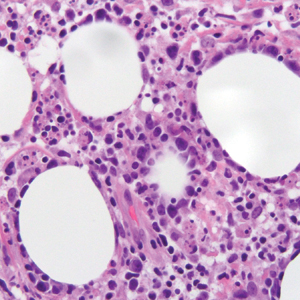
User login
Violaceous Papule With an Erythematous Rim
The Diagnosis: Targetoid Hemosiderotic Hemangioma
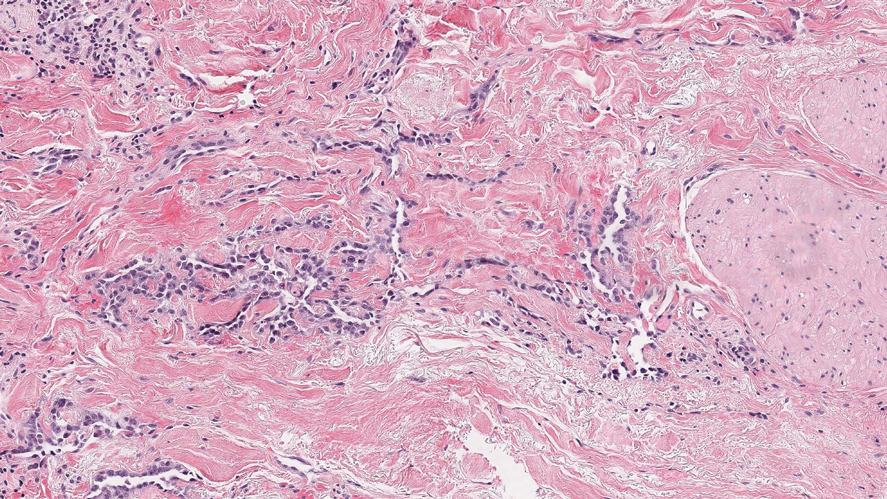
Targetoid hemosiderotic hemangioma (THH), also known as hobnail hemangioma, is a benign vascular tumor that usually occurs in young or middle-aged adults. It most commonly presents on the extremities or trunk as an isolated red-brown plaque or papule.1,2 Histologically, THH is characterized by superficial dilated ectatic vessels with underlying proliferating vascular channels lined by plump hobnail endothelial cells.1 Targetoid hemosiderotic hemangioma typically involves the dermis and spares the subcutis. The vascular channels may contain erythrocytes as well as pale eosinophilic lymph, as seen in our patient (quiz image). The deeper dermis contains vascular spaces that are more angulated and smaller and appear to be dissecting through the collagen bundles or collapsed.1,3 A variable amount of hemosiderin deposition and extravasated erythrocytes are seen.2,3 Histologic features evolve with the age of the lesion. Increasing amounts of hemosiderin deposition and erythrocyte extravasation may correspond histologically to the recent clinical color change reported by the patient.
Verrucous hemangioma is a rare congenital vascular abnormality that is characterized by dilated vessels in the papillary dermis along with acanthosis, hyperkeratosis, and irregular papillomatosis, as seen in angiokeratoma.4 However, the vascular proliferation composed of variably sized, thin-walled capillaries extends into the deep dermis as well as the subcutis (Figure 1). Verrucous hemangioma most commonly is reported on the legs and generally starts as a violaceous patch that progresses into a hyperkeratotic verrucous plaque or nodule.5,6
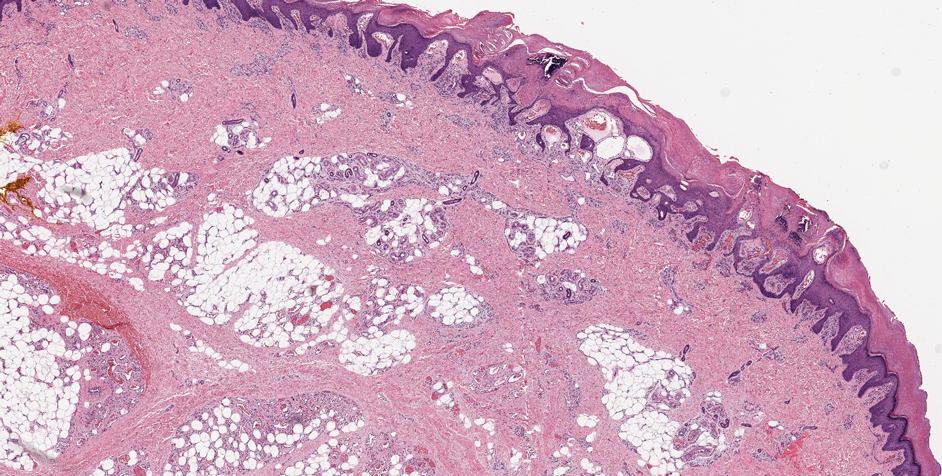
Angiokeratoma is characterized by superficial vascular ectasia of the papillary dermis in association with overlying acanthosis, hyperkeratosis, and rete elongation.7 The dilated vascular spaces appear encircled by the epidermis (Figure 2). Intravascular thrombosis can be seen within the ectatic vessels.7 In contrast to verrucous hemangioma, angiokeratoma is limited to the papillary dermis. Therefore, obtaining a biopsy of sufficient depth is necessary for differentiation.8 There are 5 clinical presentations of angiokeratoma: sporadic, angiokeratoma of Mibelli, angiokeratoma of Fordyce, angiokeratoma circumscriptum, and angiokeratoma corporis diffusum (Fabry disease). Angiokeratomas may present on the lower extremities, tongue, trunk, and scrotum as hyperkeratotic, dark red to purple or black papules.7
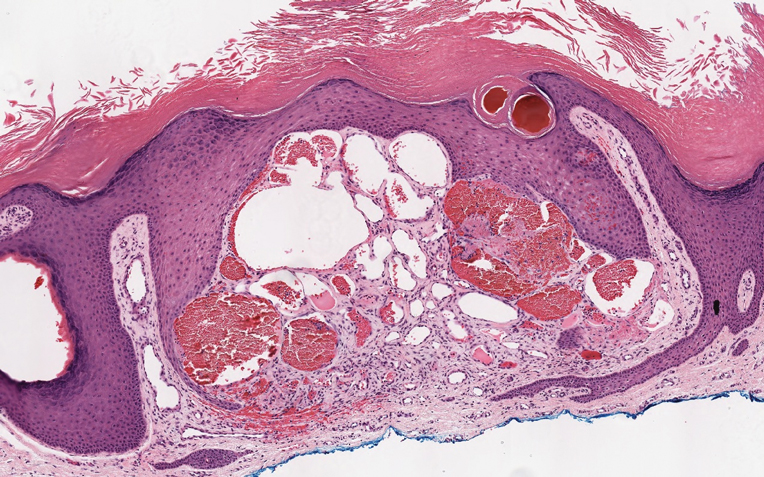
There are 3 clinical stages of Kaposi sarcoma: patch, plaque, and nodular stages. The patch stage is characterized histologically by vascular channels that dissect through the dermis and extend around native vessels (the promontory sign)(Figure 3).9,10 These features can show histologic overlap with THH. The plaque stage shows a more diffuse dermal vascular proliferation, increased cellularity of spindle cells, and possible extension into the subcutis.9,10 Focal plasma cells, hemosiderin, and extravasated red blood cells can be seen. The nodular stage is characterized by a proliferation of spindle cells with red blood cells squeezed between slitlike vascular spaces, hyaline globules, and scattered mitotic figures, but not atypical forms.10 In this stage, plasma cells and hemosiderin are more readily identifiable. A biopsy from the nodular stage is unlikely to enter the histologic differential diagnosis with THH. Clinically, there are 4 variants of Kaposi sarcoma: the classic or sporadic form, an endemic form, iatrogenic, and AIDS associated. Overall, it is more common in males and can occur at any age.10 Human herpesvirus 8 is seen in all forms, and infected cells can be highlighted by the immunohistochemical stain for latent nuclear antigen 1.9,10
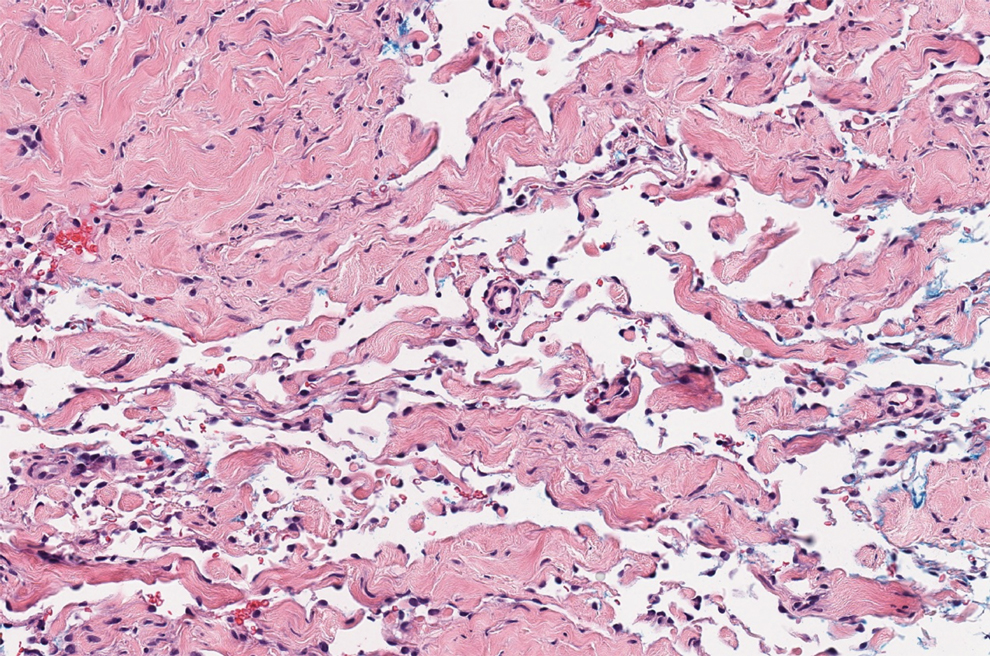
Angiosarcoma is a malignant endothelial tumor of soft tissue, skin, bone, and visceral organs.11,12 Clinically, cutaneous angiosarcoma can present in a variety of ways, including single or multiple bluish red lesions that can ulcerate or bleed; violaceous nodules or plaques; and hematomalike lesions that can mimic epithelial neoplasms including squamous cell carcinoma, basal cell carcinoma, and malignant melanoma.11,13,14 The cutaneous lesions most commonly occur on sun-exposed skin, particularly on the face and scalp.12 Other clinical variants that are important to recognize are postradiation angiosarcoma, characterized by MYC gene amplification, and lymphedema-associated angiosarcoma (Stewart-Treves syndrome). Angiosarcoma can have a variety of morphologic features, ranging from well to poorly differentiated. Classically, angiosarcoma is characterized by infiltrating vascular spaces lined by atypical endothelial cells (Figure 4). Poorly differentiated angiosarcoma can demonstrate spindle, epithelioid, or polygonal cells with increased mitotic activity, pleomorphism, and irregular vascular spaces.11 Endothelial markers such as ERG (erythroblast transformation specific-related gene)(nuclear) and CD31 (membranous) can be used to aid in the diagnosis of a poorly differentiated lesion. Epithelioid angiosarcoma also occasionally stains with cytokeratins.13,14
- Joyce JC, Keith PJ, Szabo S, et al. Superficial hemosiderotic lymphovascular malformation (hobnail hemangioma): a report of six cases. Pediatr Dermatol. 2014;31:281-285.
- Sahin MT, Demir MA, Gunduz K, et al. Targetoid haemosiderotic haemangioma: dermoscopic monitoring of three cases and review of the literature. Clin Exp Dermatol. 2005;30:672-676.
- Kakizaki P, Valente NY, Paiva DL, et al. Targetoid hemosiderotic hemangioma--case report. An Bras Dermatol. 2014;89:956-959.
- Oppermann K, Boff AL, Bonamigo RR. Verrucous hemangioma and histopathological differential diagnosis with angiokeratoma circumscriptum neviforme. An Bras Dermatol. 2018;93:712-715.
- Boccara, O, Ariche-Maman, S, Hadj-Rabia, S, et al. Verrucous hemangioma (also known as verrucous venous malformation): a vascular anomaly frequently misdiagnosed as a lymphatic malformation. Pediatr Dermatol. 2018;35:E378-E381.
- Mestre T, Amaro C, Freitas I. Verrucous haemangioma: a diagnosis to consider [published online June 4, 2014]. BMJ Case Rep. doi:10.1136/bcr-2014-204612
- Ivy H, Julian CA. Angiokeratoma circumscriptum. StatPearls. StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK549769/
- Shetty S, Geetha V, Rao R, et al. Verrucous hemangioma: importance of a deeper biopsy. Indian J Dermatopathol Diagn Dermatol. 2014;1:99-100.
- Bishop BN, Lynch DT. Cancer, Kaposi sarcoma. StatPearls. StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK534839/
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Cao J, Wang J, He C, et al. Angiosarcoma: a review of diagnosis and current treatment. Am J Cancer Res. 2019;9:2303-2313.
- Papke DJ Jr, Hornick JL. What is new in endothelial neoplasia? Virchows Arch. 2020;476:17-28.
- Ambujam S, Audhya M, Reddy A, et al. Cutaneous angiosarcoma of the head, neck, and face of the elderly in type 5 skin. J Cutan Aesthet Surg. 2013;6:45-47.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.
The Diagnosis: Targetoid Hemosiderotic Hemangioma
Targetoid hemosiderotic hemangioma (THH), also known as hobnail hemangioma, is a benign vascular tumor that usually occurs in young or middle-aged adults. It most commonly presents on the extremities or trunk as an isolated red-brown plaque or papule.1,2 Histologically, THH is characterized by superficial dilated ectatic vessels with underlying proliferating vascular channels lined by plump hobnail endothelial cells.1 Targetoid hemosiderotic hemangioma typically involves the dermis and spares the subcutis. The vascular channels may contain erythrocytes as well as pale eosinophilic lymph, as seen in our patient (quiz image). The deeper dermis contains vascular spaces that are more angulated and smaller and appear to be dissecting through the collagen bundles or collapsed.1,3 A variable amount of hemosiderin deposition and extravasated erythrocytes are seen.2,3 Histologic features evolve with the age of the lesion. Increasing amounts of hemosiderin deposition and erythrocyte extravasation may correspond histologically to the recent clinical color change reported by the patient.
Verrucous hemangioma is a rare congenital vascular abnormality that is characterized by dilated vessels in the papillary dermis along with acanthosis, hyperkeratosis, and irregular papillomatosis, as seen in angiokeratoma.4 However, the vascular proliferation composed of variably sized, thin-walled capillaries extends into the deep dermis as well as the subcutis (Figure 1). Verrucous hemangioma most commonly is reported on the legs and generally starts as a violaceous patch that progresses into a hyperkeratotic verrucous plaque or nodule.5,6
Angiokeratoma is characterized by superficial vascular ectasia of the papillary dermis in association with overlying acanthosis, hyperkeratosis, and rete elongation.7 The dilated vascular spaces appear encircled by the epidermis (Figure 2). Intravascular thrombosis can be seen within the ectatic vessels.7 In contrast to verrucous hemangioma, angiokeratoma is limited to the papillary dermis. Therefore, obtaining a biopsy of sufficient depth is necessary for differentiation.8 There are 5 clinical presentations of angiokeratoma: sporadic, angiokeratoma of Mibelli, angiokeratoma of Fordyce, angiokeratoma circumscriptum, and angiokeratoma corporis diffusum (Fabry disease). Angiokeratomas may present on the lower extremities, tongue, trunk, and scrotum as hyperkeratotic, dark red to purple or black papules.7
There are 3 clinical stages of Kaposi sarcoma: patch, plaque, and nodular stages. The patch stage is characterized histologically by vascular channels that dissect through the dermis and extend around native vessels (the promontory sign)(Figure 3).9,10 These features can show histologic overlap with THH. The plaque stage shows a more diffuse dermal vascular proliferation, increased cellularity of spindle cells, and possible extension into the subcutis.9,10 Focal plasma cells, hemosiderin, and extravasated red blood cells can be seen. The nodular stage is characterized by a proliferation of spindle cells with red blood cells squeezed between slitlike vascular spaces, hyaline globules, and scattered mitotic figures, but not atypical forms.10 In this stage, plasma cells and hemosiderin are more readily identifiable. A biopsy from the nodular stage is unlikely to enter the histologic differential diagnosis with THH. Clinically, there are 4 variants of Kaposi sarcoma: the classic or sporadic form, an endemic form, iatrogenic, and AIDS associated. Overall, it is more common in males and can occur at any age.10 Human herpesvirus 8 is seen in all forms, and infected cells can be highlighted by the immunohistochemical stain for latent nuclear antigen 1.9,10
Angiosarcoma is a malignant endothelial tumor of soft tissue, skin, bone, and visceral organs.11,12 Clinically, cutaneous angiosarcoma can present in a variety of ways, including single or multiple bluish red lesions that can ulcerate or bleed; violaceous nodules or plaques; and hematomalike lesions that can mimic epithelial neoplasms including squamous cell carcinoma, basal cell carcinoma, and malignant melanoma.11,13,14 The cutaneous lesions most commonly occur on sun-exposed skin, particularly on the face and scalp.12 Other clinical variants that are important to recognize are postradiation angiosarcoma, characterized by MYC gene amplification, and lymphedema-associated angiosarcoma (Stewart-Treves syndrome). Angiosarcoma can have a variety of morphologic features, ranging from well to poorly differentiated. Classically, angiosarcoma is characterized by infiltrating vascular spaces lined by atypical endothelial cells (Figure 4). Poorly differentiated angiosarcoma can demonstrate spindle, epithelioid, or polygonal cells with increased mitotic activity, pleomorphism, and irregular vascular spaces.11 Endothelial markers such as ERG (erythroblast transformation specific-related gene)(nuclear) and CD31 (membranous) can be used to aid in the diagnosis of a poorly differentiated lesion. Epithelioid angiosarcoma also occasionally stains with cytokeratins.13,14
The Diagnosis: Targetoid Hemosiderotic Hemangioma
Targetoid hemosiderotic hemangioma (THH), also known as hobnail hemangioma, is a benign vascular tumor that usually occurs in young or middle-aged adults. It most commonly presents on the extremities or trunk as an isolated red-brown plaque or papule.1,2 Histologically, THH is characterized by superficial dilated ectatic vessels with underlying proliferating vascular channels lined by plump hobnail endothelial cells.1 Targetoid hemosiderotic hemangioma typically involves the dermis and spares the subcutis. The vascular channels may contain erythrocytes as well as pale eosinophilic lymph, as seen in our patient (quiz image). The deeper dermis contains vascular spaces that are more angulated and smaller and appear to be dissecting through the collagen bundles or collapsed.1,3 A variable amount of hemosiderin deposition and extravasated erythrocytes are seen.2,3 Histologic features evolve with the age of the lesion. Increasing amounts of hemosiderin deposition and erythrocyte extravasation may correspond histologically to the recent clinical color change reported by the patient.
Verrucous hemangioma is a rare congenital vascular abnormality that is characterized by dilated vessels in the papillary dermis along with acanthosis, hyperkeratosis, and irregular papillomatosis, as seen in angiokeratoma.4 However, the vascular proliferation composed of variably sized, thin-walled capillaries extends into the deep dermis as well as the subcutis (Figure 1). Verrucous hemangioma most commonly is reported on the legs and generally starts as a violaceous patch that progresses into a hyperkeratotic verrucous plaque or nodule.5,6
Angiokeratoma is characterized by superficial vascular ectasia of the papillary dermis in association with overlying acanthosis, hyperkeratosis, and rete elongation.7 The dilated vascular spaces appear encircled by the epidermis (Figure 2). Intravascular thrombosis can be seen within the ectatic vessels.7 In contrast to verrucous hemangioma, angiokeratoma is limited to the papillary dermis. Therefore, obtaining a biopsy of sufficient depth is necessary for differentiation.8 There are 5 clinical presentations of angiokeratoma: sporadic, angiokeratoma of Mibelli, angiokeratoma of Fordyce, angiokeratoma circumscriptum, and angiokeratoma corporis diffusum (Fabry disease). Angiokeratomas may present on the lower extremities, tongue, trunk, and scrotum as hyperkeratotic, dark red to purple or black papules.7
There are 3 clinical stages of Kaposi sarcoma: patch, plaque, and nodular stages. The patch stage is characterized histologically by vascular channels that dissect through the dermis and extend around native vessels (the promontory sign)(Figure 3).9,10 These features can show histologic overlap with THH. The plaque stage shows a more diffuse dermal vascular proliferation, increased cellularity of spindle cells, and possible extension into the subcutis.9,10 Focal plasma cells, hemosiderin, and extravasated red blood cells can be seen. The nodular stage is characterized by a proliferation of spindle cells with red blood cells squeezed between slitlike vascular spaces, hyaline globules, and scattered mitotic figures, but not atypical forms.10 In this stage, plasma cells and hemosiderin are more readily identifiable. A biopsy from the nodular stage is unlikely to enter the histologic differential diagnosis with THH. Clinically, there are 4 variants of Kaposi sarcoma: the classic or sporadic form, an endemic form, iatrogenic, and AIDS associated. Overall, it is more common in males and can occur at any age.10 Human herpesvirus 8 is seen in all forms, and infected cells can be highlighted by the immunohistochemical stain for latent nuclear antigen 1.9,10
Angiosarcoma is a malignant endothelial tumor of soft tissue, skin, bone, and visceral organs.11,12 Clinically, cutaneous angiosarcoma can present in a variety of ways, including single or multiple bluish red lesions that can ulcerate or bleed; violaceous nodules or plaques; and hematomalike lesions that can mimic epithelial neoplasms including squamous cell carcinoma, basal cell carcinoma, and malignant melanoma.11,13,14 The cutaneous lesions most commonly occur on sun-exposed skin, particularly on the face and scalp.12 Other clinical variants that are important to recognize are postradiation angiosarcoma, characterized by MYC gene amplification, and lymphedema-associated angiosarcoma (Stewart-Treves syndrome). Angiosarcoma can have a variety of morphologic features, ranging from well to poorly differentiated. Classically, angiosarcoma is characterized by infiltrating vascular spaces lined by atypical endothelial cells (Figure 4). Poorly differentiated angiosarcoma can demonstrate spindle, epithelioid, or polygonal cells with increased mitotic activity, pleomorphism, and irregular vascular spaces.11 Endothelial markers such as ERG (erythroblast transformation specific-related gene)(nuclear) and CD31 (membranous) can be used to aid in the diagnosis of a poorly differentiated lesion. Epithelioid angiosarcoma also occasionally stains with cytokeratins.13,14
- Joyce JC, Keith PJ, Szabo S, et al. Superficial hemosiderotic lymphovascular malformation (hobnail hemangioma): a report of six cases. Pediatr Dermatol. 2014;31:281-285.
- Sahin MT, Demir MA, Gunduz K, et al. Targetoid haemosiderotic haemangioma: dermoscopic monitoring of three cases and review of the literature. Clin Exp Dermatol. 2005;30:672-676.
- Kakizaki P, Valente NY, Paiva DL, et al. Targetoid hemosiderotic hemangioma--case report. An Bras Dermatol. 2014;89:956-959.
- Oppermann K, Boff AL, Bonamigo RR. Verrucous hemangioma and histopathological differential diagnosis with angiokeratoma circumscriptum neviforme. An Bras Dermatol. 2018;93:712-715.
- Boccara, O, Ariche-Maman, S, Hadj-Rabia, S, et al. Verrucous hemangioma (also known as verrucous venous malformation): a vascular anomaly frequently misdiagnosed as a lymphatic malformation. Pediatr Dermatol. 2018;35:E378-E381.
- Mestre T, Amaro C, Freitas I. Verrucous haemangioma: a diagnosis to consider [published online June 4, 2014]. BMJ Case Rep. doi:10.1136/bcr-2014-204612
- Ivy H, Julian CA. Angiokeratoma circumscriptum. StatPearls. StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK549769/
- Shetty S, Geetha V, Rao R, et al. Verrucous hemangioma: importance of a deeper biopsy. Indian J Dermatopathol Diagn Dermatol. 2014;1:99-100.
- Bishop BN, Lynch DT. Cancer, Kaposi sarcoma. StatPearls. StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK534839/
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Cao J, Wang J, He C, et al. Angiosarcoma: a review of diagnosis and current treatment. Am J Cancer Res. 2019;9:2303-2313.
- Papke DJ Jr, Hornick JL. What is new in endothelial neoplasia? Virchows Arch. 2020;476:17-28.
- Ambujam S, Audhya M, Reddy A, et al. Cutaneous angiosarcoma of the head, neck, and face of the elderly in type 5 skin. J Cutan Aesthet Surg. 2013;6:45-47.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.
- Joyce JC, Keith PJ, Szabo S, et al. Superficial hemosiderotic lymphovascular malformation (hobnail hemangioma): a report of six cases. Pediatr Dermatol. 2014;31:281-285.
- Sahin MT, Demir MA, Gunduz K, et al. Targetoid haemosiderotic haemangioma: dermoscopic monitoring of three cases and review of the literature. Clin Exp Dermatol. 2005;30:672-676.
- Kakizaki P, Valente NY, Paiva DL, et al. Targetoid hemosiderotic hemangioma--case report. An Bras Dermatol. 2014;89:956-959.
- Oppermann K, Boff AL, Bonamigo RR. Verrucous hemangioma and histopathological differential diagnosis with angiokeratoma circumscriptum neviforme. An Bras Dermatol. 2018;93:712-715.
- Boccara, O, Ariche-Maman, S, Hadj-Rabia, S, et al. Verrucous hemangioma (also known as verrucous venous malformation): a vascular anomaly frequently misdiagnosed as a lymphatic malformation. Pediatr Dermatol. 2018;35:E378-E381.
- Mestre T, Amaro C, Freitas I. Verrucous haemangioma: a diagnosis to consider [published online June 4, 2014]. BMJ Case Rep. doi:10.1136/bcr-2014-204612
- Ivy H, Julian CA. Angiokeratoma circumscriptum. StatPearls. StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK549769/
- Shetty S, Geetha V, Rao R, et al. Verrucous hemangioma: importance of a deeper biopsy. Indian J Dermatopathol Diagn Dermatol. 2014;1:99-100.
- Bishop BN, Lynch DT. Cancer, Kaposi sarcoma. StatPearls. StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK534839/
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Cao J, Wang J, He C, et al. Angiosarcoma: a review of diagnosis and current treatment. Am J Cancer Res. 2019;9:2303-2313.
- Papke DJ Jr, Hornick JL. What is new in endothelial neoplasia? Virchows Arch. 2020;476:17-28.
- Ambujam S, Audhya M, Reddy A, et al. Cutaneous angiosarcoma of the head, neck, and face of the elderly in type 5 skin. J Cutan Aesthet Surg. 2013;6:45-47.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.

A 35-year-old man presented with a reddish brown papule on the left upper chest of 1 year’s duration that had changed color to reddish purple. Physical examination revealed a 6-mm violaceous papule with an erythematous rim.
Tender Soft Tissue Mass on the Base of the Neck
The Diagnosis: Subcutaneous Panniculitislike T-cell Lymphoma
Subcutaneous panniculitislike T-cell lymphoma (SPTCL) is a rare form of cutaneous lymphoma of mature cytotoxic T cells simulating panniculitis and preferentially infiltrating the subcutaneous tissue.1 Subcutaneous panniculitislike T-cell lymphoma can affect all ages but predominantly affects younger individuals, with 20% being younger than 20 years.2 It is a rare lymphoma that accounts for less than 1% of all non-Hodgkin lymphomas.3 It presents clinically as multiple subcutaneous masses, nodules, or plaques generally on the trunk or extremities.1,2 The skin surrounding the nodules may be erythematous, and the nodules may become necrotic; however, ulceration typically is not seen. Systemic symptoms such as fever, night sweats, and chills are present in half of cases.1 According to the World Health Organization, cytopenia and elevated liver function tests are common, and a hemophagocytic syndrome may be present in 15% to 20% of cases.3 The presence of a hemophagocytic syndrome yields a poor prognosis.1,3 Current guidelines denote that SPTCL T-cell receptor (TCR) αβ; is a distinct entity from the TCRγδ; phenotype, known as cutaneous γδ-positive T-cell lymphoma.3,4 Cutaneous γδ-positive T-cell lymphoma is associated with rapid decline and a worse prognosis.4
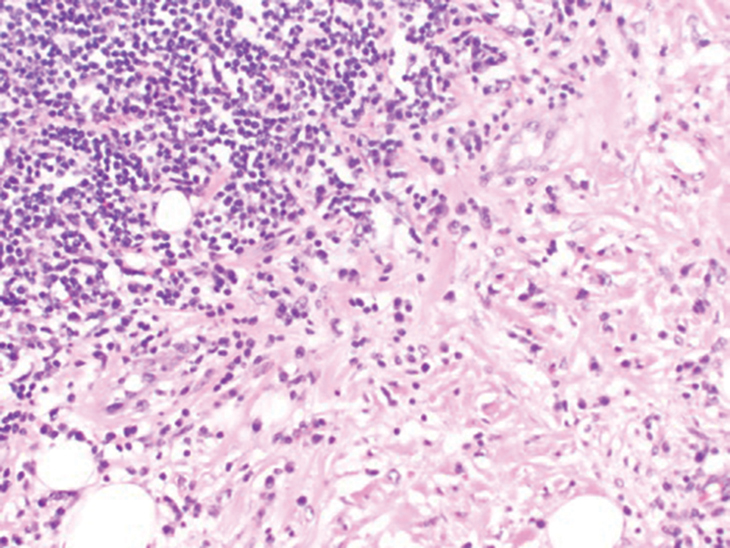
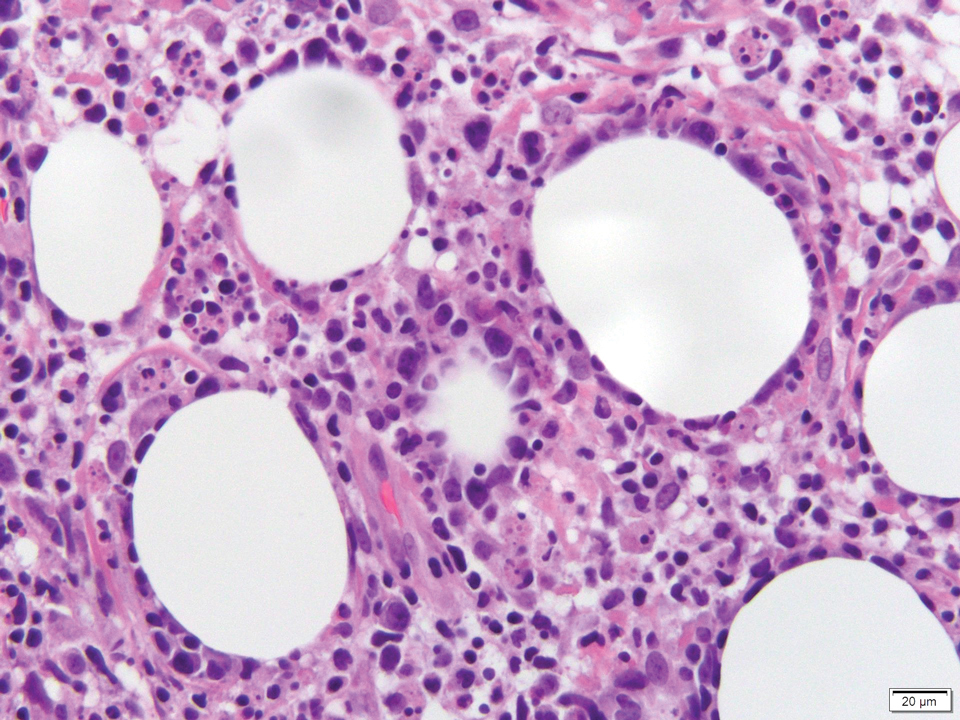
Histology of SPTCL is characteristic for a lobular panniculitislike infiltrate.1 The heavy subcutaneous lymphoid infiltrate is composed of atypical small- to medium-sized lymphocytes with mature chromatin and inconspicuous nucleoli lining adipocytes. The dense inflammatory infiltrate composed predominantly of neoplastic T cells and macrophages may diffusely invade into the subcutaneous tissue.1 Admixed histocytes and karyorrhectic debris as well as rimming of the lymphocytes around the fat cells is typical and was seen in our patient (quiz image). The T cells of SPTCL have the following immunophenotype: TCR-beta F1+, CD3+, CD4-, CD8+, CD56-. They can express numerous cytotoxic proteins, such as T1a-1, granzyme B, and perforin.2,3 Although the CD8+ T cells may be sparse, they generally surround the adipocytes in a rimming manner and may distort the adipocyte membrane.1
Lupus erythematosus profundus (LEP) is a form of chronic cutaneous lupus that affects the deep dermis and fat.5 It also can present clinically as tender plaques or nodules. It most frequently involves the upper arms, shoulders, face, or buttocks--areas that are less commonly involved in other panniculitides.6 Histologically, LEP is similar to chronic discoid lupus with features such as epidermal atrophy, interface changes, and a thickened basement membrane (Figure 1). Lupus erythematosus profundus can present as a lobular panniculitis with mucin as well as a superficial and deep lymphocytic infiltrate that can involve the septa.5 Some cases of LEP have a predominantly lobular lymphocytic panniculitis in the absence of the typical epidermal or dermal changes of lupus erythematosus. Lymphoid follicles with germinal center formation are present in half of cases and reportedly are characteristic of LEP.6,7 The lymphoid follicles often have plasma cells, can extend into the septa as well as in between collagen bundles, and may have nuclear fragmentation.5 Another characteristic feature of LEP is hyaline sclerosis of lobules with focal extension into the interlobular septa. Immunofluorescence studies usually show linear deposition of IgM and C3 at the dermoepidermal junction. Antinuclear antibodies can be present in patients who have LEP but are not entirely specific.6
Lupus erythematosus profundus and SPTCL are part of a spectrum and may have overlapping clinical and histopathologic characteristics; therefore, distinguishing them may be difficult.6-8 It is important to monitor these patients closely, as their disease may progress to lymphoma.6 Patients with SPTCL are more likely to present with advanced symptoms such as fever and hepatosplenomegaly and to succumb to hemophagocytic syndrome than patients with LEP.9
Although SPTCL usually is clonal, several cases of LEP with clonality also have been described. Clonal LEP cases generally are identified in patients who present with fever and cytopenia.8 Lymphoid atypia and morphologic abnormalities may be seen in cases of LEP, further complicating the distinction between LEP and SPTCL. An elevated Ki67 level may be seen in cases of SPTCL with periadipocytic rimming.9 LeBlanc et al10 used Ki67 "hot spots" along with CD8 immunohistochemistry to identify atypical lymphocytes associated with SPTCL. Lymphocyte rimming was defined by the presence of CD8+ lymphocytes with an elevated Ki67 index. Clinical, histopathologic, and molecular findings all should be used when dealing with challenging cases.
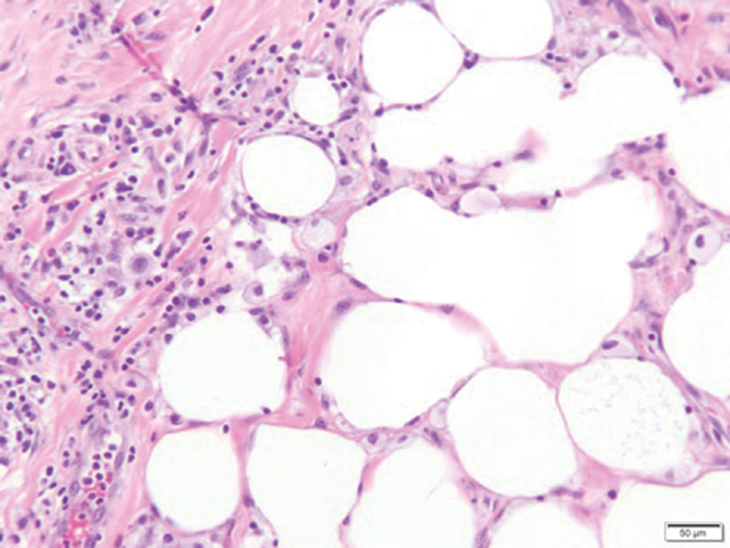
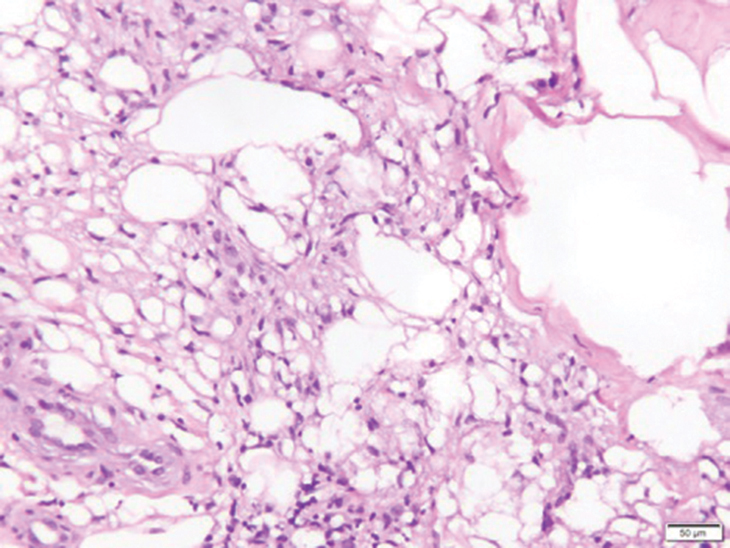
Fat necrosis can occur in any part of the body where trauma has occurred and can be associated with many disease processes. Patients typically present with a palpable mass, but a clinical history of trauma is not always present. Histopathologic findings include necrotic fat alongside lipid-laden foamy macrophages and scattered inflammatory cells (Figure 2).11 Fragments of normal as well as degenerating adipose tissue and multinucleated giant cells can be present.
Erythema nodosum (EN) is the most frequently encountered panniculitis and usually is seen in women in early adulthood.12 Patients present with several tender subcutaneous nodules and plaques that most commonly are present on the anterior surface of the legs.12,13 Patients may have a constellation of symptoms including fever and leukocytosis, but the disorder generally is self-limited.12 Erythema nodosum may be associated with a variety of diseases or infections including sarcoidosis, inflammatory bowel disease, and malignancy.14 The etiology of EN is diverse; therefore, a proper clinical workup may be necessary. Histopathology is that of a septal panniculitis with lymphocytes, histiocytes, and occasional eosinophils (Figure 3).13
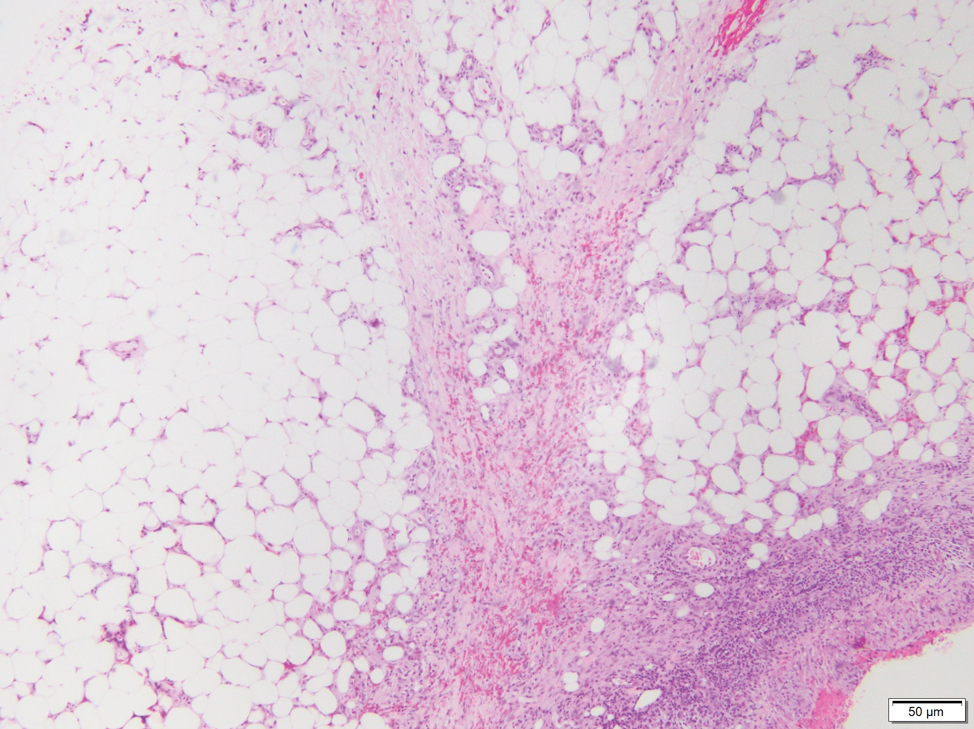
Lipodermatosclerosis also occurs on the legs, most commonly in patients with venous insufficiency.12,15 Patients present clinically with pain, induration, redness, or swelling of the legs. Histopathology predominantly is characterized by membranous fat necrosis, fibrosis, and fatty microcysts that may be lined by a thickened hyaline membrane (Figure 4). Lipodermatosclerosis lesions generally do not resolve spontaneously and may need to be treated.16
- Musick SR, Lynch DT. Subcutaneous Panniculitis Like T-cell Lymphoma. StatPearls Publishing; 2020.
- Guenova E, Schanz S, Hoetzenecker W, et al. Systemic corticosteroids for subcutaneous panniculitis-like T-cell lymphoma. Br J Dermatol. 2014;171:891-894.
- Swerdlow SH. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. International Agency for Research on Cancer; 2017.
- Bagheri F, Cervellione KL, Delgado B, et al. An illustrative case of subcutaneous panniculitis-like T-cell lymphoma [published online March 3, 2011]. J Skin Cancer. doi:10.1155/2011/824528
- Kogame T, Yamashita R, Hirata M, et al. Analysis of possible structures of inducible skin‐associated lymphoid tissue in lupus erythematosus profundus. J Dermatol. 2018;45:1117-1121.
- Arps DP, Patel RM. Lupus profundus (panniculitis): a potential mimic of subcutaneous panniculitis-like T-cell lymphoma. Arch Pathol Lab Med. 2013;137:1211-1215.
- Alberti-Violetti S, Berti E. Lymphocytic lobular panniculitis: a diagnostic challenge. Dermatopathology. 2018;5:30-33.
- Magro CM, Crowson AN, Kovatich AJ, et al. Lupus profundus, indeterminate lymphocytic lobular panniculitis and subcutaneous T-cell lymphoma: a spectrum of subcuticular T-cell lymphoid dyscrasia. J Cutan Pathol. 2001;28:235-247.
- Sitthinamsuwan P, Pattanaprichakul P, Treetipsatit J, et al. Subcutaneous panniculitis-like T-cell lymphoma versus lupus erythematosus panniculitis: distinction by means of the periadipocytic cell proliferation index. Am J Dermatopathol. 2018;40:567-574.
- LeBlanc RE, Tavallaee M, Kim YH, et al. Useful parameters for distinguishing subcutaneous panniculitis-like T-cell lymphoma from lupus erythematosus panniculitis. Am J Surg Pathol. 2016;40:745-754.
- Burkholz KJ, Roberts CC, Lidner TK. Posttraumatic pseudolipoma (fat necrosis) mimicking atypical lipoma or liposarcoma on MRI. Radiol Case Rep. 2015;2:56-60.
- Wick MR. Panniculitis: a summary. Semin Diagn Pathol. 2017;34:261-272.
- Thurber S, Kohler S. Histopathologic spectrum of erythema nodosum. J Cutan Pathol. 2006;33:18-26.
- Requena L, Requena C. Erythema nodosum. Dermatol Online J. 2002;8:4.
- Choonhakarn C, Chaowattanapanit S, Julanon N. Lipodermatosclerosis: a clinicopathologic correlation. Int J Dermatol. 2016;55:303-308.
- Huang TM, Lee JY. Lipodermatosclerosis: a clinicopathologic study of 17 cases and differential diagnosis from erythema nodosum. J Cutan Pathol. 2009;36:453-460.
The Diagnosis: Subcutaneous Panniculitislike T-cell Lymphoma
Subcutaneous panniculitislike T-cell lymphoma (SPTCL) is a rare form of cutaneous lymphoma of mature cytotoxic T cells simulating panniculitis and preferentially infiltrating the subcutaneous tissue.1 Subcutaneous panniculitislike T-cell lymphoma can affect all ages but predominantly affects younger individuals, with 20% being younger than 20 years.2 It is a rare lymphoma that accounts for less than 1% of all non-Hodgkin lymphomas.3 It presents clinically as multiple subcutaneous masses, nodules, or plaques generally on the trunk or extremities.1,2 The skin surrounding the nodules may be erythematous, and the nodules may become necrotic; however, ulceration typically is not seen. Systemic symptoms such as fever, night sweats, and chills are present in half of cases.1 According to the World Health Organization, cytopenia and elevated liver function tests are common, and a hemophagocytic syndrome may be present in 15% to 20% of cases.3 The presence of a hemophagocytic syndrome yields a poor prognosis.1,3 Current guidelines denote that SPTCL T-cell receptor (TCR) αβ; is a distinct entity from the TCRγδ; phenotype, known as cutaneous γδ-positive T-cell lymphoma.3,4 Cutaneous γδ-positive T-cell lymphoma is associated with rapid decline and a worse prognosis.4
Histology of SPTCL is characteristic for a lobular panniculitislike infiltrate.1 The heavy subcutaneous lymphoid infiltrate is composed of atypical small- to medium-sized lymphocytes with mature chromatin and inconspicuous nucleoli lining adipocytes. The dense inflammatory infiltrate composed predominantly of neoplastic T cells and macrophages may diffusely invade into the subcutaneous tissue.1 Admixed histocytes and karyorrhectic debris as well as rimming of the lymphocytes around the fat cells is typical and was seen in our patient (quiz image). The T cells of SPTCL have the following immunophenotype: TCR-beta F1+, CD3+, CD4-, CD8+, CD56-. They can express numerous cytotoxic proteins, such as T1a-1, granzyme B, and perforin.2,3 Although the CD8+ T cells may be sparse, they generally surround the adipocytes in a rimming manner and may distort the adipocyte membrane.1
Lupus erythematosus profundus (LEP) is a form of chronic cutaneous lupus that affects the deep dermis and fat.5 It also can present clinically as tender plaques or nodules. It most frequently involves the upper arms, shoulders, face, or buttocks--areas that are less commonly involved in other panniculitides.6 Histologically, LEP is similar to chronic discoid lupus with features such as epidermal atrophy, interface changes, and a thickened basement membrane (Figure 1). Lupus erythematosus profundus can present as a lobular panniculitis with mucin as well as a superficial and deep lymphocytic infiltrate that can involve the septa.5 Some cases of LEP have a predominantly lobular lymphocytic panniculitis in the absence of the typical epidermal or dermal changes of lupus erythematosus. Lymphoid follicles with germinal center formation are present in half of cases and reportedly are characteristic of LEP.6,7 The lymphoid follicles often have plasma cells, can extend into the septa as well as in between collagen bundles, and may have nuclear fragmentation.5 Another characteristic feature of LEP is hyaline sclerosis of lobules with focal extension into the interlobular septa. Immunofluorescence studies usually show linear deposition of IgM and C3 at the dermoepidermal junction. Antinuclear antibodies can be present in patients who have LEP but are not entirely specific.6
Lupus erythematosus profundus and SPTCL are part of a spectrum and may have overlapping clinical and histopathologic characteristics; therefore, distinguishing them may be difficult.6-8 It is important to monitor these patients closely, as their disease may progress to lymphoma.6 Patients with SPTCL are more likely to present with advanced symptoms such as fever and hepatosplenomegaly and to succumb to hemophagocytic syndrome than patients with LEP.9
Although SPTCL usually is clonal, several cases of LEP with clonality also have been described. Clonal LEP cases generally are identified in patients who present with fever and cytopenia.8 Lymphoid atypia and morphologic abnormalities may be seen in cases of LEP, further complicating the distinction between LEP and SPTCL. An elevated Ki67 level may be seen in cases of SPTCL with periadipocytic rimming.9 LeBlanc et al10 used Ki67 "hot spots" along with CD8 immunohistochemistry to identify atypical lymphocytes associated with SPTCL. Lymphocyte rimming was defined by the presence of CD8+ lymphocytes with an elevated Ki67 index. Clinical, histopathologic, and molecular findings all should be used when dealing with challenging cases.
Fat necrosis can occur in any part of the body where trauma has occurred and can be associated with many disease processes. Patients typically present with a palpable mass, but a clinical history of trauma is not always present. Histopathologic findings include necrotic fat alongside lipid-laden foamy macrophages and scattered inflammatory cells (Figure 2).11 Fragments of normal as well as degenerating adipose tissue and multinucleated giant cells can be present.
Erythema nodosum (EN) is the most frequently encountered panniculitis and usually is seen in women in early adulthood.12 Patients present with several tender subcutaneous nodules and plaques that most commonly are present on the anterior surface of the legs.12,13 Patients may have a constellation of symptoms including fever and leukocytosis, but the disorder generally is self-limited.12 Erythema nodosum may be associated with a variety of diseases or infections including sarcoidosis, inflammatory bowel disease, and malignancy.14 The etiology of EN is diverse; therefore, a proper clinical workup may be necessary. Histopathology is that of a septal panniculitis with lymphocytes, histiocytes, and occasional eosinophils (Figure 3).13
Lipodermatosclerosis also occurs on the legs, most commonly in patients with venous insufficiency.12,15 Patients present clinically with pain, induration, redness, or swelling of the legs. Histopathology predominantly is characterized by membranous fat necrosis, fibrosis, and fatty microcysts that may be lined by a thickened hyaline membrane (Figure 4). Lipodermatosclerosis lesions generally do not resolve spontaneously and may need to be treated.16
The Diagnosis: Subcutaneous Panniculitislike T-cell Lymphoma
Subcutaneous panniculitislike T-cell lymphoma (SPTCL) is a rare form of cutaneous lymphoma of mature cytotoxic T cells simulating panniculitis and preferentially infiltrating the subcutaneous tissue.1 Subcutaneous panniculitislike T-cell lymphoma can affect all ages but predominantly affects younger individuals, with 20% being younger than 20 years.2 It is a rare lymphoma that accounts for less than 1% of all non-Hodgkin lymphomas.3 It presents clinically as multiple subcutaneous masses, nodules, or plaques generally on the trunk or extremities.1,2 The skin surrounding the nodules may be erythematous, and the nodules may become necrotic; however, ulceration typically is not seen. Systemic symptoms such as fever, night sweats, and chills are present in half of cases.1 According to the World Health Organization, cytopenia and elevated liver function tests are common, and a hemophagocytic syndrome may be present in 15% to 20% of cases.3 The presence of a hemophagocytic syndrome yields a poor prognosis.1,3 Current guidelines denote that SPTCL T-cell receptor (TCR) αβ; is a distinct entity from the TCRγδ; phenotype, known as cutaneous γδ-positive T-cell lymphoma.3,4 Cutaneous γδ-positive T-cell lymphoma is associated with rapid decline and a worse prognosis.4
Histology of SPTCL is characteristic for a lobular panniculitislike infiltrate.1 The heavy subcutaneous lymphoid infiltrate is composed of atypical small- to medium-sized lymphocytes with mature chromatin and inconspicuous nucleoli lining adipocytes. The dense inflammatory infiltrate composed predominantly of neoplastic T cells and macrophages may diffusely invade into the subcutaneous tissue.1 Admixed histocytes and karyorrhectic debris as well as rimming of the lymphocytes around the fat cells is typical and was seen in our patient (quiz image). The T cells of SPTCL have the following immunophenotype: TCR-beta F1+, CD3+, CD4-, CD8+, CD56-. They can express numerous cytotoxic proteins, such as T1a-1, granzyme B, and perforin.2,3 Although the CD8+ T cells may be sparse, they generally surround the adipocytes in a rimming manner and may distort the adipocyte membrane.1
Lupus erythematosus profundus (LEP) is a form of chronic cutaneous lupus that affects the deep dermis and fat.5 It also can present clinically as tender plaques or nodules. It most frequently involves the upper arms, shoulders, face, or buttocks--areas that are less commonly involved in other panniculitides.6 Histologically, LEP is similar to chronic discoid lupus with features such as epidermal atrophy, interface changes, and a thickened basement membrane (Figure 1). Lupus erythematosus profundus can present as a lobular panniculitis with mucin as well as a superficial and deep lymphocytic infiltrate that can involve the septa.5 Some cases of LEP have a predominantly lobular lymphocytic panniculitis in the absence of the typical epidermal or dermal changes of lupus erythematosus. Lymphoid follicles with germinal center formation are present in half of cases and reportedly are characteristic of LEP.6,7 The lymphoid follicles often have plasma cells, can extend into the septa as well as in between collagen bundles, and may have nuclear fragmentation.5 Another characteristic feature of LEP is hyaline sclerosis of lobules with focal extension into the interlobular septa. Immunofluorescence studies usually show linear deposition of IgM and C3 at the dermoepidermal junction. Antinuclear antibodies can be present in patients who have LEP but are not entirely specific.6
Lupus erythematosus profundus and SPTCL are part of a spectrum and may have overlapping clinical and histopathologic characteristics; therefore, distinguishing them may be difficult.6-8 It is important to monitor these patients closely, as their disease may progress to lymphoma.6 Patients with SPTCL are more likely to present with advanced symptoms such as fever and hepatosplenomegaly and to succumb to hemophagocytic syndrome than patients with LEP.9
Although SPTCL usually is clonal, several cases of LEP with clonality also have been described. Clonal LEP cases generally are identified in patients who present with fever and cytopenia.8 Lymphoid atypia and morphologic abnormalities may be seen in cases of LEP, further complicating the distinction between LEP and SPTCL. An elevated Ki67 level may be seen in cases of SPTCL with periadipocytic rimming.9 LeBlanc et al10 used Ki67 "hot spots" along with CD8 immunohistochemistry to identify atypical lymphocytes associated with SPTCL. Lymphocyte rimming was defined by the presence of CD8+ lymphocytes with an elevated Ki67 index. Clinical, histopathologic, and molecular findings all should be used when dealing with challenging cases.
Fat necrosis can occur in any part of the body where trauma has occurred and can be associated with many disease processes. Patients typically present with a palpable mass, but a clinical history of trauma is not always present. Histopathologic findings include necrotic fat alongside lipid-laden foamy macrophages and scattered inflammatory cells (Figure 2).11 Fragments of normal as well as degenerating adipose tissue and multinucleated giant cells can be present.
Erythema nodosum (EN) is the most frequently encountered panniculitis and usually is seen in women in early adulthood.12 Patients present with several tender subcutaneous nodules and plaques that most commonly are present on the anterior surface of the legs.12,13 Patients may have a constellation of symptoms including fever and leukocytosis, but the disorder generally is self-limited.12 Erythema nodosum may be associated with a variety of diseases or infections including sarcoidosis, inflammatory bowel disease, and malignancy.14 The etiology of EN is diverse; therefore, a proper clinical workup may be necessary. Histopathology is that of a septal panniculitis with lymphocytes, histiocytes, and occasional eosinophils (Figure 3).13
Lipodermatosclerosis also occurs on the legs, most commonly in patients with venous insufficiency.12,15 Patients present clinically with pain, induration, redness, or swelling of the legs. Histopathology predominantly is characterized by membranous fat necrosis, fibrosis, and fatty microcysts that may be lined by a thickened hyaline membrane (Figure 4). Lipodermatosclerosis lesions generally do not resolve spontaneously and may need to be treated.16
- Musick SR, Lynch DT. Subcutaneous Panniculitis Like T-cell Lymphoma. StatPearls Publishing; 2020.
- Guenova E, Schanz S, Hoetzenecker W, et al. Systemic corticosteroids for subcutaneous panniculitis-like T-cell lymphoma. Br J Dermatol. 2014;171:891-894.
- Swerdlow SH. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. International Agency for Research on Cancer; 2017.
- Bagheri F, Cervellione KL, Delgado B, et al. An illustrative case of subcutaneous panniculitis-like T-cell lymphoma [published online March 3, 2011]. J Skin Cancer. doi:10.1155/2011/824528
- Kogame T, Yamashita R, Hirata M, et al. Analysis of possible structures of inducible skin‐associated lymphoid tissue in lupus erythematosus profundus. J Dermatol. 2018;45:1117-1121.
- Arps DP, Patel RM. Lupus profundus (panniculitis): a potential mimic of subcutaneous panniculitis-like T-cell lymphoma. Arch Pathol Lab Med. 2013;137:1211-1215.
- Alberti-Violetti S, Berti E. Lymphocytic lobular panniculitis: a diagnostic challenge. Dermatopathology. 2018;5:30-33.
- Magro CM, Crowson AN, Kovatich AJ, et al. Lupus profundus, indeterminate lymphocytic lobular panniculitis and subcutaneous T-cell lymphoma: a spectrum of subcuticular T-cell lymphoid dyscrasia. J Cutan Pathol. 2001;28:235-247.
- Sitthinamsuwan P, Pattanaprichakul P, Treetipsatit J, et al. Subcutaneous panniculitis-like T-cell lymphoma versus lupus erythematosus panniculitis: distinction by means of the periadipocytic cell proliferation index. Am J Dermatopathol. 2018;40:567-574.
- LeBlanc RE, Tavallaee M, Kim YH, et al. Useful parameters for distinguishing subcutaneous panniculitis-like T-cell lymphoma from lupus erythematosus panniculitis. Am J Surg Pathol. 2016;40:745-754.
- Burkholz KJ, Roberts CC, Lidner TK. Posttraumatic pseudolipoma (fat necrosis) mimicking atypical lipoma or liposarcoma on MRI. Radiol Case Rep. 2015;2:56-60.
- Wick MR. Panniculitis: a summary. Semin Diagn Pathol. 2017;34:261-272.
- Thurber S, Kohler S. Histopathologic spectrum of erythema nodosum. J Cutan Pathol. 2006;33:18-26.
- Requena L, Requena C. Erythema nodosum. Dermatol Online J. 2002;8:4.
- Choonhakarn C, Chaowattanapanit S, Julanon N. Lipodermatosclerosis: a clinicopathologic correlation. Int J Dermatol. 2016;55:303-308.
- Huang TM, Lee JY. Lipodermatosclerosis: a clinicopathologic study of 17 cases and differential diagnosis from erythema nodosum. J Cutan Pathol. 2009;36:453-460.
- Musick SR, Lynch DT. Subcutaneous Panniculitis Like T-cell Lymphoma. StatPearls Publishing; 2020.
- Guenova E, Schanz S, Hoetzenecker W, et al. Systemic corticosteroids for subcutaneous panniculitis-like T-cell lymphoma. Br J Dermatol. 2014;171:891-894.
- Swerdlow SH. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. International Agency for Research on Cancer; 2017.
- Bagheri F, Cervellione KL, Delgado B, et al. An illustrative case of subcutaneous panniculitis-like T-cell lymphoma [published online March 3, 2011]. J Skin Cancer. doi:10.1155/2011/824528
- Kogame T, Yamashita R, Hirata M, et al. Analysis of possible structures of inducible skin‐associated lymphoid tissue in lupus erythematosus profundus. J Dermatol. 2018;45:1117-1121.
- Arps DP, Patel RM. Lupus profundus (panniculitis): a potential mimic of subcutaneous panniculitis-like T-cell lymphoma. Arch Pathol Lab Med. 2013;137:1211-1215.
- Alberti-Violetti S, Berti E. Lymphocytic lobular panniculitis: a diagnostic challenge. Dermatopathology. 2018;5:30-33.
- Magro CM, Crowson AN, Kovatich AJ, et al. Lupus profundus, indeterminate lymphocytic lobular panniculitis and subcutaneous T-cell lymphoma: a spectrum of subcuticular T-cell lymphoid dyscrasia. J Cutan Pathol. 2001;28:235-247.
- Sitthinamsuwan P, Pattanaprichakul P, Treetipsatit J, et al. Subcutaneous panniculitis-like T-cell lymphoma versus lupus erythematosus panniculitis: distinction by means of the periadipocytic cell proliferation index. Am J Dermatopathol. 2018;40:567-574.
- LeBlanc RE, Tavallaee M, Kim YH, et al. Useful parameters for distinguishing subcutaneous panniculitis-like T-cell lymphoma from lupus erythematosus panniculitis. Am J Surg Pathol. 2016;40:745-754.
- Burkholz KJ, Roberts CC, Lidner TK. Posttraumatic pseudolipoma (fat necrosis) mimicking atypical lipoma or liposarcoma on MRI. Radiol Case Rep. 2015;2:56-60.
- Wick MR. Panniculitis: a summary. Semin Diagn Pathol. 2017;34:261-272.
- Thurber S, Kohler S. Histopathologic spectrum of erythema nodosum. J Cutan Pathol. 2006;33:18-26.
- Requena L, Requena C. Erythema nodosum. Dermatol Online J. 2002;8:4.
- Choonhakarn C, Chaowattanapanit S, Julanon N. Lipodermatosclerosis: a clinicopathologic correlation. Int J Dermatol. 2016;55:303-308.
- Huang TM, Lee JY. Lipodermatosclerosis: a clinicopathologic study of 17 cases and differential diagnosis from erythema nodosum. J Cutan Pathol. 2009;36:453-460.
A 47-year-old man presented with a tender soft tissue mass on the upper back with increasing discomfort over the last 4 weeks. He noted that he felt feverish a few times. Physical examination revealed a 3×4-cm area of induration involving the upper mid back with faint erythema of the overlying skin; no drainage was noted. A prominent left posterior cervical lymph node also was appreciated, and a punch biopsy of the mass was performed.