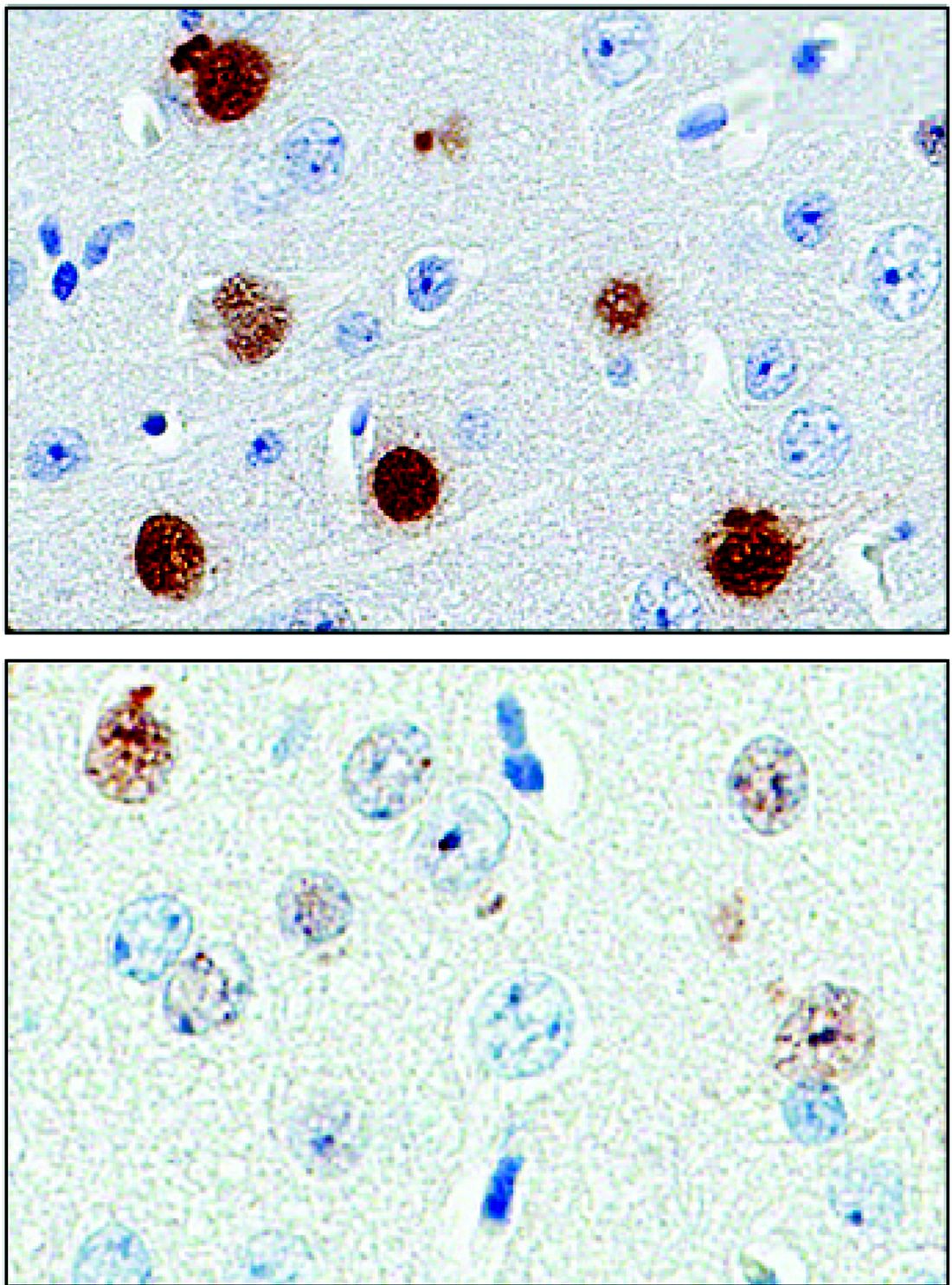
However, “A limitation in moving such treatments from bench to bedside is a lack of pharmacodynamics markers for use in clinical trials,” Dr. Gendron and her collaborators wrote. The discovery that poly(GP) tracks well with C9ORF72 means that it has the potential to serve as the kind of biomarker that’s been missing in drug development for this family of neurodegenerative diseases, they said.
“To prepare for upcoming clinical trials for c9ALS, the present study used patient CSF and several preclinical models to investigate the hypothesis that poly(GP) proteins could serve as an urgently needed pharmacodynamics marker for developing and testing therapies for treating c9ALS,” Dr. Gendron and her colleagues wrote.
They looked at CSF samples from 83 patients with c9ALS and 27 patients who were asymptomatic C9ORF72 repeat expansion carriers, as well as 24 carriers who had a neurologic disease besides ALS or FTD (total n = 134). They also examined CSF samples from 120 study participants who lacked the mutation, 48 of whom were healthy controls; the remainder had ALS (n = 57) or another neurological disease.
The investigators, who were blinded to individuals’ disease status in each study arm, found that CSF poly(GP) levels were significantly higher in patients who had the C9ORF72 mutation (P less than .0001 in unadjusted and adjusted analyses). Poly(GP) was present in both asymptomatic and symptomatic carriers of the mutation, and not significantly different between these groups when data were adjusted for multiple comparisons, age, and gender.
When Dr. Gendron and her colleagues looked at poly(GP) levels over time for patients whose longitudinal data were available, they found that levels for an individual study participant were “relatively constant,” without any significant change over the median 12.9 months that these levels were tracked (P = .84).
However, “poly(GP) is not a prognostic marker,” wrote Dr. Gendron and her colleagues. They found no consistent association between levels of the protein and disease severity of progression, age at onset, or the development of FTD. Women were more likely to have lower levels, but the significance of that finding is not known, they said. There was a trend, which lost significance after statistical adjustment, for patients with cognitive impairment to have higher poly(GP) levels (adjusted P = .12).
Treatments under investigation for ALS and FTD include the use of an antisense oligonucleotide (ASO) to bind to the repeated RNA sequences and negate their ill effects. The investigators wrote that in vitro investigations using patient-derived cell models showed that poly(GP) levels dropped when cells were exposed to an ASO for 10 days. “The data indicate that poly(GP) production mirrors expression of repeat-containing C9ORF72 transcripts in lymphoblastoid cell lines,” they wrote.
The authors reported multiple governmental and private foundation sources of support for the research. Dr. Gendron and several of her coauthors are investigators in clinical trials for an ASO to target C9ORF72. Several authors reported paid and unpaid relationships and stock positions with pharmaceutical companies, including ones developing treatments for ALS and FTD.
koakes@frontlinemedcom.com
On Twitter @karioakes
A protein found in the cerebrospinal fluid and peripheral blood cells of patients with a common type of amyotrophic lateral sclerosis may serve as a “pharmacodynamic marker,” providing a mechanism to assess RNA-based therapies that are now in clinical trials.
The protein, poly(GP), is expressed in patients who have a mutation in the gene chromosome 9 open reading frame 72 (C9ORF72), which causes one type of amyotrophic lateral sclerosis (ALS). This means that detecting poly(GP) also, eventually, may help to identify “presymptomatic individuals who are expected to benefit from early therapeutic interventions,” wrote the authors of a newly published paper (Sci Transl Med. 2017;9[383]:eaai7866).
Tania Gendron, PhD, of the department of neuroscience at the Mayo Clinic, Jacksonville, Fla., and her colleagues noted that poly(GP) was found in the cerebrospinal fluid (CSF) of both symptomatic and asymptomatic patients who carried the C9ORF72 mutation. The mutation can cause ALS, and is also associated with frontotemporal dementia (FTD) in a pattern with incomplete overlap with ALS.
However, “A limitation in moving such treatments from bench to bedside is a lack of pharmacodynamics markers for use in clinical trials,” Dr. Gendron and her collaborators wrote. The discovery that poly(GP) tracks well with C9ORF72 means that it has the potential to serve as the kind of biomarker that’s been missing in drug development for this family of neurodegenerative diseases, they said.
“To prepare for upcoming clinical trials for c9ALS, the present study used patient CSF and several preclinical models to investigate the hypothesis that poly(GP) proteins could serve as an urgently needed pharmacodynamics marker for developing and testing therapies for treating c9ALS,” Dr. Gendron and her colleagues wrote.
They looked at CSF samples from 83 patients with c9ALS and 27 patients who were asymptomatic C9ORF72 repeat expansion carriers, as well as 24 carriers who had a neurologic disease besides ALS or FTD (total n = 134). They also examined CSF samples from 120 study participants who lacked the mutation, 48 of whom were healthy controls; the remainder had ALS (n = 57) or another neurological disease.
The investigators, who were blinded to individuals’ disease status in each study arm, found that CSF poly(GP) levels were significantly higher in patients who had the C9ORF72 mutation (P less than .0001 in unadjusted and adjusted analyses). Poly(GP) was present in both asymptomatic and symptomatic carriers of the mutation, and not significantly different between these groups when data were adjusted for multiple comparisons, age, and gender.
When Dr. Gendron and her colleagues looked at poly(GP) levels over time for patients whose longitudinal data were available, they found that levels for an individual study participant were “relatively constant,” without any significant change over the median 12.9 months that these levels were tracked (P = .84).
However, “poly(GP) is not a prognostic marker,” wrote Dr. Gendron and her colleagues. They found no consistent association between levels of the protein and disease severity of progression, age at onset, or the development of FTD. Women were more likely to have lower levels, but the significance of that finding is not known, they said. There was a trend, which lost significance after statistical adjustment, for patients with cognitive impairment to have higher poly(GP) levels (adjusted P = .12).
Treatments under investigation for ALS and FTD include the use of an antisense oligonucleotide (ASO) to bind to the repeated RNA sequences and negate their ill effects. The investigators wrote that in vitro investigations using patient-derived cell models showed that poly(GP) levels dropped when cells were exposed to an ASO for 10 days. “The data indicate that poly(GP) production mirrors expression of repeat-containing C9ORF72 transcripts in lymphoblastoid cell lines,” they wrote.
The authors reported multiple governmental and private foundation sources of support for the research. Dr. Gendron and several of her coauthors are investigators in clinical trials for an ASO to target C9ORF72. Several authors reported paid and unpaid relationships and stock positions with pharmaceutical companies, including ones developing treatments for ALS and FTD.
koakes@frontlinemedcom.com
On Twitter @karioakes
A protein found in the cerebrospinal fluid and peripheral blood cells of patients with a common type of amyotrophic lateral sclerosis may serve as a “pharmacodynamic marker,” providing a mechanism to assess RNA-based therapies that are now in clinical trials.
The protein, poly(GP), is expressed in patients who have a mutation in the gene chromosome 9 open reading frame 72 (C9ORF72), which causes one type of amyotrophic lateral sclerosis (ALS). This means that detecting poly(GP) also, eventually, may help to identify “presymptomatic individuals who are expected to benefit from early therapeutic interventions,” wrote the authors of a newly published paper (Sci Transl Med. 2017;9[383]:eaai7866).
Tania Gendron, PhD, of the department of neuroscience at the Mayo Clinic, Jacksonville, Fla., and her colleagues noted that poly(GP) was found in the cerebrospinal fluid (CSF) of both symptomatic and asymptomatic patients who carried the C9ORF72 mutation. The mutation can cause ALS, and is also associated with frontotemporal dementia (FTD) in a pattern with incomplete overlap with ALS.
However, “A limitation in moving such treatments from bench to bedside is a lack of pharmacodynamics markers for use in clinical trials,” Dr. Gendron and her collaborators wrote. The discovery that poly(GP) tracks well with C9ORF72 means that it has the potential to serve as the kind of biomarker that’s been missing in drug development for this family of neurodegenerative diseases, they said.
“To prepare for upcoming clinical trials for c9ALS, the present study used patient CSF and several preclinical models to investigate the hypothesis that poly(GP) proteins could serve as an urgently needed pharmacodynamics marker for developing and testing therapies for treating c9ALS,” Dr. Gendron and her colleagues wrote.
They looked at CSF samples from 83 patients with c9ALS and 27 patients who were asymptomatic C9ORF72 repeat expansion carriers, as well as 24 carriers who had a neurologic disease besides ALS or FTD (total n = 134). They also examined CSF samples from 120 study participants who lacked the mutation, 48 of whom were healthy controls; the remainder had ALS (n = 57) or another neurological disease.
The investigators, who were blinded to individuals’ disease status in each study arm, found that CSF poly(GP) levels were significantly higher in patients who had the C9ORF72 mutation (P less than .0001 in unadjusted and adjusted analyses). Poly(GP) was present in both asymptomatic and symptomatic carriers of the mutation, and not significantly different between these groups when data were adjusted for multiple comparisons, age, and gender.
When Dr. Gendron and her colleagues looked at poly(GP) levels over time for patients whose longitudinal data were available, they found that levels for an individual study participant were “relatively constant,” without any significant change over the median 12.9 months that these levels were tracked (P = .84).
However, “poly(GP) is not a prognostic marker,” wrote Dr. Gendron and her colleagues. They found no consistent association between levels of the protein and disease severity of progression, age at onset, or the development of FTD. Women were more likely to have lower levels, but the significance of that finding is not known, they said. There was a trend, which lost significance after statistical adjustment, for patients with cognitive impairment to have higher poly(GP) levels (adjusted P = .12).
Treatments under investigation for ALS and FTD include the use of an antisense oligonucleotide (ASO) to bind to the repeated RNA sequences and negate their ill effects. The investigators wrote that in vitro investigations using patient-derived cell models showed that poly(GP) levels dropped when cells were exposed to an ASO for 10 days. “The data indicate that poly(GP) production mirrors expression of repeat-containing C9ORF72 transcripts in lymphoblastoid cell lines,” they wrote.
The authors reported multiple governmental and private foundation sources of support for the research. Dr. Gendron and several of her coauthors are investigators in clinical trials for an ASO to target C9ORF72. Several authors reported paid and unpaid relationships and stock positions with pharmaceutical companies, including ones developing treatments for ALS and FTD.
koakes@frontlinemedcom.com
On Twitter @karioakes
Key clinical point:
Major finding: Patients with a mutation predisposing them to ALS had significantly higher levels of the protein poly(GP) (P less than .0001 in unadjusted and adjusted analyses).
Data source: Prospective blinded study of CSF samples from 83 patients with c9ALS, 27 patients who were asymptomatic C9ORF72 repeat expansion carriers, 24 carriers who had a neurologic disease besides ALS or FTD, and 120 study participants who lacked the C9ORF72 mutation.
Disclosures: The authors reported multiple governmental and private foundation sources of support for their research. Dr. Gendron and several of her coauthors are investigators in clinical trials for an ASO to target C9ORF72. Several authors reported paid and unpaid relationships and stock positions with pharmaceutical companies, including ones developing treatments for ALS and FTD.
Outpatient Visits Involving CNS Polypharmacy Rising Among Elderly
The number of outpatient visits involving CNS polypharmacy by adults aged 65 and older more than doubled between 2004 and 2013, especially among those who reside in rural areas, according to research published online ahead of print February 13 in JAMA Internal Medicine.
“With each new revision of the Beers Criteria, the list of psychotropic medications considered potentially inappropriate in the elderly has grown,” said Donovan T. Maust, MD, Assistant Professor of Geriatric Psychiatry at the University of Michigan in Ann Arbor. “Opioids have recently been included in a Beers measure of CNS polypharmacy. Prescribing related drug combinations also received increased regulatory attention when the US Food and Drug Administration recently ordered a black box warning to alert patients of serious risks, including death, caused by opioids coprescribed with CNS depressants,” he said. 
Dr. Maust and his colleagues analyzed data on 97,910 outpatients age 65 and older from the National Ambulatory Medical Care Survey (NAMCS) from 2004 through 2013. Patients met Beers CNS polypharmacy criteria if three or more of the following medications were initiated or continued: antipsychotics, benzodiazepines, nonbenzodiazepine benzodiazepine receptor agonists, tricyclic antidepressants, selective serotonin reuptake inhibitors, and opioids. The researchers recorded as many as three visit diagnoses and included information collected from NAMCS such as chronic medical conditions, whether psychotherapy was provided or ordered, whether stress management or other mental health counseling services were provided or ordered, and time spent with physician.
Dr. Maust and his associates found that annual CNS polypharmacy visits by adults age 65 or older increased from 1.50 million in 2004 to 3.68 million in 2013, or from 0.6% of visits in 2004 to 1.4% in 2013 (adjusted odds ratio [AOR], 3.12). The largest increases were observed among rural visits and among visits with no mental health or pain diagnoses (AOR, 4.99 and 2.65, respectively).
More than two-thirds of polypharmacy visits (68%) were by women, and 17% were by individuals who lived in rural areas. In addition, nearly half of polypharmacy visits studied (46%) included neither mental health nor pain diagnoses. No significant demographic differences were observed between polypharmacy visits with and without opioids. “Older adults have become more open to mental health treatment,” the researchers concluded. “Because of limited access to specialty care and a preference to receive treatment in primary care settings, it is unsurprising that mental health treatment has expanded in nonpsychiatric settings.”
—Doug Brunk
Suggested Reading
Maust DT, Gerlach LB, Gibson A, et al. Trends in central nervous system-active polypharmacy among older adults seen in outpatient care in the United States. JAMA Intern Med. 2017 Feb 13 [Epub ahead of print].
The number of outpatient visits involving CNS polypharmacy by adults aged 65 and older more than doubled between 2004 and 2013, especially among those who reside in rural areas, according to research published online ahead of print February 13 in JAMA Internal Medicine.
“With each new revision of the Beers Criteria, the list of psychotropic medications considered potentially inappropriate in the elderly has grown,” said Donovan T. Maust, MD, Assistant Professor of Geriatric Psychiatry at the University of Michigan in Ann Arbor. “Opioids have recently been included in a Beers measure of CNS polypharmacy. Prescribing related drug combinations also received increased regulatory attention when the US Food and Drug Administration recently ordered a black box warning to alert patients of serious risks, including death, caused by opioids coprescribed with CNS depressants,” he said.
Dr. Maust and his colleagues analyzed data on 97,910 outpatients age 65 and older from the National Ambulatory Medical Care Survey (NAMCS) from 2004 through 2013. Patients met Beers CNS polypharmacy criteria if three or more of the following medications were initiated or continued: antipsychotics, benzodiazepines, nonbenzodiazepine benzodiazepine receptor agonists, tricyclic antidepressants, selective serotonin reuptake inhibitors, and opioids. The researchers recorded as many as three visit diagnoses and included information collected from NAMCS such as chronic medical conditions, whether psychotherapy was provided or ordered, whether stress management or other mental health counseling services were provided or ordered, and time spent with physician.
Dr. Maust and his associates found that annual CNS polypharmacy visits by adults age 65 or older increased from 1.50 million in 2004 to 3.68 million in 2013, or from 0.6% of visits in 2004 to 1.4% in 2013 (adjusted odds ratio [AOR], 3.12). The largest increases were observed among rural visits and among visits with no mental health or pain diagnoses (AOR, 4.99 and 2.65, respectively).
More than two-thirds of polypharmacy visits (68%) were by women, and 17% were by individuals who lived in rural areas. In addition, nearly half of polypharmacy visits studied (46%) included neither mental health nor pain diagnoses. No significant demographic differences were observed between polypharmacy visits with and without opioids. “Older adults have become more open to mental health treatment,” the researchers concluded. “Because of limited access to specialty care and a preference to receive treatment in primary care settings, it is unsurprising that mental health treatment has expanded in nonpsychiatric settings.”
—Doug Brunk
Suggested Reading
Maust DT, Gerlach LB, Gibson A, et al. Trends in central nervous system-active polypharmacy among older adults seen in outpatient care in the United States. JAMA Intern Med. 2017 Feb 13 [Epub ahead of print].
The number of outpatient visits involving CNS polypharmacy by adults aged 65 and older more than doubled between 2004 and 2013, especially among those who reside in rural areas, according to research published online ahead of print February 13 in JAMA Internal Medicine.
“With each new revision of the Beers Criteria, the list of psychotropic medications considered potentially inappropriate in the elderly has grown,” said Donovan T. Maust, MD, Assistant Professor of Geriatric Psychiatry at the University of Michigan in Ann Arbor. “Opioids have recently been included in a Beers measure of CNS polypharmacy. Prescribing related drug combinations also received increased regulatory attention when the US Food and Drug Administration recently ordered a black box warning to alert patients of serious risks, including death, caused by opioids coprescribed with CNS depressants,” he said.
Dr. Maust and his colleagues analyzed data on 97,910 outpatients age 65 and older from the National Ambulatory Medical Care Survey (NAMCS) from 2004 through 2013. Patients met Beers CNS polypharmacy criteria if three or more of the following medications were initiated or continued: antipsychotics, benzodiazepines, nonbenzodiazepine benzodiazepine receptor agonists, tricyclic antidepressants, selective serotonin reuptake inhibitors, and opioids. The researchers recorded as many as three visit diagnoses and included information collected from NAMCS such as chronic medical conditions, whether psychotherapy was provided or ordered, whether stress management or other mental health counseling services were provided or ordered, and time spent with physician.
Dr. Maust and his associates found that annual CNS polypharmacy visits by adults age 65 or older increased from 1.50 million in 2004 to 3.68 million in 2013, or from 0.6% of visits in 2004 to 1.4% in 2013 (adjusted odds ratio [AOR], 3.12). The largest increases were observed among rural visits and among visits with no mental health or pain diagnoses (AOR, 4.99 and 2.65, respectively).
More than two-thirds of polypharmacy visits (68%) were by women, and 17% were by individuals who lived in rural areas. In addition, nearly half of polypharmacy visits studied (46%) included neither mental health nor pain diagnoses. No significant demographic differences were observed between polypharmacy visits with and without opioids. “Older adults have become more open to mental health treatment,” the researchers concluded. “Because of limited access to specialty care and a preference to receive treatment in primary care settings, it is unsurprising that mental health treatment has expanded in nonpsychiatric settings.”
—Doug Brunk
Suggested Reading
Maust DT, Gerlach LB, Gibson A, et al. Trends in central nervous system-active polypharmacy among older adults seen in outpatient care in the United States. JAMA Intern Med. 2017 Feb 13 [Epub ahead of print].
LVADs achieve cardiac palliation in muscular dystrophies
At one time, respiratory failure was the primary cause of death in young men and boys with muscular dystrophies, but since improvements in ventilator support have addressed this problem, cardiac complications such as cardiomyopathy have become the main cause of death in this group, with the highest risk of death in people with Duchenne muscular dystrophy (DMD). Researchers from Rome have reported that the novel use of ventricular assist devices in this population can prolong life.
Gianluigi Perri, MD, PhD, of University Hospital and Bambino Gesù Children Hospital in Rome, and his coauthors, shared their experience treating seven patients with dystrophinopathies and dilated cardiomyopathy (DCM) with left ventricular assist devices (LVADs) from February 2011 to February 2016 (J Thorac Cardiovasc Surg. 2017 March;153:669-74). “Our experience indicates that the use of an LVAD as destination therapy in patients with dystrophinopathies with end-stage DCM is feasible, suggesting that it may be suitable as a palliative therapy for the treatment of these patients with no other therapeutic options,” Dr. Perri and his coauthors said.
Heart transplantation is considered the procedure of choice for children with severe advanced heart failure, but transplantation is contraindicated for children with dystrophinopathies because of the risk of respiratory failure and progression of skeletal myopathy leads to limited functional capacity. Hence, Dr. Perri and his coauthors developed their alternative treatment for end-stage heart failure in these children. They used the Jarvik 2000 LVAD (Jarvik Heart Inc., New York) as destination therapy.
Six of the seven patients they operated on had DMD and one had beta-2 sarcoglycan deficit. Their ages ranged from 14.2 to 23.4 years. Two patients had early complications: retropharyngeal bleeding and cholecystectomy; and abdominal bleeding and splenectomy. Two different patients had late complications: gastrostomy; and osteolysis and infection at the pedestal site. Three patients died after the operation: one of stroke at 15 months; one of severe bleeding about 28 months later; and one of lung infection 45 months afterward. Follow-up for the surviving patients ranged from about 2 months to 40 months. Median hospital stay was 77 days.
Dr. Perri and his coauthors noted that the DMD Care Considerations Working Group expanded acceptable therapies for DMD cardiomyopathy to include novel treatments such as mechanical circulatory support and implantable cardioverter-defibrillators.
“Although the best approach remains unclear, it does seem clear that treatment should be more aggressive,” the researchers said. The limited life expectancy of these patients makes transplantation a complicated choice when a shortage of donors is a concern. “Therefore, the alternative therapeutic option is the use of LVAD,” Dr. Perri and his coauthors said.
These patients need care at centers “with a high level of experience of patients with DMD,” the researchers stated. Common comorbidities such as severe kyphoscoliosis and respiratory muscle weakness in this population increase surgical risks.
Dr. Perri and his coauthors used a surgical technique that involved avoiding the left thoracotomy approach common in adults who undergo VAD implantation, because of respiratory insufficiency in these younger patients. They also used cardiopulmonary bypass in all but one patient who had a minimally invasive off-pump procedure through a left anterior minithoracotomy.
The researchers “strongly suggest” noninvasive ventilation after surgery to assist in pulmonary function often compromised by scoliosis and muscle weakness. “Our experience shows that postoperative care can be extremely challenging and is often burdened by unexpected complications,” they noted.
Kyphoscoliosis poses challenges when placing drains, and complications of these patients should be treated only in a specialized center. “Indeed, one of our patients died in a peripheral hospital because they underwent bronchoscopic examination with an endoscope that caused severe and intractable retropharyngeal bleeding,” they said.
The researchers no relevant financial relationships to disclose.
Almost all young men living with Duchenne muscular dystrophy will develop heart failure, but for many of these patients, continuous-flow left ventricular assist devices can provide “reliable support” for up to a decade, David L. S. Morales, MD, of the Heart Institute at Cincinnati Children’s Hospital Medical Center, said in his invited commentary (J Thorac Cardiovasc Surg. 2017;153:675-6)
“The current series demonstrates, as has been shown at our institute as well as others, that one can provide an effective therapy for certain patients with DMD and heart failure,” Dr. Morales said of the work of Dr. Perri and coauthors. Dr. Morales added that maximizing outcomes in this population hinges on finding the appropriate time point for intervention in the disease process.
While “there is still much to be learned,” Dr. Morales said, Dr. Perri and his coauthors have shown that LVAD therapy is an option in patients with DMD and heart failure who have failed other treatments. “These young men may, therefore, have the option to extend their lives and possibly have the opportunity to benefit from the impressive medical advances being made,” he said. “Perhaps they and their families have been provided hope.”
Dr. Morales disclosed relationships with Berlin Heart, HeartWare and Oregon Total Artificial Heart.
Almost all young men living with Duchenne muscular dystrophy will develop heart failure, but for many of these patients, continuous-flow left ventricular assist devices can provide “reliable support” for up to a decade, David L. S. Morales, MD, of the Heart Institute at Cincinnati Children’s Hospital Medical Center, said in his invited commentary (J Thorac Cardiovasc Surg. 2017;153:675-6)
“The current series demonstrates, as has been shown at our institute as well as others, that one can provide an effective therapy for certain patients with DMD and heart failure,” Dr. Morales said of the work of Dr. Perri and coauthors. Dr. Morales added that maximizing outcomes in this population hinges on finding the appropriate time point for intervention in the disease process.
While “there is still much to be learned,” Dr. Morales said, Dr. Perri and his coauthors have shown that LVAD therapy is an option in patients with DMD and heart failure who have failed other treatments. “These young men may, therefore, have the option to extend their lives and possibly have the opportunity to benefit from the impressive medical advances being made,” he said. “Perhaps they and their families have been provided hope.”
Dr. Morales disclosed relationships with Berlin Heart, HeartWare and Oregon Total Artificial Heart.
Almost all young men living with Duchenne muscular dystrophy will develop heart failure, but for many of these patients, continuous-flow left ventricular assist devices can provide “reliable support” for up to a decade, David L. S. Morales, MD, of the Heart Institute at Cincinnati Children’s Hospital Medical Center, said in his invited commentary (J Thorac Cardiovasc Surg. 2017;153:675-6)
“The current series demonstrates, as has been shown at our institute as well as others, that one can provide an effective therapy for certain patients with DMD and heart failure,” Dr. Morales said of the work of Dr. Perri and coauthors. Dr. Morales added that maximizing outcomes in this population hinges on finding the appropriate time point for intervention in the disease process.
While “there is still much to be learned,” Dr. Morales said, Dr. Perri and his coauthors have shown that LVAD therapy is an option in patients with DMD and heart failure who have failed other treatments. “These young men may, therefore, have the option to extend their lives and possibly have the opportunity to benefit from the impressive medical advances being made,” he said. “Perhaps they and their families have been provided hope.”
Dr. Morales disclosed relationships with Berlin Heart, HeartWare and Oregon Total Artificial Heart.
At one time, respiratory failure was the primary cause of death in young men and boys with muscular dystrophies, but since improvements in ventilator support have addressed this problem, cardiac complications such as cardiomyopathy have become the main cause of death in this group, with the highest risk of death in people with Duchenne muscular dystrophy (DMD). Researchers from Rome have reported that the novel use of ventricular assist devices in this population can prolong life.
Gianluigi Perri, MD, PhD, of University Hospital and Bambino Gesù Children Hospital in Rome, and his coauthors, shared their experience treating seven patients with dystrophinopathies and dilated cardiomyopathy (DCM) with left ventricular assist devices (LVADs) from February 2011 to February 2016 (J Thorac Cardiovasc Surg. 2017 March;153:669-74). “Our experience indicates that the use of an LVAD as destination therapy in patients with dystrophinopathies with end-stage DCM is feasible, suggesting that it may be suitable as a palliative therapy for the treatment of these patients with no other therapeutic options,” Dr. Perri and his coauthors said.
Heart transplantation is considered the procedure of choice for children with severe advanced heart failure, but transplantation is contraindicated for children with dystrophinopathies because of the risk of respiratory failure and progression of skeletal myopathy leads to limited functional capacity. Hence, Dr. Perri and his coauthors developed their alternative treatment for end-stage heart failure in these children. They used the Jarvik 2000 LVAD (Jarvik Heart Inc., New York) as destination therapy.
Six of the seven patients they operated on had DMD and one had beta-2 sarcoglycan deficit. Their ages ranged from 14.2 to 23.4 years. Two patients had early complications: retropharyngeal bleeding and cholecystectomy; and abdominal bleeding and splenectomy. Two different patients had late complications: gastrostomy; and osteolysis and infection at the pedestal site. Three patients died after the operation: one of stroke at 15 months; one of severe bleeding about 28 months later; and one of lung infection 45 months afterward. Follow-up for the surviving patients ranged from about 2 months to 40 months. Median hospital stay was 77 days.
Dr. Perri and his coauthors noted that the DMD Care Considerations Working Group expanded acceptable therapies for DMD cardiomyopathy to include novel treatments such as mechanical circulatory support and implantable cardioverter-defibrillators.
“Although the best approach remains unclear, it does seem clear that treatment should be more aggressive,” the researchers said. The limited life expectancy of these patients makes transplantation a complicated choice when a shortage of donors is a concern. “Therefore, the alternative therapeutic option is the use of LVAD,” Dr. Perri and his coauthors said.
These patients need care at centers “with a high level of experience of patients with DMD,” the researchers stated. Common comorbidities such as severe kyphoscoliosis and respiratory muscle weakness in this population increase surgical risks.
Dr. Perri and his coauthors used a surgical technique that involved avoiding the left thoracotomy approach common in adults who undergo VAD implantation, because of respiratory insufficiency in these younger patients. They also used cardiopulmonary bypass in all but one patient who had a minimally invasive off-pump procedure through a left anterior minithoracotomy.
The researchers “strongly suggest” noninvasive ventilation after surgery to assist in pulmonary function often compromised by scoliosis and muscle weakness. “Our experience shows that postoperative care can be extremely challenging and is often burdened by unexpected complications,” they noted.
Kyphoscoliosis poses challenges when placing drains, and complications of these patients should be treated only in a specialized center. “Indeed, one of our patients died in a peripheral hospital because they underwent bronchoscopic examination with an endoscope that caused severe and intractable retropharyngeal bleeding,” they said.
The researchers no relevant financial relationships to disclose.
At one time, respiratory failure was the primary cause of death in young men and boys with muscular dystrophies, but since improvements in ventilator support have addressed this problem, cardiac complications such as cardiomyopathy have become the main cause of death in this group, with the highest risk of death in people with Duchenne muscular dystrophy (DMD). Researchers from Rome have reported that the novel use of ventricular assist devices in this population can prolong life.
Gianluigi Perri, MD, PhD, of University Hospital and Bambino Gesù Children Hospital in Rome, and his coauthors, shared their experience treating seven patients with dystrophinopathies and dilated cardiomyopathy (DCM) with left ventricular assist devices (LVADs) from February 2011 to February 2016 (J Thorac Cardiovasc Surg. 2017 March;153:669-74). “Our experience indicates that the use of an LVAD as destination therapy in patients with dystrophinopathies with end-stage DCM is feasible, suggesting that it may be suitable as a palliative therapy for the treatment of these patients with no other therapeutic options,” Dr. Perri and his coauthors said.
Heart transplantation is considered the procedure of choice for children with severe advanced heart failure, but transplantation is contraindicated for children with dystrophinopathies because of the risk of respiratory failure and progression of skeletal myopathy leads to limited functional capacity. Hence, Dr. Perri and his coauthors developed their alternative treatment for end-stage heart failure in these children. They used the Jarvik 2000 LVAD (Jarvik Heart Inc., New York) as destination therapy.
Six of the seven patients they operated on had DMD and one had beta-2 sarcoglycan deficit. Their ages ranged from 14.2 to 23.4 years. Two patients had early complications: retropharyngeal bleeding and cholecystectomy; and abdominal bleeding and splenectomy. Two different patients had late complications: gastrostomy; and osteolysis and infection at the pedestal site. Three patients died after the operation: one of stroke at 15 months; one of severe bleeding about 28 months later; and one of lung infection 45 months afterward. Follow-up for the surviving patients ranged from about 2 months to 40 months. Median hospital stay was 77 days.
Dr. Perri and his coauthors noted that the DMD Care Considerations Working Group expanded acceptable therapies for DMD cardiomyopathy to include novel treatments such as mechanical circulatory support and implantable cardioverter-defibrillators.
“Although the best approach remains unclear, it does seem clear that treatment should be more aggressive,” the researchers said. The limited life expectancy of these patients makes transplantation a complicated choice when a shortage of donors is a concern. “Therefore, the alternative therapeutic option is the use of LVAD,” Dr. Perri and his coauthors said.
These patients need care at centers “with a high level of experience of patients with DMD,” the researchers stated. Common comorbidities such as severe kyphoscoliosis and respiratory muscle weakness in this population increase surgical risks.
Dr. Perri and his coauthors used a surgical technique that involved avoiding the left thoracotomy approach common in adults who undergo VAD implantation, because of respiratory insufficiency in these younger patients. They also used cardiopulmonary bypass in all but one patient who had a minimally invasive off-pump procedure through a left anterior minithoracotomy.
The researchers “strongly suggest” noninvasive ventilation after surgery to assist in pulmonary function often compromised by scoliosis and muscle weakness. “Our experience shows that postoperative care can be extremely challenging and is often burdened by unexpected complications,” they noted.
Kyphoscoliosis poses challenges when placing drains, and complications of these patients should be treated only in a specialized center. “Indeed, one of our patients died in a peripheral hospital because they underwent bronchoscopic examination with an endoscope that caused severe and intractable retropharyngeal bleeding,” they said.
The researchers no relevant financial relationships to disclose.
FROM THE JOURNAL OF THORACIC AND CARDIOVASCULAR SURGERY
Key clinical point: A left ventricular assist device can be used as destination therapy in patients with Duchenne muscular dystrophy dystrophinopathies and end-stage dilated cardiomyopathy.
Major finding: Four of seven patients who had LVAD survived long term, and survival for the three who died ranged from 15 to 44 months.
Data source: Single-center, retrospective review of seven patients with DMD who had LVAD for DCM from February 2011 to February 2016.
Disclosure: Dr. Perri and his coauthors reported having no relevant financial disclosures.
FDA approves Emflaza for Duchenne muscular dystrophy
Emflaza, a tablet and oral suspension corticosteroid, has been approved by the Food and Drug Administration for the treatment of Duchenne muscular dystrophy in patients aged 5 years and older.
The agency’s Feb. 9 announcement notes that similar corticosteroids have been used around the world to treat Duchenne muscular dystrophy (DMD), but this is the first to gain approval in the United States. Emflaza (deflazacort) works by decreasing inflammation and immune system activity.
DMD is the most common form of muscular dystrophy but is still rare, occurring in about 1 in 3,600 male infants worldwide. One study found that patients taking deflazacort had some improvements in muscle strength at 12 weeks, compared with those taking placebo, and maintained muscle strength stability through 52 weeks. A longer-term study showed that patients who took deflazacort had better average muscle strength than did those taking placebo and suggested that deflazacort helped prolong patients’ ability to walk.
Side effects experienced by patients taking Emflaza are similar to those associated with other corticosteroids, such as facial puffiness (cushingoid appearance), weight gain, increased appetite, upper respiratory tract infection, cough, extraordinary daytime urinary frequency (pollakiuria), unwanted hair growth (hirsutism), and excessive fat around the stomach (central obesity).
In the FDA’s announcement, Billy Dunn, MD, director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research, said, “We hope that this treatment option will benefit many patients with DMD.”
Emflaza, a tablet and oral suspension corticosteroid, has been approved by the Food and Drug Administration for the treatment of Duchenne muscular dystrophy in patients aged 5 years and older.
The agency’s Feb. 9 announcement notes that similar corticosteroids have been used around the world to treat Duchenne muscular dystrophy (DMD), but this is the first to gain approval in the United States. Emflaza (deflazacort) works by decreasing inflammation and immune system activity.
DMD is the most common form of muscular dystrophy but is still rare, occurring in about 1 in 3,600 male infants worldwide. One study found that patients taking deflazacort had some improvements in muscle strength at 12 weeks, compared with those taking placebo, and maintained muscle strength stability through 52 weeks. A longer-term study showed that patients who took deflazacort had better average muscle strength than did those taking placebo and suggested that deflazacort helped prolong patients’ ability to walk.
Side effects experienced by patients taking Emflaza are similar to those associated with other corticosteroids, such as facial puffiness (cushingoid appearance), weight gain, increased appetite, upper respiratory tract infection, cough, extraordinary daytime urinary frequency (pollakiuria), unwanted hair growth (hirsutism), and excessive fat around the stomach (central obesity).
In the FDA’s announcement, Billy Dunn, MD, director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research, said, “We hope that this treatment option will benefit many patients with DMD.”
Emflaza, a tablet and oral suspension corticosteroid, has been approved by the Food and Drug Administration for the treatment of Duchenne muscular dystrophy in patients aged 5 years and older.
The agency’s Feb. 9 announcement notes that similar corticosteroids have been used around the world to treat Duchenne muscular dystrophy (DMD), but this is the first to gain approval in the United States. Emflaza (deflazacort) works by decreasing inflammation and immune system activity.
DMD is the most common form of muscular dystrophy but is still rare, occurring in about 1 in 3,600 male infants worldwide. One study found that patients taking deflazacort had some improvements in muscle strength at 12 weeks, compared with those taking placebo, and maintained muscle strength stability through 52 weeks. A longer-term study showed that patients who took deflazacort had better average muscle strength than did those taking placebo and suggested that deflazacort helped prolong patients’ ability to walk.
Side effects experienced by patients taking Emflaza are similar to those associated with other corticosteroids, such as facial puffiness (cushingoid appearance), weight gain, increased appetite, upper respiratory tract infection, cough, extraordinary daytime urinary frequency (pollakiuria), unwanted hair growth (hirsutism), and excessive fat around the stomach (central obesity).
In the FDA’s announcement, Billy Dunn, MD, director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research, said, “We hope that this treatment option will benefit many patients with DMD.”
Tips for Living With Tardive Dyskinesia
Click here to download the PDF.

Click here to download the PDF.
Click here to download the PDF.

Tips for Living With Suspected CTE
Click here to download the PDF. 
Click here to download the PDF.
Click here to download the PDF.

New histopathologic marker may aid dermatomyositis diagnosis
The detection of sarcoplasmic myxovirus resistance A expression in immunohistochemical analysis of muscle biopsy in patients suspected of having dermatomyositis may add greater sensitivity for the diagnosis when compared with conventional pathologic hallmarks of the disease, according to findings from a retrospective cohort study.
Myxovirus resistance A (MxA) is one of the type 1 interferon–inducible proteins whose overexpression is believed to play a role in the pathogenesis of dermatomyositis, and MxA expression has rarely been observed in other idiopathic inflammatory myopathies, said first author Akinori Uruha, MD, PhD, of the National Center of Neurology and Psychiatry, Tokyo, and his colleagues. They compared MxA expression in muscle biopsy samples from definite, probable, and possible dermatomyositis cases as well as other idiopathic inflammatory myopathies and other control conditions to assess its value against other muscle pathologic markers of dermatomyositis, such as the presence of perifascicular atrophy (PFA) and capillary membrane attack complex (MAC) deposition (Neurology. 2016 Dec 30. doi: 10.1212/WNL.0000000000003568).
The investigators studied muscle biopsy samples collected from 154 consecutive patients with idiopathic inflammatory myopathies seen from all over Japan, including 34 with dermatomyositis (10 juvenile cases), 8 with polymyositis (1 juvenile), 16 with anti–tRNA-synthetase antibody–associated myopathy (ASM); 46 with immune-mediated necrotizing myopathy (IMNM), and 50 with inclusion body myositis. The IMNM cases involved included 24 with anti–signal recognition particle (SRP) antibodies, 6 with anti–3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) antibodies, and 16 without anti-SRP, anti-HMGCR, or anti–tRNA-synthetase antibodies (3 juvenile patients). They used 51 patients with muscular dystrophy and 26 with neuropathies as controls.
Sarcoplasmic MxA expression proved to be more sensitive for a diagnosis of dermatomyositis than PFA and capillary MAC deposition (71% vs. 47% and 35%, respectively) but still had comparable specificity to those two markers (98% vs. 98% and 93%, respectively).
Of 18 cases with probable dermatomyositis, defined as typical skin rash but a lack of PFA, 8 (44%) showed sarcoplasmic MxA expression, and its sensitivity was 90% in juvenile cases overall and 63% in adult patients. Only 3 (17%) of the 18 showed capillary MAC deposition. Sarcoplasmic MxA expression occurred in all 12 patients with definite dermatomyositis, defined by the typical skin rash plus presence of PFA, whereas only 7 (58%) showed capillary MAC deposition. Among the four patients with possible dermatomyositis (PFA present but lacking typical skin rash), all showed sarcoplasmic MxA expression, compared with just two showing capillary MAC deposition.
In all other patients without definite, probable, or possible dermatomyositis, only two were positive for sarcoplasmic MxA expression (one with ASM and one with IMNM).
Dr. Uruha and his associates said that the results are “clearly demonstrating that sarcoplasmic MxA expression should be an excellent diagnostic marker of [dermatomyositis].”
The authors noted that the study was limited by the fact that they could not obtain full information about dermatomyositis-associated antibodies, and because other proteins of type 1 interferon signature are known to be upregulated in dermatomyositis, additional studies will need to determine which of the proteins is a better diagnostic marker.
The study was supported partly by an Intramural Research Grant of the National Center of Neurology and Psychiatry and grants from the Japanese Ministry of Education, Science, Sports and Culture and the Ministry of Health, Labor and Welfare of Japan. The investigators had no relevant disclosures.
The detection of sarcoplasmic myxovirus resistance A expression in immunohistochemical analysis of muscle biopsy in patients suspected of having dermatomyositis may add greater sensitivity for the diagnosis when compared with conventional pathologic hallmarks of the disease, according to findings from a retrospective cohort study.
Myxovirus resistance A (MxA) is one of the type 1 interferon–inducible proteins whose overexpression is believed to play a role in the pathogenesis of dermatomyositis, and MxA expression has rarely been observed in other idiopathic inflammatory myopathies, said first author Akinori Uruha, MD, PhD, of the National Center of Neurology and Psychiatry, Tokyo, and his colleagues. They compared MxA expression in muscle biopsy samples from definite, probable, and possible dermatomyositis cases as well as other idiopathic inflammatory myopathies and other control conditions to assess its value against other muscle pathologic markers of dermatomyositis, such as the presence of perifascicular atrophy (PFA) and capillary membrane attack complex (MAC) deposition (Neurology. 2016 Dec 30. doi: 10.1212/WNL.0000000000003568).
The investigators studied muscle biopsy samples collected from 154 consecutive patients with idiopathic inflammatory myopathies seen from all over Japan, including 34 with dermatomyositis (10 juvenile cases), 8 with polymyositis (1 juvenile), 16 with anti–tRNA-synthetase antibody–associated myopathy (ASM); 46 with immune-mediated necrotizing myopathy (IMNM), and 50 with inclusion body myositis. The IMNM cases involved included 24 with anti–signal recognition particle (SRP) antibodies, 6 with anti–3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) antibodies, and 16 without anti-SRP, anti-HMGCR, or anti–tRNA-synthetase antibodies (3 juvenile patients). They used 51 patients with muscular dystrophy and 26 with neuropathies as controls.
Sarcoplasmic MxA expression proved to be more sensitive for a diagnosis of dermatomyositis than PFA and capillary MAC deposition (71% vs. 47% and 35%, respectively) but still had comparable specificity to those two markers (98% vs. 98% and 93%, respectively).
Of 18 cases with probable dermatomyositis, defined as typical skin rash but a lack of PFA, 8 (44%) showed sarcoplasmic MxA expression, and its sensitivity was 90% in juvenile cases overall and 63% in adult patients. Only 3 (17%) of the 18 showed capillary MAC deposition. Sarcoplasmic MxA expression occurred in all 12 patients with definite dermatomyositis, defined by the typical skin rash plus presence of PFA, whereas only 7 (58%) showed capillary MAC deposition. Among the four patients with possible dermatomyositis (PFA present but lacking typical skin rash), all showed sarcoplasmic MxA expression, compared with just two showing capillary MAC deposition.
In all other patients without definite, probable, or possible dermatomyositis, only two were positive for sarcoplasmic MxA expression (one with ASM and one with IMNM).
Dr. Uruha and his associates said that the results are “clearly demonstrating that sarcoplasmic MxA expression should be an excellent diagnostic marker of [dermatomyositis].”
The authors noted that the study was limited by the fact that they could not obtain full information about dermatomyositis-associated antibodies, and because other proteins of type 1 interferon signature are known to be upregulated in dermatomyositis, additional studies will need to determine which of the proteins is a better diagnostic marker.
The study was supported partly by an Intramural Research Grant of the National Center of Neurology and Psychiatry and grants from the Japanese Ministry of Education, Science, Sports and Culture and the Ministry of Health, Labor and Welfare of Japan. The investigators had no relevant disclosures.
The detection of sarcoplasmic myxovirus resistance A expression in immunohistochemical analysis of muscle biopsy in patients suspected of having dermatomyositis may add greater sensitivity for the diagnosis when compared with conventional pathologic hallmarks of the disease, according to findings from a retrospective cohort study.
Myxovirus resistance A (MxA) is one of the type 1 interferon–inducible proteins whose overexpression is believed to play a role in the pathogenesis of dermatomyositis, and MxA expression has rarely been observed in other idiopathic inflammatory myopathies, said first author Akinori Uruha, MD, PhD, of the National Center of Neurology and Psychiatry, Tokyo, and his colleagues. They compared MxA expression in muscle biopsy samples from definite, probable, and possible dermatomyositis cases as well as other idiopathic inflammatory myopathies and other control conditions to assess its value against other muscle pathologic markers of dermatomyositis, such as the presence of perifascicular atrophy (PFA) and capillary membrane attack complex (MAC) deposition (Neurology. 2016 Dec 30. doi: 10.1212/WNL.0000000000003568).
The investigators studied muscle biopsy samples collected from 154 consecutive patients with idiopathic inflammatory myopathies seen from all over Japan, including 34 with dermatomyositis (10 juvenile cases), 8 with polymyositis (1 juvenile), 16 with anti–tRNA-synthetase antibody–associated myopathy (ASM); 46 with immune-mediated necrotizing myopathy (IMNM), and 50 with inclusion body myositis. The IMNM cases involved included 24 with anti–signal recognition particle (SRP) antibodies, 6 with anti–3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) antibodies, and 16 without anti-SRP, anti-HMGCR, or anti–tRNA-synthetase antibodies (3 juvenile patients). They used 51 patients with muscular dystrophy and 26 with neuropathies as controls.
Sarcoplasmic MxA expression proved to be more sensitive for a diagnosis of dermatomyositis than PFA and capillary MAC deposition (71% vs. 47% and 35%, respectively) but still had comparable specificity to those two markers (98% vs. 98% and 93%, respectively).
Of 18 cases with probable dermatomyositis, defined as typical skin rash but a lack of PFA, 8 (44%) showed sarcoplasmic MxA expression, and its sensitivity was 90% in juvenile cases overall and 63% in adult patients. Only 3 (17%) of the 18 showed capillary MAC deposition. Sarcoplasmic MxA expression occurred in all 12 patients with definite dermatomyositis, defined by the typical skin rash plus presence of PFA, whereas only 7 (58%) showed capillary MAC deposition. Among the four patients with possible dermatomyositis (PFA present but lacking typical skin rash), all showed sarcoplasmic MxA expression, compared with just two showing capillary MAC deposition.
In all other patients without definite, probable, or possible dermatomyositis, only two were positive for sarcoplasmic MxA expression (one with ASM and one with IMNM).
Dr. Uruha and his associates said that the results are “clearly demonstrating that sarcoplasmic MxA expression should be an excellent diagnostic marker of [dermatomyositis].”
The authors noted that the study was limited by the fact that they could not obtain full information about dermatomyositis-associated antibodies, and because other proteins of type 1 interferon signature are known to be upregulated in dermatomyositis, additional studies will need to determine which of the proteins is a better diagnostic marker.
The study was supported partly by an Intramural Research Grant of the National Center of Neurology and Psychiatry and grants from the Japanese Ministry of Education, Science, Sports and Culture and the Ministry of Health, Labor and Welfare of Japan. The investigators had no relevant disclosures.
FROM NEUROLOGY
Key clinical point:
Major finding: Sarcoplasmic MxA expression proved to be more sensitive for a diagnosis of dermatomyositis than PFA and capillary MAC deposition (71% vs. 47% and 35%, respectively) but still had comparable specificity to those two markers (98% vs. 98% and 93%, respectively).
Data source: A retrospective cohort study of 154 patients with idiopathic inflammatory myopathies, 51 with muscular dystrophy, and 26 with neuropathies.
Disclosures: The study was supported partly by an Intramural Research Grant of the National Center of Neurology and Psychiatry and grants from the Japanese Ministry of Education, Science, Sports and Culture and the Ministry of Health, Labor and Welfare of Japan. The investigators had no relevant disclosures.