User login
Rendered speechless
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 63-year-old man at an inpatient rehabilitation center was transferred to an academic tertiary care center for evaluation of slurred speech and episodic confusion. He was accompanied by his wife, who provided the history. Three weeks earlier, the patient had fallen, sustaining a right femur fracture. He underwent surgery and was discharged to rehabilitation on postoperative day 3. During the second week of rehabilitation, he developed a cough and low-grade fevers, which prompted treatment with cefpodoxime for 5 days for presumed pneumonia. The day after completing antimicrobial therapy, he became confused and began to slur his words.
Confusion is a nonspecific symptom that typically has a diffuse or multifocal localization within the cerebral hemispheres and is unlikely to be caused by a single lesion. Slurred speech may accompany global metabolic dysfunction. However, slurred speech typically localizes to the brainstem, the cerebellum in the posterior fossa, the nuclei, or the course of cranial nerves VII, X, or XII, including where these nerves pass through the subarachnoid space.
It seems this patient’s new neurologic symptoms have some relationship to his fall. Long-bone fractures and altered mental status (AMS) lead to consideration of fat emboli, but this syndrome typically presents in the acute period after the fracture. The patient is at risk for a number of complications, related to recent surgery and hospitalization, that could affect the central nervous system (CNS), including systemic infection (possibly with associated meningeal involvement) and venous thromboembolism with concomitant stroke by paradoxical emboli. The episodic nature of the confusion leads to consideration of seizures from structural lesions in the brain. Finally, the circumstances of the fall itself should be explored to determine whether an underlying neurologic dysfunction led to imbalance and gait difficulty.
Over the next 3 days at the inpatient rehabilitation center, the patient’s slurred speech became unintelligible, and he experienced intermittent disorientation to person, place, and time. There was no concomitant fever, dizziness, headache, neck pain, weakness, dyspnea, diarrhea, dysuria, or change in hearing or vision.
Progressive dysarthria argues for an expanding lesion in the posterior fossa, worsening metabolic disturbance, or a problem affecting the cranial nerves (eg, Guillain-Barré syndrome) or neuromuscular junctions (eg, myasthenia gravis). Lack of headache makes a CNS localization less likely, though disorientation must localize to the brain itself. The transient nature of the AMS could signal an ictal phenomenon or a fluctuating toxic or metabolic condition, such as hyperammonemia, drug reaction, or healthcare–acquired delirium.
His past medical history included end-stage liver disease secondary to nonalcoholic steatohepatitis status post transjugular intrahepatic portosystemic shunt (TIPS) procedure three years prior, hepatic encephalopathy, diabetes mellitus type 2, hypertension, previous melanoma excision on his back, and recurrent Clostridium difficile colitis. Two years prior to admission he had been started on an indefinite course of metronidazole 500 mg twice daily without any recurrence. The patient’s other medications were aspirin, furosemide, insulin, lactulose, mirtazapine, pantoprazole, propranolol, spironolactone, and zinc. At the rehabilitation center, he was prescribed oral oxycodone 5 mg as needed every 4 hours for pain. He denied use of tobacco, alcohol, and recreational drugs. He previously worked as a funeral home director and embalmer.
Hyperammonemia and hepatic encephalopathy can present with a fluctuating mental state that often correlates to dietary protein intake or the frequency of bowel movements; the previous TIPS history places the patient at further risk. Use of oxycodone or another narcotic commonly leads to confusion, , especially in patients who are older, have preexisting cognitive decline, or have concomitant medical comorbidities. Mirtazapine and propranolol have been associated more rarely with encephalopathy, and therefore a careful history of adherence, drug interactions, and appropriate dosing should be obtained. Metronidazole is most often associated neurologically with a peripheral neuropathy; however, it is increasingly recognized that some patients can develop a CNS syndrome that features an AMS, which can be severe and accompanied by ataxia, dysarthria, and characteristic brain magnetic resonance imaging (MRI) findings, including hyperintensity surrounding the fourth ventricle on T2-weighted images.
Embalming fluid has a high concentration of formaldehyde, and a recent epidemiologic study suggested a link between formaldehyde exposure and increased risk for amyotrophic lateral sclerosis (ALS). ALS uncommonly presents with isolated dysarthria, but its bulbar form can, usually over a much longer course than is demonstrated here. Finally, the patient’s history of melanoma places him at risk for stroke from hypercoagulability as well as potential brain metastases or carcinomatous meningitis.
Evaluation was initiated at the rehabilitation facility at the onset of the patient’s slurred speech and confusion. Physical examination were negative for focal neurologic deficits, asterixis, and jaundice. Ammonia level was 41 µmol/L (reference range, 11-35 µmol/L). Noncontrast computed tomography (CT) of the head showed no signs of acute infarct or hemorrhage. Symptoms were attributed to hepatic encephalopathy; lactulose was up-titrated to ensure 2 or 3 bowel movements per day, and rifaximin was started.
Hyperammonemia is a cause of non-inflammatory relapsing encephalopathy, but an elevated level is neither a sensitive nor specific indicator of hepatic encephalopathy. Levels of ammonia can fluctuate widely during the day based on the frequency of bowel movements as well as dietary protein intake. In addition, proper handling of samples with prompt delivery to the laboratory is essential to minimize errors.
The ammonia level of 41 µmol/L discovered here is only modestly elevated, but given the patient’s history of TIPS as well as the clinical picture, it is reasonable to aggressively treat hepatic encephalopathy with lactulose to reduce ammonia levels. If he does not improve, an MRI of the brain to exclude a structural lesion and spinal fluid examination looking for inflammatory or infectious conditions would be important next steps. Although CT excludes a large hemorrhage or mass, this screening examination does not visualize many of the findings of the metabolic etiology and the other etiologies under consideration here.
Despite 3 days of therapy for presumed hepatic encephalopathy, the patient’s slurred speech worsened, and he was transferred to an academic tertiary care center for further evaluation. On admission, his temperature was 36.9°C, heart rate was 80 beats per minute, blood pressure was 139/67 mm Hg, respiratory rate was 10 breaths per minute, and oxygen saturation was 99% on room air. He was alert, awake, and oriented to person, place, and time. He was not jaundiced. He exhibited a moderate dysarthria characterized by monotone speech, decreased volume, decreased breath support, and a hoarse vocal quality with intact language function. Motor control of the lips, tongue, and mandible were normal. Motor strength was 5/5 bilaterally in the upper and lower extremities with the exception of right hip flexion, which was 4/5. The patient exhibited mild bilateral dysmetria on finger-to-nose examination, consistent with appendicular ataxia of the upper extremities. Reflexes were depressed throughout, and there was no asterixis. He had 2+ pulses in all extremities and 1+ pitting edema of the right lower extremity to the mid leg. Pulmonary examination revealed inspiratory crackles at the left base. The rest of the examination findings were normal.
The patient’s altered mental state appears to have resolved, and the neurological examination is now mainly characterized by signs that point to the cerebellum. The description of monotone speech typically refers to loss of prosody, the variable stress or intonation of speech, which is characteristic of a cerebellar speech pattern. The hoarseness should be explored to determine if it is a feature of the patient’s speech or is a separate process. Hoarseness may involve the vocal cord and therefore, potentially, cranial nerve X or its nuclei in the brainstem. The appendicular ataxia of the limbs points definitively to the cerebellar hemispheres or their pathways through the brainstem.
Unilateral lower extremity edema, especially in the context of a recent fracture, raises the possibility of deep vein thrombosis. If this patient has a right-to-left intracardiac or intrapulmonary shunt, embolization could lead to an ischemic stroke of the brainstem or cerebellum, potentially causing dysarthria.
Laboratory evaluation revealed hemoglobin level of 10.9 g/dL, white blood cell count of 5.3 × 10 9 /L, platelet count of 169 × 10 9 /L, glucose level of 177 mg/dL, corrected calcium level of 9.0 mg/dL, sodium level of 135 mmol/L, bicarbonate level of 30 mmol/L, creatinine level of 0.9 mg/dL, total bilirubin level of 1.3 mg/dL, direct bilirubin level of 0.4 mg/dL, alkaline phosphatase level of 503 U/L, alanine aminotransferase level of 12 U/L, aspartate aminotransferase level of 33 U/L, ammonia level of 49 µmol/L (range, 0-30 µ mol/L), international normalized ratio of 1.2, and troponin level of <0.01 ng/mL. Electrocardiogram showed normal sinus rhythm.
Some patients with bacterial meningitis do not have a leukocytosis, but patients with meningitis caused by seeding from a systemic infection nearly always do. In this patient’s case, lack of a leukocytosis makes bacterial meningitis very unlikely. The elevated alkaline phosphatase level is expected, as this level peaks about 3 weeks after a long-bone fracture and returns to normal over a few months.
Non-contrast CT scan of the head performed on admission demonstrated no large vessel cortical-based infarct, intracranial hemorrhage, hydrocephalus, mass effect, midline shift, or extra-axial fluid. There was mild cortical atrophy as well as very mild periventricular white matter hypodensity.
The atrophy and mild white-matter hypodensities seen on repeat noncontrast CT are nonspecific for any particular entity in this patient’s age group. MRI is more effective in evaluating toxic encephalopathies, including metronidazole toxicity or Wernicke encephalopathy, and in characterizing small infarcts or inflammatory conditions of the brainstem and cerebellum, which are poorly evaluated by CT due to the bone surrounded space of the posterior fossa. An urgent lumbar puncture is not necessary due to the slow pace of illness, lack of fever, nuchal rigidity, or serum elevated white blood cell count. Rather, performing MRI should be prioritized. If MRI is nondiagnostic, then spinal fluid should be evaluated for evidence of an infectious, autoimmune, paraneoplastic, or neoplastic process.
MRI was subsequently performed. It showed symmetric abnormal T2 hyperintensities involving dentate nuclei (Figure 1), left inferior olivary nuclei (Figure 2), restiform bodies, pontine tegmentum, superior cerebellar peduncles, oculomotor nuclei, and subthalamic nuclei. The most prominent hyperintensity was in the dentate nuclei.

The clinical and radiographic features confirm a diagnosis of metronidazole-associated CNS neurotoxicity. The reason for the predilection for edema in these specific areas of the brainstem and midline cerebellum is unclear but likely is related to selective neuronal vulnerability in these structures. The treatment is to stop metronidazole. In addition, the fluctuating mental status should be evaluated with electroencephalogram to ensure concomitant seizures are not occurring.
These MRI findings were consistent with metronidazole toxicity. Metronidazole was discontinued, and 2 days later the patient’s speech improved. Two weeks after medication discontinuation, his speech was normal. There were no more episodes of confusion.
DISCUSSION
Metronidazole was originally developed in France during the 1950s as an anti-parasitic medication to treat trichomonas infections. In 1962, its antibacterial properties were discovered after a patient with bacterial gingivitis improved while taking metronidazole for treatment of Trichomonas vaginalis.1 Since that time metronidazole has become a first-line treatment for anaerobic bacteria and is now recommended by the Infectious Diseases Society of America2 and the American College of Gastroenterology3 as a first-line therapy for mild and moderate C difficile infections.
Common side effects of metronidazole are nausea, vomiting, decreased appetite, diarrhea, headaches, peripheral neuropathy, and metallic taste; less common is CNS toxicity. Although the incidence of CNS toxicity is unknown, a systematic review of the literature found 64 cases reported between 1965 and 2011.4 CNS toxicity most often occurs between the fifth and sixth decades of life, and about two thirds of the people affected are men.4 CNS adverse effects characteristically fall into 4 categories: cerebellar dysfunction (eg, ataxia, dysarthria, dysmetria, nystagmus; 75%), AMS (33%), seizures (13%), and a combination of the first 3 categories.4
The exact mechanism of metronidazole CNS toxicity is unknown, but vasogenic or cytotoxic edema may be involved.5,6 Other potential etiologies are neural protein inhibition, reversible mitochondrial dysfunction, and modifications of the inhibitory neurotransmitter gamma-aminobutyric acid receptor in the cerebellum.7,8 There is no known genetic predisposition. Although the risk for CNS toxicity traditionally is thought to correlate with therapy duration and cumulative dose,7,9 in 2011 a systemic review found no significant correlation.4 In fact, 26% of patients with CNS toxicity were treated with metronidazole for less than 1 week at time of diagnosis.4
Brain CT is typically normal. On brain MRI, lesions most commonly appear as bilateral symmetric T2 hyperintensities, most often in the cerebellar dentate nuclei (85%) and less often in the midbrain (55%), the splenium of the corpus callosum (50%), the pons (35%), and the medulla (30%).4,10 Radiographic changes have been noted as early as 3 days after symptom onset. Based on damage severity and area affected (white or gray matter), vasogenic edema and cytotoxic edema may in combination be contributing to MRI abnormalities.6,10 Hyperintensities of the bilateral dentate nuclei can help in distinguishing metronidazole-induced encephalopathy from other potential disease processes, such as Wernicke encephalopathy.10
The prognosis for patients with metronidazole-induced neurotoxicity is favorable if metronidazole is discontinued. Approximately two-thirds of patients will have complete resolution of symptoms, which is more commonly observed when patients present with seizures or altered mental status. Approximately one-third will show partial improvement, particularly if the symptoms are due to cerebellar dysfunction. It is rare to experience permanent damage or death.4 Neurologic recovery usually begins within a week after medication discontinuation but may take months for complete recovery to occur.6,8,9,11 Follow-up imaging typically shows reversal of the original lesions, but this does not always correlate with symptom improvement.4,10
Despite its frequent use and long history, metronidazole can have potentially severe toxicity. When patients who are taking this medication present with new signs and symptoms of CNS dysfunction, hospitalists should include metronidazole CNS toxicity in the differential diagnosis and, if they suspect toxicity, have a brain MRI performed. Hospitalists often prescribe metronidazole because of the increasing number of patients being discharged from acute-care hospitals with a diagnosis of C difficile colitis.12 Brain MRI remains the imaging modality of choice for diagnosis. Discontinuation of metronidazole is usually salutary in reversing symptoms. Being keenly aware of this toxicity will help clinicians avoid being rendered speechless by a patient rendered speechless.
TEACHING POINTS
CNS toxicity is a rare but potentially devastating side effect of metronidazole exposure.
Metronidazole CNS adverse effects characteristically fall under 4 categories:
○ Cerebellar dysfunction, such as ataxia, dysarthria, dysmetria, or nystagmus (75%).
○ AMS (33%).
○ Seizures (13%).
○ A combination of the first 3 categories.
- Typically lesions indicating metronidazole toxicity on brain MRI are bilateral symmetric hyperintensities on T2-weighted imaging in the cerebellar dentate nuclei, corpus callosum, midbrain, pons, or medulla.
- Treatment of CNS toxicity is metronidazole discontinuation, which results in a high rate of symptom resolution.
Disclosure
Nothing to report.
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541. PubMed
2. Cohen SH, Gerding DN, Johnson S, et al; Society for Healthcare Epidemiology of America; Infectious Diseases Society of America. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol. 2010;31(5):431-455. PubMed
3. Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am J Gastroenterol. 2013;108(4):478-498. PubMed
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systemic review. Clin Neuropharmacol. 2011;34(6):241-247. PubMed
5. Graves TD, Condon M, Loucaidou M, Perry RJ. Reversible metronidazole-induced cerebellar toxicity in a multiple transplant recipient. J Neurol Sci. 2009;285(1-2):238-240. PubMed
6. Kim DW, Park JM, Yoon BW, Baek MJ, Kim JE, Kim S. Metronidazole-induced encephalopathy. J Neurol Sci. 2004;224(1-2):107-111. PubMed
7. Park KI, Chung JM, Kim JY. Metronidazole neurotoxicity: sequential neuroaxis involvement. Neurol India. 2011;59(1):104-107. PubMed
8. Patel K, Green-Hopkins I, Lu S, Tunkel AR. Cerebellar ataxia following prolonged use of metronidazole: case report and literature review. Int J Infect Dis. 2008;12(6):e111-e114. PubMed
9. Chandak S, Agarwal A, Shukla A, Joon P. A case report of metronidazole induced neurotoxicity in liver abscess patient and the usefulness of MRI for its diagnosis. J Clin Diagn Res. 2016;10(1):TD06-TD07. PubMed
10. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658. PubMed
11. Chacko J, Pramod K, Sinha S, et al. Clinical, neuroimaging and pathological features of 5-nitroimidazole-induced encephalo-neuropathy in two patients: insights into possible pathogenesis. Neurol India. 2011;59(5):743-747. PubMed
12. Peery AF, Dellon ES, Lund J, et al. Burden of gastrointestinal disease in the United States: 2012 update. Gastroenterology. 2012;143(5):1179-1187.e1-e3. PubMed
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 63-year-old man at an inpatient rehabilitation center was transferred to an academic tertiary care center for evaluation of slurred speech and episodic confusion. He was accompanied by his wife, who provided the history. Three weeks earlier, the patient had fallen, sustaining a right femur fracture. He underwent surgery and was discharged to rehabilitation on postoperative day 3. During the second week of rehabilitation, he developed a cough and low-grade fevers, which prompted treatment with cefpodoxime for 5 days for presumed pneumonia. The day after completing antimicrobial therapy, he became confused and began to slur his words.
Confusion is a nonspecific symptom that typically has a diffuse or multifocal localization within the cerebral hemispheres and is unlikely to be caused by a single lesion. Slurred speech may accompany global metabolic dysfunction. However, slurred speech typically localizes to the brainstem, the cerebellum in the posterior fossa, the nuclei, or the course of cranial nerves VII, X, or XII, including where these nerves pass through the subarachnoid space.
It seems this patient’s new neurologic symptoms have some relationship to his fall. Long-bone fractures and altered mental status (AMS) lead to consideration of fat emboli, but this syndrome typically presents in the acute period after the fracture. The patient is at risk for a number of complications, related to recent surgery and hospitalization, that could affect the central nervous system (CNS), including systemic infection (possibly with associated meningeal involvement) and venous thromboembolism with concomitant stroke by paradoxical emboli. The episodic nature of the confusion leads to consideration of seizures from structural lesions in the brain. Finally, the circumstances of the fall itself should be explored to determine whether an underlying neurologic dysfunction led to imbalance and gait difficulty.
Over the next 3 days at the inpatient rehabilitation center, the patient’s slurred speech became unintelligible, and he experienced intermittent disorientation to person, place, and time. There was no concomitant fever, dizziness, headache, neck pain, weakness, dyspnea, diarrhea, dysuria, or change in hearing or vision.
Progressive dysarthria argues for an expanding lesion in the posterior fossa, worsening metabolic disturbance, or a problem affecting the cranial nerves (eg, Guillain-Barré syndrome) or neuromuscular junctions (eg, myasthenia gravis). Lack of headache makes a CNS localization less likely, though disorientation must localize to the brain itself. The transient nature of the AMS could signal an ictal phenomenon or a fluctuating toxic or metabolic condition, such as hyperammonemia, drug reaction, or healthcare–acquired delirium.
His past medical history included end-stage liver disease secondary to nonalcoholic steatohepatitis status post transjugular intrahepatic portosystemic shunt (TIPS) procedure three years prior, hepatic encephalopathy, diabetes mellitus type 2, hypertension, previous melanoma excision on his back, and recurrent Clostridium difficile colitis. Two years prior to admission he had been started on an indefinite course of metronidazole 500 mg twice daily without any recurrence. The patient’s other medications were aspirin, furosemide, insulin, lactulose, mirtazapine, pantoprazole, propranolol, spironolactone, and zinc. At the rehabilitation center, he was prescribed oral oxycodone 5 mg as needed every 4 hours for pain. He denied use of tobacco, alcohol, and recreational drugs. He previously worked as a funeral home director and embalmer.
Hyperammonemia and hepatic encephalopathy can present with a fluctuating mental state that often correlates to dietary protein intake or the frequency of bowel movements; the previous TIPS history places the patient at further risk. Use of oxycodone or another narcotic commonly leads to confusion, , especially in patients who are older, have preexisting cognitive decline, or have concomitant medical comorbidities. Mirtazapine and propranolol have been associated more rarely with encephalopathy, and therefore a careful history of adherence, drug interactions, and appropriate dosing should be obtained. Metronidazole is most often associated neurologically with a peripheral neuropathy; however, it is increasingly recognized that some patients can develop a CNS syndrome that features an AMS, which can be severe and accompanied by ataxia, dysarthria, and characteristic brain magnetic resonance imaging (MRI) findings, including hyperintensity surrounding the fourth ventricle on T2-weighted images.
Embalming fluid has a high concentration of formaldehyde, and a recent epidemiologic study suggested a link between formaldehyde exposure and increased risk for amyotrophic lateral sclerosis (ALS). ALS uncommonly presents with isolated dysarthria, but its bulbar form can, usually over a much longer course than is demonstrated here. Finally, the patient’s history of melanoma places him at risk for stroke from hypercoagulability as well as potential brain metastases or carcinomatous meningitis.
Evaluation was initiated at the rehabilitation facility at the onset of the patient’s slurred speech and confusion. Physical examination were negative for focal neurologic deficits, asterixis, and jaundice. Ammonia level was 41 µmol/L (reference range, 11-35 µmol/L). Noncontrast computed tomography (CT) of the head showed no signs of acute infarct or hemorrhage. Symptoms were attributed to hepatic encephalopathy; lactulose was up-titrated to ensure 2 or 3 bowel movements per day, and rifaximin was started.
Hyperammonemia is a cause of non-inflammatory relapsing encephalopathy, but an elevated level is neither a sensitive nor specific indicator of hepatic encephalopathy. Levels of ammonia can fluctuate widely during the day based on the frequency of bowel movements as well as dietary protein intake. In addition, proper handling of samples with prompt delivery to the laboratory is essential to minimize errors.
The ammonia level of 41 µmol/L discovered here is only modestly elevated, but given the patient’s history of TIPS as well as the clinical picture, it is reasonable to aggressively treat hepatic encephalopathy with lactulose to reduce ammonia levels. If he does not improve, an MRI of the brain to exclude a structural lesion and spinal fluid examination looking for inflammatory or infectious conditions would be important next steps. Although CT excludes a large hemorrhage or mass, this screening examination does not visualize many of the findings of the metabolic etiology and the other etiologies under consideration here.
Despite 3 days of therapy for presumed hepatic encephalopathy, the patient’s slurred speech worsened, and he was transferred to an academic tertiary care center for further evaluation. On admission, his temperature was 36.9°C, heart rate was 80 beats per minute, blood pressure was 139/67 mm Hg, respiratory rate was 10 breaths per minute, and oxygen saturation was 99% on room air. He was alert, awake, and oriented to person, place, and time. He was not jaundiced. He exhibited a moderate dysarthria characterized by monotone speech, decreased volume, decreased breath support, and a hoarse vocal quality with intact language function. Motor control of the lips, tongue, and mandible were normal. Motor strength was 5/5 bilaterally in the upper and lower extremities with the exception of right hip flexion, which was 4/5. The patient exhibited mild bilateral dysmetria on finger-to-nose examination, consistent with appendicular ataxia of the upper extremities. Reflexes were depressed throughout, and there was no asterixis. He had 2+ pulses in all extremities and 1+ pitting edema of the right lower extremity to the mid leg. Pulmonary examination revealed inspiratory crackles at the left base. The rest of the examination findings were normal.
The patient’s altered mental state appears to have resolved, and the neurological examination is now mainly characterized by signs that point to the cerebellum. The description of monotone speech typically refers to loss of prosody, the variable stress or intonation of speech, which is characteristic of a cerebellar speech pattern. The hoarseness should be explored to determine if it is a feature of the patient’s speech or is a separate process. Hoarseness may involve the vocal cord and therefore, potentially, cranial nerve X or its nuclei in the brainstem. The appendicular ataxia of the limbs points definitively to the cerebellar hemispheres or their pathways through the brainstem.
Unilateral lower extremity edema, especially in the context of a recent fracture, raises the possibility of deep vein thrombosis. If this patient has a right-to-left intracardiac or intrapulmonary shunt, embolization could lead to an ischemic stroke of the brainstem or cerebellum, potentially causing dysarthria.
Laboratory evaluation revealed hemoglobin level of 10.9 g/dL, white blood cell count of 5.3 × 10 9 /L, platelet count of 169 × 10 9 /L, glucose level of 177 mg/dL, corrected calcium level of 9.0 mg/dL, sodium level of 135 mmol/L, bicarbonate level of 30 mmol/L, creatinine level of 0.9 mg/dL, total bilirubin level of 1.3 mg/dL, direct bilirubin level of 0.4 mg/dL, alkaline phosphatase level of 503 U/L, alanine aminotransferase level of 12 U/L, aspartate aminotransferase level of 33 U/L, ammonia level of 49 µmol/L (range, 0-30 µ mol/L), international normalized ratio of 1.2, and troponin level of <0.01 ng/mL. Electrocardiogram showed normal sinus rhythm.
Some patients with bacterial meningitis do not have a leukocytosis, but patients with meningitis caused by seeding from a systemic infection nearly always do. In this patient’s case, lack of a leukocytosis makes bacterial meningitis very unlikely. The elevated alkaline phosphatase level is expected, as this level peaks about 3 weeks after a long-bone fracture and returns to normal over a few months.
Non-contrast CT scan of the head performed on admission demonstrated no large vessel cortical-based infarct, intracranial hemorrhage, hydrocephalus, mass effect, midline shift, or extra-axial fluid. There was mild cortical atrophy as well as very mild periventricular white matter hypodensity.
The atrophy and mild white-matter hypodensities seen on repeat noncontrast CT are nonspecific for any particular entity in this patient’s age group. MRI is more effective in evaluating toxic encephalopathies, including metronidazole toxicity or Wernicke encephalopathy, and in characterizing small infarcts or inflammatory conditions of the brainstem and cerebellum, which are poorly evaluated by CT due to the bone surrounded space of the posterior fossa. An urgent lumbar puncture is not necessary due to the slow pace of illness, lack of fever, nuchal rigidity, or serum elevated white blood cell count. Rather, performing MRI should be prioritized. If MRI is nondiagnostic, then spinal fluid should be evaluated for evidence of an infectious, autoimmune, paraneoplastic, or neoplastic process.
MRI was subsequently performed. It showed symmetric abnormal T2 hyperintensities involving dentate nuclei (Figure 1), left inferior olivary nuclei (Figure 2), restiform bodies, pontine tegmentum, superior cerebellar peduncles, oculomotor nuclei, and subthalamic nuclei. The most prominent hyperintensity was in the dentate nuclei.
The clinical and radiographic features confirm a diagnosis of metronidazole-associated CNS neurotoxicity. The reason for the predilection for edema in these specific areas of the brainstem and midline cerebellum is unclear but likely is related to selective neuronal vulnerability in these structures. The treatment is to stop metronidazole. In addition, the fluctuating mental status should be evaluated with electroencephalogram to ensure concomitant seizures are not occurring.
These MRI findings were consistent with metronidazole toxicity. Metronidazole was discontinued, and 2 days later the patient’s speech improved. Two weeks after medication discontinuation, his speech was normal. There were no more episodes of confusion.
DISCUSSION
Metronidazole was originally developed in France during the 1950s as an anti-parasitic medication to treat trichomonas infections. In 1962, its antibacterial properties were discovered after a patient with bacterial gingivitis improved while taking metronidazole for treatment of Trichomonas vaginalis.1 Since that time metronidazole has become a first-line treatment for anaerobic bacteria and is now recommended by the Infectious Diseases Society of America2 and the American College of Gastroenterology3 as a first-line therapy for mild and moderate C difficile infections.
Common side effects of metronidazole are nausea, vomiting, decreased appetite, diarrhea, headaches, peripheral neuropathy, and metallic taste; less common is CNS toxicity. Although the incidence of CNS toxicity is unknown, a systematic review of the literature found 64 cases reported between 1965 and 2011.4 CNS toxicity most often occurs between the fifth and sixth decades of life, and about two thirds of the people affected are men.4 CNS adverse effects characteristically fall into 4 categories: cerebellar dysfunction (eg, ataxia, dysarthria, dysmetria, nystagmus; 75%), AMS (33%), seizures (13%), and a combination of the first 3 categories.4
The exact mechanism of metronidazole CNS toxicity is unknown, but vasogenic or cytotoxic edema may be involved.5,6 Other potential etiologies are neural protein inhibition, reversible mitochondrial dysfunction, and modifications of the inhibitory neurotransmitter gamma-aminobutyric acid receptor in the cerebellum.7,8 There is no known genetic predisposition. Although the risk for CNS toxicity traditionally is thought to correlate with therapy duration and cumulative dose,7,9 in 2011 a systemic review found no significant correlation.4 In fact, 26% of patients with CNS toxicity were treated with metronidazole for less than 1 week at time of diagnosis.4
Brain CT is typically normal. On brain MRI, lesions most commonly appear as bilateral symmetric T2 hyperintensities, most often in the cerebellar dentate nuclei (85%) and less often in the midbrain (55%), the splenium of the corpus callosum (50%), the pons (35%), and the medulla (30%).4,10 Radiographic changes have been noted as early as 3 days after symptom onset. Based on damage severity and area affected (white or gray matter), vasogenic edema and cytotoxic edema may in combination be contributing to MRI abnormalities.6,10 Hyperintensities of the bilateral dentate nuclei can help in distinguishing metronidazole-induced encephalopathy from other potential disease processes, such as Wernicke encephalopathy.10
The prognosis for patients with metronidazole-induced neurotoxicity is favorable if metronidazole is discontinued. Approximately two-thirds of patients will have complete resolution of symptoms, which is more commonly observed when patients present with seizures or altered mental status. Approximately one-third will show partial improvement, particularly if the symptoms are due to cerebellar dysfunction. It is rare to experience permanent damage or death.4 Neurologic recovery usually begins within a week after medication discontinuation but may take months for complete recovery to occur.6,8,9,11 Follow-up imaging typically shows reversal of the original lesions, but this does not always correlate with symptom improvement.4,10
Despite its frequent use and long history, metronidazole can have potentially severe toxicity. When patients who are taking this medication present with new signs and symptoms of CNS dysfunction, hospitalists should include metronidazole CNS toxicity in the differential diagnosis and, if they suspect toxicity, have a brain MRI performed. Hospitalists often prescribe metronidazole because of the increasing number of patients being discharged from acute-care hospitals with a diagnosis of C difficile colitis.12 Brain MRI remains the imaging modality of choice for diagnosis. Discontinuation of metronidazole is usually salutary in reversing symptoms. Being keenly aware of this toxicity will help clinicians avoid being rendered speechless by a patient rendered speechless.
TEACHING POINTS
CNS toxicity is a rare but potentially devastating side effect of metronidazole exposure.
Metronidazole CNS adverse effects characteristically fall under 4 categories:
○ Cerebellar dysfunction, such as ataxia, dysarthria, dysmetria, or nystagmus (75%).
○ AMS (33%).
○ Seizures (13%).
○ A combination of the first 3 categories.
- Typically lesions indicating metronidazole toxicity on brain MRI are bilateral symmetric hyperintensities on T2-weighted imaging in the cerebellar dentate nuclei, corpus callosum, midbrain, pons, or medulla.
- Treatment of CNS toxicity is metronidazole discontinuation, which results in a high rate of symptom resolution.
Disclosure
Nothing to report.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant. The bolded text represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 63-year-old man at an inpatient rehabilitation center was transferred to an academic tertiary care center for evaluation of slurred speech and episodic confusion. He was accompanied by his wife, who provided the history. Three weeks earlier, the patient had fallen, sustaining a right femur fracture. He underwent surgery and was discharged to rehabilitation on postoperative day 3. During the second week of rehabilitation, he developed a cough and low-grade fevers, which prompted treatment with cefpodoxime for 5 days for presumed pneumonia. The day after completing antimicrobial therapy, he became confused and began to slur his words.
Confusion is a nonspecific symptom that typically has a diffuse or multifocal localization within the cerebral hemispheres and is unlikely to be caused by a single lesion. Slurred speech may accompany global metabolic dysfunction. However, slurred speech typically localizes to the brainstem, the cerebellum in the posterior fossa, the nuclei, or the course of cranial nerves VII, X, or XII, including where these nerves pass through the subarachnoid space.
It seems this patient’s new neurologic symptoms have some relationship to his fall. Long-bone fractures and altered mental status (AMS) lead to consideration of fat emboli, but this syndrome typically presents in the acute period after the fracture. The patient is at risk for a number of complications, related to recent surgery and hospitalization, that could affect the central nervous system (CNS), including systemic infection (possibly with associated meningeal involvement) and venous thromboembolism with concomitant stroke by paradoxical emboli. The episodic nature of the confusion leads to consideration of seizures from structural lesions in the brain. Finally, the circumstances of the fall itself should be explored to determine whether an underlying neurologic dysfunction led to imbalance and gait difficulty.
Over the next 3 days at the inpatient rehabilitation center, the patient’s slurred speech became unintelligible, and he experienced intermittent disorientation to person, place, and time. There was no concomitant fever, dizziness, headache, neck pain, weakness, dyspnea, diarrhea, dysuria, or change in hearing or vision.
Progressive dysarthria argues for an expanding lesion in the posterior fossa, worsening metabolic disturbance, or a problem affecting the cranial nerves (eg, Guillain-Barré syndrome) or neuromuscular junctions (eg, myasthenia gravis). Lack of headache makes a CNS localization less likely, though disorientation must localize to the brain itself. The transient nature of the AMS could signal an ictal phenomenon or a fluctuating toxic or metabolic condition, such as hyperammonemia, drug reaction, or healthcare–acquired delirium.
His past medical history included end-stage liver disease secondary to nonalcoholic steatohepatitis status post transjugular intrahepatic portosystemic shunt (TIPS) procedure three years prior, hepatic encephalopathy, diabetes mellitus type 2, hypertension, previous melanoma excision on his back, and recurrent Clostridium difficile colitis. Two years prior to admission he had been started on an indefinite course of metronidazole 500 mg twice daily without any recurrence. The patient’s other medications were aspirin, furosemide, insulin, lactulose, mirtazapine, pantoprazole, propranolol, spironolactone, and zinc. At the rehabilitation center, he was prescribed oral oxycodone 5 mg as needed every 4 hours for pain. He denied use of tobacco, alcohol, and recreational drugs. He previously worked as a funeral home director and embalmer.
Hyperammonemia and hepatic encephalopathy can present with a fluctuating mental state that often correlates to dietary protein intake or the frequency of bowel movements; the previous TIPS history places the patient at further risk. Use of oxycodone or another narcotic commonly leads to confusion, , especially in patients who are older, have preexisting cognitive decline, or have concomitant medical comorbidities. Mirtazapine and propranolol have been associated more rarely with encephalopathy, and therefore a careful history of adherence, drug interactions, and appropriate dosing should be obtained. Metronidazole is most often associated neurologically with a peripheral neuropathy; however, it is increasingly recognized that some patients can develop a CNS syndrome that features an AMS, which can be severe and accompanied by ataxia, dysarthria, and characteristic brain magnetic resonance imaging (MRI) findings, including hyperintensity surrounding the fourth ventricle on T2-weighted images.
Embalming fluid has a high concentration of formaldehyde, and a recent epidemiologic study suggested a link between formaldehyde exposure and increased risk for amyotrophic lateral sclerosis (ALS). ALS uncommonly presents with isolated dysarthria, but its bulbar form can, usually over a much longer course than is demonstrated here. Finally, the patient’s history of melanoma places him at risk for stroke from hypercoagulability as well as potential brain metastases or carcinomatous meningitis.
Evaluation was initiated at the rehabilitation facility at the onset of the patient’s slurred speech and confusion. Physical examination were negative for focal neurologic deficits, asterixis, and jaundice. Ammonia level was 41 µmol/L (reference range, 11-35 µmol/L). Noncontrast computed tomography (CT) of the head showed no signs of acute infarct or hemorrhage. Symptoms were attributed to hepatic encephalopathy; lactulose was up-titrated to ensure 2 or 3 bowel movements per day, and rifaximin was started.
Hyperammonemia is a cause of non-inflammatory relapsing encephalopathy, but an elevated level is neither a sensitive nor specific indicator of hepatic encephalopathy. Levels of ammonia can fluctuate widely during the day based on the frequency of bowel movements as well as dietary protein intake. In addition, proper handling of samples with prompt delivery to the laboratory is essential to minimize errors.
The ammonia level of 41 µmol/L discovered here is only modestly elevated, but given the patient’s history of TIPS as well as the clinical picture, it is reasonable to aggressively treat hepatic encephalopathy with lactulose to reduce ammonia levels. If he does not improve, an MRI of the brain to exclude a structural lesion and spinal fluid examination looking for inflammatory or infectious conditions would be important next steps. Although CT excludes a large hemorrhage or mass, this screening examination does not visualize many of the findings of the metabolic etiology and the other etiologies under consideration here.
Despite 3 days of therapy for presumed hepatic encephalopathy, the patient’s slurred speech worsened, and he was transferred to an academic tertiary care center for further evaluation. On admission, his temperature was 36.9°C, heart rate was 80 beats per minute, blood pressure was 139/67 mm Hg, respiratory rate was 10 breaths per minute, and oxygen saturation was 99% on room air. He was alert, awake, and oriented to person, place, and time. He was not jaundiced. He exhibited a moderate dysarthria characterized by monotone speech, decreased volume, decreased breath support, and a hoarse vocal quality with intact language function. Motor control of the lips, tongue, and mandible were normal. Motor strength was 5/5 bilaterally in the upper and lower extremities with the exception of right hip flexion, which was 4/5. The patient exhibited mild bilateral dysmetria on finger-to-nose examination, consistent with appendicular ataxia of the upper extremities. Reflexes were depressed throughout, and there was no asterixis. He had 2+ pulses in all extremities and 1+ pitting edema of the right lower extremity to the mid leg. Pulmonary examination revealed inspiratory crackles at the left base. The rest of the examination findings were normal.
The patient’s altered mental state appears to have resolved, and the neurological examination is now mainly characterized by signs that point to the cerebellum. The description of monotone speech typically refers to loss of prosody, the variable stress or intonation of speech, which is characteristic of a cerebellar speech pattern. The hoarseness should be explored to determine if it is a feature of the patient’s speech or is a separate process. Hoarseness may involve the vocal cord and therefore, potentially, cranial nerve X or its nuclei in the brainstem. The appendicular ataxia of the limbs points definitively to the cerebellar hemispheres or their pathways through the brainstem.
Unilateral lower extremity edema, especially in the context of a recent fracture, raises the possibility of deep vein thrombosis. If this patient has a right-to-left intracardiac or intrapulmonary shunt, embolization could lead to an ischemic stroke of the brainstem or cerebellum, potentially causing dysarthria.
Laboratory evaluation revealed hemoglobin level of 10.9 g/dL, white blood cell count of 5.3 × 10 9 /L, platelet count of 169 × 10 9 /L, glucose level of 177 mg/dL, corrected calcium level of 9.0 mg/dL, sodium level of 135 mmol/L, bicarbonate level of 30 mmol/L, creatinine level of 0.9 mg/dL, total bilirubin level of 1.3 mg/dL, direct bilirubin level of 0.4 mg/dL, alkaline phosphatase level of 503 U/L, alanine aminotransferase level of 12 U/L, aspartate aminotransferase level of 33 U/L, ammonia level of 49 µmol/L (range, 0-30 µ mol/L), international normalized ratio of 1.2, and troponin level of <0.01 ng/mL. Electrocardiogram showed normal sinus rhythm.
Some patients with bacterial meningitis do not have a leukocytosis, but patients with meningitis caused by seeding from a systemic infection nearly always do. In this patient’s case, lack of a leukocytosis makes bacterial meningitis very unlikely. The elevated alkaline phosphatase level is expected, as this level peaks about 3 weeks after a long-bone fracture and returns to normal over a few months.
Non-contrast CT scan of the head performed on admission demonstrated no large vessel cortical-based infarct, intracranial hemorrhage, hydrocephalus, mass effect, midline shift, or extra-axial fluid. There was mild cortical atrophy as well as very mild periventricular white matter hypodensity.
The atrophy and mild white-matter hypodensities seen on repeat noncontrast CT are nonspecific for any particular entity in this patient’s age group. MRI is more effective in evaluating toxic encephalopathies, including metronidazole toxicity or Wernicke encephalopathy, and in characterizing small infarcts or inflammatory conditions of the brainstem and cerebellum, which are poorly evaluated by CT due to the bone surrounded space of the posterior fossa. An urgent lumbar puncture is not necessary due to the slow pace of illness, lack of fever, nuchal rigidity, or serum elevated white blood cell count. Rather, performing MRI should be prioritized. If MRI is nondiagnostic, then spinal fluid should be evaluated for evidence of an infectious, autoimmune, paraneoplastic, or neoplastic process.
MRI was subsequently performed. It showed symmetric abnormal T2 hyperintensities involving dentate nuclei (Figure 1), left inferior olivary nuclei (Figure 2), restiform bodies, pontine tegmentum, superior cerebellar peduncles, oculomotor nuclei, and subthalamic nuclei. The most prominent hyperintensity was in the dentate nuclei.
The clinical and radiographic features confirm a diagnosis of metronidazole-associated CNS neurotoxicity. The reason for the predilection for edema in these specific areas of the brainstem and midline cerebellum is unclear but likely is related to selective neuronal vulnerability in these structures. The treatment is to stop metronidazole. In addition, the fluctuating mental status should be evaluated with electroencephalogram to ensure concomitant seizures are not occurring.
These MRI findings were consistent with metronidazole toxicity. Metronidazole was discontinued, and 2 days later the patient’s speech improved. Two weeks after medication discontinuation, his speech was normal. There were no more episodes of confusion.
DISCUSSION
Metronidazole was originally developed in France during the 1950s as an anti-parasitic medication to treat trichomonas infections. In 1962, its antibacterial properties were discovered after a patient with bacterial gingivitis improved while taking metronidazole for treatment of Trichomonas vaginalis.1 Since that time metronidazole has become a first-line treatment for anaerobic bacteria and is now recommended by the Infectious Diseases Society of America2 and the American College of Gastroenterology3 as a first-line therapy for mild and moderate C difficile infections.
Common side effects of metronidazole are nausea, vomiting, decreased appetite, diarrhea, headaches, peripheral neuropathy, and metallic taste; less common is CNS toxicity. Although the incidence of CNS toxicity is unknown, a systematic review of the literature found 64 cases reported between 1965 and 2011.4 CNS toxicity most often occurs between the fifth and sixth decades of life, and about two thirds of the people affected are men.4 CNS adverse effects characteristically fall into 4 categories: cerebellar dysfunction (eg, ataxia, dysarthria, dysmetria, nystagmus; 75%), AMS (33%), seizures (13%), and a combination of the first 3 categories.4
The exact mechanism of metronidazole CNS toxicity is unknown, but vasogenic or cytotoxic edema may be involved.5,6 Other potential etiologies are neural protein inhibition, reversible mitochondrial dysfunction, and modifications of the inhibitory neurotransmitter gamma-aminobutyric acid receptor in the cerebellum.7,8 There is no known genetic predisposition. Although the risk for CNS toxicity traditionally is thought to correlate with therapy duration and cumulative dose,7,9 in 2011 a systemic review found no significant correlation.4 In fact, 26% of patients with CNS toxicity were treated with metronidazole for less than 1 week at time of diagnosis.4
Brain CT is typically normal. On brain MRI, lesions most commonly appear as bilateral symmetric T2 hyperintensities, most often in the cerebellar dentate nuclei (85%) and less often in the midbrain (55%), the splenium of the corpus callosum (50%), the pons (35%), and the medulla (30%).4,10 Radiographic changes have been noted as early as 3 days after symptom onset. Based on damage severity and area affected (white or gray matter), vasogenic edema and cytotoxic edema may in combination be contributing to MRI abnormalities.6,10 Hyperintensities of the bilateral dentate nuclei can help in distinguishing metronidazole-induced encephalopathy from other potential disease processes, such as Wernicke encephalopathy.10
The prognosis for patients with metronidazole-induced neurotoxicity is favorable if metronidazole is discontinued. Approximately two-thirds of patients will have complete resolution of symptoms, which is more commonly observed when patients present with seizures or altered mental status. Approximately one-third will show partial improvement, particularly if the symptoms are due to cerebellar dysfunction. It is rare to experience permanent damage or death.4 Neurologic recovery usually begins within a week after medication discontinuation but may take months for complete recovery to occur.6,8,9,11 Follow-up imaging typically shows reversal of the original lesions, but this does not always correlate with symptom improvement.4,10
Despite its frequent use and long history, metronidazole can have potentially severe toxicity. When patients who are taking this medication present with new signs and symptoms of CNS dysfunction, hospitalists should include metronidazole CNS toxicity in the differential diagnosis and, if they suspect toxicity, have a brain MRI performed. Hospitalists often prescribe metronidazole because of the increasing number of patients being discharged from acute-care hospitals with a diagnosis of C difficile colitis.12 Brain MRI remains the imaging modality of choice for diagnosis. Discontinuation of metronidazole is usually salutary in reversing symptoms. Being keenly aware of this toxicity will help clinicians avoid being rendered speechless by a patient rendered speechless.
TEACHING POINTS
CNS toxicity is a rare but potentially devastating side effect of metronidazole exposure.
Metronidazole CNS adverse effects characteristically fall under 4 categories:
○ Cerebellar dysfunction, such as ataxia, dysarthria, dysmetria, or nystagmus (75%).
○ AMS (33%).
○ Seizures (13%).
○ A combination of the first 3 categories.
- Typically lesions indicating metronidazole toxicity on brain MRI are bilateral symmetric hyperintensities on T2-weighted imaging in the cerebellar dentate nuclei, corpus callosum, midbrain, pons, or medulla.
- Treatment of CNS toxicity is metronidazole discontinuation, which results in a high rate of symptom resolution.
Disclosure
Nothing to report.
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541. PubMed
2. Cohen SH, Gerding DN, Johnson S, et al; Society for Healthcare Epidemiology of America; Infectious Diseases Society of America. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol. 2010;31(5):431-455. PubMed
3. Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am J Gastroenterol. 2013;108(4):478-498. PubMed
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systemic review. Clin Neuropharmacol. 2011;34(6):241-247. PubMed
5. Graves TD, Condon M, Loucaidou M, Perry RJ. Reversible metronidazole-induced cerebellar toxicity in a multiple transplant recipient. J Neurol Sci. 2009;285(1-2):238-240. PubMed
6. Kim DW, Park JM, Yoon BW, Baek MJ, Kim JE, Kim S. Metronidazole-induced encephalopathy. J Neurol Sci. 2004;224(1-2):107-111. PubMed
7. Park KI, Chung JM, Kim JY. Metronidazole neurotoxicity: sequential neuroaxis involvement. Neurol India. 2011;59(1):104-107. PubMed
8. Patel K, Green-Hopkins I, Lu S, Tunkel AR. Cerebellar ataxia following prolonged use of metronidazole: case report and literature review. Int J Infect Dis. 2008;12(6):e111-e114. PubMed
9. Chandak S, Agarwal A, Shukla A, Joon P. A case report of metronidazole induced neurotoxicity in liver abscess patient and the usefulness of MRI for its diagnosis. J Clin Diagn Res. 2016;10(1):TD06-TD07. PubMed
10. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658. PubMed
11. Chacko J, Pramod K, Sinha S, et al. Clinical, neuroimaging and pathological features of 5-nitroimidazole-induced encephalo-neuropathy in two patients: insights into possible pathogenesis. Neurol India. 2011;59(5):743-747. PubMed
12. Peery AF, Dellon ES, Lund J, et al. Burden of gastrointestinal disease in the United States: 2012 update. Gastroenterology. 2012;143(5):1179-1187.e1-e3. PubMed
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541. PubMed
2. Cohen SH, Gerding DN, Johnson S, et al; Society for Healthcare Epidemiology of America; Infectious Diseases Society of America. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol. 2010;31(5):431-455. PubMed
3. Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am J Gastroenterol. 2013;108(4):478-498. PubMed
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systemic review. Clin Neuropharmacol. 2011;34(6):241-247. PubMed
5. Graves TD, Condon M, Loucaidou M, Perry RJ. Reversible metronidazole-induced cerebellar toxicity in a multiple transplant recipient. J Neurol Sci. 2009;285(1-2):238-240. PubMed
6. Kim DW, Park JM, Yoon BW, Baek MJ, Kim JE, Kim S. Metronidazole-induced encephalopathy. J Neurol Sci. 2004;224(1-2):107-111. PubMed
7. Park KI, Chung JM, Kim JY. Metronidazole neurotoxicity: sequential neuroaxis involvement. Neurol India. 2011;59(1):104-107. PubMed
8. Patel K, Green-Hopkins I, Lu S, Tunkel AR. Cerebellar ataxia following prolonged use of metronidazole: case report and literature review. Int J Infect Dis. 2008;12(6):e111-e114. PubMed
9. Chandak S, Agarwal A, Shukla A, Joon P. A case report of metronidazole induced neurotoxicity in liver abscess patient and the usefulness of MRI for its diagnosis. J Clin Diagn Res. 2016;10(1):TD06-TD07. PubMed
10. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658. PubMed
11. Chacko J, Pramod K, Sinha S, et al. Clinical, neuroimaging and pathological features of 5-nitroimidazole-induced encephalo-neuropathy in two patients: insights into possible pathogenesis. Neurol India. 2011;59(5):743-747. PubMed
12. Peery AF, Dellon ES, Lund J, et al. Burden of gastrointestinal disease in the United States: 2012 update. Gastroenterology. 2012;143(5):1179-1187.e1-e3. PubMed
© 2017 Society of Hospital Medicine
The Missing Element
A 57‐year‐old man presented to an emergency department with 1 month of progressive, bilateral lower extremity pain and weakness.
The first step in evaluating weakness is to determine whether it is objective (ie, decreased muscle strength due to pathology along the neuromuscular axis) or subjective. The sensation of weakness without loss of muscle strength may result from a debilitating chronic disease (eg, congestive heart failure, anemia, or chronic obstructive pulmonary disease). In patients with true lower extremity weakness it is prudent to assess for a myelopathy with a focused history and exam that includes assessment of bowel or bladder impairment and anal reflex. The presence of pain along with weakness might suggest disease of the muscle itself. A myopathy may arise from an infectious (eg, influenza), inflammatory (eg, polymyositis), endocrine (eg, hypothyroidism), or drug‐related (eg, statin) process.
The patient described 1 month of generalized weakness and pain in his lower extremities, which had worsened progressively to the point where ambulation was difficult. He was able to rise from a seated position using his arms for assistance, but had difficulty balancing in a standing position without assistance. The pain also involved both of his knees and increased with weight bearing. He also complained of bilateral lower extremity numbness and paresthesias, which had been migrating proximally from his toes over several months. He denied any recent trauma to his legs or back.
These symmetrical, distal sensory deficits favor a peripheral neuropathy over a myopathy, with neuropathic pain and arthralgia causing his impaired ability to ambulate or remain standing. In polyneuropathy, the type of nerve involvement (sensory vs motor) and pathology (axonal vs demyelinating) helps prioritize the differential. In developed countries, the most common causes of polyneuropathy are diabetes mellitus and alcohol. However, the tempo of his disease broadens the possibilities to include acute inflammatory demyelinating polyneuropathy, paraneoplastic syndrome (eg, monoclonal gammopathy), an autoimmune process (eg, rheumatoid arthritis, vasculitis), and heavy metal toxicity such as lead poisoning.
He had no history of chronic medical illness or hospitalizations and took no medications. His social history was notable for a history of alcohol abuse. For the past several years, he had only been drinking 1 to 2 beers daily due to cost, but had a history of more significant alcohol abuse in the distant past. He smoked 1 pack of tobacco per day, and denied illicit drug use. He denied any sexual activity or recent travel. He lived in a van, and had been homeless for over 10 years.
His socioeconomic status adds a layer of complexity to the case. Human immunodeficiency virus and hepatitis C virus (HCV) are more prevalent in the homeless and are associated with polyneuropathy. His lack of funds may drive him to drink illegally distilled alcohol, which can cause polyneuropathy through lead or arsenic toxicity. Excessive smoking could be linked to a peripheral neuropathy through a paraneoplastic syndrome (eg, small cell lung cancer).
Alcohol causes polyneuropathy through toxic effects on nerves and may be playing a role in his polyneuropathy, but the rapid pace and severity suggests an additional process. Alcoholism can be associated with deficiency of various B vitamins, such as thiamine, pyridoxine, and cobalamin, which can cause polyneuropathy. In alcoholics who are hospitalized, thiamine should be administered prior to glucose to decrease risk of Wernicke encephalopathy.
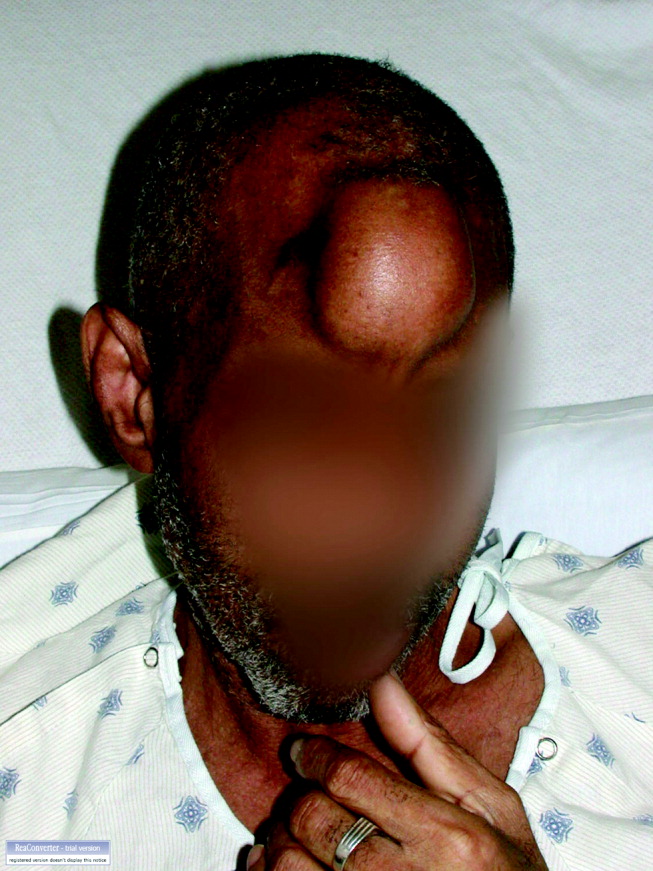
His temperature was 38.0C, heart rate 93 beats/min, blood pressure 121/60 mm Hg, respiratory rate 14/min, with an oxygen saturation of 97% on ambient air. He appeared cachectic and disheveled. He had moist mucous membranes, poor dentition with missing teeth, and no mucosal bleeding or oropharyngeal erythema. His cardiac exam revealed no murmurs, rubs, or gallops. His lungs were clear. His abdominal exam was benign, without masses or tenderness. His skin exam (Figure 1) was notable for nonpalpable petechiae on his anterior shins and thighs up to his buttocks. His extremity exam was significant for diffuse tenderness to light palpation on both lower extremities, a large indurated tender ecchymosis 15 15 cm behind the right knee, and another ecchymosis 6 8 cm behind the left knee. His dorsalis pedis and anterior tibialis pulses were appreciated by Doppler but not by palpation. He had decreased sensation to light touch of his bilateral feet to his ankles. Strength exam was challenging to assess secondary to posterior leg pain, but he demonstrated 4/5 strength of his hip flexors, quadriceps, and plantar flexors of the foot. His upper extremity strength and sensory exam were normal. Examination of the cranial nerves was normal. He had 2+ patellar and Achilles reflexes. Gait could not be adequately assessed.

Petechiae manifest as a nonblanchable rash caused by extravasated red blood cells. Common etiologies include quantitative or qualitative platelet defects, disseminated intravascular coagulopathy, trauma, and vasculitis. Cirrhosis from alcohol leading to thrombocytopenia and petechial rash is unlikely given no other stigmata of liver disease such as jaundice, spider angiomata, caput medusae, or palmar erythema. Less common causes include nutritional deficiency and light chain (AL) amyloidosis, which could explain both the neuropathy and rash.
The constellation of fever and petechial rash can represent a life‐threatening systemic process. Infectious agents that require immediate consideration with fever and petechiae include Neisseria meningitidis (meningococcemia), Rickettsia rickettsii (Rocky Mountain spotted fever), Staphylococcus, and Streptococcus. However, his normal blood pressure, dependent distribution of rash, and neuropathy make a severe bacterial infection less likely. Thrombotic thrombocytopenic purpura is possible and should prompt assessment of platelets, peripheral blood smear, and lactate dehydrogenase. Among vasculitides, the polyneuropathy, fever, and dependent distribution of petechial rash prioritize a small‐to‐medium vessel vasculitis, where the pathophysiology involves inflammation of dermal vessels and vasa nervorum (blood supply of nerves). Examples include HCV‐related cryoglobulinemic vasculitis, polyarteritis nodosa (PAN), and antineutrophilic cytoplasmic antibody (ANCA)associated vasculitis. However, ANCA‐associated vasculitis is less likely without upper or lower respiratory symptoms. Henoch‐Schonlein purpura may explain the rash but is more common in children and is not associated with neuropathy.
Posterior knee ecchymosis, in absence of trauma, raises suspicion for a ruptured Baker's cyst. However, the bilateral involvement and lack of calf manifestations makes this unlikely. The location raises concern for hemarthrosis, so a more likely explanation would be coagulopathy (eg, an acquired factor inhibitor) or a collagen defect. In developed countries, a commonly overlooked category of diseasenutritional deficiencywarrants serious consideration in alcoholics. Vitamin C deficiency (scurvy) may cause a petechial rash and ecchymosis from perifollicular hemorrhage and impaired collagen synthesis, respectively. Scurvy can masquerade as small vessel vasculitis because of its associated petechial rash. The neuropathy might be explained by concomitant thiamine or cobalamin deficiency. It is important to obtain a thorough dietary history and assess vibration and proprioception, which may be impaired from pathology of the dorsal column in cobalamin deficiency. The low‐grade fever may be a red herring, but if it becomes significant would be difficult to explain with nutritional deficiency.
In summary, a judicious evaluation for infection is mandatory, but the leading diagnoses are a small‐to‐medium vessel vasculitis (PAN or HCV‐related cryoglobulinemia), deficiency of multiple vitamins, and AL amyloidosis.
Initial labs showed white blood cell count 7800/L, hematocrit 39.2%, and platelet count of 251,000/L. Serum chemistry demonstrated a sodium of 131 mEq/L, potassium 4.7 mEq/L, chloride 93 mEq/L, bicarbonate 23 mEq/L, blood urea nitrogen 8 mg/dL, and creatinine 0.8 mg/dL. His aminotransferases, albumin, alkaline phosphatase, and coagulation studies were within normal limits. Urinalysis was remarkable for 2+ urobilinogen, 1+ ketones, and a bland sediment. Urine toxicology screen was negative.
His white blood cell count is normal, so with a heart rate of 93 beats/minute, he barely meets a single criterion of systemic inflammatory response syndrome (SIRS). The lack of SIRS and normal platelet, albumin, white blood cell, and red blood cell counts significantly reduces the likelihood of an infectious or inflammatory process. Without any clinical or biochemical evidence of HCV infection, HCV‐associated cryoglobulinemia is less likely. A normal creatinine might overestimate renal function in setting of decreased protein intake and muscle mass; nevertheless, the bland urine sediment further lowers probability of PAN and ANCA‐associated vasculitides. The normal platelet count and coagulation studies suggest either a qualitative platelet defect (eg, acquired von Willebrand disease) or impaired vessel integrity (eg, collagen defect) to explain the petechial rash. The urine ketones likely represent alcohol and/or starvation‐related ketosis. These data reduce the probability of infection and vasculitis, and prioritize vitamin deficiency and AL amyloidosis. Antibiotic therapy is not appropriate, given the absence of SIRS and subacute course. His presentation likely prompted a wide variety of tests, but most relevant would be a dietary history, cobalamin and vitamin C levels, serum free light chains, and skin biopsy. Biopsy of the rash would allow assessment for vasculitis and AL amyloidosis. The former is marked by inflammatory infiltrate of vessels, and the latter by perivascular invasion with amyloid fibrils. If the dietary history was consistent with ascorbic acid deficiency (scurvy), in addition to thiamine, he should be empirically treated with vitamin C. Patients with scurvy demonstrate rapid clinical improvement with treatment.
C‐reactive protein (CRP) was 47.9 mg/L and erythrocyte sedimentation rate (ESR) was 44 mm/hr. Human immunodeficiency antibody screen was negative. Anti‐nuclear antibodies and anti‐nuclear cytoplasmic antibody panel were negative. Computed tomography angiogram (CTA) of the lower extremities demonstrated severe stenosis of the left superficial femoral artery and severe stenosis of the right posterior tibial artery. Ankle‐brachial indices were 0.83 on the right side and 0.72 on the left, indicating mild to moderate arterial disease.
ESR and CRP are nonspecific markers of inflammation. Their elevation does not prioritize malignancy, autoimmunity, or infection. ANCA might be negative in commonly ANCA‐associated vasculitides such as eosinophilic granulomatosis with polyangiitis, microscopic polyangiitis, and granulomatosis with polyangiitis. However, the lack of respiratory and renal involvement in addition to the negative ANCA panel make such diagnoses unlikely. CTA of the patient's legs showed significant peripheral artery disease (PAD). This is unlikely to be the cause of his presentation; PAD should not cause petechiae, and his pain is disproportionate to the severity of the vascular disease reported. The additional information leaves the differential unchanged.
A dermatologist was consulted. She described and photographed a perifollicular distribution of the lower extremity petechiae with associated perifollicular hyperkeratosis and retained curled hairs (Figure 2).
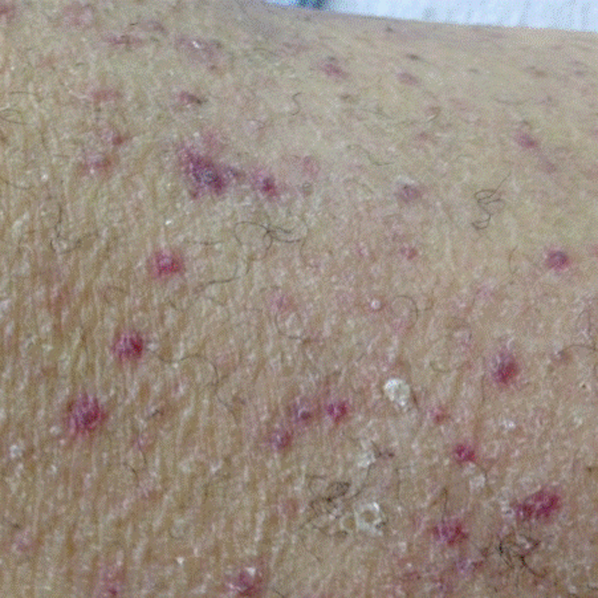
The described rash is specific for scurvy. His homelessness and alcohol intake likely made him vulnerable to ascorbic acid deficiency from lack of access to fruits and vegetables. Measurement of vitamin C level is unnecessary as the pretest probability for scurvy is very high. More relevant than a vitamin C level or skin biopsy is empiric treatment with ascorbic acid; as mentioned, patients with scurvy respond rapidly to vitamin C therapy. Given the neuropathy, he should be assessed for concomitant thiamine and/or cobalamin deficiency. His peripheral arterial disease is unlikely to be related.
His ascorbic acid level was 0.0 mg/dL (reference range, 0.22.0 mg/dL). Further history was obtained, and the patient reported exclusively eating frozen hamburgers and burritos for almost 1 year. He believed he had not had a fruit or vegetable in over 10 years. He was started on 1000 mg daily of ascorbic acid. By hospital day 2, his rash had mostly resolved and he was able to stand with some support. The patient was seen by his primary care physician 3 weeks after diagnosis, with his exercise tolerance nearly back to baseline. His rash had entirely resolved.
DISCUSSION
Unlike other mammals, humans do not have the ability to convert glucose to vitamin C and thus require an exogenous source, such as fruits and vegetables. The oft‐cited observation of scurvy in sailors during long journeys in the 18th century is a classic example of clinical disease due to vitamin C deficiency.[1] Once replete, body stores of vitamin C are usually sufficient to last over 6 months of deprivation. In some patients, symptoms of deficiency may appear within 3 months.[2] The patient in this report likely suffered years of vitamin C deficiency, resulting in the significant manifestations of scurvy reported here.
Vitamin C is a water‐soluble vitamin necessary for the biosynthesis of collagen, L‐carnitine, and neurotransmitters.[3] With deficiency, the resulting impairment in the formation of collagen affects blood vessel integrity and results in perivascular edema and erythrocyte extravasation. Clinically, this leads to hemorrhagic manifestations (eg, periosteal hemorrhage and perifollicular petechiae) and poor wound healing. Corkscrew hairs result because of vitamin C's role in disulfide bonding during hair formation. Woody edema and dyspnea are thought to be a consequence of leaky capillaries.[4]
Scurvy is still a significant cause of morbidity in at‐risk populations in the United States. Several populations have been identified as high risk for vitamin C deficiency, including the elderly, persons who live alone, alcoholics, smokers, individuals of low socioeconomic status, patients on hemodialysis, and those with psychiatric disease.[5] Specifically, the high oxidative stress associated with smoking, the history of alcohol abuse, and homelessness put this patient at an especially high risk.[6] Those with oxidative stressors have been postulated to require up to 125 mg/d of vitamin C compared to 60 to 90 mg/d of those without the same risks.[7] In a national health and nutrition survey in the United States in 2004, the prevalence of vitamin C deficiency as defined by a serum level <0.2 mg/dL was noted in 7.1% of those surveyed.[8] This study also noted a significantly higher prevalence of deficiency in smokers and individuals with low socioeconomic status.
Scurvy is a clinical diagnosis based on clinical features and dietary history. Severe manifestations of scurvy may happen quickly after the initial presentation, making early diagnosis especially important.[2] These include anemia, bone pain, ocular hemorrhage, cerebral hemorrhage, and hemopericardium.[2, 4] If needed, laboratory diagnosis can be made by demonstrating a serum ascorbic acid level <0.2 mg/dL. However, the level may be normal if the patient has had recent intake of vitamin C. In that scenario, the leukocyte vitamin C concentration may be a more accurate measure of the body stores as leukocyte levels change more slowly.[4] Biopsy of skin lesions is not necessary for the diagnosis and typically show a dilated hair follicle with keratin plugging and perifollicular hemorrhage.[9] Given the lack of adverse effects, treatment with vitamin C supplementation should begin immediately, even with low suspicion of scurvy, and response can serve as further clinical evidence and render laboratory testing unnecessary.
In this patient, the diagnosis was challenging for several reasons. The presentation was concerning for vasculitis given the dependent petechiae and elevated inflammatory markers. However, in scurvy, the petechiae are perifollicular and associated with hyperkeratosis, as opposed to the palpable purpura often described in vasculitis. Further, marked elevations in ESR and CRP have also been reported in scurvy.[10] The initial concern for vasculitis and clinician discomfort with a diagnosis based solely on a rash delayed the diagnosis. The complaint of polyneuropathy also seemed inconsistent with scurvy. Very rarely, scurvy may cause a neuropathy by hemorrhage into the nerve sheath, as seen in a case of bilateral femoral neuropathy.[11] Most likely, this patient had an underlying vitamin B deficiency explaining his polyneuropathy. Unfortunately, the patient was lost to follow‐up after his postdischarge visit with his primary physician and was not tested for other concomitant vitamin deficiencies.
Scurvy is very responsive to even small doses of vitamin C supplementation. For rapid recovery, doses ranging from 100 mg 3 times daily to 1000 mg daily of oral vitamin C are recommended for at least 1 month. Resolution of symptoms will begin within 24 hours, and complete recovery should occur by 3 months.[4] Scurvy is a classic example of how nutritional deficiencies can have a myriad of presentations and may mimic other systemic diseases. Clinicians who recall these manifestations and carefully assess patients for nutritional risks may be able to quickly identify the missing element (or elements) in a patient's diet, and initiate treatment that is often rapidly effective.
KEY TEACHING POINTS
- Vitamin C deficiency initially presents with classic dermatological findings of perifollicular petechiae with associated hyperkeratosis and corkscrew hairs.
- Scurvy is a clinical diagnosis based on history and presentation. Vitamin C serum level may not accurately reflect body stores, and a leukocyte vitamin C level may be obtained. The diagnosis may also be confirmed with observed response to vitamin C supplementation.
- Scurvy should be suspected in high‐risk populations, especially the marginally housed, the elderly, alcoholics, and smokers.
- Clinicians should screen patients with scurvy for other nutritional deficiencies including thiamine, folate, B12, and vitamin D levels.
Disclosures: Nothing to report.
- . The discovery of vitamin C. Ann Nutr Metab. 2012;61(3):259–264.
- , , , , . Clinical manifestations of ascorbic acid deficiency in man. Am J Clin Nutr. 1971;24(4):432–443.
- . New concepts in the biology and biochemistry of ascorbic acid. N Engl J Med. 1986;314(14):892–902.
- , . Adult scurvy. J Am Acad Dermatol. 1999;41(6):895–910.
- , . Be vigilant for scurvy in high‐risk groups. Practitioner. 2012;256(1755):23–5, 3.
- . Estimating ascorbic acid requirements for cigarette smokers. Ann N Y Acad Sci. 1993;686:335–346.
- , , . Vitamin C deficiency and depletion in the United States: the Third National Health and Nutrition Examination Survey, 1988 to 1994. Am J Public Health. 2004;94(5):870–875.
- , , , . Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003–2004 National Health and Nutrition Examination Survey (NHANES). Am J Clin Nutr. 2009;90(5):1252–1263.
- , , . Scurvy: a cutaneous clinical diagnosis. Australas J Dermatol. 2003;44(1):48–51.
- , . Rheumatic manifestations of scurvy: a report of three recent cases in a major urban center and a review. Semin Arthritis Rheum. 2011;41(2):286–290.
- . Femoral neuropathy in scurvy. N Engl J Med. 1969;281(23):1292–1293.
A 57‐year‐old man presented to an emergency department with 1 month of progressive, bilateral lower extremity pain and weakness.
The first step in evaluating weakness is to determine whether it is objective (ie, decreased muscle strength due to pathology along the neuromuscular axis) or subjective. The sensation of weakness without loss of muscle strength may result from a debilitating chronic disease (eg, congestive heart failure, anemia, or chronic obstructive pulmonary disease). In patients with true lower extremity weakness it is prudent to assess for a myelopathy with a focused history and exam that includes assessment of bowel or bladder impairment and anal reflex. The presence of pain along with weakness might suggest disease of the muscle itself. A myopathy may arise from an infectious (eg, influenza), inflammatory (eg, polymyositis), endocrine (eg, hypothyroidism), or drug‐related (eg, statin) process.
The patient described 1 month of generalized weakness and pain in his lower extremities, which had worsened progressively to the point where ambulation was difficult. He was able to rise from a seated position using his arms for assistance, but had difficulty balancing in a standing position without assistance. The pain also involved both of his knees and increased with weight bearing. He also complained of bilateral lower extremity numbness and paresthesias, which had been migrating proximally from his toes over several months. He denied any recent trauma to his legs or back.
These symmetrical, distal sensory deficits favor a peripheral neuropathy over a myopathy, with neuropathic pain and arthralgia causing his impaired ability to ambulate or remain standing. In polyneuropathy, the type of nerve involvement (sensory vs motor) and pathology (axonal vs demyelinating) helps prioritize the differential. In developed countries, the most common causes of polyneuropathy are diabetes mellitus and alcohol. However, the tempo of his disease broadens the possibilities to include acute inflammatory demyelinating polyneuropathy, paraneoplastic syndrome (eg, monoclonal gammopathy), an autoimmune process (eg, rheumatoid arthritis, vasculitis), and heavy metal toxicity such as lead poisoning.
He had no history of chronic medical illness or hospitalizations and took no medications. His social history was notable for a history of alcohol abuse. For the past several years, he had only been drinking 1 to 2 beers daily due to cost, but had a history of more significant alcohol abuse in the distant past. He smoked 1 pack of tobacco per day, and denied illicit drug use. He denied any sexual activity or recent travel. He lived in a van, and had been homeless for over 10 years.
His socioeconomic status adds a layer of complexity to the case. Human immunodeficiency virus and hepatitis C virus (HCV) are more prevalent in the homeless and are associated with polyneuropathy. His lack of funds may drive him to drink illegally distilled alcohol, which can cause polyneuropathy through lead or arsenic toxicity. Excessive smoking could be linked to a peripheral neuropathy through a paraneoplastic syndrome (eg, small cell lung cancer).
Alcohol causes polyneuropathy through toxic effects on nerves and may be playing a role in his polyneuropathy, but the rapid pace and severity suggests an additional process. Alcoholism can be associated with deficiency of various B vitamins, such as thiamine, pyridoxine, and cobalamin, which can cause polyneuropathy. In alcoholics who are hospitalized, thiamine should be administered prior to glucose to decrease risk of Wernicke encephalopathy.
His temperature was 38.0C, heart rate 93 beats/min, blood pressure 121/60 mm Hg, respiratory rate 14/min, with an oxygen saturation of 97% on ambient air. He appeared cachectic and disheveled. He had moist mucous membranes, poor dentition with missing teeth, and no mucosal bleeding or oropharyngeal erythema. His cardiac exam revealed no murmurs, rubs, or gallops. His lungs were clear. His abdominal exam was benign, without masses or tenderness. His skin exam (Figure 1) was notable for nonpalpable petechiae on his anterior shins and thighs up to his buttocks. His extremity exam was significant for diffuse tenderness to light palpation on both lower extremities, a large indurated tender ecchymosis 15 15 cm behind the right knee, and another ecchymosis 6 8 cm behind the left knee. His dorsalis pedis and anterior tibialis pulses were appreciated by Doppler but not by palpation. He had decreased sensation to light touch of his bilateral feet to his ankles. Strength exam was challenging to assess secondary to posterior leg pain, but he demonstrated 4/5 strength of his hip flexors, quadriceps, and plantar flexors of the foot. His upper extremity strength and sensory exam were normal. Examination of the cranial nerves was normal. He had 2+ patellar and Achilles reflexes. Gait could not be adequately assessed.
Petechiae manifest as a nonblanchable rash caused by extravasated red blood cells. Common etiologies include quantitative or qualitative platelet defects, disseminated intravascular coagulopathy, trauma, and vasculitis. Cirrhosis from alcohol leading to thrombocytopenia and petechial rash is unlikely given no other stigmata of liver disease such as jaundice, spider angiomata, caput medusae, or palmar erythema. Less common causes include nutritional deficiency and light chain (AL) amyloidosis, which could explain both the neuropathy and rash.
The constellation of fever and petechial rash can represent a life‐threatening systemic process. Infectious agents that require immediate consideration with fever and petechiae include Neisseria meningitidis (meningococcemia), Rickettsia rickettsii (Rocky Mountain spotted fever), Staphylococcus, and Streptococcus. However, his normal blood pressure, dependent distribution of rash, and neuropathy make a severe bacterial infection less likely. Thrombotic thrombocytopenic purpura is possible and should prompt assessment of platelets, peripheral blood smear, and lactate dehydrogenase. Among vasculitides, the polyneuropathy, fever, and dependent distribution of petechial rash prioritize a small‐to‐medium vessel vasculitis, where the pathophysiology involves inflammation of dermal vessels and vasa nervorum (blood supply of nerves). Examples include HCV‐related cryoglobulinemic vasculitis, polyarteritis nodosa (PAN), and antineutrophilic cytoplasmic antibody (ANCA)associated vasculitis. However, ANCA‐associated vasculitis is less likely without upper or lower respiratory symptoms. Henoch‐Schonlein purpura may explain the rash but is more common in children and is not associated with neuropathy.
Posterior knee ecchymosis, in absence of trauma, raises suspicion for a ruptured Baker's cyst. However, the bilateral involvement and lack of calf manifestations makes this unlikely. The location raises concern for hemarthrosis, so a more likely explanation would be coagulopathy (eg, an acquired factor inhibitor) or a collagen defect. In developed countries, a commonly overlooked category of diseasenutritional deficiencywarrants serious consideration in alcoholics. Vitamin C deficiency (scurvy) may cause a petechial rash and ecchymosis from perifollicular hemorrhage and impaired collagen synthesis, respectively. Scurvy can masquerade as small vessel vasculitis because of its associated petechial rash. The neuropathy might be explained by concomitant thiamine or cobalamin deficiency. It is important to obtain a thorough dietary history and assess vibration and proprioception, which may be impaired from pathology of the dorsal column in cobalamin deficiency. The low‐grade fever may be a red herring, but if it becomes significant would be difficult to explain with nutritional deficiency.
In summary, a judicious evaluation for infection is mandatory, but the leading diagnoses are a small‐to‐medium vessel vasculitis (PAN or HCV‐related cryoglobulinemia), deficiency of multiple vitamins, and AL amyloidosis.
Initial labs showed white blood cell count 7800/L, hematocrit 39.2%, and platelet count of 251,000/L. Serum chemistry demonstrated a sodium of 131 mEq/L, potassium 4.7 mEq/L, chloride 93 mEq/L, bicarbonate 23 mEq/L, blood urea nitrogen 8 mg/dL, and creatinine 0.8 mg/dL. His aminotransferases, albumin, alkaline phosphatase, and coagulation studies were within normal limits. Urinalysis was remarkable for 2+ urobilinogen, 1+ ketones, and a bland sediment. Urine toxicology screen was negative.
His white blood cell count is normal, so with a heart rate of 93 beats/minute, he barely meets a single criterion of systemic inflammatory response syndrome (SIRS). The lack of SIRS and normal platelet, albumin, white blood cell, and red blood cell counts significantly reduces the likelihood of an infectious or inflammatory process. Without any clinical or biochemical evidence of HCV infection, HCV‐associated cryoglobulinemia is less likely. A normal creatinine might overestimate renal function in setting of decreased protein intake and muscle mass; nevertheless, the bland urine sediment further lowers probability of PAN and ANCA‐associated vasculitides. The normal platelet count and coagulation studies suggest either a qualitative platelet defect (eg, acquired von Willebrand disease) or impaired vessel integrity (eg, collagen defect) to explain the petechial rash. The urine ketones likely represent alcohol and/or starvation‐related ketosis. These data reduce the probability of infection and vasculitis, and prioritize vitamin deficiency and AL amyloidosis. Antibiotic therapy is not appropriate, given the absence of SIRS and subacute course. His presentation likely prompted a wide variety of tests, but most relevant would be a dietary history, cobalamin and vitamin C levels, serum free light chains, and skin biopsy. Biopsy of the rash would allow assessment for vasculitis and AL amyloidosis. The former is marked by inflammatory infiltrate of vessels, and the latter by perivascular invasion with amyloid fibrils. If the dietary history was consistent with ascorbic acid deficiency (scurvy), in addition to thiamine, he should be empirically treated with vitamin C. Patients with scurvy demonstrate rapid clinical improvement with treatment.
C‐reactive protein (CRP) was 47.9 mg/L and erythrocyte sedimentation rate (ESR) was 44 mm/hr. Human immunodeficiency antibody screen was negative. Anti‐nuclear antibodies and anti‐nuclear cytoplasmic antibody panel were negative. Computed tomography angiogram (CTA) of the lower extremities demonstrated severe stenosis of the left superficial femoral artery and severe stenosis of the right posterior tibial artery. Ankle‐brachial indices were 0.83 on the right side and 0.72 on the left, indicating mild to moderate arterial disease.
ESR and CRP are nonspecific markers of inflammation. Their elevation does not prioritize malignancy, autoimmunity, or infection. ANCA might be negative in commonly ANCA‐associated vasculitides such as eosinophilic granulomatosis with polyangiitis, microscopic polyangiitis, and granulomatosis with polyangiitis. However, the lack of respiratory and renal involvement in addition to the negative ANCA panel make such diagnoses unlikely. CTA of the patient's legs showed significant peripheral artery disease (PAD). This is unlikely to be the cause of his presentation; PAD should not cause petechiae, and his pain is disproportionate to the severity of the vascular disease reported. The additional information leaves the differential unchanged.
A dermatologist was consulted. She described and photographed a perifollicular distribution of the lower extremity petechiae with associated perifollicular hyperkeratosis and retained curled hairs (Figure 2).
The described rash is specific for scurvy. His homelessness and alcohol intake likely made him vulnerable to ascorbic acid deficiency from lack of access to fruits and vegetables. Measurement of vitamin C level is unnecessary as the pretest probability for scurvy is very high. More relevant than a vitamin C level or skin biopsy is empiric treatment with ascorbic acid; as mentioned, patients with scurvy respond rapidly to vitamin C therapy. Given the neuropathy, he should be assessed for concomitant thiamine and/or cobalamin deficiency. His peripheral arterial disease is unlikely to be related.
His ascorbic acid level was 0.0 mg/dL (reference range, 0.22.0 mg/dL). Further history was obtained, and the patient reported exclusively eating frozen hamburgers and burritos for almost 1 year. He believed he had not had a fruit or vegetable in over 10 years. He was started on 1000 mg daily of ascorbic acid. By hospital day 2, his rash had mostly resolved and he was able to stand with some support. The patient was seen by his primary care physician 3 weeks after diagnosis, with his exercise tolerance nearly back to baseline. His rash had entirely resolved.
DISCUSSION
Unlike other mammals, humans do not have the ability to convert glucose to vitamin C and thus require an exogenous source, such as fruits and vegetables. The oft‐cited observation of scurvy in sailors during long journeys in the 18th century is a classic example of clinical disease due to vitamin C deficiency.[1] Once replete, body stores of vitamin C are usually sufficient to last over 6 months of deprivation. In some patients, symptoms of deficiency may appear within 3 months.[2] The patient in this report likely suffered years of vitamin C deficiency, resulting in the significant manifestations of scurvy reported here.
Vitamin C is a water‐soluble vitamin necessary for the biosynthesis of collagen, L‐carnitine, and neurotransmitters.[3] With deficiency, the resulting impairment in the formation of collagen affects blood vessel integrity and results in perivascular edema and erythrocyte extravasation. Clinically, this leads to hemorrhagic manifestations (eg, periosteal hemorrhage and perifollicular petechiae) and poor wound healing. Corkscrew hairs result because of vitamin C's role in disulfide bonding during hair formation. Woody edema and dyspnea are thought to be a consequence of leaky capillaries.[4]
Scurvy is still a significant cause of morbidity in at‐risk populations in the United States. Several populations have been identified as high risk for vitamin C deficiency, including the elderly, persons who live alone, alcoholics, smokers, individuals of low socioeconomic status, patients on hemodialysis, and those with psychiatric disease.[5] Specifically, the high oxidative stress associated with smoking, the history of alcohol abuse, and homelessness put this patient at an especially high risk.[6] Those with oxidative stressors have been postulated to require up to 125 mg/d of vitamin C compared to 60 to 90 mg/d of those without the same risks.[7] In a national health and nutrition survey in the United States in 2004, the prevalence of vitamin C deficiency as defined by a serum level <0.2 mg/dL was noted in 7.1% of those surveyed.[8] This study also noted a significantly higher prevalence of deficiency in smokers and individuals with low socioeconomic status.
Scurvy is a clinical diagnosis based on clinical features and dietary history. Severe manifestations of scurvy may happen quickly after the initial presentation, making early diagnosis especially important.[2] These include anemia, bone pain, ocular hemorrhage, cerebral hemorrhage, and hemopericardium.[2, 4] If needed, laboratory diagnosis can be made by demonstrating a serum ascorbic acid level <0.2 mg/dL. However, the level may be normal if the patient has had recent intake of vitamin C. In that scenario, the leukocyte vitamin C concentration may be a more accurate measure of the body stores as leukocyte levels change more slowly.[4] Biopsy of skin lesions is not necessary for the diagnosis and typically show a dilated hair follicle with keratin plugging and perifollicular hemorrhage.[9] Given the lack of adverse effects, treatment with vitamin C supplementation should begin immediately, even with low suspicion of scurvy, and response can serve as further clinical evidence and render laboratory testing unnecessary.
In this patient, the diagnosis was challenging for several reasons. The presentation was concerning for vasculitis given the dependent petechiae and elevated inflammatory markers. However, in scurvy, the petechiae are perifollicular and associated with hyperkeratosis, as opposed to the palpable purpura often described in vasculitis. Further, marked elevations in ESR and CRP have also been reported in scurvy.[10] The initial concern for vasculitis and clinician discomfort with a diagnosis based solely on a rash delayed the diagnosis. The complaint of polyneuropathy also seemed inconsistent with scurvy. Very rarely, scurvy may cause a neuropathy by hemorrhage into the nerve sheath, as seen in a case of bilateral femoral neuropathy.[11] Most likely, this patient had an underlying vitamin B deficiency explaining his polyneuropathy. Unfortunately, the patient was lost to follow‐up after his postdischarge visit with his primary physician and was not tested for other concomitant vitamin deficiencies.
Scurvy is very responsive to even small doses of vitamin C supplementation. For rapid recovery, doses ranging from 100 mg 3 times daily to 1000 mg daily of oral vitamin C are recommended for at least 1 month. Resolution of symptoms will begin within 24 hours, and complete recovery should occur by 3 months.[4] Scurvy is a classic example of how nutritional deficiencies can have a myriad of presentations and may mimic other systemic diseases. Clinicians who recall these manifestations and carefully assess patients for nutritional risks may be able to quickly identify the missing element (or elements) in a patient's diet, and initiate treatment that is often rapidly effective.
KEY TEACHING POINTS
- Vitamin C deficiency initially presents with classic dermatological findings of perifollicular petechiae with associated hyperkeratosis and corkscrew hairs.
- Scurvy is a clinical diagnosis based on history and presentation. Vitamin C serum level may not accurately reflect body stores, and a leukocyte vitamin C level may be obtained. The diagnosis may also be confirmed with observed response to vitamin C supplementation.
- Scurvy should be suspected in high‐risk populations, especially the marginally housed, the elderly, alcoholics, and smokers.
- Clinicians should screen patients with scurvy for other nutritional deficiencies including thiamine, folate, B12, and vitamin D levels.
Disclosures: Nothing to report.
A 57‐year‐old man presented to an emergency department with 1 month of progressive, bilateral lower extremity pain and weakness.
The first step in evaluating weakness is to determine whether it is objective (ie, decreased muscle strength due to pathology along the neuromuscular axis) or subjective. The sensation of weakness without loss of muscle strength may result from a debilitating chronic disease (eg, congestive heart failure, anemia, or chronic obstructive pulmonary disease). In patients with true lower extremity weakness it is prudent to assess for a myelopathy with a focused history and exam that includes assessment of bowel or bladder impairment and anal reflex. The presence of pain along with weakness might suggest disease of the muscle itself. A myopathy may arise from an infectious (eg, influenza), inflammatory (eg, polymyositis), endocrine (eg, hypothyroidism), or drug‐related (eg, statin) process.
The patient described 1 month of generalized weakness and pain in his lower extremities, which had worsened progressively to the point where ambulation was difficult. He was able to rise from a seated position using his arms for assistance, but had difficulty balancing in a standing position without assistance. The pain also involved both of his knees and increased with weight bearing. He also complained of bilateral lower extremity numbness and paresthesias, which had been migrating proximally from his toes over several months. He denied any recent trauma to his legs or back.
These symmetrical, distal sensory deficits favor a peripheral neuropathy over a myopathy, with neuropathic pain and arthralgia causing his impaired ability to ambulate or remain standing. In polyneuropathy, the type of nerve involvement (sensory vs motor) and pathology (axonal vs demyelinating) helps prioritize the differential. In developed countries, the most common causes of polyneuropathy are diabetes mellitus and alcohol. However, the tempo of his disease broadens the possibilities to include acute inflammatory demyelinating polyneuropathy, paraneoplastic syndrome (eg, monoclonal gammopathy), an autoimmune process (eg, rheumatoid arthritis, vasculitis), and heavy metal toxicity such as lead poisoning.
He had no history of chronic medical illness or hospitalizations and took no medications. His social history was notable for a history of alcohol abuse. For the past several years, he had only been drinking 1 to 2 beers daily due to cost, but had a history of more significant alcohol abuse in the distant past. He smoked 1 pack of tobacco per day, and denied illicit drug use. He denied any sexual activity or recent travel. He lived in a van, and had been homeless for over 10 years.
His socioeconomic status adds a layer of complexity to the case. Human immunodeficiency virus and hepatitis C virus (HCV) are more prevalent in the homeless and are associated with polyneuropathy. His lack of funds may drive him to drink illegally distilled alcohol, which can cause polyneuropathy through lead or arsenic toxicity. Excessive smoking could be linked to a peripheral neuropathy through a paraneoplastic syndrome (eg, small cell lung cancer).
Alcohol causes polyneuropathy through toxic effects on nerves and may be playing a role in his polyneuropathy, but the rapid pace and severity suggests an additional process. Alcoholism can be associated with deficiency of various B vitamins, such as thiamine, pyridoxine, and cobalamin, which can cause polyneuropathy. In alcoholics who are hospitalized, thiamine should be administered prior to glucose to decrease risk of Wernicke encephalopathy.
His temperature was 38.0C, heart rate 93 beats/min, blood pressure 121/60 mm Hg, respiratory rate 14/min, with an oxygen saturation of 97% on ambient air. He appeared cachectic and disheveled. He had moist mucous membranes, poor dentition with missing teeth, and no mucosal bleeding or oropharyngeal erythema. His cardiac exam revealed no murmurs, rubs, or gallops. His lungs were clear. His abdominal exam was benign, without masses or tenderness. His skin exam (Figure 1) was notable for nonpalpable petechiae on his anterior shins and thighs up to his buttocks. His extremity exam was significant for diffuse tenderness to light palpation on both lower extremities, a large indurated tender ecchymosis 15 15 cm behind the right knee, and another ecchymosis 6 8 cm behind the left knee. His dorsalis pedis and anterior tibialis pulses were appreciated by Doppler but not by palpation. He had decreased sensation to light touch of his bilateral feet to his ankles. Strength exam was challenging to assess secondary to posterior leg pain, but he demonstrated 4/5 strength of his hip flexors, quadriceps, and plantar flexors of the foot. His upper extremity strength and sensory exam were normal. Examination of the cranial nerves was normal. He had 2+ patellar and Achilles reflexes. Gait could not be adequately assessed.
Petechiae manifest as a nonblanchable rash caused by extravasated red blood cells. Common etiologies include quantitative or qualitative platelet defects, disseminated intravascular coagulopathy, trauma, and vasculitis. Cirrhosis from alcohol leading to thrombocytopenia and petechial rash is unlikely given no other stigmata of liver disease such as jaundice, spider angiomata, caput medusae, or palmar erythema. Less common causes include nutritional deficiency and light chain (AL) amyloidosis, which could explain both the neuropathy and rash.
The constellation of fever and petechial rash can represent a life‐threatening systemic process. Infectious agents that require immediate consideration with fever and petechiae include Neisseria meningitidis (meningococcemia), Rickettsia rickettsii (Rocky Mountain spotted fever), Staphylococcus, and Streptococcus. However, his normal blood pressure, dependent distribution of rash, and neuropathy make a severe bacterial infection less likely. Thrombotic thrombocytopenic purpura is possible and should prompt assessment of platelets, peripheral blood smear, and lactate dehydrogenase. Among vasculitides, the polyneuropathy, fever, and dependent distribution of petechial rash prioritize a small‐to‐medium vessel vasculitis, where the pathophysiology involves inflammation of dermal vessels and vasa nervorum (blood supply of nerves). Examples include HCV‐related cryoglobulinemic vasculitis, polyarteritis nodosa (PAN), and antineutrophilic cytoplasmic antibody (ANCA)associated vasculitis. However, ANCA‐associated vasculitis is less likely without upper or lower respiratory symptoms. Henoch‐Schonlein purpura may explain the rash but is more common in children and is not associated with neuropathy.
Posterior knee ecchymosis, in absence of trauma, raises suspicion for a ruptured Baker's cyst. However, the bilateral involvement and lack of calf manifestations makes this unlikely. The location raises concern for hemarthrosis, so a more likely explanation would be coagulopathy (eg, an acquired factor inhibitor) or a collagen defect. In developed countries, a commonly overlooked category of diseasenutritional deficiencywarrants serious consideration in alcoholics. Vitamin C deficiency (scurvy) may cause a petechial rash and ecchymosis from perifollicular hemorrhage and impaired collagen synthesis, respectively. Scurvy can masquerade as small vessel vasculitis because of its associated petechial rash. The neuropathy might be explained by concomitant thiamine or cobalamin deficiency. It is important to obtain a thorough dietary history and assess vibration and proprioception, which may be impaired from pathology of the dorsal column in cobalamin deficiency. The low‐grade fever may be a red herring, but if it becomes significant would be difficult to explain with nutritional deficiency.
In summary, a judicious evaluation for infection is mandatory, but the leading diagnoses are a small‐to‐medium vessel vasculitis (PAN or HCV‐related cryoglobulinemia), deficiency of multiple vitamins, and AL amyloidosis.
Initial labs showed white blood cell count 7800/L, hematocrit 39.2%, and platelet count of 251,000/L. Serum chemistry demonstrated a sodium of 131 mEq/L, potassium 4.7 mEq/L, chloride 93 mEq/L, bicarbonate 23 mEq/L, blood urea nitrogen 8 mg/dL, and creatinine 0.8 mg/dL. His aminotransferases, albumin, alkaline phosphatase, and coagulation studies were within normal limits. Urinalysis was remarkable for 2+ urobilinogen, 1+ ketones, and a bland sediment. Urine toxicology screen was negative.
His white blood cell count is normal, so with a heart rate of 93 beats/minute, he barely meets a single criterion of systemic inflammatory response syndrome (SIRS). The lack of SIRS and normal platelet, albumin, white blood cell, and red blood cell counts significantly reduces the likelihood of an infectious or inflammatory process. Without any clinical or biochemical evidence of HCV infection, HCV‐associated cryoglobulinemia is less likely. A normal creatinine might overestimate renal function in setting of decreased protein intake and muscle mass; nevertheless, the bland urine sediment further lowers probability of PAN and ANCA‐associated vasculitides. The normal platelet count and coagulation studies suggest either a qualitative platelet defect (eg, acquired von Willebrand disease) or impaired vessel integrity (eg, collagen defect) to explain the petechial rash. The urine ketones likely represent alcohol and/or starvation‐related ketosis. These data reduce the probability of infection and vasculitis, and prioritize vitamin deficiency and AL amyloidosis. Antibiotic therapy is not appropriate, given the absence of SIRS and subacute course. His presentation likely prompted a wide variety of tests, but most relevant would be a dietary history, cobalamin and vitamin C levels, serum free light chains, and skin biopsy. Biopsy of the rash would allow assessment for vasculitis and AL amyloidosis. The former is marked by inflammatory infiltrate of vessels, and the latter by perivascular invasion with amyloid fibrils. If the dietary history was consistent with ascorbic acid deficiency (scurvy), in addition to thiamine, he should be empirically treated with vitamin C. Patients with scurvy demonstrate rapid clinical improvement with treatment.
C‐reactive protein (CRP) was 47.9 mg/L and erythrocyte sedimentation rate (ESR) was 44 mm/hr. Human immunodeficiency antibody screen was negative. Anti‐nuclear antibodies and anti‐nuclear cytoplasmic antibody panel were negative. Computed tomography angiogram (CTA) of the lower extremities demonstrated severe stenosis of the left superficial femoral artery and severe stenosis of the right posterior tibial artery. Ankle‐brachial indices were 0.83 on the right side and 0.72 on the left, indicating mild to moderate arterial disease.
ESR and CRP are nonspecific markers of inflammation. Their elevation does not prioritize malignancy, autoimmunity, or infection. ANCA might be negative in commonly ANCA‐associated vasculitides such as eosinophilic granulomatosis with polyangiitis, microscopic polyangiitis, and granulomatosis with polyangiitis. However, the lack of respiratory and renal involvement in addition to the negative ANCA panel make such diagnoses unlikely. CTA of the patient's legs showed significant peripheral artery disease (PAD). This is unlikely to be the cause of his presentation; PAD should not cause petechiae, and his pain is disproportionate to the severity of the vascular disease reported. The additional information leaves the differential unchanged.
A dermatologist was consulted. She described and photographed a perifollicular distribution of the lower extremity petechiae with associated perifollicular hyperkeratosis and retained curled hairs (Figure 2).
The described rash is specific for scurvy. His homelessness and alcohol intake likely made him vulnerable to ascorbic acid deficiency from lack of access to fruits and vegetables. Measurement of vitamin C level is unnecessary as the pretest probability for scurvy is very high. More relevant than a vitamin C level or skin biopsy is empiric treatment with ascorbic acid; as mentioned, patients with scurvy respond rapidly to vitamin C therapy. Given the neuropathy, he should be assessed for concomitant thiamine and/or cobalamin deficiency. His peripheral arterial disease is unlikely to be related.
His ascorbic acid level was 0.0 mg/dL (reference range, 0.22.0 mg/dL). Further history was obtained, and the patient reported exclusively eating frozen hamburgers and burritos for almost 1 year. He believed he had not had a fruit or vegetable in over 10 years. He was started on 1000 mg daily of ascorbic acid. By hospital day 2, his rash had mostly resolved and he was able to stand with some support. The patient was seen by his primary care physician 3 weeks after diagnosis, with his exercise tolerance nearly back to baseline. His rash had entirely resolved.
DISCUSSION
Unlike other mammals, humans do not have the ability to convert glucose to vitamin C and thus require an exogenous source, such as fruits and vegetables. The oft‐cited observation of scurvy in sailors during long journeys in the 18th century is a classic example of clinical disease due to vitamin C deficiency.[1] Once replete, body stores of vitamin C are usually sufficient to last over 6 months of deprivation. In some patients, symptoms of deficiency may appear within 3 months.[2] The patient in this report likely suffered years of vitamin C deficiency, resulting in the significant manifestations of scurvy reported here.
Vitamin C is a water‐soluble vitamin necessary for the biosynthesis of collagen, L‐carnitine, and neurotransmitters.[3] With deficiency, the resulting impairment in the formation of collagen affects blood vessel integrity and results in perivascular edema and erythrocyte extravasation. Clinically, this leads to hemorrhagic manifestations (eg, periosteal hemorrhage and perifollicular petechiae) and poor wound healing. Corkscrew hairs result because of vitamin C's role in disulfide bonding during hair formation. Woody edema and dyspnea are thought to be a consequence of leaky capillaries.[4]
Scurvy is still a significant cause of morbidity in at‐risk populations in the United States. Several populations have been identified as high risk for vitamin C deficiency, including the elderly, persons who live alone, alcoholics, smokers, individuals of low socioeconomic status, patients on hemodialysis, and those with psychiatric disease.[5] Specifically, the high oxidative stress associated with smoking, the history of alcohol abuse, and homelessness put this patient at an especially high risk.[6] Those with oxidative stressors have been postulated to require up to 125 mg/d of vitamin C compared to 60 to 90 mg/d of those without the same risks.[7] In a national health and nutrition survey in the United States in 2004, the prevalence of vitamin C deficiency as defined by a serum level <0.2 mg/dL was noted in 7.1% of those surveyed.[8] This study also noted a significantly higher prevalence of deficiency in smokers and individuals with low socioeconomic status.
Scurvy is a clinical diagnosis based on clinical features and dietary history. Severe manifestations of scurvy may happen quickly after the initial presentation, making early diagnosis especially important.[2] These include anemia, bone pain, ocular hemorrhage, cerebral hemorrhage, and hemopericardium.[2, 4] If needed, laboratory diagnosis can be made by demonstrating a serum ascorbic acid level <0.2 mg/dL. However, the level may be normal if the patient has had recent intake of vitamin C. In that scenario, the leukocyte vitamin C concentration may be a more accurate measure of the body stores as leukocyte levels change more slowly.[4] Biopsy of skin lesions is not necessary for the diagnosis and typically show a dilated hair follicle with keratin plugging and perifollicular hemorrhage.[9] Given the lack of adverse effects, treatment with vitamin C supplementation should begin immediately, even with low suspicion of scurvy, and response can serve as further clinical evidence and render laboratory testing unnecessary.
In this patient, the diagnosis was challenging for several reasons. The presentation was concerning for vasculitis given the dependent petechiae and elevated inflammatory markers. However, in scurvy, the petechiae are perifollicular and associated with hyperkeratosis, as opposed to the palpable purpura often described in vasculitis. Further, marked elevations in ESR and CRP have also been reported in scurvy.[10] The initial concern for vasculitis and clinician discomfort with a diagnosis based solely on a rash delayed the diagnosis. The complaint of polyneuropathy also seemed inconsistent with scurvy. Very rarely, scurvy may cause a neuropathy by hemorrhage into the nerve sheath, as seen in a case of bilateral femoral neuropathy.[11] Most likely, this patient had an underlying vitamin B deficiency explaining his polyneuropathy. Unfortunately, the patient was lost to follow‐up after his postdischarge visit with his primary physician and was not tested for other concomitant vitamin deficiencies.
Scurvy is very responsive to even small doses of vitamin C supplementation. For rapid recovery, doses ranging from 100 mg 3 times daily to 1000 mg daily of oral vitamin C are recommended for at least 1 month. Resolution of symptoms will begin within 24 hours, and complete recovery should occur by 3 months.[4] Scurvy is a classic example of how nutritional deficiencies can have a myriad of presentations and may mimic other systemic diseases. Clinicians who recall these manifestations and carefully assess patients for nutritional risks may be able to quickly identify the missing element (or elements) in a patient's diet, and initiate treatment that is often rapidly effective.
KEY TEACHING POINTS
- Vitamin C deficiency initially presents with classic dermatological findings of perifollicular petechiae with associated hyperkeratosis and corkscrew hairs.
- Scurvy is a clinical diagnosis based on history and presentation. Vitamin C serum level may not accurately reflect body stores, and a leukocyte vitamin C level may be obtained. The diagnosis may also be confirmed with observed response to vitamin C supplementation.
- Scurvy should be suspected in high‐risk populations, especially the marginally housed, the elderly, alcoholics, and smokers.
- Clinicians should screen patients with scurvy for other nutritional deficiencies including thiamine, folate, B12, and vitamin D levels.
Disclosures: Nothing to report.
- . The discovery of vitamin C. Ann Nutr Metab. 2012;61(3):259–264.
- , , , , . Clinical manifestations of ascorbic acid deficiency in man. Am J Clin Nutr. 1971;24(4):432–443.
- . New concepts in the biology and biochemistry of ascorbic acid. N Engl J Med. 1986;314(14):892–902.
- , . Adult scurvy. J Am Acad Dermatol. 1999;41(6):895–910.
- , . Be vigilant for scurvy in high‐risk groups. Practitioner. 2012;256(1755):23–5, 3.
- . Estimating ascorbic acid requirements for cigarette smokers. Ann N Y Acad Sci. 1993;686:335–346.
- , , . Vitamin C deficiency and depletion in the United States: the Third National Health and Nutrition Examination Survey, 1988 to 1994. Am J Public Health. 2004;94(5):870–875.
- , , , . Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003–2004 National Health and Nutrition Examination Survey (NHANES). Am J Clin Nutr. 2009;90(5):1252–1263.
- , , . Scurvy: a cutaneous clinical diagnosis. Australas J Dermatol. 2003;44(1):48–51.
- , . Rheumatic manifestations of scurvy: a report of three recent cases in a major urban center and a review. Semin Arthritis Rheum. 2011;41(2):286–290.
- . Femoral neuropathy in scurvy. N Engl J Med. 1969;281(23):1292–1293.
- . The discovery of vitamin C. Ann Nutr Metab. 2012;61(3):259–264.
- , , , , . Clinical manifestations of ascorbic acid deficiency in man. Am J Clin Nutr. 1971;24(4):432–443.
- . New concepts in the biology and biochemistry of ascorbic acid. N Engl J Med. 1986;314(14):892–902.
- , . Adult scurvy. J Am Acad Dermatol. 1999;41(6):895–910.
- , . Be vigilant for scurvy in high‐risk groups. Practitioner. 2012;256(1755):23–5, 3.
- . Estimating ascorbic acid requirements for cigarette smokers. Ann N Y Acad Sci. 1993;686:335–346.
- , , . Vitamin C deficiency and depletion in the United States: the Third National Health and Nutrition Examination Survey, 1988 to 1994. Am J Public Health. 2004;94(5):870–875.
- , , , . Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003–2004 National Health and Nutrition Examination Survey (NHANES). Am J Clin Nutr. 2009;90(5):1252–1263.
- , , . Scurvy: a cutaneous clinical diagnosis. Australas J Dermatol. 2003;44(1):48–51.
- , . Rheumatic manifestations of scurvy: a report of three recent cases in a major urban center and a review. Semin Arthritis Rheum. 2011;41(2):286–290.
- . Femoral neuropathy in scurvy. N Engl J Med. 1969;281(23):1292–1293.
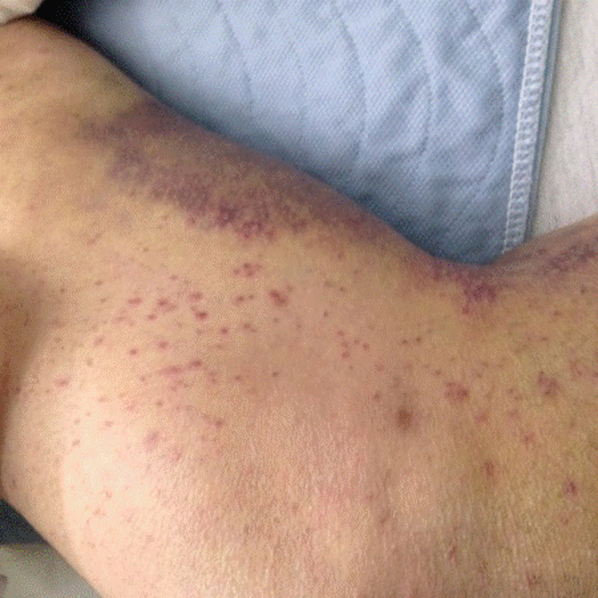
The Third Time's the Charm
A 58‐year old woman was brought to the emergency department with confusion. Her husband stated that for several hours she had been drifting in and out at home, and that he had to shout to get her attention. He described no seizure activity, weakness, incontinence, or difficulty speaking, and had noted no complaints of headache, fevers, chest pain, shortness of breath, or gastrointestinal complaints.
Altered mental status in a middle‐aged woman can result from a diverse set of etiologies. A key distinction in the neurological examination will be to assure that the complaint of confusion is accurate as opposed to aphasia; the former is usually indicative of diffuse cerebral dysfunction while the latter suggests a focal lesion in the dominant hemisphere.
The acuity of the change in mental status is important, as are the fluctuations described by the husband. Unwitnessed or nonconvulsive seizure activity can present this way. Toxic/metabolic etiologies, infectious and inflammatory disorders of the central nervous system (CNS), and vascular diseases are also important considerations. Although stroke does not typically present with global encephalopathy, intermittent large vessel occlusion, especially in the posterior circulation, can disrupt cognition in this manner. Following a physical examination, initial workup should focus on toxic/metabolic etiologies, followed rapidly by head imaging if no cause is identified.
Her past medical history was notable for type 2 diabetes mellitus, coronary artery disease, hyperlipidemia, and an unspecified seizure disorder, which according to her husband was diagnosed during a recent hospitalization for a similar presentation. She also had a remote history of venous thromboembolism and antithrombin‐III deficiency. She was unemployed, lived with her husband, and spent most of her time at home. She never smoked, and rarely drank alcohol. Her family history was unobtainable, and her husband denied that she used any illicit drugs. Her medications included pioglitazone, aspirin, simvastatin, pregabalin, ferrous sulfate, levetiracetam, warfarin, and magnesium oxide, and she was allergic to sulfa.
While the differential diagnosis remains broad, 3 elements of the history are potentially relevant. The history of epilepsy based on a similar prior presentation increases the likelihood that the current spell is ictal in nature; examination of previous records would be important in order to document whether these spells have indeed been proven to be epileptic, as many conditions can mimic seizures. Given the history of venous thromboembolism and hypercoagulability, one must consider cerebral venous sinus thrombosis, which can present with global neurologic dysfunction and seizures. Prompt identification, usually via computed tomography (CT) or magnetic resonance angiography, is vital, because anticoagulation can mitigate this potentially life‐threatening illness. Finally, although many medications can cause encephalopathy in overdose, levetiracetam has well‐described cognitive side effects even at usual doses, including encephalopathy, irritability, and depression.
The records from that recent hospitalization remarked that she had presented confused and stuporous. Her potassium had been 2.7 mmol/L, international normalized ration (INR) 3.4, and hemoglobin 8 g/dL; other routine laboratory studies were normal. CT and magnetic resonance imaging (MRI) of the brain had been negative, and electroencephalogram (EEG) reportedly was performed but specific results were unknown. She was discharged alert and oriented 1 week prior to the current presentation on the above medications, including levetiracetam for this newly‐diagnosed seizure disorder.
Previous records confirm that the current presentation is that of a relapsing acute alteration in mental status. Regardless of the EEG findings or response to antiepileptic medications, a seizure disorder should remain a primary consideration, although relapsing inflammatory, toxic/metabolic conditions, and, rarely, vascular disorders can also present in this manner.
The neurologic manifestations of hypokalemia are usually peripheral in nature, including periodic paralysis; confusion accompanying hypokalemia is usually not a result of the low potassium itself but rather due to an underlying toxic or endocrinologic cause. Various causes of anemia can lead to mental status changes; the mean corpuscular volume (MCV) will be particularly helpful given known associations between megaloblastic anemia and confusional states.
On examination, she appeared to be in good health and in no distress. She was afebrile. Her blood pressure was 93/57, pulse 90 beats per minute, respiratory rate 16 per minute, and room air oxygen saturation 100%. She was oriented to her surroundings, but slow in her responses to questioning. There were no cranial nerve, motor, or sensory deficits, or abnormal reflexes or movements. Examination of the head, skin, chest, cardiovascular system, abdomen, and extremities was normal. Serum sodium was 136 mmol/L, creatinine 1.2 mg/dL, calcium 9.3 mg/dL, and glucose 81 mg/dL; other routine blood chemistries were normal. Her white blood cell (WBC) count was 7100/L, hemoglobin 9.2 g/dL with normal MCV, and platelet count 275,000/L. INR was 3.4, and liver function tests were normal. CT of the brain demonstrated no evidence of acute pathology.
Given that her laboratory results (aside from the hemoglobin) and CT were essentially normal, the most common etiology of a recurrent encephalopathy would be a toxic exposure including drugs, alcohol, and environmental toxins or poisons. A comprehensive serum drug screen, including heavy metals, could follow a basic urinary screen for drugs of abuse; specific etiologies may be suggested by patterns of injury seen on MRI such as those seen with carbon monoxide or methanol exposure. Other recurrent metabolic processes include the porphyrias and relapsing inflammatory disorders, which could be entertained if further diagnostics are unrevealing.
An EEG is warranted at this point and is a test that is underutilized in the workup of altered mental status. Patients who have a spell and do not quickly awaken should be considered to be in nonconvulsive status epilepticus until proven otherwise. This can be easily identified on the EEG and is an important entity to recognize quickly. Additional findings on EEG may suggest focal cerebral dysfunction (such as that following a seizure or acute unilateral injury), diffuse encephalopathy (eg, triphasic waves), or fairly specific diagnoses (eg, periodic lateralized epileptiform discharges from the temporal lobes in suspected herpes simplex meningoencephalitis). While the CT of the brain is a reasonable initial screen, MRI is more sensitive for structural disease and should be obtained if no etiology is rapidly identified.
Finally, acute infectious etiologies such as abscess, encephalitis, or meningoencephalitis need to be excluded via lumbar puncture. Spinal fluid examination also can be helpful in the consideration of inflammatory and autoimmune disorders.
Over the next several hours, while still in the emergency department, she became increasingly obtunded, to the point that she was unresponsive to all stimuli. No seizure activity was witnessed, her vital signs were unchanged, and no medications had been administered. She was urgently transferred to a tertiary care center, where, at the time of arrival, she was obtunded and nonverbal, and opened her eyes only to noxious stimuli. She would withdraw all 4 extremities in response to pain. Pupils were 2 mm and symmetrically reactive. Corneal reflexes were normal, and her gag reflex was diminished. Motor tone was decreased in all 4 extremities. No fasciculations were noted. Deep tendon reflexes were present but symmetrically diminished throughout, and Babinski testing demonstrated a withdrawal response bilaterally.
Coma is a state of profound unconsciousness where the patient is unarousable and unaware of her surroundings. Coma can result either from bihemispheric dysfunction or diffuse injury to the reticular activating system in the brainstem, and the physical examination should focus on distinguishing between these 2 sites. Because the nuclei of cranial nerves III through XII (excepting XI) reside in the brainstem, the coma examination emphasizes testing the cranial nerves; although all cranial nerves are not tested in this patient, the ones that are appear to be normal, making bihemispheric dysfunction most likely. Bihemispheric coma most commonly results from diffuse toxic or metabolic etiologies such as intoxication or hepatic encephalopathy, but it can also be caused by bilateral structural lesions (including the bilateral thalami) or ongoing seizure activity.
Although an EEG remains the key test in this patient given her past history and an MRI would prove extremely useful, her deterioration warrants a workup for CNS infection. Since the head CT was negative, it would be prudent to proceed with urgent lumbar puncture (although it should never be performed in a patient with significant coagulopathy due to risks of hemorrhage leading to spinal cord injury). She should be covered empirically with broad spectrum meningeal‐dose antibiotics, including acyclovir, until the results of the spinal fluid examination are known, given that bacterial meningitis and herpes meningoencephalitis carry a high morbidity and mortality if not treated promptly.
Routine blood tests were similar to her labs at the referring emergency room. Ammonia level was 10 mol/L. Urine toxicology screen was negative, and blood tests for ethanol, salicylates, lithium, and acetaminophen were negative. Chest X‐ray and urinalysis were normal, and electrocardiogram was notable only for a sinus tachycardia. Cultures of the blood were obtained and the patient was admitted to the intensive care unit.
Levetiracetam, vancomycin, piperacillin‐tazobactam, and acyclovir were initiated. A lumbar puncture was performed without reversing the anticoagulation, and the procedure was traumatic. The cerebrospinal fluid was bloody, with a clear supernatant. Cell count demonstrated a red blood cell (RBC) count of 1250/L and a WBC count of 9/L, with a WBC differential of 42% neutrophils, 48% lymphocytes, and 8% monocytes. The cerebrospinal fluid (CSF) glucose was 62 mg/dL (with a serum glucose of 74 mg/dL) and protein 41 mg/dL. The CSF Gram stain demonstrated no organisms, and fluid was sent for routine culture and polymerase chain reaction (PCR) to detect herpes simplex virus (HSV). A neurology consultation was urgently requested.
As mentioned, it would have been more appropriate to reverse the patient's anticoagulation prior to lumbar puncture. The absence of xanthochromia suggests that the RBCs seen in the sample were introduced at the time of the lumbar puncture, arguing against a hemorrhagic disorder of the CNS (occasionally seen with herpes simplex encephalitis) or spinal fluid (eg, subarachnoid hemorrhage).
A reasonable rule of thumb to correct for the number of RBCs in a traumatic lumbar puncture is to allow 1 WBC for every 700 RBCs/L. Given this conversion, there are still too many WBCs in this sample, indicating a mild pleocytosis that is approximately one‐half neutrophilic and one‐half lymphocytic. This profile is nonspecific and can occur with a variety of conditions including stroke, seizure, inflammatory disorders, and infections, including viruses such as West Nile virus.
While coverage with acyclovir and broad‐spectrum antibacterials is appropriate, it should be noted that piperacillin‐tazobactam has poor CSF penetration and therefore is not a good choice for empiric coverage of CNS infections.
The neurologist's examination additionally noted multifocal myoclonus with noxious stimuli, most prominent in the face and toes. An urgent EEG demonstrated continuous, slow, generalized triphasic wave activity (Figures 1 and 2); no epileptiform discharges were seen. The erythrocyte sedimentation rate (ESR) was 66 mm/hour (normal, 0‐30), and tests for antinuclear antibodies, serum levetiracetam level, and thyroid function studies were ordered.
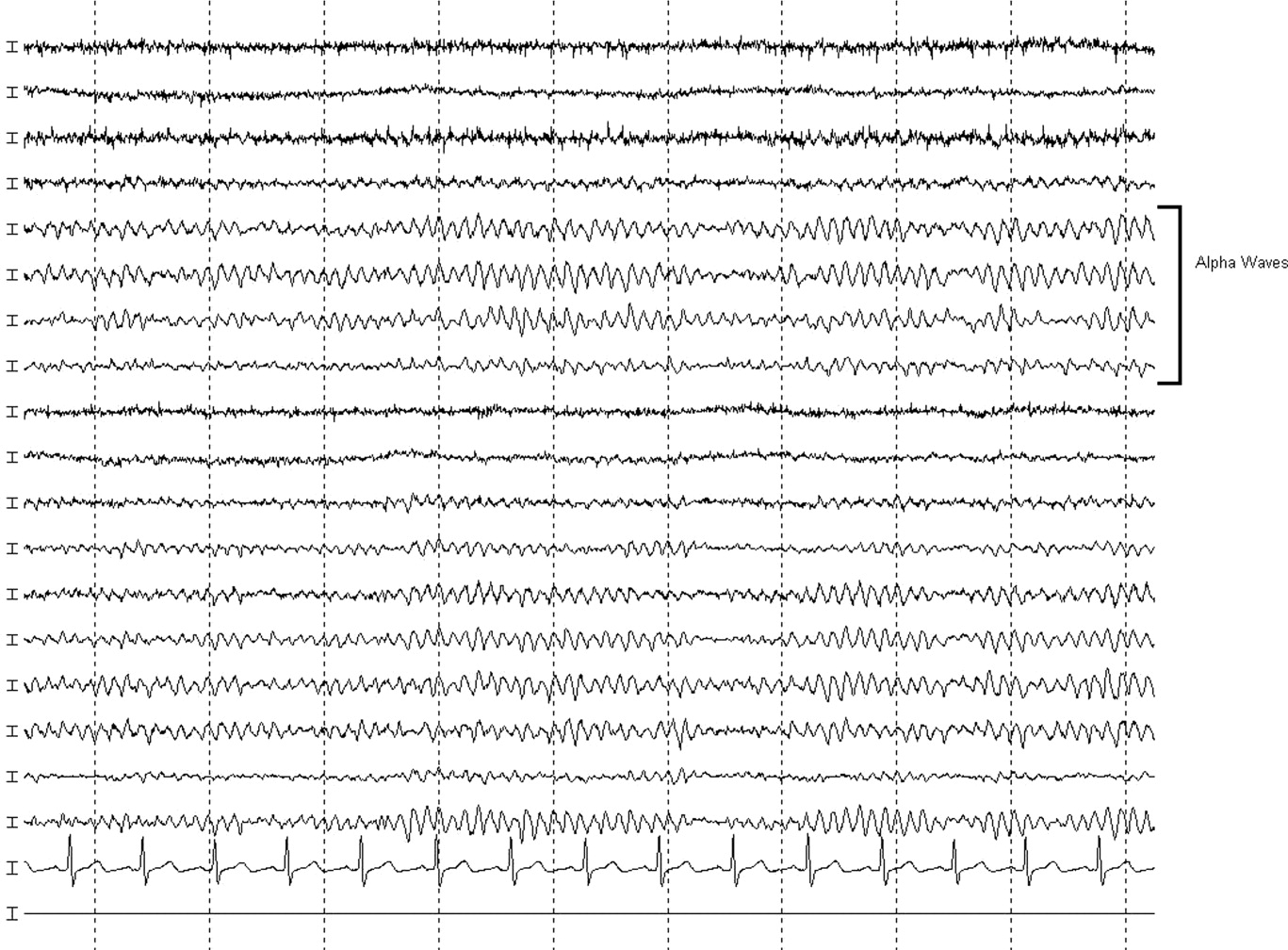
Stimulus‐evoked multifocal myoclonus is a general marker of encephalopathy found in many conditions, including hepatic and renal failure, drug intoxication (eg, opiates), neurodegenerative disorders (eg, Creutzfeldt‐Jakob disease [CJD]), and postanoxic injury, the latter of which is termed the Lance‐Adams syndrome.
Triphasic waves on EEG, while commonly associated with hepatic encephalopathy, have a similarly broad differential diagnosis, although in a comatose patient, they must first and foremost be distinguished from the repetitive discharges characteristic of nonconvulsive status epilepticus. In addition to hepatic and renal failure, triphasic waves have also been described in medication toxicity (especially with anticonvulsants, lithium, and cephalosporins), CNS infections (including Lyme disease and West Nile virus), strokes involving the bilateral thalami (usually from deep venous thrombosis), inflammatory disorders (such as Hashimoto's encephalopathy [HE]), and neurodegenerative diseases. It is important to remember that a single EEG does not exclude the possibility of an episodic ictal disorder and longer‐term monitoring would be required to definitively exclude seizures.
At this point, although the myoclonus and triphasic waves most commonly would indicate a toxic/metabolic process, the elevated ESR and CSF pleocytosis argue for an inflammatory or infectious condition. An MRI remains the next most useful test to guide further workup because many such conditions have distinct signatures on MRI.
The following day, she was noted to have periods of alertnessopening her eyes and following some commandsbut at other times she was difficult to arouse or obtunded. Tremulous movements and sporadic myoclonic jerks continued but no focal neurologic signs were found. Although there was increased muscle tone throughout, she was intermittently seen moving her limbs spontaneously, but not to command. No new findings were appreciated on routine laboratory tests. Antinuclear antibody testing was negative. Serum levetiracetam level was 23.5 g/mL (reference range, 545). Serum thyroid‐stimulating hormone was less than 0.005 U/mL, but free T3 was 3.5 pg/mL (normal, 1.8‐4.6) and free T4 was 2.0 ng/dL (normal, 0.71.8). An MRI of the brain was compromised by motion artifact but no significant abnormalities were appreciated.
At this point, a family member in another state disclosed that the patient had also been hospitalized 2 months previously while visiting him. Her chief complaint had been shortness of breath. The records were obtained; a cardiac catheterization had revealed nonobstructive coronary disease, and medical management was recommended. The notes mentioned that during the hospitalization she developed altered mental status with disorientation and shaking. CT and MRI of the brain had been unremarkable. The confusion was not explained, but she was discharged in good condition, alert and fully‐oriented.
The additional history confirms a relapsing encephalopathy, now with at least 3 occurrences. The most common etiologies in the face of a normal MRI and basic labs would be recurrent intoxication or exposures, but the inflammatory CSF profile and elevated ESR are not consistent with this. A variety of inflammatory disorders can present with recurrent encephalopathy, including demyelinating diseases and neurosarcoidosis. Some systemic rheumatologic conditions, such as systemic lupus erythematosus, can present with relapsing neurologic symptoms due to seizures, vasculitis, or cerebritis. Vasculitis would fit this picture as well, except for the normal findings on 2 MRIs. In a patient with such dramatic symptoms of neurologic dysfunction, one would expect to see changes on the MRI of cerebral inflammation with probable ischemia.
Therefore, given the CSF, ESR, clinical course, and unrevealing MRI and EEG, the most likely group of disorders responsible would be the nonvasculitic autoimmune meningoencephalitides, which present with recurrent encephalopathy and feature spontaneous remissions and/or often‐dramatic responses to corticosteroids. Key disorders in this category include Sjogren's disease, lupus, and steroid responsive encephalopathy associated with autoimmune thyroiditis (Hashimoto's encephalopathy). The latter condition is the most common of the group and is suggested by the abnormal thyroid‐stimulating hormone testing, although it may occur in the setting of normal thyroid function. The diagnosis can be confirmed with thyroperoxidase and thyroglobulin antibody testing.
Three days into the hospitalization, her mental status had gradually improved such that she was more consistently awake and oriented to person and place, and she was transferred to a regular nursing unit. Final results from the CSF and blood cultures were negative, as was PCR for HSV. The antimicrobials were discontinued. Routine serum chemistries continued to be unremarkable. Additional studies recommended by the neurologist demonstrated an antithyroperoxidase antibody concentration of 587.1 IU/mL (normal, <5), and antithyroglobulin antibody level of 52.2 IU/mL (normal, <10).
These results confirm the diagnosis of HE which, in addition to its presentation as a recurrent illness, is an important treatable cause of dementia and should be considered in young patients, those with autoimmune and thyroid disorders, and those whose dementia is rapidly progressive. Most cases are thought to be steroid‐responsive, but some studies have defined the disorder based on this responsiveness, resulting in some nonresponders likely being overlooked.
A trial of corticosteroids should be considered if the patient does not quickly return to baseline given the potential morbidities associated with prolonged altered mental status to this degree. Whether initiation of chronic immunosuppression could prevent these attacks in the future is unclear from the literature but should be considered given the recurrent, dramatic presentation in this patient.
A diagnosis of HE was made, and she was prescribed corticosteroids. Twenty‐four hours later, she was alert and fully‐oriented. She was discharged to home on prednisone and seen in follow‐up in neurology clinic 1 month later. She had had no further episodes of confusion or stupor, but because of steroid‐induced hyperglycemia, her corticosteroids were decreased and mycophenolate mofetil added for chronic immunosuppression. Four months after discharge she was neurologically stable but continued to struggle with the adverse effects of chronic corticosteroid treatment.
COMMENTARY
HE is an uncommon condition that can present with a rapidly progressive decline and should be considered in patients who present with recurrent mental status change in the setting of normal imaging studies and routine laboratory results. The entity was initially described by Lord William Russell Brain in 1966, and in the most recent terminology is known as steroid‐responsive encephalopathy associated with autoimmune thyroiditis (SREAT).1 It is characterized by an acute or subacute encephalopathy associated with thyroid autoimmunity. Patients typically present with fluctuating symptoms, episodes of confusion, alterations of consciousness, and rapid cognitive decline.2 Common features include myoclonus, tremor, ataxia, speech disturbance, stroke‐like episodes, increased muscle tone, neuropsychiatric manifestations, and seizures, that in some cases may progress to status epilepticus.3, 4
Although serum antithyroglobulin and antithyroperoxidase antibodies are elevated in HE, their presence is thought to be an epiphenomenon of the condition rather than the direct cause. Supporting this are the facts that the incidence of encephalopathy is not increased in patients with established autoimmune thyroiditis, and the presence of antithyroid antibodies ranges from 5% to 20% in the general population.2, 5 There is also no evidence that thyroid antibodies directly react with brain tissue, and the levels of these antibodies do not correlate with either neurologic manifestations or clinical improvement.2, 4, 5 As HE has been reported in patients with euthyroidism, hypothyroidism, and hyperthyroidism (with hypothyroidismeither subclinical or activemost common), it is also unlikely that the level of thyroid hormones play a role in the etiology of this disease.2, 4, 6
The etiology and pathogenesis of HE are unclear, although an immune‐mediated process is generally implicated, either from an inflammatory vasculitis or as a form of acute disseminated encephalomyelitis.7‐9 Global hypoperfusion on single‐photon emission computed tomography (SPECT) studies has also been reported.10, 11 Patients with HE may have nonspecific evidence of inflammation, including an elevated ESR, CRP, and CSF protein.12 Other laboratory abnormalities may include a mild elevation of liver aminotransferase levels; renal impairment has also been reported in a few cases of HE in the form of glomerulonephritis, and may be related to deposition of immune complexes containing thyroglobulin antigen.6, 12‐14 MRI of the brain is normal or nonspecific in most cases, and the EEG most commonly shows diffuse slowing.
The differential for a rapidly progressive cognitive decline includes CJD, CNS vasculitis, paraneoplastic syndromes, and autoimmune and subacute infectious encephalopathies. In patients with CJD, T2‐weighted imaging may show hyperintense signals in the basal ganglia, while diffusion‐weighted sequences may reveal changes in the cortical ribbon and bilateral thalami.15 In CNS vasculitis, the imaging findings are variable and range from discrete areas of vascular infarcts to hemorrhagic lesions.16 In paraneoplastic and autoimmune encephalopathies (excluding HE), MRI often shows nonenhancing signal intensity changes in the mesial temporal lobes.12 This patient had repeatedly normal MRI studies of the brain, which in combination with the history of tremor, myoclonus, seizures, and interval return to baseline status, helped point to the diagnosis of HE.
Different approaches to treatment of HE have been recommended. As the acronym SREAT suggests, patients typically respond dramatically to high‐dose steroid therapy. Although a number of patients also improve spontaneously, up to 60% of patients experience a relapsing course and require chronic immunosuppressive agents for maintenance therapy, including long‐term steroids and azathioprine.2, 17 Treatment with plasma exchange and intravenous immune globulin have also been reported, but with mixed results.18, 19 Due to her history of multiple relapses, the patient was placed on mycophenolate mofetil for additional maintenance immunosuppression, as her corticosteroid dose was reduced due to adverse effects.
Acute mental status change is a potentially emergent situation that must be evaluated with careful history and studies to exclude life‐threatening metabolic, infectious, and vascular conditions. This patient presented similarly on 2 prior occasions, and each time her physician team evaluated what appeared to be a new onset of altered consciousness, reaching a plausible but ultimately incorrect diagnosis. The patient's third presentation was finally the charm, as her physicians learned of the repeated history of a confusional state, and in particular the return to baseline status, allowing them to create a differential that focused on etiologies of relapsing encephalopathy and make the correct diagnosis.
Key Points
-
Recurrent acute or subacute cognitive deterioration invokes a differential diagnosis of toxic/metabolic disorders and unusual inflammatory conditions.
-
The nonvasculitic autoimmune encephalopathies are a group of uncommon conditions characterized by nonspecific findings of inflammation and generally unremarkable CNS imaging studies.
-
HE, or SREAT, is the most common of these conditions, and is notable for mental status changes, various findings of increased muscular tone, thyroid autoimmunity, and generally a dramatic response to corticosteroids.
- , , .Hashimoto's disease and encephalopathy.Lancet.1966;2:512–514.
- , , .Hashimoto encephalopathy: syndrome or myth?Arch Neurol.2003;60:164–171.
- , , .Pisani F. Recurrent status epilepticus as the main feature of Hashimoto's encephalopathy.Epilepsy Behav.2006;8:328–330.
- , , , et al.Steroid‐responsive encephalopathy associated with autoimmune thyroiditis.Arch Neurol.2006;63:197–202.
- , , , , .Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment.J Neurol.1996;243:585–593.
- , , , , .Hashimoto's encephalopathy: a steroid‐responsive disorder associated with high anti‐thyroid antibody titers‐report of 5 cases.Neurology.1991;41:228–233.
- , , , , .Hashimoto encephalopathy: a brainstem vasculitis?Neurology.2000;54:769–770.
- , , , , .Nonvasculitic autoimmune inflammatory meningoencephalitis (NAIM): A reversible form of encephalopathy.Neurology.1999;53:1579–1581.
- , , , .Hashimoto's encephalopathy: postmortem findings after fatal status epilepticus.Neurology.2003;61:1124–1126.
- , , .Autoimmune thyroiditis and a rapidly progressive dementia: global hypoperfusion on SPECT scanning suggests a possible mechanism.Neurology.1997;49:623–626.
- , , , , .Hashimoto's encephalopathy: clinical, SPECT and neurophysiologic data.QJM.2003;96:455–457.
- , , .Autoimmune Encephalopathies.The Neurologist.2007;13:140–147.
- , , , , , .Thyroid antigen‐antibody nephritis.Clin Immunol Immunopathol1976;6:341–346.
- , , .Immune complex glomerulonephritis mediated by thyroid antigens.Arch Pathol Lab Med1978;102:530–533.
- , , , et al.Diffusion‐weighted MR imaging of early‐stage Creutzfeldt‐Jakob disease: typical and atypical manifestations.Radiographics.2006;26:S191–S204.
- , , , , .CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.AJNR Am J Neuroradiol.1999;20:75–85.
- , .Long‐Term Treatment of Hashimoto's Encephalopathy.J Neuropsychiatry Clin Neurosci.2006;18:14–20.
- , .Hashimoto's encephalopathy: steroid resistance and response to intravenouc immunoglobulins.J Neurol Neurosurg Psychiatry.2005;76:455–456.
- , .Hashimoto's encephalopathy responding to plasmapheresis.J Neurol Neurosurg Psychiatry.2001;70:132.
A 58‐year old woman was brought to the emergency department with confusion. Her husband stated that for several hours she had been drifting in and out at home, and that he had to shout to get her attention. He described no seizure activity, weakness, incontinence, or difficulty speaking, and had noted no complaints of headache, fevers, chest pain, shortness of breath, or gastrointestinal complaints.
Altered mental status in a middle‐aged woman can result from a diverse set of etiologies. A key distinction in the neurological examination will be to assure that the complaint of confusion is accurate as opposed to aphasia; the former is usually indicative of diffuse cerebral dysfunction while the latter suggests a focal lesion in the dominant hemisphere.
The acuity of the change in mental status is important, as are the fluctuations described by the husband. Unwitnessed or nonconvulsive seizure activity can present this way. Toxic/metabolic etiologies, infectious and inflammatory disorders of the central nervous system (CNS), and vascular diseases are also important considerations. Although stroke does not typically present with global encephalopathy, intermittent large vessel occlusion, especially in the posterior circulation, can disrupt cognition in this manner. Following a physical examination, initial workup should focus on toxic/metabolic etiologies, followed rapidly by head imaging if no cause is identified.
Her past medical history was notable for type 2 diabetes mellitus, coronary artery disease, hyperlipidemia, and an unspecified seizure disorder, which according to her husband was diagnosed during a recent hospitalization for a similar presentation. She also had a remote history of venous thromboembolism and antithrombin‐III deficiency. She was unemployed, lived with her husband, and spent most of her time at home. She never smoked, and rarely drank alcohol. Her family history was unobtainable, and her husband denied that she used any illicit drugs. Her medications included pioglitazone, aspirin, simvastatin, pregabalin, ferrous sulfate, levetiracetam, warfarin, and magnesium oxide, and she was allergic to sulfa.
While the differential diagnosis remains broad, 3 elements of the history are potentially relevant. The history of epilepsy based on a similar prior presentation increases the likelihood that the current spell is ictal in nature; examination of previous records would be important in order to document whether these spells have indeed been proven to be epileptic, as many conditions can mimic seizures. Given the history of venous thromboembolism and hypercoagulability, one must consider cerebral venous sinus thrombosis, which can present with global neurologic dysfunction and seizures. Prompt identification, usually via computed tomography (CT) or magnetic resonance angiography, is vital, because anticoagulation can mitigate this potentially life‐threatening illness. Finally, although many medications can cause encephalopathy in overdose, levetiracetam has well‐described cognitive side effects even at usual doses, including encephalopathy, irritability, and depression.
The records from that recent hospitalization remarked that she had presented confused and stuporous. Her potassium had been 2.7 mmol/L, international normalized ration (INR) 3.4, and hemoglobin 8 g/dL; other routine laboratory studies were normal. CT and magnetic resonance imaging (MRI) of the brain had been negative, and electroencephalogram (EEG) reportedly was performed but specific results were unknown. She was discharged alert and oriented 1 week prior to the current presentation on the above medications, including levetiracetam for this newly‐diagnosed seizure disorder.
Previous records confirm that the current presentation is that of a relapsing acute alteration in mental status. Regardless of the EEG findings or response to antiepileptic medications, a seizure disorder should remain a primary consideration, although relapsing inflammatory, toxic/metabolic conditions, and, rarely, vascular disorders can also present in this manner.
The neurologic manifestations of hypokalemia are usually peripheral in nature, including periodic paralysis; confusion accompanying hypokalemia is usually not a result of the low potassium itself but rather due to an underlying toxic or endocrinologic cause. Various causes of anemia can lead to mental status changes; the mean corpuscular volume (MCV) will be particularly helpful given known associations between megaloblastic anemia and confusional states.
On examination, she appeared to be in good health and in no distress. She was afebrile. Her blood pressure was 93/57, pulse 90 beats per minute, respiratory rate 16 per minute, and room air oxygen saturation 100%. She was oriented to her surroundings, but slow in her responses to questioning. There were no cranial nerve, motor, or sensory deficits, or abnormal reflexes or movements. Examination of the head, skin, chest, cardiovascular system, abdomen, and extremities was normal. Serum sodium was 136 mmol/L, creatinine 1.2 mg/dL, calcium 9.3 mg/dL, and glucose 81 mg/dL; other routine blood chemistries were normal. Her white blood cell (WBC) count was 7100/L, hemoglobin 9.2 g/dL with normal MCV, and platelet count 275,000/L. INR was 3.4, and liver function tests were normal. CT of the brain demonstrated no evidence of acute pathology.
Given that her laboratory results (aside from the hemoglobin) and CT were essentially normal, the most common etiology of a recurrent encephalopathy would be a toxic exposure including drugs, alcohol, and environmental toxins or poisons. A comprehensive serum drug screen, including heavy metals, could follow a basic urinary screen for drugs of abuse; specific etiologies may be suggested by patterns of injury seen on MRI such as those seen with carbon monoxide or methanol exposure. Other recurrent metabolic processes include the porphyrias and relapsing inflammatory disorders, which could be entertained if further diagnostics are unrevealing.
An EEG is warranted at this point and is a test that is underutilized in the workup of altered mental status. Patients who have a spell and do not quickly awaken should be considered to be in nonconvulsive status epilepticus until proven otherwise. This can be easily identified on the EEG and is an important entity to recognize quickly. Additional findings on EEG may suggest focal cerebral dysfunction (such as that following a seizure or acute unilateral injury), diffuse encephalopathy (eg, triphasic waves), or fairly specific diagnoses (eg, periodic lateralized epileptiform discharges from the temporal lobes in suspected herpes simplex meningoencephalitis). While the CT of the brain is a reasonable initial screen, MRI is more sensitive for structural disease and should be obtained if no etiology is rapidly identified.
Finally, acute infectious etiologies such as abscess, encephalitis, or meningoencephalitis need to be excluded via lumbar puncture. Spinal fluid examination also can be helpful in the consideration of inflammatory and autoimmune disorders.
Over the next several hours, while still in the emergency department, she became increasingly obtunded, to the point that she was unresponsive to all stimuli. No seizure activity was witnessed, her vital signs were unchanged, and no medications had been administered. She was urgently transferred to a tertiary care center, where, at the time of arrival, she was obtunded and nonverbal, and opened her eyes only to noxious stimuli. She would withdraw all 4 extremities in response to pain. Pupils were 2 mm and symmetrically reactive. Corneal reflexes were normal, and her gag reflex was diminished. Motor tone was decreased in all 4 extremities. No fasciculations were noted. Deep tendon reflexes were present but symmetrically diminished throughout, and Babinski testing demonstrated a withdrawal response bilaterally.
Coma is a state of profound unconsciousness where the patient is unarousable and unaware of her surroundings. Coma can result either from bihemispheric dysfunction or diffuse injury to the reticular activating system in the brainstem, and the physical examination should focus on distinguishing between these 2 sites. Because the nuclei of cranial nerves III through XII (excepting XI) reside in the brainstem, the coma examination emphasizes testing the cranial nerves; although all cranial nerves are not tested in this patient, the ones that are appear to be normal, making bihemispheric dysfunction most likely. Bihemispheric coma most commonly results from diffuse toxic or metabolic etiologies such as intoxication or hepatic encephalopathy, but it can also be caused by bilateral structural lesions (including the bilateral thalami) or ongoing seizure activity.
Although an EEG remains the key test in this patient given her past history and an MRI would prove extremely useful, her deterioration warrants a workup for CNS infection. Since the head CT was negative, it would be prudent to proceed with urgent lumbar puncture (although it should never be performed in a patient with significant coagulopathy due to risks of hemorrhage leading to spinal cord injury). She should be covered empirically with broad spectrum meningeal‐dose antibiotics, including acyclovir, until the results of the spinal fluid examination are known, given that bacterial meningitis and herpes meningoencephalitis carry a high morbidity and mortality if not treated promptly.
Routine blood tests were similar to her labs at the referring emergency room. Ammonia level was 10 mol/L. Urine toxicology screen was negative, and blood tests for ethanol, salicylates, lithium, and acetaminophen were negative. Chest X‐ray and urinalysis were normal, and electrocardiogram was notable only for a sinus tachycardia. Cultures of the blood were obtained and the patient was admitted to the intensive care unit.
Levetiracetam, vancomycin, piperacillin‐tazobactam, and acyclovir were initiated. A lumbar puncture was performed without reversing the anticoagulation, and the procedure was traumatic. The cerebrospinal fluid was bloody, with a clear supernatant. Cell count demonstrated a red blood cell (RBC) count of 1250/L and a WBC count of 9/L, with a WBC differential of 42% neutrophils, 48% lymphocytes, and 8% monocytes. The cerebrospinal fluid (CSF) glucose was 62 mg/dL (with a serum glucose of 74 mg/dL) and protein 41 mg/dL. The CSF Gram stain demonstrated no organisms, and fluid was sent for routine culture and polymerase chain reaction (PCR) to detect herpes simplex virus (HSV). A neurology consultation was urgently requested.
As mentioned, it would have been more appropriate to reverse the patient's anticoagulation prior to lumbar puncture. The absence of xanthochromia suggests that the RBCs seen in the sample were introduced at the time of the lumbar puncture, arguing against a hemorrhagic disorder of the CNS (occasionally seen with herpes simplex encephalitis) or spinal fluid (eg, subarachnoid hemorrhage).
A reasonable rule of thumb to correct for the number of RBCs in a traumatic lumbar puncture is to allow 1 WBC for every 700 RBCs/L. Given this conversion, there are still too many WBCs in this sample, indicating a mild pleocytosis that is approximately one‐half neutrophilic and one‐half lymphocytic. This profile is nonspecific and can occur with a variety of conditions including stroke, seizure, inflammatory disorders, and infections, including viruses such as West Nile virus.
While coverage with acyclovir and broad‐spectrum antibacterials is appropriate, it should be noted that piperacillin‐tazobactam has poor CSF penetration and therefore is not a good choice for empiric coverage of CNS infections.
The neurologist's examination additionally noted multifocal myoclonus with noxious stimuli, most prominent in the face and toes. An urgent EEG demonstrated continuous, slow, generalized triphasic wave activity (Figures 1 and 2); no epileptiform discharges were seen. The erythrocyte sedimentation rate (ESR) was 66 mm/hour (normal, 0‐30), and tests for antinuclear antibodies, serum levetiracetam level, and thyroid function studies were ordered.
Stimulus‐evoked multifocal myoclonus is a general marker of encephalopathy found in many conditions, including hepatic and renal failure, drug intoxication (eg, opiates), neurodegenerative disorders (eg, Creutzfeldt‐Jakob disease [CJD]), and postanoxic injury, the latter of which is termed the Lance‐Adams syndrome.
Triphasic waves on EEG, while commonly associated with hepatic encephalopathy, have a similarly broad differential diagnosis, although in a comatose patient, they must first and foremost be distinguished from the repetitive discharges characteristic of nonconvulsive status epilepticus. In addition to hepatic and renal failure, triphasic waves have also been described in medication toxicity (especially with anticonvulsants, lithium, and cephalosporins), CNS infections (including Lyme disease and West Nile virus), strokes involving the bilateral thalami (usually from deep venous thrombosis), inflammatory disorders (such as Hashimoto's encephalopathy [HE]), and neurodegenerative diseases. It is important to remember that a single EEG does not exclude the possibility of an episodic ictal disorder and longer‐term monitoring would be required to definitively exclude seizures.
At this point, although the myoclonus and triphasic waves most commonly would indicate a toxic/metabolic process, the elevated ESR and CSF pleocytosis argue for an inflammatory or infectious condition. An MRI remains the next most useful test to guide further workup because many such conditions have distinct signatures on MRI.
The following day, she was noted to have periods of alertnessopening her eyes and following some commandsbut at other times she was difficult to arouse or obtunded. Tremulous movements and sporadic myoclonic jerks continued but no focal neurologic signs were found. Although there was increased muscle tone throughout, she was intermittently seen moving her limbs spontaneously, but not to command. No new findings were appreciated on routine laboratory tests. Antinuclear antibody testing was negative. Serum levetiracetam level was 23.5 g/mL (reference range, 545). Serum thyroid‐stimulating hormone was less than 0.005 U/mL, but free T3 was 3.5 pg/mL (normal, 1.8‐4.6) and free T4 was 2.0 ng/dL (normal, 0.71.8). An MRI of the brain was compromised by motion artifact but no significant abnormalities were appreciated.
At this point, a family member in another state disclosed that the patient had also been hospitalized 2 months previously while visiting him. Her chief complaint had been shortness of breath. The records were obtained; a cardiac catheterization had revealed nonobstructive coronary disease, and medical management was recommended. The notes mentioned that during the hospitalization she developed altered mental status with disorientation and shaking. CT and MRI of the brain had been unremarkable. The confusion was not explained, but she was discharged in good condition, alert and fully‐oriented.
The additional history confirms a relapsing encephalopathy, now with at least 3 occurrences. The most common etiologies in the face of a normal MRI and basic labs would be recurrent intoxication or exposures, but the inflammatory CSF profile and elevated ESR are not consistent with this. A variety of inflammatory disorders can present with recurrent encephalopathy, including demyelinating diseases and neurosarcoidosis. Some systemic rheumatologic conditions, such as systemic lupus erythematosus, can present with relapsing neurologic symptoms due to seizures, vasculitis, or cerebritis. Vasculitis would fit this picture as well, except for the normal findings on 2 MRIs. In a patient with such dramatic symptoms of neurologic dysfunction, one would expect to see changes on the MRI of cerebral inflammation with probable ischemia.
Therefore, given the CSF, ESR, clinical course, and unrevealing MRI and EEG, the most likely group of disorders responsible would be the nonvasculitic autoimmune meningoencephalitides, which present with recurrent encephalopathy and feature spontaneous remissions and/or often‐dramatic responses to corticosteroids. Key disorders in this category include Sjogren's disease, lupus, and steroid responsive encephalopathy associated with autoimmune thyroiditis (Hashimoto's encephalopathy). The latter condition is the most common of the group and is suggested by the abnormal thyroid‐stimulating hormone testing, although it may occur in the setting of normal thyroid function. The diagnosis can be confirmed with thyroperoxidase and thyroglobulin antibody testing.
Three days into the hospitalization, her mental status had gradually improved such that she was more consistently awake and oriented to person and place, and she was transferred to a regular nursing unit. Final results from the CSF and blood cultures were negative, as was PCR for HSV. The antimicrobials were discontinued. Routine serum chemistries continued to be unremarkable. Additional studies recommended by the neurologist demonstrated an antithyroperoxidase antibody concentration of 587.1 IU/mL (normal, <5), and antithyroglobulin antibody level of 52.2 IU/mL (normal, <10).
These results confirm the diagnosis of HE which, in addition to its presentation as a recurrent illness, is an important treatable cause of dementia and should be considered in young patients, those with autoimmune and thyroid disorders, and those whose dementia is rapidly progressive. Most cases are thought to be steroid‐responsive, but some studies have defined the disorder based on this responsiveness, resulting in some nonresponders likely being overlooked.
A trial of corticosteroids should be considered if the patient does not quickly return to baseline given the potential morbidities associated with prolonged altered mental status to this degree. Whether initiation of chronic immunosuppression could prevent these attacks in the future is unclear from the literature but should be considered given the recurrent, dramatic presentation in this patient.
A diagnosis of HE was made, and she was prescribed corticosteroids. Twenty‐four hours later, she was alert and fully‐oriented. She was discharged to home on prednisone and seen in follow‐up in neurology clinic 1 month later. She had had no further episodes of confusion or stupor, but because of steroid‐induced hyperglycemia, her corticosteroids were decreased and mycophenolate mofetil added for chronic immunosuppression. Four months after discharge she was neurologically stable but continued to struggle with the adverse effects of chronic corticosteroid treatment.
COMMENTARY
HE is an uncommon condition that can present with a rapidly progressive decline and should be considered in patients who present with recurrent mental status change in the setting of normal imaging studies and routine laboratory results. The entity was initially described by Lord William Russell Brain in 1966, and in the most recent terminology is known as steroid‐responsive encephalopathy associated with autoimmune thyroiditis (SREAT).1 It is characterized by an acute or subacute encephalopathy associated with thyroid autoimmunity. Patients typically present with fluctuating symptoms, episodes of confusion, alterations of consciousness, and rapid cognitive decline.2 Common features include myoclonus, tremor, ataxia, speech disturbance, stroke‐like episodes, increased muscle tone, neuropsychiatric manifestations, and seizures, that in some cases may progress to status epilepticus.3, 4
Although serum antithyroglobulin and antithyroperoxidase antibodies are elevated in HE, their presence is thought to be an epiphenomenon of the condition rather than the direct cause. Supporting this are the facts that the incidence of encephalopathy is not increased in patients with established autoimmune thyroiditis, and the presence of antithyroid antibodies ranges from 5% to 20% in the general population.2, 5 There is also no evidence that thyroid antibodies directly react with brain tissue, and the levels of these antibodies do not correlate with either neurologic manifestations or clinical improvement.2, 4, 5 As HE has been reported in patients with euthyroidism, hypothyroidism, and hyperthyroidism (with hypothyroidismeither subclinical or activemost common), it is also unlikely that the level of thyroid hormones play a role in the etiology of this disease.2, 4, 6
The etiology and pathogenesis of HE are unclear, although an immune‐mediated process is generally implicated, either from an inflammatory vasculitis or as a form of acute disseminated encephalomyelitis.7‐9 Global hypoperfusion on single‐photon emission computed tomography (SPECT) studies has also been reported.10, 11 Patients with HE may have nonspecific evidence of inflammation, including an elevated ESR, CRP, and CSF protein.12 Other laboratory abnormalities may include a mild elevation of liver aminotransferase levels; renal impairment has also been reported in a few cases of HE in the form of glomerulonephritis, and may be related to deposition of immune complexes containing thyroglobulin antigen.6, 12‐14 MRI of the brain is normal or nonspecific in most cases, and the EEG most commonly shows diffuse slowing.
The differential for a rapidly progressive cognitive decline includes CJD, CNS vasculitis, paraneoplastic syndromes, and autoimmune and subacute infectious encephalopathies. In patients with CJD, T2‐weighted imaging may show hyperintense signals in the basal ganglia, while diffusion‐weighted sequences may reveal changes in the cortical ribbon and bilateral thalami.15 In CNS vasculitis, the imaging findings are variable and range from discrete areas of vascular infarcts to hemorrhagic lesions.16 In paraneoplastic and autoimmune encephalopathies (excluding HE), MRI often shows nonenhancing signal intensity changes in the mesial temporal lobes.12 This patient had repeatedly normal MRI studies of the brain, which in combination with the history of tremor, myoclonus, seizures, and interval return to baseline status, helped point to the diagnosis of HE.
Different approaches to treatment of HE have been recommended. As the acronym SREAT suggests, patients typically respond dramatically to high‐dose steroid therapy. Although a number of patients also improve spontaneously, up to 60% of patients experience a relapsing course and require chronic immunosuppressive agents for maintenance therapy, including long‐term steroids and azathioprine.2, 17 Treatment with plasma exchange and intravenous immune globulin have also been reported, but with mixed results.18, 19 Due to her history of multiple relapses, the patient was placed on mycophenolate mofetil for additional maintenance immunosuppression, as her corticosteroid dose was reduced due to adverse effects.
Acute mental status change is a potentially emergent situation that must be evaluated with careful history and studies to exclude life‐threatening metabolic, infectious, and vascular conditions. This patient presented similarly on 2 prior occasions, and each time her physician team evaluated what appeared to be a new onset of altered consciousness, reaching a plausible but ultimately incorrect diagnosis. The patient's third presentation was finally the charm, as her physicians learned of the repeated history of a confusional state, and in particular the return to baseline status, allowing them to create a differential that focused on etiologies of relapsing encephalopathy and make the correct diagnosis.
Key Points
-
Recurrent acute or subacute cognitive deterioration invokes a differential diagnosis of toxic/metabolic disorders and unusual inflammatory conditions.
-
The nonvasculitic autoimmune encephalopathies are a group of uncommon conditions characterized by nonspecific findings of inflammation and generally unremarkable CNS imaging studies.
-
HE, or SREAT, is the most common of these conditions, and is notable for mental status changes, various findings of increased muscular tone, thyroid autoimmunity, and generally a dramatic response to corticosteroids.
A 58‐year old woman was brought to the emergency department with confusion. Her husband stated that for several hours she had been drifting in and out at home, and that he had to shout to get her attention. He described no seizure activity, weakness, incontinence, or difficulty speaking, and had noted no complaints of headache, fevers, chest pain, shortness of breath, or gastrointestinal complaints.
Altered mental status in a middle‐aged woman can result from a diverse set of etiologies. A key distinction in the neurological examination will be to assure that the complaint of confusion is accurate as opposed to aphasia; the former is usually indicative of diffuse cerebral dysfunction while the latter suggests a focal lesion in the dominant hemisphere.
The acuity of the change in mental status is important, as are the fluctuations described by the husband. Unwitnessed or nonconvulsive seizure activity can present this way. Toxic/metabolic etiologies, infectious and inflammatory disorders of the central nervous system (CNS), and vascular diseases are also important considerations. Although stroke does not typically present with global encephalopathy, intermittent large vessel occlusion, especially in the posterior circulation, can disrupt cognition in this manner. Following a physical examination, initial workup should focus on toxic/metabolic etiologies, followed rapidly by head imaging if no cause is identified.
Her past medical history was notable for type 2 diabetes mellitus, coronary artery disease, hyperlipidemia, and an unspecified seizure disorder, which according to her husband was diagnosed during a recent hospitalization for a similar presentation. She also had a remote history of venous thromboembolism and antithrombin‐III deficiency. She was unemployed, lived with her husband, and spent most of her time at home. She never smoked, and rarely drank alcohol. Her family history was unobtainable, and her husband denied that she used any illicit drugs. Her medications included pioglitazone, aspirin, simvastatin, pregabalin, ferrous sulfate, levetiracetam, warfarin, and magnesium oxide, and she was allergic to sulfa.
While the differential diagnosis remains broad, 3 elements of the history are potentially relevant. The history of epilepsy based on a similar prior presentation increases the likelihood that the current spell is ictal in nature; examination of previous records would be important in order to document whether these spells have indeed been proven to be epileptic, as many conditions can mimic seizures. Given the history of venous thromboembolism and hypercoagulability, one must consider cerebral venous sinus thrombosis, which can present with global neurologic dysfunction and seizures. Prompt identification, usually via computed tomography (CT) or magnetic resonance angiography, is vital, because anticoagulation can mitigate this potentially life‐threatening illness. Finally, although many medications can cause encephalopathy in overdose, levetiracetam has well‐described cognitive side effects even at usual doses, including encephalopathy, irritability, and depression.
The records from that recent hospitalization remarked that she had presented confused and stuporous. Her potassium had been 2.7 mmol/L, international normalized ration (INR) 3.4, and hemoglobin 8 g/dL; other routine laboratory studies were normal. CT and magnetic resonance imaging (MRI) of the brain had been negative, and electroencephalogram (EEG) reportedly was performed but specific results were unknown. She was discharged alert and oriented 1 week prior to the current presentation on the above medications, including levetiracetam for this newly‐diagnosed seizure disorder.
Previous records confirm that the current presentation is that of a relapsing acute alteration in mental status. Regardless of the EEG findings or response to antiepileptic medications, a seizure disorder should remain a primary consideration, although relapsing inflammatory, toxic/metabolic conditions, and, rarely, vascular disorders can also present in this manner.
The neurologic manifestations of hypokalemia are usually peripheral in nature, including periodic paralysis; confusion accompanying hypokalemia is usually not a result of the low potassium itself but rather due to an underlying toxic or endocrinologic cause. Various causes of anemia can lead to mental status changes; the mean corpuscular volume (MCV) will be particularly helpful given known associations between megaloblastic anemia and confusional states.
On examination, she appeared to be in good health and in no distress. She was afebrile. Her blood pressure was 93/57, pulse 90 beats per minute, respiratory rate 16 per minute, and room air oxygen saturation 100%. She was oriented to her surroundings, but slow in her responses to questioning. There were no cranial nerve, motor, or sensory deficits, or abnormal reflexes or movements. Examination of the head, skin, chest, cardiovascular system, abdomen, and extremities was normal. Serum sodium was 136 mmol/L, creatinine 1.2 mg/dL, calcium 9.3 mg/dL, and glucose 81 mg/dL; other routine blood chemistries were normal. Her white blood cell (WBC) count was 7100/L, hemoglobin 9.2 g/dL with normal MCV, and platelet count 275,000/L. INR was 3.4, and liver function tests were normal. CT of the brain demonstrated no evidence of acute pathology.
Given that her laboratory results (aside from the hemoglobin) and CT were essentially normal, the most common etiology of a recurrent encephalopathy would be a toxic exposure including drugs, alcohol, and environmental toxins or poisons. A comprehensive serum drug screen, including heavy metals, could follow a basic urinary screen for drugs of abuse; specific etiologies may be suggested by patterns of injury seen on MRI such as those seen with carbon monoxide or methanol exposure. Other recurrent metabolic processes include the porphyrias and relapsing inflammatory disorders, which could be entertained if further diagnostics are unrevealing.
An EEG is warranted at this point and is a test that is underutilized in the workup of altered mental status. Patients who have a spell and do not quickly awaken should be considered to be in nonconvulsive status epilepticus until proven otherwise. This can be easily identified on the EEG and is an important entity to recognize quickly. Additional findings on EEG may suggest focal cerebral dysfunction (such as that following a seizure or acute unilateral injury), diffuse encephalopathy (eg, triphasic waves), or fairly specific diagnoses (eg, periodic lateralized epileptiform discharges from the temporal lobes in suspected herpes simplex meningoencephalitis). While the CT of the brain is a reasonable initial screen, MRI is more sensitive for structural disease and should be obtained if no etiology is rapidly identified.
Finally, acute infectious etiologies such as abscess, encephalitis, or meningoencephalitis need to be excluded via lumbar puncture. Spinal fluid examination also can be helpful in the consideration of inflammatory and autoimmune disorders.
Over the next several hours, while still in the emergency department, she became increasingly obtunded, to the point that she was unresponsive to all stimuli. No seizure activity was witnessed, her vital signs were unchanged, and no medications had been administered. She was urgently transferred to a tertiary care center, where, at the time of arrival, she was obtunded and nonverbal, and opened her eyes only to noxious stimuli. She would withdraw all 4 extremities in response to pain. Pupils were 2 mm and symmetrically reactive. Corneal reflexes were normal, and her gag reflex was diminished. Motor tone was decreased in all 4 extremities. No fasciculations were noted. Deep tendon reflexes were present but symmetrically diminished throughout, and Babinski testing demonstrated a withdrawal response bilaterally.
Coma is a state of profound unconsciousness where the patient is unarousable and unaware of her surroundings. Coma can result either from bihemispheric dysfunction or diffuse injury to the reticular activating system in the brainstem, and the physical examination should focus on distinguishing between these 2 sites. Because the nuclei of cranial nerves III through XII (excepting XI) reside in the brainstem, the coma examination emphasizes testing the cranial nerves; although all cranial nerves are not tested in this patient, the ones that are appear to be normal, making bihemispheric dysfunction most likely. Bihemispheric coma most commonly results from diffuse toxic or metabolic etiologies such as intoxication or hepatic encephalopathy, but it can also be caused by bilateral structural lesions (including the bilateral thalami) or ongoing seizure activity.
Although an EEG remains the key test in this patient given her past history and an MRI would prove extremely useful, her deterioration warrants a workup for CNS infection. Since the head CT was negative, it would be prudent to proceed with urgent lumbar puncture (although it should never be performed in a patient with significant coagulopathy due to risks of hemorrhage leading to spinal cord injury). She should be covered empirically with broad spectrum meningeal‐dose antibiotics, including acyclovir, until the results of the spinal fluid examination are known, given that bacterial meningitis and herpes meningoencephalitis carry a high morbidity and mortality if not treated promptly.
Routine blood tests were similar to her labs at the referring emergency room. Ammonia level was 10 mol/L. Urine toxicology screen was negative, and blood tests for ethanol, salicylates, lithium, and acetaminophen were negative. Chest X‐ray and urinalysis were normal, and electrocardiogram was notable only for a sinus tachycardia. Cultures of the blood were obtained and the patient was admitted to the intensive care unit.
Levetiracetam, vancomycin, piperacillin‐tazobactam, and acyclovir were initiated. A lumbar puncture was performed without reversing the anticoagulation, and the procedure was traumatic. The cerebrospinal fluid was bloody, with a clear supernatant. Cell count demonstrated a red blood cell (RBC) count of 1250/L and a WBC count of 9/L, with a WBC differential of 42% neutrophils, 48% lymphocytes, and 8% monocytes. The cerebrospinal fluid (CSF) glucose was 62 mg/dL (with a serum glucose of 74 mg/dL) and protein 41 mg/dL. The CSF Gram stain demonstrated no organisms, and fluid was sent for routine culture and polymerase chain reaction (PCR) to detect herpes simplex virus (HSV). A neurology consultation was urgently requested.
As mentioned, it would have been more appropriate to reverse the patient's anticoagulation prior to lumbar puncture. The absence of xanthochromia suggests that the RBCs seen in the sample were introduced at the time of the lumbar puncture, arguing against a hemorrhagic disorder of the CNS (occasionally seen with herpes simplex encephalitis) or spinal fluid (eg, subarachnoid hemorrhage).
A reasonable rule of thumb to correct for the number of RBCs in a traumatic lumbar puncture is to allow 1 WBC for every 700 RBCs/L. Given this conversion, there are still too many WBCs in this sample, indicating a mild pleocytosis that is approximately one‐half neutrophilic and one‐half lymphocytic. This profile is nonspecific and can occur with a variety of conditions including stroke, seizure, inflammatory disorders, and infections, including viruses such as West Nile virus.
While coverage with acyclovir and broad‐spectrum antibacterials is appropriate, it should be noted that piperacillin‐tazobactam has poor CSF penetration and therefore is not a good choice for empiric coverage of CNS infections.
The neurologist's examination additionally noted multifocal myoclonus with noxious stimuli, most prominent in the face and toes. An urgent EEG demonstrated continuous, slow, generalized triphasic wave activity (Figures 1 and 2); no epileptiform discharges were seen. The erythrocyte sedimentation rate (ESR) was 66 mm/hour (normal, 0‐30), and tests for antinuclear antibodies, serum levetiracetam level, and thyroid function studies were ordered.
Stimulus‐evoked multifocal myoclonus is a general marker of encephalopathy found in many conditions, including hepatic and renal failure, drug intoxication (eg, opiates), neurodegenerative disorders (eg, Creutzfeldt‐Jakob disease [CJD]), and postanoxic injury, the latter of which is termed the Lance‐Adams syndrome.
Triphasic waves on EEG, while commonly associated with hepatic encephalopathy, have a similarly broad differential diagnosis, although in a comatose patient, they must first and foremost be distinguished from the repetitive discharges characteristic of nonconvulsive status epilepticus. In addition to hepatic and renal failure, triphasic waves have also been described in medication toxicity (especially with anticonvulsants, lithium, and cephalosporins), CNS infections (including Lyme disease and West Nile virus), strokes involving the bilateral thalami (usually from deep venous thrombosis), inflammatory disorders (such as Hashimoto's encephalopathy [HE]), and neurodegenerative diseases. It is important to remember that a single EEG does not exclude the possibility of an episodic ictal disorder and longer‐term monitoring would be required to definitively exclude seizures.
At this point, although the myoclonus and triphasic waves most commonly would indicate a toxic/metabolic process, the elevated ESR and CSF pleocytosis argue for an inflammatory or infectious condition. An MRI remains the next most useful test to guide further workup because many such conditions have distinct signatures on MRI.
The following day, she was noted to have periods of alertnessopening her eyes and following some commandsbut at other times she was difficult to arouse or obtunded. Tremulous movements and sporadic myoclonic jerks continued but no focal neurologic signs were found. Although there was increased muscle tone throughout, she was intermittently seen moving her limbs spontaneously, but not to command. No new findings were appreciated on routine laboratory tests. Antinuclear antibody testing was negative. Serum levetiracetam level was 23.5 g/mL (reference range, 545). Serum thyroid‐stimulating hormone was less than 0.005 U/mL, but free T3 was 3.5 pg/mL (normal, 1.8‐4.6) and free T4 was 2.0 ng/dL (normal, 0.71.8). An MRI of the brain was compromised by motion artifact but no significant abnormalities were appreciated.
At this point, a family member in another state disclosed that the patient had also been hospitalized 2 months previously while visiting him. Her chief complaint had been shortness of breath. The records were obtained; a cardiac catheterization had revealed nonobstructive coronary disease, and medical management was recommended. The notes mentioned that during the hospitalization she developed altered mental status with disorientation and shaking. CT and MRI of the brain had been unremarkable. The confusion was not explained, but she was discharged in good condition, alert and fully‐oriented.
The additional history confirms a relapsing encephalopathy, now with at least 3 occurrences. The most common etiologies in the face of a normal MRI and basic labs would be recurrent intoxication or exposures, but the inflammatory CSF profile and elevated ESR are not consistent with this. A variety of inflammatory disorders can present with recurrent encephalopathy, including demyelinating diseases and neurosarcoidosis. Some systemic rheumatologic conditions, such as systemic lupus erythematosus, can present with relapsing neurologic symptoms due to seizures, vasculitis, or cerebritis. Vasculitis would fit this picture as well, except for the normal findings on 2 MRIs. In a patient with such dramatic symptoms of neurologic dysfunction, one would expect to see changes on the MRI of cerebral inflammation with probable ischemia.
Therefore, given the CSF, ESR, clinical course, and unrevealing MRI and EEG, the most likely group of disorders responsible would be the nonvasculitic autoimmune meningoencephalitides, which present with recurrent encephalopathy and feature spontaneous remissions and/or often‐dramatic responses to corticosteroids. Key disorders in this category include Sjogren's disease, lupus, and steroid responsive encephalopathy associated with autoimmune thyroiditis (Hashimoto's encephalopathy). The latter condition is the most common of the group and is suggested by the abnormal thyroid‐stimulating hormone testing, although it may occur in the setting of normal thyroid function. The diagnosis can be confirmed with thyroperoxidase and thyroglobulin antibody testing.
Three days into the hospitalization, her mental status had gradually improved such that she was more consistently awake and oriented to person and place, and she was transferred to a regular nursing unit. Final results from the CSF and blood cultures were negative, as was PCR for HSV. The antimicrobials were discontinued. Routine serum chemistries continued to be unremarkable. Additional studies recommended by the neurologist demonstrated an antithyroperoxidase antibody concentration of 587.1 IU/mL (normal, <5), and antithyroglobulin antibody level of 52.2 IU/mL (normal, <10).
These results confirm the diagnosis of HE which, in addition to its presentation as a recurrent illness, is an important treatable cause of dementia and should be considered in young patients, those with autoimmune and thyroid disorders, and those whose dementia is rapidly progressive. Most cases are thought to be steroid‐responsive, but some studies have defined the disorder based on this responsiveness, resulting in some nonresponders likely being overlooked.
A trial of corticosteroids should be considered if the patient does not quickly return to baseline given the potential morbidities associated with prolonged altered mental status to this degree. Whether initiation of chronic immunosuppression could prevent these attacks in the future is unclear from the literature but should be considered given the recurrent, dramatic presentation in this patient.
A diagnosis of HE was made, and she was prescribed corticosteroids. Twenty‐four hours later, she was alert and fully‐oriented. She was discharged to home on prednisone and seen in follow‐up in neurology clinic 1 month later. She had had no further episodes of confusion or stupor, but because of steroid‐induced hyperglycemia, her corticosteroids were decreased and mycophenolate mofetil added for chronic immunosuppression. Four months after discharge she was neurologically stable but continued to struggle with the adverse effects of chronic corticosteroid treatment.
COMMENTARY
HE is an uncommon condition that can present with a rapidly progressive decline and should be considered in patients who present with recurrent mental status change in the setting of normal imaging studies and routine laboratory results. The entity was initially described by Lord William Russell Brain in 1966, and in the most recent terminology is known as steroid‐responsive encephalopathy associated with autoimmune thyroiditis (SREAT).1 It is characterized by an acute or subacute encephalopathy associated with thyroid autoimmunity. Patients typically present with fluctuating symptoms, episodes of confusion, alterations of consciousness, and rapid cognitive decline.2 Common features include myoclonus, tremor, ataxia, speech disturbance, stroke‐like episodes, increased muscle tone, neuropsychiatric manifestations, and seizures, that in some cases may progress to status epilepticus.3, 4
Although serum antithyroglobulin and antithyroperoxidase antibodies are elevated in HE, their presence is thought to be an epiphenomenon of the condition rather than the direct cause. Supporting this are the facts that the incidence of encephalopathy is not increased in patients with established autoimmune thyroiditis, and the presence of antithyroid antibodies ranges from 5% to 20% in the general population.2, 5 There is also no evidence that thyroid antibodies directly react with brain tissue, and the levels of these antibodies do not correlate with either neurologic manifestations or clinical improvement.2, 4, 5 As HE has been reported in patients with euthyroidism, hypothyroidism, and hyperthyroidism (with hypothyroidismeither subclinical or activemost common), it is also unlikely that the level of thyroid hormones play a role in the etiology of this disease.2, 4, 6
The etiology and pathogenesis of HE are unclear, although an immune‐mediated process is generally implicated, either from an inflammatory vasculitis or as a form of acute disseminated encephalomyelitis.7‐9 Global hypoperfusion on single‐photon emission computed tomography (SPECT) studies has also been reported.10, 11 Patients with HE may have nonspecific evidence of inflammation, including an elevated ESR, CRP, and CSF protein.12 Other laboratory abnormalities may include a mild elevation of liver aminotransferase levels; renal impairment has also been reported in a few cases of HE in the form of glomerulonephritis, and may be related to deposition of immune complexes containing thyroglobulin antigen.6, 12‐14 MRI of the brain is normal or nonspecific in most cases, and the EEG most commonly shows diffuse slowing.
The differential for a rapidly progressive cognitive decline includes CJD, CNS vasculitis, paraneoplastic syndromes, and autoimmune and subacute infectious encephalopathies. In patients with CJD, T2‐weighted imaging may show hyperintense signals in the basal ganglia, while diffusion‐weighted sequences may reveal changes in the cortical ribbon and bilateral thalami.15 In CNS vasculitis, the imaging findings are variable and range from discrete areas of vascular infarcts to hemorrhagic lesions.16 In paraneoplastic and autoimmune encephalopathies (excluding HE), MRI often shows nonenhancing signal intensity changes in the mesial temporal lobes.12 This patient had repeatedly normal MRI studies of the brain, which in combination with the history of tremor, myoclonus, seizures, and interval return to baseline status, helped point to the diagnosis of HE.
Different approaches to treatment of HE have been recommended. As the acronym SREAT suggests, patients typically respond dramatically to high‐dose steroid therapy. Although a number of patients also improve spontaneously, up to 60% of patients experience a relapsing course and require chronic immunosuppressive agents for maintenance therapy, including long‐term steroids and azathioprine.2, 17 Treatment with plasma exchange and intravenous immune globulin have also been reported, but with mixed results.18, 19 Due to her history of multiple relapses, the patient was placed on mycophenolate mofetil for additional maintenance immunosuppression, as her corticosteroid dose was reduced due to adverse effects.
Acute mental status change is a potentially emergent situation that must be evaluated with careful history and studies to exclude life‐threatening metabolic, infectious, and vascular conditions. This patient presented similarly on 2 prior occasions, and each time her physician team evaluated what appeared to be a new onset of altered consciousness, reaching a plausible but ultimately incorrect diagnosis. The patient's third presentation was finally the charm, as her physicians learned of the repeated history of a confusional state, and in particular the return to baseline status, allowing them to create a differential that focused on etiologies of relapsing encephalopathy and make the correct diagnosis.
Key Points
-
Recurrent acute or subacute cognitive deterioration invokes a differential diagnosis of toxic/metabolic disorders and unusual inflammatory conditions.
-
The nonvasculitic autoimmune encephalopathies are a group of uncommon conditions characterized by nonspecific findings of inflammation and generally unremarkable CNS imaging studies.
-
HE, or SREAT, is the most common of these conditions, and is notable for mental status changes, various findings of increased muscular tone, thyroid autoimmunity, and generally a dramatic response to corticosteroids.
- , , .Hashimoto's disease and encephalopathy.Lancet.1966;2:512–514.
- , , .Hashimoto encephalopathy: syndrome or myth?Arch Neurol.2003;60:164–171.
- , , .Pisani F. Recurrent status epilepticus as the main feature of Hashimoto's encephalopathy.Epilepsy Behav.2006;8:328–330.
- , , , et al.Steroid‐responsive encephalopathy associated with autoimmune thyroiditis.Arch Neurol.2006;63:197–202.
- , , , , .Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment.J Neurol.1996;243:585–593.
- , , , , .Hashimoto's encephalopathy: a steroid‐responsive disorder associated with high anti‐thyroid antibody titers‐report of 5 cases.Neurology.1991;41:228–233.
- , , , , .Hashimoto encephalopathy: a brainstem vasculitis?Neurology.2000;54:769–770.
- , , , , .Nonvasculitic autoimmune inflammatory meningoencephalitis (NAIM): A reversible form of encephalopathy.Neurology.1999;53:1579–1581.
- , , , .Hashimoto's encephalopathy: postmortem findings after fatal status epilepticus.Neurology.2003;61:1124–1126.
- , , .Autoimmune thyroiditis and a rapidly progressive dementia: global hypoperfusion on SPECT scanning suggests a possible mechanism.Neurology.1997;49:623–626.
- , , , , .Hashimoto's encephalopathy: clinical, SPECT and neurophysiologic data.QJM.2003;96:455–457.
- , , .Autoimmune Encephalopathies.The Neurologist.2007;13:140–147.
- , , , , , .Thyroid antigen‐antibody nephritis.Clin Immunol Immunopathol1976;6:341–346.
- , , .Immune complex glomerulonephritis mediated by thyroid antigens.Arch Pathol Lab Med1978;102:530–533.
- , , , et al.Diffusion‐weighted MR imaging of early‐stage Creutzfeldt‐Jakob disease: typical and atypical manifestations.Radiographics.2006;26:S191–S204.
- , , , , .CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.AJNR Am J Neuroradiol.1999;20:75–85.
- , .Long‐Term Treatment of Hashimoto's Encephalopathy.J Neuropsychiatry Clin Neurosci.2006;18:14–20.
- , .Hashimoto's encephalopathy: steroid resistance and response to intravenouc immunoglobulins.J Neurol Neurosurg Psychiatry.2005;76:455–456.
- , .Hashimoto's encephalopathy responding to plasmapheresis.J Neurol Neurosurg Psychiatry.2001;70:132.
- , , .Hashimoto's disease and encephalopathy.Lancet.1966;2:512–514.
- , , .Hashimoto encephalopathy: syndrome or myth?Arch Neurol.2003;60:164–171.
- , , .Pisani F. Recurrent status epilepticus as the main feature of Hashimoto's encephalopathy.Epilepsy Behav.2006;8:328–330.
- , , , et al.Steroid‐responsive encephalopathy associated with autoimmune thyroiditis.Arch Neurol.2006;63:197–202.
- , , , , .Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment.J Neurol.1996;243:585–593.
- , , , , .Hashimoto's encephalopathy: a steroid‐responsive disorder associated with high anti‐thyroid antibody titers‐report of 5 cases.Neurology.1991;41:228–233.
- , , , , .Hashimoto encephalopathy: a brainstem vasculitis?Neurology.2000;54:769–770.
- , , , , .Nonvasculitic autoimmune inflammatory meningoencephalitis (NAIM): A reversible form of encephalopathy.Neurology.1999;53:1579–1581.
- , , , .Hashimoto's encephalopathy: postmortem findings after fatal status epilepticus.Neurology.2003;61:1124–1126.
- , , .Autoimmune thyroiditis and a rapidly progressive dementia: global hypoperfusion on SPECT scanning suggests a possible mechanism.Neurology.1997;49:623–626.
- , , , , .Hashimoto's encephalopathy: clinical, SPECT and neurophysiologic data.QJM.2003;96:455–457.
- , , .Autoimmune Encephalopathies.The Neurologist.2007;13:140–147.
- , , , , , .Thyroid antigen‐antibody nephritis.Clin Immunol Immunopathol1976;6:341–346.
- , , .Immune complex glomerulonephritis mediated by thyroid antigens.Arch Pathol Lab Med1978;102:530–533.
- , , , et al.Diffusion‐weighted MR imaging of early‐stage Creutzfeldt‐Jakob disease: typical and atypical manifestations.Radiographics.2006;26:S191–S204.
- , , , , .CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.AJNR Am J Neuroradiol.1999;20:75–85.
- , .Long‐Term Treatment of Hashimoto's Encephalopathy.J Neuropsychiatry Clin Neurosci.2006;18:14–20.
- , .Hashimoto's encephalopathy: steroid resistance and response to intravenouc immunoglobulins.J Neurol Neurosurg Psychiatry.2005;76:455–456.
- , .Hashimoto's encephalopathy responding to plasmapheresis.J Neurol Neurosurg Psychiatry.2001;70:132.
A Rash Decision
A 38‐year‐old HIV+ Ohio man with a recent CD4+ count of 534 cells/mL presented to his physician with 3 weeks of fever as high as 102F. He noted mild myalgias, pruritus, and an occasional cough but no headache, sore throat, dyspnea, rash, or gastrointestinal or genitourinary complaints. He had been seen elsewhere 2 weeks previously, when he had reported a single episode of receptive oral sex with a male partner several weeks earlier. He had been prescribed ciprofloxacin and azithromycin, but a throat swab came back negative for Chlamydia and Neisseria gonorrhoeae, and he reported no change in his symptoms after the course of antibiotics. He denied smoking or using street drugs. His only medications were citalopram and trazodone for depression.
This is a HIV+ man with a mild degree of immunosuppression with a fever of unknown origin (FUO). It is not yet known if the requisite basic infectious evaluation has been completed to meet this definition, but the duration certainly qualifies, and regardless of semantics, the FUO framework is a helpful starting point. The primary considerations in FUO are infections, neoplasms, and autoimmune illnesses. Autoimmune diseases are relatively less common in HIV patients. Although pruritis is quite common in HIV alone, it may also herald renal failure, cholestasis, or a malignancy (usually hematologic). Drugs must also be considered as a cause of unexplained fever; the pruritis might suggest an allergic reaction, although I do not think of citalopram or trazodone as having this effect. The failure to respond to broad‐spectrum antimicrobials (along with the duration) lowers my suspicion for common infections such as pneumonia, urinary tract infection, or cellulitis. Among sexually transmitted diseases, syphilis can be protean and merits consideration.
On examination he appeared well. His temperature was 102.4F, pulse 111 beats/min, blood pressure 138/78 mm Hg. The head, neck, cardiovascular system, and lungs appeared normal on examination. The abdomen was soft and nontender without organomegaly; skin, extremities, and neurological system were unremarkable. Rectal examination showed small anal condylomata. Hemoglobin was 14.3 g/dL, white blood cell count 6200/cm3, and platelet count 230,000/cm3. Serum electrolytes and lactate dehydrogenase were normal. The results of his liver function tests (LFTs) demonstrated a serum aspartate transaminase of 60 U/L (normal, 7‐40 U/L), alanine transaminase of 125 U/L (normal, 5‐50 U/L), alkaline phosphatase 218 U/L (normal, 40‐150 U/L), and total bilirubin 2.1 mg/dL (normal, 0.0‐1.5 mg/dL). Urinalysis demonstrated 2+ bilirubin and was otherwise normal. His erythrocyte sedimentation rate was 32 mm/hr (normal, 0‐15 mm/hr).
After 3 weeks of illness, his CBC demonstrates no signs of chronic illness (such as anemia of a chronic disease or a reactive leukocytosis or thrombocytosis). The results of his liver function tests showed moderate elevation, slightly more cholestatic than hepatocellular. This finding may reflect a disease process involving the liver, but such abnormal findings are often nonspecific in acute and chronic illnesses. With an unremitting fever, infectious complications in the liver merit early consideration. The time course rules out common biliary disorders such as cholangitis or cholecystitis. Pyogenic or amoebic liver abscesses are possible (homosexual men are at increased risk for the latter), but the absence of pain or abdominal tenderness is atypical. This biochemical profile can also be seen in chronic (but not acute) viral infections of the liver. Chronic hepatitis B and C predispose to hepatocellular carcinoma (HCC), which can be associated with fever. Cancers that infiltrate the liver, such as lymphoma or carcinoma, could also account for this picture. Indolent infections such as tuberculosis (TB) and syphilis are also possible, so associated signs of these systemic diseases should be sought. I do not believe either of his antibiotics is commonly associated with LFT abnormalities, and his CD4 count is too high for HIV cholangiopathy. In sum, a host of liver diseases are possible, but an extrahepatic systemic disease deserves equal attention.
His CD4+ count was 537 cells/mL, and his HIV RNA viral load was 44,300 copies/mL. Radiographs of the chest were normal. Two sets of blood cultures were negative. The rapid plasma reagin (RPR) was nonreactive. The results of serologies for acute hepatitis A, B, C, and E, chronic hepatitis B and C, and toxoplasmosis were negative. Testing for both Epstein‐Barr virus and cytomegalovirus showed evidence of remote infection. Results of serologies for bartonella species, human herpesviruses 6 and 7, and parvovirus B19 were negative.
The negative RPR makes disseminated (secondary) syphilis improbable, provided the prozone phenomenon has been excluded. An extensive serological workup is common in the evaluation of fever of unknown origin, although the threat of false‐positive results always looms when many studies are sent simultaneously. This must be considered in advance here, as his relatively preserved CD4 count affords him significant protection against many opportunistic infections. His HIV infection, however, regardless of CD4 count, increases his risk for TB and lymphoma, which remain high on my list. Both may be residing primarily in the liver. In FUO, the abdominal CT is frequently a high‐yield test (primarily by demonstrating unsuspected tumors and abscesses), even in the absence of symptoms, and would certainly be of interest here given the liver function test results. Imaging could diagnose febrile tumors such as lymphoma, HCC, or renal cell carcinoma. In the event that imaging is unrevealing, causes of granulomatous hepatitis should be entertained. The constellation of cough, LFT abnormalities, and fever is compatible with Q fever. As with any FUO case, I would also carefully revisit this patient's history to discern where he was born, where he has been, and what activities or exposures he is engaged in.
He was seen 2 days later with fever of 104F and new papules over his sternal area. Over the next week, he had intermittent fevers and severe fatigue. The rash progressed, predominantly involving his chest and back, but also his legs, arms, and face (see Fig. 1). The lesions spared his palms and soles. The exanthem was intensely pruritic and maculopapular, consisting of lesions with a diameter of 0.5 cm or less, with some scaling. There were no vesicles or pustular lesions. There were no other new findings on examination. His transaminase and bilirubin had normalized, and his CBC and electrolytes were unchanged. Repeat blood cultures held for extended incubation were negative. Computerized tomography of the chest, abdomen, and pelvis demonstrated mild lymphadenopathy at the porta hepatis with increased portocaval and periaortic lymphadenopathy.
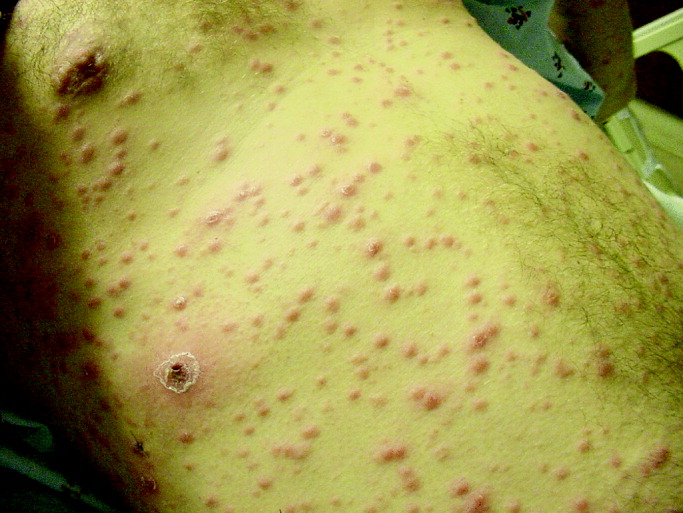
The only LFT abnormality that persists is the elevated alkaline phosphatase, which suggests (1) that liver involvement was not specific and that there is a disease process involving the bone, (2) that there is a persistent infiltrative disorder of the liver such as infection or malignancy or, less likely, amyloidosis or sarcoidosis, or (3) that the porta hepatis lymphadenopathy is causing biliary obstruction. The underlying diagnosis must explain the rash, intraabdominal lymphadenopathy, and fever. The time course does somewhat limit the extensive differential of fever and rash. After 3 weeks of illness, some of the most life‐threatening entities such as meningococcal disease, Rocky Mountain spotted fever, and toxic shock syndrome are unlikely. Concern remains for infections that are more indolent, such as mycobacteria, fungi, or spirochetes. The most striking elements of the rash are the extensive distribution, rapid progression, large number, and discreteness of the lesions, which collectively point more toward disseminated fungal (eg, histoplasmosis, as he lives in Ohio), spirochetal, rickettsial, or viral etiologies, rather than bacterial or mycobacterial entities. The absence of vesicles detracts from the diagnosis of a disseminated herpes virus such as herpes simplex or varicella. I believe that this rash is too disseminated to be caused by a common mycobacterial illness. This extent of cutaneous metastases would usually accompany a far more ill patient with an obvious primary cancer (none is seen on imaging, including the liver), and it appears too extensive to be caused by a paraneoplastic phenomenon such as Sweet's syndrome. A systemic vasculitis or another autoimmune disease remains possible, but there is minimal evidence of visceral organ involvement. All the aforementioned diseases could explain the intraabdominal lymphadenopathy, but my suspicion is highest for infection. I would biopsy and culture the skin lesions, repeat the RPR and/or send a treponemal‐specific test, place a PPD skin test, and send fungal studies (serum serologies and urine antigens) for evaluation. If the results of these noninvasive studies are unrevealing, I would consider a liver biopsy.
The patient's medications were discontinued, and a skin biopsy of the rash from his chest showed atypical lymphohistiocytic infiltrates without acute inflammatory cells and with negative Gomori methenamine silver (GMS), acid‐fast bacilli (AFB), and Fite (for Nocardia) stains. The infiltrates were predominantly T cells with a 1:1 CD4:CD8 ratio. This was read as suspicious for cytotoxic (CD8) mycosis fungoides.
I do not have reason to doubt the pathologist's impression of mycosis fungoides on histopathologic grounds, but from a clinical standpoint, I do not think mycosis fungoides is a disease that has a prolonged febrile prodrome or an explosive cutaneous onset. Rather, it is frequently preceded by nonspecific skin findings over a long period. Thinking broadly and pathophysiologically and noting that T cells are the predominant lymphocytes in skin, I wonder if they could represent a nonmalignant, immunological reaction in the skin. The stains, although not perfectly sensitive, make mycobacterial and fungal diseases less likely, although incubation of cultures is necessary.
Over the next 10 days (bringing the total duration of the patient's illness to 6 weeks), the skin lesions increased in number. In the physician's office at his next follow‐up, the patient had a temperature of 104.1F, was uncomfortable, shivering, and ill‐appearing. His blood pressure was 108/66 mm Hg, and his pulse 114 beats/min. He complained of severe shooting pains, predominantly in his pretibial regions and arms. Examination showed no other new findings, including no focal neurological findings. The results of the T‐cell rearrangement study from the skin biopsy showed evidence of a monoclonal T‐cell population. He was admitted to the hospital for further evaluation and treatment.
The extremity dysesthesias could represent a lesion of the spinal cord (including the CSF/meninges), a polyradiculopathy, or a polyneuropathy. Unfortunately, this does not add a tremendous amount of diagnostic resolution, as infection, malignancy, and autoimmune syndromes, such as vasculitis, may all involve the nervous system in these ways. In general, I associate monoclonal lymphocyte responses with hematological malignancies and polyclonal responses with the less specific inflammation that could accompany infection, autoimmunity, or solid malignancies. His age, fever, and rapid progression seem atypical for mycosis fungoides, but given the monoclonal T cells, this must now be considered. Adult T‐cell leukemia/lymphoma, with its prominent skin manifestations and its association with HLTV‐1, is an alternative T‐cell malignancy that could explain the fever, neurological symptoms, and possible visceral involvement (elevated alkaline phosphatase, which could reflect liver or bone). In cases that are diagnostic challenges, one of the highest‐yield maneuvers is to repeat the preceding evaluation, starting with the history, exam, and basic labs, and if necessary, to review or repeat the imaging or skin biopsy. Given the elevated alkaline phosphatase, disseminated rash, new neurological symptoms, and his HIV status, I remain particularly concerned about syphilis and would do further testing (accounting for the prozone phenomenon) before proceeding with the malignancy evaluation.
A lumbar puncture demonstrated clear cerebrospinal fluid, with 2 leukocytes and 195 erythrocytes/cm3, protein of 26 mg/dL, and glucose of 52 mg/dL. Bacterial and fungal cultures of the fluid were negative. The results of colonoscopy were normal. A bone marrow biopsy demonstrated ring granulomas. GMS, AFB, Fite, and Steiner (for spirochetes) stains were negative, cultures of the aspirate were negative for bacteria, and smears were negative for fungi and mycobacteria. Antibody tests for human T‐cell lymphotropic virus types I and II, Coxiella burnetii, and Bartonella henselae were negative. The dermatology consultant believed the absence of lymphadenopathy and the pruritic nature of the lesions was atypical for cytotoxic T‐cell lymphoma (CTCL). Before initiating therapy for CTCL, she suggested repeating the skin biopsy and RPR.
The repeat RPR was positive at 1:64 dilutions, and a confirmatory fluorescent treponemal antibody absorption test showed a positive result. He was prescribed intramuscular benzathine pencillin 2.4 million units weekly for 3 weeks, with almost immediate defervescence and slower resolution of his rash and shooting pains in his limbs. The repeat skin biopsy done during the hospitalization demonstrated lichenoid‐type dermatitis with interstitial and perivascular lymphohistiocytic infiltrates and granulomas. Steiner stains for spirochetes were positive. Immunohistochemical stains ruled out a lymphoproliferative process. One year later his RPR was nonreactive.
COMMENTARY
Fever of unknown origin (FUO) was first defined by Petersdorf and Beeson in 1961 as a temperature higher than 38.3C on several occasions lasting longer than 3 weeks and defying diagnosis despite 1 week of inpatient investigation.1 Dramatic changes in medical practice have rendered this definition outdated, with more recent proposals allowing thoughtful outpatient investigation to serve as a surrogate for hospitalization. Some have proposed that HIV‐associated FUO be considered a distinct entity, with the most complete North American series finding the etiology of the HIV‐associated FUO in 56 of 70 patients.2 The mean CD4+ count in this series was 58/cm3. Disseminated M. avium was the most frequently diagnosed cause, followed by P. jirovecii pneumonia, cytomegalovirus infection, disseminated histoplasmosis, and lymphoma. Of 14 patients with fever of no definable etiology, 12 eventually proved to have self‐limiting illness.
Despite numerous attempts to reduce the investigation of the patient with FUO to an algorithm, the approach must be individualized. A thorough history and careful, serial physical examinations are frequently and appropriately stressed as the foundation, followed by thoughtful selection of laboratory and imaging studies. Although FUO has a lengthy differential diagnosis, it often proves to be, as Mackowiak and Durack stress, an unusual manifestation of a common disease, rather than a typical presentation of a rare disease.3 A relatively uncommon disease in conjunction with an initially negative diagnostic test result, as was the case with this patient, may lead to a protracted diagnostic puzzle.
Syphilis is a rare cause of FUO. In 6 large studies of a total of 947 patients published over a 40‐year period, only 2 cases of syphilis (1 secondary and 1 neurosyphilis) were reported.1, 48 Syphilis as a cause of prolonged cryptic fever appears to have been seen with greater frequency in the preantibiotic era.9 In the first half of the 20th century, syphilis was known as the great imitator, with its unusual manifestations recognized and indeed expected. As a result of the dramatically lower incidence of syphilis in recent decades, these lessons have largely been forgotten, however, which may lead to diagnostic confusion when syphilis presents atypically. The manifestations of secondary syphilis are protean, including a variety of rashes, aphthous ulcers, arthralgias, pharyngitis, weight loss, fever, meningitis, ocular symptoms, cranial nerve palsies, glomerulonephritis, hepatitis, and periostitis (which afflicted this patient, who complained of severe shooting pains in his arms and shins).
After declining in the last decade of the 20th century, the rates of primary and secondary syphilis are rising in the United States.10 Oral sex is a clear risk factor for syphilis transmission, particularly for men who have sex with men.11 Because of the patient's exposure history and clinical picture, his outpatient physician considered the diagnosis of secondary syphilis early in the course of his illness. The diagnosis was not entertained further when an RPR test, highly sensitive at this stage of the disease, returned nonreactive. Likewise, when a rash subsequently appeared, the lack of palm and sole involvement dissuaded multiple clinicians from reconsidering the diagnosis of syphilis. A skin biopsy that appeared to lead in a distinctly different direction understandably confused the picture still further. Even at the time of the lumbar puncture, VDRL of the CSF was not ordered.
In retrospect, the chief confounder in the case was the false‐negative RPR test, as the discussant suspected early on. Although nontreponemal tests are generally accurate in individuals with HIV, delayed seropositivity and false‐negatives have been reported in this population.12 The false‐negative could have also been a result of the prozone phenomenon, an unusual event, occurring in fewer than 2% of cases of secondary syphilis and attributed to a mismatch between antibody and very high antigen level. The prozone reaction can be corrected for by requesting dilution of the serum prior to repeating the test. Simple lab error must be considered as well, but without access to this patient's serum from his original testing, the cause of his initial false‐negative test cannot be known with certainty.
An unusual presentation in conjunction with failure to recognize the causes of rare false‐negative testing for secondary syphilis led to a delayed diagnosis in this patient. Although syphilis and mycosis fungoides have previously been reported to mimic one another both clinically and histopathologically, the potential for secondary syphilis to be misdiagnosed in this fashion is not generally appreciated.1315 Recognition of the possibility of secondary syphilis occurred just in time to spare this patient the rash decision of treating him with cytotoxic therapy directed against CTCL.
Teaching Points
-
HIV‐associated FUO can be a diagnostic challenge, but an etiology can be found in most cases.
-
Syphilis continues to be an unusual cause of FUO and can have protean manifestations affecting nearly every organ system
-
The sensitivity of RPR is extremely high in secondary syphilis, but false‐negative tests can be seen in HIV because of both the prozone phenomenon and a delayed rise in antibodies.
- ,.Fever of unexplained origin: Report on 100 cases.Medicine.1961;40:1–30.
- ,,.Human immunodeficiency virus‐associated fever of unknown origin: A study of 70 patients in the United States and review.Clin Infect Dis.1999;28:341–345.
- ,.Fever of unknown origin. In:Mandell GL,Bennett JE,Dolin R, eds.Principles and Practice of Infectious Diseases.6th ed.Philadelphia:Elsevier Churchill Livingstone;2005:718–729.
- ,,.Fever of unknown origin: Diagnosis and follow‐up of 105 cases, 1970‐1980.Medicine.1982;61:269–292.
- ,,,.Fever of unknown origin in the 1980s: An update of the diagnostic spectrum.Arch Intern Med.1992;152:51–55.
- .Fever of unknown origin: Review of 86 patients treated in community hospitals.Clin Infect Dis.1992;15:968–973.
- ,,.Fever of unknown origin (FUO). I. A prospective multicenter study of 167 patients with FUO, using fixed epidemiologic entry criteria. The Netherlands FUO study group.Medicine.1997;76:392–400.
- ,,, et al.From prolonged febrile illness to fever of unknown origin: The challenge continues.Arch Intern Med.2003;163:1033–1041.
- ,.The diagnosis of obscure fever. II. The diagnosis of unexplained high fever.Bull Johns Hopkins Hosp.1936;58:307–331.
- Centers for Disease Control and Prevention.Primary and secondary syphilis—United States, 2003–2004.MMWR.2006;55:269–273.
- Transmission of primary and secondary syphilis by oral sex—Chicago, Illinois, 1998‐2202.MMWR.2004;53:966–968.
- ,,, et al.Seronegative secondary syphilis in 2 patients coinfected with human immunodeficiency virus.Arch Dermatol.2005;141:431–433.
- ,,,.Secondary syphilis mimicking mycosis fungoides.J Am Acad Dermatol.1980;3:92–94
- ,.A case of lues maligna in a patient with acquired immunodeficiency syndrome (AIDS).Scand J Infect Dis.2005;37:697–700.
- ,,,,.Unusual presentation of secondary syphilis in 2 HIV‐1 positive patients.Cutis.2000;66:383–389.
A 38‐year‐old HIV+ Ohio man with a recent CD4+ count of 534 cells/mL presented to his physician with 3 weeks of fever as high as 102F. He noted mild myalgias, pruritus, and an occasional cough but no headache, sore throat, dyspnea, rash, or gastrointestinal or genitourinary complaints. He had been seen elsewhere 2 weeks previously, when he had reported a single episode of receptive oral sex with a male partner several weeks earlier. He had been prescribed ciprofloxacin and azithromycin, but a throat swab came back negative for Chlamydia and Neisseria gonorrhoeae, and he reported no change in his symptoms after the course of antibiotics. He denied smoking or using street drugs. His only medications were citalopram and trazodone for depression.
This is a HIV+ man with a mild degree of immunosuppression with a fever of unknown origin (FUO). It is not yet known if the requisite basic infectious evaluation has been completed to meet this definition, but the duration certainly qualifies, and regardless of semantics, the FUO framework is a helpful starting point. The primary considerations in FUO are infections, neoplasms, and autoimmune illnesses. Autoimmune diseases are relatively less common in HIV patients. Although pruritis is quite common in HIV alone, it may also herald renal failure, cholestasis, or a malignancy (usually hematologic). Drugs must also be considered as a cause of unexplained fever; the pruritis might suggest an allergic reaction, although I do not think of citalopram or trazodone as having this effect. The failure to respond to broad‐spectrum antimicrobials (along with the duration) lowers my suspicion for common infections such as pneumonia, urinary tract infection, or cellulitis. Among sexually transmitted diseases, syphilis can be protean and merits consideration.
On examination he appeared well. His temperature was 102.4F, pulse 111 beats/min, blood pressure 138/78 mm Hg. The head, neck, cardiovascular system, and lungs appeared normal on examination. The abdomen was soft and nontender without organomegaly; skin, extremities, and neurological system were unremarkable. Rectal examination showed small anal condylomata. Hemoglobin was 14.3 g/dL, white blood cell count 6200/cm3, and platelet count 230,000/cm3. Serum electrolytes and lactate dehydrogenase were normal. The results of his liver function tests (LFTs) demonstrated a serum aspartate transaminase of 60 U/L (normal, 7‐40 U/L), alanine transaminase of 125 U/L (normal, 5‐50 U/L), alkaline phosphatase 218 U/L (normal, 40‐150 U/L), and total bilirubin 2.1 mg/dL (normal, 0.0‐1.5 mg/dL). Urinalysis demonstrated 2+ bilirubin and was otherwise normal. His erythrocyte sedimentation rate was 32 mm/hr (normal, 0‐15 mm/hr).
After 3 weeks of illness, his CBC demonstrates no signs of chronic illness (such as anemia of a chronic disease or a reactive leukocytosis or thrombocytosis). The results of his liver function tests showed moderate elevation, slightly more cholestatic than hepatocellular. This finding may reflect a disease process involving the liver, but such abnormal findings are often nonspecific in acute and chronic illnesses. With an unremitting fever, infectious complications in the liver merit early consideration. The time course rules out common biliary disorders such as cholangitis or cholecystitis. Pyogenic or amoebic liver abscesses are possible (homosexual men are at increased risk for the latter), but the absence of pain or abdominal tenderness is atypical. This biochemical profile can also be seen in chronic (but not acute) viral infections of the liver. Chronic hepatitis B and C predispose to hepatocellular carcinoma (HCC), which can be associated with fever. Cancers that infiltrate the liver, such as lymphoma or carcinoma, could also account for this picture. Indolent infections such as tuberculosis (TB) and syphilis are also possible, so associated signs of these systemic diseases should be sought. I do not believe either of his antibiotics is commonly associated with LFT abnormalities, and his CD4 count is too high for HIV cholangiopathy. In sum, a host of liver diseases are possible, but an extrahepatic systemic disease deserves equal attention.
His CD4+ count was 537 cells/mL, and his HIV RNA viral load was 44,300 copies/mL. Radiographs of the chest were normal. Two sets of blood cultures were negative. The rapid plasma reagin (RPR) was nonreactive. The results of serologies for acute hepatitis A, B, C, and E, chronic hepatitis B and C, and toxoplasmosis were negative. Testing for both Epstein‐Barr virus and cytomegalovirus showed evidence of remote infection. Results of serologies for bartonella species, human herpesviruses 6 and 7, and parvovirus B19 were negative.
The negative RPR makes disseminated (secondary) syphilis improbable, provided the prozone phenomenon has been excluded. An extensive serological workup is common in the evaluation of fever of unknown origin, although the threat of false‐positive results always looms when many studies are sent simultaneously. This must be considered in advance here, as his relatively preserved CD4 count affords him significant protection against many opportunistic infections. His HIV infection, however, regardless of CD4 count, increases his risk for TB and lymphoma, which remain high on my list. Both may be residing primarily in the liver. In FUO, the abdominal CT is frequently a high‐yield test (primarily by demonstrating unsuspected tumors and abscesses), even in the absence of symptoms, and would certainly be of interest here given the liver function test results. Imaging could diagnose febrile tumors such as lymphoma, HCC, or renal cell carcinoma. In the event that imaging is unrevealing, causes of granulomatous hepatitis should be entertained. The constellation of cough, LFT abnormalities, and fever is compatible with Q fever. As with any FUO case, I would also carefully revisit this patient's history to discern where he was born, where he has been, and what activities or exposures he is engaged in.
He was seen 2 days later with fever of 104F and new papules over his sternal area. Over the next week, he had intermittent fevers and severe fatigue. The rash progressed, predominantly involving his chest and back, but also his legs, arms, and face (see Fig. 1). The lesions spared his palms and soles. The exanthem was intensely pruritic and maculopapular, consisting of lesions with a diameter of 0.5 cm or less, with some scaling. There were no vesicles or pustular lesions. There were no other new findings on examination. His transaminase and bilirubin had normalized, and his CBC and electrolytes were unchanged. Repeat blood cultures held for extended incubation were negative. Computerized tomography of the chest, abdomen, and pelvis demonstrated mild lymphadenopathy at the porta hepatis with increased portocaval and periaortic lymphadenopathy.
The only LFT abnormality that persists is the elevated alkaline phosphatase, which suggests (1) that liver involvement was not specific and that there is a disease process involving the bone, (2) that there is a persistent infiltrative disorder of the liver such as infection or malignancy or, less likely, amyloidosis or sarcoidosis, or (3) that the porta hepatis lymphadenopathy is causing biliary obstruction. The underlying diagnosis must explain the rash, intraabdominal lymphadenopathy, and fever. The time course does somewhat limit the extensive differential of fever and rash. After 3 weeks of illness, some of the most life‐threatening entities such as meningococcal disease, Rocky Mountain spotted fever, and toxic shock syndrome are unlikely. Concern remains for infections that are more indolent, such as mycobacteria, fungi, or spirochetes. The most striking elements of the rash are the extensive distribution, rapid progression, large number, and discreteness of the lesions, which collectively point more toward disseminated fungal (eg, histoplasmosis, as he lives in Ohio), spirochetal, rickettsial, or viral etiologies, rather than bacterial or mycobacterial entities. The absence of vesicles detracts from the diagnosis of a disseminated herpes virus such as herpes simplex or varicella. I believe that this rash is too disseminated to be caused by a common mycobacterial illness. This extent of cutaneous metastases would usually accompany a far more ill patient with an obvious primary cancer (none is seen on imaging, including the liver), and it appears too extensive to be caused by a paraneoplastic phenomenon such as Sweet's syndrome. A systemic vasculitis or another autoimmune disease remains possible, but there is minimal evidence of visceral organ involvement. All the aforementioned diseases could explain the intraabdominal lymphadenopathy, but my suspicion is highest for infection. I would biopsy and culture the skin lesions, repeat the RPR and/or send a treponemal‐specific test, place a PPD skin test, and send fungal studies (serum serologies and urine antigens) for evaluation. If the results of these noninvasive studies are unrevealing, I would consider a liver biopsy.
The patient's medications were discontinued, and a skin biopsy of the rash from his chest showed atypical lymphohistiocytic infiltrates without acute inflammatory cells and with negative Gomori methenamine silver (GMS), acid‐fast bacilli (AFB), and Fite (for Nocardia) stains. The infiltrates were predominantly T cells with a 1:1 CD4:CD8 ratio. This was read as suspicious for cytotoxic (CD8) mycosis fungoides.
I do not have reason to doubt the pathologist's impression of mycosis fungoides on histopathologic grounds, but from a clinical standpoint, I do not think mycosis fungoides is a disease that has a prolonged febrile prodrome or an explosive cutaneous onset. Rather, it is frequently preceded by nonspecific skin findings over a long period. Thinking broadly and pathophysiologically and noting that T cells are the predominant lymphocytes in skin, I wonder if they could represent a nonmalignant, immunological reaction in the skin. The stains, although not perfectly sensitive, make mycobacterial and fungal diseases less likely, although incubation of cultures is necessary.
Over the next 10 days (bringing the total duration of the patient's illness to 6 weeks), the skin lesions increased in number. In the physician's office at his next follow‐up, the patient had a temperature of 104.1F, was uncomfortable, shivering, and ill‐appearing. His blood pressure was 108/66 mm Hg, and his pulse 114 beats/min. He complained of severe shooting pains, predominantly in his pretibial regions and arms. Examination showed no other new findings, including no focal neurological findings. The results of the T‐cell rearrangement study from the skin biopsy showed evidence of a monoclonal T‐cell population. He was admitted to the hospital for further evaluation and treatment.
The extremity dysesthesias could represent a lesion of the spinal cord (including the CSF/meninges), a polyradiculopathy, or a polyneuropathy. Unfortunately, this does not add a tremendous amount of diagnostic resolution, as infection, malignancy, and autoimmune syndromes, such as vasculitis, may all involve the nervous system in these ways. In general, I associate monoclonal lymphocyte responses with hematological malignancies and polyclonal responses with the less specific inflammation that could accompany infection, autoimmunity, or solid malignancies. His age, fever, and rapid progression seem atypical for mycosis fungoides, but given the monoclonal T cells, this must now be considered. Adult T‐cell leukemia/lymphoma, with its prominent skin manifestations and its association with HLTV‐1, is an alternative T‐cell malignancy that could explain the fever, neurological symptoms, and possible visceral involvement (elevated alkaline phosphatase, which could reflect liver or bone). In cases that are diagnostic challenges, one of the highest‐yield maneuvers is to repeat the preceding evaluation, starting with the history, exam, and basic labs, and if necessary, to review or repeat the imaging or skin biopsy. Given the elevated alkaline phosphatase, disseminated rash, new neurological symptoms, and his HIV status, I remain particularly concerned about syphilis and would do further testing (accounting for the prozone phenomenon) before proceeding with the malignancy evaluation.
A lumbar puncture demonstrated clear cerebrospinal fluid, with 2 leukocytes and 195 erythrocytes/cm3, protein of 26 mg/dL, and glucose of 52 mg/dL. Bacterial and fungal cultures of the fluid were negative. The results of colonoscopy were normal. A bone marrow biopsy demonstrated ring granulomas. GMS, AFB, Fite, and Steiner (for spirochetes) stains were negative, cultures of the aspirate were negative for bacteria, and smears were negative for fungi and mycobacteria. Antibody tests for human T‐cell lymphotropic virus types I and II, Coxiella burnetii, and Bartonella henselae were negative. The dermatology consultant believed the absence of lymphadenopathy and the pruritic nature of the lesions was atypical for cytotoxic T‐cell lymphoma (CTCL). Before initiating therapy for CTCL, she suggested repeating the skin biopsy and RPR.
The repeat RPR was positive at 1:64 dilutions, and a confirmatory fluorescent treponemal antibody absorption test showed a positive result. He was prescribed intramuscular benzathine pencillin 2.4 million units weekly for 3 weeks, with almost immediate defervescence and slower resolution of his rash and shooting pains in his limbs. The repeat skin biopsy done during the hospitalization demonstrated lichenoid‐type dermatitis with interstitial and perivascular lymphohistiocytic infiltrates and granulomas. Steiner stains for spirochetes were positive. Immunohistochemical stains ruled out a lymphoproliferative process. One year later his RPR was nonreactive.
COMMENTARY
Fever of unknown origin (FUO) was first defined by Petersdorf and Beeson in 1961 as a temperature higher than 38.3C on several occasions lasting longer than 3 weeks and defying diagnosis despite 1 week of inpatient investigation.1 Dramatic changes in medical practice have rendered this definition outdated, with more recent proposals allowing thoughtful outpatient investigation to serve as a surrogate for hospitalization. Some have proposed that HIV‐associated FUO be considered a distinct entity, with the most complete North American series finding the etiology of the HIV‐associated FUO in 56 of 70 patients.2 The mean CD4+ count in this series was 58/cm3. Disseminated M. avium was the most frequently diagnosed cause, followed by P. jirovecii pneumonia, cytomegalovirus infection, disseminated histoplasmosis, and lymphoma. Of 14 patients with fever of no definable etiology, 12 eventually proved to have self‐limiting illness.
Despite numerous attempts to reduce the investigation of the patient with FUO to an algorithm, the approach must be individualized. A thorough history and careful, serial physical examinations are frequently and appropriately stressed as the foundation, followed by thoughtful selection of laboratory and imaging studies. Although FUO has a lengthy differential diagnosis, it often proves to be, as Mackowiak and Durack stress, an unusual manifestation of a common disease, rather than a typical presentation of a rare disease.3 A relatively uncommon disease in conjunction with an initially negative diagnostic test result, as was the case with this patient, may lead to a protracted diagnostic puzzle.
Syphilis is a rare cause of FUO. In 6 large studies of a total of 947 patients published over a 40‐year period, only 2 cases of syphilis (1 secondary and 1 neurosyphilis) were reported.1, 48 Syphilis as a cause of prolonged cryptic fever appears to have been seen with greater frequency in the preantibiotic era.9 In the first half of the 20th century, syphilis was known as the great imitator, with its unusual manifestations recognized and indeed expected. As a result of the dramatically lower incidence of syphilis in recent decades, these lessons have largely been forgotten, however, which may lead to diagnostic confusion when syphilis presents atypically. The manifestations of secondary syphilis are protean, including a variety of rashes, aphthous ulcers, arthralgias, pharyngitis, weight loss, fever, meningitis, ocular symptoms, cranial nerve palsies, glomerulonephritis, hepatitis, and periostitis (which afflicted this patient, who complained of severe shooting pains in his arms and shins).
After declining in the last decade of the 20th century, the rates of primary and secondary syphilis are rising in the United States.10 Oral sex is a clear risk factor for syphilis transmission, particularly for men who have sex with men.11 Because of the patient's exposure history and clinical picture, his outpatient physician considered the diagnosis of secondary syphilis early in the course of his illness. The diagnosis was not entertained further when an RPR test, highly sensitive at this stage of the disease, returned nonreactive. Likewise, when a rash subsequently appeared, the lack of palm and sole involvement dissuaded multiple clinicians from reconsidering the diagnosis of syphilis. A skin biopsy that appeared to lead in a distinctly different direction understandably confused the picture still further. Even at the time of the lumbar puncture, VDRL of the CSF was not ordered.
In retrospect, the chief confounder in the case was the false‐negative RPR test, as the discussant suspected early on. Although nontreponemal tests are generally accurate in individuals with HIV, delayed seropositivity and false‐negatives have been reported in this population.12 The false‐negative could have also been a result of the prozone phenomenon, an unusual event, occurring in fewer than 2% of cases of secondary syphilis and attributed to a mismatch between antibody and very high antigen level. The prozone reaction can be corrected for by requesting dilution of the serum prior to repeating the test. Simple lab error must be considered as well, but without access to this patient's serum from his original testing, the cause of his initial false‐negative test cannot be known with certainty.
An unusual presentation in conjunction with failure to recognize the causes of rare false‐negative testing for secondary syphilis led to a delayed diagnosis in this patient. Although syphilis and mycosis fungoides have previously been reported to mimic one another both clinically and histopathologically, the potential for secondary syphilis to be misdiagnosed in this fashion is not generally appreciated.1315 Recognition of the possibility of secondary syphilis occurred just in time to spare this patient the rash decision of treating him with cytotoxic therapy directed against CTCL.
Teaching Points
-
HIV‐associated FUO can be a diagnostic challenge, but an etiology can be found in most cases.
-
Syphilis continues to be an unusual cause of FUO and can have protean manifestations affecting nearly every organ system
-
The sensitivity of RPR is extremely high in secondary syphilis, but false‐negative tests can be seen in HIV because of both the prozone phenomenon and a delayed rise in antibodies.
A 38‐year‐old HIV+ Ohio man with a recent CD4+ count of 534 cells/mL presented to his physician with 3 weeks of fever as high as 102F. He noted mild myalgias, pruritus, and an occasional cough but no headache, sore throat, dyspnea, rash, or gastrointestinal or genitourinary complaints. He had been seen elsewhere 2 weeks previously, when he had reported a single episode of receptive oral sex with a male partner several weeks earlier. He had been prescribed ciprofloxacin and azithromycin, but a throat swab came back negative for Chlamydia and Neisseria gonorrhoeae, and he reported no change in his symptoms after the course of antibiotics. He denied smoking or using street drugs. His only medications were citalopram and trazodone for depression.
This is a HIV+ man with a mild degree of immunosuppression with a fever of unknown origin (FUO). It is not yet known if the requisite basic infectious evaluation has been completed to meet this definition, but the duration certainly qualifies, and regardless of semantics, the FUO framework is a helpful starting point. The primary considerations in FUO are infections, neoplasms, and autoimmune illnesses. Autoimmune diseases are relatively less common in HIV patients. Although pruritis is quite common in HIV alone, it may also herald renal failure, cholestasis, or a malignancy (usually hematologic). Drugs must also be considered as a cause of unexplained fever; the pruritis might suggest an allergic reaction, although I do not think of citalopram or trazodone as having this effect. The failure to respond to broad‐spectrum antimicrobials (along with the duration) lowers my suspicion for common infections such as pneumonia, urinary tract infection, or cellulitis. Among sexually transmitted diseases, syphilis can be protean and merits consideration.
On examination he appeared well. His temperature was 102.4F, pulse 111 beats/min, blood pressure 138/78 mm Hg. The head, neck, cardiovascular system, and lungs appeared normal on examination. The abdomen was soft and nontender without organomegaly; skin, extremities, and neurological system were unremarkable. Rectal examination showed small anal condylomata. Hemoglobin was 14.3 g/dL, white blood cell count 6200/cm3, and platelet count 230,000/cm3. Serum electrolytes and lactate dehydrogenase were normal. The results of his liver function tests (LFTs) demonstrated a serum aspartate transaminase of 60 U/L (normal, 7‐40 U/L), alanine transaminase of 125 U/L (normal, 5‐50 U/L), alkaline phosphatase 218 U/L (normal, 40‐150 U/L), and total bilirubin 2.1 mg/dL (normal, 0.0‐1.5 mg/dL). Urinalysis demonstrated 2+ bilirubin and was otherwise normal. His erythrocyte sedimentation rate was 32 mm/hr (normal, 0‐15 mm/hr).
After 3 weeks of illness, his CBC demonstrates no signs of chronic illness (such as anemia of a chronic disease or a reactive leukocytosis or thrombocytosis). The results of his liver function tests showed moderate elevation, slightly more cholestatic than hepatocellular. This finding may reflect a disease process involving the liver, but such abnormal findings are often nonspecific in acute and chronic illnesses. With an unremitting fever, infectious complications in the liver merit early consideration. The time course rules out common biliary disorders such as cholangitis or cholecystitis. Pyogenic or amoebic liver abscesses are possible (homosexual men are at increased risk for the latter), but the absence of pain or abdominal tenderness is atypical. This biochemical profile can also be seen in chronic (but not acute) viral infections of the liver. Chronic hepatitis B and C predispose to hepatocellular carcinoma (HCC), which can be associated with fever. Cancers that infiltrate the liver, such as lymphoma or carcinoma, could also account for this picture. Indolent infections such as tuberculosis (TB) and syphilis are also possible, so associated signs of these systemic diseases should be sought. I do not believe either of his antibiotics is commonly associated with LFT abnormalities, and his CD4 count is too high for HIV cholangiopathy. In sum, a host of liver diseases are possible, but an extrahepatic systemic disease deserves equal attention.
His CD4+ count was 537 cells/mL, and his HIV RNA viral load was 44,300 copies/mL. Radiographs of the chest were normal. Two sets of blood cultures were negative. The rapid plasma reagin (RPR) was nonreactive. The results of serologies for acute hepatitis A, B, C, and E, chronic hepatitis B and C, and toxoplasmosis were negative. Testing for both Epstein‐Barr virus and cytomegalovirus showed evidence of remote infection. Results of serologies for bartonella species, human herpesviruses 6 and 7, and parvovirus B19 were negative.
The negative RPR makes disseminated (secondary) syphilis improbable, provided the prozone phenomenon has been excluded. An extensive serological workup is common in the evaluation of fever of unknown origin, although the threat of false‐positive results always looms when many studies are sent simultaneously. This must be considered in advance here, as his relatively preserved CD4 count affords him significant protection against many opportunistic infections. His HIV infection, however, regardless of CD4 count, increases his risk for TB and lymphoma, which remain high on my list. Both may be residing primarily in the liver. In FUO, the abdominal CT is frequently a high‐yield test (primarily by demonstrating unsuspected tumors and abscesses), even in the absence of symptoms, and would certainly be of interest here given the liver function test results. Imaging could diagnose febrile tumors such as lymphoma, HCC, or renal cell carcinoma. In the event that imaging is unrevealing, causes of granulomatous hepatitis should be entertained. The constellation of cough, LFT abnormalities, and fever is compatible with Q fever. As with any FUO case, I would also carefully revisit this patient's history to discern where he was born, where he has been, and what activities or exposures he is engaged in.
He was seen 2 days later with fever of 104F and new papules over his sternal area. Over the next week, he had intermittent fevers and severe fatigue. The rash progressed, predominantly involving his chest and back, but also his legs, arms, and face (see Fig. 1). The lesions spared his palms and soles. The exanthem was intensely pruritic and maculopapular, consisting of lesions with a diameter of 0.5 cm or less, with some scaling. There were no vesicles or pustular lesions. There were no other new findings on examination. His transaminase and bilirubin had normalized, and his CBC and electrolytes were unchanged. Repeat blood cultures held for extended incubation were negative. Computerized tomography of the chest, abdomen, and pelvis demonstrated mild lymphadenopathy at the porta hepatis with increased portocaval and periaortic lymphadenopathy.
The only LFT abnormality that persists is the elevated alkaline phosphatase, which suggests (1) that liver involvement was not specific and that there is a disease process involving the bone, (2) that there is a persistent infiltrative disorder of the liver such as infection or malignancy or, less likely, amyloidosis or sarcoidosis, or (3) that the porta hepatis lymphadenopathy is causing biliary obstruction. The underlying diagnosis must explain the rash, intraabdominal lymphadenopathy, and fever. The time course does somewhat limit the extensive differential of fever and rash. After 3 weeks of illness, some of the most life‐threatening entities such as meningococcal disease, Rocky Mountain spotted fever, and toxic shock syndrome are unlikely. Concern remains for infections that are more indolent, such as mycobacteria, fungi, or spirochetes. The most striking elements of the rash are the extensive distribution, rapid progression, large number, and discreteness of the lesions, which collectively point more toward disseminated fungal (eg, histoplasmosis, as he lives in Ohio), spirochetal, rickettsial, or viral etiologies, rather than bacterial or mycobacterial entities. The absence of vesicles detracts from the diagnosis of a disseminated herpes virus such as herpes simplex or varicella. I believe that this rash is too disseminated to be caused by a common mycobacterial illness. This extent of cutaneous metastases would usually accompany a far more ill patient with an obvious primary cancer (none is seen on imaging, including the liver), and it appears too extensive to be caused by a paraneoplastic phenomenon such as Sweet's syndrome. A systemic vasculitis or another autoimmune disease remains possible, but there is minimal evidence of visceral organ involvement. All the aforementioned diseases could explain the intraabdominal lymphadenopathy, but my suspicion is highest for infection. I would biopsy and culture the skin lesions, repeat the RPR and/or send a treponemal‐specific test, place a PPD skin test, and send fungal studies (serum serologies and urine antigens) for evaluation. If the results of these noninvasive studies are unrevealing, I would consider a liver biopsy.
The patient's medications were discontinued, and a skin biopsy of the rash from his chest showed atypical lymphohistiocytic infiltrates without acute inflammatory cells and with negative Gomori methenamine silver (GMS), acid‐fast bacilli (AFB), and Fite (for Nocardia) stains. The infiltrates were predominantly T cells with a 1:1 CD4:CD8 ratio. This was read as suspicious for cytotoxic (CD8) mycosis fungoides.
I do not have reason to doubt the pathologist's impression of mycosis fungoides on histopathologic grounds, but from a clinical standpoint, I do not think mycosis fungoides is a disease that has a prolonged febrile prodrome or an explosive cutaneous onset. Rather, it is frequently preceded by nonspecific skin findings over a long period. Thinking broadly and pathophysiologically and noting that T cells are the predominant lymphocytes in skin, I wonder if they could represent a nonmalignant, immunological reaction in the skin. The stains, although not perfectly sensitive, make mycobacterial and fungal diseases less likely, although incubation of cultures is necessary.
Over the next 10 days (bringing the total duration of the patient's illness to 6 weeks), the skin lesions increased in number. In the physician's office at his next follow‐up, the patient had a temperature of 104.1F, was uncomfortable, shivering, and ill‐appearing. His blood pressure was 108/66 mm Hg, and his pulse 114 beats/min. He complained of severe shooting pains, predominantly in his pretibial regions and arms. Examination showed no other new findings, including no focal neurological findings. The results of the T‐cell rearrangement study from the skin biopsy showed evidence of a monoclonal T‐cell population. He was admitted to the hospital for further evaluation and treatment.
The extremity dysesthesias could represent a lesion of the spinal cord (including the CSF/meninges), a polyradiculopathy, or a polyneuropathy. Unfortunately, this does not add a tremendous amount of diagnostic resolution, as infection, malignancy, and autoimmune syndromes, such as vasculitis, may all involve the nervous system in these ways. In general, I associate monoclonal lymphocyte responses with hematological malignancies and polyclonal responses with the less specific inflammation that could accompany infection, autoimmunity, or solid malignancies. His age, fever, and rapid progression seem atypical for mycosis fungoides, but given the monoclonal T cells, this must now be considered. Adult T‐cell leukemia/lymphoma, with its prominent skin manifestations and its association with HLTV‐1, is an alternative T‐cell malignancy that could explain the fever, neurological symptoms, and possible visceral involvement (elevated alkaline phosphatase, which could reflect liver or bone). In cases that are diagnostic challenges, one of the highest‐yield maneuvers is to repeat the preceding evaluation, starting with the history, exam, and basic labs, and if necessary, to review or repeat the imaging or skin biopsy. Given the elevated alkaline phosphatase, disseminated rash, new neurological symptoms, and his HIV status, I remain particularly concerned about syphilis and would do further testing (accounting for the prozone phenomenon) before proceeding with the malignancy evaluation.
A lumbar puncture demonstrated clear cerebrospinal fluid, with 2 leukocytes and 195 erythrocytes/cm3, protein of 26 mg/dL, and glucose of 52 mg/dL. Bacterial and fungal cultures of the fluid were negative. The results of colonoscopy were normal. A bone marrow biopsy demonstrated ring granulomas. GMS, AFB, Fite, and Steiner (for spirochetes) stains were negative, cultures of the aspirate were negative for bacteria, and smears were negative for fungi and mycobacteria. Antibody tests for human T‐cell lymphotropic virus types I and II, Coxiella burnetii, and Bartonella henselae were negative. The dermatology consultant believed the absence of lymphadenopathy and the pruritic nature of the lesions was atypical for cytotoxic T‐cell lymphoma (CTCL). Before initiating therapy for CTCL, she suggested repeating the skin biopsy and RPR.
The repeat RPR was positive at 1:64 dilutions, and a confirmatory fluorescent treponemal antibody absorption test showed a positive result. He was prescribed intramuscular benzathine pencillin 2.4 million units weekly for 3 weeks, with almost immediate defervescence and slower resolution of his rash and shooting pains in his limbs. The repeat skin biopsy done during the hospitalization demonstrated lichenoid‐type dermatitis with interstitial and perivascular lymphohistiocytic infiltrates and granulomas. Steiner stains for spirochetes were positive. Immunohistochemical stains ruled out a lymphoproliferative process. One year later his RPR was nonreactive.
COMMENTARY
Fever of unknown origin (FUO) was first defined by Petersdorf and Beeson in 1961 as a temperature higher than 38.3C on several occasions lasting longer than 3 weeks and defying diagnosis despite 1 week of inpatient investigation.1 Dramatic changes in medical practice have rendered this definition outdated, with more recent proposals allowing thoughtful outpatient investigation to serve as a surrogate for hospitalization. Some have proposed that HIV‐associated FUO be considered a distinct entity, with the most complete North American series finding the etiology of the HIV‐associated FUO in 56 of 70 patients.2 The mean CD4+ count in this series was 58/cm3. Disseminated M. avium was the most frequently diagnosed cause, followed by P. jirovecii pneumonia, cytomegalovirus infection, disseminated histoplasmosis, and lymphoma. Of 14 patients with fever of no definable etiology, 12 eventually proved to have self‐limiting illness.
Despite numerous attempts to reduce the investigation of the patient with FUO to an algorithm, the approach must be individualized. A thorough history and careful, serial physical examinations are frequently and appropriately stressed as the foundation, followed by thoughtful selection of laboratory and imaging studies. Although FUO has a lengthy differential diagnosis, it often proves to be, as Mackowiak and Durack stress, an unusual manifestation of a common disease, rather than a typical presentation of a rare disease.3 A relatively uncommon disease in conjunction with an initially negative diagnostic test result, as was the case with this patient, may lead to a protracted diagnostic puzzle.
Syphilis is a rare cause of FUO. In 6 large studies of a total of 947 patients published over a 40‐year period, only 2 cases of syphilis (1 secondary and 1 neurosyphilis) were reported.1, 48 Syphilis as a cause of prolonged cryptic fever appears to have been seen with greater frequency in the preantibiotic era.9 In the first half of the 20th century, syphilis was known as the great imitator, with its unusual manifestations recognized and indeed expected. As a result of the dramatically lower incidence of syphilis in recent decades, these lessons have largely been forgotten, however, which may lead to diagnostic confusion when syphilis presents atypically. The manifestations of secondary syphilis are protean, including a variety of rashes, aphthous ulcers, arthralgias, pharyngitis, weight loss, fever, meningitis, ocular symptoms, cranial nerve palsies, glomerulonephritis, hepatitis, and periostitis (which afflicted this patient, who complained of severe shooting pains in his arms and shins).
After declining in the last decade of the 20th century, the rates of primary and secondary syphilis are rising in the United States.10 Oral sex is a clear risk factor for syphilis transmission, particularly for men who have sex with men.11 Because of the patient's exposure history and clinical picture, his outpatient physician considered the diagnosis of secondary syphilis early in the course of his illness. The diagnosis was not entertained further when an RPR test, highly sensitive at this stage of the disease, returned nonreactive. Likewise, when a rash subsequently appeared, the lack of palm and sole involvement dissuaded multiple clinicians from reconsidering the diagnosis of syphilis. A skin biopsy that appeared to lead in a distinctly different direction understandably confused the picture still further. Even at the time of the lumbar puncture, VDRL of the CSF was not ordered.
In retrospect, the chief confounder in the case was the false‐negative RPR test, as the discussant suspected early on. Although nontreponemal tests are generally accurate in individuals with HIV, delayed seropositivity and false‐negatives have been reported in this population.12 The false‐negative could have also been a result of the prozone phenomenon, an unusual event, occurring in fewer than 2% of cases of secondary syphilis and attributed to a mismatch between antibody and very high antigen level. The prozone reaction can be corrected for by requesting dilution of the serum prior to repeating the test. Simple lab error must be considered as well, but without access to this patient's serum from his original testing, the cause of his initial false‐negative test cannot be known with certainty.
An unusual presentation in conjunction with failure to recognize the causes of rare false‐negative testing for secondary syphilis led to a delayed diagnosis in this patient. Although syphilis and mycosis fungoides have previously been reported to mimic one another both clinically and histopathologically, the potential for secondary syphilis to be misdiagnosed in this fashion is not generally appreciated.1315 Recognition of the possibility of secondary syphilis occurred just in time to spare this patient the rash decision of treating him with cytotoxic therapy directed against CTCL.
Teaching Points
-
HIV‐associated FUO can be a diagnostic challenge, but an etiology can be found in most cases.
-
Syphilis continues to be an unusual cause of FUO and can have protean manifestations affecting nearly every organ system
-
The sensitivity of RPR is extremely high in secondary syphilis, but false‐negative tests can be seen in HIV because of both the prozone phenomenon and a delayed rise in antibodies.
- ,.Fever of unexplained origin: Report on 100 cases.Medicine.1961;40:1–30.
- ,,.Human immunodeficiency virus‐associated fever of unknown origin: A study of 70 patients in the United States and review.Clin Infect Dis.1999;28:341–345.
- ,.Fever of unknown origin. In:Mandell GL,Bennett JE,Dolin R, eds.Principles and Practice of Infectious Diseases.6th ed.Philadelphia:Elsevier Churchill Livingstone;2005:718–729.
- ,,.Fever of unknown origin: Diagnosis and follow‐up of 105 cases, 1970‐1980.Medicine.1982;61:269–292.
- ,,,.Fever of unknown origin in the 1980s: An update of the diagnostic spectrum.Arch Intern Med.1992;152:51–55.
- .Fever of unknown origin: Review of 86 patients treated in community hospitals.Clin Infect Dis.1992;15:968–973.
- ,,.Fever of unknown origin (FUO). I. A prospective multicenter study of 167 patients with FUO, using fixed epidemiologic entry criteria. The Netherlands FUO study group.Medicine.1997;76:392–400.
- ,,, et al.From prolonged febrile illness to fever of unknown origin: The challenge continues.Arch Intern Med.2003;163:1033–1041.
- ,.The diagnosis of obscure fever. II. The diagnosis of unexplained high fever.Bull Johns Hopkins Hosp.1936;58:307–331.
- Centers for Disease Control and Prevention.Primary and secondary syphilis—United States, 2003–2004.MMWR.2006;55:269–273.
- Transmission of primary and secondary syphilis by oral sex—Chicago, Illinois, 1998‐2202.MMWR.2004;53:966–968.
- ,,, et al.Seronegative secondary syphilis in 2 patients coinfected with human immunodeficiency virus.Arch Dermatol.2005;141:431–433.
- ,,,.Secondary syphilis mimicking mycosis fungoides.J Am Acad Dermatol.1980;3:92–94
- ,.A case of lues maligna in a patient with acquired immunodeficiency syndrome (AIDS).Scand J Infect Dis.2005;37:697–700.
- ,,,,.Unusual presentation of secondary syphilis in 2 HIV‐1 positive patients.Cutis.2000;66:383–389.
- ,.Fever of unexplained origin: Report on 100 cases.Medicine.1961;40:1–30.
- ,,.Human immunodeficiency virus‐associated fever of unknown origin: A study of 70 patients in the United States and review.Clin Infect Dis.1999;28:341–345.
- ,.Fever of unknown origin. In:Mandell GL,Bennett JE,Dolin R, eds.Principles and Practice of Infectious Diseases.6th ed.Philadelphia:Elsevier Churchill Livingstone;2005:718–729.
- ,,.Fever of unknown origin: Diagnosis and follow‐up of 105 cases, 1970‐1980.Medicine.1982;61:269–292.
- ,,,.Fever of unknown origin in the 1980s: An update of the diagnostic spectrum.Arch Intern Med.1992;152:51–55.
- .Fever of unknown origin: Review of 86 patients treated in community hospitals.Clin Infect Dis.1992;15:968–973.
- ,,.Fever of unknown origin (FUO). I. A prospective multicenter study of 167 patients with FUO, using fixed epidemiologic entry criteria. The Netherlands FUO study group.Medicine.1997;76:392–400.
- ,,, et al.From prolonged febrile illness to fever of unknown origin: The challenge continues.Arch Intern Med.2003;163:1033–1041.
- ,.The diagnosis of obscure fever. II. The diagnosis of unexplained high fever.Bull Johns Hopkins Hosp.1936;58:307–331.
- Centers for Disease Control and Prevention.Primary and secondary syphilis—United States, 2003–2004.MMWR.2006;55:269–273.
- Transmission of primary and secondary syphilis by oral sex—Chicago, Illinois, 1998‐2202.MMWR.2004;53:966–968.
- ,,, et al.Seronegative secondary syphilis in 2 patients coinfected with human immunodeficiency virus.Arch Dermatol.2005;141:431–433.
- ,,,.Secondary syphilis mimicking mycosis fungoides.J Am Acad Dermatol.1980;3:92–94
- ,.A case of lues maligna in a patient with acquired immunodeficiency syndrome (AIDS).Scand J Infect Dis.2005;37:697–700.
- ,,,,.Unusual presentation of secondary syphilis in 2 HIV‐1 positive patients.Cutis.2000;66:383–389.
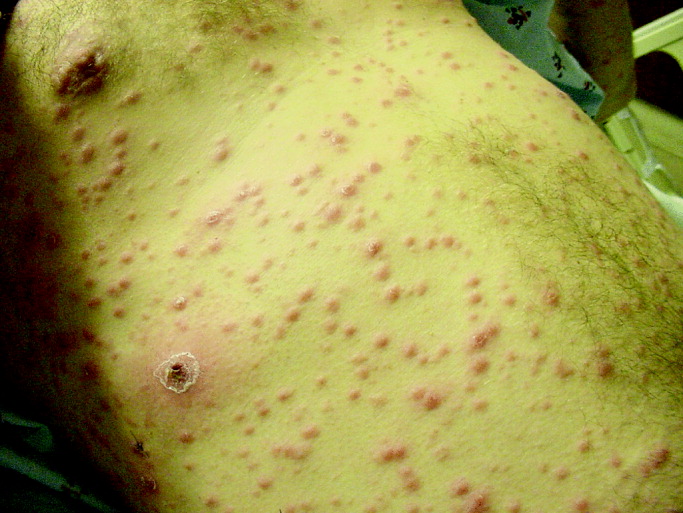
Bony Metastatic Disease
A60‐year‐old man presented with agitation and a forehead mass (Figs. 1 and 2). His wife had noticed its rapid growth over several weeks and said he had recently become extremely confused and hostile. He had a history of smoking and exposures to silica and Agent Orange. His vitals and oxygen saturation were normal. He was cachectic but in no distress, and although he was uncooperative, his examination did not demonstrate an obvious neurological deficit. Routine electrolytes were within normal limits.
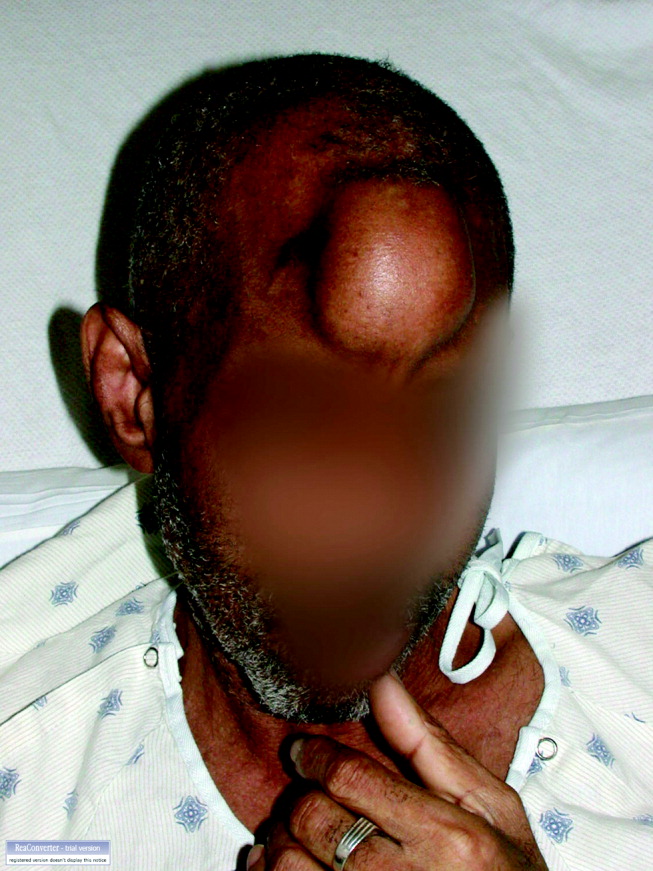
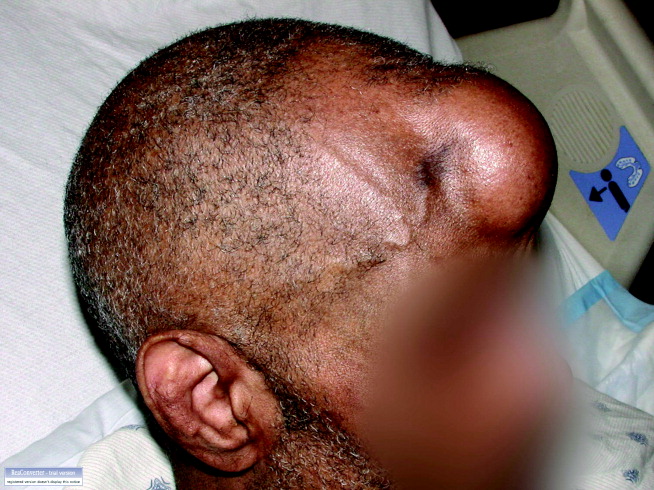
Noncontrast computed tomography of the skull revealed a 5.5 5.4 7.7cm frontal soft‐tissue density eroding his frontal bone and extending intracranially, with extensive displacement of both frontal lobes and invasion into his superior sagittal sinus (Fig. 3) and possible involvement of the brain parenchyma. A subsequent CT scan of the chest demonstrated a 7.8 7.9cm right mass in the lower lobe of the lung encasing the right pulmonary artery and 2 left renal masses.
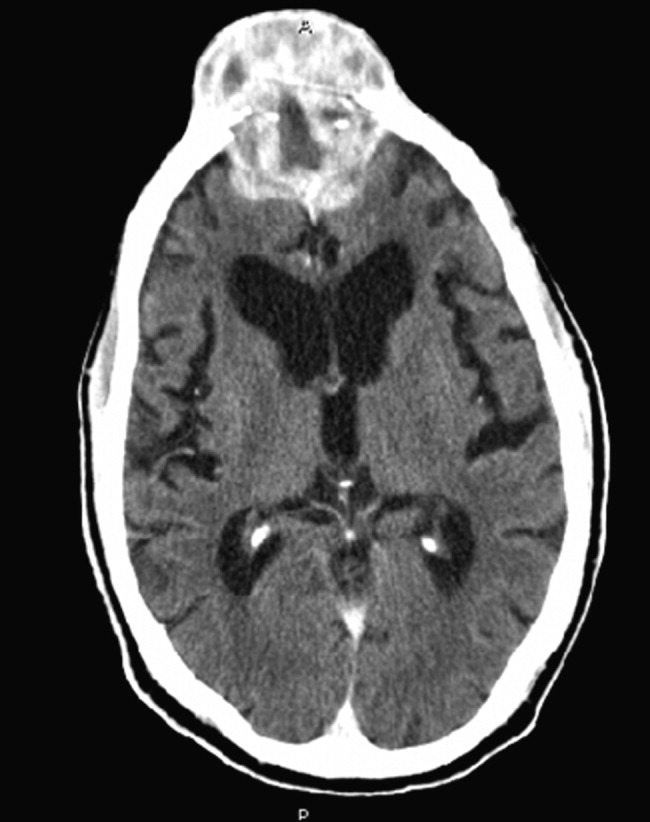
Biopsy of the forehead mass demonstrated a necrotic, poorly differentiated carcinoma with focal clear‐cell features. Immunohistochemical staining was consistent with squamous cell carcinoma.
The forehead mass was painless, and the lung mass unresectable. His behavioral changes persisted and were attributed to the tumor compressing and possibly invading both frontal lobes. At his wife's request, he was discharged on dexamethasone with home hospice and died 2 weeks later.
Although there is little literature on the epidemiology of skull metastases, the most common malignancies to metastasize to the skull are breast and lung carcinomas. Others include prostate, thyroid, myeloma, and melanoma. Symptomatic metastases, including neurological findings, are unusual, except for tumors that metastasize to the skull base and cause cranial nerve deficits. Skeletal metastasis as the first manifestation of lung cancer occurs in about 2% of lung cancer patients and is a marker of poor prognosis. Received 2 November 2006; revision received 15 December 2006; accepted 16 January 2007.
A60‐year‐old man presented with agitation and a forehead mass (Figs. 1 and 2). His wife had noticed its rapid growth over several weeks and said he had recently become extremely confused and hostile. He had a history of smoking and exposures to silica and Agent Orange. His vitals and oxygen saturation were normal. He was cachectic but in no distress, and although he was uncooperative, his examination did not demonstrate an obvious neurological deficit. Routine electrolytes were within normal limits.
Noncontrast computed tomography of the skull revealed a 5.5 5.4 7.7cm frontal soft‐tissue density eroding his frontal bone and extending intracranially, with extensive displacement of both frontal lobes and invasion into his superior sagittal sinus (Fig. 3) and possible involvement of the brain parenchyma. A subsequent CT scan of the chest demonstrated a 7.8 7.9cm right mass in the lower lobe of the lung encasing the right pulmonary artery and 2 left renal masses.
Biopsy of the forehead mass demonstrated a necrotic, poorly differentiated carcinoma with focal clear‐cell features. Immunohistochemical staining was consistent with squamous cell carcinoma.
The forehead mass was painless, and the lung mass unresectable. His behavioral changes persisted and were attributed to the tumor compressing and possibly invading both frontal lobes. At his wife's request, he was discharged on dexamethasone with home hospice and died 2 weeks later.
Although there is little literature on the epidemiology of skull metastases, the most common malignancies to metastasize to the skull are breast and lung carcinomas. Others include prostate, thyroid, myeloma, and melanoma. Symptomatic metastases, including neurological findings, are unusual, except for tumors that metastasize to the skull base and cause cranial nerve deficits. Skeletal metastasis as the first manifestation of lung cancer occurs in about 2% of lung cancer patients and is a marker of poor prognosis. Received 2 November 2006; revision received 15 December 2006; accepted 16 January 2007.
A60‐year‐old man presented with agitation and a forehead mass (Figs. 1 and 2). His wife had noticed its rapid growth over several weeks and said he had recently become extremely confused and hostile. He had a history of smoking and exposures to silica and Agent Orange. His vitals and oxygen saturation were normal. He was cachectic but in no distress, and although he was uncooperative, his examination did not demonstrate an obvious neurological deficit. Routine electrolytes were within normal limits.
Noncontrast computed tomography of the skull revealed a 5.5 5.4 7.7cm frontal soft‐tissue density eroding his frontal bone and extending intracranially, with extensive displacement of both frontal lobes and invasion into his superior sagittal sinus (Fig. 3) and possible involvement of the brain parenchyma. A subsequent CT scan of the chest demonstrated a 7.8 7.9cm right mass in the lower lobe of the lung encasing the right pulmonary artery and 2 left renal masses.
Biopsy of the forehead mass demonstrated a necrotic, poorly differentiated carcinoma with focal clear‐cell features. Immunohistochemical staining was consistent with squamous cell carcinoma.
The forehead mass was painless, and the lung mass unresectable. His behavioral changes persisted and were attributed to the tumor compressing and possibly invading both frontal lobes. At his wife's request, he was discharged on dexamethasone with home hospice and died 2 weeks later.
Although there is little literature on the epidemiology of skull metastases, the most common malignancies to metastasize to the skull are breast and lung carcinomas. Others include prostate, thyroid, myeloma, and melanoma. Symptomatic metastases, including neurological findings, are unusual, except for tumors that metastasize to the skull base and cause cranial nerve deficits. Skeletal metastasis as the first manifestation of lung cancer occurs in about 2% of lung cancer patients and is a marker of poor prognosis. Received 2 November 2006; revision received 15 December 2006; accepted 16 January 2007.